WO2012014889A1 - フタロニトリル誘導体の製造方法 - Google Patents
フタロニトリル誘導体の製造方法 Download PDFInfo
- Publication number
- WO2012014889A1 WO2012014889A1 PCT/JP2011/066961 JP2011066961W WO2012014889A1 WO 2012014889 A1 WO2012014889 A1 WO 2012014889A1 JP 2011066961 W JP2011066961 W JP 2011066961W WO 2012014889 A1 WO2012014889 A1 WO 2012014889A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound represented
- reaction
- compound
- water
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/14—Preparation of carboxylic acid nitriles by reaction of cyanides with halogen-containing compounds with replacement of halogen atoms by cyano groups
Abstract
式(I)で表される化合物を、水および臭化物塩の存在下、臭素と反応させて式(II)で表される化合物を得る工程1と、式(II)で表される化合物を、金属シアノ錯体および/または1,3-ジカルボニル化合物の存在下、シアン化合物と反応させて、式(III)で表される化合物を得る工程2を含む、式(III)で表される化合物の製造方法を提供する。式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。
Description
本発明は、フタロニトリル誘導体の製造方法に関する。より詳細には、優れたトロンビン受容体拮抗剤である2-イソインドール誘導体の合成中間体として有用なフタロニトリル誘導体の工業的製造方法に関する。
最近、トロンビン受容体に拮抗作用を有する化合物が、トロンビンが関与する疾患の治療や予防において優れた作用効果を発揮するものと期待されており、例えば血栓症、血管再狭窄、深部静脈血栓症、肺塞栓症、脳梗塞、心疾患、播種性血管内血液凝固症候群、高血圧、炎症性疾患、リウマチ、喘息、糸球体腎炎、骨粗鬆症、神経疾患、悪性腫瘍、等の治療や予防に有効であると期待することができる。そのため、薬理活性、トロンビン受容体に対する受容体特異性、安全性、投与量、経口有用性、等の点を満足させるトロンビン受容体拮抗剤の提供が待望されている。
このような状況下において、特許文献1には、優れたトロンビン受容体阻害活性を有するトロンビン受容体拮抗剤として2-イミノピロリジン誘導体およびその塩が開示されている。2-イミノピロリジン誘導体およびその塩のうち、式(A-1)で表される1-(3-tert-ブチル-4-メトキシ-5-モルホリノ-フェニル)-2-(5,6-ジエトキシ-7-フルオロ-1-イミノ-1,3-ジヒドロ-2-イソインドール-2-イル)-エタノンまたはその塩の製造方法として、式(A-2)で表される化合物と、式(A-3)で表される化合物とを、N,N-ジメチルホルムアミド(DMF)溶媒中でカップリングさせる方法が記載されている。

さらに、特許文献1には、式(A-2)で表される化合物の製造方法として、式(B-I)で表される化合物から、式(B-II)で表される化合物を経由して合成する方法が記載されている。式(B-I)で表される化合物から式(B-II)で表される化合物の具体的な方法として、アルコールやアセトニトリル等の溶媒中、氷冷から室温の条件で臭素またはN-ブロモコハク酸イミドを式(B-I)で表される化合物に作用させる方法、あるいは酢酸溶媒中酢酸ナトリウムの存在下に室温から80℃で臭素を式(B-I)で表される化合物作用させる方法が記載されている。

しかしながら、この方法では、式(B-I)で表される化合物をo-ジブロモ化する際に、副反応により、位置異性体のm-ジブロモ体、過剰に臭素化されたトリブロモ体および分解物が生成し、目的とするo-ジブロモ体を高選択的に得ることが困難である、精製が煩雑になる等の課題があった。さらに、酢酸等の溶媒を大量に必要とする等、工業的規模での製造方法としては作業面やコスト面で課題があった。
一方、特許文献2には、4-ブロモ-2-フルオロフェノールを原料にして、式(B-II)で表される化合物を位置選択的に製造する方法が記載されている。しかしながら、その製造に4工程を要することから、より短い工程で簡便に製造する方法が求められていた。
特許文献1および3には、式(B-II)で表される化合物から、式(B-III)で表される化合物の製造方法が記載されている。具体的には、特許文献1では、式(B-II)で表される化合物とシアン化銅をジメチルホルムアミド、ジメチルスルホキシド、N-メチルピペリドン、ヘキサメチルリン酸トリアミド等の溶媒あるいはそれらの混合溶媒中で反応させることにより式(B-III)で表される化合物を得ており、特許文献3では、式(B-II)で表される化合物とシアン化銅を1,3-ジメチル-2-イミダゾリジノン(以下、DMIと称することがある。)溶媒中で反応させることにより式(B-III)で表される化合物を得ている。しかしながら、これらの方法では、副生物が多く生成する、収率が低いという課題があり、工業的規模での製造方法として課題があった。

優れたトロンビン受容体阻害剤として有用である式(A-1)で表される化合物を、工業的に安価に提供するためには、その合成中間体として有用である式(A-2)で表される化合物を、より工業的に優れた方法で安価に製造することが必要であり、その前駆物質である式(B-III)で表される化合物を安価に提供することが求められている。
かかる事情に鑑み、本発明の目的は、優れたトロンビン受容体拮抗剤である2-イソインドール誘導体の工業的製造のため、その合成中間体であるフタロニトリル誘導体をより優れた製造方法で提供することにある。
本発明者らは、上記事情に鑑み鋭意研究を重ねた結果、式(I)で表される化合物を高選択的にo-ジブロモ化することにより、式(II)で表される化合物を得、さらに高選択的にニトリル化することにより、式(III)で表されるフタロニトリル誘導体を効率的に製造することが出来ることを見出し、本発明を完成した。

(式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。)
(式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。)
すなわち、本発明は、以下の[1]および[2]を提供する。
[1] 式(I)

[式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。]で表される化合物を、水および臭化物塩の存在下、臭素と反応させて式(II)
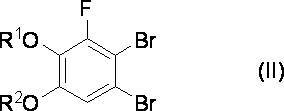
[式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。]で表される化合物を得る工程1と、
式(II)で表される化合物を、金属シアノ錯体および/または1,3-ジカルボニル化合物の存在下、シアン化合物と反応させて式(III)

[式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。]で表される化合物を得る工程2を含む、
式(III)で表される化合物の製造方法;および
[2] 前記工程2をヨウ化物塩の存在下で行うことを特徴とする[1]に記載の製造方法。
[1] 式(I)
[式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。]で表される化合物を、水および臭化物塩の存在下、臭素と反応させて式(II)
[式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。]で表される化合物を得る工程1と、
式(II)で表される化合物を、金属シアノ錯体および/または1,3-ジカルボニル化合物の存在下、シアン化合物と反応させて式(III)
[式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。]で表される化合物を得る工程2を含む、
式(III)で表される化合物の製造方法;および
[2] 前記工程2をヨウ化物塩の存在下で行うことを特徴とする[1]に記載の製造方法。
本発明によれば、トロンビン受容体拮抗剤の合成中間体として有用なフタロニトリル誘導体(III)を安価かつ効率のよい工業的製造方法で、高収率かつ高純度で得ることができる。
本発明によれば、式(I)で表される化合物の臭素化工程の選択性が向上し、生成する副生成物の量を従来より大幅に低減することが可能となり、収率および純度が向上する。さらに、式(II)で表される化合物のニトリル化の工程の収率も向上する。これにより、従来法よりも工業的規模において効率的で安価なフタロニトリル誘導体(III)の製造方法が提供でき、また同時に、優れたトロンビン受容体拮抗剤である2-イソインドール誘導体(A-1)の有用な工業的製造方法を提供できる。
以下に、本明細書において記載する記号、用語等の定義、本発明の実施の形態等を示して、本発明を詳細に説明する。なお、本発明は以下の実施の形態に限定されるものではなく、その要旨の範囲内で種々変形して実施することができる。
本明細書中においては、化合物の構造式が便宜上一定の異性体を表すことがあるが、本発明には化合物の構造上生じ得るすべての幾何異性体、立体異性体、回転異性体、互変異性体等の異性体および異性体混合物を含み、便宜上の式の記載に限定されるものではなく、いずれか一方の異性体でも混合物でもよい。また、結晶多形が存在することもあるが同様に限定されず、いずれかの単一の結晶形であっても二以上の結晶形からなる混合物であってもよい。そして、本発明に係る化合物には、化合物の塩、無水物、水和物、溶媒和物等も包含される。
本明細書において使用する「および/または」とは、「および」の場合と「または」の場合の両者を含む意味で用いられる。
本明細書において使用する「~」とは、「~」の前後の数値を含む範囲の意味で用いられる。
[工程1]

本発明に係る式(III)で表される化合物の製造方法は、式(I)で表される化合物を、水および臭化物塩の存在下、臭素と反応させて、式(I)で表される化合物を得る工程1を含む。工程1は、o-ジブロモ化工程である。
式(I)および(II)において、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。
「C1-6アルキル基」とは、炭素数1~6個の脂肪族炭化水素から任意の水素原子を1個除いて誘導される一価の基である、炭素数1~6個の直鎖状または分枝鎖状のアルキル基を意味する。
C1-6アルキル基として具体的には例えば、メチル基、エチル基、1-プロピル基、2-プロピル基、2-メチル-1-プロピル基、2-メチル-2-プロピル基、1-ブチル基、2-ブチル基、1-ペンチル基、2-ペンチル基、3-ペンチル基、2-メチル-1-ブチル基、3-メチル-1-ブチル基、2-メチル-2-ブチル基、3-メチル-2-ブチル基、2,2-ジメチル-1-プロピル基、1-へキシル基、2-へキシル基、3-へキシル基、2-メチル-1-ペンチル基、3-メチル-1-ペンチル基、4-メチル-1-ペンチル基、2-メチル-2-ペンチル基、3-メチル-2-ペンチル基、4-メチル-2-ペンチル基、2-メチル-3-ペンチル基、3-メチル-3-ペンチル基、2,3-ジメチル-1-ブチル基、3,3-ジメチル-1-ブチル基、2,2-ジメチル-1-ブチル基、2-エチル-1-ブチル基、3,3-ジメチル-2-ブチル基、2,3-ジメチル-2-ブチル基等が挙げられる。C1-6アルキル基として、メチル基またはエチル基が好ましく、エチル基がさらに好ましい。
臭化物塩としては、臭素によって酸化されない対カチオンを有する臭化物が挙げられ、好ましくはアルカリ金属の臭化物またはアルカリ土類金属の臭化物が挙げられる。具体的には、臭化リチウム、臭化ナトリウム、臭化カリウム、臭化マグネシウム、臭化カルシウムおよび臭化バリウム等が挙げられるが、コストの点から臭化ナトリウムが好ましい。
工程1は水の存在下で反応させるが、溶媒として水を用いてもよく、水以外の溶媒に水を加えてよい。水としては、蒸留水、イオン交換水、純水等の医薬品や医薬中間体の製造に通常用いられる水を用いることができる。水以外の溶媒としては、反応を阻害しないものであれば使用することができるが、特に臭素および副生する臭化水素に対して安定であるものであって、相間移動過程を阻害しないものを用いることが好ましい。このような溶媒としては、例えば、ペンタン、ヘキサン、ヘプタン等の炭化水素系溶媒;塩化メチレン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素系溶媒;臭化水素酸などの臭素化反応を阻害しない無機酸とその水溶液等が挙げられ、これらから選ばれる複数の溶媒を任意の割合に混合して用いてもよい
本発明においては、反応速度および低環境負荷の理由から、他の溶媒は用いず、水のみを用いることが好ましい。
臭化物塩は、使用する臭素1当量に対して、通常0.5当量以上、好ましくは0.8当量以上、より好ましくは1.0当量である。これより少ない場合は、臭素が水に完溶せず選択性に悪影響を与える可能性がある。また、通常10当量以下、好ましくは5当量以下、より好ましくは2当量以下である。これより多い場合は、臭化物塩を溶解するためにより多くの水が必要となり、生産性に悪影響を与える可能性がある。
臭素は、式(I)で表される化合物1当量に対して、通常2.0当量以上、好ましくは2.2当量以上、より好ましくは2.4当量以上である。これより少ない場合は、未反応の中間体が多く残存し、収率を低下させる可能性がある。また、通常、3.0当量以下、好ましくは2.8当量以下、より好ましくは2.6当量以下である。これより多い場合は、過剰な臭素化が進行し収率を低下させる可能性がある。
溶媒は、任意の量を用いることができるが、通常は式(I)で表される化合物に対して1~10倍体積量、好ましくは2~7倍体積量であり、より好ましくは3~5倍体積量である。
反応温度は、通常0℃~100℃であり、好ましくは0℃~50℃であり、より好ましくは0℃~30℃である。反応時間は、通常0.5時間~10時間であり、好ましくは1時間~5時間である。
工程1は、式(I)で表される化合物と水が相溶しないため、有機相-水相の二相系の反応となる。副生成物である臭化水素が水相に吸収されるため、式(I)で表される化合物の分解を抑制することができると考えられる。
また、臭化物塩を存在させることにより、水に難溶の臭素を可溶化することができる。これにより、相間移動反応が可能となり、位置選択性の極めて高い臭素化(o-ジブロモ化)反応が可能となると考えられる。
工程1は、例えば、臭素-臭化物塩の水溶液を調製し、これを液体状の式(I)で表される化合物に対して滴下することにより行われる。その際、水相から有機相への臭素の移動、および副生した臭化水素の有機相から水相への移動が円滑に行われるよう、十分に攪拌混合することが好ましい。
工程1の後、通常、亜硫酸ナトリウム、チオ硫酸ナトリウム等により臭素を失活させた後、トルエン、キシレン、ヘプタン、ヘキサン等の有機溶媒を添加し、水で洗浄処理することが好ましい。
工程1で得られる式(II)で表される化合物は、液体もしくは固体であり、固体の場合は相間移動過程を有効に行うために前述の有機溶媒の添加が必要となる場合もある。
式(II)で表される化合物は、必要に応じて、公知の手段、例えば蒸留、昇華、シリカゲルクロマトグラフィー、抽出や結晶化により単離精製することができ、またこれらの手段を組み合わせて用いても良い。また、特に精製が必要なければそのまま次の反応に用いることも可能である。
本発明の工程1により、反応速度が速く、位置選択性の高い臭素化(o-ジブロモ化)反応が可能となり、高い純度の式(II)で表される化合物を効率的に得ることができる。また、従来より洗浄工程を簡略化することができる。
[工程2]

本発明に係る式(III)で表される化合物の製造方法は、式(II)で表される化合物を、金属シアノ錯体および/または1,3-ジカルボニル化合物の存在下、シアン化合物と反応させて式(III)で表される化合物を得る工程2を含む。工程2はシアン化工程である。
式(II)および(III)において、R1およびR2は前記と同義である。
工程2は、
(i)金属シアノ錯体;および/または
(ii)1,3-ジカルボニル化合物
の存在下で反応させることを特徴とする。
(i)金属シアノ錯体;および/または
(ii)1,3-ジカルボニル化合物
の存在下で反応させることを特徴とする。
工程2において、シアン化合物としては、式(II)で表される化合物をシアン化する能力のあるものであって、使用する反応溶媒にある程度溶解するものであり、かつ、反応を阻害しないものであれば特に制限はなく、例えば、シアン化ナトリウム、シアン化カリウム、シアン化銅、シアン化銀、シアン化亜鉛などのシアン化物イオンをアニオンとして持つ塩などを用いることができる。また、これらから選ばれる複数のシアン化合物を任意の割合に混合して用いてもよい。好ましくは、円滑に反応が進行すること、および安価で取り扱い易いことからシアン化銅が好ましい。
シアン化合物は、式(II)で表される化合物1当量に対して、通常2当量以上、好ましくは2.5当量以上、より好ましくは3.0当量以上である。これより少ない場合は、未反応の中間体が多く残存して収率を低下させる可能性がある。また、通常10当量以下、好ましくは5当量以下、より好ましくは3.5当量以下である。これより多い場合は、銅廃棄物を除去する工程の負荷が大きくなる可能性がある。
金属シアノ錯体としては、使用する反応溶媒にある程度溶解するものであり、かつ、反応を阻害しないものであれば特に制限はなく、例えば、フェロシアン化カリウム、フェリシアン化カリウム、フェロシアン化ナトリウム、フェリシアン化ナトリウム、ヘキサシアノコバルト(III)酸カリウム、ヘキサシアノクロム(III)酸カリウム、ヘキサシアノマンガン(III)酸カリウムのようなシアノ基を含有する錯塩などを用いることができ、これらから選ばれる複数の金属シアノ錯体を任意の割合に混合して用いてもよい。中でも、副反応の抑制効果が強く、安価で毒性の小さいことから鉄シアノ錯体が好ましく、特にフェリシアン化カリウムが好ましい。金属シアノ錯体を存在させることにより、反応の選択性が向上する、副生物の生成を抑制する等の効果が得られる。
金属シアノ錯体は、式(II)で表される化合物1当量に対して、通常0.005当量以上、好ましくは0.01当量以上、より好ましくは0.03当量以上である。これより少ない場合は、副生物の抑制効果が小さくなる可能性がある。また、通常0.20当量以下、好ましくは0.15当量以下、より好ましくは0.10当量以下である。これより多い場合は、反応速度を著しく低下させる可能性がある。
また、工程2においては、金属シアノ錯体と共に、ヨウ化物塩を存在させてもよい。ヨウ化物塩を存在させることにより、反応速度が向上する、式(III)で表されるフタロニトリル誘導体の分解を抑制できる等の効果が得られる。
ヨウ化物塩としては、使用する反応溶媒にある程度溶解するものであり、かつ、反応を阻害しないものであれば特に制限はなく、例えば、ヨウ化リチウム、ヨウ化ナトリウム、ヨウ化カリウム、ヨウ化カルシウム、ヨウ化マグネシウム、ヨウ化銅、ヨウ化銀、ヨウ化亜鉛などのヨウ化物イオンをアニオンとして持つ塩などを用いることができ、これらから選ばれる複数のヨウ化物塩を任意の割合に混合して用いてもよい。中でも、円滑に反応が進行し、安価で取り扱い易いことから、ヨウ化カリウムおよび/またはヨウ化銅が好ましい。
ヨウ化物塩を用いる場合は、式(II)で表される化合物1当量に対して、通常0.01当量以上、好ましくは0.02当量以上である。これより少ない場合は、目的とする効果が得られない可能性がある。また、通常、0.2当量以下、好ましくは0.1当量以下である。これより多くしてもさらなる改善効果は認められない。
特に、ヨウ化物塩として、ヨウ化カリウムおよびヨウ化第一銅を用いる場合は、式(II)で表される化合物1当量に対して、ヨウ化カリウムを0.02当量以上0.04当量以下、およびヨウ化第一銅を0.005当量以上0.02当量以下用いることが好ましい。
反応に用いられる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、トルエン、ベンゼン、キシレン、メシチレン等の芳香族炭化水素系溶媒;ペンタン、ヘキサン、ヘプタン等の炭化水素系溶媒;塩化メチレン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテル、ジイソプロピルエーテル、ジオキサン、1,2-ジメトキシエタン、ジエチレングリコールジメチルエーテルのようなエーテル系溶媒;メタノール、エタノール、プロパノール等の水溶性低級アルコール系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、イソブチロニトリルのようなニトリル系溶媒;ジメチルホルムアミド、ジメチルアセトアミド、ヘキサメチルリン酸トリアミド、1,3-ジメチル-2-イミダゾリジノン、N-メチルピロリドンのようなアミド系溶媒;二硫化炭素、ジメチルスルホキシド等の有機溶媒;が挙げられ、これらから選ばれる複数の溶媒を任意の割合に混合して用いてもよい。
好ましくは、副生物の生成を抑制することができることからアミド系溶媒であり、より好ましくは、1,3-ジメチル-2-イミダゾリジノンである。
反応溶媒は、任意の量を用いることができるが、通常は式(II)で表される化合物に対して1~10倍体積量、好ましくは2~7倍体積量であり、より好ましくは3~5倍体積量である。
反応温度は、通常20℃~240℃であり、好ましくは100℃~160℃であり、より好ましくは120℃~140℃である。反応時間は、通常5時間~40時間であり、好ましくは10時間~30時間であり、より好ましくは15時間~25時間である。
1,3-ジカルボニル化合物としては、無水酢酸、無水プロピオン酸、無水コハク酸、無水マレイン酸、無水フタル酸等のカルボン酸無水物、コハク酸イミド、N-メチルコハク酸イミド、フタルイミド等の酸イミド、アセチルアセトン、3,3-ジメチル-2,4-ペンタンジオン等のβ-ジケトンが挙げられるが、好ましくはカルボン酸無水物であり、特に好ましくは助剤の効果およびコストの点から無水コハク酸または無水マレイン酸である。
1,3-ジカルボニル化合物を存在させることにより、副生物の生成を抑制する等の効果が得られる。また、溶媒として、N-メチルピロリドンを用いた場合も、1,3-ジメチル-2-イミダゾリジノンを用いた場合と同様に副生物の生成を抑制することができる。
1,3-ジカルボニル化合物は、式(II)で表される化合物1当量に対して、通常0.2当量以上、好ましくは0.35当量以上、より好ましくは0.5当量以上である。これより少ない場合は、副生物の抑制効果が低下する可能性がある。また、通常3.0当量以下、好ましくは2.0当量以下、より好ましくは1.0当量以下である。これより多くしてもさらなる改善効果は認められない。
上記反応は、式(II)で表される化合物、シアン化合物、金属シアノ錯体および/または1,3-ジカルボニル化合物、必要に応じてヨウ化物塩、および反応溶媒を予め混合して加熱して行なってもよいし、予め反応溶媒にシアン化合物、金属シアノ錯体および/または1,3-ジカルボニル化合物、さらに必要に応じてヨウ化物塩を溶解および/または懸濁させて加熱した液に、式(II)で表される化合物を添加してもよいが、前者の方法が工業的に安全且つ容易であり好ましい。
上記反応は、必要に応じて臭化第二銅等の二価の銅化合物の存在下で行ってもよい。これにより、後処理における反応生成物の抽出効率を向上させることが可能となる。
工程2の後、通常、アンモニア、エチレンジアミン、トリエチレンテトラミン等のアミン類、または臭化鉄(III)等の酸化剤、またはシアン化ナトリウム、シアン化カリウム等のシアン化物塩等を用いて副生する銅化合物を除去した後に、抽出、洗浄およびろ過処理することが好ましい。抽出には酢酸エチル、酢酸イソプロピル、メチル-t-ブチルエーテル(MTBE)、ジエチルエーテル、トルエン、キシレン、ヘプタン、ヘキサン等を用いることができる。また、洗浄には、水、酸性水溶液、アルカリ性水溶液または食塩水等を用いることができる。また、活性炭、珪藻土等をろ過助剤として用いてもよい。
工程2で得られる式(III)で表される化合物は、液体もしくは固体であり、必要に応じて、公知の手段、例えば蒸留、昇華、シリカゲルクロマトグラフィー、抽出や結晶化により単離精製することができ、またこれらの手段を組み合わせて用いても良い。また、特に精製が必要なければそのまま次の反応に用いることも可能である。
本発明の工程2により、公知の手段より安価かつ汎用性のある溶媒を用いた反応が可能となり、また副生物の生成を抑制することができるため、高い純度の式(III)で表される化合物を効率的に得ることができる。また、従来より洗浄工程を簡略化することができる。
また、本発明においては、ジメチルホルムアミド、ジメチルアセトアミド、ヘキサメチルリン酸トリアミド、1,3-ジメチル-2-イミダゾリジノン、N-メチルピロリドン等のアミド系溶媒、アンモニア、エチレンジアミン、トリエチレンテトラミン等のアミン類、および水を含有する溶液に、上記工程2で得られた反応液を添加して、式(III)で表される化合物を晶析させた後、洗浄、ろ過することが好ましい。単離精製時に使用するアミド系溶媒としては、工程2の反応時に使用した溶媒と同じものを用いることが好ましく、特に1,3-ジメチル-2-イミダゾリジノンまたはN-メチルピロリドンが好ましい。またアミン類としてはエチレンジアミンが好ましい。
アミド系溶媒、アミン類および水の混合比としては、式(II)で表される化合物1当量に対して、通常、アミド系溶媒:アミン類:水(重量比)=1:1~5:2~10、好ましくはアミド系溶媒:アミン類:水(重量比)=1:1.5~3:3~7である。
この方法により、洗浄液の使用量を低減することができ、また、副生物、不純物等を効率的に除去することができるため、工業的な方法として好ましい。
また、本発明においては、チオ硫酸ナトリウム、シアン化ナトリウムまたはシアン化カリウム等の一価銅イオン安定化剤、炭酸ナトリウム、炭酸リチウム、炭酸カリウム、水酸化ナトリウム、水酸化リチウムまたは水酸化カリウム等の無機塩基類、および水を含有する溶液に、上記工程2で得られた反応液を添加して、式(III)で表される化合物を晶析させた後、洗浄、ろ過することが特に好ましい。単離精製時に使用する一価銅イオン安定化剤としては、低毒性およびコストの点からチオ硫酸ナトリウムが好ましい。また無機塩基類としては炭酸ナトリウムまたは炭酸リチウムが好ましく、特に後処理の容易さおよびコストの点から炭酸ナトリウムが好ましい。
一価銅イオン安定化剤、無機塩基類および水の混合比としては、式(II)で表される化合物1当量に対して、通常、一価銅イオン安定化剤:無機塩基類:水(モル/モル/重量比)=6~14:0.5~2:6~16、好ましくは一価銅イオン安定化剤:無機塩基類:水(モル/モル/重量比)=8~12:0.8~1.4:9~13である。
この方法により、洗浄液の使用量を低減することができ、また、副生物、不純物等を効率的に除去することができる。さらに式(III)で表される化合物の分解を抑制でき、また一価銅イオンの不均化による設備の銅汚れを回避できるため、工業的な方法として好ましい。
本発明に係る化合物は、例えば以下の実施例および製造例等に記載した方法により製造することができる。ただし、これらは例示的なものであって、本発明に係る化合物は如何なる場合も以下の具体例に限定されるものではない。
以下の実施例において、高速液体クロマトグラフィー(HPLC)により生成物の純度及び反応収率を算出しているが、HPLCカラムとして株式会社ワイエムシィ社製のYMC-Pack Pro C18 RS(長さ150mm×内径4.6mm)を使用した。
[実施例1] 1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼンの製造:工程1

グラスライニング(GL)反応釜1に、市販の1,2-ジエトキシ-3-フルオロベンゼン117.0kg(635mol)および水117Lを仕込み0℃まで冷却した。GL反応釜2で臭素243.6kg(1.52kmol)、臭化ナトリウム117.6kg(1.14kmol)、水351Lを混合して臭素/臭化ナトリウム水溶液を調製し、GL反応釜1に0~9℃で4.5時間かけて滴下した。滴下終了後、GL反応釜2を水18Lで洗浄してGL反応釜1に加えた。GL反応釜1を29℃まで昇温して、さらに1.7時間攪拌し臭素化反応混合物を得た。GL反応釜3で、ヘプタン240.1kg、水酸化ナトリウム88.9kg(2.22kmol)、水356Lを混合してヘプタン/水酸化ナトリウム水溶液を調製し、1℃に冷却した。GL反応釜1の臭素化反応混合物を、GL反応釜3に25℃以下を保ちながら滴下した後、GL反応釜1をヘプタン79.6kgで洗浄してGL反応釜3に加えた。さらにGL反応釜3にチオ硫酸ナトリウム五水和物50.2kg(202mol)を25℃以下に保ちながら添加した。得られた反応混合物を25℃で静置・分液して下層を廃棄し、さらに水468Lで3回分液洗浄した。得られたヘプタン溶液をカートリッジフィルター(TCW-1-CSS;アドバンテック東洋株式会社)でろ過後、ヘプタン78.0kgで洗浄し、25℃~60℃で減圧濃縮して微黄色オイル状の粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼンを得た。(218.2kg、純度94.3重量%、601mol、収率94.7%)
[比較例1] 工程1
国際公開パンフレットWO02/085855の実施例7工程2に記載の方法に準じて、1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼンを製造した。
GL反応釜1に市販の1,2-ジエトキシ-3-フルオロベンゼン54.95kg(298mol)、酢酸289.11kg、および酢酸ナトリウム63.70kg(777mol)を仕込み25℃に調整した。内温30℃以下に保ちながら、臭素124.10kg(776mol)を2.5時間かけて滴下し、滴下終了後臭素仕込みラインを酢酸3.74kgで洗浄し反応液に加えた。反応混合物を45℃まで昇温して、さらに6.4時間攪拌した。反応終了後25℃まで冷却し、攪拌しながらヘプタン150.49kgおよび水442Lを添加し、内温30℃以下に保ちながらピロ亜硫酸ナトリウム25.06kgを添加した。攪拌・静置後、上層のヘプタン相1と下層の水相を分液して、水相をGL反応釜2へ送液した。GL反応釜2の水相にヘプタン150.53kgを添加して攪拌・静置し、上層のヘプタン相2を残して水相を廃棄した。ヘプタン相1の入ったGL反応釜1にヘプタン相2を送液後、GL反応釜2および送液ラインをヘプタン18.45kgで洗浄してGL反応釜1に加えた。得られたGL反応釜1のヘプタン溶液を、4%水酸化ナトリウム水溶液220.8kgで二回、水221Lで三回分液洗浄した。洗浄後のヘプタン溶液をカートリッジフィルター(TCW-1-CSS)でろ過して、ろ過ラインをヘプタン36.65kgで洗浄し、25℃~60℃で減圧濃縮して黄色オイル状の粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼンを得た。(101.99kg、純度85.3重量%、254mol、収率85.3%)
国際公開パンフレットWO02/085855の実施例7工程2に記載の方法に準じて、1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼンを製造した。
GL反応釜1に市販の1,2-ジエトキシ-3-フルオロベンゼン54.95kg(298mol)、酢酸289.11kg、および酢酸ナトリウム63.70kg(777mol)を仕込み25℃に調整した。内温30℃以下に保ちながら、臭素124.10kg(776mol)を2.5時間かけて滴下し、滴下終了後臭素仕込みラインを酢酸3.74kgで洗浄し反応液に加えた。反応混合物を45℃まで昇温して、さらに6.4時間攪拌した。反応終了後25℃まで冷却し、攪拌しながらヘプタン150.49kgおよび水442Lを添加し、内温30℃以下に保ちながらピロ亜硫酸ナトリウム25.06kgを添加した。攪拌・静置後、上層のヘプタン相1と下層の水相を分液して、水相をGL反応釜2へ送液した。GL反応釜2の水相にヘプタン150.53kgを添加して攪拌・静置し、上層のヘプタン相2を残して水相を廃棄した。ヘプタン相1の入ったGL反応釜1にヘプタン相2を送液後、GL反応釜2および送液ラインをヘプタン18.45kgで洗浄してGL反応釜1に加えた。得られたGL反応釜1のヘプタン溶液を、4%水酸化ナトリウム水溶液220.8kgで二回、水221Lで三回分液洗浄した。洗浄後のヘプタン溶液をカートリッジフィルター(TCW-1-CSS)でろ過して、ろ過ラインをヘプタン36.65kgで洗浄し、25℃~60℃で減圧濃縮して黄色オイル状の粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼンを得た。(101.99kg、純度85.3重量%、254mol、収率85.3%)
[実施例2] 4,5-ジエトキシ-3-フルオロフタロニトリルの製造:工程2

GL反応釜1に、実施例1で得られた粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(218.2kg、94.3重量%、601mol)、1,3-ジメチル-2-イミダゾリジノン868.9kg、シアン化銅167.00kg(1.86kmol)、フェリシアン化カリウム5.95kg(18.1mol)、ヨウ化カリウム3.00kg(18.1mol)、およびヨウ化銅1.15kg(6.04mol)を仕込んで混合物とした後、反応釜内を窒素置換した。この混合物を攪拌しながら133℃まで加熱し、21.4時間攪拌した後に冷却した。GL反応釜2にトリエチレンテトラミン343.00kg(2.35kmol)、水1726L、およびトルエン889.75kgの混合液を調製し、GL反応釜1の反応液を30℃以下に保ちながら約1時間かけて滴下した。さらにGL反応釜1をN-メチルピロリドン211.65kgで洗浄してGL反応釜2に加えた。得られた反応混合物から25℃で上層のトルエン相1と下層の水相を分液して、さらに水相をトルエン533.95kgで抽出した後、廃棄した。トルエン相1に抽出トルエン相を加え、送液ラインをトルエン17.80kgで洗浄した。得られたトルエン相に珪藻土(昭和化学工業社製、商品名:ラヂオライト)41.10kgを添加・攪拌後ろ過し、ろ過残渣と送液ラインをトルエン533.80kgで洗浄してろ過液に混合した。得られたトルエン溶液から水相を分液・廃棄後、5重量%エチレンジアミン/5重量%食塩を溶解した水溶液約825kg×2回、3.5重量%塩酸823.2kg、水825L×3回で順次分液洗浄した。洗浄後のトルエン相に、活性炭4.05kg/トルエン22.00kgの混合物を添加・攪拌後、ろ過した。反応釜と送液ラインをトルエン53.45kgで洗浄し、ろ過液に混合した。得られたトルエン溶液を50℃で減圧濃縮し、4,5-ジエトキシ-3-フルオロフタロニトリル濃度を38.3重量%に調整した。濃縮後トルエン溶液に50~55℃でヘプタン421.6kgを約1時間かけて滴下し、5℃付近まで3.5時間かけて冷却した。さらに5℃で2時間攪拌した後に遠心分離し、得られた結晶をトルエン44.45kg/ヘプタン105.70kgの混合液で洗浄して、粗4,5-ジエトキシ-3-フルオロフタロニトリル112.05kgを得た。得られた粗結晶をトルエン88.9kgに55℃で溶解し、ヘプタン70.4kgを約1時間かけて滴下した。得られた懸濁液を55℃で約1時間攪拌した後、4時間かけて5℃付近まで冷却した。さらに5℃で2時間攪拌した後に遠心分離し、得られた結晶をトルエン44.6kg/ヘプタン105.7kgの混合液で洗浄した。得られた湿体を50度以下で減圧乾燥し、微黄色結晶状の4,5-ジエトキシ-3-フルオロフタロニトリルを得た(105.70kg、HPLC純度98.0%、収率75.0%)。
[実施例3] 工程2
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.01g、92.4重量%、2.95mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)、無水酢酸0.31g(3.04mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.5時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(70.7%)。
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.01g、92.4重量%、2.95mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)、無水酢酸0.31g(3.04mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.5時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(70.7%)。
[実施例4] 工程2
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.01g、92.4重量%、2.95mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)、無水コハク酸0.30g(3.00mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.5時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(73.3%)。
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.01g、92.4重量%、2.95mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)、無水コハク酸0.30g(3.00mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.5時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(73.3%)。
[実施例5] 工程2
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.01g、92.4重量%、2.95mmol)、N-メチルピロリドン3mL、シアン化銅0.78g(8.71mmol)、無水マレイン酸0.29g(2.96mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.4時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(77.5%)。
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.01g、92.4重量%、2.95mmol)、N-メチルピロリドン3mL、シアン化銅0.78g(8.71mmol)、無水マレイン酸0.29g(2.96mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.4時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(77.5%)。
[実施例6] 工程2
試験管型反応器に、比較例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.00g、89.9重量%、2.63mmol)、1,3-ジメチル-2-イミダゾリジノン3.6mL、シアン化銅708mg(7.91mmol)、フェリシアン化カリウム41.3mg(0.13mmol)、臭化第二銅36.7mg(0.16mmol)を仕込んで、系内を窒素置換後130℃に昇温し16.2時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(71.9%)。
試験管型反応器に、比較例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.00g、89.9重量%、2.63mmol)、1,3-ジメチル-2-イミダゾリジノン3.6mL、シアン化銅708mg(7.91mmol)、フェリシアン化カリウム41.3mg(0.13mmol)、臭化第二銅36.7mg(0.16mmol)を仕込んで、系内を窒素置換後130℃に昇温し16.2時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(71.9%)。
[実施例7] 工程2
試験管型反応器に、比較例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(0.98g、89.9重量%、2.58mmol)、1,3-ジメチル-2-イミダゾリジノン3.6mL、シアン化銅707mg(7.89mmol)、フェリシアン化カリウム46.2mg(0.14mmol)、ヨウ化カリウム42.4mg(0.26mmol)、およびヨウ化銅47.4mg(0.25mmol)を仕込んで、系内を窒素置換後130℃に昇温し16.0時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(75.6%)。
試験管型反応器に、比較例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(0.98g、89.9重量%、2.58mmol)、1,3-ジメチル-2-イミダゾリジノン3.6mL、シアン化銅707mg(7.89mmol)、フェリシアン化カリウム46.2mg(0.14mmol)、ヨウ化カリウム42.4mg(0.26mmol)、およびヨウ化銅47.4mg(0.25mmol)を仕込んで、系内を窒素置換後130℃に昇温し16.0時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(75.6%)。
[実施例8] 工程2
試験管型反応器に、比較例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(0.99g、89.9重量%、2.60mmol)、1,3-ジメチル-2-イミダゾリジノン3.6mL、シアン化銅714mg(7.97mmol)、フェロシアン化カリウム三水和物59.8mg(0.14mmol)、ヨウ化カリウム45.6mg(0.27mmol)、およびヨウ化銅48.1mg(0.25mmol)を仕込んで、系内を窒素置換後130℃に昇温し16.0時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(68.0%)。
試験管型反応器に、比較例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(0.99g、89.9重量%、2.60mmol)、1,3-ジメチル-2-イミダゾリジノン3.6mL、シアン化銅714mg(7.97mmol)、フェロシアン化カリウム三水和物59.8mg(0.14mmol)、ヨウ化カリウム45.6mg(0.27mmol)、およびヨウ化銅48.1mg(0.25mmol)を仕込んで、系内を窒素置換後130℃に昇温し16.0時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(68.0%)。
[実施例9] 工程2
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.00g、92.4重量%、2.92mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)、N-メチルスクシンイミド0.34g(98重量%、2.95mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.2時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(64.1%)。
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.00g、92.4重量%、2.92mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)、N-メチルスクシンイミド0.34g(98重量%、2.95mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.2時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(64.1%)。
[実施例10] 工程2
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.00g、92.4重量%、2.92mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)、3,3-ジメチル-2,4-ペンタンジオン0.38g(2.96mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.5時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(49.9%)。
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.00g、92.4重量%、2.92mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)、3,3-ジメチル-2,4-ペンタンジオン0.38g(2.96mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.5時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(49.9%)。
[実施例11] 工程2
GL反応釜1に実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(426.97kg、96.4重量%、1.20kmol)、N-メチルピロリドン1274.75kg、シアン化銅324.00kg(3.62kmol)、フェリシアン化カリウム31.70kg(96.3mol)、および無水マレイン酸59.10kg(603mol)を仕込んだ後、反応釜内を窒素置換した。この混合物を攪拌しながら133℃まで加熱し、21.2時間攪拌した後、冷却して反応液を得た。GL反応釜2にエチレンジアミン738.6kg、水1644L、およびN-メチルピロリドン411.7kgの混合液を調製し、GL反応釜1の反応液を内温30℃以下に保ちながら約1時間かけて滴下した。GL反応釜1をN-メチルピロリドン207.45kgで洗浄して加え、得られた反応混合液を25℃で1時間攪拌後、遠心分離した。分離物を6.4重量%チオ硫酸ナトリウム水溶液1644.2kg、および水3288Lで洗浄した。洗浄後の粗結晶をトルエン1693kgに溶解し、活性炭8.25kg/トルエン89.1kgの混合物と珪藻土(昭和化学工業社製、商品名:ラヂオライト)16.40kgを添加してさらに1時間攪拌した。得られた混合物を加圧ろ過し、トルエン356.75kgで洗浄した。得られたトルエン溶液から水相を分液・廃棄後、水1645Lを添加して分液洗浄した。このトルエン溶液に、活性炭8.25kg/トルエン107kgの混合物を添加後、約1時間攪拌してろ過し、トルエン357.65kgで洗浄した。得られたトルエン溶液を50℃で減圧濃縮し、4,5-ジエトキシ-3-フルオロフタロニトリル濃度を37.7重量%に調整した。濃縮後トルエン溶液にヘプタン846.05kgを55℃以下で1時間かけて滴下し、5℃付近まで4.7時間かけて冷却した。さらに5℃で2時間攪拌した後に遠心分離し、分離物をトルエン88.6kg/ヘプタン212.0kgの混合液で洗浄した。得られた湿体を25~50℃で減圧乾燥し、白色結晶状の4,5-ジエトキシ-3-フルオロフタロニトリルを得た(213.75kg、HPLC純度98.2%、収率75.8%)。
GL反応釜1に実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(426.97kg、96.4重量%、1.20kmol)、N-メチルピロリドン1274.75kg、シアン化銅324.00kg(3.62kmol)、フェリシアン化カリウム31.70kg(96.3mol)、および無水マレイン酸59.10kg(603mol)を仕込んだ後、反応釜内を窒素置換した。この混合物を攪拌しながら133℃まで加熱し、21.2時間攪拌した後、冷却して反応液を得た。GL反応釜2にエチレンジアミン738.6kg、水1644L、およびN-メチルピロリドン411.7kgの混合液を調製し、GL反応釜1の反応液を内温30℃以下に保ちながら約1時間かけて滴下した。GL反応釜1をN-メチルピロリドン207.45kgで洗浄して加え、得られた反応混合液を25℃で1時間攪拌後、遠心分離した。分離物を6.4重量%チオ硫酸ナトリウム水溶液1644.2kg、および水3288Lで洗浄した。洗浄後の粗結晶をトルエン1693kgに溶解し、活性炭8.25kg/トルエン89.1kgの混合物と珪藻土(昭和化学工業社製、商品名:ラヂオライト)16.40kgを添加してさらに1時間攪拌した。得られた混合物を加圧ろ過し、トルエン356.75kgで洗浄した。得られたトルエン溶液から水相を分液・廃棄後、水1645Lを添加して分液洗浄した。このトルエン溶液に、活性炭8.25kg/トルエン107kgの混合物を添加後、約1時間攪拌してろ過し、トルエン357.65kgで洗浄した。得られたトルエン溶液を50℃で減圧濃縮し、4,5-ジエトキシ-3-フルオロフタロニトリル濃度を37.7重量%に調整した。濃縮後トルエン溶液にヘプタン846.05kgを55℃以下で1時間かけて滴下し、5℃付近まで4.7時間かけて冷却した。さらに5℃で2時間攪拌した後に遠心分離し、分離物をトルエン88.6kg/ヘプタン212.0kgの混合液で洗浄した。得られた湿体を25~50℃で減圧乾燥し、白色結晶状の4,5-ジエトキシ-3-フルオロフタロニトリルを得た(213.75kg、HPLC純度98.2%、収率75.8%)。
[比較例2] 工程2
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.01g、92.4重量%、2.95mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.5時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(31.5%)。
試験管型反応器に、実施例1と同様にして製造した粗1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(1.01g、92.4重量%、2.95mmol)、N-メチルピロリドン3mL、シアン化銅0.79g(8.82mmol)を仕込んで、系内を窒素置換後130℃に昇温し17.5時間攪拌した。HPLC分析結果から4,5-ジエトキシ-3-フルオロフタロニトリルの反応収率を算出した(31.5%)。
[参考例1] 国際公開パンフレットWO2006/018955の調製例1
1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(137.0g、0.401mol、含量81%)、シアン化銅(107.5g、1、200mol)および1,3-ジメチル-2-イミダゾリジノン(600mL)の混合物を減圧窒素置換後、窒素雰囲気下において130℃で17時間、140℃で4時間加熱撹拌した。
反応液を室温まで冷却後、N,N-ジメチルホルムアミド(600mL)、トルエン(1.2L)および濃アンモニア水(1.2L)を加えて分液し、得られた有機層にN,N-ジメチルホルムアミド(411mL)と濃アンモニア水(800mL)を加えて洗浄した。更に有機層を25%エチレンジアミン水(1.2L)、1N塩酸(1.2L)、水(1.2L)の順で洗浄した。活性炭を通してろ過後、50℃にて減圧濃縮して黄色シャーベット状の残渣を得た。トルエン(100mL)およびn-ヘプタン(100mL)を加えて60℃で加熱溶解後、10℃以下で冷却撹拌して析出した結晶をろ取した。得られた結晶にトルエン(50mL)およびn-ヘプタン(50mL)を加えて90℃で加熱溶解後、室温にて撹拌して結晶を析出させた。10℃以下に冷却後、結晶をろ取し、50℃にて減圧乾燥して標記化合物を白色結晶として51.5g(収率:55%)得た。
1,2-ジブロモ-4,5-ジエトキシ-3-フルオロベンゼン(137.0g、0.401mol、含量81%)、シアン化銅(107.5g、1、200mol)および1,3-ジメチル-2-イミダゾリジノン(600mL)の混合物を減圧窒素置換後、窒素雰囲気下において130℃で17時間、140℃で4時間加熱撹拌した。
反応液を室温まで冷却後、N,N-ジメチルホルムアミド(600mL)、トルエン(1.2L)および濃アンモニア水(1.2L)を加えて分液し、得られた有機層にN,N-ジメチルホルムアミド(411mL)と濃アンモニア水(800mL)を加えて洗浄した。更に有機層を25%エチレンジアミン水(1.2L)、1N塩酸(1.2L)、水(1.2L)の順で洗浄した。活性炭を通してろ過後、50℃にて減圧濃縮して黄色シャーベット状の残渣を得た。トルエン(100mL)およびn-ヘプタン(100mL)を加えて60℃で加熱溶解後、10℃以下で冷却撹拌して析出した結晶をろ取した。得られた結晶にトルエン(50mL)およびn-ヘプタン(50mL)を加えて90℃で加熱溶解後、室温にて撹拌して結晶を析出させた。10℃以下に冷却後、結晶をろ取し、50℃にて減圧乾燥して標記化合物を白色結晶として51.5g(収率:55%)得た。
Claims (2)
- 式(I)

[式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。]で表される化合物を、水および臭化物塩の存在下、臭素と反応させて式(II)

[式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。]で表される化合物を得る工程1と、
式(II)で表される化合物を、金属シアノ錯体および/または1,3-ジカルボニル化合物の存在下、シアン化合物と反応させて式(III)

[式中、R1およびR2は、それぞれ独立してC1-6アルキル基を示す。]で表される化合物を得る工程2を含む、
式(III)で表される化合物の製造方法。 - 前記工程2をヨウ化物塩の存在下で行うことを特徴とする請求項1に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012526514A JPWO2012014889A1 (ja) | 2010-07-29 | 2011-07-26 | フタロニトリル誘導体の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-170566 | 2010-07-29 | ||
| JP2010170566 | 2010-07-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012014889A1 true WO2012014889A1 (ja) | 2012-02-02 |
Family
ID=45530096
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/066961 WO2012014889A1 (ja) | 2010-07-29 | 2011-07-26 | フタロニトリル誘導体の製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2012014889A1 (ja) |
| WO (1) | WO2012014889A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002085855A1 (fr) * | 2001-04-19 | 2002-10-31 | Eisai Co., Ltd. | Derives de 2-iminopyrrolidine |
| WO2006018954A1 (ja) * | 2004-08-17 | 2006-02-23 | Eisai R & D Management Co., Ltd. | ジブロモフルオロベンゼン誘導体の製造方法 |
| WO2006018955A1 (ja) * | 2004-08-16 | 2006-02-23 | Eisai R & D Management Co., Ltd. | イソインドール誘導体の製造方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009088063A1 (ja) * | 2008-01-11 | 2009-07-16 | Eisai R & D Management Co., Ltd. | 医薬組成物、医薬組成物製造のための2-イミノピロリジン誘導体の使用および心疾患の治療用または改善用キット |
-
2011
- 2011-07-26 JP JP2012526514A patent/JPWO2012014889A1/ja active Pending
- 2011-07-26 WO PCT/JP2011/066961 patent/WO2012014889A1/ja active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002085855A1 (fr) * | 2001-04-19 | 2002-10-31 | Eisai Co., Ltd. | Derives de 2-iminopyrrolidine |
| WO2006018955A1 (ja) * | 2004-08-16 | 2006-02-23 | Eisai R & D Management Co., Ltd. | イソインドール誘導体の製造方法 |
| WO2006018954A1 (ja) * | 2004-08-17 | 2006-02-23 | Eisai R & D Management Co., Ltd. | ジブロモフルオロベンゼン誘導体の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| GEKKAN FINE CHEMICAL, vol. 35, no. 10, 2006, pages 44 - 52 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2012014889A1 (ja) | 2013-09-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2661943C (en) | Process and intermediates for preparing integrase inhibitors | |
| AU2008298943B2 (en) | Process and intermediates for preparing integrase inhibitors | |
| US8877931B2 (en) | Process and intermediates for preparing integrase inhibitors | |
| AU2005273430B2 (en) | Methods for producing isoindole derivatives | |
| KR101991148B1 (ko) | 2-아미노-4,6-디메톡시벤즈아미드 및 기타 벤즈아미드 화합물의 합성 방법 | |
| US7956222B2 (en) | Methods for producing dibromofluorobenzene derivatives | |
| WO2012014889A1 (ja) | フタロニトリル誘導体の製造方法 | |
| JP5714744B2 (ja) | 置換イソキノリンを合成するための方法 | |
| AU2008333261A1 (en) | Process for the preparation of 5-(2-ethyl-dihydro-1H-inden-2-yl)-1H-imidazole and salts thereof | |
| JP2004244362A (ja) | 1,2−ジ置換−1,4−ジヒドロ−オキソキノリン誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11812478 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012526514 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11812478 Country of ref document: EP Kind code of ref document: A1 |