WO2011149921A1 - Soluble guanylate cyclase activators - Google Patents
Soluble guanylate cyclase activators Download PDFInfo
- Publication number
- WO2011149921A1 WO2011149921A1 PCT/US2011/037718 US2011037718W WO2011149921A1 WO 2011149921 A1 WO2011149921 A1 WO 2011149921A1 US 2011037718 W US2011037718 W US 2011037718W WO 2011149921 A1 WO2011149921 A1 WO 2011149921A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- methyl
- dihydro
- amino
- pentafluorobutyl
- Prior art date
Links
- 102000007637 Soluble Guanylyl Cyclase Human genes 0.000 title claims description 28
- 108010007205 Soluble Guanylyl Cyclase Proteins 0.000 title claims description 28
- 239000003119 guanylate cyclase activator Substances 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 254
- 238000000034 method Methods 0.000 claims abstract description 73
- 150000003839 salts Chemical class 0.000 claims abstract description 69
- 239000003814 drug Substances 0.000 claims abstract description 19
- 125000000217 alkyl group Chemical group 0.000 claims description 163
- 125000003118 aryl group Chemical group 0.000 claims description 95
- 125000005843 halogen group Chemical group 0.000 claims description 87
- -1 -C3-10 cycloalkyl Chemical group 0.000 claims description 84
- 125000001072 heteroaryl group Chemical group 0.000 claims description 81
- WEVYAHXRMPXWCK-UHFFFAOYSA-N methyl cyanide Natural products CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 70
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 68
- 125000001424 substituent group Chemical group 0.000 claims description 64
- 229910052739 hydrogen Inorganic materials 0.000 claims description 61
- 125000000623 heterocyclic group Chemical group 0.000 claims description 45
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 43
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 37
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 34
- 238000011282 treatment Methods 0.000 claims description 31
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 27
- QDXGKRWDQCEABB-UHFFFAOYSA-N pyrimidine-5-carboxamide Chemical compound NC(=O)C1=CN=CN=C1 QDXGKRWDQCEABB-UHFFFAOYSA-N 0.000 claims description 20
- 229910003827 NRaRb Inorganic materials 0.000 claims description 17
- 229910052757 nitrogen Inorganic materials 0.000 claims description 15
- 238000002360 preparation method Methods 0.000 claims description 14
- 239000013543 active substance Substances 0.000 claims description 13
- UTCSSFWDNNEEBH-UHFFFAOYSA-N imidazo[1,2-a]pyridine Chemical compound C1=CC=CC2=NC=CN21 UTCSSFWDNNEEBH-UHFFFAOYSA-N 0.000 claims description 12
- 206010019280 Heart failures Diseases 0.000 claims description 11
- 229910052717 sulfur Inorganic materials 0.000 claims description 11
- 229940079593 drug Drugs 0.000 claims description 10
- DNCYBUMDUBHIJZ-UHFFFAOYSA-N 1h-pyrimidin-6-one Chemical compound O=C1C=CN=CN1 DNCYBUMDUBHIJZ-UHFFFAOYSA-N 0.000 claims description 8
- 206010020772 Hypertension Diseases 0.000 claims description 8
- 239000003795 chemical substances by application Substances 0.000 claims description 8
- 230000002265 prevention Effects 0.000 claims description 8
- 208000010125 myocardial infarction Diseases 0.000 claims description 7
- 206010002383 Angina Pectoris Diseases 0.000 claims description 6
- 201000001320 Atherosclerosis Diseases 0.000 claims description 6
- 208000007536 Thrombosis Diseases 0.000 claims description 6
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 5
- 206010048554 Endothelial dysfunction Diseases 0.000 claims description 5
- 208000010228 Erectile Dysfunction Diseases 0.000 claims description 5
- 208000006011 Stroke Diseases 0.000 claims description 5
- 206010012601 diabetes mellitus Diseases 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 230000008694 endothelial dysfunction Effects 0.000 claims description 5
- 201000001881 impotence Diseases 0.000 claims description 5
- 210000004185 liver Anatomy 0.000 claims description 5
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 4
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 claims description 4
- 206010052337 Diastolic dysfunction Diseases 0.000 claims description 4
- 206010016654 Fibrosis Diseases 0.000 claims description 4
- 206010020852 Hypertonia Diseases 0.000 claims description 4
- 208000006673 asthma Diseases 0.000 claims description 4
- 208000020832 chronic kidney disease Diseases 0.000 claims description 4
- 230000007882 cirrhosis Effects 0.000 claims description 4
- 208000019425 cirrhosis of liver Diseases 0.000 claims description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 230000002685 pulmonary effect Effects 0.000 claims description 4
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 4
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 3
- 239000003524 antilipemic agent Substances 0.000 claims description 3
- 230000000903 blocking effect Effects 0.000 claims description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 3
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 claims description 3
- 229910052705 radium Inorganic materials 0.000 claims description 3
- 239000002461 renin inhibitor Substances 0.000 claims description 3
- 229940086526 renin-inhibitors Drugs 0.000 claims description 3
- 229910052701 rubidium Inorganic materials 0.000 claims description 3
- 239000005541 ACE inhibitor Substances 0.000 claims description 2
- 229940123338 Aldosterone synthase inhibitor Drugs 0.000 claims description 2
- 229940127291 Calcium channel antagonist Drugs 0.000 claims description 2
- 230000003213 activating effect Effects 0.000 claims description 2
- 230000001800 adrenalinergic effect Effects 0.000 claims description 2
- 239000000695 adrenergic alpha-agonist Substances 0.000 claims description 2
- 239000002170 aldosterone antagonist Substances 0.000 claims description 2
- 229940083712 aldosterone antagonist Drugs 0.000 claims description 2
- 239000002333 angiotensin II receptor antagonist Substances 0.000 claims description 2
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 claims description 2
- 239000005557 antagonist Substances 0.000 claims description 2
- 239000000480 calcium channel blocker Substances 0.000 claims description 2
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 claims description 2
- 239000002934 diuretic Substances 0.000 claims description 2
- 239000002792 enkephalinase inhibitor Substances 0.000 claims description 2
- 230000002503 metabolic effect Effects 0.000 claims description 2
- 239000000810 peripheral vasodilating agent Substances 0.000 claims description 2
- 229960002116 peripheral vasodilator Drugs 0.000 claims description 2
- 239000002590 phosphodiesterase V inhibitor Substances 0.000 claims description 2
- 239000004036 potassium channel stimulating agent Substances 0.000 claims description 2
- 230000000948 sympatholitic effect Effects 0.000 claims description 2
- 229940124549 vasodilator Drugs 0.000 claims description 2
- 239000003071 vasodilator agent Substances 0.000 claims description 2
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims 1
- 101710129690 Angiotensin-converting enzyme inhibitor Proteins 0.000 claims 1
- 101710086378 Bradykinin-potentiating and C-type natriuretic peptides Proteins 0.000 claims 1
- 102000010180 Endothelin receptor Human genes 0.000 claims 1
- 108050001739 Endothelin receptor Proteins 0.000 claims 1
- 229940123333 Phosphodiesterase 5 inhibitor Drugs 0.000 claims 1
- 125000002877 alkyl aryl group Chemical group 0.000 claims 1
- 229940126317 angiotensin II receptor antagonist Drugs 0.000 claims 1
- 239000002876 beta blocker Substances 0.000 claims 1
- 230000001882 diuretic effect Effects 0.000 claims 1
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 abstract description 26
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 14
- 238000011321 prophylaxis Methods 0.000 abstract description 13
- 238000002560 therapeutic procedure Methods 0.000 abstract description 13
- 239000000825 pharmaceutical preparation Substances 0.000 abstract description 10
- 201000010099 disease Diseases 0.000 abstract description 9
- 238000004519 manufacturing process Methods 0.000 abstract description 4
- 230000008569 process Effects 0.000 abstract description 4
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 287
- 239000000243 solution Substances 0.000 description 186
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 156
- 235000019439 ethyl acetate Nutrition 0.000 description 133
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 123
- 238000006243 chemical reaction Methods 0.000 description 100
- 239000000543 intermediate Substances 0.000 description 100
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 93
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 92
- 238000005481 NMR spectroscopy Methods 0.000 description 88
- 239000000203 mixture Substances 0.000 description 77
- 239000000047 product Substances 0.000 description 74
- 238000010898 silica gel chromatography Methods 0.000 description 60
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 58
- 239000007787 solid Substances 0.000 description 57
- 239000011541 reaction mixture Substances 0.000 description 48
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 47
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 45
- 239000012267 brine Substances 0.000 description 40
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 40
- 239000002904 solvent Substances 0.000 description 40
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 39
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 38
- 238000004808 supercritical fluid chromatography Methods 0.000 description 36
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 34
- 239000012044 organic layer Substances 0.000 description 34
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 30
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 29
- 238000000926 separation method Methods 0.000 description 29
- 239000012074 organic phase Substances 0.000 description 27
- 238000003756 stirring Methods 0.000 description 26
- 238000000746 purification Methods 0.000 description 24
- 239000003921 oil Substances 0.000 description 23
- 235000019198 oils Nutrition 0.000 description 23
- ZOOGRGPOEVQQDX-UHFFFAOYSA-N cyclic GMP Natural products O1C2COP(O)(=O)OC2C(O)C1N1C=NC2=C1NC(N)=NC2=O ZOOGRGPOEVQQDX-UHFFFAOYSA-N 0.000 description 22
- 239000011734 sodium Substances 0.000 description 22
- 229920006395 saturated elastomer Polymers 0.000 description 21
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 20
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 20
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 19
- 239000010410 layer Substances 0.000 description 19
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 18
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 18
- 239000000463 material Substances 0.000 description 18
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 18
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 18
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 17
- 230000002829 reductive effect Effects 0.000 description 17
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 17
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 16
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 15
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 14
- 238000004007 reversed phase HPLC Methods 0.000 description 14
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 13
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 13
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 13
- 239000002585 base Substances 0.000 description 13
- 239000011369 resultant mixture Substances 0.000 description 13
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 12
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 12
- 0 CN(C1=I)N=C(*)C1=* Chemical compound CN(C1=I)N=C(*)C1=* 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 238000004587 chromatography analysis Methods 0.000 description 12
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 12
- 239000003480 eluent Substances 0.000 description 12
- 238000005160 1H NMR spectroscopy Methods 0.000 description 11
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 11
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 11
- 239000002253 acid Substances 0.000 description 11
- 239000003153 chemical reaction reagent Substances 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 229910052938 sodium sulfate Inorganic materials 0.000 description 11
- 235000011152 sodium sulphate Nutrition 0.000 description 11
- APWRZPQBPCAXFP-UHFFFAOYSA-N 1-(1-oxo-2H-isoquinolin-5-yl)-5-(trifluoromethyl)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrazole-4-carboxamide Chemical compound O=C1NC=CC2=C(C=CC=C12)N1N=CC(=C1C(F)(F)F)C(=O)NC1=CC(=NC=C1)C(F)(F)F APWRZPQBPCAXFP-UHFFFAOYSA-N 0.000 description 10
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 10
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 10
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 229940125782 compound 2 Drugs 0.000 description 10
- 239000000284 extract Substances 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Substances IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 9
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 9
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 9
- 238000012746 preparative thin layer chromatography Methods 0.000 description 9
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 8
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 8
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 8
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 8
- 150000001409 amidines Chemical class 0.000 description 8
- 125000004093 cyano group Chemical group *C#N 0.000 description 8
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 239000000741 silica gel Substances 0.000 description 8
- 229910002027 silica gel Inorganic materials 0.000 description 8
- 229960001866 silicon dioxide Drugs 0.000 description 8
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 8
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 8
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 7
- 125000004429 atom Chemical group 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 7
- 229910000027 potassium carbonate Inorganic materials 0.000 description 7
- 239000002002 slurry Substances 0.000 description 7
- 229910000104 sodium hydride Inorganic materials 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- IOGXOCVLYRDXLW-UHFFFAOYSA-N tert-butyl nitrite Chemical compound CC(C)(C)ON=O IOGXOCVLYRDXLW-UHFFFAOYSA-N 0.000 description 7
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 6
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 6
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 6
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 230000004913 activation Effects 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- 235000019270 ammonium chloride Nutrition 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 230000036772 blood pressure Effects 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- 125000001309 chloro group Chemical group Cl* 0.000 description 6
- 125000001153 fluoro group Chemical group F* 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- 150000002576 ketones Chemical class 0.000 description 6
- 150000002825 nitriles Chemical class 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 239000012414 tert-butyl nitrite Substances 0.000 description 6
- 238000004809 thin layer chromatography Methods 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 6
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 6
- UBDZFAGVPPMTIT-UHFFFAOYSA-N 2-aminoguanidine;hydron;chloride Chemical compound [Cl-].NC(N)=N[NH3+] UBDZFAGVPPMTIT-UHFFFAOYSA-N 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 239000005909 Kieselgur Substances 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 239000003054 catalyst Substances 0.000 description 5
- 239000012043 crude product Substances 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 5
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 5
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 5
- 125000002950 monocyclic group Chemical group 0.000 description 5
- 125000004941 pyridazin-5-yl group Chemical group N1=NC=CC(=C1)* 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 238000011699 spontaneously hypertensive rat Methods 0.000 description 5
- 230000000638 stimulation Effects 0.000 description 5
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- SRVXSISGYBMIHR-UHFFFAOYSA-N 3-[3-[3-(2-amino-2-oxoethyl)phenyl]-5-chlorophenyl]-3-(5-methyl-1,3-thiazol-2-yl)propanoic acid Chemical compound S1C(C)=CN=C1C(CC(O)=O)C1=CC(Cl)=CC(C=2C=C(CC(N)=O)C=CC=2)=C1 SRVXSISGYBMIHR-UHFFFAOYSA-N 0.000 description 4
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 4
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 4
- 101150065749 Churc1 gene Proteins 0.000 description 4
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 4
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 4
- MJBWDEQAUQTVKK-IAGOWNOFSA-N aflatoxin M1 Chemical compound C=1([C@]2(O)C=CO[C@@H]2OC=1C=C(C1=2)OC)C=2OC(=O)C2=C1CCC2=O MJBWDEQAUQTVKK-IAGOWNOFSA-N 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- HTZCNXWZYVXIMZ-UHFFFAOYSA-M benzyl(triethyl)azanium;chloride Chemical compound [Cl-].CC[N+](CC)(CC)CC1=CC=CC=C1 HTZCNXWZYVXIMZ-UHFFFAOYSA-M 0.000 description 4
- 150000003857 carboxamides Chemical class 0.000 description 4
- 150000007942 carboxylates Chemical class 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- 238000005859 coupling reaction Methods 0.000 description 4
- 239000013058 crude material Substances 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- LRBPFPZTIZSOGG-UHFFFAOYSA-N dimethyl 2-methylpropanedioate Chemical compound COC(=O)C(C)C(=O)OC LRBPFPZTIZSOGG-UHFFFAOYSA-N 0.000 description 4
- 150000003278 haem Chemical class 0.000 description 4
- 150000002367 halogens Chemical class 0.000 description 4
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 4
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 4
- 230000006698 induction Effects 0.000 description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- CUONGYYJJVDODC-UHFFFAOYSA-N malononitrile Chemical compound N#CCC#N CUONGYYJJVDODC-UHFFFAOYSA-N 0.000 description 4
- 238000004949 mass spectrometry Methods 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 238000002953 preparative HPLC Methods 0.000 description 4
- WIRTYVGMQVIVDM-UHFFFAOYSA-N pyridine-3-carbonitrile Chemical compound N#CC1=C=NC=C[CH]1 WIRTYVGMQVIVDM-UHFFFAOYSA-N 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 230000035488 systolic blood pressure Effects 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 4
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 3
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 3
- PCTZLSCYMRXUGW-UHFFFAOYSA-N 1,1,1,2,2-pentafluorobutane Chemical group [CH2]CC(F)(F)C(F)(F)F PCTZLSCYMRXUGW-UHFFFAOYSA-N 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 3
- BAZVFQBTJPBRTJ-UHFFFAOYSA-N 2-chloro-5-nitropyridine Chemical compound [O-][N+](=O)C1=CC=C(Cl)N=C1 BAZVFQBTJPBRTJ-UHFFFAOYSA-N 0.000 description 3
- WQKHERPPDYPMNX-UHFFFAOYSA-N 6-chloro-3,4-dihydro-2h-naphthalen-1-one Chemical compound O=C1CCCC2=CC(Cl)=CC=C21 WQKHERPPDYPMNX-UHFFFAOYSA-N 0.000 description 3
- 239000002083 C09CA01 - Losartan Substances 0.000 description 3
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 108010078321 Guanylate Cyclase Proteins 0.000 description 3
- 102000014469 Guanylate cyclase Human genes 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 3
- VIHYIVKEECZGOU-UHFFFAOYSA-N N-acetylimidazole Chemical compound CC(=O)N1C=CN=C1 VIHYIVKEECZGOU-UHFFFAOYSA-N 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- 239000012190 activator Substances 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 230000029936 alkylation Effects 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 229940125758 compound 15 Drugs 0.000 description 3
- 229940126214 compound 3 Drugs 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 3
- SHXJAGLEMPLMHH-UHFFFAOYSA-N ethyl 3,3-dicyano-2-methylprop-2-enoate Chemical compound CCOC(=O)C(C)=C(C#N)C#N SHXJAGLEMPLMHH-UHFFFAOYSA-N 0.000 description 3
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 239000007903 gelatin capsule Substances 0.000 description 3
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- 230000000155 isotopic effect Effects 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- WCYWZMWISLQXQU-UHFFFAOYSA-N methyl Chemical class [CH3] WCYWZMWISLQXQU-UHFFFAOYSA-N 0.000 description 3
- BIECSXCXIXHDBC-UHFFFAOYSA-N methyl 2-bromo-5-chlorobenzoate Chemical compound COC(=O)C1=CC(Cl)=CC=C1Br BIECSXCXIXHDBC-UHFFFAOYSA-N 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 3
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 3
- 239000004810 polytetrafluoroethylene Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 235000015497 potassium bicarbonate Nutrition 0.000 description 3
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 3
- 239000011736 potassium bicarbonate Substances 0.000 description 3
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 230000000069 prophylactic effect Effects 0.000 description 3
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 3
- FVGUUJNEJJPLCS-UHFFFAOYSA-N pyridine-3-carboximidamide Chemical compound NC(=N)C1=CC=CN=C1 FVGUUJNEJJPLCS-UHFFFAOYSA-N 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- UOYSTPYCLSTYPU-UHFFFAOYSA-N pyrimidine-5-carbohydrazide Chemical compound NNC(=O)C1=CN=CN=C1 UOYSTPYCLSTYPU-UHFFFAOYSA-N 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 238000007363 ring formation reaction Methods 0.000 description 3
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 235000010288 sodium nitrite Nutrition 0.000 description 3
- 159000000000 sodium salts Chemical class 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 125000001113 thiadiazolyl group Chemical group 0.000 description 3
- 125000001425 triazolyl group Chemical group 0.000 description 3
- JLTRXTDYQLMHGR-UHFFFAOYSA-N trimethylaluminium Chemical compound C[Al](C)C JLTRXTDYQLMHGR-UHFFFAOYSA-N 0.000 description 3
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 2
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 2
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 2
- POILWHVDKZOXJZ-ARJAWSKDSA-M (z)-4-oxopent-2-en-2-olate Chemical compound C\C([O-])=C\C(C)=O POILWHVDKZOXJZ-ARJAWSKDSA-M 0.000 description 2
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 2
- JFTHBDBUVHRREF-UHFFFAOYSA-N 2-acetamidopropanedioic acid Chemical compound CC(=O)NC(C(O)=O)C(O)=O JFTHBDBUVHRREF-UHFFFAOYSA-N 0.000 description 2
- CUYKNJBYIJFRCU-UHFFFAOYSA-N 3-aminopyridine Chemical compound NC1=CC=CN=C1 CUYKNJBYIJFRCU-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- XIMPGXQDHZYZOA-UHFFFAOYSA-N 4,4,5,5,5-pentafluoro-1-pyridin-2-ylpentan-1-one Chemical compound FC(F)(F)C(F)(F)CCC(=O)C1=CC=CC=N1 XIMPGXQDHZYZOA-UHFFFAOYSA-N 0.000 description 2
- HHSIXPRDVJARSI-UHFFFAOYSA-N 4,4,5,5,5-pentafluoropentanoic acid Chemical compound OC(=O)CCC(F)(F)C(F)(F)F HHSIXPRDVJARSI-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- VUZQHUVRBPILAX-UHFFFAOYSA-N 6-chloro-1h-indazole Chemical compound ClC1=CC=C2C=NNC2=C1 VUZQHUVRBPILAX-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 229940127007 Compound 39 Drugs 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 2
- RRSNDVCODIMOFX-MPKOGUQCSA-N Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O Chemical compound Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O RRSNDVCODIMOFX-MPKOGUQCSA-N 0.000 description 2
- 230000005526 G1 to G0 transition Effects 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 239000007818 Grignard reagent Substances 0.000 description 2
- RPTUSVTUFVMDQK-UHFFFAOYSA-N Hidralazin Chemical compound C1=CC=C2C(NN)=NN=CC2=C1 RPTUSVTUFVMDQK-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- MPCRDALPQLDDFX-UHFFFAOYSA-L Magnesium perchlorate Chemical compound [Mg+2].[O-]Cl(=O)(=O)=O.[O-]Cl(=O)(=O)=O MPCRDALPQLDDFX-UHFFFAOYSA-L 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 108700010041 Nicotinic acid receptor Proteins 0.000 description 2
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical group O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 2
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 description 2
- 229910019213 POCl3 Inorganic materials 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- QOSMNYMQXIVWKY-UHFFFAOYSA-N Propyl levulinate Chemical compound CCCOC(=O)CCC(C)=O QOSMNYMQXIVWKY-UHFFFAOYSA-N 0.000 description 2
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 2
- 241000720974 Protium Species 0.000 description 2
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 208000007718 Stable Angina Diseases 0.000 description 2
- 208000007814 Unstable Angina Diseases 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 2
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 2
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 2
- WEVYAHXRMPXWCK-FIBGUPNXSA-N acetonitrile-d3 Chemical compound [2H]C([2H])([2H])C#N WEVYAHXRMPXWCK-FIBGUPNXSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- HAMNKKUPIHEESI-UHFFFAOYSA-N aminoguanidine Chemical compound NNC(N)=N HAMNKKUPIHEESI-UHFFFAOYSA-N 0.000 description 2
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 2
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 2
- 150000007514 bases Chemical class 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 210000001772 blood platelet Anatomy 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 159000000007 calcium salts Chemical class 0.000 description 2
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 238000000423 cell based assay Methods 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940127573 compound 38 Drugs 0.000 description 2
- 229940125844 compound 46 Drugs 0.000 description 2
- 229940127271 compound 49 Drugs 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- UCSVJZQSZZAKLD-UHFFFAOYSA-N ethyl azide Chemical compound CCN=[N+]=[N-] UCSVJZQSZZAKLD-UHFFFAOYSA-N 0.000 description 2
- 229960000815 ezetimibe Drugs 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 150000004795 grignard reagents Chemical class 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 239000000833 heterodimer Substances 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 2
- 229960002003 hydrochlorothiazide Drugs 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- AQBLLJNPHDIAPN-LNTINUHCSA-K iron(3+);(z)-4-oxopent-2-en-2-olate Chemical compound [Fe+3].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O AQBLLJNPHDIAPN-LNTINUHCSA-K 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- PSIFNNKUMBGKDQ-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2NN=NN=2)C=C1 PSIFNNKUMBGKDQ-UHFFFAOYSA-N 0.000 description 2
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- DDNQOOHVLJSIQW-UHFFFAOYSA-N methyl 3,3-dicyano-2-methyl-2-phenylpropanoate Chemical compound COC(=O)C(C)(C(C#N)C#N)C1=CC=CC=C1 DDNQOOHVLJSIQW-UHFFFAOYSA-N 0.000 description 2
- HFLMYYLFSNEOOT-UHFFFAOYSA-N methyl 4-chloro-3-oxobutanoate Chemical compound COC(=O)CC(=O)CCl HFLMYYLFSNEOOT-UHFFFAOYSA-N 0.000 description 2
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical compound [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 2
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 2
- 229960002237 metoprolol Drugs 0.000 description 2
- 239000003094 microcapsule Substances 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000007922 nasal spray Substances 0.000 description 2
- 229960003512 nicotinic acid Drugs 0.000 description 2
- 235000001968 nicotinic acid Nutrition 0.000 description 2
- 239000011664 nicotinic acid Substances 0.000 description 2
- ZEJPMRKECMRICL-UHFFFAOYSA-N o-ethyl 2-amino-2-oxoethanethioate Chemical compound CCOC(=S)C(N)=O ZEJPMRKECMRICL-UHFFFAOYSA-N 0.000 description 2
- RNVCVTLRINQCPJ-UHFFFAOYSA-N o-toluidine Chemical compound CC1=CC=CC=C1N RNVCVTLRINQCPJ-UHFFFAOYSA-N 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 125000001715 oxadiazolyl group Chemical group 0.000 description 2
- 125000003566 oxetanyl group Chemical group 0.000 description 2
- 150000002923 oximes Chemical class 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 150000003217 pyrazoles Chemical class 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 229960002855 simvastatin Drugs 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 230000000707 stereoselective effect Effects 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000007940 sugar coated tablet Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- 125000003831 tetrazolyl group Chemical group 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- JWZZKOKVBUJMES-UHFFFAOYSA-N (+-)-Isoprenaline Chemical compound CC(C)NCC(O)C1=CC=C(O)C(O)=C1 JWZZKOKVBUJMES-UHFFFAOYSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 1
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- QIJLJZOGPPQCOG-NFAWXSAZSA-N (2s)-1-[(2s)-3-[(2r)-2-(cyclohexanecarbonylamino)propanoyl]sulfanyl-2-methylpropanoyl]pyrrolidine-2-carboxylic acid Chemical compound N([C@H](C)C(=O)SC[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)C(=O)C1CCCCC1 QIJLJZOGPPQCOG-NFAWXSAZSA-N 0.000 description 1
- BAVDEDVBIHTHJQ-UVJOBNTFSA-N (2s)-1-[(2s)-6-amino-2-[[(1s)-1-carboxy-3-phenylpropyl]amino]hexanoyl]pyrrolidine-2-carboxylic acid;hydrate Chemical compound O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 BAVDEDVBIHTHJQ-UVJOBNTFSA-N 0.000 description 1
- OJRHUICOVVSGSY-RXMQYKEDSA-N (2s)-2-chloro-3-methylbutan-1-ol Chemical compound CC(C)[C@H](Cl)CO OJRHUICOVVSGSY-RXMQYKEDSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- IYZRFOAPEUBNQP-JPZLKUPGSA-N (3s)-n-[(2s)-1-[[(5s)-5-amino-6-hydroxyhexyl]amino]-4-methyl-1-oxopentan-2-yl]-3-hydroxy-4-[[3-(1h-imidazol-5-yl)-2-[[3-naphthalen-1-yl-2-(naphthalen-1-ylmethyl)propanoyl]amino]propanoyl]amino]-6-methylheptanamide;dihydrochloride Chemical compound Cl.Cl.C=1C=CC2=CC=CC=C2C=1CC(CC=1C2=CC=CC=C2C=CC=1)C(=O)NC(C(=O)NC(CC(C)C)[C@@H](O)CC(=O)N[C@@H](CC(C)C)C(=O)NCCCC[C@H](N)CO)CC1=CN=CN1 IYZRFOAPEUBNQP-JPZLKUPGSA-N 0.000 description 1
- SYPWPWUSXPWLKW-ZQWQDMLBSA-N (3s,4s)-5-cyclohexyl-n-hexyl-3-hydroxy-4-[[(2s)-2-[[(2s)-2-[(2-morpholin-4-ylacetyl)amino]-3-naphthalen-1-ylpropanoyl]amino]-3-(1,3-thiazol-4-yl)propanoyl]amino]pentanamide Chemical compound C([C@@H]([C@@H](O)CC(=O)NCCCCCC)NC(=O)[C@H](CC=1N=CSC=1)NC(=O)[C@H](CC=1C2=CC=CC=C2C=CC=1)NC(=O)CN1CCOCC1)C1CCCCC1 SYPWPWUSXPWLKW-ZQWQDMLBSA-N 0.000 description 1
- KQJKUOYVWLBSDN-UHFFFAOYSA-N (5-chloropyridin-2-yl)methanamine Chemical compound NCC1=CC=C(Cl)C=N1 KQJKUOYVWLBSDN-UHFFFAOYSA-N 0.000 description 1
- OALKYGZLLCDVEN-UHFFFAOYSA-N (5-fluoropyridin-2-yl)methanamine Chemical compound NCC1=CC=C(F)C=N1 OALKYGZLLCDVEN-UHFFFAOYSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 1
- XHTYQFMRBQUCPX-UHFFFAOYSA-N 1,1,3,3-tetramethoxypropane Chemical compound COC(OC)CC(OC)OC XHTYQFMRBQUCPX-UHFFFAOYSA-N 0.000 description 1
- DRTQHJPVMGBUCF-UCVXFZOQSA-N 1-[(2s,3s,4s,5s)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidine-2,4-dione Chemical compound O[C@H]1[C@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UCVXFZOQSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- IVVNZDGDKPTYHK-JTQLQIEISA-N 1-cyano-2-[(2s)-3,3-dimethylbutan-2-yl]-3-pyridin-4-ylguanidine Chemical compound CC(C)(C)[C@H](C)N=C(NC#N)NC1=CC=NC=C1 IVVNZDGDKPTYHK-JTQLQIEISA-N 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- VEKHJWHWWJCKRM-UHFFFAOYSA-N 1h-pyridazine-2-carbonitrile Chemical compound N#CN1NC=CC=C1 VEKHJWHWWJCKRM-UHFFFAOYSA-N 0.000 description 1
- FQMZXMVHHKXGTM-UHFFFAOYSA-N 2-(1-adamantyl)-n-[2-[2-(2-hydroxyethylamino)ethylamino]quinolin-5-yl]acetamide Chemical compound C1C(C2)CC(C3)CC2CC13CC(=O)NC1=CC=CC2=NC(NCCNCCO)=CC=C21 FQMZXMVHHKXGTM-UHFFFAOYSA-N 0.000 description 1
- QRAZASHLGLHKEB-UHFFFAOYSA-N 2-(2,3,6-trifluorophenyl)acetic acid Chemical compound OC(=O)CC1=C(F)C=CC(F)=C1F QRAZASHLGLHKEB-UHFFFAOYSA-N 0.000 description 1
- YPZDATTWQZPZEH-UHFFFAOYSA-N 2-(dicyanomethyl)propanedioic acid Chemical compound OC(=O)C(C(O)=O)C(C#N)C#N YPZDATTWQZPZEH-UHFFFAOYSA-N 0.000 description 1
- LKOIXZIQZBMVPP-UHFFFAOYSA-N 2-(dicyanomethylidene)propanedioic acid Chemical compound OC(=O)C(C(O)=O)=C(C#N)C#N LKOIXZIQZBMVPP-UHFFFAOYSA-N 0.000 description 1
- NNJBQTDFDVLWEQ-UHFFFAOYSA-N 2-(sulfonylamino)benzoic acid Chemical class OC(=O)C1=CC=CC=C1N=S(=O)=O NNJBQTDFDVLWEQ-UHFFFAOYSA-N 0.000 description 1
- ZZRFQBQNZLFESZ-BTQNPOSSSA-N 2-[(3r)-4-[(4-chlorophenyl)methyl]-7-fluoro-5-methylsulfonyl-2,3-dihydro-1h-cyclopenta[b]indol-3-yl]acetic acid;pyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=CN=C1.C=1([C@@H](CC(O)=O)CCC=1C=1C=C(F)C=C(C2=1)S(=O)(=O)C)N2CC1=CC=C(Cl)C=C1 ZZRFQBQNZLFESZ-BTQNPOSSSA-N 0.000 description 1
- FYELSNVLZVIGTI-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-5-ethylpyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1CC)CC(=O)N1CC2=C(CC1)NN=N2 FYELSNVLZVIGTI-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 description 1
- REXUYBKPWIPONM-UHFFFAOYSA-N 2-bromoacetonitrile Chemical compound BrCC#N REXUYBKPWIPONM-UHFFFAOYSA-N 0.000 description 1
- IMRWILPUOVGIMU-UHFFFAOYSA-N 2-bromopyridine Chemical compound BrC1=CC=CC=N1 IMRWILPUOVGIMU-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- GELVZYOEQVJIRR-UHFFFAOYSA-N 2-chloropyrazine Chemical compound ClC1=CN=CC=N1 GELVZYOEQVJIRR-UHFFFAOYSA-N 0.000 description 1
- KHPAGGHFIDLUMB-UHFFFAOYSA-N 2-chloropyridine-3-carbaldehyde Chemical compound ClC1=NC=CC=C1C=O KHPAGGHFIDLUMB-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- 125000006029 2-methyl-2-butenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- APIXJSLKIYYUKG-UHFFFAOYSA-N 3 Isobutyl 1 methylxanthine Chemical compound O=C1N(C)C(=O)N(CC(C)C)C2=C1N=CN2 APIXJSLKIYYUKG-UHFFFAOYSA-N 0.000 description 1
- IFGHEEXVAVTLKH-UHFFFAOYSA-N 3,3-dicyanoprop-2-enoic acid Chemical compound OC(=O)C=C(C#N)C#N IFGHEEXVAVTLKH-UHFFFAOYSA-N 0.000 description 1
- BNBOUFHCTIFWHN-UHFFFAOYSA-N 3-bromobutan-2-one Chemical compound CC(Br)C(C)=O BNBOUFHCTIFWHN-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- BAZKUNDRKREMBW-UHFFFAOYSA-N 3-methyl-1h-pyridazin-2-amine;hydrochloride Chemical compound Cl.CC1=CC=CNN1N BAZKUNDRKREMBW-UHFFFAOYSA-N 0.000 description 1
- MPOYBFYHRQBZPM-UHFFFAOYSA-N 3h-pyridin-4-one Chemical class O=C1CC=NC=C1 MPOYBFYHRQBZPM-UHFFFAOYSA-N 0.000 description 1
- WTUCTMYLCMVYEX-UHFFFAOYSA-N 4,4,4-trifluorobutanoic acid Chemical compound OC(=O)CCC(F)(F)F WTUCTMYLCMVYEX-UHFFFAOYSA-N 0.000 description 1
- DQPRRXFKNCJAOT-UHFFFAOYSA-N 4,4,5,5,5-pentafluoro-n-methoxy-n-methylpentanamide Chemical compound CON(C)C(=O)CCC(F)(F)C(F)(F)F DQPRRXFKNCJAOT-UHFFFAOYSA-N 0.000 description 1
- QROUUECTKRZFHF-UHFFFAOYSA-N 4,4,5,5,5-pentafluoropentan-1-ol Chemical compound OCCCC(F)(F)C(F)(F)F QROUUECTKRZFHF-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- YFCIFWOJYYFDQP-PTWZRHHISA-N 4-[3-amino-6-[(1S,3S,4S)-3-fluoro-4-hydroxycyclohexyl]pyrazin-2-yl]-N-[(1S)-1-(3-bromo-5-fluorophenyl)-2-(methylamino)ethyl]-2-fluorobenzamide Chemical compound CNC[C@@H](NC(=O)c1ccc(cc1F)-c1nc(cnc1N)[C@H]1CC[C@H](O)[C@@H](F)C1)c1cc(F)cc(Br)c1 YFCIFWOJYYFDQP-PTWZRHHISA-N 0.000 description 1
- URLKLEPWQJQWMP-UHFFFAOYSA-N 4-amino-2-[5-chloro-3-(3,3,3-trifluoropropyl)indazol-1-yl]-5-methyl-5-phenyl-7h-pyrrolo[2,3-d]pyrimidin-6-one Chemical compound O=C1NC2=NC(N3C4=CC=C(Cl)C=C4C(CCC(F)(F)F)=N3)=NC(N)=C2C1(C)C1=CC=CC=C1 URLKLEPWQJQWMP-UHFFFAOYSA-N 0.000 description 1
- ZSUBGXQHVKSVKH-UHFFFAOYSA-N 4-amino-2-[6-cyano-1-(3,3,4,4,4-pentafluorobutyl)pyrazolo[3,4-b]pyridin-3-yl]-n-cyclopropyl-5-methyl-6-oxo-7h-pyrrolo[2,3-d]pyrimidine-5-carboxamide Chemical compound O=C1NC2=NC(C=3C4=CC=C(N=C4N(CCC(F)(F)C(F)(F)F)N=3)C#N)=NC(N)=C2C1(C)C(=O)NC1CC1 ZSUBGXQHVKSVKH-UHFFFAOYSA-N 0.000 description 1
- SVNIAAJRJXZBDF-UHFFFAOYSA-N 4-amino-5-methyl-2-[1-(3,3,4,4,4-pentafluorobutyl)pyrazolo[3,4-b]pyridin-3-yl]-5-(1,3,4-thiadiazol-2-yl)-7h-pyrrolo[2,3-d]pyrimidin-6-one Chemical compound O=C1NC2=NC(C=3C4=CC=CN=C4N(CCC(F)(F)C(F)(F)F)N=3)=NC(N)=C2C1(C)C1=NN=CS1 SVNIAAJRJXZBDF-UHFFFAOYSA-N 0.000 description 1
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical compound C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- LLNQWPTUJJYTTE-UHFFFAOYSA-N 4-iodopyrazole Chemical compound IC=1C=NNC=1 LLNQWPTUJJYTTE-UHFFFAOYSA-N 0.000 description 1
- 125000000590 4-methylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- WRZOMWDJOLIVQP-UHFFFAOYSA-N 5-Chloro-ortho-toluidine Chemical compound CC1=CC=C(Cl)C=C1N WRZOMWDJOLIVQP-UHFFFAOYSA-N 0.000 description 1
- PTUXQVXTJKTHSC-UHFFFAOYSA-N 5-chloro-2-(4-chlorophenyl)sulfanylaniline Chemical compound NC1=CC(Cl)=CC=C1SC1=CC=C(Cl)C=C1 PTUXQVXTJKTHSC-UHFFFAOYSA-N 0.000 description 1
- XHLAXQKXNMSVQY-UHFFFAOYSA-N 6,7-dihydro-5h-pyrrolo[2,3-d]pyrimidine-5-carboxamide Chemical compound C1=NC=C2C(C(=O)N)CNC2=N1 XHLAXQKXNMSVQY-UHFFFAOYSA-N 0.000 description 1
- UURANFWTYQPVQY-UHFFFAOYSA-N 6-chloro-1h-indazole-3-carbonitrile Chemical compound ClC1=CC=C2C(C#N)=NNC2=C1 UURANFWTYQPVQY-UHFFFAOYSA-N 0.000 description 1
- FKKMHPZZZRHKOE-UHFFFAOYSA-N 6-chloro-3-iodo-2h-indazole Chemical compound ClC1=CC=C2C(I)=NNC2=C1 FKKMHPZZZRHKOE-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- DJQOOSBJCLSSEY-UHFFFAOYSA-N Acipimox Chemical compound CC1=CN=C(C(O)=O)C=[N+]1[O-] DJQOOSBJCLSSEY-UHFFFAOYSA-N 0.000 description 1
- FHHHOYXPRDYHEZ-COXVUDFISA-N Alacepril Chemical compound CC(=O)SC[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 FHHHOYXPRDYHEZ-COXVUDFISA-N 0.000 description 1
- UXOWGYHJODZGMF-QORCZRPOSA-N Aliskiren Chemical compound COCCCOC1=CC(C[C@@H](C[C@H](N)[C@@H](O)C[C@@H](C(C)C)C(=O)NCC(C)(C)C(N)=O)C(C)C)=CC=C1OC UXOWGYHJODZGMF-QORCZRPOSA-N 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 101100262917 Arabidopsis thaliana UPP gene Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 101800001288 Atrial natriuretic factor Proteins 0.000 description 1
- 102400001282 Atrial natriuretic peptide Human genes 0.000 description 1
- 101800001890 Atrial natriuretic peptide Proteins 0.000 description 1
- XPCFTKFZXHTYIP-PMACEKPBSA-N Benazepril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C2=CC=CC=C2CC1)=O)CC1=CC=CC=C1 XPCFTKFZXHTYIP-PMACEKPBSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- TYZIQRPCDKJACL-UHFFFAOYSA-N CC(c1c(N)nc(-c2n[n](CCC(C(F)(F)F)(F)F)c3cc(I)ccc23)nc1N1)(C1=O)c1ccccc1 Chemical compound CC(c1c(N)nc(-c2n[n](CCC(C(F)(F)F)(F)F)c3cc(I)ccc23)nc1N1)(C1=O)c1ccccc1 TYZIQRPCDKJACL-UHFFFAOYSA-N 0.000 description 1
- LJVNGDIIXKSZPG-UHFFFAOYSA-N CC1(C(c2nc(NC(C3(C)c4nnc(C)[o]4)=O)c3c(NC)n2)=NN2CCC(C(F)(F)F)(F)F)C2=NC=CC1 Chemical compound CC1(C(c2nc(NC(C3(C)c4nnc(C)[o]4)=O)c3c(NC)n2)=NN2CCC(C(F)(F)F)(F)F)C2=NC=CC1 LJVNGDIIXKSZPG-UHFFFAOYSA-N 0.000 description 1
- KSFOVUSSGSKXFI-GAQDCDSVSA-N CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O Chemical compound CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O KSFOVUSSGSKXFI-GAQDCDSVSA-N 0.000 description 1
- 101100256223 Caenorhabditis elegans cho-1 gene Proteins 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- IFYLTXNCFVRALQ-OALUTQOASA-N Ceronapril Chemical compound O([C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)P(O)(=O)CCCCC1=CC=CC=C1 IFYLTXNCFVRALQ-OALUTQOASA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 229940122502 Cholesterol absorption inhibitor Drugs 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- TVZCRIROJQEVOT-CABCVRRESA-N Cromakalim Chemical compound N1([C@@H]2C3=CC(=CC=C3OC([C@H]2O)(C)C)C#N)CCCC1=O TVZCRIROJQEVOT-CABCVRRESA-N 0.000 description 1
- 238000006969 Curtius rearrangement reaction Methods 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- 101710095468 Cyclase Proteins 0.000 description 1
- 102000004654 Cyclic GMP-Dependent Protein Kinases Human genes 0.000 description 1
- 108010003591 Cyclic GMP-Dependent Protein Kinases Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 239000012594 Earle’s Balanced Salt Solution Substances 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 108010066671 Enalaprilat Proteins 0.000 description 1
- 229940118365 Endothelin receptor antagonist Drugs 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- GISRWBROCYNDME-PELMWDNLSA-N F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C Chemical compound F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C GISRWBROCYNDME-PELMWDNLSA-N 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- XQLWNAFCTODIRK-UHFFFAOYSA-N Gallopamil Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC(OC)=C(OC)C(OC)=C1 XQLWNAFCTODIRK-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 229910003803 Gold(III) chloride Inorganic materials 0.000 description 1
- 229910004039 HBF4 Inorganic materials 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 102000008015 Hemeproteins Human genes 0.000 description 1
- 108010089792 Hemeproteins Proteins 0.000 description 1
- LELOWRISYMNNSU-UHFFFAOYSA-N Hydrocyanic acid Natural products N#C LELOWRISYMNNSU-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- YQEZLKZALYSWHR-UHFFFAOYSA-N Ketamine Chemical compound C=1C=CC=C(Cl)C=1C1(NC)CCCCC1=O YQEZLKZALYSWHR-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241000270322 Lepidosauria Species 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 235000019759 Maize starch Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- ZFMITUMMTDLWHR-UHFFFAOYSA-N Minoxidil Chemical compound NC1=[N+]([O-])C(N)=CC(N2CCCCC2)=N1 ZFMITUMMTDLWHR-UHFFFAOYSA-N 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- IRLWJILLXJGJTD-UHFFFAOYSA-N Muraglitazar Chemical compound C1=CC(OC)=CC=C1OC(=O)N(CC(O)=O)CC(C=C1)=CC=C1OCCC1=C(C)OC(C=2C=CC=CC=2)=N1 IRLWJILLXJGJTD-UHFFFAOYSA-N 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- VCUFZILGIRCDQQ-KRWDZBQOSA-N N-[[(5S)-2-oxo-3-(2-oxo-3H-1,3-benzoxazol-6-yl)-1,3-oxazolidin-5-yl]methyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C1O[C@H](CN1C1=CC2=C(NC(O2)=O)C=C1)CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F VCUFZILGIRCDQQ-KRWDZBQOSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 125000000815 N-oxide group Chemical group 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 108020001621 Natriuretic Peptide Proteins 0.000 description 1
- 102000004571 Natriuretic peptide Human genes 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- ZBBHBTPTTSWHBA-UHFFFAOYSA-N Nicardipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCCN(C)CC=2C=CC=CC=2)C1C1=CC=CC([N+]([O-])=O)=C1 ZBBHBTPTTSWHBA-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- MHABMANUFPZXEB-UHFFFAOYSA-N O-demethyl-aloesaponarin I Natural products O=C1C2=CC=CC(O)=C2C(=O)C2=C1C=C(O)C(C(O)=O)=C2C MHABMANUFPZXEB-UHFFFAOYSA-N 0.000 description 1
- 239000005480 Olmesartan Substances 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 239000000026 Pentaerythritol tetranitrate Substances 0.000 description 1
- ZPHBZEQOLSRPAK-UHFFFAOYSA-N Phosphoramidon Natural products C=1NC2=CC=CC=C2C=1CC(C(O)=O)NC(=O)C(CC(C)C)NP(O)(=O)OC1OC(C)C(O)C(O)C1O ZPHBZEQOLSRPAK-UHFFFAOYSA-N 0.000 description 1
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 1
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-O Piperidinium(1+) Chemical compound C1CC[NH2+]CC1 NQRYJNQNLNOLGT-UHFFFAOYSA-O 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 241001085205 Prenanthella exigua Species 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 238000000297 Sandmeyer reaction Methods 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 108010036928 Thiorphan Proteins 0.000 description 1
- VXFJYXUZANRPDJ-WTNASJBWSA-N Trandopril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@H]2CCCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 VXFJYXUZANRPDJ-WTNASJBWSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 1
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 1
- JAWMENYCRQKKJY-UHFFFAOYSA-N [3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-ylmethyl)-1-oxa-2,8-diazaspiro[4.5]dec-2-en-8-yl]-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]methanone Chemical compound N1N=NC=2CN(CCC=21)CC1=NOC2(C1)CCN(CC2)C(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F JAWMENYCRQKKJY-UHFFFAOYSA-N 0.000 description 1
- OQQVFCKUDYMWGV-UHFFFAOYSA-N [5-[1-(phenylmethyl)-3-indazolyl]-2-furanyl]methanol Chemical compound O1C(CO)=CC=C1C(C1=CC=CC=C11)=NN1CC1=CC=CC=C1 OQQVFCKUDYMWGV-UHFFFAOYSA-N 0.000 description 1
- ZVQOOHYFBIDMTQ-UHFFFAOYSA-N [methyl(oxido){1-[6-(trifluoromethyl)pyridin-3-yl]ethyl}-lambda(6)-sulfanylidene]cyanamide Chemical compound N#CN=S(C)(=O)C(C)C1=CC=C(C(F)(F)F)N=C1 ZVQOOHYFBIDMTQ-UHFFFAOYSA-N 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- DFDGRKNOFOJBAJ-UHFFFAOYSA-N acifran Chemical compound C=1C=CC=CC=1C1(C)OC(C(O)=O)=CC1=O DFDGRKNOFOJBAJ-UHFFFAOYSA-N 0.000 description 1
- 229950000146 acifran Drugs 0.000 description 1
- 229960003526 acipimox Drugs 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 229950007884 alacepril Drugs 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229960004601 aliskiren Drugs 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- CJCSPKMFHVPWAR-JTQLQIEISA-N alpha-methyl-L-dopa Chemical compound OC(=O)[C@](N)(C)CC1=CC=C(O)C(O)=C1 CJCSPKMFHVPWAR-JTQLQIEISA-N 0.000 description 1
- 229940093740 amino acid and derivative Drugs 0.000 description 1
- 150000001413 amino acids Chemical group 0.000 description 1
- DFNYGALUNNFWKJ-UHFFFAOYSA-N aminoacetonitrile Chemical compound NCC#N DFNYGALUNNFWKJ-UHFFFAOYSA-N 0.000 description 1
- 125000005219 aminonitrile group Chemical group 0.000 description 1
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 230000000879 anti-atherosclerotic effect Effects 0.000 description 1
- 230000003276 anti-hypertensive effect Effects 0.000 description 1
- 239000000883 anti-obesity agent Substances 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 229940125708 antidiabetic agent Drugs 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 229940125710 antiobesity agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 210000002376 aorta thoracic Anatomy 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 238000006254 arylation reaction Methods 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- 229960002274 atenolol Drugs 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- FQCKMBLVYCEXJB-MNSAWQCASA-L atorvastatin calcium Chemical compound [Ca+2].C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC([O-])=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1.C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC([O-])=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 FQCKMBLVYCEXJB-MNSAWQCASA-L 0.000 description 1
- 229960001770 atorvastatin calcium Drugs 0.000 description 1
- OISFUZRUIGGTSD-LJTMIZJLSA-N azane;(2r,3r,4r,5s)-6-(methylamino)hexane-1,2,3,4,5-pentol Chemical compound N.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO OISFUZRUIGGTSD-LJTMIZJLSA-N 0.000 description 1
- YEESUBCSWGVPCE-UHFFFAOYSA-N azanylidyneoxidanium iron(2+) pentacyanide Chemical compound [Fe++].[C-]#N.[C-]#N.[C-]#N.[C-]#N.[C-]#N.N#[O+] YEESUBCSWGVPCE-UHFFFAOYSA-N 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 229960004530 benazepril Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000004622 benzoxazinyl group Chemical group O1NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 1
- 229960002781 bisoprolol Drugs 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 230000003222 cGMP degradation Effects 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- BRPQOXSCLDDYGP-UHFFFAOYSA-N calcium oxide Chemical compound [O-2].[Ca+2] BRPQOXSCLDDYGP-UHFFFAOYSA-N 0.000 description 1
- 239000000292 calcium oxide Substances 0.000 description 1
- ODINCKMPIJJUCX-UHFFFAOYSA-N calcium oxide Inorganic materials [Ca]=O ODINCKMPIJJUCX-UHFFFAOYSA-N 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- NSQLIUXCMFBZME-MPVJKSABSA-N carperitide Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(O)=O)=O)[C@@H](C)CC)C1=CC=CC=C1 NSQLIUXCMFBZME-MPVJKSABSA-N 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000017455 cell-cell adhesion Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 229940083181 centrally acting adntiadrenergic agent methyldopa Drugs 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 229950005749 ceronapril Drugs 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 229960005025 cilazapril Drugs 0.000 description 1
- JQRZBPFGBRIWSN-YOTVLOEGSA-N cilazapril monohydrate Chemical compound O.C([C@@H](C(=O)OCC)N[C@@H]1C(N2[C@@H](CCCN2CCC1)C(O)=O)=O)CC1=CC=CC=C1 JQRZBPFGBRIWSN-YOTVLOEGSA-N 0.000 description 1
- 229940090805 clavulanate Drugs 0.000 description 1
- HZZVJAQRINQKSD-PBFISZAISA-N clavulanic acid Chemical compound OC(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 HZZVJAQRINQKSD-PBFISZAISA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 229940127204 compound 29 Drugs 0.000 description 1
- 229940125807 compound 37 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 229940097499 cozaar Drugs 0.000 description 1
- 229940066901 crestor Drugs 0.000 description 1
- 229950004210 cromakalim Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 229960003280 cupric chloride Drugs 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- JMYVMOUINOAAPA-UHFFFAOYSA-N cyclopropanecarbaldehyde Chemical compound O=CC1CC1 JMYVMOUINOAAPA-UHFFFAOYSA-N 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000005595 deprotonation Effects 0.000 description 1
- 238000010537 deprotonation reaction Methods 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 229960004042 diazoxide Drugs 0.000 description 1
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 1
- WPXRJHCUCBBBDE-UHFFFAOYSA-N diethyl 2-(dicyanomethylidene)propanedioate Chemical compound CCOC(=O)C(=C(C#N)C#N)C(=O)OCC WPXRJHCUCBBBDE-UHFFFAOYSA-N 0.000 description 1
- SEKIZJHHWJROER-UHFFFAOYSA-N diethyl 2-acetamido-2-(5-chloropyridin-2-yl)propanedioate Chemical compound CCOC(=O)C(NC(C)=O)(C(=O)OCC)C1=CC=C(Cl)C=N1 SEKIZJHHWJROER-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 1
- 125000005049 dihydrooxadiazolyl group Chemical group O1N(NC=C1)* 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- SPCNPOWOBZQWJK-UHFFFAOYSA-N dimethoxy-(2-propan-2-ylsulfanylethylsulfanyl)-sulfanylidene-$l^{5}-phosphane Chemical compound COP(=S)(OC)SCCSC(C)C SPCNPOWOBZQWJK-UHFFFAOYSA-N 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229940030606 diuretics Drugs 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- 229960001484 edetic acid Drugs 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 229950005627 embonate Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 229960002680 enalaprilat Drugs 0.000 description 1
- MZYVOFLIPYDBGD-MLZQUWKJSA-N enalaprilat dihydrate Chemical compound O.O.C([C@H](N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 MZYVOFLIPYDBGD-MLZQUWKJSA-N 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 239000002308 endothelin receptor antagonist Substances 0.000 description 1
- 150000002085 enols Chemical group 0.000 description 1
- 208000002854 epidermolysis bullosa simplex superficialis Diseases 0.000 description 1
- 229950000206 estolate Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- LJQKCYFTNDAAPC-UHFFFAOYSA-N ethanol;ethyl acetate Chemical compound CCO.CCOC(C)=O LJQKCYFTNDAAPC-UHFFFAOYSA-N 0.000 description 1
- AEOCXXJPGCBFJA-UHFFFAOYSA-N ethionamide Chemical compound CCC1=CC(C(N)=S)=CC=N1 AEOCXXJPGCBFJA-UHFFFAOYSA-N 0.000 description 1
- SRCZQMGIVIYBBJ-UHFFFAOYSA-N ethoxyethane;ethyl acetate Chemical compound CCOCC.CCOC(C)=O SRCZQMGIVIYBBJ-UHFFFAOYSA-N 0.000 description 1
- NRWAZJGPHXIACF-UHFFFAOYSA-N ethyl 2-(dicyanomethyl)-2-methylbut-3-ynoate Chemical compound CCOC(=O)C(C)(C#C)C(C#N)C#N NRWAZJGPHXIACF-UHFFFAOYSA-N 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical class CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 210000001105 femoral artery Anatomy 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229960000457 gallopamil Drugs 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- RJHLTVSLYWWTEF-UHFFFAOYSA-K gold trichloride Chemical compound Cl[Au](Cl)Cl RJHLTVSLYWWTEF-UHFFFAOYSA-K 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 125000005241 heteroarylamino group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229960002474 hydralazine Drugs 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- YPGCWEMNNLXISK-UHFFFAOYSA-N hydratropic acid Chemical compound OC(=O)C(C)C1=CC=CC=C1 YPGCWEMNNLXISK-UHFFFAOYSA-N 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- DCPMPXBYPZGNDC-UHFFFAOYSA-N hydron;methanediimine;chloride Chemical compound Cl.N=C=N DCPMPXBYPZGNDC-UHFFFAOYSA-N 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- CREVBWLEPKAZBH-UHFFFAOYSA-M hydron;tetraethylazanium;sulfate Chemical compound OS([O-])(=O)=O.CC[N+](CC)(CC)CC CREVBWLEPKAZBH-UHFFFAOYSA-M 0.000 description 1
- 125000000687 hydroquinonyl group Chemical class C1(O)=C(C=C(O)C=C1)* 0.000 description 1
- TUJKJAMUKRIRHC-UHFFFAOYSA-N hydroxyl Chemical compound [OH] TUJKJAMUKRIRHC-UHFFFAOYSA-N 0.000 description 1
- 229940090022 hyzaar Drugs 0.000 description 1
- 229960001195 imidapril Drugs 0.000 description 1
- KLZWOWYOHUKJIG-BPUTZDHNSA-N imidapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1C(N(C)C[C@H]1C(O)=O)=O)CC1=CC=CC=C1 KLZWOWYOHUKJIG-BPUTZDHNSA-N 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000004857 imidazopyridinyl group Chemical group N1C(=NC2=C1C=CC=N2)* 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004245 indazol-3-yl group Chemical group [H]N1N=C(*)C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 150000002473 indoazoles Chemical class 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229940030980 inova Drugs 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 229960004903 invert sugar Drugs 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 230000002083 iodinating effect Effects 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- DXDRHHKMWQZJHT-FPYGCLRLSA-N isoliquiritigenin Chemical compound C1=CC(O)=CC=C1\C=C\C(=O)C1=CC=C(O)C=C1O DXDRHHKMWQZJHT-FPYGCLRLSA-N 0.000 description 1
- JBQATDIMBVLPRB-UHFFFAOYSA-N isoliquiritigenin Natural products OC1=CC(O)=CC=C1C1OC2=CC(O)=CC=C2C(=O)C1 JBQATDIMBVLPRB-UHFFFAOYSA-N 0.000 description 1
- 235000008718 isoliquiritigenin Nutrition 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- FMKOJHQHASLBPH-UHFFFAOYSA-N isopropyl iodide Chemical compound CC(C)I FMKOJHQHASLBPH-UHFFFAOYSA-N 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229960003299 ketamine Drugs 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-M lactobionate Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-M 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- NXFFJDQHYLNEJK-CYBMUJFWSA-N laropiprant Chemical compound C=1([C@@H](CC(O)=O)CCC=1C=1C=C(F)C=C(C2=1)S(=O)(=O)C)N2CC1=CC=C(Cl)C=C1 NXFFJDQHYLNEJK-CYBMUJFWSA-N 0.000 description 1
- 229950008292 laropiprant Drugs 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 229940095570 lescol Drugs 0.000 description 1
- 230000023404 leukocyte cell-cell adhesion Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229940002661 lipitor Drugs 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- DBTNVRCCIDISMV-UHFFFAOYSA-L lithium;magnesium;propane;dichloride Chemical compound [Li+].[Mg+2].[Cl-].[Cl-].C[CH-]C DBTNVRCCIDISMV-UHFFFAOYSA-L 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- UTEFBSAVJNEPTR-RGEXLXHISA-N loprazolam Chemical compound C1CN(C)CCN1\C=C/1C(=O)N2C3=CC=C([N+]([O-])=O)C=C3C(C=3C(=CC=CC=3)Cl)=NCC2=N\1 UTEFBSAVJNEPTR-RGEXLXHISA-N 0.000 description 1
- 229960003019 loprazolam Drugs 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 229910001623 magnesium bromide Inorganic materials 0.000 description 1
- VFZXMEQGIIWBFJ-UHFFFAOYSA-M magnesium;cyclopropane;bromide Chemical compound [Mg+2].[Br-].C1C[CH-]1 VFZXMEQGIIWBFJ-UHFFFAOYSA-M 0.000 description 1
- LROBJRRFCPYLIT-UHFFFAOYSA-M magnesium;ethyne;bromide Chemical compound [Mg+2].[Br-].[C-]#C LROBJRRFCPYLIT-UHFFFAOYSA-M 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-L malate(2-) Chemical compound [O-]C(=O)C(O)CC([O-])=O BJEPYKJPYRNKOW-UHFFFAOYSA-L 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- VPNGEIHDPSLNMU-UHFFFAOYSA-N medetomidine hydrochloride Chemical compound Cl.C=1C=CC(C)=C(C)C=1C(C)C1=CNC=N1 VPNGEIHDPSLNMU-UHFFFAOYSA-N 0.000 description 1
- 230000007087 memory ability Effects 0.000 description 1
- 230000007334 memory performance Effects 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- ZXUQEPZWVQIOJE-UHFFFAOYSA-N methyl 2-chloro-2-oxoacetate Chemical compound COC(=O)C(Cl)=O ZXUQEPZWVQIOJE-UHFFFAOYSA-N 0.000 description 1
- GWHHQWUYMDWMAY-UHFFFAOYSA-N methyl 2-cyanopropanoate Chemical compound COC(=O)C(C)C#N GWHHQWUYMDWMAY-UHFFFAOYSA-N 0.000 description 1
- LJPXXKAIOPSZEM-UHFFFAOYSA-N methyl 2-methyl-3-oxo-3-(prop-2-ynylamino)propanoate Chemical compound COC(=O)C(C)C(=O)NCC#C LJPXXKAIOPSZEM-UHFFFAOYSA-N 0.000 description 1
- DZIQUZJSNSZOCH-UHFFFAOYSA-N methyl 2-phenylpropanoate Chemical compound COC(=O)C(C)C1=CC=CC=C1 DZIQUZJSNSZOCH-UHFFFAOYSA-N 0.000 description 1
- ORAKNQSHWMHCEY-UHFFFAOYSA-N methyl 2-pyridin-2-ylacetate Chemical compound COC(=O)CC1=CC=CC=N1 ORAKNQSHWMHCEY-UHFFFAOYSA-N 0.000 description 1
- SILUMMDUAFHRKF-UHFFFAOYSA-N methyl 3,3-dicyano-2-methyl-2-pyrazin-2-ylpropanoate Chemical compound COC(=O)C(C)(C(C#N)C#N)C1=CN=CC=N1 SILUMMDUAFHRKF-UHFFFAOYSA-N 0.000 description 1
- JIBXSTBPZAAXFS-UHFFFAOYSA-N methyl 3,3-dicyano-2-methyl-2-pyridin-2-ylpropanoate Chemical compound COC(=O)C(C)(C(C#N)C#N)C1=CC=CC=N1 JIBXSTBPZAAXFS-UHFFFAOYSA-N 0.000 description 1
- 229940102396 methyl bromide Drugs 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- LRMHVVPPGGOAJQ-UHFFFAOYSA-N methyl nitrate Chemical compound CO[N+]([O-])=O LRMHVVPPGGOAJQ-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 230000001035 methylating effect Effects 0.000 description 1
- XSGHLZBESSREDT-UHFFFAOYSA-N methylenecyclopropane Chemical class C=C1CC1 XSGHLZBESSREDT-UHFFFAOYSA-N 0.000 description 1
- 229940099246 mevacor Drugs 0.000 description 1
- 229960003632 minoxidil Drugs 0.000 description 1
- 235000019426 modified starch Nutrition 0.000 description 1
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical compound CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 229950006549 moveltipril Drugs 0.000 description 1
- 229950001135 muraglitazar Drugs 0.000 description 1
- ACTNHJDHMQSOGL-UHFFFAOYSA-N n',n'-dibenzylethane-1,2-diamine Chemical compound C=1C=CC=CC=1CN(CCN)CC1=CC=CC=C1 ACTNHJDHMQSOGL-UHFFFAOYSA-N 0.000 description 1
- AEXITZJSLGALNH-UHFFFAOYSA-N n'-hydroxyethanimidamide Chemical compound CC(N)=NO AEXITZJSLGALNH-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000000692 natriuretic peptide Substances 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 229960001783 nicardipine Drugs 0.000 description 1
- LBHIOVVIQHSOQN-UHFFFAOYSA-N nicorandil Chemical compound [O-][N+](=O)OCCNC(=O)C1=CC=CN=C1 LBHIOVVIQHSOQN-UHFFFAOYSA-N 0.000 description 1
- 229960002497 nicorandil Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- VZWXXKDFACOXNT-UHFFFAOYSA-N niludipine Chemical compound CCCOCCOC(=O)C1=C(C)NC(C)=C(C(=O)OCCOCCC)C1C1=CC=CC([N+]([O-])=O)=C1 VZWXXKDFACOXNT-UHFFFAOYSA-N 0.000 description 1
- 229950000109 niludipine Drugs 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 229960002460 nitroprusside Drugs 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 235000021590 normal diet Nutrition 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000004533 oil dispersion Substances 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-M oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC([O-])=O ZQPPMHVWECSIRJ-KTKRTIGZSA-M 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- VTRAEEWXHOVJFV-UHFFFAOYSA-N olmesartan Chemical compound CCCC1=NC(C(C)(C)O)=C(C(O)=O)N1CC1=CC=C(C=2C(=CC=CC=2)C=2NN=NN=2)C=C1 VTRAEEWXHOVJFV-UHFFFAOYSA-N 0.000 description 1
- 229960005117 olmesartan Drugs 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 229940082615 organic nitrates used in cardiac disease Drugs 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- LVSJDHGRKAEGLX-UHFFFAOYSA-N oxolane;2,2,2-trifluoroacetic acid Chemical compound C1CCOC1.OC(=O)C(F)(F)F LVSJDHGRKAEGLX-UHFFFAOYSA-N 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- FAXGPCHRFPCXOO-LXTPJMTPSA-N pepstatin A Chemical class OC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)CC(C)C FAXGPCHRFPCXOO-LXTPJMTPSA-N 0.000 description 1
- 125000001151 peptidyl group Chemical group 0.000 description 1
- 229960002582 perindopril Drugs 0.000 description 1
- IPVQLZZIHOAWMC-QXKUPLGCSA-N perindopril Chemical compound C1CCC[C@H]2C[C@@H](C(O)=O)N(C(=O)[C@H](C)N[C@@H](CCC)C(=O)OCC)[C@H]21 IPVQLZZIHOAWMC-QXKUPLGCSA-N 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000003444 phase transfer catalyst Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- BWSDNRQVTFZQQD-AYVHNPTNSA-N phosphoramidon Chemical compound O([P@@](O)(=O)N[C@H](CC(C)C)C(=O)N[C@H](CC=1[C]2C=CC=CC2=NC=1)C(O)=O)[C@H]1O[C@@H](C)[C@H](O)[C@@H](O)[C@@H]1O BWSDNRQVTFZQQD-AYVHNPTNSA-N 0.000 description 1
- 108010072906 phosphoramidon Proteins 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229960002310 pinacidil Drugs 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 239000003375 plant hormone Substances 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 230000010118 platelet activation Effects 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 239000008389 polyethoxylated castor oil Substances 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 229940089484 pravachol Drugs 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- IENZQIKPVFGBNW-UHFFFAOYSA-N prazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1=CC=CO1 IENZQIKPVFGBNW-UHFFFAOYSA-N 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- JKANAVGODYYCQF-UHFFFAOYSA-N prop-2-yn-1-amine Chemical compound NCC#C JKANAVGODYYCQF-UHFFFAOYSA-N 0.000 description 1
- UZQBKCWYZBHBOW-UHFFFAOYSA-N propan-2-yl 4-cyclohexyl-2-hydroxy-3-[[3-methylsulfanyl-2-[[2-(morpholine-4-carbonylamino)-3-phenylpropanoyl]amino]propanoyl]amino]butanoate Chemical compound C1CCCCC1CC(C(O)C(=O)OC(C)C)NC(=O)C(CSC)NC(=O)C(NC(=O)N1CCOCC1)CC1=CC=CC=C1 UZQBKCWYZBHBOW-UHFFFAOYSA-N 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 229950003776 protoporphyrin Drugs 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 101150024359 pyrR gene Proteins 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- PJESVVYWPFAJCS-UHFFFAOYSA-N pyridazine-3-carbonitrile Chemical compound N#CC1=CC=CN=N1 PJESVVYWPFAJCS-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- BXCQRPSSKGRMDD-UHFFFAOYSA-N pyrimidin-5-yl carbamate Chemical compound NC(=O)OC1=CN=CN=C1 BXCQRPSSKGRMDD-UHFFFAOYSA-N 0.000 description 1
- AHRQYOSAXZQCIG-UHFFFAOYSA-N pyrimidine-5-carbonitrile Chemical compound N#CC1=C=NC=N[CH]1 AHRQYOSAXZQCIG-UHFFFAOYSA-N 0.000 description 1
- DLSOCVWDXFSIDM-UHFFFAOYSA-N pyrimidine-5-carbothioamide Chemical compound NC(=S)C1=CN=CN=C1 DLSOCVWDXFSIDM-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 229960001455 quinapril Drugs 0.000 description 1
- JSDRRTOADPPCHY-HSQYWUDLSA-N quinapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC=CC=C2C1)C(O)=O)CC1=CC=CC=C1 JSDRRTOADPPCHY-HSQYWUDLSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 230000002040 relaxant effect Effects 0.000 description 1
- 230000004648 relaxation of smooth muscle Effects 0.000 description 1
- UXIGZRQVLGFTOU-VQXQMPIVSA-N remikiren Chemical compound C([C@H](CS(=O)(=O)C(C)(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC1CCCCC1)[C@@H](O)[C@@H](O)C1CC1)C1=CC=CC=C1 UXIGZRQVLGFTOU-VQXQMPIVSA-N 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- LALFOYNTGMUKGG-BGRFNVSISA-L rosuvastatin calcium Chemical compound [Ca+2].CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O.CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O LALFOYNTGMUKGG-BGRFNVSISA-L 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 1
- 230000001235 sensitizing effect Effects 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 description 1
- 235000010378 sodium ascorbate Nutrition 0.000 description 1
- 229960005055 sodium ascorbate Drugs 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000001476 sodium potassium tartrate Substances 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical class [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 1
- 229940127296 soluble guanylate cyclase stimulator Drugs 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 229960002909 spirapril Drugs 0.000 description 1
- HRWCVUIFMSZDJS-SZMVWBNQSA-N spirapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2(C1)SCCS2)C(O)=O)CC1=CC=CC=C1 HRWCVUIFMSZDJS-SZMVWBNQSA-N 0.000 description 1
- 108700035424 spirapril Proteins 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 125000004426 substituted alkynyl group Chemical group 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 125000003375 sulfoxide group Chemical group 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 229960004084 temocapril Drugs 0.000 description 1
- FIQOFIRCTOWDOW-BJLQDIEVSA-N temocapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C[C@H](SC1)C=1SC=CC=1)=O)CC1=CC=CC=C1 FIQOFIRCTOWDOW-BJLQDIEVSA-N 0.000 description 1
- 229950002757 teoclate Drugs 0.000 description 1
- KSYCMLWDBIPVEH-UHFFFAOYSA-N tert-butyl 2-pyrazin-2-ylpropanoate Chemical compound CC(C)(C)OC(=O)C(C)C1=CN=CC=N1 KSYCMLWDBIPVEH-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- JAELLLITIZHOGQ-UHFFFAOYSA-N tert-butyl propanoate Chemical compound CCC(=O)OC(C)(C)C JAELLLITIZHOGQ-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Natural products C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- ZWZVWGITAAIFPS-UHFFFAOYSA-N thiophosgene Chemical compound ClC(Cl)=S ZWZVWGITAAIFPS-UHFFFAOYSA-N 0.000 description 1
- LJJKNPQAGWVLDQ-SNVBAGLBSA-N thiorphan Chemical compound OC(=O)CNC(=O)[C@@H](CS)CC1=CC=CC=C1 LJJKNPQAGWVLDQ-SNVBAGLBSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 229960002051 trandolapril Drugs 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- LENLQGBLVGGAMF-UHFFFAOYSA-N tributyl([1,2,4]triazolo[1,5-a]pyridin-6-yl)stannane Chemical compound C1=C([Sn](CCCC)(CCCC)CCCC)C=CC2=NC=NN21 LENLQGBLVGGAMF-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- JABYJIQOLGWMQW-UHFFFAOYSA-N undec-4-ene Chemical compound CCCCCCC=CCCC JABYJIQOLGWMQW-UHFFFAOYSA-N 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- 229960002381 vardenafil Drugs 0.000 description 1
- 230000001196 vasorelaxation Effects 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- PNAMDJVUJCJOIX-XVZWKFLSSA-N vytorin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1.N1([C@@H]([C@H](C1=O)CC[C@@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 PNAMDJVUJCJOIX-XVZWKFLSSA-N 0.000 description 1
- 229940009349 vytorin Drugs 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 229940051223 zetia Drugs 0.000 description 1
- 229940072168 zocor Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- Cyclic GMP is an important intracellular messenger which triggers a multitude of different effects via the modulation of cGMP-dependent protein kinases, phosphodiesterases and ion channels. Examples are the relaxation of smooth muscles, the inhibition of thrombocyte activation and the inhibition of the proliferation of smooth-muscle cells and of leukocyte adhesion.
- cGMP is produced by particulate and soluble guanylate cyclases as a response to a number of extracellular and intracellular stimuli. In the case of the particulate guanylate cyclases, stimulation is essentially effected by peptidic messengers, such as the atrial natriuretic peptide or the cerebral natriuretic peptide.
- sGC soluble guanylate cyclases
- sGC soluble guanylate cyclases
- the most important stimulant is nitrogen monoxide ("NO") or a closely related species.
- NO nitrogen monoxide
- the function of other factors such as carbon monoxide or the hydroxyl radical is still largely unclear.
- NO nitrogen monoxide
- the binding of NO to the heme with formation of a penta-coordinate heme-nitrosyl complex is proposed as the mechanism of the activation by NO.
- the associated release of the histidine which is bound in the basal state to the iron converts the enzyme into the active conformation.
- Active soluble guanylate cyclases are composed of an a and a ⁇ subunit each.
- subunit subtypes differ from one another with respect to sequence, tissue-specific distribution and expression in different development stages.
- the subtypes oq and ⁇ are mainly expressed in brain and lung, while ⁇ 2 is found in particular in liver and kidney.
- the subtype oc2 was shown to be present in human fetal brain.
- the subunits referred to as s and P3 were isolated from human brain and are homologous to a ⁇ and ⁇ . More recent works indicate an ⁇ x2i subunit which contains an insert in the catalytic domain. All subunits show great homologies in the region of the catalytic domain.
- the enzymes presumably contain one heme per heterodimer, which is bound via P i-Cys-78 and/or pi-His-105 and is part of the regulatory center.
- guanylate-cyclase-activating factors can be reduced, or their degradation may be promoted owing to the increased occurrence of free radicals.
- the resulting reduced activation of the sGC leads, via a weakening of the respective cGMP-mediated cellular response, for example to an increase of the blood pressure, to platelet activation or to increased cell proliferation and cell adhesion.
- a weakening of the respective cGMP-mediated cellular response for example to an increase of the blood pressure, to platelet activation or to increased cell proliferation and cell adhesion.
- endothelial dysfunction, atherosclerosis, hypertension, stable or unstable angina pectoris, thromboses, myocardial infarction, strokes or erectile dysfunction results.
- Pharmacological stimulation of sGC offers a possibility to normalize cGMP production and therefore makes possible the treatment and/or prevention of such disorders.
- the present invention relates to compounds which activate soluble guanylate cyclase and are valuable pharmaceutically active compounds for the therapy and prophylaxis of diseases, for example for cardiovascular diseases such as hypertension, heart failure, pulmonary hypertension, angina pectoris, diabetes, cardiac insufficiency, thromboses or atherosclerosis.
- cardiovascular diseases such as hypertension, heart failure, pulmonary hypertension, angina pectoris, diabetes, cardiac insufficiency, thromboses or atherosclerosis.
- the invention furthermore relates to processes for preparing compounds of Formula I, to their use for the therapy and prophylaxis of the abovementioned diseases and for preparing pharmaceuticals for this purpose, and to pharmaceutical preparations which comprise compounds of Formula I.
- the invention concerns compounds of Formula I which activate soluble guanylate cyclase:
- Each ⁇ , X ⁇ , ⁇ 3 and X ⁇ is independently N or CH, provided that no more than two of ⁇ , ⁇ 2 , x3 and X 4 is N;
- Each R x and RY are independently H, C3- 1 ⁇ cycloalkyl, or -C ⁇ -Cg alkyl;
- Each R1 is independently -H, halo, OR, -Cj-Cg alkyl, aryl, heterocyclyl, heteroaryl, -C3-1Q cycloalkyl, -CN, -NR a C(0)R b ⁇ or -C(0)NR a R b , said aryl, heteroaryl, and cycloalkyl optionally being substituted with one to three substituents selected from halo, -Cj-Cg alkyl, -OR, -CN, and -CF3;
- R2 is -(CR3 ⁇ 4)tCi-C 6 alkyl, -C 2 -C 6 alkenyl, -C 2 -C 6 alkynyl, -(CR3 ⁇ 4) t OR, -(CR ⁇ 1 ⁇ 2) t SR, -(CR d 2 ) t CF 3 , -(CR d 2 ) t C 3 - 10 cycloalkyl, -(CR d 2 ) r aryl, -(CR d 2 ) r heterocyclyl or - (CR d 2 )theteroaryl, said alkyl, cycloalkyl, aryl, heterocyclyl and heteroaryl being optionally substituted with one to three substituents selected from halo, -Cj-Cg alkyl, -CF3, -CN or -OR;
- R 3 is -(CR d 2 ) t -aryl, -(CR d 2 ) t -heteroaryl, -(CR d 2 ) t -heterocyclyl, -(CR d 2 ) t C3-i 0 cycloalkyl, -(CR d 2 ) t CN, -(CR 2 ) t -C(0)NR Rb -(CR d 2 ) t -NR C(0)Rb -(CR d 2 ) t -C(S)NR a Rb, -(CR 2 ) r C(0)OR a , -(CR d 2 ) r NR a C(0)NR b , -(CR d 2 ) t ⁇ NR C(0)0R a ⁇ (CR d 2 ) r NR R b , or-OR a , said, aryl, heteroaryl or heterocyclyl are optionally substitute
- R4 is -C ⁇ -C alkyl, C3-iQcycloalkyl, halo or CF3;
- Each R5 is independently halo, OR, CN, -(CR3 ⁇ 4) t CF 3 , S(0) p R C -(CR d 2 ) t C3-iocycloalkyl, or - Cj-Cg alkyl, said alkyl and cycloalkyl being optionally substituted with one to three substituents selected from halo or OR;
- Each 6 is independently halo, -Cj-Cg alkyl, OR, CN, CF3, aryl or heteroaryl, where said alkyl, aryl or heteroaryl are optionally subsituted with halo, Cj-Cg alkyl or CF3;
- Each R is independently -H, -Cj -Cg alkyl, -CF3, or aryl;
- Each R and R ⁇ are independently -H, -C ⁇ -Cg alkyl, aryl, heteroaryl, heterocyclyl, or
- R a and R ⁇ may be cyclized to form a C3-C6 cycloalkyl ring;
- Each R c is independently -Cj-Cg alkyl, -CF3, or aryl;
- Each R d is independently H, halo, -CF3 or -Cj-Cg alkyl;
- m is an integer selected from 1, 2, or 3;
- p is an integer independently selected from 0, 1 or 2;
- t is an integer independently selected from 0, 1, 2, 3, or 4.
- the invention is directed to compound of Formula I having structural Formula IA:
- Each ⁇ , ⁇ 2, ⁇ 3 and X ⁇ are independently N or CH, provided that no more than two of Xl, x2 X ⁇ and X 4 is N;
- Each R is independently -H, -C ⁇ -Cg alkyl, -CF3, or aryl;
- Each R a and R ⁇ are independently-H, -C ⁇ -C5 alkyl, aryl, heteroaryl, heterocyclyl, or -C3-10 cycloalkyl, wherein said alkyl, heteroaryl, heterocyclyl, and cycloalkyl are optionally substituted with one to three substituents selected R ⁇ ; optionally, when R a and R b are -C ⁇ -Cg alkyl and are attached to the same nitrogen atom, R a and R b may be cyclized to form a C3-C6 cycloalkyl ring;
- Each R c is independently -Cj-Cg alkyl, -CF3, or aryl;
- Each R d is independently H, halo, -CF3 or -C ⁇ -Cg alkyl
- Each R1 is independently -H, halo, aryl, heterocyclyl, heteroaryl, -C3-1Q cycloalkyl, -CN, -
- NR a C(0)R b or -C(0)NR a R b said aryl, heteroaryl, and cycloalkyl optionally being substituted with one to three substituents selected from halo, -C1-C6 alkyl, -OR, -CN, and -CF3;
- R2 is -C!-C 6 alkyl, -C 2 -C 6 alkenyl, -C 2 -C 6 alkynyl, -(CR3 ⁇ 4)tOR, -(CR3 ⁇ 4) t SR, -(CR3 ⁇ 4) t CF3,
- R3 is aryl, heteroaiyl, heterocyclyl, CN, -C(0)NR a R b , -NR a C(0)R b -C(S)NR a R , -C(0)OR - NR a C(0)NR b , ⁇ NR a C(0)OR a , -NR a R b , or -0R said, aryl, heteroaryl or heterocyclyl are optionally substituted with from one to three substituents selected R ⁇ ;
- R 4 is -Ci-C 6 alkyl, halo or CF3;
- Each R 5 is independently halo, OR, CN, S(0) p R c , or -Cj-Cg alkyl, said alkyl being optionally substituted with one to three substituents selected from halo or OR;
- Each R6 is independently halo, -Cj-Cg alkyl, OR, CN, CF3, aryl or heteroaryl, where said alkyl, aryl or heteroaryl are optionally subsituted with halo, C ⁇ -Cg alkyl or CF3; m is an integer selected from 1, 2, or 3;
- p is an integer independently selected from 0, 1 or 2;
- t is an integer independently selected from 0, 1, 2, 3, or 4,
- the invention is directed to compounds of Formula I having structural Formula IA:
- ⁇ , ⁇ 2 x3 a nd X 4 are independently selected from N or CH, provided that no more than one of X 1 , ⁇ 2, ⁇ 3 and X ⁇ is N; and all other variables are as previously defined in Formula ⁇ .
- said aryl, heteroaryl or heterocyclyl are optionally substituted with from one to three substituents selected from halo, OR, CN, S(0)pR c , or -Cj-Cg alkyl, said alkyl being optionally substituted with one to three substituents selected from halo or OR; and all other variables are as previously defined in Formula I.
- R ⁇ is -C ⁇ -Cg alkyl; and all other variables are as previously defined in Formula IA.
- the invention is directed to compounds of Formula I having structural Formual II:
- Each R is independently -H, -Ci -Cg alkyl, -CF3, or aryl;
- Each R a is independently -H or -Ci -C ⁇ alkyl
- Each Rk is independently-H, -Ci -C5 alkyl or -C ⁇ Q cycloalkyi, wherein said alkyl and cycloalkyi are optionally substituted with one to three substituents selected R ⁇ ;
- Each R c is independently -C ⁇ -C$ alkyl, -CF3, or aryl;
- Each R d is independently H, halo, -CF3 or -C ⁇ -Cg alkyl; Each is independently -H, CN, halo or -Cj-Cg alkyl, said alkyl optionally being substituted with one to three substituents selected from halo, -Cj-Cg alkyl, and -CF3;
- R2 is -Ci-C 6 alkyl, -(CR d 2 )tCF 3 , -(CR d 2 )rC3-iocycloalkyl, or -(CR3 ⁇ 4) t aryl, said alkyl, cycloalkyi and aryl being optionally substituted with one to three substituents selected from halo, -Cj-Cs alkyl and -CF 3 ;
- R3 is aryl, heteroaryl, heterocyclyl, CN, -C(0)NR a R ⁇ , -NR a C(0)R ⁇ s -C(0)OR a or -OR a , said aryl, heteroaryl or heterocyclyl are optionally substituted with from one to three substituent selected R ⁇ ;
- Each R 5 is independently halo, OR, CN, S(0) p R c , or -C
- Each R is independently halo, -Cj-Cg alkyl, OR, CN, CF3, aryl or heteroaryl, where said alkyl, aryl or heteroaryl are optionally subsituted with halo, C]-Cg alkyl or CF3;
- m is an integer selected from 1, 2, or 3;
- p is an integer independently selected from 0, 1 or 2;
- t is an integer independently selected from 0, 1, 2, 3, or 4.
- the invention is directed to compounds of Formula I having structural Formual III:
- X 4 is CH orN
- Each R is independently -H, -Cj-Cg alkyl, -CF3, or aryl;
- Each R a is independently -H or -Cj-Cg alkyl;
- Each Rb is independently-H, -Cj-Cg alkyl, -C3-10 cycloalkyl or heteroaryl, wherein said alkyl, cycloalkyl and heteroaryl are optionally substituted with one to three substituents selected R ⁇ ;
- Each R c is independently -Cj-Cg alkyl, -CF3, or aryl;
- Each R d is independently H, halo, -CF3 or -C1-C6 alkyl;
- Each R 1 is independently -H, OR, CN, halo or -C1-C6 alkyl, said alkyl optionally being substituted with one to three substituents selected from halo, -C ⁇ -C£ alkyl, and -CF3;
- R2 is -(CRd 2 ) t Ci ⁇ C alkyl, -(CR3 ⁇ 4) t CF 3 , ⁇ (CR ⁇ 1 ⁇ 2) R C3-i 0 cycloalkyl, or -(CR d 2 ) t aryl, said alkyl, cycloalkyl and aryl being optionally substituted with one to three substituents selected from halo, -C ⁇ -C ⁇ alkyl and -CF 3 ;
- R 3 is aryl, heteroaryl, heterocyclyl, CN, -C(0)NR a R b , -NR a C(0)R ⁇ 5 ⁇ C(0)OR a , or -OR* said alkyl, aryl, heteroaryl or heterocyclyl are optionally substituted with from one to three substituent selected R ⁇ ;
- R4 is -CH 3 or C 3 -i0cycloalkyl
- Each 5 is independently halo, OR, CN, S(0)pR c , or -Ci -Cg alkyl, said alkyl being optionally substituted with one to three substituents selected from halo, -C3-1 ocycloalkyl or OR;
- Each R is independently halo, -C ⁇ -CQ alkyl, OR, CN, CF 3 , aryl or heteroaryl, where said alkyl, aryl or heteroaryl are optionally subsituted with halo, C ⁇ -C ⁇ alkyl or CF3; m is an integer selected from 1, 2, or 3;
- p is an integer independently selected from 0, 1 or 2;
- t is an integer independently selected from 0, 1, 2, 3, or 4.
- a compound of the instant invention is:
- a compound of the instant invention is: EXAMPLE IUPAC NAME
- Rl can be attached to any available carbon atom ring.
- alkyl is intended to include both branched- and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms.
- cycloalkyl means carbocycles containing no heteroatoms. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl,
- cycloalkyl is cyclopropyl.
- alkyl groups are used throughout the specification, e.g. methyl may be represented by conventional abbreviations including "Me” or CB 3 or a symbol that is an extended bond without defined terminal group, e.g. ⁇
- ethyl may be represented by "Et” or CH 2 CH 3
- propyl may be represented by "Pr” or CH 2 CH 2 CH 3i butyl may be represented by "Bu” or CH 2 CH 2 CH 2 CH 3 , etc.
- C ⁇ -6 alkyl (or “Ci-C6 alkyl”) for example, means linear or branched chain alkyl groups, including all isomers, having the specified number of carbon atoms.
- Ci-6 alkyl includes all of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and t- butyl, n- and isopropyl, ethyl and methyl.
- Cl_4 alkyl means n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl. If no number is specified, 1-10 carbon atoms are intended for linear or branched alkyl groups.
- C ⁇ -6 alkyl wherein the alkyl group may be unsubstututed or substituted with 1-3 fluorine atoms
- alkenyl unless otherwise indicated, means carbon chains which contain at least one carbon-carbon double bond, and which may be linear or branched or combinations thereof. Examples of alkenyl include, but are not limited to, vinyl, allyl, isopropenyl, pentenyl, hexenyl, heptenyl, 1-propenyl, 2-butenyl, 2-methyl-2-butenyl, and the like,
- cycloalkenyl means carbocycles containing no heteroatoms having at least one carbon-carbon double bond.
- alkynyl refers to a hydrocarbon radical straight, branched or cyclic, containing from 2 to 10 carbon atoms and at least one carbon to carbon triple bond.
- C2-C6 alkynyl means an alkynyl radical having from 2 to 6 carbon atoms.
- Alkynyl groups include ethynyl, propynyl, butynyl, 3- methylbutynyl and so on.
- the straight, branched or cyclic portion of the alkynyl group may contain triple bonds and may be substituted if a substituted alkynyl group is indicated.
- Aryl unless otherwise indicated, means mono- and bicyclic aromatic rings containing 6-12 carbon atoms. Examples of aryl include, but are not limited to, phenyl, naphthyl, indenyl and the like. “Aryl” also includes monocyclic rings fused to an aryl group. Examples include tetrahydronaphthyl, indanyl and the like. In an embodiment, aryl is phenyl.
- Heteroaryl unless otherwise indicated, means a mono- or bicyclic aromatic ring or ring system having 5 to 10 atoms and containing at least one heteroatom selected from 0, S and N, .
- Examples include, but are not limited to, pyrrolyl, isoxazolyl, isothlazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, l,3,4-oxadiazolyl-2(3H)-one, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyridinyl, pyrimidinyl, pyrimidyl, pyridazinyl, pyrazinyl, and the like.
- Heteroaryl also includes aromatic heterocyclic groups fused to heterocycles that are non-aromatic or partially aromatic, and aromatic heterocyclic groups fused to cycloalkyl rings. Additional examples of heteroaryls include, but are not limited to, indazolyl, thienopyrazolyl, imidazopyridazinyl, pyrazolopyrazolyl, pyrazolopyridinyi, imidazopyridinyl and imidazothiazolyl. Heteroaryl also includes such groups in charged form, e.g., pyridinium.
- heteroaryl is imidazolyl, indazolyl, oxadiazolyl, l,3,4-oxadiazolyl-2(3H)-one, pyrimidinyl, pyridinyl, pyrazolyl, thiadiazolyl, triazolyl, tetrazolyl or thiazolyl.
- Heterocyclyl unless otherwise indicated, means a 5- or 6-membered
- heterocyclyl examples include, but are not limited to, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, imidazolidinyl, 2,3- dihydrofuro(2,3-b)pyridyl, benzoxazinyl, oxetanyl, tetrahydrofuranyl or tetrahydropyranyl and the like,
- the term also includes partially unsaturated monocyclic rings that are not aromatic, such as 2- or 4-pyridones attached through the nitrogen or N-substituted- (1H, 3H)-pyrimidine-2, 4-diones (N-substituted uracils).
- Heterocyclyl moreover includes such moieties in charged form, e.g., piperidinium.
- heterocyclyl is oxe
- Halogen or halo
- fluorine fluoro
- chlorine chloro
- bromine bromine
- iodine iodo
- halo is fluoro (-F) or chloro (-C1).
- substitution by a named substituent is permitted on any atom in a ring (e.g., aryl, a heteroaryl ring, or a saturated heterocyclic ring) provided such ring substitution is chemically allowed and results in a stable compound.
- a “stable” compound is a compound which can be prepared and isolated and whose structure and properties remain or can be caused to remain essentially unchanged for a period of time sufficient to allow use of the compound for the purposes described herein (e.g., therapeutic or prophylactic administration to a subject).
- variable e.g., Rl, R3 ⁇ 4, etc.
- its definition on each occurrence is independent of its definition at every other occurrence. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- Ci-5 alkylcarbonylamino Ci-6 alkyl substituent is equivalent to
- substituted shall be deemed to include multiple degrees of substitution by a named substitutent. Where multiple substituent moieties are disclosed or claimed, the substituted compound can be independently substituted by one or more of the disclosed or claimed substituent moieties, singly or plurally. By independently substituted, it is meant that the (two or more) substituents can be the same or different.
- Reference to the compounds of structural Formula I includes the compounds of other generic structural Formulae that fall within the scope of Formula I, including but not limited to Formulae I A, II and III.
- Compounds of structural Formula I may contain one or more asymmetric centers and can thus occur as racemates and racemic mixtures, single enantiomers, diastereoisomeric mixtures and individual diastereoisomers.
- the present invention is meant to comprehend all such isomeric forms of the compounds of structural Formula I.
- Compounds of structural Formula I may be separated into their individual diastereoisomers by, for example, fractional crystallization from a suitable solvent, for example methanol or ethyl acetate or a mixture thereof, or via chiral chromatography using an optically active stationary phase.
- Absolute stereochemistry may be determined by X-ray crystallography of crystalline products or crystalline intermediates which are derivatized, if necessary, with a reagent containing an asymmetric center of known absolute configuration.
- any stereoisomer or isomers of a compound of the general structural Formula I may be obtained by stereospecific synthesis using optically pure starting materials or reagents of known absolute configuration.
- racemic mixtures of the compounds may be separated so that the individual enantiomers are isolated.
- the separation can be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereoisomeric mixture, followed by separation of the individual diastereoisomers by standard methods, such as fractional crystallization or chromatography.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diasteromeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue.
- the racemic mixture of the compounds can also be separated directly by chromatographic methods utilizing chiral stationary phases, which methods are well known in the art.
- Some of the compounds described herein may exist as tautomers which have different points of attachment of hydrogen accompanied by one or more double bond shifts.
- a ketone and its enol form are keto-enol tautomers.
- the individual tautomers as well as mixtures thereof are encompassed with compounds of Formula I of the present invention.
- the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominately found in nature.
- the present invention is meant to include all suitable isotopic variations of the compounds of structural Formula I.
- different isotopic forms of hydrogen (H) include protium and deuterium ( 2 H, also denoted as D).
- Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
- Isotopically-enriched compounds within structural Formula I can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
- the present invention includes all stereoisomeric forms of the compounds of Formula I.
- Centers of asymmetry that are present in the compounds of Formula I can all independently of one another have S configuration or R configuration.
- the invention includes all possible enantiomers and diastereomers and mixtures of two or more stereoisomers, for example mixtures of enantiomers and/or diastereomers, in all ratios.
- enantiomers are a subject of the invention in enantiomerically pure form, both as levorotatory and as dextrorotatory antipodes, in the form of racemates and in the form of mixtures of the two enantiomers in all ratios.
- the invention includes both the cis form and the trans form as well as mixtures of these forms in all ratios.
- the preparation of individual stereoisomers can be carried out, if desired, by separation of a mixture by customary methods, for example by chromatography or crystallization, by the use of stereochemicaily uniform starting materials for the synthesis or by stereoselective synthesis.
- a derivatization can be carried out before a separation of stereoisomers.
- the separation of a mixture of stereoisomers can be carried out at the stage of the compounds of Formula I or at the stage of an intermediate during the synthesis.
- references to the compounds of structural Formula I are meant to also include the pharmaceutically acceptable salts, and also salts that are not pharmaceutically acceptable when they are used as precursors to the free compounds or their pharmaceutically acceptable salts or in other synthetic manipulations.
- the compounds of the present invention may be administered in the form of a pharmaceutically acceptable salt.
- pharmaceutically acceptable salt refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. Salts of basic compounds encompassed within the term “pharmaceutically acceptable salt” refer to non-toxic salts of the compounds of this invention which are generally prepared by reacting the free base with a suitable organic or inorganic acid.
- Representative salts of basic compounds of the present invention include, but are not limited to, the following: acetate, ascorbate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, butyrate, camphorate, camphorsulfonate, camsylate, carbonate, chloride, clavulanate, citrate, dihydrochloride, edetate, edisylate, estolate, esylate, fumarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, isothionate, lactate, lactobionate, laurate, malate, maleate, mandelate, mesylate, methylbromide, methylnitrate, methylsulfate, methanes
- suitable pharmaceutically acceptable salts thereof include, but are not limited to, salts derived from inorganic bases including aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, mangamous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, cyclic amines, dicyclohexyl amines and basic ion-exchange resins, such as arginine, betaine, caffeine, choline, N,N- dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion-exchange resins such as argin
- esters of carboxylic acid derivatives such as methyl, ethyl, or pivaloyloxymethyl
- acyl derivatives of alcohols such as O-acetyl, O-pivaloyl, O-benzoyl, and O-aminoacyl
- esters and acyl groups known in the art for modifying the solubility or hydrolysis characteristics for use as sustained-release or prodrug formulations.
- Solvates including but not limited to the ethyl acetate solvate, and in particular, the hydrates of the compounds of structural Formula I are included in the present invention as well.
- the invention also includes, in addition to the salt forms mentioned, inner salts or betaines (zwitterions). Salts can be obtained from the compounds of Formula I by customary methods which are known to the person skilled in the art, for example by combination with an organic or inorganic acid or base in a solvent or dispersant, or by anion exchange or cation exchange from other salts.
- the present invention also includes all salts of the compounds of Formula I which, owing to low physiological compatibility, are not directly suitable for use in pharmaceuticals but which can be used, for example, as intermediates for chemical reactions or for the preparation of physiologically acceptable salts.
- physiologically acceptable salt(s) and “pharmaceutically acceptable salt(s)” are intended to have the same meaning and are used interchangeably herein.
- the following embodiments may apply to structural Formulae I, IA, II and/or III.
- the 8- or 9-membered heteroaryl is composed of a first ring which is a 5-membered ring containing two nitrogens, fused to a second ring that optionally contains one or more heteroatoms (N, O or S).
- the two nitrogens of the first ring may be fully in the first ring, or one of the two nitrogens may be shared at a fusion point with the second ring.
- the 8- or 9-membered bicyclic heteroaryl is attached to the pyrmidinyl ring and the -CH2 ⁇ R ⁇ group of structural Formula I, IA or II via the first ring, and more specifically via each of the atoms in the first ring that are adjacent to each of the two atoms shared by both rings in the bicyclic heteroaryl
- each is independently H, halo, aryl, OR, CN, heteroaryl,
- each R is independently H, halo or -Cj-Cg alkyl, wherein said -Cj-Cg alkyl is optionally substituted with one to three substituents selected from halo or -CF3.
- each R is independently H, halo, CN, OCH3 or CH3.
- R 2 is -(CR d 2 )tCi-C 6 alkyl, -(CR d 2 )tCF 3 , -(CR d 2 )t-C 3 - lOcycloalkyl, -(CR 2)theteroaryl, or -(CR 2)taryl, said alkyl, cycloalkyl, heteroaryl, and aryl being optionally substituted with one to three substituents selected from halo, -Cj-Cg alkyl and -CF3.
- R ⁇ is -Cj-Cg alkyl, -C3-i()cycloalkyl, aryl, heteroaryl, or -C(0)Oalkyl, said alkyl, cycloalkyl, aryl, and heteroaryl being optionally substituted with one to three substituents selected from halo, -C ⁇ -Cg alkyl, -CF3, -CN and -OR.
- R 2 is -(CR d 2)tCj-Cg alkyl, or -(CR d 2 )tCF3, said alkyl being optionally substituted with one to three substituents selected from halo, -C ⁇ -Cg alkyl and -CF3.
- R 2 is -(CR d 2)tCj-Cg alkyl, or -(CR d 2 )tCF3, said alkyl being optionally substituted with one to three substituents selected from halo, -C ⁇ -Cg alkyl and -CF3.
- R 2 is -Cj-Cg alkyl or -aryl, said alkyl and aryl being optionally substituted with one to three substituents selected from halo, -Cj-Cg alkyl and -CF3.
- R 2 is CH2CF2CF3, CH 2 CH2CF3 S CH2CF2CHF 2J CH 2 CF 3 , or CH 2 CHF 2 -
- R 2 is CH2CF2CF3, or CH 2 CF 3 .
- R ⁇ is aryl, heteroaryl, CN, -C(0)NR a R b , -NR C(0)Rb - C(0)OR a , or -OR a , said alkyl, aryl, heteroaryl or heterocyclyl are optionally substituted with from one to three substituent selected R ⁇ .
- R ⁇ is -C(0)NR a R b aryl or heteroaryl, wherein said aryl and heteroaryl being optionally substituted with one to three substituents selected from halo, -C
- R ⁇ is heteroaryl,
- R ⁇ is heteroaryl, where said heteroaryl is oxadizaolyl, thiadiazolyl, dihydro-oxadiazolyl, or triazolyl, or -C(0)NR a R b , where R a and R ⁇ are independently ⁇ H, -Ci-Cg alkyl, heteroaryl, or -(CH 2 )o-3 " 3 -J O cycloalkyl.
- R4 is Cj-Cg alkyl or C3-1 ocycloalkyl.
- R ⁇ is methyl.
- R ⁇ is methyl or cyclopropyl.
- R 5 is halo, -(CR d 2)tCF 3 , -(CR d 2) t C3-i rjcycloalkyl, or -Ci-C alkyl, said alkyl and cycloalkyl being optionally substituted with one to three substituents independently selected from halo or OR.
- n is 1 or 2. More particularly, m is 1 and 1 is H, CI or F. In an embodiment, t is 0, 1, or 2.
- the present invention also relates to processes for the preparation of the compounds of Formula I which are described in the following and by which the compounds of the invention are obtainable.
- the compounds of Formula I according to the invention effect an increase of cGMP concentration via the activation of the soluble guanylate cyclase (sGC), and they are therefore useful agents for the therapy and prophylaxis of disorders which are associated with a low or decreased cGMP level or which are caused thereby, or for whose therapy or prophylaxis an increase of the present cGMP level is desired.
- the activation of the sGC by the compounds of Formula I can be examined, for example, in the activity assay described below.
- terapéuticaally effective (or efficacious ) amount and similar descriptions such as “an amount efficacious for treatment” are intended to mean that amount of a pharmaceutical drug that will elicit the biological or medical response of a tissue, a system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
- prophylacticaliy effective (or efficacious ) amount and similar
- an amount efficacious for prevention are intended to mean that amount of a pharmaceutical drug that will prevent or reduce the risk of occurrence of the biological or medical event that is sought to be prevented in a tissue, a system, animal or human by a researcher, veterinarian, medical doctor or other clinician.
- the dosage a patient receives can be selected so as to achieve the desired reduction in blood pressure; the dosage a patient receives may also be titrated over time in order to reach a target blood pressure.
- the dosage regimen utilizing a compound of the instant invention is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the potency of the compound chosen to be
- a specific daily dosage amount can simultaneously be both a therapeutically effective amount, e.g., for treatment of hypertension, and a prophylacticaliy effective amount, e.g., for prevention of myocardial infarction.
- disorders and pathological conditions which are associated with a low cGMP level or in which an increase of the cGMP level is desired and for whose therapy and prophylaxis it is possible to use compounds of Formula I are.
- cardiovascular diseases such as endothelial dysfunction, diastolic dysfunction, atherosclerosis, hypertension, heart failure, pulmonary hypertension, which includes pulmonary arterial hypertension (PAH), stable and unstable angina pectoris, thromboses, restenoses, myocardial infarction, strokes, cardiac insufficiency or pulmonary hypertonia, or, for example, erectile dysfunction, asthma bronchiale, chronic kidney insufficiency and diabetes.
- Compounds of Formula I can additionally be used in the therapy of cirrhosis of the liver and also for improving a restricted memory performance or ability to learn.
- the compounds of Formula I and their pharmaceutically acceptable salts can be administered to animals, preferably to mammals, and in particular to humans, as pharmaceuticals by themselves, in mixtures with one another or in the form of pharmaceutical preparations.
- the term "patient” includes animals, preferably mammals and especially humans, who use the instant active agents for the prevention or treatment of a medical condition. Administering of the drug to the patient includes both self-administration and administration to the patient by another person.
- the patient may be in need of, or desire, treatment for an existing disease or medical condition, or may be in need of or desire prophylactic treatment to prevent or reduce the risk of occurrence of said disease or medical condition.
- a patient "in need" of treatment of an existing condition or of prophylactic treatment encompasses both a determination of need by a medical professional as well as the desire of a patient for such treatment.
- a subject of the present invention therefore also are the compounds of Formula I and their pharmaceutically acceptable salts for use as pharmaceuticals, their use for activating soluble guanyiate cyclase, for normalizing a disturbed cGMP balance and in particular their use in the therapy and prophylaxis of the abovementioned syndromes as well as their use for preparing medicaments for these purposes.
- a therapeutically effective amount is intended to mean that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, a system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
- a prophylactically effective amount is intended to mean that amount of a
- a specific daily dosage amount can simultaneously be both a therapeutically effective amount, e.g., for treatment of hypertension, and a prophylactically effective amount, e.g., for prevention of myocardial infarction.
- a subject of the present invention are pharmaceutical preparations (or pharmaceutical compositions) which comprise as active component an effective dose of at least one compound of Formula I and/or a pharmaceutically acceptable salt thereof and a customary pharmaceutically acceptable carrier, i.e., one or more pharmaceutically acceptable carrier substances and/or additives.
- a subject of the invention are, for example, said compound and its pharmaceutically acceptable salts for use as a pharmaceutical, pharmaceutical preparations which comprise as active component an effective dose of said compound and/or a pharmaceutically acceptable salt thereof and a customary pharmaceutically acceptable carrier, and the uses of said compound and/or a pharmaceutically acceptable salt thereof in the therapy or prophylaxis of the abovementioned syndromes as well as their use for preparing medicaments for these purposes.
- the pharmaceuticals according to the invention can be administered orally, for example in the form of pills, tablets, lacquered tablets, sugar-coated tablets, granules, hard and soft gelatin capsules, aqueous, alcoholic or oily solutions, syrups, emulsions or suspensions, or rectally, for example in the form of suppositories. Administration can also be carried out parenterally, for example subcutaneously, intramuscularly or intravenously in the form of solutions for injection or infusion.
- Suitable administration forms are, for example, percutaneous or topical administration, for example in the form of ointments, tinctures, sprays or transdermal therapeutic systems, or the inhalative administration in the form of nasal sprays or aerosol mixtures, or, for example, microcapsules, implants or rods.
- the preferred administration form depends, for example, on the disease to be treated and on its severity.
- the amount of active compound of Formula I and/or its pharmaceutically acceptable salts in the pharmaceutical preparations normally is from 0.2 to 200 mg, preferably from 1 to 200 mg, per dose, but depending on the type of the pharmaceutical preparation it can also be higher.
- the pharmaceutical preparations usually comprise 0.5 to 90 percent by weight of the compounds of Formula I and/or their pharmaceutically acceptable salts.
- the preparation of the pharmaceutical preparations can be carried out in a manner known per se.
- one or more compounds of Formula I and/or their pharmaceutically acceptable salts together with one or more solid or liquid pharmaceutical carrier substances and/or additives (or auxiliary substances) and, if desired, in combination with other pharmaceutically active compounds having therapeutic or prophylactic action, are brought into a suitable administration form or dosage form which can then be used as a pharmaceutical in human or veterinary medicine.
- Carriers for soft gelatin capsules and suppositories are, for example, fats, waxes, semisolid and liquid polyols, natural or hardened oils, etc.
- Suitable carriers for the preparation of solutions, for example of solutions for injection, or of emulsions or syrups are, for example, water, physiologically sodium chloride solution, alcohols such as ethanol, glycerol, polyols, sucrose, invert sugar, glucose, mannitol, vegetable oils, etc.
- Suitable carriers for microcapsules, implants or rods are, for example, copolymers of glycolic acid and lactic acid.
- the pharmaceutical preparations can also contain customary additives, for example fillers, disintegrants, binders, lubricants, wetting agents, stabilizers, emulsifiers, dispersants, preservatives, sweeteners, colorants, flavorings, aromatizers, thickeners, diluents, buffer substances, solvents, solubilizers, agents for achieving a depot effect, salts for altering the osmotic pressure, coating agents or antioxidants.
- customary additives for example fillers, disintegrants, binders, lubricants, wetting agents, stabilizers, emulsifiers, dispersants, preservatives, sweeteners, colorants, flavorings, aromatizers, thickeners, diluents, buffer substances, solvents, solubilizers, agents for achieving a depot effect, salts for altering the osmotic pressure, coating agents or antioxidants.
- the dosage of the active compound of Formula I and/or of a pharmaceutically acceptable salt thereof to be adminstered depends on the individual case and is, as is customary, to be adapted to the individual circumstances to achieve an optimum effect. Thus, it depends on the nature and the severity of the disorder to be treated, and also on the sex, age, weight and individual responsiveness of the human or animal to be treated, on the efficacy and duration of action of the compounds used, on whether the therapy is acute or chronic or prophylactic, or on whether other active compounds are administered in addition to compounds of Formula I.
- a daily dose of approximately 0,01 to 100 mg kg, preferably 0.01 to 10 mg/kg, in particular 0.3 to 5 mg/kg (in each case mg per kg of bodyweight) is appropriate for
- the daily dose can be administered in a single dose or, in particular when larger amounts are administered, be divided into several, for example two, three or four individual doses. In some cases, depending on the individual response, it may be necessary to deviate upwards or downwards from the given daily dose. A single daily dose is preferred.
- the compounds of Formula I activate soluble guanylate cyclase.
- They can also be employed as a scientific tool or as an aid for biochemical investigations in which such an effect on soluble guanylate cyclase is intended, and also for diagnostic purposes, for example in the in vitro diagnosis of cell samples or tissue samples.
- the compounds of Formula I and salts thereof can furthermore be employed, as already mentioned above, as intermediates for the preparation of other pharmaceutically active compounds.
- One or more additional pharmacologically active agents may be administered in combination with a compound of Formula I.
- An additional active agent is intended to mean a pharmaceutically active agent (or agents) that is active in the body, including pro-drugs that convert to pharmaceutically active form after administration, which are different from the compound of Formula I, and also includes free-acid, free-base and pharmaceutically acceptable salts of said additional active agents.
- any suitable additional active agent or agents including but not limited to anti-hypertensive agents, anti-atherosclerotic agents such as a lipid modifying compound, anti-diabetic agents and/or anti -obesity agents may be used in any combination with the compound of Formula I in a single dosage formulation (a fixed dose drug combination), or may be administered to the patient in one or more separate dosage formulations which allows for concurrent or sequential administration of the active agents (co-administration of the separate active agents).
- anti-hypertensive agents such as a lipid modifying compound
- anti-diabetic agents and/or anti -obesity agents may be used in any combination with the compound of Formula I in a single dosage formulation (a fixed dose drug combination), or may be administered to the patient in one or more separate dosage formulations which allows for concurrent or sequential administration of the active agents (co-administration of the separate active agents).
- angiotensin converting enzyme inhibitors e.g, alacepril, benazepril, captopril, ceronapril, cilazapril, delapril, enalapril, enalaprilat, fosinopril, imidapril, lisinopril, moveltipril, perindopril, quinapril, ramipril, spirapril, temocapril, or trandolapril), angiotensin II receptor antagonists (e.g., losartan i.e., COZAAR ⁇ , valsartan, candesartan, olmesartan, telmesartan and any of these drugs used in combination with hydrochlorothiazide such as HYZAAR®); neutral endopeptidase inhibitors (e.g., thiorphan and phosphoramidon), ald
- angiotensin II receptor antagonists e.
- Patent 5,089,471 discloses a variety of other peptide analogs as disclosed in the following U.S. Patents 5,071,837; 5,064,965; 5,063,207; 5,036,054; 5,036,053; 5,034,512 and 4,894,437, and small molecule renin inhibitors (including diol sulfonamides and sulfinyls (U.S. Patents), and small molecule renin inhibitors (including diol sulfonamides and sulfinyls (U.S. Patent
- N-morpholino derivatives U.S. Patent 5,055,466), N-heterocyclic alcohols (U.S. Patent 4,885,292) and pyrolimidazolones (U.S. Patent 5,075,451); also, pepstatin derivatives (U.S. Patent 4,980,283) and fluoro- and chloro-derivatives of statone-containing peptides (U.S. Patent 5,066,643), enalkrein, RO 42-5892, A 65317, CP 80794, ES 1005, ES 8891, SQ 34017, aliskiren (2(S) !
- sildenafil, tadalfil and vardenafil vasodilators
- calcium channel blockers e.g., amlodipine, nifedipine, verastrial, diltiazem, gallopamil, niludipine, nimodipins, nicardipine
- potassium channel activators e.g., nicorandil, pinacidil, cromakalim, minoxidil, aprilkalim, loprazolam
- diuretics e.g., hydrochlorothiazide
- sympatholitics e.g., beta-adrenergic blocking drugs (e.g., propranolol, atenolol, bisoprolol, carvedilol, metoprolol, or metoprolol tartate), alpha adrenergic blocking drugs (e.g., doxazocin, prazocin or alpha methyldo
- lipid lowering agents e.g., HMG-CoA reductase inhibitors such as simvastatin and lovastatin which are marketed as ZOCOR® and MEVACOR® in lactone pro-drug form and function as inhibitors after administration, and pharmaceutically acceptable salts of dihydroxy open ring acid HMG-CoA reductase inhibitors such as atorvastatin (particularly the calcium salt sold in LIPITOR®), rosuvastatin (particularly the calcium salt sold in CRESTOR®), pravastatin (particularly the sodium salt sold in PRAVACHOL®), and fluvastatin (particularly the sodium salt sold in LESCOL®); a cholesterol absorption inhibitor such as ezetimibe (ZETIA®) and ezetimibe in combination with any other lipid lowering agents such as the HMG-CoA reductase inhibitors noted above and particularly with simvastatin (VYTORIN®) or with atorvastatin calcium; niacin in immediate-
- TREDAPTIVE® and/or with an HMG-CoA reductase inhibitor
- niacin receptor agonists such as acipimox and acifran, as well as niacin receptor partial agonists
- metabolic altering agents including insulin sensitizing agents and related compounds (e.g., muraglitazar, glipizide, metformin, rosiglitazone) or with other drugs beneficial for the prevention or the treatment of the above-mentioned diseases including nitroprusside and diazoxide the free-acid, free-base, and pharmaceutically acceptable salt forms of the above active agents where chemically possible.
- compounds with structure 1 may be prepared by the sequence depicted in Scheme 1.
- Ring structure Z represents a five or six membered aryl, heterocyclyl or heteroaryl ring. Reaction of compound 2 with the
- aminoguanidine hydrazone 3 in an alcohol solvent such as MeOH, n-BuOH or t-BuOH and a base such as NaOMe, NaOEt, t-BuO , K 2 C0 3 or NaHC0 3 at 90°C to 150°C gives the pyrimidine hydrazone 4.
- the reaction may also be carried out in the absence of a base. Additionally, the reaction may also be carried out on the corresponding ethyl or propyl ester of compound 2.
- Compound 1 is prepared by treating the pyrimidine hydrazone 4 with Cul and a ligand such as trans-N,N'-dimethylcyclohexane-l,2-diamine or ⁇ , ⁇ '-dimethylethylenediamine in a solvent such as DMF, DMA or NMP at ambient temperature to 160°C.
- a ligand such as trans-N,N'-dimethylcyclohexane-l,2-diamine or ⁇ , ⁇ '-dimethylethylenediamine in a solvent such as DMF, DMA or NMP at ambient temperature to 160°C.
- the reaction may also be carried out in the absence of a ligand.
- the copper mediated cyclization of hydrazones to form indazoles may also be carried out using the conditions described by Liu, R. et al Synthetic
- ester 5 The preparation of compound 2 is outlined in Scheme 2. Deprotonation of ester 5 using a base such as LiHMDS, NaHMDS, NaH or LDA in a solvent such as THF or DMF followed by treatment with methyl iodide affords the ester 6.
- the esters 5 and 6 may be prepared from the corresponding carboxylic acid by treatment with trimethylsilyl diazomethane or methanol with catalytic sulfuric acid.
- the esters 5 and 6 may be prepared by the alpha arylation/heteroarylation of esters as described by Buchwald, S. L. et al Organic Letters 2009, 11(8), 1773; or by Shen, H. C. et al Organic Letters 2006, 8(7), 1447.
- Compounds 5 and 6, where R 3 is 5-membered ring heterocyc e may be prepared using methods familiar to those skilled in the art.
- compound 5, where R 3 is a 2 ⁇ methyl-l,3-oxazol-4-yl group may be prepared by the condensation of methyl chloroacetoacetate and acetamide.
- Compound 6, where R 3 is a 3-methyl-l ,2,4-oxadiazol-5-yl group may be prepared from dimethyl methyl malonate using the procedure described by Du, W. et al Tetrahedron Letters 2006, 47(25), 4271.
- Compound 6, where R 3 is a 5-methyl-l,3-oxazol-2-yl group may be prepared from dimethyl methyl malonate using the procedure described by Hashmi, A. S. . et al Organic Letters 2004, 6(23), 4391.
- compound 6, where R 3 is a 5-methyl-l,2,4-oxadiazol-3-yl group may be prepared by the reaction of methyl 2-methylcyanoacetate with hydroxylamine and acetic anhydride.
- the compound 7 is prepared by treating compound 6 with a brominating reagent such as NBS and AIBN in a solvent such as carbon tetrachloride at refluxing
- the compound 7 may be prepared by reaction with NBS and magnesium perchlorate in acetonitrile solvent at room temperature as described by Yang, D. et al Journal of Organic Chemistry 2002, 67(21), 7429.
- Compound 7 may also be prepared by treating compound 6 with a base such as sodium hydride followed by treatment with NBS.
- Compound 2 is obtained from 7 by reaction with malononitrile and a base such as sodium hydride, t-BuOK, K2CO3 or DBU in a solvent such as THF or DMF at ambient temperature to 100°C.
- a base such as sodium hydride, t-BuOK, K2CO3 or DBU
- a solvent such as THF or DMF at ambient temperature to 100°C.
- the preparation of the aminoguanidine hydrazone 3 is outlined in Scheme 3. Formation of the dianion of carboxylic acid 9 with a base such as NaHMDS followed by treatment with ester 8 gives the ketone 10.
- the ketone 10 may be prepared using numerous methods familiar to those skilled in the art.
- Compound 3 is prepared by treatment of the ketone 10 with aminoguanidine hydrochloride and boron trifluoride etherate in an alcohol solvent such as methanol at 100°C.
- compounds with the structure 17 are prepared as outlined in Scheme 4. Reaction of fluoro aldehyde 11 with hydrazine at 100°C in a solvent such as DMA affords the indazole 12.
- a solvent such as DMA
- Pain indazole 12 may prepared from a 2- methyl aniline using the procedure described by Ruchardt, C. et al Synthesis 1972, 7, 375.
- Conversion of the nitrile 15 to the amidine 16 can be accomplished with a reagent such as amino(chloro)methylaluminum s prepared from trimethylaluminum and ammonium chloride, in a non-polar solvent such as toluene at 100°C as described by Garigipati, R. S. et al Tetrahedron Letters 1990, 31(14), 1969.
- a reagent such as amino(chloro)methylaluminum s prepared from trimethylaluminum and ammonium chloride
- a non-polar solvent such as toluene at 100°C as described by Garigipati, R. S. et al Tetrahedron Letters 1990, 31(14), 1969.
- the reaction may also be carried out on the corresponding methyl ester of compound 15.
- Compound 16 can be converted to compound 17 as described in Scheme 1 (compound 3 to 4).
- compounds with structure 20 may be prepared by the sequence depicted in Scheme 5, Conversion of the nitrile 18 to the amidine 19 can be accomplished using the conditions described for the conversion of compound 15 to 16 in Scheme 4. Reaction of amidine 19 with the compound 2 as described in Scheme 1 (compound 3 to 4) affords 20.
- Scheme 6 outlines the preparation of nitrile intermediate 18.
- Amino methyl compound 21 can be coupled with the carboxylic acid 9 and a coupling reagent such as EDC and an organic base such as DIEA or TEA in a solvent like DCM to afford the amide 22.
- This can be converted to the imidazopyridine 23 with phosphorous oxychloride in a chlorinated solvent such DCE under refluxing conditions.
- lodination of 23 to afford 24 can be accomplished with NIS in solvents like DCM or acetonitriie at ambient temperature or under reflux conditions.
- the nitrile 18 can be prepared by treatment of the iodide 24 with zinc cyanide in the presence of a suitable catalyst such as Pd(PPli 3 )4 or Pd 2 (dba) 3 and ligand such as dppf in a polar solvent such as DMF.
- a suitable catalyst such as Pd(PPli 3 )4 or Pd 2 (dba) 3 and ligand such as dppf in a polar solvent such as DMF.
- the amino methyl compound 21 may be prepared using methods familiar to those skilled in the art.
- One specific example for the preparation of compound 21A is outlined in Scheme 7.
- Pyridazine 25 can be converted to 2-cyano pyridazine 27 using the chemistry described by Dostal, W. and Heinisch, G. Heterocycles 1986, 793.
- Reduction of the nitrile 27 can be accomplished under high pressure hydrogenation conditions using a suitable catalyst such as palladium on carbon in an alcoholic solvent such as methanol or ethanol and a suitable acid such as hydrochloric acid to afford the 2-amino methyl pyridazine hydrochloride 21A.
- the amino methyl compounds 21B and 21C may be prepared as outlined in Scheme 8. Addition of diethyl acetamidomaionate to 2-chloro-5-nitropyridine affords compound 29. Reduction of 29 with hydrogen and palladium on carbon gives the amine 30. Sandmeyer reaction of 30 using the indicated conditions gives the halo (chloro or fluoro) pyridine 31.
- Compounds of the present invention may be prepared using methods familiar to those skilled in the art.
- One such method is the reduction of ring structure Z in compounds 1, 17 and 20 to the corresponding tetrahydro or dihydro compounds using hydrogen and a catalyst such as palladium or platinum. This reduction may also be carried out using a reducing agent such as triethylsilane and an acid such as TFA.
- Compounds 1, 17 and 20 bearing a halogen substituent may be converted to the corresponding cyanide as described in Scheme 4 (compound 13 to 14), or to a hydroxyl as described by Buchwald, S. L. et al Journal of the American Chemical Society 2006, 128 (33), 10694, or to another halogen as described by Arvela, R. K.
- Halogen substituents on compounds 1, 17 and 20 may be converted to aryl or heteroaryl substituents by a Suzuki coupling using conditions described by Buchwald, S. L. et al Journal of the American Chemical Society 2007, 129 (11), 3358.
- Compounds 1, 17 and 20, where R 3 is an ester may be converted to a R 3 5-membered ring heterocycle using methods mentioned for compounds 5 and 6 in Scheme 2. Additional methods for this conversion are summarized in Scheme 9.
- the ester 32 can be converted to amide 33 by treatment with amines such as ammonia, hydrazine, or methyl hydrazine in an alcohol solvent such as methanol at ambient temperature to 50°C.
- compound 2 (depicted as the ethyl ester) may also be prepared as shown in Scheme 10.
- Cycloalkyl, heteroaryl, and alkynyl magnesium halides are also suitable reagents for this reaction.
- Compound 36A R 3 is C0 2 Et
- Compound 36B (R 4 is Me) can be prepared using the procedure described by Hagiware et. al. Synthesis 1974, 9, 669.
- R substituent if not present in starting material (e.g. compound 11 in Scheme 4), may be incorporated in a late intermediate using methods familiar to those skilled in the art.
- the compound 14 R 1 is H
- R 1 bromide
- Scheme 11 Another method is depicted in Scheme 11.
- Treatment of compound ISA with wCPBA in acetic acid solvent at 75 °C affords the N-oxide which is then reacted with phosphorous oxychloride at 75 °C to give compound 15B.
- Compound 15B can be converted to 17A using the procedures described in Scheme 4.
- the chloro substituent in compound 17A may be converted to a variety of groups using methods familiar to those skilled in the art.
- the chloro substituent can be converted to methoxy, methyl and cyano substituents using the conditions summarized in Scheme 11 (compound 17B).
- compounds with structure 42 may be prepared as depicted in Scheme 12. Reaction of the ketone 37 with hydroxyl amine gives an oxime which is subsequently reduced with zinc to afford amine compound 38.
- the ketone compound 37 may be prepared using numerous methods familiar to those skilled in the art. Treatment of compound 38 with methyl oxalyl chloride affords compound 39. Cyclization of compound 39 to compound 40 can be accomplished with phosphorous oxychloride at 120 °C. Conversion of compound 40 to compound 42 is accomplished using the methods discussed in Schemes 4.
- the ester 32 in Scheme 9 can be converted to alkyl amide 33 simply by heating with an amine.
- Aryl and heteroaryl amides can be prepared by treating compound 32 with an amine and a reagent such /-PrMgCl, 2-PrMgCl with LiCl, or AlMe 3 .
- amides may be prepared from acyl hydrazide 33B as depicted in Scheme 15. Treatment of 33B with sodium nitrite or an alkyl nitrite such as tert-butyl nitrite affords the acyl azide 50. This can then be reacted with an alkyl, aryl or heteroaryl amine at ambient temperature to afford amide 33.
- the acyl azide 50 undergoes the Curtius rearrangement at elevated temperature to give an isocyanate which can then react with an amine to give a urea or an alcohol such as teri-butanol to give the carbamate 51 as depicted in Scheme 15.
- Carbamate 51 may then be treated with an acid such as trifiuoroacetic acid to give the primary amine which can then be used in a variety of reactions, such as a coupling with a carboxylic acid or a reductive animation to give compound 52, as the depicted in Scheme 15.
- the amide 33A in Scheme 9 may be converted to number of substituents using methods familiar to those skilled in the art.
- compound 33A can be converted to the corresponding nitrile by treatment with a reagent such as phosphorous oxychloride.
- the amide 33A can be reacted with Lawesson's reagent to form a thioamide which can then be converted to a number of heterocycles such as a 1 ,3-thiazole.
- a 1,2,3-triazole can be prepared by the copper catalyzed reaction of alkyne 53 and an azide as depicted in Scheme 16.
- compound 56 can be prepared by the alkylation of the mono substituted lactam 55 as depicted in Scheme 16.
- Bu butyl
- ⁇ -Bu 1 ⁇ 2rt-butyl
- t-Boc20 di-tert-butyl dicarbonate
- DIEA diisopropylethylamine
- DME 1,2-dimethoxyethane
- EDC l-Ethyl-3-(3-dimethylaminopropyl)
- HO Ac acetic acid
- HMPA hexamethylphosphoramide
- LAH Lithium aluminum hydride
- mCPBA 3-chloroperoxybenzoic acid
- NMP jV-methylpyrrolidone
- NaHMDS sodium bis(trimethylsilyl)amide
- NBS N-bromo succinmide
- PDA photodiode array
- Pd C palladium on activated carbon
- Ph phenyl
- iPrMgCl isopropylmagnesium chloride
- psig pounds per square inch gauge
- TFA trifluoroacetic acid
- THF tetrahydrofuran
- TMSCN trimethylsilyl cyanide
- TsCl 4-toluenesulfonyl chloride
- electro spray ionization was used with positive (ES+) or negative ion (ES-) detection; and diode array detection.
- Step B methyl 2-bromo-2-phenyipropanoate
- Step C methyl 3,3-dicyano-2-methyl-2-phenylpropanoate
- Step A methyl 2-( pyridm-2 ⁇ yl propanoate
- Step B methyl 2-bromo-2-(pyridin-2 ⁇ yl)propanoate
- Step C methvi 3.3-dicyano-2-methyl ⁇ 2-(pyridin-2-yl propanoate
- Step A tert-butyl 2-(pyrazin-2-vl)propanoate
- Step B methyl 2 ⁇ (pyrazin-2-yP ⁇ propanoate
- Step D methyl 33 ⁇ dicyano-2-methyl-2-(pyrazin-2-yl propanoate
- Step A ethyl (3Z)-3-amino-3-(hydroxyimino -2-methylpropanoate
- Ethyl 2-methylcyanoacetate (5 g, 39 mmol) and hydroxylamine (2.6 g, 39 mmol) were dissolved in 50 mL of MeOH. The solution was heated at the 50 °C overnight. The solution was then concentrated and the residue purified by silica gel chromatography using a hexanes/EtOAc gradient to give the indicated product.
- Step B ethyl 2-(5-memyl-L2,4-oxadiazol-3-yl propanoate
- Acetic anhydride (4.6 mL, 49 mmol) was added to a pyridine (50 mL) solution of the
- Step C ethyl 2-bromo-2-(5-methyl-L2,4-oxadiazol-3-yl propanoate
- Step D ethyl 3 -dicyano-2-methyl-2-(5-methyl-l,2.4-oxadiazol-3-yl)propanoate
- Step A methyl 2-(3-methyl-L2,4-oxadiazol-5-yl)propanoate To a screw cap pressure vessel was added acetamide oxime (0.900 g, 12.2 mmol) and dimethyl methyl malonate (3.55 g, 24.3 mmol) and the resulting mixture was heated at 140 °C for 4 hours.
- Step B methyl 2-bromo-2-(3-methyl-l,2,4-oxadiazol-5-yl propanoate
- Step C methyl 3,3-dicyano-2-methyl-2-(3-methyl-l,2,4-oxadiazol-5-yl propanoate
- Step A methyl 2-methyl-3-oxo-3-(prop-2-yn-l-ylamino)propanoate
- Step C methyl 2-bromo-2-(5-methyl-l,3-oxazol-2-yl)propanoate
- Step D methyl 3,3-dicyano-2-methyl-2-(5-methyl-l ,3-oxazol-2-yl)propanoate
- Step A methyl (2-methyl-L3-oxazol-4-yl)acetate
- Step B methyl 2-(2-methyl-l,3-oxazol-4-yl propanoate
- Step C methyl 2-bromo-2 ⁇ (2-methyl-l,3-oxazol-4-yl)propanoate
- Step D methyl 3.3-dicyano-2-methyl-2-f2-methyl-l,3-oxazol-4-yl)propanoate
- Ethyl 3,3-dicyano-2-methylprop-2-enoate was prepared according to the procedure described by Hagiware et. al. Synthesis 1974, 9, 669. The reaction was stirred for 10 min in the cooling bath then quenched with saturated aqueous NH4CI, and then diluted with water and EtOAc. The layers were separated and the organic layer was dried (sodium sulfate) and concentrated in vacuo. Purification by silica gel column
- Step A l -(2-bromo-5-chlorophenyl)-4,4,4-trifluorobutan-l-one
- Step C 4-amino-2-f (2E) ⁇ 2-[ ⁇ 1 -(2-bromo-5-chlorophenyl)-4,4,4-trifluorobutylidene1hydrazinv - 5 -methyl-5 -phenyl-5,7-dihydro ⁇ 6H-pyrrolo [2,3-d " ipyrimidin-6-one
- Step D 4-amino-2-[5-chloro-3 ⁇ (3 ,33-trifluoropropyl)-lH-indazoi-l-yl1-5-methyl-5-phenyl-5,7- dihydro-6H-pyrrolo[2,3-d pyrimidin-6-one
- tetraethylammonium hydrogen sulfate (10.21mg, 0.045 mmol).
- the solution was heated to 70 °C and an aqueous solution of sodium permanganate monohydrate (1.5M, 1.257 g, 7.86 mmol) was added over 20 minutes.
- the reaction was stirred at 70 °C for an additional 4 hours.
- the reaction mixture was filtered through CeliteTM (diatomaceous earth). The filter cake was washed with hot water (10 mL).
- Step B l-(2-bromo-5-chlorophenyl -4,4,5,5,5-pentafluoropentan-l-one
- Step C (2EV2-P -(2-bromo-5-chlorophenylV4 A5,5,5-pentafluoropentylidene "
- Step D 4-amino-2- (2E -2-[l-(2-bromo-5-chlorophenyl)-4,4,5.5,5-pentarluoropentylidene1 hydrazinvU-5-methyl-5-phenyl-5J-dihvdro-6H-pyrrolo[2»3-d]pyrimidin-6-one
- Step E 4-amino-2-r5-chloro-3-(33 ⁇ .4-pentafluorobutyl)-lH-indazol-l-yll-5-methyl-5-phenyl-- 5,7-dihvdro-6H-pyrrolor2,3-d1pyrimidin-6-one
- Step A l-(2-bromo-5-chlorophenyl)-2-(2,3,6-trifluorophenyl ethanone
- Step C 4-amino-2- ⁇ (2Z -2- 1 -(2-bromo-5-chlorophenyl)-2-(2,3,6-trifluorophenyl)ethylidenel hvdrazinyl
- Step D 4-amino-2-r5-chloro-3-(2,3,6-trifluorobenzylVlH-indazol-l-yll-5-methyl-5-phenyl-5,7- dihydro-6H-pyrrolof 2 , 3 -d]pyrimidin-6-one
- Acetic anhydride (10.0 mL, 106 mmol) was added dropwise to a benzene solution (1 10 mL) containing 5-chloro-2-methylaniline (5.0 g, 35.3 mmol) and potassium acetate (3.8 g, 38.7 mmol) at room temperature. After 10 minutes the reaction mixture, which had formed a thick white suspension, was heated to 80 °C. Tert-butyl nitrite (6.99 mL, 90%, 53.0 mmol) was added over 20 minutes. The reaction mixture was kept at 80 °C overnight. The reaction was then cooled to room temperature and concentrated. The residue was dissolved in MeOH and stirred for 10 minutes.
- Step D 6-ch3 ⁇ 4oro- 1 -(3,3 A4,4-pentafluorobutylH H-indazole-3-carbonitrile
- Step E 6-chloro-l-(3,3,4,4,4-pentafluorobutyl)-lH-indazole-3-carboximidamide
- Trimethylaluminum (2.0M in toluene, 23,17 mL, 46.3 mmol) was added dropwise to a suspension of ammonium chloride (2.49 g f 46.5 mmol) in 69 mL toluene cooled to 0 °C. The solution was then stirred at room temperature for 3 hours. This solution was then added to the intermediate from Step D (3.0 g, 9.27 mmol) and then heated at 110 °C for 6 hours, The solution was then cooled to room temperature and carefully poured to silica gel (ca 150 mL) and methanol (ca 250 mL).
- Step F 4-amino-2-[6-chloro- 1 -(3,3 ,4,4,4-pentafiuorobutyl)- 1 H-indazol-3 -yl] -5-methyl-5-phenyl- 5,7-dihydro-6i/-pyrrolor2,3-f jpyrimidin-6-one
- Step E 4-amino-2-[6-chloro- 1 -(3,3 ,4,4,4-pentafiuorobutyl)- 1 H-indazol-3 -yl] -5-methyl-5-phenyl- 5,7-dihydro-6i/-pyrrolor2,3-f jpyrimidin-6-one
- Intermediate 1 208 mg, 0,911 mmol, slower eluting enantiomer
- sodium bicarbonate (68.1 mg, 0.810 mmol
- t-butanol (12 mL).
- the reaction solution was heated at 140 °C for 75 minutes.
- the indicated compound was prepared from the intermediate from Step E in Example 58 and Intermediate 5 using the procedure described in Example 58.
- the racemic material was resolved by chiral SFC chromatography to give the indicated compound. Data is given for the more active isomer (slower eluting using an OJ column). !
- Step A 4-amino-2-r5-chloro-3-f3J,4,4,4-pentafluorobutyl)-lH-indazol-l-yl]-5-methyl-6-oxo- 6J-dihydro-5H-pyrrolo 2,3-dlpyrimidine-5-carbohydrazide
- Step B N'-acetyl-4-amino-2-r5-chloro-3-(3J.4 ⁇ ,4-pentafluorobutyl)-lH-indazol-l-yl)-5-methyl- 6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidine-5-carbohvdrazide
- Example 54 (15 mg, 0.03 mmol, racemic), 1 ,2-dichloroethane (1 mL), triethylsilane (0.7 mL) and TFA (0.3 mL).
- the reaction mixture was heated at 75 °C for 3 hours.
- the reaction solution was then concentrated and partitioned between EtOAc and IN NaOH aq.
- the organic phase was washed with brine and dried over MgS0 4 .
- the crude was purified by reverse phase HPLC using a water/acetonitrile (with 0.1% TFA) gradient to give the indicated product.
- ⁇ NMR 400 MHz, CH 3 CN-d 3 ): ⁇ 9.16 (s, 1H); 7.37-7.27 (m, 5H); 7.25-
- Example 69 (13.1 mg, 0.026 mmol), 5 mL of 4/1
- Step A diethyl facetylamino)( ' 5-nitropyridin-2-yl)propanedioate
- Step C diethyl (acetylamino)(5-chloropyridin-2-yl)propanedioate
- Step D l-(5-chloropyridin-2-yl)methanamine
- Step E l-(5-chloropyridin-2-yl)methanamine hydrochloride
- Step G 6-chloro-3-(3,3,4,4,4-pentafluorobutyl)imidazorL5-a]pyridine 8
- Step I 6-chloro-3 -(3 ,3 A4,4-pentafluorobutyl imidazo ⁇ 1 ,5-a]pyridine- 1 -carbonitriie
- Step J 6-chloro-3-(3 J,4,4,4-pentafluorobutyl)imidazorL5-alpyridine-l -carboximidainide
- Trimethyl aluminum (2.0 M toluene, 10 mL, 20 mmol) was added to ammonium chloride (1.07 g, 20 mmol) suspended in toluene (30 mL) at 0 °C. The solution was then stirred at room temperature for 2 hours to give a 0.5 M amino(chloro)methylaluminum solution in toluene.
- Step A diethyl (acetylamino)i5-fluoropyridin-2-yl)propanedioate
- Step B l-(5-fluoropyridin-2-yI)metlianamine
- Step C l-C5-fluoropyridin-2-yl methanamine hydrochloride
- Step D 4-amino ⁇ 2-r6-fluoro-3-f 3 ,3 A4,4-pentafluorobutyl imidazo[ 1 ,5-alpyridin- 1 -yll -5-(4- fluorophenvn-5-methvi-5.7-dihvdro-6H- yrrolor2,3- ⁇ j1Pyrimidin-6-one
- Step A 2-[(4-methylphenyl)sulfonyl1-2,3-dihvdropyridazine-3-carbonitrile
- Step B pyridazine-3-carbonitrile
- Step C l-fpyridazin-3-yl methanamine hydrochloride
- Step D 4-amino-5-methyl-2-f7-(3,3,4.4,4-pentafluorobutvnimidazo( " L5-6Jpyridazin-5-yl]-5- phenyl-5J-dihvdro-6H-pyrrolo 2,3-(/
- the indicated compound was prepared from the amidine intermediate derived from 2- (aminomethyl)pyridme and Intermediate 5 using the procedure described in Example 105.
- the racemic material was resolved by preparative HPLC using OD-H column to give the indicated compound (fast isomer).
- the indicated compound was prepared from the amidine derived from Example 105 Step J and Intermediate 7 using the procedure described in Example 105.
- the racemic material was resolved by preparative SFC using AD column to give the indicated compound (fast isomer).
- the racemic product can be resolved by chiral SFC chromatography using a Chiralcel OJ or Chiralpak AD column.
- Step B 3 -iodo-lH-pyrazolo [3 A ⁇ b] yridine
- a DMF (180 mL) solution containing the intermediate from Step B above (24.1 g, 98 mmol), zinc cyanide (6.93 g, 59.0 mmol), 1 , -bis(diphenylphosphino)ferrocene (4.36g, 7.87 mmol) and tris(dibenzylideneacetone)dipalladium (3.60g, 3.93 mmol) was heated at 120 °C for 1.5 hours. The solution was cooled to room temperature and diluted with water. The precipitated product was collected and dried under vacuum with a nitrogen sweep to give the title compound.
- Step D 1 -(3,3.4 A4-pentafluorobutyl)- lH-pvrazolo[3,4-fr]pyridine-3-carbomtrile
- Step E 1 -(3,3,4,4,4-pentafluorobutvD- lH-pyrazolof3,4-61pyridine-3-carboximidamide
- Trimethylaluminum (2.0M in toluene, 60 mL, 120 mmol) was added dropwise to a suspension of ammonium chloride (6.42 g, 120 mmol) in 180 mL toluene cooled to 0 °C. The solution was then stirred at room temperature for 3.5 hours. This solution (0.5M, 146 mL, 72.9mmoi) was then added to the intermediate from Step D above (4.6 g, 15.9 mmol) and then heated at 110 °C for 2.5 hours. The solution was then cooled to room temperature and carefully poured to silica gel (70 g) and methanol (750 mL). After stirring overnight the suspension was filtered and the filtered solid washed with methanol.
- Step F ethyl 4-amino-5-methyl-6-oxo-2-ri- 3.3.4,4.4-pentafluorobutyl)-lH-pyrazolor3.4- 6]p idin-3-yl]-6J-dihydro-5H-pyrrolof2,3- ⁇
- EXAMPLE 161 4-AMINO-5-METHYL-6-OXO-2-[l-(3,3 J 4,4 5 4-PENTAFLUOROBUTYL)-lH- PYRAZOLO[3,4-i?]PYRIDIN-3-YL]-6,7-DIHYDRO-5H-PYRROLO[2 s 3-Z ) ]PYRIMIDINE-5-
- reaction mixture was concentrated in vacuo and the residue purified by silica gel column chromatography using a DCM MeOH (with 0.5% NH 4 OH) gradient. Chiral separation by SFC using a Chiralpak AD column provided both enantiomers of the title compound.
- Trimethylaluminum (2.0 M in toluene, 12 mL, 2.4 mmol) was added to a toluene (8 mL) solution of aniline (248 rag, 2.66 mmol).
- Step A 4-amino-2-[6-chloro-l -(3,3,4,4,4-pentafluorobutyi)-lH-indazol-3-yl]-5-methyl-6-oxo- 6,7-dihydro-5H-pyrrolo[2,3-i/jpyrimidine-5-carbohydrazide
- Step B 4-amino-2-f6-chloro-l-(33 ⁇ ,4 ⁇ entafluorobutvn-iH-indazol-3-yll-5-methyl-6-oxo- 6J-dihydro-5H-pyrrolo[ " 2,3- i1pyrimidine-5-carbonyl azide
- Step C ethyl (4-amino-2-(6-chloro-l-(3»3,4,4,4-pentafluorobutyl)-lH-indazol-3-yl)-5-methyl-6- oxo-6J-dihydro-5H-pyrrolof2,3- ⁇ i pyrimidin-5-yl carbamate
- Step A 4-amino-2-r6-chloro-l -r33.4,4.4-pentafluorobutyl)-lH-indazol-3-yl -5-methyl-6-oxo-6,7- dihydro-5H-pyrrolof2,3-tif)pyrimidine-5-carbothioamide
- Step B 4-amino-2-i6-chloro-l -B,3.4.4.4-pentafluorobutylVlH-indazol-3-yl1-5-f4.5-dimethyl- lJ-thiazol-2-yl)-5-methyl-5J-dihydro-6H-pyrrolo[2J-iilpyrimidin-6-one
- Step A 4-amino-5-methyl-6-oxo-2-f 1 -C3,3,4.4,4-pentafluorobutyl -lH-pyrazolor3.4-61pvridin-3- yl]-6,7-dihydro-5H-pyrrolo
- Step B 4-amino-jV , -formyl-5-methyl-6-oxo-2-[ 1 -(3 ,3 A4,4-pentafluorobutyl)- 1 H-pyrazolo[3 ,4-
- Step C 4-amino-5-methyl-2-[ 1 -(3 ,3 ,4,4,4-pentafluorobutyl 1 H-pyrazolo ⁇ 3.4-61 pyr idin-3 -yll -5 - (1 ⁇ -thiadiazol-2-yl)-5J-dihvdro-6H-pyn-olof2J- ⁇ pyrimidin-6-one
- Lawesson's reagent (133 mg, 0.328 mmol) was added to a toluene (5 mL) solution of the intennediate from Step B (170 mg, 0.298 mmol).
- THF 0.5 mL was added to improve solubility and the reaction was heated to 100 °C for 1 hour in a screw cap vial. The reaction was then cooled and concentrated in vacuo.
- the crude product was purified by silica gel column chromatography using a hexanes/EtOAc gradient to give the title compound. Chiral separation using SFC on a Chiralcel OD column provided both enantiomers of the title compound.
- Step A N'-acetyl-4-amino-5-methyl-6-oxo-2-ri-(3,3,4.4.4-pentafluorobutyl)-lH-pyrazolo 3,4- ⁇ 1pyridin-3-yl]-6 -dihvdro-5H-pyrrolor2.3- ⁇ i1pyrimidine-5-carbohvdrazide
- Ste B 4-amino-5-methyl-5-(5-methyl-L3,4-oxadiazol-2-vn-2-f l-(3.3,4,4.4-pentafluorobutyl)- iH-pyrazolo[3 ⁇ -i ⁇ yridin-3 ⁇ yl]-5J-dih ⁇
- Step A 4-amino-2-[6-chloro- 1 ⁇ (33 A4,4-pentafluorobutyD- lH-indazol-3-yl]-5-methyl-5-(5- oxo-4,5-dihvdro-1 ,4-oxadiazol-2-yl)-5J-dihvdro-6H-pwolo[2 -d1pyrimidin-6-one
- Step B 4-amino-2- [6-chloro- 1 -( 3.3 ,4 ,4,4-pentafluorobutvD- 1 H-indazol-3 -yl] -5-methyl-5- [ 5 -oxo-
- Step A 4-amino-2-f 6-chloro- H3.3 A4,4-Oentafluoro
- Step B 4-amino-2-r6-chloro-l-(3 A4Apentafluoro ⁇
- Ethyl azide was prepared by adding iodoethane (59.1 ⁇ , 0.731 mmol) and sodium azide (43.2 mg, 0.665 mmol) to DMF (2.8 mL, 0.08M) in a 4 mL vial wrapped in aluminum foil.
- Step A fert-butyl (4-amino-5-methyl-6-oxo-2-f l- 3 .4.4.4-pentafluorobutylVlH-pyrazolo 3,4- b pyridin-3 -yl]-6J-dihydro-5H-pyrrolo [2 , 3 -d ⁇ pyrimidin-5 -yl ⁇ carbamate
- Step B 4,5-diamino-5-methyl-2-[l-Q ,4,4,4-pentafluorobutyl)-lH-pyrazoIo[3,4- ?1pyridin-3- yll-5,7-dihydro-6H-pyrrolo[2,3-(a pyrimidin-6-one
- Step A 4-amino-2-r6-chIoro-l- 3J ⁇ 4,4,4-pentafluorobutyl -lH-indazol-3-yl]-5-methyl-5.7- dihydro-6H-pyrrolo[2,3-( ]pyrimidin-6-one
- Step B fe -butyl 4-ammo-2-r6-chloro-l-(3,3,4.4,4-pentafluorobutyl -lH-indazol-3-yl]-5- methvi-6-oxo-5,6-dihydro-7H-pyrrolo[2,3- 1pyrimidine-7-carboxylate
- Step C ⁇ 4-amino-2-f6-chloro-l-(3,3,4,4,4-pentafluorobutyl)-lH-indazol-3-yl "
- Step A 3-cyano-l-(3,3,4,4,4-pentafluorobutyl)-lH-pyrazolo[3 t 4-&1pyridine 7-oxide
- Step B 6-chloro-l -(3 ,4,4,4-pentafluorobutylVlH-pyrazolor3,4- >]pyridine-3-carbonitrile
- Step C 6-chloro- 1 -( ,3 A4,4-pentafluorobutylV 1 H-pyrazolo[3 ,4-61 yridine-3 -carboximidamide
- Step D ethyl 4-amino-2-('6-chloro-l-(3 ,4 ⁇ ,4-pentafluorobutyl)-lH-pyrazolo[3,4- ?lpyridin-3- yl>5-methyl-6-oxo-6J-dihvdro-5H-pyrroloj ' 2,3-tflpyrimidine-5-carboxylate
- Step E 4-amino-2-(6-chloro-l-(3 ,4 ,4-pentafluorobutyl)-lH-pwazolo 3,4-j?]pyridin-3-yn-N- cyclopropyl-5-methyl-6-oxo-6J-dihvdro-5H-pyrrolor2,3-ti
- Methylmagnesium bromide (0.42 ml, 0.584 mmol, 1.4 M in THF) was added to a THF (1.2 ml) and NMP (0.3 ml) solution containing 4-amino-2-(6-chloro-l-(3 s 3 5 4 !
- Step A l-f3,3,3-trifluoropropyl -lH-pyrazolo[3,4-6]pyridine-3-carbonitrile
- Step B 1 -(3,3,3-trifluoropropyl)-l -pyrazolor3,4- ⁇ ]pyridine-3-carboximidamide
- Step C 4-amino- 5 -ethynyl-5 -methyl-2- [ 1 -(3 ,3 , 3 -trifluoropropyl - 1 H-pyrazolo [3 ,4- ⁇ pyridin-3 - yl] -5 J-dihydro-6H-pyrrolo [2,3-ci1pyrimidin-6-one
- Step D 4-amino-5-f 1 -( cvclopropylmethyl lH- 1 ,2,3-triazol-4-yll-5-methv]-2-ri - ⁇ 3.3.3- trifluoropropyiyiH-pwazoloi3 -&]py
- Step A ethyl 4-amino-5-memyl-6-oxo-2-[l-( ' 3 -trifluoropropyl -lH-pyrazolor3.4-&lpyridin- 3-yll-6,7-dihydro-5H-pyrrolof2,3-t ]pyrimidine-5-carboxylate
- Step B 4-amino-N-cvclopropyl-5-methyl-6-oxo-2-n -(3.3.3 -trifluoropropyl I H-pyrazolo [3,4- ?]pyridin-3-yll-6.7-dihydro-5H-pyrrolo[2,3-ci
- Step A 4-amino-5-metjiyl-6-oxo-2-f l-(333 ⁇ triflu ⁇
- Step B 4-amino-5 -methyl-6-oxo-N-f pyridin-3 -yi)-2- 1 -(3 ,3 ,3-trifluoropropyl)- 1 H-pyrazolo f 3 ,4-
- Step A 1-C3 ,3 ,3 -trifluoropropyl)-lH-p azolo[3,4--?1pyridine-3 -carbonitrile 7-oxide
- Step B 6-chloro-l -(3,3,3 -trifluoropropyl lH-pyrazolo [3, 4-i?]pyridine-3 -carbonitrile
- Step C 6-chloro-l -(3,3 J-trifluoropropyl)-lH-pyrazolor3 t 4- ⁇ ]pyridine-3-carboximidaniide
- Step D Ethyl 4-amino-2- 6-chloro-l-(3,3,3-trifluoropropyl)-lH-pyrazolo
- Step E 4-amino-2- 6-chloro- 1 -(3 ,3 ,3-trifluoropropyl)- 1 H-pyrazolof 3 A-b] yridin-3 -yl] -iV- cyciopropyl-5-methyl-6-oxo-6,7-dihydro-5H-pyrrolof2,3-d]pyrimidine-5-carboxamide
- Methyl magnesium bromide (2.08 mL, 2.91 mmol, 1.4 M in THF) was added to 4-amino-2-[6- chloro- 1 -(3 ,3 ,3-trifluoropropyl)- 1 H-pyr ⁇ e -dihydro-SH-pyrroloP ⁇ - ⁇ pyrimidine-S-carboxamide, as described in Example 183, (240 mg, 0.485 mmol) and iron(lll) acetylacetonate (171 mg, 0.485 mmol) in THF (2.8 mL) and NMP (0.7 mL). The solution was then stirred for 30 min. at rt. The reaction mixture was adjusted to pH 7.0 with IN HC1 at 0 °C. The reaction mixture was filtered through a plug of CeliteTM
- the title compound was prepared from 4-amino-2-(6-chloro-l -(3,3,3 -trifluoropropyl)- 1H- pyrazolo[3,4-Z>]pyridin-3-yl]-N-cyclopropyl-5-methyl-6 ⁇ oxo-6,7-dihydro-5H ⁇ yn-olo[2,3- as described in Example 183, according to the procedure described for Example 178.
- Step A 4-bromo-5-methvi-2- l-(3,3,4,4,4-pentafluorobutyl -lH-pyrazolo[3,4-61pyridin-3-yl1-5- phenyl- 5 J-dihydro-6H-pyrro lo
- Step B -methyl-4-(methylamino)-2-[ ' i-(3 ,3 t 4,4,4-pentafluorobutyi)-lH-pyrazolo[3,4- ⁇ 1pyridin-3- yl]-5-phenyl-5,7-dihydro-6H-pyrrolo 2,3-cf
- Step A ethyl 4-bromo-5-methyl-6-oxo-2- l-(3.3.4,4.4-pentafluorobutyl -lH-pyrazolor3.4- ?]pyridin-3-yl]-6J-dihydro-5H-pyrrolo[2,3- ⁇ ]pyrimidine-5-carboxylate
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pulmonology (AREA)
- Epidemiology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Reproductive Health (AREA)
- Pain & Pain Management (AREA)
- Vascular Medicine (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Hospice & Palliative Care (AREA)
- Gastroenterology & Hepatology (AREA)
- Gynecology & Obstetrics (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Saccharide Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description









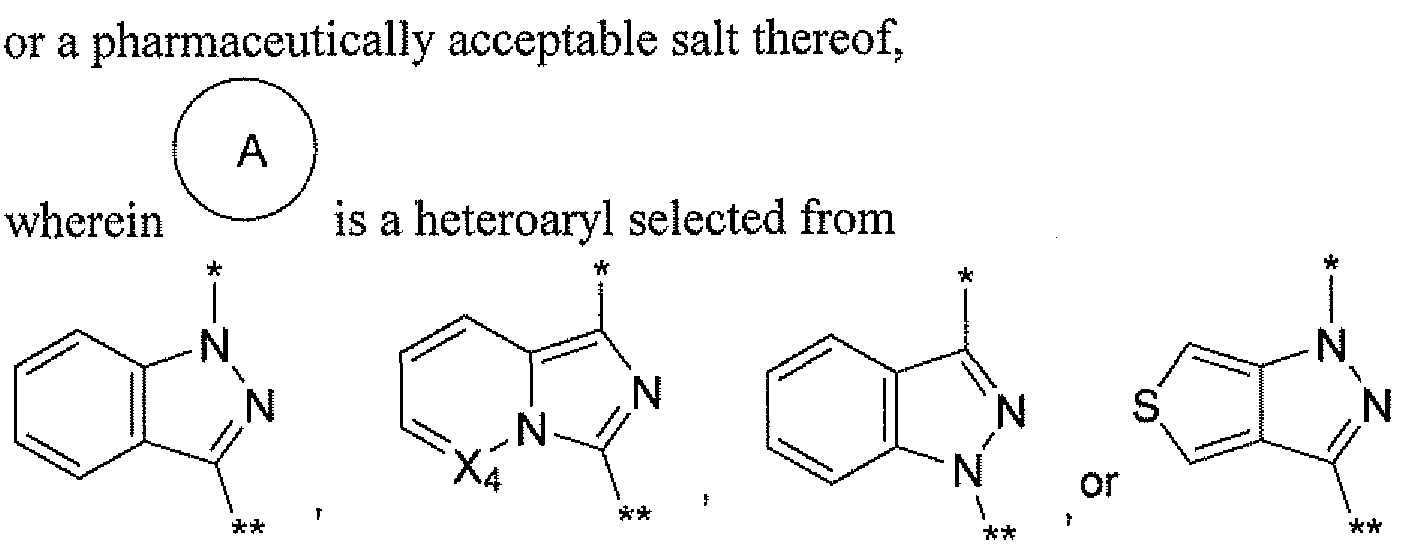


























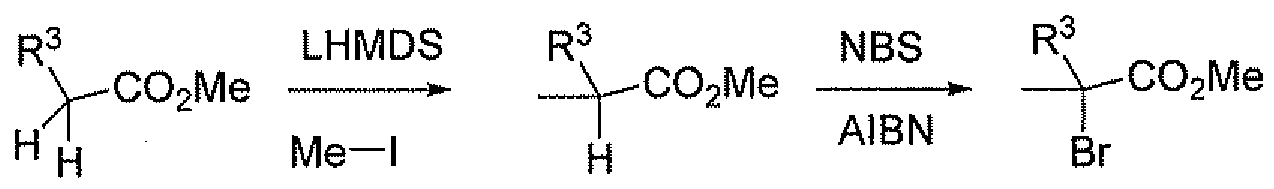







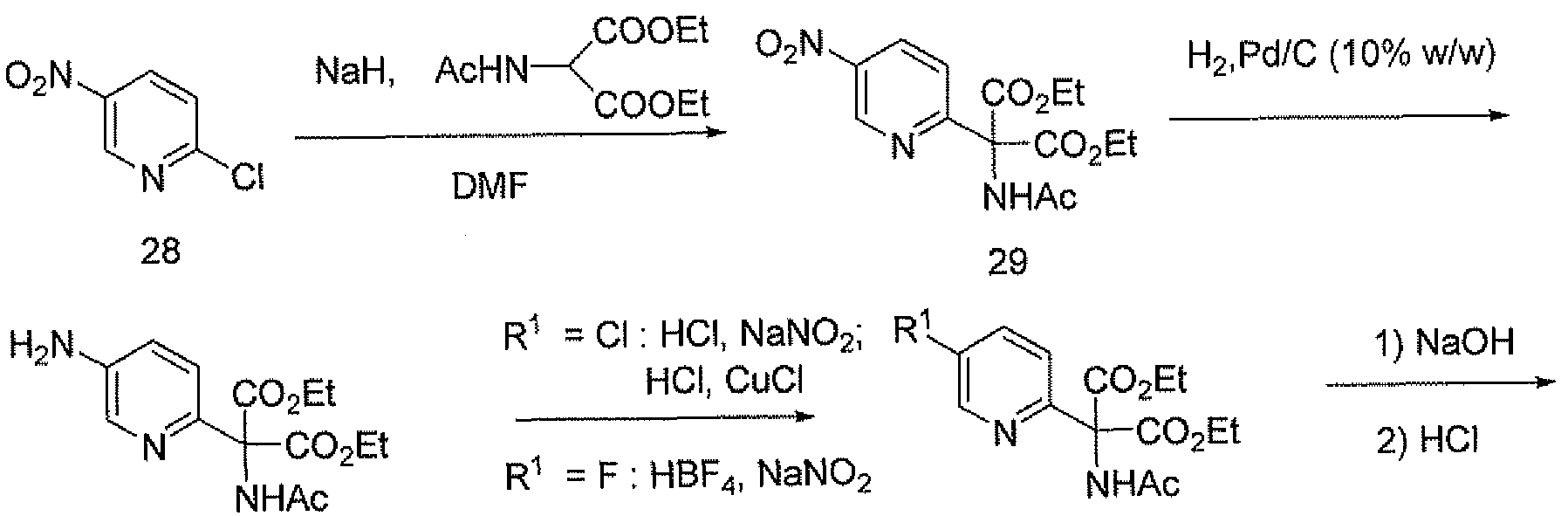
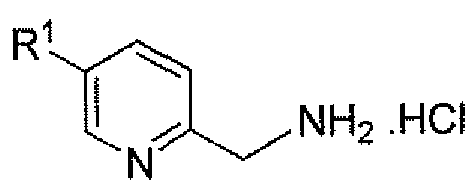


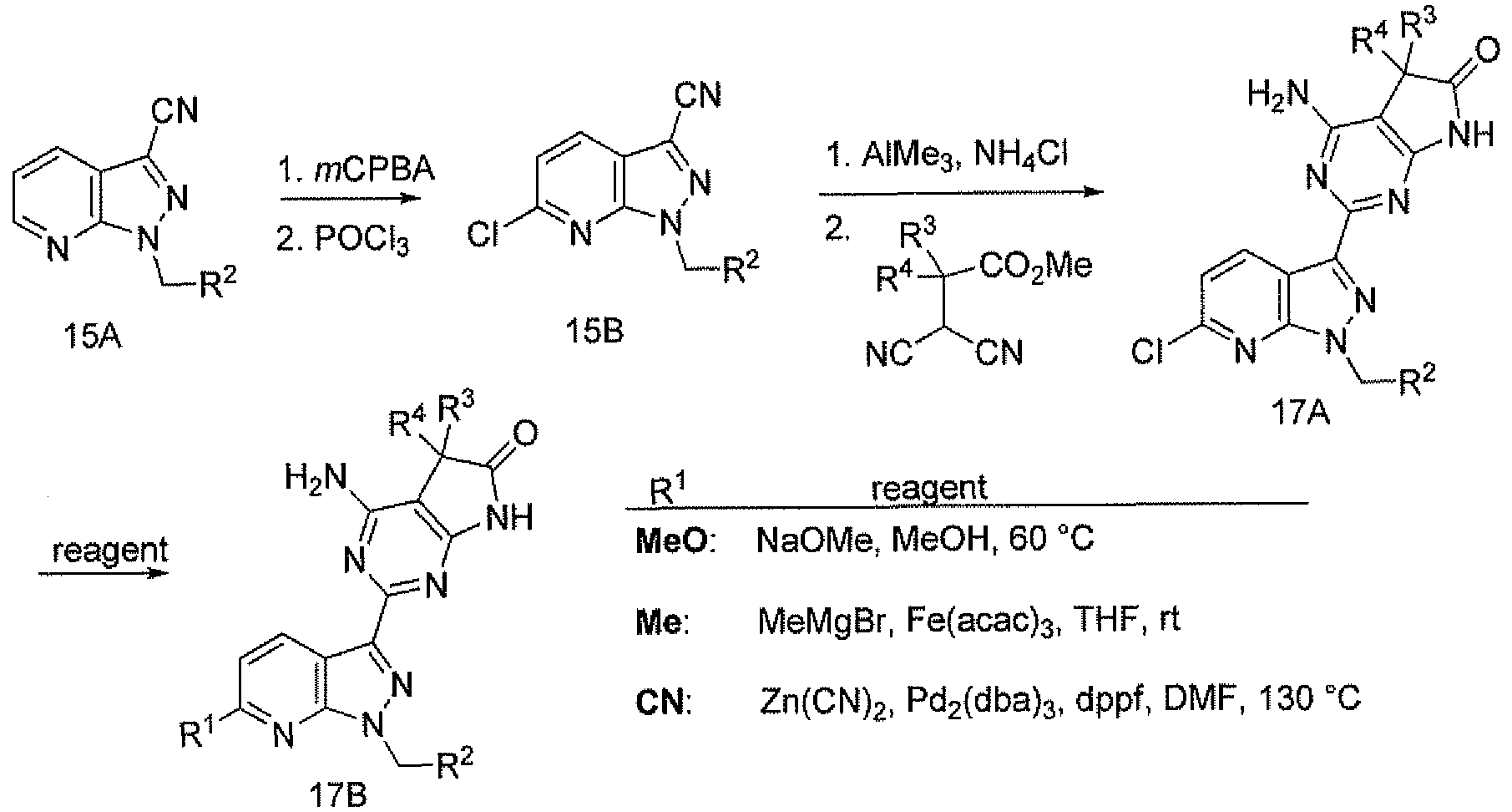






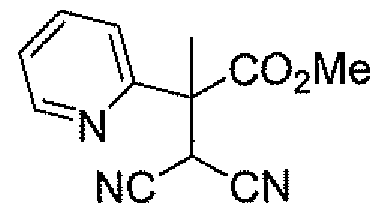












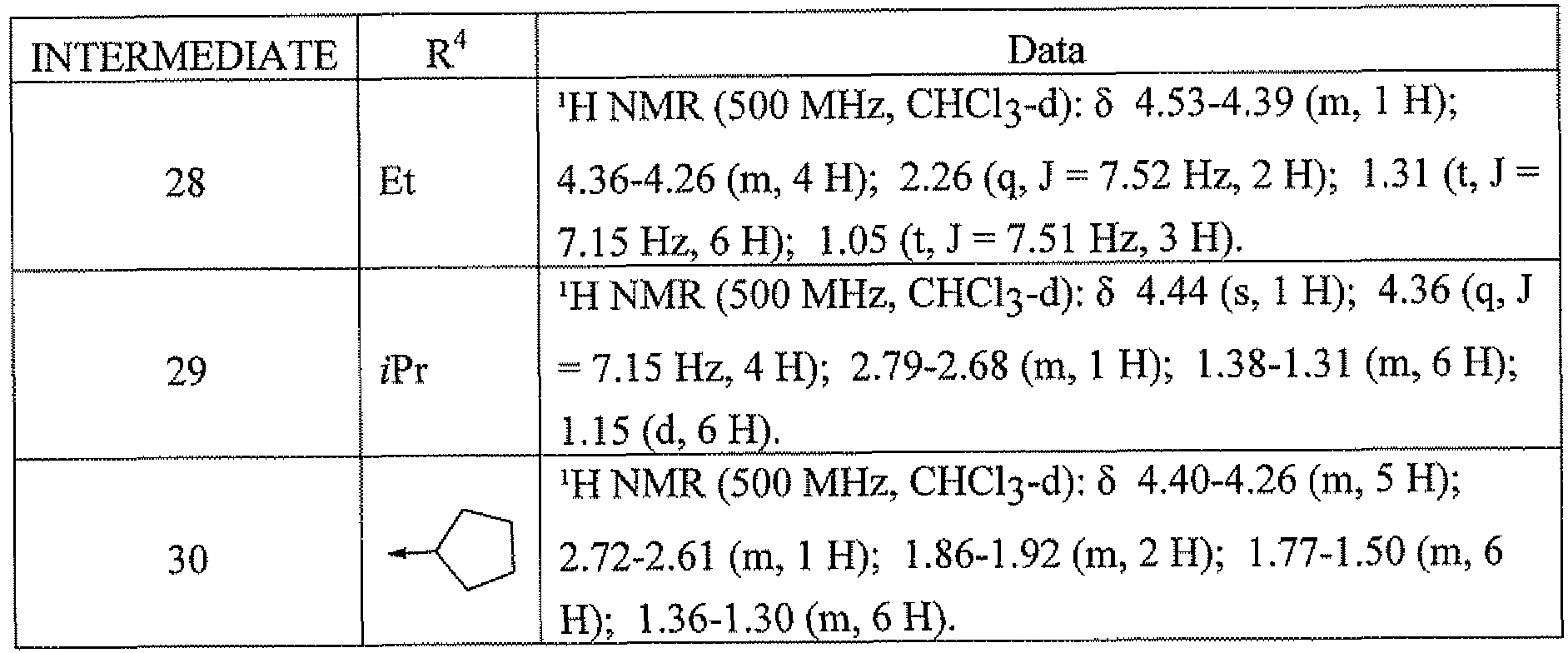

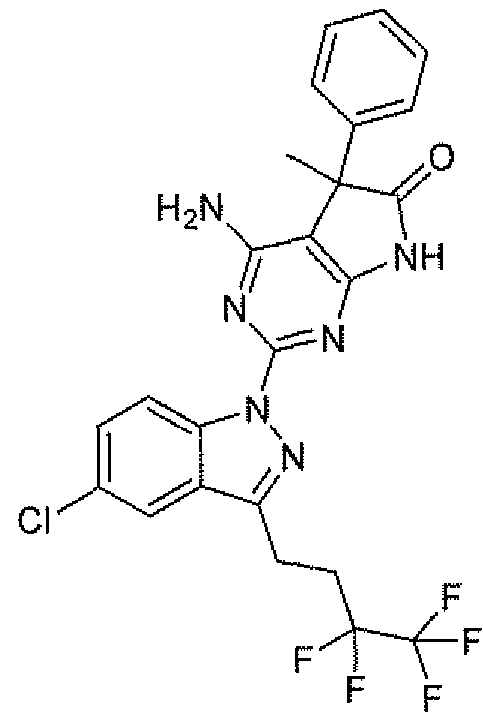









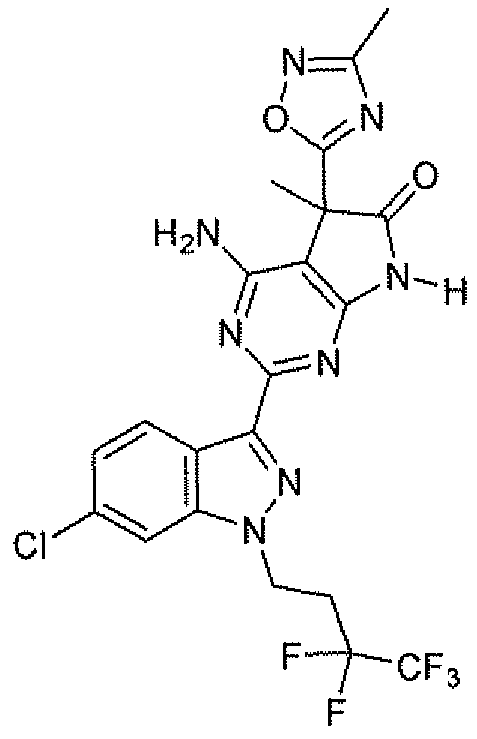























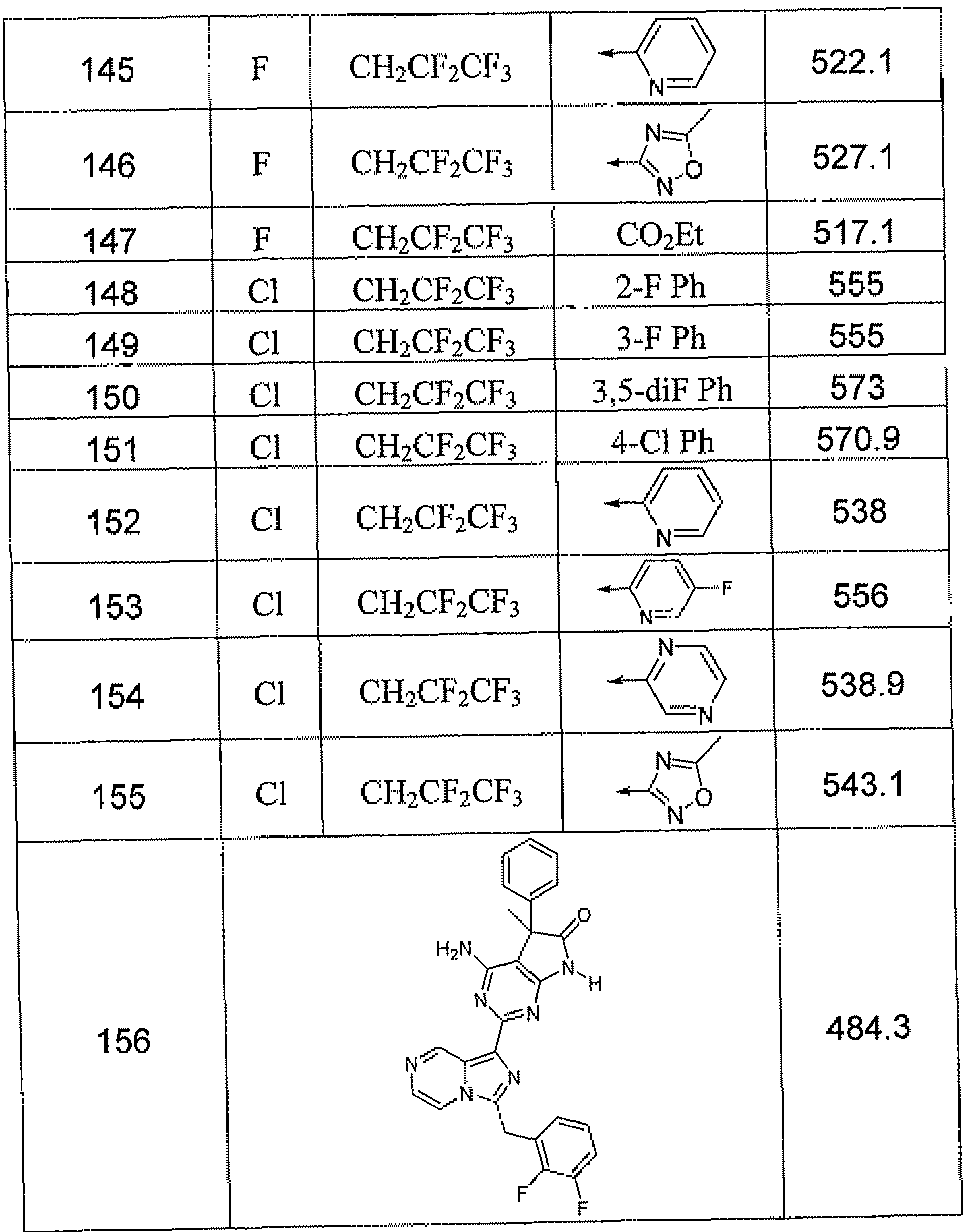



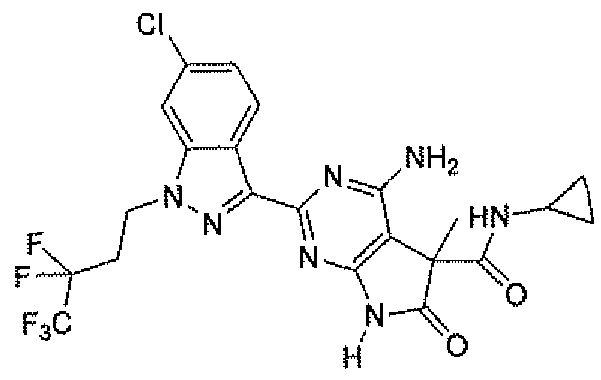


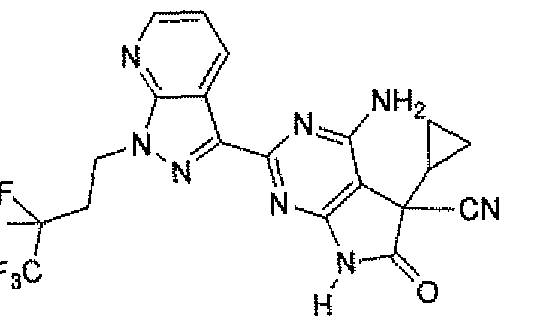




















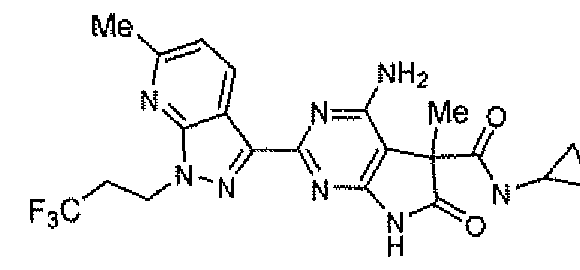



























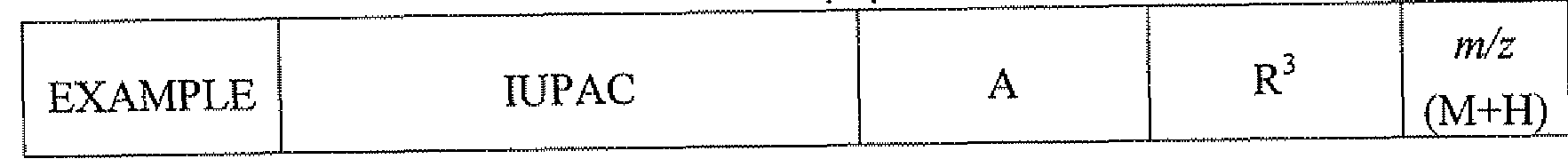



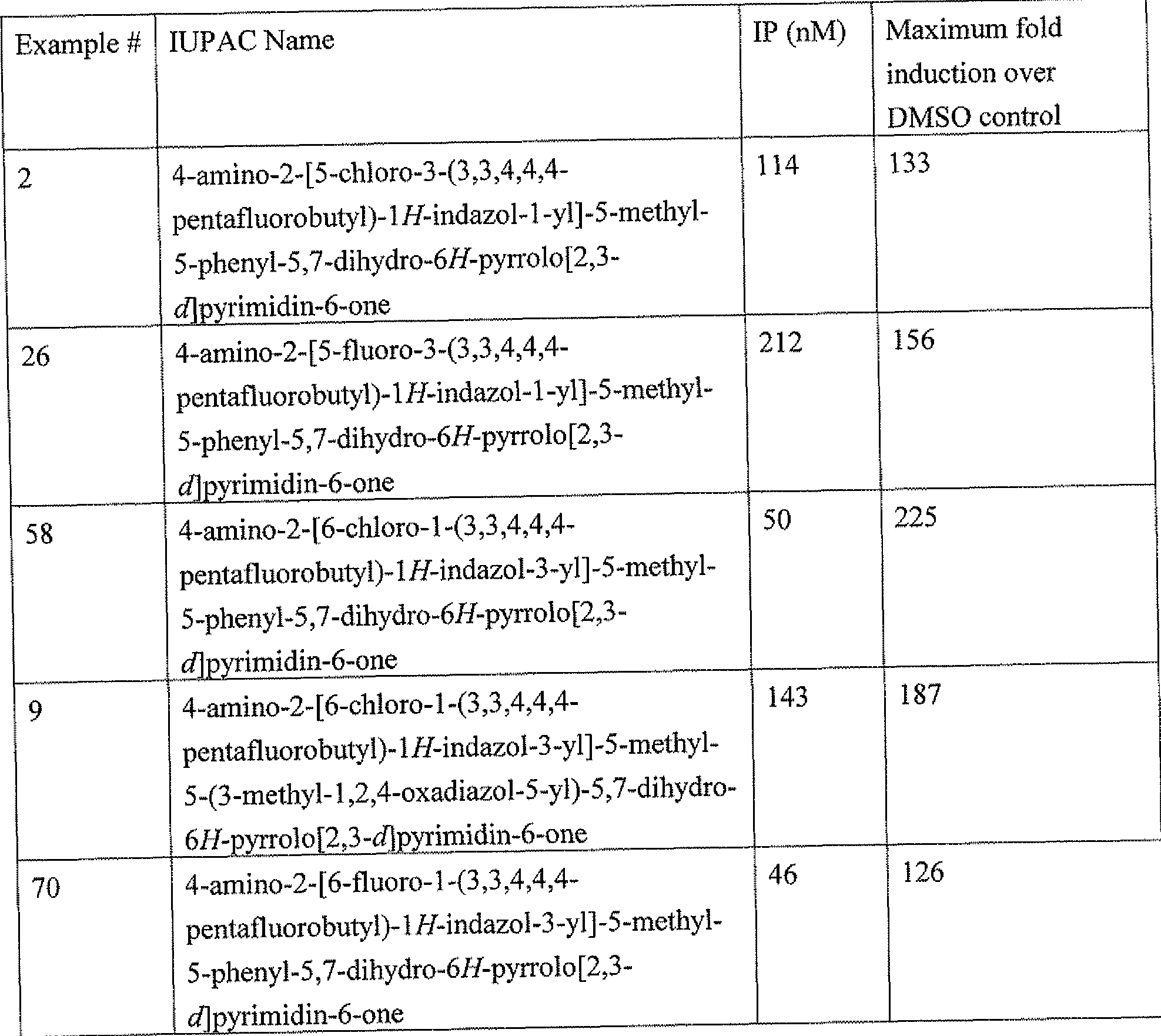

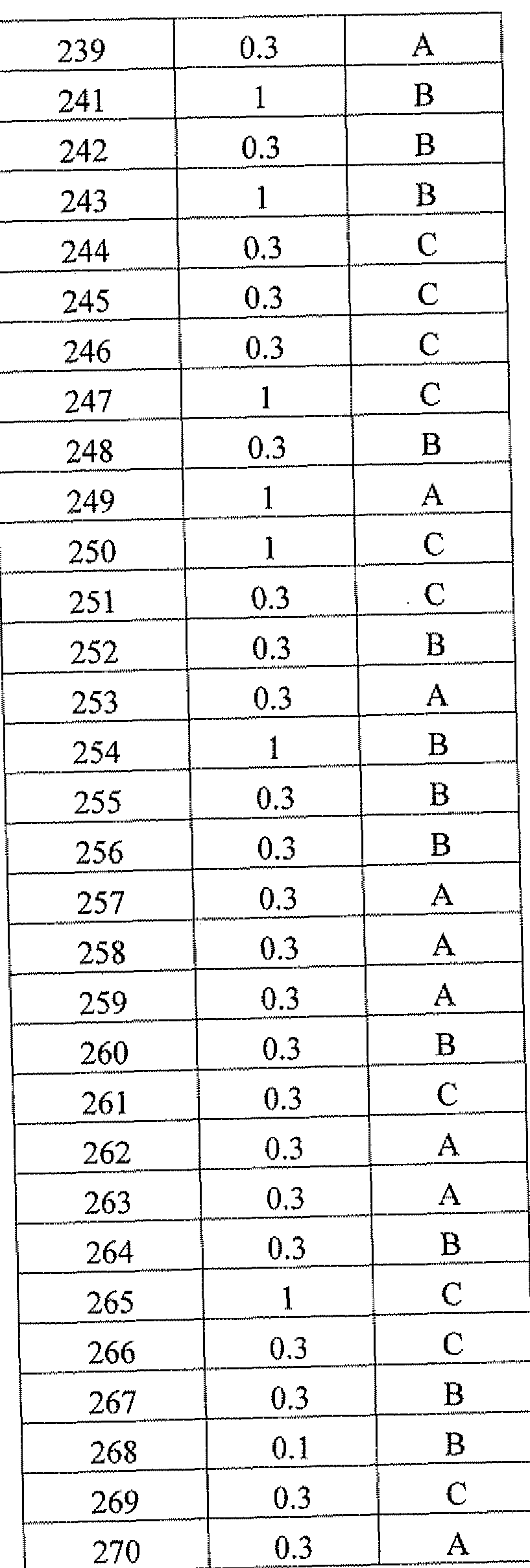
Claims





























Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NZ603884A NZ603884A (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| MA35482A MA34330B1 (en) | 2010-05-27 | 2011-05-24 | SOLUBLE GUANYLATE CYCLASE ACTIVATORS |
| KR1020127033846A KR101499308B1 (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| US13/699,046 US9365574B2 (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| EA201291348A EA023254B1 (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| BR112012030177A BR112012030177B1 (en) | 2010-05-27 | 2011-05-24 | soluble guanylate cyclase activating compound and pharmaceutical composition |
| AU2011258436A AU2011258436B2 (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| SG2012087169A SG185777A1 (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| ES11787236T ES2564503T3 (en) | 2010-05-27 | 2011-05-24 | Soluble Guanylate Cyclase Activators |
| JP2013512149A JP5409961B2 (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activator |
| MX2012013774A MX2012013774A (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators. |
| CN201180036555.6A CN103096718B (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| CA2800541A CA2800541C (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| EP11787236.6A EP2575473B1 (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| UAA201214964A UA107112C2 (en) | 2010-05-27 | 2011-05-24 | Activator of soluble guanylate cyclase |
| KR1020137034895A KR20140019004A (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
| TNP2012000544A TN2012000544A1 (en) | 2010-05-27 | 2012-11-21 | Soluble guanylate cyclase activators |
| IL223259A IL223259A0 (en) | 2010-05-27 | 2012-11-26 | Soluble guanylate cyclase activators |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US34906510P | 2010-05-27 | 2010-05-27 | |
| US61/349,065 | 2010-05-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011149921A1 true WO2011149921A1 (en) | 2011-12-01 |
Family
ID=45004331
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/037718 WO2011149921A1 (en) | 2010-05-27 | 2011-05-24 | Soluble guanylate cyclase activators |
Country Status (28)
| Country | Link |
|---|---|
| US (1) | US9365574B2 (en) |
| EP (1) | EP2575473B1 (en) |
| JP (2) | JP5409961B2 (en) |
| KR (2) | KR101499308B1 (en) |
| CN (1) | CN103096718B (en) |
| AR (1) | AR081830A1 (en) |
| AU (1) | AU2011258436B2 (en) |
| BR (1) | BR112012030177B1 (en) |
| CA (1) | CA2800541C (en) |
| CL (1) | CL2012003317A1 (en) |
| CO (1) | CO6640255A2 (en) |
| CR (1) | CR20120599A (en) |
| DO (1) | DOP2012000300A (en) |
| EA (1) | EA023254B1 (en) |
| ES (1) | ES2564503T3 (en) |
| GT (1) | GT201200320A (en) |
| HN (1) | HN2012002555A (en) |
| IL (1) | IL223259A0 (en) |
| MA (1) | MA34330B1 (en) |
| MX (1) | MX2012013774A (en) |
| NI (1) | NI201200172A (en) |
| NZ (1) | NZ603884A (en) |
| PE (1) | PE20130222A1 (en) |
| SG (1) | SG185777A1 (en) |
| TN (1) | TN2012000544A1 (en) |
| TW (1) | TW201202245A (en) |
| UA (1) | UA107112C2 (en) |
| WO (1) | WO2011149921A1 (en) |
Cited By (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102491974A (en) * | 2011-12-12 | 2012-06-13 | 南京药石药物研发有限公司 | Method for synthesizing 1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-formamidine hydrochloride |
| WO2012143510A1 (en) | 2011-04-21 | 2012-10-26 | Bayer Intellectual Property Gmbh | Fluoroalkyl-substituted pyrazolopyridines and use thereof |
| WO2012152629A1 (en) | 2011-05-06 | 2012-11-15 | Bayer Intellectual Property Gmbh | Substituted imidazopyridines and imidazopyridazines and the use thereof |
| WO2013004785A1 (en) | 2011-07-06 | 2013-01-10 | Bayer Intellectual Property Gmbh | Heteroaryl-substituted pyrazolopyridines and use thereof as soluble guanylate cyclase stimulators |
| WO2013030288A1 (en) | 2011-09-02 | 2013-03-07 | Bayer Intellectual Property Gmbh | Substituted annellated pyrimidine and the use thereof |
| DE102012200357A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | New fluoroalkyl-substituted pyrazolopyridine compounds are soluble guanylate cyclase stimulators, useful to treat e.g. heart disease, angina pectoris, hypertension, pulmonary hypertension, ischemia, vascular disease or renal insufficiency |
| DE102012200360A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituted triazines and their use |
| DE102012200354A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | New heteroaryl-substituted pyrazolopyridine compounds are soluble guanylate cyclase stimulators, useful to treat e.g. heart disease, angina pectoris, hypertension, pulmonary hypertension, ischemia, vascular disease or renal insufficiency |
| DE102012200352A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituted, fused imidazoles and pyrazoles and their use |
| DE102012200349A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituted fused pyrimidines and triazines and their use |
| DE102012200351A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Pharma Aktiengesellschaft | New substituted annellated pyrimidine compounds are guanylate cyclase stimulators useful to treat and/or prevent heart failure, hypertension, ischemia, vascular disease, renal failure, thromboembolic disorders and fibrotic diseases |
| DE102012200356A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | New substituted imidazopyrid(az)ine compounds are soluble guanylate cyclase stimulators, useful to treat e.g. heart disease, angina pectoris, hypertension, pulmonary hypertension, ischemia, vascular disease or renal insufficiency |
| WO2013131923A1 (en) * | 2012-03-06 | 2013-09-12 | Bayer Intellectual Property Gmbh | Substituted azabicycles and use thereof |
| WO2014055595A1 (en) | 2012-10-05 | 2014-04-10 | Merck Sharp & Dohme Corp. | Indoline compounds as aldosterone synthase inhibitiors related applications |
| US8741910B2 (en) | 2008-11-25 | 2014-06-03 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2014131741A1 (en) | 2013-03-01 | 2014-09-04 | Bayer Pharma Aktiengesellschaft | Benzyl-substituted pyrazolopyridines and use thereof |
| WO2014131760A1 (en) | 2013-03-01 | 2014-09-04 | Bayer Pharma Aktiengesellschaft | Trifluormethyl-substituted ring-fused pyrimidines and use thereof |
| WO2014144100A2 (en) | 2013-03-15 | 2014-09-18 | Takashi Nakai | Sgc stimulators |
| WO2015004105A1 (en) | 2013-07-10 | 2015-01-15 | Bayer Pharma Aktiengesellschaft | Benzyl-1h-pyrazolo[3,4-b]pyridines and use thereof |
| WO2015089182A1 (en) | 2013-12-11 | 2015-06-18 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2015106268A1 (en) | 2014-01-13 | 2015-07-16 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF NEUROMUSCULAR DISORDERS |
| WO2016030362A1 (en) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Substituted annulated pyrimidines and use thereof |
| WO2016030354A1 (en) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Amino-substituted annulated pyrimidines and use thereof |
| US9284301B2 (en) | 2010-03-25 | 2016-03-15 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2016044445A2 (en) | 2014-09-17 | 2016-03-24 | Ironwood Pharmaceuticals, Inc. | sGC STIMULATORS |
| WO2016044446A2 (en) | 2014-09-17 | 2016-03-24 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2016044447A1 (en) | 2014-09-17 | 2016-03-24 | Ironwood Pharmaceuticals, Inc. | Pyrazole derivatives as sgc stimulators |
| EP2927231A4 (en) * | 2012-11-30 | 2016-04-20 | Astellas Pharma Inc | Imidazopyridine compounds |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| WO2016191335A1 (en) | 2015-05-28 | 2016-12-01 | Merck Sharp & Dohme Corp. | Imidazo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| WO2017013010A1 (en) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulators and/or activators of soluble guanylate cyclase (sgc) in combination with an inhibitor of neutral endopeptidase (nep inhibitor) and/or an angiotensin aii antagonist and the use thereof |
| US9624214B2 (en) | 2012-11-05 | 2017-04-18 | Bayer Pharma Aktiengesellschaft | Amino-substituted imidazo[1,2-a]pyridinecarboxamides and their use |
| US9636399B2 (en) | 2007-11-21 | 2017-05-02 | Oregon Health & Science University | Anti-factor XI monoclonal antibodies and methods of use thereof |
| US9637550B2 (en) | 2008-12-18 | 2017-05-02 | Oregon Health & Science University | Anti-fXI antibodies and methods of use |
| WO2017106175A2 (en) | 2015-12-14 | 2017-06-22 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF GASTROINTESTINAL SPHINCTER DYSFUNCTION |
| US9688699B2 (en) | 2014-02-19 | 2017-06-27 | Bayer Pharma Aktiengesellschaft | 3-(pyrimidine-2-yl)imidazo[1,2-a]pyridines |
| WO2017112617A1 (en) * | 2015-12-22 | 2017-06-29 | Merck Sharp & Dohme Corp. | 4-amino-2-(1h-pyrazolo[3,4-b]pyridin-3-yl)-6-oxo-6,7-dihydro-5h-pyrrolo[2,3-d]pyrimidine derivatives and the respective (1h-indazol-3-yl) derivatives as cgmp modulators for treating cardiovascular diseases |
| WO2017121692A1 (en) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | Substituted sulfamides and use of same |
| WO2017121700A1 (en) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | 1,3-disubstituted 1h-pyrazolo[3,4-b]pyridine derivatives and use thereof as stimulators of soluble guanylate cyclase |
| WO2017121693A1 (en) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | Substituted thiazole and thiadiazole amides, and use thereof |
| US9776997B2 (en) | 2013-06-04 | 2017-10-03 | Bayer Pharma Aktiengesellschaft | 3-aryl-substituted imidazo[1,2-A]pyridines and their use |
| US9783614B2 (en) | 2012-05-10 | 2017-10-10 | Bayer Pharma Aktiengesellschaft | Antibodies capable of binding to the coagulation Factor XI and/or its activated form factor Xia and uses thereof |
| EP3152214A4 (en) * | 2014-06-04 | 2017-11-22 | Merck Sharp & Dohme Corp. | Imidazo-pyrazine derivatives useful as soluble guanylate cyclase activators |
| WO2017197555A1 (en) * | 2016-05-16 | 2017-11-23 | Merck Sharp & Dohme Corp. | Fused pyrazine derivatives useful as soluble guanylate cyclase stimulators |
| WO2018009609A1 (en) | 2016-07-07 | 2018-01-11 | Ironwood Pharmaceuticals, Inc. | Solid forms of an sgc stimulator |
| WO2018009596A1 (en) | 2016-07-07 | 2018-01-11 | Ironwood Pharmaceuticals, Inc. | Phosphorus prodrugs of sgc stimulators |
| WO2018045276A1 (en) | 2016-09-02 | 2018-03-08 | Ironwood Pharmaceuticals, Inc. | Fused bicyclic sgc stimulators |
| WO2018069126A1 (en) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Combination containing sgc stimulators and mineralocorticoid receptor antagonists |
| WO2018089330A2 (en) | 2016-11-08 | 2018-05-17 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2018089328A1 (en) | 2016-11-08 | 2018-05-17 | Ironwood Pharmaceuticals, Inc. | Treatment of cns diseases with sgc stimulators |
| WO2018111795A2 (en) | 2016-12-13 | 2018-06-21 | Ironwood Pharmaceuticals, Inc. | Use of sgc stimulators for the treatment of esophageal motility disorders |
| WO2018188590A1 (en) | 2017-04-11 | 2018-10-18 | Sunshine Lake Pharma Co., Ltd. | Fluorine-substituted indazole compounds and uses thereof |
| WO2019081456A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | Use of activators and stimulators of sgc comprising a beta2 subunit |
| US10292970B2 (en) | 2014-12-02 | 2019-05-21 | Bayer Pharma Aktiengesellschaft | Heteroaryl-substituted imidazo[1,2-A]pyridines and their use |
| EP3498298A1 (en) | 2017-12-15 | 2019-06-19 | Bayer AG | The use of sgc stimulators and sgc activators alone or in combination with pde5 inhibitors for the treatment of bone disorders including osteogenesis imperfecta (oi) |
| WO2019126354A1 (en) | 2017-12-19 | 2019-06-27 | Cyclerion Therapeutics, Inc. | Sgc stimulators |
| WO2019173551A1 (en) | 2018-03-07 | 2019-09-12 | Cyclerion Therapeutics, Inc. | Crystalline forms of an sgc stimulator |
| WO2019211081A1 (en) | 2018-04-30 | 2019-11-07 | Bayer Aktiengesellschaft | The use of sgc activators and sgc stimulators for the treatment of cognitive impairment |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| EP3574905A1 (en) | 2018-05-30 | 2019-12-04 | Adverio Pharma GmbH | Method of identifying a subgroup of patients suffering from dcssc which benefits from a treatment with sgc stimulators and sgc activators in a higher degree than a control group |
| WO2020014504A1 (en) | 2018-07-11 | 2020-01-16 | Cyclerion Therapeutics, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF MITOCHONRIAL DISORDERS |
| WO2020109354A1 (en) | 2018-11-28 | 2020-06-04 | Topadur Pharma Ag | Novel dual mode of action soluble guanylate cyclase activators and phosphodiesterase inhibitors and uses thereof |
| WO2020148379A1 (en) | 2019-01-17 | 2020-07-23 | Bayer Aktiengesellschaft | Methods to determine whether a subject is suitable of being treated with an agonist of soluble guanylyl cyclase (sgc) |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
| WO2021195403A1 (en) | 2020-03-26 | 2021-09-30 | Cyclerion Therapeutics, Inc. | Deuterated sgc stimulators |
| WO2021202546A1 (en) | 2020-03-31 | 2021-10-07 | Cyclerion Therapeutics, Inc. | Early drug interventions to reduce covid-19 related respiratory distress, need for ventilator assistance and death |
| WO2021245192A1 (en) | 2020-06-04 | 2021-12-09 | Topadur Pharma Ag | Novel dual mode of action soluble guanylate cyclase activators and phosphodiesterase inhibitors and uses thereof |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
| WO2022225903A1 (en) | 2021-04-20 | 2022-10-27 | Cyclerion Therapeutics, Inc. | Sgc stimulators |
| WO2022225902A1 (en) | 2021-04-20 | 2022-10-27 | Cyclerion Therapeutics, Inc. | Treatment of cns diseases with sgc stimulators |
| WO2022265984A1 (en) | 2021-06-14 | 2022-12-22 | Curtails Llc | Use of nep inhibitors for the treatment of gastrointestinal sphincter disorders |
| WO2023018795A1 (en) | 2021-08-11 | 2023-02-16 | Curtails Llc | Nep inhibitors for the treatment of laminitis |
| WO2024086179A1 (en) | 2022-10-18 | 2024-04-25 | Tisento Therapeutics, Inc. | Pyrimidine sgc stimulators |
| WO2024086182A1 (en) | 2022-10-18 | 2024-04-25 | Tisento Therapeutics Inc. | Treatment of mitochondrial diseases with sgc stimulators |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102010021637A1 (en) | 2010-05-26 | 2011-12-01 | Bayer Schering Pharma Aktiengesellschaft | Substituted 5-fluoro-1H-pyrazolopyridines and their use |
| JP5940062B2 (en) * | 2010-07-09 | 2016-06-29 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | Ring-fused 4-aminopyrimidine and its use as a stimulant of soluble guanylate cyclase |
| WO2012004258A1 (en) | 2010-07-09 | 2012-01-12 | Bayer Pharma Aktiengesellschaft | Ring-fused pyrimidines and triazines and use thereof for the treatment and/or prophylaxis of cardiovascular diseases |
| DE102010040233A1 (en) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Bicyclic aza heterocycles and their use |
| CA2901636A1 (en) | 2013-02-21 | 2014-08-28 | Adverio Pharma Gmbh | Forms of methyl {4,6-diamino-2-[1-(2-fluorobenzyl)-1h-pyrazolo[3,4-b]pyridino-3-yl]pyrimidino-5-yl}methyl carbamate |
| WO2015088885A1 (en) * | 2013-12-11 | 2015-06-18 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2015088886A1 (en) * | 2013-12-11 | 2015-06-18 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| TW201625635A (en) | 2014-11-21 | 2016-07-16 | 默沙東藥廠 | Triazolo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| WO2016191334A1 (en) | 2015-05-27 | 2016-12-01 | Merck Sharp & Dohme Corp. | Imidazo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| EP3458063A4 (en) * | 2016-05-18 | 2020-02-26 | Merck Sharp & Dohme Corp. | Methods for using triazolo-pyrazinyl soluble guanylate cyclase activators in fibrotic disorders |
| KR20210033444A (en) | 2018-06-04 | 2021-03-26 | 엑스사이언티아 엘티디 | Pyrazolopyrimidine compounds as adenosine receptor antagonists |
| US12086719B2 (en) * | 2019-10-11 | 2024-09-10 | Royal Bank Of Canada | System and method of machine learning using embedding networks |
| CN111467350B (en) * | 2020-04-20 | 2021-03-19 | 黑龙江中医药大学 | Medicine for treating mastitis and preparation method thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB876526A (en) | 1957-12-24 | 1961-09-06 | Geigy Ag J R | Process for the production of new aminobenzoic acid derivatives and their use in pest control |
| DE19744027A1 (en) | 1997-10-06 | 1999-04-08 | Hoechst Marion Roussel De Gmbh | New pyrazolo(3,4-b)pyridine derivatives useful as cGMP agonists |
| EP0908456A1 (en) | 1997-10-06 | 1999-04-14 | Hoechst Marion Roussel Deutschland GmbH | Pyrazole derivatives, their preparation and their use in drugs |
| US20040048866A1 (en) * | 2002-03-08 | 2004-03-11 | Teodozyj Kolasa | Indazole derivatives that are activators of soluble guanylate cyclase |
| US6903089B1 (en) | 2000-11-22 | 2005-06-07 | Bayer Aktiengesellschaft | Lactam-substituted pyrazolopyridine derivatives |
Family Cites Families (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4410997A1 (en) * | 1994-03-30 | 1995-10-26 | Isis Pharma Gmbh | Pharmaceutical preparations and drugs for the prevention and treatment of endothelial dysfunctions |
| US5585381A (en) | 1994-06-01 | 1996-12-17 | Kureha Chemical Industry Co., Ltd. | Pyrimidine derivatives and pharmaceutical composition |
| CN1082949C (en) * | 1995-06-06 | 2002-04-17 | 辉瑞大药厂 | Process for converting 2, 4-dichloropyridines into 2-aryloxy-4-chloropyridines |
| DE59713007D1 (en) | 1996-10-14 | 2009-06-25 | Bayer Healthcare Ag | NEW HETEROCYCLYLMETHYL-SUBSTITUTED PYRAZOLE DERIVATIVES AND THEIR USE IN THE TREATMENT OF HEART CIRCULATION DISEASES |
| US6610497B1 (en) | 1997-12-11 | 2003-08-26 | Millennium Pharmaceuticals, Inc. | Angiotensin converting enzyme homolog and therapeutic and diagnostic uses therefor |
| DE19756388A1 (en) | 1997-12-18 | 1999-06-24 | Hoechst Marion Roussel De Gmbh | New 2-aryl-4-amino-6,7-di:methoxy-quinazoline derivatives useful as guanylate cyclase activators for treating cardiovascular diseases, etc. |
| DE19834044A1 (en) * | 1998-07-29 | 2000-02-03 | Bayer Ag | New substituted pyrazole derivatives |
| DE19836697A1 (en) | 1998-08-13 | 2000-02-17 | Hoechst Marion Roussel De Gmbh | New substituted 4-amino-2-aryl-pyrimidines, are soluble guanylate cyclase activators useful e.g. for treating atherosclerosis, hypertension, angina pectoris, thrombosis, asthma or diabetes |
| GB9824310D0 (en) | 1998-11-05 | 1998-12-30 | Univ London | Activators of soluble guanylate cyclase |
| DE19904710A1 (en) | 1999-02-05 | 2000-08-10 | Aventis Pharma Gmbh | Substituted 4-amino-2-aryl-tetrahydroquinazolines, their preparation, their use and pharmaceutical compositions containing them |
| US20010044445A1 (en) | 1999-04-08 | 2001-11-22 | Bamaung Nwe Y. | Azole inhibitors of cytokine production |
| DE19943636A1 (en) | 1999-09-13 | 2001-03-15 | Bayer Ag | Novel dicarboxylic acid derivatives with pharmaceutical properties |
| DE10021069A1 (en) | 2000-04-28 | 2001-10-31 | Bayer Ag | Substituted pyrazole derivative |
| DE10054278A1 (en) | 2000-11-02 | 2002-05-08 | Bayer Ag | Use of soluble guanylate cyclase stimulators for the treatment of osteoporosis |
| AR031176A1 (en) | 2000-11-22 | 2003-09-10 | Bayer Ag | NEW DERIVATIVES OF PIRAZOLPIRIDINA SUBSTITUTED WITH PIRIDINE |
| DE10057751A1 (en) | 2000-11-22 | 2002-05-23 | Bayer Ag | New carbamate-substituted pyrazolo (3,4-b) pyridine derivatives, are soluble guanylate cyclase stimulants useful e.g. for treating cardiovascular or central nervous system diseases, sexual dysfunction or inflammation |
| DE10057754A1 (en) | 2000-11-22 | 2002-05-23 | Bayer Ag | New cyclic sulfonamido-substituted pyrazolo (3,4-b) pyridine derivatives, are soluble guanylate cyclase stimulants useful e.g. for treating cardiovascular or central nervous system diseases, sexual dysfunction or inflammation |
| MXPA03008121A (en) | 2001-03-15 | 2003-12-12 | Basf Ag | 5-phenylpyrimidine, methods and intermediate products for the production thereof and use of the same for controlling pathogenic fungi. |
| DE10122894A1 (en) | 2001-05-11 | 2002-11-14 | Bayer Ag | New pyrazolo(3,4-b)pyridin-3-yl)-5-pyrimidinyl sulfonates, are soluble guanyl cyclase stimulants useful e.g. for treating cardiovascular, thromboembolic or central nervous system diseases |
| CA2464333C (en) | 2001-10-26 | 2011-07-26 | University Of Connecticut | Heteroindanes: a new class of potent cannabimimetic ligands |
| JP4359146B2 (en) | 2002-02-13 | 2009-11-04 | エフ.ホフマン−ラ ロシュ アーゲー | Novel pyridine- and pyrimidine-derivatives |
| DE10220570A1 (en) | 2002-05-08 | 2003-11-20 | Bayer Ag | Carbamate-substituted pyrazolopyridines |
| DE10222550A1 (en) | 2002-05-17 | 2003-11-27 | Bayer Ag | New 1-benzyl-3-(2-pyrimidinyl)-pyrazolo-(3,4-b)-pyridine derivatives are soluble guanylate cyclase stimulants, useful e.g. for treating cardiovascular or thromboembolic diseases or sexual dysfunction |
| DE10232571A1 (en) | 2002-07-18 | 2004-02-05 | Bayer Ag | 4-amino substituted pyrimidine derivatives |
| US7241803B2 (en) | 2002-11-21 | 2007-07-10 | New York Blood Center | Compounds for inhibition of HIV infection by blocking HIV entry |
| US20040121994A1 (en) | 2002-12-20 | 2004-06-24 | Anderson Steven N. | Novel amides that activate soluble guanylate cyclase |
| US20050038183A1 (en) | 2003-08-14 | 2005-02-17 | Dongchan Ahn | Silicones having improved surface properties and curable silicone compositions for preparing the silicones |
| DE10351903A1 (en) | 2003-11-06 | 2005-06-09 | Bayer Healthcare Ag | New combination |
| EP1616965A1 (en) * | 2004-07-16 | 2006-01-18 | AXXAM S.r.l. | Methods and assays for detecting guanylate cyclase activity |
| CN100396678C (en) | 2004-11-16 | 2008-06-25 | 财团法人工业技术研究院 | Synthesis and uses of indole analogs of 1-benzyl-3-(5'-hydroxymethy 1-2' -furyl) indazole |
| CA2611979A1 (en) | 2005-06-15 | 2006-12-21 | Pfizer Limited | Substituted arylpyrazoles |
| BRPI0611798A2 (en) | 2005-06-15 | 2009-01-13 | Pfizer Ltd | substituted arylpyrazole compounds, their pharmaceutical composition and use |
| AU2006261845C1 (en) | 2005-06-27 | 2013-05-16 | Exelixis Patent Company Llc | Imidazole based LXR modulators |
| DE102005031575A1 (en) | 2005-07-06 | 2007-01-11 | Bayer Healthcare Ag | Use of soluble guanylate cyclase activators to promote wound healing |
| US20100016305A1 (en) | 2005-07-18 | 2010-01-21 | Bayer Healthcare Ag | novel use of activators and stimulators of soluble guanylate cyclase for the prevention or treatment of renal disorders |
| DE102006043443A1 (en) * | 2006-09-15 | 2008-03-27 | Bayer Healthcare Ag | Novel aza-bicyclic compounds and their use |
| WO2008040753A1 (en) | 2006-10-03 | 2008-04-10 | Neurosearch A/S | Indazolyl derivatives useful as potassium channel modulating agents |
| MX2009003821A (en) | 2006-10-10 | 2009-05-25 | Amgen Inc | N-aryl pyrazole compounds for use against diabetes. |
| EP1921067A1 (en) | 2006-11-08 | 2008-05-14 | Bayer Schering Pharma Aktiengesellschaft | Indole and indazole derivatives as anti-inflammatory agents |
| DE102006054757A1 (en) | 2006-11-21 | 2008-05-29 | Bayer Healthcare Ag | Novel aza-bicyclic compounds and their use |
| WO2009068652A1 (en) | 2007-11-30 | 2009-06-04 | Smithkline Beecham Corporation | 2, 6-disubstituted pyridines and 2, 4-disubstituted pyrimidines as soluble guanylate cyclase activators |
| WO2010015652A2 (en) | 2008-08-07 | 2010-02-11 | Smithkline Beecham Corporation | Thiazole compounds as activators of soluble guanylate cyclase |
| AU2009322836B2 (en) | 2008-11-25 | 2013-04-04 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| DE102009046115A1 (en) | 2009-10-28 | 2011-09-08 | Bayer Schering Pharma Aktiengesellschaft | Substituted 3-phenylpropanoic acids and their use |
| NZ602311A (en) | 2010-03-18 | 2014-08-29 | Pasteur Institut Korea | Anti-infective compounds |
| DE102010021637A1 (en) | 2010-05-26 | 2011-12-01 | Bayer Schering Pharma Aktiengesellschaft | Substituted 5-fluoro-1H-pyrazolopyridines and their use |
| WO2012004258A1 (en) * | 2010-07-09 | 2012-01-12 | Bayer Pharma Aktiengesellschaft | Ring-fused pyrimidines and triazines and use thereof for the treatment and/or prophylaxis of cardiovascular diseases |
| JP5940062B2 (en) * | 2010-07-09 | 2016-06-29 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | Ring-fused 4-aminopyrimidine and its use as a stimulant of soluble guanylate cyclase |
| DE102010040233A1 (en) * | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Bicyclic aza heterocycles and their use |
-
2011
- 2011-05-24 MA MA35482A patent/MA34330B1/en unknown
- 2011-05-24 CA CA2800541A patent/CA2800541C/en not_active Expired - Fee Related
- 2011-05-24 KR KR1020127033846A patent/KR101499308B1/en active IP Right Grant
- 2011-05-24 EP EP11787236.6A patent/EP2575473B1/en active Active
- 2011-05-24 BR BR112012030177A patent/BR112012030177B1/en not_active IP Right Cessation
- 2011-05-24 US US13/699,046 patent/US9365574B2/en active Active
- 2011-05-24 KR KR1020137034895A patent/KR20140019004A/en not_active Application Discontinuation
- 2011-05-24 UA UAA201214964A patent/UA107112C2/en unknown
- 2011-05-24 CN CN201180036555.6A patent/CN103096718B/en not_active Expired - Fee Related
- 2011-05-24 SG SG2012087169A patent/SG185777A1/en unknown
- 2011-05-24 JP JP2013512149A patent/JP5409961B2/en not_active Expired - Fee Related
- 2011-05-24 ES ES11787236T patent/ES2564503T3/en active Active
- 2011-05-24 PE PE2012002232A patent/PE20130222A1/en not_active Application Discontinuation
- 2011-05-24 WO PCT/US2011/037718 patent/WO2011149921A1/en active Application Filing
- 2011-05-24 MX MX2012013774A patent/MX2012013774A/en active IP Right Grant
- 2011-05-24 AU AU2011258436A patent/AU2011258436B2/en not_active Ceased
- 2011-05-24 NZ NZ603884A patent/NZ603884A/en not_active IP Right Cessation
- 2011-05-24 EA EA201291348A patent/EA023254B1/en not_active IP Right Cessation
- 2011-05-26 TW TW100118580A patent/TW201202245A/en unknown
- 2011-05-27 AR ARP110101824A patent/AR081830A1/en unknown
-
2012
- 2012-11-21 TN TNP2012000544A patent/TN2012000544A1/en unknown
- 2012-11-26 NI NI201200172A patent/NI201200172A/en unknown
- 2012-11-26 IL IL223259A patent/IL223259A0/en unknown
- 2012-11-26 HN HN2012002555A patent/HN2012002555A/en unknown
- 2012-11-27 CR CR20120599A patent/CR20120599A/en unknown
- 2012-11-27 CO CO12215103A patent/CO6640255A2/en active IP Right Grant
- 2012-11-27 GT GT201200320A patent/GT201200320A/en unknown
- 2012-11-27 DO DO2012000300A patent/DOP2012000300A/en unknown
- 2012-11-27 CL CL2012003317A patent/CL2012003317A1/en unknown
-
2013
- 2013-11-05 JP JP2013229137A patent/JP5789288B2/en not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB876526A (en) | 1957-12-24 | 1961-09-06 | Geigy Ag J R | Process for the production of new aminobenzoic acid derivatives and their use in pest control |
| DE19744027A1 (en) | 1997-10-06 | 1999-04-08 | Hoechst Marion Roussel De Gmbh | New pyrazolo(3,4-b)pyridine derivatives useful as cGMP agonists |
| EP0908456A1 (en) | 1997-10-06 | 1999-04-14 | Hoechst Marion Roussel Deutschland GmbH | Pyrazole derivatives, their preparation and their use in drugs |
| US6903089B1 (en) | 2000-11-22 | 2005-06-07 | Bayer Aktiengesellschaft | Lactam-substituted pyrazolopyridine derivatives |
| US20040048866A1 (en) * | 2002-03-08 | 2004-03-11 | Teodozyj Kolasa | Indazole derivatives that are activators of soluble guanylate cyclase |
Non-Patent Citations (15)
| Title |
|---|
| CHEMICAL ABDTRACTS, vol. 120, pages 41858 |
| CHEMICAL ABDTRACTS, vol. 123, pages 70224 |
| CHEMICAL ABDTRACTS, vol. 126, pages 257007 |
| CHEMICAL ABSTRACTS, vol. 119, pages 105757 |
| CHEMICAL ABSTRACTS, vol. 56, pages 15432E |
| D. L. VESELY, BIOCHEM. BIOPHYS. RES. COMM., vol. 88, 1979, pages 1244 |
| D. L. VESELY, EUR. J. CLIN. INVEST., vol. 15, 1985, pages 258 |
| IGNARRO ET AL., ADV. PHARMACOL., vol. 26, 1994, pages 35 |
| KO ET AL., BLOOD, vol. 84, 1994, pages 4226 |
| MARTIN ET AL.: "Structure of Cinaciguat (BAY 58-2667) Bound to Nostoc H-NOX Domain Reveals Insights into Heme-mimetic Activation of the Soluble Guanylyl Cyclase.", JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 285, 12 May 2010 (2010-05-12), pages 22651 - 22657 * |
| PETTIBONE ET AL., EUR. J. PHARMACOL., vol. 116, 1985, pages 307 |
| See also references of EP2575473A4 |
| WU ET AL., BRIT. J. PHARMACOL., vol. 116, 1995, pages 1973 |
| YU ET AL., BIOCHEM. J., vol. 306, 1995, pages 787 |
| YU ET AL., BRIT. J. PHARMACOL, vol. 114, 1995, pages 1587 |
Cited By (126)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9636399B2 (en) | 2007-11-21 | 2017-05-02 | Oregon Health & Science University | Anti-factor XI monoclonal antibodies and methods of use thereof |
| US10577428B2 (en) | 2007-11-21 | 2020-03-03 | Oregon Health & Science University | Anti-factor XI monoclonal antibodies and methods of use thereof |
| US8741910B2 (en) | 2008-11-25 | 2014-06-03 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| US9637550B2 (en) | 2008-12-18 | 2017-05-02 | Oregon Health & Science University | Anti-fXI antibodies and methods of use |
| US9284301B2 (en) | 2010-03-25 | 2016-03-15 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| US20140100229A1 (en) * | 2011-04-21 | 2014-04-10 | Bayer Intellectual Property Gmbh | Fluoroalkyl-substituted pyrazolopyridines and use thereof |
| WO2012143510A1 (en) | 2011-04-21 | 2012-10-26 | Bayer Intellectual Property Gmbh | Fluoroalkyl-substituted pyrazolopyridines and use thereof |
| US9090610B2 (en) * | 2011-04-21 | 2015-07-28 | Bayer Intellectual Property Gmbh | Fluoroalkyl-substituted pyrazolopyridines and use thereof |
| JP2014513112A (en) * | 2011-05-06 | 2014-05-29 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | Substituted imidazopyridines and imidazopyridazines and their use |
| WO2012152629A1 (en) | 2011-05-06 | 2012-11-15 | Bayer Intellectual Property Gmbh | Substituted imidazopyridines and imidazopyridazines and the use thereof |
| CN103906752A (en) * | 2011-07-06 | 2014-07-02 | 拜耳知识产权有限责任公司 | Heteroaryl-substituted pyrazolopyridines and use thereof as soluble guanylate cyclase stimulators |
| WO2013004785A1 (en) | 2011-07-06 | 2013-01-10 | Bayer Intellectual Property Gmbh | Heteroaryl-substituted pyrazolopyridines and use thereof as soluble guanylate cyclase stimulators |
| JP2014523895A (en) * | 2011-07-06 | 2014-09-18 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | Heteroaryl substituted pyrazolopyridines and uses thereof |
| EA026701B1 (en) * | 2011-09-02 | 2017-05-31 | Байер Интеллектуэль Проперти Гмбх | Substituted annellated pyrimidines, method of production thereof, use thereof and medicine for treating and/or preventing diseases |
| AU2012300844B2 (en) * | 2011-09-02 | 2017-03-30 | Bayer Intellectual Property Gmbh | Substituted annellated pyrimidine and the use thereof |
| WO2013030288A1 (en) | 2011-09-02 | 2013-03-07 | Bayer Intellectual Property Gmbh | Substituted annellated pyrimidine and the use thereof |
| US8957210B2 (en) | 2011-12-12 | 2015-02-17 | Pharmablock (Nanjing) R&D Co., Ltd. | Method for synthesizing 1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridine-3-formamidine hydrochloride |
| CN102491974B (en) * | 2011-12-12 | 2013-08-07 | 南京药石药物研发有限公司 | Method for synthesizing 1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-formamidine hydrochloride |
| WO2013086935A1 (en) * | 2011-12-12 | 2013-06-20 | 南京药石药物研发有限公司 | Method for synthesizing 1-(2-fluorobenzyl)-1h-pyrazolo[3,4-b]pyridin-3-formamidine hydrochloride |
| CN102491974A (en) * | 2011-12-12 | 2012-06-13 | 南京药石药物研发有限公司 | Method for synthesizing 1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-formamidine hydrochloride |
| JP2015505318A (en) * | 2012-01-11 | 2015-02-19 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | Substituted, fused imidazoles and pyrazoles and uses thereof |
| CN104812762A (en) * | 2012-01-11 | 2015-07-29 | 拜耳药业股份公司 | Substituted annulated pyrimidines and triazines, and use thereof |
| WO2013104598A3 (en) * | 2012-01-11 | 2013-10-10 | Bayer Intellectual Property Gmbh | Substituted, annulated imidazoles and pyrazoles, and use thereof |
| DE102012200357A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | New fluoroalkyl-substituted pyrazolopyridine compounds are soluble guanylate cyclase stimulators, useful to treat e.g. heart disease, angina pectoris, hypertension, pulmonary hypertension, ischemia, vascular disease or renal insufficiency |
| EA025837B1 (en) * | 2012-01-11 | 2017-02-28 | Байер Интеллектуэль Проперти Гмбх | Substituted annulated pyrimidines and triazines |
| WO2013104598A2 (en) | 2012-01-11 | 2013-07-18 | Bayer Intellectual Property Gmbh | Substituted, annulated imidazoles and pyrazoles, and use thereof |
| WO2013104703A1 (en) | 2012-01-11 | 2013-07-18 | Bayer Pharma Aktiengesellschaft | Substituted annulated pyrimidines and triazines, and use thereof |
| WO2013104597A1 (en) | 2012-01-11 | 2013-07-18 | Bayer Intellectual Property Gmbh | Substituted triazine derivatives and use thereof as stimulators of soluble guanylate cyclase |
| US9505786B2 (en) | 2012-01-11 | 2016-11-29 | Bayer Pharma Aktiengesellschaft | Substituted annulated triazines and use thereof |
| DE102012200356A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | New substituted imidazopyrid(az)ine compounds are soluble guanylate cyclase stimulators, useful to treat e.g. heart disease, angina pectoris, hypertension, pulmonary hypertension, ischemia, vascular disease or renal insufficiency |
| DE102012200351A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Pharma Aktiengesellschaft | New substituted annellated pyrimidine compounds are guanylate cyclase stimulators useful to treat and/or prevent heart failure, hypertension, ischemia, vascular disease, renal failure, thromboembolic disorders and fibrotic diseases |
| DE102012200360A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituted triazines and their use |
| DE102012200354A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | New heteroaryl-substituted pyrazolopyridine compounds are soluble guanylate cyclase stimulators, useful to treat e.g. heart disease, angina pectoris, hypertension, pulmonary hypertension, ischemia, vascular disease or renal insufficiency |
| DE102012200349A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituted fused pyrimidines and triazines and their use |
| US9133191B2 (en) | 2012-01-11 | 2015-09-15 | Bayer Intellectual Property Gmbh | Substituted triazine derivatives and use thereof as stimulators of soluble guanylate cyclase |
| DE102012200352A1 (en) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituted, fused imidazoles and pyrazoles and their use |
| WO2013131923A1 (en) * | 2012-03-06 | 2013-09-12 | Bayer Intellectual Property Gmbh | Substituted azabicycles and use thereof |
| US9498480B2 (en) | 2012-03-06 | 2016-11-22 | Bayer Intellectual Property Gmbh | Substituted azabicycles and use thereof |
| JP2015509515A (en) * | 2012-03-06 | 2015-03-30 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | Substituted azabicycles and uses thereof |
| CN104395313A (en) * | 2012-03-06 | 2015-03-04 | 拜耳知识产权有限责任公司 | Substituted azabicycles and use thereof |
| CN104395313B (en) * | 2012-03-06 | 2017-03-08 | 拜耳知识产权有限责任公司 | Azabicyclo replacing and application thereof |
| US11046783B2 (en) | 2012-05-10 | 2021-06-29 | Bayer Pharma Aktiengesellschaft | Antibodies capable of binding to the coagulation factor XIa and uses thereof |
| US10221247B2 (en) | 2012-05-10 | 2019-03-05 | Bayer Pharma Aktiengesellschaft | Antibodies capable of binding to the coagulation factor XIa and uses thereof |
| US9783614B2 (en) | 2012-05-10 | 2017-10-10 | Bayer Pharma Aktiengesellschaft | Antibodies capable of binding to the coagulation Factor XI and/or its activated form factor Xia and uses thereof |
| US10040866B2 (en) | 2012-05-10 | 2018-08-07 | Bayer Pharma Aktiengesellschaft | Nucleic acids and host cells expressing antibodies capable of binding to the coagulation factor XIa and uses thereof |
| US9745282B2 (en) | 2012-10-05 | 2017-08-29 | Merck Sharp & Dohme Corp | Indoline compounds as aldosterone synthase inhibitors |
| US9550750B2 (en) | 2012-10-05 | 2017-01-24 | Merck Sharp & Dohme Corp. | Indoline compounds as aldosterone synthase inhibitors |
| WO2014055595A1 (en) | 2012-10-05 | 2014-04-10 | Merck Sharp & Dohme Corp. | Indoline compounds as aldosterone synthase inhibitiors related applications |
| US10662185B2 (en) | 2012-11-05 | 2020-05-26 | Bayer Pharma Aktiengesellschaft | Amino-substituted imidazo[1,2-A] pyridinecarboxamides and their use |
| US10052312B2 (en) | 2012-11-05 | 2018-08-21 | Bayer Pharma Aktiengesellschaft | Amino-substituted imidazo[1,2-a]pyridinecarboxamides and their use |
| US9624214B2 (en) | 2012-11-05 | 2017-04-18 | Bayer Pharma Aktiengesellschaft | Amino-substituted imidazo[1,2-a]pyridinecarboxamides and their use |
| EP2927231A4 (en) * | 2012-11-30 | 2016-04-20 | Astellas Pharma Inc | Imidazopyridine compounds |
| WO2014131760A1 (en) | 2013-03-01 | 2014-09-04 | Bayer Pharma Aktiengesellschaft | Trifluormethyl-substituted ring-fused pyrimidines and use thereof |
| WO2014131741A1 (en) | 2013-03-01 | 2014-09-04 | Bayer Pharma Aktiengesellschaft | Benzyl-substituted pyrazolopyridines and use thereof |
| EP3998260A1 (en) | 2013-03-15 | 2022-05-18 | Cyclerion Therapeutics, Inc. | Sgc stimulators |
| WO2014144100A2 (en) | 2013-03-15 | 2014-09-18 | Takashi Nakai | Sgc stimulators |
| EP3660013A1 (en) | 2013-03-15 | 2020-06-03 | Cyclerion Therapeutics, Inc. | Sgc stimulators |
| US9776997B2 (en) | 2013-06-04 | 2017-10-03 | Bayer Pharma Aktiengesellschaft | 3-aryl-substituted imidazo[1,2-A]pyridines and their use |
| WO2015004105A1 (en) | 2013-07-10 | 2015-01-15 | Bayer Pharma Aktiengesellschaft | Benzyl-1h-pyrazolo[3,4-b]pyridines and use thereof |
| US9605008B2 (en) | 2013-07-10 | 2017-03-28 | Bayer Pharma Aktiengesellschaft | Benzyl-1H-pyrazolo[3,4-b]pyridines and use thereof |
| WO2015089182A1 (en) | 2013-12-11 | 2015-06-18 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2015106268A1 (en) | 2014-01-13 | 2015-07-16 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF NEUROMUSCULAR DISORDERS |
| US9688699B2 (en) | 2014-02-19 | 2017-06-27 | Bayer Pharma Aktiengesellschaft | 3-(pyrimidine-2-yl)imidazo[1,2-a]pyridines |
| EP3152214A4 (en) * | 2014-06-04 | 2017-11-22 | Merck Sharp & Dohme Corp. | Imidazo-pyrazine derivatives useful as soluble guanylate cyclase activators |
| CN107074883A (en) * | 2014-08-29 | 2017-08-18 | 拜耳医药股份有限公司 | Cyclic pyrimidin of amino substitution and application thereof |
| WO2016030354A1 (en) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Amino-substituted annulated pyrimidines and use thereof |
| WO2016030362A1 (en) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Substituted annulated pyrimidines and use thereof |
| WO2016044445A2 (en) | 2014-09-17 | 2016-03-24 | Ironwood Pharmaceuticals, Inc. | sGC STIMULATORS |
| WO2016044446A2 (en) | 2014-09-17 | 2016-03-24 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2016044447A1 (en) | 2014-09-17 | 2016-03-24 | Ironwood Pharmaceuticals, Inc. | Pyrazole derivatives as sgc stimulators |
| US10292970B2 (en) | 2014-12-02 | 2019-05-21 | Bayer Pharma Aktiengesellschaft | Heteroaryl-substituted imidazo[1,2-A]pyridines and their use |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| WO2016191335A1 (en) | 2015-05-28 | 2016-12-01 | Merck Sharp & Dohme Corp. | Imidazo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| EP3310782A4 (en) * | 2015-05-28 | 2018-11-21 | Merck Sharp & Dohme Corp. | Imidazo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| US11166932B2 (en) | 2015-07-23 | 2021-11-09 | Bayer Pharma Aktiengesellschaft | Stimulators and/or activators of soluble guanylate cyclase (sGC) in combination with an inhibitor of neutral endopeptidase (NEP inhibitor) and/or an angiotensin AII antagonist and the use thereof |
| WO2017013010A1 (en) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulators and/or activators of soluble guanylate cyclase (sgc) in combination with an inhibitor of neutral endopeptidase (nep inhibitor) and/or an angiotensin aii antagonist and the use thereof |
| WO2017106175A2 (en) | 2015-12-14 | 2017-06-22 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF GASTROINTESTINAL SPHINCTER DYSFUNCTION |
| EA036445B1 (en) * | 2015-12-22 | 2020-11-11 | Мерк Шарп И Доум Корп. | 4-AMINO-2-(1H-PYRAZOLO[3,4-b]PYRIDIN-3-YL)-6-OXO-6,7-DIHYDRO-5H-PYRROLO[2,3-d]PYRIMIDINE DERIVATIVES AND THE RESPECTIVE (1H-INDAZOL-3-YL) DERIVATIVES AS CGMP MODULATORS FOR TREATING CARDIOVASCULAR DISEASES |
| WO2017112617A1 (en) * | 2015-12-22 | 2017-06-29 | Merck Sharp & Dohme Corp. | 4-amino-2-(1h-pyrazolo[3,4-b]pyridin-3-yl)-6-oxo-6,7-dihydro-5h-pyrrolo[2,3-d]pyrimidine derivatives and the respective (1h-indazol-3-yl) derivatives as cgmp modulators for treating cardiovascular diseases |
| KR102191312B1 (en) | 2015-12-22 | 2020-12-15 | 머크 샤프 앤드 돔 코포레이션 | 4-amino-2-(1H-pyrazolo[3,4-B]pyridin-3-yl)-6-oxo-6,7-dihydro-5H-pyrrolo[ as a cGMP modulator for the treatment of cardiovascular disease 2,3-D]pyridine derivatives and respective (1H-indazol-3-yl) derivatives |
| US10428076B2 (en) | 2015-12-22 | 2019-10-01 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase stimulators |
| US10030027B2 (en) | 2015-12-22 | 2018-07-24 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase stimulators |
| KR20180095904A (en) * | 2015-12-22 | 2018-08-28 | 머크 샤프 앤드 돔 코포레이션 | 3-yl) -6-oxo-6,7-dihydro-5H-pyrrolo [2,3-d] pyrimidin- 2,3-D] pyridine derivatives and the respective (1H-indazol-3-yl) derivatives |
| TWI724079B (en) * | 2015-12-22 | 2021-04-11 | 美商默沙東藥廠 | Soluble guanylate cyclase stimulators |
| AU2016377364B2 (en) * | 2015-12-22 | 2019-02-14 | Merck Sharp & Dohme Llc | 4-amino-2-(1H-pyrazolo[3,4-b]pyridin-3-yl)-6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidine derivatives and the respective (1H-indazol-3-yl) derivatives as cGMP modulators for treating cardiovascular diseases |
| WO2017121692A1 (en) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | Substituted sulfamides and use of same |
| WO2017121700A1 (en) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | 1,3-disubstituted 1h-pyrazolo[3,4-b]pyridine derivatives and use thereof as stimulators of soluble guanylate cyclase |
| WO2017121693A1 (en) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | Substituted thiazole and thiadiazole amides, and use thereof |
| US10780092B2 (en) | 2016-05-16 | 2020-09-22 | Merck Sharp & Dohme Corp. | Fused pyrazine derivatives useful as soluble guanylate cyclase stimulators |
| WO2017197555A1 (en) * | 2016-05-16 | 2017-11-23 | Merck Sharp & Dohme Corp. | Fused pyrazine derivatives useful as soluble guanylate cyclase stimulators |
| WO2018009609A1 (en) | 2016-07-07 | 2018-01-11 | Ironwood Pharmaceuticals, Inc. | Solid forms of an sgc stimulator |
| WO2018009596A1 (en) | 2016-07-07 | 2018-01-11 | Ironwood Pharmaceuticals, Inc. | Phosphorus prodrugs of sgc stimulators |
| US11731977B2 (en) | 2016-09-02 | 2023-08-22 | Cyclerion Therapeutics, Inc. | SGC stimulators |
| EP3872080A1 (en) | 2016-09-02 | 2021-09-01 | Cyclerion Therapeutics, Inc. | Fused bicyclic sgc stimulators |
| US10858363B2 (en) | 2016-09-02 | 2020-12-08 | Cyclerion Therapeutics, Inc. | SGC stimulators |
| WO2018045276A1 (en) | 2016-09-02 | 2018-03-08 | Ironwood Pharmaceuticals, Inc. | Fused bicyclic sgc stimulators |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
| US10918639B2 (en) | 2016-10-11 | 2021-02-16 | Bayer Pharma Aktiengesellschaft | Combination containing SGC stimulators and mineralocorticoid receptor antagonists |
| WO2018069126A1 (en) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Combination containing sgc stimulators and mineralocorticoid receptor antagonists |
| US11684621B2 (en) | 2016-10-11 | 2023-06-27 | Bayer Pharma Aktiengesellschaft | Combination containing sGC stimulators and mineralocorticoid receptor antagonists |
| WO2018089328A1 (en) | 2016-11-08 | 2018-05-17 | Ironwood Pharmaceuticals, Inc. | Treatment of cns diseases with sgc stimulators |
| WO2018089330A2 (en) | 2016-11-08 | 2018-05-17 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2018111795A2 (en) | 2016-12-13 | 2018-06-21 | Ironwood Pharmaceuticals, Inc. | Use of sgc stimulators for the treatment of esophageal motility disorders |
| WO2018188590A1 (en) | 2017-04-11 | 2018-10-18 | Sunshine Lake Pharma Co., Ltd. | Fluorine-substituted indazole compounds and uses thereof |
| AU2018252099B2 (en) * | 2017-04-11 | 2021-08-12 | Sunshine Lake Pharma Co., Ltd. | Fluorine-substituted indazole compounds and uses thereof |
| EP3609883A4 (en) * | 2017-04-11 | 2020-12-09 | Sunshine Lake Pharma Co., Ltd. | Fluorine-substituted indazole compounds and uses thereof |
| WO2019081456A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | Use of activators and stimulators of sgc comprising a beta2 subunit |
| EP3498298A1 (en) | 2017-12-15 | 2019-06-19 | Bayer AG | The use of sgc stimulators and sgc activators alone or in combination with pde5 inhibitors for the treatment of bone disorders including osteogenesis imperfecta (oi) |
| WO2019126354A1 (en) | 2017-12-19 | 2019-06-27 | Cyclerion Therapeutics, Inc. | Sgc stimulators |
| WO2019173551A1 (en) | 2018-03-07 | 2019-09-12 | Cyclerion Therapeutics, Inc. | Crystalline forms of an sgc stimulator |
| WO2019211081A1 (en) | 2018-04-30 | 2019-11-07 | Bayer Aktiengesellschaft | The use of sgc activators and sgc stimulators for the treatment of cognitive impairment |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| EP3574905A1 (en) | 2018-05-30 | 2019-12-04 | Adverio Pharma GmbH | Method of identifying a subgroup of patients suffering from dcssc which benefits from a treatment with sgc stimulators and sgc activators in a higher degree than a control group |
| WO2020014504A1 (en) | 2018-07-11 | 2020-01-16 | Cyclerion Therapeutics, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF MITOCHONRIAL DISORDERS |
| WO2020109354A1 (en) | 2018-11-28 | 2020-06-04 | Topadur Pharma Ag | Novel dual mode of action soluble guanylate cyclase activators and phosphodiesterase inhibitors and uses thereof |
| WO2020148379A1 (en) | 2019-01-17 | 2020-07-23 | Bayer Aktiengesellschaft | Methods to determine whether a subject is suitable of being treated with an agonist of soluble guanylyl cyclase (sgc) |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
| WO2021195403A1 (en) | 2020-03-26 | 2021-09-30 | Cyclerion Therapeutics, Inc. | Deuterated sgc stimulators |
| WO2021202546A1 (en) | 2020-03-31 | 2021-10-07 | Cyclerion Therapeutics, Inc. | Early drug interventions to reduce covid-19 related respiratory distress, need for ventilator assistance and death |
| WO2021245192A1 (en) | 2020-06-04 | 2021-12-09 | Topadur Pharma Ag | Novel dual mode of action soluble guanylate cyclase activators and phosphodiesterase inhibitors and uses thereof |
| WO2022225903A1 (en) | 2021-04-20 | 2022-10-27 | Cyclerion Therapeutics, Inc. | Sgc stimulators |
| WO2022225902A1 (en) | 2021-04-20 | 2022-10-27 | Cyclerion Therapeutics, Inc. | Treatment of cns diseases with sgc stimulators |
| WO2022265984A1 (en) | 2021-06-14 | 2022-12-22 | Curtails Llc | Use of nep inhibitors for the treatment of gastrointestinal sphincter disorders |
| WO2023018795A1 (en) | 2021-08-11 | 2023-02-16 | Curtails Llc | Nep inhibitors for the treatment of laminitis |
| WO2024086179A1 (en) | 2022-10-18 | 2024-04-25 | Tisento Therapeutics, Inc. | Pyrimidine sgc stimulators |
| WO2024086182A1 (en) | 2022-10-18 | 2024-04-25 | Tisento Therapeutics Inc. | Treatment of mitochondrial diseases with sgc stimulators |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2575473B1 (en) | Soluble guanylate cyclase activators | |
| AU2009322836B2 (en) | Soluble guanylate cyclase activators | |
| US9611278B2 (en) | Soluble guanylate cyclase activators | |
| US9783552B2 (en) | Soluble guanylate cyclase activators | |
| JP6054967B2 (en) | Substituted annelated pyrimidines and uses thereof | |
| JP6123804B2 (en) | Tricyclic heterocyclic compounds and JAK inhibitors | |
| EP3152214A1 (en) | Imidazo-pyrazine derivatives useful as soluble guanylate cyclase activators | |
| JP2017535561A (en) | Triazolo-pyrazinyl derivatives useful as soluble guanylate cyclase activators | |
| EP2549875A1 (en) | Soluble guanylate cyclase activators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180036555.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11787236 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13699046 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2800541 Country of ref document: CA Ref document number: 2013512149 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9846/CHENP/2012 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12012502319 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 223259 Country of ref document: IL |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012003317 Country of ref document: CL Ref document number: 1201005814 Country of ref document: TH Ref document number: 12215103 Country of ref document: CO Ref document number: 002232-2012 Country of ref document: PE Ref document number: MX/A/2012/013774 Country of ref document: MX Ref document number: CR2012-000599 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011787236 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201291348 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12940 Country of ref document: GE |
|
| ENP | Entry into the national phase |
Ref document number: 20127033846 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2011258436 Country of ref document: AU Date of ref document: 20110524 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012030177 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012030177 Country of ref document: BR Kind code of ref document: A2 Effective date: 20121127 |