WO2011115817A1 - Methods of preparing 2'-o-substituted purine nucleosides - Google Patents
Methods of preparing 2'-o-substituted purine nucleosides Download PDFInfo
- Publication number
- WO2011115817A1 WO2011115817A1 PCT/US2011/027953 US2011027953W WO2011115817A1 WO 2011115817 A1 WO2011115817 A1 WO 2011115817A1 US 2011027953 W US2011027953 W US 2011027953W WO 2011115817 A1 WO2011115817 A1 WO 2011115817A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- group
- groups
- alkyl
- nucleoside
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/067—Pyrimidine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon, or a metal, e.g. chelates, vitamin B12
Definitions
- the methods are particularly useful in that the products are crystalline enabling purification without chromatography.
- Oligonucleotides have been used in various biological and biochemical applications. They have been used as primers and probes for the polymerase chain reaction (PCR), as antisense agents used in target validation, drug discovery and development, as ribozymes, as aptamers, and as general stimulators of the immune system. This widespread use of oligonucleotides has led to an increasing demand for rapid, inexpensive and efficient methods for their synthesis.
- PCR polymerase chain reaction
- Synthetic oligonucleotides are generally prepared through the repeated coupling reactions of nucleoside phosphoramidites to the 5'-hydroxyl group of a nucleoside monomer or the free 5'- hydroxyl group of a growing oligomer.
- the most commonly used method to perform oligomer synthesis is the phosphoramidite approach which is largely based on developments reported in the literature (see for example: Beaucage and Caruthers (1981) Tetrahedron Letters 22:1859-1862; McBride and Caruthers (1983) Tetrahedron Letters 24:245-248; Sinha et al. (1984) Nucleic Acids Res. 12:4539-4557 and Beaucage and Iyer (1992) Tetrahedron 48:2223-2311, each of which is incorporated herein by reference in its entirety).
- Oligomer synthesis can be performed using solution or solid phase chemistries.
- Solid phase oligonucleotide synthesis is the preferred method.
- SPOS Solid phase oligonucleotide synthesis
- oligonucleotides are assembled in a cyclical manner, each cycle consisting of a series of three chemical reactions.
- the first reaction is a deblocking reaction, i.e. the removal of a hydroxyl protecting group from a nucleoside monomer or an oligomer bound to a support. Generally, this requires the removal of a dimethoxytrityl protecting group to provide a free hydroxyl group.
- the second reaction is the coupling reaction, normally performed in the presence of an activator, wherein the free hydroxyl group is reacted with a nucleoside phosphoramidite to provide a phosphite triester in the presence of an activator.
- the third reaction is the oxidation of the phosphite triester to a phosphate triester.
- a capping step is included either directly before or after each oxidation reaction in order to block support bound nucleoside monomers or oligomers which failed to react in the coupling reaction and to prevent them from further chain elongation in subsequent coupling steps.
- a major limiting factor for the cost efficient synthesis of oligonucleotides is the time and cost required to prepare nucleosides, including especially modified nucleosides, and their purification and conversion into phosphoramidites.
- the standard method used in the industry for purifying nucleoside phosphoramidites and the intermediates formed during their synthesis is column chromatography, especially flash column chromatography which primarily uses silica gel. Regardless of which particular type of column chromatography is used, purification by column chromatography requires large amounts of support material (silica gel for example) and large volumes of high purity solvents. The process also requires a lot of time as it is labor-intensive. Purine nucleosides tend to be more difficult to prepare and purify than pyrimidines.
- nucleosides especially modified nucleosides such as 2'-0-substituted purine nucleosides. Provided herein are methods that fulfill this need.
- a 2'-0-substituted purine nucleoside comprising: contacting a nucleoside having Formula I:
- Pu is an unprotected purine having at least one exocyclic amino group
- Z is C C 6 alkyl or substituted C C 6 alkyl, C 2 -C 6 alkenyl, substituted C 2 -C 6 alkenyl, C 2 -C 6 alkynyl or substituted C 2 -C 6 alkynyl;
- each 31 andJ 2 is, independently, H, Cj-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, Ci-C 6 aminoalkyl or a protecting group.
- the methods provided herein further comprise contacting the 2'-0- substituted purine nucleoside with a reagent capable of protecting any exocyclic amino groups located on the purine. In certain embodiments, the methods provided herein further comprise contacting the 2'-0-substituted purine nucleoside with a reagent capable of protecting any exocyclic amino groups located on the purine and or contacting the 2'-0-substituted purine nucleoside with a reagent capable of removing the di-t-butyl silyl protecting group.
- the reagent used to protect exocyclic amino groups on the base is isobutyryl chloride. In certain embodiments, the reagent used to remove the di-t-butyl silyl protecting group is triethylamine trihydrofluoride.
- the unprotected purine is guanine, adenine or diaminopurine. In certain embodiments, the unprotected purine is guanine.
- the di-t-butyl silyl protecting reagent is tBu 2 Si(OTf) 2 .
- the alkylating agent comprises a C ! -C 6 alkyl or substituted Cj-C 6 alkyl and a leaving group. In certain embodiments, the alkylating agent comprises a substituted C ⁇ - C 6 alky group and a leaving group. In certain embodiments, the alkylating agent comprises 1-iodo- 2-methoxyethane.
- the 2'-0-substituted purine nucleoside has the configuration of Formula IV:
- the methods are particularly useful in that the products are crystalline enabling purification without chromatography.
- the methods include selecting a nucleoside having Formula I:
- Pu is an unprotected purine having at least one exocyclic amino group
- nucleoside having Formula II is further contacted with an alkylating agent under conditions to provide 2'-0-substituted purine nucleoside having Formula III:
- the alkylating agent can be selected and protected if needed to provide a desired Z group.
- each Ji and J 2 is, independently, H, Q-Q alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C ! -C 6 aminoalkyl or a protecting group.
- the methods provided herein are useful for the preparation of 2'-0-substituted purine nucleosides having at least one exocyclic amino group on the base.
- the methods provide for protection of the sugar 3' and 5' hydroxyl groups and subsequent alkylation of the 2' hydroxyl without having to protect base amino groups.
- Representative purine bases that have exocyclic amino groups include without limitation guanine, adenine and diaminopurine.
- alkylating agent will determine the 2'-0-substitutent (Z).
- the 2'-0-substituted purine nucleoside prepared using the methods provided herein can be further contacted with a reagent capable of protecting any exocyclic amino groups located on the purine.
- the 2'-0-substituted purine nucleoside prepared using the methods provided herein can be further contacted with a reagent capable of protecting any exocyclic amino groups located on the purine and or contacted with a reagent capable of removing the di-t-butyl silyl protecting group.
- the 2'-0-substituted purine nucleosides prepared here have the configuration of Formula IV:
- substituted and substituteduent group are meant to include groups that are typically added to other groups or parent compounds to enhance desired properties or provide other desired effects. Substituent groups can be protected or unprotected and can be added to one available site or to many available sites in a parent compound. Substituent groups may also be further substituted with other substituent groups and may be attached directly or via a linking group such as an alkyl or hydrocarbyl group to a parent compound.
- each R aa , R bb and R cC is, independently, H, an optionally linked chemical functional group or a further substituent group with a preferred list including without limitation, H, alkyl, alkenyl, alkynyl, aliphatic, alkoxy, acyl, aryl, aralkyl, heteroaryl, alicyclic, heterocyclic and heteroarylalkyl. Selected substituents within the compounds described herein are present to a recursive degree.
- alkyl refers to a saturated straight or branched hydrocarbon radical containing up to twenty four carbon atoms.
- alkyl groups include without limitation, methyl, ethyl, propyl, butyl, isopropyl, n-hexyl, octyl, decyl, dodecyl and the like.
- Alkyl groups typically include from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms (Q-C ⁇ alkyl) with from 1 to about 6 carbon atoms being more preferred.
- the term "lower alkyl” as used herein includes from 1 to about 6 carbon atoms.
- Alkyl groups as used herein may optionally include one or more further substituent groups.
- alkenyl refers to a straight or branched hydrocarbon chain radical containing up to twenty four carbon atoms and having at least one carbon-carbon double bond.
- alkenyl groups include without limitation, ethenyl, propenyl, butenyl, l-methyl-2- buten-l-yl, dienes such as 1,3 -butadiene and the like.
- Alkenyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred.
- Alkenyl groups as used herein may optionally include one or more further substituent groups.
- alkynyl refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms and having at least one carbon-carbon triple bond.
- alkynyl groups include, without limitation, ethynyl, 1-propynyl, 1-butynyl, and the like.
- Alkynyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred.
- Alkynyl groups as used herein may optionally include one or more further substituent groups.
- acyl refers to a radical formed by removal of a hydroxyl group from an organic acid and has the general Formula -C(0)-X where X is typically aliphatic, alicyclic or aromatic. Examples include aliphatic carbonyls, aromatic carbonyls, aliphatic sulfonyls, aromatic sulfinyls, aliphatic sulfinyls, aromatic phosphates, aliphatic phosphates and the like. Acyl groups as used herein may optionally include further substituent groups.
- alicyclic refers to a cyclic ring system wherein the ring is aliphatic.
- the ring system can comprise one or more rings wherein at least one ring is aliphatic.
- Preferred alicyclics include rings having from about 5 to about 9 carbon atoms in the ring.
- Alicyclic as used herein may optionally include further substituent groups.
- aliphatic refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms wherein the saturation between any two carbon atoms is a single, double or triple bond.
- An aliphatic group preferably contains from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms with from 1 to about 6 carbon atoms being more preferred.
- the straight or branched chain of an aliphatic group may be interrupted with one or more heteroatoms that include nitrogen, oxygen, sulfur and phosphorus.
- Such aliphatic groups interrupted by heteroatoms include without limitation, polyalkoxys, such as polyalkylene glycols, polyamines, and polyimines. Aliphatic groups as used herein may optionally include further substituent groups.
- alkoxy refers to a radical formed between an alkyl group and an oxygen atom wherein the oxygen atom is used to attach the alkoxy group to a parent molecule.
- alkoxy groups include without limitation, methoxy, ethoxy, propoxy, isopropoxy, n- butoxy, sec-butoxy, tert-butoxy, n-pentoxy, neopentoxy, n-hexoxy and the like.
- Alkoxy groups as used herein may optionally include further substituent groups.
- aminoalkyl refers to an amino substituted C ⁇ -Cn alkyl radical.
- the alkyl portion of the radical forms a covalent bond with a parent molecule.
- the amino group can be located at any position and the aminoalkyl group can be substituted with a further substituent group at the alkyl and/or amino portions.
- aralkyl and arylalkyl refer to an aromatic group that is covalently linked to a C ⁇ -Cn alkyl radical.
- the alkyl radical portion of the resulting aralkyl (or arylalkyl) group forms a covalent bond with a parent molecule. Examples include without limitation, benzyl, phenethyl and the like.
- Aralkyl groups as used herein may optionally include further substituent groups attached to the alkyl, the aryl or both groups that form the radical group.
- aryl and aromatic refer to a mono- or polycyclic carbocyclic ring system radicals having one or more aromatic rings.
- aryl groups include without limitation, phenyl, naphthyl, tetrahydronaphthyl, indanyl, idenyl and the like.
- Preferred aryl ring systems have from about 5 to about 20 carbon atoms in one or more rings.
- Aryl groups as used herein may optionally include further substituent groups.
- halo and halogen, as used herein, refer to an atom selected from fluorine, chlorine, bromine and iodine.
- heteroaryl refers to a radical comprising a mono- or poly-cyclic aromatic ring, ring system or fused ring system wherein at least one of the rings is aromatic and includes one or more heteroatoms. Heteroaryl is also meant to include fused ring systems including systems where one or more of the fused rings contain no heteroatoms.
- Heteroaryl groups typically include one ring atom selected from sulfur, nitrogen or oxygen.
- heteroaryl groups include without limitation, pyridinyl, pyrazinyl, pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isooxazolyl, thiadiazolyl, oxadiazolyl, thiophenyl, furanyl, quinolinyl, isoquinolinyl, benzimidazolyl, benzooxazolyl, quinoxalinyl and the like.
- Heteroaryl radicals can be attached to a parent molecule directly or through a linking moiety such as an aliphatic group or hetero atom.
- Heteroaryl groups as used herein may optionally include further substituent groups.
- heteroarylalkyl refers to a heteroaryl group as previously defined that further includes a covalently attached Cj-C 12 alkyl radical.
- the alkyl radical portion of the resulting heteroarylalkyl group is capable of forming a covalent bond with a parent molecule.
- heteroarylalkyl groups as used herein may optionally include further substituent groups on one or both of the heteroaryl or alkyl portions.
- Linking groups or bifunctional linking moieties such as those known in the art are useful for attachment of chemical functional groups, conjugate groups, reporter groups and other groups to selective sites in a parent compound such as for example an oligomeric compound.
- a bifunctional linking moiety comprises a hydrocarbyl moiety having two functional groups. One of the functional groups is selected to bind to a parent molecule or compound of interest and the other is selected to bind to essentially any selected group such as a chemical functional group or a conjugate group.
- the linker comprises a chain structure or a polymer of repeating units such as ethylene glycols or amino acid units.
- bifunctional linking moieties examples include without limitation, electrophiles for reacting with nucleophilic groups and nucleophiles for reacting with electrophilic groups.
- bifunctional linking moieties include amino, hydroxyl, carboxylic acid, thiol, unsaturations (e.g., double or triple bonds), and the like.
- Some nonlimiting examples of bifunctional linking moieties include 8-amino-3,6-dioxaoctanoic acid (ADO), succinimidyl 4-(N- maleimidomethyl) cyclohexane-l-carboxylate (SMCC) and 6-aminohexanoic acid (AHEX or AHA).
- linking groups include without limitation, substituted Cj-Cio alkyl, substituted or unsubstituted C 2 -C 10 alkenyl or substituted or unsubstituted C 2 -C 10 alkynyl, wherein a nonlimiting list of preferred substituent groups includes hydroxyl, amino, alkoxy, carboxy, benzyl, phenyl, nitro, thiol, thioalkoxy, halogen, alkyl, aryl, alkenyl and alkynyl.
- protecting group refers to a labile chemical moiety which is known in the art to protect reactive groups including without limitation, hydroxyl, amino and thiol groups, against undesired reactions during synthetic procedures.
- Protecting groups are typically used selectively and/or orthogonally to protect sites during reactions at other reactive sites and can then be removed to leave the unprotected group as is or available for further reactions.
- Protecting groups as known in the art are described generally in Greene's Protective Groups in Organic
- Groups can be selectively incorporated into oligomeric compounds as provided herein as precursors.
- an amino group can be placed into a compound as provided herein as an azido group that can be chemically converted to the amino group at a desired point in the synthesis.
- groups are protected or present as precursors that will be inert to reactions that modify other areas of the parent molecule for conversion into their final groups at an appropriate time. Further representative protecting or precursor groups are discussed in Agrawal et al., Protocols for Oligonucleotide Conjugates, Humana Press; New Jersey, 1994, 26, 1-72.
- orthogonal protected refers to functional groups which are protected with different classes of protecting groups, wherein each class of protecting group can be removed in any order and in the presence of all other classes (see, Barany et al, J. Am. Chem. Soc, 1977, 99, 7363- 7365; Barany et al, J. Am. Chem. Soc, 1980, 102, 3084-3095).
- Orthogonal protection is widely used in for example automated oligonucleotide synthesis.
- a functional group is deblocked in the presence of one or more other protected functional groups which is not affected by the deblocking procedure. This deblocked functional group is reacted in some manner and at some point a further orthogonal protecting group is removed under a different set of reaction conditions. This allows for selective chemistry to arrive at a desired compound or oligomeric compound.
- hydroxyl protecting groups include without limitation, acetyl, t-butyl, t- butoxymethyl, methoxymethyl, tetrahydropyranyl, 1-ethoxyethyl, l-(2-chloroethoxy)ethyl, p- chlorophenyl, 2,4-dinitrophenyl, benzyl, 2,6-dichlorobenzyl, diphenylmethyl, p-nitrobenzyl, bis(2- acetoxyethoxy)methyl (ACE), 2-trimethylsilylethyl, trimethylsilyl, triethylsilyl, t-butyldimethylsilyl, t-butyldiphenylsilyl, triphenylsilyl, [(triisopropylsilyl)oxy]methyl (TOM), benzoylformate, chloroacetyl, trichloroacetyl, trifluoroacetyl, pivaloyl, benzo
- hydroxyl protecting groups include without limitation, benzyl, 2,6-dichlorobenzyl, t-butyldimethylsilyl, t-butyldiphenylsilyl, benzoyl, mesylate, tosylate, dimethoxytrityl (DMT), 9-phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX).
- DMT dimethoxytrityl
- Pixyl 9-phenylxanthine-9-yl
- MOX 9-(p-methoxyphenyl)xanthine-9-yl
- amino protecting groups include without limitation, carbamate-protecting groups, such as 2-trimethylsilylethoxycarbonyl (Teoc), 1 -methyl- l-(4-biphenylyl)ethoxycarbonyl (Bpoc), t-butoxycarbonyl (BOC), allyloxycarbonyl (Alloc), 9-fluorenylmethyloxycarbonyl (Fmoc), and benzyloxycarbonyl (Cbz); amide-protecting groups, such as formyl, acetyl, trihaloacetyl, benzoyl, and nitrophenylacetyl; sulfonamide-protecting groups, such as 2-nitrobenzenesulfonyl; and imine- and cyclic imide-protecting groups, such as phthalimido and dithiasuccinoyl.
- carbamate-protecting groups such as 2-trimethylsilylethoxycarbonyl (Teoc), 1 -methyl- l-
- thiol protecting groups include without limitation, triphenylmethyl (trityl), benzyl (Bn), and the like.
- nucleoside is a base-sugar combination.
- the base portion of the nucleoside is normally a heterocyclic base moiety.
- the two most common classes of such heterocyclic bases are purines and pyrimidines.
- Nucleotides are nucleosides that further include a phosphate group covalently linked to the sugar portion of the nucleoside.
- the phosphate group can be linked to either the 2', 3 Or 5' hydroxyl moiety of the sugar.
- the phosphate groups covalently link adjacent nucleosides to one another to form a linear polymeric compound.
- this linear polymeric structure can be joined to form a circular structure by hybridization or by formation of a covalent bond.
- open linear structures are generally desired.
- the phosphate groups are commonly referred to as forming the internucleoside linkages of the oligonucleotide.
- the normal internucleoside linkage of RNA and DNA is a 3' to 5' phospho- diester linkage.
- modified nucleoside is meant to include all manner of modified nucleosides that can be incorporated into an oligomeric compound using oligomer synthesis.
- the term is intended to include modifications made to a nucleoside such as modified stereochemical configurations, one or more substitutions, and deletion of groups as opposed to the use of surrogate groups which are described elsewhere herein.
- the term includes nucleosides having a furanose sugar (or 4'-S analog) portion and can include a heterocyclic base or can include an abasic site.
- modified nucleosides includes without limitation, substituted nucleosides (such as 2 5 and/or 4' substituted nucleosides) 4'-S-modified nucleosides, (such as 4'-S- ribonucleosides, 4'-S-2'-deoxyribonucleosides and 4'-S-2'-substituted ribonucleosides), bicyclic modified nucleosides (such as for example, bicyclic nucleosides wherein the sugar group has a 2'-0- CHR a -4' bridging group, wherein R a is H, alkyl or substituted alkyl) and base modified nucleosides.
- substituted nucleosides such as 2 5 and/or 4' substituted nucleosides
- 4'-S-modified nucleosides such as 4'-S- ribonucleosides, 4'-S-2'-deoxyribonucleosides and 4'-S-2
- the sugar can be modified with more than one of these modifications listed such as for example a bicyclic modified nucleoside further including a 5 '-substitution or a 5' or 4' substituted nucleoside further including a 2' substituent.
- modified nucleoside also includes combinations of these modifications such as a base and sugar modified nucleosides. These modifications are meant to be illustrative and not exhaustive as other modifications are known in the art and are also envisioned as possible modifications for the modified nucleosides described herein.
- unmodified nucleobase and “naturally occurring nucleobase” include the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U).
- Modified nucleobases include other synthetic and natural nucleobases such as 5-methylcytosine (5-me-C), 5-hydroxymethyl cytosine, xanthine, hypoxanthine, 2- aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 2-thiouracil, 2-thiothymine and 2-thiocytosine, 5-halouracil and cytosine, 5-propynyl (-C ⁇ C-CH 3 ) uracil and cytosine and other alkynyl derivatives of pyrimidine bases, 6-azo uracil, cytosine and thymine, 5-uracil (pseudouracil), 4-thiouracil, 8-halo, 8-amino, 8- thiol, 8-thioalkyl, 8-hydroxyl and other 8-substituted adenines and guan
- nucleobases include tricyclic pyrimidines such as phenoxazine cytidine( [5,4-b][l,4]benzoxazin- 2(3H)-one), phenothiazine cytidine (1 H-pyrimido[5,4-b] [ 1 ,4]benzothiazin-2(3H)-one), G-clamps such as a substituted phenoxazine cytidine (e.g.
- nucleobases may also include those in which the purine or pyrimidine base is replaced with other heterocycles, for example 7-deaza-adenine, 7-deazaguanosine, 2-aminopyridine and 2-pyridone.
- nucleobases include those disclosed in United States Patent No. 3,687,808, those disclosed in The Concise Encyclopedia Of Polymer Science And Engineering, Kroschwitz, J.I., Ed., John Wiley & Sons, 1990, 858-859; those disclosed by Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613; and those disclosed by Sanghvi, Y.S., Chapter 15, Antisense Research and Applications, Crooke, S.T. and Lebleu, B., Eds., CRC Press, 1993, 273-288.
- heterocyclic base moiety of each of the 2'-0-substituted purine nucleosides can be modified with one or more substituent groups to enhance one or more properties such as affinity for a target strand or affect some other property in an advantageous manner.
- Modified nucleobases include without limitation, universal bases, hydrophobic bases, promiscuous bases, size-expanded bases, and fluorinated bases as defined herein. Certain of these nucleobases are particularly useful for increasing the binding affinity of the oligomeric compounds as provided herein.
- 5- substituted pyrimidines 6-azapyrimidines and N-2, N-6 and 0-6 substituted purines, including 2- aminopropyl adenine, 5-propynyluracil and 5-propynylcytosine.
- 5-methylcytosine substitutions have been shown to increase nucleic acid duplex stability by 0.6-1.2 °C ⁇ Antisense Research and
- bicyclic nucleic acid and “bicyclic nucleoside” refer to nucleosides wherein the sugar portion of the nucleoside is bicyclic (e.g. bicyclic sugar).
- a bicyclic nucleic acid comprises a nucleoside wherein the furanose ring comprises a bridge between two non-geminal ring carbon atoms.
- Examples of bicyclic nucleosides include without limitation nucleosides comprising a bridge between the 4' and the 2' ribosyl ring atoms.
- oligomeric compounds provided herein include one or more bicyclic nucleosides wherein the bridge comprises one of the formulae: 4'-(CH 2 )-0-2' (LNA); 4'-(CH 2 )-S-2'; 4'-(CH 2 ) 2 -0- 2' (ENA); 4'-CH(CH 3 )-0-2' and 4'-CH(CH 2 OCH 3 )-0-2' (and analogs thereof see U.S.
- Each of the foregoing bicyclic nucleosides can be prepared having one or more stereochemical sugar configurations including for example a-L-ribofuranose and ⁇ -D-ribofuranose (see PCT international application PCT/DK98/00393, published on March 25, 1999 as WO 99/14226).
- Compound 2 was prepared according to the procedures illustrated in WO 2003/091227.
- Compound 1 (15 g, 0.11 mol, 1.0 eq, commercially available) was added to a solution of sodium iodide (22.5 g, 0.15 mol, 1.36 eq) in acetone (50 mL) and stirred overnight under reflux. The reaction mixture initially turned brown, then slowly changed to dark orange. The byproduct, sodium bromide, was observed as it crashed out as a white precipitate. After stirring for 12 h, the reaction mixture was poured into ice water (200 mL) and extracted with diethyl ether (3 x 100 mL).
- Compound 2 can also be further purified by vacuum distillation.
- Example 3
- Compound 2 was prepared per the procedures illustrated in Example 1.
- Compound 4 (10 g, 0.024 mol, 1.0 eq) and tetrabutyl ammonium iodide or TBAI (2.66 g, 0.0072 mol, 0.3 eq) were dissolved in anhydrous DMF (600 mL) and the volume was reduced to 500 mL in vacuo at 50 °C.
- the reaction mixture was cooled to -20 °C with stirring under nitrogen and a solution of NaHMDS (70 mL, 1M NaHMDS in THF, 0.07 mol, 3 eq) was added dropwise at 2 mL/min.
- Isobutyryl chloride (3.5 mL, 0.033 mol, 1.5 eq) was added to a solution of Compound 5 at 0 °C which was prepared by co-evaporating Compound 5 with anhydrous pyridine (2 x 100 mL) and dissolving the coevaporated Compound 5 (10.75 g, 0.022 mol, 1.0 eq) in anhydrous pyridine (250 mL). Additional isobutyryl chloride is added until the starting material is completely consumed. After stirring under nitrogen for 2 h at 0 °C, the reaction was complete as indicated by LCMS analysis. MeOH (10 mL) was added to the reaction mixture to quench any unreacted acid chloride and the solvent was evaporated under reduced pressure.
- Triethylamine trihydro fluoride (1.2 mL, 0.0073 mol, 0.66 eq) was added to a solution of Compound 6 (6.1 g, 0.011 mol, 1.0 eq) suspended in anhydrous THF (100 mL). A minimum amount of triethylamine trihydro fluoride was added until all the starting material was consumed. The reaction was stirred at ambient temperature for 30 min after TLC analysis (10% MeOH in CH 2 C1 2 ) indicated that Compound 6 was consumed. The reaction mixture was concentrated under reduced pressure and the residue was diluted with diethyl ether (25 mL). The resulting white precipitate was collected by filtration and dried briefly under high vacuum. The crude solid was then recrystallized in acetone/H 2 0 (10:1 v/v, 100 mL) to provide pure Compound 7 (4.9 g, 94% yield with 99.67% purity) as crystals.
- Compounds 10 and 11 are prepared as per the procedures illustrated in Example 3 using Compounds 8 and 9 as starting materials (Compounds 8 and 9 are commercially available).
Abstract
Provided herein are methods for preparing 2'-O-substituted purine nucleosides without protecting exocyclic amino groups on the base during alkylation. The methods are particularly useful in that the products are crystalline enabling purification without chromatography.
Description
METHODS OF PREPARING 2*-0-SUBSTITUTED PURINE NUCLEOSIDES
FIELD OF THE INVENTION
Provided herein are methods for preparing 2'-0-substituted purine nucleosides without protecting exocyclic amino groups on the base during alkylation. The methods are particularly useful in that the products are crystalline enabling purification without chromatography.
BACKGROUND OF THE INVENTION
Oligonucleotides have been used in various biological and biochemical applications. They have been used as primers and probes for the polymerase chain reaction (PCR), as antisense agents used in target validation, drug discovery and development, as ribozymes, as aptamers, and as general stimulators of the immune system. This widespread use of oligonucleotides has led to an increasing demand for rapid, inexpensive and efficient methods for their synthesis.
Synthetic oligonucleotides are generally prepared through the repeated coupling reactions of nucleoside phosphoramidites to the 5'-hydroxyl group of a nucleoside monomer or the free 5'- hydroxyl group of a growing oligomer. The most commonly used method to perform oligomer synthesis is the phosphoramidite approach which is largely based on developments reported in the literature (see for example: Beaucage and Caruthers (1981) Tetrahedron Letters 22:1859-1862; McBride and Caruthers (1983) Tetrahedron Letters 24:245-248; Sinha et al. (1984) Nucleic Acids Res. 12:4539-4557 and Beaucage and Iyer (1992) Tetrahedron 48:2223-2311, each of which is incorporated herein by reference in its entirety).
Oligomer synthesis can be performed using solution or solid phase chemistries. Solid phase oligonucleotide synthesis (SPOS) is the preferred method. In SPOS, oligonucleotides are assembled in a cyclical manner, each cycle consisting of a series of three chemical reactions. The first reaction is a deblocking reaction, i.e. the removal of a hydroxyl protecting group from a nucleoside monomer or an oligomer bound to a support. Generally, this requires the removal of a dimethoxytrityl protecting group to provide a free hydroxyl group. The second reaction is the coupling reaction, normally performed in the presence of an activator, wherein the free hydroxyl group is reacted with a nucleoside phosphoramidite to provide a phosphite triester in the presence of an activator. The third reaction is the oxidation of the phosphite triester to a phosphate triester. Optionally, a capping step is included either directly before or after each oxidation reaction in order
to block support bound nucleoside monomers or oligomers which failed to react in the coupling reaction and to prevent them from further chain elongation in subsequent coupling steps.
A major limiting factor for the cost efficient synthesis of oligonucleotides is the time and cost required to prepare nucleosides, including especially modified nucleosides, and their purification and conversion into phosphoramidites. The standard method used in the industry for purifying nucleoside phosphoramidites and the intermediates formed during their synthesis is column chromatography, especially flash column chromatography which primarily uses silica gel. Regardless of which particular type of column chromatography is used, purification by column chromatography requires large amounts of support material (silica gel for example) and large volumes of high purity solvents. The process also requires a lot of time as it is labor-intensive. Purine nucleosides tend to be more difficult to prepare and purify than pyrimidines. Due to the difficulties inherent in the methods currently being used, there exists an ongoing need in the art for more efficient methods for the synthesis and purification of nucleosides, especially modified nucleosides such as 2'-0-substituted purine nucleosides. Provided herein are methods that fulfill this need.
SUMMARY OF THE INVENTION
Provided herein are methods of preparing a 2'-0-substituted purine nucleoside comprising: contacting a nucleoside having Formula I:

I
wherein Pu is an unprotected purine having at least one exocyclic amino group;
with a di-t-butyl silyl protecting reagent under conditions to provide a protected nucleoside having Formul

Π;
contacting the protected nucleoside having Formula II with an alkylating agent under conditions to provide 2'-0-substituted purine nucleoside having Formula III:
 wherein
wherein
Z is C C6 alkyl or substituted C C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl or substituted C2-C6 alkynyl;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, 0Jl5 SJl5 NJ]J2, N3, CN, C(=0)OJ1, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJ!J2, N(H)C(=0)NJ1J2 orN(H)C(=S)NJ1J2; and
each 31 andJ2 is, independently, H, Cj-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 aminoalkyl or a protecting group.
In certain embodiments, the methods provided herein further comprise contacting the 2'-0- substituted purine nucleoside with a reagent capable of protecting any exocyclic amino groups located on the purine. In certain embodiments, the methods provided herein further comprise contacting the 2'-0-substituted purine nucleoside with a reagent capable of protecting any exocyclic amino groups located on the purine and or contacting the 2'-0-substituted purine nucleoside with a reagent capable of removing the di-t-butyl silyl protecting group.
In certain embodiments, the reagent used to protect exocyclic amino groups on the base is isobutyryl chloride. In certain embodiments, the reagent used to remove the di-t-butyl silyl protecting group is triethylamine trihydrofluoride.
In certain embodiments, the unprotected purine is guanine, adenine or diaminopurine. In certain embodiments, the unprotected purine is guanine.
In certain embodiments, the di-t-butyl silyl protecting reagent is tBu2Si(OTf)2.
In certain embodiments, the alkylating agent comprises a C!-C6 alkyl or substituted Cj-C6 alkyl and a leaving group. In certain embodiments, the alkylating agent comprises a substituted C\- C6 alky group and a leaving group. In certain embodiments, the alkylating agent comprises 1-iodo- 2-methoxyethane.
In certain embodiments, In certain embodiments, Z is CH3, CH2CH3, (CH2)2CH3, (CH2)2-0- CH3, CH2-CH=CH2, (CH2)3-N(R,)(R2), (CH2)2-O-N(R (R2), (CH2)2-0-(CH2)2-N(Ri)(R2),
CH2C(=0)-N(Ri)(R2), CH2C(=0)-N(H)-(CH2)2-N(R1)(R2) or CH2-N(H)-C(= Ri)[N(Ri)(R2)] wherein Rj and R2 are each independently, H or C C2 alkyl.
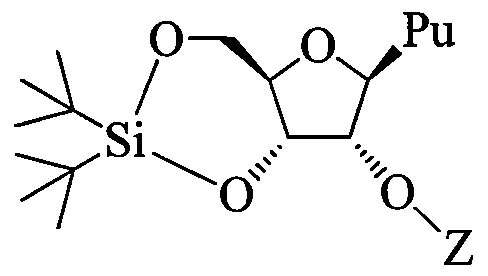
In certain embodiments, the 2'-0-substituted purine nucleoside has the configuration of Formula IV:

IV.
DETAILED DESCRIPTION OF THE INVENTION
Provided herein are methods for preparing 2'-0-substituted purine nucleosides without protecting exocyclic amino groups on the base during alkylation. The methods are particularly useful in that the products are crystalline enabling purification without chromatography.
The methods include selecting a nucleoside having Formula I:

I
wherein Pu is an unprotected purine having at least one exocyclic amino group;
and contacting this selected nucleoside with a di-t-butyl silyl protecting reagent under conditions to provide a protected nucleoside having Formula II:

II.
The nucleoside having Formula II is further contacted with an alkylating agent under conditions to provide 2'-0-substituted purine nucleoside having Formula III:

III.
The alkylating agent can be selected and protected if needed to provide a desired Z group.
Such Z groups include without limitation Ci-C6 alkyl or substituted C C6 alkyl, C2-C6 alkenyl, substituted C2-C alkenyl, C2-C6 alkynyl or substituted C2-C6 alkynyl;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, OJl5 SJj, NJ]J2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Ji, 0-C(=0)NJ!J2, N(H)C(=0)NJ]J2 orN(H)C(=S)NJ,J2; and
each Ji and J2 is, independently, H, Q-Q alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C!-C6 aminoalkyl or a protecting group.
The methods provided herein are useful for the preparation of 2'-0-substituted purine nucleosides having at least one exocyclic amino group on the base. The methods provide for protection of the sugar 3' and 5' hydroxyl groups and subsequent alkylation of the 2' hydroxyl without having to protect base amino groups. Representative purine bases that have exocyclic amino groups include without limitation guanine, adenine and diaminopurine.
The selection of the alkylating agent will determine the 2'-0-substitutent (Z). Many alkylating agents can be purchased commercially or prepared from commercial materials. Such substituents (Z) include without limitation CH3, CH2CH3, (CH2)2CH3, (CH2)2-0-CH3, CH2- CH=CH2, (CH2)3-N(R (R2), (ΟΙ2)2-0-Ν(¾)^2), (CH2)2-0-(CH2)2-N(R])(R2), CH2C(=0)- N(R])(R2), CH2C(=0)-N(H)-(CH2)2-N(R1)(R2) or CH2-N(H)-C(=NR1)[N(R1)(R2)] wherein Rj and R2 are each independently, H or Ci-C2 alkyl.
In certain embodiments, the 2'-0-substituted purine nucleoside prepared using the methods provided herein can be further contacted with a reagent capable of protecting any exocyclic amino groups located on the purine. In certain embodiments, the 2'-0-substituted purine nucleoside prepared using the methods provided herein can be further contacted with a reagent capable of protecting any exocyclic amino groups located on the purine and or contacted with a reagent capable of removing the di-t-butyl silyl protecting group.
In one embodiment, the 2'-0-substituted purine nucleosides prepared here have the configuration of Formula IV:

IV.
The terms "substituent" and "substituent group," as used herein, are meant to include groups that are typically added to other groups or parent compounds to enhance desired properties or provide other desired effects. Substituent groups can be protected or unprotected and can be added
to one available site or to many available sites in a parent compound. Substituent groups may also be further substituted with other substituent groups and may be attached directly or via a linking group such as an alkyl or hydrocarbyl group to a parent compound.
Substituent groups amenable herein include without limitation, halogen, hydroxyl, alkyl, alkenyl, alkynyl, acyl (-C(0)Raa), carboxyl (-C(0)0-Raa), aliphatic groups, alicyclic groups, alkoxy, substituted oxy (-0-Raa), aryl, aralkyl, heterocyclic radical, heteroaryl, heteroarylalkyl, amino (-N(Rbb)(Rcc)), imino(=NRbb), amido (-C(0)N(Rbb)(RcC) or -N(Rbb)C(0)Raa), azido (-N3), nitro (-N02), cyano (-CN), carbamido (-OC(0)N(Rbb)(Rcc) or -N(Rbb)C(0)ORaa), ureido (-N(Rbb)C(0)- N(Rbb)(Rcc)), thioureido (-N(Rbb)C(S)N(Rbb)(Rcc)), guanidinyl (-N(Rbb)C(=NRbb)N(Rbb)(Rcc)), amidinyl (-C(=NRbb)N(Rbb)(RcC) or -N(Rbb)C(=NRbb)(Raa)), thiol (-SRbb), sulfinyl (-S(0)Rbb), sulfonyl (-S(0)2Rb ) and sulfonamidyl (-S(0)2N(Rbb)(RcC) or -N(Rbb)S(0)2Rbb). Wherein each Raa, Rbb and RcC is, independently, H, an optionally linked chemical functional group or a further substituent group with a preferred list including without limitation, H, alkyl, alkenyl, alkynyl, aliphatic, alkoxy, acyl, aryl, aralkyl, heteroaryl, alicyclic, heterocyclic and heteroarylalkyl. Selected substituents within the compounds described herein are present to a recursive degree.
The term "alkyl," as used herein, refers to a saturated straight or branched hydrocarbon radical containing up to twenty four carbon atoms. Examples of alkyl groups include without limitation, methyl, ethyl, propyl, butyl, isopropyl, n-hexyl, octyl, decyl, dodecyl and the like. Alkyl groups typically include from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms (Q-C^ alkyl) with from 1 to about 6 carbon atoms being more preferred. The term "lower alkyl" as used herein includes from 1 to about 6 carbon atoms. Alkyl groups as used herein may optionally include one or more further substituent groups.
The term "alkenyl," as used herein, refers to a straight or branched hydrocarbon chain radical containing up to twenty four carbon atoms and having at least one carbon-carbon double bond. Examples of alkenyl groups include without limitation, ethenyl, propenyl, butenyl, l-methyl-2- buten-l-yl, dienes such as 1,3 -butadiene and the like. Alkenyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred. Alkenyl groups as used herein may optionally include one or more further substituent groups.
The term "alkynyl," as used herein, refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms and having at least one carbon-carbon triple bond.
Examples of alkynyl groups include, without limitation, ethynyl, 1-propynyl, 1-butynyl, and the
like. Alkynyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred. Alkynyl groups as used herein may optionally include one or more further substituent groups.
The term "acyl," as used herein, refers to a radical formed by removal of a hydroxyl group from an organic acid and has the general Formula -C(0)-X where X is typically aliphatic, alicyclic or aromatic. Examples include aliphatic carbonyls, aromatic carbonyls, aliphatic sulfonyls, aromatic sulfinyls, aliphatic sulfinyls, aromatic phosphates, aliphatic phosphates and the like. Acyl groups as used herein may optionally include further substituent groups.
The term "alicyclic" refers to a cyclic ring system wherein the ring is aliphatic. The ring system can comprise one or more rings wherein at least one ring is aliphatic. Preferred alicyclics include rings having from about 5 to about 9 carbon atoms in the ring. Alicyclic as used herein may optionally include further substituent groups.
The term "aliphatic," as used herein, refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms wherein the saturation between any two carbon atoms is a single, double or triple bond. An aliphatic group preferably contains from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms with from 1 to about 6 carbon atoms being more preferred. The straight or branched chain of an aliphatic group may be interrupted with one or more heteroatoms that include nitrogen, oxygen, sulfur and phosphorus. Such aliphatic groups interrupted by heteroatoms include without limitation, polyalkoxys, such as polyalkylene glycols, polyamines, and polyimines. Aliphatic groups as used herein may optionally include further substituent groups.
The term "alkoxy," as used herein, refers to a radical formed between an alkyl group and an oxygen atom wherein the oxygen atom is used to attach the alkoxy group to a parent molecule. Examples of alkoxy groups include without limitation, methoxy, ethoxy, propoxy, isopropoxy, n- butoxy, sec-butoxy, tert-butoxy, n-pentoxy, neopentoxy, n-hexoxy and the like. Alkoxy groups as used herein may optionally include further substituent groups.
The term "aminoalkyl" as used herein, refers to an amino substituted C\-Cn alkyl radical. The alkyl portion of the radical forms a covalent bond with a parent molecule. The amino group can be located at any position and the aminoalkyl group can be substituted with a further substituent group at the alkyl and/or amino portions.
The terms "aralkyl" and "arylalkyl," as used herein, refer to an aromatic group that is covalently linked to a C\-Cn alkyl radical. The alkyl radical portion of the resulting aralkyl (or
arylalkyl) group forms a covalent bond with a parent molecule. Examples include without limitation, benzyl, phenethyl and the like. Aralkyl groups as used herein may optionally include further substituent groups attached to the alkyl, the aryl or both groups that form the radical group.
The terms "aryl" and "aromatic," as used herein, refer to a mono- or polycyclic carbocyclic ring system radicals having one or more aromatic rings. Examples of aryl groups include without limitation, phenyl, naphthyl, tetrahydronaphthyl, indanyl, idenyl and the like. Preferred aryl ring systems have from about 5 to about 20 carbon atoms in one or more rings. Aryl groups as used herein may optionally include further substituent groups.
The terms "halo" and "halogen," as used herein, refer to an atom selected from fluorine, chlorine, bromine and iodine.
The terms "heteroaryl," and "heteroaromatic," as used herein, refer to a radical comprising a mono- or poly-cyclic aromatic ring, ring system or fused ring system wherein at least one of the rings is aromatic and includes one or more heteroatoms. Heteroaryl is also meant to include fused ring systems including systems where one or more of the fused rings contain no heteroatoms.
Heteroaryl groups typically include one ring atom selected from sulfur, nitrogen or oxygen.
Examples of heteroaryl groups include without limitation, pyridinyl, pyrazinyl, pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isooxazolyl, thiadiazolyl, oxadiazolyl, thiophenyl, furanyl, quinolinyl, isoquinolinyl, benzimidazolyl, benzooxazolyl, quinoxalinyl and the like. Heteroaryl radicals can be attached to a parent molecule directly or through a linking moiety such as an aliphatic group or hetero atom. Heteroaryl groups as used herein may optionally include further substituent groups.
The term "heteroarylalkyl," as used herein, refers to a heteroaryl group as previously defined that further includes a covalently attached Cj-C12 alkyl radical. The alkyl radical portion of the resulting heteroarylalkyl group is capable of forming a covalent bond with a parent molecule.
Examples include without limitation, pyridinylmethyl, pyrimidinylethyl, napthyridinylpropyl and the like. Heteroarylalkyl groups as used herein may optionally include further substituent groups on one or both of the heteroaryl or alkyl portions.
The term "oxo" refers to the group (=0).
Linking groups or bifunctional linking moieties such as those known in the art are useful for attachment of chemical functional groups, conjugate groups, reporter groups and other groups to selective sites in a parent compound such as for example an oligomeric compound. In general, a bifunctional linking moiety comprises a hydrocarbyl moiety having two functional groups. One of
the functional groups is selected to bind to a parent molecule or compound of interest and the other is selected to bind to essentially any selected group such as a chemical functional group or a conjugate group. In some embodiments, the linker comprises a chain structure or a polymer of repeating units such as ethylene glycols or amino acid units. Examples of functional groups that are routinely used in bifunctional linking moieties include without limitation, electrophiles for reacting with nucleophilic groups and nucleophiles for reacting with electrophilic groups. In some embodiments, bifunctional linking moieties include amino, hydroxyl, carboxylic acid, thiol, unsaturations (e.g., double or triple bonds), and the like. Some nonlimiting examples of bifunctional linking moieties include 8-amino-3,6-dioxaoctanoic acid (ADO), succinimidyl 4-(N- maleimidomethyl) cyclohexane-l-carboxylate (SMCC) and 6-aminohexanoic acid (AHEX or AHA). Other linking groups include without limitation, substituted Cj-Cio alkyl, substituted or unsubstituted C2-C10 alkenyl or substituted or unsubstituted C2-C10 alkynyl, wherein a nonlimiting list of preferred substituent groups includes hydroxyl, amino, alkoxy, carboxy, benzyl, phenyl, nitro, thiol, thioalkoxy, halogen, alkyl, aryl, alkenyl and alkynyl.
The term "protecting group," as used herein, refers to a labile chemical moiety which is known in the art to protect reactive groups including without limitation, hydroxyl, amino and thiol groups, against undesired reactions during synthetic procedures. Protecting groups are typically used selectively and/or orthogonally to protect sites during reactions at other reactive sites and can then be removed to leave the unprotected group as is or available for further reactions. Protecting groups as known in the art are described generally in Greene's Protective Groups in Organic
Synthesis, 4th edition, John Wiley & Sons, New York, 2007.
Groups can be selectively incorporated into oligomeric compounds as provided herein as precursors. For example an amino group can be placed into a compound as provided herein as an azido group that can be chemically converted to the amino group at a desired point in the synthesis. Generally, groups are protected or present as precursors that will be inert to reactions that modify other areas of the parent molecule for conversion into their final groups at an appropriate time. Further representative protecting or precursor groups are discussed in Agrawal et al., Protocols for Oligonucleotide Conjugates, Humana Press; New Jersey, 1994, 26, 1-72.
The term "orthogonally protected" refers to functional groups which are protected with different classes of protecting groups, wherein each class of protecting group can be removed in any order and in the presence of all other classes (see, Barany et al, J. Am. Chem. Soc, 1977, 99, 7363- 7365; Barany et al, J. Am. Chem. Soc, 1980, 102, 3084-3095). Orthogonal protection is widely
used in for example automated oligonucleotide synthesis. A functional group is deblocked in the presence of one or more other protected functional groups which is not affected by the deblocking procedure. This deblocked functional group is reacted in some manner and at some point a further orthogonal protecting group is removed under a different set of reaction conditions. This allows for selective chemistry to arrive at a desired compound or oligomeric compound.
Examples of hydroxyl protecting groups include without limitation, acetyl, t-butyl, t- butoxymethyl, methoxymethyl, tetrahydropyranyl, 1-ethoxyethyl, l-(2-chloroethoxy)ethyl, p- chlorophenyl, 2,4-dinitrophenyl, benzyl, 2,6-dichlorobenzyl, diphenylmethyl, p-nitrobenzyl, bis(2- acetoxyethoxy)methyl (ACE), 2-trimethylsilylethyl, trimethylsilyl, triethylsilyl, t-butyldimethylsilyl, t-butyldiphenylsilyl, triphenylsilyl, [(triisopropylsilyl)oxy]methyl (TOM), benzoylformate, chloroacetyl, trichloroacetyl, trifluoroacetyl, pivaloyl, benzoyl, p-phenylbenzoyl, 9-fiuorenylmethyl carbonate, mesylate, tosylate, triphenylmethyl (trityl), monomethoxytrityl, dimethoxytrityl (DMT), trimethoxytrityl, 1 (2-fluorophenyl)-4-methoxypiperidin-4-yl (FPMP), 9-phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX). Wherein more commonly used hydroxyl protecting groups include without limitation, benzyl, 2,6-dichlorobenzyl, t-butyldimethylsilyl, t-butyldiphenylsilyl, benzoyl, mesylate, tosylate, dimethoxytrityl (DMT), 9-phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX). ·
Examples of amino protecting groups include without limitation, carbamate-protecting groups, such as 2-trimethylsilylethoxycarbonyl (Teoc), 1 -methyl- l-(4-biphenylyl)ethoxycarbonyl (Bpoc), t-butoxycarbonyl (BOC), allyloxycarbonyl (Alloc), 9-fluorenylmethyloxycarbonyl (Fmoc), and benzyloxycarbonyl (Cbz); amide-protecting groups, such as formyl, acetyl, trihaloacetyl, benzoyl, and nitrophenylacetyl; sulfonamide-protecting groups, such as 2-nitrobenzenesulfonyl; and imine- and cyclic imide-protecting groups, such as phthalimido and dithiasuccinoyl.
Examples of thiol protecting groups include without limitation, triphenylmethyl (trityl), benzyl (Bn), and the like.
As is known in the art, a nucleoside is a base-sugar combination. The base portion of the nucleoside is normally a heterocyclic base moiety. The two most common classes of such heterocyclic bases are purines and pyrimidines. Nucleotides are nucleosides that further include a phosphate group covalently linked to the sugar portion of the nucleoside. For those nucleosides that include a pentofuranosyl sugar, the phosphate group can be linked to either the 2', 3 Or 5' hydroxyl moiety of the sugar. In forming oligonucleotides, the phosphate groups covalently link adjacent nucleosides to one another to form a linear polymeric compound. The respective ends of this linear
polymeric structure can be joined to form a circular structure by hybridization or by formation of a covalent bond. However, open linear structures are generally desired. Within the oligonucleotide structure, the phosphate groups are commonly referred to as forming the internucleoside linkages of the oligonucleotide. The normal internucleoside linkage of RNA and DNA is a 3' to 5' phospho- diester linkage.
As used herein the term "modified nucleoside" is meant to include all manner of modified nucleosides that can be incorporated into an oligomeric compound using oligomer synthesis. The term is intended to include modifications made to a nucleoside such as modified stereochemical configurations, one or more substitutions, and deletion of groups as opposed to the use of surrogate groups which are described elsewhere herein. The term includes nucleosides having a furanose sugar (or 4'-S analog) portion and can include a heterocyclic base or can include an abasic site. One group of representative modified nucleosides includes without limitation, substituted nucleosides (such as 2 5 and/or 4' substituted nucleosides) 4'-S-modified nucleosides, (such as 4'-S- ribonucleosides, 4'-S-2'-deoxyribonucleosides and 4'-S-2'-substituted ribonucleosides), bicyclic modified nucleosides (such as for example, bicyclic nucleosides wherein the sugar group has a 2'-0- CHRa-4' bridging group, wherein Ra is H, alkyl or substituted alkyl) and base modified nucleosides. ■ The sugar can be modified with more than one of these modifications listed such as for example a bicyclic modified nucleoside further including a 5 '-substitution or a 5' or 4' substituted nucleoside further including a 2' substituent. The term modified nucleoside also includes combinations of these modifications such as a base and sugar modified nucleosides. These modifications are meant to be illustrative and not exhaustive as other modifications are known in the art and are also envisioned as possible modifications for the modified nucleosides described herein.
As used herein the terms, "unmodified nucleobase" and "naturally occurring nucleobase" include the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U). Modified nucleobases include other synthetic and natural nucleobases such as 5-methylcytosine (5-me-C), 5-hydroxymethyl cytosine, xanthine, hypoxanthine, 2- aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 2-thiouracil, 2-thiothymine and 2-thiocytosine, 5-halouracil and cytosine, 5-propynyl (-C≡C-CH3) uracil and cytosine and other alkynyl derivatives of pyrimidine bases, 6-azo uracil, cytosine and thymine, 5-uracil (pseudouracil), 4-thiouracil, 8-halo, 8-amino, 8- thiol, 8-thioalkyl, 8-hydroxyl and other 8-substituted adenines and guanines, 5-halo particularly 5- bromo, 5-trifluoromethyl and other 5-substituted uracils and cytosines, 7-methylguanine and 7-
methyladenine, 2-F-adenine, 2-amino-adenine, 8-azaguanine and 8-azaadenine, 7-deazaguanine and 7-deazaadenine, 3-deazaguanine and 3-deazaadenine, universal bases, hydrophobic bases, promiscuous bases, size-expanded bases, and fluorinated bases as defined herein. Further modified nucleobases include tricyclic pyrimidines such as phenoxazine cytidine( [5,4-b][l,4]benzoxazin- 2(3H)-one), phenothiazine cytidine (1 H-pyrimido[5,4-b] [ 1 ,4]benzothiazin-2(3H)-one), G-clamps such as a substituted phenoxazine cytidine (e.g. 9-(2-aminoethoxy)-H-pyrimido[5,4- b][l,4]benzoxazin-2(3H)-one), carbazole cytidine (2H-pyrimido[4,5-b]indol-2-one), pyridoindole cytidine (H-pyrido[3',2,:4,5]pyrrolo[2,3-d]pyrimidin-2-one). Modified nucleobases may also include those in which the purine or pyrimidine base is replaced with other heterocycles, for example 7-deaza-adenine, 7-deazaguanosine, 2-aminopyridine and 2-pyridone. Further nucleobases include those disclosed in United States Patent No. 3,687,808, those disclosed in The Concise Encyclopedia Of Polymer Science And Engineering, Kroschwitz, J.I., Ed., John Wiley & Sons, 1990, 858-859; those disclosed by Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613; and those disclosed by Sanghvi, Y.S., Chapter 15, Antisense Research and Applications, Crooke, S.T. and Lebleu, B., Eds., CRC Press, 1993, 273-288.
The heterocyclic base moiety of each of the 2'-0-substituted purine nucleosides can be modified with one or more substituent groups to enhance one or more properties such as affinity for a target strand or affect some other property in an advantageous manner. Modified nucleobases include without limitation, universal bases, hydrophobic bases, promiscuous bases, size-expanded bases, and fluorinated bases as defined herein. Certain of these nucleobases are particularly useful for increasing the binding affinity of the oligomeric compounds as provided herein. These include 5- substituted pyrimidines, 6-azapyrimidines and N-2, N-6 and 0-6 substituted purines, including 2- aminopropyl adenine, 5-propynyluracil and 5-propynylcytosine. 5-methylcytosine substitutions have been shown to increase nucleic acid duplex stability by 0.6-1.2 °C {Antisense Research and
Applications, Sanghvi, Y.S., Crooke, S.T. and Lebleu, B., Eds., CRC Press, Boca Raton, 1993, 276- 278).
Representative United States patents that teach the preparation of certain of the above noted modified nucleobases as well as other modified nucleobases include without limitation, U.S.
3,687,808; 4,845,205; 5,130,302; 5,134,066; 5,175,273; 5,367,066; 5,432,272; 5,457,187;
5,459,255; 5,484,908; 5,502,177; 5,525,711; 5,552,540; 5,587,469; 5,594,121; 5,596,091 ;
5,614,617; 5,645,985; 5,681,941; 5,750,692; 5,763,588; 5,830,653 and 6,005,096, certain of which are commonly owned with the instant application, and each of which is herein incorporated by
reference in its entirety.
In certain embodiments, examples of substituent groups useful for modifying furanose sugar moieties (e.g., sugar substituent groups used for nucleosides), include without limitation 2'-F, 2'- allyl, 2'-amino, 2'-azido, 2*-thio, 2'-0-allyl, 2'-OCF3, 2'-O-Ci-C10 alkyl, 2'-0-CH3, OCF3, 2'-0- CH2CH3, 2'-0-(CH2)2CH3, 2'-0-(CH2)2-0-CH3 (MOE), 2'-0(CH2)2SCH3, 2,-0-CH2-CH=CH2, 2*-0- (CH2)3-N(Rm)(Rn), 2'-0-(CH2)2-0-N(Rm)(Rn), 2'-0-(CH2)2-0-(CH2)2-N(Rm)(Rn), 2'-0-CH2C(=0)- N(Rm)(R„), 2'-0-CH2C(=0)-N(H)-(CH2)2-N(Rm)(Rn) and 2^0-CH2-N(H)-C(=NRm)[N(Rm)(Rn)], 5'- vinyl, 5'-methyl (R or S) and 4'-S wherein each Rm and Rn is, independently, H, substituted or unsubstituted C\-C\o alkyl or a protecting group. Further examples of modified sugar moieties include without limitation bicyclic sugars (e.g. bicyclic nucleic acids or bicyclic nucleosides discussed below).
Combinations of these modifications are also provided for herein without limitation, such as 2'-F-5'-methyl substituted nucleosides (see PCT International Application WO 2008/101 157 Published on 8/21/08 for other disclosed 5', 2'-bis substituted nucleosides) and replacement of the ribosyl ring oxygen atom with S and further substitution at the 2'-position (see published U.S. Patent Application US2005-0130923, published on June 16, 2005) or alternatively 5'-substitution of a bicyclic nucleic acid (see PCT International Application WO 2007/134181 , published on 1 1/22/07 wherein a 4'-CH2-0-2' bicyclic nucleoside is further substituted at the 5' position with a 5'-methyl or a 5'-vinyl group).
As used herein the terms "bicyclic nucleic acid" and "bicyclic nucleoside" refer to nucleosides wherein the sugar portion of the nucleoside is bicyclic (e.g. bicyclic sugar). In certain embodiments, a bicyclic nucleic acid comprises a nucleoside wherein the furanose ring comprises a bridge between two non-geminal ring carbon atoms. Examples of bicyclic nucleosides include without limitation nucleosides comprising a bridge between the 4' and the 2' ribosyl ring atoms. In certain embodiments, oligomeric compounds provided herein include one or more bicyclic nucleosides wherein the bridge comprises one of the formulae: 4'-(CH2)-0-2' (LNA); 4'-(CH2)-S-2'; 4'-(CH2)2-0- 2' (ENA); 4'-CH(CH3)-0-2' and 4'-CH(CH2OCH3)-0-2' (and analogs thereof see U.S. Patent 7,399,845, issued on July 15, 2008); 4'-C(CH3)(CH3)-0-2' (and analogs thereof see published International Application WO/2009/006478, published January 8, 2009); 4'-CH2-N(OCH3)-2' (and analogs thereof see published International Application WO/2008/150729, published December 1 1 , 2008); 4'-CH2-0-N(CH3)-2' (see published U.S. Patent Application US2004-0171570, published September 2, 2004 ); 4'-CH2-N(R)-0-2', wherein R is H, CrCi2 alkyl, or a protecting group (see
U.S. Patent 7,427,672, issued on September 23, 2008); 4'-CH2-C(H)(CH3)-2' (see Chattopadhyaya, et al, J. Org. Chem.,2009, 74, 118-134); and 4'-CH2-C(=CH2)-2' (and analogs thereof see published International Application WO 2008/154401, published on December 8, 2008). Each of the foregoing bicyclic nucleosides can be prepared having one or more stereochemical sugar configurations including for example a-L-ribofuranose and β-D-ribofuranose (see PCT international application PCT/DK98/00393, published on March 25, 1999 as WO 99/14226).
While in certain embodiments, methods of preparing 2'-0-substituted purine nucleosides have been described with specificity, the following examples serve only to illustrate and are not intended to be limiting.
Examples (General)
1H NMR spectra were recorded on a 300 MHz Bruker spectrometer and purity was determined by HPLC/LCMS, respectively.
Example 1
Preparation of l-Iodo-2-methoxy-ethane, Compound 2
Br Nal
H3CO^^ I
H3CO^^
acetone
1 66% 2
Compound 2, was prepared according to the procedures illustrated in WO 2003/091227. Compound 1 (15 g, 0.11 mol, 1.0 eq, commercially available) was added to a solution of sodium iodide (22.5 g, 0.15 mol, 1.36 eq) in acetone (50 mL) and stirred overnight under reflux. The reaction mixture initially turned brown, then slowly changed to dark orange. The byproduct, sodium bromide, was observed as it crashed out as a white precipitate. After stirring for 12 h, the reaction mixture was poured into ice water (200 mL) and extracted with diethyl ether (3 x 100 mL). The combined organic layers were sequentially washed with saturated sodium thiosulfate (3 x 50 mL), water and brine then dried over MgS0 . The solution was filtered through a thin pad of Celite and silica gel and concentrated in vacuo to furnish 1 -iodo-2-methoxy-ethane, Compound 2 (13.31 g, 66%). To maximize shelf life, Compound 2 was stored in a freezer at -20 °C under nitrogen.
Compound 2 can also be further purified by vacuum distillation.
Example 3
Preparation of Compound 7


Compound 3 (purchased from a commercial source as a dihydrate) was dried by refluxing in toluene with a Dean-Stark trap and placed in a vacuum oven over P205 for storage. The anhydrous
Compound 3 (20 g, 0.071 mol, 1.0 eq) was dissolved in anhydrous DMF (550 mL) and the volume was reduced to 500 mL in vacuo at 50 °C. The reaction mixture was cooled to 0 °C while stirring under nitrogen and tBu2Si(OTf)2 (25 mL, 0.077mol, 1.0 eq) was added dropwise via a syringe.
After stirring for 30 min at 0 °C, the reaction was complete as determined by LCMS. The reaction mixture was carefully poured into a basic solution consisting of NaHC03 (20 g), H20 (1.5 L) and crushed ice (500 g). It is noteworthy to mention that triflic acid byproduct tends to cause depurination if the crude is over-exposed to acidic medium. The resulting precipitate was stirred for 30 min and was collected by filtration. The crude solid was re-suspended in H20 and the stirring
was continued for another 30 min. The solid was collected by filtration, dried overnight under vacuum at 45 °C and further dried by refluxing in toluene with a Dean-Stark trap to provide the crude Compound 4 (27.6 g, 93% with 94.175% purity). b) Preparation of Compound 5
The conditions used in preparing Compound 5 was in part carried out using similar procedures to those reported in the literature (Sanghvi, Y. S.; Theodorakis, E. A. et al J. Org. Chem. 2002, 67, 7887-7889 αά Nucleosides, Nucleotides and Nucleic Acids 2003, 22, 583-587).
Compound 2 was prepared per the procedures illustrated in Example 1. Compound 4 (10 g, 0.024 mol, 1.0 eq) and tetrabutyl ammonium iodide or TBAI (2.66 g, 0.0072 mol, 0.3 eq) were dissolved in anhydrous DMF (600 mL) and the volume was reduced to 500 mL in vacuo at 50 °C. The reaction mixture was cooled to -20 °C with stirring under nitrogen and a solution of NaHMDS (70 mL, 1M NaHMDS in THF, 0.07 mol, 3 eq) was added dropwise at 2 mL/min. The stirring was continued for 10 min at -20 °C followed by the addition of l-iodo-2-methoxy-ethane (13.17 g, 0.071 mol, 3 eq). The progress of the reaction was monitored by LCMS and additional NaHMDS and alkylating reagent were added until Compound 4 was completely consumed. After standing for 64 h at -20 °C, the reaction was quenched with cold MeOH (10 mL) and was poured into a solution of crushed ice (200 g) and water (1 L). The reaction mixture was diluted with EtOAc (500 mL) followed by the addition of saturated brine solution (500 mL). The organic layer was collected after partitioning and the aqueous layer was extracted twice with EtOAc (2 x 250 mL). The combined organic layers were sequentially washed with water and brine then dried (MgS04), concentrated in vacuo and dried under high vacuum to furnish the crude, Compound 5 as an orange oil, which was used without any further purification. c) Preparation of Compound 6
Isobutyryl chloride (3.5 mL, 0.033 mol, 1.5 eq) was added to a solution of Compound 5 at 0 °C which was prepared by co-evaporating Compound 5 with anhydrous pyridine (2 x 100 mL) and dissolving the coevaporated Compound 5 (10.75 g, 0.022 mol, 1.0 eq) in anhydrous pyridine (250 mL). Additional isobutyryl chloride is added until the starting material is completely consumed. After stirring under nitrogen for 2 h at 0 °C, the reaction was complete as indicated by LCMS analysis. MeOH (10 mL) was added to the reaction mixture to quench any unreacted acid chloride and the solvent was evaporated under reduced pressure. The resulting residue was further co-
evaporated with CH3CN (2 x 100 mL). The crude solid was suspended in boiling MeOH (400 mL) followed by the addition of dichloromethane (50 mL) until a solution resulted. After standing for 5 h, crystals began for form with additional crystals observed after storage overnight at 4 °C. The crystals were collected by filtration and dried over P205 in a vacuum oven at 45 °C to furnish pure Compound 6 (6.12 g, 46 % from Compound 4 with greater than 99% purity). d) Preparation of Compound 7
Triethylamine trihydro fluoride (1.2 mL, 0.0073 mol, 0.66 eq) was added to a solution of Compound 6 (6.1 g, 0.011 mol, 1.0 eq) suspended in anhydrous THF (100 mL). A minimum amount of triethylamine trihydro fluoride was added until all the starting material was consumed. The reaction was stirred at ambient temperature for 30 min after TLC analysis (10% MeOH in CH2C12) indicated that Compound 6 was consumed. The reaction mixture was concentrated under reduced pressure and the residue was diluted with diethyl ether (25 mL). The resulting white precipitate was collected by filtration and dried briefly under high vacuum. The crude solid was then recrystallized in acetone/H20 (10:1 v/v, 100 mL) to provide pure Compound 7 (4.9 g, 94% yield with 99.67% purity) as crystals.
Structural analysis for Compounds 2-7 was confirmed by 1H NMR and the purity for Compounds 4, 6 and 7 were determined by HPLC/LCMS.
We have developed and successfully demonstrated the synthesis of Compound 7 in high purity on a multigram scale with 44% overall yield employing a four-step synthetic method described herein. This method provided crystalline compounds without requiring protection of the purine base during either the protection with di-t-butyl silyl or the alkylation step. The
intermediates and the final base protected Compound 7 are purified by crystallization without the need of chromatography. Although the method is illustrated for preparing a 2'-0(CH2)2OCH3 substituted guanosine nucleoside, the method is also expected to be useful for the preparation of additional 2'-0-substituted nucleosides. Such other 2'-0-substituted nucleosides include for example adenosine and diaminopurine.
Example 4
Preparation of Compounds 10 and 11

8 X = NH2, Z = H 10 X = NH2, Z = H
9 X = NH2, Z = NH2 11 X = NH2, Z = NH2
Compounds 10 and 11 are prepared as per the procedures illustrated in Example 3 using Compounds 8 and 9 as starting materials (Compounds 8 and 9 are commercially available).
All publications, patents, and patent applications referenced herein are incorporated herein by reference. While in the foregoing specification this invention has been described in relation to certain embodiments thereof, and many details have been set forth for purposes of illustration, it will be apparent to those skilled in the art that the invention is susceptible to additional embodiments and that certain of the details described herein may be varied considerably without departing from the basic principles of the invention.
Claims
1. A method of preparing a 2'-0-substituted purine nucleoside comprising:
contacting a nucleoside having Formula I: 
I
wherein Pu is an unprotected purine having at least one exocyclic amino group;
with a di-t-butyl silyl protecting reagent under conditions to provide a protected nucleoside having Formul 
II
contacting the protected nucleoside having Formula II with an alkylating agent under conditions to provide 2'-0-substituted purine nucleoside having Formula III: 
III
wherein:
Z is Q-C6 alkyl or substituted Ci-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, C2-C6 alkynyl or substituted C2-C6 alkynyl;
wherein each substituted group is, independently, mono or poly substituted with substituent groups independently selected from halogen, OJi, SJi, NJ]J2, N3, CN, C(=0)OJi, C(=0)NJiJ2, C(=0)Jb 0-C(=0)NJ!J2, N(H)C(=0)NJiJ2 orN(H)C(=S)NJ!J2; and
each Ji and J2 is, independently, H, CrC6 alkyl, C2-C alkenyl, C2-C6 alkynyl, Q-Q aminoalkyl or a protecting group.
2. The method of claim 1 further comprising contacting the 2'-0-substituted purine nucleoside with a reagent capable of protecting any exocyclic amino groups located on the purine.
3. The method of claim 1 further comprising contacting the 2'-0-substituted purine nucleoside with a reagent capable of protecting any exocyclic amino groups located on the purine and or contacting the 2'-0-substituted purine nucleoside with a reagent capable of removing the di-t-butyl silyl protecting group.
4. The method of claim 3 wherein the reagent capable of protecting exocyclic amino groups is isobutyryl chloride.
5. The method of claim 3 wherein the reagent capable of removing the di-t-butyl silyl protecting group is triethylamine trihydrofluoride.
6. The method of claim 1 wherein the unprotected purine is guanine, adenine or diaminopurine.
7. The method of claim 4 wherein the unprotected purine is guanine.
8. The method of claim 1 wherein the di-t-butyl silyl protecting reagent is tBu2Si(OTf)2.
9. The method of claim 1 wherein the alkylating agent comprises a Q-Q alkyl or substituted C\-Ce alkyl and a leaving group.
10. The method of claim 1 wherein the alkylating agent comprises a substituted Cj-C6 alky group and a leaving group.
11. The method of claim 1 wherein the alkylating agent comprises 1 -iodo-2-methoxyethane.
12. The method of claim 1 wherein Z is CH3, CH2CH3, (CH2)2CH3, (CH2)2-0-CH3, CH2- CH=CH2, (CH2)3-N(R (R2), (CH2)2-0-N(R1)(R2), (CH2)2-0-(CH2)2-N(R,)(R2), CH2C(=0)- N(R,)(R2),  wherein R, and R2 are each independently, H or Q-Q alkyl.
wherein R, and R2 are each independently, H or Q-Q alkyl.
13. The method of claim 1 wherein the 2'-0-substituted purine nucleoside has the configuration of Formula IV:
rv.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US31444710P | 2010-03-16 | 2010-03-16 | |
| US61/314,447 | 2010-03-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011115817A1 true WO2011115817A1 (en) | 2011-09-22 |
Family
ID=44201849
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/027953 WO2011115817A1 (en) | 2010-03-16 | 2011-03-10 | Methods of preparing 2'-o-substituted purine nucleosides |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011115817A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106117289A (en) * | 2016-06-24 | 2016-11-16 | 郑州大学 | 2 ' O MOE 3 ' H thiophosphate nucleoside monomers and synthetic methods thereof |
Citations (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4845205A (en) | 1985-01-08 | 1989-07-04 | Institut Pasteur | 2,N6 -disubstituted and 2,N6 -trisubstituted adenosine-3'-phosphoramidites |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5552540A (en) | 1987-06-24 | 1996-09-03 | Howard Florey Institute Of Experimental Physiology And Medicine | Nucleoside derivatives |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5645985A (en) | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5681941A (en) | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| US5750692A (en) | 1990-01-11 | 1998-05-12 | Isis Pharmaceuticals, Inc. | Synthesis of 3-deazapurines |
| DK199800393U1 (en) | 1998-10-21 | 1998-10-21 | R E Beton A S Betonelementfabr | Roof element |
| US5830653A (en) | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| WO1999014226A2 (en) | 1997-09-12 | 1999-03-25 | Exiqon A/S | Bi- and tri-cyclic nucleoside, nucleotide and oligonucleotide analogues |
| WO2002018405A2 (en) * | 2000-09-01 | 2002-03-07 | Ribozyme Pharmaceuticals, Incorporated | Methods for synthesizing nucleosides, nucleoside derivatives and non-nucleoside derivatives |
| WO2003091227A1 (en) | 2002-04-26 | 2003-11-06 | Eli Lilly And Company | Tachykinin receptor antagonists |
| US20040171570A1 (en) | 2002-11-05 | 2004-09-02 | Charles Allerson | Polycyclic sugar surrogate-containing oligomeric compounds and compositions for use in gene modulation |
| US20050130923A1 (en) | 2003-09-18 | 2005-06-16 | Balkrishen Bhat | 4'-thionucleosides and oligomeric compounds |
| WO2007134181A2 (en) | 2006-05-11 | 2007-11-22 | Isis Pharmaceuticals, Inc. | 5'-modified bicyclic nucleic acid analogs |
| US7399845B2 (en) | 2006-01-27 | 2008-07-15 | Isis Pharmaceuticals, Inc. | 6-modified bicyclic nucleic acid analogs |
| WO2008101157A1 (en) | 2007-02-15 | 2008-08-21 | Isis Pharmaceuticals, Inc. | 5'-substituted-2'-f modified nucleosides and oligomeric compounds prepared therefrom |
| US7427672B2 (en) | 2003-08-28 | 2008-09-23 | Takeshi Imanishi | Artificial nucleic acids of n-o bond crosslinkage type |
| WO2008150729A2 (en) | 2007-05-30 | 2008-12-11 | Isis Pharmaceuticals, Inc. | N-substituted-aminomethylene bridged bicyclic nucleic acid analogs |
| WO2008154401A2 (en) | 2007-06-08 | 2008-12-18 | Isis Pharmaceuticals, Inc. | Carbocyclic bicyclic nucleic acid analogs |
| WO2009006478A2 (en) | 2007-07-05 | 2009-01-08 | Isis Pharmaceuticals, Inc. | 6-disubstituted bicyclic nucleic acid analogs |
| WO2011002207A2 (en) | 2009-07-03 | 2011-01-06 | Kim Kyung-Jo | Rotary bed which can be directed toward the position of the sun |
-
2011
- 2011-03-10 WO PCT/US2011/027953 patent/WO2011115817A1/en active Application Filing
Patent Citations (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| US4845205A (en) | 1985-01-08 | 1989-07-04 | Institut Pasteur | 2,N6 -disubstituted and 2,N6 -trisubstituted adenosine-3'-phosphoramidites |
| US5552540A (en) | 1987-06-24 | 1996-09-03 | Howard Florey Institute Of Experimental Physiology And Medicine | Nucleoside derivatives |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5750692A (en) | 1990-01-11 | 1998-05-12 | Isis Pharmaceuticals, Inc. | Synthesis of 3-deazapurines |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5681941A (en) | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| US5587469A (en) | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides containing N-2 substituted purines |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5645985A (en) | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5830653A (en) | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5763588A (en) | 1993-09-17 | 1998-06-09 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US6005096A (en) | 1993-09-17 | 1999-12-21 | Gilead Sciences, Inc. | Pyrimidine derivatives |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| WO1999014226A2 (en) | 1997-09-12 | 1999-03-25 | Exiqon A/S | Bi- and tri-cyclic nucleoside, nucleotide and oligonucleotide analogues |
| DK199800393U1 (en) | 1998-10-21 | 1998-10-21 | R E Beton A S Betonelementfabr | Roof element |
| WO2002018405A2 (en) * | 2000-09-01 | 2002-03-07 | Ribozyme Pharmaceuticals, Incorporated | Methods for synthesizing nucleosides, nucleoside derivatives and non-nucleoside derivatives |
| WO2003091227A1 (en) | 2002-04-26 | 2003-11-06 | Eli Lilly And Company | Tachykinin receptor antagonists |
| US20040171570A1 (en) | 2002-11-05 | 2004-09-02 | Charles Allerson | Polycyclic sugar surrogate-containing oligomeric compounds and compositions for use in gene modulation |
| US7427672B2 (en) | 2003-08-28 | 2008-09-23 | Takeshi Imanishi | Artificial nucleic acids of n-o bond crosslinkage type |
| US20050130923A1 (en) | 2003-09-18 | 2005-06-16 | Balkrishen Bhat | 4'-thionucleosides and oligomeric compounds |
| US7399845B2 (en) | 2006-01-27 | 2008-07-15 | Isis Pharmaceuticals, Inc. | 6-modified bicyclic nucleic acid analogs |
| WO2007134181A2 (en) | 2006-05-11 | 2007-11-22 | Isis Pharmaceuticals, Inc. | 5'-modified bicyclic nucleic acid analogs |
| WO2008101157A1 (en) | 2007-02-15 | 2008-08-21 | Isis Pharmaceuticals, Inc. | 5'-substituted-2'-f modified nucleosides and oligomeric compounds prepared therefrom |
| WO2008150729A2 (en) | 2007-05-30 | 2008-12-11 | Isis Pharmaceuticals, Inc. | N-substituted-aminomethylene bridged bicyclic nucleic acid analogs |
| WO2008154401A2 (en) | 2007-06-08 | 2008-12-18 | Isis Pharmaceuticals, Inc. | Carbocyclic bicyclic nucleic acid analogs |
| WO2009006478A2 (en) | 2007-07-05 | 2009-01-08 | Isis Pharmaceuticals, Inc. | 6-disubstituted bicyclic nucleic acid analogs |
| WO2011002207A2 (en) | 2009-07-03 | 2011-01-06 | Kim Kyung-Jo | Rotary bed which can be directed toward the position of the sun |
Non-Patent Citations (18)
| Title |
|---|
| "Antisense Research and Applications", 1993, CRC PRESS, pages: 276 - 278 |
| "Greene's Protective Groups in Organic Synthesis", 2007, JOHN WILEY & SONS |
| "The Concise Encyclopedia OfPolymer Science And Engineering", 1990, JOHN WILEY & SONS, pages: 858 - 859 |
| AGRAWAL ET AL.: "Protocols for Oligonucleotide Conjugates", vol. 26, 1994, HUMANA PRESS, pages: 1 - 72 |
| BARANY ET AL., J. AM. CHEM. SOC., vol. 102, 1980, pages 3084 - 3095 |
| BARANY ET AL., J. AM. CHEM. SOC., vol. 99, 1977, pages 7363 - 7365 |
| BEAUCAGE; CARUTHERS, TETRAHEDRON LETTERS, vol. 22, 1981, pages 1859 - 1862 |
| BEAUCAGE; IYER, TETRAHEDRON, vol. 48, 1992, pages 2223 - 2311 |
| CHATTOPADHYAYA ET AL., J. ORG. CHEM., vol. 74, 2009, pages 118 - 134 |
| ENGLISCH ET AL.: "Angewandte Chemie", vol. 30, 1991, pages: 613 |
| MCBRIDE; CARUTHERS, TETRAHEDRON LETTERS, vol. 24, 1983, pages 245 - 248 |
| MUKOBATA T ET AL: "Facile and efficient approach for the synthesis of N<2>-dimethylaminomethylene-22-O-methylguanosine", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, ELSEVIER SCIENCE, GB, vol. 20, no. 1, 1 January 2010 (2010-01-01), pages 129 - 131, XP026808788, ISSN: 0960-894X, [retrieved on 20091112] * |
| NUCLEOSIDES, NUCLEOTIDES AND NUCLEIC ACIDS, vol. 22, 2003, pages 583 - 587 |
| SANGHVI, Y. S.; THEODORAKIS, E. A. ET AL., J. ORG. CHEM., vol. 67, 2002, pages 7887 - 7889 |
| SANGHVI, Y.S.: "Antisense Research and Applications", CRC PRESS, 1993 |
| SINHA ET AL., NUCLEIC ACIDS RES., vol. 12, 1984, pages 4539 - 4557 |
| V. SEREBRYANY ET AL: "Synthesis of 2'-O-Substituted Ribonucleosides", NUCLEOSIDES, NUCLEOTIDES & NUCLEIC ACIDS, vol. 22, no. 5-8, 1 January 2003 (2003-01-01), pages 1007 - 1009, XP055001977, ISSN: 1525-7770, DOI: 10.1081/NCN-120022724 * |
| ZLATEV ET AL: "Convenient synthesis of N<2>-isobutyryl-2'-O-methyl guanosine by efficient alkylation of O<6>-trimethylsilylethyl-3',5'-di-tert-butyls ilanediyl guanosine", TETRAHEDRON, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, vol. 63, no. 45, 27 September 2007 (2007-09-27), pages 11174 - 11178, XP022274994, ISSN: 0040-4020, DOI: DOI:10.1016/J.TET.2007.08.006 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106117289A (en) * | 2016-06-24 | 2016-11-16 | 郑州大学 | 2 ' O MOE 3 ' H thiophosphate nucleoside monomers and synthetic methods thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7569686B1 (en) | Compounds and methods for synthesis of bicyclic nucleic acid analogs | |
| CA2372085C (en) | L-ribo-lna analogues | |
| WO2009143369A2 (en) | Method of preparing nucleosides and analogs thereof without using chromatography | |
| WO2019002237A1 (en) | Multiple coupling & oxidation method | |
| WO2011103468A2 (en) | Phosphoramidites for synthetic rna in the reverse direction | |
| WO2017194498A1 (en) | Enhanced coupling of stereodefined oxazaphospholidine phosphoramidite monomers to nucleoside or oligonucleotide | |
| US11400161B2 (en) | Method of conjugating oligomeric compounds | |
| JP2020512360A (en) | Orthogonal protecting groups for the preparation of sterically defined phosphorothioate oligonucleotides | |
| WO2014120861A2 (en) | Method of preparing oligomeric compounds using modified coupling protocols | |
| EP4180442A1 (en) | Method for producing nucleic acid oligomer | |
| US10450342B2 (en) | Method for solution phase detritylation of oligomeric compounds | |
| US5639867A (en) | TTTr as protective group in nucleotide synthesis | |
| KR20030081303A (en) | Synthons for oligonucleotide synthesis | |
| US8513404B2 (en) | Process for the manufacture of oligonucleotides | |
| WO2011115817A1 (en) | Methods of preparing 2'-o-substituted purine nucleosides | |
| US9758546B2 (en) | Method for solution phase detritylation of oligomeric compounds | |
| WO2006095739A1 (en) | Process for deblocking the 2'-hydroxyl groups of ribonucleosides | |
| CA2051217C (en) | Process for the chemical synthesis of oligonucleotides | |
| EP1737877B1 (en) | Process for the removal of exocyclic base protecting groups | |
| Virta | Solid-phase synthesis of base-sensitive oligonucleotides | |
| KR20220123300A (en) | Nucleic acid synthesis method using segment-type amidite | |
| WO2021080021A1 (en) | Method for producing oligonucleotide | |
| US8193337B2 (en) | Oxidation process |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11708671 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11708671 Country of ref document: EP Kind code of ref document: A1 |