WO2014007357A1 - Lithium composite metal oxide, positive electrode active substance, positive electrode, and non-aqueous electrolyte secondary battery - Google Patents
Lithium composite metal oxide, positive electrode active substance, positive electrode, and non-aqueous electrolyte secondary battery Download PDFInfo
- Publication number
- WO2014007357A1 WO2014007357A1 PCT/JP2013/068451 JP2013068451W WO2014007357A1 WO 2014007357 A1 WO2014007357 A1 WO 2014007357A1 JP 2013068451 W JP2013068451 W JP 2013068451W WO 2014007357 A1 WO2014007357 A1 WO 2014007357A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- metal oxide
- composite metal
- lithium composite
- positive electrode
- secondary battery
- Prior art date
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/40—Nickelates
- C01G53/42—Nickelates containing alkali metals, e.g. LiNiO2
- C01G53/44—Nickelates containing alkali metals, e.g. LiNiO2 containing manganese
- C01G53/50—Nickelates containing alkali metals, e.g. LiNiO2 containing manganese of the type [MnO2]n-, e.g. Li(NixMn1-x)O2, Li(MyNixMn1-x-y)O2
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/76—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by a space-group or by other symmetry indications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/80—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70
- C01P2002/85—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70 by XPS, EDX or EDAX data
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/61—Micrometer sized, i.e. from 1-100 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M2004/021—Physical characteristics, e.g. porosity, surface area
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present invention relates to a lithium composite metal oxide, a positive electrode active material, a positive electrode, and a nonaqueous electrolyte secondary battery.
- Lithium composite metal oxide is used as a positive electrode active material in nonaqueous electrolyte secondary batteries such as lithium secondary batteries.
- Lithium secondary batteries have already been put into practical use as small power sources for mobile phones and notebook computers, and are also being applied to medium and large power sources for automobiles and power storage.
- LiCoO 2 is most widely used as a positive electrode active material in commercially available lithium secondary batteries.
- Co is expensive, lithium composite metal oxidation in which the content of Co is lower than that of LiCoO 2 is used. Things are being researched.
- lithium composite metal oxides containing Ni and Mn are considered promising.
- a lithium composite metal oxide having a composition formula of LiNi 1/3 Co 1/3 Mn 1/3 O 2 is known.
- a high-capacity non-aqueous electrolyte can be obtained by a method for controlling an oxidation state or a method for controlling an average crystal structure obtained by X-ray diffraction measurement.
- Studies have been made to obtain lithium metal composite oxides suitable for secondary batteries.
- a method for obtaining a lithium metal composite oxide suitable for a high-capacity nonaqueous electrolyte secondary battery by controlling the local structure at the atomic level has not yet been sufficiently studied. The present invention has been made in view of such circumstances. By controlling the local structure at the atomic level, a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery exhibiting a higher capacity than before is obtained.
- one embodiment of the present invention is a lithium composite metal oxide containing Mn, Ni, Li, and Co, which satisfies the following (a) and (b): is there.
- (A) In a radial distribution function obtained by Fourier transform of a wide-range X-ray absorption fine structure (EXAFS) spectrum at the K absorption edge of Mn in the lithium composite metal oxide, 1.5% by oxygen atoms bonded to Mn atoms is obtained.
- the intensity I ANi when the next on the intensity of the second closest peak around 2.5 ⁇ by metal atoms near the Ni atom bonded oxygen atom to Ni atom was I BNi, the I BNi / I ANi, 1. It is 0 or more and 1.7 or less.
- a Li / A when the amount (mol) of Li is A Li and the amount (mol) of a metal other than Li is A, A Li / A is 0.7 or more and 1.4 or less. It is desirable.
- it is desirable that a product of the I BMn / I AMn and the I BNi / I ANi is 0.7 or more and 2.0 or less.
- the lithium composite metal oxide has a layered structure and is represented by Formula (1).
- M is selected from the group consisting of Al, Mg, Ti, Ca, Cu, Zn, Fe, Cr, Mo, Si, Sn, Nb and V One or more elements.
- the M is preferably Fe.
- ⁇ 0.
- Another embodiment of the present invention is a positive electrode active material including the above lithium composite metal oxide.
- Another embodiment of the present invention is a positive electrode including the above positive electrode active material.
- Another embodiment of the present invention is a nonaqueous electrolyte secondary battery including a negative electrode and the positive electrode described above.
- the separator is preferably made of a laminated film in which a heat-resistant porous layer and a porous film are laminated.
- the lithium composite metal oxide of this embodiment contains Mn, Ni, Li and Co and satisfies the following (a) and (b).
- (A) In a radial distribution function obtained by Fourier transform of a wide-range X-ray absorption fine structure (EXAFS) spectrum at the K absorption edge of Mn in the lithium composite metal oxide, 1.5% by oxygen atoms bonded to Mn atoms Near first adjacent peak A Mn Strength of I AMn , The second adjacent peak B around 2.5 ⁇ due to the metal atom next to the Mn atom next to the oxygen atom bonded to the Mn atom Mn Strength of I BMn When I BMn / I AMn Is 0.5 or more and 1.2 or less.
- EXAFS X-ray absorption fine structure
- the EXAFS spectrum used in the present embodiment is handled in the same manner as a general EXAFS spectrum.
- the measurement and principle of the EXAFS spectrum are described in, for example, “X-ray absorption spectroscopy—XAFS and its applications” (Toshiaki Ohta (2002)).
- the principle is as follows. First, when an X-ray having a specific wavelength is transmitted through a substance to be measured, the intensity of the X-ray irradiated to the substance (incident X-ray intensity: I 0 ) And the intensity of X-rays transmitted through the substance (transmitted X-ray intensity: I t ), The X-ray absorbance per unit thickness is obtained for the substance to be measured at a specific wavelength.
- the wavelength of X-rays irradiating the substance is changed (that is, the energy (eV) of incident X-rays is changed), the X-ray absorbance of each wavelength (each energy) with respect to the X-rays is measured,
- the absorption edge corresponds to the energy level of the atomic shell of the atoms constituting the material and is unique to each atom. For example, an absorption edge corresponding to the K shell of an atom is called a K absorption edge.
- EXAFS broad X-ray absorption fine structure
- the intensity of the peak of the radial distribution function is affected by the number of X-ray scattering atoms, but it also affects the isotropy of the interatomic distance between X-ray absorbing atoms and X-ray scattering atoms. Is done.
- the peak of the radial distribution function For those with high intensity, the interatomic distance between the X-ray absorbing atom and the X-ray scattering atom is isotropic with no difference in direction, and the distance distribution between the X-ray absorbing atom and the X-ray scattering atom is small. Means. Therefore, in the present embodiment, attention is paid to the peak intensity ratio of the radial distribution function obtained at the K absorption edge of Mn and Ni.
- the intensity ratio of the peak of the radial distribution function within a certain range, the atomic level local structure in the lithium composite metal oxide can be controlled to a specific condition even for samples with different composition ratios.
- a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery exhibiting a higher capacity than before can be obtained.
- the peak due to O (oxygen atom) bonded to the Mn atom in the radial distribution function of the K absorption edge of Mn is the first proximity peak A.
- Mn And First proximity peak A Mn Preferably appears in the vicinity of 1.5 to 1.9 to 1.9, more preferably 1.5 to 1.6.
- an atom X next to Mn atom next to O bonded to Mn atom (where X is Li, Mn , And a peak due to a metal atom such as Ni).
- Mn And Second adjacent peak B Mn Preferably appears in the vicinity of 2.5 mm from 2.44 mm to 2.55 mm, more preferably from 2.46 mm to 2.55 mm.
- the atom X is bonded to O bonded to the Mn atom.
- the peak due to O bonded to Ni atoms is the first proximity peak A.
- Ni And First proximity peak A Ni Preferably appears in the vicinity of 1.5 to 1.9 to 1.9, more preferably 1.5 to 1.6.
- an atom X that is closest to the Ni atom next to O bonded to the Ni atom (where X is Li, Mn , And a peak due to a metal atom such as Ni).
- Ni And Second adjacent peak B Ni Preferably appears in the vicinity of 2.5 mm from 2.44 mm to 2.55 mm, more preferably from 2.46 mm to 2.55 mm.
- atom X is bonded to O bonded to Ni atom.
- the lithium composite metal oxide of this embodiment has a peak intensity ratio of the radial distribution function, that is, I AMn And I BMn To the ratio (I BMn / I AMn ) And I ANi And I BNi To the ratio (I BNi / I ANi ) Within a specific range, the local structure at the atomic level is controlled.
- Such a lithium composite metal oxide of this embodiment is useful for a non-aqueous electrolyte secondary battery that exhibits a higher capacity than before.
- the lithium composite metal oxide of the present embodiment has a high isotropy of the interatomic distance between O and X around the Mn atom, and falls within a specific range, and thus has high characteristics as a positive electrode active material.
- the lithium composite metal oxide of the present embodiment has a high degree of isotropy in the interatomic distance between O and X around the Ni atom, and falls within a specific range, and thus has high characteristics as a positive electrode active material. .
- I like this BNi / I ANi The value of is 1.0 or more and 1.7 or less, preferably 1.1 or more and 1.7 or less, more preferably 1.2 or more and 1.7 or less.
- I BMn / I AMn Range of values and I BNi / I ANi The range of values can be arbitrarily combined. Furthermore, I BMn / I AMn And I BNi / I ANi Product with (I BMn / I AMn ⁇ I BNi / I ANi )) Has both the isotropicity of the interatomic distance between the O and the atom X around the appropriate Mn atom and the isotropicity of the interatomic distance between the O and the atom X around the proper Ni atom. 7 or more and 2.0 or less, preferably 0.9 or more and 2.0 or less, more preferably 1.1 or more and 2.0 or less.
- the crystal structure of the lithium composite metal oxide of the present embodiment is preferably a layered structure, more preferably a hexagonal crystal structure or a monoclinic crystal structure.
- the hexagonal crystal structure is P3, P3 1 , P3 2 , R3, P-3, R-3, P312, P321, P3 1 12, P3 1 21, P3 2 12, P3 2 21, R32, P3m1, P31m, P3c1, P31c, R3m, R3c, P-31m, P-31c, P-3m1, P-3c1, R-3m, R-3c, P6, P6 1 , P6 5 , P6 2 , P6 4 , P6 3 , P-6, P6 / m, P6 3 / M, P622, P6 1 22, P6 5 22, P6 2 22, P6 4 22, P6 3 22, P6mm, P6cc, P6 3 cm, P6 3 mc, P-6m2, P-6c2, P-62m, P-62c, P6 /
- the monoclinic crystal structure is P2, P2. 1 , C2, Pm, Pc, Cm, Cc, P2 / m, P2 1 / M, C2 / m, P2 / c, P2 1 / C, C2 / c is classified into any one space group selected from the group consisting of C2 / c.
- the crystal structure of the lithium composite metal oxide is a hexagonal crystal structure classified into the space group R-3m, or C2 / m.
- a monoclinic crystal structure classified as follows is particularly preferable.
- the space group of the lithium composite metal oxide of the present embodiment can be confirmed by the following method.
- X-ray powder diffraction measurement was performed on a lithium composite metal oxide using CuK ⁇ as a radiation source and a diffraction angle 2 ⁇ measurement range of 10 ° to 90 °, and then Rietveld analysis was performed based on the results. And determining a crystal structure of the lithium composite metal oxide and a space group in the crystal structure.
- Rietveld analysis is a technique for analyzing the crystal structure of a material using diffraction peak data (diffraction peak intensity, diffraction angle 2 ⁇ ) in powder X-ray diffraction measurement of the material, and is a conventionally used technique.
- the composition of the lithium composite metal oxide in this embodiment is such that the amount (mol) of Li is A Li , When the amount (mole) of metal other than Li is A, A Li / A may be 0.7 or more and 1.4 or less.
- the lithium composite metal oxide in the present embodiment preferably has a layered structure and the composition is represented by the following formula (1). Li 1 + x (Ni 1-x-y- ⁇ Mn y Co ⁇ M ⁇ ) O 2 ...
- M is preferably Fe.
- the lithium composite metal oxide particles of the present embodiment are used as a core material, and B, Al, Ga, In, Si, Ge, Sn, Mg, and the surface of the core material (lithium composite metal oxide particles)
- a compound containing one or more atoms selected from the group consisting of transition metals may be attached.
- at least one selected from the group consisting of B, Al, Mg, Co, Cr, and Mn is preferable, and Al is more preferable because a uniform coating layer can be easily formed.
- Examples of such a compound include oxides, fluorides, sulfides, hydroxides, oxyhydroxides, carbonates, nitrates, organic acid salts and mixtures thereof of the above atoms. Of these, oxides, hydroxides, oxyhydroxides or mixtures thereof are preferred.
- alumina which is an oxide of Al is preferable.
- a step of firing the product obtained in (4) (hereinafter sometimes referred to as “calcined product”) at a temperature of 600 ° C.
- the “oxygen concentration” in the step (4) refers to an average oxygen concentration in the heat treatment space when the space (heat treatment space) for heating the mixture is in the range of 200 ° C. or more and 500 ° C. or less.
- the “oxygen concentration” in step (5) refers to the average oxygen concentration in the heat treatment space when the space (heat treatment space) for calcining the calcined product is in the range of 600 ° C. or higher and 950 ° C. or lower.
- the raw material aqueous solution can be adjusted by dissolving a compound containing Ni, Mn and Co in water.
- the raw material aqueous solution is preferably an aqueous solution obtained by dissolving Ni sulfate, Mn sulfate, and Co sulfate in water.
- each raw material containing Ni, Mn and Co is difficult to dissolve in water, for example, when these raw materials are oxides, hydroxides, metal materials, these raw materials are mixed with sulfuric acid.
- the raw material aqueous solution can be obtained by dissolving in the aqueous solution.
- the alkali used in step (1) includes LiOH (lithium hydroxide), NaOH (sodium hydroxide), KOH (potassium hydroxide), Li 2 CO 3 (Lithium carbonate), Na 2 CO 3 (Sodium carbonate), K 2 CO 3 (Potassium carbonate) and (NH 4 ) 2 CO 3
- One or more salts selected from the group consisting of (ammonium carbonate) can be mentioned.
- the alkali used may be an anhydride or a hydrate.
- An anhydride and a hydrate may be used in combination.
- Aqueous ammonia can also be used as the alkaline aqueous solution.
- the alkali concentration in the alkaline aqueous solution is preferably about 0.5 to 10 M (mol / L), more preferably about 1 to 8 M. Further, from the viewpoint of production cost, NaOH or KOH is preferable as the alkali to be used. NaOH and KOH may be used in combination.
- a contact method in the step (1) (i) a method in which an aqueous alkaline solution is added and mixed, (ii) a method in which an aqueous raw material solution is added and mixed, (iii) an aqueous raw material solution in water And a method of adding and mixing an alkaline aqueous solution. In mixing, it is preferable to involve stirring.
- step (1) a method in which a raw material aqueous solution is added to and mixed with an alkaline aqueous solution is preferable because the change in pH is easily controlled.
- the pH of the alkaline aqueous solution tends to decrease as the raw material aqueous solution is added to and mixed with the alkaline aqueous solution, but the pH is adjusted to 9 or higher, preferably 10 or higher.
- it is preferable that one or both of the raw material aqueous solution and the alkaline aqueous solution are brought into contact with each other while being kept at a temperature of 40 ° C. or higher and 80 ° C.
- step (1) by bringing the raw material aqueous solution into contact with the alkali as described above, a salt containing Ni ions, Mn ions and Co ions is co-precipitated and the co-precipitate salt is dispersed. A slurry can be obtained.
- step (2) a coprecipitate is obtained from the slurry obtained in step (1).
- various methods can be adopted as the method for obtaining the coprecipitate in step (2). However, since the operation is simple, a separation operation for obtaining a solid component such as filtration is performed. Is preferred.
- the coprecipitate can also be obtained by a method of volatilizing the liquid by heating, such as spray drying of the slurry.
- a method of volatilizing the liquid by heating such as spray drying of the slurry.
- the separated coprecipitate is washed and dried in the step (2).
- the alkali remaining in the coprecipitate obtained by washing Ni sulfate, Mn sulfate, or Co sulfate is used as a raw material, SO is released into the raw material aqueous solution. 4 2-
- the amount of ions can be reduced. It is preferable to reduce these by washing because the amount of the inert flux (described later) can be easily controlled.
- a water-soluble organic solvent such as alcohol or acetone may be added to the cleaning liquid.
- the drying of the washed coprecipitate can be performed by heat treatment, but may be performed by air drying, vacuum drying, or a combination thereof.
- the heating temperature is preferably 50 to 300 ° C, more preferably about 100 to 200 ° C. (Process (3))
- the coprecipitate obtained in step (2) and the lithium compound are mixed to obtain a mixture.
- the lithium compound examples include one or more salts selected from the group consisting of lithium hydroxide, lithium chloride, lithium nitrate, and lithium carbonate.
- the lithium compound used may be an anhydride or a hydrate. Moreover, you may use an anhydride and a hydrate together. Mixing may be either dry mixing or wet mixing, but dry mixing is preferred because of the ease of operation.
- Examples of the mixing apparatus include stirring and mixing, a V-type mixer, a W-type mixer, a ribbon mixer, a drum mixer, and a ball mill. (Process (4))
- step (4) the mixture obtained in step (3) is heated at a temperature of 200 ° C. or higher and 500 ° C. or lower, preferably 250 ° C. or higher and 450 ° C.
- the heating atmosphere includes a method using air and oxygen or a mixed gas thereof, a method of mixing an inert gas such as nitrogen and argon into the air and oxygen or a mixed gas thereof, and the oxygen concentration is 5 volumes. % Of the atmosphere.
- a high-capacity lithium composite metal oxide having an intended local structure can be easily obtained, and when the obtained lithium composite metal oxide is used as a positive electrode active material, a high-capacity secondary battery can be obtained. Therefore, the oxygen concentration is preferably 7% by volume to 20% by volume, and more preferably 10% by volume to 20% by volume.
- the firing atmosphere may be an atmosphere in which air, oxygen, nitrogen, argon, or the like is mixed and the oxygen concentration is less than 5% by volume.
- a high-capacity lithium composite metal oxide having an intended local structure can be easily obtained, and when the obtained lithium composite metal oxide is used as a positive electrode active material, a high-capacity secondary battery can be obtained. Therefore, the oxygen concentration is preferably 0.5% by volume or more and less than 5% by volume, and more preferably 1% by volume or more and 3% by volume or less.
- the step (4) and the step (5) are continuously performed without lowering the temperature from the heating temperature at the end of the step (4) ( 5) is preferably performed.
- the oxygen concentration is adjusted to the step (4) while maintaining the temperature at the end of the step (4) or raising the temperature to the firing temperature of the step (5).
- the oxygen concentration in 4) is changed to the oxygen concentration in step (5).
- a method for changing the oxygen concentration a method of changing the oxygen concentration of the introduced gas is preferably used.
- the lithium composite metal oxide of the present embodiment can be produced by such steps (1) to (5).
- a mixture obtained by mixing a salt containing Ni ions, Mn ions and Co ions with a lithium compound by another method in place of steps (1) to (3) is prepared, and the obtained mixture is It is also possible to manufacture the lithium composite metal oxide of the present embodiment by heating while controlling the oxygen concentration and performing the processing corresponding to the above steps (4) and (5).
- the above-mentioned “salt containing Ni ions, Mn ions and Co ions” may be a mixture of a salt containing Ni ions, a salt containing Mn ions, and a salt containing Co ions.
- a method of mixing the above-mentioned salt in a solid phase, a slurry obtained by dispersing the above-mentioned salt in a liquid phase For example, the slurry may be spray-dried and mixed.
- Ni, Mn and Co need to be contained in the mixture in the step (4).
- the metal atom may be contained. Examples of other metal atoms include one or more atoms selected from the group consisting of Al, Mg, Ti, Ca, Cu, Zn, Fe, Cr, Mo, Si, Sn, Nb, and V.
- the mixture and calcined product may contain an inert flux.
- the inert flux is a salt that does not react with the target composite metal oxide and can be easily separated from the target. The inert flux melts at the heating temperature in step (4) and the firing temperature in step (5) to form a reaction field and promotes a uniform reaction.
- sulfate is preferable because the production process becomes simple. More preferably K 2 SO 4 It is. Two or more inert fluxes can be used in combination.
- the mixture contains an inert flux
- the reactivity at the time of heating the mixture and calcining the calcined product is improved, and thereby it may be possible to adjust the BET specific surface area of the obtained lithium composite metal oxide. is there.
- the temperature is the same, the BET specific surface area of the oxide tends to increase as the content of the inert flux increases.
- an inert fluxing agent is contained during heating or firing, a uniform reaction can be performed, so that the local structure can be controlled at the atomic level of the lithium composite metal oxide by adjusting the heating atmosphere.
- the inert flux may be mixed with the coprecipitate obtained by allowing the coprecipitate obtained by the separation operation in step (2) to contain the above inert flux solution and then drying. .
- an inert flux may be mixed with the coprecipitate obtained.
- the inert flux can be added and mixed at the time of mixing the coprecipitate and the lithium compound in the step (3). Since it is easy to control the amount of the inert flux, the method of adding the inert flux in the step (3) is preferable to the method of adding the inert flux in the step (2).
- the coprecipitate obtained in step (2) is washed, and the alkali, Ni salt, Mn salt, or Co salt remaining in the coprecipitate is washed. By reducing the amount of the derived anion, the amount of the inert flux can be easily controlled.
- the inert flux may remain in the lithium composite metal oxide or may be removed by washing.
- the inert flux is a sulfate, and when the mixture or calcined product and the sulfate are mixed, the content of the sulfate in the resulting mixture is the lithium used. It is preferable that it is 0.01 to 400 mass parts with respect to 100 mass parts of compounds. More preferably, it is 0.1 to 10 parts by mass.
- the lithium composite metal oxide obtained by the method for producing a lithium composite metal oxide of the present embodiment may be pulverized using a ball mill or a jet mill. It may be possible to adjust the BET specific surface area of the lithium composite metal oxide by grinding.
- the lithium composite metal oxide obtained by carrying out the steps (1) to (5) may be pulverized, and the steps (4) and (5) may be performed again to perform baking after the pulverization. . Furthermore, you may repeat a grinding
- the lithium composite metal oxide of the present embodiment is preferably a mixture of primary particles having a particle size of 0.05 ⁇ m or more and 1 ⁇ m or less and secondary particles having a particle size of 2 ⁇ m or more and 100 ⁇ m or less formed by aggregation of the primary particles. It consists of. The particle diameters of the primary particles and secondary particles can be measured by observing with SEM.
- the size of the secondary particles of the lithium composite metal oxide is preferably in the range of 2 ⁇ m to 50 ⁇ m, more preferably in the range of 2 ⁇ m to 10 ⁇ m, and even more preferably in the range of 3 ⁇ m to 8 ⁇ m. Especially preferably, it is the range of 3.5 micrometers or more and 7 micrometers or less. By these, the capacity
- the primary particle size of the lithium composite metal oxide is preferably in the range of 0.08 ⁇ m to 0.8 ⁇ m, more preferably in the range of 0.10 ⁇ m to 0.7 ⁇ m, and still more preferably in the range of 0.8.
- the average particle diameter of lithium composite metal oxide (D 50 ) Is preferably in the range of 1 ⁇ m to 50 ⁇ m, more preferably in the range of 1.5 ⁇ m to 30 ⁇ m, even more preferably in the range of 2 ⁇ m to 20 ⁇ m, and particularly preferably in the range of 3 ⁇ m to 10 ⁇ m. It is.
- Average particle size of lithium composite metal oxide (D 50 ) Can be measured by the following method. ⁇ Average particle diameter of lithium composite metal oxide (D 50 ) Measurement> 0.1 g of the lithium composite metal oxide powder to be measured is put into 50 ml of a 0.2 mass% sodium hexametaphosphate aqueous solution to obtain a dispersion in which the powder is dispersed.
- a particle size distribution is measured using the master sizer 2000 (laser diffraction scattering particle size distribution measuring apparatus) by Malvern, and a volume-based cumulative particle size distribution curve is obtained.
- the value of the particle size viewed from the fine particle side at the time of 50% accumulation is the average particle size (D 50 ).
- the BET specific surface area of the lithium composite metal oxide is preferably 0.1 m 2 / G or more 20m 2 / G or less, more preferably 0.5 m 2 / G or more 15m 2 / G or less, even more preferably 1 m 2 / G or more 10m 2 / G or less, particularly preferably 2 m 2 / G or more 8m 2 / G or less. These increase the discharge capacity at a high current rate of the obtained nonaqueous electrolyte secondary battery.
- the BET specific surface area of the lithium composite metal oxide can be measured by the following method. ⁇ Measurement of BET specific surface area of lithium composite metal oxide> After 1 g of the lithium composite metal oxide powder to be measured is dried at 150 ° C.
- Nonaqueous electrolyte secondary battery Next, while explaining the configuration of the nonaqueous electrolyte secondary battery, the positive electrode using the lithium composite metal oxide of the present embodiment as the positive electrode active material of the nonaqueous electrolyte secondary battery, and the nonaqueous electrolyte secondary having the positive electrode The battery will be described.
- FIG. 1 is a schematic view showing an example of the nonaqueous electrolyte secondary battery of the present embodiment.
- the cylindrical nonaqueous electrolyte secondary battery 10 of this embodiment is manufactured as follows. First, as shown in FIG. 1A, two separators 1 each having a strip shape, a strip-like positive electrode 2 having a positive electrode lead 21 at one end, and a strip-like negative electrode 3 having a negative electrode lead 31 at one end, 2, separator 1, and negative electrode 3 are laminated in this order and wound to form electrode group 4. Next, as shown in FIG.
- the nonaqueous electrolyte secondary battery 10 can be manufactured by sealing the upper part of the battery can 5 with the top insulator 7 and the sealing body 8.
- a columnar shape in which the cross-sectional shape when the electrode group 4 is cut in a direction perpendicular to the winding axis is a circle, an ellipse, a rectangle, or a rectangle with rounded corners. Can be mentioned.
- the nonaqueous electrolyte secondary battery having such an electrode group 4 a shape defined by IEC 60086 or JIS C 8500, which is a standard for batteries determined by the International Electrotechnical Commission (IEC), should be adopted. Can do. For example, cylindrical shape, square shape, etc. can be mentioned.
- the non-aqueous electrolyte secondary battery is not limited to the above-described wound type configuration, and may have a stacked type configuration in which a stacked structure of a positive electrode, a separator, a negative electrode, and a separator is repeatedly stacked.
- Examples of the laminated nonaqueous electrolyte secondary battery include so-called coin-type batteries, button-type batteries, and paper-type (or sheet-type) batteries.
- the positive electrode of the present embodiment can be manufactured by first adjusting a positive electrode mixture containing a positive electrode active material, a conductive material and a binder, and supporting the positive electrode mixture on a positive electrode current collector.
- the positive electrode active material of the present embodiment has the above-described lithium composite metal oxide.
- a carbon material can be used as a conductive material included in the positive electrode of the present embodiment.
- the carbon material examples include graphite powder, carbon black (for example, acetylene black), and a fibrous carbon material. Since carbon black is fine and has a large surface area, adding a small amount to the positive electrode mixture can improve the conductivity inside the positive electrode and improve the charge / discharge efficiency and output characteristics. Both the binding force between the positive electrode mixture and the positive electrode current collector and the binding force inside the positive electrode mixture are reduced, which causes an increase in internal resistance.
- the proportion of the conductive material in the positive electrode mixture is preferably 5 parts by mass or more and 20 parts by mass or less with respect to 100 parts by mass of the positive electrode active material. When a fibrous carbon material such as graphitized carbon fiber or carbon nanotube is used as the conductive material, this ratio can be lowered.
- thermoplastic resin can be used as the binder of the positive electrode of the present embodiment.
- thermoplastic resin include polyvinylidene fluoride (hereinafter sometimes referred to as PVdF), polytetrafluoroethylene (hereinafter sometimes referred to as PTFE), tetrafluoroethylene, hexafluoropropylene, and vinylidene fluoride.
- fluororesins such as copolymers, propylene hexafluoride / vinylidene fluoride copolymers, tetrafluoroethylene / perfluorovinyl ether copolymers; polyolefin resins such as polyethylene and polypropylene.
- thermoplastic resins may be used as a mixture of two or more.
- a fluororesin and a polyolefin resin as a binder, the ratio of the fluororesin to the whole positive electrode mixture is 1% by mass or more and 10% by mass or less, and the ratio of the polyolefin resin is 0.1% by mass or more and 2% by mass or less.
- a positive electrode mixture having both high adhesion to the current collector and high bonding strength inside the positive electrode mixture can be obtained.
- a band-shaped member made of a metal material such as Al, Ni, stainless steel or the like can be used.
- Examples of the method of supporting the positive electrode mixture on the positive electrode current collector include a method of pressure-molding the positive electrode mixture on the positive electrode current collector. Further, the positive electrode mixture is made into a paste using an organic solvent, and the obtained positive electrode mixture paste is applied to at least one surface of the positive electrode current collector, dried, pressed and fixed, whereby the positive electrode current collector is bonded to the positive electrode current collector. A mixture may be supported.
- organic solvents that can be used include amine solvents such as N, N-dimethylaminopropylamine and diethylenetriamine; ether solvents such as tetrahydrofuran; ketone solvents such as methyl ethyl ketone; methyl acetate And amide solvents such as dimethylacetamide and N-methyl-2-pyrrolidone (hereinafter sometimes referred to as NMP).
- amine solvents such as N, N-dimethylaminopropylamine and diethylenetriamine
- ether solvents such as tetrahydrofuran
- ketone solvents such as methyl ethyl ketone
- amide solvents such as dimethylacetamide and N-methyl-2-pyrrolidone
- the negative electrode included in the nonaqueous electrolyte secondary battery of this embodiment is only required to be able to dope and dedope lithium ions at a lower potential than the positive electrode, and the negative electrode mixture containing the negative electrode active material is supported on the negative electrode current collector. And an electrode composed of the negative electrode active material alone.
- Examples of the negative electrode active material possessed by the negative electrode include carbon materials, chalcogen compounds (oxides, sulfides, etc.), nitrides, metals, and alloys that can be doped and dedoped with lithium ions at a lower potential than the positive electrode. It is done.
- Examples of carbon materials that can be used as the negative electrode active material include graphites such as natural graphite and artificial graphite, cokes, carbon black, pyrolytic carbons, carbon fibers, and organic polymer compound fired bodies.
- As an oxide that can be used as a negative electrode active material SiO 2 , SiO etc. formula SiO x (Wherein x is a positive real number) silicon oxide represented by: TiO 2 TiO, formula TiO x (Where x is a positive real number) titanium oxide; V 2 O 5 , VO 2 Etc.
- VO x (Where x is a positive real number) oxide of vanadium; Fe 3 O 4 , Fe 2 O 3 FeO and other formulas FeO x (Where x is a positive real number) iron oxide; SnO 2 , SnO etc. formula SnO x (Where x is a positive real number) tin oxide represented by WO 3 , WO 2 General formula WO x (Where x is a positive real number) 4 Ti 5 O 12 , LiVO 2 And a composite metal oxide containing lithium and titanium or vanadium.
- Ti 2 S 3 TiS 2 TiS and other formula TiS x (Where x is a positive real number) titanium sulfide; V 3 S 4 , VS 2, VS formula VS x (Where x is a positive real number) Vanadium sulfide; Fe 3 S 4 , FeS 2 FeS and other formulas x (Where x is a positive real number) iron sulfide; Mo 2 S 3 , MoS 2 Etc. MoS x (Where x is a positive real number) molybdenum sulfide represented by SnS 2, SnS etc.
- carbon materials, oxides, sulfides and nitrides may be used alone or in combination of two or more. These carbon materials, oxides, sulfides and nitrides may be crystalline or amorphous.
- the metal that can be used as the negative electrode active material include lithium metal, silicon metal, and tin metal.
- alloys that can be used as the negative electrode active material include lithium alloys such as Li—Al, Li—Ni, Li—Si, Li—Sn, and Li—Sn—Ni; silicon alloys such as Si—Zn; Sn—Mn and Sn.
- -Tin alloys such as Co, Sn-Ni, Sn-Cu, Sn-La; Cu 2 Sb, La 3 Ni 2 Sn 7 And alloys such as: These metals and alloys are mainly used alone as electrodes after being processed into a foil shape, for example.
- the potential of the negative electrode hardly changes from the uncharged state to the fully charged state during charging (potential flatness is good), the average discharge potential is low, and the capacity retention rate when repeatedly charged and discharged is high.
- a carbon material mainly composed of graphite such as natural graphite or artificial graphite is preferably used.
- the shape of the carbon material may be any of a flake shape such as natural graphite, a spherical shape such as mesocarbon microbeads, a fibrous shape such as graphitized carbon fiber, or an aggregate of fine powder.
- the negative electrode mixture may contain a binder as necessary.
- the binder include thermoplastic resins, and specific examples include PVdF, thermoplastic polyimide, carboxymethyl cellulose, polyethylene, and polypropylene.
- Negative electrode current collector examples include a band-shaped member made of a metal material such as Cu, Ni, and stainless steel.
- Cu As a forming material and process it into a thin film from the viewpoint that it is difficult to make an alloy with lithium and it is easy to process.
- a method of supporting the negative electrode mixture on such a negative electrode current collector as in the case of the positive electrode, a method using pressure molding, pasting with a solvent, etc., applying to the negative electrode current collector, drying and pressing. The method of crimping is mentioned.
- Examples of the separator included in the nonaqueous electrolyte secondary battery of the present embodiment include a porous film, a nonwoven fabric, a woven fabric, and the like made of a material such as a polyolefin resin such as polyethylene and polypropylene, a fluororesin, and a nitrogen-containing aromatic polymer. A material having the following form can be used. Moreover, a separator may be formed by using two or more of these materials, or a separator may be formed by laminating these materials. Examples of the separator include separators described in JP 2000-30686 A, JP 10-324758 A, and the like.
- the thickness of the separator should be as thin as possible as long as the mechanical strength is maintained because the volume energy density of the battery is increased and the internal resistance is reduced, preferably about 5 to 200 ⁇ m, more preferably about 5 to 40 ⁇ m. is there.
- the separator preferably has a porous film containing a thermoplastic resin. In a nonaqueous electrolyte secondary battery, when an abnormal current flows in the battery due to a short circuit between the positive electrode and the negative electrode, the current at the short circuit point is interrupted to prevent an excessive current from flowing (shut down). It preferably has a function.
- the shutdown is performed by overheating the separator at the short-circuit location due to a short circuit, and when the temperature exceeds a presumed operating temperature, the porous film in the separator is softened or melted to close the micropores. And even if the temperature in a battery rises to a certain high temperature after a separator shuts down, it is preferable to maintain the shut-down state, without breaking at the temperature.
- a separator include a laminated film in which a heat resistant porous layer and a porous film are laminated. By using such a laminated film as a separator, the heat resistance of the secondary battery in this embodiment can be further increased. In the laminated film, the heat resistant porous layer may be laminated on both surfaces of the porous film.
- the heat resistant porous layer is a layer having higher heat resistance than the porous film.
- the heat resistant porous layer may be formed from an inorganic powder (first heat resistant porous layer), may be formed from a heat resistant resin (second heat resistant porous layer), and includes a heat resistant resin and a filler. (A third heat-resistant porous layer).
- first heat resistant porous layer may be formed from a heat resistant resin
- second heat resistant porous layer may be formed from a heat resistant resin
- a third heat-resistant porous layer may be formed by an easy technique such as coating.
- the heat resistant porous layer is formed of an inorganic powder
- examples of the inorganic powder used for the heat resistant porous layer include inorganic substances such as metal oxides, metal nitrides, metal carbides, metal hydroxides, carbonates, and sulfates.
- a powder made of an inorganic substance having low conductivity (insulator) is preferably used.
- Specific examples include powders made of alumina, silica, titanium dioxide, calcium carbonate, or the like.
- Such inorganic powders may be used alone or in combination of two or more.
- alumina powder is preferable because of its high chemical stability.
- all of the particles constituting the inorganic powder are alumina particles, all of the particles constituting the inorganic powder are alumina particles, and part or all of them are substantially spherical alumina particles.
- the heat resistant porous layer is formed from a heat resistant resin
- the heat resistant resin used for the heat resistant porous layer is polyamide, polyimide, polyamideimide, polycarbonate, polyacetal, polysulfone, polyphenylene sulfide, polyether ketone, aromatic polyester, polyether. Mention may be made of sulfone and polyetherimide.
- the heat-resistant resin used for the heat-resistant porous layer is a nitrogen-containing aromatic polymer such as aromatic polyamide (para-oriented aromatic polyamide, meta-oriented aromatic polyamide), aromatic polyimide, aromatic polyamideimide, Aromatic polyamides are preferred, and para-oriented aromatic polyamides (hereinafter sometimes referred to as para-aramids) are particularly preferred because they are easy to produce.
- the heat resistant resin include poly-4-methylpentene-1 and a cyclic olefin polymer.
- the heat resistance of the laminated film used as the separator of the nonaqueous electrolyte secondary battery that is, the thermal film breaking temperature of the laminated film can be further increased.
- the compatibility with the electrolytic solution that is, the liquid retention in the heat-resistant porous layer may be improved depending on the polarity in the molecule.
- the rate of impregnation with the electrolytic solution during the production of the electrolyte secondary battery is also high, and the charge / discharge capacity of the nonaqueous electrolyte secondary battery is further increased.
- the thermal film breaking temperature of such a laminated film depends on the type of heat-resistant resin, and is selected and used according to the use scene and purpose of use. More specifically, as the heat-resistant resin, when the nitrogen-containing aromatic polymer is used, the cyclic olefin polymer is about 400 ° C. When using, the thermal film breaking temperature can be controlled to about 300 ° C., respectively. In addition, when the heat resistant porous layer is made of an inorganic powder, the thermal film breaking temperature can be controlled to, for example, 500 ° C. or higher.
- the para-aramid is obtained by polycondensation of a para-oriented aromatic diamine and a para-oriented aromatic dicarboxylic acid halide, and the amide bond is in the para position of the aromatic ring or an oriented position equivalent thereto (for example, 4,4′-biphenylene, It consists essentially of repeating units that are bound together in the opposite orientation, such as 1,5-naphthalene, 2,6-naphthalene, etc., in an orientation that extends coaxially or parallelly.
- para-aramid having a structure according to the type.
- the aromatic polyimide is preferably a wholly aromatic polyimide produced by polycondensation of an aromatic dianhydride and a diamine.
- aromatic dianhydride used for the polycondensation include pyromellitic dianhydride, 3,3 ′, 4,4′-diphenylsulfonetetracarboxylic dianhydride, 3,3 ′, 4. 4,4′-benzophenone tetracarboxylic dianhydride, 2,2′-bis (3,4-dicarboxyphenyl) hexafluoropropane and 3,3 ′, 4,4′-biphenyltetracarboxylic dianhydride. It is done.
- diamines used for polycondensation include oxydianiline, paraphenylenediamine, benzophenonediamine, 3,3′-methylenedianiline, 3,3′-diaminobenzophenone, 3,3′-diaminodiphenylsulfone. And 1,5-naphthalenediamine.
- aromatic polyimide a polyimide soluble in a solvent can be suitably used. Examples of such a polyimide include a polycondensate polyimide of 3,3 ′, 4,4′-diphenylsulfonetetracarboxylic dianhydride and an aromatic diamine.
- aromatic polyamideimide examples include those obtained from polycondensation of aromatic dicarboxylic acid and aromatic diisocyanate, and those obtained from polycondensation of aromatic diacid anhydride and aromatic diisocyanate.
- aromatic dicarboxylic acid examples include isophthalic acid and terephthalic acid.
- aromatic dianhydride is trimellitic anhydride.
- aromatic diisocyanate examples include 4,4'-diphenylmethane diisocyanate, 2,4-tolylene diisocyanate, 2,6-tolylene diisocyanate, orthotolylene diisocyanate, and m-xylene diisocyanate.
- the thickness of the heat resistant porous layer of the laminated film is preferably 1 ⁇ m or more and 10 ⁇ m or less, more preferably 1 ⁇ m or more and 5 ⁇ m or less, and particularly preferably 1 ⁇ m or more and 4 ⁇ m or less.
- the heat-resistant porous layer has fine pores, and the size (diameter) of the pores is preferably 3 ⁇ m or less, more preferably 1 ⁇ m or less. (Third heat-resistant porous layer) Further, when the heat resistant porous layer is formed including a heat resistant resin and a filler, the same heat resistant resin as that used for the second heat resistant porous layer can be used.
- the filler one or more selected from the group consisting of organic powder, inorganic powder, or a mixture thereof can be used.
- the particles constituting the filler preferably have an average particle size of 0.01 ⁇ m or more and 1 ⁇ m or less.
- the organic powder that can be used as the filler include, for example, styrene, vinyl ketone, acrylonitrile, methyl methacrylate, ethyl methacrylate, glycidyl methacrylate, glycidyl acrylate, methyl acrylate, and the like, or two or more types of copolymers; Fluorine resin such as tetrafluoroethylene-6-propylene copolymer, tetrafluoroethylene-ethylene copolymer, polyvinylidene fluoride; melamine resin; urea resin; polyolefin resin; polymethacrylate; A powder is mentioned.
- Such organic powders may be used alone or in combination of two or more.
- PTFE powder is preferred because of its high chemical stability.
- examples of the inorganic powder that can be used as the filler include the same inorganic powder used in the heat-resistant porous layer.
- the filler content depends on the specific gravity of the filler material, for example, when all of the particles constituting the filler are alumina particles
- the mass of the filler is preferably 5 parts by mass or more and 95 parts by mass or less, more preferably 20 parts by mass or more and 95 parts by mass or less, Preferably they are 30 to 90 mass parts. These ranges can be appropriately set depending on the specific gravity of the filler material.
- the shape of the filler include substantially spherical, plate-like, columnar, needle-like, and fiber-like shapes, and any particle can be used.
- the substantially spherical particles include particles having a particle aspect ratio (long particle diameter / short particle diameter) of 1 or more and 1.5 or less.
- the aspect ratio of the particles can be measured by an electron micrograph.
- the porous film preferably has fine pores and has a shutdown function.
- the porous film contains a thermoplastic resin.
- the size of the micropores in the porous film is preferably 3 ⁇ m or less, more preferably 1 ⁇ m or less.
- the porosity of the porous film is preferably 30% to 80% by volume, more preferably 40% to 70% by volume.
- the porous film containing the thermoplastic resin has micropores due to softening or melting of the thermoplastic resin constituting the porous film. Can be occluded. What is necessary is just to select the thermoplastic resin used for a porous film what does not melt
- the polyethylene examples include polyethylene such as low density polyethylene, high density polyethylene, and linear polyethylene, and ultra high molecular weight polyethylene having a molecular weight of 1,000,000 or more.
- the thermoplastic resin constituting the porous film contains at least ultra high molecular weight polyethylene.
- the thermoplastic resin may preferably contain a wax made of polyolefin having a low molecular weight (weight average molecular weight of 10,000 or less).
- the thickness of the porous film in the laminated film is preferably 3 ⁇ m or more and 30 ⁇ m or less, more preferably 3 ⁇ m or more and 25 ⁇ m or less.
- the thickness of a laminated film becomes like this. Preferably it is 40 micrometers or less, More preferably, it is 30 micrometers or less. Moreover, when the thickness of the heat resistant porous layer is A ( ⁇ m) and the thickness of the porous film is B ( ⁇ m), the value of A / B is preferably 0.1 or more and 1 or less.
- the separator allows the electrolyte to permeate well when the battery is used (during charging / discharging). Or less, more preferably 50 seconds / 100 cc or more and 200 seconds / 100 cc or less.
- the porosity of the separator is preferably 30% by volume to 80% by volume, more preferably 40% by volume to 70% by volume.
- the separator may be a laminate of separators having different porosity.
- the electrolyte solution included in the nonaqueous electrolyte secondary battery of this embodiment contains an electrolyte and an organic solvent.
- As the electrolyte contained in the electrolyte LiClO 4 , LiPF 6 , LiAsF 6 , LiSbF 6 , LiBF 4 , LiCF 3 SO 3 , LiN (SO 2 CF 3 ) 2 , LiN (SO 2 C 2 F 5 ) 2 , LiN (SO 2 CF 3 ) (COCF 3 ), Li (C 4 F 9 SO 3 ), LiC (SO 2 CF 3 ) 3 , Li 2 B 10 Cl 10 , LiBOB (where BOB is bis (oxalato) borate), lower aliphatic carboxylic acid lithium salt, LiAlCl 4 And a mixture of two or more of these may be used.
- LiPF containing fluorine 6 LiAsF 6 , LiSbF 6 , LiBF 4 , LiCF 3 SO 3 , LiN (SO 2 CF 3 ) 2 And LiC (SO 2 CF 3 ) 3
- organic solvent contained in the electrolyte include propylene carbonate, ethylene carbonate, dimethyl carbonate, diethyl carbonate, ethyl methyl carbonate, 4-trifluoromethyl-1,3-dioxolan-2-one, and 1,2-di- Carbonates such as (methoxycarbonyloxy) ethane; 1,2-dimethoxyethane, 1,3-dimethoxypropane, pentafluoropropyl methyl ether, 2,2,3,3-tetrafluoropropyl difluoromethyl ether, tetrahydrofuran, 2- Ethers such as methyltetrahydr
- a mixed solvent containing carbonates is preferable, and a mixed solvent of cyclic carbonate and acyclic carbonate and a mixed solvent of cyclic carbonate and ether are more preferable.
- a mixed solvent of cyclic carbonate and acyclic carbonate a mixed solvent containing ethylene carbonate, dimethyl carbonate and ethyl methyl carbonate is preferable.
- the electrolyte using such a mixed solvent has a wide operating temperature range, hardly deteriorates even when charged and discharged at a high current rate, hardly deteriorates even when used for a long time, and natural graphite as an active material of the negative electrode.
- LiPF 6 it is preferable to use an electrolytic solution containing a lithium salt containing fluorine and an organic solvent having a fluorine substituent.
- a mixed solvent containing dimethyl carbonate and ethers having fluorine substituents such as pentafluoropropyl methyl ether and 2,2,3,3-tetrafluoropropyl difluoromethyl ether is capable of capacity even when charging / discharging at a high current rate. Since the maintenance rate is high, it is more preferable.
- a solid electrolyte may be used instead of the above electrolyte.
- the solid electrolyte for example, an organic polymer electrolyte such as a polyethylene oxide polymer compound, a polymer compound containing at least one of a polyorganosiloxane chain or a polyoxyalkylene chain can be used.
- maintained the non-aqueous electrolyte in the high molecular compound can also be used.
- An inorganic solid electrolyte containing a sulfide such as may be used.
- the safety of the nonaqueous electrolyte secondary battery may be further improved.
- the solid electrolyte secondary battery of this embodiment when a solid electrolyte is used, the solid electrolyte may serve as a separator, and in that case, the separator may not be required.
- the nonaqueous electrolyte secondary battery using the positive electrode active material can exhibit a higher capacity than before. Further, since the positive electrode has a positive electrode active material using the above-described lithium composite metal oxide of the present embodiment, the non-aqueous electrolyte secondary battery can exhibit a higher capacity than before. Furthermore, since the nonaqueous electrolyte secondary battery has the positive electrode described above, it exhibits a higher capacity than before.
- evaluation of the lithium composite metal oxide (positive electrode active material) and production evaluation of the positive electrode and the lithium secondary battery were performed as follows.
- (1) Evaluation of lithium composite metal oxide Composition analysis of lithium composite metal oxide The composition analysis of lithium composite metal oxide was conducted by dissolving the obtained lithium composite metal oxide powder in hydrochloric acid and then using an inductively coupled plasma emission spectrometer (SII Nanotechnology Inc.) Manufactured by SPS3000). 2.
- the incident X-ray intensity (I 0 ) is measured at room temperature using a 17 cm ion chamber using N 2 as a filling gas
- the transmitted X-ray intensity (I t ) is measured as a filling gas. It was measured at room temperature using a 31cm ion chamber of using N 2 as.
- the measured energy range and the number of measurement points were 4932 at an equal energy interval from 6040 eV to 7640.5 eV for the K absorption edge of Mn.
- the K absorption edge of Ni was 5333 points at equal energy intervals from 7834 eV to 9434.5 eV.
- the energy calibration is performed using a pre-edge peak (about about X-ray Absorption Near-Edge Structure) spectrum of the obtained K absorption edge when measured using copper alone as a standard sample.
- the angle of the spectral crystal at 8980 eV) was set to 12.7185 °.
- each incident X-ray energy, measured I 0, I t the following equation was determined X-ray absorbance.
- X-ray absorbance ⁇ t ⁇ ln (I 0 / I t )
- discrete X-ray absorbance is obtained corresponding to the wavelength of X-rays used for measurement (corresponding to the energy of X-rays used for measurement).
- the obtained X-ray absorbance was averaged and data interpolated as follows. First, the X-ray absorbance in the range corresponding to the K absorption edge of Mn was averaged by the following method.
- the Mn K absorption edge E 0 was set to an energy value at which the first-order differential coefficient was maximum in the vicinity of the Mn K absorption edge in the X-ray absorption spectrum.
- the K absorption edge E 0 of Ni is set to an energy value at which the first-order differential coefficient becomes maximum in the spectrum near the K absorption edge of Ni.
- the spectrum background is a spectrum in a lower energy region than the K absorption edge of Mn and the K absorption edge of Ni, and Victory's formula (A ⁇ 3 ⁇ B ⁇ 4 + C; ⁇ is the wavelength of incident X-rays, A, B and C are arbitrary constants) and determined by applying the least square method.
- the EXAFS spectrum was obtained by subtracting the background value corresponding to this Victreeen equation from the X-ray absorption spectrum. (Calculation of radial distribution function) A radial distribution function was obtained from the obtained EXAFS spectrum.
- the absorbance ( ⁇ 0 ) of isolated atoms was estimated by the Spline Smoothing method (smoothing spline method), and the EXAFS function ⁇ (k) was extracted. Note that k is the wave number of photoelectrons defined by 0.5123 ⁇ (E ⁇ E 0 ) 1/2 , and the unit of k is ⁇ 1 .
- N-methyl-2-pyrrolidone was used as the organic solvent.
- the obtained positive electrode mixture was applied to a 40 ⁇ m thick Al foil serving as a current collector and vacuum dried at 150 ° C. for 8 hours to obtain a positive electrode.
- (3) Production of nonaqueous electrolyte secondary battery (coin cell) The following operation was performed in a glove box in an argon atmosphere.
- the positive electrode created in “(2) Preparation of positive electrode” is placed on the lower lid of a coin cell (manufactured by Hosen Co., Ltd.) for coin-type battery R2032 with the aluminum foil surface facing downward, and a laminated film separator (polyethylene) is placed thereon.
- a separator (thickness 16 ⁇ m) having a heat-resistant porous layer laminated thereon was placed on the porous film. 300 ⁇ l of electrolyte was injected here.
- the electrolyte used was prepared by dissolving LiPF 6 in a 30:35:35 (volume ratio) mixture of ethylene carbonate, dimethyl carbonate, and ethyl methyl carbonate to a concentration of 1 mol / l.
- the lithium metal is placed on the upper side of the laminated film separator, covered with a gasket, and caulked with a non-aqueous electrolyte secondary battery (coin type battery R2032, hereinafter, (Sometimes referred to as a “coin-type battery”).
- Nickel sulfate hexahydrate, manganese sulfate monohydrate, and cobalt sulfate heptahydrate have a molar ratio of Ni: Mn: Co of 0.45: 0. .45: 0.10, each was weighed and dissolved in pure water to obtain an aqueous transition metal solution containing Ni ions, Mn ions, Co ions and SO 4 2- ions.
- an aqueous potassium hydroxide solution was added to perform coprecipitation to produce a precipitate, thereby obtaining a slurry.
- the obtained slurry was subjected to solid-liquid separation, washed with distilled water to obtain a coprecipitate Q 1 and dried for 8 hours at 100 ° C.. 2.
- Production of lithium composite metal oxide The amount (mol) of Li is 1.3 with respect to the obtained coprecipitate Q 1 and the total amount (mol) 1 of transition metals contained in the coprecipitate Q 1.
- the lithium carbonate weighed in this manner and potassium sulfate as an inert flux were mixed in a mortar to obtain a mixture.
- the obtained mixture was placed in an alumina firing container, and the alumina firing container was placed in an electric furnace.
- the oxygen concentration was adjusted to 8.5% by volume using the air atmosphere originally present in the electric furnace and the introduced nitrogen gas, and heated at 400 ° C.
- the crystal structure of A 1 is hexagonal, which is classified to the space group R-3m.
- a radial distribution function was obtained by Fourier transform.
- the intensity ratio I BMn / I AMn of the peak A Mn of 1.53 ⁇ and the peak B Mn of 2.49 ⁇ was 1.12.
- the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 1.69.
- I BMn / I AMn ⁇ I BNi / I ANi was 1.89.
- a coin type battery was produced by using a charge-discharge test A 1 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 170. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 178. (Example 2) 1.
- I BMn / I AMn ⁇ I BNi / I ANi was 1.82.
- a coin type battery was produced by using the charge and discharge test A 3 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 164. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 175. (Comparative Example 1) 1.
- the total amount (mol) 1 of the transition metal contained in the coprecipitate Q 1, as the amount of Li (mole) is 1.3
- Weighed lithium carbonate and potassium sulfate as an inert flux were mixed in a mortar to obtain a mixture.
- the obtained mixture was put into an alumina firing container, and fired by holding at 850 ° C. in an air atmosphere for 6 hours using an electric furnace, and cooled to room temperature to obtain a fired product.
- the obtained fired product was pulverized, washed with distilled water by decantation, filtered, and dried at 300 ° C. for 6 hours to obtain a powdered lithium composite metal oxide R 1 . 2.
- the intensity ratio I BMn / I AMn of the peak A Mn of 1.56 ⁇ and the peak B Mn of 2.49 0.9 was 0.96.
- the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 2.13.
- a coin type battery was produced by using a charge-discharge test R 1 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 154. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 162.
- a coin type battery was produced by using a charge-discharge test R 2 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 149. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 160. (Comparative Example 3) 1. Except for using the prepared coprecipitate Q 3 of the lithium composite metal oxide, the same operation as in Comparative Example 1 to obtain a powdered lithium composite metal oxide R 3. 2. Evaluation of Lithium Composite Metal Oxide When the composition analysis of R 3 was performed, the molar ratio of Li: Ni: Mn: Co was 1.10: 0.48: 0.49: 0.03.
- a coin type battery was produced by using a charge-discharge test R 3 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 148. When a discharge test at 60 ° C. was performed, the discharge capacity at 0.2 C was 159.
- the results of Examples 1 to 3 and Comparative Examples 1 to 3 are shown in Table 1 below. As a result of the evaluation, all of the non-aqueous electrolyte secondary batteries using the lithium composite metal oxides of Examples 1 to 3 as the positive electrode active material used the lithium composite metal oxides of Comparative Examples 1 to 3 as the positive electrode active material.
- a secondary battery having a higher discharge capacity and higher performance than the nonaqueous electrolyte secondary battery was obtained. Moreover, even if it is the lithium composite metal oxide of Example 3 with the least Co usage-amount among Examples, it discharges rather than the lithium composite metal oxide of Comparative Example 1 with the most Co usage-amount among Comparative Examples. Since a non-aqueous electrolyte secondary battery having a large capacity was obtained, it was found that the performance could be maintained and improved even if the amount of Co used was reduced. From the above results, it was found that the lithium composite metal oxide of the present invention is useful for a non-aqueous electrolyte secondary battery exhibiting a high capacity.
- the positive electrode active material using the lithium composite metal oxide of the present invention is useful for a high-performance non-aqueous electrolyte secondary battery, and the non-aqueous electrolyte secondary battery of the present invention has a higher capacity than before. It was found that
- a lithium composite metal oxide useful for a nonaqueous electrolyte secondary battery exhibiting a higher capacity than before.
- a positive electrode active material, a positive electrode, and a nonaqueous electrolyte secondary battery using a lithium composite metal oxide can be provided.
Landscapes
- Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Battery Electrode And Active Subsutance (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
- Cell Separators (AREA)
Abstract
A lithium composite metal oxide containing Mn, Ni, Li, and Co, and fulfilling formulas (a) and (b): (a) The ratio (IBMn/IAMn) between the intensity (IAMn) of a first proximity peak (AMn) and the intensity (IBMn) of a second proximity peak (BMn) is 0.5-1.2, in a radial distribution function obtained from an EXAFS spectrum at the K absorption end of Mn; and (b) the ratio (IBNi/IANi) between the intensity (IANi) of a first proximity peak (ANi) and the intensity (IBNi) of a second proximity peak is 1.0-1.7, in a radial distribution function obtained from an EXAFS spectrum at the K absorption end of Ni.
Description
本発明は、リチウム複合金属酸化物、正極活物質、正極および非水電解質二次電池に関するものである。
The present invention relates to a lithium composite metal oxide, a positive electrode active material, a positive electrode, and a nonaqueous electrolyte secondary battery.
リチウム複合金属酸化物は、リチウム二次電池などの非水電解質二次電池に正極活物質として用いられている。リチウム二次電池は、既に携帯電話用途やノートパソコン用途などの小型電源として実用化されており、更に自動車用途や電力貯蔵用途などの中・大型電源においても、適用が試みられている。
市販されているリチウム二次電池には、正極活物質としてLiCoO2が最も広く用いられているが、Coが高価であることから、Coの含有量をLiCoO2よりも低減させたリチウム複合金属酸化物が研究されている。なかでも、NiおよびMnを含有するリチウム複合金属酸化物が有望視されている。例えば、組成式がLiNi1/3Co1/3Mn1/3O2であるリチウム複合金属酸化物が知られている。
さらなる安価な非水電解質二次電池を提供するためには、組成式に占めるCoの比率がさらに低減することが有効である。しかしながら、例えばCoを含まないLiNi0.5Mn0.5O2では、LiNi1/3Co1/3Mn1/3O2に比べて容量が低いことからわかるように、Coの比率が低減すると相対的に容量が低くなることが知られている(特許文献1)。以上から、Coの比率がすべての遷移金属に対して少なくとも10%以下までにCoが低減した正極活物質を用いても、高容量で非水電解質二次電池に有用なリチウム複合金属酸化物が求められている。
このような背景のもと、高容量を示す非水電解質二次電池に有用なリチウム複合金属酸化物を得る方法が種々検討されてきた。例えば、Mn、NiおよびLiを含有する層状のリチウム複合金属酸化物において、MnおよびNiの酸化状態を制御することで、非水電解質二次電池用正極活物質として適したリチウム複合金属酸化物とすることが提案されている(特許文献2)。
また、リチウム複合金属酸化物において、X線回折測定による(003)面と(104)面の回折ピークの強度比を特定することにより、この酸化物中に存在するdisorder相の生成を抑制して、リチウム複合金属酸化物の構造を制御する方法が提案されており、これにより高容量な非水電解質二次電池に適したリチウム金属複合酸化物を得ている(特許文献3)。 Lithium composite metal oxide is used as a positive electrode active material in nonaqueous electrolyte secondary batteries such as lithium secondary batteries. Lithium secondary batteries have already been put into practical use as small power sources for mobile phones and notebook computers, and are also being applied to medium and large power sources for automobiles and power storage.
LiCoO 2 is most widely used as a positive electrode active material in commercially available lithium secondary batteries. However, since Co is expensive, lithium composite metal oxidation in which the content of Co is lower than that of LiCoO 2 is used. Things are being researched. Among these, lithium composite metal oxides containing Ni and Mn are considered promising. For example, a lithium composite metal oxide having a composition formula of LiNi 1/3 Co 1/3 Mn 1/3 O 2 is known.
In order to provide a further inexpensive non-aqueous electrolyte secondary battery, it is effective to further reduce the ratio of Co in the composition formula. However, for example, LiNi 0.5 Mn 0.5 O 2 that does not contain Co has a lower capacity than LiNi 1/3 Co 1/3 Mn 1/3 O 2. Then, it is known that a capacity | capacitance will become relatively low (patent document 1). From the above, even if a positive electrode active material in which the Co ratio is reduced to at least 10% or less with respect to all transition metals is used, a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery with a high capacity is obtained. It has been demanded.
Under such circumstances, various methods for obtaining a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery exhibiting a high capacity have been studied. For example, in a layered lithium composite metal oxide containing Mn, Ni and Li, by controlling the oxidation state of Mn and Ni, a lithium composite metal oxide suitable as a positive electrode active material for a non-aqueous electrolyte secondary battery and It has been proposed (Patent Document 2).
In addition, in the lithium composite metal oxide, by specifying the intensity ratio of the diffraction peaks of the (003) plane and the (104) plane by X-ray diffraction measurement, the generation of the disorder phase present in this oxide is suppressed. A method for controlling the structure of a lithium composite metal oxide has been proposed, and thus a lithium metal composite oxide suitable for a high-capacity nonaqueous electrolyte secondary battery has been obtained (Patent Document 3).
市販されているリチウム二次電池には、正極活物質としてLiCoO2が最も広く用いられているが、Coが高価であることから、Coの含有量をLiCoO2よりも低減させたリチウム複合金属酸化物が研究されている。なかでも、NiおよびMnを含有するリチウム複合金属酸化物が有望視されている。例えば、組成式がLiNi1/3Co1/3Mn1/3O2であるリチウム複合金属酸化物が知られている。
さらなる安価な非水電解質二次電池を提供するためには、組成式に占めるCoの比率がさらに低減することが有効である。しかしながら、例えばCoを含まないLiNi0.5Mn0.5O2では、LiNi1/3Co1/3Mn1/3O2に比べて容量が低いことからわかるように、Coの比率が低減すると相対的に容量が低くなることが知られている(特許文献1)。以上から、Coの比率がすべての遷移金属に対して少なくとも10%以下までにCoが低減した正極活物質を用いても、高容量で非水電解質二次電池に有用なリチウム複合金属酸化物が求められている。
このような背景のもと、高容量を示す非水電解質二次電池に有用なリチウム複合金属酸化物を得る方法が種々検討されてきた。例えば、Mn、NiおよびLiを含有する層状のリチウム複合金属酸化物において、MnおよびNiの酸化状態を制御することで、非水電解質二次電池用正極活物質として適したリチウム複合金属酸化物とすることが提案されている(特許文献2)。
また、リチウム複合金属酸化物において、X線回折測定による(003)面と(104)面の回折ピークの強度比を特定することにより、この酸化物中に存在するdisorder相の生成を抑制して、リチウム複合金属酸化物の構造を制御する方法が提案されており、これにより高容量な非水電解質二次電池に適したリチウム金属複合酸化物を得ている(特許文献3)。 Lithium composite metal oxide is used as a positive electrode active material in nonaqueous electrolyte secondary batteries such as lithium secondary batteries. Lithium secondary batteries have already been put into practical use as small power sources for mobile phones and notebook computers, and are also being applied to medium and large power sources for automobiles and power storage.
LiCoO 2 is most widely used as a positive electrode active material in commercially available lithium secondary batteries. However, since Co is expensive, lithium composite metal oxidation in which the content of Co is lower than that of LiCoO 2 is used. Things are being researched. Among these, lithium composite metal oxides containing Ni and Mn are considered promising. For example, a lithium composite metal oxide having a composition formula of LiNi 1/3 Co 1/3 Mn 1/3 O 2 is known.
In order to provide a further inexpensive non-aqueous electrolyte secondary battery, it is effective to further reduce the ratio of Co in the composition formula. However, for example, LiNi 0.5 Mn 0.5 O 2 that does not contain Co has a lower capacity than LiNi 1/3 Co 1/3 Mn 1/3 O 2. Then, it is known that a capacity | capacitance will become relatively low (patent document 1). From the above, even if a positive electrode active material in which the Co ratio is reduced to at least 10% or less with respect to all transition metals is used, a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery with a high capacity is obtained. It has been demanded.
Under such circumstances, various methods for obtaining a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery exhibiting a high capacity have been studied. For example, in a layered lithium composite metal oxide containing Mn, Ni and Li, by controlling the oxidation state of Mn and Ni, a lithium composite metal oxide suitable as a positive electrode active material for a non-aqueous electrolyte secondary battery and It has been proposed (Patent Document 2).
In addition, in the lithium composite metal oxide, by specifying the intensity ratio of the diffraction peaks of the (003) plane and the (104) plane by X-ray diffraction measurement, the generation of the disorder phase present in this oxide is suppressed. A method for controlling the structure of a lithium composite metal oxide has been proposed, and thus a lithium metal composite oxide suitable for a high-capacity nonaqueous electrolyte secondary battery has been obtained (Patent Document 3).
上述のように、Coが低減したリチウム複合金属酸化物において、酸化状態を制御する方法やX線回折測定で得られる結晶構造の平均的な構造を制御する方法により、高容量な非水電解質二次電池に適したリチウム金属複合酸化物を得る検討が成されている。しかし、原子レベルの局所的な構造を制御することにより、高容量な非水電解質二次電池に適したリチウム金属複合酸化物を得る方法については、未だ十分な検討がなされていない。
本発明はこのような事情に鑑みてなされたものであって、原子レベルの局所構造を制御することで、従来よりも高容量を示す非水電解質二次電池に有用なリチウム複合金属酸化物を提供することを目的とする。また、このようなリチウム複合金属酸化物を用いた正極活物質、正極、非水電解質二次電池を提供することを目的とする。
上記の課題を解決するため、本発明の一態様は、Mn、Ni、LiおよびCoを含有するリチウム複合金属酸化物であって、下記(a)および(b)を満たすリチウム複合金属酸化物である。
(a)前記リチウム複合金属酸化物におけるMnのK吸収端の広域X線吸収微細構造(EXAFS)スペクトルをフーリエ変換して得られる動径分布関数において、Mn原子に結合した酸素原子による1.5Å付近の第一近接ピークAMnの強度をIAMn、Mn原子に結合した酸素原子の次にMn原子に近い金属原子による2.5Å付近の第二近接ピークBMnの強度をIBMnとしたとき、IBMn/IAMnが、0.5以上1.2以下である。
(b)前記リチウム複合金属酸化物におけるNiのK吸収端のEXAFSスペクトルをフーリエ変換して得られる動径分布関数において、Ni原子に結合した酸素原子による1.5Å付近の第一近接ピークANiの強度をIANi、Ni原子に結合した酸素原子の次にNi原子に近い金属原子による2.5Å付近の第二近接ピークの強度をIBNiとしたとき、IBNi/IANiが、1.0以上1.7以下である。
本発明の一態様においては、Liの量(モル)をALi、Li以外の金属の量(モル)をAとしたとき、ALi/Aが、0.7以上、1.4以下であることが望ましい。
本発明の一態様においては、前記IBMn/IAMnと、前記IBNi/IANiとの積が、0.7以上2.0以下であることが望ましい。
本発明の一態様においては、前記リチウム複合金属酸化物が層状構造を有し、式(1)で表されることが望ましい。
Li1+x(Ni1−x−y−α−βMnyCoαMβ)O2 …(1)
(式(1)中、−0.3≦x≦0.4、0.35≦y≦0.7、0<α≦0.1、0≦β<0.1(ただし、0<α+β≦0.1)であり、−0.05≦x+y+α+β<1であり、MはAl、Mg、Ti、Ca、Cu、Zn、Fe、Cr、Mo、Si、Sn、NbおよびVからなる群より選ばれる1種類以上の元素である。)
本発明の一態様においては、前記MがFeであることが望ましい。
本発明の一態様においては、β=0であることが望ましい。
また、本発明の一態様は、上述のリチウム複合金属酸化物を有する正極活物質である。
また、本発明の一態様は、上述の正極活物質を有する正極である。
また、本発明の一態様は、負極、および上述の正極を有する非水電解質二次電池である。
本発明の一態様においては、前記負極および前記正極間に配置されたセパレータをさらに有することが望ましい。
本発明の一態様においては、前記セパレータが、耐熱多孔層と多孔質フィルムとが積層された積層フィルムからなることが望ましい。 As described above, in a lithium composite metal oxide with reduced Co, a high-capacity non-aqueous electrolyte can be obtained by a method for controlling an oxidation state or a method for controlling an average crystal structure obtained by X-ray diffraction measurement. Studies have been made to obtain lithium metal composite oxides suitable for secondary batteries. However, a method for obtaining a lithium metal composite oxide suitable for a high-capacity nonaqueous electrolyte secondary battery by controlling the local structure at the atomic level has not yet been sufficiently studied.
The present invention has been made in view of such circumstances. By controlling the local structure at the atomic level, a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery exhibiting a higher capacity than before is obtained. The purpose is to provide. It is another object of the present invention to provide a positive electrode active material, a positive electrode, and a nonaqueous electrolyte secondary battery using such a lithium composite metal oxide.
In order to solve the above problems, one embodiment of the present invention is a lithium composite metal oxide containing Mn, Ni, Li, and Co, which satisfies the following (a) and (b): is there.
(A) In a radial distribution function obtained by Fourier transform of a wide-range X-ray absorption fine structure (EXAFS) spectrum at the K absorption edge of Mn in the lithium composite metal oxide, 1.5% by oxygen atoms bonded to Mn atoms is obtained. when the first proximity peak a Mn of intensity I AMn near the intensity of the second closest peak B Mn around 2.5Å by metal atoms near the next Mn atoms of the oxygen atoms bonded to Mn atoms was I BMn , IBMn / IAMn is 0.5 or more and 1.2 or less.
(B) In the radial distribution function obtained by Fourier transforming the EXAFS spectrum at the K absorption edge of Ni in the lithium composite metal oxide, the first proximity peak A Ni near 1.5Å due to oxygen atoms bonded to Ni atoms. the intensity I ANi, when the next on the intensity of the second closest peak around 2.5Å by metal atoms near the Ni atom bonded oxygen atom to Ni atom was I BNi, the I BNi / I ANi, 1. It is 0 or more and 1.7 or less.
In one embodiment of the present invention, when the amount (mol) of Li is A Li and the amount (mol) of a metal other than Li is A, A Li / A is 0.7 or more and 1.4 or less. It is desirable.
In one aspect of the present invention, it is desirable that a product of the I BMn / I AMn and the I BNi / I ANi is 0.7 or more and 2.0 or less.
In one embodiment of the present invention, it is desirable that the lithium composite metal oxide has a layered structure and is represented by Formula (1).
Li 1 + x (Ni 1- x-y-α-β Mn y Co α M β) O 2 ... (1)
(In the formula (1), −0.3 ≦ x ≦ 0.4, 0.35 ≦ y ≦ 0.7, 0 <α ≦ 0.1, 0 ≦ β <0.1 (where 0 <α + β ≦ 0.1), −0.05 ≦ x + y + α + β <1, and M is selected from the group consisting of Al, Mg, Ti, Ca, Cu, Zn, Fe, Cr, Mo, Si, Sn, Nb and V One or more elements.)
In one aspect of the present invention, the M is preferably Fe.
In one embodiment of the present invention, it is desirable that β = 0.
Another embodiment of the present invention is a positive electrode active material including the above lithium composite metal oxide.
Another embodiment of the present invention is a positive electrode including the above positive electrode active material.
Another embodiment of the present invention is a nonaqueous electrolyte secondary battery including a negative electrode and the positive electrode described above.
In one mode of the present invention, it is desirable to further have a separator arranged between the negative electrode and the positive electrode.
In one aspect of the present invention, the separator is preferably made of a laminated film in which a heat-resistant porous layer and a porous film are laminated.
本発明はこのような事情に鑑みてなされたものであって、原子レベルの局所構造を制御することで、従来よりも高容量を示す非水電解質二次電池に有用なリチウム複合金属酸化物を提供することを目的とする。また、このようなリチウム複合金属酸化物を用いた正極活物質、正極、非水電解質二次電池を提供することを目的とする。
上記の課題を解決するため、本発明の一態様は、Mn、Ni、LiおよびCoを含有するリチウム複合金属酸化物であって、下記(a)および(b)を満たすリチウム複合金属酸化物である。
(a)前記リチウム複合金属酸化物におけるMnのK吸収端の広域X線吸収微細構造(EXAFS)スペクトルをフーリエ変換して得られる動径分布関数において、Mn原子に結合した酸素原子による1.5Å付近の第一近接ピークAMnの強度をIAMn、Mn原子に結合した酸素原子の次にMn原子に近い金属原子による2.5Å付近の第二近接ピークBMnの強度をIBMnとしたとき、IBMn/IAMnが、0.5以上1.2以下である。
(b)前記リチウム複合金属酸化物におけるNiのK吸収端のEXAFSスペクトルをフーリエ変換して得られる動径分布関数において、Ni原子に結合した酸素原子による1.5Å付近の第一近接ピークANiの強度をIANi、Ni原子に結合した酸素原子の次にNi原子に近い金属原子による2.5Å付近の第二近接ピークの強度をIBNiとしたとき、IBNi/IANiが、1.0以上1.7以下である。
本発明の一態様においては、Liの量(モル)をALi、Li以外の金属の量(モル)をAとしたとき、ALi/Aが、0.7以上、1.4以下であることが望ましい。
本発明の一態様においては、前記IBMn/IAMnと、前記IBNi/IANiとの積が、0.7以上2.0以下であることが望ましい。
本発明の一態様においては、前記リチウム複合金属酸化物が層状構造を有し、式(1)で表されることが望ましい。
Li1+x(Ni1−x−y−α−βMnyCoαMβ)O2 …(1)
(式(1)中、−0.3≦x≦0.4、0.35≦y≦0.7、0<α≦0.1、0≦β<0.1(ただし、0<α+β≦0.1)であり、−0.05≦x+y+α+β<1であり、MはAl、Mg、Ti、Ca、Cu、Zn、Fe、Cr、Mo、Si、Sn、NbおよびVからなる群より選ばれる1種類以上の元素である。)
本発明の一態様においては、前記MがFeであることが望ましい。
本発明の一態様においては、β=0であることが望ましい。
また、本発明の一態様は、上述のリチウム複合金属酸化物を有する正極活物質である。
また、本発明の一態様は、上述の正極活物質を有する正極である。
また、本発明の一態様は、負極、および上述の正極を有する非水電解質二次電池である。
本発明の一態様においては、前記負極および前記正極間に配置されたセパレータをさらに有することが望ましい。
本発明の一態様においては、前記セパレータが、耐熱多孔層と多孔質フィルムとが積層された積層フィルムからなることが望ましい。 As described above, in a lithium composite metal oxide with reduced Co, a high-capacity non-aqueous electrolyte can be obtained by a method for controlling an oxidation state or a method for controlling an average crystal structure obtained by X-ray diffraction measurement. Studies have been made to obtain lithium metal composite oxides suitable for secondary batteries. However, a method for obtaining a lithium metal composite oxide suitable for a high-capacity nonaqueous electrolyte secondary battery by controlling the local structure at the atomic level has not yet been sufficiently studied.
The present invention has been made in view of such circumstances. By controlling the local structure at the atomic level, a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery exhibiting a higher capacity than before is obtained. The purpose is to provide. It is another object of the present invention to provide a positive electrode active material, a positive electrode, and a nonaqueous electrolyte secondary battery using such a lithium composite metal oxide.
In order to solve the above problems, one embodiment of the present invention is a lithium composite metal oxide containing Mn, Ni, Li, and Co, which satisfies the following (a) and (b): is there.
(A) In a radial distribution function obtained by Fourier transform of a wide-range X-ray absorption fine structure (EXAFS) spectrum at the K absorption edge of Mn in the lithium composite metal oxide, 1.5% by oxygen atoms bonded to Mn atoms is obtained. when the first proximity peak a Mn of intensity I AMn near the intensity of the second closest peak B Mn around 2.5Å by metal atoms near the next Mn atoms of the oxygen atoms bonded to Mn atoms was I BMn , IBMn / IAMn is 0.5 or more and 1.2 or less.
(B) In the radial distribution function obtained by Fourier transforming the EXAFS spectrum at the K absorption edge of Ni in the lithium composite metal oxide, the first proximity peak A Ni near 1.5Å due to oxygen atoms bonded to Ni atoms. the intensity I ANi, when the next on the intensity of the second closest peak around 2.5Å by metal atoms near the Ni atom bonded oxygen atom to Ni atom was I BNi, the I BNi / I ANi, 1. It is 0 or more and 1.7 or less.
In one embodiment of the present invention, when the amount (mol) of Li is A Li and the amount (mol) of a metal other than Li is A, A Li / A is 0.7 or more and 1.4 or less. It is desirable.
In one aspect of the present invention, it is desirable that a product of the I BMn / I AMn and the I BNi / I ANi is 0.7 or more and 2.0 or less.
In one embodiment of the present invention, it is desirable that the lithium composite metal oxide has a layered structure and is represented by Formula (1).
Li 1 + x (Ni 1- x-y-α-β Mn y Co α M β) O 2 ... (1)
(In the formula (1), −0.3 ≦ x ≦ 0.4, 0.35 ≦ y ≦ 0.7, 0 <α ≦ 0.1, 0 ≦ β <0.1 (where 0 <α + β ≦ 0.1), −0.05 ≦ x + y + α + β <1, and M is selected from the group consisting of Al, Mg, Ti, Ca, Cu, Zn, Fe, Cr, Mo, Si, Sn, Nb and V One or more elements.)
In one aspect of the present invention, the M is preferably Fe.
In one embodiment of the present invention, it is desirable that β = 0.
Another embodiment of the present invention is a positive electrode active material including the above lithium composite metal oxide.
Another embodiment of the present invention is a positive electrode including the above positive electrode active material.
Another embodiment of the present invention is a nonaqueous electrolyte secondary battery including a negative electrode and the positive electrode described above.
In one mode of the present invention, it is desirable to further have a separator arranged between the negative electrode and the positive electrode.
In one aspect of the present invention, the separator is preferably made of a laminated film in which a heat-resistant porous layer and a porous film are laminated.
1…セパレータ、2…正極、3…負極、4…電極群、5…電池缶、6…電解液、7…トップインシュレーター、8…封口体、10…非水電解質二次電池、21…正極リード、31…負極リード
DESCRIPTION OF SYMBOLS 1 ... Separator, 2 ... Positive electrode, 3 ... Negative electrode, 4 ... Electrode group, 5 ... Battery can, 6 ... Electrolyte solution, 7 ... Top insulator, 8 ... Sealing body, 10 ... Nonaqueous electrolyte secondary battery, 21 ... Positive electrode lead 31 ... Negative electrode lead
[リチウム複合金属酸化物]
本実施形態のリチウム複合金属酸化物は、Mn、Ni、LiおよびCoを含有し、下記(a)および(b)を満たす。
(a)前記リチウム複合金属酸化物におけるMnのK吸収端の広域X線吸収微細構造(EXAFS)スペクトルをフーリエ変換して得られる動径分布関数において、Mn原子に結合した酸素原子による1.5Å付近の第一近接ピークAMnの強度をIAMn、Mn原子に結合した酸素原子の次にMn原子に近い金属原子による2.5Å付近の第二近接ピークBMnの強度をIBMnとしたとき、IBMn/IAMnが、0.5以上1.2以下である。
(b)前記リチウム複合金属酸化物におけるNiのK吸収端のEXAFSスペクトルをフーリエ変換して得られる動径分布関数において、Ni原子に結合した酸素原子による1.5Å付近の第一近接ピークANiの強度をIANi、Ni原子に結合した酸素原子の次にNi原子に近い金属原子による2.5Å付近の第二近接ピークの強度をIBNiとしたとき、IBNi/IANiが、1.0以上1.7以下である。
以下、順に説明する。
(EXAFSスペクトル)
まず、本実施形態のリチウム複合金属酸化物を規定するために用いるEXAFSスペクトルについて説明する。本実施形態で用いるEXAFSスペクトルは、一般的なEXAFSスペクトルと同様に扱われる。EXAFSスペクトルの測定および原理は、例えば、「X線吸収分光法—XAFSとその応用—」(太田俊明編(2002年))に記載されている。原理は以下の通りである。
まず、測定対象の物質に特定の波長のX線を透過させたとき、物質に照射されたX線の強度(入射X線強度:I0)と、物質を透過してきたX線の強度(透過X線強度:It)とから、特定の波長における測定対象の物質について、単位厚さあたりのX線吸光度が得られる。
物質に照射するX線の波長を変化させ(すなわち、入射X線のエネルギー(eV)を変化させ)、各波長(各エネルギー)のX線に対するX線吸光度を測定して、x軸を入射X線のエネルギー(eV)、y軸をX線吸光度とするX線吸収スペクトルを作成すると、X線吸光度が急激に増加するエネルギーがあることが分かる。このエネルギーの値を吸収端という。吸収端は、物質を構成する原子の原子殻のエネルギー準位に対応しており、各原子に固有のものである。例えば、原子のK殻に対応する吸収端はK吸収端という。
X線吸収スペクトルにおいて、この吸収端から20~1000eV程度高いエネルギー側の領域に現れる微細な振動構造を広域X線吸収微細構造(EXAFS)といい、そのスペクトルをEXAFSスペクトルという。EXAFSスペクトルについてフーリエ変換を施すと、X線吸収原子(注目する原子)を中心とした動径分布関数が得られる。この動径分布関数から、X線吸収原子とX線散乱原子(X線吸収原子近傍の原子)との距離、X線散乱原子の数などの情報を得ることができ、注目する原子近傍の情報を得ることができる。
一般的に、動径分布関数のピークの強度は、X線散乱原子の数に影響されるが、その他に、X線吸収原子とX線散乱原子との原子間距離の等方性にも影響される。例えば、ある2つのX線吸収原子について、X線散乱原子数が実質的に同数で、かつ、X線散乱原子が実質的に同等な散乱能を有すると認められる場合、動径分布関数のピーク強度が大きいものは、X線吸収原子とX線散乱原子との原子間距離が方向による違いがなく等方的であり、X線吸収原子とX線散乱原子間との距離の分布が少ないことを意味する。
そこで、本実施形態においては、MnおよびNiのK吸収端において得られた動径分布関数のピークの強度比に注目する。
すなわち、動径分布関数のピークの強度比をある一定の範囲内に制御することによって、組成比が異なる試料に対しても、リチウム複合金属酸化物における原子レベルの局所構造を特定の条件に制御でき、従来よりも高容量を示す非水電解質二次電池に有用なリチウム複合金属酸化物を得ることができる。
本実施形態のリチウム複合金属酸化物においては、MnのK吸収端の動径分布関数において、Mn原子に結合したO(酸素原子)によるピークを第一近接ピークAMnとする。第一近接ピークAMnは、好ましくは1.4Å以上1.9Å以下、より好ましくは1.5Å以上1.6Å以下の1.5Å付近に現れる。
また、本実施形態のリチウム複合金属酸化物においては、MnのK吸収端の動径分布関数において、Mn原子に結合したOの次にMn原子に近い原子X(ここで、XはLi、Mn、Niなどの金属原子となる。)によるピークを第二近接ピークBMnとする。第二近接ピークBMnは、好ましくは2.44Å以上2.55Å以下、より好ましくは2.46Å以上2.55Å以下の2.5Å付近に現れる。ここで、原子XはMn原子に結合したOに結合している。
さらに、本実施形態のリチウム複合金属酸化物においては、NiのK吸収端の動径分布関数において、Ni原子に結合したOによるピークを第一近接ピークANiとする。第一近接ピークANiは、好ましくは1.4Å以上1.9Å以下、より好ましくは1.5Å以上1.6Å以下の1.5Å付近に現れる。
そして、本実施形態のリチウム複合金属酸化物においては、NiのK吸収端の動径分布関数において、Ni原子に結合したOの次にNi原子に近い原子X(ここで、XはLi、Mn、Niなどの金属原子となる。)によるピークを第二近接ピークBNiとする。第二近接ピークBNiは、好ましくは2.44Å以上2.55Å以下、より好ましくは2.46Å以上2.55Å以下の2.5Å付近に現れる。ここで、原子XはNi原子に結合したOに結合している。
本実施形態のリチウム複合金属酸化物は、動径分布関数のピークの強度比、すなわちIAMnとIBMnとの比(IBMn/IAMn)およびIANiとIBNiとの比(IBNi/IANi)を特定の範囲内に制御することで、原子レベルの局所構造を制御している。このような本実施形態のリチウム複合金属酸化物は、従来よりも高容量を示す非水電解質二次電池に有用である。
本実施形態のリチウム複合金属酸化物は、Mn原子まわりのOおよび原子Xの原子間距離の等方性がある程度に高く、ある特定の範囲に収まるため、正極活物質としての特性が高い。このようなIBMn/IAMnの値は、0.5以上1.2以下であり、好ましくは0.6以上1.2以下であり、より好ましくは0.7以上1.2以下であり、さらにより好ましくは1.0以上1.2以下であり、特に好ましくは1.1以上1.2以下である。
また、本実施形態のリチウム複合金属酸化物は、Ni原子まわりのOおよび原子Xの原子間距離の等方性がある程度に高く、ある特定の範囲に収まるため、正極活物質としての特性が高い。このようなIBNi/IANiの値は、1.0以上1.7以下であり、好ましくは1.1以上1.7以下であり、より好ましくは1.2以上1.7以下である。
これらIBMn/IAMnの値の範囲及びIBNi/IANiの値の範囲は、任意に組み合わせることができる。
さらに、前記IBMn/IAMnとIBNi/IANiとの積(IBMn/IAMn×IBNi/IANi)は、適度なMn原子まわりのOおよび原子Xの原子間距離の等方性と、適度なNi原子まわりのOおよび原子Xの原子間距離の等方性と、を両立するため、0.7以上2.0以下であり、好ましくは0.9以上2.0以下であり、より好ましくは1.1以上2.0以下である。
本実施形態のリチウム複合金属酸化物の結晶構造は、層状構造であることが好ましく、六方晶型の結晶構造または単斜晶型の結晶構造であることがより好ましい。
六方晶型の結晶構造は、P3、P31、P32、R3、P−3、R−3、P312、P321、P3112、P3121、P3212、P3221、R32、P3m1、P31m、P3c1、P31c、R3m、R3c、P−31m、P−31c、P−3m1、P−3c1、R−3m、R−3c、P6、P61、P65、P62、P64、P63、P−6、P6/m、P63/m、P622、P6122、P6522、P6222、P6422、P6322、P6mm、P6cc、P63cm、P63mc、P−6m2、P−6c2、P−62m、P−62c、P6/mmm、P6/mcc、P63/mcm、P63/mmcからなる群から選ばれるいずれか一つの空間群に分類される。
また、単斜晶型の結晶構造は、P2、P21、C2、Pm、Pc、Cm、Cc、P2/m、P21/m、C2/m、P2/c、P21/c、C2/cからなる群から選ばれるいずれか一つの空間群に分類される。
これらのうち、得られる非水電解質二次電池の放電容量が増大するため、リチウム複合金属酸化物の結晶構造は、空間群R−3mに分類される六方晶型の結晶構造、またはC2/mに分類される単斜晶型の結晶構造であることが特に好ましい。
本実施形態のリチウム複合金属酸化物の空間群は、次の方法で確認することができる。
まず、リチウム複合金属酸化物について、CuKαを線源とし、かつ回折角2θの測定範囲を10°以上90°以下とする粉末X線回折測定を行い、次いでその結果をもとにリートベルト解析を行い、リチウム複合金属酸化物が有する結晶構造およびこの結晶構造における空間群を決定する。リートベルト解析は、材料の粉末X線回折測定における回折ピークのデータ(回折ピーク強度、回折角2θ)を用いて、材料の結晶構造を解析する手法であり、従来から使用されている手法である(例えば「粉末X線解析の実際−リートベルト法入門−」2002年2月10日発行、日本分析化学会X線分析研究懇談会編、参照)。
本実施形態におけるリチウム複合金属酸化物の組成は、Liの量(モル)をALi、Li以外の金属の量(モル)をAとしたとき、ALi/Aが、0.7以上1.4以下であることとしてもよい。
本実施形態におけるリチウム複合金属酸化物は、層状構造を有し、組成が下記式(1)で表されるものが好ましい。
Li1+x(Ni1−x−y−αMnyCoαMβ)O2 …(1)
(式(1)中、−0.3≦x≦0.4、0.35≦y≦0.7、0<α≦0.1、0≦β<0.1(ただし、0<α+β≦0.1)であり、−0.05≦x+y+α+β<1であり、MはAl、Mg、Ti、Ca、Cu、Zn、Fe、Cr、Mo、Si、Sn、NbおよびVからなる群より選ばれる1種類以上の元素である。)
式(1)中のxの値は、−0.3≦x≦0.4であり、好ましくは−0.2≦x≦0.35であり、より好ましくは−0.1≦x≦0.3である。
本実施形態のリチウム複合金属酸化物は、60℃における放電容量が高いため、MがFeであることが好ましい。
なお、本実施形態のリチウム複合金属酸化物の粒子をコア材として、コア材(リチウム複合金属酸化物の粒子)の表面に、さらにB、Al、Ga、In、Si、Ge、Sn、Mgおよび遷移金属からなる群から選ばれる1種以上の原子を含有する化合物を付着させてもよい。
上記原子の中でも、B、Al、Mg、Co、CrおよびMnからなる群から選ばれる1種以上が好ましく、均一な被覆層形成が容易であるため、Alがより好ましい。
このような化合物としては、例えば上記原子の酸化物、フッ化物、硫化物、水酸化物、オキシ水酸化物、炭酸塩、硝酸塩、有機酸塩およびこれらの混合物が挙げられる。中でも、酸化物、水酸化物、オキシ水酸化物またはこれらの混合物が好ましい。
コア材の表面に被着させる化合物としては、Alの酸化物であるアルミナが好ましい。
[リチウム複合金属酸化物の製造方法]
次に、上述したリチウム複合金属酸化物の製造方法について説明する。
本実施形態のリチウム複合金属酸化物の製造方法は、以下の(1)~(5)の工程を含む。
(1)Niイオン、MnイオンおよびCoイオンを含有する水溶液(以下、「原料水溶液」と称することがある)とアルカリとを接触させて共沈物を生成させ、スラリーを得る工程
(2)(1)で得られるスラリーから共沈物を分離する工程
(3)(2)で得られる共沈物とリチウム化合物とを混合する工程
(4)(3)で得られる混合物を、酸素濃度が5体積%以上の雰囲気において、200℃以上500℃以下の温度で加熱する工程
(5)(4)で得られる生成物(以下、「仮焼物」と称することがある)を、酸素濃度が5体積%未満の雰囲気において、600℃以上950℃以下の温度で焼成する工程。
ここで工程(4)における「酸素濃度」とは、混合物を加熱する空間(熱処理空間)が200℃以上500℃以下の範囲にある際の、熱処理空間中の平均酸素濃度のことを指す。同様に、工程(5)における「酸素濃度」とは、仮焼物を焼成する空間(熱処理空間)が600℃以上950℃以下の範囲にある際の、熱処理空間中の平均酸素濃度を指す。
(工程(1))
上記工程(1)において、原料水溶液は、Ni、MnおよびCoを含有する化合物を水に溶解させることで調整することができる。なかでも、原料水溶液は、Niの硫酸塩、Mnの硫酸塩およびCoの硫酸塩を水に溶解して得られる水溶液であることが好ましい。
また、Ni、MnおよびCoを含有するそれぞれの原料が水に溶解し難い場合、例えば、これらの原料が、酸化物、水酸化物、金属材料である場合には、これらの原料を、硫酸を含有する水溶液に溶解させて、原料水溶液を得ることができる。
工程(1)で用いられるアルカリとしては、LiOH(水酸化リチウム)、NaOH(水酸化ナトリウム)、KOH(水酸化カリウム)、Li2CO3(炭酸リチウム)、Na2CO3(炭酸ナトリウム)、K2CO3(炭酸カリウム)および(NH4)2CO3(炭酸アンモニウム)からなる群より選ばれる1種以上の塩を挙げることができる。用いるアルカリは、無水物であってもよく、水和物であってもよい。無水物と水和物とは併用してもよい。工程(1)においては、上記アルカリの水溶液(アルカリ水溶液)を用いることが好ましい。また、アルカリ水溶液として、アンモニア水を用いることもできる。
アルカリ水溶液におけるアルカリの濃度は、好ましくは0.5~10M(mol/L)程度、より好ましくは1~8M程度である。また、製造コストの面から、用いるアルカリとしてNaOHまたはKOHが好ましい。また、NaOHとKOHとは併用してもよい。
工程(1)における接触の方法としては、(i)原料水溶液にアルカリ水溶液を添加して混合する方法、(ii)アルカリ水溶液に原料水溶液を添加して混合する方法、(iii)水に原料水溶液およびアルカリ水溶液を添加して混合する方法、を挙げることができる。混合時には、攪拌を伴うことが好ましい。
また、工程(1)における接触の方法のうち、(ii)アルカリ水溶液に原料水溶液を添加して混合する方法は、pHの変化を制御しやすく好ましい。この方法の場合、アルカリ水溶液に、原料水溶液を添加し混合していくに従い、アルカリ水溶液のpHが低下していく傾向にあるが、pHが9以上、好ましくは10以上となるように調節しながら、原料水溶液を添加することが好ましい。また、原料水溶液およびアルカリ水溶液のうち、いずれか一方または両方の水溶液を40℃以上80℃以下の温度に保持しながら接触させると、より均一な組成の共沈物を得ることができ、好ましい。
工程(1)においては、上記のようにして原料水溶液とアルカリとを接触させることで、Niイオン、MnイオンおよびCoイオンを含む塩が共沈して生成し、共沈物である塩が分散したスラリーを得ることができる。
(工程(2))
工程(2)においては、工程(1)で得られたスラリーから共沈物を得る。共沈物を得ることができる限り、工程(2)では共沈物を得る方法として種々の方法を採用することができるが、操作が簡便であることから、ろ過などの固体成分を得る分離操作による方法が好ましい。スラリーを噴霧乾燥させるなどの、加熱により液体を揮発させる方法によっても共沈物を得ることができる。
工程(2)において共沈物を得る場合には、前記工程(2)では、分離した共沈物を洗浄し、乾燥させることが好ましい。洗浄することにより、得られる共沈物に残存するアルカリや、Niの硫酸塩、Mnの硫酸塩、Coの硫酸塩を原料として用いた場合に原料水溶液中に遊離するSO4 2−イオンの量を低減することができる。洗浄によりこれらを低減させると、不活性融剤(後述)の量の制御が容易となり好ましい。
共沈物を効率よく洗浄するためには、洗浄液として水を用いることが好ましい。なお、必要に応じてアルコール、アセトンなどの水溶性を有する有機溶媒を洗浄液に加えても良い。また、洗浄は2回以上行ってもよく、例えば、水洗浄を行った後、前記のような水溶性を有する有機溶媒で再度洗浄することもできる。
洗浄した共沈物の乾燥は、熱処理によって行うことができるが、送風乾燥、真空乾燥などによってもよく、さらにこれらを組み合わせてもよい。熱処理によって行う場合、加熱温度は好ましくは50~300℃であり、より好ましくは100~200℃程度である。
(工程(3))
工程(3)においては、工程(2)で得られた共沈物とリチウム化合物とを混合して混合物を得る。
リチウム化合物としては、水酸化リチウム、塩化リチウム、硝酸リチウムおよび炭酸リチウムからなる群より選ばれる1種以上の塩を挙げることができる。用いるリチウム化合物は、無水物であってもよく、水和物であってもよい。また、無水物と水和物とを併用してもよい。
混合は、乾式混合、湿式混合のいずれによってもよいが、操作が簡便であることから乾式混合が好ましい。混合装置としては、攪拌混合、V型混合機、W型混合機、リボン混合機、ドラムミキサー、ボールミルなどを挙げることができる。
(工程(4))
工程(4)においては、工程(3)で得られた混合物を、200℃以上500℃以下、好ましくは250℃以上450℃以下の温度で加熱し、仮焼物を得る。加熱の雰囲気としては、大気および酸素またはそれらの混合ガスを用いる方法、大気および酸素またはそれらの混合ガスに、窒素およびアルゴンなどの不活性ガスを混合する方法などがあるが、酸素濃度が5体積%以上である雰囲気であればよい。意図した局所構造を有する高容量のリチウム複合金属酸化物が得られやすく、得られたリチウム複合金属酸化物を正極活物質として用いた場合に高容量な二次電池とすることが可能であることから、酸素濃度は7体積%以上20体積以下であることが好ましく、10体積%以上20体積%以下であることがより好ましい。
(工程(5))
工程(5)において、工程(4)で得られた仮焼物を650℃以上950℃以下、好ましくは650℃以上900℃以下の温度で焼成する。焼成の雰囲気としては、大気、酸素、窒素およびアルゴンなどを混合し、酸素濃度が5体積%未満である雰囲気とすればよい。意図した局所構造を有する高容量のリチウム複合金属酸化物が得られやすく、得られたリチウム複合金属酸化物を正極活物質として用いた場合に高容量な二次電池とすることが可能であることから、酸素濃度は0.5体積%以上、5体積%未満であることが好ましく、1体積%以上3体積%以下であることがさらに好ましい。
上記、工程(4)および工程(5)は、得られるリチウム複合金属酸化物の組成を均一にするために、工程(4)終了時の加熱温度から温度を下げることなく、連続的に工程(5)を行うことが好ましい。工程(4)および工程(5)を連続的に行う際には、工程(4)終了時の温度を維持しながら、または工程(5)の焼成温度に昇温しながら、酸素濃度を工程(4)の酸素濃度から工程(5)の酸素濃度に変更する。酸素濃度の変更方法としては、導入するガスの酸素濃度を変化させる方法が好ましく用いられる。
このような工程(1)~工程(5)により、本実施形態のリチウム複合金属酸化物を製造することができる。
なお、本実施形態の製造方法では、工程(1)~工程(5)を有することとして説明したが、これに限られない。例えば、工程(1)~工程(3)に代わる別の方法でNiイオン、MnイオンおよびCoイオンを含む塩と、リチウム化合物とを混合して得られる混合物を用意し、得られた混合物を、酸素濃度を制御しながら加熱し、上記工程(4)および工程(5)に相当する処理を行うことでも本実施形態のリチウム複合金属酸化物を製造することが可能である。
上述の「Niイオン、MnイオンおよびCoイオンを含む塩」は、Niイオンを含む塩と、Mnイオンを含む塩と、Coイオンを含む塩との混合物であってもよい。上述の「工程(1)~工程(3)に代わる別の方法」としては、上述の塩を固相で混合する方法、上述の塩を液相中に分散してスラリーを作製し、得られたスラリーを噴霧乾燥して混合する方法などが挙げられる。
また、本実施形態のリチウム複合金属酸化物の製造方法では、工程(4)の混合物にNi、MnおよびCoが含まれている必要があるが、工程(4)で加熱する混合物に、更に他の金属原子が含まれていてもよい。他の金属原子としては、Al、Mg、Ti、Ca、Cu、Zn、Fe、Cr、Mo、Si、Sn、NbおよびVからなる群より選ばれる1種類以上の原子が挙げられる。
工程(4)で加熱する混合物に他の金属原子を含ませる方法としては、種々の方法を採用できる。なかでも、上記工程(1)において、原料水溶液に他の金属の水溶性塩を溶解させると、得られる混合物中に他の金属原子が均一に分散するため好ましい。
(不活性融剤)
工程(4)および工程(5)の際に、混合物および仮焼物は、不活性融剤を含有していてもよい。不活性融剤は、目的物である複合金属酸化物と反応せず、且つ目的物と分離が容易な塩である。不活性融剤は、工程(4)の加熱温度および工程(5)の焼成温度で溶融し反応場を形成し、均一な反応を促進する。そのため、不活性融剤を用いると、均一な組成の生成物を得やすい。
不活性融剤としては、K2SO4、Na2SO4などの硫酸塩;K2CO3、Na2CO3などの炭酸塩;NaCl、KCl、NH4Clなどの塩化物;LiF、NaF、KF、NH4Fなどのフッ化物;ホウ酸;を挙げることができる。上記、不活性融剤のなかでも、製造工程が簡便になることから、硫酸塩が好ましい。より好ましくはK2SO4である。不活性融剤は2種以上併用することもできる。
混合物が不活性融剤を含有すると、混合物の加熱時および仮焼物の焼成時の反応性が向上し、これにより、得られるリチウム複合金属酸化物のBET比表面積を調整することが可能な場合がある。温度が同じ場合には、不活性融剤の含有量が多くなればなるほど、酸化物のBET比表面積は大きくなる傾向にある。また、加熱または焼成の際に、不活性融剤を含有すると、均一な反応を行うことができるため、加熱雰囲気の調整によりリチウム複合金属酸化物の原子レベルで局所構造が制御できる。
不活性融剤は、工程(2)における分離操作で得られる共沈物に、上記不活性融剤の溶液を含ませた後、乾燥させることにより、得られる共沈物に混合されてもよい。
例えば、工程(1)において、Niの硫酸塩やMnの硫酸塩やCoの硫酸塩を原料として用いた場合、原料水溶液中にSO4 2−イオンが遊離する。このSO4 2−イオンと、共沈に用いるアルカリに含まれる金属イオン(例えば、アルカリとしてKOHを用いる場合には、Kイオン)とが、工程(2)で分離した共沈物に残存し、不活性融剤(上記例であればK2SO4)が生じることがある。そのため、工程(1)における共沈後の原料水溶液を上記「不活性融剤の溶液」として用い、工程(2)で得られる共沈物に、共沈後の原料水溶液を含ませたまま乾燥させることにより、得られる共沈物に不活性融剤が混合されてもよい。
また、不活性融剤は、工程(3)において共沈物とリチウム化合物との混合時に、添加して混合できる。不活性融剤の量の制御が容易であるため、不活性融剤を工程(2)で添加する上述の方法よりも、工程(3)で添加する方法のほうが好ましい。工程(3)において不活性融剤を添加する場合は、工程(2)において得られる共沈物を洗浄し、共沈物に残存するアルカリや、Niの塩やMnの塩やCoの塩に由来する陰イオンの量を低減しておくことにより、不活性融剤の量の制御が容易となる。
不活性融剤は、リチウム複合金属酸化物に残留していてもよいし、洗浄により除去されていてもよい。
反応の均一性向上の観点から、前記不活性融剤が硫酸塩であり、混合物または仮焼物と前記硫酸塩とを混合したときに、得られる混合物中の硫酸塩の含有量が、用いたリチウム化合物100質量部に対して0.01質量部以上400質量部以下であることが好ましい。より好ましくは、0.1質量部以上10質量部以下である。
また、本実施形態のリチウム複合金属酸化物の製造方法により得られるリチウム複合金属酸化物を、ボールミルやジェットミルなどを用いて粉砕してもよい。粉砕によって、リチウム複合金属酸化物のBET比表面積を調整することが可能な場合がある。また、工程(1)から工程(5)を実施して得られるリチウム複合金属酸化物を粉砕し、再度工程(4)と工程(5)とを行うことで、粉砕後に焼成を行ってもよい。さらに、必要に応じて、粉砕と工程(4)、(5)による焼成とを、2回以上繰り返してもよい。また、リチウム複合金属酸化物は必要に応じて洗浄あるいは分級することもできる。
本実施形態のリチウム複合金属酸化物は、好ましくは0.05μm以上1μm以下の粒径の一次粒子と、一次粒子が凝集して形成された2μm以上100μm以下の粒径の二次粒子との混合物からなる。一次粒子、二次粒子の粒径は、SEMで観察することにより、測定することができる。
リチウム複合金属酸化物の二次粒子の大きさは、好ましくは2μm以上50μm以下の範囲であり、より好ましくは2μm以上10μm以下の範囲であり、さらにより好ましくは3μm以上8μm以下の範囲であり、特に好ましくは3.5μm以上7μm以下の範囲である。これらにより、得られる非水電解質二次電池の容量がより高まる。
リチウム複合金属酸化物の一次粒子の大きさは、好ましくは0.08μm以上0.8μm以下の範囲であり、より好ましくは0.10μm以上0.7μm以下の範囲であり、さらにより好ましくは0.15μm以上0.7μm以下の範囲であり、特に好ましくは0.2μm以上0.5μm以下の範囲である。これらにより、得られる非水電解質二次電池の高い電流レートにおける放電容量が高まる。
また、リチウム複合金属酸化物の平均粒子径(D50)は、好ましくは1μm以上50μm以下の範囲であり、より好ましくは1.5μm以上30μm以下の範囲であり、さらにより好ましくは2μm以上20μm以下の範囲であり、特に好ましくは3μm以上10μm以下の範囲である。これらにより、リチウム複合金属酸化物を用いた電極の密度が高まり、高容量の非水電解質二次電池が得られる。
リチウム複合金属酸化物の平均粒子径(D50)は、以下の方法で測定できる。
<リチウム複合金属酸化物の平均粒子径(D50)の測定>
測定するリチウム複合金属酸化物の粉末0.1gを、0.2質量%ヘキサメタりん酸ナトリウム水溶液50mlに投入し、該粉末を分散させた分散液を得る。得られた分散液についてマルバーン社製マスターサイザー2000(レーザー回折散乱粒度分布測定装置)を用いて、粒度分布を測定し、体積基準の累積粒度分布曲線を得る。得られた累積粒度分布曲線において、50%累積時の微小粒子側から見た粒子径の値が、平均粒子径(D50)である。
リチウム複合金属酸化物のBET比表面積は、好ましくは0.1m2/g以上20m2/g以下の範囲であり、より好ましくは0.5m2/g以上15m2/g以下の範囲であり、さらにより好ましくは1m2/g以上10m2/g以下の範囲であり、特に好ましくは2m2/g以上8m2/g以下の範囲である。これらにより、得られる非水電解質二次電池の高い電流レートにおける放電容量が高まる。
リチウム複合金属酸化物のBET比表面積は、以下の方法で測定できる。
<リチウム複合金属酸化物のBET比表面積の測定>
測定するリチウム複合金属酸化物の粉末1gを窒素雰囲気中、150℃で15分間乾燥させた後、マイクロメリティックス製フローソーブII2300を用いて測定する。
上記リチウム複合金属酸化物を、非水電解質二次電池の正極活物質に用いた場合に、従来よりも高容量を示す非水電解質二次電池が得られる。
[非水電解質二次電池]
次いで、非水電解質二次電池の構成を説明しながら、本実施形態のリチウム複合金属酸化物を非水電解質二次電池の正極活物質として用いた正極、およびこの正極を有する非水電解質二次電池について説明する。
本実施形態の非水電解質二次電池の一例は、正極、負極、正極と負極との間に配置されるセパレータ、および電解液を有する。
図1は、本実施形態の非水電解質二次電池の一例を示す模式図である。本実施形態の円筒型の非水電解質二次電池10は、次のようにして製造する。
まず、図1(a)に示すように、帯状を呈する2つのセパレータ1、一端に正極リード21を有する帯状の正極2、および一端に負極リード31を有する帯状の負極3を、セパレータ1、正極2、セパレータ1、負極3の順に積層し、巻回することにより電極群4とする。
次いで、図1(b)に示すように、電池缶5に電極群4および不図示のインシュレーターを収容した後、缶底を封止し、電極群4に電解液6を含浸させ、正極2と負極3との間に電解質を配置する。さらに、電池缶5の上部をトップインシュレーター7および封口体8で封止することで、非水電解質二次電池10を製造することができる。
電極群4の形状としては、例えば、電極群4を巻回の軸に対して垂直方向に切断したときの断面形状が、円、楕円、長方形、角を丸めた長方形となるような柱状の形状を挙げることができる。
また、このような電極群4を有する非水電解質二次電池の形状としては、国際電気標準会議(IEC)が定めた電池に対する規格であるIEC60086、またはJIS C 8500で定められる形状を採用することができる。例えば、円筒型、角型などの形状を挙げることができる。
さらに、非水電解質二次電池は、上記巻回型の構成に限らず、正極、セパレータ、負極、セパレータの積層構造を繰り返し重ねた積層型の構成であってもよい。積層型の非水電解質二次電池としては、いわゆるコイン型電池、ボタン型電池、ペーパー型(またはシート型)電池を例示することができる。
以下、各構成について順に説明する。
(正極)
本実施形態の正極は、まず正極活物質、導電材およびバインダーを含む正極合剤を調整し、正極合剤を正極集電体に担持させることで製造することができる。
(正極活物質)
本実施形態の正極活物質は、上述のリチウム複合金属酸化物を有する。本実施形態のリチウム複合金属酸化物を、非水電解質二次電池の正極活物質として用いることで、高容量を示す非水電解質二次電池とすることができる。
(導電材)
本実施形態の正極が有する導電材としては、炭素材料を用いることができる。炭素材料として黒鉛粉末、カーボンブラック(例えばアセチレンブラック)、繊維状炭素材料などを挙げることができる。カーボンブラックは、微粒で表面積が大きいため、少量を正極合剤中に添加することにより正極内部の導電性を高め、充放電効率および出力特性を向上させることができるが、多く入れすぎるとバインダーによる正極合剤と正極集電体との結着力、および正極合剤内部の結着力がいずれも低下し、かえって内部抵抗を増加させる原因となる。
正極合剤中の導電材の割合は、正極活物質100質量部に対して5質量部以上20質量部以下であると好ましい。導電材として黒鉛化炭素繊維、カーボンナノチューブなどの繊維状炭素材料を用いる場合には、この割合を下げることも可能である。
(バインダー)
本実施形態の正極が有するバインダーとしては、熱可塑性樹脂を用いることができる。この熱可塑性樹脂としては、ポリフッ化ビニリデン(以下、PVdFということがある。)、ポリテトラフルオロエチレン(以下、PTFEということがある。)、四フッ化エチレン・六フッ化プロピレン・フッ化ビニリデン系共重合体、六フッ化プロピレン・フッ化ビニリデン系共重合体、四フッ化エチレン・パーフルオロビニルエーテル系共重合体などのフッ素樹脂;ポリエチレン、ポリプロピレンなどのポリオレフィン樹脂;を挙げることができる。
これらの熱可塑性樹脂は、2種以上を混合して用いてもよい。バインダーとしてフッ素樹脂およびポリオレフィン樹脂を用い、正極合剤全体に対するフッ素樹脂の割合を1質量%以上10質量%以下、ポリオレフィン樹脂の割合を0.1質量%以上2質量%以下とすることによって、正極集電体との密着力および正極合剤内部の結合力がいずれも高い正極合剤を得ることができる。
(正極集電体)
本実施形態の正極が有する正極集電体としては、Al、Ni、ステンレスなどの金属材料を構成材料とする帯状の部材を用いることができる。なかでも、加工しやすく、安価であるという点でAlを形成材料とし、薄膜状に加工したものが好ましい。
正極集電体に正極合剤を担持させる方法としては、正極合剤を正極集電体上で加圧成型する方法が挙げられる。また、有機溶媒を用いて正極合剤をペースト化し、得られる正極合剤のペーストを正極集電体の少なくとも一面に塗工して乾燥させ、プレスし固着することで、正極集電体に正極合剤を担持させてもよい。
正極合剤をペースト化する場合、用いることができる有機溶媒としては、N,N—ジメチルアミノプロピルアミン、ジエチレントリアミンなどのアミン系溶媒;テトラヒドロフランなどのエーテル系溶媒;メチルエチルケトンなどのケトン系溶媒;酢酸メチルなどのエステル系溶媒;ジメチルアセトアミド、N−メチル−2−ピロリドン(以下、NMPということがある。)などのアミド系溶媒;が挙げられる。
正極合剤のペーストを正極集電体へ塗工する方法としては、例えば、スリットダイ塗工法、スクリーン塗工法、カーテン塗工法、ナイフ塗工法、グラビア塗工法および静電スプレー法が挙げられる。
以上に挙げられた方法により、正極を製造することができる。
(負極)
本実施形態の非水電解質二次電池が有する負極は、正極よりも低い電位でリチウムイオンのドープかつ脱ドープが可能であればよく、負極活物質を含む負極合剤が負極集電体に担持されてなる電極、および負極活物質単独からなる電極を挙げることができる。
(負極活物質)
負極が有する負極活物質としては、炭素材料、カルコゲン化合物(酸化物、硫化物など)、窒化物、金属または合金で、正極よりも低い電位でリチウムイオンのドープかつ脱ドープが可能な材料が挙げられる。
負極活物質として使用可能な炭素材料としては、天然黒鉛、人造黒鉛などの黒鉛、コークス類、カーボンブラック、熱分解炭素類、炭素繊維および有機高分子化合物焼成体を挙げることができる。
負極活物質として使用可能な酸化物としては、SiO2、SiOなど式SiOx(ここで、xは正の実数)で表されるケイ素の酸化物;TiO2、TiOなど式TiOx(ここで、xは正の実数)で表されるチタンの酸化物;V2O5、VO2など式VOx(ここで、xは正の実数)で表されるバナジウムの酸化物;Fe3O4、Fe2O3、FeOなど式FeOx(ここで、xは正の実数)で表される鉄の酸化物;SnO2、SnOなど式SnOx(ここで、xは正の実数)で表されるスズの酸化物;WO3、WO2など一般式WOx(ここで、xは正の実数)で表されるタングステンの酸化物;Li4Ti5O12、LiVO2などのリチウムとチタンまたはバナジウムとを含有する複合金属酸化物;を挙げることができる。
負極活物質として使用可能な硫化物としては、Ti2S3、TiS2、TiSなど式TiSx(ここで、xは正の実数)で表されるチタンの硫化物;V3S4、VS2、VSなど式VSx(ここで、xは正の実数)で表されるバナジウムの硫化物;Fe3S4、FeS2、FeSなど式FeSx(ここで、xは正の実数)で表される鉄の硫化物;Mo2S3、MoS2など式MoSx(ここで、xは正の実数)で表されるモリブデンの硫化物;SnS2、SnSなど式SnSx(ここで、xは正の実数)で表されるスズの硫化物;WS2など式WSx(ここで、xは正の実数)で表されるタングステンの硫化物;Sb2S3など式SbSx(ここで、xは正の実数)で表されるアンチモンの硫化物;Se5S3、SeS2、SeSなど式SeSx(ここで、xは正の実数)で表されるセレンの硫化物;を挙げることができる。
負極活物質として使用可能な窒化物としては、Li3N、Li3−xAxN(ここで、AはNiおよびCoのいずれか一方または両方であり、0<x<3である。)などのリチウム含有窒化物を挙げることができる。
これらの炭素材料、酸化物、硫化物、窒化物は、1種のみ用いてもよく2種以上を併用して用いてもよい。また、これらの炭素材料、酸化物、硫化物、窒化物は、結晶質または非晶質のいずれでもよい。
また、負極活物質として使用可能な金属としては、リチウム金属、シリコン金属およびスズ金属などを挙げることができる。
負極活物質として使用可能な合金としては、Li−Al、Li−Ni、Li−Si、Li−Sn、Li−Sn−Niなどのリチウム合金;Si−Znなどのシリコン合金;Sn−Mn、Sn−Co、Sn−Ni、Sn−Cu、Sn−Laなどのスズ合金;Cu2Sb、La3Ni2Sn7などの合金;を挙げることもできる。
これらの金属や合金は、例えば箔状に加工された後、主に単独で電極として用いられる。
上記負極活物質の中では、充電時に未充電状態から満充電状態にかけて負極の電位がほとんど変化しない(電位平坦性が良い)、平均放電電位が低い、繰り返し充放電させたときの容量維持率が高い(サイクル特性が良い)などの理由から、天然黒鉛、人造黒鉛などの黒鉛を主成分とする炭素材料が好ましく用いられる。炭素材料の形状としては、例えば天然黒鉛のような薄片状、メソカーボンマイクロビーズのような球状、黒鉛化炭素繊維のような繊維状、または微粉末の凝集体などのいずれでもよい。
前記の負極合剤は、必要に応じて、バインダーを含有してもよい。バインダーとしては、熱可塑性樹脂を挙げることができ、具体的には、PVdF、熱可塑性ポリイミド、カルボキシメチルセルロース、ポリエチレンおよびポリプロピレンを挙げることができる。
(負極集電体)
負極が有する負極集電体としては、Cu、Ni、ステンレスなどの金属材料を構成材料とする帯状の部材を挙げることができる。なかでも、リチウムと合金を作り難く、加工しやすいという点で、Cuを形成材料とし、薄膜状に加工したものが好ましい。
このような負極集電体に負極合剤を担持させる方法としては、正極の場合と同様に、加圧成型による方法、溶媒などを用いてペースト化し負極集電体上に塗布、乾燥後プレスし圧着する方法が挙げられる。
(セパレータ)
本実施形態の非水電解質二次電池が有するセパレータとしては、例えば、ポリエチレン、ポリプロピレンなどのポリオレフィン樹脂、フッ素樹脂、含窒素芳香族重合体などの材質からなる、多孔質膜、不織布、織布などの形態を有する材料を用いることができる。また、これらの材質を2種以上用いてセパレータを形成してもよいし、これらの材料を積層してセパレータを形成してもよい。
セパレータとしては、例えば特開2000−30686号公報、特開平10−324758号公報などに記載のセパレータを挙げることができる。セパレータの厚みは電池の体積エネルギー密度が上がり、内部抵抗が小さくなるという点で、機械的強度が保たれる限り薄くした方がよく、好ましくは5~200μm程度、より好ましくは5~40μm程度である。
セパレータは、好ましくは、熱可塑性樹脂を含有する多孔質フィルムを有する。非水電解質二次電池においては、正極−負極間の短絡などが原因で電池内に異常電流が流れた際に、短絡箇所の電流を遮断して、過大電流が流れることを阻止(シャットダウン)する機能を有することが好ましい。ここで、シャットダウンは、短絡により短絡箇所のセパレータが過熱され、予め想定された使用温度を越えた場合に、セパレータにおける多孔質フィルムが軟化または融解して微細孔を閉塞することによりなされる。そして、セパレータはシャットダウンした後、ある程度の高温まで電池内の温度が上昇しても、その温度により破膜することなく、シャットダウンした状態を維持することが好ましい。
このようなセパレータとしては、耐熱多孔層と多孔質フィルムとが積層された積層フィルムが挙げられる。このような積層フィルムをセパレータとして用いることにより、本実施形態における二次電池の耐熱性をより高めることが可能となる。積層フィルムにおいては、耐熱多孔層は、多孔質フィルムの両面に積層されていてもよい。
(積層フィルム)
以下、前記の耐熱多孔層と多孔質フィルムとが互いに積層された積層フィルムについて説明する。
本実施形態の非水電解質二次電池のセパレータとして用いられる積層フィルムにおいて、耐熱多孔層は、多孔質フィルムよりも耐熱性の高い層である。耐熱多孔層は、無機粉末から形成されていてもよいし(第1の耐熱多孔層)、耐熱樹脂から形成されていてもよいし(第2の耐熱多孔層)、耐熱樹脂とフィラーとを含んで形成されていてもよい(第3の耐熱多孔層)。耐熱多孔層が、耐熱樹脂を含有することにより、塗工などの容易な手法で、耐熱多孔層を形成することができる。
(第1の耐熱多孔層)
耐熱多孔層が無機粉末から形成されている場合、耐熱多孔層に用いられる無機粉末としては、例えば、金属酸化物、金属窒化物、金属炭化物、金属水酸化物、炭酸塩、硫酸塩などの無機物からなる粉末が挙げられ、これらの中でも、導電性の低い(絶縁体の)無機物からなる粉末が好ましく用いられる。具体的に例示すると、アルミナ、シリカ、二酸化チタンまたは炭酸カルシウムなどからなる粉末が挙げられる。このような無機粉末は、単独で用いてもよいし、2種以上を混合して用いることもできる。
これらの無機粉末の中でも、化学的安定性が高いことから、アルミナ粉末が好ましい。また、無機粉末を構成する粒子のすべてがアルミナ粒子であることがより好ましく、無機粉末を構成する粒子のすべてがアルミナ粒子であり、その一部または全部が略球状のアルミナ粒子であることがさらに好ましい。
(第2の耐熱多孔層)
耐熱多孔層が耐熱樹脂から形成されている場合、耐熱多孔層に用いられる耐熱樹脂としては、ポリアミド、ポリイミド、ポリアミドイミド、ポリカーボネート、ポリアセタール、ポリサルホン、ポリフェニレンサルファイド、ポリエーテルケトン、芳香族ポリエステル、ポリエーテルサルホンおよびポリエーテルイミドを挙げることができる。積層フィルムの耐熱性をより高めるためには、ポリアミド、ポリイミド、ポリアミドイミド、ポリエーテルサルホンおよびポリエーテルイミドが好ましく、より好ましくは、ポリアミド、ポリイミドまたはポリアミドイミドである。
耐熱多孔層に用いられる耐熱樹脂としてさらに好ましくは、芳香族ポリアミド(パラ配向芳香族ポリアミド、メタ配向芳香族ポリアミド)、芳香族ポリイミド、芳香族ポリアミドイミドなどの含窒素芳香族重合体であり、とりわけ好ましくは芳香族ポリアミド、製造しやすいために特に好ましいのは、パラ配向芳香族ポリアミド(以下、パラアラミドということがある。)である。
また、耐熱樹脂として、ポリ−4−メチルペンテン−1および環状オレフィン系重合体を挙げることもできる。
これらの耐熱樹脂を用いることにより、非水電解質二次電池のセパレータとして用いられる積層フィルムの耐熱性、すなわち、積層フィルムの熱破膜温度をより高めることができる。これらの耐熱樹脂のうち、含窒素芳香族重合体を用いる場合には、その分子内の極性により、電解液との相性、すなわち、耐熱多孔層における保液性も向上する場合があり、非水電解質二次電池製造時における電解液の含浸の速度も高く、非水電解質二次電池の充放電容量もより高まる。
かかる積層フィルムの熱破膜温度は、耐熱樹脂の種類に依存し、使用場面、使用目的に応じ、選択使用される。より具体的には、耐熱樹脂として、上記含窒素芳香族重合体を用いる場合は400℃程度に、また、ポリ−4−メチルペンテン−1を用いる場合は250℃程度に、環状オレフィン系重合体を用いる場合は300℃程度に、夫々、熱破膜温度をコントロールすることができる。また、耐熱多孔層が、無機粉末からなる場合には、熱破膜温度を、例えば、500℃以上にコントロールすることも可能である。
上記パラアラミドは、パラ配向芳香族ジアミンとパラ配向芳香族ジカルボン酸ハライドとの重縮合により得られ、アミド結合が芳香族環のパラ位またはそれに準じた配向位(例えば、4,4’−ビフェニレン、1,5−ナフタレン、2,6−ナフタレンなどのような反対方向に同軸または平行に延びる配向位)で結合される繰り返し単位から実質的になる。具体的には、ポリ(パラフェニレンテレフタルアミド)、ポリ(パラベンズアミド)、ポリ(4,4’−ベンズアニリドテレフタルアミド)、ポリ(パラフェニレン−4,4’−ビフェニレンジカルボン酸アミド)、ポリ(パラフェニレン−2,6−ナフタレンジカルボン酸アミド)、ポリ(2−クロロ−パラフェニレンテレフタルアミド)、パラフェニレンテレフタルアミド/2,6−ジクロロパラフェニレンテレフタルアミド共重合体などのパラ配向型またはパラ配向型に準じた構造を有するパラアラミドが例示される。
前記の芳香族ポリイミドとしては、芳香族の二酸無水物とジアミンとの重縮合で製造される全芳香族ポリイミドが好ましい。
重縮合に用いられる芳香族の二酸無水物の具体例としては、ピロメリット酸二無水物、3,3’,4,4’−ジフェニルスルホンテトラカルボン酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、2,2’−ビス(3,4−ジカルボキシフェニル)ヘキサフルオロプロパンおよび3,3’,4,4’−ビフェニルテトラカルボン酸二無水物が挙げられる。
重縮合に用いられるジアミンの具体例としては、オキシジアニリン、パラフェニレンジアミン、ベンゾフェノンジアミン、3,3’−メチレンジアニリン、3,3’−ジアミノベンソフェノン、3,3’−ジアミノジフェニルスルフォンおよび1,5−ナフタレンジアミンが挙げられる。
また、芳香族ポリイミドとしては、溶媒に可溶なポリイミドが好適に使用できる。このようなポリイミドとしては、例えば、3,3’,4,4’−ジフェニルスルホンテトラカルボン酸二無水物と芳香族ジアミンとの重縮合物のポリイミドが挙げられる。
前記の芳香族ポリアミドイミドとしては、芳香族ジカルボン酸および芳香族ジイソシアネートの重縮合から得られるもの、芳香族二酸無水物および芳香族ジイソシアネートの重縮合から得られるものが挙げられる。芳香族ジカルボン酸の具体例としてはイソフタル酸およびテレフタル酸が挙げられる。また芳香族二酸無水物の具体例としては無水トリメリット酸が挙げられる。芳香族ジイソシアネートの具体例としては、4,4’−ジフェニルメタンジイソシアネート、2,4−トリレンジイソシアネート、2,6−トリレンジイソシアネート、オルソトリレンジイソシアネートおよびm−キシレンジイソシアネートが挙げられる。
また、イオン透過性をより高めるためには、積層フィルムが有する耐熱多孔層の厚みは、1μm以上10μm以下、さらには1μm以上5μm以下、特に1μm以上4μm以下という薄い耐熱多孔層であることが好ましい。また、耐熱多孔層は微細孔を有し、その孔のサイズ(直径)は、好ましくは3μm以下、より好ましくは1μm以下である。
(第3の耐熱多孔層)
また、耐熱多孔層が耐熱樹脂とフィラーとを含んで形成されている場合、耐熱樹脂は、上記第2の耐熱多孔層に用いられる耐熱樹脂と同じものを使用することができる。フィラーは、有機粉末、無機粉末またはこれらの混合物からなる群から選ばれる1種以上を用いることができる。フィラーを構成する粒子は、その平均粒子径が、0.01μm以上1μm以下であることが好ましい。
フィラーとして用いることができる有機粉末としては、例えば、スチレン、ビニルケトン、アクリロニトリル、メタクリル酸メチル、メタクリル酸エチル、グリシジルメタクリレート、グリシジルアクリレート、アクリル酸メチルなどの単独または2種類以上の共重合体;PTFE、4フッ化エチレン−6フッ化プロピレン共重合体、4フッ化エチレン−エチレン共重合体、ポリビニリデンフルオライドなどのフッ素系樹脂;メラミン樹脂;尿素樹脂;ポリオレフィン樹脂;ポリメタクリレート;などの有機物からなる粉末が挙げられる。このような有機粉末は、単独で用いてもよいし、2種以上を混合して用いることもできる。これらの有機粉末の中でも、化学的安定性が高いことから、PTFEの粉末が好ましい。
フィラーとして用いることができる無機粉末としては、上記耐熱多孔層に用いられる無機粉末と同じものを例示することができる。
耐熱多孔層が耐熱樹脂とフィラーとを含んで形成されている場合、フィラーの含有量としては、フィラーの材質の比重にもよるが、例えば、フィラーを構成する粒子のすべてがアルミナ粒子である場合には、耐熱多孔層の総質量を100質量部としたとき、フィラーの質量は、好ましくは5質量部以上95質量部以下であり、より好ましくは20質量部以上95質量部以下であり、さらに好ましくは30質量部以上90質量部以下である。これらの範囲は、フィラーの材質の比重により、適宜設定できる。
フィラーの形状については、略球状、板状、柱状、針状、繊維状などの形状が挙げられ、いずれの粒子も用いることができるが、均一な孔を形成しやすいことから、略球状粒子であることが好ましい。略球状粒子としては、粒子のアスペクト比(粒子の長径/粒子の短径)が1以上1.5以下である粒子が挙げられる。粒子のアスペクト比は、電子顕微鏡写真により測定することができる。
本実施形態の非水電解質二次電池のセパレータとして用いられる積層フィルムにおいて多孔質フィルムは、微細孔を有し、シャットダウン機能を有することが好ましい。この場合、多孔質フィルムは、熱可塑性樹脂を含有する。
多孔質フィルムにおける微細孔のサイズは、好ましくは3μm以下、より好ましくは1μm以下である。多孔質フィルムの空孔率は、好ましくは30体積%以上80体積%以下、より好ましくは40体積%以上70体積%以下である。非水電解質二次電池において、予め想定された使用温度を越えた場合には、熱可塑性樹脂を含有する多孔質フィルムは、多孔質フィルムを構成する熱可塑性樹脂の軟化または融解により、微細孔を閉塞することができる。
多孔質フィルムに用いられる熱可塑性樹脂は、非水電解質二次電池における電解液に溶解しないものを選択すればよい。具体的には、ポリエチレン、ポリプロピレンなどのポリオレフィン樹脂および熱可塑性ポリウレタン樹脂を挙げることができ、これらの2種以上の混合物を用いてもよい。
セパレータがより低温で軟化してシャットダウンするためには、多孔質フィルムがポリエチレンを含有することが好ましい。ポリエチレンとして、低密度ポリエチレン、高密度ポリエチレン、線状ポリエチレンなどのポリエチレンを挙げることができ、分子量が100万以上の超高分子量ポリエチレンを挙げることもできる。
多孔質フィルムの突刺し強度をより高めるためには、多孔質フィルムを構成する熱可塑性樹脂は、少なくとも超高分子量ポリエチレンを含有することが好ましい。また、多孔質フィルムの製造面において、熱可塑性樹脂は、低分子量(重量平均分子量1万以下)のポリオレフィンからなるワックスを含有することが好ましい場合もある。
また、積層フィルムにおける多孔質フィルムの厚みは、好ましくは3μm以上30μm以下であり、より好ましくは3μm以上25μm以下である。また、本実施形態において、積層フィルムの厚みは、好ましくは40μm以下、より好ましくは、30μm以下である。また、耐熱多孔層の厚みをA(μm)、多孔質フィルムの厚みをB(μm)としたときには、A/Bの値が、0.1以上1以下であることが好ましい。
本実施形態において、セパレータは、電池使用時(充放電時)に電解質を良好に透過させるため、JIS P 8117で定められるガーレー法による透気抵抗度が、50秒/100cc以上、300秒/100cc以下であることが好ましく、50秒/100cc以上、200秒/100cc以下であることがより好ましい。
また、セパレータの空孔率は、好ましくは30体積%以上80体積%以下、より好ましくは40体積%以上70体積%以下である。セパレータは空孔率の異なるセパレータを積層したものであってもよい。
(電解液)
本実施形態の非水電解質二次電池が有する電解液は、電解質および有機溶媒を含有する。
電解液に含まれる電解質としては、LiClO4、LiPF6、LiAsF6、LiSbF6、LiBF4、LiCF3SO3、LiN(SO2CF3)2、LiN(SO2C2F5)2、LiN(SO2CF3)(COCF3)、Li(C4F9SO3)、LiC(SO2CF3)3、Li2B10Cl10、LiBOB(ここで、BOBは、bis(oxalato)borateのことである。)、低級脂肪族カルボン酸リチウム塩、LiAlCl4などのリチウム塩が挙げられ、これらの2種以上の混合物を使用してもよい。なかでも電解質としては、フッ素を含むLiPF6、LiAsF6、LiSbF6、LiBF4、LiCF3SO3、LiN(SO2CF3)2およびLiC(SO2CF3)3からなる群より選ばれる少なくとも1種を含むものを用いることが好ましい。
また前記電解液に含まれる有機溶媒としては、例えばプロピレンカーボネート、エチレンカーボネート、ジメチルカーボネート、ジエチルカーボネート、エチルメチルカーボネート、4−トリフルオロメチル−1,3−ジオキソラン−2−オン、1,2−ジ(メトキシカルボニルオキシ)エタンなどのカーボネート類;1,2−ジメトキシエタン、1,3−ジメトキシプロパン、ペンタフルオロプロピルメチルエーテル、2,2,3,3−テトラフルオロプロピルジフルオロメチルエーテル、テトラヒドロフラン、2−メチルテトラヒドロフランなどのエーテル類;ギ酸メチル、酢酸メチル、γ−ブチロラクトンなどのエステル類;アセトニトリル、ブチロニトリルなどのニトリル類;N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミドなどのアミド類;3−メチル−2−オキサゾリドンなどのカーバメート類;スルホラン、ジメチルスルホキシド、1,3−プロパンサルトンなどの含硫黄化合物、またはこれらの有機溶媒にさらにフルオロ基を導入したもの(有機溶媒が有する水素原子のうち1以上をフッ素原子で置換したもの)を用いることができる。
有機溶媒としては、これらのうちの2種以上を混合して用いることが好ましい。中でもカーボネート類を含む混合溶媒が好ましく、環状カーボネートと非環状カーボネートとの混合溶媒および環状カーボネートとエーテル類との混合溶媒がさらに好ましい。環状カーボネートと非環状カーボネートとの混合溶媒としては、エチレンカーボネート、ジメチルカーボネートおよびエチルメチルカーボネートを含む混合溶媒が好ましい。このような混合溶媒を用いた電解液は、動作温度範囲が広く、高い電流レートにおける充放電を行っても劣化し難く、長時間使用しても劣化し難く、かつ負極の活物質として天然黒鉛、人造黒鉛などの黒鉛材料を用いた場合でも難分解性であるという利点を有する。
また、電解液としては、得られる非水電解質二次電池の安全性が高まるため、LiPF6などのフッ素を含むリチウム塩およびフッ素置換基を有する有機溶媒を含む電解液を用いることが好ましい。ペンタフルオロプロピルメチルエーテル、2,2,3,3−テトラフルオロプロピルジフルオロメチルエーテルなどのフッ素置換基を有するエーテル類とジメチルカーボネートとを含む混合溶媒は、高い電流レートにおける充放電を行っても容量維持率が高いため、さらに好ましい。
上記の電解液の代わりに固体電解質を用いてもよい。固体電解質としては、例えばポリエチレンオキサイド系の高分子化合物、ポリオルガノシロキサン鎖またはポリオキシアルキレン鎖の少なくとも一種以上を含む高分子化合物などの有機系高分子電解質を用いることができる。また、高分子化合物に非水電解液を保持させた、いわゆるゲルタイプのものを用いることもできる。またLi2S−SiS2、Li2S−GeS2、Li2S−P2S5、Li2S−B2S3、Li2S−SiS2−Li3PO4、Li2S−SiS2−Li2SO4などの硫化物を含む無機系固体電解質を用いてもよい。これら固体電解質を用いることで、非水電解質二次電池の安全性をより高めることができることがある。
また、本実施形態の非水電解質二次電池において、固体電解質を用いる場合には、固体電解質がセパレータの役割を果たす場合もあり、その場合には、セパレータを必要としないこともある。
上記の正極活物質は、上述した本実施形態のリチウム複合金属酸化物を用いているため、正極活物質を用いた非水電解質二次電池は、従来よりも高容量を示すことができる。
また、上記の正極は、上述した本実施形態のリチウム複合金属酸化物を用いた正極活物質を有するため、非水電解質二次電池は、従来よりも高容量を示すことができる。
さらに、上記の非水電解質二次電池は、上述した正極を有するため、従来よりも高容量を示す。 [Lithium composite metal oxide]
The lithium composite metal oxide of this embodiment contains Mn, Ni, Li and Co and satisfies the following (a) and (b).
(A) In a radial distribution function obtained by Fourier transform of a wide-range X-ray absorption fine structure (EXAFS) spectrum at the K absorption edge of Mn in the lithium composite metal oxide, 1.5% by oxygen atoms bonded to Mn atoms Near first adjacent peak AMnStrength of IAMn, The second adjacent peak B around 2.5Å due to the metal atom next to the Mn atom next to the oxygen atom bonded to the Mn atomMnStrength of IBMnWhen IBMn/ IAMnIs 0.5 or more and 1.2 or less.
(B) In the radial distribution function obtained by Fourier transforming the EXAFS spectrum at the K absorption edge of Ni in the lithium composite metal oxide, the first proximity peak A around 1.5Å due to oxygen atoms bonded to Ni atoms.NiStrength of IANi, The intensity of the second adjacent peak in the vicinity of 2.5 に よ る by the metal atom close to the Ni atom next to the oxygen atom bonded to the Ni atom isBNiWhen IBNi/ IANiIs 1.0 or more and 1.7 or less.
Hereafter, it explains in order.
(EXAFS spectrum)
First, the EXAFS spectrum used for defining the lithium composite metal oxide of this embodiment will be described. The EXAFS spectrum used in the present embodiment is handled in the same manner as a general EXAFS spectrum. The measurement and principle of the EXAFS spectrum are described in, for example, “X-ray absorption spectroscopy—XAFS and its applications” (Toshiaki Ohta (2002)). The principle is as follows.
First, when an X-ray having a specific wavelength is transmitted through a substance to be measured, the intensity of the X-ray irradiated to the substance (incident X-ray intensity: I0) And the intensity of X-rays transmitted through the substance (transmitted X-ray intensity: It), The X-ray absorbance per unit thickness is obtained for the substance to be measured at a specific wavelength.
The wavelength of X-rays irradiating the substance is changed (that is, the energy (eV) of incident X-rays is changed), the X-ray absorbance of each wavelength (each energy) with respect to the X-rays is measured, When an X-ray absorption spectrum is created with the energy of the line (eV) and the y-axis as the X-ray absorbance, it can be seen that there is energy at which the X-ray absorbance rapidly increases. This energy value is called the absorption edge. The absorption edge corresponds to the energy level of the atomic shell of the atoms constituting the material and is unique to each atom. For example, an absorption edge corresponding to the K shell of an atom is called a K absorption edge.
In the X-ray absorption spectrum, a fine vibration structure appearing in an energy side region about 20 to 1000 eV higher than the absorption edge is called a broad X-ray absorption fine structure (EXAFS), and the spectrum is called an EXAFS spectrum. When Fourier transformation is performed on the EXAFS spectrum, a radial distribution function centered on X-ray absorbing atoms (atomic atoms of interest) is obtained. From this radial distribution function, information such as the distance between X-ray absorbing atoms and X-ray scattering atoms (atoms near the X-ray absorbing atoms), the number of X-ray scattering atoms, and the like can be obtained. Can be obtained.
Generally, the intensity of the peak of the radial distribution function is affected by the number of X-ray scattering atoms, but it also affects the isotropy of the interatomic distance between X-ray absorbing atoms and X-ray scattering atoms. Is done. For example, when two X-ray absorbing atoms have substantially the same number of X-ray scattering atoms and the X-ray scattering atoms are recognized to have substantially the same scattering power, the peak of the radial distribution function For those with high intensity, the interatomic distance between the X-ray absorbing atom and the X-ray scattering atom is isotropic with no difference in direction, and the distance distribution between the X-ray absorbing atom and the X-ray scattering atom is small. Means.
Therefore, in the present embodiment, attention is paid to the peak intensity ratio of the radial distribution function obtained at the K absorption edge of Mn and Ni.
In other words, by controlling the intensity ratio of the peak of the radial distribution function within a certain range, the atomic level local structure in the lithium composite metal oxide can be controlled to a specific condition even for samples with different composition ratios. In addition, a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery exhibiting a higher capacity than before can be obtained.
In the lithium composite metal oxide of this embodiment, the peak due to O (oxygen atom) bonded to the Mn atom in the radial distribution function of the K absorption edge of Mn is the first proximity peak A.MnAnd First proximity peak AMnPreferably appears in the vicinity of 1.5 to 1.9 to 1.9, more preferably 1.5 to 1.6.
Further, in the lithium composite metal oxide of the present embodiment, in the radial distribution function of the K absorption edge of Mn, an atom X next to Mn atom next to O bonded to Mn atom (where X is Li, Mn , And a peak due to a metal atom such as Ni).MnAnd Second adjacent peak BMnPreferably appears in the vicinity of 2.5 mm from 2.44 mm to 2.55 mm, more preferably from 2.46 mm to 2.55 mm. Here, the atom X is bonded to O bonded to the Mn atom.
Furthermore, in the lithium composite metal oxide of this embodiment, in the radial distribution function of the K absorption edge of Ni, the peak due to O bonded to Ni atoms is the first proximity peak A.NiAnd First proximity peak ANiPreferably appears in the vicinity of 1.5 to 1.9 to 1.9, more preferably 1.5 to 1.6.
In the lithium composite metal oxide of this embodiment, in the radial distribution function of the K absorption edge of Ni, an atom X that is closest to the Ni atom next to O bonded to the Ni atom (where X is Li, Mn , And a peak due to a metal atom such as Ni).NiAnd Second adjacent peak BNiPreferably appears in the vicinity of 2.5 mm from 2.44 mm to 2.55 mm, more preferably from 2.46 mm to 2.55 mm. Here, atom X is bonded to O bonded to Ni atom.
The lithium composite metal oxide of this embodiment has a peak intensity ratio of the radial distribution function, that is, IAMnAnd IBMnTo the ratio (IBMn/ IAMn) And IANiAnd IBNiTo the ratio (IBNi/ IANi) Within a specific range, the local structure at the atomic level is controlled. Such a lithium composite metal oxide of this embodiment is useful for a non-aqueous electrolyte secondary battery that exhibits a higher capacity than before.
The lithium composite metal oxide of the present embodiment has a high isotropy of the interatomic distance between O and X around the Mn atom, and falls within a specific range, and thus has high characteristics as a positive electrode active material. I like thisBMn/ IAMnThe value of is 0.5 or more and 1.2 or less, preferably 0.6 or more and 1.2 or less, more preferably 0.7 or more and 1.2 or less, and even more preferably 1.0 or more. 1.2 or less, particularly preferably 1.1 or more and 1.2 or less.
In addition, the lithium composite metal oxide of the present embodiment has a high degree of isotropy in the interatomic distance between O and X around the Ni atom, and falls within a specific range, and thus has high characteristics as a positive electrode active material. . I like thisBNi/ IANiThe value of is 1.0 or more and 1.7 or less, preferably 1.1 or more and 1.7 or less, more preferably 1.2 or more and 1.7 or less.
These IBMn/ IAMnRange of values and IBNi/ IANiThe range of values can be arbitrarily combined.
Furthermore, IBMn/ IAMnAnd IBNi/ IANiProduct with (IBMn/ IAMn× IBNi/ IANi)) Has both the isotropicity of the interatomic distance between the O and the atom X around the appropriate Mn atom and the isotropicity of the interatomic distance between the O and the atom X around the proper Ni atom. 7 or more and 2.0 or less, preferably 0.9 or more and 2.0 or less, more preferably 1.1 or more and 2.0 or less.
The crystal structure of the lithium composite metal oxide of the present embodiment is preferably a layered structure, more preferably a hexagonal crystal structure or a monoclinic crystal structure.
The hexagonal crystal structure is P3, P31, P32, R3, P-3, R-3, P312, P321, P3112, P3121, P3212, P3221, R32, P3m1, P31m, P3c1, P31c, R3m, R3c, P-31m, P-31c, P-3m1, P-3c1, R-3m, R-3c, P6, P61, P65, P62, P64, P63, P-6, P6 / m, P63/ M, P622, P6122, P6522, P6222, P6422, P6322, P6mm, P6cc, P63cm, P63mc, P-6m2, P-6c2, P-62m, P-62c, P6 / mmm, P6 / mcc, P63/ Mcm, P63It is classified into any one space group selected from the group consisting of / mmc.
The monoclinic crystal structure is P2, P2.1, C2, Pm, Pc, Cm, Cc, P2 / m, P21/ M, C2 / m, P2 / c, P21/ C, C2 / c is classified into any one space group selected from the group consisting of C2 / c.
Among these, since the discharge capacity of the obtained nonaqueous electrolyte secondary battery is increased, the crystal structure of the lithium composite metal oxide is a hexagonal crystal structure classified into the space group R-3m, or C2 / m. A monoclinic crystal structure classified as follows is particularly preferable.
The space group of the lithium composite metal oxide of the present embodiment can be confirmed by the following method.
First, X-ray powder diffraction measurement was performed on a lithium composite metal oxide using CuKα as a radiation source and a diffraction angle 2θ measurement range of 10 ° to 90 °, and then Rietveld analysis was performed based on the results. And determining a crystal structure of the lithium composite metal oxide and a space group in the crystal structure. Rietveld analysis is a technique for analyzing the crystal structure of a material using diffraction peak data (diffraction peak intensity, diffraction angle 2θ) in powder X-ray diffraction measurement of the material, and is a conventionally used technique. (See, for example, “Practice of Powder X-ray Analysis—Introduction to Rietveld Method”, published on February 10, 2002, edited by the Japan Society for Analytical Chemistry X-ray Analysis Research Meeting).
The composition of the lithium composite metal oxide in this embodiment is such that the amount (mol) of Li is ALi, When the amount (mole) of metal other than Li is A, ALi/ A may be 0.7 or more and 1.4 or less.
The lithium composite metal oxide in the present embodiment preferably has a layered structure and the composition is represented by the following formula (1).
Li1 + x(Ni1-x-y-αMnyCoαMβ) O2... (1)
(In the formula (1), −0.3 ≦ x ≦ 0.4, 0.35 ≦ y ≦ 0.7, 0 <α ≦ 0.1, 0 ≦ β <0.1 (where 0 <α + β ≦ 0.1), −0.05 ≦ x + y + α + β <1, and M is selected from the group consisting of Al, Mg, Ti, Ca, Cu, Zn, Fe, Cr, Mo, Si, Sn, Nb and V One or more elements.)
The value of x in the formula (1) is −0.3 ≦ x ≦ 0.4, preferably −0.2 ≦ x ≦ 0.35, more preferably −0.1 ≦ x ≦ 0. .3.
Since the lithium composite metal oxide of the present embodiment has a high discharge capacity at 60 ° C., M is preferably Fe.
The lithium composite metal oxide particles of the present embodiment are used as a core material, and B, Al, Ga, In, Si, Ge, Sn, Mg, and the surface of the core material (lithium composite metal oxide particles) A compound containing one or more atoms selected from the group consisting of transition metals may be attached.
Among the above atoms, at least one selected from the group consisting of B, Al, Mg, Co, Cr, and Mn is preferable, and Al is more preferable because a uniform coating layer can be easily formed.
Examples of such a compound include oxides, fluorides, sulfides, hydroxides, oxyhydroxides, carbonates, nitrates, organic acid salts and mixtures thereof of the above atoms. Of these, oxides, hydroxides, oxyhydroxides or mixtures thereof are preferred.
As the compound to be deposited on the surface of the core material, alumina which is an oxide of Al is preferable.
[Method for producing lithium composite metal oxide]
Next, a method for producing the above-described lithium composite metal oxide will be described.
The method for producing a lithium composite metal oxide according to this embodiment includes the following steps (1) to (5).
(1) A step of obtaining a slurry by bringing an aqueous solution containing Ni ions, Mn ions and Co ions (hereinafter sometimes referred to as “raw material aqueous solution”) into contact with an alkali to form a coprecipitate.
(2) Separating the coprecipitate from the slurry obtained in (1)
(3) A step of mixing the coprecipitate obtained in (2) with a lithium compound.
(4) A step of heating the mixture obtained in (3) at a temperature of 200 ° C. or higher and 500 ° C. or lower in an atmosphere having an oxygen concentration of 5% by volume or higher.
(5) A step of firing the product obtained in (4) (hereinafter sometimes referred to as “calcined product”) at a temperature of 600 ° C. or higher and 950 ° C. or lower in an atmosphere having an oxygen concentration of less than 5% by volume.
Here, the “oxygen concentration” in the step (4) refers to an average oxygen concentration in the heat treatment space when the space (heat treatment space) for heating the mixture is in the range of 200 ° C. or more and 500 ° C. or less. Similarly, the “oxygen concentration” in step (5) refers to the average oxygen concentration in the heat treatment space when the space (heat treatment space) for calcining the calcined product is in the range of 600 ° C. or higher and 950 ° C. or lower.
(Process (1))
In the above step (1), the raw material aqueous solution can be adjusted by dissolving a compound containing Ni, Mn and Co in water. In particular, the raw material aqueous solution is preferably an aqueous solution obtained by dissolving Ni sulfate, Mn sulfate, and Co sulfate in water.
Further, when each raw material containing Ni, Mn and Co is difficult to dissolve in water, for example, when these raw materials are oxides, hydroxides, metal materials, these raw materials are mixed with sulfuric acid. The raw material aqueous solution can be obtained by dissolving in the aqueous solution.
The alkali used in step (1) includes LiOH (lithium hydroxide), NaOH (sodium hydroxide), KOH (potassium hydroxide), Li2CO3(Lithium carbonate), Na2CO3(Sodium carbonate), K2CO3(Potassium carbonate) and (NH4)2CO3One or more salts selected from the group consisting of (ammonium carbonate) can be mentioned. The alkali used may be an anhydride or a hydrate. An anhydride and a hydrate may be used in combination. In the step (1), it is preferable to use the above aqueous solution of alkali (alkaline aqueous solution). Aqueous ammonia can also be used as the alkaline aqueous solution.
The alkali concentration in the alkaline aqueous solution is preferably about 0.5 to 10 M (mol / L), more preferably about 1 to 8 M. Further, from the viewpoint of production cost, NaOH or KOH is preferable as the alkali to be used. NaOH and KOH may be used in combination.
As a contact method in the step (1), (i) a method in which an aqueous alkaline solution is added and mixed, (ii) a method in which an aqueous raw material solution is added and mixed, (iii) an aqueous raw material solution in water And a method of adding and mixing an alkaline aqueous solution. In mixing, it is preferable to involve stirring.
Of the contact methods in step (1), (ii) a method in which a raw material aqueous solution is added to and mixed with an alkaline aqueous solution is preferable because the change in pH is easily controlled. In the case of this method, the pH of the alkaline aqueous solution tends to decrease as the raw material aqueous solution is added to and mixed with the alkaline aqueous solution, but the pH is adjusted to 9 or higher, preferably 10 or higher. It is preferable to add a raw material aqueous solution. In addition, it is preferable that one or both of the raw material aqueous solution and the alkaline aqueous solution are brought into contact with each other while being kept at a temperature of 40 ° C. or higher and 80 ° C. or lower, whereby a coprecipitate having a more uniform composition can be obtained.
In the step (1), by bringing the raw material aqueous solution into contact with the alkali as described above, a salt containing Ni ions, Mn ions and Co ions is co-precipitated and the co-precipitate salt is dispersed. A slurry can be obtained.
(Process (2))
In step (2), a coprecipitate is obtained from the slurry obtained in step (1). As long as the coprecipitate can be obtained, various methods can be adopted as the method for obtaining the coprecipitate in step (2). However, since the operation is simple, a separation operation for obtaining a solid component such as filtration is performed. Is preferred. The coprecipitate can also be obtained by a method of volatilizing the liquid by heating, such as spray drying of the slurry.
When obtaining a coprecipitate in the step (2), it is preferable that the separated coprecipitate is washed and dried in the step (2). When the alkali remaining in the coprecipitate obtained by washing, Ni sulfate, Mn sulfate, or Co sulfate is used as a raw material, SO is released into the raw material aqueous solution.4 2-The amount of ions can be reduced. It is preferable to reduce these by washing because the amount of the inert flux (described later) can be easily controlled.
In order to efficiently wash the coprecipitate, it is preferable to use water as the washing liquid. If necessary, a water-soluble organic solvent such as alcohol or acetone may be added to the cleaning liquid. Moreover, you may perform washing | cleaning twice or more, for example, after washing with water, it can also wash | clean again with the organic solvent which has the above water solubility.
The drying of the washed coprecipitate can be performed by heat treatment, but may be performed by air drying, vacuum drying, or a combination thereof. When the heat treatment is performed, the heating temperature is preferably 50 to 300 ° C, more preferably about 100 to 200 ° C.
(Process (3))
In step (3), the coprecipitate obtained in step (2) and the lithium compound are mixed to obtain a mixture.
Examples of the lithium compound include one or more salts selected from the group consisting of lithium hydroxide, lithium chloride, lithium nitrate, and lithium carbonate. The lithium compound used may be an anhydride or a hydrate. Moreover, you may use an anhydride and a hydrate together.
Mixing may be either dry mixing or wet mixing, but dry mixing is preferred because of the ease of operation. Examples of the mixing apparatus include stirring and mixing, a V-type mixer, a W-type mixer, a ribbon mixer, a drum mixer, and a ball mill.
(Process (4))
In step (4), the mixture obtained in step (3) is heated at a temperature of 200 ° C. or higher and 500 ° C. or lower, preferably 250 ° C. or higher and 450 ° C. or lower to obtain a calcined product. The heating atmosphere includes a method using air and oxygen or a mixed gas thereof, a method of mixing an inert gas such as nitrogen and argon into the air and oxygen or a mixed gas thereof, and the oxygen concentration is 5 volumes. % Of the atmosphere. A high-capacity lithium composite metal oxide having an intended local structure can be easily obtained, and when the obtained lithium composite metal oxide is used as a positive electrode active material, a high-capacity secondary battery can be obtained. Therefore, the oxygen concentration is preferably 7% by volume to 20% by volume, and more preferably 10% by volume to 20% by volume.
(Process (5))
In step (5), the calcined product obtained in step (4) is fired at a temperature of 650 ° C. to 950 ° C., preferably 650 ° C. to 900 ° C. The firing atmosphere may be an atmosphere in which air, oxygen, nitrogen, argon, or the like is mixed and the oxygen concentration is less than 5% by volume. A high-capacity lithium composite metal oxide having an intended local structure can be easily obtained, and when the obtained lithium composite metal oxide is used as a positive electrode active material, a high-capacity secondary battery can be obtained. Therefore, the oxygen concentration is preferably 0.5% by volume or more and less than 5% by volume, and more preferably 1% by volume or more and 3% by volume or less.
In order to make the composition of the obtained lithium composite metal oxide uniform, the step (4) and the step (5) are continuously performed without lowering the temperature from the heating temperature at the end of the step (4) ( 5) is preferably performed. When performing the step (4) and the step (5) continuously, the oxygen concentration is adjusted to the step (4) while maintaining the temperature at the end of the step (4) or raising the temperature to the firing temperature of the step (5). The oxygen concentration in 4) is changed to the oxygen concentration in step (5). As a method for changing the oxygen concentration, a method of changing the oxygen concentration of the introduced gas is preferably used.
The lithium composite metal oxide of the present embodiment can be produced by such steps (1) to (5).
In addition, in the manufacturing method of this embodiment, although demonstrated as having a process (1)-a process (5), it is not restricted to this. For example, a mixture obtained by mixing a salt containing Ni ions, Mn ions and Co ions with a lithium compound by another method in place of steps (1) to (3) is prepared, and the obtained mixture is It is also possible to manufacture the lithium composite metal oxide of the present embodiment by heating while controlling the oxygen concentration and performing the processing corresponding to the above steps (4) and (5).
The above-mentioned “salt containing Ni ions, Mn ions and Co ions” may be a mixture of a salt containing Ni ions, a salt containing Mn ions, and a salt containing Co ions. As the above-mentioned “another method replacing step (1) to step (3)”, a method of mixing the above-mentioned salt in a solid phase, a slurry obtained by dispersing the above-mentioned salt in a liquid phase, For example, the slurry may be spray-dried and mixed.
Further, in the method for producing a lithium composite metal oxide according to the present embodiment, Ni, Mn and Co need to be contained in the mixture in the step (4). The metal atom may be contained. Examples of other metal atoms include one or more atoms selected from the group consisting of Al, Mg, Ti, Ca, Cu, Zn, Fe, Cr, Mo, Si, Sn, Nb, and V.
Various methods can be adopted as a method of including other metal atoms in the mixture to be heated in step (4). In particular, in the step (1), it is preferable to dissolve a water-soluble salt of another metal in the raw material aqueous solution because other metal atoms are uniformly dispersed in the resulting mixture.
(Inert flux)
In the step (4) and step (5), the mixture and calcined product may contain an inert flux. The inert flux is a salt that does not react with the target composite metal oxide and can be easily separated from the target. The inert flux melts at the heating temperature in step (4) and the firing temperature in step (5) to form a reaction field and promotes a uniform reaction. Therefore, when an inert flux is used, a product having a uniform composition is easily obtained.
As an inert flux, K2SO4, Na2SO4Sulfate such as K;2CO3, Na2CO3Carbonates such as: NaCl, KCl, NH4Chlorides such as Cl; LiF, NaF, KF, NH4Fluorides such as F; boric acid; Among the above-mentioned inert fluxes, sulfate is preferable because the production process becomes simple. More preferably K2SO4It is. Two or more inert fluxes can be used in combination.
When the mixture contains an inert flux, the reactivity at the time of heating the mixture and calcining the calcined product is improved, and thereby it may be possible to adjust the BET specific surface area of the obtained lithium composite metal oxide. is there. When the temperature is the same, the BET specific surface area of the oxide tends to increase as the content of the inert flux increases. Further, when an inert fluxing agent is contained during heating or firing, a uniform reaction can be performed, so that the local structure can be controlled at the atomic level of the lithium composite metal oxide by adjusting the heating atmosphere.
The inert flux may be mixed with the coprecipitate obtained by allowing the coprecipitate obtained by the separation operation in step (2) to contain the above inert flux solution and then drying. .
For example, when Ni sulfate, Mn sulfate, or Co sulfate is used as a raw material in step (1), SO4 2-Ions are liberated. This SO4 2-The ions and metal ions contained in the alkali used for the coprecipitation (for example, K ions when KOH is used as the alkali) remain in the coprecipitate separated in step (2), and the inert flux ( K in the above example2SO4) May occur. Therefore, the raw material aqueous solution after the coprecipitation in the step (1) is used as the “inert flux solution”, and the coprecipitate obtained in the step (2) is dried while the raw aqueous solution after the coprecipitation is included. By making it, an inert flux may be mixed with the coprecipitate obtained.
In addition, the inert flux can be added and mixed at the time of mixing the coprecipitate and the lithium compound in the step (3). Since it is easy to control the amount of the inert flux, the method of adding the inert flux in the step (3) is preferable to the method of adding the inert flux in the step (2). When an inert flux is added in step (3), the coprecipitate obtained in step (2) is washed, and the alkali, Ni salt, Mn salt, or Co salt remaining in the coprecipitate is washed. By reducing the amount of the derived anion, the amount of the inert flux can be easily controlled.
The inert flux may remain in the lithium composite metal oxide or may be removed by washing.
From the viewpoint of improving the uniformity of the reaction, the inert flux is a sulfate, and when the mixture or calcined product and the sulfate are mixed, the content of the sulfate in the resulting mixture is the lithium used. It is preferable that it is 0.01 to 400 mass parts with respect to 100 mass parts of compounds. More preferably, it is 0.1 to 10 parts by mass.
Further, the lithium composite metal oxide obtained by the method for producing a lithium composite metal oxide of the present embodiment may be pulverized using a ball mill or a jet mill. It may be possible to adjust the BET specific surface area of the lithium composite metal oxide by grinding. Further, the lithium composite metal oxide obtained by carrying out the steps (1) to (5) may be pulverized, and the steps (4) and (5) may be performed again to perform baking after the pulverization. . Furthermore, you may repeat a grinding | pulverization and baking by process (4), (5) twice or more as needed. Further, the lithium composite metal oxide can be washed or classified as necessary.
The lithium composite metal oxide of the present embodiment is preferably a mixture of primary particles having a particle size of 0.05 μm or more and 1 μm or less and secondary particles having a particle size of 2 μm or more and 100 μm or less formed by aggregation of the primary particles. It consists of. The particle diameters of the primary particles and secondary particles can be measured by observing with SEM.
The size of the secondary particles of the lithium composite metal oxide is preferably in the range of 2 μm to 50 μm, more preferably in the range of 2 μm to 10 μm, and even more preferably in the range of 3 μm to 8 μm. Especially preferably, it is the range of 3.5 micrometers or more and 7 micrometers or less. By these, the capacity | capacitance of the nonaqueous electrolyte secondary battery obtained increases more.
The primary particle size of the lithium composite metal oxide is preferably in the range of 0.08 μm to 0.8 μm, more preferably in the range of 0.10 μm to 0.7 μm, and still more preferably in the range of 0.8. It is in the range of 15 μm or more and 0.7 μm or less, and particularly preferably in the range of 0.2 μm or more and 0.5 μm or less. These increase the discharge capacity at a high current rate of the obtained nonaqueous electrolyte secondary battery.
Also, the average particle diameter of lithium composite metal oxide (D50) Is preferably in the range of 1 μm to 50 μm, more preferably in the range of 1.5 μm to 30 μm, even more preferably in the range of 2 μm to 20 μm, and particularly preferably in the range of 3 μm to 10 μm. It is. Accordingly, the density of the electrode using the lithium composite metal oxide is increased, and a high-capacity nonaqueous electrolyte secondary battery can be obtained.
Average particle size of lithium composite metal oxide (D50) Can be measured by the following method.
<Average particle diameter of lithium composite metal oxide (D50) Measurement>
0.1 g of the lithium composite metal oxide powder to be measured is put into 50 ml of a 0.2 mass% sodium hexametaphosphate aqueous solution to obtain a dispersion in which the powder is dispersed. About the obtained dispersion liquid, a particle size distribution is measured using the master sizer 2000 (laser diffraction scattering particle size distribution measuring apparatus) by Malvern, and a volume-based cumulative particle size distribution curve is obtained. In the obtained cumulative particle size distribution curve, the value of the particle size viewed from the fine particle side at the time of 50% accumulation is the average particle size (D50).
The BET specific surface area of the lithium composite metal oxide is preferably 0.1 m2/ G or more 20m2/ G or less, more preferably 0.5 m2/ G or more 15m2/ G or less, even more preferably 1 m2/ G or more 10m2/ G or less, particularly preferably 2 m2/ G or more 8m2/ G or less. These increase the discharge capacity at a high current rate of the obtained nonaqueous electrolyte secondary battery.
The BET specific surface area of the lithium composite metal oxide can be measured by the following method.
<Measurement of BET specific surface area of lithium composite metal oxide>
After 1 g of the lithium composite metal oxide powder to be measured is dried at 150 ° C. for 15 minutes in a nitrogen atmosphere, the measurement is performed using a flow sorb II 2300 manufactured by Micromeritics.
When the lithium composite metal oxide is used as a positive electrode active material for a non-aqueous electrolyte secondary battery, a non-aqueous electrolyte secondary battery showing a higher capacity than before can be obtained.
[Nonaqueous electrolyte secondary battery]
Next, while explaining the configuration of the nonaqueous electrolyte secondary battery, the positive electrode using the lithium composite metal oxide of the present embodiment as the positive electrode active material of the nonaqueous electrolyte secondary battery, and the nonaqueous electrolyte secondary having the positive electrode The battery will be described.
An example of the non-aqueous electrolyte secondary battery of the present embodiment includes a positive electrode, a negative electrode, a separator disposed between the positive electrode and the negative electrode, and an electrolytic solution.
FIG. 1 is a schematic view showing an example of the nonaqueous electrolyte secondary battery of the present embodiment. The cylindrical nonaqueous electrolytesecondary battery 10 of this embodiment is manufactured as follows.
First, as shown in FIG. 1A, twoseparators 1 each having a strip shape, a strip-like positive electrode 2 having a positive electrode lead 21 at one end, and a strip-like negative electrode 3 having a negative electrode lead 31 at one end, 2, separator 1, and negative electrode 3 are laminated in this order and wound to form electrode group 4.
Next, as shown in FIG. 1B, after the electrode group 4 and an insulator (not shown) are accommodated in the battery can 5, the bottom of the can is sealed, and the electrode group 4 is impregnated with the electrolytic solution 6, An electrolyte is disposed between the negative electrode 3 and the negative electrode 3. Furthermore, the nonaqueous electrolytesecondary battery 10 can be manufactured by sealing the upper part of the battery can 5 with the top insulator 7 and the sealing body 8.
As the shape of the electrode group 4, for example, a columnar shape in which the cross-sectional shape when the electrode group 4 is cut in a direction perpendicular to the winding axis is a circle, an ellipse, a rectangle, or a rectangle with rounded corners. Can be mentioned.
In addition, as the shape of the nonaqueous electrolyte secondary battery having such an electrode group 4, a shape defined by IEC 60086 or JIS C 8500, which is a standard for batteries determined by the International Electrotechnical Commission (IEC), should be adopted. Can do. For example, cylindrical shape, square shape, etc. can be mentioned.
Furthermore, the non-aqueous electrolyte secondary battery is not limited to the above-described wound type configuration, and may have a stacked type configuration in which a stacked structure of a positive electrode, a separator, a negative electrode, and a separator is repeatedly stacked. Examples of the laminated nonaqueous electrolyte secondary battery include so-called coin-type batteries, button-type batteries, and paper-type (or sheet-type) batteries.
Hereinafter, each configuration will be described in order.
(Positive electrode)
The positive electrode of the present embodiment can be manufactured by first adjusting a positive electrode mixture containing a positive electrode active material, a conductive material and a binder, and supporting the positive electrode mixture on a positive electrode current collector.
(Positive electrode active material)
The positive electrode active material of the present embodiment has the above-described lithium composite metal oxide. By using the lithium composite metal oxide of this embodiment as a positive electrode active material of a non-aqueous electrolyte secondary battery, a non-aqueous electrolyte secondary battery exhibiting a high capacity can be obtained.
(Conductive material)
A carbon material can be used as a conductive material included in the positive electrode of the present embodiment. Examples of the carbon material include graphite powder, carbon black (for example, acetylene black), and a fibrous carbon material. Since carbon black is fine and has a large surface area, adding a small amount to the positive electrode mixture can improve the conductivity inside the positive electrode and improve the charge / discharge efficiency and output characteristics. Both the binding force between the positive electrode mixture and the positive electrode current collector and the binding force inside the positive electrode mixture are reduced, which causes an increase in internal resistance.
The proportion of the conductive material in the positive electrode mixture is preferably 5 parts by mass or more and 20 parts by mass or less with respect to 100 parts by mass of the positive electrode active material. When a fibrous carbon material such as graphitized carbon fiber or carbon nanotube is used as the conductive material, this ratio can be lowered.
(binder)
A thermoplastic resin can be used as the binder of the positive electrode of the present embodiment. Examples of the thermoplastic resin include polyvinylidene fluoride (hereinafter sometimes referred to as PVdF), polytetrafluoroethylene (hereinafter sometimes referred to as PTFE), tetrafluoroethylene, hexafluoropropylene, and vinylidene fluoride. And fluororesins such as copolymers, propylene hexafluoride / vinylidene fluoride copolymers, tetrafluoroethylene / perfluorovinyl ether copolymers; polyolefin resins such as polyethylene and polypropylene.
These thermoplastic resins may be used as a mixture of two or more. By using a fluororesin and a polyolefin resin as a binder, the ratio of the fluororesin to the whole positive electrode mixture is 1% by mass or more and 10% by mass or less, and the ratio of the polyolefin resin is 0.1% by mass or more and 2% by mass or less. A positive electrode mixture having both high adhesion to the current collector and high bonding strength inside the positive electrode mixture can be obtained.
(Positive electrode current collector)
As the positive electrode current collector included in the positive electrode of the present embodiment, a band-shaped member made of a metal material such as Al, Ni, stainless steel or the like can be used. Among these, a material that is made of Al and formed into a thin film is preferable because it is easy to process and inexpensive.
Examples of the method of supporting the positive electrode mixture on the positive electrode current collector include a method of pressure-molding the positive electrode mixture on the positive electrode current collector. Further, the positive electrode mixture is made into a paste using an organic solvent, and the obtained positive electrode mixture paste is applied to at least one surface of the positive electrode current collector, dried, pressed and fixed, whereby the positive electrode current collector is bonded to the positive electrode current collector. A mixture may be supported.
When pasting the positive electrode mixture, organic solvents that can be used include amine solvents such as N, N-dimethylaminopropylamine and diethylenetriamine; ether solvents such as tetrahydrofuran; ketone solvents such as methyl ethyl ketone; methyl acetate And amide solvents such as dimethylacetamide and N-methyl-2-pyrrolidone (hereinafter sometimes referred to as NMP).
Examples of the method of applying the positive electrode mixture paste to the positive electrode current collector include a slit die coating method, a screen coating method, a curtain coating method, a knife coating method, a gravure coating method, and an electrostatic spray method.
The positive electrode can be manufactured by the methods mentioned above.
(Negative electrode)
The negative electrode included in the nonaqueous electrolyte secondary battery of this embodiment is only required to be able to dope and dedope lithium ions at a lower potential than the positive electrode, and the negative electrode mixture containing the negative electrode active material is supported on the negative electrode current collector. And an electrode composed of the negative electrode active material alone.
(Negative electrode active material)
Examples of the negative electrode active material possessed by the negative electrode include carbon materials, chalcogen compounds (oxides, sulfides, etc.), nitrides, metals, and alloys that can be doped and dedoped with lithium ions at a lower potential than the positive electrode. It is done.
Examples of carbon materials that can be used as the negative electrode active material include graphites such as natural graphite and artificial graphite, cokes, carbon black, pyrolytic carbons, carbon fibers, and organic polymer compound fired bodies.
As an oxide that can be used as a negative electrode active material, SiO2, SiO etc. formula SiOx(Wherein x is a positive real number) silicon oxide represented by: TiO2TiO, formula TiOx(Where x is a positive real number) titanium oxide; V2O5, VO2Etc. VOx(Where x is a positive real number) oxide of vanadium; Fe3O4, Fe2O3FeO and other formulas FeOx(Where x is a positive real number) iron oxide; SnO2, SnO etc. formula SnOx(Where x is a positive real number) tin oxide represented by WO3, WO2General formula WOx(Where x is a positive real number)4Ti5O12, LiVO2And a composite metal oxide containing lithium and titanium or vanadium.
As a sulfide that can be used as a negative electrode active material, Ti2S3TiS2TiS and other formula TiSx(Where x is a positive real number) titanium sulfide; V3S4, VS2,VS formula VSx(Where x is a positive real number) Vanadium sulfide; Fe3S4, FeS2FeS and other formulasx(Where x is a positive real number) iron sulfide; Mo2S3, MoS2Etc. MoSx(Where x is a positive real number) molybdenum sulfide represented by SnS2,SnS etc. formula SnSx(Where x is a positive real number) tin sulfide represented by WS2Formula WSx(Where x is a positive real number) tungsten sulfide represented by: Sb2S3Etc. SbSx(Where x is a positive real number) antimony sulfide; Se5S3, SeS2, SeS etc. formula SeSxSelenium sulfide represented by (where x is a positive real number).
Nitrides that can be used as negative electrode active materials include Li3N, Li3-xAxA lithium-containing nitride such as N (where A is one or both of Ni and Co, and 0 <x <3) can be given.
These carbon materials, oxides, sulfides and nitrides may be used alone or in combination of two or more. These carbon materials, oxides, sulfides and nitrides may be crystalline or amorphous.
Further, examples of the metal that can be used as the negative electrode active material include lithium metal, silicon metal, and tin metal.
Examples of alloys that can be used as the negative electrode active material include lithium alloys such as Li—Al, Li—Ni, Li—Si, Li—Sn, and Li—Sn—Ni; silicon alloys such as Si—Zn; Sn—Mn and Sn. -Tin alloys such as Co, Sn-Ni, Sn-Cu, Sn-La; Cu2Sb, La3Ni2Sn7And alloys such as:
These metals and alloys are mainly used alone as electrodes after being processed into a foil shape, for example.
Among the negative electrode active materials, the potential of the negative electrode hardly changes from the uncharged state to the fully charged state during charging (potential flatness is good), the average discharge potential is low, and the capacity retention rate when repeatedly charged and discharged is high. For reasons such as high (good cycle characteristics), a carbon material mainly composed of graphite such as natural graphite or artificial graphite is preferably used. The shape of the carbon material may be any of a flake shape such as natural graphite, a spherical shape such as mesocarbon microbeads, a fibrous shape such as graphitized carbon fiber, or an aggregate of fine powder.
The negative electrode mixture may contain a binder as necessary. Examples of the binder include thermoplastic resins, and specific examples include PVdF, thermoplastic polyimide, carboxymethyl cellulose, polyethylene, and polypropylene.
(Negative electrode current collector)
Examples of the negative electrode current collector of the negative electrode include a band-shaped member made of a metal material such as Cu, Ni, and stainless steel. In particular, it is preferable to use Cu as a forming material and process it into a thin film from the viewpoint that it is difficult to make an alloy with lithium and it is easy to process.
As a method of supporting the negative electrode mixture on such a negative electrode current collector, as in the case of the positive electrode, a method using pressure molding, pasting with a solvent, etc., applying to the negative electrode current collector, drying and pressing. The method of crimping is mentioned.
(Separator)
Examples of the separator included in the nonaqueous electrolyte secondary battery of the present embodiment include a porous film, a nonwoven fabric, a woven fabric, and the like made of a material such as a polyolefin resin such as polyethylene and polypropylene, a fluororesin, and a nitrogen-containing aromatic polymer. A material having the following form can be used. Moreover, a separator may be formed by using two or more of these materials, or a separator may be formed by laminating these materials.
Examples of the separator include separators described in JP 2000-30686 A, JP 10-324758 A, and the like. The thickness of the separator should be as thin as possible as long as the mechanical strength is maintained because the volume energy density of the battery is increased and the internal resistance is reduced, preferably about 5 to 200 μm, more preferably about 5 to 40 μm. is there.
The separator preferably has a porous film containing a thermoplastic resin. In a nonaqueous electrolyte secondary battery, when an abnormal current flows in the battery due to a short circuit between the positive electrode and the negative electrode, the current at the short circuit point is interrupted to prevent an excessive current from flowing (shut down). It preferably has a function. Here, the shutdown is performed by overheating the separator at the short-circuit location due to a short circuit, and when the temperature exceeds a presumed operating temperature, the porous film in the separator is softened or melted to close the micropores. And even if the temperature in a battery rises to a certain high temperature after a separator shuts down, it is preferable to maintain the shut-down state, without breaking at the temperature.
Examples of such a separator include a laminated film in which a heat resistant porous layer and a porous film are laminated. By using such a laminated film as a separator, the heat resistance of the secondary battery in this embodiment can be further increased. In the laminated film, the heat resistant porous layer may be laminated on both surfaces of the porous film.
(Laminated film)
Hereinafter, a laminated film in which the heat resistant porous layer and the porous film are laminated to each other will be described.
In the laminated film used as the separator of the nonaqueous electrolyte secondary battery of the present embodiment, the heat resistant porous layer is a layer having higher heat resistance than the porous film. The heat resistant porous layer may be formed from an inorganic powder (first heat resistant porous layer), may be formed from a heat resistant resin (second heat resistant porous layer), and includes a heat resistant resin and a filler. (A third heat-resistant porous layer). When the heat resistant porous layer contains a heat resistant resin, the heat resistant porous layer can be formed by an easy technique such as coating.
(First heat-resistant porous layer)
When the heat resistant porous layer is formed of an inorganic powder, examples of the inorganic powder used for the heat resistant porous layer include inorganic substances such as metal oxides, metal nitrides, metal carbides, metal hydroxides, carbonates, and sulfates. Among these, a powder made of an inorganic substance having low conductivity (insulator) is preferably used. Specific examples include powders made of alumina, silica, titanium dioxide, calcium carbonate, or the like. Such inorganic powders may be used alone or in combination of two or more.
Among these inorganic powders, alumina powder is preferable because of its high chemical stability. More preferably, all of the particles constituting the inorganic powder are alumina particles, all of the particles constituting the inorganic powder are alumina particles, and part or all of them are substantially spherical alumina particles. preferable.
(Second heat resistant porous layer)
When the heat resistant porous layer is formed from a heat resistant resin, the heat resistant resin used for the heat resistant porous layer is polyamide, polyimide, polyamideimide, polycarbonate, polyacetal, polysulfone, polyphenylene sulfide, polyether ketone, aromatic polyester, polyether. Mention may be made of sulfone and polyetherimide. In order to further increase the heat resistance of the laminated film, polyamide, polyimide, polyamideimide, polyethersulfone and polyetherimide are preferable, and polyamide, polyimide or polyamideimide is more preferable.
More preferably, the heat-resistant resin used for the heat-resistant porous layer is a nitrogen-containing aromatic polymer such as aromatic polyamide (para-oriented aromatic polyamide, meta-oriented aromatic polyamide), aromatic polyimide, aromatic polyamideimide, Aromatic polyamides are preferred, and para-oriented aromatic polyamides (hereinafter sometimes referred to as para-aramids) are particularly preferred because they are easy to produce.
Also, examples of the heat resistant resin include poly-4-methylpentene-1 and a cyclic olefin polymer.
By using these heat resistant resins, the heat resistance of the laminated film used as the separator of the nonaqueous electrolyte secondary battery, that is, the thermal film breaking temperature of the laminated film can be further increased. Among these heat-resistant resins, when a nitrogen-containing aromatic polymer is used, the compatibility with the electrolytic solution, that is, the liquid retention in the heat-resistant porous layer may be improved depending on the polarity in the molecule. The rate of impregnation with the electrolytic solution during the production of the electrolyte secondary battery is also high, and the charge / discharge capacity of the nonaqueous electrolyte secondary battery is further increased.
The thermal film breaking temperature of such a laminated film depends on the type of heat-resistant resin, and is selected and used according to the use scene and purpose of use. More specifically, as the heat-resistant resin, when the nitrogen-containing aromatic polymer is used, the cyclic olefin polymer is about 400 ° C. When using, the thermal film breaking temperature can be controlled to about 300 ° C., respectively. In addition, when the heat resistant porous layer is made of an inorganic powder, the thermal film breaking temperature can be controlled to, for example, 500 ° C. or higher.
The para-aramid is obtained by polycondensation of a para-oriented aromatic diamine and a para-oriented aromatic dicarboxylic acid halide, and the amide bond is in the para position of the aromatic ring or an oriented position equivalent thereto (for example, 4,4′-biphenylene, It consists essentially of repeating units that are bound together in the opposite orientation, such as 1,5-naphthalene, 2,6-naphthalene, etc., in an orientation that extends coaxially or parallelly. Specifically, poly (paraphenylene terephthalamide), poly (parabenzamide), poly (4,4′-benzanilide terephthalamide), poly (paraphenylene-4,4′-biphenylenedicarboxylic acid amide), poly ( Para-aligned or para-oriented such as paraphenylene-2,6-naphthalenedicarboxylic acid amide), poly (2-chloro-paraphenylene terephthalamide), paraphenylene terephthalamide / 2,6-dichloroparaphenylene terephthalamide copolymer Examples include para-aramid having a structure according to the type.
The aromatic polyimide is preferably a wholly aromatic polyimide produced by polycondensation of an aromatic dianhydride and a diamine.
Specific examples of the aromatic dianhydride used for the polycondensation include pyromellitic dianhydride, 3,3 ′, 4,4′-diphenylsulfonetetracarboxylic dianhydride, 3,3 ′, 4. 4,4′-benzophenone tetracarboxylic dianhydride, 2,2′-bis (3,4-dicarboxyphenyl) hexafluoropropane and 3,3 ′, 4,4′-biphenyltetracarboxylic dianhydride. It is done.
Specific examples of diamines used for polycondensation include oxydianiline, paraphenylenediamine, benzophenonediamine, 3,3′-methylenedianiline, 3,3′-diaminobenzophenone, 3,3′-diaminodiphenylsulfone. And 1,5-naphthalenediamine.
Also, as the aromatic polyimide, a polyimide soluble in a solvent can be suitably used. Examples of such a polyimide include a polycondensate polyimide of 3,3 ′, 4,4′-diphenylsulfonetetracarboxylic dianhydride and an aromatic diamine.
Examples of the aromatic polyamideimide include those obtained from polycondensation of aromatic dicarboxylic acid and aromatic diisocyanate, and those obtained from polycondensation of aromatic diacid anhydride and aromatic diisocyanate. Specific examples of the aromatic dicarboxylic acid include isophthalic acid and terephthalic acid. A specific example of the aromatic dianhydride is trimellitic anhydride. Specific examples of the aromatic diisocyanate include 4,4'-diphenylmethane diisocyanate, 2,4-tolylene diisocyanate, 2,6-tolylene diisocyanate, orthotolylene diisocyanate, and m-xylene diisocyanate.
In order to further enhance ion permeability, the thickness of the heat resistant porous layer of the laminated film is preferably 1 μm or more and 10 μm or less, more preferably 1 μm or more and 5 μm or less, and particularly preferably 1 μm or more and 4 μm or less. . The heat-resistant porous layer has fine pores, and the size (diameter) of the pores is preferably 3 μm or less, more preferably 1 μm or less.
(Third heat-resistant porous layer)
Further, when the heat resistant porous layer is formed including a heat resistant resin and a filler, the same heat resistant resin as that used for the second heat resistant porous layer can be used. As the filler, one or more selected from the group consisting of organic powder, inorganic powder, or a mixture thereof can be used. The particles constituting the filler preferably have an average particle size of 0.01 μm or more and 1 μm or less.
Examples of the organic powder that can be used as the filler include, for example, styrene, vinyl ketone, acrylonitrile, methyl methacrylate, ethyl methacrylate, glycidyl methacrylate, glycidyl acrylate, methyl acrylate, and the like, or two or more types of copolymers; Fluorine resin such as tetrafluoroethylene-6-propylene copolymer, tetrafluoroethylene-ethylene copolymer, polyvinylidene fluoride; melamine resin; urea resin; polyolefin resin; polymethacrylate; A powder is mentioned. Such organic powders may be used alone or in combination of two or more. Among these organic powders, PTFE powder is preferred because of its high chemical stability.
Examples of the inorganic powder that can be used as the filler include the same inorganic powder used in the heat-resistant porous layer.
When the heat-resistant porous layer is formed including a heat-resistant resin and a filler, the filler content depends on the specific gravity of the filler material, for example, when all of the particles constituting the filler are alumina particles In addition, when the total mass of the heat resistant porous layer is 100 parts by mass, the mass of the filler is preferably 5 parts by mass or more and 95 parts by mass or less, more preferably 20 parts by mass or more and 95 parts by mass or less, Preferably they are 30 to 90 mass parts. These ranges can be appropriately set depending on the specific gravity of the filler material.
Examples of the shape of the filler include substantially spherical, plate-like, columnar, needle-like, and fiber-like shapes, and any particle can be used. However, since it is easy to form uniform pores, Preferably there is. Examples of the substantially spherical particles include particles having a particle aspect ratio (long particle diameter / short particle diameter) of 1 or more and 1.5 or less. The aspect ratio of the particles can be measured by an electron micrograph.
In the laminated film used as the separator of the nonaqueous electrolyte secondary battery of this embodiment, the porous film preferably has fine pores and has a shutdown function. In this case, the porous film contains a thermoplastic resin.
The size of the micropores in the porous film is preferably 3 μm or less, more preferably 1 μm or less. The porosity of the porous film is preferably 30% to 80% by volume, more preferably 40% to 70% by volume. In a non-aqueous electrolyte secondary battery, when the presumed operating temperature is exceeded, the porous film containing the thermoplastic resin has micropores due to softening or melting of the thermoplastic resin constituting the porous film. Can be occluded.
What is necessary is just to select the thermoplastic resin used for a porous film what does not melt | dissolve in the electrolyte solution in a nonaqueous electrolyte secondary battery. Specific examples include polyolefin resins such as polyethylene and polypropylene, and thermoplastic polyurethane resins, and a mixture of two or more of these may be used.
In order for the separator to soften and shut down at a lower temperature, the porous film preferably contains polyethylene. Examples of the polyethylene include polyethylene such as low density polyethylene, high density polyethylene, and linear polyethylene, and ultra high molecular weight polyethylene having a molecular weight of 1,000,000 or more.
In order to further increase the puncture strength of the porous film, it is preferable that the thermoplastic resin constituting the porous film contains at least ultra high molecular weight polyethylene. In addition, in terms of production of the porous film, the thermoplastic resin may preferably contain a wax made of polyolefin having a low molecular weight (weight average molecular weight of 10,000 or less).
Further, the thickness of the porous film in the laminated film is preferably 3 μm or more and 30 μm or less, more preferably 3 μm or more and 25 μm or less. Moreover, in this embodiment, the thickness of a laminated film becomes like this. Preferably it is 40 micrometers or less, More preferably, it is 30 micrometers or less. Moreover, when the thickness of the heat resistant porous layer is A (μm) and the thickness of the porous film is B (μm), the value of A / B is preferably 0.1 or more and 1 or less.
In the present embodiment, the separator allows the electrolyte to permeate well when the battery is used (during charging / discharging). Or less, more preferably 50 seconds / 100 cc or more and 200 seconds / 100 cc or less.
The porosity of the separator is preferably 30% by volume to 80% by volume, more preferably 40% by volume to 70% by volume. The separator may be a laminate of separators having different porosity.
(Electrolyte)
The electrolyte solution included in the nonaqueous electrolyte secondary battery of this embodiment contains an electrolyte and an organic solvent.
As the electrolyte contained in the electrolyte, LiClO4, LiPF6, LiAsF6, LiSbF6, LiBF4, LiCF3SO3, LiN (SO2CF3)2, LiN (SO2C2F5)2, LiN (SO2CF3) (COCF3), Li (C4F9SO3), LiC (SO2CF3)3, Li2B10Cl10, LiBOB (where BOB is bis (oxalato) borate), lower aliphatic carboxylic acid lithium salt, LiAlCl4And a mixture of two or more of these may be used. Among them, as an electrolyte, LiPF containing fluorine6, LiAsF6, LiSbF6, LiBF4, LiCF3SO3, LiN (SO2CF3)2And LiC (SO2CF3)3It is preferable to use at least one selected from the group consisting of:
Examples of the organic solvent contained in the electrolyte include propylene carbonate, ethylene carbonate, dimethyl carbonate, diethyl carbonate, ethyl methyl carbonate, 4-trifluoromethyl-1,3-dioxolan-2-one, and 1,2-di- Carbonates such as (methoxycarbonyloxy) ethane; 1,2-dimethoxyethane, 1,3-dimethoxypropane, pentafluoropropyl methyl ether, 2,2,3,3-tetrafluoropropyl difluoromethyl ether, tetrahydrofuran, 2- Ethers such as methyltetrahydrofuran; Esters such as methyl formate, methyl acetate and γ-butyrolactone; Nitriles such as acetonitrile and butyronitrile; N, N-dimethylformamide, N, N-dimethyla Amides such as toamide; carbamates such as 3-methyl-2-oxazolidone; sulfur-containing compounds such as sulfolane, dimethyl sulfoxide and 1,3-propane sultone, or those obtained by further introducing a fluoro group into these organic solvents ( One obtained by substituting one or more hydrogen atoms in the organic solvent with fluorine atoms can be used.
It is preferable to use a mixture of two or more of these as the organic solvent. Among them, a mixed solvent containing carbonates is preferable, and a mixed solvent of cyclic carbonate and acyclic carbonate and a mixed solvent of cyclic carbonate and ether are more preferable. As a mixed solvent of cyclic carbonate and acyclic carbonate, a mixed solvent containing ethylene carbonate, dimethyl carbonate and ethyl methyl carbonate is preferable. The electrolyte using such a mixed solvent has a wide operating temperature range, hardly deteriorates even when charged and discharged at a high current rate, hardly deteriorates even when used for a long time, and natural graphite as an active material of the negative electrode. Even when a graphite material such as artificial graphite is used, it has the advantage of being hardly decomposable.
Also, as the electrolytic solution, since the safety of the obtained nonaqueous electrolyte secondary battery is increased, LiPF6It is preferable to use an electrolytic solution containing a lithium salt containing fluorine and an organic solvent having a fluorine substituent. A mixed solvent containing dimethyl carbonate and ethers having fluorine substituents such as pentafluoropropyl methyl ether and 2,2,3,3-tetrafluoropropyl difluoromethyl ether is capable of capacity even when charging / discharging at a high current rate. Since the maintenance rate is high, it is more preferable.
A solid electrolyte may be used instead of the above electrolyte. As the solid electrolyte, for example, an organic polymer electrolyte such as a polyethylene oxide polymer compound, a polymer compound containing at least one of a polyorganosiloxane chain or a polyoxyalkylene chain can be used. Moreover, what is called a gel type which hold | maintained the non-aqueous electrolyte in the high molecular compound can also be used. Li2S-SiS2, Li2S-GeS2, Li2SP2S5, Li2SB2S3, Li2S-SiS2-Li3PO4, Li2S-SiS2-Li2SO4An inorganic solid electrolyte containing a sulfide such as may be used. By using these solid electrolytes, the safety of the nonaqueous electrolyte secondary battery may be further improved.
In the nonaqueous electrolyte secondary battery of this embodiment, when a solid electrolyte is used, the solid electrolyte may serve as a separator, and in that case, the separator may not be required.
Since the above-described positive electrode active material uses the above-described lithium composite metal oxide of the present embodiment, the nonaqueous electrolyte secondary battery using the positive electrode active material can exhibit a higher capacity than before.
Further, since the positive electrode has a positive electrode active material using the above-described lithium composite metal oxide of the present embodiment, the non-aqueous electrolyte secondary battery can exhibit a higher capacity than before.
Furthermore, since the nonaqueous electrolyte secondary battery has the positive electrode described above, it exhibits a higher capacity than before.
本実施形態のリチウム複合金属酸化物は、Mn、Ni、LiおよびCoを含有し、下記(a)および(b)を満たす。
(a)前記リチウム複合金属酸化物におけるMnのK吸収端の広域X線吸収微細構造(EXAFS)スペクトルをフーリエ変換して得られる動径分布関数において、Mn原子に結合した酸素原子による1.5Å付近の第一近接ピークAMnの強度をIAMn、Mn原子に結合した酸素原子の次にMn原子に近い金属原子による2.5Å付近の第二近接ピークBMnの強度をIBMnとしたとき、IBMn/IAMnが、0.5以上1.2以下である。
(b)前記リチウム複合金属酸化物におけるNiのK吸収端のEXAFSスペクトルをフーリエ変換して得られる動径分布関数において、Ni原子に結合した酸素原子による1.5Å付近の第一近接ピークANiの強度をIANi、Ni原子に結合した酸素原子の次にNi原子に近い金属原子による2.5Å付近の第二近接ピークの強度をIBNiとしたとき、IBNi/IANiが、1.0以上1.7以下である。
以下、順に説明する。
(EXAFSスペクトル)
まず、本実施形態のリチウム複合金属酸化物を規定するために用いるEXAFSスペクトルについて説明する。本実施形態で用いるEXAFSスペクトルは、一般的なEXAFSスペクトルと同様に扱われる。EXAFSスペクトルの測定および原理は、例えば、「X線吸収分光法—XAFSとその応用—」(太田俊明編(2002年))に記載されている。原理は以下の通りである。
まず、測定対象の物質に特定の波長のX線を透過させたとき、物質に照射されたX線の強度(入射X線強度:I0)と、物質を透過してきたX線の強度(透過X線強度:It)とから、特定の波長における測定対象の物質について、単位厚さあたりのX線吸光度が得られる。
物質に照射するX線の波長を変化させ(すなわち、入射X線のエネルギー(eV)を変化させ)、各波長(各エネルギー)のX線に対するX線吸光度を測定して、x軸を入射X線のエネルギー(eV)、y軸をX線吸光度とするX線吸収スペクトルを作成すると、X線吸光度が急激に増加するエネルギーがあることが分かる。このエネルギーの値を吸収端という。吸収端は、物質を構成する原子の原子殻のエネルギー準位に対応しており、各原子に固有のものである。例えば、原子のK殻に対応する吸収端はK吸収端という。
X線吸収スペクトルにおいて、この吸収端から20~1000eV程度高いエネルギー側の領域に現れる微細な振動構造を広域X線吸収微細構造(EXAFS)といい、そのスペクトルをEXAFSスペクトルという。EXAFSスペクトルについてフーリエ変換を施すと、X線吸収原子(注目する原子)を中心とした動径分布関数が得られる。この動径分布関数から、X線吸収原子とX線散乱原子(X線吸収原子近傍の原子)との距離、X線散乱原子の数などの情報を得ることができ、注目する原子近傍の情報を得ることができる。
一般的に、動径分布関数のピークの強度は、X線散乱原子の数に影響されるが、その他に、X線吸収原子とX線散乱原子との原子間距離の等方性にも影響される。例えば、ある2つのX線吸収原子について、X線散乱原子数が実質的に同数で、かつ、X線散乱原子が実質的に同等な散乱能を有すると認められる場合、動径分布関数のピーク強度が大きいものは、X線吸収原子とX線散乱原子との原子間距離が方向による違いがなく等方的であり、X線吸収原子とX線散乱原子間との距離の分布が少ないことを意味する。
そこで、本実施形態においては、MnおよびNiのK吸収端において得られた動径分布関数のピークの強度比に注目する。
すなわち、動径分布関数のピークの強度比をある一定の範囲内に制御することによって、組成比が異なる試料に対しても、リチウム複合金属酸化物における原子レベルの局所構造を特定の条件に制御でき、従来よりも高容量を示す非水電解質二次電池に有用なリチウム複合金属酸化物を得ることができる。
本実施形態のリチウム複合金属酸化物においては、MnのK吸収端の動径分布関数において、Mn原子に結合したO(酸素原子)によるピークを第一近接ピークAMnとする。第一近接ピークAMnは、好ましくは1.4Å以上1.9Å以下、より好ましくは1.5Å以上1.6Å以下の1.5Å付近に現れる。
また、本実施形態のリチウム複合金属酸化物においては、MnのK吸収端の動径分布関数において、Mn原子に結合したOの次にMn原子に近い原子X(ここで、XはLi、Mn、Niなどの金属原子となる。)によるピークを第二近接ピークBMnとする。第二近接ピークBMnは、好ましくは2.44Å以上2.55Å以下、より好ましくは2.46Å以上2.55Å以下の2.5Å付近に現れる。ここで、原子XはMn原子に結合したOに結合している。
さらに、本実施形態のリチウム複合金属酸化物においては、NiのK吸収端の動径分布関数において、Ni原子に結合したOによるピークを第一近接ピークANiとする。第一近接ピークANiは、好ましくは1.4Å以上1.9Å以下、より好ましくは1.5Å以上1.6Å以下の1.5Å付近に現れる。
そして、本実施形態のリチウム複合金属酸化物においては、NiのK吸収端の動径分布関数において、Ni原子に結合したOの次にNi原子に近い原子X(ここで、XはLi、Mn、Niなどの金属原子となる。)によるピークを第二近接ピークBNiとする。第二近接ピークBNiは、好ましくは2.44Å以上2.55Å以下、より好ましくは2.46Å以上2.55Å以下の2.5Å付近に現れる。ここで、原子XはNi原子に結合したOに結合している。
本実施形態のリチウム複合金属酸化物は、動径分布関数のピークの強度比、すなわちIAMnとIBMnとの比(IBMn/IAMn)およびIANiとIBNiとの比(IBNi/IANi)を特定の範囲内に制御することで、原子レベルの局所構造を制御している。このような本実施形態のリチウム複合金属酸化物は、従来よりも高容量を示す非水電解質二次電池に有用である。
本実施形態のリチウム複合金属酸化物は、Mn原子まわりのOおよび原子Xの原子間距離の等方性がある程度に高く、ある特定の範囲に収まるため、正極活物質としての特性が高い。このようなIBMn/IAMnの値は、0.5以上1.2以下であり、好ましくは0.6以上1.2以下であり、より好ましくは0.7以上1.2以下であり、さらにより好ましくは1.0以上1.2以下であり、特に好ましくは1.1以上1.2以下である。
また、本実施形態のリチウム複合金属酸化物は、Ni原子まわりのOおよび原子Xの原子間距離の等方性がある程度に高く、ある特定の範囲に収まるため、正極活物質としての特性が高い。このようなIBNi/IANiの値は、1.0以上1.7以下であり、好ましくは1.1以上1.7以下であり、より好ましくは1.2以上1.7以下である。
これらIBMn/IAMnの値の範囲及びIBNi/IANiの値の範囲は、任意に組み合わせることができる。
さらに、前記IBMn/IAMnとIBNi/IANiとの積(IBMn/IAMn×IBNi/IANi)は、適度なMn原子まわりのOおよび原子Xの原子間距離の等方性と、適度なNi原子まわりのOおよび原子Xの原子間距離の等方性と、を両立するため、0.7以上2.0以下であり、好ましくは0.9以上2.0以下であり、より好ましくは1.1以上2.0以下である。
本実施形態のリチウム複合金属酸化物の結晶構造は、層状構造であることが好ましく、六方晶型の結晶構造または単斜晶型の結晶構造であることがより好ましい。
六方晶型の結晶構造は、P3、P31、P32、R3、P−3、R−3、P312、P321、P3112、P3121、P3212、P3221、R32、P3m1、P31m、P3c1、P31c、R3m、R3c、P−31m、P−31c、P−3m1、P−3c1、R−3m、R−3c、P6、P61、P65、P62、P64、P63、P−6、P6/m、P63/m、P622、P6122、P6522、P6222、P6422、P6322、P6mm、P6cc、P63cm、P63mc、P−6m2、P−6c2、P−62m、P−62c、P6/mmm、P6/mcc、P63/mcm、P63/mmcからなる群から選ばれるいずれか一つの空間群に分類される。
また、単斜晶型の結晶構造は、P2、P21、C2、Pm、Pc、Cm、Cc、P2/m、P21/m、C2/m、P2/c、P21/c、C2/cからなる群から選ばれるいずれか一つの空間群に分類される。
これらのうち、得られる非水電解質二次電池の放電容量が増大するため、リチウム複合金属酸化物の結晶構造は、空間群R−3mに分類される六方晶型の結晶構造、またはC2/mに分類される単斜晶型の結晶構造であることが特に好ましい。
本実施形態のリチウム複合金属酸化物の空間群は、次の方法で確認することができる。
まず、リチウム複合金属酸化物について、CuKαを線源とし、かつ回折角2θの測定範囲を10°以上90°以下とする粉末X線回折測定を行い、次いでその結果をもとにリートベルト解析を行い、リチウム複合金属酸化物が有する結晶構造およびこの結晶構造における空間群を決定する。リートベルト解析は、材料の粉末X線回折測定における回折ピークのデータ(回折ピーク強度、回折角2θ)を用いて、材料の結晶構造を解析する手法であり、従来から使用されている手法である(例えば「粉末X線解析の実際−リートベルト法入門−」2002年2月10日発行、日本分析化学会X線分析研究懇談会編、参照)。
本実施形態におけるリチウム複合金属酸化物の組成は、Liの量(モル)をALi、Li以外の金属の量(モル)をAとしたとき、ALi/Aが、0.7以上1.4以下であることとしてもよい。
本実施形態におけるリチウム複合金属酸化物は、層状構造を有し、組成が下記式(1)で表されるものが好ましい。
Li1+x(Ni1−x−y−αMnyCoαMβ)O2 …(1)
(式(1)中、−0.3≦x≦0.4、0.35≦y≦0.7、0<α≦0.1、0≦β<0.1(ただし、0<α+β≦0.1)であり、−0.05≦x+y+α+β<1であり、MはAl、Mg、Ti、Ca、Cu、Zn、Fe、Cr、Mo、Si、Sn、NbおよびVからなる群より選ばれる1種類以上の元素である。)
式(1)中のxの値は、−0.3≦x≦0.4であり、好ましくは−0.2≦x≦0.35であり、より好ましくは−0.1≦x≦0.3である。
本実施形態のリチウム複合金属酸化物は、60℃における放電容量が高いため、MがFeであることが好ましい。
なお、本実施形態のリチウム複合金属酸化物の粒子をコア材として、コア材(リチウム複合金属酸化物の粒子)の表面に、さらにB、Al、Ga、In、Si、Ge、Sn、Mgおよび遷移金属からなる群から選ばれる1種以上の原子を含有する化合物を付着させてもよい。
上記原子の中でも、B、Al、Mg、Co、CrおよびMnからなる群から選ばれる1種以上が好ましく、均一な被覆層形成が容易であるため、Alがより好ましい。
このような化合物としては、例えば上記原子の酸化物、フッ化物、硫化物、水酸化物、オキシ水酸化物、炭酸塩、硝酸塩、有機酸塩およびこれらの混合物が挙げられる。中でも、酸化物、水酸化物、オキシ水酸化物またはこれらの混合物が好ましい。
コア材の表面に被着させる化合物としては、Alの酸化物であるアルミナが好ましい。
[リチウム複合金属酸化物の製造方法]
次に、上述したリチウム複合金属酸化物の製造方法について説明する。
本実施形態のリチウム複合金属酸化物の製造方法は、以下の(1)~(5)の工程を含む。
(1)Niイオン、MnイオンおよびCoイオンを含有する水溶液(以下、「原料水溶液」と称することがある)とアルカリとを接触させて共沈物を生成させ、スラリーを得る工程
(2)(1)で得られるスラリーから共沈物を分離する工程
(3)(2)で得られる共沈物とリチウム化合物とを混合する工程
(4)(3)で得られる混合物を、酸素濃度が5体積%以上の雰囲気において、200℃以上500℃以下の温度で加熱する工程
(5)(4)で得られる生成物(以下、「仮焼物」と称することがある)を、酸素濃度が5体積%未満の雰囲気において、600℃以上950℃以下の温度で焼成する工程。
ここで工程(4)における「酸素濃度」とは、混合物を加熱する空間(熱処理空間)が200℃以上500℃以下の範囲にある際の、熱処理空間中の平均酸素濃度のことを指す。同様に、工程(5)における「酸素濃度」とは、仮焼物を焼成する空間(熱処理空間)が600℃以上950℃以下の範囲にある際の、熱処理空間中の平均酸素濃度を指す。
(工程(1))
上記工程(1)において、原料水溶液は、Ni、MnおよびCoを含有する化合物を水に溶解させることで調整することができる。なかでも、原料水溶液は、Niの硫酸塩、Mnの硫酸塩およびCoの硫酸塩を水に溶解して得られる水溶液であることが好ましい。
また、Ni、MnおよびCoを含有するそれぞれの原料が水に溶解し難い場合、例えば、これらの原料が、酸化物、水酸化物、金属材料である場合には、これらの原料を、硫酸を含有する水溶液に溶解させて、原料水溶液を得ることができる。
工程(1)で用いられるアルカリとしては、LiOH(水酸化リチウム)、NaOH(水酸化ナトリウム)、KOH(水酸化カリウム)、Li2CO3(炭酸リチウム)、Na2CO3(炭酸ナトリウム)、K2CO3(炭酸カリウム)および(NH4)2CO3(炭酸アンモニウム)からなる群より選ばれる1種以上の塩を挙げることができる。用いるアルカリは、無水物であってもよく、水和物であってもよい。無水物と水和物とは併用してもよい。工程(1)においては、上記アルカリの水溶液(アルカリ水溶液)を用いることが好ましい。また、アルカリ水溶液として、アンモニア水を用いることもできる。
アルカリ水溶液におけるアルカリの濃度は、好ましくは0.5~10M(mol/L)程度、より好ましくは1~8M程度である。また、製造コストの面から、用いるアルカリとしてNaOHまたはKOHが好ましい。また、NaOHとKOHとは併用してもよい。
工程(1)における接触の方法としては、(i)原料水溶液にアルカリ水溶液を添加して混合する方法、(ii)アルカリ水溶液に原料水溶液を添加して混合する方法、(iii)水に原料水溶液およびアルカリ水溶液を添加して混合する方法、を挙げることができる。混合時には、攪拌を伴うことが好ましい。
また、工程(1)における接触の方法のうち、(ii)アルカリ水溶液に原料水溶液を添加して混合する方法は、pHの変化を制御しやすく好ましい。この方法の場合、アルカリ水溶液に、原料水溶液を添加し混合していくに従い、アルカリ水溶液のpHが低下していく傾向にあるが、pHが9以上、好ましくは10以上となるように調節しながら、原料水溶液を添加することが好ましい。また、原料水溶液およびアルカリ水溶液のうち、いずれか一方または両方の水溶液を40℃以上80℃以下の温度に保持しながら接触させると、より均一な組成の共沈物を得ることができ、好ましい。
工程(1)においては、上記のようにして原料水溶液とアルカリとを接触させることで、Niイオン、MnイオンおよびCoイオンを含む塩が共沈して生成し、共沈物である塩が分散したスラリーを得ることができる。
(工程(2))
工程(2)においては、工程(1)で得られたスラリーから共沈物を得る。共沈物を得ることができる限り、工程(2)では共沈物を得る方法として種々の方法を採用することができるが、操作が簡便であることから、ろ過などの固体成分を得る分離操作による方法が好ましい。スラリーを噴霧乾燥させるなどの、加熱により液体を揮発させる方法によっても共沈物を得ることができる。
工程(2)において共沈物を得る場合には、前記工程(2)では、分離した共沈物を洗浄し、乾燥させることが好ましい。洗浄することにより、得られる共沈物に残存するアルカリや、Niの硫酸塩、Mnの硫酸塩、Coの硫酸塩を原料として用いた場合に原料水溶液中に遊離するSO4 2−イオンの量を低減することができる。洗浄によりこれらを低減させると、不活性融剤(後述)の量の制御が容易となり好ましい。
共沈物を効率よく洗浄するためには、洗浄液として水を用いることが好ましい。なお、必要に応じてアルコール、アセトンなどの水溶性を有する有機溶媒を洗浄液に加えても良い。また、洗浄は2回以上行ってもよく、例えば、水洗浄を行った後、前記のような水溶性を有する有機溶媒で再度洗浄することもできる。
洗浄した共沈物の乾燥は、熱処理によって行うことができるが、送風乾燥、真空乾燥などによってもよく、さらにこれらを組み合わせてもよい。熱処理によって行う場合、加熱温度は好ましくは50~300℃であり、より好ましくは100~200℃程度である。
(工程(3))
工程(3)においては、工程(2)で得られた共沈物とリチウム化合物とを混合して混合物を得る。
リチウム化合物としては、水酸化リチウム、塩化リチウム、硝酸リチウムおよび炭酸リチウムからなる群より選ばれる1種以上の塩を挙げることができる。用いるリチウム化合物は、無水物であってもよく、水和物であってもよい。また、無水物と水和物とを併用してもよい。
混合は、乾式混合、湿式混合のいずれによってもよいが、操作が簡便であることから乾式混合が好ましい。混合装置としては、攪拌混合、V型混合機、W型混合機、リボン混合機、ドラムミキサー、ボールミルなどを挙げることができる。
(工程(4))
工程(4)においては、工程(3)で得られた混合物を、200℃以上500℃以下、好ましくは250℃以上450℃以下の温度で加熱し、仮焼物を得る。加熱の雰囲気としては、大気および酸素またはそれらの混合ガスを用いる方法、大気および酸素またはそれらの混合ガスに、窒素およびアルゴンなどの不活性ガスを混合する方法などがあるが、酸素濃度が5体積%以上である雰囲気であればよい。意図した局所構造を有する高容量のリチウム複合金属酸化物が得られやすく、得られたリチウム複合金属酸化物を正極活物質として用いた場合に高容量な二次電池とすることが可能であることから、酸素濃度は7体積%以上20体積以下であることが好ましく、10体積%以上20体積%以下であることがより好ましい。
(工程(5))
工程(5)において、工程(4)で得られた仮焼物を650℃以上950℃以下、好ましくは650℃以上900℃以下の温度で焼成する。焼成の雰囲気としては、大気、酸素、窒素およびアルゴンなどを混合し、酸素濃度が5体積%未満である雰囲気とすればよい。意図した局所構造を有する高容量のリチウム複合金属酸化物が得られやすく、得られたリチウム複合金属酸化物を正極活物質として用いた場合に高容量な二次電池とすることが可能であることから、酸素濃度は0.5体積%以上、5体積%未満であることが好ましく、1体積%以上3体積%以下であることがさらに好ましい。
上記、工程(4)および工程(5)は、得られるリチウム複合金属酸化物の組成を均一にするために、工程(4)終了時の加熱温度から温度を下げることなく、連続的に工程(5)を行うことが好ましい。工程(4)および工程(5)を連続的に行う際には、工程(4)終了時の温度を維持しながら、または工程(5)の焼成温度に昇温しながら、酸素濃度を工程(4)の酸素濃度から工程(5)の酸素濃度に変更する。酸素濃度の変更方法としては、導入するガスの酸素濃度を変化させる方法が好ましく用いられる。
このような工程(1)~工程(5)により、本実施形態のリチウム複合金属酸化物を製造することができる。
なお、本実施形態の製造方法では、工程(1)~工程(5)を有することとして説明したが、これに限られない。例えば、工程(1)~工程(3)に代わる別の方法でNiイオン、MnイオンおよびCoイオンを含む塩と、リチウム化合物とを混合して得られる混合物を用意し、得られた混合物を、酸素濃度を制御しながら加熱し、上記工程(4)および工程(5)に相当する処理を行うことでも本実施形態のリチウム複合金属酸化物を製造することが可能である。
上述の「Niイオン、MnイオンおよびCoイオンを含む塩」は、Niイオンを含む塩と、Mnイオンを含む塩と、Coイオンを含む塩との混合物であってもよい。上述の「工程(1)~工程(3)に代わる別の方法」としては、上述の塩を固相で混合する方法、上述の塩を液相中に分散してスラリーを作製し、得られたスラリーを噴霧乾燥して混合する方法などが挙げられる。
また、本実施形態のリチウム複合金属酸化物の製造方法では、工程(4)の混合物にNi、MnおよびCoが含まれている必要があるが、工程(4)で加熱する混合物に、更に他の金属原子が含まれていてもよい。他の金属原子としては、Al、Mg、Ti、Ca、Cu、Zn、Fe、Cr、Mo、Si、Sn、NbおよびVからなる群より選ばれる1種類以上の原子が挙げられる。
工程(4)で加熱する混合物に他の金属原子を含ませる方法としては、種々の方法を採用できる。なかでも、上記工程(1)において、原料水溶液に他の金属の水溶性塩を溶解させると、得られる混合物中に他の金属原子が均一に分散するため好ましい。
(不活性融剤)
工程(4)および工程(5)の際に、混合物および仮焼物は、不活性融剤を含有していてもよい。不活性融剤は、目的物である複合金属酸化物と反応せず、且つ目的物と分離が容易な塩である。不活性融剤は、工程(4)の加熱温度および工程(5)の焼成温度で溶融し反応場を形成し、均一な反応を促進する。そのため、不活性融剤を用いると、均一な組成の生成物を得やすい。
不活性融剤としては、K2SO4、Na2SO4などの硫酸塩;K2CO3、Na2CO3などの炭酸塩;NaCl、KCl、NH4Clなどの塩化物;LiF、NaF、KF、NH4Fなどのフッ化物;ホウ酸;を挙げることができる。上記、不活性融剤のなかでも、製造工程が簡便になることから、硫酸塩が好ましい。より好ましくはK2SO4である。不活性融剤は2種以上併用することもできる。
混合物が不活性融剤を含有すると、混合物の加熱時および仮焼物の焼成時の反応性が向上し、これにより、得られるリチウム複合金属酸化物のBET比表面積を調整することが可能な場合がある。温度が同じ場合には、不活性融剤の含有量が多くなればなるほど、酸化物のBET比表面積は大きくなる傾向にある。また、加熱または焼成の際に、不活性融剤を含有すると、均一な反応を行うことができるため、加熱雰囲気の調整によりリチウム複合金属酸化物の原子レベルで局所構造が制御できる。
不活性融剤は、工程(2)における分離操作で得られる共沈物に、上記不活性融剤の溶液を含ませた後、乾燥させることにより、得られる共沈物に混合されてもよい。
例えば、工程(1)において、Niの硫酸塩やMnの硫酸塩やCoの硫酸塩を原料として用いた場合、原料水溶液中にSO4 2−イオンが遊離する。このSO4 2−イオンと、共沈に用いるアルカリに含まれる金属イオン(例えば、アルカリとしてKOHを用いる場合には、Kイオン)とが、工程(2)で分離した共沈物に残存し、不活性融剤(上記例であればK2SO4)が生じることがある。そのため、工程(1)における共沈後の原料水溶液を上記「不活性融剤の溶液」として用い、工程(2)で得られる共沈物に、共沈後の原料水溶液を含ませたまま乾燥させることにより、得られる共沈物に不活性融剤が混合されてもよい。
また、不活性融剤は、工程(3)において共沈物とリチウム化合物との混合時に、添加して混合できる。不活性融剤の量の制御が容易であるため、不活性融剤を工程(2)で添加する上述の方法よりも、工程(3)で添加する方法のほうが好ましい。工程(3)において不活性融剤を添加する場合は、工程(2)において得られる共沈物を洗浄し、共沈物に残存するアルカリや、Niの塩やMnの塩やCoの塩に由来する陰イオンの量を低減しておくことにより、不活性融剤の量の制御が容易となる。
不活性融剤は、リチウム複合金属酸化物に残留していてもよいし、洗浄により除去されていてもよい。
反応の均一性向上の観点から、前記不活性融剤が硫酸塩であり、混合物または仮焼物と前記硫酸塩とを混合したときに、得られる混合物中の硫酸塩の含有量が、用いたリチウム化合物100質量部に対して0.01質量部以上400質量部以下であることが好ましい。より好ましくは、0.1質量部以上10質量部以下である。
また、本実施形態のリチウム複合金属酸化物の製造方法により得られるリチウム複合金属酸化物を、ボールミルやジェットミルなどを用いて粉砕してもよい。粉砕によって、リチウム複合金属酸化物のBET比表面積を調整することが可能な場合がある。また、工程(1)から工程(5)を実施して得られるリチウム複合金属酸化物を粉砕し、再度工程(4)と工程(5)とを行うことで、粉砕後に焼成を行ってもよい。さらに、必要に応じて、粉砕と工程(4)、(5)による焼成とを、2回以上繰り返してもよい。また、リチウム複合金属酸化物は必要に応じて洗浄あるいは分級することもできる。
本実施形態のリチウム複合金属酸化物は、好ましくは0.05μm以上1μm以下の粒径の一次粒子と、一次粒子が凝集して形成された2μm以上100μm以下の粒径の二次粒子との混合物からなる。一次粒子、二次粒子の粒径は、SEMで観察することにより、測定することができる。
リチウム複合金属酸化物の二次粒子の大きさは、好ましくは2μm以上50μm以下の範囲であり、より好ましくは2μm以上10μm以下の範囲であり、さらにより好ましくは3μm以上8μm以下の範囲であり、特に好ましくは3.5μm以上7μm以下の範囲である。これらにより、得られる非水電解質二次電池の容量がより高まる。
リチウム複合金属酸化物の一次粒子の大きさは、好ましくは0.08μm以上0.8μm以下の範囲であり、より好ましくは0.10μm以上0.7μm以下の範囲であり、さらにより好ましくは0.15μm以上0.7μm以下の範囲であり、特に好ましくは0.2μm以上0.5μm以下の範囲である。これらにより、得られる非水電解質二次電池の高い電流レートにおける放電容量が高まる。
また、リチウム複合金属酸化物の平均粒子径(D50)は、好ましくは1μm以上50μm以下の範囲であり、より好ましくは1.5μm以上30μm以下の範囲であり、さらにより好ましくは2μm以上20μm以下の範囲であり、特に好ましくは3μm以上10μm以下の範囲である。これらにより、リチウム複合金属酸化物を用いた電極の密度が高まり、高容量の非水電解質二次電池が得られる。
リチウム複合金属酸化物の平均粒子径(D50)は、以下の方法で測定できる。
<リチウム複合金属酸化物の平均粒子径(D50)の測定>
測定するリチウム複合金属酸化物の粉末0.1gを、0.2質量%ヘキサメタりん酸ナトリウム水溶液50mlに投入し、該粉末を分散させた分散液を得る。得られた分散液についてマルバーン社製マスターサイザー2000(レーザー回折散乱粒度分布測定装置)を用いて、粒度分布を測定し、体積基準の累積粒度分布曲線を得る。得られた累積粒度分布曲線において、50%累積時の微小粒子側から見た粒子径の値が、平均粒子径(D50)である。
リチウム複合金属酸化物のBET比表面積は、好ましくは0.1m2/g以上20m2/g以下の範囲であり、より好ましくは0.5m2/g以上15m2/g以下の範囲であり、さらにより好ましくは1m2/g以上10m2/g以下の範囲であり、特に好ましくは2m2/g以上8m2/g以下の範囲である。これらにより、得られる非水電解質二次電池の高い電流レートにおける放電容量が高まる。
リチウム複合金属酸化物のBET比表面積は、以下の方法で測定できる。
<リチウム複合金属酸化物のBET比表面積の測定>
測定するリチウム複合金属酸化物の粉末1gを窒素雰囲気中、150℃で15分間乾燥させた後、マイクロメリティックス製フローソーブII2300を用いて測定する。
上記リチウム複合金属酸化物を、非水電解質二次電池の正極活物質に用いた場合に、従来よりも高容量を示す非水電解質二次電池が得られる。
[非水電解質二次電池]
次いで、非水電解質二次電池の構成を説明しながら、本実施形態のリチウム複合金属酸化物を非水電解質二次電池の正極活物質として用いた正極、およびこの正極を有する非水電解質二次電池について説明する。
本実施形態の非水電解質二次電池の一例は、正極、負極、正極と負極との間に配置されるセパレータ、および電解液を有する。
図1は、本実施形態の非水電解質二次電池の一例を示す模式図である。本実施形態の円筒型の非水電解質二次電池10は、次のようにして製造する。
まず、図1(a)に示すように、帯状を呈する2つのセパレータ1、一端に正極リード21を有する帯状の正極2、および一端に負極リード31を有する帯状の負極3を、セパレータ1、正極2、セパレータ1、負極3の順に積層し、巻回することにより電極群4とする。
次いで、図1(b)に示すように、電池缶5に電極群4および不図示のインシュレーターを収容した後、缶底を封止し、電極群4に電解液6を含浸させ、正極2と負極3との間に電解質を配置する。さらに、電池缶5の上部をトップインシュレーター7および封口体8で封止することで、非水電解質二次電池10を製造することができる。
電極群4の形状としては、例えば、電極群4を巻回の軸に対して垂直方向に切断したときの断面形状が、円、楕円、長方形、角を丸めた長方形となるような柱状の形状を挙げることができる。
また、このような電極群4を有する非水電解質二次電池の形状としては、国際電気標準会議(IEC)が定めた電池に対する規格であるIEC60086、またはJIS C 8500で定められる形状を採用することができる。例えば、円筒型、角型などの形状を挙げることができる。
さらに、非水電解質二次電池は、上記巻回型の構成に限らず、正極、セパレータ、負極、セパレータの積層構造を繰り返し重ねた積層型の構成であってもよい。積層型の非水電解質二次電池としては、いわゆるコイン型電池、ボタン型電池、ペーパー型(またはシート型)電池を例示することができる。
以下、各構成について順に説明する。
(正極)
本実施形態の正極は、まず正極活物質、導電材およびバインダーを含む正極合剤を調整し、正極合剤を正極集電体に担持させることで製造することができる。
(正極活物質)
本実施形態の正極活物質は、上述のリチウム複合金属酸化物を有する。本実施形態のリチウム複合金属酸化物を、非水電解質二次電池の正極活物質として用いることで、高容量を示す非水電解質二次電池とすることができる。
(導電材)
本実施形態の正極が有する導電材としては、炭素材料を用いることができる。炭素材料として黒鉛粉末、カーボンブラック(例えばアセチレンブラック)、繊維状炭素材料などを挙げることができる。カーボンブラックは、微粒で表面積が大きいため、少量を正極合剤中に添加することにより正極内部の導電性を高め、充放電効率および出力特性を向上させることができるが、多く入れすぎるとバインダーによる正極合剤と正極集電体との結着力、および正極合剤内部の結着力がいずれも低下し、かえって内部抵抗を増加させる原因となる。
正極合剤中の導電材の割合は、正極活物質100質量部に対して5質量部以上20質量部以下であると好ましい。導電材として黒鉛化炭素繊維、カーボンナノチューブなどの繊維状炭素材料を用いる場合には、この割合を下げることも可能である。
(バインダー)
本実施形態の正極が有するバインダーとしては、熱可塑性樹脂を用いることができる。この熱可塑性樹脂としては、ポリフッ化ビニリデン(以下、PVdFということがある。)、ポリテトラフルオロエチレン(以下、PTFEということがある。)、四フッ化エチレン・六フッ化プロピレン・フッ化ビニリデン系共重合体、六フッ化プロピレン・フッ化ビニリデン系共重合体、四フッ化エチレン・パーフルオロビニルエーテル系共重合体などのフッ素樹脂;ポリエチレン、ポリプロピレンなどのポリオレフィン樹脂;を挙げることができる。
これらの熱可塑性樹脂は、2種以上を混合して用いてもよい。バインダーとしてフッ素樹脂およびポリオレフィン樹脂を用い、正極合剤全体に対するフッ素樹脂の割合を1質量%以上10質量%以下、ポリオレフィン樹脂の割合を0.1質量%以上2質量%以下とすることによって、正極集電体との密着力および正極合剤内部の結合力がいずれも高い正極合剤を得ることができる。
(正極集電体)
本実施形態の正極が有する正極集電体としては、Al、Ni、ステンレスなどの金属材料を構成材料とする帯状の部材を用いることができる。なかでも、加工しやすく、安価であるという点でAlを形成材料とし、薄膜状に加工したものが好ましい。
正極集電体に正極合剤を担持させる方法としては、正極合剤を正極集電体上で加圧成型する方法が挙げられる。また、有機溶媒を用いて正極合剤をペースト化し、得られる正極合剤のペーストを正極集電体の少なくとも一面に塗工して乾燥させ、プレスし固着することで、正極集電体に正極合剤を担持させてもよい。
正極合剤をペースト化する場合、用いることができる有機溶媒としては、N,N—ジメチルアミノプロピルアミン、ジエチレントリアミンなどのアミン系溶媒;テトラヒドロフランなどのエーテル系溶媒;メチルエチルケトンなどのケトン系溶媒;酢酸メチルなどのエステル系溶媒;ジメチルアセトアミド、N−メチル−2−ピロリドン(以下、NMPということがある。)などのアミド系溶媒;が挙げられる。
正極合剤のペーストを正極集電体へ塗工する方法としては、例えば、スリットダイ塗工法、スクリーン塗工法、カーテン塗工法、ナイフ塗工法、グラビア塗工法および静電スプレー法が挙げられる。
以上に挙げられた方法により、正極を製造することができる。
(負極)
本実施形態の非水電解質二次電池が有する負極は、正極よりも低い電位でリチウムイオンのドープかつ脱ドープが可能であればよく、負極活物質を含む負極合剤が負極集電体に担持されてなる電極、および負極活物質単独からなる電極を挙げることができる。
(負極活物質)
負極が有する負極活物質としては、炭素材料、カルコゲン化合物(酸化物、硫化物など)、窒化物、金属または合金で、正極よりも低い電位でリチウムイオンのドープかつ脱ドープが可能な材料が挙げられる。
負極活物質として使用可能な炭素材料としては、天然黒鉛、人造黒鉛などの黒鉛、コークス類、カーボンブラック、熱分解炭素類、炭素繊維および有機高分子化合物焼成体を挙げることができる。
負極活物質として使用可能な酸化物としては、SiO2、SiOなど式SiOx(ここで、xは正の実数)で表されるケイ素の酸化物;TiO2、TiOなど式TiOx(ここで、xは正の実数)で表されるチタンの酸化物;V2O5、VO2など式VOx(ここで、xは正の実数)で表されるバナジウムの酸化物;Fe3O4、Fe2O3、FeOなど式FeOx(ここで、xは正の実数)で表される鉄の酸化物;SnO2、SnOなど式SnOx(ここで、xは正の実数)で表されるスズの酸化物;WO3、WO2など一般式WOx(ここで、xは正の実数)で表されるタングステンの酸化物;Li4Ti5O12、LiVO2などのリチウムとチタンまたはバナジウムとを含有する複合金属酸化物;を挙げることができる。
負極活物質として使用可能な硫化物としては、Ti2S3、TiS2、TiSなど式TiSx(ここで、xは正の実数)で表されるチタンの硫化物;V3S4、VS2、VSなど式VSx(ここで、xは正の実数)で表されるバナジウムの硫化物;Fe3S4、FeS2、FeSなど式FeSx(ここで、xは正の実数)で表される鉄の硫化物;Mo2S3、MoS2など式MoSx(ここで、xは正の実数)で表されるモリブデンの硫化物;SnS2、SnSなど式SnSx(ここで、xは正の実数)で表されるスズの硫化物;WS2など式WSx(ここで、xは正の実数)で表されるタングステンの硫化物;Sb2S3など式SbSx(ここで、xは正の実数)で表されるアンチモンの硫化物;Se5S3、SeS2、SeSなど式SeSx(ここで、xは正の実数)で表されるセレンの硫化物;を挙げることができる。
負極活物質として使用可能な窒化物としては、Li3N、Li3−xAxN(ここで、AはNiおよびCoのいずれか一方または両方であり、0<x<3である。)などのリチウム含有窒化物を挙げることができる。
これらの炭素材料、酸化物、硫化物、窒化物は、1種のみ用いてもよく2種以上を併用して用いてもよい。また、これらの炭素材料、酸化物、硫化物、窒化物は、結晶質または非晶質のいずれでもよい。
また、負極活物質として使用可能な金属としては、リチウム金属、シリコン金属およびスズ金属などを挙げることができる。
負極活物質として使用可能な合金としては、Li−Al、Li−Ni、Li−Si、Li−Sn、Li−Sn−Niなどのリチウム合金;Si−Znなどのシリコン合金;Sn−Mn、Sn−Co、Sn−Ni、Sn−Cu、Sn−Laなどのスズ合金;Cu2Sb、La3Ni2Sn7などの合金;を挙げることもできる。
これらの金属や合金は、例えば箔状に加工された後、主に単独で電極として用いられる。
上記負極活物質の中では、充電時に未充電状態から満充電状態にかけて負極の電位がほとんど変化しない(電位平坦性が良い)、平均放電電位が低い、繰り返し充放電させたときの容量維持率が高い(サイクル特性が良い)などの理由から、天然黒鉛、人造黒鉛などの黒鉛を主成分とする炭素材料が好ましく用いられる。炭素材料の形状としては、例えば天然黒鉛のような薄片状、メソカーボンマイクロビーズのような球状、黒鉛化炭素繊維のような繊維状、または微粉末の凝集体などのいずれでもよい。
前記の負極合剤は、必要に応じて、バインダーを含有してもよい。バインダーとしては、熱可塑性樹脂を挙げることができ、具体的には、PVdF、熱可塑性ポリイミド、カルボキシメチルセルロース、ポリエチレンおよびポリプロピレンを挙げることができる。
(負極集電体)
負極が有する負極集電体としては、Cu、Ni、ステンレスなどの金属材料を構成材料とする帯状の部材を挙げることができる。なかでも、リチウムと合金を作り難く、加工しやすいという点で、Cuを形成材料とし、薄膜状に加工したものが好ましい。
このような負極集電体に負極合剤を担持させる方法としては、正極の場合と同様に、加圧成型による方法、溶媒などを用いてペースト化し負極集電体上に塗布、乾燥後プレスし圧着する方法が挙げられる。
(セパレータ)
本実施形態の非水電解質二次電池が有するセパレータとしては、例えば、ポリエチレン、ポリプロピレンなどのポリオレフィン樹脂、フッ素樹脂、含窒素芳香族重合体などの材質からなる、多孔質膜、不織布、織布などの形態を有する材料を用いることができる。また、これらの材質を2種以上用いてセパレータを形成してもよいし、これらの材料を積層してセパレータを形成してもよい。
セパレータとしては、例えば特開2000−30686号公報、特開平10−324758号公報などに記載のセパレータを挙げることができる。セパレータの厚みは電池の体積エネルギー密度が上がり、内部抵抗が小さくなるという点で、機械的強度が保たれる限り薄くした方がよく、好ましくは5~200μm程度、より好ましくは5~40μm程度である。
セパレータは、好ましくは、熱可塑性樹脂を含有する多孔質フィルムを有する。非水電解質二次電池においては、正極−負極間の短絡などが原因で電池内に異常電流が流れた際に、短絡箇所の電流を遮断して、過大電流が流れることを阻止(シャットダウン)する機能を有することが好ましい。ここで、シャットダウンは、短絡により短絡箇所のセパレータが過熱され、予め想定された使用温度を越えた場合に、セパレータにおける多孔質フィルムが軟化または融解して微細孔を閉塞することによりなされる。そして、セパレータはシャットダウンした後、ある程度の高温まで電池内の温度が上昇しても、その温度により破膜することなく、シャットダウンした状態を維持することが好ましい。
このようなセパレータとしては、耐熱多孔層と多孔質フィルムとが積層された積層フィルムが挙げられる。このような積層フィルムをセパレータとして用いることにより、本実施形態における二次電池の耐熱性をより高めることが可能となる。積層フィルムにおいては、耐熱多孔層は、多孔質フィルムの両面に積層されていてもよい。
(積層フィルム)
以下、前記の耐熱多孔層と多孔質フィルムとが互いに積層された積層フィルムについて説明する。
本実施形態の非水電解質二次電池のセパレータとして用いられる積層フィルムにおいて、耐熱多孔層は、多孔質フィルムよりも耐熱性の高い層である。耐熱多孔層は、無機粉末から形成されていてもよいし(第1の耐熱多孔層)、耐熱樹脂から形成されていてもよいし(第2の耐熱多孔層)、耐熱樹脂とフィラーとを含んで形成されていてもよい(第3の耐熱多孔層)。耐熱多孔層が、耐熱樹脂を含有することにより、塗工などの容易な手法で、耐熱多孔層を形成することができる。
(第1の耐熱多孔層)
耐熱多孔層が無機粉末から形成されている場合、耐熱多孔層に用いられる無機粉末としては、例えば、金属酸化物、金属窒化物、金属炭化物、金属水酸化物、炭酸塩、硫酸塩などの無機物からなる粉末が挙げられ、これらの中でも、導電性の低い(絶縁体の)無機物からなる粉末が好ましく用いられる。具体的に例示すると、アルミナ、シリカ、二酸化チタンまたは炭酸カルシウムなどからなる粉末が挙げられる。このような無機粉末は、単独で用いてもよいし、2種以上を混合して用いることもできる。
これらの無機粉末の中でも、化学的安定性が高いことから、アルミナ粉末が好ましい。また、無機粉末を構成する粒子のすべてがアルミナ粒子であることがより好ましく、無機粉末を構成する粒子のすべてがアルミナ粒子であり、その一部または全部が略球状のアルミナ粒子であることがさらに好ましい。
(第2の耐熱多孔層)
耐熱多孔層が耐熱樹脂から形成されている場合、耐熱多孔層に用いられる耐熱樹脂としては、ポリアミド、ポリイミド、ポリアミドイミド、ポリカーボネート、ポリアセタール、ポリサルホン、ポリフェニレンサルファイド、ポリエーテルケトン、芳香族ポリエステル、ポリエーテルサルホンおよびポリエーテルイミドを挙げることができる。積層フィルムの耐熱性をより高めるためには、ポリアミド、ポリイミド、ポリアミドイミド、ポリエーテルサルホンおよびポリエーテルイミドが好ましく、より好ましくは、ポリアミド、ポリイミドまたはポリアミドイミドである。
耐熱多孔層に用いられる耐熱樹脂としてさらに好ましくは、芳香族ポリアミド(パラ配向芳香族ポリアミド、メタ配向芳香族ポリアミド)、芳香族ポリイミド、芳香族ポリアミドイミドなどの含窒素芳香族重合体であり、とりわけ好ましくは芳香族ポリアミド、製造しやすいために特に好ましいのは、パラ配向芳香族ポリアミド(以下、パラアラミドということがある。)である。
また、耐熱樹脂として、ポリ−4−メチルペンテン−1および環状オレフィン系重合体を挙げることもできる。
これらの耐熱樹脂を用いることにより、非水電解質二次電池のセパレータとして用いられる積層フィルムの耐熱性、すなわち、積層フィルムの熱破膜温度をより高めることができる。これらの耐熱樹脂のうち、含窒素芳香族重合体を用いる場合には、その分子内の極性により、電解液との相性、すなわち、耐熱多孔層における保液性も向上する場合があり、非水電解質二次電池製造時における電解液の含浸の速度も高く、非水電解質二次電池の充放電容量もより高まる。
かかる積層フィルムの熱破膜温度は、耐熱樹脂の種類に依存し、使用場面、使用目的に応じ、選択使用される。より具体的には、耐熱樹脂として、上記含窒素芳香族重合体を用いる場合は400℃程度に、また、ポリ−4−メチルペンテン−1を用いる場合は250℃程度に、環状オレフィン系重合体を用いる場合は300℃程度に、夫々、熱破膜温度をコントロールすることができる。また、耐熱多孔層が、無機粉末からなる場合には、熱破膜温度を、例えば、500℃以上にコントロールすることも可能である。
上記パラアラミドは、パラ配向芳香族ジアミンとパラ配向芳香族ジカルボン酸ハライドとの重縮合により得られ、アミド結合が芳香族環のパラ位またはそれに準じた配向位(例えば、4,4’−ビフェニレン、1,5−ナフタレン、2,6−ナフタレンなどのような反対方向に同軸または平行に延びる配向位)で結合される繰り返し単位から実質的になる。具体的には、ポリ(パラフェニレンテレフタルアミド)、ポリ(パラベンズアミド)、ポリ(4,4’−ベンズアニリドテレフタルアミド)、ポリ(パラフェニレン−4,4’−ビフェニレンジカルボン酸アミド)、ポリ(パラフェニレン−2,6−ナフタレンジカルボン酸アミド)、ポリ(2−クロロ−パラフェニレンテレフタルアミド)、パラフェニレンテレフタルアミド/2,6−ジクロロパラフェニレンテレフタルアミド共重合体などのパラ配向型またはパラ配向型に準じた構造を有するパラアラミドが例示される。
前記の芳香族ポリイミドとしては、芳香族の二酸無水物とジアミンとの重縮合で製造される全芳香族ポリイミドが好ましい。
重縮合に用いられる芳香族の二酸無水物の具体例としては、ピロメリット酸二無水物、3,3’,4,4’−ジフェニルスルホンテトラカルボン酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、2,2’−ビス(3,4−ジカルボキシフェニル)ヘキサフルオロプロパンおよび3,3’,4,4’−ビフェニルテトラカルボン酸二無水物が挙げられる。
重縮合に用いられるジアミンの具体例としては、オキシジアニリン、パラフェニレンジアミン、ベンゾフェノンジアミン、3,3’−メチレンジアニリン、3,3’−ジアミノベンソフェノン、3,3’−ジアミノジフェニルスルフォンおよび1,5−ナフタレンジアミンが挙げられる。
また、芳香族ポリイミドとしては、溶媒に可溶なポリイミドが好適に使用できる。このようなポリイミドとしては、例えば、3,3’,4,4’−ジフェニルスルホンテトラカルボン酸二無水物と芳香族ジアミンとの重縮合物のポリイミドが挙げられる。
前記の芳香族ポリアミドイミドとしては、芳香族ジカルボン酸および芳香族ジイソシアネートの重縮合から得られるもの、芳香族二酸無水物および芳香族ジイソシアネートの重縮合から得られるものが挙げられる。芳香族ジカルボン酸の具体例としてはイソフタル酸およびテレフタル酸が挙げられる。また芳香族二酸無水物の具体例としては無水トリメリット酸が挙げられる。芳香族ジイソシアネートの具体例としては、4,4’−ジフェニルメタンジイソシアネート、2,4−トリレンジイソシアネート、2,6−トリレンジイソシアネート、オルソトリレンジイソシアネートおよびm−キシレンジイソシアネートが挙げられる。
また、イオン透過性をより高めるためには、積層フィルムが有する耐熱多孔層の厚みは、1μm以上10μm以下、さらには1μm以上5μm以下、特に1μm以上4μm以下という薄い耐熱多孔層であることが好ましい。また、耐熱多孔層は微細孔を有し、その孔のサイズ(直径)は、好ましくは3μm以下、より好ましくは1μm以下である。
(第3の耐熱多孔層)
また、耐熱多孔層が耐熱樹脂とフィラーとを含んで形成されている場合、耐熱樹脂は、上記第2の耐熱多孔層に用いられる耐熱樹脂と同じものを使用することができる。フィラーは、有機粉末、無機粉末またはこれらの混合物からなる群から選ばれる1種以上を用いることができる。フィラーを構成する粒子は、その平均粒子径が、0.01μm以上1μm以下であることが好ましい。
フィラーとして用いることができる有機粉末としては、例えば、スチレン、ビニルケトン、アクリロニトリル、メタクリル酸メチル、メタクリル酸エチル、グリシジルメタクリレート、グリシジルアクリレート、アクリル酸メチルなどの単独または2種類以上の共重合体;PTFE、4フッ化エチレン−6フッ化プロピレン共重合体、4フッ化エチレン−エチレン共重合体、ポリビニリデンフルオライドなどのフッ素系樹脂;メラミン樹脂;尿素樹脂;ポリオレフィン樹脂;ポリメタクリレート;などの有機物からなる粉末が挙げられる。このような有機粉末は、単独で用いてもよいし、2種以上を混合して用いることもできる。これらの有機粉末の中でも、化学的安定性が高いことから、PTFEの粉末が好ましい。
フィラーとして用いることができる無機粉末としては、上記耐熱多孔層に用いられる無機粉末と同じものを例示することができる。
耐熱多孔層が耐熱樹脂とフィラーとを含んで形成されている場合、フィラーの含有量としては、フィラーの材質の比重にもよるが、例えば、フィラーを構成する粒子のすべてがアルミナ粒子である場合には、耐熱多孔層の総質量を100質量部としたとき、フィラーの質量は、好ましくは5質量部以上95質量部以下であり、より好ましくは20質量部以上95質量部以下であり、さらに好ましくは30質量部以上90質量部以下である。これらの範囲は、フィラーの材質の比重により、適宜設定できる。
フィラーの形状については、略球状、板状、柱状、針状、繊維状などの形状が挙げられ、いずれの粒子も用いることができるが、均一な孔を形成しやすいことから、略球状粒子であることが好ましい。略球状粒子としては、粒子のアスペクト比(粒子の長径/粒子の短径)が1以上1.5以下である粒子が挙げられる。粒子のアスペクト比は、電子顕微鏡写真により測定することができる。
本実施形態の非水電解質二次電池のセパレータとして用いられる積層フィルムにおいて多孔質フィルムは、微細孔を有し、シャットダウン機能を有することが好ましい。この場合、多孔質フィルムは、熱可塑性樹脂を含有する。
多孔質フィルムにおける微細孔のサイズは、好ましくは3μm以下、より好ましくは1μm以下である。多孔質フィルムの空孔率は、好ましくは30体積%以上80体積%以下、より好ましくは40体積%以上70体積%以下である。非水電解質二次電池において、予め想定された使用温度を越えた場合には、熱可塑性樹脂を含有する多孔質フィルムは、多孔質フィルムを構成する熱可塑性樹脂の軟化または融解により、微細孔を閉塞することができる。
多孔質フィルムに用いられる熱可塑性樹脂は、非水電解質二次電池における電解液に溶解しないものを選択すればよい。具体的には、ポリエチレン、ポリプロピレンなどのポリオレフィン樹脂および熱可塑性ポリウレタン樹脂を挙げることができ、これらの2種以上の混合物を用いてもよい。
セパレータがより低温で軟化してシャットダウンするためには、多孔質フィルムがポリエチレンを含有することが好ましい。ポリエチレンとして、低密度ポリエチレン、高密度ポリエチレン、線状ポリエチレンなどのポリエチレンを挙げることができ、分子量が100万以上の超高分子量ポリエチレンを挙げることもできる。
多孔質フィルムの突刺し強度をより高めるためには、多孔質フィルムを構成する熱可塑性樹脂は、少なくとも超高分子量ポリエチレンを含有することが好ましい。また、多孔質フィルムの製造面において、熱可塑性樹脂は、低分子量(重量平均分子量1万以下)のポリオレフィンからなるワックスを含有することが好ましい場合もある。
また、積層フィルムにおける多孔質フィルムの厚みは、好ましくは3μm以上30μm以下であり、より好ましくは3μm以上25μm以下である。また、本実施形態において、積層フィルムの厚みは、好ましくは40μm以下、より好ましくは、30μm以下である。また、耐熱多孔層の厚みをA(μm)、多孔質フィルムの厚みをB(μm)としたときには、A/Bの値が、0.1以上1以下であることが好ましい。
本実施形態において、セパレータは、電池使用時(充放電時)に電解質を良好に透過させるため、JIS P 8117で定められるガーレー法による透気抵抗度が、50秒/100cc以上、300秒/100cc以下であることが好ましく、50秒/100cc以上、200秒/100cc以下であることがより好ましい。
また、セパレータの空孔率は、好ましくは30体積%以上80体積%以下、より好ましくは40体積%以上70体積%以下である。セパレータは空孔率の異なるセパレータを積層したものであってもよい。
(電解液)
本実施形態の非水電解質二次電池が有する電解液は、電解質および有機溶媒を含有する。
電解液に含まれる電解質としては、LiClO4、LiPF6、LiAsF6、LiSbF6、LiBF4、LiCF3SO3、LiN(SO2CF3)2、LiN(SO2C2F5)2、LiN(SO2CF3)(COCF3)、Li(C4F9SO3)、LiC(SO2CF3)3、Li2B10Cl10、LiBOB(ここで、BOBは、bis(oxalato)borateのことである。)、低級脂肪族カルボン酸リチウム塩、LiAlCl4などのリチウム塩が挙げられ、これらの2種以上の混合物を使用してもよい。なかでも電解質としては、フッ素を含むLiPF6、LiAsF6、LiSbF6、LiBF4、LiCF3SO3、LiN(SO2CF3)2およびLiC(SO2CF3)3からなる群より選ばれる少なくとも1種を含むものを用いることが好ましい。
また前記電解液に含まれる有機溶媒としては、例えばプロピレンカーボネート、エチレンカーボネート、ジメチルカーボネート、ジエチルカーボネート、エチルメチルカーボネート、4−トリフルオロメチル−1,3−ジオキソラン−2−オン、1,2−ジ(メトキシカルボニルオキシ)エタンなどのカーボネート類;1,2−ジメトキシエタン、1,3−ジメトキシプロパン、ペンタフルオロプロピルメチルエーテル、2,2,3,3−テトラフルオロプロピルジフルオロメチルエーテル、テトラヒドロフラン、2−メチルテトラヒドロフランなどのエーテル類;ギ酸メチル、酢酸メチル、γ−ブチロラクトンなどのエステル類;アセトニトリル、ブチロニトリルなどのニトリル類;N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミドなどのアミド類;3−メチル−2−オキサゾリドンなどのカーバメート類;スルホラン、ジメチルスルホキシド、1,3−プロパンサルトンなどの含硫黄化合物、またはこれらの有機溶媒にさらにフルオロ基を導入したもの(有機溶媒が有する水素原子のうち1以上をフッ素原子で置換したもの)を用いることができる。
有機溶媒としては、これらのうちの2種以上を混合して用いることが好ましい。中でもカーボネート類を含む混合溶媒が好ましく、環状カーボネートと非環状カーボネートとの混合溶媒および環状カーボネートとエーテル類との混合溶媒がさらに好ましい。環状カーボネートと非環状カーボネートとの混合溶媒としては、エチレンカーボネート、ジメチルカーボネートおよびエチルメチルカーボネートを含む混合溶媒が好ましい。このような混合溶媒を用いた電解液は、動作温度範囲が広く、高い電流レートにおける充放電を行っても劣化し難く、長時間使用しても劣化し難く、かつ負極の活物質として天然黒鉛、人造黒鉛などの黒鉛材料を用いた場合でも難分解性であるという利点を有する。
また、電解液としては、得られる非水電解質二次電池の安全性が高まるため、LiPF6などのフッ素を含むリチウム塩およびフッ素置換基を有する有機溶媒を含む電解液を用いることが好ましい。ペンタフルオロプロピルメチルエーテル、2,2,3,3−テトラフルオロプロピルジフルオロメチルエーテルなどのフッ素置換基を有するエーテル類とジメチルカーボネートとを含む混合溶媒は、高い電流レートにおける充放電を行っても容量維持率が高いため、さらに好ましい。
上記の電解液の代わりに固体電解質を用いてもよい。固体電解質としては、例えばポリエチレンオキサイド系の高分子化合物、ポリオルガノシロキサン鎖またはポリオキシアルキレン鎖の少なくとも一種以上を含む高分子化合物などの有機系高分子電解質を用いることができる。また、高分子化合物に非水電解液を保持させた、いわゆるゲルタイプのものを用いることもできる。またLi2S−SiS2、Li2S−GeS2、Li2S−P2S5、Li2S−B2S3、Li2S−SiS2−Li3PO4、Li2S−SiS2−Li2SO4などの硫化物を含む無機系固体電解質を用いてもよい。これら固体電解質を用いることで、非水電解質二次電池の安全性をより高めることができることがある。
また、本実施形態の非水電解質二次電池において、固体電解質を用いる場合には、固体電解質がセパレータの役割を果たす場合もあり、その場合には、セパレータを必要としないこともある。
上記の正極活物質は、上述した本実施形態のリチウム複合金属酸化物を用いているため、正極活物質を用いた非水電解質二次電池は、従来よりも高容量を示すことができる。
また、上記の正極は、上述した本実施形態のリチウム複合金属酸化物を用いた正極活物質を有するため、非水電解質二次電池は、従来よりも高容量を示すことができる。
さらに、上記の非水電解質二次電池は、上述した正極を有するため、従来よりも高容量を示す。 [Lithium composite metal oxide]
The lithium composite metal oxide of this embodiment contains Mn, Ni, Li and Co and satisfies the following (a) and (b).
(A) In a radial distribution function obtained by Fourier transform of a wide-range X-ray absorption fine structure (EXAFS) spectrum at the K absorption edge of Mn in the lithium composite metal oxide, 1.5% by oxygen atoms bonded to Mn atoms Near first adjacent peak AMnStrength of IAMn, The second adjacent peak B around 2.5Å due to the metal atom next to the Mn atom next to the oxygen atom bonded to the Mn atomMnStrength of IBMnWhen IBMn/ IAMnIs 0.5 or more and 1.2 or less.
(B) In the radial distribution function obtained by Fourier transforming the EXAFS spectrum at the K absorption edge of Ni in the lithium composite metal oxide, the first proximity peak A around 1.5Å due to oxygen atoms bonded to Ni atoms.NiStrength of IANi, The intensity of the second adjacent peak in the vicinity of 2.5 に よ る by the metal atom close to the Ni atom next to the oxygen atom bonded to the Ni atom isBNiWhen IBNi/ IANiIs 1.0 or more and 1.7 or less.
Hereafter, it explains in order.
(EXAFS spectrum)
First, the EXAFS spectrum used for defining the lithium composite metal oxide of this embodiment will be described. The EXAFS spectrum used in the present embodiment is handled in the same manner as a general EXAFS spectrum. The measurement and principle of the EXAFS spectrum are described in, for example, “X-ray absorption spectroscopy—XAFS and its applications” (Toshiaki Ohta (2002)). The principle is as follows.
First, when an X-ray having a specific wavelength is transmitted through a substance to be measured, the intensity of the X-ray irradiated to the substance (incident X-ray intensity: I0) And the intensity of X-rays transmitted through the substance (transmitted X-ray intensity: It), The X-ray absorbance per unit thickness is obtained for the substance to be measured at a specific wavelength.
The wavelength of X-rays irradiating the substance is changed (that is, the energy (eV) of incident X-rays is changed), the X-ray absorbance of each wavelength (each energy) with respect to the X-rays is measured, When an X-ray absorption spectrum is created with the energy of the line (eV) and the y-axis as the X-ray absorbance, it can be seen that there is energy at which the X-ray absorbance rapidly increases. This energy value is called the absorption edge. The absorption edge corresponds to the energy level of the atomic shell of the atoms constituting the material and is unique to each atom. For example, an absorption edge corresponding to the K shell of an atom is called a K absorption edge.
In the X-ray absorption spectrum, a fine vibration structure appearing in an energy side region about 20 to 1000 eV higher than the absorption edge is called a broad X-ray absorption fine structure (EXAFS), and the spectrum is called an EXAFS spectrum. When Fourier transformation is performed on the EXAFS spectrum, a radial distribution function centered on X-ray absorbing atoms (atomic atoms of interest) is obtained. From this radial distribution function, information such as the distance between X-ray absorbing atoms and X-ray scattering atoms (atoms near the X-ray absorbing atoms), the number of X-ray scattering atoms, and the like can be obtained. Can be obtained.
Generally, the intensity of the peak of the radial distribution function is affected by the number of X-ray scattering atoms, but it also affects the isotropy of the interatomic distance between X-ray absorbing atoms and X-ray scattering atoms. Is done. For example, when two X-ray absorbing atoms have substantially the same number of X-ray scattering atoms and the X-ray scattering atoms are recognized to have substantially the same scattering power, the peak of the radial distribution function For those with high intensity, the interatomic distance between the X-ray absorbing atom and the X-ray scattering atom is isotropic with no difference in direction, and the distance distribution between the X-ray absorbing atom and the X-ray scattering atom is small. Means.
Therefore, in the present embodiment, attention is paid to the peak intensity ratio of the radial distribution function obtained at the K absorption edge of Mn and Ni.
In other words, by controlling the intensity ratio of the peak of the radial distribution function within a certain range, the atomic level local structure in the lithium composite metal oxide can be controlled to a specific condition even for samples with different composition ratios. In addition, a lithium composite metal oxide useful for a non-aqueous electrolyte secondary battery exhibiting a higher capacity than before can be obtained.
In the lithium composite metal oxide of this embodiment, the peak due to O (oxygen atom) bonded to the Mn atom in the radial distribution function of the K absorption edge of Mn is the first proximity peak A.MnAnd First proximity peak AMnPreferably appears in the vicinity of 1.5 to 1.9 to 1.9, more preferably 1.5 to 1.6.
Further, in the lithium composite metal oxide of the present embodiment, in the radial distribution function of the K absorption edge of Mn, an atom X next to Mn atom next to O bonded to Mn atom (where X is Li, Mn , And a peak due to a metal atom such as Ni).MnAnd Second adjacent peak BMnPreferably appears in the vicinity of 2.5 mm from 2.44 mm to 2.55 mm, more preferably from 2.46 mm to 2.55 mm. Here, the atom X is bonded to O bonded to the Mn atom.
Furthermore, in the lithium composite metal oxide of this embodiment, in the radial distribution function of the K absorption edge of Ni, the peak due to O bonded to Ni atoms is the first proximity peak A.NiAnd First proximity peak ANiPreferably appears in the vicinity of 1.5 to 1.9 to 1.9, more preferably 1.5 to 1.6.
In the lithium composite metal oxide of this embodiment, in the radial distribution function of the K absorption edge of Ni, an atom X that is closest to the Ni atom next to O bonded to the Ni atom (where X is Li, Mn , And a peak due to a metal atom such as Ni).NiAnd Second adjacent peak BNiPreferably appears in the vicinity of 2.5 mm from 2.44 mm to 2.55 mm, more preferably from 2.46 mm to 2.55 mm. Here, atom X is bonded to O bonded to Ni atom.
The lithium composite metal oxide of this embodiment has a peak intensity ratio of the radial distribution function, that is, IAMnAnd IBMnTo the ratio (IBMn/ IAMn) And IANiAnd IBNiTo the ratio (IBNi/ IANi) Within a specific range, the local structure at the atomic level is controlled. Such a lithium composite metal oxide of this embodiment is useful for a non-aqueous electrolyte secondary battery that exhibits a higher capacity than before.
The lithium composite metal oxide of the present embodiment has a high isotropy of the interatomic distance between O and X around the Mn atom, and falls within a specific range, and thus has high characteristics as a positive electrode active material. I like thisBMn/ IAMnThe value of is 0.5 or more and 1.2 or less, preferably 0.6 or more and 1.2 or less, more preferably 0.7 or more and 1.2 or less, and even more preferably 1.0 or more. 1.2 or less, particularly preferably 1.1 or more and 1.2 or less.
In addition, the lithium composite metal oxide of the present embodiment has a high degree of isotropy in the interatomic distance between O and X around the Ni atom, and falls within a specific range, and thus has high characteristics as a positive electrode active material. . I like thisBNi/ IANiThe value of is 1.0 or more and 1.7 or less, preferably 1.1 or more and 1.7 or less, more preferably 1.2 or more and 1.7 or less.
These IBMn/ IAMnRange of values and IBNi/ IANiThe range of values can be arbitrarily combined.
Furthermore, IBMn/ IAMnAnd IBNi/ IANiProduct with (IBMn/ IAMn× IBNi/ IANi)) Has both the isotropicity of the interatomic distance between the O and the atom X around the appropriate Mn atom and the isotropicity of the interatomic distance between the O and the atom X around the proper Ni atom. 7 or more and 2.0 or less, preferably 0.9 or more and 2.0 or less, more preferably 1.1 or more and 2.0 or less.
The crystal structure of the lithium composite metal oxide of the present embodiment is preferably a layered structure, more preferably a hexagonal crystal structure or a monoclinic crystal structure.
The hexagonal crystal structure is P3, P31, P32, R3, P-3, R-3, P312, P321, P3112, P3121, P3212, P3221, R32, P3m1, P31m, P3c1, P31c, R3m, R3c, P-31m, P-31c, P-3m1, P-3c1, R-3m, R-3c, P6, P61, P65, P62, P64, P63, P-6, P6 / m, P63/ M, P622, P6122, P6522, P6222, P6422, P6322, P6mm, P6cc, P63cm, P63mc, P-6m2, P-6c2, P-62m, P-62c, P6 / mmm, P6 / mcc, P63/ Mcm, P63It is classified into any one space group selected from the group consisting of / mmc.
The monoclinic crystal structure is P2, P2.1, C2, Pm, Pc, Cm, Cc, P2 / m, P21/ M, C2 / m, P2 / c, P21/ C, C2 / c is classified into any one space group selected from the group consisting of C2 / c.
Among these, since the discharge capacity of the obtained nonaqueous electrolyte secondary battery is increased, the crystal structure of the lithium composite metal oxide is a hexagonal crystal structure classified into the space group R-3m, or C2 / m. A monoclinic crystal structure classified as follows is particularly preferable.
The space group of the lithium composite metal oxide of the present embodiment can be confirmed by the following method.
First, X-ray powder diffraction measurement was performed on a lithium composite metal oxide using CuKα as a radiation source and a diffraction angle 2θ measurement range of 10 ° to 90 °, and then Rietveld analysis was performed based on the results. And determining a crystal structure of the lithium composite metal oxide and a space group in the crystal structure. Rietveld analysis is a technique for analyzing the crystal structure of a material using diffraction peak data (diffraction peak intensity, diffraction angle 2θ) in powder X-ray diffraction measurement of the material, and is a conventionally used technique. (See, for example, “Practice of Powder X-ray Analysis—Introduction to Rietveld Method”, published on February 10, 2002, edited by the Japan Society for Analytical Chemistry X-ray Analysis Research Meeting).
The composition of the lithium composite metal oxide in this embodiment is such that the amount (mol) of Li is ALi, When the amount (mole) of metal other than Li is A, ALi/ A may be 0.7 or more and 1.4 or less.
The lithium composite metal oxide in the present embodiment preferably has a layered structure and the composition is represented by the following formula (1).
Li1 + x(Ni1-x-y-αMnyCoαMβ) O2... (1)
(In the formula (1), −0.3 ≦ x ≦ 0.4, 0.35 ≦ y ≦ 0.7, 0 <α ≦ 0.1, 0 ≦ β <0.1 (where 0 <α + β ≦ 0.1), −0.05 ≦ x + y + α + β <1, and M is selected from the group consisting of Al, Mg, Ti, Ca, Cu, Zn, Fe, Cr, Mo, Si, Sn, Nb and V One or more elements.)
The value of x in the formula (1) is −0.3 ≦ x ≦ 0.4, preferably −0.2 ≦ x ≦ 0.35, more preferably −0.1 ≦ x ≦ 0. .3.
Since the lithium composite metal oxide of the present embodiment has a high discharge capacity at 60 ° C., M is preferably Fe.
The lithium composite metal oxide particles of the present embodiment are used as a core material, and B, Al, Ga, In, Si, Ge, Sn, Mg, and the surface of the core material (lithium composite metal oxide particles) A compound containing one or more atoms selected from the group consisting of transition metals may be attached.
Among the above atoms, at least one selected from the group consisting of B, Al, Mg, Co, Cr, and Mn is preferable, and Al is more preferable because a uniform coating layer can be easily formed.
Examples of such a compound include oxides, fluorides, sulfides, hydroxides, oxyhydroxides, carbonates, nitrates, organic acid salts and mixtures thereof of the above atoms. Of these, oxides, hydroxides, oxyhydroxides or mixtures thereof are preferred.
As the compound to be deposited on the surface of the core material, alumina which is an oxide of Al is preferable.
[Method for producing lithium composite metal oxide]
Next, a method for producing the above-described lithium composite metal oxide will be described.
The method for producing a lithium composite metal oxide according to this embodiment includes the following steps (1) to (5).
(1) A step of obtaining a slurry by bringing an aqueous solution containing Ni ions, Mn ions and Co ions (hereinafter sometimes referred to as “raw material aqueous solution”) into contact with an alkali to form a coprecipitate.
(2) Separating the coprecipitate from the slurry obtained in (1)
(3) A step of mixing the coprecipitate obtained in (2) with a lithium compound.
(4) A step of heating the mixture obtained in (3) at a temperature of 200 ° C. or higher and 500 ° C. or lower in an atmosphere having an oxygen concentration of 5% by volume or higher.
(5) A step of firing the product obtained in (4) (hereinafter sometimes referred to as “calcined product”) at a temperature of 600 ° C. or higher and 950 ° C. or lower in an atmosphere having an oxygen concentration of less than 5% by volume.
Here, the “oxygen concentration” in the step (4) refers to an average oxygen concentration in the heat treatment space when the space (heat treatment space) for heating the mixture is in the range of 200 ° C. or more and 500 ° C. or less. Similarly, the “oxygen concentration” in step (5) refers to the average oxygen concentration in the heat treatment space when the space (heat treatment space) for calcining the calcined product is in the range of 600 ° C. or higher and 950 ° C. or lower.
(Process (1))
In the above step (1), the raw material aqueous solution can be adjusted by dissolving a compound containing Ni, Mn and Co in water. In particular, the raw material aqueous solution is preferably an aqueous solution obtained by dissolving Ni sulfate, Mn sulfate, and Co sulfate in water.
Further, when each raw material containing Ni, Mn and Co is difficult to dissolve in water, for example, when these raw materials are oxides, hydroxides, metal materials, these raw materials are mixed with sulfuric acid. The raw material aqueous solution can be obtained by dissolving in the aqueous solution.
The alkali used in step (1) includes LiOH (lithium hydroxide), NaOH (sodium hydroxide), KOH (potassium hydroxide), Li2CO3(Lithium carbonate), Na2CO3(Sodium carbonate), K2CO3(Potassium carbonate) and (NH4)2CO3One or more salts selected from the group consisting of (ammonium carbonate) can be mentioned. The alkali used may be an anhydride or a hydrate. An anhydride and a hydrate may be used in combination. In the step (1), it is preferable to use the above aqueous solution of alkali (alkaline aqueous solution). Aqueous ammonia can also be used as the alkaline aqueous solution.
The alkali concentration in the alkaline aqueous solution is preferably about 0.5 to 10 M (mol / L), more preferably about 1 to 8 M. Further, from the viewpoint of production cost, NaOH or KOH is preferable as the alkali to be used. NaOH and KOH may be used in combination.
As a contact method in the step (1), (i) a method in which an aqueous alkaline solution is added and mixed, (ii) a method in which an aqueous raw material solution is added and mixed, (iii) an aqueous raw material solution in water And a method of adding and mixing an alkaline aqueous solution. In mixing, it is preferable to involve stirring.
Of the contact methods in step (1), (ii) a method in which a raw material aqueous solution is added to and mixed with an alkaline aqueous solution is preferable because the change in pH is easily controlled. In the case of this method, the pH of the alkaline aqueous solution tends to decrease as the raw material aqueous solution is added to and mixed with the alkaline aqueous solution, but the pH is adjusted to 9 or higher, preferably 10 or higher. It is preferable to add a raw material aqueous solution. In addition, it is preferable that one or both of the raw material aqueous solution and the alkaline aqueous solution are brought into contact with each other while being kept at a temperature of 40 ° C. or higher and 80 ° C. or lower, whereby a coprecipitate having a more uniform composition can be obtained.
In the step (1), by bringing the raw material aqueous solution into contact with the alkali as described above, a salt containing Ni ions, Mn ions and Co ions is co-precipitated and the co-precipitate salt is dispersed. A slurry can be obtained.
(Process (2))
In step (2), a coprecipitate is obtained from the slurry obtained in step (1). As long as the coprecipitate can be obtained, various methods can be adopted as the method for obtaining the coprecipitate in step (2). However, since the operation is simple, a separation operation for obtaining a solid component such as filtration is performed. Is preferred. The coprecipitate can also be obtained by a method of volatilizing the liquid by heating, such as spray drying of the slurry.
When obtaining a coprecipitate in the step (2), it is preferable that the separated coprecipitate is washed and dried in the step (2). When the alkali remaining in the coprecipitate obtained by washing, Ni sulfate, Mn sulfate, or Co sulfate is used as a raw material, SO is released into the raw material aqueous solution.4 2-The amount of ions can be reduced. It is preferable to reduce these by washing because the amount of the inert flux (described later) can be easily controlled.
In order to efficiently wash the coprecipitate, it is preferable to use water as the washing liquid. If necessary, a water-soluble organic solvent such as alcohol or acetone may be added to the cleaning liquid. Moreover, you may perform washing | cleaning twice or more, for example, after washing with water, it can also wash | clean again with the organic solvent which has the above water solubility.
The drying of the washed coprecipitate can be performed by heat treatment, but may be performed by air drying, vacuum drying, or a combination thereof. When the heat treatment is performed, the heating temperature is preferably 50 to 300 ° C, more preferably about 100 to 200 ° C.
(Process (3))
In step (3), the coprecipitate obtained in step (2) and the lithium compound are mixed to obtain a mixture.
Examples of the lithium compound include one or more salts selected from the group consisting of lithium hydroxide, lithium chloride, lithium nitrate, and lithium carbonate. The lithium compound used may be an anhydride or a hydrate. Moreover, you may use an anhydride and a hydrate together.
Mixing may be either dry mixing or wet mixing, but dry mixing is preferred because of the ease of operation. Examples of the mixing apparatus include stirring and mixing, a V-type mixer, a W-type mixer, a ribbon mixer, a drum mixer, and a ball mill.
(Process (4))
In step (4), the mixture obtained in step (3) is heated at a temperature of 200 ° C. or higher and 500 ° C. or lower, preferably 250 ° C. or higher and 450 ° C. or lower to obtain a calcined product. The heating atmosphere includes a method using air and oxygen or a mixed gas thereof, a method of mixing an inert gas such as nitrogen and argon into the air and oxygen or a mixed gas thereof, and the oxygen concentration is 5 volumes. % Of the atmosphere. A high-capacity lithium composite metal oxide having an intended local structure can be easily obtained, and when the obtained lithium composite metal oxide is used as a positive electrode active material, a high-capacity secondary battery can be obtained. Therefore, the oxygen concentration is preferably 7% by volume to 20% by volume, and more preferably 10% by volume to 20% by volume.
(Process (5))
In step (5), the calcined product obtained in step (4) is fired at a temperature of 650 ° C. to 950 ° C., preferably 650 ° C. to 900 ° C. The firing atmosphere may be an atmosphere in which air, oxygen, nitrogen, argon, or the like is mixed and the oxygen concentration is less than 5% by volume. A high-capacity lithium composite metal oxide having an intended local structure can be easily obtained, and when the obtained lithium composite metal oxide is used as a positive electrode active material, a high-capacity secondary battery can be obtained. Therefore, the oxygen concentration is preferably 0.5% by volume or more and less than 5% by volume, and more preferably 1% by volume or more and 3% by volume or less.
In order to make the composition of the obtained lithium composite metal oxide uniform, the step (4) and the step (5) are continuously performed without lowering the temperature from the heating temperature at the end of the step (4) ( 5) is preferably performed. When performing the step (4) and the step (5) continuously, the oxygen concentration is adjusted to the step (4) while maintaining the temperature at the end of the step (4) or raising the temperature to the firing temperature of the step (5). The oxygen concentration in 4) is changed to the oxygen concentration in step (5). As a method for changing the oxygen concentration, a method of changing the oxygen concentration of the introduced gas is preferably used.
The lithium composite metal oxide of the present embodiment can be produced by such steps (1) to (5).
In addition, in the manufacturing method of this embodiment, although demonstrated as having a process (1)-a process (5), it is not restricted to this. For example, a mixture obtained by mixing a salt containing Ni ions, Mn ions and Co ions with a lithium compound by another method in place of steps (1) to (3) is prepared, and the obtained mixture is It is also possible to manufacture the lithium composite metal oxide of the present embodiment by heating while controlling the oxygen concentration and performing the processing corresponding to the above steps (4) and (5).
The above-mentioned “salt containing Ni ions, Mn ions and Co ions” may be a mixture of a salt containing Ni ions, a salt containing Mn ions, and a salt containing Co ions. As the above-mentioned “another method replacing step (1) to step (3)”, a method of mixing the above-mentioned salt in a solid phase, a slurry obtained by dispersing the above-mentioned salt in a liquid phase, For example, the slurry may be spray-dried and mixed.
Further, in the method for producing a lithium composite metal oxide according to the present embodiment, Ni, Mn and Co need to be contained in the mixture in the step (4). The metal atom may be contained. Examples of other metal atoms include one or more atoms selected from the group consisting of Al, Mg, Ti, Ca, Cu, Zn, Fe, Cr, Mo, Si, Sn, Nb, and V.
Various methods can be adopted as a method of including other metal atoms in the mixture to be heated in step (4). In particular, in the step (1), it is preferable to dissolve a water-soluble salt of another metal in the raw material aqueous solution because other metal atoms are uniformly dispersed in the resulting mixture.
(Inert flux)
In the step (4) and step (5), the mixture and calcined product may contain an inert flux. The inert flux is a salt that does not react with the target composite metal oxide and can be easily separated from the target. The inert flux melts at the heating temperature in step (4) and the firing temperature in step (5) to form a reaction field and promotes a uniform reaction. Therefore, when an inert flux is used, a product having a uniform composition is easily obtained.
As an inert flux, K2SO4, Na2SO4Sulfate such as K;2CO3, Na2CO3Carbonates such as: NaCl, KCl, NH4Chlorides such as Cl; LiF, NaF, KF, NH4Fluorides such as F; boric acid; Among the above-mentioned inert fluxes, sulfate is preferable because the production process becomes simple. More preferably K2SO4It is. Two or more inert fluxes can be used in combination.
When the mixture contains an inert flux, the reactivity at the time of heating the mixture and calcining the calcined product is improved, and thereby it may be possible to adjust the BET specific surface area of the obtained lithium composite metal oxide. is there. When the temperature is the same, the BET specific surface area of the oxide tends to increase as the content of the inert flux increases. Further, when an inert fluxing agent is contained during heating or firing, a uniform reaction can be performed, so that the local structure can be controlled at the atomic level of the lithium composite metal oxide by adjusting the heating atmosphere.
The inert flux may be mixed with the coprecipitate obtained by allowing the coprecipitate obtained by the separation operation in step (2) to contain the above inert flux solution and then drying. .
For example, when Ni sulfate, Mn sulfate, or Co sulfate is used as a raw material in step (1), SO4 2-Ions are liberated. This SO4 2-The ions and metal ions contained in the alkali used for the coprecipitation (for example, K ions when KOH is used as the alkali) remain in the coprecipitate separated in step (2), and the inert flux ( K in the above example2SO4) May occur. Therefore, the raw material aqueous solution after the coprecipitation in the step (1) is used as the “inert flux solution”, and the coprecipitate obtained in the step (2) is dried while the raw aqueous solution after the coprecipitation is included. By making it, an inert flux may be mixed with the coprecipitate obtained.
In addition, the inert flux can be added and mixed at the time of mixing the coprecipitate and the lithium compound in the step (3). Since it is easy to control the amount of the inert flux, the method of adding the inert flux in the step (3) is preferable to the method of adding the inert flux in the step (2). When an inert flux is added in step (3), the coprecipitate obtained in step (2) is washed, and the alkali, Ni salt, Mn salt, or Co salt remaining in the coprecipitate is washed. By reducing the amount of the derived anion, the amount of the inert flux can be easily controlled.
The inert flux may remain in the lithium composite metal oxide or may be removed by washing.
From the viewpoint of improving the uniformity of the reaction, the inert flux is a sulfate, and when the mixture or calcined product and the sulfate are mixed, the content of the sulfate in the resulting mixture is the lithium used. It is preferable that it is 0.01 to 400 mass parts with respect to 100 mass parts of compounds. More preferably, it is 0.1 to 10 parts by mass.
Further, the lithium composite metal oxide obtained by the method for producing a lithium composite metal oxide of the present embodiment may be pulverized using a ball mill or a jet mill. It may be possible to adjust the BET specific surface area of the lithium composite metal oxide by grinding. Further, the lithium composite metal oxide obtained by carrying out the steps (1) to (5) may be pulverized, and the steps (4) and (5) may be performed again to perform baking after the pulverization. . Furthermore, you may repeat a grinding | pulverization and baking by process (4), (5) twice or more as needed. Further, the lithium composite metal oxide can be washed or classified as necessary.
The lithium composite metal oxide of the present embodiment is preferably a mixture of primary particles having a particle size of 0.05 μm or more and 1 μm or less and secondary particles having a particle size of 2 μm or more and 100 μm or less formed by aggregation of the primary particles. It consists of. The particle diameters of the primary particles and secondary particles can be measured by observing with SEM.
The size of the secondary particles of the lithium composite metal oxide is preferably in the range of 2 μm to 50 μm, more preferably in the range of 2 μm to 10 μm, and even more preferably in the range of 3 μm to 8 μm. Especially preferably, it is the range of 3.5 micrometers or more and 7 micrometers or less. By these, the capacity | capacitance of the nonaqueous electrolyte secondary battery obtained increases more.
The primary particle size of the lithium composite metal oxide is preferably in the range of 0.08 μm to 0.8 μm, more preferably in the range of 0.10 μm to 0.7 μm, and still more preferably in the range of 0.8. It is in the range of 15 μm or more and 0.7 μm or less, and particularly preferably in the range of 0.2 μm or more and 0.5 μm or less. These increase the discharge capacity at a high current rate of the obtained nonaqueous electrolyte secondary battery.
Also, the average particle diameter of lithium composite metal oxide (D50) Is preferably in the range of 1 μm to 50 μm, more preferably in the range of 1.5 μm to 30 μm, even more preferably in the range of 2 μm to 20 μm, and particularly preferably in the range of 3 μm to 10 μm. It is. Accordingly, the density of the electrode using the lithium composite metal oxide is increased, and a high-capacity nonaqueous electrolyte secondary battery can be obtained.
Average particle size of lithium composite metal oxide (D50) Can be measured by the following method.
<Average particle diameter of lithium composite metal oxide (D50) Measurement>
0.1 g of the lithium composite metal oxide powder to be measured is put into 50 ml of a 0.2 mass% sodium hexametaphosphate aqueous solution to obtain a dispersion in which the powder is dispersed. About the obtained dispersion liquid, a particle size distribution is measured using the master sizer 2000 (laser diffraction scattering particle size distribution measuring apparatus) by Malvern, and a volume-based cumulative particle size distribution curve is obtained. In the obtained cumulative particle size distribution curve, the value of the particle size viewed from the fine particle side at the time of 50% accumulation is the average particle size (D50).
The BET specific surface area of the lithium composite metal oxide is preferably 0.1 m2/ G or more 20m2/ G or less, more preferably 0.5 m2/ G or more 15m2/ G or less, even more preferably 1 m2/ G or more 10m2/ G or less, particularly preferably 2 m2/ G or more 8m2/ G or less. These increase the discharge capacity at a high current rate of the obtained nonaqueous electrolyte secondary battery.
The BET specific surface area of the lithium composite metal oxide can be measured by the following method.
<Measurement of BET specific surface area of lithium composite metal oxide>
After 1 g of the lithium composite metal oxide powder to be measured is dried at 150 ° C. for 15 minutes in a nitrogen atmosphere, the measurement is performed using a flow sorb II 2300 manufactured by Micromeritics.
When the lithium composite metal oxide is used as a positive electrode active material for a non-aqueous electrolyte secondary battery, a non-aqueous electrolyte secondary battery showing a higher capacity than before can be obtained.
[Nonaqueous electrolyte secondary battery]
Next, while explaining the configuration of the nonaqueous electrolyte secondary battery, the positive electrode using the lithium composite metal oxide of the present embodiment as the positive electrode active material of the nonaqueous electrolyte secondary battery, and the nonaqueous electrolyte secondary having the positive electrode The battery will be described.
An example of the non-aqueous electrolyte secondary battery of the present embodiment includes a positive electrode, a negative electrode, a separator disposed between the positive electrode and the negative electrode, and an electrolytic solution.
FIG. 1 is a schematic view showing an example of the nonaqueous electrolyte secondary battery of the present embodiment. The cylindrical nonaqueous electrolyte
First, as shown in FIG. 1A, two
Next, as shown in FIG. 1B, after the electrode group 4 and an insulator (not shown) are accommodated in the battery can 5, the bottom of the can is sealed, and the electrode group 4 is impregnated with the electrolytic solution 6, An electrolyte is disposed between the negative electrode 3 and the negative electrode 3. Furthermore, the nonaqueous electrolyte
As the shape of the electrode group 4, for example, a columnar shape in which the cross-sectional shape when the electrode group 4 is cut in a direction perpendicular to the winding axis is a circle, an ellipse, a rectangle, or a rectangle with rounded corners. Can be mentioned.
In addition, as the shape of the nonaqueous electrolyte secondary battery having such an electrode group 4, a shape defined by IEC 60086 or JIS C 8500, which is a standard for batteries determined by the International Electrotechnical Commission (IEC), should be adopted. Can do. For example, cylindrical shape, square shape, etc. can be mentioned.
Furthermore, the non-aqueous electrolyte secondary battery is not limited to the above-described wound type configuration, and may have a stacked type configuration in which a stacked structure of a positive electrode, a separator, a negative electrode, and a separator is repeatedly stacked. Examples of the laminated nonaqueous electrolyte secondary battery include so-called coin-type batteries, button-type batteries, and paper-type (or sheet-type) batteries.
Hereinafter, each configuration will be described in order.
(Positive electrode)
The positive electrode of the present embodiment can be manufactured by first adjusting a positive electrode mixture containing a positive electrode active material, a conductive material and a binder, and supporting the positive electrode mixture on a positive electrode current collector.
(Positive electrode active material)
The positive electrode active material of the present embodiment has the above-described lithium composite metal oxide. By using the lithium composite metal oxide of this embodiment as a positive electrode active material of a non-aqueous electrolyte secondary battery, a non-aqueous electrolyte secondary battery exhibiting a high capacity can be obtained.
(Conductive material)
A carbon material can be used as a conductive material included in the positive electrode of the present embodiment. Examples of the carbon material include graphite powder, carbon black (for example, acetylene black), and a fibrous carbon material. Since carbon black is fine and has a large surface area, adding a small amount to the positive electrode mixture can improve the conductivity inside the positive electrode and improve the charge / discharge efficiency and output characteristics. Both the binding force between the positive electrode mixture and the positive electrode current collector and the binding force inside the positive electrode mixture are reduced, which causes an increase in internal resistance.
The proportion of the conductive material in the positive electrode mixture is preferably 5 parts by mass or more and 20 parts by mass or less with respect to 100 parts by mass of the positive electrode active material. When a fibrous carbon material such as graphitized carbon fiber or carbon nanotube is used as the conductive material, this ratio can be lowered.
(binder)
A thermoplastic resin can be used as the binder of the positive electrode of the present embodiment. Examples of the thermoplastic resin include polyvinylidene fluoride (hereinafter sometimes referred to as PVdF), polytetrafluoroethylene (hereinafter sometimes referred to as PTFE), tetrafluoroethylene, hexafluoropropylene, and vinylidene fluoride. And fluororesins such as copolymers, propylene hexafluoride / vinylidene fluoride copolymers, tetrafluoroethylene / perfluorovinyl ether copolymers; polyolefin resins such as polyethylene and polypropylene.
These thermoplastic resins may be used as a mixture of two or more. By using a fluororesin and a polyolefin resin as a binder, the ratio of the fluororesin to the whole positive electrode mixture is 1% by mass or more and 10% by mass or less, and the ratio of the polyolefin resin is 0.1% by mass or more and 2% by mass or less. A positive electrode mixture having both high adhesion to the current collector and high bonding strength inside the positive electrode mixture can be obtained.
(Positive electrode current collector)
As the positive electrode current collector included in the positive electrode of the present embodiment, a band-shaped member made of a metal material such as Al, Ni, stainless steel or the like can be used. Among these, a material that is made of Al and formed into a thin film is preferable because it is easy to process and inexpensive.
Examples of the method of supporting the positive electrode mixture on the positive electrode current collector include a method of pressure-molding the positive electrode mixture on the positive electrode current collector. Further, the positive electrode mixture is made into a paste using an organic solvent, and the obtained positive electrode mixture paste is applied to at least one surface of the positive electrode current collector, dried, pressed and fixed, whereby the positive electrode current collector is bonded to the positive electrode current collector. A mixture may be supported.
When pasting the positive electrode mixture, organic solvents that can be used include amine solvents such as N, N-dimethylaminopropylamine and diethylenetriamine; ether solvents such as tetrahydrofuran; ketone solvents such as methyl ethyl ketone; methyl acetate And amide solvents such as dimethylacetamide and N-methyl-2-pyrrolidone (hereinafter sometimes referred to as NMP).
Examples of the method of applying the positive electrode mixture paste to the positive electrode current collector include a slit die coating method, a screen coating method, a curtain coating method, a knife coating method, a gravure coating method, and an electrostatic spray method.
The positive electrode can be manufactured by the methods mentioned above.
(Negative electrode)
The negative electrode included in the nonaqueous electrolyte secondary battery of this embodiment is only required to be able to dope and dedope lithium ions at a lower potential than the positive electrode, and the negative electrode mixture containing the negative electrode active material is supported on the negative electrode current collector. And an electrode composed of the negative electrode active material alone.
(Negative electrode active material)
Examples of the negative electrode active material possessed by the negative electrode include carbon materials, chalcogen compounds (oxides, sulfides, etc.), nitrides, metals, and alloys that can be doped and dedoped with lithium ions at a lower potential than the positive electrode. It is done.
Examples of carbon materials that can be used as the negative electrode active material include graphites such as natural graphite and artificial graphite, cokes, carbon black, pyrolytic carbons, carbon fibers, and organic polymer compound fired bodies.
As an oxide that can be used as a negative electrode active material, SiO2, SiO etc. formula SiOx(Wherein x is a positive real number) silicon oxide represented by: TiO2TiO, formula TiOx(Where x is a positive real number) titanium oxide; V2O5, VO2Etc. VOx(Where x is a positive real number) oxide of vanadium; Fe3O4, Fe2O3FeO and other formulas FeOx(Where x is a positive real number) iron oxide; SnO2, SnO etc. formula SnOx(Where x is a positive real number) tin oxide represented by WO3, WO2General formula WOx(Where x is a positive real number)4Ti5O12, LiVO2And a composite metal oxide containing lithium and titanium or vanadium.
As a sulfide that can be used as a negative electrode active material, Ti2S3TiS2TiS and other formula TiSx(Where x is a positive real number) titanium sulfide; V3S4, VS2,VS formula VSx(Where x is a positive real number) Vanadium sulfide; Fe3S4, FeS2FeS and other formulasx(Where x is a positive real number) iron sulfide; Mo2S3, MoS2Etc. MoSx(Where x is a positive real number) molybdenum sulfide represented by SnS2,SnS etc. formula SnSx(Where x is a positive real number) tin sulfide represented by WS2Formula WSx(Where x is a positive real number) tungsten sulfide represented by: Sb2S3Etc. SbSx(Where x is a positive real number) antimony sulfide; Se5S3, SeS2, SeS etc. formula SeSxSelenium sulfide represented by (where x is a positive real number).
Nitrides that can be used as negative electrode active materials include Li3N, Li3-xAxA lithium-containing nitride such as N (where A is one or both of Ni and Co, and 0 <x <3) can be given.
These carbon materials, oxides, sulfides and nitrides may be used alone or in combination of two or more. These carbon materials, oxides, sulfides and nitrides may be crystalline or amorphous.
Further, examples of the metal that can be used as the negative electrode active material include lithium metal, silicon metal, and tin metal.
Examples of alloys that can be used as the negative electrode active material include lithium alloys such as Li—Al, Li—Ni, Li—Si, Li—Sn, and Li—Sn—Ni; silicon alloys such as Si—Zn; Sn—Mn and Sn. -Tin alloys such as Co, Sn-Ni, Sn-Cu, Sn-La; Cu2Sb, La3Ni2Sn7And alloys such as:
These metals and alloys are mainly used alone as electrodes after being processed into a foil shape, for example.
Among the negative electrode active materials, the potential of the negative electrode hardly changes from the uncharged state to the fully charged state during charging (potential flatness is good), the average discharge potential is low, and the capacity retention rate when repeatedly charged and discharged is high. For reasons such as high (good cycle characteristics), a carbon material mainly composed of graphite such as natural graphite or artificial graphite is preferably used. The shape of the carbon material may be any of a flake shape such as natural graphite, a spherical shape such as mesocarbon microbeads, a fibrous shape such as graphitized carbon fiber, or an aggregate of fine powder.
The negative electrode mixture may contain a binder as necessary. Examples of the binder include thermoplastic resins, and specific examples include PVdF, thermoplastic polyimide, carboxymethyl cellulose, polyethylene, and polypropylene.
(Negative electrode current collector)
Examples of the negative electrode current collector of the negative electrode include a band-shaped member made of a metal material such as Cu, Ni, and stainless steel. In particular, it is preferable to use Cu as a forming material and process it into a thin film from the viewpoint that it is difficult to make an alloy with lithium and it is easy to process.
As a method of supporting the negative electrode mixture on such a negative electrode current collector, as in the case of the positive electrode, a method using pressure molding, pasting with a solvent, etc., applying to the negative electrode current collector, drying and pressing. The method of crimping is mentioned.
(Separator)
Examples of the separator included in the nonaqueous electrolyte secondary battery of the present embodiment include a porous film, a nonwoven fabric, a woven fabric, and the like made of a material such as a polyolefin resin such as polyethylene and polypropylene, a fluororesin, and a nitrogen-containing aromatic polymer. A material having the following form can be used. Moreover, a separator may be formed by using two or more of these materials, or a separator may be formed by laminating these materials.
Examples of the separator include separators described in JP 2000-30686 A, JP 10-324758 A, and the like. The thickness of the separator should be as thin as possible as long as the mechanical strength is maintained because the volume energy density of the battery is increased and the internal resistance is reduced, preferably about 5 to 200 μm, more preferably about 5 to 40 μm. is there.
The separator preferably has a porous film containing a thermoplastic resin. In a nonaqueous electrolyte secondary battery, when an abnormal current flows in the battery due to a short circuit between the positive electrode and the negative electrode, the current at the short circuit point is interrupted to prevent an excessive current from flowing (shut down). It preferably has a function. Here, the shutdown is performed by overheating the separator at the short-circuit location due to a short circuit, and when the temperature exceeds a presumed operating temperature, the porous film in the separator is softened or melted to close the micropores. And even if the temperature in a battery rises to a certain high temperature after a separator shuts down, it is preferable to maintain the shut-down state, without breaking at the temperature.
Examples of such a separator include a laminated film in which a heat resistant porous layer and a porous film are laminated. By using such a laminated film as a separator, the heat resistance of the secondary battery in this embodiment can be further increased. In the laminated film, the heat resistant porous layer may be laminated on both surfaces of the porous film.
(Laminated film)
Hereinafter, a laminated film in which the heat resistant porous layer and the porous film are laminated to each other will be described.
In the laminated film used as the separator of the nonaqueous electrolyte secondary battery of the present embodiment, the heat resistant porous layer is a layer having higher heat resistance than the porous film. The heat resistant porous layer may be formed from an inorganic powder (first heat resistant porous layer), may be formed from a heat resistant resin (second heat resistant porous layer), and includes a heat resistant resin and a filler. (A third heat-resistant porous layer). When the heat resistant porous layer contains a heat resistant resin, the heat resistant porous layer can be formed by an easy technique such as coating.
(First heat-resistant porous layer)
When the heat resistant porous layer is formed of an inorganic powder, examples of the inorganic powder used for the heat resistant porous layer include inorganic substances such as metal oxides, metal nitrides, metal carbides, metal hydroxides, carbonates, and sulfates. Among these, a powder made of an inorganic substance having low conductivity (insulator) is preferably used. Specific examples include powders made of alumina, silica, titanium dioxide, calcium carbonate, or the like. Such inorganic powders may be used alone or in combination of two or more.
Among these inorganic powders, alumina powder is preferable because of its high chemical stability. More preferably, all of the particles constituting the inorganic powder are alumina particles, all of the particles constituting the inorganic powder are alumina particles, and part or all of them are substantially spherical alumina particles. preferable.
(Second heat resistant porous layer)
When the heat resistant porous layer is formed from a heat resistant resin, the heat resistant resin used for the heat resistant porous layer is polyamide, polyimide, polyamideimide, polycarbonate, polyacetal, polysulfone, polyphenylene sulfide, polyether ketone, aromatic polyester, polyether. Mention may be made of sulfone and polyetherimide. In order to further increase the heat resistance of the laminated film, polyamide, polyimide, polyamideimide, polyethersulfone and polyetherimide are preferable, and polyamide, polyimide or polyamideimide is more preferable.
More preferably, the heat-resistant resin used for the heat-resistant porous layer is a nitrogen-containing aromatic polymer such as aromatic polyamide (para-oriented aromatic polyamide, meta-oriented aromatic polyamide), aromatic polyimide, aromatic polyamideimide, Aromatic polyamides are preferred, and para-oriented aromatic polyamides (hereinafter sometimes referred to as para-aramids) are particularly preferred because they are easy to produce.
Also, examples of the heat resistant resin include poly-4-methylpentene-1 and a cyclic olefin polymer.
By using these heat resistant resins, the heat resistance of the laminated film used as the separator of the nonaqueous electrolyte secondary battery, that is, the thermal film breaking temperature of the laminated film can be further increased. Among these heat-resistant resins, when a nitrogen-containing aromatic polymer is used, the compatibility with the electrolytic solution, that is, the liquid retention in the heat-resistant porous layer may be improved depending on the polarity in the molecule. The rate of impregnation with the electrolytic solution during the production of the electrolyte secondary battery is also high, and the charge / discharge capacity of the nonaqueous electrolyte secondary battery is further increased.
The thermal film breaking temperature of such a laminated film depends on the type of heat-resistant resin, and is selected and used according to the use scene and purpose of use. More specifically, as the heat-resistant resin, when the nitrogen-containing aromatic polymer is used, the cyclic olefin polymer is about 400 ° C. When using, the thermal film breaking temperature can be controlled to about 300 ° C., respectively. In addition, when the heat resistant porous layer is made of an inorganic powder, the thermal film breaking temperature can be controlled to, for example, 500 ° C. or higher.
The para-aramid is obtained by polycondensation of a para-oriented aromatic diamine and a para-oriented aromatic dicarboxylic acid halide, and the amide bond is in the para position of the aromatic ring or an oriented position equivalent thereto (for example, 4,4′-biphenylene, It consists essentially of repeating units that are bound together in the opposite orientation, such as 1,5-naphthalene, 2,6-naphthalene, etc., in an orientation that extends coaxially or parallelly. Specifically, poly (paraphenylene terephthalamide), poly (parabenzamide), poly (4,4′-benzanilide terephthalamide), poly (paraphenylene-4,4′-biphenylenedicarboxylic acid amide), poly ( Para-aligned or para-oriented such as paraphenylene-2,6-naphthalenedicarboxylic acid amide), poly (2-chloro-paraphenylene terephthalamide), paraphenylene terephthalamide / 2,6-dichloroparaphenylene terephthalamide copolymer Examples include para-aramid having a structure according to the type.
The aromatic polyimide is preferably a wholly aromatic polyimide produced by polycondensation of an aromatic dianhydride and a diamine.
Specific examples of the aromatic dianhydride used for the polycondensation include pyromellitic dianhydride, 3,3 ′, 4,4′-diphenylsulfonetetracarboxylic dianhydride, 3,3 ′, 4. 4,4′-benzophenone tetracarboxylic dianhydride, 2,2′-bis (3,4-dicarboxyphenyl) hexafluoropropane and 3,3 ′, 4,4′-biphenyltetracarboxylic dianhydride. It is done.
Specific examples of diamines used for polycondensation include oxydianiline, paraphenylenediamine, benzophenonediamine, 3,3′-methylenedianiline, 3,3′-diaminobenzophenone, 3,3′-diaminodiphenylsulfone. And 1,5-naphthalenediamine.
Also, as the aromatic polyimide, a polyimide soluble in a solvent can be suitably used. Examples of such a polyimide include a polycondensate polyimide of 3,3 ′, 4,4′-diphenylsulfonetetracarboxylic dianhydride and an aromatic diamine.
Examples of the aromatic polyamideimide include those obtained from polycondensation of aromatic dicarboxylic acid and aromatic diisocyanate, and those obtained from polycondensation of aromatic diacid anhydride and aromatic diisocyanate. Specific examples of the aromatic dicarboxylic acid include isophthalic acid and terephthalic acid. A specific example of the aromatic dianhydride is trimellitic anhydride. Specific examples of the aromatic diisocyanate include 4,4'-diphenylmethane diisocyanate, 2,4-tolylene diisocyanate, 2,6-tolylene diisocyanate, orthotolylene diisocyanate, and m-xylene diisocyanate.
In order to further enhance ion permeability, the thickness of the heat resistant porous layer of the laminated film is preferably 1 μm or more and 10 μm or less, more preferably 1 μm or more and 5 μm or less, and particularly preferably 1 μm or more and 4 μm or less. . The heat-resistant porous layer has fine pores, and the size (diameter) of the pores is preferably 3 μm or less, more preferably 1 μm or less.
(Third heat-resistant porous layer)
Further, when the heat resistant porous layer is formed including a heat resistant resin and a filler, the same heat resistant resin as that used for the second heat resistant porous layer can be used. As the filler, one or more selected from the group consisting of organic powder, inorganic powder, or a mixture thereof can be used. The particles constituting the filler preferably have an average particle size of 0.01 μm or more and 1 μm or less.
Examples of the organic powder that can be used as the filler include, for example, styrene, vinyl ketone, acrylonitrile, methyl methacrylate, ethyl methacrylate, glycidyl methacrylate, glycidyl acrylate, methyl acrylate, and the like, or two or more types of copolymers; Fluorine resin such as tetrafluoroethylene-6-propylene copolymer, tetrafluoroethylene-ethylene copolymer, polyvinylidene fluoride; melamine resin; urea resin; polyolefin resin; polymethacrylate; A powder is mentioned. Such organic powders may be used alone or in combination of two or more. Among these organic powders, PTFE powder is preferred because of its high chemical stability.
Examples of the inorganic powder that can be used as the filler include the same inorganic powder used in the heat-resistant porous layer.
When the heat-resistant porous layer is formed including a heat-resistant resin and a filler, the filler content depends on the specific gravity of the filler material, for example, when all of the particles constituting the filler are alumina particles In addition, when the total mass of the heat resistant porous layer is 100 parts by mass, the mass of the filler is preferably 5 parts by mass or more and 95 parts by mass or less, more preferably 20 parts by mass or more and 95 parts by mass or less, Preferably they are 30 to 90 mass parts. These ranges can be appropriately set depending on the specific gravity of the filler material.
Examples of the shape of the filler include substantially spherical, plate-like, columnar, needle-like, and fiber-like shapes, and any particle can be used. However, since it is easy to form uniform pores, Preferably there is. Examples of the substantially spherical particles include particles having a particle aspect ratio (long particle diameter / short particle diameter) of 1 or more and 1.5 or less. The aspect ratio of the particles can be measured by an electron micrograph.
In the laminated film used as the separator of the nonaqueous electrolyte secondary battery of this embodiment, the porous film preferably has fine pores and has a shutdown function. In this case, the porous film contains a thermoplastic resin.
The size of the micropores in the porous film is preferably 3 μm or less, more preferably 1 μm or less. The porosity of the porous film is preferably 30% to 80% by volume, more preferably 40% to 70% by volume. In a non-aqueous electrolyte secondary battery, when the presumed operating temperature is exceeded, the porous film containing the thermoplastic resin has micropores due to softening or melting of the thermoplastic resin constituting the porous film. Can be occluded.
What is necessary is just to select the thermoplastic resin used for a porous film what does not melt | dissolve in the electrolyte solution in a nonaqueous electrolyte secondary battery. Specific examples include polyolefin resins such as polyethylene and polypropylene, and thermoplastic polyurethane resins, and a mixture of two or more of these may be used.
In order for the separator to soften and shut down at a lower temperature, the porous film preferably contains polyethylene. Examples of the polyethylene include polyethylene such as low density polyethylene, high density polyethylene, and linear polyethylene, and ultra high molecular weight polyethylene having a molecular weight of 1,000,000 or more.
In order to further increase the puncture strength of the porous film, it is preferable that the thermoplastic resin constituting the porous film contains at least ultra high molecular weight polyethylene. In addition, in terms of production of the porous film, the thermoplastic resin may preferably contain a wax made of polyolefin having a low molecular weight (weight average molecular weight of 10,000 or less).
Further, the thickness of the porous film in the laminated film is preferably 3 μm or more and 30 μm or less, more preferably 3 μm or more and 25 μm or less. Moreover, in this embodiment, the thickness of a laminated film becomes like this. Preferably it is 40 micrometers or less, More preferably, it is 30 micrometers or less. Moreover, when the thickness of the heat resistant porous layer is A (μm) and the thickness of the porous film is B (μm), the value of A / B is preferably 0.1 or more and 1 or less.
In the present embodiment, the separator allows the electrolyte to permeate well when the battery is used (during charging / discharging). Or less, more preferably 50 seconds / 100 cc or more and 200 seconds / 100 cc or less.
The porosity of the separator is preferably 30% by volume to 80% by volume, more preferably 40% by volume to 70% by volume. The separator may be a laminate of separators having different porosity.
(Electrolyte)
The electrolyte solution included in the nonaqueous electrolyte secondary battery of this embodiment contains an electrolyte and an organic solvent.
As the electrolyte contained in the electrolyte, LiClO4, LiPF6, LiAsF6, LiSbF6, LiBF4, LiCF3SO3, LiN (SO2CF3)2, LiN (SO2C2F5)2, LiN (SO2CF3) (COCF3), Li (C4F9SO3), LiC (SO2CF3)3, Li2B10Cl10, LiBOB (where BOB is bis (oxalato) borate), lower aliphatic carboxylic acid lithium salt, LiAlCl4And a mixture of two or more of these may be used. Among them, as an electrolyte, LiPF containing fluorine6, LiAsF6, LiSbF6, LiBF4, LiCF3SO3, LiN (SO2CF3)2And LiC (SO2CF3)3It is preferable to use at least one selected from the group consisting of:
Examples of the organic solvent contained in the electrolyte include propylene carbonate, ethylene carbonate, dimethyl carbonate, diethyl carbonate, ethyl methyl carbonate, 4-trifluoromethyl-1,3-dioxolan-2-one, and 1,2-di- Carbonates such as (methoxycarbonyloxy) ethane; 1,2-dimethoxyethane, 1,3-dimethoxypropane, pentafluoropropyl methyl ether, 2,2,3,3-tetrafluoropropyl difluoromethyl ether, tetrahydrofuran, 2- Ethers such as methyltetrahydrofuran; Esters such as methyl formate, methyl acetate and γ-butyrolactone; Nitriles such as acetonitrile and butyronitrile; N, N-dimethylformamide, N, N-dimethyla Amides such as toamide; carbamates such as 3-methyl-2-oxazolidone; sulfur-containing compounds such as sulfolane, dimethyl sulfoxide and 1,3-propane sultone, or those obtained by further introducing a fluoro group into these organic solvents ( One obtained by substituting one or more hydrogen atoms in the organic solvent with fluorine atoms can be used.
It is preferable to use a mixture of two or more of these as the organic solvent. Among them, a mixed solvent containing carbonates is preferable, and a mixed solvent of cyclic carbonate and acyclic carbonate and a mixed solvent of cyclic carbonate and ether are more preferable. As a mixed solvent of cyclic carbonate and acyclic carbonate, a mixed solvent containing ethylene carbonate, dimethyl carbonate and ethyl methyl carbonate is preferable. The electrolyte using such a mixed solvent has a wide operating temperature range, hardly deteriorates even when charged and discharged at a high current rate, hardly deteriorates even when used for a long time, and natural graphite as an active material of the negative electrode. Even when a graphite material such as artificial graphite is used, it has the advantage of being hardly decomposable.
Also, as the electrolytic solution, since the safety of the obtained nonaqueous electrolyte secondary battery is increased, LiPF6It is preferable to use an electrolytic solution containing a lithium salt containing fluorine and an organic solvent having a fluorine substituent. A mixed solvent containing dimethyl carbonate and ethers having fluorine substituents such as pentafluoropropyl methyl ether and 2,2,3,3-tetrafluoropropyl difluoromethyl ether is capable of capacity even when charging / discharging at a high current rate. Since the maintenance rate is high, it is more preferable.
A solid electrolyte may be used instead of the above electrolyte. As the solid electrolyte, for example, an organic polymer electrolyte such as a polyethylene oxide polymer compound, a polymer compound containing at least one of a polyorganosiloxane chain or a polyoxyalkylene chain can be used. Moreover, what is called a gel type which hold | maintained the non-aqueous electrolyte in the high molecular compound can also be used. Li2S-SiS2, Li2S-GeS2, Li2SP2S5, Li2SB2S3, Li2S-SiS2-Li3PO4, Li2S-SiS2-Li2SO4An inorganic solid electrolyte containing a sulfide such as may be used. By using these solid electrolytes, the safety of the nonaqueous electrolyte secondary battery may be further improved.
In the nonaqueous electrolyte secondary battery of this embodiment, when a solid electrolyte is used, the solid electrolyte may serve as a separator, and in that case, the separator may not be required.
Since the above-described positive electrode active material uses the above-described lithium composite metal oxide of the present embodiment, the nonaqueous electrolyte secondary battery using the positive electrode active material can exhibit a higher capacity than before.
Further, since the positive electrode has a positive electrode active material using the above-described lithium composite metal oxide of the present embodiment, the non-aqueous electrolyte secondary battery can exhibit a higher capacity than before.
Furthermore, since the nonaqueous electrolyte secondary battery has the positive electrode described above, it exhibits a higher capacity than before.
次に、本発明を実施例によりさらに詳細に説明する。
本実施例においては、リチウム複合金属酸化物(正極活物質)の評価、正極およびリチウム二次電池の作製評価を、次のようにして行った。
(1)リチウム複合金属酸化物の評価
1.リチウム複合金属酸化物の組成分析
リチウム複合金属酸化物の組成分析は、得られたリチウム複合金属酸化物の粉末を塩酸に溶解させた後、誘導結合プラズマ発光分析装置(エスアイアイ・ナノテクノロジー株式会社製、SPS3000)を用いて行った。
2.リチウム複合金属酸化物の粉末X線回折測定
リチウム複合金属酸化物の粉末X線回折測定は、粉末X線回折装置(株式会社リガク製、RINT2500TTR、試料水平型)を用いて行った。得られたリチウム複合金属酸化物を専用の基板に充填し、CuKα線源を用いて、回折角2θ=10°~90°の範囲にて測定を行うことで、粉末X線回折図形を得た。
また、粉末X線回折図形のリートベルト解析は、解析プログラムRIETAN−2000(F.Izumi and T.Ikeda,Mater.Sci.Forum,321−324(2000)198を参照)により行い、リチウム複合金属酸化物が有する結晶構造の空間群を求めた。
3.リチウム複合金属酸化物のEXAFS測定および解析
(X線吸光度の測定、X線吸収スペクトルの作成)
リチウム複合金属酸化物のX線吸収スペクトルは、高エネルギー加速器研究機構の物質構造科学研究所が所有する放射光科学研究施設であるビームライン9C(BL−9C)のXAFS測定装置を用いてX線吸光度を測定し、得られたX線吸光度の値を用いて作成した。
X線吸光度の測定においては、Si(111)二結晶分光器を用いた。また、MnのK吸収端に対応する波長のX線の吸光度測定に関しては、高次項除去のためデチューニングを60%で行った。
X線吸光度の測定において、入射X線強度(I0)は、充填ガスとしてN2を使用した17cmのイオンチェンバーを用いて常温下で測定し、透過X線強度(It)は、充填ガスとしてN2を使用した31cmのイオンチェンバーを用いて常温下で測定した。
測定したエネルギー範囲および測定点数は、MnのK吸収端については、6040eVから7640.5eVまで等エネルギー間隔で4932点であった。また、NiのK吸収端については、7834eVから9434.5eVまで等エネルギー間隔で5333点であった。
なお、エネルギーの校正は、銅単体を標準試料として用いて測定したときに、得られるK吸収端のX線吸収端近傍構造(X−ray Absorption Near−Edge Structure)スペクトルについて、プレエッジピーク(約8980eV)における分光結晶の角度を12.7185°として行った。
X線吸光度の測定においては、各入射X線エネルギーにおいて、I0、Itを測定し、次式により、X線吸光度を求めた。
X線吸光度 μt=−ln(I0/It)
上記測定では、測定に用いるX線の波長に対応して(測定に用いるX線のエネルギーに対応して)離散的なX線吸光度が得られる。得られたX線吸光度について次のように平均処理およびデータ補間を行った。
まず、MnのK吸収端に対応する範囲のX線吸光度については、下記の方法で平均処理を行った。
6040eVから6400eVまで:21点の隣接平均処理を3回
6400eVから6700eVまで:7点のSavitzky−Golay法による重みつき平均処理を1回
6700eVから7640.5eVまで:11点の隣接平均処理を5回
次いで、得られたX線吸光度の平均値を用いて、下記のデータ間隔でデータ補間を行い、X軸をX線のエネルギー、Y軸をX吸光度とするX線吸収スペクトルを得た。
6040eVから6508eVまで:6.5eV間隔
6508eVから6609.5eVまで:0.35eV間隔
6609.5eVから6640.5eVまで:1eV間隔
6640.5eVから7040.5eVまで:2.5eV間隔
7040.5eVから7640.5eVまで:6eV間隔
また、NiのK吸収端に対応する範囲のX線吸光度については、下記の方法で平均処理を行った。
7834eVから8200eVまで:21点の隣接平均処理を3回
8200eVから8500eVまで:7点のSavitzky−Golay法による重みつき平均処理を1回
8500eVから9734.5eVまで:11点の隣接平均処理を5回
次いで、得られたX線吸光度の平均値を用いて、下記のデータ間隔でデータ補間を行い、X軸をX線のエネルギー、Y軸をX吸光度とするX線吸収スペクトルを得た。
7834eVから8302eVまで:6.5eV間隔
8302eVから8403.5eVまで:0.35eV間隔
8403.5eVから8439.5eVまで:1eV間隔
8439.5eVから8834.5eVまで:2.5eV間隔
8834.5eVから9434.5eVまで:6eV間隔
(EXAFSスペクトルの作成)
得られたX線吸収スペクトルから、次のようにして、MnのK吸収端およびNiのK吸収端のEXAFSスペクトルを得た。上記により得られたX線吸収スペクトルの解析は、解析ソフト(株式会社リガク製、REX2000)を用いて行った。
まず、MnのK吸収端E0は、X線吸収スペクトルにおけるMnのK吸収端付近において、一次微分係数が最大となるエネルギー値とした。同様に、NiのK吸収端E0は、NiのK吸収端付近のスペクトルにおいて、一次微分係数が最大となるエネルギー値とした。
また、スペクトルのバックグランドは、前記のMnのK吸収端およびNiのK吸収端よりも低エネルギー域のスペクトルにVictoreenの式(Aλ3−Bλ4+C;λは入射X線の波長、A,B,Cは任意の定数)を最小自乗法で当てはめて決定した。このVictoreenの式に対応するバックグラウンドの値を、X線吸収スペクトルから差し引くことで、EXAFSスペクトルを得た。
(動径分布関数の算出)
得られたEXAFSスペクトルから動径分布関数を得た。
まず、EXAFSスペクトルについて、Spline Smoothing法(平滑化スプライン法)により孤立原子の吸光度(μ0)を見積もり、EXAFS関数χ(k)を抽出した。なお、kは0.5123×(E−E0)1/2で定義される光電子の波数であり、kの単位はÅ−1である。
次いで、k3で重み付けしたEXAFS関数k3χ(k)について、kが3.0から11.85Å−1の範囲でフーリエ変換して動径分布関数を求めた。
(2)正極の作製
後述する製造方法で得られるリチウム複合金属酸化物(正極活物質)と導電材(アセチレンブラック:黒鉛=9:1(質量比))とバインダー(PVdF)とを、正極活物質:導電材:バインダー=87:10:3(質量比)の組成となるように加えて混練することにより、ペースト状の正極合剤を調製した。正極合剤の調製時には、N−メチル−2−ピロリドンを有機溶媒として用いた。
得られた正極合剤を、集電体となる厚さ40μmのAl箔に塗布して150℃で8時間真空乾燥を行い、正極を得た。
(3)非水電解質二次電池(コインセル)の作製
以下の操作を、アルゴン雰囲気のグローブボックス内で行った。
「(2)正極の作製」で作成した正極を、コイン型電池R2032用のコインセル(宝泉株式会社製)の下蓋にアルミ箔面を下に向けて置き、その上に積層フィルムセパレータ(ポリエチレン製多孔質フィルムの上に、耐熱多孔層を積層したセパレータ(厚み16μm))を置いた。ここに電解液を300μl注入した。用いた電解液は、エチレンカーボネートとジメチルカーボネートとエチルメチルカーボネートとの30:35:35(体積比)混合液に、LiPF6を1mol/lとなるように溶解して調製した。
次に、負極として金属リチウムを用いて、前記金属リチウムを積層フィルムセパレータの上側に置き、ガスケットを介して上蓋をし、かしめ機でかしめて非水電解質二次電池(コイン型電池R2032。以下、「コイン型電池」と称することがある。)を作製した。
(4)充放電試験
「(3)非水電解質二次電池(コインセル)の作製」で作成したコイン型電池を用いて、以下に示す条件で充放電試験を実施した。充放電試験における、充電容量および放電容量をそれぞれ以下のようにして求めた。
<充放電試験条件>
試験温度:25℃または60℃
充電時条件:充電最大電圧4.3V、充電時間10時間、充電電流0.3mA/cm2
放電時条件:放電最小電圧2.5V、放電時間10時間、放電電流0.3mA/cm2
(実施例1)
1.リチウム複合金属酸化物前駆体(共沈物)の製造
硫酸ニッケル六水和物、硫酸マンガン一水和物、硫酸コバルト七水和物を、Ni:Mn:Coのモル比が0.45:0.45:0.10となるようにそれぞれ秤量し、純水に溶解してNiイオン、Mnイオン、CoイオンおよびSO4 2−イオンを含有する遷移金属水溶液を得た。
この遷移金属水溶液に、水酸化カリウム水溶液を加えて共沈を行い、沈殿物を生成させて、スラリーを得た。得られたスラリーについて、固液分離を行い、蒸留水により洗浄し、100℃で8時間乾燥させて共沈物Q1を得た。
2.リチウム複合金属酸化物の製造
得られた共沈物Q1と、共沈物Q1に含まれる遷移金属の合計量(モル)1に対して、Liの量(モル)が1.3となるように秤量した炭酸リチウムと、不活性融剤として硫酸カリウムとを乳鉢により混合して混合物を得た。
次いで、得られた混合物をアルミナ製焼成容器に入れ、このアルミナ製焼成容器を電気炉に入れた。電気炉内部に元々存在する大気雰囲気および導入する窒素ガスを用いて、酸素濃度を8.5体積%に調整し、400℃で加熱して仮焼物を得た。さらに酸素濃度を1体積%に調整し、850℃で6時間保持して焼成を行い、室温まで冷却して焼成物を得た。
得られた焼成物を粉砕し、蒸留水に分散させた。静置後の上澄みをデカンテーションで除去した後、ろ過し、300℃で6時間乾燥して粉末状のリチウム複合金属酸化物A1を得た。
3.リチウム複合金属酸化物の評価
得られたA1の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.03:0.45:0.45:0.10であった。
A1の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、A1の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりA1のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.53ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは1.12であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは1.69であった。
さらに、IBMn/IAMn×IBNi/IANiは、1.89であった。
4.非水電解質二次電池の充放電試験
A1を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、170であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、178であった。
(実施例2)
1.リチウム複合金属酸化物前駆体(共沈物)の製造およびリチウム複合金属酸化物の製造
硫酸ニッケル六水和物、硫酸マンガン一水和物、硫酸コバルト七水和物を、Ni:Mn:Coのモル比が0.47:0.48:0.05となるようにそれぞれ秤量したこと以外は、実施例1と同様の操作を行い、共沈物Q2を得た。更に、共沈物Q2を用いたこと以外は、実施例1と同様の操作を行い、粉末状のリチウム複合金属酸化物A2を得た。
2.リチウム複合金属酸化物の評価
得られたA2の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.06:0.49:0.45:0.06であった。
A2の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、A2の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりA2のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.53ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは1.14であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは1.69であった。
さらに、IBMn/IAMn×IBNi/IANiは、1.93であった。
3.非水電解質二次電池の充放電試験
A2を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cおける放電容量(mAh/g)は、168であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、178であった。
(実施例3)
1.リチウム複合金属酸化物前駆体(共沈物)の製造およびリチウム複合金属酸化物の製造
硫酸ニッケル六水和物、硫酸マンガン一水和物、硫酸コバルト七水和物を、Ni:Mn:Coのモル比が0.48:0.49:0.03となるようにそれぞれ秤量したこと以外は、実施例1と同様の操作を行い、共沈物Q3を得た。更に、共沈物Q3を用いたこと以外は、実施例1と同様の操作を行い、粉末状のリチウム複合金属酸化物A3を得た。
2.リチウム複合金属酸化物の評価
得られたA3の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.00:0.48:0.49:0.03であった。
A3の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、A3の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりA3のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.50ÅのピークAMnと2.52ÅのピークBMnの強度比IBMn/IAMnは1.15であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは1.58であった。
さらに、IBMn/IAMn×IBNi/IANiは、1.82であった。
3.非水電解質二次電池の充放電試験
A3を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、164であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、175であった。
(比較例1)
1.リチウム複合金属酸化物の製造
前記共沈物Q1と、共沈物Q1に含まれる遷移金属の合計量(モル)1に対して、Liの量(モル)が1.3となるように秤量した炭酸リチウムと、不活性融剤として硫酸カリウムとを乳鉢により混合して混合物を得た。
次いで、得られた混合物をアルミナ製焼成容器に入れ、電気炉を用いて大気雰囲気中850℃で6時間保持して焼成を行い、室温まで冷却して焼成物を得た。
得られた焼成物を粉砕し、蒸留水でデカンテーションによる洗浄を行った後、ろ過し、300℃で6時間乾燥して粉末状のリチウム複合金属酸化物R1を得た。
2.リチウム複合金属酸化物の評価
得られたR1の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.18:0.46:0.44:0.10であった。
R1の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、R1の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりR1のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.56ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは0.96であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは2.13であった。
3.非水電解質二次電池の充放電試験
R1を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、154であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、162であった。
(比較例2)
1.リチウム複合金属酸化物の製造
共沈物Q2を用いたこと以外は、比較例1と同様の操作を行い、粉末状のリチウム複合金属酸化物R2を得た。
2.リチウム複合金属酸化物の評価
R2の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.16:0.47:0.48:0.05であった。
R2の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、R2の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりR2のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.53ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは0.94であった。
また、Niの動径分布関数において、1.60ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは2.04であった。
3.非水電解質二次電池の充放電試験
R2を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、149であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、160であった。
(比較例3)
1.リチウム複合金属酸化物の製造
共沈物Q3を用いたこと以外は、比較例1と同様の操作を行い、粉末状のリチウム複合金属酸化物R3を得た。
2.リチウム複合金属酸化物の評価
R3の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.10:0.48:0.49:0.03であった。
R3の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、R3の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりR3のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.50ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは0.94であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは2.04であった。
3.非水電解質二次電池の充放電試験
R3を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、148であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量は、159であった。
実施例1~3、および比較例1~3の結果を下記表1に示す。
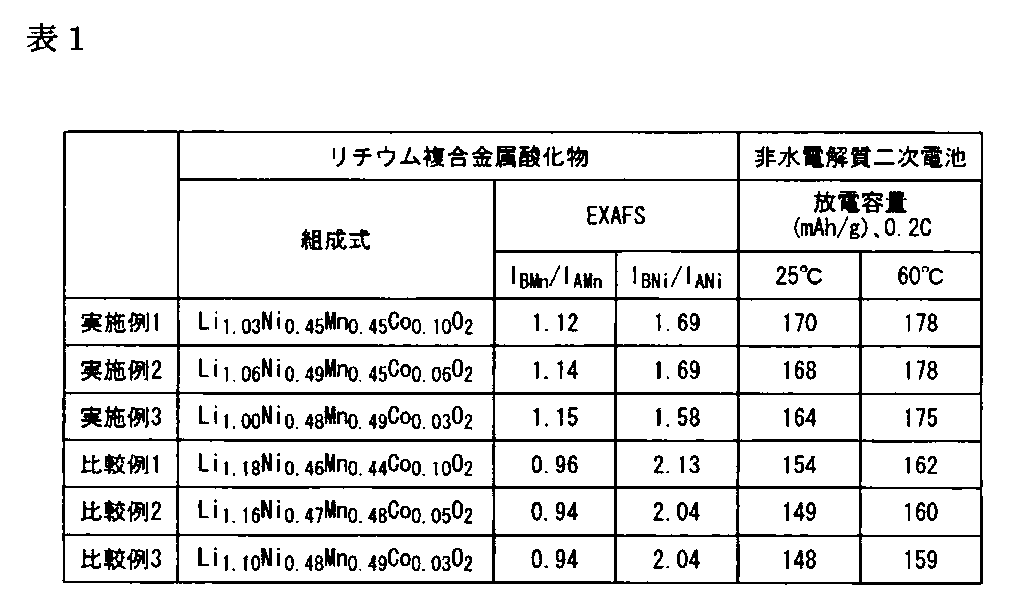
評価の結果、実施例1~3のリチウム複合金属酸化物を正極活物質として用いた非水電解質二次電池では、いずれも、比較例1~3のリチウム複合金属酸化物を正極活物質として用いた非水電解質二次電池よりも放電容量が大きく、高性能な二次電池が得られた。
また、実施例のうち最もCoの使用量が少ない実施例3のリチウム複合金属酸化物であっても、比較例のうち最もCoの使用量が多い比較例1のリチウム複合金属酸化物よりも放電容量が大きい非水電解質二次電池が得られたことから、Co使用量を低減しても性能の維持・向上が図れることが分かった。
以上の結果から、本発明のリチウム複合金属酸化物が高容量を示す非水電解質二次電池に有用であることが分かった。また、本発明のリチウム複合金属酸化物を用いた正極活物質、正極は、高性能な非水電解質二次電池に有用であり、本発明の非水電解質二次電池は、従来よりも高容量を示すことが分かった。 Next, the present invention will be described in more detail with reference to examples.
In this example, evaluation of the lithium composite metal oxide (positive electrode active material) and production evaluation of the positive electrode and the lithium secondary battery were performed as follows.
(1) Evaluation of lithium composite metal oxide Composition analysis of lithium composite metal oxide The composition analysis of lithium composite metal oxide was conducted by dissolving the obtained lithium composite metal oxide powder in hydrochloric acid and then using an inductively coupled plasma emission spectrometer (SII Nanotechnology Inc.) Manufactured by SPS3000).
2. Powder X-ray diffraction measurement of lithium composite metal oxide Powder X-ray diffraction measurement of lithium composite metal oxide was performed using a powder X-ray diffractometer (manufactured by Rigaku Corporation, RINT2500TTR, sample horizontal type). A powder X-ray diffraction pattern was obtained by filling the obtained lithium composite metal oxide into a dedicated substrate and measuring using a CuKα ray source in a diffraction angle range of 2θ = 10 ° to 90 °. .
Further, Rietveld analysis of powder X-ray diffraction patterns is performed by an analysis program RIETAN-2000 (see F. Izumi and T. Ikeda, Mater. Sci. Forum, 321-324 (2000) 198), and lithium composite metal oxidation. The space group of the crystal structure of the object was determined.
3. EXAFS measurement and analysis of lithium composite metal oxide (measurement of X-ray absorbance, creation of X-ray absorption spectrum)
The X-ray absorption spectrum of the lithium composite metal oxide is measured using the XAFS measurement device of the beamline 9C (BL-9C), a synchrotron radiation science research facility owned by the Institute for Materials Structure Science of the High Energy Accelerator Research Organization. Absorbance was measured and created using the obtained X-ray absorbance values.
In the measurement of X-ray absorbance, a Si (111) double crystal spectrometer was used. Further, regarding the X-ray absorbance measurement of the wavelength corresponding to the K absorption edge of Mn, detuning was performed at 60% in order to remove high-order terms.
In the measurement of X-ray absorbance, the incident X-ray intensity (I 0 ) is measured at room temperature using a 17 cm ion chamber using N 2 as a filling gas, and the transmitted X-ray intensity (I t ) is measured as a filling gas. It was measured at room temperature using a 31cm ion chamber of using N 2 as.
The measured energy range and the number of measurement points were 4932 at an equal energy interval from 6040 eV to 7640.5 eV for the K absorption edge of Mn. The K absorption edge of Ni was 5333 points at equal energy intervals from 7834 eV to 9434.5 eV.
The energy calibration is performed using a pre-edge peak (about about X-ray Absorption Near-Edge Structure) spectrum of the obtained K absorption edge when measured using copper alone as a standard sample. The angle of the spectral crystal at 8980 eV) was set to 12.7185 °.
In the measurement of X-ray absorbance, each incident X-ray energy, measured I 0, I t, the following equation was determined X-ray absorbance.
X-ray absorbance μt = −ln (I 0 / I t )
In the above measurement, discrete X-ray absorbance is obtained corresponding to the wavelength of X-rays used for measurement (corresponding to the energy of X-rays used for measurement). The obtained X-ray absorbance was averaged and data interpolated as follows.
First, the X-ray absorbance in the range corresponding to the K absorption edge of Mn was averaged by the following method.
From 6040 eV to 6400 eV: 3 times of adjacent average processing of 21 points From 6400 eV to 6700 eV: 1 time of weighted average processing by 7 points of Savitzky-Golay method From 6700 eV to 7640.5 eV: 5 times of adjacent average processing of 11 points Subsequently, using the average value of the obtained X-ray absorbance, data interpolation was performed at the following data intervals to obtain an X-ray absorption spectrum having the X axis as the X-ray energy and the Y axis as the X absorbance.
6040 eV to 6508 eV: 6.5 eV interval 6508 eV to 6609.5 eV: 0.35 eV interval 6609.5 eV to 6640.5 eV: 1 eV interval 6640.5 eV to 7040.5 eV: 2.5 eV interval 7040.5 eV to 7640. Up to 5 eV: 6 eV interval Further, the X-ray absorbance in the range corresponding to the K absorption edge of Ni was averaged by the following method.
From 7834 eV to 8200 eV: 3 times of adjacent average processing of 21 points From 8200 eV to 8500 eV: 1 time of weighted average processing by 7 points of Savitzky-Golay method From 8500 eV to 9734.5 eV: 5 times of adjacent average processing of 11 points Subsequently, using the average value of the obtained X-ray absorbance, data interpolation was performed at the following data intervals to obtain an X-ray absorption spectrum having the X axis as the X-ray energy and the Y axis as the X absorbance.
From 7834 eV to 8302 eV: 6.5 eV interval From 8302 eV to 8403.5 eV: 0.35 eV interval From 8403.5 eV to 8459.5 eV: 1 eV interval From 8459.5 eV to 8834.5 eV: 2.5 eV interval From 8834.5 eV to 9434. Up to 5 eV: 6 eV interval (creation of EXAFS spectrum)
From the obtained X-ray absorption spectrum, an EXAFS spectrum of the K absorption edge of Mn and the K absorption edge of Ni was obtained as follows. Analysis of the X-ray absorption spectrum obtained above was performed using analysis software (Rigaku Corporation, REX2000).
First, the Mn K absorption edge E 0 was set to an energy value at which the first-order differential coefficient was maximum in the vicinity of the Mn K absorption edge in the X-ray absorption spectrum. Similarly, the K absorption edge E 0 of Ni is set to an energy value at which the first-order differential coefficient becomes maximum in the spectrum near the K absorption edge of Ni.
The spectrum background is a spectrum in a lower energy region than the K absorption edge of Mn and the K absorption edge of Ni, and Victory's formula (Aλ 3 −Bλ 4 + C; λ is the wavelength of incident X-rays, A, B and C are arbitrary constants) and determined by applying the least square method. The EXAFS spectrum was obtained by subtracting the background value corresponding to this Victreeen equation from the X-ray absorption spectrum.
(Calculation of radial distribution function)
A radial distribution function was obtained from the obtained EXAFS spectrum.
First, regarding the EXAFS spectrum, the absorbance (μ 0 ) of isolated atoms was estimated by the Spline Smoothing method (smoothing spline method), and the EXAFS function χ (k) was extracted. Note that k is the wave number of photoelectrons defined by 0.5123 × (E−E 0 ) 1/2 , and the unit of k is −1 .
Next, the EXAFS function k 3 χ (k) weighted by k 3 was subjected to Fourier transform in the range of k from 3.0 to 11.85Å −1 to obtain a radial distribution function.
(2) Production of positive electrode A lithium composite metal oxide (positive electrode active material), a conductive material (acetylene black: graphite = 9: 1 (mass ratio)) and a binder (PVdF) obtained by the production method described later are used as a positive electrode active material. A paste-like positive electrode mixture was prepared by adding and kneading so that the composition of the substance: conductive material: binder = 87: 10: 3 (mass ratio) was obtained. When preparing the positive electrode mixture, N-methyl-2-pyrrolidone was used as the organic solvent.
The obtained positive electrode mixture was applied to a 40 μm thick Al foil serving as a current collector and vacuum dried at 150 ° C. for 8 hours to obtain a positive electrode.
(3) Production of nonaqueous electrolyte secondary battery (coin cell) The following operation was performed in a glove box in an argon atmosphere.
The positive electrode created in “(2) Preparation of positive electrode” is placed on the lower lid of a coin cell (manufactured by Hosen Co., Ltd.) for coin-type battery R2032 with the aluminum foil surface facing downward, and a laminated film separator (polyethylene) is placed thereon. A separator (thickness 16 μm) having a heat-resistant porous layer laminated thereon was placed on the porous film. 300 μl of electrolyte was injected here. The electrolyte used was prepared by dissolving LiPF 6 in a 30:35:35 (volume ratio) mixture of ethylene carbonate, dimethyl carbonate, and ethyl methyl carbonate to a concentration of 1 mol / l.
Next, using lithium metal as the negative electrode, the lithium metal is placed on the upper side of the laminated film separator, covered with a gasket, and caulked with a non-aqueous electrolyte secondary battery (coin type battery R2032, hereinafter, (Sometimes referred to as a “coin-type battery”).
(4) Charge / Discharge Test Using the coin-type battery created in “(3) Production of Nonaqueous Electrolyte Secondary Battery (Coin Cell)”, a charge / discharge test was performed under the following conditions. The charge capacity and discharge capacity in the charge / discharge test were determined as follows.
<Charge / discharge test conditions>
Test temperature: 25 ° C or 60 ° C
Charging conditions: Maximum charging voltage 4.3V, chargingtime 10 hours, charging current 0.3 mA / cm 2
Discharge conditions: discharge minimum voltage 2.5V,discharge time 10 hours, discharge current 0.3 mA / cm 2
(Example 1)
1. Production of Lithium Composite Metal Oxide Precursor (Coprecipitate) Nickel sulfate hexahydrate, manganese sulfate monohydrate, and cobalt sulfate heptahydrate have a molar ratio of Ni: Mn: Co of 0.45: 0. .45: 0.10, each was weighed and dissolved in pure water to obtain an aqueous transition metal solution containing Ni ions, Mn ions, Co ions and SO 4 2- ions.
To this transition metal aqueous solution, an aqueous potassium hydroxide solution was added to perform coprecipitation to produce a precipitate, thereby obtaining a slurry. The obtained slurry was subjected to solid-liquid separation, washed with distilled water to obtain a coprecipitate Q 1 and dried for 8 hours at 100 ° C..
2. Production of lithium composite metal oxide The amount (mol) of Li is 1.3 with respect to the obtained coprecipitate Q 1 and the total amount (mol) 1 of transition metals contained in the coprecipitate Q 1. The lithium carbonate weighed in this manner and potassium sulfate as an inert flux were mixed in a mortar to obtain a mixture.
Next, the obtained mixture was placed in an alumina firing container, and the alumina firing container was placed in an electric furnace. The oxygen concentration was adjusted to 8.5% by volume using the air atmosphere originally present in the electric furnace and the introduced nitrogen gas, and heated at 400 ° C. to obtain a calcined product. Further, the oxygen concentration was adjusted to 1% by volume, the mixture was held at 850 ° C. for 6 hours and baked, and cooled to room temperature to obtain a baked product.
The fired product obtained was pulverized and dispersed in distilled water. The supernatant after standing was removed by decantation, filtered, and dried at 300 ° C. for 6 hours to obtain a powdered lithium composite metal oxide A 1 .
3. The results of composition analysis of A 1 to evaluate the resulting lithium composite metal oxide, Li: Ni: Mn: the molar ratio of Co is 1.03: 0.45: 0.45: was 0.10 .
And by performing powder X-ray diffractometry of A 1, and a compound of layered structure type. As a result of Rietveld analysis, the crystal structure of A 1 is hexagonal, which is classified to the space group R-3m.
After obtaining EXAFS spectra of Mn and Ni of A 1 by the above-described method, a radial distribution function was obtained by Fourier transform. In the radial distribution function of Mn , the intensity ratio I BMn / I AMn of the peak A Mn of 1.53 と and the peak B Mn of 2.49 Å was 1.12.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 1.69.
Furthermore, I BMn / I AMn × I BNi / I ANi was 1.89.
4). A coin type battery was produced by using a charge-discharge test A 1 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 170. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 178.
(Example 2)
1. Preparation of lithium composite metal oxide precursor (coprecipitate) and manufacture of lithium composite metal oxide Nickel sulfate hexahydrate, manganese sulfate monohydrate, cobalt sulfate heptahydrate, Ni: Mn: Co molar ratio of 0.47: 0.48: except that each weighed so that 0.05 was obtained in the same manner as in example 1, to obtain a coprecipitate Q 2. Furthermore, except for using a coprecipitate Q 2, the procedure of Example 1 to obtain a powdered lithium composite metal oxide A 2.
2. Evaluation of Lithium Composite Metal Oxide The composition of A 2 obtained was analyzed. The molar ratio of Li: Ni: Mn: Co was 1.06: 0.49: 0.45: 0.06. .
And by performing powder X-ray diffraction measurement of A 2, and a compound of layered structure type. As a result of Rietveld analysis, the crystal structure of A 2 is a hexagonal, classified to the space group R-3m.
After obtaining EX 2 AFS spectra of Mn and Ni of A 2 by the above-mentioned method, a radial distribution function was obtained by Fourier transform. In the radial distribution function of Mn , the intensity ratio I BMn / I AMn of the peak A Mn of 1.53 と and the peak B Mn of 2.49 Å was 1.14.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 1.69.
Furthermore, I BMn / I AMn × I BNi / I ANi was 1.93.
3. A coin type battery was produced by using the charge and discharge test A 2 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., 0.2 C definitive discharge capacity (mAh / g) was 168. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 178.
(Example 3)
1. Preparation of lithium composite metal oxide precursor (coprecipitate) and manufacture of lithium composite metal oxide Nickel sulfate hexahydrate, manganese sulfate monohydrate, cobalt sulfate heptahydrate, Ni: Mn: Co molar ratio of 0.48: 0.49: except that each weighed so that 0.03 was obtained in the same manner as in example 1, to obtain a coprecipitate Q 3. Furthermore, except for using a coprecipitate Q 3, the procedure of Example 1 to obtain a powdered lithium composite metal oxide A 3.
2. The results of composition analysis of A 3 Evaluation resulting lithium composite metal oxide, Li: Ni: Mn: the molar ratio of Co is 1.00: 0.48: 0.49: was 0.03 .
And by performing powder X-ray diffraction measurement of A 3, it was a compound having a layered structure type. As a result of Rietveld analysis, the crystal structure of A 3 is hexagonal, which is classified to the space group R-3m.
After obtaining the EXAFS spectra of Mn and Ni of A 3 by the method described above to obtain the radial distribution function by applying a Fourier transform. In the Mn radial distribution function, the intensity ratio I BMn / I AMn of the peak A Mn of 1.50Å and the peak B Mn of 2.52Å was 1.15.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 1.58.
Furthermore, I BMn / I AMn × I BNi / I ANi was 1.82.
3. A coin type battery was produced by using the charge and discharge test A 3 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 164. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 175.
(Comparative Example 1)
1. And manufacturing the coprecipitate to Q 1 lithium composite metal oxide, the total amount (mol) 1 of the transition metal contained in the coprecipitate Q 1, as the amount of Li (mole) is 1.3 Weighed lithium carbonate and potassium sulfate as an inert flux were mixed in a mortar to obtain a mixture.
Subsequently, the obtained mixture was put into an alumina firing container, and fired by holding at 850 ° C. in an air atmosphere for 6 hours using an electric furnace, and cooled to room temperature to obtain a fired product.
The obtained fired product was pulverized, washed with distilled water by decantation, filtered, and dried at 300 ° C. for 6 hours to obtain a powdered lithium composite metal oxide R 1 .
2. Evaluation of lithium composite metal oxide The composition of R 1 obtained was analyzed. The molar ratio of Li: Ni: Mn: Co was 1.18: 0.46: 0.44: 0.10. .
When a powder X-ray diffraction measurement of R 1 was performed, it was a layered structure type compound. As a result of Rietveld analysis, the crystal structure of R 1 was hexagonal and was classified into the space group R-3m.
After obtaining EX1 AFS spectra of Mn and Ni of R 1 by the above method, a radial distribution function was obtained by Fourier transform. In the radial distribution function of Mn , the intensity ratio I BMn / I AMn of the peak A Mn of 1.56 と and the peak B Mn of 2.49 0.9 was 0.96.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 2.13.
3. A coin type battery was produced by using a charge-discharge test R 1 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 154. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 162.
(Comparative Example 2)
1. Except for using the prepared coprecipitate Q 2 of the lithium composite metal oxide, the same operation as in Comparative Example 1 to obtain a powdered lithium composite metal oxide R 2.
2. The results of composition analysis of the evaluation R 2 of the lithium composite metal oxide, Li: Ni: Mn: the molar ratio of Co is 1.16: 0.47: 0.48: was 0.05.
When a powder X-ray diffraction measurement of R 2 was performed, it was a layered structure type compound. As a result of Rietveld analysis, the crystal structure of R 2 was hexagonal and classified into the space group R-3m.
After obtaining EX 2 AFS spectra of Mn and Ni of R 2 by the above-described method, a radial distribution function was obtained by Fourier transform. In the Mn radial distribution function, the intensity ratio I BMn / I AMn of the peak A Mn of 1.53 Å and the peak B Mn of 2.49 Å was 0.94.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.60 と and the peak B Ni of 2.49 Å was 2.04.
3. A coin type battery was produced by using a charge-discharge test R 2 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 149. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 160.
(Comparative Example 3)
1. Except for using the prepared coprecipitate Q 3 of the lithium composite metal oxide, the same operation as in Comparative Example 1 to obtain a powdered lithium composite metal oxide R 3.
2. Evaluation of Lithium Composite Metal Oxide When the composition analysis of R 3 was performed, the molar ratio of Li: Ni: Mn: Co was 1.10: 0.48: 0.49: 0.03.
When a powder X-ray diffraction measurement of R 3 was performed, it was a layered structure type compound. As a result of Rietveld analysis, the crystal structure of R 3 was hexagonal and was classified into the space group R-3m.
After obtaining EXAFS spectra of Mn and Ni of R 3 by the above method, a radial distribution function was obtained by Fourier transform. In the Mn radial distribution function, the intensity ratio I BMn / I AMn of the peak A Mn of 1.50 Å and the peak B Mn of 2.49 Å was 0.94.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 2.04.
3. A coin type battery was produced by using a charge-discharge test R 3 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 148. When a discharge test at 60 ° C. was performed, the discharge capacity at 0.2 C was 159.
The results of Examples 1 to 3 and Comparative Examples 1 to 3 are shown in Table 1 below.
As a result of the evaluation, all of the non-aqueous electrolyte secondary batteries using the lithium composite metal oxides of Examples 1 to 3 as the positive electrode active material used the lithium composite metal oxides of Comparative Examples 1 to 3 as the positive electrode active material. A secondary battery having a higher discharge capacity and higher performance than the nonaqueous electrolyte secondary battery was obtained.
Moreover, even if it is the lithium composite metal oxide of Example 3 with the least Co usage-amount among Examples, it discharges rather than the lithium composite metal oxide of Comparative Example 1 with the most Co usage-amount among Comparative Examples. Since a non-aqueous electrolyte secondary battery having a large capacity was obtained, it was found that the performance could be maintained and improved even if the amount of Co used was reduced.
From the above results, it was found that the lithium composite metal oxide of the present invention is useful for a non-aqueous electrolyte secondary battery exhibiting a high capacity. Further, the positive electrode active material using the lithium composite metal oxide of the present invention, the positive electrode is useful for a high-performance non-aqueous electrolyte secondary battery, and the non-aqueous electrolyte secondary battery of the present invention has a higher capacity than before. It was found that
本実施例においては、リチウム複合金属酸化物(正極活物質)の評価、正極およびリチウム二次電池の作製評価を、次のようにして行った。
(1)リチウム複合金属酸化物の評価
1.リチウム複合金属酸化物の組成分析
リチウム複合金属酸化物の組成分析は、得られたリチウム複合金属酸化物の粉末を塩酸に溶解させた後、誘導結合プラズマ発光分析装置(エスアイアイ・ナノテクノロジー株式会社製、SPS3000)を用いて行った。
2.リチウム複合金属酸化物の粉末X線回折測定
リチウム複合金属酸化物の粉末X線回折測定は、粉末X線回折装置(株式会社リガク製、RINT2500TTR、試料水平型)を用いて行った。得られたリチウム複合金属酸化物を専用の基板に充填し、CuKα線源を用いて、回折角2θ=10°~90°の範囲にて測定を行うことで、粉末X線回折図形を得た。
また、粉末X線回折図形のリートベルト解析は、解析プログラムRIETAN−2000(F.Izumi and T.Ikeda,Mater.Sci.Forum,321−324(2000)198を参照)により行い、リチウム複合金属酸化物が有する結晶構造の空間群を求めた。
3.リチウム複合金属酸化物のEXAFS測定および解析
(X線吸光度の測定、X線吸収スペクトルの作成)
リチウム複合金属酸化物のX線吸収スペクトルは、高エネルギー加速器研究機構の物質構造科学研究所が所有する放射光科学研究施設であるビームライン9C(BL−9C)のXAFS測定装置を用いてX線吸光度を測定し、得られたX線吸光度の値を用いて作成した。
X線吸光度の測定においては、Si(111)二結晶分光器を用いた。また、MnのK吸収端に対応する波長のX線の吸光度測定に関しては、高次項除去のためデチューニングを60%で行った。
X線吸光度の測定において、入射X線強度(I0)は、充填ガスとしてN2を使用した17cmのイオンチェンバーを用いて常温下で測定し、透過X線強度(It)は、充填ガスとしてN2を使用した31cmのイオンチェンバーを用いて常温下で測定した。
測定したエネルギー範囲および測定点数は、MnのK吸収端については、6040eVから7640.5eVまで等エネルギー間隔で4932点であった。また、NiのK吸収端については、7834eVから9434.5eVまで等エネルギー間隔で5333点であった。
なお、エネルギーの校正は、銅単体を標準試料として用いて測定したときに、得られるK吸収端のX線吸収端近傍構造(X−ray Absorption Near−Edge Structure)スペクトルについて、プレエッジピーク(約8980eV)における分光結晶の角度を12.7185°として行った。
X線吸光度の測定においては、各入射X線エネルギーにおいて、I0、Itを測定し、次式により、X線吸光度を求めた。
X線吸光度 μt=−ln(I0/It)
上記測定では、測定に用いるX線の波長に対応して(測定に用いるX線のエネルギーに対応して)離散的なX線吸光度が得られる。得られたX線吸光度について次のように平均処理およびデータ補間を行った。
まず、MnのK吸収端に対応する範囲のX線吸光度については、下記の方法で平均処理を行った。
6040eVから6400eVまで:21点の隣接平均処理を3回
6400eVから6700eVまで:7点のSavitzky−Golay法による重みつき平均処理を1回
6700eVから7640.5eVまで:11点の隣接平均処理を5回
次いで、得られたX線吸光度の平均値を用いて、下記のデータ間隔でデータ補間を行い、X軸をX線のエネルギー、Y軸をX吸光度とするX線吸収スペクトルを得た。
6040eVから6508eVまで:6.5eV間隔
6508eVから6609.5eVまで:0.35eV間隔
6609.5eVから6640.5eVまで:1eV間隔
6640.5eVから7040.5eVまで:2.5eV間隔
7040.5eVから7640.5eVまで:6eV間隔
また、NiのK吸収端に対応する範囲のX線吸光度については、下記の方法で平均処理を行った。
7834eVから8200eVまで:21点の隣接平均処理を3回
8200eVから8500eVまで:7点のSavitzky−Golay法による重みつき平均処理を1回
8500eVから9734.5eVまで:11点の隣接平均処理を5回
次いで、得られたX線吸光度の平均値を用いて、下記のデータ間隔でデータ補間を行い、X軸をX線のエネルギー、Y軸をX吸光度とするX線吸収スペクトルを得た。
7834eVから8302eVまで:6.5eV間隔
8302eVから8403.5eVまで:0.35eV間隔
8403.5eVから8439.5eVまで:1eV間隔
8439.5eVから8834.5eVまで:2.5eV間隔
8834.5eVから9434.5eVまで:6eV間隔
(EXAFSスペクトルの作成)
得られたX線吸収スペクトルから、次のようにして、MnのK吸収端およびNiのK吸収端のEXAFSスペクトルを得た。上記により得られたX線吸収スペクトルの解析は、解析ソフト(株式会社リガク製、REX2000)を用いて行った。
まず、MnのK吸収端E0は、X線吸収スペクトルにおけるMnのK吸収端付近において、一次微分係数が最大となるエネルギー値とした。同様に、NiのK吸収端E0は、NiのK吸収端付近のスペクトルにおいて、一次微分係数が最大となるエネルギー値とした。
また、スペクトルのバックグランドは、前記のMnのK吸収端およびNiのK吸収端よりも低エネルギー域のスペクトルにVictoreenの式(Aλ3−Bλ4+C;λは入射X線の波長、A,B,Cは任意の定数)を最小自乗法で当てはめて決定した。このVictoreenの式に対応するバックグラウンドの値を、X線吸収スペクトルから差し引くことで、EXAFSスペクトルを得た。
(動径分布関数の算出)
得られたEXAFSスペクトルから動径分布関数を得た。
まず、EXAFSスペクトルについて、Spline Smoothing法(平滑化スプライン法)により孤立原子の吸光度(μ0)を見積もり、EXAFS関数χ(k)を抽出した。なお、kは0.5123×(E−E0)1/2で定義される光電子の波数であり、kの単位はÅ−1である。
次いで、k3で重み付けしたEXAFS関数k3χ(k)について、kが3.0から11.85Å−1の範囲でフーリエ変換して動径分布関数を求めた。
(2)正極の作製
後述する製造方法で得られるリチウム複合金属酸化物(正極活物質)と導電材(アセチレンブラック:黒鉛=9:1(質量比))とバインダー(PVdF)とを、正極活物質:導電材:バインダー=87:10:3(質量比)の組成となるように加えて混練することにより、ペースト状の正極合剤を調製した。正極合剤の調製時には、N−メチル−2−ピロリドンを有機溶媒として用いた。
得られた正極合剤を、集電体となる厚さ40μmのAl箔に塗布して150℃で8時間真空乾燥を行い、正極を得た。
(3)非水電解質二次電池(コインセル)の作製
以下の操作を、アルゴン雰囲気のグローブボックス内で行った。
「(2)正極の作製」で作成した正極を、コイン型電池R2032用のコインセル(宝泉株式会社製)の下蓋にアルミ箔面を下に向けて置き、その上に積層フィルムセパレータ(ポリエチレン製多孔質フィルムの上に、耐熱多孔層を積層したセパレータ(厚み16μm))を置いた。ここに電解液を300μl注入した。用いた電解液は、エチレンカーボネートとジメチルカーボネートとエチルメチルカーボネートとの30:35:35(体積比)混合液に、LiPF6を1mol/lとなるように溶解して調製した。
次に、負極として金属リチウムを用いて、前記金属リチウムを積層フィルムセパレータの上側に置き、ガスケットを介して上蓋をし、かしめ機でかしめて非水電解質二次電池(コイン型電池R2032。以下、「コイン型電池」と称することがある。)を作製した。
(4)充放電試験
「(3)非水電解質二次電池(コインセル)の作製」で作成したコイン型電池を用いて、以下に示す条件で充放電試験を実施した。充放電試験における、充電容量および放電容量をそれぞれ以下のようにして求めた。
<充放電試験条件>
試験温度:25℃または60℃
充電時条件:充電最大電圧4.3V、充電時間10時間、充電電流0.3mA/cm2
放電時条件:放電最小電圧2.5V、放電時間10時間、放電電流0.3mA/cm2
(実施例1)
1.リチウム複合金属酸化物前駆体(共沈物)の製造
硫酸ニッケル六水和物、硫酸マンガン一水和物、硫酸コバルト七水和物を、Ni:Mn:Coのモル比が0.45:0.45:0.10となるようにそれぞれ秤量し、純水に溶解してNiイオン、Mnイオン、CoイオンおよびSO4 2−イオンを含有する遷移金属水溶液を得た。
この遷移金属水溶液に、水酸化カリウム水溶液を加えて共沈を行い、沈殿物を生成させて、スラリーを得た。得られたスラリーについて、固液分離を行い、蒸留水により洗浄し、100℃で8時間乾燥させて共沈物Q1を得た。
2.リチウム複合金属酸化物の製造
得られた共沈物Q1と、共沈物Q1に含まれる遷移金属の合計量(モル)1に対して、Liの量(モル)が1.3となるように秤量した炭酸リチウムと、不活性融剤として硫酸カリウムとを乳鉢により混合して混合物を得た。
次いで、得られた混合物をアルミナ製焼成容器に入れ、このアルミナ製焼成容器を電気炉に入れた。電気炉内部に元々存在する大気雰囲気および導入する窒素ガスを用いて、酸素濃度を8.5体積%に調整し、400℃で加熱して仮焼物を得た。さらに酸素濃度を1体積%に調整し、850℃で6時間保持して焼成を行い、室温まで冷却して焼成物を得た。
得られた焼成物を粉砕し、蒸留水に分散させた。静置後の上澄みをデカンテーションで除去した後、ろ過し、300℃で6時間乾燥して粉末状のリチウム複合金属酸化物A1を得た。
3.リチウム複合金属酸化物の評価
得られたA1の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.03:0.45:0.45:0.10であった。
A1の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、A1の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりA1のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.53ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは1.12であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは1.69であった。
さらに、IBMn/IAMn×IBNi/IANiは、1.89であった。
4.非水電解質二次電池の充放電試験
A1を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、170であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、178であった。
(実施例2)
1.リチウム複合金属酸化物前駆体(共沈物)の製造およびリチウム複合金属酸化物の製造
硫酸ニッケル六水和物、硫酸マンガン一水和物、硫酸コバルト七水和物を、Ni:Mn:Coのモル比が0.47:0.48:0.05となるようにそれぞれ秤量したこと以外は、実施例1と同様の操作を行い、共沈物Q2を得た。更に、共沈物Q2を用いたこと以外は、実施例1と同様の操作を行い、粉末状のリチウム複合金属酸化物A2を得た。
2.リチウム複合金属酸化物の評価
得られたA2の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.06:0.49:0.45:0.06であった。
A2の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、A2の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりA2のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.53ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは1.14であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは1.69であった。
さらに、IBMn/IAMn×IBNi/IANiは、1.93であった。
3.非水電解質二次電池の充放電試験
A2を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cおける放電容量(mAh/g)は、168であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、178であった。
(実施例3)
1.リチウム複合金属酸化物前駆体(共沈物)の製造およびリチウム複合金属酸化物の製造
硫酸ニッケル六水和物、硫酸マンガン一水和物、硫酸コバルト七水和物を、Ni:Mn:Coのモル比が0.48:0.49:0.03となるようにそれぞれ秤量したこと以外は、実施例1と同様の操作を行い、共沈物Q3を得た。更に、共沈物Q3を用いたこと以外は、実施例1と同様の操作を行い、粉末状のリチウム複合金属酸化物A3を得た。
2.リチウム複合金属酸化物の評価
得られたA3の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.00:0.48:0.49:0.03であった。
A3の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、A3の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりA3のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.50ÅのピークAMnと2.52ÅのピークBMnの強度比IBMn/IAMnは1.15であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは1.58であった。
さらに、IBMn/IAMn×IBNi/IANiは、1.82であった。
3.非水電解質二次電池の充放電試験
A3を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、164であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、175であった。
(比較例1)
1.リチウム複合金属酸化物の製造
前記共沈物Q1と、共沈物Q1に含まれる遷移金属の合計量(モル)1に対して、Liの量(モル)が1.3となるように秤量した炭酸リチウムと、不活性融剤として硫酸カリウムとを乳鉢により混合して混合物を得た。
次いで、得られた混合物をアルミナ製焼成容器に入れ、電気炉を用いて大気雰囲気中850℃で6時間保持して焼成を行い、室温まで冷却して焼成物を得た。
得られた焼成物を粉砕し、蒸留水でデカンテーションによる洗浄を行った後、ろ過し、300℃で6時間乾燥して粉末状のリチウム複合金属酸化物R1を得た。
2.リチウム複合金属酸化物の評価
得られたR1の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.18:0.46:0.44:0.10であった。
R1の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、R1の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりR1のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.56ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは0.96であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは2.13であった。
3.非水電解質二次電池の充放電試験
R1を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、154であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、162であった。
(比較例2)
1.リチウム複合金属酸化物の製造
共沈物Q2を用いたこと以外は、比較例1と同様の操作を行い、粉末状のリチウム複合金属酸化物R2を得た。
2.リチウム複合金属酸化物の評価
R2の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.16:0.47:0.48:0.05であった。
R2の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、R2の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりR2のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.53ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは0.94であった。
また、Niの動径分布関数において、1.60ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは2.04であった。
3.非水電解質二次電池の充放電試験
R2を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、149であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、160であった。
(比較例3)
1.リチウム複合金属酸化物の製造
共沈物Q3を用いたこと以外は、比較例1と同様の操作を行い、粉末状のリチウム複合金属酸化物R3を得た。
2.リチウム複合金属酸化物の評価
R3の組成分析を行ったところ、Li:Ni:Mn:Coのモル比は、1.10:0.48:0.49:0.03であった。
R3の粉末X線回折測定を行ったところ、層状構造型の化合物であった。また、リートベルト解析の結果、R3の結晶構造は六方晶で、空間群R−3mに分類された。
上述の方法によりR3のMnおよびNiのEXAFSスペクトルを得た後、フーリエ変換して動径分布関数を求めた。Mnの動径分布関数において、1.50ÅのピークAMnと2.49ÅのピークBMnの強度比IBMn/IAMnは0.94であった。
また、Niの動径分布関数において、1.56ÅのピークANiと2.49ÅのピークBNiの強度比IBNi/IANiは2.04であった。
3.非水電解質二次電池の充放電試験
R3を用いてコイン型電池を作製し、25℃における放電試験を行ったところ、0.2Cにおける放電容量(mAh/g)は、148であった。また、60℃における放電試験を行ったところ、0.2Cにおける放電容量は、159であった。
実施例1~3、および比較例1~3の結果を下記表1に示す。
また、実施例のうち最もCoの使用量が少ない実施例3のリチウム複合金属酸化物であっても、比較例のうち最もCoの使用量が多い比較例1のリチウム複合金属酸化物よりも放電容量が大きい非水電解質二次電池が得られたことから、Co使用量を低減しても性能の維持・向上が図れることが分かった。
以上の結果から、本発明のリチウム複合金属酸化物が高容量を示す非水電解質二次電池に有用であることが分かった。また、本発明のリチウム複合金属酸化物を用いた正極活物質、正極は、高性能な非水電解質二次電池に有用であり、本発明の非水電解質二次電池は、従来よりも高容量を示すことが分かった。 Next, the present invention will be described in more detail with reference to examples.
In this example, evaluation of the lithium composite metal oxide (positive electrode active material) and production evaluation of the positive electrode and the lithium secondary battery were performed as follows.
(1) Evaluation of lithium composite metal oxide Composition analysis of lithium composite metal oxide The composition analysis of lithium composite metal oxide was conducted by dissolving the obtained lithium composite metal oxide powder in hydrochloric acid and then using an inductively coupled plasma emission spectrometer (SII Nanotechnology Inc.) Manufactured by SPS3000).
2. Powder X-ray diffraction measurement of lithium composite metal oxide Powder X-ray diffraction measurement of lithium composite metal oxide was performed using a powder X-ray diffractometer (manufactured by Rigaku Corporation, RINT2500TTR, sample horizontal type). A powder X-ray diffraction pattern was obtained by filling the obtained lithium composite metal oxide into a dedicated substrate and measuring using a CuKα ray source in a diffraction angle range of 2θ = 10 ° to 90 °. .
Further, Rietveld analysis of powder X-ray diffraction patterns is performed by an analysis program RIETAN-2000 (see F. Izumi and T. Ikeda, Mater. Sci. Forum, 321-324 (2000) 198), and lithium composite metal oxidation. The space group of the crystal structure of the object was determined.
3. EXAFS measurement and analysis of lithium composite metal oxide (measurement of X-ray absorbance, creation of X-ray absorption spectrum)
The X-ray absorption spectrum of the lithium composite metal oxide is measured using the XAFS measurement device of the beamline 9C (BL-9C), a synchrotron radiation science research facility owned by the Institute for Materials Structure Science of the High Energy Accelerator Research Organization. Absorbance was measured and created using the obtained X-ray absorbance values.
In the measurement of X-ray absorbance, a Si (111) double crystal spectrometer was used. Further, regarding the X-ray absorbance measurement of the wavelength corresponding to the K absorption edge of Mn, detuning was performed at 60% in order to remove high-order terms.
In the measurement of X-ray absorbance, the incident X-ray intensity (I 0 ) is measured at room temperature using a 17 cm ion chamber using N 2 as a filling gas, and the transmitted X-ray intensity (I t ) is measured as a filling gas. It was measured at room temperature using a 31cm ion chamber of using N 2 as.
The measured energy range and the number of measurement points were 4932 at an equal energy interval from 6040 eV to 7640.5 eV for the K absorption edge of Mn. The K absorption edge of Ni was 5333 points at equal energy intervals from 7834 eV to 9434.5 eV.
The energy calibration is performed using a pre-edge peak (about about X-ray Absorption Near-Edge Structure) spectrum of the obtained K absorption edge when measured using copper alone as a standard sample. The angle of the spectral crystal at 8980 eV) was set to 12.7185 °.
In the measurement of X-ray absorbance, each incident X-ray energy, measured I 0, I t, the following equation was determined X-ray absorbance.
X-ray absorbance μt = −ln (I 0 / I t )
In the above measurement, discrete X-ray absorbance is obtained corresponding to the wavelength of X-rays used for measurement (corresponding to the energy of X-rays used for measurement). The obtained X-ray absorbance was averaged and data interpolated as follows.
First, the X-ray absorbance in the range corresponding to the K absorption edge of Mn was averaged by the following method.
From 6040 eV to 6400 eV: 3 times of adjacent average processing of 21 points From 6400 eV to 6700 eV: 1 time of weighted average processing by 7 points of Savitzky-Golay method From 6700 eV to 7640.5 eV: 5 times of adjacent average processing of 11 points Subsequently, using the average value of the obtained X-ray absorbance, data interpolation was performed at the following data intervals to obtain an X-ray absorption spectrum having the X axis as the X-ray energy and the Y axis as the X absorbance.
6040 eV to 6508 eV: 6.5 eV interval 6508 eV to 6609.5 eV: 0.35 eV interval 6609.5 eV to 6640.5 eV: 1 eV interval 6640.5 eV to 7040.5 eV: 2.5 eV interval 7040.5 eV to 7640. Up to 5 eV: 6 eV interval Further, the X-ray absorbance in the range corresponding to the K absorption edge of Ni was averaged by the following method.
From 7834 eV to 8200 eV: 3 times of adjacent average processing of 21 points From 8200 eV to 8500 eV: 1 time of weighted average processing by 7 points of Savitzky-Golay method From 8500 eV to 9734.5 eV: 5 times of adjacent average processing of 11 points Subsequently, using the average value of the obtained X-ray absorbance, data interpolation was performed at the following data intervals to obtain an X-ray absorption spectrum having the X axis as the X-ray energy and the Y axis as the X absorbance.
From 7834 eV to 8302 eV: 6.5 eV interval From 8302 eV to 8403.5 eV: 0.35 eV interval From 8403.5 eV to 8459.5 eV: 1 eV interval From 8459.5 eV to 8834.5 eV: 2.5 eV interval From 8834.5 eV to 9434. Up to 5 eV: 6 eV interval (creation of EXAFS spectrum)
From the obtained X-ray absorption spectrum, an EXAFS spectrum of the K absorption edge of Mn and the K absorption edge of Ni was obtained as follows. Analysis of the X-ray absorption spectrum obtained above was performed using analysis software (Rigaku Corporation, REX2000).
First, the Mn K absorption edge E 0 was set to an energy value at which the first-order differential coefficient was maximum in the vicinity of the Mn K absorption edge in the X-ray absorption spectrum. Similarly, the K absorption edge E 0 of Ni is set to an energy value at which the first-order differential coefficient becomes maximum in the spectrum near the K absorption edge of Ni.
The spectrum background is a spectrum in a lower energy region than the K absorption edge of Mn and the K absorption edge of Ni, and Victory's formula (Aλ 3 −Bλ 4 + C; λ is the wavelength of incident X-rays, A, B and C are arbitrary constants) and determined by applying the least square method. The EXAFS spectrum was obtained by subtracting the background value corresponding to this Victreeen equation from the X-ray absorption spectrum.
(Calculation of radial distribution function)
A radial distribution function was obtained from the obtained EXAFS spectrum.
First, regarding the EXAFS spectrum, the absorbance (μ 0 ) of isolated atoms was estimated by the Spline Smoothing method (smoothing spline method), and the EXAFS function χ (k) was extracted. Note that k is the wave number of photoelectrons defined by 0.5123 × (E−E 0 ) 1/2 , and the unit of k is −1 .
Next, the EXAFS function k 3 χ (k) weighted by k 3 was subjected to Fourier transform in the range of k from 3.0 to 11.85Å −1 to obtain a radial distribution function.
(2) Production of positive electrode A lithium composite metal oxide (positive electrode active material), a conductive material (acetylene black: graphite = 9: 1 (mass ratio)) and a binder (PVdF) obtained by the production method described later are used as a positive electrode active material. A paste-like positive electrode mixture was prepared by adding and kneading so that the composition of the substance: conductive material: binder = 87: 10: 3 (mass ratio) was obtained. When preparing the positive electrode mixture, N-methyl-2-pyrrolidone was used as the organic solvent.
The obtained positive electrode mixture was applied to a 40 μm thick Al foil serving as a current collector and vacuum dried at 150 ° C. for 8 hours to obtain a positive electrode.
(3) Production of nonaqueous electrolyte secondary battery (coin cell) The following operation was performed in a glove box in an argon atmosphere.
The positive electrode created in “(2) Preparation of positive electrode” is placed on the lower lid of a coin cell (manufactured by Hosen Co., Ltd.) for coin-type battery R2032 with the aluminum foil surface facing downward, and a laminated film separator (polyethylene) is placed thereon. A separator (thickness 16 μm) having a heat-resistant porous layer laminated thereon was placed on the porous film. 300 μl of electrolyte was injected here. The electrolyte used was prepared by dissolving LiPF 6 in a 30:35:35 (volume ratio) mixture of ethylene carbonate, dimethyl carbonate, and ethyl methyl carbonate to a concentration of 1 mol / l.
Next, using lithium metal as the negative electrode, the lithium metal is placed on the upper side of the laminated film separator, covered with a gasket, and caulked with a non-aqueous electrolyte secondary battery (coin type battery R2032, hereinafter, (Sometimes referred to as a “coin-type battery”).
(4) Charge / Discharge Test Using the coin-type battery created in “(3) Production of Nonaqueous Electrolyte Secondary Battery (Coin Cell)”, a charge / discharge test was performed under the following conditions. The charge capacity and discharge capacity in the charge / discharge test were determined as follows.
<Charge / discharge test conditions>
Test temperature: 25 ° C or 60 ° C
Charging conditions: Maximum charging voltage 4.3V, charging
Discharge conditions: discharge minimum voltage 2.5V,
(Example 1)
1. Production of Lithium Composite Metal Oxide Precursor (Coprecipitate) Nickel sulfate hexahydrate, manganese sulfate monohydrate, and cobalt sulfate heptahydrate have a molar ratio of Ni: Mn: Co of 0.45: 0. .45: 0.10, each was weighed and dissolved in pure water to obtain an aqueous transition metal solution containing Ni ions, Mn ions, Co ions and SO 4 2- ions.
To this transition metal aqueous solution, an aqueous potassium hydroxide solution was added to perform coprecipitation to produce a precipitate, thereby obtaining a slurry. The obtained slurry was subjected to solid-liquid separation, washed with distilled water to obtain a coprecipitate Q 1 and dried for 8 hours at 100 ° C..
2. Production of lithium composite metal oxide The amount (mol) of Li is 1.3 with respect to the obtained coprecipitate Q 1 and the total amount (mol) 1 of transition metals contained in the coprecipitate Q 1. The lithium carbonate weighed in this manner and potassium sulfate as an inert flux were mixed in a mortar to obtain a mixture.
Next, the obtained mixture was placed in an alumina firing container, and the alumina firing container was placed in an electric furnace. The oxygen concentration was adjusted to 8.5% by volume using the air atmosphere originally present in the electric furnace and the introduced nitrogen gas, and heated at 400 ° C. to obtain a calcined product. Further, the oxygen concentration was adjusted to 1% by volume, the mixture was held at 850 ° C. for 6 hours and baked, and cooled to room temperature to obtain a baked product.
The fired product obtained was pulverized and dispersed in distilled water. The supernatant after standing was removed by decantation, filtered, and dried at 300 ° C. for 6 hours to obtain a powdered lithium composite metal oxide A 1 .
3. The results of composition analysis of A 1 to evaluate the resulting lithium composite metal oxide, Li: Ni: Mn: the molar ratio of Co is 1.03: 0.45: 0.45: was 0.10 .
And by performing powder X-ray diffractometry of A 1, and a compound of layered structure type. As a result of Rietveld analysis, the crystal structure of A 1 is hexagonal, which is classified to the space group R-3m.
After obtaining EXAFS spectra of Mn and Ni of A 1 by the above-described method, a radial distribution function was obtained by Fourier transform. In the radial distribution function of Mn , the intensity ratio I BMn / I AMn of the peak A Mn of 1.53 と and the peak B Mn of 2.49 Å was 1.12.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 1.69.
Furthermore, I BMn / I AMn × I BNi / I ANi was 1.89.
4). A coin type battery was produced by using a charge-discharge test A 1 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 170. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 178.
(Example 2)
1. Preparation of lithium composite metal oxide precursor (coprecipitate) and manufacture of lithium composite metal oxide Nickel sulfate hexahydrate, manganese sulfate monohydrate, cobalt sulfate heptahydrate, Ni: Mn: Co molar ratio of 0.47: 0.48: except that each weighed so that 0.05 was obtained in the same manner as in example 1, to obtain a coprecipitate Q 2. Furthermore, except for using a coprecipitate Q 2, the procedure of Example 1 to obtain a powdered lithium composite metal oxide A 2.
2. Evaluation of Lithium Composite Metal Oxide The composition of A 2 obtained was analyzed. The molar ratio of Li: Ni: Mn: Co was 1.06: 0.49: 0.45: 0.06. .
And by performing powder X-ray diffraction measurement of A 2, and a compound of layered structure type. As a result of Rietveld analysis, the crystal structure of A 2 is a hexagonal, classified to the space group R-3m.
After obtaining EX 2 AFS spectra of Mn and Ni of A 2 by the above-mentioned method, a radial distribution function was obtained by Fourier transform. In the radial distribution function of Mn , the intensity ratio I BMn / I AMn of the peak A Mn of 1.53 と and the peak B Mn of 2.49 Å was 1.14.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 1.69.
Furthermore, I BMn / I AMn × I BNi / I ANi was 1.93.
3. A coin type battery was produced by using the charge and discharge test A 2 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., 0.2 C definitive discharge capacity (mAh / g) was 168. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 178.
(Example 3)
1. Preparation of lithium composite metal oxide precursor (coprecipitate) and manufacture of lithium composite metal oxide Nickel sulfate hexahydrate, manganese sulfate monohydrate, cobalt sulfate heptahydrate, Ni: Mn: Co molar ratio of 0.48: 0.49: except that each weighed so that 0.03 was obtained in the same manner as in example 1, to obtain a coprecipitate Q 3. Furthermore, except for using a coprecipitate Q 3, the procedure of Example 1 to obtain a powdered lithium composite metal oxide A 3.
2. The results of composition analysis of A 3 Evaluation resulting lithium composite metal oxide, Li: Ni: Mn: the molar ratio of Co is 1.00: 0.48: 0.49: was 0.03 .
And by performing powder X-ray diffraction measurement of A 3, it was a compound having a layered structure type. As a result of Rietveld analysis, the crystal structure of A 3 is hexagonal, which is classified to the space group R-3m.
After obtaining the EXAFS spectra of Mn and Ni of A 3 by the method described above to obtain the radial distribution function by applying a Fourier transform. In the Mn radial distribution function, the intensity ratio I BMn / I AMn of the peak A Mn of 1.50Å and the peak B Mn of 2.52Å was 1.15.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 1.58.
Furthermore, I BMn / I AMn × I BNi / I ANi was 1.82.
3. A coin type battery was produced by using the charge and discharge test A 3 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 164. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 175.
(Comparative Example 1)
1. And manufacturing the coprecipitate to Q 1 lithium composite metal oxide, the total amount (mol) 1 of the transition metal contained in the coprecipitate Q 1, as the amount of Li (mole) is 1.3 Weighed lithium carbonate and potassium sulfate as an inert flux were mixed in a mortar to obtain a mixture.
Subsequently, the obtained mixture was put into an alumina firing container, and fired by holding at 850 ° C. in an air atmosphere for 6 hours using an electric furnace, and cooled to room temperature to obtain a fired product.
The obtained fired product was pulverized, washed with distilled water by decantation, filtered, and dried at 300 ° C. for 6 hours to obtain a powdered lithium composite metal oxide R 1 .
2. Evaluation of lithium composite metal oxide The composition of R 1 obtained was analyzed. The molar ratio of Li: Ni: Mn: Co was 1.18: 0.46: 0.44: 0.10. .
When a powder X-ray diffraction measurement of R 1 was performed, it was a layered structure type compound. As a result of Rietveld analysis, the crystal structure of R 1 was hexagonal and was classified into the space group R-3m.
After obtaining EX1 AFS spectra of Mn and Ni of R 1 by the above method, a radial distribution function was obtained by Fourier transform. In the radial distribution function of Mn , the intensity ratio I BMn / I AMn of the peak A Mn of 1.56 と and the peak B Mn of 2.49 0.9 was 0.96.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 2.13.
3. A coin type battery was produced by using a charge-discharge test R 1 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 154. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 162.
(Comparative Example 2)
1. Except for using the prepared coprecipitate Q 2 of the lithium composite metal oxide, the same operation as in Comparative Example 1 to obtain a powdered lithium composite metal oxide R 2.
2. The results of composition analysis of the evaluation R 2 of the lithium composite metal oxide, Li: Ni: Mn: the molar ratio of Co is 1.16: 0.47: 0.48: was 0.05.
When a powder X-ray diffraction measurement of R 2 was performed, it was a layered structure type compound. As a result of Rietveld analysis, the crystal structure of R 2 was hexagonal and classified into the space group R-3m.
After obtaining EX 2 AFS spectra of Mn and Ni of R 2 by the above-described method, a radial distribution function was obtained by Fourier transform. In the Mn radial distribution function, the intensity ratio I BMn / I AMn of the peak A Mn of 1.53 Å and the peak B Mn of 2.49 Å was 0.94.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.60 と and the peak B Ni of 2.49 Å was 2.04.
3. A coin type battery was produced by using a charge-discharge test R 2 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 149. When a discharge test at 60 ° C. was performed, the discharge capacity (mAh / g) at 0.2 C was 160.
(Comparative Example 3)
1. Except for using the prepared coprecipitate Q 3 of the lithium composite metal oxide, the same operation as in Comparative Example 1 to obtain a powdered lithium composite metal oxide R 3.
2. Evaluation of Lithium Composite Metal Oxide When the composition analysis of R 3 was performed, the molar ratio of Li: Ni: Mn: Co was 1.10: 0.48: 0.49: 0.03.
When a powder X-ray diffraction measurement of R 3 was performed, it was a layered structure type compound. As a result of Rietveld analysis, the crystal structure of R 3 was hexagonal and was classified into the space group R-3m.
After obtaining EXAFS spectra of Mn and Ni of R 3 by the above method, a radial distribution function was obtained by Fourier transform. In the Mn radial distribution function, the intensity ratio I BMn / I AMn of the peak A Mn of 1.50 Å and the peak B Mn of 2.49 Å was 0.94.
In the Ni radial distribution function, the intensity ratio I BNi / I ANi between the peak A Ni of 1.56 and the peak B Ni of 2.49 was 2.04.
3. A coin type battery was produced by using a charge-discharge test R 3 of the non-aqueous electrolyte secondary battery was subjected to a discharge test at 25 ° C., the discharge capacity at 0.2C (mAh / g) was 148. When a discharge test at 60 ° C. was performed, the discharge capacity at 0.2 C was 159.
The results of Examples 1 to 3 and Comparative Examples 1 to 3 are shown in Table 1 below.
Moreover, even if it is the lithium composite metal oxide of Example 3 with the least Co usage-amount among Examples, it discharges rather than the lithium composite metal oxide of Comparative Example 1 with the most Co usage-amount among Comparative Examples. Since a non-aqueous electrolyte secondary battery having a large capacity was obtained, it was found that the performance could be maintained and improved even if the amount of Co used was reduced.
From the above results, it was found that the lithium composite metal oxide of the present invention is useful for a non-aqueous electrolyte secondary battery exhibiting a high capacity. Further, the positive electrode active material using the lithium composite metal oxide of the present invention, the positive electrode is useful for a high-performance non-aqueous electrolyte secondary battery, and the non-aqueous electrolyte secondary battery of the present invention has a higher capacity than before. It was found that
本発明によれば、従来よりも高容量を示す非水電解質二次電池に有用なリチウム複合金属酸化物を提供することができる。また、リチウム複合金属酸化物を用いた正極活物質、正極、非水電解質二次電池を提供することができる。
According to the present invention, it is possible to provide a lithium composite metal oxide useful for a nonaqueous electrolyte secondary battery exhibiting a higher capacity than before. In addition, a positive electrode active material, a positive electrode, and a nonaqueous electrolyte secondary battery using a lithium composite metal oxide can be provided.
Claims (11)
- Mn、Ni、LiおよびCoを含有し、下記(a)および(b)を満たすリチウム複合金属酸化物:
(a)前記リチウム複合金属酸化物におけるMnのK吸収端の広域X線吸収微細構造(EXAFS)スペクトルをフーリエ変換して得られる動径分布関数において、Mn原子に結合した酸素原子による1.5Å付近の第一近接ピークAMnの強度をIAMn、Mn原子に結合した酸素原子の次にMn原子に近い金属原子による2.5Å付近の第二近接ピークBMnの強度をIBMnとしたとき、IBMn/IAMnが、0.5以上1.2以下である、
(b)前記リチウム複合金属酸化物におけるNiのK吸収端のEXAFSスペクトルをフーリエ変換して得られる動径分布関数において、Ni原子に結合した酸素原子による1.5Å付近の第一近接ピークANiの強度をIANi、Ni原子に結合した酸素原子の次にNi原子に近い金属原子による2.5Å付近の第二近接ピークの強度をIBNiとしたとき、IBNi/IANiが、1.0以上1.7以下である。 Lithium composite metal oxide containing Mn, Ni, Li and Co and satisfying the following (a) and (b):
(A) In a radial distribution function obtained by Fourier transform of a wide-range X-ray absorption fine structure (EXAFS) spectrum at the K absorption edge of Mn in the lithium composite metal oxide, 1.5% by oxygen atoms bonded to Mn atoms is obtained. when the first proximity peak a Mn of intensity I AMn around, the intensity of the second closest peak B Mn around 2.5Å by metal atoms near the next Mn atoms of the oxygen atoms bonded to Mn atoms was I BMn IBMn / IAMn is 0.5 or more and 1.2 or less,
(B) In the radial distribution function obtained by Fourier transforming the EXAFS spectrum at the K absorption edge of Ni in the lithium composite metal oxide, the first proximity peak A Ni near 1.5Å due to oxygen atoms bonded to Ni atoms. the intensity I ANi, when the next on the intensity of the second closest peak around 2.5Å by metal atoms near the Ni atom bonded oxygen atom to Ni atom was I BNi, the I BNi / I ANi, 1. It is 0 or more and 1.7 or less. - Liの量(モル)をALi、Li以外の金属の量(モル)をAとしたとき、ALi/Aが、0.7以上1.4以下である請求項1に記載のリチウム複合金属酸化物。 The lithium composite metal according to claim 1, wherein A Li / A is 0.7 or more and 1.4 or less, where A Li is the amount (mol) of Li and A is the amount (mol) of a metal other than Li . Oxides.
- 前記IBMn/IAMnと、前記IBNi/IANiとの積が、0.7以上2.0以下である請求項1または2に記載のリチウム複合金属酸化物。 3. The lithium mixed metal oxide according to claim 1, wherein a product of the I BMn / I AMn and the I BNi / I ANi is 0.7 or more and 2.0 or less.
- 前記リチウム複合金属酸化物が層状構造を有し、式(1)で表される請求項1から3のいずれか1項に記載のリチウム複合金属酸化物:
Li1+x(Ni1−x−y−α−βMnyCoαMβ)O2 …(1)
(式(1)中、−0.3≦x≦0.4、0.35≦y≦0.7、0<α≦0.1、0≦β<0.1(ただし、0<α+β≦0.1)であり、−0.05≦x+y+α+β<1であり、MはAl、Mg、Ti、Ca、Cu、Zn、Fe、Cr、Mo、Si、Sn、NbおよびVからなる群より選ばれる1種類以上の元素である。)。 The lithium composite metal oxide according to any one of claims 1 to 3, wherein the lithium composite metal oxide has a layered structure and is represented by the formula (1):
Li 1 + x (Ni 1- x-y-α-β Mn y Co α M β) O 2 ... (1)
(In the formula (1), −0.3 ≦ x ≦ 0.4, 0.35 ≦ y ≦ 0.7, 0 <α ≦ 0.1, 0 ≦ β <0.1 (where 0 <α + β ≦ 0.1), −0.05 ≦ x + y + α + β <1, and M is selected from the group consisting of Al, Mg, Ti, Ca, Cu, Zn, Fe, Cr, Mo, Si, Sn, Nb and V One or more elements.) - 前記MがFeである請求項4に記載のリチウム複合金属酸化物。 The lithium mixed metal oxide according to claim 4, wherein the M is Fe.
- β=0である請求項4に記載のリチウム複合金属酸化物。 The lithium composite metal oxide according to claim 4, wherein β = 0.
- 請求項1から6のいずれか1項に記載のリチウム複合金属酸化物を有する正極活物質。 A positive electrode active material comprising the lithium composite metal oxide according to any one of claims 1 to 6.
- 請求項7に記載の正極活物質を有する正極。 A positive electrode having the positive electrode active material according to claim 7.
- 負極、および請求項8に記載の正極を有する非水電解質二次電池。 A nonaqueous electrolyte secondary battery having a negative electrode and the positive electrode according to claim 8.
- 前記負極および前記正極間に配置されたセパレータをさらに有する請求項9に記載の非水電解質二次電池。 The nonaqueous electrolyte secondary battery according to claim 9, further comprising a separator disposed between the negative electrode and the positive electrode.
- 前記セパレータが、耐熱多孔層と多孔質フィルムとが積層された積層フィルムからなる請求項10に記載の非水電解質二次電池。 The non-aqueous electrolyte secondary battery according to claim 10, wherein the separator is a laminated film in which a heat-resistant porous layer and a porous film are laminated.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014523798A JPWO2014007357A1 (en) | 2012-07-06 | 2013-06-28 | Lithium composite metal oxide, positive electrode active material, positive electrode and non-aqueous electrolyte secondary battery |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-153046 | 2012-07-06 | ||
| JP2012153046 | 2012-07-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014007357A1 true WO2014007357A1 (en) | 2014-01-09 |
Family
ID=49882108
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/068451 WO2014007357A1 (en) | 2012-07-06 | 2013-06-28 | Lithium composite metal oxide, positive electrode active substance, positive electrode, and non-aqueous electrolyte secondary battery |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2014007357A1 (en) |
| WO (1) | WO2014007357A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150070626A1 (en) * | 2013-09-12 | 2015-03-12 | Lg Display Co., Ltd. | Columnar light emitting device, liquid crystal display device including the same, and method of manufacturing the same |
| JP2015528789A (en) * | 2012-07-20 | 2015-10-01 | スリーエム イノベイティブ プロパティズ カンパニー | High voltage cathode composition for lithium ion battery |
| WO2018181530A1 (en) * | 2017-03-31 | 2018-10-04 | 住友化学株式会社 | Production method for lithium metal complex oxide |
| US20210057742A1 (en) * | 2018-07-12 | 2021-02-25 | Panasonic Intellectual Property Management Co., Ltd. | Positive electrode active material and battery including the same |
| KR20210112393A (en) * | 2019-02-27 | 2021-09-14 | 미쓰이금속광업주식회사 | Active material, positive electrode mixture and solid battery using the same |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11102703A (en) * | 1997-09-26 | 1999-04-13 | Asahi Chem Ind Co Ltd | Nonaqueous secondary battery |
| JP2003346806A (en) * | 2002-05-30 | 2003-12-05 | Sony Corp | Positive electrode material for non-aqueous electrolyte secondary battery, and non-aqueous electrolyte secondary battery |
| JP2005332713A (en) * | 2004-05-20 | 2005-12-02 | Toyota Motor Corp | Lithium secondary battery and positive electrode active material for secondary battery |
| WO2009005164A1 (en) * | 2007-07-03 | 2009-01-08 | Sumitomo Chemical Company, Limited | Lithium composite metal oxide |
| JP2009152114A (en) * | 2007-12-21 | 2009-07-09 | Gs Yuasa Corporation | Active material for lithium secondary battery, lithium secondary battery, and manufacturing method thereof |
| WO2011024283A1 (en) * | 2009-08-27 | 2011-03-03 | トヨタ自動車株式会社 | Method for evaluating positive electrode active material |
-
2013
- 2013-06-28 JP JP2014523798A patent/JPWO2014007357A1/en active Pending
- 2013-06-28 WO PCT/JP2013/068451 patent/WO2014007357A1/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11102703A (en) * | 1997-09-26 | 1999-04-13 | Asahi Chem Ind Co Ltd | Nonaqueous secondary battery |
| JP2003346806A (en) * | 2002-05-30 | 2003-12-05 | Sony Corp | Positive electrode material for non-aqueous electrolyte secondary battery, and non-aqueous electrolyte secondary battery |
| JP2005332713A (en) * | 2004-05-20 | 2005-12-02 | Toyota Motor Corp | Lithium secondary battery and positive electrode active material for secondary battery |
| WO2009005164A1 (en) * | 2007-07-03 | 2009-01-08 | Sumitomo Chemical Company, Limited | Lithium composite metal oxide |
| JP2009152114A (en) * | 2007-12-21 | 2009-07-09 | Gs Yuasa Corporation | Active material for lithium secondary battery, lithium secondary battery, and manufacturing method thereof |
| WO2011024283A1 (en) * | 2009-08-27 | 2011-03-03 | トヨタ自動車株式会社 | Method for evaluating positive electrode active material |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015528789A (en) * | 2012-07-20 | 2015-10-01 | スリーエム イノベイティブ プロパティズ カンパニー | High voltage cathode composition for lithium ion battery |
| US20150070626A1 (en) * | 2013-09-12 | 2015-03-12 | Lg Display Co., Ltd. | Columnar light emitting device, liquid crystal display device including the same, and method of manufacturing the same |
| WO2018181530A1 (en) * | 2017-03-31 | 2018-10-04 | 住友化学株式会社 | Production method for lithium metal complex oxide |
| JP2018172257A (en) * | 2017-03-31 | 2018-11-08 | 住友化学株式会社 | Method for producing lithium metal composite oxide |
| US20210057742A1 (en) * | 2018-07-12 | 2021-02-25 | Panasonic Intellectual Property Management Co., Ltd. | Positive electrode active material and battery including the same |
| KR20210112393A (en) * | 2019-02-27 | 2021-09-14 | 미쓰이금속광업주식회사 | Active material, positive electrode mixture and solid battery using the same |
| KR102376785B1 (en) * | 2019-02-27 | 2022-03-21 | 미쓰이금속광업주식회사 | Active material, positive electrode mixture and solid battery using the same |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2014007357A1 (en) | 2016-06-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5701343B2 (en) | Positive electrode active material for lithium secondary battery, positive electrode and secondary battery | |
| JP5481786B2 (en) | Lithium composite metal oxide | |
| WO2018123817A1 (en) | Positive electrode active material for lithium secondary batteries, positive electrode for lithium secondary batteries, and lithium secondary battery | |
| JP6500045B2 (en) | Method for producing lithium composite metal oxide | |
| JP5640311B2 (en) | Lithium composite metal oxide and non-aqueous electrolyte secondary battery | |
| JP6669920B1 (en) | Positive electrode active material for secondary battery and method for producing the same | |
| JP6768647B2 (en) | Positive electrode active material for lithium secondary battery, positive electrode for lithium secondary battery and lithium secondary battery | |
| WO2014007360A1 (en) | Lithium composite metal oxide, production method for lithium composite metal oxide, positive electrode active substance, positive electrode, and non-aqueous electrolyte secondary battery | |
| JP5842478B2 (en) | Lithium composite metal oxide and method for producing the same | |
| JP5504800B2 (en) | Lithium composite metal oxide and positive electrode active material | |
| JP5487821B2 (en) | Lithium composite metal oxide and positive electrode active material | |
| JP2011126771A (en) | Transition metal-containing multiple hydroxide and lithium-containing multiple metal oxide | |
| JP5810497B2 (en) | Lithium composite metal oxide and non-aqueous electrolyte secondary battery | |
| JP6068530B2 (en) | Positive electrode active material for lithium secondary battery, positive electrode and secondary battery | |
| WO2014007357A1 (en) | Lithium composite metal oxide, positive electrode active substance, positive electrode, and non-aqueous electrolyte secondary battery | |
| JP5742192B2 (en) | Method for producing lithium composite metal oxide | |
| JP2013107791A (en) | Method for producing complex metal oxide, complex metal oxide, positive electrode active material, positive electrode, and nonaqueous electrolyte secondary battery | |
| JP5742193B2 (en) | Lithium composite metal oxide and non-aqueous electrolyte secondary battery | |
| JP2011153067A (en) | Method for producing composite metal hydroxide and lithium composite metal oxide, and nonaqueous electrolyte secondary battery |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13812679 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014523798 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13812679 Country of ref document: EP Kind code of ref document: A1 |