WO2013138502A1 - Salts of an epidermal growth factor receptor kinase inhibitor - Google Patents
Salts of an epidermal growth factor receptor kinase inhibitor Download PDFInfo
- Publication number
- WO2013138502A1 WO2013138502A1 PCT/US2013/030996 US2013030996W WO2013138502A1 WO 2013138502 A1 WO2013138502 A1 WO 2013138502A1 US 2013030996 W US2013030996 W US 2013030996W WO 2013138502 A1 WO2013138502 A1 WO 2013138502A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- acid
- salt
- theta
- peaks
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/02—Sulfonic acids having sulfo groups bound to acyclic carbon atoms
- C07C309/03—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C309/04—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton containing only one sulfo group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/02—Sulfonic acids having sulfo groups bound to acyclic carbon atoms
- C07C309/03—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C309/05—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton containing at least two sulfo groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/25—Sulfonic acids having sulfo groups bound to carbon atoms of rings other than six-membered aromatic rings of a carbon skeleton
- C07C309/27—Sulfonic acids having sulfo groups bound to carbon atoms of rings other than six-membered aromatic rings of a carbon skeleton containing carboxyl groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/29—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of non-condensed six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/29—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of non-condensed six-membered aromatic rings
- C07C309/30—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of non-condensed six-membered aromatic rings of six-membered aromatic rings substituted by alkyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/33—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems
- C07C309/34—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems formed by two rings
- C07C309/35—Naphthalene sulfonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C55/00—Saturated compounds having more than one carboxyl group bound to acyclic carbon atoms
- C07C55/02—Dicarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C55/00—Saturated compounds having more than one carboxyl group bound to acyclic carbon atoms
- C07C55/02—Dicarboxylic acids
- C07C55/06—Oxalic acid
- C07C55/07—Salts thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C62/00—Compounds having carboxyl groups bound to carbon atoms of rings other than six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C62/02—Saturated compounds containing hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- the present invention provides salt forms of a compound useful as mutant-selective inhibitors of epidermal growth factor receptor (EGFR) kinase, including polymorphic forms of certain salts.
- EGFR epidermal growth factor receptor
- the invention also provides pharmaceutically acceptable compositions comprising salt forms of the present invention and methods of using the compositions in the treatment of various disorders.
- Protein tyrosine kinases are a class of enzymes that catalyze the transfer of a phosphate group from ATP or GTP to a tyrosine residue located on a protein substrate. Receptor tyrosine kinases act to transmit signals from the outside of a cell to the inside by activating secondary messaging effectors via a phosphorylation event. A variety of cellular processes are promoted by these signals, including proliferation, carbohydrate utilization, protein synthesis, angiogenesis, cell growth, and cell survival.
- Activating mutations in the tyrosine kinase domain of EGFR have been identified in patients with non-small cell lung cancer (Lin, N. U.; Winer, E. P., Breast Cancer Res 6: 204-210, 2004).
- the reversible inhibitors Tarceva (erlotinib) and Iressa (gefitinib) currently are first-line therapy for non-small cell lung cancer patients with activating mutations.
- the most common activating mutations are L858R and delE746-A750.
- T790M may also be pre-existing; there may be an independent, oncogenic role for the T790M mutation.
- T790M mutations are linked with certain familial lung cancers.
- Figure 1 depicts the x-ray powder diffraction (XRPD) pattern for a bis-besylate salt of Compound 1.
- FIG. 1 depicts the thermogravimetric analysis (TGA) pattern for a bis-besylate salt of Compound 1.
- FIG. 3 depicts the thermogravimetric analysis (TGA) pattern for a further dried sample of a bis-besylate salt of Compound 1.
- Figure 4 depicts the differential scanning calorimetry (DSC) pattern for a bis- besylate salt of Compound 1.
- Figure 5 depicts the infrared (IR) spectrum of a bis-besylate salt of Compound 1.
- Figure 6 depicts the ⁇ - MR spectrum of a bis-besylate salt of Compound 1.
- Figure 7 depicts the dynamic vapor sorption (DVS) pattern of a bis-besylate salt of Compound 1.
- Figure 8 depicts the results of a hydration study of a bis-besylate salt of Compound I, as analyzed by the XRPD patterns.
- Figure 9 depicts the results of a disproportionation study of a bis-besylate salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 10 depicts the results of a stability study of a bis-besylate salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 11 depicts the results of a thermodynamic solubility study of a bis-besylate salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 12 depicts the dissolution at pH 4.5 of a compressed disc of a bis-besylate salt of Compound 1.
- Figure 13 depicts the dissolution at pH 3.0 of a compressed disc of a bis-besylate salt of Compound 1.
- Figure 14 depicts the XRPD pattern for a bis-besylate hydrate salt of Compound 1.
- Figure 15 depicts the TGA pattern for a bis-besylate hydrate salt of Compound 1.
- Figure 16 depicts the DSC pattern for a bis-besylate hydrate salt of Compound 1.
- Figure 17 depicts the IR spectrum of a bis-besylate hydrate salt of Compound 1.
- Figure 18 depicts the ⁇ - MR spectrum of a bis-besylate hydrate salt of Compound 1.
- Figure 19 depicts the DVS pattern of a bis-besylate hydrate salt of Compound 1.
- Figure 20 depicts the results of a stability study of a bis-besylate hydrate salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 21 depicts the results of a thermodynamic solubility study of a bis-besylate salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 22 depicts the dissolution at pH 4.5 of a compressed disc of a bis-besylate hydrate salt of Compound 1.
- Figure 23 depicts the dissolution at pH 3.0 of a compressed disc of a bis-besylate hydrate salt of Compound 1.
- Figure 24 depicts the XRPD pattern for a mono-maleate salt of Compound 1.
- Figure 25 depicts the TGA pattern for a mono-maleate salt of Compound 1.
- Figure 26 depicts the DSC pattern for a mono-maleate salt of Compound 1.
- Figure 27 depicts the ⁇ - MR spectrum of a mono-maleate salt of Compound 1.
- Figure 28 depicts the XRPD pattern for a bis-hydrochloride salt of Compound 1.
- Figure 29 depicts the TGA pattern for a bis-hydrochloride salt of Compound 1.
- Figure 30 depicts the DSC pattern for a bis-hydrochloride salt of Compound 1.
- Figure 31 depicts the ⁇ - MR spectrum of a bis-hydrochloride salt of Compound 1.
- Figure 32 depicts the XRPD pattern for a Form I hydrobromide salt of Compound 1.
- Figure 33 depicts the TGA pattern for a Form I hydrobromide salt of Compound 1.
- Figure 34 depicts the DSC pattern for a Form I hydrobromide salt of Compound 1.
- Figure 35 depicts the IR spectrum of a Form I hydrobromide salt of Compound 1.
- Figure 36 depicts the ⁇ - MR spectrum of a Form I hydrobromide salt of Compound 1.
- Figure 37 depicts the DVS pattern of a Form I hydrobromide salt of Compound 1.
- Figure 38 depicts the results of a hydration study of a Form I hydrobromide salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 39 depicts the results of a disproportionation study of a Form I hydrobromide salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 40 depicts the results of a stability study of a Form I hydrobromide salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 41 depicts the results of a thermodynamic solubility study of a Form I hydrobromide salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 42 depicts the dissolution at pH 4.5 of a compressed disc of a Form I hydrobromide salt of Compound 1.
- Figure 43 depicts the dissolution at pH 3.0 of a compressed disc of a Form I hydrobromide salt of Compound 1.
- Figure 44 depicts the XRPD pattern for a Form I hydrobromide salt of Compound 1.
- Figure 45 depicts the TGA pattern for a Form I hydrobromide salt of Compound 1.
- Figure 46 depicts the IR spectrum of a Form I hydrobromide salt of Compound 1.
- Figure 47 depicts the XRPD pattern for a Form I hydrobromide salt of Compound 1.
- Figure 48 depicts the ⁇ - MR spectrum for a Form I hydrobromide salt of Compound 1.
- Figure 49 depicts the DSC pattern of a Form I hydrobromide salt of Compound 1.
- Figure 50 depicts the results of a slurry experiment involving a form of the free base of Compound 1 and a bis-besylate hydrate.
- Figure 51 depicts the results of a slurry experiment involving a Form I hydrobromide salt of Compound 1 at pH 6.2.
- Figure 52 depicts the XRPD pattern for a Form I hydrobromide salt of Compoundl.
- Figure 53 depicts the XRPD pattern for a Form I hydrobromide salt of Compound 1.
- Figure 54 depicts the IR spectrum of a Form I hydrobromide salt of Compound 1.
- Figure 55 depicts the ⁇ - MR spectrum of a Form I hydrobromide salt of Compound 1.
- Figure 56 depicts the TGA pattern for a Form I hydrobromide salt of Compound 1.
- Figure 57 depicts the DSC pattern for a Form I hydrobromide salt of Compound 1.
- Figure 58 depicts the XRPD pattern for a Form I hydrobromide salt of Compound 1 after heating.
- Figure 59 depicts the XRPD pattern for a Form I hydrobromide salt of Compound 1.
- Figure 60 depicts the XRPD pattern for a Form I hydrobromide salt of Compound 1.
- Figure 61 depicts the IR spectrum of a Form I hydrobromide salt of Compound 1.
- Figure 62 depicts the ⁇ - MR spectrum of a Form I hydrobromide salt of Compound 1.
- Figure 63 depicts the TGA pattern for a Form I hydrobromide salt of Compound 1.
- Figure 64 depicts the DSC pattern for a Form I hydrobromide salt of Compound 1.
- Figure 65 depicts the results of a thermodynamic solubility study of a Form I hydrobromide salt of Compound 1, as analyzed by the XRPD patterns.
- Figure 66 depicts the XRPD pattern for a Form I hydrobromide salt of Compound 1, after storage for 1.5 months.
- Figure 67 depicts the XRPD pattern for a Form III hydrobromide salt of Compound 1.
- Figure 68 depicts theTGA pattern for a Form III hydrobromide salt of Compound 1.
- Figure 69 depicts the DSC pattern for a Form III hydrobromide salt of Compound 1.
- Figure 70 depicts the IR spectrum for a Form III hydrobromide salt of Compound 1.
- Figure 71 depicts the ⁇ - MR spectrum of a Form III hydrobromide salt of Compound 1.
- Figure 72 depicts the XRPD pattern for a Form IV hydrobromide salt of Compound 1.
- Figure 73 depicts the XRPD pattern for a Form V hydrobromide salt of Compound 1.
- Figure 74 depicts the XRPD pattern for a Form VI hydrobromide salt of Compound 1.
- Figure 75 depicts the XRPD pattern for a Form VII hydrobromide salt of Compound 1.
- Figure 76 depicts the XRPD pattern for a Form VIII hydrobromide salt of Compound 1.
- Figure 77 depicts the TGA pattern for a Form VIII hydrobromide salt of Compound 1.
- Figure 78 depicts the DSC pattern for a Form VIII hydrobromide salt of Compound 1.
- Figure 79 depicts the XRPD of an amorphous hydrobromide salt of Compound 1.
- Figure 80 depicts the XRPD of Form V hydrobromide salt of Compound 1 following desolvation conditions.
- Figure 81 depicts input material Form I hydrobromide salt of Compound 1 compared with a wet sample, and after stages of drying.
- Figure 82 depicts the XRPD analysis of hydrobromide salt Forms I and III resulting from competitive slurry experiments at ambient temperature (22 °C).
- Figure 83 depicts the XRPD analysis of hydrobromide salt Forms I and III resulting from competitive slurry experiments at 60 °C.
- Figure 84 depicts the XRPD analysis of Form I hydrobromide salt of Compound 1 slurried in EtOH:water mixtures.
- Figure 85 depicts the XRPD analysis of material slurried in IP A/acetone (9: 1): water mixtures.
- Figure 86 depicts the XRPD analysis following hydration studies at 15 °C and 35 °C.
- Figure 87 depicts the form diagram for the hydrobromide salt, including 7 different forms and the relationship between such forms.
- Compound 1 (N-(3-(2-(4-(4-acetylpiperazin-l-yl)-2-methoxyphenylamino)-5- (trifluoromethyl)pyrimidin-4-ylamino)phenyl)acrylamide)) is designated as compound number I- 4 and the synthesis of compound 1 is described in detail at Example 3 of the '061 application.
- Compound 1 is active in a variety of assays and therapeutic models demonstrating selective covalent, irreversible inhibition of mutant EGFR kinase (in enzymatic and cellular assays). Notably, compound 1 was found to inhibit human non-small cell lung cancer cell proliferation both in vitro and in vivo. Accordingly, compound 1 and its salts are useful for treating one or more disorders associated with activity of mutant EGFR kinase.
- the present invention provides a salt of compound 1, represented by compound 2:
- n 1 or 2;
- X is benzenesulfonic acid, camphor sulfonic acid, 1,2-ethane disulfonic acid,
- hydrobromic acid hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2 -sulfonic acid, 1,5 -naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid or 2,4,6- trihydroxybenzoic acid.
- compound 2 can exist in a variety of physical forms.
- compound 2 can be in solution, suspension, or in solid form.
- compound 2 is in solid form.
- said compound may be amorphous, crystalline, or a mixture thereof. Exemplary solid forms are described in more detail below.
- the present invention provides compound 2 substantially free of impurities.
- the term “substantially free of impurities” means that the compound contains no significant amount of extraneous matter. Such extraneous matter may include excess acid "X", excess compound 1, residual solvents, or any other impurities that may result from the preparation of, and/or isolation of, compound 2.
- at least about 90% by weight of compound 2 is present.
- at least about 95% by weight of compound 2 is present.
- at least about 99% by weight of compound 2 is present.
- compound 2 is present in an amount of at least about 95, 97, 97.5, 98.0, 98.5, 99, 99.5, 99.8 weight percent where the percentages are based on the total weight of the composition.
- compound 2 contains no more than about 5.0 area percent HPLC of total organic impurities and, in certain embodiments, no more than about 3.0 area percent HPLC of total organic impurities and, in certain embodiments, no more than about 1.5 area percent HPLC total organic impurities relative to the total area of the HPLC chromatogram.
- compound 2 contains no more than about 1.0 area percent HPLC of any single impurity; no more than about 0.6 area percent HPLC of any single impurity, and, in certain embodiments, no more than about 0.5 area percent HPLC of any single impurity, relative to the total area of the HPLC chromatogram.
- compound 2 can exist in a variety of solid forms. Such forms include polymorphs and amorphous forms.
- the solid forms can be solvates, hydrates and unsolvated forms of compound 2. All such forms are contemplated by the present invention.
- the present invention provides compound 2 as a mixture of one or more solid forms of compound 2.
- polymorph refers to the different crystal structures (of solvated or unsolvated forms) in which a compound can crystallize.
- solvate refers to a crystal form with either a stoichiometric or non-stoichiometric amount of solvent. For polymorphs, the solvent is incorporated into the crystal structure.
- hydrate refers to a solid form with either a stoichiometric or non-stoichiometric amount of water. For polymorphs, the water is incorporated into the crystal structure.
- the term "about”, when used in reference to a degree 2-theta value refers to the stated value ⁇ 0.3 degree 2-theta (° 2 ⁇ ). In certain embodiments, “about” refers to ⁇ 0.2 degree 2-theta or ⁇ 0.1 degree 2-theta.
- compound 2 is a crystalline solid. In other embodiments, compound 2 is a crystalline solid substantially free of amorphous compound 2. As used herein, the term "substantially free of amorphous compound 2" means that the compound contains no significant amount of amorphous compound 2. In certain embodiments, at least about 90% by weight of crystalline compound 2 is present, or at least about 95% by weight of crystalline compound 2 is present. In still other embodiments of the invention, at least about 99% by weight of crystalline compound 2 is present.
- compound 2 is a benzenesulfonic acid (besylate) salt.
- the salt can be a mono-besylate or a bis-besylate.
- a besylate salt is optionally solvated or hydrated, such as a monohydrate.
- an unsolvated bis-besylate salt has a powder X-ray diffraction pattern substantially similar to that depicted in Figure 1.
- an unsolvated bis-besylate salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 5.62, about 17.41, about 18.90, about 19.07 and about 19.52 degrees 2-theta.
- an unsolvated bis-besylate salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 5.62, about 17.41, about 18.90, about 19.07 and about 19.52 degrees 2-theta.
- an unsolvated bis-besylate salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 5.62, about 17.41, about 18.90, about 19.07 and about 19.52 degrees 2-theta.
- an unsolvated bis- besylate salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about 5.62, 7.89, 11.23, 12.64, 17.41, 18.90, 19.07, 19.52, 22.63, 23.17, 25.28 and 28.92 degrees 2-theta.
- an unsolvated bis- besylate salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- an unsolvated bis-besylate salt has a thermogravimetric analysis pattern substantially similar to that depicted in Figure 2 or 3.
- an unsolvated bis-besylate salt has a differential scanning calorimetry pattern substantially similar to that depicted in Figure 4.
- an unsolvated bis- besylate salt has an infrared spectrum substantially similar to that depicted in Figure 5.
- an unsolvated bis-besylate salt has an ⁇ - MR spectrum substantially similar to that depicted in Figure 6.
- an unsolvated bis-besylate salt has a dynamic vapour sorption pattern substantially similar to that depicted in Figure 7.
- An unsolvated bis-besylate salt can be characterized by substantial similarity to two or more of these figures simultaneously.
- a bis-besylate hydrate has a powder X-ray diffraction pattern substantially similar to that depicted in Figure 14.
- a bis- besylate hydrate salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 10.68, about 16.10, about 18.44 and about 22.36 degrees 2- theta.
- a bis-besylate hydrate salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 10.68, about 16.10, about 18.44 and about 22.36 degrees 2-theta.
- a bis-besylate hydrate salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 10.68, about 16.10, about 18.44 and about 22.36 degrees 2-theta.
- a bis-besylate hydrate salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about 9.33, 10.68, 16.10, 16.43, 16.64, 18.44, 20.05, 20.32, 20.74, 22.36 and 22.83 degrees 2-theta.
- a bis- besylate hydrate salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- a bis-besylate hydrate has a thermogravimetric analysis pattern substantially similar to that depicted in Figure 15.
- a bis- besylate hydrate has a differential scanning calorimetry pattern substantially similar to that depicted in Figure 16.
- a bis-besylate hydrate has a infrared spectrum substantially similar to that depicted in Figure 17.
- a bis-besylate hydrate has an ⁇ - MR spectrum substantially similar to that depicted in Figure 18.
- a bis-besylate hydrate has a dynamic vapour sorption pattern substantially similar to that depicted in Figure 19.
- a bis-besylate hydrate can be characterized by substantial similarity to two or more of these figures simultaneously.
- compound 2 is a camphor sulfonic acid salt (e.g., camphor- 10-sulfonic acid).
- compound 2 is a mono-camphor sulfonic acid salt.
- compound 2 is a bis-camphor sulfonic acid salt.
- compound 2 is a 1,2-ethane disulfonic acid salt. In some embodiments, compound 2 is a mono- 1,2-ethane disulfonic acid salt. In some embodiments, compound 2 is a bis- 1,2-ethane disulfonic acid salt.
- compound 2 is a hydrobromic acid salt. In some embodiments, compound 2 is an anhydrous monohydrobromic acid salt. In some embodiments, compound 2 is an anhydrous bis-hydrobromic acid salt. A hydrobromide salt is optionally solvated or hydrated. In some embodiments, compound 2 is a monohydrate hydrobromic acid salt. In some embodiments, compound 2 is a solvated hydrobromic acid salt. In some such embodiments, the solvate is selected from dimethylsulfoxide (DMSO), dimethylformamide (DMF) and 1,4-dioxane. In some embodiments, compound 2 is a hydrobromide salt selected from Form I, Form III, Form IV, Form V, Form VI, Form VII and Form VIII, each of which is described in further detail, infra.
- DMSO dimethylsulfoxide
- DMF dimethylformamide
- compound 2 is a hydrobromide salt selected from Form I, Form III, Form IV, Form V, Form VI, Form VII and Form VIII, each
- compound 2 is a Form I hydrobromide salt.
- compound 2 is an anhydrous Form I hydrobromide salt.
- a Form I hydrobromide salt is characterized by the powder X-ray diffraction pattern substantially similar to that depicted in Figure 60.
- a Form I hydrobromide salt is characterized by the powder X-ray diffraction pattern substantially similar to that depicted in Figure 59.
- a Form I mono-hydrobromide salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 17.39, about 19.45, about 21.41, about 23.56 and about 27.45 degrees 2-theta.
- a Form I mono-hydrobromide salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 17.39, about 19.45, about 21.41, about 23.56 and about 27.45 degrees 2-theta.
- a Form I mono-hydrobromide salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 17.39, about 19.45, about 21.41, about 23.56 and about 27.45 degrees 2-theta. In some embodiments, a Form I mono-hydrobromide salt is characterized by four or more peaks in its powder X-ray diffraction pattern selected from those at about 17.39, about 19.45, about 21.41, about 23.56 and about 27.45 degrees 2-theta.
- a Form I mono- hydrobromide salt is characterized by an X-ray powder diffraction pattern which includes the peaks at about 9.84, 15.62, 17.39, 19.45, 20.69, 21.41, 22.38, 23.56, 25.08 and 27.45 degrees 2- theta.
- a Form I mono-hydrobromide salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- a Form I mono-hydrobromide salt is characterized by a thermogravimetric analysis pattern substantially similar to that depicted in Figure 63.
- a Form I mono-hydrobromide salt is characterized by a differential scanning calorimetry pattern substantially similar to that depicted in Figure 64.
- a Form I mono-hydrobromide salt is characterized by an infrared spectrum substantially similar to that depicted in Figure 61.
- a Form I mono-hydrobromide salt is characterized by a ⁇ - MR spectrum substantially similar to that depicted in Figure 62.
- a Form I mono- hydrobromide salt is characterized by substantial similarity to two or more of these figures simultaneously.
- compound 2 is a Form III hydrobromide salt.
- compound 2 is an anhydrous Form III hydrobromide salt.
- a Form III hydrobromide salt is characterized by a powder X-ray diffraction pattern substantially similar to that depicted in Figure 67.
- a Form III hydrobromide salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 6.79, about 13.36, about 19.93, about 20.89, about 21.90, about 22.70, about 22.91 and about 26.34 degrees 2-theta.
- a Form III hydrobromide salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 6.79, about 13.36, about 19.93, about 20.89, about 21.90, about 22.70, about 22.91 and about 26.34 degrees 2-theta.
- a Form III hydrobromide salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 6.79, about 13.36, about 19.93, about 20.89, about 21.90, about 22.70, about 22.91 and about 26.34 degrees 2-theta.
- a Form III hydrobromide salt is characterized by four or more peaks in its powder X-ray diffraction pattern selected from those at about 6.79, about 13.36, about 19.93, about 20.89, about 21.90, about 22.70, about 22.91 and about 26.34 degrees 2-theta.
- a Form III hydrobromide salt is characterized by five or more peaks in its powder X-ray diffraction pattern selected from those at about 6.79, about 13.36, about 19.93, about 20.89, about 21.90, about 22.70, about 22.91 and about 26.34 degrees 2-theta.
- a Form III hydrobromide salt is characterized by six or more peaks in its powder X-ray diffraction pattern selected from those at about 6.79, about 13.36, about 19.93, about 20.89, about 21.90, about 22.70, about 22.91 and about 26.34 degrees 2-theta.
- a Form III hydrobromide salt is characterized by an X-ray powder diffraction pattern which includes the peaks at about 6.79, about 13.36, about 19.93, about 20.89, about 21.90, about 22.70, about 22.91 and about 26.34 degrees 2-theta.
- a Form III hydrobromide salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- a Form III hydrobromide salt is characterized by a thermogravimetric analysis pattern substantially similar to that depicted in Figure 68. In some embodiments, a Form III hydrobromide salt is characterized by a differential scanning calorimetry pattern substantially similar to that depicted in Figure 69. In some embodiments, a Form III hydrobromide salt is characterized by an infrared spectrum substantially similar to that depicted in Figure 70. In some embodiments, a Form III hydrobromide salt is characterized by a ⁇ - MR spectrum substantially similar to that depicted in Figure 71. In some embodiments, a Form III hydrobromide salt is characterized by substantial similarity to two or more of these figures simultaneously.
- compound 2 is a Form IV hydrobromide salt.
- a Form IV hydrobromide salt is a 1,4-dioxane solvate.
- a Form rv hydrobromide salt is characterized by a powder X-ray diffraction pattern substantially similar to that depicted in Figure 72.
- a Form IV hydrobromide salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 6.45, about 12.96, about 19.38, about 19.79, about 21.37 and about 21.58 degrees 2-theta.
- a Form IV hydrobromide salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 6.45, about 12.96, about 19.38, about 19.79, about 21.37 and about 21.58 degrees 2-theta.
- a Form IV hydrobromide salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 6.45, about 12.96, about 19.38, about 19.79, about 21.37 and about 21.58 degrees 2-theta.
- a Form IV hydrobromide salt is characterized by four or more peaks in its powder X-ray diffraction pattern selected from those at about 6.45, about 12.96, about 19.38, about 19.79, about 21.37 and about 21.58 degrees 2-theta. In some embodiments, a Form IV hydrobromide salt is characterized by five or more peaks in its powder X-ray diffraction pattern selected from those at about 6.45, about 12.96, about 19.38, about 19.79, about 21.37 and about 21.58 degrees 2-theta.
- a Form IV hydrobromide salt is characterized by an X-ray powder diffraction pattern which includes the peaks at about 6.45, about 12.96, about 19.38, about 19.79, about 21.37 and about 21.58 degrees 2-theta.
- a Form IV hydrobromide salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- compound 2 is a Form V hydrobromide salt.
- a Form V hydrobromide salt is a ⁇ , ⁇ -dimethylformamide (DMF) solvate.
- a Form V hydrobromide salt is characterized by a powder X-ray diffraction pattern substantially similar to that depicted in Figure 73.
- a Form V hydrobromide salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 6.17, about 6.99, about 12.50, about 14.14, about 17.72 and about 23.12 degrees 2-theta.
- a Form V hydrobromide salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 6.17, about 6.99, about 12.50, about 14.14, about 17.72 and about 23.12 degrees 2-theta.
- a Form V hydrobromide salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 6.17, about 6.99, about 12.50, about 14.14, about 17.72 and about 23.12 degrees 2-theta.
- a Form V hydrobromide salt is characterized by four or more peaks in its powder X-ray diffraction pattern selected from those at about 6.17, about 6.99, about 12.50, about 14.14, about 17.72 and about 23.12 degrees 2-theta. In some embodiments, a Form V hydrobromide salt is characterized by five or more peaks in its powder X-ray diffraction pattern selected from those at about 6.17, about 6.99, about 12.50, about 14.14, about 17.72 and about 23.12 degrees 2-theta.
- a Form V hydrobromide salt is characterized by an X-ray powder diffraction pattern which includes the peaks at about 6.17, about 6.99, about 12.50, about 14.14, about 17.72 and about 23.12 degrees 2-theta.
- a Form V hydrobromide salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- compound 2 is a Form VI hydrobromide salt.
- a Form VI hydrobromide salt is a dimethylsulfoxide (DMSO) solvate.
- DMSO dimethylsulfoxide
- a Form VI hydrobromide salt is characterized by a powder X-ray diffraction pattern substantially similar to that depicted in Figure 74.
- a Form VI hydrobromide salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 8.38, about 9.38, about 18.93, and about 21.58 degrees 2- theta.
- a Form VI hydrobromide salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 8.38, about 9.38, about 18.93, and about 21.58 degrees 2-theta.
- a Form VI hydrobromide salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 8.38, about 9.38, about 18.93, and about 21.58 degrees 2-theta.
- a Form VI hydrobromide salt is characterized by an X-ray powder diffraction pattern which includes the peaks at about 8.38, about 9.38, about 18.93, and about 21.58 degrees 2-theta.
- a Form VI hydrobromide salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- compound 2 is a Form VII hydrobromide salt.
- a Form VII hydrobromide salt is a dimethylsulfoxide (DMSO) solvate.
- DMSO dimethylsulfoxide
- a Form VII hydrobromide salt is characterized by a powder X-ray diffraction pattern substantially similar to that depicted in Figure 75.
- a Form VII hydrobromide salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 15.91, about 19.10, about 19.53, about 20.24, about 22.64 and about 25.58 degrees 2-theta.
- a Form VII hydrobromide salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 15.91, about 19.10, about 19.53, about 20.24, about 22.64 and about 25.58 degrees 2-theta.
- a Form VII hydrobromide salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 15.91, about 19.10, about 19.53, about 20.24, about 22.64 and about 25.58 degrees 2-theta.
- a Form VII hydrobromide salt is characterized by four or more peaks in its powder X-ray diffraction pattern selected from those at about 15.91, about 19.10, about 19.53, about 20.24, about 22.64 and about 25.58 degrees 2-theta. In some embodiments, a Form VII hydrobromide salt is characterized by five or more peaks in its powder X-ray diffraction pattern selected from those at about 15.91, about 19.10, about 19.53, about 20.24, about 22.64 and about 25.58 degrees 2-theta.
- a Form VII hydrobromide salt is characterized by an X-ray powder diffraction pattern which includes the peaks at about 15.91, about 19.10, about 19.53, about 20.24, about 22.64 and about 25.58 degrees 2-theta.
- a Form VII hydrobromide salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- compound 2 is a Form VIII hydrobromide salt.
- a Form VIII hydrobromide salt is a hydrate.
- a Form VIII hydrobromide salt is characterized by a powder X-ray diffraction pattern substantially similar to that depicted in Figure 76.
- a Form VIII hydrobromide salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 8.79, about 11.13, about 19.97, about 21.31, about 21.56, about 25.30 and about 26.65 degrees 2-theta.
- a Form VIII hydrobromide salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 8.79, about 11.13, about 19.97, about 21.31, about 21.56, about 25.30 and about 26.65 degrees 2-theta.
- a Form VIII hydrobromide salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 8.79, about 1 1.13, about 19.97, about 21.31, about 21.56, about 25.30 and about 26.65 degrees 2-theta.
- a Form VIII hydrobromide salt is characterized by four or more peaks in its powder X-ray diffraction pattern selected from those at about 8.79, about 1 1.13, about 19.97, about 21.31, about 21.56, about 25.30 and about 26.65 degrees 2-theta. In some embodiments, a Form VIII hydrobromide salt is characterized by five or more peaks in its powder X-ray diffraction pattern selected from those at about 8.79, about 1 1.13, about 19.97, about 21.31, about 21.56, about 25.30 and about 26.65 degrees 2-theta.
- a Form VIII hydrobromide salt is characterized by six or more peaks in its powder X-ray diffraction pattern selected from those at about 8.79, about 11.13, about 19.97, about 21.31, about 21.56, about 25.30 and about 26.65 degrees 2-theta.
- a Form VIII hydrobromide salt is characterized by an X-ray powder diffraction pattern which includes the peaks at about 8.79, about 1 1.13, about 19.97, about 21.31, about 21.56, about 25.30 and about 26.65 degrees 2-theta.
- a Form VIII hydrobromide salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- a Form VIII hydrobromide salt has a thermogravimetric analysis pattern substantially similar to that depicted in Figure 77. In some embodiments, a Form VIII hydrobromide salt has a differential scanning calorimetry pattern substantially similar to that depicted in Figure 78. In some embodiments, a Form VIII hydrobromide salt is characterized by substantial similarity to two or more of these figures simultaneously.
- compound 2 is a hydrochloric acid salt. In some embodiments, compound 2 is a mono-hydrochloric acid salt. In some embodiments, compound 2 is a bis-hydrochloric acid salt.
- a bis-hydrochloride salt has a powder X-ray diffraction pattern substantially similar to that depicted in Figure 28.
- a bis- hydrochloride salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 17.58, about 23.32, about 25.53 and about 28.37 degrees 2-theta.
- a bis-hydrochloride salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 17.58, about 23.32, about 25.53 and about 28.37 degrees 2-theta.
- a bis-hydrochloride salt is characterized by three or more peaks in its powder X-ray diffraction pattern selected from those at about 17.58, about 23.32, about 25.53 and about 28.37 degrees 2-theta.
- a bis- hydrochloride salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about 17.58, 20.13, 22.14, 23.32, 25.53, 26.60, 27.80 and 28.37 degrees 2-theta.
- a bis-hydrochloride salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- a bis-hydrochloride salt has a thermogravimetric analysis pattern substantially similar to that depicted in Figure 29.
- a bis- hydrochloride salt has a differential scanning calorimetry pattern substantially similar to that depicted in Figure 30.
- a bis -hydrochloride salt has an l H- NMR spectrum substantially similar to that depicted in Figure 31.
- compound 2 is a maleic acid salt. In some embodiments, compound 2 is a mono-maleic acid salt. In some embodiments, compound 2 is a bis-maleic acid salt.
- a mono-maleate salt has a powder X-ray diffraction pattern substantially similar to that depicted in Figure 24.
- a mono- maleate salt is characterized by one or more peaks in its powder X-ray diffraction pattern selected from those at about 8.38, about 23.59 and about 23.80 degrees 2-theta.
- a mono-maleate salt is characterized by two or more peaks in its powder X-ray diffraction pattern selected from those at about 8.38, about 23.59 and about 23.80 degrees 2- theta.
- a mono-maleate salt is characterized by three peaks in its powder X-ray diffraction pattern selected from those at about 8.38, about 23.59 and about 23.80 degrees 2-theta.
- a mono-maleate salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about 8.38, 13.74, 16.35, 16.54, 20.67, 23.15, 23.59 and 23.80 degrees 2-theta.
- a mono-maleate salt is characterized by substantially all of the peaks in its X-ray powder diffraction pattern selected from those at about
- a mono-maleate salt has a thermogravimetric analysis pattern substantially similar to that depicted in Figure 25.
- a mono- maleate salt has a differential scanning calorimetry pattern substantially similar to that depicted in Figure 26.
- a mono-maleate salt has an ⁇ - MR spectrum substantially similar to that depicted in Figure 27.
- any of the above-described polymorph forms can be characterized, for example, by reference to any of the peaks in their respective X-ray diffraction patterns. Accordingly, in some embodiments, a polymorph described herein is characterized by one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty or more XRPD peaks (°2 ⁇ ).
- compound 2 is a methanesulfonic acid salt. In some embodiments, compound 2 is a mono-methansulfonic acid salt. In some embodiments, compound 2 is a bis-methanesulfonic acid salt.
- compound 2 is a naphthalene-2 -sulfonic acid salt. In some embodiments, compound 2 is a mono-naphthalene-2-sulfonic acid salt. In some embodiments, compound 2 is a bis-naphthalene-2-sulfonic acid salt.
- compound 2 is a 1,5 -naphthalene disulfonic acid salt. In some embodiments, compound 2 is a mono- 1,5 -naphthalene disulfonic acid salt. In some embodiments, compound 2 is a bis-l,5-naphthalene disulfonic acid salt.
- compound 2 is an oxalic acid salt. In some embodiments, compound 2 is a mono-oxalic acid salt. In some embodiments, compound 2 is a bis-oxalic acid salt.
- compound 2 is a p-toluenesulfonic acid (tosylate) salt. In some embodiments, compound 2 is a mono-p-toluenesulfonic acid salt. In some embodiments, compound 2 is a bis-p-toluenesulfonic acid salt.
- compound 2 is a 2,4,6-trihydroxybenzoic acid salt. In some embodiments, compound 2 is a mono-2,4,6-trihydroxybenzoic acid salt. In some embodiments, compound 2 is a bis-2,4,6-trihydroxybenzoic acid salt.
- the present invention provides compound 2 as an amorphous solid.
- Amorphous solids are well known to one of ordinary skill in the art and are typically prepared by such methods as lyophilization, melting, and precipitation from supercritical fluid, among others.
- Compound 1 is prepared according to the methods described in detail in the '061 application, the entirety of which is hereby incorporated herein by reference.
- Compound 2 is prepared from Compound 1, according to the Scheme below.
- Compound 2 is prepared from Compound 1 by combining Compound 1 with either one or two equivalents of benzenesulfonic acid, camphor sulfonic acid, 1,2-ethane disulfonic acid, hydrobromic acid, hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2-sulfonic acid, 1,5 -naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid or 2,4,6-trihydroxybenzoic acid to form the salt thereof.
- another aspect of the present inv ntion provides a method for preparing Compound 2:
- a suitable solvent may solubilize one or more of the reaction components, or, alternatively, the suitable solvent may facilitate the agitation of a suspension of one or more of the reaction components.
- suitable solvents useful in the present invention are a protic solvent, a polar aprotic solvent, a nonpolar solvent or mixtures thereof.
- suitable solvents include water, an ether, an ester, an alcohol, a halogenated solvent, a ketone, or a mixture thereof.
- the suitable solvent is methanol, ethanol, isopropanol, ethyl acetate, isopropyl acetate, methyl ethyl ketone, methyl isobutyl ketone or acetone.
- the suitable solvent is dichloromethane.
- suitable solvents include tetrahydrofuran, dimethylformamide, dimethylsulfoxide, glyme, diglyme, methyl t-butyl ether, t-butanol, n-butanol, and acetonitrile.
- the suitable solvent is cyclohexane.
- the present invention provides a method for preparing Compound 2:
- Compound 1 is dissolved or suspended in a suitable solvent, optionally with heating. In certain embodiments Compound 1 is dissolved at about 20 to about 60 °C. In other embodiments, Compound 1 is dissolved at about 20 to about 25 °C, such as about ambient temperature. In still other embodiments, compound 1 is dissolved at the boiling temperature of the solvent. In other embodiments, compound 1 is dissolved without heating.
- benzenesulfonic acid camphor sulfonic acid, 1,2-ethane disulfonic acid, hydrobromic acid, hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2-sulfonic acid, 1,5 -naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid or 2,4,6-trihydroxybenzoic acid is added to Compound 1 to afford Compound 2.
- benzenesulfonic acid camphor sulfonic acid, 1,2-ethane disulfonic acid, hydrobromic acid, hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2-sulfonic acid, 1,5 -naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid or 2,4,6-trihydroxybenzoic acid is added to Compound 1 to afford Compound 2.
- benzenesulfonic acid camphor sulfonic acid, 1,2-ethane disulfonic acid, hydrobromic acid, hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2 -sulfonic acid, 1,5 -naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid or 2,4,6-trihydroxybenzoic acid is added to Compound 1 to afford Compound 2.
- benzenesulfonic acid camphor sulfonic acid, 1,2-ethane disulfonic acid, hydrobromic acid, hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2-sulfonic acid, 1,5 -naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid or 2,4,6-trihydroxybenzoic acid is added to Compound 1 to afford Compound 2.
- benzenesulfonic acid camphor sulfonic acid, 1,2-ethane disulfonic acid, hydrobromic acid, hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2 -sulfonic acid, 1,5- naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid or 2,4,6-trihydroxybenzoic acid is added to Compound 1 to afford Compound 2.
- about 1.8 to about 2.2 equivalents such as about 1.98 to 2.02 equivalents, of benzenesulfonic acid, camphor sulfonic acid, 1,2-ethane disulfonic acid, hydrobromic acid, hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2-sulfonic acid, 1,5 -naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid or 2,4,6-trihydroxybenzoic acid is added to Compound 1 to afford Compound 2.
- benzenesulfonic acid camphor sulfonic acid, 1,2-ethane disulfonic acid, hydrobromic acid, hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2-sulfonic acid, 1,5 -naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid or 2,4,6-trihydroxybenzoic
- the acid may be added to the mixture of Compound 1 and a suitable solvent in any suitable form.
- the acid may be added in solid form or as a solution or a suspension in a suitable solvent.
- the suitable solvent may be the same suitable solvent as that which is combined with Compound 1 or may be a different solvent.
- the acid is added in solid form.
- the acid is combined with a suitable solvent prior to adding to Compound 1.
- the acid is added as a solution in a suitable solvent.
- suitable solvents include water, an ether, an ester, an alcohol, a halogenated solvent, a ketone, or a mixture thereof.
- the suitable solvent is methanol, ethanol, isopropanol, ethyl acetate, isopropyl acetate, methyl ethyl ketone, methyl isobutyl ketone or acetone.
- the suitable solvent is dichloromethane.
- suitable solvents include tetrahydrofuran, dimethylformamide, dimethylsulfoxide, glyme, diglyme, methyl t-butyl ether, t-butanol, n-butanol, and acetonitrile.
- the suitable solvent is cyclohexane.
- the suitable solvent is selected from those above and is anhydrous.
- the resulting mixture containing Compound 2 is cooled. In other embodiments, the mixture containing Compound 2 is cooled below 20 °C, such as below 10 °C.
- Compound 2 precipitates from the mixture. In another embodiment, Compound 2 crystallizes from the mixture. In other embodiments, Compound 2 crystallizes from solution following seeding of the solution (i.e., adding crystals of Compound 2 to the solution).
- Crystalline Compound 2 can precipitate out of the reaction mixture, or be generated by removal of part or all of the solvent through methods such as evaporation, distillation, filtration (e.g., nanofiltration, ultrafiltration), reverse osmosis, absorption and reaction, by adding an anti-solvent such as water, MTBE or heptane, by cooling or by different combinations of these methods.
- Compound 2 is optionally isolated. It will be appreciated that Compound 2 may be isolated by any suitable physical means known to one of ordinary skill in the art.
- precipitated solid compound 2 is separated from the supernatant by filtration.
- precipitated solid Compound 2 is separated from the supernatant by decanting the supernatant.
- precipitated solid Compound 2 is separated from the supernatant by filtration.
- isolated Compound 2 is dried in air. In other embodiments isolated Compound 2 is dried under reduced pressure, optionally at elevated temperature.
- the invention provides a composition comprising Compound 2 and a pharmaceutically acceptable carrier, adjuvant, or vehicle.
- the amount of Compound 2 in compositions of this invention it is such that is effective to measurably inhibit a protein kinase, particularly an EGFR kinase, or a mutant thereof, in a biological sample or in a patient.
- a composition of this invention is formulated for administration to a patient in need of such composition.
- a composition of this invention is formulated for oral administration to a patient.
- patient means an animal, preferably a mammal, and most preferably a human.
- compositions of this invention refers to a nontoxic carrier, adjuvant, or vehicle that does not destroy the pharmacological activity of the compound with which it is formulated.
- Pharmaceutically acceptable carriers, adjuvants or vehicles that may be used in the compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, Vitamin E polyethylene glycol succinate (d-alpha tocopheryl polyethylene glycol 1000
- compositions of the present invention may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally or via an implanted reservoir.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, intraarticular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- the compositions are administered orally, intraperitoneally or intravenously.
- Sterile injectable forms of the compositions of this invention may be an aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
- a nontoxic parenterally acceptable diluent or solvent for example as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or di-glycerides.
- Fatty acids such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, such as carboxymethyl cellulose or similar dispersing agents that are commonly used in the formulation of pharmaceutically acceptable dosage forms including emulsions and suspensions.
- Other commonly used surfactants such as Tweens, Spans and other emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
- compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, aqueous and non-aqueous suspensions or solutions.
- carriers commonly used include lactose and corn starch.
- Lubricating agents, such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried cornstarch.
- aqueous suspensions are required for oral use, the active ingredient is typically combined with emulsifying and suspending agents. If desired, certain sweetening, flavoring or coloring agents may also be added.
- compositions of this invention may be administered in the form of suppositories for rectal administration.
- suppositories for rectal administration.
- suppositories can be prepared by mixing the agent with a suitable non-irritating excipient that is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug.
- suitable non-irritating excipient include cocoa butter, beeswax and polyethylene glycols.
- compositions of this invention may also be administered topically, especially when the target of treatment includes areas or organs readily accessible by topical application, including diseases of the eye, the skin, or the lower intestinal tract. Suitable topical formulations are readily prepared for each of these areas or organs.
- Topical application for the lower intestinal tract can be effected in a rectal suppository formulation (see above) or in a suitable enema formulation. Topically-transdermal patches may also be used.
- compositions may be formulated in a suitable ointment containing the active component suspended or dissolved in one or more carriers.
- Carriers for topical administration of Compound 2 include, but are not limited to, mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene, polyoxypropylene compound, emulsifying wax and water.
- provided pharmaceutically acceptable compositions can be formulated in a suitable lotion or cream containing the active components suspended or dissolved in one or more pharmaceutically acceptable carriers.
- Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- compositions may be formulated as micronized suspensions in isotonic, pH adjusted sterile saline, or, preferably, as solutions in isotonic, pH adjusted sterile saline, either with or without a preservative such as benzylalkonium chloride.
- the pharmaceutically acceptable compositions may be formulated in an ointment such as petrolatum.
- compositions of this invention may also be administered by nasal aerosol or inhalation. Such compositions are prepared according to techniques well- known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other conventional solubilizing or dispersing agents. [00162] In some embodiments, pharmaceutically acceptable compositions of this invention are formulated for oral administration.
- compositions are formulated so that a dosage of between 0.01 - 100 mg kg body weight/day of Compound 2 can be administered to a patient receiving these compositions.
- a specific dosage and treatment regimen for any particular patient will depend upon a variety of factors, including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, rate of excretion, drug combination, and the judgment of the treating physician and the severity of the particular disease being treated.
- Compound 2 and compositions described herein are generally useful for the inhibition of protein kinase activity of one or more enzymes.
- Examples of kinases that are inhibited by Compound 2 and compositions described herein and against which the methods described herein are useful include EGFR kinase or a mutant thereof. It has been found that Compound 2 is a selective inhibitor of at least one mutation of EGFR, as compared to wild-type ("WT") EGFR.
- WT wild-type
- an at least one mutation of EGFR is T790M.
- the at least one mutation of EGFR is a deletion mutation.
- the at least one mutation of EGFR is an activating mutation.
- Compound 2 selectively inhibits at least one resistant mutation and at least one activating mutation as compared to WT EGFR. In some embodiments, Compound 2 selectively inhibits at least one deletion mutation and/or at least one point mutation, and is sparing as to WT EGFR inhibition.
- a mutation of EGFR can be selected from T790M (resistant or oncogenic), L858R (activating), delE746-A750 (activating), G719S (activating), or a combination thereof.
- the term “selectively inhibits,” as used in comparison to inhibition of WT EGFR, means that Compound 2 inhibits at least one mutation of EGFR (i.e., at least one deletion mutation, at least one activating mutation, at least one restistant mutation, or a combination of at least one deletion mutation and at least one point mutation) in at least one assay described herein (e.g., biochemical or cellular).
- the term "selectively inhibits,” as used in comparison to WT EGFR inhibition means that Compound 2 is at least 50 times more potent, at least 45 times, at least 40, at least 35, at least 30, at least 25, or at least 20 times more potent as an inhibitor of at least one mutation of EGFR, as defined and described herein, as compared to WT EGFR.
- the term "sparing as to WT EGFR” means that a selective inhibitor of at least one mutation of EGFR, as defined and described above and herein, inhibits EGFR at the upper limit of detection of at least one assay, such as those described in the '061 application (e.g., biochemical or cellular as described in detail in Examples 56-58).
- in vitro assays include assays that determine inhibition of the phosphorylation activity and/or the subsequent functional consequences, or ATPase activity of activated EGFR (WT or mutant). Alternate in vitro assays quantitate the ability of the inhibitor to bind to EGFR (WT or mutant).
- Inhibitor binding may be measured by radiolabeling the inhibitor prior to binding, isolating the inhibitor/EGFR (WT or mutant) complex and determining the amount of radiolabel bound. Alternatively, inhibitor binding may be determined by running a competition experiment where new inhibitors are incubated with EGFR (WT or mutant) bound to known radioligands.
- the term "sparing as to WT EGFR" means that Compound 2 inhibits WT EGFR with an IC 50 of at least 10 ⁇ , at least 9 ⁇ , at least 8 ⁇ , at least 7 ⁇ , at least 6 ⁇ , at least 5 ⁇ , at least 3 ⁇ , at least 2 ⁇ , or at least 1 ⁇ .
- Compound 2 selectively inhibits (a) at least one activating mutation; and (b) T790M; and (c) is sparing as to WT.
- an at least one activating mutation is a deletion mutation.
- an at least one activating mutation is a point mutation.
- an activating mutation is delE746-A750.
- an activating mutation is L858R.
- an activating mutation is G719S.
- the at least one mutation of EGFR is L858R and/or T790M.
- the present invention provides a method for inhibiting an activating mutation in a patient comprising administering to the patient Compound 2 or composition thereof, as described herein.
- the present invention provides a method for inhibiting oncogenic T790M in a patient comprising administering to the patient a provided compound or composition thereof, as described herein.
- the amount of Compound 2 in a composition is effective to measurably inhibit at least one mutant of EGFR selectively as compared to WT EGFR and other protein kinases (e.g., ErbB2, ErbB4, a TEC-kinase, and/or JAK3), in a biological sample or in a patient.
- WT EGFR protein kinases
- other protein kinases e.g., ErbB2, ErbB4, a TEC-kinase, and/or JAK3
- treatment refers to reversing, alleviating, delaying the onset of, or inhibiting the progress of a disease or disorder, or one or more symptoms thereof, as described herein.
- treatment may be administered after one or more symptoms have developed.
- treatment may be administered in the absence of symptoms.
- treatment may be administered to a susceptible individual prior to the onset of symptoms (e.g., in light of a history of symptoms and/or in light of genetic or other susceptibility factors). Treatment may also be continued after symptoms have resolved, for example to prevent or delay their recurrence.
- Compound 2 is an inhibitor of at least one mutant of EGFR and is therefore useful for treating one or more disorders associated with activity of one of more EGFR mutants (e.g., a deletion mutation, an activating mutation, a resistant mutation, or combination thereof).
- the present invention provides a method for treating a mutant EGFR- mediated disorder comprising the step of administering to a patient in need thereof Compound 2 or pharmaceutically acceptable composition thereof.
- mutant EGFR-mediated disorders or conditions means any disease or other deleterious condition in which at least one mutant of EGFR is known to play a role.
- an at least one mutant of EGFR is T790M.
- the at least one mutant of EGFR is a deletion mutation.
- the at least one mutant of EGFR is an activating mutation.
- the at least one mutant of EGFR is L858R and/or T790M.
- a provided compound selectively inhibits (a) at least one activating mutation, (b) T790M, and (c) is sparing as to WT.
- an at least one activating mutation is a deletion mutation. In some embodiments, an at least one activating mutation is a point mutation. In some embodiments, an activating mutation is delE746-A750. In some embodiments, an activating mutation is L858R. In some embodiments, an activating mutation is G719S.
- another embodiment of the present invention relates to treating or lessening the severity of one or more diseases in which at least one mutant of EGFR is known to play a role.
- the present invention relates to a method of treating or lessening the severity of a disease or condition selected from a proliferative disorder, wherein said method comprises administering to a patient in need thereof a compound or composition according to the present invention.
- the present invention provides a method for treating or lessening the severity of one or more disorders selected from a cancer.
- the cancer is associated with a solid tumor.
- the cancer is breast cancer, glioblastoma, lung cancer, cancer of the head and neck, colorectal cancer, bladder cancer, or non-small cell lung cancer.
- the present invention provides a method for treating or lessening the severity of one or more disorders selected from squamous cell carcinoma, salivary gland carcinoma, ovarian carcinoma, or pancreatic cancer.
- the present invention provides a method for treating or lessening the severity of neurofibromatosis type I (NF1), neurofibromatosis type II (NF2) Schwann cell neoplasms (e.g. MPNST's), or Schwannomas.
- NF1 neurofibromatosis type I
- NF2 neurofibromatosis type II
- MPNST's Schwann cell neoplasms
- Schwannomas e.g. MPNST's
- Compound 2 and compositions thereof, according to the method of the present invention may be administered using any amount and any route of administration effective for treating or lessening the severity of a cancer.
- the exact amount required will vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the infection, the particular agent, its mode of administration, and the like.
- Compound 2 is preferably formulated in dosage unit form for ease of administration and uniformity of dosage.
- dosage unit form refers to a physically discrete unit of agent appropriate for the patient to be treated. It will be understood, however, that the total daily usage of the compounds and compositions of the present invention will be decided by the attending physician within the scope of sound medical judgment.
- the specific effective dose level for any particular patient or organism will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed, and like factors well known in the medical arts.
- patient means an animal, preferably a mammal, and most preferably a human.
- compositions of this invention can be administered to humans and other animals orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by powders, ointments, or drops), bucally, as an oral or nasal spray, or the like, depending on the severity of the infection being treated.
- Compound 2 may be administered orally or parenterally at dosage levels of about 0.01 mg/kg to about 60 mg/kg, or about 0.1 mg/kg to about 50 mg/kg, or about 0.25 mg/kg to about 45 mg/kg and preferably from about 0.5 mg/kg to about 25 mg/kg, of subject body weight per day, one or more times a day, to obtain the desired therapeutic effect.
- Liquid dosage forms for oral administration include, but are not limited to, pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art such as, for example, water or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, polyethylene glycol (e.g., PEG 200, PEG 400, PEG 1000, PEG 2000), propylene glycol, 1,3-butylene glycol, dimethylformamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), glycerol, tetrahydrofurfuryl alcohol, Vitamin E polyethylene glycol succinate (d-alpha tocopheryl
- the oral compositions can also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
- adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
- the liquid forms above can also be filled into a soft or hard capsule to form a solid dosage form.
- Suitable capsules can be formed from, for example, gelatin, strach and cellulose derivatives (e.g., hydroxycellulose, hydropropylmethylcellulose).
- Injectable preparations for example, sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution, suspension or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- the acceptable vehicles and solvents that may be employed are water, Ringer's solution, U.S. P. and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil can be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid are used in the preparation of injectables.
- Injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
- the rate of compound release can be controlled.
- biodegradable polymers include poly(orthoesters) and poly(anhydrides).
- Depot injectable formulations are also prepared by entrapping the compound in liposomes or microemulsions that are compatible with body tissues.
- compositions for rectal or vaginal administration are preferably suppositories which can be prepared by mixing Compound 2 of this invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules.
- Compound 2 is mixed with at least one inert, pharmaceutically acceptable excipient or carrier such as sodium citrate, Avicel, hydroxypropyl cellulose or dicalcium phosphate and/or a) fillers or extenders such as starches, lactose, sucrose, glucose, mannitol, and silicic acid, b) binders such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidinone, sucrose, PVP vinyl acetate, and acacia, c) humectants such as glycerol, d) disintegrating agents such as agar— agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, sodium croscarmellose and sodium carbonate, e) solution retarding agents such as paraffin, f) absorption accelerators such as sodium citrate, Avicel
- Solid compositions of a similar type may also be employed as fillers in soft and hard- filled capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings and other coatings well known in the pharmaceutical formulating art. They may optionally contain opacifying agents and can also be of a composition that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner. Examples of embedding compositions that can be used include polymeric substances and waxes. Solid compositions of a similar type may also be employed as fillers in soft and hard-filled capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- Compound 2 can also be in micro-encapsulated form with one or more excipients as noted above.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as cosmetic coatings, enteric coatings, release controlling coatings and other coatings well known in the pharmaceutical formulating art.
- the active compound may be admixed with at least one inert diluent such as a polymer, sucrose, lactose or starch.
- Such dosage forms may also comprise, as is normal practice, additional substances other than inert diluents, e.g., tableting lubricants and other tableting aids such a magnesium stearate and microcrystalline cellulose.
- the dosage forms may also comprise buffering agents. They may optionally contain opacifying agents and can also be of a composition that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- buffering agents include polymeric substances and waxes.
- Dosage forms for topical or transdermal administration of Compound 2 include ointments, pastes, creams, lotions, gels, powders, solutions, sprays, inhalants or patches.
- the active component is admixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives or buffers as may be required.
- Ophthalmic formulation, ear drops, and eye drops are also contemplated as being within the scope of this invention.
- the present invention contemplates the use of transdermal patches, which have the added advantage of providing controlled delivery of a compound to the body.
- Such dosage forms can be made by dissolving or dispensing the compound in the proper medium.
- Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate can be controlled by either providing a rate controlling membrane or by dispersing the compound in a polymer matrix or gel.
- the invention relates to a method of inhibiting protein kinase activity in a biological sample comprising the step of contacting said biological sample with Compound 2 or a composition comprising said compound.
- the invention relates to a method of inhibiting at least one mutant of EGFR (e.g., a deletion mutation, an activating mutation, a resistant mutations, or combination thereof) activity in a biological sample comprising the step of contacting said biological sample with Compound 2, or a composition comprising the compound.
- the invention relates to a method of irreversibly inhibiting at least one mutant of EGFR (e.g., a deletion mutation, an activating mutation, a resistant mutation, or combination thereof) activity in a biological sample comprising the step of contacting the biological sample with Compound 2, or a composition comprising the compound.
- Compound 2 selectively inhibits in a biological sample (a) at least one activating mutation, (b) T790M, and (c) is sparing as to WT.
- an at least one activating mutation is a deletion mutation.
- an at least one activating mutation is a point mutation.
- an activating mutation is delE746-A750.
- an activating mutation is L858R.
- an activating mutation is G719S.
- biological sample includes, without limitation, cell cultures or extracts thereof; biopsied material obtained from a mammal or extracts thereof; and blood, saliva, urine, feces, semen, tears, or other body fluids or extracts thereof.
- Inhibition of at least one mutant of EGFR (e.g., a deletion mutation, an activating mutation, a resistant mutation, or combination thereof) activity in a biological sample is useful for a variety of purposes that are known to one of skill in the art. Examples of such purposes include, but are not limited to, blood transfusion, organ transplantation, biological specimen storage, and biological assays.
- Another embodiment of the present invention relates to a method of inhibiting at least one mutant of EGFR (e.g., a deletion mutation, an activating mutation, a resistant mutation, or combination thereof) activity in a patient comprising the step of administering to the patient Compound 2 or a composition comprising the compound.
- the present invention provides a method for inhibiting (a) at least one activating mutation, and (b) T790M in a patient, and (c) is sparing as to WT, wherein the method comprises administering to the patient Compound 2 or composition thereof.
- an at least one activating mutation is a deletion mutation.
- an at least one activating mutation is a point mutation.
- the present invention provides a method for inhibiting at least one mutant of EGFR in a patient, wherein an activating mutation is delE746-A750. In some embodiments, the present invention provides a method for inhibiting at least one mutant of EGFR in a patient, wherein an activating mutation is L858R. In some embodiments, the present invention provides a method for inhibiting at least one mutant of EGFR in a patient, wherein an activating mutation is G719S.
- the invention relates to a method of inhibiting at least one mutant of EGFR (e.g., a deletion mutation, an activating mutation, a resistant mutation, or combination thereof) activity in a patient comprising the step of administering to the patient Compound 2 or a composition comprising the compound.
- the invention relates to a method of irreversibly inhibiting at least one mutant of EGFR activity (e.g., a deletion mutation, an activating mutation, a resistant mutation, or combination thereof) in a patient comprising the step of administering to said patient Compound 2 or a composition comprising the compound.
- the present invention provides a method for irreversibly inhibiting (a) at least one activating mutation, and (b) T790M in a patient, and (c) is sparing as to WT, wherein said method comprises administering to the patient Compound 2 or composition thereof.
- an irreversibly inhibited at least one activating mutation is a deletion mutation.
- an irreversibly inhibited at least one activating mutation is a point mutation.
- the present invention provides a method for irreversibly inhibiting at least one mutant of EGFR in a patient, wherein an activating mutation is delE746-A750.
- the present invention provides a method for irreversibly inhibiting at least one mutant of EGFR in a patient, wherein an activating mutation is L858R. In some embodiments, the present invention provides a method for irreversibly inhibiting at least one mutant of EGFR in a patient, wherein an activating mutation is G719S.
- the present invention provides a method for treating a disorder mediated by one or more of at least one mutant of EGFR (e.g., a deletion mutation, an activating mutation, a resistant mutation, or combination thereof) in a patient in need thereof, comprising the step of administering to said patient Compound 2 or pharmaceutically acceptable composition thereof.
- a disorder mediated by one or more of at least one mutant of EGFR e.g., a deletion mutation, an activating mutation, a resistant mutation, or combination thereof
- Such disorders are described in detail herein.
- additional therapeutic agents which are normally administered to treat that condition, may also be present in the compositions of this invention or as part of a treatment regimen including Compound 2.
- additional therapeutic agents that are normally administered to treat a particular disease, or condition are known as "appropriate for the disease or condition being treated.”
- Compound 2 or a pharmaceutically acceptable composition thereof is administered in combination with chemotherapeutic agents to treat proliferative diseases and cancer.
- chemotherapeutic agents include, but are not limited to, Adriamycin, dexamethasone, vincristine, cyclophosphamide, fluorouracil, topotecan, taxol, interferons, platinum derivatives, taxane (e.g., paclitaxel), vinca alkaloids (e.g., vinblastine), anthracyclines (e.g., doxorubicin), epipodophyllotoxins (e.g., etoposide), cisplatin, an mTOR inhibitor (e.g., a rapamycin), methotrexate, actinomycin D, dolastatin 10, colchicine, emetine, trimetrexate, metoprine, cyclosporine, daunorubicin, teniposide, amphoterici
- mTOR inhibitor
- Compound 2 or a pharmaceutically acceptable composition thereof is administered in combination with an antiproliferative or chemotherapeutic agent selected from any one or more of abarelix, aldesleukin, alemtuzumab, alitretinoin, allopurinol, altretamine, amifostine, anastrozole, arsenic trioxide, asparaginase, azacitidine, BCG Live, bevacuzimab, fluorouracil, bexarotene, bleomycin, bortezomib, busulfan, calusterone, capecitabine, camptothecin, carboplatin, carmustine, celecoxib, cetuximab, chlorambucil, cladribine, clofarabine, cyclophosphamide, cytarabine, dactinomycin, darbepoetin alfa, daunorubicin, denileukin,
- agents the inhibitors of this invention may also be combined with include, without limitation: treatments for Alzheimer's Disease such as donepezil hydrochloride (Aricept ® ) and rivastigmine (Exelon ® ); treatments for Parkinson's Disease such as L-DOPA/carbidopa, entacapone, ropinrole, pramipexole, bromocriptine, pergolide, trihexephendyl, and amantadine; agents for treating Multiple Sclerosis (MS) such as beta interferon (e.g., Avonex ® and Rebif ® ), glatiramer acetate (Copaxone ® ), and mitoxantrone; treatments for asthma such as albuterol and montelukast (Singulair ® ); agents for treating schizophrenia such as zyprexa, risperdal, seroquel, and haloperidol; anti-inflammatory agents such as corticosteroids, TNF block
- Compound 2 or a pharmaceutically acceptable composition thereof is administered in combination with a monoclonal antibody or an siRNA therapeutic.
- the additional agents may be administered separately from a Compound 2- containing composition, as part of a multiple dosage regimen. Alternatively, those agents may be part of a single dosage form, mixed together with Compound 2 in a single composition. If administered as part of a multiple dosage regime, the two active agents may be submitted simultaneously, sequentially or within a period of time from one another (e.g., one hour, two hours, six hours, twelve hours, one day, one week, two weeks, one month).
- the terms “combination,” “combined,” and related terms refer to the simultaneous or sequential administration of therapeutic agents in accordance with this invention.
- Compound 2 may be administered with another therapeutic agent simultaneously or sequentially in separate unit dosage forms or together in a single unit dosage form.
- the present invention provides a single unit dosage form comprising Compound 2, an additional therapeutic agent, and a pharmaceutically acceptable carrier, adjuvant, or vehicle.
- compositions of this invention should be formulated so that a dosage of between 0.01 - 100 mg/kg body weight/day of Compound 2 can be administered.
- compositions that include an additional therapeutic agent that additional therapeutic agent and Compound 2 may act synergistically. Therefore, the amount of additional therapeutic agent in such compositions may be less than that required in a monotherapy utilizing only that therapeutic agent. In such compositions, a dosage of between 0.01 - 1,000 ⁇ g/kg body weight/day of the additional therapeutic agent can be administered.
- the amount of additional therapeutic agent present in the compositions of this invention will be no more than the amount that would normally be administered in a composition comprising that therapeutic agent as the only active agent.
- the amount of additional therapeutic agent in the presently disclosed compositions will range from about 50% to 100% of the amount normally present in a composition comprising that agent as the only therapeutically active agent.
- Compound 2 or pharmaceutical compositions thereof may also be incorporated into compositions for coating an implantable medical device, such as prostheses, artificial valves, vascular grafts, stents and catheters.
- an implantable medical device such as prostheses, artificial valves, vascular grafts, stents and catheters.
- Vascular stents for example, have been used to overcome restenosis (re-narrowing of the vessel wall after injury).
- patients using stents or other implantable devices risk clot formation or platelet activation. These unwanted effects may be prevented or mitigated by pre-coating the device with a pharmaceutically acceptable composition comprising a kinase inhibitor.
- Implantable devices coated with Compound 2 are another embodiment of the present invention.
- Step 1
- DIPEA Diisopropylethylamine
- Step 3
- the counterions included benzenesulfonic acid, camphor sulfonic acid, 1,2-ethane disulfonic acid, hydrobromic acid, hydrochloric acid, maleic acid, methanesulfonic acid, naphthalene-2- sulfonic acid, 1,5 -naphthalene disulfonic acid, oxalic acid, 4-toluenesulfonic acid and 2,4,6- trihydroxybenzoic acid.
- One equivalent of each counterion was used and additional experiments with two equivalents of benzenesulfonic acid, hydrochloric acid, sulphuric acid and p-toluenesulfonic acid were performed.
- the acid solution / slurry was then added to the slurry of Compound 1 in small aliquots in order to minimize the risk of degradation.
- the pH of the reaction was then checked using universal indicator paper.
- X-Ray Powder Diffraction X-ray powder diffraction (XRPD) analysis was carried out on a Siemens D5000, scanning the samples between 3 and 30, 35 or 50 °2 ⁇ . For samples ⁇ 100 mg, ca. 5-10 mg of sample was gently compressed onto a glass slide which fitted into the sample holder. For samples >100 mg, ca. 100 mg of sample was gently compressed into a plastic sample holder, so that the sample surface was smooth and just above the level of the sample holder, Measurements were made using the following experimental conditions :
- Polarized Light Microscopy In polarized light microscopy (PLM), the presence of crystallinity (birefringence) was determined using an Olympus BX50 polarising microscope, equipped with a Motic camera and image capture software (Motic Images Plus 2.0). All images were recorded using the 20x objective, unless otherwise stated.
- PLM polarized light microscopy
- thermogravimetric analysis TGA
- TG/DTA simultaneous thermogravimetric/differential thermal analyser
- DTA differential thermal events
- the sample pan was then loaded into a Seiko DSC6200 (equipped with a cooler) cooled and held at 25°C. Once a stable heat-flow response was obtained, the sample and reference were heated to ca. 260 °C-280 °C at scan rate of 10°C/min and the resulting heat flow response monitored.
- a Sotax AT7 dissolution bath (USP 2, EP 2 apparatus) was used for the dissolution study in which paddles were used to stir the media. All tests were carried out at 37°C and a paddle speed of 100 rpm.
- the bis-besylate salt was selected to be scaled up, using acetone as the solvent.
- the hydrobromide salt was selected to be scaled up, using acetonitrile:water (90: 10) as the solvent.
- the mono-maleate and bis-hydrochloride salts were also selected for scale-up experiments to assess whether these are solvated/hydrated.
- TGA/DTA was carried after 3 days of drying at ambient under vacuum as well as after further drying for 2 days at 40 °C under vacuum and 2 days at 60 °C under vacuum. After the ambient drying process, the TGA showed a 6.7% weight loss between ca. 50-150 °C ( Figure 2) (for an acetone solvate, 1 mole equivalent of acetone would be ca. 6.3 wt%). After further drying, the TGA showed a 0.47% weight loss from the outset, likely due to unbound moisture or solvent. A further small 0.16% weight loss corresponded with the endotherm at onset ca. 142 °C ( Figure 3).
- DSC analysis ( Figure 4) indicated a broad endotherm from the outset likely due to unbound solvent. A second endotherm was present at onset ca. 139.4 °C (peak 146.1°C).
- DVS analysis ( Figure 7) showed a water uptake of ca. 2.2% between 20 and 70% RH.
- the material also appears to hydrate during DVS analysis as indicated by the change in polymorphic form seen by post DVS XRPD analysis (not shown).
- the XRPD diffractogram also showed some loss in crystallinity.
- Karl Fischer Coulometry indicated a ca. 0.77% water content (Note: due to the manual introduction of the solid material into the titration cell, measured values below 1% are generally slightly higher than the actual water content).
- HPLC purity evaluation indicated a purity of ca. 97.6% for the bis- besylate salt with the main peak eluting at a retention time of ca. 13.05 minutes.
- the bis-besylate salt was exposed to environments of 40°C/75%RH (relative humidity, open and closed vial) and 80°C (open vial) for 1 week to determine stability. Resulting solids were analysed by XRPD and HPLC to establish if any changes had occurred.
- the 1 Week stability study results from XRPD ( Figure 10) and HPLC analysis (not shown) at 40°C/ 75% RH using an open and closed vial and 80°C using an open vial are indicated Table 5. Table 5.
- TGA/DTA indicated a weight loss of ca. 2.1% between ca. 70 - 100 °C ( Figure 15). This corresponds approximately with the 2.03 wt% water required for a monohydrate. A ca. 2.2% weight loss was present from the outset to ca. 70 °C, likely due to unbound water. Although the total ca. 4.2% weight loss corresponds approximately with a dihydrate, the first weight loss occurs from the outset followed by a second clear weight loss corresponding with mono amounts of water. As the first weight loss occurs from ca. 25 °C, this would likely be due to unbound water.
- DSC analysis indicated a broad endotherm between ca. 40-115 °C. Two further endotherms were then present at onset 119.7 °C (peak 134.3 °C) and onset 153.8 °C (peak 165.1 °C) ( Figure 16).
- PLM analysis showed some birefringence, however the particle size is very small and no clear morphology could be seen (not shown). Hot-stage microscopy was carried out on a sample of the bis-besylate hydrate. No visual changes could be observed prior to the material melting and degrading (turned brown) at ca. 160 °C.
- HPLC purity determinations indicated an initial purity of ca. 98.4% and a purity of ca. 98.3% after 1 week storage at 40 °C/75% RH.
- a larger batch of the bis-besylate hydrate salt was prepared using the following procedure. Approximately 20 mL of acetone was added to ca. 14 g of Compound 1 in a round bottomed flask to form a slurry. In a separate flask, ca. 10 mL of acetone was added to 2 equivalents of benzenesulfonic acid in order to dissolve the acid. The acid solution was then added in small aliquots to the freebase slurry whilst stirring at ca. 0 °C. The resulting slurry was then allowed to stir at ambient for ca. 2 hours. It was then placed at ca. 5 °C for 2 days before stirring for a further 3 hours at ambient temperature.
- the acetone was then removed and ca. 20 mL of water was added to the material.
- the slurry was temperature cycled (0 °C - ambient temperature (ca. 22 °C)) in 2 hour cycles for ca. 1 day.
- the solid was then isolated by filtration and allowed to dry at ambient conditions under vacuum before analysis. The drying was continued for ca. 10 days.
- TGA/DTA was carried after 2 days of drying at ambient under vacuum.
- the TGA showed a 0.4% weight loss from the outset, likely due to unbound moisture or solvent.
- a large 10.9% weight loss is associated with endothermic/exothermic events in the DTA between ca. 145-185 °C, followed by further weight losses due to likely degradation (Figure
- TGA/DTA was carried after 2 days of drying at ambient under vacuum.
- the TGA showed a 2.7% gradual weight loss from the outset to ca. 180 °C.
- a further 4.3% weight loss is seen between ca. 180-210 °C, which corresponds with an endotherm in the DTA trace (Figure 29).
- DSC analysis indicated a broad endotherm between ca. 30-160 °C. A further endotherm is then present at onset 206.4°C (peak 226.5°C), directly followed by a smaller endotherm at peak 238.2°C ( Figure 30).
- Karl Fischer analysis showed ca. 3.3% water content (ca. 2.8% water required for a monohydrate).
- TGA/DTA showed a 1.01% weight loss from the outset, likely due to unbound moisture or solvent. No further weight losses were seen prior to degradation at onset ca. 230°C ( Figure 33).
- Karl Fischer Coulometry indicated a ca. 1.65% water content.
- thermodynamic solubility experiments carried out on the bis-besylate salt.
- the diffractogram appears different from the input hydrobromide material, all known forms of the
- thermodynamic solubility experiments carried out on the bis-besylate salt.
- TGA/DTA (Figure 45) showed a 1.2% weight loss from the outset to ca. 100°C, likely due to unbound moisture or solvent. No further weight losses were seen prior to degradation at onset ca. 230°C.
- the TGA/DTA is similar to the trace obtained for the 1 equivalent scaled-up form of Example 6.
- thermodynamic solubility experiments carried out on the hydrobromide salt resulted in the formation of an unknown solid form.
- the following experiments were carried out. Initially, approximately 100 mg of the hydrobromide (1 equiv.) material was slurried in a pH 6.2 aqueous solution at ambient and XRPD analysis was carried out at time points 5 min., 1 hr, 2 hrs, 4 hrs and 8 hrs. Further analysis was then also carried out on the converted material.
- the reaction was then filtered and the solid dried under vacuum at ambient temperature (ca. 22°C). The drying was continued for ca. 2 days. Due to the partially crystalline nature of the material after drying, the material was then slurried in ca. 50 mL of an acetone:water (90: 10) mixture. The reaction was temperature cycled between ca. 4-22 °C in 1 hour cycles while stirring for ca. 2 days. The reaction was then filtered and dried at ambient for ca. 4 days before being analyzed. The yield after the further slurrying was 16.4 g (63%).
- PLM (not shown) showed small particles with no defined morphology and little birefringence.
- TGA/DTA (Figure 56) showed a weight loss from the outset of ca. 0.4%, likely due to unbound moisture or solvent. No further significant weight losses were seen prior to degradation at onset ca. 230 °C.
- KF analysis determined the water content of the material to be ca. 0.76%.
- HPLC purity determination indicated a purity of ca. 98.1%.
- the content of carbon, hydrogen and nitrogen in the material was determined by placing the samples into a tin capsule, placed inside an autosampler drum of an elemental analysis system.
- the sample environment was purged by a continuous flow of helium and the samples dropped at pre-set intervals into a vertical quartz tube maintained at 900 °C.
- the mixture of combustion gases was separated and detected by a thermal conductivity detector giving a signal proportional to the concentration of the individual components of the mixture.
- the content of bromine in the material was determined by oxygen flask combustion of the sample. Once the combustion and absorption into solution had occurred, the samples were titrated using a calibrated Mercuric Nitrate solution. Elemental analysis (CHN and bromide) indicated the following percentages:
- Ion chromatography was carried out using a Metrohm 761 Compact Ion Chromatograph for the analysis of ions in aqueous solutions. Calibration standards were prepared from certified 1000 ppm stock solutions. Ion chromatography showed the presence of 12.38% bromide.
- the reactor temperature was raised to 20 °C for 2 hours.
- the reaction was then again cooled to ca. 4 °C and maintained at this temperature for a further 3 hours.
- the reaction mixture was then filtered and dried under vacuum at ambient temperature (ca. 22 °C) for 3 days. The solid was stirred periodically during the drying process.
- the yield after drying was 258.1 g (71%).
- PLM analysis showed a needle-like, fibrous morphology when wet (not shown). Upon drying and hence polymorph conversion, the needle-like morphology was lost with small particles resulting.
- TGA/DTA (Figure 63) showed a weight loss from the outset of ca. 0.4%, likely due to unbound moisture or solvent. No further significant weight losses were seen prior to degradation at onset ca. 230 °C. Thus, the material appears to retain ca. 0.5% water at ambient conditions despite extended periods of drying and therefore appears to be slightly hygroscopic.
- KF analysis determined the water content of the material to be ca. 0.74%.
- HPLC purity determination indicated a purity of ca. 99.1%.
- Crash Cooling Experiments. Crash cooling experiments were performed by placing saturated solutions of the material, in each of the 24 selected solvent systems, in environments of 2 °C and -18 °C for a minimum of 48 hours. Any solid material was then recovered for analysis.
- Rapid Evaporation Experiments. Rapid evaporation experiments were conducted by evaporating the solvents from saturated, filtered solutions of the material, in each of the 24 solvent systems, under vacuum. Any solid material was then recovered and analysed after the solvent had evaporated to dryness.
- Anti-solvent Addition Experiments. Anti-solvent addition experiments were conducted at ambient temperature by adding the selected anti-solvent to saturated, filtered solutions of the material, in each of the 24 selected solvent systems. The anti-solvent selected was heptane, with tert-butylmethyl ether and water being used for solvents immiscible with heptane. Addition of anti-solvent was continued until there was no further precipitation or until no more anti-solvent could be added. Any solid material was recovered and analysed quickly in order to prevent form changes.
- Figure 81 shows input material Form I compared with a wet sample, and after stages of drying.
- Form III an anhydrous form, was obtained from rapid evaporation of DMSO, crash cooling to 2 °C in ethanol, and anti-solvent addition from acetone, acetonitrile, and ethanol.
- Form III Hydrobromide Salt Preparation. Approximately 500 mg of amorphous Compound 2 HBr salt material was slurried in ca. 6 mL of acetonitrile. The suspension was then temperature cycled between 4 and 25 °C in four hour cycles for ca. 2 days. The secondary screen analysis was carried out on the material when it was damp, due to the instability of Form III.
- Form III From the characterisation carried out on Form III, this form was determined to be a metastable, likely anhydrous form of the hydrobromide salt. Form III was observed to be very unstable with conversion to Form I occurring upon isolation and drying of the material.
- Form I was found to be the thermodynamically most stable form in acetone, isopropanol and isopropyl acetate at both ambient and 60 °C. In acetone: water (80:20), conversion to an unidentified form resulted (labelled as Form VIII).
- Characterization of Form VIII An initial assessment of Form VIII, obtained from competitive slurry experiments of Forms I and III in acetone:water (80:20), was made in order to determine the nature of the form and evaluate whether it is consistent with freebase material or the HBr salt. The material resulting from the competitive slurry experiments appeared light yellow in colour. PLM analysis indicated birefringent material with no clearly defined morphology.
- the TGA/DTA indicated a weight loss of 5.2% from the outset followed by a second weight loss of 1.2%, with endotherms in the DTA trace at ca. 40 °C and ca. 96 °C.
- a final endotherm was observed in the DTA trace at onset ca. 184 °C (peak ca. 194 °C).
- Very little change was observed by hotstage microscopy prior to the melt at ca. 197 °C.
- DSC analysis indicated a broad endotherm starting from the outset (peak ca. 93 °C ) followed by a second endotherm at peak ca. 140 °C and a third endotherm at onset ca. 178 °C (peak ca. 193 °C).
- Ion chromatography indicated a bromide content of 12.8% (approximately 1 equivalent).
- Compound 1 free base and compound 2, as the Form I monohydrobromide (HBr) salt were evaluated in a cross-over dog PK study.
- Compound 1 free base capsule consisted of compound 1 free base in Vitamin E TPGS and PEG 400 filled into a capsule.
- the Form I hydrobromide salt capsule consisted of Form I HBr alone filled into a capsule.
- Compound 1 free base capsule and Form I HBr capsule were dosed orally at 28.5 and 24.5 mg/kg (as active) QD, respectively, to three fasted male non-nai ' ve beagle dogs (body weight range: 10.1 - 10.8 kg) with a 5-day washout period. Approximately 5 mL of tap water was orally administered to encourage swallowing and ensure delivery of capsules into the stomach. Plasma samples were collected at pre-dose and 0.5, 1, 2, 4, 6, 8, 12 and 24 hours post dose. The plasma concentrations of compound 1 were determined by a liquid chromatography-tandem mass spectrometry (LC/MS/MS) method. The results are provided in Table 20.
- LC/MS/MS liquid chromatography-tandem mass spectrometry
- compound 1 When compound 1 is administered orally to fasted dogs at 24.5 - 28.5 mg/kg QD, compound 1 exposure (based on AUC and C max ) is significantly higher when drug is administered as the Form I HBr salt compared to the free base form.
- Dog #3 had emesis at 30 min post dose. At the 1 h time point, a partial capsule was found under its cage. Thus, data from dog #3 was not included in the calculation of PK parameters.
- Compound 1 free base and compound 2, as the Form I monohydrobromide (HBr) salt were evaluated in a cross-over dog PK study in which male dogs were pre-treated with either pentagastrin (to decrease gastric pH) or famotidine (to increase gastric pH) prior to oral dosing to control gastric pH.
- pentagastrin to decrease gastric pH
- famotidine to increase gastric pH
- the effect of food on the systemic exposure to compound 1 was also evaluated in dogs receiving Form I HBr with pentagastrin pre- treatment.
- Compound 1 free base capsule consisted of compound 1 free base in Vitamin E TPGS and PEG 400 filled into a capsule.
- the Form I hydrobromide salt capsule consisted of Form I HBr alone filled into a capsule.
- the primary objective of the study is to compare the compound 1 pharmacokinetic (PK) profiles of single doses of Form I monohydrobromide formulations with that of the compound 1 free base in healthy adult males.
- One group of 12 subjects will be dosed in an effort to obtain the data described above. Each subject will receive the following formulations in a crossover investigation. Dosing will be separated by at least 7 days.
- Subjects will be provided with a light snack and then fast from all food and drink (except water) for a minimum of 8 h on the day prior to dosing until approximately 4 h post- dose at which time lunch will be provided.
- An evening meal will be provided at approximately 9 h post-dose and an evening snack at approximately 14 h post-dose. On subsequent days, meals will be provided at appropriate times.
- Venous blood samples will be withdrawn via an indwelling cannula or by venepuncture at the following times after dosing (hours): 0.5, 1, 1.5, 2, 4, 8, and 12.
- the primary endpoint of the study is to compare the PK profiles of a formulation of Form I HBr with that of compound 1 as a free base by measuring the following parameters: T lag , C max , T max , AUC (0 -iast), AUC (0 -inf), AUC% ex tra P , F re i, lambda-z, Ti/ 2 el.
- the secondary endpoint of the study is to collect information about the safety and tolerability of compound 1 (free base) and compound 2 (Form I HBr salt) by assessing: physical examinations, safety laboratory tests, vital signs, electrocardiograms (ECGs), body temperature and AEs.
- Plasma concentration data will be tabulated and plotted for each subject for whom concentrations are quantifiable.
- PK analysis of the concentration time data obtained will be performed using appropriate non-compartmental techniques to obtain estimates of the following PK parameters (where relevant).
- T 1 max the time from dosing at which C max occurs
- AUC(O-last) the area under the concentration vs time curve from time zero to the last measured time point
- AUC(O-inf) the area under the concentration vs time curve from time zero extrapolated to infinity
- AUC% extra p the percentage of AUC ( o-inf ) accounted for by extrapolation
- AUC(o -tau) the area under the concentration vs time curve within the
- AUC(0-24) the area under the concentration vs time curve from time zero to 24 hour post morning dose
- the initial 50 mg HBr dose selected for Regimen B (determined by AUC) is anticipated to be that which is expected to give a similar exposure to the 150 mg free base. This dose is also expected to provide less patient-to-patient variability.
- the Form I HBr formulation will be dosed again at the revised dose (Regimen C). There will then follow a further period of interim analysis to confirm that the Regimen C dose provides either, 1) a lower active dose as compared to Regimen A that provides an equivalent or higher exposure and/or provide less patient-to- patient variability, or 2) an active dose similar to that of Regimen A with a higher exposure and/or provide less patient-to-patient variability.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description








































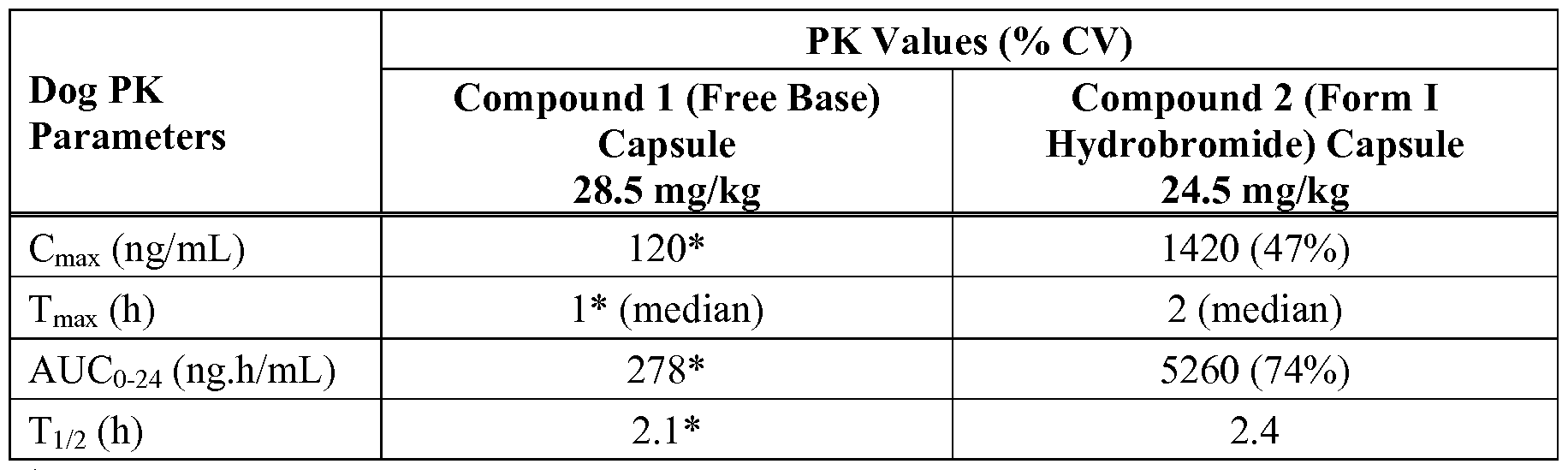

Claims


Priority Applications (22)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES13761487T ES2698298T3 (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| PL13761487T PL2825042T3 (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| SG11201405692UA SG11201405692UA (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| EP13761487.1A EP2825042B1 (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| MX2014010848A MX356179B (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor. |
| CA2866857A CA2866857C (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| SI201331233T SI2825042T1 (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| BR112014022790-0A BR112014022790B1 (en) | 2012-03-15 | 2013-03-13 | SALTS OF AN EPIDERMAL GROWTH FACTOR RECEPTOR KINASE INHIBITOR, PHARMACEUTICAL COMPOSITION AND USES THEREOF |
| RU2014136893A RU2711077C9 (en) | 2012-03-15 | 2013-03-13 | Salts of epidermal growth factor receptor kinase |
| LTEP13761487.1T LT2825042T (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| RS20181300A RS57901B1 (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| JP2015500567A JP6317320B2 (en) | 2012-03-15 | 2013-03-13 | Salt of epidermal growth factor receptor kinase inhibitor |
| CN201811096266.5A CN109053595B (en) | 2012-03-15 | 2013-03-13 | Salts of epidermal growth factor receptor kinase inhibitors |
| KR1020147028832A KR102090453B1 (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| MYPI2014002630A MY185243A (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
| CN201380025160.5A CN104284584B (en) | 2012-03-15 | 2013-03-13 | The salt of epidermal growth factor receptor kinase inhibitor |
| DK13761487.1T DK2825042T3 (en) | 2012-03-15 | 2013-03-13 | SALTS OF THE CHINASE INHIBITOR OF THE EPIDERMAL GROWTH FACTOR RECEPTOR |
| IL234426A IL234426B (en) | 2012-03-15 | 2014-09-02 | Salts of an epidermal growth factor receptor kinase inhibitor |
| PH12014502037A PH12014502037A1 (en) | 2012-03-15 | 2014-09-12 | Salts of an epidermal growth factor receptor kinase inhibitor |
| HK15106102.5A HK1205430A1 (en) | 2012-03-15 | 2015-06-26 | Salts of an epidermal growth factor receptor kinase inhibitor |
| HRP20181784TT HRP20181784T1 (en) | 2012-03-15 | 2018-10-30 | Salts of an epidermal growth factor receptor kinase inhibitor |
| CY20181101143T CY1122176T1 (en) | 2012-03-15 | 2018-11-01 | SALTS OF AN INHIBITOR OF DETERMINAL GROWTH FACTOR RECEPTOR KINASE |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261611400P | 2012-03-15 | 2012-03-15 | |
| US61/611,400 | 2012-03-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013138502A1 true WO2013138502A1 (en) | 2013-09-19 |
Family
ID=49161781
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/030996 WO2013138502A1 (en) | 2012-03-15 | 2013-03-13 | Salts of an epidermal growth factor receptor kinase inhibitor |
Country Status (28)
| Country | Link |
|---|---|
| US (5) | US9108927B2 (en) |
| EP (1) | EP2825042B1 (en) |
| JP (2) | JP6317320B2 (en) |
| KR (1) | KR102090453B1 (en) |
| CN (2) | CN104284584B (en) |
| AR (1) | AR090357A1 (en) |
| AU (3) | AU2013201519C1 (en) |
| BR (1) | BR112014022790B1 (en) |
| CA (1) | CA2866857C (en) |
| CY (1) | CY1122176T1 (en) |
| DK (1) | DK2825042T3 (en) |
| ES (1) | ES2698298T3 (en) |
| HK (1) | HK1205430A1 (en) |
| HR (1) | HRP20181784T1 (en) |
| IL (1) | IL234426B (en) |
| LT (1) | LT2825042T (en) |
| MX (1) | MX356179B (en) |
| MY (1) | MY185243A (en) |
| PH (1) | PH12014502037A1 (en) |
| PL (1) | PL2825042T3 (en) |
| PT (1) | PT2825042T (en) |
| RS (1) | RS57901B1 (en) |
| RU (1) | RU2711077C9 (en) |
| SG (2) | SG11201405692UA (en) |
| SI (1) | SI2825042T1 (en) |
| TR (1) | TR201816244T4 (en) |
| TW (1) | TWI591060B (en) |
| WO (1) | WO2013138502A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9056839B2 (en) | 2012-03-15 | 2015-06-16 | Celgene Avilomics Research, Inc. | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| WO2015117547A1 (en) * | 2014-02-04 | 2015-08-13 | Jiangsu Medolution Limited | Substituted pyrimidines useful as egfr-t790m kinase inhibitors |
| US9108927B2 (en) | 2012-03-15 | 2015-08-18 | Celgene Avilomics Research, Inc. | Salts of an epidermal growth factor receptor kinase inhibitor |
| US9145387B2 (en) | 2013-02-08 | 2015-09-29 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| CN104961688A (en) * | 2014-12-25 | 2015-10-07 | 苏州晶云药物科技有限公司 | Phosphate of epidermal growth factor receptor kinase inhibitor and preparation method thereof |
| WO2016101912A1 (en) * | 2014-12-25 | 2016-06-30 | 苏州晶云药物科技有限公司 | Crystal form of salt of epidermal growth factor receptor kinase inhibitor and preparation method thereof |
| EP2994456A4 (en) * | 2013-05-06 | 2017-01-04 | Clovis Oncology, Inc. | Salts of an epidermal growth factor receptor kinase inhibitor |
| US10005760B2 (en) | 2014-08-13 | 2018-06-26 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| EP3640246A4 (en) * | 2017-06-13 | 2020-08-05 | Korea Research Institute of Chemical Technology | N2,n4-diphenylpyrimidine-2,4-diamine derivative, method for preparing same, and pharmaceutical composition containing same as active ingredient for prevention or treatment of cancer |
| JP2021512087A (en) * | 2018-01-31 | 2021-05-13 | ディザル(ジァンスー)ファーマシューティカル・カンパニー・リミテッド | ERBB / BTK inhibitor |
| WO2023011358A1 (en) * | 2021-08-02 | 2023-02-09 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Novel pharmaceutical salts and polymorphic forms of an erbb and btk inhibitor |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11351168B1 (en) | 2008-06-27 | 2022-06-07 | Celgene Car Llc | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| NZ609957A (en) * | 2010-11-01 | 2015-08-28 | Celgene Avilomics Res Inc | Heterocyclic compounds and uses thereof |
| EP3327014A4 (en) * | 2015-07-24 | 2019-01-02 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Egfr inhibitor and pharmaceutically acceptable salt and polymorph thereof, and use thereof |
| CN105461640B (en) * | 2015-12-02 | 2017-11-21 | 芷威(上海)化学科技有限公司 | A kind of preparation method of tyrosine kinase inhibitor |
| CN105481779B (en) * | 2015-12-24 | 2019-01-25 | 南京华威医药科技集团有限公司 | Anticancer drug Rociletinib and its intermediate preparation |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6908906B2 (en) * | 2001-02-09 | 2005-06-21 | Sankyo Company, Limited | Crystalline forms of pyrimidine nucleoside derivative |
| WO2010129053A2 (en) | 2009-05-05 | 2010-11-11 | Dana Farber Cancer Institute | Egfr inhibitors and methods of treating disorders |
| WO2012021444A1 (en) * | 2010-08-10 | 2012-02-16 | Avila Therapeutics, Inc. | Besylate salt of a btk inhibitor |
| US20120149687A1 (en) | 2010-11-01 | 2012-06-14 | Avila Therapeutics, Inc. | Heterocyclic compounds and uses thereof |
Family Cites Families (162)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1062357A (en) | 1965-03-23 | 1967-03-22 | Pfizer & Co C | Quinazolone derivatives |
| US4032637A (en) | 1972-09-26 | 1977-06-28 | Sandoz Ltd. | Method of promoting sleep |
| US4337341A (en) | 1976-11-02 | 1982-06-29 | Eli Lilly And Company | 4a-Aryl-octahydro-1H-2-pyrindines |
| GB8608335D0 (en) | 1986-04-04 | 1986-05-08 | Pfizer Ltd | Pharmaceutically acceptable salts |
| US5453510A (en) | 1990-07-13 | 1995-09-26 | Burroughs Wellcome Co. | Neuromuscular blocking agents |
| JPH0741461A (en) | 1993-05-27 | 1995-02-10 | Eisai Co Ltd | Sulfonic acid ester derivative |
| WO1996028427A1 (en) | 1995-03-10 | 1996-09-19 | Berlex Laboratories, Inc. | Benzamidine derivatives their preparation and their use as anti-coagulants |
| GB9523675D0 (en) | 1995-11-20 | 1996-01-24 | Celltech Therapeutics Ltd | Chemical compounds |
| GB9619284D0 (en) | 1996-09-16 | 1996-10-30 | Celltech Therapeutics Ltd | Chemical compounds |
| GB9622363D0 (en) | 1996-10-28 | 1997-01-08 | Celltech Therapeutics Ltd | Chemical compounds |
| WO1999031073A1 (en) | 1997-12-15 | 1999-06-24 | Yamanouchi Pharmaceutical Co., Ltd. | Novel pyrimidine-5-carboxamide derivatives |
| US6303652B1 (en) | 1998-08-21 | 2001-10-16 | Hughes Institute | BTK inhibitors and methods for their identification and use |
| WO2000012485A1 (en) | 1998-08-29 | 2000-03-09 | Astrazeneca Ab | Pyrimidine compounds |
| KR100658489B1 (en) | 1998-11-10 | 2006-12-18 | 얀센 파마슈티카 엔.브이. | HIV replication inhibiting pyrimidines |
| US6262088B1 (en) | 1998-11-19 | 2001-07-17 | Berlex Laboratories, Inc. | Polyhydroxylated monocyclic N-heterocyclic derivatives as anti-coagulants |
| US6127376A (en) | 1998-12-04 | 2000-10-03 | Berlex Laboratories, Inc. | Aryl and heterocyclyl substituted pyrimidine derivatives as anti-coagulants |
| GB9828511D0 (en) | 1998-12-24 | 1999-02-17 | Zeneca Ltd | Chemical compounds |
| US6495558B1 (en) | 1999-01-22 | 2002-12-17 | Amgen Inc. | Kinase inhibitors |
| AU2871900A (en) | 1999-02-04 | 2000-08-25 | Millennium Pharmaceuticals, Inc. | G-protein coupled heptahelical receptor binding compounds and methods of use thereof |
| GB9914258D0 (en) | 1999-06-18 | 1999-08-18 | Celltech Therapeutics Ltd | Chemical compounds |
| JP2003503404A (en) | 1999-06-29 | 2003-01-28 | エギシュ ヂョヂセルヂャール エルテー | Piperazinylalkylthiopyrimidine derivative, pharmaceutical composition containing this derivative and process for producing this derivative |
| WO2001047921A1 (en) | 1999-12-28 | 2001-07-05 | Pharmacopeia, Inc. | Pyrimidine and triazine kinase inhibitors |
| AU3704101A (en) | 2000-02-17 | 2001-08-27 | Amgen Inc | Kinase inhibitors |
| GB0004888D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| GB0004890D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| GB0004887D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| WO2001085699A2 (en) | 2000-05-08 | 2001-11-15 | Janssen Pharmaceutica N.V. | Prodrugs of hiv replication inhibiting pyrimidines |
| GB0016877D0 (en) | 2000-07-11 | 2000-08-30 | Astrazeneca Ab | Chemical compounds |
| US7427689B2 (en) | 2000-07-28 | 2008-09-23 | Georgetown University | ErbB-2 selective small molecule kinase inhibitors |
| US6881737B2 (en) | 2001-04-11 | 2005-04-19 | Amgen Inc. | Substituted triazinyl acrylamide derivatives and methods of use |
| JO3429B1 (en) | 2001-08-13 | 2019-10-20 | Janssen Pharmaceutica Nv | Hiv inhibiting pyrimidines derivatives |
| US6939874B2 (en) | 2001-08-22 | 2005-09-06 | Amgen Inc. | Substituted pyrimidinyl derivatives and methods of use |
| WO2003030909A1 (en) | 2001-09-25 | 2003-04-17 | Bayer Pharmaceuticals Corporation | 2- and 4-aminopyrimidines n-substtituded by a bicyclic ring for use as kinase inhibitors in the treatment of cancer |
| HUP0402106A3 (en) | 2001-11-01 | 2009-07-28 | Janssen Pharmaceutica Nv | Heteroaryl amines as glycogen synthase kinase 3 beta inhibitors, process for their preparation and pharmaceutical compositions containing them |
| TWI329105B (en) | 2002-02-01 | 2010-08-21 | Rigel Pharmaceuticals Inc | 2,4-pyrimidinediamine compounds and their uses |
| AU2003209077A1 (en) | 2002-02-08 | 2003-09-02 | Smithkline Beecham Corporation | Pyrimidine compounds |
| GB0206215D0 (en) | 2002-03-15 | 2002-05-01 | Novartis Ag | Organic compounds |
| AU2003228345B2 (en) | 2002-03-21 | 2009-02-05 | Sunesis Pharmaceuticals, Inc. | Identification of kinase inhibitors |
| US20040002395A1 (en) | 2002-06-27 | 2004-01-01 | Poynor Raymond L. | Bridge weight for metal wood golf club |
| DE60330466D1 (en) | 2002-07-29 | 2010-01-21 | Rigel Pharmaceuticals Inc | METHOD FOR THE TREATMENT OR PREVENTION OF AUTOIMMUNE DISEASES WITH 2,4-PYRIMIDINE-IAMINE COMPOUNDS |
| CA2439440A1 (en) | 2002-09-05 | 2004-03-05 | Emory University | Treatment of tuberous sclerosis associated neoplasms |
| AU2002951853A0 (en) | 2002-10-04 | 2002-10-24 | Commonwealth Scientific And Industrial Research Organisation | Crystal structure of erbb2 and uses thereof |
| UA80767C2 (en) | 2002-12-20 | 2007-10-25 | Pfizer Prod Inc | Pyrimidine derivatives for the treatment of abnormal cell growth |
| JP5602333B2 (en) | 2003-02-07 | 2014-10-08 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Pyrimidine derivatives for prevention of HIV infection |
| WO2004074244A2 (en) | 2003-02-20 | 2004-09-02 | Smithkline Beecham Corporation | Pyrimidine compounds |
| GB0305929D0 (en) | 2003-03-14 | 2003-04-23 | Novartis Ag | Organic compounds |
| US20050014753A1 (en) | 2003-04-04 | 2005-01-20 | Irm Llc | Novel compounds and compositions as protein kinase inhibitors |
| US20050043233A1 (en) | 2003-04-29 | 2005-02-24 | Boehringer Ingelheim International Gmbh | Combinations for the treatment of diseases involving cell proliferation, migration or apoptosis of myeloma cells or angiogenesis |
| US7504396B2 (en) | 2003-06-24 | 2009-03-17 | Amgen Inc. | Substituted heterocyclic compounds and methods of use |
| NZ545270A (en) | 2003-07-30 | 2010-04-30 | Rigel Pharmaceuticals Inc | 2,4-Pyrimidinediamine compounds for use in the treatment or prevention of autoimmune diseases |
| EP1663242B1 (en) | 2003-08-07 | 2011-04-27 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and uses as anti-proliferative agents |
| EP2287156B1 (en) | 2003-08-15 | 2013-05-29 | Novartis AG | 2,4-Di(phenylamino)-pyrimidines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| GB0321710D0 (en) | 2003-09-16 | 2003-10-15 | Novartis Ag | Organic compounds |
| US20070105839A1 (en) | 2003-09-18 | 2007-05-10 | Patricia Imbach | 2, 4-Di (phenylamino) pyrimidines useful in the treatment of proliferative disorders |
| EP1694652A1 (en) | 2003-12-19 | 2006-08-30 | Rigel Pharmaceuticals, Inc. | Stereoisomers and stereoisomeric mixtures of 1-(2,4-pyrimidinediamino)-2-cyclopentanecarboxamide synthetic intermediates |
| PT1704145E (en) | 2004-01-12 | 2012-09-04 | Ym Biosciences Australia Pty | Selective kinase inhibitors |
| GT200500008A (en) | 2004-01-16 | 2005-08-16 | QUINOLINE INTERMEDIARIES AS INHIBITORS OF THIROSINE KINASE RECEPTORS AND THE SYNTHESIS OF THE SAME | |
| WO2005092836A1 (en) | 2004-03-15 | 2005-10-06 | Eli Lilly And Company | Opioid receptor antagonists |
| WO2005105988A2 (en) | 2004-04-28 | 2005-11-10 | Vertex Pharmaceuticals Incorporated | Crystal structure of human jak3 kinase domain complex and binding pockets thereof |
| JP4812763B2 (en) | 2004-05-18 | 2011-11-09 | ライジェル ファーマシューティカルズ, インコーポレイテッド | Cycloalkyl-substituted pyrimidinediamine compounds and uses thereof |
| EP1598343A1 (en) | 2004-05-19 | 2005-11-23 | Boehringer Ingelheim International GmbH | 2-Arylaminopyrimidine derivatives as PLK inhibitors |
| EP2592155B2 (en) | 2004-06-04 | 2019-09-11 | Genentech, Inc. | EGFR mutations |
| US7521457B2 (en) | 2004-08-20 | 2009-04-21 | Boehringer Ingelheim International Gmbh | Pyrimidines as PLK inhibitors |
| GB0419161D0 (en) | 2004-08-27 | 2004-09-29 | Novartis Ag | Organic compounds |
| US7622486B2 (en) | 2004-09-23 | 2009-11-24 | Reddy Us Therapeutics, Inc. | Pyridine compounds, process for their preparation and compositions containing them |
| CN101031550B (en) | 2004-09-30 | 2015-05-27 | 泰博特克药品有限公司 | HIV inhibiting 5-carbo- or heterocyclic substituted pyrimidines |
| BRPI0516597A (en) | 2004-10-13 | 2008-09-16 | Wyeth Corp | compound of the formula |
| US20060088471A1 (en) | 2004-10-20 | 2006-04-27 | Proteolix, Inc. | Compounds for enzyme inhibition |
| TW200628463A (en) | 2004-11-10 | 2006-08-16 | Synta Pharmaceuticals Corp | Heteroaryl compounds |
| GB2420559B (en) | 2004-11-15 | 2008-08-06 | Rigel Pharmaceuticals Inc | Stereoisomerically enriched 3-aminocarbonyl bicycloheptene pyrimidinediamine compounds and their uses |
| ES2380550T3 (en) | 2004-11-24 | 2012-05-16 | Rigel Pharmaceuticals, Inc. | Spiro-2,4-pyrimidinediamine compounds and their uses |
| MY169441A (en) | 2004-12-08 | 2019-04-11 | Janssen Pharmaceutica Nv | 2,4, (4,6) pyrimidine derivatives |
| JP5208516B2 (en) | 2004-12-30 | 2013-06-12 | エグゼリクシス, インコーポレイテッド | Pyrimidine derivatives as kinase modulators and methods of use |
| JP2008527007A (en) | 2005-01-14 | 2008-07-24 | ミレニアム・ファーマシューティカルズ・インコーポレイテッド | Cinnamide and hydrocinnamide derivatives having Raf-kinase inhibitory activity |
| EP2161275A1 (en) | 2005-01-19 | 2010-03-10 | Rigel Pharmaceuticals, Inc. | Prodrugs of 2,4-pyrimidinediamine compounds and their uses |
| WO2006086777A2 (en) | 2005-02-11 | 2006-08-17 | Memorial Sloan Kettering Cancer Center | Methods and compositions for detecting a drug resistant egfr mutant |
| CN101155799A (en) | 2005-03-16 | 2008-04-02 | 塔格根公司 | Pyrimidine inhibitors of kinases |
| DE102005016634A1 (en) | 2005-04-12 | 2006-10-19 | Merck Patent Gmbh | Novel aza heterocycles as kinase inhibitors |
| US20060270694A1 (en) | 2005-05-03 | 2006-11-30 | Rigel Pharmaceuticals, Inc. | JAK kinase inhibitors and their uses |
| WO2006124874A2 (en) | 2005-05-12 | 2006-11-23 | Kalypsys, Inc. | Inhibitors of b-raf kinase |
| US20070032493A1 (en) | 2005-05-26 | 2007-02-08 | Synta Pharmaceuticals Corp. | Method for treating B cell regulated autoimmune disorders |
| WO2006128129A2 (en) | 2005-05-26 | 2006-11-30 | Synta Pharmaceuticals Corp. | Method for treating cancer |
| WO2006129100A1 (en) | 2005-06-03 | 2006-12-07 | Glaxo Group Limited | Novel compounds |
| US20070203161A1 (en) | 2006-02-24 | 2007-08-30 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the jak pathway |
| KR101312225B1 (en) | 2005-06-08 | 2013-09-26 | 리겔 파마슈티칼스, 인크. | Compositions and methods for inhibition of the jak pathway |
| WO2007048064A2 (en) | 2005-10-21 | 2007-04-26 | Exelixis, Inc. | Amino-pyrimidines as casein kinase ii (ck2) modulators |
| BR122021011787B1 (en) | 2005-11-01 | 2022-01-25 | Impact Biomedicines, Inc | Biaryl metapyrimidine kinase inhibitors, pharmaceutical composition and process for preparing a pharmaceutical composition |
| JP2009514876A (en) | 2005-11-03 | 2009-04-09 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions for protein kinases |
| US7713987B2 (en) | 2005-12-06 | 2010-05-11 | Rigel Pharmaceuticals, Inc. | Pyrimidine-2,4-diamines and their uses |
| EP1968950A4 (en) | 2005-12-19 | 2010-04-28 | Genentech Inc | Pyrimidine kinase inhibitors |
| TW200736232A (en) | 2006-01-26 | 2007-10-01 | Astrazeneca Ab | Pyrimidine derivatives |
| BRPI0706747A2 (en) | 2006-01-30 | 2011-04-05 | Exelixis Inc | 4-aryl-2-amino-pyrimidines or 4-aryl-2-aminoalkyl-pyrimidines as jak-2 modulators and pharmaceutical compositions containing them |
| JP4653842B2 (en) | 2006-02-17 | 2011-03-16 | ライジェル ファーマシューティカルズ, インコーポレイテッド | 2,4-pyrimidinediamine compounds for treating or preventing autoimmune diseases |
| WO2007098507A2 (en) | 2006-02-24 | 2007-08-30 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the jak pathway |
| CA2645958C (en) | 2006-03-30 | 2014-11-04 | Tibotec Pharmaceuticals Ltd. | Hiv inhibiting 5-amido substituted pyrimidines |
| WO2007113256A1 (en) | 2006-03-30 | 2007-10-11 | Tibotec Pharmaceuticals Ltd. | Hiv inhibiting 5-(hydroxymethylene and aminomethylene) substituted pyrimidines |
| GB0608386D0 (en) | 2006-04-27 | 2006-06-07 | Senexis Ltd | Compounds |
| CA2861601A1 (en) | 2006-04-27 | 2007-11-08 | Intezyne Technologies, Inc. | Poly (ethylene glycol) containing chemically disparate endgroups |
| DE102006027156A1 (en) | 2006-06-08 | 2007-12-13 | Bayer Schering Pharma Ag | New sulfimide compounds are protein kinase inhibitors useful to treat e.g. cancer, Hodgkin's lymphoma, Kaposi's sarcoma, cardiovascular disease, Crohn's disease, endometriosis and hemangioma |
| CA2656290A1 (en) | 2006-07-05 | 2008-01-10 | Exelixis, Inc. | Methods of using igf1r and abl kinase modulators |
| MX2009000769A (en) | 2006-07-21 | 2009-01-28 | Novartis Ag | 2, 4 -di (arylaminio) -pyrimidine-5-carboxamide compounds as jak kinases inhibitors. |
| DE102006041382A1 (en) | 2006-08-29 | 2008-03-20 | Bayer Schering Pharma Ag | Carbamoyl sulfoximides as protein kinase inhibitors |
| EP2532235A1 (en) | 2006-09-22 | 2012-12-12 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| ATE497496T1 (en) | 2006-10-19 | 2011-02-15 | Rigel Pharmaceuticals Inc | 2,4-PYRIDIMEDIAMONE DERIVATIVES AS INHIBITORS OF JAKKINASES FOR THE TREATMENT OF AUTOIMMUNE DISEASES |
| ES2403546T3 (en) | 2006-11-03 | 2013-05-20 | Pharmacyclics, Inc. | Bruton tyrosine kinase activity probe and method of use |
| US8163902B2 (en) | 2006-11-21 | 2012-04-24 | Rigel Pharmaceuticals, Inc. | Prodrugs of 2,4-pyrimidinediamine compounds and their uses |
| AU2007333394C1 (en) | 2006-12-08 | 2011-08-18 | Novartis Ag | Compounds and compositions as protein kinase inhibitors |
| CA2670645A1 (en) | 2006-12-19 | 2008-07-03 | Genentech, Inc. | Pyrimidine kinase inhibitors |
| EP1939185A1 (en) | 2006-12-20 | 2008-07-02 | Bayer Schering Pharma Aktiengesellschaft | New types of hetaryl-phenylendiamin-pyrimidines as protein kinase inhibitors for the treatment of cancer |
| KR20090094073A (en) | 2006-12-29 | 2009-09-03 | 티보텍 파마슈티칼즈 리미티드 | Hiv inhibiting 6-substituted pyrimidines |
| CN106045965A (en) | 2006-12-29 | 2016-10-26 | 爱尔兰詹森科学公司 | HIV inhibiting 5,6-substituted pyrimidines |
| TW200902010A (en) | 2007-01-26 | 2009-01-16 | Smithkline Beecham Corp | Anthranilamide inhibitors of aurora kinase |
| JP5539734B2 (en) | 2007-01-31 | 2014-07-02 | ワイエム・バイオサイエンシズ・オーストラリア・ピーティーワイ・リミテッド | Thiopyrimidine-based compounds and uses thereof |
| JP4221470B2 (en) | 2007-02-01 | 2009-02-12 | トヨタ自動車株式会社 | Electric vehicle control apparatus and electric vehicle control method |
| DE102007010801A1 (en) | 2007-03-02 | 2008-09-04 | Bayer Cropscience Ag | Use of new and known 2,4-diaminopyrimidine derivatives as fungicides, especially for controlling phytopathogenic fungi |
| CN101677554A (en) | 2007-03-20 | 2010-03-24 | 史密丝克莱恩比彻姆公司 | Chemical compounds |
| JP2010522188A (en) | 2007-03-20 | 2010-07-01 | スミスクライン ビーチャム コーポレーション | Compound |
| US7947698B2 (en) | 2007-03-23 | 2011-05-24 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US7834024B2 (en) | 2007-03-26 | 2010-11-16 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US20120065201A1 (en) | 2007-03-28 | 2012-03-15 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US8809273B2 (en) | 2007-03-28 | 2014-08-19 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| WO2008151183A1 (en) | 2007-06-04 | 2008-12-11 | Avila Therapeutics, Inc. | Heterocyclic compounds and uses thereof |
| CN101903357A (en) | 2007-07-17 | 2010-12-01 | 里格尔药品股份有限公司 | Cyclic amine substituted pyrimidinediamines as PKC inhibitors |
| WO2009017838A2 (en) | 2007-08-01 | 2009-02-05 | Exelixis, Inc. | Combinations of jak-2 inhibitors and other agents |
| US20090088371A1 (en) | 2007-08-28 | 2009-04-02 | Rigel Pharmaceuticals, Inc. | Combination therapy with syk kinase inhibitor |
| WO2009032703A1 (en) | 2007-08-28 | 2009-03-12 | Irm Llc | 2- (het) arylamino-6-aminopyridine derivatives and fused forms thereof as anaplastic lymphoma kinase inhibitors |
| AU2008296545B2 (en) | 2007-08-28 | 2011-09-29 | Irm Llc | 2 -biphenylamino-4 -aminopyrimidine derivatives as kinase inhibitors |
| TWI552752B (en) | 2007-10-19 | 2016-10-11 | 賽基艾維洛米斯研究股份有限公司 | Heteroaryl compounds and uses thereof |
| US7989465B2 (en) | 2007-10-19 | 2011-08-02 | Avila Therapeutics, Inc. | 4,6-disubstituted pyrimidines useful as kinase inhibitors |
| EP2234986A2 (en) | 2007-12-20 | 2010-10-06 | Cellzome Limited | Sulfamides as zap-70 inhibitors |
| CA2715962C (en) | 2008-02-22 | 2017-03-07 | Rigel Pharmaceuticals, Inc. | Use of 2,4-pyrimidinediamines for the treatment of atherosclerosis |
| EP2276747A1 (en) | 2008-03-11 | 2011-01-26 | Cellzome Limited | Sulfonamides as zap-70 inhibitors |
| US8205348B2 (en) | 2008-03-19 | 2012-06-26 | Zashiki-Warashi Manufacturing Inc. | Tile spacer and holder therefor |
| CL2009000600A1 (en) | 2008-03-20 | 2010-05-07 | Bayer Cropscience Ag | Use of diaminopyrimidine compounds as phytosanitary agents; diaminopyrimidine compounds; its preparation procedure; agent that contains them; procedure for the preparation of said agent; and method for combating pests of animals and / or harmful plant pathogenic fungi. |
| WO2009127642A2 (en) | 2008-04-15 | 2009-10-22 | Cellzome Limited | Use of lrrk2 inhibitors for neurodegenerative diseases |
| US8138339B2 (en) | 2008-04-16 | 2012-03-20 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| CA2722326A1 (en) | 2008-04-24 | 2009-10-29 | Incyte Corporation | Macrocyclic compounds and their use as kinase inhibitors |
| EP2300013B1 (en) | 2008-05-21 | 2017-09-06 | Ariad Pharmaceuticals, Inc. | Phosphorous derivatives as kinase inhibitors |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| JP2011526299A (en) | 2008-06-27 | 2011-10-06 | アビラ セラピューティクス, インコーポレイテッド | Heteroaryl compounds and their use |
| MX2011000661A (en) | 2008-07-16 | 2011-05-25 | Pharmacyclics Inc | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors. |
| US20110245284A1 (en) | 2008-09-03 | 2011-10-06 | Bayer Cropscience Ag | Alkoxy- and Alkylthio-Substituted Anilinopyrimidines |
| US20110245156A1 (en) | 2008-12-09 | 2011-10-06 | Cytokine Pharmasciences, Inc. | Novel antiviral compounds, compositions, and methods of use |
| CA3018087C (en) | 2009-01-15 | 2019-09-24 | F. Hoffmann-La Roche Ag | Antibodies against phosphorylated tyrosines on erythropoietin receptor (epor) |
| EP2414337B1 (en) | 2009-04-03 | 2013-05-01 | Cellzome GmbH | Methods for the identification of kinase interacting molecules and for the purification of kinase proteins |
| RS54552B1 (en) | 2009-05-08 | 2016-06-30 | Astellas Pharma Inc. | Diamino heterocyclic carboxamide compound |
| US20120165332A1 (en) | 2009-06-18 | 2012-06-28 | Cellzome Limited | Sulfonamides and sulfamides as zap-70 inhibitors |
| US7718662B1 (en) | 2009-10-12 | 2010-05-18 | Pharmacyclics, Inc. | Pyrazolo-pyrimidine inhibitors of bruton's tyrosine kinase |
| WO2011079231A1 (en) | 2009-12-23 | 2011-06-30 | Gatekeeper Pharmaceutical, Inc. | Compounds that modulate egfr activity and methods for treating or preventing conditions therewith |
| WO2011140338A1 (en) | 2010-05-05 | 2011-11-10 | Gatekeeper Pharmaceuticals, Inc. | Compounds that modulate egfr activity and methods for treating or preventing conditions therewith |
| CA3240281A1 (en) | 2010-06-03 | 2011-12-08 | Pharmacyclics Llc | Use of inhibitors of bruton's tyrosine kinase (btk) in the treatment of follicular lymphoma |
| US20120071497A1 (en) | 2010-06-03 | 2012-03-22 | Pharmacyclics, Inc. | Methods of treating abc-dlbcl using inhibitors of bruton's tyrosine kinase |
| WO2011153553A2 (en) | 2010-06-04 | 2011-12-08 | The Regents Of The University Of California | Methods and compositions for kinase inhibition |
| US20130317029A1 (en) | 2010-11-01 | 2013-11-28 | Portola Pharmaceuticals, Inc. | Oxypyrimidines as syk modulators |
| EP2635285B1 (en) | 2010-11-01 | 2017-05-03 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| WO2012064706A1 (en) | 2010-11-10 | 2012-05-18 | Avila Therapeutics, Inc. | Mutant-selective egfr inhibitors and uses thereof |
| JP5999177B2 (en) | 2011-05-04 | 2016-09-28 | アリアド・ファーマシューティカルズ・インコーポレイテッド | Compound for inhibiting cell proliferation of EGFR-activated cancer |
| MX355728B (en) | 2011-05-17 | 2018-04-27 | Univ California | Kinase inhibitors. |
| BR112014022789B1 (en) | 2012-03-15 | 2022-04-19 | Celgene Car Llc | Solid forms of an epidermal growth factor receptor kinase inhibitor, pharmaceutical composition and uses thereof |
| SG11201405692UA (en) | 2012-03-15 | 2014-10-30 | Celgene Avilomics Res Inc | Salts of an epidermal growth factor receptor kinase inhibitor |
| PE20151604A1 (en) | 2012-11-02 | 2015-11-04 | Pharmacyclics Llc | ADJUVANT THERAPY WITH KINASE INHIBITORS FROM THE TEC FAMILY |
| JP2016518408A (en) | 2013-05-06 | 2016-06-23 | クロヴィス・オンコロジー,インコーポレーテッド | Salt of epidermal growth factor receptor kinase inhibitor |
-
2013
- 2013-03-13 SG SG11201405692UA patent/SG11201405692UA/en unknown
- 2013-03-13 BR BR112014022790-0A patent/BR112014022790B1/en not_active IP Right Cessation
- 2013-03-13 ES ES13761487T patent/ES2698298T3/en active Active
- 2013-03-13 TR TR2018/16244T patent/TR201816244T4/en unknown
- 2013-03-13 SG SG10201700799WA patent/SG10201700799WA/en unknown
- 2013-03-13 KR KR1020147028832A patent/KR102090453B1/en active IP Right Grant
- 2013-03-13 SI SI201331233T patent/SI2825042T1/en unknown
- 2013-03-13 LT LTEP13761487.1T patent/LT2825042T/en unknown
- 2013-03-13 CN CN201380025160.5A patent/CN104284584B/en active Active
- 2013-03-13 RS RS20181300A patent/RS57901B1/en unknown
- 2013-03-13 MX MX2014010848A patent/MX356179B/en active IP Right Grant
- 2013-03-13 DK DK13761487.1T patent/DK2825042T3/en active
- 2013-03-13 US US13/801,191 patent/US9108927B2/en active Active
- 2013-03-13 PT PT13761487T patent/PT2825042T/en unknown
- 2013-03-13 PL PL13761487T patent/PL2825042T3/en unknown
- 2013-03-13 RU RU2014136893A patent/RU2711077C9/en active
- 2013-03-13 CA CA2866857A patent/CA2866857C/en active Active
- 2013-03-13 MY MYPI2014002630A patent/MY185243A/en unknown
- 2013-03-13 EP EP13761487.1A patent/EP2825042B1/en active Active
- 2013-03-13 JP JP2015500567A patent/JP6317320B2/en active Active
- 2013-03-13 CN CN201811096266.5A patent/CN109053595B/en active Active
- 2013-03-13 WO PCT/US2013/030996 patent/WO2013138502A1/en active Application Filing
- 2013-03-14 AU AU2013201519A patent/AU2013201519C1/en not_active Ceased
- 2013-03-14 TW TW102109146A patent/TWI591060B/en not_active IP Right Cessation
- 2013-03-15 AR ARP130100852A patent/AR090357A1/en not_active Application Discontinuation
-
2014
- 2014-09-02 IL IL234426A patent/IL234426B/en active IP Right Grant
- 2014-09-12 PH PH12014502037A patent/PH12014502037A1/en unknown
-
2015
- 2015-06-26 HK HK15106102.5A patent/HK1205430A1/en not_active IP Right Cessation
- 2015-08-10 US US14/822,366 patent/US9540335B2/en active Active
-
2016
- 2016-05-23 AU AU2016203343A patent/AU2016203343A1/en not_active Abandoned
-
2017
- 2017-01-09 US US15/401,637 patent/US10005738B2/en active Active
- 2017-11-16 JP JP2017220788A patent/JP6676602B2/en not_active Expired - Fee Related
-
2018
- 2018-03-16 AU AU2018201896A patent/AU2018201896B2/en not_active Ceased
- 2018-06-22 US US16/015,626 patent/US10570099B2/en active Active
- 2018-10-30 HR HRP20181784TT patent/HRP20181784T1/en unknown
- 2018-11-01 CY CY20181101143T patent/CY1122176T1/en unknown
-
2020
- 2020-02-21 US US16/797,776 patent/US11292772B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6908906B2 (en) * | 2001-02-09 | 2005-06-21 | Sankyo Company, Limited | Crystalline forms of pyrimidine nucleoside derivative |
| WO2010129053A2 (en) | 2009-05-05 | 2010-11-11 | Dana Farber Cancer Institute | Egfr inhibitors and methods of treating disorders |
| WO2012021444A1 (en) * | 2010-08-10 | 2012-02-16 | Avila Therapeutics, Inc. | Besylate salt of a btk inhibitor |
| US20120149687A1 (en) | 2010-11-01 | 2012-06-14 | Avila Therapeutics, Inc. | Heterocyclic compounds and uses thereof |
Non-Patent Citations (3)
| Title |
|---|
| LIN, N. U.; WINER, E. P., BREAST CANCER RES, vol. 6, 2004, pages 204 - 210 |
| OGISO ET AL., CRYSTAL STRUCTURE OF THE COMPLEX OF HUMAN EPIDERMAL GROWTH FACTOR AND RECEPTOR EXTRACELLULAR DOMAINS IN CELL, vol. 110, 2002, pages 775 - 787, XP002961395 * |
| See also references of EP2825042A4 |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9540335B2 (en) | 2012-03-15 | 2017-01-10 | Celgene Avilomics Research, Inc. | Salts of an epidermal growth factor receptor kinase inhibitor |
| US11292772B2 (en) | 2012-03-15 | 2022-04-05 | Celgene Car Llc | Salts of an epidermal growth factor receptor kinase inhibitor |
| US9108927B2 (en) | 2012-03-15 | 2015-08-18 | Celgene Avilomics Research, Inc. | Salts of an epidermal growth factor receptor kinase inhibitor |
| US10946016B2 (en) | 2012-03-15 | 2021-03-16 | Celgene Car Llc | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| US10570099B2 (en) | 2012-03-15 | 2020-02-25 | Celgene Car Llc | Salts of an epidermal growth factor receptor kinase inhibitor |
| US10004741B2 (en) | 2012-03-15 | 2018-06-26 | Celgene Car Llc | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| US10005738B2 (en) | 2012-03-15 | 2018-06-26 | Celgene Car Llc | Salts of an epidermal growth factor receptor kinase inhibitor |
| US9056839B2 (en) | 2012-03-15 | 2015-06-16 | Celgene Avilomics Research, Inc. | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| US9539255B2 (en) | 2012-03-15 | 2017-01-10 | Celgene Avilomics Research, Inc. | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| US9980964B2 (en) | 2013-02-08 | 2018-05-29 | Celgene Car Llc | ERK inhibitors and uses thereof |
| US9145387B2 (en) | 2013-02-08 | 2015-09-29 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9561228B2 (en) | 2013-02-08 | 2017-02-07 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9796700B2 (en) | 2013-02-08 | 2017-10-24 | Celgene Car Llc | ERK inhibitors and uses thereof |
| US9504686B2 (en) | 2013-02-08 | 2016-11-29 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| EP2994456A4 (en) * | 2013-05-06 | 2017-01-04 | Clovis Oncology, Inc. | Salts of an epidermal growth factor receptor kinase inhibitor |
| WO2015117547A1 (en) * | 2014-02-04 | 2015-08-13 | Jiangsu Medolution Limited | Substituted pyrimidines useful as egfr-t790m kinase inhibitors |
| JP2017505319A (en) * | 2014-02-04 | 2017-02-16 | チャンスー メドリューション リミテッド | Pyrimidinium compounds substituted as EGFR-T790M kinase inhibitors |
| CN105980377A (en) * | 2014-02-04 | 2016-09-28 | 江苏迈度药物研发有限公司 | Substituted pyrimidines useful as EGFR-T790M kinase inhibitors |
| US10167264B2 (en) | 2014-02-04 | 2019-01-01 | Jiangsu Medolution Ltd | Substituted pyrimidines useful as EGFR-T790M kinase inhibitors |
| US10202364B2 (en) | 2014-08-13 | 2019-02-12 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| US10005760B2 (en) | 2014-08-13 | 2018-06-26 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| CN104961688A (en) * | 2014-12-25 | 2015-10-07 | 苏州晶云药物科技有限公司 | Phosphate of epidermal growth factor receptor kinase inhibitor and preparation method thereof |
| WO2016101912A1 (en) * | 2014-12-25 | 2016-06-30 | 苏州晶云药物科技有限公司 | Crystal form of salt of epidermal growth factor receptor kinase inhibitor and preparation method thereof |
| EP3640246A4 (en) * | 2017-06-13 | 2020-08-05 | Korea Research Institute of Chemical Technology | N2,n4-diphenylpyrimidine-2,4-diamine derivative, method for preparing same, and pharmaceutical composition containing same as active ingredient for prevention or treatment of cancer |
| JP2021512087A (en) * | 2018-01-31 | 2021-05-13 | ディザル(ジァンスー)ファーマシューティカル・カンパニー・リミテッド | ERBB / BTK inhibitor |
| EP3746424A4 (en) * | 2018-01-31 | 2021-08-25 | Dizal (Jiangsu) Pharmaceutical Co., Ltd | Erbb/btk inhibitors |
| US11504375B2 (en) | 2018-01-31 | 2022-11-22 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | ErbB/BTK inhibitors |
| JP7427597B2 (en) | 2018-01-31 | 2024-02-05 | ディザル(ジァンスー)ファーマシューティカル・カンパニー・リミテッド | ERBB/BTK inhibitor |
| EP4356975A3 (en) * | 2018-01-31 | 2024-07-24 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Erbb/btk inhibitors |
| WO2023011358A1 (en) * | 2021-08-02 | 2023-02-09 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Novel pharmaceutical salts and polymorphic forms of an erbb and btk inhibitor |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11292772B2 (en) | Salts of an epidermal growth factor receptor kinase inhibitor | |
| US10946016B2 (en) | Solid forms of an epidermal growth factor receptor kinase inhibitor | |
| US20160074399A1 (en) | Salts of an Epidermal Growth Factor Receptor Kinase Inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13761487 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2866857 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2014/010848 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2015500567 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013761487 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20147028832 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IDP00201406214 Country of ref document: ID |
|
| ENP | Entry into the national phase |
Ref document number: 2014136893 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014022790 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014022790 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140915 |