WO2012114565A1 - 無機繊維 - Google Patents
無機繊維 Download PDFInfo
- Publication number
- WO2012114565A1 WO2012114565A1 PCT/JP2011/069273 JP2011069273W WO2012114565A1 WO 2012114565 A1 WO2012114565 A1 WO 2012114565A1 JP 2011069273 W JP2011069273 W JP 2011069273W WO 2012114565 A1 WO2012114565 A1 WO 2012114565A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mass
- spinning
- inorganic fiber
- group
- raw material
- Prior art date
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/622—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/62227—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products obtaining fibres
- C04B35/62231—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products obtaining fibres based on oxide ceramics
- C04B35/62236—Fibres based on aluminium oxide
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F9/00—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments
- D01F9/08—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments of inorganic material
- D01F9/10—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments of inorganic material by decomposition of organic substances
-
- C—CHEMISTRY; METALLURGY
- C03—GLASS; MINERAL OR SLAG WOOL
- C03B—MANUFACTURE, SHAPING, OR SUPPLEMENTARY PROCESSES
- C03B37/00—Manufacture or treatment of flakes, fibres, or filaments from softened glass, minerals, or slags
- C03B37/01—Manufacture of glass fibres or filaments
- C03B37/011—Manufacture of glass fibres or filaments starting from a liquid phase reaction process, e.g. through a gel phase
-
- C—CHEMISTRY; METALLURGY
- C03—GLASS; MINERAL OR SLAG WOOL
- C03C—CHEMICAL COMPOSITION OF GLASSES, GLAZES OR VITREOUS ENAMELS; SURFACE TREATMENT OF GLASS; SURFACE TREATMENT OF FIBRES OR FILAMENTS MADE FROM GLASS, MINERALS OR SLAGS; JOINING GLASS TO GLASS OR OTHER MATERIALS
- C03C13/00—Fibre or filament compositions
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/622—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/62227—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products obtaining fibres
- C04B35/62231—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products obtaining fibres based on oxide ceramics
- C04B35/6224—Fibres based on silica
- C04B35/62245—Fibres based on silica rich in aluminium oxide
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/622—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/626—Preparing or treating the powders individually or as batches ; preparing or treating macroscopic reinforcing agents for ceramic products, e.g. fibres; mechanical aspects section B
- C04B35/63—Preparing or treating the powders individually or as batches ; preparing or treating macroscopic reinforcing agents for ceramic products, e.g. fibres; mechanical aspects section B using additives specially adapted for forming the products, e.g.. binder binders
- C04B35/632—Organic additives
- C04B35/634—Polymers
- C04B35/63404—Polymers obtained by reactions only involving carbon-to-carbon unsaturated bonds
- C04B35/63424—Polyacrylates; Polymethacrylates
-
- C—CHEMISTRY; METALLURGY
- C03—GLASS; MINERAL OR SLAG WOOL
- C03C—CHEMICAL COMPOSITION OF GLASSES, GLAZES OR VITREOUS ENAMELS; SURFACE TREATMENT OF GLASS; SURFACE TREATMENT OF FIBRES OR FILAMENTS MADE FROM GLASS, MINERALS OR SLAGS; JOINING GLASS TO GLASS OR OTHER MATERIALS
- C03C2213/00—Glass fibres or filaments
- C03C2213/02—Biodegradable glass fibres
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/30—Constituents and secondary phases not being of a fibrous nature
- C04B2235/32—Metal oxides, mixed metal oxides, or oxide-forming salts thereof, e.g. carbonates, nitrates, (oxy)hydroxides, chlorides
- C04B2235/3205—Alkaline earth oxides or oxide forming salts thereof, e.g. beryllium oxide
- C04B2235/3208—Calcium oxide or oxide-forming salts thereof, e.g. lime
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/30—Constituents and secondary phases not being of a fibrous nature
- C04B2235/32—Metal oxides, mixed metal oxides, or oxide-forming salts thereof, e.g. carbonates, nitrates, (oxy)hydroxides, chlorides
- C04B2235/3217—Aluminum oxide or oxide forming salts thereof, e.g. bauxite, alpha-alumina
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/30—Constituents and secondary phases not being of a fibrous nature
- C04B2235/34—Non-metal oxides, non-metal mixed oxides, or salts thereof that form the non-metal oxides upon heating, e.g. carbonates, nitrates, (oxy)hydroxides, chlorides
- C04B2235/3418—Silicon oxide, silicic acids, or oxide forming salts thereof, e.g. silica sol, fused silica, silica fume, cristobalite, quartz or flint
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/30—Constituents and secondary phases not being of a fibrous nature
- C04B2235/44—Metal salt constituents or additives chosen for the nature of the anions, e.g. hydrides or acetylacetonate
- C04B2235/443—Nitrates or nitrites
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/30—Constituents and secondary phases not being of a fibrous nature
- C04B2235/44—Metal salt constituents or additives chosen for the nature of the anions, e.g. hydrides or acetylacetonate
- C04B2235/444—Halide containing anions, e.g. bromide, iodate, chlorite
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/30—Constituents and secondary phases not being of a fibrous nature
- C04B2235/44—Metal salt constituents or additives chosen for the nature of the anions, e.g. hydrides or acetylacetonate
- C04B2235/448—Sulphates or sulphites
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/30—Constituents and secondary phases not being of a fibrous nature
- C04B2235/44—Metal salt constituents or additives chosen for the nature of the anions, e.g. hydrides or acetylacetonate
- C04B2235/449—Organic acids, e.g. EDTA, citrate, acetate, oxalate
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/50—Constituents or additives of the starting mixture chosen for their shape or used because of their shape or their physical appearance
- C04B2235/52—Constituents or additives characterised by their shapes
- C04B2235/5208—Fibers
- C04B2235/5264—Fibers characterised by the diameter of the fibers
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/65—Aspects relating to heat treatments of ceramic bodies such as green ceramics or pre-sintered ceramics, e.g. burning, sintering or melting processes
- C04B2235/656—Aspects relating to heat treatments of ceramic bodies such as green ceramics or pre-sintered ceramics, e.g. burning, sintering or melting processes characterised by specific heating conditions during heat treatment
- C04B2235/6562—Heating rate
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/70—Aspects relating to sintered or melt-casted ceramic products
- C04B2235/96—Properties of ceramic products, e.g. mechanical properties such as strength, toughness, wear resistance
- C04B2235/9669—Resistance against chemicals, e.g. against molten glass or molten salts
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01D—MECHANICAL METHODS OR APPARATUS IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS
- D01D5/00—Formation of filaments, threads, or the like
- D01D5/0007—Electro-spinning
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F9/00—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments
- D01F9/08—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments of inorganic material
Definitions
- the present invention relates to inorganic fibers.
- Inorganic fibers are mainly composed of fibers composed of inorganic compounds and are expected as constituent materials such as heat insulating materials and refractory materials. Particularly, those having an average fiber diameter of 1 ⁇ m or less are filter materials and sealing materials. Use as a constituent material is expected.
- asbestos (asbestos) has been conventionally known as an inorganic fiber having a small fiber diameter. Since this asbestos has a small fiber diameter and high chemical resistance to body fluids, It is said that it affects the human body by giving long-term stimulation to cells in the alveoli. Also in the inorganic fibers other than asbestos, inorganic fibers with improved heat resistance composed mainly of SiO 2, when the SiO 2 has reached the lungs inner part by generating breathing cristobalite was crystallized in a high temperature environment It is said that it affects the human body.
- an object of the present invention is to provide an inorganic fiber having a high biological dissolution rate and high heat resistance.
- the present inventors have conducted intensive studies. As a result, 35 mass% to 88 mass% Al 2 O 3 , 3 mass% to 45 mass% CaO, and 5 mass% to 40 mass%. by weight of SiO 2, found that the content ratio of the sum of the Al 2 O 3, CaO and SiO 2 may be solved by the inorganic fiber is at least 98% by weight of the total fibers, and have completed the present invention.
- the present invention (1) 35% by mass to 88% by mass of Al 2 O 3 , 3% by mass to 45% by mass of CaO and 5% by mass to 40% by mass of SiO 2 , and total of Al 2 O 3 , CaO and SiO 2
- the inorganic fiber is After dissolving a water-soluble basic aluminum acid, a water-soluble calcium compound, and a water-soluble or water-dispersible silicon compound in an aqueous medium to prepare a spinning raw material aqueous solution, A crude inorganic fiber is obtained by spinning the spinning raw material aqueous solution
- an inorganic fiber having a high biological dissolution rate and high heat resistance.
- the inorganic fiber of the present invention contains 35% by weight to 88% by weight Al 2 O 3 , 3% by weight to 45% by weight CaO and 5% by weight to 40% by weight SiO 2, and contains Al 2 O 3 , CaO and The total content of SiO 2 is 98% by mass or more of the entire fiber.
- the inorganic fiber of the present invention contains 35% to 88% by mass of Al 2 O 3 , preferably 39% to 87% by mass, and more preferably 39% to 83% by mass. 39 mass% to 66 mass% is more preferable, and 49 mass% to 66 mass% is more preferable. When the content ratio of Al 2 O 3 is within the above range, desired heat resistance can be easily obtained.
- the inorganic fiber of the present invention contains 3% to 45% by weight of CaO, more preferably 3% to 42% by weight, and still more preferably 26% to 42% by weight. .
- content ratio of CaO is within the above range, desired biological solubility can be easily obtained.
- the inorganic fiber of the present invention contains 5% by mass to 40% by mass of SiO 2 , preferably 8% by mass to 39% by mass, more preferably 8% by mass to 28% by mass, More preferably, the content is from 16% by mass to 16% by mass.
- the content ratio of SiO 2 is in the above range, desired heat resistance can be easily obtained.
- the total content of Al 2 O 3 , CaO and SiO 2 is 98% by mass or more of the whole fiber, and more preferably 99% by mass or more of the whole fiber.
- the inorganic fiber of the present invention can contain less than 2% by mass of unavoidable components, and the unavoidable component means an impurity component mixed during the preparation of the inorganic fiber.
- the inorganic fiber of the present invention contains 35% by weight to 88% by weight Al 2 O 3 , 3% by weight to 45% by weight CaO and 5% by weight to 40% by weight SiO 2, and contains Al 2 O 3 , CaO and When the total content of SiO 2 is 98% by mass or more of the entire fiber, desired biosolubility and heat resistance can be exhibited.
- the inorganic fiber of the present invention contains 39% by mass to 66% by mass of Al 2 O 3 , 26% by mass to 42% by mass of CaO and 8% by mass to 28% by mass of SiO 2, and contains Al 2 O 3 , When the total content of CaO and SiO 2 is 98% by mass or more of the entire fiber, desired biological solubility and heat resistance can be further exhibited.
- the content ratio (mass%) of each component is a powder obtained by taking a part from the spinning raw material aqueous solution used at the time of fiber production described later, drying it, and then firing it at 1000 ° C. for 2 hours. It means a value when measured using a fluorescent X-ray analyzer (RIX2000 manufactured by Rigaku) as a measurement sample.
- RIX2000 fluorescent X-ray analyzer manufactured by Rigaku
- correction calculation shall be performed so that the total value of the metal oxide excluding the balance component is 100% by mass.
- the inorganic fiber of the present invention has the composition described above, and is prepared by dissolving a water-soluble basic aluminum acid, a water-soluble calcium compound, and a water-soluble or water-dispersible silicon compound in an aqueous medium. After preparing the aqueous solution, the spinning raw material aqueous solution is preferably spun to obtain coarse inorganic fibers, and then the coarse inorganic fibers are fired.
- the water-soluble basic acid aluminum is represented by the following formula (I): Al (OH) X Y Z (I) (However, X is a positive number greater than 0 and less than 3, Y is any one selected from Cl atom, NO 3 group, SO 4 group, and RCOO group, Z is Y atom as Cl atom, NO 3 group, RCOO group 3-X, Y is SO 4 group (3-X) / 2, and R is a hydrogen atom, a hydrocarbon group having 1 to 10 carbon atoms or a hydroxyl group-containing carbon In the case where a plurality of RCOO groups are present, each R may be the same or different, and is preferably at least one selected from compounds represented by
- the inorganic fiber of the present invention is preferably one that is spun by the electrostatic spinning method described later.
- the inorganic fiber of the present invention can exhibit biosolubility even when the average fiber diameter is less than 4 ⁇ m, and the average fiber diameter of the inorganic fiber is preferably 3 ⁇ m or less, and is 1 ⁇ m or less. More preferable is 0.5 ⁇ m or less.
- the average fiber diameter of the inorganic fibers is 1 ⁇ m or less, it can be suitably used as a constituent material for the filter material and the seal material.
- Inorganic fibers having an average fiber diameter of 1 ⁇ m or less are manufactured by adjusting the atmosphere and applied voltage during electrostatic spinning by the electrostatic spinning method described later, and adjusting the concentration and viscosity of the spinning raw material aqueous solution. Can do.
- the inorganic fiber of the present invention preferably has a dissolution rate of 20 ng / cm 2 ⁇ h or more at 0 to 24 hours after the start of the test when evaluated by the biosolubility evaluation method described later. What is cm ⁇ 2 > * h or more is more suitable. Further, those having a dissolution rate of 20 ng / cm 2 ⁇ h or more in 24 to 48 hours after the start of the test are preferable, and those having a dissolution rate of 40 ng / cm 2 ⁇ h or more are more preferable.
- the solubility is low even if the dissolution rate at 24 to 48 hours after the start of the test is 20 ng / cm 2 ⁇ h or less. I can expect.
- the upper limit of the dissolution rate is not particularly limited, but is usually about 5400 ng / cm 2 ⁇ h.
- the inorganic fiber of the present invention has a high biological dissolution rate and excellent biological solubility, even if the average fiber diameter is small, the influence on the living environment is small, and it can be easily performed by the manufacturing method described later. Can be made into fine fibers. For this reason, for example, a fine fiber product having an average fiber diameter of 1 ⁇ m or less can be used in various industrial fields as a constituent material of a filter material and a sealing material.
- the average fiber diameter of the inorganic fibers is 10 to 111 fibers randomly selected from a photograph (magnification 2000 to 5000 times) taken with a scanning electron microscope (JSM-5800LV, manufactured by JEOL). Means the average value calculated from these widths.
- the inorganic fiber of the present invention preferably has a melting point of 1300 ° C. or higher, more preferably 1350 ° C. or higher, and further preferably 1367 ° C. or higher.
- fusing point of inorganic fiber means the value calculated
- various methods can be exemplified, and a method of dry spinning a spinning raw material solution having a desired composition, or a melt having a desired composition is drawn from a spinning nozzle and cooled.
- a continuous spinning method for spinning while winding a winder a spinner method (external centrifugal method) in which a melt having a desired composition is collided with a high-speed rotating body and fiberized by the centrifugal force, and a melt having a desired composition
- Examples thereof include an internal centrifugal method of discharging from a rotating body and fiberizing by centrifugal force, and a melt blowing method of converting a melt having a desired composition into fiber by compressed air.
- the dry spinning method includes a dry continuous spinning method in which a spinning raw material solution having a desired composition is discharged from a nozzle and then dried while being wound and stretched by a winder, or a spinning raw material solution having a desired composition is centrifuged by an air flow. Examples thereof include a method of drying to obtain discontinuous fibers and an electrospinning method to be described later.
- the viscosity of the spinning raw material solution it is preferable to appropriately adjust the viscosity of the spinning raw material solution.
- the addition amount of the spinning aid is changed, or concentration by heating or decompression or addition of water is added. Can be carried out by a dilution operation.
- the viscosity may be about several tens Pa ⁇ s to several hundred Pa ⁇ s. 60 Pa ⁇ s to 200 Pa ⁇ s is more preferable.
- the viscosity is less than several tens of Pa ⁇ s, the spinning solution extruded from the nozzle loses surface tension and is easily broken by a capillary, making spinning difficult.
- a spinning raw material solution is spun by spinning and spinning by spinning from a large number of holes or spinning and spinning the spinning solution by blowing air.
- the viscosity of the spinning raw material solution is preferably about several Pa ⁇ s to several tens Pa ⁇ s.
- the viscosity is less than a few Pa ⁇ s, the spinning raw material solution is not made into a fiber but splashes as a droplet, or even if it can be made into a fiber, when the spinning solution is stretched and broken, a spherical shape called a shot A large amount of particles are produced.
- the viscosity is excessive, the fiber cannot be produced because it cannot be drawn by centrifugal force or blowing.
- the organic matter in the coarse inorganic fibers can be eliminated and desired inorganic fibers can be obtained.
- a production method using a dry spinning method particularly a production method using an electrostatic spinning method, is preferable because the target inorganic fiber can be produced easily and at low cost. is there.
- the inorganic fiber of the present invention is prepared by dissolving a water-soluble basic aluminum acid, a water-soluble calcium compound, and a water-soluble or water-dispersible silicon compound in an aqueous medium to prepare a spinning raw material aqueous solution.
- the spinning raw material aqueous solution is preferably spun to obtain coarse inorganic fibers, and then the coarse inorganic fibers are preferably fired.
- the inorganic fiber of the present invention that is, 35% by weight to 88% by weight Al 2 O 3 , 3% by weight to 45% by weight CaO and 5% by weight to 40% by weight SiO 2 is contained, and Al 2
- the total content of O 3 , CaO and SiO 2 is 98% by mass or more of the whole fiber, 39% to 66% by mass of Al 2 O 3 , 26% to 42% by mass % of comprises SiO 2 of CaO and 8% to 28% by weight, may be content which is the sum of Al 2 O 3, CaO and SiO 2 to prepare inorganic fibers is at least 98% by weight of the total fiber .
- examples of the raw material include an aluminum raw material
- examples of the aluminum raw material include a water-soluble basic acid aluminum
- the water-soluble basic aluminum acid used as a raw material is represented by the following formula (I) Al (OH) X Y Z (I) (However, X is a positive number greater than 0 and less than 3, Y is any one selected from Cl atom (chlorine atom), NO 3 group, SO 4 group, RCOO group (carboxyl group), Z is 3-X when Y is a Cl atom, NO 3 group, or RCOO group, and (3-X) / 2 when Y is a SO 4 group, and the R is a hydrogen atom or a carbon number of 1 to 10 And when there are a plurality of RCOO groups, each R may be the same or different)
- X is preferably a positive number of 1 or more and less than 3, more preferably a positive number of 1 or more and 2.5 or less. 2 is more preferable.
- X can be calculated from the composition ratio of the acid added during the synthesis of the basic acid aluminum.
- the basic acid aluminum represented by the composition formula (I) is preferably a basic aluminum carboxylate (Al (OH) X (RCOO) 3 -X ), and the basic acid aluminum represented by the composition formula (I) Is a basic aluminum carboxylate, it is possible to suppress the generation of chlorine and nitric acid, which have a large environmental load when performing the baking treatment described later.
- R constituting the RCOO group is a hydrogen atom, a hydrocarbon group having 1 to 10 carbon atoms, or a hydroxyl group-containing hydrocarbon. It is a group.
- R is a hydrocarbon group or a hydroxyl group-containing hydrocarbon group
- the carbon number thereof is 1 to 10, preferably 1 to 5.
- the basic acid aluminum represented by the formula (I) is hardly water-soluble.
- the hydrocarbon group portion may be linear or branched, and may be a saturated hydrocarbon group or an unsaturated hydrocarbon group.
- R is a hydrocarbon group
- examples of the hydrocarbon group include an alkyl group, an alkenyl group, a cycloalkyl group, and an alkylcycloalkyl group.
- an alkyl group such as a methyl group, an ethyl group, a propyl group, or a butyl group (if these alkyl groups can be branched, the alkyl group may be linear or branched);
- An alkenyl group such as a propenyl group or a butenyl group (when these alkenyl groups can be branched, the alkenyl group may be linear or branched, and the position of the double bond is arbitrary);
- Examples thereof include a cycloalkyl group such as a cyclopropyl group and a cyclobutyl group; an alkylcycloalkyl group such as a methylcyclopropyl group and a methylcyclobutyl group (the substitution position of the alkyl group to the cycloalkyl group is also arbitrary), and the like.
- R is a hydroxyl group-containing hydrocarbon group
- examples of the hydroxyl group-containing hydrocarbon group include a hydroxyalkyl group, a hydroxyalkenyl group, and a hydroxycycloalkyl group.
- a hydroxyalkyl group such as a hydroxymethyl group, a hydroxyethyl group, a hydroxypropyl group, or a hydroxybutyl group (if these hydroxyalkyl groups can be branched, the alkyl group constituting the hydroxyalkyl group) May be linear or branched); a hydroxyalkenyl group such as a hydroxybutenyl group (the alkenyl group constituting the hydroxyalkenyl group may be linear or branched, and the position of the double bond is arbitrary) And a hydroxycycloalkyl group such as a hydroxycyclopropyl group and a hydroxycyclobutyl group (the substitution position of a hydroxyl group or an alkyl group to a cycloalkyl group is also arbitrary).
- the RCOO group is preferably a reaction residue of a carboxylic acid selected from formic acid, acetic acid, lactic acid, etc. (HCOO group, CH 3 COO group, CH 3 CH (OH) COO group). .
- the calcium raw material is preferably a water-soluble calcium compound.
- the calcium compound exhibits water solubility and is dissolved in a desired amount in a spinning raw material aqueous solution described later. It is not particularly limited as long as it can be used, and examples thereof include calcium carbonate, nitrate, sulfate, acetate, hydroxide, chloride, fluoride, borate, and phosphate.
- the calcium compound to be dissolved in the spinning raw material aqueous solution is a basic aluminum carboxylate
- the calcium compound is also preferably a carboxylate salt, and is soluble in the spinning raw material aqueous solution.
- calcium acetate monohydrate is more preferable because of the availability of the material.
- the silicon raw material is preferably a water-soluble or water-dispersible silicon compound, and the silicon compound is particularly suitable as long as it can be dissolved or dispersed in an aqueous spinning raw material solution.
- water-soluble silicon compounds include water-soluble silicates, water-soluble silicon alkoxides (tetramethoxysilane, tetraethoxysilane, tetrapropoxysilane, etc.), and the like.
- the functional silicon compound include silica sol (colloidal silica). Among these silicon compounds, silica sol (colloidal silica) is preferable from the viewpoint of viscosity stability of the spinning raw material aqueous solution.
- the silica sol is preferably one in which silica having a particle size of 4 to 100 nm is dispersed in a medium having a solid content of 5% by mass to 30% by mass, and the silica sol is a sol-gel method produced from alkoxysilane, It can be produced by the sodium silicate method produced from sodium acid.
- a spinning aid can be further used as necessary.
- the spinning aid is not particularly limited as long as it can produce a desired inorganic fiber, but is preferably a water-soluble organic polymer in view of ease of handling and solubility. Examples thereof include polyethylene oxide, polypropylene oxide, polyvinyl alcohol, polyvinyl ether, polyvinyl ester, polyacrylic acid ester and copolymers thereof, and among these, polyacrylic acid ester is preferable.
- the inorganic fiber of the present invention is produced by a dry spinning method, as described above, the water-soluble basic aluminum acid, the water-soluble calcium compound, the water-soluble or water-dispersible silicon compound, and, if necessary, a spinning aid. And a method in which an agent is dissolved in an aqueous medium to form a spinning raw material aqueous solution and then spinning.
- aqueous medium water is preferable, and in order to improve the stability of the solution or to improve the spinning stability, other media that are water-soluble and soluble in water, such as alcohols, ketones, amines, etc. , Amides, carboxylic acids and the like may be added. Further, an organic salt such as ammonium chloride may be added to these media.
- the concentration of the water-soluble basic aluminum acid in the spinning raw material aqueous solution is preferably 4% by mass to 83% by mass, and more preferably 4% by mass to 68% by mass.
- the abundance ratio of aluminum element to the total amount of aluminum element, calcium element, and silicon element is as follows: aluminum element is converted to Al 2 O 3 , calcium element is converted to CaO, and silicon element is converted to SiO 2 . It is preferably 35% by mass to 88% by mass in terms of Al 2 O 3 , more preferably 39% by mass to 87% by mass, still more preferably 39% by mass to 83% by mass, and 39% by mass. It is more preferably from 66% by mass, and even more preferably from 49% by mass to 66% by mass.
- the abundance ratio of the aluminum element is more than 88% by mass in terms of Al 2 O 3 with respect to the total amount of the aluminum element, the calcium element, and the silicon element, the abundance ratio of the calcium element becomes small and it is difficult to obtain desired biological solubility. If the abundance ratio of the aluminum element with respect to the total amount of the aluminum element, calcium element and silicon element is less than 35% by mass in terms of Al 2 O 3 , it becomes difficult to exhibit desired heat resistance and biological solubility.
- the concentration of the water-soluble calcium compound in the spinning raw material aqueous solution is preferably 0.4% by mass to 46% by mass, and more preferably 2% by mass to 46% by mass.
- the existing ratio of the calcium element to the total amount of aluminum element and calcium element and silicon element, CaO when the aluminum element in terms Al 2 O 3, a calcium element CaO, a silicon element in the SiO 2 It is preferably 3% by mass to 45% by mass, more preferably 3% by mass to 42% by mass, and further preferably 26% by mass to 42% by mass.
- the abundance ratio of the calcium element with respect to the total amount of the aluminum element, the calcium element, and the silicon element is more than 45% by mass in terms of CaO, the abundance ratio of the other metal elements is lowered and it is difficult to obtain desired heat resistance. If it is less than mass%, it is difficult to obtain desired biological solubility.
- the concentration of the water-soluble or water-dispersible silicon compound in the spinning raw material aqueous solution is preferably 0.4% by mass to 24% by mass, more preferably 0.4% by mass to 16% by mass, More preferably, the content is 0.4% by mass to 8% by mass.
- the abundance ratio of the silicon element with respect to the total amount of the aluminum element, the calcium element, and the silicon element is obtained when the aluminum element is converted to Al 2 O 3 , the calcium element is converted to CaO, and the silicon element is converted to SiO 2 .
- the existing ratio of silicon element to the total amount of aluminum element, calcium element, and silicon element is more than 40% by mass in terms of SiO 2 , the existing ratio of calcium element becomes small and it becomes difficult to obtain desired biological solubility. If the abundance ratio of the silicon element with respect to the total amount of the element, calcium element and silicon element is less than 5% by mass in terms of SiO 2 , it becomes difficult to exhibit desired heat resistance.
- the concentration of the spinning aid is preferably 0.1% by mass to 10% by mass, more preferably 0.1% by mass to 5% by mass.
- the spinning aid is preferably as small as possible because the fibers are densified after firing and the strength is maintained. However, since the form at the time of fiber preparation may not be stable with a small amount, the amount added may be increased as necessary. It is preferable to adjust.
- the method for preparing the spinning raw material aqueous solution is not particularly limited, and for example, an aqueous medium, a water-soluble basic aluminum acid, a water-soluble calcium compound, a water-soluble or water-dispersible silicon compound, a spinning aid, and other Arbitrary components may be prepared by mixing each component to a desired concentration, or an aqueous solution of a water-soluble basic aluminum acid, an aqueous solution of a water-soluble calcium compound, You may produce by mixing the aqueous solution of a water-dispersible silicon compound, the aqueous solution of a spinning aid, and other arbitrary components so that each component may become a desired density
- the crude inorganic fiber of the present invention it is preferable to obtain the crude inorganic fiber by spinning the above spinning raw material aqueous solution by an electrostatic spinning method.
- the electrostatic spinning method is a method of applying a voltage to a spinning raw material aqueous solution containing a fiber-forming compound and discharging the spinning raw material aqueous solution into fibers using electrostatic repulsion.
- any method can be used.
- the spinning raw material aqueous solution is supplied to an appropriate position in the electrostatic field, and the spinning raw material aqueous solution is supplied.
- a method of applying a voltage to the yarn to form a fiber using an electric field can be mentioned.
- FIG. 1 is a diagram showing an example of a spinning device used for electrostatic spinning.
- the spinning device 1 includes a syringe 2, a nozzle 3, a high voltage generator 4, and a sample collection stand 5.
- the spinning raw material aqueous solution is fed into the syringe 2 and then fed to the tip of the nozzle 3.
- the high voltage generator 4 is electrically connected to a conductive fixing part provided around the nozzle 3 and a conductive sample collection base 5, respectively, and the nozzle 3 passes through the fixing part provided around the nozzle 3.
- the spinning raw material aqueous solution is ejected from the tip of the nozzle 3 to be fiberized to obtain coarse inorganic fibers.
- the obtained crude inorganic fiber is collected on the sample collection stand 5 which is a counter electrode.
- a plurality of nozzles 3 may be used, and the nozzles 3 may be arranged in parallel to increase the production rate of the fibrous material.
- the voltage applied during electrostatic spinning is 1 to 100 kV, taking into account conditions such as the distance between the nozzle tip and the counter electrode (distance between the electrodes), the viscosity of the aqueous spinning raw material solution, and the concentration of the aqueous spinning raw material solution. Preferably, it is 3 to 30 kV.
- the distance between the electrodes depends on the charge amount, the nozzle size, the ejection amount of the spinning raw material aqueous solution from the nozzle, the concentration of the spinning raw material aqueous solution, etc., but is preferably 20 to 500 mm, more preferably 50 to 300 mm, and more preferably 100 to 200 mm. Is more preferable.
- the viscosity of the spinning raw material aqueous solution is preferably about 0.01 to 5.0 Pa ⁇ s, more preferably about 0.05 to 3.0 Pa ⁇ s.
- the spinning raw material aqueous solution may not form a filament during spinning, and spherical particles may be formed, and the spinning raw material aqueous solution has a viscosity of 5.0 Pa ⁇ s. If it is super, fiberizing treatment becomes difficult.
- the viscosity of the spinning raw material aqueous solution can be adjusted by adjusting the amount of the spinning aid added or by appropriately performing a concentration operation by heat treatment or reduced pressure treatment.
- the viscosity of the spinning raw material aqueous solution is measured using a viscoelasticity measuring device (Physica MCR301 manufactured by Anton Paar), maintaining the liquid temperature of the spinning solution at 25 ° C., and a shear rate of 10 s ⁇ 1 . Means shear viscosity.
- a viscoelasticity measuring device Physical MCR301 manufactured by Anton Paar
- the coarse inorganic fiber obtained by electrostatic spinning preferably has an average fiber diameter of 10 nm to 2000 nm, and more preferably 50 nm to 1000 nm.
- the average fiber diameter of the coarse inorganic fibers is selected at 10 to 111 locations at random from a photograph (magnification 2000 to 5000 times) taken with a scanning electron microscope (JSM-5800LV manufactured by JEOL). It means the average value calculated from the widths of the fibers measured.
- the coarse inorganic fiber obtained by the electrostatic spinning method is fired.
- the calcination temperature is preferably 500 ° C. or higher and lower than the liquid phase generation temperature, and specifically, it is preferably a temperature that is 500 ° C. or higher and 1350 ° C. or lower and does not generate a liquid phase.
- the firing temperature is less than 500 ° C.
- organic components such as organic polymers used as a spinning aid remain in the obtained inorganic fiber, and when the firing temperature exceeds 1350 ° C., crystal grains grow.
- the resulting inorganic fiber becomes very brittle or forms a liquid phase and reacts with the hearth.
- Firing can be performed using a known electric furnace or the like, and the atmosphere during firing is preferably air or an oxidizing atmosphere in order to decompose organic substances used as a spinning aid or the like.
- an inert atmosphere such as nitrogen may be used.
- the present invention in this way, even if the average fiber diameter is 1 ⁇ m or less, the influence on the human body and living environment is suppressed, and high biosolubility is exhibited. It is possible to provide an inorganic fiber exhibiting heat resistance suitable as a constituent material.
- this evaluation sample was placed on a PTFE (polytetrafluoroethylene) membrane filter having a pore diameter of 0.1 ⁇ m, and a PTFE membrane filter having a pore diameter of 1 ⁇ m was further placed on the evaluation sample and fixed as a filter unit.
- pH 5.0 physiological saline having the composition described in Table 1 was circulated at a rate of 0.15 ml / min.
- the physiological saline that has passed through the evaluation sample is collected in a tank provided at the lower part of the filter unit, but the inorganic fiber components are also eluted by the physiological saline passing through the evaluation sample.
- the physiological saline solution during the evaluation test was maintained at 37 ° C., which is the temperature of the biological fluid, and the inorganic fiber component eluate stored in the tank was taken out 24 hours and 48 hours after the start of the test. The elution amount of the component was quantified, and the solubility was calculated from the value.
- the fiber surface area is obtained by separately measuring the fiber diameter, and the measured value of the solubility, the true density of the fiber, the use of the sample.
- the solubility per unit time and unit fiber surface area (ng / cm 2 ⁇ h) was calculated from the amount and used as the dissolution rate.
- the dissolution rate is determined from 0 to 24 hours after the start of the test, and in Examples 1, 10 to 13, 16 to 20, 22 to 23, and 25, the test is performed. The rate at 24 to 48 hours after the start was also determined.
- the surface area of the inorganic fiber was calculated by determining the total area of the inorganic fiber as a cylindrical shape.
- the average fiber diameter d (m) of the inorganic fiber was measured using a scanning electron microscope (JSM-5800LV, manufactured by JEOL) as described above, and a pycnometer method was used.
- the surface area S (m 2 ) of the inorganic fiber can be calculated by measuring the true density ⁇ (kg / m 3 ) of the inorganic fiber from the pulverized inorganic fiber and substituting it into the above formula (3). .
- the dissolution rate in physiological saline obtained by the above evaluation is an index of chemical resistance to body fluids. The higher this value, the lower the chemical resistance to body fluids, and the harmfulness to living organisms is. It is said to be low.
- Example 1 As the basic aluminum carboxylate, Al (OH) X (RCOO) 3-X (where X is a value of 1.7 and R has a carbon number of 0 to 2) is used as follows. Was prepared.
- the calcium acetate aqueous solution 47.2 having a calcium concentration calculated as CaO of 7.3% by mass. Adding 2 parts by mass of 29.2 parts by mass of colloidal silica whose silicon concentration in terms of SiO 2 is 20.5% by mass, and 28.6 parts by mass of an aqueous polyacrylate ester solution prepared to a concentration of 6.0% by mass, After mixing, the solution was concentrated as appropriate to prepare a spinning raw material aqueous solution having a viscosity of 1.0 Pa ⁇ s.
- the spinning raw material aqueous solution was spun using the spinning device 1 shown in FIG.
- the above spinning raw material aqueous solution is filled into the syringe 2 and then fed to the tip of the nozzle 3, and is electrically connected to the fixed portion provided around the nozzle 3 and the sample collection base 5.
- the spinning raw material aqueous solution is ejected from the tip of the nozzle 3, fiberized, and the sample collection base 5 adjusted to a distance of 150 mm from the tip of the nozzle 3. Collected on top to obtain crude inorganic fibers.
- the obtained crude inorganic fiber was baked by raising the temperature to 1000 ° C. at 500 ° C./hour in an electric furnace in an air atmosphere and holding it for 2 hours to obtain inorganic fiber.
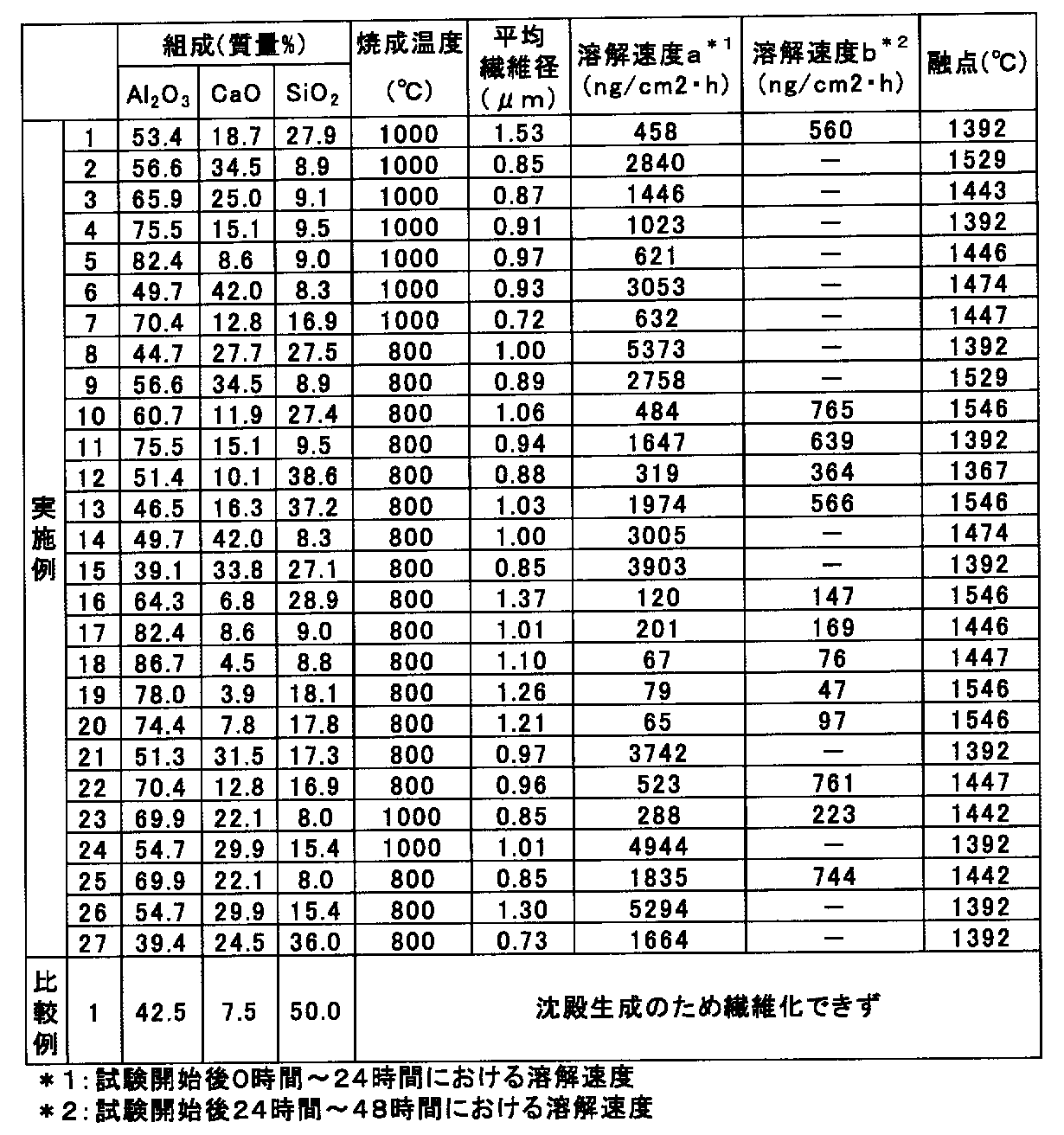
- the average fiber diameter of the obtained inorganic fiber is 1.53 ⁇ m, contains 53.4% by mass (39.6 mol%) of Al 2 O 3 , and 18.7% by mass (25.2 mol%) of CaO. SiO 2 was contained at 27.9% by mass (35.2 mol%).
- the dissolution rate of the obtained inorganic fiber was measured by the method described above.
- the dissolution rate from 0 to 48 hours after the start of the test was 458 ng / cm 2 ⁇ h.
- the dissolution rate from 24 to 48 hours was 560 ng / cm 2 ⁇ h, and the melting point was 1392 ° C.
- Table 2 shows the firing temperature and the composition (in terms of mass% and mol%) of the obtained inorganic fiber
- Table 3 shows the firing temperature and the average fiber diameter, dissolution rate, and melting point of the obtained inorganic fiber. .
- Examples 2 to 27, Comparative Example 1 The amount of basic aluminum carboxylate aqueous solution, the amount of calcium acetate aqueous solution and the colloidal silica in the spinning raw material aqueous solution so that the amount of Al 2 O 3 in the obtained inorganic fiber, the amount of CaO and the amount of SiO 2 are as shown in Table 2. While adjusting the amount, the firing temperature (temperature maintained in an electric furnace for 2 hours) was changed to the temperature shown in Table 3, and inorganic fibers were produced in the same manner as in Example 1.
- Example 13 the average fiber diameter was determined in the same manner as in Example 1, and the biosolubility was evaluated and the melting point was measured. The results are shown in Table 3. In Example 13 and the like, the dissolution rate from 24 to 48 hours after the start of the test is lower than the dissolution rate from 0 to 24 hours after the start of the test. ⁇ This is because it disappeared.
- Table 2 shows the firing temperature and the composition (in terms of mass% and mol%) of the obtained inorganic fiber
- Table 3 shows the average fiber diameter, dissolution rate, and melting point of the obtained inorganic fiber.
- spinning was not possible because a spinning raw material aqueous solution was prepared and a precipitate was formed at the stage where the viscosity was adjusted.
- the inorganic fibers obtained in Examples 1 to 27 have a dissolution rate of 65 ng / cm 2 ⁇ h or more from 0 hours to 24 hours after the start of the test, and 24 hours after the start of the test. Since the dissolution rate in 48 hours is 47 ng / cm 2 ⁇ h or more, it can be seen that it exhibits excellent biosolubility, and since the melting point is 1367 ° C. or more, it exhibits high heat resistance. I understand that.
- an inorganic fiber having a high biological dissolution rate and excellent biological solubility and high heat resistance.
Abstract
Description
(1)35質量%~88質量%のAl2O3、3質量%~45質量%のCaOおよび5質量%~40質量%のSiO2を含み、Al2O3、CaOおよびSiO2を合計した含有割合が繊維全体の98質量%以上であることを特徴とする無機繊維、
(2)39質量%~66質量%のAl2O3、26質量%~42質量%のCaOおよび8質量%~28質量%のSiO2を含み、Al2O3、CaOおよびSiO2を合計した含有割合が繊維全体の98質量%以上である上記(1)に記載の無機繊維、
(3)前記無機繊維が、
水溶性の塩基性酸アルミニウムと水溶性のカルシウム化合物と水溶性または水分散性のケイ素化合物とを水性媒体中に溶解して紡糸原料水性溶液を作製した後、
該紡糸原料水性溶液を紡糸して粗無機繊維を得、
次いで、該粗無機繊維を焼成してなるものである
上記(1)または(2)に記載の無機繊維、
(4)前記水溶性の塩基性酸アルミニウムが、下記式(I)
Al(OH)XYZ (I)
(ただし、Xは0を超え3未満の正の数であり、Yは、Cl原子、NO3基、SO4基、RCOO基から選ばれるいずれか一種であり、Zは、YがCl原子、NO3基、RCOO基である場合3-X、YがSO4基である場合(3-X)/2であり、前記Rは水素原子または炭素数1~10の炭化水素基若しくは水酸基含有炭化水素基であって、RCOO基が複数存在する場合、各Rは同一であっても異なっていてもよい)
で表される化合物から選ばれる一種以上である上記(3)に記載の無機繊維、
(5)前記紡糸が静電紡糸法により行われてなる上記(3)に記載の無機繊維、
(6)前記紡糸が静電紡糸法により行われてなる上記(4)に記載の無機繊維、
を提供するものである。
Al2O3の含有割合が上記範囲内にあることにより、所望の耐熱性を得やすくなる。
CaOの含有割合が上記範囲内にあることにより、所望の生体溶解性を得やすくなる。
SiO2の含有割合が上記範囲内にあることにより、所望の耐熱性を得やすくなる。
Al(OH)XYZ (I)
(ただし、Xは0を超え3未満の正の数であり、Yは、Cl原子、NO3基、SO4基、RCOO基から選ばれるいずれか一種であり、Zは、YがCl原子、NO3基、RCOO基である場合3-X、YがSO4基である場合(3-X)/2であり、前記Rは水素原子または炭素数1~10の炭化水素基若しくは水酸基含有炭化水素基であって、RCOO基が複数存在する場合、各Rは同一であっても異なっていてもよい)で表される化合物から選ばれる一種以上であることが好ましい。
なお、本出願書類において、無機繊維の融点は、上述した無機繊維組成を基に、熱力学平衡計算から求めた値を意味する。
上記無機繊維の製造方法のうち、乾式紡糸法を用いた製造方法、特に静電紡糸法を用いた製造方法が、目的とする無機繊維を簡便かつ低コストに製造することができるため、好適である。
本発明の無機繊維を乾式紡糸法により作製する場合、原料として用いられる水溶性の塩基性酸アルミニウムとしては、下記式(I)
Al(OH)XYZ (I)
(ただし、Xは0を超え3未満の正の数であり、Yは、Cl原子(塩素原子)、NO3基、SO4基、RCOO基(カルボキシル基)から選ばれるいずれか一種であり、Zは、YがCl原子、NO3基、RCOO基である場合3-X、YがSO4基である場合(3-X)/2であり、前記Rは水素原子または炭素数1~10の炭化水素基若しくは水酸基含有炭化水素基であって、RCOO基が複数存在する場合、各Rは同一であっても異なっていてもよい)
で表される化合物から選ばれる一種以上の化合物を挙げることができる。なお、こうした塩基性酸アルミニウムは、ヒドロキシ基で架橋された8面体配位のアルミニウム多核錯体(無機イオン性ポリマー)で、2量体やオリゴマーの形をとり得る。
また、紡糸原料水性溶液中において、アルミニウム元素とカルシウム元素とケイ素元素の総量に対するアルミニウム元素の存在割合は、アルミニウム元素をAl2O3、カルシウム元素をCaO、ケイ素元素をSiO2に換算したときのAl2O3換算で35質量%~88質量%であることが好ましく、39質量%~87質量%であることがより好ましく、39質量%~83質量%であることがさらに好ましく、39質量%~66質量%であることが一層好ましく、49質量%~66質量%であることがより一層好ましい。
また、紡糸原料水性溶液において、アルミニウム元素とカルシウム元素とケイ素元素の総量に対するカルシウム元素の存在割合は、アルミニウム元素をAl2O3、カルシウム元素をCaO、ケイ素元素をSiO2に換算したときのCaO換算で3質量%~45質量%であることが好ましく、3質量%~42質量%であることがより好ましく、26質量%~42質量%であることがさらに好ましい。
また、紡糸原料水性溶液中において、アルミニウム元素とカルシウム元素とケイ素元素の総量に対するケイ素元素の存在割合は、アルミニウム元素をAl2O3、カルシウム元素をCaO、ケイ素元素をSiO2に換算したときのSiO2換算で5質量%~40質量%であることが好ましく、8質量%~39質量%であることがより好ましく、8質量%~28質量%であることがさらに好ましく、8質量%~16質量%であることが一層好ましい。
なお、本出願書類において、紡糸原料水性溶液の粘度は、粘弾性測定装置(Anton Paar社製 Physica MCR301)を用い、紡糸液の液温を25℃に維持し、せん断速度10s-1の時のせん断粘度を意味する。以降、上記の条件で測定した粘度を本出願書類の粘度とする。
なお、以下の実施例および比較例において、生体溶解性は、以下に示す方法により評価した。
得られた無機繊維のうち、評価試料として25mgの範囲に収まる量を精秤した。
M=π×d2×L×ρ/4 (1)
また、無機繊維の表面積S(m2)は式(2)で表わされる。
S=π×d×L (2)
式(2)よりL=S/(π×d)であることから、このLを式(1)に代入してSについてまとめると、以下の式(3)のとおりとなる。
S=4M/dρ (3)

塩基性カルボン酸アルミニウムとして、Al(OH)X(RCOO)3-X(Xが1.7の値、Rの炭素数が0~2の値である)を用いて、以下のとおり紡糸原料水溶液を調製した。
上記焼成時の温度および得られた無機繊維の組成(質量%およびモル%表示)を表2に示すとともに、焼成温度および得られた無機繊維の平均繊維径、溶解速度および融点を表3に示す。
得られる無機繊維中のAl2O3量、CaO量およびSiO2量が表2に示す割合になるように、紡糸原料水性溶液中の塩基性カルボン酸アルミニウム水溶液量、酢酸カルシウム水溶液量およびコロイダルシリカ量を調整するとともに、焼成温度(電気炉中で2時間保持した温度)を表3に示す温度にして、実施例1と同様にして無機繊維を作製した。
なお、比較例1においては、紡糸原料水溶液を作製し、粘度調整した段階で沈殿が生成したため、紡糸できなかった。

2 シリンジ
3 ノズル
4 高電圧発生装置
5 試料捕集台
Claims (6)
- 35質量%~88質量%のAl2O3、3質量%~45質量%のCaOおよび5質量%~40質量%のSiO2を含み、Al2O3、CaOおよびSiO2を合計した含有割合が繊維全体の98質量%以上であることを特徴とする無機繊維。
- 39質量%~66質量%のAl2O3、26質量%~42質量%のCaOおよび8質量%~28質量%のSiO2を含み、Al2O3、CaOおよびSiO2を合計した含有割合が繊維全体の98質量%以上である請求項1に記載の無機繊維。
- 前記無機繊維が、
水溶性の塩基性酸アルミニウムと水溶性のカルシウム化合物と水溶性または水分散性のケイ素化合物とを水性媒体中に溶解して紡糸原料水性溶液を作製した後、
該紡糸原料水性溶液を紡糸して粗無機繊維を得、
次いで、該粗無機繊維を焼成してなるものである
請求項1または請求項2に記載の無機繊維。 - 前記水溶性の塩基性酸アルミニウムが、下記式(I)
Al(OH)XYZ (I)
(ただし、Xは0を超え3未満の正の数であり、Yは、Cl原子、NO3基、SO4基、RCOO基から選ばれるいずれか一種であり、Zは、YがCl原子、NO3基、RCOO基である場合3-X、YがSO4基である場合(3-X)/2であり、前記Rは水素原子または炭素数1~10の炭化水素基若しくは水酸基含有炭化水素基であって、RCOO基が複数存在する場合、各Rは同一であっても異なっていてもよい)
で表される化合物から選ばれる一種以上である請求項3に記載の無機繊維。 - 前記紡糸が静電紡糸法により行われてなる請求項3に記載の無機繊維。
- 前記紡糸が静電紡糸法により行われてなる請求項4に記載の無機繊維。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201180068507.5A CN103403238B (zh) | 2011-02-24 | 2011-08-26 | 无机纤维 |
| KR1020137022087A KR20140032377A (ko) | 2011-02-24 | 2011-08-26 | 무기 섬유 |
| US14/001,600 US9156731B2 (en) | 2010-02-25 | 2011-08-26 | Inorganic fibers |
| EP11859472.0A EP2679710B1 (en) | 2011-02-24 | 2011-08-26 | Inorganic fibers |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-038403 | 2011-02-24 | ||
| JP2011038403A JP5883227B2 (ja) | 2010-02-25 | 2011-02-24 | 無機繊維 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012114565A1 true WO2012114565A1 (ja) | 2012-08-30 |
Family
ID=46721523
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/069273 WO2012114565A1 (ja) | 2010-02-25 | 2011-08-26 | 無機繊維 |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP2679710B1 (ja) |
| KR (1) | KR20140032377A (ja) |
| CN (1) | CN103403238B (ja) |
| WO (1) | WO2012114565A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014115550A1 (ja) * | 2013-01-23 | 2014-07-31 | ニチアス株式会社 | 生体溶解性無機繊維及びその組成物 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6228461B2 (ja) * | 2012-01-31 | 2017-11-08 | ニチアス株式会社 | 無機繊維及びその製造方法 |
| JP5272103B1 (ja) | 2012-09-14 | 2013-08-28 | ニチアス株式会社 | 無機繊維及びそれを用いた成形体 |
| JP6554269B2 (ja) | 2014-07-08 | 2019-07-31 | ニチアス株式会社 | 生体溶解性無機繊維の製造方法 |
| GB2534410A (en) * | 2015-01-23 | 2016-07-27 | Morgan Advanced Mat Plc | Inorganic fibre compositions |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2756258B2 (ja) * | 1987-06-05 | 1998-05-25 | ミネソタ マイニング アンド マニユフアクチユアリング カンパニー | 耐火性固体物品及びその製法 |
| WO2006129844A1 (ja) * | 2005-05-31 | 2006-12-07 | Teijin Limited | セラミック繊維及びその製造方法 |
| JP2009515800A (ja) * | 2005-11-10 | 2009-04-16 | ザ・モーガン・クルーシブル・カンパニー・ピーエルシー | 高い温度に対して耐性である繊維 |
| JP2011106050A (ja) * | 2009-11-17 | 2011-06-02 | Nichias Corp | 無機繊維の製造方法 |
| JP2011196007A (ja) * | 2010-02-25 | 2011-10-06 | Nichias Corp | 無機繊維 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007005836A2 (en) * | 2005-06-30 | 2007-01-11 | Unifrax Corporation | Phosphate coated inorganic fiber and methods of preparation and use |
| GB0522980D0 (en) * | 2005-11-10 | 2005-12-21 | Morgan Crucible Co | High temperature resistant fibres |
-
2011
- 2011-08-26 KR KR1020137022087A patent/KR20140032377A/ko not_active Application Discontinuation
- 2011-08-26 WO PCT/JP2011/069273 patent/WO2012114565A1/ja active Application Filing
- 2011-08-26 EP EP11859472.0A patent/EP2679710B1/en not_active Not-in-force
- 2011-08-26 CN CN201180068507.5A patent/CN103403238B/zh not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2756258B2 (ja) * | 1987-06-05 | 1998-05-25 | ミネソタ マイニング アンド マニユフアクチユアリング カンパニー | 耐火性固体物品及びその製法 |
| WO2006129844A1 (ja) * | 2005-05-31 | 2006-12-07 | Teijin Limited | セラミック繊維及びその製造方法 |
| JP2009515800A (ja) * | 2005-11-10 | 2009-04-16 | ザ・モーガン・クルーシブル・カンパニー・ピーエルシー | 高い温度に対して耐性である繊維 |
| JP2011106050A (ja) * | 2009-11-17 | 2011-06-02 | Nichias Corp | 無機繊維の製造方法 |
| JP2011196007A (ja) * | 2010-02-25 | 2011-10-06 | Nichias Corp | 無機繊維 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2679710A1 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014115550A1 (ja) * | 2013-01-23 | 2014-07-31 | ニチアス株式会社 | 生体溶解性無機繊維及びその組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2679710B1 (en) | 2019-04-24 |
| CN103403238B (zh) | 2016-03-30 |
| CN103403238A (zh) | 2013-11-20 |
| EP2679710A4 (en) | 2014-09-03 |
| EP2679710A1 (en) | 2014-01-01 |
| KR20140032377A (ko) | 2014-03-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5883227B2 (ja) | 無機繊維 | |
| WO2012114565A1 (ja) | 無機繊維 | |
| US9816206B2 (en) | Carbonaceous metal/ceramic nanofibers | |
| JP5655094B2 (ja) | 無機繊維およびその製造方法 | |
| JP6228461B2 (ja) | 無機繊維及びその製造方法 | |
| JP5642956B2 (ja) | 無機繊維の製造方法 | |
| Milanović et al. | Preparation of low cost alumina nanofibers via electrospinning of aluminium chloride hydroxide/poly (vinyl alcohol) solution | |
| JP5536353B2 (ja) | 無機繊維およびその製造方法 | |
| RU2465247C2 (ru) | Волокна из поликристаллического корунда и способ их получения | |
| WO2012114566A1 (ja) | 無機繊維の製造方法 | |
| JP5649297B2 (ja) | 無機繊維 | |
| JP6361418B2 (ja) | 無機繊維、無機繊維集合体及び無機繊維成形体 | |
| CN106587645B (zh) | 具有上转换发光效应的生物玻璃纤维材料及其制备方法 | |
| CN113151932B (zh) | 一种硅酸钇纳米纤维的制备方法及其制备材料 | |
| Liang et al. | Preparation of superhydrophobic silicon-based net-like hollow nanostructure using electrospinning | |
| Dai | Calcium Phosphate Scaffolds from Electrospun PVA/inorganic Sol Precursors | |
| Uslu et al. | Fabrication And Characterization Of Boron Doped Zirconium Oxide Nanofibers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180068507.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11859472 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20137022087 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011859472 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14001600 Country of ref document: US |