WO2011106779A1 - Use of modified il-4 mutien receptor antagonists to treat dermatitis - Google Patents
Use of modified il-4 mutien receptor antagonists to treat dermatitis Download PDFInfo
- Publication number
- WO2011106779A1 WO2011106779A1 PCT/US2011/026521 US2011026521W WO2011106779A1 WO 2011106779 A1 WO2011106779 A1 WO 2011106779A1 US 2011026521 W US2011026521 W US 2011026521W WO 2011106779 A1 WO2011106779 A1 WO 2011106779A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- modified
- protein
- effective amount
- therapeutically effective
- dermatitis
- Prior art date
Links
- 239000002464 receptor antagonist Substances 0.000 title claims description 71
- 229940044551 receptor antagonist Drugs 0.000 title claims description 71
- 201000004624 Dermatitis Diseases 0.000 title claims description 47
- 102000004388 Interleukin-4 Human genes 0.000 claims abstract description 179
- 108090000978 Interleukin-4 Proteins 0.000 claims abstract description 179
- 238000000034 method Methods 0.000 claims abstract description 94
- 101001002709 Homo sapiens Interleukin-4 Proteins 0.000 claims abstract description 61
- 102000055229 human IL4 Human genes 0.000 claims abstract description 61
- 239000000203 mixture Substances 0.000 claims abstract description 35
- 208000010668 atopic eczema Diseases 0.000 claims abstract description 19
- 201000008937 atopic dermatitis Diseases 0.000 claims abstract description 15
- 206010012438 Dermatitis atopic Diseases 0.000 claims abstract description 14
- 230000000172 allergic effect Effects 0.000 claims abstract description 5
- 229920001223 polyethylene glycol Polymers 0.000 claims description 60
- 239000002202 Polyethylene glycol Substances 0.000 claims description 55
- 108090000623 proteins and genes Proteins 0.000 claims description 49
- 150000001413 amino acids Chemical class 0.000 claims description 47
- 235000001014 amino acid Nutrition 0.000 claims description 45
- 235000018102 proteins Nutrition 0.000 claims description 43
- 102000004169 proteins and genes Human genes 0.000 claims description 43
- 229940024606 amino acid Drugs 0.000 claims description 39
- 239000003795 chemical substances by application Substances 0.000 claims description 36
- 238000006467 substitution reaction Methods 0.000 claims description 32
- 235000018417 cysteine Nutrition 0.000 claims description 31
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 29
- 208000024891 symptom Diseases 0.000 claims description 25
- 229920000642 polymer Polymers 0.000 claims description 24
- 230000002401 inhibitory effect Effects 0.000 claims description 12
- 230000004048 modification Effects 0.000 claims description 12
- 238000012986 modification Methods 0.000 claims description 12
- 230000004044 response Effects 0.000 claims description 12
- 238000006243 chemical reaction Methods 0.000 claims description 10
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 claims description 9
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 claims description 9
- 239000004473 Threonine Substances 0.000 claims description 9
- 235000009582 asparagine Nutrition 0.000 claims description 9
- 229960001230 asparagine Drugs 0.000 claims description 9
- 230000003442 weekly effect Effects 0.000 claims description 9
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 claims description 8
- 206010020751 Hypersensitivity Diseases 0.000 claims description 7
- 206010012442 Dermatitis contact Diseases 0.000 claims description 6
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 claims description 6
- 208000010247 contact dermatitis Diseases 0.000 claims description 6
- 239000002552 dosage form Substances 0.000 claims description 6
- 230000000116 mitigating effect Effects 0.000 claims description 6
- 239000008194 pharmaceutical composition Substances 0.000 claims description 5
- 229920001451 polypropylene glycol Polymers 0.000 claims description 2
- 125000003275 alpha amino acid group Chemical group 0.000 claims 3
- 150000001945 cysteines Chemical group 0.000 claims 3
- 206010003645 Atopy Diseases 0.000 claims 1
- 229940028885 interleukin-4 Drugs 0.000 abstract description 156
- 102000003816 Interleukin-13 Human genes 0.000 abstract description 41
- 108090000176 Interleukin-13 Proteins 0.000 abstract description 41
- 239000005557 antagonist Substances 0.000 abstract description 35
- 208000012657 Atopic disease Diseases 0.000 abstract description 10
- 230000002757 inflammatory effect Effects 0.000 abstract description 2
- 208000017520 skin disease Diseases 0.000 abstract description 2
- 210000004027 cell Anatomy 0.000 description 69
- 230000000694 effects Effects 0.000 description 52
- 108090000765 processed proteins & peptides Proteins 0.000 description 50
- 238000011282 treatment Methods 0.000 description 41
- 102000004196 processed proteins & peptides Human genes 0.000 description 40
- 229920001184 polypeptide Polymers 0.000 description 35
- 239000013566 allergen Substances 0.000 description 28
- 208000002200 Respiratory Hypersensitivity Diseases 0.000 description 26
- 230000010085 airway hyperresponsiveness Effects 0.000 description 26
- 230000006320 pegylation Effects 0.000 description 26
- 239000013598 vector Substances 0.000 description 25
- 230000001225 therapeutic effect Effects 0.000 description 24
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 22
- 239000003814 drug Substances 0.000 description 22
- 208000026935 allergic disease Diseases 0.000 description 21
- 230000035772 mutation Effects 0.000 description 20
- 208000006673 asthma Diseases 0.000 description 19
- 229940079593 drug Drugs 0.000 description 18
- 230000014509 gene expression Effects 0.000 description 18
- 239000013543 active substance Substances 0.000 description 17
- 230000007423 decrease Effects 0.000 description 16
- 108091033319 polynucleotide Proteins 0.000 description 16
- 102000040430 polynucleotide Human genes 0.000 description 16
- 239000002157 polynucleotide Substances 0.000 description 16
- 102000005962 receptors Human genes 0.000 description 16
- 108020003175 receptors Proteins 0.000 description 16
- 241001465754 Metazoa Species 0.000 description 15
- 238000003556 assay Methods 0.000 description 15
- 210000003719 b-lymphocyte Anatomy 0.000 description 15
- 108010038486 Interleukin-4 Receptors Proteins 0.000 description 14
- 102000010787 Interleukin-4 Receptors Human genes 0.000 description 14
- 102100039078 Interleukin-4 receptor subunit alpha Human genes 0.000 description 14
- 239000000306 component Substances 0.000 description 14
- 210000001744 T-lymphocyte Anatomy 0.000 description 13
- 238000004458 analytical method Methods 0.000 description 13
- 239000000902 placebo Substances 0.000 description 13
- 229940068196 placebo Drugs 0.000 description 13
- 108020004414 DNA Proteins 0.000 description 12
- 230000001965 increasing effect Effects 0.000 description 12
- 238000002347 injection Methods 0.000 description 12
- 239000007924 injection Substances 0.000 description 12
- 230000036470 plasma concentration Effects 0.000 description 12
- 239000013612 plasmid Substances 0.000 description 12
- 108020004705 Codon Proteins 0.000 description 11
- 241000588724 Escherichia coli Species 0.000 description 11
- 101710169536 Interleukin-4 receptor subunit alpha Proteins 0.000 description 11
- 239000000872 buffer Substances 0.000 description 11
- 210000004072 lung Anatomy 0.000 description 11
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 11
- 241000244188 Ascaris suum Species 0.000 description 10
- 210000003000 inclusion body Anatomy 0.000 description 10
- NZWOPGCLSHLLPA-UHFFFAOYSA-N methacholine Chemical compound C[N+](C)(C)CC(C)OC(C)=O NZWOPGCLSHLLPA-UHFFFAOYSA-N 0.000 description 10
- 229960002329 methacholine Drugs 0.000 description 10
- 230000035755 proliferation Effects 0.000 description 10
- 238000007920 subcutaneous administration Methods 0.000 description 10
- 241000282567 Macaca fascicularis Species 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- 239000003153 chemical reaction reagent Substances 0.000 description 9
- 150000001875 compounds Chemical class 0.000 description 9
- 239000001963 growth medium Substances 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 210000003491 skin Anatomy 0.000 description 9
- 102100020941 Interleukin-4 Human genes 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 238000010521 absorption reaction Methods 0.000 description 8
- 208000037883 airway inflammation Diseases 0.000 description 8
- CKLJMWTZIZZHCS-REOHCLBHSA-N aspartic acid group Chemical group N[C@@H](CC(=O)O)C(=O)O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 8
- 230000008901 benefit Effects 0.000 description 8
- 238000004422 calculation algorithm Methods 0.000 description 8
- 238000001516 cell proliferation assay Methods 0.000 description 8
- 239000002299 complementary DNA Substances 0.000 description 8
- 238000012377 drug delivery Methods 0.000 description 8
- 210000003979 eosinophil Anatomy 0.000 description 8
- 230000006872 improvement Effects 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 239000011159 matrix material Substances 0.000 description 8
- 150000007523 nucleic acids Chemical class 0.000 description 8
- 108010010907 pitrakinra Proteins 0.000 description 8
- 229950008185 pitrakinra Drugs 0.000 description 8
- 238000003752 polymerase chain reaction Methods 0.000 description 8
- 239000013615 primer Substances 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- 102000004127 Cytokines Human genes 0.000 description 7
- 108090000695 Cytokines Proteins 0.000 description 7
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 7
- 235000003704 aspartic acid Nutrition 0.000 description 7
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- 238000005119 centrifugation Methods 0.000 description 7
- 238000011161 development Methods 0.000 description 7
- 230000018109 developmental process Effects 0.000 description 7
- -1 hyposensitization Substances 0.000 description 7
- 238000000338 in vitro Methods 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 238000013268 sustained release Methods 0.000 description 7
- 239000012730 sustained-release form Substances 0.000 description 7
- 239000004475 Arginine Substances 0.000 description 6
- 241000282693 Cercopithecidae Species 0.000 description 6
- 206010014950 Eosinophilia Diseases 0.000 description 6
- 206010061218 Inflammation Diseases 0.000 description 6
- 241000288906 Primates Species 0.000 description 6
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 6
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 230000001413 cellular effect Effects 0.000 description 6
- 230000008878 coupling Effects 0.000 description 6
- 238000010168 coupling process Methods 0.000 description 6
- 238000005859 coupling reaction Methods 0.000 description 6
- 238000010494 dissociation reaction Methods 0.000 description 6
- 230000005593 dissociations Effects 0.000 description 6
- 239000003937 drug carrier Substances 0.000 description 6
- 230000028993 immune response Effects 0.000 description 6
- 230000004054 inflammatory process Effects 0.000 description 6
- 230000003993 interaction Effects 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 102000039446 nucleic acids Human genes 0.000 description 6
- 108020004707 nucleic acids Proteins 0.000 description 6
- 230000036515 potency Effects 0.000 description 6
- 230000009696 proliferative response Effects 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 238000012384 transportation and delivery Methods 0.000 description 6
- 101100069857 Caenorhabditis elegans hil-4 gene Proteins 0.000 description 5
- 238000002965 ELISA Methods 0.000 description 5
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 5
- 239000000739 antihistaminic agent Substances 0.000 description 5
- 239000013592 cell lysate Substances 0.000 description 5
- 230000004663 cell proliferation Effects 0.000 description 5
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 5
- 238000012217 deletion Methods 0.000 description 5
- 230000037430 deletion Effects 0.000 description 5
- 238000013461 design Methods 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 238000001990 intravenous administration Methods 0.000 description 5
- 239000002085 irritant Substances 0.000 description 5
- 231100000021 irritant Toxicity 0.000 description 5
- 210000004962 mammalian cell Anatomy 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000003223 protective agent Substances 0.000 description 5
- 238000003259 recombinant expression Methods 0.000 description 5
- 230000004043 responsiveness Effects 0.000 description 5
- 238000010254 subcutaneous injection Methods 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 5
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 108010076504 Protein Sorting Signals Proteins 0.000 description 4
- 208000003251 Pruritus Diseases 0.000 description 4
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 4
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 4
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 4
- 230000003042 antagnostic effect Effects 0.000 description 4
- 239000000427 antigen Substances 0.000 description 4
- 102000036639 antigens Human genes 0.000 description 4
- 108091007433 antigens Proteins 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 230000004071 biological effect Effects 0.000 description 4
- 238000011260 co-administration Methods 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 4
- 230000009977 dual effect Effects 0.000 description 4
- 238000001502 gel electrophoresis Methods 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 230000028709 inflammatory response Effects 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 230000007803 itching Effects 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 239000002953 phosphate buffered saline Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 230000010076 replication Effects 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 238000002741 site-directed mutagenesis Methods 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- 125000003396 thiol group Chemical group [H]S* 0.000 description 4
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- ASUGWWOMVNVWAW-UHFFFAOYSA-N 1-(2-methoxyethyl)pyrrole-2,5-dione Chemical compound COCCN1C(=O)C=CC1=O ASUGWWOMVNVWAW-UHFFFAOYSA-N 0.000 description 3
- 108091026890 Coding region Proteins 0.000 description 3
- 108091035707 Consensus sequence Proteins 0.000 description 3
- 239000003155 DNA primer Substances 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 3
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 3
- 102000008300 Mutant Proteins Human genes 0.000 description 3
- 108010021466 Mutant Proteins Proteins 0.000 description 3
- 108091006006 PEGylated Proteins Proteins 0.000 description 3
- 238000012300 Sequence Analysis Methods 0.000 description 3
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 239000012620 biological material Substances 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 238000000423 cell based assay Methods 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 230000010405 clearance mechanism Effects 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 238000004590 computer program Methods 0.000 description 3
- 238000013270 controlled release Methods 0.000 description 3
- 239000003246 corticosteroid Substances 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- 150000001944 cysteine derivatives Chemical group 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- 238000003379 elimination reaction Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 239000013604 expression vector Substances 0.000 description 3
- 239000012634 fragment Substances 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000003102 growth factor Substances 0.000 description 3
- ZQDWXGKKHFNSQK-UHFFFAOYSA-N hydroxyzine Chemical compound C1CN(CCOCCO)CCN1C(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 ZQDWXGKKHFNSQK-UHFFFAOYSA-N 0.000 description 3
- 210000002865 immune cell Anatomy 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 230000001976 improved effect Effects 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 238000003780 insertion Methods 0.000 description 3
- 230000037431 insertion Effects 0.000 description 3
- 125000005647 linker group Chemical group 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000002703 mutagenesis Methods 0.000 description 3
- 231100000350 mutagenesis Toxicity 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 235000015097 nutrients Nutrition 0.000 description 3
- 230000008506 pathogenesis Effects 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 239000008363 phosphate buffer Substances 0.000 description 3
- 230000004481 post-translational protein modification Effects 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 210000001236 prokaryotic cell Anatomy 0.000 description 3
- 108091008146 restriction endonucleases Proteins 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 230000003637 steroidlike Effects 0.000 description 3
- 239000011550 stock solution Substances 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 239000007929 subcutaneous injection Substances 0.000 description 3
- 238000012385 systemic delivery Methods 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- 102220471101 Aryl hydrocarbon receptor_F82D_mutation Human genes 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 150000008574 D-amino acids Chemical class 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 102100031780 Endonuclease Human genes 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- 108091034117 Oligonucleotide Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 229920002732 Polyanhydride Polymers 0.000 description 2
- 229920001710 Polyorthoester Polymers 0.000 description 2
- 102220471486 Protein MON2 homolog_S36C_mutation Human genes 0.000 description 2
- PLXBWHJQWKZRKG-UHFFFAOYSA-N Resazurin Chemical compound C1=CC(=O)C=C2OC3=CC(O)=CC=C3[N+]([O-])=C21 PLXBWHJQWKZRKG-UHFFFAOYSA-N 0.000 description 2
- 102000013968 STAT6 Transcription Factor Human genes 0.000 description 2
- 108010011005 STAT6 Transcription Factor Proteins 0.000 description 2
- 102220492463 Selenoprotein V_E19A_mutation Human genes 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 2
- 201000009961 allergic asthma Diseases 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 230000001387 anti-histamine Effects 0.000 description 2
- 229940125715 antihistaminic agent Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 238000005341 cation exchange Methods 0.000 description 2
- 238000005277 cation exchange chromatography Methods 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical compound C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 210000003191 femoral vein Anatomy 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 229960000789 guanidine hydrochloride Drugs 0.000 description 2
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 239000000833 heterodimer Substances 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229960002591 hydroxyproline Drugs 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 108700025647 major vault Proteins 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 238000002483 medication Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 238000004806 packaging method and process Methods 0.000 description 2
- 239000000816 peptidomimetic Substances 0.000 description 2
- 210000005259 peripheral blood Anatomy 0.000 description 2
- 239000011886 peripheral blood Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 150000008300 phosphoramidites Chemical class 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 230000004983 pleiotropic effect Effects 0.000 description 2
- 229920001308 poly(aminoacid) Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000011809 primate model Methods 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 102220105250 rs879254386 Human genes 0.000 description 2
- 239000012146 running buffer Substances 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 229960001967 tacrolimus Drugs 0.000 description 2
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 229920001169 thermoplastic Polymers 0.000 description 2
- 210000000115 thoracic cavity Anatomy 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000000451 tissue damage Effects 0.000 description 2
- 231100000827 tissue damage Toxicity 0.000 description 2
- 238000004448 titration Methods 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 235000012431 wafers Nutrition 0.000 description 2
- IQFYYKKMVGJFEH-OFKYTIFKSA-N 1-[(2r,4s,5r)-4-hydroxy-5-(tritiooxymethyl)oxolan-2-yl]-5-methylpyrimidine-2,4-dione Chemical compound C1[C@H](O)[C@@H](CO[3H])O[C@H]1N1C(=O)NC(=O)C(C)=C1 IQFYYKKMVGJFEH-OFKYTIFKSA-N 0.000 description 1
- FUFLCEKSBBHCMO-UHFFFAOYSA-N 11-dehydrocorticosterone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 FUFLCEKSBBHCMO-UHFFFAOYSA-N 0.000 description 1
- CIVCELMLGDGMKZ-UHFFFAOYSA-N 2,4-dichloro-6-methylpyridine-3-carboxylic acid Chemical compound CC1=CC(Cl)=C(C(O)=O)C(Cl)=N1 CIVCELMLGDGMKZ-UHFFFAOYSA-N 0.000 description 1
- QVSACOUKYXVIGR-UHFFFAOYSA-N 2-(3,5-dimethyl-1,2-oxazol-4-yl)ethanol Chemical compound CC1=NOC(C)=C1CCO QVSACOUKYXVIGR-UHFFFAOYSA-N 0.000 description 1
- PMUNIMVZCACZBB-UHFFFAOYSA-N 2-hydroxyethylazanium;chloride Chemical compound Cl.NCCO PMUNIMVZCACZBB-UHFFFAOYSA-N 0.000 description 1
- BRMWTNUJHUMWMS-UHFFFAOYSA-N 3-Methylhistidine Natural products CN1C=NC(CC(N)C(O)=O)=C1 BRMWTNUJHUMWMS-UHFFFAOYSA-N 0.000 description 1
- 125000004042 4-aminobutyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])N([H])[H] 0.000 description 1
- 229940117976 5-hydroxylysine Drugs 0.000 description 1
- 206010001343 Adrenal cortical hypofunctions Diseases 0.000 description 1
- 206010001367 Adrenal insufficiency Diseases 0.000 description 1
- 201000004384 Alopecia Diseases 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 102100039339 Atrial natriuretic peptide receptor 1 Human genes 0.000 description 1
- 101710102163 Atrial natriuretic peptide receptor 1 Proteins 0.000 description 1
- 241000193830 Bacillus <bacterium> Species 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 238000012492 Biacore method Methods 0.000 description 1
- 101150013553 CD40 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- DBAKFASWICGISY-BTJKTKAUSA-N Chlorpheniramine maleate Chemical compound OC(=O)\C=C/C(O)=O.C=1C=CC=NC=1C(CCN(C)C)C1=CC=C(Cl)C=C1 DBAKFASWICGISY-BTJKTKAUSA-N 0.000 description 1
- 206010010744 Conjunctivitis allergic Diseases 0.000 description 1
- MFYSYFVPBJMHGN-ZPOLXVRWSA-N Cortisone Chemical compound O=C1CC[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MFYSYFVPBJMHGN-ZPOLXVRWSA-N 0.000 description 1
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 206010012434 Dermatitis allergic Diseases 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 108010042407 Endonucleases Proteins 0.000 description 1
- 208000031637 Erythroblastic Acute Leukemia Diseases 0.000 description 1
- 208000036566 Erythroleukaemia Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 208000004262 Food Hypersensitivity Diseases 0.000 description 1
- 206010016946 Food allergy Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 206010020112 Hirsutism Diseases 0.000 description 1
- 101000746373 Homo sapiens Granulocyte-macrophage colony-stimulating factor Proteins 0.000 description 1
- 101001003135 Homo sapiens Interleukin-13 receptor subunit alpha-1 Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 102000018682 Interleukin Receptor Common gamma Subunit Human genes 0.000 description 1
- 108010066719 Interleukin Receptor Common gamma Subunit Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- YQEZLKZALYSWHR-UHFFFAOYSA-N Ketamine Chemical compound C=1C=CC=C(Cl)C=1C1(NC)CCCCC1=O YQEZLKZALYSWHR-UHFFFAOYSA-N 0.000 description 1
- 241000235058 Komagataella pastoris Species 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 206010024438 Lichenification Diseases 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- HOKDBMAJZXIPGC-UHFFFAOYSA-N Mequitazine Chemical compound C12=CC=CC=C2SC2=CC=CC=C2N1CC1C(CC2)CCN2C1 HOKDBMAJZXIPGC-UHFFFAOYSA-N 0.000 description 1
- JDHILDINMRGULE-LURJTMIESA-N N(pros)-methyl-L-histidine Chemical compound CN1C=NC=C1C[C@H](N)C(O)=O JDHILDINMRGULE-LURJTMIESA-N 0.000 description 1
- JJIHLJJYMXLCOY-BYPYZUCNSA-N N-acetyl-L-serine Chemical compound CC(=O)N[C@@H](CO)C(O)=O JJIHLJJYMXLCOY-BYPYZUCNSA-N 0.000 description 1
- PYUSHNKNPOHWEZ-YFKPBYRVSA-N N-formyl-L-methionine Chemical compound CSCC[C@@H](C(O)=O)NC=O PYUSHNKNPOHWEZ-YFKPBYRVSA-N 0.000 description 1
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- BYPFEZZEUUWMEJ-UHFFFAOYSA-N Pentoxifylline Chemical compound O=C1N(CCCCC(=O)C)C(=O)N(C)C2=C1N(C)C=N2 BYPFEZZEUUWMEJ-UHFFFAOYSA-N 0.000 description 1
- 208000008469 Peptic Ulcer Diseases 0.000 description 1
- 208000037581 Persistent Infection Diseases 0.000 description 1
- 241001662443 Phemeranthus parviflorus Species 0.000 description 1
- 241000233805 Phoenix Species 0.000 description 1
- 108010047620 Phytohemagglutinins Proteins 0.000 description 1
- 208000012641 Pigmentation disease Diseases 0.000 description 1
- 229920001244 Poly(D,L-lactide) Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 241001415846 Procellariidae Species 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 108091034057 RNA (poly(A)) Proteins 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 230000010799 Receptor Interactions Effects 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 206010039793 Seborrhoeic dermatitis Diseases 0.000 description 1
- 206010070834 Sensitisation Diseases 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 108091081024 Start codon Proteins 0.000 description 1
- 108010006785 Taq Polymerase Proteins 0.000 description 1
- 101800001703 Thymopentin Proteins 0.000 description 1
- 102400000160 Thymopentin Human genes 0.000 description 1
- 108060008539 Transglutaminase Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 1
- 206010045240 Type I hypersensitivity Diseases 0.000 description 1
- 244000000188 Vaccinium ovalifolium Species 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 208000021841 acute erythroid leukemia Diseases 0.000 description 1
- 208000017515 adrenocortical insufficiency Diseases 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 229920003232 aliphatic polyester Polymers 0.000 description 1
- 230000002009 allergenic effect Effects 0.000 description 1
- 208000002205 allergic conjunctivitis Diseases 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 231100000360 alopecia Toxicity 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 239000003945 anionic surfactant Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 229940124623 antihistamine drug Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 150000001483 arginine derivatives Chemical class 0.000 description 1
- 229940065779 atarax Drugs 0.000 description 1
- 208000024998 atopic conjunctivitis Diseases 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 229940088007 benadryl Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 229960001102 betamethasone dipropionate Drugs 0.000 description 1
- CIWBQSYVNNPZIQ-XYWKZLDCSA-N betamethasone dipropionate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O CIWBQSYVNNPZIQ-XYWKZLDCSA-N 0.000 description 1
- 239000012867 bioactive agent Substances 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 238000010241 blood sampling Methods 0.000 description 1
- 239000008366 buffered solution Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- UHBYWPGGCSDKFX-UHFFFAOYSA-N carboxyglutamic acid Chemical compound OC(=O)C(N)CC(C(O)=O)C(O)=O UHBYWPGGCSDKFX-UHFFFAOYSA-N 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 description 1
- 238000012219 cassette mutagenesis Methods 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000006041 cell recruitment Effects 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000002975 chemoattractant Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229940046978 chlorpheniramine maleate Drugs 0.000 description 1
- 235000019504 cigarettes Nutrition 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 229960004703 clobetasol propionate Drugs 0.000 description 1
- CBGUOGMQLZIXBE-XGQKBEPLSA-N clobetasol propionate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CCl)(OC(=O)CC)[C@@]1(C)C[C@@H]2O CBGUOGMQLZIXBE-XGQKBEPLSA-N 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 229940124558 contraceptive agent Drugs 0.000 description 1
- 239000003433 contraceptive agent Substances 0.000 description 1
- 239000000599 controlled substance Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- BMCQMVFGOVHVNG-TUFAYURCSA-N cortisol 17-butyrate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)CO)(OC(=O)CCC)[C@@]1(C)C[C@@H]2O BMCQMVFGOVHVNG-TUFAYURCSA-N 0.000 description 1
- 229960004544 cortisone Drugs 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 230000001351 cycling effect Effects 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- JJCFRYNCJDLXIK-UHFFFAOYSA-N cyproheptadine Chemical compound C1CN(C)CCC1=C1C2=CC=CC=C2C=CC2=CC=CC=C21 JJCFRYNCJDLXIK-UHFFFAOYSA-N 0.000 description 1
- 229960001140 cyproheptadine Drugs 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000003412 degenerative effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- YSMODUONRAFBET-UHFFFAOYSA-N delta-DL-hydroxylysine Natural products NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 229950006825 dexamethasone valerate Drugs 0.000 description 1
- SNHRLVCMMWUAJD-OMPPIWKSSA-N dexamethasone valerate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(OC(=O)CCCC)[C@@]1(C)C[C@@H]2O SNHRLVCMMWUAJD-OMPPIWKSSA-N 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 235000020805 dietary restrictions Nutrition 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 229960000520 diphenhydramine Drugs 0.000 description 1
- 229960000525 diphenhydramine hydrochloride Drugs 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 229910000397 disodium phosphate Inorganic materials 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000007071 enzymatic hydrolysis Effects 0.000 description 1
- 238000006047 enzymatic hydrolysis reaction Methods 0.000 description 1
- YSMODUONRAFBET-UHNVWZDZSA-N erythro-5-hydroxy-L-lysine Chemical compound NC[C@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-UHNVWZDZSA-N 0.000 description 1
- WEMXGZLXCDDUSY-UHFFFAOYSA-N ethane-1,2-diol;pyrrole-2,5-dione Chemical class OCCO.O=C1NC(=O)C=C1 WEMXGZLXCDDUSY-UHFFFAOYSA-N 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 208000024711 extrinsic asthma Diseases 0.000 description 1
- 238000000855 fermentation Methods 0.000 description 1
- 230000004151 fermentation Effects 0.000 description 1
- 239000012997 ficoll-paque Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000020932 food allergy Nutrition 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229940084910 gliadel Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 102000046157 human CSF2 Human genes 0.000 description 1
- 230000000887 hydrating effect Effects 0.000 description 1
- 229960001524 hydrocortisone butyrate Drugs 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 229960000930 hydroxyzine Drugs 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000008105 immune reaction Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000006749 inflammatory damage Effects 0.000 description 1
- 238000002664 inhalation therapy Methods 0.000 description 1
- 230000003434 inspiratory effect Effects 0.000 description 1
- 238000012482 interaction analysis Methods 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 230000018711 interleukin-13 production Effects 0.000 description 1
- 230000017307 interleukin-4 production Effects 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 1
- 230000005722 itchiness Effects 0.000 description 1
- 210000004731 jugular vein Anatomy 0.000 description 1
- 229940015418 ketaset Drugs 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 150000002605 large molecules Chemical class 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 150000002617 leukotrienes Chemical class 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000013178 mathematical model Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229960005042 mequitazine Drugs 0.000 description 1
- VUZPPFZMUPKLLV-UHFFFAOYSA-N methane;hydrate Chemical compound C.O VUZPPFZMUPKLLV-UHFFFAOYSA-N 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- AYLRODJJLADBOB-QMMMGPOBSA-N methyl (2s)-2,6-diisocyanatohexanoate Chemical compound COC(=O)[C@@H](N=C=O)CCCCN=C=O AYLRODJJLADBOB-QMMMGPOBSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000001823 molecular biology technique Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000000302 molecular modelling Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 230000003562 morphometric effect Effects 0.000 description 1
- 238000013425 morphometry Methods 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 239000003887 narcotic antagonist Substances 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 238000002663 nebulization Methods 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 229940053934 norethindrone Drugs 0.000 description 1
- VIKNJXKGJWUCNN-XGXHKTLJSA-N norethisterone Chemical compound O=C1CC[C@@H]2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 VIKNJXKGJWUCNN-XGXHKTLJSA-N 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 238000001821 nucleic acid purification Methods 0.000 description 1
- 238000001668 nucleic acid synthesis Methods 0.000 description 1
- 229940124624 oral corticosteroid Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229960001476 pentoxifylline Drugs 0.000 description 1
- 208000011906 peptic ulcer disease Diseases 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 210000001322 periplasm Anatomy 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 239000007981 phosphate-citrate buffer Substances 0.000 description 1
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 1
- BZQFBWGGLXLEPQ-REOHCLBHSA-N phosphoserine Chemical compound OC(=O)[C@@H](N)COP(O)(O)=O BZQFBWGGLXLEPQ-REOHCLBHSA-N 0.000 description 1
- 238000001126 phototherapy Methods 0.000 description 1
- 230000001885 phytohemagglutinin Effects 0.000 description 1
- 108010086652 phytohemagglutinin-P Proteins 0.000 description 1
- 230000019612 pigmentation Effects 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- UVTKHPSJNFFIDG-UHFFFAOYSA-L potassium tetrathionate Chemical compound [K+].[K+].[O-]S(=O)(=O)SSS([O-])(=O)=O UVTKHPSJNFFIDG-UHFFFAOYSA-L 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 210000004986 primary T-cell Anatomy 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- XXPDBLUZJRXNNZ-UHFFFAOYSA-N promethazine hydrochloride Chemical compound Cl.C1=CC=C2N(CC(C)N(C)C)C3=CC=CC=C3SC2=C1 XXPDBLUZJRXNNZ-UHFFFAOYSA-N 0.000 description 1
- 229960002244 promethazine hydrochloride Drugs 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 230000030788 protein refolding Effects 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 229940112971 protopic Drugs 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 230000036647 reaction Effects 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000001525 receptor binding assay Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 208000008742 seborrheic dermatitis Diseases 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 239000000932 sedative agent Substances 0.000 description 1
- 230000001624 sedative effect Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000008313 sensitization Effects 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 230000007781 signaling event Effects 0.000 description 1
- 229920000260 silastic Polymers 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 238000004513 sizing Methods 0.000 description 1
- 230000008591 skin barrier function Effects 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000011537 solubilization buffer Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 238000000528 statistical test Methods 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 229920002994 synthetic fiber Polymers 0.000 description 1
- 239000012209 synthetic fiber Substances 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000011269 tar Substances 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 239000004416 thermosoftening plastic Substances 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- PSWFFKRAVBDQEG-YGQNSOCVSA-N thymopentin Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 PSWFFKRAVBDQEG-YGQNSOCVSA-N 0.000 description 1
- 229960004517 thymopentin Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000011426 transformation method Methods 0.000 description 1
- 102000003601 transglutaminase Human genes 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 238000003211 trypan blue cell staining Methods 0.000 description 1
- 238000009192 ultraviolet light therapy Methods 0.000 description 1
- 229940079707 vistaril Drugs 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- BPICBUSOMSTKRF-UHFFFAOYSA-N xylazine Chemical compound CC1=CC=CC(C)=C1NC1=NCCCS1 BPICBUSOMSTKRF-UHFFFAOYSA-N 0.000 description 1
- 229960001600 xylazine Drugs 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4702—Regulators; Modulating activity
- C07K14/4703—Inhibitors; Suppressors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
- C07K14/5406—IL-4
Definitions
- the present invention relates to methods for treating atopic diseases, including atopic dermatitis and other inflammatory or allergic skin disorders by administering mutant human Interleukin-4 (IL-4) compositions that act as antagonists to IL-4 and IL-13.
- IL-4 human Interleukin-4
- Interleukin-4 is a pleiotropic cytokine with a broad spectrum of biological effects on several target cells, including activation, proliferation and differentiation of T and B cells.
- IL-4 is increasingly appreciated as a pivotal cytokine initiating the "Th2-type" inflammatory response, whereas IL-13 is now appreciated as the more probable downstream effector cytokine.
- IL4 acts as a differentiation factor by regulating class switching to the IgGl and IgE isotypes.
- Atopic diseases are characterized by formation of IgE antibodies, which results in immediate hypersensitivity reactions upon exposure to specific allergens.
- the frequent and chronic infections occurring on the skin of atopic disease patients result from the impaired immune response and from the skin barrier breaking down.
- Known treatments of atopic diseases include, hydrating the skin, dietary restrictions, avoidance of irritants and allergens in the environment, tars, antihistamines, hyposensitization, corticosteroids, antibacterials, antifungals, ultraviolet light, leukotriene blockers, inhibitors of mast cell content release, pentoxifylline, azathioprine, cyclosporin A, cyclophosphamide, tacrolimus, interferon gamma, thymopentin and phosphodiesterase inhibitors.
- anti-histamine and steroidal agents are used as therapeutic treatments for atopic diseases.
- Anti-histamine agents typically reduce the itchiness of the allergic response and include diphenhydramine hydrochloride, mequitazine, promethazine hydrochloride, and chlorpheniramine maleate.
- Steroidal agents including prednisolone, hydrocortisone butyrate, dexamethasone valerate, betamethasone dipropionate, clobetasol propionate and the like have also been used to control the itching.
- anti-histamine and steroidal agents relieve the itching, they are not desirable therapeutic agents because they cause other adverse side affects including infection, secondary adrenal cortical insufficiency, diabetes, peptic ulcer, hirsutism, alopecia, and pigmentation.
- the invention provides a method for inhibiting a dermatitis response in a subject.
- the method entails: administering to a subject in need thereof a therapeutically effective amount of a modified IL-4 mutein receptor antagonist, wherein the modified IL-4 mutein receptor antagonist includes at least the following modifications:
- the modified IL-4 mutein receptor antagonist is co-administered with a therapeutically effective amount of an additional agent that mitigates a symptom of dermatitis.
- the invention provides pharmaceutical composition, which includes: a therapeutically effective amount of a modified IL-4 mutein receptor antagonist, wherein the modified IL-4 mutein receptor antagonist includes at least the following modifications:
- a therapeutically effective amount of an additional agent that is useful for mitigating a symptom of dermatitis is sufficient to mitigate a symptom of dermatitis, and the therapeutically effective amount of the additional agent is sufficient to mitigate a symptom of dermatitis.
- the invention provides a kit that includes:
- At least one unit dosage form comprising a therapeutically effective amount of a modified IL-4 mutein receptor antagonist, wherein the modified IL-4 mutein receptor antagonist comprises at least the following modifications:
- At least one unit dosage form comprising a therapeutically effective amount of an additional agent that is useful for mitigating a symptom of dermatitis.
- the therapeutically effective amount of the modified IL-4 mutein receptor antagonist is sufficient to mitigate a symptom of dermatitis, and the therapeutically effective amount of the additional agent is sufficient to mitigate a symptom of dermatitis.
- Figure 1 shows a schematic representation of the chemistry of a
- Figure 2 is a graphical diagram showing data from BIAcore binding to IL-4Ra comparing the IL-4 double mutein (IL-4DM) to the same molecule with a 30kD linear or a 40kD branched PEG at position 38C.
- IL-4DM IL-4 double mutein
- Figure 3 is a graphical diagram showing data from inhibition of TF-1 growth with IL-4 stimulation revealing that PEGylated IL-4TM (T13D/R121D/Y124D) is more potent than PEGylated IL-4DM and is equally potent to IL-4DM
- FIG. 4A-4B AER 003 Model. Model of AER 003 derived from the structure of human IL-4 is shown.
- A Residues implicated in the antagonist activity are highlighted in yellow (AER 001). The mutation T13D (red) results in increased binding to IL-4R a, while N38C (green) serves as an attachment point for PEG.
- B Pegylated AER 003.
- FIG. 5 Study design. AER 003 biological activity was evaluated in a primate model of AHR and inflammation using a double-blind, 2-period cross-over design (A). All animals were rested at least 8 weeks between studies to allow airway responsiveness and inflammation to return to baseline (pre-allergen) levels. Studies were performed using the 7 day primate asthma model (B). Airway responsiveness to inhaled MCh and airway cellular composition by BAL were determined 2 days before (Day 0) and 2 days after (Day 7) three consecutive-day (Days 3, 4, 5) inhalations of Ascaris suum extract. AER 003 (2 mg/kg) or vehicle was administered subcutaneously 48 h prior to the first antigen challenge (Day 1).
- Figure 6 Effect of AER 003 on allergen-induced AHR. Treatment-by- treatment scatterplot of the effect of AER 003 on Day 7 PCioo relative to placebo. Each symbol represents the outcome of a single subject. Points lying above the reference line of no treatment effect indicate an improvement with AER 003 relative to placebo.
- Figure 7 Effect of AER 003 on allergen-induced airway eosnophilia. Treatment-by-treatment scatterplot of the effect of AER 003 on Day 7 lung eosinophil count relative to placebo. Each symbol represents the outcome of a single subject. Points lying below the reference line of no treatment effect indicate an improvement with AER 003 relative to placebo.
- Figure 8 PK profile of AER 003 during monkey efficacy studies.
- the invention provides for methods of treatment of atopic diseases (AD), in particular, atopic dermatitis, by administering a therapeutic effective amount of mutant human IL-4 compositions.
- AD atopic diseases
- Human IL-4 mutant proteins used as antagonists or partial agonists of human IL-4 are also described in U.S. Pat. No. 6,130,318 to Wild et al., the entire contents of which is incorporated herein by reference, in particular, for its description of human IL-4 mutant proteins.
- the methods of the invention can be used to treat typical atopic diseases or allergic dermatitis including contact dermatitis, atopic dermatitis (i.e., eczema), psoriasis, seborrheic dermatitis, and the like. Definitions
- dermatitis is defined generally as an inflammation of the skin. Stedman's Medical Dictionary, 27th edition, Lippincott Williams & Wilkins (2000).
- contact dermatitis is an inflammatory response of the skin to an antigen (or allergen) or irritant (Stedman's Medical
- Irritants are substances that directly affect the skin or cause direct tissue damage, while allergens induce an immunologic reaction that causes
- Some common irritants include wool and synthetic fibers, soaps and detergents, perfumes and cosmetics, dust and sand, cigarette smoke, and substances such as chlorine, mineral oil or solvents.
- allergens are substances typically from foods, plants, or animals that inflame the skin and cause an immune reaction. Initially, allergens typically illicit inflammatory response, including recruitment of cells, for example T cells,
- the contact dermatitis can then develop into eczema accompanied with lichenification and infiltration of the cells.
- atopic dermatitis As used herein, the term "atopic dermatitis,” “atopic eczema,” or “eczema” and related terms are used interchangeably and represent a complex disease primarily caused by cellular immune deficiency and elevated immunoglobulin E (IgE). Allergens that are also irritants to the skin are believed to predispose an individual to develop dermatitis more often than simply exposure to an allergenic trigger. Anxiety, stress and depression may all play a role in the exacerbation of eczema. Further, those with atopic eczema may be discovered to have an increased eosinophil count.
- IgE immunoglobulin E
- wild type IL-4" or "wtIL-4" and equivalents thereof are used interchangeably and refer to human Interleukin-4, native or recombinant, having the 129 normally occurring amino acid sequence of native human IL-4, as disclosed in U.S. Pat. No. 5,017,691, which is incorporated herein by reference.
- the modified human IL-4 receptor antagonists described herein may have various insertions and/or deletions and/or couplings to a non-protein polymer, and are numbered in accordance with the wtIL-4.
- amino acids at positions may be shifted in a mutein.
- the threonine at position 13 in which an amino acid is added to the N-terminus of IL-4
- the threonine at position 13 is actually the 14th amino acid in the mutein sequence; however, this amino acid is still termed "theonine 13" or "T13.”
- "numbered according to wild type IL-4" mean identifying a chosen amino acid with reference to the position at which that amino acid normally occurs in wild type IL-4.
- mutant human IL-4 protein As used herein, the terms "mutant human IL-4 protein,” “modified human IL-4 receptor antagonist,” “mhIL-4,” “IL-4 mutein,” “IL-4 antagonist,” and equivalents thereof are used interchangeably. These polypeptides and functional fragments thereof refer to polypeptides wherein specific amino acid substitutions to the mature human IL-4 protein have been made. These polypeptides include the mIL-4 compositions of the present invention, which are administered to a subject in need of treatment for dermititis. In particular, the mhIL-4 described herein include at least the R121D/Y124D pair of substitutions (“IL-4RA”) (SEQ ID NO: 31). The modification of hIL-4 and of mhIL-4 are described in US Patent Publication No. 20090010874 (published Jan. 8, 2009) and International Publication No. WO 2009065007 (published May 22, 2009), the entire contents of both of which are incorporated herein by reference, in particular, for their description of mhIL-4.
- identity refers to a relationship between the sequences of two or more polypeptide molecules or two or more nucleic acid molecules, as determined by comparing the sequences.
- identity also means the degree of sequence relatedness between nucleic acid molecules or polypeptides, as the case may be, as determined by the match between strings of two or more nucleotide or two or more amino acid sequences.
- Identity measures the percent of identical matches between the smaller of two or more sequences with gap alignments (if any) addressed by a particular mathematical model or computer program (i.e., "algorithms”).
- Preferred methods to determine identity are designed to give the largest match between the sequences tested. Methods to determine identity are described in publicly available computer programs. Preferred computer program methods to determine identity between two sequences include, but are not limited to, the GCG program package, including GAP (Devereux et al, 1984, Nucl. Acid. Res., 12:387; Genetics Computer Group, University of Wisconsin, Madison, WI), BLASTP, BLASTN, and FASTA (Altschul et al, 1990, J. Mol Biol, 215:403-410).
- GCG program package including GAP (Devereux et al, 1984, Nucl. Acid. Res., 12:387; Genetics Computer Group, University of Wisconsin, Madison, WI), BLASTP, BLASTN, and FASTA (Altschul et al, 1990, J. Mol Biol, 215:403-410).
- the BLASTX program is publicly available from the National Center for Biotechnology Information (NCBI) and other sources (BLAST Manual, Altschul et al NCB/NLM/NIH Bethesda, MD 20894; Altschul et al, 1990, supra).
- NCBI National Center for Biotechnology Information
- the well-known Smith Waterman algorithm may also be used to determine identity.
- Certain alignment schemes for aligning two amino acid sequences may result in matching of only a short region of the two sequences, and this small aligned region may have very high sequence identity even though there is no significant relationship between the two full-length sequences. Accordingly, in certain
- the selected alignment method (GAP program) will result in an alignment that spans at least 50 contiguous amino acids of the target polypeptide.
- a gap opening penalty (which is calculated as three-times the average diagonal; where the "average diagonal” is the average of the diagonal of the comparison matrix being used; the “diagonal” is the score or number assigned to each perfect amino acid match by the particular comparison matrix) and a gap extension penalty (which is usually one-tenth of the gap opening penalty), as well as a
- comparison matrix such as PAM250 or BLOSUM 62 are used in conjunction with the algorithm.
- a standard comparison matrix see Dayhoff et al, 1978, Atlas of Protein Sequence and Structure, 5:345-352 for the PAM 250 comparison matrix; Henikoff et al, 1992, Proc. Natl. Acad. Sci USA, 89:10915-10919 for the BLOSUM 62 comparison matrix) is also used by the algorithm.
- the parameters for a polypeptide sequence comparison include the following:
- the GAP program may be useful with the above parameters.
- the aforementioned parameters are the default parameters for polypeptide comparisons (along with no penalty for end gaps) using the GAP algorithm.
- Examples of unconventional amino acids include: 4-hydroxyproline, ⁇ - carboxyglutamate, ⁇ - ⁇ , ⁇ , ⁇ -trimethyllysine, ⁇ - ⁇ -acetyllysine, O-phosphoserine, N- acetylserine, N-formylmethionine, 3-methylhistidine, 5 -hydroxy lysine, ⁇ - ⁇ - methylarginine, and other similar amino acids and imino acids (e.g., 4-hydroxyproline).
- the left-hand direction is the amino terminal direction and the right-hand direction is the carboxyl-terminal direction, in accordance with standard usage and convention.
- Peptide analogs are commonly used in the pharmaceutical industry as non-peptide drugs with properties analogous to those of the template peptide. These types of non-peptide compound are termed "peptide mimetics” or "peptidomimetics". See Fauchere, 1986, Adv. Drug Res. 15:29; Veber & Freidinger, 1985, TINS p.392; and Evans et al, 1987, J. Med. Chem. 30: 1229, which are incorporated herein by reference for their descriptions of peptide mimetics. Such compounds are often developed with the aid of computerized molecular modeling. Peptide mimetics that are structurally similar to therapeutically useful peptides may be used to produce a similar therapeutic or prophylactic effect. Generally, peptidomimetics are structurally similar to a paradigm polypeptide (i.e., a polypeptide that has a biochemical property or
- pharmacological activity such as human antibody
- Systematic substitution of one or more amino acids of a consensus sequence with a D-amino acid of the same type may be used in certain embodiments to generate more stable peptides.
- constrained peptides comprising a consensus sequence or a substantially identical consensus sequence variation may be generated by methods known in the art (Rizo & Gierasch, 1992, Ann. Rev. Biochem. 61 :387, incorporated herein by reference for any purpose); for example, by adding internal cysteine residues capable of forming intramolecular disulfide bridges which cyclize the peptide. Treatment Method In General
- a modified human IL-4 mutien receptor antagonist is useful for treating various conditions associated with one of the pleiotropic effects of IL-4 and IL-13.
- antagonists of IL-4 and IL-13 are useful in treating conditions exacerbated by IL-4 and IL-13 production including asthma, allergy, dermatitis or other inflammatory response-related conditions.
- Some uses of the modified human IL-4 mutein receptor antagonists are described in U.S. Pat. No. 6,130,318 and in U.S. Publication No. 20070009479 (published Jan. 11, 2007), the entire contents of both of which are incorporated herein by reference.
- modified human IL-4 mutein receptor antagonists are employed to treat dermatitis, such as that resulting from a hypersensitivity reaction.
- the hypersensitivity reaction includes contact dermatitis and/or atopic dermatitis.
- the modified human IL-4 mutien receptor antagonist is used to treat eczema.
- the subject of the method can be any organism that exhibits (i.e., is experiencing or at risk for) an allergic response, such as dermatitis. Examples of suitable subjects include research animals or pets, such as mice, rats, guinea pigs, rabbits, cats, dogs, as well as monkeys and other primates, and humans.
- Modified IL-4 Mutein Receptor Antagonists include mice, rats, guinea pigs, rabbits, cats, dogs, as well as monkeys and other primates, and humans.
- Modified IL-4 mutein receptor antagonists includes the IL-4RA mutein described in US Pat. Nos. 6,028,176 & 6,313,272 (hereby incorporated by reference in their entirety), with additional amino acid substitutions at one or more positions of the mature IL-4 protein.
- Exemplary triple muteins include, but are not limited to a substitution of arginine by aspartic acid at position 121, of tyrosine by aspartic acid at position 124, and of serine by aspartic acid at position 125 in the D-helix; and a substitution of threonine by aspartic acid at position 13, of arginine by aspartic acid at position 121, and of tyrosine by aspartic acid at position 124 in the D-helix.
- the triple muteins further comprise an N-terminal methionine. Variations in this section of the D helix positively correlate with changes in interactions at the second binding region.
- the modified IL-4 mutein receptor antagonist may further include one or more substitutions wherein said substitutions enable the site-specific coupling of at least one non-protein polymer, such as polypropylene glycol, polyoxyalkylene, or polyethylene glycol (PEG) molecule to the mutein.
- Site-specific coupling of PEG allows the generation of a modified mutein which possesses the benefits of a polyethylene-glycosylated
- the PEG moiety is linear.
- Linear PEG moieties are limited in size by the manufacturing process because the amount of PEG-diol increases as PEG molecular weight increases. With linear moieties, increases in PEG-mutein molecular size is typically accomplished by increasing the number of PEG attachment sites on the mutein. This often results in suboptimal pharmacological profiles.
- the PEG moiety is branched from a single attachment site. Branched PEG moieties have the advantage of increasing the size of the PEG molecule without increasing the number of site attachments.
- the polyethylene glycol (PEG) moiety has a molecular weight ranging from about 2 kD to about 50 kD, e.g, about: 3, 5, 10, 15, 20, 25, 30, 35, 40, 45, or any molecular weight falling within a range bound by any of these values. In one embodiment, the PEG moiety is about 40 kD.
- Covalent attachment of the PEG to the drug may be accomplished by known chemical reactions and/or synthesis techniques.
- the PEGylation of protein may be accomplished by reacting NHS-activated PEG with the mutein under suitable reaction conditions.
- the PEGylation of protein may be accomplished by reacting a maleimide activated PEG with the sulhydryl group of the cytsteine residue in the protein under suitable reaction conditions.
- PEG-mutein conjugates can be created in at least three different ways:
- a single large PEG moiety can be attached at a single site on the mutein; a branched PEG moiety (i.e., two or more medium PEG chains joined together via a linker) can be attached at a single site on the mutein; or several small chains may be attached at multiple sites on the mutein.
- monosite PEGylated muteins have higher activity because the PEG attachment is less likely to occur at or near receptor-binding domains.
- PTMs post-translational modifications
- Techniques and reagents for PEGylation include, for example: (i) specialized linkers and coupling chemistries; (ii) branched PEGs which effectively allow additional PEG groups to be attached to a single conjugation site; (iii) site-specific PEGylation, including site-specific monoPEGylation; and (iv) site-directed enzymatic PEGylation (e.g. using a transglutaminase reaction).
- site-directed enzymatic PEGylation e.g. using a transglutaminase reaction
- Nektar/Shearwater on the world wide web at nektar.com
- Sunbio on the world wide web at sunbio.com and sunbio.com/peg-shop
- Celares GmbH on the world wide web at celares.com
- NOF Corporation on the world wide web at peg-drug.com
- Modified IL-4 mutein receptor antagonists bind to IL-4 and IL-13 with an affinity loss not greater than 10-fold relative to that of IL-4RA. Modified IL-4 mutein receptor antagonists inhibit IL-4 and IL-13 mediated activity with a loss of potency not greater than 10-fold relative to that of IL-4RA. In addition, modified IL-4 mutein receptor antagonists possess a plasma half-life which is at least 2 to 10-fold greater than that of unmodified IL-4RA.
- the IL-4 muteins of this invention may also be characterized by amino acid insertions, deletions, substitutions and modifications at one or more sites in or at the other residues of the native IL-4 polypeptide chain. In accordance with this invention any such insertions, deletions, substitutions and modifications should result in an IL-4 mutein that retains its IL-4 antagonist activity.
- Example 2 An additional aspect of this invention is provided in the method with which the protein is expressed and refolded, as depicted in Example 2.
- the IL-4 mutein is preferably purified so as to allow efficient PEGylation.
- An exemplary method for purification is described in Example 2 below.
- sulfhydryl protecting agent dithiothreitol DTT
- DTT dithiothreitol
- IL-4 muteins purified after refolding in the presence of a sulfhydryl protecting agent can react with the PEG reagent if treated with DTT, but a mixture of monoPEGylated and
- the Ka of modified IL-4 mutein receptor antagonists to the IL-4 receptor can be assayed using any method known in the art, including technologies such as realtime Bimolecular Interaction Analysis (BIA) outlined in Example 4.
- BIA is a technology for studying biospecific interactions in real time, without labeling any of the interactants (e.g., BIAcoreTM). Changes in the optical phenomenon surface plasmon resonance (SPR) can be used as an indication of real-time reactions between biological molecules.
- the capacity of modified IL-4 mutein receptor antagonists to inhibit the proliferative response of immune cells can be assessed using proliferative assays as outlined in Example 5 and this capacity expressed as an Inhibitory Concentration 50% (IC 50 ).
- modified IL-4 mutein receptor antagonists of the present invention specifically bind to the human IL-4 receptor with a preferred Ka in the range of from about 1.0 nM to about 100 nM. More preferred embodiments of the present invention bind to human IL-4 receptor with a Kj of approximately 0.5 nM to about 1.0 uM. Still more preferred embodiments of the present invention bind to human IL-4 receptor with a K d of approximately 0.1 nM to about 10 ⁇ .
- modified IL-4 mutein receptor antagonists of the present invention will bind to human IL-4 receptor and neutralize its capacity to promote immune cell proliferation with a preferred IC 50 ranging from about 1.0 nM to about 100 nM. More preferred human antagonists bind IL-4 receptor and neutralize its immune cell proliferation capacity with an IC 50 ranging from approximately 0.5 nM to 1 ⁇ , with the most preferred antagonists of this invention binding and inhibiting IL-4 receptor with an IC 50 of approximately 0.1 nM to about 10 ⁇ .
- modified IL-4 mutein receptor antagonists described herein exhibit enhanced bioavailability, compared to unmodified, e.g., non-PEGylated, forms.
- modified IL-4 mutein receptor antagonists described herein also exhibit a plasma half-life that is preferably at least 2 to 10-fold greater than that of unmodified IL4RA with the most preferred embodiments of the present invention exhibiting a plasma half- life which is 10-100-fold greater than that of unmodified IL-4RA (see Example 7).
- Modified IL-4 mutein receptor antagonists can be produced using any method capable of producing polypeptides of having the desired amino acid sequence. Typically, recombinant expression will be the most convenient method. Polynucleotides Encoding Modified IL-4 Mutein Receptor
- mutein receptor antagonists 4 mutein receptor antagonists. These polynucleotides can be used, for example, to produce quantities of the antagonists for therapeutic use. Methods of constructing and expressing degenerative DNA sequences capable of expressing the same amino acid sequence as a given polynucleotide sequence are known in the art.
- a polynucleotide of the invention can be readily obtained in a variety of ways including, without limitation, cDNA or genomic library screening, expression library screening, and/or PCR amplification of cDNA. Such methods are well known and include those set forth in Sambrook et al, Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Laboratory Press, 1989) and/or Current Protocols in Molecular Biology (Ausubel et al, eds., Green Publishers Inc. and Wiley and Sons 1994).
- PCR polymerase chain reaction
- poly(A)+RNA or total RNA using the enzyme reverse transcriptase Two primers, typically complementary to two separate regions of a modified IL-4 mutein receptor antagonist cDNA, are then added to the cDNA along with a polymerase such as Taq polymerase, and the polymerase amplifies the cDNA region between the two primers.
- a polymerase such as Taq polymerase
- nucleic acid molecule of the invention Another means of preparing a nucleic acid molecule of the invention is chemical synthesis using methods well known to the skilled artisan such as those described by Engels et al, 1989, Angew. Chem. Intl. Ed. 28:716-34. These methods include, inter alia, the phosphotriester, phosphoramidite, and H-phosphonate methods for nucleic acid synthesis.
- a preferred method for such chemical synthesis is polymer- supported synthesis using standard phosphoramidite chemistry.
- the DNA will be several hundred nucleotides in length. Nucleic acids larger than about 100 nucleotides can be synthesized as several fragments using these methods. The fragments can then be ligated together.
- a polynucleotide can be incorporated into a vector for propagation and/or expression in a host cell.
- Such vectors typically contain a replication sequence capable of effecting replication of the vector in a suitable host cell (i.e., an origin of replication) as well as sequences encoding a selectable marker, such as an antibiotic resistance gene.
- a suitable host cell i.e., an origin of replication
- the vector can replicate and function independently of the host genome or integrate into the host genome.
- Vector design depends, among other things, on the intended use and host cell for the vector, and the design of a vector of the invention for a particular use and host cell is within the level of skill in the art.
- the vector includes one or more control sequences capable of effecting and/or enhancing the expression of an operably linked polypeptide coding sequence.
- Control sequences that are suitable for expression in prokaryotes include a promoter sequence, an operator sequence, and a ribosome binding site.
- Control sequences for expression in eukaryotic cells include a promoter, an enhancer, and a transcription termination sequence (i.e., a polyadenylation signal).
- An expression vector according to the invention can also include other sequences, such as, for example, nucleic acid sequences encoding a signal sequence or an amplifiable gene.
- a signal sequence can direct the secretion of a polypeptide fused thereto from a cell expressing the protein.
- nucleic acid encoding a signal sequence is linked to a polypeptide coding sequence so as to preserve the reading frame of the polypeptide coding sequence.
- the inclusion in a vector of a gene complementing an auxotrophic deficiency in the chosen host cell allows for the selection of host cells transformed with the vector.
- Vectors are typically produced by linking desired elements by ligation at convenient restriction sites. If such sites do not exist, suitable sites can be introduced by standard mutagenesis (e.g., site-directed or cassette mutagenesis) or synthetic oligonucleotide adaptors or linkers can be used in accordance with conventional practice.
- suitable sites can be introduced by standard mutagenesis (e.g., site-directed or cassette mutagenesis) or synthetic oligonucleotide adaptors or linkers can be used in accordance with conventional practice.
- a wide variety of host cells are available for propagation and/or expression of vectors. Examples include prokaryotic cells (such as E. coli and strains of Bacillus, Pseudomonas, and other bacteria), yeast or other fungal cells (including S. cerevesiae and P. pastoris), insect cells, plant cells, and phage, as well as higher eukaryotic cells (such as human embryonic kidney cells and other mammalian cells).
- prokaryotic cells such as E. coli and strains of Bacillus, Pseudomonas, and other bacteria
- yeast or other fungal cells including S. cerevesiae and P. pastoris
- insect cells such as human embryonic kidney cells and other mammalian cells.
- a vector can be introduced into a host cell by any convenient method, which will vary depending on the vector-host system employed. Generally, a vector is introduced into a host cell by transformation (also known as "transfection") or infection with a virus (e.g., phage) bearing the vector. If the host cell is a prokaryotic cell (or other cell having a cell wall), convenient transformation methods include the calcium treatment method described by Cohen, et al. (1972) Proc. Natl. Acad. Sci., USA, 69:2110-14. If a prokaryotic cell is used as the host and the vector is a phagemid vector, the vector can be introduced into the host cell by infection.
- Yeast cells can be transformed using polyethylene glycol, for example, as taught by Hinnen (1978) Proc. Natl. Acad. Sci, USA, 75: 1929-33. Mammalian cells are conveniently transformed using the calcium phosphate precipitation method described by Graham, et al. (1978) Virology, 52:546 and by Gorman, et al. (1990) DNA and Prot. Eng. Tech., 2:3-10. However, other known methods for introducing DNA into host cells, such as nuclear injection, electroporation, and protoplast fusion also are acceptable for use in the invention.
- Host cells transformed with expression vectors can be used to express the polypeptides encoded by the polynucleotides of the invention.
- Expression entails culturing the host cells under conditions suitable for cell growth and expression and recovering the expressed polypeptides from a cell lysate or, if the polypeptides are secreted, from the culture medium.
- the culture medium contains appropriate nutrients and growth factors for the host cell employed. The nutrients and growth factors are, in many cases, well known or can be readily determined empirically by those skilled in the art. Suitable culture conditions for mammalian host cells, for instance, are described in Mammalian Cell Culture (Mather ed., Plenum Press 1984) and in Barnes and Sato (1980) Cell 22:649.
- the culture conditions should allow transcription, translation, and protein transport between cellular compartments.
- Factors that affect these processes are well-known and include, for example, DNA/RNA copy number; factors that stabilize DNA; nutrients, supplements, and transcriptional inducers or repressors present in the culture medium; temperature, pH and osmolality of the culture; and cell density.
- the adjustment of these factors to promote expression in a particular vector- host cell system is within the level of skill in the art. Principles and practical techniques for maximizing the productivity of in vitro mammalian cell cultures, for example, can be found in Mammalian Cell Biotechnology: a Practical Approach (Butler ed., IRL Press (1991).
- any of a number of well-known techniques for large- or small-scale production of proteins can be employed in expressing the polypeptides of the invention. These include, but are not limited to, the use of a shaken flask, a fluidized bed bioreactor, a roller bottle culture system, and a stirred tank bioreactor system. Cell culture can be carried out in a batch, fed-batch, or continuous mode.
- a polypeptide including a signal sequence can be recovered from the culture medium or the periplasm. Polypeptides can also be expressed intracellularly and recovered from cell lysates.
- the expressed polypeptides can be purified from culture medium or a cell lysate by any method capable of separating the polypeptide from one or more components of the host cell or culture medium. Typically, the polypeptide is separated from host cell and/or culture medium components that would interfere with the intended use of the polypeptide.
- the culture medium or cell lysate can be centrifuged or filtered to remove cellular debris. The supernatant is then typically concentrated or diluted to a desired volume or diafiltered into a suitable buffer to condition the preparation for further purification.
- the polypeptide can then be further purified using well-known techniques. The technique chosen will vary depending on the properties of the expressed polypeptide.
- Polynucleotides of the invention present in a host cell can be isolated free of other cellular components such as membrane components, proteins, and lipids.
- Polynucleotides can be isolated from cells using standard nucleic acid purification techniques, or synthesized using an amplification technique, such as the polymerase chain reaction (PCR), or by using an automatic synthesizer. Methods for isolating polynucleotides are routine and are known in the art. Any such technique for obtaining a polynucleotide can be used to obtain isolated polynucleotides encoding antagonists of the invention. For example, restriction enzymes and probes can be used to isolate polynucleotides which encode the antagonists.
- isolated polynucleotides are in preparations that are free or at least 70, 80, or 90% free of other molecules.
- modified IL-4 mutein receptor antagonist cDNA molecules are produced, using mR A as a template. Thereafter, cDNA molecules can be replicated using molecular biology techniques known in the art and described in Example 1.
- Example 2 describes the specific recombinant expression and purification techniques employed in generating exemplary modified mutein
- the antagonist can be tested in vitro in cell proliferation assays as detailed in Examples 5 and 6.
- the plasma half-life of the modified IL-4 mutein receptor antagonist can be measured in vivo with a rat pharmacokinetic study according to Example 6.
- modified IL-4 mutein receptor antagonist is co-administered with an additional agent that is useful for mitigating a symptom of an allergic response, such as dermatitis.
- the amount of additional agent administered is sufficient to produce a beneficial effect (e.g., mitigation of dermatitis) in the subject when co-administered with the selected modified IL-4 mutein receptor antagonist.
- any additional agent that mitigates a symptom of the allergic response being treated e.g., dermatitis
- the additional agent can be one that acts by the same, or a different, mechanism than the modified IL-4 mutein receptor antagonist with which it is co-administered.
- additional agents suitable for use this embodiment include steroids.
- Corticosteroid creams are sometimes prescribed to decrease the inflammatory reaction in the skin. These may be mild-, medium-, or high-potency corticosteroid creams depending upon the severity of the symptoms. If itching is severe, oral antihistamines may be prescribed.
- the sedative type antihistamine drugs such as, e.g., diphenhydramine (Benadryl), hydroxyzine (Atarax, Vistaril), and cyproheptadine
- a short course of oral corticosteroids such as prednisone
- the oral immunosuppressant drug cyclosporine has also been used to treat some cases of eczema.
- Ultraviolet light therapy phototherapy
- two topical (cream) medications have been approved by the U.S. FDA for the treatment of eczema: tacrolimus (Protopic) and pimecrolimus (Elidel).
- compositions such as are described in Remington's Pharmaceutical Sciences (1980) 16th editions, Osol, ed., 1980.
- Pharmaceutically acceptable carriers can contain one or more physiologically acceptable compound(s) that act, for example, to stabilize the composition or to increase or decrease the absorption of the active agent(s).
- a pharmaceutically acceptable carrier suitable for use in the invention is non-toxic to cells, tissues, or subjects at the dosages employed, and can include a buffer (such as a phosphate buffer, citrate buffer, and buffers made from other organic acids), an antioxidant (e.g., ascorbic acid), a low-molecular weight (less than about 10 residues) peptide, a polypeptide (such as serum albumin, gelatin, and an immunoglobulin), a hydrophilic polymer (such as polyvinylpyrrolidone), an amino acid (such as glycine, glutamine, asparagine, arginine, and/or lysine), a monosaccharide, a disaccharide, and/or other carbohydrates (including glucose, mannose, and dextrins), a chelating agent (e.g., ethylenediammetetratacetic acid [EDTA]), a sugar alcohol (such as mannitol and sorbitol), a salt-forming counterion
- compositions include wetting agents, emulsifying agents, dispersing agents or preservatives that are particularly useful for preventing the growth or action of microorganisms.
- preservatives include, for example, phenol and ascorbic acid.
- pharmaceutically acceptable carrier(s) including a physiologically acceptable compound depends, for example, on the route of administration of the active agent(s) and on the particular physio-chemical
- compositions of the invention can be stored in any standard form, including, e.g., an aqueous solution or a lyophilized cake. Such compositions are typically sterile when administered to subjects. Sterilization of an aqueous solution is readily accomplished by filtration through a sterile filtration membrane. If the composition is stored in lyophilized form, the composition can be filtered before or after lyophilization and reconstitution. Administration
- the active agents identified herein are useful for parenteral (e.g., subcutaneously, intravenously, intra-arterially, intramuscularly, intraperitoneally, intradermally), nasal (or otherwise inhaled), oral, sublingual, rectal, topical, or local administration, such as by aerosol aerosol (e.g., nebulization, dry powder or metered dose inhalation), or transdermally, for prophylactic and/or therapeutic treatment of one or more of the pathologies/indications described herein (e.g., to mitigate one or more symptoms of an allergic response, such as dermatitis).
- parenteral e.g., subcutaneously, intravenously, intra-arterially, intramuscularly, intraperitoneally, intradermally
- nasal or otherwise inhaled
- oral sublingual, rectal, topical, or local administration
- aerosol aerosol e.g., nebulization, dry powder or metered dose inhalation
- transdermally for prophylactic and/or
- a modified IL-4 mutein receptor antagonist can be administered parenterally (e.g., subcutaneously) no more than about twice per week, once per week, every two weeks, every three weeks, once per month, or once every 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 months.
- parenterally e.g., subcutaneously
- about 10 mg/kg can be administered no more than about once per week
- about 20 mg/kg can be administered no more than about every two weeks
- about 40 mg/kg can be administered no more than about once per month.
- the active agents described herein can be administered orally, in which case delivery can be enhanced by the use of protective excipients. This is typically accomplished either by complexing the active agent(s) with a composition to render them resistant to acidic and enzymatic hydrolysis or by packaging the agents in an appropriately resistant carrier, e.g. a liposome.
- protective excipients e.g. a composition to render them resistant to acidic and enzymatic hydrolysis or by packaging the agents in an appropriately resistant carrier, e.g. a liposome.
- Means of protecting agents for oral delivery are well known in the art (see, e.g., U.S. Patent No. 5,391,377).
- Elevated serum half-life can be maintained by the use of sustained- release "packaging" systems.
- sustained release systems are well known to those of skill in the art (see, e.g., Tracy (1998) Biotechnol. Prog. 14: 108; Johnson et al. (1996), Nature Med. 2: 795; Herbert et al. (1998), Pharmaceut. Res. 15, 357).
- the pharmaceutical compositions can be administered in a variety of unit dosage forms depending upon the method of administration. Suitable unit dosage forms, include, but are not limited to powders, tablets, pills, capsules, lozenges, suppositories, patches, nasal sprays, injectibles, implantable sustained-release formulations, lipid complexes, etc.
- one or more components of a solution can be provided as a "concentrate,” e.g., in a storage container (e.g., in a premeasured volume) ready for dilution or in a soluble capsule ready for addition to a volume of water.
- one or more active agents described herein are administered alone or in combination with other therapeutics in implantable (e.g., subcutaneous) matrices, termed "depot formulations.”
- drugs e.g., the active agents described herein
- drugs embedded, for example, in polymer beads or in polymer wafers have several advantages. First, most systems allow slow release of the drug, thus creating a continuous dosing of the body with small levels of drug. This typically prevents side effects associated with high burst levels of normal injected or pill-based drugs. Secondly, since these polymers can be made to release over hours to months, the therapeutic span of the drug is markedly increased. Often, by mixing different ratios of the same polymer components, polymers of different degradation rates can be made, allowing remarkable flexibility depending on the agent being used.
- a long rate of drug release is beneficial for people who might have trouble staying on regular dosage, such as the elderly, but also represents an ease of use improvement that everyone can appreciate.
- Most polymers can be made to degrade and be cleared by the body over time, so they will not remain in the body after the therapeutic interval.
- polymer-based drug delivery Another advantage of polymer-based drug delivery is that the polymers often can stabilize or solubilize proteins, peptides, and other large molecules that would otherwise be unusable as medications. Finally, many drug/polymer mixes can be placed directly in the disease area, allowing specific targeting of the medication where it is needed without losing drug to the "first pass" effect.
- a wide variety of approaches to designing depot formulations that provide sustained release of an active agent are known and are suitable for use in the invention.
- the components of such formulations are biocompatible and may be biodegradable.
- Biocompatible polymeric materials have been used extensively in therapeutic drug delivery and medical implant applications to effect a localized and sustained release. See Leong et al, "Polymeric Controlled Drug Delivery,” Advanced Drug Delivery Rev., 1 : 199-233 (1987); Langer, "New Methods of Drug Delivery,”
- polyesters Patent et al, "Biodegradable Drug Delivery Systems Based on Aliphatic Polyesters: Applications to Contraceptives and Narcotic Antagonists," Controlled Release of Bioactive Materials, 19-44 (Richard Baker ed., 1980); poly(amino acids) and pseudo-poly(amino acids) (Pulapura et al. "Trends in the Development of Bioresorbable Polymers for Medical Applications," J.
- the active agent(s) can be incorporated into a biocompatible polymeric composition and formed into the desired shape outside the body. This solid implant is then typically inserted into the body of the subject through an incision. Alternatively, small discrete particles composed of these polymeric compositions can be injected into the body, e.g., using a syringe.
- the active agent(s) can be encapsulated in microspheres of poly (D,L- lactide) polymer suspended in a diluent of water, mannitol, carboxymethyl-cellulose, and polysorbate 80. The polylactide polymer is gradually metabolized to carbon dioxide and water, releasing the active agent(s) into the system.
- depot formulations can be injected via syringe as a liquid polymeric composition.
- Liquid polymeric compositions useful for biodegradable controlled release drug delivery systems are described, e.g., in U.S. Patent Nos. 4,938,763; 5,702,716; 5,744,153; 5,990,194; and 5,324,519. After injection in a liquid state or, alternatively, as a solution, the composition coagulates into a solid.
- One type of polymeric composition suitable for this application includes a nonreactive thermoplastic polymer or copolymer dissolved in a body fluid-dispersible solvent. This polymeric solution is placed into the body where the polymer congeals or precipitates and solidifies upon the dissipation or diffusion of the solvent into the surrounding body tissues. See, e.g., Dunn et al, U.S. Patent Nos. 5,278,201;
- thermoplastic drug delivery system in which a solid, linear-chain, biodegradable polymer or copolymer is dissolved in a solvent to form a liquid solution.
- the active agent(s) can also be adsorbed onto a membrane, such as a silastic membrane, which can be implanted, as described in International Publication No. WO 91/04014.
- a membrane such as a silastic membrane
- Other exemplary implantable sustained release systems include, but are not limited to Re-Gel®, SQ2Gel®, and Oligosphere® by MacroMed,
- Modified IL-4 mutien receptor antagonist compounds can be co- administered with additional agents that mitigate a symptom of an allergic response, such as dermititis.
- Co-administration of an additional agent can be useful, for example, to control symptoms of dermatitis prior to the point at which the modified IL-4 mutien receptor antagonists begins to reduce such symptoms. More specifically, in certain embodiments, there will be a delay between the administration of the antagonist and the time at which the subject experiences diminution of symptoms.
- Treatment with an additional agent such as a topical or oral cortisone can provide relief from symptoms during this period.
- treatment with the additional agent can be initiated before, at the same time as, or after treatment with the antagonist.
- Treatment with the additional agent can be continued or discontinued, if it does not provide additional therapeutic benefit beyond that of the antagonist.
- treatment with an additional agent is initiated before, or at the same time as, the initiation of treatment with the antagonist, and the additional agent is administered for about 1 week, about 2 week, about 3 weeks, or about 1 month from the time treatment with the antagonist is initiated.
- Additional agents can be administered by a route that is the same as, or different from, the route of administration of the modified IL-4 mutien receptor antagonist. Where possible, it is generally desirable to administer these agents by the same route of administration, preferably in the same composition. However, differences in pharmacodynamics, pharmacokinetics, or other considerations may dictate the co-administration of modified IL-4 mutien receptor antagonist compound and additional agent in separate compositions. Additional agents can be administered according to standard practice.
- compositions of this invention are administered, for example, to a subject experiencing, or at risk for, an allergic response, such as dermatitis, to mitigate at least one symptom of this response.
- An amount adequate to accomplish this is defined as a "therapeutically effective dose.” Amounts effective for this use will depend upon the severity of the condition and the general state of the subject's health. Single or multiple doses of the compositions may be administered depending on the dosage and frequency as required and tolerated by the subject. In any event, the composition should provide a sufficient quantity of the active agent(s) of the composition(s) of this invention to effectively treat the condition.
- the concentration of active agent(s) can vary widely and will be selected primarily based on fluid volumes, viscosities, body weight and the like in accordance with the particular mode of administration selected and the subject's needs.
- the clinician can titer the dosage and modify the route of administration as required to obtain the optimal therapeutic effect. Generally, the clinician begins with a low dose and increases the dosage until the desired therapeutic effect is achieved. Starting doses for a given active agent can, for example be extrapolated from in vitro and/or animal data.
- concentrations of modified IL-4 mutein receptor antagonists will typically be selected to provide dosages of about 0.1 mg/kg, about 0.2 mg/kg, about 0.3 mg/kg, about 0.4 mg/kg, about 0.5 mg/kg, about 0.6 mg/kg, about 0.7 mg/kg, about 0.8 mg/kg, about 0.9 mg/kg, about 1 mg/kg, about 2 mg/kg, about 5 mg/kg, about 0.7 mg/kg, about 10 mg/kg, about 15 mg/kg, about 20 mg/kg, about 25 mg/kg, about 30 mg/kg, about 35 mg/kg, about 40 mg/kg about 45 mg/kg, about 50 mg/kg and sometimes higher.
- compositions and administration methods are intended to be illustrative and not limiting. It will be appreciated that, using the teaching provided herein, other suitable compositions and modes of administration can be readily devised.
- the immune response of a subject being treated as described herein can be monitored by making periodic, e.g., daily observation of the existing dermatitis and plasma levels of IgE before and after dosing.
- periodic, e.g., daily observation of the existing dermatitis and plasma levels of IgE before and after dosing.
- those subjects receiving the drug have a pronounced reduction in the immune response, they can, in certain
- embodiments be taken off the modified IL-4 mutien receptor antagonist and monitored for the length of remission.
- Administration of the antagonist to subjects is expected to reduce or eliminate early and/or latent immune responses.
- kits useful in practicing the methods of the invention include a modified IL-4 mutien receptor antagonist in a suitable container.
- the modified IL-4 mutien receptor antagonist is formulated in a pharmaceutically acceptable carrier.
- the kit preferably includes instructions for administering the antagonis to a subject to inhibit a dermatitis response, such as a hypersensitivity reaction, e.g., contact dermatitis or atopic dermatitis.
- the instructions are directed to the treatment of eczema.
- kits of the invention can be affixed to packaging material or can be included as a package insert. While the instructions are typically written or printed materials they are not limited to such. Any medium capable of storing such instructions and communicating them to an end user is contemplated by this invention. Such media include, but are not limited to, electronic storage media (e.g., magnetic discs, tapes, cartridges, chips), optical media (e.g., CD ROM), and the like. As used herein, the term "instructions" can include the address of an internet site that provides the instructions.
- the pET Directional TOPO® expression system (Invitrogen) was selected for recombinant expression of IL-4.
- the system uses a highly efficient one- step "TOPO® Cloning" strategy to directionally clone a blunt-end PCR product and a
- TTlac promoter for high-level and IPTG-inducible expression of the gene of interest in E.coli. Additional features include a lacl gene to reduce basal transcription, a pBR322 origin for replication and maintenance of the plasmid and an ampicillin resistance gene for selection.
- IL-4 was cloned into pET101/D-TOPO vector for production of recombinant IL-4 protein.
- the oligonucleotide primers are shown in Table 3.
- the forward PCR primer was designed with a 5'CACC overhang to facilitate directional cloning, followed by a unique Ndel restriction enzyme site for subcloning and the initial ATG start codon.
- the reverse PCR primer included two stop codons to make sure no c-terminal tags were incorporated and a unique BamHI restriction enzyme site for subcloning.
- a blunt-end IL-4 PCR product was generated using previously cloned human IL-4 as a template. The product was gel purified and incubated with salt solution and TOPO® vector for 5 minutes at room temperature to allow for
- the recombinant vector was transformed into chemically competent One Shot TOP 10 E.coli.
- the recombinant plasmid DNA was sent out for DNA sequencing to confirm the correct sequence.
- IL-4/pET 101/D-TOPO served as a template for producing IL-4 RE cysteine muteins with the QuikChange® Site-Directed Mutagenesis Kit from
- Each cysteine mutein was made using two oligonucleotide primers, each complementary to opposite strands of the vector and containing the codon TGC or GC A to incorporate the desired cysteine mutation.
- Table 4 lists the primers used for producing the IL-4 RE muteins.
- a mutated plasmid containing staggered nicks was generated using cycling parameters and conditions defined in the manufacturer's protocol. The product was treated with Dpnl endonuclease for 1 hour at 37°C to digest the methylated, non-mutated parental DNA template. The Dpnl -treated DNA was transformed into XL-1 Blue supercompetent cells where nicks in the mutated plasmid were repaired. The mutagenic 5 plasmid DNA was analysed according to standard sequencing techniques to confirm the correct sequence.
- BL21 Star (DE3) One Shot cells (Invitrogen) transformed with the protein containing plasmids were characterized for optimal expression and grown at 37°C until OD 6 oo reached approximately 0.4 and induced by lmM IPTG (Invitrogen) for 3 hours at 37°C.
- One liter of cells were pelleted at 13,000 rpm for 10 minutes, weighed and stored at -80°C.
- the frozen cell pellet was resuspended in 8 ml cell disruption buffer (0.1M phosphate buffer pH 7.3, 0.1% Triton X100, lmM EDTA) per gram of cells and sonicated 4x for 1 minute with 1 minute intervals.
- the cell lysate was removed by centrifugation at 35,000g for 10 minutes.
- the cell pellets were then washed 2-3x by resuspension in 30 ml of cell disruption buffer, by sonication for 1 minute, followed by centrifugation.
- the final cell pellet, inclusion bodies was stored at -20°C.
- Inclusion bodies were resuspended in 5 ml solubilization buffer (0.2M Tris pH 9, 7M guanidine hydrochloride) per gram of cells.
- Sulphotolysis reagents (0.16 grams sodium sulfite, 0.08 gram potassium tetrathionate per gram of cells) were added and the inclusion bodies were stirred at room temperature for 2 hours.
- the mixture was diluted 5x with water and subjected to dialysis into 4.5L 3mM NaH 2 P0 4 , 7mM Na 2 HP0 4 , 2mM KC1, 120mM NaCl. Dialysis was continued for 3-4 days with fresh buffer change at least 3 times. The dialyzed material was then filtered through an 0.2 ⁇ filter and the pH was adjusted to 5 with acetic acid. The column was equilibrated with 10CV of Buffer 1 (25mM Ammonium Acetate pH5) followed by a 20 minute gradient to 100% buffer B (25mM Ammonium Acetate pH5/lM NaCl) post injection.
- Buffer 1 25mM Ammonium Acetate pH5
- buffer B 25mM Ammonium Acetate pH5/lM NaCl
- Peak fractions (0.5 ml each) were collected and screened by 12% or 4-20% Bis-Tris-SDS gel electrophoresis. Product containing fractions were pooled and diluted 2x into Buffer A (0.1% TF A/water). The protein was then chromatographed on C4 Reverse Phase-HPLC (Beckman system Gold), using a 5 ml loop and flow rate of 1 ml/min with the following program: 10% Buffer A for duration of injection, 10 minute gradient to 40% Buffer B (0.1% TFA/ACN), 30 minute gradient to 50% Buffer B, and 5 minute gradient to 100% Buffer B. Peak fractions (0.5 ml each) were collected and screened by 12% or 4-20% Bis-Tris-SDS gel electrophoresis. Protein containing fractions were dried down and resuspended in 0.1 M MES pH 6.1 for analysis and assays.
- a protocol was established to PEGylate the cysteine containing IL4 RA muteins via a stable thioether linkage between the sulfhydryl of the protein and the maleimide group of a linear 22kD methoxy -poly ethylene glycol-maleimide derivative (Nektar Therapeutics).
- a 2-fold molar excess of mPEG-MAL 22kD reagent was added to 60 ⁇ of protein dissolved in reaction buffer, 0.1M MES, pH 6. After 0.5 hour at room temperature, the reaction was terminated with 2-fold molar excess of cysteine over mPEG-MAL 22kD ( Figure 1).
- PEGylated protein was purified away from unreacted mPEG-MAL 22kD (quenched with cysteine) and unreacted IL4 RA cysteine mutein by cation exchange and size exclusion chromatography.
- Crude reaction mixtures were applied to Vivapure Mini S cation exchange columns (Vivascience) equilibrated with 0.4mL of 0.1 M MES, pH 6. The columns were washed twice with 0.4mL of 0.1M MES, pH 6 followed by centrifugation at 2,000 x g after each wash. The samples were eluted by centrifugation from the column with 0.4mL of 0.6M NaCl/O. lM MES, pH6. The 0.4mL elutions were loaded onto a TSK-GEL
- IL-4 receptor was immobilized on a BIAcore CM5 research grade sensor chip through amine coupling.
- the sensor surface was activated with an EDC/NHS pulse.
- IL-4 receptor was dissolved in lOmM acetate buffer (pH 5.0) and injected into flowcell 2 followed by a pulse of 1.0M ethanolamine-HCL to deactivate the surface.
- the immobilization level for the receptor was -300RU.
- Flowcell 1 was also activated without a ligand to function as a blank.
- the Biacore Wizard was used to perform kinetics analysis.
- Candidate IL4RE antagonists were diluted in HBS-EP (running buffer) and injected at 30ul/minute flow rate for 3 minutes and a dissociation time of 15 minutes. Regeneration of the chip was performed by two 30 second injections of lOmM Glycine pH2.5 (flow lOOul/min) to baseline prior to next injection in the concentration series. Dissociation constant (K D ) values were calculated for each candidate based on direct binding kinetics (Table 5). Results show constructs IL4- RE-A104C, IL4-RE-N105C, and IL4-RE-Q106C all yielded dissociation constants below 0.6nM.
- TF-1 cells were cultured for 2-4 days in 96 well plates (lxl0 4 /well, 100 ⁇ volume) in RPMI + 10% serum with or without IL-4 or IL-13 and IL-4RE molecules.
- GM-CSF treatment was used as a positive control. Twenty- four hours before the final reading, 10 ⁇ AlamarBlue (10%> vol) was added to each well. Fluorescence was determined at 530/590 nm using a WALLAC Victor 2.
- IL-4 was also evaluated following IL-4RE molecule pre-treatment.
- Peripheral blood mononuclear cells PBMCs
- PBMCs Peripheral blood mononuclear cells
- PBMCs peripheral blood mononuclear cells
- anti-CD40 to activate B cell activity and used immediately.
- the cells were seeded in 96-well plates (10 5 cells per well).
- PHA T-cell blasts and B cells preparations were stimulated for 3 days with IL-4 (10 ng/ml, 0.667 nM) in the presence of varying concentrations of IL-4RE molecules.
- the incorporation of tritiated thymidine in the last 20 hours of incubation was used as an indicator of proliferation.
- the results of these assays are shown in Table 6. Results indicate that all
- PEGylated constructs demonstrated an IC 50 less than 5 -fold greater than that of IL- 4RA for both primary cell assays.
- Table 6 PEG-IL4RE bioactivity evaluation in B-cell and T-cell blast proliferation assays.
- the rats were given either IL-4RA or a modified IL-4 mutein receptor antagonist at doses of 1 and 0.5 mg/kg, respectively. Both IV and SC (subcutaneous) routes of administration were used. The IV dose was given by injection directly into the indwelling femoral vein catheter. The SC dose was given by injection into the dorsal thoracic region. Three rats were used for each dose group.
- Plasma concentrations of IL-4RA and modified mutein were quantified with an enzyme-linked immunoassay.
- Anti-IL-4 antibody was used as coating and detection reagents. The lower limit of quantification for this assay was 0.2 ng/ml.
- Pharmacokinetic parameters were derived by non-compartmental analysis using WinNonlin (Pharsight, Mountain view, CA). Of particular interest is the assessment of absorption and elimination kinetics, distribution volumes as well as the amount absorbed.
- IL-4 triple mutein molecules can be made in the laboratory by including an IL-4 triple mutein construct utilizing human DNA codons into an appropriate plasmid vector suitable for expression in E.coli. Although small quantities of inclusion bodies can be obtained from an IL-4 triple mutein construct using human codons, the E.coli fermentation process is not robust. It was observed that expression of an IL-4 triple mutein construct using human codons can be toxic to E.coli cells causing lysis, resulting in low yields of inclusion 5 bodies. Without being limited to certain theories, this observation is likely due to
- E.coli-codon IL-4RA plasmid was used to make E.co //-encoded IL4-RE-T13D with QuickChange (Strategene). Briefly, IL-4RA plasmid was altered by QuickChange and suitable primers to alter the codons at either position alanine 104 (codon GCT) or 10 asparagine 38 (codon AAC) to a cysteine (codon TGC).
- GCT position alanine 104
- AAC asparagine 38
- TGC cysteine
- T13D plasmids were made that coded for IL-4TM (IL4-RE-T13D) molecules (Table 7).
- IL-4TM-N38C or IL-4TM-A104C is PEGylated with a maleimide-PEG (20 kDa linear from Nektar, Dow or NOF; 30 kDa linear from Nektar, Dow or NOF; 40 kDa linear from Nektar, Dow or NOF; or a 40 kDa branched from Necktar or NOF), the activity decreases significantly when assayed using the BIAcore method, TF-1 cell-based assay or primary B or T lymphocytes from peripheral blood ( Figure 2).
- T13D mutation was included onto the IL-4TM-N38C using QuikChange, appropriate primers, and the E.co/z ' -codon N38C plasmid.
- This T13D mutation has previously been shown to increase binding affinity to the IL-4Ra (see U.S. Pat. No. 6,028,176, incorporated herein by reference).
- PEGylated T13D-N38C with 40kDa branched PEG has equal BIAcore as the unPEGylated IL-4DM (R121D/Y124D), but the data cannot accurately predict the cell-based assay since PEGylation affects the on- rate of all PEGylated IL-4TM molecules.
- PEGylated T13D-N38C with 40kDa branched PEG has equal activity as IL-4DM.
- IL-4TM (T13D/R121D/Y124D) contains a 7 th cysteine residue in the protein sequence at either position 38C or 104C. This odd cysteine reduces the efficiently of refolding IL-4TM from inclusion bodies.
- glutathione to the IL-4TM refold buffer optimizing the yield of IL-4TM refolding while still preserving the ability of 38C or 104C to be PEGylated by a maleimide-linked PEG molecule.
- glutathione to the refolding mixture, there is a time- dependency of refolding to optimize the yields of IL-4TM for PEGylation.
- Soluble IL-4 receptor inhibits airway inflammation following allergen challenge in a mouse model of asthma. J. Immunol. 164: 1086-1095.
- AER 003 a PEGylated IL-4/IL-13 antagonist for the treatment of allergic diseases Abstract
- IL-4 and IL-13 are key cytokines in the pathogenesis of allergic diseases.
- AER 003 is a PEGylated recombinant protein derived from human IL-4.
- T13D, R121D, Y124D Three point mutations (T13D, R121D, Y124D) result in a protein that binds with high affinity to the IL-4 receptor a chain and acts as an antagonist of IL-4 and IL-13, while PEGylation (40kDa at N38C) extends circulating half life.
- PEGylation 40kDa at N38C
- methacholine and bronchoalveolar lavage cell composition were determined 3 days before (Day 0) and 2 days after (Day 7) 3 consecutive-day (Days 3-5) inhalations of Ascaris suum extract.
- Plasma samples for pharmacokinetic (PK) analysis were collected to 34 days post-dose.
- the PK data indicate a 400-fold reduction in clearance/bioavailability relative to AER 001 (AERO V ANTTM), a non-PEGylated analog of AER 003. These data demonstrate the therapeutic potential of AER 003 for the treatment of allergic diseases, and support once weekly dosing in the clinic.
- Allergic diseases such as atopic dermatitis and allergic asthma are complex genetic diseases with major environmental influences. Despite the disease heterogeneity, clinical and genetic studies imply that both interleuin-4 (IL-4) and interleukin-13 (IL-13) are central to the pathogenesis of atopic diseases 1 14 . Both cytokines have multiple and overlapping biological functions considered essential to the development of allergic responses 15 " 17 18 " 22 23 26. Over-expression or administration of either IL-4 or IL-13 in the skin or airways can induce phenotypic characteristics of atopic dermatitis and allergic asthma while the use of inhibitors of either cytokine confirm the principle role of IL-4 and IL-13 in the initial antigen sensitization and
- IL-4 is capable of inducing the allergic phenotype and that a therapeutic strategy targeting both IL-4 and IL-13 may be more beneficial 40 .
- IL-4Ra chain and STAT6 signal transduction pathway are common to both the IL-4 activated type I receptor heterodimer (IL-4Ra chain/ common ⁇ -chain), and the IL-4/IL-13 activated type II receptor heterodimer (IL-13Ral chain/IL-4Ra chain) 41 ' 42 .
- IL-4 is a 15KDa four a-helix-bundle protein with two binding epitopes. The binding epitope for the high affinity IL-4Ra chain is located in the A and C helices, while the binding epitope for the low affinity ⁇ - or IL-13Ral chains is located in the A and D helices 44"46 .
- [pitrakinra or AER 001] inhibits allergen-induced airway hyper-responsiveness (AHR) in the cynomolgus monkey 52"55 .
- Pitrakinra administered either once daily by subcutaneous injection or twice daily by inhalation (nebulizer) inhibited the allergen- induced late-phase fall in FEVlin two independent Phase Ila studies with mild to moderate asthmatics (Wenzel).
- the relatively short half-life of pitrakinra ( ⁇ 3h) requires multiple daily administration and precludes systemic delivery.
- the successful inhibition of the allergen-induced late asthmatic response following local delivery has lead to the further development of pitrakinra as an inhaled dry powder.
- a Phase Ila proof of concept clinical trial in eczema suggested potential therapeutic benefit when
- PEG polyethylene glycol
- Covalent attachment of PEG to a protein increases the protein's effective size and reduces its rate of clearance from the body.
- biological activity is often decreased in PEGylated conjugates due to a decrease in observed target association rates.
- the most commonly used method for PEGylating proteins attaches PEG to amine groups, typically at lysine residues and/or at the N-terminal amino acid. A limiting factor in this approach is potential attachment to multiple sites. Variations in the position and number of PEG adducts gives rise to variations in characteristics relevant to manufacturing and clinical effects.
- a forth mutation (N38C) (AER 003) was introduced to allow attachment of a large 40 kDa-PEG moiety to a single, non-essential site in the protein to extend the circulating half life.
- N38C A forth mutation
- Tyr 124 located on the D helix of IL-4 4 have been previously shown to play a role in interaction of IL-4 with Class I, IL-4Ra/IL-2Ry, and Class II, IL-4Ra/IL-13Ra receptor complexes.
- Tyr 13 located on the A helix, has been shown to play a role in interaction of IL-4 to the IL-4Ra chain.
- Asp 38 a glycosylation site in human IL-4, does not affect the binding of IL-4 to any of the receptor subunits.
- AER 001 (Aerovant, Pitrakinra) containing the double mutation R121D, Y124D, was used as the scaffold for AER 004 (R121D, Y124D, and T13D) and AER 003 (R121D, Y124D, T13D, and N38C).
- AER 003 binds the IL-4Rq
- AER 004 (R121D, Y124D, 1.5 0.037 0.026 0.2
- AER 003 (R121D, Y124D, 1.3 0.036 0.027 0.2
- AER 003 (R121D, Y124D, 14 0.028 0.2 1.3
- AER 003 inhibits IL-4 and IL-13-induced TF-1 cell proliferation
- AER 003 inhibits IL-4-induced proliferation of human T- and B- lymphocytes
- AER 001 (R121D, Y124D) 1.1 ⁇ 0.7 1 1.7 ⁇ 0.6 1
- AER 004 (R121D, Y124D, 0.7 ⁇ 0.2 0.6 0.5 ⁇ 0.1 0.3 T13D)
- AER 003 (R121D, Y124D, 3.3 ⁇ 1.4 3 2.4 ⁇ 1.1 1.4 T13D, and N38C) + 40B PEG
- PC100 improvement with AER 003 over placebo was generally greater in animals with increased sensitivity to MCh.
- AER 003 pharmacokinetics following a single subcutaneous bolus injection were described by a 1 -compartment model with l st -order absorption and parallel l st -order and Michaelis-Menten clearance processes (Table 13, Figure 5).
- AER 001 (R121D, Y124D) 0.13 1 1.3 1
- AER 004 (R121D, Y124D,
- AER 003 (R121D, Y124D, 0.14 1.1 1.3 1 T13D, and N38C) + 40B PEG
- AER 003 concentration with slower decline (t 2 ⁇ 5.4 days) at plasma concentrations well above the C50 of the saturable clearance component (401 ng/mL), and more rapid decline (t 1/2 - 18 hours) when plasma concentrations fall below the C 50 value.
- V/F (niL) a 860-(WT/7.9 kg) 860 35%
- Th2 cytokines IL-4 and IL-13 are thought to be central to the pathogenesis of allergic diseases such as allergic rhinitis, food allergy, allergic conjunctivitis, atopic dermatitis and asthma. Thus targeting both cytokines may have significant therapeutic potential.
- AER 003, a dual IL- 4/IL-13 antagonist was assessed on allergen-induced AHR and airway inflammation in the Ascaris suum sensitive cynomolgus monkey.
- Our results indicate that inhibition of IL-4 and IL-13 using AER 003 effectively inhibited the onset of allergen-induced AHR.
- the favorable pharmacokinetic profile of AER 003 observed in the current study indicates that once weekly dosing of AER 003 would be sufficient in the clinic.
- the affinity of the triple mutein for the IL-4Ra was demonstrated to be 5 times greater than that of either native IL-4 or the double mutein (R121D/Y124D) confirming that the addition of the T13D mutation to the two point mutations
- T13D/R121D/Y124D was shown to potently inhibit the proliferative response of TF-1 cells to both IL-4 and IL-13, demonstrating the dual antagonistic nature was retained in the IL-4 triple mutein.
- PEG polyethylene glycol
- PEGylation may confer several advantages including reduced immunogenicity, reduced proteolysis, reduced toxicity and improved solubility, although the most exploited property is the improved pharmacokinetic profile that can be achieved " .
- a site-specific PEGylation strategy was utilized to improve the pharmacokinetic profile of the IL-4 triple mutein, T13D/R121D/Y 124D.
- N38C cysteine at position 38
- the N38 position was selected to limit the interference of the PEG with the IL-4Ra binding epitope and reduce the potential loss of bioactivity, a common problem associated with PEGylation 57"60 .
- the dissociation rate of the PEGylated AER 003 molecule from the IL-4Ra subunit retained a similar rate to the triple IL-4 mutein, and approximately 15 -fold greater that wild type IL-4 and the double mutein.
- assessment of the PEGylated mutein, AER 003, in the TF-1 cell assay confirmed inhibitory activity against both IL-4 and IL-13 induced proliferation, comparable to that of the double mutein (R121D/Y124D).
- the inhibitory activity of the PEGylated mutein (T13D/N38C/R121D/Y124D), AER 003, was assessed utilizing primary T and B lymphocytes, cell types considered to be central to the pathogeneisis of allergic disease.
- AER 003 significantly inhibited IL-4 induced proliferation of both T and B lymphocytes, with activity comparable to that of the double mutein (R121D/Y 124D).
- AER 003 may be a promising dual IL-4/IL-13 antagonist for the treatment of allergic disease
- translation of this activity in vivo is dependent upon the pharmacokinetic profile of the molecule.
- PEGylation may increase systemic exposure by reducing clearance, absorption and distribution may be negatively impacted.
- the effect of PEGylation on the pharmacokinetics and efficacy of AER 003 was evaluated, in a model of allergen- induced airway hyperresponsiveness and airway inflammation, cardinal features of asthma.
- Ascaris suum sensitive cynomolgus monkeys studies were performed to evaluate the effects of AER 003 on the induction of AHR and airway inflammation.
- a single subcutaneous dose of AER 003 administered 2 days prior to three successive days of allergen challenge significantly prevented the development of AHR 2 days post allergen challenge.
- AER 003 has no significant evidence for an effect of AER 003 on lung eosinophilia.
- the effect of AER 003 on AHR was comparable to that previously observed with the double mutein (R121D/Y124D) in this model, although some effect, albeit small, was previously noted on airway
- a single one-compartment systemic model with parallel first-order and saturable clearance mechanisms describes AER 003 plasma PK in cynomolgus monkeys following s.c. injection. Absorption of AER 003 was rapid relative to elimination, with absorption complete within the first day after dosing. Although the maximum rate of the saturable component of clearance is estimated to be more than five times greater than the non-saturable component, the contribution of the saturable clearance mechanism at the peak concentrations observed in this study (-10,000 ng/mL) is less than 5%. Therefore, decline in AER 003 plasma concentration from peak values initially occurs with a half-life of approximately 5.4 days.
- AER 003 and AER 004 were expressed in E. coli as insoluble protein within inclusion bodies.
- AER 003 and AER 004 intermediates were refolded from inclusion bodies and purified using a two-step chromatographic process consisting of hydrophobic interaction and cation exchange.
- AER 003 was PEGylated using Sunbright GL2-400MA 40kDa Branched PEG (NOF Corporation) and PEGylated product isolated using cation exchange chromatography.
- Ascaris suum extract (lyophilized cake, Lot # XPB33-X10 Greer Labs Inc., Lenoir, NC, USA) stock solution was prepared by adding 50 ml of sterile water to lyophilized cake.
- IL-4R IL-4 Receptor
- human IL-4 The extracellular domain of IL-4 Receptor (IL-4R) containing an Fc fusion, and human IL-4, were purchased from R&D Systems. All kinetic experiments were performed using a BIAcore 2000 instrument (GE Healthcare) at 25 (C with 10 mM
- HEPES 150 mM NaCl, 3 mM EDTA, 0.005% Surfactant P20, 0.005% BSA, pH 7.4.
- IL-4R/Fc was covalently attached to a CM5 chip via amine coupling to achieve a binding surface density of 150-250 RU.
- Six different concentrations of analyte in running buffer were evaluated in duplicate on the receptor surface using a flow rate of
- TF-1 cells erythroleukemia cell line
- human (h) IL-4 R&D Systems, Minneapolis, MN, USA
- human (h) IL-13 R&D Systems, Minneapolis, MN, USA
- TF-1 cells were cultured for 3 days in 96 well plates (lxl04/well, 100 ⁇ , volume) in RPMI (Invitrogen, Carlsbad, CA, USA) + 10% serum (Mediatech, Herndon, VA) with or without hIL-4 or hIL-13 and pitrakinra.
- AER 003 inhibits TF-1 cell proliferation by interfering with IL-4 signaling alone
- human GM-CSF R&D Systems, Minneapolis, MN, USA
- an alternative growth factor for TF-1 cells that does not bind to the IL-4 receptor was used as a specificity control.
- IC50 was calculated based on dose titration of AER 003 (71.4 nM to 0.033 nM). T- and B- Lymphocyte proliferation
- PBMC Peripheral blood mononuclear cells
- AER 003 biological activity was evaluated in a primate model of airway hyperresponsiveness (AHR) and inflammation using a double-blind, 2-period crossover design (Figure 1 A). All animals were rested at least 8 weeks between studies to allow airway responsiveness and inflammation to return to baseline (pre-allergen) levels. Studies were performed using the 7 day primate asthma model ( Figure IB). Airway responsiveness to inhaled methacholine (MCh) and airway cellular composition by bronchoalveolar lavage (BAL) were determined 2 days before (Day 0) and 2 days after (Day 7) three consecutive-day (Days 3, 4, 5) inhalations of Ascaris suum extract. AER 003 (2 mg/kg) or vehicle was administered subcutaneously 48 h prior to the first antigen challenge (Day 1 of Study).
- Ascaris suum solution (1 : 1 stock solution:PBS) was aerosolized using a
- Bird micronebulizer and Mark 7A respirator (Viasys Health Care, Palm Springs, CA, USA) delivering 15 breaths/min for 2 minutes.
- AHR to aerosolized methacholine (MCh; 0.1-100 mg/ml) was determined in anesthetized ventilated animals.
- MCh was administered using a Bird micronebulizer and Bird Mark 7A respirator set to deliver 15 breaths/min with each inspiratory breath terminating at 20 cm H 2 0.
- Lung resistance ((RL), cmH 2 0/ml/s) was measured continuously for 10 minutes after each dose while animals were ventilated using a Harvard ventilator with parameters set to 40
- Airflow was measured by a Fleisch pneumotachograph (Hans Rudolph Model 8420, Kansas City, MO, USA) and thoracic pressure by a Validyne pressure transducer. Lung resistance was computed (Buxco analyzer) using the primary signals of flow and pressure. The peak response was recorded for each dose of MCh. Dosing continued until a 100% increase in baseline resistance was achieved. Data were reported as the dose (by interpolation) of MCh required to produce a 100% increase in baseline RL (PC 100). Segmental bronchoscopic bronchoalveolar lavage (BAL) was performed after measurement of AHR.
- a pediatric fiberoptic bronchoscope (Olympus, model BF3C30, New York, NY, USA) was inserted down the endotracheal tube and guided into the distal lung until the tip of the bronchoscope became wedged.
- Normal saline (15 ml, room temperature) was instilled and then slowly aspirated.
- BAL samples were analyzed for total leukocyte count (Coulter counter, Beckman, Fullerton, CA, USA), and differential cell counts were performed by counting at least 200 cells on
- cytocentrifuged slide preparations (Cytospin 2, Cytospin Shandon, Pittsburgh, PA, USA), stained with Diff-Quick (Dade Behring Inc., Newark, DE, USA),differentiated using standard morphometric criteria.
- Dose volume was calculated based on the monkey weights on Day 0 of the study period in order to achieve a final dose of 2 mg/kg.
- AER 003 was formulated at a concentration of 6.26 mg/ml (0.32 ml/kg).
- the animals' backs were shaved and wiped with 70%> ethanol.
- the solution was injected to the subcutaneous space between the shoulder blades. All syringes were weighed before and after injection to enable accurate calculation of the dose administered.
- PK samples were collected from all animals in each period predose and at 2, 3, 4, 6, 9, 12, 16, 23 and 34 days postdose. For each sample, whole blood (2 mL) was collected into K 2 EDTA tubes, the plasma collected and frozen on dry ice and stored at -70°C for subsequent analysis. An additional sample was collected 1 day postdose during Period 2 after analysis of the first set of samples revealed higher than expected circulating AER 003 levels 2 days postdose. ELISA detection of AER 003 in plasma
- pitrakinra was detected by ELISA, using commercially available antibodies against wild type-human IL-4. Therefore, the ELISA was unable to distinguish between wild type human IL-4 and AER 003.
- ELISA 96 well plates (Maxisorp, Nunc, Rochester, NY, USA) were coated overnight (4°C) with mouse anti- human IL-4 monoclonal 8D4-8 (BD Biosciences, Franklin Lakes, NJ, USA) and blocked with 1% BSA for 1 h at room temperature. Samples and standards were added to the plate and incubated for 1 h prior to detection using biotinylated rat anti-human IL-4 monoclonal MP4-25D2 (BD Biosciences, Franklin Lakes, NJ, USA).
- Reactions were amplified using HRP-Avidin (KPL, Gaithersburg, MD, USA). Plates were read using a SpectraMAX (Molecular Devices, Sunnyvale, CA) plate reader at 450 nm with reference at 570 nm.
- AER 003 is a PEGylated recombinant protein derived from human IL-4.
- the mutated IL-4 protein (T13D, N38C, R121D, Y124D) was expressed and purified from E. coli.
- the mutein was subsequently PEGylated (position N38C) using a 40KDa branched PEG.
- AER 001 a mutated (Arg-121-Asp and Tyr-124-Asp) recombinant protein derived from hIL-4, was expressed in E. coli.
- Ascaris suum extract (lyophilized cake, Lot # XPB33-X10 Greer Labs Inc., Lenoir, NC, USA) stock solution was prepared by adding 50 ml of sterile water to lyophilized cake.
- lung eosinophil count was not a significant determinant of the Day 7 value, in agreement with historical lung eosinophil data from the 7- or 10-day monkey asthma model. Lung eosinophil counts were log- transformed prior to analysis.
- Tung AS, et al. IL-4 is required to generate and sustain in vivo IgE responses. J Immunol 1988; 141 :2335-41.
- Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci U S A 1993; 90:3730-4.
- Interleukin- 13 alters the activation state of murine macrophages in vitro:
- IL-13Ralpha2 reverses the effects of IL-13 and IL-4 on bronchial reactivity and acetylcholine-induced Ca+ signaling. Int Arch Allergy Immunol 2007; 142: 199-210.
- Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest 1999; 103:779-88.
- mice hyperreactivity in mice can occur independently of IL-4 and allergen-specific immunoglobulins. J Clin Invest 1997; 99: 1329-39.
- IL-4 receptor alpha is an important modulator of IL-4 and IL-13 receptor binding:
- Duschl A A murine interleukin-4 antagonistic mutant protein completely inhibits interleukin-4-induced cell proliferation, differentiation, and signal transduction. J Biol Chem 1997; 272: 1480-3. [0229] 62. Tomkinson A, Duez C, Cieslewicz G, Pratt JC, Joetham A,
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Public Health (AREA)
- Biophysics (AREA)
- Veterinary Medicine (AREA)
- Toxicology (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
The present invention provides methods for treating atopic diseases, including atopic dermatitis and other inflammatory or allergic skin disorders by administering mutant human Interleukin-4 (IL-4) compositions that act as antagonists to IL 4 and IL-13.
Description
USE OF MODIFIED IL-4 MUTIEN RECEPTOR ANTAGONISTS TO TREAT DERMATITIS
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. provisional application no. 61/339,073, filed February 26, 2010, which is hereby incorporated by reference in its entirety.
FIELD OF THE INVENTION
[0002] The present invention relates to methods for treating atopic diseases, including atopic dermatitis and other inflammatory or allergic skin disorders by administering mutant human Interleukin-4 (IL-4) compositions that act as antagonists to IL-4 and IL-13.
BACKGROUND OF THE INVENTION
[0003] Interleukin-4 (IL-4) is a pleiotropic cytokine with a broad spectrum of biological effects on several target cells, including activation, proliferation and differentiation of T and B cells. IL-4 is increasingly appreciated as a pivotal cytokine initiating the "Th2-type" inflammatory response, whereas IL-13 is now appreciated as the more probable downstream effector cytokine. During proliferation of B- lymphocytes, IL4 acts as a differentiation factor by regulating class switching to the IgGl and IgE isotypes.
[0004] Atopic diseases are characterized by formation of IgE antibodies, which results in immediate hypersensitivity reactions upon exposure to specific allergens. The frequent and chronic infections occurring on the skin of atopic disease patients result from the impaired immune response and from the skin barrier breaking down. Known treatments of atopic diseases include, hydrating the skin, dietary restrictions, avoidance of irritants and allergens in the environment, tars, antihistamines, hyposensitization, corticosteroids, antibacterials, antifungals, ultraviolet light, leukotriene blockers, inhibitors of mast cell content release, pentoxifylline, azathioprine, cyclosporin A, cyclophosphamide, tacrolimus, interferon gamma, thymopentin and phosphodiesterase inhibitors.
[0005] Generally, anti-histamine and steroidal agents are used as therapeutic treatments for atopic diseases. Anti-histamine agents typically reduce the itchiness of the allergic response and include diphenhydramine hydrochloride, mequitazine, promethazine hydrochloride, and chlorpheniramine maleate. Steroidal agents including prednisolone, hydrocortisone butyrate, dexamethasone valerate, betamethasone dipropionate, clobetasol propionate and the like have also been used to control the itching. While anti-histamine and steroidal agents relieve the itching, they are not desirable therapeutic agents because they cause other adverse side affects including infection, secondary adrenal cortical insufficiency, diabetes, peptic ulcer, hirsutism, alopecia, and pigmentation.
SUMMARY OF THE INVENTION
[0006] In certain embodiments, the invention provides a method for inhibiting a dermatitis response in a subject. The method entails: administering to a subject in need thereof a therapeutically effective amount of a modified IL-4 mutein receptor antagonist, wherein the modified IL-4 mutein receptor antagonist includes at least the following modifications:
(1) substitution of each of the amino acids occurring in the wild-type human IL-4 protein at positions 121 and 124 with different amino acids;
(2) substitution of the threonine occurring in the wild-type human IL 4 protein at position 13 with a different amino acid;
(3) substitution of the asparagine occurring in the wild-type human IL 4 protein at position 38 with a cysteine; and
(4) a non-protein polymer covalently attached to the substituted cysteine at position 38.
[0007] In particular embodiments of this method, the modified IL-4 mutein receptor antagonist is co-administered with a therapeutically effective amount of an additional agent that mitigates a symptom of dermatitis.
[0008] In other embodiments, the invention provides pharmaceutical composition, which includes:
a therapeutically effective amount of a modified IL-4 mutein receptor antagonist, wherein the modified IL-4 mutein receptor antagonist includes at least the following modifications:
(1) substitution of each of the amino acids occurring in the wild- type human IL-4 protein at positions 121 and 124 with different amino acids;
(2) substitution of the threonine occurring in the wild-type human IL 4 protein at position 13 with a different amino acid;
(3) substitution of the asparagine occurring in the wild-type human IL 4 protein at position 38 with a cysteine; and
(4) a non-protein polymer covalently attached to the substituted cysteine at position 38; and
a therapeutically effective amount of an additional agent that is useful for mitigating a symptom of dermatitis. The therapeutically effective amount of the modified IL-4 mutein receptor antagonist is sufficient to mitigate a symptom of dermatitis, and the therapeutically effective amount of the additional agent is sufficient to mitigate a symptom of dermatitis.
[0009] In yet other embodiments, the invention provides a kit that includes:
at least one unit dosage form comprising a therapeutically effective amount of a modified IL-4 mutein receptor antagonist, wherein the modified IL-4 mutein receptor antagonist comprises at least the following modifications:
(1) substitution of each of the amino acids occurring in the wild- type human IL-4 protein at positions 121 and 124 with different amino acids;
(2) substitution of the threonine occurring in the wild-type human IL 4 protein at position 13 with a different amino acid;
(3) substitution of the asparagine occurring in the wild-type human IL 4 protein at position 38 with a cysteine; and
(4) a non-protein polymer covalently attached to the substituted cysteine at position 38; and
at least one unit dosage form comprising a therapeutically effective amount of an additional agent that is useful for mitigating a symptom of dermatitis.
The therapeutically effective amount of the modified IL-4 mutein receptor antagonist is sufficient to mitigate a symptom of dermatitis, and the therapeutically effective amount of the additional agent is sufficient to mitigate a symptom of dermatitis.
BRIEF DESCRIPTION OF THE DRAWINGS
[0010] Figure 1 shows a schematic representation of the chemistry of a
PEGylation reaction.
[0011] Figure 2 is a graphical diagram showing data from BIAcore binding to IL-4Ra comparing the IL-4 double mutein (IL-4DM) to the same molecule with a 30kD linear or a 40kD branched PEG at position 38C.
[0012] Figure 3 is a graphical diagram showing data from inhibition of TF-1 growth with IL-4 stimulation revealing that PEGylated IL-4TM (T13D/R121D/Y124D) is more potent than PEGylated IL-4DM and is equally potent to IL-4DM
(R121D/Y124D).
[0013] Figure 4A-4B: AER 003 Model. Model of AER 003 derived from the structure of human IL-4 is shown. (A) Residues implicated in the antagonist activity are highlighted in yellow (AER 001). The mutation T13D (red) results in increased binding to IL-4R a, while N38C (green) serves as an attachment point for PEG. (B) Pegylated AER 003.
[0014] Figure 5: Study design. AER 003 biological activity was evaluated in a primate model of AHR and inflammation using a double-blind, 2-period cross-over design (A). All animals were rested at least 8 weeks between studies to allow airway responsiveness and inflammation to return to baseline (pre-allergen) levels. Studies were performed using the 7 day primate asthma model (B). Airway responsiveness to inhaled MCh and airway cellular composition by BAL were determined 2 days before (Day 0) and 2 days after (Day 7) three consecutive-day (Days 3, 4, 5) inhalations of Ascaris suum extract. AER 003 (2 mg/kg) or vehicle was administered subcutaneously 48 h prior to the first antigen challenge (Day 1).
[0015] Figure 6: Effect of AER 003 on allergen-induced AHR. Treatment-by- treatment scatterplot of the effect of AER 003 on Day 7 PCioo relative to placebo. Each symbol represents the outcome of a single subject. Points lying above the reference line of no treatment effect indicate an improvement with AER 003 relative to placebo.
[0016] Figure 7: Effect of AER 003 on allergen-induced airway eosnophilia. Treatment-by-treatment scatterplot of the effect of AER 003 on Day 7 lung eosinophil count relative to placebo. Each symbol represents the outcome of a single subject.
Points lying below the reference line of no treatment effect indicate an improvement with AER 003 relative to placebo.
[0017] Figure 8: PK profile of AER 003 during monkey efficacy studies.
Observed and predicted AER 003 plasma concentrations following a single 2-mg/kg subcutaneous injection to cynomolgus monkeys. Solid lines represent the population prediction for a 7.9-kg monkey derived from the pharmacokinetic model. Symbols represent individual observations, with results below detection assigned to half the analytical method's lower limit of quantitation (LLOQ) and indicated with an "x" for graphical display. The LLOQ is indicated by a horizontal dotted line (1.5 ng/mL). DETAILED DESCRIPTION
[0018] In certain embodiments, the invention provides for methods of treatment of atopic diseases (AD), in particular, atopic dermatitis, by administering a therapeutic effective amount of mutant human IL-4 compositions. Human IL-4 mutant proteins used as antagonists or partial agonists of human IL-4 are also described in U.S. Pat. No. 6,130,318 to Wild et al., the entire contents of which is incorporated herein by reference, in particular, for its description of human IL-4 mutant proteins. The methods of the invention can be used to treat typical atopic diseases or allergic dermatitis including contact dermatitis, atopic dermatitis (i.e., eczema), psoriasis, seborrheic dermatitis, and the like. Definitions
[0019] Terms used in the claims and specification are defined as set forth below unless otherwise specified.
[0020] As used herein, the term "dermatitis" is defined generally as an inflammation of the skin. Stedman's Medical Dictionary, 27th edition, Lippincott Williams & Wilkins (2000).
[0021] As used herein, the term "contact dermatitis" is an inflammatory response of the skin to an antigen (or allergen) or irritant (Stedman's Medical
Dictionary, supra). Irritants are substances that directly affect the skin or cause direct tissue damage, while allergens induce an immunologic reaction that causes
inflammation and tissue damage. Some common irritants include wool and synthetic
fibers, soaps and detergents, perfumes and cosmetics, dust and sand, cigarette smoke, and substances such as chlorine, mineral oil or solvents.
[0022] Allergens are substances typically from foods, plants, or animals that inflame the skin and cause an immune reaction. Initially, allergens typically illicit inflammatory response, including recruitment of cells, for example T cells,
macrophages and the like. Upon repeated contact with the allergen, the contact dermatitis can then develop into eczema accompanied with lichenification and infiltration of the cells.
[0023] As used herein, the term "atopic dermatitis," "atopic eczema," or "eczema" and related terms are used interchangeably and represent a complex disease primarily caused by cellular immune deficiency and elevated immunoglobulin E (IgE). Allergens that are also irritants to the skin are believed to predispose an individual to develop dermatitis more often than simply exposure to an allergenic trigger. Anxiety, stress and depression may all play a role in the exacerbation of eczema. Further, those with atopic eczema may be discovered to have an increased eosinophil count.
[0024] As used herein, "wild type IL-4" or "wtIL-4" and equivalents thereof are used interchangeably and refer to human Interleukin-4, native or recombinant, having the 129 normally occurring amino acid sequence of native human IL-4, as disclosed in U.S. Pat. No. 5,017,691, which is incorporated herein by reference. Further, the modified human IL-4 receptor antagonists described herein may have various insertions and/or deletions and/or couplings to a non-protein polymer, and are numbered in accordance with the wtIL-4. Accordingly, one skilled in the art will appreciate that the amino acids at positions, e.g., 13 (threonine), 38 (asparagine), 121 (arginine), and/or 124 (tyrosine), may be shifted in a mutein. For example, in a mutein in which an amino acid is added to the N-terminus of IL-4, the threonine at position 13 (in the wild type sequence) is actually the 14th amino acid in the mutein sequence; however, this amino acid is still termed "theonine 13" or "T13." Thus, "numbered according to wild type IL-4" mean identifying a chosen amino acid with reference to the position at which that amino acid normally occurs in wild type IL-4.
[0025] As used herein, the terms "mutant human IL-4 protein," "modified human IL-4 receptor antagonist," "mhIL-4," "IL-4 mutein," "IL-4 antagonist," and equivalents thereof are used interchangeably. These polypeptides and functional
fragments thereof refer to polypeptides wherein specific amino acid substitutions to the mature human IL-4 protein have been made. These polypeptides include the mIL-4 compositions of the present invention, which are administered to a subject in need of treatment for dermititis. In particular, the mhIL-4 described herein include at least the R121D/Y124D pair of substitutions ("IL-4RA") (SEQ ID NO: 31). The modification of hIL-4 and of mhIL-4 are described in US Patent Publication No. 20090010874 (published Jan. 8, 2009) and International Publication No. WO 2009065007 (published May 22, 2009), the entire contents of both of which are incorporated herein by reference, in particular, for their description of mhIL-4.
[0026] As used with respect to sequences, the term "identity" refers to a relationship between the sequences of two or more polypeptide molecules or two or more nucleic acid molecules, as determined by comparing the sequences. In the art, "identity" also means the degree of sequence relatedness between nucleic acid molecules or polypeptides, as the case may be, as determined by the match between strings of two or more nucleotide or two or more amino acid sequences. "Identity" measures the percent of identical matches between the smaller of two or more sequences with gap alignments (if any) addressed by a particular mathematical model or computer program (i.e., "algorithms").
[0027] Identity two nucleic acids or two polypeptides can be readily calculated by known methods. Such methods include, but are not limited to, those described in COMPUTATIONAL MOLECULAR BIOLOGY, (Lesk, A.M., ed.), 1988, Oxford University Press, New York; BIOCOMPUTING: INFORMATICS AND GENOME PROJECTS, (Smith, D.W., ed.), 1993, Academic Press, New York; COMPUTER ANALYSIS OF SEQUENCE DATA, Part 1, (Griffin, A.M., and Griffin, H.G., eds.), 1994, Humana Press, New Jersey; von Heinje, G., SEQUENCE ANALYSIS IN MOLECULAR BIOLOGY, 1987, Academic Press; SEQUENCE ANALYSIS
PRIMER, (Gribskov, M. and Devereux, J., eds.), 1991, M. Stockton Press, New York; Carillo et al, 1988, SIAM J. Applied Math., 48:1073; and Durbin et al, 1998,
BIOLOGICAL SEQUENCE ANALYSIS, Cambridge University Press.
[0028] Preferred methods to determine identity are designed to give the largest match between the sequences tested. Methods to determine identity are described in publicly available computer programs. Preferred computer program methods to
determine identity between two sequences include, but are not limited to, the GCG program package, including GAP (Devereux et al, 1984, Nucl. Acid. Res., 12:387; Genetics Computer Group, University of Wisconsin, Madison, WI), BLASTP, BLASTN, and FASTA (Altschul et al, 1990, J. Mol Biol, 215:403-410). The BLASTX program is publicly available from the National Center for Biotechnology Information (NCBI) and other sources (BLAST Manual, Altschul et al NCB/NLM/NIH Bethesda, MD 20894; Altschul et al, 1990, supra). The well-known Smith Waterman algorithm may also be used to determine identity.
[0029] Certain alignment schemes for aligning two amino acid sequences may result in matching of only a short region of the two sequences, and this small aligned region may have very high sequence identity even though there is no significant relationship between the two full-length sequences. Accordingly, in certain
embodiments, the selected alignment method (GAP program) will result in an alignment that spans at least 50 contiguous amino acids of the target polypeptide.
[0030] For example, using the computer algorithm GAP (Genetics Computer
Group, University of Wisconsin, Madison, WI), two polypeptides for which the percent sequence identity is to be determined are aligned for optimal matching of their respective amino acids (the "matched span", as determined by the algorithm). In certain embodiments, a gap opening penalty (which is calculated as three-times the average diagonal; where the "average diagonal" is the average of the diagonal of the comparison matrix being used; the "diagonal" is the score or number assigned to each perfect amino acid match by the particular comparison matrix) and a gap extension penalty (which is usually one-tenth of the gap opening penalty), as well as a
comparison matrix such as PAM250 or BLOSUM 62 are used in conjunction with the algorithm. In certain embodiments, a standard comparison matrix (see Dayhoff et al, 1978, Atlas of Protein Sequence and Structure, 5:345-352 for the PAM 250 comparison matrix; Henikoff et al, 1992, Proc. Natl. Acad. Sci USA, 89:10915-10919 for the BLOSUM 62 comparison matrix) is also used by the algorithm.
[0031] In certain embodiments, the parameters for a polypeptide sequence comparison include the following:
Algorithm: Needleman et al, 1970, J. Mol Biol, 48:443-453;
Comparison matrix: BLOSUM 62 from Henikoff et al, 1992, supra;
Gap Penalty: 12
Gap Length Penalty: 4
Threshold of Similarity: 0
The GAP program may be useful with the above parameters. In certain embodiments, the aforementioned parameters are the default parameters for polypeptide comparisons (along with no penalty for end gaps) using the GAP algorithm.
[0032] As used herein, the twenty conventional amino acids and their abbreviations follow conventional usage. See IMMUNOLOGY-A SYNTHESIS, 2nd Edition, (E. S. Golub and D. R. Gren, Eds.), Sinauer Associates: Sunderland, MA, 1991, incorporated herein by reference for any purpose. Stereoisomers (e.g., D-amino acids) of the twenty conventional amino acids; unnatural amino acids such as α-, α- disubstituted amino acids, N-alkyl amino acids, lactic acid, and other unconventional amino acids may also be suitable components for polypeptides described herein.
Examples of unconventional amino acids include: 4-hydroxyproline, γ- carboxyglutamate, ε-Ν,Ν,Ν-trimethyllysine, ε-Ν-acetyllysine, O-phosphoserine, N- acetylserine, N-formylmethionine, 3-methylhistidine, 5 -hydroxy lysine, σ-Ν- methylarginine, and other similar amino acids and imino acids (e.g., 4-hydroxyproline). In the polypeptide notation used herein, the left-hand direction is the amino terminal direction and the right-hand direction is the carboxyl-terminal direction, in accordance with standard usage and convention.
[0033] Peptide analogs are commonly used in the pharmaceutical industry as non-peptide drugs with properties analogous to those of the template peptide. These types of non-peptide compound are termed "peptide mimetics" or "peptidomimetics". See Fauchere, 1986, Adv. Drug Res. 15:29; Veber & Freidinger, 1985, TINS p.392; and Evans et al, 1987, J. Med. Chem. 30: 1229, which are incorporated herein by reference for their descriptions of peptide mimetics. Such compounds are often developed with the aid of computerized molecular modeling. Peptide mimetics that are structurally similar to therapeutically useful peptides may be used to produce a similar therapeutic or prophylactic effect. Generally, peptidomimetics are structurally similar to a paradigm polypeptide (i.e., a polypeptide that has a biochemical property or
pharmacological activity), such as human antibody, but have one or more peptide linkages optionally replaced by a linkage selected from: -CH2-NH-, -CH2-S-, -CH2-
CH2-, -CH=CH-(cw and trans), -COCH2-, -CH(OH)CH2-, and -CH2SO-, by methods well known in the art. Systematic substitution of one or more amino acids of a consensus sequence with a D-amino acid of the same type (e.g., D-lysine in place of L- lysine) may be used in certain embodiments to generate more stable peptides. In addition, constrained peptides comprising a consensus sequence or a substantially identical consensus sequence variation may be generated by methods known in the art (Rizo & Gierasch, 1992, Ann. Rev. Biochem. 61 :387, incorporated herein by reference for any purpose); for example, by adding internal cysteine residues capable of forming intramolecular disulfide bridges which cyclize the peptide. Treatment Method In General
[0034] In one embodiment of the invention, a modified human IL-4 mutien receptor antagonist is useful for treating various conditions associated with one of the pleiotropic effects of IL-4 and IL-13. For instance, antagonists of IL-4 and IL-13 are useful in treating conditions exacerbated by IL-4 and IL-13 production including asthma, allergy, dermatitis or other inflammatory response-related conditions. Some uses of the modified human IL-4 mutein receptor antagonists are described in U.S. Pat. No. 6,130,318 and in U.S. Publication No. 20070009479 (published Jan. 11, 2007), the entire contents of both of which are incorporated herein by reference. In an exemplary embodiment, modified human IL-4 mutein receptor antagonists are employed to treat dermatitis, such as that resulting from a hypersensitivity reaction. In variations of this embodiment, the hypersensitivity reaction includes contact dermatitis and/or atopic dermatitis. In an illustrative embodiment, the modified human IL-4 mutien receptor antagonist is used to treat eczema. [0035] The subject of the method can be any organism that exhibits (i.e., is experiencing or at risk for) an allergic response, such as dermatitis. Examples of suitable subjects include research animals or pets, such as mice, rats, guinea pigs, rabbits, cats, dogs, as well as monkeys and other primates, and humans.
Modified IL-4 Mutein Receptor Antagonists
[0036] "Modified IL-4 mutein receptor antagonists," as used herein, includes the IL-4RA mutein described in US Pat. Nos. 6,028,176 & 6,313,272 (hereby incorporated by reference in their entirety), with additional amino acid substitutions at one or more positions of the mature IL-4 protein. Exemplary triple muteins include, but are not limited to a substitution of arginine by aspartic acid at position 121, of tyrosine by aspartic acid at position 124, and of serine by aspartic acid at position 125 in the D-helix; and a substitution of threonine by aspartic acid at position 13, of arginine by aspartic acid at position 121, and of tyrosine by aspartic acid at position 124 in the D-helix. In one embodiment, the triple muteins further comprise an N-terminal methionine. Variations in this section of the D helix positively correlate with changes in interactions at the second binding region. The modified IL-4 mutein receptor antagonist may further include one or more substitutions wherein said substitutions enable the site-specific coupling of at least one non-protein polymer, such as polypropylene glycol, polyoxyalkylene, or polyethylene glycol (PEG) molecule to the mutein. Site-specific coupling of PEG, for example, allows the generation of a modified mutein which possesses the benefits of a polyethylene-glycosylated
(PEGylated) molecule, namely increased plasma half life and decreased
immunogenicity while maintaining greater potency over non-specific PEGylation strategies such as N-terminal and lysine side-chain PEGylation.
[0037] It should be understood that the structure of the attached PEG moieties should be considered in optimizing the PEGylation of the muteins of the invention. In one embodiment, the PEG moiety is linear. Linear PEG moieties are limited in size by the manufacturing process because the amount of PEG-diol increases as PEG molecular weight increases. With linear moieties, increases in PEG-mutein molecular size is typically accomplished by increasing the number of PEG attachment sites on the mutein. This often results in suboptimal pharmacological profiles. In another embodiment, the PEG moiety is branched from a single attachment site. Branched PEG moieties have the advantage of increasing the size of the PEG molecule without increasing the number of site attachments.
[0038] Thus, in various embodiments, the polyethylene glycol (PEG) moiety has a molecular weight ranging from about 2 kD to about 50 kD, e.g, about: 3, 5, 10,
15, 20, 25, 30, 35, 40, 45, or any molecular weight falling within a range bound by any of these values. In one embodiment, the PEG moiety is about 40 kD. Covalent attachment of the PEG to the drug (known as "PEGylation") may be accomplished by known chemical reactions and/or synthesis techniques. For example, in one aspect of the present invention, the PEGylation of protein may be accomplished by reacting NHS-activated PEG with the mutein under suitable reaction conditions. In another aspect of the present invention, the PEGylation of protein may be accomplished by reacting a maleimide activated PEG with the sulhydryl group of the cytsteine residue in the protein under suitable reaction conditions.
[0039] PEG-mutein conjugates can be created in at least three different ways:
A single large PEG moiety can be attached at a single site on the mutein; a branched PEG moiety (i.e., two or more medium PEG chains joined together via a linker) can be attached at a single site on the mutein; or several small chains may be attached at multiple sites on the mutein. Theoretically, monosite PEGylated muteins have higher activity because the PEG attachment is less likely to occur at or near receptor-binding domains.
[0040] Improvements in PEGylation and other types of post-translational modifications (PTMs) have been extensive and there are now a large number of PTM techniques and reagents known in the art, and new ones are regularly being developed. Techniques and reagents for PEGylation include, for example: (i) specialized linkers and coupling chemistries; (ii) branched PEGs which effectively allow additional PEG groups to be attached to a single conjugation site; (iii) site-specific PEGylation, including site-specific monoPEGylation; and (iv) site-directed enzymatic PEGylation (e.g. using a transglutaminase reaction). There are also additional technologies and reagents available from an increasing number of commercial suppliers (see, e.g.,
Nektar/Shearwater (on the world wide web at nektar.com), Sunbio (on the world wide web at sunbio.com and sunbio.com/peg-shop), Celares GmbH (on the world wide web at celares.com), NOF Corporation (on the world wide web at peg-drug.com), and others).
[0041] Also included in this invention is the selection of the specific site of amino acid substitution which enables proper folding of the molecule following expression. Modified IL-4 mutein receptor antagonists bind to IL-4 and IL-13 with an
affinity loss not greater than 10-fold relative to that of IL-4RA. Modified IL-4 mutein receptor antagonists inhibit IL-4 and IL-13 mediated activity with a loss of potency not greater than 10-fold relative to that of IL-4RA. In addition, modified IL-4 mutein receptor antagonists possess a plasma half-life which is at least 2 to 10-fold greater than that of unmodified IL-4RA.
[0042] The IL-4 muteins of this invention may also be characterized by amino acid insertions, deletions, substitutions and modifications at one or more sites in or at the other residues of the native IL-4 polypeptide chain. In accordance with this invention any such insertions, deletions, substitutions and modifications should result in an IL-4 mutein that retains its IL-4 antagonist activity.
[0043] An additional aspect of this invention is provided in the method with which the protein is expressed and refolded, as depicted in Example 2. The IL-4 mutein is preferably purified so as to allow efficient PEGylation. An exemplary method for purification is described in Example 2 below. When the mutein is refolded in the presence of a sulfhydryl protecting agent, a covalent disulfide bond is formed between the IL-4 mutein' s free cysteine and the protecting agent. In contrast, the use of the sulfhydryl protecting agent dithiothreitol (DTT), which oxidizes to form a stable disulfide bond, will not form a covalent bond with the IL-4 mutein' s free cysteine, thus leaving its sulfydryl group free to react with the PEG maleimide reagent. IL-4 muteins purified after refolding in the presence of a sulfhydryl protecting agent can react with the PEG reagent if treated with DTT, but a mixture of monoPEGylated and
multiPEGylated products are generated, suggesting that existing IL-4 cysteines are also PEGylated. PEGylation of existing cysteines would lead to misfolded products that are inactive. [0044] The Ka of modified IL-4 mutein receptor antagonists to the IL-4 receptor can be assayed using any method known in the art, including technologies such as realtime Bimolecular Interaction Analysis (BIA) outlined in Example 4. BIA is a technology for studying biospecific interactions in real time, without labeling any of the interactants (e.g., BIAcore™). Changes in the optical phenomenon surface plasmon resonance (SPR) can be used as an indication of real-time reactions between biological molecules.
[0045] The capacity of modified IL-4 mutein receptor antagonists to inhibit the proliferative response of immune cells can be assessed using proliferative assays as outlined in Example 5 and this capacity expressed as an Inhibitory Concentration 50% (IC50).
[0046] In a BIAcore™ assay, modified IL-4 mutein receptor antagonists of the present invention specifically bind to the human IL-4 receptor with a preferred Ka in the range of from about 1.0 nM to about 100 nM. More preferred embodiments of the present invention bind to human IL-4 receptor with a Kj of approximately 0.5 nM to about 1.0 uM. Still more preferred embodiments of the present invention bind to human IL-4 receptor with a Kd of approximately 0.1 nM to about 10μΜ. Additionally, modified IL-4 mutein receptor antagonists of the present invention, as envisioned, will bind to human IL-4 receptor and neutralize its capacity to promote immune cell proliferation with a preferred IC50 ranging from about 1.0 nM to about 100 nM. More preferred human antagonists bind IL-4 receptor and neutralize its immune cell proliferation capacity with an IC50 ranging from approximately 0.5 nM to 1 μΜ, with the most preferred antagonists of this invention binding and inhibiting IL-4 receptor with an IC50 of approximately 0.1 nM to about 10 μΜ.
[0047] In particular embodiments, modified IL-4 mutein receptor antagonists described herein, such as PEGylated forms, exhibit enhanced bioavailability, compared to unmodified, e.g., non-PEGylated, forms.
[0048] Certain embodiments of modified IL-4 mutein receptor antagonists described herein also exhibit a plasma half-life that is preferably at least 2 to 10-fold greater than that of unmodified IL4RA with the most preferred embodiments of the present invention exhibiting a plasma half- life which is 10-100-fold greater than that of unmodified IL-4RA (see Example 7).
[0049] A number of modified IL-4 mutein receptor antagonists with the characteristics described above have been identified by screening candidates with the above assays. The embodiments of the present invention have the polypeptide sequences shown in Table 1 (SEQ ID NOS: 10-16 and 32-33).
Table 1: Polypeptide Sequences

[0050] Modified IL-4 mutein receptor antagonists can be produced using any method capable of producing polypeptides of having the desired amino acid sequence. Typically, recombinant expression will be the most convenient method. Polynucleotides Encoding Modified IL-4 Mutein Receptor
Antagonists
[0051] Recombinant expression requires polynucleotides encoding modified IL-
4 mutein receptor antagonists. These polynucleotides can be used, for example, to produce quantities of the antagonists for therapeutic use. Methods of constructing and expressing degenerative DNA sequences capable of expressing the same amino acid sequence as a given polynucleotide sequence are known in the art.
[0052] A polynucleotide of the invention can be readily obtained in a variety of ways including, without limitation, cDNA or genomic library screening, expression library screening, and/or PCR amplification of cDNA. Such methods are well known and include those set forth in Sambrook et al, Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Laboratory Press, 1989) and/or Current Protocols in Molecular Biology (Ausubel et al, eds., Green Publishers Inc. and Wiley and Sons 1994).
[0053] One method for obtaining a suitable nucleic acid sequence is the polymerase chain reaction (PCR). In this method, cDNA is prepared from
poly(A)+RNA or total RNA using the enzyme reverse transcriptase. Two primers, typically complementary to two separate regions of a modified IL-4 mutein receptor antagonist cDNA, are then added to the cDNA along with a polymerase such as Taq polymerase, and the polymerase amplifies the cDNA region between the two primers.
[0054] Another means of preparing a nucleic acid molecule of the invention is chemical synthesis using methods well known to the skilled artisan such as those described by Engels et al, 1989, Angew. Chem. Intl. Ed. 28:716-34. These methods include, inter alia, the phosphotriester, phosphoramidite, and H-phosphonate methods for nucleic acid synthesis. A preferred method for such chemical synthesis is polymer- supported synthesis using standard phosphoramidite chemistry. Typically, the DNA will be several hundred nucleotides in length. Nucleic acids larger than about 100
nucleotides can be synthesized as several fragments using these methods. The fragments can then be ligated together.
[0055] Polynucleotides that encode illustrative modified IL-4 mutein receptor antagonists are shown in Table 2 (SEQ ID NOs: 2-8 and 31).
Table 2: Polynucleotide Sequences
SEQ ID Name Sequence
NO
1 IL-4RA ATGCACAAGTGCGATATCACCTTACAGGAGATC
ATCAAAACTTTGAACAGCCTCACAGAGCAGAA
GACTCTGTGCACCGAGTTGACCGTAACAGACAT
CTTTGCTGCCTCCAAGAACACAACTGAGAAGGA
AACCTTCTGCAGGGCTGCGACTGTGCTCCGGCA
GTTCTACAGCCACCATGAGAAGGACACTCGCTG
CCTGGGTGCGACTGCACAGCAGTTCCACAGGCA
CAAGCAGCTGATCCGATTCCTGAAACGGCTCGA
CAGGAACCTCTGGGGCCTGGCGGGCTTGAATTC
CTGTCCTGTGAAGGAAGCCAACCAGAGTACGTT
GGAAAACTTCTTGGAAAGGCTAAAGACGATCAT
GGACGAGAAAGACTCAAAGTGTTCGAGCTAAT
AA
2 IL4-RE-T28C ATGCACAAGTGCGATATCACCTTACAGGAGAT
CATCAAAACTTTGAACAGCCTCACAGAGCAGA
AGACTCTGTGCACCGAGTTGTGCGTAACAGAC
ATCTTTGCTGCCTCCAAGAACACAACTGAGAA
GGAAACCTTCTGCAGGGCTGCGACTGTGCTCC
GGCAGTTCTACAGCCACCATGAGAAGGACACT
CGCTGCCTGGGTGCGACTGCACAGCAGTTCCA
CAGGCACAAGCAGCTGATCCGATTCCTGAAAC
GGCTCGACAGGAACCTCTGGGGCCTGGCGGGC
TTGAATTCCTGTCCTGTGAAGGAAGCCAACCA
GAGTACGTTGGAAAACTTCTTGGAAAGGCTAA
AGACGATCATGGACGAGAAAGACTCAAAGTGT
TCGAGCTAATAA
3 IL4-RE-S36C ATGCACAAGTGCGATATCACCTTACAGGAGAT
CATCAAAACTTTGAACAGCCTCACAGAGCAGA
AGACTCTGTGCACCGAGTTGACCGTAACAGAC
ATCTTTGCTGCCTGCAAGAACACAACTGAGAA
GGAAACCTTCTGCAGGGCTGCGACTGTGCTCC
GGCAGTTCTACAGCCACCATGAGAAGGACACT
CGCTGCCTGGGTGCGACTGCACAGCAGTTCCA
CAGGCACAAGCAGCTGATCCGATTCCTGAAAC
GGCTCGACAGGAACCTCTGGGGCCTGGCGGGC
TTGAATTCCTGTCCTGTGAAGGAAGCCAACCA
GAGTACGTTGGAAAACTTCTTGGAAAGGCTAA
AGACGATCATGGACGAGAAAGACTCAAAGTGT
TCGAGCTAATAA
IL4-RE-K37C ATGCACAAGTGCGATATCACCTTACAGGAGAT
CATCAAAACTTTGAACAGCCTCACAGAGCAGA
AGACTCTGTGCACCGAGTTGACCGTAACAGAC
ATCTTTGCTGCCTCCTGCAACACAACTGAGAA
GGAAACCTTCTGCAGGGCTGCGACTGTGCTCC
GGCAGTTCTACAGCCACCATGAGAAGGACACT
CGCTGCCTGGGTGCGACTGCACAGCAGTTCCA
CAGGCACAAGCAGCTGATCCGATTCCTGAAAC
GGCTCGACAGGAACCTCTGGGGCCTGGCGGGC
TTGAATTCCTGTCCTGTGAAGGAAGCCAACCA
GAGTACGTTGGAAAACTTCTTGGAAAGGCTAA
AGACGATCATGGACGAGAAAGACTCAAAGTGT
TCGAGCTAATAA
IL4-RE-N38C ATGCACAAGTGCGATATCACCTTACAGGAGAT
CATCAAAACTTTGAACAGCCTCACAGAGCAGA
AGACTCTGTGCACCGAGTTGACCGTAACAGAC
ATCTTTGCTGCCTCCAAGTGCACAACTGAGAA
GGAAACCTTCTGCAGGGCTGCGACTGTGCTCC
GGCAGTTCTACAGCCACCATGAGAAGGACACT
CGCTGCCTGGGTGCGACTGCACAGCAGTTCCA
CAGGCACAAGCAGCTGATCCGATTCCTGAAAC
GGCTCGACAGGAACCTCTGGGGCCTGGCGGGC
TTGAATTCCTGTCCTGTGAAGGAAGCCAACCA
GAGTACGTTGGAAAACTTCTTGGAAAGGCTAA
AGACGATCATGGACGAGAAAGACTCAAAGTGT
TCGAGCTAATAA
IL4-RE-A104C ATGCACAAGTGCGATATCACCTTACAGGAGAT
CATCAAAACTTTGAACAGCCTCACAGAGCAGA
AGACTCTGTGCACCGAGTTGACCGTAACAGAC
ATCTTTGCTGCCTCCAAGAACACAACTGAGAA
GGAAACCTTCTGCAGGGCTGCGACTGTGCTCC
GGCAGTTCTACAGCCACCATGAGAAGGACACT
CGCTGCCTGGGTGCGACTGCACAGCAGTTCCA
CAGGCACAAGCAGCTGATCCGATTCCTGAAAC
GGCTCGACAGGAACCTCTGGGGCCTGGCGGGC
TTGAATTCCTGTCCTGTGAAGGAATGCAACCA
GAGTACGTTGGAAAACTTCTTGGAAAGGCTAA
AGACGATCATGGACGAGAAAGACTCAAAGTGT
TCGAGCTAATAA
IL4-RE-N105C ATGCACAAGTGCGATATCACCTTACAGGAGAT
CATCAAAACTTTGAACAGCCTCACAGAGCAGA
AGACTCTGTGCACCGAGTTGACCGTAACAGAC
ATCTTTGCTGCCTCCAAGAACACAACTGAGAA
GGAAACCTTCTGCAGGGCTGCGACTGTGCTCC
GGCAGTTCTACAGCCACCATGAGAAGGACACT
CGCTGCCTGGGTGCGACTGCACAGCAGTTCCA
CAGGCACAAGCAGCTGATCCGATTCCTGAAAC
GGCTCGACAGGAACCTCTGGGGCCTGGCGGGC
TTGAATTCCTGTCCTGTGAAGGAAGCCTGCCA
GAGTACGTTGGAAAACTTCTTGGAAAGGCTAA
AGACGATCATGGACGAGAAAGACTCAAAGTGT
TCGAGCTAATAA
8 IL4-RE-Q106C ATGCACAAGTGCGATATCACCTTACAGGAGAT
CATCAAAACTTTGAACAGCCTCACAGAGCAGA
AGACTCTGTGCACCGAGTTGACCGTAACAGAC
ATCTTTGCTGCCTCCAAGAACACAACTGAGAA
GGAAACCTTCTGCAGGGCTGCGACTGTGCTCC
GGCAGTTCTACAGCCACCATGAGAAGGACACT
CGCTGCCTGGGTGCGACTGCACAGCAGTTCCA
CAGGCACAAGCAGCTGATCCGATTCCTGAAAC
GGCTCGACAGGAACCTCTGGGGCCTGGCGGGC
TTGAATTCCTGTCCTGTGAAGGAAGCCAACTG
CAGTACGTTGGAAAACTTCTTGGAAAGGCTAA
31 IL4-RE-T13D- ATGCACAAATGCGATATCACCCTGCAGGAAATC
ATCAAAGACCTGAATTCTCTGACCGAACAGAAA
N38C
ACCCTGTGCACCGAACTGACCGTTACCGACATC
(IL-4TM-N38C) TTCGCTGCTTCGAAATGCACCACCGAAAAAGAA
ACCTTCTGCCGTGCTGCTACCGTTCTGCGTCAGT
TCTACTCTCACCACGAAAAAGACACCCGTTGCC
TGGGTGCTACCGCTCAGCAGTTCCACCGTCACA
AACAGCTGATCCGTTTCCTGAAACGTCTGGACC
GTAACCTGTGGGGTCTGGCTGGTCTGAACAGCT
GCCCGGTTAAAGAAGCTAACCAGTCTACCCTGG
AAAACTTCCTGGAACGTCTGAAAACCATCATGG
ACGAAAAAGACTCTAAATGCTCTTCT
Vectors
[0056] A polynucleotide can be incorporated into a vector for propagation and/or expression in a host cell. Such vectors typically contain a replication sequence capable of effecting replication of the vector in a suitable host cell (i.e., an origin of replication) as well as sequences encoding a selectable marker, such as an antibiotic resistance gene. Upon transformation of a suitable host, the vector can replicate and function independently of the host genome or integrate into the host genome. Vector design depends, among other things, on the intended use and host cell for the vector, and the design of a vector of the invention for a particular use and host cell is within the level of skill in the art.
[0057] If the vector is intended for expression of a polypeptide, the vector includes one or more control sequences capable of effecting and/or enhancing the expression of an operably linked polypeptide coding sequence. Control sequences that
are suitable for expression in prokaryotes, for example, include a promoter sequence, an operator sequence, and a ribosome binding site. Control sequences for expression in eukaryotic cells include a promoter, an enhancer, and a transcription termination sequence (i.e., a polyadenylation signal).
[0058] An expression vector according to the invention can also include other sequences, such as, for example, nucleic acid sequences encoding a signal sequence or an amplifiable gene. A signal sequence can direct the secretion of a polypeptide fused thereto from a cell expressing the protein. In the expression vector, nucleic acid encoding a signal sequence is linked to a polypeptide coding sequence so as to preserve the reading frame of the polypeptide coding sequence. The inclusion in a vector of a gene complementing an auxotrophic deficiency in the chosen host cell allows for the selection of host cells transformed with the vector.
[0059] Vectors are typically produced by linking desired elements by ligation at convenient restriction sites. If such sites do not exist, suitable sites can be introduced by standard mutagenesis (e.g., site-directed or cassette mutagenesis) or synthetic oligonucleotide adaptors or linkers can be used in accordance with conventional practice.
Host Cells
[0060] A wide variety of host cells are available for propagation and/or expression of vectors. Examples include prokaryotic cells (such as E. coli and strains of Bacillus, Pseudomonas, and other bacteria), yeast or other fungal cells (including S. cerevesiae and P. pastoris), insect cells, plant cells, and phage, as well as higher eukaryotic cells (such as human embryonic kidney cells and other mammalian cells).
[0061] A vector can be introduced into a host cell by any convenient method, which will vary depending on the vector-host system employed. Generally, a vector is introduced into a host cell by transformation (also known as "transfection") or infection with a virus (e.g., phage) bearing the vector. If the host cell is a prokaryotic cell (or other cell having a cell wall), convenient transformation methods include the calcium treatment method described by Cohen, et al. (1972) Proc. Natl. Acad. Sci., USA, 69:2110-14. If a prokaryotic cell is used as the host and the vector is a phagemid vector, the vector can be introduced into the host cell by infection. Yeast cells can be
transformed using polyethylene glycol, for example, as taught by Hinnen (1978) Proc. Natl. Acad. Sci, USA, 75: 1929-33. Mammalian cells are conveniently transformed using the calcium phosphate precipitation method described by Graham, et al. (1978) Virology, 52:546 and by Gorman, et al. (1990) DNA and Prot. Eng. Tech., 2:3-10. However, other known methods for introducing DNA into host cells, such as nuclear injection, electroporation, and protoplast fusion also are acceptable for use in the invention.
Recombinant Production Methods
[0062] Host cells transformed with expression vectors can be used to express the polypeptides encoded by the polynucleotides of the invention. Expression entails culturing the host cells under conditions suitable for cell growth and expression and recovering the expressed polypeptides from a cell lysate or, if the polypeptides are secreted, from the culture medium. In particular, the culture medium contains appropriate nutrients and growth factors for the host cell employed. The nutrients and growth factors are, in many cases, well known or can be readily determined empirically by those skilled in the art. Suitable culture conditions for mammalian host cells, for instance, are described in Mammalian Cell Culture (Mather ed., Plenum Press 1984) and in Barnes and Sato (1980) Cell 22:649.
[0063] In addition, the culture conditions should allow transcription, translation, and protein transport between cellular compartments. Factors that affect these processes are well-known and include, for example, DNA/RNA copy number; factors that stabilize DNA; nutrients, supplements, and transcriptional inducers or repressors present in the culture medium; temperature, pH and osmolality of the culture; and cell density. The adjustment of these factors to promote expression in a particular vector- host cell system is within the level of skill in the art. Principles and practical techniques for maximizing the productivity of in vitro mammalian cell cultures, for example, can be found in Mammalian Cell Biotechnology: a Practical Approach (Butler ed., IRL Press (1991).
[0064] Any of a number of well-known techniques for large- or small-scale production of proteins can be employed in expressing the polypeptides of the invention. These include, but are not limited to, the use of a shaken flask, a fluidized bed
bioreactor, a roller bottle culture system, and a stirred tank bioreactor system. Cell culture can be carried out in a batch, fed-batch, or continuous mode.
[0065] Methods for recovery of recombinant proteins produced as described above are well-known and vary depending on the expression system employed. A polypeptide including a signal sequence can be recovered from the culture medium or the periplasm. Polypeptides can also be expressed intracellularly and recovered from cell lysates.
[0066] The expressed polypeptides can be purified from culture medium or a cell lysate by any method capable of separating the polypeptide from one or more components of the host cell or culture medium. Typically, the polypeptide is separated from host cell and/or culture medium components that would interfere with the intended use of the polypeptide. As a first step, the culture medium or cell lysate can be centrifuged or filtered to remove cellular debris. The supernatant is then typically concentrated or diluted to a desired volume or diafiltered into a suitable buffer to condition the preparation for further purification. The polypeptide can then be further purified using well-known techniques. The technique chosen will vary depending on the properties of the expressed polypeptide.
[0067] Polynucleotides of the invention present in a host cell can be isolated free of other cellular components such as membrane components, proteins, and lipids. Polynucleotides can be isolated from cells using standard nucleic acid purification techniques, or synthesized using an amplification technique, such as the polymerase chain reaction (PCR), or by using an automatic synthesizer. Methods for isolating polynucleotides are routine and are known in the art. Any such technique for obtaining a polynucleotide can be used to obtain isolated polynucleotides encoding antagonists of the invention. For example, restriction enzymes and probes can be used to isolate polynucleotides which encode the antagonists. Preferably, isolated polynucleotides are in preparations that are free or at least 70, 80, or 90% free of other molecules.
[0068] In an illustrative embodiment, modified IL-4 mutein receptor antagonist cDNA molecules are produced, using mR A as a template. Thereafter, cDNA molecules can be replicated using molecular biology techniques known in the art and described in Example 1. Example 2 describes the specific recombinant expression and
purification techniques employed in generating exemplary modified mutein
antagonists.
Confirmation of Therapeutic Utility of Modified IL-4 Mutein Receptor Antagonists
[0069] To assess the therapeutic utility of a particular modified IL-4 mutein receptor antagonists in allergic therapy, e.g., treatment of dermatitis, the antagonist can be tested in vitro in cell proliferation assays as detailed in Examples 5 and 6. In addition, the plasma half-life of the modified IL-4 mutein receptor antagonist can be measured in vivo with a rat pharmacokinetic study according to Example 6. Co-administration of Modified IL-4 Mutein Receptor Antagonists with
Additional Agents
[0070] In a particular embodiment of the method, modified IL-4 mutein receptor antagonist is co-administered with an additional agent that is useful for mitigating a symptom of an allergic response, such as dermatitis. In this embodiment, the amount of additional agent administered is sufficient to produce a beneficial effect (e.g., mitigation of dermatitis) in the subject when co-administered with the selected modified IL-4 mutein receptor antagonist.
[0071] Any additional agent that mitigates a symptom of the allergic response being treated (e.g., dermatitis) and is tolerated by the subject can be employed in the method of the invention. The additional agent can be one that acts by the same, or a different, mechanism than the modified IL-4 mutein receptor antagonist with which it is co-administered. Examples of additional agents suitable for use this embodiment include steroids. Corticosteroid creams are sometimes prescribed to decrease the inflammatory reaction in the skin. These may be mild-, medium-, or high-potency corticosteroid creams depending upon the severity of the symptoms. If itching is severe, oral antihistamines may be prescribed. To control itching, the sedative type antihistamine drugs, such as, e.g., diphenhydramine (Benadryl), hydroxyzine (Atarax, Vistaril), and cyproheptadine) can be helpful. In some cases, a short course of oral corticosteroids (such as prednisone) is prescribed to control an acute outbreak of eczema, although their long-term use is discouraged in the treatment of this non life- threatening condition because of unpleasant and potentially harmful side effects. The
oral immunosuppressant drug cyclosporine has also been used to treat some cases of eczema. Ultraviolet light therapy (phototherapy) is another treatment option. In addition, two topical (cream) medications have been approved by the U.S. FDA for the treatment of eczema: tacrolimus (Protopic) and pimecrolimus (Elidel). Pharmaceutical Compositions
[0072] The active agents described are typically combined with a
pharmaceutically acceptable carrier (excipient), such as are described in Remington's Pharmaceutical Sciences (1980) 16th editions, Osol, ed., 1980. Pharmaceutically acceptable carriers can contain one or more physiologically acceptable compound(s) that act, for example, to stabilize the composition or to increase or decrease the absorption of the active agent(s). A pharmaceutically acceptable carrier suitable for use in the invention is non-toxic to cells, tissues, or subjects at the dosages employed, and can include a buffer (such as a phosphate buffer, citrate buffer, and buffers made from other organic acids), an antioxidant (e.g., ascorbic acid), a low-molecular weight (less than about 10 residues) peptide, a polypeptide (such as serum albumin, gelatin, and an immunoglobulin), a hydrophilic polymer (such as polyvinylpyrrolidone), an amino acid (such as glycine, glutamine, asparagine, arginine, and/or lysine), a monosaccharide, a disaccharide, and/or other carbohydrates (including glucose, mannose, and dextrins), a chelating agent (e.g., ethylenediammetetratacetic acid [EDTA]), a sugar alcohol (such as mannitol and sorbitol), a salt-forming counterion (e.g., sodium), and/or an anionic surfactant (such as Tween™, Pluronics™, and PEG). In one embodiment, the pharmaceutically acceptable carrier is an aqueous pH-buffered solution.
[0073] Other pharmaceutically acceptable compounds include wetting agents, emulsifying agents, dispersing agents or preservatives that are particularly useful for preventing the growth or action of microorganisms. Various preservatives are well known and include, for example, phenol and ascorbic acid. One skilled in the art would appreciate that the choice of pharmaceutically acceptable carrier(s), including a physiologically acceptable compound depends, for example, on the route of administration of the active agent(s) and on the particular physio-chemical
characteristics of the active agent(s).
[0074] Pharmaceutical compositions of the invention can be stored in any standard form, including, e.g., an aqueous solution or a lyophilized cake. Such
compositions are typically sterile when administered to subjects. Sterilization of an aqueous solution is readily accomplished by filtration through a sterile filtration membrane. If the composition is stored in lyophilized form, the composition can be filtered before or after lyophilization and reconstitution. Administration
[0075] The active agents identified herein are useful for parenteral (e.g., subcutaneously, intravenously, intra-arterially, intramuscularly, intraperitoneally, intradermally), nasal (or otherwise inhaled), oral, sublingual, rectal, topical, or local administration, such as by aerosol aerosol (e.g., nebulization, dry powder or metered dose inhalation), or transdermally, for prophylactic and/or therapeutic treatment of one or more of the pathologies/indications described herein (e.g., to mitigate one or more symptoms of an allergic response, such as dermatitis).
[0076] In illustrative embodiments, a modified IL-4 mutein receptor antagonist can be administered parenterally (e.g., subcutaneously) no more than about twice per week, once per week, every two weeks, every three weeks, once per month, or once every 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 months. For example, about 10 mg/kg can be administered no more than about once per week; about 20 mg/kg can be administered no more than about every two weeks; and about 40 mg/kg can be administered no more than about once per month.
[0077] In various embodiments, the active agents described herein can be administered orally, in which case delivery can be enhanced by the use of protective excipients. This is typically accomplished either by complexing the active agent(s) with a composition to render them resistant to acidic and enzymatic hydrolysis or by packaging the agents in an appropriately resistant carrier, e.g. a liposome. Means of protecting agents for oral delivery are well known in the art (see, e.g., U.S. Patent No. 5,391,377).
[0078] Elevated serum half-life can be maintained by the use of sustained- release "packaging" systems. Such sustained release systems are well known to those of skill in the art (see, e.g., Tracy (1998) Biotechnol. Prog. 14: 108; Johnson et al. (1996), Nature Med. 2: 795; Herbert et al. (1998), Pharmaceut. Res. 15, 357).
[0079] The pharmaceutical compositions can be administered in a variety of unit dosage forms depending upon the method of administration. Suitable unit dosage forms, include, but are not limited to powders, tablets, pills, capsules, lozenges, suppositories, patches, nasal sprays, injectibles, implantable sustained-release formulations, lipid complexes, etc. In another embodiment, one or more components of a solution can be provided as a "concentrate," e.g., in a storage container (e.g., in a premeasured volume) ready for dilution or in a soluble capsule ready for addition to a volume of water.
[0080] In certain embodiments, one or more active agents described herein are administered alone or in combination with other therapeutics in implantable (e.g., subcutaneous) matrices, termed "depot formulations."
[0081] In particular embodiments, drugs (e.g., the active agents described herein) imbedded in various matrix materials for sustained release. Drugs embedded, for example, in polymer beads or in polymer wafers have several advantages. First, most systems allow slow release of the drug, thus creating a continuous dosing of the body with small levels of drug. This typically prevents side effects associated with high burst levels of normal injected or pill-based drugs. Secondly, since these polymers can be made to release over hours to months, the therapeutic span of the drug is markedly increased. Often, by mixing different ratios of the same polymer components, polymers of different degradation rates can be made, allowing remarkable flexibility depending on the agent being used. A long rate of drug release is beneficial for people who might have trouble staying on regular dosage, such as the elderly, but also represents an ease of use improvement that everyone can appreciate. Most polymers can be made to degrade and be cleared by the body over time, so they will not remain in the body after the therapeutic interval.
[0082] Another advantage of polymer-based drug delivery is that the polymers often can stabilize or solubilize proteins, peptides, and other large molecules that would otherwise be unusable as medications. Finally, many drug/polymer mixes can be placed directly in the disease area, allowing specific targeting of the medication where it is needed without losing drug to the "first pass" effect.
[0083] A wide variety of approaches to designing depot formulations that provide sustained release of an active agent are known and are suitable for use in the
invention. Generally, the components of such formulations are biocompatible and may be biodegradable. Biocompatible polymeric materials have been used extensively in therapeutic drug delivery and medical implant applications to effect a localized and sustained release. See Leong et al, "Polymeric Controlled Drug Delivery," Advanced Drug Delivery Rev., 1 : 199-233 (1987); Langer, "New Methods of Drug Delivery,"
Science, 249:1527-33 (1990); Chien et al, Novel Drug Delivery Systems (1982). Such delivery systems offer the potential of enhanced therapeutic efficacy and reduced overall toxicity.
[0084] Examples of classes of synthetic polymers that have been studied as possible solid biodegradable materials include polyesters (Pitt et al, "Biodegradable Drug Delivery Systems Based on Aliphatic Polyesters: Applications to Contraceptives and Narcotic Antagonists," Controlled Release of Bioactive Materials, 19-44 (Richard Baker ed., 1980); poly(amino acids) and pseudo-poly(amino acids) (Pulapura et al. "Trends in the Development of Bioresorbable Polymers for Medical Applications," J. Biomaterials AppL, 6:1 , 216-50 (1992); polyurethanes (Bruin et al, "Biodegradable Lysine Diisocyanate-based Poly(Glycolide-co-.epsilon. Caprolactone)-Urethane Network in Artificial Skin," Biomaterials, 11 :4, 291-95 (1990); polyorthoesters (Heller et al., "Release of Norethindrone from Poly(Ortho Esters)," Polymer Engineering Sci., 21 : 11, 727-31 (1981); and polyanhydrides (Leong et al, "Polyanhydrides for
Controlled Release of Bioactive Agents," Biomaterials 7:5, 364-71 (1986).
[0085] Thus, for example, the active agent(s) can be incorporated into a biocompatible polymeric composition and formed into the desired shape outside the body. This solid implant is then typically inserted into the body of the subject through an incision. Alternatively, small discrete particles composed of these polymeric compositions can be injected into the body, e.g., using a syringe. In an exemplary embodiment, the active agent(s) can be encapsulated in microspheres of poly (D,L- lactide) polymer suspended in a diluent of water, mannitol, carboxymethyl-cellulose, and polysorbate 80. The polylactide polymer is gradually metabolized to carbon dioxide and water, releasing the active agent(s) into the system.
[0086] In yet another approach, depot formulations can be injected via syringe as a liquid polymeric composition. Liquid polymeric compositions useful for biodegradable controlled release drug delivery systems are described, e.g., in U.S.
Patent Nos. 4,938,763; 5,702,716; 5,744,153; 5,990,194; and 5,324,519. After injection in a liquid state or, alternatively, as a solution, the composition coagulates into a solid.
[0087] One type of polymeric composition suitable for this application includes a nonreactive thermoplastic polymer or copolymer dissolved in a body fluid-dispersible solvent. This polymeric solution is placed into the body where the polymer congeals or precipitates and solidifies upon the dissipation or diffusion of the solvent into the surrounding body tissues. See, e.g., Dunn et al, U.S. Patent Nos. 5,278,201;
5,278,202; and 5,340,849 (disclosing a thermoplastic drug delivery system in which a solid, linear-chain, biodegradable polymer or copolymer is dissolved in a solvent to form a liquid solution).
[0088] The active agent(s) can also be adsorbed onto a membrane, such as a silastic membrane, which can be implanted, as described in International Publication No. WO 91/04014. Other exemplary implantable sustained release systems include, but are not limited to Re-Gel®, SQ2Gel®, and Oligosphere® by MacroMed,
ProLease® and Medisorb® by Alkermes, Paclimer® and Gliadel® Wafer by Guilford pharmaceuticals, the Duros implant by Alza, acoustic biSpheres by Point Biomedical, the Intelsite capsule by Scintipharma, Inc., and the like.
[0089] Modified IL-4 mutien receptor antagonist compounds can be co- administered with additional agents that mitigate a symptom of an allergic response, such as dermititis. Co-administration of an additional agent can be useful, for example, to control symptoms of dermatitis prior to the point at which the modified IL-4 mutien receptor antagonists begins to reduce such symptoms. More specifically, in certain embodiments, there will be a delay between the administration of the antagonist and the time at which the subject experiences diminution of symptoms. Treatment with an additional agent, such as a topical or oral cortisone can provide relief from symptoms during this period. In particular embodiments, treatment with the additional agent can be initiated before, at the same time as, or after treatment with the antagonist.
Treatment with the additional agent can be continued or discontinued, if it does not provide additional therapeutic benefit beyond that of the antagonist. In illustrative embodiments, treatment with an additional agent is initiated before, or at the same time as, the initiation of treatment with the antagonist, and the additional agent is
administered for about 1 week, about 2 week, about 3 weeks, or about 1 month from the time treatment with the antagonist is initiated.
[0090] Additional agents can be administered by a route that is the same as, or different from, the route of administration of the modified IL-4 mutien receptor antagonist. Where possible, it is generally desirable to administer these agents by the same route of administration, preferably in the same composition. However, differences in pharmacodynamics, pharmacokinetics, or other considerations may dictate the co-administration of modified IL-4 mutien receptor antagonist compound and additional agent in separate compositions. Additional agents can be administered according to standard practice.
Dose
[0091] In therapeutic applications, the compositions of this invention are administered, for example, to a subject experiencing, or at risk for, an allergic response, such as dermatitis, to mitigate at least one symptom of this response. An amount adequate to accomplish this is defined as a "therapeutically effective dose." Amounts effective for this use will depend upon the severity of the condition and the general state of the subject's health. Single or multiple doses of the compositions may be administered depending on the dosage and frequency as required and tolerated by the subject. In any event, the composition should provide a sufficient quantity of the active agent(s) of the composition(s) of this invention to effectively treat the condition.
[0092] The concentration of active agent(s) can vary widely and will be selected primarily based on fluid volumes, viscosities, body weight and the like in accordance with the particular mode of administration selected and the subject's needs. In accordance with standard practice, the clinician can titer the dosage and modify the route of administration as required to obtain the optimal therapeutic effect. Generally, the clinician begins with a low dose and increases the dosage until the desired therapeutic effect is achieved. Starting doses for a given active agent can, for example be extrapolated from in vitro and/or animal data.
[0093] In particular embodiments, concentrations of modified IL-4 mutein receptor antagonists will typically be selected to provide dosages of about 0.1 mg/kg, about 0.2 mg/kg, about 0.3 mg/kg, about 0.4 mg/kg, about 0.5 mg/kg, about 0.6 mg/kg,
about 0.7 mg/kg, about 0.8 mg/kg, about 0.9 mg/kg, about 1 mg/kg, about 2 mg/kg, about 5 mg/kg, about 0.7 mg/kg, about 10 mg/kg, about 15 mg/kg, about 20 mg/kg, about 25 mg/kg, about 30 mg/kg, about 35 mg/kg, about 40 mg/kg about 45 mg/kg, about 50 mg/kg and sometimes higher. For example, lower dosages can be employed with more frequent dosing, and higher dosages can be employed with more infrequent dosing. It will be appreciated that such dosages may be varied to optimize a therapeutic regimen in a particular subject or group of subjects, and thus any of these values can represent the upper or lower limit of a suitable dosage range according to the invention.
[0094] In embodiments of the method in which an additional agent that mitigates a symptom of an allergic response is co-administered with the modified IL-4 mutien receptor antagonist, suitable doses of additional agents are known and can be adjusted by the clinician for co-administration with an modified IL-4 mutien receptor antagonist described herein.
[0095] The foregoing compositions and administration methods are intended to be illustrative and not limiting. It will be appreciated that, using the teaching provided herein, other suitable compositions and modes of administration can be readily devised.
Monitoring of Response
[0096] The immune response of a subject being treated as described herein can be monitored by making periodic, e.g., daily observation of the existing dermatitis and plasma levels of IgE before and after dosing. When those subjects receiving the drug have a pronounced reduction in the immune response, they can, in certain
embodiments, be taken off the modified IL-4 mutien receptor antagonist and monitored for the length of remission. Administration of the antagonist to subjects is expected to reduce or eliminate early and/or latent immune responses.
[0097] Administration of the antagonist to human subjects should produce a reduced immune response, as compared to that in other animals, because fewer antibodies against the antagonists are expected. Fewer antibodies to the modified IL-4 mutien receptor antagonist and its epitopes imply that there are fewer inhibiting substances or agents binding to the antagonist. Fewer inhibiting substances and agents binding directly or indirectly to the antagonist allows the drug to bind to the IL-4 receptor and thus inhibit the IL-4- and IL- 13 -induced response and inactivating the
cascade of downstream events, e.g. release of various interleukins, chemokines, and chemoattractants involved in an immune response.
Kits
[0098] The invention also provides kits useful in practicing the methods of the invention. In one embodiment, a kit of the invention includes a modified IL-4 mutien receptor antagonist in a suitable container. In a variation of this embodiment, the modified IL-4 mutien receptor antagonist is formulated in a pharmaceutically acceptable carrier. The kit preferably includes instructions for administering the antagonis to a subject to inhibit a dermatitis response, such as a hypersensitivity reaction, e.g., contact dermatitis or atopic dermatitis. In an illustrative embodiment, the instructions are directed to the treatment of eczema.
[0099] Instructions included in kits of the invention can be affixed to packaging material or can be included as a package insert. While the instructions are typically written or printed materials they are not limited to such. Any medium capable of storing such instructions and communicating them to an end user is contemplated by this invention. Such media include, but are not limited to, electronic storage media (e.g., magnetic discs, tapes, cartridges, chips), optical media (e.g., CD ROM), and the like. As used herein, the term "instructions" can include the address of an internet site that provides the instructions.
[0100] The present invention is more particularly described in the following examples which are intended as illustrative only since numerous modifications and variations therein will be apparent to those skilled in the art. The following examples are intended to illustrate but not limit the invention.
EXAMPLES
EXAMPLE 1
Recombinant production of IL-4-RA and IL-4-RE cysteine muteins
[0101] The pET Directional TOPO® expression system (Invitrogen) was selected for recombinant expression of IL-4. The system uses a highly efficient one- step "TOPO® Cloning" strategy to directionally clone a blunt-end PCR product and a
TTlac promoter for high-level and IPTG-inducible expression of the gene of interest in
E.coli. Additional features include a lacl gene to reduce basal transcription, a pBR322 origin for replication and maintenance of the plasmid and an ampicillin resistance gene for selection.
[0102] IL-4 was cloned into pET101/D-TOPO vector for production of recombinant IL-4 protein. The oligonucleotide primers are shown in Table 3. The forward PCR primer was designed with a 5'CACC overhang to facilitate directional cloning, followed by a unique Ndel restriction enzyme site for subcloning and the initial ATG start codon. The reverse PCR primer included two stop codons to make sure no c-terminal tags were incorporated and a unique BamHI restriction enzyme site for subcloning. A blunt-end IL-4 PCR product was generated using previously cloned human IL-4 as a template. The product was gel purified and incubated with salt solution and TOPO® vector for 5 minutes at room temperature to allow for
directionally cloning into the pET101/D-TOPO vector. The recombinant vector was transformed into chemically competent One Shot TOP 10 E.coli. The recombinant plasmid DNA was sent out for DNA sequencing to confirm the correct sequence.
Table 3: Oligonucleotide primers for generating IL-4 RE cysteine muteins
SEQ ID NO Oligos for IL4 Sequence
RE
17 T28C Fwd GAAGACTCTGTGCACCGAGTTGTGCGTAACA
GACATCTTTGC
18 T28C Rev GCAAAGATGTCTGTTACGCACAACTCGGTGC
ACAGAGTCTTC
19 S36C Fwd GTAACAGACATCTTTGCTGCCTGCAAGAACA
CAACTGAG
20 S36C Rev CTCAGTTGTGTTCTTGCAGGCAGCAAAGATGT
CTGTTAC
21 K37C Fwd CCGTAACAGACATCTTTGCTGCCTCCTGCAAC
ACAACTGAGAAGG
22 K37C Rev CCTTCTCAGTTGTGTTGCAGGAGGCAGCAAA
GATGTCTGTTACGG
23 N38C Fwd GACATCTTTGCTGCCTCCAAGTGCACAACTGA
GAAGGAAACC
24 N38C Rev GGTTTCCTTCTCAGTTGTGCACTTGGAGGCAG
CAAAGATGTC
25 A104C Fwd GAATTCCTGTCCTGTGAAGGAATGCAACCAG
AGTACGTTGG
26 A104CRev CCAACGTACTCTGGTTGCATTCCTTCACAGGA
CAGGAATTC
27 N105C Fwd CCTGTGAAGGAAGCCTGCCAGAGTACGTTGG
AAAACTTC
28 N105C Rev GAAGTTTTCCAACGTACTCTGGCAGGCTTCCT
TCACAGG
29 Q106CFwd CCTGTCCTGTGAAGGAAGCCAACTGCAGTAC
GTTGGAAAACTTC
30 Q106C Rev GAAGTTTTCCAACGTACTGCAGTTGGCTTCCT
TCACAGGACAGG
[0103] IL-4/pET 101/D-TOPO served as a template for producing IL-4 RE cysteine muteins with the QuikChange® Site-Directed Mutagenesis Kit from
Strategene. Each cysteine mutein was made using two oligonucleotide primers, each complementary to opposite strands of the vector and containing the codon TGC or GC A to incorporate the desired cysteine mutation. Table 4 lists the primers used for producing the IL-4 RE muteins. A mutated plasmid containing staggered nicks was generated using cycling parameters and conditions defined in the manufacturer's protocol. The product was treated with Dpnl endonuclease for 1 hour at 37°C to digest the methylated, non-mutated parental DNA template. The Dpnl -treated DNA was transformed into XL-1 Blue supercompetent cells where nicks in the mutated plasmid were repaired. The mutagenic 5 plasmid DNA was analysed according to standard sequencing techniques to confirm the correct sequence.
EXAMPLE 2
Recombinant Expression & Purification
[0104] BL21 Star (DE3) One Shot cells (Invitrogen) transformed with the protein containing plasmids were characterized for optimal expression and grown at 37°C until OD6oo reached approximately 0.4 and induced by lmM IPTG (Invitrogen) for 3 hours at 37°C. One liter of cells were pelleted at 13,000 rpm for 10 minutes, weighed and stored at -80°C. The frozen cell pellet was resuspended in 8 ml cell disruption buffer (0.1M phosphate buffer pH 7.3, 0.1% Triton X100, lmM EDTA) per gram of cells and sonicated 4x for 1 minute with 1 minute intervals. The cell lysate was removed by centrifugation at 35,000g for 10 minutes. The cell pellets were then washed 2-3x by resuspension in 30 ml of cell disruption buffer, by sonication for 1 minute, followed by centrifugation. The final cell pellet, inclusion bodies, was stored at -20°C. Inclusion bodies were resuspended in 5 ml solubilization buffer (0.2M Tris pH 9, 7M guanidine hydrochloride) per gram of cells. Sulphotolysis reagents (0.16 grams sodium sulfite, 0.08 gram potassium tetrathionate per gram of cells) were added and the
inclusion bodies were stirred at room temperature for 2 hours. Undissolved constituents were then removed by centrifugation at 35,000g for 20 minutes leaving solubilized inclusion bodies. The inclusion bodies were then run on a Superdex200 size exclusion column (Akta) to isolate the protein. The column was equilibrated with 2 column volume (CV) of 6M guanidine hydrochloride/PBS pH 7 at a flow rate of lml/min and the protein was eluted in 1.5CV. Peak fractions (1.5 ml each) were collected and screened by 12% or 4-20% Bis-Tris-SDS gel electrophoresis. Fractions containing the protein were pooled and a final concentration of 7.5mM DTT was added in order to reduce the protein molecules. Following a 2 hour incubation at room temperature, the mixture was diluted 5x with water and subjected to dialysis into 4.5L 3mM NaH2P04, 7mM Na2HP04, 2mM KC1, 120mM NaCl. Dialysis was continued for 3-4 days with fresh buffer change at least 3 times. The dialyzed material was then filtered through an 0.2μιη filter and the pH was adjusted to 5 with acetic acid. The column was equilibrated with 10CV of Buffer 1 (25mM Ammonium Acetate pH5) followed by a 20 minute gradient to 100% buffer B (25mM Ammonium Acetate pH5/lM NaCl) post injection. Peak fractions (0.5 ml each) were collected and screened by 12% or 4-20% Bis-Tris-SDS gel electrophoresis. Product containing fractions were pooled and diluted 2x into Buffer A (0.1% TF A/water). The protein was then chromatographed on C4 Reverse Phase-HPLC (Beckman system Gold), using a 5 ml loop and flow rate of 1 ml/min with the following program: 10% Buffer A for duration of injection, 10 minute gradient to 40% Buffer B (0.1% TFA/ACN), 30 minute gradient to 50% Buffer B, and 5 minute gradient to 100% Buffer B. Peak fractions (0.5 ml each) were collected and screened by 12% or 4-20% Bis-Tris-SDS gel electrophoresis. Protein containing fractions were dried down and resuspended in 0.1 M MES pH 6.1 for analysis and assays.
EXAMPLE 3
Site-Specific Cysteine PEGylation and Purification
[0105] A protocol was established to PEGylate the cysteine containing IL4 RA muteins via a stable thioether linkage between the sulfhydryl of the protein and the maleimide group of a linear 22kD methoxy -poly ethylene glycol-maleimide derivative (Nektar Therapeutics). A 2-fold molar excess of mPEG-MAL 22kD reagent was added to 60 μΜ of protein dissolved in reaction buffer, 0.1M MES, pH 6. After 0.5 hour at room temperature, the reaction was terminated with 2-fold molar excess of cysteine
over mPEG-MAL 22kD (Figure 1). PEGylated protein was purified away from unreacted mPEG-MAL 22kD (quenched with cysteine) and unreacted IL4 RA cysteine mutein by cation exchange and size exclusion chromatography. Crude reaction mixtures were applied to Vivapure Mini S cation exchange columns (Vivascience) equilibrated with 0.4mL of 0.1 M MES, pH 6. The columns were washed twice with 0.4mL of 0.1M MES, pH 6 followed by centrifugation at 2,000 x g after each wash. The samples were eluted by centrifugation from the column with 0.4mL of 0.6M NaCl/O. lM MES, pH6. The 0.4mL elutions were loaded onto a TSK-GEL
G2000SWXL HPLC sizing column (Tosoh Biosep) using a Beckman HPLC system Gold. The samples were resolved using a Phosphate Buffered Saline (Dulbecco's PBS) mobile phase at a flow rate of lml/min for 30min. Peak fractions (0.5ml) were collected and evaluated by 4-12% Bis-Tris-SDS gel electrophoresis for PEGylated protein. Fractions containing the product were pooled and concentrated using an Ultrafree Biomax-5 device (Millipore) per manufacturer's protocol to approximately 60μΜ (or ~lmg/ml) for analysis and in vitro assays. Final concentrations for the PEGylated proteins were determined by amino acid analysis. Final yields are depicted in Table 4.
Table 4: Purification yields for PEGylated IL4RA cysteine muteins
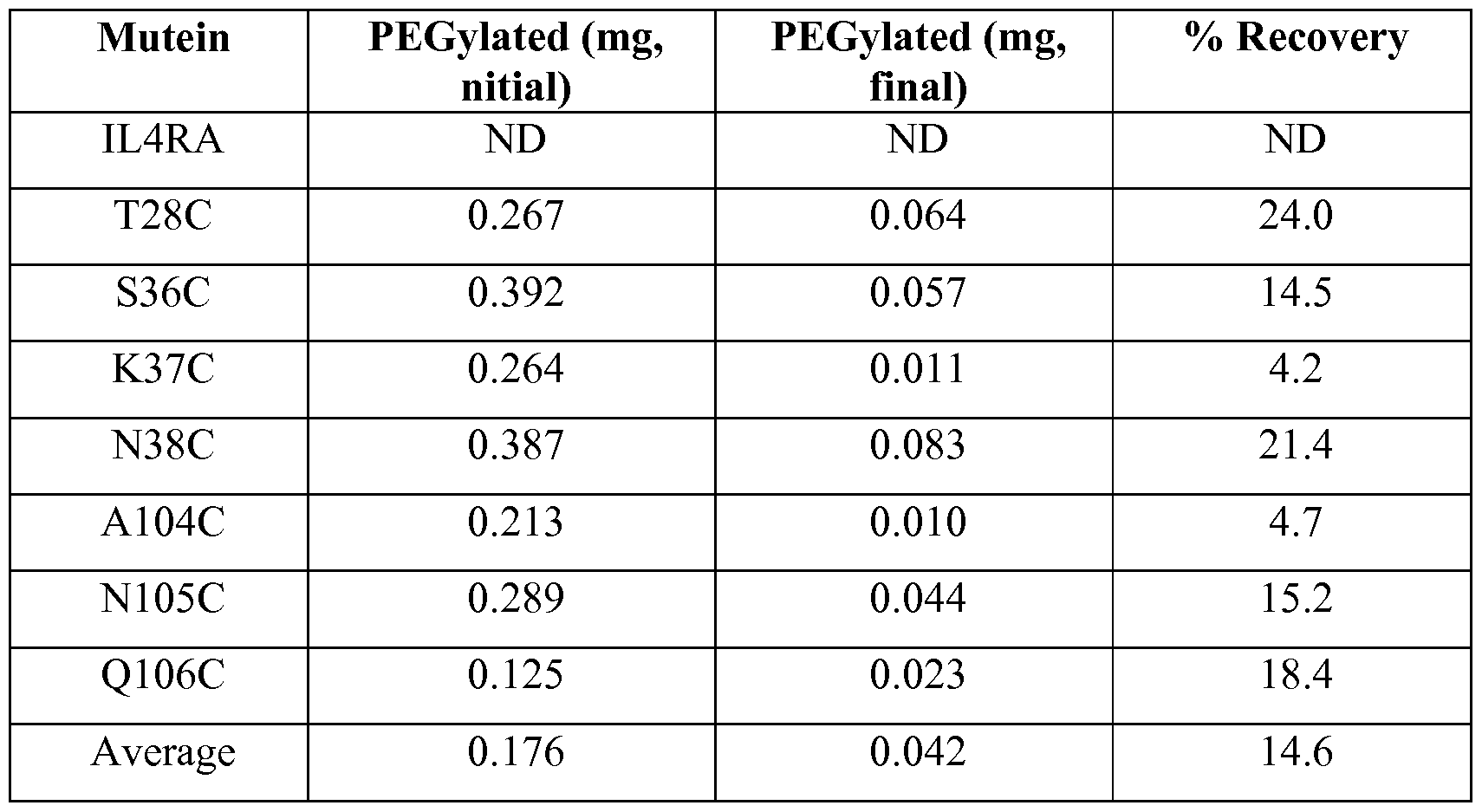
BiaCore IL-4 Receptor Binding Assay
[0106] IL-4 receptor was immobilized on a BIAcore CM5 research grade sensor chip through amine coupling. The sensor surface was activated with an EDC/NHS pulse. IL-4 receptor was dissolved in lOmM acetate buffer (pH 5.0) and injected into flowcell 2 followed by a pulse of 1.0M ethanolamine-HCL to deactivate the surface. The immobilization level for the receptor was -300RU. Flowcell 1 was also activated without a ligand to function as a blank. The Biacore Wizard was used to perform kinetics analysis. Candidate IL4RE antagonists were diluted in HBS-EP (running buffer) and injected at 30ul/minute flow rate for 3 minutes and a dissociation time of 15 minutes. Regeneration of the chip was performed by two 30 second injections of lOmM Glycine pH2.5 (flow lOOul/min) to baseline prior to next injection in the concentration series. Dissociation constant (KD) values were calculated for each candidate based on direct binding kinetics (Table 5). Results show constructs IL4- RE-A104C, IL4-RE-N105C, and IL4-RE-Q106C all yielded dissociation constants below 0.6nM.
EXAMPLE 5
TF-1 Cell Proliferation Assay
[0107] The proliferative response of TF-1 cells to IL-4 (0.5 ng/ml, 0.033 nM) or IL-13 (5 ng/ml, 0.416 nM), was used to assess the functional antagonistic activity of IL-4RE molecules. In this assay, TF-1 cells were cultured for 2-4 days in 96 well plates (lxl04/well, 100 μΐ volume) in RPMI + 10% serum with or without IL-4 or IL-13 and IL-4RE molecules. GM-CSF treatment was used as a positive control. Twenty- four hours before the final reading, 10 μΐ AlamarBlue (10%> vol) was added to each well. Fluorescence was determined at 530/590 nm using a WALLAC Victor 2. Inhibitory Concentration 50% (IC50) was calculated based on dose titration of the candidate IL-4RE molecules. A summary of the TF-1 bioassay results for IL-4 and IL-13 inhibition are shown in Table 5. Results indicate that constructs IL4-RE-K37C, IL4-RE-N38C and IL4-RE-A104C demonstrated comparable IC50 values to that of IL-4-RA in the presence of IL-4 or IL-13.
Table 5: PEG-IL4RE BIAcore binding assay and bioactivity evaluation of PEGylated muteins versus IL4RA in TF-1 cell proliferation assays.

EXAMPLE 6
Primary Cell Proliferation Assay
[0108] The proliferative response of human primary cells (T- and B-cells) to
IL-4 was also evaluated following IL-4RE molecule pre-treatment. Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood and some were treated with PHA for 4 days to induce T cell blast formation. PBMCs were also treated with anti-CD40 to activate B cell activity and used immediately. The cells were seeded in 96-well plates (105 cells per well). PHA T-cell blasts and B cells preparations were stimulated for 3 days with IL-4 (10 ng/ml, 0.667 nM) in the presence of varying concentrations of IL-4RE molecules. The incorporation of tritiated thymidine in the last 20 hours of incubation was used as an indicator of proliferation. The results of these assays are shown in Table 6. Results indicate that all
PEGylated constructs demonstrated an IC50 less than 5 -fold greater than that of IL- 4RA for both primary cell assays.
Table 6: PEG-IL4RE bioactivity evaluation in B-cell and T-cell blast proliferation assays.

IL4-RE-A104C 3.29 +1.46 (n=2) 4.67 + 4.65 (n=2)
EXAMPLE 7
Rat Pharmacokinetics Studies
[0109] Adult male Sprague-Dawley rats weighing 250 to 300 grams were used. The rats were cannulated with jugular vein catheter for blood sample collection. In addition, the rats of intravenous (IV) dose group were cannulated with femoral vein catheters for drug administration.
[0110] The rats were given either IL-4RA or a modified IL-4 mutein receptor antagonist at doses of 1 and 0.5 mg/kg, respectively. Both IV and SC (subcutaneous) routes of administration were used. The IV dose was given by injection directly into the indwelling femoral vein catheter. The SC dose was given by injection into the dorsal thoracic region. Three rats were used for each dose group.
[0111] Following a single bolus injection (IV or SC), blood samples were collected at predose and at predetermined times up to 168 hours post dose.
Centrifugation for samples began within 1 hour of collection. Plasma was harvested and placed on dry ice prior to storage at approximately -70°C.
[0112] Plasma concentrations of IL-4RA and modified mutein were quantified with an enzyme-linked immunoassay. Anti-IL-4 antibody was used as coating and detection reagents. The lower limit of quantification for this assay was 0.2 ng/ml. Pharmacokinetic parameters were derived by non-compartmental analysis using WinNonlin (Pharsight, Mountain view, CA). Of particular interest is the assessment of absorption and elimination kinetics, distribution volumes as well as the amount absorbed.
EXAMPLE 8
E.coli-encoded IL-4 Mutein
[0113] High purity and high yield inclusion body expression directly affects manufacturing ability and costs. It has been shown that IL-4 triple mutein molecules can be made in the laboratory by including an IL-4 triple mutein construct utilizing human DNA codons into an appropriate plasmid vector suitable for expression in
E.coli. Although small quantities of inclusion bodies can be obtained from an IL-4 triple mutein construct using human codons, the E.coli fermentation process is not robust. It was observed that expression of an IL-4 triple mutein construct using human codons can be toxic to E.coli cells causing lysis, resulting in low yields of inclusion 5 bodies. Without being limited to certain theories, this observation is likely due to
limited quantities of human codons in E.coli. In order to improve the process, the E.coli-codon IL-4RA plasmid was used to make E.co //-encoded IL4-RE-T13D with QuickChange (Strategene). Briefly, IL-4RA plasmid was altered by QuickChange and suitable primers to alter the codons at either position alanine 104 (codon GCT) or 10 asparagine 38 (codon AAC) to a cysteine (codon TGC). Two E.co//-codon IL4-RE-
T13D plasmids were made that coded for IL-4TM (IL4-RE-T13D) molecules (Table 7).
Table 7: E.coli-codon IL-4TM Plasmids

T13D
caccatgagtgacgactgaatccggtgagaatggcaaaagcttatgcatttctttccagacttgttcaacaggccagc
N38C
cattacgctcgtcatcaaaatcactcgcatcaaccaaaccgttattcattcgtgattgcgcctgagcgagacgaaatac sequence gcgatcgctgttaaaaggacaattacaaacaggaatcgaatgcaaccggcgcaggaacactgccagcgcatcaac aatattttcacctgaatcaggatattcttctaatacctggaatgctgttttcccggggatcgcagtggtgagtaaccatgc atcatcaggagtacggataaaatgcttgatggtcggaagaggcataaattccgtcagccagtttagtctgaccatctca tctgtaacatcattggcaacgctacctttgccatgtttcagaaacaactctggcgcatcgggcttcccatacaatcgata gattgtcgcacctgattgcccgacattatcgcgagcccatttatacccatataaatcagcatccatgttggaatttaatcg cggcctcgagcaagacgtttcccgttgaatatggctcataacaccccttgtattactgtttatgtaagcagacagttttatt gttcatgaccaaaatcccttaacgtgagttttcgttccactgagcgtcagaccccgtagaaaagatcaaaggatcttctt gagatcctttttttctgcgcgtaatctgctgcttgcaaacaaaaaaaccaccgctaccagcggtggtttgtttgccggatc aagagctaccaactctttttccgaaggtaactggcttcagcagagcgcagataccaaatactgtccttctagtgtagcc gtagttaggccaccacttcaagaactctgtagcaccgcctacatacctcgctctgctaatcctgttaccagtggctgctg ccagtggcgataagtcgtgtcttaccgggttggactcaagacgatagttaccggataaggcgcagcggtcgggctga acggggggttcgtgcacacagcccagcttggagcgaacgacctacaccgaactgagatacctacagcgtgagctat gagaaagcgccacgcttcccgaagggagaaaggcggacaggtatccggtaagcggcagggtcggaacaggaga gcgcacgagggagcttccagggggaaacgcctggtatctttatagtcctgtcgggtttcgccacctctgacttgagcgt cgatttttgtgatgctcgtcaggggggcggagcctatggaaaaacgccagcaacgcggcctttttacggttcctggcct tttgctggccttttgctcacatgttctttcctgcgttatcccctgattctgtggataaccgtattaccgcctttgagtgagctga taccgctcgccgcagccgaacgaccgagcgcagcgagtcagtgagcgaggaagcggaagagcgcctgatgcggt attttctccttacgcatctgtgcggtatttcacaccgcaatggtgcactctcagtacaatctgctctgatgccgcatagttaa gccagtatacactccgctatcgctacgtgactgggtcatggctgcgccccgacacccgccaacacccgctgacgcgc cctgacgggcttgtctgctcccggcatccgcttacagacaagctgtgaccgtctccgggagctgcatgtgtcagaggtt ttcaccgtcatcaccgaaacgcgcgaggcagctgcggtaaagctcatcagcgtggtcgtgaagcgattcacagatgtc tgcctgttcatccgcgtccagctcgttgagtttctccagaagcgttaatgtctggcttctgataaagcgggccatgttaag ggcggttttttcctgtttggtcactgatgcctccgtgtaagggggatttctgttcatgggggtaatgataccgatgaaacga gagaggatgctcacgatacgggttactgatgatgaacatgcccggttactggaacgttgtgagggtaaacaactggcg gtatggatgcggcgggaccagagaaaaatcactcagggtcaatgccagcgctcatgagcccgaagtggcgagccc gatcttccccatcggtgatgtcggcgatataggcgccagcaaccgcacctgtggcgccggtgatgccggccacgatg cgtccggcgtagaggatcgagatccatttacgttgacaccatcgaatggtgcaaaacctttcgcggtatggcatgatag cgcccggaagagagtcaattcagggtggtgaatgtgaaaccagtaacgttatacgatgtcgcagagtatgccggtgtc tcttatcagaccgtttcccgcgtggtgaaccaggccagccacgtttctgcgaaaacgcgggaaaaagtggaagcggc gatggcggagctgaattacattcccaaccgcgtggcacaacaactggcgggcaaacagtcgttgctgattggcgttgc cacctccagtctggccctgcacgcgccgtcgcaaattgtcgcggcgattaaatctcgcgccgatcaactgggtgccag cgtggtggtgtcgatggtagaacgaagcggcgtcgaagcctgtaaagcggcggtgcacaatcttctcgcgcaacgcg tcagtgggctgatcattaactatccgctggatgaccaggatgccattgctgtggaagctgcctgcactaatgttccggcg ttatttcttgatgtctctgaccagacacccatcaacagtattattttctcccatgaagacggtacgcgactgggcgtggagc atctggtcgcattgggtcaccagcaaatcgcgctgttagcgggcccattaagttctgtctcggcgcgtctgcgtctggct ggctggcataaatatctcactcgcaatcaaattcagccgatagcggaacgggaaggcgactggagtgccatgtccgg ttttcaacaaaccatgcaaatgctgaatgagggcatcgttcccactgcgatgctggttgccaacgatcagatggcgctg ggcgcaatgcgcgccattaccgagtccgggctgcgcgttggtgcggatatctcggtagtgggatacgacgataccga agacagctcatgttatatcccgccgttaaccaccatcaaacaggattttcgcctgctggggcaaaccagcgtggaccgc ttgctgcaactctctcagggccaggcggtgaagggcaatcagctgttgcccgtctcactggtgaaaagaaaaaccacc ctggctcgagaaatcataaaaaatttatttgctttgtgagcggataacaattataatagattcaattgtgagcggataacaat ttcacacatctagaaataattttatttaactttaagaaggagatatacatatgcacaaatgcgatatcaccctgcaggaaat catcaaagacctgaattctctgaccgaacagaaaaccctgtgcaccgaactgaccgttaccgacatcttcgctgcttcg aaatgcaccaccgaaaaagaaaccttctgccgtgctgctaccgttctgcgtcagttctactctcaccacgaaaaagaca cccgttgcctgggtgctaccgctcagcagttccaccgtcacaaacagctgatccgtttcctgaaacgtctggaccgtaa cctgtggggtctggctggtctgaacagctgcccggttaaagaagctaaccagtctaccctggaaaacttcctggaacg tctgaaaaccatcatggacgaaaaagactctaaatgctcttcttaataaggatccggctgctaacaaagcccgaaagg aagctgag
EXAMPLE 9
PEGylated IL-4/IL-13 Inhibitor
[0114] When IL-4TM-N38C or IL-4TM-A104C is PEGylated with a maleimide-PEG (20 kDa linear from Nektar, Dow or NOF; 30 kDa linear from Nektar, Dow or NOF; 40 kDa linear from Nektar, Dow or NOF; or a 40 kDa branched from Necktar or NOF), the activity decreases significantly when assayed using the BIAcore method, TF-1 cell-based assay or primary B or T lymphocytes from peripheral blood (Figure 2).
[0115] Additionally, it was observed that the position in which the additional cysteine is placed is important. As shown in Table 8, PEGylated 104C looses about 2- fold activity more than PEGylated 38C in the TF-1 activity assay.
Table 8: Comparison of activities of IL-4DM to PEGylated IL-4TM
Molecule IL-4DM N38C-40B 104C-40B

[0116] To compensate for this loss of activity an additional T13D mutation was included onto the IL-4TM-N38C using QuikChange, appropriate primers, and the E.co/z'-codon N38C plasmid. This T13D mutation has previously been shown to increase binding affinity to the IL-4Ra (see U.S. Pat. No. 6,028,176, incorporated herein by reference). As shown in Figure 3, PEGylated T13D-N38C with 40kDa branched PEG has equal BIAcore as the unPEGylated IL-4DM (R121D/Y124D), but the data cannot accurately predict the cell-based assay since PEGylation affects the on- rate of all PEGylated IL-4TM molecules. It is the off-rate that is generally potency-
determining in a bioassay. Using the TF-1 cell-based assay, PEGylated T13D-N38C with 40kDa branched PEG has equal activity as IL-4DM.
[0117] Similar results were observed in a primary T cell assay. In this assay whole blood from 10 donors was collected and PBMCs isolated. PBMCs were incubated for 4 days with PHA-P, and these "blasts" were then used for the assay. Blasts were incubated for 3 days with IL-4 plus one of 12 dilutions of IL-4TM test compound. In the last 18 hours, 3H-thymidine was added to the culture. Cells were harvested and DNA that incorporated 3H-thymidine was quantified on a b-counter. Table 9 demonstrates the loss of potency with PEGylation and the improvement in potency gained from PEGylating at 38C compared to 104C.
Table 9

[0118] Studies were also done in primary B cells. Similarly, whole blood from
5 donors was collected and PBMCs isolated. PBMCs were incubated for 3 days with mouse anti-human CD40, IL-4, and one of 12 dilutions of IL-4TM test compound. In the last 18 hours, 3H-thymidine was added to the culture. Cells were harvested and DNA that incorporated 3H-thymidine was quantified on a b-counter (Table 10).
Table 10

[0119] Again the improvement of PEGylation at the 38C position compared to the 104C position was observed as well as the increased or similar potency of the T13D and the T13D 40B constructs, respectively, compared to IL-4DM (R121D/Y124D).
EXAMPLE 10
Improving the Refolding Yields of an IL-4/IL-13 Inhibitor
[0120] IL-4TM (T13D/R121D/Y124D) contains a 7th cysteine residue in the protein sequence at either position 38C or 104C. This odd cysteine reduces the efficiently of refolding IL-4TM from inclusion bodies. The addition of glutathione to the IL-4TM refold buffer optimizing the yield of IL-4TM refolding while still preserving the ability of 38C or 104C to be PEGylated by a maleimide-linked PEG molecule. In addition to adding glutathione to the refolding mixture, there is a time- dependency of refolding to optimize the yields of IL-4TM for PEGylation. Maximized protein refolding yields are monitored by the BIAcore assay. Once the maximum refolding yield is achieved, the pH of the refold solution is lowered with an acid to a pH where cysteine oxidation chemistries are no longer efficient. Low pH is maintained until IL-4TM is reacted with maleimide-PEG at a more neutral pH where the cysteine- maleimide bond forms.
References
[0121] 1. Ying, S., M. Humbert, J. Barkans, C.J. Corrigan, R. Pfister, G.
Menz, M. Larche, D.S. Robinson, S.R. Durham and A.B. Kay. 1997. Expression of
IL-4 and IL-5 mRNA and protein product by CD4+ and CD8+ T cells, eosinophils, and mast cells in bronchial biopsies obtained from atopic and nonatopic (intrinsic) asthmatics. J. Immunol. 158: 3539-3544.
[0122] 2. Huang, S.K., H.Q. Xiao, J. Kleine-Tebbe, G. Paciotti, D.G Marsh, L.M. Lichtenstein and M.C. Liu. 1995. IL-13 expression at the sites of allergen challenge in patients with asthma. J. Immunol. 155: 2688-2694.
[0123] 3. Zhu, Z., Rj. Homer, Z. Wang, Q. Chen, G.P. Geba, J. Wang, Y.
Zhang and J.A. Elias. 1999. Pulmonary expression of interleukin- 13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic
abnormalities, and eotaxin production. J. Clin. Invest. 103: 779-788.
[0124] 4. Henderson, W.R.J., E.Y. Chi and C.R. Maliszewski. 2000.
Soluble IL-4 receptor inhibits airway inflammation following allergen challenge in a mouse model of asthma. J. Immunol. 164: 1086-1095.
[0125] 5. Sambrook (1989) CURRENT PROTOCOLS IN MOLECULAR
BIOLOGY, John Wiley & Sons, New York, N.Y., 1995.
EXAMPLE 11
AER 003 a PEGylated IL-4/IL-13 antagonist for the treatment of allergic diseases Abstract
[0126] IL-4 and IL-13 are key cytokines in the pathogenesis of allergic diseases. AER 003 is a PEGylated recombinant protein derived from human IL-4.
Three point mutations (T13D, R121D, Y124D) result in a protein that binds with high affinity to the IL-4 receptor a chain and acts as an antagonist of IL-4 and IL-13, while PEGylation (40kDa at N38C) extends circulating half life. A double-blind, randomized, two-period cross-over study investigated the effect of a single
subcutaneous dose of AER 003 or placebo on allergen-induced AHR and airway eosinophilia in the cynomolgus monkey. Airway responsiveness to inhaled
methacholine and bronchoalveolar lavage cell composition were determined 3 days before (Day 0) and 2 days after (Day 7) 3 consecutive-day (Days 3-5) inhalations of Ascaris suum extract. Plasma samples for pharmacokinetic (PK) analysis were collected to 34 days post-dose. AER 003 significantly reduced the development of allergen-induced AHR relative to placebo (p = 0.0003), with an improvement in PC 100
of 2.24-fold (AER 003 / placebo ratio of ANCOVA-adjusted geometric mean PCIOO values, with 95% CI of 1.52 to 3.30). In contrast, there was no significant effect of AER 003 on lung eosinophilia (p = 0.69). The PK data indicate a 400-fold reduction in clearance/bioavailability relative to AER 001 (AERO V ANT™), a non-PEGylated analog of AER 003. These data demonstrate the therapeutic potential of AER 003 for the treatment of allergic diseases, and support once weekly dosing in the clinic.
Introduction
[0127] Allergic diseases such as atopic dermatitis and allergic asthma are complex genetic diseases with major environmental influences. Despite the disease heterogeneity, clinical and genetic studies imply that both interleuin-4 (IL-4) and interleukin-13 (IL-13) are central to the pathogenesis of atopic diseases 1 14. Both cytokines have multiple and overlapping biological functions considered essential to the development of allergic responses 15 " 17 18 " 22 23 26. Over-expression or administration of either IL-4 or IL-13 in the skin or airways can induce phenotypic characteristics of atopic dermatitis and allergic asthma while the use of inhibitors of either cytokine confirm the principle role of IL-4 and IL-13 in the initial antigen sensitization and
25 27 32 29 33 39
secondary allergic response respectively ' ' " . However, despite the apparent dominant role of IL-13 during the effector phase, it is clear that in the absence of IL-13, IL-4 is capable of inducing the allergic phenotype and that a therapeutic strategy targeting both IL-4 and IL-13 may be more beneficial 40 .
[0128] The IL-4Ra chain and STAT6 signal transduction pathway are common to both the IL-4 activated type I receptor heterodimer (IL-4Ra chain/ common γ-chain), and the IL-4/IL-13 activated type II receptor heterodimer (IL-13Ral chain/IL-4Ra chain) 41' 42. IL-4 is a 15KDa four a-helix-bundle protein with two binding epitopes. The binding epitope for the high affinity IL-4Ra chain is located in the A and C helices, while the binding epitope for the low affinity γ- or IL-13Ral chains is located in the A and D helices44"46. Mutagenesis studies have identified that mutations at residues arginine 121, tyrosine 124, and serine 125 in the C terminus of the D helix of human IL-4 do not interfere with binding to the IL-4Ra chain, but do prevent formation of either the type I or type II heterodimeric receptors, and thus generate antagonists of both human IL-4- and IL-13 -induced in vitro responses 47-49. In contrast, single
mutations in the A and C helices (T13D, E19A, F82D) can increase binding affinity of IL-4 for the IL-4Ra chain increasing agonist activity50' 51.
[0129] Previously, we have demonstrated that twice daily pulmonary or subcutaneous administration of the human IL-4 double mutein (R121D, Y124D)
[pitrakinra or AER 001] inhibits allergen-induced airway hyper-responsiveness (AHR) in the cynomolgus monkey52"55. Pitrakinra administered either once daily by subcutaneous injection or twice daily by inhalation (nebulizer) inhibited the allergen- induced late-phase fall in FEVlin two independent Phase Ila studies with mild to moderate asthmatics (Wenzel). The relatively short half-life of pitrakinra (~3h), requires multiple daily administration and precludes systemic delivery. However, the successful inhibition of the allergen-induced late asthmatic response following local delivery has lead to the further development of pitrakinra as an inhaled dry powder. In addition to the clinical development of pitrakinra for asthma, a Phase Ila proof of concept clinical trial in eczema suggested potential therapeutic benefit when
administered twice daily by subcutaneous injection(REF). However, the potential requirement for systemic delivery of an IL4/IL13 antagonist for the treatment of eczema would favor a clinical candidate with an extended half life to allow for once weekly injection.
[0130] Several second-generation therapeutic proteins with extended half-lives and improved potencies in vivo have been created by modifying the proteins with polymers such as polyethylene glycol (PEG). Covalent attachment of PEG to a protein increases the protein's effective size and reduces its rate of clearance from the body. However, biological activity is often decreased in PEGylated conjugates due to a decrease in observed target association rates. The most commonly used method for PEGylating proteins attaches PEG to amine groups, typically at lysine residues and/or at the N-terminal amino acid. A limiting factor in this approach is potential attachment to multiple sites. Variations in the position and number of PEG adducts gives rise to variations in characteristics relevant to manufacturing and clinical effects. Specific chemistries utilizing thiol-selective PEG (such as maleimide-PEG) have been successfully used in native proteins containing a free cysteine, or in proteins with a site- directed mutation of an amino acid to cysteine in an area of the protein not associated with the active site.
[0131] In the present study, we have utilized the primate asthma model to evaluate the therapeutic potential of AER 003, a novel PEGylated human IL-4 mutein. Three point mutations (T13D, R121D, Y124D) (AER 004) were integrated to generate a variant that binds with high affinity to the IL-4 receptor a chain and acts as a pure antagonist of IL-4 and IL-13. A forth mutation (N38C) (AER 003) was introduced to allow attachment of a large 40 kDa-PEG moiety to a single, non-essential site in the protein to extend the circulating half life. Here we demonstrate the therapeutic potential of AER 003 for the treatment of allergic diseases, with the potential for weekly dosing in the clinic. Results
[0132] We modified human IL-4 by site-directed mutagenesis (Figure 1). Arg
121 and Tyr 124 (located on the D helix of IL-4) have been previously shown to play a role in interaction of IL-4 with Class I, IL-4Ra/IL-2Ry, and Class II, IL-4Ra/IL-13Ra receptor complexes. Tyr 13, located on the A helix, has been shown to play a role in interaction of IL-4 to the IL-4Ra chain. Asp 38, a glycosylation site in human IL-4, does not affect the binding of IL-4 to any of the receptor subunits. AER 001 (Aerovant, Pitrakinra) containing the double mutation R121D, Y124D, was used as the scaffold for AER 004 (R121D, Y124D, and T13D) and AER 003 (R121D, Y124D, T13D, and N38C). AER 003 binds the IL-4Rq
[0133] The binding kinetics of AER 001, AER 003, both pre and post
PEGylation, and AER 004 were compared to that of human IL-4 using the IL-4Ra subunit by surface Plasmon resonance (Table 11). The affinity of IL-4 to the IL-4Ra subunit was similar to AER 001, as shown in previous studies (Sebald). The T13D mutation (AER 004) led to a 5 -fold increase in affinity for IL-4Ra primarily through a decrease in the dissociation-rate (!¾) constant. The addition of the N38C mutation resulted in a protein largely unaffected in binding to the IL-4Ra subunit. PEGylation of the N38C site led to an overall 6.5-fold decrease in affinity for IL-4Ra, relative to the pre-PEGylated intermediate, primarily through an increase in the association-rate (ka) constant. The dissociation rate of the AER 003/ IL-4Ra complex was not affected by the 40 kDa branched PEG.
Table 11. Kinetic binding properties at 25°C of human IL-4 muteins to IL-4Ra subunit
IL-4 Mutein ka O l ) D(nM) Fold increase in KD x 107 x 10"3 relative to AER 001
IL-4 5.8 8.7 0.15 1
AER 001 (R121D, Y124D) 4.3 ± 1.7 6.4 ± 2.1 0.15 1
AER 004 (R121D, Y124D, 1.5 0.037 0.026 0.2
T13D)
AER 003 (R121D, Y124D, 1.3 0.036 0.027 0.2
T13D, and N38C)
AER 003 (R121D, Y124D, 14 0.028 0.2 1.3
T13D, and N38C) + 40B PEG
AER 003 inhibits IL-4 and IL-13-induced TF-1 cell proliferation AER 003 inhibits IL-4-induced proliferation of human T- and B- lymphocytes
[0134] Cells were stimulated with human IL-4 required to increase proliferation of cells by 50%. IC50 values represent the amount of AER 001 or AER 003 required to inhibit 50% activity of human IL-4. The results are shown in Table 12.
Table 12. Inhibitory effects of IL-4 muteins on Human T and B lymphocyte
proliferation stimulated with human IL-4
T-Cell B-Cell
IL-4 Mutein IC50 (nM) Fold increase in IC50 (nM) Fold increase in
IC50 relative to IC50 relative to AER 001 AER 001
AER 001 (R121D, Y124D) 1.1 ± 0.7 1 1.7 ± 0.6 1
AER 004 (R121D, Y124D, 0.7 ± 0.2 0.6 0.5 ± 0.1 0.3 T13D)
AER 003 (R121D, Y124D, 3.3 ± 1.4 3 2.4 ± 1.1 1.4 T13D, and N38C) + 40B PEG
Effects of AER 003 on AHR and airways esosinphilia
[0135] The effects of dual inhibition of IL-4 and IL- 13 using AER 003, on allergen induced AHR and airway inflammation was assessed. From the ANCOVA model with effects for treatment, period and subject and a baseline covariate, Day 7 adjusted geometric mean PC100 values of 32.9 and 14.7 mg/mL were estimated for the AER 003 and placebo treatments, respectively. The ratio (AER 003 / placebo) of 2.24 (95% CI, 1.52, 3.30) was statistically significant (p = 0.0003). The effect of subject was also significant (p = 0.026), although the effects of period (p = 0.806) and baseline PCioo (p = 0.157) were not. The effect of AER 003 treatment relative to placebo on
Day 7 PC100 is shown for each subject in Figure 2. PC100 improvement with AER 003 over placebo was generally greater in animals with increased sensitivity to MCh.
[0136] Despite a significant inhibitory effect of AER 003 on AHR, effects on airway eosinophilia were less apparent. Day 7 adjusted geometric mean lung
eosinophil counts of 2.27 x 105 and 2.45 x 105 cells/mL were estimated for the AER
003 and placebo treatments, respectively. The ratio (AER 003 / placebo) of 0.93 (95%> CI, 0.62, 1.38) was not statistically significant (p = 0.69, Figure 3). The effect of subject was significant (p = 0.002), and there was a marginally significant effect of
period (p = 0.05). This model accounted for 81% of the observed variability in Day 7 lung eosinophil number (R2 = 81%).
Plasma concentration and effect of AER 003 on AHR and airway
inflammation
[0137] AER 003 pharmacokinetics following a single subcutaneous bolus injection were described by a 1 -compartment model with lst-order absorption and parallel lst-order and Michaelis-Menten clearance processes (Table 13, Figure 5).
Clearance and volume parameters were observed to increase with body weight. Day 0 /ogyo(PCioo), antibody status, animal source and AER 003 treatment period were not observed to influence AER 003 pharmacokinetics.
Table 13. Inhibitory effects of human IL-4 muteins on IL-4 and IL-13 TF-1 cell proliferation
IL-4 IL-13
IL-4 Mutein IC50 (nM) Fold increase in IC50 (nM) Fold increase in
IC50 relative to IC50 relative to AER 001 AER 001
AER 001 (R121D, Y124D) 0.13 1 1.3 1
AER 004 (R121D, Y124D,
T13D)
AER 003 (R121D, Y124D, 0.14 1.1 1.3 1 T13D, and N38C) + 40B PEG
[0138] Following a single 2-mg/kg subcutaneous dose, peak AER 003 plasma concentrations of 6.8 to 22.8 μg/mL were observed, typically at the first post-dose sampling occasion (24 to 48 hours post-dose). Average AER 003 plasma concentration over the 168-hr period of efficacy evaluation (Cavg o-i68hr) was 9.1 μg/mL (24%)
(geometric mean and geometric coefficient of variation). There was no correlation observed between Cavg o-i68hr and magnitude of effect, suggesting the 2-mg/kg dose was supra-maximal.
[0139] The data indicates that the rate of AER 003 plasma decline varies with
AER 003 concentration, with slower decline (t 2 ~ 5.4 days) at plasma concentrations well above the C50 of the saturable clearance component (401 ng/mL), and more rapid decline (t1/2 - 18 hours) when plasma concentrations fall below the C50 value.
[0140] The final model parameters (Table 14) indicate that absorption is rapid relative to elimination, with absorption complete within the first day after dosing. Although the maximum rate of the saturable component of clearance is estimated to be more than five times greater than the non-saturable component, the contribution of the saturable clearance mechanism at the peak concentrations observed in this study (-10,000 ng/mL) is less than 5%. Therefore, decline in AER 003 plasma concentration from peak values initially occurs with a half-life of approximately 5.4 days (estimated as ti/2 = n(2)/(CL/V). As AER 003 concentrations fall to around and below the C50 value (401 ng/mL), the rate of decline increases such that the half-life of decline between 16 and 23 days post-dosing is approximately 18 hours (estimated as ti/2 = /n(2)/([CL+CLsat]/V).
[0141] Following a single 2-mg/kg subcutaneous dose, peak AER 003 plasma concentrations of 6.8 to 22.8 μg/mL were observed, typically at the first post-dose sampling occasion (24 to 48 hours post-dose). Average AER 003 plasma concentration over the 168-hr period of efficacy evaluation (Cavg o-i68hr) was 9.1 μg/mL (24%) (geometric mean and geometric coefficient of variation). There was no correlation observed between Cavg o-i68hr and magnitude of effect (Figure 7-2), suggesting the 2- mg/kg dose was supra-maximal.
Table 14. Parameters of the pharmacokinetic model. Parameter Population Inter-Individual
Value, Θ Variability, ω
V/F (niL) a = 860-(WT/7.9 kg) 860 35%
CLsat/F (mIJhr) e = 27.9-(WT/7.9 kg)
401 2.12 a Population values are centered around a body weight (WT) of 7.9 kg, with relative systemic availability following s.c. injection defined as 100%.
Discussion
[0142] The Th2 cytokines IL-4 and IL-13 are thought to be central to the pathogenesis of allergic diseases such as allergic rhinitis, food allergy, allergic conjunctivitis, atopic dermatitis and asthma. Thus targeting both cytokines may have significant therapeutic potential. In the present study, the effect of AER 003, a dual IL- 4/IL-13 antagonist, was assessed on allergen-induced AHR and airway inflammation in the Ascaris suum sensitive cynomolgus monkey. Our results indicate that inhibition of IL-4 and IL-13 using AER 003 effectively inhibited the onset of allergen-induced AHR. Moreover, the favorable pharmacokinetic profile of AER 003 observed in the current study indicates that once weekly dosing of AER 003 would be sufficient in the clinic.
[0143] Mutagenesis studies have identified the regions of IL-4 important for interaction with the IL-4Ra chain, a common component of both type I and II receptors. Mutations of arginine 121, and tyrosine 124, to aspartic acid in the D helix of the human protein results in an IL-4 mutant that has the same capacity to bind to the IL-4Ra chain as native IL-4, but has no signaling activity and is an antagonist of both human IL-4- and IL-13 -induced responses in vitro 47-49. In addition, single mutations in the A and C helices at positions T13D, E19A and F82D can increase binding affinity
of IL-4 for the IL-4Ra50' 51. In the present study, It was hypothesized that combining the double mutations R121D/Y124D, with the T13D mutation would generate an IL-4 mutein that would bind to the IL-4Ra chain with greater affinity than native IL-4 or the R121D/Y124D double mutant and would be a potent IL-4 and IL-13 antagonist. [0144] Initial studies were performed to assess the binding affinity of the triple mutein (T13D/R121D/Y124D) for the IL-4Ra and confirm the antagonistic activities of the triple mutein in vitro against both IL-4 and IL-13. Using surface plasmon resonance, the affinity of the triple mutein for the IL-4Ra was demonstrated to be 5 times greater than that of either native IL-4 or the double mutein (R121D/Y124D) confirming that the addition of the T13D mutation to the two point mutations
(R121D/Y124D) increased the binding affinity of the resultant triple mutein for the receptor47' 49. This increase in affinity was achieved through a decrease in the dissociation rate constant. In subsequent studies, the triple mutein
(T13D/R121D/Y124D) was shown to potently inhibit the proliferative response of TF-1 cells to both IL-4 and IL-13, demonstrating the dual antagonistic nature was retained in the IL-4 triple mutein.
[0145] Previously we have shown that twice daily inhalation administration of the IL-4 double mutein (R121D/Y124D) inhibits allergen-induced AHR in the cynomolgus monkey indicating localized delivery to the lung was sufficient to inhibit the asthma phenotype and demonstrating the therapeutic potential of the inhaled mutein for the treatment of asthma55 . Indeed, twice daily aerosol administration of the double mutein to asthmatics effectively inhibited the allergen-induced late phase response, and is currently being developed as an inhalation therapy for the treatment of asthma56. Although systemic delivery of the double mutein (R121D/Y124D) was also effective in the primate asthma model, the pharmacokinetic profile of the 15KDa molecule requires frequent delivery (twice daily) via the systemic route, and would be unfavorable for clinical application in asthma, or other related allergic diseases such as atopic dermatitis 52-55.
[0146] Polyethylene glycol (PEG) modification, PEGylation, is now a well established technique for the modification of therapeutic peptides and proteins.
PEGylation may confer several advantages including reduced immunogenicity, reduced proteolysis, reduced toxicity and improved solubility, although the most exploited
property is the improved pharmacokinetic profile that can be achieved " . In the current study, a site-specific PEGylation strategy was utilized to improve the pharmacokinetic profile of the IL-4 triple mutein, T13D/R121D/Y 124D. To achieve this, a further single point mutation to a cysteine at position 38 (N38C) was introduced into the IL-4 variant, to which a 40KDa Branched PEG was covalently linked. The N38 position was selected to limit the interference of the PEG with the IL-4Ra binding epitope and reduce the potential loss of bioactivity, a common problem associated with PEGylation57"60.
[0147] Assessment of receptor binding affinity revealed that the PEGylated IL- 4 mutein (T13D/N38C/R121D/Y124D), also known as AER 003, was lower in overall affinity than the triple IL-4 mutein (T13D/R121D/Y124D), reflected in an increase in the association rate of the PEGylated mutein for IL-4Ra. AER 003 showed a receptor affinity comparable to the double mutein (R121D/Y124D) and wild type IL-4.
Importantly, the dissociation rate of the PEGylated AER 003 molecule from the IL-4Ra subunit retained a similar rate to the triple IL-4 mutein, and approximately 15 -fold greater that wild type IL-4 and the double mutein. Similarly, assessment of the PEGylated mutein, AER 003, in the TF-1 cell assay confirmed inhibitory activity against both IL-4 and IL-13 induced proliferation, comparable to that of the double mutein (R121D/Y124D). Subsequently, the inhibitory activity of the PEGylated mutein (T13D/N38C/R121D/Y124D), AER 003, was assessed utilizing primary T and B lymphocytes, cell types considered to be central to the pathogeneisis of allergic disease. AER 003 significantly inhibited IL-4 induced proliferation of both T and B lymphocytes, with activity comparable to that of the double mutein (R121D/Y 124D).
[0148] Although our in vitro data indicate that AER 003 may be a promising dual IL-4/IL-13 antagonist for the treatment of allergic disease, translation of this activity in vivo is dependent upon the pharmacokinetic profile of the molecule. Whilst PEGylation may increase systemic exposure by reducing clearance, absorption and distribution may be negatively impacted. Thus the effect of PEGylation on the pharmacokinetics and efficacy of AER 003 was evaluated, in a model of allergen- induced airway hyperresponsiveness and airway inflammation, cardinal features of asthma.
[0149] Using Ascaris suum sensitive cynomolgus monkeys, studies were performed to evaluate the effects of AER 003 on the induction of AHR and airway inflammation. A single subcutaneous dose of AER 003 administered 2 days prior to three successive days of allergen challenge, significantly prevented the development of AHR 2 days post allergen challenge. In contrast, there was no significant evidence for an effect of AER 003 on lung eosinophilia. The effect of AER 003 on AHR was comparable to that previously observed with the double mutein (R121D/Y124D) in this model, although some effect, albeit small, was previously noted on airway
eosinophilia54' 55.
[0150] A single one-compartment systemic model with parallel first-order and saturable clearance mechanisms describes AER 003 plasma PK in cynomolgus monkeys following s.c. injection. Absorption of AER 003 was rapid relative to elimination, with absorption complete within the first day after dosing. Although the maximum rate of the saturable component of clearance is estimated to be more than five times greater than the non-saturable component, the contribution of the saturable clearance mechanism at the peak concentrations observed in this study (-10,000 ng/mL) is less than 5%. Therefore, decline in AER 003 plasma concentration from peak values initially occurs with a half-life of approximately 5.4 days. As AER 003 concentrations fall to around and below the C50 value, the rate of decline increases such that the half- life of decline between 16 and 23 days post-dosing is approximately 18 hours. Scaling of the PK to humans using indicates that a dosing regimen of no more than once a week may be achieved.
[0151] Thee observations of this study are consistent with several rodent studies. Indeed, either deletion of IL-4Ra or STAT6 result in significant inhibition of allergen-induced AHR and airway inflammation in mice 29' 43. More specifically, murine IL-4 mutant proteins that bind with high affinity to the IL-4Ra chain without inducing signal transduction have also been developed 61 ' 62. Both the murine IL-4 double mutein (Ql 16D/Y119D) and truncated protein (CI 18 deletion) significantly attenuate allergen induced AHR, airway eosinophilia, mucus hyperproduction and IgE levels 27' 62.
[0152] In summary, we demonstrate the effect of AER 003, a PEGylated human
IL-4 mutant (T13D/N38C/R121D/Y124D) receptor antagonist, on allergen-induced
AHR and airway inflammation in the Ascaris suum sensitive cynomolgus monkey. Our results indicate that inhibition of IL-4 and IL-13 using AER 001 prevents the onset of allergen-induced AHR a cardinal feature of asthma. Moreover, the favorable pharmacokinetic profile of AER 003 observed in the current study is sufficient to support once weekly dosing in the clinic. Overall, these data indicate the therapeutic potential of AER 003 for the treatment of allergic disease.
Methods
Reagents
[0153] Site-directed mutagenesis of human IL-4 followed the method of Kunkel et al. (Methods Enzymol 154, 367-382 (1987)). AER 003 and AER 004 were expressed in E. coli as insoluble protein within inclusion bodies. AER 003 and AER 004 intermediates were refolded from inclusion bodies and purified using a two-step chromatographic process consisting of hydrophobic interaction and cation exchange. AER 003 was PEGylated using Sunbright GL2-400MA 40kDa Branched PEG (NOF Corporation) and PEGylated product isolated using cation exchange chromatography. Ascaris suum extract (lyophilized cake, Lot # XPB33-X10 Greer Labs Inc., Lenoir, NC, USA) stock solution was prepared by adding 50 ml of sterile water to lyophilized cake.
Receptor Interaction Studies
[0154] The extracellular domain of IL-4 Receptor (IL-4R) containing an Fc fusion, and human IL-4, were purchased from R&D Systems. All kinetic experiments were performed using a BIAcore 2000 instrument (GE Healthcare) at 25 (C with 10 mM
HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% Surfactant P20, 0.005% BSA, pH 7.4.
IL-4R/Fc was covalently attached to a CM5 chip via amine coupling to achieve a binding surface density of 150-250 RU. Six different concentrations of analyte in running buffer were evaluated in duplicate on the receptor surface using a flow rate of
50 μΕ/ηιίη. A blank channel was used as the control channel. Data were fit using the global analysis software provided by BIAevaluation.
TF-1 proliferation assay
[0155] The functional antagonistic activity of AER 003 was confirmed against the proliferative response of TF-1 cells (erythroleukemia cell line) to human (h) IL-4 (R&D Systems, Minneapolis, MN, USA) (0.5 ng/ml, 0.033 nM) or human (h) IL-13 (R&D Systems, Minneapolis, MN, USA) (5 ng/ml, 0.416nM). TF-1 cells were cultured for 3 days in 96 well plates (lxl04/well, 100 μΐ, volume) in RPMI (Invitrogen, Carlsbad, CA, USA) + 10% serum (Mediatech, Herndon, VA) with or without hIL-4 or hIL-13 and pitrakinra. To show that AER 003 inhibits TF-1 cell proliferation by interfering with IL-4 signaling alone, human GM-CSF (R&D Systems, Minneapolis, MN, USA), an alternative growth factor for TF-1 cells that does not bind to the IL-4 receptor, was used as a specificity control. Twenty-four hours before the final reading, 10 μΐ, AlamarBlue (10%> volume, Invitrogen, Camarillo, CA, USA) was added to each well. Fluorescence was determined at 530/590 nm using a WALLAC Victor 2. IC50 was calculated based on dose titration of AER 003 (71.4 nM to 0.033 nM). T- and B- Lymphocyte proliferation
[0156] The ability of AER 003 to effectively antagonize human IL-4 responses was examined using the proliferative response of primary T- and B- lymphocytes. For the T-cell and B-cell proliferation assays, 20 and 10 human donors were required, respectively. Peripheral blood mononuclear cells (PBMC) from heparinized human blood were purified by centrifugation through Ficoll-Paque PLUS (Sigma, St Louis, MO) gradient. Cells were washed and resuspended in culture medium (RPMI- 1640 containing 10% FBS, 1% Pen-Strep and 10 mM HEPES buffer), and the number of viable cells was determined by trypan blue dye exclusion.
[0157] For T cell assays, PBMCS were incubated in complete RPMI containing 10% FCS with phytohemagglutinin (PHA, Sigma, St Louis, MO, USA) (5 μg/ml) for 4 days at 37°C. PHA blasts were washed, resuspended in complete RPMI with 10%> FCS
6
(1x10 /well, 96 well plates), and incubated for a further 72 h in the presence of either hIL-4 alone, or hIL-4 plus AER 003. 3H-thyimidine (1 μΟΛνεΙΙ, Amersham,
Piscataway, NJ, USA) was added for the last 24 h and thymidine incorporation measured by betaplate scintillation (Wallac, Gaithersburg, MD).
[0158] On the same day of blood collection, isolated PBMC's were incubated with anti-CD40 MAb (1 μg/ml). Then PBMCs (in the presence of anti-CD40) were used for the proliferation assay with recombinant human IL-4. The cells were seeded in 96-well plates (lxl 06 cells per well) and stimulated for 3 days with IL-4 (10 ng/ml, 0.667 nM) in the presence of varying concentrations of IL-4RE molecules. The incorporation of 3H-thyimidine (1 μΟΛνεΙΙ, Amersham, Piscataway, NJ, USA) in the last 18 h of incubation was used as an indicator of proliferation.
Evaluation of AER 003 activity in vivo
[0159] AER 003 biological activity was evaluated in a primate model of airway hyperresponsiveness (AHR) and inflammation using a double-blind, 2-period crossover design (Figure 1 A). All animals were rested at least 8 weeks between studies to allow airway responsiveness and inflammation to return to baseline (pre-allergen) levels. Studies were performed using the 7 day primate asthma model (Figure IB). Airway responsiveness to inhaled methacholine (MCh) and airway cellular composition by bronchoalveolar lavage (BAL) were determined 2 days before (Day 0) and 2 days after (Day 7) three consecutive-day (Days 3, 4, 5) inhalations of Ascaris suum extract. AER 003 (2 mg/kg) or vehicle was administered subcutaneously 48 h prior to the first antigen challenge (Day 1 of Study).
Animals
[0160] Twenty four Ascaris suum sensitive, adult (6-11kg) male cynomolgus monkeys (Macaca fascicularis) were used in these studies. The primates were housed in individual, open meshed cages and maintained at constant temperature and humidity with a 12 hour light cycle. Monkeys were fed twice daily, except on experimental days when food was withheld the night before a procedure. Water was available ad libitum except during procedures. Experiments were performed in compliance with the Valley Biosystems Institutional Animal Care and Use Committee. All procedures, except subcutaneous administration of AER 003, were performed in intubated animals under anesthesia (7mg/kg, Ketaset, Ft Dodge, IN, USA: Xylazine 1.2 mg/kg i.m., Phoenix Scientific Inc., St Joseph, MO, USA).
Allergen-induced AHR and airway inflammation
[0161] Ascaris suum solution (1 : 1 stock solution:PBS) was aerosolized using a
Bird micronebulizer and Mark 7A respirator (Viasys Health Care, Palm Springs, CA, USA) delivering 15 breaths/min for 2 minutes. AHR to aerosolized methacholine (MCh; 0.1-100 mg/ml) was determined in anesthetized ventilated animals. MCh was administered using a Bird micronebulizer and Bird Mark 7A respirator set to deliver 15 breaths/min with each inspiratory breath terminating at 20 cm H20. Lung resistance ((RL), cmH20/ml/s) was measured continuously for 10 minutes after each dose while animals were ventilated using a Harvard ventilator with parameters set to 40
breaths/minute, a 40/60 I:E ratio, and a tidal volume of 35 ml. Airflow was measured by a Fleisch pneumotachograph (Hans Rudolph Model 8420, Kansas City, MO, USA) and thoracic pressure by a Validyne pressure transducer. Lung resistance was computed (Buxco analyzer) using the primary signals of flow and pressure. The peak response was recorded for each dose of MCh. Dosing continued until a 100% increase in baseline resistance was achieved. Data were reported as the dose (by interpolation) of MCh required to produce a 100% increase in baseline RL (PC 100). Segmental bronchoscopic bronchoalveolar lavage (BAL) was performed after measurement of AHR. The distal end of a pediatric fiberoptic bronchoscope (Olympus, model BF3C30, New York, NY, USA) was inserted down the endotracheal tube and guided into the distal lung until the tip of the bronchoscope became wedged. Normal saline (15 ml, room temperature) was instilled and then slowly aspirated. BAL samples were analyzed for total leukocyte count (Coulter counter, Beckman, Fullerton, CA, USA), and differential cell counts were performed by counting at least 200 cells on
cytocentrifuged slide preparations (Cytospin 2, Cytospin Shandon, Pittsburgh, PA, USA), stained with Diff-Quick (Dade Behring Inc., Newark, DE, USA),differentiated using standard morphometric criteria.
Preparation and administration of AER 003
[0162] Dose volume was calculated based on the monkey weights on Day 0 of the study period in order to achieve a final dose of 2 mg/kg. AER 003 was formulated at a concentration of 6.26 mg/ml (0.32 ml/kg). On the day of dosing, the animals' backs were shaved and wiped with 70%> ethanol. The solution was injected to the
subcutaneous space between the shoulder blades. All syringes were weighed before and after injection to enable accurate calculation of the dose administered.
Blood sampling for pharmacokinetics
[0163] PK samples were collected from all animals in each period predose and at 2, 3, 4, 6, 9, 12, 16, 23 and 34 days postdose. For each sample, whole blood (2 mL) was collected into K2 EDTA tubes, the plasma collected and frozen on dry ice and stored at -70°C for subsequent analysis. An additional sample was collected 1 day postdose during Period 2 after analysis of the first set of samples revealed higher than expected circulating AER 003 levels 2 days postdose. ELISA detection of AER 003 in plasma
[0164] pitrakinra was detected by ELISA, using commercially available antibodies against wild type-human IL-4. Therefore, the ELISA was unable to distinguish between wild type human IL-4 and AER 003. ELISA 96 well plates (Maxisorp, Nunc, Rochester, NY, USA) were coated overnight (4°C) with mouse anti- human IL-4 monoclonal 8D4-8 (BD Biosciences, Franklin Lakes, NJ, USA) and blocked with 1% BSA for 1 h at room temperature. Samples and standards were added to the plate and incubated for 1 h prior to detection using biotinylated rat anti-human IL-4 monoclonal MP4-25D2 (BD Biosciences, Franklin Lakes, NJ, USA). Reactions were amplified using HRP-Avidin (KPL, Gaithersburg, MD, USA). Plates were read using a SpectraMAX (Molecular Devices, Sunnyvale, CA) plate reader at 450 nm with reference at 570 nm.
[0165] Nonlinear mixed-effects modeling of the plasma concentration-time data was conducted using NONMEM® Version VI 2.0 (licensed and distributed by
GloboMax, ICON Development Solutions, Ellicot City, MD) and PLT Tools Version 1.7.0 (PLTsoft, San Francisco, CA).
Materials
[0166] AER 003 is a PEGylated recombinant protein derived from human IL-4.
The mutated IL-4 protein (T13D, N38C, R121D, Y124D) was expressed and purified from E. coli. The mutein was subsequently PEGylated (position N38C) using a 40KDa branched PEG. AER 001, a mutated (Arg-121-Asp and Tyr-124-Asp) recombinant
protein derived from hIL-4, was expressed in E. coli. Ascaris suum extract (lyophilized cake, Lot # XPB33-X10 Greer Labs Inc., Lenoir, NC, USA) stock solution was prepared by adding 50 ml of sterile water to lyophilized cake.
Statistical Analysis
[0167] All statistical tests were two-sided and were performed using a 5% significance level, leading to 95% (two-sided) confidence intervals. The effect of treatment on Day 7 PCioo was assessed using analysis of covariance (ANCOVA). The model included effects for treatment (the main variable of interest), period and subject. In addition, a covariate for baseline (Day 0) PCioo was included. PCioo values were log-transformed prior to analysis. The effect of treatment on Day 7 lung eosinophil count was assessed using a general linear analysis of variance model with effects for treatment, period and subject. Baseline (Day 0) lung eosinophil count was not a significant determinant of the Day 7 value, in agreement with historical lung eosinophil data from the 7- or 10-day monkey asthma model. Lung eosinophil counts were log- transformed prior to analysis.
REFERENCES
[0168] 1. Ying S, Humbert M, Barkans J, Corrigan CJ, Pfister R, Menz G, et al. Expression of IL-4 and IL-5 mRNA and protein product by CD4+ and CD8+ T cells, eosinophils, and mast cells in bronchial biopsies obtained from atopic and nonatopic (intrinsic) asthmatics. J Immunol 1997; 158:3539-44.
[0169] 2. Kotsimbos TC, Ernst P, Hamid QA. Interleukin-13 and interleukin-4 are coexpressed in atopic asthma. Proc Assoc Am Physicians 1996;
108:368-73.
[0170] 3. Truyen E, Coteur L, Dilissen E, Overbergh L, Dupont LJ, Ceuppens JL, et al. Evaluation of airway inflammation by quantitative Thl/Th2 cytokine mRNA measurement in sputum of asthma patients. Thorax 2006; 61 :202-8.
[0171] 4. Kroegel C, Julius P, Matthys H, Virchow JC, Jr., Luttmann W.
Endobronchial secretion of interleukin-13 following local allergen challenge in atopic asthma: relationship to interleukin-4 and eosinophil counts. Eur Respir J 1996; 9:899- 904.
[0172] 5. Beghe B, Barton S, Rorke S, Peng Q, Sayers I, Gaunt T, et al.
Polymorphisms in the interleukin-4 and interleukin-4 receptor alpha chain genes confer susceptibility to asthma and atopy in a Caucasian population. Clin Exp Allergy 2003; 33: 1111-7.
[0173] 6. Heinzmann A, Mao XQ, Akaiwa M, Kreomer RT, Gao PS,
Ohshima K, et al. Genetic variants of IL-13 signalling and human asthma and atopy. Hum Mol Genet 2000; 9:549-59.
[0174] 7. Tamura K, Suzuki M, Arakawa H, Tokuyama K, Morikawa A.
Linkage and association studies of STAT6 gene polymorphisms and allergic diseases. Int Arch Allergy Immunol 2003; 131 :33-8.
[0175] 8. Antunez C, Torres MJ, Mayorga C, Cornejo-Garcia JA,
Santamaria-Babi LF, Blanca M. Different cytokine production and activation marker profiles in circulating cutaneous-lymphocyte-associated antigen T cells from patients with acute or chronic atopic dermatitis. Clin Exp Allergy 2004; 34:559-66.
[0176] 9. Chien YH, Hwu WL, Chiang BL. The genetics of atopic dermatitis. Clin Rev Allergy Immunol 2007; 33: 178-90.
[0177] 10. Hamid Q, Boguniewicz M, Leung DY. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest 1994; 94:870-6.
[0178] 11. He JQ, Chan- Yeung M, Becker AB, Dimich-Ward H, Ferguson
AC, Manfreda J, et al. Genetic variants of the IL13 and IL4 genes and atopic diseases in at-risk children. Genes Immun 2003; 4:385-9.
[0179] 12. Kaminishi K, Soma Y, Kawa Y, Mizoguchi M. Flow cytometric analysis of IL-4, IL-13 and IFN-gamma expression in peripheral blood mononuclear cells and detection of circulating IL-13 in patients with atopic dermatitis provide evidence for the involvement of type 2 cytokines in the disease. J Dermatol Sci 2002; 29: 19-25.
[0180] 13. Tazawa T, Sugiura H, Sugiura Y, Uehara M. Relative importance of IL-4 and IL-13 in lesional skin of atopic dermatitis. Arch Dermatol Res 2004; 295:459-64.
[0181] 14. Yamada N, Wakugawa M, Kuwata S, Yoshida T, Nakagawa H.
Chronologic analysis of in situ cytokine expression in mite allergen-induced dermatitis in atopic subjects. J Allergy Clin Immunol 1995; 96: 1069-75.
[0182] 15. Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Kohler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature 1993; 362:245-8.
[0183] 16. Finkelman FD, Katona IM, Urban JF, Jr., Holmes J, Ohara J,
Tung AS, et al. IL-4 is required to generate and sustain in vivo IgE responses. J Immunol 1988; 141 :2335-41.
[0184] 17. Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S,
Zurawski G, et al. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci U S A 1993; 90:3730-4.
[0185] 18. Bochner BS, Klunk DA, Sterbinsky SA, Coffman RL, Schleimer RP. IL-13 selectively induces vascular cell adhesion molecule- 1 expression in human endothelial cells. J Immunol 1995; 154:799-803.
[0186] 19. Doyle AG, Herbein G, Montaner LJ, Minty AJ, Caput D, Ferrara
P, et al. Interleukin- 13 alters the activation state of murine macrophages in vitro:
comparison with interleukin-4 and interferon-gamma. Eur J Immunol 1994; 24: 1441-5.
[0187] 20. Fukuda T, Fukushima Y, Numao T, Ando N, Arima M,
Nakajima H, et al. Role of interleukin-4 and vascular cell adhesion molecule- 1 in selective eosinophil migration into the airways in allergic asthma. Am J Respir Cell Mol Biol 1996; 14:84-94.
[0188] 21. McKenzie AN, Culpepper JA, de Waal Malefyt R, Briere F, Punnonen J, Aversa G, et al. Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc Natl Acad Sci U S A 1993; 90:3735-9.
[0189] 22. Webb DC, Cai Y, Matthaei KI, Foster PS. Comparative roles of
IL-4, IL-13, and IL-4Ralpha in dendritic cell maturation and CD4+ Th2 cell function. J Immunol 2007; 178:219-27.
[0190] 23. Kellner J, Gamarra F, Welsch U, Jorres RA, Huber RM, Bergner
A. IL-13Ralpha2 reverses the effects of IL-13 and IL-4 on bronchial reactivity and acetylcholine-induced Ca+ signaling. Int Arch Allergy Immunol 2007; 142: 199-210.
[0191] 24. Temann UA, Prasad B, Gallup MW, Basbaum C, Ho SB, Flavell RA, et al. A novel role for murine IL-4 in vivo: induction of MUC5AC gene expression and mucin hypersecretion. Am J Respir Cell Mol Biol 1997; 16:471-8.
[0192] 25. Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al.
Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest 1999; 103:779-88.
[0193] 26. Kuperman DA, Huang X, Nguyenvu L, Holscher C, Brombacher
F, Erie DJ. IL-4 receptor signaling in Clara cells is required for allergen-induced mucus production. J Immunol 2005; 175:3746-52.
[0194] 27. Zavorotinskaya T, Tomkinson A, Murphy JE. Treatment of experimental asthma by long-term gene therapy directed against IL-4 and IL-13. Mol Ther 2003; 7: 155-62.
[0195] 28. Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY,
Karp CL, et al. Interleukin-13: central mediator of allergic asthma. Science 1998;
282:2258-61.
[0196] 29. Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F,
Rennick DM, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science 1998; 282:2261-3.
[0197] 30. Shi HZ, Deng JM, Xu H, Nong ZX, Xiao CQ, Liu ZM, et al.
Effect of inhaled interleukin-4 on airway hyperreactivity in asthmatics. Am J Respir Crit Care Med 1998; 157: 1818-21.
[0198] 31. Chen L, Martinez O, Overbergh L, Mathieu C, Prabhakar BS,
Chan LS. Early up-regulation of Th2 cytokines and late surge of Thl cytokines in an atopic dermatitis model. Clin Exp Immunol 2004; 138:375-87.
[0199] 32. Lee GR, Flavell RA. Transgenic mice which overproduce Th2 cytokines develop spontaneous atopic dermatitis and asthma. Int Immunol 2004;
16: 1155-60.
[0200] 33. Coyle AJ, Le Gros G, Bertrand C, Tsuyuki S, Heusser CH, Kopf
M, et al. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am J Respir Cell Mol Biol 1995; 13:54-9.
[0201] 34. Cony DB, Folkesson HG, Warnock ML, Erie DJ, Matthay MA, Wiener-Kronish JP, et al. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med 1996; 183: 109-17.
[0202] 35. Hogan SP, Mould A, Kikutani H, Ramsay AJ, Foster PS.
Aeroallergen-induced eosinophilic inflammation, lung damage, and airways
hyperreactivity in mice can occur independently of IL-4 and allergen-specific immunoglobulins. J Clin Invest 1997; 99: 1329-39.
[0203] 36. Henderson WR, Jr., Chi EY, Maliszewski CR. Soluble IL-4 receptor inhibits airway inflammation following allergen challenge in a mouse model of asthma. J Immunol 2000; 164: 1086-95.
[0204] 37. Yang G, Li L, Volk A, Emmell E, Petley T, Giles-Komar J, et al. Therapeutic dosing with anti-interleukin-13 monoclonal antibody inhibits asthma progression in mice. J Pharmacol Exp Ther 2005; 313:8-15.
[0205] 38. Kasaian MT, Donaldson DD, Tchistiakova L, Marquette K, Tan
XY, Ahmed A, et al. Efficacy of IL-13 neutralization in a sheep model of experimental asthma. Am J Respir Cell Mol Biol 2007; 36:368-76.
[0206] 39. Bree A, Schlerman FJ, Wadanoli M, Tchistiakova L, Marquette
K, Tan XY, et al. IL-13 blockade reduces lung inflammation after Ascaris suum challenge in cynomolgus monkeys. J Allergy Clin Immunol 2007; 119: 1251-7.
[0207] 40. Perkins C, Wills-Karp M, Finkelman FD. IL-4 induces IL-13- independent allergic airway inflammation. J Allergy Clin Immunol 2006; 118:410-9.
[0208] 41. Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol 1999; 17:701-38.
[0209] 42. de Vries JE. The role of IL-13 and its receptor in allergy and inflammatory responses. J Allergy Clin Immunol 1998; 102: 165-9.
[0210] 43. Tomkinson A, Kanehiro A, Rabinovitch N, Joetham A,
Cieslewicz G, Gelfand EW. The failure of STAT6-deficient mice to develop airway eosinophilia and airway hyperresponsiveness is overcome by interleukin-5. Am J Respir Crit Care Med 1999; 160: 1283-91.
[0211] 44. Kruse N, Shen BJ, Arnold S, Tony HP, Muller T, Sebald W.
Two distinct functional sites of human interleukin 4 are identified by variants impaired in either receptor binding or receptor activation. Embo J 1993; 12:5121-9.
[0212] 45. Letzelter F, Wang Y, Sebald W. The interleukin-4 site-2 epitope determining binding of the common receptor gamma chain. Eur J Biochem 1998; 257: 11-20.
[0213] 46. Shen BJ, Hage T, Sebald W. Global and local determinants for the kinetics of interleukin-4/interleukin-4 receptor alpha chain interaction. A biosensor study employing recombinant interleukin-4-binding protein. Eur J Biochem 1996; 240:252-61.
[0214] 47. Tony HP, Shen BJ, Reusch P, Sebald W. Design of human interleukin-4 antagonists inhibiting interleukin-4-dependent and interleukin- 13- dependent responses in T-cells and B-cells with high efficiency. Eur J Biochem 1994; 225:659-65.
[0215] 48. Mueller TD, Zhang JL, Sebald W, Duschl A. Structure, binding, and antagonists in the IL-4/IL-13 receptor system. Biochim Biophys Acta 2002;
1592:237-50.
[0216] 49. Andrews AL, Holloway JW, Holgate ST, Davies DE. IL-4 receptor alpha is an important modulator of IL-4 and IL-13 receptor binding:
implications for the development of therapeutic targets. J Immunol 2006; 176:7456-61.
[0217] 50. Kraich M, Klein M, Patino E, Harrer H, Nickel J, Sebald W, et al. A modular interface of IL-4 allows for scalable affinity without affecting specificity for the IL-4 receptor. BMC Biol 2006; 4: 13.
[0218] 51. Wang Y, Shen BJ, Sebald W. A mixed-charge pair in human interleukin 4 dominates high-affinity interaction with the receptor alpha chain. Proc Natl Acad Sci U S A 1997; 94: 1657-62.
[0219] 52. Harris P, Lindell D, Fitch N, Gundel R. The IL-4 receptor antagonist (BAY 16-9996) reverses airway hyperresponsiveness in a primate model of asthma (Abstract). Am J Respir Crit Care Med 1999; 159:A230.
[0220] 53. Morton M, Harris P, Xu J, Gorina S, Roczniak S, Fitch N, et al. The effects of an IL-4 receptor antagonist (BAY 16-9996) on antigen-induced cutaneous wheal and flare reactions in the primate (Abstract). Am J Respir Crit Care Med 1999; 159:A660.
[0221] 54. Fitch N, Morton M, Bowden A, Shanafelt MC, Shanafelt A,
Wetzel G, et al. Preclinical Evaluation of Bay 16-9996 a Dual I1-4/I1-13 Receptor Antagonist (Abstract) . J Allergy Clin Immunol 2001 ; 107.
[0222] 55. Tomkinson A, Stevens L, Morton M, Chen J, Wong T,
Burmeister Getz E, et al. Therapeutic effects of inhaled AER 001, an IL-4/IL-13 antagonist, on allergen-induced airway hyperresponsiveness (AHR) and airway inflammation (Abstract). Am J Respir Crit Care Med 2006:A521.
[0223] 56. Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet 2007; 370:1422-31.
[0224] 57. Francis GE, Fisher D, Delgado C, Malik F, Gardiner A, Neale D.
PEGylation of cytokines and other therapeutic proteins and peptides: the importance of biological optimisation of coupling techniques. Int J Hematol 1998; 68:1-18.
[0225] 58. Gaberc-Porekar V, Zore I, Podobnik B, Menart V. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr Opin Drug Discov Devel 2008; 11 :242-50.
[0226] 59. Greenwald RB. PEG drugs: an overview. J Control Release 2001; 74: 159-71.
[0227] 60. Hamidi M, Azadi A, Rafiei P. Pharmacokinetic consequences of pegylation. Drug Deliv 2006; 13:399-409.
[0228] 61. Grunewald SM, Kunzmann S, Schnarr B, Ezernieks J, Sebald W,
Duschl A. A murine interleukin-4 antagonistic mutant protein completely inhibits interleukin-4-induced cell proliferation, differentiation, and signal transduction. J Biol Chem 1997; 272: 1480-3.
[0229] 62. Tomkinson A, Duez C, Cieslewicz G, Pratt JC, Joetham A,
Shanafelt MC, et al. A murine IL-4 receptor antagonist that inhibits IL-4- and IL-13- induced responses prevents antigen-induced airway eosinophilia and airway hyperresponsiveness. J Immunol 2001; 166:5792-800.
Claims
1. A method for inhibiting a dermatitis response in a subject, the method comprising: administering to a subject in need thereof a therapeutically effective amount of a modified IL-4 mutein receptor antagonist, wherein the modified IL-4 mutein receptor antagonist comprises at least the following modifications:
(1) substitution of each of the amino acids occurring in the wild- type human IL-4 protein at positions 121 and 124 with different amino acids;
(2) substitution of the threonine occurring in the wild-type human IL-4 protein at position 13 with a different amino acid;
(3) substitution of the asparagine occurring in the wild-type human IL-4 protein at position 38 with a cysteine; and
(4) a non-protein polymer covalently attached to the substituted cysteine at position 38.
2. The method of claim 1, wherein the modified IL-4 mutein receptor antagonist has 95% percent sequence identity with the wild-type human IL-4 protein.
3. The method of claim 2, wherein the modified IL-4 mutein receptor antagonist has 99% percent sequence identity with the wild-type human IL-4 protein.
4. The method of any of the preceding claims, wherein said therapeutically effective amount is administered systemically.
5. The method of claim 4, wherein said therapeutically effective amount is administered subcutaneously.
6. The method of any of the preceding claims, wherein said therapeutically effective amount is administered no more than about once per week.
7. The method of any of the preceding claims, wherein said therapeutically effective amount is administered no more than about once every two weeks.
8. The method of any of the preceding claims, wherein said therapeutically effective amount is administered no more than about once per month.
9. The method of any of the preceding claims the therapeutically effective amount is from about 2 mg/kg to about 40 mg/kg weekly.
10. The method of any of the preceding claims the therapeutically effective amount is from about 5 mg/kg to about 30 mg/kg weekly.
11. The method of any of the preceding claims the therapeutically effective amount is from about 7 mg/kg to about 20 mg/kg weekly.
12. The method of any of the preceding claims the therapeutically effective amount is about 10 mg/kg weekly.
13. The method of any of the preceding claims, wherein modified IL-4 mutein receptor antagonist comprises one or more of the following amino acid substitutions: R121D, Y124D, T13D, and N38C.
14. The method of claim 13, wherein modified IL-4 mutein receptor antagonist comprises at least the following amino acid substitutions: R121D and Y124D.
15. The method of claim 14, wherein modified IL-4 mutein receptor antagonist additionally comprises the amino acid substitution T13D.
16. The method of any of the preceding claims, wherein the nonprotein polymer is selected from the group consisting of polyethylene glycol (PEG), polypropylene glycol and polyoxyalkylenes.
17. The method of any of the preceding claims, wherein the nonprotein polymer comprises polyethylene glycol (PEG),
18. The method of claim 17, wherein the PEG comprises a branched
PEG.
19. The method of claim 17, wherein the PEG has an average molecular weight between about 2 KDa and about 50 KDa.
20. The method of claim 19, wherein the PEG has an average molecular weight of about 40 KDa.
21. The method of any of the preceding claims, wherein the dermatitis comprises an allergic or atopic reaction.
22. The method of any of the preceding claims, wherein the dermatitis comprises a hypersensitivity reaction.
23. The method of claim 22, wherein the hypersensitivity reaction comprises contact dermatitis.
24. The method of claim 22, wherein the hypersensitivity reaction comprises atopic dermatitis.
25. The method of any of the preceding claims, wherein the dermatitis comprises exzema.
26. The method of any of the preceding claims, wherein the subject is human.
27. The method of any of the preceding claims, wherein the modified IL-4 mutein receptor antagonist is co-administered with a therapeutically effective amount of an additional agent that mitigates a symptom of dermatitis.
28. A pharmaceutical composition, wherein the composition comprises:
a therapeutically effective amount of a modified IL-4 mutein receptor antagonist, wherein the modified IL-4 mutein receptor antagonist comprises at least the following modifications: (1) substitution of each of the amino acids occurring in the wild-type human IL-4 protein at positions 121 and 124 with different amino acids;
(2) substitution of the threonine occurring in the wild- type human IL-4 protein at position 13 with a different amino acid;
(3) substitution of the asparagine occurring in the wild- type human IL-4 protein at position 38 with a cysteine; and
(4) a non-protein polymer covalently attached to the substituted cysteine at position 38; and
a therapeutically effective amount of an additional agent that is useful for mitigating a symptom of dermatitis;
wherein the therapeutically effective amount of the modified IL- 4 mutein receptor antagonist mitigates a symptom of dermatitis; and
wherein the therapeutically effective amount of the additional agent mitigates a symptom of dermatitis.
29. A kit comprising:
at least one unit dosage form comprising a therapeutically effective amount of a modified IL-4 mutein receptor antagonist, wherein the modified IL-4 mutein receptor antagonist comprises at least the following modifications:
(1) substitution of each of the amino acids occurring in the wild-type human IL-4 protein at positions 121 and 124 with different amino acids;
(2) substitution of the threonine occurring in the wild- type human IL-4 protein at position 13 with a different amino acid;
(3) substitution of the asparagine occurring in the wild- type human IL-4 protein at position 38 with a cysteine; and
(4) a non-protein polymer covalently attached to the substituted cysteine at position 38; and
at least one unit dosage form comprising a therapeutically effective amount of an additional agent that is useful for mitigating a symptom of dermatitis;
wherein the therapeutically effective amount of the a modified
IL-4 mutein receptor antagonist mitigates a symptom of dermatitis; and wherein the therapeutically effective amount of the additional agent mitigates a symptom of dermatitis.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US33907310P | 2010-02-26 | 2010-02-26 | |
| US61/339,073 | 2010-02-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011106779A1 true WO2011106779A1 (en) | 2011-09-01 |
Family
ID=44507258
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/026521 WO2011106779A1 (en) | 2010-02-26 | 2011-02-28 | Use of modified il-4 mutien receptor antagonists to treat dermatitis |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011106779A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2882449A4 (en) * | 2012-08-09 | 2016-03-09 | Univ Leland Stanford Junior | SUPERKINES AND SYNTHÉKINES: REHABILITABLE CYTOKINES HAVING NEW AND IMPROVED SIGNALING ACTIVITIES |
| EP3553078A1 (en) | 2018-04-11 | 2019-10-16 | Julius-Maximilians-Universität Würzburg | Glyco-engineered interleukin-4 based antagonists |
| US11352402B2 (en) | 2013-09-24 | 2022-06-07 | Medicenna Therapeutics, Inc. | Interleukin-4 receptor-binding fusion proteins and uses thereof |
| EP3946412A4 (en) * | 2019-04-05 | 2023-01-04 | Azitra, Inc. | Methods and compositions for treating atopic dermatitis with recombinant microorganisms |
| US11993639B2 (en) | 2012-01-27 | 2024-05-28 | The Board Of Trustees Of The Leland Stanford Junior University | Therapeutic IL-13 polypeptides |
| US12274735B2 (en) | 2017-10-10 | 2025-04-15 | Medicenna Therapeutics Inc. | IL-4-fusion formulations for treatment of central nervous system (CNS) tumors |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090010874A1 (en) * | 2003-08-29 | 2009-01-08 | Aerovance, Inc. | Modified il-4 mutein receptor antagonists |
| US20090104662A1 (en) * | 2000-05-26 | 2009-04-23 | Immunex Corporation | Use of interleukin-4 antagonists and compositions thereof |
-
2011
- 2011-02-28 WO PCT/US2011/026521 patent/WO2011106779A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090104662A1 (en) * | 2000-05-26 | 2009-04-23 | Immunex Corporation | Use of interleukin-4 antagonists and compositions thereof |
| US20090010874A1 (en) * | 2003-08-29 | 2009-01-08 | Aerovance, Inc. | Modified il-4 mutein receptor antagonists |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11993639B2 (en) | 2012-01-27 | 2024-05-28 | The Board Of Trustees Of The Leland Stanford Junior University | Therapeutic IL-13 polypeptides |
| EP2882449A4 (en) * | 2012-08-09 | 2016-03-09 | Univ Leland Stanford Junior | SUPERKINES AND SYNTHÉKINES: REHABILITABLE CYTOKINES HAVING NEW AND IMPROVED SIGNALING ACTIVITIES |
| US9738696B2 (en) | 2012-08-09 | 2017-08-22 | The Board Of Trustees Of The Leland Stanford Junior University | Superkines and synthekines: repurposed cytokines with new and enhanced signaling activities |
| US12187771B2 (en) | 2012-08-09 | 2025-01-07 | The Board Of Trustees Of The Leland Stanford Junior University | Method of producing Th9 T-cells |
| US11352402B2 (en) | 2013-09-24 | 2022-06-07 | Medicenna Therapeutics, Inc. | Interleukin-4 receptor-binding fusion proteins and uses thereof |
| US12274735B2 (en) | 2017-10-10 | 2025-04-15 | Medicenna Therapeutics Inc. | IL-4-fusion formulations for treatment of central nervous system (CNS) tumors |
| EP3553078A1 (en) | 2018-04-11 | 2019-10-16 | Julius-Maximilians-Universität Würzburg | Glyco-engineered interleukin-4 based antagonists |
| WO2019197510A1 (en) | 2018-04-11 | 2019-10-17 | Julius-Maximilians Universität Würzburg | Glyco-engineered interleukin-4 based antagonists |
| EP3946412A4 (en) * | 2019-04-05 | 2023-01-04 | Azitra, Inc. | Methods and compositions for treating atopic dermatitis with recombinant microorganisms |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7227951B2 (en) | Interleukin-2 muteins for proliferation of regulatory T cells | |
| AU2009255994B2 (en) | Use of pegylated Type III Interferons for the treatment of hepatitis C | |
| US7232562B2 (en) | E38N interferon gamma polypeptide variants | |
| WO2011106779A1 (en) | Use of modified il-4 mutien receptor antagonists to treat dermatitis | |
| US20120277143A1 (en) | IL4/IL13 Binding Repeat Proteins and Uses | |
| CA2281794A1 (en) | Antibody fragment-polymer conjugates and humanized anti-il-8 monoclonal antibodies | |
| CA2343094A1 (en) | Interferon-beta fusion proteins and uses | |
| BRPI1006443B1 (en) | FUSION PROTEIN OR PEPTIDE AND METHOD TO INCREASE THE IN VIVO HALF-LIFE OF A PROTEIN OR PEPTIDE | |
| KR20080019619A (en) | Improved Interferon-alpha Polypeptides | |
| EP3431507A1 (en) | Fusion protein comprising nerve growth factor and preparation method and use thereof | |
| CA2250570A1 (en) | Formulations of ob protein | |
| US7230081B1 (en) | Interferon gamma conjugates | |
| MXPA06002285A (en) | Modified il-4 mutein receptor antagonists. | |
| WO2011156000A2 (en) | Use of il-4/il-13 antagonists to treat eosinophilic disorders | |
| CN118679178A (en) | IL10 variants and uses thereof | |
| RU2268749C2 (en) | Interferon-gamma conjugates | |
| JP7707071B2 (en) | Systems and methods for producing collagen 7 compositions | |
| AU782635B2 (en) | Interferon gamma conjugates | |
| US20070009479A1 (en) | Methods for treating dermatitis using mutant human IL-4 compositions | |
| BR112015023145B1 (en) | POLYPEPTIDES CONTAINING AGLYCOSYLATED FC AND ANTIBODY OR FC FUSION PROTEIN |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11748234 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11748234 Country of ref document: EP Kind code of ref document: A1 |