WO2014064706A1 - Processes for the preparation of agomelatine using novel intermediates - Google Patents
Processes for the preparation of agomelatine using novel intermediates Download PDFInfo
- Publication number
- WO2014064706A1 WO2014064706A1 PCT/IN2012/000694 IN2012000694W WO2014064706A1 WO 2014064706 A1 WO2014064706 A1 WO 2014064706A1 IN 2012000694 W IN2012000694 W IN 2012000694W WO 2014064706 A1 WO2014064706 A1 WO 2014064706A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- hydroxy
- solvent
- salt
- group
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 216
- YJYPHIXNFHFHND-UHFFFAOYSA-N agomelatine Chemical compound C1=CC=C(CCNC(C)=O)C2=CC(OC)=CC=C21 YJYPHIXNFHFHND-UHFFFAOYSA-N 0.000 title claims abstract description 102
- 229960002629 agomelatine Drugs 0.000 title claims abstract description 86
- 238000002360 preparation method Methods 0.000 title claims abstract description 77
- 239000000543 intermediate Substances 0.000 title abstract description 45
- 150000003839 salts Chemical class 0.000 claims abstract description 211
- 239000002904 solvent Substances 0.000 claims description 337
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 180
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 171
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 153
- 238000006243 chemical reaction Methods 0.000 claims description 149
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 132
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 126
- 150000001875 compounds Chemical class 0.000 claims description 125
- 239000002253 acid Substances 0.000 claims description 120
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 117
- 239000000203 mixture Substances 0.000 claims description 117
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 110
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 95
- -1 alkyl carboxylic acid Chemical class 0.000 claims description 95
- 238000005899 aromatization reaction Methods 0.000 claims description 93
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 93
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 82
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 82
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 81
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 77
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 72
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 72
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 72
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 claims description 71
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 69
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 68
- 238000010992 reflux Methods 0.000 claims description 67
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 66
- 239000003153 chemical reaction reagent Substances 0.000 claims description 65
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 63
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 claims description 60
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 claims description 59
- 239000002585 base Substances 0.000 claims description 56
- CUWKIYJRLCRYCG-UHFFFAOYSA-N 8-(2-aminoethyl)-5,6-dihydronaphthalen-2-ol Chemical compound C1=C(O)C=C2C(CCN)=CCCC2=C1 CUWKIYJRLCRYCG-UHFFFAOYSA-N 0.000 claims description 50
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 50
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 48
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 48
- 239000008096 xylene Substances 0.000 claims description 48
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 47
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 46
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 43
- 229910052717 sulfur Inorganic materials 0.000 claims description 43
- 239000011593 sulfur Substances 0.000 claims description 43
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 41
- ZXZAVAINGDXZPV-UHFFFAOYSA-N 8-(2-aminoethyl)-2-naphthalenol Chemical compound C1=C(O)C=C2C(CCN)=CC=CC2=C1 ZXZAVAINGDXZPV-UHFFFAOYSA-N 0.000 claims description 40
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 40
- GGIZWHGYBIOVIK-UHFFFAOYSA-N 2-(7-hydroxy-3,4-dihydronaphthalen-1-yl)acetonitrile Chemical compound C1CC=C(CC#N)C2=CC(O)=CC=C21 GGIZWHGYBIOVIK-UHFFFAOYSA-N 0.000 claims description 39
- 239000004215 Carbon black (E152) Substances 0.000 claims description 38
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 claims description 38
- 229930195733 hydrocarbon Natural products 0.000 claims description 38
- 150000002430 hydrocarbons Chemical class 0.000 claims description 38
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 37
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 37
- 239000003638 chemical reducing agent Substances 0.000 claims description 36
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims description 35
- 239000003054 catalyst Substances 0.000 claims description 35
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 claims description 34
- MLIREBYILWEBDM-UHFFFAOYSA-N cyanoacetic acid Chemical compound OC(=O)CC#N MLIREBYILWEBDM-UHFFFAOYSA-N 0.000 claims description 34
- 150000002148 esters Chemical class 0.000 claims description 34
- UNTZQBYXDYYXIY-UHFFFAOYSA-N n-[2-(7-hydroxynaphthalen-1-yl)ethyl]acetamide Chemical compound C1=C(O)C=C2C(CCNC(=O)C)=CC=CC2=C1 UNTZQBYXDYYXIY-UHFFFAOYSA-N 0.000 claims description 33
- 239000012345 acetylating agent Substances 0.000 claims description 32
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 claims description 32
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 claims description 31
- 239000007868 Raney catalyst Substances 0.000 claims description 31
- 229910000564 Raney nickel Inorganic materials 0.000 claims description 31
- 150000008282 halocarbons Chemical class 0.000 claims description 31
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 30
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 claims description 30
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 claims description 30
- 229940011051 isopropyl acetate Drugs 0.000 claims description 30
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 claims description 30
- HZNVUJQVZSTENZ-UHFFFAOYSA-N 2,3-dichloro-5,6-dicyano-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O HZNVUJQVZSTENZ-UHFFFAOYSA-N 0.000 claims description 29
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 claims description 29
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 claims description 29
- 150000001412 amines Chemical class 0.000 claims description 28
- HIJPGWKHVWYNRF-UHFFFAOYSA-N 2-(7-hydroxynaphthalen-1-yl)acetonitrile Chemical compound C1=CC=C(CC#N)C2=CC(O)=CC=C21 HIJPGWKHVWYNRF-UHFFFAOYSA-N 0.000 claims description 27
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 27
- 239000012022 methylating agents Substances 0.000 claims description 27
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 claims description 26
- 229910021529 ammonia Inorganic materials 0.000 claims description 26
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 claims description 26
- 239000003880 polar aprotic solvent Substances 0.000 claims description 25
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 claims description 24
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 claims description 24
- 239000003795 chemical substances by application Substances 0.000 claims description 24
- 239000002841 Lewis acid Substances 0.000 claims description 23
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 23
- 150000007517 lewis acids Chemical class 0.000 claims description 23
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 23
- 235000011181 potassium carbonates Nutrition 0.000 claims description 23
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 claims description 22
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 claims description 22
- UGNWTBMOAKPKBL-UHFFFAOYSA-N tetrachloro-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(Cl)=C(Cl)C1=O UGNWTBMOAKPKBL-UHFFFAOYSA-N 0.000 claims description 22
- 150000002576 ketones Chemical class 0.000 claims description 20
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 claims description 19
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 claims description 18
- 150000004678 hydrides Chemical class 0.000 claims description 18
- 150000002825 nitriles Chemical class 0.000 claims description 18
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 18
- 235000017550 sodium carbonate Nutrition 0.000 claims description 18
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 claims description 17
- 229910003446 platinum oxide Inorganic materials 0.000 claims description 17
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 claims description 16
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 claims description 16
- 230000000397 acetylating effect Effects 0.000 claims description 16
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 claims description 16
- 239000000347 magnesium hydroxide Substances 0.000 claims description 16
- 229910001862 magnesium hydroxide Inorganic materials 0.000 claims description 16
- 229910052751 metal Inorganic materials 0.000 claims description 16
- 239000002184 metal Substances 0.000 claims description 16
- SPQRYIZGYFGODL-UHFFFAOYSA-N n-[2-(7-hydroxy-3,4-dihydronaphthalen-1-yl)ethyl]acetamide Chemical compound C1=C(O)C=C2C(CCNC(=O)C)=CCCC2=C1 SPQRYIZGYFGODL-UHFFFAOYSA-N 0.000 claims description 16
- 239000011736 potassium bicarbonate Substances 0.000 claims description 16
- 229910000028 potassium bicarbonate Inorganic materials 0.000 claims description 16
- 235000015497 potassium bicarbonate Nutrition 0.000 claims description 16
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 claims description 16
- 239000001632 sodium acetate Substances 0.000 claims description 16
- 235000017281 sodium acetate Nutrition 0.000 claims description 16
- WBQTXTBONIWRGK-UHFFFAOYSA-N sodium;propan-2-olate Chemical compound [Na+].CC(C)[O-] WBQTXTBONIWRGK-UHFFFAOYSA-N 0.000 claims description 16
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 claims description 15
- 239000000920 calcium hydroxide Substances 0.000 claims description 15
- 229910001861 calcium hydroxide Inorganic materials 0.000 claims description 15
- 238000005984 hydrogenation reaction Methods 0.000 claims description 15
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 claims description 15
- 229910052808 lithium carbonate Inorganic materials 0.000 claims description 15
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 claims description 15
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 claims description 14
- LGFSAJZSDNYVCW-UHFFFAOYSA-N 7-hydroxy-3,4-dihydro-2h-naphthalen-1-one Chemical compound C1CCC(=O)C2=CC(O)=CC=C21 LGFSAJZSDNYVCW-UHFFFAOYSA-N 0.000 claims description 14
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 claims description 14
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 14
- 239000003960 organic solvent Substances 0.000 claims description 14
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 14
- LWHDQPLUIFIFFT-UHFFFAOYSA-N 2,3,5,6-tetrabromocyclohexa-2,5-diene-1,4-dione Chemical compound BrC1=C(Br)C(=O)C(Br)=C(Br)C1=O LWHDQPLUIFIFFT-UHFFFAOYSA-N 0.000 claims description 13
- PYJMGUQHJINLLD-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)acetonitrile Chemical compound C1=CC=C(CC#N)C2=CC(OC)=CC=C21 PYJMGUQHJINLLD-UHFFFAOYSA-N 0.000 claims description 13
- 230000021736 acetylation Effects 0.000 claims description 13
- 238000006640 acetylation reaction Methods 0.000 claims description 13
- 229910000085 borane Inorganic materials 0.000 claims description 13
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 claims description 13
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 claims description 12
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 12
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 claims description 12
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 claims description 12
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 claims description 12
- 150000004059 quinone derivatives Chemical class 0.000 claims description 12
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 11
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 claims description 11
- 239000012346 acetyl chloride Substances 0.000 claims description 11
- 239000012280 lithium aluminium hydride Substances 0.000 claims description 11
- 239000012279 sodium borohydride Substances 0.000 claims description 11
- 229910000033 sodium borohydride Inorganic materials 0.000 claims description 11
- YXDUMIVUPSVYLB-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)ethanamine Chemical compound C1=CC=C(CCN)C2=CC(OC)=CC=C21 YXDUMIVUPSVYLB-UHFFFAOYSA-N 0.000 claims description 10
- VQQCENVCUZJCJL-UHFFFAOYSA-N 8-(2-aminoethyl)-5,6-dihydronaphthalen-2-ol;hydrochloride Chemical compound Cl.C1=C(O)C=C2C(CCN)=CCCC2=C1 VQQCENVCUZJCJL-UHFFFAOYSA-N 0.000 claims description 10
- 150000003974 aralkylamines Chemical class 0.000 claims description 10
- 239000007858 starting material Substances 0.000 claims description 10
- 150000004055 1,2-benzoquinones Chemical class 0.000 claims description 9
- DXKHBLYQXDEINJ-UHFFFAOYSA-N 3,4,5,6-tetrabromocyclohexa-3,5-diene-1,2-dione Chemical compound BrC1=C(Br)C(=O)C(=O)C(Br)=C1Br DXKHBLYQXDEINJ-UHFFFAOYSA-N 0.000 claims description 9
- VRGCYEIGVVTZCC-UHFFFAOYSA-N 3,4,5,6-tetrachlorocyclohexa-3,5-diene-1,2-dione Chemical compound ClC1=C(Cl)C(=O)C(=O)C(Cl)=C1Cl VRGCYEIGVVTZCC-UHFFFAOYSA-N 0.000 claims description 9
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 claims description 9
- GWQMMPDGVHNIQK-UHFFFAOYSA-N 8-(2-aminoethyl)naphthalen-2-ol;hydrochloride Chemical compound Cl.C1=C(O)C=C2C(CCN)=CC=CC2=C1 GWQMMPDGVHNIQK-UHFFFAOYSA-N 0.000 claims description 9
- FXXACINHVKSMDR-UHFFFAOYSA-N acetyl bromide Chemical compound CC(Br)=O FXXACINHVKSMDR-UHFFFAOYSA-N 0.000 claims description 9
- LEKJTGQWLAUGQA-UHFFFAOYSA-N acetyl iodide Chemical compound CC(I)=O LEKJTGQWLAUGQA-UHFFFAOYSA-N 0.000 claims description 9
- JVXNCJLLOUQYBF-UHFFFAOYSA-N cyclohex-4-ene-1,3-dione Chemical class O=C1CC=CC(=O)C1 JVXNCJLLOUQYBF-UHFFFAOYSA-N 0.000 claims description 9
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 claims description 9
- 229910003445 palladium oxide Inorganic materials 0.000 claims description 9
- JQPTYAILLJKUCY-UHFFFAOYSA-N palladium(ii) oxide Chemical compound [O-2].[Pd+2] JQPTYAILLJKUCY-UHFFFAOYSA-N 0.000 claims description 9
- DNXUGBMARDFRGG-UHFFFAOYSA-N 3,6-dioxocyclohexa-1,4-diene-1,2-dicarbonitrile Chemical compound O=C1C=CC(=O)C(C#N)=C1C#N DNXUGBMARDFRGG-UHFFFAOYSA-N 0.000 claims description 8
- HDEXQZFQDZGFLQ-UHFFFAOYSA-N 4,5-dibromo-3,6-dioxocyclohexa-1,4-diene-1,2-dicarbonitrile Chemical compound BrC1=C(Br)C(=O)C(C#N)=C(C#N)C1=O HDEXQZFQDZGFLQ-UHFFFAOYSA-N 0.000 claims description 8
- JEDZLBFUGJTJGQ-UHFFFAOYSA-N [Na].COCCO[AlH]OCCOC Chemical compound [Na].COCCO[AlH]OCCOC JEDZLBFUGJTJGQ-UHFFFAOYSA-N 0.000 claims description 8
- 230000011987 methylation Effects 0.000 claims description 8
- 238000007069 methylation reaction Methods 0.000 claims description 8
- 229910052763 palladium Inorganic materials 0.000 claims description 8
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 claims description 8
- 229910052703 rhodium Inorganic materials 0.000 claims description 8
- 239000010948 rhodium Substances 0.000 claims description 8
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 claims description 8
- 239000012419 sodium bis(2-methoxyethoxy)aluminum hydride Substances 0.000 claims description 8
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 claims description 7
- 229910052697 platinum Inorganic materials 0.000 claims description 7
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 claims description 7
- 239000012448 Lithium borohydride Substances 0.000 claims description 6
- CYTYCFOTNPOANT-UHFFFAOYSA-N Perchloroethylene Chemical group ClC(Cl)=C(Cl)Cl CYTYCFOTNPOANT-UHFFFAOYSA-N 0.000 claims description 6
- 150000003973 alkyl amines Chemical class 0.000 claims description 6
- AUHZEENZYGFFBQ-UHFFFAOYSA-N mesitylene Substances CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 claims description 6
- 125000001827 mesitylenyl group Chemical group [H]C1=C(C(*)=C(C([H])=C1C([H])([H])[H])C([H])([H])[H])C([H])([H])[H] 0.000 claims description 6
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 claims description 6
- 229920006395 saturated elastomer Polymers 0.000 claims description 6
- 125000003107 substituted aryl group Chemical group 0.000 claims description 6
- 229950011008 tetrachloroethylene Drugs 0.000 claims description 6
- WFMXLYNKBOYPRA-UHFFFAOYSA-N 8-(2-aminoethyl)-5,6-dihydronaphthalen-2-ol;hydrobromide Chemical compound Br.C1=C(O)C=C2C(CCN)=CCCC2=C1 WFMXLYNKBOYPRA-UHFFFAOYSA-N 0.000 claims description 5
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 claims description 5
- 239000012044 organic layer Substances 0.000 claims description 5
- FOKMSWFEISQKHX-UHFFFAOYSA-N 8-(2-aminoethyl)naphthalen-2-ol;hydrobromide Chemical compound Br.C1=C(O)C=C2C(CCN)=CC=CC2=C1 FOKMSWFEISQKHX-UHFFFAOYSA-N 0.000 claims description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 239000011591 potassium Substances 0.000 claims description 3
- 229910052700 potassium Inorganic materials 0.000 claims description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 2
- XWBDWHCCBGMXKG-UHFFFAOYSA-N ethanamine;hydron;chloride Chemical class Cl.CCN XWBDWHCCBGMXKG-UHFFFAOYSA-N 0.000 claims description 2
- CHPCGRFPKVEFJJ-UHFFFAOYSA-N n-[2-(7-hydroxy-1,2,3,4-tetrahydronaphthalen-1-yl)ethyl]acetamide Chemical compound C1=C(O)C=C2C(CCNC(=O)C)CCCC2=C1 CHPCGRFPKVEFJJ-UHFFFAOYSA-N 0.000 claims description 2
- 238000001556 precipitation Methods 0.000 claims description 2
- 239000011734 sodium Substances 0.000 claims description 2
- 229910052708 sodium Inorganic materials 0.000 claims description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims 2
- 239000000010 aprotic solvent Substances 0.000 claims 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 claims 1
- 239000007787 solid Substances 0.000 description 29
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 28
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 26
- 238000002844 melting Methods 0.000 description 23
- 230000008018 melting Effects 0.000 description 23
- 230000035484 reaction time Effects 0.000 description 23
- 238000004128 high performance liquid chromatography Methods 0.000 description 22
- 150000007529 inorganic bases Chemical class 0.000 description 20
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 18
- 239000000047 product Substances 0.000 description 17
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 16
- 235000011121 sodium hydroxide Nutrition 0.000 description 16
- 238000010626 work up procedure Methods 0.000 description 16
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 235000011114 ammonium hydroxide Nutrition 0.000 description 15
- 238000001704 evaporation Methods 0.000 description 15
- 230000008020 evaporation Effects 0.000 description 14
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 14
- 235000011118 potassium hydroxide Nutrition 0.000 description 13
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 13
- 125000003944 tolyl group Chemical group 0.000 description 13
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 12
- 238000002955 isolation Methods 0.000 description 12
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 12
- 238000007796 conventional method Methods 0.000 description 11
- 238000001816 cooling Methods 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 238000010438 heat treatment Methods 0.000 description 11
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 10
- 238000001914 filtration Methods 0.000 description 10
- 231100001261 hazardous Toxicity 0.000 description 10
- 239000011541 reaction mixture Substances 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 9
- 150000007530 organic bases Chemical class 0.000 description 9
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 8
- 239000012296 anti-solvent Substances 0.000 description 8
- 239000010410 layer Substances 0.000 description 8
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 8
- 238000010899 nucleation Methods 0.000 description 8
- FBCQUCJYYPMKRO-UHFFFAOYSA-N prop-2-enyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC=C FBCQUCJYYPMKRO-UHFFFAOYSA-N 0.000 description 8
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 8
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 8
- 238000005292 vacuum distillation Methods 0.000 description 8
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 8
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 7
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 7
- 239000008188 pellet Substances 0.000 description 7
- 238000001953 recrystallisation Methods 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 6
- MGJHVZKRAOYORH-UHFFFAOYSA-N 2-(7-methoxy-3,4-dihydronaphthalen-1-yl)acetonitrile Chemical compound C1CC=C(CC#N)C2=CC(OC)=CC=C21 MGJHVZKRAOYORH-UHFFFAOYSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 238000000605 extraction Methods 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- 239000012535 impurity Substances 0.000 description 6
- 238000010979 pH adjustment Methods 0.000 description 6
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 6
- GABLTKRIYDNDIN-UHFFFAOYSA-N 7-methoxy-3,4-dihydro-2h-naphthalen-1-one Chemical compound C1CCC(=O)C2=CC(OC)=CC=C21 GABLTKRIYDNDIN-UHFFFAOYSA-N 0.000 description 5
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 239000003513 alkali Substances 0.000 description 5
- 150000001342 alkaline earth metals Chemical class 0.000 description 5
- 125000000217 alkyl group Chemical group 0.000 description 5
- 125000003118 aryl group Chemical group 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 238000001035 drying Methods 0.000 description 5
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 5
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 4
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 150000004703 alkoxides Chemical class 0.000 description 4
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 4
- 125000003710 aryl alkyl group Chemical group 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 description 4
- 239000001110 calcium chloride Substances 0.000 description 4
- 229910001628 calcium chloride Inorganic materials 0.000 description 4
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 4
- 150000001735 carboxylic acids Chemical class 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- 239000012153 distilled water Substances 0.000 description 4
- 150000004679 hydroxides Chemical class 0.000 description 4
- 230000001035 methylating effect Effects 0.000 description 4
- 238000011946 reduction process Methods 0.000 description 4
- ODZPKZBBUMBTMG-UHFFFAOYSA-N sodium amide Chemical compound [NH2-].[Na+] ODZPKZBBUMBTMG-UHFFFAOYSA-N 0.000 description 4
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 4
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 4
- 239000011592 zinc chloride Substances 0.000 description 4
- 235000005074 zinc chloride Nutrition 0.000 description 4
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 3
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 3
- HVHZEKKZMFRULH-UHFFFAOYSA-N 2,6-ditert-butyl-4-methylpyridine Chemical compound CC1=CC(C(C)(C)C)=NC(C(C)(C)C)=C1 HVHZEKKZMFRULH-UHFFFAOYSA-N 0.000 description 3
- MFZBSWSCIWCRKS-UHFFFAOYSA-N 2,9-dimethyl-1,10-phenanthroline;hydrate Chemical compound O.C1=C(C)N=C2C3=NC(C)=CC=C3C=CC2=C1 MFZBSWSCIWCRKS-UHFFFAOYSA-N 0.000 description 3
- KWMBADTWRIGGGG-UHFFFAOYSA-N 2-diethoxyphosphorylacetonitrile Chemical compound CCOP(=O)(CC#N)OCC KWMBADTWRIGGGG-UHFFFAOYSA-N 0.000 description 3
- IGDLZDCWMRPMGL-UHFFFAOYSA-N 2-ethenylisoindole-1,3-dione Chemical compound C1=CC=C2C(=O)N(C=C)C(=O)C2=C1 IGDLZDCWMRPMGL-UHFFFAOYSA-N 0.000 description 3
- ZUYNLEPNQXVMKO-UHFFFAOYSA-N 8-(2-aminoethyl)-5,6,7,8-tetrahydronaphthalen-2-ol Chemical compound C1=C(O)C=C2C(CCN)CCCC2=C1 ZUYNLEPNQXVMKO-UHFFFAOYSA-N 0.000 description 3
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- 235000011054 acetic acid Nutrition 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 238000011097 chromatography purification Methods 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 238000006356 dehydrogenation reaction Methods 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 229910052744 lithium Inorganic materials 0.000 description 3
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 3
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 3
- 229910000510 noble metal Inorganic materials 0.000 description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 3
- 230000002035 prolonged effect Effects 0.000 description 3
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 2
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 2
- FEZNCCWLSSNPLW-UHFFFAOYSA-N 2-(7-methoxy-3,4-dihydronaphthalen-1-yl)ethanamine Chemical compound C1CC=C(CCN)C2=CC(OC)=CC=C21 FEZNCCWLSSNPLW-UHFFFAOYSA-N 0.000 description 2
- HPYGZUDDGWEYDQ-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)ethanamine;hydrochloride Chemical compound Cl.C1=CC=C(CCN)C2=CC(OC)=CC=C21 HPYGZUDDGWEYDQ-UHFFFAOYSA-N 0.000 description 2
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 2
- OAIQRWGXAFDMLY-UHFFFAOYSA-N 8-(1-aminoethyl)naphthalen-2-ol Chemical compound OC1=CC=C2C=CC=C(C2=C1)C(C)N OAIQRWGXAFDMLY-UHFFFAOYSA-N 0.000 description 2
- RZFRIHXIWWHFSC-UHFFFAOYSA-N Br.OC1=CC=C2C=CC=C(C2=C1)C(C)N Chemical compound Br.OC1=CC=C2C=CC=C(C2=C1)C(C)N RZFRIHXIWWHFSC-UHFFFAOYSA-N 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- 239000001358 L(+)-tartaric acid Substances 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- 229940092714 benzenesulfonic acid Drugs 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000002894 chemical waste Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 238000010908 decantation Methods 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- VPXDDUIYFMKQBI-UHFFFAOYSA-N ethyl 2-(7-methoxynaphthalen-1-yl)acetate Chemical compound C1=C(OC)C=C2C(CC(=O)OCC)=CC=CC2=C1 VPXDDUIYFMKQBI-UHFFFAOYSA-N 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 239000001630 malic acid Substances 0.000 description 2
- 235000011090 malic acid Nutrition 0.000 description 2
- VYQNQWIPOKLVHL-UHFFFAOYSA-N n-[2-(7-methoxy-3,4-dihydronaphthalen-1-yl)ethyl]acetamide Chemical compound C1CC=C(CCNC(C)=O)C2=CC(OC)=CC=C21 VYQNQWIPOKLVHL-UHFFFAOYSA-N 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- 230000002085 persistent effect Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 235000011007 phosphoric acid Nutrition 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- 239000013557 residual solvent Substances 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- XHLHPRDBBAGVEG-UHFFFAOYSA-N 1-tetralone Chemical compound C1=CC=C2C(=O)CCCC2=C1 XHLHPRDBBAGVEG-UHFFFAOYSA-N 0.000 description 1
- GTVWMPFUOTWFBA-UHFFFAOYSA-N 2-(7-ethoxynaphthalen-1-yl)acetonitrile Chemical compound C1=CC=C(CC#N)C2=CC(OCC)=CC=C21 GTVWMPFUOTWFBA-UHFFFAOYSA-N 0.000 description 1
- LGYBVRIYYBHYCX-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)acetamide Chemical compound C1=CC=C(CC(N)=O)C2=CC(OC)=CC=C21 LGYBVRIYYBHYCX-UHFFFAOYSA-N 0.000 description 1
- CRIAVNMNHHQBGQ-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)acetyl chloride Chemical compound C1=CC=C(CC(Cl)=O)C2=CC(OC)=CC=C21 CRIAVNMNHHQBGQ-UHFFFAOYSA-N 0.000 description 1
- BRZANYWZXQNWSW-UHFFFAOYSA-N 2-(7-methoxynaphthalen-1-yl)ethanamine;hydrobromide Chemical compound Br.C1=CC=C(CCN)C2=CC(OC)=CC=C21 BRZANYWZXQNWSW-UHFFFAOYSA-N 0.000 description 1
- YSTUXESYWJJTQI-UHFFFAOYSA-N 2-[1-(2-aminoethyl)-7-methoxy-1H-naphthalen-2-ylidene]acetonitrile methanol Chemical compound NCCC1C(C=CC2=CC=C(C=C12)OC)=CC#N.CO YSTUXESYWJJTQI-UHFFFAOYSA-N 0.000 description 1
- NYPYHUZRZVSYKL-UHFFFAOYSA-N 2-azaniumyl-3-(4-hydroxy-3,5-diiodophenyl)propanoate Chemical compound OC(=O)C(N)CC1=CC(I)=C(O)C(I)=C1 NYPYHUZRZVSYKL-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- LPSGHPWFGVBIMR-UHFFFAOYSA-N 4-(7-hydroxy-3,4-dihydronaphthalen-1-yl)butanamide Chemical compound C1=C(O)C=C2C(CCCC(=O)N)=CCCC2=C1 LPSGHPWFGVBIMR-UHFFFAOYSA-N 0.000 description 1
- NSWTUNAPARHDSP-UHFFFAOYSA-N 8-(2-aminoethyl)-5,6,7,8-tetrahydronaphthalen-2-ol;hydrochloride Chemical compound Cl.C1=C(O)C=C2C(CCN)CCCC2=C1 NSWTUNAPARHDSP-UHFFFAOYSA-N 0.000 description 1
- XAJIRHYFGJXWHB-UHFFFAOYSA-N C(C)#N.C(C)(=O)N.COC1=CC=C2C=CC=C(C2=C1)C(C(=O)O)(C1=CC=CC2=CC=C(C=C12)OC)C1=CC=CC2=CC=C(C=C12)OC Chemical compound C(C)#N.C(C)(=O)N.COC1=CC=C2C=CC=C(C2=C1)C(C(=O)O)(C1=CC=CC2=CC=C(C=C12)OC)C1=CC=CC2=CC=C(C=C12)OC XAJIRHYFGJXWHB-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- UDQUTZIDSGXXFP-UHFFFAOYSA-N acetamide acetonitrile 2,2,2-tris(7-methoxynaphthalen-1-yl)acetyl chloride Chemical compound CC#N.CC(N)=O.COc1ccc2cccc(c2c1)C(C(Cl)=O)(c1cccc2ccc(OC)cc12)c1cccc2ccc(OC)cc12 UDQUTZIDSGXXFP-UHFFFAOYSA-N 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 238000004176 ammonification Methods 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 230000000561 anti-psychotic effect Effects 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 239000002249 anxiolytic agent Substances 0.000 description 1
- 230000000949 anxiolytic effect Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 150000008280 chlorinated hydrocarbons Chemical class 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 238000004042 decolorization Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- FDPIMTJIUBPUKL-UHFFFAOYSA-N dimethylacetone Natural products CCC(=O)CC FDPIMTJIUBPUKL-UHFFFAOYSA-N 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000003912 environmental pollution Methods 0.000 description 1
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 1
- AMANDCZTVNQSNB-UHFFFAOYSA-N glyoxamide Chemical compound NC(=O)C=O AMANDCZTVNQSNB-UHFFFAOYSA-N 0.000 description 1
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 description 1
- 239000000383 hazardous chemical Substances 0.000 description 1
- ZYCMDWDFIQDPLP-UHFFFAOYSA-N hbr bromine Chemical compound Br.Br ZYCMDWDFIQDPLP-UHFFFAOYSA-N 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000002519 immonomodulatory effect Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 208000024714 major depressive disease Diseases 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000002790 naphthalenes Chemical class 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 230000016087 ovulation Effects 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000009738 saturating Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/02—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions involving the formation of amino groups from compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/10—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C215/00—Compounds containing amino and hydroxy groups bound to the same carbon skeleton
- C07C215/02—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C215/22—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being unsaturated
- C07C215/26—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being unsaturated and containing rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/02—Preparation of carboxylic acid amides from carboxylic acids or from esters, anhydrides, or halides thereof by reaction with ammonia or amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/16—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
- C07C233/17—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/22—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom having the carbon atom of the carboxamide group bound to an acyclic carbon atom of a carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/30—Preparation of carboxylic acid nitriles by reactions not involving the formation of cyano groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/32—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms having cyano groups bound to acyclic carbon atoms of a carbon skeleton containing at least one six-membered aromatic ring
- C07C255/36—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms having cyano groups bound to acyclic carbon atoms of a carbon skeleton containing at least one six-membered aromatic ring the carbon skeleton being further substituted by hydroxy groups
Definitions
- the present invention relates to novel, commercially viable and industrially advantageous processes for the preparation of agomelatine or a salt thereof, in high yield and purity, using novel intermediates.
- U.S. Patent No. 5,225,442 discloses 1- alkoxy-2-(acylaminoethyl)naphthalene derivatives, processes for their preparation, pharmaceutical compositions comprising the derivatives, and method of use thereof. These compounds possess valuable pharmacological properties with regard to the central nervous system, particularly anxiolytic, antipsychotic and analgesic properties, and with regard to ovulation, cerebral circulation and immunomodulation.
- Agomelatine chemically named N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide, is an important antidepressant and useful for the treatment of major depressive episodes in adults.
- Agomelatine has dual pharmacological effects, which is not only the agonist of melatonergic system receptor, but also the antagonist of 5HT 2 c receptor.
- Agomelatine is represented by the following structural formula I:
- Agomelatine is marketed by Servier (Ireland) Industries Ltd. in Europe under the brand names VALDOXAN ® and THYMANAX ® , and it is orally administered as tablets containing 25 mg of agomelatine.
- agomelatine was first described in the '442 patent and its corresponding European equivalent Patent No. EP 0447285 Bl .
- Various pjOcesses_fot-the--- preparation of agomelatine, its intermediates, and related compounds are described in U.S. Patent Nos. US 5,420,158, US 7,476,751, US 7,544,839, US 7,999,129, US 8,212,077 and US 8,143,449; PCT Publication Nos.
- N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide (Agomelatine) is prepared by a process as depicted in scheme 1 :
- agomelatine As per the process described in the '442 patent, agomelatine is prepared by reacting
- agomelatine disclosed in the '442 patent suffers from various disadvantages such as the use of highly corrosive and toxic reagents like thionyl chloride, benzene, chloroform and pyridine; and involves the use of tedious and cumbersome procedures since it requires eight steps to synthesize agomelatine, thereby rendering the overall yield being less than 30% and making the process commercially unfeasible.
- Agomelatine obtained by the process described in the '442 patent does not have the satisfactory purity for pharmaceutical use. Unacceptable amounts of impurities are generally formed along with agomelatine.
- Agomelatine is prepared by a process as depicted in scheme 2:
- agomelatine is prepared by reacting 7-methoxy-l-tetralone with cyanoacetic acid in the presence of heptanoic acid and benzylamine to produce (7-methoxy-3,4-dihydro-l-naphthalenyl)acetonitrile, followed by dehydrogenation in the presence of hydrogenation catalyst Pd/C with allyl methacrylate as the dehydrogenating agent to produce (7-methoxy-l-naphthyl)acetonitrile, which is then subjected to reduction with Raney Nickel under hydrogen pressure in the presence of ammonium hydroxide to produce 2-(7-methoxy-l-naphthyl)ethanamine hydrochloride, followed by acetylation to produce agomelatine.
- the reported overall yield of the product is 72%.
- the methoxy intermediates obtained according to the process described in the '839 patent suffer from disadvantages since these methoxy intermediates are characterized by having low melting points.
- the reported melting point for (7-methoxy-3,4-dihydro-l-naphthalenyl)acetonitrile is 48-50°C
- the reported melting point for (7-methoxy-i-naphthyl)acetonitrile is 83°C.
- the known methoxy intermediate compounds are not stable and they decompose at higher temperatures due to their low melting points, for example, when sulfur (which is a cheaper aromatizing agent) is employed as reagent for aromatization of (7-methoxy-3,4-dihydro-l- naphthalenyl)acetonitrile (this reaction requires heating the reactants at higher temperature 180-200°C) to produce (7-methoxy-l-naphthyl)acetonitrile, thus leading to the formation of unacceptable amounts of impurities, thereby decreasing the yield and purity of the product.
- sulfur which is a cheaper aromatizing agent
- agomelatine is prepared by reacting 7-methoxy-l-tetralone with acetonitrile in the presence of sodium hexamethyldisilazide in tetrahydrofuran to provide 2-(l-hydroxy-7-methoxy-l,2,3,4-tetrahydronaphthalen-l-yl)acetonitrile, which is further crystallized from cyclohexane to produce pure compound, which is then treated with hydrogen gas in the presence of Raney Ni, in aqueous methanol in the presence of ammonia at 45-60°C to provide 2-(7-methoxy-3,4-dihydronaphthalen-l-yl)ethanamine, followed by treatment with hydrochloric acid in ethylacetate-to-produee-its-hydrochloride ⁇ salt, which is then acetylated with acetyl chlor
- Agomelatine is prepared by a process as depicted in scheme 4:
- Agomelatine 2-oxoacetamide According to Journal of Medicinal Chemistry 1992, 35(8), 1484-1486 (hereinafter referred to as the 'JMC article'), Agomelatine is prepared by a process as depicted in scheme 6:
- N-[2-(7-methoxy-l- naphthalenyl)ethyl]acetamide (Agomelatine) described in the aforementioned prior art suffer from several disadvantages such as the use of highly flammable, corrosive and hazardous reagents like thionyl chloride, pyridine, triflic anhydride, allyl methacrylate, sodium hydride, sodamide, n-butyl lithium and lithium aluminium hydride; use of expensive reagents such as diethyl cyanomethyl phosphonate, 2,6-di-tert-butyl-4-methyl- pyridine, N-vinylphthalimide, Palladium tetrakis(triphenylphosphine), hydrogenation catalysts such as Pd/C, acrylamide, Neocuproine hydrate, 18-crown-6-ether, propylphosphonic anhydride; use of additional and excessive amounts of reagents; use of multiple solvents and
- the major drawback of the processes for the preparation of agomelatine described in the aforementioned prior art is that the processes involve the use of highly flammable, corrosive and hazardous reagents like thionyl chloride, pyridine, triflic anhydride, allyl methacrylate, sodium hydride, sodamide, n-butyl lithium and lithium aluminium hydride, thereby requiring very strict control of reaction conditions at low temperatures. Handling of these reagents is very difficult at lab scale and in commercial scale operations. Moreover, the yields and purities of the product obtained according to the prior art processes are low to moderate.
- the methoxy intermediates decompose at higher temperatures due to their low melting points, hence, the aromatization of methoxy intermediates such as (7-methoxy-3,4- dihydro- 1 -naphthalenyl)acetonitrile and N-[2-(7-methoxy-3 ,4-dihydronaphthalen- 1 - yl)ethyl]acetamide must be carried out at low temperatures and requires the use of expensive noble metal catalysts like palladium on carbon and additional hazardous reagents like allyl methacrylate, and expensive reagents like DDQ;
- methoxy intermediates such as (7-methoxy-3,4- dihydro- 1 -naphthalenyl)acetonitrile and N-[2-(7-methoxy-3 ,4-dihydronaphthalen- 1 - yl)ethyl]acetamide
- the processes involve the use of highly flammable, corrosive and hazardous reagents like thionyl chloride, pyridine, trifilc anhydride, allyl methacrylate, sodium hydride, sodamide, n-butyl lithium and lithium alumimium hydride;
- the processes involve the use of expensive reagents such as diethyl cyanomethyl phosphonate, 2,6-di-tert-butyl-4-methyl-pyridine, N-vinylphthalimide, Palladium tetrakis(triphenylphosphine), hydrogenation catalysts such as Pd/C, acrylamide, Neocuproine hydrate, 18-crown-6-ether, propylphosphonic anhydride;
- expensive reagents such as diethyl cyanomethyl phosphonate, 2,6-di-tert-butyl-4-methyl-pyridine, N-vinylphthalimide, Palladium tetrakis(triphenylphosphine), hydrogenation catalysts such as Pd/C, acrylamide, Neocuproine hydrate, 18-crown-6-ether, propylphosphonic anhydride;
- Desirable process properties include non-hazardous conditions, environmentally friendly and easy to handle reagents, reduced cost, greater simplicity, increased purity, and increased yield of the product, thereby enabling the production of Agomelatine, in high purity and with high yield.
- Agomelatine, N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide, of formula I or a salt thereof can be prepared in high purity and with high yield by reacting 7-hydroxy-l-tetralone with cyanoacetic acid in the presence of a suitable reagent to produce (7-hydroxy-3,4-dihydro- l-naphthalenyl)acetonitrile or a salt thereof, which is_then_subjected-to-reduction ⁇ ⁇ itir suitable reducing agent to produce 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine or a salt thereof, followed by aromatization with a suitable reagent to produce 2-(7- hydroxy-l-naphthyl)ethanamine or an acid addition salt thereof, which is then subjected to acetylation with a suitable acetylating agent to produce N
- provided herein are efficient, industrially advantageous and environmentally friendly processes for the preparation of agomelatine in high yield and with high purity using novel intermediates.
- the processes disclosed herein avoid the tedious and cumbersome procedures of the prior processes, thereby resolving the problems associated with the processes described in the prior art, which are more convenient to operate at lab scale and in commercial scale operations.
- the present invention also encompasses the use of the novel compounds of formulae III, IV, V, VII, VIII, XII and XIII disclosed herein for preparing Agomelatine.
- the novel hydroxy intermediate compounds are characterized by having higher melting points when compared with that of the known methoxy intermediates;
- the processes avoid the use of highly flammable, corrosive and hazardous reagents like thionyl chloride, pyridine, triflic anhydride, allyl methacrylate, sodium hydride, sodamide, n-butyl lithium and lithium aluminium hydride;
- the processes avoid the use of highly toxic or hazardous chemicals like benzene, pyridine and chloroform; ix) the processes avoid the use of tedious and cumbersome procedures like prolonged reaction time periods, very low temperatures, multiple process steps, column chromatographic purifications, multiple isolations, use of additional and excess amounts of solvents;
- the processes avoid the use of expensive reagents such as diethyl cyanomethyl phosphonate, 2,6-di-tert-butyl-4-methyl-pyridine, N-vinylphthalimide, Palladium tetrakis(triphenylphosphine), hydrogenation catalysts such as Pd/C, acrylamide, Neocuproine hydrate, 18-crown-6-ether, propylphosphonic anhydride;
- expensive reagents such as diethyl cyanomethyl phosphonate, 2,6-di-tert-butyl-4-methyl-pyridine, N-vinylphthalimide, Palladium tetrakis(triphenylphosphine), hydrogenation catalysts such as Pd/C, acrylamide, Neocuproine hydrate, 18-crown-6-ether, propylphosphonic anhydride;
- novel intermediate compounds of Agomelatine disclosed herein are obtained as solid state forms in substantially pure form.
- substantially pure refers to the solid state form of agomelatine intermediates, disclosed herein, having a purity of greater than about 97 wt%, specifically greater than about 98 wt%, more specifically greater than about 99 wt%, and still more specifically greater than about 99.5 wt%.
- the purity is preferably measured by High Performance Liquid Chromatography (HPLC).
- HPLC High Performance Liquid Chromatography
- the purity of solid state form of agomelatine intermediates obtained by the processes disclosed herein can be about 97% to about 99.5%, or about 98% to about 99.9%, as measured by HPLC.
- salts may include acid addition salts and base addition salts.
- Acid addition salts include the salts that are derived from organic and inorganic acids.
- the acid addition salts are derived from a therapeutically acceptable acid such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, oxalic acid, acetic acid, propionic acid, phosphoric acid, succinic acid, maleic acid, fumaric acid, citric acid, glutaric acid, tartaric acid, benzenesulfonic acid, toluenesulfonic acid, di- p-toluoyl-L-(+)-tartaric acid, malic acid, ascorbic acid, and the like.
- a therapeutically acceptable acid such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, oxalic acid, acetic acid, propionic acid, phosphoric acid, succinic acid, maleic acid, fumaric acid, citric acid, glutaric acid, tartaric acid, benzenesul
- Base addition salts may be derived from an organic or an inorganic base.
- the base addition salts are derived from alkali or alkaline earth metals such as sodium, calcium, potassium and magnesium; ammonium salt, organic amines such as ethylamine, tert-butylamine, diethylamine, diisopropylamine, and the like.
- Exemplary acid addition salts of the compounds of formulae IV and V include, but are not limited to, hydrochloride, hydrobromide, ⁇ uJphate,_nitrate,--phosphate7-acetate propionate, oxalate, succinate, maleate, fumarate, benzenesulfonate, toluenesulfonate, citrate, tartrate, and the like.
- Specific acid addition salts are hydrochloride and hydrobromide, and most specifically hydrochloride salt.
- Base addition salts of the compound of formula X as used herein include the salts that are derived from an organic or an inorganic base.
- Exemplary base addition salts of the compound of formula X include, but are not limited to, sodium salt, calcium salt, potassium salt, magnesium salt and ammonium salt.
- the reaction in step-(a) is carried out in the presence or absence of a solvent. In one embodiment, the reaction in step-(a) is carried out in the presence of a first solvent.
- solvent also includes mixture of solvents.
- Exemplary first solvents used in step-(a) include, but are not limited to, a hydrocarbon solvent, a halogenated hydrocarbon solvent, an ether, and mixtures thereof.
- the first solvent used in step-(a) is a reaction solvent that has a boiling temperature higher than or equal to that of water, and more preferably that forms an azeotrope with water.
- the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; more specifically the first solvent is toluene or xylene; and a most specific solvent is toluene.
- reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine.
- the carboxylic acid and the organic amine compounds are employed in catalytic amounts.
- Exemplary carboxylic acids used in step-(a) include, but are not limited to, a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid.
- Exemplary organic amines used in step-(a) include, but are not limited to, a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine.
- alkyl denotes an aliphatic hydrocarbon group which may be straight or branched having 1 to 12 carbon atoms in the chain. Preferred alkyl groups have 3 to 10 carbon atoms in the chain.
- the alkyL may be substituted with one or more "cycloalkyl groups". Exemplary alkyl groups include methyl, ethyl, n-propyl, iso- propyl, n-butyl, iso-butyl, t-butyl, n-pentyl, hexyl and heptyl.
- cycloalkyl denotes a non-aromatic mono- or multicyclic ring system of 3 to 10 carbon atoms, preferably of about 5 to about 10 carbon atoms.
- exemplary monocyclic cycloalkyl groups include cyclopentyl, cyclohexyl, cycloheptyl and the like.
- aryl denotes an aromatic monocyclic or multicyclic ring system of 6 to 10 carbon atoms.
- the aryl is optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined herein.
- exemplary aryl groups include phenyl, tolyl, naphthyl or biphenyl group.
- aralkyl denotes an aryl-alkyl group wherein the aryl and alkyl are as herein described. Preferred aralkyls contain a lower alkyl moiety. Exemplary aralkyl groups include benzyl, 2-phenethyl and naphthalenemethyl.
- the carboxylic acid used in step-(a) is an unsubstituted or substituted aryl carboxylic acid; and a most specific carboxylic acid is 4-hydroxybenzoic acid.
- the organic amine used in step-(a) is an unsubstituted or substituted linear or branched aralkyl amine; more specifically the organic amine is morpholine or benzyl amine; and a most specific organic amine is benzyl amine.
- reaction temperature and time period will ordinarily depend on the starting compounds and the solvent employed in the reaction.
- the reaction in step-(a) is carried out at a temperature of about 25 °C to the reflux temperature of the solvent used, specifically at a temperature of about 60°C to the reflux temperature of the solvent used, and more specifically at the reflux temperature of the solvent used.
- the reaction time may vary between about 10 hours to about 40 hours, and most specifically about 25 hours to about 30 hours.
- the cyanoacetic acid of formula X is used in a molar ratio of about 1 to 3 equivalents, specifically about .1 to 1.5 equivalents, with respect to the 7- hydroxy-l-tetralone of formula II in order to ensure a proper course of the reaction.
- the reaction mass containing the (7-hydroxy-3,4-dihydro-l- naphthalenyl)acetonitrile of formula III obtained in step-(a) may subjected to usual work up such as a washing, an extraction, a pH adjustment, jm_evaporation, a layer separation, ⁇ a decolorization, or a combination thereof.
- the reaction mass may be used directly in the next step to produce the amine compound of formula IV or the compound of formula III may be isolated and/or recrystallized and then used in the next step.
- the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated in the form of a solid.
- the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated and/or re-crystallized from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
- the solvent used for isolating and/or recrystallizing the pure (7-hydroxy-3,4- dihydro-l-naphthalenyl)acetonitrile of formula III is selected from the group consisting of water, an alcohol, a ketone, an ether, an ester, a hydrocarbon solvent, a halogenated hydrocarbon, and mixtures thereof.
- the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- the reduction in step-(b) is carried out in the presence of a second solvent.
- solvent also includes mixture of solvents.
- Exemplary second solvents used in step-(b) include, but are not limited to, water, an alcohol, an ester, a hydrocarbon solvent, an ether, and mixtures thereof.
- the second solvent used in step-(b) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; more specifically, the second solvent is selected from the group consisting of methanol, ethanol, isopropanol, and mixtures thereof; and a most specific second solvent is methanol,
- Exemplary reducing agents used in step-(b) include, but are not limited to, hydrogenation catalysts (metal catalysts) such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like; hydride agents such as lithium aluminum hydride, sodium borohydride/Lewis acid, lithium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2-methoxyethoxy)aluminium hydride (Vitride), and the like; and other reducing agents such as borane, borane-THF complex, and the like.
- metal catalysts such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like
- hydride agents such as lithium aluminum hydride,
- step-(b) is advantageously and efficiently carried out by employing the less expensive hydride agents such as sodium borohydride when the reaction is performed in the presence of a Lewis acid.
- hydride agents such as sodium borohydride
- the use of hydride agents in the presence of Lewis acid in the reduction process leads to the resulting product with high purity and in good yield.
- Exemplary Lewis acids used in step-(b) include, but are not limited to, aluminium chloride, calcium chloride, boron triflouride and zinc chloride, nickel chloride, and the like. Specific Lewis acids are aluminium chloride and nickel chloride.
- step-(b) may be carried out in the presence or absence of hydrogen gas.
- a most specific reducing agent used in step-(b) is Raney-Nickel.
- ammonia used may be in the form of aqueous ammonia or in the form of ammonia gas or ammonia saturated in an organic solvent.
- the organic solvent used for saturating ammonia is selected from the group consisting of ethanol, methanol, isopropyl alcohol and ethyl acetate.
- the reduction in step-(b) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and more specifically at about 25°C to about 65°C.
- the reaction time may vary between about 3 hours to about 8 hours, and most specifically about 5 hours to about 6 hours.
- the reducing agent is used in an amount of about 5%w/w to 50%w/w, specifically about 5%w/w to 10%w/w, with respect to the (7-hydroxy-3,4- dihydro-l-naphthalenyl)acetonitrile of formula III in order to ensure a proper course of the reaction.
- reaction mass containing the 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV obtained in step-(b) may subjected to usual work up methods as described above.
- the reaction mass may be used directly in the next step to produce the amine compound of formula V or the compound of formula IV may be isolated and/or recrystallized and then used in the next step.
- the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV obtained in step-(b) is converted into its acid addition salts by treatment with a suitable acid.
- acids suitable for forming acid addition salts include, but are not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, oxalic acid, acetic acid, propionic acid, phosphoric acid, succinic acid, maleic acid, fumaric acid, citric acid, glutaric acid, tartaric acid, benzenesulfonic acid, toluenesulfonic acid, di-p-toluoyl-L-(+)- tartaric acid, malic acid, ascorbic acid, and the like. Most specific acids are hydrochloric acid and hydrobromic acid.
- hydrochloric acid used may be in the form of aqueous hydrochloric acid or in the form of hydrogen chloride gas or hydrogen chloride dissolved in an organic solvent.
- the organic solvent used for dissolving hydrogen chloride gas or hydrogen chloride is selected from the group consisting of ethanol, methanol, isopropyl alcohol, ethyl acetate, diethyl ether, dimethyl ether and acetone.
- the treatment of 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV with a suitable acid is carried out in a solvent selected from the group consisting of water, an ester, an alcohol, a ketone, a chlorinated hydrocarbon, a hydrocarbon, an ether, and mixtures thereof.
- the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2- methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof; and most specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, ethyl acetate, and mixtures thereof.
- the treatment of 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV with a suitable acid is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, and most specifically at a temperature of about 20°C to the reflux temperature of the solvent used.
- the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof is isolated in the form of a solid.
- the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
- the solvent used for isolating and/or recrystallizing the pure 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV is selected from the group as described above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-peritane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- a most specific acid addition salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV prepared by the process described herein is 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrochloride salt.
- a most specific acid addition salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV prepared by the process described herein is 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrobromide salt.
- Exemplary reagents suitable for facilitating the aromatization reaction in step-(c) include, but are not limited to, sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, metal catalysts such as palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, and the like; quinone derivatives such as 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), p-bromanil, p-floranil, 2,3- dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6-dicyano benzoquinone, 2,3- dicyano-4-chlorobenzoquinone, 2,3-dicyanobenzoquinone; and other ⁇ quinone derfvatives ⁇ such as 1,2-benzoquinones, 1,3-benzoquinones, o-chloranil
- aromatization reagents are sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ) and raney nickel; and most specifically sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil).
- the starting material 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV, in step-(c), is employed in the form of an acid addition salt, and most specifically in the form of its hydrochloride salt.
- the novel hydroxy intermediate compounds of formulae III, IV, V, VII and VIII, and their salts, disclosed herein are characterized by having higher melting points when compared with that of the known methoxy intermediates.
- the hydrochloride salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV obtained by the processes disclosed, is characterized by having melting range at about 195-200°C, whereas the corresponding methoxy analogue, i.e., the hydrochloride salt of 2-(7-methoxy-3,4-dihydro-l- naphthalenyl)ethanamine is characterized by having melting point at about 151.12°C.
- This high melting range of the novel hydroxy intermediate compounds disclosed herein is advantageous since this property makes these compounds stable even at higher temperature (e.g. 185-190°C), thereby making it possible to advantageously employ the cheaper aromatization reagents such as sulfur for aromatizing the respective dihydro intermediates (e.g., the compound of formula IV) at higher temperature, for example, at 185-190°C.
- the corresponding methoxy intermediates decompose at such higher temperatures due to their low melting points when aromatizing with sulfur thereby effecting purity and yield of the resulting products.
- step-(c) is advantageously carried out by using cheaper aromatization reagent sulfur in the absence of a solvent.
- the aromatization reaction in step-(c) is carried out in the presence or absence of a solvent.
- the aromatization in step-(c) is carried out as a neat reaction in the absence of a solvent.
- the aromatization in step-(c) is optionally carried out in the presence of a third solvent.
- solvent also includes mixture of solvents.
- Exemplary third solvents suitable for facilitating the aromatization in step-(c) include, but are not limited to, water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
- the third solvent is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, ⁇ , ⁇ -dimethylacetamide, dimethylsulfoxide, and mixtures thereof; and a most specific third solvent is toluene.
- the aromatization in step-(c) is carried out as a neat reaction, in the absence of solvents, by heating the contents at a temperature of about 160°C to about 200°C, and specifically at a temperature of about 180°C to about 190°C.
- the reaction time may vary between about 30 minutes to about 5 hours, and most specifically about 1 hour to about 2 hours.
- the aromatization in step-(c) is carried out in the presenceof the third solvent at a temperature of about 20°C to the reflux temperature of the solvent used, specifically at a temperature of about 50°C to the reflux temperature of the solvent used, and more specifically at about 60°C to about 120°C.
- the reaction time may vary between about 3 hours to about 30 hours, and most specifically about 5 hours to about 25 hours.
- the metal catalysts suitable for facilitating aromatization is used in a amount of about 5%w/w to about 50%w/w, specifically about 5%w/w to 20%w/w, with respect to the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof in order to ensure a proper course of the reaction.
- the sulfur is used in a molar ratio of about 1 to 5 equivalents, specifically about 1 to 2 equivalents, with respect to the 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof in order to ensure a proper course of the reaction.
- the 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p- chloranil) is used in a molar ratio of about 1 to 5 equivalents, specifically about 1 to 2 equivalents, with respect to the 2-(7-hydroxy-3,4-dihydro-l-naphthalehyl)ethanamine of formula IV or an acid addition salt thereof in order to ensure a proper course of the reaction.
- the aqueous hydrobromic acid is used in an amount of about 1 to 5 times, specifically about 1 to 2 times, with respect to the 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof in order to ensure a proper course of the reaction.
- reaction mass containing the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof obtained in step-(c) may subjected to usual work up methods as described above.
- the reaction mass may be used directly in the next step to produce the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI or the compound of formula V may be isolated and/or recrystallized and then used in the next step.
- the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V obtained in step-(c) is converted into its acid addition salts by treatment with a suitable acid as per the methods described hereinabove.
- the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof obtained in step-(c) is isolated in the form of a solid.
- the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by the methods as described hereinabove.
- the solvent used for isolating and/or recrystallizing the pure 2-(7-hydroxy-l- naphthyl)ethanamine of formula V or an acid addition salt thereof is selected from the group as described above.
- the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2- methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyXejher, regularlyethyTaGetate n-penfane n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- a most specific acid addition salt of 2-(7-hydroxy-l- naphthyl)ethanamine of formula V prepared by the process described herein is 2-(7- hydroxy- l-naphthyl)ethanamine hydrochloride salt.
- a most specific acid addition salt of 2-(7-hydroxy-l- naphthyl)ethanamine of formula V prepared by the process described herein is 2-(7- hydroxy-l-naphthyl)ethanamine hydrobromide salt.
- Exemplary acetylating agents used in step-(d) include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium acetate, and the like, or a combination thereof.
- acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide
- acetic anhydride sodium acetate, and the like, or a combination thereof.
- a most specific acetylating agent is acetic anhydride.
- the reaction in step-(d) is optionally carried out in the presence of a base.
- the base is an organic or inorganic base, and most specifically an inorganic base.
- Exemplary bases include, but are not limited to, hydroxides, alkoxides, bicarbonates and carbonates of alkali or alkaline earth metals; ammonia, collidine, trimethylamine, tributylamine, triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4-(N,N-dimethylamino)pyridine and 1-alkylimidazole.
- Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically sodium hydroxide and potassium hydroxide.
- the acetylation in step-(d) is carried out in the presence of a fourth solvent.
- Exemplary fourth solvents used in step-(d) include, but are not limited to, water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
- the fourth solvent used in step-(d) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanoI,_eth-yl— acetate, ⁇ methyT acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, ⁇ , ⁇ -dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichlor
- the acetylating agent in step-(d) is used in a ratio of about 1 to 3 equivalents, specifically about 1 to 1.5 equivalents, with respect to the 2-(7 -hydroxy- 1- naphthyl)ethanamine of formula V or an acid addition salt thereof in order to ensure a proper course of the reaction.
- the reaction in step-(d) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used.
- the reaction time may vary between about 20 minutes to about 3 hours, and specifically about 30 minutes to about 2 hours.
- reaction mass containing the N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide of formula VI obtained in step-(d) may be subjected to usual work up methods as described above.
- the reaction mass may be used directly in the next step to produce the compound of formula I, or the compound of formula VI may be isolated and/or recrystallized from a suitable solvent by conventional methods, as described hereinabove, and then used in the next step.
- the solvent used for isolating and/or recrystallizing the N-[2-(7-hydroxy-l- naphthyl)ethyl]acetamide of formula VI is selected from the group as described hereinabove for such purpose.
- methylating agents used in step-(e) include, but are not limited to, dimethyl sulfate, methyl iodide, and the like.
- a most specific methylating agent is dimethyl sulfate.
- the reaction in step-(e) is optionally carried out in the presence of a base.
- the base is an organic or inorganic base, and most specifically an inorganic base, Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically potassium carbonate.
- the methylation in step-(e) is carried out in the presence of a fifth solvent.
- Exemplary fifth solvents used in step-(e) include, but are not limited to, water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
- the fifth solvent used in step-(e) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, ⁇ , ⁇ -dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- Most specific fifth solvents are water, acetone, and mixture
- the methylating agent in step-(e) is used in a ratio of about 1 to 3 equivalents, specifically about 1 to 1.5 equivalents, with respect to the N-[2-(7-hydroxy- l-naphthyl)ethyl]acetamide of formula VI in order to ensure a proper course of the reaction.
- the reaction in step-(e) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used.
- the reaction time may vary between about 30 minutes to about 6 hours, and specifically about 3 hours to about 5 hours.
- the reaction mass containing the agomelatine of formula I obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
- the solvent used for isolating/recrystallizing the pure agomelatine of formula I is selected from the group as described herein above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- the isolation is carried out by cooling the reaction mass at a temperature of below about 35°C, followed by the addition of water at a temperature of about 10°C to about 35°C, and more specifically at a temperature of about 20°C to about 30°C.
- the resulting mass is optionally stirred at a temperature of about 10°C to about 35°C for at least 10 minutes, and most specifically at a temperature of about 20°C to about 30°C for about 15 minutes to about 2 hours.
- a process for the preparation of Agomelatine, N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide, of formula I: or a salt thereof comprising reacting N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide of formula VI: or a salt thereof, with a methylating agent to produce the agomelatine of formula I.
- the reaction in step-(a) is carried out in the presence or absence of a solvent.
- the reaction in step-(a) is carried out in the presence of a first solvent selected from the group as described above.
- the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; more specifically the first solvent is toluene or xylene; and a most specific solvent is toluene.
- reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine.
- the carboxylic acid and the organic amine compounds are employed in catalytic amounts.
- Exemplary carboxylic acids used in step-(a) include, but are not limited to, a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid.
- Exemplary organic amines used in step-(a) include, but are not limited to, a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine.
- the carboxylic acid used in step-(a) is an unsubstituted or substituted aryl carboxylic acid; and a most specific carboxylic acid is 4-hydroxybenzoic acid.
- the organic amine used in step-(a) is an unsubstituted or substituted linear or branched aralkyl amine; more specifically the organic amine is morpholine or benzyl amine; and a most specific organic amine is benzyl amine.
- the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated in the.form of a solid.
- the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula HI is isolated and/or re-crystallized from a suitable solvent by the methods described hereinabove.
- the solvent used for isolating and/or recrystallizing the pure (7-hydroxy-3,4- dihydro-l-naphthalenyl)acetonitrile of formula III is selected from the group as described hereinabove.
- step-(b) is carried out in the presence of a second solvent selected from the group as described above.
- the second solvent used in step-(b) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; more specifically, the second solvent is selected from the group consisting of methanol, ethanol, isopropanol, and mixtures thereof; and a most specific second solvent is methanol,
- Exemplary reducing agents used in step-(b) include, but are not limited to, hydrogenation catalysts (metal catalysts) such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like; hydride agents such as lithium aluminum hydride, sodium borohydride/Lewis acid, lithium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2-methoxyethoxy)aluminium hydride (Vitride), and the like; and other reducing agents such as borane, borane-THF complex, and the like.
- metal catalysts such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like
- hydride agents such as lithium aluminum hydride,
- step-(b) is advantageously and efficiently carried out by employing the less expensive hydride agents-such-as sodiunT borohydride when the reaction is performed in the presence of a Lewis acid.
- hydride agents such-as sodiunT borohydride
- the use of hydride agents in the presence of Lewis acid in the reduction process leads to the resulting product with high purity and in good yield.
- Exemplary Lewis acids used in step-(b) include, but are not limited to, aluminium chloride, calcium chloride, boron triflouride and zinc chloride, nickel chloride, and the like. Specific Lewis acids are aluminium chloride and nickel chloride.
- step-(b) may be carried out in the presence or absence of hydrogen gas.
- a most specific reducing agent used in step-(b) is Raney-Nickel.
- step-(b) is optionally carried out in the presence of ammonia as per the methods described hereinbefore.
- the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV obtained in step-(b) is converted into its acid addition salts by treatment with a suitable acid selected from the group described hereinbefore.
- a suitable acid selected from the group described hereinbefore. Most specific acids are hydrochloric acid and hydrobromic acid.
- the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof is isolated in the form of a solid.
- the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by the methods described hereinbefore.
- the solvent used for isolating and/or recrystallizing the pure 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV is selected from the group as described above.
- a most specific acid addition salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV prepared by the process described herein is 2-(7-hydroxy-3,4-dihydro-l -naphthalenyl)ethanamine hydrochloride salt.
- a most specific acid addition salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV prepared by the process described herein is 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrobromide salt.
- Exemplary acetylating agents used in step-(c) include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium acetate, and the like, or a combination thereof.
- acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide
- acetic anhydride sodium acetate, and the like, or a combination thereof.
- a most specific acetylating agent is acetic anhydride.
- the reaction in step-(c) is optionally carried out in the presence of a base.
- the base is an organic or inorganic base, and most specifically an inorganic base.
- Exemplary bases include, but are not limited to, hydroxides, alkoxides, bicarbonates and carbonates of alkali or alkaline earth metals; ammonia, collidine, trimethylamine, tributylamine, triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4-(N,N-dimethylamino)pyridine and 1-alkylimidazole.
- Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically sodium hydroxide and potassium hydroxide.
- the acetylation in step-(c) is carried out in the presence of a fourth solvent selected from the group as described hereinbefore.
- Exemplary fourth solvents used in step-(c) include, but are not limited to, water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
- the fourth solvent used in step-(c) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, ⁇ , ⁇ -dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chlor
- the reaction in step-(c) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most ventilatspecifically-at " th3 ⁇ 4 ⁇ fluj temperature of the solvent used.
- the reaction time may vary between about 20 minutes to about 3 hours, and specifically about 30 minutes to about 2 hours.
- reaction mass containing the N-[2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethyl]acetamide, of formula VIII obtained in step-(c) may be subjected to usual work up methods as described above.
- the reaction mass may be used directly in the next step to produce the compound of formula VI, or the compound of formula VIII may be isolated and/or recrystallized from a suitable solvent by conventional methods, as described hereinabove, and then used in the next step.
- the solvent used for isolating and/or recrystallizing the N-[2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethyl]acetamide, of formula VIII is selected from the group as described hereinabove for such purpose.
- Exemplary reagents suitable for facilitating the aromatization reaction in step-(d) include, but are not limited to, sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, hydrogenation catalysts such as palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, and the like; quinone derivatives such as 2,3,5,6-tetrachlorocyclohexa-2,5rdiene-l,4-dione (p-Chloranil), p- bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6- dicyano benzoquinone, 2,3- dicyano-4-chlorobenzoquinone, 2,3-dicyanobenzoquinone; and other quinone derivatives such as 1 ,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o
- aromatization reagents are sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ) and raney nickel; and most specifically sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil).
- step-(d) is advantageously carried out by using cheaper aromatization reagent sulfur in the absence of a solvent.
- step-(d) The aromatization reaction in step-(d) is carried out in the presence or absence of a solvent.
- the aromatization in step-(d) is carried out as a neat reaction in the absence of a solvent. In another embodiment, the aromatization in step-(d) is optionally carried out in the presence of a third solvent selected from the group as described hereinbefore.
- Exemplary third solvents suitable for facilitating the aromatization in step-(d) include, but are not limited to, water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
- the third solvent is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, ⁇ , ⁇ -dimethylacetamide, dimethylsulfoxide, and mixtures thereof; and a most specific third solvent is toluene.
- the aromatization in step-(d) is carried out as a neat reaction, in the absence of solvents, by heating the contents at a temperature' of about 160°C to about 200°C, and specifically at a temperature of about 180°C to about 190°C.
- the reaction time may vary between about 30 minutes to about 5 hours, and most specifically about 1 hour to about 2 hours.
- the aromatization in step-(d) is carried out in the presence of the third solvent at a temperature of about 20°C to the reflux temperature of the solvent used, specifically at a temperature of about 50°C to the reflux temperature of the solvent used, and more specifically at about 60°C to about 120°C.
- the reaction time may vary between about 3 hours to about 30 hours, and most specifically about 5 hours to about 25 hours.
- reaction mass containing the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI or a salt thereof obtained in step-(d) may subjected to usual work up methods as described above.
- the reaction mass may be used directly in the next step to produce the agomelatine of formula I or the compound of formula VI may be isolated and/or recrystallized and then used in the next step.
- the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI obtained in step-(d) is isolated in the form of a solid.
- the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI is isolated and/or re-crystallized from a suitable solvent by the methods as described hereinabove.
- the solvent used for isolating and/or recrystallizing the pure N-[2-(7-Hydroxy-l- naphthyl)ethyl]acetamide of formula VI is selected from the group as described above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- methylating agents used in step-(e) include, but are not limited to, dimethyl sulfate, methyl iodide, and the like.
- a most specific methylating agent is dimethyl sulfate.
- the reaction in step-(e) is optionally carried out in the presence of a base.
- the base is an organic or inorganic base, and most specifically an inorganic base, selected from the group as described hereinabove.
- Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically potassium carbonate.
- the methylation in step-(e) is carried out in the presence of a fifth solvent.
- Exemplary fifth solvents used in step-(e) include, but are not limited to, water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
- the fifth solvent used in step-(e) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-hutyLether ⁇ monogLym ⁇ ⁇ , ⁇ -dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- Most specific fifth solvents are water, acetone, and mixtures thereof.
- the reaction in step-(e) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used.
- the reaction time may vary between about 30 minutes to about 6 hours, and specifically about 3 hours to about 5 hours.
- the reaction mass containing the agomelatine of formula I obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
- the solvent used for isolating/recrystallizing the pure agomelatine of formula I is selected from the group as described herein above.
- the reaction in step-(a) is carried out in the presence or absence of a solvent.
- the reaction in step-(a) is carried out in the presence of a first solvent selected from the group as described above.
- the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; more specifically the first solvent is toluene or xylene; and a most specific solvent is toluene.
- reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine.
- the carboxylic acid and the organic amine compounds are employed in catalytic amounts.
- Exemplary carboxylic acids used in step-(a) include, but are not limited to, a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid.
- Exemplary organic amines used in step-(a) include, but are not limited to, a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine.
- the carboxylic acid used in step-(a) is an unsubstituted or substituted aryl carboxylic acid; and a most specific carboxylic acid is 4-hydroxybenzoic acid.
- the organic amine used in step-(a) is an unsubstituted or substituted linear or branched aralkyl amine; more specifically the organic amine is morpholine or benzyl amine; and a most specific organic amine is benzyl amine.
- the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitriIe of formula III is isolated in the form of a solid.
- the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated and/or re-crystallized from a suitable solvent by the methods described hereinabove.
- the solvent used for isolating and/or recrystallizing the pure (7-hydroxy-3,4- dihydro-l-naphthalenyl)acetonitrile of formula III is selected from the group as described hereinabove.
- Exemplary reagents suitable for facilitating the aromatization reaction in step-(b) include, but are not limited to, sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, hydrogenation catalysts such as palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, and the like; quinone derivatives such as 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), p- bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6- dicyano benzoquinone, 2,3- dicyano-4-chlorobenzoquinone, 2,3-dicyanobenzpquinone; and other quinone derivatives such as 1,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bro
- aromatization reagents are sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ) and raney nickel; and most specifically sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil).
- step-(b) is advantageously carried out by using cheaper aromatization reagent sulfur in the absence of a solvent.
- step-(b) The aromatization reaction in step-(b) is carried out in the presence or absence of a solvent.
- the aromatization in step-(b) is carried out as a neat reaction in _the_absence_of.a-sol-vent In another embodiment, the aromatization in step-(b) is optionally carried out in the presence of a third solvent selected from the group as described hereinbefore.
- Exemplary third solvents suitable for facilitating the aromatization in step-(b) include, but are not limited to, water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
- the third solvent is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, ⁇ , ⁇ -dimethylacetamide, dimethylsulfoxide, and mixtures thereof; and a most specific third solvent is toluene.
- the aromatization in step-(b) is carried out as a neat reaction, in the absence of solvents, by heating the contents at a temperature of about 160°C to about 200°C, and specifically at a temperature of about 180°C to about 190°C.
- the reaction time may vary between about 30 minutes to about 5 hours, and most specifically about 1 hour to about 2 hours.
- the aromatization in step-(b) is carried out in the presence of the third solvent at a temperature of about 20°C to the reflux temperature of the solvent used, specifically at a temperature of about 50°C to the reflux temperature of the solvent used, and more specifically at about 60°C to about 120°C.
- the reaction time may vary between about 3 hours to about 30 hours, and most specifically about 5 hours to about 25 hours.
- reaction mass containing the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII or a salt thereof obtained in step-(b) may subjected to usual work up methods as described above.
- the reaction mass may be used directly in the next step to produce the 2- (7-hydroxy-l-naphthyl)ethanamine of formula V or the compound of formula VII may be isolated and/or recrystallized and then used in the next step.
- the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII obtained in step-(b) is isolated in the form of a solid.
- the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII is isolated and/or re-crystallized from a suitable solvent by the methods as described hereinabove.
- the solvent used for isolating and/or recrystallizing the pure (7 -hydroxy- 1- naphthyl)acetonitrile, of formula VII is selected from the group as described above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- step-(c) is carried out in the presence of a second solvent selected from the group as described above.
- the second solvent used in step-(c) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; more specifically, the second solvent is selected from the group consisting of methanol, ethanol, isopropanol, and mixtures thereof; and a most specific second solvent is methanol,
- Exemplary reducing agents used in step-(c) include, but are not limited to, hydrogenation catalysts (metal catalysts) such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like; hydride agents such as lithium aluminum hydride, sodium borohydride/Lewis acid, lithium ⁇ borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2-methoxyethoxy)aluminium hydride (Vitride), and the like; and other reducing agents such as borane, borane-THF complex, and the like.
- metal catalysts such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like
- hydride agents such as lithium aluminum hydr
- step-(c) is advantageously and efficiently carried out by employing the less expensive hydride agents such as sodium borohydride whenjhej-eactiojLis ⁇ performed- in-the-presenee-of-a-bewis ' acid: he " u " se ⁇ " of hydride agents in the presence of Lewis acid in the reduction process leads to the resulting product with high purity and in good yield.
- the less expensive hydride agents such as sodium borohydride whenjhej-eactiojLis ⁇ performed- in-the-presenee-of-a-bewis ' acid: he " u " se ⁇ " of hydride agents in the presence of Lewis acid in the reduction process leads to the resulting product with high purity and in good yield.
- Exemplary Lewis acids used in step-(c) include, but are not limited to, aluminium chloride, calcium chloride, boron triflouride and zinc chloride, nickel chloride, and the like. Specific Lewis acids are aluminium chloride and nickel chloride.
- step-(c) may be carried out in the presence or absence of hydrogen gas.
- a most specific reducing agent used in step-(c) is Raney-Nickel.
- step-(c) is optionally carried out in the presence of ammonia as per the methods described hereinbefore.
- the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V obtained in step-(c) is converted into its acid addition salts by treatment with a suitable acid selected from the group described hereinbefore.
- a suitable acid selected from the group described hereinbefore.
- Most specific acids are hydrochloric acid and hydrobromic acid.
- the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof is isolated in the form of a solid.
- the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by the methods described hereinbefore.
- the solvent used for isolating and/or recrystallizing the pure 2-(7-hydroxy-l- naphthyl)ethanamine of formula V is selected from the group as described above.
- a most specific acid addition salt of 2-(7-hydroxy-l- naphthyl)ethanamine of formula V prepared by the process described herein is 2-(7- hydroxy-l-naphthyl)ethanamine hydrochloride salt.
- a most specific acid addition salt of 2-(7-hydroxy-l- naphthyl)ethanamine of formula V prepared by the process described herein is 2-(7- hydroxy- 1 -naphthyl)ethanamine hydrobromide salt.
- Exemplary acetylating agents used in step-(d) include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium acetate, and the like, or a combination thereof.
- a most specific acetylating agent is acetic anhydride.
- the reaction in step-(d) is optionally carried out in the presence of a base.
- the base is an organic or inorganic base, and most specifically an inorganic base.
- Exemplary bases include, but are not limited to, hydroxides, alkoxides, bicarbonates and carbonates of alkali or alkaline earth metals; ammonia, collidine, trimethylamine, tributylamine, triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4-(N,N-dimethylamino)pyridine and 1-alkylimidazole.
- Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically sodium hydroxide and potassium hydroxide.
- the acetylation in step-(d) is carried out in the presence of a fourth solvent selected from the group as described hereinbefore.
- Exemplary fourth solvents used in step-(d) include, but are not limited to, water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
- the fourth solvent used in step-(d) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, ⁇ ⁇ 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, ⁇ , ⁇ -dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroe
- the reaction in step-(d) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used.
- the reaction time may vary between about 20 minutes to about 3 hours, and specifically about 30 minutes to aJ?mrt 2_ho_urs.
- the reaction mass containing the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI obtained in step-(d) may be subjected to usual work up methods as described above.
- the reaction mass may be used directly in the next step to produce the agomelatine of formula I, or the compound of formula VI may be isolated and/or recrystallized from a suitable solvent by conventional methods, as described hereinabove, and then used in the next step.
- the solvent used for isolating and/or recrystallizing the N-[2-(7-Hydroxy-l- naphthyl)ethyl]acetamide of formula VI is selected from the group as described hereinabove for such purpose.
- methylating agents used in step-(e) include, but are not limited to, dimethyl sulfate, methyl iodide, and the like.
- a most specific methylating agent is dimethyl sulfate.
- the reaction in step-(e) is optionally carried out in the presence of a base.
- the base is an organic or inorganic base, and most specifically an inorganic base, selected from the group as described hereinabove.
- Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically potassium carbonate.
- the methylation in step-(e) is carried out in the presence of a fifth solvent.
- Exemplary fifth solvents used in step-(e) include, but are not limited to, water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
- the fifth solvent used in step-(e) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyI tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, ⁇ , ⁇ -dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- Most specific fifth solvents are water, acetone
- the reaction in step-(e) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used.
- the reaction time may vary between about 30 minutes to about 6 hours, and specifically about 3 hours to about 5 hours.
- the reaction mass containing the agomelatine of formula I obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
- the solvent used for isolating/recrystallizing the pure agomelatine of formula I is selected from the group as described herein above.
- V disclosed herein is carried out by the methods and parameters as described hereinabove.
- the reaction in step-(a) is carried out in the presence or absence of a solvent.
- the reaction in step-(a) is carried out in the presence of a first solvent selected from the group as described above.
- the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; more specifically the first solvent is toluene or xylene; and a most specific solvent is toluene.
- reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine.
- the carboxylic acid and the organic amine compounds are employed in catalytic amounts.
- Exemplary carboxylic acids used in step-(a) include, but are not limited to, a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid.
- Exemplary organic amines used in step-(a) include, but are not limited to, a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine.
- the carboxylic acid used in step-(a) is an unsubstituted or substituted aryl carboxylic acid; and a most specific carboxylic acid is 4-hydroxybenzoic acid.
- the organic amine used in step-(a) is an unsubstituted or substituted linear or branched aralkyl amine; more specifically the organic amine is morpholine or benzyl amine; and a most specific organic amine is benzyl amine.
- the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated in the form of a solid.
- the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated and/or re-crystallized from a suitable solvent by the methods described hereinabove.
- the solvent used for isolating and/or recrystallizing the pure (7-hydroxy-3,4- dihydro-l-naphthalenyl)acetonitrile of formula III is selected from the group as described hereinabove.
- Exemplary reagents suitable for facilitating the aromatization reaction in step-(b) include, but are not limited to, sulfur or its derivatives, ' selenium metal, aqueous hydrobromic acid, hydrogenation catalysts such as palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, and the like; quinone derivatives such as 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), p- bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6- dicyano benzoquinone, 2,3- dicyano-4-chlorobenzoquinone, 2,3-dicyanobenzoquinone; and other quinone derivatives such as 1 ,2-benzoquinones, 1,3-benzoquinones, o-chloranil,
- aromatization reagents are sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano : benzoquinone (DDQ) and raney nickel; and most specifically sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil).
- step-(b) is advantageously carried out by using cheaper aromatization reagent sulfur in the absence of a solvent.
- step-(b) The aromatization reaction in step-(b) is carried out in the presence or absence of a solvent.
- the aromatization in step-(b) is carried out as a neat reaction in the gripabsencej>f a solvent.
- the aromatization in step-(b) is optionally carried out in the presence of a third solvent selected from the group as described hereinbefore.
- Exemplary third solvents suitable for facilitating the aromatization in step-(b) include, but are not limited to, water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
- the third solvent is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, ⁇ , ⁇ -dimethylacetamide, dimethylsulfoxide, and mixtures thereof; and a most specific third solvent is toluene.
- the aromatization in step-(b) is carried out as a neat reaction, in the absence of solvents, by heating the contents at a temperature of about 160°C to about 200°C, and specifically at a temperature of about 180°C to about 190°C.
- the reaction time may vary between about 30 minutes to about 5 hours, and most specifically about 1 hour to about 2 hours.
- the aromatization in step-(b) is carried out in the presence of the third solvent at a temperature of about 20°C to the reflux temperature of the solvent used, specifically at a temperature of about 50°C to the reflux temperature of the solvent used, and more specifically at about 60°C to about 120°C.
- the reaction time may vary between about 3 hours to about 30 hours, and most specifically about 5 hours to about 25 hours.
- reaction mass containing the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII or a salt thereof obtained in step-(b) may subjected to usual work up methods as described above.
- the reaction mass may be used directly in the next step to produce the 2- (7-hydroxy-l-naphthyl)ethanamine of formula V or the compound of formula VII may be isolated and/or recrystallized and then used in the next step.
- the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII obtained in step-(b) is isolated in the form of a solid.
- the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII is isolated and/or re-crystallized from a suitable solvent by the methods as described hereinabove.
- the solvent used for isolating and/or recrystallizing the pure (7-hydroxy-l- naphthyl)acetonitrile, of formula VII is selected from the group as described above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- methylating agents used in step-(c) include, but are not limited to, dimethyl sulfate, methyl iodide, and the like.
- a most specific methylating agent is dimethyl sulfate.
- the reaction in step-(c) is optionally carried out in the presence of a base.
- the base is an organic or inorganic base, and most specifically an inorganic base, selected from the group as described hereinabove.
- Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically potassium carbonate.
- the methylation in step-(c) is carried out in the presence of a fifth solvent selected from the group as described above.
- Exemplary fifth solvents used in step-(c) include, but are not limited to, water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
- the fifth solvent used in step-(c) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitnle, contextprQpionitrile,-N T N-dimethylfom ⁇ , ⁇ -dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
- Most specific fifth solvents are water,
- the reaction in step-(c) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used.
- the reaction time may vary between about 30 minutes to about 6 hours, and specifically about 3 hours to about 5 hours.
- reaction mass containing the (7-methoxy-l-naphthyl)acetonitrile of formula IX obtained in step-(c) may be subjected to usual work up methods as described above.
- the reaction mass may be used directly in the next step to produce the 2-(7-methoxy-l- naphthyl)ethanamine of formula XI, or the compound of formula IX may be isolated and/or recrystallized from a suitable solvent by conventional methods, as described hereinabove, and then used in the next step.
- the solvent used for isolating and/or recrystallizing the (7-methoxy-l- naphthyl)acetonitrile of formula IX is selected from the group as described hereinabove for such purpose.
- step-(d) is carried out in the presence of a second solvent selected from the group as described above.
- the second solvent used in step-(d) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; more specifically, the second solvent is selected from the group consisting of methanol, ethanol, isopropanol, and mixtures thereof; and a most specific second solvent is methanol,
- Exemplary reducing agents used in step-(d) include, but are not limited to, hydrogenation catalysts (metal catalysts) such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like; hydride agents such as lithium aluminum hydride, sodium borohydride/Lewis acid, lithium b ⁇ rohy ⁇ ng ⁇ ,_sodlum ⁇ cyanoboroh-ydr-ide diisobutylalum ⁇ i tri-tert-butoxyaluminum hydride, sodium bis(2-methoxyethoxy)aluminium hydride (Vitride), and the like; and other reducing agents such as borane, borane-THF complex, and the like.
- metal catalysts such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like
- hydride agents such as
- step-(d) is advantageously and efficiently carried out by employing the less expensive hydride agents such as sodium borohydride when the reaction is performed in the presence of a Lewis acid.
- hydride agents such as sodium borohydride
- the use of hydride agents in the presence of Lewis acid in the reduction process leads to the resulting product with high purity and in good yield.
- Exemplary Lewis acids used in step-(d) include, but are not limited to, aluminium chloride, calcium chloride, boron triflouride and zinc chloride, nickel chloride, and the like. Specific Lewis acids are aluminium chloride and nickel chloride.
- step-(d) may be carried out in the presence or absence of hydrogen gas.
- a most specific reducing agent used in step-(d) is Raney-Nickel.
- step-(d) is optionally carried out in the presence of ammonia as per the methods described hereinbefore.
- the 2-(7-methoxy-l-naphthyl)ethanamine of formula -XI obtained in step-(d) is converted into its acid addition salts by treatment with a suitable acid selected from the group described hereinbefore.
- a suitable acid selected from the group described hereinbefore. Most specific acids are hydrochloric acid and hydrobromic acid.
- the 2-(7-methoxy-l-naphthyl)ethanamine of formula XI or an acid addition salt thereof is isolated in the form of a solid.
- the 2-(7-methoxy-l-naphthyl)ethanamine of formula XI or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by the methods described hereinbefore.
- the solvent used for isolating and/or recrystallizing the pure 2-(7-methoxy-l- naphthyl)ethanamine of formula XI is selected from the group as described above.
- a most specific acid addition salt of 2-(7-methoxy-l- naphthyl)ethanamine of formula XI prepared by the process described herein is 2-(7- methoxy-l-naphthyl)ethanamine hydrochloride salt.
- a most specific acid addition salt of 2-(7-methoxy-l- naphthyl)ethanamine of formula XI prepared by the process described herein is 2-(7- methoxy- 1 -naphthyl)ethanamine hydrobromide salt.
- Exemplary acetylating agents used in step-(e) include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium acetate, and the like, or a combination thereof.
- acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide
- acetic anhydride sodium acetate, and the like, or a combination thereof.
- a most specific acetylating agent is acetic anhydride.
- the reaction in step-(e) is optionally carried out in the presence of a base.
- the base is an organic or inorganic base, and most specifically an inorganic base.
- Exemplary bases include, but are not limited to, hydroxides, alkoxides, bicarbonates and carbonates of alkali or alkaline earth metals; ammonia, collidine, trimethylamine, tributylamine, triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4-(N,N-dimethylamino)pyridine and 1-alkylimidazole.
- Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically sodium hydroxide and potassium hydroxide.
- the acetylation in step-(e) is carried out in the presence of a fourth solvent selected from the group as described hereinbefore.
- Exemplary fourth solvents used in step-(e) include, but are not limited to, water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
- the fourth solvent used in step-(e) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, ⁇ , ⁇ -dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichlorom£thane,-dichloroe
- the reaction in step-(e) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used.
- the reaction time may vary between about 20 minutes to about 3 hours, and specifically about 30 minutes to about 2 hours.
- the reaction mass containing the agomelatirie of formula I obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
- the solvent used for isolating/recrystallizing the pure agomelatine of formula I is selected from the group as described herein above.
- the process for the preparation of (7-methoxy-l-naphthyl)acetonitrile of formula IX disclosed herein is carried out by the methods and parameters as described hereinabove.
- the solids obtained in any of the above process steps described hereinabove may be collected by filtration, filtration under vacuum, decantation, centrifugation, filtration employing a filtration media of a silica gel or celite, or a combination thereof.
- novel hydroxyl intermediates of formulae III, IV, V, VII, VIII, XII and XIII disclosed employed for the preparation of agomelatine disclosed herein allows the product to be easily isolated and purified, thereby producing a product with 72-80% overall yield.
- the highly pure agomelatine, or a pharmaceutically acceptable salt thereof, obtained by the above processes may be further dried in, for example, a Vacuum Tray Dryer, a Rotocon Vacuum Dryer, a Vacuum Paddle Dryer or a pilot plant Rota vapor, to further lower residual solvents. Drying can be carried out under reduced pressure until the residual solvent content reduces to the desired amount such as an amount that is within the limits given by the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (“ICH”) guidelines.
- ICH International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use
- the drying is carried out at atmospheric pressure or reduced pressures, such as below about 200 mm Hg, or below about 50 mm Hg, at temperatures such as about 35°C to about 90°C, and specifically at about 50°C to about 85°C.
- the drying can be carried out for any desired time period that achieves the desired result, such as times about 1 to 20 hours. Drying may also be carried out for shorter or longer periods of time depending on the product specifications. Temperatures and pressures will be chosen based on the volatility of the solvent being used and the foregoing should be considered as only a general guidance. Drying can be suitably carried out in a tray dryer, vacuum oven, air oven, or using a fluidized bed dryer, spin flash dryer, flash dryer, and the like.
- the highly pure agomelatine or a salt thereof obtained by the process disclosed herein has a purity of greater than about 99%, specifically greater than about 99.5%, more specifically greater than about 99.9%, and most specifically greater than about 99.95% as measured by HPLC.
- the purity of the agomelatine or a salt thereof can be about 99% to about 99.95%, or about 99.5% to about 99.99%.
- Hydrobromic acid can also be used as a reagent for aromatization of 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV:
- step-(c) optionally, extracting the 2-(7-hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine of formula XII obtained in step-(c) into an organic solvent;
- the reaction in step-(a) is carried out at a temperature of about 25°C to the reflux temperature of the solvent used, specifically at a temperature of about 60°C to the reflux temperature of the solvent used ? _andjriQr.e_specifieaily-at _ theTOflu) ⁇ temperature of the solvent used.
- the reaction time may vary between about 2 hours to about 5 hours.
- reaction in step-(a) is optionally carried out in the presence of water-miscible organic solvents.
- the base used in step-(c) is an organic or inorganic base selected from the group as described above.
- the base is an inorganic base, and most specifically, the base is aqueous ammonia, sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, or a combination thereof.
- step-(c) the pH of the mother liquors in step-(c) is adjusted to above 8, and more specifically between 8 and 9.
- Exemplary organic solvents used in step-(d) include, but are not limited to, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a halogenated hydrocarbon solvent, and mixtures thereof.
- the organic solvent used in step-(d) is selected from the group consisting of ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, dichloromethane, dichloroethane, chloroform, and mixtures thereof. Most specific organic solvents are ethyl acetate, toluene, dichloromethane, and mixtures thereof.
- the reagents suitable for facilitating the aromatization reaction in step-(f) is selected from the group as described hereinbefore.
- Specific aromatization reagents are sulfur or its derivatives, selenium metal, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ) and Pd/C; and most specifically sulfur, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil) and 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ).
- the isolation in step-(b) and step-(e) is carried out by methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
- the recovery in step-(b) and step-(e) is accomplished by filtration, filtration under vacuum, decantation, centrifugation, filtration employing a filtration media of a silica gel or celite, or a combination thereof.
- Exemplary acetylating agents include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium acetate, and the like, or a combination thereof.
- acetylating agent is acetic anhydride.
- the acetylation is optionally carried out in the presence of a base selected from the group as described hereinbefore.
- the acetylation is carried out in the presence of a fourth solvent selected from the group as described hereinbefore.
- the acetylation is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used.
- the reaction mass containing the N-[2-(7-hydroxy-l,2,3,4-tetrahydro-l- naphthyl)ethyl]acetamide of formula XIII obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
- the present invention also encompasses the use of the novel compounds of formulae III, IV, IVa, IVb, V, Va, VII, VIII, XII, Xlla and XIII disclosed herein for preparing Agomelatine.
- novel compounds of formulae III, IV, IVa, IVb, V, Va, VII, VIII, XII, Xlla and XIII disclosed herein for preparing Agomelatine.
- the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrochloride (30 gm) was dissolved in methanol (150 ml), followed by the addition of sodium acetate (22 gm) and acetic anhydride (21 gm), and then heating the resulting mixture for 30 minutes at reflux temperature (65-68°C).
- 15% Sodium hydroxide solution (90 ml) was added to the reaction mass under reflux temperature, followed by maintainingJhe-resuliiag-mtxture ⁇ fbT ⁇ rio aT reflux temperature.
- the reaction mass was cooled to room temperature, followed by the addition of distilled water (150 ml) and then cooling the mass to 0-10°C.
- the pH of the reaction mass was adjusted to below 2 with hydrochloric acid, followed by extracting the mass three times with dichloromethane (3 x 50 ml).
- the dichloromethane layer was washed with distilled water (50 ml) and the resulting dichloromethane layer was distilled to produce 30 gm of N-[2-(7-hydroxy-3,4-dihydro-l-naphthyl)ethyl]acetamide (Yield: 97.7%; Melting Range: 115-120°C; Purity by HPLC: 99.3%).
- N-[2-(7-Hydroxy-3,4-dihydro-l-naphthyl)ethyl]acetamide (5 gm) and sulfur (15 gm) were heated to 185-190°C for about 1-2 hours.
- the reaction mass was cooled to 55-60°C, followed by the addition of methanol (500 ml) and then refluxing for 1 hour.
- step-1 The filtrate (mother liquors) obtained in step-1 was neutralized with 20% sodium hydroxide solution, followed by adjusting the pH to 8.9 with aqueous ammonia solution.
- the resulting aqueous layer was extracted into dichloromethane (3 x 150 ml) and the dichloromethane layer was washed with water (100 ml).
- the solvent was distilled off from the resulting organic layer to produce crude solid (20 gm).
- the resulting solid was taken in ethyl acetate (100 ml), followed by adjusting the pH to below 2.0 with isopropanolic-HCl solution at room temperature.
- the obtained clear solution was distilled under vacuum to produce 20 gm of the titled compound as hydrochloride salt (Purity by HPLC: 99.4%).
- Step-3 Preparation of (7-Hydroxy-l-naphthyl)ethanamine hydrochloride salt
- N-[2-(7-hydroxy-l ,2,3,4-tetrahydro-l -naphthyl)ethyl]acetamide (10 gm) was dissolved in toluene (100 ml), followed by the addition of Pd/C (2.5 gm). The resulting mixture was heated to reflux temperature and then maintained for 20 hours. After completion of the reaction, the reaction mass was filtered and the filtrate was distilled under vacuum to produce 9.1 gm of pure N-[2-(7-Hydroxy-l -naphthyl)ethyl]acetamide.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Provided herein are novel, commercially viable and industrially advantageous processes for the preparation of Agomelatine or a salt thereof, in high yield and purity, using novel intermediates.
Description
PROCESSES FOR THE PREPARATION OF AGOMELATINE USING NOVEL
INTERMEDIATES
FIELD OF THE INVENTION
The present invention relates to novel, commercially viable and industrially advantageous processes for the preparation of agomelatine or a salt thereof, in high yield and purity, using novel intermediates.
BACKGROUND OF THE INVENTION
U.S. Patent No. 5,225,442 (hereinafter referred to as the '442 patent) discloses 1- alkoxy-2-(acylaminoethyl)naphthalene derivatives, processes for their preparation, pharmaceutical compositions comprising the derivatives, and method of use thereof. These compounds possess valuable pharmacological properties with regard to the central nervous system, particularly anxiolytic, antipsychotic and analgesic properties, and with regard to ovulation, cerebral circulation and immunomodulation. Among them, Agomelatine, chemically named N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide, is an important antidepressant and useful for the treatment of major depressive episodes in adults. Agomelatine has dual pharmacological effects, which is not only the agonist of melatonergic system receptor, but also the antagonist of 5HT2c receptor. Agomelatine is represented by the following structural formula I:

Agomelatine is marketed by Servier (Ireland) Industries Ltd. in Europe under the brand names VALDOXAN® and THYMANAX®, and it is orally administered as tablets containing 25 mg of agomelatine.
The synthesis of agomelatine was first described in the '442 patent and its corresponding European equivalent Patent No. EP 0447285 Bl . Various pjOcesses_fot-the---
preparation of agomelatine, its intermediates, and related compounds are described in U.S. Patent Nos. US 5,420,158, US 7,476,751, US 7,544,839, US 7,999,129, US 8,212,077 and US 8,143,449; PCT Publication Nos. WO 2011/054917, WO 201 1/153939, WO 2011/154140, WO2012/046253, WO2012/093402, WO2012/127483, WO 2012/093225, WO 2012/1 13999 and WO 2012/070025; and Journal of Medicinal Chemistry 1992, 35(8), 1484-1486; and Synthetic Communications 2001, 31(4), 621-629.
According to the '442 patent, N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide (Agomelatine) is prepared by a process as depicted in scheme 1 :
Scheme 1:

7- ethoxy-1 -tetrlone Et ylbromo acetate HCI , Pj05
Ethyl (7-methoxy-1 ,2,3,4-tetrahydro- 1 -na hthylidene)acetate

Ethyl (7-methoxy- 1-naphthyl)acetate (7- ethoxy-1 -naphthyl)
acetic acid

(7-Methoxy-1-naphthyl) (7-Methoxy-1 -naphthyl) (7-Methoxy-1 -naphthyl) acetyl chloride acetamide acetonitrile
Ethanolic ammonia
Raney Ni, H2
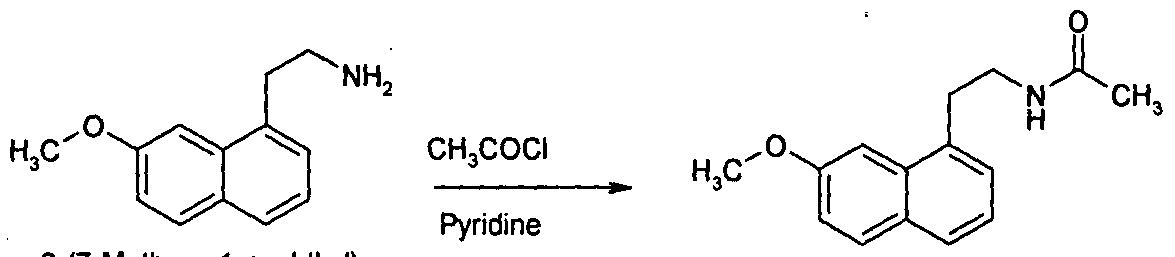
2-(7- ethoxy-1 -naphthyl) N-[2-(7-Methoxynaphth-1-yl)
ethylamine ethyrjacetamide
(Agomelatine) As per the process described in the '442 patent, agomelatine is prepared by reacting
7-methoxy-l-tetralone with ethylbromo acetate in the presence of activated zinc filings and benzene to produce ethyl (7-methoxy-l,2,3,4-tetrahydro-l-naphthylidene)acetate,_which_is-
then subjected to dehydro-aromatization with sulfur at 215°C to produce ethyl (7-methoxy- l-naphthyl)acetate, followed by hydrolysis and subsequent acyl chlorination with thionyl chloride in chloroform to produce (7-methoxy-l-naphthyl)acetyl chloride, and then ammonification with aqueous ammonia to produce (7-methoxy-l-naphthyl)acetamide, which is then reacted with triflic anhydride in the presence of triethylamine to produce (7- methoxy-l-naphthyl)acetonitrile, followed by reduction with Raney Nickel under hydrogen pressure to produce 2-(7-methoxy-l-naphthyl)ethyl amine, which is finally acetylated with acetyl chloride in pyridine to produce agomelatine.
The process for the preparation of agomelatine disclosed in the '442 patent suffers from various disadvantages such as the use of highly corrosive and toxic reagents like thionyl chloride, benzene, chloroform and pyridine; and involves the use of tedious and cumbersome procedures since it requires eight steps to synthesize agomelatine, thereby rendering the overall yield being less than 30% and making the process commercially unfeasible. Agomelatine obtained by the process described in the '442 patent does not have the satisfactory purity for pharmaceutical use. Unacceptable amounts of impurities are generally formed along with agomelatine. Most of the methoxy intermediates compounds, obtained according to the process described in the '442 patent, exist in the form of residues, which are not isolated as solids, and thus leading to the formation of unacceptable amounts of impurities which are persistent impurities and cannot be removed at final stage.
According to U.S. Patent No. 7,544,839 (hereinafter referred to as the '839 patent),
Agomelatine is prepared by a process as depicted in scheme 2:
Scheme 2:

The process described in the '839 patent requires the use of expensive noble metal catalysts like Palladium on carbon and hazardous reagents like allyl methacrylate, which causes a lot of environmental pollution, for dehydrogenation of (7-methoxy-3,4-dihydro-l- naphthalenyl)acetonitrile to produce (7-methoxy-l-naphthyl)acetonitrile. Moreover, this step of dehydrogenation is difficult to reproduce and the yield of the resulting amine compound is very low.
Furthermore, the methoxy intermediates obtained according to the process described in the '839 patent suffer from disadvantages since these methoxy intermediates are characterized by having low melting points. For example, the reported melting point for (7-methoxy-3,4-dihydro-l-naphthalenyl)acetonitrile is 48-50°C, and the reported melting point for (7-methoxy-i-naphthyl)acetonitrile is 83°C. Hence, the known methoxy intermediate compounds are not stable and they decompose at higher temperatures due to their low melting points, for example, when sulfur (which is a cheaper aromatizing agent) is employed as reagent for aromatization of (7-methoxy-3,4-dihydro-l- naphthalenyl)acetonitrile (this reaction requires heating the reactants at higher temperature 180-200°C) to produce (7-methoxy-l-naphthyl)acetonitrile, thus leading to the formation of unacceptable amounts of impurities, thereby decreasing the yield and purity of the product. Hence, the aromatization of (7-methoxy-3,4-dihydro-l-naphthalenyl)acetonitrile must be carried out at low temperatures and requires the use of expensive noble metal catalysts like palladium on carbon and additional hazardous reagents like allyl methacrylate. Therefore, the cheaper aromatizing agents like sulfur cannot be used for aromatization of known methoxy intennedjate _compounds The— time— periods—for
completion of the reaction between 7-methoxy-l-tetralone and cyanoacetic acid takes around 30 hours at reflux temperature.
According to U.S. Patent No. 8,212,077 (hereinafter referred to as the Ό77 patent), Agomelatine is prepared by a process as depicted in scheme 3:
Scheme 3:

2-(7-Methoxy-1 -naphthyl) Agomelatine
ethanamine
According to PCT Publication No. WO 2012/046253 (hereinafter referred to as the '253 application), agomelatine is prepared by reacting 7-methoxy-l-tetralone with acetonitrile in the presence of sodium hexamethyldisilazide in tetrahydrofuran to provide 2-(l-hydroxy-7-methoxy-l,2,3,4-tetrahydronaphthalen-l-yl)acetonitrile, which is further crystallized from cyclohexane to produce pure compound, which is then treated with hydrogen gas in the presence of Raney Ni, in aqueous methanol in the presence of ammonia at 45-60°C to provide 2-(7-methoxy-3,4-dihydronaphthalen-l-yl)ethanamine, followed by treatment with hydrochloric acid in ethylacetate-to-produee-its-hydrochloride^
salt, which is then acetylated with acetyl chloride in the presence of potassium carbonate in aqueous ethyl acetate to produce N-(2-(7-methoxy-3,4-dihydronaphthalen-l- yl)ethyl)acetamide, isolating the compound as a solid from cyclohexane, followed by aromatizing the compound with DDQ in dichloromethane to produce agomelatine.
According to PCT Publication No. WO 2011/154140 (hereinafter referred to as the ' 140 application), Agomelatine is prepared by a process as depicted in scheme 4:
Scheme 4:
MeO. -A, Sodium hydride
Diethyl cyanomethyl

tetralone Ph°sPn°na>e NaBH<
Tetrahydrofuran 7-Methoxy-1,2,3,4-tetrahydro Methanol 1 -(2-Aminoethyl)-7-methoxy- naphthylidene acetonitrile Q0nc 1 ,2,3,4-tetrahydro naphthalene
Acetic anhydride
Methanol

naphthalene
According to U.S. Patent No. 8,143,449 (hereinafter referred to as the '449 patent), Agomelatine is prepared by a process as depicted in scheme 5:
Scheme 5:

3-Methoxyacenaphtho
(7-Methoxy-1 -naphthyl) Acetonitrile
quinone
(oxo)acetic acid

2-(7-Methoxy-1 -naphthyl)- ethanamine Agomelatine 2-oxoacetamide
According to Journal of Medicinal Chemistry 1992, 35(8), 1484-1486 (hereinafter referred to as the 'JMC article'), Agomelatine is prepared by a process as depicted in scheme 6:
Scheme 6:

(7-Methoxy-1-Naphthyl) (7-Methoxy-1-Naphthyl) (7-Methoxy-1-Naphthyl) acetic acid acetamide acetonitrile
LiAIH4, Ether a) Hj, Raney Ni
b) HCI, Ether

The processes for the preparation of N-[2-(7-methoxy-l- naphthalenyl)ethyl]acetamide (Agomelatine) described in the aforementioned prior art suffer from several disadvantages such as the use of highly flammable, corrosive and hazardous reagents like thionyl chloride, pyridine, triflic anhydride, allyl methacrylate, sodium hydride, sodamide, n-butyl lithium and lithium aluminium hydride; use of expensive reagents such as diethyl cyanomethyl phosphonate, 2,6-di-tert-butyl-4-methyl- pyridine, N-vinylphthalimide, Palladium tetrakis(triphenylphosphine), hydrogenation catalysts such as Pd/C, acrylamide, Neocuproine hydrate, 18-crown-6-ether, propylphosphonic anhydride; use of additional and excessive amounts of reagents; use of multiple solvents and in excess amounts; use of highly flammable and/or hazardous solvents like benzene, pyridine, chloroform and dimethylsulfoxide; and involve the use of tedious and cumbersome procedures like prolonged reaction time periods, very low temperature conditions, multiple process steps, column chromatographic purifications, multiple isolations/ re-crystallizations, and thus resulting in a poor product yield and quality Methods involving column chromatographic plifificafions are generally-
undesirable for large-scale operations, thereby making the process commercially unfeasible.
The major drawback of the processes for the preparation of agomelatine described in the aforementioned prior art is that the processes involve the use of highly flammable, corrosive and hazardous reagents like thionyl chloride, pyridine, triflic anhydride, allyl methacrylate, sodium hydride, sodamide, n-butyl lithium and lithium aluminium hydride, thereby requiring very strict control of reaction conditions at low temperatures. Handling of these reagents is very difficult at lab scale and in commercial scale operations. Moreover, the yields and purities of the product obtained according to the prior art processes are low to moderate.
The processes for the preparation of Agomelatine, N-[2-(7-methoxy-l- naphthalenyl)ethyl]acetamide, of formula I described in the above mentioned prior art have the following disadvantages and limitations:
a) the melting points of the known methoxy intermediate compounds are low;
b) the methoxy intermediates decompose at higher temperatures due to their low melting points, hence, the aromatization of methoxy intermediates such as (7-methoxy-3,4- dihydro- 1 -naphthalenyl)acetonitrile and N-[2-(7-methoxy-3 ,4-dihydronaphthalen- 1 - yl)ethyl]acetamide must be carried out at low temperatures and requires the use of expensive noble metal catalysts like palladium on carbon and additional hazardous reagents like allyl methacrylate, and expensive reagents like DDQ;
c) the cheaper aromatizing agents like sulfur cannot be employed for aromatization of known methoxy intermediate compounds;
d) the reaction between 7-methoxy-l-tetralone and cyanoacetic acid takes around 30 hours at reflux temperature for reaction completion;
e) most of the known methoxy intermediates compounds exist in the form of residues, which are not isolated as solids, and thus leading to the formation of impurities which are persistent impurities, thereby requiring additional purification steps which effects the yield of the product;
f) longer reaction times, low yields and low purities of the products;
g) the processes involve the use of highly flammable, corrosive and hazardous reagents like thionyl chloride, pyridine, trifilc anhydride, allyl methacrylate, sodium hydride, sodamide, n-butyl lithium and lithium alumimium hydride;
h) the processes involve the use of expensive reagents such as diethyl cyanomethyl phosphonate, 2,6-di-tert-butyl-4-methyl-pyridine, N-vinylphthalimide, Palladium tetrakis(triphenylphosphine), hydrogenation catalysts such as Pd/C, acrylamide, Neocuproine hydrate, 18-crown-6-ether, propylphosphonic anhydride;
i) the processes involve the use of highly flammable and hazardous solvents like benzene, pyridine, chloroform arid dimethylsulfoxide;
j) the processes involve the use of tedious and cumbersome procedures like prolonged reaction time periods, very low temperature conditions, multiple process steps, column chromatographic purifications, multiple isolations/ re-crystallizations;
k) the overall processes generate a large quantity of chemical waste which is difficult to treat.
Based on the aforementioned drawbacks, the prior art processes have been found to be unsuitable for the preparation of N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide (Agomelatine) at lab scale and in commercial scale operations.
A need remains for an improved, commercially viable and environmentally friendly process of preparing Agomelatine with high yield and purity, to resolve the problems associated with the processes described in the prior art, and that will be suitable for large- scale preparation. Desirable process properties include non-hazardous conditions, environmentally friendly and easy to handle reagents, reduced cost, greater simplicity, increased purity, and increased yield of the product, thereby enabling the production of Agomelatine, in high purity and with high yield.
SUMMARY OF THE INVENTION
The present inventors have surprisingly and unexpectedly found that Agomelatine, N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide, of formula I or a salt thereof can be prepared in high purity and with high yield by reacting 7-hydroxy-l-tetralone with cyanoacetic acid in the presence of a suitable reagent to produce (7-hydroxy-3,4-dihydro- l-naphthalenyl)acetonitrile or a salt thereof, which is_then_subjected-to-reduction^ ¥itir
suitable reducing agent to produce 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine or a salt thereof, followed by aromatization with a suitable reagent to produce 2-(7- hydroxy-l-naphthyl)ethanamine or an acid addition salt thereof, which is then subjected to acetylation with a suitable acetylating agent to produce N-[2-(7-hydroxy-l- naphthyl)ethyl]acetamide, followed by reaction with a methylating agent to produce agomelatine.
In one aspect, provided herein are efficient, industrially advantageous and environmentally friendly processes for the preparation of agomelatine in high yield and with high purity using novel intermediates. The processes disclosed herein avoid the tedious and cumbersome procedures of the prior processes, thereby resolving the problems associated with the processes described in the prior art, which are more convenient to operate at lab scale and in commercial scale operations.
In another aspect, provided herein is a novel intermediate compound, (7-hydroxy- 3,4-dihydro-l-naphthalenyl)acetonitrile, of formula III:

In another aspect, provided herein is a novel intermediate compound, 2-(7- hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine, of formula IV:


In another aspect, provided herein is a novel intermediate compound, (7-hydroxy- l-naphthyl)acetonitrile, of formula VII:

or a salt thereof.
In another aspect, provided herein is a novel intermediate compound, N-[2-(7- hydroxy-3,4-dihydro-l-naphthalenyl)ethyl]acetamide, of formula VIII:

In another aspect, provided herein is a novel intermediate compound, 2-(7- hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine, of formula XII:

In another aspect, provided herein is a novel intermediate compound, N-[2-(7- hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethyl]acetamide, of formula XIII:

or a salt thereof.
In another aspect, the present invention also encompasses the use of the novel compounds of formulae III, IV, V, VII, VIII, XII and XIII disclosed herein for preparing Agomelatine.
The process for the preparation of Agomelatine disclosed herein have the following advantages over the processes described in the prior art:
i) the processes involve the use of novel hydroxy intermediates of formulae III, IV, V, . VII, VIII, XII and XIII;
ii) the novel hydroxy intermediate compounds are characterized by having higher melting points when compared with that of the known methoxy intermediates;
iii) the processes involve the use of less expensive and/or cheaper reagents;
iv) the novel hydroxy intermediate compounds are isolated as solid state forms in substantially pure form;
v) the reaction between 7-hydroxy-l-tetralone and cyanoacetic acid takes around 10 hours for completion of the reaction;
vi) the overall processes involve a reduced number of process steps and shorter reactions times;
vii) the processes avoid the use of highly flammable, corrosive and hazardous reagents like thionyl chloride, pyridine, triflic anhydride, allyl methacrylate, sodium hydride, sodamide, n-butyl lithium and lithium aluminium hydride;
viii) the processes avoid the use of highly toxic or hazardous chemicals like benzene, pyridine and chloroform;
ix) the processes avoid the use of tedious and cumbersome procedures like prolonged reaction time periods, very low temperatures, multiple process steps, column chromatographic purifications, multiple isolations, use of additional and excess amounts of solvents;
x) the processes avoid the use of expensive reagents such as diethyl cyanomethyl phosphonate, 2,6-di-tert-butyl-4-methyl-pyridine, N-vinylphthalimide, Palladium tetrakis(triphenylphosphine), hydrogenation catalysts such as Pd/C, acrylamide, Neocuproine hydrate, 18-crown-6-ether, propylphosphonic anhydride;
xi) the processes involve easy work-up methods and simple isolation processes, and there is a reduction in chemical waste;
xii) the purity of the product is increased without additional purifications; and
xiii) the overall yield of the product is increased.
DETAILED DESCRIPTION OF THE INVENTION
According to one aspect, there is provided a novel process for the preparation of Agomelatine, N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide, of formula I:

a) reacting 7-hydroxy- 1 -tetralone of formula II:


or a salt thereof;
b) reducing the compound of formula III with a suitable reducing agent to produce 2-(7- hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV:

c) subjecting the compound of formula IV or an acid addition salt thereof to aromatization by reacting with a suitable reagent to produce 2-(7-hydroxy-l-naphthyl)ethanamine of formula V:
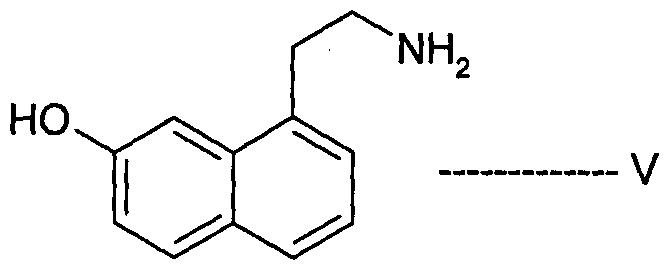
d) acetylating the compound of formula V with a suitable acetylating agent to produce N- [2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI:

e) reacting the compound of formula VI with a methylating agent to produce the agomelatine of formula I.
The compounds of formulae III, IV, V, VII, VIII, XII and XIII disclosed herein are novel and constitute another aspect of the present invention.
The use of the novel intermediate compounds of formulae III, IV, V, VII, VIII, XII and XIII, or a salt thereof, in the preparation of agomelatine of formula I is novel and forms further aspect of the present invention.
Advantageously, the novel intermediate compounds of Agomelatine disclosed herein are obtained as solid state forms in substantially pure form.
The term "substantially pure" as used herein refers to the solid state form of agomelatine intermediates, disclosed herein, having a purity of greater than about 97 wt%, specifically greater than about 98 wt%, more specifically greater than about 99 wt%, and still more specifically greater than about 99.5 wt%. The purity is preferably measured by High Performance Liquid Chromatography (HPLC). For example, the purity of solid state form of agomelatine intermediates obtained by the processes disclosed herein can be about 97% to about 99.5%, or about 98% to about 99.9%, as measured by HPLC.
Unless otherwise specified, the term 'salt' as used herein may include acid addition salts and base addition salts.
Acid addition salts, as used herein, include the salts that are derived from organic and inorganic acids. For example, the acid addition salts are derived from a therapeutically acceptable acid such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, oxalic acid, acetic acid, propionic acid, phosphoric acid, succinic acid, maleic acid, fumaric acid, citric acid, glutaric acid, tartaric acid, benzenesulfonic acid, toluenesulfonic acid, di- p-toluoyl-L-(+)-tartaric acid, malic acid, ascorbic acid, and the like.
Base addition salts may be derived from an organic or an inorganic base. For example, the base addition salts are derived from alkali or alkaline earth metals such as sodium, calcium, potassium and magnesium; ammonium salt, organic amines such as ethylamine, tert-butylamine, diethylamine, diisopropylamine, and the like.
Exemplary acid addition salts of the compounds of formulae IV and V include, but are not limited to, hydrochloride, hydrobromide,^uJphate,_nitrate,--phosphate7-acetate
propionate, oxalate, succinate, maleate, fumarate, benzenesulfonate, toluenesulfonate, citrate, tartrate, and the like. Specific acid addition salts are hydrochloride and hydrobromide, and most specifically hydrochloride salt.
Base addition salts of the compound of formula X as used herein include the salts that are derived from an organic or an inorganic base. Exemplary base addition salts of the compound of formula X include, but are not limited to, sodium salt, calcium salt, potassium salt, magnesium salt and ammonium salt.
The reaction in step-(a) is carried out in the presence or absence of a solvent. In one embodiment, the reaction in step-(a) is carried out in the presence of a first solvent. The term solvent also includes mixture of solvents.
Exemplary first solvents used in step-(a) include, but are not limited to, a hydrocarbon solvent, a halogenated hydrocarbon solvent, an ether, and mixtures thereof.
In one embodiment, the first solvent used in step-(a) is a reaction solvent that has a boiling temperature higher than or equal to that of water, and more preferably that forms an azeotrope with water.
Specifically, the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; more specifically the first solvent is toluene or xylene; and a most specific solvent is toluene.
In one embodiment, the reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine. In another embodiment, the carboxylic acid and the organic amine compounds are employed in catalytic amounts.
Exemplary carboxylic acids used in step-(a) include, but are not limited to, a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid.
Exemplary organic amines used in step-(a) include, but are not limited to, a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine.
The term "alkyl", as used herein, denotes an aliphatic hydrocarbon group which may be straight or branched having 1 to 12 carbon atoms in the chain. Preferred alkyl groups have 3 to 10 carbon atoms in the chain. The alkyLmay be substituted with one or
more "cycloalkyl groups". Exemplary alkyl groups include methyl, ethyl, n-propyl, iso- propyl, n-butyl, iso-butyl, t-butyl, n-pentyl, hexyl and heptyl.
The term "cycloalkyl", as used herein, denotes a non-aromatic mono- or multicyclic ring system of 3 to 10 carbon atoms, preferably of about 5 to about 10 carbon atoms. Exemplary monocyclic cycloalkyl groups include cyclopentyl, cyclohexyl, cycloheptyl and the like.
The term "aryl", as used herein, denotes an aromatic monocyclic or multicyclic ring system of 6 to 10 carbon atoms. The aryl is optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined herein. Exemplary aryl groups include phenyl, tolyl, naphthyl or biphenyl group.
The term "aralkyl", as used herein, denotes an aryl-alkyl group wherein the aryl and alkyl are as herein described. Preferred aralkyls contain a lower alkyl moiety. Exemplary aralkyl groups include benzyl, 2-phenethyl and naphthalenemethyl.
Specifically, the carboxylic acid used in step-(a) is an unsubstituted or substituted aryl carboxylic acid; and a most specific carboxylic acid is 4-hydroxybenzoic acid.
Specifically, the organic amine used in step-(a) is an unsubstituted or substituted linear or branched aralkyl amine; more specifically the organic amine is morpholine or benzyl amine; and a most specific organic amine is benzyl amine.
The reaction temperature and time period will ordinarily depend on the starting compounds and the solvent employed in the reaction.
In one embodiment, the reaction in step-(a) is carried out at a temperature of about 25 °C to the reflux temperature of the solvent used, specifically at a temperature of about 60°C to the reflux temperature of the solvent used, and more specifically at the reflux temperature of the solvent used. The reaction time may vary between about 10 hours to about 40 hours, and most specifically about 25 hours to about 30 hours.
In another embodiment, the cyanoacetic acid of formula X is used in a molar ratio of about 1 to 3 equivalents, specifically about .1 to 1.5 equivalents, with respect to the 7- hydroxy-l-tetralone of formula II in order to ensure a proper course of the reaction.
The reaction mass containing the (7-hydroxy-3,4-dihydro-l- naphthalenyl)acetonitrile of formula III obtained in step-(a) may subjected to usual work up such as a washing, an extraction, a pH adjustment, jm_evaporation, a layer separation,~a
decolorization, or a combination thereof. The reaction mass may be used directly in the next step to produce the amine compound of formula IV or the compound of formula III may be isolated and/or recrystallized and then used in the next step.
In one embodiment, the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated in the form of a solid.
In one embodiment, the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated and/or re-crystallized from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
The solvent used for isolating and/or recrystallizing the pure (7-hydroxy-3,4- dihydro-l-naphthalenyl)acetonitrile of formula III is selected from the group consisting of water, an alcohol, a ketone, an ether, an ester, a hydrocarbon solvent, a halogenated hydrocarbon, and mixtures thereof. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
In one embodiment, the reduction in step-(b) is carried out in the presence of a second solvent. The term solvent also includes mixture of solvents.
Exemplary second solvents used in step-(b) include, but are not limited to, water, an alcohol, an ester, a hydrocarbon solvent, an ether, and mixtures thereof.
Specifically, the second solvent used in step-(b) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; more specifically, the second solvent is selected from the group consisting of methanol, ethanol, isopropanol, and mixtures thereof; and a most specific second solvent is methanol or ethanol.
Exemplary reducing agents used in step-(b) include, but are not limited to, hydrogenation catalysts (metal catalysts) such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like; hydride agents such as lithium aluminum hydride, sodium borohydride/Lewis acid, lithium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2-methoxyethoxy)aluminium hydride (Vitride), and the like; and other reducing agents such as borane, borane-THF complex, and the like.
It has been surprisingly found that the reduction in step-(b) is advantageously and efficiently carried out by employing the less expensive hydride agents such as sodium borohydride when the reaction is performed in the presence of a Lewis acid. The use of hydride agents in the presence of Lewis acid in the reduction process leads to the resulting product with high purity and in good yield.
Exemplary Lewis acids used in step-(b) include, but are not limited to, aluminium chloride, calcium chloride, boron triflouride and zinc chloride, nickel chloride, and the like. Specific Lewis acids are aluminium chloride and nickel chloride.
The reduction in step-(b) may be carried out in the presence or absence of hydrogen gas. A most specific reducing agent used in step-(b) is Raney-Nickel.
The reduction in step-(b) is optionally carried out in the presence of ammonia. In one embodiment, ammonia used may be in the form of aqueous ammonia or in the form of ammonia gas or ammonia saturated in an organic solvent. The organic solvent used for saturating ammonia is selected from the group consisting of ethanol, methanol, isopropyl alcohol and ethyl acetate.
In one embodiment, the reduction in step-(b) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and more specifically at about 25°C to about 65°C. The reaction time may vary between about 3 hours to about 8 hours, and most specifically about 5 hours to about 6 hours.
In another embodiment, the reducing agent is used in an amount of about 5%w/w to 50%w/w, specifically about 5%w/w to 10%w/w, with respect to the (7-hydroxy-3,4-
dihydro-l-naphthalenyl)acetonitrile of formula III in order to ensure a proper course of the reaction.
The reaction mass containing the 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV obtained in step-(b) may subjected to usual work up methods as described above. The reaction mass may be used directly in the next step to produce the amine compound of formula V or the compound of formula IV may be isolated and/or recrystallized and then used in the next step.
In one embodiment, the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV obtained in step-(b) is converted into its acid addition salts by treatment with a suitable acid.
Exemplary acids suitable for forming acid addition salts include, but are not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, oxalic acid, acetic acid, propionic acid, phosphoric acid, succinic acid, maleic acid, fumaric acid, citric acid, glutaric acid, tartaric acid, benzenesulfonic acid, toluenesulfonic acid, di-p-toluoyl-L-(+)- tartaric acid, malic acid, ascorbic acid, and the like. Most specific acids are hydrochloric acid and hydrobromic acid.
For example, hydrochloric acid used may be in the form of aqueous hydrochloric acid or in the form of hydrogen chloride gas or hydrogen chloride dissolved in an organic solvent. The organic solvent used for dissolving hydrogen chloride gas or hydrogen chloride is selected from the group consisting of ethanol, methanol, isopropyl alcohol, ethyl acetate, diethyl ether, dimethyl ether and acetone.
The treatment of 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV with a suitable acid is carried out in a solvent selected from the group consisting of water, an ester, an alcohol, a ketone, a chlorinated hydrocarbon, a hydrocarbon, an ether, and mixtures thereof. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2- methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof; and most specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, ethyl acetate, and mixtures thereof.
The treatment of 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV with a suitable acid is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, and most specifically at a temperature of about 20°C to the reflux temperature of the solvent used.
In one embodiment, the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof is isolated in the form of a solid.
In one embodiment, the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
The solvent used for isolating and/or recrystallizing the pure 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV is selected from the group as described above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-peritane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
In another embodiment, a most specific acid addition salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV prepared by the process described herein is 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrochloride salt.
In another embodiment, a most specific acid addition salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV prepared by the process described herein is 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrobromide salt.
Exemplary reagents suitable for facilitating the aromatization reaction in step-(c) include, but are not limited to, sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, metal catalysts such as palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, and the like; quinone derivatives such as 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), p-bromanil, p-floranil, 2,3- dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6-dicyano benzoquinone, 2,3- dicyano-4-chlorobenzoquinone, 2,3-dicyanobenzoquinone; and other^quinone derfvatives^
such as 1,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil, o-floranil, and the like.
Specific aromatization reagents are sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ) and raney nickel; and most specifically sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil).
Specifically, the starting material 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV, in step-(c), is employed in the form of an acid addition salt, and most specifically in the form of its hydrochloride salt.
It has been surprisingly and unexpectedly found that the novel hydroxy intermediate compounds of formulae III, IV, V, VII and VIII, and their salts, disclosed herein, are characterized by having higher melting points when compared with that of the known methoxy intermediates. For example, the hydrochloride salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV, obtained by the processes disclosed, is characterized by having melting range at about 195-200°C, whereas the corresponding methoxy analogue, i.e., the hydrochloride salt of 2-(7-methoxy-3,4-dihydro-l- naphthalenyl)ethanamine is characterized by having melting point at about 151.12°C. This high melting range of the novel hydroxy intermediate compounds disclosed herein is advantageous since this property makes these compounds stable even at higher temperature (e.g. 185-190°C), thereby making it possible to advantageously employ the cheaper aromatization reagents such as sulfur for aromatizing the respective dihydro intermediates (e.g., the compound of formula IV) at higher temperature, for example, at 185-190°C. Whereas, the corresponding methoxy intermediates decompose at such higher temperatures due to their low melting points when aromatizing with sulfur thereby effecting purity and yield of the resulting products.
Therefore, the aromatization reaction in step-(c) is advantageously carried out by using cheaper aromatization reagent sulfur in the absence of a solvent.
The aromatization reaction in step-(c) is carried out in the presence or absence of a solvent.
In one embodiment, the aromatization in step-(c) is carried out as a neat reaction in the absence of a solvent.
In another embodiment, the aromatization in step-(c) is optionally carried out in the presence of a third solvent. The term solvent also includes mixture of solvents.
Exemplary third solvents suitable for facilitating the aromatization in step-(c) include, but are not limited to, water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
Specifically, the third solvent is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, Ν,Ν-dimethylacetamide, dimethylsulfoxide, and mixtures thereof; and a most specific third solvent is toluene.
In one embodiment, the aromatization in step-(c) is carried out as a neat reaction, in the absence of solvents, by heating the contents at a temperature of about 160°C to about 200°C, and specifically at a temperature of about 180°C to about 190°C. The reaction time may vary between about 30 minutes to about 5 hours, and most specifically about 1 hour to about 2 hours.
In another embodiment, the aromatization in step-(c) is carried out in the presenceof the third solvent at a temperature of about 20°C to the reflux temperature of the solvent used, specifically at a temperature of about 50°C to the reflux temperature of the solvent used, and more specifically at about 60°C to about 120°C. The reaction time may vary between about 3 hours to about 30 hours, and most specifically about 5 hours to about 25 hours.
In another embodiment, the metal catalysts suitable for facilitating aromatization is used in a amount of about 5%w/w to about 50%w/w, specifically about 5%w/w to 20%w/w, with respect to the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof in order to ensure a proper course of the reaction.
In another embodiment, the sulfur is used in a molar ratio of about 1 to 5 equivalents, specifically about 1 to 2 equivalents, with respect to the 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof in order to ensure a proper course of the reaction.
In another embodiment, the 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p- chloranil) is used in a molar ratio of about 1 to 5 equivalents, specifically about 1 to 2 equivalents, with respect to the 2-(7-hydroxy-3,4-dihydro-l-naphthalehyl)ethanamine of formula IV or an acid addition salt thereof in order to ensure a proper course of the reaction.
In another embodiment, the aqueous hydrobromic acid is used in an amount of about 1 to 5 times, specifically about 1 to 2 times, with respect to the 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof in order to ensure a proper course of the reaction.
The reaction mass containing the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof obtained in step-(c) may subjected to usual work up methods as described above. The reaction mass may be used directly in the next step to produce the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI or the compound of formula V may be isolated and/or recrystallized and then used in the next step.
In one embodiment, the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V obtained in step-(c) is converted into its acid addition salts by treatment with a suitable acid as per the methods described hereinabove.
In another embodiment, the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof obtained in step-(c) is isolated in the form of a solid.
In another embodiment, the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by the methods as described hereinabove.
The solvent used for isolating and/or recrystallizing the pure 2-(7-hydroxy-l- naphthyl)ethanamine of formula V or an acid addition salt thereof is selected from the group as described above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2- methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyXejher,„ethyTaGetate n-penfane
n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
In another embodiment, a most specific acid addition salt of 2-(7-hydroxy-l- naphthyl)ethanamine of formula V prepared by the process described herein is 2-(7- hydroxy- l-naphthyl)ethanamine hydrochloride salt.
In another embodiment, a most specific acid addition salt of 2-(7-hydroxy-l- naphthyl)ethanamine of formula V prepared by the process described herein is 2-(7- hydroxy-l-naphthyl)ethanamine hydrobromide salt.
Exemplary acetylating agents used in step-(d) include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium acetate, and the like, or a combination thereof. A most specific acetylating agent is acetic anhydride.
In another embodiment, the reaction in step-(d) is optionally carried out in the presence of a base. Specifically, the base is an organic or inorganic base, and most specifically an inorganic base.
Exemplary bases include, but are not limited to, hydroxides, alkoxides, bicarbonates and carbonates of alkali or alkaline earth metals; ammonia, collidine, trimethylamine, tributylamine, triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4-(N,N-dimethylamino)pyridine and 1-alkylimidazole. Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically sodium hydroxide and potassium hydroxide.
In one embodiment, the acetylation in step-(d) is carried out in the presence of a fourth solvent.
Exemplary fourth solvents used in step-(d) include, but are not limited to, water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
Specifically, the fourth solvent used in step-(d) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanoI,_eth-yl— acetate,~methyT
acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, Ν,Ν-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof. Most specific fourth solvents are methanol, ethanol, isopropanol, and mixtures thereof.
In one embodiment, the acetylating agent in step-(d) is used in a ratio of about 1 to 3 equivalents, specifically about 1 to 1.5 equivalents, with respect to the 2-(7 -hydroxy- 1- naphthyl)ethanamine of formula V or an acid addition salt thereof in order to ensure a proper course of the reaction.
In one embodiment, the reaction in step-(d) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used. The reaction time may vary between about 20 minutes to about 3 hours, and specifically about 30 minutes to about 2 hours.
The reaction mass containing the N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide of formula VI obtained in step-(d) may be subjected to usual work up methods as described above. The reaction mass may be used directly in the next step to produce the compound of formula I, or the compound of formula VI may be isolated and/or recrystallized from a suitable solvent by conventional methods, as described hereinabove, and then used in the next step.
The solvent used for isolating and/or recrystallizing the N-[2-(7-hydroxy-l- naphthyl)ethyl]acetamide of formula VI is selected from the group as described hereinabove for such purpose.
Exemplary methylating agents used in step-(e) include, but are not limited to, dimethyl sulfate, methyl iodide, and the like. A most specific methylating agent is dimethyl sulfate.
In another embodiment, the reaction in step-(e) is optionally carried out in the presence of a base. Specifically, the base is an organic or inorganic base, and most specifically an inorganic base,
 Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically potassium carbonate.
Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically potassium carbonate.
In one embodiment, the methylation in step-(e) is carried out in the presence of a fifth solvent.
Exemplary fifth solvents used in step-(e) include, but are not limited to, water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
Specifically, the fifth solvent used in step-(e) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, Ν,Ν-dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof. Most specific fifth solvents are water, acetone, and mixtures thereof.
In one embodiment, the methylating agent in step-(e) is used in a ratio of about 1 to 3 equivalents, specifically about 1 to 1.5 equivalents, with respect to the N-[2-(7-hydroxy- l-naphthyl)ethyl]acetamide of formula VI in order to ensure a proper course of the reaction.
In one embodiment, the reaction in step-(e) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used. The reaction time may vary between about 30 minutes to about 6 hours, and specifically about 3 hours to about 5 hours.
The reaction mass containing the agomelatine of formula I obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the
solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
The solvent used for isolating/recrystallizing the pure agomelatine of formula I is selected from the group as described herein above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
In one embodiment, the isolation is carried out by cooling the reaction mass at a temperature of below about 35°C, followed by the addition of water at a temperature of about 10°C to about 35°C, and more specifically at a temperature of about 20°C to about 30°C. After completion of addition process, the resulting mass is optionally stirred at a temperature of about 10°C to about 35°C for at least 10 minutes, and most specifically at a temperature of about 20°C to about 30°C for about 15 minutes to about 2 hours.
According to another aspect, there is provided a process for the preparation of (7- hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III:

or a salt thereof, comprising reacting 7-hydroxy-l-tetralone of formula II:


According to another aspect, there is provided a process for the preparation of 2-(7- hydroxy-3,4-dihydro-l -naphthalenyl)ethanamine of formula IV:


The process for the preparation of 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV disclosed herein is carried out by the methods and parameters as described hereinabove.
According to another aspect, there is provided a process for the preparation of 2-(7- hydroxy- 1 -naphthyl)ethanamine of formula V:

or an acid addition salt thereof, comprising subjecting 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV:
 or an acid addition salt thereof, to aromatization by reacting with a suitable reagent to produce the compound of formula V or an acid addition salt thereof.
or an acid addition salt thereof, to aromatization by reacting with a suitable reagent to produce the compound of formula V or an acid addition salt thereof.
The process for the preparation of 2-(7-hydroxy-l-naphthyl)ethanamine of formula V disclosed herein is carried out by the methods and parameters as described hereinabove.
According to another aspect, there is provided a process for the preparation of N- [2-(7-hydroxy-l-naphthyl)ethyl]acetamide of formula VI:

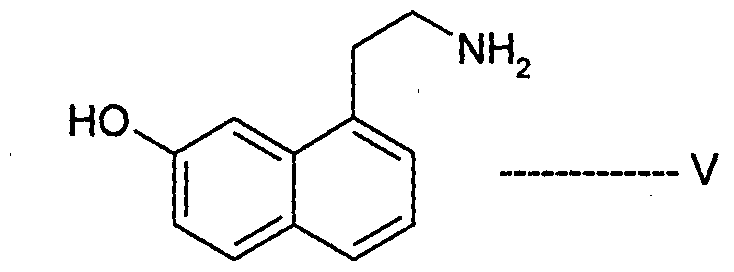
The process for the preparation of N-[2-(7-hydroxy-l-naphthyl)ethyl]aeetamide of formula VI disclosed herein is carried out by the methods and parameters as described hereinabove.
According to another aspect, there is provided a process for the preparation of Agomelatine, N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide, of formula I:
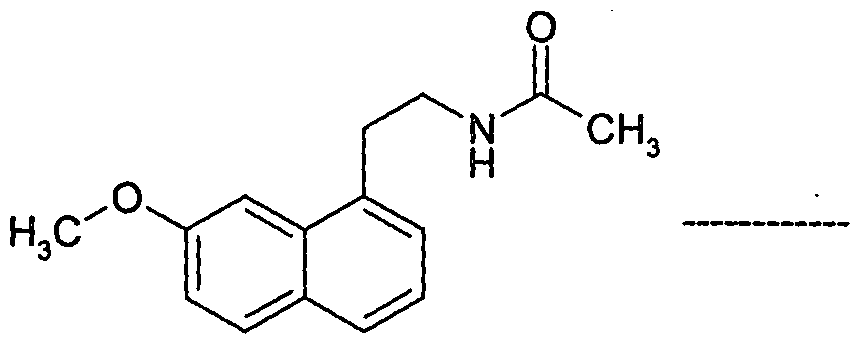 or a salt thereof, comprising reacting N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide of formula VI:
or a salt thereof, comprising reacting N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide of formula VI:
 or a salt thereof, with a methylating agent to produce the agomelatine of formula I.
or a salt thereof, with a methylating agent to produce the agomelatine of formula I.
The process for the preparation of Agomelatine, N-[2-(7-methoxy-l - naphthalenyl)ethyl]acetamide, of formula I disclosed herein is carried out by the methods and parameters as described hereinabove.
According to another aspect, there is provided a novel process for the preparation of Agomelatine, N-[2- 7-methoxy-l-naphthalenyl)ethyl]acetamide, of formula I:
 or a salt thereof, comprising:
or a salt thereof, comprising:
a) reacting 7-hydroxy-l -tetralone of formula II:

HO
CN X
O or a salt thereof, to produce (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III:

b) reducing the compound of formula III with a suitable reducing agent to produce 2-(7- hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV:

acetylating the compound of formula IV with a suitable "acetylating agent to prodi [2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethyl]acetamide, of formula VIII:
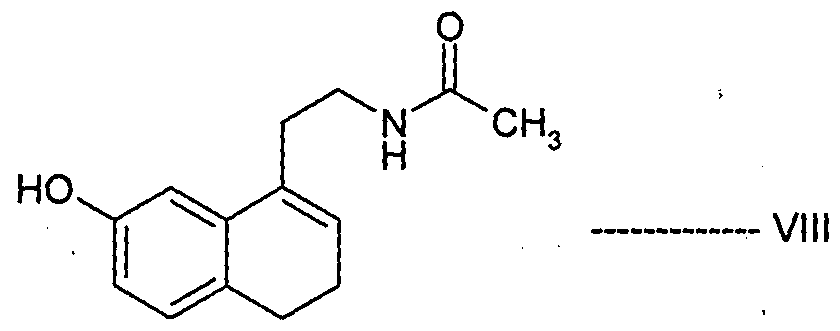
d) subjecting the compound of formula VIII to aromatization by reacting with a suitable reagent to produce N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI: ...
 or a salt thereof; and
or a salt thereof; and
e) reacting the compound of formula VI with a methylating agent to produce the agomelatine of formula I.
Unless otherwise specified, the reactions in the above process steps-(a), (b) and (e) are carried out by the methods as described hereinabove.
The reaction in step-(a) is carried out in the presence or absence of a solvent. In one embodiment, the reaction in step-(a) is carried out in the presence of a first solvent selected from the group as described above.
Specifically, the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; more specifically the first solvent is toluene or xylene; and a most specific solvent is toluene.
In one embodiment, the reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine. In another embodiment, the carboxylic acid and the organic amine compounds are employed in catalytic amounts.
Exemplary carboxylic acids used in step-(a) include, but are not limited to, a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid.
Exemplary organic amines used in step-(a) include, but are not limited to, a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine.
Specifically, the carboxylic acid used in step-(a) is an unsubstituted or substituted aryl carboxylic acid; and a most specific carboxylic acid is 4-hydroxybenzoic acid.
Specifically, the organic amine used in step-(a) is an unsubstituted or substituted linear or branched aralkyl amine; more specifically the organic amine is morpholine or benzyl amine; and a most specific organic amine is benzyl amine.
In one embodiment, the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated in the.form of a solid.
In one embodiment, the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula HI is isolated and/or re-crystallized from a suitable solvent by the methods described hereinabove.
The solvent used for isolating and/or recrystallizing the pure (7-hydroxy-3,4- dihydro-l-naphthalenyl)acetonitrile of formula III is selected from the group as described hereinabove.
In one embodiment, the reduction in step-(b) is carried out in the presence of a second solvent selected from the group as described above.
Specifically, the second solvent used in step-(b) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; more specifically, the second solvent is selected from the group consisting of methanol, ethanol, isopropanol, and mixtures thereof; and a most specific second solvent is methanol or ethanol.
Exemplary reducing agents used in step-(b) include, but are not limited to, hydrogenation catalysts (metal catalysts) such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like; hydride agents such as lithium aluminum hydride, sodium borohydride/Lewis acid, lithium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2-methoxyethoxy)aluminium hydride (Vitride), and the like; and other reducing agents such as borane, borane-THF complex, and the like.
It has been surprisingly found that the reduction in step-(b) is advantageously and efficiently carried out by employing the less expensive hydride agents-such-as sodiunT
borohydride when the reaction is performed in the presence of a Lewis acid. The use of hydride agents in the presence of Lewis acid in the reduction process leads to the resulting product with high purity and in good yield.
Exemplary Lewis acids used in step-(b) include, but are not limited to, aluminium chloride, calcium chloride, boron triflouride and zinc chloride, nickel chloride, and the like. Specific Lewis acids are aluminium chloride and nickel chloride.
The reduction in step-(b) may be carried out in the presence or absence of hydrogen gas. A most specific reducing agent used in step-(b) is Raney-Nickel.
The reduction in step-(b) is optionally carried out in the presence of ammonia as per the methods described hereinbefore.
In one embodiment, the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV obtained in step-(b) is converted into its acid addition salts by treatment with a suitable acid selected from the group described hereinbefore. Most specific acids are hydrochloric acid and hydrobromic acid.
In one embodiment, the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof is isolated in the form of a solid.
In one embodiment, the 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by the methods described hereinbefore.
The solvent used for isolating and/or recrystallizing the pure 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV is selected from the group as described above.
In another embodiment, a most specific acid addition salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV prepared by the process described herein is 2-(7-hydroxy-3,4-dihydro-l -naphthalenyl)ethanamine hydrochloride salt.
In another embodiment, a most specific acid addition salt of 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine of formula IV prepared by the process described herein is 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrobromide salt.
Exemplary acetylating agents used in step-(c) include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium
acetate, and the like, or a combination thereof. A most specific acetylating agent is acetic anhydride.
In another embodiment, the reaction in step-(c) is optionally carried out in the presence of a base. Specifically, the base is an organic or inorganic base, and most specifically an inorganic base.
Exemplary bases include, but are not limited to, hydroxides, alkoxides, bicarbonates and carbonates of alkali or alkaline earth metals; ammonia, collidine, trimethylamine, tributylamine, triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4-(N,N-dimethylamino)pyridine and 1-alkylimidazole. Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically sodium hydroxide and potassium hydroxide.
In one embodiment, the acetylation in step-(c) is carried out in the presence of a fourth solvent selected from the group as described hereinbefore.
Exemplary fourth solvents used in step-(c) include, but are not limited to, water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
Specifically, the fourth solvent used in step-(c) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, Ν,Ν-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof. Most specific fourth solvents are methanol, ethanol, isopropanol, and mixtures thereof.
In one embodiment, the reaction in step-(c) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most„specifically-at"th¾^fluj
temperature of the solvent used. The reaction time may vary between about 20 minutes to about 3 hours, and specifically about 30 minutes to about 2 hours.
The reaction mass containing the N-[2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethyl]acetamide, of formula VIII obtained in step-(c) may be subjected to usual work up methods as described above. The reaction mass may be used directly in the next step to produce the compound of formula VI, or the compound of formula VIII may be isolated and/or recrystallized from a suitable solvent by conventional methods, as described hereinabove, and then used in the next step.
The solvent used for isolating and/or recrystallizing the N-[2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethyl]acetamide, of formula VIII is selected from the group as described hereinabove for such purpose.
Exemplary reagents suitable for facilitating the aromatization reaction in step-(d) include, but are not limited to, sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, hydrogenation catalysts such as palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, and the like; quinone derivatives such as 2,3,5,6-tetrachlorocyclohexa-2,5rdiene-l,4-dione (p-Chloranil), p- bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6- dicyano benzoquinone, 2,3- dicyano-4-chlorobenzoquinone, 2,3-dicyanobenzoquinone; and other quinone derivatives such as 1 ,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil, o-floranil, and the like.
Specific aromatization reagents are sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ) and raney nickel; and most specifically sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil).
Therefore, the aromatization reaction in step-(d) is advantageously carried out by using cheaper aromatization reagent sulfur in the absence of a solvent.
The aromatization reaction in step-(d) is carried out in the presence or absence of a solvent.
In one embodiment, the aromatization in step-(d) is carried out as a neat reaction in the absence of a solvent.
In another embodiment, the aromatization in step-(d) is optionally carried out in the presence of a third solvent selected from the group as described hereinbefore.
Exemplary third solvents suitable for facilitating the aromatization in step-(d) include, but are not limited to, water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
Specifically, the third solvent is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, Ν,Ν-dimethylacetamide, dimethylsulfoxide, and mixtures thereof; and a most specific third solvent is toluene.
In one embodiment, the aromatization in step-(d) is carried out as a neat reaction, in the absence of solvents, by heating the contents at a temperature' of about 160°C to about 200°C, and specifically at a temperature of about 180°C to about 190°C. The reaction time may vary between about 30 minutes to about 5 hours, and most specifically about 1 hour to about 2 hours.
In another embodiment, the aromatization in step-(d) is carried out in the presence of the third solvent at a temperature of about 20°C to the reflux temperature of the solvent used, specifically at a temperature of about 50°C to the reflux temperature of the solvent used, and more specifically at about 60°C to about 120°C. The reaction time may vary between about 3 hours to about 30 hours, and most specifically about 5 hours to about 25 hours.
The reaction mass containing the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI or a salt thereof obtained in step-(d) may subjected to usual work up methods as described above. The reaction mass may be used directly in the next step to produce the agomelatine of formula I or the compound of formula VI may be isolated and/or recrystallized and then used in the next step.
In another embodiment, the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI obtained in step-(d) is isolated in the form of a solid.
In another embodiment, the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI is isolated and/or re-crystallized from a suitable solvent by the methods as described hereinabove.
The solvent used for isolating and/or recrystallizing the pure N-[2-(7-Hydroxy-l- naphthyl)ethyl]acetamide of formula VI is selected from the group as described above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
Exemplary methylating agents used in step-(e) include, but are not limited to, dimethyl sulfate, methyl iodide, and the like. A most specific methylating agent is dimethyl sulfate.
In another embodiment, the reaction in step-(e) is optionally carried out in the presence of a base. Specifically, the base is an organic or inorganic base, and most specifically an inorganic base, selected from the group as described hereinabove.
Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically potassium carbonate.
In one embodiment, the methylation in step-(e) is carried out in the presence of a fifth solvent.
Exemplary fifth solvents used in step-(e) include, but are not limited to, water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
Specifically, the fifth solvent used in step-(e) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-hutyLether^monogLym^
Ν,Ν-dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof. Most specific fifth solvents are water, acetone, and mixtures thereof.
In one embodiment, the reaction in step-(e) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used. The reaction time may vary between about 30 minutes to about 6 hours, and specifically about 3 hours to about 5 hours.
The reaction mass containing the agomelatine of formula I obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
The solvent used for isolating/recrystallizing the pure agomelatine of formula I is selected from the group as described herein above.
According to another aspect, there is provided a process for the preparation of N- [2-(7-hydroxy-3,4-dihydro-l-na hthalenyl)ethyl]acetamide of formula VIII:


According to another aspect, there is provided a process for the preparation of N- [2-(7-hydroxy-l-naphthyl)ethyl]acetamide of formula VI:


The process for the preparation of N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide of formula VI disclosed herein is carried out by the methods and parameters as described hereinabove.
According to another aspect, there is provided a novel process for the preparation of Agomelatine, N-[2-(7-methoxy- 1 -naphthalenyl)ethyl]acetamide, of formula I:

-a)— reacting 7-hydroxy- tetralOne of "formula"!!!
 with cyanoacetic acid of formula X:
with cyanoacetic acid of formula X:
HO
CN X or a salt thereof, to produce (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III:

or a salt thereof;
b) subjecting the compound of formula III to aromatization by reacting with a suitable reagent to produce (7-h droxy-l-naphthyl)acetonitrile, of formula VII:

or a salt thereof; and
c) reducing the compound of formula VII with a suitable reducing agent to produce 2-(7- hydroxy-l-naphthyl)ethanamine of formula V:


or a salt thereof; and
e) reacting the compound of formula VI with a methylating agent to produce the agomelatine of formula I.
Unless otherwise specified, the reactions in the above process steps-(a), (d) and (e) are carried out by the methods as described hereinabove.
The reaction in step-(a) is carried out in the presence or absence of a solvent. In one embodiment, the reaction in step-(a) is carried out in the presence of a first solvent selected from the group as described above.
Specifically, the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; more specifically the first solvent is toluene or xylene; and a most specific solvent is toluene.
In one embodiment, the reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine. In another embodiment, the carboxylic acid and the organic amine compounds are employed in catalytic amounts.
Exemplary carboxylic acids used in step-(a) include, but are not limited to, a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid.
Exemplary organic amines used in step-(a) include, but are not limited to, a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine.
Specifically, the carboxylic acid used in step-(a) is an unsubstituted or substituted aryl carboxylic acid; and a most specific carboxylic acid is 4-hydroxybenzoic acid.
Specifically, the organic amine used in step-(a) is an unsubstituted or substituted linear or branched aralkyl amine; more specifically the organic amine is morpholine or benzyl amine; and a most specific organic amine is benzyl amine.
In one embodiment, the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitriIe of formula III is isolated in the form of a solid.
In one embodiment, the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated and/or re-crystallized from a suitable solvent by the methods described hereinabove.
The solvent used for isolating and/or recrystallizing the pure (7-hydroxy-3,4- dihydro-l-naphthalenyl)acetonitrile of formula III is selected from the group as described hereinabove.
Exemplary reagents suitable for facilitating the aromatization reaction in step-(b) include, but are not limited to, sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, hydrogenation catalysts such as palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, and the like; quinone derivatives such as 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), p- bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6- dicyano benzoquinone, 2,3- dicyano-4-chlorobenzoquinone, 2,3-dicyanobenzpquinone; and other quinone derivatives such as 1,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil, o-floranil, and the like.
Specific aromatization reagents are sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ) and raney nickel; and most specifically sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil).
Therefore, the aromatization reaction in step-(b) is advantageously carried out by using cheaper aromatization reagent sulfur in the absence of a solvent.
The aromatization reaction in step-(b) is carried out in the presence or absence of a solvent.
In one embodiment, the aromatization in step-(b) is carried out as a neat reaction in _the_absence_of.a-sol-vent
In another embodiment, the aromatization in step-(b) is optionally carried out in the presence of a third solvent selected from the group as described hereinbefore.
Exemplary third solvents suitable for facilitating the aromatization in step-(b) include, but are not limited to, water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
Specifically, the third solvent is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, Ν,Ν-dimethylacetamide, dimethylsulfoxide, and mixtures thereof; and a most specific third solvent is toluene.
In one embodiment, the aromatization in step-(b) is carried out as a neat reaction, in the absence of solvents, by heating the contents at a temperature of about 160°C to about 200°C, and specifically at a temperature of about 180°C to about 190°C. The reaction time may vary between about 30 minutes to about 5 hours, and most specifically about 1 hour to about 2 hours.
In another embodiment, the aromatization in step-(b) is carried out in the presence of the third solvent at a temperature of about 20°C to the reflux temperature of the solvent used, specifically at a temperature of about 50°C to the reflux temperature of the solvent used, and more specifically at about 60°C to about 120°C. The reaction time may vary between about 3 hours to about 30 hours, and most specifically about 5 hours to about 25 hours.
The reaction mass containing the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII or a salt thereof obtained in step-(b) may subjected to usual work up methods as described above. The reaction mass may be used directly in the next step to produce the 2- (7-hydroxy-l-naphthyl)ethanamine of formula V or the compound of formula VII may be isolated and/or recrystallized and then used in the next step.
In another embodiment, the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII obtained in step-(b) is isolated in the form of a solid.
In another embodiment, the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII is isolated and/or re-crystallized from a suitable solvent by the methods as described hereinabove.
The solvent used for isolating and/or recrystallizing the pure (7 -hydroxy- 1- naphthyl)acetonitrile, of formula VII is selected from the group as described above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
In one embodiment, the reduction in step-(c) is carried out in the presence of a second solvent selected from the group as described above.
Specifically, the second solvent used in step-(c) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; more specifically, the second solvent is selected from the group consisting of methanol, ethanol, isopropanol, and mixtures thereof; and a most specific second solvent is methanol or ethanol.
Exemplary reducing agents used in step-(c) include, but are not limited to, hydrogenation catalysts (metal catalysts) such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like; hydride agents such as lithium aluminum hydride, sodium borohydride/Lewis acid, lithium · borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2-methoxyethoxy)aluminium hydride (Vitride), and the like; and other reducing agents such as borane, borane-THF complex, and the like.
It has been surprisingly found that the reduction in step-(c) is advantageously and efficiently carried out by employing the less expensive hydride agents such as sodium borohydride whenjhej-eactiojLis ^performed- in-the-presenee-of-a-bewis'acid: he"u"se~"of
hydride agents in the presence of Lewis acid in the reduction process leads to the resulting product with high purity and in good yield.
Exemplary Lewis acids used in step-(c) include, but are not limited to, aluminium chloride, calcium chloride, boron triflouride and zinc chloride, nickel chloride, and the like. Specific Lewis acids are aluminium chloride and nickel chloride.
The reduction in step-(c) may be carried out in the presence or absence of hydrogen gas. A most specific reducing agent used in step-(c) is Raney-Nickel.
The reduction in step-(c) is optionally carried out in the presence of ammonia as per the methods described hereinbefore.
In one embodiment, the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V obtained in step-(c) is converted into its acid addition salts by treatment with a suitable acid selected from the group described hereinbefore. Most specific acids are hydrochloric acid and hydrobromic acid.
In one embodiment, the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof is isolated in the form of a solid.
In one embodiment, the 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by the methods described hereinbefore.
The solvent used for isolating and/or recrystallizing the pure 2-(7-hydroxy-l- naphthyl)ethanamine of formula V is selected from the group as described above.
In another embodiment, a most specific acid addition salt of 2-(7-hydroxy-l- naphthyl)ethanamine of formula V prepared by the process described herein is 2-(7- hydroxy-l-naphthyl)ethanamine hydrochloride salt.
In another embodiment, a most specific acid addition salt of 2-(7-hydroxy-l- naphthyl)ethanamine of formula V prepared by the process described herein is 2-(7- hydroxy- 1 -naphthyl)ethanamine hydrobromide salt.
Exemplary acetylating agents used in step-(d) include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium acetate, and the like, or a combination thereof. A most specific acetylating agent is acetic anhydride.
In another embodiment, the reaction in step-(d) is optionally carried out in the presence of a base. Specifically, the base is an organic or inorganic base, and most specifically an inorganic base.
Exemplary bases include, but are not limited to, hydroxides, alkoxides, bicarbonates and carbonates of alkali or alkaline earth metals; ammonia, collidine, trimethylamine, tributylamine, triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4-(N,N-dimethylamino)pyridine and 1-alkylimidazole. Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically sodium hydroxide and potassium hydroxide.
In one embodiment, the acetylation in step-(d) is carried out in the presence of a fourth solvent selected from the group as described hereinbefore.
Exemplary fourth solvents used in step-(d) include, but are not limited to, water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
Specifically, the fourth solvent used in step-(d) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, ■< 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, Ν,Ν-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof. Most specific fourth solvents are methanol, ethanol, isopropanol, and mixtures thereof.
In one embodiment, the reaction in step-(d) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used. The reaction time may vary between about 20 minutes to about 3 hours, and specifically about 30 minutes to aJ?mrt 2_ho_urs.-
The reaction mass containing the N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI obtained in step-(d) may be subjected to usual work up methods as described above. The reaction mass may be used directly in the next step to produce the agomelatine of formula I, or the compound of formula VI may be isolated and/or recrystallized from a suitable solvent by conventional methods, as described hereinabove, and then used in the next step.
The solvent used for isolating and/or recrystallizing the N-[2-(7-Hydroxy-l- naphthyl)ethyl]acetamide of formula VI is selected from the group as described hereinabove for such purpose.
Exemplary methylating agents used in step-(e) include, but are not limited to, dimethyl sulfate, methyl iodide, and the like. A most specific methylating agent is dimethyl sulfate.
In another embodiment, the reaction in step-(e) is optionally carried out in the presence of a base. Specifically, the base is an organic or inorganic base, and most specifically an inorganic base, selected from the group as described hereinabove.
Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically potassium carbonate.
In one embodiment, the methylation in step-(e) is carried out in the presence of a fifth solvent.
Exemplary fifth solvents used in step-(e) include, but are not limited to, water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
Specifically, the fifth solvent used in step-(e) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyI tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide,
Ν,Ν-dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof. Most specific fifth solvents are water, acetone, and mixtures thereof.
In one embodiment, the reaction in step-(e) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used. The reaction time may vary between about 30 minutes to about 6 hours, and specifically about 3 hours to about 5 hours.
The reaction mass containing the agomelatine of formula I obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
The solvent used for isolating/recrystallizing the pure agomelatine of formula I is selected from the group as described herein above.
According to another aspect, there is provided a process for the preparation of (7- hydroxy-l-naphthyl)acetonitrile of formula VII:

or a salt thereof, comprising subjecting (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III:

According to another aspect, there is provided a process for the preparation of 2-(7- hydroxy- 1 -naphthyl)ethanamine of formula V:


The process for the preparation of 2-(7-hydroxy- l-naphthyl)ethanamine of formula
V disclosed herein is carried out by the methods and parameters as described hereinabove.
According to another aspect, there is provided a novel process for the preparation of Agomelatine, N-[2-(7-methoxy-l-naphthalenyl)ethyl]acetamide, of formula I:



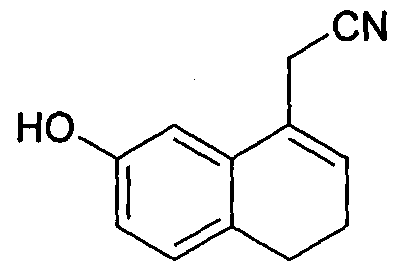
or a salt thereof;
b) subjecting the compound of formula III to aromatization by reacting with a suitable reagent to produce (7-hydroxy-l-naphthyl)acetonitrile, of formula VII:

or a salt thereof;
c) reacting the compound of formula VII with a methylating agent to produce (7- methoxy-l-naphthyl)acetonitrile of formula IX:


e) acetylating the compound of formula XI with a suitable acetylating agent to produce the agomelatine of formula I.
Unless otherwise specified, the reactions in the above process steps-(a), (b), (d) and (e) are carried out by the methods as described hereinabove.
The reaction in step-(a) is carried out in the presence or absence of a solvent. In one embodiment, the reaction in step-(a) is carried out in the presence of a first solvent selected from the group as described above.
Specifically, the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; more specifically the first solvent is toluene or xylene; and a most specific solvent is toluene.
In one embodiment, the reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine. In another embodiment, the carboxylic acid and the organic amine compounds are employed in catalytic amounts.
Exemplary carboxylic acids used in step-(a) include, but are not limited to, a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid.
Exemplary organic amines used in step-(a) include, but are not limited to, a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine.
Specifically, the carboxylic acid used in step-(a) is an unsubstituted or substituted aryl carboxylic acid; and a most specific carboxylic acid is 4-hydroxybenzoic acid.
Specifically, the organic amine used in step-(a) is an unsubstituted or substituted linear or branched aralkyl amine; more specifically the organic amine is morpholine or benzyl amine; and a most specific organic amine is benzyl amine.
In one embodiment, the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated in the form of a solid.
In one embodiment, the (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III is isolated and/or re-crystallized from a suitable solvent by the methods described hereinabove.
The solvent used for isolating and/or recrystallizing the pure (7-hydroxy-3,4- dihydro-l-naphthalenyl)acetonitrile of formula III is selected from the group as described hereinabove.
Exemplary reagents suitable for facilitating the aromatization reaction in step-(b) include, but are not limited to, sulfur or its derivatives, ' selenium metal, aqueous hydrobromic acid, hydrogenation catalysts such as palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, and the like; quinone derivatives such as 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), p- bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6- dicyano benzoquinone, 2,3- dicyano-4-chlorobenzoquinone, 2,3-dicyanobenzoquinone; and other quinone derivatives such as 1 ,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil, o-floranil, and the like.
Specific aromatization reagents are sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano:benzoquinone (DDQ) and raney nickel; and most specifically sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil).
Therefore, the aromatization reaction in step-(b) is advantageously carried out by using cheaper aromatization reagent sulfur in the absence of a solvent.
The aromatization reaction in step-(b) is carried out in the presence or absence of a solvent.
In one embodiment, the aromatization in step-(b) is carried out as a neat reaction in the„absencej>f a solvent.-
In another embodiment, the aromatization in step-(b) is optionally carried out in the presence of a third solvent selected from the group as described hereinbefore.
Exemplary third solvents suitable for facilitating the aromatization in step-(b) include, but are not limited to, water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
Specifically, the third solvent is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, Ν,Ν-dimethylacetamide, dimethylsulfoxide, and mixtures thereof; and a most specific third solvent is toluene.
In one embodiment, the aromatization in step-(b) is carried out as a neat reaction, in the absence of solvents, by heating the contents at a temperature of about 160°C to about 200°C, and specifically at a temperature of about 180°C to about 190°C. The reaction time may vary between about 30 minutes to about 5 hours, and most specifically about 1 hour to about 2 hours.
In another embodiment, the aromatization in step-(b) is carried out in the presence of the third solvent at a temperature of about 20°C to the reflux temperature of the solvent used, specifically at a temperature of about 50°C to the reflux temperature of the solvent used, and more specifically at about 60°C to about 120°C. The reaction time may vary between about 3 hours to about 30 hours, and most specifically about 5 hours to about 25 hours.
The reaction mass containing the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII or a salt thereof obtained in step-(b) may subjected to usual work up methods as described above. The reaction mass may be used directly in the next step to produce the 2- (7-hydroxy-l-naphthyl)ethanamine of formula V or the compound of formula VII may be isolated and/or recrystallized and then used in the next step.
In another embodiment, the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII obtained in step-(b) is isolated in the form of a solid.
In "another embodiment, the (7-hydroxy-l-naphthyl)acetonitrile, of formula VII is isolated and/or re-crystallized from a suitable solvent by the methods as described hereinabove.
The solvent used for isolating and/or recrystallizing the pure (7-hydroxy-l- naphthyl)acetonitrile, of formula VII is selected from the group as described above. Specifically, the solvent is selected from the group consisting of water, methanol, ethanol, n-propanol, isopropyl alcohol, acetone, tetrahydrofuran, 2-methyl-tetrahydrofuran, diisopropyl ether, methyl tert-butyl ether, ethyl acetate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
Exemplary methylating agents used in step-(c) include, but are not limited to, dimethyl sulfate, methyl iodide, and the like. A most specific methylating agent is dimethyl sulfate.
In another embodiment, the reaction in step-(c) is optionally carried out in the presence of a base. Specifically, the base is an organic or inorganic base, and most specifically an inorganic base, selected from the group as described hereinabove.
Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically potassium carbonate.
In one embodiment, the methylation in step-(c) is carried out in the presence of a fifth solvent selected from the group as described above.
Exemplary fifth solvents used in step-(c) include, but are not limited to, water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
Specifically, the fifth solvent used in step-(c) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitnle,„prQpionitrile,-NTN-dimethylfom
Ν,Ν-dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof. Most specific fifth solvents are water, acetone, and mixtures thereof.
In one embodiment, the reaction in step-(c) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used. The reaction time may vary between about 30 minutes to about 6 hours, and specifically about 3 hours to about 5 hours.
The reaction mass containing the (7-methoxy-l-naphthyl)acetonitrile of formula IX obtained in step-(c) may be subjected to usual work up methods as described above. The reaction mass may be used directly in the next step to produce the 2-(7-methoxy-l- naphthyl)ethanamine of formula XI, or the compound of formula IX may be isolated and/or recrystallized from a suitable solvent by conventional methods, as described hereinabove, and then used in the next step.
The solvent used for isolating and/or recrystallizing the (7-methoxy-l- naphthyl)acetonitrile of formula IX is selected from the group as described hereinabove for such purpose.
In one embodiment, the reduction in step-(d) is carried out in the presence of a second solvent selected from the group as described above.
Specifically, the second solvent used in step-(d) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; more specifically, the second solvent is selected from the group consisting of methanol, ethanol, isopropanol, and mixtures thereof; and a most specific second solvent is methanol or ethanol.
Exemplary reducing agents used in step-(d) include, but are not limited to, hydrogenation catalysts (metal catalysts) such as platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, and the like; hydride agents such as lithium aluminum hydride, sodium borohydride/Lewis acid, lithium b^rohy^ng^,_sodlum^cyanoboroh-ydr-ide diisobutylalum^i
tri-tert-butoxyaluminum hydride, sodium bis(2-methoxyethoxy)aluminium hydride (Vitride), and the like; and other reducing agents such as borane, borane-THF complex, and the like.
It has been surprisingly found that the reduction in step-(d) is advantageously and efficiently carried out by employing the less expensive hydride agents such as sodium borohydride when the reaction is performed in the presence of a Lewis acid. The use of hydride agents in the presence of Lewis acid in the reduction process leads to the resulting product with high purity and in good yield.
Exemplary Lewis acids used in step-(d) include, but are not limited to, aluminium chloride, calcium chloride, boron triflouride and zinc chloride, nickel chloride, and the like. Specific Lewis acids are aluminium chloride and nickel chloride.
The reduction in step-(d) may be carried out in the presence or absence of hydrogen gas. A most specific reducing agent used in step-(d) is Raney-Nickel.
The reduction in step-(d) is optionally carried out in the presence of ammonia as per the methods described hereinbefore.
In one embodiment, the 2-(7-methoxy-l-naphthyl)ethanamine of formula -XI obtained in step-(d) is converted into its acid addition salts by treatment with a suitable acid selected from the group described hereinbefore. Most specific acids are hydrochloric acid and hydrobromic acid.
In one embodiment, the 2-(7-methoxy-l-naphthyl)ethanamine of formula XI or an acid addition salt thereof is isolated in the form of a solid.
In one embodiment, the 2-(7-methoxy-l-naphthyl)ethanamine of formula XI or an acid addition salt thereof is isolated and/or re-crystallized from a suitable solvent by the methods described hereinbefore.
The solvent used for isolating and/or recrystallizing the pure 2-(7-methoxy-l- naphthyl)ethanamine of formula XI is selected from the group as described above.
In another embodiment, a most specific acid addition salt of 2-(7-methoxy-l- naphthyl)ethanamine of formula XI prepared by the process described herein is 2-(7- methoxy-l-naphthyl)ethanamine hydrochloride salt.
In another embodiment, a most specific acid addition salt of 2-(7-methoxy-l- naphthyl)ethanamine of formula XI prepared by the process described herein is 2-(7- methoxy- 1 -naphthyl)ethanamine hydrobromide salt.
Exemplary acetylating agents used in step-(e) include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium acetate, and the like, or a combination thereof. A most specific acetylating agent is acetic anhydride.
In another embodiment, the reaction in step-(e) is optionally carried out in the presence of a base. Specifically, the base is an organic or inorganic base, and most specifically an inorganic base.
Exemplary bases include, but are not limited to, hydroxides, alkoxides, bicarbonates and carbonates of alkali or alkaline earth metals; ammonia, collidine, trimethylamine, tributylamine, triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4-(N,N-dimethylamino)pyridine and 1-alkylimidazole. Specific bases are aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and most specifically sodium hydroxide and potassium hydroxide.
In one embodiment, the acetylation in step-(e) is carried out in the presence of a fourth solvent selected from the group as described hereinbefore.
Exemplary fourth solvents used in step-(e) include, but are not limited to, water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
Specifically, the fourth solvent used in step-(e) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, Ν,Ν-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichlorom£thane,-dichloroethane chlorofom7 afld_
mixtures thereof. Most specific fourth solvents are methanol, ethanol, isopropanol, and mixtures thereof.
In one embodiment, the reaction in step-(e) is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used. The reaction time may vary between about 20 minutes to about 3 hours, and specifically about 30 minutes to about 2 hours.
The reaction mass containing the agomelatirie of formula I obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
The solvent used for isolating/recrystallizing the pure agomelatine of formula I is selected from the group as described herein above.
According to another aspect, there is provided a process for the preparation of (7- methoxy- 1 -naphthyl)acetonitrile of formula IX:

or a salt thereof, comprising reacting (7-hydroxy-l-naphthyl)acetonitrile of formula VII:

The process for the preparation of (7-methoxy-l-naphthyl)acetonitrile of formula IX disclosed herein is carried out by the methods and parameters as described hereinabove.
The solids obtained in any of the above process steps described hereinabove may be collected by filtration, filtration under vacuum, decantation, centrifugation, filtration employing a filtration media of a silica gel or celite, or a combination thereof.
The novel hydroxyl intermediates of formulae III, IV, V, VII, VIII, XII and XIII disclosed employed for the preparation of agomelatine disclosed herein allows the product to be easily isolated and purified, thereby producing a product with 72-80% overall yield.
The highly pure agomelatine, or a pharmaceutically acceptable salt thereof, obtained by the above processes may be further dried in, for example, a Vacuum Tray Dryer, a Rotocon Vacuum Dryer, a Vacuum Paddle Dryer or a pilot plant Rota vapor, to further lower residual solvents. Drying can be carried out under reduced pressure until the residual solvent content reduces to the desired amount such as an amount that is within the limits given by the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use ("ICH") guidelines.
In one embodiment, the drying is carried out at atmospheric pressure or reduced pressures, such as below about 200 mm Hg, or below about 50 mm Hg, at temperatures such as about 35°C to about 90°C, and specifically at about 50°C to about 85°C. The drying can be carried out for any desired time period that achieves the desired result, such as times about 1 to 20 hours. Drying may also be carried out for shorter or longer periods of time depending on the product specifications. Temperatures and pressures will be chosen based on the volatility of the solvent being used and the foregoing should be considered as only a general guidance. Drying can be suitably carried out in a tray dryer, vacuum oven, air oven, or using a fluidized bed dryer, spin flash dryer, flash dryer, and the like.
In another embodiment, the highly pure agomelatine or a salt thereof obtained by the process disclosed herein has a purity of greater than about 99%, specifically greater than about 99.5%, more specifically greater than about 99.9%, and most specifically greater than about 99.95% as measured by HPLC. For example, the purity of the agomelatine or a salt thereof can be about 99% to about 99.95%, or about 99.5% to about 99.99%.
According to another aspect, there is provided a novel intermediate compound, (7- hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile, of formula III:

or a salt thereof.
According to another aspect, there is provided a novel intermediate compound, 2- (7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine, of formula IV:

or an acid addition salt thereof.
According to another aspect, there is provided 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine hydrochloride salt of formula IVa:

According to another aspect, there is provided 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine hydrobromide salt of formula IVb:
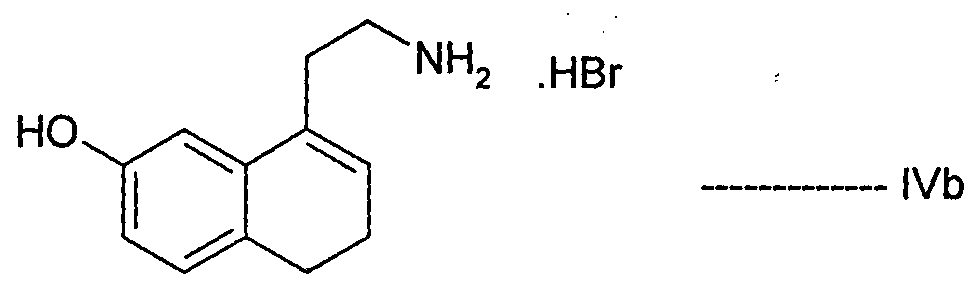
According to another aspect, there is provided a novel intermediate compound, 2- (7-hydroxy-l-naphthyl)ethanamine, of formula V:

According to another aspect, there is provided 2-(7-hydroxy-l- naphthyl)ethanamine hydrochloride salt of formula Va:

According to another aspect, there is provided a novel intermediate compound, (7- hydroxy- 1 -naphthyl)acetonitrile, of formula VII:

According to another aspect, there is provided a novel intermediate compound, N- [2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethyl]acetamide, of formula VIII:

Based on the extensive research and experimentation carried out by the present inventors, it has been surprisingly and unexpectedly found that the Hydrobromic acid (HBr) can also be used as a reagent for aromatization of 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV:

or an acid addition salt thereof, to produce 2-(7-hydroxy-l-naphthyl)ethanamine of formula V:


According to another aspect, there is provided a process for the preparation of 2-(7- hydroxy-l-naphthyl)ethanamine of formula V:

a) reacting 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof with aqueous hydrobromic acid to produce a reaction mass containing 2-(7-hydroxy-l-naphthyl)ethanamine of formula V and 2-(7-hydroxy- l,2,3,4-tetrahydro-l-naphthyl)ethanamine of formula XII:

(V) (XII) or an acid addition salt thereof;
b) isolating and/or recovering the pure 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof from the reaction mass obtained in step-(a);
c) treating the mother liquors with a suitable base to adjust the pH to produce cause precipitation;
d) optionally, extracting the 2-(7-hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine of formula XII obtained in step-(c) into an organic solvent;
e) isolating and/or recovering the pure 2-(7-hydroxy- 1,2,3, 4-tetrahydro-l- naphthyl)ethanamine of formula XII or an acid addition salt thereof from the reaction mass obtained in step-(c) or from the organic layer obtained in step-(d); and
f) subjecting the compound of formula XII or an acid addition salt to aromatization by reacting with a suitable reagent to produce the compound of formula V.
In one embodiment, the reaction in step-(a) is carried out at a temperature of about 25°C to the reflux temperature of the solvent used, specifically at a temperature of about 60°C to the reflux temperature of the solvent used?_andjriQr.e_specifieaily-at_theTOflu) ^
temperature of the solvent used. The reaction time may vary between about 2 hours to about 5 hours.
In another embodiment, the reaction in step-(a) is optionally carried out in the presence of water-miscible organic solvents.
The base used in step-(c) is an organic or inorganic base selected from the group as described above.
Specifically, the base is an inorganic base, and most specifically, the base is aqueous ammonia, sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, or a combination thereof.
Specifically, the pH of the mother liquors in step-(c) is adjusted to above 8, and more specifically between 8 and 9.
Exemplary organic solvents used in step-(d) include, but are not limited to, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a halogenated hydrocarbon solvent, and mixtures thereof.
Specifically, the organic solvent used in step-(d) is selected from the group consisting of ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, dichloromethane, dichloroethane, chloroform, and mixtures thereof. Most specific organic solvents are ethyl acetate, toluene, dichloromethane, and mixtures thereof.
The reagents suitable for facilitating the aromatization reaction in step-(f) is selected from the group as described hereinbefore. Specific aromatization reagents are sulfur or its derivatives, selenium metal, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ) and Pd/C; and most specifically sulfur, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil) and 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ).
In one embodiment, the isolation in step-(b) and step-(e) is carried out by methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
The recovery in step-(b) and step-(e) is accomplished by filtration, filtration under vacuum, decantation, centrifugation, filtration employing a filtration media of a silica gel or celite, or a combination thereof.
According to another aspect, there is provided a process for the preparation of N- [2-(7-hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethyl]acetamide of formula XIII:

or a salt thereof, comprising acetylating 2-(7-hydroxy-l,2,3,4-tetrahydro-l- naphthyl)ethanamine, of formula XII:

Exemplary acetylating agents include, but are not limited to, acetyl halide such as acetyl chloride, acetyl bromide, acetyl iodide; acetic anhydride, sodium acetate, and the like, or a combination thereof. A most specific acetylating agent is acetic anhydride.
In another embodiment, the acetylation is optionally carried out in the presence of a base selected from the group as described hereinbefore.
In one embodiment, the acetylation is carried out in the presence of a fourth solvent selected from the group as described hereinbefore.
In one embodiment, the acetylation is carried out at a temperature of about 0°C to the reflux temperature of the solvent used, specifically at a temperature of about 20°C to the reflux temperature of the solvent used, and most specifically at the reflux temperature of the solvent used.
The reaction mass containing the N-[2-(7-hydroxy-l,2,3,4-tetrahydro-l- naphthyl)ethyl]acetamide of formula XIII obtained may be subjected to usual work up such as a washing, an extraction, an evaporation, a pH adjustment etc., followed by isolation and/or recrystallization from a suitable solvent by conventional methods such as cooling, seeding, partial removal of the solvent from the solution, by adding an anti-solvent to the solution, evaporation, vacuum distillation, or a combination thereof.
According to another aspect, there is provided a novel intermediate compound, 2- (7-hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine, of formula XII:

or an acid addition salt thereof.
According to another aspect, there is provided a novel intermediate compound, 2- (7-hydroxy- 1 ,2,3,4-tetrahydro- 1 -naphthyl)ethanamine hydrochloride salt, of formula Xlla:

According to another aspect, there is provided a novel intermediate compound, N- [2-(7-hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethyl]acetamide, of formula XIII:

or a salt thereof.
According to another aspect, the present invention also encompasses the use of the novel compounds of formulae III, IV, IVa, IVb, V, Va, VII, VIII, XII, Xlla and XIII disclosed herein for preparing Agomelatine.
The following examples are given for the purpose of illustrating the present invention and should not be considered as limitation on the scope or spirit of the invention.
EXAMPLES
Example 1
Preparation of (7-Hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile
7-Hydroxy-l-tetralone (100 gm), cyanoacetic acid (75 gm), 4-hydroxybenzoic acid (25.5 gm) and benzylamine (20 gm) were added to toluene (2 L) at room temperature (25-30°C). The mixture was heated to reflux temperature (108-1 10°C) for about 6 hours. The reaction mass was cooled to room temperature and then filtered. The resulting precipitate was washed with toluene (100 ml), followed by washing the filtrate with 10% sodium carbonate solution (2 x 100 ml) and subsequently with distilled water (100 ml). The resulting filtrate was subjected to evaporation to remove the solvent to produce 1 13 gm of (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile as a solid (Melting Range: 120 - 120°C; Yield: 99%; Purity by HPLC: 99%).
Infra-red (FT-IR) Data (KBr pellet): 3346 cm"1 (-OH); 2262 cm'1 (-CN); 1613 cm-1 (-C=C, Ar); 2827-2918 cm"1 (-CH2).
Ή-NMR (DMSO-d6) δ: 2.193-2.260 (m, 2H), 2.574-2.627 (t, 2H), 3.732-3.735 (s, 2H), 6.147-6.177 (m, 1H), 6.595-6.995 (m, 3H), 9.279 (s, OH); Mass (m/z): 184 (M-l).
Example 2
Preparation of 2-(7-Hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine
(7-Hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile (100 gm) was added to a mixture of Raney nickel (50 gm), methanol (1000 ml) and ammonia solution (500 ml) at room temperature (25-30°C) in an autoclave. The reaction mixture was stirred under 5 kg m2 hydrogen pressure at 40-45 °C for 5-6 hours. After completion of the reaction, the autoclave was cooled to 25-30°C. The reaction mass was filtered and washed with methanol. The solvent was distilled completely from the resulting filtrate, followed by co- distillation twice with methanol (200 ml). Ethyl acetate (500 ml) was added to the resulting residue at 25-30°C, followed by cooling the resulting solution to 0-5°C for 1 hour. The obtained solid was filtered and then dried to produce 97 gm_2-(7^hydroxy-"374^ihydro-l-
naphthalenyl)ethanamine (Yield: 95.1%; and Melting Range: 140-145°C; Purity by HPLC: 99.1%).
Infra-red (FT-IR) Data (KBr pellet): 3340, 3287 cm"1 (-NH2); 1602 cm-1 (-C=C, Ar); 2826- 2928 cm-1 (-CH2).
Example 3
Preparation of 2-(7-Hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrochloride
Ethyl acetate (200 ml) was added to 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine (50 gm), followed by adjusting the pH of the resulting mixture to below 2 with IPA-HC1 solution at 10-15°C and hexane (200 ml) was added. The reaction mass was stirred for 30 minutes at 0-10°C. The resulting solid was filtered and washed with n-hexane (50 ml) and then dried to yield 58 gm of 2-(7-hydroxy-3,4-dihydro- l-naphthalenyl)ethanamine hydrochloride (Yield: 97.3%; Melting Range: 195-200°C; Purity by HPLC: 99.5%).
Infra-red (FT-IR) Data (KBr pellet): 3380 on 1 (-OH); 3027 cm"1 (-NH3 +); 2824-2892 cm"1 (-CH2).
Ή-NMR (DMSO-d6) δ: 2.129-2.195 (m, 2H), 2.551-2.604 (m, 2H), 2.636-2.686 (m, 2H), 2.887-2.938 (m, 2H), 5-910-5.939 (t, 1H), 6.576-6.964 (m, 3H), 7.971 (s, -NH3), 9.242 (s, OH); Mass (m/z): 190 (M+l). Example 4
Preparation of 2-(7-Hydroxy-l-naphthyl)ethanamine hydrochloride
Sulfur (8.5 gm) was added to 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrochloride (50 gm), followed by heating the mixture to 185-190°C for 1-2 hours. The resulting mass was cooled to 50-55°C and then methanol (500 ml) was added. The reaction mixture was refluxed for 30 minutes, followed by treating with activated carbon (5 gm). The methanolic layer was distilled under vacuum to produce 48 gm of 2-(7-Hydroxy-l- naphthyl)ethanamine hydrochloride (Yield: 97%; Melting Range: 200-205°C; Purity by HPLC: 99.4%).
Infra-red (FT-IR) Data (KBr pellet): 3240-3460 cm-1 (-OH); 2480-2720 cm-1 (-NH3 +); 2950-3060 cm'1 (-CH).
Example 5
Preparation of N-[2-(7-Hydroxy-l-naphthyI)ethyl]acetamide
2-(7-Hydroxy-l-naphthyl)ethanamine hydrochloride (50 gm), sodium acetate (36.5 gm) and acetic anhydride (34 gm) were added to methanol (250 ml). The reaction mixture was heated to reflux temperature (65-68°C) for 15-20 minutes, followed by the addition of 10% sodium hydroxide solution (200 ml) under reflux. The reaction mixture was cooled to room temperature (25-30°C), followed by the addition of distilled water (250 ml). The resulting mixture was cooled to 0-10°C, followed by adjusting the pH to below 2 with concentrated hydrochloric acid. The resulting solid was filtered and then dried to produce 50 gm of the title compound, which was re-crystallized form water (500 ml) to yield 46 gm of the pure N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide (Yield: 90%; Melting Range: 123-125°C; Purity by HPLC: 99.6%).
Infra-red (FT-IR) Data (KBr pellet): 3347 cm-1 (-OH); 1641 cm"1 (-CO); 1626 crn 1 (- C=C).
Example 6
Preparation of N-[2-(7-methoxy-l-naphthaIenyl)ethyI]acetamide (Agomelatine)
N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide (35 gm), potassium carbonate (30 gm) and dimethyl sulfate (20 gm) were added to acetone (175 ml) at room temperature (25-30°C). The reaction mixture was refluxed for 5 hours. After completion of the reaction, the reaction mass was cooled to room temperature and then added into water (200 ml). The resulting solid was filtered and then dried to produce 35 gm of agomelatine (Yield: 94.5%; Purity by HPLC: 99.93%). Example 7
Preparation of N-[2-(7-hydroxy-3,4-dihydro-l-naphthyl)ethyl]acetamide
The 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrochloride (30 gm) was dissolved in methanol (150 ml), followed by the addition of sodium acetate (22 gm) and acetic anhydride (21 gm), and then heating the resulting mixture for 30 minutes at reflux temperature (65-68°C). 15% Sodium hydroxide solution (90 ml) was added to the reaction mass under reflux temperature, followed by maintainingJhe-resuliiag-mtxture^fbT^ rio aT
reflux temperature. The reaction mass was cooled to room temperature, followed by the addition of distilled water (150 ml) and then cooling the mass to 0-10°C. The pH of the reaction mass was adjusted to below 2 with hydrochloric acid, followed by extracting the mass three times with dichloromethane (3 x 50 ml). The dichloromethane layer was washed with distilled water (50 ml) and the resulting dichloromethane layer was distilled to produce 30 gm of N-[2-(7-hydroxy-3,4-dihydro-l-naphthyl)ethyl]acetamide (Yield: 97.7%; Melting Range: 115-120°C; Purity by HPLC: 99.3%).
Infra-red (FT-IR) Data (KBr pellet): 3331 cnf1 (-OH); 2824-2897 cm"1 (-CH2); 1642 cm (-CO-); 1606 cm"1 (-C=C-).
Ή-NMR (DMSO-d6) δ: 1.795 (s, 3H), 2.101-2.164 (m, 2H), 2.430-2.454 (m, 2H), 2.546- 2.573 (m, 2H), 3.124-3.192 (m, 2H), 5.835-5.868 (t, 1H), 6.521-6.941 (m, 3H), 7.877- 7.926 (t, -NH), 9.124 (s, OH); Mass (m/z): 232 (M+ 1).
Example 8
Preparation of N-[2-(7-Hydroxy-l-naphthyl)ethyI]acetamide
N-[2-(7-Hydroxy-3,4-dihydro-l-naphthyl)ethyl]acetamide (5 gm) and sulfur (15 gm) were heated to 185-190°C for about 1-2 hours. The reaction mass .was cooled to 55-60°C, followed by the addition of methanol (500 ml) and then refluxing for 1 hour. The resulting hot methanolic layer was treated with activated carbon (5 gm), followed by distillation under vacuum to produce 50 gm of the crude N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide, which was then recrystallized from water (500 ml) to yield 45 gm of the pure compound (Yield: 90.9%; Melting Range: 123-125°C; Purity by HPLC: 99.6%).
Example 9
Preparation of (7-Hydroxy-l-naphthyI)acetonitrile
(7-Hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile (10 gm) was dissolved in toluene (100 ml), followed by the addition of Pd/C (2.5 gm) and then heating the resulting mixture at reflux temperature for 30 hours. After completion of reaction, the mixture was filtered and the filtrate was distilled under vacuum to produce 9.5 gm of (7-hydroxy-l- naphthyl)acetonitrile (Yield: 96.9%; Melting Range: 110-115°C; Purity by HPLC: 99.2%).
Infra-red (FT-IR) Data (KBr pellet): 3394 cm"1 (-0H); 2267 cm"1 (-CN); 1613 cm"1 (-C=C, Ar).
Example 10
Preparation of (7-Hydroxy-l-naphthyl)acetonitrile
(7-Hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile (5 gm) and sulfur (1.5 gm) were heated to 185-190°C for 1 hour. The reaction mixture was cooled to 55-60°C, followed by the addition of methanol (50 ml) and then heating the resulting mass at reflux for 30 minutes. Activated carbon (1.0 gm) was added to the reaction mass and then filtered through celite bed. The resulting filtrate was distilled under vacuum to produce 4.5 gm of (7-hydroxy-l-naphthyl)acetonitrile (Yield: 91.8%; Melting Range: 1 10-145°C; Purity by HPLC: 99.1%).
Example 11
Preparation of (7-hydroxy-l-naphthyl)acetonitriIe
(7-Hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile (10 gm) was dissolved in toluene (100 ml), followed by the addition of chloranil (18.5 gm) and then heating the mixture at reflux temperature for about 20-24 hours. After completion of the reaction, the mixture was filtered and the filtrate was washed with saturated aqueous sodium carbonate solution (2 x 50 ml), and subsequently with water (50 ml). The resulting organic layer was distilled under vacuum to produce 9.5 gm of (7-hydroxy-l-naphthyl)acetonitrile (Yield: 96.9%; Melting Range: 1 10-115°C; Purity by HPLC: 98.7%).
Example 12
Preparation of (7-Hydroxy-l-naphthyl)ethanamine
(7-Hydroxy-l-naphthyl)acetonitrile (5 gm) was dissolved in methanol (500 ml), followed by the addition of aqueous ammonia solution (25 ml), Raney Nickel (5 gm). The reaction mixture was hydrogenated with hydrogen gas under 5 kg/m2 pressure at 40°C until completion of the reaction. The reaction mass was cooled to room temperature, followed by filtration and then concentrating the mass to remove methanol to produce 5 gm of (7- hydroxy- l-naphthyl)ethanamine (Yield: 98%; Purity by HPLC: 99.3%).
Example 13
Preparation of (7-Hydroxy-l-naphthyl)ethanamine hydrochloride salt
Step-1: Preparation of (7-Hydroxy-l-naphthyl)ethanamine hydrobromide
A mixture of 2-(7-Hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrochloride (50 gm) and 48% aqueous hydrobromic acid (100 ml) was heated at 95-100°C for 5 hours. After completion of the reaction, the reaction mass was cooled to 0-10°C, followed by maintaining the reaction mass at the same temperature for 1 hour. The resulting solid was filtered and washed with cold water (50 ml) and then dried to produce 25 gm of (7- hydroxy-l-naphthyl)ethanamine hydrobromide (Yield: 42%; Purity by HPLC: 99.5%). Step-2: Isolation of (7-Hydroxy-l,2,3i4-tetrahydro-l-naphthyl)ethanamine from Mother Liquors
The filtrate (mother liquors) obtained in step-1 was neutralized with 20% sodium hydroxide solution, followed by adjusting the pH to 8.9 with aqueous ammonia solution. The resulting aqueous layer was extracted into dichloromethane (3 x 150 ml) and the dichloromethane layer was washed with water (100 ml). The solvent was distilled off from the resulting organic layer to produce crude solid (20 gm). The resulting solid was taken in ethyl acetate (100 ml), followed by adjusting the pH to below 2.0 with isopropanolic-HCl solution at room temperature. The obtained clear solution was distilled under vacuum to produce 20 gm of the titled compound as hydrochloride salt (Purity by HPLC: 99.4%). Step-3: Preparation of (7-Hydroxy-l-naphthyl)ethanamine hydrochloride salt
Sulfur (6 gm) was added to (7-Hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine (20 g, obtained in step-2), followed by heating the mixture at 190-195°C for 10 hours. The resulting mass was cooled to 25-30°C and then methanol (100 ml) was added. The resulting mixture was heated at reflux temperature (65-70°C) for about 30 minutes. Charcoal (2 gm) was added to the reaction mass, followed by filtration through celite pad. The obtained filtrate was distilled under vacuum to produce 19 gm of (7-Hydroxy-l- naphthyl)ethanamine hydrochloride salt (Yield: 96.9%; Purity by HPLC: 99.5%).
Example 14
Preparation of (7-Methoxy-l-naphthyl)acetonitrile
(7-Hydroxy-l-naphthyl)acetonitrile (5 gm) was dissolved in acetone (50 ml), followed by the addition of potassium
Example 15
Preparation of N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide
N-[2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethyl]acetamide (10 gm) was dissolved in toluene (100 ml), followed by the addition of Pd/C (2.5 gm). The resulting mixture was heated to reflux temperature and then maintained for 20-25 hours. After completion of reaction, the reaction mass was filtered and the filtrate was distilled under vacuum to produce 9 gm ofpure N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide (Yield: 90.9%; Melting Point: 123-125°C; Purity by HPLC: 98.9%). Example 16
Preparation of N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide
N-[2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethyl]acetamide (10 gm) was dissolved in toluene (100 ml), followed by the addition of chloranil (12 gm).. The resulting mixture was heated to reflux temperature and then maintained for 20 hours. After completion of the reaction, the reaction mass was filtered and the filtrate was washed with saturated sodium carbonate solution (2 x 50 ml), and subsequently with water (50 ml). The resulting organic layer was distilled under vacuum to produce 8 gm of N-[2-(7 -hydroxy- 1- naphthyl)ethyl]acetamide (Yield: 80.8%; Purity by HPLC: 99.2%). Example 17
Preparation of N-[2-(7-hydroxy-l,2,3,4-tetrahydro-l-naphthyI)ethyl]acetamide
2-(7-Hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine hydrochloride (10 gm), sodium acetate (8 gm) and acetic anhydride (6 gm) were added to methanol (100 ml). The reaction mixture was heated to reflux temperature (65-68°C) for 30 minutes. The reaction mixture was cooled to room temperature (25-30°C) and then added into water (150 ml). The resulting mixture was extracted into dichloromethane (3_x-5.0-ml)^d-the esulting^fganic
Layer was washed with water (50 ml), the solvent was evaporated under vacuum to produce 12 gm of the pure N-[2-(7-hydroxy- 1 ,2,3, 4-tetrahydro-l -naphthyl)ethyl]acetamide (Yield: 98.4%; Purity by HPLC: 99.6%).
Example 18
Preparation of N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide
N-[2-(7-hydroxy-l ,2,3,4-tetrahydro-l -naphthyl)ethyl]acetamide (10 gm) was dissolved in toluene (100 ml), followed by the addition of Pd/C (2.5 gm). The resulting mixture was heated to reflux temperature and then maintained for 20 hours. After completion of the reaction, the reaction mass was filtered and the filtrate was distilled under vacuum to produce 9.1 gm of pure N-[2-(7-Hydroxy-l -naphthyl)ethyl]acetamide.
All ranges disclosed herein are inclusive and combinable. While the invention has been described with reference to a preferred embodiment, it will be understood by those skilled in the art that various changes may be made and equivalents may be substituted for elements thereof without departing from the scope of the invention. In addition, many modifications may be made to adapt a particular situation or material to the teachings of the invention without departing from essential scope thereof. Therefore, it is intended that the invention not be limited to the particular embodiment disclosed as the best mode contemplated for carrying out this invention, but that the invention will include all embodiments falling within the scope of the appended claims.
Claims
1. A process for the preparation of Agomelatine, N-[2-(7-methoxy-l- naphthalenyl)ethyl]acetamide, of formula I:

a) reacting 7-hydroxy-l-tetralone of formula II:



or a salt thereof;
25 b) reducing the compound of formula III with a suitable reducing agent to produce 2- (7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV:
_30.
 or an acid addition salt thereof;
or an acid addition salt thereof;
c) subjecting the compound of formula IV or an acid addition salt thereof to aromatization by reacting with a suitable reagent to produce 2-(7-hydroxy-l- naphthyl)ethanamine of formula V:

d) acetylating the compound of formula V with a suitable acetylating agent to produce N-[2-(7-Hydroxy- 1 -naphthyl)ethyl]acetamide of formula VI:

e) reacting the compound of formula VI with a methylating agent to produce the agomelatine of formula I.
2. The process of claim 1, wherein the reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine; wherein the reaction in step-(a) is carried out in the presence of a first solvent; and wherein the reaction in step-(a) is carried out at a temperature of about 25°C to the reflux temperature of the solvent used.
3. The process of claim 2, wherein the carboxylic acid used in step-(a) is a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid; wherein the organic amine used in step-(a) is a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine; wherein the first solvent used in step-(a) is selected from the group consisting of a hydrocarbon solvent, a^iajogenated-hydrocarbon-solventr-an-etherrand mixtwes'
thereof; and wherein the reaction in step-(a) is carried out at a temperature of about 60°C to the reflux temperature of the solvent used.
The process of claim 3, wherein the carboxylic acid used in step-(a) is 4- hydroxybenzoic acid and wherein the organic amine used in step-(a) is benzyl amine or morpholine; wherein the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; and wherein the reaction in step-(a) is carried out at the reflux temperature of the solvent used.
The process of claim 1, wherein the reduction in step-(b) is carried out in the presence of a second solvent; wherein the reducing agent used in step-(b) is selected from the group consisting of hydrogenation catalysts (metal catalysts), hydride agents, borane and borane-THF complex; and wherein the reduction in step-(b) is optionally carried out in the presence of ammonia.
The process of claim 5, wherein the second solvent used in step-(b) is selected from the group consisting of water, an alcohol, an ester, a hydrocarbon solvent, an ether, and mixtures thereof; wherein the reducing agent used in step-(b) is selected from the group consisting of platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, lithium aluminum hydride, sodium borohydride/Lewis acid, lithium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2- methoxyethoxy)aluminium hydride (Vitride), borane and borane-THF complex; and wherein the ammonia is used in the form of aqueous ammonia or in the form of ammonia gas or ammonia saturated in an organic solvent.
The process of claim 6, wherein the second solvent used in step-(b) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n- pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; and wherein the reducing agent used in step-(b) is Raney-Nickel.
8. The process of claim 1, wherein the acid addition salts of 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV prepared by the process are 2-(7-hydroxy-3,4- dihydro-l-naphthalenyl)ethanamine hydrochloride salt and 2-(7-hydroxy-3,4-dihydro- l-naphthalenyl)ethanamine hydrobromide salt.
9. The process of claim 1, wherein the reagent suitable for facilitating the aromatization reaction in step-(c) is selected from the group consisting of sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, metal catalysts and quinone derivatives; wherein the starting material 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV in step-(c) is employed in the form of an acid addition salt; and wherein the aromatization in step-(c) is optionally carried out in the presence of a third solvent.
10. The process of claim 9, wherein the reagent suitable for facilitating the aromatization reaction in step-(c) is selected from the group consisting of sulfur, selenium metal, aqueous hydrobromic acid, palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), p-bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6-dicyano benzoquinone, 2,3-dicyano-4-chlorobenzoquinone, 2,3- dicyanobenzoquinone, 1,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil and o-floranil; wherein the starting material 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV in step-(c) is employed in the form of its hydrochloride salt; and wherein the third solvent used in step-(c) is selected from the group consisting of water, a halogenated hydrocarbon, ah ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
11. The process of claim 10, wherein the reagent suitable for facilitating the aromatization reaction in step-(c) is selected from the group consisting of sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil); and wherein the third solvent used in step-(c) is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n- heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme,
acetonitrile, propionitrile, N,N-dimethylformamide, N,N-dimethylacetamide, dimethylsulfoxide, and mixtures thereof.
12. The process of claim 1, wherein the aromatization in step-(c) is carried out as a neat reaction, in the absence of solvent; and wherein the acid addition salts of 2-(7-hydroxy- l-naphthyl)ethanamine of formula V prepared are 2-(7-hydroxy-l- naphthyl)ethanamine hydrochloride salt and 2-(7-hydroxy-l-naphthyl)ethanamine hydrobromide salt.
13. The process of claim 1, wherein the acetylating agent used in step-(d) is selected from the group consisting of acetyl chloride, acetyl bromide, acetyl iodide, acetic anhydride, sodium acetate, or a combination thereof; wherein the reaction in step-(d) is optionally carried out in the presence of a base; and wherein the acetylation in step-(d),is carried out in the presence of a fourth solvent selected from the group consisting of .water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar$aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
14. The process of claim 13, wherein the acetylating agent used in step-(d) is acetic anhydride or a combination with sodium acetate; wherein the base used in step-(d) is selected from the group consisting of aqueous ammonia, sodium hydroxide^ealcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide,-isodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate;llithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butox e; and wherein the fourth solvent used in step-(d) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, Ν,Ν-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
15. The process of claim 1, wherein the methylating agent used in step-(e) is dimethyl sulfate or methyl iodide; wherein the reaction in step-(e) is optionally carried out in the presence of a base; and_wherein^the-methy-lation4n-step-(e)-is-carried -out in the
presence of a fifth solvent selected from the group consisting of water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
16. The process of claim 15, wherein the methylating agent used in step-(e) is dimethyl sulfate; wherein the base used in step-(e) is selected from the group consisting of aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and wherein the fifth solvent used in step-(e) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, Ν,Ν-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
17. A process for the preparation of (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III:



18. The process of claim 17, wherein the reaction is carried out in the presence of a carboxylic acid and an organic amine; wherein the reaction is carried out in the presence of a first solvent selected from the group consisting of a hydrocarbon solvent, a halogenated hydrocarbon solvent, an ether, and mixtures thereof; and wherein the reaction is carried out at a temperature of about 25°C to the reflux temperature of the solvent used.
19. A process for the preparation of 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV:

with a suitable reducing agent to produce the compound of formula IV or an acid addition salt thereof.
20. The process of claim 19, wherein the reduction is carried out in the presence of a second solvent; wherein the reducing agent is selected from the group consisting of hydrbgenation catalysts (metal catalysts), hydride agents, borane and borane-THF complex; and wherein the reduction is optionally carried out in the presence of ammonia.
21. The process of claim 20, wherein the second solvent is selected from the group consisting of water, an alcohol, an ester, a hydrocarbon solvent, an ether, and mixtures
-thereof; wherein-the-redueing-is-seleeted~-fr^
palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, lithium aluminum hydride, sodium borohydride/Lewis acid, lithium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2-methoxyethoxy)aluminium hydride (Vitride), borane and borane-THF complex; and wherein the ammonia is used in the form of aqueous ammonia or in the form of ammonia gas or ammonia saturated in an organic solvent.
22. A process for the preparation of 2-(7-hydroxy-l-naphthyl)ethanamine of formula V:

or an acid addition salt thereof, comprising subjecting 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV:

23. The process of claim 22, wherein the reagent suitable for facilitating the aromatization reaction is selected from the group consisting of sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, metal catalysts and quinone derivatives; wherein the starting material 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV is employed in the form of an acid addition salt; and wherein the aromatization is optionally carried out in the presence of a third solvent.
24. The process of claim 23, wherein the reagent suitable for facilitating the aromatization reaction is selected from the group consisting of sulfur, selenium metal, aqueous hydrobromic acid, palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-
 dibromo-5,6-dicyano benzoquinone, 2,3-dicyano-4-chlorobenzoquinone, 2,3- dicyanobenzoquinone, 1,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil and o-floranil; wherein the starting material 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV is employed in the form of its hydrochloride salt; and wherein the third solvent is selected from the group consisting of water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
dibromo-5,6-dicyano benzoquinone, 2,3-dicyano-4-chlorobenzoquinone, 2,3- dicyanobenzoquinone, 1,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil and o-floranil; wherein the starting material 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV is employed in the form of its hydrochloride salt; and wherein the third solvent is selected from the group consisting of water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
25. The process of claim 24, wherein the reagent suitable for facilitating the aromatization reaction is selected from the group consisting of sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil); and wherein the third solvent is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N- dimethylformamide, Ν,Ν-dimethylacetamide, dimethylsulfoxide, and mixtures thereof.
26. The process of claim 22, wherein the aromatization is carried out as a neat reaction, in the absence of solvent; and wherein the acid addition salts of 2-(7-hydroxy-l- naphthyl)ethanamine of formula V prepared are 2-(7-hydroxy-l-naphthyl)ethanamine hydrochloride salt and 2-(7-hydroxy-l-naphthyl)ethanamine hydrobromide salt.
27. A process for the preparation of N-[2-(7 -hydroxy- l-naphthyl)ethyl]acetamide of formula VI:


28. The process of claim 27, wherein the acetylating is selected from the group consisting of acetyl chloride, acetyl bromide, acetyl iodide, acetic anhydride, sodium acetate, or a combination thereof; wherein the reaction is optionally carried out in the presence of a base; and wherein the acetylation is carried out in the presence of a fourth solvent selected from the group consisting of water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
29. The process of claim 28, wherein the acetylating agent is acetic anhydride or a combination with sodium acetate; wherein the base is selected from the group consisting of aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert- butoxide, sodium isopropoxide and potassium tert-butoxide; and wherein the fourth solvent is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N- dimethylformamide, Ν,Ν-dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
30. A process for the preparation of Agomelatine, N-[2-(7-methoxy-l-naphthalenyl) ethyl] acetamide, of formula I:


31. The process of claim 30, wherein the methylating agent is dimethyl sulfate or methyl iodide; wherein the reaction is optionally carried out in the presence of a base; and wherein the methylation is carried out in the presence of a fifth solvent selected from the group consisting of water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
32. The process of claim 31, wherein the methylating agent is dimethyl sulfate; wherein the base is selected from the group consisting of aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert- butoxide; and wherein the fifth solvent is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-
dimethylformamide, Ν,Ν-dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
33. A process for the preparation of agomelatine, N-[2-(7-methoxy-l-naphthalenyl) ethyl] acetamide, of formula I:

a) reacting 7-hydroxy-l-tetralone of formula II:


or a salt thereof, to produce (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III:

or a salt thereof;
b) reducing the compound of formula III with a suitable reducing agent to produce 2- (7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV:
 or an acid addition salt thereof;
or an acid addition salt thereof;
c) acetylating the compound of formula IV with a suitable acetylating agent to produce N-[2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethyl]acetamide, of formula VIII:

d) subjecting the compound of formula VIII to aromatization by reacting with a suitable reagent to produce N-[2-(7-Hydroxy-l-naphthyl)ethyl]acetamide of formula VI:

e) reacting the compound of formula VI with a methylating agent to produce the
agomelatine of formula I.
34. The process of claim 33, wherein the reaction in step-(a) is carried out in the presence of a carboxylic acid and an organic amine; wherein the reaction in step-(a) is carried out in the presence of a first solvent; and wherein the reaction in step-(a) is carried out at a temperature of about 25°C to the reflux temperature of the solvent used.
35. The process of claim 34, wherein the carboxylic acid used in step-(a) is a linear or branched alkyl carboxylic acid, an unsubstituted or substituted aryl carboxylic acid, or an unsubstituted or substituted linear or branched aralkyl carboxylic acid; wherein the organic amine used in step-(a) is a linear or branched alkyl amine, an unsubstituted or substituted aryl amine, or an unsubstituted or substituted linear or branched aralkyl amine; wherein the first solvent used in step-(a) is selected from the group consisting of a hydrocarbon solvent, a halogenated hydrocarbon solvent, an ether, and mixtures thereof; and wherein the reaction in step-(a) is carried out at a temperature of about 60°C to the reflux temperature of the solvent used.
36. The process of claim 35, wherein the carboxylic acid used in step-(a) is 4- hydroxybenzoic acid and wherein the organic amine used in step-(a) is benzyl amine or morpholine; wherein the first solvent used in step-(a) is selected from the group consisting of toluene, xylene, anisole, ethylbenzene, tetrachloroethylene, cyclohexene, mesitylene, and mixtures thereof; and wherein the reaction in step-(a) is carried out at the reflux temperature of the solvent used.
37. The process of claim 33, wherein the reduction in step-(b) is carried out in the presence of a second solvent; wherein the reducing agent used in step-(b) is selected from the group consisting of hydrogenation catalysts (metal catalysts), hydride agents, borane and borane-THF complex; and wherein the reduction in step-(b) is optionally carried out in the presence of ammonia.
38. The process of claim 37, wherein the second solvent used in step-(b) is selected from the group consisting of water, an alcohol, an ester, a hydrocarbon solvent, an ether, and mixtures thereof; wherein the reducing agent used in step-(b) is selected from the group consisting of platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, lithium aluminum hydride, sodium borohydride/Lewis acid, lithium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2- methoxyethoxy)aluminium hydride (Vitride), borane and borane-THF complex; and wherein the ammonia is used in the form of aqueous ammonia or in the form of ammonia gas or ammonia saturated in an organic solvent.
39. The process of claim 38, wherein the second solvent used in step-(b) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, tert-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2- methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, and mixtures thereof; and wherein the reducing agent used in step-(b) is Raney-Nickel.
40. The process of claim 33, wherein the acetylating agent used in step-(c) is selected from the group consisting of acetyl chloride, acetyl bromide, acetyl iodide, acetic anhydride, sodium acetate, or a combination thereof; wherein the reaction in step-(c) is optionally carried out in the presence of a base; and wherein the acetylation in step-(c) is carried out in the presence of a fourth solvent selected from the group consisting of water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
41. The process of claim 40, wherein the acetylating agent used in step-(c) is acetic anhydride or a combination with sodium acetate; wherein the base used in step-(c) is selected from the group consisting of aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and wherein the fourth solvent used in step-(c) is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, Ν,Ν-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
42. The process of claim 33, wherein the reagent suitable for facilitating the aromatization reaction in step-(d) is selected from the group consisting of. sulfur or its derivatives,
 wherein the starting material 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV in step-(d) is employed in the form of an acid addition salt; and wherein the aromatization in step-(d) is optionally carried out in the presence of a third solvent.
wherein the starting material 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV in step-(d) is employed in the form of an acid addition salt; and wherein the aromatization in step-(d) is optionally carried out in the presence of a third solvent.
43. The process of claim 42, wherein the reagent suitable for facilitating the aromatization reaction in step-(d) is selected from the group consisting of sulfur, selenium metal, aqueous hydrobromic acid, palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), p-bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6-dicyano benzoquinone, 2,3-dicyano-4-chlorobenzoquinone, 2,3- dicyanobenzoquinone, 1,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil and o-floranil; wherein the starting material 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV in step-(d) is employed in the form of its hydrochloride salt; and wherein the third solvent used in step-(d) is selected from the group consisting of water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
44. The process of claim 43, wherein the reagent suitable for facilitating the aromatization reaction in step-(d) is selected from the group consisting of sulfur, aqueous hydrobromic acid and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil); and wherein the third solvent used in step-(d) is selected from the group consisting of water, dichloromethane, dichloroethane, chloroform, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, n-pentane, n-hexane, n- heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, N,N-dimethylacetamide, dimethylsulfoxide, and mixtures thereof.
45. The process of claim 33, wherein the aromatization in step-(d) is carried out as a neat reaction, in the absence of solvent.
46. The process of claim 33, wherein the methylating agent used in step-(e) is dimethyl sulfate or methyl iodide; wherein the reaction- in step-(e) is optionally carried out in the presence of a base; and wherein the methylation in step-(e) is carried out in the presence of a fifth solvent selected from the group consisting of water, a hydrocarbon
solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
47. The process of claim 46, wherein the methylating agent used in step-(e) is dimethyl sulfate; wherein the base used in step-(e) is selected from the group consisting of aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and wherein the fifth solvent used in step-(e) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, Ν,Ν-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
48. A process for the preparation of N-[2-(7-hydroxy-3,4-dihydro-l-naphthalenyl) ethyl]acetamide of formula VIII:


or an acid addition salt thereof, with a suitable acetylating agent to produce the compound of formula VIII or a salt thereof.
49. The process of claim 48, wherein the acetylating is selected from the group consisting of acetyl chloride, acetyl bromide, acetyl iodide, acetic anhydride, sodium acetate, or a combination thereof; wherein the reaction is optionally carried out in the presence of a base; and wherein the acetylation is carried out in the presence of a fourth solvent selected from the group consisting of water, an alcohol, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof
50. The process of claim 49, wherein the acetylating agent is acetic anhydride or a combination with sodium acetate; wherein the base is selected from the group consisting of aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert- butoxide, sodium isopropoxide and potassium tert-butoxide; and wherein the fourth solvent is selected from the group consisting of water, methanol, ethanol, isopropanol, n-butanol, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n-hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N- dimethylformamide, Ν,Ν-dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
51. A process for the preparation of N-[2-(7-hydroxy-l-naphthyl)ethyl]acetamide of formula VI:


52. The process of claim 51, wherein the reagent suitable for facilitating the aromatization reaction is selected from the group consisting of sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, metal catalysts and quinone derivatives; wherein the starting material 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV is employed in the form of an acid addition salt; and wherein the aromatization is optionally carried out in the presence of a third solvent.
53. The process of claim 52, wherein the reagent suitable for facilitating the aromatization reaction is selected from the group consisting of sulfur, selenium metal, aqueous hydrobromic acid, palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p- Chloranil), p-bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3- dibromo-5,6-dicyano benzoquinone, 2,3-dicyano-4-chlorobenzoquinone, 2,3- dicyanobenzoquinone, 1 ,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil and o-floranil; wherein the starting material 2-(7-hydroxy-3,4-dihydro-l- naphthalenyl)ethanamine of formula IV is employed in the form of its hydrochloride salt; and wherein the third solvent is selected from the group consisting of water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
54. The process of claim 51, wherein the aromatization is carried out as a neat reaction, in the absence of solvent.
55. A process for the preparation of agomelatine, N-[2-(7-methoxy-l- naphthalenyl)ethyl]acetamide, of formula I:

a) reacting 7-hydroxy- 1 -tetralone of formula II :

HO.
CN or a salt thereof, to produce (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III:

or a salt thereof;
b) subjecting the compound of formula III to aromatization by reacting with a suitable reagent to produce (7-hydroxy- l-naphthyl)acetonitrile, of formula VII:

or a salt thereof; and
c) reducing the compound of formula VII with a suitable reducing agent to produce 2- (7-hydroxy- 1 -naphthyl)ethanamine of formula V:

d) acetylating the compound of formula V with a suitable acetylating agent to produce N-[2-(7-Hydroxy- 1 -naphthyl)ethyl]acetamide of formula VI:

or a salt thereof; and
e) reacting the compound of formula VI with a methylating agent to produce the agomelatine of formula I.
56. The process of claim 55, wherein the reagent suitable for facilitating the aromatization reaction in step-(b) is selected from the group consisting of sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, metal catalysts and quinone derivatives; and wherein the aromatization in step-(b) is optionally carried out in the presence of a third solvent.
57. The process of claim 56, wherein the reagent suitable for facilitating the aromatization reaction in step-(b) is selected from the group consisting of sulfur, selenium metal, aqueous hydrobromic acid, palladium on carbon in various percentages, platinum oxide, raney nickel, palladium oxide, 2,3,5,6-tetrachlorocyclohexa-2,5-diene-l,4-dione (p-Chloranil), p-bromanil, p-floranil, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ), 2,3-dibromo-5,6-dicyano benzoquinone, 2,3-dicyano-4-chlorobenzoquinone, 2,3- dicyanobenzoquinone, 1,2-benzoquinones, 1,3-benzoquinones, o-chloranil, o-bromanil
_ _and Q-flQranil;_and_wherein he hird-solvent-used n-step-(b)-is seleeted-from the group
consisting of water, a halogenated hydrocarbon, an ester, a hydrocarbon, an ether, a polar aprotic solvent, and mixtures thereof.
58. The process of claim 55, wherein the aromatization in step-(b) is carried out as a neat reaction, in the absence of solvent.
59. The process of claim 55, wherein the reduction in step-(c) is carried out in the presence of a second solvent; wherein the reducing agent used in step-(c) is selected from the group consisting of hydrogenation catalysts (metal catalysts), hydride agents, borane and borane-THF complex; and wherein the reduction in step-(c) is optionally carried out in the presence of ammonia.
60. The process of claim 59, wherein the second solvent used in step-(c) is selected from the group consisting of water, an alcohol, an ester, a hydrocarbon solvent, an ether, and mixtures thereof; wherein the reducing agent used in step-(c) is selected from the group consisting of platinum, palladium, palladium hydroxide, palladium on carbon, platinum oxide, rhodium, Raney-Nickel, lithium aluminum hydride, sodium borohydride/Lewis acid, lithium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride (DIBAL-H), lithium tri-tert-butoxyaluminum hydride, sodium bis(2- methoxyethoxy)aluminium hydride (Vitride), borane and borane-THF complex; and wherein the ammonia is used in the form of aqueous ammonia or in the form of ammonia gas or ammonia saturated in an organic solvent.
61. A process for the preparation of (7-hydroxy-l-naphthyl)acetonitrile of formula VII:

or a salt thereof, comprising subjecting (7-hydroxy-3,4-dihydro-l- naphthalenyl)acetonitrile of formula III:

62. The process of claim 61, wherein the reagent suitable for facilitating the aromatization reaction is selected from the group consisting of sulfur or its derivatives, selenium metal, aqueous hydrobromic acid, metal catalysts and quinone derivatives; and wherein the aromatization is optionally carried out in the presence of a third solvent.
63. A process for the preparation of 2- 7-hydroxy-l-naphthyl)ethanamine of formula V:

or an acid addition salt thereof, comprising reducing (7-hydroxy-l- naphthyl)acetonitrile of formula VII:

with a suitable reducing agent to produce the compound of formula V or an acid addition salt thereof.
64. The process of claim 63, wherein the reduction is carried out in the presence of a second solvent; wherein the reducing agent is selected from the group consisting of hydrogenation catalysts (metal catalysts), hydride agents, borane and borane-THF complex; and wherein the reduction is optionally carried out in the presence of ammonia.
65. A process for the preparation of agomelatine, N-[2-(7-methoxy-l- naphthalenyl)ethyl]acetamide, of formula I:

a) reacting 7-hydroxy- 1 -tetralone of formula II:


or a salt thereof, to produce (7-hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile of formula III:

or a salt thereof;
b) subjecting the compound of formula III to aromatization by reacting with a
reagent to produce (7-hydroxy- l-naphthyl)acetonitrile, of formula VII:

or a salt thereof;
c) reacting the compound of formula VII with a methylating agent to produce (7- methoxy- 1 -naphthyl)acetonitrile of formula IX:

d) reducing the compound of formula IX with a suitable reducing agent to produce 2- (7-methoxy-l-naphthyl)ethanamine of formula XI:

e) acetylating the compound of formula XI with a suitable acetylating agent to produce the agomelatine of formula I.
66. The process of claim 65, wherein the methylating agent used in step-(c) is dimethyl sulfate or methyl iodide; wherein the reaction in step-(c) is optionally carried out in the presence of a base; and wherein the methylation in step-(c) is carried out in the presence of a fifth solvent selected from the group consisting of water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
67. The process of claim 66, wherein the methylating agent used in step-(c) is dimethyl sulfate; wherein the base used in step-(c) is selected from the group consisting of aqueous ammonia, sodium hydroxide, calcium hydroxide, magnesium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, lithium carbonate, sodium tert-butoxide, sodium isopropoxide and potassium tert-butoxide; and wherein the fifth solvent used in step-(c) is selected from the group consisting of water, ethyl acetate, methyl acetate, isopropyl acetate, tert-butyl methyl acetate, ethyl formate, acetone, n-pentane, n- hexane, n-heptane, cyclohexane, toluene, xylene, tetrahydrofuran, 2-methyl tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, methyl tert-butyl ether, monoglyme, diglyme, acetonitrile, propionitrile, N,N-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, dichloromethane, dichloroethane, chloroform, and mixtures thereof.
68. -A-process-for he-prepar-ation-of-(-7-methoxy^naphthyl)ae
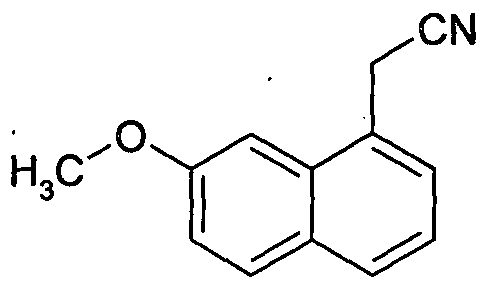
or a salt thereof, comprising reacting (7-hydroxy-l-naphthyl)acetonitrile of formula VII:
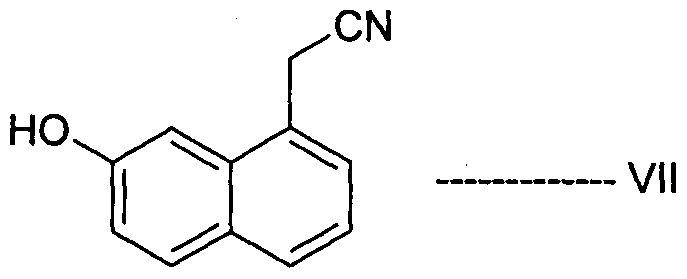
69. The process of claim 68, wherein the methylating agent is dimethyl sulfate or methyl iodide; wherein the reaction is optionally carried out in the presence of a base; and wherein the methylation is carried out in the presence of a fifth solvent selected from the group consisting of water, a hydrocarbon solvent, an ester, a ketone, an ether, a nitrile, a polar aprotic solvent, a halogenated hydrocarbon solvent, and mixtures thereof.
70. A compound, (7-Hydroxy-3,4-dihydro-l-naphthalenyl)acetonitrile, of formula III:
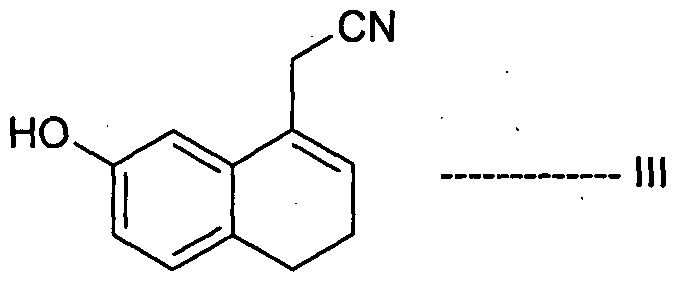
or a salt thereof.
71. A compound, 2-(7-Hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine, of formula IV:

or an acid addition salt thereof.
72. A compound, 2-(7-Hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrochloride salt, of formulajVaL

73. A compound, 2-(7-Hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine hydrobromide salt, of formula IVb:

74. A compound, 2-(7-Hydroxy-l-naphthyl)ethanamine, of formula V:

75. A compound, 2-(7-hydroxy-l-naphthyl)ethanamine hydrochloride salt, of formula Va:

76. A compound, (7-hydroxy-l-naphthyl)acetonitrile, of formula VII:

A compound, N-[2-(7-hydroxy-3,4-dihydro- 1 -naphthalenyl)ethyl]acetamide, formula VIII:

78. Use of the compounds of formulae III, IV, IVa, IVb, V, Va, VII and VIII as claimed in claims 70 to 77, in the process for manufacture of agomelatine or a salt thereof.
79. A compound, 2-(7-hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine, of formula XII:

80. A compound, 2-(7-hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine hydrochloride salt, of formula Xlla:

81. A compound, N-[2-(7-hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethyl]acetamide, of formula XIII:

82. Use of the compounds of formulae XII, Xlla and XIII as claimed in claims 79 to 81, in the process for manufacture of agomelatine or a salt thereof.
83. A process for the preparation of 2-(7-hydroxy-l-naphthyl)ethanamine of formula V:

a) reacting 2-(7-hydroxy-3,4-dihydro-l-naphthalenyl)ethanamine of formula IV or an acid addition salt thereof with aqueous hydrobromic acid to produce a reaction mass containing 2-(7-hydroxy-l-naphthyl)ethanamine of formula V and 2-(7- hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine of formula XII:

(V) (XII) or an acid addition salt thereof;
b) isolating and/or recovering the pure 2-(7-hydroxy-l-naphthyl)ethanamine of formula V or an acid addition salt thereof from the reaction mass obtained in step- (a);
c) treating the mother liquors with a suitable base to adjust the pH to produce cause precipitation;
d) optionally, extracting the 2-(7-hydroxy-l,2,3,4-tetrahydro-l-naphthyl)ethanamine of formula XII obtained in step-(c) into an organic solvent;
e) isolating and/or recovering the pure 2-(7-hydroxy-l,2,3,4-tetrahydro-l- naphthyl)ethanamine of formula XII or an acid addition salt thereof from the reaction mass obtained in step-(c) or from the organic layer obtained in step-(d); and
f) subjecting the compound of formula XII or an acid addition salt to aromatization by reacting with a suitable reagent to produce the compound of formula V.
84. A process for the preparation of N-[2-(7 -hydroxy- 1,2,3, 4-tetrahydro-l-naphthyl)ethyl] acetamide of formula XIII:
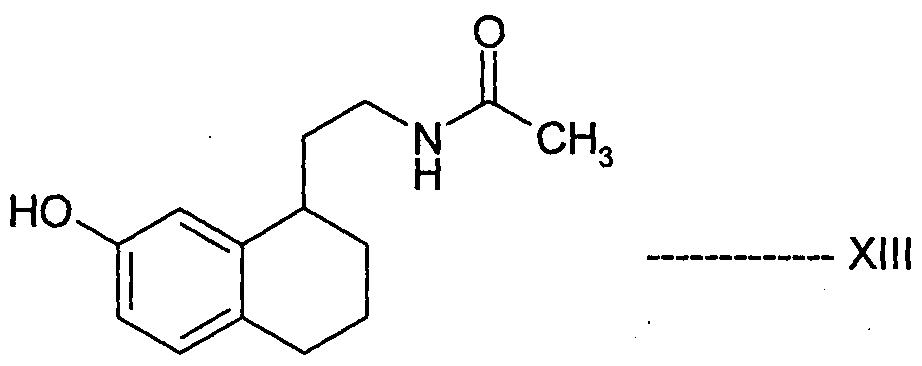
or a salt thereof, comprising acetylating 2-(7-hydroxy-l,2,3,4-tetrahydro-l- naphthyl)ethanamine, of formula XII:

Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12887041.7A EP2909166A4 (en) | 2012-10-22 | 2012-10-22 | Processes for the preparation of agomelatine using novel intermediates |
| PCT/IN2012/000694 WO2014064706A1 (en) | 2012-10-22 | 2012-10-22 | Processes for the preparation of agomelatine using novel intermediates |
| ARP130103817A AR093245A1 (en) | 2012-10-22 | 2013-10-22 | PROCESSES FOR THE PREPARATION OF AGOMELATIN USING INTERMEDIARIES |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IN2012/000694 WO2014064706A1 (en) | 2012-10-22 | 2012-10-22 | Processes for the preparation of agomelatine using novel intermediates |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014064706A1 true WO2014064706A1 (en) | 2014-05-01 |
Family
ID=50544131
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2012/000694 WO2014064706A1 (en) | 2012-10-22 | 2012-10-22 | Processes for the preparation of agomelatine using novel intermediates |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP2909166A4 (en) |
| AR (1) | AR093245A1 (en) |
| WO (1) | WO2014064706A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016046833A3 (en) * | 2014-09-24 | 2016-05-19 | Symed Labs Limited | Improved processes for the preparation of agomelatine using novel intermediates |
| CN107353229A (en) * | 2017-08-08 | 2017-11-17 | 许昌恒生制药有限公司 | A kind of preparation method of agomelatine intermediate body |
| CN113527139A (en) * | 2020-04-17 | 2021-10-22 | 上海法默生物科技有限公司 | Method for synthesizing 7-methoxy-1-naphthylacetonitrile and intermediate |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5071875A (en) * | 1989-09-25 | 1991-12-10 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5420158A (en) * | 1992-03-27 | 1995-05-30 | Adir Et Compagnie | Naphthylalkylamines |
| US5449689A (en) * | 1991-08-23 | 1995-09-12 | Adir Et Compagnie | Naphthylethylureas and naphthylethylthioureas |
| US7250531B2 (en) * | 2004-02-13 | 2007-07-31 | Les Laboratoires Servier | Process for the synthesis and crystalline form of agomelatine |
| US20100004340A1 (en) * | 2007-05-01 | 2010-01-07 | Concert Pharmaceuticals, Inc. | Naphthyl(ethyl) acetamides |
| WO2012093402A1 (en) * | 2011-01-04 | 2012-07-12 | Symed Labs Limited | Processes for the preparation of n-[2-(7-methoxy-1-naphthyl)ethyl]acetamide |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2658818B1 (en) * | 1990-02-27 | 1993-12-31 | Adir Cie | NOVEL DERIVATIVES WITH NAPHTHALENIC STRUCTURE, PROCESS FOR THEIR PREPARATION AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| FR2734814B1 (en) * | 1995-05-31 | 1997-07-04 | Adir | NOVEL ALKOXY-ARYL COMPOUNDS, PROCESS FOR THEIR PREPARATION AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
-
2012
- 2012-10-22 EP EP12887041.7A patent/EP2909166A4/en not_active Withdrawn
- 2012-10-22 WO PCT/IN2012/000694 patent/WO2014064706A1/en active Application Filing
-
2013
- 2013-10-22 AR ARP130103817A patent/AR093245A1/en unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5071875A (en) * | 1989-09-25 | 1991-12-10 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5449689A (en) * | 1991-08-23 | 1995-09-12 | Adir Et Compagnie | Naphthylethylureas and naphthylethylthioureas |
| US5420158A (en) * | 1992-03-27 | 1995-05-30 | Adir Et Compagnie | Naphthylalkylamines |
| US7250531B2 (en) * | 2004-02-13 | 2007-07-31 | Les Laboratoires Servier | Process for the synthesis and crystalline form of agomelatine |
| US20100004340A1 (en) * | 2007-05-01 | 2010-01-07 | Concert Pharmaceuticals, Inc. | Naphthyl(ethyl) acetamides |
| WO2012093402A1 (en) * | 2011-01-04 | 2012-07-12 | Symed Labs Limited | Processes for the preparation of n-[2-(7-methoxy-1-naphthyl)ethyl]acetamide |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2909166A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016046833A3 (en) * | 2014-09-24 | 2016-05-19 | Symed Labs Limited | Improved processes for the preparation of agomelatine using novel intermediates |
| CN107353229A (en) * | 2017-08-08 | 2017-11-17 | 许昌恒生制药有限公司 | A kind of preparation method of agomelatine intermediate body |
| CN113527139A (en) * | 2020-04-17 | 2021-10-22 | 上海法默生物科技有限公司 | Method for synthesizing 7-methoxy-1-naphthylacetonitrile and intermediate |
Also Published As
| Publication number | Publication date |
|---|---|
| AR093245A1 (en) | 2015-05-27 |
| EP2909166A1 (en) | 2015-08-26 |
| EP2909166A4 (en) | 2016-10-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2005200616B8 (en) | New process for the synthesis and new crystalline form of agomelatine and pharmaceutical compositions containing it | |
| WO2010128518A2 (en) | Novel process for the preparation of cisatracurium besylate | |
| AU2005273430B2 (en) | Methods for producing isoindole derivatives | |
| WO2016055015A1 (en) | Method for preparing sitagliptin intermediate via asymmetrical reduction method | |
| WO2008035380A2 (en) | An improved process for the preparation of high purity formoterol and its pharmaceutically acceptable salts | |
| EP2909166A1 (en) | Processes for the preparation of agomelatine using novel intermediates | |
| NZ578381A (en) | New process for the synthesis of agomelatine or N-[2-(7-methoxy-1-naphthyl)ethyl]acetamide | |
| EP3413891A1 (en) | Processes for the preparation of highly pure prucalopride succinate and its intermediates | |
| TW200831478A (en) | Chromane derivatives, synthesis thereof, and intermediates thereto | |
| IL223180A (en) | Agomelatine intermediates and preparation method thereof | |
| WO2018010651A1 (en) | Method for manufacturing obeticholic acid and intermediate thereof | |
| CN103450027B (en) | The preparation method of cinacalcet hydrochloride | |
| JPH05112525A (en) | Process for manufacturing 4-hydroxy-2-oxopyrrolidin-1-yl acetamide | |
| WO2014001939A1 (en) | Process for preparation of agomelatine and crystalline form i thereof | |
| WO2016046833A2 (en) | Improved processes for the preparation of agomelatine using novel intermediates | |
| JPS6121621B2 (en) | ||
| US20050033088A1 (en) | Process for the preparation of phenethylamine derivative, an intermediate of Venlafaxine hydrochloride | |
| MXPA06007686A (en) | Process for obtaining tolterodine tartrate. | |
| JP5564060B2 (en) | Method for producing olopatadine and / or a pharmaceutically acceptable salt thereof | |
| TW202019872A (en) | Preparation method for fused tricyclic [gamma]-amino acid derivative and intermediate thereof | |
| EP2984070B1 (en) | N-haloalkylindoline intermediates, their process and use in preparation of silodosin and its derivatives | |
| CA2484585A1 (en) | A process for the preparation of benazepril hydrochloride | |
| PL82685B1 (en) | Dibenzazepine derivatives and their preparation[ie34441b1] | |
| GB1568976A (en) | Cis-4-phenyl-isoquinolines | |
| US8674120B2 (en) | Process for manufacturing zeranol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12887041 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012887041 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012887041 Country of ref document: EP |