WO2015138986A1 - Fxr agonists and methods for making and using - Google Patents
Fxr agonists and methods for making and using Download PDFInfo
- Publication number
- WO2015138986A1 WO2015138986A1 PCT/US2015/020582 US2015020582W WO2015138986A1 WO 2015138986 A1 WO2015138986 A1 WO 2015138986A1 US 2015020582 W US2015020582 W US 2015020582W WO 2015138986 A1 WO2015138986 A1 WO 2015138986A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- aliphatic
- subject
- alkyl
- heteroaliphatic
- Prior art date
Links
- 0 CC(C)c1c(C(*)OC(C=C([C@@](C2)C=Cc3cccc(C(O)=O)c3)Cl)=C2F)c(-c(c(Cl)ccc2)c2Cl)n[o]1 Chemical compound CC(C)c1c(C(*)OC(C=C([C@@](C2)C=Cc3cccc(C(O)=O)c3)Cl)=C2F)c(-c(c(Cl)ccc2)c2Cl)n[o]1 0.000 description 10
- JDCSUGXPJNJRJY-UHFFFAOYSA-N CC(C(CC1)C2SC2CC=C1C(c1nnn[n]1-c1c(C)cccc1C)N1CCCC1)=C Chemical compound CC(C(CC1)C2SC2CC=C1C(c1nnn[n]1-c1c(C)cccc1C)N1CCCC1)=C JDCSUGXPJNJRJY-UHFFFAOYSA-N 0.000 description 1
- LOZDPQHJLODBSV-UHFFFAOYSA-N CC(C(CC1)CC=C1C(c1ncn[n]1-c1c(C)cccc1C)N1CCCC1)=C Chemical compound CC(C(CC1)CC=C1C(c1ncn[n]1-c1c(C)cccc1C)N1CCCC1)=C LOZDPQHJLODBSV-UHFFFAOYSA-N 0.000 description 1
- JTQOUVXRRYIHJU-UHFFFAOYSA-N CC(C(CC1)CC=C1C(c1ncn[n]1-c1c(C)cccc1C)N1CCCCC1)=C Chemical compound CC(C(CC1)CC=C1C(c1ncn[n]1-c1c(C)cccc1C)N1CCCCC1)=C JTQOUVXRRYIHJU-UHFFFAOYSA-N 0.000 description 1
- YAAADGCGUXKBAD-UHFFFAOYSA-N CC(C(CC1)CC=C1C1N(CCCCC2)C2c(cccc2C)c2-[n]2nnnc12)=C Chemical compound CC(C(CC1)CC=C1C1N(CCCCC2)C2c(cccc2C)c2-[n]2nnnc12)=C YAAADGCGUXKBAD-UHFFFAOYSA-N 0.000 description 1
- KCZFBLNQOSFGSH-UHFFFAOYSA-N CC(C)(C)OC(Nc1ccccc1N)=O Chemical compound CC(C)(C)OC(Nc1ccccc1N)=O KCZFBLNQOSFGSH-UHFFFAOYSA-N 0.000 description 1
- CLDPAJYNRMTDDM-UHFFFAOYSA-N CC(C)C(CC1)CC=C1C(c1ncn[n]1-c1c(C)cccc1C)N1C(C)CCC1 Chemical compound CC(C)C(CC1)CC=C1C(c1ncn[n]1-c1c(C)cccc1C)N1C(C)CCC1 CLDPAJYNRMTDDM-UHFFFAOYSA-N 0.000 description 1
- JEYHUCCAEAOGCL-UHFFFAOYSA-N CC(C)C(CC1)CC=C1C1N(CCCCC2)C2c(cccc2C)c2-[n]2ncnc12 Chemical compound CC(C)C(CC1)CC=C1C1N(CCCCC2)C2c(cccc2C)c2-[n]2ncnc12 JEYHUCCAEAOGCL-UHFFFAOYSA-N 0.000 description 1
- PLTHOFAZXCKNKS-SNCIUUNGSA-N CC(C)[C@@H](CC1)SCC=C1C1N(CCCC2)C2c(cccc2C)c2-[n]2nnnc12 Chemical compound CC(C)[C@@H](CC1)SCC=C1C1N(CCCC2)C2c(cccc2C)c2-[n]2nnnc12 PLTHOFAZXCKNKS-SNCIUUNGSA-N 0.000 description 1
- BYTNEISLBIENSA-MDZDMXLPSA-N CC(C)c1c(COc2ccc(/C=C/c3cccc(C(O)=O)c3)c(Cl)c2)c(-c(c(Cl)ccc2)c2Cl)n[o]1 Chemical compound CC(C)c1c(COc2ccc(/C=C/c3cccc(C(O)=O)c3)c(Cl)c2)c(-c(c(Cl)ccc2)c2Cl)n[o]1 BYTNEISLBIENSA-MDZDMXLPSA-N 0.000 description 1
- WZRUEECWFKKBJG-UHFFFAOYSA-N CC(CCC1)N1C(c1nnn[n]1-c1c(C)cccc1C)C(CC1)=CCC2SC2C1C(C)=C Chemical compound CC(CCC1)N1C(c1nnn[n]1-c1c(C)cccc1C)C(CC1)=CCC2SC2C1C(C)=C WZRUEECWFKKBJG-UHFFFAOYSA-N 0.000 description 1
- VLQTUNDJHLEFEQ-KGENOOAVSA-N CN(C)c(cc1)ccc1-c1ccc(CN(C(C2CCCCC2)=O)c2cccc(/C=C/C(OC)=O)c2)cc1 Chemical compound CN(C)c(cc1)ccc1-c1ccc(CN(C(C2CCCCC2)=O)c2cccc(/C=C/C(OC)=O)c2)cc1 VLQTUNDJHLEFEQ-KGENOOAVSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/422—Oxazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/18—Benzimidazoles; Hydrogenated benzimidazoles with aryl radicals directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/04—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles
- C07D249/06—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles with aryl radicals directly attached to ring atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D257/04—Five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Definitions
- This disclosure concerns new FXR agonists and a method for using the agonists, such as to treat or prevent gastrointestinal (GI) inflammatory conditions and metabolic disorders, including obesity and diabetes.
- GI gastrointestinal
- Metabolic syndrome a western diet-induced, pro-inflammatory disease affecting up to 25% of Americans, is characterized by central obesity, impaired glucose tolerance, dyslipidemia, insulin resistance, and type II diabetes. Secondary complications associated with metabolic syndrome include atherosclerosis, stroke, fatty liver disease, blindness, gallbladder disease, cancer, polycystic ovary disease and others. Consequently there is interest in reducing food intake, losing weight, and reducing elevated blood glucose. There is also an interest in combating obesity and related conditions using methods that do not require drastic lifestyle or dietary changes. In addition, inflammatory gastrointestinal conditions resulting from various types of pathology affect millions of people. Thus, effective and targeted treatments for various inflammatory gastrointestinal (GI) conditions are also needed.
- GI inflammatory gastrointestinal
- Farnesoid X receptor is a ligand-activated transcriptional receptor expressed in diverse tissues including the adrenal gland, kidney, stomach, duodenum, jejunum, ileum, colon, gall bladder, liver, macrophages, and white and brown adipose tissue (Forman et al., Cell 81:687-693 (1995). FXR has been reported to contribute to the regulation of whole body metabolism including bile acid/cholesterol, glucose and lipid metabolism. Synthetic ligands for FXR have been identified and applied to animal models of metabolic disorders, but these known synthetic ligands have shown limited efficacy and, in certain cases, exacerbated phenotypes.
- Bile acids function as endogenous ligands for FXR such that enteric and systemic release of BAs induces FXR-directed changes in gene expression networks (Lee et al., Trends
- FXR activation suppresses hepatic BA synthesis, alters BA composition, reduces the BA pool size (Wang et al, Dev Cell 2:721-731, 2002; Fang et al, Mol Cell Biol 27: 1407-1424, 2007; Lu et al, Mol Cell 6:507-515, 2000), and contributes to liver regeneration (Huang et al, Science 312:233-236, 2006) as well as lipid and cholesterol homeostasis (Zhang et al, Genes Dev 18: 157-169, 2004; Ma et al, J Clin Invest 116: 1102-1109, 2006).
- hepatic FXR by the synthetic bile acid 6cc-ethyl chenodeoxycholic acid (6-eCDCA) is beneficial in the treatment of diabetes, non-alcoholic fatty liver disease (NAFLD), and primary biliary cirrhosis (PBC) (Stanimirov et al, Acta Gastroenterol Belg 75:389-398, 2012; Mudaliar et al, Gastroenterology 145:574-582 e571, 2013).
- NASH non-alcoholic fatty liver disease
- PBC primary biliary cirrhosis
- FXR is also widely expressed in the intestine where it regulates production of the endocrine hormone FGF15 (FGF19 in humans), which, in conjunction with hepatic FXR, is thought to control BA synthesis, transport and metabolism (Kim et al, J Lipid Res 48:2664- 2672, 2007; Song et al.,Hepatology 49,:97-305, 2009; Inagak et al, Cell Metab 2:217-225, 2005). Intestinal FXR activity is also known to be involved in reducing overgrowth of the microbiome during feeding (Li et al, Nat Commun 4:2384, 2013; Inagaki et al, Proc Natl Acad Sci U SA 103:3920-3925, 2006).
- R ⁇ -R 15 independently are selected from hydrogen, deuterium, halogen, CF3, NO2, OH, amino, acyl, carboxyl, carboxyl ester, cyano, aminocarbonyl, aminosulfonyl, aliphatic, D- aliphatic, hetero aliphatic, D-heteroaliphatic, or -(CH2)ni-R 150 -(CH2) n 2-R 151 , wherein nl and n2 are independently selected from the group consisting of 0, 1, 2, 3, and 4, R 150 is O, NR 16 , or absent, and R 151 is carboxyl ester or amino; R 16 is selected from hydrogen, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic; R a and R b are independently hydrogen, deuterium, aliphatic or D-aliphatic, or together form a bond, such as
- the com ound has a formula
- the com ound has a formula
- R -R is or comprises deuterium.
- R 7 is alkyl or deuterated alkyl, such as isopropyl or a deuterated isopropyl group comprising from 1 to 7 deuterium atoms.
- at least one of R x -R 5 is a halogen, such as fluoro.
- R 16 is hydrogen.
- R 10 and R 11 independently are alkyl or deuterated alkyl, such as methyl or deuterated methyl, wherein the deuterated alkyl group comprises from 1 to n halogen
- R 21 -R 34 independently are selected from hydrogen, deuterium, halogen, CX3, where X is a halogen, such as fluorine, with CF3 being a particular example, NO2, OH, amino, acyl, carboxyl, carboxyl ester, cyano, aminocarbonyl, aminosulfonyl, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic; R 35 is aliphatic, D-aliphatic, heteroaliphatic or D- heteroaliphatic; R 36 is hydrogen, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic; X is N or CR 37 ; and R 37 is hydrogen, deuterium, halogen, CF3, NO2, OH, amino, acyl, carboxyl, carboxyl ester, cyano, aminocarbonyl, aminos
- the compound has a formula
- the compound has a formula
- R is alkyl, cycloalkyl, deuterated alkyl or deuterated cycloalkyl, such as cyclohexyl or deuterated cyclohexyl comprising 1 to 11 deuterium atoms.
- R 36 is hydrogen;
- R 34 is CF 3 ;
- R 23 is halogen, such as fluorine or chlorine.
- Certain compounds are chiral, and all stereoisomers are included in this disclosure.
- the compound is the most biologically active stereoisomer, such as the ⁇ -stereoisomer.
- Exemplary compounds according to this formula include
- R -R and R 52 -R 55 independently are selected from hydrogen, deuterium, halogen, CF 3 , N0 2 , OH, amino, acyl, carboxyl, carboxyl ester, cyano, aminocarbonyl, aminosulfonyl, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic;
- R 49 -R 51 independently are selected from hydrogen, deuterium, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic;
- R 56 is amino, cycloamino or substituted cycloamino;
- Y and Z are independently N or CR ; and each R independently is selected from deuterium, halogen, CF3, NO2, OH, amino, acyl, carboxyl, carboxyl ester, cyano, aminocarbonyl, aminosulfonyl, aliphatic,
- R 51 is aliphatic or D-aliphatic, such as methyl or deuterated methyl having from 1 to 3 deuterium atoms.
- R 49 and R 50 independently are hydrogen or deuterium; and R 41 and R 45 independently are aliphatic or D- aliphatic, such as methyl or deuterated methyl having from 1 to 3 deuterium atoms.
- R 56 is a cycloamino or substituted cycloamino, such as pyrrolidine, 2- methylpyrrolidine, morpholine, 4-methylpiperazine, piperidine, or azepane. Exemplary compounds having this formula include
- R ⁇ -R 57 is -R x -L x -R x2 , where R x is selected from O, NR x3 , sulfonyl or S; R x3 is selected from H, aliphatic, or aryl; L x is selected from a bond, aliphatic, heteroaliphatic, aryl, heteroaryl or CR x4 R x5 ; R x4 and R x5 are each independently selected from H, D, halogen, aliphatic, -C(0)OR x6 , or -C(0)NR x6 R x7 ; R x6 and R x7 are each independently selected from H, aliphatic; R x2 is selected from -C(0)L x2 R x8 or a carboxyl bioisostere; L x2 is a bond or NR x3 ; R x8 is H, aliphatic, -
- compositions comprising any such compound, or compounds, and at least one additional component, such as a pharmaceutically exceptable excipient, an additional therapeutic, or combinations thereof, also are disclosed.
- the compositions may include an enteric coating.
- Such methods can include administering to the subject a therapeutically effective amount of one or more of the disclosed compounds and/or
- compositions such as 1, 2, 3, 4, or 5 of such compounds and/or compositions.
- certain disclosed embodiments concerning compounds that are substantially absorbed in the gastrointestinal tract, thereby activating FXR receptors in the intestines to treat or prevent a metabolic disorder in the subject.
- Certain method embodiments also may improve glucose and/or lipid homeostasis in the subject.
- the method further includes administering to the subject a statin, an insulin sensitizing drug, (such as sitagliptin, vildagliptin, saxagliptin, linagliptin, anaglptin, teneligliptin, alogliptin, gemiglptin, or dutoglpitin), meglitinide, sulfonylurea, peroxisome proliferator-activated receptor (alpha-glucosidase inhibitor, amylin agonist, dipeptidyl-peptidase 4 (DPP-4) inhibitor PPAR)-gamma agonist (e.g., a thiazolidinedione (TZD) [such as ioglitazone, rosiglitazone, rivoglitazone, or troglitazone], aleglitazar, farglitazar, muraglitazar, or tesaglitazar), a statin, an insulin
- absorption of the compounds is substantially limited to the intestines.
- the compound substantially enhances FXR target gene expression in the intestines while not substantially enhancing FXR target gene expression in the liver or kidney.
- administering the compounds reduces or prevents diet-induced weight gain and/or increases a metabolic rate in the subject.
- Increasing the metabolic rate may include enhancing oxidative phosphorylation in the subject.
- administering the compounds results in no substantial change in food intake and/or fat consumption in the subject, and/or no substantial change in appetite in the subject.
- Administering the compounds can protect against diet-induced weight gain, reduce inflammation, enhance thermogenesis, enhance insulin sensitivity in the liver, reduce hepatic steatosis, promote browning of white adipose tissue (WAT), promote activation of brown adipose tissue (BAT), decrease blood glucose, increase weight loss, or any combination thereof.
- administering the compounds enhances insulin sensitivity in the liver and promotes BAT activation.
- Exemplary metabolic disorders include but are not limited to: obesity, diabetes (such as a BMI of greater than 25, at least 30, at least 35, or at least 40, such as 25 to 30, 35 to 40, or over 40), insulin resistance, dyslipidemia (such as an elevated serum lipids and/or triglycerides, such as a serum LDL of at least 100 mg/dL, such as at least 130 mg/dL, at least 160 mg/dL or at least 200 mg/dL, such as 100 to 129 mg/dL, 130 to 159 mg/dL, 160 to 199 mg/dL or greater than 200 mg/dL, and/or such as a serum triglyceride of at least of at least 151 mg/dL, such as at least 200 mg/dL, or at least 500 mg/dL, such as 151 to 199 mg/dL, 200 to 499 mg/dL or greater than 499 mg/dL) or any combination thereof.
- the metabolic disorder is non- insulin dependent diabetes mellitus.
- Embodiments of a method for treating or preventing inflammation, such as inflammation in an intestinal region of a subject are also disclosed. Administering to a subject a
- the method further includes administering a therapeutically effective amount of an antibiotic (such as metronidazole, vancomycin, and/or fidaxomicin) to the subject, such as to treat or substantially prevent inflammation associated with pseudomembranous colitis in the subject.
- an antibiotic such as metronidazole, vancomycin, and/or fidaxomicin
- the method comprises administering to the subject a
- an oral corticosteroid and/or other anti-inflammatory or immunomodulatory therapy in combination with the compound, and/or in combination with an antibiotic.
- Inflammation may be associated with a clinical condition selected from necrotizing enterocolitis, gastritis, ulcerative colitis, Crohn's disease, inflammatory bowel disease, irritable bowel syndrome, gastroenteritis, radiation induced enteritis, pseudomembranous colitis, chemotherapy induced enteritis, gastro-esophageal reflux disease (GERD), peptic ulcer, non- ulcer dyspepsia (NUD), celiac disease, intestinal celiac disease, post-surgical inflammation, gastric carcinogenesis or any combination thereof.
- the one or more FXR target genes comprises IBABP, OSTcc, Perl, FGF15, FGF19, or combinations thereof.
- Embodiments of a method for treating or preventing a cell proliferation disease e.g., cancer, such as adenocarcinoma, such as cancer of the colon, jejunum, and/or ileum
- a cell proliferation disease e.g., cancer, such as adenocarcinoma, such as cancer of the colon, jejunum, and/or ileum
- Administering to a subject a therapeutically effective amount of one or more of the disclosed compounds, or one or more of the disclosed compositions, such as 1, 2, 3, 4, or 5 of such compounds and/or compositions activates FXR receptors in the intestines, thereby treating or substantially preventing a cell proliferation disease, for example in the intestinal region of the subject.
- the method further includes administering a therapeutically effective amount of another therapeutic agent, (such as a chemo therapeutic, a biologic, a radiotherapeutic, or combinations thereof) to the subject, such as to treat or substantially prevent a cell proliferation disease in the subject.
- a therapeutically effective amount of another therapeutic agent such as a chemo therapeutic, a biologic, a radiotherapeutic, or combinations thereof
- the method may increase HSL phosphorylation and p3-adrenergic receptor expression (such as an increase of at least 20%, at least 25%, at least 30%, at least 40%, at least 50%, at least 75%, or at least 100%). Additionally, the serum concentration of the compound in the subject may remain below its EC50 following
- FIGS. 1A-1C are a comparative expression chart and two bar charts, respectively, illustrating increased levels of FXR target gene expression in the intestine relative to expression in the liver and kidney.
- 8 week-old C57BL/6J mice were treated with vehicle or fexaramine (100 mg/kg) via oral (PO) or intraperitoneal (IP) injection for three days (FIGS. 1A-1B) or five days (FIG. 1C).
- FIG. 1A shows FXR target SHP gene expression in FXR abundant tissues including liver, kidney and intestine from 8 week-old mice that were treated with vehicle or fexaramine (100 mg/kg) via oral (PO) or intraperitoneal (IP) injection for three days.
- FXR target gene expression was analyzed by qPCR. Gene expression was normalized against a vehicle-treated group.
- FIG. IB shows that PO administration of fexaramine (solid bars), but not vehicle (open bars), substantially enhances FXR target gene expression in the intestine, and not in the liver or kidney.
- FIG. 1C shows that IP injection of fexaramine increases FXR target gene expression in the liver and kidney, in addition to the intestines. Data represent the mean + SD. Statistical analysis was performed with the Student's t test. *p ⁇ 0.05, **p ⁇ 0.01
- FIG. ID is a schematic diagram illustrating an experimental procedure used to evaluate fexaramine, where mice were treated with vehicle or fexaramine (100 mg/kg) via PO or IP injection, and LC/MS quantification of serum fexaramine was conducted five days later.
- FIG. IE is a bar chart illustrating serum fexaramine concentrations after administration as described in FIG. ID. Data represent mean values + STD. Statistical analysis was performed with the Student's t test (*p ⁇ 0.05, **p ⁇ 0.01).
- FIG. IF is a bar chart illustrating that orally delivered fexaramine is intestinally- restricted. Mice received vehicle or Fexaramine (lOOmg/kg) via per os (PO) or intraperitoneal (IP) injection for 5 days. Expression of the FXR target gene SHP after PO or IP injection in selected tissues is shown.
- FIGS. 2A-2G are graphs illustrating the reduction of diet-induced obesity and improvement in metabolic homeostasis with fexaramine.
- Mice were fed a high fat diet (HFD) for 14 weeks and then administered daily oral injections of vehicle (open boxes) or fexaramine (100 mg/kg) (solid boxes) for 5 weeks with HFD. Data represent the mean + STD.
- Statistical analysis was performed with the Student's t test (*p ⁇ 0.05, **p ⁇ 0.01).
- FIG. 2B shows mice body weight composition by MRI at the completion of the study.
- FIG. 2C shows the wet weight of inguinal fat (iWAT), gonadal fat (gWAT), mesenteric fat (mWAT), liver, kidney, heart and spleen at the completion of the study.
- iWAT inguinal fat
- gWAT gonadal fat
- mWAT mesenteric fat
- FIG. 2D shows the serum levels (samples were collected after 8 hours-fasting for parameter analysis) of insulin, cholesterol, leptin, resistin and triglycerides.
- FIG. 2E shows the serum levels of cytokines at the completion of the study.
- FIG. 2F is a line graph representing glucose tolerance testing (GTT), which revealed that fexaramine treatment improved glucose clearance.
- FIG. 2G is a line graph representing insulin tolerance testing (ITT), which showed that fexaramine treatment improved insulin sensitivity.
- FIGS. 3A-3D are line graphs and a bar graph showing the effects of fexaramine administration in normal chow-fed mice.
- the mice were treated with vehicle (left bar) or fexaramine (100 mg/kg) (right bar) via PO for 5 weeks. Data represent the mean + STD.
- FIG. 3A is a line graph showing hourly composite carbon dioxide production.
- FIG. 3B is a line graph showing hourly composite oxygen consumption.
- FIG. 3C is a glucose tolerance test.
- FIG. 3D is a bar graph showing core body temperature.
- FIG. 4A is a line graph showing the effects of fexaramine at various dosage levels on the body weight of mice fed a HFD for 14 weeks and then administered daily oral injections of vehicle or fexaramine (10, 50 or 100 mg/kg) for 5 weeks with HFD. Data represent the mean + STD. Statistical analysis was performed with the Student's t test (*p ⁇ 0.05, **p ⁇ 0.01).
- FIG. 4B is a set of digital images showing histological analysis of the ileum and colon following treatment with fexaramine or vehicle. Mice were fed on HFD for 14 weeks, and then administered daily oral injections of vehicle or fexaramine (100 mg/kg) for 5 weeks with HFD.
- FIG. 4C is a line graph showing glucose tolerance tests in mice fed a HFD for 14 weeks and then administered daily oral injections of vehicle or fexaramine (10, 50 or 100 mg/kg) for 5 weeks with HFD. Data represent the mean + STD. Statistical analysis was performed with the Student's t test (*p ⁇ 0.05, **p ⁇ 0.01).
- FIG. 4D is a line graph showing fasting glucose levels in 14 week HFD-fed mice treated with vehicle or fexaramine (lOOmg/kg/day os for 5 week). Data represent the mean + STD. Statistical analysis was performed with the Student's t test (*p ⁇ 0.05, **p ⁇ 0.01).
- FIGS. 5A-5I show that FXR is required for fexaramine's effects
- A Body weights
- B glucose tolerance test
- C insulin tolerance test
- D oxygen consumption
- E carbon dioxide production
- F core body temperature
- G brown adipose tissue gene expression
- H liver gene expression
- I FXR target gene expressions in ileum of 14 week HFD fed FXR-null mice treated with vehicle or fexaramine (lOOmg/kg) for 5 week with HFD.
- Data represent the mean + SD.
- Statistical analysis was performed with the Student's t test. *p ⁇ 0.05, **p ⁇ 0.01.
- FIGS. 6A-6J demonstrate that fexaramine increases OXPHOS to enhance metabolic rate in brown adipose tissue.
- Mice were fed HFD for 14 weeks and then administered vehicle or fexaramine (100 mg/kg) daily by oral administration for 5 weeks with HFD. Data represent the mean + STD.
- Statistical analysis was performed with the Student's t test (*p ⁇ 0.05, **p ⁇ 0.01).
- FIG. 6A is a bar chart showing daily food intake during the first week treatment.
- FIG. 6B is a line chart showing carbon dioxide production.
- FIG. 6C is a line chart showing oxygen consumption.
- FIG. 6D is a bar chart showing daytime and nighttime cumulative ambulatory counts.
- FIG. 6E is a bar chart showing core body temperature.
- FIG. 6F shows hematoxyin and eosin staining of brown adipose tissue (BAT) for histological analysis.
- FIG. 6G is a bar chart showing relative gene expression of nuclear receptors and other genes encoding proteins involved in mitochondrial biogenesis, glucose transport and FA oxidation in BAT.
- FIG. 6H is a set of digital images of gel electrophoreses showing protein expression levels of total and phosphorylated p38 in BAT. RalA levels are shown as a loading control.
- FIG. 61 is a bar chart showing the relative levels of phosphorylated p38 in BAT after vehicle (open bar) or Fexaramine administration (solid bar).
- FIG. 6J is a chart showing changes in relative expression of OXPHOS genes based on RN A- sequencing transcriptomic analysis in inguinal fat (iWAT), gonadal fat (gWAT) and brown fat (BAT) after vehicle or fexaramine treatment.
- iWAT inguinal fat
- gWAT gonadal fat
- BAT brown fat
- FIG. 6K is a heatmap depiction of changes in genes involved in chemokine and cytokine signaling in BAT after vehicle or fexaramine treatment.
- FIG. 6L is a bar graph showing PKA activity in BAT. Data represent the mean + SD. Statistical analysis was performed with the Student's t test. *p ⁇ 0.05, **p ⁇ 0.01.
- FIG. 6M is a bar chart showing the effect of fexaramine on respiratory exchange ratio (RER). Mice were fed on HFD for 14 weeks, and then administered daily oral injections of vehicle (solid bar) or fexaramine (100 mg/kg) (open bar) for 5 weeks with HFD. No changes were observed in respiratory exchange ratio by fexaramine treatment.
- FIG. 6N is a bar graph showing the effect of fexaramine administration on serum lactate concentrations. Mice were fed on HFD for 14 weeks, and then administered daily oral injections of vehicle (left bar) or fexaramine (100 mg/kg) (right bar) for 5 weeks with HFD. Serum lactate levels were found to be significantly decreased with fexaramine treatment. Data represent the mean + STD. Statistical analysis was performed with the Student's t test (*p ⁇ 0.05, **p ⁇ 0.01).
- FIGS. 7A-7H show a comparative expression chart and bar charts illustrating that fexaramine increased endogenous FGF15 signaling and changes in BA composition.
- mice were fed HFD for 14 weeks and then administered daily oral injections of vehicle or fexaramine (100 mg/kg) for 5 weeks with HFD.
- open bars represent vehicle treatment and solid bars represent fexaramine treatment, and data represent the mean + STD.
- Statistical analysis was performed with the Student's t test (*p ⁇ 0.05, **p ⁇ 0.01).
- FIG. 7A is a heatmap depicting changes in expression of ileal FXR target genes following PO fexaramine administration.
- FIG. 7B is a bar chart showing FGF15 protein levels from ileal extract.
- FIG. 7C is a bar chart showing FGF15 protein levels in the serum.
- FIG. 7D is a bar chart showing changes in the expression of hepatic genes involved in bile acid metabolism.
- FIG. 7E is a bar chart showing total serum bile acid (BA) levels.
- FIG. 7F is a bar chart showing composition ratios of bile acids. The ratio of
- FIG. 7G is a bar chart showing changes in intestinal permeability.
- FIG. 7H is a bar chart showing changes in expression of intestinal genes involved in mucosal defense.
- FIG. 8 is a bar graph showing hepatic Cyp7al levels determined by ELISA. Data represent the mean + SD. Statistical analysis was performed with the Student's t test. *p ⁇ 0.05, **p ⁇ 0.01.
- FIG. 9 is a bar graph showing that fexaramine fails to activate TGR5.
- HEK293 cells were transfected with expression vectors for cAMP-response element luciferase, ⁇ -galactosidase and human TGR5. 24 hours after transfection, cells were treated with fexaramine or INT-777 (a TGR5 agonist).
- FIGS. 10A-10F show that systemic TGR5 activation is required to affect glucose homeostasis.
- HFD-fed mice were treated with vehicle, the intestinally-restricted TGR5 ligand L755-0379 (A, L755, lOOmg/kg, EC50 300nM) or the systemic ligand R05527239 (B, RO, lOOmg/kg. EC50 70nM) via per os for 14 days.
- C Plasma L755 concentrations in portal and tail veins after PO administration.
- D Body weight curve.
- E Glucose tolerance test.
- F Serum insulin levels after a glucose challenge (vehicle left bar, RO middle bar, L755 right bar). Data represent the mean + SD. Statistical analysis was performed with the Student's t test. *p ⁇ 0.05, **p ⁇ 0.01.
- FIGS. 1 lA-1 IN show that TGR5 is required for a subset of fexaramine's effects.
- A Ileal FXR target gene expressions
- B Serum BA levels
- C Fasting glucose levels
- D Glucose tolerance test
- E Core body temperature
- F Oxygen consumption rate
- G Carbon dioxide production
- H Gene expression in BAT
- I Body weight curve
- J Body composition by MRI
- K Insulin Tolerance Test
- L Hepatic gene expression
- N Hepatic TG levels
- FIGS. 12A-12H demonstrate that fexaramine reduces inflammation and increases lipolysis in adipose tissues. Mice were fed on HFD for 14 weeks and subsequently subjected to daily PO injection of vehicle or fexaramine (100 mg/kg) for 5 weeks with HFD. In the bar graphs, open bars are vehicle, solid bars of fexaramine, and data represent the mean + STD.
- FIG. 12A shows histological sections of mesenteric white adipose tissues from vehicle and fexaramine-treated mice.
- FIG. 12B is a set of photographs of gel electrophoreses showing protein expression levels of TBK1, and total and phosphorylated ⁇ and S6K, in gonadal adipose tissues
- mice (gWAT) from vehicle or fexaramine-treated mice.
- FIG. 12C is a bar chart showing relative gene expression levels of ⁇ -3-adrenergic receptor and various cytokines in gonadal adipose tissue. Vehicle open bar, Fex solid bar.
- FIG. 12D is a set of photographs of gel electrophoreses showing protein expression levels of total and phosphorylated HSL (p-HSL) and p65 in gonadal and inguinal adipose tissues.
- FIG. 12E is a bar chart showing serum levels of catecholamines, in vehicle or fexaramine-treated mice. Vehicle open bar, Fex solid bar.
- FIG. 12F is a bar chart showing serum glycerol levels, in vehicle or fexaramine-treated mice. Isoproterenol ( ⁇ g/kg) was injected at 0 minutes and free glycerol levels were measured at the indicated time points. Vehicle left bar, Fex right bar.
- FIG. 12G is a bar chart showing serum levels of free fatty acids in vehicle or fexaramine- treated mice. Data represent the mean + STD. Statistical analysis was performed with the Student's t test (*p ⁇ 0.05, **p ⁇ 0.01). Vehicle open bar, Fex solid bar.
- FIG. 12H shows UCP1 staining of brown fat-like cells in inguinal adipose tissues (iWAT) from vehicle or fexaramine-treated mice (Magnification: 100X).
- FIGS. 121 and 12J show that fexaramine enhances OXPHOS in iWAT.
- Mice fed a HFD for 14 weeks were maintained on a HFD and treated with vehicle or fexaramine (lOOmg/kg/day os for 5 week).
- (I) Changes in genes associated with the browning of adipose tissue and
- Statistical analysis was performed with the Student's t test. *p ⁇ 0.05, **p ⁇ 0.01.
- FIG. 13 is a set of digital images of gel electrophoreses (Western blots) showing the level of expression of various proteins in gonadal white adipose tissue (gWAT). Mice fed a HFD for 14 weeks were maintained on a HFD and treated with vehicle or fexaramine (50mg or lOOmg/kg/day os for 5 week).
- FIG. 14 is a bar chart showing that fexaramine reduces brown adipose tissue (BAT) inflammation.
- Mice fed a HFD for 14 weeks were maintained on a HFD and treated with vehicle or fexaramine (lOOmg/kg/day os for 5 week).
- Data represent the mean + SD.
- Statistical analysis was performed with the Student's t test. *p ⁇ 0.05, **p ⁇ 0.01.
- FIGS. 15A-15H are a set of histology stains and bar charts demonstrating that fexaramine induced less weight gain and improved glucose homeostasis relative to mice that did not receive fexaramine. Mice were fed HFD for 14 weeks and then subjected to daily PO injection of vehicle (open bar in bar graphs) or fexaramine (100 mg/kg) (solid bar in bar graphs) for 5 weeks with HFD.
- FIG. 15A is a bar chart showing basal hepatic glucose production (HGP).
- FIG. 15B is a bar chart showing glucose disposal rate (GDR).
- FIG. 15C is a bar chart showing percentage free fatty acid (FFA) suppression by insulin.
- FIG. 15D is a bar chart showing HGP suppression by insulin, as measured by hyperinsulinemic-euglycemic clamps.
- FIG. 15E shows hematoxylin and eosin staining for liver histology.
- FIG. 15F is a bar chart showing triglyceride levels in the liver.
- FIG. 15G is a bar chart showing hepatic gene expression levels for genes involved in gluconeogenesis and lipogenesis.
- FIG. 15H is a bar chart showing serum levels of alanine aminotransferase (ALT).
- FIGS. 151- 15K are a line graph and two bar graphs showing the effect of fexaramine treatment on body weight, insulin-stimulated GDR, and fasting insulin levels.
- Mice were fed HFD for 14 weeks, and then administered daily oral injections of vehicle or fexaramine (100 mg/kg) for 3 weeks with HFD. The mice treated with fexaramine were initially heavier (by 2-3 grams). Three weeks after treatment, a clamp study was performed on the mice. Data represent the mean + STD. Statistical analysis was performed with the Student's t test (*p ⁇ 0.05,
- FIG. 151 is a line graph showing the changes in body weight for the two groups of mice. Vehicle bottom line, Fex, top line.
- FIG. 15J is a bar chart showing the insulin-stimulated GDR (IS-GDR). Vehicle left bar, Fex, right bar.
- FIG. 15K is a bar chart showing the fasting insulin levels. Vehicle left bar, Fex, right bar.
- amino acid sequences are shown using standard three letter code for amino acids, as defined in 37 C.F.R. 1.822.
- SEQ ID NO. 1 is a protein sequence of GLP-l-(7-36).
- SEQ ID NO. 2 is a protein sequence of GLP-2.
- Aliphatic refers to a substantially hydrocarbon-based compound, or a radical thereof (e.g., C6Hi3, for a hexane radical), including alkanes, alkenes, alkynes, including cyclic versions thereof, such as alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, and cycloalkynyl, and further including straight- and branched-chain arrangements, and all stereo and position isomers as well.
- an aliphatic group contains from one to at least twenty-five carbon atoms; for example, from one to fifteen, from one to ten, from one to six, or from one to four carbon atoms.
- lower aliphatic refers to an aliphatic group comprising from one to ten carbon atoms.
- An aliphatic chain may be substituted or unsubstituted. Unless expressly referred to as an "unsubstituted aliphatic," an aliphatic group can either be unsubstituted or substituted.
- Exemplary aliphatic substituents include, for instance, amino, amide, sulfonamide, halo, cyano, carboxy, hydroxyl, mercapto, trifluoromethyl, alkyl, alkoxy, acetoxy, alkylthio, thioalkoxy, arylalkyl, heteroaryl, alkylamino, dialkylamino, or other functionality.
- D-aliphatic refers to an aliphatic group where at least one hydrogen has been substituted by deuterium.
- Amino refers to the group -NR'R", wherein R' and R" independently are selected from hydrogen, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic, or where R' and R" are optionally joined together with the nitrogen bound thereto to form a cycloamino group such as a heterocyclic, deuterated heterocyclic, heteroaryl or deuterated heteroaryl group comprising at least one ring nitrogen.
- Exemplary cycloamino groups include, but are not limited to, pyrrolidine, pyrrole, imidazole, triazole, tetrazole, piperidine, triazinane, piperazine, morpholine, azepane, diazepane, azocane, diazocane, azonane or azecane.
- a primary aminocarbonyl is -CONH2.
- cyano refers to the chemical functional group -CN.
- Carboxyl refers to the chemical functional group -CO2H.
- carboxyl ester refers to the chemical functional group -CO2R where R is aliphatic, D-aliphatic, heteroaliphatic or D- heteroaliphatic.
- amino sulfonyl refers to a chemical function group -SC -amino, where amino is as defined herein.
- a primary aminosulfonyl is -SO2NH2.
- acyl means, unless otherwise stated, -C(0)R where R is aliphatic, D- aliphatic, heteroaliphatic or D-heteroaliphatic.
- aryl refers to a monovalent aromatic carbocyclic group of from 6 to 15 carbon atoms having a single ring (e.g., phenyl) or multiple condensed rings (e.g., naphthyl or anthryl) in which at least one of the condensed rings is aromatic (e.g., 2-benzoxazolinone, 2H-l,4-benzoxazin-3(4H)-one-7-yl, 9,10-dihydrophenanthrene, and the like), provided that the point of attachment is through an atom of the aromatic aryl group. Unless otherwise specified, the aryl group may be optionally substituted. Preferred aryl groups include phenyl and naphthyl.
- Heteroaliphatic refers to an aliphatic compound or group having at least one heteroatom, i.e., one or more carbon atoms has been replaced with an atom having at least one lone pair of electrons, typically nitrogen, oxygen, phosphorus, silicon, or sulfur. Heteroaliphatic compounds or groups may be substituted or unsubstituted, branched or unbranched, cyclic or acyclic, and include “heterocycle”, “heterocyclyl”, “heterocycloaliphatic", or “heterocyclic” groups. Examples of heterocycles include morpholine and piperidine. "D-heteroaliphatic” refers to a heteroaliphatic group where at least one hydrogen has been substituted by a deuterium.
- Halo refers to fluoro, chloro, bromo, and iodo, and is preferably fluoro or chloro.
- Heteroaryl refers to an aromatic group having from 1 to 15 carbon atoms and at least one, and more typically 1 to 4, heteroatoms selected from oxygen, nitrogen or sulfur within the ring. Unless otherwise specified, the heteroaryl group may be optionally substituted.
- Such heteroaryl groups can have a single ring (e.g., pyridinyl, imidazolyl or furyl) or multiple condensed rings (e.g., indolizinyl, quinolinyl, benzimidazolyl, benzopyrazolyl or benzothienyl), wherein at least one of the condensed rings is aromatic and may or may not contain a heteroatom, provided that the point of attachment is through an atom of an aromatic ring.
- the nitrogen and/or sulfur ring atom(s) of the heteroaryl group are optionally oxidized to provide N-oxide (N ⁇ 0), sulfinyl, or sulfonyl moieties.
- Preferred heteroaryls include pyridinyl, pyrrolyl, indolyl, thiophenyl, benzopyrazolyl and furanyl.
- “Sulfonyl” refers to the group -SO2-, and includes -SC -aliphatic, -SC -aryl,
- Sulfonyl includes groups such as methyl-S0 2 -, phenyl-S0 2 -, and 4- methylphenyl-SC -.
- carboxyl bioisosteric refers to a group with similar physical or chemical properties to a carboxyl groupthat produce broadly similar biological properties, but which may reduce toxicity or modify the activity of the compound, and may alter the metabolism of the compound.
- exemplary carboxyl bioisosteres include, but are not limited where X , Y , and Z are each independently
- a group that is substituted has 1 substituent, 1 or 2 substituents, 1, 2, or 3 substituents or 1, 2, 3 or 4 substituents.
- impermissible substitution patterns are understood by a person having ordinary skill in the art.
- “Pharmaceutically acceptable salt” refers to pharmaceutically acceptable salts of a compound, which salts are derived from a variety of organic and inorganic counter ions well known in the art and include, by way of example only, sodium, potassium, calcium, magnesium, ammonium, tetraalkylammonium, and the like. If the molecule contains a basic functionality, pharmaceutically acceptable salts include salts of organic or inorganic acids, such as
- hydrochloride hydrobromide, tartrate, mesylate, acetate, maleate, oxalate, and the like.
- “Pharmaceutically acceptable excipient” refers to a substantially physiologically inert substance that is used as an additive in a pharmaceutical composition. As used herein, an excipient may be incorporated within particles of a pharmaceutical composition, or it may be physically mixed with particles of a pharmaceutical composition. An excipient can be used, for example, as a carrier, flavoring agent, thickener, diluent, buffer, preservative, or surface active agent and/or to modify properties of a pharmaceutical composition.
- excipients include, but are not limited, to polyvinylpyrrolidone (PVP), tocopheryl polyethylene glycol 1000 succinate (also known as vitamin E TPGS, or TPGS), dipalmitoyl phosphatidyl choline (DPPC), trehalose, sodium bicarbonate, glycine, sodium citrate, and lactose.
- PVP polyvinylpyrrolidone
- DPPC dipalmitoyl phosphatidyl choline
- trehalose sodium bicarbonate
- glycine sodium citrate
- lactose lactose
- Enteric coating refers to a coating such as may be applied to disclosed compounds or compositions comprising the compounds to help protect drugs from disintegration, digestion etc. in the stomach, such as by enzymes or the pH of the stomach. Typically, the coating helps prevent the drug from being digested in the stomach, and allows delivery of the medication to the intestine.
- administer refers to methods that may be used to enable delivery of agents or compositions to the desired site of biological action. These methods include, but are not limited to oral routes, intraduodenal routes and rectal administration. Administration techniques that are optionally employed with the agents and methods described herein are found in sources e.g., Goodman and Gilman, The Pharmacological Basis of Therapeutics, current ed.; Pergamon; and Remington's,
- agents and compositions described herein are administered orally.
- calorie refers to the amount of energy, e.g. heat, required to raise the temperature of 1 gram of water by 1 °C.
- the term “calorie” is often used to describe a kilocalorie.
- a kilocalorie is the amount of energy needed to increase the temperature of 1 kilogram of water by 1 °C.
- One kilocalorie equals 1000 calories.
- the kilocalorie is abbreviated as kc, kcal or Cal, whereas the calorie or gram calorie is abbreviated as cal.
- food intake in the subject is measured in terms of overall calorie consumption.
- fat intake can be measured in terms of calories from fat.
- co-administration are meant to encompass administration of the selected therapeutic agents to a single patient, and are intended to include treatment regimens in which the agents are administered by the same or different route of administration or at the same or different times.
- the agents described herein will be co-administered with other agents.
- These terms encompass administration of two or more agents to the subject so that both agents and/or their metabolites are present in the subject at the same time. They include simultaneous administration in separate compositions, administration at different times in separate
- compositions and/or administration in a composition in which both agents are present.
- the agents described herein and the other agent(s) are administered in a single composition.
- the agents described herein and the other agent(s) are admixed in the composition.
- an “effective amount,” “pharmaceutically effective amount” or “therapeutically effective amount” as used herein, refer to a sufficient amount of at least one agent being administered to achieve a desired result, e.g., to relieve to some extent one or more symptoms of a disease or condition being treated. In certain instances, the result is a reduction and/or alleviation of the signs, symptoms, or causes of a disease, or any other desired alteration of a biological system. In certain instances, an “effective amount” for therapeutic uses is the amount of the composition comprising an agent as set forth herein required to provide a clinically significant decrease in a disease. An appropriate "effective" amount in any individual case can be determined using any suitable technique, such as a dose escalation study.
- Enhancing enteroendocrine peptide secretion refers to a sufficient increase in the level of the enteroendocrine peptide agent to, for example, decrease hunger in a subject, to curb appetite in a subject and/or decrease the food intake of a subject or individual and/or treat any disease or disorder described herein.
- FXR farnesoid X receptor (also known as nuclear receptor subfamily 1, group H, member 4 (NR1H4)) (OMIM: 603826): This protein functions as a receptor for bile acids, and when bound to bile acids, regulates the expression of genes involved in bile acid synthesis and transport. FXR is expressed at high levels in the liver and intestine. Chenodeoxycholic acid and other bile acids are natural ligands for FXR. Similar to other nuclear receptors, when activated, FXR translocates to the cell nucleus, forms a dimer (in this case a heterodimer with RXR) and binds to hormone response elements on DNA, which up- or down-regulates the expression of certain genes.
- NR1H4 nuclear receptor subfamily 1, group H, member 4
- FXR activation is the suppression of cholesterol 7 alpha-hydroxylase (CYP7A1), the rate-limiting enzyme in bile acid synthesis from cholesterol.
- CYP7A1 cholesterol 7 alpha-hydroxylase
- FXR does not directly bind to the CYP7A1 promoter. Rather, FXR induces expression of small heterodimer partner (SHP), which then functions to inhibit transcription of the CYP7A1 gene. In this way, a negative feedback pathway is established in which synthesis of bile acids is inhibited when cellular levels are already high.
- FXR sequences are publically available, for example from GenBank® sequence database (e.g., accession numbers NP_001193906 (human, protein) and NP_001156976 (mouse, protein), and NM_001206977 (human, nucleic acid) and NM_001163504 (mouse, nucleic acid)).
- GenBank® sequence database e.g., accession numbers NP_001193906 (human, protein) and NP_001156976 (mouse, protein), and NM_001206977 (human, nucleic acid) and NM_001163504 (mouse, nucleic acid)).
- metabolic disorder refers to any disorder that involves an alteration in the normal metabolism of carbohydrates, lipids, proteins, nucleic acids or a combination thereof.
- a metabolic disorder is associated with either a deficiency or excess in a metabolic pathway resulting in an imbalance in metabolism of nucleic acids, proteins, lipids, and/or carbohydrates.
- Factors affecting metabolism include, but are not limited to, the endocrine (hormonal) control system (e.g., the insulin pathway, the enteroendocrine hormones including GLP-1, GLP-2, oxyntomodulin, PYY or the like), the neural control system (e.g. , GLP-1 in the brain) or the like.
- Examples of metabolic disorders include and are not limited to diabetes, insulin resistance, dyslipidemia, metabolic syndrome, or the like.
- the term "metabolic rate” refers to the rate at which the subject uses energy. This is also known as the rate of metabolism, or the rate of energy consumption, and reflects the overall activity of the individual's metabolism.
- basal metabolism refers to the minimum amount of energy required to maintain vital functions in an individual at complete rest, measured by the basal metabolic rate in a fasting individual who is awake and resting in a comfortably warm environment.
- basal metabolic rate refers to the rate at which energy is used by an individual at rest. Basal metabolic rate is measured in humans by the heat given off per unit time, and expressed as the calories released per kilogram of body weight or per square meter of body surface per hour. The heart beating, breathing, maintaining body temperature, and other basic bodily functions all contribute to basal metabolic rate.
- Basal metabolic rate can be determined to be the stable rate of energy metabolism measured in individuals under conditions of minimum environmental and physiological stress, or essentially at rest with no temperature change.
- the basal metabolic rate among individuals can vary widely.
- One example of an average value for basal metabolic rate is about 1 calorie per hour per kilogram of body weight.
- non- systemic or “minimally absorbed” as used herein refer to low systemic bioavailability and/or absorption of an administered compound.
- a non- systemic compound is a compound that is substantially not absorbed systemically.
- FXR agonist compositions described herein deliver an FXR agonist to the distal ileum, colon, and/or rectum and not systemically (e.g., a substantial portion of the FXR agonist administered is not systemically absorbed).
- the systemic absorption of a non-systemic compound is ⁇ 0.1 , ⁇ 0.3 , ⁇ 0.5%, ⁇ 0.6%, ⁇ 0.7%, ⁇ 0.8%, ⁇ 0.9%, ⁇ %, ⁇ 1.5 , ⁇ 2 , ⁇ 3 , or ⁇ 5 of the administered dose (wt. % or mol %).
- the systemic absorption of a non-systemic compound is ⁇ 15 of the administered dose.
- the systemic absorption of a non-systemic compound is ⁇ 25 of the administered dose.
- a non-systemic FXR agonist is a compound that has lower systemic bioavailability relative to the systemic bioavailability of a systemic FXR agonist.
- the bioavailability of a non-systemic FXR agonist described herein is ⁇ 30 , ⁇ 40 , ⁇ 50 , ⁇ 60 , or ⁇ 70 of the bioavailability of a systemic FXR agonist.
- the serum concentration of the FXR agonist in the subject remains below the compound's EC50 following administration.
- prevent include preventing additional symptoms, preventing the underlying metabolic causes of symptoms, inhibiting the disease or condition, e.g., arresting the development of the disease or condition and are intended to include prophylaxis.
- the terms further include achieving a prophylactic benefit.
- the compositions are optionally administered to a patient at risk of developing a particular disease, to a patient reporting one or more of the physiological symptoms of a disease, or to a patient at risk of reoccurrence of the disease.
- subject may be used interchangeably herein and refer to mammals and non-mammals, e.g., suffering from a disorder described herein.
- mammals include, but are not limited to, any member of the mammalian class: humans, non- human primates such as chimpanzees, and other apes and monkey species; farm animals such as cattle, horses, sheep, goats, swine; domestic animals such as rabbits, dogs, and cats; laboratory animals including rodents, such as rats, mice and guinea pigs, and the like.
- non- mammals include, but are not limited to, birds, fish, amphibians, and the like.
- the mammal is a human.
- treat include alleviating, inhibiting or reducing symptoms, reducing or inhibiting severity of, reducing incidence of, prophylactic treatment of, reducing or inhibiting recurrence of, preventing, delaying onset of, delaying recurrence of, abating or ameliorating a disease or condition symptoms, ameliorating the underlying metabolic causes of symptoms, inhibiting the disease or condition, e.g., arresting the development of the disease or condition, relieving the disease or condition, causing regression of the disease or condition, relieving a condition caused by the disease or condition, or stopping the symptoms of the disease or condition.
- the terms further include achieving a therapeutic benefit.
- Therapeutic benefit means eradication or amelioration of the underlying disorder being treated, and/or the eradication or amelioration of one or more of the physiological symptoms associated with the underlying disorder, such that an improvement is observed in the patient.
- a cell proliferative disorder such as cancer
- an FXR agonist such as one of the novel FXR agonists disclosed herein.
- the absorption of these FXR agonists may be substantially restricted to the intestinal lumen when delivered orally.
- administration of one or more of the disclosed FXR agonists may result in activation of FXR transcriptional activity in the intestine, without substantially affecting other target tissues, such as liver or kidney.
- target tissues such as liver or kidney.
- chronic administration with these agonists may lead to beneficial body- wide effects in obese subjects.
- the disclosed FXR agonists may have potent anti-obesity and glucose lowering effects in vivo. These effects have not been observed with systemically-acting FXR ligands and may include reductions in weight gain, hyperglycemia, and/or insulin resistance.
- administration of these FXR agonists may produce a beneficial, anti-inflammatory effect in the intestines.
- a compound that may have activity as an FXR agonist include compounds of Formula I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, XIII, XIV, XV, XVI and XVII. Certain compounds are chiral, and all stereoisomers are included in this disclosure, as well as all geometric and structural isomers such as cis and trans isomers.
- R -R independently are selected from hydrogen, deuterium, halogen, CF3, NO2, OH, amino, acyl, carboxyl, carboxyl ester, cyano, aminocarbonyl, aminosulfonyl, aliphatic, D-aliphatic, heteroaliphatic, D-heteroaliphatic, or -(CH 2 )ni-R 150 -
- R 150 is O, NR 16 , or absent, and R 151 is carboxyl ester or amino;
- R 16 is selected from hydrogen, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic;
- R a and R b are
- R x -R 16 is -R x -L x -R x2 , where R x is selected from
- R x3 is selected from H, aliphatic, or aryl
- L x is selected from a bond, aliphatic, heteroaliphatic, aryl, heteroaryl or CR x4 R x5
- R x4 and R" 5 are each independently selected from H, D, halogen, aliphatic, -C(0)OR x6 , or -C(0)NR x6 R x7
- R x6 and R x7 are each independently selected from H, aliphatic
- R x2 is selected from -C(0)L x2 R x8 or a carboxyl bioisostere
- L x2 is a bond or NR x3
- R x8 is H, aliphatic, -OR x9 , N(R x9 ) 2 , -C(0)R x9 , -S(0) 2 R x9 , - C(0)OR x
- At least one of R x -R 16 is or comprises deuterium.
- R 7 may be H, aliphatic, heteroaliphatic or D-heteroaliphatic.
- R 7 is alkyl or deuterated alkyl, and in certain embodiments, R 7 is isopropyl or deuterated isopropyl, having from 1 to 7 deuterium atoms.
- At least one of R x -R 5 is a halogen.
- R 2 and R 3 are both fluoro.
- R 16 is hydrogen
- R 10 and R 11 independently are alkyl or deuterated alkyl, and in certain examples, R 10 and R 11 independently are methyl or deuterated methyl, having from 1 to 3 deuterium atoms.
- R a and R b together form a pi-bond, leading to compounds have formula II
- R ⁇ -R 16 are as defined above with respect to formula I, and at least one of R x -R 15 is or comprises deuterium.
- R a and R b are both hydrogen, leading to compounds having a formula III
- R 21 -R 34 independently are selected from hydrogen, deuterium, halogen, CF3, NO2, OH, amino, acyl, carboxyl, carboxyl ester, cyano, aminocarbonyl, aminosulfonyl, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic;
- R 35 is aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic;
- R 36 is hydrogen, aliphatic, D- aliphatic, heteroaliphatic or D-heteroaliphatic;
- R 37 is hydrogen, deuterium, halogen, CF3, NO2, OH, amino, acyl, carboxyl, carboxyl ester, cyano, aminocarbonyl, aminosulfonyl, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic.
- at least one of R 21 -R 34 independently are selected from hydrogen, deuterium, hal
- R 21 -R 37 is -R x -L x -R x2 , where R x is selected from O, NR x3 , sulfonyl or S; R x3 is selected from H, aliphatic, or aryl; L x is selected from a bond, aliphatic, heteroaliphatic, aryl, heteroaryl or CR ⁇ R* 5 ; R x4 and R" 5 are each independently selected from H, D, halogen, aliphatic, -C(0)OR x6 , or -C(0)NR x6 R x7 ; R x6 and R x7 are each independently selected from H, aliphatic; R x2 is selected from -C(0)L x2 R x8 or a carboxyl bioisostere; L x2 is a bond or NR x3 ; R x8 is H, aliphatic, -OR x9 , N
- R 35 is alkyl, cycloalkyl, deuterated alkyl or deuterated cycloalkyl.
- R 35 is cycloalkyl or deuterated cycloalkyl, typically cyclohexyl or deuterated cyclohexyl, having from 1 to 11 deuterium atoms.
- R 36 is hydrogen
- R 32 is carboxyl and/or R 34 is CF3.
- R 23 is halogen, and in certain embodiments R 23 is chloro.
- the compound is chiral, and in certain embodiments, the compound is the S- stereoisomer.
- X is N, leading to compounds having a formula V
- R 21 -R 36 is as defined above with respect to formula IV, and at least one of R 2 comprises deuterium.
- X is CH, leading to compounds having formula VI
- R 21 -R 36 is as defined above with respect to formula IV.
- Exemplary compounds having formula IV include:
- R 41 -R 48 and R 52 -R 55 independently are selected from hydrogen, deuterium, halogen, CF 3 , N0 2 , OH, amino, acyl, carboxyl, carboxyl ester, cyano,
- R 49 - R 51 independently are selected from hydrogen, deuterium, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic;
- R 56 is amino, cycloamino or substituted cycloamino, such as 5-, 6-, or 7- membered cycloamino;
- Y and Z are independently N or CR ; and each R independently is selected from deuterium, halogen, CF3, NO2, OH, amino, acyl, carboxyl, carboxyl ester, cyano, aminocarbonyl, aminosulfonyl, aliphatic, D-aliphatic, heteroaliphatic or D-heteroaliphatic.
- R 41 -R 57 is -R x -L x -R x2 , where R x is selected from O, NR x3 , sulfonyl or S; R x3 is selected from H, aliphatic, or aryl; L x is selected from a bond, aliphatic, heteroaliphatic, aryl, heteroaryl or CR ⁇ R" 5 ; R x4 and R* 5 are each independently selected from H, D, halogen, aliphatic, -C(0)OR x6 , or -C(0)NR x6 R x7 ; R x6 and R x7 are each independently selected from H, aliphatic; R x2 is selected from -C(0)L x2 R x8 or a carboxyl bioisostere; L x2 is a bond or NR x3 ; R x8 is H, aliphatic, -OR x9 ,
- At least one of R 41 -R 56 is or comprises deuterium.
- R 51 is an aliphatic or D-aliphatic, and in certain embodiments, R 5 is a methyl or deuterated methyl, having from 1 to 3 deuterium atoms.
- R 49 and R 50 independently are hydrogen or deuterium.
- R 41 and R 45 independently are aliphatic or D-aliphatic, and in particular embodiments, R 41 and R 45 are methyl or deuterated methyl, having from 1 to 3 deuterium atoms.
- R 56 is a cycloamino or substituted cycloamino, such as pyrrolidine, 2-methylpyrrolidine, morpholine, 4-methylpiperazine, piperidine, or azepane (homopiperidine) .
- Y is N and Z is N leadin to compounds having a formula VIII
- Y is CH and Z is CH leading to compounds having a formula IX
- Y is N and Z is CH leadin to compounds having a formula X
- R 41 -R 56 are as defined for formula VII.
- Exemplary compounds having formula VII include: Also disclosed herein are com ounds having formula XII,
- R 100 and R 101 are independently H, D, lower alkyl, halogen, or CF 3 ;
- R 102 is lower alkyl;
- R 103 and R 104 are independently H, D, lower alkyl, halogen, CF 3 , OH, O-alkyl, or O-polyhaloalkyl;
- R 105 and R 106 are each independently H, D, halogen, alkyl or deuterated alkyl;
- R 107 and R 108 are each independently H, D, alkyl, deuterated alkyl or halogen.
- At least one of R 100 , R 101 , R 102 , R 103 , R 104 , R 105 , R 106 , R 107 and R 108 is or comprises deuterium. In some embodiments, at least one of R 105 , R 106 , R 107 and R 108 is or comprises deuterium. In other embodiments, at least one of R 107 and R 108 is halogen, and may be fluoro.
- the compound has a formula XIII
- G 1 is CH or N; G 2 is O or NH; R 100 and R 101 are independently H, lower alkyl, halogen, or CF 3 ; R 102 is lower alkyl; R 103 and R 104 are independently H, lower alkyl, halogen, CF 3 , OH, O-alkyl, or O-polyhaloalkyl.
- Exemplary compounds having formula XII or formula XIII include
- R 205 is selected from the group consisting of COOR 210 , CONR 211 R 212 , tetrazolyl, S0 2 NR 211 R 212 , Ci-6 alkyl, S0 2 -Ci- 6 alkyl and H, with R 210 independently selected from the group consisting of H or Ci-6 alkyl, and R 211 and R 212 independently from each other selected from the group consisting of H, Ci-6 alkyl, halo-Ci-6 alkyl, Ci-6 alkylene-R 213 , S0 2 -Ci-6 alkyl, wherein R 213 is selected from the group consisting of COOH, OH and SO3H;
- R 206 is selected from the group consisting of phenyl, pyridyl, pyrimidyl, pyrazolyl, indolyl, thienyl, benzothienyl, indazolyl, benzisoxazolyl, benzofuranyl, benzotriazolyl, furanyl, benzothiazolyl, thiazolyl, oxadiazolyl, each optionally substituted with one or two groups independently selected from the group consisting of OH, O-Ci-6 alkyl, O-halo-Ci-6 alkyl, Ci-6 alkyl, halo-Ci-6 alkyl, C3-6 cycloalkyl, D and halogen;
- R 207 is selected from N or CH;
- R 208 is selected from the group consisting of phenyl, pyridyl, thiazolyl, thiophenyl, pyrimidyl, each optionally substituted with one or two groups independently selected from the group consisting of D, Ci-6 alkyl, halo-Ci-6 alkyl, halogen and CF3;
- R 209 is selected from
- R CH, N, NO, CD
- R 215 is selected from the group consisting of hydrogen, C1-3 alkyl, C3.6 cylcoalkyl, C4.5 alkylcycloalkyl, wherein C1-3 alkyl is optionally substituted with 1 to 3 substituents independently selected from halogen, hydroxy or Ci-6 alkoxy;
- R 216 and R 217 are independently selected from the group consisting of hydrogen, D, Ci- 3 alkyl, C1-3 haloalkyl, C1-3 alkoxy, C1-3 haloalkoxy, D-aliphatic and halogen.
- R 218 and R 219 are each independently H or D. In some embodiments, R 218 and R 219 are both H. In other embodiments, at least one of R 218 and R 219 is D. In some embodiments, the compound comprises at least one deuterium. In some embodiments, R 206 and/or R 208 comprise at least one deuterium. In other embodiments, R 214 is CD. In certain embodiments, at least one of R 216 and R 217 is or comprises deuterium.
- R 318 is selected from the group consisting of COOR 322 , CONR 323 R 324 , tetrazolyl or H, with R independently selected from the group consisting of H, or lower alkyl, and R and R 324 independently from each other selected from the group consisting of H, lower alkyl, Ci-6 haloalkyl, Ci-6 alkylene-R 325 , SO2-C1-6 alkyl wherein R 325 is selected from the group consisting of COOH, OH, or S0 3 H;
- R 320 is selected from the group consisting of phenyl, pyridyl, thiazolyl, thiophenyl, pyrimidyl, each optionally substituted with one or two groups independently selected from the group consisting of lower alkyl, halogen, D or CF3;
- R is CH, N, NO;
- R327 is selected from the group consisting of hydrogen, C1-C3 alkyl, C3-C6 cylcoalkyl, C4-C5 alkylcycloalkyl, wherein C1-3 alkyl is optionally substituted with 1 to 3 substituents independently selected from halogen, hydroxy or Ci-6 alkoxy,
- R 328 and R 329 are independently selected from the group consisting of hydrogen, Ci- C3 alkyl, C1-C3 haloalkyl, C1-C3 alkoxy, C1-C3 haloalkoxy and halogen.
- R 334 and R 335 are each independently H or D. In some embodiments, at least one of R 33 and R 335 are D.
- R 320 is substituted with at least one halogen or deuterium.
- R 318 is selected from the group consisting of COOR 322 , CONR 323 R 324 , tetrazolyl or H, with R 322 , R 323 and R 324 independently selected from the group consisting of H, lower alkyl;
- R 319 is selected from the group consisting of phenyl, pyridyl, indolyl, thienyl, benzothienyl, indazolyl, benzisoxazolyl, benzofuranyl, benzotriazolyl, furanyl, benzothiazolyl, thiazolyl, each optionally substituted with one or two groups independently selected from the group consisting of OH, lower alkyl, lower cycloalkyl;
- R 320 is selected from the group consisting of phenyl, pyridyl, thiazolyl, thiophenyl, pyrimidyl, each optionally substituted with one or two groups independently selected from the group consisting of lower alkyl, halogen, D or CF3;
- R is CH, N, NO;
- R 327 is selected from the group consisting of hydrogen, C1-C3 alkyl, C1-C3 haloalkyl, C3 Ce cylcoalkyl, C4-C5 alkylcycloalkyl;
- R 328 and R 329 are independently selected from the group consisting of hydrogen, Ci- C3 alkyl, C1-C3 haloalkyl, C1-C3 alkoxy, C1-C3 haloalkoxy and halogen.
- compounds having formula XV may also have formula XVI
- compounds having formula XV may also have the formula XVII,
- R 332 is CH, CD or N;
- R 330 and R 331 are independently selected from the group consisting of H, D, lower alkyl, halogen and CF3;
- R 3i8_ R 3i9 is se i ectec i f rom
- R 327 is selected from the group consisting of isopropyl, t-butyl and cyclopropyl;
- R 328 and R 329 are independently selected from the group consisting of halogen, Ci- C 3 alkyl, methoxy and trifluoromethoxy;
- R 334 and R 335 are each independently H or D. In some embodiments, at least one of R 33 ' and R 335 are D.
- R 320 is optionally substituted phenyl, preferably substituted with one substituent, preferably halogen, or two substituents, preferably both halogen or one halogen one deuterium;
- R 326 is CH
- R 327 is cycloalkyl
- R 328 and R 329 each are halogen.
- Exemplary compounds having formula XV, XVI or XVII include:
- kits that include any FXR agonist (or composition containing such an agonist) described herein and a device for localized delivery within a region of the intestines, such as the ileum or colon.
- the device is a syringe, bag, or a pressurized container.
- compositions comprising at least one compound having formulas I-III.
- Pharmaceutical compositions comprising at least one of the disclosed compounds can be formulated for use in human or veterinary medicine. Particular formulations of a disclosed pharmaceutical composition may depend, for example, on the mode of administration (e.g., oral).
- disclosed pharmaceutical compositions include a pharmaceutically acceptable carrier in addition to at least one or two or more active ingredients, such as a compound or compounds disclosed herein.
- one or more of the disclosed compounds can be formulated with one or more of (such as 1, 2, 3, 4, or 5 of) an antibiotic (e.g., metronidazole, vancomycin, and/or fidaxomicin), statin, alpha-glucosidase inhibitor, amylin agonist, dipeptidyl-peptidase 4 (DPP-4) inhibitor (such as sitagliptin, vildagliptin, saxagliptin, linagliptin, anaglptin, teneligliptin, alogliptin, gemiglptin, or dutoglpitin), meglitinide, sulfonylurea, peroxisome proliferator- activated receptor (PPAR)-
- an antibiotic e.g., metronidazole, vancomycin, and/or fidaxomicin
- statin e.g., metronidazole, vancomycin, and/or fida
- rivoglitazone or troglitazone
- aleglitazar farglitazar
- muraglitazar or tesaglitazar
- antiinflammatory agent e.g., oral corticosteroid
- chemo therapeutic biologic, radiotherapeutic, nicotinamide ribonucleoside, analogs of nicotinamide ribonucleoside (such as those that promote NAD+ production of which is a substrate for many enzymatic reactions such as p450s which are a target of FXR, for example see Yang et al., J. Med Chem. 50:6458-61, 2007, herein incorporated by reference), and the like.
- compositions e.g., powder, pill, tablet, or capsule forms
- conventional non-toxic solid carriers can include, without limitation, pharmaceutical grades of sugars, such as mannitol or lactose, polysaccharides, such as starch, or salts of organic acids, such as magnesium stearate.
- pharmaceutical compositions can optionally contain amounts of auxiliary substances (e.g. , excipients), such as wetting or emulsifying agents, preservatives, and pH buffering agents and the like; for example, sodium acetate or sorbitan monolaurate.
- the pharmaceutical composition comprises a sufficient amount of a disclosed compound to have a desired therapeutic effect.
- the disclosed compound constitutes greater than 0% to less than 100% of the pharmaceutical composition, such as 10% or less, 20% or less, 30% or less, 40% or less, 50% or less, 60% or less, 70% or less, 80% or less, 90% or less, or 90% to less than 100% of the pharmaceutical composition.
- the disclosed pharmaceutical compositions may be formulated as a pharmaceutically acceptable salt, solvate, hydrate, N-oxide or combination thereof, of a disclosed compound. Additionally, the pharmaceutical composition may comprise one or more polymorph of the disclosed compound.
- Pharmaceutically acceptable salts are salts of a free base form of a compound that possesses the desired pharmacological activity of the free base. These salts may be derived from inorganic or organic acids. Non-limiting examples of suitable inorganic acids include hydrochloric acid, nitric acid, hydrobromic acid, sulfuric acid, hydriodic acid, and phosphoric acid.
- Non-limiting examples of suitable organic acids include acetic acid, propionic acid, glycolic acid, lactic acid, pyruvic acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid,
- methanesulfonic acid methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, methyl sulfonic acid, salicylic acid, formic acid, trichloroacetic acid, trifluoroacetic acid, gluconic acid, asparagic acid, aspartic acid, benzenesulfonic acid, p-toluenesulfonic acid, naphthalenesulfonic acid, and the like.
- the compounds disclosed herein may be formulated to have a suitable particle size.
- a suitable particle size may be one which reduces or substantially precludes separation of the components of the composition, e.g. , no separation between the drug and any other components of the composition, such as a second drug, a pharmaceutically acceptable excipient, a corticosteroid, an antibiotic or any combination thereof. Additionally, the particle size may be selected to ensure the composition is suitable for delivery, such as oral delivery.
- the composition further includes an enteric coating.
- an enteric coating is a polymer barrier applied to an oral medication to help protect the drug from the acidity and/or enzymes of the stomach, esophagus and/or mouth.
- this coating can reduce or substantially prevent systemic delivery of the disclosed compound, thereby allowing substantially selective delivery to the intestines.
- the enteric coating will not dissolve in the acid environment of the stomach, which has an acidic, pH of about 3, but will dissolve in the alkaline environments of the small intestine, with, for example, a pH of about 7 to 9.
- Materials used for enteric coating include, but are not limited to, fatty acids, waxes, shellac, plastics and plant fibers.
- the coating may comprise methyl acrylate-methacrylic acid copolymers, cellulose acetate succinate, hydroxy propyl methyl cellulose phthalate, hydroxy propyl methyl cellulose acetate succinate (hypromellose acetate succinate), polyvinyl acetate phthalate (PVAP), methyl methacrylate-methacrylic acid copolymers, shellac, cellulose acetate trimellitate, sodium alginate, or any combination thereof.
- an indole acetonitrile 1 is treated with a suitable protecting Scheme 1 illustrates using di-ie/t-butyl dicarbonate, in the presence of a base and in a suitable solvent, to form a BOC-protected indole (not shown).
- suitable solvents include, but are not limited to, aprotic solvents, such as dichloromethane, dichloroethane, THF, chloroform, or combinations thereof.
- Suitable bases include, but are not limited to, triethylamine, 4- dimethylaminopyridine (DMAP), diiospropylethylamine, or combinations thereof.
- the BOC- protected indole is further reacted with lithium bis(trimethylsilyl)amide (LiHMDS) in a suitable, aprotic solvent such as THF or ether, and at a temperature effective to facilitate a reaction, to form compound 2.
- the effective temperature is from about -100 °C to about -50 °C, such as from about -80 °C to about -60 °C.
- a suitable alkyl halide is then added to the reaction mixture, and the reaction mixture is warmed, or allowed to warm, to room temperature, such as to from about 20 °C to 25 °C.
- alkyl portion of the alkyl halide will correspond to the desired R a and/or R b group.
- R a and/or R b is methyl
- a suitable alkyl halide may be methyl iodide.
- R a and R b are alkyl
- an excess of LiHMDS and alkyl halide are used in the reaction, such as about 2.5 equivalents.
- only one of R a or R b is alkyl, and the other is hydrogen, then only 1 equivalent of LiHMDS and alkyl halide is used.
- Compound 2 is then deprotected, such as by removal of the BOC group, to form the deprotected indole compound (not shown).
- Suitable deprotection methods are known to persons of ordinary skill in the art and typically include reacting with an acid or acidic solution, including, but not limited to, trifluoroacetic acid or hydrochloric acid.
- the cyano group on the deprotected indole compound is then reduced by a suitable reducing agent, such as lithium aluminum hydride (LAH, LiAlH 4 ), at a temperature effective to facilitate a reaction, to form compound 3.
- suitable solvents for the reduction reaction include any aprotic solvent that will not react with the reducing agent, such as THF and ethers.
- the effective temperature is from about 20 °C to greater than 100 °C, such as from about 40 °C to about 80 °C.
- Compound 3 is then reacted with a halopyruvate, such as R c -bromopyruvate, where R c is the desired ester.
- the reaction is conducted in the presence of an acid, and in a suitable solvent and at an effective temperature, to form compound 4.
- a halopyruvate such as R c -bromopyruvate, where R c is the desired ester.
- the reaction is conducted in the presence of an acid, and in a suitable solvent and at an effective temperature, to form compound 4.
- Suitable bromopyruvates include ethyl bromopyruvate and isopropyl bromopyruvate.
- Suitable acids include aqueous acid such as hydrochloric acid.
- Suitable solvents include pro tic solvents, such as alcohols. In some embodiments, ethanol is used as the solvent.
- the effective temperature is from about 20 °C to greater than 100 °C, such as from about 50 °C
- Compound 4 is then reacted with a base at a temperature effective to form compound 5.
- Suitable bases include, but are not limited to, triethylamine, diisopropylethylamine, pyridine or combinations thereof.
- the effective temperature is from about 20 °C to greater than 120 °C, such as from about 50 °C to about 110 °C.
- Compound 5 is then reacted with a suitable acid or activated acid derivative, such as an acid chloride, to form the desired compound 6.
- a suitable solvent include, but are not limited to, halogenated solvents such as chloroform, dichloroethane and dichloromethane, aprotic solvents such as DMF, DMSO, THF, acetonitrile, pyridine, toluene, or combinations thereof.
- Suitable bases include, but are not limited to, triethylamine, diisopropylethylamine, pyridine, potassium carbonate, sodium carbonate or sodium hydrogen carbonate.
- the reaction is conducted at a temperature effective to facilitate a reaction. In some embodiments, the effective temperature is from greater than 20 °C to greater than 120 °C, such as from about 50 °C to about 100 °C.
- Scheme 2 Another exemplary embodiment of a general method of making a compound having formula I is shown in Scheme 2. This method is a modification of the method disclosed by Wang, et al. Tetrahedron Letters, 2011, 52, 3295-3297, which is incorporated herein in its entirety.
- a pyrroloindoline 7 is reacted with an acetylene ester 8 in a suitable solvent, and at a temperature effective to facilitate a reaction, to form compound 9.
- the reaction is performed under an inert atmosphere, such as nitrogen or argon.
- Suitable solvents include, but are not limited to, polar, aprotic solvents such as DMF, DMSO or acetonitrile.
- the effective temperature is from greater than 0 °C to greater than about 100 °C, such as from about 10 °C to about 50 °C, or about 20 °C to about 30 °C.
- the reaction proceeds in the presence of a catalyst.
- Suitable catalysts include, but are not limited to, copper halides, such as copper iodide, copper bromide, or copper chloride, salts of vitamin C such as sodium salt, potassium salt or lithium salt, or combinations thereof.
- R e can be hydrogen or methyl.
- compound 9 is demethylated prior to acylation (not shown).
- the demethylation can be performed by any suitable method such as by reacting the tertiary amine with 1- chloroethylchloroformate in a suitable solvent.
- Solvents suitable for the demethylation include, but are not limited to, halogenated solvents such as dichloromethane, dichloroethane and chloroform, or THF.
- the reaction mixture is evaporated and then heated with an alcohol such as methanol for a time effective to form the secondary amine.
- the effective time is from greater than 1 minute to greater than 1 hour, such as from about 10 minutes to about 30 minutes.
- Compound 9, or the demethylated compound 9, is then reacted with a suitable acid or activated acid derivative, such as an acid chloride, to form the desired compound 10.
- a suitable solvent include, but are not limited to, halogenated solvents such as chloroform, dichloroethane and dichloromethane, aprotic solvents such as DMF, DMSO, THF, acetonitrile, pyridine, toluene, or combinations thereof.
- Suitable bases include, but are not limited to, triethylamine, diisopropylethylamine, pyridine, potassium carbonate, sodium carbonate or sodium hydrogen carbonate.
- the reaction is conducted at a temperature effective to facilitate a reaction. In some embodiments, the effective temperature is from greater than 20 °C to greater than 120 °C, such as from about 50 °C to about 100 °C.
- a protected diamine 21, such as a BOC-protected diamine is reacted with an aldehyde 22 in a suitable solvent for from about 10 minutes to greater than 60 minutes, such as from about 20 minutes to about 40 minutes.
- suitable solvents include, but are not limited to, alcohols, such as methanol or ethanol, water or polar, aprotic solvents such as DMF or DMSO, or combinations thereof.
- Acid 23 and isocyanide 24 are then added. After an amount of time effective to allow the reaction to proceed, the resulting product is deprotected, such as by adding a suitable acid 25 for removing the BOC protecting group.
- the effective amount of time is from about 30 minutes to greater than 12 hours, such as from about 1 hour to about 4 hours.
- Suitable acids are those known to a person of ordinary skill in the art to remove the protecting group, and include, but are not limited to, hydrochloric acid and trifluoroacetic acid.
- the reaction mixture is left for an amount of time effective to facilitate a reaction to form compound 26, such as from about 6 hours to greater than 24 hours, such as from about 12 hours to about 20 hours.
- the reaction mixture is agitated, such as by stirring or shaking, for at least some of the reaction time, and in some embodiments, for substantially all of the reaction time.
- the reaction is conducted at a temperature effective to facilitate a reaction, such as from about 10 °C to greater than about 50 °C, typically from about 20 °C to about 40 °C.
- Scheme 4 Another exemplary method of making a compound having formula IV is shown in Scheme 4. The method is a modification of the method disclosed in WO2004087714, which is incorporated herein in its entirety.
- a haloindole 27, such as a bromo indole, is reacted with an ester compound 27a, which comprises a desired R group and a leaving group LG, to form compound 28.
- the leaving group can be any suitable leaving group, such as a halide, triflate, mesalate or tosylate.
- the reaction is performed in the presence of a base, such as sodium hydride, and in a suitable solvent, such as DMF or THF.
- Compound 28 is typically saponified to an acid (not shown) by any suitable method known to a person of ordinary skill in the art, such as by reacting the acid with a hydroxide base, or by treatment with an aqueous acid, such as hydrochloric acid.
- the acid is then typically activated, such as by forming an acid chloride, and then reacted with aniline to form compound 29.
- the reaction is conducted in a suitable solvent, and in the presence of a suitable base.
- Suitable solvents include, but are not limited to, halogenated solvents such as chloroform, dichloroethane and dichloromethane, aprotic solvents such as DMF, DMSO, THF, acetonitrile, pyridine, toluene, or combinations thereof.
- Suitable bases include, but are not limited to, triethylamine, diisopropylethylamine, pyridine, potassium carbonate, sodium carbonate or sodium hydrogen carbonate.
- the reaction is conducted at a temperature effective to facilitate a reaction. In some embodiments, the effective temperature is from greater than 20 °C to greater than 120 °C, such as from about 50 °C to about 100 °C.
- Compound 29 is then reacted with a boronic acid (not shown) in a Suzuki-type coupling to form compound 30.
- the boronic acid is an aromatic boronic acid.
- the coupling is performed in the presence of a catalyst effective to facilitate the coupling reaction, and optionally in the presence of one or more additional compounds.
- Typical catalysts for a Suzuki coupling are palladium or nickel catalysts, including but not limited to, NiCl 2 (dppf), NiCl 2 (dppp), Pd(PPh 3 ) 4 , Pd(OAC) 2 or PdCl 2 (PPh 3 ) 4 .
- Typical additional compounds include, but are not limited to, triphenylphosphine (PPh 3 ), and/or bases such as potassium carbonate, sodium carbonate, cesium carbonate, sodium hydroxide, potassium hydroxide, triethylamine, sodium ethoxide, sodium methoxide, tripotassium phosphate or any combination thereof.
- the coupling reaction is performed in any suitable solvent, such as DMF, ethanol, methanol, isopropanol, propanol, benzene, toluene, THF, dioxane, water or any combination thereof.
- Scheme 5 One exemplary embodiment of a method of making a compound having formula VII is shown in Scheme 5. A person of ordinary skill in the art will appreciate that other suitable methods for making compounds having formula VII can be determined.
- an amine 31 is reacted with an aldehyde 32.
- the reaction typically is conducted in a suitable solvent, such as an alcohol, such as methanol or ethanol, water, or polar, aprotic solvents such as DMF or DMSO, or combinations thereof, for from about 10 minutes to greater than 60 minutes, such as from about 20 minutes to about 40 minutes.
- An isocyanide 33 and a suitable azide 34 are then added, and the reaction mixture is left for an amount of time effective to facilitate a reaction to form compound 35, such as from about 6 hours to greater than 48 hours, such as from about 12 hours to about 24 hours.
- One possible suitable azide is trimethylsilyl azide.
- Scheme 6 provides one possible reaction mechanism for the reaction described in Scheme 5.
- the amine 31 reacts with the aldehyde 32 with the loss of water, to form an imine 36.
- the imine 36 then reacts with the isocyanide 33 to form an intermediate 37, which then reacts with the azide compound 34, to form an intermediate 38.
- the intermediate 38 then cyclizes to form the desired compound 35.
- Scheme 7 Another exemplary embodiment of a method of making a compound having formula VII is shown in Scheme 7. The method is a modification of the method disclosed by Chen, et al. Synthesis, 2010, No. 9, 1505-1511, which is incorporated herein in its entirety.
- an aromatic halide compound 40 is reacted with an imidazole compound 41 in the presence of a copper catalyst, such as copper (I) bromide and an additional compound 42.
- a copper catalyst such as copper (I) bromide and an additional compound 42.
- the reaction is performed in a suitable solvent and in the presence of a suitable base.
- suitable solvents include aprotic solvents such as DMSO or DMF.
- Suitable bases include any base that will facilitate the reaction, such as sodium carbonate, potassium carbonate, lithium carbonate or cesium carbonate.
- the reaction is conducted at a temperature effective to facilitate a reaction. In some embodiments, the effective temperature is from greater than 20 °C to greater than 120 °C, such as from about 50 °C to about 80 °C.
- Fex Orally delivered fexaramine (Fex) (Downes et al, Mol Cell 11: 1079-1092, 2003) is poorly absorbed, resulting in intestinally-restricted FXR activation. It is shown herein that despite this restricted activation, Fex treatment of diet-induced obesity (DIO) mice produces a novel metabolic profile that includes reduced weight gain, decreased inflammation, browning of white adipose tissue and increased insulin sensitization. The beneficial systemic efficacy achieved with Fex suggests intestinal FXR therapy as a potentially safer approach in the treatment of insulin resistance and metabolic syndrome.
- DIO diet-induced obesity
- the gut-biased FXR agonist fexaramine has profound metabolic benefits in a mouse model of obesity. Fex protects against diet-induced weight gain by promoting the expression of genes involved in thermogenesis, mitochondrial biogenesis, and fatty acid oxidation. Linked to the unexpected browning of white adipose, Fex lowers inflammatory cytokine levels while up-regulating ⁇ -adrenergic signaling. These changes appear to be mediated in part by a change in bile acid levels and composition. In addition, intestinal- specific FXR activation corrected numerous obesity-related defects, enhanced glucose tolerance, and lowered hepatic glucose production. Notably, these physiologic changes are dependent on FXR expression and result in hepatic insulin sensitization and BAT activation, properties not formerly associated with this class of drug.
- FGF15 a key regulator of energy expenditure reported to increase metabolic rate, and improve glucose and lipid homeostasis without significant changes in food intake (Fu et ah,
- Treatment of subjects, including diet-induced obesity (DIO) subjects, with one or more of the disclosed FXR agonists (such as two or more, three or more, four or more, or five or more of the disclosed FXR agonists, such as 2, 3, 4, or 5 of the disclosed FXR agonists) may produce beneficial body- wide metabolic effects such as reduced weight gain, decreased inflammation, browning of white adipose tissue, activation of BAT, improved insulin sensitization, or combinations thereof.
- intestinally-restricted FXR administration is superior to systemic FXR therapy for body- wide metabolic disorders including obesity and metabolic syndrome.
- One or more of the FXR agonists disclosed herein may be administered to a gastrointestinal (GI) tract of the subject to activate FXR receptors in the intestines, and thereby treat or prevent a metabolic disorder in the subject.
- the FXR agonist(s) can be administered to, without limitation, the mouth (such as by injection or by ingestion by the subject), the esophagus, the stomach or the intestines themselves. Orally delivered, these agonists may in some examples be ineffectively absorbed, resulting in intestinally-restricted FXR activation.
- FXR activation is completely limited to the intestine.
- administration of one or more of the disclosed agonists does not result in significant activation in the liver or kidney.
- the FXR agonist is minimally absorbed.
- the FXR agonist is directly administered to the intestines (such as to the distal ileum) of an individual in need thereof.
- the FXR agonist is directly administered to the colon or the rectum of an individual in need thereof.
- the FXR agonist is administered orally, and less than 50%, less than 40%, less than 30%, less than 20%, less than 10%, less than 9%, less than 8%, less than 7%, less than 6%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% of the FXR agonist is systemically absorbed.
- the subject to be treated is one who is diabetic (for example has type
- the subject is obese, for example has a body mass index (BMI) of 25 of higher, 30 or greater, 35 or greater, 40 or greater, such as a BMI of 25 to 29, 30 to 34, or 35 to 40.
- BMI body mass index
- the disclosed methods may reduce weight gain in a subject (such as a human), such as diet-induced weight gain.
- a subject such as a human
- such methods reduce weight gain in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30% or even at least 50% (such as 5% to 50%, 5% to 25%, 10% to 20%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- the disclosed methods reduce the BMI of a subject (such as a human).
- such methods reduce the BMI of a subject by at least 5%, at least 10%, at least 15%, at least 20%, or at least 30% (such as 5% to 30%, 5% to 25%, 10% to 20%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- the disclosed methods may increase browning of white adipose tissue in a subject (such as a human). In some examples, such methods increase browning of white adipose tissue in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30% or even at least 50% (such as 5% to 50%, 5% to 25%, 10% to 20%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies. In some embodiments, the method may reduce or prevent diet-induced weight gain, for example in a mammalian subject, such as a human. In some embodiments, the one or more FXR agonists are administered to an obese subject whose obesity is diet-related (i.e., diet- induced obesity).
- diet-related i.e., diet- induced obesity
- the one or more FXR agonists can be administered to an obese subject whose obesity is not diet-related (such as an individual with familial/genetic obesity or obesity resulting from medication use). In other embodiments, the one or more FXR agonists can be administered to a subject who is overweight (but not obese) or a subject that is neither overweight nor obese. Thus, in some embodiments, the one or more FXR agonists can be used to prevent obesity from developing. In some embodiments, the targeting of the therapy to the intestines reduces the chance of side effects which can result from systemic action, thus improving the safety profile of the therapy.
- the one or more FXR agonists are administered to an obese or non-obese subject for a metabolic disorder or condition other than obesity or weight gain.
- the metabolic disorder is insulin resistance, including non-insulin- dependent diabetes mellitus (NIDDM) (i.e., type II diabetes).
- NIDDM non-insulin- dependent diabetes mellitus
- the administration of the one or more FXR agonists can result in increased insulin sensitivity to insulin in the liver, leading to increased uptake of glucose into hepatic cells.
- the metabolic disorder is dyslipidemia, including hyperlipidemia (elevated LDL, VLDL or triglycerides) or low HDL levels.
- administration of one or more FXR agonists can result in improved glucose and/or lipid homeostasis in the subject.
- administration of the one or more FXR agonists results in a decrease in the amount of serum lipids and/or triglycerides, decrease liver free fatty acids, decrease liver cholesterol, increase liver glycogen, decrease muscle free fatty acids, decrease muscle cholesterol, or combinations thereof, in the subject.
- the disclosed methods decrease the amount of serum lipids and/or triglycerides in a subject (such as a human).
- such methods decrease serum lipids and/or triglycerides in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to levels observed in a subject not treated with the disclosed therapies.
- such methods decrease liver free fatty acids in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to levels observed in a subject not treated with the disclosed therapies.
- such methods decrease liver cholesterol in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to levels observed in a subject not treated with the disclosed therapies.
- such methods increase liver glycogen in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50%, at least 75%, at least 90%, at least 100%, or at least 200% (such as 5% to 50%, 5% to 25%, 100% to 200%, 10% to 100%, or 10% to 200%), for example relative to levels observed in a subject not treated with the disclosed therapies.
- such methods decrease muscle free fatty acids in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to levels observed in a subject not treated with the disclosed therapies.
- such methods decrease muscle cholesterol in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to levels observed in a subject not treated with the disclosed therapies.
- the disclosed embodiments may increase insulin sensitivity to insulin in the liver of a subject (such as a human).
- such methods increase insulin sensitivity to insulin in the liver of the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30% or even at least 50% (such as 5% to 50%, 5% to 25%, 10% to 20%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- administration of the one or more FXR agonists results in no substantial change in food intake and/or fat consumption in the subject.
- food intake and/or fat consumption is reduced minimally, such as by less than 15%, less than 10%, or less than 5%.
- no substantial change in appetite in the subject results.
- reduction in appetite is minimal as reported by the subject.
- administration of the one or more FXR agonists results in an increase in the metabolic rate in the subject.
- the disclosed methods may increase the metabolic rate in a subject (such as a human).
- such methods increase the metabolic rate in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- this increase in metabolism results from enhanced oxidative phosphorylation in the subject, which in turn can lead to increased energy expenditure in tissues (such as BAT).
- the disclosed methods may increase BAT activity in a subject (such as a human).
- a subject such as a human
- such methods increase BAT activity in a subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- administration of the one or more FXR agonists results in a decrease in the amount of serum insulin in the subject.
- the disclosed methods decrease the amount of serum insulin in a subject (such as a human).
- such methods decrease serum insulin in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to levels observed in a subject not treated with the disclosed therapies.
- administration of the one or more FXR agonists results in a decrease in the amount of serum glucose in the subject.
- the disclosed methods decrease the amount of serum glucose in a subject (such as a human).
- such methods decrease serum glucose in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to levels observed in a subject not treated with the disclosed therapies.
- Embodiments of a method are provided for lowering elevations in blood glucose resulting from food intake in a subject.
- such methods decrease blood glucose in a subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- Such methods can include orally administering to the subject a therapeutically effective amount of one of the disclosed minimally absorbed FXR agonists.
- a method for lowering elevated body weight in a subject is provided, wherein the method includes orally administering to said subject a therapeutically effective amount of one of the disclosed minimally absorbed FXR agonists.
- such methods decrease the body weight of a subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, or at least 50% (such as 5% to 50%, 5% to 25%, 5% to 20%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- the elevated body weight and/or elevated glucose levels resulted from a particular pattern of food intake, such as a high fat diet and/or a high calorie diet.
- the one or more FXR agonists are co-administered with one or more additional compounds or therapies, for treatment or prevention of a metabolic disorder.
- one or more FXR agonists can be administered with an insulin sensitizing drug, an insulin secretagogue, an alpha-glucosidase inhibitor, a glucagon-like peptide (GLP) agonist, a DPP-4 inhibitor (such as sitagliptin, vildagliptin, saxagliptin, linagliptin, anaglptin, teneligliptin, alogliptin, gemiglptin, or dutoglpitin), a catecholamine (such as epinephrine, norepinephrine, or dopamine), peroxisome proliferator-activated receptor (PPAR)-gamma agonist (e.g., a thiazolidinedione (TZD) [such as
- one or more FXR agonists can be administered with a statin, HMG-CoA reductase inhibitor, fish oil, fibrate, niacin or other treatment for dyslipidemia.
- a method for treating a metabolic disorder in a subject comprising orally co-administering to said subject a therapeutically effective amount of a disclosed minimally absorbed FXR agonist and retinoic acid.
- 9 cis-retinoic acid is the ligand for retinoic acid receptor (RXR), the heterodimeric partner of FXR.
- the method includes also administering nicotinamide ribonucleoside and/or an analog of nicotinamide ribonucleoside (such as those that promote NAD+ production of which is a substrate for many enzymatic reactions such as p450s which are a target of FXR, for example see Yang et al., J. Med Chem. 50:6458-61, 2007, herein incorporated by reference).
- nicotinamide ribonucleoside and/or an analog of nicotinamide ribonucleoside such as those that promote NAD+ production of which is a substrate for many enzymatic reactions such as p450s which are a target of FXR, for example see Yang et al., J. Med Chem. 50:6458-61, 2007, herein incorporated by reference).
- GLP-1 Glucagon-like peptide- 1
- the major source of GLP-1 in the body is the intestinal L cell that secretes GLP-1 as a gut hormone.
- the biologically active forms of GLP-1 include GLP-1 -(7- 37) and GLP-l-(7-36)NH 2 (HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR; SEQ ID NO: 1), which result from selective cleavage of the proglucagon molecule.
- GLP-2 is a 33 amino acid peptide (HADGSFSDEMNTILDNLAARDFINWLIQTKITD; SEQ ID NO: 2) in humans.
- GLP-2 is created by specific post-translational proteolytic cleavage of proglucagon in a process that also liberates GLP-1.
- GLP agonists are a class of drugs ("incretin mimetics") that can be used to treattype 2 diabetes. Examples include, but are not limited to: exenatide
- the FXR agonist enhances the secretion of glucagon-like peptide- 1 (GLP-1) and/or glucagon-like peptide-2 (GLP-2). In some embodiments, the FXR agonist enhances the secretion of a pancreatic polypeptide-fold such as peptide YY (PYY). In certain embodiments, the FXR agonist enhances the activity of FGF15 or FGF19. In certain embodiments, the FXR agonist enhances secretion of an enteroendocrine peptide and/or is administered in combination with an agent that enhances secretion or activity of an
- the disclosed methods may increase the secretion of one or more of GLP-1, GLP-2, and PYY in a subject (such as a human).
- a subject such as a human
- such methods increase the secretion of one or more of GLP-1, GLP-2, and PYY in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- the disclosed methods increase the secretion of one or more of GLP-1, GLP-2, and PYY in a subject (such as a human).
- a subject such as a human
- such methods increase the activity of one or more of FGF15 and FGF19 in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- the gut-biased FXR agonists disclosed herein can have profound metabolic benefits with respect to obesity.
- the gut-biased FXR agonists can protect against diet-induced weight gain by, for example, promoting the expression of genes involved in thermogenesis, mitochondrial biogenesis, and/or fatty acid oxidation.
- the disclosed gut-biased FXR agonists linked to the unexpected browning of white adipose, can lower inflammatory cytokine levels while up-regulating ⁇ -adrenergic signaling. These changes can be mediated, at least in part, by a change in bile acid levels and composition.
- a prandial activation of intestinal FXR is triggered by administering to a subject one of the FXR agonists disclosed herein, such as synthetic FXR agonist fexaramine (Fex).
- the intestinal- specific FXR activation disclosed herein can be utilized to enhance glucose tolerance and lower hepatic glucose production.
- such methods may decrease hepatic glucose production in a subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30%, at least 50% or even at least 75% (such as 5% to 50%, 5% to 25%, 10% to 20%, 10% to 70%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- These physiologic changes can result in hepatic insulin sensitization and/or BAT activation - properties not previously associated with FXR agonists.
- intestinal FXR in contrast to the effects of system- wide drugs (including systemic FXR agonists), selective activation of intestinal FXR as disclosed herein can mimic the restricted bile acid response linked to feeding.
- the FXR agonists disclosed herein may be gut-specific and robustly induce enteral FGF15, leading to alterations in bile acid composition without activating hepatic FXR target genes.
- these gut-specific FXR agonists may protect against diet-induced weight gain, reduce body- wide inflammation, enhance thermo genesis, promote browning of white adipose tissue, promote activation of BAT, and suppress hepatic glucose production.
- the initial event triggering systemic metabolic activation is coordinated by FGF15 (the mouse equivalent of human FGF19) or FGF19.
- administration of the FXR agonist results in activation of FGF15 or FGF19 (such as an increase in FGF15 or FGF19 activity of at least 25%, at least 50%, at least 75%, at least 90%, or at least 95%, relative to no treatment with an FXR agonist), which in turn can regulate energy expenditure, such as by increasing metabolic rate, improving glucose homeostasis (such as by improving insulin sensitivity), and/or improving lipid homeostasis without requiring significant changes in food intake.
- treatment with one or more of the disclosed FXR agonists can produce a change in the bile acid pool, such as a dramatic increase in the level of deoxycholic acid (such as an increase of at least 25%, at least 50%, at least 75%, at least 90%, or at least 100%, relative to no treatment with an FXR agonist), a potent ligand for the G protein-coupled bile acid receptor TGR5.
- Fex treatment was observed to induce DI02, a downstream target of TGR5, in brown adipose tissue (BAT), thus implicating this additional pathway in the observed increase in energy expenditure.
- the coordinate "browning" of white adipose tissue provides an independent yet complementary contribution to increased thermogenic capacity.
- Certain disclosed embodiments can include administering a therapeutically effective amount of one or more FXR agonists to an individual in need thereof, such as one or more of the novel FXR agonists disclosed herein (such as 1, 2, 3, 4 or 5 such agonists).
- the disclosed embodiments may reduce inflammation in a subject (such as a human), such as inflammation in the intestine.
- a subject such as a human
- inflammation in the intestine may reduce inflammation (such as intestinal inflammation) in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30% or even at least 50% (such as 5% to 50%, 5% to 25%, 10% to 20%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- the inflammatory condition can be necrotizing enterocolitis (NEC), gastritis, ulcerative colitis, inflammatory bowel disease, irritable bowel syndrome, pseudomembranous colitis, gastroenteritis, radiation induced enteritis, chemotherapy induced enteritis, gastro-esophageal reflux disease (GERD), peptic ulcer, non-ulcer dyspepsia (NUD), celiac disease, intestinal celiac disease, gastrointestinal complications following bariatric surgery, gastric carcinogenesis, or gastric carcinogenesis following gastric or bowel resection.
- the inflammatory condition is NEC and the subject is a newborn or prematurely born infant.
- the subject is enterally-fed infant or formula- fed infant.
- the one or more FXR agonists are co-administered with one or more additional compounds or therapies, for treatment or prevention of an inflammatory intestinal condition.
- the one or more FXR agonists are co-administered with an oral corticosteroid and/or other anti-inflammatory or immuno-modulatory therapy.
- the FXR agonist can be administered to the subject in conjunction with one or more antibiotics (e.g., metronidazole, vancomycin, and/or fidaxomicin) to treat or prevent the inflammatory condition.
- the FXR agonist can be administered to the subject in conjunction with or following antibiotic therapy to treat or prevent
- the FXR agonist can be administered to the subject in conjunction with metronidazole or other indicated therapy to treat inflammation associated with bacterial overgrowth in an intestinal area.
- the FXR agonist can be administered to the subject in conjunction with the ingestion of foods or other substances predicted to induce inflammation in the gastro-intestinal system of the subject (such as in a subject with celiac disease).
- the method includes also administering nicotinamide ribonucleoside and/or an analog of nicotinamide ribonucleoside (such as those that promote NAD+ production of which is a substrate for many enzymatic reactions such as p450s which are a target of FXR, for example see Yang et al., J. Med Chem. 50:6458-61, 2007, herein incorporated by reference) .
- nicotinamide ribonucleoside and/or an analog of nicotinamide ribonucleoside such as those that promote NAD+ production of which is a substrate for many enzymatic reactions such as p450s which are a target of FXR, for example see Yang et al., J. Med Chem. 50:6458-61, 2007, herein incorporated by reference
- Certain disclosed embodiments can include administering a therapeutically effective amount of one or more FXR agonists to an individual in need thereof, such as one or more of the novel FXR agonists disclosed herein (such as 1, 2, 3, 4 or 5 such agonists).
- the compounds disclosed herein may be used in the prevention or treatment of adenocarcinomas, i.e. carcinoma derived from glandular tissue or in which the tumor cells form recognizable glandular structures.
- Adenocarcinomas can be classified according to the predominant pattern of cell arrangement, as papillary, alveolar, etc., or according to a particular product of the cells, as mucinous adenocarcinoma.
- Adenocarcinomas arise in several tissues, including the colon, kidney, breast, cervix, esophagus, gastric, pancreas, prostate and lung.
- the compounds disclosed herein may be used in the prevention or treatment of a cancer of the intestine, such as colon cancer, i.e. cancer that forms in the tissues of the colon (the longest part of the large intestine), or a cancer of another part of the intestine, such as the jejunum, and/or ileum.
- Colon cancer is also referred to as "colorectal cancer.”
- Most colon cancers are adenocarcinomas (cancers that begin in cells that may line internal organs and have gland-like properties). Cancer progression is characterized by stages, or the extent of cancer in the body. Staging is usually based on the size of the tumor, whether lymph nodes contain cancer, and whether the cancer has spread from the original site to other parts of the body.
- Stages of colon cancer include stage I, stage II, stage III and stage IV.
- the colon adenocarcinoma is from any stage.
- the colon adenocarcinoma is a stage I cancer, a stage II cancer or a stage III cancer.
- the disclosed embodiments reduce tumor burden in a subject (such as a human).
- disclosed embodiments reduce tumor burden (such as colon tumor burden) in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30% or even at least 50% (such as 5% to 50%, 5% to 25%, 10% to 20%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- the disclosed embodiments reduce tumor size and/or volume in a subject (such as a human).
- a subject such as a human
- disclosed embodiments reduce tumor size and/or volume (such as a colon tumor) in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30% or even at least 50% (such as 5% to 50%, 5% to 25%, 10% to 20%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- the disclosed embodiments reduce effects of cachexia due to a tumor in a subject (such as a human).
- a subject such as a human
- disclosed embodiments reduce effects of cachexia (such as due to a colon tumor) in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30% or even at least 50% (such as 5% to 50%, 5% to 25%, 10% to 20%, or 10% to 30%), for example relative to a subject not treated with the disclosed
- the disclosed embodiments increase survival rates of a subject (such as a human) with a tumor.
- disclosed embodiments increase survival rates of a subject (such as a human) with a tumor (such as a colon cancer) in the subject by at least 5%, at least 10%, at least 15%, at least 20%, at least 30% or even at least 50% (such as 5% to 50%, 5% to 25%, 10% to 20%, or 10% to 30%), for example relative to a subject not treated with the disclosed therapies.
- the compounds disclosed herein may be administered in combination with one or more additional anticancer therapies (such as a biologic [e.g., antibody, for example bevacizumab, cetuximab, or panitumumab], chemo therapeutic, or radiologic, for example FOLFOX, FOLFIRI, CapeOX, 5-FU, leucovorin, regorafenib, irinotecan, and oxaliplatin), to prevent or treat a cell proliferation disease.
- a biologic e.g., antibody, for example bevacizumab, cetuximab, or panitumumab
- chemo therapeutic e.g., chemo therapeutic
- radiologic for example FOLFOX, FOLFIRI, CapeOX, 5-FU, leucovorin, regorafenib, irinotecan, and oxaliplatin
- the method includes also administering nicotinamide ribonucleoside and/or an analog of nicotinamide ribonucleoside (such as those that promote NAD+ production of which is a substrate for many enzymatic reactions such as p450s which are a target of FXR, for example see Yang et al., J. Med Chem. 50:6458-61, 2007, herein incorporated by reference).
- nicotinamide ribonucleoside and/or an analog of nicotinamide ribonucleoside such as those that promote NAD+ production of which is a substrate for many enzymatic reactions such as p450s which are a target of FXR, for example see Yang et al., J. Med Chem. 50:6458-61, 2007, herein incorporated by reference).
- the particular mode of administration and the dosage regimen will be selected by the attending clinician, taking into account the particulars of the case (e.g. the subject, the disease, the disease state involved, the particular treatment, and whether the treatment is to be selected by the attending clinician, taking into account the particulars of the case (e.g. the subject, the disease, the disease state involved, the particular treatment, and whether the treatment is to be selected by the attending clinician, taking into account the particulars of the case (e.g. the subject, the disease, the disease state involved, the particular treatment, and whether the treatment is
- Treatment can involve daily or multi-daily or less than daily (such as weekly or monthly etc.) doses over a period of a few days to months, or even years.
- a therapeutically effective amount of one or more compounds disclosed herein can be
- treatment involves once daily dose or twice daily dose.
- the FXR agonist(s) is administered orally.
- the FXR agonist is administered as an ileal-pH sensitive release formulation that delivers the FXR agonist to the intestines, such as to the ileum of an individual.
- the FXR agonist is administered as an enterically coated formulation.
- oral delivery of an FXR agonist provided herein can include formulations, as are well known in the art, to provide prolonged or sustained delivery of the drug to the gastrointestinal tract by any number of mechanisms.
- enteric-coated and enteric-coated controlled release formulations are within the scope of the present disclosure.
- Suitable enteric coatings include cellulose acetate phthalate, polyvinylacetate phthalate, hydroxypropylmethylcellulose phthalate and anionic polymers of methacrylic acid and methacrylic acid methyl ester.
- the FXR agonist is administered before ingestion of food, such as at least 10 minutes, at least 15 minutes, at least 20 minutes, or at least 30 minutes before ingestion of food (such as 10-60 minutes or 10-30 minutes before ingesting food). In some embodiments of the methods described herein, the FXR agonist is administered less than about 60 minutes before ingestion of food. In some embodiments of the methods described above, the FXR agonist is administered less than about 30 minutes before ingestion of food. In some embodiments of the methods described herein, the FXR agonist is administered after ingestion of food.
- the methods further comprise administration of a DPP-IV inhibitor, a TGR5 agonist, a biguanide, an incretin mimetic, or GLP- 1 or an analog thereof.
- the methods further comprise administration of a steroid or other antiinflammatory compound which may have an effect in the gut.
- the methods further include co-administration of an antibiotic therapy, and the FXR agonist treats or prevents inflammation, such as inflammation associated with antibiotic-induced colitis.
- the composition administered can include at least one of a spreading agent or a wetting agent.
- the absorption inhibitor is a mucoadhesive agent (e.g., a mucoadhesive polymer).
- the mucoadhesive agent is selected from methyl cellulose, polycarbophil, polyvinylpyrrolidone, sodium carboxymethyl cellulose, and a combination thereof.
- a pharmaceutical composition administered further includes an enteroendocrine peptide and/or an agent that enhances secretion or activity of an enteroendocrine peptide.
- compositions that comprise one or more compounds disclosed herein can be formulated in unit dosage form, suitable for individual administration of precise dosages.
- a unit dosage contains from about 1 mg to about 50 g of one or more compounds disclosed herein, such as about 10 mg to about 10 g, about 100 mg to about 10 g, about 100 mg to about 1 g, about 500 mg to about 5 g, or about 500 mg to about 1 g.
- a therapeutically effective amount of one or more compounds disclosed herein is from about 0.01 mg/kg to about 500 mg/kg, for example, about 0.5 mg/kg to about
- a therapeutically effective amount of one or more compounds disclosed herein is from about 50 mg/kg to about 250 mg/kg, for example about 100 mg/kg.
- Fexaramine prevents diet-induced obesity weight gain
- mice were subjected to chronic fexaramine (100 mg/kg Fex) PO treatment for 5 weeks.
- Chronically treated chow-fed mice were indistinguishable from vehicle-treated mice in terms of weight gain, basal metabolic activity and glucose tolerance (FIGS. 3A-3D).
- GTTs glucose tolerance tests
- ITTs insulin tolerance tests
- Fexaramine enhances energy expenditure in brown adipose tissue
- Fex treatment increased the core body temperature approximately 1.5 °C (FIG. 6E).
- BAT brown adipose tissue
- Fex-treated mice FIG. 6F.
- Gene expression analysis confirmed the induction of ERRy, PGC- la, and PGC- ⁇ , as well as a number of their target genes involved in thermogenesis, mitochondrial biogenesis, and fatty acid oxidation in BAT (FIG. 6G).
- Fex treatment increased the phosphorylation level of p38 (FIG.
- thermogenic transcriptional program a key coactivator of the thermogenic transcriptional program in BAT.
- a comparison of the transcriptional changes induced by Fex in inguinal, gonadal and brown adipose depots revealed coordinated changes that selectively enhance OXPHOS activity only in BAT, indicating that BAT is a key contributor to the increased energy expenditure and thermogenesis (FIG. 6J).
- KEGG pathway analysis of Fex-induced transcriptional changes from RNA-sequence analysis in BAT identified oxidative phosphorylation as significantly changed (Table 1), and increased PKA activity was seen in Fex-treated mice (FIG. 6L).
- RNA-Seq of intestinal tissues was used to explore the mechanisms through which Fex might contribute to systemic changes in energy expenditure and metabolic rate.
- Mice were fed on HFD for 14 weeks, and then subjected to daily oral injection of vehicle or fexaramine (100 mg/kg) for 5 weeks with HFD.
- KEGG pathway analysis revealed the induction of multiple cellular metabolic pathways including PPAR and adipocytokine signaling in both ileum and colon (Tables 2 and 3).
- FGF15 corresponds to FGF19 in humans
- Perl genes exhibiting regulation by FXR were identified including Perl (FIG. 7A).
- FGF15 induction is of interest since it activates the thermogenic program in BAT, as well as negatively regulate BA synthesis through suppression of hepatic CYP7A1, the rate-limiting enzyme for BA synthesis.
- An increase in circulating FGF15 accompanied the increase in mRNA expression in ileum (FIGS. 7B and 7C) (such as an increase of at least 100%, at least 125%, or at least 150%).
- hepatic CYP7A1 expression was significantly repressed at both the mRNA and protein level after chronic Fex treatment, while the expression of CYP8B1 and CYP27A1 (enzymes not regulated by FGF15) were not affected (FIG.
- FXR transgene in intestine constitutively activated FXR transgene in intestine, as well as after injection of FGF19, the human analogue of FGF15 (Wu et al. PloS one 6, el7868, 2011). Furthermore, changes in bile acid synthesis away from cholic acid towards chenodeoxycholic acid and its derivatives, which includes lithocholic acid, were observed upon FGF19 treatment, consistent with a reduction in hepatic CYP7A1 and an increase in CYP7B1 expression.
- FXR activation has been reported to enhance mucosal defense gene expression and intestinal barrier function (Inagaki et al., Proc Natl Acad Sci U SA 103:3920-3925, 2006;
- mice showed reduced intestinal permeability, as measured by FITC-dextran leakage into the serum, and increased expression of mucosal defense genes Occludin and Muc2, after chronic Fex-treatment (FIGS. 7G and 7H).
- TGR5 null mice were chronically treated with Fex (100 mg/kg/day PO for 5 weeks).
- Fex treatment induced multiple FXR target genes in the ileum of TGR5 null mice including FGF15, resulting in lowered serum BA levels (FIGS. 11 A, 1 IB).
- Fex treatment induced moderate improvements in fasting glucose levels and glucose tolerance (FIGS. 11C, 11D).
- FIGS. 11E-11H somewhat blunted increases in core body temperature and metabolic rate, correlating with the induction of thermogenic genes in BAT, were observed (FIGS. 11E-11H), indicating that these effects do not require TGR5 activation.
- Fexaramine induces browning of white adipose tissue
- adipose tissue expands by hyperplastic and/or hypertrophic growth, is chronically inflamed, and produces inflammatory cytokines that ultimately contribute to systemic metabolic dysregulation.
- the cross-sectional area of adipocytes in visceral depots including gonadal and mesenteric was markedly reduced (FIG. 12A).
- Investigation of signaling pathways implicated in diet-induced inflammation identified reduced levels of ⁇ - ⁇ and TANK-binding kinase 1 (TBK1) in Fex-treated DIO mice (FIGS. 12B, 13).
- FIG. 12B Consistent with reduced adiposity, expression of the inflammatory cytokines TNFcc, MCP-1 and IL-lcc, as well as the macrophage marker F4/80, were reduced in visceral and brown adipose depots of Fex-treated mice (FIGS. 12C and 14).
- Brown adipose-driven adaptive thermogenesis is fueled by mitochondrial oxidation of free fatty acids (FFAs) released from triglyceride stores into the circulation predominantly by the action of hormone-sensitive lipase (HSL).
- FFAs free fatty acids
- HSL hormone-sensitive lipase
- Low levels of HSL phosphorylation were seen in visceral and subcutaneous adipose depots from control mice, as expected, due to desensitization of the ⁇ -adrenergic pathway in WAT during obesity (Carmen & Victor, Cell Signal 18:401- 408, 2006; Song et al. Nature 468:933-9, 2010).
- Fexaramine improves insulin sensitivity and glucose tolerance
- liver insulin resistance has been linked to obesity-induced hepatic steatosis (Cohen et al., Science 332: 1519-1523, 2011). Histological examination of liver tissue from Fex-treated
- DIO mice revealed a reduction in lipid droplets compared to controls indicating amelioration of hepatic steatosis (FIG. 15E). Consistent with this histology, a marked decrease in hepatic triglycerides (such as a reduction of at least 10%, or at least 20%) and reduced hepatic expression of gluconeogenic and lipogenic genes (such as a reduction of at least 20%, or at least 30%, or at least 50%) were seen after chronic Fex treatment (FIGS.15F and 15G). Furthermore, decreased serum alanine aminotransferase (ALT) levels were measured in Fex-treated mice, indicating reduced HFD-induced liver damage (FIG. 15H). Thus, in DIO mice Fex promotes hepatic insulin sensitization, reduced steatosis, improved metabolic markers, decreased ALT and enhanced BAT activity.
- Example 7 Example 7
- FXR activity screen for determining EC50 determination Cell Culture and Transfection: CV-1 cells were grown in DMEM+10 charcoal stripped FCS. Cells were seeded into 384- well plates the day before transfection to give a confluency of 50-80% at transfection. A total of 0.8 grams DNA containing 0.32 micrograms pCMX-hFXRfl, 0.32 micrograms pCMX-hRXRfl, 0.1 micrograms pCMX.beta.Gal, 0.08 micrograms pGLFXRE reporter and 0.02 micrograms pCMX empty vector was transfected per well using FuGene transfection reagent according to the manufacturer's instructions (Roche). Cells were allowed to express protein for 48 hours followed by addition of compound.
- Plasmids Human FXR full length and RXR full length was obtained from Ronald Evans' laboratory and PCR amplification of the hFXR cDNA and the hRXR cDNA was performed. The amplified cDNAs was cloned into the vector pCMX generating the plasmids pCMX- hFXRfl and pCMX-hRXRfl. Ensuing fusions were verified by sequencing. The
- pCMXMH2004 luciferase reporter contains multiple copies of the GAL4 DNA response element under a minimal eukaryotic promoter (Hollenberg and Evans, 1988).
- pCMX.beta.Gal was generated in the Evans laboratory, Salk Institute.
- Compounds All compounds were dissolved in DMSO and diluted 1: 1000 upon addition to the cells. Compounds were tested in quadruple in concentrations ranging from 0.001 to 100 ⁇ . Cells were treated with compound for 24 hours followed by luciferase assay. Each compound was tested in at least two separate experiments. Luciferase assay: Medium including test compound was aspirated and washed with PBS.
- luciferase assay 50 ⁇ PBS including 1 mM Mg 2+ and Ca 2+ were then added to each well.
- the luciferase assay was performed using the LucLite kit according to the manufacturer's instructions (Packard Instruments). Light emission was quantified by counting on a Perkin Elmer Envision reader. To measure 3-galactosidase activity 25 supernatant from each transfection lysate was transferred to a new 384 microplate. Beta-galactosidase assays were performed in the microwell plates using a kit from Promega and read in a Perkin Elmer Envision reader. The beta-galactosidase data were used to normalize (transfection efficiency, cell growth etc.) the luciferase data.
- the activity of a compound is calculated as fold induction compared to an untreated sample. For each compound the efficacy (maximal activity) is given as a relative activity compared to Fexaramine, a FXR agonist.
- the EC50 is the concentration giving 50% of maximal observed activity. EC50 values were calculated via non-linear regression using GraphPad PRISM (GraphPad Software, San Diego, Calif.).
Abstract
Description

































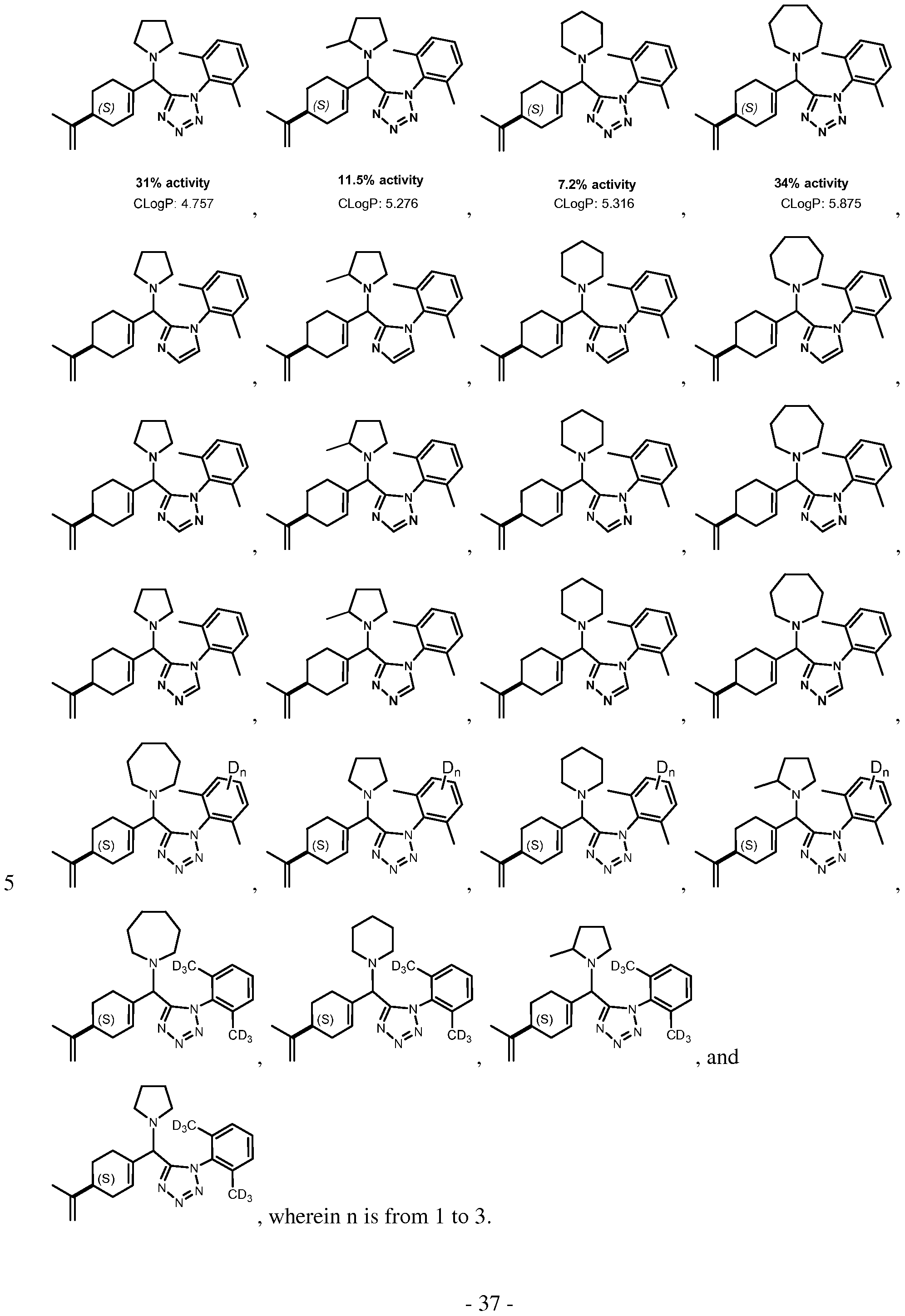



























Claims


















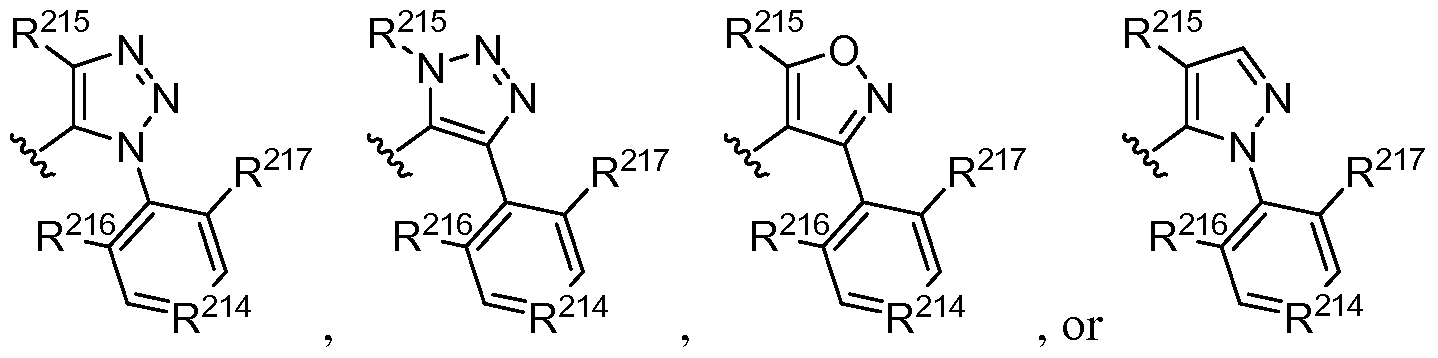


Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016556962A JP2017510572A (en) | 2014-03-13 | 2015-03-13 | FXR agonists and methods for making and using |
| CA2942403A CA2942403A1 (en) | 2014-03-13 | 2015-03-13 | Fxr agonists and methods for making and using |
| EP15761517.0A EP3116878A4 (en) | 2014-03-13 | 2015-03-13 | Fxr agonists and methods for making and using |
| KR1020167028569A KR20160132111A (en) | 2014-03-13 | 2015-03-13 | Fxr agonists and methods for making and using |
| AU2015229072A AU2015229072A1 (en) | 2014-03-13 | 2015-03-13 | FXR agonists and methods for making and using |
| US15/263,048 US10077268B2 (en) | 2014-03-13 | 2016-09-12 | FXR agonists and methods for making and using |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461952754P | 2014-03-13 | 2014-03-13 | |
| US61/952,754 | 2014-03-13 | ||
| US201462061463P | 2014-10-08 | 2014-10-08 | |
| US62/061,463 | 2014-10-08 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/263,048 Continuation-In-Part US10077268B2 (en) | 2014-03-13 | 2016-09-12 | FXR agonists and methods for making and using |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015138986A1 true WO2015138986A1 (en) | 2015-09-17 |
Family
ID=54072502
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/020582 WO2015138986A1 (en) | 2014-03-13 | 2015-03-13 | Fxr agonists and methods for making and using |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP3116878A4 (en) |
| JP (1) | JP2017510572A (en) |
| KR (1) | KR20160132111A (en) |
| AU (1) | AU2015229072A1 (en) |
| CA (1) | CA2942403A1 (en) |
| WO (1) | WO2015138986A1 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017078928A1 (en) * | 2015-11-06 | 2017-05-11 | Salk Institute For Biological Studies | Fxr agonists and methods for making and using |
| WO2017143134A1 (en) * | 2016-02-19 | 2017-08-24 | Alios Biopharma, Inc. | Fxr modulators and methods of their use |
| EP3257847A1 (en) * | 2016-06-13 | 2017-12-20 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| WO2018051230A1 (en) * | 2016-09-14 | 2018-03-22 | Novartis Ag | Combination of fxr agonists |
| WO2018153933A1 (en) | 2017-02-21 | 2018-08-30 | Genfit | Combination of a ppar agonist with a fxr agonist |
| US10077268B2 (en) | 2014-03-13 | 2018-09-18 | Salk Institute For Biological Studies | FXR agonists and methods for making and using |
| US10183917B2 (en) | 2015-02-13 | 2019-01-22 | Sunshine Lake Pharma Co., Ltd. | Tricyclic compounds and uses thereof in medicine |
| US10189872B2 (en) | 2015-03-09 | 2019-01-29 | W. R. Grace & Co.-Conn | Crystalline form of nicotinamide riboside |
| CN109311849A (en) * | 2016-06-13 | 2019-02-05 | 吉利德科学公司 | Adjust the compound of FXR (NR1H4) |
| US10220027B2 (en) | 2011-07-13 | 2019-03-05 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US10233207B2 (en) | 2014-07-24 | 2019-03-19 | W. R. Grace & Co.—Conn. | Crystalline form of nicotinamide riboside |
| US10301268B2 (en) | 2014-03-13 | 2019-05-28 | The Salk Institute For Biological Studies | Analogs of fexaramine and methods of making and using |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| WO2021014350A1 (en) | 2019-07-23 | 2021-01-28 | Novartis Ag | Combination treatment of liver diseases using fxr agonists |
| WO2021014349A1 (en) | 2019-07-23 | 2021-01-28 | Novartis Ag | Treatment comprising fxr agonists |
| WO2021044287A1 (en) | 2019-09-03 | 2021-03-11 | Novartis Ag | Treatment of liver disease or disorder comprising actrii receptor antagonists |
| WO2021053618A1 (en) | 2019-09-19 | 2021-03-25 | Novartis Ag | Treatment comprising fxr agonists |
| WO2021064575A1 (en) | 2019-09-30 | 2021-04-08 | Novartis Ag | Treatment comprising the use of fxr agonists |
| WO2021127466A1 (en) | 2019-12-20 | 2021-06-24 | Novartis Ag | Combination treatment of liver diseases using integrin inhibitors |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| US11214538B2 (en) | 2015-09-16 | 2022-01-04 | Metacrine, Inc. | Farnesoid X receptor agonists and uses thereof |
| US11225473B2 (en) | 2019-01-15 | 2022-01-18 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| WO2022101853A1 (en) | 2020-11-16 | 2022-05-19 | Novartis Ag | Method of determining liver fibrosis |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| US11773094B2 (en) | 2018-09-18 | 2023-10-03 | Organovo, Inc. | Farnesoid X receptor agonists and uses thereof |
| US11833150B2 (en) | 2017-03-28 | 2023-12-05 | Gilead Sciences, Inc. | Methods of treating liver disease |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003099821A1 (en) * | 2002-05-24 | 2003-12-04 | X-Ceptor Therapeutics, Inc. | Azepinoindole and pyridoindole derivatives as pharmaceutical agents |
| WO2007070796A1 (en) * | 2005-12-15 | 2007-06-21 | Exelixis, Inc. | Azepinoindole derivatives as pharmaceutical agents |
| US20080300235A1 (en) * | 2007-06-01 | 2008-12-04 | Wyeth | FXR Agonists for Reducing LOX-1 Expression |
| US20090163474A1 (en) * | 2007-10-19 | 2009-06-25 | Wyeth | FXR Agonists for the Treatment of Nonalcoholic Fatty Liver and Cholesterol Gallstone Diseases |
| US20090215748A1 (en) * | 2007-12-20 | 2009-08-27 | Wyeth | FXR agonists for treating vitamin D associated diseases |
| WO2010036362A1 (en) * | 2008-09-26 | 2010-04-01 | Wyeth | 1,2,3,6-tetrahydroazepino[4,5-b]indole-5-carboxylate nuclear receptor inhibitors |
| WO2011150286A2 (en) * | 2010-05-26 | 2011-12-01 | Satiogen Pharmaceuticals,Inc. | Bile acid recycling inhibitors and satiogens for treatment of diabetes, obesity, and inflammatory gastrointestinal conditions |
| WO2013020108A2 (en) * | 2011-08-04 | 2013-02-07 | Lumena Pharmaceuticals, Inc. | Bile acid recycling inhibitors for treatment of pancreatitis |
-
2015
- 2015-03-13 CA CA2942403A patent/CA2942403A1/en not_active Abandoned
- 2015-03-13 WO PCT/US2015/020582 patent/WO2015138986A1/en active Application Filing
- 2015-03-13 JP JP2016556962A patent/JP2017510572A/en active Pending
- 2015-03-13 KR KR1020167028569A patent/KR20160132111A/en unknown
- 2015-03-13 EP EP15761517.0A patent/EP3116878A4/en not_active Withdrawn
- 2015-03-13 AU AU2015229072A patent/AU2015229072A1/en not_active Abandoned
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003099821A1 (en) * | 2002-05-24 | 2003-12-04 | X-Ceptor Therapeutics, Inc. | Azepinoindole and pyridoindole derivatives as pharmaceutical agents |
| WO2007070796A1 (en) * | 2005-12-15 | 2007-06-21 | Exelixis, Inc. | Azepinoindole derivatives as pharmaceutical agents |
| US20080300235A1 (en) * | 2007-06-01 | 2008-12-04 | Wyeth | FXR Agonists for Reducing LOX-1 Expression |
| US20090163474A1 (en) * | 2007-10-19 | 2009-06-25 | Wyeth | FXR Agonists for the Treatment of Nonalcoholic Fatty Liver and Cholesterol Gallstone Diseases |
| US20090215748A1 (en) * | 2007-12-20 | 2009-08-27 | Wyeth | FXR agonists for treating vitamin D associated diseases |
| WO2010036362A1 (en) * | 2008-09-26 | 2010-04-01 | Wyeth | 1,2,3,6-tetrahydroazepino[4,5-b]indole-5-carboxylate nuclear receptor inhibitors |
| WO2011150286A2 (en) * | 2010-05-26 | 2011-12-01 | Satiogen Pharmaceuticals,Inc. | Bile acid recycling inhibitors and satiogens for treatment of diabetes, obesity, and inflammatory gastrointestinal conditions |
| WO2013020108A2 (en) * | 2011-08-04 | 2013-02-07 | Lumena Pharmaceuticals, Inc. | Bile acid recycling inhibitors for treatment of pancreatitis |
Non-Patent Citations (4)
| Title |
|---|
| HAMBRUCH ET AL.: "Synthetic Farnesoid X Receptor Agonists Induce High-Density Lipoprotein-Mediated Transhepatic Cholesterol Efflux in Mice and Monkeys and Prevent Atherosclerosis in Cholesteryl Ester Transfer Protein Transgenic Low-Density Lipoprotein Receptor (-/-) Mice", JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, vol. 343, no. 3, 2012, pages 556 - 567, XP055158296 * |
| LUNDQUIST ET AL.: "Improvement of Physiochemical Properties of the Tetrahydroazepinoindole Series of Farnesoid X Receptor (FXR) Agonists: Beneficial Modulation of Lipids in Primates", JOURNAL OF MEDICINAL CHEMISTRY, vol. 53, no. 4, 2010, pages 1774 - 1787, XP055223735, ISSN: 0022-2623 * |
| SCHUSTER ET AL.: "Pharmacophore-based discovery of FXR agonists. Part I: Model development and experimental validation", BIOORGANIC & MEDICINAL CHEMISTRY, vol. 19, no. 23, 2011, pages 7168 - 7180, XP028104545, ISSN: 0968-0896 * |
| See also references of EP3116878A4 * |
Cited By (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10485795B2 (en) | 2011-07-13 | 2019-11-26 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US10220027B2 (en) | 2011-07-13 | 2019-03-05 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US10077268B2 (en) | 2014-03-13 | 2018-09-18 | Salk Institute For Biological Studies | FXR agonists and methods for making and using |
| US11440889B2 (en) | 2014-03-13 | 2022-09-13 | The Salk Institute For Biological Studies | Analogs of fexaramine and methods of making and using |
| US10815203B2 (en) | 2014-03-13 | 2020-10-27 | Salk Institute For Biological Studies | Analogs of fexaramine and methods of making and using |
| US10450277B2 (en) | 2014-03-13 | 2019-10-22 | The Salk Institute For Biological Studies | Analogs of fexaramine and methods of making and using |
| US10301268B2 (en) | 2014-03-13 | 2019-05-28 | The Salk Institute For Biological Studies | Analogs of fexaramine and methods of making and using |
| US10323058B2 (en) | 2014-07-24 | 2019-06-18 | W. R. Grace & Co.-Conn. | Crystalline form of nicotinamide riboside |
| US10233207B2 (en) | 2014-07-24 | 2019-03-19 | W. R. Grace & Co.—Conn. | Crystalline form of nicotinamide riboside |
| US10183917B2 (en) | 2015-02-13 | 2019-01-22 | Sunshine Lake Pharma Co., Ltd. | Tricyclic compounds and uses thereof in medicine |
| US10189872B2 (en) | 2015-03-09 | 2019-01-29 | W. R. Grace & Co.-Conn | Crystalline form of nicotinamide riboside |
| US11214538B2 (en) | 2015-09-16 | 2022-01-04 | Metacrine, Inc. | Farnesoid X receptor agonists and uses thereof |
| WO2017078928A1 (en) * | 2015-11-06 | 2017-05-11 | Salk Institute For Biological Studies | Fxr agonists and methods for making and using |
| WO2017143134A1 (en) * | 2016-02-19 | 2017-08-24 | Alios Biopharma, Inc. | Fxr modulators and methods of their use |
| IL263493A (en) * | 2016-06-13 | 2019-01-31 | Gilead Sciences Inc | Fxr (nr1h4) modulating compounds |
| AU2017284109B2 (en) * | 2016-06-13 | 2020-01-23 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| CN109476636A (en) * | 2016-06-13 | 2019-03-15 | 吉利德科学公司 | Adjust the compound of FXR (NR1H4) |
| CN109311849A (en) * | 2016-06-13 | 2019-02-05 | 吉利德科学公司 | Adjust the compound of FXR (NR1H4) |
| JP2019517568A (en) * | 2016-06-13 | 2019-06-24 | ギリアード サイエンシーズ, インコーポレイテッド | FXR (NR1 H4) modulating compound |
| JP2019517569A (en) * | 2016-06-13 | 2019-06-24 | ギリアード サイエンシーズ, インコーポレイテッド | FXR (NR1 H4) modulating compound |
| US10329286B2 (en) | 2016-06-13 | 2019-06-25 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10421730B2 (en) | 2016-06-13 | 2019-09-24 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| EA037694B1 (en) * | 2016-06-13 | 2021-05-04 | Джилид Сайэнс, Инк. | Fxr (nr1h4) modulating compounds |
| US11739065B2 (en) | 2016-06-13 | 2023-08-29 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| EP3587412A1 (en) * | 2016-06-13 | 2020-01-01 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| EP4089072A1 (en) * | 2016-06-13 | 2022-11-16 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| US10774054B2 (en) | 2016-06-13 | 2020-09-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| CN109476636B (en) * | 2016-06-13 | 2024-01-09 | 吉利德科学公司 | Compounds that modulate FXR (NR 1H 4) |
| WO2017218330A1 (en) * | 2016-06-13 | 2017-12-21 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| JP7057460B2 (en) | 2016-06-13 | 2022-04-19 | ギリアード サイエンシーズ, インコーポレイテッド | FXR (NR1H4) regulatory compound |
| US11247986B2 (en) | 2016-06-13 | 2022-02-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| KR102361996B1 (en) | 2016-06-13 | 2022-02-14 | 길리애드 사이언시즈, 인코포레이티드 | Fxr (nr1h4) modulating compounds |
| CN109311849B (en) * | 2016-06-13 | 2021-02-26 | 吉利德科学公司 | Compounds that modulate FXR (NR1H4) |
| EP3257847A1 (en) * | 2016-06-13 | 2017-12-20 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| JP2021113198A (en) * | 2016-06-13 | 2021-08-05 | ギリアード サイエンシーズ, インコーポレイテッド | Fxr (nr1h4) modulating compounds |
| KR20210088748A (en) * | 2016-06-13 | 2021-07-14 | 길리애드 사이언시즈, 인코포레이티드 | Fxr (nr1h4) modulating compounds |
| US10981881B2 (en) | 2016-06-13 | 2021-04-20 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| CN109689105A (en) * | 2016-09-14 | 2019-04-26 | 诺华股份有限公司 | The combination of FXR agonist |
| WO2018051230A1 (en) * | 2016-09-14 | 2018-03-22 | Novartis Ag | Combination of fxr agonists |
| KR102218498B1 (en) | 2016-09-14 | 2021-02-22 | 노파르티스 아게 | Combination of FXR agonists |
| KR20190044666A (en) * | 2016-09-14 | 2019-04-30 | 노파르티스 아게 | Combinations of FXR agonists |
| WO2018153933A1 (en) | 2017-02-21 | 2018-08-30 | Genfit | Combination of a ppar agonist with a fxr agonist |
| US11833150B2 (en) | 2017-03-28 | 2023-12-05 | Gilead Sciences, Inc. | Methods of treating liver disease |
| US11773094B2 (en) | 2018-09-18 | 2023-10-03 | Organovo, Inc. | Farnesoid X receptor agonists and uses thereof |
| US11225473B2 (en) | 2019-01-15 | 2022-01-18 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| WO2021014349A1 (en) | 2019-07-23 | 2021-01-28 | Novartis Ag | Treatment comprising fxr agonists |
| WO2021014350A1 (en) | 2019-07-23 | 2021-01-28 | Novartis Ag | Combination treatment of liver diseases using fxr agonists |
| WO2021044287A1 (en) | 2019-09-03 | 2021-03-11 | Novartis Ag | Treatment of liver disease or disorder comprising actrii receptor antagonists |
| WO2021053618A1 (en) | 2019-09-19 | 2021-03-25 | Novartis Ag | Treatment comprising fxr agonists |
| WO2021064575A1 (en) | 2019-09-30 | 2021-04-08 | Novartis Ag | Treatment comprising the use of fxr agonists |
| WO2021127466A1 (en) | 2019-12-20 | 2021-06-24 | Novartis Ag | Combination treatment of liver diseases using integrin inhibitors |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| WO2022101853A1 (en) | 2020-11-16 | 2022-05-19 | Novartis Ag | Method of determining liver fibrosis |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3116878A4 (en) | 2018-02-14 |
| EP3116878A1 (en) | 2017-01-18 |
| JP2017510572A (en) | 2017-04-13 |
| KR20160132111A (en) | 2016-11-16 |
| AU2015229072A1 (en) | 2016-09-29 |
| CA2942403A1 (en) | 2015-09-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015138986A1 (en) | Fxr agonists and methods for making and using | |
| US10077268B2 (en) | FXR agonists and methods for making and using | |
| AU2015229055B2 (en) | Analogs of fexaramine and methods of making and using | |
| WO2017078928A1 (en) | Fxr agonists and methods for making and using | |
| US11440889B2 (en) | Analogs of fexaramine and methods of making and using | |
| Xu | Recent progress on bile acid receptor modulators for treatment of metabolic diseases | |
| JP2021113198A (en) | Fxr (nr1h4) modulating compounds | |
| JP5420400B2 (en) | G protein-coupled receptor agonist | |
| EP3267991A1 (en) | Treating latent autoimmune diabetes of adults with farnesoid x receptor agonists to activate intestinal receptors | |
| US10183015B2 (en) | Heterocyclic compounds and methods of use | |
| Shah et al. | GPR119 agonists for the potential treatment of type 2 diabetes and related metabolic disorders | |
| US20150258052A1 (en) | Methods of using fexaramine and agents that increase sympathetic nervous system activity to promote browning of white adipose tissue | |
| BR112019017312A2 (en) | combination of a ppar agonist with an fxr agonist | |
| WO2017078927A1 (en) | Analogs of fexaramine and methods of making and using | |
| WO2014117292A1 (en) | Amide compounds and preparation methods, pharmaceutical compositions and uses thereof | |
| WO2021204755A1 (en) | Pharmaceutical combination for the treatment of liver diseases | |
| TW202122078A (en) | Methods of treating liver disease using lta4h inhibitors | |
| WO2021037702A1 (en) | Pharmaceutical combination of a specific thienopyridone derivative with an fxr agonist for the treatment of liver diseases | |
| WO2023242810A1 (en) | Mchr1 antagonists for the treatment of prader-willi syndrome |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15761517 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2942403 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2016556962 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2015229072 Country of ref document: AU Date of ref document: 20150313 Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015761517 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015761517 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20167028569 Country of ref document: KR Kind code of ref document: A |