WO2012108478A1 - Monocyclic compound - Google Patents
Monocyclic compound Download PDFInfo
- Publication number
- WO2012108478A1 WO2012108478A1 PCT/JP2012/052899 JP2012052899W WO2012108478A1 WO 2012108478 A1 WO2012108478 A1 WO 2012108478A1 JP 2012052899 W JP2012052899 W JP 2012052899W WO 2012108478 A1 WO2012108478 A1 WO 2012108478A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- optionally substituted
- reaction
- substituted
- Prior art date
Links
- 0 C*1*CCC1 Chemical compound C*1*CCC1 0.000 description 6
- OOZNVLRXXYRESF-XYDAIERRSA-N CCCOC(C)([C@H](CCc(cc1)ccc1OCC1C=CC(OCC=C)=CC1)NC(OCC1c2ccccc2-c2c1cccc2)=O)O Chemical compound CCCOC(C)([C@H](CCc(cc1)ccc1OCC1C=CC(OCC=C)=CC1)NC(OCC1c2ccccc2-c2c1cccc2)=O)O OOZNVLRXXYRESF-XYDAIERRSA-N 0.000 description 1
- XAZKFISIRYLAEE-ULUSZKPHSA-N C[C@H]1CC(C)CC1 Chemical compound C[C@H]1CC(C)CC1 XAZKFISIRYLAEE-ULUSZKPHSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/16—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
- C07C233/17—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/18—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom having the carbon atom of the carboxamide group bound to a hydrogen atom or to a carbon atom of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/30—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by doubly-bound oxygen atoms
- C07C233/31—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by doubly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/34—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by amino groups
- C07C233/35—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by amino groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/36—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by amino groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom having the carbon atom of the carboxamide group bound to a hydrogen atom or to a carbon atom of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/54—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and etherified hydroxy groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/16—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/04—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms
- C07C275/06—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton
- C07C275/10—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton being further substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/26—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C317/28—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to acyclic carbon atoms of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/39—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton at least one of the nitrogen atoms being part of any of the groups, X being a hetero atom, Y being any atom
- C07C323/40—Y being a hydrogen or a carbon atom
- C07C323/41—Y being a hydrogen or an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/69—Two or more oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/10—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D261/14—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Definitions
- the present invention has an inhibitory action on acetyl-CoA carboxylase (which may be abbreviated as ACC in the present specification), and is obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complications, metabolic syndrome Further, the present invention relates to a monocyclic compound useful for prevention / treatment of sarcopenia, cancer and the like.
- acetyl-CoA carboxylase which may be abbreviated as ACC in the present specification
- ACC acetyl-CoA carboxylase
- ACC is an enzyme that converts acetyl-CoA to malonyl-CoA and catalyzes the rate-limiting reaction in fatty acid metabolism or synthesis.
- Malonyl-CoA the product of the ACC catalytic reaction, inhibits mitochondrial fatty acid oxidation by feedback inhibition of carnitine palmitoyltransferase-1 (CPT-1).
- CPT-1 carnitine palmitoyltransferase-1
- Decreased malonyl-CoA levels due to ACC inhibition include increased fatty acid oxidation, inhibition of fatty acid synthesis, decreased triglyceride (TG) rich lipoprotein (VLDL) secretion in the liver, regulation of insulin secretion in the pancreas, and May improve insulin sensitivity in liver, skeletal muscle and adipose tissue.
- long-term administration of a compound having an ACC inhibitory action by promoting fatty acid oxidation and suppressing fatty acid de novo synthesis is a TG of liver and adipose tissue in obese subjects ingesting a low-fat diet.
- the content can be greatly reduced and body fat can be selectively reduced.
- a compound having an ACC inhibitory action is extremely useful for the prevention and treatment of metabolic syndrome, obesity, hypertension, diabetes, cardiovascular diseases related to atherosclerosis, and the like.
- Patent Document 1 includes the following compounds:
- Patent Document 2 includes the following compounds:
- a 1 , A 4 and A 5 independently represent N, C (R 1 ) (R 1 represents H, alkyl, halogen or haloalkyl); A 2 and A 3 independently represent C (—L 2 —R 2 ) (L 2 represents O or the like, R 2 represents alkyl or the like); L 1 represents O, N (R X ), S, S (O), S (O) 2 or C (R y R z ) m; m represents 1, 2 or 3; A represents phenyl or monocyclic heteroaryl; X represents —O— (CR y R z ) p—NR d R e or the like; R d represents —C (O) alkyl or the like; Re represents H or the like] Has been reported as a compound having an ACC inhibitory action.
- Non-patent Document 1 Journal of Medicinal Chemistry, 2010, 53, 8679-8687 (Non-patent Document 1) includes the following compounds:
- Patent Document 3 includes the following compounds:
- A represents an acyl group or an optionally substituted 5- to 6-membered aromatic ring group
- Ring M represents a 5- to 7-membered ring which may be further substituted and optionally condensed
- Ring P and Ring Q are (1) Ring P represents a 5-membered heterocyclic ring which may be further substituted, Ring Q represents a 6-membered ring which may be further substituted, and further condensed by condensing ring P and ring Q Forming a bicyclic aromatic heterocycle which may be (2)
- Ring P represents an optionally substituted 5-membered non-aromatic ring, Ring Q represents an optionally further substituted 6-membered aromatic ring, and ring P and ring Q are condensed Forming an optionally substituted bicyclic non-aromatic ring;
- R 1 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 3-6 cycloalkyl group;
- L 1 and L 2 are (1) independently represents optionally substituted methylene, O,
- A represents H, lower alkyl, cycloalkyl or —NR 3 R 4 (R 3 and R 4 independently represent H, lower alkyl, etc.);
- Ar 1 represents arylene or heteroarylene;
- Ar 2 represents an optionally substituted aryl, an optionally substituted aralkyl, an optionally substituted heteroaryl, or an optionally substituted heteroarylalkyl;
- R 1 represents H, optionally substituted lower alkyl, or optionally substituted lower alkenyl;
- R 2a is
- JP 2008-031064 A discloses the following compounds:
- R 41 and R 42 independently represent H or lower alkyl; R 1 represents —R 00 -optionally substituted aryl (R 00 : lower alkylene) or the like] has been reported as a compound having a DPP4 inhibitory action.
- Patent Document 6 includes the following compounds:
- Has ACC inhibitory action is useful for the prevention and treatment of obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complications, metabolic syndrome, sarcopenia, cancer, etc. Development of the compound which has is desired.
- R 1 represents a group represented by the formula: —COR 2 (R 2 represents a hydrogen atom or a substituent), or an optionally substituted 5- or 6-membered aromatic ring group;
- R 3 represents a C 1-6 alkyl group which may be substituted with a halogen atom, or a C 3-6 cycloalkyl group which may be substituted;
- R 4a and R 4b independently represent a hydrogen atom or a substituent, or R 4a and R 4b may be bonded to each other to form an optionally substituted 3- or 4-membered ring;
- R 5a and R 5b independently represent a hydrogen atom or a substituent, or R 5a and R 5b may be bonded to each other to form an optionally substituted 3- or 4-membered ring;
- R 6 represents an optionally substituted C 1-6 alkyl group, or an optionally substituted C 3-6 cycloalkyl group;
- X is O, CO, CR 7a R 7b (R
- R 1 is represented by the formula: —COR 2 (R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, a substituted
- R 1 is substituted with the formula: —COR 2 (R 2 is substituted with a hydrogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group, or 1 to 2 C 1-6 alkyl groups.
- a compound represented by the above-mentioned [1] or a salt thereof which is a group represented by: [4] The compound or a salt thereof according to the above [1], [2] or [3], wherein R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms; [5] The compound or salt thereof according to [1], [2], [3] or [4] above, wherein R 4a and R 4b are hydrogen atoms; [6] The compound or salt thereof according to the above [1], [2], [3], [4] or [5], wherein R 5a and R 5b are hydrogen atoms; [7] The compound according to the above [1], [2], [3], [4], [5] or [6], wherein R 6 is an optionally substituted C 1-6 alkyl group or Its salt; [8] R 6 is (a) a halogen atom, (b) a C 3-6 cycloalkyl group optionally substituted with 1 to 3
- R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms
- R 4a and R 4b are hydrogen atoms
- R 5a and R 5b are hydrogen atoms
- R 6 is (a) a halogen atom, (b) a C 3-6 cycloalkyl group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups, (c) a 4- to 7-membered heterocyclic group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups, and (d) substituted with 1 to 7 substituents selected from 1 to 3 halogen atoms or C 6-14 aryl group optionally substituted with 1 to 3 C 1-6 alkyl groups; Or a C 1-6 alkyl group;
- X is O, CO, CH 2 , NR 7c (R 7c represents a hydrogen atom or a
- Compound (I) has an ACC inhibitory action and is useful for the prevention and treatment of obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complications, metabolic syndrome, sarcopenia, cancer, etc. And has excellent medicinal properties.
- halogen atom in the present specification means a fluorine atom, a chlorine atom, a bromine atom, or an iodine atom unless otherwise specified.
- C 1-3 alkylenedioxy group in the present specification means methylenedioxy, ethylenedioxy and the like unless otherwise specified.
- C 1-6 alkyl group means methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, 1-ethyl unless otherwise specified. It means propyl, hexyl, isohexyl, 1,1-dimethylbutyl, 2,2-dimethylbutyl, 3,3-dimethylbutyl, 2-ethylbutyl and the like.
- C 1-6 alkoxy group in the present specification means methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy and the like unless otherwise specified.
- C 1-6 alkoxy-carbonyl group in the present specification means methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert-butoxycarbonyl and the like, unless otherwise specified.
- C 1-6 alkyl-carbonyl group in the present specification means acetyl, propanoyl, butanoyl, isobutanoyl, pentanoyl, isopentanoyl, hexanoyl and the like, unless otherwise specified.
- examples of the “ optionally substituted C 6-14 arylsulfonyloxy group” include a benzenesulfonyloxy group and a p-toluenesulfonyloxy group.
- examples of the “optionally substituted C 1-6 alkylsulfonyloxy group” include a methanesulfonyloxy group and a trifluoromethanesulfonyloxy group.
- R 1 represents a group represented by the formula: —COR 2 (R 2 represents a hydrogen atom or a substituent), or an optionally substituted 5- or 6-membered aromatic ring group.
- Examples of the “substituent” represented by R 2 include “optionally substituted hydrocarbon group”, “optionally substituted heterocyclic group”, “optionally substituted hydroxy group”, “substituted” And optionally substituted amino group, “optionally substituted sulfanyl group”, “acyl group”, “halogen atom”, “cyano group”, “nitro group” and the like.
- hydrocarbon group in the “optionally substituted hydrocarbon group” include a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 2-10 alkynyl group, and a C 3-10 cycloalkyl group.
- examples of the C 1-10 alkyl group include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, 1-ethylpropyl, hexyl, isohexyl, 1 , 1-dimethylbutyl, 2,2-dimethylbutyl, 3,3-dimethylbutyl, 2-ethylbutyl, heptyl, octyl, nonyl, decyl and the like.
- a C 1-6 alkyl group is preferable.
- Examples of the C 2-10 alkenyl group include ethenyl, 1-propenyl, 2-propenyl, 2-methyl-1-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, 3-methyl-2-butenyl, 1 -Pentenyl, 2-pentenyl, 3-pentenyl, 4-pentenyl, 4-methyl-3-pentenyl, 1-hexenyl, 3-hexenyl, 5-hexenyl, 1-heptenyl, 1-octenyl and the like. Of these, a C 2-6 alkenyl group is preferable.
- Examples of the C 2-10 alkynyl group include ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1-pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1 -Hexynyl, 2-hexynyl, 3-hexynyl, 4-hexynyl, 5-hexynyl, 1-heptynyl, 1-octynyl and the like. Of these, a C 2-6 alkynyl group is preferable.
- Examples of the C 3-10 cycloalkyl group include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl and the like. Of these, a C 3-6 cycloalkyl group is preferable.
- Examples of the C 3-10 cycloalkenyl group include 2-cyclopenten-1-yl, 3-cyclopenten-1-yl, 2-cyclohexen-1-yl, 3-cyclohexen-1-yl and the like. Of these, a C 3-6 cycloalkenyl group is preferable.
- Examples of the C 4-10 cycloalkadienyl group include 2,4-cyclopentadien-1-yl, 2,4-cyclohexadien-1-yl, 2,5-cyclohexadien-1-yl, and the like. . Of these, a C 4-6 cycloalkadienyl group is preferable.
- the above C 3-10 cycloalkyl group, C 3-10 cycloalkenyl group and C 4-10 cycloalkadienyl group may each be condensed with a benzene ring to form a condensed ring group.
- Examples of the condensed ring group include indanyl, dihydronaphthyl, tetrahydronaphthyl, fluorenyl and the like.
- the C 3-10 cycloalkyl group, the C 3-10 cycloalkenyl group and the C 4-10 cycloalkadienyl group may be a C 7-10 bridged hydrocarbon group.
- Examples of the C 7-10 bridged hydrocarbon group include bicyclo [2.2.1] heptyl (norbornyl), bicyclo [2.2.2] octyl, bicyclo [3.2.1] octyl, bicyclo [3. 2.2] nonyl, bicyclo [3.3.1] nonyl, bicyclo [4.2.1] nonyl, bicyclo [4.3.1] decyl, adamantyl and the like.
- C 3-10 cycloalkyl group, C 3-10 cycloalkenyl group and C 4-10 cycloalkadienyl group are respectively C 3-10 cycloalkane, C 3-10 cycloalkene or C 4-10.
- a cycloalkadiene may form a spiro ring group.
- C 3-10 cycloalkane, C 3-10 cycloalkene and C 4-10 cycloalkadiene the above C 3-10 cycloalkyl group, C 3-10 cycloalkenyl group and C 4-10 cycloalkane are mentioned. Examples include rings corresponding to alkadienyl groups. Examples of such a spiro ring group include spiro [4.5] decan-8-yl.
- Examples of the C 6-14 aryl group include phenyl, naphthyl, anthryl, phenanthryl, acenaphthylenyl, biphenylyl and the like. Of these, a C 6-12 aryl group is preferable.
- Examples of the C 7-13 aralkyl group include benzyl, phenethyl, naphthylmethyl, biphenylylmethyl and the like.
- Examples of the C 8-13 arylalkenyl group include styryl and the like.
- the C 1-10 alkyl group, the C 2-10 alkenyl group and the C 2-10 alkynyl group exemplified as the “hydrocarbon group” have 1 to 7 (preferably 1 to 3) substituted positions. It may have a substituent.
- a substituent for example, (1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl); (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally substituted with 1 to 3 halogen atoms, and (d) a C 6- optionally substituted with 1 to 3 substituents selected from halogen atoms.
- a C 3-10 cycloalkyl group eg, cyclopropyl, cyclohexyl
- (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally substituted with 1 to 3 halogen atoms, and (d) a C 6- optionally substituted with 1 to 3 substituents selected from hal
- aryl groups eg, phenyl, naphthyl
- (3) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, and (d) an aromatic complex which may be substituted with 1 to 3 substituents selected from halogen atoms.
- a cyclic group (eg, thienyl, furyl, pyridyl, pyrazolyl, imidazolyl, tetrazolyl, oxazolyl, thiazolyl, oxadiazolyl, thiadiazolyl); (4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, (d) a non-aromatic heterocyclic group (eg, tetrahydrofuryl, morpholinyl, thiomorpholinyl, piperidinyl, pyrrolidinyl, piperazinyl) optionally substituted with 1 to 3 substituents selected from halogen atoms and (e) oxo groups ); (5) (a) a C 1-6 alkyl group which may be substituted with 1 to 3
- a C 1-6 alkoxy-carbonyl group optionally substituted by 1 to 3 substituents selected from: (8) a C 1-6 alkylsulfonyl group which may be substituted with 1 to 3 halogen atoms (eg, methylsulfonyl, ethylsulfonyl, isopropylsulfonyl); (9) a carbamoyl group which may be mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms; (10) a thiocarbamoyl group optionally mono- or disubstituted with a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms; (11) a sulfamoyl group optionally mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms; (12) a carboxy group; (1
- a C 1-6 alkoxy group which may be substituted with 1 to 3 substituents selected from: (15) a C 2-6 alkenyloxy group (eg, ethenyloxy) optionally substituted by 1 to 3 halogen atoms; (16) C 7-13 aralkyloxy group (eg, benzyloxy); (17) C 6-14 aryloxy group (eg, phenyloxy, naphthyloxy); (18) C 1-6 alkyl-carbonyloxy group (eg, acetyloxy, tert-butylcarbonyloxy); (19) optionally substituted with 1 to 3 substituents selected from (a) a halogen atom, and (b) a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms A C 6-14 aryl-carbonyl group (eg, benzoyl); (20) a non-aromatic heterocyclic carbonyl group optionally substituted with 1 to 3 substituents selected from C
- the group and the C 8-13 arylalkenyl group may have 1 to 3 substituents at substitutable positions.
- substituents for example, (1) groups exemplified as substituents in the aforementioned C 1-10 alkyl group and the like; (2) (a) a halogen atom, (b) a carboxy group, (c) a hydroxy group, (d) a C 1-6 alkoxy-carbonyl group, (e) a C 1-6 alkoxy group, and (f) a C 1-6 alkyl group optionally substituted with 1 to 3 substituents selected from amino groups optionally mono- or di-substituted.
- a 1-6 alkyl group (3) (a) a halogen atom, (b) a carboxy group, (c) a hydroxy group, (d) a C 1-6 alkoxy-carbonyl group, (e) a C 1-6 alkoxy group, and (f) a C 1-6 alkyl group optionally substituted with 1 to 3 substituents selected from amino groups optionally mono- or di-substituted.
- 2-6 alkenyl groups eg, ethenyl, 1-propenyl
- (4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group, and (d) a C 7-13 aralkyl group (eg, benzyl) optionally substituted with 1 to 3 substituents selected from halogen atoms; Etc.
- each substituent may be the same or different.
- heterocyclic group in the “optionally substituted heterocyclic group” includes an aromatic heterocyclic group and a non-aromatic heterocyclic group.
- the aromatic heterocyclic group is, for example, a 4 to 7 member (preferably 5 or 5) containing 1 to 4 heteroatoms selected from an oxygen atom, a sulfur atom and a nitrogen atom in addition to a carbon atom as a ring constituent atom.
- 6-membered) monocyclic aromatic heterocyclic group and condensed aromatic heterocyclic group examples include a ring corresponding to the 4- to 7-membered monocyclic aromatic heterocyclic group and a 5- or 6-membered aromatic heterocyclic ring containing 1 or 2 nitrogen atoms.
- Furyl eg, 2-furyl, 3-furyl
- thienyl eg, 2-thienyl, 3-thienyl
- pyridyl eg, 2-pyridyl, 3-pyridyl, 4-pyridyl
- pyrimidinyl eg, 2-pyrimidinyl
- 5-pyrimidinyl pyridazinyl
- pyridazinyl eg, 3-pyridazinyl, 4-pyridazinyl
- pyrazinyl eg, 2-pyrazinyl
- pyrrolyl eg, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl
- imidazolyl Eg, 1-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl
- pyrazolyl eg, 1-pyrazolyl, 3-pyrazolyl, 4-pyrazolyl
- thiazolyl e
- non-aromatic heterocyclic group examples include 4 to 7 members (preferably 5 or 6 members) containing 1 to 4 heteroatoms selected from oxygen atoms, sulfur atoms and nitrogen atoms in addition to carbon atoms as ring constituent atoms.
- Monocyclic non-aromatic heterocyclic group and condensed non-aromatic heterocyclic group examples include a ring corresponding to the 4- to 7-membered monocyclic non-aromatic heterocyclic group, and a 5- or 6-membered aromatic containing 1 or 2 nitrogen atoms.
- 1 or 2 rings selected from a heterocycle (eg, pyrrole, imidazole, pyrazole, pyrazine, pyridine, pyrimidine), a 5-membered aromatic heterocycle containing one sulfur atom (eg, thiophene) and a benzene ring
- a heterocycle eg, pyrrole, imidazole, pyrazole, pyrazine, pyridine, pyrimidine
- a 5-membered aromatic heterocycle containing one sulfur atom eg, thiophene
- benzene ring examples thereof include a group derived from a condensed ring and a group obtained by partial saturation of the group.
- Azetidinyl eg, 1-azetidinyl, 2-azetidinyl, 3-azetidinyl
- pyrrolidinyl eg, 1-pyrrolidinyl, 2-pyrrolidinyl
- piperidinyl eg, piperidino, 2-piperidinyl, 3-piperidinyl, 4-piperidinyl
- morpholinyl eg, morpholino
- thiomorpholinyl eg, thiomorpholino
- piperazinyl eg, 1-piperazinyl, 2-piperazinyl, 3-piperazinyl
- hexamethyleneiminyl eg, hexamethyleneimin-1-yl
- oxazolidinyl eg, Oxazolidin-2-yl
- thiazolidinyl eg, thiazolidin-2-yl
- imidazolidinyl eg,
- heterocyclic group in the “optionally substituted heterocyclic group” may have 1 to 3 substituents at substitutable positions. Examples of such substituents are the same as the substituents that the C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” may have. Can be mentioned.
- the heterocyclic group is a “non-aromatic heterocyclic group”
- an oxo group is further included as a substituent.
- each substituent may be the same or different.
- Examples of the “optionally substituted hydroxy group” include, for example, a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 3-10 cycloalkyl group, and a C 3-10 which may each be substituted. It may be substituted with a substituent selected from a cycloalkenyl group, a C 6-14 aryl group, a C 7-13 aralkyl group, a C 8-13 arylalkenyl group, a C 1-6 alkyl-carbonyl group, a heterocyclic group and the like. Good hydroxy groups are mentioned.
- Examples of the 13 arylalkenyl group include those exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group”.
- heterocyclic group examples include “aromatic heterocyclic group” and “non-aromatic heterocyclic group” exemplified as the “heterocyclic group” in the “optionally substituted heterocyclic group”.
- the arylalkenyl group, C 1-6 alkyl-carbonyl group and heterocyclic group each may have 1 to 3 substituents at substitutable positions. When there are two or more substituents, each substituent may be the same or different.
- the substituent of the C 1-10 alkyl group, the C 2-10 alkenyl group and the C 1-6 alkyl-carbonyl group is the “hydrocarbon group” in the “optionally substituted hydrocarbon group”. Examples thereof include the same substituents as the exemplified C 1-10 alkyl group and the like.
- examples of the substituent for the C 3-10 cycloalkyl group, the C 3-10 cycloalkenyl group, the C 6-14 aryl group, the C 7-13 aralkyl group, and the C 8-13 arylalkenyl group include the above-mentioned “substituted Examples thereof include the same substituents that the C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optional hydrocarbon group” may have.
- Examples of the substituent of the heterocyclic group include the same substituents that the “heterocyclic group” in the “optionally substituted heterocyclic group” may have.
- Examples of the “optionally substituted sulfanyl group” include, for example, a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 3-10 cycloalkyl group, and a C 3-10 which may each be substituted. It may be substituted with a substituent selected from a cycloalkenyl group, a C 6-14 aryl group, a C 7-13 aralkyl group, a C 8-13 arylalkenyl group, a C 1-6 alkyl-carbonyl group, a heterocyclic group and the like. A good sulfanyl group is mentioned.
- substituents examples include those exemplified as the substituent in the “optionally substituted hydroxy group”.
- Examples of the “optionally substituted amino group” include, for example, a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 3-10 cycloalkyl group, and a C 3-10 which may each be substituted.
- a cycloalkenyl group, a C 6-14 aryl group, a C 7-13 aralkyl group, a C 8-13 arylalkenyl group and a heterocyclic group; an amino group which may be mono- or di-substituted with a substituent selected from an acyl group and the like Is mentioned.
- Examples of the 13 arylalkenyl group include those exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group”.
- heterocyclic group examples include the “aromatic heterocyclic group” and the “non-aromatic heterocyclic group” exemplified as the “heterocyclic group” in the “optionally substituted heterocyclic group”. Is a 5- to 7-membered monocyclic aromatic heterocyclic group.
- the alkenyl group and the heterocyclic group each may have 1 to 3 substituents at substitutable positions. When there are two or more substituents, each substituent may be the same or different.
- examples of the substituent of the C 1-10 alkyl group and the C 2-10 alkenyl group include the C 1-10 alkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” and the like. The same thing as the substituent which may have is mentioned.
- examples of the substituent for the C 3-10 cycloalkyl group, the C 3-10 cycloalkenyl group, the C 6-14 aryl group, the C 7-13 aralkyl group, and the C 8-13 arylalkenyl group include the above-mentioned “substituted Examples thereof include the same substituents that the C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optional hydrocarbon group” may have.
- Examples of the substituent of the heterocyclic group include the same substituents that the “heterocyclic group” in the “optionally substituted heterocyclic group” may have.
- acyl group exemplified as the substituent of the “optionally substituted amino group” include those similar to the “acyl group” exemplified as the “substituent” represented by R 2 shown below.
- the “acyl group” exemplified as the “substituent” represented by R 2 includes, for example, the formula: —COR A , —CO—OR A , —SO 3 R A , —S (O) 2 R A , —SOR A , -CO-NR A 'R B ', -CS-NR A 'R B ', -S (O) 2 NR A 'R B ' [wherein R A is a hydrogen atom, substituted A good hydrocarbon group or an optionally substituted heterocyclic group is shown.
- R A ′ and R B ′ are the same or different and each represents a hydrogen atom, an optionally substituted hydrocarbon group, or an optionally substituted heterocyclic group, or R A ′ and R B ′ are A nitrogen-containing heterocyclic ring which may be substituted with an adjacent nitrogen atom may be formed], and the like.
- R A , R A ′ or R B ′ are the “substituent” represented by R 2 , respectively. Examples thereof are the same as the exemplified “optionally substituted hydrocarbon group” and “optionally substituted heterocyclic group”.
- the “nitrogen-containing heterocycle” in the “optionally substituted nitrogen-containing heterocycle” formed by R A ′ and R B ′ together with the adjacent nitrogen atom includes, for example, at least one ring-constituting atom other than a carbon atom And a 5- to 7-membered nitrogen-containing heterocyclic ring which may further contain 1 to 2 heteroatoms selected from oxygen, sulfur and nitrogen atoms.
- the nitrogen-containing heterocycle include pyrrolidine, imidazolidine, pyrazolidine, piperidine, piperazine, morpholine, thiomorpholine and the like.
- the nitrogen-containing heterocycle may have 1 to 5 (preferably 1 or 2) substituents at substitutable positions.
- substituents are the same as the substituent that the “heterocyclic group” in the “optionally substituted heterocyclic group” exemplified as the “substituent” represented by R 2 may have. Can be mentioned. When there are two or more substituents, each substituent may be the same or different.
- acyl group (1) formyl group; (2) a carboxy group; (3) a C 1-6 alkyl-carbonyl group (eg acetyl) optionally substituted by 1 to 3 halogen atoms; (4) a C 1-6 alkoxy-carbonyl group (eg, methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert-butoxycarbonyl) optionally substituted by 1 to 3 halogen atoms; (5) C 3-10 cycloalkyl-carbonyl group (eg, cyclopropylcarbonyl, cyclopentylcarbonyl, cyclohexylcarbonyl); (6) a C 6-14 aryl-carbonyl group (eg, benzoyl, 1-naphthoyl, 2-naphthoyl) optionally substituted with 1 to 3 halogen atoms; (7) (a) a C 1-6 alkyl group which may be substituted with 1 to 3
- R 2 is preferably a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, an optionally substituted C 1-6 alkoxy group, Or it is an amino group which may be substituted.
- R 2 is more preferably a hydrogen atom, an optionally substituted C 1-6 alkyl group, a optionally substituted C 1-6 alkoxy group or an optionally substituted amino group.
- R 2 is more preferably a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups (eg, An amino group which may be substituted with (methyl).
- R 2 is particularly preferably a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups (eg, methyl). An amino group which may be substituted.
- the “group represented by the formula: —COR 2 ” represented by R 1 is preferably —COR 2 (wherein R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, substituted An optionally substituted C 3-6 cycloalkyl group, an optionally substituted C 1-6 alkoxy group, or an optionally substituted amino group.
- the “group represented by the formula: —COR 2 ” represented by R 1 is more preferably —COR 2 (wherein R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, A C 1-6 alkoxy group which may be substituted, or an amino group which may be substituted.
- the “group represented by the formula: —COR 2 ” represented by R 1 is more preferably —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), C And a group represented by a 1-6 alkoxy group (eg, methoxy) or an amino group optionally substituted by 1 to 2 C 1-6 alkyl groups (eg, methyl).
- the “group represented by the formula: —COR 2 ” represented by R 1 is particularly preferably —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), C 1-6 And a group represented by an alkoxy group (eg, methoxy) or an amino group optionally substituted by 1 to 2 C 1-6 alkyl groups (eg, methyl).
- Examples of the “5- or 6-membered aromatic ring group” in the “optionally substituted 5- or 6-membered aromatic ring group” represented by R 1 include, for example, phenyl, pyrrolyl, pyrazolyl, imidazolyl, triazolyl (1, 2, 3 -Triazolyl, 1,2,4-triazolyl, 1,3,4-triazolyl), tetrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, oxadiazolyl, thiadiazolyl, furyl, thienyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, etc. It is done.
- the “5- or 6-membered aromatic ring group” is preferably a 5-membered aromatic heterocyclic group, more preferably pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, etc., particularly preferably isoxazolyl. .
- the “5- or 6-membered aromatic ring group” in the “optionally substituted 5- or 6-membered aromatic ring group” represented by R 1 may have 1 to 3 substituents at substitutable positions. Good. Examples of such a substituent include a C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 and the like. The same thing as the substituent which may have is mentioned.
- the “optionally substituted 5- or 6-membered aromatic ring group” represented by R 1 is preferably substituted with 1 to 3 substituents selected from C 1-6 alkyl groups (eg, methyl). And may be a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl).
- the “optionally substituted 5- or 6-membered aromatic ring group” represented by R 1 is more preferably a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably Isoxazolyl).
- a 5-membered aromatic heterocyclic group eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably Isoxazolyl.
- R 1 is preferably (1) —COR 2 (wherein R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, or optionally substituted) A C 1-6 alkoxy group, or an optionally substituted amino group.); Or (2) an optionally substituted 5- or 6-membered aromatic ring group; It is.
- R 1 is more preferably (1) -COR 2 (wherein, R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, optionally substituted C 1-6 alkoxy group or may be substituted, A group represented by: an amino group; or (2) an optionally substituted 5- or 6-membered aromatic ring group; It is.
- R 1 is more preferably (1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or (2) an optionally substituted 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl); It is.
- R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or (2) an optionally substituted 5-membered aromatic heterocyclic
- R 1 is even more preferably (1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or (2) a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl); It is.
- R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally
- R 1 is particularly preferably (1) —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or (2) a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl); It is.
- R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or (2) a 5-membered aromatic heterocyclic group (eg,
- R 1 is particularly preferably (1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or (2) a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl); It is.
- R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or (2) a 5-membered aromatic heterocyclic group (eg,
- R 3 represents a C 1-6 alkyl group which may be substituted with a halogen atom, or a C 3-6 cycloalkyl group which may be substituted.
- the “C 1-6 alkyl group” in the “C 1-6 alkyl group optionally substituted with a halogen atom” for R 3 is preferably 1 to 7, more preferably 1 to 3, in the substitutable position. May have one halogen atom.
- Examples of the “C 3-6 cycloalkyl group” in the “optionally substituted C 3-6 cycloalkyl group” represented by R 3 include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl. Represented by R 3 "C 3-6 cycloalkyl group" of the "optionally substituted C 3-6 cycloalkyl group” may have 1 to 3 substituents at substitutable position . Examples of such a substituent include a C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 and the like. The same thing as the substituent which may have is mentioned.
- R 3 is preferably a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms.
- R 3 is more preferably a C 1-6 alkyl group (eg, methyl, ethyl).
- R 4a and R 4b independently represent a hydrogen atom or a substituent, or R 4a and R 4b are bonded to each other to form an optionally substituted 3- or 4-membered ring.
- Examples of the “substituent” represented by R 4a or R 4b include those similar to the “substituent” represented by R 2 .
- the “3- or 4-membered ring” in the “optionally substituted 3- or 4-membered ring” formed by R 4a and R 4b bonded to each other has 1 to 6 substituents at substitutable positions. It may be. Examples of such a substituent are the same as the substituent that the “heterocyclic group” in the “optionally substituted heterocyclic group” exemplified as the “substituent” represented by R 2 may have. Can be mentioned.
- R 4a and R 4b are preferably both hydrogen atoms.
- R 5a and R 5b independently represent a hydrogen atom or a substituent, or R 5a and R 5b are bonded to each other to form an optionally substituted 3- or 4-membered ring.
- Examples of the “substituent” represented by R 5a or R 5b include the same “substituent” as represented by R 2 .
- the “3- or 4-membered ring” in the “optionally substituted 3- or 4-membered ring” formed by R 5a or R 5b bonded to each other has 1 to 6 substituents at substitutable positions. It may be. Examples of such a substituent are the same as the substituent that the “heterocyclic group” in the “optionally substituted heterocyclic group” exemplified as the “substituent” represented by R 2 may have. Can be mentioned.
- R 5a or R 5b is preferably independently a hydrogen atom, a carboxy group or a C 1-6 alkoxy-carbonyl group.
- R 5a or R 5b is more preferably both a hydrogen atom.
- R 6 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 3-6 cycloalkyl group.
- the “C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” for R 6 may have 1 to 3 substituents at substitutable positions.
- substituents include a C 1-10 alkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 and the like. Examples thereof are the same as the substituents that may be present.
- R 6 is more preferably (a) a halogen atom (eg, fluorine atom), (b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl), (c) a 4- to 7-membered heterocyclic group (preferably substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl)) Is a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic hetero group (eg tetradofryl), and (d) a C 6-14 aryl group (eg, phenyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (
- R 6 is more preferably (a) a halogen atom (eg, fluorine atom), (b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl), (c) a 4- to 7-membered heterocyclic group (preferably a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic heterocyclic group (eg, tetradrofuryl)), and (d) C 6-14 aryl group (eg, phenyl) A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted by 1 to 7 (preferably 1 to 3) substituents selected from
- X is O, CO, CR 7a R 7b (R 7a and R 7b independently represent a hydrogen atom or a substituent), NR 7c (R 7c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 .
- R 7a or R 7b examples include the same “substituent” represented by R 2 .
- R 7a and R 7b are preferably independently a hydrogen atom or a halogen atom (eg, fluorine atom).
- R 7a and R 7b are more preferably a hydrogen atom.
- R 7c examples include the same “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 .
- R 7c is preferably a hydrogen atom or an optionally substituted C 1-6 alkyl group, and more preferably a C 1-6 alkyl group (eg, methyl).
- X is preferably O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a hydrogen atom) Or an optionally substituted C 1-6 alkyl group), S, SO or S (O) 2 .
- X is more preferably O, CO, CR 7a R 7b (R 7a and R 7b are independently a hydrogen atom or a halogen atom (eg, fluorine atom)), NR 7c (R 7c is C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 .
- X is more preferably O, CO, CH 2 , NR 7c (R 7c is a hydrogen atom or a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 .
- X is particularly preferably O, CO, CH 2 , NR 7c (R 7c is a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 .
- Y is O, CO, CR 8a R 8b (R 8a and R 8b independently represent a hydrogen atom or a substituent), NR 8c (R 8c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 .
- R 8a or R 8b examples include those similar to the “substituent” represented by R 2 .
- R 8a and R 8b are preferably independently a hydrogen atom or a halogen atom (eg, fluorine atom).
- R 8a and R 8b are more preferably a hydrogen atom.
- R 8c is preferably a hydrogen atom or an optionally substituted C 1-6 alkyl group.
- Y is preferably O, CO, or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, a fluorine atom)).
- Y is more preferably O, CO or CH 2 .
- Y is more preferably O.
- Ring P represents a 3- to 7-membered ring which may be further substituted.
- Examples of the “3- to 7-membered ring” in the “optionally substituted 3- to 7-membered ring” represented by ring P include benzene, C 3-7 cycloalkane, C 3-7 cycloalkene, C 4-7. And cycloalkadienes and 3- to 7-membered heterocycles (preferably 5- or 6-membered aromatic heterocycles and 3- to 7-membered non-aromatic heterocycles).
- the C 3-7 cycloalkane includes cyclopropane, cyclobutane, cyclopentane, cyclohexane and cycloheptane, preferably C 4-6 cycloalkane, and particularly preferably cyclobutane and cyclohexane.
- Examples of C 3-7 cycloalkene include cyclopentene, cyclobutene, cyclopentene, cyclohexene, and cycloheptene, and C 4-6 cycloalkene is preferable.
- C 4-7 cycloalkadiene examples include 2,4-cyclopentadiene, 2,4-cyclohexadiene, 2,5-cyclohexadiene, and preferably C 4-6 cycloalkadiene.
- the 5- or 6-membered aromatic heterocycle include pyrrole, pyrazole, imidazole, triazole (1,2,3-triazole, 1,2,4-triazole, 1,3,4-triazole), tetrazole, oxazole, Examples include isoxazole, thiazole, isothiazole, oxadiazole, thiadiazole, furan, thiophene, pyridine, pyridazine, pyrimidine, pyrazine, triazine and the like, preferably a 6-membered nitrogen-containing aromatic heterocyclic ring (preferably pyridine, pyridazine , Pyrimidine, pyrazine, particularly preferably pyridine).
- the “3- to 7-membered ring” in the “optionally substituted 3- to 7-membered ring” represented by ring P is a group —XC (R 4a ) (R 4b ) CH (R 3 ) —NH—R 1
- —YC (R 5a ) (R 5b ) — it may have 1 to 9 substituents at substitutable positions. Examples of such a substituent are the same as the substituent that the “heterocyclic group” in the “optionally substituted heterocyclic group” exemplified as the “substituent” represented by R 2 may have. Can be mentioned.
- Ring P is preferably benzene, C 3-7 cycloalkane or 3- to 7-membered heterocycle (preferably 5- or 6-membered aromatic heterocycle or 3- to 7-membered non-aromatic, each of which may be further substituted. Family heterocycle).
- Ring P is more preferably benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocyclic ring (preferably 5- or 6-membered (preferably Is a 6-membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or a 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle (preferably , Pyrrolidine, piperidine, piperazine, particularly preferably piperidine)).
- C 4-6 cycloalkane preferably cyclobutane, cyclohexane
- 3- to 7-membered heterocyclic ring preferably 5- or 6-membered (preferably Is a 6-membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyr
- Ring P is more preferably 1 to 4 (preferably selected from a halogen atom (eg, fluorine atom), a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy).
- a halogen atom eg, fluorine atom
- C 1-6 alkyl group eg, methyl
- C 1-6 alkoxy group eg, methoxy
- Benzene, C 4-6 cycloalkane preferably cyclobutane, cyclohexane
- 3- to 7-membered heterocycle preferably 5- or 6-membered (preferably 5 to 6-membered) each optionally further substituted with 1 to 3 substituents.
- 6-membered) nitrogen-containing aromatic heterocycle preferably pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine
- 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle preferably Is pyrrolidine, piperidine, piperazine, particularly preferably piperidine
- Ring P is more preferably 1 to 4 (preferably 1 to 3) substituents selected from a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy).
- Ring P is particularly preferably 1 to 4 (preferably 1 to 3) substituents selected from a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy). They are each optionally further substituted benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane), pyridine or piperidine.
- substituents selected from a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy). They are each optionally further substituted benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane), pyridine or piperidine.
- Ring Q represents a 5- or 6-membered aromatic ring which may be further substituted.
- Examples of the “5- or 6-membered aromatic ring” in the “optionally substituted 5- or 6-membered aromatic ring” represented by ring Q include benzene, pyrrole, pyrazole, imidazole, triazole (1,2,3-triazole, 1,2,4-triazole, 1,3,4-triazole), tetrazole, oxazole, isoxazole, thiazole, isothiazole, oxadiazole, thiadiazole, furan, thiophene, pyridine, pyridazine, pyrimidine, pyrazine, triazine, etc.
- a 6-membered aromatic ring eg, benzene, pyridine, pyridazine, pyrimidine, pyrazine, triazine
- benzene and pyridine are particularly preferable.
- the “5- or 6-membered aromatic ring” in the “optionally substituted 5- or 6-membered aromatic ring” represented by ring Q is a group —O—R 6 and —C (R 5a ) (R 5b ) Y— In addition, it may have 1 to 4 substituents at substitutable positions. Examples of such a substituent include a C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 and the like. The same thing as the substituent which may have is mentioned.
- the further substituent of the “5- or 6-membered aromatic ring” in the “optionally substituted 5- or 6-membered aromatic ring” represented by ring Q is preferably a halogen atom (eg, fluorine atom, chlorine atom). is there.
- Ring Q is preferably (a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom), (b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), (c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and (d) benzene or a 5- or 6-membered aromatic heterocycle (eg, pyridine, pyridazine) which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a cyano group, respectively.
- a halogen atom eg, fluorine atom, chlorine atom, bromine atom
- a C 1-6 alkyl group eg, methyl
- a C 1-6 alkoxy group eg, methoxy
- Ring Q is more preferably (a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom), (b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), (c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and (d) a cyano group, Benzene, pyridine or isoxazole, each of which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from
- Preferred examples of compound (I) include the following compounds.
- R 1 is (1) a group represented by —COR 2 (wherein R 2 is an optionally substituted C 1-6 alkyl group or an optionally substituted amino group); or (2) an optionally substituted 5- or 6-membered aromatic ring group; Is; R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms; R 4a and R 4b are both hydrogen atoms; R 5a and R 5b are independently a hydrogen atom, a carboxy group or a C 1-6 alkoxy-carbonyl group; R 6 is an optionally substituted C 1-6 alkyl group; X is O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a hydrogen atom or a substituted atom) Is an optionally substituted C 1-6 alkyl group), S, SO or S (O)
- R 1 is (1) a group represented by —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl) or an amino group); or (2) 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl) Is; R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms; R 4a and R 4b are both hydrogen atoms; R 5a and R 5b are both hydrogen atoms; R 6 may be substituted with 1 to 5 halogen atoms (eg, fluorine atom), or may be substituted with 1 to 3 C 3-6 cycloalkyl groups (eg, cycloalkyl
- a C 1-6 alkyl group (eg, methyl, ethyl, propyl);
- X is O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a C 1-6 alkyl) A group (eg, methyl)), S, SO or S (O) 2 ;
- Y is O, CO or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, fluorine atom));
- Ring P is further substituted with 1 to 4 substituents selected from a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy) and a halogen atom (eg, fluorine atom), respectively.
- halogen atoms eg, fluorine atom, chlorine atom
- R 1 is (1) a group represented by —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl) or an amino group); or (2) 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl) Is; R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms; R 4a and R 4b are both hydrogen atoms; R 5a and R 5b are both hydrogen atoms; R 6 is (a) a C 3-6 cycloalkyl group (eg, cyclopropyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine
- Benzene, C 4-6 cycloalkane preferably cyclohexane
- 5 or 6 membered preferably 6 membered
- nitrogen-containing aromatic heterocycle preferably pyridine, pyridazine, pyrimidine, pyrazine, Pyridine
- a 5- or 6-membered preferably 6-membered
- nitrogen-containing non-aromatic heterocyclic ring preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine
- ring Q is 1 to Compound which is benzene or pyridine, each of which may be further substituted with 4 halogen atoms (eg, fluorine atom, chlorine atom) I).
- R 1 is (1) a group represented by —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl) or an amino group); or (2) 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl) Is; R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms; R 4a and R 4b are both hydrogen atoms; R 5a and R 5b are both hydrogen atoms; R 6 is (a) a C 3-6 cycloalkyl group (eg, cyclopropyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine
- Benzene, C 4-6 cycloalkane preferably cyclohexane
- 5 or 6 membered preferably 6 membered
- nitrogen-containing aromatic heterocycle preferably pyridine, pyridazine, pyrimidine, pyrazine, Pyridine
- a 5- or 6-membered preferably 6-membered
- nitrogen-containing non-aromatic heterocyclic ring preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine
- ring Q is 1 to Compound which is benzene or pyridine, each of which may be further substituted with 4 halogen atoms (eg, fluorine atom, chlorine atom) I).
- R 1 is (1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or (2) an optionally substituted 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl); Is; R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms; R 4a and R 4b are both hydrogen atoms; R 5a and R 5b are both hydrogen atoms; R 6 is (a) a halogen atom (eg, fluorine atom),
- R 1 is (1) —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or (2) 5-membered aromatic heterocyclic group optionally substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl) (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably Isoxazolyl); Is; R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms; R 4a and R 4b are both hydrogen atoms; R 5a and R 5b are both hydrogen atoms; R 6 is (a) a halogen atom (eg, methyl),
- R 1 is (1) —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or (2) 5-membered aromatic heterocyclic group optionally substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl) (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably Isoxazolyl); Is; R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms; R 4a and R 4b are both hydrogen atoms; R 5a and R 5b are both hydrogen atoms; R 6 is (a) a halogen atom (eg, methyl),
- the salt of the compound represented by formula (I) is preferably a pharmacologically acceptable salt.
- a salt with an inorganic base examples include a salt with an inorganic base, a salt with an organic base, and a salt with an inorganic acid.
- the salt with an organic base include trimethylamine, triethylamine, pyridine, picoline, ethanolamine, diethanolamine, triethanolamine, tromethamine [tris (hydroxymethyl) methylamine], tert-butylamine, cyclohexylamine, benzylamine, And salts with dicyclohexylamine, N, N-dibenzylethylenediamine and the like.
- salt with inorganic acid examples include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
- salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, and benzenesulfonic acid And salts with p-toluenesulfonic acid and the like.
- salts with basic amino acids include salts with arginine, lysine, ornithine and the like.
- salt with acidic amino acid include salts with aspartic acid, glutamic acid and the like.
- a prodrug of compound (I) is a compound that is converted to compound (I) by a reaction with an enzyme, gastric acid, or the like under physiological conditions in vivo, that is, compound (I) that is enzymatically oxidized, reduced, hydrolyzed, etc. Or a compound that undergoes hydrolysis or the like by gastric acid or the like and changes to compound (I).
- Compound (I) may be labeled with an isotope (eg, 3 H, 13 C, 14 C, 18 F, 35 S, 125 I) or the like.
- compound (I) may be a hydrate, a non-hydrate, a solvate or a solvate.
- a deuterium converter obtained by converting 1 H into 2 H (D) is also encompassed in compound (I).
- Compound (I) may be a pharmaceutically acceptable cocrystal or cocrystal salt.
- co-crystals or co-crystal salts are two or more unique at room temperature, each having different physical properties (eg structure, melting point, heat of fusion, hygroscopicity, solubility and stability). It means a crystalline substance composed of a simple solid.
- the cocrystal or cocrystal salt can be produced according to a cocrystallization method known per se.
- Compound (I) or a prodrug thereof (hereinafter, sometimes simply abbreviated as the compound of the present invention) has low toxicity and should be used as it is or mixed with a pharmacologically acceptable carrier to form a pharmaceutical composition.
- a pharmaceutical composition e.g, a preventive or therapeutic agent for various diseases described below for mammals (eg, humans, mice, rats, rabbits, dogs, cats, cows, horses, pigs, monkeys).
- the pharmacologically acceptable carrier various conventional organic or inorganic carrier substances are used as a pharmaceutical material, and excipients, lubricants, binders, disintegrants in solid preparations; solvents in liquid preparations , Solubilizing agents, suspending agents, isotonic agents, buffers, soothing agents and the like. If necessary, preparation additives such as preservatives, antioxidants, colorants, sweeteners and the like can also be used.
- excipients include lactose, sucrose, D-mannitol, D-sorbitol, starch, pregelatinized starch, dextrin, crystalline cellulose, low-substituted hydroxypropylcellulose, sodium carboxymethylcellulose, gum arabic, pullulan, light
- excipients include anhydrous silicic acid, synthetic aluminum silicate, and magnesium aluminate metasilicate.
- lubricant examples include magnesium stearate, calcium stearate, talc and colloidal silica.
- Preferred examples of the binder include pregelatinized starch, sucrose, gelatin, gum arabic, methylcellulose, carboxymethylcellulose, sodium carboxymethylcellulose, crystalline cellulose, sucrose, D-mannitol, trehalose, dextrin, pullulan, hydroxypropylcellulose, hydroxy Examples include propylmethylcellulose and polyvinylpyrrolidone.
- disintegrant examples include lactose, sucrose, starch, carboxymethyl cellulose, carboxymethyl cellulose calcium, croscarmellose sodium, carboxymethyl starch sodium, light anhydrous silicic acid, and low-substituted hydroxypropyl cellulose.
- Suitable examples of the solvent include water for injection, physiological saline, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, sesame oil, corn oil, olive oil, and cottonseed oil.
- solubilizer examples include polyethylene glycol, propylene glycol, D-mannitol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, sodium salicylate, sodium acetate. Is mentioned.
- Preferable examples of the isotonic agent include sodium chloride, glycerin, D-mannitol, D-sorbitol and glucose.
- buffers such as phosphate, acetate, carbonate and citrate.
- a preferred example of the soothing agent is benzyl alcohol.
- Preferable examples of the preservative include p-hydroxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid and sorbic acid.
- Preferable examples of the antioxidant include sulfite and ascorbate.
- the colorant examples include water-soluble edible tar dyes (eg, edible dyes such as edible red Nos. 2 and 3, edible yellows Nos. 4 and 5, edible blue Nos. 1 and 2, etc.), water-insoluble lake dyes (Eg, the aluminum salt of the water-soluble edible tar dye) and natural dyes (eg, ⁇ -carotene, chlorophyll, bengara).
- water-soluble edible tar dyes eg, edible dyes such as edible red Nos. 2 and 3, edible yellows Nos. 4 and 5, edible blue Nos. 1 and 2, etc.
- water-insoluble lake dyes Eg, the aluminum salt of the water-soluble edible tar dye
- natural dyes eg, ⁇ -carotene, chlorophyll, bengara
- Suitable examples of sweeteners include saccharin sodium, dipotassium glycyrrhizinate, aspartame, and stevia.
- the medicament containing the compound of the present invention can be used alone or mixed with a pharmacologically acceptable carrier according to a method known per se as a method for producing a pharmaceutical preparation (eg, a method described in the Japanese Pharmacopoeia).
- tablets including sugar-coated tablets, film-coated tablets, sublingual tablets, orally disintegrating tablets, buccal tablets, etc.
- pills powders, granules, capsules (including soft capsules and microcapsules), troches Agent, syrup, solution, emulsion, suspension, controlled release formulation (eg, immediate release formulation, sustained release formulation, sustained release microcapsule), aerosol, film agent (eg, orally disintegrating film, Oral mucosa adhesive film), injection (eg, subcutaneous injection, intravenous injection, intramuscular injection, intraperitoneal injection), drip, transdermal preparation, ointment, lotion, patch, sitting Suppositories (eg, rectal suppositories) Vaginal suppositories), pellets,
- compositions may be controlled-release preparations (eg, sustained-release microcapsules) such as immediate-release preparations or sustained-release preparations.
- the pharmaceutical composition can be produced by a method commonly used in the field of pharmaceutical technology, for example, a method described in the Japanese Pharmacopoeia.
- the content of the compound of the present invention in the pharmaceutical composition varies depending on the dosage form, the dose of the compound of the present invention, etc., but is, for example, about 0.1 to 100% by weight.
- coating base used for coating examples include sugar coating base, water-soluble film coating base, enteric film coating base and sustained-release film coating base.
- sucrose is used, and one or more selected from talc, precipitated calcium carbonate, gelatin, gum arabic, pullulan, carnauba wax and the like may be used in combination.
- water-soluble film coating base examples include cellulose polymers such as hydroxypropylcellulose, hydroxypropylmethylcellulose, hydroxyethylcellulose, and methylhydroxyethylcellulose; polyvinyl acetal diethylaminoacetate, aminoalkyl methacrylate copolymer E [Eudragit E (trade name) ], Synthetic polymers such as polyvinylpyrrolidone; polysaccharides such as pullulan.
- enteric film coating bases include cellulose polymers such as hydroxypropylmethylcellulose phthalate, hydroxypropylmethylcellulose acetate succinate, carboxymethylethylcellulose, and cellulose acetate phthalate; methacrylic acid copolymer L [Eudragit L (trade name) ] Acrylic acid polymers such as methacrylic acid copolymer LD [Eudragit L-30D55 (trade name)], methacrylic acid copolymer S [Eudragit S (trade name)]; natural products such as shellac.
- cellulose polymers such as hydroxypropylmethylcellulose phthalate, hydroxypropylmethylcellulose acetate succinate, carboxymethylethylcellulose, and cellulose acetate phthalate
- methacrylic acid copolymer L (Eudragit L (trade name) ]
- Acrylic acid polymers such as methacrylic acid copolymer LD [Eudragit L-30D55 (trade name)], methacrylic acid copolymer
- sustained-release film coating base examples include cellulose polymers such as ethyl cellulose; aminoalkyl methacrylate copolymer RS [Eudragit RS (trade name)], ethyl acrylate-methyl methacrylate copolymer suspension [Eudragit Acrylic polymer such as NE (trade name)].
- cellulose polymers such as ethyl cellulose; aminoalkyl methacrylate copolymer RS [Eudragit RS (trade name)], ethyl acrylate-methyl methacrylate copolymer suspension [Eudragit Acrylic polymer such as NE (trade name)].
- the above-mentioned coating bases may be used by mixing two or more of them in an appropriate ratio. Moreover, you may use light-shielding agents, such as a titanium oxide, ferric oxide, etc. in the case of coating.
- the compound of the present invention has low toxicity (eg, acute toxicity, chronic toxicity, genotoxicity, reproductive toxicity, pulmonary toxicity, carcinogenicity), has few side effects, and prevents or treats or diagnoses various diseases for mammals. It can be used as a medicine.
- the compound of the present invention has an excellent ACC (acetyl-CoA carboxylase) inhibitory action.
- examples of ACC include liver, adipose tissue, pancreas-specific isozyme (ACC1); muscle-specific isozyme (ACC2).
- the compound of the present invention is excellent in metabolic stability, and has advantages such as a long half-life of the compound and difficulty in being metabolized in vivo.
- the compounds of the present invention have ACC2 selectivity, and in particular, the example compounds of the present invention have high ACC2 selectivity.
- the compound of the present invention is excellent in pharmacokinetics (eg, oral absorbability, bioavailability).
- the compound of the present invention contains obesity, diabetes (eg, type 1 diabetes, type 2 diabetes, gestational diabetes, obesity type diabetes), hyperlipidemia (eg, hypertriglyceridemia, hypercholesterolemia, high LDL cholesterolemia). , Low HDL cholesterolemia, postprandial hyperlipidemia), hypertension, heart failure, diabetic complications [eg, neuropathy, nephropathy, retinopathy, diabetic cardiomyopathy, cataract, macrovascular disorder, osteopenia, Diabetic hyperosmotic coma, infections (eg, respiratory infections, urinary tract infections, gastrointestinal infections, soft skin tissue infections, lower limb infections), diabetic gangrene, xerostomia, hearing loss, Cerebrovascular disorder, peripheral blood circulation disorder], metabolic syndrome (high triglyceride (TG) emia, low HDL cholesterol (HDL-C) emia, hypertension, abdominal obesity and glucose intolerance) State), it can be used as an agent for preventing or treating sarcopenia or cancer, or the like.
- diabetes
- diabetes is a fasting blood glucose level (glucose concentration in venous plasma) of 126 mg / dl or higher, and a 75 g oral glucose tolerance test (75 gOGTT) 2-hour value (glucose concentration in venous plasma) of 200 mg / dl or higher.
- 75 gOGTT 75 g oral glucose tolerance test
- a fasting blood glucose level (glucose concentration in venous plasma) is less than 110 mg / dl or a 75 g oral glucose tolerance test (75 g OGTT) 2 hour value (glucose concentration in venous plasma) is 140 mg / dl.
- a state that is not “a state indicating less than dl” (normal type) is referred to as a “boundary type”.
- diabetes is a fasting blood glucose level (glucose concentration in venous plasma) of 126 mg / dl or more, and a 75 g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma) is 200 mg / dl. This is a state showing dl or more.
- glucose intolerance is a fasting blood glucose level (glucose concentration in venous plasma) of less than 126 mg / dl, and a 75-g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma). Is a state showing 140 mg / dl or more and less than 200 mg / dl. Furthermore, according to the report of ADA, the state where the fasting blood glucose level (glucose concentration in venous plasma) is 110 mg / dl or more and less than 126 mg / dl is called IFG (Impaired Fasting Glucose).
- the IFG is a state where the 75 g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma) is less than 140 mg / dl as IFG (Impaired Fasting Glycemia). Call.
- the compound of the present invention is also used as a prophylactic / therapeutic agent for diabetes, borderline type, glucose intolerance, IFG (Impaired Fasting Glucose) and IFG (Impaired Fasting Glycemia) determined by the above-mentioned new criteria. Furthermore, the compound of the present invention can also prevent progression from borderline type, impaired glucose tolerance, IFG (Impaired Fasting Glucose) or IFG (Impaired Fasting Glycemia) to diabetes.
- the mammal to be applied may be any mammal that wishes to avoid weight gain, may be a mammal that is genetically at risk of weight gain, and may have diabetes, hypertension and / or hyperlipidemia.
- Weight gain may be due to excessive dietary intake or a diet lacking nutritional balance, and has a PPAR ⁇ agonist-like action such as concomitant drugs (eg, troglitazone, rosiglitazone, englitazone, ciglitazone, pioglitazone) It may be a weight gain derived from an insulin resistance improving agent or the like. The weight gain may be a weight gain before reaching obesity or may be a weight gain of an obese patient.
- concomitant drugs eg, troglitazone, rosiglitazone, englitazone, ciglitazone, pioglitazone
- the weight gain may be a weight gain before reaching obesity or may be a weight gain of an obese patient.
- the compound of the present invention is also useful as a prophylactic / therapeutic agent for metabolic syndrome (metabolic syndrome).
- metabolic syndrome a prophylactic / therapeutic agent for metabolic syndrome.
- Patients with metabolic syndrome have a significantly higher rate of developing cardiovascular disease than patients with a single lifestyle-related disease, so preventing or treating metabolic syndrome prevents cardiovascular disease It is extremely important to do.
- Criteria for metabolic syndrome were published by WHO in 1999 and NCEP in 2001. According to WHO criteria, metabolic syndrome is diagnosed in patients with visceral obesity, dyslipidemia (high TG or low HDL), or hypertension based on hyperinsulinemia or impaired glucose tolerance. (World Health Organization: Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. Part I: Diagnosis and Classification of Diabetes Mellitus, World Health Organization, Geneva, 1999).
- the compound of the present invention is, for example, osteoporosis, cachexia (eg, cancer cachexia, tuberculosis cachexia, diabetic cachexia, hematological cachexia, endocrine disease cachexia, infectious cachexia or acquired cachexia).
- cachexia eg, cancer cachexia, tuberculosis cachexia, diabetic cachexia, hematological cachexia, endocrine disease cachexia, infectious cachexia or acquired cachexia.
- Cachexia due to immunodeficiency syndrome fatty liver, polycystic ovary syndrome, kidney disease (eg, diabetic nephropathy, glomerulonephritis, glomerulosclerosis, nephrotic syndrome, hypertensive nephrosclerosis, end-stage renal disease), Muscular dystrophy, myocardial infarction, angina pectoris, cerebrovascular disorder (eg, cerebral infarction, stroke), Alzheimer's disease, Parkinson's disease, anxiety, dementia, insulin resistance syndrome, syndrome X, hyperinsulinemia, hyperinsulinemia Sensory impairment, acute or chronic diarrhea, inflammatory diseases (eg, rheumatoid arthritis, osteoarthritis spondylitis, osteoarthritis, low back pain, gout, postoperative or traumatic inflammation, swelling, nerves Pharyngopharyngitis, cystitis, hepatitis (including non-alcoholic steatohepatitis), pneumonia, pancreatitis, enteritis, inflammatory bowel disease
- the compound of the present invention is used for various cancers (among others breast cancer (for example, invasive breast cancer, non-invasive breast cancer, inflammatory breast cancer, etc.)), prostate cancer (for example, hormone-dependent prostate cancer, hormone-independent). Prostate cancer, etc.), pancreatic cancer (eg, pancreatic duct cancer, etc.), stomach cancer (eg, papillary adenocarcinoma, mucinous adenocarcinoma, adenosquamous carcinoma, etc.), lung cancer (eg, non-small cell lung cancer, small cell lung cancer, malignant mesothelioma) ), Colon cancer (eg, gastrointestinal stromal tumor), rectal cancer (eg, gastrointestinal stromal tumor), colorectal cancer (eg, familial colorectal cancer, hereditary non-polyposis colorectal cancer, gastrointestinal tract) Tumor, etc.), small intestine cancer (eg, non-Hodgkin lymphoma, gastrointestinal strom
- the compound of the present invention is also used for secondary prevention and progression suppression of the various diseases described above (eg, cardiovascular events such as myocardial infarction).
- cardiovascular events such as myocardial infarction
- the dose of the compound of the present invention varies depending on the administration subject, administration route, target disease, symptom, etc. For example, when administered orally to an adult obese patient, it is usually about 0.01 to 100 mg / kg body weight as a single dose.
- the dose is preferably 0.05 to 30 mg / kg body weight, more preferably 0.5 to 10 mg / kg body weight, and this amount is desirably administered once to three times a day.
- the compound of the present invention is used for the purpose of enhancing the action of the compound or reducing the dose of the compound, etc., a therapeutic agent for diabetes, a therapeutic agent for diabetic complications, a therapeutic agent for hyperlipidemia, an antihypertensive agent, an antiobesity agent, a diuretic It can be used in combination with a drug such as an agent or an antithrombotic drug (hereinafter abbreviated as a concomitant drug).
- a concomitant drug a drug such as an agent or an antithrombotic drug
- the administration time of the compound of the present invention and the concomitant drug is not limited, and these concomitant drugs may be low molecular compounds, and may be high molecular proteins, polypeptides, antibodies, vaccines and the like.
- the compound of the present invention and the concomitant drug may be administered as two types of preparations containing each active ingredient, or may be administered as a single preparation containing both active ingredients.
- the dose of the concomitant drug can be appropriately selected based on the clinically used dose.
- the compounding ratio of the compound of the present invention and the concomitant drug can be appropriately selected depending on the administration subject, administration route, target disease, symptom, combination and the like.
- the concomitant drug may be used in an amount of 0.01 to 100 parts by weight per 1 part by weight of the compound of the present invention.
- insulin preparations eg, animal insulin preparations extracted from bovine and porcine pancreas; human insulin preparations genetically engineered using Escherichia coli or yeast; insulin zinc; protamine insulin zinc; insulin Fragment or derivative (eg, INS-1), oral insulin preparation
- insulin resistance improving agent eg, pioglitazone or a salt thereof (preferably hydrochloride), rosiglitazone or a salt thereof (preferably maleate), metaglidacene (Metaglidasen), AMG-131, Balaglitazone, MBX-2044, Riboglitazone, Aleglitazar, Chiglitazar, Lobeglitazone, PLX-204, PN-2034, GFT -505, THR-0921, WO2007 / 013694, WO2007 / 018314, WO2008 / 093639 or WO2008 / 09979 4), ⁇ -glucosidase inhibitors (eg, voglib
- Examples of the therapeutic agent for diabetic complications include aldose reductase inhibitors (eg, tolrestat, epalrestat, zopolrestat, fidarestat, CT-112, ranirestat (AS-3201), ridressat), neurotrophic factor and its increasing drug (Eg, NGF, NT-3, BDNF, neurotrophin production / secretion promoter described in WO01 / 14372 (for example, 4- (4-chlorophenyl) -2- (2-methyl-1-imidazolyl) -5 [3- (2-methylphenoxy) propyl] oxazole), compounds described in WO2004 / 039365), nerve regeneration promoters (eg, Y-128), PKC inhibitors (eg, ruboxistaurin mesylate) AGE inhibitors (eg, ALT946, pyratoxatin, N-phenacylthiazolium bromide (ALT766), ALT-711, EXO-22 6, pyridoline (Py
- statin compounds eg, pravastatin, simvastatin, lovastatin, atorvastatin, fluvastatin, rosuvastatin, pitavastatin or salts thereof (eg, sodium salt, calcium salt)
- squalene synthase inhibitors Eg, compounds described in WO97 / 10224, for example, N-[[(3R, 5S) -1- (3-acetoxy-2,2-dimethylpropyl) -7-chloro-5- (2,3-dimethoxy Phenyl) -2-oxo-1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl] acetyl] piperidine-4-acetic acid), fibrate compounds (eg, bezafibrate, clofibrate, Simfibrate, clinofibrate), anion exchange resin (eg, cholestyramine), probucol, nicotin
- statin compounds eg
- antihypertensive agent examples include angiotensin converting enzyme inhibitors (eg, captopril, enalapril, delapril, etc.), angiotensin II antagonists (eg, candesartan cilexetil, candesartan, losartan, losartan potassium, eprosartan, valsartan, telmisartan, irbesartan, tasosartan , Olmesartan, olmesartan medoxomil, azilsartan, azilsartan medoxomil), calcium antagonists (eg, manidipine, nifedipine, amlodipine, nifodipine, nicardipine, amlodipine, cilnidipine, etc.), ⁇ -blockers (eg, metoprolol, atenolol, propranolol, propranolol, propranolol, proprano
- anti-obesity agents include monoamine uptake inhibitors (eg, phentermine, sibutramine, mazindol, floxetine, tesofensin), serotonin 2C receptor agonists (eg, lorcaserin), serotonin 6 receptor antagonists, histamine H3 receptor , GABA modulators (eg, topiramate), MCH receptor antagonists (eg, SB-568849; SNAP-7941; compounds described in WO01 / 82925 or WO01 / 87834), neuropeptide Y antagonists (eg, Berneperit) , Cannabinoid receptor antagonists (eg, rimonabant, taranaban), ghrelin antagonists, ghrelin receptor antagonists, ghrelin acylase inhibitors, opioid receptor antagonists (eg, GSK-1521498), orexin receptor antagonists, Melanocortin 4 receptor agonist, 11 ⁇ -hydroxysteroid dehydrogenase inhibitors
- FGF21 preparation eg, animal FGF21 preparation extracted from bovine and porcine pancreas; human FGF21 preparation genetically engineered using E. coli and yeast; FGF21 fragment or derivative)
- Naltrexone hydrochloride sustained-release preparation and bupropion hydrochloride sustained-release preparation feeding inhibitors (eg, P-57), and the like.
- diuretics examples include xanthine derivatives (eg, sodium salicylate theobromine, calcium salicylate theobromine), thiazide preparations (eg, etiazide, cyclopentiazide, trichloromethiazide, hydrochlorothiazide, hydroflumethiazide, benchylhydrochlorothiazide, penfluthiazide.
- xanthine derivatives eg, sodium salicylate theobromine, calcium salicylate theobromine
- thiazide preparations eg, etiazide, cyclopentiazide, trichloromethiazide, hydrochlorothiazide, hydroflumethiazide, benchylhydrochlorothiazide, penfluthiazide.
- Polythiazide, meticlotiazide), anti-aldosterone formulations eg, spironolactone, triamterene
- carbonic anhydrase inhibitors eg, acetazolamide
- chlorobenzenesulfonamides eg, chlorthalidone, mefluside, indapamide
- azosemide isosorbide, etacrine
- Examples include acid, piretanide, bumetanide, furosemide and the like.
- Antithrombotic agents include, for example, heparin (eg, heparin sodium, heparin calcium, enoxaparin sodium, dalteparinsodium), warfarin (eg, warfarin potassium), antithrombin drugs (eg, argatroban) ), Dabigatran), thrombolytic drugs (eg, urokinase, tisokinase,reteplase, nateplase, monteplase, pamiteplase), platelet aggregation inhibitor ( Examples, ticlopidine hydrochloride, clopidogrel, E5555, SHC530348, cilostazol, ethyl icosapentate, beraprost sodium, sarpogrelate hydrochloride, prasugrel, E5555, SH C530348), FXa inhibitors (eg, compounds described in rivaroxaban, apixaban, edoxaban, YM150
- the administration time of the aforementioned concomitant drug is not limited, and the compound of the present invention and the concomitant drug may be administered to the administration subject at the same time or may be administered with a time difference.
- the dose of the concomitant drug may be determined according to the dose used clinically, and can be appropriately selected depending on the administration subject, administration route, disease, combination and the like.
- the administration form of the concomitant drug is not particularly limited, as long as the compound of the present invention and the concomitant drug are combined at the time of administration.
- Examples of such dosage forms include: 1) administration of a single preparation obtained by simultaneously formulating the compound of the present invention and a concomitant drug, 2) Simultaneous administration by the same route of administration of two kinds of preparations obtained by separately formulating the compound of the present invention and a concomitant drug, 3) Administration of the two compounds obtained by formulating the compound of the present invention and the concomitant drug separately with a time difference in the same administration route, 4) Simultaneous administration by different administration routes of two preparations obtained by separately formulating the compound of the present invention and a concomitant drug, 5) Administration of two preparations obtained by separately formulating the compound of the present invention and a concomitant drug at different time intervals in different administration routes (for example, administration in the order of the compound of the present invention and concomitant drug, or vice versa) Administration in this order).
- the compounding ratio of the compound of the present invention and the concomitant drug can be appropriately selected depending on the administration subject, administration route, disease and the like.
- Each raw material compound may form a salt as long as it does not inhibit the reaction.
- a salt those exemplified as the salt of the compound represented by the aforementioned formula (I) are used.
- a raw material compound can be easily obtained and used commercially, or can be produced according to a method known per se or a method analogous thereto.
- the product of each reaction can be used in the next reaction as a reaction mixture or as a crude product, but can be isolated from the reaction mixture according to a conventional method, such as recrystallization, distillation, chromatography and HPLC. It can be purified by separation means.
- a separation means such as a diastereomeric salt method, chromatography, HPLC, or SFC (supercritical fluid chromatography). For example, it can be carried out by the method described in the examples or a method analogous thereto.
- Reagents and reagents used in each reaction can be used as they are when commercially available, or can be produced according to a method known per se or a method analogous thereto, or a method described in Examples. it can.
- the reagents and reagents described in the examples can be used.
- the solvent for each reaction is not particularly limited as long as the reaction proceeds, and the reaction can be performed in a solvent inert to the reaction or in the absence of a solvent. You may mix and use.
- the solvents described in the examples can be used.
- the equivalent amounts of reagents and reagents used in each reaction are 0.001 to 100 equivalents relative to the substrate of each reaction.
- equivalent amounts of reagents and reactants described in the examples can be used.
- reaction time for each reaction is usually from 5 minutes to 72 hours.
- the reaction can be performed with the reaction times described in the examples.
- reaction temperature of each reaction is from ice-cooling to heating under reflux. For example, it can be carried out at the reaction temperature described in the examples.
- a protective group generally used in peptide chemistry or the like is introduced into these functional groups.
- the target compound can be obtained by removing the protecting group as necessary after the reaction.
- the reaction for introducing a protecting group into these functional groups was described as “protection reaction”, and the reaction for removing the protecting group was described as “deprotection reaction”.
- the protecting group introduction method (protection reaction) and the protecting group removal method (deprotection reaction) are known per se, for example, Greens Protective Groups in Organic Synthesis in ORGANIC SYNTHESIS, 4th edition. The methods described in Wiley-Interscience, 2006, etc., or the methods described in the examples can be used.
- examples of the “amino-protecting group” include a formyl group, a C 1-6 alkyl-carbonyl group, a C 1-6 alkoxy-carbonyl group, a benzoyl group, a C 7-10 aralkyl-carbonyl group ( Example, benzylcarbonyl), C 7-14 aralkyloxy-carbonyl group (eg, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl), trityl group, phthaloyl group, N, N-dimethylaminomethylene group, substituted silyl group ( Examples, trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), C 2-6 alkenyl groups (eg, 1-allyl), substituted C 7-10 aralkyl groups (eg, 2, 4-dimethoxybenzyl), a
- examples of the “carboxy group protecting group” include a C 1-6 alkyl group, a C 7-11 aralkyl group (eg, benzyl), a phenyl group, a trityl group, a substituted silyl group (eg, trimethylsilyl, Triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), C 2-6 alkenyl groups (eg, 1-allyl) and the like. These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkoxy group and a nitro group.
- examples of the “protecting group for hydroxy group” include C 1-6 alkyl group, phenyl group, trityl group, C 7-10 aralkyl group (eg, benzyl), formyl group, C 1-6 alkyl.
- -Carbonyl group benzoyl group, C 7-10 aralkyl-carbonyl group (eg, benzylcarbonyl), 2-tetrahydropyranyl group, 2-tetrahydrofuranyl group, substituted silyl group (eg, trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), C 2-6 alkenyl groups (eg, 1-allyl) and the like. These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group or a nitro group.
- substituents selected from a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group or a nitro group.
- examples of the “carbonyl-protecting group” include cyclic acetals (eg, 1,3-dioxane), acyclic acetals (eg, di-C 1-6 alkylacetal) and the like.
- examples of the “protecting group for sulfanyl group” include a C 1-6 alkyl group, a phenyl group, a trityl group, a C 7-10 aralkyl group (eg, benzyl), a C 1-6 alkyl-carbonyl group.
- Benzoyl group C 7-10 aralkyl-carbonyl group (eg, benzylcarbonyl), C 1-6 alkoxy-carbonyl group, C 6-14 aryloxy-carbonyl group (eg, phenyloxycarbonyl), C 7-14 aralkyl Oxy-carbonyl groups (eg, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl), 2-tetrahydropyranyl groups, C 1-6 alkylamino-carbonyl groups (eg, methylaminocarbonyl, ethylaminocarbonyl), etc. Can be mentioned. These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group or a nitro group.
- a 1 is a protecting group for a hydroxy group
- L 1 is a hydroxy group, a halogen atom, an optionally substituted C 6-14 arylsulfonyloxy group, or an optionally substituted C 1-6 alkylsulfonyl group
- Compound (1) can be produced, for example, according to the methods described in Reaction Schemes 5 and 8, a method known per se, or a method analogous thereto.
- Compound (2) can be produced, for example, by deprotection of compound (1).
- compound (I) can be produced, for example, by an alkylation reaction of compound (2) and compound (3).
- the alkylation reaction is carried out, for example, by reacting compound (2) and compound (3) with a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene). Etc.) in the presence of an inert solvent (for example, DMF (dimethylformamide), acetonitrile, THF (tetrahydrofuran), toluene, water, etc.).
- a base for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene).
- Etc. in the presence of an inert solvent (for example, DMF (dimethylformamide), acetonitrile, THF (tetrahydrofuran), toluene, water, etc.).
- an inert solvent
- a phase transfer catalyst for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.
- compound (3) is an alcohol
- compound (I) can be produced, for example, by Mitsunobu reaction between compound (2) and compound (3).
- Mitsunobu reaction can be carried out, for example, by converting compound (2) and compound (3) into a hydroxy group activator (eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl).
- a hydroxy group activator eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl
- the reaction is carried out in the presence of phosphine, ADDP (1,1 ′-(azodicarbonyl) dipiperidine and tributylphosphine), etc.) in an inert solvent (for example, toluene, THF, etc.).
- an inert solvent for example, toluene, THF, etc.
- a compound in which X or Y of compound (I) is an oxidized sulfur atom that is, a sulfone derivative or a sulfoxide derivative can be produced by an oxidation reaction of compound (I) in which X or Y is a sulfur atom.
- an oxidation reaction for example, the method described in 4th edition Experimental Chemistry Course 20 (Edited by The Chemical Society of Japan), pages 276 to 278, 503 or a method analogous thereto is used.
- Y 1 represents a sulfur atom, an oxygen atom, or a nitrogen atom which may be substituted with a substituent R 8c
- L 2 is a halogen atom or an optionally substituted C 6-14 arylsulfonyloxy group Or an optionally substituted C 1-6 alkylsulfonyloxy group, and other symbols are as defined above.
- Compound (5) can be produced, for example, according to the method described in Reaction Formula 6, Reaction Formula 9, Reaction Formula 10, and Reaction Formula 11, a method known per se, or a method analogous thereto.
- Compound (I-1) can be produced, for example, by Mitsunobu reaction between compound (4) and compound (5).
- compound (4) and compound (5) are converted into a hydroxy group activator (for example, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). Phosphine, etc.) in the presence of an inert solvent (for example, toluene, THF, etc.).
- a hydroxy group activator for example, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl.
- Phosphine, etc. in the presence of an inert solvent (for example, toluene, THF, etc.).
- Compound (6) can be produced, for example, by halogenation reaction or sulfonylation reaction of compound (4). This reaction is performed, for example, by reacting compound (4) with a halogenating agent (eg, thionyl chloride, phosphorus tribromide, etc.) in an inert solvent (eg, THF, toluene, diethyl ether, etc.).
- a halogenating agent eg, thionyl chloride, phosphorus tribromide, etc.
- an inert solvent eg, THF, toluene, diethyl ether, etc.
- this reaction may be carried out, for example, by reacting compound (4) and a sulfonylating agent (eg, methanesulfonyl chloride, p-toluenesulfonyl chloride, etc.) in the presence of a base (eg, triethylamine, pyridine, etc.) in an inert solvent (eg, THF). , Toluene, diethyl ether, etc.).
- a sulfonylating agent eg, methanesulfonyl chloride, p-toluenesulfonyl chloride, etc.
- a base eg, triethylamine, pyridine, etc.
- an inert solvent eg, THF.
- Compound (I-1) can also be produced, for example, by an alkylation reaction of compound (5) and compound (6).
- compound (5) and compound (6) are reacted with an inert solvent (eg, DMF, acetonitrile, etc.) in the presence of a base (eg, potassium carbonate, cesium carbonate, sodium hydroxide, sodium hydride, DBU, etc.).
- a base eg, potassium carbonate, cesium carbonate, sodium hydroxide, sodium hydride, DBU, etc.
- a phase transfer catalyst for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.
- phase transfer catalyst for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.
- a 2 represents an amino-protecting group, and other symbols are as defined above.
- Compound (7) can be produced, for example, according to the method described in Reaction Formula 15 and Reaction Formula 16, a method known per se, or a method analogous thereto.
- Compound (8) can be produced by, for example, deprotection reaction of compound (7).
- Compound (I-2) can be produced, for example, by an acylation reaction of compound (8).
- the “acylation reaction” includes, for example, a synthesis reaction of an amide derivative, a carbamate derivative or a urea derivative. This reaction is performed, for example, by reacting compound (8) with an acylating agent in an inert solvent (eg, DMF, acetonitrile, dichloromethane, THF, etc.).
- an acylating agent include carboxylic acids, reactive derivatives of carboxylic acids (eg, acid chloride, acid anhydrides, mixed acid anhydrides, active esters, active amides, etc.), dicarbonates, chloroformates, isocyanates, And carbamoyl chloride derivatives.
- a dehydrating condensing agent for example, dicyclohexylcarbodiimide (DCC), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (WSC), N-[(dimethylamino) (3H- [1,2 , 3] triazolo [4,5-b] pyridin-3-yloxy) methylidene] -N-methylmethanaminium hexafluorophosphate (HATU), etc., additives (for example, 1-hydroxybenzotriazole (HOBt), etc. ) Or a base (for example, triethylamine, pyridine, etc.) may be reacted.
- DCC dicyclohexylcarbodiimide
- WSC 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride
- Compound (9) can be produced, for example, according to the method described in Reaction Scheme 4, Reaction Formula 7, and Reaction Formula 14, a method known per se, or a method analogous thereto.
- Compound (10) can be produced, for example, by halogenation or sulfonylation reaction of compound (9). This reaction is performed, for example, in the same manner as in the method for producing compound (6) of reaction formula 2.
- Compound (11) can be produced, for example, by subjecting compound (10) to an azidation reaction.
- the compound (10) and an azide for example, sodium azide, diphenylphosphoryl azide (DPPA), trimethylsilyl azide, etc.
- an inert solvent for example, DMF, acetonitrile, THF, etc.
- the reaction is carried out at 0 ° C. to 150 ° C. for 5 minutes to 72 hours. If necessary, the reaction may be performed in the presence of a base (for example, triethylamine, pyridine, DBU, etc.).
- a base for example, triethylamine, pyridine, DBU, etc.
- Compound (8) can also be produced, for example, by a reduction reaction of compound (11).
- the compound (11) is reacted with an inert solvent (eg, ethanol, for example) in the presence of a metal catalyst (eg, palladium-carbon, platinum oxide, etc.) and a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.).
- an inert solvent eg, ethanol, for example
- a metal catalyst eg, palladium-carbon, platinum oxide, etc.
- a hydrogen source eg, hydrogen gas, formic acid, ammonium formate, etc.
- This reaction can also be carried out, for example, by reacting compound (11), triphenylphosphine and water in an inert solvent (for example, THF).
- R 1a and R 9 each independently represents an optionally substituted 5- or 6-membered aromatic ring group, and other symbols are as defined above.
- Compound (12) can be produced, for example, according to the method described in Reaction Scheme 7, a method known per se, or a method analogous thereto.
- Compound (I-3) can be produced, for example, by a reductive amination reaction of compound (12) and compound (13).
- compound (12) and compound (13) are reacted with an inert solvent (for example, sodium borohydride, sodium cyanoborohydride, sodium triacetoxyborohydride, decaborane, etc.) in the presence of a reducing agent.
- an inert solvent for example, sodium borohydride, sodium cyanoborohydride, sodium triacetoxyborohydride, decaborane, etc.
- a reducing agent for example, sodium borohydride, sodium cyanoborohydride, sodium triacetoxyborohydride, decaborane, etc.
- Compound (9) can be produced, for example, by a reduction reaction of compound (12).
- compound (12) is converted into an inert solvent (for example, ethanol, methanol, etc.) in the presence of a reducing agent (for example, sodium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride, lithium aluminum hydride, etc.).
- a reducing agent for example, sodium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride, lithium aluminum hydride, etc.
- Compound (15) can be produced, for example, by Mitsunobu reaction between compound (9) and compound (14).
- compound (9) and compound (14) are converted into a hydroxy group activator (for example, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). Phosphine, etc.) in the presence of an inert solvent (for example, toluene, THF, etc.).
- a hydroxy group activator for example, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl.
- Phosphine, etc. in the presence of an inert solvent (for example, toluene, THF, etc.).
- Compound (I-3) can also be produced, for example, by a desulfonylation reaction of compound (15). This reaction is carried out, for example, in the presence of a compound (15), a base (for example, triethylamine, lithium hydroxide dihydrate, etc.) and an organic mercaptan (for example, sulfanyl acetic acid, etc.) and an inert solvent (for example, THF, dichloromethane, In DMF etc.).
- a base for example, triethylamine, lithium hydroxide dihydrate, etc.
- an organic mercaptan for example, sulfanyl acetic acid, etc.
- an inert solvent for example, THF, dichloromethane, In DMF etc.
- Compound (18) can be produced, for example, by alkylation of compound (16) and compound (17) or Mitsunobu reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
- Compound (19) can be produced, for example, by deprotection reaction of compound (18).
- Compound (1-1) can be produced, for example, by an acylation reaction of compound (19). This reaction is performed, for example, in the same manner as in the production of (I-2) in Reaction Scheme 3.
- Compound (21) can be produced, for example, by an alkylation reaction or Mitsunobu reaction between compound (20) and compound (17). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
- Compound (22) can be produced, for example, by deprotection of compound (21).
- Compound (23) can be produced, for example, by deprotection of compound (22).
- Compound (5-1) can be produced, for example, by acylation reaction of compound (23). This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
- compound (5-1) can also be produced by changing the order of the method described in Reaction Scheme 6 (deprotection reaction and acylation reaction of A 2 and A 3 ).
- aromatic ring P of compound (21), compound (22), compound (23) or compound (5-1) is aromatic ring P 1 (5- and 6-membered aromatic ring which may be substituted)
- aromatic ring P A compound having a corresponding saturated ring P 2 (optionally substituted 5-membered and 6-membered saturated ring) can be produced by a reduction reaction of 1 .
- the compound (21), the compound (22), the compound (23) or the compound (5-1) is converted into a metal catalyst (for example, rhodium-carbon, palladium-carbon, platinum oxide, etc.) and a hydrogen source (for example, , Hydrogen gas, formic acid, ammonium formate, etc.) in the presence of an inert solvent (for example, ethanol, methanol, acetic acid, THF, etc.).
- a metal catalyst for example, rhodium-carbon, palladium-carbon, platinum oxide, etc.
- a hydrogen source for example, Hydrogen gas, formic acid, ammonium formate, etc.
- an inert solvent for example, ethanol, methanol, acetic acid, THF, etc.
- hydrogen pressure is usually 1 to 10 atmospheres.
- R 3 -M represents an organometallic reagent
- R 10a and R 10b independently represent a substituent
- R 10a and R 10b may combine with each other to form a ring.
- the symbols of the above are as defined above, wherein compound (9-1) is included in compound (9) and compound (12-1) is included in compound (12).
- Compound (25) can be produced, for example, by an alkylation reaction or Mitsunobu reaction between compound (24) and compound (6). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
- Compound (26) can be produced, for example, by subjecting compound (25) to deprotection.
- Compound (26) can also be produced, for example, according to the method described in Reaction Scheme 12, a method known per se, or a method analogous thereto.
- Compound (28) can be produced, for example, by an alkylation reaction or Mitsunobu reaction of compound (26) and compound (27). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
- Compound (12-1) can be produced, for example, by subjecting compound (28) to an organometallic reagent R 3 -M.
- organometallic reagent R 3 -M eg, methylmagnesium chloride, methylmagnesium bromide, methyllithium, etc.
- an inert solvent eg, toluene, THF, diethyl ether, etc.
- the reaction is performed at ⁇ 78 ° C. to 100 ° C.
- Compound (9-1) can be produced, for example, by a reduction reaction of compound (12-1). This reaction is performed, for example, in the same manner as in the method for producing compound (9) of reaction formula 4.
- Compound (30) can be produced, for example, by an alkylation reaction or Mitsunobu reaction of compound (29) and compound (17). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
- Compound (31) can be produced, for example, by subjecting compound (30) to deprotection.
- Compound (32) can be produced, for example, by subjecting compound (31) to an acylation reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
- Compound (34) can be produced, for example, by a coupling reaction of compound (32) and compound (33).
- compound (32) and compound (33) are present in the presence of a metal catalyst (eg, bistriphenylphosphinedichloropalladium (II) and copper (I) iodide) and a base (eg, triethylamine, pyridine, etc.).
- a metal catalyst eg, bistriphenylphosphinedichloropalladium (II) and copper (I) iodide
- a base eg, triethylamine, pyridine, etc.
- the reaction is usually carried out in an inert solvent (eg, toluene, THF, DMF, etc.) under an inert gas (eg, argon, nitrogen, etc.) atmosphere.
- an inert solvent eg, toluene, THF, DMF, etc.
- an inert gas eg,
- Compound (1-2) can be produced, for example, by a reduction reaction of compound (34).
- the compound (34) is reacted with an inert solvent (eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.) and a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.).
- an inert solvent eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.
- a hydrogen source eg, hydrogen gas, formic acid, ammonium formate, etc.
- the reaction is performed in ethanol, methanol, acetic acid, THF, or the like.
- hydrogen pressure is usually 1 to 10 atmospheres.
- the metal catalyst is used in the same amount (weight) or more as the compound (34).
- a reduction reaction of the aromatic ring P 1 from the compound (30), the compound (31), the compound (32), the compound (34) or the compound (1-2) results in the corresponding saturated ring P 2 (optionally substituted 5 And 6-membered saturated rings) can be prepared.
- compound (30), compound (31), compound (32), compound (34) or compound (1-2) is converted to a metal catalyst (eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.) And a reaction in an inert solvent (eg, ethanol, methanol, acetic acid, THF, etc.) in the presence of a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.).
- a hydrogen source eg, hydrogen gas, formic acid, ammonium formate, etc.
- Compound (37) can be produced, for example, by a coupling reaction of compound (35) and compound (36). This reaction is performed, for example, in the same manner as in the method for producing compound (34) of reaction formula 8.
- Compound (38) can be produced, for example, by subjecting compound (37) to deprotection.
- Compound (39) can be produced, for example, by subjecting compound (38) to an acylation reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
- Compound (40) can be produced, for example, by subjecting compound (39) to a reduction reaction. This reaction is performed, for example, in the same manner as in the method for producing compound (1-2) of reaction formula 8.
- Compound (5-2) can be produced, for example, by deprotection of compound (40).
- compound (5-2) can also be produced by changing the order of the method described in Reaction Scheme 9 (deprotection reaction of A 2 and A 3 , coupling reaction and reduction reaction). .
- Reduction of the aromatic ring P 1 from the compound (37), compound (38), compound (39), compound (40) or compound (5-2) results in the corresponding saturated ring P 2 (optionally substituted 5 And 6-membered saturated rings) can be prepared.
- compound (37), compound (38), compound (39), compound (40) or compound (5-2) is converted into a metal catalyst (eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.).
- a reaction in an inert solvent eg, ethanol, methanol, acetic acid, THF, etc.
- a hydrogen source eg, hydrogen gas, formic acid, ammonium formate, etc.
- hydrogen pressure is usually 1 to 10 atmospheres.
- Compound (43) can be produced, for example, by an addition reaction of compound (41) and compound (42). This reaction is performed, for example, by reacting compound (41) with compound (42) in an inert solvent (eg, ethanol, methanol, THF, etc.). If necessary, it is carried out in the presence of a metal salt (for example, bismuth nitrate, etc.).
- an inert solvent eg, ethanol, methanol, THF, etc.
- a metal salt for example, bismuth nitrate, etc.
- Compound (44) can be produced, for example, by subjecting compound (43) to deprotection.
- Compound (5-3) can be produced, for example, by subjecting compound (44) to an acylation reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
- Compound (47) can be produced, for example, by an amidation reaction of compound (45) and compound (46).
- compound (45) and compound (46) are combined with an inert solvent (eg, DMF, acetonitrile, etc.) in the presence of a dehydrating condensing agent (eg, WSC, HATU, etc.) and an additive (eg, HOBt, etc.). Reaction in dichloromethane, THF, etc.).
- compound (46) may be used after being converted into a reactive derivative of carboxylic acid (for example, acid chloride, acid anhydride, mixed acid anhydride, active ester, active amide, etc.).
- a base for example, a triethylamine, a pyridine, etc.
- Compound (48) can be produced, for example, by subjecting compound (47) to a reduction reaction.
- This reaction is carried out, for example, by reacting compound (47) in the presence of a reducing agent (eg, borane, lithium aluminum hydride) in an inert solvent (eg, toluene, THF, etc.).
- a reducing agent eg, borane, lithium aluminum hydride
- an inert solvent eg, toluene, THF, etc.
- Compound (49) can be produced, for example, by subjecting compound (48) to deprotection.
- Compound (50) can be produced, for example, by subjecting compound (49) to an acylation reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
- Compound (5-4) can be produced, for example, by subjecting compound (50) to deprotection.
- R 11a and R 11b independently represent a hydroxyl group or a substituent, and these may be bonded to each other to form a ring. Each symbol has the same meaning as described above. 26-1 is encompassed by compound (26).)
- Compound (53) can be produced, for example, by a nucleophilic substitution reaction of compound (51) and compound (52).
- compound (51) and compound (52) are reacted with an inert solvent (eg, DMF, acetonitrile, dichloromethane, THF, etc.) in the presence of a base (eg, sodium hydride, potassium carbonate, triethylamine, pyridine, etc.).
- an inert solvent eg, DMF, acetonitrile, dichloromethane, THF, etc.
- a base eg, sodium hydride, potassium carbonate, triethylamine, pyridine, etc.
- Compound (54) can be produced, for example, by a boronation reaction of compound (53).
- a halogen atom is converted into a metal atom with an alkyl metal (eg, butyllithium, isopropylmagnesium bromide, etc.) in compound (53) in an inert solvent (eg, diethyl ether, toluene, THF, etc.).
- an organic boron compound for example, trimethoxyborane etc.
- Compound (26-1) can be produced, for example, by subjecting the boron atom of compound (54) to an oxidation reaction.
- compound (54) is reacted with an oxidizing agent (eg, oxygen, hydrogen peroxide, m-chloroperbenzoic acid, sodium perborate, etc.) in an inert solvent (eg, water, THF, etc.).
- an oxidizing agent eg, oxygen, hydrogen peroxide, m-chloroperbenzoic acid, sodium perborate, etc.
- an inert solvent eg, water, THF, etc.
- a base for example, sodium hydroxide
- Compound (56) can be produced, for example, by an alkylation reaction or Mitsunobu reaction of compound (55) and compound (17). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
- Compound (57) can be produced, for example, by a boronation reaction of compound (56). This reaction is performed, for example, in the same manner as in the method for producing compound (54) of reaction formula 12.
- Compound (22-1) can be produced, for example, by subjecting the boron atom of compound (57) to an oxidation reaction. This reaction is performed, for example, in the same manner as in the method for producing compound (26-1) of reaction formula 12.
- Compound (60) can be produced, for example, by an alkylation reaction or Mitsunobu reaction between compound (58) and compound (59). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
- Compound (61) can be produced, for example, by a reduction reaction of compound (60). This reaction is performed, for example, in the same manner as in the method for producing compound (9) of reaction formula 4.
- Compound (62) can be produced, for example, by deprotection of compound (61).
- Compound (9-2) can also be produced, for example, by an alkylation reaction of compound (62) and compound (6). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
- Compound (64) can be produced, for example, by an alkylation reaction or Mitsunobu reaction between compound (63) and compound (17). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
- Compound (65) can be produced, for example, by a reduction reaction of aromatic ring P 1 of compound (64).
- compound (64) is reacted with an inert solvent (eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.) and a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.).
- an inert solvent eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.
- a hydrogen source eg, hydrogen gas, formic acid, ammonium formate, etc.
- the reaction is performed in ethanol, methanol, acetic acid, THF, or the like.
- hydrogen pressure is usually 1 to 10 atmospheres.
- Compound (70) can also be produced, for example, by an alkylation reaction of compound (68) and compound (69).
- compound (68) and compound (69) are reacted in the presence of a base (for example, potassium carbonate, cesium carbonate, sodium hydroxide, sodium hydride, DBU, etc.) in an inert solvent (for example, DMF, acetonitrile, (Ethanol, THF, water, etc.).
- a phase transfer catalyst for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc. may be used as necessary.
- Compound (16-1) can be produced, for example, by subjecting compound (70) to deprotection.
- Compound (73) can be produced, for example, by a coupling reaction of compound (71) and compound (72).
- compound (71) and compound (72) are present in the presence of a metal catalyst (for example, bistriphenylphosphinedichloropalladium (II) and copper iodide (I)) and a base (for example, triethylamine, pyridine, etc.).
- a metal catalyst for example, bistriphenylphosphinedichloropalladium (II) and copper iodide (I)
- a base for example, triethylamine, pyridine, etc.
- the reaction is usually carried out in an inert solvent (eg, toluene, THF, DMF, etc.) under an inert gas (eg, argon, nitrogen, etc.) atmosphere.
- an inert solvent eg, toluene, THF, DMF, etc.
- an inert gas eg, argon
- Compound (74) can be produced, for example, by a reduction reaction of compound (73). This reaction is performed, for example, in the same manner as in the method for producing compound (1-2) of reaction formula 8.
- Compound (16-2) can be produced, for example, by subjecting compound (74) to deprotection.
- the compound (16-2) can be produced by performing the reaction leading from the compound (73) to the compound (16-2), that is, the reduction reaction and the deprotection reaction at the same time or by changing the order.
- compound (77) can be produced, for example, by an alkylation reaction of compound (75) and compound (76).
- the alkylation reaction is carried out, for example, by converting the compound (75) and the compound (76) into a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene). Etc.) in the presence of an inert solvent (for example, DMF, acetonitrile, THF, toluene, water, etc.).
- a base for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene).
- Etc. in the presence of an inert solvent (for example, DMF, acetonitrile, THF, toluene, water, etc.).
- a phase transfer catalyst for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.
- compound (76) is an alcohol
- compound (77) can be produced, for example, by Mitsunobu reaction between compound (75) and compound (76).
- Mitsunobu reaction can be carried out, for example, by converting compound (75) and compound (76) into a hydroxy group activator (eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl).
- a hydroxy group activator eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl.
- an inert solvent for example,
- Compound (78) can be produced, for example, by a reduction reaction of compound (77).
- a metal hydride compound for example, diisobutylaluminum hydride
- a metal hydride complex compound for example, sodium borohydride, sodium cyanoborohydride, lithium aluminum hydride, sodium aluminum hydride, hydrogen
- a reducing agent such as bis (2-methoxyethoxy) aluminum chloride
- an inert solvent for example, ethanol, methanol, THF, etc.
- Compound (1-3) is produced, for example, using compound (78), for example, in the same manner as in the production of compound (I-1) from compound (4) in Reaction Scheme 2.
- Compound (1-3) is produced, for example, using compound (79), for example, in the same manner as in the production of compound (I-1) from compound (6) in Reaction Scheme 2.
- compound (81) can be produced, for example, by an alkylation reaction of compound (80) and compound (3).
- the alkylation reaction is carried out, for example, by converting the compound (80) and the compound (3) into a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene). Etc.) in the presence of an inert solvent (for example, DMF, acetonitrile, THF, toluene, water, etc.).
- a base for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene).
- Etc. in the presence of an inert solvent (for example, DMF, acetonitrile, THF, toluene, water, etc.).
- a phase transfer catalyst for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.
- compound (3) is an alcohol
- compound (81) can be produced, for example, by Mitsunobu reaction between compound (80) and compound (3).
- Mitsunobu reaction can be achieved, for example, by converting compound (80) and compound (3) into a hydroxy group activator (eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl).
- a hydroxy group activator eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl.
- an inert solvent for example, toluene, T
- Compound (83) can be produced, for example, by subjecting compound (82) to an alkoxylation reaction.
- the compound (82) and an alcohol represented by the chemical formula R 6 —OH can be converted into a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4. 0] undecaker 7-ene) and the like in the presence of an inert solvent (eg, DMF, acetonitrile, THF, toluene, water, etc.).
- a base for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4. 0] undecaker 7-ene
- an inert solvent eg, DMF, acetonitrile, THF, toluene, water, etc.
- the alcohol represented by the chemical formula R 6 —OH may be used in excess of the compound (82), or a phase transfer catalyst (eg, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used. Good.
- a phase transfer catalyst eg, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.
- Compound (81) can also be produced, for example, by a formylation reaction of compound (83).
- compound (83) is reacted with an alkyl metal (eg, butyl lithium) or a metal amide (eg, lithium diisopropylamide, lithium) in an inert solvent (eg, diethyl ether, toluene, THF, etc.).
- an alkyl metal eg, butyl lithium
- a metal amide eg, lithium diisopropylamide, lithium
- an inert solvent eg, diethyl ether, toluene, THF, etc.
- a formylating agent eg, DMF, ethyl formate, etc.
- Compound (21) can be produced, for example, by subjecting compound (22) to a protection reaction.
- Compound (84) can be produced, for example, by subjecting compound (21) to deprotection.
- Compound (5-1) can be produced, for example, by subjecting compound (85) to deprotection.
- the functional group in the molecule can be converted to the target functional group by combining a chemical reaction known per se.
- the chemical reaction include an oxidation reaction, a reduction reaction, an alkylation reaction, an acylation reaction, a urealation reaction, a hydrolysis reaction, an amination reaction, an esterification reaction, an aryl coupling reaction, and a deprotection reaction.
- Compound (I) obtained by the above production method can be isolated and purified by a known means such as solvent extraction, liquid conversion, phase transfer, crystallization, recrystallization, chromatography and the like.
- compound (I) contains optical isomers, stereoisomers, positional isomers, and rotational isomers, these are also included as compound (I), and are synthesized by known synthesis methods and separation methods, respectively. Can be obtained as a single product.
- compound (I) has an optical isomer
- the optical isomer resolved from the compound is also encompassed in compound (I).
- the optical isomer can be produced by a method known per se.
- Compound (I) may be a crystal. Crystals of compound (I) (hereinafter sometimes abbreviated as crystals of the present invention) can be produced by crystallization by applying a crystallization method known per se to compound (I). In the present specification, the melting point is measured using, for example, a trace melting point measuring device (Yanako, MP-500D type or Buchi, B-545 type) or a DSC (differential scanning calorimetry) apparatus (SEIKO, EXSTAR6000). Mean melting point. In general, the melting point may vary depending on measurement equipment, measurement conditions, and the like. The crystal in the present specification may be a crystal having a value different from the melting point described in the present specification as long as it is within a normal error range.
- the crystals of the present invention are excellent in physicochemical properties (eg, melting point, solubility, stability) and biological properties (eg, pharmacokinetics (absorbability, distribution, metabolism, excretion), expression of medicinal properties), and are extremely useful as pharmaceuticals. Useful.
- MS mass spectrum
- LC / MS liquid chromatograph mass spectrometer
- ESI ElectroSpray Ionization
- APCI Adtomospheric Pressure Chemical Ionization
- ESI + positive mode
- ESI- negative mode
- a molecular ion peak is observed, but in the case of a compound having a tert-butoxycarbonyl group (-Boc), a peak from which a tert-butoxycarbonyl group or a tert-butyl group is eliminated should be observed as a fragment ion. There is also. Depending on the compound, a peak in which sodium ion (+ Na) is added to the molecular ion peak may be observed as a fragment ion. In the case of a compound having a hydroxyl group (—OH), a peak from which H 2 O is eliminated may be observed as a fragment ion.
- the solid obtained was dissolved in ethyl acetate (70 mL), 4 M hydrogen chloride / ethyl acetate (100 mL) was added, and the mixture was stirred at room temperature for 1.5 hr. did.
- the reaction mixture was concentrated under reduced pressure, the residue was dissolved in pyridine (100 mL), acetic anhydride (30 mL) was added, and the mixture was stirred at room temperature for 16 hr.
- the reaction mixture was concentrated under reduced pressure, and the residue was washed with diisopropyl ether.
- the obtained solid was purified by NH silica gel column chromatography (THF) to obtain the title compound (7.36 g).
- reaction mixture was concentrated under reduced pressure, the residue was dissolved in pyridine (20 mL), acetic anhydride (0.345 mL) was added, and the mixture was stirred at room temperature for 3 hr.
- 1 M aqueous sodium hydroxide solution 10 mL
- the reaction mixture was concentrated under reduced pressure, and the residue was diluted with ethyl acetate and saturated brine.
- the organic layer was separated, washed with 1 M hydrochloric acid, saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (0.88 g).
- the precipitate was filtered off, and the filtrate was neutralized with a saturated aqueous sodium hydrogen carbonate solution and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- the obtained residue (645 mg) was dissolved in DMF (4.7 mL), sodium azide (0.462 g) was added at room temperature, and the mixture was stirred at 80 ° C. for 3 hr. After evaporating the solvent under reduced pressure, water was added and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- CHIRALPAK AD trade name
- Example 5b 1- ⁇ 2-[(trans-4- ⁇ [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ urea optically active substance
- Example 6 N-[(1S) -2- (4- ⁇ [4- (cyclopropylmethoxy) benzyl] oxy ⁇ phenoxy) -1-methylethyl] acetamide [4- (cyclopropylmethoxy) phenyl] methanol and N-[( Using 1S) -2- (4-hydroxyphenoxy) -1-methylethyl] acetamide, the title compound was obtained in the same manner as in Step E of Example 1 or a method analogous thereto.
- Example 9 N- ⁇ (1S) -2-[(4- ⁇ [4- (cyclopropylmethoxy) benzyl] oxy ⁇ phenyl) sulfinyl] -1-methylethyl ⁇ acetamide N- ⁇ (1S) -2-[(4- Add 3-chloroperoxybenzoic acid (112 mg) to a solution of ⁇ [4- (cyclopropylmethoxy) benzyl] oxy ⁇ phenyl) sulfanyl] -1-methylethyl ⁇ acetamide (250 mg) in THF (5 mL) at room temperature. For 15 minutes. The precipitated solid was collected by filtration and washed with ethyl acetate to give the title compound (118 mg).
- Example 10 N- ⁇ (1S) -2-[(4- ⁇ [4- (cyclopropylmethoxy) benzyl] oxy ⁇ phenyl) sulfonyl] -1-methylethyl ⁇ acetamide N- ⁇ (1S) -2-[(4- Add 3-chloroperoxybenzoic acid (280 mg) to a solution of ⁇ [4- (cyclopropylmethoxy) benzyl] oxy ⁇ phenyl) sulfanyl] -1-methylethyl ⁇ acetamide (250 mg) in THF (5 mL) at room temperature. For 15 minutes. The reaction mixture was passed through a NH silica gel short column (ethyl acetate), the solvent was distilled off under reduced pressure, and the obtained solid was washed with ethyl acetate to obtain the title compound (223 mg).
- the reaction mixture was allowed to cool to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH silica gel column chromatography (hexane / ethyl acetate). The solid obtained was dissolved in ethanol (40 mL), 10% palladium-carbon (50% water content, 2.0 g) was added, and the mixture was added under a hydrogen atmosphere at room temperature. For 16 hours. After removing the catalyst by Celite filtration, the obtained filtrate was concentrated under reduced pressure.
- Example 12 N-[(1S) -3- (4- ⁇ [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ phenyl) -1-methylpropyl] acetamide N-[(1S) -3- (4- Using hydroxyphenyl) -1-methylpropyl] acetamide and [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol, the title compound was obtained in the same manner as in Step E of Example 1 or a method analogous thereto. .
- Example 14 N-[(1S) -3- ⁇ 4-[(4-Ethoxy-2-fluorobenzyl) oxy] phenyl ⁇ -1-methylpropyl] acetamide (4-ethoxy-2-fluorophenyl) methanol (118 mg) Thionyl chloride (0.084 mL) was added to a toluene (1 mL) solution, and the mixture was stirred at room temperature for 30 minutes. The reaction mixture was concentrated under reduced pressure to give DMF (1 mL), N-[(1S) -3- (4-hydroxyphenyl) -1-methylpropyl] acetamide (120 mg) and cesium carbonate (120 mg).
- Methyl 2-fluoro-4-propoxybenzoate Methyl 2-fluoro-4-hydroxybenzoate (2.85 g), 1-iodopropane (3.12 g) and cesium carbonate (8.19 g) in DMF (17 mL) suspension The solution was stirred at 50 ° C. for 20 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed 3 times with saturated brine and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to obtain the title compound (3.39 g).
- Example 18 Performed with N- ⁇ 1-[(4- ⁇ [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ phenoxy) methyl] propyl ⁇ acetamide tert-butyl [1- (hydroxymethyl) propyl] carbamate
- the title compound was obtained in the same manner as in Step A of Example 1, Step B of Example 1, Step C of Example 1 and Step E of Example 1 or a method analogous thereto.
- Example 20 (4- ⁇ [(2S) -2- (acetylamino) propyl] oxy ⁇ phenoxy) [4- (cyclopropylmethoxy) phenyl] acetic acid ethyl (4- ⁇ [(2S) -2- (acetylamino) propyl]
- 1 M aqueous sodium hydroxide solution 1.2 mL
- methanol 2 mL
- the reaction mixture was neutralized with 1 M hydrochloric acid and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (174 mg).
- Trimethyl borate (0.600 mL) was added to the reaction mixture, and the resulting mixture was stirred at 0 ° C. for 30 minutes.
- 8 M aqueous sodium hydroxide solution (0.719 mL) and 30% aqueous hydrogen peroxide (2 mL)
- the reaction mixture was acidified with 1 M hydrochloric acid, and extracted with ethyl acetate. The obtained organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate.
- the residue obtained by evaporating the solvent under reduced pressure was acidified with 0.5 M hydrochloric acid, washed with ethyl acetate, and the aqueous layer was basified with saturated sodium bicarbonate. Ethyl acetate (10 mL) and acetic anhydride (10 mL) were added to this aqueous layer and stirred for 30 minutes. The reaction mixture was extracted with ethyl acetate and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.11 g).
- Example 26 N- ⁇ (1S) -2-[(trans-4- ⁇ [4- (cyclopropylmethoxy) -3-fluorobenzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [4- (cyclopropylmethoxy ) -3-Fluorophenyl] methanol was used to obtain the title compound by a method similar to or similar to Step A and Step F of Example 2.
- Example 27 N- ⁇ (1S) -2-[(trans-4- ⁇ [3-chloro-4- (cyclopropylmethoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [3-chloro-4- The title compound was obtained in the same manner as in Step A and Step F of Example 2 or a method analogous thereto using (cyclopropylmethoxy) phenyl] methanol.
- Example 30 N-[(1S) -2- (3- ⁇ [4- (cyclopropylmethoxy) benzyl] oxy ⁇ phenoxy) -1-methylethyl] acetamide Step A of Example 22 with 3- (benzyloxy) phenol The title compound was obtained in the same manner as in Step B of Example 7, Step C of Example 1 and Step E of Example 1, or a method analogous thereto.
- Example 32 N-[(1S) -2- (4- ⁇ [3- (cyclopropylmethoxy) benzyl] oxy ⁇ phenoxy) -1-methylethyl] acetamide [3- (cyclopropylmethoxy) phenyl] methanol and N-[( 1S) -2- (4-Hydroxyphenoxy) -1-methylethyl] acetamide was used to give the title compound by the same method as in Step E of Example 1 or a method analogous thereto.
- Example 33 N-[(1S) -3- (4- ⁇ [3- (cyclopropylmethoxy) benzyl] oxy ⁇ phenyl) -1-methylpropyl] acetamide [3- (cyclopropylmethoxy) phenyl] methanol and N-[( 1S) -3- (4-Hydroxyphenyl) -1-methylpropyl] acetamide was used to give the title compound in the same manner as in Step E of Example 1 or a method analogous thereto.
- Example 40a N-[(1S) -2- ⁇ [trans-4-( ⁇ 2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl ⁇ oxy) cyclohexyl] oxy ⁇ -1-methylethyl] acetamide Optically active form of
- Example 41 N-[(1S) -2- ⁇ [trans-4-( ⁇ 4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl ⁇ oxy) cyclohexyl] oxy ⁇ -1-methylethyl] acetamide N-[(1S) -2-( ⁇ trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl ⁇ oxy) -1-methylethyl] acetamide (400 mg) in DMF (5 mL) To the mixture were added potassium carbonate (489 mg) and (2,2-difluorocyclopropyl) methyl methanesulfonate (658 mL), and the mixture was heated and stirred at 60 ° C.
- Example 42 N-[(1S) -2- ⁇ [trans-4-( ⁇ 4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl ⁇ oxy) cyclohexyl] oxy ⁇ -1-methylethyl] acetamide
- the optically active form of N-[(1S) -2- ⁇ [trans-4-( ⁇ 4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl ⁇ oxy) cyclohexyl] oxy ⁇ -1- Methylethyl] acetamide diastereomer mixture (430 mg) was analyzed by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID ⁇ 500 mmL, Daicel Chemical Industries, mobile phase: hexane / 2-propanol 70: 30).
- Example 43 N-[(1S) -2- ⁇ [trans-4-( ⁇ 4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl ⁇ oxy) cyclohexyl] oxy ⁇ -1-methylethyl] acetamide
- the optically active form of N-[(1S) -2- ⁇ [trans-4-( ⁇ 4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl ⁇ oxy) cyclohexyl] oxy ⁇ -1- Methylethyl] acetamide diastereomer mixture (430 mg) was analyzed by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID ⁇ 500 mmL, Daicel Chemical Industries, mobile phase: hexane / 2-propanol 70: 30).
- Example 45 N-[(1S) -2- ⁇ [trans-4-( ⁇ 4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl ⁇ oxy) cyclohexyl] oxy ⁇ -1-methylethyl
- Example 46 N-[(1S) -2- ⁇ [trans-4-( ⁇ 4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl ⁇ oxy) cyclohexyl] oxy ⁇ -1-methylethyl
- Example 47 N- ⁇ (1S) -2-[(trans-4- ⁇ [5- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide 2-fluoro-5-hydroxy
- the title compound was obtained in the same manner as in Step 15 of Example 15, Step B of Example 15, Step D of Example 36 and Step B of Example 7 using benzoic acid and bromomethylcyclopropane.
- Example 50 N- ⁇ (1S) -2-[(trans-4- ⁇ [4- (cyclopropylmethoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [4- (cyclopropylmethoxy) phenyl] methanol
- the title compound was obtained in the same manner as in Steps A and F of Example 2.
- Example 51 N- ⁇ (1S) -2-[(trans-4- ⁇ [6- (cyclopropylmethoxy) -4-fluoropyridin-3-yl] methoxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [6- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -4-fluoropyridin-3-yl] methanol.
- Example 52 N- ⁇ (1S) -2-[(trans-4- ⁇ [4- (cyclopropylmethoxy) -2,6-difluorobenzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [4- (cyclo The title compound was obtained in the same manner as in Steps A and F of Example 2 using (propylmethoxy) -2,6-difluorophenyl] methanol.
- Example 53 N- ⁇ (1S) -2-[(trans-4- ⁇ [6- (cyclopropylmethoxy) -4-methylpyridin-3-yl] methoxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [6- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -4-methylpyridin-3-yl] methanol.
- Example 54 Optically active form of methyl ⁇ 2-[(trans-4- ⁇ [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ carbamate
- Example 56 N- ⁇ (1S) -2-[(trans-4- ⁇ [2-cyano-4- (cyclopropylmethoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide 2- (bromomethyl) -5
- the title compound was obtained in the same manner as in Step F of Example 2 using-(cyclopropylmethoxy) benzonitrile.
- Example 57 N-[(1S) -2-( ⁇ trans-4-[(2-chloro-4-ethoxybenzyl) oxy] cyclohexyl ⁇ oxy) -1-methylethyl] acetamide 1- (bromomethyl) -2-chloro-4
- the title compound was obtained in the same manner as in Step F of Example 2 using -ethoxybenzene.
- Example 58 N- ⁇ (1S) -2-[(trans-4- ⁇ [2-chloro-4- (1-methylethoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [2-chloro-4 The title compound was obtained in the same manner as in Steps A and F of Example 2 using-(1-methylethoxy) phenyl] methanol.
- Example 59 N- ⁇ (1S) -2-[(trans-4- ⁇ [5- (cyclopropylmethoxy) -3-fluoropyridin-2-yl] methoxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [5- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -3-fluoropyridin-2-yl] methanol.
- Example 61 N- ⁇ (1S) -2-[(trans-4- ⁇ [4- (cyclopropylmethoxy) -2-methylbenzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [4- (cyclopropylmethoxy ) -2-Methylphenyl] methanol was used to obtain the title compound in the same manner as in Steps A and F of Example 2.
- Example 62 N- ⁇ (1S) -2-[(trans-4- ⁇ [4- (cyclopropylmethoxy) -3- (trifluoromethoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide [4- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -3- (trifluoromethoxy) phenyl] methanol.
- Example 64 N-[(1S) -3- (trans-4- ⁇ [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ cyclohexyl) -1-methylpropyl] acetamide [4- (cyclopropylmethoxy) -2 The title compound was obtained in the same manner as in Step D of Example 34 using [-fluorophenyl] methanol.
- Example 68 N- ⁇ (1S) -2-[(trans-4- ⁇ [2-fluoro-4- (2-methylpropoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide 1-bromo-2- The title compound was obtained in the same manner as in Step D of Example 35 using methylpropane.
- Example 70 N- ⁇ (1S) -2-[(cis-4- ⁇ [3- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide 2-fluoro-3-hydroxy After performing the same operation as in Step C of Example 34 using benzaldehyde and bromomethylcyclopropane, N- ⁇ (1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl ⁇ The title compound was obtained in the same manner as in Step D of Example 36 using acetamide.
- Example 71 N- ⁇ (1S) -2-[(cis-4- ⁇ [5- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide (5- (cyclopropylmethoxy ) -2-Fluorophenyl) methanol and N- ⁇ (1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl ⁇ acetamide as in step D of Example 36 Gave the title compound.
- Example 75 N- ⁇ (1S) -2-[(trans-4- ⁇ [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ -4-methylcyclohexyl) oxy] -1-methylethyl ⁇ acetamide N- ⁇ (1S) -2-[(4-Hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl ⁇ acetamide was used to obtain N- ⁇ (1S) -2 obtained by the same method as in Step B of Example 3.
- Example 76 N- ⁇ (1S) -2-[(cis-4- ⁇ [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy ⁇ -4-methylcyclohexyl) oxy] -1-methylethyl ⁇ acetamide N- ⁇ (1S) -2-[(4-Hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl ⁇ acetamide was used to obtain N- ⁇ (1S) -2 obtained by the same method as in Step B of Example 3.
- reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine.
- the organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- the resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (71.5 mg) as a white solid.
- Triethylamine (1.90 mL) and methanesulfonyl chloride (0.791 mL) were added to a THF (10 mL) solution of cyclopropylethanol (587 mg) and stirred at room temperature for 10 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
- Example 80 N- ⁇ (1S) -2-[(trans-4- ⁇ [3- (cyclobutylmethoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide (bromomethyl) cyclobutane
- the title compound was obtained in the same manner as in Step B.
- Example 82 N- ⁇ (1S) -2-[(trans-4- ⁇ [4- (2,2-difluoropropoxy) -2-fluorobenzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide N- ⁇ ( 1S) -2-[(trans-4- ⁇ [2-fluoro-4- (2-oxopropoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇ acetamide (177 mg), bis (2-methoxy A mixture of ethyl) aminosulfur trifluoride (297 mg) and toluene (20 mL) was stirred at room temperature for 1 hour, followed by 80 ° C.
- Example 83 N-[(1S) -2- ⁇ [trans-4-( ⁇ 4-[(3,3-difluorocyclobutyl) methoxy] -2-fluorobenzyl ⁇ oxy) cyclohexyl] oxy ⁇ -1-methylethyl] acetamide N-[(1S) -2-( ⁇ trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl ⁇ oxy) -1-methylethyl] acetamide and (3,3-difluorocyclobutyl) methyl The title compound was obtained in the same manner as in Step E of Example 7 using methanesulfonate.
- reaction mixture was passed through silica gel, and the organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- a THF solution 0.5 M, 30.9 mL
- 2-methylallylmagnesium chloride was added to a THF (40 mL) solution of the obtained residue, and the mixture was stirred at room temperature for 20 minutes.
- To the reaction mixture was added aqueous ammonium chloride solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure.
- Example 87 N-[(1S) -2-( ⁇ trans-4-[(2-fluoro-4-propoxybenzyl) oxy] cyclohexyl ⁇ oxy) -1-methylethyl] acetamide N-[(1S) -2-( ⁇ trans-4-[(2-Fluoro-4-hydroxybenzyl) oxy] cyclohexyl ⁇ oxy) -1-methylethyl] acetamide and 1-iodopropane and the title compound by a method similar to Example 7, step E. Obtained.
- Example 88 N-[(1S) -2-( ⁇ trans-4-[(4-butoxy-2-fluorobenzyl) oxy] cyclohexyl ⁇ oxy) -1-methylethyl] acetamide N-[(1S) -2-( ⁇ The title compound was obtained in the same manner as in Step E of Example 7 using trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl ⁇ oxy) -1-methylethyl] acetamide and 1-iodobutane. It was.
- Example 90 N- ⁇ (1S) -2-[(trans-4- ⁇ [2-fluoro-4- (2,2,3,3-tetrafluoropropoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methylethyl ⁇
- the title compound was obtained in the same manner as in Step D of Example 35 using acetamide 2,2,3,3-tetrafluoropropyl trifluoromethanesulfonate.
- Example 91 N- ⁇ (1S) -2-[(trans-4- ⁇ [2-fluoro-4- (2,2,3,3,3-pentafluoropropoxy) benzyl] oxy ⁇ cyclohexyl) oxy] -1-methyl
- the title compound was obtained in the same manner as in Step D of Example 35 using ethyl ⁇ acetamide 2,2,3,3,3-pentafluoropropyl trifluoromethanesulfonate.
- the resulting residue was mixed with THF (10 mL), methanol (10 mL), and 1 M aqueous sodium hydroxide solution (10 mL), and the resulting mixture was stirred at room temperature for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (498 mg) as a colorless oily compound.
- N- (2- ⁇ [trans-3- (benzyloxy) cyclobutyl] oxy ⁇ -1-methylethyl) acetamide (65.5 mg, mixture with impurities), 20% palladium hydroxide / carbon (50%
- a mixture of water (65 mg) and ethanol (2 mL) was stirred at room temperature for 4 hours.
- the catalyst was removed by filtration, and the obtained filtrate was concentrated under reduced pressure.
- the obtained residue was 1- (bromomethyl) -4- (cyclopropylmethoxy) -2-fluorobenzene (306 mg), tetrabutylammonium hydrogen sulfate (8.02 mg), 50% aqueous sodium hydroxide solution (2 mL), and Mix with toluene (4 mL) and stir the resulting mixture at 100 ° C. for 3 days. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- Tables 1 to 13 show the measured values of MS or NMR spectrum data in addition to the compound names and structural formulas of the example compounds.
- the measured value of MS usually indicates the measured value in the positive mode (ESI +), and the measured value in the negative mode (ESI-) is indicated with [MH] - . Further, in the positive mode (ESI +), when a fragment peak in which sodium ion (+ Na) was added to the molecular ion peak was observed, [M + Na] + was also written.
- Test example 1 The ACC2 inhibitory action of the compound of the present invention was evaluated by the following method. (1) Cloning of human ACC2 gene and preparation of recombinant baculovirus The human ACC2 gene was cloned by PCR using Primer 1 and Primer 2 shown below using a human skeletal muscle cDNA library (Clontech) as a template. Primer 1 and Primer 2 were prepared by adding SalI and XbaI restriction enzyme recognition sequences based on the information on the base sequence of the human ACC2 gene (Genbank Accession U89344).
- Primer 1 5'-AAAAGTCGACCCACCATGGTCTTGCTTCTTTGTCTATCTTG-3 '(SEQ ID NO: 1)
- Primer 2 5'-TTTTTCTAGATCAGGTAGAGGCCGGGCTGTCCATG-3 '(SEQ ID NO: 2)
- PCR was performed using Pyrobest DNA polymerase (Takara Bio Inc.). The obtained PCR product was cloned into pT7 Blue vector (Novagen), and after confirming the base sequence, digested with restriction enzymes SalI and XbaI.
- the obtained DNA fragment was inserted into pFAST-BacHTa (Invitrogen) digested with restriction enzymes SalI and XbaI to prepare an expression plasmid ACC2 / pFAST-BacHTa.
- pFAST-BacHTa Invitrogen digested with restriction enzymes SalI and XbaI to prepare an expression plasmid ACC2 / pFAST-BacHTa.
- PCR using Primer 3 (added with a SalI restriction enzyme recognition sequence) and Primer 4 prepared from information on the base sequence of the human ACC2 gene (Genbank Accession U89344) and ACC2 from which the mitochondrial translocation sequence was removed A plasmid for expression was prepared.
- Primer 3 5'-CCAGGTCGACCCGCCAACGGGACTGGGACACAAGG-3 '(SEQ ID NO: 3)
- Primer 4 5'-CGCACTCTCAGTTTCCCGGATTCCC-3 '(SEQ ID NO: 4)
- PCR was performed using Pyrobest-DNA polymerase (Takara Bio Inc.). The obtained PCR product was cloned into pT7 Blue vector (Novagen). After confirming the nucleotide sequence, it was digested with restriction enzymes SalI and AflII.
- the obtained DNA fragment was inserted into ACC2 / pFAST-BacHTa digested with restriction enzymes SalI and AflII to prepare an expression plasmid ACC2mito7 / pFAST-BacHTa.
- an expression plasmid ACC2mito7 / pFAST-BacHTa was prepared using the expression plasmids ACC2mito7 / pFAST-BacHTa and BAC-TO-BAC Baculovirus Expression System (Invitrogen), a recombinant baculovirus virus stock BAC-ACC2 (N terminal deletion (hereinafter referred to as Nd)) was prepared.
- ACC2 (Nd) protein SF-9 cells were cultured in insect cell medium (5% fetal bovine serum (Trace), 50 mg / L Gentamicin (Wako Pure Chemical Industries), 0.1% Pluronic F- Sf-900IISFM medium containing 68 (Invitrogen Corp.) (Invitrogen Corp.)) 10 L, seeded at 1.0 ⁇ 10 6 cells / mL, using a Wave bioreactor (GE Healthcare Corp.), 27 ° C., 20 rpm The shaking culture was performed at a rocking angle of 10 degrees and an oxygen concentration of 30%. Recombinant baculovirus BAC-ACC2 (Nd) was added on the second day of culture and cultured for 3 days.
- insect cell medium 5% fetal bovine serum (Trace), 50 mg / L Gentamicin (Wako Pure Chemical Industries), 0.1% Pluronic F- Sf-900IISFM medium containing 68 (Invitrogen Corp.) (Invitrogen Corp.)) 10 L,
- the culture solution was centrifuged at 1000 ⁇ g for 10 minutes to obtain virus-infected cells.
- the cells were washed with phosphate physiological buffer (Invitrogen) and centrifuged under the same conditions, and the obtained cells were stored frozen at -80 ° C. After thawing the cryopreserved cells in ice, 25 mM HEPES buffer containing 10% Glycerol, 0.3 M NaCl, 1 mM EDTA, 25 mM Sodium ⁇ -Glycerophosphate, and 1 mM Sodium Orthovanadate with Complete Protease Inhibitor (Roche) added ( It was suspended in 900 mL of pH 7.5).
- phosphate physiological buffer Invitrogen
- the obtained suspension was homogenized three times using a Polytron homogenizer (Kinematica) at 20,000 rpm for 30 seconds.
- the obtained cell lysate was clarified by centrifugation at 186,000 ⁇ g for 60 minutes.
- 5 mL of AF-chelate 650M Ni chelate carrier (Tosoh Corporation) was added and rotated at 4 ° C. for 1 hour.
- the support was transferred to an open column by centrifugation at 1000 ⁇ g for 5 minutes.
- ACC2 (Nd) (1.1 mg / ml) obtained in (2) above was used as a buffer for enzyme reaction (50 mM HEPES (pH 7.5), 10 mM MgCl 2 , 10 mM Tripottasium Citrate, 2 After dilution to a concentration of 6.4 ⁇ g / ml with mM Dithiothreitol, 0.75 mg / ml Fatty acid free BSA), 10 ⁇ l was added to each well of a 384 well assay plate (Nunc 265196).
- the reaction was stopped by adding 5 ⁇ l of malachite green solution to each reaction solution thus obtained and stirring.
- the obtained reaction solution was allowed to stand at room temperature for 20 minutes, and then the absorbance (620 nm) was measured using wallac1420 (Perkin Elmer).
- the ACC2 inhibition rate (%) was determined according to the following formula. (1 ⁇ (absorbance of test compound added group ⁇ absorbance of control group) ⁇ (absorbance of test compound non-added group ⁇ absorbance of control group)) ⁇ 100
- IC 50 value the inhibitory activity
- the compound concentration showing a 50% inhibition rate was calculated from the dose-response curve using XLfit.
- a compound with an inhibition rate of less than 50% at a compound concentration of 10 ⁇ M was expressed as> 10 ⁇ M.
- Table 14 and Table 15 show the inhibition rate (%) against ACC2 at 10 ⁇ M of the test compound.
- Test example 2 The ACC1 inhibitory action of the compound of the present invention was evaluated by the following method. (1) Cloning of human ACC1 gene and preparation of recombinant baculovirus The human ACC1 gene was cloned by PCR using Primer 1 and Primer 2 shown below using a human liver cDNA library (Clontech) as a template. Primer1 and Primer2 were prepared by adding SalI and NotI restriction enzyme recognition sequences based on the information on the base sequence of the human ACC1 gene (Genbank Accession U19822).
- Primer 1 5'-AAAAGTCGACCCACCATGGATGAACCTTCTCCCTTGGCCC-3 '(SEQ ID NO: 5)
- Primer 2 5'-AAAAGCGGCCGCCTACGTAGAAGGGGAGTCCATAGTG-3 '(SEQ ID NO: 6)
- PCR was performed using Pyrobest DNA polymerase (Takara Bio Inc.). The obtained PCR product was cloned into pT7 Blue vector (Novagen), and after confirming the nucleotide sequence, it was digested with restriction enzymes SalI and NotI.
- the obtained DNA fragment was inserted into pFAST-BacHTc (Invitrogen) digested with restriction enzymes SalI and NotI to prepare an expression plasmid ACC1 / pFAST-BacHTc.
- pFAST-BacHTc Invitrogen
- restriction enzymes SalI and NotI restriction enzymes SalI and NotI
- a recombinant baculovirus virus stock BAC-ACC1 was prepared.
- the cells were washed with phosphate physiological buffer (Invitrogen) and centrifuged under the same conditions, and the obtained cells were stored frozen at -80 ° C. After thawing the cryopreserved cells in ice, 25 mM HEPES buffer solution (pH 7. 5) Suspended in 100 mL. The obtained suspension was homogenized three times using a Polytron homogenizer (Kinematica) at 20,000 rpm for 30 seconds. The obtained cell lysate was clarified by centrifugation at 185700 ⁇ g for 50 minutes, followed by filtration using a 0.45 ⁇ m filter.
- phosphate physiological buffer Invitrogen
- 25 mM HEPES buffer solution pH 7.5
- the obtained suspension was homogenized three times using a Polytron homogenizer (Kinematica) at 20,000 rpm for 30 seconds.
- the obtained cell lysate was clarified by centrifugation at 185700 ⁇ g for 50 minutes, followed by filtration using
- the filtrate was passed through a column packed with 12 mL of Ni-NTA Super Flow Gel (Qiagen) at a flow rate of about 5 mL / min.
- the column was washed with buffer A (50 mM HEPES (pH 7.5) containing 0.3 M NaCl), further washed with buffer A containing 20 mM Imidazole, and then eluted with buffer A containing 100 mM Imidazole.
- the eluate was concentrated with Vivapine 20 (Viva Science) having a molecular weight cut off of 30K.
- the resulting concentrated solution was dialyzed using 358 mL of Sephadex G-25 (Amersham Biosciences) equilibrated with 50 mM HEPES (pH 7.5) containing 10 mM MgCl 2 , 2 mM Dithiothreitol, 10 mM Tripotassium Citrate, 0.3 M NaCl.
- the dialyzed internal solution was concentrated with Vivaspin 20 (Vivascience) having a molecular weight cut off of 30K, and then the concentrated solution was filtered with a 0.22 ⁇ m filter to obtain ACC1.
- the obtained ACC1 was stored frozen at ⁇ 80 ° C.
- test compound addition group 5 ⁇ l each of a test compound dissolved in dimethyl sulfoxide (DMSO) diluted with an enzyme reaction buffer was added to each well and incubated at 30 ° C. for 60 minutes.
- DMSO dimethyl sulfoxide
- a substrate solution 50 mM KHCO 3 , 200 ⁇ M ATP, 200 ⁇ M Acetyl-CoA
- the reaction was stopped by adding 5 ⁇ l of malachite green solution to each reaction solution thus obtained and stirring.
- the obtained reaction solution was allowed to stand at room temperature for 20 minutes, and then the absorbance (620 nm) was measured using wallac1420 (Perkin Elmer).
- the ACC1 inhibition rate (%) and the inhibitory activity (IC 50 value) were determined.
- Table 16 shows the ACC2 inhibitory activity and ACC1 inhibitory activity (IC 50 value) of the test compounds.
- Test example 3 Male F344 / Jcl rats (CLEA Japan, Tokyo) that arrived at 5 weeks old were fed Western diet (D12079B, Research Diets) and acclimatized for more than 3 weeks, and then tested for malonyl-CoA content at 8-10 weeks of age. Used for. In the experiment for measuring the malonyl-CoA content, acclimation was administered 2-5 days before the test compound administration, and the groups were divided so that there was no difference between the administration groups based on the body weight of the previous week.
- the test compound was prepared in advance as a 0.5% Methyl Cellulose (Wako) suspension at a dose of 5 mL / kg, and PM5: 00 based on the body weight (measured at AM8: 00-11: 00) on the day of test compound administration.
- the thigh muscle was quickly removed, put into 80-150 mg strips, placed in an Eppendorf tube, and quickly frozen in liquid nitrogen to measure malonyl-CoA content.
- the tissue was stored at ⁇ 80 ° C. until use.
- MBP-DBAA di-n-butylammonium acetate aqueous solution
- the total amount of the extract was centrifuged (13,000 rpm, 2 min), and the supernatant was added to a solid phase extraction cartridge (OASIS HLB 1 cc / 30 mg, WAT05882, Waters) for solid phase extraction.
- the solution was reduced to 5%, and the solution was fed under the same conditions until the analysis end time (10 minutes).
- the eluate having an analysis time of 2.0 to 6.0 minutes was introduced into MS / MS.
- the analysis was performed at a column temperature of 40 ° C. and a sample injection volume of 10 ⁇ L.
- the mass spectrometer used was API5000 (AB Sciex), turbo ion spray was used as the ionization mode, and ions were detected by selected reaction monitoring (SRM) in the negative ion mode. Ion spray was performed using zero air and the voltage was 4.5 kV. Nitrogen was used for collision-induced decomposition of ions.
- a precursor ion and a fragment ion were set to malonyl CoA; m / z 852.0 ⁇ m / z 808.0 and [ 13 C 3 ] -malonyl CoA; m / z 855.0 ⁇ m / z 810.0 Da, respectively.
- the internal area ([ 13 C 3 ] -malonyl-CoA) and the peak area ratio of malonyl-CoA were used.
- All values are shown as mean ⁇ standard deviation, and Welch test was used for statistical analysis. The results are shown in Table 17.
- Formulation Example 1 (Manufacture of capsules) 1) 30 mg of the compound of Example 1 2) Fine powder cellulose 10 mg 3) Lactose 19 mg 4) Magnesium stearate 1 mg 60 mg total 1), 2), 3) and 4) are mixed and filled into gelatin capsules.
- Formulation Example 2 Manufacture of tablets
- the compound of the present invention has an ACC (acetyl-CoA carboxylase) inhibitory action and prevents obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complications, metabolic syndrome, sarcopenia, cancer, etc. Useful for treatment.
- ACC acetyl-CoA carboxylase
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Heart & Thoracic Surgery (AREA)
- Diabetes (AREA)
- Cardiology (AREA)
- Ophthalmology & Optometry (AREA)
- Physical Education & Sports Medicine (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Neurology (AREA)
- Child & Adolescent Psychology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Biomedical Technology (AREA)
- Vascular Medicine (AREA)
- Neurosurgery (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention is a compound represented by formula (I) (in the formula, each of the reference numerals is as recited in the specification) or a salt thereof.
Description
本発明は、アセチル-CoAカルボキシラーゼ(本明細書中、ACCと略記することがある)阻害作用を有し、肥満症、糖尿病、高血圧症、高脂血症、心不全、糖尿病性合併症、メタボリックシンドローム、筋肉減少症、癌等の予防・治療に有用な単環化合物に関する。
The present invention has an inhibitory action on acetyl-CoA carboxylase (which may be abbreviated as ACC in the present specification), and is obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complications, metabolic syndrome Further, the present invention relates to a monocyclic compound useful for prevention / treatment of sarcopenia, cancer and the like.
[発明の背景]
ACCは、アセチル-CoAをマロニル-CoAに変換する酵素であり、脂肪酸代謝または合成での律速反応を触媒する。ACC触媒反応の産物であるマロニル-CoAは、カルニチンパルミトイルトランスフェラーゼ-1(CPT-1)のフィードバック阻害により、ミトコンドリアの脂肪酸酸化を阻害する。従って、ACCは肝臓と骨格筋での炭水化物と脂肪酸利用のバランスを制御する際、また、肝臓、骨格筋、脂肪組織でのインスリン感受性を制御する際に鍵となる役割を演ずる。 [Background of the invention]
ACC is an enzyme that converts acetyl-CoA to malonyl-CoA and catalyzes the rate-limiting reaction in fatty acid metabolism or synthesis. Malonyl-CoA, the product of the ACC catalytic reaction, inhibits mitochondrial fatty acid oxidation by feedback inhibition of carnitine palmitoyltransferase-1 (CPT-1). Thus, ACC plays a key role in controlling the balance of carbohydrate and fatty acid utilization in the liver and skeletal muscle, and in controlling insulin sensitivity in the liver, skeletal muscle and adipose tissue.
ACCは、アセチル-CoAをマロニル-CoAに変換する酵素であり、脂肪酸代謝または合成での律速反応を触媒する。ACC触媒反応の産物であるマロニル-CoAは、カルニチンパルミトイルトランスフェラーゼ-1(CPT-1)のフィードバック阻害により、ミトコンドリアの脂肪酸酸化を阻害する。従って、ACCは肝臓と骨格筋での炭水化物と脂肪酸利用のバランスを制御する際、また、肝臓、骨格筋、脂肪組織でのインスリン感受性を制御する際に鍵となる役割を演ずる。 [Background of the invention]
ACC is an enzyme that converts acetyl-CoA to malonyl-CoA and catalyzes the rate-limiting reaction in fatty acid metabolism or synthesis. Malonyl-CoA, the product of the ACC catalytic reaction, inhibits mitochondrial fatty acid oxidation by feedback inhibition of carnitine palmitoyltransferase-1 (CPT-1). Thus, ACC plays a key role in controlling the balance of carbohydrate and fatty acid utilization in the liver and skeletal muscle, and in controlling insulin sensitivity in the liver, skeletal muscle and adipose tissue.
ACC阻害によるマロニル-CoAレベルの低下は、脂肪酸酸化の増加、脂肪酸合成の抑制、肝臓におけるトリグリセライド(TG)豊富な(rich)リポ蛋白質(VLDL)の分泌低下、膵臓におけるインスリン分泌の制御、さらには肝臓、骨格筋、脂肪組織でのインスリン感受性の改善を促し得る。
Decreased malonyl-CoA levels due to ACC inhibition include increased fatty acid oxidation, inhibition of fatty acid synthesis, decreased triglyceride (TG) rich lipoprotein (VLDL) secretion in the liver, regulation of insulin secretion in the pancreas, and May improve insulin sensitivity in liver, skeletal muscle and adipose tissue.
また、脂肪酸酸化を促進し、脂肪酸のデノボ(de novo)合成を抑制することによって、ACC阻害作用を有する化合物の長期投与は、低脂肪食事を摂取する肥満被験体において、肝臓と脂肪組織のTG含量を大きく減少させ、体脂肪を選択的に減少させ得る。
In addition, long-term administration of a compound having an ACC inhibitory action by promoting fatty acid oxidation and suppressing fatty acid de novo synthesis is a TG of liver and adipose tissue in obese subjects ingesting a low-fat diet. The content can be greatly reduced and body fat can be selectively reduced.
従って、ACC阻害作用を有する化合物は、メタボリックシンドローム、肥満症、高血圧症、糖尿病、アテローム性動脈硬化と関連する心臓血管系疾患等の予防および治療に極めて有用である。
Therefore, a compound having an ACC inhibitory action is extremely useful for the prevention and treatment of metabolic syndrome, obesity, hypertension, diabetes, cardiovascular diseases related to atherosclerosis, and the like.
一方、WO2010/003624(特許文献1)には、以下の化合物
On the other hand, WO2010 / 003624 (Patent Document 1) includes the following compounds:

[式中、
A、B、DおよびEは、独立して、C(R5)(式中、R5はClまたはFを示す)またはN(但し、A、B、DおよびEのうちの2以上はNではない。)を示し;
G、L、RおよびTは、独立して、=C(R6)-、-C(R6)(R7)-、=N-、-N(R8)-またはO(但し、G、L、RおよびTのうちの2以上は、=N-、-N(R8)-およびOのいずれでもない。)を示し;
(但し、A、B、D、E、G、L、R、Tおよびそれらに結合したC原子は同時にフェニルを形成しない)
Mは、=C-、-C(R9)-またはNを示し;
Wは、O、S、CH(R10)を示し;
Yは、(C2-C10)アルキレン(1または2個のCH2がO,S,N(R10a)等に置き換わってもよい)を示し;
R1は、(C1-C6)アルキレン-(C3-C12)シクロアルキル等を示し;
R2は、-COR4(式中、R4は、(C1-C6)アルキル等を示す)等を示し;
R3は、H等を示す]
がACC阻害作用を有する化合物として報告されている。 [Where:
A, B, D and E are independently C (R5) (wherein R5 represents Cl or F) or N (provided that two or more of A, B, D and E are not N). .);
G, L, R and T are independently ═C (R6) —, —C (R6) (R7) —, ═N—, —N (R8) — or O (provided that G, L, R And two or more of T and T represent ═N—, —N (R8) — or O.);
(However, A, B, D, E, G, L, R, T and C atoms bonded to them do not simultaneously form phenyl)
M represents = C-, -C (R9)-or N;
W represents O, S, CH (R10);
Y represents (C 2 -C 10 ) alkylene (1 or 2 CH 2 may be replaced by O, S, N (R10a), etc.);
R1 represents (C 1 -C 6 ) alkylene- (C 3 -C 12 ) cycloalkyl or the like;
R2 represents —COR4 (wherein R4 represents (C 1 -C 6 ) alkyl or the like);
R3 represents H or the like]
Has been reported as a compound having an ACC inhibitory action.
A、B、DおよびEは、独立して、C(R5)(式中、R5はClまたはFを示す)またはN(但し、A、B、DおよびEのうちの2以上はNではない。)を示し;
G、L、RおよびTは、独立して、=C(R6)-、-C(R6)(R7)-、=N-、-N(R8)-またはO(但し、G、L、RおよびTのうちの2以上は、=N-、-N(R8)-およびOのいずれでもない。)を示し;
(但し、A、B、D、E、G、L、R、Tおよびそれらに結合したC原子は同時にフェニルを形成しない)
Mは、=C-、-C(R9)-またはNを示し;
Wは、O、S、CH(R10)を示し;
Yは、(C2-C10)アルキレン(1または2個のCH2がO,S,N(R10a)等に置き換わってもよい)を示し;
R1は、(C1-C6)アルキレン-(C3-C12)シクロアルキル等を示し;
R2は、-COR4(式中、R4は、(C1-C6)アルキル等を示す)等を示し;
R3は、H等を示す]
がACC阻害作用を有する化合物として報告されている。 [Where:
A, B, D and E are independently C (R5) (wherein R5 represents Cl or F) or N (provided that two or more of A, B, D and E are not N). .);
G, L, R and T are independently ═C (R6) —, —C (R6) (R7) —, ═N—, —N (R8) — or O (provided that G, L, R And two or more of T and T represent ═N—, —N (R8) — or O.);
(However, A, B, D, E, G, L, R, T and C atoms bonded to them do not simultaneously form phenyl)
M represents = C-, -C (R9)-or N;
W represents O, S, CH (R10);
Y represents (C 2 -C 10 ) alkylene (1 or 2 CH 2 may be replaced by O, S, N (R10a), etc.);
R1 represents (C 1 -C 6 ) alkylene- (C 3 -C 12 ) cycloalkyl or the like;
R2 represents —COR4 (wherein R4 represents (C 1 -C 6 ) alkyl or the like);
R3 represents H or the like]
Has been reported as a compound having an ACC inhibitory action.
また、WO2008/079610(特許文献2)には、以下の化合物
In addition, WO2008 / 079610 (Patent Document 2) includes the following compounds:

[式中、
A1、A4およびA5は、独立して、N、C(R1)(R1は、H、アルキル、ハロゲンまたはハロアルキルを示す)を示し;
A2およびA3は、独立して、C(-L2-R2)(L2は、O等を、R2は、アルキル等を示す)を示し;
L1は、O、N(RX)、S、S(O)、S(O)2またはC(RyRz)mを示し;
mは、1、2または3を示し;
Aは、フェニルまたは単環のヘテロアリールを示し;
Xは、-O-(CRyRz)p-NRdRe等を示し;
Rdは、-C(O)アルキル等を示し;
Reは、H等を示す]
がACC阻害作用を有する化合物として報告されている。 [Where:
A 1 , A 4 and A 5 independently represent N, C (R 1 ) (R 1 represents H, alkyl, halogen or haloalkyl);
A 2 and A 3 independently represent C (—L 2 —R 2 ) (L 2 represents O or the like, R 2 represents alkyl or the like);
L 1 represents O, N (R X ), S, S (O), S (O) 2 or C (R y R z ) m;
m represents 1, 2 or 3;
A represents phenyl or monocyclic heteroaryl;
X represents —O— (CR y R z ) p—NR d R e or the like;
R d represents —C (O) alkyl or the like;
Re represents H or the like]
Has been reported as a compound having an ACC inhibitory action.
A1、A4およびA5は、独立して、N、C(R1)(R1は、H、アルキル、ハロゲンまたはハロアルキルを示す)を示し;
A2およびA3は、独立して、C(-L2-R2)(L2は、O等を、R2は、アルキル等を示す)を示し;
L1は、O、N(RX)、S、S(O)、S(O)2またはC(RyRz)mを示し;
mは、1、2または3を示し;
Aは、フェニルまたは単環のヘテロアリールを示し;
Xは、-O-(CRyRz)p-NRdRe等を示し;
Rdは、-C(O)アルキル等を示し;
Reは、H等を示す]
がACC阻害作用を有する化合物として報告されている。 [Where:
A 1 , A 4 and A 5 independently represent N, C (R 1 ) (R 1 represents H, alkyl, halogen or haloalkyl);
A 2 and A 3 independently represent C (—L 2 —R 2 ) (L 2 represents O or the like, R 2 represents alkyl or the like);
L 1 represents O, N (R X ), S, S (O), S (O) 2 or C (R y R z ) m;
m represents 1, 2 or 3;
A represents phenyl or monocyclic heteroaryl;
X represents —O— (CR y R z ) p—NR d R e or the like;
R d represents —C (O) alkyl or the like;
Re represents H or the like]
Has been reported as a compound having an ACC inhibitory action.
また、Journal of Medicinal Chemistry,2010,53,8679-8687(非特許文献1)には、以下の化合物
In addition, Journal of Medicinal Chemistry, 2010, 53, 8679-8687 (Non-patent Document 1) includes the following compounds:



で表される化合物等がACC阻害作用を有する化合物として報告されている。
Have been reported as compounds having an ACC inhibitory action.
さらに、WO2010/050445(特許文献3)には、以下の化合物
Furthermore, WO2010 / 050445 (Patent Document 3) includes the following compounds:

[式中、
Aは、アシル基または置換されていてもよい5ないし6員芳香環基を;
環Mは、さらに置換されていてもよく、縮合していてもよい5ないし7員環を;
環Pおよび環Qは、
(1)環Pはさらに置換されていてもよい5員複素環を示し、環Qはさらに置換されていてもよい6員環を示し、かつ、環Pと環Qとで縮合してさらに置換されていてもよい二環性芳香族複素環を形成するか、または、
(2)環Pはさらに置換されていてもよい5員非芳香環を示し、環Qはさらに置換されていてもよい6員芳香環を示し、かつ、環Pと環Qとで縮合してさらに置換されていてもよい二環性非芳香環を形成し;
R1は、置換されていてもよいC1-6アルキル基または置換されていてもよいC3-6シクロアルキル基を;
L1およびL2は、
(1)独立して、置換されていてもよいメチレン、O、S、SOまたはSO2を示すか、または、
(2)L1とL2とで、置換されていてもよいビニレン、またはエチニレンを形成する。但し、以下の化合物を除く:
(a)Aが、α-アミノイソブチロイル基である化合物;および
(b)Aが、式:-CO-(CH2)3-COORA1(式中、RA1は、水素原子またはC1-6アルキル基を示す)で表される基、または式:-CO-NRA2-CRA3RA4-CRA5RA6-COORA7(式中、RA2、RA3、RA4、RA5およびRA7は、独立して、水素原子またはC1-6アルキル基を示し;RA6は、水素原子、C1-6アルキル基またはヒドロキシ基を示す)で表される基で置換された5または6員芳香環基である化合物。]
がACC阻害作用を有する化合物として報告されている。 [Where:
A represents an acyl group or an optionally substituted 5- to 6-membered aromatic ring group;
Ring M represents a 5- to 7-membered ring which may be further substituted and optionally condensed;
Ring P and Ring Q are
(1) Ring P represents a 5-membered heterocyclic ring which may be further substituted, Ring Q represents a 6-membered ring which may be further substituted, and further condensed by condensing ring P and ring Q Forming a bicyclic aromatic heterocycle which may be
(2) Ring P represents an optionally substituted 5-membered non-aromatic ring, Ring Q represents an optionally further substituted 6-membered aromatic ring, and ring P and ring Q are condensed Forming an optionally substituted bicyclic non-aromatic ring;
R 1 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 3-6 cycloalkyl group;
L 1 and L 2 are
(1) independently represents optionally substituted methylene, O, S, SO or SO 2 , or
(2) L 1 and L 2 form optionally substituted vinylene or ethynylene. Except for the following compounds:
(A) a compound in which A is an α-aminoisobutyroyl group; and (b) A is represented by the formula: —CO— (CH 2 ) 3 —COOR A1 wherein R A1 is a hydrogen atom or C 1 -6 groups represented by alkyl group shows a) or formula: in -CO-NR A2 -CR A3 R A4 -CR A5 R A6 -COOR A7 ( wherein, R A2, R A3, R A4, R A5 and R A7 independently represents a hydrogen atom or a C 1-6 alkyl group; R A6 represents a hydrogen atom, a C 1-6 alkyl group or a hydroxy group) A compound which is a 6-membered aromatic ring group. ]
Has been reported as a compound having an ACC inhibitory action.
Aは、アシル基または置換されていてもよい5ないし6員芳香環基を;
環Mは、さらに置換されていてもよく、縮合していてもよい5ないし7員環を;
環Pおよび環Qは、
(1)環Pはさらに置換されていてもよい5員複素環を示し、環Qはさらに置換されていてもよい6員環を示し、かつ、環Pと環Qとで縮合してさらに置換されていてもよい二環性芳香族複素環を形成するか、または、
(2)環Pはさらに置換されていてもよい5員非芳香環を示し、環Qはさらに置換されていてもよい6員芳香環を示し、かつ、環Pと環Qとで縮合してさらに置換されていてもよい二環性非芳香環を形成し;
R1は、置換されていてもよいC1-6アルキル基または置換されていてもよいC3-6シクロアルキル基を;
L1およびL2は、
(1)独立して、置換されていてもよいメチレン、O、S、SOまたはSO2を示すか、または、
(2)L1とL2とで、置換されていてもよいビニレン、またはエチニレンを形成する。但し、以下の化合物を除く:
(a)Aが、α-アミノイソブチロイル基である化合物;および
(b)Aが、式:-CO-(CH2)3-COORA1(式中、RA1は、水素原子またはC1-6アルキル基を示す)で表される基、または式:-CO-NRA2-CRA3RA4-CRA5RA6-COORA7(式中、RA2、RA3、RA4、RA5およびRA7は、独立して、水素原子またはC1-6アルキル基を示し;RA6は、水素原子、C1-6アルキル基またはヒドロキシ基を示す)で表される基で置換された5または6員芳香環基である化合物。]
がACC阻害作用を有する化合物として報告されている。 [Where:
A represents an acyl group or an optionally substituted 5- to 6-membered aromatic ring group;
Ring M represents a 5- to 7-membered ring which may be further substituted and optionally condensed;
Ring P and Ring Q are
(1) Ring P represents a 5-membered heterocyclic ring which may be further substituted, Ring Q represents a 6-membered ring which may be further substituted, and further condensed by condensing ring P and ring Q Forming a bicyclic aromatic heterocycle which may be
(2) Ring P represents an optionally substituted 5-membered non-aromatic ring, Ring Q represents an optionally further substituted 6-membered aromatic ring, and ring P and ring Q are condensed Forming an optionally substituted bicyclic non-aromatic ring;
R 1 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 3-6 cycloalkyl group;
L 1 and L 2 are
(1) independently represents optionally substituted methylene, O, S, SO or SO 2 , or
(2) L 1 and L 2 form optionally substituted vinylene or ethynylene. Except for the following compounds:
(A) a compound in which A is an α-aminoisobutyroyl group; and (b) A is represented by the formula: —CO— (CH 2 ) 3 —COOR A1 wherein R A1 is a hydrogen atom or C 1 -6 groups represented by alkyl group shows a) or formula: in -CO-NR A2 -CR A3 R A4 -CR A5 R A6 -COOR A7 ( wherein, R A2, R A3, R A4, R A5 and R A7 independently represents a hydrogen atom or a C 1-6 alkyl group; R A6 represents a hydrogen atom, a C 1-6 alkyl group or a hydroxy group) A compound which is a 6-membered aromatic ring group. ]
Has been reported as a compound having an ACC inhibitory action.
さらに、WO03/022801(特許文献4)には、以下の化合物
Furthermore, WO03 / 022801 (Patent Document 4) includes the following compounds:

[式中、
Aは、H、低級アルキル、シクロアルキルまたは-NR3R4(R3およびR4は、独立して、H、低級アルキル等を示す)を示し;
Ar1は、アリーレンまたはヘテロアリーレンを示し;
Ar2は、置換されていてもよいアリール、置換されていてもよいアラルキル、置換されていてもよいヘテロアリール、または置換されていてもよいヘテロアリールアルキルを示し;
R1は、H、置換されていてもよい低級アルキル、または置換されていてもよい低級アルケニルを示し;
R2aは、 [Where:
A represents H, lower alkyl, cycloalkyl or —NR 3 R 4 (R 3 and R 4 independently represent H, lower alkyl, etc.);
Ar 1 represents arylene or heteroarylene;
Ar 2 represents an optionally substituted aryl, an optionally substituted aralkyl, an optionally substituted heteroaryl, or an optionally substituted heteroarylalkyl;
R 1 represents H, optionally substituted lower alkyl, or optionally substituted lower alkenyl;
R 2a is
Aは、H、低級アルキル、シクロアルキルまたは-NR3R4(R3およびR4は、独立して、H、低級アルキル等を示す)を示し;
Ar1は、アリーレンまたはヘテロアリーレンを示し;
Ar2は、置換されていてもよいアリール、置換されていてもよいアラルキル、置換されていてもよいヘテロアリール、または置換されていてもよいヘテロアリールアルキルを示し;
R1は、H、置換されていてもよい低級アルキル、または置換されていてもよい低級アルケニルを示し;
R2aは、 [Where:
A represents H, lower alkyl, cycloalkyl or —NR 3 R 4 (R 3 and R 4 independently represent H, lower alkyl, etc.);
Ar 1 represents arylene or heteroarylene;
Ar 2 represents an optionally substituted aryl, an optionally substituted aralkyl, an optionally substituted heteroaryl, or an optionally substituted heteroarylalkyl;
R 1 represents H, optionally substituted lower alkyl, or optionally substituted lower alkenyl;
R 2a is

等を示す]
がTNF-α変換酵素阻害作用を有する化合物として報告されている。 Etc.]
Has been reported as a compound having an inhibitory action on TNF-α converting enzyme.
がTNF-α変換酵素阻害作用を有する化合物として報告されている。 Etc.]
Has been reported as a compound having an inhibitory action on TNF-α converting enzyme.
さらに、特開2008-031064号公報(特許文献5)には、以下の化合物
Furthermore, JP 2008-031064 A (Patent Document 5) discloses the following compounds:

[式中、
R41およびR42は、独立して、Hまたは低級アルキルを示し;
R1は、-R00-置換されていてもよいアリール(R00:低級アルキレン)等を示す]
がDPP4阻害薬作用を有する化合物として報告されている。 [Where:
R 41 and R 42 independently represent H or lower alkyl;
R 1 represents —R 00 -optionally substituted aryl (R 00 : lower alkylene) or the like]
Has been reported as a compound having a DPP4 inhibitory action.
R41およびR42は、独立して、Hまたは低級アルキルを示し;
R1は、-R00-置換されていてもよいアリール(R00:低級アルキレン)等を示す]
がDPP4阻害薬作用を有する化合物として報告されている。 [Where:
R 41 and R 42 independently represent H or lower alkyl;
R 1 represents —R 00 -optionally substituted aryl (R 00 : lower alkylene) or the like]
Has been reported as a compound having a DPP4 inhibitory action.
さらに、以下の化合物が開示されている。
Furthermore, the following compounds are disclosed.












さらに、US2011/0263562(特許文献6)には、以下の化合物
Furthermore, US2011 / 0263562 (Patent Document 6) includes the following compounds:

[式中、
R1は、式:-COR2(R2は水素原子または置換基を示す)で表される基、置換されていてもよい5または6員の芳香族複素環基、または置換されていてもよいフェニル基を示し;
R3は、ハロゲン原子で置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示し;
R4は、水素原子または置換基を示し;
Xは、O、CO、CR5aR5b(R5aおよびR5bは、独立してそれぞれ、水素原子、ハロゲン原子、または置換されていてもよいC1-6アルキル基を示す)、NR5c(R5cは、水素原子、または置換されていてもよいC1-6アルキル基を示す)、S、SO、またはS(O)2を示し;
環Aは、さらに置換されていてもよい4ないし7員の非芳香環(該環は架橋されていてもよい)を示し;
環Pは、5員の芳香族複素環を示し、かつ
環Qは、さらに置換されていてもよい6員環を示し、
環Pと環Qとが縮合して、さらに置換されていてもよい二環性芳香族複素環を形成し;
R6は、置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示す]
がACC阻害作用を有する化合物として報告されている。 [Where:
R 1 is a group represented by the formula: —COR 2 (R 2 represents a hydrogen atom or a substituent), an optionally substituted 5- or 6-membered aromatic heterocyclic group, or an optionally substituted group. Indicates a good phenyl group;
R 3 represents a C 1-6 alkyl group which may be substituted with a halogen atom, or a C 3-6 cycloalkyl group which may be substituted;
R 4 represents a hydrogen atom or a substituent;
X is O, CO, CR 5a R 5b (R 5a and R 5b each independently represents a hydrogen atom, a halogen atom, or an optionally substituted C 1-6 alkyl group), NR 5c ( R 5c represents a hydrogen atom or an optionally substituted C 1-6 alkyl group), S, SO, or S (O) 2 ;
Ring A represents a 4-7 membered non-aromatic ring which may be further substituted, which ring may be bridged;
Ring P represents a 5-membered aromatic heterocycle, and Ring Q represents an optionally substituted 6-membered ring,
Ring P and Ring Q are condensed to form an optionally substituted bicyclic aromatic heterocycle;
R 6 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 3-6 cycloalkyl group.
Has been reported as a compound having an ACC inhibitory action.
R1は、式:-COR2(R2は水素原子または置換基を示す)で表される基、置換されていてもよい5または6員の芳香族複素環基、または置換されていてもよいフェニル基を示し;
R3は、ハロゲン原子で置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示し;
R4は、水素原子または置換基を示し;
Xは、O、CO、CR5aR5b(R5aおよびR5bは、独立してそれぞれ、水素原子、ハロゲン原子、または置換されていてもよいC1-6アルキル基を示す)、NR5c(R5cは、水素原子、または置換されていてもよいC1-6アルキル基を示す)、S、SO、またはS(O)2を示し;
環Aは、さらに置換されていてもよい4ないし7員の非芳香環(該環は架橋されていてもよい)を示し;
環Pは、5員の芳香族複素環を示し、かつ
環Qは、さらに置換されていてもよい6員環を示し、
環Pと環Qとが縮合して、さらに置換されていてもよい二環性芳香族複素環を形成し;
R6は、置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示す]
がACC阻害作用を有する化合物として報告されている。 [Where:
R 1 is a group represented by the formula: —COR 2 (R 2 represents a hydrogen atom or a substituent), an optionally substituted 5- or 6-membered aromatic heterocyclic group, or an optionally substituted group. Indicates a good phenyl group;
R 3 represents a C 1-6 alkyl group which may be substituted with a halogen atom, or a C 3-6 cycloalkyl group which may be substituted;
R 4 represents a hydrogen atom or a substituent;
X is O, CO, CR 5a R 5b (R 5a and R 5b each independently represents a hydrogen atom, a halogen atom, or an optionally substituted C 1-6 alkyl group), NR 5c ( R 5c represents a hydrogen atom or an optionally substituted C 1-6 alkyl group), S, SO, or S (O) 2 ;
Ring A represents a 4-7 membered non-aromatic ring which may be further substituted, which ring may be bridged;
Ring P represents a 5-membered aromatic heterocycle, and Ring Q represents an optionally substituted 6-membered ring,
Ring P and Ring Q are condensed to form an optionally substituted bicyclic aromatic heterocycle;
R 6 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 3-6 cycloalkyl group.
Has been reported as a compound having an ACC inhibitory action.
ACC阻害作用を有し、肥満症、糖尿病、高血圧症、高脂血症、心不全、糖尿病性合併症、メタボリックシンドローム、筋肉減少症、癌等の予防・治療に有用であり、かつ優れた薬効を有する化合物の開発が望まれている。
Has ACC inhibitory action, is useful for the prevention and treatment of obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complications, metabolic syndrome, sarcopenia, cancer, etc. Development of the compound which has is desired.
本発明者らは、式(I):
We have the formula (I):

[式中、
R1は、式:-COR2(R2は、水素原子または置換基を示す)で表される基、または置換されていてもよい5または6員芳香環基を示し;
R3は、ハロゲン原子で置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示し;
R4aおよびR4bは、独立して、水素原子または置換基を示し、またはR4aおよびR4bは、互いに結合して置換されていてもよい3または4員環を形成してもよく;
R5aおよびR5bは、独立して、水素原子または置換基を示し、またはR5aおよびR5bは、互いに結合して置換されていてもよい3または4員環を形成してもよく;
R6は、置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示し;
Xは、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子または置換基を示す)、NR7c(R7cは、水素原子または置換されていてもよい炭化水素基を示す)、S、SOまたはS(O)2を示し;
Yは、O、CO、CR8aR8b(R8aおよびR8bは、独立して、水素原子または置換基を示す)、NR8c(R8cは、水素原子または置換されていてもよい炭化水素基を示す)、S、SOまたはS(O)2を示し;
環Pは、さらに置換されていてもよい3ないし7員環を示し;
環Qは、さらに置換されていてもよい5または6員芳香環を示す]
で表される化合物またはその塩[以下、化合物(I)と称する場合がある]が、優れたACC阻害作用を有し、肥満症、糖尿病、高血圧症、高脂血症、心不全、糖尿病性合併症、メタボリックシンドローム、筋肉減少症、癌等の予防・治療に有用であり、かつ優れた薬効を有することを初めて見いだした。この知見に基づいて、本発明者らは、鋭意研究を行い、本発明を完成するに至った。 [Where:
R 1 represents a group represented by the formula: —COR 2 (R 2 represents a hydrogen atom or a substituent), or an optionally substituted 5- or 6-membered aromatic ring group;
R 3 represents a C 1-6 alkyl group which may be substituted with a halogen atom, or a C 3-6 cycloalkyl group which may be substituted;
R 4a and R 4b independently represent a hydrogen atom or a substituent, or R 4a and R 4b may be bonded to each other to form an optionally substituted 3- or 4-membered ring;
R 5a and R 5b independently represent a hydrogen atom or a substituent, or R 5a and R 5b may be bonded to each other to form an optionally substituted 3- or 4-membered ring;
R 6 represents an optionally substituted C 1-6 alkyl group, or an optionally substituted C 3-6 cycloalkyl group;
X is O, CO, CR 7a R 7b (R 7a and R 7b independently represent a hydrogen atom or a substituent), NR 7c (R 7c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 ;
Y is O, CO, CR 8a R 8b (R 8a and R 8b independently represent a hydrogen atom or a substituent), NR 8c (R 8c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 ;
Ring P represents a 3- to 7-membered ring which may be further substituted;
Ring Q represents a 5- or 6-membered aromatic ring which may be further substituted]
Or a salt thereof [hereinafter sometimes referred to as compound (I)] has an excellent ACC inhibitory action, obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complication It has been found for the first time that it is useful for the prevention and treatment of infectious diseases, metabolic syndrome, sarcopenia, cancer, etc. and has excellent medicinal effects. Based on this knowledge, the present inventors have conducted intensive studies and completed the present invention.
R1は、式:-COR2(R2は、水素原子または置換基を示す)で表される基、または置換されていてもよい5または6員芳香環基を示し;
R3は、ハロゲン原子で置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示し;
R4aおよびR4bは、独立して、水素原子または置換基を示し、またはR4aおよびR4bは、互いに結合して置換されていてもよい3または4員環を形成してもよく;
R5aおよびR5bは、独立して、水素原子または置換基を示し、またはR5aおよびR5bは、互いに結合して置換されていてもよい3または4員環を形成してもよく;
R6は、置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示し;
Xは、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子または置換基を示す)、NR7c(R7cは、水素原子または置換されていてもよい炭化水素基を示す)、S、SOまたはS(O)2を示し;
Yは、O、CO、CR8aR8b(R8aおよびR8bは、独立して、水素原子または置換基を示す)、NR8c(R8cは、水素原子または置換されていてもよい炭化水素基を示す)、S、SOまたはS(O)2を示し;
環Pは、さらに置換されていてもよい3ないし7員環を示し;
環Qは、さらに置換されていてもよい5または6員芳香環を示す]
で表される化合物またはその塩[以下、化合物(I)と称する場合がある]が、優れたACC阻害作用を有し、肥満症、糖尿病、高血圧症、高脂血症、心不全、糖尿病性合併症、メタボリックシンドローム、筋肉減少症、癌等の予防・治療に有用であり、かつ優れた薬効を有することを初めて見いだした。この知見に基づいて、本発明者らは、鋭意研究を行い、本発明を完成するに至った。 [Where:
R 1 represents a group represented by the formula: —COR 2 (R 2 represents a hydrogen atom or a substituent), or an optionally substituted 5- or 6-membered aromatic ring group;
R 3 represents a C 1-6 alkyl group which may be substituted with a halogen atom, or a C 3-6 cycloalkyl group which may be substituted;
R 4a and R 4b independently represent a hydrogen atom or a substituent, or R 4a and R 4b may be bonded to each other to form an optionally substituted 3- or 4-membered ring;
R 5a and R 5b independently represent a hydrogen atom or a substituent, or R 5a and R 5b may be bonded to each other to form an optionally substituted 3- or 4-membered ring;
R 6 represents an optionally substituted C 1-6 alkyl group, or an optionally substituted C 3-6 cycloalkyl group;
X is O, CO, CR 7a R 7b (R 7a and R 7b independently represent a hydrogen atom or a substituent), NR 7c (R 7c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 ;
Y is O, CO, CR 8a R 8b (R 8a and R 8b independently represent a hydrogen atom or a substituent), NR 8c (R 8c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 ;
Ring P represents a 3- to 7-membered ring which may be further substituted;
Ring Q represents a 5- or 6-membered aromatic ring which may be further substituted]
Or a salt thereof [hereinafter sometimes referred to as compound (I)] has an excellent ACC inhibitory action, obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complication It has been found for the first time that it is useful for the prevention and treatment of infectious diseases, metabolic syndrome, sarcopenia, cancer, etc. and has excellent medicinal effects. Based on this knowledge, the present inventors have conducted intensive studies and completed the present invention.
即ち、本発明は、
[1]化合物(I);
[1A]上記[1]記載の化合物またはそのプロドラッグ;
[2]R1が、式:-COR2(R2は、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC3-6シクロアルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基を示す)で表される基、または置換されていてもよい5または6員芳香環基である、上記[1]記載の化合物またはその塩;
[3]R1が、式:-COR2(R2は、水素原子、C1-6アルキル基、C1-6アルコキシ基、または1ないし2個のC1-6アルキル基で置換されていてもよいアミノ基を示す)で表される基、または5員の芳香族複素環基である、上記[1]記載の化合物またはその塩;
[4]R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、上記[1]、[2]または[3]記載の化合物またはその塩;
[5]R4aおよびR4bが水素原子である、上記[1]、[2]、[3]または[4]記載の化合物またはその塩;
[6]R5aおよびR5bが水素原子である、上記[1]、[2]、[3]、[4]または[5]記載の化合物またはその塩;
[7]R6が、置換されていてもよいC1-6アルキル基である、上記[1]、[2]、[3]、[4]、[5]または[6]記載の化合物またはその塩;
[8]R6が、
(a) ハロゲン原子、
(b) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC3-6シクロアルキル基、
(c) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよい4ないし7員の複素環基、および
(d) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC6-14アリール基
から選ばれる1ないし7個の置換基で置換されていてもよいC1-6アルキル基である、上記[1]、[2]、[3]、[4]、[5]、[6]または[7]記載の化合物またはその塩;
[9]Xが、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基を示す)、S、SOまたはS(O)2である、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]または[8]記載の化合物またはその塩;
[10]Yが、O、COまたはCH2である、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]または[9]記載の化合物またはその塩;
[10A]Yが、Oである、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]または[9]記載の化合物またはその塩;
[11]環Pが、ハロゲン原子、C1-6アルキル基およびC1-6アルコキシ基から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C3-6シクロアルカンまたは3ないし7員の複素環である、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]または[10A]記載の化合物またはその塩;
[12]環Qが、
(a) ハロゲン原子、
(b) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(c) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d) シアノ基
から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環である、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]または[11]記載の化合物またはその塩;
[13]
R1が、式:-COR2(R2は、水素原子、C1-6アルキル基、C1-6アルコキシ基、または1ないし2個のC1-6アルキル基で置換されていてもよいアミノ基を示す)で表される基、または5員の芳香族複素環基であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基であり;
R4aおよびR4bが水素原子であり;
R5aおよびR5bが水素原子であり;
R6が、
(a) ハロゲン原子、
(b) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC3-6シクロアルキル基、
(c) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよい4ないし7員の複素環基、および
(d) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC6-14アリール基
から選ばれる1ないし7個の置換基で置換されていてもよいC1-6アルキル基であり;
Xが、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基を示す)、S、SOまたはS(O)2であり;
Yが、O、COまたはCH2であり;
環Pが、ハロゲン原子、C1-6アルキル基およびC1-6アルコキシ基から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C3-6シクロアルカンまたは3ないし7員の複素環であり;かつ
環Qが、
(a) ハロゲン原子、
(b) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(c) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d) シアノ基
から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環である、上記[1]記載の化合物。
[14]N-{(1S)-2-[(trans-4-{[2-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド;
[15]N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド;
[16]N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド;
[17]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物を含有してなる医薬;
[17A]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグを含有してなる医薬;
[18]アセチル-CoAカルボキシラーゼ阻害剤である、上記[17]または[17A]記載の医薬;
[19]肥満症または糖尿病の予防または治療剤である、上記[17]または[17A]記載の医薬;
[20]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物を哺乳動物に有効量投与することを特徴とする、アセチル-CoAカルボキシラーゼを阻害する方法;
[20A]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグを哺乳動物に有効量投与することを特徴とする、アセチル-CoAカルボキシラーゼを阻害する方法;
[21]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物を哺乳動物に有効量投与することを特徴とする、該哺乳動物における肥満症または糖尿病の予防または治療方法;
[21A]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグを哺乳動物に有効量投与することを特徴とする、該哺乳動物における肥満症または糖尿病の予防または治療方法;
[22]肥満症または糖尿病の予防または治療剤として使用するための上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物;
[22A]肥満症または糖尿病の予防または治療剤として使用するための上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグ;
[23]肥満症または糖尿病の予防または治療剤を製造するための上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物の使用;
[23A]肥満症または糖尿病の予防または治療剤を製造するための上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグの使用;
等に関する。 That is, the present invention
[1] Compound (I);
[1A] The compound according to [1] above or a prodrug thereof;
[2] R 1 is represented by the formula: —COR 2 (R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, a substituted The above-mentioned [1], which is a group represented by a C 1-6 alkoxy group which may be substituted or an amino group which may be substituted, or a 5- or 6-membered aromatic ring group which may be substituted Or a salt thereof;
[3] R 1 is substituted with the formula: —COR 2 (R 2 is substituted with a hydrogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group, or 1 to 2 C 1-6 alkyl groups. A compound represented by the above-mentioned [1] or a salt thereof, which is a group represented by:
[4] The compound or a salt thereof according to the above [1], [2] or [3], wherein R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
[5] The compound or salt thereof according to [1], [2], [3] or [4] above, wherein R 4a and R 4b are hydrogen atoms;
[6] The compound or salt thereof according to the above [1], [2], [3], [4] or [5], wherein R 5a and R 5b are hydrogen atoms;
[7] The compound according to the above [1], [2], [3], [4], [5] or [6], wherein R 6 is an optionally substituted C 1-6 alkyl group or Its salt;
[8] R 6 is
(a) a halogen atom,
(b) a C 3-6 cycloalkyl group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups,
(c) a 4- to 7-membered heterocyclic group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups, and
(d) substituted with 1 to 7 substituents selected from 1 to 3 halogen atoms or C 6-14 aryl group optionally substituted with 1 to 3 C 1-6 alkyl groups; A compound or a salt thereof according to the above [1], [2], [3], [4], [5], [6] or [7], which is a C 1-6 alkyl group;
[9] The above [1], wherein X is O, CO, CH 2 , NR 7c (R 7c represents a hydrogen atom or a C 1-6 alkyl group), S, SO or S (O) 2 [2], [3], [4], [5], [6], [7] or [8] compound or salt thereof;
[10] The above [1], [2], [3], [4], [5], [6], [7], [8] or [9] wherein Y is O, CO or CH 2 Or a salt thereof;
[10A] The compound according to [1], [2], [3], [4], [5], [6], [7], [8] or [9], wherein Y is O or Its salt;
[11] Ring P may be further substituted with 1 to 3 substituents each selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group, benzene, C 3-6 cyclo [1], [2], [3], [4], [5], [6], [7], [8], [9], [9], which is an alkane or a 3- to 7-membered heterocyclic ring 10] or [10A] or a salt thereof;
[12] Ring Q is
(a) a halogen atom,
(b) a C 1-6 alkyl group optionally substituted with 1 to 5 halogen atoms,
(c) a C 1-6 alkoxy group optionally substituted with 1 to 5 halogen atoms, and
(d) The above [1], [2], [3], which is benzene or a 5- or 6-membered aromatic heterocyclic ring, each of which may be further substituted with 1 to 3 substituents selected from a cyano group ], [4], [5], [6], [7], [8], [9], [10], [10A] or [11], or a salt thereof;
[13]
R 1 may be substituted with the formula: —COR 2 (R 2 is a hydrogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group, or 1 to 2 C 1-6 alkyl groups. An amino group), or a 5-membered aromatic heterocyclic group;
R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
R 4a and R 4b are hydrogen atoms;
R 5a and R 5b are hydrogen atoms;
R 6 is
(a) a halogen atom,
(b) a C 3-6 cycloalkyl group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups,
(c) a 4- to 7-membered heterocyclic group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups, and
(d) substituted with 1 to 7 substituents selected from 1 to 3 halogen atoms or C 6-14 aryl group optionally substituted with 1 to 3 C 1-6 alkyl groups; Or a C 1-6 alkyl group;
X is O, CO, CH 2 , NR 7c (R 7c represents a hydrogen atom or a C 1-6 alkyl group), S, SO or S (O) 2 ;
Y is O, CO or CH 2 ;
Ring P may be further substituted with 1 to 3 substituents each selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group, benzene, C 3-6 cycloalkane or 3 To a 7-membered heterocycle; and ring Q is
(a) a halogen atom,
(b) a C 1-6 alkyl group optionally substituted with 1 to 5 halogen atoms,
(c) a C 1-6 alkoxy group optionally substituted with 1 to 5 halogen atoms, and
(d) The compound according to the above [1], which is benzene or a 5- or 6-membered aromatic heterocyclic ring, each of which may be further substituted with 1 to 3 substituents selected from a cyano group.
[14] N-{(1S) -2-[(trans-4-{[2-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide;
[15] N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methyl Ethyl] acetamide;
[16] N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1 -Methylethyl] acetamide;
[17] The above [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 ], A medicament comprising the compound according to [12] or [13];
[17A] [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 ] A pharmaceutical comprising the compound of [12] or [13] or a prodrug thereof;
[18] The medicament according to [17] or [17A] above, which is an acetyl-CoA carboxylase inhibitor;
[19] The medicament according to [17] or [17A] above, which is a prophylactic or therapeutic agent for obesity or diabetes;
[20] The above [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 A method for inhibiting acetyl-CoA carboxylase, which comprises administering an effective amount of the compound according to [12] or [13] to a mammal;
[20A] [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11] A method for inhibiting acetyl-CoA carboxylase, which comprises administering an effective amount of the compound according to [12] or [13] or a prodrug thereof to a mammal;
[21] Above [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 ] A method for preventing or treating obesity or diabetes in a mammal, which comprises administering an effective amount of the compound of [12] or [13] to the mammal;
[21A] [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 ] A method for preventing or treating obesity or diabetes in a mammal, which comprises administering an effective amount of the compound according to [12] or [13] or a prodrug thereof to the mammal;
[22] The above [1], [2], [3], [4], [5], [6], [7], [8], for use as a preventive or therapeutic agent for obesity or diabetes, [9], [10], [10A], [11], [12] or a compound according to [13];
[22A] The above [1], [2], [3], [4], [5], [6], [7], [8], for use as a preventive or therapeutic agent for obesity or diabetes, [9], [10], [10A], [11], [12] or a compound according to [13] or a prodrug thereof;
[23] The above [1], [2], [3], [4], [5], [6], [7], [8], for producing an agent for preventing or treating obesity or diabetes, Use of a compound according to [9], [10], [10A], [11], [12] or [13];
[23A] The above [1], [2], [3], [4], [5], [6], [7], [8], for producing an agent for preventing or treating obesity or diabetes, [9], [10], [10A], [11], [12] Use of the compound or prodrug thereof according to [13];
Etc.
[1]化合物(I);
[1A]上記[1]記載の化合物またはそのプロドラッグ;
[2]R1が、式:-COR2(R2は、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC3-6シクロアルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基を示す)で表される基、または置換されていてもよい5または6員芳香環基である、上記[1]記載の化合物またはその塩;
[3]R1が、式:-COR2(R2は、水素原子、C1-6アルキル基、C1-6アルコキシ基、または1ないし2個のC1-6アルキル基で置換されていてもよいアミノ基を示す)で表される基、または5員の芳香族複素環基である、上記[1]記載の化合物またはその塩;
[4]R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、上記[1]、[2]または[3]記載の化合物またはその塩;
[5]R4aおよびR4bが水素原子である、上記[1]、[2]、[3]または[4]記載の化合物またはその塩;
[6]R5aおよびR5bが水素原子である、上記[1]、[2]、[3]、[4]または[5]記載の化合物またはその塩;
[7]R6が、置換されていてもよいC1-6アルキル基である、上記[1]、[2]、[3]、[4]、[5]または[6]記載の化合物またはその塩;
[8]R6が、
(a) ハロゲン原子、
(b) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC3-6シクロアルキル基、
(c) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよい4ないし7員の複素環基、および
(d) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC6-14アリール基
から選ばれる1ないし7個の置換基で置換されていてもよいC1-6アルキル基である、上記[1]、[2]、[3]、[4]、[5]、[6]または[7]記載の化合物またはその塩;
[9]Xが、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基を示す)、S、SOまたはS(O)2である、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]または[8]記載の化合物またはその塩;
[10]Yが、O、COまたはCH2である、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]または[9]記載の化合物またはその塩;
[10A]Yが、Oである、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]または[9]記載の化合物またはその塩;
[11]環Pが、ハロゲン原子、C1-6アルキル基およびC1-6アルコキシ基から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C3-6シクロアルカンまたは3ないし7員の複素環である、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]または[10A]記載の化合物またはその塩;
[12]環Qが、
(a) ハロゲン原子、
(b) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(c) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d) シアノ基
から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環である、上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]または[11]記載の化合物またはその塩;
[13]
R1が、式:-COR2(R2は、水素原子、C1-6アルキル基、C1-6アルコキシ基、または1ないし2個のC1-6アルキル基で置換されていてもよいアミノ基を示す)で表される基、または5員の芳香族複素環基であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基であり;
R4aおよびR4bが水素原子であり;
R5aおよびR5bが水素原子であり;
R6が、
(a) ハロゲン原子、
(b) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC3-6シクロアルキル基、
(c) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよい4ないし7員の複素環基、および
(d) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC6-14アリール基
から選ばれる1ないし7個の置換基で置換されていてもよいC1-6アルキル基であり;
Xが、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基を示す)、S、SOまたはS(O)2であり;
Yが、O、COまたはCH2であり;
環Pが、ハロゲン原子、C1-6アルキル基およびC1-6アルコキシ基から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C3-6シクロアルカンまたは3ないし7員の複素環であり;かつ
環Qが、
(a) ハロゲン原子、
(b) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(c) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d) シアノ基
から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環である、上記[1]記載の化合物。
[14]N-{(1S)-2-[(trans-4-{[2-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド;
[15]N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド;
[16]N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド;
[17]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物を含有してなる医薬;
[17A]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグを含有してなる医薬;
[18]アセチル-CoAカルボキシラーゼ阻害剤である、上記[17]または[17A]記載の医薬;
[19]肥満症または糖尿病の予防または治療剤である、上記[17]または[17A]記載の医薬;
[20]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物を哺乳動物に有効量投与することを特徴とする、アセチル-CoAカルボキシラーゼを阻害する方法;
[20A]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグを哺乳動物に有効量投与することを特徴とする、アセチル-CoAカルボキシラーゼを阻害する方法;
[21]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物を哺乳動物に有効量投与することを特徴とする、該哺乳動物における肥満症または糖尿病の予防または治療方法;
[21A]上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグを哺乳動物に有効量投与することを特徴とする、該哺乳動物における肥満症または糖尿病の予防または治療方法;
[22]肥満症または糖尿病の予防または治療剤として使用するための上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物;
[22A]肥満症または糖尿病の予防または治療剤として使用するための上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグ;
[23]肥満症または糖尿病の予防または治療剤を製造するための上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物の使用;
[23A]肥満症または糖尿病の予防または治療剤を製造するための上記[1]、[2]、[3]、[4]、[5]、[6]、[7]、[8]、[9]、[10]、[10A]、[11]、[12]または[13]記載の化合物またはそのプロドラッグの使用;
等に関する。 That is, the present invention
[1] Compound (I);
[1A] The compound according to [1] above or a prodrug thereof;
[2] R 1 is represented by the formula: —COR 2 (R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, a substituted The above-mentioned [1], which is a group represented by a C 1-6 alkoxy group which may be substituted or an amino group which may be substituted, or a 5- or 6-membered aromatic ring group which may be substituted Or a salt thereof;
[3] R 1 is substituted with the formula: —COR 2 (R 2 is substituted with a hydrogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group, or 1 to 2 C 1-6 alkyl groups. A compound represented by the above-mentioned [1] or a salt thereof, which is a group represented by:
[4] The compound or a salt thereof according to the above [1], [2] or [3], wherein R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
[5] The compound or salt thereof according to [1], [2], [3] or [4] above, wherein R 4a and R 4b are hydrogen atoms;
[6] The compound or salt thereof according to the above [1], [2], [3], [4] or [5], wherein R 5a and R 5b are hydrogen atoms;
[7] The compound according to the above [1], [2], [3], [4], [5] or [6], wherein R 6 is an optionally substituted C 1-6 alkyl group or Its salt;
[8] R 6 is
(a) a halogen atom,
(b) a C 3-6 cycloalkyl group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups,
(c) a 4- to 7-membered heterocyclic group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups, and
(d) substituted with 1 to 7 substituents selected from 1 to 3 halogen atoms or C 6-14 aryl group optionally substituted with 1 to 3 C 1-6 alkyl groups; A compound or a salt thereof according to the above [1], [2], [3], [4], [5], [6] or [7], which is a C 1-6 alkyl group;
[9] The above [1], wherein X is O, CO, CH 2 , NR 7c (R 7c represents a hydrogen atom or a C 1-6 alkyl group), S, SO or S (O) 2 [2], [3], [4], [5], [6], [7] or [8] compound or salt thereof;
[10] The above [1], [2], [3], [4], [5], [6], [7], [8] or [9] wherein Y is O, CO or CH 2 Or a salt thereof;
[10A] The compound according to [1], [2], [3], [4], [5], [6], [7], [8] or [9], wherein Y is O or Its salt;
[11] Ring P may be further substituted with 1 to 3 substituents each selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group, benzene, C 3-6 cyclo [1], [2], [3], [4], [5], [6], [7], [8], [9], [9], which is an alkane or a 3- to 7-membered heterocyclic ring 10] or [10A] or a salt thereof;
[12] Ring Q is
(a) a halogen atom,
(b) a C 1-6 alkyl group optionally substituted with 1 to 5 halogen atoms,
(c) a C 1-6 alkoxy group optionally substituted with 1 to 5 halogen atoms, and
(d) The above [1], [2], [3], which is benzene or a 5- or 6-membered aromatic heterocyclic ring, each of which may be further substituted with 1 to 3 substituents selected from a cyano group ], [4], [5], [6], [7], [8], [9], [10], [10A] or [11], or a salt thereof;
[13]
R 1 may be substituted with the formula: —COR 2 (R 2 is a hydrogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group, or 1 to 2 C 1-6 alkyl groups. An amino group), or a 5-membered aromatic heterocyclic group;
R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
R 4a and R 4b are hydrogen atoms;
R 5a and R 5b are hydrogen atoms;
R 6 is
(a) a halogen atom,
(b) a C 3-6 cycloalkyl group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups,
(c) a 4- to 7-membered heterocyclic group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups, and
(d) substituted with 1 to 7 substituents selected from 1 to 3 halogen atoms or C 6-14 aryl group optionally substituted with 1 to 3 C 1-6 alkyl groups; Or a C 1-6 alkyl group;
X is O, CO, CH 2 , NR 7c (R 7c represents a hydrogen atom or a C 1-6 alkyl group), S, SO or S (O) 2 ;
Y is O, CO or CH 2 ;
Ring P may be further substituted with 1 to 3 substituents each selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group, benzene, C 3-6 cycloalkane or 3 To a 7-membered heterocycle; and ring Q is
(a) a halogen atom,
(b) a C 1-6 alkyl group optionally substituted with 1 to 5 halogen atoms,
(c) a C 1-6 alkoxy group optionally substituted with 1 to 5 halogen atoms, and
(d) The compound according to the above [1], which is benzene or a 5- or 6-membered aromatic heterocyclic ring, each of which may be further substituted with 1 to 3 substituents selected from a cyano group.
[14] N-{(1S) -2-[(trans-4-{[2-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide;
[15] N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methyl Ethyl] acetamide;
[16] N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1 -Methylethyl] acetamide;
[17] The above [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 ], A medicament comprising the compound according to [12] or [13];
[17A] [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 ] A pharmaceutical comprising the compound of [12] or [13] or a prodrug thereof;
[18] The medicament according to [17] or [17A] above, which is an acetyl-CoA carboxylase inhibitor;
[19] The medicament according to [17] or [17A] above, which is a prophylactic or therapeutic agent for obesity or diabetes;
[20] The above [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 A method for inhibiting acetyl-CoA carboxylase, which comprises administering an effective amount of the compound according to [12] or [13] to a mammal;
[20A] [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11] A method for inhibiting acetyl-CoA carboxylase, which comprises administering an effective amount of the compound according to [12] or [13] or a prodrug thereof to a mammal;
[21] Above [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 ] A method for preventing or treating obesity or diabetes in a mammal, which comprises administering an effective amount of the compound of [12] or [13] to the mammal;
[21A] [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [10A], [11 ] A method for preventing or treating obesity or diabetes in a mammal, which comprises administering an effective amount of the compound according to [12] or [13] or a prodrug thereof to the mammal;
[22] The above [1], [2], [3], [4], [5], [6], [7], [8], for use as a preventive or therapeutic agent for obesity or diabetes, [9], [10], [10A], [11], [12] or a compound according to [13];
[22A] The above [1], [2], [3], [4], [5], [6], [7], [8], for use as a preventive or therapeutic agent for obesity or diabetes, [9], [10], [10A], [11], [12] or a compound according to [13] or a prodrug thereof;
[23] The above [1], [2], [3], [4], [5], [6], [7], [8], for producing an agent for preventing or treating obesity or diabetes, Use of a compound according to [9], [10], [10A], [11], [12] or [13];
[23A] The above [1], [2], [3], [4], [5], [6], [7], [8], for producing an agent for preventing or treating obesity or diabetes, [9], [10], [10A], [11], [12] Use of the compound or prodrug thereof according to [13];
Etc.
化合物(I)は、ACC阻害作用を有し、肥満症、糖尿病、高血圧症、高脂血症、心不全、糖尿病性合併症、メタボリックシンドローム、筋肉減少症、癌等の予防・治療に有用であり、かつ優れた薬効を有する。
Compound (I) has an ACC inhibitory action and is useful for the prevention and treatment of obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complications, metabolic syndrome, sarcopenia, cancer, etc. And has excellent medicinal properties.
[発明の詳細な説明]
以下、式(I)中の各記号の定義について詳述する。
本明細書中の「ハロゲン原子」は、特に断りのない限り、フッ素原子、塩素原子、臭素原子、ヨウ素原子を意味する。
本明細書中の「C1-3アルキレンジオキシ基」は、特に断りのない限り、メチレンジオキシ、エチレンジオキシ等を意味する。
本明細書中の「C1-6アルキル基」は、特に断りのない限り、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、イソペンチル、ネオペンチル、1-エチルプロピル、ヘキシル、イソヘキシル、1,1-ジメチルブチル、2,2-ジメチルブチル、3,3-ジメチルブチル、2-エチルブチル等を意味する。 Detailed Description of the Invention
Hereinafter, the definition of each symbol in formula (I) will be described in detail.
The “halogen atom” in the present specification means a fluorine atom, a chlorine atom, a bromine atom, or an iodine atom unless otherwise specified.
The “C 1-3 alkylenedioxy group” in the present specification means methylenedioxy, ethylenedioxy and the like unless otherwise specified.
In the present specification, “C 1-6 alkyl group” means methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, 1-ethyl unless otherwise specified. It means propyl, hexyl, isohexyl, 1,1-dimethylbutyl, 2,2-dimethylbutyl, 3,3-dimethylbutyl, 2-ethylbutyl and the like.
以下、式(I)中の各記号の定義について詳述する。
本明細書中の「ハロゲン原子」は、特に断りのない限り、フッ素原子、塩素原子、臭素原子、ヨウ素原子を意味する。
本明細書中の「C1-3アルキレンジオキシ基」は、特に断りのない限り、メチレンジオキシ、エチレンジオキシ等を意味する。
本明細書中の「C1-6アルキル基」は、特に断りのない限り、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、イソペンチル、ネオペンチル、1-エチルプロピル、ヘキシル、イソヘキシル、1,1-ジメチルブチル、2,2-ジメチルブチル、3,3-ジメチルブチル、2-エチルブチル等を意味する。 Detailed Description of the Invention
Hereinafter, the definition of each symbol in formula (I) will be described in detail.
The “halogen atom” in the present specification means a fluorine atom, a chlorine atom, a bromine atom, or an iodine atom unless otherwise specified.
The “C 1-3 alkylenedioxy group” in the present specification means methylenedioxy, ethylenedioxy and the like unless otherwise specified.
In the present specification, “C 1-6 alkyl group” means methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, 1-ethyl unless otherwise specified. It means propyl, hexyl, isohexyl, 1,1-dimethylbutyl, 2,2-dimethylbutyl, 3,3-dimethylbutyl, 2-ethylbutyl and the like.
本明細書中の「C1-6アルコキシ基」は、特に断りのない限り、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソブトキシ、sec-ブトキシ、tert-ブトキシ等を意味する。
本明細書中の「C1-6アルコキシ-カルボニル基」は、特に断りのない限り、メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、tert-ブトキシカルボニル等を意味する。
本明細書中の「C1-6アルキル-カルボニル基」は、特に断りのない限り、アセチル、プロパノイル、ブタノイル、イソブタノイル、ペンタノイル、イソペンタノイル、ヘキサノイル等を意味する。 The “C 1-6 alkoxy group” in the present specification means methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy and the like unless otherwise specified.
The “C 1-6 alkoxy-carbonyl group” in the present specification means methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert-butoxycarbonyl and the like, unless otherwise specified.
The “C 1-6 alkyl-carbonyl group” in the present specification means acetyl, propanoyl, butanoyl, isobutanoyl, pentanoyl, isopentanoyl, hexanoyl and the like, unless otherwise specified.
本明細書中の「C1-6アルコキシ-カルボニル基」は、特に断りのない限り、メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、tert-ブトキシカルボニル等を意味する。
本明細書中の「C1-6アルキル-カルボニル基」は、特に断りのない限り、アセチル、プロパノイル、ブタノイル、イソブタノイル、ペンタノイル、イソペンタノイル、ヘキサノイル等を意味する。 The “C 1-6 alkoxy group” in the present specification means methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy and the like unless otherwise specified.
The “C 1-6 alkoxy-carbonyl group” in the present specification means methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert-butoxycarbonyl and the like, unless otherwise specified.
The “C 1-6 alkyl-carbonyl group” in the present specification means acetyl, propanoyl, butanoyl, isobutanoyl, pentanoyl, isopentanoyl, hexanoyl and the like, unless otherwise specified.
本明細書中、「置換されていてもよいC6-14アリールスルホニルオキシ基」としては、ベンゼンスルホニルオキシ基、p-トルエンスルホニルオキシ基等が挙げられる。
本明細書中、「置換されていてもよいC1-6アルキルスルホニルオキシ基」としては、メタンスルホニルオキシ基、トリフルオロメタンスルホニルオキシ基等が挙げられる。 In the present specification, examples of the “ optionally substituted C 6-14 arylsulfonyloxy group” include a benzenesulfonyloxy group and a p-toluenesulfonyloxy group.
In the present specification, examples of the “optionally substituted C 1-6 alkylsulfonyloxy group” include a methanesulfonyloxy group and a trifluoromethanesulfonyloxy group.
本明細書中、「置換されていてもよいC1-6アルキルスルホニルオキシ基」としては、メタンスルホニルオキシ基、トリフルオロメタンスルホニルオキシ基等が挙げられる。 In the present specification, examples of the “ optionally substituted C 6-14 arylsulfonyloxy group” include a benzenesulfonyloxy group and a p-toluenesulfonyloxy group.
In the present specification, examples of the “optionally substituted C 1-6 alkylsulfonyloxy group” include a methanesulfonyloxy group and a trifluoromethanesulfonyloxy group.
R1は、式:-COR2(R2は、水素原子または置換基を示す)で表される基、または置換されていてもよい5または6員芳香環基を示す。
R 1 represents a group represented by the formula: —COR 2 (R 2 represents a hydrogen atom or a substituent), or an optionally substituted 5- or 6-membered aromatic ring group.
R2で示される「置換基」としては、「置換されていてもよい炭化水素基」、「置換されていてもよい複素環基」、「置換されていてもよいヒドロキシ基」、「置換されていてもよいアミノ基」、「置換されていてもよいスルファニル基」、「アシル基」、「ハロゲン原子」、「シアノ基」、「ニトロ基」等が挙げられる。
Examples of the “substituent” represented by R 2 include “optionally substituted hydrocarbon group”, “optionally substituted heterocyclic group”, “optionally substituted hydroxy group”, “substituted” And optionally substituted amino group, “optionally substituted sulfanyl group”, “acyl group”, “halogen atom”, “cyano group”, “nitro group” and the like.
前記「置換されていてもよい炭化水素基」における「炭化水素基」としては、例えば、C1-10アルキル基、C2-10アルケニル基、C2-10アルキニル基、C3-10シクロアルキル基、C3-10シクロアルケニル基、C4-10シクロアルカジエニル基、C6-14アリール基、C7-13アラルキル基、C8-13アリールアルケニル基等が挙げられる。
Examples of the “hydrocarbon group” in the “optionally substituted hydrocarbon group” include a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 2-10 alkynyl group, and a C 3-10 cycloalkyl group. Group, C 3-10 cycloalkenyl group, C 4-10 cycloalkadienyl group, C 6-14 aryl group, C 7-13 aralkyl group, C 8-13 arylalkenyl group and the like.
ここで、C1-10アルキル基としては、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、イソペンチル、ネオペンチル、1-エチルプロピル、ヘキシル、イソヘキシル、1,1-ジメチルブチル、2,2-ジメチルブチル、3,3-ジメチルブチル、2-エチルブチル、ヘプチル、オクチル、ノニル、デシル等が挙げられる。なかでも、C1-6アルキル基が好ましい。
Here, examples of the C 1-10 alkyl group include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, 1-ethylpropyl, hexyl, isohexyl, 1 , 1-dimethylbutyl, 2,2-dimethylbutyl, 3,3-dimethylbutyl, 2-ethylbutyl, heptyl, octyl, nonyl, decyl and the like. Of these, a C 1-6 alkyl group is preferable.
C2-10アルケニル基としては、例えば、エテニル、1-プロペニル、2-プロペニル、2-メチル-1-プロペニル、1-ブテニル、2-ブテニル、3-ブテニル、3-メチル-2-ブテニル、1-ペンテニル、2-ペンテニル、3-ペンテニル、4-ペンテニル、4-メチル-3-ペンテニル、1-ヘキセニル、3-ヘキセニル、5-ヘキセニル、1-ヘプテニル、1-オクテニル等が挙げられる。なかでも、C2-6アルケニル基が好ましい。
Examples of the C 2-10 alkenyl group include ethenyl, 1-propenyl, 2-propenyl, 2-methyl-1-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, 3-methyl-2-butenyl, 1 -Pentenyl, 2-pentenyl, 3-pentenyl, 4-pentenyl, 4-methyl-3-pentenyl, 1-hexenyl, 3-hexenyl, 5-hexenyl, 1-heptenyl, 1-octenyl and the like. Of these, a C 2-6 alkenyl group is preferable.
C2-10アルキニル基としては、例えば、エチニル、1-プロピニル、2-プロピニル、1-ブチニル、2-ブチニル、3-ブチニル、1-ペンチニル、2-ペンチニル、3-ペンチニル、4-ペンチニル、1-ヘキシニル、2-ヘキシニル、3-ヘキシニル、4-ヘキシニル、5-ヘキシニル、1-ヘプチニル、1-オクチニル等が挙げられる。なかでも、C2-6アルキニル基が好ましい。
Examples of the C 2-10 alkynyl group include ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1-pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1 -Hexynyl, 2-hexynyl, 3-hexynyl, 4-hexynyl, 5-hexynyl, 1-heptynyl, 1-octynyl and the like. Of these, a C 2-6 alkynyl group is preferable.
C3-10シクロアルキル基としては、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル等が挙げられる。なかでも、C3-6シクロアルキル基が好ましい。
Examples of the C 3-10 cycloalkyl group include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl and the like. Of these, a C 3-6 cycloalkyl group is preferable.
C3-10シクロアルケニル基としては、例えば、2-シクロペンテン-1-イル、3-シクロペンテン-1-イル、2-シクロヘキセン-1-イル、3-シクロヘキセン-1-イル等が挙げられる。なかでも、C3-6シクロアルケニル基が好ましい。
Examples of the C 3-10 cycloalkenyl group include 2-cyclopenten-1-yl, 3-cyclopenten-1-yl, 2-cyclohexen-1-yl, 3-cyclohexen-1-yl and the like. Of these, a C 3-6 cycloalkenyl group is preferable.
C4-10シクロアルカジエニル基としては、例えば、2,4-シクロペンタジエン-1-イル、2,4-シクロヘキサジエン-1-イル、2,5-シクロヘキサジエン-1-イル等が挙げられる。なかでも、C4-6シクロアルカジエニル基が好ましい。
Examples of the C 4-10 cycloalkadienyl group include 2,4-cyclopentadien-1-yl, 2,4-cyclohexadien-1-yl, 2,5-cyclohexadien-1-yl, and the like. . Of these, a C 4-6 cycloalkadienyl group is preferable.
上記のC3-10シクロアルキル基、C3-10シクロアルケニル基およびC4-10シクロアルカジエニル基は、それぞれベンゼン環と縮合して縮合環基を形成していてもよく、このような縮合環基としては、例えば、インダニル、ジヒドロナフチル、テトラヒドロナフチル、フルオレニル等が挙げられる。
The above C 3-10 cycloalkyl group, C 3-10 cycloalkenyl group and C 4-10 cycloalkadienyl group may each be condensed with a benzene ring to form a condensed ring group. Examples of the condensed ring group include indanyl, dihydronaphthyl, tetrahydronaphthyl, fluorenyl and the like.
また、上記のC3-10シクロアルキル基、C3-10シクロアルケニル基およびC4-10シクロアルカジエニル基は、C7-10橋かけ式炭化水素基であってもよい。C7-10橋かけ式炭化水素基としては、ビシクロ[2.2.1]ヘプチル(ノルボルニル)、ビシクロ[2.2.2]オクチル、ビシクロ[3.2.1]オクチル、ビシクロ[3.2.2]ノニル、ビシクロ[3.3.1]ノニル、ビシクロ[4.2.1]ノニル、ビシクロ[4.3.1]デシル、アダマンチル等が挙げられる。
The C 3-10 cycloalkyl group, the C 3-10 cycloalkenyl group and the C 4-10 cycloalkadienyl group may be a C 7-10 bridged hydrocarbon group. Examples of the C 7-10 bridged hydrocarbon group include bicyclo [2.2.1] heptyl (norbornyl), bicyclo [2.2.2] octyl, bicyclo [3.2.1] octyl, bicyclo [3. 2.2] nonyl, bicyclo [3.3.1] nonyl, bicyclo [4.2.1] nonyl, bicyclo [4.3.1] decyl, adamantyl and the like.
さらに、上記のC3-10シクロアルキル基、C3-10シクロアルケニル基およびC4-10シクロアルカジエニル基は、それぞれC3-10シクロアルカン、C3-10シクロアルケンまたはC4-10シクロアルカジエンとスピロ環基を形成していてもよい。ここで、C3-10シクロアルカン、C3-10シクロアルケンおよびC4-10シクロアルカジエンとしては、上記のC3-10シクロアルキル基、C3-10シクロアルケニル基およびC4-10シクロアルカジエニル基に対応する環が挙げられる。このようなスピロ環基としては、スピロ[4.5]デカン-8-イル等が挙げられる。
Further, the above C 3-10 cycloalkyl group, C 3-10 cycloalkenyl group and C 4-10 cycloalkadienyl group are respectively C 3-10 cycloalkane, C 3-10 cycloalkene or C 4-10. A cycloalkadiene may form a spiro ring group. Here, as C 3-10 cycloalkane, C 3-10 cycloalkene and C 4-10 cycloalkadiene, the above C 3-10 cycloalkyl group, C 3-10 cycloalkenyl group and C 4-10 cycloalkane are mentioned. Examples include rings corresponding to alkadienyl groups. Examples of such a spiro ring group include spiro [4.5] decan-8-yl.
C6-14アリール基としては、例えば、フェニル、ナフチル、アントリル、フェナントリル、アセナフチレニル、ビフェニリル等が挙げられる。なかでも、C6-12アリール基が好ましい。
Examples of the C 6-14 aryl group include phenyl, naphthyl, anthryl, phenanthryl, acenaphthylenyl, biphenylyl and the like. Of these, a C 6-12 aryl group is preferable.
C7-13アラルキル基としては、例えば、ベンジル、フェネチル、ナフチルメチル、ビフェニリルメチル等が挙げられる。
Examples of the C 7-13 aralkyl group include benzyl, phenethyl, naphthylmethyl, biphenylylmethyl and the like.
C8-13アリールアルケニル基としては、例えば、スチリル等が挙げられる。
Examples of the C 8-13 arylalkenyl group include styryl and the like.
前記「炭化水素基」として例示したC1-10アルキル基、C2-10アルケニル基およびC2-10アルキニル基は、置換可能な位置に1ないし7個(好ましくは、1ないし3個)の置換基を有していてもよい。
The C 1-10 alkyl group, the C 2-10 alkenyl group and the C 2-10 alkynyl group exemplified as the “hydrocarbon group” have 1 to 7 (preferably 1 to 3) substituted positions. It may have a substituent.
このような置換基としては、例えば、
(1)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル);
(2)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール基(例、フェニル、ナフチル);
(3)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよい芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(d)ハロゲン原子、および
(e)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい非芳香族複素環基(例、テトラヒドロフリル、モルホリニル、チオモルホリニル、ピペリジニル、ピロリジニル、ピペラジニル);
(5)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基、
(d)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル)、
(e)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基、および
(f)芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル)
から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基;
(6)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基;
(7)(a)ハロゲン原子、
(b)C1-6アルコキシ基、
(c)C6-14アリール基(例、フェニル)、および
(d)複素環基(例、テトラヒドロフリル)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ-カルボニル基;
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基;
(10)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいチオカルバモイル基;
(11)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいスルファモイル基;
(12)カルボキシ基;
(13)ヒドロキシ基;
(14)(a)ハロゲン原子、
(b)カルボキシ基、
(c)C1-6アルコキシ基、
(d)1ないし3個のC6-14アリール基(例、フェニル)で置換されていてもよいC1-6アルコキシ-カルボニル基、
(e)C1-6アルキル基およびC1-6アルコキシ-カルボニル基から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基、
(f)複素環基(例、テトラヒドロフリル)、および
(g)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ基;
(15)1ないし3個のハロゲン原子で置換されていてもよいC2-6アルケニルオキシ基(例、エテニルオキシ);
(16)C7-13アラルキルオキシ基(例、ベンジルオキシ);
(17)C6-14アリールオキシ基(例、フェニルオキシ、ナフチルオキシ);
(18)C1-6アルキル-カルボニルオキシ基(例、アセチルオキシ、tert-ブチルカルボニルオキシ);
(19)(a)ハロゲン原子、および
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル);
(20)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい非芳香族複素環カルボニル基(例、ピロリジニルカルボニル、モルホリニルカルボニル);
(21)スルファニル基;
(22)(a)ハロゲン原子、および
(b)C1-6アルコキシ-カルボニル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキルスルファニル基(例、メチルスルファニル、エチルスルファニル);
(23)C7-13アラルキルスルファニル基(例、ベンジルスルファニル);
(24)C6-14アリールスルファニル基(例、フェニルスルファニル、ナフチルスルファニル);
(25)シアノ基;
(26)ニトロ基;
(27)ハロゲン原子;
(28)C1-3アルキレンジオキシ基;
(29)C1-3アルキレンオキシ基(例、メチレンオキシ、エチレンオキシ);
(30)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
から選ばれる1ないし3個の置換基で置換されていてもよい芳香族複素環カルボニル基(例、ピラゾリルカルボニル、ピラジニルカルボニル、イソキサゾリルカルボニル、ピリジルカルボニル、チアゾリルカルボニル);
(31)(a)ハロゲン原子(例、フッ素原子)、および
(b)C1-6アルコキシ基(例、メトキシ)
から選ばれる1ないし3個の置換基で置換されていてもよいC3-10シクロアルコキシ基(例、シクロプロポキシ、シクロペンチルオキシ)
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。 As such a substituent, for example,
(1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl);
(2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms,
(b) a hydroxy group,
(c) a C 1-6 alkoxy group optionally substituted with 1 to 3 halogen atoms, and (d) a C 6- optionally substituted with 1 to 3 substituents selected from halogen atoms. 14 aryl groups (eg, phenyl, naphthyl);
(3) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms,
(b) a hydroxy group,
(c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, and (d) an aromatic complex which may be substituted with 1 to 3 substituents selected from halogen atoms. A cyclic group (eg, thienyl, furyl, pyridyl, pyrazolyl, imidazolyl, tetrazolyl, oxazolyl, thiazolyl, oxadiazolyl, thiadiazolyl);
(4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms,
(b) a hydroxy group,
(c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms,
(d) a non-aromatic heterocyclic group (eg, tetrahydrofuryl, morpholinyl, thiomorpholinyl, piperidinyl, pyrrolidinyl, piperazinyl) optionally substituted with 1 to 3 substituents selected from halogen atoms and (e) oxo groups );
(5) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms,
(b) a C 1-6 alkyl-carbonyl group optionally substituted by 1 to 3 halogen atoms,
(c) a C 1-6 alkoxy-carbonyl group optionally substituted by 1 to 3 halogen atoms,
(d) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl) optionally substituted with 1 to 3 halogen atoms,
(e) a carbamoyl group optionally mono- or di-substituted with a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms, and (f) an aromatic heterocyclic group (eg, thienyl, Furyl, pyridyl, pyrazolyl, imidazolyl, tetrazolyl, oxazolyl, thiazolyl, oxadiazolyl, thiadiazolyl)
An amino group which may be mono- or di-substituted with a substituent selected from:
(6) a C 1-6 alkyl-carbonyl group optionally substituted with 1 to 3 halogen atoms;
(7) (a) a halogen atom,
(b) a C 1-6 alkoxy group,
(c) a C 6-14 aryl group (eg, phenyl), and (d) a heterocyclic group (eg, tetrahydrofuryl).
A C 1-6 alkoxy-carbonyl group optionally substituted by 1 to 3 substituents selected from:
(8) a C 1-6 alkylsulfonyl group which may be substituted with 1 to 3 halogen atoms (eg, methylsulfonyl, ethylsulfonyl, isopropylsulfonyl);
(9) a carbamoyl group which may be mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
(10) a thiocarbamoyl group optionally mono- or disubstituted with a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms;
(11) a sulfamoyl group optionally mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
(12) a carboxy group;
(13) a hydroxy group;
(14) (a) a halogen atom,
(b) a carboxy group,
(c) a C 1-6 alkoxy group,
(d) a C 1-6 alkoxy-carbonyl group optionally substituted by 1 to 3 C 6-14 aryl groups (eg, phenyl),
(e) an amino group which may be mono- or di-substituted with a substituent selected from a C 1-6 alkyl group and a C 1-6 alkoxy-carbonyl group,
(f) a heterocyclic group (eg, tetrahydrofuryl), and (g) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl).
A C 1-6 alkoxy group which may be substituted with 1 to 3 substituents selected from:
(15) a C 2-6 alkenyloxy group (eg, ethenyloxy) optionally substituted by 1 to 3 halogen atoms;
(16) C 7-13 aralkyloxy group (eg, benzyloxy);
(17) C 6-14 aryloxy group (eg, phenyloxy, naphthyloxy);
(18) C 1-6 alkyl-carbonyloxy group (eg, acetyloxy, tert-butylcarbonyloxy);
(19) optionally substituted with 1 to 3 substituents selected from (a) a halogen atom, and (b) a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms A C 6-14 aryl-carbonyl group (eg, benzoyl);
(20) a non-aromatic heterocyclic carbonyl group optionally substituted with 1 to 3 substituents selected from C 1-6 alkyl groups optionally substituted with 1 to 3 halogen atoms (eg, Pyrrolidinylcarbonyl, morpholinylcarbonyl);
(21) a sulfanyl group;
(22) a C 1-6 alkylsulfanyl group (eg, methylsulfanyl) optionally substituted with 1 to 3 substituents selected from (a) a halogen atom, and (b) a C 1-6 alkoxy-carbonyl group Ethylsulfanyl);
(23) C 7-13 aralkylsulfanyl group (eg, benzylsulfanyl);
(24) C 6-14 arylsulfanyl group (eg, phenylsulfanyl, naphthylsulfanyl);
(25) a cyano group;
(26) a nitro group;
(27) a halogen atom;
(28) a C 1-3 alkylenedioxy group;
(29) C 1-3 alkyleneoxy group (eg, methyleneoxy, ethyleneoxy);
(30) an aromatic heterocyclic carbonyl group optionally substituted with 1 to 3 substituents selected from C 1-6 alkyl groups optionally substituted with 1 to 3 halogen atoms (eg, pyrazolyl) Carbonyl, pyrazinylcarbonyl, isoxazolylcarbonyl, pyridylcarbonyl, thiazolylcarbonyl);
(31) (a) a halogen atom (eg, fluorine atom), and (b) a C 1-6 alkoxy group (eg, methoxy)
C 3-10 cycloalkoxy group optionally substituted by 1 to 3 substituents selected from (eg, cyclopropoxy, cyclopentyloxy)
Etc. When there are two or more substituents, each substituent may be the same or different.
(1)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル);
(2)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール基(例、フェニル、ナフチル);
(3)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよい芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(d)ハロゲン原子、および
(e)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい非芳香族複素環基(例、テトラヒドロフリル、モルホリニル、チオモルホリニル、ピペリジニル、ピロリジニル、ピペラジニル);
(5)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基、
(d)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル)、
(e)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基、および
(f)芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル)
から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基;
(6)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基;
(7)(a)ハロゲン原子、
(b)C1-6アルコキシ基、
(c)C6-14アリール基(例、フェニル)、および
(d)複素環基(例、テトラヒドロフリル)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ-カルボニル基;
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基;
(10)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいチオカルバモイル基;
(11)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいスルファモイル基;
(12)カルボキシ基;
(13)ヒドロキシ基;
(14)(a)ハロゲン原子、
(b)カルボキシ基、
(c)C1-6アルコキシ基、
(d)1ないし3個のC6-14アリール基(例、フェニル)で置換されていてもよいC1-6アルコキシ-カルボニル基、
(e)C1-6アルキル基およびC1-6アルコキシ-カルボニル基から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基、
(f)複素環基(例、テトラヒドロフリル)、および
(g)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ基;
(15)1ないし3個のハロゲン原子で置換されていてもよいC2-6アルケニルオキシ基(例、エテニルオキシ);
(16)C7-13アラルキルオキシ基(例、ベンジルオキシ);
(17)C6-14アリールオキシ基(例、フェニルオキシ、ナフチルオキシ);
(18)C1-6アルキル-カルボニルオキシ基(例、アセチルオキシ、tert-ブチルカルボニルオキシ);
(19)(a)ハロゲン原子、および
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル);
(20)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい非芳香族複素環カルボニル基(例、ピロリジニルカルボニル、モルホリニルカルボニル);
(21)スルファニル基;
(22)(a)ハロゲン原子、および
(b)C1-6アルコキシ-カルボニル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキルスルファニル基(例、メチルスルファニル、エチルスルファニル);
(23)C7-13アラルキルスルファニル基(例、ベンジルスルファニル);
(24)C6-14アリールスルファニル基(例、フェニルスルファニル、ナフチルスルファニル);
(25)シアノ基;
(26)ニトロ基;
(27)ハロゲン原子;
(28)C1-3アルキレンジオキシ基;
(29)C1-3アルキレンオキシ基(例、メチレンオキシ、エチレンオキシ);
(30)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
から選ばれる1ないし3個の置換基で置換されていてもよい芳香族複素環カルボニル基(例、ピラゾリルカルボニル、ピラジニルカルボニル、イソキサゾリルカルボニル、ピリジルカルボニル、チアゾリルカルボニル);
(31)(a)ハロゲン原子(例、フッ素原子)、および
(b)C1-6アルコキシ基(例、メトキシ)
から選ばれる1ないし3個の置換基で置換されていてもよいC3-10シクロアルコキシ基(例、シクロプロポキシ、シクロペンチルオキシ)
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。 As such a substituent, for example,
(1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl);
(2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms,
(b) a hydroxy group,
(c) a C 1-6 alkoxy group optionally substituted with 1 to 3 halogen atoms, and (d) a C 6- optionally substituted with 1 to 3 substituents selected from halogen atoms. 14 aryl groups (eg, phenyl, naphthyl);
(3) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms,
(b) a hydroxy group,
(c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, and (d) an aromatic complex which may be substituted with 1 to 3 substituents selected from halogen atoms. A cyclic group (eg, thienyl, furyl, pyridyl, pyrazolyl, imidazolyl, tetrazolyl, oxazolyl, thiazolyl, oxadiazolyl, thiadiazolyl);
(4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms,
(b) a hydroxy group,
(c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms,
(d) a non-aromatic heterocyclic group (eg, tetrahydrofuryl, morpholinyl, thiomorpholinyl, piperidinyl, pyrrolidinyl, piperazinyl) optionally substituted with 1 to 3 substituents selected from halogen atoms and (e) oxo groups );
(5) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms,
(b) a C 1-6 alkyl-carbonyl group optionally substituted by 1 to 3 halogen atoms,
(c) a C 1-6 alkoxy-carbonyl group optionally substituted by 1 to 3 halogen atoms,
(d) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl) optionally substituted with 1 to 3 halogen atoms,
(e) a carbamoyl group optionally mono- or di-substituted with a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms, and (f) an aromatic heterocyclic group (eg, thienyl, Furyl, pyridyl, pyrazolyl, imidazolyl, tetrazolyl, oxazolyl, thiazolyl, oxadiazolyl, thiadiazolyl)
An amino group which may be mono- or di-substituted with a substituent selected from:
(6) a C 1-6 alkyl-carbonyl group optionally substituted with 1 to 3 halogen atoms;
(7) (a) a halogen atom,
(b) a C 1-6 alkoxy group,
(c) a C 6-14 aryl group (eg, phenyl), and (d) a heterocyclic group (eg, tetrahydrofuryl).
A C 1-6 alkoxy-carbonyl group optionally substituted by 1 to 3 substituents selected from:
(8) a C 1-6 alkylsulfonyl group which may be substituted with 1 to 3 halogen atoms (eg, methylsulfonyl, ethylsulfonyl, isopropylsulfonyl);
(9) a carbamoyl group which may be mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
(10) a thiocarbamoyl group optionally mono- or disubstituted with a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms;
(11) a sulfamoyl group optionally mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
(12) a carboxy group;
(13) a hydroxy group;
(14) (a) a halogen atom,
(b) a carboxy group,
(c) a C 1-6 alkoxy group,
(d) a C 1-6 alkoxy-carbonyl group optionally substituted by 1 to 3 C 6-14 aryl groups (eg, phenyl),
(e) an amino group which may be mono- or di-substituted with a substituent selected from a C 1-6 alkyl group and a C 1-6 alkoxy-carbonyl group,
(f) a heterocyclic group (eg, tetrahydrofuryl), and (g) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl).
A C 1-6 alkoxy group which may be substituted with 1 to 3 substituents selected from:
(15) a C 2-6 alkenyloxy group (eg, ethenyloxy) optionally substituted by 1 to 3 halogen atoms;
(16) C 7-13 aralkyloxy group (eg, benzyloxy);
(17) C 6-14 aryloxy group (eg, phenyloxy, naphthyloxy);
(18) C 1-6 alkyl-carbonyloxy group (eg, acetyloxy, tert-butylcarbonyloxy);
(19) optionally substituted with 1 to 3 substituents selected from (a) a halogen atom, and (b) a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms A C 6-14 aryl-carbonyl group (eg, benzoyl);
(20) a non-aromatic heterocyclic carbonyl group optionally substituted with 1 to 3 substituents selected from C 1-6 alkyl groups optionally substituted with 1 to 3 halogen atoms (eg, Pyrrolidinylcarbonyl, morpholinylcarbonyl);
(21) a sulfanyl group;
(22) a C 1-6 alkylsulfanyl group (eg, methylsulfanyl) optionally substituted with 1 to 3 substituents selected from (a) a halogen atom, and (b) a C 1-6 alkoxy-carbonyl group Ethylsulfanyl);
(23) C 7-13 aralkylsulfanyl group (eg, benzylsulfanyl);
(24) C 6-14 arylsulfanyl group (eg, phenylsulfanyl, naphthylsulfanyl);
(25) a cyano group;
(26) a nitro group;
(27) a halogen atom;
(28) a C 1-3 alkylenedioxy group;
(29) C 1-3 alkyleneoxy group (eg, methyleneoxy, ethyleneoxy);
(30) an aromatic heterocyclic carbonyl group optionally substituted with 1 to 3 substituents selected from C 1-6 alkyl groups optionally substituted with 1 to 3 halogen atoms (eg, pyrazolyl) Carbonyl, pyrazinylcarbonyl, isoxazolylcarbonyl, pyridylcarbonyl, thiazolylcarbonyl);
(31) (a) a halogen atom (eg, fluorine atom), and (b) a C 1-6 alkoxy group (eg, methoxy)
C 3-10 cycloalkoxy group optionally substituted by 1 to 3 substituents selected from (eg, cyclopropoxy, cyclopentyloxy)
Etc. When there are two or more substituents, each substituent may be the same or different.
また、前記「炭化水素基」として例示した、C3-10シクロアルキル基、C3-10シクロアルケニル基、C4-10シクロアルカジエニル基、C6-14アリール基、C7-13アラルキル基およびC8-13アリールアルケニル基は、置換可能な位置に1ないし3個の置換基を有していてもよい。
In addition, the C 3-10 cycloalkyl group, C 3-10 cycloalkenyl group, C 4-10 cycloalkadienyl group, C 6-14 aryl group, C 7-13 aralkyl exemplified as the “hydrocarbon group” The group and the C 8-13 arylalkenyl group may have 1 to 3 substituents at substitutable positions.
このような置換基としては、例えば、
(1)前記したC1-10アルキル基等における置換基として例示した基;
(2)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基;
(3)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC2-6アルケニル基(例、エテニル、1-プロペニル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)C1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC7-13アラルキル基(例、ベンジル);
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。 As such a substituent, for example,
(1) groups exemplified as substituents in the aforementioned C 1-10 alkyl group and the like;
(2) (a) a halogen atom,
(b) a carboxy group,
(c) a hydroxy group,
(d) a C 1-6 alkoxy-carbonyl group,
(e) a C 1-6 alkoxy group, and (f) a C 1-6 alkyl group optionally substituted with 1 to 3 substituents selected from amino groups optionally mono- or di-substituted. A 1-6 alkyl group;
(3) (a) a halogen atom,
(b) a carboxy group,
(c) a hydroxy group,
(d) a C 1-6 alkoxy-carbonyl group,
(e) a C 1-6 alkoxy group, and (f) a C 1-6 alkyl group optionally substituted with 1 to 3 substituents selected from amino groups optionally mono- or di-substituted. 2-6 alkenyl groups (eg, ethenyl, 1-propenyl);
(4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms,
(b) a hydroxy group,
(c) a C 1-6 alkoxy group, and (d) a C 7-13 aralkyl group (eg, benzyl) optionally substituted with 1 to 3 substituents selected from halogen atoms;
Etc. When there are two or more substituents, each substituent may be the same or different.
(1)前記したC1-10アルキル基等における置換基として例示した基;
(2)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基;
(3)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC2-6アルケニル基(例、エテニル、1-プロペニル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)C1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC7-13アラルキル基(例、ベンジル);
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。 As such a substituent, for example,
(1) groups exemplified as substituents in the aforementioned C 1-10 alkyl group and the like;
(2) (a) a halogen atom,
(b) a carboxy group,
(c) a hydroxy group,
(d) a C 1-6 alkoxy-carbonyl group,
(e) a C 1-6 alkoxy group, and (f) a C 1-6 alkyl group optionally substituted with 1 to 3 substituents selected from amino groups optionally mono- or di-substituted. A 1-6 alkyl group;
(3) (a) a halogen atom,
(b) a carboxy group,
(c) a hydroxy group,
(d) a C 1-6 alkoxy-carbonyl group,
(e) a C 1-6 alkoxy group, and (f) a C 1-6 alkyl group optionally substituted with 1 to 3 substituents selected from amino groups optionally mono- or di-substituted. 2-6 alkenyl groups (eg, ethenyl, 1-propenyl);
(4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms,
(b) a hydroxy group,
(c) a C 1-6 alkoxy group, and (d) a C 7-13 aralkyl group (eg, benzyl) optionally substituted with 1 to 3 substituents selected from halogen atoms;
Etc. When there are two or more substituents, each substituent may be the same or different.
前記「置換されていてもよい複素環基」における「複素環基」としては、芳香族複素環基および非芳香族複素環基が挙げられる。
The “heterocyclic group” in the “optionally substituted heterocyclic group” includes an aromatic heterocyclic group and a non-aromatic heterocyclic group.
ここで、芳香族複素環基としては、例えば、環構成原子として炭素原子以外に酸素原子、硫黄原子および窒素原子から選ばれるヘテロ原子を1ないし4個含有する4ないし7員(好ましくは5または6員)の単環式芳香族複素環基および縮合芳香族複素環基が挙げられる。該縮合芳香族複素環基としては、例えば、これら4ないし7員の単環式芳香族複素環基に対応する環と、1または2個の窒素原子を含む5または6員の芳香族複素環(例、ピロール、イミダゾール、ピラゾール、ピラジン、ピリジン、ピリミジン)、1個の硫黄原子を含む5員の芳香族複素環(例、チオフェン)およびベンゼン環から選ばれる1または2個が縮合した環から誘導される基等が挙げられる。
Here, the aromatic heterocyclic group is, for example, a 4 to 7 member (preferably 5 or 5) containing 1 to 4 heteroatoms selected from an oxygen atom, a sulfur atom and a nitrogen atom in addition to a carbon atom as a ring constituent atom. 6-membered) monocyclic aromatic heterocyclic group and condensed aromatic heterocyclic group. Examples of the condensed aromatic heterocyclic group include a ring corresponding to the 4- to 7-membered monocyclic aromatic heterocyclic group and a 5- or 6-membered aromatic heterocyclic ring containing 1 or 2 nitrogen atoms. (Eg, pyrrole, imidazole, pyrazole, pyrazine, pyridine, pyrimidine) From a ring in which 1 or 2 selected from a 5-membered aromatic heterocycle containing one sulfur atom (eg, thiophene) and a benzene ring are condensed Examples include groups to be derived.
芳香族複素環基の好適な例としては、
フリル(例、2-フリル、3-フリル)、チエニル(例、2-チエニル、3-チエニル)、ピリジル(例、2-ピリジル、3-ピリジル、4-ピリジル)、ピリミジニル(例、2-ピリミジニル、4-ピリミジニル、5-ピリミジニル)、ピリダジニル(例、3-ピリダジニル、4-ピリダジニル)、ピラジニル(例、2-ピラジニル)、ピロリル(例、1-ピロリル、2-ピロリル、3-ピロリル)、イミダゾリル(例、1-イミダゾリル、2-イミダゾリル、4-イミダゾリル、5-イミダゾリル)、ピラゾリル(例、1-ピラゾリル、3-ピラゾリル、4-ピラゾリル)、チアゾリル(例、2-チアゾリル、4-チアゾリル、5-チアゾリル)、イソチアゾリル(例、3-イソチアゾリル、4-イソチアゾリル、5-イソチアゾリル)、オキサゾリル(例、2-オキサゾリル、4-オキサゾリル、5-オキサゾリル)、イソオキサゾリル(例、3-イソオキサゾリル、4-イソオキサゾリル、5-イソオキサゾリル)、オキサジアゾリル(例、1,2,4-オキサジアゾール-5-イル、1,3,4-オキサジアゾール-2-イル)、チアジアゾリル(例、1,3,4-チアジアゾール-2-イル)、トリアゾリル(例、1,2,4-トリアゾール-1-イル、1,2,4-トリアゾール-3-イル、1,2,3-トリアゾール-1-イル、1,2,3-トリアゾール-2-イル、1,2,3-トリアゾール-4-イル)、テトラゾリル(例、テトラゾール-1-イル、テトラゾール-5-イル)、トリアジニル(例、1,2,4-トリアジン-1-イル、1,2,4-トリアジン-3-イル)等の単環式芳香族複素環基;
キノリル(例、2-キノリル、3-キノリル、4-キノリル、6-キノリル)、イソキノリル(例、3-イソキノリル)、キナゾリル(例、2-キナゾリル、4-キナゾリル)、キノキサリル(例、2-キノキサリル、6-キノキサリル)、ベンゾフラニル(例、2-ベンゾフラニル、3-ベンゾフラニル)、ベンゾチエニル(例、2-ベンゾチエニル、3-ベンゾチエニル)、ベンズオキサゾリル(例、2-ベンズオキサゾリル)、ベンズイソオキサゾリル(例、7-ベンズイソオキサゾリル)、ベンゾチアゾリル(例、2-ベンゾチアゾリル)、ベンズイミダゾリル(例、ベンズイミダゾール-1-イル、ベンズイミダゾール-2-イル、ベンズイミダゾール-5-イル)、ベンゾトリアゾリル(例、1H-1,2,3-ベンゾトリアゾール-5-イル)、インドリル(例、インドール-1-イル、インドール-2-イル、インドール-3-イル、インドール-5-イル)、インダゾリル(例、1H-インダゾール-3-イル)、ピロロピラジニル(例、1H-ピロロ[2,3-b]ピラジン-2-イル、1H-ピロロ[2,3-b]ピラジン-6-イル)、イミダゾピリジニル(例、1H-イミダゾ[4,5-b]ピリジン-2-イル、1H-イミダゾ[4,5-c]ピリジン-2-イル、2H-イミダゾ[1,2-a]ピリジン-3-イル)、チエノピリジニル(例、チエノ[2,3-b]ピリジン-3-イル)、イミダゾピラジニル(例、1H-イミダゾ[4,5-b]ピラジン-2-イル)、ピラゾロピリジニル(例、1H-ピラゾロ[4,3-c]ピリジン-3-イル)、ピラゾロチエニル(例、2H-ピラゾロ[3,4-b]チオフェン-2-イル)、ピラゾロトリアジニル(例、ピラゾロ[5,1-c][1,2,4]トリアジン-3-イル)、ピリドピリジニル(例、ピリド[2,3-b]ピリジン-3-イル)、チエノピリジル(例、チエノ[2,3-b]ピリジン-3-イル)等の縮合芳香族複素環基;
等が挙げられる。 As preferable examples of the aromatic heterocyclic group,
Furyl (eg, 2-furyl, 3-furyl), thienyl (eg, 2-thienyl, 3-thienyl), pyridyl (eg, 2-pyridyl, 3-pyridyl, 4-pyridyl), pyrimidinyl (eg, 2-pyrimidinyl) 4-pyrimidinyl, 5-pyrimidinyl), pyridazinyl (eg, 3-pyridazinyl, 4-pyridazinyl), pyrazinyl (eg, 2-pyrazinyl), pyrrolyl (eg, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl), imidazolyl (Eg, 1-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl), pyrazolyl (eg, 1-pyrazolyl, 3-pyrazolyl, 4-pyrazolyl), thiazolyl (eg, 2-thiazolyl, 4-thiazolyl, 5 -Thiazolyl), isothiazolyl (eg, 3-isothiazolyl, 4-isothiazolyl, 5-isothiazolyl) Oxazolyl (eg, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl), isoxazolyl (eg, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl), oxadiazolyl (eg, 1,2,4-oxadiazole-5- Yl, 1,3,4-oxadiazol-2-yl), thiadiazolyl (eg, 1,3,4-thiadiazol-2-yl), triazolyl (eg, 1,2,4-triazol-1-yl), 1,2,4-triazol-3-yl, 1,2,3-triazol-1-yl, 1,2,3-triazol-2-yl, 1,2,3-triazol-4-yl), tetrazolyl (Eg, tetrazol-1-yl, tetrazol-5-yl), triazinyl (eg, 1,2,4-triazin-1-yl, 1,2,4-tri 3-yl) monocyclic aromatic heterocyclic group and the like;
Quinolyl (eg, 2-quinolyl, 3-quinolyl, 4-quinolyl, 6-quinolyl), isoquinolyl (eg, 3-isoquinolyl), quinazolyl (eg, 2-quinazolyl, 4-quinazolyl), quinoxalyl (eg, 2-quinoxalyl) , 6-quinoxalyl), benzofuranyl (eg, 2-benzofuranyl, 3-benzofuranyl), benzothienyl (eg, 2-benzothienyl, 3-benzothienyl), benzoxazolyl (eg, 2-benzoxazolyl), Benzisoxazolyl (eg, 7-benzisoxazolyl), benzothiazolyl (eg, 2-benzothiazolyl), benzimidazolyl (eg, benzimidazol-1-yl, benzimidazol-2-yl, benzimidazol-5- Yl), benzotriazolyl (eg, 1H-1,2,3-benzotriazo Ruyl-5-yl), indolyl (eg, indol-1-yl, indol-2-yl, indol-3-yl, indol-5-yl), indazolyl (eg, 1H-indazol-3-yl), pyrrolopyrazinyl (Eg, 1H-pyrrolo [2,3-b] pyrazin-2-yl, 1H-pyrrolo [2,3-b] pyrazin-6-yl), imidazopyridinyl (eg, 1H-imidazo [4,5 -B] pyridin-2-yl, 1H-imidazo [4,5-c] pyridin-2-yl, 2H-imidazo [1,2-a] pyridin-3-yl), thienopyridinyl (eg, thieno [2, 3-b] pyridin-3-yl), imidazopyrazinyl (eg, 1H-imidazo [4,5-b] pyrazin-2-yl), pyrazolopyridinyl (eg, 1H-pyrazolo [4,3 -C] pyridine-3-i ), Pyrazolothienyl (eg, 2H-pyrazolo [3,4-b] thiophen-2-yl), pyrazolotriazinyl (eg, pyrazolo [5,1-c] [1,2,4] triazine-3- Yl), pyridopyridinyl (eg, pyrido [2,3-b] pyridin-3-yl), thienopyridyl (eg, thieno [2,3-b] pyridin-3-yl) and the like;
Etc.
フリル(例、2-フリル、3-フリル)、チエニル(例、2-チエニル、3-チエニル)、ピリジル(例、2-ピリジル、3-ピリジル、4-ピリジル)、ピリミジニル(例、2-ピリミジニル、4-ピリミジニル、5-ピリミジニル)、ピリダジニル(例、3-ピリダジニル、4-ピリダジニル)、ピラジニル(例、2-ピラジニル)、ピロリル(例、1-ピロリル、2-ピロリル、3-ピロリル)、イミダゾリル(例、1-イミダゾリル、2-イミダゾリル、4-イミダゾリル、5-イミダゾリル)、ピラゾリル(例、1-ピラゾリル、3-ピラゾリル、4-ピラゾリル)、チアゾリル(例、2-チアゾリル、4-チアゾリル、5-チアゾリル)、イソチアゾリル(例、3-イソチアゾリル、4-イソチアゾリル、5-イソチアゾリル)、オキサゾリル(例、2-オキサゾリル、4-オキサゾリル、5-オキサゾリル)、イソオキサゾリル(例、3-イソオキサゾリル、4-イソオキサゾリル、5-イソオキサゾリル)、オキサジアゾリル(例、1,2,4-オキサジアゾール-5-イル、1,3,4-オキサジアゾール-2-イル)、チアジアゾリル(例、1,3,4-チアジアゾール-2-イル)、トリアゾリル(例、1,2,4-トリアゾール-1-イル、1,2,4-トリアゾール-3-イル、1,2,3-トリアゾール-1-イル、1,2,3-トリアゾール-2-イル、1,2,3-トリアゾール-4-イル)、テトラゾリル(例、テトラゾール-1-イル、テトラゾール-5-イル)、トリアジニル(例、1,2,4-トリアジン-1-イル、1,2,4-トリアジン-3-イル)等の単環式芳香族複素環基;
キノリル(例、2-キノリル、3-キノリル、4-キノリル、6-キノリル)、イソキノリル(例、3-イソキノリル)、キナゾリル(例、2-キナゾリル、4-キナゾリル)、キノキサリル(例、2-キノキサリル、6-キノキサリル)、ベンゾフラニル(例、2-ベンゾフラニル、3-ベンゾフラニル)、ベンゾチエニル(例、2-ベンゾチエニル、3-ベンゾチエニル)、ベンズオキサゾリル(例、2-ベンズオキサゾリル)、ベンズイソオキサゾリル(例、7-ベンズイソオキサゾリル)、ベンゾチアゾリル(例、2-ベンゾチアゾリル)、ベンズイミダゾリル(例、ベンズイミダゾール-1-イル、ベンズイミダゾール-2-イル、ベンズイミダゾール-5-イル)、ベンゾトリアゾリル(例、1H-1,2,3-ベンゾトリアゾール-5-イル)、インドリル(例、インドール-1-イル、インドール-2-イル、インドール-3-イル、インドール-5-イル)、インダゾリル(例、1H-インダゾール-3-イル)、ピロロピラジニル(例、1H-ピロロ[2,3-b]ピラジン-2-イル、1H-ピロロ[2,3-b]ピラジン-6-イル)、イミダゾピリジニル(例、1H-イミダゾ[4,5-b]ピリジン-2-イル、1H-イミダゾ[4,5-c]ピリジン-2-イル、2H-イミダゾ[1,2-a]ピリジン-3-イル)、チエノピリジニル(例、チエノ[2,3-b]ピリジン-3-イル)、イミダゾピラジニル(例、1H-イミダゾ[4,5-b]ピラジン-2-イル)、ピラゾロピリジニル(例、1H-ピラゾロ[4,3-c]ピリジン-3-イル)、ピラゾロチエニル(例、2H-ピラゾロ[3,4-b]チオフェン-2-イル)、ピラゾロトリアジニル(例、ピラゾロ[5,1-c][1,2,4]トリアジン-3-イル)、ピリドピリジニル(例、ピリド[2,3-b]ピリジン-3-イル)、チエノピリジル(例、チエノ[2,3-b]ピリジン-3-イル)等の縮合芳香族複素環基;
等が挙げられる。 As preferable examples of the aromatic heterocyclic group,
Furyl (eg, 2-furyl, 3-furyl), thienyl (eg, 2-thienyl, 3-thienyl), pyridyl (eg, 2-pyridyl, 3-pyridyl, 4-pyridyl), pyrimidinyl (eg, 2-pyrimidinyl) 4-pyrimidinyl, 5-pyrimidinyl), pyridazinyl (eg, 3-pyridazinyl, 4-pyridazinyl), pyrazinyl (eg, 2-pyrazinyl), pyrrolyl (eg, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl), imidazolyl (Eg, 1-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl), pyrazolyl (eg, 1-pyrazolyl, 3-pyrazolyl, 4-pyrazolyl), thiazolyl (eg, 2-thiazolyl, 4-thiazolyl, 5 -Thiazolyl), isothiazolyl (eg, 3-isothiazolyl, 4-isothiazolyl, 5-isothiazolyl) Oxazolyl (eg, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl), isoxazolyl (eg, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl), oxadiazolyl (eg, 1,2,4-oxadiazole-5- Yl, 1,3,4-oxadiazol-2-yl), thiadiazolyl (eg, 1,3,4-thiadiazol-2-yl), triazolyl (eg, 1,2,4-triazol-1-yl), 1,2,4-triazol-3-yl, 1,2,3-triazol-1-yl, 1,2,3-triazol-2-yl, 1,2,3-triazol-4-yl), tetrazolyl (Eg, tetrazol-1-yl, tetrazol-5-yl), triazinyl (eg, 1,2,4-triazin-1-yl, 1,2,4-tri 3-yl) monocyclic aromatic heterocyclic group and the like;
Quinolyl (eg, 2-quinolyl, 3-quinolyl, 4-quinolyl, 6-quinolyl), isoquinolyl (eg, 3-isoquinolyl), quinazolyl (eg, 2-quinazolyl, 4-quinazolyl), quinoxalyl (eg, 2-quinoxalyl) , 6-quinoxalyl), benzofuranyl (eg, 2-benzofuranyl, 3-benzofuranyl), benzothienyl (eg, 2-benzothienyl, 3-benzothienyl), benzoxazolyl (eg, 2-benzoxazolyl), Benzisoxazolyl (eg, 7-benzisoxazolyl), benzothiazolyl (eg, 2-benzothiazolyl), benzimidazolyl (eg, benzimidazol-1-yl, benzimidazol-2-yl, benzimidazol-5- Yl), benzotriazolyl (eg, 1H-1,2,3-benzotriazo Ruyl-5-yl), indolyl (eg, indol-1-yl, indol-2-yl, indol-3-yl, indol-5-yl), indazolyl (eg, 1H-indazol-3-yl), pyrrolopyrazinyl (Eg, 1H-pyrrolo [2,3-b] pyrazin-2-yl, 1H-pyrrolo [2,3-b] pyrazin-6-yl), imidazopyridinyl (eg, 1H-imidazo [4,5 -B] pyridin-2-yl, 1H-imidazo [4,5-c] pyridin-2-yl, 2H-imidazo [1,2-a] pyridin-3-yl), thienopyridinyl (eg, thieno [2, 3-b] pyridin-3-yl), imidazopyrazinyl (eg, 1H-imidazo [4,5-b] pyrazin-2-yl), pyrazolopyridinyl (eg, 1H-pyrazolo [4,3 -C] pyridine-3-i ), Pyrazolothienyl (eg, 2H-pyrazolo [3,4-b] thiophen-2-yl), pyrazolotriazinyl (eg, pyrazolo [5,1-c] [1,2,4] triazine-3- Yl), pyridopyridinyl (eg, pyrido [2,3-b] pyridin-3-yl), thienopyridyl (eg, thieno [2,3-b] pyridin-3-yl) and the like;
Etc.
非芳香族複素環基としては、例えば、環構成原子として炭素原子以外に酸素原子、硫黄原子および窒素原子から選ばれるヘテロ原子を1ないし4個含有する4ないし7員(好ましくは5または6員)の単環式非芳香族複素環基および縮合非芳香族複素環基が挙げられる。該縮合非芳香族複素環基としては、例えば、これら4ないし7員の単環式非芳香族複素環基に対応する環と、1または2個の窒素原子を含む5または6員の芳香族複素環(例、ピロール、イミダゾール、ピラゾール、ピラジン、ピリジン、ピリミジン)、1個の硫黄原子を含む5員の芳香族複素環(例、チオフェン)およびベンゼン環から選ばれる1または2個の環が縮合した環から誘導される基、ならびに該基の部分飽和により得られる基等が挙げられる。
Examples of the non-aromatic heterocyclic group include 4 to 7 members (preferably 5 or 6 members) containing 1 to 4 heteroatoms selected from oxygen atoms, sulfur atoms and nitrogen atoms in addition to carbon atoms as ring constituent atoms. ) Monocyclic non-aromatic heterocyclic group and condensed non-aromatic heterocyclic group. Examples of the condensed non-aromatic heterocyclic group include a ring corresponding to the 4- to 7-membered monocyclic non-aromatic heterocyclic group, and a 5- or 6-membered aromatic containing 1 or 2 nitrogen atoms. 1 or 2 rings selected from a heterocycle (eg, pyrrole, imidazole, pyrazole, pyrazine, pyridine, pyrimidine), a 5-membered aromatic heterocycle containing one sulfur atom (eg, thiophene) and a benzene ring Examples thereof include a group derived from a condensed ring and a group obtained by partial saturation of the group.
非芳香族複素環基の好適な例としては、
アゼチジニル(例、1-アゼチジニル、2-アゼチジニル、3-アゼチジニル)、ピロリジニル(例、1-ピロリジニル、2-ピロリジニル)、ピペリジニル(例、ピペリジノ、2-ピペリジニル、3-ピペリジニル、4-ピペリジニル)、モルホリニル(例、モルホリノ)、チオモルホリニル(例、チオモルホリノ)、ピペラジニル(例、1-ピペラジニル、2-ピペラジニル、3-ピペラジニル)、ヘキサメチレンイミニル(例、ヘキサメチレンイミン-1-イル)、オキサゾリジニル(例、オキサゾリジン-2-イル)、チアゾリジニル(例、チアゾリジン-2-イル)、イミダゾリジニル(例、イミダゾリジン-2-イル、イミダゾリジン-3-イル)、オキサゾリニル(例、オキサゾリン-2-イル)、チアゾリニル(例、チアゾリン-2-イル)、イミダゾリニル(例、イミダゾリン-2-イル、イミダゾリン-3-イル)、ジオキソリル(例、1,3-ジオキソール-4-イル)、ジオキソラニル(例、1,3-ジオキソラン-4-イル)、ジヒドロオキサジアゾリル(例、4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)、ピラニル(例、4-ピラニル)、テトラヒドロピラニル(例、2-テトラヒドロピラニル、3-テトラヒドロピラニル、4-テトラヒドロピラニル)、チオピラニル(例、4-チオピラニル)、テトラヒドロチオピラニル(例、2-テトラヒドロチオピラニル、3-テトラヒドロチオピラニル、4-テトラヒドロチオピラニル)、テトラヒドロフリル(例、テトラヒドロフラン-3-イル、テトラヒドロフラン-2-イル)、ピラゾリジニル(例、ピラゾリジン-1-イル、ピラゾリジン-3-イル)、ピラゾリニル(例、ピラゾリン-1-イル)、テトラヒドロピリミジニル(例、テトラヒドロピリミジン-1-イル)、ジヒドロトリアゾリル(例、2,3-ジヒドロ-1H-1,2,3-トリアゾール-1-イル)、テトラヒドロトリアゾリル(例、2,3,4,5-テトラヒドロ-1H-1,2,3-トリアゾール-1-イル)等の単環式非芳香族複素環基;
ジヒドロインドリル(例、2,3-ジヒドロ-1H-インドール-1-イル)、ジヒドロイソインドリル(例、1,3-ジヒドロ-2H-イソインドール-2-イル)、ジヒドロベンゾフラニル(例、2,3-ジヒドロ-1-ベンゾフラン-5-イル)、ジヒドロベンゾジオキシニル(例、2,3-ジヒドロ-1,4-ベンゾジオキシニル)、ジヒドロベンゾジオキセピニル(例、3,4-ジヒドロ-2H-1,5-ベンゾジオキセピニル)、テトラヒドロベンゾフラニル(例、4,5,6,7-テトラヒドロ-1-ベンゾフラン-3-イル)、クロメニル(例、4H-クロメン-2-イル、2H-クロメン-3-イル)、ジヒドロクロメニル(例、3,4-ジヒドロ-2H-クロメン-2-イル)、ジヒドロキノリニル(例、1,2-ジヒドロキノリン-4-イル)、テトラヒドロキノリニル(例、1,2,3,4-テトラヒドロキノリン-4-イル)、ジヒドロイソキノリニル(例、1,2-ジヒドロイソキノリン-4-イル)、テトラヒドロイソキノリニル(例、1,2,3,4-テトラヒドロイソキノリン-4-イル)、ジヒドロフタラジニル(例、1,4-ジヒドロフタラジン-4-イル)等の縮合非芳香族複素環基;
等が挙げられる。 As a suitable example of a non-aromatic heterocyclic group,
Azetidinyl (eg, 1-azetidinyl, 2-azetidinyl, 3-azetidinyl), pyrrolidinyl (eg, 1-pyrrolidinyl, 2-pyrrolidinyl), piperidinyl (eg, piperidino, 2-piperidinyl, 3-piperidinyl, 4-piperidinyl), morpholinyl (Eg, morpholino), thiomorpholinyl (eg, thiomorpholino), piperazinyl (eg, 1-piperazinyl, 2-piperazinyl, 3-piperazinyl), hexamethyleneiminyl (eg, hexamethyleneimin-1-yl), oxazolidinyl (eg, Oxazolidin-2-yl), thiazolidinyl (eg, thiazolidin-2-yl), imidazolidinyl (eg, imidazolidin-2-yl, imidazolidin-3-yl), oxazolinyl (eg, oxazolin-2-yl), thiazolinyl (Eg, thiazo -2-yl), imidazolinyl (eg, imidazolin-2-yl, imidazolin-3-yl), dioxolyl (eg, 1,3-dioxol-4-yl), dioxolanyl (eg, 1,3-dioxolane-4) -Yl), dihydrooxadiazolyl (eg, 4,5-dihydro-1,2,4-oxadiazol-3-yl), pyranyl (eg, 4-pyranyl), tetrahydropyranyl (eg, 2-tetrahydro) Pyranyl, 3-tetrahydropyranyl, 4-tetrahydropyranyl), thiopyranyl (eg, 4-thiopyranyl), tetrahydrothiopyranyl (eg, 2-tetrahydrothiopyranyl, 3-tetrahydrothiopyranyl, 4-tetrahydrothio) Pyranyl), tetrahydrofuryl (eg, tetrahydrofuran-3-yl, tetrahydrofuran-2-yl), Lazolidinyl (eg, pyrazolidin-1-yl, pyrazolidin-3-yl), pyrazolinyl (eg, pyrazolin-1-yl), tetrahydropyrimidinyl (eg, tetrahydropyrimidin-1-yl), dihydrotriazolyl (eg, 2, 3-dihydro-1H-1,2,3-triazol-1-yl), tetrahydrotriazolyl (eg, 2,3,4,5-tetrahydro-1H-1,2,3-triazol-1-yl) Monocyclic non-aromatic heterocyclic groups such as;
Dihydroindolyl (eg, 2,3-dihydro-1H-indol-1-yl), dihydroisoindolyl (eg, 1,3-dihydro-2H-isoindol-2-yl), dihydrobenzofuranyl (eg, 2,3-dihydro-1-benzofuran-5-yl), dihydrobenzodioxinyl (eg, 2,3-dihydro-1,4-benzodioxinyl), dihydrobenzodioxepinyl (eg, 3 , 4-dihydro-2H-1,5-benzodioxepinyl), tetrahydrobenzofuranyl (eg, 4,5,6,7-tetrahydro-1-benzofuran-3-yl), chromenyl (eg, 4H- Chromen-2-yl, 2H-chromen-3-yl), dihydrochromenyl (eg, 3,4-dihydro-2H-chromen-2-yl), dihydroquinolinyl (eg, 1,2-dihydro) Roquinolin-4-yl), tetrahydroquinolinyl (eg, 1,2,3,4-tetrahydroquinolin-4-yl), dihydroisoquinolinyl (eg, 1,2-dihydroisoquinolin-4-yl), Condensed non-aromatic heterocycles such as tetrahydroisoquinolinyl (eg, 1,2,3,4-tetrahydroisoquinolin-4-yl), dihydrophthalazinyl (eg, 1,4-dihydrophthalazin-4-yl) A ring group;
Etc.
アゼチジニル(例、1-アゼチジニル、2-アゼチジニル、3-アゼチジニル)、ピロリジニル(例、1-ピロリジニル、2-ピロリジニル)、ピペリジニル(例、ピペリジノ、2-ピペリジニル、3-ピペリジニル、4-ピペリジニル)、モルホリニル(例、モルホリノ)、チオモルホリニル(例、チオモルホリノ)、ピペラジニル(例、1-ピペラジニル、2-ピペラジニル、3-ピペラジニル)、ヘキサメチレンイミニル(例、ヘキサメチレンイミン-1-イル)、オキサゾリジニル(例、オキサゾリジン-2-イル)、チアゾリジニル(例、チアゾリジン-2-イル)、イミダゾリジニル(例、イミダゾリジン-2-イル、イミダゾリジン-3-イル)、オキサゾリニル(例、オキサゾリン-2-イル)、チアゾリニル(例、チアゾリン-2-イル)、イミダゾリニル(例、イミダゾリン-2-イル、イミダゾリン-3-イル)、ジオキソリル(例、1,3-ジオキソール-4-イル)、ジオキソラニル(例、1,3-ジオキソラン-4-イル)、ジヒドロオキサジアゾリル(例、4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)、ピラニル(例、4-ピラニル)、テトラヒドロピラニル(例、2-テトラヒドロピラニル、3-テトラヒドロピラニル、4-テトラヒドロピラニル)、チオピラニル(例、4-チオピラニル)、テトラヒドロチオピラニル(例、2-テトラヒドロチオピラニル、3-テトラヒドロチオピラニル、4-テトラヒドロチオピラニル)、テトラヒドロフリル(例、テトラヒドロフラン-3-イル、テトラヒドロフラン-2-イル)、ピラゾリジニル(例、ピラゾリジン-1-イル、ピラゾリジン-3-イル)、ピラゾリニル(例、ピラゾリン-1-イル)、テトラヒドロピリミジニル(例、テトラヒドロピリミジン-1-イル)、ジヒドロトリアゾリル(例、2,3-ジヒドロ-1H-1,2,3-トリアゾール-1-イル)、テトラヒドロトリアゾリル(例、2,3,4,5-テトラヒドロ-1H-1,2,3-トリアゾール-1-イル)等の単環式非芳香族複素環基;
ジヒドロインドリル(例、2,3-ジヒドロ-1H-インドール-1-イル)、ジヒドロイソインドリル(例、1,3-ジヒドロ-2H-イソインドール-2-イル)、ジヒドロベンゾフラニル(例、2,3-ジヒドロ-1-ベンゾフラン-5-イル)、ジヒドロベンゾジオキシニル(例、2,3-ジヒドロ-1,4-ベンゾジオキシニル)、ジヒドロベンゾジオキセピニル(例、3,4-ジヒドロ-2H-1,5-ベンゾジオキセピニル)、テトラヒドロベンゾフラニル(例、4,5,6,7-テトラヒドロ-1-ベンゾフラン-3-イル)、クロメニル(例、4H-クロメン-2-イル、2H-クロメン-3-イル)、ジヒドロクロメニル(例、3,4-ジヒドロ-2H-クロメン-2-イル)、ジヒドロキノリニル(例、1,2-ジヒドロキノリン-4-イル)、テトラヒドロキノリニル(例、1,2,3,4-テトラヒドロキノリン-4-イル)、ジヒドロイソキノリニル(例、1,2-ジヒドロイソキノリン-4-イル)、テトラヒドロイソキノリニル(例、1,2,3,4-テトラヒドロイソキノリン-4-イル)、ジヒドロフタラジニル(例、1,4-ジヒドロフタラジン-4-イル)等の縮合非芳香族複素環基;
等が挙げられる。 As a suitable example of a non-aromatic heterocyclic group,
Azetidinyl (eg, 1-azetidinyl, 2-azetidinyl, 3-azetidinyl), pyrrolidinyl (eg, 1-pyrrolidinyl, 2-pyrrolidinyl), piperidinyl (eg, piperidino, 2-piperidinyl, 3-piperidinyl, 4-piperidinyl), morpholinyl (Eg, morpholino), thiomorpholinyl (eg, thiomorpholino), piperazinyl (eg, 1-piperazinyl, 2-piperazinyl, 3-piperazinyl), hexamethyleneiminyl (eg, hexamethyleneimin-1-yl), oxazolidinyl (eg, Oxazolidin-2-yl), thiazolidinyl (eg, thiazolidin-2-yl), imidazolidinyl (eg, imidazolidin-2-yl, imidazolidin-3-yl), oxazolinyl (eg, oxazolin-2-yl), thiazolinyl (Eg, thiazo -2-yl), imidazolinyl (eg, imidazolin-2-yl, imidazolin-3-yl), dioxolyl (eg, 1,3-dioxol-4-yl), dioxolanyl (eg, 1,3-dioxolane-4) -Yl), dihydrooxadiazolyl (eg, 4,5-dihydro-1,2,4-oxadiazol-3-yl), pyranyl (eg, 4-pyranyl), tetrahydropyranyl (eg, 2-tetrahydro) Pyranyl, 3-tetrahydropyranyl, 4-tetrahydropyranyl), thiopyranyl (eg, 4-thiopyranyl), tetrahydrothiopyranyl (eg, 2-tetrahydrothiopyranyl, 3-tetrahydrothiopyranyl, 4-tetrahydrothio) Pyranyl), tetrahydrofuryl (eg, tetrahydrofuran-3-yl, tetrahydrofuran-2-yl), Lazolidinyl (eg, pyrazolidin-1-yl, pyrazolidin-3-yl), pyrazolinyl (eg, pyrazolin-1-yl), tetrahydropyrimidinyl (eg, tetrahydropyrimidin-1-yl), dihydrotriazolyl (eg, 2, 3-dihydro-1H-1,2,3-triazol-1-yl), tetrahydrotriazolyl (eg, 2,3,4,5-tetrahydro-1H-1,2,3-triazol-1-yl) Monocyclic non-aromatic heterocyclic groups such as;
Dihydroindolyl (eg, 2,3-dihydro-1H-indol-1-yl), dihydroisoindolyl (eg, 1,3-dihydro-2H-isoindol-2-yl), dihydrobenzofuranyl (eg, 2,3-dihydro-1-benzofuran-5-yl), dihydrobenzodioxinyl (eg, 2,3-dihydro-1,4-benzodioxinyl), dihydrobenzodioxepinyl (eg, 3 , 4-dihydro-2H-1,5-benzodioxepinyl), tetrahydrobenzofuranyl (eg, 4,5,6,7-tetrahydro-1-benzofuran-3-yl), chromenyl (eg, 4H- Chromen-2-yl, 2H-chromen-3-yl), dihydrochromenyl (eg, 3,4-dihydro-2H-chromen-2-yl), dihydroquinolinyl (eg, 1,2-dihydro) Roquinolin-4-yl), tetrahydroquinolinyl (eg, 1,2,3,4-tetrahydroquinolin-4-yl), dihydroisoquinolinyl (eg, 1,2-dihydroisoquinolin-4-yl), Condensed non-aromatic heterocycles such as tetrahydroisoquinolinyl (eg, 1,2,3,4-tetrahydroisoquinolin-4-yl), dihydrophthalazinyl (eg, 1,4-dihydrophthalazin-4-yl) A ring group;
Etc.
前記「置換されていてもよい複素環基」における「複素環基」は、置換可能な位置に1ないし3個の置換基を有していてもよい。このような置換基としては、例えば、前記「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC3-10シクロアルキル基等が有していてもよい置換基と同様のものが挙げられる。また該複素環基が「非芳香族複素環基」である場合、置換基としてオキソ基がさらに含まれる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
The “heterocyclic group” in the “optionally substituted heterocyclic group” may have 1 to 3 substituents at substitutable positions. Examples of such substituents are the same as the substituents that the C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” may have. Can be mentioned. When the heterocyclic group is a “non-aromatic heterocyclic group”, an oxo group is further included as a substituent. When there are two or more substituents, each substituent may be the same or different.
前記「置換されていてもよいヒドロキシ基」としては、例えば、それぞれ置換されていてもよい、C1-10アルキル基、C2-10アルケニル基、C3-10シクロアルキル基、C3-10シクロアルケニル基、C6-14アリール基、C7-13アラルキル基、C8-13アリールアルケニル基、C1-6アルキル-カルボニル基、複素環基等から選ばれる置換基で置換されていてもよいヒドロキシ基が挙げられる。
Examples of the “optionally substituted hydroxy group” include, for example, a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 3-10 cycloalkyl group, and a C 3-10 which may each be substituted. It may be substituted with a substituent selected from a cycloalkenyl group, a C 6-14 aryl group, a C 7-13 aralkyl group, a C 8-13 arylalkenyl group, a C 1-6 alkyl-carbonyl group, a heterocyclic group and the like. Good hydroxy groups are mentioned.
ここで、C1-10アルキル基、C2-10アルケニル基、C3-10シクロアルキル基、C3-10シクロアルケニル基、C6-14アリール基、C7-13アラルキル基およびC8-13アリールアルケニル基としては、それぞれ前記「置換されていてもよい炭化水素基」における「炭化水素基」として例示したものが挙げられる。
Here, a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 3-10 cycloalkyl group, a C 3-10 cycloalkenyl group, a C 6-14 aryl group, a C 7-13 aralkyl group and a C 8- Examples of the 13 arylalkenyl group include those exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group”.
複素環基としては、前記「置換されていてもよい複素環基」における「複素環基」として例示した「芳香族複素環基」および「非芳香族複素環基」が挙げられる。
Examples of the heterocyclic group include “aromatic heterocyclic group” and “non-aromatic heterocyclic group” exemplified as the “heterocyclic group” in the “optionally substituted heterocyclic group”.
前記したC1-10アルキル基、C2-10アルケニル基、C3-10シクロアルキル基、C3-10シクロアルケニル基、C6-14アリール基、C7-13アラルキル基、C8-13アリールアルケニル基、C1-6アルキル-カルボニル基および複素環基は、それぞれ置換可能な位置に1ないし3個の置換基を有していてもよい。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
C 1-10 alkyl group, C 2-10 alkenyl group, C 3-10 cycloalkyl group, C 3-10 cycloalkenyl group, C 6-14 aryl group, C 7-13 aralkyl group, C 8-13 The arylalkenyl group, C 1-6 alkyl-carbonyl group and heterocyclic group each may have 1 to 3 substituents at substitutable positions. When there are two or more substituents, each substituent may be the same or different.
ここで、C1-10アルキル基、C2-10アルケニル基およびC1-6アルキル-カルボニル基の置換基としては、前記「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC1-10アルキル基等が有していてもよい置換基と同様のものが挙げられる。
Here, the substituent of the C 1-10 alkyl group, the C 2-10 alkenyl group and the C 1-6 alkyl-carbonyl group is the “hydrocarbon group” in the “optionally substituted hydrocarbon group”. Examples thereof include the same substituents as the exemplified C 1-10 alkyl group and the like.
また、C3-10シクロアルキル基、C3-10シクロアルケニル基、C6-14アリール基、C7-13アラルキル基およびC8-13アリールアルケニル基の置換基としては、前記「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC3-10シクロアルキル基等が有していてもよい置換基と同様のものが挙げられる。複素環基の置換基としては、前記「置換されていてもよい複素環基」における「複素環基」が有していてもよい置換基と同様のものが挙げられる。
In addition, examples of the substituent for the C 3-10 cycloalkyl group, the C 3-10 cycloalkenyl group, the C 6-14 aryl group, the C 7-13 aralkyl group, and the C 8-13 arylalkenyl group include the above-mentioned “substituted Examples thereof include the same substituents that the C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optional hydrocarbon group” may have. Examples of the substituent of the heterocyclic group include the same substituents that the “heterocyclic group” in the “optionally substituted heterocyclic group” may have.
前記「置換されていてもよいスルファニル基」としては、例えば、それぞれ置換されていてもよい、C1-10アルキル基、C2-10アルケニル基、C3-10シクロアルキル基、C3-10シクロアルケニル基、C6-14アリール基、C7-13アラルキル基、C8-13アリールアルケニル基、C1-6アルキル-カルボニル基、複素環基等から選ばれる置換基で置換されていてもよいスルファニル基が挙げられる。
Examples of the “optionally substituted sulfanyl group” include, for example, a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 3-10 cycloalkyl group, and a C 3-10 which may each be substituted. It may be substituted with a substituent selected from a cycloalkenyl group, a C 6-14 aryl group, a C 7-13 aralkyl group, a C 8-13 arylalkenyl group, a C 1-6 alkyl-carbonyl group, a heterocyclic group and the like. A good sulfanyl group is mentioned.
該置換基としては、それぞれ、前記「置換されていてもよいヒドロキシ基」における置換基として例示したものが挙げられる。
Examples of the substituent include those exemplified as the substituent in the “optionally substituted hydroxy group”.
前記「置換されていてもよいアミノ基」としては、例えば、それぞれ置換されていてもよい、C1-10アルキル基、C2-10アルケニル基、C3-10シクロアルキル基、C3-10シクロアルケニル基、C6-14アリール基、C7-13アラルキル基、C8-13アリールアルケニル基および複素環基;アシル基等から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基が挙げられる。
Examples of the “optionally substituted amino group” include, for example, a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 3-10 cycloalkyl group, and a C 3-10 which may each be substituted. A cycloalkenyl group, a C 6-14 aryl group, a C 7-13 aralkyl group, a C 8-13 arylalkenyl group and a heterocyclic group; an amino group which may be mono- or di-substituted with a substituent selected from an acyl group and the like Is mentioned.
ここで、C1-10アルキル基、C2-10アルケニル基、C3-10シクロアルキル基、C3-10シクロアルケニル基、C6-14アリール基、C7-13アラルキル基およびC8-13アリールアルケニル基としては、それぞれ前記「置換されていてもよい炭化水素基」における「炭化水素基」として例示したものが挙げられる。
Here, a C 1-10 alkyl group, a C 2-10 alkenyl group, a C 3-10 cycloalkyl group, a C 3-10 cycloalkenyl group, a C 6-14 aryl group, a C 7-13 aralkyl group and a C 8- Examples of the 13 arylalkenyl group include those exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group”.
複素環基としては、前記「置換されていてもよい複素環基」における「複素環基」として例示した「芳香族複素環基」および「非芳香族複素環基」が挙げられ、なかでも好ましくは、5~7員の単環式芳香族複素環基である。
Examples of the heterocyclic group include the “aromatic heterocyclic group” and the “non-aromatic heterocyclic group” exemplified as the “heterocyclic group” in the “optionally substituted heterocyclic group”. Is a 5- to 7-membered monocyclic aromatic heterocyclic group.
これらC1-10アルキル基、C2-10アルケニル基、C3-10シクロアルキル基、C3-10シクロアルケニル基、C6-14アリール基、C7-13アラルキル基、C8-13アリールアルケニル基および複素環基は、それぞれ置換可能な位置に1ないし3個の置換基を有していてもよい。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
These C 1-10 alkyl group, C 2-10 alkenyl group, C 3-10 cycloalkyl group, C 3-10 cycloalkenyl group, C 6-14 aryl group, C 7-13 aralkyl group, C 8-13 aryl The alkenyl group and the heterocyclic group each may have 1 to 3 substituents at substitutable positions. When there are two or more substituents, each substituent may be the same or different.
ここで、C1-10アルキル基およびC2-10アルケニル基の置換基としては、前記「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC1-10アルキル基等が有していてもよい置換基と同様のものが挙げられる。
Here, examples of the substituent of the C 1-10 alkyl group and the C 2-10 alkenyl group include the C 1-10 alkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” and the like. The same thing as the substituent which may have is mentioned.
また、C3-10シクロアルキル基、C3-10シクロアルケニル基、C6-14アリール基、C7-13アラルキル基およびC8-13アリールアルケニル基の置換基としては、前記「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC3-10シクロアルキル基等が有していてもよい置換基と同様のものが挙げられる。複素環基の置換基としては、前記「置換されていてもよい複素環基」における「複素環基」が有していてもよい置換基と同様のものが挙げられる。
In addition, examples of the substituent for the C 3-10 cycloalkyl group, the C 3-10 cycloalkenyl group, the C 6-14 aryl group, the C 7-13 aralkyl group, and the C 8-13 arylalkenyl group include the above-mentioned “substituted Examples thereof include the same substituents that the C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optional hydrocarbon group” may have. Examples of the substituent of the heterocyclic group include the same substituents that the “heterocyclic group” in the “optionally substituted heterocyclic group” may have.
「置換されていてもよいアミノ基」の置換基として例示した「アシル基」としては、次に示すR2で示される「置換基」として例示する「アシル基」と同様のものが挙げられる。
Examples of the “acyl group” exemplified as the substituent of the “optionally substituted amino group” include those similar to the “acyl group” exemplified as the “substituent” represented by R 2 shown below.
R2で示される「置換基」として例示した「アシル基」としては、例えば、式:-CORA、-CO-ORA、-SO3RA、-S(O)2RA、-SORA、-CO-NRA’RB’、-CS-NRA’RB’、-S(O)2NRA’RB’[式中、RAは、水素原子、置換されていてもよい炭化水素基、または置換されていてもよい複素環基を示す。RA’およびRB’は、同一または異なって、水素原子、置換されていてもよい炭化水素基、または置換されていてもよい複素環基を示すか、RA’およびRB’は、隣接する窒素原子とともに、置換されていてもよい含窒素複素環を形成していてもよい]で表される基等が挙げられる。
The “acyl group” exemplified as the “substituent” represented by R 2 includes, for example, the formula: —COR A , —CO—OR A , —SO 3 R A , —S (O) 2 R A , —SOR A , -CO-NR A 'R B ', -CS-NR A 'R B ', -S (O) 2 NR A 'R B ' [wherein R A is a hydrogen atom, substituted A good hydrocarbon group or an optionally substituted heterocyclic group is shown. R A ′ and R B ′ are the same or different and each represents a hydrogen atom, an optionally substituted hydrocarbon group, or an optionally substituted heterocyclic group, or R A ′ and R B ′ are A nitrogen-containing heterocyclic ring which may be substituted with an adjacent nitrogen atom may be formed], and the like.
RA、RA’またはRB’で示される「置換されていてもよい炭化水素基」および「置換されていてもよい複素環基」としては、それぞれR2で示される「置換基」として例示した「置換されていてもよい炭化水素基」、「置換されていてもよい複素環基」と同様のものが挙げられる。
The “optionally substituted hydrocarbon group” and the “optionally substituted heterocyclic group” represented by R A , R A ′ or R B ′ are the “substituent” represented by R 2 , respectively. Examples thereof are the same as the exemplified “optionally substituted hydrocarbon group” and “optionally substituted heterocyclic group”.
RA’およびRB’が隣接する窒素原子とともに形成する「置換されていてもよい含窒素複素環」における「含窒素複素環」としては、例えば、環構成原子として炭素原子以外に少なくとも1個の窒素原子を含み、さらに酸素原子、硫黄原子および窒素原子から選ばれるヘテロ原子を1ないし2個含有していてもよい5~7員の含窒素複素環が挙げられる。該含窒素複素環の好適な例としては、ピロリジン、イミダゾリジン、ピラゾリジン、ピペリジン、ピペラジン、モルホリン、チオモルホリン等が挙げられる。
The “nitrogen-containing heterocycle” in the “optionally substituted nitrogen-containing heterocycle” formed by R A ′ and R B ′ together with the adjacent nitrogen atom includes, for example, at least one ring-constituting atom other than a carbon atom And a 5- to 7-membered nitrogen-containing heterocyclic ring which may further contain 1 to 2 heteroatoms selected from oxygen, sulfur and nitrogen atoms. Preferable examples of the nitrogen-containing heterocycle include pyrrolidine, imidazolidine, pyrazolidine, piperidine, piperazine, morpholine, thiomorpholine and the like.
該含窒素複素環は、置換可能な位置に1ないし5個(好ましくは1または2個)の置換基を有していてもよい。このような置換基としては、例えば、R2で示される「置換基」として例示した「置換されていてもよい複素環基」における「複素環基」が有していてもよい置換基と同様のものが挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
The nitrogen-containing heterocycle may have 1 to 5 (preferably 1 or 2) substituents at substitutable positions. Examples of such a substituent are the same as the substituent that the “heterocyclic group” in the “optionally substituted heterocyclic group” exemplified as the “substituent” represented by R 2 may have. Can be mentioned. When there are two or more substituents, each substituent may be the same or different.
「アシル基」の好適な例としては、
(1)ホルミル基;
(2)カルボキシ基;
(3)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基(例、アセチル);
(4)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基(例、メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、tert-ブトキシカルボニル);
(5)C3-10シクロアルキル-カルボニル基(例、シクロプロピルカルボニル、シクロペンチルカルボニル、シクロヘキシルカルボニル);
(6)1ないし3個のハロゲン原子で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル、1-ナフトイル、2-ナフトイル);
(7)(a)ハロゲン原子、C1-6アルコキシ基、C1-6アルコキシ-カルボニル基およびカルボキシ基から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基、および
(b)C1-6アルコキシ-カルボニル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる置換基でモノまたはジ置換されていてもよいカルバモイル基;
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)C6-14アリールスルホニル基(例、ベンゼンスルホニル);
(10)スルファモイル基;
(11)チオカルバモイル基;
(12)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい芳香族複素環カルボニル基(例、フリルカルボニル、チエニルカルボニル);
(13)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい非芳香族複素環カルボニル基(例、テトラヒドロフリルカルボニル、ピロリジノカルボニル);
等が挙げられる。 As preferable examples of the “acyl group”,
(1) formyl group;
(2) a carboxy group;
(3) a C 1-6 alkyl-carbonyl group (eg acetyl) optionally substituted by 1 to 3 halogen atoms;
(4) a C 1-6 alkoxy-carbonyl group (eg, methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert-butoxycarbonyl) optionally substituted by 1 to 3 halogen atoms;
(5) C 3-10 cycloalkyl-carbonyl group (eg, cyclopropylcarbonyl, cyclopentylcarbonyl, cyclohexylcarbonyl);
(6) a C 6-14 aryl-carbonyl group (eg, benzoyl, 1-naphthoyl, 2-naphthoyl) optionally substituted with 1 to 3 halogen atoms;
(7) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkoxy group, a C 1-6 alkoxy-carbonyl group and a carboxy group And (b) a carbamoyl group optionally mono- or di-substituted with a substituent selected from an amino group optionally mono- or di-substituted with a C 1-6 alkoxy-carbonyl group;
(8) a C 1-6 alkylsulfonyl group which may be substituted with 1 to 3 halogen atoms (eg, methylsulfonyl, ethylsulfonyl, isopropylsulfonyl);
(9) C 6-14 arylsulfonyl group (eg, benzenesulfonyl);
(10) a sulfamoyl group;
(11) a thiocarbamoyl group;
(12) an aromatic heterocyclic carbonyl group optionally substituted with 1 to 3 substituents selected from C 1-6 alkyl groups optionally substituted with 1 to 3 halogen atoms (eg, furyl Carbonyl, thienylcarbonyl);
(13) A non-aromatic heterocyclic carbonyl group optionally substituted with 1 to 3 substituents selected from C 1-6 alkyl groups optionally substituted with 1 to 3 halogen atoms (eg, Tetrahydrofurylcarbonyl, pyrrolidinocarbonyl);
Etc.
(1)ホルミル基;
(2)カルボキシ基;
(3)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基(例、アセチル);
(4)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基(例、メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、tert-ブトキシカルボニル);
(5)C3-10シクロアルキル-カルボニル基(例、シクロプロピルカルボニル、シクロペンチルカルボニル、シクロヘキシルカルボニル);
(6)1ないし3個のハロゲン原子で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル、1-ナフトイル、2-ナフトイル);
(7)(a)ハロゲン原子、C1-6アルコキシ基、C1-6アルコキシ-カルボニル基およびカルボキシ基から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基、および
(b)C1-6アルコキシ-カルボニル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる置換基でモノまたはジ置換されていてもよいカルバモイル基;
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)C6-14アリールスルホニル基(例、ベンゼンスルホニル);
(10)スルファモイル基;
(11)チオカルバモイル基;
(12)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい芳香族複素環カルボニル基(例、フリルカルボニル、チエニルカルボニル);
(13)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい非芳香族複素環カルボニル基(例、テトラヒドロフリルカルボニル、ピロリジノカルボニル);
等が挙げられる。 As preferable examples of the “acyl group”,
(1) formyl group;
(2) a carboxy group;
(3) a C 1-6 alkyl-carbonyl group (eg acetyl) optionally substituted by 1 to 3 halogen atoms;
(4) a C 1-6 alkoxy-carbonyl group (eg, methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert-butoxycarbonyl) optionally substituted by 1 to 3 halogen atoms;
(5) C 3-10 cycloalkyl-carbonyl group (eg, cyclopropylcarbonyl, cyclopentylcarbonyl, cyclohexylcarbonyl);
(6) a C 6-14 aryl-carbonyl group (eg, benzoyl, 1-naphthoyl, 2-naphthoyl) optionally substituted with 1 to 3 halogen atoms;
(7) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkoxy group, a C 1-6 alkoxy-carbonyl group and a carboxy group And (b) a carbamoyl group optionally mono- or di-substituted with a substituent selected from an amino group optionally mono- or di-substituted with a C 1-6 alkoxy-carbonyl group;
(8) a C 1-6 alkylsulfonyl group which may be substituted with 1 to 3 halogen atoms (eg, methylsulfonyl, ethylsulfonyl, isopropylsulfonyl);
(9) C 6-14 arylsulfonyl group (eg, benzenesulfonyl);
(10) a sulfamoyl group;
(11) a thiocarbamoyl group;
(12) an aromatic heterocyclic carbonyl group optionally substituted with 1 to 3 substituents selected from C 1-6 alkyl groups optionally substituted with 1 to 3 halogen atoms (eg, furyl Carbonyl, thienylcarbonyl);
(13) A non-aromatic heterocyclic carbonyl group optionally substituted with 1 to 3 substituents selected from C 1-6 alkyl groups optionally substituted with 1 to 3 halogen atoms (eg, Tetrahydrofurylcarbonyl, pyrrolidinocarbonyl);
Etc.
R2は、好ましくは、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC3-6シクロアルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基である。
R2は、より好ましくは、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基である。
R2は、さらに好ましくは、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。
R2は、特に好ましくは、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。 R 2 is preferably a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, an optionally substituted C 1-6 alkoxy group, Or it is an amino group which may be substituted.
R 2 is more preferably a hydrogen atom, an optionally substituted C 1-6 alkyl group, a optionally substituted C 1-6 alkoxy group or an optionally substituted amino group.
R 2 is more preferably a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups (eg, An amino group which may be substituted with (methyl).
R 2 is particularly preferably a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups (eg, methyl). An amino group which may be substituted.
R2は、より好ましくは、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基である。
R2は、さらに好ましくは、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。
R2は、特に好ましくは、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。 R 2 is preferably a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, an optionally substituted C 1-6 alkoxy group, Or it is an amino group which may be substituted.
R 2 is more preferably a hydrogen atom, an optionally substituted C 1-6 alkyl group, a optionally substituted C 1-6 alkoxy group or an optionally substituted amino group.
R 2 is more preferably a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups (eg, An amino group which may be substituted with (methyl).
R 2 is particularly preferably a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups (eg, methyl). An amino group which may be substituted.
R1で示される「式:-COR2で表される基」は、好ましくは、-COR2(式中、R2が、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC3-6シクロアルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基である。)で表される基である。
The “group represented by the formula: —COR 2 ” represented by R 1 is preferably —COR 2 (wherein R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, substituted An optionally substituted C 3-6 cycloalkyl group, an optionally substituted C 1-6 alkoxy group, or an optionally substituted amino group.
R1で示される「式:-COR2で表される基」は、より好ましくは、-COR2(式中、R2が、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基である。)で表される基である。
The “group represented by the formula: —COR 2 ” represented by R 1 is more preferably —COR 2 (wherein R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, A C 1-6 alkoxy group which may be substituted, or an amino group which may be substituted.
R1で示される「式:-COR2で表される基」は、さらに好ましくは、-COR2(式中、R2が、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基である。
The “group represented by the formula: —COR 2 ” represented by R 1 is more preferably —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), C And a group represented by a 1-6 alkoxy group (eg, methoxy) or an amino group optionally substituted by 1 to 2 C 1-6 alkyl groups (eg, methyl).
R1で示される「式:-COR2で表される基」は、特に好ましくは、-COR2(式中、R2が、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基である。
The “group represented by the formula: —COR 2 ” represented by R 1 is particularly preferably —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), C 1-6 And a group represented by an alkoxy group (eg, methoxy) or an amino group optionally substituted by 1 to 2 C 1-6 alkyl groups (eg, methyl).
R1で示される「置換されていてもよい5または6員芳香環基」における「5または6員芳香環基」としては、例えば、フェニル、ピロリル、ピラゾリル、イミダゾリル、トリアゾリル(1,2,3-トリアゾリル、1,2,4-トリアゾリル、1,3,4-トリアゾリル)、テトラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、オキサジアゾリル、チアジアゾリル、フリル、チエニル、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、トリアジニル等が挙げられる。
Examples of the “5- or 6-membered aromatic ring group” in the “optionally substituted 5- or 6-membered aromatic ring group” represented by R 1 include, for example, phenyl, pyrrolyl, pyrazolyl, imidazolyl, triazolyl (1, 2, 3 -Triazolyl, 1,2,4-triazolyl, 1,3,4-triazolyl), tetrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, oxadiazolyl, thiadiazolyl, furyl, thienyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, etc. It is done.
当該「5または6員芳香環基」は、好ましくは、5員芳香族複素環基であり、より好ましくは、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル等であり、特に好ましくはイソオキサゾリルである。
The “5- or 6-membered aromatic ring group” is preferably a 5-membered aromatic heterocyclic group, more preferably pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, etc., particularly preferably isoxazolyl. .
R1で示される「置換されていてもよい5または6員芳香環基」における「5または6員芳香環基」は、置換可能な位置に1ないし3個の置換基を有していてもよい。このような置換基としては、例えば、R2で示される「置換基」として例示した「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC3-10シクロアルキル基等が有していてもよい置換基と同様のものが挙げられる。
The “5- or 6-membered aromatic ring group” in the “optionally substituted 5- or 6-membered aromatic ring group” represented by R 1 may have 1 to 3 substituents at substitutable positions. Good. Examples of such a substituent include a C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 and the like. The same thing as the substituent which may have is mentioned.
R1で示される「置換されていてもよい5または6員芳香環基」は、好ましくは、C1-6アルキル基(例、メチル)から選ばれる1ないし3個の置換基で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル)である。
The “optionally substituted 5- or 6-membered aromatic ring group” represented by R 1 is preferably substituted with 1 to 3 substituents selected from C 1-6 alkyl groups (eg, methyl). And may be a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl).
R1で示される「置換されていてもよい5または6員芳香環基」は、より好ましくは、5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル)である。
The “optionally substituted 5- or 6-membered aromatic ring group” represented by R 1 is more preferably a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably Isoxazolyl).
R1は、好ましくは、
(1) -COR2(式中、R2が、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC3-6シクロアルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5または6員芳香環基;
である。 R 1 is preferably
(1) —COR 2 (wherein R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, or optionally substituted) A C 1-6 alkoxy group, or an optionally substituted amino group.); Or
(2) an optionally substituted 5- or 6-membered aromatic ring group;
It is.
(1) -COR2(式中、R2が、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC3-6シクロアルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5または6員芳香環基;
である。 R 1 is preferably
(1) —COR 2 (wherein R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, or optionally substituted) A C 1-6 alkoxy group, or an optionally substituted amino group.); Or
(2) an optionally substituted 5- or 6-membered aromatic ring group;
It is.
R1は、より好ましくは、
(1) -COR2(式中、R2が、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5または6員芳香環基;
である。 R 1 is more preferably
(1) -COR 2 (wherein, R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, optionally substituted C 1-6 alkoxy group or may be substituted, A group represented by: an amino group; or
(2) an optionally substituted 5- or 6-membered aromatic ring group;
It is.
(1) -COR2(式中、R2が、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5または6員芳香環基;
である。 R 1 is more preferably
(1) -COR 2 (wherein, R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, optionally substituted C 1-6 alkoxy group or may be substituted, A group represented by: an amino group; or
(2) an optionally substituted 5- or 6-membered aromatic ring group;
It is.
R1は、さらに好ましくは、
(1) -COR2(式中、R2が、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5員の芳香環複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
である。 R 1 is more preferably
(1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or
(2) an optionally substituted 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl);
It is.
(1) -COR2(式中、R2が、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5員の芳香環複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
である。 R 1 is more preferably
(1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or
(2) an optionally substituted 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl);
It is.
R1は、さらにより好ましくは、
(1) -COR2(式中、R2が、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
である。 R 1 is even more preferably
(1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or
(2) a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl);
It is.
(1) -COR2(式中、R2が、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
である。 R 1 is even more preferably
(1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or
(2) a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl);
It is.
R1は、特に好ましくは、、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
である。 R 1 is particularly preferably
(1) —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or
(2) a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl);
It is.
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
である。 R 1 is particularly preferably
(1) —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or
(2) a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl);
It is.
あるいは、R1は、特に好ましくは、
(1) -COR2(式中、R2が、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
である。 Alternatively, R 1 is particularly preferably
(1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or
(2) a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl);
It is.
(1) -COR2(式中、R2が、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
である。 Alternatively, R 1 is particularly preferably
(1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or
(2) a 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl);
It is.
R3は、ハロゲン原子で置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示す。
R3で示される「ハロゲン原子で置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」は置換可能な位置に好ましくは1ないし7個、より好ましくは1ないし3個のハロゲン原子を有していてもよい。 R 3 represents a C 1-6 alkyl group which may be substituted with a halogen atom, or a C 3-6 cycloalkyl group which may be substituted.
The “C 1-6 alkyl group” in the “C 1-6 alkyl group optionally substituted with a halogen atom” for R 3 is preferably 1 to 7, more preferably 1 to 3, in the substitutable position. May have one halogen atom.
R3で示される「ハロゲン原子で置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」は置換可能な位置に好ましくは1ないし7個、より好ましくは1ないし3個のハロゲン原子を有していてもよい。 R 3 represents a C 1-6 alkyl group which may be substituted with a halogen atom, or a C 3-6 cycloalkyl group which may be substituted.
The “C 1-6 alkyl group” in the “C 1-6 alkyl group optionally substituted with a halogen atom” for R 3 is preferably 1 to 7, more preferably 1 to 3, in the substitutable position. May have one halogen atom.
R3で示される「置換されていてもよいC3-6シクロアルキル基」における「C3-6シクロアルキル基」としては、シクロプロピル、シクロブチル、シクロペンチルおよびシクロヘキシルが挙げられる。
R3で示される「置換されていてもよいC3-6シクロアルキル基」における「C3-6シクロアルキル基」は置換可能な位置に1ないし3個の置換基を有していてもよい。このような置換基としては、例えば、R2で示される「置換基」として例示した「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC3-10シクロアルキル基等が有していてもよい置換基と同様のものが挙げられる。 Examples of the “C 3-6 cycloalkyl group” in the “optionally substituted C 3-6 cycloalkyl group” represented by R 3 include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
Represented by R 3 "C 3-6 cycloalkyl group" of the "optionally substituted C 3-6 cycloalkyl group" may have 1 to 3 substituents at substitutable position . Examples of such a substituent include a C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 and the like. The same thing as the substituent which may have is mentioned.
R3で示される「置換されていてもよいC3-6シクロアルキル基」における「C3-6シクロアルキル基」は置換可能な位置に1ないし3個の置換基を有していてもよい。このような置換基としては、例えば、R2で示される「置換基」として例示した「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC3-10シクロアルキル基等が有していてもよい置換基と同様のものが挙げられる。 Examples of the “C 3-6 cycloalkyl group” in the “optionally substituted C 3-6 cycloalkyl group” represented by R 3 include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
Represented by R 3 "C 3-6 cycloalkyl group" of the "optionally substituted C 3-6 cycloalkyl group" may have 1 to 3 substituents at substitutable position . Examples of such a substituent include a C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 and the like. The same thing as the substituent which may have is mentioned.
R3は、好ましくは、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)である。
R3は、より好ましくは、C1-6アルキル基(例、メチル、エチル)である。 R 3 is preferably a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms.
R 3 is more preferably a C 1-6 alkyl group (eg, methyl, ethyl).
R3は、より好ましくは、C1-6アルキル基(例、メチル、エチル)である。 R 3 is preferably a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms.
R 3 is more preferably a C 1-6 alkyl group (eg, methyl, ethyl).
R4aおよびR4bは、独立して、水素原子または置換基を示すか、またはR4aおよびR4bは、互いに結合して置換されていてもよい3または4員環を形成する。
R 4a and R 4b independently represent a hydrogen atom or a substituent, or R 4a and R 4b are bonded to each other to form an optionally substituted 3- or 4-membered ring.
R4aまたはR4bで示される「置換基」としては、R2で示される「置換基」と同様のものが挙げられる。
Examples of the “substituent” represented by R 4a or R 4b include those similar to the “substituent” represented by R 2 .
R4aおよびR4bが互いに結合して形成する「置換されていてもよい3または4員環」における「3または4員環」としては、シクロプロパン、シクロブタン、シクロブテン、アジリジン、アゼチジン、オキシラン、オキセタン等が挙げられる。
As the “3- or 4-membered ring” in the “optionally substituted 3- or 4-membered ring” formed by R 4a and R 4b bonded to each other, cyclopropane, cyclobutane, cyclobutene, aziridine, azetidine, oxirane, oxetane Etc.
R4aおよびR4bが互いに結合して形成する「置換されていてもよい3または4員環」における「3または4員環」は、置換可能な位置に1ないし6個の置換基を有していてもよい。このような置換基としては、例えば、R2で示される「置換基」として例示した「置換されていてもよい複素環基」における「複素環基」が有していてもよい置換基と同様のものが挙げられる。
The “3- or 4-membered ring” in the “optionally substituted 3- or 4-membered ring” formed by R 4a and R 4b bonded to each other has 1 to 6 substituents at substitutable positions. It may be. Examples of such a substituent are the same as the substituent that the “heterocyclic group” in the “optionally substituted heterocyclic group” exemplified as the “substituent” represented by R 2 may have. Can be mentioned.
R4aおよびR4bは、好ましくは、共に水素原子である。
R 4a and R 4b are preferably both hydrogen atoms.
R5aおよびR5bは、独立して、水素原子または置換基を示すか、またはR5aおよびR5bは、互いに結合して置換されていてもよい3または4員環を形成する。
R 5a and R 5b independently represent a hydrogen atom or a substituent, or R 5a and R 5b are bonded to each other to form an optionally substituted 3- or 4-membered ring.
R5aまたはR5bで示される「置換基」としては、R2で示される「置換基」と同様のものが挙げられる。
Examples of the “substituent” represented by R 5a or R 5b include the same “substituent” as represented by R 2 .
R5aまたはR5bが互いに結合して形成する「置換されていてもよい3または4員環」における「3または4員環」としては、シクロプロパン、シクロブタン、シクロブテン、アジリジン、アゼチジン、オキシラン、オキセタン等が挙げられる。
As the “3- or 4-membered ring” in the “optionally substituted 3- or 4-membered ring” formed by R 5a or R 5b bonded to each other, cyclopropane, cyclobutane, cyclobutene, aziridine, azetidine, oxirane, oxetane Etc.
R5aまたはR5bが互いに結合して形成する「置換されていてもよい3または4員環」における「3または4員環」は、置換可能な位置に1ないし6個の置換基を有していてもよい。このような置換基としては、例えば、R2で示される「置換基」として例示した「置換されていてもよい複素環基」における「複素環基」が有していてもよい置換基と同様のものが挙げられる。
The “3- or 4-membered ring” in the “optionally substituted 3- or 4-membered ring” formed by R 5a or R 5b bonded to each other has 1 to 6 substituents at substitutable positions. It may be. Examples of such a substituent are the same as the substituent that the “heterocyclic group” in the “optionally substituted heterocyclic group” exemplified as the “substituent” represented by R 2 may have. Can be mentioned.
R5aまたはR5bは、好ましくは、独立して、水素原子、カルボキシ基またはC1-6アルコキシ-カルボニル基である。
R5aまたはR5bは、より好ましくは、共に水素原子である。 R 5a or R 5b is preferably independently a hydrogen atom, a carboxy group or a C 1-6 alkoxy-carbonyl group.
R 5a or R 5b is more preferably both a hydrogen atom.
R5aまたはR5bは、より好ましくは、共に水素原子である。 R 5a or R 5b is preferably independently a hydrogen atom, a carboxy group or a C 1-6 alkoxy-carbonyl group.
R 5a or R 5b is more preferably both a hydrogen atom.
R6は、置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示す。
R 6 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 3-6 cycloalkyl group.
R6で示される「置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」は置換可能な位置に1ないし3個の置換基を有していてもよい。このような置換基としては、例えば、R2で示される「置換基」として例示した「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC1-10アルキル基等が有していてもよい置換基と同様のものが挙げられる。
The “C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” for R 6 may have 1 to 3 substituents at substitutable positions. Examples of such a substituent include a C 1-10 alkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 and the like. Examples thereof are the same as the substituents that may be present.
R6で示される「置換されていてもよいC3-6シクロアルキル基」としては、R3で示される「置換されていてもよいC3-6シクロアルキル基」と同様のものが挙げられる。
As the "optionally substituted C 3-6 cycloalkyl group" represented by R 6, include those similar to the "optionally substituted C 3-6 cycloalkyl group" represented by R 3 .
R6は、好ましくは、置換されていてもよいC1-6アルキル基である。
R 6 is preferably an optionally substituted C 1-6 alkyl group.
R6は、より好ましくは、
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)である。 R 6 is more preferably
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl)) Is a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic hetero group (eg tetradofryl), and
(d) a C 6-14 aryl group (eg, phenyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl) )
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted by 1 to 7 (preferably 1 to 3) substituents selected from
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)である。 R 6 is more preferably
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl)) Is a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic hetero group (eg tetradofryl), and
(d) a C 6-14 aryl group (eg, phenyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl) )
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted by 1 to 7 (preferably 1 to 3) substituents selected from
R6は、さらに好ましくは、
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) C6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)である。 R 6 is more preferably
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic heterocyclic group (eg, tetradrofuryl)), and
(d) C 6-14 aryl group (eg, phenyl)
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted by 1 to 7 (preferably 1 to 3) substituents selected from
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) C6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)である。 R 6 is more preferably
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic heterocyclic group (eg, tetradrofuryl)), and
(d) C 6-14 aryl group (eg, phenyl)
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted by 1 to 7 (preferably 1 to 3) substituents selected from
Xは、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子または置換基を示す)、NR7c(R7cは、水素原子または置換されていてもよい炭化水素基を示す)、S、SOまたはS(O)2を示す。
X is O, CO, CR 7a R 7b (R 7a and R 7b independently represent a hydrogen atom or a substituent), NR 7c (R 7c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 .
R7aまたはR7bで示される「置換基」としては、R2で示される「置換基」と同様のものが挙げられる。
R7aおよびR7bは、好ましくは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である。
R7aおよびR7bは、より好ましくは、水素原子である。 Examples of the “substituent” represented by R 7a or R 7b include the same “substituent” represented by R 2 .
R 7a and R 7b are preferably independently a hydrogen atom or a halogen atom (eg, fluorine atom).
R 7a and R 7b are more preferably a hydrogen atom.
R7aおよびR7bは、好ましくは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である。
R7aおよびR7bは、より好ましくは、水素原子である。 Examples of the “substituent” represented by R 7a or R 7b include the same “substituent” represented by R 2 .
R 7a and R 7b are preferably independently a hydrogen atom or a halogen atom (eg, fluorine atom).
R 7a and R 7b are more preferably a hydrogen atom.
R7cで示される「置換されていてもよい炭化水素基」としては、R2で示される「置換基」として例示した「置換されていてもよい炭化水素基」と同様のものが挙げられる。
R7cは、好ましくは、水素原子または置換されていてもよいC1-6アルキル基であり、より好ましくは、C1-6アルキル基(例、メチル)である。 Examples of the “ optionally substituted hydrocarbon group” represented by R 7c include the same “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 .
R 7c is preferably a hydrogen atom or an optionally substituted C 1-6 alkyl group, and more preferably a C 1-6 alkyl group (eg, methyl).
R7cは、好ましくは、水素原子または置換されていてもよいC1-6アルキル基であり、より好ましくは、C1-6アルキル基(例、メチル)である。 Examples of the “ optionally substituted hydrocarbon group” represented by R 7c include the same “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 .
R 7c is preferably a hydrogen atom or an optionally substituted C 1-6 alkyl group, and more preferably a C 1-6 alkyl group (eg, methyl).
Xは、好ましくは、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)、NR7c(R7cは、水素原子または置換されていてもよいC1-6アルキル基である)、S、SOまたはS(O)2である。
Xは、より好ましくは、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2である。
Xは、さらに好ましくは、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2である。
Xは、特に好ましくは、O、CO、CH2、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2である。 X is preferably O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a hydrogen atom) Or an optionally substituted C 1-6 alkyl group), S, SO or S (O) 2 .
X is more preferably O, CO, CR 7a R 7b (R 7a and R 7b are independently a hydrogen atom or a halogen atom (eg, fluorine atom)), NR 7c (R 7c is C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 .
X is more preferably O, CO, CH 2 , NR 7c (R 7c is a hydrogen atom or a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 .
X is particularly preferably O, CO, CH 2 , NR 7c (R 7c is a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 .
Xは、より好ましくは、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2である。
Xは、さらに好ましくは、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2である。
Xは、特に好ましくは、O、CO、CH2、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2である。 X is preferably O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a hydrogen atom) Or an optionally substituted C 1-6 alkyl group), S, SO or S (O) 2 .
X is more preferably O, CO, CR 7a R 7b (R 7a and R 7b are independently a hydrogen atom or a halogen atom (eg, fluorine atom)), NR 7c (R 7c is C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 .
X is more preferably O, CO, CH 2 , NR 7c (R 7c is a hydrogen atom or a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 .
X is particularly preferably O, CO, CH 2 , NR 7c (R 7c is a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 .
Yは、O、CO、CR8aR8b(R8aおよびR8bは、独立して、水素原子または置換基を示す)、NR8c(R8cは、水素原子または置換されていてもよい炭化水素基を示す)、S、SOまたはS(O)2を示す。
Y is O, CO, CR 8a R 8b (R 8a and R 8b independently represent a hydrogen atom or a substituent), NR 8c (R 8c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 .
R8aまたはR8bで示される「置換基」としては、R2で示される「置換基」と同様のものが挙げられる。
R8aおよびR8bは、好ましくは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である。
R8aおよびR8bは、より好ましくは、水素原子である。 Examples of the “substituent” represented by R 8a or R 8b include those similar to the “substituent” represented by R 2 .
R 8a and R 8b are preferably independently a hydrogen atom or a halogen atom (eg, fluorine atom).
R 8a and R 8b are more preferably a hydrogen atom.
R8aおよびR8bは、好ましくは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である。
R8aおよびR8bは、より好ましくは、水素原子である。 Examples of the “substituent” represented by R 8a or R 8b include those similar to the “substituent” represented by R 2 .
R 8a and R 8b are preferably independently a hydrogen atom or a halogen atom (eg, fluorine atom).
R 8a and R 8b are more preferably a hydrogen atom.
R8cで示される「置換されていてもよい炭化水素基」としては、R2で示される「置換基」として例示した「置換されていてもよい炭化水素基」と同様のものが挙げられる。
R8cは、好ましくは、水素原子または置換されていてもよいC1-6アルキル基である。 As the “ optionally substituted hydrocarbon group” for R 8c , the same as the “ optionally substituted hydrocarbon group” exemplified as the “substituent” for R 2 can be mentioned.
R 8c is preferably a hydrogen atom or an optionally substituted C 1-6 alkyl group.
R8cは、好ましくは、水素原子または置換されていてもよいC1-6アルキル基である。 As the “ optionally substituted hydrocarbon group” for R 8c , the same as the “ optionally substituted hydrocarbon group” exemplified as the “substituent” for R 2 can be mentioned.
R 8c is preferably a hydrogen atom or an optionally substituted C 1-6 alkyl group.
Yは、好ましくは、O、COまたはCR8aR8b(R8aおよびR8bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)である。
Yは、より好ましくは、O、COまたはCH2である。
Yは、さらに好ましくは、Oである。 Y is preferably O, CO, or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, a fluorine atom)).
Y is more preferably O, CO or CH 2 .
Y is more preferably O.
Yは、より好ましくは、O、COまたはCH2である。
Yは、さらに好ましくは、Oである。 Y is preferably O, CO, or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, a fluorine atom)).
Y is more preferably O, CO or CH 2 .
Y is more preferably O.
環Pは、さらに置換されていてもよい3ないし7員環を示す。
環Pで示される「さらに置換されていてもよい3ないし7員環」における「3ないし7員環」としては、ベンゼン、C3-7シクロアルカン、C3-7シクロアルケン、C4-7シクロアルカジエンおよび3ないし7員複素環(好ましくは、5または6員芳香族複素環および3ないし7員非芳香族複素環)が挙げられる。 Ring P represents a 3- to 7-membered ring which may be further substituted.
Examples of the “3- to 7-membered ring” in the “optionally substituted 3- to 7-membered ring” represented by ring P include benzene, C 3-7 cycloalkane, C 3-7 cycloalkene, C 4-7. And cycloalkadienes and 3- to 7-membered heterocycles (preferably 5- or 6-membered aromatic heterocycles and 3- to 7-membered non-aromatic heterocycles).
環Pで示される「さらに置換されていてもよい3ないし7員環」における「3ないし7員環」としては、ベンゼン、C3-7シクロアルカン、C3-7シクロアルケン、C4-7シクロアルカジエンおよび3ないし7員複素環(好ましくは、5または6員芳香族複素環および3ないし7員非芳香族複素環)が挙げられる。 Ring P represents a 3- to 7-membered ring which may be further substituted.
Examples of the “3- to 7-membered ring” in the “optionally substituted 3- to 7-membered ring” represented by ring P include benzene, C 3-7 cycloalkane, C 3-7 cycloalkene, C 4-7. And cycloalkadienes and 3- to 7-membered heterocycles (preferably 5- or 6-membered aromatic heterocycles and 3- to 7-membered non-aromatic heterocycles).
C3-7シクロアルカンとしては、シクロプロパン、シクロブタン、シクロペンタン、シクロヘキサンおよびシクロヘプタンが挙げられ、好ましくは、C4-6シクロアルカンであり、特に好ましくは、シクロブタンおよびシクロヘキサンである。
C3-7シクロアルケンとしては、シクロペンテン、シクロブテン、シクロペンテン、シクロヘキセンおよびシクロヘプテンが挙げられ、好ましくは、C4-6シクロアルケンである。
C4-7シクロアルカジエンとしては、2,4-シクロペンタジエン、2,4-シクロヘキサジエン、2,5-シクロヘキサジエン等が挙げられ、好ましくは、C4-6シクロアルカジエンである。
5または6員芳香族複素環としては、例えば、ピロール、ピラゾール、イミダゾール、トリアゾール(1,2,3-トリアゾール、1,2,4-トリアゾール、1,3,4-トリアゾール)、テトラゾール、オキサゾール、イソオキサゾール、チアゾール、イソチアゾール、オキサジアゾール、チアジアゾール、フラン、チオフェン、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン等が挙げられ、好ましくは、6員含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)である。
3ないし7員非芳香族複素環としては、オキシラン、オキセタン、アジリジン、アゼチジン、ピロリジン、ピペリジン、モルホリン、チオモルホリン、ピペラジン、ヘキサメチレンイミン、オキサゾリジン、チアゾリジン、イミダゾリジン、オキサゾリン、チアゾリン、イミダゾリン、ジオキソール、ジオキソラン、ジヒドロオキサジアゾール、ピラン、テトラヒドロピラン、チオピラン、テトラヒドロチオピラン、テトラヒドロフラン、ピラゾリジン、ピラゾリン、テトラヒドロピリミジン、ジヒドロトリアゾール、テトラヒドロトリアゾール等が挙げられ、好ましくは、5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン)である。 The C 3-7 cycloalkane includes cyclopropane, cyclobutane, cyclopentane, cyclohexane and cycloheptane, preferably C 4-6 cycloalkane, and particularly preferably cyclobutane and cyclohexane.
Examples of C 3-7 cycloalkene include cyclopentene, cyclobutene, cyclopentene, cyclohexene, and cycloheptene, and C 4-6 cycloalkene is preferable.
Examples of C 4-7 cycloalkadiene include 2,4-cyclopentadiene, 2,4-cyclohexadiene, 2,5-cyclohexadiene, and preferably C 4-6 cycloalkadiene.
Examples of the 5- or 6-membered aromatic heterocycle include pyrrole, pyrazole, imidazole, triazole (1,2,3-triazole, 1,2,4-triazole, 1,3,4-triazole), tetrazole, oxazole, Examples include isoxazole, thiazole, isothiazole, oxadiazole, thiadiazole, furan, thiophene, pyridine, pyridazine, pyrimidine, pyrazine, triazine and the like, preferably a 6-membered nitrogen-containing aromatic heterocyclic ring (preferably pyridine, pyridazine , Pyrimidine, pyrazine, particularly preferably pyridine).
Examples of the 3- to 7-membered non-aromatic heterocycle include oxirane, oxetane, aziridine, azetidine, pyrrolidine, piperidine, morpholine, thiomorpholine, piperazine, hexamethyleneimine, oxazolidine, thiazolidine, imidazolidine, oxazoline, thiazoline, imidazoline, dioxol, Examples thereof include dioxolane, dihydrooxadiazole, pyran, tetrahydropyran, thiopyran, tetrahydrothiopyran, tetrahydrofuran, pyrazolidine, pyrazoline, tetrahydropyrimidine, dihydrotriazole, tetrahydrotriazole, and preferably 5 or 6 members (preferably 6 members) A nitrogen-containing non-aromatic heterocyclic ring (preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine).
C3-7シクロアルケンとしては、シクロペンテン、シクロブテン、シクロペンテン、シクロヘキセンおよびシクロヘプテンが挙げられ、好ましくは、C4-6シクロアルケンである。
C4-7シクロアルカジエンとしては、2,4-シクロペンタジエン、2,4-シクロヘキサジエン、2,5-シクロヘキサジエン等が挙げられ、好ましくは、C4-6シクロアルカジエンである。
5または6員芳香族複素環としては、例えば、ピロール、ピラゾール、イミダゾール、トリアゾール(1,2,3-トリアゾール、1,2,4-トリアゾール、1,3,4-トリアゾール)、テトラゾール、オキサゾール、イソオキサゾール、チアゾール、イソチアゾール、オキサジアゾール、チアジアゾール、フラン、チオフェン、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン等が挙げられ、好ましくは、6員含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)である。
3ないし7員非芳香族複素環としては、オキシラン、オキセタン、アジリジン、アゼチジン、ピロリジン、ピペリジン、モルホリン、チオモルホリン、ピペラジン、ヘキサメチレンイミン、オキサゾリジン、チアゾリジン、イミダゾリジン、オキサゾリン、チアゾリン、イミダゾリン、ジオキソール、ジオキソラン、ジヒドロオキサジアゾール、ピラン、テトラヒドロピラン、チオピラン、テトラヒドロチオピラン、テトラヒドロフラン、ピラゾリジン、ピラゾリン、テトラヒドロピリミジン、ジヒドロトリアゾール、テトラヒドロトリアゾール等が挙げられ、好ましくは、5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン)である。 The C 3-7 cycloalkane includes cyclopropane, cyclobutane, cyclopentane, cyclohexane and cycloheptane, preferably C 4-6 cycloalkane, and particularly preferably cyclobutane and cyclohexane.
Examples of C 3-7 cycloalkene include cyclopentene, cyclobutene, cyclopentene, cyclohexene, and cycloheptene, and C 4-6 cycloalkene is preferable.
Examples of C 4-7 cycloalkadiene include 2,4-cyclopentadiene, 2,4-cyclohexadiene, 2,5-cyclohexadiene, and preferably C 4-6 cycloalkadiene.
Examples of the 5- or 6-membered aromatic heterocycle include pyrrole, pyrazole, imidazole, triazole (1,2,3-triazole, 1,2,4-triazole, 1,3,4-triazole), tetrazole, oxazole, Examples include isoxazole, thiazole, isothiazole, oxadiazole, thiadiazole, furan, thiophene, pyridine, pyridazine, pyrimidine, pyrazine, triazine and the like, preferably a 6-membered nitrogen-containing aromatic heterocyclic ring (preferably pyridine, pyridazine , Pyrimidine, pyrazine, particularly preferably pyridine).
Examples of the 3- to 7-membered non-aromatic heterocycle include oxirane, oxetane, aziridine, azetidine, pyrrolidine, piperidine, morpholine, thiomorpholine, piperazine, hexamethyleneimine, oxazolidine, thiazolidine, imidazolidine, oxazoline, thiazoline, imidazoline, dioxol, Examples thereof include dioxolane, dihydrooxadiazole, pyran, tetrahydropyran, thiopyran, tetrahydrothiopyran, tetrahydrofuran, pyrazolidine, pyrazoline, tetrahydropyrimidine, dihydrotriazole, tetrahydrotriazole, and preferably 5 or 6 members (preferably 6 members) A nitrogen-containing non-aromatic heterocyclic ring (preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine).
環Pで示される「さらに置換されていてもよい3ないし7員環」における「3ないし7員環」は、基-XC(R4a)(R4b)CH(R3)-NH-R1および-YC(R5a)(R5b)-に加えて、置換可能な位置に1ないし9個の置換基を有していてもよい。このような置換基としては、例えば、R2で示される「置換基」として例示した「置換されていてもよい複素環基」における「複素環基」が有していてもよい置換基と同様のものが挙げられる。
The “3- to 7-membered ring” in the “optionally substituted 3- to 7-membered ring” represented by ring P is a group —XC (R 4a ) (R 4b ) CH (R 3 ) —NH—R 1 In addition to —YC (R 5a ) (R 5b ) —, it may have 1 to 9 substituents at substitutable positions. Examples of such a substituent are the same as the substituent that the “heterocyclic group” in the “optionally substituted heterocyclic group” exemplified as the “substituent” represented by R 2 may have. Can be mentioned.
環Pは、好ましくは、それぞれさらに置換されていてもよい、ベンゼン、C3-7シクロアルカンまたは3ないし7員複素環(好ましくは、5または6員芳香族複素環または3ないし7員非芳香族複素環)である。
Ring P is preferably benzene, C 3-7 cycloalkane or 3- to 7-membered heterocycle (preferably 5- or 6-membered aromatic heterocycle or 3- to 7-membered non-aromatic, each of which may be further substituted. Family heterocycle).
環Pは、より好ましくは、それぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)または3ないし7員複素環(好ましくは、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン))である。
Ring P is more preferably benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocyclic ring (preferably 5- or 6-membered (preferably Is a 6-membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or a 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle (preferably , Pyrrolidine, piperidine, piperazine, particularly preferably piperidine)).
環Pは、さらに好ましくは、ハロゲン原子(例、フッ素原子)、C1-6アルキル基(例、メチル)およびC1-6アルコキシ基(例、メトキシ)から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)または3ないし7員複素環(好ましくは、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン))である。
Ring P is more preferably 1 to 4 (preferably selected from a halogen atom (eg, fluorine atom), a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy). Benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocycle (preferably 5- or 6-membered (preferably 5 to 6-membered) each optionally further substituted with 1 to 3 substituents. Preferably 6-membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle (preferably Is pyrrolidine, piperidine, piperazine, particularly preferably piperidine)).
環Pは、さらにより好ましくは、C1-6アルキル基(例、メチル)およびC1-6アルコキシ基(例、メトキシ)から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)または3ないし7員複素環(好ましくは、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン))である。
Ring P is more preferably 1 to 4 (preferably 1 to 3) substituents selected from a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy). Benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocycle (preferably 5- or 6-membered (preferably 6-membered) nitrogen-containing aromatic Group heterocycles (preferably pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycles (preferably pyrrolidine, piperidine, piperazine, Particularly preferred is piperidine)).
環Pは、特に好ましくは、C1-6アルキル基(例、メチル)およびC1-6アルコキシ基(例、メトキシ)から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)、ピリジンまたはピペリジンである。
Ring P is particularly preferably 1 to 4 (preferably 1 to 3) substituents selected from a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy). They are each optionally further substituted benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane), pyridine or piperidine.
環Qは、さらに置換されていてもよい5または6員芳香環を示す。
環Qで示される「さらに置換されていてもよい5または6員芳香環」における「5または6員芳香環」としては、ベンゼン、ピロール、ピラゾール、イミダゾール、トリアゾール(1,2,3-トリアゾール、1,2,4-トリアゾール、1,3,4-トリアゾール)、テトラゾール、オキサゾール、イソオキサゾール、チアゾール、イソチアゾール、オキサジアゾール、チアジアゾール、フラン、チオフェン、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン等が挙げられ、中でも、6員芳香環(例、ベンゼン、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン)が好ましく、ベンゼン、ピリジンが特に好ましい。 Ring Q represents a 5- or 6-membered aromatic ring which may be further substituted.
Examples of the “5- or 6-membered aromatic ring” in the “optionally substituted 5- or 6-membered aromatic ring” represented by ring Q include benzene, pyrrole, pyrazole, imidazole, triazole (1,2,3-triazole, 1,2,4-triazole, 1,3,4-triazole), tetrazole, oxazole, isoxazole, thiazole, isothiazole, oxadiazole, thiadiazole, furan, thiophene, pyridine, pyridazine, pyrimidine, pyrazine, triazine, etc. Among them, a 6-membered aromatic ring (eg, benzene, pyridine, pyridazine, pyrimidine, pyrazine, triazine) is preferable, and benzene and pyridine are particularly preferable.
環Qで示される「さらに置換されていてもよい5または6員芳香環」における「5または6員芳香環」としては、ベンゼン、ピロール、ピラゾール、イミダゾール、トリアゾール(1,2,3-トリアゾール、1,2,4-トリアゾール、1,3,4-トリアゾール)、テトラゾール、オキサゾール、イソオキサゾール、チアゾール、イソチアゾール、オキサジアゾール、チアジアゾール、フラン、チオフェン、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン等が挙げられ、中でも、6員芳香環(例、ベンゼン、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン)が好ましく、ベンゼン、ピリジンが特に好ましい。 Ring Q represents a 5- or 6-membered aromatic ring which may be further substituted.
Examples of the “5- or 6-membered aromatic ring” in the “optionally substituted 5- or 6-membered aromatic ring” represented by ring Q include benzene, pyrrole, pyrazole, imidazole, triazole (1,2,3-triazole, 1,2,4-triazole, 1,3,4-triazole), tetrazole, oxazole, isoxazole, thiazole, isothiazole, oxadiazole, thiadiazole, furan, thiophene, pyridine, pyridazine, pyrimidine, pyrazine, triazine, etc. Among them, a 6-membered aromatic ring (eg, benzene, pyridine, pyridazine, pyrimidine, pyrazine, triazine) is preferable, and benzene and pyridine are particularly preferable.
環Qで示される「さらに置換されていてもよい5または6員芳香環」における「5または6員芳香環」は、基-O-R6および-C(R5a)(R5b)Y-に加えて、置換可能な位置に1ないし4個の置換基を有していてもよい。このような置換基としては、例えば、R2で示される「置換基」として例示した「置換されていてもよい炭化水素基」における「炭化水素基」として例示したC3-10シクロアルキル基等が有していてもよい置換基と同様のものが挙げられる。
The “5- or 6-membered aromatic ring” in the “optionally substituted 5- or 6-membered aromatic ring” represented by ring Q is a group —O—R 6 and —C (R 5a ) (R 5b ) Y— In addition, it may have 1 to 4 substituents at substitutable positions. Examples of such a substituent include a C 3-10 cycloalkyl group exemplified as the “hydrocarbon group” in the “optionally substituted hydrocarbon group” exemplified as the “substituent” represented by R 2 and the like. The same thing as the substituent which may have is mentioned.
環Qで示される「さらに置換されていてもよい5または6員芳香環」における「5または6員芳香環」のさらなる置換基は、好ましくは、ハロゲン原子(例、フッ素原子、塩素原子)である。
The further substituent of the “5- or 6-membered aromatic ring” in the “optionally substituted 5- or 6-membered aromatic ring” represented by ring Q is preferably a halogen atom (eg, fluorine atom, chlorine atom). is there.
環Qは、好ましくは、
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基
から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環(例、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン、イソオキサゾール)である。
環Qは、より好ましくは、
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基、
から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼン、ピリジンまたはイソオキサゾールである。 Ring Q is preferably
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) benzene or a 5- or 6-membered aromatic heterocycle (eg, pyridine, pyridazine) which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a cyano group, respectively. , Pyrimidine, pyrazine, triazine, isoxazole).
Ring Q is more preferably
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) a cyano group,
Benzene, pyridine or isoxazole, each of which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基
から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環(例、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン、イソオキサゾール)である。
環Qは、より好ましくは、
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基、
から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼン、ピリジンまたはイソオキサゾールである。 Ring Q is preferably
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) benzene or a 5- or 6-membered aromatic heterocycle (eg, pyridine, pyridazine) which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a cyano group, respectively. , Pyrimidine, pyrazine, triazine, isoxazole).
Ring Q is more preferably
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) a cyano group,
Benzene, pyridine or isoxazole, each of which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from
化合物(I)の好適な例としては、以下の化合物が挙げられる。
Preferred examples of compound (I) include the following compounds.
[化合物A]
R1が、
(1) -COR2(式中、R2が、置換されていてもよいC1-6アルキル基、または置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5または6員芳香環基;
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、独立して、水素原子、カルボキシ基またはC1-6アルコキシ-カルボニル基であり;
R6が、置換されていてもよいC1-6アルキル基であり;
Xが、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)、NR7c(R7cは、水素原子または置換されていてもよいC1-6アルキル基である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCR8aR8b(R8aおよびR8bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)であり;
環Pが、それぞれさらに置換されていてもよい、ベンゼン、C3-7シクロアルカン、5または6員芳香族複素環または3ないし7員非芳香族複素環であり;かつ
環Qが、1ないし3個のハロゲン原子(例、フッ素原子、塩素原子)でさらに置換されていてもよい6員芳香環(例、ベンゼン、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン)である、化合物(I)。 [Compound A]
R 1 is
(1) a group represented by —COR 2 (wherein R 2 is an optionally substituted C 1-6 alkyl group or an optionally substituted amino group); or
(2) an optionally substituted 5- or 6-membered aromatic ring group;
Is;
R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are independently a hydrogen atom, a carboxy group or a C 1-6 alkoxy-carbonyl group;
R 6 is an optionally substituted C 1-6 alkyl group;
X is O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a hydrogen atom or a substituted atom) Is an optionally substituted C 1-6 alkyl group), S, SO or S (O) 2 ;
Y is O, CO or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, fluorine atom));
Ring P is benzene, C 3-7 cycloalkane, 5 or 6 membered aromatic heterocycle or 3 to 7 membered non-aromatic heterocycle, each optionally further substituted; and Ring Q is 1 to Compound (I) which is a 6-membered aromatic ring (eg, benzene, pyridine, pyridazine, pyrimidine, pyrazine, triazine) which may be further substituted with 3 halogen atoms (eg, fluorine atom, chlorine atom).
R1が、
(1) -COR2(式中、R2が、置換されていてもよいC1-6アルキル基、または置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5または6員芳香環基;
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、独立して、水素原子、カルボキシ基またはC1-6アルコキシ-カルボニル基であり;
R6が、置換されていてもよいC1-6アルキル基であり;
Xが、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)、NR7c(R7cは、水素原子または置換されていてもよいC1-6アルキル基である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCR8aR8b(R8aおよびR8bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)であり;
環Pが、それぞれさらに置換されていてもよい、ベンゼン、C3-7シクロアルカン、5または6員芳香族複素環または3ないし7員非芳香族複素環であり;かつ
環Qが、1ないし3個のハロゲン原子(例、フッ素原子、塩素原子)でさらに置換されていてもよい6員芳香環(例、ベンゼン、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン)である、化合物(I)。 [Compound A]
R 1 is
(1) a group represented by —COR 2 (wherein R 2 is an optionally substituted C 1-6 alkyl group or an optionally substituted amino group); or
(2) an optionally substituted 5- or 6-membered aromatic ring group;
Is;
R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are independently a hydrogen atom, a carboxy group or a C 1-6 alkoxy-carbonyl group;
R 6 is an optionally substituted C 1-6 alkyl group;
X is O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a hydrogen atom or a substituted atom) Is an optionally substituted C 1-6 alkyl group), S, SO or S (O) 2 ;
Y is O, CO or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, fluorine atom));
Ring P is benzene, C 3-7 cycloalkane, 5 or 6 membered aromatic heterocycle or 3 to 7 membered non-aromatic heterocycle, each optionally further substituted; and Ring Q is 1 to Compound (I) which is a 6-membered aromatic ring (eg, benzene, pyridine, pyridazine, pyrimidine, pyrazine, triazine) which may be further substituted with 3 halogen atoms (eg, fluorine atom, chlorine atom).
[化合物B]
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)またはアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル)
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよい、1ないし3個のC3-6シクロアルキル基(例、シクロプロピル)で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル)であり;
Xが、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCR8aR8b(R8aおよびR8bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)であり;
環Pが、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)およびハロゲン原子(例、フッ素原子)から選ばれる1ないし4個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロヘキサン)、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン)であり;かつ
環Qが、1ないし4個のハロゲン原子(例、フッ素原子、塩素原子)でさらに置換されていてもよいベンゼンである、化合物(I)。 [Compound B]
R 1 is
(1) a group represented by —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl) or an amino group); or
(2) 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl)
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 may be substituted with 1 to 5 halogen atoms (eg, fluorine atom), or may be substituted with 1 to 3 C 3-6 cycloalkyl groups (eg, cyclopropyl). A C 1-6 alkyl group (eg, methyl, ethyl, propyl);
X is O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a C 1-6 alkyl) A group (eg, methyl)), S, SO or S (O) 2 ;
Y is O, CO or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, fluorine atom));
Ring P is further substituted with 1 to 4 substituents selected from a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy) and a halogen atom (eg, fluorine atom), respectively. Benzene, C 4-6 cycloalkane (preferably cyclohexane), 5 or 6 membered (preferably 6 membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, Pyridine), or a 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocyclic ring (preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine); and ring Q is 1 to Compound (I), which is benzene which may be further substituted with four halogen atoms (eg, fluorine atom, chlorine atom).
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)またはアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル)
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよい、1ないし3個のC3-6シクロアルキル基(例、シクロプロピル)で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル)であり;
Xが、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCR8aR8b(R8aおよびR8bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)であり;
環Pが、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)およびハロゲン原子(例、フッ素原子)から選ばれる1ないし4個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロヘキサン)、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン)であり;かつ
環Qが、1ないし4個のハロゲン原子(例、フッ素原子、塩素原子)でさらに置換されていてもよいベンゼンである、化合物(I)。 [Compound B]
R 1 is
(1) a group represented by —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl) or an amino group); or
(2) 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl)
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 may be substituted with 1 to 5 halogen atoms (eg, fluorine atom), or may be substituted with 1 to 3 C 3-6 cycloalkyl groups (eg, cyclopropyl). A C 1-6 alkyl group (eg, methyl, ethyl, propyl);
X is O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a C 1-6 alkyl) A group (eg, methyl)), S, SO or S (O) 2 ;
Y is O, CO or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, fluorine atom));
Ring P is further substituted with 1 to 4 substituents selected from a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy) and a halogen atom (eg, fluorine atom), respectively. Benzene, C 4-6 cycloalkane (preferably cyclohexane), 5 or 6 membered (preferably 6 membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, Pyridine), or a 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocyclic ring (preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine); and ring Q is 1 to Compound (I), which is benzene which may be further substituted with four halogen atoms (eg, fluorine atom, chlorine atom).
[化合物C]
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)またはアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル)
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル)、および
(b) ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル)であり;
Xが、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCR8aR8b(R8aおよびR8bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)であり;
環Pが、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)およびハロゲン原子(例、フッ素原子)から選ばれる1ないし4個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロヘキサン)、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン)であり;かつ
環Qが、1ないし4個のハロゲン原子(例、フッ素原子、塩素原子)でさらにそれぞれ置換されていてもよい、ベンゼンまたはピリジンである、化合物(I)。 [Compound C]
R 1 is
(1) a group represented by —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl) or an amino group); or
(2) 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl)
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a C 3-6 cycloalkyl group (eg, cyclopropyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(b) Halogen atoms (eg, fluorine atoms)
A C 1-6 alkyl group (eg, methyl, ethyl, propyl) optionally substituted with 1 to 3 substituents selected from:
X is O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a C 1-6 alkyl) A group (eg, methyl)), S, SO or S (O) 2 ;
Y is O, CO or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, fluorine atom));
Ring P is further substituted with 1 to 4 substituents selected from a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy) and a halogen atom (eg, fluorine atom), respectively. Benzene, C 4-6 cycloalkane (preferably cyclohexane), 5 or 6 membered (preferably 6 membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, Pyridine), or a 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocyclic ring (preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine); and ring Q is 1 to Compound which is benzene or pyridine, each of which may be further substituted with 4 halogen atoms (eg, fluorine atom, chlorine atom) I).
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)またはアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル)
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル)、および
(b) ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル)であり;
Xが、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCR8aR8b(R8aおよびR8bは、独立して、水素原子またはハロゲン原子(例、フッ素原子)である)であり;
環Pが、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)およびハロゲン原子(例、フッ素原子)から選ばれる1ないし4個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロヘキサン)、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン)であり;かつ
環Qが、1ないし4個のハロゲン原子(例、フッ素原子、塩素原子)でさらにそれぞれ置換されていてもよい、ベンゼンまたはピリジンである、化合物(I)。 [Compound C]
R 1 is
(1) a group represented by —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl) or an amino group); or
(2) 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl)
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a C 3-6 cycloalkyl group (eg, cyclopropyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(b) Halogen atoms (eg, fluorine atoms)
A C 1-6 alkyl group (eg, methyl, ethyl, propyl) optionally substituted with 1 to 3 substituents selected from:
X is O, CO, CR 7a R 7b (R 7a and R 7b are each independently a hydrogen atom or a halogen atom (eg, a fluorine atom)), NR 7c (R 7c is a C 1-6 alkyl) A group (eg, methyl)), S, SO or S (O) 2 ;
Y is O, CO or CR 8a R 8b (R 8a and R 8b are independently a hydrogen atom or a halogen atom (eg, fluorine atom));
Ring P is further substituted with 1 to 4 substituents selected from a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy) and a halogen atom (eg, fluorine atom), respectively. Benzene, C 4-6 cycloalkane (preferably cyclohexane), 5 or 6 membered (preferably 6 membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, Pyridine), or a 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocyclic ring (preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine); and ring Q is 1 to Compound which is benzene or pyridine, each of which may be further substituted with 4 halogen atoms (eg, fluorine atom, chlorine atom) I).
[化合物D]
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)またはアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル)
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル)、および
(b) ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル)であり;
Xが、O、CO、CR7aR7b(R7aおよびR7bは、水素原子である)、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCR8aR8b(R8aおよびR8bは、水素原子である)であり;
環Pが、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)およびハロゲン原子(例、フッ素原子)から選ばれる1ないし4個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロヘキサン)、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン)であり;かつ
環Qが、1ないし4個のハロゲン原子(例、フッ素原子、塩素原子)でさらにそれぞれ置換されていてもよい、ベンゼンまたはピリジンである、化合物(I)。 [Compound D]
R 1 is
(1) a group represented by —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl) or an amino group); or
(2) 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl)
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a C 3-6 cycloalkyl group (eg, cyclopropyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(b) Halogen atoms (eg, fluorine atoms)
A C 1-6 alkyl group (eg, methyl, ethyl, propyl) optionally substituted with 1 to 3 substituents selected from:
X is O, CO, CR 7a R 7b (R 7a and R 7b are hydrogen atoms), NR 7c (R 7c is a C 1-6 alkyl group (eg, methyl)), S, SO Or S (O) 2 ;
Y is O, CO or CR 8a R 8b (R 8a and R 8b are hydrogen atoms);
Ring P is further substituted with 1 to 4 substituents selected from a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy) and a halogen atom (eg, fluorine atom), respectively. Benzene, C 4-6 cycloalkane (preferably cyclohexane), 5 or 6 membered (preferably 6 membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, Pyridine), or a 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocyclic ring (preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine); and ring Q is 1 to Compound which is benzene or pyridine, each of which may be further substituted with 4 halogen atoms (eg, fluorine atom, chlorine atom) I).
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)またはアミノ基である。)で表される基;または
(2) 1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員芳香族複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル)
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル)、および
(b) ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル)であり;
Xが、O、CO、CR7aR7b(R7aおよびR7bは、水素原子である)、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCR8aR8b(R8aおよびR8bは、水素原子である)であり;
環Pが、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)およびハロゲン原子(例、フッ素原子)から選ばれる1ないし4個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロヘキサン)、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン)であり;かつ
環Qが、1ないし4個のハロゲン原子(例、フッ素原子、塩素原子)でさらにそれぞれ置換されていてもよい、ベンゼンまたはピリジンである、化合物(I)。 [Compound D]
R 1 is
(1) a group represented by —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl) or an amino group); or
(2) 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl), preferably Isoxazolyl)
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a C 3-6 cycloalkyl group (eg, cyclopropyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(b) Halogen atoms (eg, fluorine atoms)
A C 1-6 alkyl group (eg, methyl, ethyl, propyl) optionally substituted with 1 to 3 substituents selected from:
X is O, CO, CR 7a R 7b (R 7a and R 7b are hydrogen atoms), NR 7c (R 7c is a C 1-6 alkyl group (eg, methyl)), S, SO Or S (O) 2 ;
Y is O, CO or CR 8a R 8b (R 8a and R 8b are hydrogen atoms);
Ring P is further substituted with 1 to 4 substituents selected from a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy) and a halogen atom (eg, fluorine atom), respectively. Benzene, C 4-6 cycloalkane (preferably cyclohexane), 5 or 6 membered (preferably 6 membered) nitrogen-containing aromatic heterocycle (preferably pyridine, pyridazine, pyrimidine, pyrazine, Pyridine), or a 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocyclic ring (preferably pyrrolidine, piperidine, piperazine, particularly preferably piperidine); and ring Q is 1 to Compound which is benzene or pyridine, each of which may be further substituted with 4 halogen atoms (eg, fluorine atom, chlorine atom) I).
[化合物E]
R1が、
(1) -COR2(式中、R2が、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5員の芳香環複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)であり;
Xが、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCH2であり;
環Pが、ハロゲン原子(例、フッ素原子)、C1-6アルキル基(例、メチル)およびC1-6アルコキシ基(例、メトキシ)から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)または3ないし7員複素環(好ましくは、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン))であり;かつ
環Qが、
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基
から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環(例、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン、イソオキサゾール)である、化合物(I)。 [Compound E]
R 1 is
(1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or
(2) an optionally substituted 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl);
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl)) Is a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic hetero group (eg tetradofryl), and
(d) a C 6-14 aryl group (eg, phenyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl) )
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted with 1 to 7 (preferably 1 to 3) substituents selected from:
X is O, CO, CH 2 , NR 7c (R 7c is a hydrogen atom or a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 ;
Y is O, CO or CH 2 ;
Ring P is further substituted with 1 to 3 substituents each selected from a halogen atom (eg, fluorine atom), a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy). Benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocycle (preferably 5- or 6-membered (preferably 6-membered) nitrogen-containing aromatic heterocycle ( Preferably, pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle (preferably pyrrolidine, piperidine, piperazine, particularly preferably Piperidine)); and ring Q is
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) benzene or a 5- or 6-membered aromatic heterocyclic ring (eg, pyridine, pyridazine, pyrimidine, pyrazine, triazine, iso-azo, which may be further substituted with 1 to 3 substituents selected from cyano groups. Compound (I), which is oxazole).
R1が、
(1) -COR2(式中、R2が、水素原子、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2) 置換されていてもよい5員の芳香環複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)であり;
Xが、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCH2であり;
環Pが、ハロゲン原子(例、フッ素原子)、C1-6アルキル基(例、メチル)およびC1-6アルコキシ基(例、メトキシ)から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)または3ないし7員複素環(好ましくは、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン))であり;かつ
環Qが、
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基
から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環(例、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン、イソオキサゾール)である、化合物(I)。 [Compound E]
R 1 is
(1) —COR 2 (wherein R 2 is a hydrogen atom, a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-1 A group represented by a 6 alkyl group (eg, an amino group optionally substituted by methyl); or
(2) an optionally substituted 5-membered aromatic heterocyclic group (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably isoxazolyl);
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl)) Is a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic hetero group (eg tetradofryl), and
(d) a C 6-14 aryl group (eg, phenyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl) )
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted with 1 to 7 (preferably 1 to 3) substituents selected from:
X is O, CO, CH 2 , NR 7c (R 7c is a hydrogen atom or a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 ;
Y is O, CO or CH 2 ;
Ring P is further substituted with 1 to 3 substituents each selected from a halogen atom (eg, fluorine atom), a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy). Benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocycle (preferably 5- or 6-membered (preferably 6-membered) nitrogen-containing aromatic heterocycle ( Preferably, pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle (preferably pyrrolidine, piperidine, piperazine, particularly preferably Piperidine)); and ring Q is
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) benzene or a 5- or 6-membered aromatic heterocyclic ring (eg, pyridine, pyridazine, pyrimidine, pyrazine, triazine, iso-azo, which may be further substituted with 1 to 3 substituents selected from cyano groups. Compound (I), which is oxazole).
[化合物F]
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2)1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員の芳香環複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)であり;
Xが、O、CO、CH2、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCH2であり;
環Pが、C1-6アルキル基(例、メチル)、およびC1-6アルコキシ基(例、メトキシ)から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)または3ないし7員複素環(好ましくは、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン))であり;かつ
環Qが、
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基
から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環(例、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン、イソオキサゾール)である、化合物(I)。 [Compound F]
R 1 is
(1) —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or
(2) 5-membered aromatic heterocyclic group optionally substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl) (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably Isoxazolyl);
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl)) Is a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic hetero group (eg tetradofryl), and
(d) a C 6-14 aryl group (eg, phenyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl) )
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted with 1 to 7 (preferably 1 to 3) substituents selected from:
X is O, CO, CH 2 , NR 7c (R 7c is a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 ;
Y is O, CO or CH 2 ;
Ring P is further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy), respectively. Benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocycle (preferably 5- or 6-membered (preferably 6-membered) nitrogen-containing aromatic heterocycle ( Preferably, pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle (preferably pyrrolidine, piperidine, piperazine, particularly preferably Piperidine)); and ring Q is
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) benzene or a 5- or 6-membered aromatic heterocyclic ring (eg, pyridine, pyridazine), each of which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a cyano group , Pyrimidine, pyrazine, triazine, isoxazole).
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2)1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員の芳香環複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)であり;
Xが、O、CO、CH2、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、O、COまたはCH2であり;
環Pが、C1-6アルキル基(例、メチル)、およびC1-6アルコキシ基(例、メトキシ)から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)または3ないし7員複素環(好ましくは、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン))であり;かつ
環Qが、
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基
から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環(例、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン、イソオキサゾール)である、化合物(I)。 [Compound F]
R 1 is
(1) —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or
(2) 5-membered aromatic heterocyclic group optionally substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl) (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably Isoxazolyl);
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl)) Is a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic hetero group (eg tetradofryl), and
(d) a C 6-14 aryl group (eg, phenyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl) )
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted with 1 to 7 (preferably 1 to 3) substituents selected from:
X is O, CO, CH 2 , NR 7c (R 7c is a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 ;
Y is O, CO or CH 2 ;
Ring P is further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy), respectively. Benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocycle (preferably 5- or 6-membered (preferably 6-membered) nitrogen-containing aromatic heterocycle ( Preferably, pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle (preferably pyrrolidine, piperidine, piperazine, particularly preferably Piperidine)); and ring Q is
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) benzene or a 5- or 6-membered aromatic heterocyclic ring (eg, pyridine, pyridazine), each of which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a cyano group , Pyrimidine, pyrazine, triazine, isoxazole).
[化合物G]
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2)1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員の芳香環複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)であり;
Xが、O、CO、CH2、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、Oであり;
環Pが、C1-6アルキル基(例、メチル)、およびC1-6アルコキシ基(例、メトキシ)から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)または3ないし7員複素環(好ましくは、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン))であり;かつ
環Qが、
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基
から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環(例、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン、イソオキサゾール)である、化合物(I)。 [Compound G]
R 1 is
(1) —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or
(2) 5-membered aromatic heterocyclic group optionally substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl) (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably Isoxazolyl);
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl)) Is a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic hetero group (eg tetradofryl), and
(d) a C 6-14 aryl group (eg, phenyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl) )
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted with 1 to 7 (preferably 1 to 3) substituents selected from:
X is O, CO, CH 2 , NR 7c (R 7c is a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 ;
Y is O;
Ring P is further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy), respectively. Benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocycle (preferably 5- or 6-membered (preferably 6-membered) nitrogen-containing aromatic heterocycle ( Preferably, pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle (preferably pyrrolidine, piperidine, piperazine, particularly preferably Piperidine)); and ring Q is
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) benzene or a 5- or 6-membered aromatic heterocyclic ring (eg, pyridine, pyridazine), each of which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a cyano group , Pyrimidine, pyrazine, triazine, isoxazole).
R1が、
(1) -COR2(式中、R2が、C1-6アルキル基(例、メチル)、C1-6アルコキシ基(例、メトキシ)、または1ないし2個のC1-6アルキル基(例、メチル)で置換されていてもよいアミノ基である。)で表される基;または
(2)1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい5員の芳香環複素環基(例、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、チアジアゾリル、好ましくはイソオキサゾリル);
であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基(例、メチル、エチル)であり;
R4aおよびR4bが、共に水素原子であり;
R5aおよびR5bが、共に水素原子であり;
R6が、
(a) ハロゲン原子(例、フッ素原子)、
(b) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC3-6シクロアルキル基(例、シクロプロピル、シクロブチル)、
(c) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよい4ないし7員の複素環基(好ましくは、4ないし7員(好ましくは5または6員)の非芳香族複素基(例、テトラドロフリル))、および
(d) 1ないし3個のハロゲン原子(例、フッ素原子)または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいC6-14アリール基(例、フェニル)
から選ばれる1ないし7個(好ましくは1ないし3個)の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル)であり;
Xが、O、CO、CH2、NR7c(R7cは、C1-6アルキル基(例、メチル)である)、S、SOまたはS(O)2であり;
Yが、Oであり;
環Pが、C1-6アルキル基(例、メチル)、およびC1-6アルコキシ基(例、メトキシ)から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C4-6シクロアルカン(好ましくは、シクロブタン、シクロヘキサン)または3ないし7員複素環(好ましくは、5または6員(好ましくは6員)含窒素芳香族複素環(好ましくは、ピリジン、ピリダジン、ピリミジン、ピラジン、特に好ましくは、ピリジン)、または5または6員(好ましくは6員)含窒素非芳香族複素環(好ましくは、ピロリジン、ピペリジン、ピペラジン、特に好ましくは、ピペリジン))であり;かつ
環Qが、
(a) ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)、
(b) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(c) 1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ)、および
(d) シアノ基
から選ばれる1ないし4個(好ましくは1ないし3個)の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環(例、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン、イソオキサゾール)である、化合物(I)。 [Compound G]
R 1 is
(1) —COR 2 (wherein R 2 is a C 1-6 alkyl group (eg, methyl), a C 1-6 alkoxy group (eg, methoxy), or 1 to 2 C 1-6 alkyl groups) (Eg, an amino group optionally substituted with methyl)) or
(2) 5-membered aromatic heterocyclic group optionally substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl) (eg, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, preferably Isoxazolyl);
Is;
R 3 is a C 1-6 alkyl group (eg, methyl, ethyl) optionally substituted with 1 to 3 halogen atoms;
R 4a and R 4b are both hydrogen atoms;
R 5a and R 5b are both hydrogen atoms;
R 6 is
(a) a halogen atom (eg, fluorine atom),
(b) C 3-6 cycloalkyl group (eg, optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl group (eg, methyl)) Cyclopropyl, cyclobutyl),
(c) a 4- to 7-membered heterocyclic group (preferably substituted with 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl)) Is a 4- to 7-membered (preferably 5- or 6-membered) non-aromatic hetero group (eg tetradofryl), and
(d) a C 6-14 aryl group (eg, phenyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom) or 1 to 3 C 1-6 alkyl groups (eg, methyl) )
A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, isobutyl) optionally substituted with 1 to 7 (preferably 1 to 3) substituents selected from:
X is O, CO, CH 2 , NR 7c (R 7c is a C 1-6 alkyl group (eg, methyl)), S, SO or S (O) 2 ;
Y is O;
Ring P is further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a C 1-6 alkyl group (eg, methyl) and a C 1-6 alkoxy group (eg, methoxy), respectively. Benzene, C 4-6 cycloalkane (preferably cyclobutane, cyclohexane) or 3- to 7-membered heterocycle (preferably 5- or 6-membered (preferably 6-membered) nitrogen-containing aromatic heterocycle ( Preferably, pyridine, pyridazine, pyrimidine, pyrazine, particularly preferably pyridine), or 5- or 6-membered (preferably 6-membered) nitrogen-containing non-aromatic heterocycle (preferably pyrrolidine, piperidine, piperazine, particularly preferably Piperidine)); and ring Q is
(a) a halogen atom (eg, fluorine atom, chlorine atom, bromine atom),
(b) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom),
(c) a C 1-6 alkoxy group (eg, methoxy) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom), and
(d) benzene or a 5- or 6-membered aromatic heterocyclic ring (eg, pyridine, pyridazine), each of which may be further substituted with 1 to 4 (preferably 1 to 3) substituents selected from a cyano group , Pyrimidine, pyrazine, triazine, isoxazole).
[化合物H]
N-{(1S)-2-[(trans-4-{[2-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド;
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド;または
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド。 [Compound H]
N-{(1S) -2-[(trans-4-{[2-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide;
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide Or
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Acetamide.
N-{(1S)-2-[(trans-4-{[2-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド;
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド;または
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド。 [Compound H]
N-{(1S) -2-[(trans-4-{[2-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide;
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide Or
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Acetamide.
式(I)で表される化合物の塩としては、薬理学的に許容される塩が好ましく、このような塩としては、例えば、無機塩基との塩、有機塩基との塩、無機酸との塩、有機酸との塩、塩基性または酸性アミノ酸との塩等が挙げられる。
The salt of the compound represented by formula (I) is preferably a pharmacologically acceptable salt. Examples of such a salt include a salt with an inorganic base, a salt with an organic base, and a salt with an inorganic acid. And a salt with an organic acid, a salt with a basic or acidic amino acid, and the like.
無機塩基との塩の好適な例としては、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アルミニウム塩;アンモニウム塩等が挙げられる。
Preferable examples of the salt with an inorganic base include alkali metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as calcium salt and magnesium salt; aluminum salt; ammonium salt and the like.
有機塩基との塩の好適な例としては、トリメチルアミン、トリエチルアミン、ピリジン、ピコリン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、トロメタミン[トリス(ヒドロキシメチル)メチルアミン]、tert-ブチルアミン、シクロヘキシルアミン、ベンジルアミン、ジシクロヘキシルアミン、N,N-ジベンジルエチレンジアミン等との塩が挙げられる。
Preferable examples of the salt with an organic base include trimethylamine, triethylamine, pyridine, picoline, ethanolamine, diethanolamine, triethanolamine, tromethamine [tris (hydroxymethyl) methylamine], tert-butylamine, cyclohexylamine, benzylamine, And salts with dicyclohexylamine, N, N-dibenzylethylenediamine and the like.
無機酸との塩の好適な例としては、塩酸、臭化水素酸、硝酸、硫酸、リン酸等との塩が挙げられる。
Preferable examples of the salt with inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
有機酸との塩の好適な例としては、ギ酸、酢酸、トリフルオロ酢酸、フタル酸、フマル酸、シュウ酸、酒石酸、マレイン酸、クエン酸、コハク酸、リンゴ酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸等との塩が挙げられる。
Preferable examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, and benzenesulfonic acid And salts with p-toluenesulfonic acid and the like.
塩基性アミノ酸との塩の好適な例としては、アルギニン、リジン、オルニチン等との塩が挙げられる。
Preferable examples of salts with basic amino acids include salts with arginine, lysine, ornithine and the like.
酸性アミノ酸との塩の好適な例としては、アスパラギン酸、グルタミン酸等との塩が挙げられる。
Preferable examples of the salt with acidic amino acid include salts with aspartic acid, glutamic acid and the like.
化合物(I)のプロドラッグは、生体内における生理条件下で酵素や胃酸等による反応により化合物(I)に変換する化合物、すなわち酵素的に酸化、還元、加水分解等を起こして化合物(I)に変化する化合物、胃酸等により加水分解等を起こして化合物(I)に変化する化合物である。
A prodrug of compound (I) is a compound that is converted to compound (I) by a reaction with an enzyme, gastric acid, or the like under physiological conditions in vivo, that is, compound (I) that is enzymatically oxidized, reduced, hydrolyzed, etc. Or a compound that undergoes hydrolysis or the like by gastric acid or the like and changes to compound (I).
化合物(I)のプロドラッグとしては、
化合物(I)のアミノ基がアシル化、アルキル化またはリン酸化された化合物(例、化合物(I)のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化、ピバロイルオキシメチル化またはtert-ブチル化された化合物);
化合物(I)のヒドロキシ基がアシル化、アルキル化、リン酸化またはホウ酸化された化合物(例、化合物(I)のヒドロキシ基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化またはジメチルアミノメチルカルボニル化された化合物);
化合物(I)のカルボキシ基がエステル化またはアミド化された化合物(例、化合物(I)のカルボキシ基がエチルエステル化、フェニルエステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、エトキシカルボニルオキシエチルエステル化、フタリジルエステル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メチルエステル化、シクロヘキシルオキシカルボニルエチルエステル化またはメチルアミド化された化合物);
等が挙げられる。これらの化合物は自体公知の方法によって化合物(I)から製造することができる。 As a prodrug of compound (I),
Compounds wherein the amino group of compound (I) is acylated, alkylated or phosphorylated (eg, the amino group of compound (I) is eicosanoylated, alanylated, pentylaminocarbonylated, (5-methyl-2-oxo- 1,3-dioxolen-4-yl) methoxycarbonylated, tetrahydrofuranylated, pyrrolidylmethylated, pivaloyloxymethylated or tert-butylated compounds);
Compounds in which the hydroxy group of compound (I) is acylated, alkylated, phosphorylated or borated (eg, hydroxy group of compound (I) is acetylated, palmitoylated, propanoylated, pivaloylated, succinylated, fumarylated Alanylated or dimethylaminomethylcarbonylated compounds);
Compound in which carboxy group of compound (I) is esterified or amidated (eg, carboxy group of compound (I) is ethyl esterified, phenyl esterified, carboxymethyl esterified, dimethylaminomethyl esterified, pivaloyloxy Methyl esterification, ethoxycarbonyloxyethyl esterification, phthalidyl esterification, (5-methyl-2-oxo-1,3-dioxolen-4-yl) methyl esterification, cyclohexyloxycarbonylethyl esterification or methylamidation Compound);
Etc. These compounds can be produced from compound (I) by a method known per se.
化合物(I)のアミノ基がアシル化、アルキル化またはリン酸化された化合物(例、化合物(I)のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化、ピバロイルオキシメチル化またはtert-ブチル化された化合物);
化合物(I)のヒドロキシ基がアシル化、アルキル化、リン酸化またはホウ酸化された化合物(例、化合物(I)のヒドロキシ基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化またはジメチルアミノメチルカルボニル化された化合物);
化合物(I)のカルボキシ基がエステル化またはアミド化された化合物(例、化合物(I)のカルボキシ基がエチルエステル化、フェニルエステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、エトキシカルボニルオキシエチルエステル化、フタリジルエステル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メチルエステル化、シクロヘキシルオキシカルボニルエチルエステル化またはメチルアミド化された化合物);
等が挙げられる。これらの化合物は自体公知の方法によって化合物(I)から製造することができる。 As a prodrug of compound (I),
Compounds wherein the amino group of compound (I) is acylated, alkylated or phosphorylated (eg, the amino group of compound (I) is eicosanoylated, alanylated, pentylaminocarbonylated, (5-methyl-2-oxo- 1,3-dioxolen-4-yl) methoxycarbonylated, tetrahydrofuranylated, pyrrolidylmethylated, pivaloyloxymethylated or tert-butylated compounds);
Compounds in which the hydroxy group of compound (I) is acylated, alkylated, phosphorylated or borated (eg, hydroxy group of compound (I) is acetylated, palmitoylated, propanoylated, pivaloylated, succinylated, fumarylated Alanylated or dimethylaminomethylcarbonylated compounds);
Compound in which carboxy group of compound (I) is esterified or amidated (eg, carboxy group of compound (I) is ethyl esterified, phenyl esterified, carboxymethyl esterified, dimethylaminomethyl esterified, pivaloyloxy Methyl esterification, ethoxycarbonyloxyethyl esterification, phthalidyl esterification, (5-methyl-2-oxo-1,3-dioxolen-4-yl) methyl esterification, cyclohexyloxycarbonylethyl esterification or methylamidation Compound);
Etc. These compounds can be produced from compound (I) by a method known per se.
また、化合物(I)のプロドラッグは、廣川書店1990年刊「医薬品の開発」第7巻分子設計163頁から198頁に記載されているような、生理的条件で化合物(I)に変化するものであってもよい。
本明細書において、プロドラッグは塩を形成していてもよく、かかる塩としては、前述の式(I)で示される化合物の塩として例示したものが挙げられる。 In addition, the prodrug of compound (I) is a compound that changes to compound (I) under physiological conditions as described in Yodogawa Shoten 1990, “Development of Drugs”, Volume 7, pages 163 to 198. It may be.
In the present specification, the prodrug may form a salt, and examples of the salt include those exemplified as the salt of the compound represented by the aforementioned formula (I).
本明細書において、プロドラッグは塩を形成していてもよく、かかる塩としては、前述の式(I)で示される化合物の塩として例示したものが挙げられる。 In addition, the prodrug of compound (I) is a compound that changes to compound (I) under physiological conditions as described in Yodogawa Shoten 1990, “Development of Drugs”, Volume 7, pages 163 to 198. It may be.
In the present specification, the prodrug may form a salt, and examples of the salt include those exemplified as the salt of the compound represented by the aforementioned formula (I).
また、化合物(I)は、同位元素(例、3H、13C、14C、18F、35S、125I)等で標識されていてもよい。
さらに、化合物(I)は、水和物であっても、非水和物であっても、無溶媒和物であっても、溶媒和物であってもよい。
さらに、1Hを2H(D)に変換した重水素変換体も、化合物(I)に包含される。
さらに、化合物(I)は、薬学的に許容され得る共結晶または共結晶塩であってもよい。ここで、共結晶または共結晶塩とは、各々が異なる物理的特性(例えば、構造、融点、融解熱、吸湿性、溶解性および安定性等)を持つ、室温で二種またはそれ以上の独特な固体から構成される結晶性物質を意味する。共結晶または共結晶塩は、自体公知の共結晶化法に従い製造することができる。 Compound (I) may be labeled with an isotope (eg, 3 H, 13 C, 14 C, 18 F, 35 S, 125 I) or the like.
Furthermore, compound (I) may be a hydrate, a non-hydrate, a solvate or a solvate.
Further, a deuterium converter obtained by converting 1 H into 2 H (D) is also encompassed in compound (I).
Furthermore, Compound (I) may be a pharmaceutically acceptable cocrystal or cocrystal salt. Here, co-crystals or co-crystal salts are two or more unique at room temperature, each having different physical properties (eg structure, melting point, heat of fusion, hygroscopicity, solubility and stability). It means a crystalline substance composed of a simple solid. The cocrystal or cocrystal salt can be produced according to a cocrystallization method known per se.
さらに、化合物(I)は、水和物であっても、非水和物であっても、無溶媒和物であっても、溶媒和物であってもよい。
さらに、1Hを2H(D)に変換した重水素変換体も、化合物(I)に包含される。
さらに、化合物(I)は、薬学的に許容され得る共結晶または共結晶塩であってもよい。ここで、共結晶または共結晶塩とは、各々が異なる物理的特性(例えば、構造、融点、融解熱、吸湿性、溶解性および安定性等)を持つ、室温で二種またはそれ以上の独特な固体から構成される結晶性物質を意味する。共結晶または共結晶塩は、自体公知の共結晶化法に従い製造することができる。 Compound (I) may be labeled with an isotope (eg, 3 H, 13 C, 14 C, 18 F, 35 S, 125 I) or the like.
Furthermore, compound (I) may be a hydrate, a non-hydrate, a solvate or a solvate.
Further, a deuterium converter obtained by converting 1 H into 2 H (D) is also encompassed in compound (I).
Furthermore, Compound (I) may be a pharmaceutically acceptable cocrystal or cocrystal salt. Here, co-crystals or co-crystal salts are two or more unique at room temperature, each having different physical properties (eg structure, melting point, heat of fusion, hygroscopicity, solubility and stability). It means a crystalline substance composed of a simple solid. The cocrystal or cocrystal salt can be produced according to a cocrystallization method known per se.
化合物(I)またはそのプロドラッグ(以下、単に本発明化合物と略記することがある)は、毒性が低く、そのまま、または薬理学的に許容し得る担体等と混合して医薬組成物とすることにより、哺乳動物(例、ヒト、マウス、ラット、ウサギ、イヌ、ネコ、ウシ、ウマ、ブタ、サル)に対して、後述する各種疾患の予防または治療剤として用いることができる。
Compound (I) or a prodrug thereof (hereinafter, sometimes simply abbreviated as the compound of the present invention) has low toxicity and should be used as it is or mixed with a pharmacologically acceptable carrier to form a pharmaceutical composition. Thus, it can be used as a preventive or therapeutic agent for various diseases described below for mammals (eg, humans, mice, rats, rabbits, dogs, cats, cows, horses, pigs, monkeys).
ここにおいて、薬理学的に許容し得る担体としては、製剤素材として慣用の各種有機または無機担体物質が用いられ、固形製剤における賦形剤、滑沢剤、結合剤、崩壊剤;液状製剤における溶剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、無痛化剤等として配合される。また必要に応じて、防腐剤、抗酸化剤、着色剤、甘味剤等の製剤添加物を用いることもできる。
Here, as the pharmacologically acceptable carrier, various conventional organic or inorganic carrier substances are used as a pharmaceutical material, and excipients, lubricants, binders, disintegrants in solid preparations; solvents in liquid preparations , Solubilizing agents, suspending agents, isotonic agents, buffers, soothing agents and the like. If necessary, preparation additives such as preservatives, antioxidants, colorants, sweeteners and the like can also be used.
賦形剤の好適な例としては、乳糖、白糖、D-マンニトール、D-ソルビトール、デンプン、α化デンプン、デキストリン、結晶セルロース、低置換度ヒドロキシプロピルセルロース、カルボキシメチルセルロースナトリウム、アラビアゴム、プルラン、軽質無水ケイ酸、合成ケイ酸アルミニウム、メタケイ酸アルミン酸マグネシウムが挙げられる。
Preferable examples of excipients include lactose, sucrose, D-mannitol, D-sorbitol, starch, pregelatinized starch, dextrin, crystalline cellulose, low-substituted hydroxypropylcellulose, sodium carboxymethylcellulose, gum arabic, pullulan, light Examples thereof include anhydrous silicic acid, synthetic aluminum silicate, and magnesium aluminate metasilicate.
滑沢剤の好適な例としては、ステアリン酸マグネシウム、ステアリン酸カルシウム、タルク、コロイドシリカが挙げられる。
Preferable examples of the lubricant include magnesium stearate, calcium stearate, talc and colloidal silica.
結合剤の好適な例としては、α化デンプン、ショ糖、ゼラチン、アラビアゴム、メチルセルロース、カルボキシメチルセルロース、カルボキシメチルセルロースナトリウム、結晶セルロース、白糖、D-マンニトール、トレハロース、デキストリン、プルラン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドンが挙げられる。
Preferred examples of the binder include pregelatinized starch, sucrose, gelatin, gum arabic, methylcellulose, carboxymethylcellulose, sodium carboxymethylcellulose, crystalline cellulose, sucrose, D-mannitol, trehalose, dextrin, pullulan, hydroxypropylcellulose, hydroxy Examples include propylmethylcellulose and polyvinylpyrrolidone.
崩壊剤の好適な例としては、乳糖、白糖、デンプン、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム、軽質無水ケイ酸、低置換度ヒドロキシプロピルセルロースが挙げられる。
Preferable examples of the disintegrant include lactose, sucrose, starch, carboxymethyl cellulose, carboxymethyl cellulose calcium, croscarmellose sodium, carboxymethyl starch sodium, light anhydrous silicic acid, and low-substituted hydroxypropyl cellulose.
溶剤の好適な例としては、注射用水、生理的食塩水、リンゲル液、アルコール、プロピレングリコール、ポリエチレングリコール、ゴマ油、トウモロコシ油、オリーブ油、綿実油が挙げられる。
Suitable examples of the solvent include water for injection, physiological saline, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, sesame oil, corn oil, olive oil, and cottonseed oil.
溶解補助剤の好適な例としては、ポリエチレングリコール、プロピレングリコール、D-マンニトール、トレハロース、安息香酸ベンジル、エタノール、トリスアミノメタン、コレステロール、トリエタノールアミン、炭酸ナトリウム、クエン酸ナトリウム、サリチル酸ナトリウム、酢酸ナトリウムが挙げられる。
Preferable examples of the solubilizer include polyethylene glycol, propylene glycol, D-mannitol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, sodium salicylate, sodium acetate. Is mentioned.
懸濁化剤の好適な例としては、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、塩化ベンザルコニウム、塩化ベンゼトニウム、モノステアリン酸グリセリン等の界面活性剤;ポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース等の親水性高分子;ポリソルベート類、ポリオキシエチレン硬化ヒマシ油が挙げられる。
Suitable examples of the suspending agent include surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glyceryl monostearate; polyvinyl alcohol, polyvinylpyrrolidone , Hydrophilic polymers such as sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose, hydroxypropylcellulose; polysorbates, and polyoxyethylene hydrogenated castor oil.
等張化剤の好適な例としては、塩化ナトリウム、グリセリン、D-マンニトール、D-ソルビトール、ブドウ糖が挙げられる。
Preferable examples of the isotonic agent include sodium chloride, glycerin, D-mannitol, D-sorbitol and glucose.
緩衝剤の好適な例としては、リン酸塩、酢酸塩、炭酸塩、クエン酸塩等の緩衝液が挙げられる。
無痛化剤の好適な例としては、ベンジルアルコールが挙げられる。 Preferable examples of the buffer include buffers such as phosphate, acetate, carbonate and citrate.
A preferred example of the soothing agent is benzyl alcohol.
無痛化剤の好適な例としては、ベンジルアルコールが挙げられる。 Preferable examples of the buffer include buffers such as phosphate, acetate, carbonate and citrate.
A preferred example of the soothing agent is benzyl alcohol.
防腐剤の好適な例としては、パラオキシ安息香酸エステル類、クロロブタノール、ベンジルアルコール、フェネチルアルコール、デヒドロ酢酸、ソルビン酸が挙げられる。
抗酸化剤の好適な例としては、亜硫酸塩、アスコルビン酸塩等が挙げられる。 Preferable examples of the preservative include p-hydroxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid and sorbic acid.
Preferable examples of the antioxidant include sulfite and ascorbate.
抗酸化剤の好適な例としては、亜硫酸塩、アスコルビン酸塩等が挙げられる。 Preferable examples of the preservative include p-hydroxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid and sorbic acid.
Preferable examples of the antioxidant include sulfite and ascorbate.
着色剤の好適な例としては、水溶性食用タール色素(例、食用赤色2号および3号、食用黄色4号および5号、食用青色1号および2号等の食用色素)、水不溶性レーキ色素(例、前記水溶性食用タール色素のアルミニウム塩)、天然色素(例、β-カロチン、クロロフィル、ベンガラ)が挙げられる。
Preferred examples of the colorant include water-soluble edible tar dyes (eg, edible dyes such as edible red Nos. 2 and 3, edible yellows Nos. 4 and 5, edible blue Nos. 1 and 2, etc.), water-insoluble lake dyes (Eg, the aluminum salt of the water-soluble edible tar dye) and natural dyes (eg, β-carotene, chlorophyll, bengara).
甘味剤の好適な例としては、サッカリンナトリウム、グリチルリチン酸二カリウム、アスパルテーム、ステビアが挙げられる。
Suitable examples of sweeteners include saccharin sodium, dipotassium glycyrrhizinate, aspartame, and stevia.
本発明化合物を含有する医薬は、医薬製剤の製造法として自体公知の方法(例、日本薬局方記載の方法等)に従って、本発明化合物を単独で、または薬理学的に許容される担体と混合して、例えば錠剤(糖衣錠、フィルムコーティング錠、舌下錠、口腔内崩壊錠、バッカル錠等を含む)、丸剤、散剤、顆粒剤、カプセル剤(ソフトカプセル剤、マイクロカプセル剤を含む)、トローチ剤、シロップ剤、液剤、乳剤、懸濁剤、放出制御製剤(例、速放性製剤、徐放性製剤、徐放性マイクロカプセル剤)、エアゾール剤、フィルム剤(例、口腔内崩壊フィルム、口腔粘膜貼付フィルム)、注射剤(例、皮下注射剤、静脈内注射剤、筋肉内注射剤、腹腔内注射剤)、点滴剤、経皮吸収型製剤、軟膏剤、ローション剤、貼付剤、坐剤(例、肛門坐剤、膣坐剤)、ペレット、経鼻剤、経肺剤(吸入剤)、点眼剤等として、経口的または非経口的(例、静脈内、筋肉内、皮下、臓器内、鼻腔内、皮内、点眼、脳内、直腸内、膣内、腹腔内、腫瘍内部、腫瘍の近位等への投与および直接的な病巣への投与)に安全に投与することができる。
The medicament containing the compound of the present invention can be used alone or mixed with a pharmacologically acceptable carrier according to a method known per se as a method for producing a pharmaceutical preparation (eg, a method described in the Japanese Pharmacopoeia). For example, tablets (including sugar-coated tablets, film-coated tablets, sublingual tablets, orally disintegrating tablets, buccal tablets, etc.), pills, powders, granules, capsules (including soft capsules and microcapsules), troches Agent, syrup, solution, emulsion, suspension, controlled release formulation (eg, immediate release formulation, sustained release formulation, sustained release microcapsule), aerosol, film agent (eg, orally disintegrating film, Oral mucosa adhesive film), injection (eg, subcutaneous injection, intravenous injection, intramuscular injection, intraperitoneal injection), drip, transdermal preparation, ointment, lotion, patch, sitting Suppositories (eg, rectal suppositories) Vaginal suppositories), pellets, nasal preparations, pulmonary preparations (inhalants), eye drops, etc., orally or parenterally (eg, intravenous, intramuscular, subcutaneous, intraorgan, intranasal, intradermal, Ophthalmic, intracerebral, intrarectal, intravaginal, intraperitoneal, intratumoral, proximal to the tumor, etc. and direct administration to the lesion).
これらの製剤は、速放性製剤または徐放性製剤等の放出制御製剤(例、徐放性マイクロカプセル)であってもよい。
These preparations may be controlled-release preparations (eg, sustained-release microcapsules) such as immediate-release preparations or sustained-release preparations.
医薬組成物は、製剤技術分野において慣用の方法、例えば、日本薬局方に記載の方法等により製造することができる。
The pharmaceutical composition can be produced by a method commonly used in the field of pharmaceutical technology, for example, a method described in the Japanese Pharmacopoeia.
なお、医薬組成物中の本発明化合物の含量は、剤形、本発明化合物の投与量等により異なるが、例えば、約0.1~100重量%である。
The content of the compound of the present invention in the pharmaceutical composition varies depending on the dosage form, the dose of the compound of the present invention, etc., but is, for example, about 0.1 to 100% by weight.
経口剤を製造する際には、必要により、味のマスキング、腸溶性または持続性を目的として、コーティングを行ってもよい。
When manufacturing an oral preparation, if necessary, coating may be performed for the purpose of taste masking, enteric properties or sustainability.
コーティングに用いられるコーティング基剤としては、例えば、糖衣基剤、水溶性フィルムコーティング基剤、腸溶性フィルムコーティング基剤、徐放性フィルムコーティング基剤が挙げられる。
Examples of the coating base used for coating include sugar coating base, water-soluble film coating base, enteric film coating base and sustained-release film coating base.
糖衣基剤としては、白糖が用いられ、さらに、タルク、沈降炭酸カルシウム、ゼラチン、アラビアゴム、プルラン、カルナバロウ等から選ばれる1種または2種以上を併用してもよい。
As the sugar coating base, sucrose is used, and one or more selected from talc, precipitated calcium carbonate, gelatin, gum arabic, pullulan, carnauba wax and the like may be used in combination.
水溶性フィルムコーティング基剤としては、例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、メチルヒドロキシエチルセルロース等のセルロース系高分子;ポリビニルアセタールジエチルアミノアセテート、アミノアルキルメタアクリレートコポリマーE〔オイドラギットE(商品名)〕、ポリビニルピロリドン等の合成高分子;プルラン等の多糖類が挙げられる。
Examples of the water-soluble film coating base include cellulose polymers such as hydroxypropylcellulose, hydroxypropylmethylcellulose, hydroxyethylcellulose, and methylhydroxyethylcellulose; polyvinyl acetal diethylaminoacetate, aminoalkyl methacrylate copolymer E [Eudragit E (trade name) ], Synthetic polymers such as polyvinylpyrrolidone; polysaccharides such as pullulan.
腸溶性フィルムコーティング基剤としては、例えば、ヒドロキシプロピルメチルセルロース フタレート、ヒドロキシプロピルメチルセルロース アセテートサクシネート、カルボキシメチルエチルセルロース、酢酸フタル酸セルロース等のセルロース系高分子;メタアクリル酸コポリマーL〔オイドラギットL(商品名)〕、メタアクリル酸コポリマーLD〔オイドラギットL-30D55(商品名)〕、メタアクリル酸コポリマーS〔オイドラギットS(商品名)〕等のアクリル酸系高分子;セラック等の天然物が挙げられる。
Examples of enteric film coating bases include cellulose polymers such as hydroxypropylmethylcellulose phthalate, hydroxypropylmethylcellulose acetate succinate, carboxymethylethylcellulose, and cellulose acetate phthalate; methacrylic acid copolymer L [Eudragit L (trade name) ] Acrylic acid polymers such as methacrylic acid copolymer LD [Eudragit L-30D55 (trade name)], methacrylic acid copolymer S [Eudragit S (trade name)]; natural products such as shellac.
徐放性フィルムコーティング基剤としては、例えば、エチルセルロース等のセルロース系高分子;アミノアルキルメタアクリレートコポリマーRS〔オイドラギットRS(商品名)〕、アクリル酸エチル-メタクリル酸メチル共重合体懸濁液〔オイドラギットNE(商品名)〕等のアクリル酸系高分子が挙げられる。
Examples of the sustained-release film coating base include cellulose polymers such as ethyl cellulose; aminoalkyl methacrylate copolymer RS [Eudragit RS (trade name)], ethyl acrylate-methyl methacrylate copolymer suspension [Eudragit Acrylic polymer such as NE (trade name)].
上記したコーティング基剤は、その2種以上を適宜の割合で混合して用いてもよい。また、コーティングの際に、例えば、酸化チタン、三二酸化鉄等のような遮光剤を用いてもよい。
The above-mentioned coating bases may be used by mixing two or more of them in an appropriate ratio. Moreover, you may use light-shielding agents, such as a titanium oxide, ferric oxide, etc. in the case of coating.
本発明化合物は、毒性(例、急性毒性、慢性毒性、遺伝毒性、生殖毒性、肺毒性、癌原性)が低く、副作用も少なく、哺乳動物に対し、各種疾患の予防または治療剤、または診断薬として用いることができる。
The compound of the present invention has low toxicity (eg, acute toxicity, chronic toxicity, genotoxicity, reproductive toxicity, pulmonary toxicity, carcinogenicity), has few side effects, and prevents or treats or diagnoses various diseases for mammals. It can be used as a medicine.
本発明化合物は、優れたACC(アセチル-CoAカルボキシラーゼ)阻害作用を有する。ここで、ACCとしては、例えば、肝臓、脂肪組織、膵臓特異的アイソザイム(ACC1);筋肉特異的アイソザイム(ACC2)が挙げられる。
また本発明化合物は、代謝安定性に優れ、例えば、化合物の半減期が長い、生体内で代謝され難い等の利点を有する。
本発明化合物はACC2選択性を有し、特に本発明の実施例化合物はACC2高選択性を有する。
さらに本発明化合物は、体内動態(例、経口吸収性、バイオアベイラビリティー)に優れている。 The compound of the present invention has an excellent ACC (acetyl-CoA carboxylase) inhibitory action. Here, examples of ACC include liver, adipose tissue, pancreas-specific isozyme (ACC1); muscle-specific isozyme (ACC2).
The compound of the present invention is excellent in metabolic stability, and has advantages such as a long half-life of the compound and difficulty in being metabolized in vivo.
The compounds of the present invention have ACC2 selectivity, and in particular, the example compounds of the present invention have high ACC2 selectivity.
Furthermore, the compound of the present invention is excellent in pharmacokinetics (eg, oral absorbability, bioavailability).
また本発明化合物は、代謝安定性に優れ、例えば、化合物の半減期が長い、生体内で代謝され難い等の利点を有する。
本発明化合物はACC2選択性を有し、特に本発明の実施例化合物はACC2高選択性を有する。
さらに本発明化合物は、体内動態(例、経口吸収性、バイオアベイラビリティー)に優れている。 The compound of the present invention has an excellent ACC (acetyl-CoA carboxylase) inhibitory action. Here, examples of ACC include liver, adipose tissue, pancreas-specific isozyme (ACC1); muscle-specific isozyme (ACC2).
The compound of the present invention is excellent in metabolic stability, and has advantages such as a long half-life of the compound and difficulty in being metabolized in vivo.
The compounds of the present invention have ACC2 selectivity, and in particular, the example compounds of the present invention have high ACC2 selectivity.
Furthermore, the compound of the present invention is excellent in pharmacokinetics (eg, oral absorbability, bioavailability).
本発明化合物は、肥満症、糖尿病(例、1型糖尿病、2型糖尿病、妊娠糖尿病、肥満型糖尿病)、高脂血症(例、高トリグリセリド血症、高コレステロール血症、高LDLコレステロール血症、低HDLコレステロール血症、食後高脂血症)、高血圧症、心不全、糖尿病性合併症[例、神経障害、腎症、網膜症、糖尿病性心筋症、白内障、大血管障害、骨減少症、糖尿病性高浸透圧昏睡、感染症(例、呼吸器感染症、尿路感染症、消化器感染症、皮膚軟部組織感染症、下肢感染症)、糖尿病性壊疽、口腔乾燥症、聴覚の低下、脳血管障害、末梢血行障害]、メタボリックシンドローム(高トリグリセライド(TG)血症、低HDLコレステロール(HDL-C)血症、高血圧症、腹部肥満および耐糖能不全から選ばれる3つ以上を保有する病態)、筋肉減少症、または癌等の予防・治療剤として用いることができる。
The compound of the present invention contains obesity, diabetes (eg, type 1 diabetes, type 2 diabetes, gestational diabetes, obesity type diabetes), hyperlipidemia (eg, hypertriglyceridemia, hypercholesterolemia, high LDL cholesterolemia). , Low HDL cholesterolemia, postprandial hyperlipidemia), hypertension, heart failure, diabetic complications [eg, neuropathy, nephropathy, retinopathy, diabetic cardiomyopathy, cataract, macrovascular disorder, osteopenia, Diabetic hyperosmotic coma, infections (eg, respiratory infections, urinary tract infections, gastrointestinal infections, soft skin tissue infections, lower limb infections), diabetic gangrene, xerostomia, hearing loss, Cerebrovascular disorder, peripheral blood circulation disorder], metabolic syndrome (high triglyceride (TG) emia, low HDL cholesterol (HDL-C) emia, hypertension, abdominal obesity and glucose intolerance) State), it can be used as an agent for preventing or treating sarcopenia or cancer, or the like.
糖尿病の判定基準については、1999年に日本糖尿病学会から新たな判定基準が報告されている。
Regarding the criteria for determining diabetes, a new criterion was reported in 1999 by the Japan Diabetes Society.
この報告によれば、糖尿病とは、空腹時血糖値(静脈血漿におけるグルコース濃度)が126mg/dl以上、75g経口ブドウ糖負荷試験(75gOGTT)2時間値(静脈血漿におけるグルコース濃度)が200mg/dl以上、随時血糖値(静脈血漿におけるグルコース濃度)が200mg/dl以上のいずれかを示す状態である。また、上記糖尿病に該当せず、かつ、「空腹時血糖値(静脈血漿におけるグルコース濃度)が110mg/dl未満または75g経口ブドウ糖負荷試験(75gOGTT)2時間値(静脈血漿におけるグルコース濃度)が140mg/dl未満を示す状態」(正常型)でない状態を、「境界型」と呼ぶ。
According to this report, diabetes is a fasting blood glucose level (glucose concentration in venous plasma) of 126 mg / dl or higher, and a 75 g oral glucose tolerance test (75 gOGTT) 2-hour value (glucose concentration in venous plasma) of 200 mg / dl or higher. This is a state in which the blood glucose level (glucose concentration in venous plasma) is at least 200 mg / dl as needed. In addition, it does not correspond to the above-mentioned diabetes, and “a fasting blood glucose level (glucose concentration in venous plasma) is less than 110 mg / dl or a 75 g oral glucose tolerance test (75 g OGTT) 2 hour value (glucose concentration in venous plasma) is 140 mg / dl. A state that is not “a state indicating less than dl” (normal type) is referred to as a “boundary type”.
また、糖尿病の判定基準については、1997年にADA(米国糖尿病学会)から、1998年にWHO(世界保健機構)から、新たな判定基準が報告されている。
As for the criteria for determining diabetes, new criteria were reported from ADA (American Diabetes Association) in 1997 and from WHO (World Health Organization) in 1998.
これらの報告によれば、糖尿病とは、空腹時血糖値(静脈血漿におけるグルコース濃度)が126mg/dl以上であり、かつ、75g経口ブドウ糖負荷試験2時間値(静脈血漿におけるグルコース濃度)が200mg/dl以上を示す状態である。
According to these reports, diabetes is a fasting blood glucose level (glucose concentration in venous plasma) of 126 mg / dl or more, and a 75 g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma) is 200 mg / dl. This is a state showing dl or more.
また、上記報告によれば、耐糖能不全とは、空腹時血糖値(静脈血漿におけるグルコース濃度)が126mg/dl未満であり、かつ、75g経口ブドウ糖負荷試験2時間値(静脈血漿におけるグルコース濃度)が140mg/dl以上200mg/dl未満を示す状態である。さらに、ADAの報告によれば、空腹時血糖値(静脈血漿におけるグルコース濃度)が110mg/dl以上126mg/dl未満の状態をIFG(Impaired Fasting Glucose)と呼ぶ。一方、WHOの報告によれば、該IFG(Impaired Fasting Glucose)のうち、75g経口ブドウ糖負荷試験2時間値(静脈血漿におけるグルコース濃度)が140mg/dl未満である状態をIFG(Impaired Fasting Glycemia)と呼ぶ。
According to the above report, glucose intolerance is a fasting blood glucose level (glucose concentration in venous plasma) of less than 126 mg / dl, and a 75-g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma). Is a state showing 140 mg / dl or more and less than 200 mg / dl. Furthermore, according to the report of ADA, the state where the fasting blood glucose level (glucose concentration in venous plasma) is 110 mg / dl or more and less than 126 mg / dl is called IFG (Impaired Fasting Glucose). On the other hand, according to the report of WHO, the IFG (Impaired Fasting Glucose) is a state where the 75 g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma) is less than 140 mg / dl as IFG (Impaired Fasting Glycemia). Call.
本発明化合物は、上記した新たな判定基準により決定される糖尿病、境界型、耐糖能不全、IFG(Impaired Fasting Glucose)およびIFG(Impaired Fasting Glycemia)の予防・治療剤としても用いられる。さらに、本発明化合物は、境界型、耐糖能不全、IFG(Impaired Fasting Glucose)またはIFG(Impaired Fasting Glycemia)から糖尿病への進展を防止することもできる。
The compound of the present invention is also used as a prophylactic / therapeutic agent for diabetes, borderline type, glucose intolerance, IFG (Impaired Fasting Glucose) and IFG (Impaired Fasting Glycemia) determined by the above-mentioned new criteria. Furthermore, the compound of the present invention can also prevent progression from borderline type, impaired glucose tolerance, IFG (Impaired Fasting Glucose) or IFG (Impaired Fasting Glycemia) to diabetes.
本発明化合物は体重増加を抑制する作用を有していることから、哺乳動物に対し体重増加抑制剤として使用することができる。適用対象の哺乳動物は体重増加を回避したい哺乳動物であればよく、遺伝的に体重増加のリスクを有している哺乳動物であってもよいし、糖尿病、高血圧症および/または高脂血症等の生活習慣病を患っている哺乳動物であってもよい。体重増加は食事摂取の過多または栄養バランスを欠いた食生活に起因するものであってもよいし、併用薬剤(例えば、トログリタゾン、ロシグリタゾン、エングリタゾン、シグリタゾン、ピオグリタゾン等のPPARγアゴニスト様作用を有するインスリン抵抗性改善剤等)に由来する体重増加であってもよい。また、体重増加は肥満症に至る前の体重増加であってもよいし、肥満患者の体重増加であってもよい。ここで、肥満症とは、日本人ではBMI(ボディー・マス・インデックス:体重(kg)÷[身長(m)]2)が25以上(日本肥満学会の基準による)、欧米人ではBMIが30以上(WHOの基準による)と定義される。
Since the compound of the present invention has an action of suppressing weight gain, it can be used as a weight gain inhibitor for mammals. The mammal to be applied may be any mammal that wishes to avoid weight gain, may be a mammal that is genetically at risk of weight gain, and may have diabetes, hypertension and / or hyperlipidemia. It may be a mammal suffering from a lifestyle-related disease such as Weight gain may be due to excessive dietary intake or a diet lacking nutritional balance, and has a PPARγ agonist-like action such as concomitant drugs (eg, troglitazone, rosiglitazone, englitazone, ciglitazone, pioglitazone) It may be a weight gain derived from an insulin resistance improving agent or the like. The weight gain may be a weight gain before reaching obesity or may be a weight gain of an obese patient. Here, obesity means that BMI (body mass index: body weight (kg) ÷ [height (m)] 2 ) is 25 or more (according to the standards of the Japanese Obesity Society) in Japanese, and BMI is 30 in Westerners. It is defined as above (according to WHO standards).
本発明化合物は、代謝症候群(メタボリックシンドローム)の予防・治療剤としても有用である。代謝症候群の患者では、単一の生活習慣病を発症している患者に比べて心血管系疾患を発症する率が著しく高いことから、代謝症候群を予防・治療することは心血管系疾患を予防するために極めて重要である。
代謝症候群の判定基準が、1999年にWHOから、2001年にNCEPから発表されている。WHOの判定基準によれば、高インスリン血症または耐糖能異常を基本条件に、内臓肥満、異常脂質血症(高TGまたは低HDL)、高血圧のうち2つ以上を持つ場合に代謝症候群と診断される(World Health Organization: Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. Part I: Diagnosis and Classification of Diabetes Mellitus, World Health Organization, Geneva, 1999)。米国の虚血性心疾患の管理指標であるNational Cholesterol Education Program のAdult Treatment Panel IIIの判定基準によれば、内臓肥満、高中性脂肪血症、低HDLコレステロール血症、高血圧、耐糖能異常のうち3つ以上を持つ場合に代謝症候群と診断される(National Cholesterol Education Program: Executive Summary of the Third Report of National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adults Treatment Panel III). The Journal of the American Medical Association, Vol. 285, 2486-2497, 2001)。 The compound of the present invention is also useful as a prophylactic / therapeutic agent for metabolic syndrome (metabolic syndrome). Patients with metabolic syndrome have a significantly higher rate of developing cardiovascular disease than patients with a single lifestyle-related disease, so preventing or treating metabolic syndrome prevents cardiovascular disease It is extremely important to do.
Criteria for metabolic syndrome were published by WHO in 1999 and NCEP in 2001. According to WHO criteria, metabolic syndrome is diagnosed in patients with visceral obesity, dyslipidemia (high TG or low HDL), or hypertension based on hyperinsulinemia or impaired glucose tolerance. (World Health Organization: Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. Part I: Diagnosis and Classification of Diabetes Mellitus, World Health Organization, Geneva, 1999). According to the criteria of the Adult Treatment Panel III of the National Cholesterol Education Program, which is a management index for ischemic heart disease in the United States, it is 3 (National Cholesterol Education Program: Executive Summary of the Third Report of National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adults Treatment Panel III). The Journal of the American Medical Association, Vol. 285, 2486-2497, 2001).
代謝症候群の判定基準が、1999年にWHOから、2001年にNCEPから発表されている。WHOの判定基準によれば、高インスリン血症または耐糖能異常を基本条件に、内臓肥満、異常脂質血症(高TGまたは低HDL)、高血圧のうち2つ以上を持つ場合に代謝症候群と診断される(World Health Organization: Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. Part I: Diagnosis and Classification of Diabetes Mellitus, World Health Organization, Geneva, 1999)。米国の虚血性心疾患の管理指標であるNational Cholesterol Education Program のAdult Treatment Panel IIIの判定基準によれば、内臓肥満、高中性脂肪血症、低HDLコレステロール血症、高血圧、耐糖能異常のうち3つ以上を持つ場合に代謝症候群と診断される(National Cholesterol Education Program: Executive Summary of the Third Report of National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adults Treatment Panel III). The Journal of the American Medical Association, Vol. 285, 2486-2497, 2001)。 The compound of the present invention is also useful as a prophylactic / therapeutic agent for metabolic syndrome (metabolic syndrome). Patients with metabolic syndrome have a significantly higher rate of developing cardiovascular disease than patients with a single lifestyle-related disease, so preventing or treating metabolic syndrome prevents cardiovascular disease It is extremely important to do.
Criteria for metabolic syndrome were published by WHO in 1999 and NCEP in 2001. According to WHO criteria, metabolic syndrome is diagnosed in patients with visceral obesity, dyslipidemia (high TG or low HDL), or hypertension based on hyperinsulinemia or impaired glucose tolerance. (World Health Organization: Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. Part I: Diagnosis and Classification of Diabetes Mellitus, World Health Organization, Geneva, 1999). According to the criteria of the Adult Treatment Panel III of the National Cholesterol Education Program, which is a management index for ischemic heart disease in the United States, it is 3 (National Cholesterol Education Program: Executive Summary of the Third Report of National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adults Treatment Panel III). The Journal of the American Medical Association, Vol. 285, 2486-2497, 2001).
本発明化合物は、例えば、骨粗鬆症、悪液質(例、癌性悪液質、結核性悪液質、糖尿病性悪液質、血液疾患性悪液質、内分泌疾患性悪液質、感染症性悪液質または後天性免疫不全症候群による悪液質)、脂肪肝、多嚢胞性卵巣症候群、腎臓疾患(例、糖尿病性ネフロパシー、糸球体腎炎、糸球体硬化症、ネフローゼ症候群、高血圧性腎硬化症、末期腎臓疾患)、筋ジストロフィー、心筋梗塞、狭心症、脳血管障害(例、脳梗塞、脳卒中)、アルツハイマー病、パーキンソン病、不安症、痴呆症、インスリン抵抗性症候群、シンドロームX、高インスリン血症、高インスリン血症における知覚障害、急性または慢性下痢、炎症性疾患(例、慢性関節リウマチ、変形性脊椎炎、変形性関節炎、腰痛、痛風、手術または外傷後の炎症、腫脹、神経痛、咽喉頭炎、膀胱炎、肝炎(非アルコール性脂肪性肝炎を含む)、肺炎、膵炎、腸炎、炎症性腸疾患(炎症性大腸疾患を含む)、潰瘍性大腸炎、胃粘膜損傷(アスピリンにより引き起こされた胃粘膜損傷を含む))、小腸粘膜損傷、吸収不良、精巣機能障害、内臓肥満症候群、筋肉減少症の予防・治療剤としても用いることができる。
The compound of the present invention is, for example, osteoporosis, cachexia (eg, cancer cachexia, tuberculosis cachexia, diabetic cachexia, hematological cachexia, endocrine disease cachexia, infectious cachexia or acquired cachexia). Cachexia due to immunodeficiency syndrome), fatty liver, polycystic ovary syndrome, kidney disease (eg, diabetic nephropathy, glomerulonephritis, glomerulosclerosis, nephrotic syndrome, hypertensive nephrosclerosis, end-stage renal disease), Muscular dystrophy, myocardial infarction, angina pectoris, cerebrovascular disorder (eg, cerebral infarction, stroke), Alzheimer's disease, Parkinson's disease, anxiety, dementia, insulin resistance syndrome, syndrome X, hyperinsulinemia, hyperinsulinemia Sensory impairment, acute or chronic diarrhea, inflammatory diseases (eg, rheumatoid arthritis, osteoarthritis spondylitis, osteoarthritis, low back pain, gout, postoperative or traumatic inflammation, swelling, nerves Pharyngopharyngitis, cystitis, hepatitis (including non-alcoholic steatohepatitis), pneumonia, pancreatitis, enteritis, inflammatory bowel disease (including inflammatory bowel disease), ulcerative colitis, gastric mucosal damage (due to aspirin) Including gastric mucosal damage caused)), small intestinal mucosal damage, malabsorption, testicular dysfunction, visceral obesity syndrome, and sarcopenia.
更に、本発明化合物は、種々の癌(なかでも乳癌(例えば、浸潤性乳管癌、非浸潤性乳管癌、炎症性乳癌等)、前立腺癌(例えば、ホルモン依存性前立腺癌、ホルモン非依存性前立腺癌等)、膵癌(例えば、膵管癌等)、胃癌(例えば、乳頭腺癌、粘液性腺癌、腺扁平上皮癌等)、肺癌(例えば、非小細胞肺癌、小細胞肺癌、悪性中皮腫等)、結腸癌(例えば、消化管間質腫瘍等)、直腸癌(例えば、消化管間質腫瘍等)、大腸癌(例えば、家族性大腸癌、遺伝性非ポリポーシス大腸癌、消化管間質腫瘍等)、小腸癌(例えば、非ホジキンリンパ腫、消化管間質腫瘍等)、食道癌、十二指腸癌、舌癌、咽頭癌(例えば、上咽頭癌、中咽頭癌、下咽頭癌等)、唾液腺癌、脳腫瘍(例えば、松果体星細胞腫瘍、毛様細胞性星細胞腫、びまん性星細胞腫、退形成性星細胞腫等)、神経鞘腫、肝臓癌(例えば、原発性肝癌、肝外胆管癌等)、腎臓癌(例えば、腎細胞癌、腎盂と尿管の移行上皮癌等)、胆管癌、子宮内膜癌、子宮頸癌、卵巣癌(例えば、上皮性卵巣癌、性腺外胚細胞腫瘍、卵巣性胚細胞腫瘍、卵巣低悪性度腫瘍等)、膀胱癌、尿道癌、皮膚癌(例えば、眼内(眼)黒色腫、メルケル細胞癌等)、血管腫、悪性リンパ腫、悪性黒色腫、甲状腺癌(例えば、甲状腺髄様癌等)、副甲状腺癌、鼻腔癌、副鼻腔癌、骨腫瘍(例えば、骨肉腫、ユーイング腫瘍、子宮肉腫、軟部組織肉腫等)、血管線維腫、網膜肉腫、陰茎癌、精巣腫瘍、小児固形癌(例えば、ウィルムス腫瘍、小児腎腫瘍等)、カポジ肉腫、AIDSに起因するカポジ肉腫、上顎洞腫瘍、線維性組織球腫、平滑筋肉腫、横紋筋肉腫、白血病(例えば、急性骨髄性白血病、急性リンパ芽球性白血病等)等)の予防・治療剤としても用いることができる。
Furthermore, the compound of the present invention is used for various cancers (among others breast cancer (for example, invasive breast cancer, non-invasive breast cancer, inflammatory breast cancer, etc.)), prostate cancer (for example, hormone-dependent prostate cancer, hormone-independent). Prostate cancer, etc.), pancreatic cancer (eg, pancreatic duct cancer, etc.), stomach cancer (eg, papillary adenocarcinoma, mucinous adenocarcinoma, adenosquamous carcinoma, etc.), lung cancer (eg, non-small cell lung cancer, small cell lung cancer, malignant mesothelioma) ), Colon cancer (eg, gastrointestinal stromal tumor), rectal cancer (eg, gastrointestinal stromal tumor), colorectal cancer (eg, familial colorectal cancer, hereditary non-polyposis colorectal cancer, gastrointestinal tract) Tumor, etc.), small intestine cancer (eg, non-Hodgkin lymphoma, gastrointestinal stromal tumor), esophageal cancer, duodenal cancer, tongue cancer, pharyngeal cancer (eg, nasopharyngeal cancer, oropharyngeal cancer, hypopharyngeal cancer, etc.), Salivary gland cancer, brain tumors (eg pineal astrocytoma, ciliary cell astrocytoma, diffuse star Cystoma, anaplastic astrocytoma, etc.), schwannoma, liver cancer (eg, primary liver cancer, extrahepatic bile duct cancer, etc.), kidney cancer (eg, renal cell carcinoma, transitional cell carcinoma of the renal pelvis and ureter, etc.) , Cholangiocarcinoma, endometrial cancer, cervical cancer, ovarian cancer (eg epithelial ovarian cancer, extragonadal germ cell tumor, ovarian germ cell tumor, ovarian low grade tumor, etc.), bladder cancer, urethral cancer, skin Cancer (eg, intraocular (eye) melanoma, Merkel cell carcinoma, etc.), hemangioma, malignant lymphoma, malignant melanoma, thyroid cancer (eg, medullary thyroid cancer, etc.), parathyroid cancer, nasal cavity cancer, sinus cancer , Bone tumors (eg, osteosarcoma, Ewing tumor, uterine sarcoma, soft tissue sarcoma, etc.), angiofibroma, retinal sarcoma, penile cancer, testicular tumor, childhood solid cancer (eg, Wilms tumor, childhood kidney tumor, etc.), Kaposi Sarcoma, Kaposi sarcoma caused by AIDS, maxillary sinus tumor, fibrous histiocytoma, leiomyosarcoma Rhabdomyosarcoma, leukemia (e.g., acute myeloid leukemia, acute lymphoblastic leukemia, etc.) can be used as an agent for preventing or treating, etc.).
本発明化合物は、上記した各種疾患(例、心筋梗塞等の心血管イベント)の2次予防および進展抑制にも用いられる。
The compound of the present invention is also used for secondary prevention and progression suppression of the various diseases described above (eg, cardiovascular events such as myocardial infarction).
本発明化合物の投与量は、投与対象、投与ルート、対象疾患、症状等によっても異なるが、例えば、成人の肥満患者に経口投与する場合、通常1回量として約0.01~100mg/kg体重、好ましくは0.05~30mg/kg体重、さらに好ましくは0.5~10mg/kg体重であり、この量を1日1回~3回投与するのが望ましい。
The dose of the compound of the present invention varies depending on the administration subject, administration route, target disease, symptom, etc. For example, when administered orally to an adult obese patient, it is usually about 0.01 to 100 mg / kg body weight as a single dose. The dose is preferably 0.05 to 30 mg / kg body weight, more preferably 0.5 to 10 mg / kg body weight, and this amount is desirably administered once to three times a day.
本発明化合物は、該化合物の作用の増強または該化合物の投与量の低減等を目的として、糖尿病治療剤、糖尿病性合併症治療剤、高脂血症治療剤、降圧剤、抗肥満剤、利尿剤、抗血栓剤等の薬剤(以下、併用薬剤と略記する)と組み合わせて用いることができる。この際、本発明化合物と併用薬剤の投与時期は限定されず、これらの併用薬剤は低分子化合物であってもよく、また高分子のタンパク質、ポリペプチド、抗体、ワクチン等であってもよい。これらを投与対象に対し、同時に投与してもよいし、時間差をおいて投与してもよい。さらに、本発明化合物と併用薬剤とは、それぞれの活性成分を含む2種類の製剤として投与されてもよいし、両方の活性成分を含む単一の製剤として投与されてもよい。
The compound of the present invention is used for the purpose of enhancing the action of the compound or reducing the dose of the compound, etc., a therapeutic agent for diabetes, a therapeutic agent for diabetic complications, a therapeutic agent for hyperlipidemia, an antihypertensive agent, an antiobesity agent, a diuretic It can be used in combination with a drug such as an agent or an antithrombotic drug (hereinafter abbreviated as a concomitant drug). In this case, the administration time of the compound of the present invention and the concomitant drug is not limited, and these concomitant drugs may be low molecular compounds, and may be high molecular proteins, polypeptides, antibodies, vaccines and the like. These may be administered to the administration subject at the same time, or may be administered with a time difference. Furthermore, the compound of the present invention and the concomitant drug may be administered as two types of preparations containing each active ingredient, or may be administered as a single preparation containing both active ingredients.
併用薬剤の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、本発明化合物と併用薬剤の配合比は、投与対象、投与ルート、対象疾患、症状、組み合わせ等により適宜選択することができる。例えば、投与対象がヒトである場合、本発明化合物1重量部に対し、併用薬剤を0.01~100重量部用いればよい。
The dose of the concomitant drug can be appropriately selected based on the clinically used dose. The compounding ratio of the compound of the present invention and the concomitant drug can be appropriately selected depending on the administration subject, administration route, target disease, symptom, combination and the like. For example, when the administration subject is a human, the concomitant drug may be used in an amount of 0.01 to 100 parts by weight per 1 part by weight of the compound of the present invention.
糖尿病治療剤としては、例えば、インスリン製剤(例、ウシ、ブタの膵臓から抽出された動物インスリン製剤;大腸菌またはイーストを用い、遺伝子工学的に合成したヒトインスリン製剤;インスリン亜鉛;プロタミンインスリン亜鉛;インスリンのフラグメントまたは誘導体(例、INS-1)、経口インスリン製剤)、インスリン抵抗性改善剤(例、ピオグリタゾンまたはその塩(好ましくは塩酸塩)、ロシグリタゾンまたはその塩(好ましくはマレイン酸塩)、メタグリダセン(Metaglidasen)、AMG-131、バラグリタゾン(Balaglitazone)、MBX-2044、リボグリタゾン(Rivoglitazone)、アレグリタザール(Aleglitazar)、チグリタザール(Chiglitazar)、ロベグリタゾン(Lobeglitazone)、PLX-204、PN-2034、GFT-505、THR-0921、WO2007/013694、WO2007/018314、WO2008/093639またはWO2008/099794記載の化合物)、α-グルコシダーゼ阻害剤(例、ボグリボース、アカルボース、ミグリトール、エミグリテート)、ビグアナイド剤(例、メトホルミン、ブホルミンまたはその塩(例、塩酸塩、フマル酸塩、コハク酸塩))、インスリン分泌促進剤[スルホニルウレア剤(例、トルブタミド、グリベンクラミド、グリクラジド、クロルプロパミド、トラザミド、アセトヘキサミド、グリクロピラミド、グリメピリド、グリピザイド、グリブゾール)、レパグリニド、ナテグリニド、ミチグリニドまたはそのカルシウム塩水和物]、ジペプチジルペプチダーゼIV阻害剤(例、アログリプチン(Alogliptin)、ヴィルダグリプチン(Vildagliptin)、シタグリプチン(Sitagliptin)、サクサグリプチン(Saxagliptin)、BI1356、GRC8200、MP-513、PF-00734200、PHX1149、SK-0403、ALS2-0426、TA-6666、TS-021、KRP-104、2-[[6-[(3R)-3-アミノ-1-ピペリジニル]-3,4-ジヒドロ-3-メチル-2,4-ジオキソ-1(2H)-ピリミジニル]メチル]-4-フルオロベンゾニトリルまたはその塩)、β3アゴニスト(例、N-5984)、GPR40アゴニスト(例、WO2004/041266、WO2004/106276、WO2005/063729、WO2005/063725、WO2005/087710、WO2005/095338、WO2007/013689またはWO2008/001931記載の化合物)、GLP-1受容体アゴニスト[例、GLP-1、GLP-1MR剤、リラグルチド(Liraglutide)、エキセナチド(Exenatide)、AVE-0010、BIM-51077、Aib(8,35)hGLP-1(7,37)NH2、CJC-1131、Albiglutide]、アミリンアゴニスト(例、プラムリンチド)、ホスホチロシンホスファターゼ阻害剤(例、バナジン酸ナトリウム)、糖新生阻害剤(例、グリコーゲンホスホリラーゼ阻害剤、グルコース-6-ホスファターゼ阻害剤、グルカゴン拮抗剤、FBPase阻害薬)、SGLT2(sodium-glucose cotransporter 2)阻害剤(例、Depagliflozin、AVE2268、TS-033、YM543、TA-7284、Remogliflozin、ASP1941)、SGLT1阻害薬、11β-ヒドロキシステロイドデヒドロゲナーゼ阻害薬(例、BVT-3498)、アジポネクチンまたはその作動薬、IKK阻害薬(例、AS-2868)、レプチン抵抗性改善薬、ソマトスタチン受容体作動薬、グルコキナーゼ活性化薬(例、Piragliatin、AZD1656、AZD6370、TTP-355、WO2006/112549、WO2007/028135、WO2008/047821、WO2008/050821、WO2008/136428またはWO2008/156757記載の化合物)、GIP(Glucose-dependent insulinotropic peptide)等が挙げられる。
Examples of the therapeutic agent for diabetes include insulin preparations (eg, animal insulin preparations extracted from bovine and porcine pancreas; human insulin preparations genetically engineered using Escherichia coli or yeast; insulin zinc; protamine insulin zinc; insulin Fragment or derivative (eg, INS-1), oral insulin preparation), insulin resistance improving agent (eg, pioglitazone or a salt thereof (preferably hydrochloride), rosiglitazone or a salt thereof (preferably maleate), metaglidacene (Metaglidasen), AMG-131, Balaglitazone, MBX-2044, Riboglitazone, Aleglitazar, Chiglitazar, Lobeglitazone, PLX-204, PN-2034, GFT -505, THR-0921, WO2007 / 013694, WO2007 / 018314, WO2008 / 093639 or WO2008 / 09979 4), α-glucosidase inhibitors (eg, voglibose, acarbose, miglitol, emiglitate), biguanides (eg, metformin, buformin or salts thereof (eg, hydrochloride, fumarate, succinate)), Insulin secretion promoter [sulfonylurea (eg, tolbutamide, glibenclamide, gliclazide, chlorpropamide, tolazamide, acetohexamide, glyclopyramide, glimepiride, glipizide, glybazole), repaglinide, nateglinide, mitiglinide or its calcium salt hydrate], Dipeptidyl peptidase IV inhibitor (eg, alogliptin, vildagliptin, sitagliptin, saxagliptin, BI1356, GRC8200, MP-513, PF-00734200, PHX1149, SK-0403, ALS2 -0426, TA-6666, TS-021, KRP-104, 2-[[6-[(3R) -3-amino-1-piperidinyl] -3,4-dihydro-3-methyl-2,4-dioxo -1 (2H) -pyrimidinyl] methyl] -4-fluorobenzonitrile or a salt thereof), β3 agonist (eg, N-5984), GPR40 agonist (eg, WO2004 / 041266, WO2004 / 106276, WO2005 / 063729, WO2005 / 063725, WO2005 / 087710, WO2005 / 095338, WO2007 / 013689 or WO2008 / 001931), GLP-1 receptor agonist [eg, GLP-1, GLP-1MR agent, Liraglutide, Exenatide, AVE-0010, BIM-51077, Aib (8,35) hGLP-1 (7,37) NH 2 , CJC-1131, Albiglutide], amylin agonist (eg, pramlintide), phosphotyrosine phosphatase inhibitor (eg, sodium vanadate) ), Gluconeogenesis inhibitors (eg, glycogen phosphorylase inhibitors, glucose-6-phosphatase inhibitors, glucagon antagonists) , FBPase inhibitor), SGLT2 (sodium-glucose cotransporter 2) inhibitor (eg, Depagliflozin, AVE2268, TS-033, YM543, TA-7284, Remogliflozin, ASP1941), SGLT1 inhibitor, 11β-hydroxysteroid dehydrogenase inhibitor ( E.g., BVT-3498), adiponectin or agonist thereof, IKK inhibitor (e.g., AS-2868), leptin resistance ameliorating agent, somatostatin receptor agonist, glucokinase activator (e.g., Piragliatin, AZD1656, AZD6370, TTP-355, WO2006 / 112549, WO2007 / 028135, WO2008 / 047821, WO2008 / 050821, WO2008 / 136428 or WO2008 / 156757), GIP (Glucose-dependent insulinotropic peptide) and the like.
糖尿病性合併症治療剤としては、例えば、アルドース還元酵素阻害剤(例、トルレスタット、エパルレスタット、ゾポルレスタット、フィダレスタット、CT-112、ラニレスタット(AS-3201)、リドレスタット)、神経栄養因子およびその増加薬(例、NGF、NT-3、BDNF、WO01/14372に記載のニューロトロフィン産生・分泌促進剤(例えば、4-(4-クロロフェニル)-2-(2-メチル-1-イミダゾリル)-5-[3-(2-メチルフェノキシ)プロピル]オキサゾール)、WO2004/039365記載の化合物)、神経再生促進薬(例、Y-128)、PKC阻害剤(例、ルボキシスタウリン メシレート(ruboxistaurin mesylate))、AGE阻害剤(例、ALT946、ピラトキサチン、N-フェナシルチアゾリウム ブロマイド(ALT766)、ALT-711、EXO-226、ピリドリン(Pyridorin)、ピリドキサミン)、GABA受容体作動薬(例、ギャバペンチン、プレギャバリン)、セロトニン・ノルアドレナリン再取込み阻害薬(例、デュロキセチン)、ナトリウムチャンネル阻害薬(例、ラコサミド)、活性酸素消去薬(例、チオクト酸)、脳血管拡張剤(例、チアプリド、メキシレチン)、ソマトスタチン受容体作動薬(例、BIM23190)、アポトーシスシグナルレギュレーティングキナーゼ-1(ASK-1)阻害薬等が挙げられる。
Examples of the therapeutic agent for diabetic complications include aldose reductase inhibitors (eg, tolrestat, epalrestat, zopolrestat, fidarestat, CT-112, ranirestat (AS-3201), ridressat), neurotrophic factor and its increasing drug (Eg, NGF, NT-3, BDNF, neurotrophin production / secretion promoter described in WO01 / 14372 (for example, 4- (4-chlorophenyl) -2- (2-methyl-1-imidazolyl) -5 [3- (2-methylphenoxy) propyl] oxazole), compounds described in WO2004 / 039365), nerve regeneration promoters (eg, Y-128), PKC inhibitors (eg, ruboxistaurin mesylate) AGE inhibitors (eg, ALT946, pyratoxatin, N-phenacylthiazolium bromide (ALT766), ALT-711, EXO-22 6, pyridoline (Pyridorin), pyridoxamine), GABA receptor agonist (eg, gabapentin, pregabalin), serotonin and noradrenaline reuptake inhibitor (eg, duloxetine), sodium channel inhibitor (eg, lacosamide), reactive oxygen scavenger (Eg, thioctic acid), cerebral vasodilators (eg, thioprid, mexiletine), somatostatin receptor agonists (eg, BIM23190), apoptosis signal regulating kinase-1 (ASK-1) inhibitors and the like.
高脂血症治療剤としては、例えば、スタチン系化合物(例、プラバスタチン、シンバスタチン、ロバスタチン、アトルバスタチン、フルバスタチン、ロスバスタチン、ピタバスタチンまたはその塩(例、ナトリウム塩、カルシウム塩))、スクアレン合成酵素阻害剤(例、WO97/10224に記載の化合物、例えば、N-[[(3R,5S)-1-(3-アセトキシ-2,2-ジメチルプロピル)-7-クロロ-5-(2,3-ジメトキシフェニル)-2-オキソ-1,2,3,5-テトラヒドロ-4,1-ベンゾオキサゼピン-3-イル]アセチル]ピペリジン-4-酢酸)、フィブラート系化合物(例、ベザフィブラート、クロフィブラート、シムフィブラート、クリノフィブラート)、陰イオン交換樹脂(例、コレスチラミン)、プロブコール、ニコチン酸系薬剤(例、ニコモール(nicomol)、ニセリトロール(niceritrol)、ナイアスパン(niaspan))、イコサペント酸エチル、植物ステロール(例、ソイステロール(soysterol)、ガンマオリザノール(γ-oryzanol))、コレステロール吸収阻害剤 (例、ゼチア)、CETP阻害剤(例、ダルセトラピブ(dalcetrapib)、アナセトラピブ(anacetrapib))、ω-3脂肪酸製剤(例、ω-3-acid ethyl esters 90)等が挙げられる。
Examples of therapeutic agents for hyperlipidemia include statin compounds (eg, pravastatin, simvastatin, lovastatin, atorvastatin, fluvastatin, rosuvastatin, pitavastatin or salts thereof (eg, sodium salt, calcium salt)), squalene synthase inhibitors (Eg, compounds described in WO97 / 10224, for example, N-[[(3R, 5S) -1- (3-acetoxy-2,2-dimethylpropyl) -7-chloro-5- (2,3-dimethoxy Phenyl) -2-oxo-1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl] acetyl] piperidine-4-acetic acid), fibrate compounds (eg, bezafibrate, clofibrate, Simfibrate, clinofibrate), anion exchange resin (eg, cholestyramine), probucol, nicotinic acid drugs (eg, nicomol, niceritrol, nia) Span (niaspan), ethyl icosapentate, plant sterols (eg, soysterol, gamma-oryzanol), cholesterol absorption inhibitors (eg, zetia), CETP inhibitors (eg, dalcetrapib) , Anacetrapib), ω-3 fatty acid preparations (eg, ω-3-acid ethyl esters 90), and the like.
降圧剤としては、例えば、アンジオテンシン変換酵素阻害剤(例、カプトプリル、エナラプリル、デラプリル等)、アンジオテンシンII拮抗剤(例、カンデサルタン シレキセチル、カンデサルタン、ロサルタン、ロサルタン カリウム、エプロサルタン、バルサルタン、テルミサルタン、イルベサルタン、タソサルタン、オルメサルタン、オルメサルタン メドキソミル、アジルサルタン、アジルサルタン メドキソミル)、カルシウム拮抗剤(例、マニジピン、ニフェジピン、アムロジピン、エホニジピン、ニカルジピン、アムロジピン、シルニジピン等)、βブロッカー(例、メトプロロール、アテノロール、プロプラノロール、カルベジロール、ピンドロール)、クロニジン等が挙げられる。
Examples of the antihypertensive agent include angiotensin converting enzyme inhibitors (eg, captopril, enalapril, delapril, etc.), angiotensin II antagonists (eg, candesartan cilexetil, candesartan, losartan, losartan potassium, eprosartan, valsartan, telmisartan, irbesartan, tasosartan , Olmesartan, olmesartan medoxomil, azilsartan, azilsartan medoxomil), calcium antagonists (eg, manidipine, nifedipine, amlodipine, nifodipine, nicardipine, amlodipine, cilnidipine, etc.), β-blockers (eg, metoprolol, atenolol, propranolol, propranolol, propranolol, propranolol, ), Clonidine and the like.
抗肥満剤としては、例えば、モノアミン取り込み阻害薬(例、フェンテルミン、シブトラミン、マジンドール、フロキセチン、テソフェンシン)、セロトニン2C受容体作動薬(例、ロルカセリン)、セロトニン6受容体拮抗薬、ヒスタミンH3受容体、GABA調節薬(例、トピラメイト)、MCH受容体拮抗薬(例、SB-568849;SNAP-7941;WO01/82925またはWO01/87834に記載の化合物)、ニューロペプチドY拮抗薬(例、ベルネペリット)、カンナビノイド受容体拮抗薬(例、リモナバン、タラナバン)、グレリン拮抗薬、グレリン受容体拮抗薬、グレリンアシル化酵素阻害薬、オピオイド受容体拮抗薬(例、GSK-1521498)、オレキシン受容体拮抗薬、メラノコルチン4受容体作動薬、11β-ヒドロキシステロイドデヒドロゲナーゼ阻害薬(例、AZD-4017)、膵リパーゼ阻害薬(例、オルリスタット、セティリスタット(cetilistat))、β3アゴニスト(例、N-5984)、ジアシルグリセロールアシルトランスフェラーゼ1(DGAT1)阻害薬、アセチルCoAカルボキシラーゼ(ACC)阻害薬、ステアリン酸CoA脱飽和酵素阻害、ミクロソームトリグリセリド転送蛋白阻害薬(例、R-256918)、Na-グルコース共輸送担体阻害薬(例、JNJ-28431754、レモグリフロジン)、NFκ阻害(例、HE-3286)、PPARアゴニスト(例、GFT-505、DRF-11605)、ホスホチロシンホスファターゼ阻害剤(例、バナジン酸ナトリウム、トロダスケミン(Trodusquemin))、GPR119作動薬(例、PSN-821)、グルコキナーゼ活性化薬(例、AZD-1656)、レプチン、レプチン誘導体(例、メトレレプチン)、CNTF(毛様体神経栄養因子)、BDNF(脳由来神経栄養因子)、コレシストキニンアゴニスト、グルカゴン様ペプチド-1(GLP-1)製剤(例、ウシ、ブタの膵臓から抽出された動物GLP-1製剤;大腸菌、イーストを用い遺伝子工学的に合成したヒトGLP-1製剤;GLP-1のフラグメントまたは誘導体(例、エクセナチド、リラグルチド))、アミリン製剤(例、プラムリンタイド、AC-2307)、ニューロペプチドYアゴニスト(例、PYY3-36、PYY3-36の誘導体、オビネプタイド、TM-30339、TM-30335)、オキシントモジュリン製剤:FGF21製剤(例、ウシ、ブタの膵臓から抽出された動物FGF21製剤;大腸菌、イーストを用い遺伝子工学的に合成したヒトFGF21製剤;FGF21のフラグメントまたは誘導体))、ナルトレキソン塩酸塩徐放製剤とブプロピオン塩酸塩徐放製剤の合剤、摂食抑制薬(例、P-57)等が挙げられる。
Examples of anti-obesity agents include monoamine uptake inhibitors (eg, phentermine, sibutramine, mazindol, floxetine, tesofensin), serotonin 2C receptor agonists (eg, lorcaserin), serotonin 6 receptor antagonists, histamine H3 receptor , GABA modulators (eg, topiramate), MCH receptor antagonists (eg, SB-568849; SNAP-7941; compounds described in WO01 / 82925 or WO01 / 87834), neuropeptide Y antagonists (eg, Berneperit) , Cannabinoid receptor antagonists (eg, rimonabant, taranaban), ghrelin antagonists, ghrelin receptor antagonists, ghrelin acylase inhibitors, opioid receptor antagonists (eg, GSK-1521498), orexin receptor antagonists, Melanocortin 4 receptor agonist, 11β-hydroxysteroid dehydrogenase inhibitor (eg, AZD-4017), pancreatic lipase Pesticides (eg, orlistat, cetilistat), β3 agonists (eg, N-5984), diacylglycerol acyltransferase 1 (DGAT1) inhibitors, acetyl CoA carboxylase (ACC) inhibitors, stearic acid CoA desaturation Enzyme inhibition, microsomal triglyceride transfer protein inhibitor (eg, R-256918), Na-glucose cotransport carrier inhibitor (eg, JNJ-28431754, remogliflozin), NFκ inhibition (eg, HE-3286), PPAR agonist (eg, GFT-505, DRF-11605), phosphotyrosine phosphatase inhibitors (eg, sodium vanadate, Trodusquemin), GPR119 agonists (eg, PSN-821), glucokinase activators (eg, AZD-1656), Leptin, leptin derivatives (eg, metreleptin), CNTF (ciliary neurotrophic factor), BDNF (brain-derived neurotrophic factor), cholecystokini Agonist, glucagon-like peptide-1 (GLP-1) preparation (eg, animal GLP-1 preparation extracted from bovine and porcine pancreas; human GLP-1 preparation genetically engineered using E. coli and yeast; GLP- 1 fragment or derivative (eg, exenatide, liraglutide)), amylin preparation (eg, pramlintide, AC-2307), neuropeptide Y agonist (eg, PYY3-36, derivative of PYY3-36, obineptide, TM-30339 , TM-30335), oxyntomodulin preparation: FGF21 preparation (eg, animal FGF21 preparation extracted from bovine and porcine pancreas; human FGF21 preparation genetically engineered using E. coli and yeast; FGF21 fragment or derivative) ), Naltrexone hydrochloride sustained-release preparation and bupropion hydrochloride sustained-release preparation, feeding inhibitors (eg, P-57), and the like.
利尿剤としては、例えば、キサンチン誘導体(例、サリチル酸ナトリウムテオブロミン、サリチル酸カルシウムテオブロミン)、チアジド系製剤(例、エチアジド、シクロペンチアジド、トリクロルメチアジド、ヒドロクロロチアジド、ヒドロフルメチアジド、ベンチルヒドロクロロチアジド、ペンフルチアジド、ポリチアジド、メチクロチアジド)、抗アルドステロン製剤(例、スピロノラクトン、トリアムテレン)、炭酸脱水酵素阻害剤(例、アセタゾラミド)、クロルベンゼンスルホンアミド系製剤(例、クロルタリドン、メフルシド、インダパミド)、アゾセミド、イソソルビド、エタクリン酸、ピレタニド、ブメタニド、フロセミド等が挙げられる。
Examples of diuretics include xanthine derivatives (eg, sodium salicylate theobromine, calcium salicylate theobromine), thiazide preparations (eg, etiazide, cyclopentiazide, trichloromethiazide, hydrochlorothiazide, hydroflumethiazide, benchylhydrochlorothiazide, penfluthiazide. , Polythiazide, meticlotiazide), anti-aldosterone formulations (eg, spironolactone, triamterene), carbonic anhydrase inhibitors (eg, acetazolamide), chlorobenzenesulfonamides (eg, chlorthalidone, mefluside, indapamide), azosemide, isosorbide, etacrine Examples include acid, piretanide, bumetanide, furosemide and the like.
抗血栓剤としては、例えば、ヘパリン(例、ヘパリンナトリウム、ヘパリンカルシウム、エノキサパリンナトリウム(enoxaparin sodium)、ダルテパリンナトリウム(dalteparinsodium))、ワルファリン(例、ワルファリンカリウム)、抗トロンビン薬(例、アルガトロバン(argatroban)、ダビガトラン(dabigatran))、血栓溶解薬(例、ウロキナーゼ(urokinase)、チソキナーゼ(tisokinase)、アルテプラーゼ(alteplase)、ナテプラーゼ(nateplase)、モンテプラーゼ(monteplase)、パミテプラーゼ(pamiteplase))、血小板凝集抑制薬(例、塩酸チクロピジン(ticlopidine hydrochloride)、クロピドグレル、E5555、SHC530348、シロスタゾール(cilostazol)、イコサペント酸エチル、ベラプロストナトリウム(beraprost sodium)、塩酸サルポグレラート(sarpogrelate hydrochloride)、プラスグレル(prasugrel)、E5555、SHC530348)、FXa阻害薬(例、リバロキサバン(rivaroxaban)、アピキサバン(apixaban)、エドキサバン(edoxaban)、YM150、WO02/06234、WO2004/048363、WO2005/030740、WO2005/058823またはWO2005/113504記載の化合物)等が挙げられる。
Antithrombotic agents include, for example, heparin (eg, heparin sodium, heparin calcium, enoxaparin sodium, dalteparinsodium), warfarin (eg, warfarin potassium), antithrombin drugs (eg, argatroban) ), Dabigatran), thrombolytic drugs (eg, urokinase, tisokinase, alteplase, nateplase, monteplase, pamiteplase), platelet aggregation inhibitor ( Examples, ticlopidine hydrochloride, clopidogrel, E5555, SHC530348, cilostazol, ethyl icosapentate, beraprost sodium, sarpogrelate hydrochloride, prasugrel, E5555, SH C530348), FXa inhibitors (eg, compounds described in rivaroxaban, apixaban, edoxaban, YM150, WO02 / 06234, WO2004 / 048363, WO2005 / 030740, WO2005 / 058823 or WO2005 / 113504), etc. Is mentioned.
前記した併用薬剤の投与時期は限定されず、本発明化合物と併用薬剤とを、投与対象に対し、同時に投与してもよいし、時間差をおいて投与してもよい。併用薬剤の投与量は、臨床上用いられている投与量に準ずればよく、投与対象、投与ルート、疾患、組み合わせ等により適宜選択することができる。
併用薬剤の投与形態は、特に限定されず、投与時に、本発明化合物と併用薬剤とが組み合わされていればよい。このような投与形態としては、例えば、
1)本発明化合物と併用薬剤とを同時に製剤化して得られる単一の製剤の投与、
2)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の同一投与経路での同時投与、
3)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の同一投与経路での時間差をおいての投与、
4)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の異なる投与経路での同時投与、
5)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の異なる投与経路での時間差をおいての投与(例えば、本発明化合物、併用薬剤の順序での投与、または逆の順序での投与)等が挙げられる。
本発明化合物と併用薬剤との配合比は、投与対象、投与ルート、疾患等により適宜選択することができる。 The administration time of the aforementioned concomitant drug is not limited, and the compound of the present invention and the concomitant drug may be administered to the administration subject at the same time or may be administered with a time difference. The dose of the concomitant drug may be determined according to the dose used clinically, and can be appropriately selected depending on the administration subject, administration route, disease, combination and the like.
The administration form of the concomitant drug is not particularly limited, as long as the compound of the present invention and the concomitant drug are combined at the time of administration. Examples of such dosage forms include:
1) administration of a single preparation obtained by simultaneously formulating the compound of the present invention and a concomitant drug,
2) Simultaneous administration by the same route of administration of two kinds of preparations obtained by separately formulating the compound of the present invention and a concomitant drug,
3) Administration of the two compounds obtained by formulating the compound of the present invention and the concomitant drug separately with a time difference in the same administration route,
4) Simultaneous administration by different administration routes of two preparations obtained by separately formulating the compound of the present invention and a concomitant drug,
5) Administration of two preparations obtained by separately formulating the compound of the present invention and a concomitant drug at different time intervals in different administration routes (for example, administration in the order of the compound of the present invention and concomitant drug, or vice versa) Administration in this order).
The compounding ratio of the compound of the present invention and the concomitant drug can be appropriately selected depending on the administration subject, administration route, disease and the like.
併用薬剤の投与形態は、特に限定されず、投与時に、本発明化合物と併用薬剤とが組み合わされていればよい。このような投与形態としては、例えば、
1)本発明化合物と併用薬剤とを同時に製剤化して得られる単一の製剤の投与、
2)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の同一投与経路での同時投与、
3)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の同一投与経路での時間差をおいての投与、
4)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の異なる投与経路での同時投与、
5)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の異なる投与経路での時間差をおいての投与(例えば、本発明化合物、併用薬剤の順序での投与、または逆の順序での投与)等が挙げられる。
本発明化合物と併用薬剤との配合比は、投与対象、投与ルート、疾患等により適宜選択することができる。 The administration time of the aforementioned concomitant drug is not limited, and the compound of the present invention and the concomitant drug may be administered to the administration subject at the same time or may be administered with a time difference. The dose of the concomitant drug may be determined according to the dose used clinically, and can be appropriately selected depending on the administration subject, administration route, disease, combination and the like.
The administration form of the concomitant drug is not particularly limited, as long as the compound of the present invention and the concomitant drug are combined at the time of administration. Examples of such dosage forms include:
1) administration of a single preparation obtained by simultaneously formulating the compound of the present invention and a concomitant drug,
2) Simultaneous administration by the same route of administration of two kinds of preparations obtained by separately formulating the compound of the present invention and a concomitant drug,
3) Administration of the two compounds obtained by formulating the compound of the present invention and the concomitant drug separately with a time difference in the same administration route,
4) Simultaneous administration by different administration routes of two preparations obtained by separately formulating the compound of the present invention and a concomitant drug,
5) Administration of two preparations obtained by separately formulating the compound of the present invention and a concomitant drug at different time intervals in different administration routes (for example, administration in the order of the compound of the present invention and concomitant drug, or vice versa) Administration in this order).
The compounding ratio of the compound of the present invention and the concomitant drug can be appropriately selected depending on the administration subject, administration route, disease and the like.
以下、本発明化合物の製造法について説明する。
化合物(I)、(I-1)、(I-2)および(I-3)の製造法の例として、代表的な製造法を以下に述べるが、製造法はこれらに限定されない。化合物(I)、(I-1)、(I-2)および(I-3)は下記の反応式1~4で示される方法またはそれに準じた方法等により製造することもできる。なお、化合物(I-1)、(I-2)および(I-3)はすべて化合物(I)に包含される。 Hereafter, the manufacturing method of this invention compound is demonstrated.
As examples of the production methods of the compounds (I), (I-1), (I-2) and (I-3), typical production methods are described below, but the production methods are not limited thereto. Compounds (I), (I-1), (I-2) and (I-3) can also be produced by the method shown in the following reaction formulas 1 to 4 or a method analogous thereto. Compounds (I-1), (I-2) and (I-3) are all encompassed in compound (I).
化合物(I)、(I-1)、(I-2)および(I-3)の製造法の例として、代表的な製造法を以下に述べるが、製造法はこれらに限定されない。化合物(I)、(I-1)、(I-2)および(I-3)は下記の反応式1~4で示される方法またはそれに準じた方法等により製造することもできる。なお、化合物(I-1)、(I-2)および(I-3)はすべて化合物(I)に包含される。 Hereafter, the manufacturing method of this invention compound is demonstrated.
As examples of the production methods of the compounds (I), (I-1), (I-2) and (I-3), typical production methods are described below, but the production methods are not limited thereto. Compounds (I), (I-1), (I-2) and (I-3) can also be produced by the method shown in the following reaction formulas 1 to 4 or a method analogous thereto. Compounds (I-1), (I-2) and (I-3) are all encompassed in compound (I).
各原料化合物は反応を阻害しないのであれば、塩を形成していてもよく、かかる塩としては、前述の式(I)で示される化合物の塩として例示したものが用いられる。
原料化合物は具体的製法を述べない場合、市販されているものを容易に入手して用いることができるか、または自体公知の方法、またはそれに準ずる方法に従って製造することもできる。 Each raw material compound may form a salt as long as it does not inhibit the reaction. As such a salt, those exemplified as the salt of the compound represented by the aforementioned formula (I) are used.
In the case where a specific production method is not described, a raw material compound can be easily obtained and used commercially, or can be produced according to a method known per se or a method analogous thereto.
原料化合物は具体的製法を述べない場合、市販されているものを容易に入手して用いることができるか、または自体公知の方法、またはそれに準ずる方法に従って製造することもできる。 Each raw material compound may form a salt as long as it does not inhibit the reaction. As such a salt, those exemplified as the salt of the compound represented by the aforementioned formula (I) are used.
In the case where a specific production method is not described, a raw material compound can be easily obtained and used commercially, or can be produced according to a method known per se or a method analogous thereto.
各反応の生成物は反応混合物のまま、または粗生成物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、再結晶、蒸留、クロマトグラフィーおよびHPLC等の分離手段により精製することができる。また、生成物が立体異性体の混合物である場合、ジアステレオマー塩法、クロマトグラフィー、HPLCあるいはSFC(超臨界流体クロマトグラフィー)等の分離手段により精製することができる。例えば、実施例に記載の方法、またはそれに準じた方法等により行うことができる。
The product of each reaction can be used in the next reaction as a reaction mixture or as a crude product, but can be isolated from the reaction mixture according to a conventional method, such as recrystallization, distillation, chromatography and HPLC. It can be purified by separation means. When the product is a mixture of stereoisomers, it can be purified by a separation means such as a diastereomeric salt method, chromatography, HPLC, or SFC (supercritical fluid chromatography). For example, it can be carried out by the method described in the examples or a method analogous thereto.
各反応で用いる試薬類および反応剤類は、市販されている場合は市販品をそのまま用いることもでき、自体公知の方法またはこれらに準じた方法、または実施例に記載の方法に従って製造することもできる。例えば、実施例に記載の試薬類および反応剤類を用いることができる。
Reagents and reagents used in each reaction can be used as they are when commercially available, or can be produced according to a method known per se or a method analogous thereto, or a method described in Examples. it can. For example, the reagents and reagents described in the examples can be used.
特に明記している場合を除き、各反応の該溶媒は反応が進行する限り特に限定されず、反応に不活性な溶媒中、または無溶媒下行うことができ、二種以上を適宜の割合で混合して用いてもよい。例えば、実施例に記載の溶媒を用いることができる。
特に明記している場合を除き、各反応で用いる試薬類および反応剤類の当量は、各反応の基質に対し、0.001当量~100当量である。例えば、実施例に記載の試薬類および反応剤類の当量を用いることができる。 Unless otherwise specified, the solvent for each reaction is not particularly limited as long as the reaction proceeds, and the reaction can be performed in a solvent inert to the reaction or in the absence of a solvent. You may mix and use. For example, the solvents described in the examples can be used.
Unless otherwise specified, the equivalent amounts of reagents and reagents used in each reaction are 0.001 to 100 equivalents relative to the substrate of each reaction. For example, equivalent amounts of reagents and reactants described in the examples can be used.
特に明記している場合を除き、各反応で用いる試薬類および反応剤類の当量は、各反応の基質に対し、0.001当量~100当量である。例えば、実施例に記載の試薬類および反応剤類の当量を用いることができる。 Unless otherwise specified, the solvent for each reaction is not particularly limited as long as the reaction proceeds, and the reaction can be performed in a solvent inert to the reaction or in the absence of a solvent. You may mix and use. For example, the solvents described in the examples can be used.
Unless otherwise specified, the equivalent amounts of reagents and reagents used in each reaction are 0.001 to 100 equivalents relative to the substrate of each reaction. For example, equivalent amounts of reagents and reactants described in the examples can be used.
特に明記している場合を除き、各反応の反応時間は通常5分~72時間である。例えば、実施例に記載の反応時間で行うことができる。
特に明記している場合を除き、各反応の反応温度は氷冷下から加熱還流下である。例えば、実施例に記載の反応温度で行うことができる。 Unless otherwise specified, the reaction time for each reaction is usually from 5 minutes to 72 hours. For example, the reaction can be performed with the reaction times described in the examples.
Unless otherwise specified, the reaction temperature of each reaction is from ice-cooling to heating under reflux. For example, it can be carried out at the reaction temperature described in the examples.
特に明記している場合を除き、各反応の反応温度は氷冷下から加熱還流下である。例えば、実施例に記載の反応温度で行うことができる。 Unless otherwise specified, the reaction time for each reaction is usually from 5 minutes to 72 hours. For example, the reaction can be performed with the reaction times described in the examples.
Unless otherwise specified, the reaction temperature of each reaction is from ice-cooling to heating under reflux. For example, it can be carried out at the reaction temperature described in the examples.
以下の反応式において、アルキル化反応、加水分解反応、アミノ化反応、エステル化反応、アミド化反応、エステル化反応、エーテル化反応、酸化反応、還元反応等を行う場合、これらの反応は、自体公知の方法に従って行われる。このような方法としては、例えば、オーガニック ファンクショナル グループ プレパレーションズ(ORGANIC FUNCTIONAL GROUP PREPARATIONS)、第2版、アカデミックプレス社(Academic Press Inc.)、1989年刊、または、コンプリヘンシブ オーガニック トランスフォーメーションズ:ア ガイド トゥー ファンクショナル プレパレーションズ(Comprehensive Organic Transformations: A Guide to Functional Group Preparations)、第2版、ワイリー-VCH社(Wiley-VCH)、1999年刊等に記載の方法等が挙げられる。
In the following reaction formula, when performing alkylation reaction, hydrolysis reaction, amination reaction, esterification reaction, amidation reaction, esterification reaction, etherification reaction, oxidation reaction, reduction reaction, etc., these reactions are themselves This is performed according to a known method. Such methods include, for example, Organic Functional Group Preparations (ORGANIC FUNCTIONAL GROUP PREPARATIONS), 2nd Edition, Academic Press Inc., 1989, or Comprehensive Organic Transformations: Guide to Functional Preparations (Comprehensive Organic Transformations: A Guide to Functional Group Preparations), 2nd edition, Wiley-VCH (Wiley-VCH), published in 1999, etc.
以下の各反応において、原料化合物がアミノ基、カルボキシ基、ヒドロキシ基、カルボニル基またはスルファニル基を有する場合、これらの官能基にペプチド化学等で一般的に用いられるような保護基が導入されていてもよく、反応後に必要に応じて保護基を除去することにより目的化合物を得ることができる。これらの官能基に保護基を導入する反応を「保護反応」と記載し、保護基を除去する反応を「脱保護反応」と記載した。保護基の導入方法(保護反応)や保護基の除去方法(脱保護反応)は、自体公知の方法、例えば、グリーンズ プロテクティブ グループス イン オーガニック シンセシス(Greene‘s PROTECTIVE GROUPS in ORGANIC SYNTHESIS)、第4版、ワイリー-インターサイエンス社(Wiley-Interscience)、2006年刊等に記載の方法、または、実施例に記載の方法等に準じて行うことができる。
In each of the following reactions, when the raw material compound has an amino group, a carboxy group, a hydroxy group, a carbonyl group, or a sulfanyl group, a protective group generally used in peptide chemistry or the like is introduced into these functional groups. Alternatively, the target compound can be obtained by removing the protecting group as necessary after the reaction. The reaction for introducing a protecting group into these functional groups was described as “protection reaction”, and the reaction for removing the protecting group was described as “deprotection reaction”. The protecting group introduction method (protection reaction) and the protecting group removal method (deprotection reaction) are known per se, for example, Greens Protective Groups in Organic Synthesis in ORGANIC SYNTHESIS, 4th edition. The methods described in Wiley-Interscience, 2006, etc., or the methods described in the examples can be used.
本明細書中、「アミノ基の保護基」としては、例えば、ホルミル基、C1-6アルキル-カルボニル基、C1-6アルコキシ-カルボニル基、ベンゾイル基、C7-10アラルキル-カルボニル基(例、ベンジルカルボニル)、C7-14アラルキルオキシ-カルボニル基(例、ベンジルオキシカルボニル、9-フルオレニルメトキシカルボニル)、トリチル基、フタロイル基、N,N-ジメチルアミノメチレン基、置換シリル基(例、トリメチルシリル、トリエチルシリル、ジメチルフェニルシリル、tert-ブチルジメチルシリル、tert-ブチルジエチルシリル)、C2-6アルケニル基(例、1-アリル)、置換C7-10アラルキル基(例、2,4-ジメトキシベンジル)等が挙げられる。これらの基は、ハロゲン原子、C1-6アルコキシ基およびニトロ基から選ばれる1ないし3個の置換基で置換されていてもよい。
In the present specification, examples of the “amino-protecting group” include a formyl group, a C 1-6 alkyl-carbonyl group, a C 1-6 alkoxy-carbonyl group, a benzoyl group, a C 7-10 aralkyl-carbonyl group ( Example, benzylcarbonyl), C 7-14 aralkyloxy-carbonyl group (eg, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl), trityl group, phthaloyl group, N, N-dimethylaminomethylene group, substituted silyl group ( Examples, trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), C 2-6 alkenyl groups (eg, 1-allyl), substituted C 7-10 aralkyl groups (eg, 2, 4-dimethoxybenzyl) and the like. These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkoxy group and a nitro group.
本明細書中、「カルボキシ基の保護基」としては、例えば、C1-6アルキル基、C7-11アラルキル基(例、ベンジル)、フェニル基、トリチル基、置換シリル基(例、トリメチルシリル、トリエチルシリル、ジメチルフェニルシリル、tert-ブチルジメチルシリル、tert-ブチルジエチルシリル)、C2-6アルケニル基(例、1-アリル)等が挙げられる。これらの基は、ハロゲン原子、C1-6アルコキシ基およびニトロ基から選ばれる1ないし3個の置換基で置換されていてもよい。
In the present specification, examples of the “carboxy group protecting group” include a C 1-6 alkyl group, a C 7-11 aralkyl group (eg, benzyl), a phenyl group, a trityl group, a substituted silyl group (eg, trimethylsilyl, Triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), C 2-6 alkenyl groups (eg, 1-allyl) and the like. These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkoxy group and a nitro group.
本明細書中、「ヒドロキシ基の保護基」としては、例えば、C1-6アルキル基、フェニル基、トリチル基、C7-10アラルキル基(例、ベンジル)、ホルミル基、C1-6アルキル-カルボニル基、ベンゾイル基、C7-10アラルキル-カルボニル基(例、ベンジルカルボニル)、2-テトラヒドロピラニル基、2-テトラヒドロフラニル基、置換シリル基(例、トリメチルシリル、トリエチルシリル、ジメチルフェニルシリル、tert-ブチルジメチルシリル、tert-ブチルジエチルシリル)、C2-6アルケニル基(例、1-アリル)等が挙げられる。これらの基は、ハロゲン原子、C1-6アルキル基、C1-6アルコキシ基またはニトロ基から選ばれる1ないし3個の置換基で置換されていてもよい。
In the present specification, examples of the “protecting group for hydroxy group” include C 1-6 alkyl group, phenyl group, trityl group, C 7-10 aralkyl group (eg, benzyl), formyl group, C 1-6 alkyl. -Carbonyl group, benzoyl group, C 7-10 aralkyl-carbonyl group (eg, benzylcarbonyl), 2-tetrahydropyranyl group, 2-tetrahydrofuranyl group, substituted silyl group (eg, trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), C 2-6 alkenyl groups (eg, 1-allyl) and the like. These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group or a nitro group.
本明細書中、「カルボニル基の保護基」としては、例えば、環状アセタール(例、1,3-ジオキサン)、非環状アセタール(例、ジ-C1-6アルキルアセタール)等が挙げられる。
In the present specification, examples of the “carbonyl-protecting group” include cyclic acetals (eg, 1,3-dioxane), acyclic acetals (eg, di-C 1-6 alkylacetal) and the like.
本明細書中、「スルファニル基の保護基」としては、例えば、C1-6アルキル基、フェニル基、トリチル基、C7-10アラルキル基(例、ベンジル)、C1-6アルキル-カルボニル基、ベンゾイル基、C7-10アラルキル-カルボニル基(例、ベンジルカルボニル)、C1-6アルコキシ-カルボニル基、C6-14アリールオキシ-カルボニル基(例、フェニルオキシカルボニル)、C7-14アラルキルオキシ-カルボニル基(例、ベンジルオキシカルボニル、9-フルオレニルメトキシカルボニル)、2-テトラヒドロピラニル基、C1-6アルキルアミノ-カルボニル基(例、メチルアミノカルボニル、エチルアミノカルボニル)、等が挙げられる。これらの基は、ハロゲン原子、C1-6アルキル基、C1-6アルコキシ基またはニトロ基から選ばれる1ないし3個の置換基で置換されていてもよい。
In the present specification, examples of the “protecting group for sulfanyl group” include a C 1-6 alkyl group, a phenyl group, a trityl group, a C 7-10 aralkyl group (eg, benzyl), a C 1-6 alkyl-carbonyl group. Benzoyl group, C 7-10 aralkyl-carbonyl group (eg, benzylcarbonyl), C 1-6 alkoxy-carbonyl group, C 6-14 aryloxy-carbonyl group (eg, phenyloxycarbonyl), C 7-14 aralkyl Oxy-carbonyl groups (eg, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl), 2-tetrahydropyranyl groups, C 1-6 alkylamino-carbonyl groups (eg, methylaminocarbonyl, ethylaminocarbonyl), etc. Can be mentioned. These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group or a nitro group.
<反応式1>
<Reaction Formula 1>

(式中、A1はヒドロキシ基の保護基、L1はヒドロキシ基、ハロゲン原子、置換されていてもよいC6-14アリールスルホニルオキシ基、または置換されていてもよいC1-6アルキルスルホニルオキシ基を示し、その他の記号は前記と同意義を示す。)
(Wherein A 1 is a protecting group for a hydroxy group, L 1 is a hydroxy group, a halogen atom, an optionally substituted C 6-14 arylsulfonyloxy group, or an optionally substituted C 1-6 alkylsulfonyl group) Represents an oxy group, and other symbols are as defined above.)
化合物(1)は、例えば、反応式5および8に記載の方法または自体公知の方法またはこれらに準じた方法に従って製造することができる。
Compound (1) can be produced, for example, according to the methods described in Reaction Schemes 5 and 8, a method known per se, or a method analogous thereto.
化合物(2)は、例えば、化合物(1)の脱保護反応によって製造することができる。
Compound (2) can be produced, for example, by deprotection of compound (1).
化合物(3)がハライドまたはスルホナートの場合、化合物(I)は、例えば、化合物(2)と化合物(3)のアルキル化反応によって製造することができる。
アルキル化反応は、例えば、化合物(2)と化合物(3)を塩基(例えば、炭酸カリウム、水素化ナトリウム、水酸化ナトリウム、DBU(1,8-ジアザビシクロ[5.4.0]ウンデカー7-エン)等)存在下、不活性溶媒(例えば、DMF(ジメチルホルムアミド)、アセトニトリル、THF(テトラヒドロフラン)、トルエン、水等)中で反応させることによって行われる。必要に応じて、相間移動触媒(例えば、テトラブチルアンモニウムブロミド、硫酸水素テトラブチルアンモニウム等)を用いてもよい。
化合物(3)がアルコールの場合、化合物(I)は、例えば、化合物(2)と化合物(3)の光延反応によって製造することができる。
光延反応は、例えば、化合物(2)と化合物(3)をヒドロキシ基活性化剤(例えば、シアノメチレントリ-n-ブチルホスホラン、アゾジカルボン酸ジイソプロピルとトリフェニルホスフィン、アゾジカルボン酸ジエチルとトリフェニルホスフィン、ADDP(1,1’-(アゾジカルボニル)ジピペリジンとトリブチルホスフィン)等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。 When compound (3) is a halide or sulfonate, compound (I) can be produced, for example, by an alkylation reaction of compound (2) and compound (3).
The alkylation reaction is carried out, for example, by reacting compound (2) and compound (3) with a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene). Etc.) in the presence of an inert solvent (for example, DMF (dimethylformamide), acetonitrile, THF (tetrahydrofuran), toluene, water, etc.). A phase transfer catalyst (for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used as necessary.
When compound (3) is an alcohol, compound (I) can be produced, for example, by Mitsunobu reaction between compound (2) and compound (3).
Mitsunobu reaction can be carried out, for example, by converting compound (2) and compound (3) into a hydroxy group activator (eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). The reaction is carried out in the presence of phosphine, ADDP (1,1 ′-(azodicarbonyl) dipiperidine and tributylphosphine), etc.) in an inert solvent (for example, toluene, THF, etc.).
アルキル化反応は、例えば、化合物(2)と化合物(3)を塩基(例えば、炭酸カリウム、水素化ナトリウム、水酸化ナトリウム、DBU(1,8-ジアザビシクロ[5.4.0]ウンデカー7-エン)等)存在下、不活性溶媒(例えば、DMF(ジメチルホルムアミド)、アセトニトリル、THF(テトラヒドロフラン)、トルエン、水等)中で反応させることによって行われる。必要に応じて、相間移動触媒(例えば、テトラブチルアンモニウムブロミド、硫酸水素テトラブチルアンモニウム等)を用いてもよい。
化合物(3)がアルコールの場合、化合物(I)は、例えば、化合物(2)と化合物(3)の光延反応によって製造することができる。
光延反応は、例えば、化合物(2)と化合物(3)をヒドロキシ基活性化剤(例えば、シアノメチレントリ-n-ブチルホスホラン、アゾジカルボン酸ジイソプロピルとトリフェニルホスフィン、アゾジカルボン酸ジエチルとトリフェニルホスフィン、ADDP(1,1’-(アゾジカルボニル)ジピペリジンとトリブチルホスフィン)等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。 When compound (3) is a halide or sulfonate, compound (I) can be produced, for example, by an alkylation reaction of compound (2) and compound (3).
The alkylation reaction is carried out, for example, by reacting compound (2) and compound (3) with a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene). Etc.) in the presence of an inert solvent (for example, DMF (dimethylformamide), acetonitrile, THF (tetrahydrofuran), toluene, water, etc.). A phase transfer catalyst (for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used as necessary.
When compound (3) is an alcohol, compound (I) can be produced, for example, by Mitsunobu reaction between compound (2) and compound (3).
Mitsunobu reaction can be carried out, for example, by converting compound (2) and compound (3) into a hydroxy group activator (eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). The reaction is carried out in the presence of phosphine, ADDP (1,1 ′-(azodicarbonyl) dipiperidine and tributylphosphine), etc.) in an inert solvent (for example, toluene, THF, etc.).
化合物(I)のXまたはYが酸化された硫黄原子である化合物、すなわちスルホン誘導体またはスルホキシド誘導体は、XまたはYが硫黄原子の化合物(I)の酸化反応によって製造することができる。本反応は、例えば、第4版実験化学講座20(日本化学会編)、276ないし278頁、503項記載の方法またはそれに準じる方法等が用いられる。
A compound in which X or Y of compound (I) is an oxidized sulfur atom, that is, a sulfone derivative or a sulfoxide derivative can be produced by an oxidation reaction of compound (I) in which X or Y is a sulfur atom. For this reaction, for example, the method described in 4th edition Experimental Chemistry Course 20 (Edited by The Chemical Society of Japan), pages 276 to 278, 503 or a method analogous thereto is used.
<反応式2>
<Reaction Formula 2>

(式中、Y1は硫黄原子、酸素原子、または置換基R8cで置換されていてもよい窒素原子を示し、L2はハロゲン原子、置換されていてもよいC6-14アリールスルホニルオキシ基、または置換されていてもよいC1-6アルキルスルホニルオキシ基を示し、その他の記号は前記と同意義を示す。)
Wherein Y 1 represents a sulfur atom, an oxygen atom, or a nitrogen atom which may be substituted with a substituent R 8c , and L 2 is a halogen atom or an optionally substituted C 6-14 arylsulfonyloxy group Or an optionally substituted C 1-6 alkylsulfonyloxy group, and other symbols are as defined above.)
化合物(5)は、例えば、反応式6、反応式9、反応式10および反応式11に記載の方法または自体公知の方法またはこれらに準じた方法に従って製造することができる。
Compound (5) can be produced, for example, according to the method described in Reaction Formula 6, Reaction Formula 9, Reaction Formula 10, and Reaction Formula 11, a method known per se, or a method analogous thereto.
化合物(I-1)は、例えば、化合物(4)と化合物(5)の光延反応によって製造することができる。本反応は、例えば、化合物(4)と化合物(5)をヒドロキシ基活性化剤(例えば、シアノメチレントリ-n-ブチルホスホラン、アゾジカルボン酸ジイソプロピルとトリフェニルホスフィン、アゾジカルボン酸ジエチルとトリフェニルホスフィン等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。
Compound (I-1) can be produced, for example, by Mitsunobu reaction between compound (4) and compound (5). In this reaction, for example, compound (4) and compound (5) are converted into a hydroxy group activator (for example, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). Phosphine, etc.) in the presence of an inert solvent (for example, toluene, THF, etc.).
化合物(6)は、例えば、化合物(4)のハロゲン化反応またはスルホニル化反応によって製造することができる。本反応は、例えば、化合物(4)とハロゲン化剤(例えば、塩化チオニル、三臭化リン等)を不活性溶媒(例えば、THF、トルエン、ジエチルエーテル等)中で反応させることによって行われる。または、本反応は、例えば、化合物(4)とスルホニル化剤(例えば、塩化メタンスルホニル、塩化p-トルエンスルホニル等)を塩基(例えば、トリエチルアミン、ピリジン等)存在下、不活性溶媒(例えば、THF、トルエン、ジエチルエーテル等)中で反応させることによって行われる。
Compound (6) can be produced, for example, by halogenation reaction or sulfonylation reaction of compound (4). This reaction is performed, for example, by reacting compound (4) with a halogenating agent (eg, thionyl chloride, phosphorus tribromide, etc.) in an inert solvent (eg, THF, toluene, diethyl ether, etc.). Alternatively, this reaction may be carried out, for example, by reacting compound (4) and a sulfonylating agent (eg, methanesulfonyl chloride, p-toluenesulfonyl chloride, etc.) in the presence of a base (eg, triethylamine, pyridine, etc.) in an inert solvent (eg, THF). , Toluene, diethyl ether, etc.).
化合物(I-1)は、例えば、化合物(5)と化合物(6)のアルキル化反応によって製造することもできる。本反応は、例えば、化合物(5)と化合物(6)を塩基(例えば、炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水素化ナトリウム、DBU等)存在下、不活性溶媒(例えば、DMF、アセトニトリル、エタノール、THF、水等)中で反応させることによって行われる。必要に応じて、相間移動触媒(例えば、テトラブチルアンモニウムブロミド、硫酸水素テトラブチルアンモニウム等)を用いてもよい。
Compound (I-1) can also be produced, for example, by an alkylation reaction of compound (5) and compound (6). In this reaction, for example, compound (5) and compound (6) are reacted with an inert solvent (eg, DMF, acetonitrile, etc.) in the presence of a base (eg, potassium carbonate, cesium carbonate, sodium hydroxide, sodium hydride, DBU, etc.). (Ethanol, THF, water, etc.). A phase transfer catalyst (for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used as necessary.
<反応式3>
<Reaction Formula 3>

(式中、A2はアミノ基の保護基を示し、その他の記号は前記と同意義を示す。)
(In the formula, A 2 represents an amino-protecting group, and other symbols are as defined above.)
化合物(7)は、例えば、反応式15および反応式16に記載の方法または自体公知の方法またはこれらに準じた方法に従って製造することができる。
Compound (7) can be produced, for example, according to the method described in Reaction Formula 15 and Reaction Formula 16, a method known per se, or a method analogous thereto.
化合物(8)は、例えば、化合物(7)の脱保護反応によって製造することができる。
Compound (8) can be produced by, for example, deprotection reaction of compound (7).
化合物(I-2)は、例えば、化合物(8)のアシル化反応によって製造することができる。上記「アシル化反応」には、例えば、アミド誘導体、カルバマート誘導体またはウレア誘導体の合成反応等が含まれる。本反応は、例えば、化合物(8)とアシル化剤を不活性溶媒(例えば、DMF、アセトニトリル、ジクロロメタン、THF等)中で反応させることによって行われる。アシル化剤としては、カルボン酸、カルボン酸の反応性誘導体(例えば、酸クロリド、酸無水物、混合酸無水物、活性エステル、活性アミド等)、二炭酸エステル、クロロギ酸エステル、イソシアン酸エステル、カルバモイルクロリド誘導体等が挙げられる。必要に応じ、脱水縮合剤(例えば、ジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(WSC)、N-[(ジメチルアミノ)(3H-[1,2,3]トリアゾロ[4,5-b]ピリジン-3-イルオキシ)メチリデン]-N-メチルメタンアミニウム ヘキサフルオロホスファート(HATU)等)、添加剤(例えば、1-ヒドロキシベンゾトリアゾール(HOBt)等)、塩基(例えば、トリエチルアミン、ピリジン等)存在下に反応を行ってもよい。
Compound (I-2) can be produced, for example, by an acylation reaction of compound (8). The “acylation reaction” includes, for example, a synthesis reaction of an amide derivative, a carbamate derivative or a urea derivative. This reaction is performed, for example, by reacting compound (8) with an acylating agent in an inert solvent (eg, DMF, acetonitrile, dichloromethane, THF, etc.). Examples of acylating agents include carboxylic acids, reactive derivatives of carboxylic acids (eg, acid chloride, acid anhydrides, mixed acid anhydrides, active esters, active amides, etc.), dicarbonates, chloroformates, isocyanates, And carbamoyl chloride derivatives. If necessary, a dehydrating condensing agent (for example, dicyclohexylcarbodiimide (DCC), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (WSC), N-[(dimethylamino) (3H- [1,2 , 3] triazolo [4,5-b] pyridin-3-yloxy) methylidene] -N-methylmethanaminium hexafluorophosphate (HATU), etc., additives (for example, 1-hydroxybenzotriazole (HOBt), etc. ) Or a base (for example, triethylamine, pyridine, etc.) may be reacted.
化合物(9)は、例えば、反応式4、反応式7および反応式14に記載の方法または自体公知の方法またはこれらに準じた方法に従って製造することができる。
Compound (9) can be produced, for example, according to the method described in Reaction Scheme 4, Reaction Formula 7, and Reaction Formula 14, a method known per se, or a method analogous thereto.
化合物(10)は、例えば、化合物(9)のハロゲン化またはスルホニル化反応により製造することができる。本反応は、例えば、反応式2の化合物(6)を製造する方法と同様にして行われる。
Compound (10) can be produced, for example, by halogenation or sulfonylation reaction of compound (9). This reaction is performed, for example, in the same manner as in the method for producing compound (6) of reaction formula 2.
化合物(11)は、例えば、化合物(10)のアジド化反応により製造することができる。
本反応は、例えば、化合物(10)とアジ化物(例えば、アジ化ナトリウム、ジフェニルホスホリルアジド(DPPA)、アジ化トリメチルシリル等)を不活性溶媒(例えば、DMF、アセトニトリル、THF等)中、通常、0℃ないし150℃で5分間ないし72時間反応させることによって行われる。必要に応じ、塩基(例えば、トリエチルアミン、ピリジン、DBU等)存在下に反応を行ってもよい。 Compound (11) can be produced, for example, by subjecting compound (10) to an azidation reaction.
In this reaction, for example, the compound (10) and an azide (for example, sodium azide, diphenylphosphoryl azide (DPPA), trimethylsilyl azide, etc.) in an inert solvent (for example, DMF, acetonitrile, THF, etc.) are usually The reaction is carried out at 0 ° C. to 150 ° C. for 5 minutes to 72 hours. If necessary, the reaction may be performed in the presence of a base (for example, triethylamine, pyridine, DBU, etc.).
本反応は、例えば、化合物(10)とアジ化物(例えば、アジ化ナトリウム、ジフェニルホスホリルアジド(DPPA)、アジ化トリメチルシリル等)を不活性溶媒(例えば、DMF、アセトニトリル、THF等)中、通常、0℃ないし150℃で5分間ないし72時間反応させることによって行われる。必要に応じ、塩基(例えば、トリエチルアミン、ピリジン、DBU等)存在下に反応を行ってもよい。 Compound (11) can be produced, for example, by subjecting compound (10) to an azidation reaction.
In this reaction, for example, the compound (10) and an azide (for example, sodium azide, diphenylphosphoryl azide (DPPA), trimethylsilyl azide, etc.) in an inert solvent (for example, DMF, acetonitrile, THF, etc.) are usually The reaction is carried out at 0 ° C. to 150 ° C. for 5 minutes to 72 hours. If necessary, the reaction may be performed in the presence of a base (for example, triethylamine, pyridine, DBU, etc.).
化合物(8)は、例えば、化合物(11)の還元反応によって製造することもできる。
本反応は、例えば、化合物(11)を金属触媒(例えば、パラジウム-炭素、酸化白金等)および水素源(例えば、水素ガス、ギ酸、ギ酸アンモニウム等)存在下、不活性溶媒(例えば、エタノール、メタノール、酢酸エチル、THF等)中で反応させることによって行うことができる。また、本反応は、例えば、化合物(11)、トリフェニルホスフィンおよび水を不活性溶媒(例えば、THF等)中で反応させることによっても行うことができる。 Compound (8) can also be produced, for example, by a reduction reaction of compound (11).
In this reaction, for example, the compound (11) is reacted with an inert solvent (eg, ethanol, for example) in the presence of a metal catalyst (eg, palladium-carbon, platinum oxide, etc.) and a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.). (Methanol, ethyl acetate, THF, etc.). This reaction can also be carried out, for example, by reacting compound (11), triphenylphosphine and water in an inert solvent (for example, THF).
本反応は、例えば、化合物(11)を金属触媒(例えば、パラジウム-炭素、酸化白金等)および水素源(例えば、水素ガス、ギ酸、ギ酸アンモニウム等)存在下、不活性溶媒(例えば、エタノール、メタノール、酢酸エチル、THF等)中で反応させることによって行うことができる。また、本反応は、例えば、化合物(11)、トリフェニルホスフィンおよび水を不活性溶媒(例えば、THF等)中で反応させることによっても行うことができる。 Compound (8) can also be produced, for example, by a reduction reaction of compound (11).
In this reaction, for example, the compound (11) is reacted with an inert solvent (eg, ethanol, for example) in the presence of a metal catalyst (eg, palladium-carbon, platinum oxide, etc.) and a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.). (Methanol, ethyl acetate, THF, etc.). This reaction can also be carried out, for example, by reacting compound (11), triphenylphosphine and water in an inert solvent (for example, THF).
<反応式4>
<Reaction Formula 4>

(式中、R1aおよびR9は、独立して、置換されていてもよい5または6員芳香環基を示し、その他の記号は前記と同意義を示す。)
(In the formula, R 1a and R 9 each independently represents an optionally substituted 5- or 6-membered aromatic ring group, and other symbols are as defined above.)
化合物(12)は、例えば、反応式7に記載の方法または自体公知の方法またはこれらに準じた方法に従って製造することができる。
Compound (12) can be produced, for example, according to the method described in Reaction Scheme 7, a method known per se, or a method analogous thereto.
化合物(I-3)は、例えば、化合物(12)と化合物(13)の還元アミノ化反応によって製造することができる。本反応は、例えば、化合物(12)と化合物(13)を還元剤(例えば、水素化ホウ素ナトリウム、シアノ水素化ホウ素ナトリウム、トリアセトキシ水素化ホウ素ナトリウム、デカボラン等)存在下、不活性溶媒(例えば、メタノール、THF、ジクロロエタン、ジクロロメタン等)中で反応させることによって行われる。
Compound (I-3) can be produced, for example, by a reductive amination reaction of compound (12) and compound (13). In this reaction, for example, compound (12) and compound (13) are reacted with an inert solvent (for example, sodium borohydride, sodium cyanoborohydride, sodium triacetoxyborohydride, decaborane, etc.) in the presence of a reducing agent. , Methanol, THF, dichloroethane, dichloromethane, etc.).
化合物(9)は、例えば、化合物(12)の還元反応によって製造することができる。本反応は、例えば、化合物(12)を還元剤(例えば、水素化ホウ素ナトリウム、シアノ水素化ホウ素ナトリウム、水素化ジイソブチルアルミニウム、水素化アルミニウムリチウム等)存在下、不活性溶媒(例えば、エタノール、メタノール、トルエン、THF、酢酸等)中で反応させることによって行われる。
Compound (9) can be produced, for example, by a reduction reaction of compound (12). In this reaction, for example, compound (12) is converted into an inert solvent (for example, ethanol, methanol, etc.) in the presence of a reducing agent (for example, sodium borohydride, sodium cyanoborohydride, diisobutylaluminum hydride, lithium aluminum hydride, etc.). , Toluene, THF, acetic acid, etc.).
化合物(15)は、例えば、化合物(9)と化合物(14)の光延反応によって製造することができる。本反応は、例えば、化合物(9)と化合物(14)をヒドロキシ基活性化剤(例えば、シアノメチレントリ-n-ブチルホスホラン、アゾジカルボン酸ジイソプロピルとトリフェニルホスフィン、アゾジカルボン酸ジエチルとトリフェニルホスフィン等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。
Compound (15) can be produced, for example, by Mitsunobu reaction between compound (9) and compound (14). In this reaction, for example, compound (9) and compound (14) are converted into a hydroxy group activator (for example, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). Phosphine, etc.) in the presence of an inert solvent (for example, toluene, THF, etc.).
化合物(I-3)は、例えば、化合物(15)の脱スルホニル化反応によって製造することもできる。本反応は、例えば、化合物(15)と塩基(例えば、トリエチルアミン、水酸化リチウム二水和物等)および有機メルカプタン類(例えば、スルファニル酢酸等)存在下、不活性溶媒(例えば、THF、ジクロロメタン、DMF等)中で反応させることによって行われる。
Compound (I-3) can also be produced, for example, by a desulfonylation reaction of compound (15). This reaction is carried out, for example, in the presence of a compound (15), a base (for example, triethylamine, lithium hydroxide dihydrate, etc.) and an organic mercaptan (for example, sulfanyl acetic acid, etc.) and an inert solvent (for example, THF, dichloromethane, In DMF etc.).
<反応式5>
<Reaction Formula 5>

(式中、X1は硫黄原子、酸素原子、または置換基R7cで置換されていてもよい窒素原子を示し、その他の記号は前記と同意義を示す。なお、化合物(1-1)は化合物(1)に包含される。)
(Wherein X 1 represents a sulfur atom, an oxygen atom, or a nitrogen atom which may be substituted with a substituent R 7c , and other symbols are as defined above. Compound (1-1) is (Included in compound (1))
化合物(18)は、例えば、化合物(16)と化合物(17)のアルキル化または光延反応によって製造することができる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (18) can be produced, for example, by alkylation of compound (16) and compound (17) or Mitsunobu reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
化合物(19)は、例えば、化合物(18)の脱保護反応によって製造することができる。
Compound (19) can be produced, for example, by deprotection reaction of compound (18).
化合物(1-1)は、例えば、化合物(19)のアシル化反応によって製造することができる。本反応は、例えば、反応式3の(I-2)を製造する方法と同様にして行われる。
Compound (1-1) can be produced, for example, by an acylation reaction of compound (19). This reaction is performed, for example, in the same manner as in the production of (I-2) in Reaction Scheme 3.
<反応式6>
<Reaction Scheme 6>

(式中、A3はヒドロキシ基、アミノ基またはスルファニル基の保護基を示し、その他の記号は前記と同意義を示す。なお、化合物(5-1)は化合物(5)に包含される。)
(Wherein A 3 represents a protecting group for a hydroxy group, an amino group or a sulfanyl group, and other symbols have the same meanings as described above. Compound (5-1) is encompassed in compound (5). )
化合物(21)は、例えば、化合物(20)と化合物(17)のアルキル化反応または光延反応によって製造することができる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (21) can be produced, for example, by an alkylation reaction or Mitsunobu reaction between compound (20) and compound (17). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
化合物(22)は、例えば、化合物(21)の脱保護反応によって製造することができる。
Compound (22) can be produced, for example, by deprotection of compound (21).
化合物(23)は、例えば、化合物(22)の脱保護反応によって製造することができる。
Compound (23) can be produced, for example, by deprotection of compound (22).
化合物(5-1)は、例えば、化合物(23)のアシル化反応によって製造することができる。本反応は、例えば、反応式3の化合物(I-2)を製造する方法と同様にして行われる。
Compound (5-1) can be produced, for example, by acylation reaction of compound (23). This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
必要に応じ、反応式6に記載の方法(A2およびA3の脱保護反応およびアシル化反応)の順序を変えて行うことによっても化合物(5-1)を製造することができる。
If necessary, compound (5-1) can also be produced by changing the order of the method described in Reaction Scheme 6 (deprotection reaction and acylation reaction of A 2 and A 3 ).
化合物(21)、化合物(22)、化合物(23)または化合物(5-1)の環Pが芳香環P1(置換されていてもよい5員および6員芳香環)の場合、芳香環P1の還元反応によって対応する飽和環P2(置換されていてもよい5員および6員飽和環)を有する化合物を製造することができる。本反応は、例えば、化合物(21)、化合物(22)、化合物(23)または化合物(5-1)を金属触媒(例えば、ロジウム-炭素、パラジウム-炭素、酸化白金等)および水素源(例えば、水素ガス、ギ酸、ギ酸アンモニウム等)存在下、不活性溶媒(例えば、エタノール、メタノール、酢酸、THF等)中で反応させることによって行われる。水素源として水素ガスを用いる場合、通常、水素圧は1気圧ないし10気圧である。
When ring P of compound (21), compound (22), compound (23) or compound (5-1) is aromatic ring P 1 (5- and 6-membered aromatic ring which may be substituted), aromatic ring P A compound having a corresponding saturated ring P 2 (optionally substituted 5-membered and 6-membered saturated ring) can be produced by a reduction reaction of 1 . In this reaction, for example, the compound (21), the compound (22), the compound (23) or the compound (5-1) is converted into a metal catalyst (for example, rhodium-carbon, palladium-carbon, platinum oxide, etc.) and a hydrogen source (for example, , Hydrogen gas, formic acid, ammonium formate, etc.) in the presence of an inert solvent (for example, ethanol, methanol, acetic acid, THF, etc.). When hydrogen gas is used as the hydrogen source, the hydrogen pressure is usually 1 to 10 atmospheres.
<反応式7>
<Reaction Scheme 7>

(式中、R3-Mは有機金属試薬を示し、R10aおよびR10bは、独立して、置換基を示し、R10aおよびR10bは互いに結合して環を形成してもよい。その他の記号は前記と同意義を示す。なお、化合物(9-1)は化合物(9)に、化合物(12-1)は化合物(12)に包含される。)
(Wherein R 3 -M represents an organometallic reagent, R 10a and R 10b independently represent a substituent, and R 10a and R 10b may combine with each other to form a ring. The symbols of the above are as defined above, wherein compound (9-1) is included in compound (9) and compound (12-1) is included in compound (12).
化合物(25)は、例えば、化合物(24)と化合物(6)のアルキル化反応または光延反応によって製造することができる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (25) can be produced, for example, by an alkylation reaction or Mitsunobu reaction between compound (24) and compound (6). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
化合物(26)は、例えば、化合物(25)の脱保護反応によって製造することができる。
Compound (26) can be produced, for example, by subjecting compound (25) to deprotection.
化合物(26)は、例えば、反応式12に記載の方法または自体公知の方法またはこれらに準じた方法に従って製造することもできる。
Compound (26) can also be produced, for example, according to the method described in Reaction Scheme 12, a method known per se, or a method analogous thereto.
化合物(28)は、例えば、化合物(26)と化合物(27)のアルキル化反応または光延反応によって製造することができる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (28) can be produced, for example, by an alkylation reaction or Mitsunobu reaction of compound (26) and compound (27). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
化合物(12-1)は、例えば、化合物(28)と有機金属試薬R3-Mの置換反応によって製造することができる。本反応は、例えば、化合物(28)と有機金属試薬R3-M(例えば、メチルマグネシウムクロリド、メチルマグネシウムブロミド、メチルリチウム等)を不活性溶媒(例えば、トルエン、THF、ジエチルエーテル等)中、通常、-78℃ないし100℃で反応させることによって行われる。
Compound (12-1) can be produced, for example, by subjecting compound (28) to an organometallic reagent R 3 -M. In this reaction, for example, compound (28) and organometallic reagent R 3 -M (eg, methylmagnesium chloride, methylmagnesium bromide, methyllithium, etc.) in an inert solvent (eg, toluene, THF, diethyl ether, etc.) Usually, the reaction is performed at −78 ° C. to 100 ° C.
化合物(9-1)は、例えば、化合物(12-1)の還元反応によって製造することができる。本反応は、例えば、反応式4の化合物(9)を製造する方法と同様にして行われる。
Compound (9-1) can be produced, for example, by a reduction reaction of compound (12-1). This reaction is performed, for example, in the same manner as in the method for producing compound (9) of reaction formula 4.
<反応式8>
<Reaction Scheme 8>

(式中、P1は置換されていてもよい5員および6員芳香環を示し、その他の記号は前記と同意義を示す。なお、化合物(1-2)は化合物(1)に包含される。)
(In the formula, P 1 represents an optionally substituted 5-membered and 6-membered aromatic ring, and other symbols have the same meanings as described above. Compound (1-2) is included in Compound (1). )
化合物(30)は、例えば、化合物(29)と化合物(17)のアルキル化反応または光延反応によって製造することができる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (30) can be produced, for example, by an alkylation reaction or Mitsunobu reaction of compound (29) and compound (17). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
化合物(31)は、例えば、化合物(30)の脱保護反応によって製造することができる。
Compound (31) can be produced, for example, by subjecting compound (30) to deprotection.
化合物(32)は、例えば、化合物(31)のアシル化反応によって製造することができる。本反応は、例えば、反応式3の化合物(I-2)を製造する方法と同様にして行われる。
Compound (32) can be produced, for example, by subjecting compound (31) to an acylation reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
化合物(34)は、例えば、化合物(32)と化合物(33)のカップリング反応によって製造することができる。本反応は、例えば、化合物(32)と化合物(33)を、金属触媒(例えば、ビストリフェニルホスフィンジクロロパラジウム(II)およびヨウ化銅(I)等)および塩基(例えば、トリエチルアミン、ピリジン等)存在下、不活性溶媒(例えば、トルエン、THF、DMF等)中、通常、不活性ガス(例えば、アルゴン、窒素等)雰囲気下で反応させることによって行われる。
Compound (34) can be produced, for example, by a coupling reaction of compound (32) and compound (33). In this reaction, for example, compound (32) and compound (33) are present in the presence of a metal catalyst (eg, bistriphenylphosphinedichloropalladium (II) and copper (I) iodide) and a base (eg, triethylamine, pyridine, etc.). The reaction is usually carried out in an inert solvent (eg, toluene, THF, DMF, etc.) under an inert gas (eg, argon, nitrogen, etc.) atmosphere.
化合物(1-2)は、例えば、化合物(34)の還元反応によって製造することができる。本反応は、例えば、化合物(34)を金属触媒(例えば、ロジウム-炭素、パラジウム-炭素、酸化白金等)および水素源(例えば、水素ガス、ギ酸、ギ酸アンモニウム等)存在下、不活性溶媒(例えば、エタノール、メタノール、酢酸、THF等)中で反応させることによって行われる。水素源として水素を用いる場合、通常、水素圧は1気圧ないし10気圧である。必要に応じ、金属触媒は化合物(34)と同量(重量)以上用いられる。
Compound (1-2) can be produced, for example, by a reduction reaction of compound (34). In this reaction, for example, the compound (34) is reacted with an inert solvent (eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.) and a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.). For example, the reaction is performed in ethanol, methanol, acetic acid, THF, or the like). When hydrogen is used as a hydrogen source, the hydrogen pressure is usually 1 to 10 atmospheres. If necessary, the metal catalyst is used in the same amount (weight) or more as the compound (34).
必要に応じ、反応式8に記載の方法(A2の脱保護反応、アシル化反応、カップリング反応および還元反応)の順序を変えて行うことによっても化合物(1-2)を製造することができる。
If necessary, the method described in Scheme 8 can also be produced a compound (1-2) by performing a different order of (deprotection reaction of A 2, acylation reaction, coupling reaction and reduction) it can.
化合物(30)、化合物(31)、化合物(32)、化合物(34)または化合物(1-2)から芳香環P1の還元反応により、対応する飽和環P2(置換されていてもよい5員および6員飽和環)を有する化合物を製造することができる。本反応は、例えば、化合物(30)、化合物(31)、化合物(32)、化合物(34)または化合物(1-2)を金属触媒(例えば、ロジウム-炭素、パラジウム-炭素、酸化白金等)および水素源(例えば、水素ガス、ギ酸、ギ酸アンモニウム等)存在下、不活性溶媒(例えば、エタノール、メタノール、酢酸、THF等)中で反応させることによって行われる。水素源として水素を用いる場合、通常、水素圧は1気圧ないし10気圧である。
A reduction reaction of the aromatic ring P 1 from the compound (30), the compound (31), the compound (32), the compound (34) or the compound (1-2) results in the corresponding saturated ring P 2 (optionally substituted 5 And 6-membered saturated rings) can be prepared. In this reaction, for example, compound (30), compound (31), compound (32), compound (34) or compound (1-2) is converted to a metal catalyst (eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.) And a reaction in an inert solvent (eg, ethanol, methanol, acetic acid, THF, etc.) in the presence of a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.). When hydrogen is used as the hydrogen source, the hydrogen pressure is usually 1 to 10 atmospheres.
<反応式9>
<Reaction Scheme 9>

(式中、L3はハロゲン原子を示し、その他の記号は前記と同意義を示す。なお、化合物(5-2)は化合物(5)に包含される。)
(Wherein L 3 represents a halogen atom, and other symbols are as defined above. Compound (5-2) is encompassed in compound (5).)
化合物(37)は、例えば、化合物(35)と化合物(36)のカップリング反応によって製造することができる。本反応は、例えば、反応式8の化合物(34)を製造する方法と同様にして行われる。
Compound (37) can be produced, for example, by a coupling reaction of compound (35) and compound (36). This reaction is performed, for example, in the same manner as in the method for producing compound (34) of reaction formula 8.
化合物(38)は、例えば、化合物(37)の脱保護反応によって製造することができる。
Compound (38) can be produced, for example, by subjecting compound (37) to deprotection.
化合物(39)は、例えば、化合物(38)のアシル化反応によって製造することができる。本反応は、例えば、反応式3の化合物(I-2)を製造する方法と同様にして行われる。
Compound (39) can be produced, for example, by subjecting compound (38) to an acylation reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
化合物(40)は、例えば、化合物(39)の還元反応によって製造することができる。本反応は、例えば、反応式8の化合物(1-2)を製造する方法と同様にして行われる。
Compound (40) can be produced, for example, by subjecting compound (39) to a reduction reaction. This reaction is performed, for example, in the same manner as in the method for producing compound (1-2) of reaction formula 8.
化合物(5-2)は、例えば、化合物(40)の脱保護反応によって製造することができる。
Compound (5-2) can be produced, for example, by deprotection of compound (40).
必要に応じ、反応式9に記載の方法(A2およびA3の脱保護反応、カップリング反応および還元反応)の順序を変えて行うことによっても化合物(5-2)を製造することができる。
If necessary, compound (5-2) can also be produced by changing the order of the method described in Reaction Scheme 9 (deprotection reaction of A 2 and A 3 , coupling reaction and reduction reaction). .
化合物(37)、化合物(38)、化合物(39)、化合物(40)または化合物(5-2)から芳香環P1の還元反応により、対応する飽和環P2(置換されていてもよい5員および6員飽和環)を有する化合物を製造することができる。本反応は、例えば、化合物(37)、化合物(38)、化合物(39)、化合物(40)または化合物(5-2)を金属触媒(例えば、ロジウム-炭素、パラジウム-炭素、酸化白金等)および水素源(例えば、水素ガス、ギ酸、ギ酸アンモニウム等)存在下、不活性溶媒(例えば、エタノール、メタノール、酢酸、THF等)中で反応させることによって行われる。水素源として水素ガスを用いる場合、通常、水素圧は1気圧ないし10気圧である。
Reduction of the aromatic ring P 1 from the compound (37), compound (38), compound (39), compound (40) or compound (5-2) results in the corresponding saturated ring P 2 (optionally substituted 5 And 6-membered saturated rings) can be prepared. In this reaction, for example, compound (37), compound (38), compound (39), compound (40) or compound (5-2) is converted into a metal catalyst (eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.). And a reaction in an inert solvent (eg, ethanol, methanol, acetic acid, THF, etc.) in the presence of a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.). When hydrogen gas is used as the hydrogen source, the hydrogen pressure is usually 1 to 10 atmospheres.
<反応式10>
<Reaction Scheme 10>

(式中、各記号は前記と同意義を示す。なお、化合物(5-3)は化合物(5)に包含される。)
(In the formula, each symbol is as defined above. Compound (5-3) is included in compound (5).)
化合物(43)は、例えば、化合物(41)と化合物(42)の付加反応によって製造することができる。本反応は、例えば、化合物(41)と化合物(42)を不活性溶媒(例えば、エタノール、メタノール、THF等)中で反応させることによって行われる。必要に応じ、金属塩類(例えば、硝酸ビスマス等)存在下で行われる。
Compound (43) can be produced, for example, by an addition reaction of compound (41) and compound (42). This reaction is performed, for example, by reacting compound (41) with compound (42) in an inert solvent (eg, ethanol, methanol, THF, etc.). If necessary, it is carried out in the presence of a metal salt (for example, bismuth nitrate, etc.).
化合物(44)は、例えば、化合物(43)の脱保護反応によって製造することができる。
Compound (44) can be produced, for example, by subjecting compound (43) to deprotection.
化合物(5-3)は、例えば、化合物(44)のアシル化反応によって製造することができる。本反応は、例えば、反応式3の化合物(I-2)を製造する方法と同様にして行われる。
Compound (5-3) can be produced, for example, by subjecting compound (44) to an acylation reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
<反応式11>
<Reaction Scheme 11>

(式中、各記号は前記と同意義を示す。なお、化合物(5-4)は化合物(5)に包含される。)
(In the formula, each symbol is as defined above. Compound (5-4) is encompassed in compound (5).)
化合物(47)は、例えば、化合物(45)と化合物(46)のアミド化反応によって製造することができる。本反応は、例えば、化合物(45)と化合物(46)を、脱水縮合剤(例えば、WSC、HATU等)および添加剤(例えば、HOBt等)存在下、不活性溶媒(例えば、DMF、アセトニトリル、ジクロロメタン、THF等)中で反応させることによって行われる。必要に応じ、化合物(46)をカルボン酸の反応性誘導体(例えば、酸クロリド、酸無水物、混合酸無水物、活性エステル、活性アミド等)に変換した後に用いてもよい。また、必要に応じ、塩基(例えば、トリエチルアミン、ピリジン等)存在下に反応を行ってもよい。
Compound (47) can be produced, for example, by an amidation reaction of compound (45) and compound (46). In this reaction, for example, compound (45) and compound (46) are combined with an inert solvent (eg, DMF, acetonitrile, etc.) in the presence of a dehydrating condensing agent (eg, WSC, HATU, etc.) and an additive (eg, HOBt, etc.). Reaction in dichloromethane, THF, etc.). If necessary, compound (46) may be used after being converted into a reactive derivative of carboxylic acid (for example, acid chloride, acid anhydride, mixed acid anhydride, active ester, active amide, etc.). Moreover, you may react in presence of a base (for example, a triethylamine, a pyridine, etc.) as needed.
化合物(48)は、例えば、化合物(47)の還元反応によって製造することができる。
本反応は、例えば、化合物(47)を還元剤(例えば、ボラン、水素化アルミニウムリチウム等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。 Compound (48) can be produced, for example, by subjecting compound (47) to a reduction reaction.
This reaction is carried out, for example, by reacting compound (47) in the presence of a reducing agent (eg, borane, lithium aluminum hydride) in an inert solvent (eg, toluene, THF, etc.).
本反応は、例えば、化合物(47)を還元剤(例えば、ボラン、水素化アルミニウムリチウム等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。 Compound (48) can be produced, for example, by subjecting compound (47) to a reduction reaction.
This reaction is carried out, for example, by reacting compound (47) in the presence of a reducing agent (eg, borane, lithium aluminum hydride) in an inert solvent (eg, toluene, THF, etc.).
化合物(49)は、例えば、化合物(48)の脱保護反応によって製造することができる。
Compound (49) can be produced, for example, by subjecting compound (48) to deprotection.
化合物(50)は、例えば、化合物(49)のアシル化反応によって製造することができる。本反応は、例えば、反応式3の化合物(I-2)を製造する方法と同様にして行われる。
Compound (50) can be produced, for example, by subjecting compound (49) to an acylation reaction. This reaction is performed, for example, in the same manner as in the production of compound (I-2) of reaction formula 3.
化合物(5-4)は、例えば、化合物(50)の脱保護反応によって製造することができる。
Compound (5-4) can be produced, for example, by subjecting compound (50) to deprotection.
<反応式12>
<Reaction Scheme 12>

(式中、R11aおよびR11bは、独立して、水酸基または置換基を示し、これらは互いに結合して環を形成してもよい。各記号は前記と同意義を示す。なお、化合物(26-1)は化合物(26)に包含される。)
(Wherein R 11a and R 11b independently represent a hydroxyl group or a substituent, and these may be bonded to each other to form a ring. Each symbol has the same meaning as described above. 26-1) is encompassed by compound (26).)
化合物(53)は、例えば、化合物(51)と化合物(52)の求核置換反応によって製造することができる。本反応は、例えば、化合物(51)と化合物(52)を、塩基(例えば、水素化ナトリウム、炭酸カリウム、トリエチルアミン、ピリジン等)存在下、不活性溶媒(例えば、DMF、アセトニトリル、ジクロロメタン、THF等)中で反応させることによって行われる。
Compound (53) can be produced, for example, by a nucleophilic substitution reaction of compound (51) and compound (52). In this reaction, for example, compound (51) and compound (52) are reacted with an inert solvent (eg, DMF, acetonitrile, dichloromethane, THF, etc.) in the presence of a base (eg, sodium hydride, potassium carbonate, triethylamine, pyridine, etc.). ) In the reaction.
化合物(54)は、例えば、化合物(53)のホウ素化反応によって製造することができる。本反応は、例えば、化合物(53)を不活性溶媒(例えば、ジエチルエーテル、トルエン、THF等)中でアルキル金属類(例えば、ブチルリチウム、イソプロピルマグネシウムブロミド等)によってハロゲン原子を金属原子に変換した後に、有機ホウ素化合物(例えば、トリメトキシボラン等)と、通常、-100℃ないし100℃で反応させることによって行われる。
Compound (54) can be produced, for example, by a boronation reaction of compound (53). In this reaction, for example, a halogen atom is converted into a metal atom with an alkyl metal (eg, butyllithium, isopropylmagnesium bromide, etc.) in compound (53) in an inert solvent (eg, diethyl ether, toluene, THF, etc.). Thereafter, it is carried out by reacting with an organic boron compound (for example, trimethoxyborane etc.) usually at −100 ° C. to 100 ° C.
化合物(26-1)は、例えば、化合物(54)のホウ素原子の酸化反応によって製造することができる。本反応は、例えば、化合物(54)を酸化剤(例えば、酸素、過酸化水素、m-クロロ過安息香酸、過ホウ酸ナトリウム等)と不活性溶媒(例えば、水、THF等)中で反応させることによって行われる。必要に応じて、塩基(例えば、水酸化ナトリウム等)を用いてもよい。
Compound (26-1) can be produced, for example, by subjecting the boron atom of compound (54) to an oxidation reaction. In this reaction, for example, compound (54) is reacted with an oxidizing agent (eg, oxygen, hydrogen peroxide, m-chloroperbenzoic acid, sodium perborate, etc.) in an inert solvent (eg, water, THF, etc.). Is done by letting If necessary, a base (for example, sodium hydroxide) may be used.
<反応式13>
<Reaction Formula 13>

(式中、各記号は前記と同意義を示す。なお、化合物(22-1)は化合物(22)に包含される。)
(In the formula, each symbol is as defined above. Compound (22-1) is included in compound (22).)
化合物(56)は、例えば、化合物(55)と化合物(17)のアルキル化反応または光延反応によって製造することができる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (56) can be produced, for example, by an alkylation reaction or Mitsunobu reaction of compound (55) and compound (17). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
化合物(57)は、例えば、化合物(56)のホウ素化反応によって製造することができる。本反応は、例えば、反応式12の化合物(54)を製造する方法と同様にして行われる。
Compound (57) can be produced, for example, by a boronation reaction of compound (56). This reaction is performed, for example, in the same manner as in the method for producing compound (54) of reaction formula 12.
化合物(22-1)は、例えば、化合物(57)のホウ素原子の酸化反応によって製造することができる。本反応は、例えば、反応式12の化合物(26-1)を製造する方法と同様にして行われる。
Compound (22-1) can be produced, for example, by subjecting the boron atom of compound (57) to an oxidation reaction. This reaction is performed, for example, in the same manner as in the method for producing compound (26-1) of reaction formula 12.
<反応式14>
<Reaction Scheme 14>

(式中、各記号は前記と同意義を示す。なお、化合物(9-2)は化合物(9)に包含される。)
(In the formula, each symbol is as defined above. Compound (9-2) is included in compound (9).)
化合物(60)は、例えば、化合物(58)と化合物(59)のアルキル化反応または光延反応によって製造することができる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (60) can be produced, for example, by an alkylation reaction or Mitsunobu reaction between compound (58) and compound (59). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
化合物(61)は、例えば、化合物(60)の還元反応によって製造することができる。本反応は、例えば、反応式4の化合物(9)を製造する方法と同様にして行われる。
Compound (61) can be produced, for example, by a reduction reaction of compound (60). This reaction is performed, for example, in the same manner as in the method for producing compound (9) of reaction formula 4.
化合物(62)は、例えば、化合物(61)の脱保護反応によって製造することができる。
Compound (62) can be produced, for example, by deprotection of compound (61).
化合物(9-2)は、例えば、化合物(62)と化合物(6)のアルキル化反応によって製造することもできる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (9-2) can also be produced, for example, by an alkylation reaction of compound (62) and compound (6). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
<反応式15>
<Reaction Scheme 15>

(式中、環P2は置換されていてもよい5員および6員環の飽和環を示し、その他の記号は前記と同意義を示す。なお、化合物(7-1)は化合物(7)に包含される。)
(Wherein ring P 2 represents an optionally substituted 5-membered and 6-membered saturated ring, and other symbols are as defined above. Compound (7-1) is compound (7). Included.)
化合物(64)は、例えば、化合物(63)と化合物(17)のアルキル化反応または光延反応によって製造することができる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (64) can be produced, for example, by an alkylation reaction or Mitsunobu reaction between compound (63) and compound (17). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
化合物(65)は、例えば、化合物(64)の芳香環P1の還元反応によって製造することができる。本反応は、例えば、化合物(64)を金属触媒(例えば、ロジウム-炭素、パラジウム-炭素、酸化白金等)および水素源(例えば、水素ガス、ギ酸、ギ酸アンモニウム等)存在下、不活性溶媒(例えば、エタノール、メタノール、酢酸、THF等)中で反応させることによって行われる。水素源として水素ガスを用いる場合、通常、水素圧は1気圧ないし10気圧である。
Compound (65) can be produced, for example, by a reduction reaction of aromatic ring P 1 of compound (64). In this reaction, for example, compound (64) is reacted with an inert solvent (eg, rhodium-carbon, palladium-carbon, platinum oxide, etc.) and a hydrogen source (eg, hydrogen gas, formic acid, ammonium formate, etc.). For example, the reaction is performed in ethanol, methanol, acetic acid, THF, or the like). When hydrogen gas is used as the hydrogen source, the hydrogen pressure is usually 1 to 10 atmospheres.
化合物(7-1)は、例えば、化合物(65)と化合物(66)のアミド化反応によって製造することができる。本反応は、例えば、反応式11の化合物(47)を製造する方法と同様にして行われる。
Compound (7-1) can be produced, for example, by an amidation reaction of compound (65) and compound (66). This reaction is performed, for example, in the same manner as in the method for producing compound (47) of reaction formula 11.
<反応式16>
<Reaction Scheme 16>

(式中、各記号は前記と同意義を示す。なお、化合物(7-2)は化合物(7)に包含される。)
(In the formula, each symbol is as defined above. Compound (7-2) is included in compound (7).)
化合物(7-2)は、例えば、化合物(4)と化合物(67)、または化合物(6)と化合物(67)のアルキル化反応または光延反応によって製造することができる。本反応は、例えば、反応式2の化合物(I-1)を製造する方法と同様にして行われる。
Compound (7-2) can be produced, for example, by an alkylation reaction or Mitsunobu reaction of compound (4) and compound (67), or compound (6) and compound (67). This reaction is performed, for example, in the same manner as in the production of compound (I-1) of reaction formula 2.
<反応式17>
<Reaction Scheme 17>

(式中、各記号は前記と同意義を示す。なお、化合物(16-1)は化合物(16)に包含される。)
(In the formula, each symbol is as defined above. Compound (16-1) is encompassed in Compound (16).)
化合物(70)は、例えば、化合物(68)と化合物(69)のアルキル化反応によって製造することもできる。本反応は、例えば、化合物(68)と化合物(69)を塩基(例えば、炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水素化ナトリウム、DBU等)存在下、不活性溶媒(例えば、DMF、アセトニトリル、エタノール、THF、水等)中で反応させることによって行われる。必要に応じて、相間移動触媒(例えば、テトラブチルアンモニウムブロミド、硫酸水素テトラブチルアンモニウム等)を用いてもよい。
Compound (70) can also be produced, for example, by an alkylation reaction of compound (68) and compound (69). In this reaction, for example, compound (68) and compound (69) are reacted in the presence of a base (for example, potassium carbonate, cesium carbonate, sodium hydroxide, sodium hydride, DBU, etc.) in an inert solvent (for example, DMF, acetonitrile, (Ethanol, THF, water, etc.). A phase transfer catalyst (for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used as necessary.
化合物(16-1)は、例えば、化合物(70)の脱保護反応によって製造することができる。
Compound (16-1) can be produced, for example, by subjecting compound (70) to deprotection.
<反応式18>
<Reaction Scheme 18>

(式中、各記号は前記と同意義を示す。なお、化合物(16-2)は化合物(16)に包含される。)
(In the formula, each symbol is as defined above. Compound (16-2) is encompassed in compound (16).)
化合物(73)は、例えば、化合物(71)と化合物(72)のカップリング反応によって製造することができる。本反応は、例えば、化合物(71)と化合物(72)を、金属触媒(例えば、ビストリフェニルホスフィンジクロロパラジウム(II)およびヨウ化銅(I)等)および塩基(例えば、トリエチルアミン、ピリジン等)存在下、不活性溶媒(例えば、トルエン、THF、DMF等)中、通常、不活性ガス(例えば、アルゴン、窒素等)雰囲気下で反応させることによって行われる。
Compound (73) can be produced, for example, by a coupling reaction of compound (71) and compound (72). In this reaction, for example, compound (71) and compound (72) are present in the presence of a metal catalyst (for example, bistriphenylphosphinedichloropalladium (II) and copper iodide (I)) and a base (for example, triethylamine, pyridine, etc.). The reaction is usually carried out in an inert solvent (eg, toluene, THF, DMF, etc.) under an inert gas (eg, argon, nitrogen, etc.) atmosphere.
化合物(74)は、例えば、化合物(73)の還元反応によって製造することができる。本反応は、例えば、反応式8の化合物(1-2)を製造する方法と同様にして行われる。
Compound (74) can be produced, for example, by a reduction reaction of compound (73). This reaction is performed, for example, in the same manner as in the method for producing compound (1-2) of reaction formula 8.
化合物(16-2)は、例えば、化合物(74)の脱保護反応によって製造することができる。
Compound (16-2) can be produced, for example, by subjecting compound (74) to deprotection.
化合物(73)から化合物(16-2)に導く反応、すなわち還元反応および脱保護反応は同時に行っても、あるいは順序を変えて行っても、化合物(16-2)を製造することができる。
The compound (16-2) can be produced by performing the reaction leading from the compound (73) to the compound (16-2), that is, the reduction reaction and the deprotection reaction at the same time or by changing the order.
<反応式19>
<Reaction Formula 19>

(式中、各記号は前記と同意義を示す。なお、化合物(1-3)は化合物(1)に包含される。)
(In the formula, each symbol is as defined above. Compound (1-3) is encompassed in compound (1).)
化合物(76)がハライドまたはスルホナートの場合、化合物(77)は、例えば、化合物(75)と化合物(76)のアルキル化反応によって製造することができる。アルキル化反応は、例えば、化合物(75)と化合物(76)を塩基(例えば、炭酸カリウム、水素化ナトリウム、水酸化ナトリウム、DBU(1,8-ジアザビシクロ[5.4.0]ウンデカー7-エン)等)存在下、不活性溶媒(例えば、DMF、アセトニトリル、THF、トルエン、水等)中で反応させることによって行われる。必要に応じて、相間移動触媒(例えば、テトラブチルアンモニウムブロミド、硫酸水素テトラブチルアンモニウム等)を用いてもよい。
化合物(76)がアルコールの場合、化合物(77)は、例えば、化合物(75)と化合物(76)の光延反応によって製造することができる。光延反応は、例えば、化合物(75)と化合物(76)をヒドロキシ基活性化剤(例えば、シアノメチレントリ-n-ブチルホスホラン、アゾジカルボン酸ジイソプロピルとトリフェニルホスフィン、アゾジカルボン酸ジエチルとトリフェニルホスフィン、ADDPとトリブチルホスフィン等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。 When compound (76) is a halide or sulfonate, compound (77) can be produced, for example, by an alkylation reaction of compound (75) and compound (76). The alkylation reaction is carried out, for example, by converting the compound (75) and the compound (76) into a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene). Etc.) in the presence of an inert solvent (for example, DMF, acetonitrile, THF, toluene, water, etc.). A phase transfer catalyst (for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used as necessary.
When compound (76) is an alcohol, compound (77) can be produced, for example, by Mitsunobu reaction between compound (75) and compound (76). Mitsunobu reaction can be carried out, for example, by converting compound (75) and compound (76) into a hydroxy group activator (eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). (Phosphine, ADDP and tributylphosphine, etc.) in the presence of an inert solvent (for example, toluene, THF, etc.).
化合物(76)がアルコールの場合、化合物(77)は、例えば、化合物(75)と化合物(76)の光延反応によって製造することができる。光延反応は、例えば、化合物(75)と化合物(76)をヒドロキシ基活性化剤(例えば、シアノメチレントリ-n-ブチルホスホラン、アゾジカルボン酸ジイソプロピルとトリフェニルホスフィン、アゾジカルボン酸ジエチルとトリフェニルホスフィン、ADDPとトリブチルホスフィン等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。 When compound (76) is a halide or sulfonate, compound (77) can be produced, for example, by an alkylation reaction of compound (75) and compound (76). The alkylation reaction is carried out, for example, by converting the compound (75) and the compound (76) into a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene). Etc.) in the presence of an inert solvent (for example, DMF, acetonitrile, THF, toluene, water, etc.). A phase transfer catalyst (for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used as necessary.
When compound (76) is an alcohol, compound (77) can be produced, for example, by Mitsunobu reaction between compound (75) and compound (76). Mitsunobu reaction can be carried out, for example, by converting compound (75) and compound (76) into a hydroxy group activator (eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). (Phosphine, ADDP and tributylphosphine, etc.) in the presence of an inert solvent (for example, toluene, THF, etc.).
化合物(78)は、例えば、化合物(77)の還元反応によって製造することができる。本反応は、化合物(77)と金属水素化合物(例えば、水素化ジイソブチルアルミニウム)、金属水素錯化合物(例えば、水素化ホウ素ナトリウム、シアノ水素化ホウ素ナトリウム、水素化アルミニウムリチウム、水素化アルミニウムナトリウム、水素化ビス(2-メトキシエトキシ)アルミニウムナトリウム)等の還元剤を不活性溶媒中(例えば、エタノール、メタノール、THF等)で反応させることによって行われる。
Compound (78) can be produced, for example, by a reduction reaction of compound (77). In this reaction, the compound (77) and a metal hydride compound (for example, diisobutylaluminum hydride), a metal hydride complex compound (for example, sodium borohydride, sodium cyanoborohydride, lithium aluminum hydride, sodium aluminum hydride, hydrogen It is carried out by reacting a reducing agent such as bis (2-methoxyethoxy) aluminum chloride) in an inert solvent (for example, ethanol, methanol, THF, etc.).
化合物(1-3)は、例えば、化合物(78)を用いて、例えば、反応式2の化合物(4)から化合物(I-1)を製造する方法と同様にして行われる。
Compound (1-3) is produced, for example, using compound (78), for example, in the same manner as in the production of compound (I-1) from compound (4) in Reaction Scheme 2.
化合物(79)は、例えば、化合物(78)を用いて、例えば、反応式2の化合物(4)から化合物(6)を製造する方法と同様にして行われる。
Compound (79) is produced, for example, using compound (78), for example, in the same manner as in the production of compound (6) from compound (4) in Reaction Scheme 2.
化合物(1-3)は、例えば、化合物(79)を用いて、例えば、反応式2の化合物(6)から化合物(I-1)を製造する方法と同様にして行われる。
Compound (1-3) is produced, for example, using compound (79), for example, in the same manner as in the production of compound (I-1) from compound (6) in Reaction Scheme 2.
<反応式20>
<Reaction Scheme 20>

(式中、各記号は前記と同意義を示す。なお、化合物(4-1)は化合物(4)に包含される。)
(In the formula, each symbol is as defined above. Compound (4-1) is encompassed in compound (4).)
化合物(3)がハライドまたはスルホナートの場合、化合物(81)は、例えば、化合物(80)と化合物(3)のアルキル化反応によって製造することができる。アルキル化反応は、例えば、化合物(80)と化合物(3)を塩基(例えば、炭酸カリウム、水素化ナトリウム、水酸化ナトリウム、DBU(1,8-ジアザビシクロ[5.4.0]ウンデカー7-エン)等)存在下、不活性溶媒(例えば、DMF、アセトニトリル、THF、トルエン、水等)中で反応させることによって行われる。必要に応じて、相間移動触媒(例えば、テトラブチルアンモニウムブロミド、硫酸水素テトラブチルアンモニウム等)を用いてもよい。
化合物(3)がアルコールの場合、化合物(81)は、例えば、化合物(80)と化合物(3)の光延反応によって製造することができる。光延反応は、例えば、化合物(80)と化合物(3)をヒドロキシ基活性化剤(例えば、シアノメチレントリ-n-ブチルホスホラン、アゾジカルボン酸ジイソプロピルとトリフェニルホスフィン、アゾジカルボン酸ジエチルとトリフェニルホスフィン、ADDPとトリブチルホスフィン等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。 When compound (3) is a halide or sulfonate, compound (81) can be produced, for example, by an alkylation reaction of compound (80) and compound (3). The alkylation reaction is carried out, for example, by converting the compound (80) and the compound (3) into a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene). Etc.) in the presence of an inert solvent (for example, DMF, acetonitrile, THF, toluene, water, etc.). A phase transfer catalyst (for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used as necessary.
When compound (3) is an alcohol, compound (81) can be produced, for example, by Mitsunobu reaction between compound (80) and compound (3). Mitsunobu reaction can be achieved, for example, by converting compound (80) and compound (3) into a hydroxy group activator (eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). (Phosphine, ADDP and tributylphosphine, etc.) in the presence of an inert solvent (for example, toluene, THF, etc.).
化合物(3)がアルコールの場合、化合物(81)は、例えば、化合物(80)と化合物(3)の光延反応によって製造することができる。光延反応は、例えば、化合物(80)と化合物(3)をヒドロキシ基活性化剤(例えば、シアノメチレントリ-n-ブチルホスホラン、アゾジカルボン酸ジイソプロピルとトリフェニルホスフィン、アゾジカルボン酸ジエチルとトリフェニルホスフィン、ADDPとトリブチルホスフィン等)存在下、不活性溶媒(例えば、トルエン、THF等)中で反応させることによって行われる。 When compound (3) is a halide or sulfonate, compound (81) can be produced, for example, by an alkylation reaction of compound (80) and compound (3). The alkylation reaction is carried out, for example, by converting the compound (80) and the compound (3) into a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4.0] undeca-7-ene). Etc.) in the presence of an inert solvent (for example, DMF, acetonitrile, THF, toluene, water, etc.). A phase transfer catalyst (for example, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used as necessary.
When compound (3) is an alcohol, compound (81) can be produced, for example, by Mitsunobu reaction between compound (80) and compound (3). Mitsunobu reaction can be achieved, for example, by converting compound (80) and compound (3) into a hydroxy group activator (eg, cyanomethylene tri-n-butylphosphorane, diisopropyl azodicarboxylate and triphenylphosphine, diethyl azodicarboxylate and triphenyl). (Phosphine, ADDP and tributylphosphine, etc.) in the presence of an inert solvent (for example, toluene, THF, etc.).
化合物(4-1)は、例えば、化合物(81)の還元反応によって製造することができる。本反応は、例えば、反応式19の化合物(77)から化合物(78)を製造する方法と同様にして行われる。
Compound (4-1) can be produced, for example, by a reduction reaction of compound (81). This reaction is performed, for example, in the same manner as in the production of compound (78) from compound (77) in Reaction Scheme 19.
化合物(83)は、例えば、化合物(82)のアルコキシ化反応によって製造することができる。アルコキシ化反応は、例えば、化合物(82)と化学式R6-OHで表されるアルコールを塩基(例えば、炭酸カリウム、水素化ナトリウム、水酸化ナトリウム、DBU(1,8-ジアザビシクロ[5.4.0]ウンデカー7-エン)等)存在下、不活性溶媒(例えば、DMF、アセトニトリル、THF、トルエン、水等)中で反応させることによって行われる。必要に応じて、化学式R6-OHで表されるアルコールを化合物(82)より過剰に用いても、あるいは相間移動触媒(例えば、テトラブチルアンモニウムブロミド、硫酸水素テトラブチルアンモニウム等)を用いてもよい。
Compound (83) can be produced, for example, by subjecting compound (82) to an alkoxylation reaction. In the alkoxylation reaction, for example, the compound (82) and an alcohol represented by the chemical formula R 6 —OH can be converted into a base (for example, potassium carbonate, sodium hydride, sodium hydroxide, DBU (1,8-diazabicyclo [5.4. 0] undecaker 7-ene) and the like in the presence of an inert solvent (eg, DMF, acetonitrile, THF, toluene, water, etc.). If necessary, the alcohol represented by the chemical formula R 6 —OH may be used in excess of the compound (82), or a phase transfer catalyst (eg, tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, etc.) may be used. Good.
化合物(81)は、例えば、化合物(83)のホルミル化反応によって製造することもできる。本反応は、例えば、化合物(83)を不活性溶媒(例えば、ジエチルエーテル、トルエン、THF等)中でアルキル金属類(例えば、ブチルリチウム等)あるいは、金属アミド類(例えば、リチウム ジイソプロピルアミド、リチウムヘキサメチルジシラジド等)によって水素原子を金属原子に変換した後に、ホルミル化剤(例えば、DMF、ギ酸エチル等)を、通常-100℃ないし100℃で反応させることによって行われる。
Compound (81) can also be produced, for example, by a formylation reaction of compound (83). In this reaction, for example, compound (83) is reacted with an alkyl metal (eg, butyl lithium) or a metal amide (eg, lithium diisopropylamide, lithium) in an inert solvent (eg, diethyl ether, toluene, THF, etc.). This is carried out by converting a hydrogen atom into a metal atom with hexamethyldisilazide, etc., and then reacting with a formylating agent (eg, DMF, ethyl formate, etc.) usually at −100 ° C. to 100 ° C.
また、化合物(81)は、例えば、化合物(83)のビルスマイヤー反応によっても製造することができる。本反応は、例えば、化合物(83)、塩化ホスホリルおよびDMFを不活性溶媒(例えば、トルエン、THF等)中で、通常-100℃ないし150℃で反応させることによって行われる。
Compound (81) can also be produced, for example, by the Vilsmeier reaction of compound (83). This reaction is carried out, for example, by reacting compound (83), phosphoryl chloride and DMF in an inert solvent (eg, toluene, THF, etc.) usually at −100 ° C. to 150 ° C.
<反応式21>
<Reaction Formula 21>

(式中、各記号は前記と同意義を示す。なお、化合物(5-1)は化合物(5)に包含される。)
(In the formula, each symbol is as defined above. Compound (5-1) is included in compound (5).)
化合物(21)は、例えば、化合物(22)の保護反応によって製造することができる。
Compound (21) can be produced, for example, by subjecting compound (22) to a protection reaction.
化合物(84)は、例えば、化合物(21)の脱保護反応によって製造することができる。
Compound (84) can be produced, for example, by subjecting compound (21) to deprotection.
化合物(85)は、例えば、化合物(84)のアシル化反応によって製造することができる。本反応は、例えば、反応式3の(I-2)を製造する方法と同様にして行われる。
Compound (85) can be produced, for example, by subjecting compound (84) to an acylation reaction. This reaction is performed, for example, in the same manner as in the production of (I-2) in Reaction Scheme 3.
化合物(5-1)は、例えば、化合物(85)の脱保護反応によって製造することができる。
Compound (5-1) can be produced, for example, by subjecting compound (85) to deprotection.
このようにして得られた化合物(I)において、分子内の官能基は、自体公知の化学反応を組み合わせることにより目的の官能基に変換することもできる。ここで、化学反応の例としては、酸化反応、還元反応、アルキル化反応、アシル化反応、ウレア化反応、加水分解反応、アミノ化反応、エステル化反応、アリールカップリング反応、脱保護反応等が挙げられる。
In the compound (I) thus obtained, the functional group in the molecule can be converted to the target functional group by combining a chemical reaction known per se. Here, examples of the chemical reaction include an oxidation reaction, a reduction reaction, an alkylation reaction, an acylation reaction, a urealation reaction, a hydrolysis reaction, an amination reaction, an esterification reaction, an aryl coupling reaction, and a deprotection reaction. Can be mentioned.
上記製造法により得られた化合物(I)は、公知の手段、例えば、溶媒抽出、液性変換、転溶、晶出、再結晶、クロマトグラフィー等によって単離精製することができる。
Compound (I) obtained by the above production method can be isolated and purified by a known means such as solvent extraction, liquid conversion, phase transfer, crystallization, recrystallization, chromatography and the like.
化合物(I)が、光学異性体、立体異性体、位置異性体、回転異性体を含有する場合には、これらも化合物(I)として含有されるとともに、自体公知の合成手法、分離手法によりそれぞれを単品として得ることができる。例えば、化合物(I)に光学異性体が存在する場合には、該化合物から分割された光学異性体も化合物(I)に包含される。
ここで、光学異性体は自体公知の方法により製造することができる。 When compound (I) contains optical isomers, stereoisomers, positional isomers, and rotational isomers, these are also included as compound (I), and are synthesized by known synthesis methods and separation methods, respectively. Can be obtained as a single product. For example, when compound (I) has an optical isomer, the optical isomer resolved from the compound is also encompassed in compound (I).
Here, the optical isomer can be produced by a method known per se.
ここで、光学異性体は自体公知の方法により製造することができる。 When compound (I) contains optical isomers, stereoisomers, positional isomers, and rotational isomers, these are also included as compound (I), and are synthesized by known synthesis methods and separation methods, respectively. Can be obtained as a single product. For example, when compound (I) has an optical isomer, the optical isomer resolved from the compound is also encompassed in compound (I).
Here, the optical isomer can be produced by a method known per se.
化合物(I)は、結晶であってもよい。
化合物(I)の結晶(以下、本発明の結晶と略記することがある)は、化合物(I)に自体公知の結晶化法を適用して、結晶化することによって製造することができる。
本明細書中、融点は、例えば、微量融点測定器(ヤナコ、MP-500D型またはBuchi、B-545型)またはDSC(示差走査熱量分析)装置(SEIKO、EXSTAR6000)等を用いて測定される融点を意味する。
一般に、融点は、測定機器、測定条件等によって変動する場合がある。本明細書中の結晶は、通常の誤差範囲内であれば、本明細書に記載の融点と異なる値を示す結晶であってもよい。
本発明の結晶は、物理化学的性質(例、融点、溶解度、安定性)および生物学的性質(例、体内動態(吸収性、分布、代謝、排泄)、薬効発現)に優れ、医薬として極めて有用である。 Compound (I) may be a crystal.
Crystals of compound (I) (hereinafter sometimes abbreviated as crystals of the present invention) can be produced by crystallization by applying a crystallization method known per se to compound (I).
In the present specification, the melting point is measured using, for example, a trace melting point measuring device (Yanako, MP-500D type or Buchi, B-545 type) or a DSC (differential scanning calorimetry) apparatus (SEIKO, EXSTAR6000). Mean melting point.
In general, the melting point may vary depending on measurement equipment, measurement conditions, and the like. The crystal in the present specification may be a crystal having a value different from the melting point described in the present specification as long as it is within a normal error range.
The crystals of the present invention are excellent in physicochemical properties (eg, melting point, solubility, stability) and biological properties (eg, pharmacokinetics (absorbability, distribution, metabolism, excretion), expression of medicinal properties), and are extremely useful as pharmaceuticals. Useful.
化合物(I)の結晶(以下、本発明の結晶と略記することがある)は、化合物(I)に自体公知の結晶化法を適用して、結晶化することによって製造することができる。
本明細書中、融点は、例えば、微量融点測定器(ヤナコ、MP-500D型またはBuchi、B-545型)またはDSC(示差走査熱量分析)装置(SEIKO、EXSTAR6000)等を用いて測定される融点を意味する。
一般に、融点は、測定機器、測定条件等によって変動する場合がある。本明細書中の結晶は、通常の誤差範囲内であれば、本明細書に記載の融点と異なる値を示す結晶であってもよい。
本発明の結晶は、物理化学的性質(例、融点、溶解度、安定性)および生物学的性質(例、体内動態(吸収性、分布、代謝、排泄)、薬効発現)に優れ、医薬として極めて有用である。 Compound (I) may be a crystal.
Crystals of compound (I) (hereinafter sometimes abbreviated as crystals of the present invention) can be produced by crystallization by applying a crystallization method known per se to compound (I).
In the present specification, the melting point is measured using, for example, a trace melting point measuring device (Yanako, MP-500D type or Buchi, B-545 type) or a DSC (differential scanning calorimetry) apparatus (SEIKO, EXSTAR6000). Mean melting point.
In general, the melting point may vary depending on measurement equipment, measurement conditions, and the like. The crystal in the present specification may be a crystal having a value different from the melting point described in the present specification as long as it is within a normal error range.
The crystals of the present invention are excellent in physicochemical properties (eg, melting point, solubility, stability) and biological properties (eg, pharmacokinetics (absorbability, distribution, metabolism, excretion), expression of medicinal properties), and are extremely useful as pharmaceuticals. Useful.
本発明は、更に以下の実施例、試験例および製剤例によって詳しく説明されるが、これらは本発明を限定するものではなく、また本発明の範囲を逸脱しない範囲で変化させてもよい。
以下の実施例中の「室温」は通常約10℃ないし約35℃を示す。混合溶媒において示した比は、特に断らない限り容量比を示す。%は、特に断らない限り重量%を示す。
シリカゲルカラムクロマトグラフィーにおいて、NHと記載した場合は、アミノプロピルシラン結合シリカゲルを用いた。HPLC(高速液体クロマトグラフィー)において、C18と記載した場合は、オクタデシル結合シリカゲルを用いた。溶出溶媒の比は、特に断らない限り容量比を示す。 The present invention is further explained in detail by the following examples, test examples and formulation examples, which are not intended to limit the present invention, and may be changed without departing from the scope of the present invention.
“Room temperature” in the following examples usually indicates about 10 ° C. to about 35 ° C. The ratio shown in the mixed solvent is a volume ratio unless otherwise specified. % Indicates% by weight unless otherwise specified.
In the silica gel column chromatography, when described as NH, aminopropylsilane-bonded silica gel was used. In HPLC (high performance liquid chromatography), when it was described as C18, octadecyl-bonded silica gel was used. The ratio of elution solvent indicates a volume ratio unless otherwise specified.
以下の実施例中の「室温」は通常約10℃ないし約35℃を示す。混合溶媒において示した比は、特に断らない限り容量比を示す。%は、特に断らない限り重量%を示す。
シリカゲルカラムクロマトグラフィーにおいて、NHと記載した場合は、アミノプロピルシラン結合シリカゲルを用いた。HPLC(高速液体クロマトグラフィー)において、C18と記載した場合は、オクタデシル結合シリカゲルを用いた。溶出溶媒の比は、特に断らない限り容量比を示す。 The present invention is further explained in detail by the following examples, test examples and formulation examples, which are not intended to limit the present invention, and may be changed without departing from the scope of the present invention.
“Room temperature” in the following examples usually indicates about 10 ° C. to about 35 ° C. The ratio shown in the mixed solvent is a volume ratio unless otherwise specified. % Indicates% by weight unless otherwise specified.
In the silica gel column chromatography, when described as NH, aminopropylsilane-bonded silica gel was used. In HPLC (high performance liquid chromatography), when it was described as C18, octadecyl-bonded silica gel was used. The ratio of elution solvent indicates a volume ratio unless otherwise specified.
以下の実施例においては下記の略号を使用する。
mp: 融点
THF: テトラヒドロフラン
DMF: ジメチルホルムアミド
WSC: 1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩
HOBt: 1-ヒドロキシベンゾトリアゾール一水和物
1H NMR(プロトン核磁気共鳴スペクトル)はフーリエ変換型NMRで測定した。解析にはACD/SpecManager(商品名)などを用いた。水酸基やアミノ基などのプロトンが非常に緩やかなピークについては記載していない。
その他の本文中で用いられている略号は下記の意味を示す。
s: シングレット(singlet)
d: ダブレット(doublet)
t: トリプレット(triplet)
q: クァルテット(quartet)
m: マルチプレット(multiplet)
br: ブロード(broad)
J: カップリング定数(coupling constant)
Hz: ヘルツ(Hertz)
CDCl3: 重クロロホルム
DMSO-d6: d6-ジメチルスルホキシド
1H-NMR: プロトン核磁気共鳴
TFA: トリフルオロ酢酸 The following abbreviations are used in the following examples.
mp: melting point
THF: tetrahydrofuran
DMF: Dimethylformamide
WSC: 1- (3-Dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride
HOBt: 1-hydroxybenzotriazole monohydrate 1 H NMR (proton nuclear magnetic resonance spectrum) was measured by Fourier transform NMR. For the analysis, ACD / SpecManager (trade name) was used. Peaks with very gentle protons such as hydroxyl groups and amino groups are not described.
Other abbreviations used in the text have the following meanings.
s: singlet
d: Doublet
t: triplet
q: quartet
m: multiplet
br: broad
J: coupling constant
Hz: Hertz
CDCl 3 : Deuterated chloroform
DMSO-d 6 : d 6 -dimethyl sulfoxide
1 H-NMR: Proton nuclear magnetic resonance
TFA: trifluoroacetic acid
mp: 融点
THF: テトラヒドロフラン
DMF: ジメチルホルムアミド
WSC: 1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩
HOBt: 1-ヒドロキシベンゾトリアゾール一水和物
1H NMR(プロトン核磁気共鳴スペクトル)はフーリエ変換型NMRで測定した。解析にはACD/SpecManager(商品名)などを用いた。水酸基やアミノ基などのプロトンが非常に緩やかなピークについては記載していない。
その他の本文中で用いられている略号は下記の意味を示す。
s: シングレット(singlet)
d: ダブレット(doublet)
t: トリプレット(triplet)
q: クァルテット(quartet)
m: マルチプレット(multiplet)
br: ブロード(broad)
J: カップリング定数(coupling constant)
Hz: ヘルツ(Hertz)
CDCl3: 重クロロホルム
DMSO-d6: d6-ジメチルスルホキシド
1H-NMR: プロトン核磁気共鳴
TFA: トリフルオロ酢酸 The following abbreviations are used in the following examples.
mp: melting point
THF: tetrahydrofuran
DMF: Dimethylformamide
WSC: 1- (3-Dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride
HOBt: 1-hydroxybenzotriazole monohydrate 1 H NMR (proton nuclear magnetic resonance spectrum) was measured by Fourier transform NMR. For the analysis, ACD / SpecManager (trade name) was used. Peaks with very gentle protons such as hydroxyl groups and amino groups are not described.
Other abbreviations used in the text have the following meanings.
s: singlet
d: Doublet
t: triplet
q: quartet
m: multiplet
br: broad
J: coupling constant
Hz: Hertz
CDCl 3 : Deuterated chloroform
DMSO-d 6 : d 6 -dimethyl sulfoxide
1 H-NMR: Proton nuclear magnetic resonance
TFA: trifluoroacetic acid
MS(マススペクトル)は、LC/MS(液体クロマトグラフ質量分析計)により測定した。イオン化法としては、ESI(ElectroSpray Ionization、エレクトロスプレーイオン化)法、または、APCI(Atomospheric Pressure Chemical Ionization、大気圧化学イオン化)法を用いた。イオン化モードは、ポジティブモード(ESI+)とネガティブモード(ESI-)の両方、または一方を用い、いずれかのデータを記載した。データは実測値(found)を記載した。通常、分子イオンピークが観測されるが、tert-ブトキシカルボニル基(-Boc)を有する化合物の場合、フラグメントイオンとして、tert-ブトキシカルボニル基またはtert-ブチル基が脱離したピークが観測されることもある。また、化合物によっては、フラグメントイオンとして、分子イオンピークにナトリウムイオン(+Na)が付加したピークが観測されることもある。また、水酸基(-OH)を有する化合物の場合、フラグメントイオンとして、H2Oが脱離したピークが観測されることもある。塩の場合は、通常、フリー体の分子イオンピークもしくはフラグメントイオンピークが観測される。
旋光度([α]D)における試料濃度(c)の単位はg/100 mLである。
元素分析値(Anal.)は、計算値(Calcd)と実測値(Found)を記載した。 MS (mass spectrum) was measured by LC / MS (liquid chromatograph mass spectrometer). As an ionization method, an ESI (ElectroSpray Ionization) method or an APCI (Atomospheric Pressure Chemical Ionization) method was used. As the ionization mode, positive mode (ESI +) and / or negative mode (ESI-) was used, and either data was described. The data described the actual measurement (found). Usually, a molecular ion peak is observed, but in the case of a compound having a tert-butoxycarbonyl group (-Boc), a peak from which a tert-butoxycarbonyl group or a tert-butyl group is eliminated should be observed as a fragment ion. There is also. Depending on the compound, a peak in which sodium ion (+ Na) is added to the molecular ion peak may be observed as a fragment ion. In the case of a compound having a hydroxyl group (—OH), a peak from which H 2 O is eliminated may be observed as a fragment ion. In the case of a salt, a free molecular ion peak or a fragment ion peak is usually observed.
The unit of sample concentration (c) in optical rotation ([α] D ) is g / 100 mL.
As the elemental analysis value (Anal.), A calculated value (Calcd) and an actual measurement value (Found) are described.
旋光度([α]D)における試料濃度(c)の単位はg/100 mLである。
元素分析値(Anal.)は、計算値(Calcd)と実測値(Found)を記載した。 MS (mass spectrum) was measured by LC / MS (liquid chromatograph mass spectrometer). As an ionization method, an ESI (ElectroSpray Ionization) method or an APCI (Atomospheric Pressure Chemical Ionization) method was used. As the ionization mode, positive mode (ESI +) and / or negative mode (ESI-) was used, and either data was described. The data described the actual measurement (found). Usually, a molecular ion peak is observed, but in the case of a compound having a tert-butoxycarbonyl group (-Boc), a peak from which a tert-butoxycarbonyl group or a tert-butyl group is eliminated should be observed as a fragment ion. There is also. Depending on the compound, a peak in which sodium ion (+ Na) is added to the molecular ion peak may be observed as a fragment ion. In the case of a compound having a hydroxyl group (—OH), a peak from which H 2 O is eliminated may be observed as a fragment ion. In the case of a salt, a free molecular ion peak or a fragment ion peak is usually observed.
The unit of sample concentration (c) in optical rotation ([α] D ) is g / 100 mL.
As the elemental analysis value (Anal.), A calculated value (Calcd) and an actual measurement value (Found) are described.
実施例1
N-[(1S)-2-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)-1-メチルエチル]アセトアミド Example 1
N-[(1S) -2- (4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] acetamide
N-[(1S)-2-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)-1-メチルエチル]アセトアミド Example 1
N-[(1S) -2- (4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] acetamide
A) (2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル 4-メチルベンゼンスルホナート
tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(11.8 g)のピリジン(45 mL)溶液に氷冷下塩化4-メチルベンゼンスルホニル(12.8 g)を加え、室温で15時間撹拌した。反応混合物に酢酸エチルおよび1 M塩酸を加え、水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(11.8 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.15 (3H, d, J = 6.8 Hz), 1.40 (9H, s), 2.45 (3H, s), 3.74-4.12 (3H, m), 4.57 (1H, brs), 7.35 (2H, d, J = 7.9 Hz), 7.79 (2H, d, J = 8.3 Hz). A) (2S) -2-[(tert-butoxycarbonyl) amino] propyl 4-methylbenzenesulfonate tert-butyl [(1S) -2-hydroxy-1-methylethyl] carbamate (11.8 g) in pyridine (45 To the solution was added 4-methylbenzenesulfonyl chloride (12.8 g) under ice-cooling, and the mixture was stirred at room temperature for 15 hours. Ethyl acetate and 1 M hydrochloric acid were added to the reaction mixture, washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (11.8 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.15 (3H, d, J = 6.8 Hz), 1.40 (9H, s), 2.45 (3H, s), 3.74-4.12 (3H, m), 4.57 (1H, brs), 7.35 (2H, d, J = 7.9 Hz), 7.79 (2H, d, J = 8.3 Hz).
tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(11.8 g)のピリジン(45 mL)溶液に氷冷下塩化4-メチルベンゼンスルホニル(12.8 g)を加え、室温で15時間撹拌した。反応混合物に酢酸エチルおよび1 M塩酸を加え、水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(11.8 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.15 (3H, d, J = 6.8 Hz), 1.40 (9H, s), 2.45 (3H, s), 3.74-4.12 (3H, m), 4.57 (1H, brs), 7.35 (2H, d, J = 7.9 Hz), 7.79 (2H, d, J = 8.3 Hz). A) (2S) -2-[(tert-butoxycarbonyl) amino] propyl 4-methylbenzenesulfonate tert-butyl [(1S) -2-hydroxy-1-methylethyl] carbamate (11.8 g) in pyridine (45 To the solution was added 4-methylbenzenesulfonyl chloride (12.8 g) under ice-cooling, and the mixture was stirred at room temperature for 15 hours. Ethyl acetate and 1 M hydrochloric acid were added to the reaction mixture, washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (11.8 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.15 (3H, d, J = 6.8 Hz), 1.40 (9H, s), 2.45 (3H, s), 3.74-4.12 (3H, m), 4.57 (1H, brs), 7.35 (2H, d, J = 7.9 Hz), 7.79 (2H, d, J = 8.3 Hz).
B) N-{(1S)-2-[4-(ベンジルオキシ)フェノキシ]-1-メチルエチル}アセトアミド
水素化ナトリウム(60%油性, 2.16 g)のDMF(40 mL)懸濁液に4-(ベンジルオキシ)フェノール(8.99 g)のDMF(40 mL)溶液を滴下し、窒素雰囲気下0℃で30分間撹拌した。反応混合物に、(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル 4-メチルベンゼンスルホナート(14.8 g)のDMF(40 mL)溶液を氷冷下で滴下し、窒素雰囲気下85℃で16時間撹拌した。反応混合物を酢酸エチルで希釈し、飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して得られた固体を酢酸エチル(70 mL)に溶解させ、4 M塩化水素/酢酸エチル(100 mL)を加え室温で1.5時間撹拌した。反応混合物を減圧下で濃縮した後、残渣をピリジン(100 mL)に溶解させ、無水酢酸(30 mL)を加え室温で16時間撹拌した。反応混合物を減圧下濃縮し、残渣をジイソプロピルエーテルを用いて洗浄した。得られた固体をNHシリカゲルカラムクロマトグラフィー(THF)で精製して標題化合物(7.36 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.86 (1H, dd, J = 9.2, 3.6 Hz), 3.94 (1H, dd, J = 9.0, 4.1 Hz), 4.30-4.40 (1H, m), 5.02 (2H, s), 5.72 (1H, brs), 6.71-7.02 (4H, m), 7.28-7.49 (5H, m). B) N-{(1S) -2- [4- (Benzyloxy) phenoxy] -1-methylethyl} acetamide To a suspension of sodium hydride (60% oily, 2.16 g) in DMF (40 mL) A solution of (benzyloxy) phenol (8.99 g) in DMF (40 mL) was added dropwise, and the mixture was stirred at 0 ° C. for 30 minutes under a nitrogen atmosphere. To the reaction mixture, a solution of (2S) -2-[(tert-butoxycarbonyl) amino] propyl 4-methylbenzenesulfonate (14.8 g) in DMF (40 mL) was added dropwise under ice-cooling, and the mixture was added at 85 ° C. under a nitrogen atmosphere. For 16 hours. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH silica gel column chromatography (hexane / ethyl acetate). The solid obtained was dissolved in ethyl acetate (70 mL), 4 M hydrogen chloride / ethyl acetate (100 mL) was added, and the mixture was stirred at room temperature for 1.5 hr. did. The reaction mixture was concentrated under reduced pressure, the residue was dissolved in pyridine (100 mL), acetic anhydride (30 mL) was added, and the mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure, and the residue was washed with diisopropyl ether. The obtained solid was purified by NH silica gel column chromatography (THF) to obtain the title compound (7.36 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.86 (1H, dd, J = 9.2, 3.6 Hz), 3.94 (1H, dd, J = 9.0, 4.1 Hz), 4.30-4.40 (1H, m), 5.02 (2H, s), 5.72 (1H, brs), 6.71-7.02 (4H, m), 7.28-7.49 (5H, m).
水素化ナトリウム(60%油性, 2.16 g)のDMF(40 mL)懸濁液に4-(ベンジルオキシ)フェノール(8.99 g)のDMF(40 mL)溶液を滴下し、窒素雰囲気下0℃で30分間撹拌した。反応混合物に、(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル 4-メチルベンゼンスルホナート(14.8 g)のDMF(40 mL)溶液を氷冷下で滴下し、窒素雰囲気下85℃で16時間撹拌した。反応混合物を酢酸エチルで希釈し、飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して得られた固体を酢酸エチル(70 mL)に溶解させ、4 M塩化水素/酢酸エチル(100 mL)を加え室温で1.5時間撹拌した。反応混合物を減圧下で濃縮した後、残渣をピリジン(100 mL)に溶解させ、無水酢酸(30 mL)を加え室温で16時間撹拌した。反応混合物を減圧下濃縮し、残渣をジイソプロピルエーテルを用いて洗浄した。得られた固体をNHシリカゲルカラムクロマトグラフィー(THF)で精製して標題化合物(7.36 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.86 (1H, dd, J = 9.2, 3.6 Hz), 3.94 (1H, dd, J = 9.0, 4.1 Hz), 4.30-4.40 (1H, m), 5.02 (2H, s), 5.72 (1H, brs), 6.71-7.02 (4H, m), 7.28-7.49 (5H, m). B) N-{(1S) -2- [4- (Benzyloxy) phenoxy] -1-methylethyl} acetamide To a suspension of sodium hydride (60% oily, 2.16 g) in DMF (40 mL) A solution of (benzyloxy) phenol (8.99 g) in DMF (40 mL) was added dropwise, and the mixture was stirred at 0 ° C. for 30 minutes under a nitrogen atmosphere. To the reaction mixture, a solution of (2S) -2-[(tert-butoxycarbonyl) amino] propyl 4-methylbenzenesulfonate (14.8 g) in DMF (40 mL) was added dropwise under ice-cooling, and the mixture was added at 85 ° C. under a nitrogen atmosphere. For 16 hours. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH silica gel column chromatography (hexane / ethyl acetate). The solid obtained was dissolved in ethyl acetate (70 mL), 4 M hydrogen chloride / ethyl acetate (100 mL) was added, and the mixture was stirred at room temperature for 1.5 hr. did. The reaction mixture was concentrated under reduced pressure, the residue was dissolved in pyridine (100 mL), acetic anhydride (30 mL) was added, and the mixture was stirred at room temperature for 16 hr. The reaction mixture was concentrated under reduced pressure, and the residue was washed with diisopropyl ether. The obtained solid was purified by NH silica gel column chromatography (THF) to obtain the title compound (7.36 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.86 (1H, dd, J = 9.2, 3.6 Hz), 3.94 (1H, dd, J = 9.0, 4.1 Hz), 4.30-4.40 (1H, m), 5.02 (2H, s), 5.72 (1H, brs), 6.71-7.02 (4H, m), 7.28-7.49 (5H, m).
C) N-[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]アセトアミド
水素雰囲気下、N-{(1S)-2-[4-(ベンジルオキシ)フェノキシ]-1-メチルエチル}アセトアミド(7.36 g)、10%パラジウム-炭素(50%含水、8.0 g)およびメタノール(120 mL)の混合物を室温で1時間撹拌した。濾過により触媒を除去した後、得られた濾液を減圧下濃縮して標題化合物(5.10 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.79-3.98 (2H, m), 4.23-4.46 (1H, m), 4.67 (1H, brs), 5.71 (1H, brs), 6.67-6.87 (4H, m). C) N-[(1S) -2- (4-Hydroxyphenoxy) -1-methylethyl] acetamide Under hydrogen atmosphere, N-{(1S) -2- [4- (benzyloxy) phenoxy] -1-methyl A mixture of ethyl} acetamide (7.36 g), 10% palladium-carbon (containing 50% water, 8.0 g) and methanol (120 mL) was stirred at room temperature for 1 hour. After removing the catalyst by filtration, the obtained filtrate was concentrated under reduced pressure to obtain the title compound (5.10 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.79-3.98 (2H, m), 4.23-4.46 (1H, m), 4.67 ( 1H, brs), 5.71 (1H, brs), 6.67-6.87 (4H, m).
水素雰囲気下、N-{(1S)-2-[4-(ベンジルオキシ)フェノキシ]-1-メチルエチル}アセトアミド(7.36 g)、10%パラジウム-炭素(50%含水、8.0 g)およびメタノール(120 mL)の混合物を室温で1時間撹拌した。濾過により触媒を除去した後、得られた濾液を減圧下濃縮して標題化合物(5.10 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.79-3.98 (2H, m), 4.23-4.46 (1H, m), 4.67 (1H, brs), 5.71 (1H, brs), 6.67-6.87 (4H, m). C) N-[(1S) -2- (4-Hydroxyphenoxy) -1-methylethyl] acetamide Under hydrogen atmosphere, N-{(1S) -2- [4- (benzyloxy) phenoxy] -1-methyl A mixture of ethyl} acetamide (7.36 g), 10% palladium-carbon (containing 50% water, 8.0 g) and methanol (120 mL) was stirred at room temperature for 1 hour. After removing the catalyst by filtration, the obtained filtrate was concentrated under reduced pressure to obtain the title compound (5.10 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.79-3.98 (2H, m), 4.23-4.46 (1H, m), 4.67 ( 1H, brs), 5.71 (1H, brs), 6.67-6.87 (4H, m).
D) [4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール
4-(シクロプロピルメトキシ)-2-フルオロ安息香酸(5.00 g)のTHF(100 mL)溶液にボラン-THF錯体のTHF溶液(1.0 M, 54.7 mL)を氷冷下で加え、3時間撹拌した。反応混合物に水(100 mL)をゆっくり滴下し、酢酸エチルで抽出した。有機層を水、10%炭酸カリウム水溶液および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。この溶液をNHシリカゲルクロマトグラフィー(酢酸エチル)で精製して濃縮した。残渣をヘキサンで固化して、標題化合物(4.08 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.32-0.37 (2H, m), 0.62-0.68 (2H, m), 1.21-1.31 (1H, m), 1.66-1.70 (1H, m), 3.77 (2H, d, J = 6.9 Hz), 4.66 (2H, d, J = 6.6 Hz), 6.58-6.69 (2H, m), 7.23-7.29 (1H, m). D) [4- (Cyclopropylmethoxy) -2-fluorophenyl] methanol 4- (cyclopropylmethoxy) -2-fluorobenzoic acid (5.00 g) in THF (100 mL) solution in borane-THF complex in THF ( 1.0 M, 54.7 mL) was added under ice cooling, and the mixture was stirred for 3 hours. Water (100 mL) was slowly added dropwise to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water, 10% aqueous potassium carbonate solution and saturated brine, and then dried over anhydrous sodium sulfate. The solution was purified by NH silica gel chromatography (ethyl acetate) and concentrated. The residue was solidified with hexane to give the title compound (4.08 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.32-0.37 (2H, m), 0.62-0.68 (2H, m), 1.21-1.31 (1H, m), 1.66-1.70 (1H, m), 3.77 (2H , d, J = 6.9 Hz), 4.66 (2H, d, J = 6.6 Hz), 6.58-6.69 (2H, m), 7.23-7.29 (1H, m).
4-(シクロプロピルメトキシ)-2-フルオロ安息香酸(5.00 g)のTHF(100 mL)溶液にボラン-THF錯体のTHF溶液(1.0 M, 54.7 mL)を氷冷下で加え、3時間撹拌した。反応混合物に水(100 mL)をゆっくり滴下し、酢酸エチルで抽出した。有機層を水、10%炭酸カリウム水溶液および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。この溶液をNHシリカゲルクロマトグラフィー(酢酸エチル)で精製して濃縮した。残渣をヘキサンで固化して、標題化合物(4.08 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.32-0.37 (2H, m), 0.62-0.68 (2H, m), 1.21-1.31 (1H, m), 1.66-1.70 (1H, m), 3.77 (2H, d, J = 6.9 Hz), 4.66 (2H, d, J = 6.6 Hz), 6.58-6.69 (2H, m), 7.23-7.29 (1H, m). D) [4- (Cyclopropylmethoxy) -2-fluorophenyl] methanol 4- (cyclopropylmethoxy) -2-fluorobenzoic acid (5.00 g) in THF (100 mL) solution in borane-THF complex in THF ( 1.0 M, 54.7 mL) was added under ice cooling, and the mixture was stirred for 3 hours. Water (100 mL) was slowly added dropwise to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water, 10% aqueous potassium carbonate solution and saturated brine, and then dried over anhydrous sodium sulfate. The solution was purified by NH silica gel chromatography (ethyl acetate) and concentrated. The residue was solidified with hexane to give the title compound (4.08 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.32-0.37 (2H, m), 0.62-0.68 (2H, m), 1.21-1.31 (1H, m), 1.66-1.70 (1H, m), 3.77 (2H , d, J = 6.9 Hz), 4.66 (2H, d, J = 6.6 Hz), 6.58-6.69 (2H, m), 7.23-7.29 (1H, m).
E) N-[(1S)-2-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)-1-メチルエチル]アセトアミド
窒素雰囲気下、[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール(8.68 g)のTHF(65 mL)溶液に塩化チオニル(3.88 mL)とDMF(1滴)を加え、室温で30分間撹拌した。反応混合物を減圧下濃縮し、得られた残渣をDMF(25 mL)に溶解させた。この溶液を、N-[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]アセトアミド(6.67 g)、炭酸カリウム(8.15 g)およびDMF(40 mL)の混合物に加え、60℃で15時間撹拌した。反応混合物を酢酸エチルで希釈し、飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル)で精製した後、得られた固体を再結晶(ヘキサン/酢酸エチル)し、標題化合物を白色結晶(8.6 g)として得た。
1H NMR (300 MHz, CDCl3) δ 0.26-0.43 (2H, m), 0.56-0.73 (2H, m), 1.19-1.37 (4H, m), 1.98 (3H, s), 3.78 (2H, d, J = 6.8 Hz), 3.82-3.89 (1H, m), 3.89-3.98 (1H, m), 4.27-4.43 (1H, m), 4.98 (2H, s), 5.75 (1H, brs), 6.57-6.75 (2H, m), 6.76-6.97 (4H, m), 7.33 (1H, t, J = 8.5 Hz).
mp 113-116℃
Anal. Calcd for C22H26FNO4: C, 68.20; H, 6.76; N, 3.62. Found: C, 68.27; H, 6.84; N, 3.50. E) N-[(1S) -2- (4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] acetamide [4- (cyclopropyl To a solution of (methoxy) -2-fluorophenyl] methanol (8.68 g) in THF (65 mL) were added thionyl chloride (3.88 mL) and DMF (1 drop), and the mixture was stirred at room temperature for 30 minutes. The reaction mixture was concentrated under reduced pressure, and the resulting residue was dissolved in DMF (25 mL). This solution was added to a mixture of N-[(1S) -2- (4-hydroxyphenoxy) -1-methylethyl] acetamide (6.67 g), potassium carbonate (8.15 g) and DMF (40 mL) at 60 ° C. For 15 hours. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound as white crystals (8.6 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.26-0.43 (2H, m), 0.56-0.73 (2H, m), 1.19-1.37 (4H, m), 1.98 (3H, s), 3.78 (2H, d , J = 6.8 Hz), 3.82-3.89 (1H, m), 3.89-3.98 (1H, m), 4.27-4.43 (1H, m), 4.98 (2H, s), 5.75 (1H, brs), 6.57- 6.75 (2H, m), 6.76-6.97 (4H, m), 7.33 (1H, t, J = 8.5 Hz).
mp 113-116 ℃
Anal. Calcd for C 22 H 26 FNO 4 : C, 68.20; H, 6.76; N, 3.62. Found: C, 68.27; H, 6.84; N, 3.50.
窒素雰囲気下、[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール(8.68 g)のTHF(65 mL)溶液に塩化チオニル(3.88 mL)とDMF(1滴)を加え、室温で30分間撹拌した。反応混合物を減圧下濃縮し、得られた残渣をDMF(25 mL)に溶解させた。この溶液を、N-[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]アセトアミド(6.67 g)、炭酸カリウム(8.15 g)およびDMF(40 mL)の混合物に加え、60℃で15時間撹拌した。反応混合物を酢酸エチルで希釈し、飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル)で精製した後、得られた固体を再結晶(ヘキサン/酢酸エチル)し、標題化合物を白色結晶(8.6 g)として得た。
1H NMR (300 MHz, CDCl3) δ 0.26-0.43 (2H, m), 0.56-0.73 (2H, m), 1.19-1.37 (4H, m), 1.98 (3H, s), 3.78 (2H, d, J = 6.8 Hz), 3.82-3.89 (1H, m), 3.89-3.98 (1H, m), 4.27-4.43 (1H, m), 4.98 (2H, s), 5.75 (1H, brs), 6.57-6.75 (2H, m), 6.76-6.97 (4H, m), 7.33 (1H, t, J = 8.5 Hz).
mp 113-116℃
Anal. Calcd for C22H26FNO4: C, 68.20; H, 6.76; N, 3.62. Found: C, 68.27; H, 6.84; N, 3.50. E) N-[(1S) -2- (4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] acetamide [4- (cyclopropyl To a solution of (methoxy) -2-fluorophenyl] methanol (8.68 g) in THF (65 mL) were added thionyl chloride (3.88 mL) and DMF (1 drop), and the mixture was stirred at room temperature for 30 minutes. The reaction mixture was concentrated under reduced pressure, and the resulting residue was dissolved in DMF (25 mL). This solution was added to a mixture of N-[(1S) -2- (4-hydroxyphenoxy) -1-methylethyl] acetamide (6.67 g), potassium carbonate (8.15 g) and DMF (40 mL) at 60 ° C. For 15 hours. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound as white crystals (8.6 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.26-0.43 (2H, m), 0.56-0.73 (2H, m), 1.19-1.37 (4H, m), 1.98 (3H, s), 3.78 (2H, d , J = 6.8 Hz), 3.82-3.89 (1H, m), 3.89-3.98 (1H, m), 4.27-4.43 (1H, m), 4.98 (2H, s), 5.75 (1H, brs), 6.57- 6.75 (2H, m), 6.76-6.97 (4H, m), 7.33 (1H, t, J = 8.5 Hz).
mp 113-116 ℃
Anal. Calcd for C 22 H 26 FNO 4 : C, 68.20; H, 6.76; N, 3.62. Found: C, 68.27; H, 6.84; N, 3.50.
実施例2
N-{(1S)-2-[(trans-4-{[2-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 2
N-{(1S) -2-[(trans-4-{[2-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[2-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 2
N-{(1S) -2-[(trans-4-{[2-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) 1-(ブロモメチル)-2-クロロ-4-(シクロプロピルメトキシ)ベンゼン
[2-クロロ-4-(シクロプロピルメトキシ)フェニル]メタノール(1.00 g)、三臭化リン(0.532 mL)、およびトルエン(10 mL)の混合物を0℃で2時間撹拌した。反応混合物を0℃の飽和炭酸水素ナトリウム水溶液に注ぎ、酢酸エチルで抽出した。有機層を分離し、水および飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.00 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.24-0.39 (2H, m), 0.47-0.65 (2H, m), 1.12-1.28 (1H, m), 3.85 (2H, d, J = 7.2 Hz), 4.72 (2H, s), 6.92 (1H, dd, J = 8.7, 2.7 Hz), 7.05 (1H, d, J = 2.7 Hz), 7.51 (1H, d, J = 8.7 Hz). A) 1- (Bromomethyl) -2-chloro-4- (cyclopropylmethoxy) benzene [2-chloro-4- (cyclopropylmethoxy) phenyl] methanol (1.00 g), phosphorus tribromide (0.532 mL), and A mixture of toluene (10 mL) was stirred at 0 ° C. for 2 hours. The reaction mixture was poured into a saturated aqueous sodium hydrogen carbonate solution at 0 ° C. and extracted with ethyl acetate. The organic layer was separated, washed with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.00 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.24-0.39 (2H, m), 0.47-0.65 (2H, m), 1.12-1.28 (1H, m), 3.85 (2H, d, J = 7.2 Hz ), 4.72 (2H, s), 6.92 (1H, dd, J = 8.7, 2.7 Hz), 7.05 (1H, d, J = 2.7 Hz), 7.51 (1H, d, J = 8.7 Hz).
[2-クロロ-4-(シクロプロピルメトキシ)フェニル]メタノール(1.00 g)、三臭化リン(0.532 mL)、およびトルエン(10 mL)の混合物を0℃で2時間撹拌した。反応混合物を0℃の飽和炭酸水素ナトリウム水溶液に注ぎ、酢酸エチルで抽出した。有機層を分離し、水および飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.00 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.24-0.39 (2H, m), 0.47-0.65 (2H, m), 1.12-1.28 (1H, m), 3.85 (2H, d, J = 7.2 Hz), 4.72 (2H, s), 6.92 (1H, dd, J = 8.7, 2.7 Hz), 7.05 (1H, d, J = 2.7 Hz), 7.51 (1H, d, J = 8.7 Hz). A) 1- (Bromomethyl) -2-chloro-4- (cyclopropylmethoxy) benzene [2-chloro-4- (cyclopropylmethoxy) phenyl] methanol (1.00 g), phosphorus tribromide (0.532 mL), and A mixture of toluene (10 mL) was stirred at 0 ° C. for 2 hours. The reaction mixture was poured into a saturated aqueous sodium hydrogen carbonate solution at 0 ° C. and extracted with ethyl acetate. The organic layer was separated, washed with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.00 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.24-0.39 (2H, m), 0.47-0.65 (2H, m), 1.12-1.28 (1H, m), 3.85 (2H, d, J = 7.2 Hz ), 4.72 (2H, s), 6.92 (1H, dd, J = 8.7, 2.7 Hz), 7.05 (1H, d, J = 2.7 Hz), 7.51 (1H, d, J = 8.7 Hz).
B) 4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)フェニル アセタート
4-ヒドロキシフェニル アセタート(8.68 g)、tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(10.0 g)およびトリフェニルホスフィン(22.45 g)のトルエン(200 mL)溶液に、アゾジカルボン酸ジイソプロピルのトルエン溶液(1.9 M, 45.1 mL)を滴下して、室温で16 時間撹拌した。反応混合物を減圧下濃縮した後に、残渣をジエチルエーテル中で懸濁させ、沈殿物を濾過により除去した。濾液を濃縮した後に、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)およびNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(10.5 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.11 (3H, d, J = 6.4 Hz), 1.38 (9H, s), 2.23 (3H, s), 3.59-3.97 (3H, m), 6.87 (1H, d, J = 6.8 Hz), 6.88-6.97 (2H, m), 6.99-7.08 (2H, m). B) 4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl} oxy) phenyl acetate 4-hydroxyphenyl acetate (8.68 g), tert-butyl [(1S) -2-hydroxy-1 -Methylethyl] carbamate (10.0 g) and triphenylphosphine (22.45 g) in toluene (200 mL) were added dropwise with a solution of diisopropyl azodicarboxylate in toluene (1.9 M, 45.1 mL) and stirred at room temperature for 16 hours. did. After the reaction mixture was concentrated under reduced pressure, the residue was suspended in diethyl ether and the precipitate was removed by filtration. After the filtrate was concentrated, the residue was purified by silica gel column chromatography (hexane / ethyl acetate) and NH silica gel column chromatography (hexane / ethyl acetate) to give the title compound (10.5 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.11 (3H, d, J = 6.4 Hz), 1.38 (9H, s), 2.23 (3H, s), 3.59-3.97 (3H, m), 6.87 ( 1H, d, J = 6.8 Hz), 6.88-6.97 (2H, m), 6.99-7.08 (2H, m).
4-ヒドロキシフェニル アセタート(8.68 g)、tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(10.0 g)およびトリフェニルホスフィン(22.45 g)のトルエン(200 mL)溶液に、アゾジカルボン酸ジイソプロピルのトルエン溶液(1.9 M, 45.1 mL)を滴下して、室温で16 時間撹拌した。反応混合物を減圧下濃縮した後に、残渣をジエチルエーテル中で懸濁させ、沈殿物を濾過により除去した。濾液を濃縮した後に、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)およびNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(10.5 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.11 (3H, d, J = 6.4 Hz), 1.38 (9H, s), 2.23 (3H, s), 3.59-3.97 (3H, m), 6.87 (1H, d, J = 6.8 Hz), 6.88-6.97 (2H, m), 6.99-7.08 (2H, m). B) 4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl} oxy) phenyl acetate 4-hydroxyphenyl acetate (8.68 g), tert-butyl [(1S) -2-hydroxy-1 -Methylethyl] carbamate (10.0 g) and triphenylphosphine (22.45 g) in toluene (200 mL) were added dropwise with a solution of diisopropyl azodicarboxylate in toluene (1.9 M, 45.1 mL) and stirred at room temperature for 16 hours. did. After the reaction mixture was concentrated under reduced pressure, the residue was suspended in diethyl ether and the precipitate was removed by filtration. After the filtrate was concentrated, the residue was purified by silica gel column chromatography (hexane / ethyl acetate) and NH silica gel column chromatography (hexane / ethyl acetate) to give the title compound (10.5 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.11 (3H, d, J = 6.4 Hz), 1.38 (9H, s), 2.23 (3H, s), 3.59-3.97 (3H, m), 6.87 ( 1H, d, J = 6.8 Hz), 6.88-6.97 (2H, m), 6.99-7.08 (2H, m).
C) tert-ブチル[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]カルバマート
1 M水酸化ナトリウム水溶液(30 mL)を4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)フェニル アセタート(6.00 g)のTHF(10 mL)/メタノール(10 mL)の混合溶液に室温で加えた。反応混合物を室温で2時間撹拌した後に、水で希釈してジイソプロピルエーテルで洗浄した。得られた水溶液を1 M塩酸で中和した後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して標題化合物(4.79 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.09 (3H, d, J = 6.4 Hz), 1.38 (9H, s), 3.58-3.71 (1H, m), 3.71-3.89 (2H, m), 6.60-6.69 (2H, m), 6.70-6.78 (2H, m), 6.81 (1H, d, J = 6.4 Hz), 8.88 (1H, s). C) tert-Butyl [(1S) -2- (4-hydroxyphenoxy) -1-methylethyl] carbamate 1 M aqueous sodium hydroxide solution (30 mL) was added to 4-({(2S) -2-[(tert- To a mixed solution of butoxycarbonyl) amino] propyl} oxy) phenyl acetate (6.00 g) in THF (10 mL) / methanol (10 mL) was added at room temperature. The reaction mixture was stirred at room temperature for 2 hours, then diluted with water and washed with diisopropyl ether. The resulting aqueous solution was neutralized with 1 M hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (4.79 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.09 (3H, d, J = 6.4 Hz), 1.38 (9H, s), 3.58-3.71 (1H, m), 3.71-3.89 (2H, m), 6.60-6.69 (2H, m), 6.70-6.78 (2H, m), 6.81 (1H, d, J = 6.4 Hz), 8.88 (1H, s).
1 M水酸化ナトリウム水溶液(30 mL)を4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)フェニル アセタート(6.00 g)のTHF(10 mL)/メタノール(10 mL)の混合溶液に室温で加えた。反応混合物を室温で2時間撹拌した後に、水で希釈してジイソプロピルエーテルで洗浄した。得られた水溶液を1 M塩酸で中和した後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して標題化合物(4.79 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.09 (3H, d, J = 6.4 Hz), 1.38 (9H, s), 3.58-3.71 (1H, m), 3.71-3.89 (2H, m), 6.60-6.69 (2H, m), 6.70-6.78 (2H, m), 6.81 (1H, d, J = 6.4 Hz), 8.88 (1H, s). C) tert-Butyl [(1S) -2- (4-hydroxyphenoxy) -1-methylethyl] carbamate 1 M aqueous sodium hydroxide solution (30 mL) was added to 4-({(2S) -2-[(tert- To a mixed solution of butoxycarbonyl) amino] propyl} oxy) phenyl acetate (6.00 g) in THF (10 mL) / methanol (10 mL) was added at room temperature. The reaction mixture was stirred at room temperature for 2 hours, then diluted with water and washed with diisopropyl ether. The resulting aqueous solution was neutralized with 1 M hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (4.79 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.09 (3H, d, J = 6.4 Hz), 1.38 (9H, s), 3.58-3.71 (1H, m), 3.71-3.89 (2H, m), 6.60-6.69 (2H, m), 6.70-6.78 (2H, m), 6.81 (1H, d, J = 6.4 Hz), 8.88 (1H, s).
D) tert-ブチル{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマート
tert-ブチル[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]カルバマート(5.0 g)および5%ロジウム-炭素(50%含水、1.0 g)のメタノール(200 mL)懸濁液を水素雰囲気下(5 気圧)60℃で5時間撹拌した。濾過により触媒を除去し、濾液を濃縮してtert-ブチル{(1S)-2-[(4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマートのトランス/シス混合物を得た。この混合物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.03 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.98 (3H, d, J = 6.8 Hz), 1.06-1.28 (4H, m), 1.37 (9H, s), 1.65-1.95 (4H, m), 3.08-3.24 (2H, m), 3.24-3.30 (1H, m), 3.34-3.67 (2H, m), 4.46 (1H, d, J = 4.1 Hz), 6.57 (1H, d, J = 7.9 Hz). D) tert-butyl {(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate tert-butyl [(1S) -2- (4-hydroxyphenoxy) -1-methyl A suspension of ethyl] carbamate (5.0 g) and 5% rhodium-carbon (containing 50% water, 1.0 g) in methanol (200 mL) was stirred under a hydrogen atmosphere (5 atm) at 60 ° C. for 5 hours. The catalyst was removed by filtration and the filtrate was concentrated to give a trans / cis mixture of tert-butyl {(1S) -2-[(4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate. The mixture was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.03 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.98 (3H, d, J = 6.8 Hz), 1.06-1.28 (4H, m), 1.37 (9H, s), 1.65-1.95 (4H, m), 3.08-3.24 (2H, m), 3.24-3.30 (1H, m), 3.34-3.67 (2H, m), 4.46 (1H, d, J = 4.1 Hz), 6.57 (1H, d, J = 7.9 Hz) .
tert-ブチル[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]カルバマート(5.0 g)および5%ロジウム-炭素(50%含水、1.0 g)のメタノール(200 mL)懸濁液を水素雰囲気下(5 気圧)60℃で5時間撹拌した。濾過により触媒を除去し、濾液を濃縮してtert-ブチル{(1S)-2-[(4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマートのトランス/シス混合物を得た。この混合物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.03 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.98 (3H, d, J = 6.8 Hz), 1.06-1.28 (4H, m), 1.37 (9H, s), 1.65-1.95 (4H, m), 3.08-3.24 (2H, m), 3.24-3.30 (1H, m), 3.34-3.67 (2H, m), 4.46 (1H, d, J = 4.1 Hz), 6.57 (1H, d, J = 7.9 Hz). D) tert-butyl {(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate tert-butyl [(1S) -2- (4-hydroxyphenoxy) -1-methyl A suspension of ethyl] carbamate (5.0 g) and 5% rhodium-carbon (containing 50% water, 1.0 g) in methanol (200 mL) was stirred under a hydrogen atmosphere (5 atm) at 60 ° C. for 5 hours. The catalyst was removed by filtration and the filtrate was concentrated to give a trans / cis mixture of tert-butyl {(1S) -2-[(4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate. The mixture was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.03 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.98 (3H, d, J = 6.8 Hz), 1.06-1.28 (4H, m), 1.37 (9H, s), 1.65-1.95 (4H, m), 3.08-3.24 (2H, m), 3.24-3.30 (1H, m), 3.34-3.67 (2H, m), 4.46 (1H, d, J = 4.1 Hz), 6.57 (1H, d, J = 7.9 Hz) .
E) N-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
tert-ブチル{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマート(1.0 g)を酢酸エチル(20 mL)に溶解させ、4 M塩化水素/酢酸エチル(10 mL)を加え室温で3時間撹拌した。反応混合物を減圧下で濃縮した後、残渣をピリジン(20 mL)に溶解させ、無水酢酸(0.345 mL)を加え室温で3時間撹拌した。反応混合物に1 M水酸化ナトリウム水溶液(10 mL)を加え、室温で15時間撹拌した。反応混合物を減圧下濃縮し、残渣を酢酸エチルおよび飽和食塩水で希釈した。有機層を分離し、1 M塩酸、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して標題化合物(0.88 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.92-1.05 (3H, m), 1.06-1.27 (4H, m), 1.60-1.95 (7H, m), 3.02-3.24 (1H, m), 3.24-3.55 (3H, m), 3.70-3.89 (1H, m), 4.48 (1H, brs), 7.64 (1H, d, J = 7.6 Hz). E) N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide tert-butyl {(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate (1.0 g) was dissolved in ethyl acetate (20 mL), 4 M hydrogen chloride / ethyl acetate (10 mL) was added, and the mixture was stirred at room temperature for 3 hr. The reaction mixture was concentrated under reduced pressure, the residue was dissolved in pyridine (20 mL), acetic anhydride (0.345 mL) was added, and the mixture was stirred at room temperature for 3 hr. To the reaction mixture was added 1 M aqueous sodium hydroxide solution (10 mL), and the mixture was stirred at room temperature for 15 hours. The reaction mixture was concentrated under reduced pressure, and the residue was diluted with ethyl acetate and saturated brine. The organic layer was separated, washed with 1 M hydrochloric acid, saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (0.88 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.92-1.05 (3H, m), 1.06-1.27 (4H, m), 1.60-1.95 (7H, m), 3.02-3.24 (1H, m), 3.24 -3.55 (3H, m), 3.70-3.89 (1H, m), 4.48 (1H, brs), 7.64 (1H, d, J = 7.6 Hz).
tert-ブチル{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマート(1.0 g)を酢酸エチル(20 mL)に溶解させ、4 M塩化水素/酢酸エチル(10 mL)を加え室温で3時間撹拌した。反応混合物を減圧下で濃縮した後、残渣をピリジン(20 mL)に溶解させ、無水酢酸(0.345 mL)を加え室温で3時間撹拌した。反応混合物に1 M水酸化ナトリウム水溶液(10 mL)を加え、室温で15時間撹拌した。反応混合物を減圧下濃縮し、残渣を酢酸エチルおよび飽和食塩水で希釈した。有機層を分離し、1 M塩酸、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して標題化合物(0.88 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.92-1.05 (3H, m), 1.06-1.27 (4H, m), 1.60-1.95 (7H, m), 3.02-3.24 (1H, m), 3.24-3.55 (3H, m), 3.70-3.89 (1H, m), 4.48 (1H, brs), 7.64 (1H, d, J = 7.6 Hz). E) N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide tert-butyl {(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate (1.0 g) was dissolved in ethyl acetate (20 mL), 4 M hydrogen chloride / ethyl acetate (10 mL) was added, and the mixture was stirred at room temperature for 3 hr. The reaction mixture was concentrated under reduced pressure, the residue was dissolved in pyridine (20 mL), acetic anhydride (0.345 mL) was added, and the mixture was stirred at room temperature for 3 hr. To the reaction mixture was added 1 M aqueous sodium hydroxide solution (10 mL), and the mixture was stirred at room temperature for 15 hours. The reaction mixture was concentrated under reduced pressure, and the residue was diluted with ethyl acetate and saturated brine. The organic layer was separated, washed with 1 M hydrochloric acid, saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (0.88 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.92-1.05 (3H, m), 1.06-1.27 (4H, m), 1.60-1.95 (7H, m), 3.02-3.24 (1H, m), 3.24 -3.55 (3H, m), 3.70-3.89 (1H, m), 4.48 (1H, brs), 7.64 (1H, d, J = 7.6 Hz).
F) N-{(1S)-2-[(trans-4-{[2-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(217 mg)、1-(ブロモメチル)-2-クロロ-4-(シクロプロピルメトキシ)ベンゼン(556 mg)、硫酸水素テトラブチルアンモニウム(34.2 mg)、50%水酸化ナトリウム水溶液(5 mL)、およびトルエン(10 mL)の混合物を100℃で終夜撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、得られた固体を再結晶(ヘキサン/酢酸エチル)し、標題化合物(86.0 mg)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.22-0.38 (2H, m), 0.46-0.63 (2H, m), 1.01 (3H, d, J = 6.8 Hz), 1.10-1.43 (5H, m), 1.77 (3H, s), 1.80-2.03 (4H, m), 3.07-3.47 (4H, m), 3.72-3.90 (3H, m), 4.45 (2H, s), 6.89 (1H, dd, J = 8.6, 2.4 Hz), 6.99 (1H, d, J = 2.4 Hz), 7.36 (1H, d, J = 8.6 Hz), 7.65 (1H, d, J = 7.9 Hz).
mp 85-86 ℃
Anal. Calcd for C22H32NO4Cl:C,64.46;H,7.87;N,3.42.Found:C,64.51;H,7.88;N,3.41. F) N-{(1S) -2-[(trans-4-{[2-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide N-{(1S ) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (217 mg), 1- (bromomethyl) -2-chloro-4- (cyclopropylmethoxy) benzene (556 mg), A mixture of tetrabutylammonium hydrogen sulfate (34.2 mg), 50% aqueous sodium hydroxide solution (5 mL), and toluene (10 mL) was stirred at 100 ° C. overnight. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound (86.0 mg) as white crystals.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.22-0.38 (2H, m), 0.46-0.63 (2H, m), 1.01 (3H, d, J = 6.8 Hz), 1.10-1.43 (5H, m ), 1.77 (3H, s), 1.80-2.03 (4H, m), 3.07-3.47 (4H, m), 3.72-3.90 (3H, m), 4.45 (2H, s), 6.89 (1H, dd, J = 8.6, 2.4 Hz), 6.99 (1H, d, J = 2.4 Hz), 7.36 (1H, d, J = 8.6 Hz), 7.65 (1H, d, J = 7.9 Hz).
mp 85-86 ℃
Anal.Calcd for C 22 H 32 NO 4 Cl: C, 64.46; H, 7.87; N, 3.42.Found: C, 64.51; H, 7.88; N, 3.41.
N-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(217 mg)、1-(ブロモメチル)-2-クロロ-4-(シクロプロピルメトキシ)ベンゼン(556 mg)、硫酸水素テトラブチルアンモニウム(34.2 mg)、50%水酸化ナトリウム水溶液(5 mL)、およびトルエン(10 mL)の混合物を100℃で終夜撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、得られた固体を再結晶(ヘキサン/酢酸エチル)し、標題化合物(86.0 mg)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.22-0.38 (2H, m), 0.46-0.63 (2H, m), 1.01 (3H, d, J = 6.8 Hz), 1.10-1.43 (5H, m), 1.77 (3H, s), 1.80-2.03 (4H, m), 3.07-3.47 (4H, m), 3.72-3.90 (3H, m), 4.45 (2H, s), 6.89 (1H, dd, J = 8.6, 2.4 Hz), 6.99 (1H, d, J = 2.4 Hz), 7.36 (1H, d, J = 8.6 Hz), 7.65 (1H, d, J = 7.9 Hz).
mp 85-86 ℃
Anal. Calcd for C22H32NO4Cl:C,64.46;H,7.87;N,3.42.Found:C,64.51;H,7.88;N,3.41. F) N-{(1S) -2-[(trans-4-{[2-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide N-{(1S ) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (217 mg), 1- (bromomethyl) -2-chloro-4- (cyclopropylmethoxy) benzene (556 mg), A mixture of tetrabutylammonium hydrogen sulfate (34.2 mg), 50% aqueous sodium hydroxide solution (5 mL), and toluene (10 mL) was stirred at 100 ° C. overnight. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound (86.0 mg) as white crystals.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.22-0.38 (2H, m), 0.46-0.63 (2H, m), 1.01 (3H, d, J = 6.8 Hz), 1.10-1.43 (5H, m ), 1.77 (3H, s), 1.80-2.03 (4H, m), 3.07-3.47 (4H, m), 3.72-3.90 (3H, m), 4.45 (2H, s), 6.89 (1H, dd, J = 8.6, 2.4 Hz), 6.99 (1H, d, J = 2.4 Hz), 7.36 (1H, d, J = 8.6 Hz), 7.65 (1H, d, J = 7.9 Hz).
mp 85-86 ℃
Anal.Calcd for C 22 H 32 NO 4 Cl: C, 64.46; H, 7.87; N, 3.42.Found: C, 64.51; H, 7.88; N, 3.41.
実施例3
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 3
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 3
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) 1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2-フルオロベンゼン
三臭化リン(1.73 mL)を[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール(3.0 g)のジエチルエーテル(50 mL)溶液に室温で滴下し、反応混合物を室温で1時間撹拌した。反応混合物を氷水に注ぎ、ジエチルエーテルで抽出した。有機層を分離して、水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、シリカゲルショートカラム(酢酸エチル)に通した後、減圧下濃縮して標題化合物(4.00 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.19-0.41 (2H, m), 0.48-0.68 (2H, m), 1.13-1.36 (1H, m), 3.84 (2H, d, J = 6.8 Hz), 4.67 (2H, s), 6.68-6.87 (2H, m), 7.24-7.59 (1H, m). A) 1- (Bromomethyl) -4- (cyclopropylmethoxy) -2-fluorobenzene Phosphorus tribromide (1.73 mL) was added to [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol (3.0 g) in diethyl To the ether (50 mL) solution was added dropwise at room temperature, and the reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was poured into ice water and extracted with diethyl ether. The organic layer was separated, washed with water and saturated brine, dried over anhydrous magnesium sulfate, passed through a silica gel short column (ethyl acetate), and concentrated under reduced pressure to give the title compound (4.00 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.19-0.41 (2H, m), 0.48-0.68 (2H, m), 1.13-1.36 (1H, m), 3.84 (2H, d, J = 6.8 Hz ), 4.67 (2H, s), 6.68-6.87 (2H, m), 7.24-7.59 (1H, m).
三臭化リン(1.73 mL)を[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール(3.0 g)のジエチルエーテル(50 mL)溶液に室温で滴下し、反応混合物を室温で1時間撹拌した。反応混合物を氷水に注ぎ、ジエチルエーテルで抽出した。有機層を分離して、水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、シリカゲルショートカラム(酢酸エチル)に通した後、減圧下濃縮して標題化合物(4.00 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.19-0.41 (2H, m), 0.48-0.68 (2H, m), 1.13-1.36 (1H, m), 3.84 (2H, d, J = 6.8 Hz), 4.67 (2H, s), 6.68-6.87 (2H, m), 7.24-7.59 (1H, m). A) 1- (Bromomethyl) -4- (cyclopropylmethoxy) -2-fluorobenzene Phosphorus tribromide (1.73 mL) was added to [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol (3.0 g) in diethyl To the ether (50 mL) solution was added dropwise at room temperature, and the reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was poured into ice water and extracted with diethyl ether. The organic layer was separated, washed with water and saturated brine, dried over anhydrous magnesium sulfate, passed through a silica gel short column (ethyl acetate), and concentrated under reduced pressure to give the title compound (4.00 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.19-0.41 (2H, m), 0.48-0.68 (2H, m), 1.13-1.36 (1H, m), 3.84 (2H, d, J = 6.8 Hz ), 4.67 (2H, s), 6.68-6.87 (2H, m), 7.24-7.59 (1H, m).
B) N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2-フルオロベンゼン(1.37 g)、N-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(570 mg)、50%水酸化ナトリウム水溶液(20 mL)、硫酸水素テトラブチルアンモニウム(70 mg)およびトルエン(40 mL)の混合物を100℃で15時間撹拌した。反応混合物を室温に放冷した後、水およびジエチルエーテルで希釈した。有機層を分離し、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、得られた固体を再結晶(ヘキサン/酢酸エチル)し、標題化合物(350 mg)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.21-0.38 (2H, m), 0.48-0.67 (2H, m), 1.01 (3H, d, J = 6.4 Hz), 1.08-1.38 (5H, m), 1.77 (3H, s), 1.80-1.96 (4H, m), 3.08-3.32 (4H, m), 3.71-3.92 (3H, m), 4.41 (2H, s), 6.61-6.85 (2H, m), 7.29 (1H, t, J = 8.7 Hz), 7.65 (1H, d, J = 8.0 Hz).
mp 93-94 ℃
Anal. Calcd for C22H32NO4F:C,67.15;H,8.20;N,3.56.Found:C,67.10;H,8.30;N,3.54. B) N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 1- (bromomethyl) -4- (cyclopropylmethoxy) -2-fluorobenzene (1.37 g), N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (570 mg), A mixture of 50% aqueous sodium hydroxide solution (20 mL), tetrabutylammonium hydrogen sulfate (70 mg) and toluene (40 mL) was stirred at 100 ° C. for 15 hours. The reaction mixture was allowed to cool to room temperature and then diluted with water and diethyl ether. The organic layer was separated, washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound (350 mg) as white crystals.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.21-0.38 (2H, m), 0.48-0.67 (2H, m), 1.01 (3H, d, J = 6.4 Hz), 1.08-1.38 (5H, m ), 1.77 (3H, s), 1.80-1.96 (4H, m), 3.08-3.32 (4H, m), 3.71-3.92 (3H, m), 4.41 (2H, s), 6.61-6.85 (2H, m ), 7.29 (1H, t, J = 8.7 Hz), 7.65 (1H, d, J = 8.0 Hz).
mp 93-94 ℃
Anal.Calcd for C 22 H 32 NO 4 F: C, 67.15; H, 8.20; N, 3.56.Found: C, 67.10; H, 8.30; N, 3.54.
1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2-フルオロベンゼン(1.37 g)、N-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(570 mg)、50%水酸化ナトリウム水溶液(20 mL)、硫酸水素テトラブチルアンモニウム(70 mg)およびトルエン(40 mL)の混合物を100℃で15時間撹拌した。反応混合物を室温に放冷した後、水およびジエチルエーテルで希釈した。有機層を分離し、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、得られた固体を再結晶(ヘキサン/酢酸エチル)し、標題化合物(350 mg)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.21-0.38 (2H, m), 0.48-0.67 (2H, m), 1.01 (3H, d, J = 6.4 Hz), 1.08-1.38 (5H, m), 1.77 (3H, s), 1.80-1.96 (4H, m), 3.08-3.32 (4H, m), 3.71-3.92 (3H, m), 4.41 (2H, s), 6.61-6.85 (2H, m), 7.29 (1H, t, J = 8.7 Hz), 7.65 (1H, d, J = 8.0 Hz).
mp 93-94 ℃
Anal. Calcd for C22H32NO4F:C,67.15;H,8.20;N,3.56.Found:C,67.10;H,8.30;N,3.54. B) N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 1- (bromomethyl) -4- (cyclopropylmethoxy) -2-fluorobenzene (1.37 g), N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (570 mg), A mixture of 50% aqueous sodium hydroxide solution (20 mL), tetrabutylammonium hydrogen sulfate (70 mg) and toluene (40 mL) was stirred at 100 ° C. for 15 hours. The reaction mixture was allowed to cool to room temperature and then diluted with water and diethyl ether. The organic layer was separated, washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound (350 mg) as white crystals.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.21-0.38 (2H, m), 0.48-0.67 (2H, m), 1.01 (3H, d, J = 6.4 Hz), 1.08-1.38 (5H, m ), 1.77 (3H, s), 1.80-1.96 (4H, m), 3.08-3.32 (4H, m), 3.71-3.92 (3H, m), 4.41 (2H, s), 6.61-6.85 (2H, m ), 7.29 (1H, t, J = 8.7 Hz), 7.65 (1H, d, J = 8.0 Hz).
mp 93-94 ℃
Anal.Calcd for C 22 H 32 NO 4 F: C, 67.15; H, 8.20; N, 3.56.Found: C, 67.10; H, 8.30; N, 3.54.
実施例4
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素((1S)体と(1R)体のラセミ体) Example 4
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea ((1S) and (1R) forms (Racemic)
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素((1S)体と(1R)体のラセミ体) Example 4
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea ((1S) and (1R) forms (Racemic)
A) trans-4-{[4-(シクロプロピルメトキシ)-3-フルオロベンジル]オキシ}シクロヘキサノール
[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール(28.8 g)のジエチルエーテル(500 mL)溶液に三臭化リン(17.54 mL)を0℃で加え、室温で3時間撹拌した。反応混合物を氷水に加え、ジエチルエーテルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、シリカゲルショートカラム(酢酸エチル)に通した後、減圧下濃縮した。得られた残渣(38.0 g)、trans-4-{[tert-ブチル(ジメチル)シリル]オキシ}シクロヘキサノール(17.5 g)、硫酸水素テトラブチルアンモニウム(2.58 g)、トルエン(500 mL)および50%水酸化ナトリウム水溶液(250 mL)の混合物を100℃で終夜撹拌した。放冷した後に、反応混合物に水を加え酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(5.09 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.27-0.34 (2H, m), 0.52-0.61 (2H, m),1.07-1.29 (5H,m), 1.70-1.81 (2H, m), 1.86-1.97 (2H, m), 3.23-3.29 (1H, m), 3.36-3.49 (1H, m), 3.81 (2H, d, J = 7.2 Hz), 4.41 (2H, s), 4.47 (1H, d, J = 4.2 Hz), 6.61-6.88 (2H, m), 7.28 (1H, t, J = 8.9 Hz). A) trans-4-{[4- (cyclopropylmethoxy) -3-fluorobenzyl] oxy} cyclohexanol [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol (28.8 g) in diethyl ether (500 mL ) Phosphorus tribromide (17.54 mL) was added to the solution at 0 ° C., and the mixture was stirred at room temperature for 3 hours. The reaction mixture was added to ice water and extracted with diethyl ether. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, passed through a silica gel short column (ethyl acetate), and concentrated under reduced pressure. The resulting residue (38.0 g), trans-4-{[tert-butyl (dimethyl) silyl] oxy} cyclohexanol (17.5 g), tetrabutylammonium hydrogen sulfate (2.58 g), toluene (500 mL) and 50% A mixture of aqueous sodium hydroxide (250 mL) was stirred at 100 ° C. overnight. After allowing to cool, water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (5.09 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.27-0.34 (2H, m), 0.52-0.61 (2H, m), 1.07-1.29 (5H, m), 1.70-1.81 (2H, m), 1.86 -1.97 (2H, m), 3.23-3.29 (1H, m), 3.36-3.49 (1H, m), 3.81 (2H, d, J = 7.2 Hz), 4.41 (2H, s), 4.47 (1H, d , J = 4.2 Hz), 6.61-6.88 (2H, m), 7.28 (1H, t, J = 8.9 Hz).
[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール(28.8 g)のジエチルエーテル(500 mL)溶液に三臭化リン(17.54 mL)を0℃で加え、室温で3時間撹拌した。反応混合物を氷水に加え、ジエチルエーテルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、シリカゲルショートカラム(酢酸エチル)に通した後、減圧下濃縮した。得られた残渣(38.0 g)、trans-4-{[tert-ブチル(ジメチル)シリル]オキシ}シクロヘキサノール(17.5 g)、硫酸水素テトラブチルアンモニウム(2.58 g)、トルエン(500 mL)および50%水酸化ナトリウム水溶液(250 mL)の混合物を100℃で終夜撹拌した。放冷した後に、反応混合物に水を加え酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(5.09 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.27-0.34 (2H, m), 0.52-0.61 (2H, m),1.07-1.29 (5H,m), 1.70-1.81 (2H, m), 1.86-1.97 (2H, m), 3.23-3.29 (1H, m), 3.36-3.49 (1H, m), 3.81 (2H, d, J = 7.2 Hz), 4.41 (2H, s), 4.47 (1H, d, J = 4.2 Hz), 6.61-6.88 (2H, m), 7.28 (1H, t, J = 8.9 Hz). A) trans-4-{[4- (cyclopropylmethoxy) -3-fluorobenzyl] oxy} cyclohexanol [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol (28.8 g) in diethyl ether (500 mL ) Phosphorus tribromide (17.54 mL) was added to the solution at 0 ° C., and the mixture was stirred at room temperature for 3 hours. The reaction mixture was added to ice water and extracted with diethyl ether. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, passed through a silica gel short column (ethyl acetate), and concentrated under reduced pressure. The resulting residue (38.0 g), trans-4-{[tert-butyl (dimethyl) silyl] oxy} cyclohexanol (17.5 g), tetrabutylammonium hydrogen sulfate (2.58 g), toluene (500 mL) and 50% A mixture of aqueous sodium hydroxide (250 mL) was stirred at 100 ° C. overnight. After allowing to cool, water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (5.09 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.27-0.34 (2H, m), 0.52-0.61 (2H, m), 1.07-1.29 (5H, m), 1.70-1.81 (2H, m), 1.86 -1.97 (2H, m), 3.23-3.29 (1H, m), 3.36-3.49 (1H, m), 3.81 (2H, d, J = 7.2 Hz), 4.41 (2H, s), 4.47 (1H, d , J = 4.2 Hz), 6.61-6.88 (2H, m), 7.28 (1H, t, J = 8.9 Hz).
B) 1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オン
trans-4-{[4-(シクロプロピルメトキシ)-3-フルオロベンジル]オキシ}シクロヘキサノール(4.5 g)およびカリウム tert-ブトキシド(5.15 g)のTHF(75 mL)懸濁液に4-(クロロアセチル)モルホリン(5.97 mL)を室温で加え、60℃で終夜撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し油状物(7.66 g)を得た。得られた油状物の一部(40 mg)のTHF(0.4 mL)溶液をメチルマグネシウムブロミドのTHF溶液(1.0 M, 2.0 mL)に窒素雰囲気下室温で加え、室温で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し標題化合物(24.6 mg)を得た。
MS (ESI-): [M-H]- 349.0. B) 1-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-one trans-4-{[4- (cyclopropylmethoxy)- 4- (Chloroacetyl) morpholine (5.97 mL) was added to a suspension of 3-fluorobenzyl] oxy} cyclohexanol (4.5 g) and potassium tert-butoxide (5.15 g) in THF (75 mL) at room temperature. And stirred overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give an oil (7.66 g). A solution of a portion (40 mg) of the obtained oily substance in THF (0.4 mL) was added to a THF solution of methylmagnesium bromide (1.0 M, 2.0 mL) at room temperature under a nitrogen atmosphere, and the mixture was stirred at room temperature for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (24.6 mg).
MS (ESI-): [MH] - 349.0.
trans-4-{[4-(シクロプロピルメトキシ)-3-フルオロベンジル]オキシ}シクロヘキサノール(4.5 g)およびカリウム tert-ブトキシド(5.15 g)のTHF(75 mL)懸濁液に4-(クロロアセチル)モルホリン(5.97 mL)を室温で加え、60℃で終夜撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し油状物(7.66 g)を得た。得られた油状物の一部(40 mg)のTHF(0.4 mL)溶液をメチルマグネシウムブロミドのTHF溶液(1.0 M, 2.0 mL)に窒素雰囲気下室温で加え、室温で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し標題化合物(24.6 mg)を得た。
MS (ESI-): [M-H]- 349.0. B) 1-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-one trans-4-{[4- (cyclopropylmethoxy)- 4- (Chloroacetyl) morpholine (5.97 mL) was added to a suspension of 3-fluorobenzyl] oxy} cyclohexanol (4.5 g) and potassium tert-butoxide (5.15 g) in THF (75 mL) at room temperature. And stirred overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give an oil (7.66 g). A solution of a portion (40 mg) of the obtained oily substance in THF (0.4 mL) was added to a THF solution of methylmagnesium bromide (1.0 M, 2.0 mL) at room temperature under a nitrogen atmosphere, and the mixture was stirred at room temperature for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (24.6 mg).
MS (ESI-): [MH] - 349.0.
C) 1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オール
1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オン(1.07 g)をTHF(15 mL)/メタノール(7.5 mL)の混合溶液に溶解させた後水素化ホウ素ナトリウム(0.116 g)を加え、室温で3時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(0.520 g)を得た。
MS (ESI-): [M-H]- 351.1. C) 1-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-ol 1-[(trans-4-{[4- (cyclo Propylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-one (1.07 g) was dissolved in a mixed solution of THF (15 mL) / methanol (7.5 mL), and then sodium borohydride (0.116 g) was added and stirred at room temperature for 3 hours. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (0.520 g).
MS (ESI-): [MH] - 351.1.
1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オン(1.07 g)をTHF(15 mL)/メタノール(7.5 mL)の混合溶液に溶解させた後水素化ホウ素ナトリウム(0.116 g)を加え、室温で3時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(0.520 g)を得た。
MS (ESI-): [M-H]- 351.1. C) 1-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-ol 1-[(trans-4-{[4- (cyclo Propylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-one (1.07 g) was dissolved in a mixed solution of THF (15 mL) / methanol (7.5 mL), and then sodium borohydride (0.116 g) was added and stirred at room temperature for 3 hours. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (0.520 g).
MS (ESI-): [MH] - 351.1.
D) 1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素
1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オール(500 mg)およびトリエチルアミン(9.5 mL)のTHF(9.5 mL)溶液に、塩化メタンスルホニル(0.165 mL)を室温で加え、2時間撹拌した。析出物を濾別し、濾液を飽和炭酸水素ナトリウム水溶液で中和した後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣(645 mg)をDMF(4.7 mL)に溶解させ、アジ化ナトリウム(0.462 g)を室温で加え、80℃で3時間撹拌した。減圧下溶媒を留去した後、水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣(536 mg)およびトリフェニルホスフィン(520 mg)をTHF(3 mL)/水(1 mL)の混合溶媒に室温で加え、60℃で3時間撹拌した。減圧下溶媒を留去した後、水を加え、酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣(220 mg)およびトリエチルアミン(0.174 mL)のTHF(5 mL)溶液にイソシアナト(トリメチル)シラン(0.096 mL)を室温で加え、3時間撹拌した。撹拌後、再度イソシアナト(トリメチル)シラン(0.050 mL)を加え、室温で終夜撹拌した。溶媒を減圧下留去した後、得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(197 mg)を得た。
MS (ESI+): [M+H]+ 395.3. D) 1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea 1-[(trans-4- { To a solution of [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-ol (500 mg) and triethylamine (9.5 mL) in THF (9.5 mL) was added methanesulfonyl chloride (0.165 mL). ) Was added at room temperature and stirred for 2 hours. The precipitate was filtered off, and the filtrate was neutralized with a saturated aqueous sodium hydrogen carbonate solution and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue (645 mg) was dissolved in DMF (4.7 mL), sodium azide (0.462 g) was added at room temperature, and the mixture was stirred at 80 ° C. for 3 hr. After evaporating the solvent under reduced pressure, water was added and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue (536 mg) and triphenylphosphine (520 mg) were added to a mixed solvent of THF (3 mL) / water (1 mL) at room temperature, and the mixture was stirred at 60 ° C. for 3 hours. After evaporating the solvent under reduced pressure, water was added and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. To a solution of the obtained residue (220 mg) and triethylamine (0.174 mL) in THF (5 mL) was added isocyanato (trimethyl) silane (0.096 mL) at room temperature, and the mixture was stirred for 3 hours. After stirring, isocyanato (trimethyl) silane (0.050 mL) was added again and stirred overnight at room temperature. The solvent was evaporated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (197 mg).
MS (ESI +): [M + H] + 395.3.
1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オール(500 mg)およびトリエチルアミン(9.5 mL)のTHF(9.5 mL)溶液に、塩化メタンスルホニル(0.165 mL)を室温で加え、2時間撹拌した。析出物を濾別し、濾液を飽和炭酸水素ナトリウム水溶液で中和した後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣(645 mg)をDMF(4.7 mL)に溶解させ、アジ化ナトリウム(0.462 g)を室温で加え、80℃で3時間撹拌した。減圧下溶媒を留去した後、水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣(536 mg)およびトリフェニルホスフィン(520 mg)をTHF(3 mL)/水(1 mL)の混合溶媒に室温で加え、60℃で3時間撹拌した。減圧下溶媒を留去した後、水を加え、酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣(220 mg)およびトリエチルアミン(0.174 mL)のTHF(5 mL)溶液にイソシアナト(トリメチル)シラン(0.096 mL)を室温で加え、3時間撹拌した。撹拌後、再度イソシアナト(トリメチル)シラン(0.050 mL)を加え、室温で終夜撹拌した。溶媒を減圧下留去した後、得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(197 mg)を得た。
MS (ESI+): [M+H]+ 395.3. D) 1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea 1-[(trans-4- { To a solution of [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-ol (500 mg) and triethylamine (9.5 mL) in THF (9.5 mL) was added methanesulfonyl chloride (0.165 mL). ) Was added at room temperature and stirred for 2 hours. The precipitate was filtered off, and the filtrate was neutralized with a saturated aqueous sodium hydrogen carbonate solution and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue (645 mg) was dissolved in DMF (4.7 mL), sodium azide (0.462 g) was added at room temperature, and the mixture was stirred at 80 ° C. for 3 hr. After evaporating the solvent under reduced pressure, water was added and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue (536 mg) and triphenylphosphine (520 mg) were added to a mixed solvent of THF (3 mL) / water (1 mL) at room temperature, and the mixture was stirred at 60 ° C. for 3 hours. After evaporating the solvent under reduced pressure, water was added and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. To a solution of the obtained residue (220 mg) and triethylamine (0.174 mL) in THF (5 mL) was added isocyanato (trimethyl) silane (0.096 mL) at room temperature, and the mixture was stirred for 3 hours. After stirring, isocyanato (trimethyl) silane (0.050 mL) was added again and stirred overnight at room temperature. The solvent was evaporated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (197 mg).
MS (ESI +): [M + H] + 395.3.
実施例5a
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素の光学活性体
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素の(1S)体と(1R)体のラセミ体(194 mg)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=80:20)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(69.0 mg)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.27-0.34 (2H, m), 0.52-0.61 (2H, m), 1.00 (3H, d, J = 6.4 Hz), 1.12-1.34 (5H, m), 1.82-1.97 (4H, m), 3.13-3.27 (2H, m), 3.27-3.41 (2H, m), 3.63 (1H, s), 3.81 (2H, d, J = 7.2 Hz), 4.41 (2H, s), 5.38 (2H, s), 5.73 (1H, d, J = 8.3 Hz), 6.70-6.81 (2H, m), 7.24-7.34 (1H, m).
mp 142-143 ℃
Anal. Calcd for C21H31N2O4F: C, 63.94; H, 7.92; N, 7.10. Found: C, 63.85; H, 8.01; N, 7.08.
保持時間 6.802 分
光学純度 >99.9%ee. Example 5a
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea optically active substance 1- {2-[( trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea (1S) and (1R) racemates (194 mg) Fractionated by HPLC (column: CHIRALPAK AD (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 80: 20) Recrystallization gave the title compound (69.0 mg) as white crystals.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.27-0.34 (2H, m), 0.52-0.61 (2H, m), 1.00 (3H, d, J = 6.4 Hz), 1.12-1.34 (5H, m ), 1.82-1.97 (4H, m), 3.13-3.27 (2H, m), 3.27-3.41 (2H, m), 3.63 (1H, s), 3.81 (2H, d, J = 7.2 Hz), 4.41 ( 2H, s), 5.38 (2H, s), 5.73 (1H, d, J = 8.3 Hz), 6.70-6.81 (2H, m), 7.24-7.34 (1H, m).
mp 142-143 ℃
Anal.Calcd for C 21 H 31 N 2 O 4 F: C, 63.94; H, 7.92; N, 7.10. Found: C, 63.85; H, 8.01; N, 7.08.
Retention time 6.802 Spectroscopy> 99.9% ee.
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素の光学活性体
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素の(1S)体と(1R)体のラセミ体(194 mg)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=80:20)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(69.0 mg)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.27-0.34 (2H, m), 0.52-0.61 (2H, m), 1.00 (3H, d, J = 6.4 Hz), 1.12-1.34 (5H, m), 1.82-1.97 (4H, m), 3.13-3.27 (2H, m), 3.27-3.41 (2H, m), 3.63 (1H, s), 3.81 (2H, d, J = 7.2 Hz), 4.41 (2H, s), 5.38 (2H, s), 5.73 (1H, d, J = 8.3 Hz), 6.70-6.81 (2H, m), 7.24-7.34 (1H, m).
mp 142-143 ℃
Anal. Calcd for C21H31N2O4F: C, 63.94; H, 7.92; N, 7.10. Found: C, 63.85; H, 8.01; N, 7.08.
保持時間 6.802 分
光学純度 >99.9%ee. Example 5a
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea optically active substance 1- {2-[( trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea (1S) and (1R) racemates (194 mg) Fractionated by HPLC (column: CHIRALPAK AD (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 80: 20) Recrystallization gave the title compound (69.0 mg) as white crystals.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.27-0.34 (2H, m), 0.52-0.61 (2H, m), 1.00 (3H, d, J = 6.4 Hz), 1.12-1.34 (5H, m ), 1.82-1.97 (4H, m), 3.13-3.27 (2H, m), 3.27-3.41 (2H, m), 3.63 (1H, s), 3.81 (2H, d, J = 7.2 Hz), 4.41 ( 2H, s), 5.38 (2H, s), 5.73 (1H, d, J = 8.3 Hz), 6.70-6.81 (2H, m), 7.24-7.34 (1H, m).
mp 142-143 ℃
Anal.Calcd for C 21 H 31 N 2 O 4 F: C, 63.94; H, 7.92; N, 7.10. Found: C, 63.85; H, 8.01; N, 7.08.
Retention time 6.802 Spectroscopy> 99.9% ee.
実施例5b
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素の光学活性体
実施例5aと同様にして、1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素の(1S)体と(1R)体のラセミ体(194 mg)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=80:20)にて分取し、保持時間がより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(63.0 mg)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.27-0.35 (2H, m), 0.52-0.60 (2H, m), 1.00 (3H, d, J = 6.8 Hz), 1.12-1.35 (5H, m), 1.81-1.97 (4H, m), 3.14-3.27 (2H, m), 3.27-3.42 (2H, m), 3.54-3.72(1H, m), 3.81 (2H, d, J=7.2 Hz), 4.41 (2H, s), 5.38 (2H, s), 5.73 (1H, d, J=8.0 Hz), 6.68-6.84 (2H, m), 7.21-7.37 (1H, m).
mp 142-143 ℃.
Anal. Calcd for C21H31N2O4F: C, 63.94; H, 7.92; N, 7.10. Found: C, 63.80; H, 8.03; N, 7.06.
保持時間 7.721 分
光学純度 99.6%ee. Example 5b
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea optically active substance In the same manner as in Example 5a , 1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea (1S) and (1R) isomers The racemate (194 mg) was fractionated by HPLC (column: CHIRALPAK AD (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 80: 20). The large compound was recrystallized from hexane / ethyl acetate to give the title compound (63.0 mg) as white crystals.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.27-0.35 (2H, m), 0.52-0.60 (2H, m), 1.00 (3H, d, J = 6.8 Hz), 1.12-1.35 (5H, m ), 1.81-1.97 (4H, m), 3.14-3.27 (2H, m), 3.27-3.42 (2H, m), 3.54-3.72 (1H, m), 3.81 (2H, d, J = 7.2 Hz), 4.41 (2H, s), 5.38 (2H, s), 5.73 (1H, d, J = 8.0 Hz), 6.68-6.84 (2H, m), 7.21-7.37 (1H, m).
mp 142-143 ° C.
Anal.Calcd for C 21 H 31 N 2 O 4 F: C, 63.94; H, 7.92; N, 7.10. Found: C, 63.80; H, 8.03; N, 7.06.
Retention time 7.721 Minute optical purity 99.6% ee.
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素の光学活性体
実施例5aと同様にして、1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}尿素の(1S)体と(1R)体のラセミ体(194 mg)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=80:20)にて分取し、保持時間がより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(63.0 mg)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.27-0.35 (2H, m), 0.52-0.60 (2H, m), 1.00 (3H, d, J = 6.8 Hz), 1.12-1.35 (5H, m), 1.81-1.97 (4H, m), 3.14-3.27 (2H, m), 3.27-3.42 (2H, m), 3.54-3.72(1H, m), 3.81 (2H, d, J=7.2 Hz), 4.41 (2H, s), 5.38 (2H, s), 5.73 (1H, d, J=8.0 Hz), 6.68-6.84 (2H, m), 7.21-7.37 (1H, m).
mp 142-143 ℃.
Anal. Calcd for C21H31N2O4F: C, 63.94; H, 7.92; N, 7.10. Found: C, 63.80; H, 8.03; N, 7.06.
保持時間 7.721 分
光学純度 99.6%ee. Example 5b
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea optically active substance In the same manner as in Example 5a , 1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} urea (1S) and (1R) isomers The racemate (194 mg) was fractionated by HPLC (column: CHIRALPAK AD (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 80: 20). The large compound was recrystallized from hexane / ethyl acetate to give the title compound (63.0 mg) as white crystals.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.27-0.35 (2H, m), 0.52-0.60 (2H, m), 1.00 (3H, d, J = 6.8 Hz), 1.12-1.35 (5H, m ), 1.81-1.97 (4H, m), 3.14-3.27 (2H, m), 3.27-3.42 (2H, m), 3.54-3.72 (1H, m), 3.81 (2H, d, J = 7.2 Hz), 4.41 (2H, s), 5.38 (2H, s), 5.73 (1H, d, J = 8.0 Hz), 6.68-6.84 (2H, m), 7.21-7.37 (1H, m).
mp 142-143 ° C.
Anal.Calcd for C 21 H 31 N 2 O 4 F: C, 63.94; H, 7.92; N, 7.10. Found: C, 63.80; H, 8.03; N, 7.06.
Retention time 7.721 Minute optical purity 99.6% ee.
実施例6
N-[(1S)-2-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]アセトアミド
[4-(シクロプロピルメトキシ)フェニル]メタノールとN-[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]アセトアミドを用いて、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 6
N-[(1S) -2- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] acetamide [4- (cyclopropylmethoxy) phenyl] methanol and N-[( Using 1S) -2- (4-hydroxyphenoxy) -1-methylethyl] acetamide, the title compound was obtained in the same manner as in Step E of Example 1 or a method analogous thereto.
N-[(1S)-2-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]アセトアミド
[4-(シクロプロピルメトキシ)フェニル]メタノールとN-[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]アセトアミドを用いて、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 6
N-[(1S) -2- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] acetamide [4- (cyclopropylmethoxy) phenyl] methanol and N-[( Using 1S) -2- (4-hydroxyphenoxy) -1-methylethyl] acetamide, the title compound was obtained in the same manner as in Step E of Example 1 or a method analogous thereto.
実施例7
N-[(1S)-2-(4-{2-[4-(シクロプロピルメトキシ)フェニル]エチル}フェノキシ)-1-メチルエチル]アセトアミド Example 7
N-[(1S) -2- (4- {2- [4- (cyclopropylmethoxy) phenyl] ethyl} phenoxy) -1-methylethyl] acetamide
N-[(1S)-2-(4-{2-[4-(シクロプロピルメトキシ)フェニル]エチル}フェノキシ)-1-メチルエチル]アセトアミド Example 7
N-[(1S) -2- (4- {2- [4- (cyclopropylmethoxy) phenyl] ethyl} phenoxy) -1-methylethyl] acetamide
A) tert-ブチル[(1S)-2-(4-ヨードフェノキシ)-1-メチルエチル]カルバマート
4-ヨードフェノール(1.27 g)、tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(1.01 g)およびトリフェニルホスフィン(2.27 g)のTHF(20 mL)溶液に、アゾジカルボン酸ジエチルのトルエン溶液(2.2 M, 3.94 mL)を滴下して、50℃で3日間撹拌した。反応混合物を減圧下濃縮した後、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して、標題化合物 (230 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.27 (3H, d, J = 6.8 Hz), 1.45 (9H, s), 3.86 (2H, d, J = 4.5 Hz), 3.94-4.10 (1H, m), 4.41-4.92 (1H, m), 6.66 (2H, d, J = 8.7 Hz), 7.54 (2H, d, J= 9.0 Hz). A) tert-butyl [(1S) -2- (4-iodophenoxy) -1-methylethyl] carbamate 4-iodophenol (1.27 g), tert-butyl [(1S) -2-hydroxy-1-methylethyl ] To a solution of carbamate (1.01 g) and triphenylphosphine (2.27 g) in THF (20 mL) was added dropwise a solution of diethyl azodicarboxylate in toluene (2.2 M, 3.94 mL), and the mixture was stirred at 50 ° C for 3 days. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (230 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.27 (3H, d, J = 6.8 Hz), 1.45 (9H, s), 3.86 (2H, d, J = 4.5 Hz), 3.94-4.10 (1H, m) , 4.41-4.92 (1H, m), 6.66 (2H, d, J = 8.7 Hz), 7.54 (2H, d, J = 9.0 Hz).
4-ヨードフェノール(1.27 g)、tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(1.01 g)およびトリフェニルホスフィン(2.27 g)のTHF(20 mL)溶液に、アゾジカルボン酸ジエチルのトルエン溶液(2.2 M, 3.94 mL)を滴下して、50℃で3日間撹拌した。反応混合物を減圧下濃縮した後、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して、標題化合物 (230 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.27 (3H, d, J = 6.8 Hz), 1.45 (9H, s), 3.86 (2H, d, J = 4.5 Hz), 3.94-4.10 (1H, m), 4.41-4.92 (1H, m), 6.66 (2H, d, J = 8.7 Hz), 7.54 (2H, d, J= 9.0 Hz). A) tert-butyl [(1S) -2- (4-iodophenoxy) -1-methylethyl] carbamate 4-iodophenol (1.27 g), tert-butyl [(1S) -2-hydroxy-1-methylethyl ] To a solution of carbamate (1.01 g) and triphenylphosphine (2.27 g) in THF (20 mL) was added dropwise a solution of diethyl azodicarboxylate in toluene (2.2 M, 3.94 mL), and the mixture was stirred at 50 ° C for 3 days. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (230 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.27 (3H, d, J = 6.8 Hz), 1.45 (9H, s), 3.86 (2H, d, J = 4.5 Hz), 3.94-4.10 (1H, m) , 4.41-4.92 (1H, m), 6.66 (2H, d, J = 8.7 Hz), 7.54 (2H, d, J = 9.0 Hz).
B) N-[(1S)-2-(4-ヨードフェノキシ)-1-メチルエチル]アセトアミド
tert-ブチル[(1S)-2-(4-ヨードフェノキシ)-1-メチルエチル]カルバマート(338 mg)を酢酸エチル(2 mL)に溶解させ、4 M塩化水素/酢酸エチル(3 mL)を加え室温で15分間撹拌した。反応混合物を減圧下で濃縮した後、残渣をピリジン(3 mL)に溶解させ、無水酢酸(1 mL)を加え室温で1時間撹拌した。反応混合物を減圧下濃縮して、残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(249 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.82-4.02 (2H, m), 4.25-4.45 (1H, m), 5.53-5.76 (1H, m), 6.69 (2H, d, J = 9.0 Hz), 7.56 (2H, d, J = 8.7 Hz). B) N-[(1S) -2- (4-Iodophenoxy) -1-methylethyl] acetamide tert-butyl [(1S) -2- (4-iodophenoxy) -1-methylethyl] carbamate (338 mg ) Was dissolved in ethyl acetate (2 mL), 4 M hydrogen chloride / ethyl acetate (3 mL) was added, and the mixture was stirred at room temperature for 15 min. The reaction mixture was concentrated under reduced pressure, the residue was dissolved in pyridine (3 mL), acetic anhydride (1 mL) was added, and the mixture was stirred at room temperature for 1 hr. The reaction mixture was concentrated under reduced pressure, and the residue was purified by NH silica gel column chromatography (hexane / ethyl acetate) to give the title compound (249 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.82-4.02 (2H, m), 4.25-4.45 (1H, m), 5.53- 5.76 (1H, m), 6.69 (2H, d, J = 9.0 Hz), 7.56 (2H, d, J = 8.7 Hz).
tert-ブチル[(1S)-2-(4-ヨードフェノキシ)-1-メチルエチル]カルバマート(338 mg)を酢酸エチル(2 mL)に溶解させ、4 M塩化水素/酢酸エチル(3 mL)を加え室温で15分間撹拌した。反応混合物を減圧下で濃縮した後、残渣をピリジン(3 mL)に溶解させ、無水酢酸(1 mL)を加え室温で1時間撹拌した。反応混合物を減圧下濃縮して、残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(249 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.82-4.02 (2H, m), 4.25-4.45 (1H, m), 5.53-5.76 (1H, m), 6.69 (2H, d, J = 9.0 Hz), 7.56 (2H, d, J = 8.7 Hz). B) N-[(1S) -2- (4-Iodophenoxy) -1-methylethyl] acetamide tert-butyl [(1S) -2- (4-iodophenoxy) -1-methylethyl] carbamate (338 mg ) Was dissolved in ethyl acetate (2 mL), 4 M hydrogen chloride / ethyl acetate (3 mL) was added, and the mixture was stirred at room temperature for 15 min. The reaction mixture was concentrated under reduced pressure, the residue was dissolved in pyridine (3 mL), acetic anhydride (1 mL) was added, and the mixture was stirred at room temperature for 1 hr. The reaction mixture was concentrated under reduced pressure, and the residue was purified by NH silica gel column chromatography (hexane / ethyl acetate) to give the title compound (249 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.30 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 3.82-4.02 (2H, m), 4.25-4.45 (1H, m), 5.53- 5.76 (1H, m), 6.69 (2H, d, J = 9.0 Hz), 7.56 (2H, d, J = 8.7 Hz).
C) N-[(1S)-2-(4-{[4-(ベンジルオキシ)フェニル]エチニル}フェノキシ)-1-メチルエチル]アセトアミド
窒素雰囲気下、N-[(1S)-2-(4-ヨードフェノキシ)-1-メチルエチル]アセトアミド(249 mg)、1-(ベンジルオキシ)-4-エチニルベンゼン(194 mg)、トリエチルアミン(0.32 mL)のTHF(4 mL)溶液に、ビストリフェニルホスフィンジクロロパラジウム(II)(27.2 mg)およびヨウ化銅(I)(14.8 mg)を加え、室温で30分間撹拌した。反応混合物を酢酸エチルで希釈し、水および飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去した。残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し、標題化合物(194 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.32 (3H, d, J = 6.8 Hz), 2.00 (3H, s), 3.97 (2H, qd, J = 9.4, 4.1 Hz), 4.27-4.48 (1H, m), 5.08 (2H, s), 5.68 (1H, brs), 6.87 (2H, d, J = 9.0 Hz), 6.94 (2H, d, J = 8.7 Hz), 7.29-7.51 (9H, m). C) N-[(1S) -2- (4-{[4- (Benzyloxy) phenyl] ethynyl} phenoxy) -1-methylethyl] acetamide N-[(1S) -2- (4 -Iodophenoxy) -1-methylethyl] acetamide (249 mg), 1- (benzyloxy) -4-ethynylbenzene (194 mg), triethylamine (0.32 mL) in THF (4 mL) was added to bistriphenylphosphine dichloromethane. Palladium (II) (27.2 mg) and copper (I) iodide (14.8 mg) were added, and the mixture was stirred at room temperature for 30 minutes. The reaction mixture was diluted with ethyl acetate, washed with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH silica gel column chromatography (hexane / ethyl acetate) to give the title compound (194 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.32 (3H, d, J = 6.8 Hz), 2.00 (3H, s), 3.97 (2H, qd, J = 9.4, 4.1 Hz), 4.27-4.48 (1H, m), 5.08 (2H, s), 5.68 (1H, brs), 6.87 (2H, d, J = 9.0 Hz), 6.94 (2H, d, J = 8.7 Hz), 7.29-7.51 (9H, m).
窒素雰囲気下、N-[(1S)-2-(4-ヨードフェノキシ)-1-メチルエチル]アセトアミド(249 mg)、1-(ベンジルオキシ)-4-エチニルベンゼン(194 mg)、トリエチルアミン(0.32 mL)のTHF(4 mL)溶液に、ビストリフェニルホスフィンジクロロパラジウム(II)(27.2 mg)およびヨウ化銅(I)(14.8 mg)を加え、室温で30分間撹拌した。反応混合物を酢酸エチルで希釈し、水および飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去した。残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し、標題化合物(194 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.32 (3H, d, J = 6.8 Hz), 2.00 (3H, s), 3.97 (2H, qd, J = 9.4, 4.1 Hz), 4.27-4.48 (1H, m), 5.08 (2H, s), 5.68 (1H, brs), 6.87 (2H, d, J = 9.0 Hz), 6.94 (2H, d, J = 8.7 Hz), 7.29-7.51 (9H, m). C) N-[(1S) -2- (4-{[4- (Benzyloxy) phenyl] ethynyl} phenoxy) -1-methylethyl] acetamide N-[(1S) -2- (4 -Iodophenoxy) -1-methylethyl] acetamide (249 mg), 1- (benzyloxy) -4-ethynylbenzene (194 mg), triethylamine (0.32 mL) in THF (4 mL) was added to bistriphenylphosphine dichloromethane. Palladium (II) (27.2 mg) and copper (I) iodide (14.8 mg) were added, and the mixture was stirred at room temperature for 30 minutes. The reaction mixture was diluted with ethyl acetate, washed with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH silica gel column chromatography (hexane / ethyl acetate) to give the title compound (194 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.32 (3H, d, J = 6.8 Hz), 2.00 (3H, s), 3.97 (2H, qd, J = 9.4, 4.1 Hz), 4.27-4.48 (1H, m), 5.08 (2H, s), 5.68 (1H, brs), 6.87 (2H, d, J = 9.0 Hz), 6.94 (2H, d, J = 8.7 Hz), 7.29-7.51 (9H, m).
D) N-[(1S)-2-{4-[2-(4-ヒドロキシフェニル)エチル]フェノキシ}-1-メチルエチル]アセトアミド
N-[(1S)-2-(4-{[4-(ベンジルオキシ)フェニル]エチニル}フェノキシ)-1-メチルエチル]アセトアミドを用い、実施例1の工程Cと同様の方法またはそれに準ずる方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 1.31 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 2.81 (4H, s), 3.93 (2H, qd, J = 10.6, 3.8 Hz), 4.25-4.49 (1H, m), 4.70 (1H, s), 5.74 (1H, brs), 6.74 (2H, d, J = 8.3 Hz), 6.81 (2H, d, J = 8.3 Hz), 7.01 (2H, d, J = 8.3 Hz),7.06 (2H, d, J = 8.7 Hz). D) N-[(1S) -2- {4- [2- (4-hydroxyphenyl) ethyl] phenoxy} -1-methylethyl] acetamide N-[(1S) -2- (4-{[4- Using (benzyloxy) phenyl] ethynyl} phenoxy) -1-methylethyl] acetamide, the title compound was obtained in the same manner as in Step C of Example 1 or a method analogous thereto.
1 H NMR (300 MHz, CDCl 3 ) δ 1.31 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 2.81 (4H, s), 3.93 (2H, qd, J = 10.6, 3.8 Hz) , 4.25-4.49 (1H, m), 4.70 (1H, s), 5.74 (1H, brs), 6.74 (2H, d, J = 8.3 Hz), 6.81 (2H, d, J = 8.3 Hz), 7.01 ( 2H, d, J = 8.3 Hz), 7.06 (2H, d, J = 8.7 Hz).
N-[(1S)-2-(4-{[4-(ベンジルオキシ)フェニル]エチニル}フェノキシ)-1-メチルエチル]アセトアミドを用い、実施例1の工程Cと同様の方法またはそれに準ずる方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 1.31 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 2.81 (4H, s), 3.93 (2H, qd, J = 10.6, 3.8 Hz), 4.25-4.49 (1H, m), 4.70 (1H, s), 5.74 (1H, brs), 6.74 (2H, d, J = 8.3 Hz), 6.81 (2H, d, J = 8.3 Hz), 7.01 (2H, d, J = 8.3 Hz),7.06 (2H, d, J = 8.7 Hz). D) N-[(1S) -2- {4- [2- (4-hydroxyphenyl) ethyl] phenoxy} -1-methylethyl] acetamide N-[(1S) -2- (4-{[4- Using (benzyloxy) phenyl] ethynyl} phenoxy) -1-methylethyl] acetamide, the title compound was obtained in the same manner as in Step C of Example 1 or a method analogous thereto.
1 H NMR (300 MHz, CDCl 3 ) δ 1.31 (3H, d, J = 6.8 Hz), 1.99 (3H, s), 2.81 (4H, s), 3.93 (2H, qd, J = 10.6, 3.8 Hz) , 4.25-4.49 (1H, m), 4.70 (1H, s), 5.74 (1H, brs), 6.74 (2H, d, J = 8.3 Hz), 6.81 (2H, d, J = 8.3 Hz), 7.01 ( 2H, d, J = 8.3 Hz), 7.06 (2H, d, J = 8.7 Hz).
E) N-[(1S)-2-(4-{2-[4-(シクロプロピルメトキシ)フェニル]エチル}フェノキシ)-1-メチルエチル]アセトアミド
N-[(1S)-2-{4-[2-(4-ヒドロキシフェニル)エチル]フェノキシ}-1-メチルエチル]アセトアミド(115 mg)、炭酸カリウム(101 mg)およびDMF(2 mL)の混合物に、(ブロモメチル)シクロプロパン(0.053 mL)を加え、60℃で16時間撹拌した。反応混合物に(ブロモメチル)シクロプロパン(0.053 mL)を加え、60℃で1時間および80℃で2時間撹拌した。反応混合物を酢酸エチルで希釈し、飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、再結晶(ヘキサン/酢酸エチル)を行い、標題化合物(44.2 mg)を得た。 E) N-[(1S) -2- (4- {2- [4- (cyclopropylmethoxy) phenyl] ethyl} phenoxy) -1-methylethyl] acetamide N-[(1S) -2- {4- To a mixture of [2- (4-hydroxyphenyl) ethyl] phenoxy} -1-methylethyl] acetamide (115 mg), potassium carbonate (101 mg) and DMF (2 mL), (bromomethyl) cyclopropane (0.053 mL) And stirred at 60 ° C. for 16 hours. (Bromomethyl) cyclopropane (0.053 mL) was added to the reaction mixture, and the mixture was stirred at 60 ° C. for 1 hour and at 80 ° C. for 2 hours. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH silica gel column chromatography (hexane / ethyl acetate) and recrystallized (hexane / ethyl acetate) to give the title compound (44.2 mg).
N-[(1S)-2-{4-[2-(4-ヒドロキシフェニル)エチル]フェノキシ}-1-メチルエチル]アセトアミド(115 mg)、炭酸カリウム(101 mg)およびDMF(2 mL)の混合物に、(ブロモメチル)シクロプロパン(0.053 mL)を加え、60℃で16時間撹拌した。反応混合物に(ブロモメチル)シクロプロパン(0.053 mL)を加え、60℃で1時間および80℃で2時間撹拌した。反応混合物を酢酸エチルで希釈し、飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、再結晶(ヘキサン/酢酸エチル)を行い、標題化合物(44.2 mg)を得た。 E) N-[(1S) -2- (4- {2- [4- (cyclopropylmethoxy) phenyl] ethyl} phenoxy) -1-methylethyl] acetamide N-[(1S) -2- {4- To a mixture of [2- (4-hydroxyphenyl) ethyl] phenoxy} -1-methylethyl] acetamide (115 mg), potassium carbonate (101 mg) and DMF (2 mL), (bromomethyl) cyclopropane (0.053 mL) And stirred at 60 ° C. for 16 hours. (Bromomethyl) cyclopropane (0.053 mL) was added to the reaction mixture, and the mixture was stirred at 60 ° C. for 1 hour and at 80 ° C. for 2 hours. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH silica gel column chromatography (hexane / ethyl acetate) and recrystallized (hexane / ethyl acetate) to give the title compound (44.2 mg).
実施例8
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルファニル]-1-メチルエチル}アセトアミド Example 8
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfanyl] -1-methylethyl} acetamide
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルファニル]-1-メチルエチル}アセトアミド Example 8
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfanyl] -1-methylethyl} acetamide
A) tert-ブチル{(1S)-2-[(4-ヒドロキシフェニル)スルファニル]-1-メチルエチル}カルバマート
4-スルファニルフェノール(766 mg)と(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル 4-メチルベンゼンスルホナート(1.00 g)と炭酸カリウム(841 mg)のDMF(10 mL)懸濁液を室温で1時間撹拌した。反応混合物に酢酸エチルを加え、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(920 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.20 (3H, d, J = 6.8 Hz), 1.43 (9H, s), 2.82-2.98 (2H, m), 3.75-3.83 (1H, m), 4.62 (1H, brs), 5.94 (1H, brs), 6.75 (2H, d, J = 8.7 Hz), 7.32 (2H, d, J = 8.7 Hz). A) tert-butyl {(1S) -2-[(4-hydroxyphenyl) sulfanyl] -1-methylethyl} carbamate 4-sulfanylphenol (766 mg) and (2S) -2-[(tert-butoxycarbonyl) A suspension of amino] propyl 4-methylbenzenesulfonate (1.00 g) and potassium carbonate (841 mg) in DMF (10 mL) was stirred at room temperature for 1 hour. Ethyl acetate was added to the reaction mixture, washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure and the obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (920 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.20 (3H, d, J = 6.8 Hz), 1.43 (9H, s), 2.82-2.98 (2H, m), 3.75-3.83 (1H, m), 4.62 ( 1H, brs), 5.94 (1H, brs), 6.75 (2H, d, J = 8.7 Hz), 7.32 (2H, d, J = 8.7 Hz).
4-スルファニルフェノール(766 mg)と(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル 4-メチルベンゼンスルホナート(1.00 g)と炭酸カリウム(841 mg)のDMF(10 mL)懸濁液を室温で1時間撹拌した。反応混合物に酢酸エチルを加え、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(920 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.20 (3H, d, J = 6.8 Hz), 1.43 (9H, s), 2.82-2.98 (2H, m), 3.75-3.83 (1H, m), 4.62 (1H, brs), 5.94 (1H, brs), 6.75 (2H, d, J = 8.7 Hz), 7.32 (2H, d, J = 8.7 Hz). A) tert-butyl {(1S) -2-[(4-hydroxyphenyl) sulfanyl] -1-methylethyl} carbamate 4-sulfanylphenol (766 mg) and (2S) -2-[(tert-butoxycarbonyl) A suspension of amino] propyl 4-methylbenzenesulfonate (1.00 g) and potassium carbonate (841 mg) in DMF (10 mL) was stirred at room temperature for 1 hour. Ethyl acetate was added to the reaction mixture, washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure and the obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (920 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.20 (3H, d, J = 6.8 Hz), 1.43 (9H, s), 2.82-2.98 (2H, m), 3.75-3.83 (1H, m), 4.62 ( 1H, brs), 5.94 (1H, brs), 6.75 (2H, d, J = 8.7 Hz), 7.32 (2H, d, J = 8.7 Hz).
B) N-{(1S)-2-[(4-ヒドロキシフェニル)スルファニル]-1-メチルエチル}アセトアミド
tert-ブチル{(1S)-2-[(4-ヒドロキシフェニル)スルファニル]-1-メチルエチル}カルバマート(920 mg)に4 M塩化水素/酢酸エチル(5 mL)を加え、室温で15分間撹拌し、濃縮した。残渣にピリジン(5 mL)と無水酢酸(5 mL)を加え、室温で10分間撹拌した。濃縮後、残渣にメタノール(10 mL)と炭酸カリウム(2.25 g)を加えて室温で15分間撹拌した。反応混合物を1 M塩酸で中和した後、酢酸エチルで抽出し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(730 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.24 (3H, d, J = 6.8 Hz), 1.90 (3H, s), 2.92 (2H, d, J = 6.0 Hz), 4.10-4.19 (1H, m), 5.53 (1H, brs), 6.81 (2H, d, J = 8.7 Hz), 7.35 (2H, d, J = 8.7 Hz), 8.08 (1H, brs). B) N-{(1S) -2-[(4-hydroxyphenyl) sulfanyl] -1-methylethyl} acetamide tert-butyl {(1S) -2-[(4-hydroxyphenyl) sulfanyl] -1-methyl Ethyl} carbamate (920 mg) was added 4 M hydrogen chloride / ethyl acetate (5 mL), stirred at room temperature for 15 minutes, and concentrated. To the residue were added pyridine (5 mL) and acetic anhydride (5 mL), and the mixture was stirred at room temperature for 10 minutes. After concentration, methanol (10 mL) and potassium carbonate (2.25 g) were added to the residue, and the mixture was stirred at room temperature for 15 min. The reaction mixture was neutralized with 1 M hydrochloric acid, extracted with ethyl acetate, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure and the obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (730 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.24 (3H, d, J = 6.8 Hz), 1.90 (3H, s), 2.92 (2H, d, J = 6.0 Hz), 4.10-4.19 (1H, m) , 5.53 (1H, brs), 6.81 (2H, d, J = 8.7 Hz), 7.35 (2H, d, J = 8.7 Hz), 8.08 (1H, brs).
tert-ブチル{(1S)-2-[(4-ヒドロキシフェニル)スルファニル]-1-メチルエチル}カルバマート(920 mg)に4 M塩化水素/酢酸エチル(5 mL)を加え、室温で15分間撹拌し、濃縮した。残渣にピリジン(5 mL)と無水酢酸(5 mL)を加え、室温で10分間撹拌した。濃縮後、残渣にメタノール(10 mL)と炭酸カリウム(2.25 g)を加えて室温で15分間撹拌した。反応混合物を1 M塩酸で中和した後、酢酸エチルで抽出し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(730 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.24 (3H, d, J = 6.8 Hz), 1.90 (3H, s), 2.92 (2H, d, J = 6.0 Hz), 4.10-4.19 (1H, m), 5.53 (1H, brs), 6.81 (2H, d, J = 8.7 Hz), 7.35 (2H, d, J = 8.7 Hz), 8.08 (1H, brs). B) N-{(1S) -2-[(4-hydroxyphenyl) sulfanyl] -1-methylethyl} acetamide tert-butyl {(1S) -2-[(4-hydroxyphenyl) sulfanyl] -1-methyl Ethyl} carbamate (920 mg) was added 4 M hydrogen chloride / ethyl acetate (5 mL), stirred at room temperature for 15 minutes, and concentrated. To the residue were added pyridine (5 mL) and acetic anhydride (5 mL), and the mixture was stirred at room temperature for 10 minutes. After concentration, methanol (10 mL) and potassium carbonate (2.25 g) were added to the residue, and the mixture was stirred at room temperature for 15 min. The reaction mixture was neutralized with 1 M hydrochloric acid, extracted with ethyl acetate, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure and the obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (730 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.24 (3H, d, J = 6.8 Hz), 1.90 (3H, s), 2.92 (2H, d, J = 6.0 Hz), 4.10-4.19 (1H, m) , 5.53 (1H, brs), 6.81 (2H, d, J = 8.7 Hz), 7.35 (2H, d, J = 8.7 Hz), 8.08 (1H, brs).
C) N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルファニル]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-ヒドロキシフェニル)スルファニル]-1-メチルエチル}アセトアミドおよび[4-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfanyl] -1-methylethyl} acetamide N-{(1S) -2-[( The title compound was obtained in the same manner as in Step E of Example 1 or a method analogous thereto using 4-hydroxyphenyl) sulfanyl] -1-methylethyl} acetamide and [4- (cyclopropylmethoxy) phenyl] methanol.
N-{(1S)-2-[(4-ヒドロキシフェニル)スルファニル]-1-メチルエチル}アセトアミドおよび[4-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfanyl] -1-methylethyl} acetamide N-{(1S) -2-[( The title compound was obtained in the same manner as in Step E of Example 1 or a method analogous thereto using 4-hydroxyphenyl) sulfanyl] -1-methylethyl} acetamide and [4- (cyclopropylmethoxy) phenyl] methanol.
実施例9
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルフィニル]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルファニル]-1-メチルエチル}アセトアミド(250 mg)のTHF(5 mL)溶液に3-クロロペルオキシ安息香酸(112 mg)を加えて室温で15分間撹拌した。析出した固体を濾取し、酢酸エチルで洗浄して標題化合物(118 mg)を得た。 Example 9
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfinyl] -1-methylethyl} acetamide N-{(1S) -2-[(4- Add 3-chloroperoxybenzoic acid (112 mg) to a solution of {[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfanyl] -1-methylethyl} acetamide (250 mg) in THF (5 mL) at room temperature. For 15 minutes. The precipitated solid was collected by filtration and washed with ethyl acetate to give the title compound (118 mg).
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルフィニル]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルファニル]-1-メチルエチル}アセトアミド(250 mg)のTHF(5 mL)溶液に3-クロロペルオキシ安息香酸(112 mg)を加えて室温で15分間撹拌した。析出した固体を濾取し、酢酸エチルで洗浄して標題化合物(118 mg)を得た。 Example 9
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfinyl] -1-methylethyl} acetamide N-{(1S) -2-[(4- Add 3-chloroperoxybenzoic acid (112 mg) to a solution of {[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfanyl] -1-methylethyl} acetamide (250 mg) in THF (5 mL) at room temperature. For 15 minutes. The precipitated solid was collected by filtration and washed with ethyl acetate to give the title compound (118 mg).
実施例10
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルホニル]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルファニル]-1-メチルエチル}アセトアミド(250 mg)のTHF(5 mL)溶液に3-クロロペルオキシ安息香酸(280 mg)を加えて室温で15分間撹拌した。反応混合物をNHシリカゲルショートカラム(酢酸エチル)に通して、溶媒を減圧下留去して得られた固体を酢酸エチルで洗浄して標題化合物(223 mg)を得た。 Example 10
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfonyl] -1-methylethyl} acetamide N-{(1S) -2-[(4- Add 3-chloroperoxybenzoic acid (280 mg) to a solution of {[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfanyl] -1-methylethyl} acetamide (250 mg) in THF (5 mL) at room temperature. For 15 minutes. The reaction mixture was passed through a NH silica gel short column (ethyl acetate), the solvent was distilled off under reduced pressure, and the obtained solid was washed with ethyl acetate to obtain the title compound (223 mg).
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルホニル]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)スルファニル]-1-メチルエチル}アセトアミド(250 mg)のTHF(5 mL)溶液に3-クロロペルオキシ安息香酸(280 mg)を加えて室温で15分間撹拌した。反応混合物をNHシリカゲルショートカラム(酢酸エチル)に通して、溶媒を減圧下留去して得られた固体を酢酸エチルで洗浄して標題化合物(223 mg)を得た。 Example 10
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfonyl] -1-methylethyl} acetamide N-{(1S) -2-[(4- Add 3-chloroperoxybenzoic acid (280 mg) to a solution of {[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) sulfanyl] -1-methylethyl} acetamide (250 mg) in THF (5 mL) at room temperature. For 15 minutes. The reaction mixture was passed through a NH silica gel short column (ethyl acetate), the solvent was distilled off under reduced pressure, and the obtained solid was washed with ethyl acetate to obtain the title compound (223 mg).
実施例11
N-[(1S)-3-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)-1-メチルプロピル]アセトアミド Example 11
N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methylpropyl] acetamide
N-[(1S)-3-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)-1-メチルプロピル]アセトアミド Example 11
N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methylpropyl] acetamide
A) 4-[(3S)-3-アミノブチル]フェノール塩酸塩
1-(ベンジルオキシ)-4-ヨードベンゼン(1.90 g)、tert-ブチル [(1S)-1-メチルプロパ-2-イン-1-イル]カルバマート(1.55 g)およびトリエチルアミン(1.85 g)のTHF(30 mL)溶液に、ビストリフェニルホスフィンジクロロパラジウム(II)(215 mg)とヨウ化銅(I)(117 mg)を加え、アルゴン雰囲気下60℃で3時間撹拌した。反応混合物を室温に放冷した後、水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去した。残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して得た固体をエタノール(40 mL)に溶解させ、10%パラジウム-炭素(50%含水、2.0 g)を加え、水素雰囲気下室温で16時間撹拌した。セライト濾過により触媒を除去した後、得られた濾液を減圧下濃縮した。残渣をエタノール(40 mL)溶液に溶解した後に10%パラジウム-炭素(50%含水、2.0 g)を加え、水素雰囲気下室温で5時間撹拌した。セライト濾過して触媒を除去した後、得られた濾液を減圧下濃縮した。得られた油状物に4 M塩化水素/酢酸エチル(40 mL)を加え室温で2時間撹拌した。反応混合物を減圧下濃縮した後、残渣をヘキサンで洗浄して標題化合物(1.37 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.21 (3H, d, J = 6.8 Hz), 1.58-1.74 (1H, m), 1.79-1.91 (1H, m), 2.43-2.63 (2H, m), 3.02-3.17 (1H, m), 6.69 (2H, d, J = 8.7 Hz), 6.99 (2H, d, J = 8.3 Hz), 8.07 (3H, brs), 9.24 (1H, brs). A) 4-[(3S) -3-Aminobutyl] phenol hydrochloride 1- (benzyloxy) -4-iodobenzene (1.90 g), tert-butyl [(1S) -1-methylprop-2-yne-1 To a solution of -yl] carbamate (1.55 g) and triethylamine (1.85 g) in THF (30 mL), bistriphenylphosphinedichloropalladium (II) (215 mg) and copper (I) iodide (117 mg) were added. The mixture was stirred at 60 ° C. for 3 hours under an atmosphere. The reaction mixture was allowed to cool to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH silica gel column chromatography (hexane / ethyl acetate). The solid obtained was dissolved in ethanol (40 mL), 10% palladium-carbon (50% water content, 2.0 g) was added, and the mixture was added under a hydrogen atmosphere at room temperature. For 16 hours. After removing the catalyst by Celite filtration, the obtained filtrate was concentrated under reduced pressure. The residue was dissolved in an ethanol (40 mL) solution, 10% palladium-carbon (containing 50% water, 2.0 g) was added, and the mixture was stirred at room temperature for 5 hours in a hydrogen atmosphere. After removing the catalyst by Celite filtration, the obtained filtrate was concentrated under reduced pressure. To the obtained oil was added 4 M hydrogen chloride / ethyl acetate (40 mL), and the mixture was stirred at room temperature for 2 hr. The reaction mixture was concentrated under reduced pressure, and the residue was washed with hexane to give the title compound (1.37 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.21 (3H, d, J = 6.8 Hz), 1.58-1.74 (1H, m), 1.79-1.91 (1H, m), 2.43-2.63 (2H, m ), 3.02-3.17 (1H, m), 6.69 (2H, d, J = 8.7 Hz), 6.99 (2H, d, J = 8.3 Hz), 8.07 (3H, brs), 9.24 (1H, brs).
1-(ベンジルオキシ)-4-ヨードベンゼン(1.90 g)、tert-ブチル [(1S)-1-メチルプロパ-2-イン-1-イル]カルバマート(1.55 g)およびトリエチルアミン(1.85 g)のTHF(30 mL)溶液に、ビストリフェニルホスフィンジクロロパラジウム(II)(215 mg)とヨウ化銅(I)(117 mg)を加え、アルゴン雰囲気下60℃で3時間撹拌した。反応混合物を室温に放冷した後、水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去した。残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して得た固体をエタノール(40 mL)に溶解させ、10%パラジウム-炭素(50%含水、2.0 g)を加え、水素雰囲気下室温で16時間撹拌した。セライト濾過により触媒を除去した後、得られた濾液を減圧下濃縮した。残渣をエタノール(40 mL)溶液に溶解した後に10%パラジウム-炭素(50%含水、2.0 g)を加え、水素雰囲気下室温で5時間撹拌した。セライト濾過して触媒を除去した後、得られた濾液を減圧下濃縮した。得られた油状物に4 M塩化水素/酢酸エチル(40 mL)を加え室温で2時間撹拌した。反応混合物を減圧下濃縮した後、残渣をヘキサンで洗浄して標題化合物(1.37 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.21 (3H, d, J = 6.8 Hz), 1.58-1.74 (1H, m), 1.79-1.91 (1H, m), 2.43-2.63 (2H, m), 3.02-3.17 (1H, m), 6.69 (2H, d, J = 8.7 Hz), 6.99 (2H, d, J = 8.3 Hz), 8.07 (3H, brs), 9.24 (1H, brs). A) 4-[(3S) -3-Aminobutyl] phenol hydrochloride 1- (benzyloxy) -4-iodobenzene (1.90 g), tert-butyl [(1S) -1-methylprop-2-yne-1 To a solution of -yl] carbamate (1.55 g) and triethylamine (1.85 g) in THF (30 mL), bistriphenylphosphinedichloropalladium (II) (215 mg) and copper (I) iodide (117 mg) were added. The mixture was stirred at 60 ° C. for 3 hours under an atmosphere. The reaction mixture was allowed to cool to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH silica gel column chromatography (hexane / ethyl acetate). The solid obtained was dissolved in ethanol (40 mL), 10% palladium-carbon (50% water content, 2.0 g) was added, and the mixture was added under a hydrogen atmosphere at room temperature. For 16 hours. After removing the catalyst by Celite filtration, the obtained filtrate was concentrated under reduced pressure. The residue was dissolved in an ethanol (40 mL) solution, 10% palladium-carbon (containing 50% water, 2.0 g) was added, and the mixture was stirred at room temperature for 5 hours in a hydrogen atmosphere. After removing the catalyst by Celite filtration, the obtained filtrate was concentrated under reduced pressure. To the obtained oil was added 4 M hydrogen chloride / ethyl acetate (40 mL), and the mixture was stirred at room temperature for 2 hr. The reaction mixture was concentrated under reduced pressure, and the residue was washed with hexane to give the title compound (1.37 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.21 (3H, d, J = 6.8 Hz), 1.58-1.74 (1H, m), 1.79-1.91 (1H, m), 2.43-2.63 (2H, m ), 3.02-3.17 (1H, m), 6.69 (2H, d, J = 8.7 Hz), 6.99 (2H, d, J = 8.3 Hz), 8.07 (3H, brs), 9.24 (1H, brs).
B) N-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミド
4-[(3S)-3-アミノブチル]フェノール塩酸塩(1.37 g)をピリジン(30 mL)に溶解させ、無水酢酸(15 mL)を加え室温で3日間撹拌した。反応混合物を減圧下濃縮して得られた油状物をメタノール(35 mL)に溶解させ、炭酸カリウム(2.82 g)を加え、室温で18時間撹拌した。反応混合物を減圧下濃縮し、残渣に水を加え酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/メタノール)で精製して標題化合物(1.07 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.02 (3H, d, J = 6.8 Hz), 1.51-1.66 (2H, m), 1.79 (3H, s), 2.39-2.47 (2H, m), 3.65-3.79 (1H, m), 6.65 (2H, d, J = 8.3 Hz), 6.96 (2H, d, J = 8.3 Hz), 7.68 (1H, d, J = 8.0 Hz), 9.09 (1H, s). B) N-[(1S) -3- (4-hydroxyphenyl) -1-methylpropyl] acetamide 4-[(3S) -3-aminobutyl] phenol hydrochloride (1.37 g) in pyridine (30 mL) After dissolution, acetic anhydride (15 mL) was added and the mixture was stirred at room temperature for 3 days. The oil obtained by concentrating the reaction mixture under reduced pressure was dissolved in methanol (35 mL), potassium carbonate (2.82 g) was added, and the mixture was stirred at room temperature for 18 hr. The reaction mixture was concentrated under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate / methanol) to give the title compound (1.07 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.02 (3H, d, J = 6.8 Hz), 1.51-1.66 (2H, m), 1.79 (3H, s), 2.39-2.47 (2H, m), 3.65-3.79 (1H, m), 6.65 (2H, d, J = 8.3 Hz), 6.96 (2H, d, J = 8.3 Hz), 7.68 (1H, d, J = 8.0 Hz), 9.09 (1H, s ).
4-[(3S)-3-アミノブチル]フェノール塩酸塩(1.37 g)をピリジン(30 mL)に溶解させ、無水酢酸(15 mL)を加え室温で3日間撹拌した。反応混合物を減圧下濃縮して得られた油状物をメタノール(35 mL)に溶解させ、炭酸カリウム(2.82 g)を加え、室温で18時間撹拌した。反応混合物を減圧下濃縮し、残渣に水を加え酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/メタノール)で精製して標題化合物(1.07 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.02 (3H, d, J = 6.8 Hz), 1.51-1.66 (2H, m), 1.79 (3H, s), 2.39-2.47 (2H, m), 3.65-3.79 (1H, m), 6.65 (2H, d, J = 8.3 Hz), 6.96 (2H, d, J = 8.3 Hz), 7.68 (1H, d, J = 8.0 Hz), 9.09 (1H, s). B) N-[(1S) -3- (4-hydroxyphenyl) -1-methylpropyl] acetamide 4-[(3S) -3-aminobutyl] phenol hydrochloride (1.37 g) in pyridine (30 mL) After dissolution, acetic anhydride (15 mL) was added and the mixture was stirred at room temperature for 3 days. The oil obtained by concentrating the reaction mixture under reduced pressure was dissolved in methanol (35 mL), potassium carbonate (2.82 g) was added, and the mixture was stirred at room temperature for 18 hr. The reaction mixture was concentrated under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate / methanol) to give the title compound (1.07 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.02 (3H, d, J = 6.8 Hz), 1.51-1.66 (2H, m), 1.79 (3H, s), 2.39-2.47 (2H, m), 3.65-3.79 (1H, m), 6.65 (2H, d, J = 8.3 Hz), 6.96 (2H, d, J = 8.3 Hz), 7.68 (1H, d, J = 8.0 Hz), 9.09 (1H, s ).
C) N-[(1S)-3-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)-1-メチルプロピル]アセトアミド
N-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミドおよび[4-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methylpropyl] acetamide N-[(1S) -3- (4-hydroxyphenyl) ) -1-Methylpropyl] acetamide and [4- (cyclopropylmethoxy) phenyl] methanol were used in the same manner as in Step E of Example 1 or a method analogous thereto to give the title compound.
N-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミドおよび[4-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methylpropyl] acetamide N-[(1S) -3- (4-hydroxyphenyl) ) -1-Methylpropyl] acetamide and [4- (cyclopropylmethoxy) phenyl] methanol were used in the same manner as in Step E of Example 1 or a method analogous thereto to give the title compound.
実施例12
N-[(1S)-3-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェニル)-1-メチルプロピル]アセトアミド
N-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミドおよび[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノールを用いて、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 12
N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenyl) -1-methylpropyl] acetamide N-[(1S) -3- (4- Using hydroxyphenyl) -1-methylpropyl] acetamide and [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol, the title compound was obtained in the same manner as in Step E of Example 1 or a method analogous thereto. .
N-[(1S)-3-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェニル)-1-メチルプロピル]アセトアミド
N-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミドおよび[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノールを用いて、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 12
N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenyl) -1-methylpropyl] acetamide N-[(1S) -3- (4- Using hydroxyphenyl) -1-methylpropyl] acetamide and [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol, the title compound was obtained in the same manner as in Step E of Example 1 or a method analogous thereto. .
実施例13
N-[3-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)-1-メチル-3-オキソプロピル]アセトアミド Example 13
N- [3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methyl-3-oxopropyl] acetamide
N-[3-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)-1-メチル-3-オキソプロピル]アセトアミド Example 13
N- [3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methyl-3-oxopropyl] acetamide
A) ベンジル [3-(4-メトキシフェニル)-1-メチル-3-オキソプロピル]カルバマート
(2E)-1-(4-メトキシフェニル)ブタ-2-エン-1-オン(6.00 g)、カルバミン酸ベンジル(5.15 g)の混合物に硝酸ビスマス五水和物(3.30 g)を加え、反応混合物を室温で16時間撹拌した。反応混合物を酢酸エチルで希釈し、NHシリカゲルカラムクロマトグラフィー(酢酸エチル)に通した。溶媒を減圧下留去した後、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して、得られた油状物をジイソプロピルエーテルで固化することにより、標題化合物(6.63 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.29 (3H, d, J = 6.9 Hz), 2.95-3.02 (1H, m), 3.29-3.35 (1H, m), 3.86 (3H, s), 4.18-4.26 (1H, m), 5.03-5.12 (2H, m) 5.33 (1H, brs), 6.91 (2H, d, J = 8.4 Hz), 7.29-7.34 (5H, m), 7.92 (2H, d, J = 8.4 Hz). A) Benzyl [3- (4-methoxyphenyl) -1-methyl-3-oxopropyl] carbamate (2E) -1- (4-methoxyphenyl) but-2-en-1-one (6.00 g), carbamine Bismuth nitrate pentahydrate (3.30 g) was added to a mixture of benzyl acid (5.15 g) and the reaction mixture was stirred at room temperature for 16 hours. The reaction mixture was diluted with ethyl acetate and passed through NH silica gel column chromatography (ethyl acetate). The solvent was evaporated under reduced pressure, the residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained oil was solidified with diisopropyl ether to give the title compound (6.63 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.29 (3H, d, J = 6.9 Hz), 2.95-3.02 (1H, m), 3.29-3.35 (1H, m), 3.86 (3H, s), 4.18- 4.26 (1H, m), 5.03-5.12 (2H, m) 5.33 (1H, brs), 6.91 (2H, d, J = 8.4 Hz), 7.29-7.34 (5H, m), 7.92 (2H, d, J = 8.4 Hz).
(2E)-1-(4-メトキシフェニル)ブタ-2-エン-1-オン(6.00 g)、カルバミン酸ベンジル(5.15 g)の混合物に硝酸ビスマス五水和物(3.30 g)を加え、反応混合物を室温で16時間撹拌した。反応混合物を酢酸エチルで希釈し、NHシリカゲルカラムクロマトグラフィー(酢酸エチル)に通した。溶媒を減圧下留去した後、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して、得られた油状物をジイソプロピルエーテルで固化することにより、標題化合物(6.63 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.29 (3H, d, J = 6.9 Hz), 2.95-3.02 (1H, m), 3.29-3.35 (1H, m), 3.86 (3H, s), 4.18-4.26 (1H, m), 5.03-5.12 (2H, m) 5.33 (1H, brs), 6.91 (2H, d, J = 8.4 Hz), 7.29-7.34 (5H, m), 7.92 (2H, d, J = 8.4 Hz). A) Benzyl [3- (4-methoxyphenyl) -1-methyl-3-oxopropyl] carbamate (2E) -1- (4-methoxyphenyl) but-2-en-1-one (6.00 g), carbamine Bismuth nitrate pentahydrate (3.30 g) was added to a mixture of benzyl acid (5.15 g) and the reaction mixture was stirred at room temperature for 16 hours. The reaction mixture was diluted with ethyl acetate and passed through NH silica gel column chromatography (ethyl acetate). The solvent was evaporated under reduced pressure, the residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained oil was solidified with diisopropyl ether to give the title compound (6.63 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.29 (3H, d, J = 6.9 Hz), 2.95-3.02 (1H, m), 3.29-3.35 (1H, m), 3.86 (3H, s), 4.18- 4.26 (1H, m), 5.03-5.12 (2H, m) 5.33 (1H, brs), 6.91 (2H, d, J = 8.4 Hz), 7.29-7.34 (5H, m), 7.92 (2H, d, J = 8.4 Hz).
B) N-[3-(4-ヒドロキシフェニル)-1-メチル-3-オキソプロピル]アセトアミド
ベンジル [3-(4-メトキシフェニル)-1-メチル-3-オキソプロピル]カルバマート(1.00 g)と48%臭化水素酸(15 mL)の混合物を4時間加熱還流した。放冷後、水で希釈し、酢酸エチルで洗浄した。水層に炭酸カリウムを少しずつ加え、塩基性(pH 9)にした後、酢酸エチル(200 mL)を加えた。無水酢酸(11.5 mL)を加え、混合物を30分間撹拌した。1 M塩酸を加え、pH 5にした後、塩化ナトリウムを加え酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧下濃縮した。得られた残渣をメタノール(10 mL)に溶解し、2 M水酸化ナトリウム水溶液(10 mL)を加え、反応混合物を20分間撹拌した。1 M塩酸を加え、pH 1-2にした後、塩化ナトリウムを加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して、得られた油状物をジイソプロピルエーテルで固化することにより、標題化合物(287 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.07 (3H, d, J = 6.6 Hz), 1.75 (3H, s), 2.80-2.88 (1H, m), 3.10-3.17 (1H, m), 4.16-4.20 (1H, m), 6.82-6.86 (2H, m), 7.78-7.85 (3H, m), 10.33 (1H, s). B) N- [3- (4-hydroxyphenyl) -1-methyl-3-oxopropyl] acetamide benzyl [3- (4-methoxyphenyl) -1-methyl-3-oxopropyl] carbamate (1.00 g) and A mixture of 48% hydrobromic acid (15 mL) was heated to reflux for 4 hours. After cooling, it was diluted with water and washed with ethyl acetate. To the aqueous layer was added potassium carbonate little by little to make it basic (pH 9), and then ethyl acetate (200 mL) was added. Acetic anhydride (11.5 mL) was added and the mixture was stirred for 30 minutes. 1 M hydrochloric acid was added to adjust to pH 5, sodium chloride was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained residue was dissolved in methanol (10 mL), 2 M aqueous sodium hydroxide solution (10 mL) was added, and the reaction mixture was stirred for 20 min. 1 M hydrochloric acid was added to adjust to pH 1-2, sodium chloride was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained oil was solidified with diisopropyl ether to give the title compound (287 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.07 (3H, d, J = 6.6 Hz), 1.75 (3H, s), 2.80-2.88 (1H, m), 3.10-3.17 (1H, m), 4.16-4.20 (1H, m), 6.82-6.86 (2H, m), 7.78-7.85 (3H, m), 10.33 (1H, s).
ベンジル [3-(4-メトキシフェニル)-1-メチル-3-オキソプロピル]カルバマート(1.00 g)と48%臭化水素酸(15 mL)の混合物を4時間加熱還流した。放冷後、水で希釈し、酢酸エチルで洗浄した。水層に炭酸カリウムを少しずつ加え、塩基性(pH 9)にした後、酢酸エチル(200 mL)を加えた。無水酢酸(11.5 mL)を加え、混合物を30分間撹拌した。1 M塩酸を加え、pH 5にした後、塩化ナトリウムを加え酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧下濃縮した。得られた残渣をメタノール(10 mL)に溶解し、2 M水酸化ナトリウム水溶液(10 mL)を加え、反応混合物を20分間撹拌した。1 M塩酸を加え、pH 1-2にした後、塩化ナトリウムを加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して、得られた油状物をジイソプロピルエーテルで固化することにより、標題化合物(287 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.07 (3H, d, J = 6.6 Hz), 1.75 (3H, s), 2.80-2.88 (1H, m), 3.10-3.17 (1H, m), 4.16-4.20 (1H, m), 6.82-6.86 (2H, m), 7.78-7.85 (3H, m), 10.33 (1H, s). B) N- [3- (4-hydroxyphenyl) -1-methyl-3-oxopropyl] acetamide benzyl [3- (4-methoxyphenyl) -1-methyl-3-oxopropyl] carbamate (1.00 g) and A mixture of 48% hydrobromic acid (15 mL) was heated to reflux for 4 hours. After cooling, it was diluted with water and washed with ethyl acetate. To the aqueous layer was added potassium carbonate little by little to make it basic (pH 9), and then ethyl acetate (200 mL) was added. Acetic anhydride (11.5 mL) was added and the mixture was stirred for 30 minutes. 1 M hydrochloric acid was added to adjust to pH 5, sodium chloride was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained residue was dissolved in methanol (10 mL), 2 M aqueous sodium hydroxide solution (10 mL) was added, and the reaction mixture was stirred for 20 min. 1 M hydrochloric acid was added to adjust to pH 1-2, sodium chloride was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained oil was solidified with diisopropyl ether to give the title compound (287 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.07 (3H, d, J = 6.6 Hz), 1.75 (3H, s), 2.80-2.88 (1H, m), 3.10-3.17 (1H, m), 4.16-4.20 (1H, m), 6.82-6.86 (2H, m), 7.78-7.85 (3H, m), 10.33 (1H, s).
C) N-[3-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)-1-メチル-3-オキソプロピル]アセトアミド
N-[3-(4-ヒドロキシフェニル)-1-メチル-3-オキソプロピル]アセトアミド(120 mg)、1-(クロロメチル)-4-(シクロプロピルメトキシ)ベンゼン(128 mg)、炭酸カリウム(150 mg)およびDMF(2 mL)の混合物を60℃で16時間撹拌した。反応混合物を酢酸エチルおよび10%炭酸カリウム水溶液で希釈して、有機層を分離した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶液をNHシリカゲルショートカラム(酢酸エチル)に通した。溶媒を減圧下留去した後、ジイソプロピルエーテルで粉末にすることにより、標題化合物(163 mg)を得た。 C) N- [3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methyl-3-oxopropyl] acetamide N- [3- (4-hydroxyphenyl) -1- Methyl-3-oxopropyl] acetamide (120 mg), 1- (chloromethyl) -4- (cyclopropylmethoxy) benzene (128 mg), potassium carbonate (150 mg) and DMF (2 mL) For 16 hours. The reaction mixture was diluted with ethyl acetate and 10% aqueous potassium carbonate solution, and the organic layer was separated. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solution was passed through NH silica gel short column (ethyl acetate). After evaporating the solvent under reduced pressure, the title compound (163 mg) was obtained by triturating with diisopropyl ether.
N-[3-(4-ヒドロキシフェニル)-1-メチル-3-オキソプロピル]アセトアミド(120 mg)、1-(クロロメチル)-4-(シクロプロピルメトキシ)ベンゼン(128 mg)、炭酸カリウム(150 mg)およびDMF(2 mL)の混合物を60℃で16時間撹拌した。反応混合物を酢酸エチルおよび10%炭酸カリウム水溶液で希釈して、有機層を分離した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶液をNHシリカゲルショートカラム(酢酸エチル)に通した。溶媒を減圧下留去した後、ジイソプロピルエーテルで粉末にすることにより、標題化合物(163 mg)を得た。 C) N- [3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methyl-3-oxopropyl] acetamide N- [3- (4-hydroxyphenyl) -1- Methyl-3-oxopropyl] acetamide (120 mg), 1- (chloromethyl) -4- (cyclopropylmethoxy) benzene (128 mg), potassium carbonate (150 mg) and DMF (2 mL) For 16 hours. The reaction mixture was diluted with ethyl acetate and 10% aqueous potassium carbonate solution, and the organic layer was separated. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solution was passed through NH silica gel short column (ethyl acetate). After evaporating the solvent under reduced pressure, the title compound (163 mg) was obtained by triturating with diisopropyl ether.
実施例14
N-[(1S)-3-{4-[(4-エトキシ-2-フルオロベンジル)オキシ]フェニル}-1-メチルプロピル]アセトアミド
(4-エトキシ-2-フルオロフェニル)メタノール(118 mg)のトルエン(1 mL)溶液に塩化チオニル(0.084 mL)を加え、室温で30分間撹拌した。反応混合物を減圧下濃縮して得られた残渣に、DMF(1 mL)、N-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミド(120 mg)および炭酸セシウム(377 mg)を加え、室温で5時間撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製し、得られた固体をジイソプロピルエーテルで洗浄して標題化合物(132 mg)を得た。 Example 14
N-[(1S) -3- {4-[(4-Ethoxy-2-fluorobenzyl) oxy] phenyl} -1-methylpropyl] acetamide (4-ethoxy-2-fluorophenyl) methanol (118 mg) Thionyl chloride (0.084 mL) was added to a toluene (1 mL) solution, and the mixture was stirred at room temperature for 30 minutes. The reaction mixture was concentrated under reduced pressure to give DMF (1 mL), N-[(1S) -3- (4-hydroxyphenyl) -1-methylpropyl] acetamide (120 mg) and cesium carbonate (120 mg). 377 mg) was added, and the mixture was stirred at room temperature for 5 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate), and the obtained solid was washed with diisopropyl ether to give the title compound (132 mg).
N-[(1S)-3-{4-[(4-エトキシ-2-フルオロベンジル)オキシ]フェニル}-1-メチルプロピル]アセトアミド
(4-エトキシ-2-フルオロフェニル)メタノール(118 mg)のトルエン(1 mL)溶液に塩化チオニル(0.084 mL)を加え、室温で30分間撹拌した。反応混合物を減圧下濃縮して得られた残渣に、DMF(1 mL)、N-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミド(120 mg)および炭酸セシウム(377 mg)を加え、室温で5時間撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製し、得られた固体をジイソプロピルエーテルで洗浄して標題化合物(132 mg)を得た。 Example 14
N-[(1S) -3- {4-[(4-Ethoxy-2-fluorobenzyl) oxy] phenyl} -1-methylpropyl] acetamide (4-ethoxy-2-fluorophenyl) methanol (118 mg) Thionyl chloride (0.084 mL) was added to a toluene (1 mL) solution, and the mixture was stirred at room temperature for 30 minutes. The reaction mixture was concentrated under reduced pressure to give DMF (1 mL), N-[(1S) -3- (4-hydroxyphenyl) -1-methylpropyl] acetamide (120 mg) and cesium carbonate (120 mg). 377 mg) was added, and the mixture was stirred at room temperature for 5 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate), and the obtained solid was washed with diisopropyl ether to give the title compound (132 mg).
実施例15
N-[(1S)-3-{4-[(2-フルオロ-4-プロポキシベンジル)オキシ]フェニル}-1-メチルプロピル]アセトアミド Example 15
N-[(1S) -3- {4-[(2-Fluoro-4-propoxybenzyl) oxy] phenyl} -1-methylpropyl] acetamide
N-[(1S)-3-{4-[(2-フルオロ-4-プロポキシベンジル)オキシ]フェニル}-1-メチルプロピル]アセトアミド Example 15
N-[(1S) -3- {4-[(2-Fluoro-4-propoxybenzyl) oxy] phenyl} -1-methylpropyl] acetamide
A) メチル 2-フルオロ-4-プロポキシベンゾアート
メチル 2-フルオロ-4-ヒドロキシベンゾアート(2.85 g)、1-ヨードプロパン(3.12 g)および炭酸セシウム(8.19 g)のDMF(17 mL)懸濁液を50℃で20分間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で3度洗浄し、無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去し、標題化合物(3.39 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.04 (3H, t, J = 7.5 Hz), 1.82 (2H, qd, J = 7.5, 6.6 Hz), 3.89 (3H, s), 3.95 (2H, t, J = 6.6 Hz), 6.62 (1H, dd, J = 12.8, 2.3 Hz), 6.71 (1H, dd, J = 8.9, 2.4 Hz), 7.83-7.93 (1H, m). A) Methyl 2-fluoro-4-propoxybenzoate Methyl 2-fluoro-4-hydroxybenzoate (2.85 g), 1-iodopropane (3.12 g) and cesium carbonate (8.19 g) in DMF (17 mL) suspension The solution was stirred at 50 ° C. for 20 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed 3 times with saturated brine and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to obtain the title compound (3.39 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.04 (3H, t, J = 7.5 Hz), 1.82 (2H, qd, J = 7.5, 6.6 Hz), 3.89 (3H, s), 3.95 (2H, t, J = 6.6 Hz), 6.62 (1H, dd, J = 12.8, 2.3 Hz), 6.71 (1H, dd, J = 8.9, 2.4 Hz), 7.83-7.93 (1H, m).
メチル 2-フルオロ-4-ヒドロキシベンゾアート(2.85 g)、1-ヨードプロパン(3.12 g)および炭酸セシウム(8.19 g)のDMF(17 mL)懸濁液を50℃で20分間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で3度洗浄し、無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去し、標題化合物(3.39 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.04 (3H, t, J = 7.5 Hz), 1.82 (2H, qd, J = 7.5, 6.6 Hz), 3.89 (3H, s), 3.95 (2H, t, J = 6.6 Hz), 6.62 (1H, dd, J = 12.8, 2.3 Hz), 6.71 (1H, dd, J = 8.9, 2.4 Hz), 7.83-7.93 (1H, m). A) Methyl 2-fluoro-4-propoxybenzoate Methyl 2-fluoro-4-hydroxybenzoate (2.85 g), 1-iodopropane (3.12 g) and cesium carbonate (8.19 g) in DMF (17 mL) suspension The solution was stirred at 50 ° C. for 20 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed 3 times with saturated brine and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to obtain the title compound (3.39 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.04 (3H, t, J = 7.5 Hz), 1.82 (2H, qd, J = 7.5, 6.6 Hz), 3.89 (3H, s), 3.95 (2H, t, J = 6.6 Hz), 6.62 (1H, dd, J = 12.8, 2.3 Hz), 6.71 (1H, dd, J = 8.9, 2.4 Hz), 7.83-7.93 (1H, m).
B) (2-フルオロ-4-プロポキシフェニル)メタノール
メチル 2-フルオロ-4-プロポキシベンゾアート(3.39 g)のTHF(50 mL)溶液に水素化リチウムアルミニウム(956 mg)を0℃で加え、0℃で30分間撹拌した。反応混合物に水(0.95 mL)、15%水酸化ナトリウム水溶液(0.95 mL)および水(2.9 mL)を順次加え、室温で30分間撹拌した。生じた固体をセライト濾過により除去し、溶媒を減圧下留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物 (2.71 g) を得た。
1H NMR (300 MHz, CDCl3) δ 1.03 (3H, t, J = 7.5 Hz), 1.68 (1H, t, J = 6.0 Hz), 1.73-1.87 (2H, m), 3.90 (2H, t, J = 6.6 Hz), 4.66 (2H, d, J = 6.0 Hz), 6.61 (1H, dd, J = 11.7, 2.3 Hz), 6.68 (1H, dd, J = 8.7, 2.6 Hz), 7.20-7.33(1H, m). B) (2-Fluoro-4-propoxyphenyl) methanol methyl Lithium aluminum hydride (956 mg) was added to a THF (50 mL) solution of 2-fluoro-4-propoxybenzoate (3.39 g) at 0 ° C. Stir at 30 ° C. for 30 minutes. Water (0.95 mL), 15% aqueous sodium hydroxide solution (0.95 mL) and water (2.9 mL) were sequentially added to the reaction mixture, and the mixture was stirred at room temperature for 30 min. The resulting solid was removed by Celite filtration, the solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (2.71 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.03 (3H, t, J = 7.5 Hz), 1.68 (1H, t, J = 6.0 Hz), 1.73-1.87 (2H, m), 3.90 (2H, t, J = 6.6 Hz), 4.66 (2H, d, J = 6.0 Hz), 6.61 (1H, dd, J = 11.7, 2.3 Hz), 6.68 (1H, dd, J = 8.7, 2.6 Hz), 7.20-7.33 ( 1H, m).
メチル 2-フルオロ-4-プロポキシベンゾアート(3.39 g)のTHF(50 mL)溶液に水素化リチウムアルミニウム(956 mg)を0℃で加え、0℃で30分間撹拌した。反応混合物に水(0.95 mL)、15%水酸化ナトリウム水溶液(0.95 mL)および水(2.9 mL)を順次加え、室温で30分間撹拌した。生じた固体をセライト濾過により除去し、溶媒を減圧下留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物 (2.71 g) を得た。
1H NMR (300 MHz, CDCl3) δ 1.03 (3H, t, J = 7.5 Hz), 1.68 (1H, t, J = 6.0 Hz), 1.73-1.87 (2H, m), 3.90 (2H, t, J = 6.6 Hz), 4.66 (2H, d, J = 6.0 Hz), 6.61 (1H, dd, J = 11.7, 2.3 Hz), 6.68 (1H, dd, J = 8.7, 2.6 Hz), 7.20-7.33(1H, m). B) (2-Fluoro-4-propoxyphenyl) methanol methyl Lithium aluminum hydride (956 mg) was added to a THF (50 mL) solution of 2-fluoro-4-propoxybenzoate (3.39 g) at 0 ° C. Stir at 30 ° C. for 30 minutes. Water (0.95 mL), 15% aqueous sodium hydroxide solution (0.95 mL) and water (2.9 mL) were sequentially added to the reaction mixture, and the mixture was stirred at room temperature for 30 min. The resulting solid was removed by Celite filtration, the solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (2.71 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.03 (3H, t, J = 7.5 Hz), 1.68 (1H, t, J = 6.0 Hz), 1.73-1.87 (2H, m), 3.90 (2H, t, J = 6.6 Hz), 4.66 (2H, d, J = 6.0 Hz), 6.61 (1H, dd, J = 11.7, 2.3 Hz), 6.68 (1H, dd, J = 8.7, 2.6 Hz), 7.20-7.33 ( 1H, m).
C) N-[(1S)-3-{4-[(2-フルオロ-4-プロポキシベンジル)オキシ]フェニル}-1-メチルプロピル]アセトアミド
(2-フルオロ-4-プロポキシフェニル)メタノールおよびN-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミドを用いて実施例14と同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N-[(1S) -3- {4-[(2-fluoro-4-propoxybenzyl) oxy] phenyl} -1-methylpropyl] acetamide (2-fluoro-4-propoxyphenyl) methanol and N- The title compound was obtained in the same manner as in Example 14 or a method analogous thereto using [(1S) -3- (4-hydroxyphenyl) -1-methylpropyl] acetamide.
(2-フルオロ-4-プロポキシフェニル)メタノールおよびN-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミドを用いて実施例14と同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N-[(1S) -3- {4-[(2-fluoro-4-propoxybenzyl) oxy] phenyl} -1-methylpropyl] acetamide (2-fluoro-4-propoxyphenyl) methanol and N- The title compound was obtained in the same manner as in Example 14 or a method analogous thereto using [(1S) -3- (4-hydroxyphenyl) -1-methylpropyl] acetamide.
実施例16
N-[(1S)-3-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}-2-メトキシフェニル)-1-メチルプロピル]アセトアミド Example 16
N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} -2-methoxyphenyl) -1-methylpropyl] acetamide
N-[(1S)-3-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}-2-メトキシフェニル)-1-メチルプロピル]アセトアミド Example 16
N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} -2-methoxyphenyl) -1-methylpropyl] acetamide
A) 5-(ベンジルオキシ)-2-ヨードフェノール
3-(ベンジルオキシ)フェノール(5.00 g)とトリフルオロ酢酸銀(5.52 g)のトルエン(25 mL)混合液にヨウ素(6.35 g)のトルエン(75 mL)溶液を氷冷下で30分間かけて滴下した。反応混合物に飽和チオ硫酸ナトリウム水溶液と飽和食塩水を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(7.26 g)を得た。
1H NMR (300 MHz, CDCl3) δ 5.03 (2H, s), 5.23 (1H, s), 6.40 (1H, dd, J = 8.7, 3.0 Hz), 6.67 (1H, d, J = 3.0 Hz), 7.28-7.44 (5H, m), 7.49 (1H, d, J = 9.1 Hz). A) 5- (benzyloxy) -2-iodophenol 3- (benzyloxy) phenol (5.00 g) and silver trifluoroacetate (5.52 g) in toluene (25 mL) mixed with iodine (6.35 g) in toluene (6.35 g) 75 mL) was added dropwise over 30 minutes under ice cooling. A saturated aqueous sodium thiosulfate solution and saturated brine were added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure. The residue was purified by silica gel chromatography (hexane / ethyl acetate) to give the title compound (7.26 g).
1 H NMR (300 MHz, CDCl 3 ) δ 5.03 (2H, s), 5.23 (1H, s), 6.40 (1H, dd, J = 8.7, 3.0 Hz), 6.67 (1H, d, J = 3.0 Hz) , 7.28-7.44 (5H, m), 7.49 (1H, d, J = 9.1 Hz).
3-(ベンジルオキシ)フェノール(5.00 g)とトリフルオロ酢酸銀(5.52 g)のトルエン(25 mL)混合液にヨウ素(6.35 g)のトルエン(75 mL)溶液を氷冷下で30分間かけて滴下した。反応混合物に飽和チオ硫酸ナトリウム水溶液と飽和食塩水を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(7.26 g)を得た。
1H NMR (300 MHz, CDCl3) δ 5.03 (2H, s), 5.23 (1H, s), 6.40 (1H, dd, J = 8.7, 3.0 Hz), 6.67 (1H, d, J = 3.0 Hz), 7.28-7.44 (5H, m), 7.49 (1H, d, J = 9.1 Hz). A) 5- (benzyloxy) -2-iodophenol 3- (benzyloxy) phenol (5.00 g) and silver trifluoroacetate (5.52 g) in toluene (25 mL) mixed with iodine (6.35 g) in toluene (6.35 g) 75 mL) was added dropwise over 30 minutes under ice cooling. A saturated aqueous sodium thiosulfate solution and saturated brine were added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure. The residue was purified by silica gel chromatography (hexane / ethyl acetate) to give the title compound (7.26 g).
1 H NMR (300 MHz, CDCl 3 ) δ 5.03 (2H, s), 5.23 (1H, s), 6.40 (1H, dd, J = 8.7, 3.0 Hz), 6.67 (1H, d, J = 3.0 Hz) , 7.28-7.44 (5H, m), 7.49 (1H, d, J = 9.1 Hz).
B) 4-(ベンジルオキシ)-1-ヨード-2-メトキシベンゼン
5-(ベンジルオキシ)-2-ヨードフェノール(2.00 g)と炭酸カリウム(1.28 g)のDMF(20 mL)混合液にヨウ化メチル(0.460 mL)を加えて30分間撹拌した。反応混合物に酢酸エチルを加え、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.34 g)を得た。
1H NMR (300 MHz, CDCl3) δ 3.83 (3H, s), 5.04 (2H, s), 6.39 (1H, dd, J = 8.7, 2.7 Hz), 6.51 (1H, d, J = 2.7 Hz), 7.31-7.49 (5H, m), 7.61 (1H, d, J = 8.3 Hz). B) 4- (Benzyloxy) -1-iodo-2-methoxybenzene Iodinated into a mixture of 5- (benzyloxy) -2-iodophenol (2.00 g) and potassium carbonate (1.28 g) in DMF (20 mL) Methyl (0.460 mL) was added and stirred for 30 minutes. Ethyl acetate was added to the reaction mixture, washed with saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.34 g).
1 H NMR (300 MHz, CDCl 3 ) δ 3.83 (3H, s), 5.04 (2H, s), 6.39 (1H, dd, J = 8.7, 2.7 Hz), 6.51 (1H, d, J = 2.7 Hz) , 7.31-7.49 (5H, m), 7.61 (1H, d, J = 8.3 Hz).
5-(ベンジルオキシ)-2-ヨードフェノール(2.00 g)と炭酸カリウム(1.28 g)のDMF(20 mL)混合液にヨウ化メチル(0.460 mL)を加えて30分間撹拌した。反応混合物に酢酸エチルを加え、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.34 g)を得た。
1H NMR (300 MHz, CDCl3) δ 3.83 (3H, s), 5.04 (2H, s), 6.39 (1H, dd, J = 8.7, 2.7 Hz), 6.51 (1H, d, J = 2.7 Hz), 7.31-7.49 (5H, m), 7.61 (1H, d, J = 8.3 Hz). B) 4- (Benzyloxy) -1-iodo-2-methoxybenzene Iodinated into a mixture of 5- (benzyloxy) -2-iodophenol (2.00 g) and potassium carbonate (1.28 g) in DMF (20 mL) Methyl (0.460 mL) was added and stirred for 30 minutes. Ethyl acetate was added to the reaction mixture, washed with saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.34 g).
1 H NMR (300 MHz, CDCl 3 ) δ 3.83 (3H, s), 5.04 (2H, s), 6.39 (1H, dd, J = 8.7, 2.7 Hz), 6.51 (1H, d, J = 2.7 Hz) , 7.31-7.49 (5H, m), 7.61 (1H, d, J = 8.3 Hz).
C) tert-ブチル{(1S)-3-[4-(ベンジルオキシ)-2-メトキシフェニル]-1-メチルプロパ-2-イン-1-イル}カルバマート
4-(ベンジルオキシ)-1-ヨード-2-メトキシベンゼン(600 mg)、tert-ブチル [(1S)-1-メチルプロパ-2-イン-1-イル]カルバマート(442 mg)およびトリエチルアミン(0.494 mL)のTHF(10 mL)溶液に、ビストリフェニルホスフィンジクロロパラジウム(II)(62.2 mg)とヨウ化銅(I)(33.8 mg)を加え、アルゴン雰囲気下室温で20分間撹拌した。反応混合物に飽和食塩水を加え、酢酸エチルで抽出し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(660 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.45-1.49 (12H, m), 3.82 (3H, s), 4.72-4.81 (2H, m), 5.05 (2H, s), 6.48-6.52 (2H, m), 7.27-7.44 (6H, m). C) tert-butyl {(1S) -3- [4- (benzyloxy) -2-methoxyphenyl] -1-methylprop-2-yn-1-yl} carbamate 4- (benzyloxy) -1-iodo- To a solution of 2-methoxybenzene (600 mg), tert-butyl [(1S) -1-methylprop-2-yn-1-yl] carbamate (442 mg) and triethylamine (0.494 mL) in THF (10 mL), Phenylphosphinedichloropalladium (II) (62.2 mg) and copper iodide (I) (33.8 mg) were added, and the mixture was stirred at room temperature for 20 minutes under an argon atmosphere. To the reaction mixture was added saturated brine, extracted with ethyl acetate, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure and the obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (660 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.45-1.49 (12H, m), 3.82 (3H, s), 4.72-4.81 (2H, m), 5.05 (2H, s), 6.48-6.52 (2H, m ), 7.27-7.44 (6H, m).
4-(ベンジルオキシ)-1-ヨード-2-メトキシベンゼン(600 mg)、tert-ブチル [(1S)-1-メチルプロパ-2-イン-1-イル]カルバマート(442 mg)およびトリエチルアミン(0.494 mL)のTHF(10 mL)溶液に、ビストリフェニルホスフィンジクロロパラジウム(II)(62.2 mg)とヨウ化銅(I)(33.8 mg)を加え、アルゴン雰囲気下室温で20分間撹拌した。反応混合物に飽和食塩水を加え、酢酸エチルで抽出し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(660 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 1.45-1.49 (12H, m), 3.82 (3H, s), 4.72-4.81 (2H, m), 5.05 (2H, s), 6.48-6.52 (2H, m), 7.27-7.44 (6H, m). C) tert-butyl {(1S) -3- [4- (benzyloxy) -2-methoxyphenyl] -1-methylprop-2-yn-1-yl} carbamate 4- (benzyloxy) -1-iodo- To a solution of 2-methoxybenzene (600 mg), tert-butyl [(1S) -1-methylprop-2-yn-1-yl] carbamate (442 mg) and triethylamine (0.494 mL) in THF (10 mL), Phenylphosphinedichloropalladium (II) (62.2 mg) and copper iodide (I) (33.8 mg) were added, and the mixture was stirred at room temperature for 20 minutes under an argon atmosphere. To the reaction mixture was added saturated brine, extracted with ethyl acetate, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure and the obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (660 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.45-1.49 (12H, m), 3.82 (3H, s), 4.72-4.81 (2H, m), 5.05 (2H, s), 6.48-6.52 (2H, m ), 7.27-7.44 (6H, m).
D) N-[(1S)-3-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}-2-メトキシフェニル)-1-メチルプロピル]アセトアミド
tert-ブチル{(1S)-3-[4-(ベンジルオキシ)-2-メトキシフェニル]-1-メチルプロパ-2-イン-1-イル}カルバマートを用いて実施例1の工程C、実施例8の工程B、実施例13の工程Cと同様の方法またはそれに準ずる方法により標題化合物を得た。 D) N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} -2-methoxyphenyl) -1-methylpropyl] acetamide tert-butyl {(1S) -3- Step C of Example 1, Step B of Example 8, Step C of Example 13 using [4- (benzyloxy) -2-methoxyphenyl] -1-methylprop-2-yn-1-yl} carbamate The title compound was obtained by a method similar to that described above or a method analogous thereto.
tert-ブチル{(1S)-3-[4-(ベンジルオキシ)-2-メトキシフェニル]-1-メチルプロパ-2-イン-1-イル}カルバマートを用いて実施例1の工程C、実施例8の工程B、実施例13の工程Cと同様の方法またはそれに準ずる方法により標題化合物を得た。 D) N-[(1S) -3- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} -2-methoxyphenyl) -1-methylpropyl] acetamide tert-butyl {(1S) -3- Step C of Example 1, Step B of Example 8, Step C of Example 13 using [4- (benzyloxy) -2-methoxyphenyl] -1-methylprop-2-yn-1-yl} carbamate The title compound was obtained by a method similar to that described above or a method analogous thereto.
実施例17
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)(メチル)アミノ]-1-メチルエチル}アセトアミド Example 17
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) (methyl) amino] -1-methylethyl} acetamide
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)(メチル)アミノ]-1-メチルエチル}アセトアミド Example 17
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) (methyl) amino] -1-methylethyl} acetamide
A) tert-ブチル[(1S)-2-{[4-(ベンジルオキシ)フェニル](メチル)アミノ}-1-メチル-2-オキソエチル]カルバマート
WSC(1.02 g)を4-(ベンジルオキシ)-N-メチルアニリン(0.950 g)、Boc-L-アラニン(1.02 g)およびHOBt(0.723 g)のDMF(20 mL)溶液に加えて、室温で1時間撹拌した。反応混合物を酢酸エチルで希釈した後、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.34 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.11 (3H, d, J = 7.2 Hz), 1.41 (9H, s), 3.24 (3H, s), 4.30-4.38 (1H, m), 5.07 (2H, s), 5.28 (1H, d, J = 7.5 Hz), 7.01 (2H, d, J = 9.0 Hz), 7.18 (2H, d, J = 9.0 Hz), 7.35-7.46 (5H, m). A) tert-butyl [(1S) -2-{[4- (benzyloxy) phenyl] (methyl) amino} -1-methyl-2-oxoethyl] carbamate WSC (1.02 g) was converted to 4- (benzyloxy)- N-methylaniline (0.950 g), Boc-L-alanine (1.02 g) and HOBt (0.723 g) were added to a DMF (20 mL) solution, and the mixture was stirred at room temperature for 1 hour. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.34 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.11 (3H, d, J = 7.2 Hz), 1.41 (9H, s), 3.24 (3H, s), 4.30-4.38 (1H, m), 5.07 (2H, s), 5.28 (1H, d, J = 7.5 Hz), 7.01 (2H, d, J = 9.0 Hz), 7.18 (2H, d, J = 9.0 Hz), 7.35-7.46 (5H, m).
WSC(1.02 g)を4-(ベンジルオキシ)-N-メチルアニリン(0.950 g)、Boc-L-アラニン(1.02 g)およびHOBt(0.723 g)のDMF(20 mL)溶液に加えて、室温で1時間撹拌した。反応混合物を酢酸エチルで希釈した後、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.34 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.11 (3H, d, J = 7.2 Hz), 1.41 (9H, s), 3.24 (3H, s), 4.30-4.38 (1H, m), 5.07 (2H, s), 5.28 (1H, d, J = 7.5 Hz), 7.01 (2H, d, J = 9.0 Hz), 7.18 (2H, d, J = 9.0 Hz), 7.35-7.46 (5H, m). A) tert-butyl [(1S) -2-{[4- (benzyloxy) phenyl] (methyl) amino} -1-methyl-2-oxoethyl] carbamate WSC (1.02 g) was converted to 4- (benzyloxy)- N-methylaniline (0.950 g), Boc-L-alanine (1.02 g) and HOBt (0.723 g) were added to a DMF (20 mL) solution, and the mixture was stirred at room temperature for 1 hour. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.34 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.11 (3H, d, J = 7.2 Hz), 1.41 (9H, s), 3.24 (3H, s), 4.30-4.38 (1H, m), 5.07 (2H, s), 5.28 (1H, d, J = 7.5 Hz), 7.01 (2H, d, J = 9.0 Hz), 7.18 (2H, d, J = 9.0 Hz), 7.35-7.46 (5H, m).
B) tert-ブチル[(1S)-2-{[4-(ベンジルオキシ)フェニル](メチル)アミノ}-1-メチルエチル]カルバマート
tert-ブチル[(1S)-2-{[4-(ベンジルオキシ)フェニル](メチル)アミノ}-1-メチル-2-オキソエチル]カルバマート(1.75 g)のTHF(20 mL)溶液にボラン-THF錯体のTHF溶液(1.0 M, 9.1 mL)を加えて15分間撹拌した。反応混合物に1 M水酸化ナトリウム水溶液を加えて5分間撹拌した後、酢酸エチルで抽出し、得られた有機層を飽和食塩水で洗浄した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.16 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.16 (3H, d, J = 7.2 Hz), 1.42 (9H, s), 2.90 (3H, s), 3.04 (1H, dd, J = 14.3, 7.2 Hz), 3.37 (1H, dd, J = 14.3, 6.4 Hz), 3.66-3.95 (1H, m), 4.41 (1H, brs), 5.00 (2H, s), 6.73 (2H, d, J = 9.0 Hz), 6.90 (2H, d, J = 9.4 Hz), 7.28-7.44 (5H, m). B) tert-butyl [(1S) -2-{[4- (benzyloxy) phenyl] (methyl) amino} -1-methylethyl] carbamate tert-butyl [(1S) -2-{[4- (benzyl Oxy) phenyl] (methyl) amino} -1-methyl-2-oxoethyl] carbamate (1.75 g) in THF (20 mL) was added borane-THF complex in THF (1.0 M, 9.1 mL) for 15 minutes. Stir. A 1 M aqueous sodium hydroxide solution was added to the reaction mixture, and the mixture was stirred for 5 minutes, extracted with ethyl acetate, and the obtained organic layer was washed with saturated brine. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.16 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.16 (3H, d, J = 7.2 Hz), 1.42 (9H, s), 2.90 (3H, s), 3.04 (1H, dd, J = 14.3, 7.2 Hz) , 3.37 (1H, dd, J = 14.3, 6.4 Hz), 3.66-3.95 (1H, m), 4.41 (1H, brs), 5.00 (2H, s), 6.73 (2H, d, J = 9.0 Hz), 6.90 (2H, d, J = 9.4 Hz), 7.28-7.44 (5H, m).
tert-ブチル[(1S)-2-{[4-(ベンジルオキシ)フェニル](メチル)アミノ}-1-メチル-2-オキソエチル]カルバマート(1.75 g)のTHF(20 mL)溶液にボラン-THF錯体のTHF溶液(1.0 M, 9.1 mL)を加えて15分間撹拌した。反応混合物に1 M水酸化ナトリウム水溶液を加えて5分間撹拌した後、酢酸エチルで抽出し、得られた有機層を飽和食塩水で洗浄した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.16 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.16 (3H, d, J = 7.2 Hz), 1.42 (9H, s), 2.90 (3H, s), 3.04 (1H, dd, J = 14.3, 7.2 Hz), 3.37 (1H, dd, J = 14.3, 6.4 Hz), 3.66-3.95 (1H, m), 4.41 (1H, brs), 5.00 (2H, s), 6.73 (2H, d, J = 9.0 Hz), 6.90 (2H, d, J = 9.4 Hz), 7.28-7.44 (5H, m). B) tert-butyl [(1S) -2-{[4- (benzyloxy) phenyl] (methyl) amino} -1-methylethyl] carbamate tert-butyl [(1S) -2-{[4- (benzyl Oxy) phenyl] (methyl) amino} -1-methyl-2-oxoethyl] carbamate (1.75 g) in THF (20 mL) was added borane-THF complex in THF (1.0 M, 9.1 mL) for 15 minutes. Stir. A 1 M aqueous sodium hydroxide solution was added to the reaction mixture, and the mixture was stirred for 5 minutes, extracted with ethyl acetate, and the obtained organic layer was washed with saturated brine. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.16 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.16 (3H, d, J = 7.2 Hz), 1.42 (9H, s), 2.90 (3H, s), 3.04 (1H, dd, J = 14.3, 7.2 Hz) , 3.37 (1H, dd, J = 14.3, 6.4 Hz), 3.66-3.95 (1H, m), 4.41 (1H, brs), 5.00 (2H, s), 6.73 (2H, d, J = 9.0 Hz), 6.90 (2H, d, J = 9.4 Hz), 7.28-7.44 (5H, m).
C) N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)(メチル)アミノ]-1-メチルエチル}アセトアミド
tert-ブチル[(1S)-2-{[4-(ベンジルオキシ)フェニル](メチル)アミノ}-1-メチルエチル]カルバマートを用いて実施例7の工程B、実施例1の工程Cおよび実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) (methyl) amino] -1-methylethyl} acetamide tert-butyl [(1S)- Similar to Step B of Example 7, Step C of Example 1, and Step E of Example 1 using 2-{[4- (benzyloxy) phenyl] (methyl) amino} -1-methylethyl] carbamate The title compound was obtained by a method or a method analogous thereto.
tert-ブチル[(1S)-2-{[4-(ベンジルオキシ)フェニル](メチル)アミノ}-1-メチルエチル]カルバマートを用いて実施例7の工程B、実施例1の工程Cおよび実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenyl) (methyl) amino] -1-methylethyl} acetamide tert-butyl [(1S)- Similar to Step B of Example 7, Step C of Example 1, and Step E of Example 1 using 2-{[4- (benzyloxy) phenyl] (methyl) amino} -1-methylethyl] carbamate The title compound was obtained by a method or a method analogous thereto.
実施例18
N-{1-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)メチル]プロピル}アセトアミド
tert-ブチル[1-(ヒドロキシメチル)プロピル]カルバマートを用い、実施例1の工程A、実施例1の工程B、実施例1の工程Cおよび実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 18
Performed with N- {1-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) methyl] propyl} acetamide tert-butyl [1- (hydroxymethyl) propyl] carbamate The title compound was obtained in the same manner as in Step A of Example 1, Step B of Example 1, Step C of Example 1 and Step E of Example 1 or a method analogous thereto.
N-{1-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)メチル]プロピル}アセトアミド
tert-ブチル[1-(ヒドロキシメチル)プロピル]カルバマートを用い、実施例1の工程A、実施例1の工程B、実施例1の工程Cおよび実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 18
Performed with N- {1-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) methyl] propyl} acetamide tert-butyl [1- (hydroxymethyl) propyl] carbamate The title compound was obtained in the same manner as in Step A of Example 1, Step B of Example 1, Step C of Example 1 and Step E of Example 1 or a method analogous thereto.
実施例19
エチル (4-{[(2S)-2-(アセチルアミノ)プロピル]オキシ}フェノキシ)[4-(シクロプロピルメトキシ)フェニル]アセタート Example 19
Ethyl (4-{[(2S) -2- (acetylamino) propyl] oxy} phenoxy) [4- (cyclopropylmethoxy) phenyl] acetate
エチル (4-{[(2S)-2-(アセチルアミノ)プロピル]オキシ}フェノキシ)[4-(シクロプロピルメトキシ)フェニル]アセタート Example 19
Ethyl (4-{[(2S) -2- (acetylamino) propyl] oxy} phenoxy) [4- (cyclopropylmethoxy) phenyl] acetate
A) エチル [4-(シクロプロピルメトキシ)フェニル](ヒドロキシ)アセタート
エチル ヒドロキシ(4-ヒドロキシフェニル)アセタート(1.0 g)、(ブロモメチル)シクロプロパン(1.37 g)および炭酸カリウム(1.41 g)のDMF(5 mL)懸濁液を室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(935 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.42 (2H, m), 0.59-0.73 (2H, m), 1.18-1.36 (4H, m), 3.36 (1H, d, J = 5.7 Hz), 3.80 (2H, d, J = 7.2 Hz), 4.07-4.40 (2H, m), 5.09 (1H, d, J = 5.7 Hz), 6.89 (2H, d, J = 8.7 Hz), 7.31 (2H, d, J = 8.7 Hz). A) ethyl [4- (cyclopropylmethoxy) phenyl] (hydroxy) acetate ethyl hydroxy (4-hydroxyphenyl) acetate (1.0 g), (bromomethyl) cyclopropane (1.37 g) and potassium carbonate (1.41 g) in DMF ( The suspension was stirred overnight at room temperature. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure and the obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (935 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.42 (2H, m), 0.59-0.73 (2H, m), 1.18-1.36 (4H, m), 3.36 (1H, d, J = 5.7 Hz), 3.80 (2H, d, J = 7.2 Hz), 4.07-4.40 (2H, m), 5.09 (1H, d, J = 5.7 Hz), 6.89 (2H, d, J = 8.7 Hz), 7.31 (2H, d , J = 8.7 Hz).
エチル ヒドロキシ(4-ヒドロキシフェニル)アセタート(1.0 g)、(ブロモメチル)シクロプロパン(1.37 g)および炭酸カリウム(1.41 g)のDMF(5 mL)懸濁液を室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(935 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.42 (2H, m), 0.59-0.73 (2H, m), 1.18-1.36 (4H, m), 3.36 (1H, d, J = 5.7 Hz), 3.80 (2H, d, J = 7.2 Hz), 4.07-4.40 (2H, m), 5.09 (1H, d, J = 5.7 Hz), 6.89 (2H, d, J = 8.7 Hz), 7.31 (2H, d, J = 8.7 Hz). A) ethyl [4- (cyclopropylmethoxy) phenyl] (hydroxy) acetate ethyl hydroxy (4-hydroxyphenyl) acetate (1.0 g), (bromomethyl) cyclopropane (1.37 g) and potassium carbonate (1.41 g) in DMF ( The suspension was stirred overnight at room temperature. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure and the obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (935 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.42 (2H, m), 0.59-0.73 (2H, m), 1.18-1.36 (4H, m), 3.36 (1H, d, J = 5.7 Hz), 3.80 (2H, d, J = 7.2 Hz), 4.07-4.40 (2H, m), 5.09 (1H, d, J = 5.7 Hz), 6.89 (2H, d, J = 8.7 Hz), 7.31 (2H, d , J = 8.7 Hz).
B) エチル (4-{[(2S)-2-(アセチルアミノ)プロピル]オキシ}フェノキシ)[4-(シクロプロピルメトキシ)フェニル]アセタート
エチル [4-(シクロプロピルメトキシ)フェニル](ヒドロキシ)アセタートを用いて実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 B) Ethyl (4-{[(2S) -2- (acetylamino) propyl] oxy} phenoxy) [4- (cyclopropylmethoxy) phenyl] acetate ethyl [4- (cyclopropylmethoxy) phenyl] (hydroxy) acetate Was used to give the title compound in the same manner as in Step E of Example 1 or a method analogous thereto.
エチル [4-(シクロプロピルメトキシ)フェニル](ヒドロキシ)アセタートを用いて実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 B) Ethyl (4-{[(2S) -2- (acetylamino) propyl] oxy} phenoxy) [4- (cyclopropylmethoxy) phenyl] acetate ethyl [4- (cyclopropylmethoxy) phenyl] (hydroxy) acetate Was used to give the title compound in the same manner as in Step E of Example 1 or a method analogous thereto.
実施例20
(4-{[(2S)-2-(アセチルアミノ)プロピル]オキシ}フェノキシ)[4-(シクロプロピルメトキシ)フェニル]酢酸
エチル (4-{[(2S)-2-(アセチルアミノ)プロピル]オキシ}フェノキシ)[4-(シクロプロピルメトキシ)フェニル]アセタート(175 mg)と1 M水酸化ナトリウム水溶液(1.2 mL)とメタノール(2 mL)の混合物を50℃で2時間撹拌した。反応混合物を1 M塩酸で中和し、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去して標題化合物(174 mg)を得た。 Example 20
(4-{[(2S) -2- (acetylamino) propyl] oxy} phenoxy) [4- (cyclopropylmethoxy) phenyl] acetic acid ethyl (4-{[(2S) -2- (acetylamino) propyl] A mixture of (oxy} phenoxy) [4- (cyclopropylmethoxy) phenyl] acetate (175 mg), 1 M aqueous sodium hydroxide solution (1.2 mL) and methanol (2 mL) was stirred at 50 ° C. for 2 hours. The reaction mixture was neutralized with 1 M hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (174 mg).
(4-{[(2S)-2-(アセチルアミノ)プロピル]オキシ}フェノキシ)[4-(シクロプロピルメトキシ)フェニル]酢酸
エチル (4-{[(2S)-2-(アセチルアミノ)プロピル]オキシ}フェノキシ)[4-(シクロプロピルメトキシ)フェニル]アセタート(175 mg)と1 M水酸化ナトリウム水溶液(1.2 mL)とメタノール(2 mL)の混合物を50℃で2時間撹拌した。反応混合物を1 M塩酸で中和し、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去して標題化合物(174 mg)を得た。 Example 20
(4-{[(2S) -2- (acetylamino) propyl] oxy} phenoxy) [4- (cyclopropylmethoxy) phenyl] acetic acid ethyl (4-{[(2S) -2- (acetylamino) propyl] A mixture of (oxy} phenoxy) [4- (cyclopropylmethoxy) phenyl] acetate (175 mg), 1 M aqueous sodium hydroxide solution (1.2 mL) and methanol (2 mL) was stirred at 50 ° C. for 2 hours. The reaction mixture was neutralized with 1 M hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (174 mg).
実施例21
N-{2-[(6-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-3-イル)オキシ]-1-メチルエチル}アセトアミド Example 21
N- {2-[(6-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-3-yl) oxy] -1-methylethyl} acetamide
N-{2-[(6-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-3-イル)オキシ]-1-メチルエチル}アセトアミド Example 21
N- {2-[(6-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-3-yl) oxy] -1-methylethyl} acetamide
A) 5-ブロモ-2-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン [4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール(1.08 g)のDMF(20 mL)溶液に水素化ナトリウム(60%油性, 0.219 g)を加えた後、室温で15分間撹拌した。反応混合物に2,5-ジブロモピリジン(1.00 g)を加えた後、100℃で1時間撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.35 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.30-0.39 (2H, m), 0.60-0.70 (2H, m), 1.18-1.36 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 5.31 (2H, s), 6.59-6.73 (3H, m), 7.35 (1H, t, J = 8.3 Hz), 7.63 (1H, dd, J = 8.9, 2.5 Hz), 8.21 (1H, d, J = 2.7 Hz). A) 5-Bromo-2-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridine [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol (1.08 g) in DMF (20 mL ) Sodium hydride (60% oily, 0.219 g) was added to the solution, followed by stirring at room temperature for 15 minutes. 2,5-Dibromopyridine (1.00 g) was added to the reaction mixture, and the mixture was stirred at 100 ° C. for 1 hr. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.35 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.30-0.39 (2H, m), 0.60-0.70 (2H, m), 1.18-1.36 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 5.31 (2H, s), 6.59-6.73 (3H, m), 7.35 (1H, t, J = 8.3 Hz), 7.63 (1H, dd, J = 8.9, 2.5 Hz), 8.21 (1H, d, J = 2.7 Hz).
1H NMR (300 MHz, CDCl3) δ 0.30-0.39 (2H, m), 0.60-0.70 (2H, m), 1.18-1.36 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 5.31 (2H, s), 6.59-6.73 (3H, m), 7.35 (1H, t, J = 8.3 Hz), 7.63 (1H, dd, J = 8.9, 2.5 Hz), 8.21 (1H, d, J = 2.7 Hz). A) 5-Bromo-2-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridine [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol (1.08 g) in DMF (20 mL ) Sodium hydride (60% oily, 0.219 g) was added to the solution, followed by stirring at room temperature for 15 minutes. 2,5-Dibromopyridine (1.00 g) was added to the reaction mixture, and the mixture was stirred at 100 ° C. for 1 hr. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.35 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.30-0.39 (2H, m), 0.60-0.70 (2H, m), 1.18-1.36 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 5.31 (2H, s), 6.59-6.73 (3H, m), 7.35 (1H, t, J = 8.3 Hz), 7.63 (1H, dd, J = 8.9, 2.5 Hz), 8.21 (1H, d, J = 2.7 Hz).
B) 6-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-3-オール
アルゴン雰囲気下、5-ブロモ-2-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン(1.35 g)のTHF(15 mL)溶液にn-ブチルリチウムのヘキサン溶液(1.6 M, 3.11 mL)を-78℃で滴下し、得られた混合物を-78℃で15分間撹拌した。反応混合物にトリメチル ボラート(0.600 mL)を加え、得られた混合物を0℃で30分間撹拌した。反応混合物に8 M水酸化ナトリウム水溶液(0.719 mL)および30%過酸化水素水(2 mL)を加えた後、室温で1時間撹拌した。反応混合物に1 M塩酸を加え酸性にした後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(0.580 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.31-0.38 (2H, m), 0.61-0.69 (2H, m), 1.20-1.33 (1H, m), 3.78 (2H, d, J = 7.2 Hz), 5.27 (2H, s), 6.57-6.74 (3H, m), 7.17 (1H, dd, J = 8.7, 3.0 Hz), 7.36 (1H, t, J = 8.5 Hz), 7.79 (1H, d, J = 3.0 Hz). B) 6-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-3-ol, 5-bromo-2-{[4- (cyclopropylmethoxy) -2-fluorobenzyl under argon atmosphere ] To a solution of oxy} pyridine (1.35 g) in THF (15 mL), hexane solution of n-butyllithium (1.6 M, 3.11 mL) was added dropwise at −78 ° C., and the resulting mixture was stirred at −78 ° C. for 15 minutes. did. Trimethyl borate (0.600 mL) was added to the reaction mixture, and the resulting mixture was stirred at 0 ° C. for 30 minutes. To the reaction mixture were added 8 M aqueous sodium hydroxide solution (0.719 mL) and 30% aqueous hydrogen peroxide (2 mL), and the mixture was stirred at room temperature for 1 hr. The reaction mixture was acidified with 1 M hydrochloric acid, and extracted with ethyl acetate. The obtained organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (0.580 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.31-0.38 (2H, m), 0.61-0.69 (2H, m), 1.20-1.33 (1H, m), 3.78 (2H, d, J = 7.2 Hz), 5.27 (2H, s), 6.57-6.74 (3H, m), 7.17 (1H, dd, J = 8.7, 3.0 Hz), 7.36 (1H, t, J = 8.5 Hz), 7.79 (1H, d, J = 3.0 Hz).
アルゴン雰囲気下、5-ブロモ-2-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン(1.35 g)のTHF(15 mL)溶液にn-ブチルリチウムのヘキサン溶液(1.6 M, 3.11 mL)を-78℃で滴下し、得られた混合物を-78℃で15分間撹拌した。反応混合物にトリメチル ボラート(0.600 mL)を加え、得られた混合物を0℃で30分間撹拌した。反応混合物に8 M水酸化ナトリウム水溶液(0.719 mL)および30%過酸化水素水(2 mL)を加えた後、室温で1時間撹拌した。反応混合物に1 M塩酸を加え酸性にした後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(0.580 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.31-0.38 (2H, m), 0.61-0.69 (2H, m), 1.20-1.33 (1H, m), 3.78 (2H, d, J = 7.2 Hz), 5.27 (2H, s), 6.57-6.74 (3H, m), 7.17 (1H, dd, J = 8.7, 3.0 Hz), 7.36 (1H, t, J = 8.5 Hz), 7.79 (1H, d, J = 3.0 Hz). B) 6-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-3-ol, 5-bromo-2-{[4- (cyclopropylmethoxy) -2-fluorobenzyl under argon atmosphere ] To a solution of oxy} pyridine (1.35 g) in THF (15 mL), hexane solution of n-butyllithium (1.6 M, 3.11 mL) was added dropwise at −78 ° C., and the resulting mixture was stirred at −78 ° C. for 15 minutes. did. Trimethyl borate (0.600 mL) was added to the reaction mixture, and the resulting mixture was stirred at 0 ° C. for 30 minutes. To the reaction mixture were added 8 M aqueous sodium hydroxide solution (0.719 mL) and 30% aqueous hydrogen peroxide (2 mL), and the mixture was stirred at room temperature for 1 hr. The reaction mixture was acidified with 1 M hydrochloric acid, and extracted with ethyl acetate. The obtained organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (0.580 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.31-0.38 (2H, m), 0.61-0.69 (2H, m), 1.20-1.33 (1H, m), 3.78 (2H, d, J = 7.2 Hz), 5.27 (2H, s), 6.57-6.74 (3H, m), 7.17 (1H, dd, J = 8.7, 3.0 Hz), 7.36 (1H, t, J = 8.5 Hz), 7.79 (1H, d, J = 3.0 Hz).
C) 1-[(6-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-3-イル)オキシ]プロパン-2-オール
6-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-3-オールを用いて実施例4の工程Bおよび工程Cと同様の方法またはそれに準ずる方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.40 (2H, m), 0.59-0.72 (2H, m), 1.15-1.30 (1H, m), 1.33 (3H, d, J = 6.4 Hz), 3.78 (2H, d, J = 6.8 Hz), 3.83-4.03 (3H, m), 5.29 (2H, s), 6.59-6.78 (3H, m), 7.19-7.26 (1H, m), 7.37 (1H, t, J = 8.5 Hz), 7.82 (1H, d, J = 3.0 Hz). C) 1-[(6-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-3-yl) oxy] propan-2-ol 6-{[4- (cyclopropylmethoxy)- The title compound was obtained in the same manner as in Step B and Step C of Example 4 or a method analogous thereto using 2-fluorobenzyl] oxy} pyridin-3-ol.
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.40 (2H, m), 0.59-0.72 (2H, m), 1.15-1.30 (1H, m), 1.33 (3H, d, J = 6.4 Hz), 3.78 (2H, d, J = 6.8 Hz), 3.83-4.03 (3H, m), 5.29 (2H, s), 6.59-6.78 (3H, m), 7.19-7.26 (1H, m), 7.37 (1H, t, J = 8.5 Hz), 7.82 (1H, d, J = 3.0 Hz).
6-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-3-オールを用いて実施例4の工程Bおよび工程Cと同様の方法またはそれに準ずる方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.40 (2H, m), 0.59-0.72 (2H, m), 1.15-1.30 (1H, m), 1.33 (3H, d, J = 6.4 Hz), 3.78 (2H, d, J = 6.8 Hz), 3.83-4.03 (3H, m), 5.29 (2H, s), 6.59-6.78 (3H, m), 7.19-7.26 (1H, m), 7.37 (1H, t, J = 8.5 Hz), 7.82 (1H, d, J = 3.0 Hz). C) 1-[(6-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-3-yl) oxy] propan-2-ol 6-{[4- (cyclopropylmethoxy)- The title compound was obtained in the same manner as in Step B and Step C of Example 4 or a method analogous thereto using 2-fluorobenzyl] oxy} pyridin-3-ol.
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.40 (2H, m), 0.59-0.72 (2H, m), 1.15-1.30 (1H, m), 1.33 (3H, d, J = 6.4 Hz), 3.78 (2H, d, J = 6.8 Hz), 3.83-4.03 (3H, m), 5.29 (2H, s), 6.59-6.78 (3H, m), 7.19-7.26 (1H, m), 7.37 (1H, t, J = 8.5 Hz), 7.82 (1H, d, J = 3.0 Hz).
D) 5-(2-アジドプロポキシ)-2-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン
1-[(6-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-3-イル)オキシ]プロパン-2-オール(2.07 g)およびトリエチルアミン(1.66 mL)のTHF(15 mL)溶液に、塩化メタンスルホニル(0.692 mL)を室温で加え、室温で10分間撹拌した。反応混合物に飽和重炭酸ナトリウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をDMF(10 mL)に溶解させ、アジ化ナトリウム(1.94 g)を室温で加え、100℃で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(酢酸エチル)で精製して標題化合物(2.11 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.40 (2H, m), 0.59-0.72 (2H, m), 1.15-1.30 (1H, m), 1.33 (3H, d, J = 6.4 Hz), 3.78 (2H, d, J = 6.8 Hz), 3.83-4.03 (3H, m), 5.29 (2H, s), 6.59-6.78 (3H, m), 7.19-7.26 (1H, m), 7.37 (1H, t, J = 8.5 Hz), 7.82 (1H, d, J = 3.0 Hz). D) 5- (2-Azidopropoxy) -2-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridine 1-[(6-{[4- (cyclopropylmethoxy) -2-fluoro To a solution of (benzyl) oxy} pyridin-3-yl) oxy] propan-2-ol (2.07 g) and triethylamine (1.66 mL) in THF (15 mL) at room temperature was added methanesulfonyl chloride (0.692 mL). Stir for 10 minutes. Saturated aqueous sodium bicarbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was dissolved in DMF (10 mL), sodium azide (1.94 g) was added at room temperature, and the mixture was stirred at 100 ° C. for 3 hr. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel chromatography (ethyl acetate) to obtain the title compound (2.11 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.40 (2H, m), 0.59-0.72 (2H, m), 1.15-1.30 (1H, m), 1.33 (3H, d, J = 6.4 Hz), 3.78 (2H, d, J = 6.8 Hz), 3.83-4.03 (3H, m), 5.29 (2H, s), 6.59-6.78 (3H, m), 7.19-7.26 (1H, m), 7.37 (1H, t, J = 8.5 Hz), 7.82 (1H, d, J = 3.0 Hz).
1-[(6-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-3-イル)オキシ]プロパン-2-オール(2.07 g)およびトリエチルアミン(1.66 mL)のTHF(15 mL)溶液に、塩化メタンスルホニル(0.692 mL)を室温で加え、室温で10分間撹拌した。反応混合物に飽和重炭酸ナトリウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をDMF(10 mL)に溶解させ、アジ化ナトリウム(1.94 g)を室温で加え、100℃で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(酢酸エチル)で精製して標題化合物(2.11 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.40 (2H, m), 0.59-0.72 (2H, m), 1.15-1.30 (1H, m), 1.33 (3H, d, J = 6.4 Hz), 3.78 (2H, d, J = 6.8 Hz), 3.83-4.03 (3H, m), 5.29 (2H, s), 6.59-6.78 (3H, m), 7.19-7.26 (1H, m), 7.37 (1H, t, J = 8.5 Hz), 7.82 (1H, d, J = 3.0 Hz). D) 5- (2-Azidopropoxy) -2-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridine 1-[(6-{[4- (cyclopropylmethoxy) -2-fluoro To a solution of (benzyl) oxy} pyridin-3-yl) oxy] propan-2-ol (2.07 g) and triethylamine (1.66 mL) in THF (15 mL) at room temperature was added methanesulfonyl chloride (0.692 mL). Stir for 10 minutes. Saturated aqueous sodium bicarbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was dissolved in DMF (10 mL), sodium azide (1.94 g) was added at room temperature, and the mixture was stirred at 100 ° C. for 3 hr. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel chromatography (ethyl acetate) to obtain the title compound (2.11 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.40 (2H, m), 0.59-0.72 (2H, m), 1.15-1.30 (1H, m), 1.33 (3H, d, J = 6.4 Hz), 3.78 (2H, d, J = 6.8 Hz), 3.83-4.03 (3H, m), 5.29 (2H, s), 6.59-6.78 (3H, m), 7.19-7.26 (1H, m), 7.37 (1H, t, J = 8.5 Hz), 7.82 (1H, d, J = 3.0 Hz).
E) N-{2-[(6-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-3-イル)オキシ]-1-メチルエチル}アセトアミド
5-(2-アジドプロポキシ)-2-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン(2.11 g)のTHF(15 mL)溶液に、トリフェニルホスフィン(1.78 g)と水(1 mL)を加えた後、60℃で15時間撹拌した。溶媒を減圧下留去して得られた残渣を0.5 M 塩酸で酸性とした後、酢酸エチルで洗浄し、水層を飽和炭酸水素ナトリウムで塩基性とした。この水層に酢酸エチル(10 mL)と無水酢酸(10 mL)を加え、30分間撹拌した。反応混合物を酢酸エチルで抽出し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.11 g)を得た。 E) N- {2-[(6-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-3-yl) oxy] -1-methylethyl} acetamide 5- (2-azidopropoxy ) -2-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridine (2.11 g) in THF (15 mL) was added triphenylphosphine (1.78 g) and water (1 mL). After that, the mixture was stirred at 60 ° C. for 15 hours. The residue obtained by evaporating the solvent under reduced pressure was acidified with 0.5 M hydrochloric acid, washed with ethyl acetate, and the aqueous layer was basified with saturated sodium bicarbonate. Ethyl acetate (10 mL) and acetic anhydride (10 mL) were added to this aqueous layer and stirred for 30 minutes. The reaction mixture was extracted with ethyl acetate and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.11 g).
5-(2-アジドプロポキシ)-2-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン(2.11 g)のTHF(15 mL)溶液に、トリフェニルホスフィン(1.78 g)と水(1 mL)を加えた後、60℃で15時間撹拌した。溶媒を減圧下留去して得られた残渣を0.5 M 塩酸で酸性とした後、酢酸エチルで洗浄し、水層を飽和炭酸水素ナトリウムで塩基性とした。この水層に酢酸エチル(10 mL)と無水酢酸(10 mL)を加え、30分間撹拌した。反応混合物を酢酸エチルで抽出し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.11 g)を得た。 E) N- {2-[(6-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-3-yl) oxy] -1-methylethyl} acetamide 5- (2-azidopropoxy ) -2-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridine (2.11 g) in THF (15 mL) was added triphenylphosphine (1.78 g) and water (1 mL). After that, the mixture was stirred at 60 ° C. for 15 hours. The residue obtained by evaporating the solvent under reduced pressure was acidified with 0.5 M hydrochloric acid, washed with ethyl acetate, and the aqueous layer was basified with saturated sodium bicarbonate. Ethyl acetate (10 mL) and acetic anhydride (10 mL) were added to this aqueous layer and stirred for 30 minutes. The reaction mixture was extracted with ethyl acetate and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (1.11 g).
実施例22
N-{(1S)-2-[(5-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-2-イル)オキシ]-1-メチルエチル}アセトアミド Example 22
N-{(1S) -2-[(5-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-2-yl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(5-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-2-イル)オキシ]-1-メチルエチル}アセトアミド Example 22
N-{(1S) -2-[(5-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-2-yl) oxy] -1-methylethyl} acetamide
A) tert-ブチル{(1S)-2-[(5-ブロモピリジン-2-イル)オキシ]-1-メチルエチル}カルバマート
5-ブロモピリジン-2-オール(5.88 g)、tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(8.88 g)およびトリフェニルホスフィン(13.3 g)のTHF(50 mL)溶液にアゾジカルボン酸ジイソプロピルのトルエン溶液(1.9 M、26.7 mL)を0℃で滴下して、室温で15時間撹拌した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(10.0 g)を得た。
MS (ESI+): [M+H]+ 332.9. A) tert-butyl {(1S) -2-[(5-bromopyridin-2-yl) oxy] -1-methylethyl} carbamate 5-bromopyridin-2-ol (5.88 g), tert-butyl [( 1S) -2-Hydroxy-1-methylethyl] carbamate (8.88 g) and triphenylphosphine (13.3 g) in THF (50 mL) were added diisopropyl azodicarboxylate in toluene (1.9 M, 26.7 mL) at 0 ° C. Was added dropwise and stirred at room temperature for 15 hours. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (10.0 g).
MS (ESI +): [M + H] + 332.9.
5-ブロモピリジン-2-オール(5.88 g)、tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(8.88 g)およびトリフェニルホスフィン(13.3 g)のTHF(50 mL)溶液にアゾジカルボン酸ジイソプロピルのトルエン溶液(1.9 M、26.7 mL)を0℃で滴下して、室温で15時間撹拌した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(10.0 g)を得た。
MS (ESI+): [M+H]+ 332.9. A) tert-butyl {(1S) -2-[(5-bromopyridin-2-yl) oxy] -1-methylethyl} carbamate 5-bromopyridin-2-ol (5.88 g), tert-butyl [( 1S) -2-Hydroxy-1-methylethyl] carbamate (8.88 g) and triphenylphosphine (13.3 g) in THF (50 mL) were added diisopropyl azodicarboxylate in toluene (1.9 M, 26.7 mL) at 0 ° C. Was added dropwise and stirred at room temperature for 15 hours. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate) to obtain the title compound (10.0 g).
MS (ESI +): [M + H] + 332.9.
B) N-{(1S)-2-[(5-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}ピリジン-2-イル)オキシ]-1-メチルエチル}アセトアミド
tert-ブチル{(1S)-2-[(5-ブロモピリジン-2-イル)オキシ]-1-メチルエチル}カルバマートを用いて実施例21の工程B、実施例2の工程E、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 B) N-{(1S) -2-[(5-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-2-yl) oxy] -1-methylethyl} acetamide tert-butyl Step 21 of Example 21, Step E of Example 2, Step E of Example 1 using {(1S) -2-[(5-bromopyridin-2-yl) oxy] -1-methylethyl} carbamate The title compound was obtained by a method similar to that described above or a method analogous thereto.
tert-ブチル{(1S)-2-[(5-ブロモピリジン-2-イル)オキシ]-1-メチルエチル}カルバマートを用いて実施例21の工程B、実施例2の工程E、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 B) N-{(1S) -2-[(5-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} pyridin-2-yl) oxy] -1-methylethyl} acetamide tert-butyl Step 21 of Example 21, Step E of Example 2, Step E of Example 1 using {(1S) -2-[(5-bromopyridin-2-yl) oxy] -1-methylethyl} carbamate The title compound was obtained by a method similar to that described above or a method analogous thereto.
実施例23
N-{(1S)-2-[(1-{[4-(シクロプロピルメトキシ)フェニル]アセチル}ピペリジン-4-イル)オキシ]-1-メチルエチル}アセトアミド Example 23
N-{(1S) -2-[(1-{[4- (cyclopropylmethoxy) phenyl] acetyl} piperidin-4-yl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(1-{[4-(シクロプロピルメトキシ)フェニル]アセチル}ピペリジン-4-イル)オキシ]-1-メチルエチル}アセトアミド Example 23
N-{(1S) -2-[(1-{[4- (cyclopropylmethoxy) phenyl] acetyl} piperidin-4-yl) oxy] -1-methylethyl} acetamide
A) tert-ブチル[(1S)-1-メチル-2-(ピリジン-4-イルオキシ)エチル]カルバマート
ピリジン-4-オール(1.00 g)、tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(1.84 g)、1,1'-(アゾジカルボニル)ジピペリジン(3.18 g)、トリブチルホスフィン(2.55 g)、THF(50 mL)およびDMF(5 mL)の混合物を70℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(890 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.11 (3H, d, J = 6.8 Hz), 1.38 (9H, s), 3.72-4.02 (3H, m), 6.82-7.06 (3H, m), 8.27-8.49 (2H, m). A) tert-butyl [(1S) -1-methyl-2- (pyridin-4-yloxy) ethyl] carbamate pyridin-4-ol (1.00 g), tert-butyl [(1S) -2-hydroxy-1- Methylethyl] carbamate (1.84 g), 1,1 ′-(azodicarbonyl) dipiperidine (3.18 g), tributylphosphine (2.55 g), THF (50 mL) and DMF (5 mL) at 70 ° C. overnight. Stir. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (890 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.11 (3H, d, J = 6.8 Hz), 1.38 (9H, s), 3.72-4.02 (3H, m), 6.82-7.06 (3H, m), 8.27-8.49 (2H, m).
ピリジン-4-オール(1.00 g)、tert-ブチル[(1S)-2-ヒドロキシ-1-メチルエチル]カルバマート(1.84 g)、1,1'-(アゾジカルボニル)ジピペリジン(3.18 g)、トリブチルホスフィン(2.55 g)、THF(50 mL)およびDMF(5 mL)の混合物を70℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(890 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.11 (3H, d, J = 6.8 Hz), 1.38 (9H, s), 3.72-4.02 (3H, m), 6.82-7.06 (3H, m), 8.27-8.49 (2H, m). A) tert-butyl [(1S) -1-methyl-2- (pyridin-4-yloxy) ethyl] carbamate pyridin-4-ol (1.00 g), tert-butyl [(1S) -2-hydroxy-1- Methylethyl] carbamate (1.84 g), 1,1 ′-(azodicarbonyl) dipiperidine (3.18 g), tributylphosphine (2.55 g), THF (50 mL) and DMF (5 mL) at 70 ° C. overnight. Stir. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (890 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.11 (3H, d, J = 6.8 Hz), 1.38 (9H, s), 3.72-4.02 (3H, m), 6.82-7.06 (3H, m), 8.27-8.49 (2H, m).
B) tert-ブチル [(1S)-1-メチル-2-(ピペリジン-4-イルオキシ)エチル]カルバマート
水素雰囲気下、tert-ブチル [(1S)-1-メチル-2-(ピリジン-4-イルオキシ)エチル]カルバマート(890 mg)、酸化白金(90 mg)、エタノール(10 mL)および酢酸(10 mL)の混合物を室温で5時間、続いて50℃で終夜撹拌した。触媒を濾過して除去した後、濾液を減圧下濃縮した。残渣に水を加え、酢酸エチルで洗浄した。得られた水層に飽和炭酸カリウム水溶液を加え塩基性とした後に、酢酸エチル/THF(4/1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して標題化合物(520 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.99 (3H, d, J = 6.4 Hz), 1.24-1.46 (11H, m), 1.66-1.83 (2H, m), 2.30-2.46 (2H, m), 2.71-2.97 (2H, m), 3.07-3.66 (5H, m), 6.60 (1H, d, J = 7.9 Hz). B) tert-butyl [(1S) -1-methyl-2- (piperidin-4-yloxy) ethyl] carbamate Under hydrogen atmosphere, tert-butyl [(1S) -1-methyl-2- (pyridin-4-yloxy) ) Ethyl] carbamate (890 mg), platinum oxide (90 mg), ethanol (10 mL) and acetic acid (10 mL) were stirred at room temperature for 5 h followed by 50 ° C. overnight. After removing the catalyst by filtration, the filtrate was concentrated under reduced pressure. Water was added to the residue and washed with ethyl acetate. The resulting aqueous layer was made basic by adding saturated aqueous potassium carbonate solution, and then extracted with ethyl acetate / THF (4/1). The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (520 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.99 (3H, d, J = 6.4 Hz), 1.24-1.46 (11H, m), 1.66-1.83 (2H, m), 2.30-2.46 (2H, m ), 2.71-2.97 (2H, m), 3.07-3.66 (5H, m), 6.60 (1H, d, J = 7.9 Hz).
水素雰囲気下、tert-ブチル [(1S)-1-メチル-2-(ピリジン-4-イルオキシ)エチル]カルバマート(890 mg)、酸化白金(90 mg)、エタノール(10 mL)および酢酸(10 mL)の混合物を室温で5時間、続いて50℃で終夜撹拌した。触媒を濾過して除去した後、濾液を減圧下濃縮した。残渣に水を加え、酢酸エチルで洗浄した。得られた水層に飽和炭酸カリウム水溶液を加え塩基性とした後に、酢酸エチル/THF(4/1)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して標題化合物(520 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.99 (3H, d, J = 6.4 Hz), 1.24-1.46 (11H, m), 1.66-1.83 (2H, m), 2.30-2.46 (2H, m), 2.71-2.97 (2H, m), 3.07-3.66 (5H, m), 6.60 (1H, d, J = 7.9 Hz). B) tert-butyl [(1S) -1-methyl-2- (piperidin-4-yloxy) ethyl] carbamate Under hydrogen atmosphere, tert-butyl [(1S) -1-methyl-2- (pyridin-4-yloxy) ) Ethyl] carbamate (890 mg), platinum oxide (90 mg), ethanol (10 mL) and acetic acid (10 mL) were stirred at room temperature for 5 h followed by 50 ° C. overnight. After removing the catalyst by filtration, the filtrate was concentrated under reduced pressure. Water was added to the residue and washed with ethyl acetate. The resulting aqueous layer was made basic by adding saturated aqueous potassium carbonate solution, and then extracted with ethyl acetate / THF (4/1). The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (520 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.99 (3H, d, J = 6.4 Hz), 1.24-1.46 (11H, m), 1.66-1.83 (2H, m), 2.30-2.46 (2H, m ), 2.71-2.97 (2H, m), 3.07-3.66 (5H, m), 6.60 (1H, d, J = 7.9 Hz).
C) tert-ブチル{(1S)-2-[(1-{[4-(シクロプロピルメトキシ)フェニル]アセチル}ピペリジン-4-イル)オキシ]-1-メチルエチル}カルバマート
[4-(シクロプロピルメトキシ)フェニル]酢酸(120 mg)、tert-ブチル [(1S)-1-メチル-2-(ピペリジン-4-イルオキシ)エチル]カルバマート(150 mg)、WSC(167 mg)、HOBt(118 mg)、トリエチルアミン(0.121 mL)、およびDMF(2 mL)の混合物を室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(214 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.21-0.38 (2H, m), 0.45-0.61 (2H, m), 0.98 (3H, d, J = 6.4 Hz), 1.10-1.47 (12H, m), 1.53-1.81 (2H, m), 3.00-3.88 (12H, m), 6.55-6.73 (1H, m), 6.76-6.93 (2H, m), 7.03-7.19 (2H, m). C) tert-butyl {(1S) -2-[(1-{[4- (cyclopropylmethoxy) phenyl] acetyl} piperidin-4-yl) oxy] -1-methylethyl} carbamate [4- (cyclopropyl Methoxy) phenyl] acetic acid (120 mg), tert-butyl [(1S) -1-methyl-2- (piperidin-4-yloxy) ethyl] carbamate (150 mg), WSC (167 mg), HOBt (118 mg) , Triethylamine (0.121 mL), and DMF (2 mL) were stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (214 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.21-0.38 (2H, m), 0.45-0.61 (2H, m), 0.98 (3H, d, J = 6.4 Hz), 1.10-1.47 (12H, m ), 1.53-1.81 (2H, m), 3.00-3.88 (12H, m), 6.55-6.73 (1H, m), 6.76-6.93 (2H, m), 7.03-7.19 (2H, m).
[4-(シクロプロピルメトキシ)フェニル]酢酸(120 mg)、tert-ブチル [(1S)-1-メチル-2-(ピペリジン-4-イルオキシ)エチル]カルバマート(150 mg)、WSC(167 mg)、HOBt(118 mg)、トリエチルアミン(0.121 mL)、およびDMF(2 mL)の混合物を室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(214 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.21-0.38 (2H, m), 0.45-0.61 (2H, m), 0.98 (3H, d, J = 6.4 Hz), 1.10-1.47 (12H, m), 1.53-1.81 (2H, m), 3.00-3.88 (12H, m), 6.55-6.73 (1H, m), 6.76-6.93 (2H, m), 7.03-7.19 (2H, m). C) tert-butyl {(1S) -2-[(1-{[4- (cyclopropylmethoxy) phenyl] acetyl} piperidin-4-yl) oxy] -1-methylethyl} carbamate [4- (cyclopropyl Methoxy) phenyl] acetic acid (120 mg), tert-butyl [(1S) -1-methyl-2- (piperidin-4-yloxy) ethyl] carbamate (150 mg), WSC (167 mg), HOBt (118 mg) , Triethylamine (0.121 mL), and DMF (2 mL) were stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (214 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.21-0.38 (2H, m), 0.45-0.61 (2H, m), 0.98 (3H, d, J = 6.4 Hz), 1.10-1.47 (12H, m ), 1.53-1.81 (2H, m), 3.00-3.88 (12H, m), 6.55-6.73 (1H, m), 6.76-6.93 (2H, m), 7.03-7.19 (2H, m).
D) N-{(1S)-2-[(1-{[4-(シクロプロピルメトキシ)フェニル]アセチル}ピペリジン-4-イル)オキシ]-1-メチルエチル}アセトアミド
tert-ブチル{(1S)-2-[(1-{[4-(シクロプロピルメトキシ)フェニル]アセチル}ピペリジン-4-イル)オキシ]-1-メチルエチル}カルバマートを用い、実施例7の工程Bと同様の方法またはそれに準ずる方法により標題化合物を得た。 D) N-{(1S) -2-[(1-{[4- (cyclopropylmethoxy) phenyl] acetyl} piperidin-4-yl) oxy] -1-methylethyl} acetamide tert-butyl {(1S) -2-[(1-{[4- (cyclopropylmethoxy) phenyl] acetyl} piperidin-4-yl) oxy] -1-methylethyl} carbamate, or a method similar to or similar to step B of Example 7. The title compound was obtained by a similar method.
tert-ブチル{(1S)-2-[(1-{[4-(シクロプロピルメトキシ)フェニル]アセチル}ピペリジン-4-イル)オキシ]-1-メチルエチル}カルバマートを用い、実施例7の工程Bと同様の方法またはそれに準ずる方法により標題化合物を得た。 D) N-{(1S) -2-[(1-{[4- (cyclopropylmethoxy) phenyl] acetyl} piperidin-4-yl) oxy] -1-methylethyl} acetamide tert-butyl {(1S) -2-[(1-{[4- (cyclopropylmethoxy) phenyl] acetyl} piperidin-4-yl) oxy] -1-methylethyl} carbamate, or a method similar to or similar to step B of Example 7. The title compound was obtained by a similar method.
実施例24
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
実施例2の工程Dと同様の方法またはそれに準ずる方法により得たtert-ブチル {(1S)-2-[(4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマートのトランス/シス混合物を用いて、実施例3の工程Bおよび実施例2の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 24
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide Same as step D of Example 2 Using a trans / cis mixture of tert-butyl {(1S) -2-[(4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate obtained by the method or a method analogous thereto; The title compound was obtained by a method similar to that in Step E of Example 2 or a method analogous thereto.
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
実施例2の工程Dと同様の方法またはそれに準ずる方法により得たtert-ブチル {(1S)-2-[(4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマートのトランス/シス混合物を用いて、実施例3の工程Bおよび実施例2の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 24
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide Same as step D of Example 2 Using a trans / cis mixture of tert-butyl {(1S) -2-[(4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate obtained by the method or a method analogous thereto; The title compound was obtained by a method similar to that in Step E of Example 2 or a method analogous thereto.
実施例25
N-{(1S)-2-[(cis-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 25
N-{(1S) -2-[(cis-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(cis-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 25
N-{(1S) -2-[(cis-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) tert-ブチル{(1S)-2-[(cis-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマート
実施例2の工程Dと同様の方法またはそれに準ずる方法により得たtert-ブチル {(1S)-2-[(4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマートのトランス/シス混合物を、シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.00 (3H, d, J = 6.3 Hz), 1.32-1.54 (15H, m), 1.60-1.75 (2H, m), 3.02-3.20 (1H, m), 3.21-3.34 (2H, m), 3.43-3.64 (2H, m), 4.35 (1H, d, J = 4.2 Hz), 6.57 (1H, d, J = 7.8 Hz). A) tert-butyl {(1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate tert-butyl obtained by a method similar to or equivalent to step D of Example 2 A trans / cis mixture of {(1S) -2-[(4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.00 (3H, d, J = 6.3 Hz), 1.32-1.54 (15H, m), 1.60-1.75 (2H, m), 3.02-3.20 (1H, m ), 3.21-3.34 (2H, m), 3.43-3.64 (2H, m), 4.35 (1H, d, J = 4.2 Hz), 6.57 (1H, d, J = 7.8 Hz).
実施例2の工程Dと同様の方法またはそれに準ずる方法により得たtert-ブチル {(1S)-2-[(4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマートのトランス/シス混合物を、シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.00 (3H, d, J = 6.3 Hz), 1.32-1.54 (15H, m), 1.60-1.75 (2H, m), 3.02-3.20 (1H, m), 3.21-3.34 (2H, m), 3.43-3.64 (2H, m), 4.35 (1H, d, J = 4.2 Hz), 6.57 (1H, d, J = 7.8 Hz). A) tert-butyl {(1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate tert-butyl obtained by a method similar to or equivalent to step D of Example 2 A trans / cis mixture of {(1S) -2-[(4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.00 (3H, d, J = 6.3 Hz), 1.32-1.54 (15H, m), 1.60-1.75 (2H, m), 3.02-3.20 (1H, m ), 3.21-3.34 (2H, m), 3.43-3.64 (2H, m), 4.35 (1H, d, J = 4.2 Hz), 6.57 (1H, d, J = 7.8 Hz).
B) N-{(1S)-2-[(cis-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
tert-ブチル{(1S)-2-[(cis-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマートを用いて、実施例3の工程Bおよび実施例2の工程Eと同様の方法またはそれに準ずる方法により、標題化合物を得た。 B) N-{(1S) -2-[(cis-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide tert-butyl {( 1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate by a method similar to or equivalent to Step B of Example 3 and Step E of Example 2, The title compound was obtained.
tert-ブチル{(1S)-2-[(cis-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマートを用いて、実施例3の工程Bおよび実施例2の工程Eと同様の方法またはそれに準ずる方法により、標題化合物を得た。 B) N-{(1S) -2-[(cis-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide tert-butyl {( 1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate by a method similar to or equivalent to Step B of Example 3 and Step E of Example 2, The title compound was obtained.
実施例26
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-3-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)-3-フルオロフェニル]メタノールを用いて実施例2の工程Aおよび工程Fと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 26
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -3-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (cyclopropylmethoxy ) -3-Fluorophenyl] methanol was used to obtain the title compound by a method similar to or similar to Step A and Step F of Example 2.
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-3-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)-3-フルオロフェニル]メタノールを用いて実施例2の工程Aおよび工程Fと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 26
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -3-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (cyclopropylmethoxy ) -3-Fluorophenyl] methanol was used to obtain the title compound by a method similar to or similar to Step A and Step F of Example 2.
実施例27
N-{(1S)-2-[(trans-4-{[3-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[3-クロロ-4-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例2の工程Aおよび工程Fと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 27
N-{(1S) -2-[(trans-4-{[3-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [3-chloro-4- The title compound was obtained in the same manner as in Step A and Step F of Example 2 or a method analogous thereto using (cyclopropylmethoxy) phenyl] methanol.
N-{(1S)-2-[(trans-4-{[3-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[3-クロロ-4-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例2の工程Aおよび工程Fと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 27
N-{(1S) -2-[(trans-4-{[3-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [3-chloro-4- The title compound was obtained in the same manner as in Step A and Step F of Example 2 or a method analogous thereto using (cyclopropylmethoxy) phenyl] methanol.
実施例28
N-[2-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)-1-メチルエチル]-3,4-ジメチルイソオキサゾール-5-アミン Example 28
N- [2- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] -3,4-dimethylisoxazol-5-amine
N-[2-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)-1-メチルエチル]-3,4-ジメチルイソオキサゾール-5-アミン Example 28
N- [2- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] -3,4-dimethylisoxazol-5-amine
A) 4-(2-ヒドロキシプロポキシ)フェノール
4-(ベンジルオキシ)フェノール(8.0 g)、クロロアセトン(7.6 mL)および炭酸セシウム(30 g)のDMF(40 mL)懸濁液を80℃で5時間撹拌した。さらにヨウ化ナトリウム(6.6 g)、クロロアセトン(3.8 mL)および炭酸セシウム(6.6 g)を加え、80℃で終夜撹拌した。水を加え、酢酸エチルで2度抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を留去し得られた残渣にメタノール(4 mL)およびTHF(40 mL)を加え、水素化ホウ素ナトリウム(3.0 g)を少量ずつ加えた。反応混合物を室温で30分間撹拌した。反応混合物に水を加え、酢酸エチルで2度抽出し、合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去した後に、ジイソプロピルエーテルで洗浄して得た固体、10%パラジウム-炭素(50%含水、2 g)、メタノール(20 mL)およびTHF(20 mL)の混合物を水素雰囲気下室温で2時間撹拌した。反応混合物をセライト濾過した後、溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(2.16 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.27 (3H, d, J = 6.2 Hz), 2.38 (1H, brs), 3.73 (1H, dd, J = 9.2, 7.7 Hz), 3.82-3.95 (1H, m), 4.05-4.24 (1H, m), 4.69 (1H, s), 6.69-6.90 (4H, m). A) 4- (2-hydroxypropoxy) phenol 4- (benzyloxy) phenol (8.0 g), chloroacetone (7.6 mL) and cesium carbonate (30 g) in DMF (40 mL) Stir for hours. Further, sodium iodide (6.6 g), chloroacetone (3.8 mL) and cesium carbonate (6.6 g) were added, and the mixture was stirred at 80 ° C. overnight. Water was added and extracted twice with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. Methanol (4 mL) and THF (40 mL) were added to the residue obtained by evaporating the solvent, and sodium borohydride (3.0 g) was added little by little. The reaction mixture was stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The combined organic layers were washed with saturated brine and dried over anhydrous magnesium sulfate. After evaporating the solvent under reduced pressure, a mixture of solid obtained by washing with diisopropyl ether, 10% palladium-carbon (50% water content, 2 g), methanol (20 mL) and THF (20 mL) was placed under a hydrogen atmosphere. Stir at room temperature for 2 hours. The reaction mixture was filtered through celite, the solvent was evaporated under reduced pressure, and the obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to give the title compound (2.16 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.27 (3H, d, J = 6.2 Hz), 2.38 (1H, brs), 3.73 (1H, dd, J = 9.2, 7.7 Hz), 3.82-3.95 (1H, m), 4.05-4.24 (1H, m), 4.69 (1H, s), 6.69-6.90 (4H, m).
4-(ベンジルオキシ)フェノール(8.0 g)、クロロアセトン(7.6 mL)および炭酸セシウム(30 g)のDMF(40 mL)懸濁液を80℃で5時間撹拌した。さらにヨウ化ナトリウム(6.6 g)、クロロアセトン(3.8 mL)および炭酸セシウム(6.6 g)を加え、80℃で終夜撹拌した。水を加え、酢酸エチルで2度抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を留去し得られた残渣にメタノール(4 mL)およびTHF(40 mL)を加え、水素化ホウ素ナトリウム(3.0 g)を少量ずつ加えた。反応混合物を室温で30分間撹拌した。反応混合物に水を加え、酢酸エチルで2度抽出し、合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去した後に、ジイソプロピルエーテルで洗浄して得た固体、10%パラジウム-炭素(50%含水、2 g)、メタノール(20 mL)およびTHF(20 mL)の混合物を水素雰囲気下室温で2時間撹拌した。反応混合物をセライト濾過した後、溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(2.16 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.27 (3H, d, J = 6.2 Hz), 2.38 (1H, brs), 3.73 (1H, dd, J = 9.2, 7.7 Hz), 3.82-3.95 (1H, m), 4.05-4.24 (1H, m), 4.69 (1H, s), 6.69-6.90 (4H, m). A) 4- (2-hydroxypropoxy) phenol 4- (benzyloxy) phenol (8.0 g), chloroacetone (7.6 mL) and cesium carbonate (30 g) in DMF (40 mL) Stir for hours. Further, sodium iodide (6.6 g), chloroacetone (3.8 mL) and cesium carbonate (6.6 g) were added, and the mixture was stirred at 80 ° C. overnight. Water was added and extracted twice with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. Methanol (4 mL) and THF (40 mL) were added to the residue obtained by evaporating the solvent, and sodium borohydride (3.0 g) was added little by little. The reaction mixture was stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The combined organic layers were washed with saturated brine and dried over anhydrous magnesium sulfate. After evaporating the solvent under reduced pressure, a mixture of solid obtained by washing with diisopropyl ether, 10% palladium-carbon (50% water content, 2 g), methanol (20 mL) and THF (20 mL) was placed under a hydrogen atmosphere. Stir at room temperature for 2 hours. The reaction mixture was filtered through celite, the solvent was evaporated under reduced pressure, and the obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to give the title compound (2.16 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.27 (3H, d, J = 6.2 Hz), 2.38 (1H, brs), 3.73 (1H, dd, J = 9.2, 7.7 Hz), 3.82-3.95 (1H, m), 4.05-4.24 (1H, m), 4.69 (1H, s), 6.69-6.90 (4H, m).
B) 1-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)プロパン-2-オール
[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール(1.14 g)のトルエン(7 mL)混合物に塩化チオニル(0.736 mL)を加え、室温で30分間撹拌した。溶媒を留去し得られた残渣に、DMF(3.4 mL)、4-(2-ヒドロキシプロポキシ)フェノール(570 mg)および炭酸カリウム(936 mg)を加え、室温で終夜撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、NHシリカゲルカラムショートカラム(酢酸エチル)に通した後、溶媒を減圧下留去した。得られた固体をヘキサンで洗浄し標題化合物(1.03 g)を得た。
MS (ESI-): [M-H]- 345.2. B) 1- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) propan-2-ol [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol (1.14 g) Thionyl chloride (0.736 mL) was added to a toluene (7 mL) mixture, and the mixture was stirred at room temperature for 30 minutes. To the residue obtained by evaporating the solvent, DMF (3.4 mL), 4- (2-hydroxypropoxy) phenol (570 mg) and potassium carbonate (936 mg) were added, and the mixture was stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, passed through an NH silica gel column short column (ethyl acetate), and the solvent was evaporated under reduced pressure. The obtained solid was washed with hexane to obtain the title compound (1.03 g).
MS (ESI-): [MH] - 345.2.
[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノール(1.14 g)のトルエン(7 mL)混合物に塩化チオニル(0.736 mL)を加え、室温で30分間撹拌した。溶媒を留去し得られた残渣に、DMF(3.4 mL)、4-(2-ヒドロキシプロポキシ)フェノール(570 mg)および炭酸カリウム(936 mg)を加え、室温で終夜撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、NHシリカゲルカラムショートカラム(酢酸エチル)に通した後、溶媒を減圧下留去した。得られた固体をヘキサンで洗浄し標題化合物(1.03 g)を得た。
MS (ESI-): [M-H]- 345.2. B) 1- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) propan-2-ol [4- (cyclopropylmethoxy) -2-fluorophenyl] methanol (1.14 g) Thionyl chloride (0.736 mL) was added to a toluene (7 mL) mixture, and the mixture was stirred at room temperature for 30 minutes. To the residue obtained by evaporating the solvent, DMF (3.4 mL), 4- (2-hydroxypropoxy) phenol (570 mg) and potassium carbonate (936 mg) were added, and the mixture was stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, passed through an NH silica gel column short column (ethyl acetate), and the solvent was evaporated under reduced pressure. The obtained solid was washed with hexane to obtain the title compound (1.03 g).
MS (ESI-): [MH] - 345.2.
C) N-[2-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)-1-メチルエチル]-N-(3,4-ジメチルイソオキサゾール-5-イル)-4-ニトロベンゼンスルホンアミド
1-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)プロパン-2-オール(346 mg)、N-(3,4-ジメチルイソオキサゾール-5-イル)-4-ニトロベンゼンスルホンアミド(297 mg)およびトリフェニルホスフィン(314 mg)のTHF(10 mL)溶液に、アゾジカルボン酸ジイソプロピルのトルエン溶液(1.9 M、0.60 mL)を加え、室温で2日間撹拌した。溶媒を減圧下留去し、得られた残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(376 mg)を得た。
MS (ESI+): [M+H]+ 626.3. C) N- [2- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] -N- (3,4-dimethylisoxazol-5-yl ) -4-Nitrobenzenesulfonamide 1- (4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) propan-2-ol (346 mg), N- (3,4-dimethyliso To a solution of oxazol-5-yl) -4-nitrobenzenesulfonamide (297 mg) and triphenylphosphine (314 mg) in THF (10 mL) was added a toluene solution of diisopropyl azodicarboxylate (1.9 M, 0.60 mL). Stir at room temperature for 2 days. The solvent was evaporated under reduced pressure, and the obtained residue was purified by NH silica gel column chromatography (hexane / ethyl acetate) to give the title compound (376 mg).
MS (ESI +): [M + H] + 626.3.
1-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)プロパン-2-オール(346 mg)、N-(3,4-ジメチルイソオキサゾール-5-イル)-4-ニトロベンゼンスルホンアミド(297 mg)およびトリフェニルホスフィン(314 mg)のTHF(10 mL)溶液に、アゾジカルボン酸ジイソプロピルのトルエン溶液(1.9 M、0.60 mL)を加え、室温で2日間撹拌した。溶媒を減圧下留去し、得られた残渣をNHシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(376 mg)を得た。
MS (ESI+): [M+H]+ 626.3. C) N- [2- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] -N- (3,4-dimethylisoxazol-5-yl ) -4-Nitrobenzenesulfonamide 1- (4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) propan-2-ol (346 mg), N- (3,4-dimethyliso To a solution of oxazol-5-yl) -4-nitrobenzenesulfonamide (297 mg) and triphenylphosphine (314 mg) in THF (10 mL) was added a toluene solution of diisopropyl azodicarboxylate (1.9 M, 0.60 mL). Stir at room temperature for 2 days. The solvent was evaporated under reduced pressure, and the obtained residue was purified by NH silica gel column chromatography (hexane / ethyl acetate) to give the title compound (376 mg).
MS (ESI +): [M + H] + 626.3.
D) N-[2-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)-1-メチルエチル]-3,4-ジメチルイソオキサゾール-5-アミン
N-[2-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)-1-メチルエチル]-N-(3,4-ジメチルイソオキサゾール-5-イル)-4-ニトロベンゼンスルホンアミド(376 mg)のDMF(2 mL)溶液に、スルファニル酢酸(0.21 mL)と水酸化リチウム二水和物(126 mg)を加え、60℃で終夜撹拌した。飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をNHシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して得られた固体をジイソプロピルエーテルで洗浄して標題化合物(84.6 mg)を得た。 D) N- [2- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] -3,4-dimethylisoxazol-5-amine N- [ 2- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] -N- (3,4-dimethylisoxazol-5-yl) -4-nitrobenzene To a solution of sulfonamide (376 mg) in DMF (2 mL) were added sulfanylacetic acid (0.21 mL) and lithium hydroxide dihydrate (126 mg), and the mixture was stirred at 60 ° C. overnight. Saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by NH silica gel chromatography (hexane / ethyl acetate), and the resulting solid was washed with diisopropyl ether to give the title compound (84.6 mg).
N-[2-(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}フェノキシ)-1-メチルエチル]-N-(3,4-ジメチルイソオキサゾール-5-イル)-4-ニトロベンゼンスルホンアミド(376 mg)のDMF(2 mL)溶液に、スルファニル酢酸(0.21 mL)と水酸化リチウム二水和物(126 mg)を加え、60℃で終夜撹拌した。飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をNHシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して得られた固体をジイソプロピルエーテルで洗浄して標題化合物(84.6 mg)を得た。 D) N- [2- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] -3,4-dimethylisoxazol-5-amine N- [ 2- (4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} phenoxy) -1-methylethyl] -N- (3,4-dimethylisoxazol-5-yl) -4-nitrobenzene To a solution of sulfonamide (376 mg) in DMF (2 mL) were added sulfanylacetic acid (0.21 mL) and lithium hydroxide dihydrate (126 mg), and the mixture was stirred at 60 ° C. overnight. Saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by NH silica gel chromatography (hexane / ethyl acetate), and the resulting solid was washed with diisopropyl ether to give the title compound (84.6 mg).
実施例29
N-[2-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]-3,4-ジメチルイソオキサゾール-5-アミン Example 29
N- [2- (4-{[4- (Cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] -3,4-dimethylisoxazol-5-amine
N-[2-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]-3,4-ジメチルイソオキサゾール-5-アミン Example 29
N- [2- (4-{[4- (Cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] -3,4-dimethylisoxazol-5-amine
A) 1-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)プロパン-2-オール
[4-(シクロプロピルメトキシ)フェニル]メタノールおよび4-(2-ヒドロキシプロポキシ)フェノールを用いて実施例28の工程Bと同様の方法またはそれに準ずる方法により標題化合物を得た。
MS (ESI-): [M-H]- 327.4. A) Using 1- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) propan-2-ol [4- (cyclopropylmethoxy) phenyl] methanol and 4- (2-hydroxypropoxy) phenol The title compound was obtained by a method similar to that in Step B of Example 28 or a method analogous thereto.
MS (ESI-): [MH] - 327.4.
[4-(シクロプロピルメトキシ)フェニル]メタノールおよび4-(2-ヒドロキシプロポキシ)フェノールを用いて実施例28の工程Bと同様の方法またはそれに準ずる方法により標題化合物を得た。
MS (ESI-): [M-H]- 327.4. A) Using 1- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) propan-2-ol [4- (cyclopropylmethoxy) phenyl] methanol and 4- (2-hydroxypropoxy) phenol The title compound was obtained by a method similar to that in Step B of Example 28 or a method analogous thereto.
MS (ESI-): [MH] - 327.4.
B) N-[2-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]-N-(3,4-ジメチルイソオキサゾール-5-イル)-4-ニトロベンゼンスルホンアミド
1-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)プロパン-2-オールを用いて実施例28の工程Cと同様の方法またはそれに準ずる方法により標題化合物を得た。
MS (ESI+): [M+H]+ 608.5. B) N- [2- (4-{[4- (Cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] -N- (3,4-dimethylisoxazol-5-yl) -4- Nitrobenzenesulfonamide 1- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) propan-2-ol is used to give the title compound in the same manner as in Step 28 of Example 28 or a method analogous thereto. It was.
MS (ESI +): [M + H] + 608.5.
1-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)プロパン-2-オールを用いて実施例28の工程Cと同様の方法またはそれに準ずる方法により標題化合物を得た。
MS (ESI+): [M+H]+ 608.5. B) N- [2- (4-{[4- (Cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] -N- (3,4-dimethylisoxazol-5-yl) -4- Nitrobenzenesulfonamide 1- (4-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) propan-2-ol is used to give the title compound in the same manner as in Step 28 of Example 28 or a method analogous thereto. It was.
MS (ESI +): [M + H] + 608.5.
C) N-[2-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]-3,4-ジメチルイソオキサゾール-5-アミン
N-[2-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]-N-(3,4-ジメチルイソオキサゾール-5-イル)-4-ニトロベンゼンスルホンアミドを用いて実施例28の工程Dと同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N- [2- (4-{[4- (Cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] -3,4-dimethylisoxazol-5-amine N- [2- (4 Example 28 using-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] -N- (3,4-dimethylisoxazol-5-yl) -4-nitrobenzenesulfonamide The title compound was obtained by a method similar to that in Step D or a method analogous thereto.
N-[2-(4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]-N-(3,4-ジメチルイソオキサゾール-5-イル)-4-ニトロベンゼンスルホンアミドを用いて実施例28の工程Dと同様の方法またはそれに準ずる方法により標題化合物を得た。 C) N- [2- (4-{[4- (Cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] -3,4-dimethylisoxazol-5-amine N- [2- (4 Example 28 using-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] -N- (3,4-dimethylisoxazol-5-yl) -4-nitrobenzenesulfonamide The title compound was obtained by a method similar to that in Step D or a method analogous thereto.
実施例30
N-[(1S)-2-(3-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]アセトアミド
3-(ベンジルオキシ)フェノールを用いて実施例22の工程A、実施例7の工程B、実施例1の工程Cおよび実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 30
N-[(1S) -2- (3-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] acetamide Step A of Example 22 with 3- (benzyloxy) phenol The title compound was obtained in the same manner as in Step B of Example 7, Step C of Example 1 and Step E of Example 1, or a method analogous thereto.
N-[(1S)-2-(3-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]アセトアミド
3-(ベンジルオキシ)フェノールを用いて実施例22の工程A、実施例7の工程B、実施例1の工程Cおよび実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 30
N-[(1S) -2- (3-{[4- (cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] acetamide Step A of Example 22 with 3- (benzyloxy) phenol The title compound was obtained in the same manner as in Step B of Example 7, Step C of Example 1 and Step E of Example 1, or a method analogous thereto.
実施例31
N-[(1S)-3-(6-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}ピリジン-3-イル)-1-メチルプロピル]アセトアミド Example 31
N-[(1S) -3- (6-{[3- (cyclopropylmethoxy) benzyl] oxy} pyridin-3-yl) -1-methylpropyl] acetamide
N-[(1S)-3-(6-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}ピリジン-3-イル)-1-メチルプロピル]アセトアミド Example 31
N-[(1S) -3- (6-{[3- (cyclopropylmethoxy) benzyl] oxy} pyridin-3-yl) -1-methylpropyl] acetamide
A) 5-ヨードピリジン-2-イル アセタート
5-ヨードピリジン-2-オール(10 g)およびトリエチルアミン(18.82 mL)のTHF(100 mL)溶液に塩化アセチル(9.65 mL)を室温で加え、室温で2日間撹拌した。反応混合物に0℃で水を加え酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(5.20 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 2.30 (3H, s), 7.10 (1H, d, J = 9.1 Hz), 8.29 (1H, dd, J = 8.5, 2.5 Hz), 8.62 (1H, d, J = 2.3 Hz). A) 5-Iodopyridin-2-yl acetate To a solution of 5-iodopyridin-2-ol (10 g) and triethylamine (18.82 mL) in THF (100 mL) was added acetyl chloride (9.65 mL) at room temperature. Stir for 2 days. Water was added to the reaction mixture at 0 ° C., and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to give the title compound (5.20 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 2.30 (3H, s), 7.10 (1H, d, J = 9.1 Hz), 8.29 (1H, dd, J = 8.5, 2.5 Hz), 8.62 (1H, d, J = 2.3 Hz).
5-ヨードピリジン-2-オール(10 g)およびトリエチルアミン(18.82 mL)のTHF(100 mL)溶液に塩化アセチル(9.65 mL)を室温で加え、室温で2日間撹拌した。反応混合物に0℃で水を加え酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(5.20 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 2.30 (3H, s), 7.10 (1H, d, J = 9.1 Hz), 8.29 (1H, dd, J = 8.5, 2.5 Hz), 8.62 (1H, d, J = 2.3 Hz). A) 5-Iodopyridin-2-yl acetate To a solution of 5-iodopyridin-2-ol (10 g) and triethylamine (18.82 mL) in THF (100 mL) was added acetyl chloride (9.65 mL) at room temperature. Stir for 2 days. Water was added to the reaction mixture at 0 ° C., and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel chromatography (hexane / ethyl acetate) to give the title compound (5.20 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 2.30 (3H, s), 7.10 (1H, d, J = 9.1 Hz), 8.29 (1H, dd, J = 8.5, 2.5 Hz), 8.62 (1H, d, J = 2.3 Hz).
B) 5-{(3S)-3-[(tert-ブトキシカルボニル)アミノ]ブタ-1-イン-1-イル}ピリジン-2-イルアセタート
5-ヨードピリジン-2-イル アセタート(1.0 g)、tert-ブチル [(1S)-1-メチルプロパ-2-イン-1-イル]カルバマート(1.287 g)およびトリエチルアミン(1.581 mL)のDMF(26 mL)溶液に、ビストリフェニルホスフィンジクロロパラジウム(II)(0.267 g)およびヨウ化銅(I)(0.072 g)を室温で加え、窒素雰囲気下60℃で5時間撹拌した。溶媒を減圧下留去させた後、残渣に水を加え酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し、標題化合物(0.556 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.37 (3H, d, J = 7.2 Hz), 1.40 (9H, s), 2.30 (3H, s), 4.43-4.68 (1H, m), 7.23 (1H, d, J = 7.9 Hz), 7.47 (1H, d, J = 6.0 Hz), 7.96 (1H, dd, J = 8.3, 2.3 Hz), 8.40 (1H, d, J = 2.3 Hz). B) 5-{(3S) -3-[(tert-Butoxycarbonyl) amino] but-1-in-1-yl} pyridin-2-yl acetate 5-iodopyridin-2-yl acetate (1.0 g), tert 2-Butyl [(1S) -1-methylprop-2-in-1-yl] carbamate (1.287 g) and triethylamine (1.581 mL) in DMF (26 mL) were added to bistriphenylphosphine dichloropalladium (II) (0.267 g ) And copper (I) iodide (0.072 g) were added at room temperature, and the mixture was stirred at 60 ° C. for 5 hours under a nitrogen atmosphere. The solvent was evaporated under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (0.556 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.37 (3H, d, J = 7.2 Hz), 1.40 (9H, s), 2.30 (3H, s), 4.43-4.68 (1H, m), 7.23 ( 1H, d, J = 7.9 Hz), 7.47 (1H, d, J = 6.0 Hz), 7.96 (1H, dd, J = 8.3, 2.3 Hz), 8.40 (1H, d, J = 2.3 Hz).
5-ヨードピリジン-2-イル アセタート(1.0 g)、tert-ブチル [(1S)-1-メチルプロパ-2-イン-1-イル]カルバマート(1.287 g)およびトリエチルアミン(1.581 mL)のDMF(26 mL)溶液に、ビストリフェニルホスフィンジクロロパラジウム(II)(0.267 g)およびヨウ化銅(I)(0.072 g)を室温で加え、窒素雰囲気下60℃で5時間撹拌した。溶媒を減圧下留去させた後、残渣に水を加え酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し、標題化合物(0.556 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.37 (3H, d, J = 7.2 Hz), 1.40 (9H, s), 2.30 (3H, s), 4.43-4.68 (1H, m), 7.23 (1H, d, J = 7.9 Hz), 7.47 (1H, d, J = 6.0 Hz), 7.96 (1H, dd, J = 8.3, 2.3 Hz), 8.40 (1H, d, J = 2.3 Hz). B) 5-{(3S) -3-[(tert-Butoxycarbonyl) amino] but-1-in-1-yl} pyridin-2-yl acetate 5-iodopyridin-2-yl acetate (1.0 g), tert 2-Butyl [(1S) -1-methylprop-2-in-1-yl] carbamate (1.287 g) and triethylamine (1.581 mL) in DMF (26 mL) were added to bistriphenylphosphine dichloropalladium (II) (0.267 g ) And copper (I) iodide (0.072 g) were added at room temperature, and the mixture was stirred at 60 ° C. for 5 hours under a nitrogen atmosphere. The solvent was evaporated under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (0.556 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.37 (3H, d, J = 7.2 Hz), 1.40 (9H, s), 2.30 (3H, s), 4.43-4.68 (1H, m), 7.23 ( 1H, d, J = 7.9 Hz), 7.47 (1H, d, J = 6.0 Hz), 7.96 (1H, dd, J = 8.3, 2.3 Hz), 8.40 (1H, d, J = 2.3 Hz).
C) tert-ブチル[(1S)-3-(6-ヒドロキシピリジン-3-イル)-1-メチルプロピル]カルバマート
5-{(3S)-3-[(tert-ブトキシカルボニル)アミノ]ブタ-1-イン-1-イル}ピリジン-2-イル アセタート(960 mg)および10%パラジウム-炭素(50%含水、960 mg)のメタノール(17 mL)懸濁液を、水素雰囲気下室温で2時間撹拌した。反応混合物を濾過して触媒を除去した後に、濾液に1 M水酸化ナトリウム水溶液(9.0 mL)を加え、室温で3時間撹拌した。反応混合物に0℃で1 M塩酸を加えて中和した後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下濃縮して標題化合物(500 mg)を得た。
MS (ESI-): [M-H]- 265.0. C) tert-butyl [(1S) -3- (6-hydroxypyridin-3-yl) -1-methylpropyl] carbamate 5-{(3S) -3-[(tert-butoxycarbonyl) amino] buta-1 -In-1-yl} pyridin-2-yl acetate (960 mg) and 10% palladium-carbon (50% water content, 960 mg) in methanol (17 mL) were stirred in a hydrogen atmosphere at room temperature for 2 hours. did. The reaction mixture was filtered to remove the catalyst, 1 M aqueous sodium hydroxide solution (9.0 mL) was added to the filtrate, and the mixture was stirred at room temperature for 3 hr. The reaction mixture was neutralized with 1 M hydrochloric acid at 0 ° C., and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to give the title compound (500 mg).
MS (ESI-): [MH] - 265.0.
5-{(3S)-3-[(tert-ブトキシカルボニル)アミノ]ブタ-1-イン-1-イル}ピリジン-2-イル アセタート(960 mg)および10%パラジウム-炭素(50%含水、960 mg)のメタノール(17 mL)懸濁液を、水素雰囲気下室温で2時間撹拌した。反応混合物を濾過して触媒を除去した後に、濾液に1 M水酸化ナトリウム水溶液(9.0 mL)を加え、室温で3時間撹拌した。反応混合物に0℃で1 M塩酸を加えて中和した後、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下濃縮して標題化合物(500 mg)を得た。
MS (ESI-): [M-H]- 265.0. C) tert-butyl [(1S) -3- (6-hydroxypyridin-3-yl) -1-methylpropyl] carbamate 5-{(3S) -3-[(tert-butoxycarbonyl) amino] buta-1 -In-1-yl} pyridin-2-yl acetate (960 mg) and 10% palladium-carbon (50% water content, 960 mg) in methanol (17 mL) were stirred in a hydrogen atmosphere at room temperature for 2 hours. did. The reaction mixture was filtered to remove the catalyst, 1 M aqueous sodium hydroxide solution (9.0 mL) was added to the filtrate, and the mixture was stirred at room temperature for 3 hr. The reaction mixture was neutralized with 1 M hydrochloric acid at 0 ° C., and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to give the title compound (500 mg).
MS (ESI-): [MH] - 265.0.
D) N-[(1S)-3-(6-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}ピリジン-3-イル)-1-メチルプロピル]アセトアミド
tert-ブチル[(1S)-3-(6-ヒドロキシピリジン-3-イル)-1-メチルプロピル]カルバマートおよび[3-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例22の工程Aおよび実施例2の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 D) N-[(1S) -3- (6-{[3- (cyclopropylmethoxy) benzyl] oxy} pyridin-3-yl) -1-methylpropyl] acetamide tert-butyl [(1S) -3- A method similar to step A of Example 22 and Step E of Example 2 using (6-hydroxypyridin-3-yl) -1-methylpropyl] carbamate and [3- (cyclopropylmethoxy) phenyl] methanol or The title compound was obtained by a method analogous thereto.
tert-ブチル[(1S)-3-(6-ヒドロキシピリジン-3-イル)-1-メチルプロピル]カルバマートおよび[3-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例22の工程Aおよび実施例2の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 D) N-[(1S) -3- (6-{[3- (cyclopropylmethoxy) benzyl] oxy} pyridin-3-yl) -1-methylpropyl] acetamide tert-butyl [(1S) -3- A method similar to step A of Example 22 and Step E of Example 2 using (6-hydroxypyridin-3-yl) -1-methylpropyl] carbamate and [3- (cyclopropylmethoxy) phenyl] methanol or The title compound was obtained by a method analogous thereto.
実施例32
N-[(1S)-2-(4-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]アセトアミド
[3-(シクロプロピルメトキシ)フェニル]メタノールおよびN-[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]アセトアミドを用い、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 32
N-[(1S) -2- (4-{[3- (cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] acetamide [3- (cyclopropylmethoxy) phenyl] methanol and N-[( 1S) -2- (4-Hydroxyphenoxy) -1-methylethyl] acetamide was used to give the title compound by the same method as in Step E of Example 1 or a method analogous thereto.
N-[(1S)-2-(4-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}フェノキシ)-1-メチルエチル]アセトアミド
[3-(シクロプロピルメトキシ)フェニル]メタノールおよびN-[(1S)-2-(4-ヒドロキシフェノキシ)-1-メチルエチル]アセトアミドを用い、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 32
N-[(1S) -2- (4-{[3- (cyclopropylmethoxy) benzyl] oxy} phenoxy) -1-methylethyl] acetamide [3- (cyclopropylmethoxy) phenyl] methanol and N-[( 1S) -2- (4-Hydroxyphenoxy) -1-methylethyl] acetamide was used to give the title compound by the same method as in Step E of Example 1 or a method analogous thereto.
実施例33
N-[(1S)-3-(4-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)-1-メチルプロピル]アセトアミド
[3-(シクロプロピルメトキシ)フェニル]メタノールおよびN-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミドを用い、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 33
N-[(1S) -3- (4-{[3- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methylpropyl] acetamide [3- (cyclopropylmethoxy) phenyl] methanol and N-[( 1S) -3- (4-Hydroxyphenyl) -1-methylpropyl] acetamide was used to give the title compound in the same manner as in Step E of Example 1 or a method analogous thereto.
N-[(1S)-3-(4-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}フェニル)-1-メチルプロピル]アセトアミド
[3-(シクロプロピルメトキシ)フェニル]メタノールおよびN-[(1S)-3-(4-ヒドロキシフェニル)-1-メチルプロピル]アセトアミドを用い、実施例1の工程Eと同様の方法またはそれに準ずる方法により標題化合物を得た。 Example 33
N-[(1S) -3- (4-{[3- (cyclopropylmethoxy) benzyl] oxy} phenyl) -1-methylpropyl] acetamide [3- (cyclopropylmethoxy) phenyl] methanol and N-[( 1S) -3- (4-Hydroxyphenyl) -1-methylpropyl] acetamide was used to give the title compound in the same manner as in Step E of Example 1 or a method analogous thereto.
実施例34
N-[(1S)-3-(trans-4-{[4-(シクロプロピルメトキシ)-2,3-ジフルオロベンジル]オキシ}シクロヘキシル)-1-メチルプロピル]アセトアミド Example 34
N-[(1S) -3- (trans-4-{[4- (cyclopropylmethoxy) -2,3-difluorobenzyl] oxy} cyclohexyl) -1-methylpropyl] acetamide
N-[(1S)-3-(trans-4-{[4-(シクロプロピルメトキシ)-2,3-ジフルオロベンジル]オキシ}シクロヘキシル)-1-メチルプロピル]アセトアミド Example 34
N-[(1S) -3- (trans-4-{[4- (cyclopropylmethoxy) -2,3-difluorobenzyl] oxy} cyclohexyl) -1-methylpropyl] acetamide
A) tert-ブチル [(1S)-3-(trans-4-ヒドロキシシクロヘキシル)-1-メチルプロピル]カルバマート
tert-ブチル{(1S)-3-[4-(ベンジルオキシ)フェニル]-1-メチルプロパ-2-イン-1-イル}カルバマート(6.54 g)、10%パラジウム-炭素(50%含水、6.54 g)、メタノール(150 mL)およびTHF(50 mL)の混合物を水素雰囲気下(1気圧)、室温で3日間撹拌した。触媒をろ過により除去し、ろ液を減圧下濃縮した。得られた油状物、10%ロジウム-炭素(50%含水、1.0 g)およびメタノール(50 mL)の混合物を水素雰囲気下(5気圧)、60℃で6時間撹拌した。触媒を濾過により除去し、濾液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(0.95 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.74-0.92 (2H, m), 0.97 (3H, d, J = 6.4 Hz), 1.00-1.25 (5H, m), 1.25-1.49 (11H, m), 1.64 (2H, d, J = 13.2 Hz), 1.78 (2H, d, J = 9.0 Hz), 3.04-3.52 (2H, m), 4.42 (1H, d, J = 4.1 Hz), 6.57 (1H, d, J = 8.3 Hz). A) tert-butyl [(1S) -3- (trans-4-hydroxycyclohexyl) -1-methylpropyl] carbamate tert-butyl {(1S) -3- [4- (benzyloxy) phenyl] -1-methylprop -2-In-1-yl} carbamate (6.54 g), 10% palladium-carbon (50% water content, 6.54 g), methanol (150 mL) and THF (50 mL) in a hydrogen atmosphere (1 atm) And stirred at room temperature for 3 days. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. A mixture of the obtained oil, 10% rhodium-carbon (containing 50% water, 1.0 g) and methanol (50 mL) was stirred at 60 ° C. for 6 hours under a hydrogen atmosphere (5 atm). The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (0.95 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.74-0.92 (2H, m), 0.97 (3H, d, J = 6.4 Hz), 1.00-1.25 (5H, m), 1.25-1.49 (11H, m ), 1.64 (2H, d, J = 13.2 Hz), 1.78 (2H, d, J = 9.0 Hz), 3.04-3.52 (2H, m), 4.42 (1H, d, J = 4.1 Hz), 6.57 (1H , d, J = 8.3 Hz).
tert-ブチル{(1S)-3-[4-(ベンジルオキシ)フェニル]-1-メチルプロパ-2-イン-1-イル}カルバマート(6.54 g)、10%パラジウム-炭素(50%含水、6.54 g)、メタノール(150 mL)およびTHF(50 mL)の混合物を水素雰囲気下(1気圧)、室温で3日間撹拌した。触媒をろ過により除去し、ろ液を減圧下濃縮した。得られた油状物、10%ロジウム-炭素(50%含水、1.0 g)およびメタノール(50 mL)の混合物を水素雰囲気下(5気圧)、60℃で6時間撹拌した。触媒を濾過により除去し、濾液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(0.95 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.74-0.92 (2H, m), 0.97 (3H, d, J = 6.4 Hz), 1.00-1.25 (5H, m), 1.25-1.49 (11H, m), 1.64 (2H, d, J = 13.2 Hz), 1.78 (2H, d, J = 9.0 Hz), 3.04-3.52 (2H, m), 4.42 (1H, d, J = 4.1 Hz), 6.57 (1H, d, J = 8.3 Hz). A) tert-butyl [(1S) -3- (trans-4-hydroxycyclohexyl) -1-methylpropyl] carbamate tert-butyl {(1S) -3- [4- (benzyloxy) phenyl] -1-methylprop -2-In-1-yl} carbamate (6.54 g), 10% palladium-carbon (50% water content, 6.54 g), methanol (150 mL) and THF (50 mL) in a hydrogen atmosphere (1 atm) And stirred at room temperature for 3 days. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. A mixture of the obtained oil, 10% rhodium-carbon (containing 50% water, 1.0 g) and methanol (50 mL) was stirred at 60 ° C. for 6 hours under a hydrogen atmosphere (5 atm). The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (0.95 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.74-0.92 (2H, m), 0.97 (3H, d, J = 6.4 Hz), 1.00-1.25 (5H, m), 1.25-1.49 (11H, m ), 1.64 (2H, d, J = 13.2 Hz), 1.78 (2H, d, J = 9.0 Hz), 3.04-3.52 (2H, m), 4.42 (1H, d, J = 4.1 Hz), 6.57 (1H , d, J = 8.3 Hz).
B) N-[(1S)-3-(trans-4-ヒドロキシシクロヘキシル)-1-メチルプロピル]アセトアミド
4 M 塩化水素/酢酸エチル(5 mL)をtert-ブチル[(1S)-3-(trans-4-ヒドロキシシクロヘキシル)-1-メチルプロピル]カルバマート(0.60 g)の酢酸エチル溶液(10 mL)に滴下した。混合物を室温で3時間撹拌した後に、減圧下濃縮した。残渣をピリジン(10 mL)に溶解し、無水酢酸(0.23 g)を加えて、混合物を室温で1時間撹拌した。反応混合物を減圧下濃縮して得られた残渣に、メタノール(10 mL)および1 M 水酸化ナトリウム水溶液(10 mL)を加え、混合物を室温で15時間撹拌した。混合物を1 M 塩酸で中和した後に酢酸エチル/THF(5/1)混合溶媒で抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して標題化合物を白色固体(0.58 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.68-0.93 (2H, m), 0.98 (3H, d, J = 6.4 Hz), 1.00-1.19 (5H, m), 1.23-1.40 (2H, m), 1.53-1.72 (2H, m), 1.70-1.89 (5H, m), 3.12-3.44 (1H, m), 3.47 (1H, brs), 3.56-3.95 (1H, m), 7.59 (1H, d, J = 8.0 Hz). B) N-[(1S) -3- (trans-4-hydroxycyclohexyl) -1-methylpropyl] acetamide 4 M hydrogen chloride / ethyl acetate (5 mL) was added to tert-butyl [(1S) -3- (trans 4-Hydroxycyclohexyl) -1-methylpropyl] carbamate (0.60 g) was added dropwise to an ethyl acetate solution (10 mL). The mixture was stirred at room temperature for 3 hours and then concentrated under reduced pressure. The residue was dissolved in pyridine (10 mL), acetic anhydride (0.23 g) was added and the mixture was stirred at room temperature for 1 hour. Methanol (10 mL) and 1 M aqueous sodium hydroxide solution (10 mL) were added to the residue obtained by concentrating the reaction mixture under reduced pressure, and the mixture was stirred at room temperature for 15 hours. The mixture was neutralized with 1 M hydrochloric acid and extracted with a mixed solvent of ethyl acetate / THF (5/1). The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound as a white solid (0.58 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.68-0.93 (2H, m), 0.98 (3H, d, J = 6.4 Hz), 1.00-1.19 (5H, m), 1.23-1.40 (2H, m ), 1.53-1.72 (2H, m), 1.70-1.89 (5H, m), 3.12-3.44 (1H, m), 3.47 (1H, brs), 3.56-3.95 (1H, m), 7.59 (1H, d , J = 8.0 Hz).
4 M 塩化水素/酢酸エチル(5 mL)をtert-ブチル[(1S)-3-(trans-4-ヒドロキシシクロヘキシル)-1-メチルプロピル]カルバマート(0.60 g)の酢酸エチル溶液(10 mL)に滴下した。混合物を室温で3時間撹拌した後に、減圧下濃縮した。残渣をピリジン(10 mL)に溶解し、無水酢酸(0.23 g)を加えて、混合物を室温で1時間撹拌した。反応混合物を減圧下濃縮して得られた残渣に、メタノール(10 mL)および1 M 水酸化ナトリウム水溶液(10 mL)を加え、混合物を室温で15時間撹拌した。混合物を1 M 塩酸で中和した後に酢酸エチル/THF(5/1)混合溶媒で抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して標題化合物を白色固体(0.58 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.68-0.93 (2H, m), 0.98 (3H, d, J = 6.4 Hz), 1.00-1.19 (5H, m), 1.23-1.40 (2H, m), 1.53-1.72 (2H, m), 1.70-1.89 (5H, m), 3.12-3.44 (1H, m), 3.47 (1H, brs), 3.56-3.95 (1H, m), 7.59 (1H, d, J = 8.0 Hz). B) N-[(1S) -3- (trans-4-hydroxycyclohexyl) -1-methylpropyl] acetamide 4 M hydrogen chloride / ethyl acetate (5 mL) was added to tert-butyl [(1S) -3- (trans 4-Hydroxycyclohexyl) -1-methylpropyl] carbamate (0.60 g) was added dropwise to an ethyl acetate solution (10 mL). The mixture was stirred at room temperature for 3 hours and then concentrated under reduced pressure. The residue was dissolved in pyridine (10 mL), acetic anhydride (0.23 g) was added and the mixture was stirred at room temperature for 1 hour. Methanol (10 mL) and 1 M aqueous sodium hydroxide solution (10 mL) were added to the residue obtained by concentrating the reaction mixture under reduced pressure, and the mixture was stirred at room temperature for 15 hours. The mixture was neutralized with 1 M hydrochloric acid and extracted with a mixed solvent of ethyl acetate / THF (5/1). The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound as a white solid (0.58 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.68-0.93 (2H, m), 0.98 (3H, d, J = 6.4 Hz), 1.00-1.19 (5H, m), 1.23-1.40 (2H, m ), 1.53-1.72 (2H, m), 1.70-1.89 (5H, m), 3.12-3.44 (1H, m), 3.47 (1H, brs), 3.56-3.95 (1H, m), 7.59 (1H, d , J = 8.0 Hz).
C) [4-(シクロプロピルメトキシ)-2,3-ジフルオロフェニル]メタノール
2,3-ジフルオロ-4-ヒドロキシベンズアルデヒド(1.00 g)のDMF(10 mL)溶液に炭酸カリウム(1.75 g)と(ブロモメチル)シクロプロパン(0.920 mL)を加えて、70℃で1時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣のTHF(10 mL)溶液にメタノール(5 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(107 mg)を加えて、20分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(1.05 g)として得た。
1H NMR (300 MHz, CDCl3) δ 0.30-0.41 (2H, m), 0.59-0.71 (2H, m), 1.21-1.39 (1H, m), 1.71 (1H, t, J = 6.1 Hz), 3.89 (2H, d, J = 6.8 Hz), 4.70 (2H, d, J = 6.1 Hz), 6.64-6.77 (1H, m), 6.99-7.12 (1H, m). C) [4- (Cyclopropylmethoxy) -2,3-difluorophenyl] methanol In a DMF (10 mL) solution of 2,3-difluoro-4-hydroxybenzaldehyde (1.00 g), potassium carbonate (1.75 g) and (bromomethyl) ) Cyclopropane (0.920 mL) was added, and the mixture was stirred with heating at 70 ° C. for 1 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. After drying the organic layer over anhydrous magnesium sulfate, methanol (5 mL) was added to a THF (10 mL) solution of the residue obtained by evaporating the solvent under reduced pressure, and sodium tetrahydroborate (107 mg) was further added. And stirred for 20 minutes. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound as a white solid (1.05 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.30-0.41 (2H, m), 0.59-0.71 (2H, m), 1.21-1.39 (1H, m), 1.71 (1H, t, J = 6.1 Hz), 3.89 (2H, d, J = 6.8 Hz), 4.70 (2H, d, J = 6.1 Hz), 6.64-6.77 (1H, m), 6.99-7.12 (1H, m).
2,3-ジフルオロ-4-ヒドロキシベンズアルデヒド(1.00 g)のDMF(10 mL)溶液に炭酸カリウム(1.75 g)と(ブロモメチル)シクロプロパン(0.920 mL)を加えて、70℃で1時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣のTHF(10 mL)溶液にメタノール(5 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(107 mg)を加えて、20分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(1.05 g)として得た。
1H NMR (300 MHz, CDCl3) δ 0.30-0.41 (2H, m), 0.59-0.71 (2H, m), 1.21-1.39 (1H, m), 1.71 (1H, t, J = 6.1 Hz), 3.89 (2H, d, J = 6.8 Hz), 4.70 (2H, d, J = 6.1 Hz), 6.64-6.77 (1H, m), 6.99-7.12 (1H, m). C) [4- (Cyclopropylmethoxy) -2,3-difluorophenyl] methanol In a DMF (10 mL) solution of 2,3-difluoro-4-hydroxybenzaldehyde (1.00 g), potassium carbonate (1.75 g) and (bromomethyl) ) Cyclopropane (0.920 mL) was added, and the mixture was stirred with heating at 70 ° C. for 1 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. After drying the organic layer over anhydrous magnesium sulfate, methanol (5 mL) was added to a THF (10 mL) solution of the residue obtained by evaporating the solvent under reduced pressure, and sodium tetrahydroborate (107 mg) was further added. And stirred for 20 minutes. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound as a white solid (1.05 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.30-0.41 (2H, m), 0.59-0.71 (2H, m), 1.21-1.39 (1H, m), 1.71 (1H, t, J = 6.1 Hz), 3.89 (2H, d, J = 6.8 Hz), 4.70 (2H, d, J = 6.1 Hz), 6.64-6.77 (1H, m), 6.99-7.12 (1H, m).
D) N-[(1S)-3-(trans-4-{[4-(シクロプロピルメトキシ)-2,3-ジフルオロベンジル]オキシ}シクロヘキシル)-1-メチルプロピル]アセトアミド
[4-(シクロプロピルメトキシ)-2,3-ジフルオロフェニル]メタノール(1.05 g)のジエチルエーテル(15 mL)溶液に、三臭化リン(0.532 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下、溶媒を留去して得られた残渣とN-[(1S)-3-(trans-4-ヒドロキシシクロヘキシル)-1-メチルプロピル]アセトアミド(200 mg)、硫酸水素テトラブチルアンモニウム(21.3 mg)、50%水酸化ナトリウム水溶液(4.5 mL)、およびトルエン(9 mL)の混合物を100℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(105 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 0.30-0.40 (2H, m), 0.58-0.71 (2H, m), 0.79-1.00 (2H, m), 1.11 (3H, d, J = 6.4 Hz), 1.14-1.46 (8H, m), 1.71-1.84 (2H, m), 1.95 (3H, s), 2.00-2.14 (2H, m), 3.08-3.35 (1H, m), 3.87 (2H, d, J = 6.8 Hz), 3.89-4.01 (1H, m), 4.53 (2H, s), 5.15 (1H, d, J = 8.7 Hz), 6.64-6.74 (1H, m), 6.97-7.12 (1H, m). D) N-[(1S) -3- (trans-4-{[4- (cyclopropylmethoxy) -2,3-difluorobenzyl] oxy} cyclohexyl) -1-methylpropyl] acetamide [4- (cyclopropyl To a solution of (methoxy) -2,3-difluorophenyl] methanol (1.05 g) in diethyl ether (15 mL) was added phosphorus tribromide (0.532 mL), and the mixture was stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), the solvent was distilled off under reduced pressure, and the obtained residue and N-[(1S) -3- (trans-4-hydroxycyclohexyl) -1-methylpropyl ] A mixture of acetamide (200 mg), tetrabutylammonium hydrogen sulfate (21.3 mg), 50% aqueous sodium hydroxide (4.5 mL), and toluene (9 mL) was stirred at 100 ° C. overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (105 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.30-0.40 (2H, m), 0.58-0.71 (2H, m), 0.79-1.00 (2H, m), 1.11 (3H, d, J = 6.4 Hz), 1.14-1.46 (8H, m), 1.71-1.84 (2H, m), 1.95 (3H, s), 2.00-2.14 (2H, m), 3.08-3.35 (1H, m), 3.87 (2H, d, J = 6.8 Hz), 3.89-4.01 (1H, m), 4.53 (2H, s), 5.15 (1H, d, J = 8.7 Hz), 6.64-6.74 (1H, m), 6.97-7.12 (1H, m) .
[4-(シクロプロピルメトキシ)-2,3-ジフルオロフェニル]メタノール(1.05 g)のジエチルエーテル(15 mL)溶液に、三臭化リン(0.532 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下、溶媒を留去して得られた残渣とN-[(1S)-3-(trans-4-ヒドロキシシクロヘキシル)-1-メチルプロピル]アセトアミド(200 mg)、硫酸水素テトラブチルアンモニウム(21.3 mg)、50%水酸化ナトリウム水溶液(4.5 mL)、およびトルエン(9 mL)の混合物を100℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(105 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 0.30-0.40 (2H, m), 0.58-0.71 (2H, m), 0.79-1.00 (2H, m), 1.11 (3H, d, J = 6.4 Hz), 1.14-1.46 (8H, m), 1.71-1.84 (2H, m), 1.95 (3H, s), 2.00-2.14 (2H, m), 3.08-3.35 (1H, m), 3.87 (2H, d, J = 6.8 Hz), 3.89-4.01 (1H, m), 4.53 (2H, s), 5.15 (1H, d, J = 8.7 Hz), 6.64-6.74 (1H, m), 6.97-7.12 (1H, m). D) N-[(1S) -3- (trans-4-{[4- (cyclopropylmethoxy) -2,3-difluorobenzyl] oxy} cyclohexyl) -1-methylpropyl] acetamide [4- (cyclopropyl To a solution of (methoxy) -2,3-difluorophenyl] methanol (1.05 g) in diethyl ether (15 mL) was added phosphorus tribromide (0.532 mL), and the mixture was stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), the solvent was distilled off under reduced pressure, and the obtained residue and N-[(1S) -3- (trans-4-hydroxycyclohexyl) -1-methylpropyl ] A mixture of acetamide (200 mg), tetrabutylammonium hydrogen sulfate (21.3 mg), 50% aqueous sodium hydroxide (4.5 mL), and toluene (9 mL) was stirred at 100 ° C. overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (105 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.30-0.40 (2H, m), 0.58-0.71 (2H, m), 0.79-1.00 (2H, m), 1.11 (3H, d, J = 6.4 Hz), 1.14-1.46 (8H, m), 1.71-1.84 (2H, m), 1.95 (3H, s), 2.00-2.14 (2H, m), 3.08-3.35 (1H, m), 3.87 (2H, d, J = 6.8 Hz), 3.89-4.01 (1H, m), 4.53 (2H, s), 5.15 (1H, d, J = 8.7 Hz), 6.64-6.74 (1H, m), 6.97-7.12 (1H, m) .
実施例35
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(3,3,3-トリフルオロプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 35
N-{(1S) -2-[(trans-4-{[2-fluoro-4- (3,3,3-trifluoropropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(3,3,3-トリフルオロプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 35
N-{(1S) -2-[(trans-4-{[2-fluoro-4- (3,3,3-trifluoropropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) [4-(ベンジルオキシ)-2-フルオロフェニル]メタノール
2-フルオロ-4-ヒドロキシベンズアルデヒド(3.0 g)のDMF(70 mL)溶液に炭酸カリウム(5.92 g)と(ブロモメチル)ベンゼン(3.06 mL)を加えて、70℃で30分加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣のTHF(30 mL)溶液にメタノール(15 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(405 mg)を加えて、20分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(5.46 g)として得た。
1H NMR (300 MHz, CDCl3) δ 4.67 (2H, d, J = 6.0 Hz), 5.05 (2H, s), 6.58-6.80 (2H, m), 7.22-7.49 (6H, m). A) [4- (Benzyloxy) -2-fluorophenyl] methanol 2-fluoro-4-hydroxybenzaldehyde (3.0 g) in DMF (70 mL) with potassium carbonate (5.92 g) and (bromomethyl) benzene (3.06 mL) ) Was added and stirred at 70 ° C. for 30 minutes. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. After drying the organic layer over anhydrous magnesium sulfate, methanol (15 mL) was added to a THF (30 mL) solution of the residue obtained by evaporating the solvent under reduced pressure, and sodium tetrahydroborate (405 mg) was further added. And stirred for 20 minutes. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (5.46 g).
1 H NMR (300 MHz, CDCl 3 ) δ 4.67 (2H, d, J = 6.0 Hz), 5.05 (2H, s), 6.58-6.80 (2H, m), 7.22-7.49 (6H, m).
2-フルオロ-4-ヒドロキシベンズアルデヒド(3.0 g)のDMF(70 mL)溶液に炭酸カリウム(5.92 g)と(ブロモメチル)ベンゼン(3.06 mL)を加えて、70℃で30分加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣のTHF(30 mL)溶液にメタノール(15 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(405 mg)を加えて、20分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(5.46 g)として得た。
1H NMR (300 MHz, CDCl3) δ 4.67 (2H, d, J = 6.0 Hz), 5.05 (2H, s), 6.58-6.80 (2H, m), 7.22-7.49 (6H, m). A) [4- (Benzyloxy) -2-fluorophenyl] methanol 2-fluoro-4-hydroxybenzaldehyde (3.0 g) in DMF (70 mL) with potassium carbonate (5.92 g) and (bromomethyl) benzene (3.06 mL) ) Was added and stirred at 70 ° C. for 30 minutes. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. After drying the organic layer over anhydrous magnesium sulfate, methanol (15 mL) was added to a THF (30 mL) solution of the residue obtained by evaporating the solvent under reduced pressure, and sodium tetrahydroborate (405 mg) was further added. And stirred for 20 minutes. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (5.46 g).
1 H NMR (300 MHz, CDCl 3 ) δ 4.67 (2H, d, J = 6.0 Hz), 5.05 (2H, s), 6.58-6.80 (2H, m), 7.22-7.49 (6H, m).
B) N-{(1S)-2-[(trans-4-{[4-(ベンジルオキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(ベンジルオキシ)-2-フルオロフェニル]メタノール(1.08 g)のジエチルエーテル(15 mL)溶液に、三臭化リン(0.661 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下、溶媒を留去して得られた残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(400 mg)、硫酸水素テトラブチルアンモニウム(42.2 mg)、50%水酸化ナトリウム水溶液(9.0 mL)、およびトルエン(18 mL)の混合物を100℃で15時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(512 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.23-1.46 (4H, m), 1.91-2.11 (7H, m), 3.24-3.33 (1H, m), 3.34-3.48 (3H, m), 4.03-4.20 (1H, m), 4.50 (2H, s), 5.04 (2H, s), 5.66 (1H, d, J = 7.2 Hz), 6.67 (1H, dd, J = 11.7, 2.3 Hz), 6.75 (1H, dd, J = 8.3, 2.6 Hz), 7.28-7.46 (6H, m). B) N-{(1S) -2-[(trans-4-{[4- (benzyloxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (benzyloxy ) -2-Fluorophenyl] methanol (1.08 g) in diethyl ether (15 mL) was added phosphorus tribromide (0.661 mL) and stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), and the solvent was distilled off under reduced pressure. The resulting residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1 A mixture of -methylethyl} acetamide (400 mg), tetrabutylammonium hydrogen sulfate (42.2 mg), 50% aqueous sodium hydroxide solution (9.0 mL), and toluene (18 mL) was stirred at 100 ° C. for 15 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (512 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.23-1.46 (4H, m), 1.91-2.11 (7H, m), 3.24-3.33 (1H, m), 3.34-3.48 (3H, m), 4.03-4.20 (1H, m), 4.50 (2H, s), 5.04 (2H, s), 5.66 (1H, d, J = 7.2 Hz), 6.67 (1H, dd, J = 11.7, 2.3 Hz), 6.75 (1H, dd, J = 8.3, 2.6 Hz), 7.28-7.46 (6H, m).
[4-(ベンジルオキシ)-2-フルオロフェニル]メタノール(1.08 g)のジエチルエーテル(15 mL)溶液に、三臭化リン(0.661 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下、溶媒を留去して得られた残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(400 mg)、硫酸水素テトラブチルアンモニウム(42.2 mg)、50%水酸化ナトリウム水溶液(9.0 mL)、およびトルエン(18 mL)の混合物を100℃で15時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(512 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.23-1.46 (4H, m), 1.91-2.11 (7H, m), 3.24-3.33 (1H, m), 3.34-3.48 (3H, m), 4.03-4.20 (1H, m), 4.50 (2H, s), 5.04 (2H, s), 5.66 (1H, d, J = 7.2 Hz), 6.67 (1H, dd, J = 11.7, 2.3 Hz), 6.75 (1H, dd, J = 8.3, 2.6 Hz), 7.28-7.46 (6H, m). B) N-{(1S) -2-[(trans-4-{[4- (benzyloxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (benzyloxy ) -2-Fluorophenyl] methanol (1.08 g) in diethyl ether (15 mL) was added phosphorus tribromide (0.661 mL) and stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), and the solvent was distilled off under reduced pressure. The resulting residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1 A mixture of -methylethyl} acetamide (400 mg), tetrabutylammonium hydrogen sulfate (42.2 mg), 50% aqueous sodium hydroxide solution (9.0 mL), and toluene (18 mL) was stirred at 100 ° C. for 15 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (512 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.23-1.46 (4H, m), 1.91-2.11 (7H, m), 3.24-3.33 (1H, m), 3.34-3.48 (3H, m), 4.03-4.20 (1H, m), 4.50 (2H, s), 5.04 (2H, s), 5.66 (1H, d, J = 7.2 Hz), 6.67 (1H, dd, J = 11.7, 2.3 Hz), 6.75 (1H, dd, J = 8.3, 2.6 Hz), 7.28-7.46 (6H, m).
C) N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
水素雰囲気下、N-{(1S)-2-[(trans-4-{[4-(ベンジルオキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(500 mg)、10%パラジウム-炭素(50%含水、300 mg)および酢酸エチル(10 mL)の混合物を室温で終夜撹拌した。濾過により触媒を除去した後、得られた濾液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(455 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.29-1.47 (4H, m), 1.89-2.03 (7H, m), 3.19-3.49 (4H, m), 4.49 (2H, s), 5.75 (1H, d, J = 8.3 Hz), 6.50-6.65 (2H, m), 6.90 (1H, s), 7.21 (1H, t, J = 8.5 Hz). C) N-[(1S) -2-({trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide under hydrogen atmosphere, N-{(1S ) -2-[(trans-4-{[4- (benzyloxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (500 mg), 10% palladium-carbon (50% A mixture of water (300 mg) and ethyl acetate (10 mL) was stirred at room temperature overnight. After removing the catalyst by filtration, the obtained filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (455 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.29-1.47 (4H, m), 1.89-2.03 (7H, m), 3.19-3.49 (4H, m), 4.49 (2H, s), 5.75 (1H, d, J = 8.3 Hz), 6.50-6.65 (2H, m), 6.90 (1H, s), 7.21 (1H, t, J = 8.5 Hz).
水素雰囲気下、N-{(1S)-2-[(trans-4-{[4-(ベンジルオキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(500 mg)、10%パラジウム-炭素(50%含水、300 mg)および酢酸エチル(10 mL)の混合物を室温で終夜撹拌した。濾過により触媒を除去した後、得られた濾液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(455 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.29-1.47 (4H, m), 1.89-2.03 (7H, m), 3.19-3.49 (4H, m), 4.49 (2H, s), 5.75 (1H, d, J = 8.3 Hz), 6.50-6.65 (2H, m), 6.90 (1H, s), 7.21 (1H, t, J = 8.5 Hz). C) N-[(1S) -2-({trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide under hydrogen atmosphere, N-{(1S ) -2-[(trans-4-{[4- (benzyloxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (500 mg), 10% palladium-carbon (50% A mixture of water (300 mg) and ethyl acetate (10 mL) was stirred at room temperature overnight. After removing the catalyst by filtration, the obtained filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (455 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.29-1.47 (4H, m), 1.89-2.03 (7H, m), 3.19-3.49 (4H, m), 4.49 (2H, s), 5.75 (1H, d, J = 8.3 Hz), 6.50-6.65 (2H, m), 6.90 (1H, s), 7.21 (1H, t, J = 8.5 Hz).
D) N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(3,3,3-トリフルオロプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(110 mg)のDMF(1 mL)溶液に炭酸カリウム(4.48 g)と1,1,1-トリフルオロ-3-ヨードプロパン(3.68 mL)を加えて、60℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(45 mg)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.95-1.08 (3H, m), 1.12-1.37 (4H, m), 1.77 (3H, s), 1.89 (4H, brs), 2.78 (2H, qt, J = 11.4, 5.8 Hz), 3.11-3.31 (3H, m), 3.33-3.44 (1H, m), 3.82 (1H, dt, J = 13.3, 6.8 Hz), 4.21 (2H, t, J = 5.9 Hz), 4.43 (2H, s), 6.70-6.92 (2H, m), 7.33 (1H, t, J = 8.5 Hz), 7.65 (1H, d, J = 7.9 Hz). D) N-{(1S) -2-[(trans-4-{[2-fluoro-4- (3,3,3-trifluoropropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} Acetamide N-[(1S) -2-({trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (110 mg) in DMF (1 mL) Potassium carbonate (4.48 g) and 1,1,1-trifluoro-3-iodopropane (3.68 mL) were added to the solution, and the mixture was stirred with heating at 60 ° C. for 2 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, the solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (45 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.95-1.08 (3H, m), 1.12-1.37 (4H, m), 1.77 (3H, s), 1.89 (4H, brs), 2.78 (2H, qt , J = 11.4, 5.8 Hz), 3.11-3.31 (3H, m), 3.33-3.44 (1H, m), 3.82 (1H, dt, J = 13.3, 6.8 Hz), 4.21 (2H, t, J = 5.9 Hz), 4.43 (2H, s), 6.70-6.92 (2H, m), 7.33 (1H, t, J = 8.5 Hz), 7.65 (1H, d, J = 7.9 Hz).
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(110 mg)のDMF(1 mL)溶液に炭酸カリウム(4.48 g)と1,1,1-トリフルオロ-3-ヨードプロパン(3.68 mL)を加えて、60℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(45 mg)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.95-1.08 (3H, m), 1.12-1.37 (4H, m), 1.77 (3H, s), 1.89 (4H, brs), 2.78 (2H, qt, J = 11.4, 5.8 Hz), 3.11-3.31 (3H, m), 3.33-3.44 (1H, m), 3.82 (1H, dt, J = 13.3, 6.8 Hz), 4.21 (2H, t, J = 5.9 Hz), 4.43 (2H, s), 6.70-6.92 (2H, m), 7.33 (1H, t, J = 8.5 Hz), 7.65 (1H, d, J = 7.9 Hz). D) N-{(1S) -2-[(trans-4-{[2-fluoro-4- (3,3,3-trifluoropropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} Acetamide N-[(1S) -2-({trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (110 mg) in DMF (1 mL) Potassium carbonate (4.48 g) and 1,1,1-trifluoro-3-iodopropane (3.68 mL) were added to the solution, and the mixture was stirred with heating at 60 ° C. for 2 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, the solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (45 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.95-1.08 (3H, m), 1.12-1.37 (4H, m), 1.77 (3H, s), 1.89 (4H, brs), 2.78 (2H, qt , J = 11.4, 5.8 Hz), 3.11-3.31 (3H, m), 3.33-3.44 (1H, m), 3.82 (1H, dt, J = 13.3, 6.8 Hz), 4.21 (2H, t, J = 5.9 Hz), 4.43 (2H, s), 6.70-6.92 (2H, m), 7.33 (1H, t, J = 8.5 Hz), 7.65 (1H, d, J = 7.9 Hz).
実施例36
N-{(1S)-2-[(trans-4-{[6-(シクロプロピルメトキシ)-2-フルオロピリジン-3-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 36
N-{(1S) -2-[(trans-4-{[6- (cyclopropylmethoxy) -2-fluoropyridin-3-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[6-(シクロプロピルメトキシ)-2-フルオロピリジン-3-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 36
N-{(1S) -2-[(trans-4-{[6- (cyclopropylmethoxy) -2-fluoropyridin-3-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) 2-(シクロプロピルメトキシ)-6-フルオロピリジン
シクロプロピルメタノール(19.4 mL)のDMF(300 mL)溶液に水素化ナトリウム(60%油状、9.56 g)を加えて、室温で15分間撹拌した。反応混合物に2,6-ジフルオロピリジン(25.0 g)を加えて、室温で30分間撹拌した。反応混合物に水を加えた後、ジエチルエーテルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(27.3 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.28-0.40 (2H, m), 0.55-0.66 (2H, m), 1.18-1.35 (1H, m), 4.10 (2H, d, J = 7.2 Hz), 6.44 (1H, dd, J = 7.7, 2.5 Hz), 6.62 (1H, dd, J = 8.1, 1.3 Hz), 7.63 (1H, q, J = 8.2 Hz). A) To a solution of 2- (cyclopropylmethoxy) -6-fluoropyridine cyclopropylmethanol (19.4 mL) in DMF (300 mL) was added sodium hydride (60% oil, 9.56 g), and the mixture was stirred at room temperature for 15 minutes. . 2,6-Difluoropyridine (25.0 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (27.3 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.28-0.40 (2H, m), 0.55-0.66 (2H, m), 1.18-1.35 (1H, m), 4.10 (2H, d, J = 7.2 Hz), 6.44 (1H, dd, J = 7.7, 2.5 Hz), 6.62 (1H, dd, J = 8.1, 1.3 Hz), 7.63 (1H, q, J = 8.2 Hz).
シクロプロピルメタノール(19.4 mL)のDMF(300 mL)溶液に水素化ナトリウム(60%油状、9.56 g)を加えて、室温で15分間撹拌した。反応混合物に2,6-ジフルオロピリジン(25.0 g)を加えて、室温で30分間撹拌した。反応混合物に水を加えた後、ジエチルエーテルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(27.3 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.28-0.40 (2H, m), 0.55-0.66 (2H, m), 1.18-1.35 (1H, m), 4.10 (2H, d, J = 7.2 Hz), 6.44 (1H, dd, J = 7.7, 2.5 Hz), 6.62 (1H, dd, J = 8.1, 1.3 Hz), 7.63 (1H, q, J = 8.2 Hz). A) To a solution of 2- (cyclopropylmethoxy) -6-fluoropyridine cyclopropylmethanol (19.4 mL) in DMF (300 mL) was added sodium hydride (60% oil, 9.56 g), and the mixture was stirred at room temperature for 15 minutes. . 2,6-Difluoropyridine (25.0 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (27.3 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.28-0.40 (2H, m), 0.55-0.66 (2H, m), 1.18-1.35 (1H, m), 4.10 (2H, d, J = 7.2 Hz), 6.44 (1H, dd, J = 7.7, 2.5 Hz), 6.62 (1H, dd, J = 8.1, 1.3 Hz), 7.63 (1H, q, J = 8.2 Hz).
B) 6-(シクロプロピルメトキシ)-2-フルオロピリジン-3-カルバルデヒド
2-(シクロプロピルメトキシ)-6-フルオロピリジン(27.3 g)のTHF(200 mL)溶液にリチウム ジイソプロピルアミド(2 M THF溶液、98 mL)を-78℃で加えて、同温度で30分間撹拌した。反応混合物にギ酸エチルを同温度で加えた後、室温で30分間撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.08 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.45 (2H, m), 0.55-0.72 (2H, m), 1.20-1.39 (1H, m), 4.21 (2H, d, J = 7.6 Hz), 6.73 (1H, d, J = 8.3 Hz), 8.16 (1H, dd, J = 9.5, 8.3 Hz), 10.16 (1H, s). B) 6- (Cyclopropylmethoxy) -2-fluoropyridine-3-carbaldehyde 2- (cyclopropylmethoxy) -6-fluoropyridine (27.3 g) in THF (200 mL) in lithium diisopropylamide (2 M THF Solution, 98 mL) was added at −78 ° C., and the mixture was stirred at the same temperature for 30 minutes. Ethyl formate was added to the reaction mixture at the same temperature, followed by stirring at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.08 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.45 (2H, m), 0.55-0.72 (2H, m), 1.20-1.39 (1H, m), 4.21 (2H, d, J = 7.6 Hz), 6.73 (1H, d, J = 8.3 Hz), 8.16 (1H, dd, J = 9.5, 8.3 Hz), 10.16 (1H, s).
2-(シクロプロピルメトキシ)-6-フルオロピリジン(27.3 g)のTHF(200 mL)溶液にリチウム ジイソプロピルアミド(2 M THF溶液、98 mL)を-78℃で加えて、同温度で30分間撹拌した。反応混合物にギ酸エチルを同温度で加えた後、室温で30分間撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.08 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.45 (2H, m), 0.55-0.72 (2H, m), 1.20-1.39 (1H, m), 4.21 (2H, d, J = 7.6 Hz), 6.73 (1H, d, J = 8.3 Hz), 8.16 (1H, dd, J = 9.5, 8.3 Hz), 10.16 (1H, s). B) 6- (Cyclopropylmethoxy) -2-fluoropyridine-3-carbaldehyde 2- (cyclopropylmethoxy) -6-fluoropyridine (27.3 g) in THF (200 mL) in lithium diisopropylamide (2 M THF Solution, 98 mL) was added at −78 ° C., and the mixture was stirred at the same temperature for 30 minutes. Ethyl formate was added to the reaction mixture at the same temperature, followed by stirring at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.08 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.45 (2H, m), 0.55-0.72 (2H, m), 1.20-1.39 (1H, m), 4.21 (2H, d, J = 7.6 Hz), 6.73 (1H, d, J = 8.3 Hz), 8.16 (1H, dd, J = 9.5, 8.3 Hz), 10.16 (1H, s).
C) [6-(シクロプロピルメトキシ)-2-フルオロピリジン-3-イル]メタノール
6-(シクロプロピルメトキシ)-2-フルオロピリジン-3-カルバルデヒド(1.08 g)のTHF(10 mL)溶液にメタノール(10 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(209 mg)を加えて、30分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物 (391 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 0.30-0.40 (2H, m), 0.57-0.68 (2H, m), 1.18-1.37 (1H, m), 4.10 (2H, d, J = 7.2 Hz), 4.66 (2H, d, J = 5.3 Hz), 6.64 (1H, d, J = 8.0 Hz), 7.72 (1H, dd, J = 10.0, 8.1 Hz). C) [6- (Cyclopropylmethoxy) -2-fluoropyridin-3-yl] methanol 6- (Cyclopropylmethoxy) -2-fluoropyridine-3-carbaldehyde (1.08 g) in THF (10 mL) solution Methanol (10 mL) was added, sodium tetrahydroborate (209 mg) was further added, and the mixture was stirred for 30 min. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, the solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (391 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.30-0.40 (2H, m), 0.57-0.68 (2H, m), 1.18-1.37 (1H, m), 4.10 (2H, d, J = 7.2 Hz), 4.66 (2H, d, J = 5.3 Hz), 6.64 (1H, d, J = 8.0 Hz), 7.72 (1H, dd, J = 10.0, 8.1 Hz).
6-(シクロプロピルメトキシ)-2-フルオロピリジン-3-カルバルデヒド(1.08 g)のTHF(10 mL)溶液にメタノール(10 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(209 mg)を加えて、30分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物 (391 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 0.30-0.40 (2H, m), 0.57-0.68 (2H, m), 1.18-1.37 (1H, m), 4.10 (2H, d, J = 7.2 Hz), 4.66 (2H, d, J = 5.3 Hz), 6.64 (1H, d, J = 8.0 Hz), 7.72 (1H, dd, J = 10.0, 8.1 Hz). C) [6- (Cyclopropylmethoxy) -2-fluoropyridin-3-yl] methanol 6- (Cyclopropylmethoxy) -2-fluoropyridine-3-carbaldehyde (1.08 g) in THF (10 mL) solution Methanol (10 mL) was added, sodium tetrahydroborate (209 mg) was further added, and the mixture was stirred for 30 min. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, the solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (391 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.30-0.40 (2H, m), 0.57-0.68 (2H, m), 1.18-1.37 (1H, m), 4.10 (2H, d, J = 7.2 Hz), 4.66 (2H, d, J = 5.3 Hz), 6.64 (1H, d, J = 8.0 Hz), 7.72 (1H, dd, J = 10.0, 8.1 Hz).
D) N-{(1S)-2-[(trans-4-{[6-(シクロプロピルメトキシ)-2-フルオロピリジン-3-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[6-(シクロプロピルメトキシ)-2-フルオロピリジン-3-イル]メタノール(391 mg)のジエチルエーテル(10 mL)溶液に、三臭化リン(0.207 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通し、減圧下溶媒を留去して得られた残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(200 mg)、硫酸水素テトラブチルアンモニウム(21.1 mg)、50%水酸化ナトリウム水溶液(4.5 mL)、およびトルエン(9 mL)の混合物を100℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(226 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 0.27-0.40 (2H, m), 0.56-0.66 (2H, m), 1.17 (3H, d, J = 6.8 Hz), 1.21-1.47 (5H, m), 1.86-2.11 (7H, m), 3.21-3.34 (1H, m), 3.35-3.47 (3H, m), 4.03-4.18 (3H, m), 4.47 (2H, s), 5.57-5.71 (1H, m), 6.63 (1H, dd, J = 8.3, 0.8 Hz), 7.71 (1H, dd, J = 9.8, 7.9 Hz). D) N-{(1S) -2-[(trans-4-{[6- (cyclopropylmethoxy) -2-fluoropyridin-3-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide [ Phosphorus tribromide (0.207 mL) was added to a solution of 6- (cyclopropylmethoxy) -2-fluoropyridin-3-yl] methanol (391 mg) in diethyl ether (10 mL), and the mixture was stirred at room temperature for 30 min. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), the solvent was distilled off under reduced pressure, and the obtained residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1- A mixture of methylethyl} acetamide (200 mg), tetrabutylammonium hydrogen sulfate (21.1 mg), 50% aqueous sodium hydroxide solution (4.5 mL), and toluene (9 mL) was stirred at 100 ° C. overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (226 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.27-0.40 (2H, m), 0.56-0.66 (2H, m), 1.17 (3H, d, J = 6.8 Hz), 1.21-1.47 (5H, m), 1.86-2.11 (7H, m), 3.21-3.34 (1H, m), 3.35-3.47 (3H, m), 4.03-4.18 (3H, m), 4.47 (2H, s), 5.57-5.71 (1H, m ), 6.63 (1H, dd, J = 8.3, 0.8 Hz), 7.71 (1H, dd, J = 9.8, 7.9 Hz).
[6-(シクロプロピルメトキシ)-2-フルオロピリジン-3-イル]メタノール(391 mg)のジエチルエーテル(10 mL)溶液に、三臭化リン(0.207 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通し、減圧下溶媒を留去して得られた残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(200 mg)、硫酸水素テトラブチルアンモニウム(21.1 mg)、50%水酸化ナトリウム水溶液(4.5 mL)、およびトルエン(9 mL)の混合物を100℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(226 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 0.27-0.40 (2H, m), 0.56-0.66 (2H, m), 1.17 (3H, d, J = 6.8 Hz), 1.21-1.47 (5H, m), 1.86-2.11 (7H, m), 3.21-3.34 (1H, m), 3.35-3.47 (3H, m), 4.03-4.18 (3H, m), 4.47 (2H, s), 5.57-5.71 (1H, m), 6.63 (1H, dd, J = 8.3, 0.8 Hz), 7.71 (1H, dd, J = 9.8, 7.9 Hz). D) N-{(1S) -2-[(trans-4-{[6- (cyclopropylmethoxy) -2-fluoropyridin-3-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide [ Phosphorus tribromide (0.207 mL) was added to a solution of 6- (cyclopropylmethoxy) -2-fluoropyridin-3-yl] methanol (391 mg) in diethyl ether (10 mL), and the mixture was stirred at room temperature for 30 min. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), the solvent was distilled off under reduced pressure, and the obtained residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1- A mixture of methylethyl} acetamide (200 mg), tetrabutylammonium hydrogen sulfate (21.1 mg), 50% aqueous sodium hydroxide solution (4.5 mL), and toluene (9 mL) was stirred at 100 ° C. overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (226 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.27-0.40 (2H, m), 0.56-0.66 (2H, m), 1.17 (3H, d, J = 6.8 Hz), 1.21-1.47 (5H, m), 1.86-2.11 (7H, m), 3.21-3.34 (1H, m), 3.35-3.47 (3H, m), 4.03-4.18 (3H, m), 4.47 (2H, s), 5.57-5.71 (1H, m ), 6.63 (1H, dd, J = 8.3, 0.8 Hz), 7.71 (1H, dd, J = 9.8, 7.9 Hz).
実施例37
N-[(1S)-2-({trans-4-[(2-クロロ-4-プロポキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド Example 37
N-[(1S) -2-({trans-4-[(2-chloro-4-propoxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide
N-[(1S)-2-({trans-4-[(2-クロロ-4-プロポキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド Example 37
N-[(1S) -2-({trans-4-[(2-chloro-4-propoxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide
A) 2-クロロ-4-プロポキシベンズアルデヒド
2-クロロ-4-ヒドロキシベンズアルデヒド(2.00 g)、1-ヨードプロパン(2.48 mL)、炭酸カリウム(3.53 g)、およびDMF(20 mL)の混合物を60℃で終夜撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を淡黄色油状化合物(2.51 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.98 (3H, t, J = 7.6 Hz), 1.68-1.84 (2H, m), 4.08 (2H, t, J = 6.6 Hz), 7.03-7.13 (1H, m), 7.18 (1H, d, J = 2.3 Hz), 7.83 (1H, d, J = 8.7 Hz), 10.19 (1H, d, J = 0.8 Hz). A) A mixture of 2-chloro-4-propoxybenzaldehyde 2-chloro-4-hydroxybenzaldehyde (2.00 g), 1-iodopropane (2.48 mL), potassium carbonate (3.53 g), and DMF (20 mL) at 60 ° C. And stirred overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a pale yellow oily compound (2.51 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.98 (3H, t, J = 7.6 Hz), 1.68-1.84 (2H, m), 4.08 (2H, t, J = 6.6 Hz), 7.03-7.13 ( 1H, m), 7.18 (1H, d, J = 2.3 Hz), 7.83 (1H, d, J = 8.7 Hz), 10.19 (1H, d, J = 0.8 Hz).
2-クロロ-4-ヒドロキシベンズアルデヒド(2.00 g)、1-ヨードプロパン(2.48 mL)、炭酸カリウム(3.53 g)、およびDMF(20 mL)の混合物を60℃で終夜撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を淡黄色油状化合物(2.51 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.98 (3H, t, J = 7.6 Hz), 1.68-1.84 (2H, m), 4.08 (2H, t, J = 6.6 Hz), 7.03-7.13 (1H, m), 7.18 (1H, d, J = 2.3 Hz), 7.83 (1H, d, J = 8.7 Hz), 10.19 (1H, d, J = 0.8 Hz). A) A mixture of 2-chloro-4-propoxybenzaldehyde 2-chloro-4-hydroxybenzaldehyde (2.00 g), 1-iodopropane (2.48 mL), potassium carbonate (3.53 g), and DMF (20 mL) at 60 ° C. And stirred overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a pale yellow oily compound (2.51 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.98 (3H, t, J = 7.6 Hz), 1.68-1.84 (2H, m), 4.08 (2H, t, J = 6.6 Hz), 7.03-7.13 ( 1H, m), 7.18 (1H, d, J = 2.3 Hz), 7.83 (1H, d, J = 8.7 Hz), 10.19 (1H, d, J = 0.8 Hz).
B) (2-クロロ-4-プロポキシフェニル)メタノール
2-クロロ-4-プロポキシベンズアルデヒド(2.51 g)およびメタノール(25 mL)の混合物にテトラヒドロホウ酸ナトリウム(717 mg)を0℃で加え、混合物を0℃で1時間、続いて室温で1時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(2.26 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.96 (3H, t, J = 7.6 Hz), 1.56-1.80 (2H, m), 3.93 (2H, t, J = 6.6 Hz), 4.48 (2H, d, J = 5.7 Hz), 5.22 (1H, t, J = 5.7 Hz), 6.83-7.01 (2H, m), 7.40 (1H, d, J = 8.3 Hz). B) (2-Chloro-4-propoxyphenyl) methanol Sodium tetrahydroborate (717 mg) was added to a mixture of 2-chloro-4-propoxybenzaldehyde (2.51 g) and methanol (25 mL) at 0 ° C. Stir at 0 ° C. for 1 hour, then at room temperature for 1 hour. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oily compound (2.26 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.96 (3H, t, J = 7.6 Hz), 1.56-1.80 (2H, m), 3.93 (2H, t, J = 6.6 Hz), 4.48 (2H, d, J = 5.7 Hz), 5.22 (1H, t, J = 5.7 Hz), 6.83-7.01 (2H, m), 7.40 (1H, d, J = 8.3 Hz).
2-クロロ-4-プロポキシベンズアルデヒド(2.51 g)およびメタノール(25 mL)の混合物にテトラヒドロホウ酸ナトリウム(717 mg)を0℃で加え、混合物を0℃で1時間、続いて室温で1時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(2.26 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.96 (3H, t, J = 7.6 Hz), 1.56-1.80 (2H, m), 3.93 (2H, t, J = 6.6 Hz), 4.48 (2H, d, J = 5.7 Hz), 5.22 (1H, t, J = 5.7 Hz), 6.83-7.01 (2H, m), 7.40 (1H, d, J = 8.3 Hz). B) (2-Chloro-4-propoxyphenyl) methanol Sodium tetrahydroborate (717 mg) was added to a mixture of 2-chloro-4-propoxybenzaldehyde (2.51 g) and methanol (25 mL) at 0 ° C. Stir at 0 ° C. for 1 hour, then at room temperature for 1 hour. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oily compound (2.26 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.96 (3H, t, J = 7.6 Hz), 1.56-1.80 (2H, m), 3.93 (2H, t, J = 6.6 Hz), 4.48 (2H, d, J = 5.7 Hz), 5.22 (1H, t, J = 5.7 Hz), 6.83-7.01 (2H, m), 7.40 (1H, d, J = 8.3 Hz).
C) 1-(ブロモメチル)-2-クロロ-4-プロポキシベンゼン
(2-クロロ-4-プロポキシフェニル)メタノール(2.26 g)、三臭化リン(1.06 mL)、およびトルエン(25 mL)の混合物を室温で1時間撹拌した。反応混合物を飽和炭酸水素ナトリウム水溶液に0℃で加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状物(1.76 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.92-1.01 (3H, m), 1.61-1.81 (2H, m), 3.96 (2H, t, J = 6.4 Hz), 4.72 (2H, s), 6.92 (1H, dd, J = 8.7, 2.6 Hz), 7.06 (1H, d, J = 2.6 Hz), 7.52 (1H, d, J = 8.7 Hz). C) A mixture of 1- (bromomethyl) -2-chloro-4-propoxybenzene (2-chloro-4-propoxyphenyl) methanol (2.26 g), phosphorus tribromide (1.06 mL), and toluene (25 mL). Stir at room temperature for 1 hour. The reaction mixture was added to saturated aqueous sodium hydrogen carbonate solution at 0 ° C., and extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oil (1.76 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.92-1.01 (3H, m), 1.61-1.81 (2H, m), 3.96 (2H, t, J = 6.4 Hz), 4.72 (2H, s), 6.92 (1H, dd, J = 8.7, 2.6 Hz), 7.06 (1H, d, J = 2.6 Hz), 7.52 (1H, d, J = 8.7 Hz).
(2-クロロ-4-プロポキシフェニル)メタノール(2.26 g)、三臭化リン(1.06 mL)、およびトルエン(25 mL)の混合物を室温で1時間撹拌した。反応混合物を飽和炭酸水素ナトリウム水溶液に0℃で加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状物(1.76 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.92-1.01 (3H, m), 1.61-1.81 (2H, m), 3.96 (2H, t, J = 6.4 Hz), 4.72 (2H, s), 6.92 (1H, dd, J = 8.7, 2.6 Hz), 7.06 (1H, d, J = 2.6 Hz), 7.52 (1H, d, J = 8.7 Hz). C) A mixture of 1- (bromomethyl) -2-chloro-4-propoxybenzene (2-chloro-4-propoxyphenyl) methanol (2.26 g), phosphorus tribromide (1.06 mL), and toluene (25 mL). Stir at room temperature for 1 hour. The reaction mixture was added to saturated aqueous sodium hydrogen carbonate solution at 0 ° C., and extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oil (1.76 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.92-1.01 (3H, m), 1.61-1.81 (2H, m), 3.96 (2H, t, J = 6.4 Hz), 4.72 (2H, s), 6.92 (1H, dd, J = 8.7, 2.6 Hz), 7.06 (1H, d, J = 2.6 Hz), 7.52 (1H, d, J = 8.7 Hz).
D) N-[(1S)-2-({trans-4-[(2-クロロ-4-プロポキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
N-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(200 mg)、1-(ブロモメチル)-2-クロロ-4-プロポキシベンゼン(979 mg)、硫酸水素テトラブチルアンモニウム(31.5 mg)、50%水酸化ナトリウム水溶液(5 mL)、およびトルエン(10 mL)の混合物を100℃で3日間撹拌した。反応混合物に水を加えた後、ジエチルエーテルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、再結晶(酢酸エチル/ヘキサン)を行うことにより標題化合物を白色結晶(136 mg)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.91-1.05 (6H, m), 1.14-1.40 (4H, m), 1.64-1.76 (2H, m), 1.77 (3H, s), 1.82-1.98 (4H, m), 3.10-3.48 (4H, m), 3.74-3.89 (1H, m), 3.93 (2H, t, J = 6.4 Hz), 4.45 (2H, s), 6.90 (1H, dd, J = 8.7, 2.6 Hz), 7.00 (1H, d, J = 2.3 Hz), 7.36 (1H, d, J = 8.3 Hz), 7.64 (1H, d, J = 7.9 Hz).
Anal. Calcd for C21H32NO4Cl:C,63.38;H,8.11;N,3.52.Found:C,63.37;H,8.15;N,3.36. D) N-[(1S) -2-({trans-4-[(2-chloro-4-propoxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide N-{(1S) -2- [(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (200 mg), 1- (bromomethyl) -2-chloro-4-propoxybenzene (979 mg), tetrabutylammonium hydrogen sulfate (31.5 mg ), 50% aqueous sodium hydroxide solution (5 mL), and toluene (10 mL) were stirred at 100 ° C. for 3 days. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) and recrystallized (ethyl acetate / hexane) to give the title compound as white crystals (136 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.91-1.05 (6H, m), 1.14-1.40 (4H, m), 1.64-1.76 (2H, m), 1.77 (3H, s), 1.82-1.98 (4H, m), 3.10-3.48 (4H, m), 3.74-3.89 (1H, m), 3.93 (2H, t, J = 6.4 Hz), 4.45 (2H, s), 6.90 (1H, dd, J = 8.7, 2.6 Hz), 7.00 (1H, d, J = 2.3 Hz), 7.36 (1H, d, J = 8.3 Hz), 7.64 (1H, d, J = 7.9 Hz).
Anal.Calcd for C 21 H 32 NO 4 Cl: C, 63.38; H, 8.11; N, 3.52.Found: C, 63.37; H, 8.15; N, 3.36.
N-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(200 mg)、1-(ブロモメチル)-2-クロロ-4-プロポキシベンゼン(979 mg)、硫酸水素テトラブチルアンモニウム(31.5 mg)、50%水酸化ナトリウム水溶液(5 mL)、およびトルエン(10 mL)の混合物を100℃で3日間撹拌した。反応混合物に水を加えた後、ジエチルエーテルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、再結晶(酢酸エチル/ヘキサン)を行うことにより標題化合物を白色結晶(136 mg)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.91-1.05 (6H, m), 1.14-1.40 (4H, m), 1.64-1.76 (2H, m), 1.77 (3H, s), 1.82-1.98 (4H, m), 3.10-3.48 (4H, m), 3.74-3.89 (1H, m), 3.93 (2H, t, J = 6.4 Hz), 4.45 (2H, s), 6.90 (1H, dd, J = 8.7, 2.6 Hz), 7.00 (1H, d, J = 2.3 Hz), 7.36 (1H, d, J = 8.3 Hz), 7.64 (1H, d, J = 7.9 Hz).
Anal. Calcd for C21H32NO4Cl:C,63.38;H,8.11;N,3.52.Found:C,63.37;H,8.15;N,3.36. D) N-[(1S) -2-({trans-4-[(2-chloro-4-propoxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide N-{(1S) -2- [(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (200 mg), 1- (bromomethyl) -2-chloro-4-propoxybenzene (979 mg), tetrabutylammonium hydrogen sulfate (31.5 mg ), 50% aqueous sodium hydroxide solution (5 mL), and toluene (10 mL) were stirred at 100 ° C. for 3 days. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) and recrystallized (ethyl acetate / hexane) to give the title compound as white crystals (136 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.91-1.05 (6H, m), 1.14-1.40 (4H, m), 1.64-1.76 (2H, m), 1.77 (3H, s), 1.82-1.98 (4H, m), 3.10-3.48 (4H, m), 3.74-3.89 (1H, m), 3.93 (2H, t, J = 6.4 Hz), 4.45 (2H, s), 6.90 (1H, dd, J = 8.7, 2.6 Hz), 7.00 (1H, d, J = 2.3 Hz), 7.36 (1H, d, J = 8.3 Hz), 7.64 (1H, d, J = 7.9 Hz).
Anal.Calcd for C 21 H 32 NO 4 Cl: C, 63.38; H, 8.11; N, 3.52.Found: C, 63.37; H, 8.15; N, 3.36.
実施例38
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 38
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 38
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
A) (2,2-ジフルオロシクロプロピル)メチル メタンスルホナート
(2,2-ジフルオロシクロプロピル)メタノール(10.0 g)およびトリエチルアミン(19.34 ml)のTHF(150 ml)溶液に、メタンスルホニルクロリド(7.88 ml)を室温で加え、3時間撹拌した。反応液に飽和重炭酸ナトリウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮して標題化合物を茶褐色油状物(17.22 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.20-1.51 (1H, m), 1.64 (1H, tdd, J = 11.5, 8.2, 5.1 Hz), 1.97-2.18 (1H, m), 3.05 (3H, s), 4.08-4.53 (2H, m). A) (2,2-difluorocyclopropyl) methyl methanesulfonate (2,2-difluorocyclopropyl) methanol (10.0 g) and triethylamine (19.34 ml) in THF (150 ml) solution with methanesulfonyl chloride (7.88 ml ) Was added at room temperature and stirred for 3 hours. A saturated aqueous sodium bicarbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound as a brown oil (17.22 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.20-1.51 (1H, m), 1.64 (1H, tdd, J = 11.5, 8.2, 5.1 Hz), 1.97-2.18 (1H, m), 3.05 (3H, s ), 4.08-4.53 (2H, m).
(2,2-ジフルオロシクロプロピル)メタノール(10.0 g)およびトリエチルアミン(19.34 ml)のTHF(150 ml)溶液に、メタンスルホニルクロリド(7.88 ml)を室温で加え、3時間撹拌した。反応液に飽和重炭酸ナトリウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮して標題化合物を茶褐色油状物(17.22 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.20-1.51 (1H, m), 1.64 (1H, tdd, J = 11.5, 8.2, 5.1 Hz), 1.97-2.18 (1H, m), 3.05 (3H, s), 4.08-4.53 (2H, m). A) (2,2-difluorocyclopropyl) methyl methanesulfonate (2,2-difluorocyclopropyl) methanol (10.0 g) and triethylamine (19.34 ml) in THF (150 ml) solution with methanesulfonyl chloride (7.88 ml ) Was added at room temperature and stirred for 3 hours. A saturated aqueous sodium bicarbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound as a brown oil (17.22 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.20-1.51 (1H, m), 1.64 (1H, tdd, J = 11.5, 8.2, 5.1 Hz), 1.97-2.18 (1H, m), 3.05 (3H, s ), 4.08-4.53 (2H, m).
B) 2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンズアルデヒド
2-クロロ-4-ヒドロキシベンズアルデヒド (9.40 g)のDMF(80 mL)溶液に炭酸カリウム(12.45 g)および(2,2-ジフルオロシクロプロピル)メチル メタンスルホナート(16.77 g)を加えて、60℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を黄色油状物(14.84 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.31 (1H, dtd, J = 13.0, 7.7, 4.0 Hz), 1.53-1.75 (1H, m), 2.09 (1H, ddq, J = 12.7, 11.3, 7.5 Hz), 3.91-4.25 (2H, m), 6.73-7.04 (2H, m), 7.90 (1H, d, J = 8.7 Hz), 10.33 (1H, d, J = 0.8 Hz). B) 2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzaldehyde To a solution of 2-chloro-4-hydroxybenzaldehyde (9.40 g) in DMF (80 mL) was added potassium carbonate (12.45 g) and (2 , 2-Difluorocyclopropyl) methyl methanesulfonate (16.77 g) was added, and the mixture was stirred with heating at 60 ° C. for 2 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a yellow oil (14.84 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.31 (1H, dtd, J = 13.0, 7.7, 4.0 Hz), 1.53-1.75 (1H, m), 2.09 (1H, ddq, J = 12.7, 11.3, 7.5 Hz ), 3.91-4.25 (2H, m), 6.73-7.04 (2H, m), 7.90 (1H, d, J = 8.7 Hz), 10.33 (1H, d, J = 0.8 Hz).
2-クロロ-4-ヒドロキシベンズアルデヒド (9.40 g)のDMF(80 mL)溶液に炭酸カリウム(12.45 g)および(2,2-ジフルオロシクロプロピル)メチル メタンスルホナート(16.77 g)を加えて、60℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を黄色油状物(14.84 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.31 (1H, dtd, J = 13.0, 7.7, 4.0 Hz), 1.53-1.75 (1H, m), 2.09 (1H, ddq, J = 12.7, 11.3, 7.5 Hz), 3.91-4.25 (2H, m), 6.73-7.04 (2H, m), 7.90 (1H, d, J = 8.7 Hz), 10.33 (1H, d, J = 0.8 Hz). B) 2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzaldehyde To a solution of 2-chloro-4-hydroxybenzaldehyde (9.40 g) in DMF (80 mL) was added potassium carbonate (12.45 g) and (2 , 2-Difluorocyclopropyl) methyl methanesulfonate (16.77 g) was added, and the mixture was stirred with heating at 60 ° C. for 2 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a yellow oil (14.84 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.31 (1H, dtd, J = 13.0, 7.7, 4.0 Hz), 1.53-1.75 (1H, m), 2.09 (1H, ddq, J = 12.7, 11.3, 7.5 Hz ), 3.91-4.25 (2H, m), 6.73-7.04 (2H, m), 7.90 (1H, d, J = 8.7 Hz), 10.33 (1H, d, J = 0.8 Hz).
C) {2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]フェニル}メタノール
2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンズアルデヒド(894 mg) のTHF(4 mL)溶液にメタノール(2 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(165 mg)を加えて、30分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下、溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(842 mg)として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28-1.87 (2H, m), 2.03-2.34 (1H, m), 3.87-4.08 (1H, m), 4.17 (1H, ddd, J = 10.3, 6.7, 3.0 Hz), 4.49 (2H, d, J = 5.3 Hz), 5.10-5.34 (1H, m), 6.86-7.10 (2H, m), 7.42 (1H, d, J = 8.3 Hz). C) {2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] phenyl} methanol 2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzaldehyde (894 mg) in THF ( 4 mL) To the solution was added methanol (2 mL), sodium tetrahydroborate (165 mg) was further added, and the mixture was stirred for 30 minutes. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oily compound (842 mg). It was.
1 H NMR (300 MHz, DMSO-d6) δ 1.28-1.87 (2H, m), 2.03-2.34 (1H, m), 3.87-4.08 (1H, m), 4.17 (1H, ddd, J = 10.3, 6.7 , 3.0 Hz), 4.49 (2H, d, J = 5.3 Hz), 5.10-5.34 (1H, m), 6.86-7.10 (2H, m), 7.42 (1H, d, J = 8.3 Hz).
2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンズアルデヒド(894 mg) のTHF(4 mL)溶液にメタノール(2 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(165 mg)を加えて、30分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下、溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(842 mg)として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28-1.87 (2H, m), 2.03-2.34 (1H, m), 3.87-4.08 (1H, m), 4.17 (1H, ddd, J = 10.3, 6.7, 3.0 Hz), 4.49 (2H, d, J = 5.3 Hz), 5.10-5.34 (1H, m), 6.86-7.10 (2H, m), 7.42 (1H, d, J = 8.3 Hz). C) {2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] phenyl} methanol 2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzaldehyde (894 mg) in THF ( 4 mL) To the solution was added methanol (2 mL), sodium tetrahydroborate (165 mg) was further added, and the mixture was stirred for 30 minutes. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oily compound (842 mg). It was.
1 H NMR (300 MHz, DMSO-d6) δ 1.28-1.87 (2H, m), 2.03-2.34 (1H, m), 3.87-4.08 (1H, m), 4.17 (1H, ddd, J = 10.3, 6.7 , 3.0 Hz), 4.49 (2H, d, J = 5.3 Hz), 5.10-5.34 (1H, m), 6.86-7.10 (2H, m), 7.42 (1H, d, J = 8.3 Hz).
D) N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
{2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]フェニル}メタノール(590 mg)のジエチルエーテル(6 mL)溶液に、三臭化リン(0.291 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下溶媒を留去した。残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(204 mg)、硫酸水素テトラブチルアンモニウム(21.5 mg)、50%水酸化ナトリウム水溶液(3.0 mL)、およびトルエン(6 mL)の混合物を100℃で11時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(266 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.49 (5H, m), 1.50-1.62 (1H, m), 1.90-2.17 (8H, m), 3.20-3.54 (4H, m), 3.92-4.24 (3H, m), 4.54 (2H, s), 5.67 (1H, d, J = 6.1 Hz), 6.80 (1H, dd, J = 8.7, 2.7 Hz), 6.91 (1H, d, J = 2.7 Hz), 7.37 (1H, d, J = 8.7 Hz). D) N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl ] Acetamide {2-Chloro-4-[(2,2-difluorocyclopropyl) methoxy] phenyl} methanol (590 mg) in diethyl ether (6 mL) was added phosphorus tribromide (0.291 mL) at room temperature. For 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), and the solvent was distilled off under reduced pressure. Residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (204 mg), tetrabutylammonium hydrogen sulfate (21.5 mg), 50% aqueous sodium hydroxide solution (3.0 mL) and a mixture of toluene (6 mL) were stirred at 100 ° C. for 11 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (266 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.49 (5H, m), 1.50-1.62 (1H, m), 1.90-2.17 (8H, m), 3.20-3.54 (4H, m), 3.92-4.24 (3H, m), 4.54 (2H, s), 5.67 (1H, d, J = 6.1 Hz), 6.80 (1H, dd, J = 8.7, 2.7 Hz) , 6.91 (1H, d, J = 2.7 Hz), 7.37 (1H, d, J = 8.7 Hz).
{2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]フェニル}メタノール(590 mg)のジエチルエーテル(6 mL)溶液に、三臭化リン(0.291 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下溶媒を留去した。残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(204 mg)、硫酸水素テトラブチルアンモニウム(21.5 mg)、50%水酸化ナトリウム水溶液(3.0 mL)、およびトルエン(6 mL)の混合物を100℃で11時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(266 mg)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.49 (5H, m), 1.50-1.62 (1H, m), 1.90-2.17 (8H, m), 3.20-3.54 (4H, m), 3.92-4.24 (3H, m), 4.54 (2H, s), 5.67 (1H, d, J = 6.1 Hz), 6.80 (1H, dd, J = 8.7, 2.7 Hz), 6.91 (1H, d, J = 2.7 Hz), 7.37 (1H, d, J = 8.7 Hz). D) N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl ] Acetamide {2-Chloro-4-[(2,2-difluorocyclopropyl) methoxy] phenyl} methanol (590 mg) in diethyl ether (6 mL) was added phosphorus tribromide (0.291 mL) at room temperature. For 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), and the solvent was distilled off under reduced pressure. Residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (204 mg), tetrabutylammonium hydrogen sulfate (21.5 mg), 50% aqueous sodium hydroxide solution (3.0 mL) and a mixture of toluene (6 mL) were stirred at 100 ° C. for 11 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (266 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.49 (5H, m), 1.50-1.62 (1H, m), 1.90-2.17 (8H, m), 3.20-3.54 (4H, m), 3.92-4.24 (3H, m), 4.54 (2H, s), 5.67 (1H, d, J = 6.1 Hz), 6.80 (1H, dd, J = 8.7, 2.7 Hz) , 6.91 (1H, d, J = 2.7 Hz), 7.37 (1H, d, J = 8.7 Hz).
実施例39
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(243 mg)をHPLC(カラム:CHIRALPAK OJ(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=90:10)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(80.3 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.50 (5H, m), 1.56-1.66 (1H, m), 1.92-2.15 (8H, m), 3.21-3.53 (4H, m), 3.90-4.22 (3H, m), 4.54 (2H, s), 5.64 (1H, brs), 6.80 (1H, dd, J = 8.5, 2.4 Hz), 6.91 (1H, d, J = 2.6 Hz), 7.37 (1H, d, J = 8.7 Hz).
保持時間 16.9 分
光学純度 >99.9 %ee. Example 39
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide diastereomeric mixture (243 mg) was fractionated by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 90: 10) The compound with a shorter retention time was recrystallized from hexane / ethyl acetate to give the title compound (80.3 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.50 (5H, m), 1.56-1.66 (1H, m), 1.92-2.15 (8H, m), 3.21-3.53 (4H, m), 3.90-4.22 (3H, m), 4.54 (2H, s), 5.64 (1H, brs), 6.80 (1H, dd, J = 8.5, 2.4 Hz), 6.91 (1H, d, J = 2.6 Hz), 7.37 (1H, d, J = 8.7 Hz).
Retention time 16.9 minutes Optical purity> 99.9% ee.
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(243 mg)をHPLC(カラム:CHIRALPAK OJ(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=90:10)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(80.3 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.50 (5H, m), 1.56-1.66 (1H, m), 1.92-2.15 (8H, m), 3.21-3.53 (4H, m), 3.90-4.22 (3H, m), 4.54 (2H, s), 5.64 (1H, brs), 6.80 (1H, dd, J = 8.5, 2.4 Hz), 6.91 (1H, d, J = 2.6 Hz), 7.37 (1H, d, J = 8.7 Hz).
保持時間 16.9 分
光学純度 >99.9 %ee. Example 39
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide diastereomeric mixture (243 mg) was fractionated by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 90: 10) The compound with a shorter retention time was recrystallized from hexane / ethyl acetate to give the title compound (80.3 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.50 (5H, m), 1.56-1.66 (1H, m), 1.92-2.15 (8H, m), 3.21-3.53 (4H, m), 3.90-4.22 (3H, m), 4.54 (2H, s), 5.64 (1H, brs), 6.80 (1H, dd, J = 8.5, 2.4 Hz), 6.91 (1H, d, J = 2.6 Hz), 7.37 (1H, d, J = 8.7 Hz).
Retention time 16.9 minutes Optical purity> 99.9% ee.
実施例40
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(243 mg)をHPLC(カラム:CHIRALPAK OJ(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=90:10)にて分取し、保持時間がより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(80.4 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.48 (5H, m), 1.56-1.66 (1H, m), 1.93-2.12 (8H, m), 3.24-3.49 (4H, m), 3.93-4.19 (3H, m), 4.54 (2H, s), 5.58-5.71 (1H, m), 6.80 (1H, dd, J = 8.5, 2.4 Hz), 6.91 (1H, d, J = 2.6 Hz), 7.37 (1H, d, J = 8.7 Hz).
保持時間 20.4 分
光学純度 98.7 %ee. Example 40
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide diastereomeric mixture (243 mg) was fractionated by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 90: 10) The compound with a longer retention time was recrystallized from hexane / ethyl acetate to give the title compound (80.4 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.48 (5H, m), 1.56-1.66 (1H, m), 1.93-2.12 (8H, m), 3.24-3.49 (4H, m), 3.93-4.19 (3H, m), 4.54 (2H, s), 5.58-5.71 (1H, m), 6.80 (1H, dd, J = 8.5, 2.4 Hz), 6.91 ( 1H, d, J = 2.6 Hz), 7.37 (1H, d, J = 8.7 Hz).
Retention time 20.4 min Optical purity 98.7% ee.
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(243 mg)をHPLC(カラム:CHIRALPAK OJ(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=90:10)にて分取し、保持時間がより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(80.4 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.48 (5H, m), 1.56-1.66 (1H, m), 1.93-2.12 (8H, m), 3.24-3.49 (4H, m), 3.93-4.19 (3H, m), 4.54 (2H, s), 5.58-5.71 (1H, m), 6.80 (1H, dd, J = 8.5, 2.4 Hz), 6.91 (1H, d, J = 2.6 Hz), 7.37 (1H, d, J = 8.7 Hz).
保持時間 20.4 分
光学純度 98.7 %ee. Example 40
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide diastereomeric mixture (243 mg) was fractionated by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 90: 10) The compound with a longer retention time was recrystallized from hexane / ethyl acetate to give the title compound (80.4 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.48 (5H, m), 1.56-1.66 (1H, m), 1.93-2.12 (8H, m), 3.24-3.49 (4H, m), 3.93-4.19 (3H, m), 4.54 (2H, s), 5.58-5.71 (1H, m), 6.80 (1H, dd, J = 8.5, 2.4 Hz), 6.91 ( 1H, d, J = 2.6 Hz), 7.37 (1H, d, J = 8.7 Hz).
Retention time 20.4 min Optical purity 98.7% ee.
実施例40a
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体 Example 40a
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide Optically active form of
N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体 Example 40a
N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide Optically active form of
A) 4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)シクロヘキシル ベンゾアートのトランス/シス混合物
tert-ブチル{(1S)-2-[(4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマート(66 g)、安息香酸(35.4 g)、ジシクロヘキシルカルボジイミド(62.1 g)、4-ジメチルアミノピリジン(2.99 g)およびジクロロメタン(764 mL)の混合物を窒素雰囲気下室温で終夜撹拌した。反応混合物に水を加え、析出した固体を濾過により除去した。濾液の有機層を分離し、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧下濃縮して、4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)シクロヘキシル ベンゾアートのトランス/シス混合物(81 g)を得た。
1H NMR (400 MHz, CDCl3) δ 1.16 (3H, s), 1.44 (9H, s), 1.51-1.61 (4H, m), 1.98-2.07 (4H, m), 3.39 (3H, s), 3.69 (1H, s), 4.7 (1H, s), 5.04 (1H, s), 3.40-7.44 (2H, m), 7.52-7.54 (1H, m), 8.01-8.03 (2H, m). A) 4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl} oxy) cyclohexyl benzoate trans / cis mixture tert-butyl {(1S) -2-[(4-hydroxycyclohexyl) Oxy] -1-methylethyl} carbamate (66 g), benzoic acid (35.4 g), dicyclohexylcarbodiimide (62.1 g), 4-dimethylaminopyridine (2.99 g) and dichloromethane (764 mL) at room temperature under a nitrogen atmosphere. And stirred overnight. Water was added to the reaction mixture, and the precipitated solid was removed by filtration. The organic layer of the filtrate was separated, washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give 4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl} oxy ) A trans / cis mixture of cyclohexyl benzoate (81 g) was obtained.
1 H NMR (400 MHz, CDCl 3 ) δ 1.16 (3H, s), 1.44 (9H, s), 1.51-1.61 (4H, m), 1.98-2.07 (4H, m), 3.39 (3H, s), 3.69 (1H, s), 4.7 (1H, s), 5.04 (1H, s), 3.40-7.44 (2H, m), 7.52-7.54 (1H, m), 8.01-8.03 (2H, m).
tert-ブチル{(1S)-2-[(4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマート(66 g)、安息香酸(35.4 g)、ジシクロヘキシルカルボジイミド(62.1 g)、4-ジメチルアミノピリジン(2.99 g)およびジクロロメタン(764 mL)の混合物を窒素雰囲気下室温で終夜撹拌した。反応混合物に水を加え、析出した固体を濾過により除去した。濾液の有機層を分離し、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧下濃縮して、4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)シクロヘキシル ベンゾアートのトランス/シス混合物(81 g)を得た。
1H NMR (400 MHz, CDCl3) δ 1.16 (3H, s), 1.44 (9H, s), 1.51-1.61 (4H, m), 1.98-2.07 (4H, m), 3.39 (3H, s), 3.69 (1H, s), 4.7 (1H, s), 5.04 (1H, s), 3.40-7.44 (2H, m), 7.52-7.54 (1H, m), 8.01-8.03 (2H, m). A) 4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl} oxy) cyclohexyl benzoate trans / cis mixture tert-butyl {(1S) -2-[(4-hydroxycyclohexyl) Oxy] -1-methylethyl} carbamate (66 g), benzoic acid (35.4 g), dicyclohexylcarbodiimide (62.1 g), 4-dimethylaminopyridine (2.99 g) and dichloromethane (764 mL) at room temperature under a nitrogen atmosphere. And stirred overnight. Water was added to the reaction mixture, and the precipitated solid was removed by filtration. The organic layer of the filtrate was separated, washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give 4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl} oxy ) A trans / cis mixture of cyclohexyl benzoate (81 g) was obtained.
1 H NMR (400 MHz, CDCl 3 ) δ 1.16 (3H, s), 1.44 (9H, s), 1.51-1.61 (4H, m), 1.98-2.07 (4H, m), 3.39 (3H, s), 3.69 (1H, s), 4.7 (1H, s), 5.04 (1H, s), 3.40-7.44 (2H, m), 7.52-7.54 (1H, m), 8.01-8.03 (2H, m).
B) trans-4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)シクロヘキシル ベンゾアート
4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)シクロヘキシル ベンゾアートのトランス/シス混合物(300 g)を、SFC(超臨界流体クロマトグラフィー)(装置:Thar analytical SFC、カラム:CHIRALPAK AD(商品名)、250 × 50 mm I.D.、移動相:二酸化炭素/メタノール=75:25、流速:150 mL/min、圧力:100 bar、カラム温度:38℃、検出波長:220 nm)にて分取し、保持時間がより大きい標題化合物(90 g)を得た。
精製時保持時間 5.55 分
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 8.4 Hz), 1.45 (9H, s), 1.53-1.63 (4H, m), 1.98-2.08 (4H, m), 3.39-3.41 (3H, m), 3.70-3.80 (1H, m), 4.65-4.75 (1H, m), 5.00-5.10 (1H, m), 7.41-7.45 (2H, m,), 7.53-7.57 (1H, m), 8.02-8.04 (2H, m). B) trans-4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl} oxy) cyclohexyl benzoate 4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl } Oxy) cyclohexyl Trans / cis mixture of benzoate (300 g), SFC (supercritical fluid chromatography) (instrument: Thar analytical SFC, column: CHIRALPAK AD (trade name), 250 x 50 mm ID, mobile phase: The title compound (90 g) with a larger retention time was fractionated at carbon dioxide / methanol = 75:25, flow rate: 150 mL / min, pressure: 100 bar, column temperature: 38 ° C, detection wavelength: 220 nm) Got.
Retention time during purification 5.55 minutes
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 8.4 Hz), 1.45 (9H, s), 1.53-1.63 (4H, m), 1.98-2.08 (4H, m), 3.39- 3.41 (3H, m), 3.70-3.80 (1H, m), 4.65-4.75 (1H, m), 5.00-5.10 (1H, m), 7.41-7.45 (2H, m,), 7.53-7.57 (1H, m), 8.02-8.04 (2H, m).
4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)シクロヘキシル ベンゾアートのトランス/シス混合物(300 g)を、SFC(超臨界流体クロマトグラフィー)(装置:Thar analytical SFC、カラム:CHIRALPAK AD(商品名)、250 × 50 mm I.D.、移動相:二酸化炭素/メタノール=75:25、流速:150 mL/min、圧力:100 bar、カラム温度:38℃、検出波長:220 nm)にて分取し、保持時間がより大きい標題化合物(90 g)を得た。
精製時保持時間 5.55 分
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 8.4 Hz), 1.45 (9H, s), 1.53-1.63 (4H, m), 1.98-2.08 (4H, m), 3.39-3.41 (3H, m), 3.70-3.80 (1H, m), 4.65-4.75 (1H, m), 5.00-5.10 (1H, m), 7.41-7.45 (2H, m,), 7.53-7.57 (1H, m), 8.02-8.04 (2H, m). B) trans-4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl} oxy) cyclohexyl benzoate 4-({(2S) -2-[(tert-butoxycarbonyl) amino] propyl } Oxy) cyclohexyl Trans / cis mixture of benzoate (300 g), SFC (supercritical fluid chromatography) (instrument: Thar analytical SFC, column: CHIRALPAK AD (trade name), 250 x 50 mm ID, mobile phase: The title compound (90 g) with a larger retention time was fractionated at carbon dioxide / methanol = 75:25, flow rate: 150 mL / min, pressure: 100 bar, column temperature: 38 ° C, detection wavelength: 220 nm) Got.
Retention time during purification 5.55 minutes
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 8.4 Hz), 1.45 (9H, s), 1.53-1.63 (4H, m), 1.98-2.08 (4H, m), 3.39- 3.41 (3H, m), 3.70-3.80 (1H, m), 4.65-4.75 (1H, m), 5.00-5.10 (1H, m), 7.41-7.45 (2H, m,), 7.53-7.57 (1H, m), 8.02-8.04 (2H, m).
C) N-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
trans-4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)シクロヘキシル ベンゾアート (10 g)に4 M塩化水素/酢酸エチル(62.5 mL)を加え、混合物を室温で1時間撹拌した後に減圧下濃縮した。残渣をピリジン(38 mL)に溶解し、混合物に無水酢酸(3.47 g)を氷冷下で滴下し、混合物を1時間撹拌した後に減圧下濃縮した。残渣をTHF(15.5 mL)およびメタノール(15.5 mL)の混合溶媒に溶解し、2 M水酸化ナトリウム水溶液(39.15 mL)を加え、混合物を室温で15時間撹拌した。反応混合物に飽和食塩水を加え、酢酸エチルで5回抽出した。合わせた有機層を無水硫酸ナトリウムで乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し標題化合物(3.0 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.16 (3H, d, J = 6.8 Hz), 1.29-1.34 (4H, m), 1.68 (1H, s), 1.95-1.97 (6H, m), 3.21-3.31 (1H, m), 3.24 (1H, s), 3.26-3.45 (2H, m), 3.68 (1H, s), 4.08-4.12 (1H, m), 5.70 (1H, s). C) N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide trans-4-({(2S) -2-[(tert-butoxycarbonyl) amino] 4M Hydrogen chloride / ethyl acetate (62.5 mL) was added to (propyl} oxy) cyclohexyl benzoate (10 g), and the mixture was stirred at room temperature for 1 hour and then concentrated under reduced pressure. The residue was dissolved in pyridine (38 mL), acetic anhydride (3.47 g) was added dropwise to the mixture under ice-cooling, and the mixture was stirred for 1 hour and then concentrated under reduced pressure. The residue was dissolved in a mixed solvent of THF (15.5 mL) and methanol (15.5 mL), 2 M aqueous sodium hydroxide solution (39.15 mL) was added, and the mixture was stirred at room temperature for 15 hours. Saturated brine was added to the reaction mixture, and the mixture was extracted 5 times with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (3.0 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.16 (3H, d, J = 6.8 Hz), 1.29-1.34 (4H, m), 1.68 (1H, s), 1.95-1.97 (6H, m), 3.21- 3.31 (1H, m), 3.24 (1H, s), 3.26-3.45 (2H, m), 3.68 (1H, s), 4.08-4.12 (1H, m), 5.70 (1H, s).
trans-4-({(2S)-2-[(tert-ブトキシカルボニル)アミノ]プロピル}オキシ)シクロヘキシル ベンゾアート (10 g)に4 M塩化水素/酢酸エチル(62.5 mL)を加え、混合物を室温で1時間撹拌した後に減圧下濃縮した。残渣をピリジン(38 mL)に溶解し、混合物に無水酢酸(3.47 g)を氷冷下で滴下し、混合物を1時間撹拌した後に減圧下濃縮した。残渣をTHF(15.5 mL)およびメタノール(15.5 mL)の混合溶媒に溶解し、2 M水酸化ナトリウム水溶液(39.15 mL)を加え、混合物を室温で15時間撹拌した。反応混合物に飽和食塩水を加え、酢酸エチルで5回抽出した。合わせた有機層を無水硫酸ナトリウムで乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し標題化合物(3.0 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.16 (3H, d, J = 6.8 Hz), 1.29-1.34 (4H, m), 1.68 (1H, s), 1.95-1.97 (6H, m), 3.21-3.31 (1H, m), 3.24 (1H, s), 3.26-3.45 (2H, m), 3.68 (1H, s), 4.08-4.12 (1H, m), 5.70 (1H, s). C) N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide trans-4-({(2S) -2-[(tert-butoxycarbonyl) amino] 4M Hydrogen chloride / ethyl acetate (62.5 mL) was added to (propyl} oxy) cyclohexyl benzoate (10 g), and the mixture was stirred at room temperature for 1 hour and then concentrated under reduced pressure. The residue was dissolved in pyridine (38 mL), acetic anhydride (3.47 g) was added dropwise to the mixture under ice-cooling, and the mixture was stirred for 1 hour and then concentrated under reduced pressure. The residue was dissolved in a mixed solvent of THF (15.5 mL) and methanol (15.5 mL), 2 M aqueous sodium hydroxide solution (39.15 mL) was added, and the mixture was stirred at room temperature for 15 hours. Saturated brine was added to the reaction mixture, and the mixture was extracted 5 times with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (3.0 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.16 (3H, d, J = 6.8 Hz), 1.29-1.34 (4H, m), 1.68 (1H, s), 1.95-1.97 (6H, m), 3.21- 3.31 (1H, m), 3.24 (1H, s), 3.26-3.45 (2H, m), 3.68 (1H, s), 4.08-4.12 (1H, m), 5.70 (1H, s).
D) (2,2-ジフルオロシクロプロピル)メチル 4-ニトロベンゼンスルホナート
(2,2-ジフルオロシクロプロピル)メタノール(20.0 g)およびトリエチルアミン(32.2 mL)のTHF(300 mL)溶液に、4-ニトロベンゼンスルホニル クロリド(41.0 g)を0℃で加え、4時間撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(47.2 g)を白色固体として得た。
1H NMR (300 MHz, CDCl3) δ 1.18-1.36 (1H, m), 1.58-1.70 (1H, m), 1.86-2.11 (1H, m), 4.07-4.22 (1H, m), 4.24-4.37 (1H, m), 8.07-8.17 (2H, m), 8.37-8.49 (2H, m). D) (2,2-Difluorocyclopropyl) methyl 4-nitrobenzenesulfonate (2,2-difluorocyclopropyl) methanol (20.0 g) and triethylamine (32.2 mL) in THF (300 mL) in 4-nitrobenzenesulfonyl Chloride (41.0 g) was added at 0 ° C. and stirred for 4 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (47.2 g) as a white solid.
1 H NMR (300 MHz, CDCl 3 ) δ 1.18-1.36 (1H, m), 1.58-1.70 (1H, m), 1.86-2.11 (1H, m), 4.07-4.22 (1H, m), 4.24-4.37 (1H, m), 8.07-8.17 (2H, m), 8.37-8.49 (2H, m).
(2,2-ジフルオロシクロプロピル)メタノール(20.0 g)およびトリエチルアミン(32.2 mL)のTHF(300 mL)溶液に、4-ニトロベンゼンスルホニル クロリド(41.0 g)を0℃で加え、4時間撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(47.2 g)を白色固体として得た。
1H NMR (300 MHz, CDCl3) δ 1.18-1.36 (1H, m), 1.58-1.70 (1H, m), 1.86-2.11 (1H, m), 4.07-4.22 (1H, m), 4.24-4.37 (1H, m), 8.07-8.17 (2H, m), 8.37-8.49 (2H, m). D) (2,2-Difluorocyclopropyl) methyl 4-nitrobenzenesulfonate (2,2-difluorocyclopropyl) methanol (20.0 g) and triethylamine (32.2 mL) in THF (300 mL) in 4-nitrobenzenesulfonyl Chloride (41.0 g) was added at 0 ° C. and stirred for 4 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (47.2 g) as a white solid.
1 H NMR (300 MHz, CDCl 3 ) δ 1.18-1.36 (1H, m), 1.58-1.70 (1H, m), 1.86-2.11 (1H, m), 4.07-4.22 (1H, m), 4.24-4.37 (1H, m), 8.07-8.17 (2H, m), 8.37-8.49 (2H, m).
E) (2,2-ジフルオロシクロプロピル)メチル4-ニトロベンゼンスルホナートの光学活性体
(2,2-ジフルオロシクロプロピル)メチル 4-ニトロベンゼンスルホナートのラセミ体(48.4 g)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=50:50)にて分取し、保持時間がより小さい標題化合物(18.4 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.18-1.32 (1H, m), 1.56-1.69 (1H, m), 1.90-2.09 (1H, m), 4.10-4.21 (1H, m), 4.23-4.35 (1H, m), 8.09-8.17 (2H, m), 8.38-8.48 (2H, m).
分析保持時間 15.4 分
光学純度 >99.9% ee E) Optically active form of (2,2-difluorocyclopropyl) methyl 4-nitrobenzenesulfonate Racemic form (48.4 g) of (2,2-difluorocyclopropyl) methyl 4-nitrobenzenesulfonate was analyzed by HPLC (column: CHIRALPAK AD (Trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 50: 50) to obtain the title compound (18.4 g) having a shorter retention time.
1 H NMR (300 MHz, CDCl 3 ) δ 1.18-1.32 (1H, m), 1.56-1.69 (1H, m), 1.90-2.09 (1H, m), 4.10-4.21 (1H, m), 4.23-4.35 (1H, m), 8.09-8.17 (2H, m), 8.38-8.48 (2H, m).
Analysis retention time 15.4 minutes Optical purity> 99.9% ee
(2,2-ジフルオロシクロプロピル)メチル 4-ニトロベンゼンスルホナートのラセミ体(48.4 g)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=50:50)にて分取し、保持時間がより小さい標題化合物(18.4 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.18-1.32 (1H, m), 1.56-1.69 (1H, m), 1.90-2.09 (1H, m), 4.10-4.21 (1H, m), 4.23-4.35 (1H, m), 8.09-8.17 (2H, m), 8.38-8.48 (2H, m).
分析保持時間 15.4 分
光学純度 >99.9% ee E) Optically active form of (2,2-difluorocyclopropyl) methyl 4-nitrobenzenesulfonate Racemic form (48.4 g) of (2,2-difluorocyclopropyl) methyl 4-nitrobenzenesulfonate was analyzed by HPLC (column: CHIRALPAK AD (Trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 50: 50) to obtain the title compound (18.4 g) having a shorter retention time.
1 H NMR (300 MHz, CDCl 3 ) δ 1.18-1.32 (1H, m), 1.56-1.69 (1H, m), 1.90-2.09 (1H, m), 4.10-4.21 (1H, m), 4.23-4.35 (1H, m), 8.09-8.17 (2H, m), 8.38-8.48 (2H, m).
Analysis retention time 15.4 minutes Optical purity> 99.9% ee
F) 2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンズアルデヒドの光学活性体
2-クロロ-4-ヒドロキシベンズアルデヒド (854 mg)のDMF(20 mL)溶液に炭酸カリウム(1.13 g)および(2,2-ジフルオロシクロプロピル)メチル 4-ニトロベンゼンスルホナートの光学活性体(保持時間の小さい光学活性体、2.40 g)を加えて、60℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物 (1.26 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.31 (1H, dtd, J = 13.1, 7.8, 4.0 Hz), 1.56-1.73 (1H, m), 2.00-2.17 (1H, m), 4.03-4.19 (2H, m), 6.85-6.98 (2H, m), 7.84-7.96 (1H, m), 10.33-10.35 (1H, m). F) 2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzaldehyde optically active substance Potassium carbonate (1.13 g) in DMF (20 mL) solution of 2-chloro-4-hydroxybenzaldehyde (854 mg) ) And (2,2-difluorocyclopropyl) methyl 4-nitrobenzenesulfonate optically active substance (optically active substance having a short retention time, 2.40 g) was added, and the mixture was heated and stirred at 60 ° C. for 2 hours. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.26 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.31 (1H, dtd, J = 13.1, 7.8, 4.0 Hz), 1.56-1.73 (1H, m), 2.00-2.17 (1H, m), 4.03-4.19 (2H , m), 6.85-6.98 (2H, m), 7.84-7.96 (1H, m), 10.33-10.35 (1H, m).
2-クロロ-4-ヒドロキシベンズアルデヒド (854 mg)のDMF(20 mL)溶液に炭酸カリウム(1.13 g)および(2,2-ジフルオロシクロプロピル)メチル 4-ニトロベンゼンスルホナートの光学活性体(保持時間の小さい光学活性体、2.40 g)を加えて、60℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物 (1.26 g)を得た。
1H NMR (300 MHz, CDCl3) δ 1.31 (1H, dtd, J = 13.1, 7.8, 4.0 Hz), 1.56-1.73 (1H, m), 2.00-2.17 (1H, m), 4.03-4.19 (2H, m), 6.85-6.98 (2H, m), 7.84-7.96 (1H, m), 10.33-10.35 (1H, m). F) 2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzaldehyde optically active substance Potassium carbonate (1.13 g) in DMF (20 mL) solution of 2-chloro-4-hydroxybenzaldehyde (854 mg) ) And (2,2-difluorocyclopropyl) methyl 4-nitrobenzenesulfonate optically active substance (optically active substance having a short retention time, 2.40 g) was added, and the mixture was heated and stirred at 60 ° C. for 2 hours. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.26 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.31 (1H, dtd, J = 13.1, 7.8, 4.0 Hz), 1.56-1.73 (1H, m), 2.00-2.17 (1H, m), 4.03-4.19 (2H , m), 6.85-6.98 (2H, m), 7.84-7.96 (1H, m), 10.33-10.35 (1H, m).
G) N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンズアルデヒドの光学活性体(1.26 g) のTHF(20 mL)溶液にメタノール(5 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(290 mg)を加えて、室温で90分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下、溶媒を留去した。残渣のジエチルエーテル(25 mL)溶液に、三臭化リン(0.723 mL)を加えて室温で60分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下溶媒を留去した。残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(1.10 g)、硫酸水素テトラブチルアンモニウム(173 mg)、50%水酸化ナトリウム水溶液(17 mL)、およびトルエン(35 mL)の混合物を100℃で11時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、得られた固体を再結晶(ヘキサン/酢酸エチル)し、標題化合物(709 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.28-1.47 (5H, m), 1.58-1.63 (1H, m), 1.93-2.14 (8H, m), 3.22-3.49 (4H, m), 3.91-4.18 (3H, m), 4.54 (2H, s), 5.60-5.73 (1H, m), 6.81 (1H, dd, J = 8.5, 2.5 Hz), 6.91 (1H, d, J = 2.5 Hz), 7.37 (1H, d, J = 8.5 Hz).
Anal. Calcd for C22H30NO4ClF2:C,59.26;H,6.78;N,3.14. Found:C,59.44;H,6.96;N,3.08.
mp. 86.1-87.1℃ G) N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl Acetamide optically active substance 2-Chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzaldehyde Optically active substance (1.26 g) in THF (20 mL) was added methanol (5 mL), and Sodium tetrahydroborate (290 mg) was added and stirred at room temperature for 90 minutes. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. To a solution of the residue in diethyl ether (25 mL) was added phosphorus tribromide (0.723 mL), and the mixture was stirred at room temperature for 60 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), and the solvent was distilled off under reduced pressure. Residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (1.10 g), tetrabutylammonium hydrogen sulfate (173 mg), 50% aqueous sodium hydroxide solution A mixture of (17 mL) and toluene (35 mL) was stirred at 100 ° C. for 11 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound (709 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.28-1.47 (5H, m), 1.58-1.63 (1H, m), 1.93-2.14 (8H, m), 3.22-3.49 (4H, m), 3.91-4.18 (3H, m), 4.54 (2H, s), 5.60-5.73 (1H, m), 6.81 (1H, dd, J = 8.5, 2.5 Hz), 6.91 ( 1H, d, J = 2.5 Hz), 7.37 (1H, d, J = 8.5 Hz).
Anal.Calcd for C 22 H 30 NO 4 ClF 2 : C, 59.26; H, 6.78; N, 3.14.Found: C, 59.44; H, 6.96; N, 3.08.
mp. 86.1-87.1 ℃
2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンズアルデヒドの光学活性体(1.26 g) のTHF(20 mL)溶液にメタノール(5 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(290 mg)を加えて、室温で90分間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下、溶媒を留去した。残渣のジエチルエーテル(25 mL)溶液に、三臭化リン(0.723 mL)を加えて室温で60分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下溶媒を留去した。残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(1.10 g)、硫酸水素テトラブチルアンモニウム(173 mg)、50%水酸化ナトリウム水溶液(17 mL)、およびトルエン(35 mL)の混合物を100℃で11時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、得られた固体を再結晶(ヘキサン/酢酸エチル)し、標題化合物(709 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.28-1.47 (5H, m), 1.58-1.63 (1H, m), 1.93-2.14 (8H, m), 3.22-3.49 (4H, m), 3.91-4.18 (3H, m), 4.54 (2H, s), 5.60-5.73 (1H, m), 6.81 (1H, dd, J = 8.5, 2.5 Hz), 6.91 (1H, d, J = 2.5 Hz), 7.37 (1H, d, J = 8.5 Hz).
Anal. Calcd for C22H30NO4ClF2:C,59.26;H,6.78;N,3.14. Found:C,59.44;H,6.96;N,3.08.
mp. 86.1-87.1℃ G) N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl Acetamide optically active substance 2-Chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzaldehyde Optically active substance (1.26 g) in THF (20 mL) was added methanol (5 mL), and Sodium tetrahydroborate (290 mg) was added and stirred at room temperature for 90 minutes. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. To a solution of the residue in diethyl ether (25 mL) was added phosphorus tribromide (0.723 mL), and the mixture was stirred at room temperature for 60 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), and the solvent was distilled off under reduced pressure. Residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (1.10 g), tetrabutylammonium hydrogen sulfate (173 mg), 50% aqueous sodium hydroxide solution A mixture of (17 mL) and toluene (35 mL) was stirred at 100 ° C. for 11 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound (709 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.28-1.47 (5H, m), 1.58-1.63 (1H, m), 1.93-2.14 (8H, m), 3.22-3.49 (4H, m), 3.91-4.18 (3H, m), 4.54 (2H, s), 5.60-5.73 (1H, m), 6.81 (1H, dd, J = 8.5, 2.5 Hz), 6.91 ( 1H, d, J = 2.5 Hz), 7.37 (1H, d, J = 8.5 Hz).
Anal.Calcd for C 22 H 30 NO 4 ClF 2 : C, 59.26; H, 6.78; N, 3.14.Found: C, 59.44; H, 6.96; N, 3.08.
mp. 86.1-87.1 ℃
実施例41
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(400 mg)のDMF(5 mL)溶液に炭酸カリウム(489 mg)および(2,2-ジフルオロシクロプロピル)メチル メタンスルホナート(658 mL)を加えて、60℃で終夜加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(430 mg)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.95-1.08 (3H, m), 1.18-1.35 (4H, m), 1.37-1.58 (1H, m), 1.62-1.76 (1H, m), 1.77 (3H, s), 1.89 (4H, brs), 2.10-2.33 (1H, m), 3.09-3.31 (3H, m), 3.36 (1H, d, J = 4.2 Hz), 3.82 (1H, dt, J = 13.3, 6.8 Hz), 3.92-4.03 (1H, m), 4.09-4.24 (1H, m), 4.42 (2H, s), 6.65-6.93 (2H, m), 7.32 (1H, t, J = 8.7 Hz), 7.65 (1H, d, J = 7.9 Hz). Example 41
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide N-[(1S) -2-({trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (400 mg) in DMF (5 mL) To the mixture were added potassium carbonate (489 mg) and (2,2-difluorocyclopropyl) methyl methanesulfonate (658 mL), and the mixture was heated and stirred at 60 ° C. overnight. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (430 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.95-1.08 (3H, m), 1.18-1.35 (4H, m), 1.37-1.58 (1H, m), 1.62-1.76 (1H, m), 1.77 (3H, s), 1.89 (4H, brs), 2.10-2.33 (1H, m), 3.09-3.31 (3H, m), 3.36 (1H, d, J = 4.2 Hz), 3.82 (1H, dt, J = 13.3, 6.8 Hz), 3.92-4.03 (1H, m), 4.09-4.24 (1H, m), 4.42 (2H, s), 6.65-6.93 (2H, m), 7.32 (1H, t, J = 8.7 Hz), 7.65 (1H, d, J = 7.9 Hz).
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(400 mg)のDMF(5 mL)溶液に炭酸カリウム(489 mg)および(2,2-ジフルオロシクロプロピル)メチル メタンスルホナート(658 mL)を加えて、60℃で終夜加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(430 mg)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.95-1.08 (3H, m), 1.18-1.35 (4H, m), 1.37-1.58 (1H, m), 1.62-1.76 (1H, m), 1.77 (3H, s), 1.89 (4H, brs), 2.10-2.33 (1H, m), 3.09-3.31 (3H, m), 3.36 (1H, d, J = 4.2 Hz), 3.82 (1H, dt, J = 13.3, 6.8 Hz), 3.92-4.03 (1H, m), 4.09-4.24 (1H, m), 4.42 (2H, s), 6.65-6.93 (2H, m), 7.32 (1H, t, J = 8.7 Hz), 7.65 (1H, d, J = 7.9 Hz). Example 41
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide N-[(1S) -2-({trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (400 mg) in DMF (5 mL) To the mixture were added potassium carbonate (489 mg) and (2,2-difluorocyclopropyl) methyl methanesulfonate (658 mL), and the mixture was heated and stirred at 60 ° C. overnight. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (430 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.95-1.08 (3H, m), 1.18-1.35 (4H, m), 1.37-1.58 (1H, m), 1.62-1.76 (1H, m), 1.77 (3H, s), 1.89 (4H, brs), 2.10-2.33 (1H, m), 3.09-3.31 (3H, m), 3.36 (1H, d, J = 4.2 Hz), 3.82 (1H, dt, J = 13.3, 6.8 Hz), 3.92-4.03 (1H, m), 4.09-4.24 (1H, m), 4.42 (2H, s), 6.65-6.93 (2H, m), 7.32 (1H, t, J = 8.7 Hz), 7.65 (1H, d, J = 7.9 Hz).
実施例42
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(430 mg)をHPLC(カラム:CHIRALPAK OJ(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/2-プロパノール=70:30)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(187 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.4 Hz), 1.21-1.47 (5H, m), 1.57-1.71 (1H, m), 1.85-2.14 (8H, m), 3.17-3.53 (4H, m), 3.84-4.26 (3H, m), 4.50 (2H, s), 5.66 (1H, brs), 6.54-6.72 (2H, m), 7.27-7.35 (1H, m).
保持時間 7.2 分
光学純度 98.6 %ee. Example 42
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide The optically active form of N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide diastereomer mixture (430 mg) was analyzed by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / 2-propanol = 70: 30). The compound having a smaller retention time was recrystallized from hexane / ethyl acetate to give the title compound (187 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.4 Hz), 1.21-1.47 (5H, m), 1.57-1.71 (1H, m), 1.85-2.14 (8H, m), 3.17-3.53 (4H, m), 3.84-4.26 (3H, m), 4.50 (2H, s), 5.66 (1H, brs), 6.54-6.72 (2H, m), 7.27-7.35 (1H, m).
Retention time 7.2 minutes Optical purity 98.6% ee.
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(430 mg)をHPLC(カラム:CHIRALPAK OJ(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/2-プロパノール=70:30)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(187 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.4 Hz), 1.21-1.47 (5H, m), 1.57-1.71 (1H, m), 1.85-2.14 (8H, m), 3.17-3.53 (4H, m), 3.84-4.26 (3H, m), 4.50 (2H, s), 5.66 (1H, brs), 6.54-6.72 (2H, m), 7.27-7.35 (1H, m).
保持時間 7.2 分
光学純度 98.6 %ee. Example 42
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide The optically active form of N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide diastereomer mixture (430 mg) was analyzed by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / 2-propanol = 70: 30). The compound having a smaller retention time was recrystallized from hexane / ethyl acetate to give the title compound (187 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.4 Hz), 1.21-1.47 (5H, m), 1.57-1.71 (1H, m), 1.85-2.14 (8H, m), 3.17-3.53 (4H, m), 3.84-4.26 (3H, m), 4.50 (2H, s), 5.66 (1H, brs), 6.54-6.72 (2H, m), 7.27-7.35 (1H, m).
Retention time 7.2 minutes Optical purity 98.6% ee.
実施例43
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(430 mg)をHPLC(カラム:CHIRALPAK OJ(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/2-プロパノール=70:30)にて分取し、保持時間がより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(190 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.20-1.47 (6H, m), 1.58-1.69 (1H, m), 1.96-2.16 (7H, m), 3.21-3.49 (4H, m), 3.90-4.20 (3H, m), 4.51 (2H, s), 5.64 (1H, brs), 6.50-6.74 (2H, m), 7.30 (1H, t, J = 8.5 Hz).
保持時間 8.00 分
光学純度 96.8 %ee. Example 43
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide The optically active form of N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide diastereomer mixture (430 mg) was analyzed by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / 2-propanol = 70: 30). The compound with longer retention time was recrystallized with hexane / ethyl acetate to give the title compound (190 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.20-1.47 (6H, m), 1.58-1.69 (1H, m), 1.96-2.16 (7H, m), 3.21-3.49 (4H, m), 3.90-4.20 (3H, m), 4.51 (2H, s), 5.64 (1H, brs), 6.50-6.74 (2H, m), 7.30 (1H, t, J = 8.5 Hz).
Retention time 8.00 min Optical purity 96.8% ee.
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(430 mg)をHPLC(カラム:CHIRALPAK OJ(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/2-プロパノール=70:30)にて分取し、保持時間がより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(190 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.20-1.47 (6H, m), 1.58-1.69 (1H, m), 1.96-2.16 (7H, m), 3.21-3.49 (4H, m), 3.90-4.20 (3H, m), 4.51 (2H, s), 5.64 (1H, brs), 6.50-6.74 (2H, m), 7.30 (1H, t, J = 8.5 Hz).
保持時間 8.00 分
光学純度 96.8 %ee. Example 43
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide The optically active form of N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide diastereomer mixture (430 mg) was analyzed by HPLC (column: CHIRALPAK OJ (trade name), 50 mm ID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / 2-propanol = 70: 30). The compound with longer retention time was recrystallized with hexane / ethyl acetate to give the title compound (190 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.20-1.47 (6H, m), 1.58-1.69 (1H, m), 1.96-2.16 (7H, m), 3.21-3.49 (4H, m), 3.90-4.20 (3H, m), 4.51 (2H, s), 5.64 (1H, brs), 6.50-6.74 (2H, m), 7.30 (1H, t, J = 8.5 Hz).
Retention time 8.00 min Optical purity 96.8% ee.
実施例44
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 44
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Acetamide
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 44
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Acetamide
A) 4-(ベンジルオキシ)-2,6-ジフルオロベンズアルデヒド
2,6-ジフルオロ-4-ヒドロキシベンズアルデヒド(1.50 g)のDMF(15 mL)溶液に炭酸カリウム(2.62 g)と(ブロモメチル)ベンゼン(1.35 mL)を加えて、70℃で1時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(2.34 g)として得た。
1H NMR (300 MHz, CDCl3) δ 5.11 (2H, s), 6.45-6.68 (2H, m), 7.30-7.50 (5H, m), 10.19 (1H, s). A) 4- (Benzyloxy) -2,6-difluorobenzaldehyde 2,6-difluoro-4-hydroxybenzaldehyde (1.50 g) in DMF (15 mL) was added to potassium carbonate (2.62 g) and (bromomethyl) benzene (1.35 mL) was added and stirred with heating at 70 ° C. for 1 hour. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (2.34 g).
1 H NMR (300 MHz, CDCl 3 ) δ 5.11 (2H, s), 6.45-6.68 (2H, m), 7.30-7.50 (5H, m), 10.19 (1H, s).
2,6-ジフルオロ-4-ヒドロキシベンズアルデヒド(1.50 g)のDMF(15 mL)溶液に炭酸カリウム(2.62 g)と(ブロモメチル)ベンゼン(1.35 mL)を加えて、70℃で1時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(2.34 g)として得た。
1H NMR (300 MHz, CDCl3) δ 5.11 (2H, s), 6.45-6.68 (2H, m), 7.30-7.50 (5H, m), 10.19 (1H, s). A) 4- (Benzyloxy) -2,6-difluorobenzaldehyde 2,6-difluoro-4-hydroxybenzaldehyde (1.50 g) in DMF (15 mL) was added to potassium carbonate (2.62 g) and (bromomethyl) benzene (1.35 mL) was added and stirred with heating at 70 ° C. for 1 hour. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (2.34 g).
1 H NMR (300 MHz, CDCl 3 ) δ 5.11 (2H, s), 6.45-6.68 (2H, m), 7.30-7.50 (5H, m), 10.19 (1H, s).
B) [4-(ベンジルオキシ)-2,6-ジフルオロフェニル]メタノール
4-(ベンジルオキシ)-2,6-ジフルオロベンズアルデヒド(2.34 g) のTHF(20 mL)溶液にメタノール(5 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(535 mg)を加えて、1時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下、溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(2.28 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.71 (1H, t, J = 6.4 Hz), 4.70 (2H, d, J = 6.4 Hz), 5.03 (2H, s), 6.43-6.67 (2H, m), 7.29-7.48 (5H, m). B) [4- (Benzyloxy) -2,6-difluorophenyl] methanol Add methanol (5 mL) to a solution of 4- (benzyloxy) -2,6-difluorobenzaldehyde (2.34 g) in THF (20 mL). Further, sodium tetrahydroborate (535 mg) was added and stirred for 1 hour. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (2.28 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.71 (1H, t, J = 6.4 Hz), 4.70 (2H, d, J = 6.4 Hz), 5.03 (2H, s), 6.43-6.67 (2H, m) , 7.29-7.48 (5H, m).
4-(ベンジルオキシ)-2,6-ジフルオロベンズアルデヒド(2.34 g) のTHF(20 mL)溶液にメタノール(5 mL)を加え、さらにテトラヒドロホウ酸ナトリウム(535 mg)を加えて、1時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥後、減圧下、溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(2.28 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.71 (1H, t, J = 6.4 Hz), 4.70 (2H, d, J = 6.4 Hz), 5.03 (2H, s), 6.43-6.67 (2H, m), 7.29-7.48 (5H, m). B) [4- (Benzyloxy) -2,6-difluorophenyl] methanol Add methanol (5 mL) to a solution of 4- (benzyloxy) -2,6-difluorobenzaldehyde (2.34 g) in THF (20 mL). Further, sodium tetrahydroborate (535 mg) was added and stirred for 1 hour. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (2.28 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.71 (1H, t, J = 6.4 Hz), 4.70 (2H, d, J = 6.4 Hz), 5.03 (2H, s), 6.43-6.67 (2H, m) , 7.29-7.48 (5H, m).
C) N-{(1S)-2-[(trans-4-{[4-(ベンジルオキシ)-2,6-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(ベンジルオキシ)-2,6-ジフルオロフェニル]メタノール(2.28 g)のジエチルエーテル(30 mL)溶液に、三臭化リン(1.117 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下溶媒を留去した。残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(1.32 g)、硫酸水素テトラブチルアンモニウム(209 mg)、50%水酸化ナトリウム水溶液(15 mL)、およびトルエン(30 mL)の混合物を100℃で11時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(1.38 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.4 Hz), 1.22-1.46 (4H, m), 1.88-2.10 (7H, m), 3.16-3.32 (1H, m), 3.40 (3H, qd, J = 9.3, 4.0 Hz), 4.05-4.24 (1H, m), 4.50 (2H, s), 5.02 (2H, s), 5.66 (1H, d, J = 6.4 Hz), 6.41-6.60 (2H, m), 7.30-7.48 (5H, m). C) N-{(1S) -2-[(trans-4-{[4- (benzyloxy) -2,6-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- ( To a solution of (benzyloxy) -2,6-difluorophenyl] methanol (2.28 g) in diethyl ether (30 mL) was added phosphorus tribromide (1.117 mL), and the mixture was stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), and the solvent was distilled off under reduced pressure. Residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (1.32 g), tetrabutylammonium hydrogen sulfate (209 mg), 50% aqueous sodium hydroxide solution A mixture of (15 mL) and toluene (30 mL) was stirred at 100 ° C. for 11 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (1.38 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.4 Hz), 1.22-1.46 (4H, m), 1.88-2.10 (7H, m), 3.16-3.32 (1H, m), 3.40 (3H, qd, J = 9.3, 4.0 Hz), 4.05-4.24 (1H, m), 4.50 (2H, s), 5.02 (2H, s), 5.66 (1H, d, J = 6.4 Hz), 6.41 -6.60 (2H, m), 7.30-7.48 (5H, m).
[4-(ベンジルオキシ)-2,6-ジフルオロフェニル]メタノール(2.28 g)のジエチルエーテル(30 mL)溶液に、三臭化リン(1.117 mL)を加えて室温で30分間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。有機層をシリカゲルカラムクロマトグラフィー(酢酸エチル)に通じ、減圧下溶媒を留去した。残渣とN-{(1S)-2-[(trans-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(1.32 g)、硫酸水素テトラブチルアンモニウム(209 mg)、50%水酸化ナトリウム水溶液(15 mL)、およびトルエン(30 mL)の混合物を100℃で11時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(1.38 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.4 Hz), 1.22-1.46 (4H, m), 1.88-2.10 (7H, m), 3.16-3.32 (1H, m), 3.40 (3H, qd, J = 9.3, 4.0 Hz), 4.05-4.24 (1H, m), 4.50 (2H, s), 5.02 (2H, s), 5.66 (1H, d, J = 6.4 Hz), 6.41-6.60 (2H, m), 7.30-7.48 (5H, m). C) N-{(1S) -2-[(trans-4-{[4- (benzyloxy) -2,6-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- ( To a solution of (benzyloxy) -2,6-difluorophenyl] methanol (2.28 g) in diethyl ether (30 mL) was added phosphorus tribromide (1.117 mL), and the mixture was stirred at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The organic layer was passed through silica gel column chromatography (ethyl acetate), and the solvent was distilled off under reduced pressure. Residue and N-{(1S) -2-[(trans-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide (1.32 g), tetrabutylammonium hydrogen sulfate (209 mg), 50% aqueous sodium hydroxide solution A mixture of (15 mL) and toluene (30 mL) was stirred at 100 ° C. for 11 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (1.38 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.4 Hz), 1.22-1.46 (4H, m), 1.88-2.10 (7H, m), 3.16-3.32 (1H, m), 3.40 (3H, qd, J = 9.3, 4.0 Hz), 4.05-4.24 (1H, m), 4.50 (2H, s), 5.02 (2H, s), 5.66 (1H, d, J = 6.4 Hz), 6.41 -6.60 (2H, m), 7.30-7.48 (5H, m).
D) N-[(1S)-2-({trans-4-[(2,6-ジフルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
水素雰囲気下、N-{(1S)-2-[(trans-4-{[4-(ベンジルオキシ)-2,6-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(1.38 g)、10%パラジウム-炭素(50%含水、820 mg)および酢酸エチル(20 mL)の混合物を室温で終夜撹拌した。濾過により触媒を除去した後、得られた濾液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(1.04 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.45 (4H, m), 1.89-2.10 (7H, m), 3.19-3.33 (1H, m), 3.41 (3H, qd, J = 9.5, 4.0 Hz), 4.05-4.19 (1H, m), 4.50 (2H, s), 5.70 (1H, d, J = 6.0 Hz), 6.32-6.43 (2H, m), 6.52 (1H, brs). D) N-[(1S) -2-({trans-4-[(2,6-difluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide under hydrogen atmosphere, N- { (1S) -2-[(trans-4-{[4- (benzyloxy) -2,6-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (1.38 g), 10% palladium- A mixture of carbon (50% water content, 820 mg) and ethyl acetate (20 mL) was stirred at room temperature overnight. After removing the catalyst by filtration, the obtained filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (1.04 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.45 (4H, m), 1.89-2.10 (7H, m), 3.19-3.33 (1H, m), 3.41 (3H, qd, J = 9.5, 4.0 Hz), 4.05-4.19 (1H, m), 4.50 (2H, s), 5.70 (1H, d, J = 6.0 Hz), 6.32-6.43 (2H, m) , 6.52 (1H, brs).
水素雰囲気下、N-{(1S)-2-[(trans-4-{[4-(ベンジルオキシ)-2,6-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(1.38 g)、10%パラジウム-炭素(50%含水、820 mg)および酢酸エチル(20 mL)の混合物を室温で終夜撹拌した。濾過により触媒を除去した後、得られた濾液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(1.04 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.45 (4H, m), 1.89-2.10 (7H, m), 3.19-3.33 (1H, m), 3.41 (3H, qd, J = 9.5, 4.0 Hz), 4.05-4.19 (1H, m), 4.50 (2H, s), 5.70 (1H, d, J = 6.0 Hz), 6.32-6.43 (2H, m), 6.52 (1H, brs). D) N-[(1S) -2-({trans-4-[(2,6-difluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide under hydrogen atmosphere, N- { (1S) -2-[(trans-4-{[4- (benzyloxy) -2,6-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (1.38 g), 10% palladium- A mixture of carbon (50% water content, 820 mg) and ethyl acetate (20 mL) was stirred at room temperature overnight. After removing the catalyst by filtration, the obtained filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (1.04 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.21-1.45 (4H, m), 1.89-2.10 (7H, m), 3.19-3.33 (1H, m), 3.41 (3H, qd, J = 9.5, 4.0 Hz), 4.05-4.19 (1H, m), 4.50 (2H, s), 5.70 (1H, d, J = 6.0 Hz), 6.32-6.43 (2H, m) , 6.52 (1H, brs).
E) N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
N-[(1S)-2-({trans-4-[(2,6-ジフルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(810 mg)のDMF(8 mL)溶液に炭酸カリウム(940 mg)と2-(ブロモメチル)-1,1-ジフルオロシクロプロパン(1.163 g)を加えて、60℃で1時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(1.10 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.18 (3H, s), 1.21-1.44 (5H, m), 1.59-1.67 (1H, m), 1.90-2.13 (8H, m), 3.18-3.47 (4H, m), 3.90-4.22 (3H, m), 4.51 (2H, s), 5.66 (1H, d, J = 6.4 Hz), 6.39-6.50 (2H, m). E) N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide N-[(1S) -2-({trans-4-[(2,6-difluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (810 mg) To a DMF (8 mL) solution were added potassium carbonate (940 mg) and 2- (bromomethyl) -1,1-difluorocyclopropane (1.163 g), and the mixture was stirred with heating at 60 ° C. for 1 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, the solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (1.10 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.18 (3H, s), 1.21-1.44 (5H, m), 1.59-1.67 (1H, m), 1.90-2.13 (8H, m), 3.18-3.47 (4H , m), 3.90-4.22 (3H, m), 4.51 (2H, s), 5.66 (1H, d, J = 6.4 Hz), 6.39-6.50 (2H, m).
N-[(1S)-2-({trans-4-[(2,6-ジフルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(810 mg)のDMF(8 mL)溶液に炭酸カリウム(940 mg)と2-(ブロモメチル)-1,1-ジフルオロシクロプロパン(1.163 g)を加えて、60℃で1時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を白色固体(1.10 g)として得た。
1H NMR (300 MHz, CDCl3) δ 1.18 (3H, s), 1.21-1.44 (5H, m), 1.59-1.67 (1H, m), 1.90-2.13 (8H, m), 3.18-3.47 (4H, m), 3.90-4.22 (3H, m), 4.51 (2H, s), 5.66 (1H, d, J = 6.4 Hz), 6.39-6.50 (2H, m). E) N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1- Methylethyl] acetamide N-[(1S) -2-({trans-4-[(2,6-difluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (810 mg) To a DMF (8 mL) solution were added potassium carbonate (940 mg) and 2- (bromomethyl) -1,1-difluorocyclopropane (1.163 g), and the mixture was stirred with heating at 60 ° C. for 1 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, the solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a white solid (1.10 g).
1 H NMR (300 MHz, CDCl 3 ) δ 1.18 (3H, s), 1.21-1.44 (5H, m), 1.59-1.67 (1H, m), 1.90-2.13 (8H, m), 3.18-3.47 (4H , m), 3.90-4.22 (3H, m), 4.51 (2H, s), 5.66 (1H, d, J = 6.4 Hz), 6.39-6.50 (2H, m).
実施例45
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(127 mg)をHPLC(カラム:CHIRALPAK AS(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=90:10)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(29.3 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.24-1.44 (5H, m), 1.58-1.68 (1H, m), 1.93-2.15 (8H, m), 3.21-3.32 (1H, m), 3.34-3.48 (3H, m), 3.91-4.19 (3H, m), 4.51 (2H, s), 5.57-5.73 (1H, m), 6.39-6.50 (2H, m).
保持時間 32.9 分
光学純度 96.2 %ee. Example 45
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Optically active form of acetamide N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy } -1-Methylethyl] acetamide diastereomer mixture (127 mg) on HPLC (column: CHIRALPAK AS (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 90: 10) The compound having a smaller retention time was recrystallized from hexane / ethyl acetate to give the title compound (29.3 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.24-1.44 (5H, m), 1.58-1.68 (1H, m), 1.93-2.15 (8H, m), 3.21-3.32 (1H, m), 3.34-3.48 (3H, m), 3.91-4.19 (3H, m), 4.51 (2H, s), 5.57-5.73 (1H, m), 6.39-6.50 (2H, m ).
Retention time 32.9 minutes Optical purity 96.2% ee.
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(127 mg)をHPLC(カラム:CHIRALPAK AS(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=90:10)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(29.3 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.24-1.44 (5H, m), 1.58-1.68 (1H, m), 1.93-2.15 (8H, m), 3.21-3.32 (1H, m), 3.34-3.48 (3H, m), 3.91-4.19 (3H, m), 4.51 (2H, s), 5.57-5.73 (1H, m), 6.39-6.50 (2H, m).
保持時間 32.9 分
光学純度 96.2 %ee. Example 45
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Optically active form of acetamide N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy } -1-Methylethyl] acetamide diastereomer mixture (127 mg) on HPLC (column: CHIRALPAK AS (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 90: 10) The compound having a smaller retention time was recrystallized from hexane / ethyl acetate to give the title compound (29.3 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.24-1.44 (5H, m), 1.58-1.68 (1H, m), 1.93-2.15 (8H, m), 3.21-3.32 (1H, m), 3.34-3.48 (3H, m), 3.91-4.19 (3H, m), 4.51 (2H, s), 5.57-5.73 (1H, m), 6.39-6.50 (2H, m ).
Retention time 32.9 minutes Optical purity 96.2% ee.
実施例45a
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体 Example 45a
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Optically active form of acetamide
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体 Example 45a
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Optically active form of acetamide
N-[(1S)-2-({trans-4-[(2,6-ジフルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(1.00 g)のDMF(15 mL)溶液に炭酸カリウム(580 mg)と(2,2-ジフルオロシクロプロピル)メチル 4-ニトロベンゼンスルホナートの光学活性体 (保持時間の小さい光学活性体、1.23 g)を加えて、70℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製した後、得られた固体を再結晶(ヘキサン/酢酸エチル)し、標題化合物(973 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.23-1.45 (5H, m), 1.58-1.68 (1H, m), 1.93-2.10 (8H, m), 3.23-3.32 (1H, m), 3.35-3.47 (3H, m),3.93-4.20 (3H, m), 4.51 (2H, s), 5.65 (1H, brs), 6.39-6.49 (2H, m).
Anal. Calcd for C22H29NO4F4:C,59.05;H,6.53;N,3.13. Found:C,59.15;H,6.76;N,3.02.
mp. 71.3-71.6℃ N-[(1S) -2-({trans-4-[(2,6-difluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (1.00 g) in DMF (15 mL ) Add potassium carbonate (580 mg) and (2,2-difluorocyclopropyl) methyl 4-nitrobenzenesulfonate optically active substance (optically active substance with a short retention time, 1.23 g) to the solution and add it at 70 ° C for 2 hours. Stir with heating. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound (973 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.23-1.45 (5H, m), 1.58-1.68 (1H, m), 1.93-2.10 (8H, m), 3.23-3.32 (1H, m), 3.35-3.47 (3H, m), 3.93-4.20 (3H, m), 4.51 (2H, s), 5.65 (1H, brs), 6.39-6.49 (2H, m).
Anal.Calcd for C 22 H 29 NO 4 F 4 : C, 59.05; H, 6.53; N, 3.13. Found: C, 59.15; H, 6.76; N, 3.02.
mp. 71.3-71.6 ℃
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.23-1.45 (5H, m), 1.58-1.68 (1H, m), 1.93-2.10 (8H, m), 3.23-3.32 (1H, m), 3.35-3.47 (3H, m),3.93-4.20 (3H, m), 4.51 (2H, s), 5.65 (1H, brs), 6.39-6.49 (2H, m).
Anal. Calcd for C22H29NO4F4:C,59.05;H,6.53;N,3.13. Found:C,59.15;H,6.76;N,3.02.
mp. 71.3-71.6℃ N-[(1S) -2-({trans-4-[(2,6-difluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (1.00 g) in DMF (15 mL ) Add potassium carbonate (580 mg) and (2,2-difluorocyclopropyl) methyl 4-nitrobenzenesulfonate optically active substance (optically active substance with a short retention time, 1.23 g) to the solution and add it at 70 ° C for 2 hours. Stir with heating. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the obtained solid was recrystallized (hexane / ethyl acetate) to give the title compound (973 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.23-1.45 (5H, m), 1.58-1.68 (1H, m), 1.93-2.10 (8H, m), 3.23-3.32 (1H, m), 3.35-3.47 (3H, m), 3.93-4.20 (3H, m), 4.51 (2H, s), 5.65 (1H, brs), 6.39-6.49 (2H, m).
Anal.Calcd for C 22 H 29 NO 4 F 4 : C, 59.05; H, 6.53; N, 3.13. Found: C, 59.15; H, 6.76; N, 3.02.
mp. 71.3-71.6 ℃
実施例46
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(127 mg)をHPLC(カラム:CHIRALPAK AS(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=90:10)にて分取し、保持時間がより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(27.8 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.24-1.44 (5H, m), 1.58-1.69 (1H, m), 1.93-2.16 (8H, m), 3.28 (1H, brs), 3.40 (3H, qd, J = 9.2, 4.0 Hz), 3.90-4.04 (2H, m), 4.11 (1H, dt, J = 7.3, 3.7 Hz), 4.51 (2H, s), 5.64 (1H, brs), 6.32-6.56 (2H, m).
保持時間 37.0 分
光学純度 97.0 %ee. Example 46
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Optically active form of acetamide N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy } -1-Methylethyl] acetamide diastereomer mixture (127 mg) on HPLC (column: CHIRALPAK AS (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 90: 10) The compound with a longer retention time was recrystallized from hexane / ethyl acetate to give the title compound (27.8 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.24-1.44 (5H, m), 1.58-1.69 (1H, m), 1.93-2.16 (8H, m), 3.28 (1H, brs), 3.40 (3H, qd, J = 9.2, 4.0 Hz), 3.90-4.04 (2H, m), 4.11 (1H, dt, J = 7.3, 3.7 Hz), 4.51 (2H, s) , 5.64 (1H, brs), 6.32-6.56 (2H, m).
Retention time 37.0 minutes Optical purity 97.0% ee.
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドの光学活性体
N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミドのジアステレオ混合物(127 mg)をHPLC(カラム:CHIRALPAK AS(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=90:10)にて分取し、保持時間がより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(27.8 mg)を白色結晶として得た。
1H NMR (300 MHz, CDCl3) δ 1.17 (3H, d, J = 6.8 Hz), 1.24-1.44 (5H, m), 1.58-1.69 (1H, m), 1.93-2.16 (8H, m), 3.28 (1H, brs), 3.40 (3H, qd, J = 9.2, 4.0 Hz), 3.90-4.04 (2H, m), 4.11 (1H, dt, J = 7.3, 3.7 Hz), 4.51 (2H, s), 5.64 (1H, brs), 6.32-6.56 (2H, m).
保持時間 37.0 分
光学純度 97.0 %ee. Example 46
N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Optically active form of acetamide N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy } -1-Methylethyl] acetamide diastereomer mixture (127 mg) on HPLC (column: CHIRALPAK AS (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 90: 10) The compound with a longer retention time was recrystallized from hexane / ethyl acetate to give the title compound (27.8 mg) as white crystals.
1 H NMR (300 MHz, CDCl 3 ) δ 1.17 (3H, d, J = 6.8 Hz), 1.24-1.44 (5H, m), 1.58-1.69 (1H, m), 1.93-2.16 (8H, m), 3.28 (1H, brs), 3.40 (3H, qd, J = 9.2, 4.0 Hz), 3.90-4.04 (2H, m), 4.11 (1H, dt, J = 7.3, 3.7 Hz), 4.51 (2H, s) , 5.64 (1H, brs), 6.32-6.56 (2H, m).
Retention time 37.0 minutes Optical purity 97.0% ee.
実施例47
N-{(1S)-2-[(trans-4-{[5-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2-フルオロ-5-ヒドロキシ安息香酸およびブロモメチルシクロプロパンを用いて、実施例15の工程A、実施例15の工程B、実施例36の工程D、実施例7の工程Bと同様の方法により標題化合物を得た。 Example 47
N-{(1S) -2-[(trans-4-{[5- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2-fluoro-5-hydroxy The title compound was obtained in the same manner as in Step 15 of Example 15, Step B of Example 15, Step D of Example 36 and Step B of Example 7 using benzoic acid and bromomethylcyclopropane.
N-{(1S)-2-[(trans-4-{[5-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2-フルオロ-5-ヒドロキシ安息香酸およびブロモメチルシクロプロパンを用いて、実施例15の工程A、実施例15の工程B、実施例36の工程D、実施例7の工程Bと同様の方法により標題化合物を得た。 Example 47
N-{(1S) -2-[(trans-4-{[5- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2-fluoro-5-hydroxy The title compound was obtained in the same manner as in Step 15 of Example 15, Step B of Example 15, Step D of Example 36 and Step B of Example 7 using benzoic acid and bromomethylcyclopropane.
実施例48
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素の光学活性体 Example 48
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea optically active form
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素の光学活性体 Example 48
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea optically active form
A) 1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素
1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オールおよびメチルイソシアナートを用いて実施例4の工程Dと同様の方法により標題化合物を得た。
MS (ESI+): [M+H]+ 409.3. A) 1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea 1-[(trans -4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-ol and methyl isocyanate by a method similar to that of Example 4, Step D. Obtained.
MS (ESI +): [M + H] + 409.3.
1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オールおよびメチルイソシアナートを用いて実施例4の工程Dと同様の方法により標題化合物を得た。
MS (ESI+): [M+H]+ 409.3. A) 1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea 1-[(trans -4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-ol and methyl isocyanate by a method similar to that of Example 4, Step D. Obtained.
MS (ESI +): [M + H] + 409.3.
B) 1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素の光学活性体
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素のラセミ体(198 mg)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=80:20)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(70.2 mg)を得た。
保持時間 5.6 分
光学純度 >99.9% ee B) Optically active substance of 1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea 1 -{2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea racemate (198 mg) Fractionated by HPLC (column: CHIRALPAK AD (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 80: 20) Recrystallization gave the title compound (70.2 mg).
Retention time 5.6 minutes Optical purity> 99.9% ee
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素のラセミ体(198 mg)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール=80:20)にて分取し、保持時間がより小さい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(70.2 mg)を得た。
保持時間 5.6 分
光学純度 >99.9% ee B) Optically active substance of 1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea 1 -{2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea racemate (198 mg) Fractionated by HPLC (column: CHIRALPAK AD (trade name), 50 mmID × 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 80: 20) Recrystallization gave the title compound (70.2 mg).
Retention time 5.6 minutes Optical purity> 99.9% ee
実施例49
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素の光学活性体
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素のラセミ体(198 mg)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール = 80:20)にて分取し、保持時間のより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(72.2 mg)を得た。
保持時間 6.7 分
光学純度 >99.9% ee Example 49
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea 1- { A racemate (198 mg) of 2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea was analyzed by HPLC ( Column: CHIRALPAK AD (trade name), 50 mmID x 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 80:20), fractionated and recrystallized from compound with longer retention time in hexane / ethyl acetate To give the title compound (72.2 mg).
Retention time 6.7 minutes Optical purity> 99.9% ee
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素の光学活性体
1-{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}-3-メチル尿素のラセミ体(198 mg)をHPLC(カラム:CHIRALPAK AD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/エタノール = 80:20)にて分取し、保持時間のより大きい化合物をヘキサン/酢酸エチルで再結晶して標題化合物(72.2 mg)を得た。
保持時間 6.7 分
光学純度 >99.9% ee Example 49
1- {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea 1- { A racemate (198 mg) of 2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} -3-methylurea was analyzed by HPLC ( Column: CHIRALPAK AD (trade name), 50 mmID x 500 mmL, Daicel Chemical Industries, mobile phase: hexane / ethanol = 80:20), fractionated and recrystallized from compound with longer retention time in hexane / ethyl acetate To give the title compound (72.2 mg).
Retention time 6.7 minutes Optical purity> 99.9% ee
実施例50
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 50
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (cyclopropylmethoxy) phenyl] methanol The title compound was obtained in the same manner as in Steps A and F of Example 2.
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 50
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (cyclopropylmethoxy) phenyl] methanol The title compound was obtained in the same manner as in Steps A and F of Example 2.
実施例51
N-{(1S)-2-[(trans-4-{[6-(シクロプロピルメトキシ)-4-フルオロピリジン-3-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[6-(シクロプロピルメトキシ)-4-フルオロピリジン-3-イル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 51
N-{(1S) -2-[(trans-4-{[6- (cyclopropylmethoxy) -4-fluoropyridin-3-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide [6- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -4-fluoropyridin-3-yl] methanol.
N-{(1S)-2-[(trans-4-{[6-(シクロプロピルメトキシ)-4-フルオロピリジン-3-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[6-(シクロプロピルメトキシ)-4-フルオロピリジン-3-イル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 51
N-{(1S) -2-[(trans-4-{[6- (cyclopropylmethoxy) -4-fluoropyridin-3-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide [6- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -4-fluoropyridin-3-yl] methanol.
実施例52
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2,6-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)-2,6-ジフルオロフェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 52
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,6-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (cyclo The title compound was obtained in the same manner as in Steps A and F of Example 2 using (propylmethoxy) -2,6-difluorophenyl] methanol.
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2,6-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)-2,6-ジフルオロフェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 52
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,6-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (cyclo The title compound was obtained in the same manner as in Steps A and F of Example 2 using (propylmethoxy) -2,6-difluorophenyl] methanol.
実施例53
N-{(1S)-2-[(trans-4-{[6-(シクロプロピルメトキシ)-4-メチルピリジン-3-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[6-(シクロプロピルメトキシ)-4-メチルピリジン-3-イル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 53
N-{(1S) -2-[(trans-4-{[6- (cyclopropylmethoxy) -4-methylpyridin-3-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide [6- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -4-methylpyridin-3-yl] methanol.
N-{(1S)-2-[(trans-4-{[6-(シクロプロピルメトキシ)-4-メチルピリジン-3-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[6-(シクロプロピルメトキシ)-4-メチルピリジン-3-イル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 53
N-{(1S) -2-[(trans-4-{[6- (cyclopropylmethoxy) -4-methylpyridin-3-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide [6- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -4-methylpyridin-3-yl] methanol.
実施例54
{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチルの光学活性体 Example 54
Optically active form of methyl {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} carbamate
{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチルの光学活性体 Example 54
Optically active form of methyl {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} carbamate
A) {2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチル
1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オールおよびクロロギ酸メチルを用いて実施例4の工程Dと同様の方法により標題化合物(50 mg)を得た。
MS (ESI+): [M+H]+ 410.3. A) Methyl {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} carbamate 1-[(trans-4- { The title compound (50 mg) was prepared in the same manner as in Step D of Example 4 using [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-ol and methyl chloroformate. Obtained.
MS (ESI +): [M + H] + 410.3.
1-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]プロパン-2-オールおよびクロロギ酸メチルを用いて実施例4の工程Dと同様の方法により標題化合物(50 mg)を得た。
MS (ESI+): [M+H]+ 410.3. A) Methyl {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} carbamate 1-[(trans-4- { The title compound (50 mg) was prepared in the same manner as in Step D of Example 4 using [4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] propan-2-ol and methyl chloroformate. Obtained.
MS (ESI +): [M + H] + 410.3.
B) {2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチルの光学活性体
{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチルのラセミ体(50 mg)をHPLC(カラム:CHIRALPAK OD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/イソプロピルアルコール = 90:10)にて分取し、保持時間の小さい標題化合物(19.7 mg)を得た。
保持時間 8.0 分
光学純度 >99.9% ee B) {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} methyl carbamate optically active substance {2-[( trans-4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} racemate of methyl carbamate (50 mg) was HPLC (column: CHIRALPAK OD (product) No.), 50 mm ID × 500 mm L, Daicel Chemical Industries, mobile phase: hexane / isopropyl alcohol = 90: 10) to give the title compound (19.7 mg) having a short retention time.
Retention time 8.0 minutes Optical purity> 99.9% ee
{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチルのラセミ体(50 mg)をHPLC(カラム:CHIRALPAK OD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/イソプロピルアルコール = 90:10)にて分取し、保持時間の小さい標題化合物(19.7 mg)を得た。
保持時間 8.0 分
光学純度 >99.9% ee B) {2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} methyl carbamate optically active substance {2-[( trans-4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} racemate of methyl carbamate (50 mg) was HPLC (column: CHIRALPAK OD (product) No.), 50 mm ID × 500 mm L, Daicel Chemical Industries, mobile phase: hexane / isopropyl alcohol = 90: 10) to give the title compound (19.7 mg) having a short retention time.
Retention time 8.0 minutes Optical purity> 99.9% ee
実施例55
{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチルの光学活性体
{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチルのラセミ体(50 mg)をHPLC(カラム:CHIRALPAK OD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/イソプロピルアルコール=90:10)にて分取し、保持時間の大きい標題化合物(19.6 mg)を得た。
保持時間 16.2 分
光学純度 >99.9% ee Example 55
{2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} methyl carbamate optically active substance {2-[(trans- Racemate of methyl 4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} carbamate (50 mg) by HPLC (column: CHIRALPAK OD (trade name) , 50 mm ID × 500 mm L, Daicel Chemical Industries, mobile phase: hexane / isopropyl alcohol = 90: 10) to give the title compound (19.6 mg) having a long retention time.
Retention time 16.2 Spectroscopy> 99.9% ee
{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチルの光学活性体
{2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}カルバミン酸メチルのラセミ体(50 mg)をHPLC(カラム:CHIRALPAK OD(商品名)、50 mmID×500 mmL、ダイセル化学工業、移動相:ヘキサン/イソプロピルアルコール=90:10)にて分取し、保持時間の大きい標題化合物(19.6 mg)を得た。
保持時間 16.2 分
光学純度 >99.9% ee Example 55
{2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} methyl carbamate optically active substance {2-[(trans- Racemate of methyl 4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} carbamate (50 mg) by HPLC (column: CHIRALPAK OD (trade name) , 50 mm ID × 500 mm L, Daicel Chemical Industries, mobile phase: hexane / isopropyl alcohol = 90: 10) to give the title compound (19.6 mg) having a long retention time.
Retention time 16.2 Spectroscopy> 99.9% ee
実施例56
N-{(1S)-2-[(trans-4-{[2-シアノ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2-(ブロモメチル)-5-(シクロプロピルメトキシ)ベンゾニトリルを用いて実施例2の工程Fと同様の方法により標題化合物を得た。 Example 56
N-{(1S) -2-[(trans-4-{[2-cyano-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2- (bromomethyl) -5 The title compound was obtained in the same manner as in Step F of Example 2 using-(cyclopropylmethoxy) benzonitrile.
N-{(1S)-2-[(trans-4-{[2-シアノ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2-(ブロモメチル)-5-(シクロプロピルメトキシ)ベンゾニトリルを用いて実施例2の工程Fと同様の方法により標題化合物を得た。 Example 56
N-{(1S) -2-[(trans-4-{[2-cyano-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2- (bromomethyl) -5 The title compound was obtained in the same manner as in Step F of Example 2 using-(cyclopropylmethoxy) benzonitrile.
実施例57
N-[(1S)-2-({trans-4-[(2-クロロ-4-エトキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
1-(ブロモメチル)-2-クロロ-4-エトキシベンゼンを用いて実施例2の工程Fと同様の方法により標題化合物を得た。 Example 57
N-[(1S) -2-({trans-4-[(2-chloro-4-ethoxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide 1- (bromomethyl) -2-chloro-4 The title compound was obtained in the same manner as in Step F of Example 2 using -ethoxybenzene.
N-[(1S)-2-({trans-4-[(2-クロロ-4-エトキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
1-(ブロモメチル)-2-クロロ-4-エトキシベンゼンを用いて実施例2の工程Fと同様の方法により標題化合物を得た。 Example 57
N-[(1S) -2-({trans-4-[(2-chloro-4-ethoxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide 1- (bromomethyl) -2-chloro-4 The title compound was obtained in the same manner as in Step F of Example 2 using -ethoxybenzene.
実施例58
N-{(1S)-2-[(trans-4-{[2-クロロ-4-(1-メチルエトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[2-クロロ-4-(1-メチルエトキシ)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 58
N-{(1S) -2-[(trans-4-{[2-chloro-4- (1-methylethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [2-chloro-4 The title compound was obtained in the same manner as in Steps A and F of Example 2 using-(1-methylethoxy) phenyl] methanol.
N-{(1S)-2-[(trans-4-{[2-クロロ-4-(1-メチルエトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[2-クロロ-4-(1-メチルエトキシ)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 58
N-{(1S) -2-[(trans-4-{[2-chloro-4- (1-methylethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [2-chloro-4 The title compound was obtained in the same manner as in Steps A and F of Example 2 using-(1-methylethoxy) phenyl] methanol.
実施例59
N-{(1S)-2-[(trans-4-{[5-(シクロプロピルメトキシ)-3-フルオロピリジン-2-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[5-(シクロプロピルメトキシ)-3-フルオロピリジン-2-イル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 59
N-{(1S) -2-[(trans-4-{[5- (cyclopropylmethoxy) -3-fluoropyridin-2-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide [5- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -3-fluoropyridin-2-yl] methanol.
N-{(1S)-2-[(trans-4-{[5-(シクロプロピルメトキシ)-3-フルオロピリジン-2-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[5-(シクロプロピルメトキシ)-3-フルオロピリジン-2-イル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 59
N-{(1S) -2-[(trans-4-{[5- (cyclopropylmethoxy) -3-fluoropyridin-2-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide [5- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -3-fluoropyridin-2-yl] methanol.
実施例60
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 60
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 60
N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide
A) tert-ブチル {(1S)-1-メチル-2-[(4-オキソシクロヘキシル)オキシ]エチル}カルバマート
三酸化硫黄-ピリジン錯体(5.24 g)およびジメチルスルホキシド(30 mL)の混合物を、tert-ブチル {(1S)-2-[(cis-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマート(3.00 g)、トリエチルアミン(7.65 mL)、およびジメチルスルホキシド(30 mL)の混合物に室温でゆっくり滴下し、混合物を室温で2時間撹拌した。反応混合物に0℃で水を加えた後、酢酸エチルで抽出した。得られた有機層を1 M塩酸、続いて飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(2.55 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.03 (3H, d, J = 6.8 Hz), 1.37 (9H, s), 1.82-1.95 (4H, m), 2.09-2.47 (4H, m), 3.18-3.30 (1H, m), 3.32-3.45 (1H, m), 3.51-3.74 (2H, m), 6.68 (1H, d, J = 8.3 Hz). A) tert-butyl {(1S) -1-methyl-2-[(4-oxocyclohexyl) oxy] ethyl} carbamate A mixture of sulfur trioxide-pyridine complex (5.24 g) and dimethyl sulfoxide (30 mL) -Butyl ((1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate (3.00 g), triethylamine (7.65 mL), and dimethyl sulfoxide (30 mL) at room temperature. It was slowly added dropwise and the mixture was stirred at room temperature for 2 hours. Water was added to the reaction mixture at 0 ° C., and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with 1 M hydrochloric acid and then with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oily compound (2.55 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.03 (3H, d, J = 6.8 Hz), 1.37 (9H, s), 1.82-1.95 (4H, m), 2.09-2.47 (4H, m), 3.18-3.30 (1H, m), 3.32-3.45 (1H, m), 3.51-3.74 (2H, m), 6.68 (1H, d, J = 8.3 Hz).
三酸化硫黄-ピリジン錯体(5.24 g)およびジメチルスルホキシド(30 mL)の混合物を、tert-ブチル {(1S)-2-[(cis-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}カルバマート(3.00 g)、トリエチルアミン(7.65 mL)、およびジメチルスルホキシド(30 mL)の混合物に室温でゆっくり滴下し、混合物を室温で2時間撹拌した。反応混合物に0℃で水を加えた後、酢酸エチルで抽出した。得られた有機層を1 M塩酸、続いて飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(2.55 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.03 (3H, d, J = 6.8 Hz), 1.37 (9H, s), 1.82-1.95 (4H, m), 2.09-2.47 (4H, m), 3.18-3.30 (1H, m), 3.32-3.45 (1H, m), 3.51-3.74 (2H, m), 6.68 (1H, d, J = 8.3 Hz). A) tert-butyl {(1S) -1-methyl-2-[(4-oxocyclohexyl) oxy] ethyl} carbamate A mixture of sulfur trioxide-pyridine complex (5.24 g) and dimethyl sulfoxide (30 mL) -Butyl ((1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl} carbamate (3.00 g), triethylamine (7.65 mL), and dimethyl sulfoxide (30 mL) at room temperature. It was slowly added dropwise and the mixture was stirred at room temperature for 2 hours. Water was added to the reaction mixture at 0 ° C., and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with 1 M hydrochloric acid and then with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oily compound (2.55 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.03 (3H, d, J = 6.8 Hz), 1.37 (9H, s), 1.82-1.95 (4H, m), 2.09-2.47 (4H, m), 3.18-3.30 (1H, m), 3.32-3.45 (1H, m), 3.51-3.74 (2H, m), 6.68 (1H, d, J = 8.3 Hz).
B) tert-ブチル {(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}カルバマート
tert-ブチル {(1S)-1-メチル-2-[(4-オキソシクロヘキシル)オキシ]エチル}カルバマート(2.55 g)のTHF(40 mL)溶液にメチルマグネシウムブロミドのTHF溶液(1.0 M、9.40 mL)を0℃でゆっくり滴下し、混合物を0℃で2時間撹拌した。反応混合物に1 M塩酸を加えた後、酢酸エチルで抽出した。得られた有機層を1 M塩酸、続いて飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(2.21 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.99 (3H, d, J = 6.4 Hz), 1.04-1.12 (3H, m), 1.13-1.81 (17H, m), 3.03-3.63 (4H, m), 4.00-4.07 (1H, m), 6.58 (1H, d, J = 7.9 Hz). B) tert-butyl {(1S) -2-[(4-hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} carbamate tert-butyl {(1S) -1-methyl-2-[(4- To a solution of (oxocyclohexyl) oxy] ethyl} carbamate (2.55 g) in THF (40 mL) was slowly added methyl magnesium bromide in THF (1.0 M, 9.40 mL) at 0 ° C., and the mixture was stirred at 0 ° C. for 2 hours. . 1 M hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with 1 M hydrochloric acid and then with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oily compound (2.21 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.99 (3H, d, J = 6.4 Hz), 1.04-1.12 (3H, m), 1.13-1.81 (17H, m), 3.03-3.63 (4H, m ), 4.00-4.07 (1H, m), 6.58 (1H, d, J = 7.9 Hz).
tert-ブチル {(1S)-1-メチル-2-[(4-オキソシクロヘキシル)オキシ]エチル}カルバマート(2.55 g)のTHF(40 mL)溶液にメチルマグネシウムブロミドのTHF溶液(1.0 M、9.40 mL)を0℃でゆっくり滴下し、混合物を0℃で2時間撹拌した。反応混合物に1 M塩酸を加えた後、酢酸エチルで抽出した。得られた有機層を1 M塩酸、続いて飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(2.21 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.99 (3H, d, J = 6.4 Hz), 1.04-1.12 (3H, m), 1.13-1.81 (17H, m), 3.03-3.63 (4H, m), 4.00-4.07 (1H, m), 6.58 (1H, d, J = 7.9 Hz). B) tert-butyl {(1S) -2-[(4-hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} carbamate tert-butyl {(1S) -1-methyl-2-[(4- To a solution of (oxocyclohexyl) oxy] ethyl} carbamate (2.55 g) in THF (40 mL) was slowly added methyl magnesium bromide in THF (1.0 M, 9.40 mL) at 0 ° C., and the mixture was stirred at 0 ° C. for 2 hours. . 1 M hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with 1 M hydrochloric acid and then with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oily compound (2.21 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.99 (3H, d, J = 6.4 Hz), 1.04-1.12 (3H, m), 1.13-1.81 (17H, m), 3.03-3.63 (4H, m ), 4.00-4.07 (1H, m), 6.58 (1H, d, J = 7.9 Hz).
C) N-{(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
tert-ブチル {(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}カルバマートを用い、実施例7の工程Bと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.01 (3H, d, J = 6.8 Hz), 1.04-1.11 (3H, m), 1.15-1.72 (8H, m), 1.77 (3H, s), 3.04-3.40 (3H, m), 3.70-4.18 (2H, m), 7.64 (1H, d, J = 7.9 Hz). C) N-{(1S) -2-[(4-hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} acetamide tert-butyl {(1S) -2-[(4-hydroxy-4-methyl The title compound was obtained in the same manner as in Step B of Example 7 using (cyclohexyl) oxy] -1-methylethyl} carbamate.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.01 (3H, d, J = 6.8 Hz), 1.04-1.11 (3H, m), 1.15-1.72 (8H, m), 1.77 (3H, s), 3.04-3.40 (3H, m), 3.70-4.18 (2H, m), 7.64 (1H, d, J = 7.9 Hz).
tert-ブチル {(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}カルバマートを用い、実施例7の工程Bと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.01 (3H, d, J = 6.8 Hz), 1.04-1.11 (3H, m), 1.15-1.72 (8H, m), 1.77 (3H, s), 3.04-3.40 (3H, m), 3.70-4.18 (2H, m), 7.64 (1H, d, J = 7.9 Hz). C) N-{(1S) -2-[(4-hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} acetamide tert-butyl {(1S) -2-[(4-hydroxy-4-methyl The title compound was obtained in the same manner as in Step B of Example 7 using (cyclohexyl) oxy] -1-methylethyl} carbamate.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.01 (3H, d, J = 6.8 Hz), 1.04-1.11 (3H, m), 1.15-1.72 (8H, m), 1.77 (3H, s), 3.04-3.40 (3H, m), 3.70-4.18 (2H, m), 7.64 (1H, d, J = 7.9 Hz).
D) N-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用い、実施例3の工程Bと同様の方法により標題化合物を得た。 D) N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide N- { The title compound was obtained in the same manner as in Step B of Example 3 using (1S) -2-[(4-hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} acetamide.
N-{(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用い、実施例3の工程Bと同様の方法により標題化合物を得た。 D) N-{(1S) -2-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide N- { The title compound was obtained in the same manner as in Step B of Example 3 using (1S) -2-[(4-hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} acetamide.
実施例61
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-メチルベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)-2-メチルフェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 61
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2-methylbenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (cyclopropylmethoxy ) -2-Methylphenyl] methanol was used to obtain the title compound in the same manner as in Steps A and F of Example 2.
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-メチルベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)-2-メチルフェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 61
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2-methylbenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (cyclopropylmethoxy ) -2-Methylphenyl] methanol was used to obtain the title compound in the same manner as in Steps A and F of Example 2.
実施例62
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-3-(トリフルオロメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)-3-(トリフルオロメトキシ)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 62
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -3- (trifluoromethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -3- (trifluoromethoxy) phenyl] methanol.
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-3-(トリフルオロメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)-3-(トリフルオロメトキシ)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 62
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -3- (trifluoromethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- The title compound was obtained in the same manner as in Steps A and F of Example 2 using (cyclopropylmethoxy) -3- (trifluoromethoxy) phenyl] methanol.
実施例63
N-{(1S)-2-[(trans-4-{[4-(ベンジルオキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(ベンジルオキシ)-2-フルオロフェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 63
N-{(1S) -2-[(trans-4-{[4- (benzyloxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (benzyloxy)- The title compound was obtained in the same manner as in Steps A and F of Example 2 using 2-fluorophenyl] methanol.
N-{(1S)-2-[(trans-4-{[4-(ベンジルオキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(ベンジルオキシ)-2-フルオロフェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 63
N-{(1S) -2-[(trans-4-{[4- (benzyloxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [4- (benzyloxy)- The title compound was obtained in the same manner as in Steps A and F of Example 2 using 2-fluorophenyl] methanol.
実施例64
N-[(1S)-3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-1-メチルプロピル]アセトアミド
[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノールを用いて実施例34の工程Dと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.29-0.40 (2H, m), 0.57-0.70 (2H, m), 0.81-0.99 (2H, m), 1.11 (3H, d, J = 6.4 Hz), 1.15-1.33 (6H, m), 1.35-1.47 (2H, m), 1.70-1.82 (2H, m), 1.95 (3H, s), 2.01-2.11 (2H, m), 3.20-3.34 (1H, m), 3.77 (2H, d, J = 6.8 Hz), 3.87-4.00 (1H, m), 4.52 (2H, s), 5.16 (1H, d, J = 8.0 Hz), 6.58 (1H, dd, J = 11.7, 2.7 Hz), 6.67 (1H, dd, J = 8.5, 2.5 Hz), 7.28 (1H, t, J = 8.5 Hz). Example 64
N-[(1S) -3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methylpropyl] acetamide [4- (cyclopropylmethoxy) -2 The title compound was obtained in the same manner as in Step D of Example 34 using [-fluorophenyl] methanol.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.29-0.40 (2H, m), 0.57-0.70 (2H, m), 0.81-0.99 (2H, m), 1.11 (3H, d, J = 6.4 Hz ), 1.15-1.33 (6H, m), 1.35-1.47 (2H, m), 1.70-1.82 (2H, m), 1.95 (3H, s), 2.01-2.11 (2H, m), 3.20-3.34 (1H , m), 3.77 (2H, d, J = 6.8 Hz), 3.87-4.00 (1H, m), 4.52 (2H, s), 5.16 (1H, d, J = 8.0 Hz), 6.58 (1H, dd, J = 11.7, 2.7 Hz), 6.67 (1H, dd, J = 8.5, 2.5 Hz), 7.28 (1H, t, J = 8.5 Hz).
N-[(1S)-3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-1-メチルプロピル]アセトアミド
[4-(シクロプロピルメトキシ)-2-フルオロフェニル]メタノールを用いて実施例34の工程Dと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.29-0.40 (2H, m), 0.57-0.70 (2H, m), 0.81-0.99 (2H, m), 1.11 (3H, d, J = 6.4 Hz), 1.15-1.33 (6H, m), 1.35-1.47 (2H, m), 1.70-1.82 (2H, m), 1.95 (3H, s), 2.01-2.11 (2H, m), 3.20-3.34 (1H, m), 3.77 (2H, d, J = 6.8 Hz), 3.87-4.00 (1H, m), 4.52 (2H, s), 5.16 (1H, d, J = 8.0 Hz), 6.58 (1H, dd, J = 11.7, 2.7 Hz), 6.67 (1H, dd, J = 8.5, 2.5 Hz), 7.28 (1H, t, J = 8.5 Hz). Example 64
N-[(1S) -3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methylpropyl] acetamide [4- (cyclopropylmethoxy) -2 The title compound was obtained in the same manner as in Step D of Example 34 using [-fluorophenyl] methanol.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.29-0.40 (2H, m), 0.57-0.70 (2H, m), 0.81-0.99 (2H, m), 1.11 (3H, d, J = 6.4 Hz ), 1.15-1.33 (6H, m), 1.35-1.47 (2H, m), 1.70-1.82 (2H, m), 1.95 (3H, s), 2.01-2.11 (2H, m), 3.20-3.34 (1H , m), 3.77 (2H, d, J = 6.8 Hz), 3.87-4.00 (1H, m), 4.52 (2H, s), 5.16 (1H, d, J = 8.0 Hz), 6.58 (1H, dd, J = 11.7, 2.7 Hz), 6.67 (1H, dd, J = 8.5, 2.5 Hz), 7.28 (1H, t, J = 8.5 Hz).
実施例65
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2,3-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 65
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,3-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2,3-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 65
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,3-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) 4-(シクロプロピルメトキシ)-2,3-ジフルオロベンズアルデヒド
2,3-ジフルオロ-4-ヒドロキシベンズアルデヒドおよび(ブロモメチル)シクロプロパンを用い、実施例37の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.25-0.44 (2H, m), 0.53-0.73 (2H, m), 1.18-1.37 (1H, m), 4.07 (2H, d, J = 7.2 Hz), 7.01-7.35 (1H, m), 7.47-7.83 (1H, m), 10.04 (1H, s). A) The title compound was obtained in the same manner as in Step A of Example 37, using 4- (cyclopropylmethoxy) -2,3-difluorobenzaldehyde 2,3-difluoro-4-hydroxybenzaldehyde and (bromomethyl) cyclopropane. It was.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.25-0.44 (2H, m), 0.53-0.73 (2H, m), 1.18-1.37 (1H, m), 4.07 (2H, d, J = 7.2 Hz ), 7.01-7.35 (1H, m), 7.47-7.83 (1H, m), 10.04 (1H, s).
2,3-ジフルオロ-4-ヒドロキシベンズアルデヒドおよび(ブロモメチル)シクロプロパンを用い、実施例37の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.25-0.44 (2H, m), 0.53-0.73 (2H, m), 1.18-1.37 (1H, m), 4.07 (2H, d, J = 7.2 Hz), 7.01-7.35 (1H, m), 7.47-7.83 (1H, m), 10.04 (1H, s). A) The title compound was obtained in the same manner as in Step A of Example 37, using 4- (cyclopropylmethoxy) -2,3-difluorobenzaldehyde 2,3-difluoro-4-hydroxybenzaldehyde and (bromomethyl) cyclopropane. It was.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.25-0.44 (2H, m), 0.53-0.73 (2H, m), 1.18-1.37 (1H, m), 4.07 (2H, d, J = 7.2 Hz ), 7.01-7.35 (1H, m), 7.47-7.83 (1H, m), 10.04 (1H, s).
B) [4-(シクロプロピルメトキシ)-2,3-ジフルオロフェニル]メタノール
4-(シクロプロピルメトキシ)-2,3-ジフルオロベンズアルデヒドを用い、実施例37の工程Bと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.26-0.42 (2H, m), 0.49-0.64 (2H, m), 1.12-1.35 (1H, m), 3.91 (2H, d, J = 7.2 Hz), 4.47 (2H, d, J = 5.8 Hz), 5.23 (1H, t, J = 5.8 Hz), 6.89-7.04 (1H, m), 7.05-7.22 (1H, m). B) [4- (Cyclopropylmethoxy) -2,3-difluorophenyl] methanol Using 4- (cyclopropylmethoxy) -2,3-difluorobenzaldehyde, the title compound was prepared in the same manner as in Step B of Example 37. Obtained.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.26-0.42 (2H, m), 0.49-0.64 (2H, m), 1.12-1.35 (1H, m), 3.91 (2H, d, J = 7.2 Hz ), 4.47 (2H, d, J = 5.8 Hz), 5.23 (1H, t, J = 5.8 Hz), 6.89-7.04 (1H, m), 7.05-7.22 (1H, m).
4-(シクロプロピルメトキシ)-2,3-ジフルオロベンズアルデヒドを用い、実施例37の工程Bと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.26-0.42 (2H, m), 0.49-0.64 (2H, m), 1.12-1.35 (1H, m), 3.91 (2H, d, J = 7.2 Hz), 4.47 (2H, d, J = 5.8 Hz), 5.23 (1H, t, J = 5.8 Hz), 6.89-7.04 (1H, m), 7.05-7.22 (1H, m). B) [4- (Cyclopropylmethoxy) -2,3-difluorophenyl] methanol Using 4- (cyclopropylmethoxy) -2,3-difluorobenzaldehyde, the title compound was prepared in the same manner as in Step B of Example 37. Obtained.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.26-0.42 (2H, m), 0.49-0.64 (2H, m), 1.12-1.35 (1H, m), 3.91 (2H, d, J = 7.2 Hz ), 4.47 (2H, d, J = 5.8 Hz), 5.23 (1H, t, J = 5.8 Hz), 6.89-7.04 (1H, m), 7.05-7.22 (1H, m).
C) 1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2,3-ジフルオロベンゼン
[4-(シクロプロピルメトキシ)-2,3-ジフルオロフェニル]メタノールを用い、実施例37の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.23-0.46 (2H, m), 0.52-0.74 (2H, m), 1.09-1.39 (1H, m), 3.95 (2H, d, J = 7.2 Hz), 4.71 (2H, s), 6.88-7.05 (1H, m), 7.20-7.36 (1H, m). C) 1- (Bromomethyl) -4- (cyclopropylmethoxy) -2,3-difluorobenzene [4- (Cyclopropylmethoxy) -2,3-difluorophenyl] methanol as in Step 37 of Example 37 To give the title compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.23-0.46 (2H, m), 0.52-0.74 (2H, m), 1.09-1.39 (1H, m), 3.95 (2H, d, J = 7.2 Hz ), 4.71 (2H, s), 6.88-7.05 (1H, m), 7.20-7.36 (1H, m).
[4-(シクロプロピルメトキシ)-2,3-ジフルオロフェニル]メタノールを用い、実施例37の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.23-0.46 (2H, m), 0.52-0.74 (2H, m), 1.09-1.39 (1H, m), 3.95 (2H, d, J = 7.2 Hz), 4.71 (2H, s), 6.88-7.05 (1H, m), 7.20-7.36 (1H, m). C) 1- (Bromomethyl) -4- (cyclopropylmethoxy) -2,3-difluorobenzene [4- (Cyclopropylmethoxy) -2,3-difluorophenyl] methanol as in Step 37 of Example 37 To give the title compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.23-0.46 (2H, m), 0.52-0.74 (2H, m), 1.09-1.39 (1H, m), 3.95 (2H, d, J = 7.2 Hz ), 4.71 (2H, s), 6.88-7.05 (1H, m), 7.20-7.36 (1H, m).
D) N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2,3-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2,3-ジフルオロベンゼンを用い、実施例3の工程Bと同様の方法により標題化合物を得た。 D) N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,3-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 1- ( The title compound was obtained in the same manner as in Step B of Example 3 using bromomethyl) -4- (cyclopropylmethoxy) -2,3-difluorobenzene.
1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2,3-ジフルオロベンゼンを用い、実施例3の工程Bと同様の方法により標題化合物を得た。 D) N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,3-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 1- ( The title compound was obtained in the same manner as in Step B of Example 3 using bromomethyl) -4- (cyclopropylmethoxy) -2,3-difluorobenzene.
実施例66
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2,5-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 66
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,5-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2,5-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 66
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,5-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) 4-(シクロプロピルメトキシ)-2,5-ジフルオロベンズアルデヒド
2,5-ジフルオロ-4-ヒドロキシベンズアルデヒドおよび(ブロモメチル)シクロプロパンを用い、実施例37の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.24-0.45 (2H, m), 0.53-0.75 (2H, m), 1.15-1.47 (1H, m), 4.04 (2H, d, J = 7.2 Hz), 7.28 (1H, dd, J = 12.3, 6.6 Hz), 7.60 (1H, dd, J = 11.1, 6.6 Hz), 10.05 (1H, d, J = 2.6 Hz). A) 4- (Cyclopropylmethoxy) -2,5-difluorobenzaldehyde The title compound was obtained in the same manner as in Step A of Example 37, using 2,5-difluoro-4-hydroxybenzaldehyde and (bromomethyl) cyclopropane. It was.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.24-0.45 (2H, m), 0.53-0.75 (2H, m), 1.15-1.47 (1H, m), 4.04 (2H, d, J = 7.2 Hz ), 7.28 (1H, dd, J = 12.3, 6.6 Hz), 7.60 (1H, dd, J = 11.1, 6.6 Hz), 10.05 (1H, d, J = 2.6 Hz).
2,5-ジフルオロ-4-ヒドロキシベンズアルデヒドおよび(ブロモメチル)シクロプロパンを用い、実施例37の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.24-0.45 (2H, m), 0.53-0.75 (2H, m), 1.15-1.47 (1H, m), 4.04 (2H, d, J = 7.2 Hz), 7.28 (1H, dd, J = 12.3, 6.6 Hz), 7.60 (1H, dd, J = 11.1, 6.6 Hz), 10.05 (1H, d, J = 2.6 Hz). A) 4- (Cyclopropylmethoxy) -2,5-difluorobenzaldehyde The title compound was obtained in the same manner as in Step A of Example 37, using 2,5-difluoro-4-hydroxybenzaldehyde and (bromomethyl) cyclopropane. It was.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.24-0.45 (2H, m), 0.53-0.75 (2H, m), 1.15-1.47 (1H, m), 4.04 (2H, d, J = 7.2 Hz ), 7.28 (1H, dd, J = 12.3, 6.6 Hz), 7.60 (1H, dd, J = 11.1, 6.6 Hz), 10.05 (1H, d, J = 2.6 Hz).
B) [4-(シクロプロピルメトキシ)-2,5-ジフルオロフェニル]メタノール
4-(シクロプロピルメトキシ)-2,5-ジフルオロベンズアルデヒドを用い、実施例37の工程Bと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.17-0.44 (2H, m), 0.50-0.67 (2H, m), 1.10-1.32 (1H, m), 3.88 (2H, d, J = 7.2 Hz), 4.43 (2H, d, J = 5.7 Hz), 5.23 (1H, t, J = 5.7 Hz), 7.03 (1H, dd, J = 11.5, 7.0 Hz), 7.22 (1H, dd, J = 11.7, 7.2 Hz). B) [4- (Cyclopropylmethoxy) -2,5-difluorophenyl] methanol Using 4- (cyclopropylmethoxy) -2,5-difluorobenzaldehyde, the title compound was prepared in the same manner as in Step B of Example 37. Obtained.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.17-0.44 (2H, m), 0.50-0.67 (2H, m), 1.10-1.32 (1H, m), 3.88 (2H, d, J = 7.2 Hz ), 4.43 (2H, d, J = 5.7 Hz), 5.23 (1H, t, J = 5.7 Hz), 7.03 (1H, dd, J = 11.5, 7.0 Hz), 7.22 (1H, dd, J = 11.7, 7.2 Hz).
4-(シクロプロピルメトキシ)-2,5-ジフルオロベンズアルデヒドを用い、実施例37の工程Bと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.17-0.44 (2H, m), 0.50-0.67 (2H, m), 1.10-1.32 (1H, m), 3.88 (2H, d, J = 7.2 Hz), 4.43 (2H, d, J = 5.7 Hz), 5.23 (1H, t, J = 5.7 Hz), 7.03 (1H, dd, J = 11.5, 7.0 Hz), 7.22 (1H, dd, J = 11.7, 7.2 Hz). B) [4- (Cyclopropylmethoxy) -2,5-difluorophenyl] methanol Using 4- (cyclopropylmethoxy) -2,5-difluorobenzaldehyde, the title compound was prepared in the same manner as in Step B of Example 37. Obtained.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.17-0.44 (2H, m), 0.50-0.67 (2H, m), 1.10-1.32 (1H, m), 3.88 (2H, d, J = 7.2 Hz ), 4.43 (2H, d, J = 5.7 Hz), 5.23 (1H, t, J = 5.7 Hz), 7.03 (1H, dd, J = 11.5, 7.0 Hz), 7.22 (1H, dd, J = 11.7, 7.2 Hz).
C) 1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2,5-ジフルオロベンゼン
[4-(シクロプロピルメトキシ)-2,5-ジフルオロフェニル]メタノールを用い、実施例37の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.25-0.41 (2H, m), 0.50-0.70 (2H, m), 1.11-1.33 (1H, m), 3.92 (2H, d, J = 7.2 Hz), 4.63 (2H, s), 7.12 (1H, dd, J = 11.7, 7.2 Hz), 7.44 (1H, dd, J = 11.5, 7.3 Hz). C) 1- (Bromomethyl) -4- (cyclopropylmethoxy) -2,5-difluorobenzene [4- (Cyclopropylmethoxy) -2,5-difluorophenyl] methanol as in Step 37 of Example 37 To give the title compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.25-0.41 (2H, m), 0.50-0.70 (2H, m), 1.11-1.33 (1H, m), 3.92 (2H, d, J = 7.2 Hz ), 4.63 (2H, s), 7.12 (1H, dd, J = 11.7, 7.2 Hz), 7.44 (1H, dd, J = 11.5, 7.3 Hz).
[4-(シクロプロピルメトキシ)-2,5-ジフルオロフェニル]メタノールを用い、実施例37の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.25-0.41 (2H, m), 0.50-0.70 (2H, m), 1.11-1.33 (1H, m), 3.92 (2H, d, J = 7.2 Hz), 4.63 (2H, s), 7.12 (1H, dd, J = 11.7, 7.2 Hz), 7.44 (1H, dd, J = 11.5, 7.3 Hz). C) 1- (Bromomethyl) -4- (cyclopropylmethoxy) -2,5-difluorobenzene [4- (Cyclopropylmethoxy) -2,5-difluorophenyl] methanol as in Step 37 of Example 37 To give the title compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.25-0.41 (2H, m), 0.50-0.70 (2H, m), 1.11-1.33 (1H, m), 3.92 (2H, d, J = 7.2 Hz ), 4.63 (2H, s), 7.12 (1H, dd, J = 11.7, 7.2 Hz), 7.44 (1H, dd, J = 11.5, 7.3 Hz).
D) N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2,5-ジフルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2,5-ジフルオロベンゼンを用い、実施例3の工程Bと同様の方法により標題化合物を得た。 D) N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,5-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 1- ( The title compound was obtained in the same manner as in Step B of Example 3 using bromomethyl) -4- (cyclopropylmethoxy) -2,5-difluorobenzene.
1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2,5-ジフルオロベンゼンを用い、実施例3の工程Bと同様の方法により標題化合物を得た。 D) N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2,5-difluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 1- ( The title compound was obtained in the same manner as in Step B of Example 3 using bromomethyl) -4- (cyclopropylmethoxy) -2,5-difluorobenzene.
実施例67
N-{(1S)-2-[(trans-4-{[3-(シクロプロピルメトキシ)イソオキサゾール-5-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 67
N-{(1S) -2-[(trans-4-{[3- (cyclopropylmethoxy) isoxazol-5-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[3-(シクロプロピルメトキシ)イソオキサゾール-5-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 67
N-{(1S) -2-[(trans-4-{[3- (cyclopropylmethoxy) isoxazol-5-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) メチル 3-(シクロプロピルメトキシ)イソオキサゾール-5-カルボキシラート
メチル 3-ヒドロキシイソオキサゾール-5-カルボキシラートおよび(ブロモメチル)シクロプロパンを用い、実施例37の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.23-0.42 (2H, m), 0.50-0.73 (2H, m), 1.20-1.39 (1H, m), 3.88 (3H, s), 4.08 (2H, d, J = 7.2 Hz), 7.08 (1H, s). A) Methyl 3- (cyclopropylmethoxy) isoxazole-5-carboxylate The title compound was prepared in the same manner as in Step A of Example 37 using methyl 3-hydroxyisoxazole-5-carboxylate and (bromomethyl) cyclopropane. Got.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.23-0.42 (2H, m), 0.50-0.73 (2H, m), 1.20-1.39 (1H, m), 3.88 (3H, s), 4.08 (2H , d, J = 7.2 Hz), 7.08 (1H, s).
メチル 3-ヒドロキシイソオキサゾール-5-カルボキシラートおよび(ブロモメチル)シクロプロパンを用い、実施例37の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.23-0.42 (2H, m), 0.50-0.73 (2H, m), 1.20-1.39 (1H, m), 3.88 (3H, s), 4.08 (2H, d, J = 7.2 Hz), 7.08 (1H, s). A) Methyl 3- (cyclopropylmethoxy) isoxazole-5-carboxylate The title compound was prepared in the same manner as in Step A of Example 37 using methyl 3-hydroxyisoxazole-5-carboxylate and (bromomethyl) cyclopropane. Got.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.23-0.42 (2H, m), 0.50-0.73 (2H, m), 1.20-1.39 (1H, m), 3.88 (3H, s), 4.08 (2H , d, J = 7.2 Hz), 7.08 (1H, s).
B) [3-(シクロプロピルメトキシ)イソオキサゾール-5-イル]メタノール
テトラヒドロホウ酸ナトリウム(3.30 g)、塩化カルシウム(4.84 g)、およびメタノール(30 mL)の混合物を0℃で1時間撹拌した。反応混合物にメチル 3-(シクロプロピルメトキシ)イソオキサゾール-5-カルボキシラート(2.15 g)を加え、得られた混合物を0℃で1時間、続いて室温で終夜撹拌した。反応混合物に1 M塩酸を加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(1.75 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.18-0.40 (2H, m), 0.46-0.69 (2H, m), 1.19-1.39 (1H, m), 3.98 (2H, d, J = 7.6 Hz), 4.43 (2H, d, J = 5.3 Hz), 5.60 (1H, t, J = 6.0 Hz), 6.09 (1H, s). B) A mixture of [3- (cyclopropylmethoxy) isoxazol-5-yl] methanol sodium tetrahydroborate (3.30 g), calcium chloride (4.84 g), and methanol (30 mL) was stirred at 0 ° C. for 1 hour. . To the reaction mixture was added methyl 3- (cyclopropylmethoxy) isoxazole-5-carboxylate (2.15 g) and the resulting mixture was stirred at 0 ° C. for 1 hour, then at room temperature overnight. 1 M hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oil (1.75 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.18-0.40 (2H, m), 0.46-0.69 (2H, m), 1.19-1.39 (1H, m), 3.98 (2H, d, J = 7.6 Hz ), 4.43 (2H, d, J = 5.3 Hz), 5.60 (1H, t, J = 6.0 Hz), 6.09 (1H, s).
テトラヒドロホウ酸ナトリウム(3.30 g)、塩化カルシウム(4.84 g)、およびメタノール(30 mL)の混合物を0℃で1時間撹拌した。反応混合物にメチル 3-(シクロプロピルメトキシ)イソオキサゾール-5-カルボキシラート(2.15 g)を加え、得られた混合物を0℃で1時間、続いて室温で終夜撹拌した。反応混合物に1 M塩酸を加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物を無色油状化合物(1.75 g)として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.18-0.40 (2H, m), 0.46-0.69 (2H, m), 1.19-1.39 (1H, m), 3.98 (2H, d, J = 7.6 Hz), 4.43 (2H, d, J = 5.3 Hz), 5.60 (1H, t, J = 6.0 Hz), 6.09 (1H, s). B) A mixture of [3- (cyclopropylmethoxy) isoxazol-5-yl] methanol sodium tetrahydroborate (3.30 g), calcium chloride (4.84 g), and methanol (30 mL) was stirred at 0 ° C. for 1 hour. . To the reaction mixture was added methyl 3- (cyclopropylmethoxy) isoxazole-5-carboxylate (2.15 g) and the resulting mixture was stirred at 0 ° C. for 1 hour, then at room temperature overnight. 1 M hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound as a colorless oil (1.75 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.18-0.40 (2H, m), 0.46-0.69 (2H, m), 1.19-1.39 (1H, m), 3.98 (2H, d, J = 7.6 Hz ), 4.43 (2H, d, J = 5.3 Hz), 5.60 (1H, t, J = 6.0 Hz), 6.09 (1H, s).
C) 5-(ブロモメチル)-3-(シクロプロピルメトキシ)イソオキサゾール
[3-(シクロプロピルメトキシ)イソオキサゾール-5-イル]メタノールを用い、実施例37の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.21-0.42 (2H, m), 0.47-0.66 (2H, m), 1.02-1.40 (1H, m), 4.00 (2H, d, J = 7.2 Hz), 4.69 (2H, s), 6.35 (1H, s). C) 5- (Bromomethyl) -3- (cyclopropylmethoxy) isoxazole [3- (cyclopropylmethoxy) isoxazol-5-yl] methanol was used to prepare the title compound in the same manner as in Step C of Example 37. Obtained.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.21-0.42 (2H, m), 0.47-0.66 (2H, m), 1.02-1.40 (1H, m), 4.00 (2H, d, J = 7.2 Hz ), 4.69 (2H, s), 6.35 (1H, s).
[3-(シクロプロピルメトキシ)イソオキサゾール-5-イル]メタノールを用い、実施例37の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.21-0.42 (2H, m), 0.47-0.66 (2H, m), 1.02-1.40 (1H, m), 4.00 (2H, d, J = 7.2 Hz), 4.69 (2H, s), 6.35 (1H, s). C) 5- (Bromomethyl) -3- (cyclopropylmethoxy) isoxazole [3- (cyclopropylmethoxy) isoxazol-5-yl] methanol was used to prepare the title compound in the same manner as in Step C of Example 37. Obtained.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.21-0.42 (2H, m), 0.47-0.66 (2H, m), 1.02-1.40 (1H, m), 4.00 (2H, d, J = 7.2 Hz ), 4.69 (2H, s), 6.35 (1H, s).
D) N-{(1S)-2-[(trans-4-{[3-(シクロプロピルメトキシ)イソオキサゾール-5-イル]メトキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
5-(ブロモメチル)-3-(シクロプロピルメトキシ)イソオキサゾールを用い、実施例3の工程Bと同様の方法により標題化合物を得た。 D) N-{(1S) -2-[(trans-4-{[3- (cyclopropylmethoxy) isoxazol-5-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide 5- (bromomethyl ) -3- (Cyclopropylmethoxy) isoxazole was used to give the title compound in the same manner as in Step B of Example 3.
5-(ブロモメチル)-3-(シクロプロピルメトキシ)イソオキサゾールを用い、実施例3の工程Bと同様の方法により標題化合物を得た。 D) N-{(1S) -2-[(trans-4-{[3- (cyclopropylmethoxy) isoxazol-5-yl] methoxy} cyclohexyl) oxy] -1-methylethyl} acetamide 5- (bromomethyl ) -3- (Cyclopropylmethoxy) isoxazole was used to give the title compound in the same manner as in Step B of Example 3.
実施例68
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(2-メチルプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
1-ブロモ-2-メチルプロパンを用いて実施例35の工程Dと同様の方法により標題化合物を得た。 Example 68
N-{(1S) -2-[(trans-4-{[2-fluoro-4- (2-methylpropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 1-bromo-2- The title compound was obtained in the same manner as in Step D of Example 35 using methylpropane.
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(2-メチルプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
1-ブロモ-2-メチルプロパンを用いて実施例35の工程Dと同様の方法により標題化合物を得た。 Example 68
N-{(1S) -2-[(trans-4-{[2-fluoro-4- (2-methylpropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 1-bromo-2- The title compound was obtained in the same manner as in Step D of Example 35 using methylpropane.
実施例69
N-{(1S)-2-[(trans-4-{[4-(2-シクロプロピルエトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 69
N-{(1S) -2-[(trans-4-{[4- (2-cyclopropylethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[4-(2-シクロプロピルエトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 69
N-{(1S) -2-[(trans-4-{[4- (2-cyclopropylethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) 2-シクロプロピルエチル メタンスルホナート
2-シクロプロピルエタノールを用い、実施例38の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.07-0.17 (2H, m), 0.47-0.57 (2H, m), 0.68-0.86 (1H, m), 1.65 (2H, q, J = 6.8 Hz), 2.90-3.16 (3H, m), 4.29 (2H, t, J = 6.6 Hz). A) 2-Cyclopropylethyl methanesulfonate The title compound was obtained in the same manner as in Step A of Example 38 using 2-cyclopropylethanol.
1 H NMR (300 MHz, CDCl 3 ) δ 0.07-0.17 (2H, m), 0.47-0.57 (2H, m), 0.68-0.86 (1H, m), 1.65 (2H, q, J = 6.8 Hz), 2.90-3.16 (3H, m), 4.29 (2H, t, J = 6.6 Hz).
2-シクロプロピルエタノールを用い、実施例38の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.07-0.17 (2H, m), 0.47-0.57 (2H, m), 0.68-0.86 (1H, m), 1.65 (2H, q, J = 6.8 Hz), 2.90-3.16 (3H, m), 4.29 (2H, t, J = 6.6 Hz). A) 2-Cyclopropylethyl methanesulfonate The title compound was obtained in the same manner as in Step A of Example 38 using 2-cyclopropylethanol.
1 H NMR (300 MHz, CDCl 3 ) δ 0.07-0.17 (2H, m), 0.47-0.57 (2H, m), 0.68-0.86 (1H, m), 1.65 (2H, q, J = 6.8 Hz), 2.90-3.16 (3H, m), 4.29 (2H, t, J = 6.6 Hz).
B) N-{(1S)-2-[(trans-4-{[4-(2-シクロプロピルエトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2-シクロプロピルエチル メタンスルホナートを用い、実施例35の工程Dと同様の方法により標題化合物を得た。 B) N-{(1S) -2-[(trans-4-{[4- (2-cyclopropylethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2-cyclo The title compound was obtained in the same manner as in Step D of Example 35 using propylethyl methanesulfonate.
2-シクロプロピルエチル メタンスルホナートを用い、実施例35の工程Dと同様の方法により標題化合物を得た。 B) N-{(1S) -2-[(trans-4-{[4- (2-cyclopropylethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2-cyclo The title compound was obtained in the same manner as in Step D of Example 35 using propylethyl methanesulfonate.
実施例70
N-{(1S)-2-[(cis-4-{[3-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2-フルオロ-3-ヒドロキシベンズアルデヒドおよびブロモメチルシクロプロパンを用いて、実施例34の工程Cと同様の操作を行った後に、N-{(1S)-2-[(cis-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用いて実施例36の工程Dと同様の方法により標題化合物を得た。 Example 70
N-{(1S) -2-[(cis-4-{[3- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2-fluoro-3-hydroxy After performing the same operation as in Step C of Example 34 using benzaldehyde and bromomethylcyclopropane, N-{(1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl } The title compound was obtained in the same manner as in Step D of Example 36 using acetamide.
N-{(1S)-2-[(cis-4-{[3-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2-フルオロ-3-ヒドロキシベンズアルデヒドおよびブロモメチルシクロプロパンを用いて、実施例34の工程Cと同様の操作を行った後に、N-{(1S)-2-[(cis-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用いて実施例36の工程Dと同様の方法により標題化合物を得た。 Example 70
N-{(1S) -2-[(cis-4-{[3- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2-fluoro-3-hydroxy After performing the same operation as in Step C of Example 34 using benzaldehyde and bromomethylcyclopropane, N-{(1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl } The title compound was obtained in the same manner as in Step D of Example 36 using acetamide.
実施例71
N-{(1S)-2-[(cis-4-{[5-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
(5-(シクロプロピルメトキシ)-2-フルオロフェニル)メタノールおよびN-{(1S)-2-[(cis-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用いて、実施例36の工程Dと同様の方法により標題化合物を得た。 Example 71
N-{(1S) -2-[(cis-4-{[5- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (5- (cyclopropylmethoxy ) -2-Fluorophenyl) methanol and N-{(1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide as in step D of Example 36 Gave the title compound.
N-{(1S)-2-[(cis-4-{[5-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
(5-(シクロプロピルメトキシ)-2-フルオロフェニル)メタノールおよびN-{(1S)-2-[(cis-4-ヒドロキシシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用いて、実施例36の工程Dと同様の方法により標題化合物を得た。 Example 71
N-{(1S) -2-[(cis-4-{[5- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (5- (cyclopropylmethoxy ) -2-Fluorophenyl) methanol and N-{(1S) -2-[(cis-4-hydroxycyclohexyl) oxy] -1-methylethyl} acetamide as in step D of Example 36 Gave the title compound.
実施例72
N-{(1S)-2-[(trans-4-{[2-ブロモ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 72
N-{(1S) -2-[(trans-4-{[2-bromo-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[2-ブロモ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 72
N-{(1S) -2-[(trans-4-{[2-bromo-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) 2-ブロモ-4-(シクロプロピルメトキシ)-1-メチルベンゼン
3-ブロモ-4-メチルフェノールを用いて実施例7の工程Eと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.22-0.42 (2H, m), 0.42-0.67 (2H, m), 1.07-1.30 (1H, m), 2.25 (3H, s), 3.79 (2H, d, J = 6.8 Hz), 6.86 (1H, dd, J = 8.3, 2.6 Hz), 7.13 (1H, d, J = 2.6 Hz), 7.23 (1H, d, J = 8.3 Hz). A) 2-Bromo-4- (cyclopropylmethoxy) -1-methylbenzene The title compound was obtained in the same manner as in Step E of Example 7 using 3-bromo-4-methylphenol.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.22-0.42 (2H, m), 0.42-0.67 (2H, m), 1.07-1.30 (1H, m), 2.25 (3H, s), 3.79 (2H , d, J = 6.8 Hz), 6.86 (1H, dd, J = 8.3, 2.6 Hz), 7.13 (1H, d, J = 2.6 Hz), 7.23 (1H, d, J = 8.3 Hz).
3-ブロモ-4-メチルフェノールを用いて実施例7の工程Eと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.22-0.42 (2H, m), 0.42-0.67 (2H, m), 1.07-1.30 (1H, m), 2.25 (3H, s), 3.79 (2H, d, J = 6.8 Hz), 6.86 (1H, dd, J = 8.3, 2.6 Hz), 7.13 (1H, d, J = 2.6 Hz), 7.23 (1H, d, J = 8.3 Hz). A) 2-Bromo-4- (cyclopropylmethoxy) -1-methylbenzene The title compound was obtained in the same manner as in Step E of Example 7 using 3-bromo-4-methylphenol.
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.22-0.42 (2H, m), 0.42-0.67 (2H, m), 1.07-1.30 (1H, m), 2.25 (3H, s), 3.79 (2H , d, J = 6.8 Hz), 6.86 (1H, dd, J = 8.3, 2.6 Hz), 7.13 (1H, d, J = 2.6 Hz), 7.23 (1H, d, J = 8.3 Hz).
B) 2-ブロモ-1-(ブロモメチル)-4-(シクロプロピルメトキシ)ベンゼン
2-ブロモ-4-(シクロプロピルメトキシ)-1-メチルベンゼン(1 g)の酢酸エチル(21 mL)溶液に2,2'-アゾビス(2-メチルプロピオニトリル) (31 mg)とN-ブロモスクシンイミド(0.89 g)を加え、80℃で2日間撹拌した。反応混合物に水を加え酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し、標題化合物(400 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.26-0.39 (2H, m), 0.51-0.64 (2H, m), 1.11-1.29 (1H, m), 3.85 (2H, d, J = 7.2 Hz), 4.72 (2H, s), 6.96 (1H, dd, J = 8.7, 2.7 Hz), 7.20 (1H, d, J = 2.3 Hz), 7.53 (1H, d, J = 8.3 Hz). B) 2-Bromo-1- (bromomethyl) -4- (cyclopropylmethoxy) benzene 2-bromo-4- (cyclopropylmethoxy) -1-methylbenzene (1 g) in a solution of ethyl acetate (21 mL) , 2′-azobis (2-methylpropionitrile) (31 mg) and N-bromosuccinimide (0.89 g) were added, and the mixture was stirred at 80 ° C. for 2 days. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (400 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.26-0.39 (2H, m), 0.51-0.64 (2H, m), 1.11-1.29 (1H, m), 3.85 (2H, d, J = 7.2 Hz ), 4.72 (2H, s), 6.96 (1H, dd, J = 8.7, 2.7 Hz), 7.20 (1H, d, J = 2.3 Hz), 7.53 (1H, d, J = 8.3 Hz).
2-ブロモ-4-(シクロプロピルメトキシ)-1-メチルベンゼン(1 g)の酢酸エチル(21 mL)溶液に2,2'-アゾビス(2-メチルプロピオニトリル) (31 mg)とN-ブロモスクシンイミド(0.89 g)を加え、80℃で2日間撹拌した。反応混合物に水を加え酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し、標題化合物(400 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.26-0.39 (2H, m), 0.51-0.64 (2H, m), 1.11-1.29 (1H, m), 3.85 (2H, d, J = 7.2 Hz), 4.72 (2H, s), 6.96 (1H, dd, J = 8.7, 2.7 Hz), 7.20 (1H, d, J = 2.3 Hz), 7.53 (1H, d, J = 8.3 Hz). B) 2-Bromo-1- (bromomethyl) -4- (cyclopropylmethoxy) benzene 2-bromo-4- (cyclopropylmethoxy) -1-methylbenzene (1 g) in a solution of ethyl acetate (21 mL) , 2′-azobis (2-methylpropionitrile) (31 mg) and N-bromosuccinimide (0.89 g) were added, and the mixture was stirred at 80 ° C. for 2 days. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (400 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 0.26-0.39 (2H, m), 0.51-0.64 (2H, m), 1.11-1.29 (1H, m), 3.85 (2H, d, J = 7.2 Hz ), 4.72 (2H, s), 6.96 (1H, dd, J = 8.7, 2.7 Hz), 7.20 (1H, d, J = 2.3 Hz), 7.53 (1H, d, J = 8.3 Hz).
C) N-{(1S)-2-[(trans-4-{[2-ブロモ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2-ブロモ-1-(ブロモメチル)-4-(シクロプロピルメトキシ)ベンゼンを用いて実施例2の工程Fと同様の方法により標題化合物を得た。 C) N-{(1S) -2-[(trans-4-{[2-bromo-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2-bromo-1 The title compound was obtained in the same manner as in Step F of Example 2 using-(bromomethyl) -4- (cyclopropylmethoxy) benzene.
2-ブロモ-1-(ブロモメチル)-4-(シクロプロピルメトキシ)ベンゼンを用いて実施例2の工程Fと同様の方法により標題化合物を得た。 C) N-{(1S) -2-[(trans-4-{[2-bromo-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide 2-bromo-1 The title compound was obtained in the same manner as in Step F of Example 2 using-(bromomethyl) -4- (cyclopropylmethoxy) benzene.
実施例73
N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(1-メチルシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 73
N-[(1S) -2-{[trans-4-({2-fluoro-4-[(1-methylcyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(1-メチルシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 73
N-[(1S) -2-{[trans-4-({2-fluoro-4-[(1-methylcyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
A) N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(2-メチルプロパ-2-エン-1-イル)オキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
3-ブロモ-2-メチルプロパ-1-エンを用い、実施例35の工程Dと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ1.01 (3H, d, J = 6.8 Hz), 1.13-1.35 (4H, m), 1.71-1.81 (6H, m), 1.81-1.97 (4H, m), 3.10-3.29 (2H, m), 3.32-3.43 (2H, m), 3.82 (1H, dt, J = 13.4, 6.5 Hz), 4.37-4.52 (4H, m), 4.91-5.08 (2H, m), 6.73-6.85 (2H, m), 7.30 (1H, t, J = 8.9 Hz), 7.63 (1H, d, J = 7.9 Hz). A) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2-methylprop-2-en-1-yl) oxy] benzyl} oxy) cyclohexyl] oxy}- 1-methylethyl] acetamide The title compound was obtained in the same manner as in Step D of Example 35 using 3-bromo-2-methylprop-1-ene.
1 H NMR (300 MHz, DMSO-d 6 ) δ1.01 (3H, d, J = 6.8 Hz), 1.13-1.35 (4H, m), 1.71-1.81 (6H, m), 1.81-1.97 (4H, m), 3.10-3.29 (2H, m), 3.32-3.43 (2H, m), 3.82 (1H, dt, J = 13.4, 6.5 Hz), 4.37-4.52 (4H, m), 4.91-5.08 (2H, m), 6.73-6.85 (2H, m), 7.30 (1H, t, J = 8.9 Hz), 7.63 (1H, d, J = 7.9 Hz).
3-ブロモ-2-メチルプロパ-1-エンを用い、実施例35の工程Dと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ1.01 (3H, d, J = 6.8 Hz), 1.13-1.35 (4H, m), 1.71-1.81 (6H, m), 1.81-1.97 (4H, m), 3.10-3.29 (2H, m), 3.32-3.43 (2H, m), 3.82 (1H, dt, J = 13.4, 6.5 Hz), 4.37-4.52 (4H, m), 4.91-5.08 (2H, m), 6.73-6.85 (2H, m), 7.30 (1H, t, J = 8.9 Hz), 7.63 (1H, d, J = 7.9 Hz). A) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2-methylprop-2-en-1-yl) oxy] benzyl} oxy) cyclohexyl] oxy}- 1-methylethyl] acetamide The title compound was obtained in the same manner as in Step D of Example 35 using 3-bromo-2-methylprop-1-ene.
1 H NMR (300 MHz, DMSO-d 6 ) δ1.01 (3H, d, J = 6.8 Hz), 1.13-1.35 (4H, m), 1.71-1.81 (6H, m), 1.81-1.97 (4H, m), 3.10-3.29 (2H, m), 3.32-3.43 (2H, m), 3.82 (1H, dt, J = 13.4, 6.5 Hz), 4.37-4.52 (4H, m), 4.91-5.08 (2H, m), 6.73-6.85 (2H, m), 7.30 (1H, t, J = 8.9 Hz), 7.63 (1H, d, J = 7.9 Hz).
B) N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(1-メチルシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
アルゴン雰囲気下、ジエチル亜鉛のヘキサン溶液 (1.0 M、0.77 mL)にトルエン (0.5 mL)を加えた溶液にジヨードメタン(0.10 mL) を室温で滴下し、得られた混合物を白濁するまで室温で撹拌した。反応混合物にN-[(1S)-2-{[trans-4-({2-フルオロ-4-[(2-メチルプロパ-2-エン-1-イル)オキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド (90.0 mg) を加え、得られた混合物を室温で48時間撹拌した。反応混合物に1 M塩酸を加え酸性にした後、酢酸エチルで抽出した。得られた有機層を飽和炭酸水素ナトリウム水溶液で洗浄した後に飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー (ヘキサン/酢酸エチル) で精製した後、HPLC(カラム:L-Column 2 ODS、移動相:アセトニトリル/水=50/50)でさらに精製し、標題化合物 (10.0 mg) を得た。 B) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(1-methylcyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide Under an argon atmosphere, diiodomethane (0.10 mL) was added dropwise at room temperature to a solution of diethylzinc in hexane (1.0 M, 0.77 mL) and toluene (0.5 mL), and the resulting mixture was stirred at room temperature until cloudy. . N-[(1S) -2-{[trans-4-({2-fluoro-4-[(2-methylprop-2-en-1-yl) oxy] benzyl} oxy) cyclohexyl] oxy} was added to the reaction mixture. -1-Methylethyl] acetamide (90.0 mg) was added and the resulting mixture was stirred at room temperature for 48 hours. The reaction mixture was acidified with 1 M hydrochloric acid, and extracted with ethyl acetate. The obtained organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, washed with saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate), and further purified by HPLC (column: L-Column 2 ODS, mobile phase: acetonitrile / water = 50/50). Purification gave the title compound (10.0 mg).
アルゴン雰囲気下、ジエチル亜鉛のヘキサン溶液 (1.0 M、0.77 mL)にトルエン (0.5 mL)を加えた溶液にジヨードメタン(0.10 mL) を室温で滴下し、得られた混合物を白濁するまで室温で撹拌した。反応混合物にN-[(1S)-2-{[trans-4-({2-フルオロ-4-[(2-メチルプロパ-2-エン-1-イル)オキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド (90.0 mg) を加え、得られた混合物を室温で48時間撹拌した。反応混合物に1 M塩酸を加え酸性にした後、酢酸エチルで抽出した。得られた有機層を飽和炭酸水素ナトリウム水溶液で洗浄した後に飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた残渣をシリカゲルクロマトグラフィー (ヘキサン/酢酸エチル) で精製した後、HPLC(カラム:L-Column 2 ODS、移動相:アセトニトリル/水=50/50)でさらに精製し、標題化合物 (10.0 mg) を得た。 B) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(1-methylcyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide Under an argon atmosphere, diiodomethane (0.10 mL) was added dropwise at room temperature to a solution of diethylzinc in hexane (1.0 M, 0.77 mL) and toluene (0.5 mL), and the resulting mixture was stirred at room temperature until cloudy. . N-[(1S) -2-{[trans-4-({2-fluoro-4-[(2-methylprop-2-en-1-yl) oxy] benzyl} oxy) cyclohexyl] oxy} was added to the reaction mixture. -1-Methylethyl] acetamide (90.0 mg) was added and the resulting mixture was stirred at room temperature for 48 hours. The reaction mixture was acidified with 1 M hydrochloric acid, and extracted with ethyl acetate. The obtained organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution, washed with saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel chromatography (hexane / ethyl acetate), and further purified by HPLC (column: L-Column 2 ODS, mobile phase: acetonitrile / water = 50/50). Purification gave the title compound (10.0 mg).
実施例74
N-{(1S)-2-[(trans-4-{[3-(ベンジルオキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[3-(ベンジルオキシ)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 74
N-{(1S) -2-[(trans-4-{[3- (benzyloxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [3- (benzyloxy) phenyl] methanol In the same manner as in Steps A and F of Example 2, the title compound was obtained.
N-{(1S)-2-[(trans-4-{[3-(ベンジルオキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[3-(ベンジルオキシ)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 Example 74
N-{(1S) -2-[(trans-4-{[3- (benzyloxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [3- (benzyloxy) phenyl] methanol In the same manner as in Steps A and F of Example 2, the title compound was obtained.
実施例75
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用い、実施例3の工程Bと同様の方法により得たN-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドをHPLC(カラム:CHIRALPAK AD、50 mmID×500 mmL、移動相:ヘキサン/エタノール=500/500)にて分取し、保持時間がより小さい標題化合物を得た。
分析保持時間 6.91 分 Example 75
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide N- { (1S) -2-[(4-Hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} acetamide was used to obtain N-{(1S) -2 obtained by the same method as in Step B of Example 3. -[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide by HPLC (column: CHIRALPAK AD, 50 mmID × 500 mmL , Mobile phase: hexane / ethanol = 500/500) to give the title compound having a shorter retention time.
Analysis retention time 6.91 minutes
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用い、実施例3の工程Bと同様の方法により得たN-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドをHPLC(カラム:CHIRALPAK AD、50 mmID×500 mmL、移動相:ヘキサン/エタノール=500/500)にて分取し、保持時間がより小さい標題化合物を得た。
分析保持時間 6.91 分 Example 75
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide N- { (1S) -2-[(4-Hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} acetamide was used to obtain N-{(1S) -2 obtained by the same method as in Step B of Example 3. -[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide by HPLC (column: CHIRALPAK AD, 50 mmID × 500 mmL , Mobile phase: hexane / ethanol = 500/500) to give the title compound having a shorter retention time.
Analysis retention time 6.91 minutes
実施例76
N-{(1S)-2-[(cis-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用い、実施例3の工程Bと同様の方法により得たN-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドをHPLC(カラム:CHIRALPAK AD、50 mmID×500 mmL、移動相:ヘキサン/エタノール=500/500)にて分取し、保持時間がより大きい標題化合物を得た。
分析保持時間 8.81 分 Example 76
N-{(1S) -2-[(cis-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide N- { (1S) -2-[(4-Hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} acetamide was used to obtain N-{(1S) -2 obtained by the same method as in Step B of Example 3. -[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide by HPLC (column: CHIRALPAK AD, 50 mmID × 500 mmL , Mobile phase: hexane / ethanol = 500/500) to give the title compound having a longer retention time.
Analysis retention time 8.81 minutes
N-{(1S)-2-[(cis-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(4-ヒドロキシ-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用い、実施例3の工程Bと同様の方法により得たN-{(1S)-2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-4-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミドをHPLC(カラム:CHIRALPAK AD、50 mmID×500 mmL、移動相:ヘキサン/エタノール=500/500)にて分取し、保持時間がより大きい標題化合物を得た。
分析保持時間 8.81 分 Example 76
N-{(1S) -2-[(cis-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide N- { (1S) -2-[(4-Hydroxy-4-methylcyclohexyl) oxy] -1-methylethyl} acetamide was used to obtain N-{(1S) -2 obtained by the same method as in Step B of Example 3. -[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -4-methylcyclohexyl) oxy] -1-methylethyl} acetamide by HPLC (column: CHIRALPAK AD, 50 mmID × 500 mmL , Mobile phase: hexane / ethanol = 500/500) to give the title compound having a longer retention time.
Analysis retention time 8.81 minutes
実施例77
N-{(1S)-2-[(trans-4-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 77
N-{(1S) -2-[(trans-4-{[3- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 77
N-{(1S) -2-[(trans-4-{[3- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) N-[(1S)-2-({trans-4-[(3-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
N-{(1S)-2-[(trans-4-{[3-(ベンジルオキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用いて実施例1の工程Cと同様の方法により標題化合物を得た。
MS (ESI+): [M+Na]+ 348.9 A) N-[(1S) -2-({trans-4-[(3-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide N-{(1S) -2-[(trans- The title compound was obtained in the same manner as in Step C of Example 1 using 4-{[3- (benzyloxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide.
MS (ESI +): [M + Na] + 348.9
N-{(1S)-2-[(trans-4-{[3-(ベンジルオキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミドを用いて実施例1の工程Cと同様の方法により標題化合物を得た。
MS (ESI+): [M+Na]+ 348.9 A) N-[(1S) -2-({trans-4-[(3-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide N-{(1S) -2-[(trans- The title compound was obtained in the same manner as in Step C of Example 1 using 4-{[3- (benzyloxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide.
MS (ESI +): [M + Na] + 348.9
B) N-{(1S)-2-[(trans-4-{[3-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-[(1S)-2-({trans-4-[(3-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(110 mg)のDMF(5 mL)溶液に炭酸カリウム(70.9 mg)と(ブロモメチル)シクロプロパン(0.100 mL)を加えて、70℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られる残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(71.5 mg)を白色固体として得た。 B) N-{(1S) -2-[(trans-4-{[3- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide N-[(1S) -2- ({trans-4-[(3-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (110 mg) in DMF (5 mL) solution in potassium carbonate (70.9 mg) and (bromomethyl) cyclopropane (0.100 mL) was added, and the mixture was stirred with heating at 70 ° C. for 2 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (71.5 mg) as a white solid.
N-[(1S)-2-({trans-4-[(3-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(110 mg)のDMF(5 mL)溶液に炭酸カリウム(70.9 mg)と(ブロモメチル)シクロプロパン(0.100 mL)を加えて、70℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られる残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(71.5 mg)を白色固体として得た。 B) N-{(1S) -2-[(trans-4-{[3- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide N-[(1S) -2- ({trans-4-[(3-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (110 mg) in DMF (5 mL) solution in potassium carbonate (70.9 mg) and (bromomethyl) cyclopropane (0.100 mL) was added, and the mixture was stirred with heating at 70 ° C. for 2 hr. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (71.5 mg) as a white solid.
実施例78
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-(トリフルオロメチル)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 78
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2- (trifluoromethyl) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-(トリフルオロメチル)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 78
N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2- (trifluoromethyl) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
A) [4-(シクロプロピルメトキシ)-2-(トリフルオロメチル)フェニル]メタノール
シクロプロピルメタノール(1.29 g)のDMF(80 mL)溶液に水素化ナトリウム(60%油性、0.72 g)を0℃で加え、30分間撹拌した。反応混合物にメチル4-フルオロ-2-(トリフルオロメチル)ベンゾアート(3.61 g)を加え、60℃で18時間撹拌した後、シクロプロピルメタノール(2.34 g)と水素化ナトリウム(60%油性、1.30 g)を加え、60℃で5時間撹拌した。反応混合物を室温に放冷した後、水を加え、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して無色油状物を得た。得られた油状物のTHF(20 mL)溶液を水素化アルミニウムリチウム(1.30 g)のTHF(60 mL)懸濁液に0℃で滴下した後、反応混合物を室温で1時間攪拌した。反応混合物に、水(2 mL)、1M水酸化ナトリウム(2 mL)、水(6 mL)を0℃で順次加え、セライト濾過した後、得られた濾液を減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.49 g)を無色油状物として得た。
1H NMR (300 MHz, CDCl3) δ0.32-0.41 (2H, m), 0.61-0.71 (2H, m), 1.20-1.34 (1H, m), 1.71 (1H, t, J = 6.2 Hz), 3.84 (2H, d, J = 6.8 Hz), 4.78 (2H, d, J = 6.0 Hz), 7.06 (1H, dd, J = 8.3, 2.6 Hz), 7.18 (1H, d, J = 2.6 Hz), 7.56 (1H, d, J = 8.7 Hz). A) [4- (Cyclopropylmethoxy) -2- (trifluoromethyl) phenyl] methanol Sodium hydride (60% oily, 0.72 g) was added to a solution of cyclopropylmethanol (1.29 g) in DMF (80 mL) at 0 ° C. And stirred for 30 minutes. To the reaction mixture was added methyl 4-fluoro-2- (trifluoromethyl) benzoate (3.61 g), and the mixture was stirred at 60 ° C. for 18 hours. g) was added and stirred at 60 ° C. for 5 hours. The reaction mixture was allowed to cool to room temperature, water was added, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give a colorless oil. A solution of the obtained oily substance in THF (20 mL) was added dropwise to a suspension of lithium aluminum hydride (1.30 g) in THF (60 mL) at 0 ° C., and the reaction mixture was stirred at room temperature for 1 hr. To the reaction mixture, water (2 mL), 1M sodium hydroxide (2 mL), and water (6 mL) were sequentially added at 0 ° C., and the mixture was filtered through celite. Then, the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.49 g) as a colorless oil.
1 H NMR (300 MHz, CDCl 3 ) δ0.32-0.41 (2H, m), 0.61-0.71 (2H, m), 1.20-1.34 (1H, m), 1.71 (1H, t, J = 6.2 Hz) , 3.84 (2H, d, J = 6.8 Hz), 4.78 (2H, d, J = 6.0 Hz), 7.06 (1H, dd, J = 8.3, 2.6 Hz), 7.18 (1H, d, J = 2.6 Hz) , 7.56 (1H, d, J = 8.7 Hz).
シクロプロピルメタノール(1.29 g)のDMF(80 mL)溶液に水素化ナトリウム(60%油性、0.72 g)を0℃で加え、30分間撹拌した。反応混合物にメチル4-フルオロ-2-(トリフルオロメチル)ベンゾアート(3.61 g)を加え、60℃で18時間撹拌した後、シクロプロピルメタノール(2.34 g)と水素化ナトリウム(60%油性、1.30 g)を加え、60℃で5時間撹拌した。反応混合物を室温に放冷した後、水を加え、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して無色油状物を得た。得られた油状物のTHF(20 mL)溶液を水素化アルミニウムリチウム(1.30 g)のTHF(60 mL)懸濁液に0℃で滴下した後、反応混合物を室温で1時間攪拌した。反応混合物に、水(2 mL)、1M水酸化ナトリウム(2 mL)、水(6 mL)を0℃で順次加え、セライト濾過した後、得られた濾液を減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.49 g)を無色油状物として得た。
1H NMR (300 MHz, CDCl3) δ0.32-0.41 (2H, m), 0.61-0.71 (2H, m), 1.20-1.34 (1H, m), 1.71 (1H, t, J = 6.2 Hz), 3.84 (2H, d, J = 6.8 Hz), 4.78 (2H, d, J = 6.0 Hz), 7.06 (1H, dd, J = 8.3, 2.6 Hz), 7.18 (1H, d, J = 2.6 Hz), 7.56 (1H, d, J = 8.7 Hz). A) [4- (Cyclopropylmethoxy) -2- (trifluoromethyl) phenyl] methanol Sodium hydride (60% oily, 0.72 g) was added to a solution of cyclopropylmethanol (1.29 g) in DMF (80 mL) at 0 ° C. And stirred for 30 minutes. To the reaction mixture was added methyl 4-fluoro-2- (trifluoromethyl) benzoate (3.61 g), and the mixture was stirred at 60 ° C. for 18 hours. g) was added and stirred at 60 ° C. for 5 hours. The reaction mixture was allowed to cool to room temperature, water was added, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give a colorless oil. A solution of the obtained oily substance in THF (20 mL) was added dropwise to a suspension of lithium aluminum hydride (1.30 g) in THF (60 mL) at 0 ° C., and the reaction mixture was stirred at room temperature for 1 hr. To the reaction mixture, water (2 mL), 1M sodium hydroxide (2 mL), and water (6 mL) were sequentially added at 0 ° C., and the mixture was filtered through celite. Then, the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.49 g) as a colorless oil.
1 H NMR (300 MHz, CDCl 3 ) δ0.32-0.41 (2H, m), 0.61-0.71 (2H, m), 1.20-1.34 (1H, m), 1.71 (1H, t, J = 6.2 Hz) , 3.84 (2H, d, J = 6.8 Hz), 4.78 (2H, d, J = 6.0 Hz), 7.06 (1H, dd, J = 8.3, 2.6 Hz), 7.18 (1H, d, J = 2.6 Hz) , 7.56 (1H, d, J = 8.7 Hz).
B) N-{(1S)-2-[(trans-4-{[4-(シクロプロピルメトキシ)-2-(トリフルオロメチル)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
[4-(シクロプロピルメトキシ)-2-(トリフルオロメチル)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 B) N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2- (trifluoromethyl) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [ The title compound was obtained in the same manner as in Steps A and F of Example 2 using 4- (cyclopropylmethoxy) -2- (trifluoromethyl) phenyl] methanol.
[4-(シクロプロピルメトキシ)-2-(トリフルオロメチル)フェニル]メタノールを用いて実施例2の工程A、Fと同様の方法により標題化合物を得た。 B) N-{(1S) -2-[(trans-4-{[4- (cyclopropylmethoxy) -2- (trifluoromethyl) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide [ The title compound was obtained in the same manner as in Steps A and F of Example 2 using 4- (cyclopropylmethoxy) -2- (trifluoromethyl) phenyl] methanol.
実施例79
N-{(1S)-2-[(trans-4-{[3-(2-シクロプロピルエトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 79
N-{(1S) -2-[(trans-4-{[3- (2-cyclopropylethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
N-{(1S)-2-[(trans-4-{[3-(2-シクロプロピルエトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 79
N-{(1S) -2-[(trans-4-{[3- (2-cyclopropylethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide
シクロプロピルエタノール(587 mg)のTHF(10 mL)溶液にトリエチルアミン(1.90 mL)とメタンスルホニルクロリド(0.791 mL)を加え、室温で10分間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣のDMF(5 mL)溶液にN-[(1S)-2-({trans-4-[(3-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド(110 mg)と炭酸カリウム(70.9 mg)を加えて、70℃で2時間加熱撹拌した。反応混合物を室温に放冷後、酢酸エチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(81.9 mg)を白色固体として得た。
Triethylamine (1.90 mL) and methanesulfonyl chloride (0.791 mL) were added to a THF (10 mL) solution of cyclopropylethanol (587 mg) and stirred at room temperature for 10 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. N-[(1S) -2-({trans-4-[(3-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide (110 mg) and carbonic acid were added to a DMF (5 mL) solution of the residue. Potassium (70.9 mg) was added and stirred with heating at 70 ° C. for 2 hours. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane / ethyl acetate) to obtain the title compound (81.9 mg) as a white solid.
実施例80
N-{(1S)-2-[(trans-4-{[3-(シクロブチルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
(ブロモメチル)シクロブタンを用いて実施例77の工程Bと同様の方法により標題化合物を得た。 Example 80
N-{(1S) -2-[(trans-4-{[3- (cyclobutylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (bromomethyl) cyclobutane The title compound was obtained in the same manner as in Step B.
N-{(1S)-2-[(trans-4-{[3-(シクロブチルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
(ブロモメチル)シクロブタンを用いて実施例77の工程Bと同様の方法により標題化合物を得た。 Example 80
N-{(1S) -2-[(trans-4-{[3- (cyclobutylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (bromomethyl) cyclobutane The title compound was obtained in the same manner as in Step B.
実施例81
N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(1-フルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 81
N-[(1S) -2-{[trans-4-({2-fluoro-4-[(1-fluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(1-フルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 81
N-[(1S) -2-{[trans-4-({2-fluoro-4-[(1-fluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
A) (1-フルオロシクロプロピル)メチル メタンスルホナート
(1-フルオロシクロプロピル)メタノールを用い、実施例38の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.76-0.96 (2H, m), 1.30-1.53 (2H, m), 3.14 (3H, d, J = 7.2 Hz), 4.30-4.63 (2H, m). A) (1-Fluorocyclopropyl) methyl methanesulfonate The title compound was obtained in the same manner as in Step A of Example 38 using (1-fluorocyclopropyl) methanol.
1 H NMR (300 MHz, CDCl 3 ) δ 0.76-0.96 (2H, m), 1.30-1.53 (2H, m), 3.14 (3H, d, J = 7.2 Hz), 4.30-4.63 (2H, m).
(1-フルオロシクロプロピル)メタノールを用い、実施例38の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.76-0.96 (2H, m), 1.30-1.53 (2H, m), 3.14 (3H, d, J = 7.2 Hz), 4.30-4.63 (2H, m). A) (1-Fluorocyclopropyl) methyl methanesulfonate The title compound was obtained in the same manner as in Step A of Example 38 using (1-fluorocyclopropyl) methanol.
1 H NMR (300 MHz, CDCl 3 ) δ 0.76-0.96 (2H, m), 1.30-1.53 (2H, m), 3.14 (3H, d, J = 7.2 Hz), 4.30-4.63 (2H, m).
B) N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(1-フルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
(1-フルオロシクロプロピル)メチル メタンスルホナートを用い、実施例35の工程Dと同様の方法により標題化合物を得た。 B) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(1-fluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide The title compound was obtained in the same manner as in Step D of Example 35 using (1-fluorocyclopropyl) methyl methanesulfonate.
(1-フルオロシクロプロピル)メチル メタンスルホナートを用い、実施例35の工程Dと同様の方法により標題化合物を得た。 B) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(1-fluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide The title compound was obtained in the same manner as in Step D of Example 35 using (1-fluorocyclopropyl) methyl methanesulfonate.
実施例82
N-{(1S)-2-[(trans-4-{[4-(2,2-ジフルオロプロポキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(2-オキソプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(177 mg)、ビス(2-メトキシエチル)アミノサルファートリフルオリド(297 mg)、およびトルエン(20 mL)の混合物を室温で1時間、続いて80℃で1時間撹拌した。ビス(2-メトキシエチル)アミノサルファートリフルオリド(297 mg)をさらに加え、混合物を80℃で1時間撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(37.7 mg)を淡黄色固体として得た。 Example 82
N-{(1S) -2-[(trans-4-{[4- (2,2-difluoropropoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide N-{( 1S) -2-[(trans-4-{[2-fluoro-4- (2-oxopropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (177 mg), bis (2-methoxy A mixture of ethyl) aminosulfur trifluoride (297 mg) and toluene (20 mL) was stirred at room temperature for 1 hour, followed by 80 ° C. for 1 hour. Bis (2-methoxyethyl) aminosulfur trifluoride (297 mg) was further added and the mixture was stirred at 80 ° C. for 1 hour. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (37.7 mg) as a pale yellow solid.
N-{(1S)-2-[(trans-4-{[4-(2,2-ジフルオロプロポキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(2-オキソプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド(177 mg)、ビス(2-メトキシエチル)アミノサルファートリフルオリド(297 mg)、およびトルエン(20 mL)の混合物を室温で1時間、続いて80℃で1時間撹拌した。ビス(2-メトキシエチル)アミノサルファートリフルオリド(297 mg)をさらに加え、混合物を80℃で1時間撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加えた後、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(37.7 mg)を淡黄色固体として得た。 Example 82
N-{(1S) -2-[(trans-4-{[4- (2,2-difluoropropoxy) -2-fluorobenzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide N-{( 1S) -2-[(trans-4-{[2-fluoro-4- (2-oxopropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide (177 mg), bis (2-methoxy A mixture of ethyl) aminosulfur trifluoride (297 mg) and toluene (20 mL) was stirred at room temperature for 1 hour, followed by 80 ° C. for 1 hour. Bis (2-methoxyethyl) aminosulfur trifluoride (297 mg) was further added and the mixture was stirred at 80 ° C. for 1 hour. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (37.7 mg) as a pale yellow solid.
実施例83
N-[(1S)-2-{[trans-4-({4-[(3,3-ジフルオロシクロブチル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミドおよび(3,3-ジフルオロシクロブチル)メチル メタンスルホナートを用い、実施例7の工程Eと同様の方法により標題化合物を得た。 Example 83
N-[(1S) -2-{[trans-4-({4-[(3,3-difluorocyclobutyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide N-[(1S) -2-({trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide and (3,3-difluorocyclobutyl) methyl The title compound was obtained in the same manner as in Step E of Example 7 using methanesulfonate.
N-[(1S)-2-{[trans-4-({4-[(3,3-ジフルオロシクロブチル)メトキシ]-2-フルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミドおよび(3,3-ジフルオロシクロブチル)メチル メタンスルホナートを用い、実施例7の工程Eと同様の方法により標題化合物を得た。 Example 83
N-[(1S) -2-{[trans-4-({4-[(3,3-difluorocyclobutyl) methoxy] -2-fluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide N-[(1S) -2-({trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide and (3,3-difluorocyclobutyl) methyl The title compound was obtained in the same manner as in Step E of Example 7 using methanesulfonate.
実施例84
N-[3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-1-メチル-3-オキソプロピル]アセトアミド Example 84
N- [3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methyl-3-oxopropyl] acetamide
N-[3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-1-メチル-3-オキソプロピル]アセトアミド Example 84
N- [3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methyl-3-oxopropyl] acetamide
A) trans-4-({[tert-ブチル(ジフェニル)シリル]オキシ}メチル)シクロヘキサノール
trans-4-(ヒドロキシメチル)シクロヘキサノール(5.00 g)、イミダゾール(3.92 g)、tert-ブチル(クロロ)ジフェニルシラン(10.6 g)およびDMF(80 mL)の混合物を室温で終夜撹拌した。減圧下溶媒を留去した後、残渣に水を加え酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(13.1 g)を淡褐色油状物として得た。
1H NMR (300 MHz, CDCl3) δ 0.87-1.12 (11H, m), 1.15-1.35 (2H, m), 1.40-1.54 (1H, m), 1.83 (2H, d, J = 12.9 Hz), 1.99 (2H, dd, J = 12.7, 4.0 Hz), 3.46 (2H, d, J = 6.4 Hz), 3.48-3.61 (1H, m), 7.32-7.47 (6H, m), 7.65 (4H, dd, J = 7.8, 1.7 Hz). A) trans-4-({[tert-butyl (diphenyl) silyl] oxy} methyl) cyclohexanol trans-4- (hydroxymethyl) cyclohexanol (5.00 g), imidazole (3.92 g), tert-butyl (chloro) A mixture of diphenylsilane (10.6 g) and DMF (80 mL) was stirred at room temperature overnight. After evaporating the solvent under reduced pressure, water was added to the residue and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (13.1 g) as a pale brown oil.
1 H NMR (300 MHz, CDCl 3 ) δ 0.87-1.12 (11H, m), 1.15-1.35 (2H, m), 1.40-1.54 (1H, m), 1.83 (2H, d, J = 12.9 Hz), 1.99 (2H, dd, J = 12.7, 4.0 Hz), 3.46 (2H, d, J = 6.4 Hz), 3.48-3.61 (1H, m), 7.32-7.47 (6H, m), 7.65 (4H, dd, J = 7.8, 1.7 Hz).
trans-4-(ヒドロキシメチル)シクロヘキサノール(5.00 g)、イミダゾール(3.92 g)、tert-ブチル(クロロ)ジフェニルシラン(10.6 g)およびDMF(80 mL)の混合物を室温で終夜撹拌した。減圧下溶媒を留去した後、残渣に水を加え酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(13.1 g)を淡褐色油状物として得た。
1H NMR (300 MHz, CDCl3) δ 0.87-1.12 (11H, m), 1.15-1.35 (2H, m), 1.40-1.54 (1H, m), 1.83 (2H, d, J = 12.9 Hz), 1.99 (2H, dd, J = 12.7, 4.0 Hz), 3.46 (2H, d, J = 6.4 Hz), 3.48-3.61 (1H, m), 7.32-7.47 (6H, m), 7.65 (4H, dd, J = 7.8, 1.7 Hz). A) trans-4-({[tert-butyl (diphenyl) silyl] oxy} methyl) cyclohexanol trans-4- (hydroxymethyl) cyclohexanol (5.00 g), imidazole (3.92 g), tert-butyl (chloro) A mixture of diphenylsilane (10.6 g) and DMF (80 mL) was stirred at room temperature overnight. After evaporating the solvent under reduced pressure, water was added to the residue and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (13.1 g) as a pale brown oil.
1 H NMR (300 MHz, CDCl 3 ) δ 0.87-1.12 (11H, m), 1.15-1.35 (2H, m), 1.40-1.54 (1H, m), 1.83 (2H, d, J = 12.9 Hz), 1.99 (2H, dd, J = 12.7, 4.0 Hz), 3.46 (2H, d, J = 6.4 Hz), 3.48-3.61 (1H, m), 7.32-7.47 (6H, m), 7.65 (4H, dd, J = 7.8, 1.7 Hz).
B) tert-ブチル[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)メトキシ]ジフェニルシラン
trans-4-({[tert-ブチル(ジフェニル)シリル]オキシ}メチル)シクロヘキサノールのDMF(80 mL)溶液に水素化ナトリウム(60%油状、1.57 g)を加え、窒素雰囲気下0℃で20分撹拌した。反応混合物に、1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2-フルオロベンゼン(11.1 g)を加え、窒素雰囲気下室温で1時間撹拌した。反応混合物を酢酸エチルで希釈し、飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(16.3 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.27-0.40 (2H, m), 0.55-0.70 (2H, m), 1.05 (9H, s), 1.17-1.40 (6H, m), 1.45-1.54 (1H, m), 1.74-2.15 (3H, m), 3.42-3.49 (2H, m), 3.53 (1H, q, J = 6.8 Hz), 3.77 (2H, d, J = 6.8 Hz), 4.50 (2H, d, J = 14.0 Hz), 6.53-6.77 (2H, m), 7.24-7.32 (1H, m), 7.33-7.48 (6H, m), 7.64 (4H, d, J = 1.5 Hz). B) tert-butyl [(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) methoxy] diphenylsilane trans-4-({[tert-butyl (diphenyl) silyl] oxy } To a solution of methyl) cyclohexanol in DMF (80 mL) was added sodium hydride (60% oil, 1.57 g), and the mixture was stirred at 0 ° C. for 20 minutes under a nitrogen atmosphere. 1- (Bromomethyl) -4- (cyclopropylmethoxy) -2-fluorobenzene (11.1 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 1 hour under a nitrogen atmosphere. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (16.3 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.27-0.40 (2H, m), 0.55-0.70 (2H, m), 1.05 (9H, s), 1.17-1.40 (6H, m), 1.45-1.54 (1H , m), 1.74-2.15 (3H, m), 3.42-3.49 (2H, m), 3.53 (1H, q, J = 6.8 Hz), 3.77 (2H, d, J = 6.8 Hz), 4.50 (2H, d, J = 14.0 Hz), 6.53-6.77 (2H, m), 7.24-7.32 (1H, m), 7.33-7.48 (6H, m), 7.64 (4H, d, J = 1.5 Hz).
trans-4-({[tert-ブチル(ジフェニル)シリル]オキシ}メチル)シクロヘキサノールのDMF(80 mL)溶液に水素化ナトリウム(60%油状、1.57 g)を加え、窒素雰囲気下0℃で20分撹拌した。反応混合物に、1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2-フルオロベンゼン(11.1 g)を加え、窒素雰囲気下室温で1時間撹拌した。反応混合物を酢酸エチルで希釈し、飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(16.3 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.27-0.40 (2H, m), 0.55-0.70 (2H, m), 1.05 (9H, s), 1.17-1.40 (6H, m), 1.45-1.54 (1H, m), 1.74-2.15 (3H, m), 3.42-3.49 (2H, m), 3.53 (1H, q, J = 6.8 Hz), 3.77 (2H, d, J = 6.8 Hz), 4.50 (2H, d, J = 14.0 Hz), 6.53-6.77 (2H, m), 7.24-7.32 (1H, m), 7.33-7.48 (6H, m), 7.64 (4H, d, J = 1.5 Hz). B) tert-butyl [(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) methoxy] diphenylsilane trans-4-({[tert-butyl (diphenyl) silyl] oxy } To a solution of methyl) cyclohexanol in DMF (80 mL) was added sodium hydride (60% oil, 1.57 g), and the mixture was stirred at 0 ° C. for 20 minutes under a nitrogen atmosphere. 1- (Bromomethyl) -4- (cyclopropylmethoxy) -2-fluorobenzene (11.1 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 1 hour under a nitrogen atmosphere. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (16.3 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.27-0.40 (2H, m), 0.55-0.70 (2H, m), 1.05 (9H, s), 1.17-1.40 (6H, m), 1.45-1.54 (1H , m), 1.74-2.15 (3H, m), 3.42-3.49 (2H, m), 3.53 (1H, q, J = 6.8 Hz), 3.77 (2H, d, J = 6.8 Hz), 4.50 (2H, d, J = 14.0 Hz), 6.53-6.77 (2H, m), 7.24-7.32 (1H, m), 7.33-7.48 (6H, m), 7.64 (4H, d, J = 1.5 Hz).
C) (trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)メタノール
tert-ブチル[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)メトキシ]ジフェニルシラン(16.3 g) のTHF(80 mL)溶液に、テトラブチルアンモニウムフルオリドのTHF溶液(1.0 M, 44.8 mL)を室温で加え1時間撹拌した。反応混合物に水を加え酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(3.18 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.40 (2H, m), 0.57-0.70 (2H, m), 0.85-1.08 (2H, m), 1.19-1.38 (4H, m), 1.40-1.56 (1H, m), 1.85 (2H, d, J = 12.5 Hz), 2.07-2.19 (2H, m), 3.20-3.36 (1H, m), 3.40-3.50 (2H, m), 3.77 (2H, d, J = 7.2 Hz), 4.53 (2H, s), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.67 (1H, dd, J = 8.3, 2.7 Hz), 7.23-7.33 (1H, m). C) (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) methanol tert-butyl [(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl To a solution of [oxy} cyclohexyl) methoxy] diphenylsilane (16.3 g) in THF (80 mL) was added tetrabutylammonium fluoride in THF (1.0 M, 44.8 mL) at room temperature, and the mixture was stirred for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (3.18 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.40 (2H, m), 0.57-0.70 (2H, m), 0.85-1.08 (2H, m), 1.19-1.38 (4H, m), 1.40-1.56 (1H, m), 1.85 (2H, d, J = 12.5 Hz), 2.07-2.19 (2H, m), 3.20-3.36 (1H, m), 3.40-3.50 (2H, m), 3.77 (2H, d , J = 7.2 Hz), 4.53 (2H, s), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.67 (1H, dd, J = 8.3, 2.7 Hz), 7.23-7.33 (1H, m) .
tert-ブチル[(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)メトキシ]ジフェニルシラン(16.3 g) のTHF(80 mL)溶液に、テトラブチルアンモニウムフルオリドのTHF溶液(1.0 M, 44.8 mL)を室温で加え1時間撹拌した。反応混合物に水を加え酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(3.18 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.29-0.40 (2H, m), 0.57-0.70 (2H, m), 0.85-1.08 (2H, m), 1.19-1.38 (4H, m), 1.40-1.56 (1H, m), 1.85 (2H, d, J = 12.5 Hz), 2.07-2.19 (2H, m), 3.20-3.36 (1H, m), 3.40-3.50 (2H, m), 3.77 (2H, d, J = 7.2 Hz), 4.53 (2H, s), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.67 (1H, dd, J = 8.3, 2.7 Hz), 7.23-7.33 (1H, m). C) (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) methanol tert-butyl [(trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl To a solution of [oxy} cyclohexyl) methoxy] diphenylsilane (16.3 g) in THF (80 mL) was added tetrabutylammonium fluoride in THF (1.0 M, 44.8 mL) at room temperature, and the mixture was stirred for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (3.18 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.29-0.40 (2H, m), 0.57-0.70 (2H, m), 0.85-1.08 (2H, m), 1.19-1.38 (4H, m), 1.40-1.56 (1H, m), 1.85 (2H, d, J = 12.5 Hz), 2.07-2.19 (2H, m), 3.20-3.36 (1H, m), 3.40-3.50 (2H, m), 3.77 (2H, d , J = 7.2 Hz), 4.53 (2H, s), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.67 (1H, dd, J = 8.3, 2.7 Hz), 7.23-7.33 (1H, m) .
D) 1-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-3-メチルブタ-3-エン-1-オール
(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)メタノール(3.18 g)、モレキュラーシーブズ4A(4 g)、テトラプロピルアンモニウム ペルルテナート(362 mg)、4-メチルモルホリンN-オキシド(1.81 g)およびアセトニトリル(40 mL) の混合物を室温で20分間撹拌した。反応混合物をシリカゲルに通じた後、有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣のTHF(40 mL)溶液に、2-メチルアリルマグネシウム クロリドのTHF溶液(0.5 M、30.9 mL)を加えて、室温で20分間撹拌した。反応混合物に塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し標題化合物(1.26 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.24-0.42 (2H, m), 0.56-0.72 (2H, m), 0.98-1.43 (7H, m), 1.69-1.85 (4H, m), 1.88-2.00 (1H, m), 2.01-2.29 (3H, m), 3.20-3.35 (1H, m), 3.44-3.55 (1H, m), 3.77 (2H, d, J = 6.8 Hz), 4.53 (2H, s), 4.80 (1H, s), 4.90 (1H, s), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.67 (1H, dd, J = 8.5, 2.5 Hz), 7.22-7.33 (1H, m). D) 1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-methylbut-3-en-1-ol (trans-4-{[4- ( Cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) methanol (3.18 g), molecular sieves 4A (4 g), tetrapropylammonium perruthenate (362 mg), 4-methylmorpholine N-oxide (1.81 g) and acetonitrile (40 mL) was stirred at room temperature for 20 minutes. The reaction mixture was passed through silica gel, and the organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. A THF solution (0.5 M, 30.9 mL) of 2-methylallylmagnesium chloride was added to a THF (40 mL) solution of the obtained residue, and the mixture was stirred at room temperature for 20 minutes. To the reaction mixture was added aqueous ammonium chloride solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.26 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.24-0.42 (2H, m), 0.56-0.72 (2H, m), 0.98-1.43 (7H, m), 1.69-1.85 (4H, m), 1.88-2.00 (1H, m), 2.01-2.29 (3H, m), 3.20-3.35 (1H, m), 3.44-3.55 (1H, m), 3.77 (2H, d, J = 6.8 Hz), 4.53 (2H, s ), 4.80 (1H, s), 4.90 (1H, s), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.67 (1H, dd, J = 8.5, 2.5 Hz), 7.22-7.33 (1H, m).
(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)メタノール(3.18 g)、モレキュラーシーブズ4A(4 g)、テトラプロピルアンモニウム ペルルテナート(362 mg)、4-メチルモルホリンN-オキシド(1.81 g)およびアセトニトリル(40 mL) の混合物を室温で20分間撹拌した。反応混合物をシリカゲルに通じた後、有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣のTHF(40 mL)溶液に、2-メチルアリルマグネシウム クロリドのTHF溶液(0.5 M、30.9 mL)を加えて、室温で20分間撹拌した。反応混合物に塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し標題化合物(1.26 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.24-0.42 (2H, m), 0.56-0.72 (2H, m), 0.98-1.43 (7H, m), 1.69-1.85 (4H, m), 1.88-2.00 (1H, m), 2.01-2.29 (3H, m), 3.20-3.35 (1H, m), 3.44-3.55 (1H, m), 3.77 (2H, d, J = 6.8 Hz), 4.53 (2H, s), 4.80 (1H, s), 4.90 (1H, s), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.67 (1H, dd, J = 8.5, 2.5 Hz), 7.22-7.33 (1H, m). D) 1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-methylbut-3-en-1-ol (trans-4-{[4- ( Cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) methanol (3.18 g), molecular sieves 4A (4 g), tetrapropylammonium perruthenate (362 mg), 4-methylmorpholine N-oxide (1.81 g) and acetonitrile (40 mL) was stirred at room temperature for 20 minutes. The reaction mixture was passed through silica gel, and the organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. A THF solution (0.5 M, 30.9 mL) of 2-methylallylmagnesium chloride was added to a THF (40 mL) solution of the obtained residue, and the mixture was stirred at room temperature for 20 minutes. To the reaction mixture was added aqueous ammonium chloride solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.26 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.24-0.42 (2H, m), 0.56-0.72 (2H, m), 0.98-1.43 (7H, m), 1.69-1.85 (4H, m), 1.88-2.00 (1H, m), 2.01-2.29 (3H, m), 3.20-3.35 (1H, m), 3.44-3.55 (1H, m), 3.77 (2H, d, J = 6.8 Hz), 4.53 (2H, s ), 4.80 (1H, s), 4.90 (1H, s), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.67 (1H, dd, J = 8.5, 2.5 Hz), 7.22-7.33 (1H, m).
E) tert-ブチル{[1-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-3-メチルブタ-3-エン-1-イル]オキシ}ジメチルシラン
1-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-3-メチルブタ-3-エン-1-オールとtert-ブチル(クロロ)ジメチルシランを用いて実施例84の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.03 (6H, s), 0.31-0.40 (2H, m), 0.59-0.70 (2H, m), 0.89 (9H, s), 1.05-1.45 (6H, m), 1.58-1.84 (5H, m), 2.00-2.27 (4H, m), 3.08-3.36 (1H, m), 3.59-3.72 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 4.53 (2H, s), 4.66-4.82 (2H, m), 6.60 (1H, dd, J = 11.7, 2.3 Hz), 6.68 (1H, dd, J = 8.3, 2.7 Hz), 7.24-7.35 (1H, m). E) tert-butyl {[1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-methylbut-3-en-1-yl] oxy} dimethylsilane Using 1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-methylbut-3-en-1-ol and tert-butyl (chloro) dimethylsilane The title compound was obtained in the same manner as in Step A of Example 84.
1 H NMR (300 MHz, CDCl 3 ) δ 0.03 (6H, s), 0.31-0.40 (2H, m), 0.59-0.70 (2H, m), 0.89 (9H, s), 1.05-1.45 (6H, m ), 1.58-1.84 (5H, m), 2.00-2.27 (4H, m), 3.08-3.36 (1H, m), 3.59-3.72 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 4.53 (2H, s), 4.66-4.82 (2H, m), 6.60 (1H, dd, J = 11.7, 2.3 Hz), 6.68 (1H, dd, J = 8.3, 2.7 Hz), 7.24-7.35 (1H, m).
1-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-3-メチルブタ-3-エン-1-オールとtert-ブチル(クロロ)ジメチルシランを用いて実施例84の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.03 (6H, s), 0.31-0.40 (2H, m), 0.59-0.70 (2H, m), 0.89 (9H, s), 1.05-1.45 (6H, m), 1.58-1.84 (5H, m), 2.00-2.27 (4H, m), 3.08-3.36 (1H, m), 3.59-3.72 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 4.53 (2H, s), 4.66-4.82 (2H, m), 6.60 (1H, dd, J = 11.7, 2.3 Hz), 6.68 (1H, dd, J = 8.3, 2.7 Hz), 7.24-7.35 (1H, m). E) tert-butyl {[1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-methylbut-3-en-1-yl] oxy} dimethylsilane Using 1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-methylbut-3-en-1-ol and tert-butyl (chloro) dimethylsilane The title compound was obtained in the same manner as in Step A of Example 84.
1 H NMR (300 MHz, CDCl 3 ) δ 0.03 (6H, s), 0.31-0.40 (2H, m), 0.59-0.70 (2H, m), 0.89 (9H, s), 1.05-1.45 (6H, m ), 1.58-1.84 (5H, m), 2.00-2.27 (4H, m), 3.08-3.36 (1H, m), 3.59-3.72 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 4.53 (2H, s), 4.66-4.82 (2H, m), 6.60 (1H, dd, J = 11.7, 2.3 Hz), 6.68 (1H, dd, J = 8.3, 2.7 Hz), 7.24-7.35 (1H, m).
F) 4-{[tert-ブチル(ジメチル)シリル]オキシ}-4-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)ブタン-2-オール
tert-ブチル{[1-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-3-メチルブタ-3-エン-1-イル]オキシ}ジメチルシラン(1.47 g)の酢酸エチル(20 mL)溶液にオゾンを-78℃で10分間通じた。反応混合物に窒素を5分間通じた後、水素化ホウ素ナトリウム(117 mg)とメタノール(5 mL)を加えて、室温で10分間撹拌した。反応混合物に塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し標題化合物(1.14 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.05-0.16 (6H, m), 0.31-0.40 (2H, m), 0.61-0.70 (2H, m), 0.91 (9H, s), 1.11-1.21 (4H, m), 1.27 (3H, t, J = 7.2 Hz), 1.37-1.98 (6H, m), 2.07-2.23 (2H, m), 3.21-3.32 (1H, m), 3.59-3.97 (4H, m), 4.53 (2H, s), 6.60 (1H, dd, J = 11.9, 2.5 Hz), 6.68 (1H, dd, J = 8.5, 2.5 Hz), 7.19-7.34 (1H, m). F) 4-{[tert-Butyl (dimethyl) silyl] oxy} -4- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) butan-2-ol tert- Butyl {[1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-methylbut-3-en-1-yl] oxy} dimethylsilane (1.47 g) Ozone in ethyl acetate (20 mL) was passed at −78 ° C. for 10 minutes. After passing nitrogen through the reaction mixture for 5 minutes, sodium borohydride (117 mg) and methanol (5 mL) were added, and the mixture was stirred at room temperature for 10 minutes. To the reaction mixture was added aqueous ammonium chloride solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.14 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.05-0.16 (6H, m), 0.31-0.40 (2H, m), 0.61-0.70 (2H, m), 0.91 (9H, s), 1.11-1.21 (4H , m), 1.27 (3H, t, J = 7.2 Hz), 1.37-1.98 (6H, m), 2.07-2.23 (2H, m), 3.21-3.32 (1H, m), 3.59-3.97 (4H, m ), 4.53 (2H, s), 6.60 (1H, dd, J = 11.9, 2.5 Hz), 6.68 (1H, dd, J = 8.5, 2.5 Hz), 7.19-7.34 (1H, m).
tert-ブチル{[1-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-3-メチルブタ-3-エン-1-イル]オキシ}ジメチルシラン(1.47 g)の酢酸エチル(20 mL)溶液にオゾンを-78℃で10分間通じた。反応混合物に窒素を5分間通じた後、水素化ホウ素ナトリウム(117 mg)とメタノール(5 mL)を加えて、室温で10分間撹拌した。反応混合物に塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥した後、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製し標題化合物(1.14 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.05-0.16 (6H, m), 0.31-0.40 (2H, m), 0.61-0.70 (2H, m), 0.91 (9H, s), 1.11-1.21 (4H, m), 1.27 (3H, t, J = 7.2 Hz), 1.37-1.98 (6H, m), 2.07-2.23 (2H, m), 3.21-3.32 (1H, m), 3.59-3.97 (4H, m), 4.53 (2H, s), 6.60 (1H, dd, J = 11.9, 2.5 Hz), 6.68 (1H, dd, J = 8.5, 2.5 Hz), 7.19-7.34 (1H, m). F) 4-{[tert-Butyl (dimethyl) silyl] oxy} -4- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) butan-2-ol tert- Butyl {[1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-methylbut-3-en-1-yl] oxy} dimethylsilane (1.47 g) Ozone in ethyl acetate (20 mL) was passed at −78 ° C. for 10 minutes. After passing nitrogen through the reaction mixture for 5 minutes, sodium borohydride (117 mg) and methanol (5 mL) were added, and the mixture was stirred at room temperature for 10 minutes. To the reaction mixture was added aqueous ammonium chloride solution, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.14 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.05-0.16 (6H, m), 0.31-0.40 (2H, m), 0.61-0.70 (2H, m), 0.91 (9H, s), 1.11-1.21 (4H , m), 1.27 (3H, t, J = 7.2 Hz), 1.37-1.98 (6H, m), 2.07-2.23 (2H, m), 3.21-3.32 (1H, m), 3.59-3.97 (4H, m ), 4.53 (2H, s), 6.60 (1H, dd, J = 11.9, 2.5 Hz), 6.68 (1H, dd, J = 8.5, 2.5 Hz), 7.19-7.34 (1H, m).
G) [3-アジド-1-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)ブトキシ](tert-ブチル)ジメチルシラン
4-{[tert-ブチル(ジメチル)シリル]オキシ}-4-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)ブタン-2-オールを用いて実施例21の工程Dと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.03-0.11 (6H, m), 0.30-0.41 (2H, m), 0.59-0.71 (2H, m), 0.90 (9H, s), 0.97-1.52 (11H, m), 1.60-1.87 (2H, m), 2.08-2.22 (2H, m), 3.14-3.34 (1H, m), 3.41-3.75 (2H, m), 3.78 (2H, d, J = 6.8 Hz), 4.53 (2H, s), 6.60 (1H, dd, J = 11.7, 2.6 Hz), 6.68 (1H, dd, J = 8.5, 2.4 Hz), 7.28-7.33 (1H, m). G) [3-Azido-1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) butoxy] (tert-butyl) dimethylsilane 4-{[tert-butyl ( Dimethyl) silyl] oxy} -4- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) butan-2-ol as in step D of Example 21 The title compound was obtained by the method.
1 H NMR (300 MHz, CDCl 3 ) δ 0.03-0.11 (6H, m), 0.30-0.41 (2H, m), 0.59-0.71 (2H, m), 0.90 (9H, s), 0.97-1.52 (11H , m), 1.60-1.87 (2H, m), 2.08-2.22 (2H, m), 3.14-3.34 (1H, m), 3.41-3.75 (2H, m), 3.78 (2H, d, J = 6.8 Hz ), 4.53 (2H, s), 6.60 (1H, dd, J = 11.7, 2.6 Hz), 6.68 (1H, dd, J = 8.5, 2.4 Hz), 7.28-7.33 (1H, m).
4-{[tert-ブチル(ジメチル)シリル]オキシ}-4-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)ブタン-2-オールを用いて実施例21の工程Dと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.03-0.11 (6H, m), 0.30-0.41 (2H, m), 0.59-0.71 (2H, m), 0.90 (9H, s), 0.97-1.52 (11H, m), 1.60-1.87 (2H, m), 2.08-2.22 (2H, m), 3.14-3.34 (1H, m), 3.41-3.75 (2H, m), 3.78 (2H, d, J = 6.8 Hz), 4.53 (2H, s), 6.60 (1H, dd, J = 11.7, 2.6 Hz), 6.68 (1H, dd, J = 8.5, 2.4 Hz), 7.28-7.33 (1H, m). G) [3-Azido-1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) butoxy] (tert-butyl) dimethylsilane 4-{[tert-butyl ( Dimethyl) silyl] oxy} -4- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) butan-2-ol as in step D of Example 21 The title compound was obtained by the method.
1 H NMR (300 MHz, CDCl 3 ) δ 0.03-0.11 (6H, m), 0.30-0.41 (2H, m), 0.59-0.71 (2H, m), 0.90 (9H, s), 0.97-1.52 (11H , m), 1.60-1.87 (2H, m), 2.08-2.22 (2H, m), 3.14-3.34 (1H, m), 3.41-3.75 (2H, m), 3.78 (2H, d, J = 6.8 Hz ), 4.53 (2H, s), 6.60 (1H, dd, J = 11.7, 2.6 Hz), 6.68 (1H, dd, J = 8.5, 2.4 Hz), 7.28-7.33 (1H, m).
H) N-[3-{[tert-ブチル(ジメチル)シリル]オキシ}-3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-1-メチルプロピル]アセトアミド
水素雰囲気下、[3-アジド-1-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)ブトキシ](tert-ブチル)ジメチルシラン(1.13 g)、10%パラジウム-炭素(50%含水、1.0 g)および酢酸エチル(10 mL)の混合物を室温で10分間撹拌した。濾過により触媒を除去した後、得られた濾液を減圧下濃縮して得られる残渣をピリジン(5 mL)に溶解させ、無水酢酸(5 mL)を加え室温で10分間撹拌した。反応混合物を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(0.960 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.02-0.13 (6H, m), 0.30-0.40 (2H, m), 0.59-0.71 (2H, m), 0.85-1.34 (17H, m), 1.36-1.62 (3H, m), 1.65-1.81 (2H, m), 1.87-2.00 (3H, m), 2.07-2.19 (2H, m), 3.09-3.34 (1H, m), 3.47-3.69 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 3.94-4.07 (1H, m), 4.52 (2H, s), 5.27-5.98 (1H, m), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.68 (1H, dd, J = 8.3, 2.3 Hz), 7.29 (1H, t, J = 8.5 Hz). H) N- [3-{[tert-butyl (dimethyl) silyl] oxy} -3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methyl Propyl] acetamide Under a hydrogen atmosphere, [3-azido-1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) butoxy] (tert-butyl) dimethylsilane (1.13 g ), 10% palladium-carbon (50% water content, 1.0 g) and ethyl acetate (10 mL) were stirred at room temperature for 10 minutes. After removing the catalyst by filtration, the obtained filtrate was concentrated under reduced pressure, the resulting residue was dissolved in pyridine (5 mL), acetic anhydride (5 mL) was added, and the mixture was stirred at room temperature for 10 min. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (0.960 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.02-0.13 (6H, m), 0.30-0.40 (2H, m), 0.59-0.71 (2H, m), 0.85-1.34 (17H, m), 1.36-1.62 (3H, m), 1.65-1.81 (2H, m), 1.87-2.00 (3H, m), 2.07-2.19 (2H, m), 3.09-3.34 (1H, m), 3.47-3.69 (1H, m) , 3.78 (2H, d, J = 6.8 Hz), 3.94-4.07 (1H, m), 4.52 (2H, s), 5.27-5.98 (1H, m), 6.59 (1H, dd, J = 11.9, 2.5 Hz ), 6.68 (1H, dd, J = 8.3, 2.3 Hz), 7.29 (1H, t, J = 8.5 Hz).
水素雰囲気下、[3-アジド-1-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)ブトキシ](tert-ブチル)ジメチルシラン(1.13 g)、10%パラジウム-炭素(50%含水、1.0 g)および酢酸エチル(10 mL)の混合物を室温で10分間撹拌した。濾過により触媒を除去した後、得られた濾液を減圧下濃縮して得られる残渣をピリジン(5 mL)に溶解させ、無水酢酸(5 mL)を加え室温で10分間撹拌した。反応混合物を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(0.960 g)を得た。
1H NMR (300 MHz, CDCl3) δ 0.02-0.13 (6H, m), 0.30-0.40 (2H, m), 0.59-0.71 (2H, m), 0.85-1.34 (17H, m), 1.36-1.62 (3H, m), 1.65-1.81 (2H, m), 1.87-2.00 (3H, m), 2.07-2.19 (2H, m), 3.09-3.34 (1H, m), 3.47-3.69 (1H, m), 3.78 (2H, d, J = 6.8 Hz), 3.94-4.07 (1H, m), 4.52 (2H, s), 5.27-5.98 (1H, m), 6.59 (1H, dd, J = 11.9, 2.5 Hz), 6.68 (1H, dd, J = 8.3, 2.3 Hz), 7.29 (1H, t, J = 8.5 Hz). H) N- [3-{[tert-butyl (dimethyl) silyl] oxy} -3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methyl Propyl] acetamide Under a hydrogen atmosphere, [3-azido-1- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) butoxy] (tert-butyl) dimethylsilane (1.13 g ), 10% palladium-carbon (50% water content, 1.0 g) and ethyl acetate (10 mL) were stirred at room temperature for 10 minutes. After removing the catalyst by filtration, the obtained filtrate was concentrated under reduced pressure, the resulting residue was dissolved in pyridine (5 mL), acetic anhydride (5 mL) was added, and the mixture was stirred at room temperature for 10 min. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (0.960 g).
1 H NMR (300 MHz, CDCl 3 ) δ 0.02-0.13 (6H, m), 0.30-0.40 (2H, m), 0.59-0.71 (2H, m), 0.85-1.34 (17H, m), 1.36-1.62 (3H, m), 1.65-1.81 (2H, m), 1.87-2.00 (3H, m), 2.07-2.19 (2H, m), 3.09-3.34 (1H, m), 3.47-3.69 (1H, m) , 3.78 (2H, d, J = 6.8 Hz), 3.94-4.07 (1H, m), 4.52 (2H, s), 5.27-5.98 (1H, m), 6.59 (1H, dd, J = 11.9, 2.5 Hz ), 6.68 (1H, dd, J = 8.3, 2.3 Hz), 7.29 (1H, t, J = 8.5 Hz).
I) N-[3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-3-ヒドロキシ-1-メチルプロピル]アセトアミド
N-[3-{[tert-ブチル(ジメチル)シリル]オキシ}-3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-1-メチルプロピル]アセトアミドを用いて、実施例84の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.27-0.40 (2H, m), 0.57-0.72 (2H, m), 0.93-1.38 (8H, m), 1.48-1.78 (3H, m), 1.90-2.19 (7H, m), 3.17-3.33 (2H, m), 3.77 (2H, d, J = 6.8 Hz), 4.06-4.12 (1H, m), 4.16-4.32 (1H, m), 4.52 (2H, s), 5.35 (1H, d, J = 8.7 Hz), 6.58 (1H, dd, J = 11.9, 2.5 Hz), 6.66 (1H, dd, J = 8.5, 2.5 Hz), 7.28 (1H, t, J = 8.5 Hz). I) N- [3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-hydroxy-1-methylpropyl] acetamide N- [3-{[tert -Butyl (dimethyl) silyl] oxy} -3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methylpropyl] acetamide Example 84 The title compound was obtained in the same manner as in Step C.
1 H NMR (300 MHz, CDCl 3 ) δ 0.27-0.40 (2H, m), 0.57-0.72 (2H, m), 0.93-1.38 (8H, m), 1.48-1.78 (3H, m), 1.90-2.19 (7H, m), 3.17-3.33 (2H, m), 3.77 (2H, d, J = 6.8 Hz), 4.06-4.12 (1H, m), 4.16-4.32 (1H, m), 4.52 (2H, s ), 5.35 (1H, d, J = 8.7 Hz), 6.58 (1H, dd, J = 11.9, 2.5 Hz), 6.66 (1H, dd, J = 8.5, 2.5 Hz), 7.28 (1H, t, J = 8.5 Hz).
N-[3-{[tert-ブチル(ジメチル)シリル]オキシ}-3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-1-メチルプロピル]アセトアミドを用いて、実施例84の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 0.27-0.40 (2H, m), 0.57-0.72 (2H, m), 0.93-1.38 (8H, m), 1.48-1.78 (3H, m), 1.90-2.19 (7H, m), 3.17-3.33 (2H, m), 3.77 (2H, d, J = 6.8 Hz), 4.06-4.12 (1H, m), 4.16-4.32 (1H, m), 4.52 (2H, s), 5.35 (1H, d, J = 8.7 Hz), 6.58 (1H, dd, J = 11.9, 2.5 Hz), 6.66 (1H, dd, J = 8.5, 2.5 Hz), 7.28 (1H, t, J = 8.5 Hz). I) N- [3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-hydroxy-1-methylpropyl] acetamide N- [3-{[tert -Butyl (dimethyl) silyl] oxy} -3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methylpropyl] acetamide Example 84 The title compound was obtained in the same manner as in Step C.
1 H NMR (300 MHz, CDCl 3 ) δ 0.27-0.40 (2H, m), 0.57-0.72 (2H, m), 0.93-1.38 (8H, m), 1.48-1.78 (3H, m), 1.90-2.19 (7H, m), 3.17-3.33 (2H, m), 3.77 (2H, d, J = 6.8 Hz), 4.06-4.12 (1H, m), 4.16-4.32 (1H, m), 4.52 (2H, s ), 5.35 (1H, d, J = 8.7 Hz), 6.58 (1H, dd, J = 11.9, 2.5 Hz), 6.66 (1H, dd, J = 8.5, 2.5 Hz), 7.28 (1H, t, J = 8.5 Hz).
J) N-[3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-1-メチル-3-オキソプロピル]アセトアミド
N-[3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-3-ヒドロキシ-1-メチルプロピル]アセトアミド(370 mg)のアセトニトリル(5 mL)溶液にデスマーチン試薬(578 mg)を加えて、室温で30分間撹拌した。反応混合物に亜硫酸ナトリウム水溶液を加えて、酢酸エチルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して得られた固体をジエチルエーテルで洗浄して、標題化合物(310 mg)を得た。 J) N- [3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methyl-3-oxopropyl] acetamide N- [3- (trans- 4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-hydroxy-1-methylpropyl] acetamide (370 mg) in acetonitrile (5 mL) in a desmartin reagent (578 mg ) Was added and stirred at room temperature for 30 minutes. A sodium sulfite aqueous solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and dried over anhydrous magnesium sulfate, and then the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the resulting solid was washed with diethyl ether to give the title compound (310 mg).
N-[3-(trans-4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロヘキシル)-3-ヒドロキシ-1-メチルプロピル]アセトアミド(370 mg)のアセトニトリル(5 mL)溶液にデスマーチン試薬(578 mg)を加えて、室温で30分間撹拌した。反応混合物に亜硫酸ナトリウム水溶液を加えて、酢酸エチルで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して得られた固体をジエチルエーテルで洗浄して、標題化合物(310 mg)を得た。 J) N- [3- (trans-4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -1-methyl-3-oxopropyl] acetamide N- [3- (trans- 4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclohexyl) -3-hydroxy-1-methylpropyl] acetamide (370 mg) in acetonitrile (5 mL) in a desmartin reagent (578 mg ) Was added and stirred at room temperature for 30 minutes. A sodium sulfite aqueous solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and dried over anhydrous magnesium sulfate, and then the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate), and the resulting solid was washed with diethyl ether to give the title compound (310 mg).
実施例85
N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(2S)-テトラヒドロフラン-2-イルメトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 85
N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2S) -tetrahydrofuran-2-ylmethoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(2S)-テトラヒドロフラン-2-イルメトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 85
N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2S) -tetrahydrofuran-2-ylmethoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
A) (2S)-テトラヒドロフラン-2-イルメチル メタンスルホナート
(2S)-テトラヒドロフラン-2-イルメタノールを用い、実施例38の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 1.60-1.82 (1H, m), 1.86-2.13 (3H, m), 3.07 (3H, s), 3.85 (2H, d, J = 17.0 Hz), 4.04-4.36 (3H, m). A) (2S) -Tetrahydrofuran-2-ylmethyl methanesulfonate Using (2S) -tetrahydrofuran-2-ylmethanol, the title compound was obtained in the same manner as in Step A of Example 38.
1 H NMR (300 MHz, CDCl 3 ) δ 1.60-1.82 (1H, m), 1.86-2.13 (3H, m), 3.07 (3H, s), 3.85 (2H, d, J = 17.0 Hz), 4.04- 4.36 (3H, m).
(2S)-テトラヒドロフラン-2-イルメタノールを用い、実施例38の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 1.60-1.82 (1H, m), 1.86-2.13 (3H, m), 3.07 (3H, s), 3.85 (2H, d, J = 17.0 Hz), 4.04-4.36 (3H, m). A) (2S) -Tetrahydrofuran-2-ylmethyl methanesulfonate Using (2S) -tetrahydrofuran-2-ylmethanol, the title compound was obtained in the same manner as in Step A of Example 38.
1 H NMR (300 MHz, CDCl 3 ) δ 1.60-1.82 (1H, m), 1.86-2.13 (3H, m), 3.07 (3H, s), 3.85 (2H, d, J = 17.0 Hz), 4.04- 4.36 (3H, m).
B) N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(2S)-テトラヒドロフラン-2-イルメトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
(2S)-テトラヒドロフラン-2-イルメチル メタンスルホナートを用い、実施例35の工程Dと同様の方法により標題化合物を得た。 B) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2S) -tetrahydrofuran-2-ylmethoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] The title compound was obtained in the same manner as in Step D of Example 35 using acetamide (2S) -tetrahydrofuran-2-ylmethyl methanesulfonate.
(2S)-テトラヒドロフラン-2-イルメチル メタンスルホナートを用い、実施例35の工程Dと同様の方法により標題化合物を得た。 B) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2S) -tetrahydrofuran-2-ylmethoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] The title compound was obtained in the same manner as in Step D of Example 35 using acetamide (2S) -tetrahydrofuran-2-ylmethyl methanesulfonate.
実施例86
N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(2R)-テトラヒドロフラン-2-イルメトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 86
N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2R) -tetrahydrofuran-2-ylmethoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(2R)-テトラヒドロフラン-2-イルメトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド Example 86
N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2R) -tetrahydrofuran-2-ylmethoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide
A) (2R)-テトラヒドロフラン-2-イルメチル メタンスルホナート
(2R)-テトラヒドロフラン-2-イルメタノールを用い、実施例38の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 1.60-1.78 (1H, m), 1.83-2.14 (3H, m), 3.04-3.11 (3H, m), 3.64-3.97 (2H, m), 4.06-4.37 (3H, m). A) (2R) -Tetrahydrofuran-2-ylmethyl methanesulfonate The title compound was obtained in the same manner as in Step A of Example 38 using (2R) -tetrahydrofuran-2-ylmethanol.
1 H NMR (300 MHz, CDCl 3 ) δ 1.60-1.78 (1H, m), 1.83-2.14 (3H, m), 3.04-3.11 (3H, m), 3.64-3.97 (2H, m), 4.06-4.37 (3H, m).
(2R)-テトラヒドロフラン-2-イルメタノールを用い、実施例38の工程Aと同様の方法により標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 1.60-1.78 (1H, m), 1.83-2.14 (3H, m), 3.04-3.11 (3H, m), 3.64-3.97 (2H, m), 4.06-4.37 (3H, m). A) (2R) -Tetrahydrofuran-2-ylmethyl methanesulfonate The title compound was obtained in the same manner as in Step A of Example 38 using (2R) -tetrahydrofuran-2-ylmethanol.
1 H NMR (300 MHz, CDCl 3 ) δ 1.60-1.78 (1H, m), 1.83-2.14 (3H, m), 3.04-3.11 (3H, m), 3.64-3.97 (2H, m), 4.06-4.37 (3H, m).
B) N-[(1S)-2-{[trans-4-({2-フルオロ-4-[(2R)-テトラヒドロフラン-2-イルメトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド
(2R)-テトラヒドロフラン-2-イルメチル メタンスルホナートを用い、実施例35の工程Dと同様の方法により標題化合物を得た。 B) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2R) -tetrahydrofuran-2-ylmethoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] The title compound was obtained in the same manner as in Step D of Example 35 using acetamide (2R) -tetrahydrofuran-2-ylmethyl methanesulfonate.
(2R)-テトラヒドロフラン-2-イルメチル メタンスルホナートを用い、実施例35の工程Dと同様の方法により標題化合物を得た。 B) N-[(1S) -2-{[trans-4-({2-Fluoro-4-[(2R) -tetrahydrofuran-2-ylmethoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] The title compound was obtained in the same manner as in Step D of Example 35 using acetamide (2R) -tetrahydrofuran-2-ylmethyl methanesulfonate.
実施例87
N-[(1S)-2-({trans-4-[(2-フルオロ-4-プロポキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミドおよび1-ヨードプロパンを用い、実施例7の工程Eと同様の方法により標題化合物を得た。 Example 87
N-[(1S) -2-({trans-4-[(2-fluoro-4-propoxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide N-[(1S) -2-({ trans-4-[(2-Fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide and 1-iodopropane and the title compound by a method similar to Example 7, step E. Obtained.
N-[(1S)-2-({trans-4-[(2-フルオロ-4-プロポキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミドおよび1-ヨードプロパンを用い、実施例7の工程Eと同様の方法により標題化合物を得た。 Example 87
N-[(1S) -2-({trans-4-[(2-fluoro-4-propoxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide N-[(1S) -2-({ trans-4-[(2-Fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide and 1-iodopropane and the title compound by a method similar to Example 7, step E. Obtained.
実施例88
N-[(1S)-2-({trans-4-[(4-ブトキシ-2-フルオロベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミドおよび1-ヨードブタンを用い、実施例7の工程Eと同様の方法により標題化合物を得た。 Example 88
N-[(1S) -2-({trans-4-[(4-butoxy-2-fluorobenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide N-[(1S) -2-({ The title compound was obtained in the same manner as in Step E of Example 7 using trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide and 1-iodobutane. It was.
N-[(1S)-2-({trans-4-[(4-ブトキシ-2-フルオロベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミド
N-[(1S)-2-({trans-4-[(2-フルオロ-4-ヒドロキシベンジル)オキシ]シクロヘキシル}オキシ)-1-メチルエチル]アセトアミドおよび1-ヨードブタンを用い、実施例7の工程Eと同様の方法により標題化合物を得た。 Example 88
N-[(1S) -2-({trans-4-[(4-butoxy-2-fluorobenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide N-[(1S) -2-({ The title compound was obtained in the same manner as in Step E of Example 7 using trans-4-[(2-fluoro-4-hydroxybenzyl) oxy] cyclohexyl} oxy) -1-methylethyl] acetamide and 1-iodobutane. It was.
実施例89
N-{2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-1-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 89
N- {2-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -1-methylcyclohexyl) oxy] -1-methylethyl} acetamide
N-{2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-1-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド Example 89
N- {2-[(4-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -1-methylcyclohexyl) oxy] -1-methylethyl} acetamide
A) エチル [(8-メチル-1,4-ジオキサスピロ[4.5]デカ-8-イル)オキシ]アセタート
8-メチル-1,4-ジオキサスピロ[4.5]デカン-8-オール(5.51 g)およびロジウム(II) ジアセタート二量体(1.42 g)のトルエン(80 mL)溶液にエチル ジアゾアセタート(36.5 g)を80℃でゆっくり加えた。反応混合物を110℃で1時間撹拌後、室温に放冷し、水を加えて酢酸エチルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(6.08 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.08 (3H, s), 1.17-1.22 (3H, m), 1.33-1.55 (4H, m), 1.61-1.80 (4H, m), 3.83 (4H, s), 3.97 (2H, s), 4.10 (2H, q, J = 7.1 Hz). A) Ethyl [(8-methyl-1,4-dioxaspiro [4.5] dec-8-yl) oxy] acetate 8-methyl-1,4-dioxaspiro [4.5] decan-8-ol (5.51 g) and rhodium ( II) Ethyl diazoacetate (36.5 g) was slowly added to a solution of diacetate dimer (1.42 g) in toluene (80 mL) at 80 ° C. The reaction mixture was stirred at 110 ° C. for 1 hr, allowed to cool to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound (6.08 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.08 (3H, s), 1.17-1.22 (3H, m), 1.33-1.55 (4H, m), 1.61-1.80 (4H, m), 3.83 (4H , s), 3.97 (2H, s), 4.10 (2H, q, J = 7.1 Hz).
8-メチル-1,4-ジオキサスピロ[4.5]デカン-8-オール(5.51 g)およびロジウム(II) ジアセタート二量体(1.42 g)のトルエン(80 mL)溶液にエチル ジアゾアセタート(36.5 g)を80℃でゆっくり加えた。反応混合物を110℃で1時間撹拌後、室温に放冷し、水を加えて酢酸エチルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(6.08 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.08 (3H, s), 1.17-1.22 (3H, m), 1.33-1.55 (4H, m), 1.61-1.80 (4H, m), 3.83 (4H, s), 3.97 (2H, s), 4.10 (2H, q, J = 7.1 Hz). A) Ethyl [(8-methyl-1,4-dioxaspiro [4.5] dec-8-yl) oxy] acetate 8-methyl-1,4-dioxaspiro [4.5] decan-8-ol (5.51 g) and rhodium ( II) Ethyl diazoacetate (36.5 g) was slowly added to a solution of diacetate dimer (1.42 g) in toluene (80 mL) at 80 ° C. The reaction mixture was stirred at 110 ° C. for 1 hr, allowed to cool to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound (6.08 g).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.08 (3H, s), 1.17-1.22 (3H, m), 1.33-1.55 (4H, m), 1.61-1.80 (4H, m), 3.83 (4H , s), 3.97 (2H, s), 4.10 (2H, q, J = 7.1 Hz).
B) 4-メチル-4-[2-(モルホリン-4-イル)-2-オキソエトキシ]シクロヘキサノン
エチル [(8-メチル-1,4-ジオキサスピロ[4.5]デカ-8-イル)オキシ]アセタート(6.08 g)および6N塩酸(15.7 mL)のTHF(60 mL)溶液を60℃で4時間撹拌した。反応混合物を室温に放冷後、酢酸エチルと水で抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣にモルホリン(2.05 mL)、WSC(4.51 g)、HOBt (3.61 g)およびDMF(50 mL)を加えた。反応混合物を室温で終夜撹拌し、酢酸エチルと水で抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(438 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.21 (3H, s), 1.59-1.85 (4H, m), 1.94-2.14 (4H, m), 3.35-3.64 (8H, m), 4.12 (2H, s). B) 4-Methyl-4- [2- (morpholin-4-yl) -2-oxoethoxy] cyclohexanone ethyl [(8-methyl-1,4-dioxaspiro [4.5] dec-8-yl) oxy] acetate ( A solution of 6.08 g) and 6N hydrochloric acid (15.7 mL) in THF (60 mL) was stirred at 60 ° C. for 4 hours. The reaction mixture was allowed to cool to room temperature, and extracted with ethyl acetate and water. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. To the obtained residue, morpholine (2.05 mL), WSC (4.51 g), HOBt (3.61 g) and DMF (50 mL) were added. The reaction mixture was stirred at room temperature overnight and extracted with ethyl acetate and water. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound (438 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.21 (3H, s), 1.59-1.85 (4H, m), 1.94-2.14 (4H, m), 3.35-3.64 (8H, m), 4.12 (2H , s).
エチル [(8-メチル-1,4-ジオキサスピロ[4.5]デカ-8-イル)オキシ]アセタート(6.08 g)および6N塩酸(15.7 mL)のTHF(60 mL)溶液を60℃で4時間撹拌した。反応混合物を室温に放冷後、酢酸エチルと水で抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣にモルホリン(2.05 mL)、WSC(4.51 g)、HOBt (3.61 g)およびDMF(50 mL)を加えた。反応混合物を室温で終夜撹拌し、酢酸エチルと水で抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(438 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.21 (3H, s), 1.59-1.85 (4H, m), 1.94-2.14 (4H, m), 3.35-3.64 (8H, m), 4.12 (2H, s). B) 4-Methyl-4- [2- (morpholin-4-yl) -2-oxoethoxy] cyclohexanone ethyl [(8-methyl-1,4-dioxaspiro [4.5] dec-8-yl) oxy] acetate ( A solution of 6.08 g) and 6N hydrochloric acid (15.7 mL) in THF (60 mL) was stirred at 60 ° C. for 4 hours. The reaction mixture was allowed to cool to room temperature, and extracted with ethyl acetate and water. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. To the obtained residue, morpholine (2.05 mL), WSC (4.51 g), HOBt (3.61 g) and DMF (50 mL) were added. The reaction mixture was stirred at room temperature overnight and extracted with ethyl acetate and water. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound (438 mg).
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.21 (3H, s), 1.59-1.85 (4H, m), 1.94-2.14 (4H, m), 3.35-3.64 (8H, m), 4.12 (2H , s).
C) 4-メチル-4-[2-(モルホリン-4-イル)-2-オキソエトキシ]シクロヘキサノール
4-メチル-4-[2-(モルホリン-4-イル)-2-オキソエトキシ]シクロヘキサノン(438 mg)を用い、実施例4の工程Cと同様の手法により、標題化合物(85 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.06 (3H, s), 1.25-1.80 (8H, m), 3.33-3.68 (9H, m), 3.90-3.98 (2H, m), 4.29-4.48 (1H, m). C) 4-methyl-4- [2- (morpholin-4-yl) -2-oxoethoxy] cyclohexanol 4-methyl-4- [2- (morpholin-4-yl) -2-oxoethoxy] cyclohexanone ( 438 mg) and the title compound (85 mg) was obtained in the same manner as in Step C of Example 4.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.06 (3H, s), 1.25-1.80 (8H, m), 3.33-3.68 (9H, m), 3.90-3.98 (2H, m), 4.29-4.48 (1H, m).
4-メチル-4-[2-(モルホリン-4-イル)-2-オキソエトキシ]シクロヘキサノン(438 mg)を用い、実施例4の工程Cと同様の手法により、標題化合物(85 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.06 (3H, s), 1.25-1.80 (8H, m), 3.33-3.68 (9H, m), 3.90-3.98 (2H, m), 4.29-4.48 (1H, m). C) 4-methyl-4- [2- (morpholin-4-yl) -2-oxoethoxy] cyclohexanol 4-methyl-4- [2- (morpholin-4-yl) -2-oxoethoxy] cyclohexanone ( 438 mg) and the title compound (85 mg) was obtained in the same manner as in Step C of Example 4.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.06 (3H, s), 1.25-1.80 (8H, m), 3.33-3.68 (9H, m), 3.90-3.98 (2H, m), 4.29-4.48 (1H, m).
D) 4-{[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-1-メチルシクロヘキシル)オキシ]アセチル}モルホリン
4-メチル-4-[2-(モルホリン-4-イル)-2-オキソエトキシ]シクロヘキサノール(85 mg)を用い、実施例2の工程Fと同様の手法により、標題化合物(43 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 0.27-0.41 (2H, m), 0.56-0.74 (2H, m), 1.10-1.20 (3H, m), 1.27-1.38 (2H, m), 1.49-1.97 (7H, m), 2.33-2.52 (1H, m), 3.23-3.39 (1H, m), 3.55-3.73 (8H, m), 3.77 (2H, d, J = 7.2 Hz), 3.98-4.07 (2H, m), 4.51 (1H, s), 6.53-6.72 (2H, m), 7.26-7.32 (1H, m). D) 4-{[(4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} -1-methylcyclohexyl) oxy] acetyl} morpholine 4-methyl-4- [2- (morpholine-4 The title compound (43 mg) was obtained in the same manner as in Step F of Example 2 using -yl) -2-oxoethoxy] cyclohexanol (85 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.27-0.41 (2H, m), 0.56-0.74 (2H, m), 1.10-1.20 (3H, m), 1.27-1.38 (2H, m), 1.49-1.97 (7H, m), 2.33-2.52 (1H, m), 3.23-3.39 (1H, m), 3.55-3.73 (8H, m), 3.77 (2H, d, J = 7.2 Hz), 3.98-4.07 (2H , m), 4.51 (1H, s), 6.53-6.72 (2H, m), 7.26-7.32 (1H, m).
4-メチル-4-[2-(モルホリン-4-イル)-2-オキソエトキシ]シクロヘキサノール(85 mg)を用い、実施例2の工程Fと同様の手法により、標題化合物(43 mg)を得た。
1H NMR (300 MHz, CDCl3) δ 0.27-0.41 (2H, m), 0.56-0.74 (2H, m), 1.10-1.20 (3H, m), 1.27-1.38 (2H, m), 1.49-1.97 (7H, m), 2.33-2.52 (1H, m), 3.23-3.39 (1H, m), 3.55-3.73 (8H, m), 3.77 (2H, d, J = 7.2 Hz), 3.98-4.07 (2H, m), 4.51 (1H, s), 6.53-6.72 (2H, m), 7.26-7.32 (1H, m). D) 4-{[(4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} -1-methylcyclohexyl) oxy] acetyl} morpholine 4-methyl-4- [2- (morpholine-4 The title compound (43 mg) was obtained in the same manner as in Step F of Example 2 using -yl) -2-oxoethoxy] cyclohexanol (85 mg).
1 H NMR (300 MHz, CDCl 3 ) δ 0.27-0.41 (2H, m), 0.56-0.74 (2H, m), 1.10-1.20 (3H, m), 1.27-1.38 (2H, m), 1.49-1.97 (7H, m), 2.33-2.52 (1H, m), 3.23-3.39 (1H, m), 3.55-3.73 (8H, m), 3.77 (2H, d, J = 7.2 Hz), 3.98-4.07 (2H , m), 4.51 (1H, s), 6.53-6.72 (2H, m), 7.26-7.32 (1H, m).
E) N-{2-[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-1-メチルシクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
4-{[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-1-メチルシクロヘキシル)オキシ]アセチル}モルホリン(43 mg)とTHF(4 mL)の混合物に、氷冷下でメチルマグネシウムブロミドのTHF溶液(1 M, 0.2 mL)を加えた。氷冷下で1時間撹拌後、反応混合物を酢酸エチルと1M塩酸で抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣を用い、実施例4の工程C、実施例21の工程Dおよび工程Eと同様に手法により、標題化合物(3.5 mg)を得た。 E) N- {2-[(4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} -1-methylcyclohexyl) oxy] -1-methylethyl} acetamide 4-{[(4- To a mixture of {[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -1-methylcyclohexyl) oxy] acetyl} morpholine (43 mg) and THF (4 mL) was added methylmagnesium bromide under ice-cooling. A THF solution (1 M, 0.2 mL) was added. After stirring for 1 hour under ice cooling, the reaction mixture was extracted with ethyl acetate and 1M hydrochloric acid. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The title compound (3.5 mg) was obtained by the same method as in Step C of Example 4, Step D and Step E of Example 21 using the obtained residue.
4-{[(4-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}-1-メチルシクロヘキシル)オキシ]アセチル}モルホリン(43 mg)とTHF(4 mL)の混合物に、氷冷下でメチルマグネシウムブロミドのTHF溶液(1 M, 0.2 mL)を加えた。氷冷下で1時間撹拌後、反応混合物を酢酸エチルと1M塩酸で抽出した。有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣を用い、実施例4の工程C、実施例21の工程Dおよび工程Eと同様に手法により、標題化合物(3.5 mg)を得た。 E) N- {2-[(4-{[4- (Cyclopropylmethoxy) -2-fluorobenzyl] oxy} -1-methylcyclohexyl) oxy] -1-methylethyl} acetamide 4-{[(4- To a mixture of {[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} -1-methylcyclohexyl) oxy] acetyl} morpholine (43 mg) and THF (4 mL) was added methylmagnesium bromide under ice-cooling. A THF solution (1 M, 0.2 mL) was added. After stirring for 1 hour under ice cooling, the reaction mixture was extracted with ethyl acetate and 1M hydrochloric acid. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The title compound (3.5 mg) was obtained by the same method as in Step C of Example 4, Step D and Step E of Example 21 using the obtained residue.
実施例90
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(2,2,3,3-テトラフルオロプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2,2,3,3-テトラフルオロプロピル トリフルオロメタンスルホナートを用いて実施例35の工程Dと同様の方法により標題化合物を得た。 Example 90
N-{(1S) -2-[(trans-4-{[2-fluoro-4- (2,2,3,3-tetrafluoropropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} The title compound was obtained in the same manner as in Step D of Example 35 using acetamide 2,2,3,3-tetrafluoropropyl trifluoromethanesulfonate.
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(2,2,3,3-テトラフルオロプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2,2,3,3-テトラフルオロプロピル トリフルオロメタンスルホナートを用いて実施例35の工程Dと同様の方法により標題化合物を得た。 Example 90
N-{(1S) -2-[(trans-4-{[2-fluoro-4- (2,2,3,3-tetrafluoropropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} The title compound was obtained in the same manner as in Step D of Example 35 using acetamide 2,2,3,3-tetrafluoropropyl trifluoromethanesulfonate.
実施例91
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(2,2,3,3,3-ペンタフルオロプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2,2,3,3,3-ペンタフルオロプロピル トリフルオロメタンスルホナートを用いて実施例35の工程Dと同様の方法により標題化合物を得た。 Example 91
N-{(1S) -2-[(trans-4-{[2-fluoro-4- (2,2,3,3,3-pentafluoropropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methyl The title compound was obtained in the same manner as in Step D of Example 35 using ethyl} acetamide 2,2,3,3,3-pentafluoropropyl trifluoromethanesulfonate.
N-{(1S)-2-[(trans-4-{[2-フルオロ-4-(2,2,3,3,3-ペンタフルオロプロポキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド
2,2,3,3,3-ペンタフルオロプロピル トリフルオロメタンスルホナートを用いて実施例35の工程Dと同様の方法により標題化合物を得た。 Example 91
N-{(1S) -2-[(trans-4-{[2-fluoro-4- (2,2,3,3,3-pentafluoropropoxy) benzyl] oxy} cyclohexyl) oxy] -1-methyl The title compound was obtained in the same manner as in Step D of Example 35 using ethyl} acetamide 2,2,3,3,3-pentafluoropropyl trifluoromethanesulfonate.
実施例92
N-{2-[(trans-3-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロブチル)オキシ]-1-メチルエチル}アセトアミド Example 92
N- {2-[(trans-3-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclobutyl) oxy] -1-methylethyl} acetamide
N-{2-[(trans-3-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロブチル)オキシ]-1-メチルエチル}アセトアミド Example 92
N- {2-[(trans-3-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclobutyl) oxy] -1-methylethyl} acetamide
A) 3-(ベンジルオキシ)-N-メトキシ-N-メチルシクロブタンカルボキサミド
3-(ベンジルオキシ)シクロブタンカルボン酸(1.76 g)、N,O-ジメチルヒドロキシアミン塩酸塩(1.25 g)、WSC(2.45 g)、HOBt(1.73 g)、トリエチルアミン(3.57 mL)、およびDMF(30 mL)の混合物を室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.93 g)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.91-2.41 (4H, m), 2.88-3.48 (4H, m), 3.56-3.68 (3H, m), 3.85-4.17 (1H, m), 4.37 (2H, s), 7.17-7.40 (5H, m). A) 3- (Benzyloxy) -N-methoxy-N-methylcyclobutanecarboxamide 3- (Benzyloxy) cyclobutanecarboxylic acid (1.76 g), N, O-dimethylhydroxyamine hydrochloride (1.25 g), WSC (2.45 g ), HOBt (1.73 g), triethylamine (3.57 mL), and DMF (30 mL) were stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.93 g) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.91-2.41 (4H, m), 2.88-3.48 (4H, m), 3.56-3.68 (3H, m), 3.85-4.17 (1H, m), 4.37 (2H, s), 7.17-7.40 (5H, m).
3-(ベンジルオキシ)シクロブタンカルボン酸(1.76 g)、N,O-ジメチルヒドロキシアミン塩酸塩(1.25 g)、WSC(2.45 g)、HOBt(1.73 g)、トリエチルアミン(3.57 mL)、およびDMF(30 mL)の混合物を室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(1.93 g)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.91-2.41 (4H, m), 2.88-3.48 (4H, m), 3.56-3.68 (3H, m), 3.85-4.17 (1H, m), 4.37 (2H, s), 7.17-7.40 (5H, m). A) 3- (Benzyloxy) -N-methoxy-N-methylcyclobutanecarboxamide 3- (Benzyloxy) cyclobutanecarboxylic acid (1.76 g), N, O-dimethylhydroxyamine hydrochloride (1.25 g), WSC (2.45 g ), HOBt (1.73 g), triethylamine (3.57 mL), and DMF (30 mL) were stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (1.93 g) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.91-2.41 (4H, m), 2.88-3.48 (4H, m), 3.56-3.68 (3H, m), 3.85-4.17 (1H, m), 4.37 (2H, s), 7.17-7.40 (5H, m).
B) 1-[trans-3-(ベンジルオキシ)シクロブチル]エタノン
3-(ベンジルオキシ)-N-メトキシ-N-メチルシクロブタンカルボキサミド(1.93 g)のTHF(20 mL)溶液にメチルマグネシウムブロミド(1.0 M THF溶液、6.82 mL)を0℃で加え、得られた混合物を室温で1時間撹拌した。反応混合物に1 M塩酸を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(764 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.00-2.18 (5H, m), 2.30-2.41 (2H, m), 3.11-3.25 (1H, m), 3.87-4.08 (1H, m), 4.35 (2H, s), 7.16-7.45 (5H, m). B) 1- [trans-3- (Benzyloxy) cyclobutyl] ethanone 3- (Benzyloxy) -N-methoxy-N-methylcyclobutanecarboxamide (1.93 g) in THF (20 mL) in methylmagnesium bromide (1.0 M THF solution, 6.82 mL) was added at 0 ° C., and the resulting mixture was stirred at room temperature for 1 hour. 1 M hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (764 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 2.00-2.18 (5H, m), 2.30-2.41 (2H, m), 3.11-3.25 (1H, m), 3.87-4.08 (1H, m), 4.35 (2H, s), 7.16-7.45 (5H, m).
3-(ベンジルオキシ)-N-メトキシ-N-メチルシクロブタンカルボキサミド(1.93 g)のTHF(20 mL)溶液にメチルマグネシウムブロミド(1.0 M THF溶液、6.82 mL)を0℃で加え、得られた混合物を室温で1時間撹拌した。反応混合物に1 M塩酸を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(764 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.00-2.18 (5H, m), 2.30-2.41 (2H, m), 3.11-3.25 (1H, m), 3.87-4.08 (1H, m), 4.35 (2H, s), 7.16-7.45 (5H, m). B) 1- [trans-3- (Benzyloxy) cyclobutyl] ethanone 3- (Benzyloxy) -N-methoxy-N-methylcyclobutanecarboxamide (1.93 g) in THF (20 mL) in methylmagnesium bromide (1.0 M THF solution, 6.82 mL) was added at 0 ° C., and the resulting mixture was stirred at room temperature for 1 hour. 1 M hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (764 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 2.00-2.18 (5H, m), 2.30-2.41 (2H, m), 3.11-3.25 (1H, m), 3.87-4.08 (1H, m), 4.35 (2H, s), 7.16-7.45 (5H, m).
C) trans-3-(ベンジルオキシ)シクロブタノール
1-[trans-3-(ベンジルオキシ)シクロブチル]エタノン(764 mg)、m-クロロ過安息香酸(4.48 g)、およびトルエン(20 mL)の混合物を40℃で終夜撹拌した。沈殿物をろ過により除去した後、ろ液に飽和炭酸水素ナトリウム水溶液および飽和チオ硫酸ナトリウム水溶液加え、得られた混合物を酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をTHF(10 mL)、メタノール(10 mL)、および1 M水酸化ナトリウム水溶液(10 mL)と混合し、得られた混合物を室温で2時間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(498 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.95-2.08 (2H, m), 2.11-2.24 (2H, m), 4.07-4.19 (1H, m), 4.21-4.31 (1H, m), 4.33 (2H, s), 4.97 (1H, d, J = 5.3 Hz), 7.20-7.39 (5H, m). C) A mixture of trans-3- (benzyloxy) cyclobutanol 1- [trans-3- (benzyloxy) cyclobutyl] ethanone (764 mg), m-chloroperbenzoic acid (4.48 g), and toluene (20 mL) Was stirred at 40 ° C. overnight. The precipitate was removed by filtration, saturated aqueous sodium hydrogen carbonate solution and saturated aqueous sodium thiosulfate solution were added to the filtrate, and the resulting mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was mixed with THF (10 mL), methanol (10 mL), and 1 M aqueous sodium hydroxide solution (10 mL), and the resulting mixture was stirred at room temperature for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (498 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.95-2.08 (2H, m), 2.11-2.24 (2H, m), 4.07-4.19 (1H, m), 4.21-4.31 (1H, m), 4.33 (2H, s), 4.97 (1H, d, J = 5.3 Hz), 7.20-7.39 (5H, m).
1-[trans-3-(ベンジルオキシ)シクロブチル]エタノン(764 mg)、m-クロロ過安息香酸(4.48 g)、およびトルエン(20 mL)の混合物を40℃で終夜撹拌した。沈殿物をろ過により除去した後、ろ液に飽和炭酸水素ナトリウム水溶液および飽和チオ硫酸ナトリウム水溶液加え、得られた混合物を酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をTHF(10 mL)、メタノール(10 mL)、および1 M水酸化ナトリウム水溶液(10 mL)と混合し、得られた混合物を室温で2時間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(498 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.95-2.08 (2H, m), 2.11-2.24 (2H, m), 4.07-4.19 (1H, m), 4.21-4.31 (1H, m), 4.33 (2H, s), 4.97 (1H, d, J = 5.3 Hz), 7.20-7.39 (5H, m). C) A mixture of trans-3- (benzyloxy) cyclobutanol 1- [trans-3- (benzyloxy) cyclobutyl] ethanone (764 mg), m-chloroperbenzoic acid (4.48 g), and toluene (20 mL) Was stirred at 40 ° C. overnight. The precipitate was removed by filtration, saturated aqueous sodium hydrogen carbonate solution and saturated aqueous sodium thiosulfate solution were added to the filtrate, and the resulting mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was mixed with THF (10 mL), methanol (10 mL), and 1 M aqueous sodium hydroxide solution (10 mL), and the resulting mixture was stirred at room temperature for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (498 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.95-2.08 (2H, m), 2.11-2.24 (2H, m), 4.07-4.19 (1H, m), 4.21-4.31 (1H, m), 4.33 (2H, s), 4.97 (1H, d, J = 5.3 Hz), 7.20-7.39 (5H, m).
D) ({[trans-3-(プロパ-2-エン-1-イルオキシ)シクロブチル]オキシ}メチル)ベンゼン
trans-3-(ベンジルオキシ)シクロブタノール(177 mg)、3-ブロモプロパ-1-エン(180 mg)、t-ブトキシカリウム(167 mg)、およびTHF(4 mL)の混合物を0℃で30分間、続いて室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(181 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.04-2.24 (4H, m), 3.79-3.88 (2H, m), 4.02-4.22 (2H, m), 4.36 (2H, s), 4.97-5.31 (2H, m), 5.69-5.99 (1H, m), 7.19-7.40 (5H, m). D) ({[trans-3- (prop-2-en-1-yloxy) cyclobutyl] oxy} methyl) benzene trans-3- (benzyloxy) cyclobutanol (177 mg), 3-bromoprop-1-ene ( A mixture of 180 mg), t-butoxypotassium (167 mg), and THF (4 mL) was stirred at 0 ° C. for 30 minutes, followed by overnight at room temperature. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (181 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 2.04-2.24 (4H, m), 3.79-3.88 (2H, m), 4.02-4.22 (2H, m), 4.36 (2H, s), 4.97-5.31 (2H, m), 5.69-5.99 (1H, m), 7.19-7.40 (5H, m).
trans-3-(ベンジルオキシ)シクロブタノール(177 mg)、3-ブロモプロパ-1-エン(180 mg)、t-ブトキシカリウム(167 mg)、およびTHF(4 mL)の混合物を0℃で30分間、続いて室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(181 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.04-2.24 (4H, m), 3.79-3.88 (2H, m), 4.02-4.22 (2H, m), 4.36 (2H, s), 4.97-5.31 (2H, m), 5.69-5.99 (1H, m), 7.19-7.40 (5H, m). D) ({[trans-3- (prop-2-en-1-yloxy) cyclobutyl] oxy} methyl) benzene trans-3- (benzyloxy) cyclobutanol (177 mg), 3-bromoprop-1-ene ( A mixture of 180 mg), t-butoxypotassium (167 mg), and THF (4 mL) was stirred at 0 ° C. for 30 minutes, followed by overnight at room temperature. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (181 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 2.04-2.24 (4H, m), 3.79-3.88 (2H, m), 4.02-4.22 (2H, m), 4.36 (2H, s), 4.97-5.31 (2H, m), 5.69-5.99 (1H, m), 7.19-7.40 (5H, m).
E) 1-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}プロパン-2-オール
ボラン-THF錯体(1.2 M THF溶液、3.45 mL)を({[trans-3-(プロパ-2-エン-1-イルオキシ)シクロブチル]オキシ}メチル)ベンゼン(181 mg)のTHF(5 mL)溶液に0℃で加え、得られた混合物を0℃で2時間、続いて室温で終夜撹拌した。反応混合物を0℃に冷却した後、8 M水酸化ナトリウム水溶液(5 mL)および30%過酸化水素水(5 mL)を加え、得られた混合物を室温で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(55.4 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.01 (3H, d, J = 6.0 Hz), 2.00-2.29 (4H, m), 2.95-3.23 (2H, m), 3.59-3.78 (1H, m), 4.02-4.22 (2H, m), 4.35 (2H, s), 4.52 (1H, d, J = 4.9 Hz), 7.15-7.44 (5H, m). E) 1-{[trans-3- (Benzyloxy) cyclobutyl] oxy} propan-2-ol borane-THF complex (1.2 M THF solution, 3.45 mL) ({[trans-3- (prop-2-ene To a solution of 1-yloxy) cyclobutyl] oxy} methyl) benzene (181 mg) in THF (5 mL) at 0 ° C., the resulting mixture was stirred at 0 ° C. for 2 hours and then at room temperature overnight. After the reaction mixture was cooled to 0 ° C., 8 M aqueous sodium hydroxide solution (5 mL) and 30% aqueous hydrogen peroxide (5 mL) were added, and the resulting mixture was stirred at room temperature for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (55.4 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.01 (3H, d, J = 6.0 Hz), 2.00-2.29 (4H, m), 2.95-3.23 (2H, m), 3.59-3.78 (1H, m ), 4.02-4.22 (2H, m), 4.35 (2H, s), 4.52 (1H, d, J = 4.9 Hz), 7.15-7.44 (5H, m).
ボラン-THF錯体(1.2 M THF溶液、3.45 mL)を({[trans-3-(プロパ-2-エン-1-イルオキシ)シクロブチル]オキシ}メチル)ベンゼン(181 mg)のTHF(5 mL)溶液に0℃で加え、得られた混合物を0℃で2時間、続いて室温で終夜撹拌した。反応混合物を0℃に冷却した後、8 M水酸化ナトリウム水溶液(5 mL)および30%過酸化水素水(5 mL)を加え、得られた混合物を室温で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(55.4 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.01 (3H, d, J = 6.0 Hz), 2.00-2.29 (4H, m), 2.95-3.23 (2H, m), 3.59-3.78 (1H, m), 4.02-4.22 (2H, m), 4.35 (2H, s), 4.52 (1H, d, J = 4.9 Hz), 7.15-7.44 (5H, m). E) 1-{[trans-3- (Benzyloxy) cyclobutyl] oxy} propan-2-ol borane-THF complex (1.2 M THF solution, 3.45 mL) ({[trans-3- (prop-2-ene To a solution of 1-yloxy) cyclobutyl] oxy} methyl) benzene (181 mg) in THF (5 mL) at 0 ° C., the resulting mixture was stirred at 0 ° C. for 2 hours and then at room temperature overnight. After the reaction mixture was cooled to 0 ° C., 8 M aqueous sodium hydroxide solution (5 mL) and 30% aqueous hydrogen peroxide (5 mL) were added, and the resulting mixture was stirred at room temperature for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (55.4 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.01 (3H, d, J = 6.0 Hz), 2.00-2.29 (4H, m), 2.95-3.23 (2H, m), 3.59-3.78 (1H, m ), 4.02-4.22 (2H, m), 4.35 (2H, s), 4.52 (1H, d, J = 4.9 Hz), 7.15-7.44 (5H, m).
F) 2-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル メタンスルホナート
1-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}プロパン-2-オール(87.8 mg)、メタンスルホニルクロリド(0.086 mL)、トリエチルアミン(0.155 mL)、およびTHF(2 mL)の混合物を0℃で1時間、続いて室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(95.1 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, d, J = 6.4 Hz), 2.02-2.27 (4H, m), 3.14 (3H, s), 3.33-3.47 (2H, m), 4.06-4.22 (2H, m), 4.36 (2H, s), 4.63-4.88 (1H, m), 7.20-7.43 (5H, m). F) 2-{[trans-3- (Benzyloxy) cyclobutyl] oxy} -1-methylethyl methanesulfonate 1-{[trans-3- (benzyloxy) cyclobutyl] oxy} propan-2-ol (87.8 mg ), Methanesulfonyl chloride (0.086 mL), triethylamine (0.155 mL), and THF (2 mL) were stirred at 0 ° C. for 1 hour, followed by overnight at room temperature. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (95.1 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.28 (3H, d, J = 6.4 Hz), 2.02-2.27 (4H, m), 3.14 (3H, s), 3.33-3.47 (2H, m), 4.06-4.22 (2H, m), 4.36 (2H, s), 4.63-4.88 (1H, m), 7.20-7.43 (5H, m).
1-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}プロパン-2-オール(87.8 mg)、メタンスルホニルクロリド(0.086 mL)、トリエチルアミン(0.155 mL)、およびTHF(2 mL)の混合物を0℃で1時間、続いて室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(95.1 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, d, J = 6.4 Hz), 2.02-2.27 (4H, m), 3.14 (3H, s), 3.33-3.47 (2H, m), 4.06-4.22 (2H, m), 4.36 (2H, s), 4.63-4.88 (1H, m), 7.20-7.43 (5H, m). F) 2-{[trans-3- (Benzyloxy) cyclobutyl] oxy} -1-methylethyl methanesulfonate 1-{[trans-3- (benzyloxy) cyclobutyl] oxy} propan-2-ol (87.8 mg ), Methanesulfonyl chloride (0.086 mL), triethylamine (0.155 mL), and THF (2 mL) were stirred at 0 ° C. for 1 hour, followed by overnight at room temperature. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (95.1 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.28 (3H, d, J = 6.4 Hz), 2.02-2.27 (4H, m), 3.14 (3H, s), 3.33-3.47 (2H, m), 4.06-4.22 (2H, m), 4.36 (2H, s), 4.63-4.88 (1H, m), 7.20-7.43 (5H, m).
G) N-{2-[(trans-3-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロブチル)オキシ]-1-メチルエチル}アセトアミド
2-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル メタンスルホナート(95.1 mg)、アジ化ナトリウム(39.3 mg)、およびジメチルスルホキシド(2 mL)の混合物を100℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をトリフェニルホスフィン(95 mg)、THF(3 mL)、および水(1 mL)と混合し、得られた混合物を90℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣を無水酢酸(0.5 mL)、トリエチルアミン(0.5 mL)、および酢酸エチル(2 mL)と混合し、得られた混合物を60℃で1時間撹拌した。反応混合物を減圧下濃縮した後、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製してN-(2-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル)アセトアミド(95.1 mg、不純物との混合物)を得た。水素雰囲気下、N-(2-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル)アセトアミド(65.5 mg、不純物との混合物)、20%水酸化パラジウム/炭素(50%含水、65 mg)、およびエタノール(2 mL)の混合物を室温で4時間撹拌した。触媒をろ過により除去し、得られたろ液を減圧下濃縮した。得られた残渣を1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2-フルオロベンゼン(306 mg)、硫酸水素テトラブチルアンモニウム(8.02 mg)、50%水酸化ナトリウム水溶液(2 mL)、およびトルエン(4 mL)と混合し、得られた混合物を100℃で3日間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)により得られた残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(24.7 mg)を無色油状化合物として得た。 G) N- {2-[(trans-3-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclobutyl) oxy] -1-methylethyl} acetamide 2-{[trans-3- ( A mixture of (benzyloxy) cyclobutyl] oxy} -1-methylethyl methanesulfonate (95.1 mg), sodium azide (39.3 mg), and dimethyl sulfoxide (2 mL) was stirred at 100 ° C. for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was mixed with triphenylphosphine (95 mg), THF (3 mL), and water (1 mL), and the resulting mixture was stirred at 90 ° C. for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was mixed with acetic anhydride (0.5 mL), triethylamine (0.5 mL), and ethyl acetate (2 mL), and the resulting mixture was stirred at 60 ° C. for 1 hour. After the reaction mixture was concentrated under reduced pressure, the residue was purified by silica gel column chromatography (hexane / ethyl acetate) and purified by N- (2-{[trans-3- (benzyloxy) cyclobutyl] oxy} -1-methylethyl) Acetamide (95.1 mg, mixture with impurities) was obtained. Under hydrogen atmosphere, N- (2-{[trans-3- (benzyloxy) cyclobutyl] oxy} -1-methylethyl) acetamide (65.5 mg, mixture with impurities), 20% palladium hydroxide / carbon (50% A mixture of water (65 mg) and ethanol (2 mL) was stirred at room temperature for 4 hours. The catalyst was removed by filtration, and the obtained filtrate was concentrated under reduced pressure. The obtained residue was 1- (bromomethyl) -4- (cyclopropylmethoxy) -2-fluorobenzene (306 mg), tetrabutylammonium hydrogen sulfate (8.02 mg), 50% aqueous sodium hydroxide solution (2 mL), and Mix with toluene (4 mL) and stir the resulting mixture at 100 ° C. for 3 days. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue obtained by silica gel column chromatography (hexane / ethyl acetate) was fractionated by HPLC (C18, mobile phase: water / acetonitrile (containing 0.1% TFA)), and the obtained fraction was saturated with sodium bicarbonate. Aqueous solution was added and extracted with ethyl acetate. The extract was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound (24.7 mg) as a colorless oily compound.
2-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル メタンスルホナート(95.1 mg)、アジ化ナトリウム(39.3 mg)、およびジメチルスルホキシド(2 mL)の混合物を100℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をトリフェニルホスフィン(95 mg)、THF(3 mL)、および水(1 mL)と混合し、得られた混合物を90℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣を無水酢酸(0.5 mL)、トリエチルアミン(0.5 mL)、および酢酸エチル(2 mL)と混合し、得られた混合物を60℃で1時間撹拌した。反応混合物を減圧下濃縮した後、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製してN-(2-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル)アセトアミド(95.1 mg、不純物との混合物)を得た。水素雰囲気下、N-(2-{[trans-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル)アセトアミド(65.5 mg、不純物との混合物)、20%水酸化パラジウム/炭素(50%含水、65 mg)、およびエタノール(2 mL)の混合物を室温で4時間撹拌した。触媒をろ過により除去し、得られたろ液を減圧下濃縮した。得られた残渣を1-(ブロモメチル)-4-(シクロプロピルメトキシ)-2-フルオロベンゼン(306 mg)、硫酸水素テトラブチルアンモニウム(8.02 mg)、50%水酸化ナトリウム水溶液(2 mL)、およびトルエン(4 mL)と混合し、得られた混合物を100℃で3日間撹拌した。反応混合物に水を加え、ジエチルエーテルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)により得られた残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(24.7 mg)を無色油状化合物として得た。 G) N- {2-[(trans-3-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclobutyl) oxy] -1-methylethyl} acetamide 2-{[trans-3- ( A mixture of (benzyloxy) cyclobutyl] oxy} -1-methylethyl methanesulfonate (95.1 mg), sodium azide (39.3 mg), and dimethyl sulfoxide (2 mL) was stirred at 100 ° C. for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was mixed with triphenylphosphine (95 mg), THF (3 mL), and water (1 mL), and the resulting mixture was stirred at 90 ° C. for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was mixed with acetic anhydride (0.5 mL), triethylamine (0.5 mL), and ethyl acetate (2 mL), and the resulting mixture was stirred at 60 ° C. for 1 hour. After the reaction mixture was concentrated under reduced pressure, the residue was purified by silica gel column chromatography (hexane / ethyl acetate) and purified by N- (2-{[trans-3- (benzyloxy) cyclobutyl] oxy} -1-methylethyl) Acetamide (95.1 mg, mixture with impurities) was obtained. Under hydrogen atmosphere, N- (2-{[trans-3- (benzyloxy) cyclobutyl] oxy} -1-methylethyl) acetamide (65.5 mg, mixture with impurities), 20% palladium hydroxide / carbon (50% A mixture of water (65 mg) and ethanol (2 mL) was stirred at room temperature for 4 hours. The catalyst was removed by filtration, and the obtained filtrate was concentrated under reduced pressure. The obtained residue was 1- (bromomethyl) -4- (cyclopropylmethoxy) -2-fluorobenzene (306 mg), tetrabutylammonium hydrogen sulfate (8.02 mg), 50% aqueous sodium hydroxide solution (2 mL), and Mix with toluene (4 mL) and stir the resulting mixture at 100 ° C. for 3 days. Water was added to the reaction mixture, and the mixture was extracted with diethyl ether. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue obtained by silica gel column chromatography (hexane / ethyl acetate) was fractionated by HPLC (C18, mobile phase: water / acetonitrile (containing 0.1% TFA)), and the obtained fraction was saturated with sodium bicarbonate. Aqueous solution was added and extracted with ethyl acetate. The extract was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound (24.7 mg) as a colorless oily compound.
実施例93
N-{2-[(cis-3-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロブチル)オキシ]-1-メチルエチル}アセトアミド Example 93
N- {2-[(cis-3-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclobutyl) oxy] -1-methylethyl} acetamide
N-{2-[(cis-3-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロブチル)オキシ]-1-メチルエチル}アセトアミド Example 93
N- {2-[(cis-3-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclobutyl) oxy] -1-methylethyl} acetamide
A) 1-[cis-3-(ベンジルオキシ)シクロブチル]エタノン
3-(ベンジルオキシ)-N-メトキシ-N-メチルシクロブタンカルボキサミド(1.93 g)のTHF(20 mL)溶液にメチルマグネシウムブロミド(1.0 M THF溶液、6.82 mL)を0℃で加え、得られた混合物を室温で1時間撹拌した。反応混合物に1 M塩酸を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(682 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.84-2.01 (2H, m), 2.04 (3H, s), 2.26-2.43 (2H, m), 2.71-2.94 (1H, m), 3.84-4.02 (1H, m), 4.35 (2H, s), 7.17-7.42 (5H, m). A) 1- [cis-3- (Benzyloxy) cyclobutyl] ethanone 3- (Benzyloxy) -N-methoxy-N-methylcyclobutanecarboxamide (1.93 g) in THF (20 mL) in methylmagnesium bromide (1.0 M THF solution, 6.82 mL) was added at 0 ° C., and the resulting mixture was stirred at room temperature for 1 hour. 1 M hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (682 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.84-2.01 (2H, m), 2.04 (3H, s), 2.26-2.43 (2H, m), 2.71-2.94 (1H, m), 3.84-4.02 (1H, m), 4.35 (2H, s), 7.17-7.42 (5H, m).
3-(ベンジルオキシ)-N-メトキシ-N-メチルシクロブタンカルボキサミド(1.93 g)のTHF(20 mL)溶液にメチルマグネシウムブロミド(1.0 M THF溶液、6.82 mL)を0℃で加え、得られた混合物を室温で1時間撹拌した。反応混合物に1 M塩酸を加え、酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)で精製して標題化合物(682 mg)を無色油状化合物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.84-2.01 (2H, m), 2.04 (3H, s), 2.26-2.43 (2H, m), 2.71-2.94 (1H, m), 3.84-4.02 (1H, m), 4.35 (2H, s), 7.17-7.42 (5H, m). A) 1- [cis-3- (Benzyloxy) cyclobutyl] ethanone 3- (Benzyloxy) -N-methoxy-N-methylcyclobutanecarboxamide (1.93 g) in THF (20 mL) in methylmagnesium bromide (1.0 M THF solution, 6.82 mL) was added at 0 ° C., and the resulting mixture was stirred at room temperature for 1 hour. 1 M hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (682 mg) as a colorless oily compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.84-2.01 (2H, m), 2.04 (3H, s), 2.26-2.43 (2H, m), 2.71-2.94 (1H, m), 3.84-4.02 (1H, m), 4.35 (2H, s), 7.17-7.42 (5H, m).
B) cis-3-(ベンジルオキシ)シクロブタノール
1-[cis-3-(ベンジルオキシ)シクロブチル]エタノンを用い、実施例92の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.61-1.81 (2H, m), 2.46-2.61 (2H, m), 3.47-3.60 (1H, m), 3.61-3.77 (1H, m), 4.33 (2H, s), 5.00 (1H, d, J = 6.8 Hz), 7.17-7.44 (5H, m). B) cis-3- (Benzyloxy) cyclobutanol The title compound was obtained in the same manner as in Step C of Example 92 using 1- [cis-3- (benzyloxy) cyclobutyl] ethanone.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.61-1.81 (2H, m), 2.46-2.61 (2H, m), 3.47-3.60 (1H, m), 3.61-3.77 (1H, m), 4.33 (2H, s), 5.00 (1H, d, J = 6.8 Hz), 7.17-7.44 (5H, m).
1-[cis-3-(ベンジルオキシ)シクロブチル]エタノンを用い、実施例92の工程Cと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.61-1.81 (2H, m), 2.46-2.61 (2H, m), 3.47-3.60 (1H, m), 3.61-3.77 (1H, m), 4.33 (2H, s), 5.00 (1H, d, J = 6.8 Hz), 7.17-7.44 (5H, m). B) cis-3- (Benzyloxy) cyclobutanol The title compound was obtained in the same manner as in Step C of Example 92 using 1- [cis-3- (benzyloxy) cyclobutyl] ethanone.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.61-1.81 (2H, m), 2.46-2.61 (2H, m), 3.47-3.60 (1H, m), 3.61-3.77 (1H, m), 4.33 (2H, s), 5.00 (1H, d, J = 6.8 Hz), 7.17-7.44 (5H, m).
C) ({[cis-3-(プロパ-2-エン-1-イルオキシ)シクロブチル]オキシ}メチル)ベンゼン
cis-3-(ベンジルオキシ)シクロブタノールを用い、実施例92の工程Dと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.59-1.81 (2H, m), 2.52-2.67 (2H, m), 3.51-3.72 (2H, m), 3.78-3.90 (2H, m), 4.35 (2H, s), 5.07-5.16 (1H, m), 5.17-5.32 (1H, m), 5.76-5.96 (1H, m), 7.20-7.40 (5H, m). C) ({[cis-3- (prop-2-en-1-yloxy) cyclobutyl] oxy} methyl) benzene The same method as step D of Example 92 using cis-3- (benzyloxy) cyclobutanol Gave the title compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.59-1.81 (2H, m), 2.52-2.67 (2H, m), 3.51-3.72 (2H, m), 3.78-3.90 (2H, m), 4.35 (2H, s), 5.07-5.16 (1H, m), 5.17-5.32 (1H, m), 5.76-5.96 (1H, m), 7.20-7.40 (5H, m).
cis-3-(ベンジルオキシ)シクロブタノールを用い、実施例92の工程Dと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.59-1.81 (2H, m), 2.52-2.67 (2H, m), 3.51-3.72 (2H, m), 3.78-3.90 (2H, m), 4.35 (2H, s), 5.07-5.16 (1H, m), 5.17-5.32 (1H, m), 5.76-5.96 (1H, m), 7.20-7.40 (5H, m). C) ({[cis-3- (prop-2-en-1-yloxy) cyclobutyl] oxy} methyl) benzene The same method as step D of Example 92 using cis-3- (benzyloxy) cyclobutanol Gave the title compound.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.59-1.81 (2H, m), 2.52-2.67 (2H, m), 3.51-3.72 (2H, m), 3.78-3.90 (2H, m), 4.35 (2H, s), 5.07-5.16 (1H, m), 5.17-5.32 (1H, m), 5.76-5.96 (1H, m), 7.20-7.40 (5H, m).
D) 1-{[cis-3-(ベンジルオキシ)シクロブチル]オキシ}プロパン-2-オール
({[cis-3-(プロパ-2-エン-1-イルオキシ)シクロブチル]オキシ}メチル)ベンゼンを用い、実施例92の工程Eと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.01 (3H, d, J = 6.0 Hz), 1.53-1.86 (2H, m), 2.52-2.64 (2H, m), 2.95-3.19 (2H, m), 3.44-3.76 (3H, m), 4.35 (2H, s), 4.53 (1H, d, J = 4.5 Hz), 7.23-7.38 (5H, m). D) Using 1-{[cis-3- (benzyloxy) cyclobutyl] oxy} propan-2-ol ({[cis-3- (prop-2-en-1-yloxy) cyclobutyl] oxy} methyl) benzene The title compound was obtained in the same manner as in Step E of Example 92.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.01 (3H, d, J = 6.0 Hz), 1.53-1.86 (2H, m), 2.52-2.64 (2H, m), 2.95-3.19 (2H, m ), 3.44-3.76 (3H, m), 4.35 (2H, s), 4.53 (1H, d, J = 4.5 Hz), 7.23-7.38 (5H, m).
({[cis-3-(プロパ-2-エン-1-イルオキシ)シクロブチル]オキシ}メチル)ベンゼンを用い、実施例92の工程Eと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.01 (3H, d, J = 6.0 Hz), 1.53-1.86 (2H, m), 2.52-2.64 (2H, m), 2.95-3.19 (2H, m), 3.44-3.76 (3H, m), 4.35 (2H, s), 4.53 (1H, d, J = 4.5 Hz), 7.23-7.38 (5H, m). D) Using 1-{[cis-3- (benzyloxy) cyclobutyl] oxy} propan-2-ol ({[cis-3- (prop-2-en-1-yloxy) cyclobutyl] oxy} methyl) benzene The title compound was obtained in the same manner as in Step E of Example 92.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.01 (3H, d, J = 6.0 Hz), 1.53-1.86 (2H, m), 2.52-2.64 (2H, m), 2.95-3.19 (2H, m ), 3.44-3.76 (3H, m), 4.35 (2H, s), 4.53 (1H, d, J = 4.5 Hz), 7.23-7.38 (5H, m).
E) 2-{[cis-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル メタンスルホナート
1-{[cis-3-(ベンジルオキシ)シクロブチル]オキシ}プロパン-2-オールを用い、実施例92の工程Fと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, d, J = 6.4 Hz), 1.66-2.88 (4H, m), 3.14 (3H, s), 3.31-3.82 (4H, m), 4.36 (2H, s), 4.70-4.86 (1H, m), 7.13-7.45 (5H, m). E) 2-{[cis-3- (benzyloxy) cyclobutyl] oxy} -1-methylethyl methanesulfonate 1-{[cis-3- (benzyloxy) cyclobutyl] oxy} propan-2-ol The title compound was obtained in the same manner as in Step 92 of Example 92.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.28 (3H, d, J = 6.4 Hz), 1.66-2.88 (4H, m), 3.14 (3H, s), 3.31-3.82 (4H, m), 4.36 (2H, s), 4.70-4.86 (1H, m), 7.13-7.45 (5H, m).
1-{[cis-3-(ベンジルオキシ)シクロブチル]オキシ}プロパン-2-オールを用い、実施例92の工程Fと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, d, J = 6.4 Hz), 1.66-2.88 (4H, m), 3.14 (3H, s), 3.31-3.82 (4H, m), 4.36 (2H, s), 4.70-4.86 (1H, m), 7.13-7.45 (5H, m). E) 2-{[cis-3- (benzyloxy) cyclobutyl] oxy} -1-methylethyl methanesulfonate 1-{[cis-3- (benzyloxy) cyclobutyl] oxy} propan-2-ol The title compound was obtained in the same manner as in Step 92 of Example 92.
1 H NMR (300 MHz, DMSO-d 6 ) δ 1.28 (3H, d, J = 6.4 Hz), 1.66-2.88 (4H, m), 3.14 (3H, s), 3.31-3.82 (4H, m), 4.36 (2H, s), 4.70-4.86 (1H, m), 7.13-7.45 (5H, m).
F) N-{2-[(cis-3-{[4-(シクロプロピルメトキシ)-2-フルオロベンジル]オキシ}シクロブチル)オキシ]-1-メチルエチル}アセトアミド
2-{[cis-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル メタンスルホナートを用い、実施例92の工程Gと同様の方法により標題化合物を得た。 F) N- {2-[(cis-3-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclobutyl) oxy] -1-methylethyl} acetamide 2-{[cis-3- ( The title compound was obtained in the same manner as in Step G of Example 92 using [benzyloxy) cyclobutyl] oxy} -1-methylethyl methanesulfonate.
2-{[cis-3-(ベンジルオキシ)シクロブチル]オキシ}-1-メチルエチル メタンスルホナートを用い、実施例92の工程Gと同様の方法により標題化合物を得た。 F) N- {2-[(cis-3-{[4- (cyclopropylmethoxy) -2-fluorobenzyl] oxy} cyclobutyl) oxy] -1-methylethyl} acetamide 2-{[cis-3- ( The title compound was obtained in the same manner as in Step G of Example 92 using [benzyloxy) cyclobutyl] oxy} -1-methylethyl methanesulfonate.
表1ないし表13に、実施例化合物の化合物名と構造式に加えて、MSの実測値またはNMRスペクトルのデータを示す。MSの実測値は、通常、ポジティブモード(ESI+)における実測値を示し、ネガティブモード(ESI-)における実測値には、[M-H]-を併記した。また、ポジティブモード(ESI+)において、分子イオンピークにナトリウムイオン(+Na)が付加したフラグメントピークが観測された場合、[M+Na]+を併記した。
Tables 1 to 13 show the measured values of MS or NMR spectrum data in addition to the compound names and structural formulas of the example compounds. The measured value of MS usually indicates the measured value in the positive mode (ESI +), and the measured value in the negative mode (ESI-) is indicated with [MH] - . Further, in the positive mode (ESI +), when a fragment peak in which sodium ion (+ Na) was added to the molecular ion peak was observed, [M + Na] + was also written.


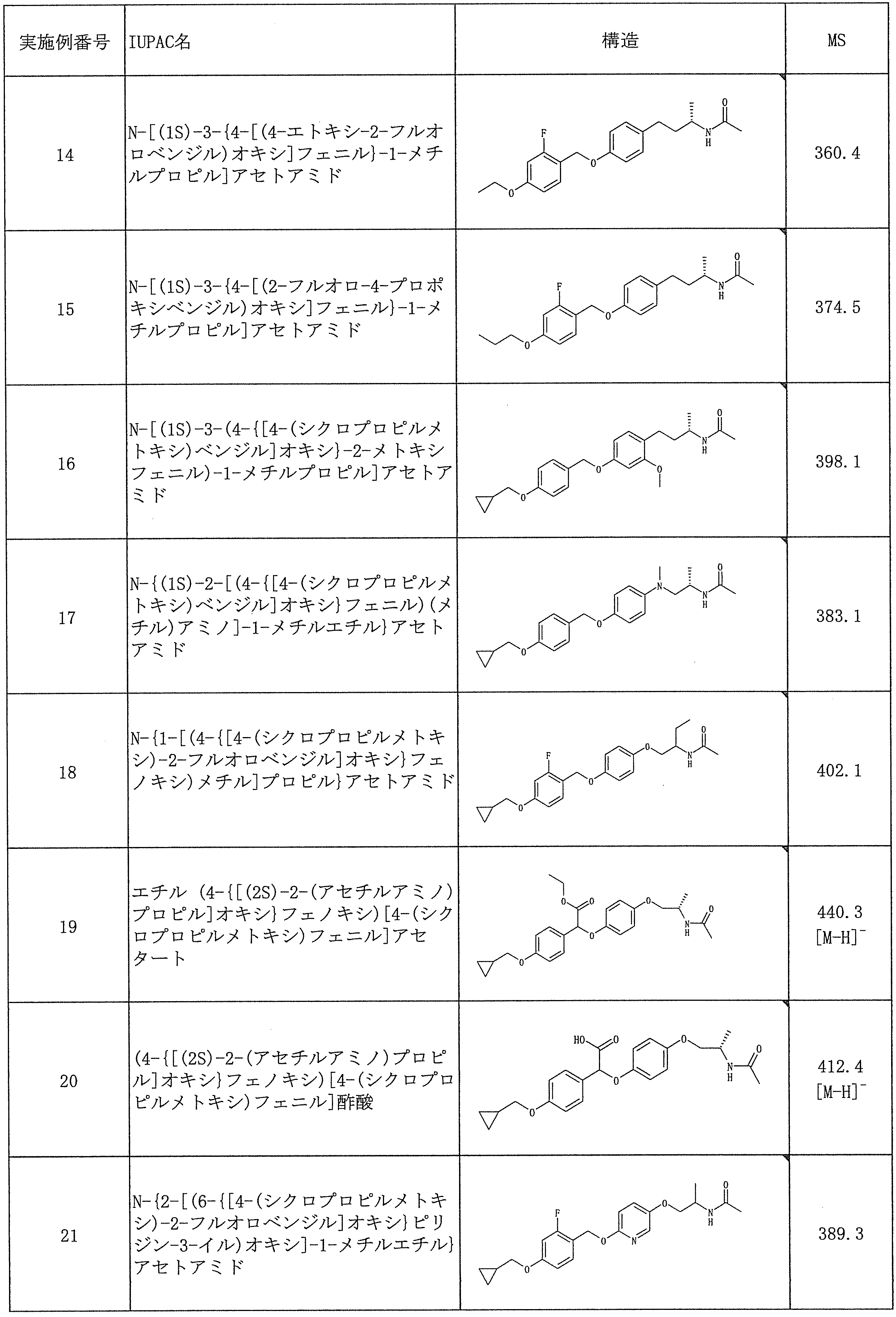










試験例1
以下の方法により、本発明化合物のACC2阻害作用を評価した。
(1)ヒトACC2遺伝子のクローニングと組換えバキュロウイルスの調製
ヒトACC2遺伝子は、ヒト骨格筋cDNAライブラリー(Clontech社)を鋳型とし、以下に示すPrimer 1およびPrimer 2を用いたPCRによりクローニングした。Primer 1およびPrimer 2は、ヒトACC2遺伝子の塩基配列(Genbank Accession U89344)情報より、SalI、XbaI制限酵素認識配列を加えて作製した。
Primer 1: 5’-AAAAGTCGACCCACCATGGTCTTGCTTCTTTGTCTATCTTG-3’(配列番号:1)
Primer 2: 5’-TTTTTCTAGATCAGGTAGAGGCCGGGCTGTCCATG-3’(配列番号:2)
PCRはPyrobest DNA polymerase(タカラバイオ株式会社)を用いて実施した。得られたPCR産物をpT7 Blue vector(Novagen社)にクローニングし、塩基配列を確認後、制限酵素SalI、XbaIで消化した。得られたDNA断片を、制限酵素SalI、XbaIで消化したpFAST-BacHTa(インビトロジェン社)へ挿入し、発現プラスミドACC2/pFAST-BacHTaを作製した。
該発現プラスミドを鋳型とし、ヒトACC2遺伝子の塩基配列(Genbank Accession U89344)情報より作製したPrimer 3(SalI制限酵素認識配列を付加)およびPrimer 4を用いたPCRにより、ミトコンドリア移行配列を除去したACC2を発現させるためのプラスミドを作製した。
Primer 3: 5’-CCAGGTCGACCCGCCAACGGGACTGGGACACAAGG-3’(配列番号:3)
Primer 4: 5’-CGCACTCTCAGTTTCCCGGATTCCC-3’(配列番号:4)
PCRはPyrobest-DNA polymerase(タカラバイオ株式会社)を用いて実施した。得られたPCR産物をpT7 Blue vector(Novagen)にクローニングし、塩基配列を確認後、制限酵素SalI、AflIIで消化した。得られたDNA断片を、制限酵素SalI、AflIIで消化したACC2/pFAST-BacHTaへ挿入し、発現プラスミドACC2mito7/pFAST-BacHTaを作製した。
該発現プラスミドACC2mito7/pFAST-BacHTaおよびBAC-TO-BAC Baculovirus Expression System (インビトロジェン社)を用いて、組換えバキュロウイルスのウイルスストックBAC-ACC2(N terminal deletion(以下Nd))を調製した。 Test example 1
The ACC2 inhibitory action of the compound of the present invention was evaluated by the following method.
(1) Cloning of human ACC2 gene and preparation of recombinant baculovirus The human ACC2 gene was cloned by PCR using Primer 1 and Primer 2 shown below using a human skeletal muscle cDNA library (Clontech) as a template. Primer 1 and Primer 2 were prepared by adding SalI and XbaI restriction enzyme recognition sequences based on the information on the base sequence of the human ACC2 gene (Genbank Accession U89344).
Primer 1: 5'-AAAAGTCGACCCACCATGGTCTTGCTTCTTTGTCTATCTTG-3 '(SEQ ID NO: 1)
Primer 2: 5'-TTTTTCTAGATCAGGTAGAGGCCGGGCTGTCCATG-3 '(SEQ ID NO: 2)
PCR was performed using Pyrobest DNA polymerase (Takara Bio Inc.). The obtained PCR product was cloned into pT7 Blue vector (Novagen), and after confirming the base sequence, digested with restriction enzymes SalI and XbaI. The obtained DNA fragment was inserted into pFAST-BacHTa (Invitrogen) digested with restriction enzymes SalI and XbaI to prepare an expression plasmid ACC2 / pFAST-BacHTa.
Using the expression plasmid as a template, PCR using Primer 3 (added with a SalI restriction enzyme recognition sequence) and Primer 4 prepared from information on the base sequence of the human ACC2 gene (Genbank Accession U89344) and ACC2 from which the mitochondrial translocation sequence was removed A plasmid for expression was prepared.
Primer 3: 5'-CCAGGTCGACCCGCCAACGGGACTGGGACACAAGG-3 '(SEQ ID NO: 3)
Primer 4: 5'-CGCACTCTCAGTTTCCCGGATTCCC-3 '(SEQ ID NO: 4)
PCR was performed using Pyrobest-DNA polymerase (Takara Bio Inc.). The obtained PCR product was cloned into pT7 Blue vector (Novagen). After confirming the nucleotide sequence, it was digested with restriction enzymes SalI and AflII. The obtained DNA fragment was inserted into ACC2 / pFAST-BacHTa digested with restriction enzymes SalI and AflII to prepare an expression plasmid ACC2mito7 / pFAST-BacHTa.
Using the expression plasmids ACC2mito7 / pFAST-BacHTa and BAC-TO-BAC Baculovirus Expression System (Invitrogen), a recombinant baculovirus virus stock BAC-ACC2 (N terminal deletion (hereinafter referred to as Nd)) was prepared.
以下の方法により、本発明化合物のACC2阻害作用を評価した。
(1)ヒトACC2遺伝子のクローニングと組換えバキュロウイルスの調製
ヒトACC2遺伝子は、ヒト骨格筋cDNAライブラリー(Clontech社)を鋳型とし、以下に示すPrimer 1およびPrimer 2を用いたPCRによりクローニングした。Primer 1およびPrimer 2は、ヒトACC2遺伝子の塩基配列(Genbank Accession U89344)情報より、SalI、XbaI制限酵素認識配列を加えて作製した。
Primer 1: 5’-AAAAGTCGACCCACCATGGTCTTGCTTCTTTGTCTATCTTG-3’(配列番号:1)
Primer 2: 5’-TTTTTCTAGATCAGGTAGAGGCCGGGCTGTCCATG-3’(配列番号:2)
PCRはPyrobest DNA polymerase(タカラバイオ株式会社)を用いて実施した。得られたPCR産物をpT7 Blue vector(Novagen社)にクローニングし、塩基配列を確認後、制限酵素SalI、XbaIで消化した。得られたDNA断片を、制限酵素SalI、XbaIで消化したpFAST-BacHTa(インビトロジェン社)へ挿入し、発現プラスミドACC2/pFAST-BacHTaを作製した。
該発現プラスミドを鋳型とし、ヒトACC2遺伝子の塩基配列(Genbank Accession U89344)情報より作製したPrimer 3(SalI制限酵素認識配列を付加)およびPrimer 4を用いたPCRにより、ミトコンドリア移行配列を除去したACC2を発現させるためのプラスミドを作製した。
Primer 3: 5’-CCAGGTCGACCCGCCAACGGGACTGGGACACAAGG-3’(配列番号:3)
Primer 4: 5’-CGCACTCTCAGTTTCCCGGATTCCC-3’(配列番号:4)
PCRはPyrobest-DNA polymerase(タカラバイオ株式会社)を用いて実施した。得られたPCR産物をpT7 Blue vector(Novagen)にクローニングし、塩基配列を確認後、制限酵素SalI、AflIIで消化した。得られたDNA断片を、制限酵素SalI、AflIIで消化したACC2/pFAST-BacHTaへ挿入し、発現プラスミドACC2mito7/pFAST-BacHTaを作製した。
該発現プラスミドACC2mito7/pFAST-BacHTaおよびBAC-TO-BAC Baculovirus Expression System (インビトロジェン社)を用いて、組換えバキュロウイルスのウイルスストックBAC-ACC2(N terminal deletion(以下Nd))を調製した。 Test example 1
The ACC2 inhibitory action of the compound of the present invention was evaluated by the following method.
(1) Cloning of human ACC2 gene and preparation of recombinant baculovirus The human ACC2 gene was cloned by PCR using Primer 1 and Primer 2 shown below using a human skeletal muscle cDNA library (Clontech) as a template. Primer 1 and Primer 2 were prepared by adding SalI and XbaI restriction enzyme recognition sequences based on the information on the base sequence of the human ACC2 gene (Genbank Accession U89344).
Primer 1: 5'-AAAAGTCGACCCACCATGGTCTTGCTTCTTTGTCTATCTTG-3 '(SEQ ID NO: 1)
Primer 2: 5'-TTTTTCTAGATCAGGTAGAGGCCGGGCTGTCCATG-3 '(SEQ ID NO: 2)
PCR was performed using Pyrobest DNA polymerase (Takara Bio Inc.). The obtained PCR product was cloned into pT7 Blue vector (Novagen), and after confirming the base sequence, digested with restriction enzymes SalI and XbaI. The obtained DNA fragment was inserted into pFAST-BacHTa (Invitrogen) digested with restriction enzymes SalI and XbaI to prepare an expression plasmid ACC2 / pFAST-BacHTa.
Using the expression plasmid as a template, PCR using Primer 3 (added with a SalI restriction enzyme recognition sequence) and Primer 4 prepared from information on the base sequence of the human ACC2 gene (Genbank Accession U89344) and ACC2 from which the mitochondrial translocation sequence was removed A plasmid for expression was prepared.
Primer 3: 5'-CCAGGTCGACCCGCCAACGGGACTGGGACACAAGG-3 '(SEQ ID NO: 3)
Primer 4: 5'-CGCACTCTCAGTTTCCCGGATTCCC-3 '(SEQ ID NO: 4)
PCR was performed using Pyrobest-DNA polymerase (Takara Bio Inc.). The obtained PCR product was cloned into pT7 Blue vector (Novagen). After confirming the nucleotide sequence, it was digested with restriction enzymes SalI and AflII. The obtained DNA fragment was inserted into ACC2 / pFAST-BacHTa digested with restriction enzymes SalI and AflII to prepare an expression plasmid ACC2mito7 / pFAST-BacHTa.
Using the expression plasmids ACC2mito7 / pFAST-BacHTa and BAC-TO-BAC Baculovirus Expression System (Invitrogen), a recombinant baculovirus virus stock BAC-ACC2 (N terminal deletion (hereinafter referred to as Nd)) was prepared.
(2)ACC2(Nd)タンパクの調製
SF-9細胞(インビトロジェン社)を昆虫細胞用培地(5%ウシ胎児血清(トレース社)、50 mg/L Gentamicin(和光純薬)、0.1% Pluronic F-68(インビトロジェン社)を含むSf-900IISFM培地(インビトロジェン社))10 Lに1.0×106cells/mLとなるように播種し、Waveバイオリアクター(GEヘルスケア社)を用いて27℃、20 rpm、揺動角度10度、酸素濃度30%で振盪培養した。
培養2日目に組換えバキュロウイルスBAC-ACC2(Nd)を添加し、3日間の培養を行った。培養液を1000×gで10分間遠心分離し、ウィルス感染細胞を得た。該細胞をリン酸生理緩衝液(インビトロジェン社)で洗浄して同条件で遠心分離後、得られた細胞を-80℃で凍結保存した。
凍結保存した細胞を氷中で融解後、Complete Protease Inhibitor(ロシュ社)を添加した10% Glycerol、0.3 M NaCl、1 mM EDTA、25 mM Sodium β-Glycerophosphate、1mM Sodium Orthovanadate を含む25mM HEPES緩衝液 (pH 7.5)900 mLに懸濁した。得られた懸濁液をポリトロンホモジナイザー(キネマティカ社)を用いて20,000 rpm, 30秒の条件で3回ホモジナイズした。得られた細胞破砕液を186,000×g, 60分間の遠心分離により清澄化を行った。上清にAF-chelate 650M Niキレート担体(トーソー社) 5mLを添加し、4℃で1時間ローテートした。1000×gで5分間遠心分離し担体をオープンカラムに移した。50mLの緩衝液A(0.3 M NaClを含む50 mM HEPES(pH 7.5))で洗浄し、更に20 mM Imidazoleを含む緩衝液Aで洗浄した後、250 mM Imidazoleを含む緩衝液Aで溶出した。溶出液を分画分子量50Kのアミコンウルトラ15(ミリポア社)で濃縮した。得られた濃縮液を10m M MgCl2、2 mM Dithiothreitol、10 mM Tripotassium Citrate、0.3 M NaClを含む50 mM HEPES緩衝液(pH 7.5)でHiLoad 26/60 Superdex200 prep gradeゲルろ過カラム(GEヘルスケア社)を用い、ゲルろ過することでACC2(Nd)を得た。得られたACC2(Nd)は-80℃で凍結保存した。 (2) Preparation of ACC2 (Nd) protein SF-9 cells (Invitrogen) were cultured in insect cell medium (5% fetal bovine serum (Trace), 50 mg / L Gentamicin (Wako Pure Chemical Industries), 0.1% Pluronic F- Sf-900IISFM medium containing 68 (Invitrogen Corp.) (Invitrogen Corp.)) 10 L, seeded at 1.0 × 10 6 cells / mL, using a Wave bioreactor (GE Healthcare Corp.), 27 ° C., 20 rpm The shaking culture was performed at a rocking angle of 10 degrees and an oxygen concentration of 30%.
Recombinant baculovirus BAC-ACC2 (Nd) was added on the second day of culture and cultured for 3 days. The culture solution was centrifuged at 1000 × g for 10 minutes to obtain virus-infected cells. The cells were washed with phosphate physiological buffer (Invitrogen) and centrifuged under the same conditions, and the obtained cells were stored frozen at -80 ° C.
After thawing the cryopreserved cells in ice, 25 mM HEPES buffer containing 10% Glycerol, 0.3 M NaCl, 1 mM EDTA, 25 mM Sodium β-Glycerophosphate, and 1 mM Sodium Orthovanadate with Complete Protease Inhibitor (Roche) added ( It was suspended in 900 mL of pH 7.5). The obtained suspension was homogenized three times using a Polytron homogenizer (Kinematica) at 20,000 rpm for 30 seconds. The obtained cell lysate was clarified by centrifugation at 186,000 × g for 60 minutes. To the supernatant, 5 mL of AF-chelate 650M Ni chelate carrier (Tosoh Corporation) was added and rotated at 4 ° C. for 1 hour. The support was transferred to an open column by centrifugation at 1000 × g for 5 minutes. After washing with 50 mL of buffer A (50 mM HEPES (pH 7.5) containing 0.3 M NaCl), and further washing with buffer A containing 20 mM Imidazole, elution was performed with buffer A containing 250 mM Imidazole. The eluate was concentrated with Amicon Ultra 15 (Millipore) having a fractional molecular weight of 50K. The obtained concentrated solution was subjected to HiLoad 26/60 Superdex200 prep grade gel filtration column (GE Healthcare) with 50 mM HEPES buffer (pH 7.5) containing 10 mM MgCl 2 , 2 mM Dithiothreitol, 10 mM Tripotassium Citrate, 0.3 M NaCl. ) To obtain ACC2 (Nd) by gel filtration. The obtained ACC2 (Nd) was stored frozen at −80 ° C.
SF-9細胞(インビトロジェン社)を昆虫細胞用培地(5%ウシ胎児血清(トレース社)、50 mg/L Gentamicin(和光純薬)、0.1% Pluronic F-68(インビトロジェン社)を含むSf-900IISFM培地(インビトロジェン社))10 Lに1.0×106cells/mLとなるように播種し、Waveバイオリアクター(GEヘルスケア社)を用いて27℃、20 rpm、揺動角度10度、酸素濃度30%で振盪培養した。
培養2日目に組換えバキュロウイルスBAC-ACC2(Nd)を添加し、3日間の培養を行った。培養液を1000×gで10分間遠心分離し、ウィルス感染細胞を得た。該細胞をリン酸生理緩衝液(インビトロジェン社)で洗浄して同条件で遠心分離後、得られた細胞を-80℃で凍結保存した。
凍結保存した細胞を氷中で融解後、Complete Protease Inhibitor(ロシュ社)を添加した10% Glycerol、0.3 M NaCl、1 mM EDTA、25 mM Sodium β-Glycerophosphate、1mM Sodium Orthovanadate を含む25mM HEPES緩衝液 (pH 7.5)900 mLに懸濁した。得られた懸濁液をポリトロンホモジナイザー(キネマティカ社)を用いて20,000 rpm, 30秒の条件で3回ホモジナイズした。得られた細胞破砕液を186,000×g, 60分間の遠心分離により清澄化を行った。上清にAF-chelate 650M Niキレート担体(トーソー社) 5mLを添加し、4℃で1時間ローテートした。1000×gで5分間遠心分離し担体をオープンカラムに移した。50mLの緩衝液A(0.3 M NaClを含む50 mM HEPES(pH 7.5))で洗浄し、更に20 mM Imidazoleを含む緩衝液Aで洗浄した後、250 mM Imidazoleを含む緩衝液Aで溶出した。溶出液を分画分子量50Kのアミコンウルトラ15(ミリポア社)で濃縮した。得られた濃縮液を10m M MgCl2、2 mM Dithiothreitol、10 mM Tripotassium Citrate、0.3 M NaClを含む50 mM HEPES緩衝液(pH 7.5)でHiLoad 26/60 Superdex200 prep gradeゲルろ過カラム(GEヘルスケア社)を用い、ゲルろ過することでACC2(Nd)を得た。得られたACC2(Nd)は-80℃で凍結保存した。 (2) Preparation of ACC2 (Nd) protein SF-9 cells (Invitrogen) were cultured in insect cell medium (5% fetal bovine serum (Trace), 50 mg / L Gentamicin (Wako Pure Chemical Industries), 0.1% Pluronic F- Sf-900IISFM medium containing 68 (Invitrogen Corp.) (Invitrogen Corp.)) 10 L, seeded at 1.0 × 10 6 cells / mL, using a Wave bioreactor (GE Healthcare Corp.), 27 ° C., 20 rpm The shaking culture was performed at a rocking angle of 10 degrees and an oxygen concentration of 30%.
Recombinant baculovirus BAC-ACC2 (Nd) was added on the second day of culture and cultured for 3 days. The culture solution was centrifuged at 1000 × g for 10 minutes to obtain virus-infected cells. The cells were washed with phosphate physiological buffer (Invitrogen) and centrifuged under the same conditions, and the obtained cells were stored frozen at -80 ° C.
After thawing the cryopreserved cells in ice, 25 mM HEPES buffer containing 10% Glycerol, 0.3 M NaCl, 1 mM EDTA, 25 mM Sodium β-Glycerophosphate, and 1 mM Sodium Orthovanadate with Complete Protease Inhibitor (Roche) added ( It was suspended in 900 mL of pH 7.5). The obtained suspension was homogenized three times using a Polytron homogenizer (Kinematica) at 20,000 rpm for 30 seconds. The obtained cell lysate was clarified by centrifugation at 186,000 × g for 60 minutes. To the supernatant, 5 mL of AF-chelate 650M Ni chelate carrier (Tosoh Corporation) was added and rotated at 4 ° C. for 1 hour. The support was transferred to an open column by centrifugation at 1000 × g for 5 minutes. After washing with 50 mL of buffer A (50 mM HEPES (pH 7.5) containing 0.3 M NaCl), and further washing with buffer A containing 20 mM Imidazole, elution was performed with buffer A containing 250 mM Imidazole. The eluate was concentrated with Amicon Ultra 15 (Millipore) having a fractional molecular weight of 50K. The obtained concentrated solution was subjected to HiLoad 26/60 Superdex200 prep grade gel filtration column (GE Healthcare) with 50 mM HEPES buffer (pH 7.5) containing 10 mM MgCl 2 , 2 mM Dithiothreitol, 10 mM Tripotassium Citrate, 0.3 M NaCl. ) To obtain ACC2 (Nd) by gel filtration. The obtained ACC2 (Nd) was stored frozen at −80 ° C.
(3)ACC2阻害活性の測定
上記(2)で得られたACC2(Nd)(1.1 mg/ml)を酵素反応用緩衝液(50 mM HEPES(pH 7.5), 10mM MgCl2, 10mM Tripottasium Citrate, 2 mM Dithiothreitol, 0.75mg/ml Fatty acid free BSA)で6.4μg/mlの濃度に希釈後、384 well assay plate(Nunc 265196)の各ウェルに10μlずつ添加した。ついで、各ウェルに、ジメチルスルホキシド(DMSO)に溶解した試験化合物を酵素反応用緩衝液で希釈した溶液5μlずつを添加し、30℃で20分間インキュベーションした。ついで、各ウェルに、基質溶液(50mM KHCO3, 200μM ATP, 200μM Acetyl-CoA)5μlずつを添加し、30℃で20分間反応させた(試験化合物添加群)。
また、試験化合物を添加しないことを除いて、上記と同様の反応を行った(試験化合物非添加群)。
さらに、試験化合物およびAcetyl-CoAを添加しないことを除いて、上記と同様の反応を行った(コントロール群)。
このようにして得られた各反応液にマラカイトグリーン液5μlずつを添加し攪拌することにより反応を停止させた。得られた反応液を室温で20分間放置した後、wallac1420(Perkin Elmer社)を用いて吸光度(620 nm)を測定した。なお、前記マラカイトグリーン液は、A液(0.12%マラカイトグリーン溶液。5 N H2SO4で調製、遮光し4℃で保存)、B液(7.5%アンモニウムモリブデート水溶液。用時調製)およびC液(11% Tween 20水溶液。室温保存)を、A液:B液:C液=100:25:2の割合(容積比)で混合することにより調製した。
ACC2阻害率(%)は以下の計算式に従って求めた。
(1-(試験化合物添加群の吸光度-コントロール群の吸光度)÷(試験化合物非添加群の吸光度-コントロール群の吸光度))×100
阻害活性(IC50値)は、XLfitを用いて用量反応曲線から50%の阻害率を示す化合物濃度を算出した。化合物濃度10μMにおける阻害率が50%に満たない化合物については、>10μMと表記した。
試験化合物10μMにおけるACC2に対する阻害率(%)を表14および表15に示す。 (3) Measurement of ACC2 inhibitory activity ACC2 (Nd) (1.1 mg / ml) obtained in (2) above was used as a buffer for enzyme reaction (50 mM HEPES (pH 7.5), 10 mM MgCl 2 , 10 mM Tripottasium Citrate, 2 After dilution to a concentration of 6.4 μg / ml with mM Dithiothreitol, 0.75 mg / ml Fatty acid free BSA), 10 μl was added to each well of a 384 well assay plate (Nunc 265196). Subsequently, 5 μl each of a solution obtained by diluting a test compound dissolved in dimethyl sulfoxide (DMSO) with an enzyme reaction buffer was added to each well and incubated at 30 ° C. for 20 minutes. Next, 5 μl of a substrate solution (50 mM KHCO 3 , 200 μM ATP, 200 μM Acetyl-CoA) was added to each well and reacted at 30 ° C. for 20 minutes (test compound addition group).
Moreover, except having not added a test compound, reaction similar to the above was performed (test compound non-addition group).
Furthermore, a reaction similar to the above was performed except that the test compound and Acetyl-CoA were not added (control group).
The reaction was stopped by adding 5 μl of malachite green solution to each reaction solution thus obtained and stirring. The obtained reaction solution was allowed to stand at room temperature for 20 minutes, and then the absorbance (620 nm) was measured using wallac1420 (Perkin Elmer). The malachite green solution is solution A (0.12% malachite green solution. Prepared with 5 N H 2 SO 4 , protected from light and stored at 4 ° C.), solution B (7.5% ammonium molybdate aqueous solution, prepared before use) and solution C. (11% Tween 20 aqueous solution, stored at room temperature) was prepared by mixing at a ratio (volume ratio) of liquid A: liquid B: liquid C = 100: 25: 2.
The ACC2 inhibition rate (%) was determined according to the following formula.
(1− (absorbance of test compound added group−absorbance of control group) ÷ (absorbance of test compound non-added group−absorbance of control group)) × 100
For the inhibitory activity (IC 50 value), the compound concentration showing a 50% inhibition rate was calculated from the dose-response curve using XLfit. A compound with an inhibition rate of less than 50% at a compound concentration of 10 μM was expressed as> 10 μM.
Table 14 and Table 15 show the inhibition rate (%) against ACC2 at 10 μM of the test compound.
上記(2)で得られたACC2(Nd)(1.1 mg/ml)を酵素反応用緩衝液(50 mM HEPES(pH 7.5), 10mM MgCl2, 10mM Tripottasium Citrate, 2 mM Dithiothreitol, 0.75mg/ml Fatty acid free BSA)で6.4μg/mlの濃度に希釈後、384 well assay plate(Nunc 265196)の各ウェルに10μlずつ添加した。ついで、各ウェルに、ジメチルスルホキシド(DMSO)に溶解した試験化合物を酵素反応用緩衝液で希釈した溶液5μlずつを添加し、30℃で20分間インキュベーションした。ついで、各ウェルに、基質溶液(50mM KHCO3, 200μM ATP, 200μM Acetyl-CoA)5μlずつを添加し、30℃で20分間反応させた(試験化合物添加群)。
また、試験化合物を添加しないことを除いて、上記と同様の反応を行った(試験化合物非添加群)。
さらに、試験化合物およびAcetyl-CoAを添加しないことを除いて、上記と同様の反応を行った(コントロール群)。
このようにして得られた各反応液にマラカイトグリーン液5μlずつを添加し攪拌することにより反応を停止させた。得られた反応液を室温で20分間放置した後、wallac1420(Perkin Elmer社)を用いて吸光度(620 nm)を測定した。なお、前記マラカイトグリーン液は、A液(0.12%マラカイトグリーン溶液。5 N H2SO4で調製、遮光し4℃で保存)、B液(7.5%アンモニウムモリブデート水溶液。用時調製)およびC液(11% Tween 20水溶液。室温保存)を、A液:B液:C液=100:25:2の割合(容積比)で混合することにより調製した。
ACC2阻害率(%)は以下の計算式に従って求めた。
(1-(試験化合物添加群の吸光度-コントロール群の吸光度)÷(試験化合物非添加群の吸光度-コントロール群の吸光度))×100
阻害活性(IC50値)は、XLfitを用いて用量反応曲線から50%の阻害率を示す化合物濃度を算出した。化合物濃度10μMにおける阻害率が50%に満たない化合物については、>10μMと表記した。
試験化合物10μMにおけるACC2に対する阻害率(%)を表14および表15に示す。 (3) Measurement of ACC2 inhibitory activity ACC2 (Nd) (1.1 mg / ml) obtained in (2) above was used as a buffer for enzyme reaction (50 mM HEPES (pH 7.5), 10 mM MgCl 2 , 10 mM Tripottasium Citrate, 2 After dilution to a concentration of 6.4 μg / ml with mM Dithiothreitol, 0.75 mg / ml Fatty acid free BSA), 10 μl was added to each well of a 384 well assay plate (Nunc 265196). Subsequently, 5 μl each of a solution obtained by diluting a test compound dissolved in dimethyl sulfoxide (DMSO) with an enzyme reaction buffer was added to each well and incubated at 30 ° C. for 20 minutes. Next, 5 μl of a substrate solution (50 mM KHCO 3 , 200 μM ATP, 200 μM Acetyl-CoA) was added to each well and reacted at 30 ° C. for 20 minutes (test compound addition group).
Moreover, except having not added a test compound, reaction similar to the above was performed (test compound non-addition group).
Furthermore, a reaction similar to the above was performed except that the test compound and Acetyl-CoA were not added (control group).
The reaction was stopped by adding 5 μl of malachite green solution to each reaction solution thus obtained and stirring. The obtained reaction solution was allowed to stand at room temperature for 20 minutes, and then the absorbance (620 nm) was measured using wallac1420 (Perkin Elmer). The malachite green solution is solution A (0.12% malachite green solution. Prepared with 5 N H 2 SO 4 , protected from light and stored at 4 ° C.), solution B (7.5% ammonium molybdate aqueous solution, prepared before use) and solution C. (11% Tween 20 aqueous solution, stored at room temperature) was prepared by mixing at a ratio (volume ratio) of liquid A: liquid B: liquid C = 100: 25: 2.
The ACC2 inhibition rate (%) was determined according to the following formula.
(1− (absorbance of test compound added group−absorbance of control group) ÷ (absorbance of test compound non-added group−absorbance of control group)) × 100
For the inhibitory activity (IC 50 value), the compound concentration showing a 50% inhibition rate was calculated from the dose-response curve using XLfit. A compound with an inhibition rate of less than 50% at a compound concentration of 10 μM was expressed as> 10 μM.
Table 14 and Table 15 show the inhibition rate (%) against ACC2 at 10 μM of the test compound.
試験例2
以下の方法により、本発明化合物のACC1阻害作用を評価した。
(1)ヒトACC1遺伝子のクローニングと組換えバキュロウイルスの調製
ヒトACC1遺伝子は、ヒト肝臓cDNAライブラリー(Clontech社)を鋳型とし、以下に示すPrimer 1およびPrimer 2を用いたPCRによりクローニングした。Primer1およびPrimer2は、ヒトACC1遺伝子の塩基配列(Genbank Accession U19822)情報より、SalI、NotI制限酵素認識配列を加えて作製した。
Primer 1: 5’-AAAAGTCGACCCACCATGGATGAACCTTCTCCCTTGGCCC-3’(配列番号:5)
Primer 2: 5’-AAAAGCGGCCGCCTACGTAGAAGGGGAGTCCATAGTG-3’(配列番号:6)
PCRはPyrobest DNA polymerase(タカラバイオ株式会社)を用いて実施した。得られたPCR産物をpT7 Blue vector(Novagen社)にクローニングし、塩基配列を確認後、制限酵素SalI、NotIで消化した。得られたDNA断片を、制限酵素SalI、NotIで消化したpFAST-BacHTc(インビトロジェン社)へ挿入し、発現プラスミドACC1/pFAST-BacHTcを作製した。
該発現プラスミドACC1/pFAST-BacHTcおよびBAC-TO-BAC Baculovirus Expression System (インビトロジェン社)を用いて、組換えバキュロウイルスのウイルスストックBAC-ACC1を調製した。 Test example 2
The ACC1 inhibitory action of the compound of the present invention was evaluated by the following method.
(1) Cloning of human ACC1 gene and preparation of recombinant baculovirus The human ACC1 gene was cloned by PCR using Primer 1 and Primer 2 shown below using a human liver cDNA library (Clontech) as a template. Primer1 and Primer2 were prepared by adding SalI and NotI restriction enzyme recognition sequences based on the information on the base sequence of the human ACC1 gene (Genbank Accession U19822).
Primer 1: 5'-AAAAGTCGACCCACCATGGATGAACCTTCTCCCTTGGCCC-3 '(SEQ ID NO: 5)
Primer 2: 5'-AAAAGCGGCCGCCTACGTAGAAGGGGAGTCCATAGTG-3 '(SEQ ID NO: 6)
PCR was performed using Pyrobest DNA polymerase (Takara Bio Inc.). The obtained PCR product was cloned into pT7 Blue vector (Novagen), and after confirming the nucleotide sequence, it was digested with restriction enzymes SalI and NotI. The obtained DNA fragment was inserted into pFAST-BacHTc (Invitrogen) digested with restriction enzymes SalI and NotI to prepare an expression plasmid ACC1 / pFAST-BacHTc.
Using the expression plasmids ACC1 / pFAST-BacHTc and BAC-TO-BAC Baculovirus Expression System (Invitrogen), a recombinant baculovirus virus stock BAC-ACC1 was prepared.
以下の方法により、本発明化合物のACC1阻害作用を評価した。
(1)ヒトACC1遺伝子のクローニングと組換えバキュロウイルスの調製
ヒトACC1遺伝子は、ヒト肝臓cDNAライブラリー(Clontech社)を鋳型とし、以下に示すPrimer 1およびPrimer 2を用いたPCRによりクローニングした。Primer1およびPrimer2は、ヒトACC1遺伝子の塩基配列(Genbank Accession U19822)情報より、SalI、NotI制限酵素認識配列を加えて作製した。
Primer 1: 5’-AAAAGTCGACCCACCATGGATGAACCTTCTCCCTTGGCCC-3’(配列番号:5)
Primer 2: 5’-AAAAGCGGCCGCCTACGTAGAAGGGGAGTCCATAGTG-3’(配列番号:6)
PCRはPyrobest DNA polymerase(タカラバイオ株式会社)を用いて実施した。得られたPCR産物をpT7 Blue vector(Novagen社)にクローニングし、塩基配列を確認後、制限酵素SalI、NotIで消化した。得られたDNA断片を、制限酵素SalI、NotIで消化したpFAST-BacHTc(インビトロジェン社)へ挿入し、発現プラスミドACC1/pFAST-BacHTcを作製した。
該発現プラスミドACC1/pFAST-BacHTcおよびBAC-TO-BAC Baculovirus Expression System (インビトロジェン社)を用いて、組換えバキュロウイルスのウイルスストックBAC-ACC1を調製した。 Test example 2
The ACC1 inhibitory action of the compound of the present invention was evaluated by the following method.
(1) Cloning of human ACC1 gene and preparation of recombinant baculovirus The human ACC1 gene was cloned by PCR using Primer 1 and Primer 2 shown below using a human liver cDNA library (Clontech) as a template. Primer1 and Primer2 were prepared by adding SalI and NotI restriction enzyme recognition sequences based on the information on the base sequence of the human ACC1 gene (Genbank Accession U19822).
Primer 1: 5'-AAAAGTCGACCCACCATGGATGAACCTTCTCCCTTGGCCC-3 '(SEQ ID NO: 5)
Primer 2: 5'-AAAAGCGGCCGCCTACGTAGAAGGGGAGTCCATAGTG-3 '(SEQ ID NO: 6)
PCR was performed using Pyrobest DNA polymerase (Takara Bio Inc.). The obtained PCR product was cloned into pT7 Blue vector (Novagen), and after confirming the nucleotide sequence, it was digested with restriction enzymes SalI and NotI. The obtained DNA fragment was inserted into pFAST-BacHTc (Invitrogen) digested with restriction enzymes SalI and NotI to prepare an expression plasmid ACC1 / pFAST-BacHTc.
Using the expression plasmids ACC1 / pFAST-BacHTc and BAC-TO-BAC Baculovirus Expression System (Invitrogen), a recombinant baculovirus virus stock BAC-ACC1 was prepared.
(2)ACC1タンパクの調製
SF-9細胞(インビトロジェン社)を昆虫細胞用培地(10%ウシ胎児血清(トレース社)、50mg/L Gentamicin(インビトロジェン社)、0.1% Pluronic F-68(インビトロジェン社)を含むSf-900IISFM培地(インビトロジェン社))1Lに1×106 cells/mLとなるように播種し、2L容マイヤーを用いて27℃、100rpmで振盪培養した。
培養24時間後に組換えバキュロウイルスBAC-ACC1を10mL添加し、さらに3日間の培養を行った。培養液を1000×gで5分間遠心分離し、ウィルス感染細胞を得た。該細胞をリン酸生理緩衝液(インビトロジェン社)で洗浄して同条件で遠心分離後、得られた細胞を-80℃で凍結保存した。
凍結保存した細胞を氷中で融解後、Complete Protease Inhibitor(ベーリンガー社)を添加した10% Glycerol、0.13M NaCl、1mM EDTA、25mM Sodium β-Glycerophosphate、1mM Sodium Orthovanadate を含む25mM HEPES緩衝液 (pH7.5)100 mLに懸濁した。得られた懸濁液をポリトロンホモジナイザー(キネマティカ社)を用いて20,000 rpm, 30秒の条件で3回ホモジナイズした。得られた細胞破砕液を185700×g, 50分間の遠心分離により清澄化後、0.45μmフィルターを用いたろ過を行った。ろ過液をNi-NTA Super Flow Gel (キアゲン社) 12mLを詰めたカラムに流速約5 mL/minで通した。カラムを緩衝液A(0.3M NaClを含む50mM HEPES(pH7.5))で洗浄し、更に20mM Imidazoleを含む緩衝液Aで洗浄した後、100mM Imidazoleを含む緩衝液Aで溶出した。溶出液を分画分子量30Kのビバスピン20(ビバサイエンス社)で濃縮した。得られた濃縮液を10mM MgCl2、2mM Dithiothreitol、10mM Tripotassium Citrate、0.3M NaClを含む50mM HEPES(pH7.5)で平衡化したSephadexG-25(アマシャムバイオサイエンス社)358mLを用いて透析した。透析内液を分画分子量30Kのビバスピン20(ビバサイエンス社)で濃縮した後、濃縮液を0.22μmフィルターでろ過し、ACC1を得た。得られたACC1は-80℃で凍結保存した。 (2) Preparation of ACC1 protein SF-9 cells (Invitrogen) were added to insect cell culture medium (10% fetal bovine serum (Trace), 50 mg / L Gentamicin (Invitrogen), 0.1% Pluronic F-68 (Invitrogen)) Sf-900IISFM medium (Invitrogen) containing 1) was seeded at 1 × 10 6 cells / mL and cultured with shaking at 27 ° C. and 100 rpm using a 2 L Meyer.
After 24 hours of culture, 10 mL of recombinant baculovirus BAC-ACC1 was added, and further cultured for 3 days. The culture solution was centrifuged at 1000 × g for 5 minutes to obtain virus-infected cells. The cells were washed with phosphate physiological buffer (Invitrogen) and centrifuged under the same conditions, and the obtained cells were stored frozen at -80 ° C.
After thawing the cryopreserved cells in ice, 25 mM HEPES buffer solution (pH 7. 5) Suspended in 100 mL. The obtained suspension was homogenized three times using a Polytron homogenizer (Kinematica) at 20,000 rpm for 30 seconds. The obtained cell lysate was clarified by centrifugation at 185700 × g for 50 minutes, followed by filtration using a 0.45 μm filter. The filtrate was passed through a column packed with 12 mL of Ni-NTA Super Flow Gel (Qiagen) at a flow rate of about 5 mL / min. The column was washed with buffer A (50 mM HEPES (pH 7.5) containing 0.3 M NaCl), further washed with buffer A containing 20 mM Imidazole, and then eluted with buffer A containing 100 mM Imidazole. The eluate was concentrated with Vivapine 20 (Viva Science) having a molecular weight cut off of 30K. The resulting concentrated solution was dialyzed using 358 mL of Sephadex G-25 (Amersham Biosciences) equilibrated with 50 mM HEPES (pH 7.5) containing 10 mM MgCl 2 , 2 mM Dithiothreitol, 10 mM Tripotassium Citrate, 0.3 M NaCl. The dialyzed internal solution was concentrated with Vivaspin 20 (Vivascience) having a molecular weight cut off of 30K, and then the concentrated solution was filtered with a 0.22 μm filter to obtain ACC1. The obtained ACC1 was stored frozen at −80 ° C.
SF-9細胞(インビトロジェン社)を昆虫細胞用培地(10%ウシ胎児血清(トレース社)、50mg/L Gentamicin(インビトロジェン社)、0.1% Pluronic F-68(インビトロジェン社)を含むSf-900IISFM培地(インビトロジェン社))1Lに1×106 cells/mLとなるように播種し、2L容マイヤーを用いて27℃、100rpmで振盪培養した。
培養24時間後に組換えバキュロウイルスBAC-ACC1を10mL添加し、さらに3日間の培養を行った。培養液を1000×gで5分間遠心分離し、ウィルス感染細胞を得た。該細胞をリン酸生理緩衝液(インビトロジェン社)で洗浄して同条件で遠心分離後、得られた細胞を-80℃で凍結保存した。
凍結保存した細胞を氷中で融解後、Complete Protease Inhibitor(ベーリンガー社)を添加した10% Glycerol、0.13M NaCl、1mM EDTA、25mM Sodium β-Glycerophosphate、1mM Sodium Orthovanadate を含む25mM HEPES緩衝液 (pH7.5)100 mLに懸濁した。得られた懸濁液をポリトロンホモジナイザー(キネマティカ社)を用いて20,000 rpm, 30秒の条件で3回ホモジナイズした。得られた細胞破砕液を185700×g, 50分間の遠心分離により清澄化後、0.45μmフィルターを用いたろ過を行った。ろ過液をNi-NTA Super Flow Gel (キアゲン社) 12mLを詰めたカラムに流速約5 mL/minで通した。カラムを緩衝液A(0.3M NaClを含む50mM HEPES(pH7.5))で洗浄し、更に20mM Imidazoleを含む緩衝液Aで洗浄した後、100mM Imidazoleを含む緩衝液Aで溶出した。溶出液を分画分子量30Kのビバスピン20(ビバサイエンス社)で濃縮した。得られた濃縮液を10mM MgCl2、2mM Dithiothreitol、10mM Tripotassium Citrate、0.3M NaClを含む50mM HEPES(pH7.5)で平衡化したSephadexG-25(アマシャムバイオサイエンス社)358mLを用いて透析した。透析内液を分画分子量30Kのビバスピン20(ビバサイエンス社)で濃縮した後、濃縮液を0.22μmフィルターでろ過し、ACC1を得た。得られたACC1は-80℃で凍結保存した。 (2) Preparation of ACC1 protein SF-9 cells (Invitrogen) were added to insect cell culture medium (10% fetal bovine serum (Trace), 50 mg / L Gentamicin (Invitrogen), 0.1% Pluronic F-68 (Invitrogen)) Sf-900IISFM medium (Invitrogen) containing 1) was seeded at 1 × 10 6 cells / mL and cultured with shaking at 27 ° C. and 100 rpm using a 2 L Meyer.
After 24 hours of culture, 10 mL of recombinant baculovirus BAC-ACC1 was added, and further cultured for 3 days. The culture solution was centrifuged at 1000 × g for 5 minutes to obtain virus-infected cells. The cells were washed with phosphate physiological buffer (Invitrogen) and centrifuged under the same conditions, and the obtained cells were stored frozen at -80 ° C.
After thawing the cryopreserved cells in ice, 25 mM HEPES buffer solution (pH 7. 5) Suspended in 100 mL. The obtained suspension was homogenized three times using a Polytron homogenizer (Kinematica) at 20,000 rpm for 30 seconds. The obtained cell lysate was clarified by centrifugation at 185700 × g for 50 minutes, followed by filtration using a 0.45 μm filter. The filtrate was passed through a column packed with 12 mL of Ni-NTA Super Flow Gel (Qiagen) at a flow rate of about 5 mL / min. The column was washed with buffer A (50 mM HEPES (pH 7.5) containing 0.3 M NaCl), further washed with buffer A containing 20 mM Imidazole, and then eluted with buffer A containing 100 mM Imidazole. The eluate was concentrated with Vivapine 20 (Viva Science) having a molecular weight cut off of 30K. The resulting concentrated solution was dialyzed using 358 mL of Sephadex G-25 (Amersham Biosciences) equilibrated with 50 mM HEPES (pH 7.5) containing 10 mM MgCl 2 , 2 mM Dithiothreitol, 10 mM Tripotassium Citrate, 0.3 M NaCl. The dialyzed internal solution was concentrated with Vivaspin 20 (Vivascience) having a molecular weight cut off of 30K, and then the concentrated solution was filtered with a 0.22 μm filter to obtain ACC1. The obtained ACC1 was stored frozen at −80 ° C.
(3)ACC1阻害活性の測定
上記(2)で得られたACC1(0.93mg/ml)を酵素反応用緩衝液(50mM HEPES (pH7.5), 10mM MgCl2, 10mM Tripottasium Citrate, 2mM Dithiothreitol, 0.75mg/ml Fatty acid free BSA)で8μg/mlの濃度に希釈後、384 well assay plate(Nunc 265196)の各ウェルに10μlずつ添加した。ついで、各ウェルに、ジメチルスルホキシド(DMSO)に溶解した試験化合物を酵素反応用緩衝液で希釈した溶液5μlずつを添加し、30℃で60分間インキュベーションした。ついで、各ウェルに、基質溶液(50mM KHCO3, 200μM ATP, 200μM Acetyl-CoA)5μlずつを添加し、30℃で20分間反応させた(試験化合物添加群)。
また、試験化合物を添加しないことを除いて、上記と同様の反応を行った(試験化合物非添加群)。
さらに、試験化合物およびAcetyl-CoAを添加しないことを除いて、上記と同様の反応を行った(コントロール群)。
このようにして得られた各反応液にマラカイトグリーン液5μlずつを添加し攪拌することにより反応を停止させた。得られた反応液を室温で20分間放置した後、wallac1420(Perkin Elmer社)を用いて吸光度(620nm)を測定した。なお、前記マラカイトグリーン液は、A液(0.12%マラカイトグリーン溶液。5N H2SO4で調製、遮光し4℃で保存)、B液(7.5%アンモニウムモリブデート水溶液。用時調製)およびC液(11% Tween 20水溶液。室温保存)を、A液:B液:C液=100:25:2の割合(容積比)で混合することにより調製した。
前述の実験例1(3)と同様にして、ACC1阻害率(%)および阻害活性(IC50値)を求めた。 (3) Measurement of ACC1 inhibitory activity ACC1 (0.93 mg / ml) obtained in (2) above was used as a buffer for enzyme reaction (50 mM HEPES (pH 7.5), 10 mM MgCl 2 , 10 mM Tripottasium Citrate, 2 mM Dithiothreitol, 0.75 mg / ml Fatty acid free BSA) was diluted to a concentration of 8 μg / ml, and 10 μl was added to each well of a 384 well assay plate (Nunc 265196). Next, 5 μl each of a test compound dissolved in dimethyl sulfoxide (DMSO) diluted with an enzyme reaction buffer was added to each well and incubated at 30 ° C. for 60 minutes. Next, 5 μl of a substrate solution (50 mM KHCO 3 , 200 μM ATP, 200 μM Acetyl-CoA) was added to each well and reacted at 30 ° C. for 20 minutes (test compound addition group).
Moreover, except having not added a test compound, reaction similar to the above was performed (test compound non-addition group).
Furthermore, a reaction similar to the above was performed except that the test compound and Acetyl-CoA were not added (control group).
The reaction was stopped by adding 5 μl of malachite green solution to each reaction solution thus obtained and stirring. The obtained reaction solution was allowed to stand at room temperature for 20 minutes, and then the absorbance (620 nm) was measured using wallac1420 (Perkin Elmer). The malachite green solution is a solution A (0.12% malachite green solution. Prepared with 5N H 2 SO 4 and stored at 4 ° C. protected from light), solution B (7.5% ammonium molybdate aqueous solution, prepared at the time of use) and solution C. (11% Tween 20 aqueous solution, stored at room temperature) was prepared by mixing at a ratio (volume ratio) of liquid A: liquid B: liquid C = 100: 25: 2.
In the same manner as in Experimental Example 1 (3) described above, the ACC1 inhibition rate (%) and the inhibitory activity (IC 50 value) were determined.
上記(2)で得られたACC1(0.93mg/ml)を酵素反応用緩衝液(50mM HEPES (pH7.5), 10mM MgCl2, 10mM Tripottasium Citrate, 2mM Dithiothreitol, 0.75mg/ml Fatty acid free BSA)で8μg/mlの濃度に希釈後、384 well assay plate(Nunc 265196)の各ウェルに10μlずつ添加した。ついで、各ウェルに、ジメチルスルホキシド(DMSO)に溶解した試験化合物を酵素反応用緩衝液で希釈した溶液5μlずつを添加し、30℃で60分間インキュベーションした。ついで、各ウェルに、基質溶液(50mM KHCO3, 200μM ATP, 200μM Acetyl-CoA)5μlずつを添加し、30℃で20分間反応させた(試験化合物添加群)。
また、試験化合物を添加しないことを除いて、上記と同様の反応を行った(試験化合物非添加群)。
さらに、試験化合物およびAcetyl-CoAを添加しないことを除いて、上記と同様の反応を行った(コントロール群)。
このようにして得られた各反応液にマラカイトグリーン液5μlずつを添加し攪拌することにより反応を停止させた。得られた反応液を室温で20分間放置した後、wallac1420(Perkin Elmer社)を用いて吸光度(620nm)を測定した。なお、前記マラカイトグリーン液は、A液(0.12%マラカイトグリーン溶液。5N H2SO4で調製、遮光し4℃で保存)、B液(7.5%アンモニウムモリブデート水溶液。用時調製)およびC液(11% Tween 20水溶液。室温保存)を、A液:B液:C液=100:25:2の割合(容積比)で混合することにより調製した。
前述の実験例1(3)と同様にして、ACC1阻害率(%)および阻害活性(IC50値)を求めた。 (3) Measurement of ACC1 inhibitory activity ACC1 (0.93 mg / ml) obtained in (2) above was used as a buffer for enzyme reaction (50 mM HEPES (pH 7.5), 10 mM MgCl 2 , 10 mM Tripottasium Citrate, 2 mM Dithiothreitol, 0.75 mg / ml Fatty acid free BSA) was diluted to a concentration of 8 μg / ml, and 10 μl was added to each well of a 384 well assay plate (Nunc 265196). Next, 5 μl each of a test compound dissolved in dimethyl sulfoxide (DMSO) diluted with an enzyme reaction buffer was added to each well and incubated at 30 ° C. for 60 minutes. Next, 5 μl of a substrate solution (50 mM KHCO 3 , 200 μM ATP, 200 μM Acetyl-CoA) was added to each well and reacted at 30 ° C. for 20 minutes (test compound addition group).
Moreover, except having not added a test compound, reaction similar to the above was performed (test compound non-addition group).
Furthermore, a reaction similar to the above was performed except that the test compound and Acetyl-CoA were not added (control group).
The reaction was stopped by adding 5 μl of malachite green solution to each reaction solution thus obtained and stirring. The obtained reaction solution was allowed to stand at room temperature for 20 minutes, and then the absorbance (620 nm) was measured using wallac1420 (Perkin Elmer). The malachite green solution is a solution A (0.12% malachite green solution. Prepared with 5N H 2 SO 4 and stored at 4 ° C. protected from light), solution B (7.5% ammonium molybdate aqueous solution, prepared at the time of use) and solution C. (11% Tween 20 aqueous solution, stored at room temperature) was prepared by mixing at a ratio (volume ratio) of liquid A: liquid B: liquid C = 100: 25: 2.
In the same manner as in Experimental Example 1 (3) described above, the ACC1 inhibition rate (%) and the inhibitory activity (IC 50 value) were determined.
試験化合物のACC2阻害活性およびACC1阻害活性(IC50値)を表16に示す。
Table 16 shows the ACC2 inhibitory activity and ACC1 inhibitory activity (IC 50 value) of the test compounds.



表14、表15および表16から明らかなように、本発明化合物は優れたACC2阻害作用を示した。
As is clear from Tables 14, 15 and 16, the compounds of the present invention showed an excellent ACC2 inhibitory action.
試験例3
5週齢で入荷した雄性F344/Jclラット(日本クレア、東京)にWestern diet (D12079B、Research Diets社)を給餌して3週間以上馴化してから8-10週齢でマロニルCoA含量測定の実験に用いた。マロニルCoA含量測定の実験では、試験化合物投与2-5日前から馴化投与を行い前週の体重を元に投与群間に差が無いように群分けした。試験化合物は予め5 mL/kgの用量となるように0.5% Methyl Cellulose(和光)懸濁液として調製し、試験化合物投与当日の体重(AM8:00-11:00に測定)に基づきPM5:00に強制経口投与した。Vehicle群には溶媒(0.5% Methyl Cellulose)のみを強制経口投与した。試験化合物の投与16時間後(翌日AM9:00)に大腿筋を速やかに摘出し、80-150 mgの細片にしてエッペンドルフチューブに入れて液体窒素で速やかに凍結し、マロニルCoA含量の測定に供するまで組織のまま-80℃で保存した。
マロニルCoA測定用のサンプルホモジネートは、重量を測定した凍結組織をマルチビーズショッカー(MB400U, 安井機械)にかけて破砕し、内部標準物質である[13C3]-マロニルCoAリチウム塩(100 pmol/mL, シグマアルドリッチ)を含む抽出液(6%過塩素酸:酢酸ジ-n-ブチルアンモニウム水溶液(IPC-DBAA, 0.5 mol/L, 東京化成)=1000:1) 500μLを添加し、再度マルチビーズショッカーにかけて作成した。抽出液全量を遠心分離(13,000 rpm, 2 min)し、上清を固相抽出カートリッジ(OASIS HLB 1 cc/30 mg, WAT05882, Waters)に添加して固相抽出を行った。固相抽出カートリッジは、事前に500μLの溶液A (50%アセトニトリル)による活性化、さらに1 mLの溶液B (6%過塩素酸:IPC-DBAA=1000:1)による平衡化を2回行なったものを用いた。試料を固相カートリッジに添加後、超純水1 mLによる洗浄を2回行い、500μLの溶液Aにより溶出した。この溶出液を直接LC/MS/MS分析に使用した。検量線用の試料は、100μLの4%BSA(w/v)水溶液を組織100 mgと見なして上記濃度のマロニルCoAリチウム塩(シグマアルドリッチ)を添加し、組織サンプルと同様に内部標準物質を加えて固相抽出を行った。
LC/MS/MS分析のHPLCはProminence(島津製作所)を分離カラムはCAPCELL PAK C18 AQ (粒子径;3 μm、内径;2.0 mm,長さ;35 mm、資生堂株式会社)を用い、移動相(A) 10 mmol/L 酢酸アンモニウム/IPC-DBAA(100:1, v/v, pH 9.0)と、移動相(B)アセトニトリルのグラジエントモードで実施した。グラジエントプログラムは移動相(B)濃度を0分(分析開始時)5%から2分間で35%、その後2.1分までに100%まで直線的に上昇させ6分後まで100%で送液し、6.1分後に5%まで低下させ、分析終了時間(10分)まで同条件で送液した。スイッチングバルブを用い、分析時間2.0-6.0分の溶出液をMS/MSに導入した。カラム温度は40℃、サンプル注入量は10 μLで分析を実施した。質量分析計はAPI5000(AB Sciex社)を使用し、イオン化モードとしてターボイオンスプレーを用い、陰イオンモードでのselected reaction monitoring(SRM)によりイオンを検出した。イオンスプレーにはゼロエアーを用い、電圧は4.5 kVでイオン化を行った。イオンの衝突誘起分解には窒素を用いた。モニターイオンとしてプリカーサーイオン、フラグメントイオンをそれぞれマロニルCoA;m/z 852.0→m/z 808.0、[13C3]-マロニルCoA;m/z 855.0→m/z 810.0 Daに設定した。定量計算には、内部標準([13C3]-マロニルCoA)およびマロニルCoAのピーク面積比を用い、各濃度の標準検量線の回帰式(1/濃度の重み付け)から各試料のマロニルCoA濃度を算出した。すべての値は平均値±標準偏差で示し、統計解析にはWelch検定を用いた。その結果を表17に示す。 Test example 3
Male F344 / Jcl rats (CLEA Japan, Tokyo) that arrived at 5 weeks old were fed Western diet (D12079B, Research Diets) and acclimatized for more than 3 weeks, and then tested for malonyl-CoA content at 8-10 weeks of age. Used for. In the experiment for measuring the malonyl-CoA content, acclimation was administered 2-5 days before the test compound administration, and the groups were divided so that there was no difference between the administration groups based on the body weight of the previous week. The test compound was prepared in advance as a 0.5% Methyl Cellulose (Wako) suspension at a dose of 5 mL / kg, and PM5: 00 based on the body weight (measured at AM8: 00-11: 00) on the day of test compound administration. Was administered by oral gavage. Only vehicle (0.5% Methyl Cellulose) was forcibly orally administered to the Vehicle group. At 16 hours after administration of the test compound (9:00 AM the next day), the thigh muscle was quickly removed, put into 80-150 mg strips, placed in an Eppendorf tube, and quickly frozen in liquid nitrogen to measure malonyl-CoA content. The tissue was stored at −80 ° C. until use.
The sample homogenate for measuring malonyl CoA was crushed by weighing the frozen tissue on a multi-bead shocker (MB400U, Yasui Machine), and [ 13 C 3 ] -malonyl CoA lithium salt (100 pmol / mL, Add 500μL of extract (6% perchloric acid: di-n-butylammonium acetate aqueous solution (IPC-DBAA, 0.5 mol / L, Tokyo Kasei) = 1000: 1) containing Sigma-Aldrich) Created. The total amount of the extract was centrifuged (13,000 rpm, 2 min), and the supernatant was added to a solid phase extraction cartridge (OASIS HLB 1 cc / 30 mg, WAT05882, Waters) for solid phase extraction. The solid-phase extraction cartridge was previously activated with 500 μL of solution A (50% acetonitrile) and further equilibrated twice with 1 mL of solution B (6% perchloric acid: IPC-DBAA = 1000: 1). A thing was used. After the sample was added to the solid phase cartridge, it was washed twice with 1 mL of ultrapure water and eluted with 500 μL of solution A. This eluate was directly used for LC / MS / MS analysis. For the sample for the calibration curve, 100 μL of 4% BSA (w / v) aqueous solution is regarded as 100 mg of tissue, malonyl-CoA lithium salt (Sigma Aldrich) with the above concentration is added, and internal standard substance is added in the same manner as the tissue sample. The solid phase extraction was performed.
For HPLC of LC / MS / MS analysis, Prominence (Shimadzu Corporation) was used, and the separation column was CAPCELL PAK C18 AQ (particle size: 3 μm, inner diameter: 2.0 mm, length: 35 mm, Shiseido Co., Ltd.), and mobile phase ( A) Gradient mode of 10 mmol / L ammonium acetate / IPC-DBAA (100: 1, v / v, pH 9.0) and mobile phase (B) acetonitrile. The gradient program increases the mobile phase (B) concentration from 0 minutes (at the start of analysis) from 5% to 35% in 2 minutes, then increases linearly to 100% by 2.1 minutes, and then delivers it at 100% until 6 minutes. After 6.1 minutes, the solution was reduced to 5%, and the solution was fed under the same conditions until the analysis end time (10 minutes). Using a switching valve, the eluate having an analysis time of 2.0 to 6.0 minutes was introduced into MS / MS. The analysis was performed at a column temperature of 40 ° C. and a sample injection volume of 10 μL. The mass spectrometer used was API5000 (AB Sciex), turbo ion spray was used as the ionization mode, and ions were detected by selected reaction monitoring (SRM) in the negative ion mode. Ion spray was performed using zero air and the voltage was 4.5 kV. Nitrogen was used for collision-induced decomposition of ions. As a monitor ion, a precursor ion and a fragment ion were set to malonyl CoA; m / z 852.0 → m / z 808.0 and [ 13 C 3 ] -malonyl CoA; m / z 855.0 → m / z 810.0 Da, respectively. For quantitative calculation, the internal area ([ 13 C 3 ] -malonyl-CoA) and the peak area ratio of malonyl-CoA were used. Was calculated. All values are shown as mean ± standard deviation, and Welch test was used for statistical analysis. The results are shown in Table 17.
5週齢で入荷した雄性F344/Jclラット(日本クレア、東京)にWestern diet (D12079B、Research Diets社)を給餌して3週間以上馴化してから8-10週齢でマロニルCoA含量測定の実験に用いた。マロニルCoA含量測定の実験では、試験化合物投与2-5日前から馴化投与を行い前週の体重を元に投与群間に差が無いように群分けした。試験化合物は予め5 mL/kgの用量となるように0.5% Methyl Cellulose(和光)懸濁液として調製し、試験化合物投与当日の体重(AM8:00-11:00に測定)に基づきPM5:00に強制経口投与した。Vehicle群には溶媒(0.5% Methyl Cellulose)のみを強制経口投与した。試験化合物の投与16時間後(翌日AM9:00)に大腿筋を速やかに摘出し、80-150 mgの細片にしてエッペンドルフチューブに入れて液体窒素で速やかに凍結し、マロニルCoA含量の測定に供するまで組織のまま-80℃で保存した。
マロニルCoA測定用のサンプルホモジネートは、重量を測定した凍結組織をマルチビーズショッカー(MB400U, 安井機械)にかけて破砕し、内部標準物質である[13C3]-マロニルCoAリチウム塩(100 pmol/mL, シグマアルドリッチ)を含む抽出液(6%過塩素酸:酢酸ジ-n-ブチルアンモニウム水溶液(IPC-DBAA, 0.5 mol/L, 東京化成)=1000:1) 500μLを添加し、再度マルチビーズショッカーにかけて作成した。抽出液全量を遠心分離(13,000 rpm, 2 min)し、上清を固相抽出カートリッジ(OASIS HLB 1 cc/30 mg, WAT05882, Waters)に添加して固相抽出を行った。固相抽出カートリッジは、事前に500μLの溶液A (50%アセトニトリル)による活性化、さらに1 mLの溶液B (6%過塩素酸:IPC-DBAA=1000:1)による平衡化を2回行なったものを用いた。試料を固相カートリッジに添加後、超純水1 mLによる洗浄を2回行い、500μLの溶液Aにより溶出した。この溶出液を直接LC/MS/MS分析に使用した。検量線用の試料は、100μLの4%BSA(w/v)水溶液を組織100 mgと見なして上記濃度のマロニルCoAリチウム塩(シグマアルドリッチ)を添加し、組織サンプルと同様に内部標準物質を加えて固相抽出を行った。
LC/MS/MS分析のHPLCはProminence(島津製作所)を分離カラムはCAPCELL PAK C18 AQ (粒子径;3 μm、内径;2.0 mm,長さ;35 mm、資生堂株式会社)を用い、移動相(A) 10 mmol/L 酢酸アンモニウム/IPC-DBAA(100:1, v/v, pH 9.0)と、移動相(B)アセトニトリルのグラジエントモードで実施した。グラジエントプログラムは移動相(B)濃度を0分(分析開始時)5%から2分間で35%、その後2.1分までに100%まで直線的に上昇させ6分後まで100%で送液し、6.1分後に5%まで低下させ、分析終了時間(10分)まで同条件で送液した。スイッチングバルブを用い、分析時間2.0-6.0分の溶出液をMS/MSに導入した。カラム温度は40℃、サンプル注入量は10 μLで分析を実施した。質量分析計はAPI5000(AB Sciex社)を使用し、イオン化モードとしてターボイオンスプレーを用い、陰イオンモードでのselected reaction monitoring(SRM)によりイオンを検出した。イオンスプレーにはゼロエアーを用い、電圧は4.5 kVでイオン化を行った。イオンの衝突誘起分解には窒素を用いた。モニターイオンとしてプリカーサーイオン、フラグメントイオンをそれぞれマロニルCoA;m/z 852.0→m/z 808.0、[13C3]-マロニルCoA;m/z 855.0→m/z 810.0 Daに設定した。定量計算には、内部標準([13C3]-マロニルCoA)およびマロニルCoAのピーク面積比を用い、各濃度の標準検量線の回帰式(1/濃度の重み付け)から各試料のマロニルCoA濃度を算出した。すべての値は平均値±標準偏差で示し、統計解析にはWelch検定を用いた。その結果を表17に示す。 Test example 3
Male F344 / Jcl rats (CLEA Japan, Tokyo) that arrived at 5 weeks old were fed Western diet (D12079B, Research Diets) and acclimatized for more than 3 weeks, and then tested for malonyl-CoA content at 8-10 weeks of age. Used for. In the experiment for measuring the malonyl-CoA content, acclimation was administered 2-5 days before the test compound administration, and the groups were divided so that there was no difference between the administration groups based on the body weight of the previous week. The test compound was prepared in advance as a 0.5% Methyl Cellulose (Wako) suspension at a dose of 5 mL / kg, and PM5: 00 based on the body weight (measured at AM8: 00-11: 00) on the day of test compound administration. Was administered by oral gavage. Only vehicle (0.5% Methyl Cellulose) was forcibly orally administered to the Vehicle group. At 16 hours after administration of the test compound (9:00 AM the next day), the thigh muscle was quickly removed, put into 80-150 mg strips, placed in an Eppendorf tube, and quickly frozen in liquid nitrogen to measure malonyl-CoA content. The tissue was stored at −80 ° C. until use.
The sample homogenate for measuring malonyl CoA was crushed by weighing the frozen tissue on a multi-bead shocker (MB400U, Yasui Machine), and [ 13 C 3 ] -malonyl CoA lithium salt (100 pmol / mL, Add 500μL of extract (6% perchloric acid: di-n-butylammonium acetate aqueous solution (IPC-DBAA, 0.5 mol / L, Tokyo Kasei) = 1000: 1) containing Sigma-Aldrich) Created. The total amount of the extract was centrifuged (13,000 rpm, 2 min), and the supernatant was added to a solid phase extraction cartridge (OASIS HLB 1 cc / 30 mg, WAT05882, Waters) for solid phase extraction. The solid-phase extraction cartridge was previously activated with 500 μL of solution A (50% acetonitrile) and further equilibrated twice with 1 mL of solution B (6% perchloric acid: IPC-DBAA = 1000: 1). A thing was used. After the sample was added to the solid phase cartridge, it was washed twice with 1 mL of ultrapure water and eluted with 500 μL of solution A. This eluate was directly used for LC / MS / MS analysis. For the sample for the calibration curve, 100 μL of 4% BSA (w / v) aqueous solution is regarded as 100 mg of tissue, malonyl-CoA lithium salt (Sigma Aldrich) with the above concentration is added, and internal standard substance is added in the same manner as the tissue sample. The solid phase extraction was performed.
For HPLC of LC / MS / MS analysis, Prominence (Shimadzu Corporation) was used, and the separation column was CAPCELL PAK C18 AQ (particle size: 3 μm, inner diameter: 2.0 mm, length: 35 mm, Shiseido Co., Ltd.), and mobile phase ( A) Gradient mode of 10 mmol / L ammonium acetate / IPC-DBAA (100: 1, v / v, pH 9.0) and mobile phase (B) acetonitrile. The gradient program increases the mobile phase (B) concentration from 0 minutes (at the start of analysis) from 5% to 35% in 2 minutes, then increases linearly to 100% by 2.1 minutes, and then delivers it at 100% until 6 minutes. After 6.1 minutes, the solution was reduced to 5%, and the solution was fed under the same conditions until the analysis end time (10 minutes). Using a switching valve, the eluate having an analysis time of 2.0 to 6.0 minutes was introduced into MS / MS. The analysis was performed at a column temperature of 40 ° C. and a sample injection volume of 10 μL. The mass spectrometer used was API5000 (AB Sciex), turbo ion spray was used as the ionization mode, and ions were detected by selected reaction monitoring (SRM) in the negative ion mode. Ion spray was performed using zero air and the voltage was 4.5 kV. Nitrogen was used for collision-induced decomposition of ions. As a monitor ion, a precursor ion and a fragment ion were set to malonyl CoA; m / z 852.0 → m / z 808.0 and [ 13 C 3 ] -malonyl CoA; m / z 855.0 → m / z 810.0 Da, respectively. For quantitative calculation, the internal area ([ 13 C 3 ] -malonyl-CoA) and the peak area ratio of malonyl-CoA were used. Was calculated. All values are shown as mean ± standard deviation, and Welch test was used for statistical analysis. The results are shown in Table 17.

製剤例1(カプセルの製造)
1)実施例1の化合物 30 mg
2)微粉末セルロース 10 mg
3)乳糖 19 mg
4)ステアリン酸マグネシウム 1 mg
計 60 mg
1)、2)、3)および4)を混合して、ゼラチンカプセルに充填する。 Formulation Example 1 (Manufacture of capsules)
1) 30 mg of the compound of Example 1
2) Fine powder cellulose 10 mg
3) Lactose 19 mg
4) Magnesium stearate 1 mg
60 mg total
1), 2), 3) and 4) are mixed and filled into gelatin capsules.
1)実施例1の化合物 30 mg
2)微粉末セルロース 10 mg
3)乳糖 19 mg
4)ステアリン酸マグネシウム 1 mg
計 60 mg
1)、2)、3)および4)を混合して、ゼラチンカプセルに充填する。 Formulation Example 1 (Manufacture of capsules)
1) 30 mg of the compound of Example 1
2) Fine powder cellulose 10 mg
3) Lactose 19 mg
4) Magnesium stearate 1 mg
60 mg total
1), 2), 3) and 4) are mixed and filled into gelatin capsules.
製剤例2(錠剤の製造)
1)実施例1の化合物 30 g
2)乳糖 50 g
3)トウモロコシデンプン 15 g
4)カルボキシメチルセルロースカルシウム 44 g
5)ステアリン酸マグネシウム 1 g
1000錠 計 140 g
1)、2)、3)の全量および30gの4)を水で練合し、真空乾燥後、整粒を行う。この整粒末に14gの4)および1gの5)を混合し、打錠機により打錠する。このようにして、1錠あたり実施例1の化合物30mgを含有する錠剤1000錠を得る。 Formulation Example 2 (Manufacture of tablets)
1) Compound of Example 1 30 g
2) Lactose 50 g
3) Corn starch 15 g
4) Carboxymethylcellulose calcium 44 g
5) Magnesium stearate 1 g
1000 tablets total 140 g
The total amount of 1), 2) and 3) and 30 g of 4) are kneaded with water, and after vacuum drying, the particles are sized. 14 g of 4) and 1 g of 5) are mixed with the sized powder, and tableted with a tableting machine. In this way, 1000 tablets containing 30 mg of the compound of Example 1 per tablet are obtained.
1)実施例1の化合物 30 g
2)乳糖 50 g
3)トウモロコシデンプン 15 g
4)カルボキシメチルセルロースカルシウム 44 g
5)ステアリン酸マグネシウム 1 g
1000錠 計 140 g
1)、2)、3)の全量および30gの4)を水で練合し、真空乾燥後、整粒を行う。この整粒末に14gの4)および1gの5)を混合し、打錠機により打錠する。このようにして、1錠あたり実施例1の化合物30mgを含有する錠剤1000錠を得る。 Formulation Example 2 (Manufacture of tablets)
1) Compound of Example 1 30 g
2) Lactose 50 g
3) Corn starch 15 g
4) Carboxymethylcellulose calcium 44 g
5) Magnesium stearate 1 g
1000 tablets total 140 g
The total amount of 1), 2) and 3) and 30 g of 4) are kneaded with water, and after vacuum drying, the particles are sized. 14 g of 4) and 1 g of 5) are mixed with the sized powder, and tableted with a tableting machine. In this way, 1000 tablets containing 30 mg of the compound of Example 1 per tablet are obtained.
本発明化合物は、ACC(アセチル-CoAカルボキシラーゼ)阻害作用を有し、肥満症、糖尿病、高血圧症、高脂血症、心不全、糖尿病性合併症、メタボリックシンドローム、筋肉減少症、癌等の予防・治療に有用である。
The compound of the present invention has an ACC (acetyl-CoA carboxylase) inhibitory action and prevents obesity, diabetes, hypertension, hyperlipidemia, heart failure, diabetic complications, metabolic syndrome, sarcopenia, cancer, etc. Useful for treatment.
本出願は、日本で出願された特願2011-26342および特願2011-218594を基礎としており、その内容は本明細書にすべて包含されるものである。
This application is based on Japanese Patent Application Nos. 2011-26342 and 2011-218594 filed in Japan, the contents of which are incorporated in full herein.
Claims (23)
- 式(I):

R1は、式:-COR2(R2は、水素原子または置換基を示す)で表される基、または置換されていてもよい5または6員芳香環基を示し;
R3は、ハロゲン原子で置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示し;
R4aおよびR4bは、独立して、水素原子または置換基を示し、またはR4aおよびR4bは、互いに結合して置換されていてもよい3または4員環を形成してもよく;
R5aおよびR5bは、独立して、水素原子または置換基を示し、またはR5aおよびR5bは、互いに結合して置換されていてもよい3または4員環を形成してもよく;
R6は、置換されていてもよいC1-6アルキル基、または置換されていてもよいC3-6シクロアルキル基を示し;
Xは、O、CO、CR7aR7b(R7aおよびR7bは、独立して、水素原子または置換基を示す)、NR7c(R7cは、水素原子または置換されていてもよい炭化水素基を示す)、S、SOまたはS(O)2を示し;
Yは、O、CO、CR8aR8b(R8aおよびR8bは、独立して、水素原子または置換基を示す)、NR8c(R8cは、水素原子または置換されていてもよい炭化水素基を示す)、S、SOまたはS(O)2を示し;
環Pは、さらに置換されていてもよい3ないし7員環を示し;
環Qは、さらに置換されていてもよい5または6員芳香環を示す]
で表される化合物またはその塩。 Formula (I):
R 1 represents a group represented by the formula: —COR 2 (R 2 represents a hydrogen atom or a substituent), or an optionally substituted 5- or 6-membered aromatic ring group;
R 3 represents a C 1-6 alkyl group which may be substituted with a halogen atom, or a C 3-6 cycloalkyl group which may be substituted;
R 4a and R 4b independently represent a hydrogen atom or a substituent, or R 4a and R 4b may be bonded to each other to form an optionally substituted 3- or 4-membered ring;
R 5a and R 5b independently represent a hydrogen atom or a substituent, or R 5a and R 5b may be bonded to each other to form an optionally substituted 3- or 4-membered ring;
R 6 represents an optionally substituted C 1-6 alkyl group, or an optionally substituted C 3-6 cycloalkyl group;
X is O, CO, CR 7a R 7b (R 7a and R 7b independently represent a hydrogen atom or a substituent), NR 7c (R 7c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 ;
Y is O, CO, CR 8a R 8b (R 8a and R 8b independently represent a hydrogen atom or a substituent), NR 8c (R 8c is a hydrogen atom or an optionally substituted hydrocarbon) Group), S, SO or S (O) 2 ;
Ring P represents a 3- to 7-membered ring which may be further substituted;
Ring Q represents a 5- or 6-membered aromatic ring which may be further substituted]
Or a salt thereof. - R1が、式:-COR2(R2は、水素原子、置換されていてもよいC1-6アルキル基、置換されていてもよいC3-6シクロアルキル基、置換されていてもよいC1-6アルコキシ基、または置換されていてもよいアミノ基を示す)で表される基、または置換されていてもよい5または6員芳香環基である、請求項1記載の化合物またはその塩。 R 1 is represented by the formula: —COR 2 (R 2 is a hydrogen atom, an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-6 cycloalkyl group, or optionally substituted. Or a 5- or 6-membered aromatic ring group which may be substituted, or a compound thereof, which is a C 1-6 alkoxy group or an optionally substituted amino group) salt.
- R1が、式:-COR2(R2は、水素原子、C1-6アルキル基、C1-6アルコキシ基、または1ないし2個のC1-6アルキル基で置換されていてもよいアミノ基を示す)で表される基、または5員の芳香族複素環基である、請求項1記載の化合物またはその塩。 R 1 may be substituted with the formula: —COR 2 (R 2 is a hydrogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group, or 1 to 2 C 1-6 alkyl groups. The compound or a salt thereof according to claim 1, which is a group represented by (A), or a 5-membered aromatic heterocyclic group.
- R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、請求項1記載の化合物またはその塩。 The compound or a salt thereof according to claim 1, wherein R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms.
- R4aおよびR4bが水素原子である、請求項1記載の化合物またはその塩。 The compound or a salt thereof according to claim 1, wherein R 4a and R 4b are hydrogen atoms.
- R5aおよびR5bが水素原子である、請求項1記載の化合物またはその塩。 The compound or a salt thereof according to claim 1, wherein R 5a and R 5b are hydrogen atoms.
- R6が、置換されていてもよいC1-6アルキル基である、請求項1記載の化合物またはその塩。 The compound or a salt thereof according to claim 1, wherein R 6 is an optionally substituted C 1-6 alkyl group.
- R6が、
(a) ハロゲン原子、
(b) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC3-6シクロアルキル基、
(c) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよい4ないし7員の複素環基、および
(d) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC6-14アリール基
から選ばれる1ないし7個の置換基で置換されていてもよいC1-6アルキル基である、請求項1記載の化合物またはその塩。 R 6 is
(a) a halogen atom,
(b) a C 3-6 cycloalkyl group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups,
(c) a 4- to 7-membered heterocyclic group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups, and
(d) substituted with 1 to 7 substituents selected from 1 to 3 halogen atoms or C 6-14 aryl group optionally substituted with 1 to 3 C 1-6 alkyl groups; The compound or a salt thereof according to claim 1, wherein said compound is a C 1-6 alkyl group. - Xが、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基を示す)、S、SOまたはS(O)2である、請求項1記載の化合物またはその塩。 The compound according to claim 1, wherein X is O, CO, CH 2 , NR 7c (R 7c represents a hydrogen atom or a C 1-6 alkyl group), S, SO or S (O) 2 , or a compound thereof salt.
- Yが、O、COまたはCH2である、請求項1記載の化合物またはその塩。 Y is, O, is CO or CH 2, the compound or salt thereof according to claim 1.
- 環Pが、ハロゲン原子、C1-6アルキル基およびC1-6アルコキシ基から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C3-6シクロアルカンまたは3ないし7員の複素環である、請求項1記載の化合物またはその塩。 Ring P may be further substituted with 1 to 3 substituents each selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group, benzene, C 3-6 cycloalkane or 3 The compound or a salt thereof according to claim 1, which is a 7-membered heterocyclic ring.
- 環Qが、
(a) ハロゲン原子、
(b) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(c) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)シアノ基
から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環である、請求項1記載の化合物またはその塩。 Ring Q is
(a) a halogen atom,
(b) a C 1-6 alkyl group optionally substituted with 1 to 5 halogen atoms,
(c) a C 1-6 alkoxy group optionally substituted with 1 to 5 halogen atoms, and
(d) The compound according to claim 1 or a salt thereof, which is benzene or a 5- or 6-membered aromatic heterocyclic ring, each of which may be further substituted with 1 to 3 substituents selected from a cyano group. - R1が、式:-COR2(R2は、水素原子、C1-6アルキル基、C1-6アルコキシ基、または1ないし2個のC1-6アルキル基で置換されていてもよいアミノ基を示す)で表される基、または5員の芳香族複素環基であり;
R3が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基であり;
R4aおよびR4bが水素原子であり;
R5aおよびR5bが水素原子であり;
R6が、
(a) ハロゲン原子、
(b) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC3-6シクロアルキル基、
(c) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよい4ないし7員の複素環基、および
(d) 1ないし3個のハロゲン原子または1ないし3個のC1-6アルキル基で置換されていてもよいC6-14アリール基
から選ばれる1ないし7個の置換基で置換されていてもよいC1-6アルキル基であり;
Xが、O、CO、CH2、NR7c(R7cは、水素原子またはC1-6アルキル基を示す)、S、SOまたはS(O)2であり;
Yが、O、COまたはCH2であり;
環Pが、ハロゲン原子、C1-6アルキル基およびC1-6アルコキシ基から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼン、C3-6シクロアルカンまたは3ないし7員の複素環であり;かつ
環Qが、
(a) ハロゲン原子、
(b) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(c) 1ないし5個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d) シアノ基
から選ばれる1ないし3個の置換基でそれぞれさらに置換されていてもよい、ベンゼンまたは5または6員の芳香族複素環である、請求項1記載の化合物またはその塩。 R 1 may be substituted with the formula: —COR 2 (R 2 is a hydrogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group, or 1 to 2 C 1-6 alkyl groups. An amino group), or a 5-membered aromatic heterocyclic group;
R 3 is a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms;
R 4a and R 4b are hydrogen atoms;
R 5a and R 5b are hydrogen atoms;
R 6 is
(a) a halogen atom,
(b) a C 3-6 cycloalkyl group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups,
(c) a 4- to 7-membered heterocyclic group optionally substituted with 1 to 3 halogen atoms or 1 to 3 C 1-6 alkyl groups, and
(d) substituted with 1 to 7 substituents selected from 1 to 3 halogen atoms or C 6-14 aryl group optionally substituted with 1 to 3 C 1-6 alkyl groups; Or a C 1-6 alkyl group;
X is O, CO, CH 2 , NR 7c (R 7c represents a hydrogen atom or a C 1-6 alkyl group), S, SO or S (O) 2 ;
Y is O, CO or CH 2 ;
Ring P may be further substituted with 1 to 3 substituents each selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group, benzene, C 3-6 cycloalkane or 3 To a 7-membered heterocycle; and ring Q is
(a) a halogen atom,
(b) a C 1-6 alkyl group optionally substituted with 1 to 5 halogen atoms,
(c) a C 1-6 alkoxy group optionally substituted with 1 to 5 halogen atoms, and
(d) The compound or a salt thereof according to claim 1, which is benzene or a 5- or 6-membered aromatic heterocyclic ring, each of which may be further substituted with 1 to 3 substituents selected from a cyano group. - N-{(1S)-2-[(trans-4-{[2-クロロ-4-(シクロプロピルメトキシ)ベンジル]オキシ}シクロヘキシル)オキシ]-1-メチルエチル}アセトアミド。 N-{(1S) -2-[(trans-4-{[2-chloro-4- (cyclopropylmethoxy) benzyl] oxy} cyclohexyl) oxy] -1-methylethyl} acetamide.
- N-[(1S)-2-{[trans-4-({2-クロロ-4-[(2,2-ジフルオロシクロプロピル)メトキシ]ベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド。 N-[(1S) -2-{[trans-4-({2-chloro-4-[(2,2-difluorocyclopropyl) methoxy] benzyl} oxy) cyclohexyl] oxy} -1-methylethyl] acetamide .
- N-[(1S)-2-{[trans-4-({4-[(2,2-ジフルオロシクロプロピル)メトキシ]-2,6-ジフルオロベンジル}オキシ)シクロヘキシル]オキシ}-1-メチルエチル]アセトアミド。 N-[(1S) -2-{[trans-4-({4-[(2,2-difluorocyclopropyl) methoxy] -2,6-difluorobenzyl} oxy) cyclohexyl] oxy} -1-methylethyl Acetamide.
- 請求項1記載の化合物またはその塩を含有してなる医薬。 A medicament comprising the compound according to claim 1 or a salt thereof.
- アセチル-CoAカルボキシラーゼ阻害剤である、請求項17記載の医薬。 The medicament according to claim 17, which is an acetyl-CoA carboxylase inhibitor.
- 肥満症または糖尿病の予防または治療剤である、請求項17記載の医薬。 The medicament according to claim 17, which is a prophylactic or therapeutic agent for obesity or diabetes.
- 請求項1記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、アセチル-CoAカルボキシラーゼを阻害する方法。 A method for inhibiting acetyl-CoA carboxylase, which comprises administering an effective amount of the compound or salt thereof according to claim 1 to a mammal.
- 請求項1記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における肥満症または糖尿病の予防または治療方法。 A method for preventing or treating obesity or diabetes in a mammal, comprising administering an effective amount of the compound according to claim 1 or a salt thereof to the mammal.
- 肥満症または糖尿病の予防または治療剤として使用するための請求項1記載の化合物またはその塩。 The compound according to claim 1 or a salt thereof for use as a preventive or therapeutic agent for obesity or diabetes.
- 肥満症または糖尿病の予防または治療剤を製造するための請求項1記載の化合物またはその塩の使用。 Use of the compound according to claim 1 or a salt thereof for producing a prophylactic or therapeutic agent for obesity or diabetes.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| TW101128195A TW201332945A (en) | 2012-02-08 | 2012-08-06 | Monocyclic compound |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011026342 | 2011-02-09 | ||
| JP2011-026342 | 2011-02-09 | ||
| JP2011218594 | 2011-09-30 | ||
| JP2011-218594 | 2011-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012108478A1 true WO2012108478A1 (en) | 2012-08-16 |
Family
ID=46638691
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/052899 WO2012108478A1 (en) | 2011-02-09 | 2012-02-08 | Monocyclic compound |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2012108478A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013061962A1 (en) | 2011-10-24 | 2013-05-02 | 武田薬品工業株式会社 | Bicyclic compound |
| WO2013092976A1 (en) * | 2011-12-23 | 2013-06-27 | Boehringer Ingelheim International Gmbh | New piperidine derivatives, pharmaceutical compositions and uses thereof |
| US8703758B2 (en) | 2010-04-27 | 2014-04-22 | Takeda Pharmaceutical Company Limited | Bicyclic compound |
| US8729102B2 (en) | 2010-11-30 | 2014-05-20 | Takeda Pharmaceutical Company Limited | Bicyclic compound |
| WO2015036892A1 (en) | 2013-09-12 | 2015-03-19 | Pfizer Inc. | Use of acetyl-coa carboxylase inhibitors for treating acne vulgaris |
| WO2015056782A1 (en) * | 2013-10-17 | 2015-04-23 | 塩野義製薬株式会社 | Novel alkylene derivative |
| WO2015097713A1 (en) | 2013-11-14 | 2015-07-02 | Cadila Healthcare Limited | Novel heterocyclic compounds |
| JP2016037462A (en) * | 2014-08-07 | 2016-03-22 | 長谷川香料株式会社 | Method for producing solanone, and synthetic intermediate thereof |
| WO2016159049A1 (en) * | 2015-03-31 | 2016-10-06 | 武田薬品工業株式会社 | Monocyclic compound |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005194191A (en) * | 2001-12-28 | 2005-07-21 | Ajinomoto Co Inc | Drug against obesity and therapeutic drug for fatty liver |
| US20070167435A1 (en) * | 2005-12-21 | 2007-07-19 | Schering Corporation | Phenoxypiperidines and analogs thereof useful as histamine H3 antagonists |
| WO2008079610A2 (en) * | 2006-12-21 | 2008-07-03 | Abbott Laboratories | Novel acetyl-coa carboxylase (acc) inhibitors and their use in diabetes, obesity and metabolic syndrome |
| WO2010003624A2 (en) * | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010050445A1 (en) * | 2008-10-27 | 2010-05-06 | 武田薬品工業株式会社 | Bicyclic compound |
| WO2011067306A1 (en) * | 2009-12-03 | 2011-06-09 | Novartis Ag | Cyclohexane derivatives as inhibitors of acetyl-coa carboxylase (acc) |
-
2012
- 2012-02-08 WO PCT/JP2012/052899 patent/WO2012108478A1/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005194191A (en) * | 2001-12-28 | 2005-07-21 | Ajinomoto Co Inc | Drug against obesity and therapeutic drug for fatty liver |
| US20070167435A1 (en) * | 2005-12-21 | 2007-07-19 | Schering Corporation | Phenoxypiperidines and analogs thereof useful as histamine H3 antagonists |
| WO2008079610A2 (en) * | 2006-12-21 | 2008-07-03 | Abbott Laboratories | Novel acetyl-coa carboxylase (acc) inhibitors and their use in diabetes, obesity and metabolic syndrome |
| WO2010003624A2 (en) * | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010050445A1 (en) * | 2008-10-27 | 2010-05-06 | 武田薬品工業株式会社 | Bicyclic compound |
| WO2011067306A1 (en) * | 2009-12-03 | 2011-06-09 | Novartis Ag | Cyclohexane derivatives as inhibitors of acetyl-coa carboxylase (acc) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8703758B2 (en) | 2010-04-27 | 2014-04-22 | Takeda Pharmaceutical Company Limited | Bicyclic compound |
| US8729102B2 (en) | 2010-11-30 | 2014-05-20 | Takeda Pharmaceutical Company Limited | Bicyclic compound |
| CN103998432A (en) * | 2011-10-24 | 2014-08-20 | 武田药品工业株式会社 | Bicyclic compound |
| WO2013061962A1 (en) | 2011-10-24 | 2013-05-02 | 武田薬品工業株式会社 | Bicyclic compound |
| US9133129B2 (en) | 2011-10-24 | 2015-09-15 | Takeda Pharmaceutical Company Limited | Bicyclic compound |
| WO2013092976A1 (en) * | 2011-12-23 | 2013-06-27 | Boehringer Ingelheim International Gmbh | New piperidine derivatives, pharmaceutical compositions and uses thereof |
| US8765959B2 (en) | 2011-12-23 | 2014-07-01 | Boehringer Ingelheim International Gmbh | Piperidine derivatives |
| WO2015036892A1 (en) | 2013-09-12 | 2015-03-19 | Pfizer Inc. | Use of acetyl-coa carboxylase inhibitors for treating acne vulgaris |
| US10150728B2 (en) | 2013-10-17 | 2018-12-11 | Shionogi & Co., Ltd. | Alkylene derivatives |
| WO2015056782A1 (en) * | 2013-10-17 | 2015-04-23 | 塩野義製薬株式会社 | Novel alkylene derivative |
| WO2015097713A1 (en) | 2013-11-14 | 2015-07-02 | Cadila Healthcare Limited | Novel heterocyclic compounds |
| US10011609B2 (en) | 2013-11-14 | 2018-07-03 | Cadila Healthcare Limited | Heterocyclic compounds |
| US10246470B2 (en) | 2013-11-14 | 2019-04-02 | Cadila Healthcare Limited | Heterocyclic compounds |
| JP2016037462A (en) * | 2014-08-07 | 2016-03-22 | 長谷川香料株式会社 | Method for producing solanone, and synthetic intermediate thereof |
| WO2016159049A1 (en) * | 2015-03-31 | 2016-10-06 | 武田薬品工業株式会社 | Monocyclic compound |
| US10252997B2 (en) | 2015-03-31 | 2019-04-09 | Takeda Pharmaceutical Company Limited | Monocyclic compound |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5824517B2 (en) | Bicyclic compound | |
| JP5693611B2 (en) | Bicyclic compound | |
| WO2012108478A1 (en) | Monocyclic compound | |
| JP6106179B2 (en) | Aromatic ring compounds | |
| US9133129B2 (en) | Bicyclic compound | |
| WO2011102514A1 (en) | Aromatic ring compound | |
| EP2583965B1 (en) | Heterocyclic ring compound | |
| JPWO2011055770A1 (en) | Fused heterocyclic compounds | |
| US9073864B2 (en) | Aromatic ring compound | |
| TW201332945A (en) | Monocyclic compound | |
| TW201350491A (en) | Bicyclic compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12745009 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12745009 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |