WO2013007387A1 - Novel fxr (nr1h4) binding and activity modulating compounds - Google Patents
Novel fxr (nr1h4) binding and activity modulating compounds Download PDFInfo
- Publication number
- WO2013007387A1 WO2013007387A1 PCT/EP2012/002941 EP2012002941W WO2013007387A1 WO 2013007387 A1 WO2013007387 A1 WO 2013007387A1 EP 2012002941 W EP2012002941 W EP 2012002941W WO 2013007387 A1 WO2013007387 A1 WO 2013007387A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- liver
- alkyl
- fxr
- compound
- group
- Prior art date
Links
- 0 **C1C(*OCN)C1 Chemical compound **C1C(*OCN)C1 0.000 description 4
- GZLDBBCVYWWGKR-ZBGNCUAUSA-N C/C=C\C(\Cl)=C(/C(Cl)=C)\c1n[o]c(C2CC2)c1COc1ccc([C@](C2)(C[C@H]2c2c(C)ccc(C(O)=O)c2)O)c(Cl)c1 Chemical compound C/C=C\C(\Cl)=C(/C(Cl)=C)\c1n[o]c(C2CC2)c1COc1ccc([C@](C2)(C[C@H]2c2c(C)ccc(C(O)=O)c2)O)c(Cl)c1 GZLDBBCVYWWGKR-ZBGNCUAUSA-N 0.000 description 1
- VOEXUHSXLWZXMO-UHFFFAOYSA-N CS(c1cc(C(C2)CC2(c(c(Cl)c2)ccc2OCc2c(C3CC3)[o]nc2-c(c(Cl)ccc2)c2Cl)O)ccc1)(=O)=O Chemical compound CS(c1cc(C(C2)CC2(c(c(Cl)c2)ccc2OCc2c(C3CC3)[o]nc2-c(c(Cl)ccc2)c2Cl)O)ccc1)(=O)=O VOEXUHSXLWZXMO-UHFFFAOYSA-N 0.000 description 1
- FJKMAESQKWYCNU-NJTBPKAJSA-N Cc(c([C@H](C1)C[C@@]1(c(ccc(OCc1c(C2CC2)[o]nc1-c(c(Cl)ccc1)c1Cl)c1)c1Cl)O)c1)ccc1C(O)=O Chemical compound Cc(c([C@H](C1)C[C@@]1(c(ccc(OCc1c(C2CC2)[o]nc1-c(c(Cl)ccc1)c1Cl)c1)c1Cl)O)c1)ccc1C(O)=O FJKMAESQKWYCNU-NJTBPKAJSA-N 0.000 description 1
- OSRYLUZWSNQSMF-NJTBPKAJSA-N Cc(ccc([C@H](C1)C[C@@]1(c(ccc(OCc1c(C2CC2)[o]nc1-c(c(Cl)ccc1)c1Cl)c1)c1Cl)O)c1)c1C(O)=O Chemical compound Cc(ccc([C@H](C1)C[C@@]1(c(ccc(OCc1c(C2CC2)[o]nc1-c(c(Cl)ccc1)c1Cl)c1)c1Cl)O)c1)c1C(O)=O OSRYLUZWSNQSMF-NJTBPKAJSA-N 0.000 description 1
- OSRYLUZWSNQSMF-HQLWIIKZSA-N Cc(ccc([C@H](C1)C[C@]1(c(ccc(OCc1c(C2CC2)[o]nc1-c(c(Cl)ccc1)c1Cl)c1)c1Cl)O)c1)c1C(O)=O Chemical compound Cc(ccc([C@H](C1)C[C@]1(c(ccc(OCc1c(C2CC2)[o]nc1-c(c(Cl)ccc1)c1Cl)c1)c1Cl)O)c1)c1C(O)=O OSRYLUZWSNQSMF-HQLWIIKZSA-N 0.000 description 1
- RPSCCXCTBCXPTP-ONQVFSDLSA-N Cc1cc(C(O)=O)cc([C@H](C2)C[C@@]2(c(ccc(OC/C(/C(c(c(Cl)ccc2)c2Cl)=N)=C(\C2CC2)/O)c2)c2Cl)O)c1 Chemical compound Cc1cc(C(O)=O)cc([C@H](C2)C[C@@]2(c(ccc(OC/C(/C(c(c(Cl)ccc2)c2Cl)=N)=C(\C2CC2)/O)c2)c2Cl)O)c1 RPSCCXCTBCXPTP-ONQVFSDLSA-N 0.000 description 1
- MAJJMEAYYRQVFN-SOYRUXDNSA-N Cc1cc(C(O)=O)cc([C@H](C2)C[C@]2(c(c(Cl)c2)ccc2OCc2c(C3CC3)[o]nc2-c(c(Cl)ccc2)c2Cl)O)c1 Chemical compound Cc1cc(C(O)=O)cc([C@H](C2)C[C@]2(c(c(Cl)c2)ccc2OCc2c(C3CC3)[o]nc2-c(c(Cl)ccc2)c2Cl)O)c1 MAJJMEAYYRQVFN-SOYRUXDNSA-N 0.000 description 1
- YVDIWFNHPWYPDR-UHFFFAOYSA-N NS(c1cccc(C(C2)CC2(c(c(Cl)c2)ccc2OCc2c(C3CC3)[o]nc2-c(c(Cl)ccc2)c2Cl)O)c1)(=O)=O Chemical compound NS(c1cccc(C(C2)CC2(c(c(Cl)c2)ccc2OCc2c(C3CC3)[o]nc2-c(c(Cl)ccc2)c2Cl)O)c1)(=O)=O YVDIWFNHPWYPDR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/422—Oxazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to compounds which bind to the NR1 H4 receptor (FXR) and act as agonists or modulators of FXR.
- the invention further relates to the use of the compounds for the treatment and/or prophylaxis of diseases and/or conditions through binding of said nuclear receptor by said compounds.
- Multicellular organisms are dependent on advanced mechanisms of information transfer between cells and body compartments.
- the information that is transmitted can be highly complex and can result in the alteration of genetic programs involved in cellular differentiation, proliferation, or reproduction.
- the signals, or hormones are often low molecular weight molecules, such as peptides, fatty acid, or cholesterol derivatives.
- NR nuclear receptors
- Orphan receptors may be indicative of unknown signalling pathways in the cell or may be nuclear receptors that function without ligand activation. The activation of transcription by some of these orphan receptors may occur in the absence of an exogenous ligand and/or through signal transduction pathways originating from the cell surface (D. J. Mangelsdorf et al., Cell 1995, 83, 835; R. M. Evans, Mol. Endocrinol. 2005, 19, 1429).
- DBD DNA-binding domain
- HRE Hormone Responsive Element
- Coactivators or transcriptional activators are proposed to bridge between sequence specific transcription factors, the basal transcription machinery and in addition to influence the chromatin structure of a target cell.
- proteins like SRC-1 , ACTR, and Gripl interact with NRs in a ligand enhanced manner (D. M. Heery et al., Nature 1997, 387, 733; T. Heinzel et al., Nature 1997, 387, 43; K. W. Nettles and G. L. Greene, Annu. Rev. Physiol. 2005, 67, 309).
- Nuclear receptor modulators like steroid hormones affect the growth and function of specific cells by binding to intracellular receptors and forming nuclear receptor-ligand complexes. Nuclear receptor-hormone complexes then interact with a HRE in the control region of specific genes and alter specific gene expression (A. Aranda and A. Pascual, Physiol. Rev. 2001 , 81 , 1269).
- the Famesoid X Receptor alpha (hereinafter also often referred to as NR1 H4 when referring to the human receptor) is a prototypical type 2 nuclear receptor which activates genes upon binding to promoter region of target genes in a heterodimeric fashion with Retinoid X Receptor (B. M. Forman et al., Cell 1995, 81 , 687).
- the relevant physiological ligands of NR1 H4 are bile acids (D. J. Parks et al., Science 1999, 284, 1365; M. Makishima et al., Science 1999, 284, 1362).
- chenodeoxycholic acid which regulates the expression of several genes that participate in bile acid homeostasis.
- Farnesol and derivatives, together called farnesoids, are originally described to activate the rat orthologue at high concentration but they do not activate the human or mouse receptor.
- FXR is expressed in the liver, throughout the entire gastrointestinal tract including the esophagus, stomach, duodenum, small intestine, colon, ovary, adrenal gland and kidney. Beyond controlling intracellular gene expression, FXR seems to be also involved in paracrine and endocrine signalling by upregulating the expression of the cytokine Fibroblast Growth Factor 15 (rodents) or 9 (monkeys, humans, J. A. Holt et al., Genes Dev. 2003, 17, 1581 ; T. Inagaki et al., Cell Metab. 2005, 2, 217).
- Small molecule compounds which act as FXR modulators have been disclosed in the following publications: WO 2000/037077, WO 2003/015771 , WO 2004/048349, WO 2007/076260, WO 2007/092751 , WO 2007/140174, WO 2007/140183, WO 2008/051942, WO 2008/157270, WO 2009/005998, WO 2009/012125, WO 2008/025539 and WO 2008/025540. Further small molecule FXR modulators have been recently reviewed (M. L. Crawley, Expert Opin Ther. Pat. 2010, 20,1047; D. Merk et al., Future Med. Chem. 2012, 4, 1015).
- the problem underlying the present invention is to generate FXR-agonists with improved physicochemical properties in general, and reduced hydrophobicity, improved aqueous solubility and better membrane permeability, in particular, compared to compounds claimed in WO 2011/020615.
- R is selected from the group consisting of COORe, CONR7R8, tetrazolyl, SO2NR7R8, C1-6 alkyl, S0 2 -Cve alkyl and H, with R 6 independently selected from the group consisting of H or d-e alkyl, and R 7 and R 8 independently from each other selected from the group consisting of H, alkyl, halo-Ci-6 alkyl, Ci. 6 alkylene-R 9 , S0 2 -C 1 . 6 alkyl, wherein R 9 is selected from the group consisting of COOH, OH and S0 3 H;
- A is selected from the group consisting of phenyl, pyridyl, pyrimidyl, pyrazolyl, indolyl, thienyl, benzothienyl, indazolyl, benzisoxazolyl, benzofuranyl, benzotriazolyl, furanyl, benzothiazolyl, thiazolyl, oxadiazolyl, each optionally substituted with one or two groups independently selected from the group consisting of OH, alkyl, O-halo-d-e alkyl, d. 6 alkyl, halo-Ci. 6 alkyl, C3.6 cycloalkyl and halogen;
- Q is selected from the group consisting of phenyl, pyridyl, thiazolyl, thiophenyl, pyrimidyl, each optionally substituted with one or two groups independently selected from the group consisting of d-e alkyl, halo-Ci- 6 alkyl, halogen and CF 3 ;
- Y is selected from N or CH
- Ri is selected from the group consisting of hydrogen, d. 3 alkyl, C 3 ⁇ cylcoalkyl, C4-5 alkylcycloalkyl, wherein d. 3 alkyl is optionally substituted with 1 to 3 substituents independently selected from halogen, hydroxy or d ⁇ alkoxy;
- R 2 and R 3 are independently selected from the group consisting of hydrogen, C 1 -3 alkyl, d. 3 haloalkyl, d- 3 alkoxy, C1.3 haloalkoxy and halogen.
- R-A in the compound according to Formula (1) is selected from
- Z in the compound according to Formula (1) is
- the compound according to Formula (1) is selected from
- R is selected from the group consisting of C0 2 H, CONHS0 2 Me, and tetrazolyl.
- the present invention is directed to a compound according to Formula (1) for use as a medicament.
- the present invention is directed to a compound according to Formula (1) for use in the prophylaxis and/or treatment of diseases mediated by FXR.
- the present invention is directed to the use of a compound according to Formula (1) for the preparation of a medicament for the prophylaxis and/or treatment of diseases mediated by FXR.
- the disease is selected from chronic intrahepatic or some forms of extrahepatic cholestatic conditions; liver fibrosis; obstructive or chronic inflammatory disorders of the liver; liver cirrhosis; liver steatosis and associated syndromes, cholestatic or fibrotic effects that are associated with alcohol-induced cirrhosis or with viral-borne forms of hepatitis; liver failure or liver ischemia after major liver resection; chemotherapy associated steatohepatitis (CASH); acute liver failure; and/or Inflammatory Bowel Diseases.
- CASH chemotherapy associated steatohepatitis
- the disease is selected from lipid and lipoprotein disorders; Type II Diabetes and clinical complications of Type I and Type II Diabetes, including diabetic nephropathy, diabetic neuropathy, diabetic retinopathy and other observed effects of clinically manifest long term Diabetes; conditions and diseases which result from chronic fatty and fibrotic degeneration of organs due to enforced lipid and specifically triglyceride accumulation and subsequent activation of profibrotic pathways, such as Non- Alcoholic Fatty Liver Disease (NAFLD), or Non- Alcoholic Steatohepatitis (NASH); obesity or metabolic syndrome (combined conditions of dyslipidemia, diabetes or abnormally high body-mass index); and/or cute myocardial infarction, acute stroke or thrombosis which occurs as an endpoint of chronic obstructive atherosclerosis.
- NAFLD Non- Alcoholic Fatty Liver Disease
- NASH Non- Alcoholic Steatohepatitis
- obesity or metabolic syndrome combined conditions of dyslipidemia, diabetes or abnormally high body-mass index
- the disease is selected from non-malignant hyperproliferative disorders and malignant hyperproliferative disorders, specifically of hepatocellular carcinoma, colon adenoma and polyposis, colon adenocarcinoma, breast cancer, pancreas adenocarcinoma, Barrett's esophagus or other forms of neoplastic diseases of the gastrointestinal tract and the liver.
- the improved physico-chemical properties have been achieved by the introduction of a polar hydroxyl group on a 1 ,3-cyclobutyltdene or 1 ,3-azetidinylidene group replacing the former 1 ,2- cyclopropylidene ring.
- the resulting compounds maintained their activity on the FXR receptor but demonstrated improved physico-chemical properties, such as higher aqueous solubility and/or membrane permeability.
- the compounds of the present invention share a common chemical structure according to Formula (1) in claim 1.
- the present invention is directed to an enantiomer, diastereomer or pharmaceutically acceptable salt of a compound according to Formula (1).
- R in Formula (1) is selected from the group consisting of COOR 6 , CONR 7 R 8 , S0 2 NR 7 R 8 , and S0 2 -Ci. 6 alkyl.
- R 6 in Formula (1) is H.
- R 7 and R 8 in Formula (1) are independently from each other selected from the group consisting of H and S0 2 -Ci. 6 alkyl.
- R 7 in Formula (1) is H.
- R 8 in Formula (1) is SC C ⁇ alkyl.
- A is selected from the group consisting of phenyl, pyridyl, pyrimidyl, pyrazolyl, indazolyl, and oxadiazolyl.
- A is substituted with one or two groups independently selected from C 1 .6 alkyl, more preferably C 1 .3 alkyl. In another preferred embodiment in combination with any of the above or below embodiments, A is unsubstituted.
- Q is phenyl
- Q is substituted with one or two groups independently selected from halogen, more preferably one group selected from halogen, in particular CI.
- Z is
- X CH.
- Ri is C 3 -6 cylcoalkyl, in particular cyclopropyl.
- R 2 and R 3 are independently selected from halogen, in particular CI.
- the compounds of the present invention can be in the form of a prodrug compound.
- Prodrug compound means a derivative that is converted into a compound according to the present invention by a reaction with an enzyme, gastric acid or the like under a physiological condition in the living body, e.g. by oxidation, reduction, hydrolysis or the like, each of which is carried out enzymatically.
- prodrug examples include compounds, wherein the amino group in a compound of the present invention is acylated, alkylated or phosphorylated to form, e.g., eicosanoylamino, alanylamino, pivaloyloxymethylamino or wherein the hydroxyl group is acylated, alkylated, phosphorylated or converted into the borate, e.g. acetyloxy, palmitoyloxy, pivaloyloxy, succinyloxy, fumaryloxy, alanyloxy or wherein the carboxyl group is esterified or amidated.
- these compounds can be produced from compounds of the present invention according to well-known methods.
- prodrug examples are compounds, wherein the carboxylate in a compound of the present invention is, for example, converted into an alkyl-, aryl-, choline-, amino, acyloxymethylester, linolenoylester.
- Metabolites of compounds of the present invention are also within the scope of the present invention.
- tautomerism like e.g. keto-enol tautomerism
- the individual forms like e.g. the keto and enol form, are each within the scope of the invention as well as their mixtures in any ratio. Same applies for stereoisomers, like e.g. enantiomers, cis/trans isomers, conformers and the like.
- isomers can be separated by methods well known in the art, e.g. by liquid chromatography. Same applies for enantiomers by using e.g. chiral stationary phases. Additionally, enantiomers may be isolated by converting them into diastereomers, i.e. coupling with an enantiomerically pure auxiliary compound, subsequent separation of the resulting diastereomers and cleavage of the auxiliary residue. Alternatively, any enantiomer of a compound of the present invention may be obtained from stereoselective synthesis using optically pure starting materials. Another way to obtain pure enantiomers from racemic mixtures would use enantioselective crystallization with chiral counterions.
- the compounds of the present invention can be in the form of a pharmaceutically acceptable salt or a solvate.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids, including inorganic bases or acids and organic bases or acids.
- the invention also comprises their corresponding pharmaceutically or toxicologically acceptable salts, in particular their pharmaceutically utilizable salts.
- the compounds of the present invention which contain acidic groups can be present on these groups and can be used according to the invention, for example, as alkali metal salts, alkaline earth metal salts or ammonium salts.
- salts include sodium salts, potassium salts, calcium salts, magnesium salts or salts with ammonia or organic amines such as, for example, ethylamine, ethanolamine, triethanotamine or amino acids.
- the compounds of the present invention which contain one or more basic groups, i.e. groups which can be protonated, can be present and can be used according to the invention in the form of their addition salts with inorganic or organic acids.
- suitable acids include hydrogen chloride, hydrogen bromide, phosphoric acid, sulfuric acid, nitric acid, methanesulfonic acid, p- toluenesulfonic acid, naphthalenedisulfonic acids, oxalic acid, acetic acid, tartaric acid, lactic acid, salicylic acid, benzoic acid, formic acid, propionic acid, pivalic acid, diethylacetic acid, malonic acid, succinic acid, pimelic acid, fumaric acid, maleic acid, malic acid, sulfaminic acid, phenylpropionic acid, gluconic acid, ascorbic acid, isonicotinic acid, citric acid, adipic acid, and other acids known to the person skilled in the art.
- the invention also includes, in addition to the salt forms mentioned, inner salts or betaines (zwitterions).
- inner salts or betaines can be obtained by customary methods which are known to the person skilled in the art like, for example, by contacting these with an organic or inorganic acid or base in a solvent or dispersant, or by anion exchange or cation exchange with other salts.
- the present invention also includes all salts of the compounds of the present invention which, owing to low physiological compatibility, are not directly suitable for use in pharmaceuticals but which can be used, for example, as intermediates for chemical reactions or for the preparation of pharmaceutically acceptable salts.
- solvates such as those which include as solvate water, or pharmaceutically acceptable solvates, such as alcohols, in particular ethanol.
- the present invention provides pharmaceutical compositions comprising at least one compound of the present invention, or a prodrug compound thereof, or a pharmaceutically acceptable salt or solvate thereof as active ingredient together with a pharmaceutically acceptable carrier.
- “Pharmaceutical composition” means one or more active ingredients, and one or more inert ingredients that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- the pharmaceutical compositions of the present invention encompass any composition made by admixing at least one compound of the present invention and a pharmaceutically acceptable carrier.
- the pharmaceutical composition of the present invention may additionally comprise one or more other compounds as active ingredients like a prodrug compound or other nuclear receptor modulators.
- compositions are suitable for oral, rectal, topical, parenteral (including subcutaneous, intramuscular, and intravenous), ocular (ophthalmic), pulmonary (nasal or buccal inhalation) or nasal administration, although the most suitable route in any given case will depend on the nature and severity of the conditions being treated and on the nature of the active ingredient. They may be conveniently presented in unit dosage form and prepared by any of the methods well-known in the art of pharmacy.
- the compounds of the present invention can be prepared by a combination of methods described in Schemes I to III.
- M metal, e.g. Li
- Y two isomers can form (A and Q transannular cis or trans to each other). Under optimized conditions the formation of mainly one of the two isomers can be achieved.
- the two isomers can be separated by appropriate methods known in the art like e.g. silica gel chromatography or preparative RP-HPLC.
- dehalogenation e.g. with Zn in acetic acid under reflux, the desired 3-substituted cyclobutanones are obtained.
- the vinyl-intermediates can react with in situ generated unsubtituted ketene to afford in one step the desired cyclobutanone intermediates.
- 3-methylenecyclobutanecarbonitrile is used as starting material.
- Substituted heterocycles can be built up from the cyano group in several steps by methods known to those skilled in the art.
- the desired cyclobutanones can be obtained by oxidative cleavage of the exocyclic double bond using conditions and reagents known to those skilled in the art, e.g. by the use of Os0 4 , ozone or RhCi NalC ⁇ as oxidants.
- Option c) shows the methods used to prepare the substituted azetidinones.
- HetAr heteroaromatic ring c) Cu or Pd catalysed
- Scheme III illustrates some possibilities to perform modifications of the substituents at the A group after the formation of the 4-membered hydroxy-bearing rings.
- a leaving group X e.g. bromide
- a cyano group e.g. carboxylic ester, methylsulfonyl or thioether by transition metal catalysed cross coupling reactions.
- the obtained derivatives can be further transformed into other derivatives by methods known to those skilled in the art.
- the cyano and the ester group can be hydrolysed under basic conditions to the afford a carboxylic acid which in turn can be transformed into acyl-sulfonamides.
- a benzyl thioether can be chlorinated to afford the chlorosulfonyl intermediate which reacts with ammonia to the corresponding sulfonamides.
- the present invention relates to compounds according to the general Formula (1) which bind to FXR and act as agonists or modulators of FXR.
- the invention further relates to the use of said compounds for the treatment and/or prophylaxis of diseases and/or conditions through binding of said nuclear receptor by said compounds. Further the present invention relates to the use of said compounds for the preparation of a medicament for the treatment and/or prophylaxis of diseases and/or conditions through binding of said nuclear receptor by said compounds.
- the present invention relates to the use of compounds according to Formula (1) in the preparation of a medicament for the prophylaxis and/or treatment of chronic intrahepatic or some forms of extrahepatic cholestatic conditions, of liver fibrosis, of acute intraheptic cholestatic conditions, of obstructive or chronic inflammatory disorders that arise out of improper bile composition, of gastrointestinal conditions with a reduced uptake of dietary fat and fat-soluble dietary vitamins, of inflammatory bowel diseases, of lipid and lipoprotein disorders, of Type II Diabetes and clinical complications of Type I and Type II Diabetes, of conditions and diseases which result from chronic fatty and fibrotic degeneration of organs due to enforced lipid and specifically triglyceride accumulation and subsequent activation of profibrotic pathways, of obesity and metabolic syndrome (combined conditions of dyslipidemia, diabetes and abnormally high body-mass index), of acute myocardial infarction, of acute stroke, of thrombosis which occurs as an endpoint of chronic obstructive atherosclerosis,
- Medicaments as referred to herein may be prepared by conventional processes, including the combination of a compound according to the present invention and a pharmaceutically acceptable carrier.
- FXR is proposed to be a nuclear bile acid sensor. As a result, it modulates both, the synthetic output of bile acids in the liver and their recycling in the intestine (by regulating bile acid binding proteins). But beyond bile acid physiology, FXR seems to be involved in the regulation of many diverse physiological processes which are relevant in the etiology and for the treatment of diseases as diverse as cholesterol gallstones, metabolic disorders such as Type II Diabetes, dyslipidemias or obesity, chronic inflammatory diseases such as Inflammatory Bowel Diseases or chronic intrahepatic forms of cholestasis and many others diseases (T. Claudel et al., Arterioscler. Thromb. Vase. Biol. 2005, 25, 2020; Y. D. Wang et al., Cell Res. 2008, 18, 1087.
- FXR regulates a complex pattern of response genes in the liver and in the gastrointestinal tract.
- the gene products have impact on diverse physiological processes.
- the first regulatory network that was analyzed was the regulation of bile acid synthesis. While the LXRs induce the key enzyme of the conversion of cholesterol into bile acids, Cyp7A1 , via the induction of the regulatory nuclear receptor LRH-1 , FXR represses the induction of Cyp7A1 via the upregulation of mRNA encoding SHP, a further nuclear receptor that is dominant repressive over LRH-1.
- FXR binds the end products of this pathway, primary bile acids such as cholic acid (CA) or CDCA, this can be regarded as an example of feedback inhibition on the gene expression level (B. Goodwin et al., Mol. Cell 2000, 6, 517; T. T. Lu et al., Mol. Cell 2000, 6, 507).
- CA cholic acid
- CDCA CDCA
- ABC ATP-binding cassette
- MRP-2 (ABCC4)
- MDR-3 (ABCB4)
- FXR farnesoid receptor
- FXR seems to be the major metabolite sensor and regulator for the synthesis, export and re-circulation of bile acids
- FXR ligands to induce bile flow and change bile acid composition towards more hydrophilic composition.
- This hepatoprotective effect was further narrowed down to an anti-fibrotic effect that results from the repression of Tissue Inhibitors of Matrix-Metalloproteinases, TIMP-1 and 2, the induction of collagen-deposit resolving Matrix- Metalloproteinase 2 in hepatic stellate cells and the subsequent reduction of alpha-collagen mRNA and Transforming growth factor beta (TGF-beta) mRNA which are both pro-fibrotic factors by FXR agonists (S. Fiorucci et al., Gastroenterology 2004, 127, 1497; S. Fiorucci et al., J. Pharmacol. Exp. Ther. 2005, 314, 584).
- TGF-beta Transforming growth factor beta
- FXR binding compounds will demonstrate substantial clinical utility in the therapeutic regimen of chronic cholestatic conditions such as Primary Biliary Cirrhosis (PBC) or Primary Sclerosing Cholangitis (PSC) (reviewed in: G. Rizzo et al., Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 289; G. Zollner et al., Mol. Pharm. 2006, 3, 231 ; S. Y. Cai et al., Expert Opin. Ther. Targets 2006, 10, 409).
- PBC Primary Biliary Cirrhosis
- PSC Primary Sclerosing Cholangitis
- FXR activation has on bile acid metabolism and excretion is not only relevant for cholestatic syndromes but even more directly for a therapy against gallstone formation.
- FXR polymorphisms map as quantitative trait loci as one factor contributing to gallstone disease (H. Wittenburg, Gastroenterology 2003, 125, 868).
- FXR small molecule agonists that can be used to prevent cholesterol gallstone formation or to prevent re-formation of gallstones after surgical removal or Shockwave lithotripsy (discussed in: S. A. Doggrell, Curr. Opin. Investig. Drugs 2006, 7, 344).
- the compound according to Formula (1) and pharmaceutical compositions comprising said compound is used for the prophylaxis and/or treatment of obstructive or chronic inflammatory disorders that arise out of improper bile composition such as cholelithiasis also known as cholesterol gallstones.
- the compounds according to the invention and pharmaceutical compositions comprising said compounds are used in the treatment of liver diseases such as HCC, stimulation of liver regrowth and amelioration of side effects associated with major liver resection, liver cirrhosis independent of the etiology and prevention or treatment of liver ischemia in the course of liver transplantation or major liver surgery.
- Urizar et al. Science 2002, 296, 1703; G. Lambert et al., J. Biol. Chem. 2003, 278, 2563; M. Watanabe et al., J. Clin. Invest. 2004, 113, 1408; A. Figge et al., J. Biol. Chem. 2004, 279, 2790; S. Bilz et al., Am. J. Physiol. Endocrinol. Metab. 2006, 290, E716).
- FXR binding compounds are thought to be good candidates for the treatment of Type II Diabetes because of their insulin sensitization, glycogenogenic, and lipid lowering effects.
- the compounds according to the invention and pharmaceutical compositions comprising said compounds are used in the prophylaxis and/or treatment of Type II Diabetes which can be overcome by FXR-mediated upregulation of systemic insulin sensitivity and intracellular insulin signalling in liver, increased peripheral glucose uptake and metabolisation, increased glycogen storage in liver, decreased output of glucose into serum from liver-borne gluconeogenesis.
- said compounds and pharmaceutical compositions are used for the prophylaxis and/or treatment of chronic intrahepatic, such as PBC, PSC, progressive familiar cholestasis (PFIC), alcohol-induced cirrhosis and associated cholestasis, and some forms of extrahepatic cholestatic conditions, or liver fibrosis.
- chronic intrahepatic such as PBC, PSC, progressive familiar cholestasis (PFIC), alcohol-induced cirrhosis and associated cholestasis, and some forms of extrahepatic cholestatic conditions, or liver fibrosis.
- the invention also relates to a compound of Formula (1) or to a pharmaceutical composition comprising said compound for the prophylaxis and/or treatment of gastrointestinal conditions with a reduced uptake of dietary fat and fat-soluble dietary vitamins which can be overcome by increased intestinal levels of bile acids and phospholipids.
- said compound or pharmaceutical composition is used for preventing and/or treating a disease selected from the group consisting of lipid and lipoprotein disorders such as hypercholesterolemia, hypertriglyceridemia, and atherosclerosis as a clinically manifest condition which can be ameliorated by FXR ' s beneficial effect on lowering total plasma cholesterol, lowering serum triglycerides, increasing conversion of liver cholesterol into bile acids and increased clearance and metabolic conversion of VLDL and other lipoproteins in the liver.
- lipid and lipoprotein disorders such as hypercholesterolemia, hypertriglyceridemia, and atherosclerosis
- said compound and pharmaceutical composition are used for the prophylaxis and/or treatment of diseases where the combined lipid lowering, anti-cholestatic and anti-fibrotic effects of FXR-targeted medicaments can be exploited for the treatment of liver steatosis and associated syndromes such as NASH, or for the treatment of cholestatic and fibrotic effects that are associated with alcohol-induced cirrhosis, or with viral-borne forms of hepatitis.
- FXR agonists might have clinical utility as anti-atherosclerotic and cardioprotective drugs.
- the downregulation of Endothelin-1 in Vascular Smooth Muscle Cells might also contribute to such beneficial therapeutic effects (F. He et al., Circ. Res. 2006, 98, 192).
- the invention also relates to a compound according to Formula (1) or a pharmaceutical composition comprising said compound for preventive and posttraumatic treatment of cardiovascular disorders such as acute myocardial infarction, acute stroke, or thrombosis which occur as an endpoint of chronic obstructive atherosclerosis.
- cardiovascular disorders such as acute myocardial infarction, acute stroke, or thrombosis which occur as an endpoint of chronic obstructive atherosclerosis.
- FXR seems to be expressed in breast cancer tissue and cell lines but not in healthy breast tissue and seems to interact with the Estrogen Receptor in ER positive breast cancer cells (K. E. Swales et al., Cancer Res. 2006, 66, 10120 and F. Journe et al., Breast Cancer Res. Treat. 2009, 1 15, 523).
- FXR also as a potential target for the treatment of proliferative diseases, especially metastasizing cancer forms that express a small molecule responsive form of FXR.
- said compounds and pharmaceutical compositions are used for the prophylaxis and/or treatment of malignant hyperproliferative disorders such as different forms of cancer, specifically certain forms of breast, liver or colon cancer where interference with an FXR ligand will have a beneficial impact.
- FXR seems also to be involved in the control of antibacterial defense in the intestine (T. Inagaki et al., PNAS. 2006, 103, 3920) although an exact mechanism is not provided. From these published data, however, one can conclude that treatment with FXR agonists might have a beneficial impact in the therapy of Inflammatory Bowel Disorders (IBD), in particular those forms where the upper (ileal) part of the intestine is affected (e.g. ileal Crohn ' s disease) because this seems to be the site of action of FXR ' s control on bacterial growth. In IBD the desensitization of the adaptive immune response is somehow impaired in the intestinal immune system.
- IBD Inflammatory Bowel Disorders
- the invention also relates to a compound according to Formula (1) or a pharmaceutical composition comprising said compound for preventing and/or treating a disease related to Inflammatory Bowel Diseases such as Crohn ' s disease or Colitis ulcerosa.
- FXR-mediated restoration of intestinal barrier function and reduction in non-commensal bacterial load is believed to be helpful in reducing the exposure of bacterial antigens to the intestinal immune system and can therefore reduce inflammatory responses.
- the invention further relates to a compound or pharmaceutical composition for the prophylaxis and/or treatment of obesity and associated disorders such as metabolic syndrome (combined conditions of dyslipidemias, diabetes and abnormally high body-mass index) which can be overcome by FXR-mediated lowering of serum triglycerides, blood glucose and increased insulin sensitivity and FXR-mediated weight loss.
- metabolic syndrome combined conditions of dyslipidemias, diabetes and abnormally high body-mass index
- the compounds or pharmaceutical composition of the present invention are useful in preventing and/or treating clinical complications of Type I and Type II Diabetes.
- Such complications include Diabetic Nephropathy, Diabetic Retinopathy, Diabetic Neuropathies, or Peripheral Arterial Occlusive Disease (PAOD).
- PAOD Peripheral Arterial Occlusive Disease
- Other clinical complications of Diabetes are also encompassed by the present invention.
- conditions and diseases which result from chronic fatty and fibrotic degeneration of organs due to enforced lipid and specifically triglyceride accumulation and subsequent activation of profibrotic pathways may also be prevented and/or treated by applying the compounds or pharmaceutical composition of the present invention.
- Such conditions and diseases encompass NASH and chronic cholestatic conditions in the liver, Glomerulosclerosis and Diabetic Nephropathy in the kidney, Macula Degeneration and Diabetic Retinopathy in the eye and Neurodegenerative diseases such as Alzheimer ' s Disease in the brain, or Diabetic Neuropathies in the peripheral nervous system.
- the compounds of the present invention can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous).
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as, for example, suspensions, elixirs and solutions; or carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparations. Because of their ease of administration, tablets and capsules represent the most advantageous oral dosage unit form in which case solid pharmaceutical carriers are obviously employed.
- tablets may be coated by standard aqueous or non-aqueous techniques.
- Such compositions and preparations should contain at least 0.1 percent of active compound.
- the percentage of active compound in these compositions may, of course, be varied and may conveniently be between about 2 percent to about 60 percent of the weight of the unit.
- the amount of active compound in such therapeutically useful compositions is such that an effective dosage will be obtained.
- the active compounds can also be administered intranasally as, for example, liquid drops or spray.
- the tablets, pills, capsules, and the like may also contain a binder such as gum tragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin.
- a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
- tablets may be coated with shellac, sugar or both.
- a syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor.
- the compounds of the present invention may also be used as salts with various countercations to yield an orally available formulation.
- Such pharmaceutically acceptable cations may be amongst others mono- or bivalent ions such as ammonium, the alkaline metals sodium or potassium or the alkaline earth metals magnesium or calcium, certain pharmaceutically acceptable amines such as tris(hydroxymethyl)aminomethane, ethylendiamine, diethylamine, piperazine or others, or certain cationic amino acids such as lysine or arginine.
- the compounds of the present invention may also be administered parenterally. Solutions or suspensions of these active compounds can be prepared in water suitably mixed with a surfactant such as hydroxy-propylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form must be sterile and must be fluid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g., glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
- Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dose of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed.
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
- compounds of the present invention are administered orally.
- the effective dosage of active ingredient employed may vary depending on the particular compound employed, the mode of administration, the condition being treated and the severity of the condition being treated. Such dosage may be ascertained readily by a person skilled in the art.
- the compounds of the present invention are administered at a daily dosage of from about 0.1 milligram to about 100 milligram per kilogram of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 milligrams to about 1000 milligrams, preferably from about 1 milligram to about 50 milligrams.
- the total daily dose will generally be from about 7 milligrams to about 350 milligrams. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds of the present invention can be prepared according to the procedures of the following Schemes and Examples, using appropriate materials and are further exemplified by the following specific examples. Moreover, by utilizing the procedures described herein, in conjunction with ordinary skills in the art, additional compounds of the present invention claimed herein can be readily prepared.
- the compounds illustrated in the examples are not, however, to be construed as forming the only genus that is considered as the invention.
- the examples further illustrate details for the preparation of the compounds of the present invention. Those skilled in the art will readily understand that known variations of the conditions and processes of the following preparative procedures can be used to prepare these compounds.
- the instant compounds are generally isolated in the form of their pharmaceutically acceptable salts, such as those described above.
- the amine-free bases corresponding to the isolated salts can be generated by neutralization with a suitable base, such as aqueous sodium hydrogen carbonate, sodium carbonate, sodium hydroxide and potassium hydroxide, and extraction of the liberated amine-free base into an organic solvent, followed by evaporation.
- a suitable base such as aqueous sodium hydrogen carbonate, sodium carbonate, sodium hydroxide and potassium hydroxide
- the amine-free base, isolated in this manner can be further converted into another pharmaceutically acceptable salt by dissolution in an organic solvent, followed by addition of the appropriate acid and subsequent evaporation, precipitation or crystallization.
- the carboxylic free acids corresponding to the isolated salts can be generated by neutralization with a suitable acid, such as aqueous hydrochloric acid, sodium hydrogen sulfate, sodium dihydrogen phosphate, and extraction of the liberated carboxylic-free acid into an organic solvent, followed by evaporation.
- a suitable acid such as aqueous hydrochloric acid, sodium hydrogen sulfate, sodium dihydrogen phosphate
- the carboxylic acid, isolated in this manner can be further converted into another pharmaceutically acceptable salt by dissolution in an organic solvent, followed by addition of the appropriate base and subsequent evaporation, precipitation or crystallization.
- Step 1 4-((4-Bromo-3-chlorophenoxy)methvn-5-cvclopropyl-3-(2,6-dichlorophenyl)-isoxazole (la)
- the filtrate was concentrated to give the crude product as a pale yellow oil.
- Step 3 Methyl 3-(3-(2-chloro-4-((5-cvclopropyl-3-(2.6-dichlorophenvnisoxazol-4- yl)methoxy)phenyl)-3-hvdroxycvclobutyl)benzoate (1 )
- A/,AADimethylacetamide (9.0 g, 103 mmol) was dissolved in 1 ,2-dichloroethane (200 mL). The solution was cooled to 0°C before trifluoromethanesulfonic anhydride (63 g, 223 mmol) was added. The reaction was stirred for an additional 60 min at 0°C. Then 1-bromo-3-vinylbenzene (15 g, 81.9 mmol) and 2,4,6-collidine (10.5 g, 86.6 mmol) were added. The reaction was heated to reflux overnight, quenched by addition of water (300 mL) and stirred for 2 hr at rt.
- Step 2 3-(3-Bromophenyl)-1-(2-chloro-4-((5-cvclopropyl-3- 2,6-dichlorophenyl)isoxazol-4- yl)methoxy)phenyl)cvclobutanol (2b)
- Step 3 3-(3-Cvanophenyl)-1 -(2-chloro-4-((5-cvclopropyl-3-(2,6-dichlorophenyl)isoxazol-4- vOmethoxy)phenvOcvclobutanol (2c)
- Step 4 3-((1s.3s)-3-(2-Chloro-4-((5-cvclopropyl-3-(2.6-dichlorophenyl)isoxazol-4- yl)methoxy)phenyl)-3-hvdroxycvclobutyl)benzoic acid (2)
- Step 1 3-(3-(Benzylthio)phenyl)-1 -(2-chloro-4-((5-cvclopropyl-3-(2.6-dichlorophenvnisoxazol-4- yl)methoxy)phenyl)cvclobutanol (5a)
- Step 2 3-(3-(2-Chloro-4-((5-cvclopropyl-3-(2.6-dichlorophenyl)isoxazol-4-yl)methoxy)phenyl)-3- hvdroxycvclobutyl)benzene-1 -sulfonyl chloride (5b)
- compound 5a 34 mg, 0.05 mmol
- CH 3 CN/HOAc/H 2 0 1 mL/37 ⁇ _/25 ⁇
- 2,4-dichloro-5,5-dimethylhydantion (20 mg, 0.1 mmol). The mixture was stirred at 0- 5°C for 2 h.
- Step 3 3-(3-(2-Chloro-4-((5-cvclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl)methoxy)phenyl)-3- hydroxycvclobutvObenzenesulfonamide (5)
- Step 3 Methyl 5-f(1s.3s)-3-(2-chloro-4-((5-cvclODropyl-3-(2.6-dichlorophenvnisoxazol-4- yl)methoxy)phenyl)-3-hvdroxycvclobutyl)- 1 -isopropyl- 1 H-pyrazole-3-carboxylate (7)
- Example 8 5-((1s,3s)-3-(2-Chloro-4-((5-cvclopropyl-3-(2.6-dichlorophenyl)isoxazol-4- yl)methoxy)phenyl)-3-hvdroxycvclobutyl)-1 -isopropyl- 1 H-pyrazole-3-carboxylic acid (8)
- Ester 7 (98.3 mg, 0.156 mmol) was dissolved in a mixture of THF (7.5 mL), MeOH (2.5 mL) and water (2.5 mL) and LiOH H 2 0 (65 mg, 1.56 mmol) was added at rt. The mixture was stirred at rt for 18 h. The mixture was partitioned between diluted aq. NH 4 CI solution and EA and the organic layer was washed once with water. The combined aq. layer was extracted twice with EA. The combined organic layer was dried (Na 2 S0 4 ), filtered and concentrated to give 103 mg of an almost white solid.
- Step 5 4-((4-Bromo-3-chlorophenoxy)methyl)-5-cvclopropyl-3-(2,6-dichlorophenyl)isoxazole (8e)
- Step 6 Ethyl 5-((1s.3s)-3-(2-chloro-4-((5-cvclopropyl-3-(2.6-dichlorophenvnisoxazol-4- yl)methoxy)phenyl)-3-hvdroxycvclobutyl)-1-isopropyl-1 H-pyrazole-3-carboxylate (8f)
- Step 7 5-((1s.3s)-3-(2-Chloro-4-((5-cvclopropyl-3-(2.6-dichlorophenyl)isoxazol-4- yl)methoxy)phenyl)-3-hvdroxycvclobutylH-isopropyl-1 H-pyrazole-3-carboxylic acid (8)
- Example 8A 5-((1 r.3r)-3-(2-Chloro-4-((5-cvclopropyl-3-(2.6-dichlorophenylVisoxazol-4- yl)methoxy)phenyl)-3-hvdroxycvclobutylH -isopropyl-1 H-pyrazole-3-carboxylate (8A)
- Example 8A can be prepared by subjecting the crude product 8f to the ester hydrolysis as described for 8 and isolation from the crude product 8 as a minor isomer by preparative RP- HPLC.
- ⁇ -NMR (CDCI 3 ), ⁇ (ppm): 7.42-7.30 (m, 2H), 7.11 (d, J 8.0 Hz, 1 H), 6.75-6.65 (m, 1 H), 6.57 (s.
- Step 3 Methyl 6-(3-(2-chloro-4-((5-cvclopropyl-3-(2.6-dichlorophenyl)isoxazol-4- yl)methoxy)phenyl)-3-hvdroxycvclobutyl)-1-methyl-1 H-indazole-3-carboxylate (9)
- Determination of a ligand mediated cofactor peptide interaction to quantify ligand binding to the nuclear receptor FXR was performed as follows: Preparation of human FXR alpha ligand binding domain: The human FXRalpha LBD was expressed in E coli strain BL21(DE3) as an N- terminally GST tagged fusion protein. The DNA encoding the FXR ligand binding domain was cloned into vector pDEST15 (Invitrogen). Expression was under control of an IPTG inducible T7 promoter. The amino acid boundaries of the ligand binding domain were amino acids 187-472 of Database entry NM_005123 (RefSeq).
- Glutathione 4B sepharose beads were pelleted by centrifugation (2000 x g, 15 sec, 4°C) and washed twice in wash buffer (25 mM Tris, 50 mM KCI, 4 mM MgCI 2 and 1 M NaCI). The pellet was resuspended in 3 mL elution buffer per liter of original culture (elution buffer: 20 mM Tris, 60 mM KCI, 5 mM MgCI 2 and 80 mM glutathione added immediately prior to use as powder). The suspension was left rotating for 15 min at 4°C, the beads pelleted and eluted again with half the volume of elution buffer than the first time.
- the eluates were pooled and dialysed overnight in 20 mM Hepes buffer (pH 7.5) containing 60 mM KCI, 5 mM MgCI 2 as well as 1 mM dithiothreitol and 10% (v/v) glycerol.
- the protein was analysed by SDS- Page.
- the method measures the ability of putative ligands to modulate the interaction between the purified bacterial expressed FXR ligand binding domain (LBD) and a synthetic biotinylated peptide based on residues 676-700 of SRC-1 (LCD2, 676-700).
- the sequence of the peptide used was B-CPSSHSSLTERHKILHRLLQEGSPS-COOH where the N-terminus was biotinylated (B).
- the ligand binding domain (LBD) of FXR was expressed as fusion protein with GST in BL- 21 cells using the vector pDEST15. Cells were lysed by sonication, and the fusion proteins purified over glutathione sepharose (Pharmacia) according to the manufacturers instructions.
- the Perkin Elmer LANCE technology was applied. This method relies on the binding dependent energy transfer from a donor to an acceptor fluorophor attached to the binding partner of interest.
- This method relies on the binding dependent energy transfer from a donor to an acceptor fluorophor attached to the binding partner of interest.
- For ease of handling and reduction of background from compound fluorescence LANCE technology makes use of generic fluorophore labels and time resolved detection Assays were done in a final volume of 25 pL in a 384 well plate, in a Tris-based buffer (20 mM Tris-HCI pH 7.5; 60 mM KCI, 5 mM MgCI 2 ; 35 ng/pL BSA), containing 20-60 ng well recombinantly expressed FXR-LBD fused to GST, 200-600 nM N-terminally biotinylated peptide, representing SRC1 aminoacids 676-700, 200 ng/well Streptavidin-xlAPC conjugate(Prozyme)
- DMSO content of the samples was kept at 1%.
- the assay was equilibrated for 1 h in the dark at rt in FIA-plates black 384 well (Greiner).
- the LANCE signal was detected by a Perkin Elmer VICTOR2VTM Multilabel Counter. The results were visualized by plotting the ratio between the emitted light at 665 and 615 nm. A basal level of FXR-peptide formation is observed in the absence of added ligand. Ligands that promote the complex formation induce a concentration-dependent increase in time-resolved fluorescent signal.
- Mammalian one hybrid (M1H) assay Determination of a ligand mediated Gal4 promoter driven transactivation to quantify ligand binding mediated activation of FXR was performed as follows: The cDNA part encoding the FXR ligand binding domain was cloned into vector pCMV-BD (Stratagene) as a fusion to the yeast GAL4 DNA binding domain under the control of the CMV promoter. The amino acid boundaries of the ligand binding domain were amino acids 187-472 of Database entry NM_005123 (RefSeq).
- the plasmid pFR-Luc (Stratagene) was used as the reporter plasmid, containing a synthetic promoter with five tandem repeats of the yeast GAL4 binding sites, driving the expression of the Photinus pyralis (American firefly) luciferase gene as the reporter gene.
- the plasmid pRL-CMV (Promega) was cotransfected.
- pRL-CMV contains the constitutive CMV promoter, controlling the expression of the Renilla reniformis luciferase.
- Gal4 reporter gene assays were done in HEK293 cells (obtained from DSMZ, Braunschweig, Germany) grown in MEM with L-Glutamine and Earle's BSS supplemented with 10% fetal bovine serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 100 units Penicilin/Streptavidin per ml_ at 37°C in 5% C0 2 . Medium and supplements were obtained from Invitrogen.
- 5 x 10 5 cells were plated per well in 96well plates in 100 ⁇ _ per well MEM without Phenol Red and L-Glutamine and with Earle's BSS supplemented with 10% charcoal/dextran treated FBS (HyClone, South Logan, Utah), 0.1 mM nonessential amino acids, 2 mM glutamine, 1 mM sodium pyruvate, and 100 units Penicilin/ Streptavidin per mL, incubated at 37 °C in 5% C0 2 . The following day the cells were >90% confluence.
- OptiMEM polyethylene-imine-based transfection-reagent
- MEM with the same composition as used for plating cells was added 2-4 h after addition of transfection mixture. Then compound stocks, prediluted in MEM were added (final vehicle concentration not exceeding 0.1%). Cells were incubated for additional 16 h before firefly and renilla luciferase activities were measured sequentially in the same cell extract using a Dual- Light-Luciferase- Assay system (Dyer et al., Anal. Biochem. 2000, 282, 158-161). All experiments were done in triplicates.
- the aq. solubility in PBS, pH 7.4 was determined as follows. A 10 mM compound stock solution in DMSO was added to PBS (pH 7.4) to reach a theoretical final concentration of 200 ⁇ . The resulting solution/suspension was shaken at 1250 rpm for 1 h and then stored in the dark at rt for 23 h. At this time any precipitate is separated from the solution by centrifugation at 3900 rpm for 30 min. The aq. solubility was determined by comparing the peak area of the principle peak in a calibration standard (200 ⁇ ) in an organic solvent (methanol/water 60:40, v/v) with the peak area of the corresponding peak in the buffer sample. As detection method was used HPLC-UV VIS at 230 nm.
- Permeation experiments were carried out in a Multiscreen 96 well tray (donor) covered by a 96-well Multiscreen Immobilon (acceptor).
- the hydrophobic filter material of the Immobilon plate was pre-wetted with 70% ethanol and treated with a solution of lipids (lecithin dissolved in dodecane).
- the donor plate was filled with test compounds and reference compounds and both plates were inserted into each other and placed onto an orbital shaker for 15 min at 100 rpm.
- the transport study was started by applying 150 ⁇ _ PBS-buffer containing the test and reference compounds to the donor plate.
- the contents of the acceptor and donor plate were collected and quantified using LC/MS-detection (test items) or by UV spectroscopy using a Spectramax Plus 384 (Molecular Devices) (reference items).
- the absorption maxima for the reference items ceftriaxone, guanabenz and carbamazepine were 240 nm, 270 nm and 286 nm, respectively.
- Recovery samples were prepared as described for the permeation assay samples and were incubated in representative vials during the permeation period under the same conditions.
- test item samples from the lipid layer were extracted by flushing each well two times with 150 pL EA. The solutions were collected in 1.5 mL reaction tubes and the solvent was evaporated. The dried residues were resuspended in a PBS/DMSO/ACN mixture reflecting the composition of the acceptor and donor samples (i.e. 100 pL buffer supplemented with 5% DMSO, 200 pL ACN+ISTD). The final solvent content of each sample was 66% ACN.
- Samples from donor and acceptor compartments and calibration standards were precipitated by addition of 200 pL ACN/ISTD or 400 pL ACN/ISTD, respectively. After vigorous shaking (10 seconds) and centrifugation (5 min at 4800 x g, rt), the particle free supernatants were subjected to LC-MS/MS. Membrane compartments were extracted as described above. After reconstitution, samples were vigorously shaken (10 seconds) and spun down (5 min at 4800 x g, rt). The particle free supernatants were subjected to LC-MS/MS.
- the HPLC system consisted of an Accela U-HPLC pump and an Accela auto sampler (Thermo Fisher Scientific, USA). Mass spectrometry was performed on an Exactive mass spectrometer (orbitrap technology with accurate mass) equipped with an heated electrospray (H-ESI2) interface (Thermo Fisher Scientific, USA) connected to a PC running the standard software Xcalibur 2.1.
- H-ESI2 heated electrospray
- the LC was performed in the gradient mode (Table 3) using ACN/0.1% formic acid as organic phase (A) and 10 mM ammonium formate/0.1% formic acid as aq. phase (B); and the pump flow rate was set to 500 pL min. Separation was performed on a Gemini C6-Phenyl, 3 pm, 50x2.0 mm (Phenomenex, Germany) analytical column with a pre-column (Gemini C6-Phenyl, 3 pm, 4x2.0 mm).
- Analyte was acquired by scanning ⁇ 1 Thomson around the expected mass of the monoisotopic [M+H] + or [M-H] ⁇ ion.
- the mass resolution of the Orbitrap was set to 50,000.
- the accurate mass of each analyte was used for peak integration. Further instruments settings were as follows: HCD-Gas off, AGC high dynamic range, max. trap injection time 100 ms, sheath gas 30, aux gas 8, sweep gas 2, spray voltage 4 kV, capillary temperature 250°C, ESI 2 heater temperature 250°C.
- the objective of the present invention was to generate FXR-agonists with improved physico- chemical properties compared to compounds claimed in WO 201 1/020615. This was achieved by the introduction of a polar hydroxyl group on a 1 ,3-cyclobutylidene or 1 ,3-azetidinylidene group replacing the former 1 ,2-cyclopropylidene ring.
- aqueous solubility or the PAMPA membrane permeability or both are significantly improved by the introduction of the hydroxy-cyclobutyl or hydroxy-azetidyl moiety.
- FXR agonists are generally very lipophilic (M. L Crawley, Expert Opin. Ther. Patents 2010, 20, 1047). Therefore, better aqeous solubility and membrane permeability are supposed to result in a higher oral bioavailability and in general in a better suitability for clinical development of those compounds as drugs (L. Huang, J. Dong, S. Karki in Evaluation of drug candidates for preclinical development (Eds. C. Han, C. B. Davis, B. Wang), Wiley & Sons, Hoboken 2010, 187-217).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Rheumatology (AREA)
- Gastroenterology & Hepatology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Pain & Pain Management (AREA)
- Cardiology (AREA)
- Child & Adolescent Psychology (AREA)
- Heart & Thoracic Surgery (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Medicinal Preparation (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
































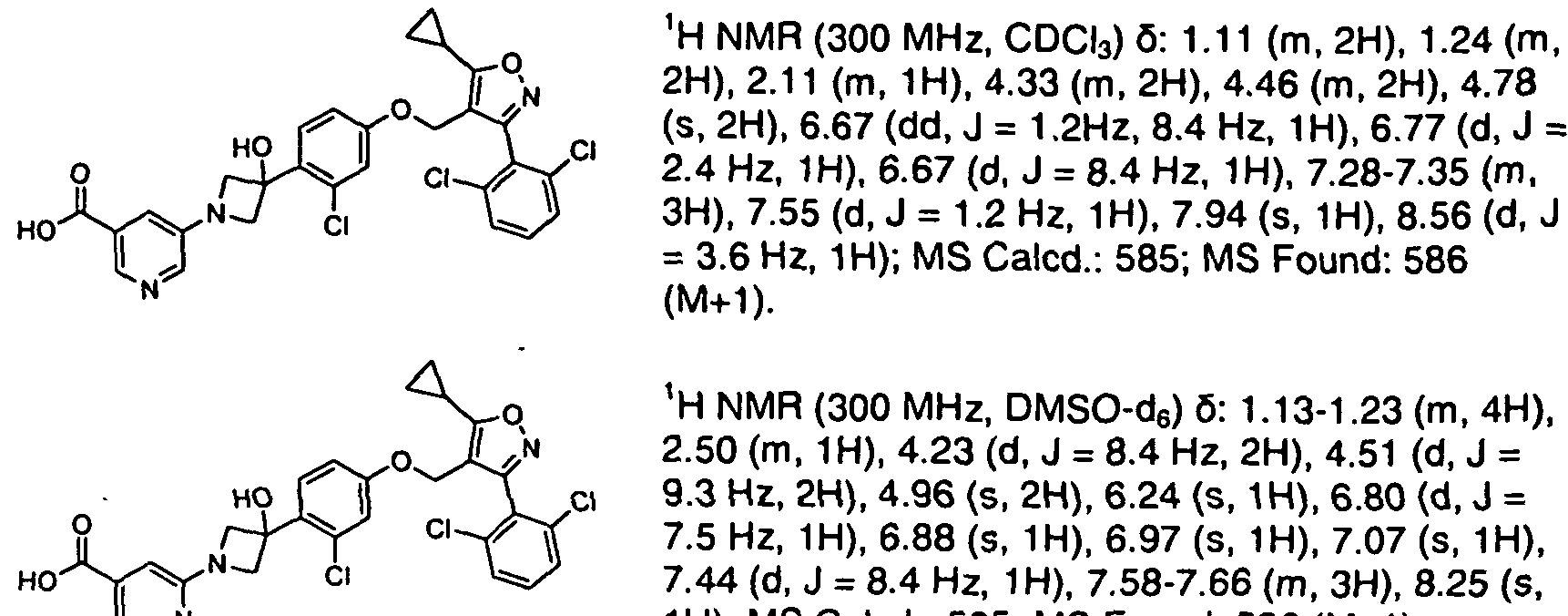









Claims








Priority Applications (39)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2014000534A MX368371B (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds. |
| CA2839357A CA2839357C (en) | 2011-07-13 | 2012-07-12 | Farnesoid x receptor (fxr) binding and activity modulating compounds |
| US14/232,118 US9139539B2 (en) | 2011-07-13 | 2012-07-12 | FXR (NR1H4) binding and activity modulating compounds |
| KR1020177021651A KR101859533B1 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds |
| RS20160266A RS54786B1 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) modulating compounds |
| BR112014000260-6A BR112014000260B1 (en) | 2011-07-13 | 2012-07-12 | FXR AGONIST COMPOUNDS, THEIR USE AND PHARMACEUTICAL COMPOSITION |
| EP12735100.5A EP2731676B1 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) modulating compounds |
| JP2014519453A JP5986633B2 (en) | 2011-07-13 | 2012-07-12 | Novel FXR (NR1H4) binding and activity modulating compounds |
| AU2012283387A AU2012283387C1 (en) | 2011-07-13 | 2012-07-12 | Novel FXR (NR1H4) binding and activity modulating compounds |
| EP17000383.4A EP3246070B1 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating azoles |
| SI201230535A SI2731676T1 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) modulating compounds |
| EA201391674A EA024843B1 (en) | 2011-07-13 | 2012-07-12 | Farnesoid x receptor (nr1h4) binding and activity modulating compounds |
| MEP-2016-81A ME02434B (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds |
| KR1020187013304A KR101934335B1 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds |
| DK12735100.5T DK2731676T3 (en) | 2011-07-13 | 2012-07-12 | New FXR (NR1H4) modulating compounds |
| BR122020008872-9A BR122020008872B1 (en) | 2011-07-13 | 2012-07-12 | Use of a compound in the treatment of diseases and/or conditions by binding said nuclear receptor |
| BR122019026062-1A BR122019026062B1 (en) | 2011-07-13 | 2012-07-12 | COMPOUNDS 3- (4 - ((3- (PIRIDIN-4-IL) ISOXAZOL-4-IL) METOXI) PHENYL) -3-HYDROXY-AZETIDIN-1-IL, 3- (4 - ((1- (PIRIDIN- ) 4-IL) -1H-1,2,3-TRIAZOL-5-IL) METOXI) PHENYL) -3-HYDROXY-AZETIDIN-1-IL, 3- (4 - ((4- (PIRIDIN-4-IL ) -1H-1,2,3-TRIAZOL-5-IL) METOXI) PHENYL) -3-HYDROXY-AZETIDIN-1-IL, E 3- (4 - ((1- (PIRIDIN-4-IL) -1H -PIRAZOL-5-IL) METOXI) PHENYL) -3-HYDROXY-AZETIDIN- 1-IL REPLACED AND OXIDES OF THE SAME |
| EP24164624.9A EP4400503A3 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds |
| ES12735100.5T ES2569718T3 (en) | 2011-07-13 | 2012-07-12 | New compounds that modulate the activity of FXR (NR1H4) |
| CN201280033148.4A CN103702719B (en) | 2011-07-13 | 2012-07-12 | New FXR(NR1H4)With reference to and activity modulating compounds |
| KR1020177000763A KR101766323B1 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds |
| PL12735100T PL2731676T3 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) modulating compounds |
| NZ620177A NZ620177B2 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds |
| KR1020147002937A KR101722410B1 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds |
| IL229944A IL229944A (en) | 2011-07-13 | 2013-12-17 | Fxr (nr1h4) binding and activity modulating compounds |
| ZA2013/09521A ZA201309521B (en) | 2011-07-13 | 2013-12-17 | Novel fxr (nr1h4)binding and activity modulating compounds |
| HK14105521.1A HK1192181A1 (en) | 2011-07-13 | 2014-06-11 | Novel fxr (nr1h4) modulating compounds fxr(nr1h4) |
| US14/824,971 US9539244B2 (en) | 2011-07-13 | 2015-08-12 | FXR (NR1H4) binding and activity modulating compounds |
| HRP20160442TT HRP20160442T1 (en) | 2011-07-13 | 2016-04-25 | Novel fxr (nr1h4) modulating compounds |
| SM201600169T SMT201600169B (en) | 2011-07-13 | 2016-06-10 | NEW COMPOUNDS FOR FXR MODULATION (NR1 H4) |
| AU2016265993A AU2016265993B2 (en) | 2011-07-13 | 2016-11-29 | Novel FXR (NR1H4) binding and activity modulating compounds |
| US15/369,521 US9820979B2 (en) | 2011-07-13 | 2016-12-05 | FXR (NR1H4) binding and activity modulating compounds |
| IL253437A IL253437A0 (en) | 2011-07-13 | 2017-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds |
| US15/783,530 US10220027B2 (en) | 2011-07-13 | 2017-10-13 | FXR (NR1H4) binding and activity modulating compounds |
| AU2018203613A AU2018203613B2 (en) | 2011-07-13 | 2018-05-22 | Novel FXR (NR1H4) binding and activity modulating compounds |
| US16/248,178 US10485795B2 (en) | 2011-07-13 | 2019-01-15 | FXR (NR1H4) binding and activity modulating compounds |
| IL265973A IL265973B (en) | 2011-07-13 | 2019-04-11 | Novel fxr (nr1h4) binding and activity modulating compounds |
| AU2019261667A AU2019261667B2 (en) | 2011-07-13 | 2019-11-04 | Novel FXR (NR1H4) binding and activity modulating compounds |
| IL277570A IL277570A (en) | 2011-07-13 | 2020-09-24 | Novel fxr (nr1h4) binding and activity modulating compounds |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161507153P | 2011-07-13 | 2011-07-13 | |
| EP11005722A EP2545964A1 (en) | 2011-07-13 | 2011-07-13 | Novel FXR (NR1H4) binding and activity modulating compounds |
| EP11005722.1 | 2011-07-13 | ||
| US61/507,153 | 2011-07-13 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/232,118 A-371-Of-International US9139539B2 (en) | 2011-07-13 | 2012-07-12 | FXR (NR1H4) binding and activity modulating compounds |
| US14/824,971 Continuation US9539244B2 (en) | 2011-07-13 | 2015-08-12 | FXR (NR1H4) binding and activity modulating compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013007387A1 true WO2013007387A1 (en) | 2013-01-17 |
Family
ID=44513243
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2012/002941 WO2013007387A1 (en) | 2011-07-13 | 2012-07-12 | Novel fxr (nr1h4) binding and activity modulating compounds |
Country Status (31)
| Country | Link |
|---|---|
| US (5) | US9139539B2 (en) |
| EP (5) | EP2545964A1 (en) |
| JP (2) | JP5986633B2 (en) |
| KR (4) | KR101722410B1 (en) |
| CN (2) | CN107252424B (en) |
| AR (1) | AR087127A1 (en) |
| AU (4) | AU2012283387C1 (en) |
| BR (3) | BR122019026062B1 (en) |
| CA (1) | CA2839357C (en) |
| CY (1) | CY1117453T1 (en) |
| DK (2) | DK3246070T3 (en) |
| EA (1) | EA024843B1 (en) |
| ES (3) | ES2632492T3 (en) |
| FI (1) | FI3246070T3 (en) |
| HK (2) | HK1192181A1 (en) |
| HR (2) | HRP20240422T1 (en) |
| HU (2) | HUE027931T2 (en) |
| IL (1) | IL229944A (en) |
| LT (1) | LT3246070T (en) |
| ME (1) | ME02434B (en) |
| MX (2) | MX368371B (en) |
| MY (1) | MY161158A (en) |
| PL (3) | PL2731676T3 (en) |
| PT (2) | PT2987532T (en) |
| RS (1) | RS54786B1 (en) |
| SI (3) | SI2987532T1 (en) |
| SM (1) | SMT201600169B (en) |
| TW (1) | TWI439455B (en) |
| UY (1) | UY34196A (en) |
| WO (1) | WO2013007387A1 (en) |
| ZA (1) | ZA201309521B (en) |
Cited By (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3006939A1 (en) | 2014-10-06 | 2016-04-13 | Gilead Sciences, Inc. | Histidine-rich Glycoprotein as a marker for hepatic Farnesoid X receptor activation |
| WO2016086218A1 (en) | 2014-11-26 | 2016-06-02 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as fxr/tgr5 agonists and methods of use thereof |
| EP3034501A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| EP3034499A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Novel FXR (NR1H4) modulating compounds |
| WO2016127924A1 (en) | 2015-02-13 | 2016-08-18 | Sunshine Lake Pharma Co., Ltd. | Tricyclic compounds and uses thereof in medicine |
| CN106588804A (en) * | 2016-12-09 | 2017-04-26 | 都创(上海)医药科技有限公司 | Preparation method of compound serving as farnesoid X receptor (FXR) |
| WO2017201150A1 (en) * | 2016-05-18 | 2017-11-23 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as fxr agonists and methods of use thereof |
| WO2017218337A1 (en) | 2016-06-13 | 2017-12-21 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| WO2018153933A1 (en) | 2017-02-21 | 2018-08-30 | Genfit | Combination of a ppar agonist with a fxr agonist |
| US10080742B2 (en) | 2016-04-26 | 2018-09-25 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10080743B2 (en) | 2016-04-26 | 2018-09-25 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10080741B2 (en) | 2016-04-26 | 2018-09-25 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| WO2018178260A1 (en) | 2017-03-30 | 2018-10-04 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for reducing persistence and expression of episomal viruses |
| US20180333401A1 (en) * | 2017-04-12 | 2018-11-22 | Gilead Sciences, Inc. | Methods of treating liver disease |
| US10138228B2 (en) | 2016-05-18 | 2018-11-27 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use therof |
| US10149835B2 (en) | 2016-05-18 | 2018-12-11 | Elmore Patent Law Group, P.C. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| WO2018231851A1 (en) | 2017-06-13 | 2018-12-20 | Gilead Sciences, Inc. | Methods of treating liver fibrosis |
| KR20190017028A (en) * | 2016-06-13 | 2019-02-19 | 길리애드 사이언시즈, 인코포레이티드 | FXR (NR1H4) adjusting compound |
| US10220027B2 (en) | 2011-07-13 | 2019-03-05 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| WO2019094777A1 (en) | 2017-11-13 | 2019-05-16 | Gilead Sciences, Inc. | Compositions and methods for identifying and treating liver diseases and monitoring treatment outcomes |
| WO2019149158A1 (en) | 2018-02-02 | 2019-08-08 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous tricyclic compounds and uses thereof in medicine |
| US10450306B2 (en) | 2016-10-04 | 2019-10-22 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| CN110461328A (en) * | 2017-03-28 | 2019-11-15 | 吉利德科学公司 | The therapeutic combination for treating liver disease |
| JP2019535811A (en) * | 2016-10-22 | 2019-12-12 | へパジーン セラピューティクス インコーポレイテッド | Heterocyclic FXR modulator |
| US10597391B2 (en) | 2016-10-26 | 2020-03-24 | Enanta Pharmaceuticals, Inc. | Urea-containing isoxazole derivatives as FXR agonists and methods of use thereof |
| WO2020065597A1 (en) | 2018-09-28 | 2020-04-02 | Richter Gedeon Nyrt. | Bicyclic derivatives as gabaa α5 receptor modulators |
| US10676500B2 (en) | 2017-04-07 | 2020-06-09 | Enanta Pharmaceuticals, Inc. | Process for preparation of sulfonyl carbamate bile acid derivatives |
| US10689391B2 (en) | 2017-12-12 | 2020-06-23 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| EP3711762A1 (en) | 2013-09-11 | 2020-09-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | A farnesoid x receptor agonsits foruse and pharmaceutical compositions for the treatment of chronic hepatitis b virus infection |
| US10829486B2 (en) | 2018-02-14 | 2020-11-10 | Enanta Pharmacueticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| US10988449B2 (en) | 2017-04-12 | 2021-04-27 | Il Dong Pharmaceutical Co., Ltd. | Isoxazole derivatives as nuclear receptor agonists and uses thereof |
| US11040998B2 (en) | 2015-03-31 | 2021-06-22 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| KR20210114457A (en) * | 2019-01-15 | 2021-09-23 | 길리애드 사이언시즈, 인코포레이티드 | FXR (NR1H4) modulating compound |
| WO2021191837A1 (en) | 2020-03-26 | 2021-09-30 | Richter Gedeon Nyrt. | 1,3-dihydro-2h-pyrrolo[3,4-c]pyridine derivatives as gabaa α5 receptor modulators |
| CN113573700A (en) * | 2019-03-11 | 2021-10-29 | 吉利德科学公司 | Formulations of compounds and uses thereof |
| US11168079B2 (en) | 2017-11-01 | 2021-11-09 | Bristol-Myers Squibb Company | Alkene compounds as farnesoid x receptor modulators |
| US11286252B2 (en) | 2017-11-01 | 2022-03-29 | Bristol-Myers Squibb Company | Alkene spirocyclic compounds as farnesoid X receptor modulators |
| US11370785B2 (en) | 2017-11-01 | 2022-06-28 | Bristol-Myers Squibb Company | Multicyclic compounds as farnesoid X receptor modulators |
| WO2022134146A1 (en) * | 2020-12-22 | 2022-06-30 | 江苏天士力帝益药业有限公司 | Novel fxr agonist having pyrazine structure, and preparation method and use |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| US11478533B2 (en) | 2020-04-27 | 2022-10-25 | Novo Nordisk A/S | Semaglutide for use in medicine |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| WO2022266444A1 (en) | 2021-06-18 | 2022-12-22 | Gilead Sciences, Inc. | Il-31 modulators for treating fxr-induced pruritis |
| US11555032B2 (en) | 2019-05-13 | 2023-01-17 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US11919879B2 (en) | 2021-06-16 | 2024-03-05 | Celgene Corporation | Carboxylic acid containing azetidinyl compounds for the treatment of neurodegenerative diseases |
| WO2024089582A1 (en) | 2022-10-25 | 2024-05-02 | Assia Chemical Industries Ltd. | Solid state forms of cilofexor salts |
| US12030835B2 (en) | 2019-02-15 | 2024-07-09 | Bristol-Myers Squibb Company | Substituted amide compounds useful as farnesoid X receptor modulators |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI598347B (en) | 2009-07-13 | 2017-09-11 | 基利科學股份有限公司 | Apoptosis signal-regulating kinase inhibitors |
| IL253437A0 (en) * | 2011-07-13 | 2017-09-28 | Gilead Sciences Inc | Novel fxr (nr1h4) binding and activity modulating compounds |
| EP3524243A1 (en) | 2014-09-24 | 2019-08-14 | Gilead Sciences, Inc. | Methods of treating liver disease |
| EP3233083A1 (en) * | 2014-12-18 | 2017-10-25 | Novartis AG | Azabicyclooctane derivatives as fxr agonists for use in the treatment of liver and gastrointestinal diseases |
| MA41252A (en) * | 2014-12-23 | 2017-10-31 | Gilead Sciences Inc | SOLID FORMS OF AN ASK 1 INHIBITOR |
| PT3237404T (en) | 2014-12-23 | 2021-01-19 | Gilead Sciences Inc | Processes for preparing ask1 inhibitors |
| KR20240008398A (en) | 2015-07-06 | 2024-01-18 | 길리애드 사이언시즈, 인코포레이티드 | Cot modulators and methods of use thereof |
| CN106995416A (en) * | 2016-01-26 | 2017-08-01 | 上海翰森生物医药科技有限公司 | FXR activators and its preparation method and application |
| WO2017128896A1 (en) * | 2016-01-26 | 2017-08-03 | 江苏豪森药业集团有限公司 | Fxr agonist and preparation method and use thereof |
| TW201741307A (en) * | 2016-02-22 | 2017-12-01 | 艾洛斯生物製藥公司 | FXR modulators and methods of their use |
| WO2017152062A1 (en) | 2016-03-04 | 2017-09-08 | Gilead Sciences, Inc. | Compositions and combinations of autotaxin inhibitors |
| CA3019003A1 (en) | 2016-04-08 | 2017-10-12 | Gilead Sciences, Inc. | Compositions and methods for treating cancer, inflammatory diseases and autoimmune diseases |
| AR108711A1 (en) * | 2016-06-13 | 2018-09-19 | Gilead Sciences Inc | FXR MODULATING COMPOUNDS (NR1H4) |
| TW201808283A (en) | 2016-08-05 | 2018-03-16 | 廣東東陽光藥業有限公司 | Nitrogen-containing tricyclic compounds and uses thereof in medicine |
| CN106237332A (en) * | 2016-08-11 | 2016-12-21 | 河南大学 | Nuclear receptor FXR application in liver-cancer stem cell targeted therapy |
| JP2019533706A (en) * | 2016-11-11 | 2019-11-21 | ギリアード サイエンシーズ, インコーポレイテッド | How to treat liver disease |
| CN108218852A (en) * | 2016-12-15 | 2018-06-29 | 宁波百纳西药业有限公司 | A kind of spiro-compound, preparation method, composition and purposes |
| WO2018133730A1 (en) * | 2017-01-20 | 2018-07-26 | 四川科伦博泰生物医药股份有限公司 | Heterocyclic compound, preparation method and use therefor |
| JOP20180017A1 (en) | 2017-03-14 | 2019-01-30 | Gilead Sciences Inc | Apoptosis signal-regulating kinase inhibitor |
| JP7029583B2 (en) | 2017-05-31 | 2022-03-04 | パナソニックIpマネジメント株式会社 | washing machine |
| CN111182904A (en) | 2017-10-06 | 2020-05-19 | 吉利德科学公司 | Combination therapy comprising ACC inhibitors |
| KR20200081434A (en) | 2017-11-01 | 2020-07-07 | 브리스톨-마이어스 스큅 컴퍼니 | Crosslinked bicyclic compounds as farnesoid X receptor modulators |
| MX2020004400A (en) | 2017-11-01 | 2020-08-06 | Bristol Myers Squibb Co | Spirocyclic compounds as farnesoid x receptor modulators. |
| CN110818704B (en) * | 2018-08-08 | 2023-08-01 | 广州市恒诺康医药科技有限公司 | Spiro bridged ring compounds, pharmaceutical compositions thereof and uses thereof |
| DK3833434T3 (en) | 2018-08-08 | 2022-02-14 | Inorbit Therapeutics Ab | COMPOUNDS WHICH ARE USEFUL IN MODULATING THE FARNESOID-X RECEPTOR, AS WELL AS PROCEDURES FOR MANUFACTURE AND USE |
| BR112021004919A2 (en) | 2018-09-18 | 2021-06-01 | Metacrine, Inc. | farnesoid x receptor agonists and uses thereof |
| AR118050A1 (en) | 2019-02-15 | 2021-09-15 | Bristol Myers Squibb Co | BICYCLIC COMPOUNDS REPLACED AS MODULATORS OF THE FARNESOID X RECEIVER |
| TWI770527B (en) | 2019-06-14 | 2022-07-11 | 美商基利科學股份有限公司 | Cot modulators and methods of use thereof |
| US20230105984A1 (en) | 2019-12-23 | 2023-04-06 | Svetlana Marukian | Compositions and methods for the treatment of liver diseases and disorders |
| CN115666559A (en) * | 2020-03-18 | 2023-01-31 | 梅塔科林公司 | Farnesoin X receptor agonists for the treatment of disease |
| WO2021202224A1 (en) | 2020-03-30 | 2021-10-07 | Gilead Sciences, Inc. | Solid forms of (s)-6-(((1-(bicyclo[1.1.1]pentan-1-yl)-1h-1,2,3-triazol-4-yl)2-methyl-1-oxo-1,2- dihydroisoquinolin-5-yl)methyl)))amino)8-chloro-(neopentylamino)quinoline-3-carb onitrile a cot inhibitor compound |
| US11845737B2 (en) | 2020-04-02 | 2023-12-19 | Gilead Sciences, Inc. | Process for preparing a Cot inhibitor compound |
| WO2022192428A1 (en) | 2021-03-11 | 2022-09-15 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| US20230079863A1 (en) | 2021-03-29 | 2023-03-16 | Gilead Sciences, Inc. | Khk inhibitors |
| TW202304435A (en) | 2021-06-04 | 2023-02-01 | 美商基利科學股份有限公司 | Methods of treating nash |
| WO2023125904A1 (en) * | 2021-12-30 | 2023-07-06 | 苏州晶云药物科技股份有限公司 | Crystal form of azacyclobutyl nicotinic acid compound and preparation method therefor |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000037077A1 (en) | 1998-12-23 | 2000-06-29 | Glaxo Group Limited | Assays for ligands for nuclear receptors |
| WO2003015771A1 (en) | 2001-08-13 | 2003-02-27 | Lion Bioscience Ag | Fxr nr1h4 nuclear receptor binding compounds |
| WO2003080803A2 (en) | 2002-03-21 | 2003-10-02 | Smithkline Beecham Corporation | Methods of using farnesoid x receptor (fxr) agonists |
| WO2004048349A1 (en) | 2002-11-22 | 2004-06-10 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2004087076A2 (en) | 2003-03-31 | 2004-10-14 | The Rockefeller University | Methods for inhibiting adipogenesis and for treating type 2 diabetes |
| WO2007076260A2 (en) | 2005-12-19 | 2007-07-05 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2007092751A2 (en) | 2006-02-03 | 2007-08-16 | Eli Lilly And Company | Compounds and methods for modulating fx-receptors |
| WO2007140174A2 (en) | 2006-05-24 | 2007-12-06 | Eli Lilly And Company | Compounds and methods for modulating fxr |
| WO2007140183A1 (en) | 2006-05-24 | 2007-12-06 | Eli Lilly And Company | Fxr agonists |
| EP1894924A1 (en) * | 2006-08-29 | 2008-03-05 | Phenex Pharmaceuticals AG | Heterocyclic FXR binding compounds |
| WO2008025539A1 (en) | 2006-08-29 | 2008-03-06 | Phenex Pharmaceuticals Ag | Heterocyclic fxr binding compounds |
| WO2008051942A2 (en) | 2006-10-24 | 2008-05-02 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2008157270A1 (en) | 2007-06-13 | 2008-12-24 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2009005998A1 (en) | 2007-07-02 | 2009-01-08 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2009012125A1 (en) | 2007-07-16 | 2009-01-22 | Eli Lilly And Company | Compounds and methods for modulating fxr |
| WO2011020615A1 (en) | 2009-08-19 | 2011-02-24 | Phenex Pharmaceuticals Ag | Novel fxr (nr1h4 ) binding and activity modulating compounds |
Family Cites Families (170)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ193011A (en) | 1979-03-19 | 1983-03-15 | Ici Australia Ltd | Diarylamine derivatives intermediates herbicidal compositions |
| ES2054725T3 (en) | 1987-04-21 | 1994-08-16 | Basf Ag | RING HETEROAROMATES OF FIVE MEMBERS OF P-PHENOXY-PHENOXIMETHYL. |
| JP3121061B2 (en) | 1991-10-04 | 2000-12-25 | 塩野義製薬株式会社 | Method for producing intermediate for producing alkoxyiminoacetamides and intermediate used therein |
| DE4137940A1 (en) | 1991-11-18 | 1993-05-19 | Basf Ag | 3-ISOXAZOLYLPHENYL COMPOUNDS, THEIR PRODUCTION AND THEIR USE |
| US5258551A (en) | 1991-12-18 | 1993-11-02 | Shionogi & Co., Ltd. | Process for producing α-ketoamide derivative |
| WO1994017059A1 (en) | 1993-01-29 | 1994-08-04 | Nippon Soda Co., Ltd. | Heterocyclic derivative |
| WO1994024095A1 (en) | 1993-04-16 | 1994-10-27 | Abbott Laboratories | Immunosuppressive agents |
| IL112721A0 (en) | 1994-03-10 | 1995-05-26 | Zeneca Ltd | Azole derivatives |
| WO1996004274A1 (en) | 1994-08-02 | 1996-02-15 | Merck Sharp & Dohme Limited | Azetidine, pyrrolidine and piperidine derivatives |
| GB9501865D0 (en) | 1995-01-31 | 1995-03-22 | Merck Sharp & Dohme | Therapeutic agents |
| US5633272A (en) | 1995-02-13 | 1997-05-27 | Talley; John J. | Substituted isoxazoles for the treatment of inflammation |
| DE19536811A1 (en) | 1995-10-02 | 1997-04-03 | Basf Ag | Intermediates and processes for the production of substituted salicylic acid derivatives as crop protection agents |
| ATE223732T1 (en) | 1996-02-13 | 2002-09-15 | Searle & Co | DRUG COMBINATIONS WITH IMMUNOSUPPRESSIVE EFFECTS WHICH CONTAIN CYCLOOXYGENASE-2 INHIBITORS AND LEUCOTRIEN LTA4 HYDRASE INHIBITORS |
| AU774538B2 (en) | 1999-06-11 | 2004-07-01 | Allergan, Inc. | Organosilyl compounds having nuclear hormone receptor modulating activity |
| EP1340749A4 (en) | 2000-11-17 | 2007-09-05 | Takeda Pharmaceutical | Isoxazole derivatives |
| US20040105883A1 (en) | 2001-04-17 | 2004-06-03 | Ping Gao | Pharmaceutical dosage form capable of maintaining stable dissolution profile upon storage |
| US20040131670A1 (en) | 2001-04-17 | 2004-07-08 | Ping Gao | Pellicle-resistant gelatin capsule |
| US20040105884A1 (en) | 2001-04-17 | 2004-06-03 | Ping Gao | Pharmaceutical dosage form comprising a sulfite compound |
| US20040105885A1 (en) | 2001-04-17 | 2004-06-03 | Ping Gao | Gelatin capsule exhibiting reduced cross-linking |
| WO2003000249A1 (en) | 2001-06-26 | 2003-01-03 | Takeda Chemical Industries, Ltd. | Function regulator for retinoid relative receptor |
| WO2003016280A1 (en) | 2001-08-13 | 2003-02-27 | Lion Bioscience Ag | Nr1h4 nuclear receptor binding compounds |
| WO2003015777A1 (en) | 2001-08-13 | 2003-02-27 | Lion Bioscience Ag | Nr1h4 nuclear receptor binding compounds |
| US7595311B2 (en) | 2002-05-24 | 2009-09-29 | Exelixis, Inc. | Azepinoindole derivatives as pharmaceutical agents |
| AU2003264018A1 (en) | 2002-08-09 | 2004-02-25 | Astrazeneca Ab | Compounds having an activity at metabotropic glutamate receptors |
| TW200812986A (en) | 2002-08-09 | 2008-03-16 | Nps Pharma Inc | New compounds |
| CN101723941A (en) | 2002-08-09 | 2010-06-09 | 阿斯利康(瑞典)有限公司 | “1,2,4” oxadiazole as a metabotropic glutamate receptor-5 modulator |
| EP1407774A1 (en) | 2002-09-10 | 2004-04-14 | LION Bioscience AG | 2-Amino-4-quinazolinones as LXR nuclear receptor binding compounds |
| AU2003290796A1 (en) | 2002-11-14 | 2004-06-15 | The Scripps Research Institute | Non-steroidal fxr agonists |
| US20050143449A1 (en) | 2002-11-15 | 2005-06-30 | The Salk Institute For Biological Studies | Non-steroidal farnesoid X receptor modulators and methods for the use thereof |
| WO2005077373A2 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Treatment of gastro-esophageal reflux disease (gerd) |
| WO2005077345A1 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Compounds for the treatment of gastro-esophageal reflux disease |
| US7585881B2 (en) | 2004-02-18 | 2009-09-08 | Astrazeneca Ab | Additional heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| CA2564429A1 (en) | 2004-05-14 | 2005-12-01 | Irm Llc | Compounds and compositions as ppar modulators |
| MY144903A (en) | 2004-06-17 | 2011-11-30 | Novartis Ag | Pyrrolopyridine derivatives and their use as crth2 antagonists |
| EP3067053A1 (en) | 2004-10-13 | 2016-09-14 | PTC Therapeutics, Inc. | Compounds for nonsense suppression, and methods for their use |
| JP2008137894A (en) | 2005-03-22 | 2008-06-19 | Nippon Kayaku Co Ltd | New acetylene derivative |
| US8952176B2 (en) | 2005-06-07 | 2015-02-10 | Shionogi & Co., Ltd. | Heterocyclic compound having type I 11 β hydroxysteroid dehydrogenase inhibitory activity |
| RU2008128823A (en) | 2005-12-15 | 2010-01-20 | Экселиксис, Инк. (Us) | AZEPINOINDOL DERIVATIVES AS PHARMACEUTICAL PRODUCTS |
| US7560551B2 (en) | 2006-01-23 | 2009-07-14 | Amgen Inc. | Aurora kinase modulators and method of use |
| EP1987051A2 (en) * | 2006-02-14 | 2008-11-05 | Intercept Pharmaceuticals, Inc. | Bile acid derivatives as fxr ligands for the prevention or treatment of fxr-mediated diseases or conditions |
| EP2007759A4 (en) | 2006-04-17 | 2010-12-22 | Neuromed Pharmaceuticals Ltd | Isoxazole derivatives as calcium channel blockers |
| CN101448798A (en) * | 2006-05-24 | 2009-06-03 | 伊莱利利公司 | Compounds and methods for modulating FXR |
| ATE549338T1 (en) | 2006-05-24 | 2012-03-15 | Boehringer Ingelheim Int | SUBSTITUTED PTERIDINES SUBSTITUTED WITH A FOUR-MEMBER HETEROCYCLE |
| ES2523591T3 (en) | 2006-06-27 | 2014-11-27 | Intercept Pharmaceuticals Inc. | Bile acid derivatives as FXR ligands for the prevention or treatment of diseases or conditions mediated by FXR |
| CA2656290A1 (en) | 2006-07-05 | 2008-01-10 | Exelixis, Inc. | Methods of using igf1r and abl kinase modulators |
| WO2008013660A2 (en) | 2006-07-07 | 2008-01-31 | Biostratum, Inc. | Inhibitors of advanced glycation end products |
| UA95298C2 (en) | 2006-07-07 | 2011-07-25 | Бьёрингер Ингельхайм Интернациональ Гмбх | Phenyl substituted heteroaryl-derivatives and use thereof as anti-tumor agents |
| US8193225B2 (en) | 2006-10-13 | 2012-06-05 | The Board Of Regents Of The University Of Texas System | Isoxazole amides, derivatives and methods of chemical induction of neurogenesis |
| US8501933B2 (en) | 2006-11-09 | 2013-08-06 | Roche Palo Alto Llc | Thiazole and oxazole-substituted arylamides as P2X3 and P2X2/3 antagonists |
| CN101679297B (en) | 2006-12-08 | 2012-01-11 | 埃克塞利希斯股份有限公司 | LXR and FXR modulators |
| GB0625842D0 (en) | 2006-12-22 | 2007-02-07 | Argenta Discovery Ltd | Indolizine derivatives |
| US20090105251A1 (en) | 2007-01-25 | 2009-04-23 | Benjamin Jones | Renin inhibitors |
| WO2008097235A1 (en) | 2007-02-09 | 2008-08-14 | Dow Agrosciences Llc | Process for the oxidation of certain substituted sulfilimines to insecticidal sulfoximines |
| US7511149B2 (en) | 2007-02-09 | 2009-03-31 | Dow Agrosciences Llc | Process for the oxidation of certain substituted sulfilimines to insecticidal sulfoximines |
| EP2114886B1 (en) | 2007-02-26 | 2014-03-26 | Dow AgroSciences LLC | Process for the preparation of certain substituted sulfilimines |
| JP2008308448A (en) | 2007-06-15 | 2008-12-25 | Sankyo Agro Kk | (3-sulfur atom-substituted phenyl)heteroaryl derivative |
| WO2008155054A1 (en) | 2007-06-20 | 2008-12-24 | F. Hoffmann-La Roche Ag | Farnesoid-x-receptor mutants, and crystallisation thereof |
| CA2692379A1 (en) | 2007-07-02 | 2009-01-08 | Boehringer Ingelheim International Gmbh | Antiproliferative compounds based on 5-membered heterocycles |
| US20090197880A1 (en) | 2007-07-13 | 2009-08-06 | Genelabs Technologies, Inc. | Anti-viral compounds, compositions, and methods of use |
| TW200920372A (en) | 2007-07-13 | 2009-05-16 | Genelabs Tech Inc | Anti-viral compounds, compositions, and methods of use |
| MX2010001171A (en) | 2007-08-01 | 2010-03-01 | Lundbeck & Co As H | Use of kncq potassium channel openers for reducing symptoms of or treating disorders or conditions wherein the dopaminergic system is disrupted. |
| US8188080B2 (en) | 2007-10-17 | 2012-05-29 | Sanford-Burnham Medical Research Institute | VHR protein tyrosine phosphatase inhibitors, compositions and methods of use |
| US20090143451A1 (en) | 2007-11-14 | 2009-06-04 | Andrews William H | Compounds that increase telomerase reverse transcriptase (tert) expression and methods for using the same |
| EP2110374A1 (en) | 2008-04-18 | 2009-10-21 | Merck Sante | Benzofurane, benzothiophene, benzothiazol derivatives as FXR modulators |
| EP2283001A2 (en) | 2008-05-13 | 2011-02-16 | Boehringer Ingelheim International GmbH | Sulfone compounds which modulate the cb2 receptor |
| CA2723424A1 (en) | 2008-05-19 | 2009-11-26 | Burnham Institute For Medical Research | Intestinal alkaline phosphatase modulators and uses thereof |
| WO2009143018A2 (en) | 2008-05-19 | 2009-11-26 | Plexxikon, Inc. | Compounds and methods for kinase modulation, and indications therefor |
| EP2128158A1 (en) | 2008-05-26 | 2009-12-02 | Phenex Pharmaceuticals AG | Heterocyclic cyclopropyl-substituted FXR binding compounds |
| JP5767965B2 (en) | 2008-06-10 | 2015-08-26 | プレキシコン インコーポレーテッドPlexxikon Inc. | 5H-pyrrolo [2,3-B] pyrazine derivatives that regulate kinases and indications thereof |
| US8822513B2 (en) | 2010-03-01 | 2014-09-02 | Gtx, Inc. | Compounds for treatment of cancer |
| EP2307370B1 (en) | 2008-06-23 | 2012-01-04 | Basf Se | Sulfoximinamide compounds for combating animal pests |
| WO2010006096A1 (en) | 2008-07-11 | 2010-01-14 | Smithkline Beecham Corporation | Processes for the preparation of anti-viral compounds and compositions containing them |
| WO2010025035A1 (en) | 2008-08-25 | 2010-03-04 | Dow Global Technologies Inc. | Process for preparing isoxazole compounds |
| WO2010033906A2 (en) | 2008-09-19 | 2010-03-25 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| CA2736434A1 (en) | 2008-09-25 | 2010-04-01 | F. Hoffmann-La Roche Ag | 3-amino-indazole or 3-amino-4,5,6,7-tetrahydro-indazole derivatives |
| WO2010034649A1 (en) | 2008-09-25 | 2010-04-01 | F. Hoffmann-La Roche Ag | 2,3-substituted indazole or 4,5,6,7-tetrahydro-indazoles as fxr modulators against dyslipidemia and related diseases |
| WO2010036362A1 (en) | 2008-09-26 | 2010-04-01 | Wyeth | 1,2,3,6-tetrahydroazepino[4,5-b]indole-5-carboxylate nuclear receptor inhibitors |
| BRPI0920237A2 (en) | 2008-10-21 | 2015-12-29 | Metabolex Inc | aril gpr120 receptor agonists and their respective uses |
| EP2393807B1 (en) | 2009-02-04 | 2013-08-14 | Boehringer Ingelheim International GmbH | Cyclic inhibitors of 11 -hydroxysteroid dehydrogenase 1 |
| KR20100092909A (en) | 2009-02-13 | 2010-08-23 | 주식회사 엘지생명과학 | Novel compounds effective as xanthine oxidase inhibitors, method for preparing the same, and pharmaceutical composition containing the same |
| FR2943059A1 (en) | 2009-03-16 | 2010-09-17 | Sanofi Aventis | N-6-AZA-BICYCLO® 3.2.1.0-OCT-5-YL) -ARYL-METHYL-HETEROBENZAMIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE |
| ES2461618T3 (en) | 2009-07-06 | 2014-05-20 | Basf Se | Pyridazine compounds for the control of invertebrate pests |
| US8883832B2 (en) | 2009-07-06 | 2014-11-11 | Aerpio Therapeutics Inc. | Compounds, compositions, and methods for preventing metastasis of cancer cells |
| CN102469785A (en) | 2009-07-24 | 2012-05-23 | 巴斯夫欧洲公司 | Pyridine derivatives compounds for controlling invertebrate pests |
| US9212177B2 (en) | 2009-08-05 | 2015-12-15 | Versitech Limited | Antiviral compounds and methods of making and using thereof |
| WO2011026241A1 (en) | 2009-09-04 | 2011-03-10 | Zalicus Pharmaceuticals Ltd. | Substituted heterocyclic derivatives for the treatment of pain and epilepsy |
| WO2011047129A1 (en) | 2009-10-15 | 2011-04-21 | Southern Research Institute | Treatment of neurodegenerative diseases, causation of memory enhancement, and assay for screening compounds for such |
| AU2010347233B2 (en) | 2010-03-01 | 2015-06-18 | Oncternal Therapeutics, Inc. | Compounds for treatment of cancer |
| US20130231348A1 (en) | 2010-06-09 | 2013-09-05 | Afraxis, Inc. | 8-(HETEROARYLMETHYL)PYRIDO[2,3-d]PYRIMIDIN-7(8H)-ONES FOR THE TREATMENT OF CNS DISORDERS |
| WO2012058531A2 (en) | 2010-10-29 | 2012-05-03 | North Carolina State University | Modulation of response regulators by imidazole derivatives |
| TWI408128B (en) | 2010-12-03 | 2013-09-11 | Nat Univ Tsing Hua | M-terphenyl compound derivatives and application for organic light emitting diode |
| CU24152B1 (en) | 2010-12-20 | 2016-02-29 | Irm Llc | 1,2 OXAZOL-8-AZABICICLO [3,2,1] OCTANO 8 IL AS FXR MODULATORS |
| EP2655369A1 (en) | 2010-12-20 | 2013-10-30 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| EP2655368A1 (en) | 2010-12-20 | 2013-10-30 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| EP2545964A1 (en) * | 2011-07-13 | 2013-01-16 | Phenex Pharmaceuticals AG | Novel FXR (NR1H4) binding and activity modulating compounds |
| WO2013037482A1 (en) | 2011-09-15 | 2013-03-21 | Phenex Pharmaceuticals Ag | Farnesoid x receptor agonists for cancer treatment and prevention |
| SI3336097T1 (en) | 2012-06-19 | 2021-07-30 | Intercept Pharmaceuticals, Inc. | Preparation of the non-crystalline form of obeticholic acid |
| TWI621618B (en) | 2013-03-13 | 2018-04-21 | 比利時商健生藥品公司 | Substituted 2-azabicycles and their use as orexin receptor modulators |
| WO2014181287A1 (en) | 2013-05-09 | 2014-11-13 | Piramal Enterprises Limited | Heterocyclyl compounds and uses thereof |
| PL2997035T3 (en) | 2013-05-14 | 2018-10-31 | Intercept Pharmaceuticals, Inc. | 11-hydroxyl-6-substituted-derivatives of bile acids and amino acid conjugates thereof as farnesoid x receptor modulators |
| JP6556129B2 (en) | 2013-08-01 | 2019-08-07 | アメリカ合衆国 | Farnesoid X receptor inhibitor and pharmaceutical use |
| RS61540B1 (en) | 2013-09-11 | 2021-04-29 | Inst Nat Sante Rech Med | Methods and pharmaceutical compositions for the treatment of hepatitis b virus infection |
| US20150082981A1 (en) | 2013-09-20 | 2015-03-26 | E I Du Pont De Nemours And Company | Capture of trifluoromethane using ionic liquids |
| CN104513213A (en) | 2013-09-28 | 2015-04-15 | 山东亨利医药科技有限责任公司 | Fxr agonist |
| US20150119345A1 (en) | 2013-10-29 | 2015-04-30 | Lumena Pharmaceuticals, Inc. | Bile acid recycling inhibitors for treatment of gastrointestinal infections |
| PE20160682A1 (en) | 2013-11-05 | 2016-07-23 | Novartis Ag | COMPOSITIONS AND METHODS TO MODULATE X PHARNESOID RECEPTORS |
| WO2015116856A2 (en) | 2014-01-29 | 2015-08-06 | City Of Hope | Farnesoid x receptor antagonists |
| EP3116851B1 (en) | 2014-03-13 | 2023-07-26 | Salk Institute for Biological Studies | Analogs of fexaramine and methods of making and using |
| US10077268B2 (en) | 2014-03-13 | 2018-09-18 | Salk Institute For Biological Studies | FXR agonists and methods for making and using |
| WO2015138986A1 (en) | 2014-03-13 | 2015-09-17 | Salk Institute For Biological Studies | Fxr agonists and methods for making and using |
| EA032581B1 (en) | 2014-04-14 | 2019-06-28 | Грюненталь Гмбх | Heteroaryl substituted heterocyclyl sulfones |
| WO2015162538A1 (en) | 2014-04-21 | 2015-10-29 | Lupin Limited | Heterocyclic compounds as calcium sensing receptor modulators for the treatment of hyperparathyroidism, chronic renal failure and chronic kidney disease |
| WO2015162244A1 (en) | 2014-04-25 | 2015-10-29 | Basf Se | N-acylamidine compounds |
| WO2015165960A1 (en) | 2014-04-30 | 2015-11-05 | Basf Se | N-acylamidine compounds |
| ES2768718T3 (en) | 2014-05-29 | 2020-06-23 | Bar Pharmaceuticals S R L | Collane derivatives for use in the treatment and / or prevention of diseases mediated by FXR and TGR5 / GPBAR1 |
| CN104045635A (en) | 2014-06-23 | 2014-09-17 | 华东理工大学 | 3,4,5-tri-substituted isoxazole compounds and applications thereof |
| US9855249B2 (en) | 2014-10-02 | 2018-01-02 | Flatley Discovery Lab, Llc | Isoxazole compounds and methods for the treatment of cystic fibrosis |
| EP3006939A1 (en) | 2014-10-06 | 2016-04-13 | Gilead Sciences, Inc. | Histidine-rich Glycoprotein as a marker for hepatic Farnesoid X receptor activation |
| WO2016068585A1 (en) | 2014-10-27 | 2016-05-06 | 주식회사 엘지화학 | Organic electroluminescence device |
| SG11201703717SA (en) | 2014-11-06 | 2017-06-29 | Enanta Pharm Inc | Bile acid analogs an fxr/tgr5 agonists and methods of use thereof |
| MY192927A (en) | 2014-11-21 | 2022-09-15 | Akarna Therapeutics Ltd | Fused bicyclic compounds for the treatment of disease |
| WO2016086134A1 (en) | 2014-11-26 | 2016-06-02 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as fxr/tgr5 agonists and methods of use thereof |
| US10208081B2 (en) | 2014-11-26 | 2019-02-19 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
| US10519191B2 (en) | 2014-11-26 | 2019-12-31 | Enanta Pharmaceuticals, Inc. | Bile acid analogs as FXR/TGR5 agonists and methods of use thereof |
| WO2016086115A1 (en) | 2014-11-26 | 2016-06-02 | Enanta Pharmaceuticals, Inc. | Tetrazole derivatives of bile acids as fxr/tgr5 agonists and methods of use thereof |
| EP3034499A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Novel FXR (NR1H4) modulating compounds |
| EP3034501A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| EP3233083A1 (en) | 2014-12-18 | 2017-10-25 | Novartis AG | Azabicyclooctane derivatives as fxr agonists for use in the treatment of liver and gastrointestinal diseases |
| EA201892625A1 (en) | 2015-01-09 | 2019-07-31 | Джилид Аполло, Ллс | COMBINED THERAPY WITH ACETYL-COA-CARBOXYLASE (ACC) INHIBITOR FOR THE TREATMENT OF A NON-ALCOHOLIC FATAL LIVER DISEASE DISEASE |
| CN107257793A (en) | 2015-01-20 | 2017-10-17 | 梅里亚股份有限公司 | Anti- worm compound, composition and its application method |
| TWI698430B (en) | 2015-02-13 | 2020-07-11 | 南北兄弟藥業投資有限公司 | Tricyclic compounds and uses thereof in medicine |
| US10100285B2 (en) | 2015-04-03 | 2018-10-16 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| CN106146483A (en) | 2015-04-23 | 2016-11-23 | 上海迪诺医药科技有限公司 | Heterocyclic method Buddhist nun's ester derivant X receptor modulators |
| GB201507340D0 (en) | 2015-04-29 | 2015-06-10 | Univ St Andrews | Light emitting devices and compounds |
| ES2550374B1 (en) * | 2015-06-30 | 2016-09-08 | Universidad De La Rioja | Analog photoprotective compounds of MAA, synthesis procedure and composition comprising the same |
| WO2017011466A1 (en) | 2015-07-13 | 2017-01-19 | Zwiebel Laurence J | Thermal volatilization of orco agonists |
| EP3347450B1 (en) | 2015-09-11 | 2021-03-17 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US11390616B2 (en) | 2015-12-04 | 2022-07-19 | Bristol-Myers Squibb Company | Apelin receptor agonists and methods of use |
| TW201734002A (en) | 2015-12-11 | 2017-10-01 | 拜耳作物科學股份有限公司 | Substituted malonamides as insecticides |
| CN106946867B (en) | 2016-01-06 | 2019-11-12 | 广州市恒诺康医药科技有限公司 | FXR receptor modulators and its preparation method and application |
| EP3190103A1 (en) | 2016-01-08 | 2017-07-12 | Rijksuniversiteit Groningen | Inhibitors of the pd-1/pd-l1 protein/protein interaction |
| EP3400229B1 (en) | 2016-01-10 | 2024-03-06 | Provincial Health Services Authority | 18/19f-labelled compounds which target the prostate specific membrane antigen |
| WO2017122209A2 (en) | 2016-01-12 | 2017-07-20 | Yeda Research And Development Co. Ltd. | NF-kappaB INHIBITORS |
| WO2017128896A1 (en) | 2016-01-26 | 2017-08-03 | 江苏豪森药业集团有限公司 | Fxr agonist and preparation method and use thereof |
| CN107021958A (en) | 2016-02-01 | 2017-08-08 | 山东轩竹医药科技有限公司 | FXR receptor stimulating agents |
| CN108602811B (en) | 2016-02-01 | 2021-11-16 | 轩竹生物科技有限公司 | FXR receptor agonists |
| CN107021957A (en) | 2016-02-01 | 2017-08-08 | 山东轩竹医药科技有限公司 | FXR receptor stimulating agents |
| TW201741307A (en) | 2016-02-22 | 2017-12-01 | 艾洛斯生物製藥公司 | FXR modulators and methods of their use |
| CN107224583A (en) | 2016-03-24 | 2017-10-03 | 中美华世通生物医药科技(武汉)有限公司 | Medical composition and its use |
| WO2017189651A1 (en) | 2016-04-26 | 2017-11-02 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| WO2017189663A1 (en) | 2016-04-26 | 2017-11-02 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| WO2017189652A1 (en) | 2016-04-26 | 2017-11-02 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| US10144729B2 (en) | 2016-05-18 | 2018-12-04 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| US10149835B2 (en) | 2016-05-18 | 2018-12-11 | Elmore Patent Law Group, P.C. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10138228B2 (en) | 2016-05-18 | 2018-11-27 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use therof |
| KR102444366B1 (en) | 2016-06-03 | 2022-09-19 | 케모센트릭스, 인크. | How to treat liver fibrosis |
| EP3730487B1 (en) | 2016-06-13 | 2022-04-27 | Gilead Sciences, Inc. | Azetidine derivatives as fxr (nr1h4) modulators |
| CA2968836A1 (en) | 2016-06-13 | 2017-12-13 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| TW201808283A (en) | 2016-08-05 | 2018-03-16 | 廣東東陽光藥業有限公司 | Nitrogen-containing tricyclic compounds and uses thereof in medicine |
| US11091482B2 (en) | 2016-08-23 | 2021-08-17 | Ardelyx, Inc. | Isoxazolyl-carbonyloxy azabicyclo[3.2.1]octanyl compounds as FXR activators |
| EP3504205B1 (en) | 2016-08-23 | 2021-08-04 | Ardelyx, Inc. | Hormone receptor modulators for treating metabolic conditions and disorders |
| WO2018059314A1 (en) | 2016-09-28 | 2018-04-05 | 四川科伦博泰生物医药股份有限公司 | Azabicycle derivatives and preparation method and use thereof |
| US20200045972A1 (en) | 2016-09-29 | 2020-02-13 | Bayer Cropscience Aktiengesellschaft | Novel 5-substituted imidazolylmethyl derivatives |
| KR20190056436A (en) | 2016-10-04 | 2019-05-24 | 이난타 파마슈티칼스, 인코포레이티드 | Isoxazole analogs as FXR agonists and methods for their use |
| CN107973790A (en) | 2016-10-22 | 2018-05-01 | 合帕吉恩治疗公司 | Heterocyclic FXR conditioning agent |
| MA47558A (en) | 2016-11-10 | 2019-09-18 | Galmed Res And Development Ltd | FIBROSIS TREATMENT |
| JP2019533706A (en) | 2016-11-11 | 2019-11-21 | ギリアード サイエンシーズ, インコーポレイテッド | How to treat liver disease |
| CN106588804B (en) | 2016-12-09 | 2018-11-09 | 都创(上海)医药科技有限公司 | A kind of preparation method of compound as Farnesoid X receptor (FXR) |
| CN106632294A (en) | 2016-12-15 | 2017-05-10 | 宁波百纳西药业有限公司 | Spiro compound and medicinal use thereof |
| CN106748922B (en) | 2017-01-12 | 2019-02-01 | 中国药科大学 | A kind of novel sulfone acid derivative, preparation method and its purposes as drug |
-
2011
- 2011-07-13 EP EP11005722A patent/EP2545964A1/en not_active Withdrawn
-
2012
- 2012-07-02 TW TW101123785A patent/TWI439455B/en active
- 2012-07-11 AR ARP120102511A patent/AR087127A1/en active IP Right Grant
- 2012-07-12 AU AU2012283387A patent/AU2012283387C1/en active Active
- 2012-07-12 PL PL12735100T patent/PL2731676T3/en unknown
- 2012-07-12 EP EP17000383.4A patent/EP3246070B1/en active Active
- 2012-07-12 MY MYPI2013004373A patent/MY161158A/en unknown
- 2012-07-12 US US14/232,118 patent/US9139539B2/en active Active
- 2012-07-12 ME MEP-2016-81A patent/ME02434B/en unknown
- 2012-07-12 ES ES15002478.4T patent/ES2632492T3/en active Active
- 2012-07-12 KR KR1020147002937A patent/KR101722410B1/en active IP Right Grant
- 2012-07-12 MX MX2014000534A patent/MX368371B/en active IP Right Grant
- 2012-07-12 PL PL15002478T patent/PL2987532T3/en unknown
- 2012-07-12 HU HUE12735100A patent/HUE027931T2/en unknown
- 2012-07-12 CA CA2839357A patent/CA2839357C/en active Active
- 2012-07-12 KR KR1020177000763A patent/KR101766323B1/en active IP Right Grant
- 2012-07-12 RS RS20160266A patent/RS54786B1/en unknown
- 2012-07-12 EP EP15002478.4A patent/EP2987532B1/en active Active
- 2012-07-12 HU HUE17000383A patent/HUE066734T2/en unknown
- 2012-07-12 PT PT150024784T patent/PT2987532T/en unknown
- 2012-07-12 PL PL17000383.4T patent/PL3246070T3/en unknown
- 2012-07-12 BR BR122019026062-1A patent/BR122019026062B1/en active IP Right Grant
- 2012-07-12 SI SI201230969A patent/SI2987532T1/en unknown
- 2012-07-12 SI SI201230535A patent/SI2731676T1/en unknown
- 2012-07-12 CN CN201710308892.5A patent/CN107252424B/en active Active
- 2012-07-12 DK DK17000383.4T patent/DK3246070T3/en active
- 2012-07-12 SI SI201232062T patent/SI3246070T1/en unknown
- 2012-07-12 FI FIEP17000383.4T patent/FI3246070T3/en active
- 2012-07-12 PT PT170003834T patent/PT3246070T/en unknown
- 2012-07-12 DK DK12735100.5T patent/DK2731676T3/en active
- 2012-07-12 CN CN201280033148.4A patent/CN103702719B/en active Active
- 2012-07-12 BR BR122020008872-9A patent/BR122020008872B1/en active IP Right Grant
- 2012-07-12 LT LTEP17000383.4T patent/LT3246070T/en unknown
- 2012-07-12 JP JP2014519453A patent/JP5986633B2/en active Active
- 2012-07-12 HR HRP20240422TT patent/HRP20240422T1/en unknown
- 2012-07-12 ES ES17000383T patent/ES2978166T3/en active Active
- 2012-07-12 EP EP12735100.5A patent/EP2731676B1/en active Active
- 2012-07-12 KR KR1020187013304A patent/KR101934335B1/en active IP Right Grant
- 2012-07-12 EA EA201391674A patent/EA024843B1/en unknown
- 2012-07-12 KR KR1020177021651A patent/KR101859533B1/en active IP Right Grant
- 2012-07-12 UY UY0001034196A patent/UY34196A/en active IP Right Grant
- 2012-07-12 ES ES12735100.5T patent/ES2569718T3/en active Active
- 2012-07-12 EP EP24164624.9A patent/EP4400503A3/en active Pending
- 2012-07-12 WO PCT/EP2012/002941 patent/WO2013007387A1/en active Application Filing
- 2012-07-12 BR BR112014000260-6A patent/BR112014000260B1/en active IP Right Grant
-
2013
- 2013-12-17 IL IL229944A patent/IL229944A/en active IP Right Grant
- 2013-12-17 ZA ZA2013/09521A patent/ZA201309521B/en unknown
-
2014
- 2014-01-13 MX MX2019011629A patent/MX2019011629A/en unknown
- 2014-06-11 HK HK14105521.1A patent/HK1192181A1/en unknown
- 2014-06-11 HK HK16109776.3A patent/HK1221680A1/en unknown
-
2015
- 2015-08-12 US US14/824,971 patent/US9539244B2/en active Active
-
2016
- 2016-04-25 HR HRP20160442TT patent/HRP20160442T1/en unknown
- 2016-04-28 CY CY20161100369T patent/CY1117453T1/en unknown
- 2016-06-10 SM SM201600169T patent/SMT201600169B/en unknown
- 2016-08-05 JP JP2016154511A patent/JP6321097B2/en active Active
- 2016-11-29 AU AU2016265993A patent/AU2016265993B2/en active Active
- 2016-12-05 US US15/369,521 patent/US9820979B2/en active Active
-
2017
- 2017-10-13 US US15/783,530 patent/US10220027B2/en active Active
-
2018
- 2018-05-22 AU AU2018203613A patent/AU2018203613B2/en active Active
-
2019
- 2019-01-15 US US16/248,178 patent/US10485795B2/en active Active
- 2019-11-04 AU AU2019261667A patent/AU2019261667B2/en active Active
Patent Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000037077A1 (en) | 1998-12-23 | 2000-06-29 | Glaxo Group Limited | Assays for ligands for nuclear receptors |
| WO2003015771A1 (en) | 2001-08-13 | 2003-02-27 | Lion Bioscience Ag | Fxr nr1h4 nuclear receptor binding compounds |
| WO2003080803A2 (en) | 2002-03-21 | 2003-10-02 | Smithkline Beecham Corporation | Methods of using farnesoid x receptor (fxr) agonists |
| WO2004048349A1 (en) | 2002-11-22 | 2004-06-10 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2004087076A2 (en) | 2003-03-31 | 2004-10-14 | The Rockefeller University | Methods for inhibiting adipogenesis and for treating type 2 diabetes |
| WO2007076260A2 (en) | 2005-12-19 | 2007-07-05 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2007092751A2 (en) | 2006-02-03 | 2007-08-16 | Eli Lilly And Company | Compounds and methods for modulating fx-receptors |
| WO2007140174A2 (en) | 2006-05-24 | 2007-12-06 | Eli Lilly And Company | Compounds and methods for modulating fxr |
| WO2007140183A1 (en) | 2006-05-24 | 2007-12-06 | Eli Lilly And Company | Fxr agonists |
| EP1894924A1 (en) * | 2006-08-29 | 2008-03-05 | Phenex Pharmaceuticals AG | Heterocyclic FXR binding compounds |
| WO2008025540A1 (en) | 2006-08-29 | 2008-03-06 | Phenex Pharmaceuticals Ag | Heterocyclic fxr binding compounds |
| WO2008025539A1 (en) | 2006-08-29 | 2008-03-06 | Phenex Pharmaceuticals Ag | Heterocyclic fxr binding compounds |
| WO2008051942A2 (en) | 2006-10-24 | 2008-05-02 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2008157270A1 (en) | 2007-06-13 | 2008-12-24 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2009005998A1 (en) | 2007-07-02 | 2009-01-08 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2009012125A1 (en) | 2007-07-16 | 2009-01-22 | Eli Lilly And Company | Compounds and methods for modulating fxr |
| WO2011020615A1 (en) | 2009-08-19 | 2011-02-24 | Phenex Pharmaceuticals Ag | Novel fxr (nr1h4 ) binding and activity modulating compounds |
Non-Patent Citations (73)
| Title |
|---|
| "March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure", JOHN WILEY & SONS |
| A. ARANDA; A. PASCUAL, PHYSIOL. REV., vol. 81, 2001, pages 1269 |
| A. FIGGE ET AL., J. BIOL. CHEM., vol. 279, 2004, pages 2790 |
| A. M. BRZOZOWSKI ET AL., NATURE, vol. 389, 1997, pages 753 |
| A. MOSCHETTA ET AL., NATURE MEDICINE, vol. 10, 2004, pages 1352 |
| B. CARIOU ET AL., J. BIOL. CHEM., vol. 281, 2006, pages 11039 |
| B. GOODWIN ET AL., MOL. CELL, vol. 6, 2000, pages 517 |
| B. M. FORMAN ET AL., CELL, vol. 81, 1995, pages 687 |
| C. J. SINAL ET AL., CELL, vol. 102, 2000, pages 731 |
| C. LIHONG ET AL., AMERICAN DIABETES ASSOCIATION (ADA) 66TH ANNUAL SCIENTIFIC SESSIONS, June 2006 (2006-06-01) |
| D. DURAN-SANDOVAL ET AL., BIOCHIMIE, vol. 87, 2005, pages 93 |
| D. J. MANGELSDORF ET AL., CELL, vol. 83, 1995, pages 835 |
| D. J. PARKS ET AL., SCIENCE, vol. 284, 1999, pages 1365 |
| D. M. HEERY ET AL., NATURE, vol. 387, 1997, pages 733 |
| D. MERK ET AL., FUTURE MED. CHEM., vol. 4, 2012, pages 1015 |
| DYER ET AL., ANAL. BIOCHEM., vol. 282, 2000, pages 158 - 161 |
| E. A. HANNIMAN ET AL., J. LIPID RES., vol. 46, 2005, pages 2595 |
| E. TOMLINSON ET AL., ENDOCRINOLOGY, vol. 143, 2002, pages 1741 |
| F. CHEN ET AL., GASTROENTEROLOGY, vol. 126, 2004, pages 756 |
| F. HE ET AL., CIRC. RES., vol. 98, 2006, pages 192 |
| F. JOURNE ET AL., BREAST CANCER RES. TREAT., vol. 115, 2009, pages 523 |
| F. YANG ET AL., CANCER RES., vol. 67, 2007, pages 863 |
| FIESER ET AL.: "Fiesers' Reagents for organic Synthesis", 2000, JOHN WILEY & SONS |
| G. LAMBERT ET AL., J. BIOL. CHEM., vol. 278, 2003, pages 2563 |
| G. RIZZO ET AL., CURR. DRUG TARGETS IMMUNE ENDOCR. METABOL. DISORD., vol. 5, 2005, pages 289 |
| G. RIZZO ET AL., CURR. DRUG TARGETS IMMUNE ENDOER. METABOL. DISORD., vol. 5, 2005, pages 289 |
| G. ZOLLNER ET AL., MOL. PHARM., vol. 3, 2006, pages 231 |
| H. R. KAST ET AL., J. BIOL. CHEM., vol. 277, 2002, pages 2908 |
| H. R. KAST ET AL., MOL. ENDOCRINOL., vol. 15, 2001, pages 1720 |
| H. WITTENBURG, GASTROENTEROLOGY, vol. 125, 2003, pages 868 |
| I. KIM ET AL., CARCINOGENESIS, vol. 28, 2007, pages 940 |
| J. A. HOLT ET AL., GENES DEV., vol. 17, 2003, pages 1581 |
| J. HOLT ET AL., GENES DEV., vol. 17, 2003, pages 1581 |
| J. R. PLASS ET AL., HEPATOLOGY, vol. 35, 2002, pages 589 |
| K. E. SWALES ET AL., CANCER RES., vol. 66, 2006, pages 10120 |
| K. MA ET AL., J. CLIN. INVEST., vol. 116, 2006, pages 1102 |
| K. R. STAYROOK ET AL., ENDOCRINOLOGY, vol. 146, 2005, pages 984 |
| K. W. NETTLES; G. L. GREENE, ANNU. REV. PHYSIOL., vol. 67, 2005, pages 309 |
| L. ALVAREZ ET AL., HUM. MOL. GENET., vol. 13, 2004, pages 2451 |
| L. HUANG ET AL., J. BIOL. CHEM., vol. 278, 2003, pages 51085 |
| L. HUANG; J. DONG; S. KARKI: "Evaluation of drug candidates for preclinical development", 2010, WILEY & SONS, pages: 187 - 217 |
| M. ANANTHANARAYANAN ET AL., J. BIOL. CHEM., vol. 276, 2001, pages 28857 |
| M. L. CRAWLEY, EXPERT OPIN THER. PAT., vol. 20, 2010, pages 1047 |
| M. MAKISHIMA ET AL., SCIENCE, vol. 284, 1999, pages 1362 |
| M. MIYATA, J. PHARMACOL. EXP. THER., vol. 312, 2005, pages 759 |
| M. SCHENA; K. R. YAMAMOTO, SCIENCE, vol. 241, 1988, pages 965 |
| M. WATANABE ET AL., J. CLIN. INVEST., vol. 113, 2004, pages 1408 |
| N. L. URIZAR ET AL., J. BIOL. CHEM., vol. 275, 2000, pages 39313 |
| N. L. URIZAR ET AL., SCIENCE, vol. 296, 2002, pages 1703 |
| P. MALONEY ET AL., J. MED. CHEM., vol. 43, 2000, pages 2971 |
| P. R. MALONEY ET AL., J. MED. CHEM., vol. 43, 2000, pages 2971 |
| R. M. EVANS, MOL. ENDOCRINOL., vol. 19, 2005, pages 1429 |
| R. PELLICCIARI ET AL., J. MED. CHEM., vol. 45, 2002, pages 3569 |
| R. R. MARAN ET AL., J. PHARMACOL. EXP. THER., vol. 328, 2009, pages 469 |
| S. A. DOGGRELL, CURR. OPIN. INVESTIG. DRUGS, vol. 7, 2006, pages 344 |
| S. BILZ ET AL., AM. J. PHYSIOL. ENDOCRINOL. METAB., vol. 290, 2006, pages 716 |
| S. FIORUCCI ET AL., GASTROENTEROLOGY, vol. 127, 2004, pages 1497 |
| S. FIORUCCI ET AL., J. PHARMACOL. EXP. THER., vol. 313, 2005, pages 604 |
| S. FIORUCCI ET AL., J. PHARMACOL. EXP. THER., vol. 314, 2005, pages 584 |
| S. MODICA ET AL., CANCER RES., vol. 68, 2008, pages 9589 |
| S. Y. CAI ET AL., EXPERT OPIN. THER. TARGETS, vol. 10, 2006, pages 409 |
| T. CLAUDEL ET AL., ARTERIOSCLER. THROMB. VASC. BIOL., vol. 25, 2005, pages 2020 |
| T. EICHER; S. HAUPTMANN: "The Chemistry of Heterocycles; Structures, Reactions, Synthesis and Application", 2003, WILEY-VCH |
| T. HEINZEL ET AL., NATURE, vol. 387, 1997, pages 43 |
| T. INAGAKI ET AL., CELL METAB., vol. 2, 2005, pages 217 |
| T. INAGAKI ET AL., PNAS, vol. 103, 2006, pages 3920 |
| T. M. WILLSON ET AL., MED. RES. REV., vol. 21, 2001, pages 513 |
| T. T. LU ET AL., MOL. CELL, vol. 6, 2000, pages 507 |
| T. WILLSON ET AL., MED. RES. REV., vol. 21, 2001, pages 513 |
| W. HUANG ET AL., SCIENCE, vol. 312, 2006, pages 233 |
| Y. D. WANG ET AL., CELL RES., vol. 18, 2008, pages 1087 |
| Y. LIU ET AL., J. CLIN. INVEST., vol. 112, 2003, pages 1678 |
| Y. ZHANG ET AL., PNAS, vol. 103, 2006, pages 1006 |
Cited By (93)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10485795B2 (en) | 2011-07-13 | 2019-11-26 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US10220027B2 (en) | 2011-07-13 | 2019-03-05 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| EP3711762A1 (en) | 2013-09-11 | 2020-09-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | A farnesoid x receptor agonsits foruse and pharmaceutical compositions for the treatment of chronic hepatitis b virus infection |
| EP3006939A1 (en) | 2014-10-06 | 2016-04-13 | Gilead Sciences, Inc. | Histidine-rich Glycoprotein as a marker for hepatic Farnesoid X receptor activation |
| US10968249B2 (en) | 2014-11-26 | 2021-04-06 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
| WO2016086218A1 (en) | 2014-11-26 | 2016-06-02 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as fxr/tgr5 agonists and methods of use thereof |
| US11718641B2 (en) | 2014-11-26 | 2023-08-08 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
| CN108064223B (en) * | 2014-12-17 | 2021-06-01 | 吉利德科学公司 | Fxr (nr1h4) modulating compounds containing hydroxy groups |
| AU2015365945B2 (en) * | 2014-12-17 | 2018-09-27 | Gilead Sciences, Inc. | Novel FXR (NR1H4) modulating compounds |
| KR20170095955A (en) * | 2014-12-17 | 2017-08-23 | 길리애드 사이언시즈, 인코포레이티드 | Hydroxy containing fxr (nr1h4) modulating compounds |
| CN107108595A (en) * | 2014-12-17 | 2017-08-29 | 吉利德科学公司 | New FXR (NR1H4) modulating compound |
| US9751874B2 (en) | 2014-12-17 | 2017-09-05 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| KR101998007B1 (en) | 2014-12-17 | 2019-07-08 | 길리애드 사이언시즈, 인코포레이티드 | Hydroxy containing fxr (nr1h4) modulating compounds |
| CN107108595B (en) * | 2014-12-17 | 2021-04-06 | 吉利德科学公司 | Novel FXR (NR1H4) modulating compounds |
| JP2018500304A (en) * | 2014-12-17 | 2018-01-11 | ギリアード サイエンシーズ, インコーポレイテッド | Hydroxy-containing FXR (NR1H4) modulating compound |
| JP2018500305A (en) * | 2014-12-17 | 2018-01-11 | ギリアード サイエンシーズ, インコーポレイテッド | Novel FXR (NR1H4) modulating compound |
| US9938278B2 (en) | 2014-12-17 | 2018-04-10 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| CN108064223A (en) * | 2014-12-17 | 2018-05-22 | 吉利德科学公司 | FXR (NR1H4) modulating compound of hydroxyl |
| EA033642B1 (en) * | 2014-12-17 | 2019-11-12 | Gilead Sciences Inc | Fxr (nr1h4) modulating compounds |
| EP3034501A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| KR101955840B1 (en) | 2014-12-17 | 2019-03-07 | 길리애드 사이언시즈, 인코포레이티드 | Novel fxr (nr1h4) modulating compounds |
| EP3034499A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Novel FXR (NR1H4) modulating compounds |
| KR20170093969A (en) * | 2014-12-17 | 2017-08-16 | 길리애드 사이언시즈, 인코포레이티드 | Novel fxr (nr1h4) modulating compounds |
| WO2016096116A1 (en) * | 2014-12-17 | 2016-06-23 | Gilead Sciences, Inc. | Novel fxr (nr1h4) modulating compounds |
| TWI697483B (en) * | 2014-12-17 | 2020-07-01 | 美商基利科學股份有限公司 | Hydroxy containing fxr (nr1h4) modulating compounds |
| WO2016096115A1 (en) * | 2014-12-17 | 2016-06-23 | Gilead Sciences, Inc. | Hydroxy containing fxr (nr1h4) modulating compounds |
| AU2015365944B2 (en) * | 2014-12-17 | 2019-01-17 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| WO2016127924A1 (en) | 2015-02-13 | 2016-08-18 | Sunshine Lake Pharma Co., Ltd. | Tricyclic compounds and uses thereof in medicine |
| US11040998B2 (en) | 2015-03-31 | 2021-06-22 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
| US11958879B2 (en) | 2015-03-31 | 2024-04-16 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
| US10080741B2 (en) | 2016-04-26 | 2018-09-25 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10080743B2 (en) | 2016-04-26 | 2018-09-25 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10080742B2 (en) | 2016-04-26 | 2018-09-25 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10149835B2 (en) | 2016-05-18 | 2018-12-11 | Elmore Patent Law Group, P.C. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10144729B2 (en) | 2016-05-18 | 2018-12-04 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| US10138228B2 (en) | 2016-05-18 | 2018-11-27 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use therof |
| WO2017201150A1 (en) * | 2016-05-18 | 2017-11-23 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as fxr agonists and methods of use thereof |
| KR20190017028A (en) * | 2016-06-13 | 2019-02-19 | 길리애드 사이언시즈, 인코포레이티드 | FXR (NR1H4) adjusting compound |
| EP3730487A1 (en) | 2016-06-13 | 2020-10-28 | Gilead Sciences, Inc. | Azetidine derivatives as fxr (nr1h4) modulators |
| US10981881B2 (en) | 2016-06-13 | 2021-04-20 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10329286B2 (en) | 2016-06-13 | 2019-06-25 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| WO2017218337A1 (en) | 2016-06-13 | 2017-12-21 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| US11739065B2 (en) | 2016-06-13 | 2023-08-29 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US20190315729A1 (en) * | 2016-06-13 | 2019-10-17 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| EA037744B1 (en) * | 2016-06-13 | 2021-05-17 | Джилид Сайэнс, Инк. | Fxr (nr1h4) modulating compounds |
| AU2017284160B2 (en) * | 2016-06-13 | 2019-12-19 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10774054B2 (en) | 2016-06-13 | 2020-09-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11247986B2 (en) | 2016-06-13 | 2022-02-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| KR102276042B1 (en) * | 2016-06-13 | 2021-07-14 | 길리애드 사이언시즈, 인코포레이티드 | FXR (NR1H4) modulating compound |
| KR102269305B1 (en) * | 2016-06-13 | 2021-06-25 | 길리애드 사이언시즈, 인코포레이티드 | FXR (NR1H4) modulating compound |
| KR20190017027A (en) * | 2016-06-13 | 2019-02-19 | 길리애드 사이언시즈, 인코포레이티드 | FXR (NR1H4) adjusting compound |
| US11034684B2 (en) | 2016-10-04 | 2021-06-15 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| US10450306B2 (en) | 2016-10-04 | 2019-10-22 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| JP2019535811A (en) * | 2016-10-22 | 2019-12-12 | へパジーン セラピューティクス インコーポレイテッド | Heterocyclic FXR modulator |
| JP7065107B2 (en) | 2016-10-22 | 2022-05-11 | へパジーン セラピューティクス (エイチケイ) リミテッド | Heterocyclic FXR modulator |
| US10597391B2 (en) | 2016-10-26 | 2020-03-24 | Enanta Pharmaceuticals, Inc. | Urea-containing isoxazole derivatives as FXR agonists and methods of use thereof |
| CN106588804A (en) * | 2016-12-09 | 2017-04-26 | 都创(上海)医药科技有限公司 | Preparation method of compound serving as farnesoid X receptor (FXR) |
| WO2018153933A1 (en) | 2017-02-21 | 2018-08-30 | Genfit | Combination of a ppar agonist with a fxr agonist |
| US11833150B2 (en) | 2017-03-28 | 2023-12-05 | Gilead Sciences, Inc. | Methods of treating liver disease |
| CN110461328A (en) * | 2017-03-28 | 2019-11-15 | 吉利德科学公司 | The therapeutic combination for treating liver disease |
| WO2018178260A1 (en) | 2017-03-30 | 2018-10-04 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for reducing persistence and expression of episomal viruses |
| US10961272B2 (en) | 2017-04-07 | 2021-03-30 | Enanta Pharmaceuticals, Inc. | Process for preparation of sulfonyl carbamate bile acid derivatives |
| US10676500B2 (en) | 2017-04-07 | 2020-06-09 | Enanta Pharmaceuticals, Inc. | Process for preparation of sulfonyl carbamate bile acid derivatives |
| US10988449B2 (en) | 2017-04-12 | 2021-04-27 | Il Dong Pharmaceutical Co., Ltd. | Isoxazole derivatives as nuclear receptor agonists and uses thereof |
| US20180333401A1 (en) * | 2017-04-12 | 2018-11-22 | Gilead Sciences, Inc. | Methods of treating liver disease |
| US10495648B2 (en) | 2017-06-13 | 2019-12-03 | Gilead Sciences, Inc. | Methods of treating liver fibrosis |
| WO2018231851A1 (en) | 2017-06-13 | 2018-12-20 | Gilead Sciences, Inc. | Methods of treating liver fibrosis |
| US11168079B2 (en) | 2017-11-01 | 2021-11-09 | Bristol-Myers Squibb Company | Alkene compounds as farnesoid x receptor modulators |
| US11370785B2 (en) | 2017-11-01 | 2022-06-28 | Bristol-Myers Squibb Company | Multicyclic compounds as farnesoid X receptor modulators |
| US11286252B2 (en) | 2017-11-01 | 2022-03-29 | Bristol-Myers Squibb Company | Alkene spirocyclic compounds as farnesoid X receptor modulators |
| WO2019094777A1 (en) | 2017-11-13 | 2019-05-16 | Gilead Sciences, Inc. | Compositions and methods for identifying and treating liver diseases and monitoring treatment outcomes |
| US10689391B2 (en) | 2017-12-12 | 2020-06-23 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| WO2019149158A1 (en) | 2018-02-02 | 2019-08-08 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous tricyclic compounds and uses thereof in medicine |
| US10829486B2 (en) | 2018-02-14 | 2020-11-10 | Enanta Pharmacueticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| WO2020065597A1 (en) | 2018-09-28 | 2020-04-02 | Richter Gedeon Nyrt. | Bicyclic derivatives as gabaa α5 receptor modulators |
| US11225473B2 (en) | 2019-01-15 | 2022-01-18 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| KR20210114457A (en) * | 2019-01-15 | 2021-09-23 | 길리애드 사이언시즈, 인코포레이티드 | FXR (NR1H4) modulating compound |
| KR102692309B1 (en) | 2019-01-15 | 2024-08-07 | 길리애드 사이언시즈, 인코포레이티드 | FXR (NR1H4) coordination compound |
| US12030835B2 (en) | 2019-02-15 | 2024-07-09 | Bristol-Myers Squibb Company | Substituted amide compounds useful as farnesoid X receptor modulators |
| US12102625B2 (en) | 2019-02-19 | 2024-10-01 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| CN113573700A (en) * | 2019-03-11 | 2021-10-29 | 吉利德科学公司 | Formulations of compounds and uses thereof |
| US11555032B2 (en) | 2019-05-13 | 2023-01-17 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| WO2021191837A1 (en) | 2020-03-26 | 2021-09-30 | Richter Gedeon Nyrt. | 1,3-dihydro-2h-pyrrolo[3,4-c]pyridine derivatives as gabaa α5 receptor modulators |
| US11478533B2 (en) | 2020-04-27 | 2022-10-25 | Novo Nordisk A/S | Semaglutide for use in medicine |
| WO2022134146A1 (en) * | 2020-12-22 | 2022-06-30 | 江苏天士力帝益药业有限公司 | Novel fxr agonist having pyrazine structure, and preparation method and use |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
| US11919879B2 (en) | 2021-06-16 | 2024-03-05 | Celgene Corporation | Carboxylic acid containing azetidinyl compounds for the treatment of neurodegenerative diseases |
| WO2022266444A1 (en) | 2021-06-18 | 2022-12-22 | Gilead Sciences, Inc. | Il-31 modulators for treating fxr-induced pruritis |
| WO2024089582A1 (en) | 2022-10-25 | 2024-05-02 | Assia Chemical Industries Ltd. | Solid state forms of cilofexor salts |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2018203613B2 (en) | Novel FXR (NR1H4) binding and activity modulating compounds | |
| JP2019214611A (en) | Novel FXR (NR1H4) binding and activity modulating compounds | |
| NZ710770B2 (en) | Novel fxr (nr1h4) binding and activity modulating compounds | |
| NZ620177B2 (en) | Novel fxr (nr1h4) binding and activity modulating compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12735100 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201391674 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 2839357 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2012283387 Country of ref document: AU Date of ref document: 20120712 Kind code of ref document: A Ref document number: 2014519453 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2014/000534 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012735100 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20147002937 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14232118 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014000260 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2016/0266 Country of ref document: RS |
|
| ENP | Entry into the national phase |
Ref document number: 112014000260 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140106 |