WO2011133751A2 - Process of producing cycloalkylcarboxamido-indole compounds - Google Patents
Process of producing cycloalkylcarboxamido-indole compounds Download PDFInfo
- Publication number
- WO2011133751A2 WO2011133751A2 PCT/US2011/033396 US2011033396W WO2011133751A2 WO 2011133751 A2 WO2011133751 A2 WO 2011133751A2 US 2011033396 W US2011033396 W US 2011033396W WO 2011133751 A2 WO2011133751 A2 WO 2011133751A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- aliphatic
- organic solvent
- ring
- Prior art date
Links
- 0 C1=C=*1c1cc(cccc2)c2cc1 Chemical compound C1=C=*1c1cc(cccc2)c2cc1 0.000 description 4
- UWUKUFGWTFKPMT-UHFFFAOYSA-N CC(C)(C)COCc1ccccc1 Chemical compound CC(C)(C)COCc1ccccc1 UWUKUFGWTFKPMT-UHFFFAOYSA-N 0.000 description 1
- KDUGDKUMCVFJHS-UHFFFAOYSA-N CC(O1)=C(C)OC1(F)F Chemical compound CC(O1)=C(C)OC1(F)F KDUGDKUMCVFJHS-UHFFFAOYSA-N 0.000 description 1
- NGKKNENXNZRLAT-UHFFFAOYSA-N CCOC(C(c(cc1O2)ccc1OC2(F)F)C#N)=O Chemical compound CCOC(C(c(cc1O2)ccc1OC2(F)F)C#N)=O NGKKNENXNZRLAT-UHFFFAOYSA-N 0.000 description 1
- FVNYSBKXILOVTD-UHFFFAOYSA-N O=C(C1(CC1)c(cc1)cc(O2)c1OC2(F)F)Cl Chemical compound O=C(C1(CC1)c(cc1)cc(O2)c1OC2(F)F)Cl FVNYSBKXILOVTD-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/12—Radicals substituted by oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
- A61K31/4045—Indole-alkylamines; Amides thereof, e.g. serotonin, melatonin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
- A61K31/405—Indole-alkanecarboxylic acids; Derivatives thereof, e.g. tryptophan, indomethacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/10—Expectorants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/12—Mucolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/68—Preparation of compounds containing amino groups bound to a carbon skeleton from amines, by reactions not involving amino groups, e.g. reduction of unsaturated amines, aromatisation, or substitution of the carbon skeleton
- C07C209/74—Preparation of compounds containing amino groups bound to a carbon skeleton from amines, by reactions not involving amino groups, e.g. reduction of unsaturated amines, aromatisation, or substitution of the carbon skeleton by halogenation, hydrohalogenation, dehalogenation, or dehydrohalogenation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C225/00—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones
- C07C225/02—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton
- C07C225/04—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being saturated
- C07C225/06—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being saturated and acyclic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/40—Nitrogen atoms, not forming part of a nitro radical, e.g. isatin semicarbazone
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/50—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to atoms of the carbocyclic ring
- C07D317/60—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention features processes for preparing compounds useful for treating CFTR mediated diseases such as cystic fibrosis.
- CFTR is a cAMP/ATP -mediated anion channel that is expressed in a variety of cells types, including absorptive and secretory epithelia cells, where it regulates anion flux across the membrane, as well as the activity of other ion channels and proteins. In epithelia cells, normal functioning of CFTR is critical for the maintenance of electrolyte transport throughout the body, including respiratory and digestive tissue.
- CFTR is composed of approximately 1480 amino acids that encode a protein made up of a tandem repeat of transmembrane domains, each containing six transmembrane helices and a nucleotide binding domain. The two transmembrane domains are linked by a large, polar, regulatory (R)-domain with multiple phosphorylation sites that regulate channel activity and cellular trafficking.
- CFTR cystic fibrosis
- a defect in this gene causes mutations in CFTR resulting in cystic fibrosis ("CF"), the most common fatal genetic disease in humans. Cystic fibrosis affects approximately one in every 2,500 infants in the United States. Within the general United States population, up to 10 million people carry a single copy of the defective gene without apparent ill effects. In contrast, individuals with two copies of the CF associated gene suffer from the debilitating and fatal effects of CF, including chronic lung disease.
- CFTR transports a variety of molecules in addition to anions
- this role represents one element in an important mechanism of transporting ions and water across the epithelium.
- the other elements include the epithelial Na + channel, ENaC, Na + /2C17K + co-transporter, Na + -K + -ATPase pump and the basolateral membrane K + channels, that are responsible for the uptake of chloride into the cell.
- the present invention provides processes for preparing CFTR correctors useful in the treatment of CFTR mediated diseases, such as cystic fibrosis.
- Such compounds include (i?)-l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)-N-(l-(2,3- dihydroxypropyl)-6-fluoro-2-( 1 -hydroxy-2-methylpropan-2-yl)- 1 H-indol-5 - yl)cyclopropanecarboxamide (hereinafter "Compound 1”) which has the structure below:
- Compound 1 and pharmaceutically acceptable compositions thereof are useful for treating or lessening the severity of CFTR mediated diseases such as, for example, cystic fibrosis.
- Compound 1 may exist in several different solid forms such as substantially crystalline forms or amorphous forms. DETAILED DESCRIPTION OF THE INVENTION
- CFTR cystic fibrosis transmembrane conductance regulator or a mutation thereof capable of regulator activity, including, but not limited to, AF508 CFTR and G551D CFTR (see, e.g., http://www.genet.sickkids.on.ca/cftr/, for CFTR mutations).
- modulating means increasing or decreasing, e.g.
- the term "chemically stable”, as used herein, means that the solid form of Compound 1 does not decompose into one or more different chemical compounds when subjected to specified conditions, e.g., 40 °C/75 % relative humidity, for a specific period of time. e.g. 1 day, 2 days, 3 days, 1 week, 2 weeks, or longer. In some embodiments, less than 25% of the solid form of Compound 1 decomposes, in some embodiments, less than about 20%>, less than about 15%, less than about 10%, less than about 5%, less than about 3%, less than about 1%), less than about 0.5% of the form of Compound 1 decomposes under the conditions specified. In some embodiments, no detectable amount of the solid form of Compound 1 decomposes.
- the term "physically stable”, as used herein, means that the solid form of Compound 1 does not change into one or more different physical forms of Compound 1 (e.g. different solid forms as measured by XRPD, DSC, etc.) when subjected to specific conditions, e.g., 40 °C/75 % relative humidity, for a specific period of time. e.g. 1 day, 2 days, 3 days, 1 week, 2 weeks, or longer. In some embodiments, less than 25% of the solid form of Compound 1 changes into one or more different physical forms when subjected to specified conditions.
- specific conditions e.g. 40 °C/75 % relative humidity
- less than about 20%, less than about 15%, less than about 10%, less than about 5%), less than about 3%, less than about 1%, less than about 0.5% of the solid form of Compound 1 changes into one or more different physical forms of Compound 1 when subjected to specified conditions. In some embodiments, no detectable amount of the solid form of Compound 1 changes into one or more physically different solid forms of Compound 1.
- the term “about” or “approximately” means an acceptable error for a particular value as determined by one of ordinary skill in the art, which depends in part on how the value is measured or determined.
- the term “about” or “approximately” means within 1, 2, 3, or 4 standard deviations.
- the term "about” or “approximately” means within 30%, 25%, 20%>, 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.5%, 0.1%, or 0.05% of a given value or range.
- structures depicted herein are also meant to include all isomeric (e.g., enantiomeric, diastereomeric, and geometric (or conformational)) forms of the structure; for example, the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers. Therefore, single isomeric (e.g., enantiomeric, diastereomeric, and geometric (or conformational)) forms of the structure; for example, the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers. Therefore, single isomeric (e.g., enantiomeric, diastereomeric, and geometric (or conformational)) forms of the structure; for example, the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers
- structures depicted herein are also meant to include compounds that differ only in the presence of one or more isotopically enriched atoms.
- Compound 1, wherein one or more hydrogen atoms are replaced deuterium or tritium, or one or more carbon atoms are replaced by a 13 C- or 14 C-enriched carbon are within the scope of this invention.
- Such compounds are useful, for example, as analytical tools, probes in biological assays, or compounds with improved therapeutic profile.
- protecting group abbreviated as P, as used herein refers to any chemical group introduced into a molecule by chemical modification of a functional group in order to obtain chemoselectivity in a subsequent chemical reaction.
- Non-limiting examples of alcohol protecting groups include acetyl (Ac), benzoyl (Bz), benzyl (Bn), ⁇ - methoxyethoxymethyl ether (MEM), dimethoxytrityl (DMT), methoxymethyl ether (MOM), methoxytrityl (MMT), /?-methoxybenzyl ether (PMB), pivaloyl (Piv), tetrahydropyranyl (THP), trityl (Tr), and trimethylsilyl (TMS).
- the protecting group is Bn which has the structure -CH 2 C 6 H 5 .
- the abbreviation "DCM” stands for dichloromethane.
- the abbreviation “IP A” stands for isopropyl alcohol.
- the abbreviation “DMSO” stands for dimethylsulfoxide.
- the abbreviation “MTBE” stands for methyl t-butyl ether.
- the abbreviation “THF” stands for tetrahydrofuran.
- the abbreviation “TEA” stands for triethylamine.
- the abbreviation “dba” as in Pd(dba) 2 stands for dibenzylideneacetone.
- the abbreviation “dpp ' as in Pd(dppf)Cl 2 stands for stands for l,l '-bis(diphenylphosphino) ferrocene.
- the invention features a method for preparing a compound of formula I:
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN, -Ci_ 4 haloalkyl,
- -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , - COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic
- X is CN or C0 2 R
- R is Ci_6 aliphatic or aryl; and m is an integer from 0 to 3 inclusive; comprising the steps of
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic; m is an integer from 0 to 3 inclusive; and
- Hal is a halide
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic
- X is CN or C0 2 R
- R is R is Ci_6 aliphatic or aryl; and m is an integer from 0 to 3 inclusive;
- the invention features the above method wherein ring A is a fused heterocycloalkyl or heteroaryl. In another embodiment, ring A is selected from
- the invention features the above method wherein X is CN. In another embodiment, X is C0 2 Et.
- the invention features the above method wherein m is 0.
- the invention features the above method wherein R J is a Ci_6 aliphatic. In another embodiment, R J is -CH 2 CH 3 .
- the invention features the above method wherein Hal is
- the invention features the above method wherein the first organic solvent is an aprotic solvent.
- the first organic solvent is selected from 1,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t- butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N-dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or
- the first organic solvent is selected from
- the first organic solvent is toluene.
- the invention features the above method wherein step a) is carried out in the presence of a transition metal catalyst. In another embodiment, step a) is carried out in the presence of a palladium catalyst. In another embodiment, step a) is carried out in the presence of a palladium catalyst selected from palladium(II)acetate, Pd(dppf)Cl 2 , Pd(dba) 2 , tetrakis(triphenylphosphine)palladium(0) or
- step a) is carried out in the presence of Pd(dba) 2 .
- the invention features the above method wherein step a) is carried out at about 50 °C to 90 °C. In another embodiment, step a) is carried out at about 60 °C to 80 °C. In another embodiment, step a) is carried out at about 70 °C.
- the invention features the above method wherein the second organic solvent is an aprotic solvent.
- the second organic solvent is selected from 1,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N- dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate,
- the second organic solvent is dimethylsulfoxide .
- the invention features the above method wherein step b) is carried out in the presence of an inorganic acid.
- step b) is carried out in the presence of an inorganic acid selected from hydrochloric, sulfuric, nitric, phosphoric, or boric acid.
- step b) is carried out in the presence of hydrochloric acid.
- the invention features the above method wherein step b) is carried out at about 55 °C to 95 °C.
- step b) is carried out at about 65 °C to 85 °C.
- step b) is carried out at about 75 °C.
- the invention features a method for preparing a compound of formula II:
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Hal is a halide;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic
- n is an integer from 0 to 3 inclusive
- n is an integer from 1 to 4 inclusive; comprising the steps of
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic; m is an integer from 0 to 3 inclusive; and
- Hal is a halide
- X is CN or C0 2 R
- R is Ci_6 aliphatic or aryl
- R J is hydrogen or Ci_ 6 aliphatic, to form a compound of formula IIC:
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic;
- X is CN or C0 2 R;
- R is Ci_6 aliphatic or aryl
- n is an integer from 0 to 3 inclusive; b) removing the -C0 2 R J group from compound IIC in a second organic solvent to form a compound of formula I:
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic
- X is CN or C0 2 R
- R is Ci_6 aliphatic or aryl; and m is an integer from 0 to 3 inclusive;
- Hal is a halide
- q is an integer from 0 to 3 inclusive;
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic
- n is an integer from 0 to 3 inclusive
- X is CN or C0 2 R
- R is Ci_6 aliphatic or aryl
- n is an integer from 1 to 4 inclusive;
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic
- n is an integer from 0 to 3 inclusive
- n is an integer from 1 to 4 inclusive;
- the invention features the above method wherein in step a), the first organic solvent is an aprotic solvent.
- the first organic solvent is selected from 1 ,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N- dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or dimethylsulfoxide.
- the first organic solvent is toluene.
- the invention features the above method wherein in step a), m is 0.
- the invention features the above method wherein in step a), Hal is Br.
- the invention features the above method wherein in step a), ring A is a fused heterocyclic or heteroaryl ring. In another embodiment, ring A is selected In another embodiment, ring
- the invention features the above method wherein in step a), X is CN. In another embodiment, X is C0 2 Et.
- the invention features the above method wherein in step a) R J is Et.
- the invention features the above method wherein in
- the invention features the above method wherein in step b), the second solvent is an aprotic solvent.
- the second solvent is selected from 1,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t- butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N-dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or
- the second solvent is dimethylsulfoxide.
- the second solvent is dimethylsulfoxide.
- the invention features the above method wherein in
- the invention features the above method wherein in step c), the base is an inorganic base.
- the base is a hydroxide.
- the base is NaOH.
- the invention features the above method wherein in formula IID, q is 1. [0049] In another embodiment, the invention features the above method wherein in formula IID, one Hal is CI and the other Hal is Br.
- the invention features the above method wherein in step d), the base is NaOH. In another embodiment, in step d), the acid is HC1.
- the invention features the above method wherein in step d), reaction with a hydroxide base takes place at about 60 °C to 100 °C.
- reaction with a hydroxide takes place at about 70 °C to 90 °C. In another embodiment, reaction with a hydroxide takes place at about 80 °C.
- the invention features the above method wherein in
- the invention features the above method wherein in step e), the third organic solvent is an aprotic solvent.
- the third organic solvent is selected from 1,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N- dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or dimethylsulfoxide.
- the third organic solvent is toluene.
- the invention features the above method wherein in step e), the halogenating agent is SOCl 2 .
- the invention features the above method wherein step e) takes place at about 40 °C to 80 °C. In another embodiment, step e) takes place at about 50 °C to 70 °C. In another embodiment, step e) takes place at about 60 °C.
- the invention features the above method wherein in
- the invention features a method of preparing a compound of formula III:
- R 2 is -R J , -OR J , -N(R J ) 2 , -NO 2 , halogen, -CN, -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- R3 is Ci_6 aliphatic optionally substituted with OH, OP, -0-Ci_ 6 aliphatic, aryl, heteroaryl, -O-aryl, or -O-heteroaryl;
- P is a protecting group; and o is an integer from 0 to 3;
- R 2 is -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN, -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- o is an integer from 0 to 3; with a halogenating reagent in a first organic solvent to form a compound of formula IIIB:
- R 2 is -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN, -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- o is an integer from 0 to 3;
- Hal is a halide
- R 2 is -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN, -C haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- o is an integer from 0 to 3;
- Hal is a halide
- P is a protecting group
- A is an anion; c) neutralizing a compound of formula HID in the presence of a base to form a compound of formula IIID-a:
- R 2 is -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN, -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- o is an integer from 0 to 3;
- Hal is a halide;
- P is a protecting group
- P 3 is a Ci_6 aliphatic optionally substituted with OH, OP, -0-Ci_ 6 aliphatic, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; in the presence of a catalyst to form a compound of formula III.
- the invention features the above method wherein in formula IIIA, o is 1. In another embodiment, o is 1 and R 2 is F.
- the invention features the above method wherein in step a), the halogenating reagent is N-bromosuccinimide.
- the invention features the above method wherein in step a), the first organic solvent is an aprotic solvent.
- the first organic solvent is selected from 1 ,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N- dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or dimethylsulfoxide.
- the first organic solvent is ethyl acetate.
- the invention features the above method wherein step a) takes place at about 2 °C to 42 °C. In another embodiment, step a) takes place at about 12 °C to 32 °C. In another embodiment, step a) takes place at about 22 °C. [0063] In another embodiment, the invention features the above method wherein in formula IIIB, o is 1, R 2 is F, and Hal is Br.
- the invention features the above method wherein in formula IIIC, P is benzyl.
- the invention features the above method wherein in step b), the second organic solvent is an aprotic solvent.
- the second organic solvent is selected from 1 ,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N-dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or dimethylsulfoxide.
- the second organic solvent is toluene.
- the invention features the above method wherein in step b), the reaction with a compound of formula IIIC takes place at about 60 °C to 100 °C. In another embodiment, in step b), the reaction with a compound of formula IIIC takes place at about 70 °C to 90 °C. In another embodiment, in step b), the reaction with a compound of formula IIIC takes place at about 80 °C.
- the invention features the above method wherein in step b), reduction is carried out with hydrogen.
- the invention features the above method wherein in step b) , the acid is p-toluenesulfonic acid.
- the invention features the above method wherein in formula HID, o is 1, R 2 is F, Hal is Br, A " is Tos " , and P is benzyl.
- the invention features the above method wherein in formula HIE, R 3 is C(CH 3 ) 2 CH 2 0(benzyl).
- the invention features the above method wherein in step c) , the base is an inorganic base.
- the invention features the above method wherein in step c), the base is NaHC0 3 .
- the invention features the above method wherein in step d), the third organic solvent is an aprotic solvent.
- the third organic solvent is selected from 1,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N- dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or dimethylsulfoxide.
- the third organic solvent is acetonitrile.
- step d) takes place at about 60 °C to 100 °C. In another embodiment, step d) takes place at about 70 °C to 90 °C. In another embodiment, step d) takes place at about 80 °C.
- the invention features the above method wherein in step d), the catalyst is a palladium catalyst.
- the catalyst in step d), is selected from palladium(II)acetate, Pd(dppf)Cl 2 , Pd(dba) 2 , (MeCN) 2 PdCl 2 ,
- the catalyst in step d), is palladium(II)acetate.
- the invention features a method of preparing a compound of formula IV:
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Ri and R 2 is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN, -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic
- P 3 is a Ci_6 aliphatic optionally substituted with OH, OP, -0-Ci_ 6 aliphatic, aryl, heteroaryl, -O-aryl, or -O-heteroaryl;
- P is a protecting group
- n is an integer from 0 to 3 inclusive
- n is an integer from 1 to 4 inclusive;
- o is an integer from 1 to 3 inclusive
- Pv 2 is -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN, -C haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- o is an integer from 0 to 3;
- R 2 is -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN, -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- o is an integer from 0 to 3; and Hal is a halide;
- R 2 is -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN, -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- o is an integer from 0 to 3;
- Hal is a halide
- P is a protecting group
- A is an anion; c) neutralizing a compound of formula HID in the presence of a base to form a compound of formula IIID-a:
- R 2 is -R J , -OR J , -N(R J ) 2 , -NO 2 , halogen, -CN, -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- o is an integer from 0 to 3;
- Hal is a halide
- R3 is a Ci_6 aliphatic optionally substituted with OH, OP, -0-Ci_ 6 aliphatic, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; in the presence of a catalyst to form a compound of formula III:
- R 2 is -R J , -OR J , -N(R J ) 2 , -NO 2 , halogen, -CN, -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy,
- R J is hydrogen or Ci_ 6 aliphatic
- R3 is Ci_6 aliphatic optionally substituted with OH, OP, -0-Ci_ 6 aliphatic, aryl, heteroaryl, -O-aryl, or -O-heteroaryl;
- P is a protecting group
- o is an integer from 0 to 3; e) reacting the compound of formula III in a fourth organic solvent with a compound of formula II:
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Hal is a halide;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic;
- m is an integer from 0 to 3 inclusive; and
- n is an integer from 1 to 4 inclusive; to form the compound of formula IV.
- the invention features the above method wherein in
- the invention features the above method wherein in formula IV, m is 0. In another embodiment, in formula IV, n is 1. In another embodiment, in formula IV, o is 1 and R 2 is F.
- the invention features the above method wherein in formula IV, P is benzyl.
- the invention features the above method wherein in formula IV, R 3 is a C 4 aliphatic optionally substituted with OP. In another embodiment, in In another embodiment, in formula IV, R 3 is [0081] In another embodiment, the invention features the above method wherein in
- the invention features the above method wherein in step a), the halogenating reagent is N-bromosuccinimide.
- the invention features the above method wherein in step a) , the first organic solvent is an aprotic solvent.
- the first organic solvent is selected from 1,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N- dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or dimethylsulfoxide.
- the first organic solvent is ethyl acetate.
- the invention features the above method wherein step a) takes place at about 2 °C to 42 °C. In another embodiment, step a) takes place at about 12 °C to 32 °C. In another embodiment, step a) takes place at about 22 °C.
- the invention features the above method wherein in formula IIIB, o is 1, R 2 is F, and Hal is Br.
- the invention features the above method wherein in formula IIIC, P is benzyl.
- the invention features the above method wherein in step b) , the second organic solvent is an aprotic solvent.
- the second organic solvent is selected from 1 ,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N-dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or dimethylsulfoxide.
- the second organic solvent is toluene.
- the invention features the above method wherein in step b), the reaction with a compound of formula IIIC takes place at about 60 °C to 100 °C. In another embodiment, in step b), the reaction with a compound of formula IIIC takes place at about 70 °C to 90 °C. In another embodiment, in step b), the reaction with a compound of formula IIIC takes place at about 80 °C.
- the invention features the above method wherein in step b), reduction is carried out with hydrogen.
- the invention features the above method wherein in step b) , the acid is p-toluenesulfonic acid.
- the invention features the above method wherein in formula HID, o is 1, R 2 is F, Hal is Br, A " is Tos " , and P is benzyl.
- the invention features the above method wherein in formula HIE, R 3 is C(CH 3 ) 2 CH 2 0(benzyl).
- the invention features the above method wherein in step c) , the base is an inorganic base.
- the invention features the above method wherein in step c) , the base is NaHC0 3 .
- the invention features the above method wherein in step d) , the third organic solvent is an aprotic solvent.
- the third organic solvent is selected from 1,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N- dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or dimethylsulfoxide.
- step d) in step d), the third organic solvent is acetonitrile.
- the invention features the above method wherein step d) takes place at about 60 °C to 100 °C. In another embodiment, step d) takes place at about 70 °C to 90 °C. In another embodiment, step d) takes place at about 80 °C.
- the invention features the above method wherein in step d), the catalyst is a palladium catalyst.
- the catalyst in step d), is selected from palladium(II)acetate, Pd(dppf)Cl2, Pd(dba) 2 ,
- the catalyst in step d), is palladium(II)acetate.
- the invention features the above method wherein in step
- the invention features the above method wherein in step e), the fourth organic solvent is an aprotic solvent.
- the fourth organic solvent is selected from 1,2-dimethoxyethane, dioxane, acetonitrile, toluene, benzene, xylenes, methyl t-butyl ether, methyl ethyl ketone, methyl isobutyl ketone, acetone, N,N-dimethylformamide, N,N-dimethylacetamide, N-methylpyrrolidinone, ethyl acetate, dichloromethane, or dimethylsulfoxide.
- the fourth organic solvent is dichloromethane.
- the invention features the above method wherein step e) takes place at about -20 °C to 20 °C. In another embodiment, step e) takes place at about -10 °C to 10 °C. In another embodiment, step e) takes place at about 0 °C.
- the invention features the above method wherein in step e), the compound of formula II is prepared in situ by halogenating the acid precursor and reacted with the compound of formula III without isolation.
- the invention features the above method further comprising removing the two protecting groups from the compound of formula IV to form a compound of formula IVA:
- the protecting groups are removed by hydrogenation.
- the invention features a method of preparing Compound 1 :
- the invention features the above method wherein in step a), the brominating agent is N-bromosuccinimide.
- the invention features the above method wherein in setp b), the reduction is carried out with hydrogen.
- the invention features the above method wherein in setp b), the base is an inorganic base.
- the invention features the above method wherein in setp b), the base is NaHC0 3 .
- the invention features the above method wherein in step c), the catalyst is a palladium catalyst.
- the catalyst in step c), is selected from palladium(II)acetate, Pd(dppf)Cl2, Pd(dba) 2 ,
- the catalyst is palladium(II)acetate.
- the invention features the above method wherein in step d), compound 8 is made in situ by halogenating the acid precursor without isolation.
- the invention features the above method wherein in step e), the Bn protecting groups are removed by hydrogenation.
- the invention features a compound of formula 23:
- ring A is a fused cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Ri is independently selected from -R J , -OR J , -N(R J ) 2 , -N0 2 , halogen, -CN,
- -Ci_ 4 haloalkyl, -Ci_ 4 haloalkoxy, -C(0)N(R J ) 2 , -NR J C(0)R J , -SOR J , -S0 2 R J , -S0 2 N(R J ) 2 , -NR J S0 2 R J , -COR J , -C0 2 R J , -NR J S0 2 N(R J ) 2 , -COCOR J ;
- R J is hydrogen or Ci_ 6 aliphatic
- X is CN or C0 2 R
- R is Ci_6 aliphatic or aryl; and m is an integer from 0 to 3 inclusive.
- the invention features a compound of formula 23 and the attendant definitions, wherein ring A is a fused heterocycloalkyl or heteroaryl.
- ring A is .
- the invention features a compound of formula 23 and the attendant definitions, wherein X is CN. In another embodiment, X is C0 2 Et. [00115] In another embodiment, the invention features a compound of formula 23 and the attendant definitions, wherein m is 0.
- the invention features a compound of formula 23 and the attendant definitions, wherein R J is Ci_ 6 aliphatic. In another embodiment, R J is -CH 2 CH 3 .
- the invention features the compound
- the invention features the compound
- aryl halide IA is reacted with ester IB in the presence of a transition metal catalyst in a suitable solvent (e.g. toluene) to produce ester IC.
- a suitable solvent e.g. toluene
- X can either be CN or C0 2 R.
- Treatment of IC with an acid in a suitable solvent e.g.
- IA is commercially available.
- ring A is a 5 membered dioxyl ring.
- Hal in IA is Br.
- the reaction of IA and IIB takes place in toluene in the presence of a Pd(0) caystalyst, e.g. Pd(dba) 2 .
- the reaction takes place in the presence of an alkyl phosphine, e.g. t- Bu 3 P and phosphate salt, e.g. Na 3 P0 4 .
- the reaction of IA and IIB takes place at about 70 °C.
- R J is Et.
- the de-esterification of IC to I is done with an inorganic acid.
- the inorganic acid is HC1.
- the conversion takes place in an appropriate aprotic solvent (e.g. DMSO) at about 75 °C.
- I is reacted with NaOH and an alkyl dihalide to yield the cycloalkylidene in a suitable solvent (e.g. MTBE).
- a suitable solvent e.g. MTBE
- the process is adaptable to several spirocyclic rings by choosing the appropriate alkyl dihalide.
- a spirocylic butane ring can be produced by reacting I with, for example, l-bromo-3-chloropropane. It has been found that a mixed bromo and chloro dihalide works best on an economic scale as it is believed that the thermodynamics of the reaction are more favorable.
- HE is hydrolized to the carboxylic acid IIF in the presence of water and a base (e.g. NaOH) in a suitable solvent (e.g. ethanol). Subseqent treatment with an acid such as HC1 yields IIF. In another embodiment, IIF is worked up by recrystallizing it from toluene.
- a base e.g. NaOH
- a suitable solvent e.g. ethanol
- the halogenating agent that converts IIF to II is thionyl chloride.
- the thionyl chloride is added to IIF in toluene at about 60 °C. In one embodiment, this step directly proceeds the coupling between II and amine III (see below) and is carried out in the same reaction vessel.
- a halogenating agent
- b Zn(II) catalyst
- c H 2 , Pt
- d acid
- e base
- f Pd(II) catalyst
- R 2 , o, Hal, A , and P are defined as above.
- R 2 is F and is meta to the amine group.
- IIIA is brominated with N-bromosuccinimide in a suitable solvent (e.g.
- IIIB is reacted with epoxide IIIC effecting a ring opening reaction with the amine group of IIIB to form HID.
- the protecting group, P, in IIIC is benzyl (Bn).
- epoxide IIIC is chiral.
- IIIC is (R) IIIC.
- IIIC is (S) IIIC.
- the ring opening reaction is carried out in a suitable solvent (e.g. toluene) at about 80 °C.
- the ring opening reaction takes place in the presence of a Zn(II) catalyst (e.g.
- the conversion from IIIB to HID comprises the ring opening reaction with epoxide IIIC, followed by hydrogenation, and then treatment with an acid to form HID.
- hydrogenation is carried out with H2/Pt(S)/C.
- the acid is toluene sulfonic acid, such that A is a tosylate anion.
- alkyne HIE is coupled with HID in a suitable solvent (e.g. acetonitrile) at about 80 °C.
- the coupling reaction takes place in the presence of a Pd(II) catalyst, such as Pd(OAc) 2 .
- the initial reaction does not result in ring closure, only replacement of the halide on HID.
- Ring closure is accomplished through reaction with another Pd(II) catalyst, such as (MeCN) 2 PdCl 2 in a suitable solvent (e.g. acetonitrile).
- ring closure takes place at about 80 °C.
- R 3 in alkyne HIE is -C(CH 3 ) 2 CH 2 OBn.
- the product from the coupling reaction is not isolated but taken up in acetonitrile and reacted with (MeCN) 2 PdCl 2 .
- a halogenating agent
- b aprotic solvent
- dichloromethane (DCM)) yields the protected analog of Compound 1.
- the acid halide II is prepared from IIF as depicted in Scheme 1 in the same reaction vessel and is not isolated.
- the acid-based reaction is carried out in the presence of a base such as triethylamine (TEA).
- TEA triethylamine
- the amount of TEA is 2 equivalents relative to II.
- water is added to the mixture and stirred for an additional 30 minutes.
- the organic phase is separated and IV is isolated by distilling off the reaction solvent.
- IV is collected by silica pad filtration.
- compounds of formula IV may be deprotected to form compounds of formula IVa according to Scheme 4.
- hydrogen pressurization is 3 Bars. In another embodiment, hydrogen pressurization is 3 Bars. In another
- the hydrogenation agitation rate is increased to 800 rpm.
- the hydrogenation vessel is heated to about 50 °C for 2 days.
- more catalyst is added and hydrogenation continues for another 4 days.
- IV is dissolved in a suitable solvent (e.g. THF).
- Compound 1 may be prepared by coupling the acid halide moiety 7 with the amine moiety 8 to form compound 9 followed by deprotection according to Scheme 5.
- compositions comprising Compound 1 Form A or amorphous Compound 1 as described herein, and optionally comprise a pharmaceutically acceptable carrier, adjuvant or vehicle. In certain embodiments, these compositions optionally further comprise one or more additional therapeutic agents.
- compositions of the present invention additionally comprise a pharmaceutically acceptable carrier, adjuvant, or vehicle, which, as used herein, includes any and all solvents, diluents, or other liquid vehicle, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired.
- a pharmaceutically acceptable carrier, adjuvant, or vehicle which, as used herein, includes any and all solvents, diluents, or other liquid vehicle, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired.
- materials which can serve as pharmaceutically acceptable carriers include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, or potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, polyacrylates, waxes, polyethylene -polyoxypropylene -block polymers, wool fat, sugars such as lactose, glucose and sucrose; starches such as corn starch and potato starch; cellulose and its derivatives such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; powdered tragacanth; malt; gelatin;
- glycols such a propylene glycol or polyethylene glycol
- esters such as ethyl oleate and ethyl laurate
- agar buffering agents such as magnesium hydroxide and aluminum hydroxide; alginic acid; pyrogen-free water; isotonic saline; Ringer's solution; ethyl alcohol, and phosphate buffer solutions, as well as other non-toxic compatible lubricants such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, releasing agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants can also be present in the composition, according to the judgment of the formulator.
- the present invention provides a method of treating a condition, disease, or disorder implicated by CFTR.
- the present invention provides a method of treating a condition, disease, or disorder implicated by a deficiency of CFTR activity, the method comprising administering a composition comprising a Compound 1 described herein to a subject, preferably a mammal, in need thereof.
- a "CFTR-mediated disease” as used herein is a disease selected from cystic fibrosis, asthma, smoke induced COPD, chronic bronchitis, rhinosinusitis, constipation, pancreatitis, pancreatic insufficiency, male infertility caused by congenital bilateral absence of the vas deferens (CBAVD), mild pulmonary disease, idiopathic pancreatitis, allergic bronchopulmonary aspergillosis (ABPA), liver disease, hereditary emphysema, hereditary hemochromatosis, coagulation-fibrinolysis deficiencies, such as protein C deficiency, Type 1 hereditary angioedema, lipid processing deficiencies, such as familial hypercholesterolemia, Type 1 chylomicronemia, abetalipoproteinemia, lysosomal storage diseases, such as I-cell disease/pseudo-Hurler, mucopolysaccharidoses, Sandhof
- the present invention provides a method of treating a CFTR-mediated disease in a human comprising the step of administering to said human an effective amount of a composition comprising Compound 1 described herein.
- an "effective amount" of Compound 1 Form A or amorphous Compound 1 or a pharmaceutically acceptable composition thereof is that amount effective for treating or lessening the severity of any of the diseases recited above.
- Compound 1 or a pharmaceutically acceptable composition thereof may be administered using any amount and any route of administration effective for treating or lessening the severity of one or more of the diseases recited above.
- compositions thereof are useful for treating or lessening the severity of cystic fibrosis in patients who exhibit residual CFTR activity in the apical membrane of respiratory and non-respiratory epithelia.
- the presence of residual CFTR activity at the epithelial surface can be readily detected using methods known in the art, e.g., standard electrophysiological, biochemical, or histochemical techniques. Such methods identify CFTR activity using in vivo or ex vivo electrophysiological techniques, measurement of sweat or salivary CI " concentrations, or ex vivo biochemical or histochemical techniques to monitor cell surface density. Using such methods, residual CFTR activity can be readily detected in patients heterozygous or homozygous for a variety of different mutations, including patients homozygous or heterozygous for the most common mutation, AF508.
- Compound 1 described herein or a pharmaceutically acceptable composition thereof is useful for treating or lessening the severity of cystic fibrosis in patients within certain genotypes exhibiting residual CFTR activity, e.g., class III mutations (impaired regulation or gating), class IV mutations (altered conductance), or class V mutations (reduced synthesis) (Lee R. Choo-Kang, Pamela L., Zeitlin, Type I, II, III, IV, and V cystic fibrosis Tansmembrane Conductance Regulator Defects and Opportunities of Therapy; Current Opinion in Pulmonary Medicine 6:521 - 529, 2000).
- Other patient genotypes that exhibit residual CFTR activity include patients homozygous for one of these classes or heterozygous with any other class of mutations, including class I mutations, class II mutations, or a mutation that lacks classification.
- Compound 1 described herein or a pharmaceutically acceptable composition thereof is useful for treating or lessening the severity of cystic fibrosis in patients within certain clinical phenotypes, e.g., a moderate to mild clinical phenotype that typically correlates with the amount of residual CFTR activity in the apical membrane of epithelia.
- phenotypes include patients exhibiting pancreatic insufficiency or patients diagnosed with idiopathic pancreatitis and congenital bilateral absence of the vas deferens, or mild lung disease.
- the exact amount required will vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the infection, the particular agent, its mode of administration, and the like.
- the compounds of the invention are preferably formulated in dosage unit form for ease of administration and uniformity of dosage.
- dosage unit form refers to a physically discrete unit of agent appropriate for the patient to be treated. It will be understood, however, that the total daily usage of the compounds and compositions of the present invention will be decided by the attending physician within the scope of sound medical judgment.
- the specific effective dose level for any particular patient or organism will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed, and like factors well known in the medical arts.
- patient or "subject”, as used herein, means an animal, preferably a mammal, and most preferably a human.
- compositions of this invention can be administered to humans and other animals orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by powders, ointments, or drops), bucally, as an oral or nasal spray, or the like, depending on the severity of the infection being treated.
- the compounds of the invention may be administered orally or parenterally at dosage levels of about 0.01 mg/kg to about 50 mg/kg and preferably from about 1 mg/kg to about 25 mg/kg, of subject body weight per day, one or more times a day, to obtain the desired therapeutic effect.
- the dosage amount of Compound 1 in the dosage unit form is from 100 mg to 1,000 mg. In another embodiment, the dosage amount of
- Compound 1 is from 200 mg to 900 mg. In another embodiment, the dosage amount of Compound 1 is from 300 mg to 800 mg. In another embodiment, the dosage amount of Compound 1 is from 400 mg to 700 mg. In another embodiment, the dosage amount of Compound 1 is from 500 mg to 600 mg.
- Injectable preparations for example, sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution, suspension or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- the acceptable vehicles and solvents that may be employed are water, Ringer's solution, U.S. P. and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil can be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid are used in the preparation of injectables.
- the injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
- compositions for rectal or vaginal administration are preferably
- suppositories which can be prepared by mixing the compounds of this invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules.
- the active compound is mixed with at least one inert, pharmaceutically acceptable excipient or carrier such as sodium citrate or dicalcium phosphate and/or a) fillers or extenders such as starches, lactose, sucrose, glucose, mannitol, and silicic acid, b) binders such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidinone, sucrose, and acacia, c) humectants such as glycerol, d) disintegrating agents such as agar—agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate, e) solution retarding agents such as paraffin, f) absorption accelerators such as quaternary ammonium compounds, g) wetting agents such as, for example, cetyl alcohol
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings and other coatings well known in the pharmaceutical formulating art. They may optionally contain opacifying agents and can also be of a composition that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner. Examples of embedding compositions that can be used include polymeric substances and waxes. Solid compositions of a similar type may also be employed as fillers in soft and hard- filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polethylene glycols and the like.
- the active compounds can also be in microencapsulated form with one or more excipients as noted above.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings, release controlling coatings and other coatings well known in the pharmaceutical formulating art.
- the active compound may be admixed with at least one inert diluent such as sucrose, lactose or starch.
- Such dosage forms may also comprise, as is normal practice, additional substances other than inert diluents, e.g., tableting lubricants and other tableting aids such a magnesium stearate and microcrystalline cellulose.
- the dosage forms may also comprise buffering agents. They may optionally contain opacifying agents and can also be of a composition that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- buffering agents include polymeric substances and waxes.
- Compound 1 described herein or a pharmaceutically acceptable composition thereof can be employed in combination therapies, that is, Compound 1 can be administered concurrently with, prior to, or subsequent to, one or more other desired therapeutics or medical procedures.
- the particular combination of therapies (therapeutics or procedures) to employ in a combination regimen will take into account compatibility of the desired therapeutics and/or procedures and the desired therapeutic effect to be achieved.
- the therapies employed may achieve a desired effect for the same disorder (for example, an inventive compound may be administered concurrently with another agent used to treat the same disorder), or they may achieve different effects (e.g., control of any adverse effects).
- additional therapeutic agents that are normally administered to treat or prevent a particular disease, or condition are known as "appropriate for the disease, or condition, being treated”.
- the additional agent is selected from a mucolytic agent, bronchodialator, an anti-biotic, an anti-infective agent, an anti-inflammatory agent, a CFTR modulator other than a compound of the present invention, or a nutritional agent.
- the additional therapeutic agent is an antibiotic.
- antibiotics useful herein include tobramycin, including tobramycin inhaled powder (TIP), azithromycin, aztreonam, including the aerosolized form of aztreonam, amikacin, including liposomal formulations thereof, ciprofloxacin, including formulations thereof suitable for administration by inhalation, levoflaxacin, including aerosolized formulations thereof, and combinations of two antibiotics, e.g., fosfomycin and tobramycin.
- TIP tobramycin inhaled powder
- azithromycin aztreonam
- aztreonam including the aerosolized form of aztreonam
- amikacin including liposomal formulations thereof
- ciprofloxacin including formulations thereof suitable for administration by inhalation
- levoflaxacin including aerosolized formulations thereof
- combinations of two antibiotics e.g., fosfomycin and tobramycin.
- the additional agent is a mucolyte.
- exemplary mucolytes useful herein includes Pulmozyme®.
- the additional agent is a bronchodialator.
- bronchodialtors include albuterol, metaprotenerol sulfate, pirbuterol acetate, salmeterol, or tetrabuline sulfate.
- the additional agent is effective in restoring lung airway surface liquid. Such agents improve the movement of salt in and out of cells, allowing mucus in the lung airway to be more hydrated and, therefore, cleared more easily.
- Exemplary such agents include hypertonic saline, denufosol tetrasodium ([[(3S, 5R)-5-(4-amino-2- oxopyrimidin-l-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4R,5R)-5- (2,4-dioxopyrimidin- 1 -yl)-3 , 4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy- hydroxyphosphoryl] hydrogen phosphate), or bronchitol (inhaled formulation of mannitol).
- the additional agent is an anti-inflammatory agent, i.e., an agent that can reduce the inflammation in the lungs.
- agents useful herein include ibuprofen, docosahexanoic acid (DHA), sildenafil, inhaled glutathione, pioglitazone, hydroxychloroquine, or simavastatin.
- the additional agent is a CFTR modulator other than Compound 1, i.e., an agent that has the effect of modulating CFTR activity.
- CFTR modulator other than Compound 1, i.e., an agent that has the effect of modulating CFTR activity.
- agents include ataluren ("PTC 124®"; 3-[5-(2-fluorophenyl)-l,2,4-oxadiazol-3-yl]benzoic acid), sinapultide, lancovutide, depelestat (a human recombinant neutrophil elastase inhibitor), cobiprostone (7- ⁇ (2R, 4aR, 5R, 7aR)-2-[(3S)-l,l-difluoro-3-methylpentyl]-2-hydroxy-6- oxooctahydrocyclopenta[b]pyran-5-yl ⁇ heptanoic acid), and N-(5-hydroxy-2,4-ditert-butyl
- the additional agent is a nutritional agent.
- Exemplary nutritional agents include pancrelipase (pancreating enzyme replacement), including Pancrease®, Pancreacarb®, Ultrase®, or Creon®, Liprotomase® (formerly Trizytek®), Aquadeks®, or glutathione inhalation.
- the additional nutritional agent is pancrelipase.
- the additional agent is a compound selected from gentamicin, curcumin, cyclophosphamide, 4-phenylbutyrate, miglustat, felodipine, nimodipine, Philoxin B, geniestein, Apigenin, cAMP/cGMP modulators such as rolipram, sildenafil, milrinone, tadalafil, amrinone, isoproterenol, albuterol, and almeterol, deoxyspergualin, HSP 90 inhibitors, HSP 70 inhibitors, proteosome inhibitors such as epoxomicin, lactacystin, etc.
- the additional agent is a compound disclosed in WO 2004028480, WO 2004110352, WO 2005094374, WO 2005120497, or WO 2006101740.
- the additiona agent is a benzo(c)quinolizinium derivative that exhibits CFTR modulation activity or a benzopyran derivative that exhibits CFTR modulation activity.
- the addditional agent is a compound disclosed in US7202262, US6992096, US20060148864, US20060148863, US20060035943,
- the additional agent is a compound disclosed in WO2004080972, WO2004111014, WO2005035514, WO2005049018, WO2006099256, WO2006127588, or WO2007044560.
- the amount of additional therapeutic agent present in the compositions of this invention will be no more than the amount that would normally be administered in a composition comprising that therapeutic agent as the only active agent.
- the amount of additional therapeutic agent in the presently disclosed compositions will range from about 50% to 100% of the amount normally present in a composition comprising that agent as the only therapeutically active agent.
- Vitride® sodium bis(2-methoxyethoxy)aluminum hydride
- NaAlH 2 (OCH 2 CH 2 OCFl 3 ) 2 ], 65 wgt% solution in toluene) was purchased from Aldrich Chemicals.
- 3-Fluoro-4-nitroaniline was purchased from Capot Chemicals.
- 5-Bromo-2,2- difluoro- 1 ,3-benzodioxole was purchased from Alfa Aesar.
- 2,2-Difluoro- 1 ,3-benzodioxole-5- carboxylic acid was purchased from Saltigo (an affiliate of the Lanxess Corporation).
- a reactor was purged with nitrogen and charged with 900 mL of toluene. The solvent was degassed via nitrogen sparge for no less than 16 h. To the reactor was then charged Na 3 P0 4 (155.7 g, 949.5 mmol), followed by bis(dibenzylideneacetone) palladium (0) (7.28 g, 12.66 mmol). A 10% w/w solution of tert-butylphosphine in hexanes (51.23 g, 25.32 mmol) was charged over 10 min at 23 °C from a nitrogen purged addition funnel.
- a stock solution of 50% w/w NaOH was degassed via nitrogen sparge for no less than 16 h.
- An appropriate amount of MTBE was similarly degassed for several hours.
- To a reactor purged with nitrogen was charged degassed MTBE (143 mL) followed by (2,2- difluoro- l,3-benzodioxol-5-yl)-acetonitrile (40.95 g, 207.7 mmol) and tetrabutylammonium bromide (2.25 g, 10.38 mmol).
- the volume of the mixture was noted and the mixture was degassed via nitrogen sparge for 30 min. Enough degassed MTBE is charged to return the mixture to the original volume prior to degassing.
- the aqueous phase was extracted with MTBE (123 mL), and the combined organic phase was washed with 1 N HC1 (163mL) and 5% NaCl (163 mL).
- the solution of (2,2- difluoro-l,3-benzodioxol-5-yl)-cyclopropanecarbonitrile in MTBE was concentrated to 164 mL under vacuum at 40 - 50 °C.
- the solution was charged with ethanol (256 mL) and again concentrated to 164 mL under vacuum at 50 - 60 °C.
- Ethanol (256 mL) was charged and the mixture concentrated to 164 mL under vacuum at 50 - 60 °C.
- Toluene (328 mL) was charged and the mixture condensed to 164 mL at 70 - 75 °C.
- the mixture was cooled to 45 °C, charged with MTBE (364 mL) and stirred at 60 °C for 20 min.
- the solution was cooled to 25 °C and polish filtered to remove residual inorganic salts.
- MTBE (123 mL) was used to rinse the reactor and the collected solids. The combined organics were transferred to a clean reactor in preparation for the next step.
- the hydrogenator was charged with 5wt% Pt(S)/C (1.5 mol%>) and the mixture was stirred under N 2 at 30 °C (internal temperature). The reaction was flushed with N 2 followed by hydrogen. The hydrogenator pressure was adjusted to 1 Bar of hydrogen and the mixture was stirred rapidly (>1200 rpm). At the end of the reaction, the catalyst was filtered through a pad of Celite and washed with dichloromethane (10 vol). The filtrate was concentrated in vacuo. Any remaining isopropyl acetate was chased with dichloromethane (2 vol) and concentrated on a rotavap to dryness.
- the aqueous phase (pH 9) was drained off and discarded. The remaining organic phase was washed with water (2 L, 2 vol). The organic phase was concentrated in vacuo using a 22 L rotary evaporator, providing the crude product as an orange oil.
- Method B Magnesium turnings (106 g, 4.35 mol, 1.0 eq) were charged to a 22 L reactor and then suspended in THF (760 mL, 1 vol). The vessel was cooled in an ice-water bath such that the batch temperature reached 2 °C. A solution of the propargyl chloride (760 g, 4.35 mol, 1.0 equiv) in THF (4.5 L, 6 vol) was added slowly to the reactor. After 100 mL was added, the addition was stopped and the mixture stirred until a 13 °C exotherm was observed, indicating the Grignard reagent initiation.
- the mixture is filtered through Celite and the cake is washed with acetonitrile.
- a solvent swap into ethyl acetate (7.5 vol) is performed.
- the ethyl acetate solution is washed with aqueous NH 3 -NH 4 CI solution (2 x 2.5 vol) followed by 10% brine (2.5 vol).
- the ethyl acetate solution is then stirred with silica gel (1.8 wt eq) and Si-TMT (0.1 wt eq) for 6 h. After filtration, the resulting solution is concentrated down.
- the residual oil is dissolved in DCM / heptane (4 vol) and then purified by column chromatography.
- the acid chloride solution in toluene (1 vol) is added while maintaining the batch temperature below 10 °C.
- the reaction progress is monitored by HPLC, and the reaction is usually complete within minutes. After warming to 25 °C, the reaction mixture is washed with 5% NaHC0 3 (3.5 vol), 1 M NaOH (3.5 vol) and 1 M HCl (5 vol).
- Compound 1 may also be prepared by one of several synthetic routes disclosed in US published patent application US20090131492, incorporated herein by reference.
- the optical membrane potential assay utilized voltage-sensitive FRET sensors described by Gonzalez and Tsien (See, Gonzalez, J. E. and R. Y. Tsien (1995) "Voltage sensing by fluorescence resonance energy transfer in single cells” Biophys J 69(4): 1272-80, and Gonzalez, J. E. and R. Y. Tsien (1997) “Improved indicators of cell membrane potential that use fluorescence resonance energy transfer” Chem Biol 4(4): 269-77) in combination with instrumentation for measuring fluorescence changes such as the Voltage/Ion Probe Reader (VIPR) (See, Gonzalez, J. E., K. Oades, et al. (1999) "Cell-based assays and instrumentation for screening ion-channel targets” Drug Discov Today 4(9): 431-439).
- VIPR Voltage/Ion Probe Reader
- Bath Solution #1 (in mM) NaCl 160, KC1 4.5, CaCl 2 2, MgCl 2 1,
- Chloride-free bath solution Chloride salts in Bath Solution #1 are substituted with gluconate salts.
- CC2-DMPE Prepared as a 10 mM stock solution in
- DiSBAC 2 (3) Prepared as a 10 mM stock in DMSO and stored at -20°C.
- NIH3T3 mouse fibroblasts stably expressing AF508-CFTR are used for optical measurements of membrane potential.
- the cells are maintained at 37 °C in 5% C0 2 and 90 % humidity in Dulbecco's modified Eagle's medium supplemented with 2 mM glutamine, 10 % fetal bovine serum, 1 X NEAA, ⁇ - ⁇ , 1 X pen/strep, and 25 mM HEPES in 175 cm 2 culture flasks.
- the cells were seeded at 30,000/well in 384-well matrigel-coated plates and cultured for 2 hrs at 37 °C before culturing at 27 °C for 24 hrs for the potentiator assay.
- the cells are cultured at 27 °C or 37 °C with and without compounds for 16 - 24 hours.
- the FRT epithelia demonstrated resistances of 4 ⁇ / cm 2 or more.
- the solutions were maintained at 27 °C and bubbled with air.
- the electrode offset potential and fluid resistance were corrected using a cell-free insert.
- the current reflects the flow of CI " through AF508-CFTR expressed in the apical membrane.
- the Isc was digitally acquired using an MP100A-CE interface and AcqKnowledge software (v3.2.6; BIOPAC Systems, Santa Barbara, CA).
- Typical protocol utilized a basolateral to apical membrane CI " concentration gradient.
- normal ringers was used on the basolateral membrane and was permeabilized with nystatin (360 ⁇ g/ml), whereas apical NaCl was replaced by equimolar sodium gluconate (titrated to pH 7.4 with NaOH) to give a large CI " concentration gradient across the epithelium.
- All experiments were performed 30 min after nystatin permeabilization. Forskolin (10 ⁇ ) and all test compounds were added to both sides of the cell culture inserts. The efficacy of the putative AF508-CFTR potentiators was compared to that of the known potentiator, genistein.
- Basolateral solution in mM: NaCl (135), CaCl 2 (1.2), MgCl 2 (1.2), K 2 HP0 4
- Fisher rat epithelial (FRT) cells expressing AF508-CFTR wereg used for Ussing chamber experiments for the putative AF508-CFTR modulators identified from our optical assays.
- the cells were cultured on Costar Snapwell cell culture inserts and cultured for five days at 37 °C and 5% C0 2 in Coon's modified Ham's F-12 medium supplemented with 5% fetal calf serum, 100 U/ml penicillin, and 100 ⁇ g/ml streptomycin. Prior to use for characterizing the potentiator activity of compounds, the cells were incubated at 27 °C for 16 - 48 hrs to correct for the AF508-CFTR. To determine the activity of corrections compounds, the cells were incubated at 27 °C or 37 °C with and without the compounds for 24 hours.
- I AFSOS The macroscopic AF508-CFTR current (I AFSOS ) in temperature- and test compound- corrected NIH3T3 cells stably expressing AF508-CFTR were monitored using the perforated- patch, whole-cell recording.
- voltage-clamp recordings of I AFSOS were performed at room temperature using an Axopatch 200B patch-clamp amplifier (Axon Instruments Inc., Foster City, CA). All recordings were acquired at a sampling frequency of 10 kHz and low- pass filtered at 1 kHz. Pipettes had a resistance of 5 - 6 ⁇ when filled with the intracellular solution. Under these recording conditions, the calculated reversal potential for CI " (Eci) at room temperature was -28 mV.
- the cells were incubated with 10 ⁇ of the test compound for 24 hours at 37°C and the current density was compared to the 27°C and 37°C controls (% activity). Prior to recording, the cells were washed 3X with extracellular recording medium to remove any remaining test compound. Preincubation with 10 ⁇ of correction compounds significantly increased the cAMP- and genistein-dependent current compared to the 37°C controls.
- AF508-CFTR potentiators to increase the macroscopic AF508- CFTR CI " current (I AFSOS ) in NIH3T3 cells stably expressing AF508-CFTR was also investigated using perforated-patch-recording techniques.
- the potentiators identified from the optical assays evoked a dose-dependent increase in I AFSOS with similar potency and efficacy observed in the optical assays.
- the reversal potential before and during potentiator application was around -30 mV, which is the calculated EQ (-28 mV).
- Intracellular solution in mM: Cs-aspartate (90), CsCl (50), MgCl 2 (1), HEPES
- NIH3T3 mouse fibroblasts stably expressing AF508-CFTR are used for whole- cell recordings.
- the cells are maintained at 37 °C in 5% C0 2 and 90 % humidity in Dulbecco's modified Eagle's medium supplemented with 2 mM glutamine, 10 % fetal bovine serum, 1 X NEAA, ⁇ - ⁇ , 1 X pen/strep, and 25 mM HEPES in 175 cm 2 culture flasks.
- 2,500 - 5,000 cells were seeded on poly-L-lysine-coated glass coverslips and cultured for 24 - 48 hrs at 27 °C before use to test the activity of potentiators; and incubated with or without the correction compound at 37 °C for measuring the activity of correctors.
- the inflow was placed adjacent to the patch, resulting in complete solution exchange within 1 - 2 sec.
- the nonspecific phosphatase inhibitor F " (10 mM NaF) was added to the bath solution. Under these recording conditions, channel activity remained constant throughout the duration of the patch recording (up to 60 min). Currents produced by positive charge moving from the intra- to extracellular solutions (anions moving in the opposite direction) are shown as positive currents.
- the pipette potential (V p ) was maintained at 80 mV.
- Intracellular solution in mM: NMDG-C1 (150), MgCl 2 (2), EGTA (5), TES
- NIH3T3 mouse fibroblasts stably expressing AF508-CFTR are used for excised-membrane patch-clamp recordings.
- the cells are maintained at 37 °C in 5% C0 2 and 90 % humidity in Dulbecco's modified Eagle's medium supplemented with 2 mM glutamine, 10 % fetal bovine serum, 1 X NEAA, ⁇ - ⁇ , 1 X pen/strep, and 25 mM HEPES in 175 cm 2 culture flasks.
- 2,500 - 5,000 cells were seeded on poly-L-lysine- coated glass coverslips and cultured for 24 - 48 hrs at 27 °C before use.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Physical Education & Sports Medicine (AREA)
- Diabetes (AREA)
- Neurosurgery (AREA)
- Rheumatology (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pain & Pain Management (AREA)
- Psychology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Pregnancy & Childbirth (AREA)
- Otolaryngology (AREA)
- Gynecology & Obstetrics (AREA)
- Emergency Medicine (AREA)
- Gastroenterology & Hepatology (AREA)
- Ophthalmology & Optometry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Indole Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description





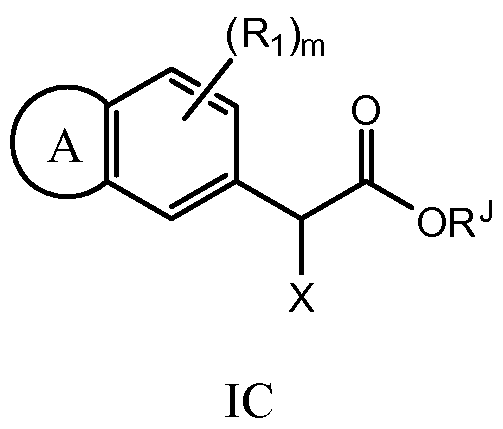




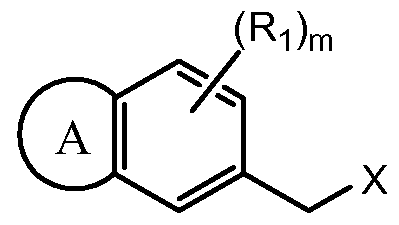
































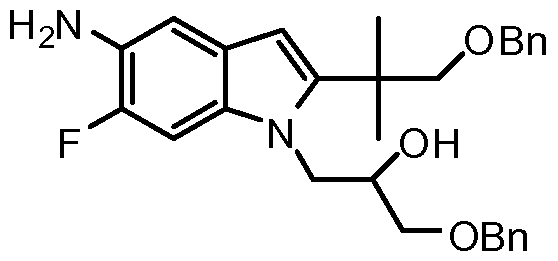


































Claims






















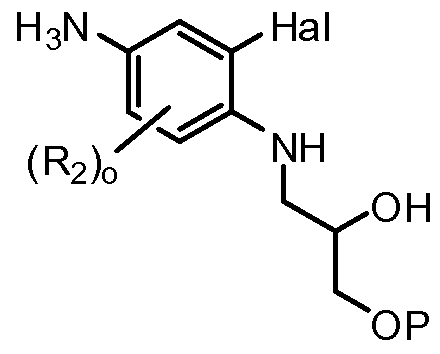

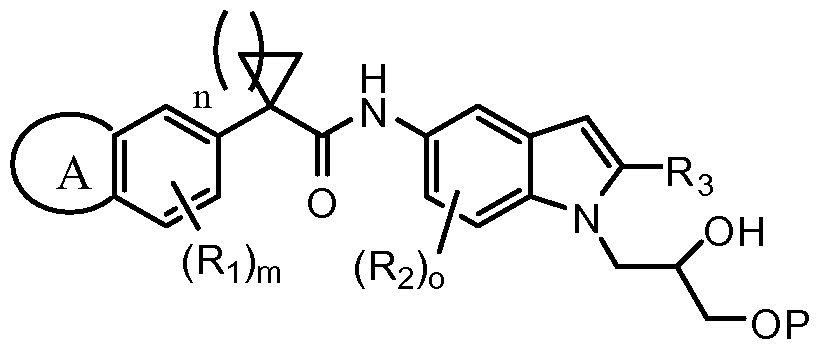























Priority Applications (26)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18173910.3A EP3381899B1 (en) | 2010-04-22 | 2011-04-21 | Intermediate compound for process of producing cycloalkylcarboxamido-indole compounds |
| US13/642,642 US9035072B2 (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| KR1020127030452A KR20130056244A (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| KR1020197015032A KR20190061096A (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| CA2797118A CA2797118C (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| CN201180031319.5A CN103038214B (en) | 2010-04-22 | 2011-04-21 | Prepare the method for cycloalkyl carboxamides base-benzazolyl compounds |
| CA3108488A CA3108488A1 (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| AU2011242712A AU2011242712B2 (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| JP2013506299A JP2013525371A (en) | 2010-04-22 | 2011-04-21 | Method for producing cycloalkylcarboxamide-indole compound |
| SG2012078374A SG184987A1 (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| MX2016009018A MX353408B (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds. |
| NZ603721A NZ603721A (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| RU2012149691/04A RU2569678C2 (en) | 2010-04-22 | 2011-04-21 | Method for producing cycloalkylcarboxamido-indole compounds |
| EP11729195.5A EP2560954B1 (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| ES11729195.5T ES2608474T3 (en) | 2010-04-22 | 2011-04-21 | Production process of indole compounds cycloalkylcarboxamido |
| KR1020167010394A KR101984225B1 (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| BR112012027056A BR112012027056B8 (en) | 2010-04-22 | 2011-04-21 | Process of production of cycloalkylcarboxamido-indole compounds |
| MX2015003252A MX342288B (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds. |
| MX2012012204A MX2012012204A (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds. |
| IL222539A IL222539A0 (en) | 2010-04-22 | 2012-10-18 | Process of producing cycloalkylcarboxamido-indole compounds |
| IL236209A IL236209B (en) | 2010-04-22 | 2014-12-11 | Process of producing cycloalkylcarboxamido-indole compounds |
| US14/687,286 US10071979B2 (en) | 2010-04-22 | 2015-04-15 | Process of producing cycloalkylcarboxamido-indole compounds |
| AU2016202569A AU2016202569B2 (en) | 2010-04-22 | 2016-04-22 | Process of producing cycloalkylcarboxamido-indole compounds |
| US16/059,724 US20190210991A1 (en) | 2010-04-22 | 2018-08-09 | Process of producing cycloalkylcarboxamido-indole compounds |
| IL268953A IL268953B (en) | 2010-04-22 | 2019-08-27 | Process of producing cycloalkylcarboxamido-indole compounds |
| US17/003,051 US20210238158A1 (en) | 2010-04-22 | 2020-08-26 | Process of producing cycloalkylcarboxamido-indole compounds |
Applications Claiming Priority (16)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US32709910P | 2010-04-22 | 2010-04-22 | |
| US32709110P | 2010-04-22 | 2010-04-22 | |
| US32709510P | 2010-04-22 | 2010-04-22 | |
| US32705710P | 2010-04-22 | 2010-04-22 | |
| US61/327,095 | 2010-04-22 | ||
| US61/327,091 | 2010-04-22 | ||
| US61/327,099 | 2010-04-22 | ||
| US61/327,057 | 2010-04-22 | ||
| US32951010P | 2010-04-29 | 2010-04-29 | |
| US32950010P | 2010-04-29 | 2010-04-29 | |
| US32949310P | 2010-04-29 | 2010-04-29 | |
| US61/329,500 | 2010-04-29 | ||
| US61/329,493 | 2010-04-29 | ||
| US61/329,510 | 2010-04-29 | ||
| US33387010P | 2010-05-12 | 2010-05-12 | |
| US61/333,870 | 2010-05-12 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/642,642 A-371-Of-International US9035072B2 (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
| US14/687,286 Division US10071979B2 (en) | 2010-04-22 | 2015-04-15 | Process of producing cycloalkylcarboxamido-indole compounds |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2011133751A2 true WO2011133751A2 (en) | 2011-10-27 |
| WO2011133751A3 WO2011133751A3 (en) | 2012-01-12 |
| WO2011133751A9 WO2011133751A9 (en) | 2012-02-16 |
Family
ID=44278819
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/033396 WO2011133751A2 (en) | 2010-04-22 | 2011-04-21 | Process of producing cycloalkylcarboxamido-indole compounds |
Country Status (18)
| Country | Link |
|---|---|
| US (4) | US9035072B2 (en) |
| EP (3) | EP3381899B1 (en) |
| JP (4) | JP2013525371A (en) |
| KR (3) | KR20190061096A (en) |
| CN (2) | CN103038214B (en) |
| AR (1) | AR081333A1 (en) |
| AU (2) | AU2011242712B2 (en) |
| BR (1) | BR112012027056B8 (en) |
| CA (2) | CA3108488A1 (en) |
| ES (2) | ES2608474T3 (en) |
| HK (2) | HK1218419A1 (en) |
| IL (3) | IL222539A0 (en) |
| MX (3) | MX2012012204A (en) |
| NZ (2) | NZ734535A (en) |
| RU (2) | RU2569678C2 (en) |
| SG (3) | SG184987A1 (en) |
| TW (3) | TWI518082B (en) |
| WO (1) | WO2011133751A2 (en) |
Cited By (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014534236A (en) * | 2011-11-08 | 2014-12-18 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Modifiers for ATP-binding cassette transporters |
| US8937178B2 (en) | 2013-03-13 | 2015-01-20 | Flatley Discovery Lab | Phthalazinone compounds and methods for the treatment of cystic fibrosis |
| WO2016160945A1 (en) | 2015-03-31 | 2016-10-06 | Concert Pharmaceuticals, Inc. | Deuterated vx-661 |
| US10047077B2 (en) | 2016-04-13 | 2018-08-14 | Skyline Antiinfectives, Inc. | Deuterated O-sulfated beta-lactam hydroxamic acids and deuterated N-sulfated beta-lactams |
| WO2018227049A1 (en) | 2017-06-08 | 2018-12-13 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US10167278B2 (en) | 2014-12-31 | 2019-01-01 | Auspex Pharmaceuticals, Inc. | Cyclopropanecarboxamide modulators of cystic fibrosis transmembrane conductance regulator |
| WO2019010092A1 (en) | 2017-07-01 | 2019-01-10 | Vertex Pharmaceuticals Incorporated | Compositions and methods for treatment of cystic fibrosis |
| WO2019018395A1 (en) | 2017-07-17 | 2019-01-24 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2019018353A1 (en) | 2017-07-17 | 2019-01-24 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2020102346A1 (en) | 2018-11-14 | 2020-05-22 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US10751363B2 (en) | 2015-03-23 | 2020-08-25 | Algipharma As | Use of aliginate oligomers and CFTR modulators in treatment of conditions associated with CFTR dysfunction |
| US10758534B2 (en) | 2014-10-06 | 2020-09-01 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US10793547B2 (en) | 2016-12-09 | 2020-10-06 | Vertex Pharmaceuticals Incorporated | Modulator of the cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| WO2020214921A1 (en) | 2019-04-17 | 2020-10-22 | Vertex Pharmaceuticals Incorporated | Solid forms of modulators of cftr |
| US10875846B2 (en) | 2019-01-15 | 2020-12-29 | Apotex Inc. | Processes for the preparation of Tezacaftor and intermediates thereof |
| US10906891B2 (en) | 2010-03-25 | 2021-02-02 | Vertex Pharmaceuticals Incoporated | Solid forms of (R)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| WO2021030556A1 (en) | 2019-08-14 | 2021-02-18 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2021030555A1 (en) | 2019-08-14 | 2021-02-18 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2021030552A1 (en) | 2019-08-14 | 2021-02-18 | Vertex Pharmaceuticals Incorporated | Crystalline forms of cftr modulators |
| US10975061B2 (en) | 2006-04-07 | 2021-04-13 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10980746B2 (en) | 2014-04-15 | 2021-04-20 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| EP3835297A1 (en) * | 2010-03-25 | 2021-06-16 | Vertex Pharmaceuticals Incorporated | Synthesis and intermediates of (r)-1(2,2 -difluorobenzo[d][1,3]dioxol-5yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2yl)-1h-indol-5yl)cyclopropanecarboxamide |
| US11066417B2 (en) | 2018-02-15 | 2021-07-20 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulators |
| US11155533B2 (en) | 2017-10-19 | 2021-10-26 | Vertex Pharmaceuticals Incorporated | Crystalline forms and compositions of CFTR modulators |
| US11179367B2 (en) | 2018-02-05 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for treating cystic fibrosis |
| US11186566B2 (en) | 2016-09-30 | 2021-11-30 | Vertex Pharmaceuticals Incorporated | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| WO2022032068A1 (en) | 2020-08-07 | 2022-02-10 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022036060A1 (en) | 2020-08-13 | 2022-02-17 | Vertex Pharmaceuticals Incorporated | Crystalline forms of cftr modulators |
| WO2022076628A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076629A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076626A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076624A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076625A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076621A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076622A2 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076618A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076627A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076620A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022125826A1 (en) | 2020-12-10 | 2022-06-16 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US11413306B2 (en) | 2015-10-06 | 2022-08-16 | Algipharma As | Alginate oligomers for the treatment or prevention of microbial overgrowth in the intestinal tract |
| US11414439B2 (en) | 2018-04-13 | 2022-08-16 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| US11434201B2 (en) | 2017-08-02 | 2022-09-06 | Vertex Pharmaceuticals Incorporated | Processes for preparing pyrrolidine compounds |
| US11465985B2 (en) | 2017-12-08 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| US11584761B2 (en) | 2019-08-14 | 2023-02-21 | Vertex Pharmaceuticals Incorporated | Process of making CFTR modulators |
| WO2023150237A1 (en) | 2022-02-03 | 2023-08-10 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2023150236A1 (en) | 2022-02-03 | 2023-08-10 | Vertex Pharmaceuticals Incorporated | Methods of preparing and crystalline forms of (6a,12a)-17-amino-12-methyl-6,15-bis(trifluoromethyl)-13,19-dioxa-3,4,18-triazatricyclo[ 12.3.1.12,5]nonadeca-1(18),2,4,14,16-pentaen-6-ol |
| WO2023154291A1 (en) | 2022-02-08 | 2023-08-17 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2023196429A1 (en) | 2022-04-06 | 2023-10-12 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2023224924A1 (en) | 2022-05-16 | 2023-11-23 | Vertex Pharmaceuticals Incorporated | Solid forms of a macrocyclic compounds as cftr modulators and their preparation |
| WO2023224931A1 (en) | 2022-05-16 | 2023-11-23 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US11992553B2 (en) | 2014-08-29 | 2024-05-28 | Algipharma As | Inhalable powder formulations of alginate oligomers |
| US12122788B2 (en) | 2023-01-04 | 2024-10-22 | Vertex Pharmaceuticals Incorporated | Process of making CFTR modulators |
Families Citing this family (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100074949A1 (en) | 2008-08-13 | 2010-03-25 | William Rowe | Pharmaceutical composition and administration thereof |
| RU2006111093A (en) | 2003-09-06 | 2007-10-27 | Вертекс Фармасьютикалз Инкорпорейтед (Us) | MODULATORS OF ATR-BINDING CASSETTE TRANSPORTERS |
| US7977322B2 (en) | 2004-08-20 | 2011-07-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| BR122018075478B8 (en) | 2004-06-24 | 2023-10-31 | Vertex Pharma | atp link cassette carrier modulators |
| JP5143738B2 (en) | 2005-08-11 | 2013-02-13 | バーテックス ファーマシューティカルズ インコーポレイテッド | Modulator of cystic fibrosis membrane conductance regulator |
| ES2439736T3 (en) | 2005-11-08 | 2014-01-24 | Vertex Pharmaceuticals Incorporated | Heterocyclic modulators of ATP binding cassette transporters |
| HUE049976T2 (en) | 2005-12-28 | 2020-11-30 | Vertex Pharma | Pharmaceutical compositions of the amorphous form of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| US7645789B2 (en) | 2006-04-07 | 2010-01-12 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
| US10022352B2 (en) | 2006-04-07 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US8563573B2 (en) | 2007-11-02 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| CN104447716A (en) | 2007-05-09 | 2015-03-25 | 沃泰克斯药物股份有限公司 | Modulators of CFTR |
| WO2009038913A2 (en) | 2007-08-24 | 2009-03-26 | Vertex Pharmaceuticals Incorporated | Isothiazolopyridinones useful for the treatment of (inter alia) cystic fibrosis |
| CA2705562C (en) | 2007-11-16 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Isoquinoline modulators of atp-binding cassette transporters |
| CN101910156B (en) | 2007-12-07 | 2013-12-04 | 沃泰克斯药物股份有限公司 | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3] dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| CA2989620C (en) | 2007-12-07 | 2022-05-03 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| US20100036130A1 (en) | 2007-12-07 | 2010-02-11 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| EP2271622B1 (en) | 2008-02-28 | 2017-10-04 | Vertex Pharmaceuticals Incorporated | Heteroaryl derivatives as CFTR Modulators |
| PT2615085E (en) | 2008-03-31 | 2015-10-09 | Vertex Pharma | Pyridyl derivatives as cftr modulators |
| CN102164587A (en) | 2008-09-29 | 2011-08-24 | 沃泰克斯药物股份有限公司 | Dosage units of 3-(6-(1-(2,2-difluorobenzo [D] [1,3] dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| SG10201504084QA (en) | 2009-03-20 | 2015-06-29 | Vertex Pharma | Process for making modulators of cystic fibrosis transmembrane conductance regulator |
| DK3150198T3 (en) | 2010-04-07 | 2021-11-01 | Vertex Pharma | PHARMACEUTICAL COMPOSITIONS OF 3- (6- (1- (2,2-DIFLUOROBENZO [D] [1,3] DIOXOL-5-YL) -CYCLOPROPANCARBOXAMIDO) -3-METHYLPYRIODIN-2-YL) BENZOIC ACID AND ADMINISTRATION |
| BR112012026257A2 (en) | 2010-04-07 | 2017-03-14 | Vertex Pharma | solid forms of 3- (6- (1- (2-, 2-difluorbenzo [d] [1,3] dioxol-5-yl) cyclopropanecarboxamido) -3-methylpyridin-2-yl) benzoic acid |
| MX2012012204A (en) * | 2010-04-22 | 2012-12-05 | Vertex Pharma | Process of producing cycloalkylcarboxamido-indole compounds. |
| CN109966264A (en) | 2012-02-27 | 2019-07-05 | 沃泰克斯药物股份有限公司 | Pharmaceutical composition and its application |
| AR092857A1 (en) | 2012-07-16 | 2015-05-06 | Vertex Pharma | PHARMACEUTICAL COMPOSITIONS OF (R) -1- (2,2-DIFLUOROBENZO [D] [1,3] DIOXOL-5-IL) -N- (1- (2,3-DIHYDROXIPROPIL) -6-FLUORO-2- ( 1-HYDROXI-2-METHYLPROPAN-2-IL) -1H-INDOL-5-IL) CYCLOPROPANCARBOXAMIDE AND ADMINISTRATION OF THE SAME |
| US10231932B2 (en) | 2013-11-12 | 2019-03-19 | Vertex Pharmaceuticals Incorporated | Process of preparing pharmaceutical compositions for the treatment of CFTR mediated diseases |
| CN107250113B (en) | 2014-10-07 | 2019-03-29 | 弗特克斯药品有限公司 | Co-crystals of modulators of cystic fibrosis transmembrane conductance regulator |
| JP6494757B2 (en) | 2014-11-18 | 2019-04-03 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Process for high-throughput high performance liquid chromatography |
| US10604492B2 (en) | 2015-12-24 | 2020-03-31 | The Regents Of The Universtiy Of California | CFTR regulators and methods of use thereof |
| WO2018183367A1 (en) | 2017-03-28 | 2018-10-04 | Van Goor Fredrick F | Methods of treating cystic fibrosis in patients with residual function mutations |
| CN111629730A (en) | 2017-08-24 | 2020-09-04 | 加利福尼亚大学董事会 | Ophthalmic pharmaceutical composition |
| EP3971186B1 (en) * | 2019-06-19 | 2023-08-02 | LG Chem, Ltd. | Preparation method for indole or indazole compound |
| WO2020256430A1 (en) * | 2019-06-19 | 2020-12-24 | 주식회사 엘지화학 | Method for producing indole or indazole compound |
| CN110437125B (en) * | 2019-09-06 | 2021-03-12 | 苏州旺山旺水生物医药有限公司 | Preparation method of Tezacaftor intermediate II |
| CN113121358A (en) * | 2019-12-31 | 2021-07-16 | 阜新金特莱氟化学有限责任公司 | Preparation method of 2-bromo-5-fluoro-4-nitroaniline |
| CN111187197B (en) * | 2020-01-13 | 2021-10-01 | 苏州旺山旺水生物医药有限公司 | Synthesis method of Tezacaftor intermediate |
| EP4099991A4 (en) * | 2020-02-05 | 2024-06-26 | Laurus Labs Limited | Novel processes for preparation of tezacaftor |
| CN113657599B (en) * | 2021-08-20 | 2024-05-28 | 北京航空航天大学 | Accident cause and effect reasoning method, device, electronic equipment and readable storage medium |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004028480A2 (en) | 2002-09-30 | 2004-04-08 | The Regents Of The University Of California | Cystic fibrosis transmembrane conductance regulator protein inhibitors and uses thereof |
| WO2004080972A1 (en) | 2003-03-12 | 2004-09-23 | Vertex Pharmaceuticals Incorporated | Pirazole modulators of atp-binding cassette transporters |
| WO2004091502A2 (en) | 2003-04-11 | 2004-10-28 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds |
| WO2004110352A2 (en) | 2003-05-16 | 2004-12-23 | The Regents Of The University Of California | Compounds having activity in increasing ion transport by mutant-cftr and uses thereof |
| WO2004111014A1 (en) | 2003-06-06 | 2004-12-23 | Vertex Pharmaceuticals Incorporated | Pyrimidine derivatives as modulators of atp-binding cassette transporters |
| WO2005035514A2 (en) | 2003-10-08 | 2005-04-21 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters containing cycloalkyl or pyranyl groups |
| WO2005049018A1 (en) | 2003-11-14 | 2005-06-02 | Vertex Pharmaceuticals Incorporated | Thiazoles and oxazoles useful as modulators of atp-binding cassette transporters |
| WO2005094374A2 (en) | 2004-03-30 | 2005-10-13 | The Regents Of The University Of California | Hydrazide-containing cftr inhibitor compounds and uses thereof |
| WO2005120497A2 (en) | 2004-06-04 | 2005-12-22 | The Regents Of The University Of California | Compounds having activity in increasing ion transport by mutant-cftr and uses thereof |
| WO2006044456A1 (en) | 2004-10-13 | 2006-04-27 | Ptc Therapeutics, Inc. | Compounds for nonsense suppression, and methods for their use |
| WO2006099256A2 (en) | 2005-03-11 | 2006-09-21 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2006101740A2 (en) | 2005-03-18 | 2006-09-28 | The Regents Of The University Of California | Compounds having activity in correcting mutant-cftr processing and uses thereof |
| WO2006110483A1 (en) | 2005-04-08 | 2006-10-19 | Ptc Therapeutics, Inc. | Compositions of an orally active 1,2,4-oxadiazole for nonsense mutation suppression therapy |
| WO2006127588A2 (en) | 2005-05-24 | 2006-11-30 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2007044560A2 (en) | 2005-10-06 | 2007-04-19 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| US20090131492A1 (en) | 2006-04-07 | 2009-05-21 | Ruah Sara S Hadida | Indole derivatives as CFTR modulators |
Family Cites Families (312)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL7605382A (en) * | 1975-06-04 | 1976-12-07 | Sumitomo Chemical Co | PROCESS FOR PREPARING INDOLE DERIVATIVES. |
| JPS5328173A (en) * | 1976-08-06 | 1978-03-16 | Zenyaku Kogyo Kk | Novel phenylmethanenitro compound and its preparation |
| DE2735133C2 (en) | 1977-08-04 | 1981-10-15 | Universität Karlsruhe Institut für Elektrotechnische Grundlagen der Informatik, 7500 Karlsruhe | Memory cell for non-destructive reading with 2 Josephson contacts |
| US4138397A (en) | 1978-02-27 | 1979-02-06 | Richardson-Merrell Inc. | 6-(2,3-Dihydro-5-benzofuranyl)acetamido penicillin derivatives |
| GB8524157D0 (en) | 1984-10-19 | 1985-11-06 | Ici America Inc | Heterocyclic amides |
| JPS61103861A (en) * | 1984-10-27 | 1986-05-22 | Nitto Kasei Kk | Preparation of aryl-substituted cyanoacetic acid ester |
| US5304121A (en) | 1990-12-28 | 1994-04-19 | Boston Scientific Corporation | Drug delivery system making use of a hydrogel polymer coating |
| US5716981A (en) | 1993-07-19 | 1998-02-10 | Angiogenesis Technologies, Inc. | Anti-angiogenic compositions and methods of use |
| GB9317764D0 (en) | 1993-08-26 | 1993-10-13 | Pfizer Ltd | Therapeutic compound |
| US5510379A (en) | 1994-12-19 | 1996-04-23 | Warner-Lambert Company | Sulfonate ACAT inhibitors |
| US6099562A (en) | 1996-06-13 | 2000-08-08 | Schneider (Usa) Inc. | Drug coating with topcoat |
| JPH10213820A (en) | 1997-01-31 | 1998-08-11 | Canon Inc | Liquid crystal element and liquid crystal device |
| WO1998047868A1 (en) | 1997-04-18 | 1998-10-29 | Smithkline Beecham Plc | Heterocycle-containing urea derivatives as 5ht1a, 5ht1b and 5ht1d receptor antagonists |
| KR20010014020A (en) | 1997-06-21 | 2001-02-26 | 로셰 디아그노스틱스 게엠베하 | Barbituric acid derivatives with antimetastatic and antitumor activity |
| EP0901786B1 (en) | 1997-08-11 | 2007-06-13 | Pfizer Products Inc. | Solid pharmaceutical dispersions with enhanced bioavailability |
| US20020017295A1 (en) | 2000-07-07 | 2002-02-14 | Weers Jeffry G. | Phospholipid-based powders for inhalation |
| JP2001525365A (en) | 1997-12-11 | 2001-12-11 | バイオケム・フアーマ・インコーポレーテツド | Antiviral compounds |
| US6426331B1 (en) | 1998-07-08 | 2002-07-30 | Tularik Inc. | Inhibitors of STAT function |
| AUPP609198A0 (en) | 1998-09-22 | 1998-10-15 | Curtin University Of Technology | Use of non-peptidyl compounds for the treatment of insulin related ailments |
| ATE496032T1 (en) | 1999-02-24 | 2011-02-15 | Hoffmann La Roche | 4-PHENYLPYRIDINE DERIVATIVES AND THEIR USE AS NK-1 RECEPTOR ANTAGONISTS |
| DE60028791T2 (en) | 1999-09-10 | 2007-05-24 | Novo Nordisk A/S | MODULATORS OF THE PROTEIN TYROSINE PHOSPHATASE (PTPASES) |
| JP2003509430A (en) | 1999-09-10 | 2003-03-11 | ノボ ノルディスク アクティーゼルスカブ | Modulator of protein tyrosine phosphatase (PTPase) |
| AUPQ288499A0 (en) | 1999-09-16 | 1999-10-07 | Biota Scientific Management Pty Ltd | Antiviral agents |
| US6514964B1 (en) | 1999-09-27 | 2003-02-04 | Amgen Inc. | Fused cycloheptane and fused azacycloheptane compounds and their methods of use |
| IT1315267B1 (en) | 1999-12-23 | 2003-02-03 | Novuspharma Spa | DERIVATIVES OF 2- (1H-INDOL-3-IL) -2-OXO-ACETAMIDES FOR ANTI-TUMOR ACTIVITY |
| TWI284639B (en) | 2000-01-24 | 2007-08-01 | Shionogi & Co | A compound having thrombopoietin receptor agonistic effect |
| DE10038019A1 (en) * | 2000-08-04 | 2002-02-14 | Bayer Ag | Substituted triazolopyride (az) ine |
| US6777400B2 (en) | 2000-08-05 | 2004-08-17 | Smithkline Beecham Corporation | Anti-inflammatory androstane derivative compositions |
| US6562989B2 (en) * | 2000-08-07 | 2003-05-13 | Yale University | Catalyst for aromatic C—O, C—N, and C—C bond formation |
| TWI259180B (en) | 2000-08-08 | 2006-08-01 | Hoffmann La Roche | 4-Phenyl-pyridine derivatives |
| AR030357A1 (en) | 2000-08-18 | 2003-08-20 | Lundbeck & Co As H | DERIVATIVES 4 -, 5 -, 6 - AND 7-INDOL |
| US6550895B1 (en) | 2000-10-20 | 2003-04-22 | Silverbrook Research Pty Ltd | Moving nozzle ink jet with inlet restriction |
| KR100600550B1 (en) | 2000-10-20 | 2006-07-13 | 에자이 가부시키가이샤 | Nitrogenous aromatic ring compounds |
| EP1217000A1 (en) | 2000-12-23 | 2002-06-26 | Aventis Pharma Deutschland GmbH | Inhibitors of factor Xa and factor VIIa |
| GB0102109D0 (en) | 2001-01-26 | 2001-03-14 | Syngenta Ltd | Chemical process |
| US20100074949A1 (en) | 2008-08-13 | 2010-03-25 | William Rowe | Pharmaceutical composition and administration thereof |
| CA2442654A1 (en) | 2001-04-10 | 2002-10-10 | Transtech Pharma, Inc. | Probes, systems, and methods for drug discovery |
| CN1300116C (en) | 2001-04-16 | 2007-02-14 | 卫材株式会社 | Novel 1H-indazole compound |
| CN1522246B (en) | 2001-06-28 | 2010-04-21 | 辉瑞产品公司 | Triamide-substituted indoles, benzofuranes and benzothiophenes |
| US6841566B2 (en) | 2001-07-20 | 2005-01-11 | Boehringer Ingelheim, Ltd. | Viral polymerase inhibitors |
| AUPR738301A0 (en) | 2001-08-30 | 2001-09-20 | Starpharma Limited | Chemotherapeutic agents |
| WO2003041649A2 (en) | 2001-11-13 | 2003-05-22 | Merck Frosst Canada & Co. | Cyanoalkylamino derivatives as protease inhibitors |
| JP2005518391A (en) | 2001-12-21 | 2005-06-23 | ノボ ノルディスク アクティーゼルスカブ | Amide derivatives as GK activators |
| US7074805B2 (en) | 2002-02-20 | 2006-07-11 | Abbott Laboratories | Fused azabicyclic compounds that inhibit vanilloid receptor subtype 1 (VR1) receptor |
| US20030158188A1 (en) | 2002-02-20 | 2003-08-21 | Chih-Hung Lee | Fused azabicyclic compounds that inhibit vanilloid receptor subtype 1 (VR1) receptor |
| WO2003093498A1 (en) | 2002-04-29 | 2003-11-13 | The Ohio State University | Inhibition of protein tyrosine phosphatases and sh2 domains by a neutral phosphotyrosine mimetic |
| MXPA05000130A (en) | 2002-06-27 | 2005-02-17 | Novo Nordisk As | Aryl carbonyl derivatives as therapeutic agents. |
| US20060004010A1 (en) | 2002-07-10 | 2006-01-05 | Hiromu Habashita | Ccr4 antagonist and medical use thereof |
| TWI244393B (en) | 2002-08-06 | 2005-12-01 | Idenix Pharmaceuticals Inc | Crystalline and amorphous forms of beta-L-2'-deoxythymidine |
| TW200413372A (en) | 2002-09-18 | 2004-08-01 | Ono Pharmaceutical Co | Derivatives of triazaspiro [5.5] undecane and medicants using such derivatives as effective ingredient |
| JP2004131393A (en) | 2002-10-08 | 2004-04-30 | Kowa Co | Readily eluting pharmaceutical preparation |
| CA2501547A1 (en) | 2002-10-15 | 2004-04-29 | Rigel Pharmaceuticals, Inc. | Substituted indoles and their use as hcv inhibitors |
| FR2846327B1 (en) | 2002-10-25 | 2006-03-24 | Merck Sante Sas | N-BENZODIOXOLYL, N-BENZODIOXANYL AND N-BENZODIOXEPINYL ARYLCARBOXAMIDE DERIVATIVES USEFUL IN THE TREATMENT OF DYSLIPIDEMIA AND PHARMACEUTICAL COMPOSITIONS CONTAINING SAME |
| WO2004041277A1 (en) | 2002-11-01 | 2004-05-21 | Merck & Co., Inc. | Carbonylamino-benzimidazole derivatives as androgen receptor modulators |
| RS20050294A (en) | 2002-11-02 | 2007-11-15 | Sanofi-Aventis Deutschland Gmbh., | Novel pyrimidine-4,6-dicarboxamides for the selective inhibition of collagenases |
| DE10251019A1 (en) | 2002-11-02 | 2004-05-19 | Aventis Pharma Deutschland Gmbh | New pyrimidine-4,6-dicarboxamide derivatives, are selective collagenase inhibitors useful e.g. for treating degenerative joint diseases, connective tissue disorders or cancer |
| US20040146941A1 (en) | 2002-11-04 | 2004-07-29 | Biliang Zhang | Chemical encoding technology for combinatorial synthesis |
| WO2004056744A1 (en) | 2002-12-23 | 2004-07-08 | Janssen Pharmaceutica N.V. | Adamantyl acetamides as hydroxysteroid dehydrogenase inhibitors |
| DE10300017A1 (en) | 2003-01-03 | 2004-07-15 | Aventis Pharma Deutschland Gmbh | Selective MMP 13 inhibitors |
| KR101145252B1 (en) | 2003-01-08 | 2012-05-24 | 유니버시티 오브 워싱톤 | Antibacterial agents |
| US6933311B2 (en) | 2003-02-11 | 2005-08-23 | Abbott Laboratories | Fused azabicyclic compounds that inhibit vanilloid receptor subtype 1 (VR1) receptor |
| US7531558B2 (en) | 2003-02-14 | 2009-05-12 | Glaxo Group Limited | Carboxamide derivatives |
| DE10306941A1 (en) | 2003-02-18 | 2004-08-26 | Merck Patent Gmbh | New indolyl-substituted benzofuranyloxy-alkylamine derivatives, are 5-hydroxytryptamine reuptake inhibitors useful e.g. as anxiolytic, antidepressant, neuroleptic and/or antihypertensive agents |
| EP1720844B1 (en) | 2003-04-03 | 2009-04-29 | MERCK PATENT GmbH | Pyrrolidino-1,2-dicarboxy-1-(phenylamide)-2-(4-(3-oxo-morpholino-4-yl)-phenylamide) derivatives and related compounds for use as inhibitors of coagulation factor xa in the treatment of thrombo-embolic diseases |
| US20050164951A1 (en) | 2003-04-03 | 2005-07-28 | The Regents Of The University Of California | Inhibitors for the soluble epoxide hydrolase |
| DE10315377A1 (en) | 2003-04-03 | 2004-10-14 | Merck Patent Gmbh | New carbonyl-substituted carbocyclic or heterocyclic compounds, are factor Xa and factor VIIa inhibitors useful e.g. for treating thrombosis, myocardial infarction, arteriosclerosis, inflammation or tumors |
| JP4629657B2 (en) | 2003-04-11 | 2011-02-09 | ハイ・ポイント・ファーマスーティカルズ、エルエルシー | 11β-hydroxysteroid dehydrogenase type 1 active compound |
| WO2005002519A2 (en) | 2003-06-27 | 2005-01-13 | Henry M.Jackson Foundation For The Advancement Of Military Medicine, Inc. | Amphiphilic pyridinium compounds, method of making and use thereof |
| US20050113576A1 (en) | 2003-08-05 | 2005-05-26 | Chih-Hung Lee | Fused azabicyclic compounds that inhibit vanilloid receptor subtype 1 (VR1) receptor |
| AU2004265020B8 (en) | 2003-08-15 | 2009-11-05 | H. Lundbeck A/S | Cyclopropyl derivatives as NK3 receptor antagonists |
| RU2006111093A (en) | 2003-09-06 | 2007-10-27 | Вертекс Фармасьютикалз Инкорпорейтед (Us) | MODULATORS OF ATR-BINDING CASSETTE TRANSPORTERS |
| US20050070718A1 (en) | 2003-09-30 | 2005-03-31 | Abbott Gmbh & Co. Kg | Heteroaryl-substituted 1,3-dihydroindol-2-one derivatives and medicaments containing them |
| WO2005037802A1 (en) * | 2003-10-16 | 2005-04-28 | Sankyo Company, Limited | 5-arylpyrimidine derivative |
| EP1679309A4 (en) | 2003-10-24 | 2007-03-28 | Ono Pharmaceutical Co | Antistress drug and medical use thereof |
| GB0330043D0 (en) | 2003-12-24 | 2004-01-28 | Pharmacia Italia Spa | Pyrrolo [2,3-b] pyridine derivatives active as kinase inhibitors process for their preparation and pharmaceutical compositions comprising them |
| BRPI0507278A (en) | 2004-01-30 | 2007-06-26 | Vertex Pharma | modulators of atp-binding cassette transporters |
| US7977322B2 (en) | 2004-08-20 | 2011-07-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US8906676B2 (en) | 2004-02-02 | 2014-12-09 | Ambrx, Inc. | Modified human four helical bundle polypeptides and their uses |
| US20050222271A1 (en) | 2004-03-31 | 2005-10-06 | Le Huang | Novel amorphous form of memantine hydrochloride |
| FR2868417B1 (en) | 2004-04-02 | 2006-06-23 | Rhodia Chimie Sa | PROCESS FOR FORMING CARBON-CARBON BOND |
| AU2005249154B2 (en) | 2004-06-01 | 2011-02-10 | Luminex Molecular Diagnostics, Inc. | Method of detecting cystic fibrosis associated mutations |
| US8354427B2 (en) | 2004-06-24 | 2013-01-15 | Vertex Pharmaceutical Incorporated | Modulators of ATP-binding cassette transporters |
| US20140343098A1 (en) | 2004-06-24 | 2014-11-20 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| BR122018075478B8 (en) | 2004-06-24 | 2023-10-31 | Vertex Pharma | atp link cassette carrier modulators |
| WO2006014427A1 (en) | 2004-07-02 | 2006-02-09 | Advancis Pharmaceutical Corporation | Tablet for pulsed delivery |
| GB0416730D0 (en) | 2004-07-27 | 2004-09-01 | Novartis Ag | Organic compounds |
| MX2007001215A (en) | 2004-08-06 | 2007-04-17 | Otsuka Pharma Co Ltd | Aromatic compounds. |
| DE102004047254A1 (en) | 2004-09-29 | 2006-04-13 | Merck Patent Gmbh | carbonyl |
| NZ554555A (en) | 2004-10-20 | 2011-09-30 | Univ California | Cyclohexyl-urea derivatives as improved inhibitors for the soluble epoxide hydrolase |
| JPWO2006057448A1 (en) | 2004-11-26 | 2008-06-05 | 武田薬品工業株式会社 | Arylalkanoic acid derivatives |
| CA2599348C (en) | 2005-02-25 | 2013-07-23 | Ono Pharmaceutical Co., Ltd. | Indole compound and use thereof |
| US20100120789A1 (en) | 2005-03-24 | 2010-05-13 | Nigel Vicker | Compound |
| US7297700B2 (en) | 2005-03-24 | 2007-11-20 | Renovis, Inc. | Bicycloheteroaryl compounds as P2X7 modulators and uses thereof |
| GB0506133D0 (en) | 2005-03-24 | 2005-05-04 | Sterix Ltd | Compound |
| NZ561672A (en) * | 2005-03-28 | 2010-04-30 | Toyama Chemical Co Ltd | Process for production of 1-(3-(2-(1-benzothiophen-5-yl)-ethoxy)propyl)azetidin-3-ol or salts thereof |
| JP2006282534A (en) | 2005-03-31 | 2006-10-19 | Fuji Photo Film Co Ltd | Preparation method of amides |
| CA2604920A1 (en) | 2005-04-15 | 2006-10-26 | Elan Pharmaceuticals, Inc. | Novel compounds useful for bradykinin b1 receptor antagonism |
| ES2367844T3 (en) | 2005-08-11 | 2011-11-10 | Vertex Pharmaceuticals, Inc. | MODULATORS OF THE REGULATOR OF THE TRANSMEMBRANE CONDUCTANCE OF THE CHYSICAL FIBROSIS. |
| JP5143738B2 (en) | 2005-08-11 | 2013-02-13 | バーテックス ファーマシューティカルズ インコーポレイテッド | Modulator of cystic fibrosis membrane conductance regulator |
| CA2624220A1 (en) * | 2005-09-29 | 2007-04-12 | Wyeth | 1- (1h- indol- 1-yl) -3- (methylamino) -1- phenylpropan-2-ol derivatives and related compounds as modulators of the monoamine reuptake for the treatment of vasomotor symptoms (vms) |
| EP1942732A2 (en) | 2005-11-02 | 2008-07-16 | Cytokinetics, Inc. | Certain chemical entities, compositions, and methods |
| ES2439736T3 (en) | 2005-11-08 | 2014-01-24 | Vertex Pharmaceuticals Incorporated | Heterocyclic modulators of ATP binding cassette transporters |
| US20120232059A1 (en) | 2005-11-08 | 2012-09-13 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-Binding Cassette Transporters |
| GB0525144D0 (en) | 2005-12-09 | 2006-01-18 | Novartis Ag | Organic compounds |
| BRPI0620292B1 (en) | 2005-12-21 | 2021-08-24 | Janssen Pharmaceutica N. V. | TRIAZOLOPYRIDAZINE COMPOUNDS AS KINASE MODULATORS, COMPOSITION, USE, COMBINATION AND PREPARATION PROCESS OF SUCH COMPOUND |
| US20090105272A1 (en) | 2005-12-24 | 2009-04-23 | Grootenhuis Peter D J | Prodrugs of modulators of ABC transporters |
| CA2635214A1 (en) | 2005-12-27 | 2007-07-05 | Vertex Pharmaceuticals Incorporated | Compounds useful in cftr assays and methods therewith |
| HUE049976T2 (en) | 2005-12-28 | 2020-11-30 | Vertex Pharma | Pharmaceutical compositions of the amorphous form of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| CA2856037C (en) | 2005-12-28 | 2017-03-07 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| US7671221B2 (en) | 2005-12-28 | 2010-03-02 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-Binding Cassette transporters |
| TW200808723A (en) | 2006-03-13 | 2008-02-16 | Univ California | Conformationally restricted urea inhibitors of soluble epoxide hydrolase |
| KR20140107691A (en) | 2006-03-20 | 2014-09-04 | 버텍스 파마슈티칼스 인코포레이티드 | Pharmaceutical compositions |
| CN101494979A (en) | 2006-03-20 | 2009-07-29 | 沃泰克斯药物股份有限公司 | Pharmaceutical compositions |
| JP2009530399A (en) | 2006-03-22 | 2009-08-27 | シンデクサ ファーマシューティカルズ コーポレーション | Compounds and methods for the treatment of diseases associated with ER stress |
| RU2451018C2 (en) | 2006-04-07 | 2012-05-20 | Вертекс Фармасьютикалз Инкорпорейтед | Modulators of atp-binding cassette transporters |
| US10022352B2 (en) | 2006-04-07 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| AU2007249269A1 (en) | 2006-05-12 | 2007-11-22 | Vertex Pharmaceuticals Incorporated | Compositions of N-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl)-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| AU2007257959A1 (en) | 2006-06-09 | 2007-12-21 | Kemia, Inc. | Therapy using cytokine inhibitors |
| WO2008020227A2 (en) | 2006-08-17 | 2008-02-21 | Astrazeneca Ab | Antibacterial pyrrolecarboxamides |
| WO2008029168A2 (en) | 2006-09-08 | 2008-03-13 | Summit Corporation Plc | Treatment of duchenne muscular dystrophy |
| WO2008029152A2 (en) | 2006-09-08 | 2008-03-13 | Summit Corporation Plc | Treatment of duchenne muscular dystrophy |
| PL2081937T3 (en) * | 2006-10-23 | 2013-01-31 | Sgx Pharmaceuticals Inc | Triazolopyridazine protein kinase modulators |
| US8563573B2 (en) | 2007-11-02 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| EP2118103B1 (en) | 2006-11-03 | 2014-04-23 | Vertex Pharmaceuticals Inc. | Azaindole derivatives as cftr modulators |
| US7754739B2 (en) | 2007-05-09 | 2010-07-13 | Vertex Pharmaceuticals Incorporated | Modulators of CFTR |
| US20080132560A1 (en) | 2006-11-21 | 2008-06-05 | San-Laung Chow | Solid dispersion composition |
| WO2008065732A1 (en) | 2006-11-27 | 2008-06-05 | Nippon Polyurethane Industry Co., Ltd. | Process for production of modified isocyanate mixture containing allophanate bond and isocyanurate bond |
| US20080260820A1 (en) | 2007-04-19 | 2008-10-23 | Gilles Borrelly | Oral dosage formulations of protease-resistant polypeptides |
| CN104447716A (en) | 2007-05-09 | 2015-03-25 | 沃泰克斯药物股份有限公司 | Modulators of CFTR |
| ES2548292T3 (en) | 2007-05-25 | 2015-10-15 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US8557823B2 (en) | 2007-06-18 | 2013-10-15 | Advanced Cancer Therapeutics, Llc | Family of PFKFB3 inhibitors with anti-neoplastic activities |
| WO2008156783A2 (en) | 2007-06-18 | 2008-12-24 | University Of Louisville Research Foundation, Inc. | Family of pfkfb3 inhibitors with anti-neoplastic activities |
| PT2178865E (en) | 2007-07-19 | 2015-11-16 | Lundbeck & Co As H | 5-membered heterocyclic amides and related compounds |
| US20110177999A1 (en) | 2007-08-09 | 2011-07-21 | Vertex Pharmaceuticals Incorporated | Therapeutic Combinations Useful in Treating CFTR Related Diseases |
| WO2009038913A2 (en) | 2007-08-24 | 2009-03-26 | Vertex Pharmaceuticals Incorporated | Isothiazolopyridinones useful for the treatment of (inter alia) cystic fibrosis |
| CN101821266B (en) | 2007-09-14 | 2014-03-12 | 沃泰克斯药物股份有限公司 | Modulators of cystic fibrosis transmembrane conductance regulator |
| NZ600865A (en) | 2007-09-14 | 2014-01-31 | Vertex Pharma | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| EP2217588A4 (en) | 2007-11-02 | 2013-12-04 | Methylgene Inc | Inhibitors of histone deacetylase |
| CA2705562C (en) | 2007-11-16 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Isoquinoline modulators of atp-binding cassette transporters |
| AU2008327095B2 (en) | 2007-11-22 | 2013-07-25 | Ohara Pharmaceutical Co., Ltd. | Amorphous form of heterocyclic compound, solid dispersion and medicinal preparation each comprising the same, and process for production of the same |
| WO2009076141A2 (en) | 2007-12-07 | 2009-06-18 | Vertex Pharmaceuticals Incorporated | Formulations of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cycklopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| US20100036130A1 (en) | 2007-12-07 | 2010-02-11 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| CN101910156B (en) | 2007-12-07 | 2013-12-04 | 沃泰克斯药物股份有限公司 | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3] dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| CA2989620C (en) | 2007-12-07 | 2022-05-03 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| JP5637859B2 (en) | 2007-12-13 | 2014-12-10 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Modulator of cystic fibrosis membrane conductance regulator |
| WO2009086426A2 (en) | 2007-12-28 | 2009-07-09 | Arete Therapeutics, Inc. | Soluble epoxide hydrolase inhibitors for the treatment of endothelial dysfunction |
| EP2271622B1 (en) | 2008-02-28 | 2017-10-04 | Vertex Pharmaceuticals Incorporated | Heteroaryl derivatives as CFTR Modulators |
| PT2615085E (en) | 2008-03-31 | 2015-10-09 | Vertex Pharma | Pyridyl derivatives as cftr modulators |
| US20110071197A1 (en) | 2008-04-16 | 2011-03-24 | Peter Nilsson | Bis-aryl compounds for use as medicaments |
| WO2009129501A1 (en) | 2008-04-18 | 2009-10-22 | Arete Therapeutics, Inc. | Use of soluble epoxide hydrolase inhibitors in the treatment of smooth muscle disorders |
| BRPI0911482A2 (en) | 2008-04-24 | 2017-08-29 | Bristol Myers Squibb Co | USE OF EPOTHYLONE D IN THE TREATMENT OF TAU-ASSOCIATED DISEASES INCLUDING ALZHEIMER DISEASES |
| US20110112193A1 (en) | 2008-05-14 | 2011-05-12 | Peter Nilsson | Bis-aryl compounds for use as medicaments |
| US9447049B2 (en) | 2010-03-01 | 2016-09-20 | University Of Tennessee Research Foundation | Compounds for treatment of cancer |
| US8822513B2 (en) | 2010-03-01 | 2014-09-02 | Gtx, Inc. | Compounds for treatment of cancer |
| US20100256184A1 (en) | 2008-08-13 | 2010-10-07 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administrations thereof |
| JP5575768B2 (en) | 2008-08-13 | 2014-08-20 | バーテックス ファーマシューティカルズ インコーポレイテッド | Pharmaceutical composition and its administration |
| US8895781B2 (en) | 2008-09-04 | 2014-11-25 | Georgetown University | Transition metal-catalyzed C—H amination using unactivated amines |
| TW201011009A (en) | 2008-09-15 | 2010-03-16 | Priaxon Ag | Novel pyrrolidin-2-ones |
| CN102164587A (en) | 2008-09-29 | 2011-08-24 | 沃泰克斯药物股份有限公司 | Dosage units of 3-(6-(1-(2,2-difluorobenzo [D] [1,3] dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| WO2010048149A2 (en) | 2008-10-20 | 2010-04-29 | Kalypsys, Inc. | Heterocyclic modulators of gpr119 for treatment of disease |
| JP5645834B2 (en) | 2008-10-23 | 2014-12-24 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Modulator of cystic fibrosis membrane conductance regulator |
| US20110257223A1 (en) * | 2008-10-23 | 2011-10-20 | Vertex Pharmaceuticals Incorporated | Modulators of Cystic Fibrosis Transmembrane Conductance Regulator |
| EA018891B1 (en) * | 2008-10-23 | 2013-11-29 | Вертекс Фармасьютикалз, Инкорпорейтед | Modulators of cystic fibrosis transmembrane conductance regulator |
| JP5645835B2 (en) | 2008-10-23 | 2014-12-24 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | N- (4- (7-azabicyclo [2.2.1] heptane-7-yl) -2- (trifluoromethyl) phenyl) -4-oxo-5- (trifluoromethyl) -1,4-dihydro Solid form of quinoline-3-carboxamide |
| UA121188C2 (en) | 2008-11-06 | 2020-04-27 | Вертекс Фармасьютікалз Інкорпорейтед | ATV-CONNECTING CASSETTE CONVEYOR MODULATORS |
| UA104876C2 (en) | 2008-11-06 | 2014-03-25 | Вертекс Фармасьютікалз Інкорпорейтед | Modulators of atp-binding cassette transporters |
| AR074231A1 (en) | 2008-11-27 | 2010-12-29 | Boehringer Ingelheim Int | DERIVATIVES OF 6, 7, 8, 9-TETRAHIDRO-5H-1, 4, 7, 10A-TETRAAZA-CICLOHEPT [F] INDENO, PHARMACEUTICAL COMPOSITIONS CONTAINING THESE COMPOUNDS, THEIR USE IN THE 5-HT2C RECEPTOR AGONISM AND PROCESSES FOR PREPARE THEM. |
| WO2010065668A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| EP2382197B1 (en) | 2008-12-30 | 2016-10-05 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US8946419B2 (en) | 2009-02-23 | 2015-02-03 | Mallinckrodt Llc | (+)-6-hydroxy-morphinan or (+)-6-amino-morphinan derivatives |
| KR20100101054A (en) | 2009-03-07 | 2010-09-16 | 주식회사 메디젠텍 | Composition for treating or preventing nuclear export of gsk3- mediated disease including compound for inhibiting nuclear export of gsk3 |
| SG10201504084QA (en) | 2009-03-20 | 2015-06-29 | Vertex Pharma | Process for making modulators of cystic fibrosis transmembrane conductance regulator |
| CA2755969C (en) | 2009-03-20 | 2018-05-08 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| GB0905641D0 (en) | 2009-04-01 | 2009-05-13 | Serodus As | Compounds |
| ES2475091T3 (en) | 2009-04-16 | 2014-07-10 | Centro Nacional De Investigaciones Oncol�Gicas (Cnio) | Imidazopyrazines as protein kinase inhibitors |
| WO2011005355A1 (en) | 2009-05-07 | 2011-01-13 | Achaogen, Inc. | Combinations comprising a lpxc inhibitor and an antibiotic for use in the treatment of infections caused by gram-negative bacteria |
| CA2761639C (en) | 2009-05-29 | 2016-06-07 | Raqualia Pharma Inc. | Aryl substituted carboxamide derivatives as calcium or sodium channel blockers |
| EP2264012A1 (en) | 2009-06-03 | 2010-12-22 | Bayer CropScience AG | Heteroarylamidines and their use as fungicides |
| KR101256018B1 (en) | 2009-08-20 | 2013-04-18 | 한국과학기술연구원 | 1,3,6-Substituted indole derivatives having inhibitory activity for protein kinase |
| WO2011029832A1 (en) | 2009-09-09 | 2011-03-17 | Vifor (International) Ag | Novel thiazol and oxazol hepcidine antagonists |
| KR20120083416A (en) | 2009-09-17 | 2012-07-25 | 버텍스 파마슈티칼스 인코포레이티드 | Process for preparing azabicyclic compounds |
| US9334230B2 (en) | 2009-09-18 | 2016-05-10 | Nanyang Technological University | Process of forming an amide |
| EP2490687A1 (en) | 2009-10-22 | 2012-08-29 | Vertex Pharmaceuticals Incorporated | Compositions for treatment of cystic fibrosis and other chronic diseases |
| CN102648182A (en) | 2009-10-23 | 2012-08-22 | 沃泰克斯药物股份有限公司 | Process for preparing modulators of cystic fibrosis transmembrane conductance regulator |
| CA2778493A1 (en) | 2009-10-23 | 2011-04-28 | Vertex Pharmaceuticals Incorporated | Solid forms of n-(4-(7-azabicyclo[2.2.1]heptan-7-yl)-2-trifluoromethyl)phenyl)-4-oxo-5-(trifluoromethyl)-1,4-dihydroquinoline-3-carboxamide |
| WO2011094890A1 (en) | 2010-02-02 | 2011-08-11 | Argusina Inc. | Phenylalanine derivatives and their use as non-peptide glp-1 receptor modulators |
| CN102883607B (en) | 2010-03-01 | 2015-07-22 | Gtx公司 | Compounds for treatment of cancer |
| WO2011115892A1 (en) | 2010-03-15 | 2011-09-22 | Griffin Patrick R | Modulators of the retinoic acid receptor-related orphan receptors |
| CN103180298A (en) | 2010-03-19 | 2013-06-26 | 沃泰克斯药物股份有限公司 | Solid form of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydr o-4-oxoquinoline-3-carboxamide |
| PT2826776T (en) * | 2010-03-25 | 2021-02-01 | Vertex Pharma | Solid amorphous form of (r)-1(2,2-difluorobenzo(d)(1,3)dioxol-5-yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl)-cyclopropanecarboxamide |
| US8802868B2 (en) * | 2010-03-25 | 2014-08-12 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxo1-5-yl)-N-(1-(2,3-dihydroxypropyl-6-fluoro-2-(1-hydroxy-2-methylpropan2-yl)-1H-Indol-5-yl)-Cyclopropanecarboxamide |
| BR112012026257A2 (en) | 2010-04-07 | 2017-03-14 | Vertex Pharma | solid forms of 3- (6- (1- (2-, 2-difluorbenzo [d] [1,3] dioxol-5-yl) cyclopropanecarboxamido) -3-methylpyridin-2-yl) benzoic acid |
| DK3150198T3 (en) | 2010-04-07 | 2021-11-01 | Vertex Pharma | PHARMACEUTICAL COMPOSITIONS OF 3- (6- (1- (2,2-DIFLUOROBENZO [D] [1,3] DIOXOL-5-YL) -CYCLOPROPANCARBOXAMIDO) -3-METHYLPYRIODIN-2-YL) BENZOIC ACID AND ADMINISTRATION |
| EP2560649A1 (en) * | 2010-04-22 | 2013-02-27 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions and administrations thereof |
| MX2012012204A (en) * | 2010-04-22 | 2012-12-05 | Vertex Pharma | Process of producing cycloalkylcarboxamido-indole compounds. |
| EP2560651A1 (en) * | 2010-04-22 | 2013-02-27 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions and administrations thereof |
| NZ603043A (en) * | 2010-04-22 | 2015-02-27 | Vertex Pharma | Pharmaceutical compositions comprising cftr modulators and administrations thereof |
| EP2386606B1 (en) | 2010-04-30 | 2018-03-21 | Total Marketing Services | Use of organogelator derivatives in bituminous compositions to improve the resistance thereof to chemical attacks |
| US8404849B2 (en) | 2010-05-20 | 2013-03-26 | Vertex Pharmaceuticals | Processes for producing modulators of cystic fibrosis transmembrane conductance regulator |
| US9051346B2 (en) | 2010-05-20 | 2015-06-09 | Cempra Pharmaceuticals, Inc. | Process for preparing triazole-containing ketolide antibiotics |
| AU2011255237A1 (en) | 2010-05-20 | 2012-11-29 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions and administrations thereof |
| US8563593B2 (en) * | 2010-06-08 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Formulations of (R)-1-(2,2-difluorobenzo[D] [1,3] dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| US20120149708A1 (en) | 2010-06-17 | 2012-06-14 | George Mason University | Modulators of viral transcription, and methods and compositions therewith |
| UA110113C2 (en) | 2010-07-29 | 2015-11-25 | BICYCLIC AZAGETEROCYCLIC CARBOXAMIDES | |
| WO2012016133A2 (en) | 2010-07-29 | 2012-02-02 | President And Fellows Of Harvard College | Ros1 kinase inhibitors for the treatment of glioblastoma and other p53-deficient cancers |
| MX2013002035A (en) * | 2010-08-23 | 2013-03-25 | Vertex Pharma | Pharmaceutical composition of (r)-1-(2,2-difluorobenzo[d][1,3]dio xol-5-yl)-n-(1-(2,3-dihydroxy propyl)-6-fluoro-2-(1-hydroxy-2-met hylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration therof. |
| RU2013113627A (en) | 2010-08-27 | 2014-10-10 | Вертекс Фармасьютикалз Инкорпорейтед | PHARMACEUTICAL COMPOSITION AND ITS INTRODUCTION |
| WO2012049555A1 (en) | 2010-10-13 | 2012-04-19 | Lupin Limited | Spirocyclic compounds as voltage-gated sodium channel modulators |
| US9394290B2 (en) | 2010-10-21 | 2016-07-19 | Universitaet Des Saarlandes Campus Saarbruecken | Selective CYP11B1 inhibitors for the treatment of cortisol dependent diseases |
| US8802700B2 (en) | 2010-12-10 | 2014-08-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-Binding Cassette transporters |
| WO2012079583A1 (en) | 2010-12-15 | 2012-06-21 | Aarhus Universitet | System providing controlled delivery of gaseous co for carbonylation reactions |
| WO2012116135A2 (en) | 2011-02-24 | 2012-08-30 | Emory University | Noggin blocking compositions for ossification and methods related thereto |
| US9464065B2 (en) | 2011-03-24 | 2016-10-11 | The Scripps Research Institute | Compounds and methods for inducing chondrogenesis |
| CN102731492B (en) | 2011-03-30 | 2016-06-29 | 江苏恒瑞医药股份有限公司 | Cyclohexanes derivant, its preparation method and in application pharmaceutically |
| HUE047354T2 (en) | 2011-05-18 | 2020-04-28 | Vertex Pharmaceuticals Europe Ltd | Deuterated derivatives of ivacaftor |
| US8945605B2 (en) | 2011-06-07 | 2015-02-03 | Parion Sciences, Inc. | Aerosol delivery systems, compositions and methods |
| WO2013005057A1 (en) | 2011-07-07 | 2013-01-10 | Centro Nacional De Investigaciones Oncológicas (Cnio) | New compounds |
| CN104159890B (en) | 2011-09-09 | 2018-04-10 | 蓝瑟斯医学影像公司 | Composition, method and system for synthesizing and using developer |
| WO2013038381A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine/pyrazine amide derivatives |
| EP2755652B1 (en) | 2011-09-16 | 2021-06-02 | Novartis AG | N-substituted heterocyclyl carboxamides |
| CN103946221B (en) | 2011-09-16 | 2016-08-03 | 诺华股份有限公司 | For treating the heterocyclic compound of cystic fibrosis |
| WO2013038373A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| WO2013038378A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| GB201116559D0 (en) | 2011-09-26 | 2011-11-09 | Univ Leuven Kath | Novel viral replication inhibitors |
| AU2012332225A1 (en) | 2011-11-02 | 2014-05-15 | Vertex Pharmaceuticals Incorporated | Use of (N- [2, 4 -bis (1, 1 -dimethylethyl) - 5 - hydroxyphenyl] - 1, 4 - dihydro - 4 - oxoquinoline - 3 - ca rboxamide) for treating CFTR mediated diseases |
| ME02650B (en) * | 2011-11-08 | 2017-06-20 | Vertex Pharma | Modulators of atp-binding cassette transporters |
| US20140127901A1 (en) * | 2012-11-08 | 2014-05-08 | Taiwan Semiconductor Manufacturing Company, Ltd. | Low-k damage free integration scheme for copper interconnects |
| WO2013086131A1 (en) | 2011-12-06 | 2013-06-13 | The Trustees Of The University Of Pennsylvania | Inhibitors targeting drug-resistant influenza a |
| US8772541B2 (en) | 2011-12-15 | 2014-07-08 | University of Pittsburgh—of the Commonwealth System of Higher Education | Cannabinoid receptor 2 (CB2) inverse agonists and therapeutic potential for multiple myeloma and osteoporosis bone diseases |
| EP2606726A1 (en) | 2011-12-21 | 2013-06-26 | Bayer CropScience AG | N-Arylamidine-substituted trifluoroethylsulfide derivatives as acaricides and insecticides |
| CA2862859C (en) | 2012-01-25 | 2022-08-02 | Vertex Pharmaceuticals Incorporated | Formulations of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| CN109966264A (en) | 2012-02-27 | 2019-07-05 | 沃泰克斯药物股份有限公司 | Pharmaceutical composition and its application |
| JP2015518504A (en) | 2012-04-03 | 2015-07-02 | スリーエム イノベイティブ プロパティズ カンパニー | Crosslinkable composition containing photobase generator |
| US8674108B2 (en) | 2012-04-20 | 2014-03-18 | Vertex Pharmaceuticals Incorporated | Solid forms of N-[2,4-bis(1,1-dimethylethy)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| US8888905B2 (en) | 2012-04-26 | 2014-11-18 | Xerox Corporation | Fast crystallizing crystalline-amorphous ink compositions and methods for making the same |
| US9528016B2 (en) | 2012-04-26 | 2016-12-27 | Xerox Corporation | Phase change inks comprising crystalline amides |
| US20130284054A1 (en) | 2012-04-26 | 2013-10-31 | Xerox Corporation | Rapid solidifying crystalline-amorphous inks |
| CA2813472A1 (en) | 2012-04-26 | 2013-10-26 | Xerox Corporation | Rapid solidifying crystalline-amorphous inks |
| US9228101B2 (en) | 2012-04-26 | 2016-01-05 | Xerox Corporation | Rapidly crystallizing phase change inks and methods for forming the same |
| GB2502624A (en) | 2012-06-01 | 2013-12-04 | Univ East Anglia | Phosphoramide and phosphoramide-based catalysts and their use in intermolecular aziridination |
| WO2013184198A1 (en) | 2012-06-08 | 2013-12-12 | Massachusetts Institute Of Technology | Phosphine-ligated palladium sulfonate palladacycles |
| AU2013270681A1 (en) | 2012-06-08 | 2014-12-18 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of CFTR -mediated disorders |
| FR2992317B1 (en) | 2012-06-22 | 2016-05-13 | Diverchim | PROCESS FOR THE PREPARATION OF CHIRAL PEPTIDES |
| WO2014002106A1 (en) | 2012-06-25 | 2014-01-03 | Cadila Healthcare Limited | Novel compounds for the treatment of dyslipidemia and related diseases |
| US20150209448A1 (en) | 2012-07-12 | 2015-07-30 | Proqr Therapeutics N.V. | Exon replacement with stabilized artificial rnas |
| EP2852668B1 (en) | 2012-07-12 | 2016-04-27 | ProQR Therapeutics II B.V. | Oligonucleotides for making a change in the sequence of a target rna molecule present in a living cell |
| AR092857A1 (en) | 2012-07-16 | 2015-05-06 | Vertex Pharma | PHARMACEUTICAL COMPOSITIONS OF (R) -1- (2,2-DIFLUOROBENZO [D] [1,3] DIOXOL-5-IL) -N- (1- (2,3-DIHYDROXIPROPIL) -6-FLUORO-2- ( 1-HYDROXI-2-METHYLPROPAN-2-IL) -1H-INDOL-5-IL) CYCLOPROPANCARBOXAMIDE AND ADMINISTRATION OF THE SAME |
| CN104812382A (en) | 2012-09-20 | 2015-07-29 | 坦普尔大学 | Substituted alkyl diaryl derivatives, methods of preparation and uses |
| CN104769465A (en) | 2012-10-29 | 2015-07-08 | 柯尼卡美能达株式会社 | Phase difference film, circularly polarizing plate, and image forming device |
| IL283276B2 (en) | 2012-11-02 | 2024-05-01 | Vertex Pharma | Compositions comprising 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid and n-(5-hydroxy-2,4-ditert-butyl-phenyl)-4-oxo-1h-quinoline-3-carboxamide and uses thereof |
| EP2919770A4 (en) | 2012-11-14 | 2017-03-08 | The Board of Regents of The University of Texas System | Inhibition of hif-2 heterodimerization with hif1 (arnt) |
| JP6146990B2 (en) | 2012-11-16 | 2017-06-14 | コンサート ファーマシューティカルズ インコーポレイテッド | Deuterated CFTR enhancer |
| BR112015011441A2 (en) | 2012-11-20 | 2017-07-11 | Merial Inc | anthelmintic compositions and compounds and methods of their use |
| ITMI20122065A1 (en) | 2012-12-03 | 2014-06-04 | Univ Padova | USE OF CFTR CORRECTORS IN THE TREATMENT OF STRUCTURAL MUSCLE PATHOLOGIES |
| EP2929346A1 (en) | 2012-12-05 | 2015-10-14 | Institut National de la Santé et de la Recherche Médicale (INSERM) | Diagnosis of cystic fibrosis |
| EP2928532A4 (en) | 2012-12-07 | 2016-06-29 | Parion Sciences Inc | Nasal cannula for delivery of aerosolized medicaments |
| US20140221424A1 (en) | 2013-01-30 | 2014-08-07 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for use in the treatment of cystic fibrosis |
| US9452139B2 (en) | 2013-03-14 | 2016-09-27 | Novartis Ag | Respirable agglomerates of porous carrier particles and micronized drug |
| US20140296164A1 (en) | 2013-03-29 | 2014-10-02 | Calista Therapeutics, Inc. | Compositions and methods of use for cell targeted inhibitors of the Cystic Fibrosis transmembrane regulator associated ligand |
| AU2014264936B2 (en) | 2013-05-07 | 2018-09-27 | Galapagos Nv | Novel compounds and pharmaceutical compositions thereof for the treatment of cystic fibrosis |
| AU2014268477A1 (en) | 2013-05-24 | 2015-11-12 | The California Institute For Biomedical Research | Compounds for treatment of drug resistant and persistent tuberculosis |
| JP2014232188A (en) | 2013-05-29 | 2014-12-11 | コニカミノルタ株式会社 | Cellulose acylate film, circularly polarizing plate and image display device |
| EP3013341A4 (en) | 2013-06-26 | 2017-02-08 | Proteostasis Therapeutics, Inc. | Methods of modulating cftr activity |
| RU2708690C2 (en) | 2013-08-08 | 2019-12-11 | Галапагос Нв | Derivatives of thieno[2,3-c]pyrans as cftr modulators |
| WO2015036552A1 (en) | 2013-09-12 | 2015-03-19 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for treatment of cystic fibrosis |
| JP6564380B2 (en) | 2013-09-20 | 2019-08-21 | ユニバーシティ オブ ピッツバーグ − オブ ザ コモンウェルス システム オブ ハイヤー エデュケイション | Compounds for treating prostate cancer |
| WO2015050984A1 (en) | 2013-10-01 | 2015-04-09 | New York University | Amino, amido, and heterocyclic compounds as modulators of rage activity and uses thereof |
| WO2015054337A1 (en) | 2013-10-09 | 2015-04-16 | Emory University | Heterocyclic coupling catalysts and methods related thereto |
| US10231932B2 (en) | 2013-11-12 | 2019-03-19 | Vertex Pharmaceuticals Incorporated | Process of preparing pharmaceutical compositions for the treatment of CFTR mediated diseases |
| KR20150062652A (en) | 2013-11-29 | 2015-06-08 | 삼성전자주식회사 | Sonosensitive liposome, a pharmaceutical composition comprising the same and a method of delivering an active agent into a subject using the same |
| JP6759100B2 (en) | 2013-12-30 | 2020-09-23 | ライフサイ ファーマシューティカルズ,インク. | Therapeutic inhibitor compound |
| AU2015226881A1 (en) | 2014-03-07 | 2016-09-29 | Intonation Research Laboratories | Inhibitors of histone lysine specific demethylase (LSD1) and histone deacetylases (HDACs) |
| JP2015172005A (en) | 2014-03-11 | 2015-10-01 | 国立大学法人 東京大学 | Method of producing coupling compound by iron catalyst |
| EP3116870A1 (en) | 2014-03-13 | 2017-01-18 | Proteostasis Therapeutics, Inc. | Compounds, compositions, and methods for increasing cftr activity |
| WO2015138909A1 (en) | 2014-03-13 | 2015-09-17 | Proteostasis Therapeutics, Inc. | Compounds, compositions, and methods for increasing cftr activity |
| ES2957761T3 (en) | 2014-04-15 | 2024-01-25 | Vertex Pharma | Pharmaceutical compositions for the treatment of diseases mediated by the cystic fibrosis transmembrane conductance regulator |
| AU2015255752B2 (en) | 2014-05-08 | 2020-07-23 | Immunoforge Co., Ltd. | Methods and compositions for treating Cystic Fibrosis |
| JP5932881B2 (en) * | 2014-05-08 | 2016-06-08 | 株式会社フジクラ | Multi-core fiber and method for producing the multi-core fiber |
| PL3142701T3 (en) | 2014-05-12 | 2018-11-30 | Verona Pharma Plc | New treatment |
| BR112016026378B8 (en) | 2014-05-16 | 2020-06-09 | Liqwd Inc | method for bleaching hair and using an active agent that is maleic acid to reduce or prevent damage to hair |
| AU2015264336B2 (en) | 2014-05-19 | 2018-08-30 | Boehringer Ingelheim Animal Health USA Inc. | Anthelmintic compounds |
| AU2015269598B2 (en) | 2014-06-05 | 2019-11-14 | Merck Patent Gmbh | Novel quinoline derivatives and their use in neurodegenerative diseases |
| WO2015196071A1 (en) | 2014-06-19 | 2015-12-23 | Proteostasis Therapeutics, Inc. | Compounds, compositions and methods of increasing cftr activity |
| WO2016025448A2 (en) | 2014-08-13 | 2016-02-18 | Akeso Biomedical, Inc. | Antimicrobial compounds and compositions, and uses thereof |
| GB201415381D0 (en) | 2014-08-29 | 2014-10-15 | Algipharma As | Inhalable powder formulations of alginate oligomers |
| SG10201901192TA (en) | 2014-09-10 | 2019-03-28 | Epizyme Inc | Smyd inhibitors |
| CN104725628B (en) | 2014-10-01 | 2018-04-17 | 厦门赛诺邦格生物科技股份有限公司 | A kind of single functionalization branched polyethylene glycol, preparation method and its bio-related substance containing degradable group |
| WO2016050208A1 (en) | 2014-10-01 | 2016-04-07 | 厦门赛诺邦格生物科技有限公司 | Bio-related substance modified by multifunctionalized polyethylene glycol derivative |
| WO2016050209A1 (en) | 2014-10-01 | 2016-04-07 | 厦门赛诺邦格生物科技有限公司 | Heterofunctionalized polyethylene glycol derivative, preparation method, and bio-related substance |
| WO2016050210A1 (en) | 2014-10-01 | 2016-04-07 | 厦门赛诺邦格生物科技有限公司 | Multifunctionalized polyethylene glycol derivative and preparation method therefor |
| WO2016054560A1 (en) | 2014-10-02 | 2016-04-07 | Flatley Discovery Lab | Isoxazole compounds and methods for the treatment of cystic fibrosis |
| SG10201913603QA (en) | 2014-10-06 | 2020-02-27 | Vertex Pharma | Modulators of cystic fibrosis transmembrane conductance regulator |
| US9573948B2 (en) | 2014-10-06 | 2017-02-21 | Flatley Discovery Lab | Triazolopyridine compounds and methods for the treatment of cystic fibrosis |
| CN107250113B (en) | 2014-10-07 | 2019-03-29 | 弗特克斯药品有限公司 | Co-crystals of modulators of cystic fibrosis transmembrane conductance regulator |
| US20160108406A1 (en) | 2014-10-08 | 2016-04-21 | University Of Iowa Research Foundation | Method of regulating cftr expression and processing |
| US20170281612A1 (en) | 2014-10-08 | 2017-10-05 | Nivalis Therapeutics, Inc. | Methods for the Treatment of Cystic Fibrosis |
| GB201418892D0 (en) | 2014-10-23 | 2014-12-10 | Proqr Therapeutics B V | DNA editing |
| EP3212201B1 (en) | 2014-10-28 | 2022-10-19 | BCI Pharma | Nucleoside kinase inhibitors |
| BR112017009194A2 (en) | 2014-10-31 | 2017-12-26 | Abbvie Sarl | substituted tetrahydropyran and method of use |
| PL3212189T3 (en) | 2014-10-31 | 2021-03-08 | AbbVie Overseas S.à r.l. | Substituted chromanes and method of use |
| WO2016086015A1 (en) | 2014-11-25 | 2016-06-02 | University Of Rochester | Myoglobin-based catalysts for carbene transfer reactions |
| MA41031A (en) | 2014-11-26 | 2017-10-03 | Catabasis Pharmaceuticals Inc | CYSTEAMINE-FATTY ACID CONJUGATES AND THEIR USE AS AUTOPHAGIC ACTIVATORS |
| WO2016086136A1 (en) | 2014-11-26 | 2016-06-02 | Catabasis Pharmaceuticals, Inc. | Fatty acid cysteamine conjugates of cftr modulators and their use in treating medical disorders |
| US10392378B2 (en) | 2014-12-23 | 2019-08-27 | Proteostasis Therapeutics, Inc. | Derivatives of 5-phenyl- or 5-heteroarylathiazol-2-carboxylic amide useful for the treatment of inter alia cystic fibrosis |
| CA2971855A1 (en) | 2014-12-23 | 2016-06-30 | Proteostasis Therapeutics, Inc. | Derivatives of 5-(hetero)arylpyrazol-3-carboxylic amide or 1-(hetero)aryltriazol-4-carboxylic amide useful for the treatment of inter alia cystic fibrosis |
| MA41253A (en) | 2014-12-23 | 2017-10-31 | Proteostasis Therapeutics Inc | COMPOUNDS, COMPOSITIONS AND PROCESSES TO INCREASE THE ACTIVITY OF CFTR |
| WO2016105468A1 (en) | 2014-12-23 | 2016-06-30 | Proteostasis Therapeutics, Inc. | Derivatives of 3-heteroarylisoxazol-5-carboxylic amide useful for the treatment of inter alia cystic fibrosis |
| AU2015370463B2 (en) | 2014-12-24 | 2020-12-24 | Kither Biotech S.R.L. | Novel PI3K gamma inhibitor peptide for treatment of respiratory system diseases |
| CA2970948A1 (en) | 2014-12-31 | 2016-07-07 | Auspex Pharmaceuticals, Inc. | Cyclopropanecarboxamide modulators of cystic fibrosis transmembrane conductance regulator |
| CN105753814A (en) | 2015-01-01 | 2016-07-13 | 成都贝斯凯瑞生物科技有限公司 | Substituted nitrogen heterocyclic derivative and application thereof |
| US20180147187A1 (en) | 2015-01-12 | 2018-05-31 | Proteostasis Therapeutics, Inc. | Compounds, compositions, and methods for increasing cftr activity |
| CA2981495C (en) | 2015-03-31 | 2023-09-26 | Vertex Pharmaceuticals (Europe) Limited | Deuterated vx-661 |
-
2011
- 2011-04-21 MX MX2012012204A patent/MX2012012204A/en active IP Right Grant
- 2011-04-21 CA CA3108488A patent/CA3108488A1/en not_active Abandoned
- 2011-04-21 MX MX2016009018A patent/MX353408B/en unknown
- 2011-04-21 EP EP18173910.3A patent/EP3381899B1/en active Active
- 2011-04-21 ES ES11729195.5T patent/ES2608474T3/en active Active
- 2011-04-21 CA CA2797118A patent/CA2797118C/en active Active
- 2011-04-21 NZ NZ734535A patent/NZ734535A/en unknown
- 2011-04-21 CN CN201180031319.5A patent/CN103038214B/en active Active
- 2011-04-21 SG SG2012078374A patent/SG184987A1/en unknown
- 2011-04-21 US US13/642,642 patent/US9035072B2/en active Active
- 2011-04-21 ES ES18173910T patent/ES2858351T3/en active Active
- 2011-04-21 BR BR112012027056A patent/BR112012027056B8/en not_active IP Right Cessation
- 2011-04-21 RU RU2012149691/04A patent/RU2569678C2/en active
- 2011-04-21 KR KR1020197015032A patent/KR20190061096A/en not_active Application Discontinuation
- 2011-04-21 KR KR1020167010394A patent/KR101984225B1/en active IP Right Grant
- 2011-04-21 RU RU2015145784A patent/RU2745977C2/en active
- 2011-04-21 JP JP2013506299A patent/JP2013525371A/en active Pending
- 2011-04-21 CN CN201510526116.3A patent/CN105130948A/en active Pending
- 2011-04-21 EP EP16154612.2A patent/EP3045452A1/en not_active Withdrawn
- 2011-04-21 EP EP11729195.5A patent/EP2560954B1/en active Active
- 2011-04-21 AU AU2011242712A patent/AU2011242712B2/en active Active
- 2011-04-21 WO PCT/US2011/033396 patent/WO2011133751A2/en active Application Filing
- 2011-04-21 SG SG10201505700QA patent/SG10201505700QA/en unknown
- 2011-04-21 MX MX2015003252A patent/MX342288B/en unknown
- 2011-04-21 KR KR1020127030452A patent/KR20130056244A/en not_active Application Discontinuation
- 2011-04-21 NZ NZ603721A patent/NZ603721A/en unknown
- 2011-04-21 SG SG10201913594UA patent/SG10201913594UA/en unknown
- 2011-04-22 TW TW100114198A patent/TWI518082B/en active
- 2011-04-22 TW TW104142162A patent/TWI561518B/en active
- 2011-04-22 TW TW105122115A patent/TWI620744B/en active
- 2011-04-25 AR ARP110101412A patent/AR081333A1/en not_active Application Discontinuation
-
2012
- 2012-10-18 IL IL222539A patent/IL222539A0/en unknown
-
2014
- 2014-12-11 IL IL236209A patent/IL236209B/en active IP Right Grant
-
2015
- 2015-04-15 US US14/687,286 patent/US10071979B2/en active Active
- 2015-05-22 JP JP2015104812A patent/JP2015166382A/en not_active Withdrawn
-
2016
- 2016-04-22 AU AU2016202569A patent/AU2016202569B2/en active Active
- 2016-06-07 HK HK16106480.6A patent/HK1218419A1/en unknown
- 2016-12-07 HK HK16113950A patent/HK1225721A1/en unknown
-
2017
- 2017-01-25 JP JP2017011298A patent/JP6484652B2/en active Active
-
2018
- 2018-08-09 US US16/059,724 patent/US20190210991A1/en not_active Abandoned
- 2018-11-09 JP JP2018210982A patent/JP6714673B2/en active Active
-
2019
- 2019-08-27 IL IL268953A patent/IL268953B/en active IP Right Grant
-
2020
- 2020-08-26 US US17/003,051 patent/US20210238158A1/en not_active Abandoned
Patent Citations (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004028480A2 (en) | 2002-09-30 | 2004-04-08 | The Regents Of The University Of California | Cystic fibrosis transmembrane conductance regulator protein inhibitors and uses thereof |
| WO2004080972A1 (en) | 2003-03-12 | 2004-09-23 | Vertex Pharmaceuticals Incorporated | Pirazole modulators of atp-binding cassette transporters |
| US20060148864A1 (en) | 2003-04-11 | 2006-07-06 | Ptc Therapeutics, Inc. | 3-[5-(2-Fluoro-phenyl)-[1,2,4]oxadiazol-3-yl]-benzoic acid, compositions, and methods for the use thereof |
| WO2004091502A2 (en) | 2003-04-11 | 2004-10-28 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds |
| US20050164973A1 (en) | 2003-04-11 | 2005-07-28 | Pct Therapeutics, Inc. | 1,2,4-Oxadiazole benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| US7202262B2 (en) | 2003-04-11 | 2007-04-10 | Ptc Therapeutics, Inc. | Benzoic acid or benzoate substituted 1,2,4-oxadiazole compounds and their use for the treatment of disease |
| US20060148863A1 (en) | 2003-04-11 | 2006-07-06 | Ptc Therapeutics, Inc. | Substituted 1,2,4-oxadiazoles, compositions and methods of use |
| US6992096B2 (en) | 2003-04-11 | 2006-01-31 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| US20060035943A1 (en) | 2003-04-11 | 2006-02-16 | Ptc Therapeutics, Inc. | 1,2,4-Oxadiazole benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| WO2004110352A2 (en) | 2003-05-16 | 2004-12-23 | The Regents Of The University Of California | Compounds having activity in increasing ion transport by mutant-cftr and uses thereof |
| WO2004111014A1 (en) | 2003-06-06 | 2004-12-23 | Vertex Pharmaceuticals Incorporated | Pyrimidine derivatives as modulators of atp-binding cassette transporters |
| WO2005035514A2 (en) | 2003-10-08 | 2005-04-21 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters containing cycloalkyl or pyranyl groups |
| WO2005049018A1 (en) | 2003-11-14 | 2005-06-02 | Vertex Pharmaceuticals Incorporated | Thiazoles and oxazoles useful as modulators of atp-binding cassette transporters |
| WO2005094374A2 (en) | 2004-03-30 | 2005-10-13 | The Regents Of The University Of California | Hydrazide-containing cftr inhibitor compounds and uses thereof |
| WO2005120497A2 (en) | 2004-06-04 | 2005-12-22 | The Regents Of The University Of California | Compounds having activity in increasing ion transport by mutant-cftr and uses thereof |
| WO2006044503A2 (en) | 2004-10-13 | 2006-04-27 | Ptc Therapeutics, Inc. | Compounds for nonsense suppression, use of these compounds for the manufacture of a medicament for treating somatic mutation-related diseases |
| WO2006044682A1 (en) | 2004-10-13 | 2006-04-27 | Ptc Therapeutics, Inc. | Compounds for nonsense suppression, and methods for their use |
| WO2006044505A2 (en) | 2004-10-13 | 2006-04-27 | Ptc Therapeutics, Inc. | Compounds for nonsense suppression, and methods for their use |
| WO2006044456A1 (en) | 2004-10-13 | 2006-04-27 | Ptc Therapeutics, Inc. | Compounds for nonsense suppression, and methods for their use |
| WO2006044502A2 (en) | 2004-10-13 | 2006-04-27 | Ptc Therapeutics, Inc. | Pyrazole or triazole compounds and their use for the manufacture of a medicament for treating somatic mutation-related diseases |
| WO2006099256A2 (en) | 2005-03-11 | 2006-09-21 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2006101740A2 (en) | 2005-03-18 | 2006-09-28 | The Regents Of The University Of California | Compounds having activity in correcting mutant-cftr processing and uses thereof |
| WO2006110483A1 (en) | 2005-04-08 | 2006-10-19 | Ptc Therapeutics, Inc. | Compositions of an orally active 1,2,4-oxadiazole for nonsense mutation suppression therapy |
| WO2006127588A2 (en) | 2005-05-24 | 2006-11-30 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2007044560A2 (en) | 2005-10-06 | 2007-04-19 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| US20090131492A1 (en) | 2006-04-07 | 2009-05-21 | Ruah Sara S Hadida | Indole derivatives as CFTR modulators |
Non-Patent Citations (20)
| Title |
|---|
| ARIDOR M ET AL., NATURE MED., vol. 5, no. 7, 1999, pages 745 - 751 |
| BROSS P. ET AL., HUMAN MUT., vol. 14, 1999, pages 186 - 198 |
| CUTTING, G. R. ET AL., NATURE, vol. 346, 1990, pages 366 - 369 |
| DALEMANS ET AL., NATURE LOND., vol. 354, 1991, pages 526 - 528 |
| DEAN, M. ET AL., CELL, vol. 61, 1990 |
| E. W. MARTIN: "Remington's Pharmaceutical Sciences", 1980, MACK PUBLISHING CO. |
| GONZALEZ, J. E., K. OADES ET AL.: "Cell-based assays and instrumentation for screening ion-channel targets", DRUG DISCOV TODAY, vol. 4, no. 9, 1999, pages 431 - 439, XP001026838, DOI: doi:10.1016/S1359-6446(99)01383-5 |
| GONZALEZ, J. E., R. Y. TSIEN: "Improved indicators of cell membrane potential that use fluorescence resonance energy transfer", CHEM BIOL, vol. 4, no. 4, 1997, pages 269 - 77, XP000961796, DOI: doi:10.1016/S1074-5521(97)90070-3 |
| GONZALEZ, J. E., R. Y. TSIEN: "Voltage sensing by fluorescence resonance energy transfer in single cells", BIOPHYS J, vol. 69, no. 4, 1995, pages 1272 - 80, XP000961694 |
| GREGORY, R. J. ET AL., NATURE, vol. 347, 1990, pages 382 - 386 |
| KEREM, B-S ET AL., PROC. NATL. ACAD. SCI. USA, vol. 87, 1990, pages 8447 - 8451 |
| KEREM, B-S. ET AL., SCIENCE, vol. 245, 1989, pages 1073 - 1080 |
| LEE R. CHOO-KANG, PAMELA L.: "Zeitlin, Type l, II, IlL IV, and V cystic fibrosis Tansmembrane Conductance Regulator Defects and Opportunities of Therapy", CURRENT OPINION IN PULMONARY MEDICINE, vol. 6, 2000, pages 521 - 529 |
| MORELLO, JP ET AL., TIPS, vol. 21, 2000, pages 466 - 469 |
| PASYK, FOSKETT, J. CELL. BIOCHEM., vol. 270, 1995, pages 12347 - 50 |
| QUINTON, P. M., FASEB J., vol. 4, 1990, pages 2709 - 2727 |
| RICH, D. P. ET AL., NATURE, vol. 347, 1990, pages 358 - 362 |
| RIORDAN, J. R. ET AL., SCIENCE, vol. 245, 1989, pages 1066 - 1073 |
| RUTISHAUSER, J. ET AL., SWISS MED WKLY, vol. 132, 2002, pages 211 - 222 |
| SHASTRY, B.S. ET AL., NEUROCHEM. INTERNATIONAL, vol. 43, 2003, pages 1 - 7 |
Cited By (67)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10975061B2 (en) | 2006-04-07 | 2021-04-13 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US11639347B2 (en) | 2006-04-07 | 2023-05-02 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| EP3835297A1 (en) * | 2010-03-25 | 2021-06-16 | Vertex Pharmaceuticals Incorporated | Synthesis and intermediates of (r)-1(2,2 -difluorobenzo[d][1,3]dioxol-5yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2yl)-1h-indol-5yl)cyclopropanecarboxamide |
| US11578062B2 (en) | 2010-03-25 | 2023-02-14 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| US10906891B2 (en) | 2010-03-25 | 2021-02-02 | Vertex Pharmaceuticals Incoporated | Solid forms of (R)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| JP2014534236A (en) * | 2011-11-08 | 2014-12-18 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Modifiers for ATP-binding cassette transporters |
| US8937178B2 (en) | 2013-03-13 | 2015-01-20 | Flatley Discovery Lab | Phthalazinone compounds and methods for the treatment of cystic fibrosis |
| US9783529B2 (en) | 2013-03-13 | 2017-10-10 | Flatley Discovery Lab, Llc | Pyridazinone compounds and methods for the treatment of cystic fibrosis |
| US9790215B2 (en) | 2013-03-13 | 2017-10-17 | Flatley Discovery Lab, Llc | Pyridazinone compounds and methods for the treatment of cystic fibrosis |
| US11951212B2 (en) | 2014-04-15 | 2024-04-09 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US10980746B2 (en) | 2014-04-15 | 2021-04-20 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US11992553B2 (en) | 2014-08-29 | 2024-05-28 | Algipharma As | Inhalable powder formulations of alginate oligomers |
| US11426407B2 (en) | 2014-10-06 | 2022-08-30 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US10758534B2 (en) | 2014-10-06 | 2020-09-01 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US10689370B2 (en) | 2014-12-31 | 2020-06-23 | Auspex Pharmaceuticals, Inc. | Cyclopropane carboxamide modulators of cystic fibrosis transmembrane conductance regulator |
| US10167278B2 (en) | 2014-12-31 | 2019-01-01 | Auspex Pharmaceuticals, Inc. | Cyclopropanecarboxamide modulators of cystic fibrosis transmembrane conductance regulator |
| US10751363B2 (en) | 2015-03-23 | 2020-08-25 | Algipharma As | Use of aliginate oligomers and CFTR modulators in treatment of conditions associated with CFTR dysfunction |
| WO2016160945A1 (en) | 2015-03-31 | 2016-10-06 | Concert Pharmaceuticals, Inc. | Deuterated vx-661 |
| US11413306B2 (en) | 2015-10-06 | 2022-08-16 | Algipharma As | Alginate oligomers for the treatment or prevention of microbial overgrowth in the intestinal tract |
| US10093666B2 (en) | 2016-04-13 | 2018-10-09 | Arixa Pharmaceuticals, Inc. | Deuterated O-sulfated beta lactam hydroxamic acids and deuterated N-sulfated beta lactams |
| US10047077B2 (en) | 2016-04-13 | 2018-08-14 | Skyline Antiinfectives, Inc. | Deuterated O-sulfated beta-lactam hydroxamic acids and deuterated N-sulfated beta-lactams |
| US11186566B2 (en) | 2016-09-30 | 2021-11-30 | Vertex Pharmaceuticals Incorporated | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| US10793547B2 (en) | 2016-12-09 | 2020-10-06 | Vertex Pharmaceuticals Incorporated | Modulator of the cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| US11453655B2 (en) | 2016-12-09 | 2022-09-27 | Vertex Pharmaceuticals Incorporated | Modulator of the cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| WO2018227049A1 (en) | 2017-06-08 | 2018-12-13 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US11253509B2 (en) | 2017-06-08 | 2022-02-22 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2019010092A1 (en) | 2017-07-01 | 2019-01-10 | Vertex Pharmaceuticals Incorporated | Compositions and methods for treatment of cystic fibrosis |
| US11517564B2 (en) | 2017-07-17 | 2022-12-06 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2019018353A1 (en) | 2017-07-17 | 2019-01-24 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2019018395A1 (en) | 2017-07-17 | 2019-01-24 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US11434201B2 (en) | 2017-08-02 | 2022-09-06 | Vertex Pharmaceuticals Incorporated | Processes for preparing pyrrolidine compounds |
| US11155533B2 (en) | 2017-10-19 | 2021-10-26 | Vertex Pharmaceuticals Incorporated | Crystalline forms and compositions of CFTR modulators |
| US11465985B2 (en) | 2017-12-08 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| US11179367B2 (en) | 2018-02-05 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for treating cystic fibrosis |
| US11066417B2 (en) | 2018-02-15 | 2021-07-20 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulators |
| US11866450B2 (en) | 2018-02-15 | 2024-01-09 | Vertex Pharmaceuticals Incorporated | Modulators of Cystic Fibrosis Transmembrane Conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulators |
| US11414439B2 (en) | 2018-04-13 | 2022-08-16 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| WO2020102346A1 (en) | 2018-11-14 | 2020-05-22 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| EP4218754A2 (en) | 2018-11-14 | 2023-08-02 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US10875846B2 (en) | 2019-01-15 | 2020-12-29 | Apotex Inc. | Processes for the preparation of Tezacaftor and intermediates thereof |
| WO2020214921A1 (en) | 2019-04-17 | 2020-10-22 | Vertex Pharmaceuticals Incorporated | Solid forms of modulators of cftr |
| US11591350B2 (en) | 2019-08-14 | 2023-02-28 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2021030556A1 (en) | 2019-08-14 | 2021-02-18 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2021030555A1 (en) | 2019-08-14 | 2021-02-18 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US11584761B2 (en) | 2019-08-14 | 2023-02-21 | Vertex Pharmaceuticals Incorporated | Process of making CFTR modulators |
| US11873300B2 (en) | 2019-08-14 | 2024-01-16 | Vertex Pharmaceuticals Incorporated | Crystalline forms of CFTR modulators |
| WO2021030552A1 (en) | 2019-08-14 | 2021-02-18 | Vertex Pharmaceuticals Incorporated | Crystalline forms of cftr modulators |
| WO2022032068A1 (en) | 2020-08-07 | 2022-02-10 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022036060A1 (en) | 2020-08-13 | 2022-02-17 | Vertex Pharmaceuticals Incorporated | Crystalline forms of cftr modulators |
| WO2022076618A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076624A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076620A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076627A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076622A2 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076621A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076625A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076628A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076629A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076626A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022125826A1 (en) | 2020-12-10 | 2022-06-16 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2023150236A1 (en) | 2022-02-03 | 2023-08-10 | Vertex Pharmaceuticals Incorporated | Methods of preparing and crystalline forms of (6a,12a)-17-amino-12-methyl-6,15-bis(trifluoromethyl)-13,19-dioxa-3,4,18-triazatricyclo[ 12.3.1.12,5]nonadeca-1(18),2,4,14,16-pentaen-6-ol |
| WO2023150237A1 (en) | 2022-02-03 | 2023-08-10 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2023154291A1 (en) | 2022-02-08 | 2023-08-17 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2023196429A1 (en) | 2022-04-06 | 2023-10-12 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2023224924A1 (en) | 2022-05-16 | 2023-11-23 | Vertex Pharmaceuticals Incorporated | Solid forms of a macrocyclic compounds as cftr modulators and their preparation |
| WO2023224931A1 (en) | 2022-05-16 | 2023-11-23 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US12122788B2 (en) | 2023-01-04 | 2024-10-22 | Vertex Pharmaceuticals Incorporated | Process of making CFTR modulators |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2016202569B2 (en) | Process of producing cycloalkylcarboxamido-indole compounds | |
| EP2555754B1 (en) | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid | |
| AU2015221470B2 (en) | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180031319.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11729195 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2013506299 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2012/012204 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2797118 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3253/KOLNP/2012 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2011242712 Country of ref document: AU Date of ref document: 20110421 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20127030452 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2012149691 Country of ref document: RU Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011729195 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011729195 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012027056 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13642642 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 112012027056 Country of ref document: BR Kind code of ref document: A2 Effective date: 20121022 |