WO2013185116A1 - Lipids for therapeutic agent delivery formulations - Google Patents
Lipids for therapeutic agent delivery formulations Download PDFInfo
- Publication number
- WO2013185116A1 WO2013185116A1 PCT/US2013/044849 US2013044849W WO2013185116A1 WO 2013185116 A1 WO2013185116 A1 WO 2013185116A1 US 2013044849 W US2013044849 W US 2013044849W WO 2013185116 A1 WO2013185116 A1 WO 2013185116A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- dcm
- diyl
- ethane
- bis
- Prior art date
Links
- 150000002632 lipids Chemical class 0.000 title claims abstract description 65
- 239000003814 drug Substances 0.000 title claims abstract description 38
- 239000000203 mixture Substances 0.000 title claims description 154
- 229940124597 therapeutic agent Drugs 0.000 title abstract description 27
- 238000009472 formulation Methods 0.000 title description 63
- 239000002502 liposome Substances 0.000 claims abstract description 42
- -1 lipid compound Chemical class 0.000 claims description 129
- 150000001875 compounds Chemical class 0.000 claims description 89
- 108020004459 Small interfering RNA Proteins 0.000 claims description 38
- 238000000034 method Methods 0.000 claims description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 30
- 239000008194 pharmaceutical composition Substances 0.000 claims description 25
- 150000004492 retinoid derivatives Chemical class 0.000 claims description 24
- 150000003839 salts Chemical group 0.000 claims description 20
- 239000003937 drug carrier Substances 0.000 claims description 18
- 239000002202 Polyethylene glycol Substances 0.000 claims description 13
- 239000007924 injection Substances 0.000 claims description 13
- 238000002347 injection Methods 0.000 claims description 13
- 229920001223 polyethylene glycol Polymers 0.000 claims description 13
- 239000002609 medium Substances 0.000 claims description 12
- 239000007788 liquid Substances 0.000 claims description 10
- 239000003085 diluting agent Substances 0.000 claims description 8
- 229940079593 drug Drugs 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 6
- 210000004500 stellate cell Anatomy 0.000 claims description 6
- 239000012736 aqueous medium Substances 0.000 claims description 5
- 239000003960 organic solvent Substances 0.000 claims description 5
- 125000005647 linker group Chemical group 0.000 claims description 4
- 125000001117 oleyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 3
- 229910052760 oxygen Inorganic materials 0.000 claims description 3
- 150000003904 phospholipids Chemical class 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 230000008685 targeting Effects 0.000 claims description 2
- 125000005645 linoleyl group Chemical group 0.000 claims 1
- 230000002708 enhancing effect Effects 0.000 abstract description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 322
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 132
- 238000002360 preparation method Methods 0.000 description 59
- 238000003756 stirring Methods 0.000 description 43
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 42
- 210000004027 cell Anatomy 0.000 description 41
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 36
- 239000002924 silencing RNA Substances 0.000 description 35
- 238000010898 silica gel chromatography Methods 0.000 description 33
- 238000000746 purification Methods 0.000 description 32
- 239000011541 reaction mixture Substances 0.000 description 32
- 239000000243 solution Substances 0.000 description 32
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 30
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 30
- 239000012074 organic phase Substances 0.000 description 30
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 28
- 239000008346 aqueous phase Substances 0.000 description 25
- 125000000217 alkyl group Chemical group 0.000 description 24
- 125000001821 azanediyl group Chemical group [H]N(*)* 0.000 description 24
- 230000015572 biosynthetic process Effects 0.000 description 21
- 125000002947 alkylene group Chemical group 0.000 description 20
- 238000006243 chemical reaction Methods 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 18
- 238000003786 synthesis reaction Methods 0.000 description 18
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 17
- 239000012044 organic layer Substances 0.000 description 17
- 229910000027 potassium carbonate Inorganic materials 0.000 description 17
- 239000000725 suspension Substances 0.000 description 16
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 14
- 229910052786 argon Inorganic materials 0.000 description 14
- 239000002953 phosphate buffered saline Substances 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- 238000000338 in vitro Methods 0.000 description 11
- 239000000463 material Substances 0.000 description 11
- 239000003921 oil Substances 0.000 description 11
- 235000019198 oils Nutrition 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- 125000004432 carbon atom Chemical group C* 0.000 description 10
- HCUYBXPSSCRKRF-UHFFFAOYSA-N diphosgene Chemical compound ClC(=O)OC(Cl)(Cl)Cl HCUYBXPSSCRKRF-UHFFFAOYSA-N 0.000 description 10
- 239000010410 layer Substances 0.000 description 10
- UMFJAHHVKNCGLG-UHFFFAOYSA-N n-Nitrosodimethylamine Chemical compound CN(C)N=O UMFJAHHVKNCGLG-UHFFFAOYSA-N 0.000 description 10
- OYHQOLUKZRVURQ-UHFFFAOYSA-M octadeca-9,12-dienoate Chemical compound CCCCCC=CCC=CCCCCCCCC([O-])=O OYHQOLUKZRVURQ-UHFFFAOYSA-M 0.000 description 10
- 239000003381 stabilizer Substances 0.000 description 10
- YWWVWXASSLXJHU-UHFFFAOYSA-N tetradec-9-enoic acid Chemical compound CCCCC=CCCCCCCCC(O)=O YWWVWXASSLXJHU-UHFFFAOYSA-N 0.000 description 10
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 125000003342 alkenyl group Chemical group 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 230000014509 gene expression Effects 0.000 description 9
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 239000007789 gas Substances 0.000 description 8
- 239000011261 inert gas Substances 0.000 description 8
- 239000000546 pharmaceutical excipient Substances 0.000 description 8
- 238000001890 transfection Methods 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 7
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 7
- 206010028980 Neoplasm Diseases 0.000 description 7
- 101800001271 Surface protein Proteins 0.000 description 7
- 101800000385 Transmembrane protein Proteins 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 7
- 201000011510 cancer Diseases 0.000 description 7
- 239000000284 extract Substances 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 239000007821 HATU Substances 0.000 description 6
- 101000836383 Homo sapiens Serpin H1 Proteins 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 102100027287 Serpin H1 Human genes 0.000 description 6
- 230000037396 body weight Effects 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 6
- 108020004999 messenger RNA Proteins 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 238000003757 reverse transcription PCR Methods 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 239000000375 suspending agent Substances 0.000 description 6
- HQYVJRZFQDKZCJ-UHFFFAOYSA-N tert-butyl n,n-bis(3-hydroxypropyl)carbamate Chemical compound CC(C)(C)OC(=O)N(CCCO)CCCO HQYVJRZFQDKZCJ-UHFFFAOYSA-N 0.000 description 6
- 231100000419 toxicity Toxicity 0.000 description 6
- 230000001988 toxicity Effects 0.000 description 6
- 230000003612 virological effect Effects 0.000 description 6
- CXMYWOCYTPKBPP-UHFFFAOYSA-N 3-(3-hydroxypropylamino)propan-1-ol Chemical compound OCCCNCCCO CXMYWOCYTPKBPP-UHFFFAOYSA-N 0.000 description 5
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 5
- 206010016654 Fibrosis Diseases 0.000 description 5
- 102100031181 Glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 230000001668 ameliorated effect Effects 0.000 description 5
- 238000010936 aqueous wash Methods 0.000 description 5
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 5
- 125000002091 cationic group Chemical group 0.000 description 5
- 229940106189 ceramide Drugs 0.000 description 5
- 239000013058 crude material Substances 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 230000004761 fibrosis Effects 0.000 description 5
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 230000002195 synergetic effect Effects 0.000 description 5
- WLKADMLMRXJDKL-UHFFFAOYSA-N 2-[2-(dimethylazaniumyl)ethylsulfanyl]acetate Chemical compound CN(C)CCSCC(O)=O WLKADMLMRXJDKL-UHFFFAOYSA-N 0.000 description 4
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical class Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 4
- FFDGPVCHZBVARC-UHFFFAOYSA-N N,N-dimethylglycine Chemical compound CN(C)CC(O)=O FFDGPVCHZBVARC-UHFFFAOYSA-N 0.000 description 4
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 230000036470 plasma concentration Effects 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 238000010791 quenching Methods 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 238000002390 rotary evaporation Methods 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- KMUNFRBJXIEULW-UHFFFAOYSA-N tert-butyl n,n-bis(2-hydroxyethyl)carbamate Chemical compound CC(C)(C)OC(=O)N(CCO)CCO KMUNFRBJXIEULW-UHFFFAOYSA-N 0.000 description 4
- IFABLCIRROMTAN-MDZDMXLPSA-N (e)-1-chlorooctadec-9-ene Chemical compound CCCCCCCC\C=C\CCCCCCCCCl IFABLCIRROMTAN-MDZDMXLPSA-N 0.000 description 3
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 3
- XCJLSNCDAIYFHX-UHFFFAOYSA-N 2-(2-tetradecanoyloxyethylamino)ethyl tetradecanoate Chemical compound CCCCCCCCCCCCCC(=O)OCCNCCOC(=O)CCCCCCCCCCCCC XCJLSNCDAIYFHX-UHFFFAOYSA-N 0.000 description 3
- DENMGZODXQRYAR-UHFFFAOYSA-N 2-(dimethylamino)ethanethiol Chemical compound CN(C)CCS DENMGZODXQRYAR-UHFFFAOYSA-N 0.000 description 3
- 239000000232 Lipid Bilayer Substances 0.000 description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- VYGQUTWHTHXGQB-FFHKNEKCSA-N Retinol Palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C VYGQUTWHTHXGQB-FFHKNEKCSA-N 0.000 description 3
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 238000010171 animal model Methods 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 229920006317 cationic polymer Polymers 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 235000012000 cholesterol Nutrition 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 108700003601 dimethylglycine Proteins 0.000 description 3
- 238000004821 distillation Methods 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 201000005202 lung cancer Diseases 0.000 description 3
- 208000020816 lung neoplasm Diseases 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- YWWVWXASSLXJHU-AATRIKPKSA-N (9E)-tetradecenoic acid Chemical compound CCCC\C=C\CCCCCCCC(O)=O YWWVWXASSLXJHU-AATRIKPKSA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- KILNVBDSWZSGLL-KXQOOQHDSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCC KILNVBDSWZSGLL-KXQOOQHDSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- ZEGDFCCYTFPECB-UHFFFAOYSA-N 2,3-dimethoxynaphthalene-1,4-dione Chemical compound C1=CC=C2C(=O)C(OC)=C(OC)C(=O)C2=C1 ZEGDFCCYTFPECB-UHFFFAOYSA-N 0.000 description 2
- BHQDXEHJSJWKMQ-UHFFFAOYSA-N 2-[2-(dimethylamino)ethylsulfanyl]acetic acid;hydrochloride Chemical compound Cl.CN(C)CCSCC(O)=O BHQDXEHJSJWKMQ-UHFFFAOYSA-N 0.000 description 2
- UNSAJINGUOTTRA-UHFFFAOYSA-N 3-(3-bromophenyl)prop-2-yn-1-ol Chemical compound OCC#CC1=CC=CC(Br)=C1 UNSAJINGUOTTRA-UHFFFAOYSA-N 0.000 description 2
- 102100028104 39S ribosomal protein L19, mitochondrial Human genes 0.000 description 2
- OXOWTLDONRGYOT-UHFFFAOYSA-N 4-(dimethylamino)butanoic acid Chemical compound CN(C)CCCC(O)=O OXOWTLDONRGYOT-UHFFFAOYSA-N 0.000 description 2
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- UYZSNVLEDLCWGU-UHFFFAOYSA-N 5-(dimethylazaniumyl)pentanoate Chemical compound CN(C)CCCCC(O)=O UYZSNVLEDLCWGU-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- 201000003883 Cystic fibrosis Diseases 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 206010019668 Hepatic fibrosis Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101001079803 Homo sapiens 39S ribosomal protein L19, mitochondrial Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 206010067125 Liver injury Diseases 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- JMOXSQYGVIXBBZ-UHFFFAOYSA-N N,N-dimethyl-beta-alanine Chemical compound CN(C)CCC(O)=O JMOXSQYGVIXBBZ-UHFFFAOYSA-N 0.000 description 2
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- 229920002873 Polyethylenimine Polymers 0.000 description 2
- WUGQZFFCHPXWKQ-UHFFFAOYSA-N Propanolamine Chemical compound NCCCO WUGQZFFCHPXWKQ-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 239000008366 buffered solution Substances 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 238000002784 cytotoxicity assay Methods 0.000 description 2
- 231100000263 cytotoxicity test Toxicity 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 230000003176 fibrotic effect Effects 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 238000003633 gene expression assay Methods 0.000 description 2
- 231100000234 hepatic damage Toxicity 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 230000008818 liver damage Effects 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 239000000178 monomer Substances 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 239000008024 pharmaceutical diluent Substances 0.000 description 2
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 239000001103 potassium chloride Substances 0.000 description 2
- 235000011164 potassium chloride Nutrition 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 235000020944 retinol Nutrition 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 239000004055 small Interfering RNA Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 231100000027 toxicology Toxicity 0.000 description 2
- 231100000041 toxicology testing Toxicity 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 210000001260 vocal cord Anatomy 0.000 description 2
- QIJRTFXNRTXDIP-UHFFFAOYSA-N (1-carboxy-2-sulfanylethyl)azanium;chloride;hydrate Chemical compound O.Cl.SCC(N)C(O)=O QIJRTFXNRTXDIP-UHFFFAOYSA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- GRZHHTYDZVRPIC-UHFFFAOYSA-N (benzyloxy)acetic acid Chemical compound OC(=O)COCC1=CC=CC=C1 GRZHHTYDZVRPIC-UHFFFAOYSA-N 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- QFMZQPDHXULLKC-UHFFFAOYSA-N 1,2-bis(diphenylphosphino)ethane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCP(C=1C=CC=CC=1)C1=CC=CC=C1 QFMZQPDHXULLKC-UHFFFAOYSA-N 0.000 description 1
- SLKDGVPOSSLUAI-PGUFJCEWSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine zwitterion Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OCCN)OC(=O)CCCCCCCCCCCCCCC SLKDGVPOSSLUAI-PGUFJCEWSA-N 0.000 description 1
- NRJAVPSFFCBXDT-HUESYALOSA-N 1,2-distearoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCCCC NRJAVPSFFCBXDT-HUESYALOSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- AHIKFYZDDRDNCK-UHFFFAOYSA-N 2-[4-(dimethylamino)butanoyl-(2-tetradecanoyloxyethyl)amino]ethyl tetradecanoate Chemical compound CCCCCCCCCCCCCC(=O)OCCN(C(=O)CCCN(C)C)CCOC(=O)CCCCCCCCCCCCC AHIKFYZDDRDNCK-UHFFFAOYSA-N 0.000 description 1
- XTOXGMHQNFKTAF-WRBBJXAJSA-N 2-[[2-(dimethylamino)acetyl]-[2-[(z)-octadec-9-enoyl]oxyethyl]amino]ethyl (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCCN(C(=O)CN(C)C)CCOC(=O)CCCCCCC\C=C/CCCCCCCC XTOXGMHQNFKTAF-WRBBJXAJSA-N 0.000 description 1
- LYVCKXIOCZNKRV-UHFFFAOYSA-N 2-[[2-[2-(dimethylamino)ethylsulfanyl]acetyl]-(2-tetradecanoyloxyethyl)amino]ethyl tetradecanoate Chemical compound CCCCCCCCCCCCCC(=O)OCCN(C(=O)CSCCN(C)C)CCOC(=O)CCCCCCCCCCCCC LYVCKXIOCZNKRV-UHFFFAOYSA-N 0.000 description 1
- LSTRKXWIZZZYAS-UHFFFAOYSA-N 2-bromoacetyl bromide Chemical compound BrCC(Br)=O LSTRKXWIZZZYAS-UHFFFAOYSA-N 0.000 description 1
- PYSGFFTXMUWEOT-UHFFFAOYSA-N 3-(dimethylamino)propan-1-ol Chemical compound CN(C)CCCO PYSGFFTXMUWEOT-UHFFFAOYSA-N 0.000 description 1
- JTNKXYWGZCNBCH-UHFFFAOYSA-N 3-(dimethylamino)propanoic acid;hydron;chloride Chemical compound Cl.CN(C)CCC(O)=O JTNKXYWGZCNBCH-UHFFFAOYSA-N 0.000 description 1
- RDTALXUBMCLWBB-UHFFFAOYSA-N 4-(dimethylamino)butanoic acid;hydron;chloride Chemical compound Cl.CN(C)CCCC(O)=O RDTALXUBMCLWBB-UHFFFAOYSA-N 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- OVVWZQMHXPQDPL-UIMRCOCASA-N CC1(C)C(/C=C/C(/C)=C/C=C/C(/C)=C/C(NCCCC[C@@H](C(NCCCOCCOCCOCCCNC(CCOCCOCCOCCOCCOCCC(NCCCOCCOCCOCCCNC([C@H](CCCCNC(/C=C(\C)/C=C/C=C(\C)/C=C/C2=C(C)CCCC2(C)C)=O)NC(/C=C(\C)/C=C/C=C(\C)/C=C/C2=C(C)CCCC2(C)C)=O)=O)=O)=O)=O)NC(/C=C(\C)/C=C/C=C(\C)/C=C/C2=C(C)CCCC2(C)C)=O)=O)=C(C)CCC1 Chemical compound CC1(C)C(/C=C/C(/C)=C/C=C/C(/C)=C/C(NCCCC[C@@H](C(NCCCOCCOCCOCCCNC(CCOCCOCCOCCOCCOCCC(NCCCOCCOCCOCCCNC([C@H](CCCCNC(/C=C(\C)/C=C/C=C(\C)/C=C/C2=C(C)CCCC2(C)C)=O)NC(/C=C(\C)/C=C/C=C(\C)/C=C/C2=C(C)CCCC2(C)C)=O)=O)=O)=O)=O)NC(/C=C(\C)/C=C/C=C(\C)/C=C/C2=C(C)CCCC2(C)C)=O)=O)=C(C)CCC1 OVVWZQMHXPQDPL-UIMRCOCASA-N 0.000 description 1
- FOFAMHACECNGNX-PGUFJCEWSA-N CCCCCCCCCCCCCC(=O)OCCN(CCOC(=O)CCCCCCCCCCCCC)C(=O)[C@H]1CCCN1C Chemical compound CCCCCCCCCCCCCC(=O)OCCN(CCOC(=O)CCCCCCCCCCCCC)C(=O)[C@H]1CCCN1C FOFAMHACECNGNX-PGUFJCEWSA-N 0.000 description 1
- BKVZHRXSCJGLOK-UHFFFAOYSA-N CCCCCCCCCCCCCC(OCCN(CCOC(CCCCCCCCCCCCC)=O)C(CO)=O)=O Chemical compound CCCCCCCCCCCCCC(OCCN(CCOC(CCCCCCCCCCCCC)=O)C(CO)=O)=O BKVZHRXSCJGLOK-UHFFFAOYSA-N 0.000 description 1
- MFQGVTOMYMUFPY-UHFFFAOYSA-N CCCCCCCCCCCCCC(OCCN(CCOC(CCCCCCCCCCCCC)=O)C(COCc1ccccc1)=O)=O Chemical compound CCCCCCCCCCCCCC(OCCN(CCOC(CCCCCCCCCCCCC)=O)C(COCc1ccccc1)=O)=O MFQGVTOMYMUFPY-UHFFFAOYSA-N 0.000 description 1
- 101150041968 CDC13 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 238000003734 CellTiter-Glo Luminescent Cell Viability Assay Methods 0.000 description 1
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000012594 Earle’s Balanced Salt Solution Substances 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 238000010268 HPLC based assay Methods 0.000 description 1
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 1
- 208000014919 IgG4-related retroperitoneal fibrosis Diseases 0.000 description 1
- 231100000750 In vitro toxicology Toxicity 0.000 description 1
- OYHQOLUKZRVURQ-HZJYTTRNSA-N Linoleic acid Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(O)=O OYHQOLUKZRVURQ-HZJYTTRNSA-N 0.000 description 1
- 101100317378 Mus musculus Wnt3 gene Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- CWLQUGTUXBXTLF-YFKPBYRVSA-N N-methylproline Chemical compound CN1CCC[C@H]1C(O)=O CWLQUGTUXBXTLF-YFKPBYRVSA-N 0.000 description 1
- 208000003510 Nephrogenic Fibrosing Dermopathy Diseases 0.000 description 1
- 206010067467 Nephrogenic systemic fibrosis Diseases 0.000 description 1
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 206010038979 Retroperitoneal fibrosis Diseases 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930182558 Sterol Natural products 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- FPIPGXGPPPQFEQ-BOOMUCAASA-N Vitamin A Natural products OC/C=C(/C)\C=C\C=C(\C)/C=C/C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-BOOMUCAASA-N 0.000 description 1
- SBCVJZHHHVYKNM-UHFFFAOYSA-M [2-[bis(2-tetradecanoyloxyethyl)amino]-2-oxoethyl]-(2-hydroxyethyl)-dimethylazanium;bromide Chemical compound [Br-].CCCCCCCCCCCCCC(=O)OCCN(C(=O)C[N+](C)(C)CCO)CCOC(=O)CCCCCCCCCCCCC SBCVJZHHHVYKNM-UHFFFAOYSA-M 0.000 description 1
- YKTSYUJCYHOUJP-UHFFFAOYSA-N [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] Chemical compound [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] YKTSYUJCYHOUJP-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- WJGAPUXHSQQWQF-UHFFFAOYSA-N acetic acid;hydrochloride Chemical compound Cl.CC(O)=O WJGAPUXHSQQWQF-UHFFFAOYSA-N 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 229940050528 albumin Drugs 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 239000011717 all-trans-retinol Substances 0.000 description 1
- 229940100609 all-trans-retinol Drugs 0.000 description 1
- 235000019169 all-trans-retinol Nutrition 0.000 description 1
- 150000004347 all-trans-retinol derivatives Chemical class 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000013011 aqueous formulation Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 125000005019 carboxyalkenyl group Chemical group 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001733 carboxylic acid esters Chemical group 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 210000005056 cell body Anatomy 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- FOCAUTSVDIKZOP-UHFFFAOYSA-N chloroacetic acid Chemical compound OC(=O)CCl FOCAUTSVDIKZOP-UHFFFAOYSA-N 0.000 description 1
- 229940106681 chloroacetic acid Drugs 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 229940124301 concurrent medication Drugs 0.000 description 1
- 238000011340 continuous therapy Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 229960001305 cysteine hydrochloride Drugs 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 210000001787 dendrite Anatomy 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- PXEDJBXQKAGXNJ-QTNFYWBSSA-L disodium L-glutamate Chemical compound [Na+].[Na+].[O-]C(=O)[C@@H](N)CCC([O-])=O PXEDJBXQKAGXNJ-QTNFYWBSSA-L 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 208000002854 epidermolysis bullosa simplex superficialis Diseases 0.000 description 1
- SUPCQIBBMFXVTL-UHFFFAOYSA-N ethyl 2-methylprop-2-enoate Chemical compound CCOC(=O)C(C)=C SUPCQIBBMFXVTL-UHFFFAOYSA-N 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229960001031 glucose Drugs 0.000 description 1
- 210000005161 hepatic lobe Anatomy 0.000 description 1
- 210000004024 hepatic stellate cell Anatomy 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 231100000334 hepatotoxic Toxicity 0.000 description 1
- 230000003082 hepatotoxic effect Effects 0.000 description 1
- 125000004404 heteroalkyl group Chemical group 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 210000001926 inhibitory interneuron Anatomy 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 229960001375 lactose Drugs 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 235000020778 linoleic acid Nutrition 0.000 description 1
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 description 1
- 125000003473 lipid group Chemical group 0.000 description 1
- 239000002479 lipoplex Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 238000005461 lubrication Methods 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- NRVFDGZJTPCULU-UHFFFAOYSA-N meda Chemical compound Cl.CN(C)CCS NRVFDGZJTPCULU-UHFFFAOYSA-N 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 150000002762 monocarboxylic acid derivatives Chemical class 0.000 description 1
- 235000013923 monosodium glutamate Nutrition 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 229940078490 n,n-dimethylglycine Drugs 0.000 description 1
- DXASQZJWWGZNSF-UHFFFAOYSA-N n,n-dimethylmethanamine;sulfur trioxide Chemical group CN(C)C.O=S(=O)=O DXASQZJWWGZNSF-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002086 nanomaterial Substances 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 230000004768 organ dysfunction Effects 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920002246 poly[2-(dimethylamino)ethyl methacrylate] polymer Polymers 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000000955 prescription drug Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 235000020945 retinal Nutrition 0.000 description 1
- 150000003726 retinal derivatives Chemical class 0.000 description 1
- 230000002207 retinal effect Effects 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- 150000004508 retinoic acid derivatives Chemical class 0.000 description 1
- 229960003471 retinol Drugs 0.000 description 1
- 239000011607 retinol Substances 0.000 description 1
- 229940108325 retinyl palmitate Drugs 0.000 description 1
- 235000019172 retinyl palmitate Nutrition 0.000 description 1
- 239000011769 retinyl palmitate Substances 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229940073490 sodium glutamate Drugs 0.000 description 1
- HJHVQCXHVMGZNC-JCJNLNMISA-M sodium;(2z)-2-[(3r,4s,5s,8s,9s,10s,11r,13r,14s,16s)-16-acetyloxy-3,11-dihydroxy-4,8,10,14-tetramethyl-2,3,4,5,6,7,9,11,12,13,15,16-dodecahydro-1h-cyclopenta[a]phenanthren-17-ylidene]-6-methylhept-5-enoate Chemical compound [Na+].O[C@@H]([C@@H]12)C[C@H]3\C(=C(/CCC=C(C)C)C([O-])=O)[C@@H](OC(C)=O)C[C@]3(C)[C@@]2(C)CC[C@@H]2[C@]1(C)CC[C@@H](O)[C@H]2C HJHVQCXHVMGZNC-JCJNLNMISA-M 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 238000007711 solidification Methods 0.000 description 1
- 230000008023 solidification Effects 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 150000003432 sterols Chemical class 0.000 description 1
- 235000003702 sterols Nutrition 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 150000003505 terpenes Chemical group 0.000 description 1
- QERYCTSHXKAMIS-UHFFFAOYSA-N thiophene-2-carboxylic acid Chemical compound OC(=O)C1=CC=CS1 QERYCTSHXKAMIS-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000003151 transfection method Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 235000019155 vitamin A Nutrition 0.000 description 1
- 239000011719 vitamin A Substances 0.000 description 1
- 229940045997 vitamin a Drugs 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
- C07C237/12—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atom of at least one of the carboxamide groups bound to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/20—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing sulfur, e.g. dimethyl sulfoxide [DMSO], docusate, sodium lauryl sulfate or aminosulfonic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/22—Heterocyclic compounds, e.g. ascorbic acid, tocopherol or pyrrolidones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes, liposomes coated with polymers
- A61K9/1272—Non-conventional liposomes, e.g. PEGylated liposomes, liposomes coated with polymers with substantial amounts of non-phosphatidyl, i.e. non-acylglycerophosphate, surfactants as bilayer-forming substances, e.g. cationic lipids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C235/08—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atom of at least one of the carboxamide groups bound to an acyclic carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
- C07C237/08—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atom of at least one of the carboxamide groups bound to an acyclic carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/16—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and unsaturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/16—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/18—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by doubly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C321/00—Thiols, sulfides, hydropolysulfides or polysulfides
- C07C321/12—Sulfides, hydropolysulfides, or polysulfides having thio groups bound to acyclic carbon atoms
- C07C321/14—Sulfides, hydropolysulfides, or polysulfides having thio groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/60—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton with the carbon atom of at least one of the carboxyl groups bound to nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C333/00—Derivatives of thiocarbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C333/02—Monothiocarbamic acids; Derivatives thereof
- C07C333/04—Monothiocarbamic acids; Derivatives thereof having nitrogen atoms of thiocarbamic groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D333/40—Thiophene-2-carboxylic acid
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
- Y10T428/2991—Coated
- Y10T428/2998—Coated including synthetic resin or polymer
Definitions
- the description is directed to ionizable lipids for enhancing the delivery of therapeutic agents.
- Non-viral transfection systems can include, for example, polymers, lipids, liposomes, micelles, dendrimers, and nanomaterials.
- Polymers that have been studies for cell transfection include cationic polymers such as, for example, poly(L-lysine) ("PLL"), polyethyleneimine (“PEI”), chitosan, and poly(2-dimethylamino)ethyl methacrylate
- viral and non- viral transfection techniques have drawbacks, however.
- viral systems can yield high transfection efficiency, but may not be entirely safe.
- viral systems can be complicated and/or expensive to prepare.
- Non-viral transfection systems for example, those employing cationic polymers, have been reported to transfer plasmid DNA into cells.
- Cationic polymers however, can be unstable and can be toxic to cells.
- Cio-18 alkyl or C 12-1 8 alkenyl is -CH 2 -, S, O, N, or absent;
- L is C 1-4 alkylene; -S-C 1-4 alkylene; -
- compositions, pharmaceutical formulations, drug carriers, and methods of using the compounds of formula I are also described.
- FIG. 1 depicts in vitro synergistic efficacy of ionizable lipid:ionizable lipid embodiments of the present description
- FIG. 2 depicts in vitro synergistic efficacy of ionizable lipid:ionizable lipid embodiments of the present description
- FIG. 3 depicts in vitro synergistic efficacy of ionizable lipid: cationic lipid embodiments of the present description
- FIG. 4 depicts in vivo efficacy of ionizable lipid: cationic lipid embodiments of the present description
- the present description is directed to ionizable lipid compounds, as well as their uses in delivering therapeutic agents to cells, tissues, and organisms.
- n and m are independently 1, 2, 3, or 4;
- Ri and R 2 are independently Cio-is alkyl or C 12-18 alkenyl
- X is -CH 2 -, S, O, N, or absent;
- L is Ci-4 alkylene; -S-C 1-4 alkylene; -0-C 1-4 alkylene; -0-C(0)-Ci
- n and m can be the same or different. In preferred embodiments, n and m are the same. Particularly preferred are those embodiments wherein n and m are both 1 or n and m are both 2.
- X is a bond. In other embodiments, X is -CH 2 -. In still others, X is S. Also preferred, are embodiments wherein X is O. Embodiments wherein X is N are also within the scope of the description.
- L is C 1-4 alkylene.
- L is
- L is -S-Ci-4 alkylene. In yet other embodiments, L is -O-C1-4 alkylene. In still other embodiments, L is
- L is -S(0) 2 -C 1-4 alkylene. Also within the scope of the description are embodiments wherein L is
- L is preferably C 1-4 alkylene.
- L include -CH 2 -, -CH 2 -CH 2 -, -CH 2 CH 2 CH 2 -, and -CH 2 CH 2 CH 2 CH 2 -.
- L is S C-
- X is -CH 2 -
- L is preferably -S-C 1-4 alkylene.
- L include -S-CH 2 -, -S-CH 2 -CH 2 -, -S-CH 2 CH 2 CH 2 -, and -S-CH 2
- L is preferably -S(0) 2 -C 1-4 alkylene.
- L include -S(0) 2 -CH 2 , -S(0) 2 -CH 2 -CH 2 , -S(0) 2 -CH 2 -CH 2 -, and S(0) 2 -CH 2 -CH 2 -CH 2 -CH 2 -.
- L is -0-C 1-4 alkylene.
- L include -0-CH 2 -, -0-CH 2 -CH 2 -, -0-CH 2 CH 2 CH 2 -, and -0-CH 2 CH 2 CH 2 CH 2 -.
- L is preferably Ci_ 4 alkylene.
- L include -CH 2 -, -CH 2 -CH 2 -, -CH 2 CH 2 CH 2 -, and -CH 2
- L is preferably C 1-4 alkylene.
- L include -CH 2 -, -CH 2 -CH 2 -, -CH 2 CH 2 CH 2 -, and -CH 2 CH 2 CH 2 CH 2 -.
- Ri and R 2 can be the same or different.
- Ri and R 2 are the same.
- Ri and R 2 are Cw-is alkyl.
- the Cio-is alkyl is a straight-chain C 10 -i 8 alkyl. More preferred are embodiments wherein Ri and R 2 are C 12-18 alkyl. Also preferred are embodiments wherein Ri and R 2 are
- Ri and R 2 are both C 13 alkyl.
- Ri and R 2 are C 12-18 alkenyl.
- Ri and R 2 are and R 2 are each oleyl:
- Ri and R 2 are C 12-18 alkenyl.
- Ri and R 2 are C 13- linoleoyl.
- compositions comprising a compound of formula I in a liposome, wherein the liposome comprises a bilayer of lipid molecules.
- the compound of formula I can comprise any mole percentage of the lipid molecules in such compositions, it is preferred that the compound of formula I is about 5 to about 50 mol % of the lipid molecules of the compositions of the description.
- compositions of the description comprising a compound of formula I in a liposome can contain more than one compound of formula I.
- such compositions of the description include two compounds of formula I.
- it is preferred that the molar ratio of the two compounds of formula I is about 10:30 to about 30: 10.
- compositions of the description comprising a compound of formula I in a liposome may further comprise a cationic lipid.
- the cationic lipid is about 5 to about 40 mol % of the lipid molecules of the composition.
- the molar ratio of the compound of formula I to the cationic lipid is about 5:35 to about 35:5. More preferable, the ratio is about 10:30 to about 30: 10.
- any compositions of the description may further comprise a liquid medium.
- the liquid medium is suitable for injection into a living organism.
- the liquid medium comprises an organic solvent.
- the liquid medium used in certain embodiments of the description comprises water and an organic solvent.
- the liquid medium may further comprise a non-aqueous medium.
- compositions of the description may further comprise at least one phospholipid.
- any compositions of the description may further comprise at least one PEG-conjugated lipid.
- the drug carriers of the description will further comprising a siRNA molecule.
- compositions within the scope of the description include any of the
- the siRNA is encapsulated by the liposome of the compositions of the description.
- kits for delivering a drug to a patient in need of treatment comprise providing a pharmaceutical formulation within the scope of the description and administering the pharmaceutical formulation to the patient.
- cationic lipid refers to a compound that includes at least one lipid moiety and a positively charged quaternary nitrogen associated with a counterion.
- Lipids are understood in the art to be comprises of a hydrophobic alkyl or alkenyl moiety and a carboxylic acid or ester moiety. Preferred cationic lipids for use in the present description include:
- ionizable lipid refers to a compound of formula I within the scope of the description. These compounds are capable of forming charged species when contacted with an appropriate counterion species, for example, a species that includes an ionizable hydrogen atom.
- alkyl refers to a straight or branched fully saturated (no double or triple bonds) hydrocarbon group, for example, a group having the general formula - C n H2 n+1 .
- the alkyl group may have 1 to 50 carbon atoms (whenever it appears herein, a numerical range such as “1 to 50” refers to each integer in the given range; e.g., "1 to 50 carbon atoms” means that the alkyl group may consist of 1 carbon atom, 2 carbon atoms, 3 carbon atoms, etc., up to and including 50 carbon atoms, although the present definition also covers the occurrence of the term "alkyl" where no numerical range is designated).
- the alkyl group may also be a medium size alkyl having 1 to 30 carbon atoms.
- the alkyl group could also be a lower alkyl having 1 to 5 carbon atoms.
- the alkyl group of the compounds may be designated as "C 1-4 alkyl” or similar designations.
- “C 1-4 alkyl” indicates that there are one to four carbon atoms in the alkyl chain, i.e., the alkyl chain is selected from the group consisting of methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, sec -butyl, and t-butyl.
- Typical alkyl groups include, but are in no way limited to, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tertiary butyl, pentyl, hexyl and the like.
- alkylene refers to an alkanediyl functional group, for example, -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CH 2 -, -CH 2 CH 2 CH 2 CH 2 -, and the like.
- alkenyl refers to an alkyl group that contains in the straight or branched hydrocarbon chain one or more double bonds.
- An alkenyl group may be unsubstituted or substituted. When substituted, the substituent(s) may be selected from the same groups disclosed above with regard to alkyl group substitution unless otherwise indicated.
- Oleyl is an example of such an alkenyl group.
- DMSO dimethyl sulfoxide
- the term "diluent” refers to chemical compounds diluted in water that will dissolve the formulation of interest (e.g., the formulation that can include a compound, a retinoid, a second lipid, a stabilizing agent, and/or a therapeutic agent) as well as stabilize the biologically active form of the formulation.
- Salts dissolved in buffered solutions are utilized as diluents in the art.
- One commonly used buffered solution is phosphate buffered saline because it mimics the salt conditions of human blood. Since buffer salts can control the pH of a solution at low concentrations, a buffered diluent rarely modifies the biological activity of the formulation.
- an “excipient” refers to an inert substance that is added to a formulation to provide, without limitation, bulk, consistency, stability, binding ability, lubrication, disintegrating ability, etc., to the composition.
- a “diluent” is a type of excipient.
- Organic solvents used within the scope of the description are known in the art, per se. and include, for example, alcohols, dimethyl sulfoxide (“DMSO”), and the like.
- therapeutic agent refers to a compound that, upon activation of a cell or tissue.
- a therapeutic agent may be referred to herein as a drug.
- a therapeutic agent is not limited to drugs that have received regulatory approval.
- a “therapeutic agent” can be operatively associated with a compound as described herein, a retinoid, and/or a second lipid.
- a second lipid as described herein can form a liposome, and the therapeutic agent can be operatively associated with the liposome, e.g., as described herein.
- retinoid is a member of the class of compounds consisting of four isoprenoid units joined in a head-to-tail manner, see G. P. Moss, "Biochemical
- retinoid refers to natural and synthetic retinoids including first generation, second generation, and third generation retinoids. Examples of naturally occurring retinoids include, but are not limited to, (1) 1 1-cis-retinal, (2) all-trans retinol, (3) retinyl palmitate, (4) all- trans retinoic acid, and (5) 13-cis-retinoic acids. Furthermore, the term "retinoid” encompasses retinols, retinals, retinoic acids, retinoids, and derivatives thereof.
- retinoid conjugate refers to a molecule that includes at least one retinoid moiety.
- the retinoid conjugate will be present at a concentration of about 0.3 to about 30 weight percent, based on the total weight of the composition or formulation, which is equivalent to about 0.1 to about 10 mol.%, which is equivalent to a molar ratio of about 0.1 to about 10.
- the retinoid conjugate is a retinoid-linker-lipid molecule or a retinoid-linker-retinoid molecule.
- retinoid conjugates include those compounds of formula II:
- q, r, and s are each independently 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10, and enantiomers and diastereomers thereof.
- Preferred compounds of formula II include those wherein q, r, and s are each independently 1, 2, 3, 4, 5, 6, or 7. More preferred are those compounds of formula II wherein q, r, and s are each independently 3, 4, or 5. Most preferred are those compounds of formula II wherein q is 3, r is 5, and s is 3.
- One example of a compound of formula II is
- DiVA-PEG-DiVA includes stereocenters and all enantiomers and diastereomers are considered to be within the scope of the description.
- retinoid-linker-lipid molecule refers to a molecule that includes at least one retinoid moiety attached to at least one lipid moiety through at least one linker such as, for example, a PEG moiety.
- retinoid linker-retinoid molecule refers to a molecule that includes at least one retinoid moiety attached to at least one other retinoid moiety (which may be the same or different) through at least one linker such as, for example, a PEG moiety.
- lipid and lipophilic are used herein in their ordinary meanings as understood by those skilled in the art.
- Non-limiting examples of lipids and lipophilic groups include fatty acids, sterols, C2-C50 alkyl, C2-C50 heteroalkyl, C2-C50 alkenyl, C2-C50 heteroalkenyl, C2-C50 aryl, C2-C50 heteroaryl, C2-C50 alkynyl, C2-C50 heteroalkynyl, C2-C50 carboxyalkenyl, and C2-C50 carboxyheteroalkenyl.
- a fatty acid is a saturated or unsaturated long-chain monocarboxylic acid that contains, for example, 12 to 24 carbon atoms
- a lipid is characterized as being essentially water insoluble, having a solubility in water of less than about 0.01% (weight basis).
- the terms "lipid moiety” and "lipophilic moiety” refers to a lipid or portion thereof that has become attached to another group.
- a lipid group may become attached to another compound (e.g., a monomer) by a chemical reaction between a functional group (such as a carboxylic acid group) of the lipid and an appropriate functional group of a monomer.
- tellate cell refers to neurons with several dendrites radiating from the cell body. Examples of stellate cells are inhibitory interneurons.
- siRNA refers to small interfering RNA, also known in the art as short interfering RNA or silencing RNA. siRNA is a class of double stranded RNA molecules that have a variety of effects known in the art, the most notable being the interference with the expression of specific genes and protein expression.
- the term "liposome” is used herein in its ordinary meaning as understood by those skilled in the art, and refers to a lipid bilayer structure that contains lipids attached to polar, hydrophilic groups which form a substantially closed structure in aqueous media. In some embodiments, the liposome can be operatively associated with one or more compounds, such as a therapeutic agent and a retinoid.
- a liposome may be comprised of a single lipid bilayer (i.e., unilamellar) or it may comprised of two or more lipid bilayers (i.e., multilamellar). While the interior of a liposome may consist of a variety of compounds, the exterior of the liposome is accessible to the aqueous formulation comprising the liposome.
- a liposome can be
- the siRNA will be encapsulated by the liposome so that the siRNA is inaccessible to the aqueous medium.
- the liposome will have a solid core; such liposomes encapsulating siRNA and having a solid core are termed "lipid nanoparticles" herein.
- the siRNA will not be encapsulated by the liposome.
- the siRNA can be complexed on the outer surface of the liposome by mixing preformed liposomes with RNA in an aqueous solution. In these
- the siRNA is accessible to the aqueous medium.
- Liposomes having siRNA bound only on their outer surface are termed "lipoplexes" herein.
- the formulations of the description can also include PEG-conjugated lipids.
- PEG-conjugated lipids within the scope of the description are known in the art per se.
- Suitable PEG-lipids include PEG-phospholipids and PEG-ceramides such as, for example, PEG2000- DSPE, PEG2000-DPPE, PEG2000-DMPE, PEG2000-DOPE, PEG1000-DSPE, PEG1000- DPPE, PEG1000-DMPE, PEG1000-DOPE, PEG550-DSPE, PEG550-DPPE, PEG-550DMPE, PEG-1000DOPE, PEG-BML, PEG-Cholesterol.
- compositions of the description can include one or more phospholipids such as, for example, l,2-distearoyl-sn-glycero-3-phosphocholine (“DSPC”), dipalmitoylphosphatidylcholine (“DPPC”), 1 ,2-dipalmitoyl-sn-glycero-3 -phosphoethanolamine (“DPPE”), and l,2-dioleoyl-sn-glycero-3-phosphoethanolamine (“DOPE”).
- the helper lipid is DOPE.
- compositions that include any of the aforementioned compositions in addition to a pharmaceutically acceptable carrier or diluent.
- Pharmaceutical formulations of the description will include at least one therapeutic agent.
- the therapeutic agent is an siRNA.
- siRNA may include: Sense (5'->3') GGACAGGCCUCUACAACUATT (SEQ. ID. NO. 1) Antisense (3'->5') TTCCUGUCCGGAGAUGUUGAU (SEQ. ID. NO. 2)
- the siRNA is encapsulated by the liposome.
- the siRNA can be outside of the liposome.
- the siRNA can be complexed to the outside of the liposome.
- kits for delivering a therapeutic agent to a patient comprise providing a pharmaceutical formulation including any of the foregoing compositions and a pharmaceutically acceptable carrier or diluent; and administering the pharmaceutical formulation to the patient.
- the present disclosure relates to a pharmaceutical formulation comprising one or more physiologically acceptable surface active agents, pharmaceutical carriers, diluents, excipients, and suspension agents, or a combination thereof; and a formulation (e.g., the formulation that can include a compound, a retinoid, a second lipid, a stabilizing agent, and/or a therapeutic agent) disclosed herein.
- a formulation e.g., the formulation that can include a compound, a retinoid, a second lipid, a stabilizing agent, and/or a therapeutic agent
- Acceptable additional pharmaceutical carriers or diluents for therapeutic use are well known in the pharmaceutical art, and are described, for example, in Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), which is incorporated herein by reference in its entirety.
- Preservatives may be provided in the pharmaceutical formulation.
- sodium benzoate, ascorbic acid and esters of p-hydroxybenzoic acid may be added as preservatives.
- antioxidants and suspending agents may be used.
- alcohols, esters, sulfated aliphatic alcohols, and the like may be used as surface active agents; sucrose, glucose, lactose, starch, crystallized cellulose, mannitol, light anhydrous silicate, magnesium aluminate, magnesium metasilicate aluminate, synthetic aluminum silicate, calcium carbonate, sodium acid carbonate, calcium hydrogen phosphate, calcium carboxymethyl cellulose, and the like may be used as excipients; coconut oil, olive oil, sesame oil, peanut oil, soya may be used as suspension agents or lubricants; cellulose acetate phthalate as a derivative of a carbohydrate such as cellulose or sugar, or methylacetate-methacrylate copolymer as a derivative of polyvinyl may be used as suspension agents; and plasticizers such as ester phthalates and the like may be used as suspension agents.
- compositions described herein can be administered to a human patient per se, or in pharmaceutical formulations where they are mixed with other active ingredients, as in combination therapy, or suitable pharmaceutical carriers or excipient(s).
- Suitable routes of administration may include, for example, parenteral delivery, including intramuscular, subcutaneous, intravenous, intramedullary injections, as well as intrathecal, direct intraventricular, intraperitoneal, intranasal, or intraocular injections.
- the formulation e.g., the formulation that can include a compound, a retinoid, a second lipid, a stabilizing agent, and/or a therapeutic agent
- the route of administration may be local or systemic.
- the pharmaceutical formulations may be manufactured in a manner that is itself known, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or tableting processes.
- compositions may be formulated in any conventional manner using one or more physiologically acceptable pharmaceutical carriers comprising excipients and auxiliaries which facilitate processing of the active compounds into preparations which can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen. Any of the well-known techniques, pharmaceutical carriers, and excipients may be used as suitable and as understood in the art; e.g., in Remington's Pharmaceutical Sciences, above.
- injectables can be prepared in conventional forms, either as liquid solutions or suspensions, solid forms suitable for solution or suspension in liquid prior to injection, or as emulsions.
- Suitable excipients are, for example, water, saline, dextrose, mannitol, lactose, lecithin, albumin, sodium glutamate, cysteine hydrochloride, and the like.
- the injectable pharmaceutical formulations may contain minor amounts of nontoxic auxiliary substances, such as wetting agents, pH buffering agents, and the like.
- Physiologically compatible buffers include, but are not limited to, Hanks's solution, Ringer's solution, or physiological saline buffer. If desired, absorption enhancing preparations may be utilized.
- compositions for parenteral administration include aqueous solutions of the active formulation (e.g., the formulation that can include a compound, a retinoid, a second lipid, a stabilizing agent, and/or a therapeutic agent) in water-soluble form.
- suspensions of the active compounds may be prepared as appropriate oily injection suspensions.
- Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran.
- the suspension may also contain suitable stabilizers or agents that increase the solubility of the compounds to allow for the preparation of highly concentrated solutions.
- Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative.
- the formulations may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- the active ingredient may be in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
- the formulations may also be formulated as a depot preparation. Such long acting formulations may be administered by intramuscular injection.
- the formulations e.g., the formulation that can include a compound, a retinoid, a second lipid, a stabilizing agent, and/or a therapeutic agent
- the formulations may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- compositions and formulations of the description may also be formulated for topical delivery and may be applied to the subject's skin using any suitable process for application of topical delivery vehicle.
- the formulation may be applied manually, using an applicator, or by a process that involves both.
- the formulation may be worked into the subject's skin, e.g., by rubbing.
- Application may be performed multiple times daily or on a once-daily basis.
- the formulation may be applied to a subject's skin once a day, twice a day, or multiple times a day, or may be applied once every two days, once every three days, or about once every week, once every two weeks, or once every several weeks.
- Some embodiments herein are directed to a method of delivering a therapeutic agent to a cell.
- some embodiments are directed to a method of delivering a therapeutic agent such as siRNA into a cell.
- Suitable cells for use according to the methods described herein include prokaryotes, yeast, or higher eukaryotic cells, including plant and animal cells (e.g., mammalian cells).
- the cells can be human fibrosarcoma cells (e.g., HT1080 cell line).
- the cells can be hepatic stellate cells (LX2 cell line).
- the cells can be cancer cells.
- the cells can be stem cells (pHSC cell line).
- Cell lines which are model systems for cancer may be used, including but not limited to breast cancer (MCF-7, MDA-MB-438 cell lines), U87 glioblastoma cell line, B 16F0 cells (melanoma), HeLa cells (cervical cancer), A549 cells (lung cancer), and rat tumor cell lines GH3 and 9L.
- the formulations described herein can be used to transfect a cell. These embodiments may include contacting the cell with a formulation described herein that includes a therapeutic agent, to thereby deliver a therapeutic agent to the cell.
- Conditions characterized by abnormal fibrosis may include cancer and/or a fibrotic disease.
- Types of cancer that may be treated or ameliorated by a formulation described herein include, but are not limited to, lung cancer, pancreatic cancer, breast cancer, liver cancer, stomach cancer, and colon cancer.
- the cancer that may be treated or ameliorated is pancreatic cancer.
- the cancer that may be treated or ameliorated is lung cancer.
- Types of fibrotic disease that may be treated or ameliorated by a formulation described herein include, but are not limited to, hepatic fibrosis, hepatic cirrhosis, pancreatitis, pancreatic fibrosis, cystic fibrosis, vocal cord scarring, vocal cord mucosal fibrosis, laryngeal fibrosis, pulmonary fibrosis, idiopathic pulmonary fibrosis, cystic fibrosis,
- the condition that may be treated or ameliorated is hepatic fibrosis.
- compositions or pharmaceutical compositions described herein may be administered to the subject by any suitable means.
- methods of administration include, among others, (a) administration via injection, subcutaneously, intraperitoneally, intravenously, intramuscularly, intradermally, intraorbitally, intracapsularly, intraspinally, intrasternally, or the like, including infusion pump delivery; (b) administration locally such as by injection directly in the renal or cardiac area, e.g., by depot implantation; as well as as deemed appropriate by those of skill in the art for bringing the active compound into contact with living tissue.
- compositions suitable for administration include formulations
- a therapeutically effective amount of the compounds disclosed herein required as a dose will depend on the route of administration, the type of animal, including human, being treated, and the physical characteristics of the specific animal under consideration. The dose can be tailored to achieve a desired effect, but will depend on such factors as weight, diet, concurrent medication and other factors which those skilled in the medical arts will recognize. More specifically, a therapeutically effective amount means an amount of compound effective to prevent, alleviate or ameliorate symptoms of disease or prolong the survival of the subject being treated. Determination of a therapeutically effective amount is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
- the useful in vivo dosage to be administered and the particular mode of administration will vary depending upon the age, weight and mammalian species treated, the particular compounds employed, and the specific use for which these compounds are employed.
- the determination of effective dosage levels can be accomplished by one skilled in the art using routine pharmacological methods. Typically, human clinical applications of products are commenced at lower dosage levels, with dosage level being increased until the desired effect is achieved. Alternatively, acceptable in vitro studies can be used to establish useful doses and routes of administration of the compositions identified by the present methods using established pharmacological methods.
- dosages may range broadly, depending upon the desired effects and the therapeutic indication. Typically, dosages may be about 10 microgram/kg to about 100 mg/kg body weight, preferably about 100 microgram/kg to about 10 mg/kg body weight. Alternatively dosages may be based and calculated upon the surface area of the patient, as understood by those of skill in the art.
- compositions can be chosen by the individual physician in view of the patient's condition. (See e.g., Fingl et al. 1975, in "The Pharmacological Basis of Therapeutics", which is hereby incorporated herein by reference in its entirety, with particular reference to Ch. 1, p. 1).
- the dose range of the composition administered to the patient can be from about 0.5 to about 1000 mg/kg of the patient's body weight.
- the dosage may be a single one or a series of two or more given in the course of one or more days, as is needed by the patient.
- the dosages will be about the same, or dosages that are about 0.1% to about 500%, more preferably about 25% to about 250% of the established human dosage.
- a suitable human dosage can be inferred from ED50 or ID50 values, or other appropriate values derived from in vitro or in vivo studies, as qualified by toxicity studies and efficacy studies in animals.
- the attending physician would know how to and when to terminate, interrupt, or adjust administration due to toxicity or organ dysfunctions. Conversely, the attending physician would also know to adjust treatment to higher levels if the clinical response were not adequate (precluding toxicity).
- the magnitude of an administrated dose in the management of the disorder of interest will vary with the severity of the condition to be treated and to the route of administration. The severity of the condition may, for example, be evaluated, in part, by standard prognostic evaluation methods. Further, the dose and perhaps dose frequency, will also vary according to the age, body weight, and response of the individual patient. A program comparable to that discussed above may be used in veterinary medicine.
- the daily dosage regimen for an adult human patient may be, for example, a dose of about 0.1 mg to 2000 mg of each active ingredient, preferably about 1 mg to about 500 mg, e.g. 5 to 200 mg.
- an intravenous, subcutaneous, or intramuscular dose of each active ingredient of about 0.01 mg to about 100 mg, preferably about 0.1 mg to about 60 mg, e.g. about 1 to about 40 mg is used.
- dosages may be calculated as the free base.
- the formulation is administered 1 to 4 times per day.
- the formulations may be administered by continuous intravenous infusion, preferably at a dose of each active ingredient up to about 1000 mg per day.
- each active ingredient up to about 1000 mg per day.
- the formulations will be administered for a period of continuous therapy, for example for a week or more, or for months or years.
- Dosage amount and interval may be adjusted individually to provide plasma levels of the active moiety which are sufficient to maintain the modulating effects, or minimal effective concentration (MEC).
- MEC minimal effective concentration
- the MEC will vary for each compound but can be estimated from in vitro data. Dosages necessary to achieve the MEC will depend on individual characteristics and route of administration. However, HPLC assays or bioassays can be used to determine plasma concentrations.
- Dosage intervals can also be determined using MEC value. Compositions should be administered using a regimen which maintains plasma levels above the MEC for 10- 90% of the time, preferably between 30-90% and most preferably between 50-90%.
- the effective local concentration of the drug may not be related to plasma concentration.
- the amount of formulation administered may be dependent on the subject being treated, on the subject's weight, the severity of the affliction, the manner of administration and the judgment of the prescribing physician.
- Formulations disclosed herein can be evaluated for efficacy and toxicity using known methods.
- the toxicology of a particular compound, or of a subset of the compounds, sharing certain chemical moieties may be established by determining in vitro toxicity towards a cell line, such as a mammalian, and preferably human, cell line. The results of such studies are often predictive of toxicity in animals, such as mammals, or more specifically, humans.
- the toxicity of particular compounds in an animal model such as mice, rats, rabbits, or monkeys, may be determined using known methods.
- the efficacy of a particular compound may be established using several recognized methods, such as in vitro methods, animal models, or human clinical trials.
- the formulations may, if desired, be presented in a pack or dispenser device which may contain one or more unit dosage forms containing the active ingredient.
- the pack may for example comprise metal or plastic foil, such as a blister pack.
- the pack or dispenser device may be accompanied by instructions for administration.
- the pack or dispenser may also be accompanied with a notice associated with the container in form prescribed by a
- compositions comprising a compound formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labeled for treatment of an indicated condition.
- each center may independently be of R-configuration or S-configuration or a mixture thereof.
- the compounds provided herein may be enantiomerically pure or be stereoisomeric mixtures.
- each double bond may independently be E or Z a mixture thereof.
- all tautomeric forms are also intended to be included.
- Step 1 Preparation of Intermediate 1: 3,3 '-azanediylbis(propan-l-ol)
- Step 3 Preparation of Intermediate 3: ((tert-butoxycarbonyl)azanediyl)bis(propane-3,l-diyl) ditetradecanoate [0093] tert-butyl bis(3-hydroxypropyl)carbamate (4.00 g, 17.3 mmol), Et3N (4.8 mL, 34.6 mmol) and DMAP (529 mg, 4.33 mmol) were dissolved in chloroform (50 mL). While being stirred in an ice-bath, a solution of myristoyl chloride was added over 15 min. The addition was carried out in such a way that the temperature of the reaction did not exceed 30°C. The reaction was allowed to stir at room temperature overnight.
- Step 4 Preparation of Intermediate 4: azanediylbis(propane-3,l-diyl) ditetradecanoate TFA salt
- Step 5 Preparation ofi-Pr-DC: ((2-(dimethylamino)acetyl)azanediyl)bis(propane-3,l-diyl) ditetradecanoate
- Azanediylbis(propane-3,l-diyl) ditetradecanoate TFA salt (750 mg, 1.35 mmol) was diluted with DCM (5 mL) and added to a pre-activated mixture of N,N-dimethylglycine (154 mg, 1.49 mmol), HATU (616 mg, 1.62 mmol) and DIEA (495 uL, 2.84 mmol) in DCM (5 mL). Flask was flushed with argon and allowed to stir at room temperature overnight. The reaction mixture was concentrated.
- Step 2 Prep : tert-butyl bis(3-hydroxypropyl)carbamate '
- Step 3 Preparation of Intermediate 3: (Z)-((tert-butoxycarbonyl)azanediyl)bis(propane-3,l- diyl) dioleate [0099] tert-butyl bis(3-hydroxypropyl)carbamate, triethylamine and DMAP were dissolved in chloroform. While being stirred in an ice-bath, a solution of oleyl chloride was added over 15 minutes. The addition was carried out in such a way that the temperature of the reaction did not exceed 30°C. The reaction was allowed to stir at room temperature overnight. Next day, MeOH (50 mL) and 0.9% saline solution (50mL) was added to quench the reaction.
- MeOH 50 mL
- 0.9% saline solution 50mL
- Step 5 Preparation of i-Pr-DODC: (Z)-((2-(dimethylamino)acetyl)azanediyl)bis(propane-3,l-
- Step 1 Preparation of Intermediate 1: ((tert-butoxycarbonyl)azanediyl)bis(ethane-2,l-diyl) ditetradecanoate
- N-Boc diethanolamine (MW 205.25; 8.4 g, 0.041 mole), triethylamine (MW 101.19; 11.5 ml, 0.083 mol) and 4-(dimethylamino)pyridine (MW 122.17; 1.3 g, 0.01 1 mole) were dissolved in chloroform (170 mL). While being stirred in an ice/water bath, a solution of myristoyl chloride (MW 246.82; 22 mL, 80.9 mmol) in 100 mL of chloroform was added dropwise. The reaction mixture was then taken out of the cold bath, and the stirring was continued at room temperature for 2 h.
- Step 2 Preparation of Intermediate 2: azanediylbis(ethane-2,l-diyl) ditetradecanoate TFA salt
- Azanediylbis(ethane-2, l-diyl) ditetradecanoate TFA salt(10g, 16 mmol) was diluted with dimethylglycine (42.5g, 25 mmol), DCC (4.7 g, 23 mmol) and DIEA (6.33 mL, 40 mmol) in Pyridine (20 mL). The round-bottomed flask was flushed with argon gas and the reaction mixture was heated at 55°C overnight. Next day, the reaction mixture was concentrated.
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt (1.50 g, 2.85 mmol) was diluted with DCM (10 mL) and added to a pre-activated mixture of 3-(dimethylamino)propionic acid HCl salt (482 mg, 3.14 mmol), HATU (1.30 g, 3.42 mmol) and DIEA (1.04 mL, 5.98 mmol) in DCM (10 mL).
- the round-bottomed flask was flushed with argon gas and the reaction mixture was allowed to stir at room temperature overnight.
- the reaction mixture was concentrated. After purification by silica gel chromatography eluating with a DCM/MeOH, the pooled fractions were concentrated and stirred in DCM (20 niL) and 10% K 2 C0 3 (20 niL) at 0-5°C for 30 min. The organic layer was isolated and the aqueous layer further extracted with DCM (2 xlO rnL).
- Step 1 Preparation of Intermediate 1: (Z)-((tert-butoxycarbonyl)azanediyl)bis(ethane-2,l-
- N-Boc diethanolamine (17.8 g, 0.087 mole), triethylamine (24.4 mL, 0.176 mole) and 4- (dimethylamino)pyridine (2.76 g, 0.023 mole) were dissolved in 350 ml of chloroform. While being stirred, a solution of oleyl chloride (61.6 g, 0.174 mole) in 100 ml of chloroform was added over 10 min (Alternatively, the chloroform solution of N-Boc
- Step 3 Preparation ofi-Et-DODC: (Z)-((3-(dimethylamino)propanoyl)azanediyl)bis(ethane- 2, -diyl) dioleate
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt (1.00 g, 1.90 mmol) was diluted with DCM (5 mL) and added to a pre-activated mixture of 4-(Dimethylamino) butyric acid HCl salt (382 mg, 2.28mmol), HATU (867 mg, 2.28 mmol) and DIEA (728 uL, 4.18 mmol) in DCM (5 mL).
- the round-bottomed flask was flushed with argon gas and the reaction mixture was allowed to stir at room temperature overnight.
- the reaction mixture was concentrated. After purification by silica gel chromatography eluating with a DCM/MeOH, the pooled fractions were concentrated and stirred in DCM (20 mL) and 10% K 2 C0 3 (20 mL) at 0-5°C for 30 min. The organic layer was isolated and the aqueous layer further extracted with DCM (2 xlO mL).
- the round-bottomed flask was flushed with argon gas and the reaction mixture was allowed to stir at room temperature overnight.
- the reaction mixture was concentrated. After purification by silica gel chromatography eluating with a DCM/MeOH, the pooled fractions were concentrated and stirred in DCM (20 mL) and 10% K 2 C0 3 (20 mL) at 0-5°C for 30 min. The organic layer was isolated and the aqueous layer further extracted with DCM (2 xlO mL).
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt previously described.
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt (730 mg, 1.14 mmol) was stirred in DCM (20 mL) with 10% K 2 C0 3 (10 mL) at 0-5 °C. After 30 min, the organic phase was separated and the aqueous phase was further extracted with DCM (10 mL). The combined organic phases are stirred with MgS0 4 for a period of 30 minutes at 0-5°C, filtered and washed with DCM (10 mL).
- Trichloromethyl chloroformate (aka diphosgene) (257 uL, 2.13 mmol) was added to a solution of 2-(dimethylamino)ethanethiol HC1 salt (302 mg, 2.13 mmol) in dry DCM (20 mL) and stirred under a blanket of argon at room temperature for 4 h. Afterwards, DCM and excess diphosgene were removed in vacuo. Azanediylbis(ethane-2, 1-diyl) ditetradecanoate free base (1068 mg, 2.03 mmol), DCM (20 mL) and triethylamine (580 uL, 4.16 mmol) were then added.
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt previously described.
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt (1500mg, 2.34mmol) was dissolved in DCM (20mL) and placed in an ice-bath.
- Bromoacetyl bromide (214uL, 2.46mmol) was added followed by triethylamine(685uL, 4.91 mmol).
- the ice-bath was removed and the reaction was allowed to stir overnight at room temperature under a blanket of inert gas. Next day, diluted with DCM to lOOmL.
- Step 2 Preparation of O104: ((2-(2-(dimethylamino)ethoxy)acetyl)azanediyl)bis(ethane-2,l-
- Step 1 Preparation of Intermediate 1: ((2-(benzyloxy)acetyl)azanediyl)bis(ethane-2,l-diyl) ditetradecan oate
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt was stirred in DCM (25 mL) with 10% K2CO 3 (12.5 mL) at 0-5°C. After 30 min, the organic layer was isolated and the aqueous layer was further extracted with DCM (12 mL). The combined organic phases are stirred with MgS0 4 for 30 min at 0-5°C, filtered, washed with DCM (12 mL).
- Step 2 Preparation of Intermediate 2: ((2-hydroxyacetyl)azanediyl)bis(ethane-2,l-diyl) ditetradecanoate
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt previously described.
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt (1004 mg, 1.57 mmol) was stirred in DCM (20 mL) with 10% K 2 C0 3 (20mL) at 0-5°C. After 30 min, the organic phase was isolated and the aqueous phase was further extracted with DCM (10 mL). The combined organics are dried with MgS0 4 for 30 min at 0-5°C, filtered and washed with DCM (10 mL).
- Step 1 Preparation of Intermediate 1: (((4-nitrophenoxy)carbonyl)azanediyl)bis(ethane-2,l- diyl) ditetradeca-noate
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt was dissolved in dry DCM (10 mL) and triethylamine (654 uL, 4.69 mmol) was added. The reaction vessel was flushed with inert gas and 4-nitrophenyl chloroformate was added. Material was allowed to stir at RT overnight. The reaction mixture was quenched with water (50 mL) and DCM (50 mL). The organic layer was isolated, and the aqueous layer was extracted further with DCM (2 x 50 mL).
- Step 2 Preparation of CA104: (((2-(dimethylamino)ethoxy)carbonyl)azanediyl)bis(ethane- -diyl) ditetra-decanoate
- Step 1 Preparation of intermediate 1: 2-((2-(dimethylamino)ethyl)thio) acetic acid hydrochloride:
- Ethanol 500 mL was degassed by three times evacuation to 60 mBar for 1-2 minutes and repressurizing with nitrogen. Chloroacetic acid (36.9 g, 0.391 mol) was added and a clear solution was formed after having stirred for 5 minutes at 17-20°C. 2-(Dimethylamino)- ethanethiol hydrochloride (52.7 g, 0.372 mol) was added, and a clear solution was formed after having stirred for 20 minutes at 25°C. Solid sodium hydroxide (47.7 g, 1.19 mol) was added in portions over a period of 20 minutes with cooling in order to keep the temperature below 35°C - a short period at 44°C was observed.
- the aqueous phase was washed with DCM (3x 100 mL) to remove the disulphide impurity (all 3 washes required). Concentrated HCl was added to the aqueous phase (pH 10.7, temp. 22°C) until the pH was 1.4 (57.5 mL added, temp. 35°C). The aqueous phase was washed with DCM (100 mL) and then concentrated to dryness (bath temperature 55°C). Toluene (250 mL) was added, the mixture was concentrated to dryness (bath temperature 55°C) and this was repeated once to give a wet white solid (98 g).
- Acetonitrile (750 niL) was added to the solid, the mixture was stirred at 55°C for a period of 45 minutes, and then filtered through a G3 glass-sintered plate.
- Acetonitrile (250 mL) was added to the filter cake, the mixture was stirred at 55°C for a period of 25 minutes and then filtered through a G3 glass-sintered plate, washing with acetonitrile (50 mL).
- the combined filtrates are concentrated to 300 mL, resulting in a heavy, white precipitate.
- the mixture was cooled under nitrogen and stirred at 0°C for a period of 30 minutes.
- Step 2 Preparation ofS104: ((2-((2-(dimethylamino)ethyl)thio)acetyl)azanediyl)bis(ethane- -diyl) ditetradecanoate
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt (152 g, 238 mmol) was stirred with DCM (2.3 L) and 10% potassium bicarbonate (1.15 L) at 0-5°C. The organic phase was separated and the aqueous phase was further extracted with DCM (1.15 L). The combined organic phases were stirred with magnesium sulfate hydrate (236 g) for a period of 30 minutes at
- Tripotassium phosphate (85 g, 0.40 mol) and dipotassium hydrogen phosphate (226 g, 1.30 mol) were added to purified water (1.7 L), and the solution formed with pH 10.9 was cooled to 18-20°C.
- DCM (1.3 L) and recrystallized S104 hydrochloride (133.0 g, 0.188 mol) are added, and the mixture was stirred for a period of 10 min minutes. A clear organic phase was separated at moderate rate (over a period of 35 minutes), and the turbid aqueous phase was further extracted with DCM (650 mL).
- Step 1 Preparation of Intermediate 1: (9Z,9'Z)-((tert-butoxycarbonyl)azanediyl)bis(ethane- -diyl) bis( tetradec-9-enoate)
- N-Boc-diethanolamine (454 mg, 2.21 mmol), myristoleic acid (1000 mg, 4.42 mmol) and DMAP (54 mg, 0.442 mmol) was dissolved in DCM (25 mL) and placed in a water bath at ambient temperature in a round-bottom flask was flushed with inert gas.
- EDC HC1 salt (932 mg, 4.86 mmol) was added in 3 portions over 5 min. The reaction was allowed to stir overnight at room temperature under a blanket of inert gas. Next day, added FLO (25 mL) and stirred for 10 min. The organic layer was isolated and the aqueous layer was further extracted with DCM (50 mL).
- Step 2 Preparation of Intermediate 2: (9Z,9'Z)-azanediylbis(ethane-2,l-diyl) bis(tetradec-9- enoate
- the turbid aqueous phase was extracted with DCM (2 x 20 rnL).
- the combined organic extracts are dried with MgS0 4 , filtered and concentrated.
- Step 1 Preparation of Pro-DC: (R)-((l-methylpyrrolidine-2-carbonyl)azanediyl)bis(ethane-
- Azanediylbis(ethane-2, 1-diyl) ditetradecanoate TFA salt 1000 mg, 1.56 mmol
- DCM 10 mL
- N-Methyl-L-Proline 228 mg, 1.77 mmol
- HOBtH 2 0 239 mg, 1.77 mmol
- NMM 365 uL, 3.32 mmol
- N-Boc-diethanolamine (5 g, 24.4 mmol), linoleic acid (14.4 g, 51.2 mmol) and was dissolved in DCM (100 mL).
- EDC HC1 salt (10.3 g, 53.7 mmol) was added followed by DMAP (596 mg, 4.88 mmol).
- the reaction was allowed to stir for about 12 hours at room temperature under a blanket of inert gas. Thereafter, 50 mL of water and 50 mL of methanol were added, and the mixture was stirred for 10 min. The organic layer was isolated and the aqueous layer was further extracted with DCM (150 mL). The combined organics were dried (MgS0 4 ) for 10 min, filtered and concentrated.
- Step 2 Preparation of Intermediate 2: (9Z,9'Z,12Z,12'Z)-azanediylbis(ethane-2,l-diyl)
- Step 1 Preparation of TU104-Dlin: (9Z,9'Z,12Z,12'Z)-((2-((2- azanediyl)bis(ethan [0160] Synthesis of (9Z,9'Z,12Z, 12'Z)-azanediylbis(ethane-2,l-diyl) bis(octadeca-9, 12- dienoate) previously described.
- Trichloromethyl chloroformate (aka diphosgene) (740 uL, ) was added to a solution (9Z,9'Z, 12Z, 12'Z)-azanediylbis(ethane-2,l-diyl) bis(octadeca-9, 12-dienoate) (2.6 g) in dry DCM (40 mL) and stirred under a blanket of argon at room temperature for 12 hourrs. DCM and excess diphosgene were removed in vacuo. 2-(dimethylamino)ethane thiol HC1 salt (2.9 g), DCM (40 mL) and triethylamine (3.7 mL) were then added.
- Non-diVA siRNA containing liposomes lonizable lipid, DOPE, cholesterol, and a PEG-conjugated lipid were solubilized in absolute EtOH (200 proof) at a final weight concentration of ⁇ 4.4 mg/mL.
- the siRNA was solubilized in citrate buffer at a concentration -0.26 mg/mL and the temperature was adjusted to 35-40°C.
- the ethanol/lipid mixture was then added to the siRNA-containing buffer while stirring to spontaneously form siRNA loaded liposomes. Lipids were combined with siRNA to reach a final total lipid to siRNA ratio of 7: 1 to 14: 1 (w wt).
- the siRNA loaded liposomes were diafiltered against lOx volumes of PBS to remove ethanol and exchange the buffer. Final product was filtered through 0.22 ⁇ , sterilizing grade, filter for bioburden reduction. This process yielded liposomes with a mean particle diameter of 40-100 nm, PDI ⁇ 0.2, and >85% RNA encapsulation (entrapment) efficiency.
- siRNA containing liposomes co-solubilized with diVA ⁇ siRNA- diVA-Liposome formulations were prepared using the method described above.
- diVA-PEG- diVA was co-solubilized in absolute ethanol with the other lipids (ionizable lipid, DOPE, cholesterol, and PEG-conjugated lipids) prior to addition to the siRNA containing buffer.
- Molar content of diVA-PEG-diVA ranged from 0.1 to 5 mol%. This process yielded liposomes with a mean particle diameter of 40-100 nm, PDI ⁇ 0.2, and >85% entrapment efficiency.
- siRNA-diVA-Liposome formulations and siRNA-Liposome formulations were prepared using the method described above.
- Cationic lipid was co-solubilized in absolute ethanol with the other lipids (ionizable lipid, DOPE, cholesterol, PEG-conjugated lipids, and diVA-PEG-diVA) prior to addition to the siRNA containing buffer.
- Molar content of cationic lipid ranged from 5 to 40 mol%. This process yielded liposomes with a mean particle diameter of 40-100 nm, PDI ⁇ 0.2, and >85% entrapment efficiency.
- DMNQ short-term liver damage model
- a hepatotoxic agent such as dimethylnitrosamine (DMN)
- DMN dimethylnitrosamine
- liver lobes were excised and both gp46 and MRPL19 mRNA levels were determined by quantitative RT-PCR (TaqMan) assay. mRNA levels for gp46 were normalized to MRPL19 levels. Results are depicted in Figure 4.
- Several combinations of ionizable:cationic lipids achieved 50% reduction in gene expression expression for a single, 0.5 mg per kg of animal body weight, dose of encapsulated siRNA.
- in vitro toxicology data in vitro cytotoxicity (HepG2 @ 200 nM)
- HepG2 cells an adherent cell line derived from human hepatocellular carcinoma, was cultured in MEM/EBSS (Hyclone, Logan, Utah, Cat# SH30024.01)
- Chemiluminescent signal were measured on Clarity Luminescence Microplate Reader (502- Biotek, Winooski, Vermont). Viability was calculated based on % of chemiluminescent signal in formulation treated well normalized against mock treated wells.
- HEDC:S104 (20:20) formulation of the present description is exceptionally well tolerated as shown in toxicity studies. No toxicity was observed when the formulation was injected intravenously in rats and monkey at doses up to 25 mg/kg and 12 mg/kg, respectively, which is considered by those skilled in the art to be superior.
- Transfection method is same for LX-2 and pHSCs.
- the liposome formulations or lipoplex formulations of the description were mixed with growth medium at desired concentrations. 100 ⁇ of the mixture was added to the cells in 96-well plate and cells were incubated for 30 min at 37 °C in the incubator with 5% C0 2 . After 30 min, medium was replaced with fresh growth medium. After 48 h of transfection, cells were processed using Cell-to-Ct lysis reagents (Applied Biosystems) according to the manufacturer's instructions. Quantitatve (q) RT-PCR for measuring HSP47 mRNA expression
- HSP47 and GAPDH TaqMan® ® assays and One-Step RT-PCR master mix were purchased from Applied Biosystems. Each PCR reaction contained the following composition: One-step RT-PCR mix 5 ⁇ , TaqMan® RT enzyme mix 0.25 ⁇ , TaqMan® gene expression assay probe (HSP47) 0.25 ⁇ , TaqMan® gene expression assay probe (GAPDH) 0.5 ⁇ , RNase free water 3.25 ⁇ , Cell lysate 0.75 ⁇ , Total volume of 10 ⁇ . GAPDH was used as endogenous control for the relative quantification of HSP47 mRNA levels. Quantitative RT-PCR was performed in ViiA 7 realtime PCR system (Applied Biosciences). All values were normalized to the average HSP47 expression of the mock transfected cells and expressed as percentage of HSP47 expression compared to mock.
Abstract
Description












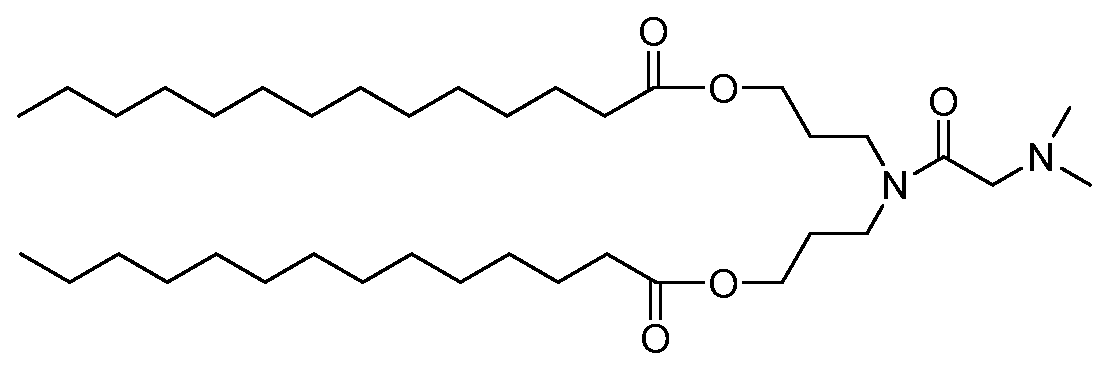
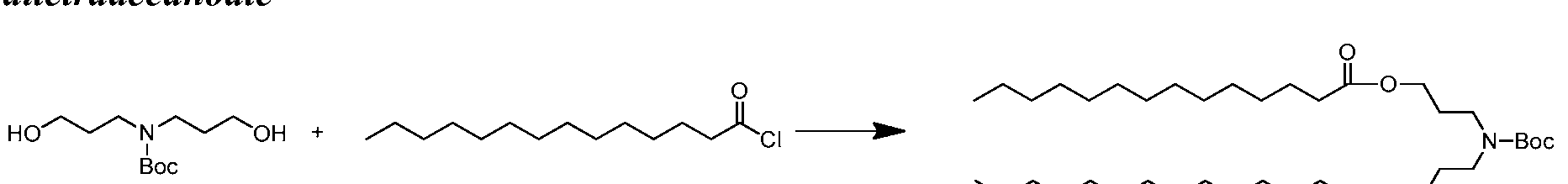














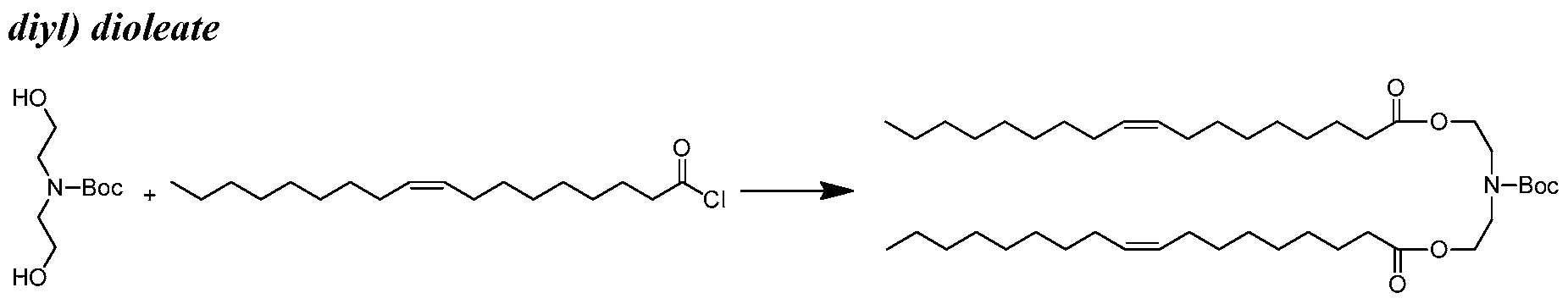




































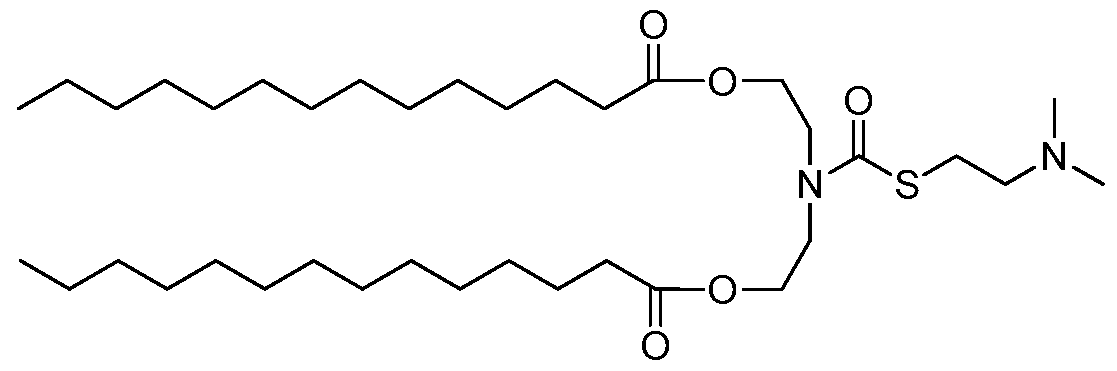


Claims





Priority Applications (19)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13729241.3A EP2858974B9 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| AU2013270685A AU2013270685B2 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| EP18200656.9A EP3489220B9 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| DK13729241.3T DK2858974T3 (en) | 2012-06-08 | 2013-06-07 | LIPIDS FOR THERAPEUTIC MEDICINE DELIVERY FORMULATIONS |
| RS20181385A RS58108B9 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| ES13729241T ES2697680T3 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent supply formulations |
| SI201331245T SI2858974T1 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| CA2876148A CA2876148C (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| BR112014030714-8A BR112014030714B1 (en) | 2012-06-08 | 2013-06-07 | ionizable lipid compound, composition, drug carrier and pharmaceutical formulation |
| PL18200656T PL3489220T3 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| LTEP13729241.3T LT2858974T (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| CN201380041854.8A CN104640841B (en) | 2012-06-08 | 2013-06-07 | Lipid for therapeutic agent delivering preparation |
| JP2015516267A JP5873604B2 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| PL13729241T PL2858974T3 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| KR1020157000454A KR102102801B1 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| RU2014153658A RU2658007C2 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
| HRP20181855TT HRP20181855T1 (en) | 2012-06-08 | 2018-11-07 | Lipids for therapeutic agent delivery formulations |
| CY181101255T CY1120929T1 (en) | 2012-06-08 | 2018-11-27 | LIPIDS FOR DISTRIBUTION RECOMMENDATIONS |
| CY20211100818T CY1124499T1 (en) | 2012-06-08 | 2021-09-17 | LIPIDS FOR THERAPEUTIC AGENT DELIVERY COMPOSITIONS |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261657480P | 2012-06-08 | 2012-06-08 | |
| US61/657,480 | 2012-06-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013185116A1 true WO2013185116A1 (en) | 2013-12-12 |
Family
ID=48626705
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/044849 WO2013185116A1 (en) | 2012-06-08 | 2013-06-07 | Lipids for therapeutic agent delivery formulations |
Country Status (22)
| Country | Link |
|---|---|
| US (3) | US9308267B2 (en) |
| EP (2) | EP3489220B9 (en) |
| JP (1) | JP5873604B2 (en) |
| KR (1) | KR102102801B1 (en) |
| CN (1) | CN104640841B (en) |
| AU (1) | AU2013270685B2 (en) |
| BR (1) | BR112014030714B1 (en) |
| CA (1) | CA2876148C (en) |
| CY (2) | CY1120929T1 (en) |
| DK (2) | DK2858974T3 (en) |
| ES (2) | ES2897216T3 (en) |
| HR (2) | HRP20211424T2 (en) |
| HU (2) | HUE056014T2 (en) |
| LT (2) | LT3489220T (en) |
| PL (2) | PL3489220T3 (en) |
| PT (2) | PT3489220T (en) |
| RS (2) | RS58108B9 (en) |
| RU (1) | RU2658007C2 (en) |
| SI (2) | SI3489220T1 (en) |
| TR (1) | TR201816986T4 (en) |
| TW (1) | TWI654997B (en) |
| WO (1) | WO2013185116A1 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016081029A1 (en) * | 2014-11-18 | 2016-05-26 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for rna delivery |
| US9365610B2 (en) | 2013-11-18 | 2016-06-14 | Arcturus Therapeutics, Inc. | Asymmetric ionizable cationic lipid for RNA delivery |
| US20160376229A1 (en) * | 2015-06-24 | 2016-12-29 | Nitto Denko Corporation | Ionizable compounds and compositions and uses thereof |
| US9567296B2 (en) | 2013-11-18 | 2017-02-14 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| WO2018084168A1 (en) | 2016-11-02 | 2018-05-11 | 日東電工株式会社 | Skin fibrosis treatment agent |
| US9976142B2 (en) | 2014-04-02 | 2018-05-22 | Nitto Denko Corporation | Targeting molecule and a use thereof |
| WO2019074071A1 (en) | 2017-10-11 | 2019-04-18 | 日東電工株式会社 | Regulation of nucleic acid molecule expression |
| US10383952B2 (en) | 2016-12-21 | 2019-08-20 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US10441659B2 (en) | 2012-06-08 | 2019-10-15 | Nitto Denko Corporation | Lipids for therapeutic agent delivery formulations |
| US10526284B2 (en) | 2016-12-21 | 2020-01-07 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| WO2022037652A1 (en) * | 2020-08-20 | 2022-02-24 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2022136641A1 (en) * | 2020-12-23 | 2022-06-30 | Etherna Immunotherapies Nv | Ionizable lipids |
| WO2023044333A1 (en) * | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Cyclic lipids and methods of use thereof |
| WO2023222870A1 (en) * | 2022-05-20 | 2023-11-23 | Etherna Immunotherapies Nv | Ionizable lipids |
Families Citing this family (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120141576A1 (en) * | 2007-03-15 | 2012-06-07 | Benjamin Johnson | Treatment of Dermatologic Skin Disorders |
| EP3578205A1 (en) | 2010-08-06 | 2019-12-11 | ModernaTX, Inc. | A pharmaceutical formulation comprising engineered nucleic acids and medical use thereof |
| CA3162352A1 (en) | 2010-10-01 | 2012-04-05 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| DE12722942T1 (en) | 2011-03-31 | 2021-09-30 | Modernatx, Inc. | RELEASE AND FORMULATION OF MANIPULATED NUCLEIC ACIDS |
| US9464124B2 (en) | 2011-09-12 | 2016-10-11 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| PT3682905T (en) | 2011-10-03 | 2022-04-07 | Modernatx Inc | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| LT2791160T (en) | 2011-12-16 | 2022-06-10 | Modernatx, Inc. | Modified mrna compositions |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US9303079B2 (en) | 2012-04-02 | 2016-04-05 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| CA2868996A1 (en) | 2012-04-02 | 2013-10-10 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins |
| US9283287B2 (en) | 2012-04-02 | 2016-03-15 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of nuclear proteins |
| PL2922554T3 (en) | 2012-11-26 | 2022-06-20 | Modernatx, Inc. | Terminally modified rna |
| WO2014152211A1 (en) | 2013-03-14 | 2014-09-25 | Moderna Therapeutics, Inc. | Formulation and delivery of modified nucleoside, nucleotide, and nucleic acid compositions |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| EP3052106A4 (en) | 2013-09-30 | 2017-07-19 | ModernaTX, Inc. | Polynucleotides encoding immune modulating polypeptides |
| JP2016538829A (en) | 2013-10-03 | 2016-12-15 | モデルナ セラピューティクス インコーポレイテッドModerna Therapeutics,Inc. | Polynucleotide encoding low density lipoprotein receptor |
| RU2021109685A (en) | 2014-04-23 | 2021-04-13 | МОДЕРНАТиЭкс, ИНК. | VACCINES BASED ON NUCLEIC ACIDS |
| US20180002702A1 (en) | 2014-12-26 | 2018-01-04 | Nitto Denko Corporation | Methods and compositions for treating malignant tumors associated with kras mutation |
| US10264976B2 (en) | 2014-12-26 | 2019-04-23 | The University Of Akron | Biocompatible flavonoid compounds for organelle and cell imaging |
| US10792299B2 (en) | 2014-12-26 | 2020-10-06 | Nitto Denko Corporation | Methods and compositions for treating malignant tumors associated with kras mutation |
| US20180235995A1 (en) * | 2015-02-19 | 2018-08-23 | Nitto Denko Corporation | Rna interference therapeutics against ebola virus |
| CA2990202A1 (en) * | 2015-06-29 | 2017-01-05 | Acuitas Therapeutics Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| JP6875373B2 (en) | 2015-07-22 | 2021-05-26 | 日東電工株式会社 | Compositions and Methods for Nanoparticle Freeze-Dried Forms |
| US11564893B2 (en) | 2015-08-17 | 2023-01-31 | Modernatx, Inc. | Methods for preparing particles and related compositions |
| AU2016324310B2 (en) | 2015-09-17 | 2021-04-08 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| DK3386484T3 (en) | 2015-12-10 | 2022-07-04 | Modernatx Inc | COMPOSITIONS AND METHODS OF DELIVERING THERAPY PRODUCTS |
| DK3394030T3 (en) | 2015-12-22 | 2022-03-28 | Modernatx Inc | COMPOUNDS AND COMPOSITIONS FOR INTRACELLULAR RELEASE OF FUNDS |
| AU2016379454B2 (en) | 2015-12-23 | 2021-01-28 | The University Of British Columbia | Lipid-linked prodrugs |
| US9834510B2 (en) * | 2015-12-30 | 2017-12-05 | Arcturus Therapeutics, Inc. | Aromatic ionizable cationic lipid |
| WO2018089540A1 (en) | 2016-11-08 | 2018-05-17 | Modernatx, Inc. | Stabilized formulations of lipid nanoparticles |
| EP3554546A4 (en) | 2016-12-14 | 2021-04-14 | Ligandal, Inc. | Methods and compositions for nucleic acid and protein payload delivery |
| JP7220154B2 (en) | 2017-03-15 | 2023-02-09 | モデルナティエックス インコーポレイテッド | Crystalline forms of amino lipids |
| HUE060693T2 (en) | 2017-03-15 | 2023-04-28 | Modernatx Inc | Compound and compositions for intracellular delivery of therapeutic agents |
| MA48047A (en) | 2017-04-05 | 2020-02-12 | Modernatx Inc | REDUCTION OR ELIMINATION OF IMMUNE RESPONSES TO NON-INTRAVENOUS THERAPEUTIC PROTEINS, FOR EXAMPLE SUBCUTANEOUSLY |
| WO2018232357A1 (en) | 2017-06-15 | 2018-12-20 | Modernatx, Inc. | Rna formulations |
| JP7275111B2 (en) | 2017-08-31 | 2023-05-17 | モデルナティエックス インコーポレイテッド | Method for producing lipid nanoparticles |
| WO2019090359A1 (en) | 2017-11-06 | 2019-05-09 | Nitto Denko Corporation | Fusogenic compounds for delivery of biologically active molecules |
| CN108148870B (en) * | 2018-01-12 | 2021-09-21 | 中国人民解放军第四军医大学 | Method for improving siRNA transferring efficiency in vitro of chitosan |
| AU2019359299B2 (en) | 2018-10-09 | 2022-04-21 | The University Of British Columbia | Compositions and systems comprising transfection-competent vesicles free of organic-solvents and detergents and methods related thereto |
| CA3154618A1 (en) | 2019-09-19 | 2021-03-25 | Modernatx, Inc. | Branched tail lipid compounds and compositions for intracellular delivery of therapeutic agents |
| US20230250056A1 (en) * | 2020-06-24 | 2023-08-10 | Bristol-Myers Squibb Company | Process for synthesizing lipids |
| KR20230042022A (en) * | 2020-06-24 | 2023-03-27 | 브리스톨-마이어스 스큅 컴퍼니 | Methods for synthesizing cationic lipids |
| US11524023B2 (en) | 2021-02-19 | 2022-12-13 | Modernatx, Inc. | Lipid nanoparticle compositions and methods of formulating the same |
| WO2023205424A2 (en) * | 2022-04-21 | 2023-10-26 | Greenlight Biosciences, Inc. | Lipid compositions and methods for nucleic acid delivery |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2800818A1 (en) * | 2010-04-28 | 2011-11-03 | Kyowa Hakko Kirin Co., Ltd. | Cationic lipid |
| WO2012170952A2 (en) * | 2011-06-08 | 2012-12-13 | Nitto Denko Corporation | Compounds for targeting drug delivery and enhancing sirna activity |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5264618A (en) * | 1990-04-19 | 1993-11-23 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| US5650386A (en) * | 1995-03-31 | 1997-07-22 | Emisphere Technologies, Inc. | Compositions for oral delivery of active agents |
| US5994317A (en) * | 1996-04-09 | 1999-11-30 | Vical Incorporated | Quaternary cytofectins |
| CN1277024A (en) * | 1999-06-11 | 2000-12-20 | 克林诺斯生物药品工业有限公司 | Application of cationic liposome and polydeoxyribonucleosides composite using as medicament |
| CN101163796B (en) * | 2004-06-07 | 2012-08-29 | 普洛体维生物治疗公司 | Cationic lipids and methods of use |
| US9393315B2 (en) | 2011-06-08 | 2016-07-19 | Nitto Denko Corporation | Compounds for targeting drug delivery and enhancing siRNA activity |
| SI2730277T1 (en) | 2004-12-22 | 2020-07-31 | Nitto Denko Corporation | Drug carrier and drug carrier kit for inhibiting fibrosis |
| JP2006254877A (en) * | 2005-03-18 | 2006-09-28 | Fukuoka Prefecture | Carrier containing glycolipid and method for transferring gene using the same |
| US10196637B2 (en) | 2011-06-08 | 2019-02-05 | Nitto Denko Corporation | Retinoid-lipid drug carrier |
| TWI658830B (en) | 2011-06-08 | 2019-05-11 | 日東電工股份有限公司 | Retinoid-liposomes for enhancing modulation of hsp47 expression |
| US9011903B2 (en) | 2011-06-08 | 2015-04-21 | Nitto Denko Corporation | Cationic lipids for therapeutic agent delivery formulations |
| EP2773328A2 (en) | 2011-11-04 | 2014-09-10 | Nitto Denko Corporation | Method of producing lipid nanoparticles for drug delivery |
| US9579338B2 (en) | 2011-11-04 | 2017-02-28 | Nitto Denko Corporation | Method of producing lipid nanoparticles for drug delivery |
| TWI594723B (en) | 2011-12-19 | 2017-08-11 | 愛爾康眼科手術激光股份有限公司 | Image processor for intra-surgical optical coherence tomographic imaging of laser cataract procedures |
| EP3489220B9 (en) | 2012-06-08 | 2021-11-10 | Nitto Denko Corporation | Lipids for therapeutic agent delivery formulations |
-
2013
- 2013-06-07 EP EP18200656.9A patent/EP3489220B9/en active Active
- 2013-06-07 RU RU2014153658A patent/RU2658007C2/en active
- 2013-06-07 JP JP2015516267A patent/JP5873604B2/en active Active
- 2013-06-07 LT LTEP18200656.9T patent/LT3489220T/en unknown
- 2013-06-07 PT PT182006569T patent/PT3489220T/en unknown
- 2013-06-07 EP EP13729241.3A patent/EP2858974B9/en active Active
- 2013-06-07 SI SI201331928T patent/SI3489220T1/en unknown
- 2013-06-07 DK DK13729241.3T patent/DK2858974T3/en active
- 2013-06-07 TR TR2018/16986T patent/TR201816986T4/en unknown
- 2013-06-07 ES ES18200656T patent/ES2897216T3/en active Active
- 2013-06-07 AU AU2013270685A patent/AU2013270685B2/en active Active
- 2013-06-07 HU HUE18200656A patent/HUE056014T2/en unknown
- 2013-06-07 SI SI201331245T patent/SI2858974T1/en unknown
- 2013-06-07 KR KR1020157000454A patent/KR102102801B1/en active IP Right Grant
- 2013-06-07 HR HRP20211424TT patent/HRP20211424T2/en unknown
- 2013-06-07 ES ES13729241T patent/ES2697680T3/en active Active
- 2013-06-07 PL PL18200656T patent/PL3489220T3/en unknown
- 2013-06-07 PL PL13729241T patent/PL2858974T3/en unknown
- 2013-06-07 BR BR112014030714-8A patent/BR112014030714B1/en active IP Right Grant
- 2013-06-07 LT LTEP13729241.3T patent/LT2858974T/en unknown
- 2013-06-07 DK DK18200656.9T patent/DK3489220T3/en active
- 2013-06-07 HU HUE13729241A patent/HUE041882T2/en unknown
- 2013-06-07 RS RS20181385A patent/RS58108B9/en unknown
- 2013-06-07 TW TW102120444A patent/TWI654997B/en active
- 2013-06-07 WO PCT/US2013/044849 patent/WO2013185116A1/en active Application Filing
- 2013-06-07 PT PT13729241T patent/PT2858974T/en unknown
- 2013-06-07 CA CA2876148A patent/CA2876148C/en active Active
- 2013-06-07 CN CN201380041854.8A patent/CN104640841B/en active Active
- 2013-06-07 RS RS20211054A patent/RS62288B9/en unknown
- 2013-06-10 US US13/913,918 patent/US9308267B2/en active Active
-
2015
- 2015-10-01 US US14/872,951 patent/US10441659B2/en active Active
-
2018
- 2018-11-07 HR HRP20181855TT patent/HRP20181855T1/en unknown
- 2018-11-27 CY CY181101255T patent/CY1120929T1/en unknown
-
2019
- 2019-07-02 US US16/460,812 patent/US11103583B2/en active Active
-
2021
- 2021-09-17 CY CY20211100818T patent/CY1124499T1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2800818A1 (en) * | 2010-04-28 | 2011-11-03 | Kyowa Hakko Kirin Co., Ltd. | Cationic lipid |
| WO2012170952A2 (en) * | 2011-06-08 | 2012-12-13 | Nitto Denko Corporation | Compounds for targeting drug delivery and enhancing sirna activity |
Non-Patent Citations (3)
| Title |
|---|
| "Remington's Pharmaceutical Sciences, 18th Ed.,", 1990, MACK PUBLISHING CO. |
| FINGL ET AL.: "The Pharmacological Basis of Therapeutics", 1975, pages: 1 |
| G. P. MOSS: "Biochemical Nomenclature and Related Documents," 2nd Ed.", 1992, PORTLAND PRESS, pages: 247 - 251 |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11103583B2 (en) | 2012-06-08 | 2021-08-31 | Nitto Denko Corporation | Lipids for therapeutic agent delivery formulations |
| US10441659B2 (en) | 2012-06-08 | 2019-10-15 | Nitto Denko Corporation | Lipids for therapeutic agent delivery formulations |
| US9567296B2 (en) | 2013-11-18 | 2017-02-14 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US9850202B2 (en) | 2013-11-18 | 2017-12-26 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US9580711B2 (en) | 2013-11-18 | 2017-02-28 | Arcturus Therapeutics, Inc. | Lipid particles with asymmetric cationic lipids for RNA delivery |
| US9670152B2 (en) | 2013-11-18 | 2017-06-06 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US9365610B2 (en) | 2013-11-18 | 2016-06-14 | Arcturus Therapeutics, Inc. | Asymmetric ionizable cationic lipid for RNA delivery |
| USRE49233E1 (en) | 2013-11-18 | 2022-10-04 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US10781169B2 (en) | 2013-11-18 | 2020-09-22 | Arcturus Therapeutics, Inc. | Method of synthesis of an ionizable cationic lipid |
| US10556861B2 (en) | 2013-11-18 | 2020-02-11 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US9896413B2 (en) | 2013-11-18 | 2018-02-20 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US10399937B2 (en) | 2013-11-18 | 2019-09-03 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US9976142B2 (en) | 2014-04-02 | 2018-05-22 | Nitto Denko Corporation | Targeting molecule and a use thereof |
| AU2015350561B2 (en) * | 2014-11-18 | 2020-01-30 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| JP2017535610A (en) * | 2014-11-18 | 2017-11-30 | アークトゥルス セラピューティクス, インコーポレイテッド | Ionizable cationic lipids for RNA delivery |
| CN107207428A (en) * | 2014-11-18 | 2017-09-26 | 阿克丘勒斯治疗公司 | Ionizable cation lipid for RNA delivery |
| KR102380363B1 (en) * | 2014-11-18 | 2022-03-29 | 아크투루스 쎄라퓨틱스, 인크. | Ionizable cationic lipid for rna delivery |
| WO2016081029A1 (en) * | 2014-11-18 | 2016-05-26 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for rna delivery |
| KR20170095241A (en) * | 2014-11-18 | 2017-08-22 | 아크투루스 쎄라퓨틱스, 인크. | Ionizable cationic lipid for rna delivery |
| US10167253B2 (en) * | 2015-06-24 | 2019-01-01 | Nitto Denko Corporation | Ionizable compounds and compositions and uses thereof |
| US11384051B2 (en) | 2015-06-24 | 2022-07-12 | Nitto Denko Corporation | Ionizable compounds and compositions and uses thereof |
| EP3686184A3 (en) * | 2015-06-24 | 2020-08-26 | Nitto Denko Corporation | Ionizable compounds and compositions and uses thereof |
| AU2016281685B2 (en) * | 2015-06-24 | 2021-08-12 | Nitto Denko Corporation | Ionizable compounds and compositions and uses thereof |
| US20190127318A1 (en) * | 2015-06-24 | 2019-05-02 | Nitto Denko Corporation | Ionizable compounds and compositions and uses thereof |
| US20160376229A1 (en) * | 2015-06-24 | 2016-12-29 | Nitto Denko Corporation | Ionizable compounds and compositions and uses thereof |
| WO2018084168A1 (en) | 2016-11-02 | 2018-05-11 | 日東電工株式会社 | Skin fibrosis treatment agent |
| US10526284B2 (en) | 2016-12-21 | 2020-01-07 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US10980895B2 (en) | 2016-12-21 | 2021-04-20 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| US10383952B2 (en) | 2016-12-21 | 2019-08-20 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| WO2019074071A1 (en) | 2017-10-11 | 2019-04-18 | 日東電工株式会社 | Regulation of nucleic acid molecule expression |
| US10961188B2 (en) | 2017-12-20 | 2021-03-30 | Arcturus Therapeutics, Inc. | Ionizable cationic lipid for RNA delivery |
| WO2022037652A1 (en) * | 2020-08-20 | 2022-02-24 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2022136641A1 (en) * | 2020-12-23 | 2022-06-30 | Etherna Immunotherapies Nv | Ionizable lipids |
| WO2023044333A1 (en) * | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Cyclic lipids and methods of use thereof |
| US11773061B2 (en) | 2021-09-14 | 2023-10-03 | Renagade Therapeutics Management Inc. | Cyclic lipids and methods of use thereof |
| WO2023222870A1 (en) * | 2022-05-20 | 2023-11-23 | Etherna Immunotherapies Nv | Ionizable lipids |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11103583B2 (en) | Lipids for therapeutic agent delivery formulations | |
| US11084779B2 (en) | Cationic lipids for therapeutic agent delivery formulations | |
| US10669229B2 (en) | Compounds for targeting drug delivery and enhancing siRNA activity | |
| WO2012170952A9 (en) | Compounds for targeting drug delivery and enhancing sirna activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13729241 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2876148 Country of ref document: CA Ref document number: 2015516267 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013729241 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20157000454 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2014153658 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2013270685 Country of ref document: AU Date of ref document: 20130607 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014030714 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014030714 Country of ref document: BR Kind code of ref document: A2 Effective date: 20141208 |