WO2010127152A2 - Compounds and compositions as microsomal prostaglandin e synthase-1 inhibitors - Google Patents
Compounds and compositions as microsomal prostaglandin e synthase-1 inhibitors Download PDFInfo
- Publication number
- WO2010127152A2 WO2010127152A2 PCT/US2010/033022 US2010033022W WO2010127152A2 WO 2010127152 A2 WO2010127152 A2 WO 2010127152A2 US 2010033022 W US2010033022 W US 2010033022W WO 2010127152 A2 WO2010127152 A2 WO 2010127152A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fluorophenyl
- chloro
- imidazole
- imidazol
- carbonitrile
- Prior art date
Links
- 0 CC(*C1=CC=C*)=*C1=S Chemical compound CC(*C1=CC=C*)=*C1=S 0.000 description 30
- SPNAUURQVBIITF-UHFFFAOYSA-N CC(C)(C)c1n[o]c(C(C)(C)C)n1 Chemical compound CC(C)(C)c1n[o]c(C(C)(C)C)n1 SPNAUURQVBIITF-UHFFFAOYSA-N 0.000 description 1
- LOHQDABXBSTGKM-UHFFFAOYSA-N CC(CS)c1cc(cccc2)c2[nH]1 Chemical compound CC(CS)c1cc(cccc2)c2[nH]1 LOHQDABXBSTGKM-UHFFFAOYSA-N 0.000 description 1
- BHNHHSOHWZKFOX-UHFFFAOYSA-N Cc1cc2ccccc2[nH]1 Chemical compound Cc1cc2ccccc2[nH]1 BHNHHSOHWZKFOX-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N OC(c1ccccc1)=O Chemical compound OC(c1ccccc1)=O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- mPGES-1 microsomal prostaglandin E synthase-1
- Inflammation is a common cause of pain, with inflammatory pain resulting from a variety of causes, such as infection, surgery or other trauma.
- Other inflammatory diseases include asthma, chronic obstructive pulmonary disease (COPD), inflammatory bowel disease, rheumatoid arthritis, osteoarthritis, rhinitis, conjunctivitis and dermatitis.
- COX chronic obstructive pulmonary disease
- PGs prostaglandins
- TX A2 thromboxane
- COX cyclooxygenase
- the cyclooxygenase (COX) enzyme activity originates from two distinct and independently regulated isozymes: COX-I which is constitutively expressed in many cells and tissues and catalyzes the formation of prostaglandins that subserve housekeeping functions, such as the maintenance of gastrointestinal (GI) integrity; and COX-2 which is induced by pro-inflammatory stimuli, such as cytokines, during an inflammatory response and is the dominant source of prostaglandins which mediate pain and inflammation.
- GI gastrointestinal
- COX-2 which is induced by pro-inflammatory stimuli, such as cytokines, during an inflammatory response and is the dominant source of prostaglandins which mediate pain and inflammation.
- These enzymes metabolize arachidonic acid to the unstable intermediate prostaglandin H 2 (PGH 2 ).
- PGH 2 is further metabolized to other prostaglandins including PGE 2 , PGF 2 CC, PGD 2 , prostacyclin and thromboxane A 2 . These arachidonic acid metabolites are known to have pronounced physiological and pathophysiological activity including pro-inflammatory effects.
- Prostaglandin H 2 (PGH 2 ) is transformed to PGE 2 by prostaglandin E synthases (PGES).
- PGES prostaglandin E synthases
- Three prostaglandin E synthases (PGES) are known: two microsomal prostaglandin E synthases (mPGES-1 and mPGES-2), and one cytosolic prostaglandin E synthase (cPGES).
- Microsomal prostaglandin E synthase-1 belongs to the membrane- associated proteins in the eicosanoid and glutathione metabolism (MAPEG) family. Other members of this family include the microsomal glutathione S-transferases (MGSTl, MGST2 and MGST3).
- PGE 2 in particular is a strong pro-inflammatory mediator, and is known to induce fever and pain. PGE 2 is also involved in arthritis and inflammation. The inhibition of PGE 2 formation by inhibiting the COX enzymes underlies the effectiveness of a variety of antiinflammatory drugs.
- numerous drugs including non-steroidal anti-inflammatory drugs (NSAIDs) and coxibs (selective COX-2 inhibitors), have been developed for inhibiting the formation of PGE 2 .
- NSAIDs non-steroidal anti-inflammatory drugs
- coxibs selective COX-2 inhibitors
- Such drugs act predominantly by inhibition of COX-I and/or COX-2, thereby reducing the formation of PGE 2 .
- the inhibition of COX-I and/or COX-2 also results in the reduction of the formation of all metabolites of arachidonic acid, some of which are known to have beneficial properties.
- PGI 2 prostacyclin synthase
- PGI 2 the dominant product of arachidonic acid in macrovascular endothelial cells, is formed by prostacyclin synthase (PGIS) action on prostaglandin endoperoxide intermediates, which are produced catalytically by COX-2.
- PGIS prostacyclin synthase
- PGI 2 exhibits properties of potential relevance to atheroprotection by inhibiting platelet aggregation, vascular smooth muscle contraction and proliferation, leukocyte-endothelial cell interactions and cholesteryl ester hydrolase. PGI 2 also activates reverse cholesterol transport.
- PGI 2 protects against oxidant- induced tissue injury.
- IP PGI 2 receptor
- PGI 2 also limits the cardiovascular effects of thromboxane A 2 (TxA 2 ), the major PGHS-I product of platelets.
- TxA 2 thromboxane A 2
- the cardiovascular effects of TxA 2 include: platelet aggregation, elevation of blood pressure and acceleration of atherogenesis.
- Inhibition of the transformation of PGH 2 to the pro-inflammatory mediator PGE 2 might be expected to reduce the inflammatory response, in the absence of a corresponding reduction of the formation of other, beneficial arachidonic acid metabolites, and thereby alleviate the undesirable side-effects mentioned above.
- agents that are capable of inhibiting the action of in PGES in particular inhibitors of microsomal prostaglandin E synthase-1 (mPGES-1), and thus reducing the formation of the specific arachidonic acid metabolite PGE 2 , are likely to be of benefit in the treatment of inflammation and pain.
- mPGES-1 microsomal prostaglandin E synthase-1
- mPGES-1 useful inhibitors of microsomal prostaglandin E synthase-1
- methods of using such compound and pharmaceutical compositions for treating diseases or disorders associated with mPGES-1 activity are also provided herein.
- such compounds and the pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, have a structure according to Formula (I): wherein, each R 1 is independently selected from H, CrC ⁇ alkyl, or -(CH 2 XC(O)ORg, ;
- R 2 is H, Ci-Cealkyl, aryl, halogen, -CN, -C(O)OR 1 or -C(O)NR 1 R 1 ;
- R 3 is -CN, CrCealkylA-C ⁇ haloalkyl or halogen
- R 4 is H, -CN, Ci-Cealkyl , CrCehaloalkyl or halogen;
- R 5 is H, halogen, CrC ⁇ alkyl, CrC ⁇ alkoxy or 4-6 membered heterocycloalkyl containing
- L 1 is a bond, C 2 -C 6 alkenylene, C 2 -Coalkynylene, -C(O)NRs-,
- X 1 is CR 15 or N
- X 2 is O, S or NR 8 ;
- X 3 is S, O or NR 8 ;
- X 4 is CR 15 or N
- X 5 is CR 15 or N; each Re and each R 7 are independently selected from H, halogen, -CN, CrCealkyl, Ci.Cehaloalkyl, -OQ-Cealkyl, -OCi.Cehaloalkyl, -Q 2 , -C(O)OR 8 , -C(O)N(Rg) 2 , -NR 8 C(O)R 8 , -N(Rg) 2 , -S(O) 2 R 8 , - NR 8 C(O)Q 2 and -OR 8 ; each R 8 is independently selected from H and CrC ⁇ alkyl; each R 45 is independently selected from H, halogen, -CN, CrC ⁇ alkyl, Ci-C ⁇ haloalkyl, -OCi.C ⁇ alkyl, -OCi.C ⁇ haloalkyl, -Q 2 , -C(O)OR 8 , -C(O)N(Rg)
- heteroatoms independently selected from N, O and S, wherein:
- X 6 is C(R 9 ) 2 , NR 9 , O or S; X 7 is N or CR 9 ; X 8 is CR 9 , NR 9 , O or S; X 9 is N or CR 9 ; X 10 is S or O; X 11 is CR 9 or N; each R 9 is independently H or Ci-C ⁇ alkyl; and wherein the C 2 -C 6 alkenylene, C 2 -Coalkynylene, arylenes and heterocycloalkylenes of L 2 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, CN, -OC 1 - C ⁇ alkyl, halogen and OH; Q 1 is phenyl, Cioaryl, C 14 aryl, 5-6 membered heteroaryl containing 1-2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S
- the phenyl, aryls, heteroaryls, heterocycloalkyls and C 3 -Cgcycloalkyls of Qi are optionally substituted with 1-4 substituents independently selected from Q.C ⁇ alkyl, Q.C ⁇ haloalkyl, halogen, -CN, -Q 2 , -0(CRioRii)i- 4 ORio, -O(CRi 0 Rn)i- 4 D , -0(CRI O RII)I- 4 RI O , -OCi_C 6 alkyl, -OCi_C 6 haloalkyl, -C(O)ORi 4 , -N(Rw) 2 , -C(O)N(Ri4) 2 , - NRi 4 C(O)Ri 4 , -NRi 4 C(O)Q 2 , -S(O) 2 Ri 4 , -SRi 4 and -0R
- Q 2 is phenyl, Cioaryl, Ci 4 aryl, 5-6 membered heteroaryl containing 1- 2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S or C 3 -C 8 cycloalkyl, each of which is optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, -CN, - N(Rw) 2 , -OCi_C 6 alkyl, halogen and -OH;
- L 3 is C 1 -C ⁇ alkylene, C 2 -C ⁇ alkenylene or C 2 -C ⁇ alkynylene;
- X 12 is NR 12 , S or O;
- X 13 is (CH 2 ) q or O(CH 2 ) P ;
- R 12 is H, Ci-Cealkyl or -C(O)OR 14 ;
- the phenyl, aryls, heteroaryls, heterocycloalkyls and C 3 -Cgcycloalkyls of Q 1 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, halogen, -CN, -Q 2 , -OCCRIORIOMORIO, -0(CRi 0 RiI)I -4 D , -0(CR 10 Rii)i- 4 Rio, -OCi-Cealkyl, -OCi-C ⁇ haloalkyl, -C(O)OR 14 , -N(R 14 ) 2 , -C(O)N(R 14 ) 2 , - NR 14 C(O)R 14 , -NR 14 C(O)Q 2 , -S(O) 2 R 14 , -SR 14 and -OR 14 , and provided that when the combination of L 1 ,
- each R 1 is independently selected from H and Ci-C ⁇ alkyl
- R 2 is H, halogen, -CN, -C(O)OR 1 or -C(O)NR 1 R 1
- R 3 is Ci-C ⁇ haloalkyl or halogen
- R 4 is H, CrC ⁇ haloalkyl or halogen
- X 1 is CR 15 or N;
- X 2 is O, S or NR 8 ;
- X 3 is S, O or NR 8 ;
- X 4 is CR 15 or N
- X 5 is CR 15 or N; each Re and each R 7 are independently selected from H, halogen,
- L 2 is a bond, C 2 -C 6 alkynylene, phenylene, -0-, -C(O)-, -C(O)NR 8 -, -
- X 6 is C(R 9 ) 2 , NR 9 , O or S; X 7 is N or CR 9 ; X 8 is CR 9 , NR 9 , O or S; X 9 is N or CR 9 ; X 11 is CR 9 or N; each R 9 is independently H or CrC ⁇ alkyl; and wherein the C 2 -C 6 alkynylene and phenylene of L 2 are each optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, CN, -OCi-C ⁇ alkyl, halogen and OH;
- the phenyl, heteroaryl, C 2 -C 6 heterocycloalkyl and Cs-Cscycloalkyl groups of Q 1 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, halogen, -CN, -Q 2 , -O(CRi 0 Rn)i- 4 ORi 0 , -O(CRi 0 Rn)i- 4 D , - 0(CRioRii)i-4Rio, -OCi-C ⁇ alkyl, -OCi-C ⁇ haloalkyl, -C(O)OR 14 , -N(R 14 ) 2 , -C(O)N(R 14 ) 2 , - NR 14 C(O)R 14 , -NR 14 C(O)Q 2 , - S(O) 2 R 14 , -SR 14 and -OR 14 ; each R 10 and each R 11
- Q 2 is phenyl, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S or C 3 - Cgcycloalkyl, each of which is optionally substituted with 1-4 substituents independently selected from halogen and C 1- C 6 haloalkyl;
- X 12 is NR 12 , S or O;
- Xi 3 is -(CH 2 ),- or -O(CH 2 ) P -;
- R 12 is H, Ci-Cealkyl or -C(O)OR 14 ; each R 14 is independently selected from H and C 1 -C ⁇ alkyl; n is O, 1, 2 or 3; p is O, 1, 2 or 3; q is 1, 2 or 3; s is 0 or 1, provided that when the combination of L 1 , L 2 and Q 1 is, L 1 is pyridyl and L 2 is
- R 2 is -CN, -C(O)OR 1 or -C(O)NR 1 R 1
- L 2 is a bond, -O-, -C(O)-, C 2 alkynylene, phenylene,
- phenyl, pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl of Q 1 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, halogen, -CN, -Q 2 , -0(CRioRn)i- 4OR10, -0(CRioR 11 ) 1 - 4 D , -0(CRiOR 11 )WR 1 O, -OCi_C 6 alkyl, -OCi_C 6 haloalkyl, -C(O)ORi 4 , N(Rw) 2 , -C(O)N(RM) 2 , - -
- Li is or anc j Q 1 j s phenyl optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, halogen, -CN, -Q 2 , -0(CRioRn)i-4 ⁇ Rio, -O(CRi 0 Rn)wD , -0(CRi 0 Rn)i- 4 Rio, -OCi_C 6 alkyl, -OCi- C ⁇ haloalkyl, -C(O)OR 14 , -N(Rw) 2 , -C(O)N(R 14 ) 2 , - NR 14 C(O)R 145 -NR 14 C(O)Q 2 , - S(O) 2 R 14 , -SR 14 and -OR 14 .
- L 1 is , L 2 is C 2 alkynylene and Q 1 is phenyl optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, C 1- C ⁇ haloalkyl, halogen, -CN, -Q 2 , -O(CRi 0 R 11 ) 1 - 4 ORi 0 , -O(CRi 0 R 11 ) 1 - 4 D , -0(CRi 0 R 1 i)i- 4 Rio, - OCi-C ⁇ alkyl, -OCi-Cehaloalkyl, -C(O)ORi 4 , -N(Rw) 2 , -C(O)N(Rw) 2 , - NRi 4 C(O)Rw, - NRwC(O)Q 2 , -S(O) 2 Rw, -SRw and -ORi 4 .
- Li is or , L 2 is a bond and Qi is phenyl optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, halogen, -CN, -Q 2 , -0(CRi 0 Ri i)i- 4 ORio, -O(CRi 0 Rn)i_ 4 D , -O(CRi 0 Rn)i_ 4 Ri 0 , -OCi_C 6 alkyl, -OCi_C 6 haloalkyl, -C(O)ORw, -N(Rw) 2 , -C(O)N(Rw) 2 , - NRWC(O)RW 5 -NRWC(O)Q 2 , - S(O) 2 Rw, -SRw and -ORw.
- pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl of Qi are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-Cehaloalkyl, halogen, -CN, -Q 2 , -0(CRi 0 Rii)i- 4 ORio, -O(CRi 0 Rn)i- 4 D , -O(CRi 0 Rn)i- 4R10, -OCi-C ⁇ alkyl, -OCi.C 6 haloalkyl, -C(O)ORi 4 , -N(Rw) 2 , -C(O)N(Ri 4 ) 2 , - NR
- phenyl, pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl of Q 1 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, C 1 .
- each R 1 is independently selected from H, methyl and ethyl.
- pharmaceutically acceptable solvates e.g. hydrates
- R 2 is H, Br, -CN, -C(O)OR 1 or -C(O)NR 1 R 1 ; provided that when the combination of L 1 , L 2 and Q 1 is, L 1 is pyridyl and L 2 is C 2 alkynylene and Q 1 is phenyl, pyridyl, thiazolyl, cyclohexyl or cyclopropyl, then R 2 is -CN, -C(O)OR 1 or -C(O)NR 1 R 1 .
- R 3 is -CF 3 , Cl or F.
- pharmaceutically acceptable solvates e.g. hydrates
- R 4 is H, -CF 3 , Cl or F.
- pharmaceutically acceptable solvates e.g. hydrates
- Q 2 is phenyl, 5 optionally substituted with 1-4 halogen groups.
- Certain embodiments of such compounds of Formula (Ia) are selected from: 2- (2- chloro-6-fluorophenyl)-5-[6-(2-phenylethynyl)pyridin-3-yl]-lH-imidazole-4-carbonitrile; 2- (2-chloro-6-fluorophenyl)-5-[6-(2-phenylethynyl)pyridin-3-yl]-lH-imidazole-4- carboxamide; ethyl 2-(2-chloro-6-fluorophenyl)-4-[6-(2-phenylethynyl)pyridin-3-yl]- IH- imidazole-5-carboxylate; 2-(2-chloro-6-fluorophenyl)-5-[6-(2-phenylethynyl)pyridin-3-yl]- lH-imidazole-4-carboxylic acid; 2-(2-chloro-6-fluorophenyl)
- Another aspect provided herein is a method for treating a disease or disorder associated with mPGES-1 activity, wherein the method comprises administering to a subject in need thereof a therapeutically effective amount of a compound of Formula (Ia).
- the diseases or disorder is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease.
- the compound of Formula (Ia) is an inhibitor of mPGES-1.
- Another aspect provided herein is the use of a compound of Formula (I) in the manufacture of a medicament for treating a disease or disorder associated with mPGES-1 activity, wherein the disease or disorder is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease.
- Another aspect provided herein is a medicament for treating a disease or disorder associated with mPGES-1 activity, wherein the medicament comprises a therapeutically effective amount of a compound of Formula (Ia).
- the disease or disorder associated with mPGES-1 activity is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease.
- a compound of Formula (Ia) for use in a method of medical treatment wherein the method of medical treatment is for treating a disease or disorder associated with mPGES-1 activity, wherein the disease or disorder is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease.
- mPGES-1-associated diseases such as pain, including but not limited to, inflammatory pain, dental pain, postdental procedure pain, osteoarthritic pain, nociceptive pain, neuropathic pain muscoskeletal pain, low back pain, headaches, sinus headaches, migraines, post-surgical pain, surgical pain, burn injury and pain associated with bacterial, fungal or viral illnesses (by way of example only, influenza, the common cold, a viral infections (such as, by way of example only, influenza, common cold, herpes zoster, hepatitis C and AIDS), a bacterial infection or a fungal infection) and periodontal disease; inflammatory diseases, inflammation, arthritis, rheumatoid arthritis (RA), juvenile arthritis, inflammatory arthritis, arthritis, atherosis, tendonitis, bursitis, gouty arthritis, poly
- alkene or "alkenyl”, as used herein, refers to a partially unsaturated branched or straight chain hydrocarbon having at least one carbon-carbon double bond. Atoms oriented about the double bond are in either the cis (Z) or trans (E) conformation.
- C 2 -C 4 alkenyl refers to an alkenyl group containing at least 2, and at most 4, 5, 6, 7 or 8 carbon atoms, respectively.
- alkenyl groups include ethenyl, propenyl, allyl (2-propenyl), butenyl, pentenyl, hexenyl, heptenyl, octenyl, nonenyl, decenyl and the like. In certain embodiments an alkene or alkenyl group is optionally substituted.
- alkenylene refers to a saturated branched or straight chain divalent hydrocarbon radical derived from an alkenyl group. In certain embodiments an alkenylene group is optionally substituted.
- C 2 -C 3 alkenylene refers to an alkenylene group containing at least 2, and at most 3, 4, 5, 6, 7 or 8 carbon atoms respectively.
- alkenylene groups as used herein include, ethenlene, n-propenylene, isopropenylene, n-butenylene, isobutenylene, sec- butenylene, t-butenylene, n-pentenylene, isopentenylene, hexenylene and the like.
- alkyl refers to a saturated branched or straight chain hydrocarbon. An alkyl group can be optionally substituted.
- C 1 - C 3 alkyl refers to an alkyl group containing at least 1, and at most 3, 4, 5, 6, 7 or 8 carbon atoms, respectively.
- alkyl groups as used herein include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec -butyl, t-butyl, n-pentyl, isopentyl, hexyl, heptyl, octyl, nonyl, decyl and the like.
- an alkyl group is optionally substituted.
- alkylene refers to a saturated branched or straight chain divalent hydrocarbon radical derived from an alkyl group. In certain embodiments an alkylene group is optionally substituted.
- CrCsalkylene refers to an alkylene group containing at least 1, and at most 3, 4, 5, 6, 7 or 8 carbon atoms respectively.
- alkylene groups include, methylene, ethylene, n-propylene, isopropylene, n-butylene, isobutylene, sec-butylene, t-butylene, n- pentylene, isopentylene, hexylene and the like.
- alkynyl refers to a partially unsaturated branched or straight chain hydrocarbon radical having at least one carbon-carbon triple bond.
- C 2 -C 4 alkynyl refers to an alkynyl group containing at least 2, and at most 4, 5, 6, 7 or 8 carbon atoms, respectively.
- alkynyl groups include ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, octynyl, nonynyl, decynyl and the like.
- an alkynyl group is optionally substituted.
- alkynylene refers to a saturated branched or straight chain divalent hydrocarbon radical derived from an alkynyl group. In certain embodiments an alkynylene group is optionally substituted.
- C 2 -C 3 alkynylene refers to an alkynylene group containing at least 2, and at most 3, 4, 5, 6, 7 or 8 carbon atoms respectively.
- alkenylene groups include, ethynlene, propynylene, butynylene, pentynylene, hexynylene and the like.
- alkoxy refers to the group -OR a , where R a is an alkyl group as defined herein.
- R a is an alkyl group as defined herein.
- An alkoxy group can be optionally substituted.
- the terms "Ci-C 3 alkoxy”, “C 1 -C 4 alkoxy”, “Ci-C 5 alkoxy”, “Ci-C 6 alkoxy”, “Ci-C 7 alkoxy” and "Ci-Cgalkoxy” refer to an alkoxy group wherein the alkyl moiety contains at least 1, and at most 3, 4, 5, 6, 7 or 8, carbon atoms.
- Non-limiting examples of alkoxy groups include methoxy, ethoxy, n-propoxy, isopropoxy, n-butyloxy, t-butyloxy, pentyloxy, hexyloxy, heptyloxy, octyloxy, nonyloxy, decyloxy and the like.
- aryl refers to monocyclic, fused bicyclic, and fused tricyclic ring systems having a total of six to fourteen ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains 3 to 7 ring members. In certain embodiments an aryl group is optionally substituted.
- arylene as used means a divalent radical derived from an aryl group. In certain embodiments an arylene group is optionally substituted.
- cyano refers to a -CN group.
- cycloalkyl refers to a saturated or partially unsaturated, monocyclic, fused bicyclic, fused tricyclic or bridged polycyclic ring assembly.
- C 3 -C 5 cycloalkyl refers to a saturated or partially unsaturated, monocyclic, fused bicyclic, fused tricyclic or bridged polycyclic ring assembly.
- C 3 -C 5 cycloalkyl refers to a saturated or partially unsaturated, monocyclic, fused bicyclic, fused tricyclic or bridged polycyclic ring assembly.
- C 3 -C 5 cycloalkyl refers to a saturated or partially unsaturated, monocyclic, fused bicyclic or bridged polycyclic ring assembly contain at least 3, and at most 5, 6, 7, 8, 9 or 10, carbon atoms.
- a cycloalkyl group is optionally substituted.
- cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cyclopentenyl, cyclohexenyl, decahydronaphthalenyl, 2,3,4,5,6,7-hexahydro-lH-indenyl and the like.
- cycloalkylene as used means a divalent radical derived from a cycloalkyl group.
- a cycloalkylene group is optionally substituted.
- halogen refers to fluorine (F), chlorine (Cl), bromine (Br), or iodine (I).
- halo refers to the halogen radicals: fluoro (-F), chloro (- Cl), bromo (-Br), and iodo (-1).
- haloalkyl or halo-substituted alkyl,” as used herein, refers to an alkyl group as defined herein, substituted with one or more halogen groups, wherein the halogen groups are the same or different.
- a haloalkyl group can be optionally substituted.
- Non- limiting examples of such branched or straight chained haloalkyl groups include methyl, ethyl, propyl, isopropyl, isobutyl and n-butyl substituted with one or more halogen groups, wherein the halogen groups are the same or different, including, but not limited to, trifluoromethyl, pentafluoroethyl, and the like.
- halo-substituted alkenyl refers to an alkenyl group as defined above, substituted with at least one halo group or combinations thereof.
- Non- limiting examples of such branched or straight chained haloalkenyl groups, as used herein, include ethenyl, propenyl, allyl (2-propenyl), butenyl, pentenyl, hexenyl, heptenyl, octenyl, nonenyl, decenyl and the like substituted with one or more halo groups or combinations thereof.
- halo-substituted alkynyl refers to an alkynyl group as defined above, substituted with at least one halo group or combinations thereof.
- Non- limiting examples of such branched or straight chained haloalkynyl groups, as used herein, include ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, octynyl, nonynyl, decynyl and the like substituted with one or more halo groups or combinations thereof.
- haloalkoxy or "halo-substituted -alkoxy,” as used herein, refers to an alkoxy group as defined herein, substituted with one or more halogen groups, wherein the halogen groups are the same or different.
- a haloalkoxy group can be optionally substituted.
- Non-limiting examples of such branched or straight chained haloalkynyl groups include methoxy, ethoxy, n-propoxy, isopropoxy, n-butyloxy, t-butyloxy, pentyloxy, hexyloxy, heptyloxy, octyloxy, nonyloxy, decyloxy and the like, substituted with one or more halogen groups, wherein the halogen groups are the same or different.
- heteroalkyl refers to an alkyl group as defined herein wherein one or more carbon atoms are independently replaced by one or more of oxygen, sulfur, nitrogen, or combinations thereof.
- heteroaryl refers to monocyclic, fused bicyclic, and fused tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic, at least one ring in the system contains one or more heteroatoms selected from nitrogen, oxygen and sulfur, and wherein each ring in a multi-ring system contains 3 to 7 ring members.
- a heteroaryl group may contain one or more substituents. In certain embodiments a heteroaryl group is optionally substituted.
- heteroaryl groups include benzofuranyl, benzofurazanyl, benzoxazolyl, benzopyranyl, benzthiazolyl, benzothienyl, benzazepinyl, benzimidazolyl, benzothiopyranyl, benzo[l,3]dioxole, benzo[b]furyl, benzo[b]thienyl, cinnolinyl, furazanyl, furyl, furopyridinyl, imidazolyl, indolyl, indolizinyl, indolin-2-one, indazolyl, isoindolyl, isoquinolinyl, isoxazolyl, isothiazolyl, 1,8-naphthyridinyl, oxazolyl, oxaindolyl, oxadiazolyl, pyrazolyl, pyrrolyl, phthal
- a heterocycloalkyl group is optionally substituted.
- heterocycloalkyl groups include morpholino, pyrrolidinyl, pyrrolidinyl-2-one, piperazinyl, piperidinyl, piperidinylone, l,4-dioxa-8-aza- spiro[4.5]dec-8-yl, 2H-pyrrolyl, 2-pyrrolinyl, 3-pyrrolinyl, 1,3-dioxolanyl, 2-imidazolinyl, imidazolidinyl, 2-pyrazolinyl, pyrazolidinyl, 1,4-dioxanyl, 1,4-dithianyl, thiomorpholinyl, azepanyl, hexahydro-l,4-diazepinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl,
- terai refers to one or more of oxygen, sulfur, nitrogen, phosphorus, or silicon.
- heterocycloalkylene refers to a divalent radical derived from a heterocycloalkyl group. In certain embodiments a heterocycloalkylene group is optionally substituted.
- hydroxyl refers to the group -OH.
- hydroxyalkyl or hydroxyl-substituted-alkyl, as used herein, refers to an alkyl group as defined herein substituted with one or more hydroxyl group.
- Non-limiting examples of branched or straight chained "C 1 -C O hydroxyalkyl groups as used herein include methyl, ethyl, propyl, isopropyl, isobutyl and n-butyl groups substituted with one or more hydroxyl groups.
- optionally substituted means that the referenced group may or may not be substituted with one or more additional group(s) individually and independently selected from alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocycloalkyl, hydroxyl, alkoxy, mercaptyl, cyano, halo, carbonyl, thiocarbonyl, isocyanato, thiocyanato, isothiocyanato, nitro, perhaloalkyl, perfluoroalkyl, and amino, including mono- and di- substituted amino groups, and the protected derivatives thereof.
- solvate refers to a complex of variable stoichiometry formed by a solute (by way of example, a compound of Formula (I), or a salt thereof, as described herein) and a solvent.
- a solvent are water, acetone, methanol, ethanol and acetic acid.
- administering means providing a compound of Formula (I), a pharmaceutically acceptable salt, a pharmaceutically acceptable solvate, or prodrug thereof to a subject in need of treatment.
- agent includes any substance, molecule, element, compound, entity, or a combination thereof. It includes, but is not limited to a protein, a polypeptide, a small organic molecule, a polysaccharide, a polynucleotide, and the like. It can be a natural product, a synthetic compound, or a chemical compound, or a combination of two or more substances. Unless otherwise specified, the terms "agent”,
- carrier refers to chemical compounds or agents that facilitate the incorporation of a compound described herein into cells or tissues.
- co-administration or “combined administration” or the like as used herein are meant to encompass administration of the selected therapeutic agents to a single patient, and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time.
- dilute a compound described herein prior to delivery refers to chemical compounds that are used to dilute a compound described herein prior to delivery. Diluents can also be used to stabilize compounds described herein.
- an "effective amount" for therapeutic uses is the amount of the composition comprising a compound as disclosed herein required to provide a clinically significant decrease in disease symptoms.
- An appropriate "effective" amount in any individual case may be determined using techniques, such as a dose escalation study.
- the terms “enhance” or “enhancing,” as used herein, means to increase or prolong either in potency or duration a desired effect.
- the term “enhancing” refers to the ability to increase or prolong, either in potency or duration, the effect of other therapeutic agents on a system.
- An “enhancing- effective amount,” as used herein, refers to an amount adequate to enhance the effect of another therapeutic agent in a desired system.
- iatrogenic means a condition, disorder, or disease created or worsened by medical or surgical therapy.
- inflammation includes any condition characterized by a localized or a systemic protective response, which may be elicited by physical trauma, infection, chronic diseases, such as those mentioned herein, and/or chemical and/or physiological reactions to external stimuli (for example, as part of an allergic response). Any such response, which may serve to destroy, dilute or sequester both the injurious agent and the injured tissue, may be manifest by, for example, heat, swelling, pain, redness, dilation of blood vessels and/or increased blood flow, invasion of the affected area by white blood cells, loss of function and/or any other symptoms known to be associated with inflammatory conditions.
- inflammation also includes any inflammatory disease, disorder or condition per se, any condition that has an inflammatory component associated with it, and/or any condition characterized by inflammation as a symptom, including inter alia acute, chronic, ulcerative, specific, allergic and necrotic inflammation, and other forms of inflammation known to those skilled in the art.
- the term also includes inflammatory pain, pain generally and/or fever.
- inflammatory disorders refers to those diseases or conditions that are characterized by one or more of the signs of pain (dolor, from the generation of noxious substances and the stimulation of nerves), heat (calor, from vasodilatation), redness (rubor, from vasodilatation and increased blood flow), swelling (tumor, from excessive inflow or restricted outflow of fluid), and loss of function (functio laesa, which may be partial or complete, temporary or permanent).
- Inflammation takes many forms and includes, but is not limited to, inflammation that is one or more of the following: acute, adhesive, atrophic, catarrhal, chronic, cirrhotic, diffuse, disseminated, exudative, fibrinous, fibrosing, focal, granulomatous, hyperplastic, hypertrophic, interstitial, metastatic, necrotic, obliterative, parenchymatous, plastic, productive, proliferous, pseudomembranous, purulent, sclerosing, seroplastic, serous, simple, specific, subacute, suppurative, toxic, traumatic, and/or ulcerative.
- Inflammatory disorders further include, without being limited to those affecting the blood vessels (polyarteritis, temporarl arteritis); joints (arthritis: crystalline, osteo-, psoriatic, reactive, rheumatoid, Reiter's); gastrointestinal tract; skin (dermatitis); or multiple organs and tissues (systemic lupus erythematosus).
- the terms "inhibiting” or “inhibition,” as used herein in the context of modulation of enzymatic activities, inhibition relates to reversible suppression or reduction of an enzymatic activity including competitive, uncompetitive, and noncompetitive inhibition.
- pharmaceutically acceptable refers a material, such as a carrier or diluent, which does not abrogate the biological activity or properties of the compounds described herein. Such materials are administered to an individual without causing undesirable biological effects or interacting in a deleterious manner with any of the components of the composition in which it is contained.
- pharmaceutically acceptable salt refers to a formulation of a compound that does not cause significant irritation to an organism to which it is administered and does not abrogate the biological activity and properties of the compounds described herein.
- prevent refers to a complete inhibition of development of primary or secondary tumors or any secondary effects of disease.
- the terms “combination” or “pharmaceutical combination,” as used herein mean a product that results from the mixing or combining of more than one active ingredient and includes both fixed and non-fixed combinations of the active ingredients.
- the term “fixed combination” means that the active ingredients, by way of example, a compound of Formula (I) and an additional therapeutic agent, are both administered to a patient simultaneously in the form of a single entity or dosage.
- the term “non-fixed combination” means that the active ingredients, by way of example, a compound of Formula (I) and an additional therapeutic agent, are both administered to a patient as separate entities either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the 2 compounds in the body of the patient.
- cocktail therapy e.g. the administration of 3 or more active ingredients.
- composition refers to a mixture of at least one compound of Formula (I) described herein with other chemical components, such as carriers, stabilizers, diluents, dispersing agents, suspending agents, thickening agents, and/or excipients.
- prodrug refers to an agent that is converted into the parent drug in vivo.
- a non-limiting example of a prodrug of the compounds described herein is a compound described herein administered as an ester which is then metabolically hydrolyzed to a carboxylic acid, the active entity, once inside the cell.
- a further example of a prodrug is a short peptide bonded to an acid group where the peptide is metabolized to reveal the active moiety.
- subject or "patient”, as used herein, encompasses mammals and non- mammals.
- mammals include, but are not limited to, humans, chimpanzees, apes, monkeys, cattle, horses, sheep, goats, swine; rabbits, dogs, cats, rats, mice, guinea pigs, and the like.
- non-mammals include, but are not limited to, birds, fish and the like.
- terapéuticaally effective amount refers to any amount of a compound which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder.
- the term also includes within its scope amounts effective to enhance normal physiological function.
- treat refers to methods of alleviating, abating or ameliorating a disease or condition symptoms, preventing additional symptoms, ameliorating or preventing or delaying the underlying metabolic causes of symptoms, inhibiting the disease or condition, arresting the development of the disease or condition, relieving the disease or condition, causing regression of the disease or condition, relieving a condition caused by the disease or condition, or stopping the symptoms of the disease or condition either prophylactically (prevent or delay the onset of the disease, or to prevent the manifestation of clinical or subclinical symptoms thereof) and/or therapeutically.
- mPGES-1 microsomal prostaglandin E synthase-1
- mPGES-2 microsomal prostaglandin E synthase-1
- GSTM2-2 mPGES-2
- GSTM3-3 mPGES/p23
- cPGES/p23 cPGES/p23
- such compounds, pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, and such pharmaceutical compositions provided herein are inhibitors of microsomal prostaglandin E synthase-1 (mPGES-1).
- prostaglandin E synthases inhibitors are compounds having the structure of Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof:
- each R 1 is independently selected from H, Ci-C ⁇ alkyl, or -(CH 2 XC(O)ORg, ;
- R 2 is H, Ci-Cealkyl, aryl, halogen, -CN, -C(O)OR 1 or -C(O)NR 1 R 1 ;
- R 3 is -CN, Ci-CealkylA-Cehaloalkyl or halogen;
- R 4 is H, -CN, Ci-Cealkyl , d-Cehaloalkyl or halogen;
- R 5 is H, halogen, Ci-C ⁇ alkyl, Ci-C ⁇ alkoxy or 4-6 membered heterocycloalkyl containing
- L 1 is a bond, C 2 -C6alkenylene, C 2 -Coalkynylene, -C(O)NRs-,
- X 1 is CR 15 or N;
- X 2 is O, S or NR 8 ;
- X 3 is S, O or NR 8 ;
- X 4 is CR 15 or N
- X 5 is CR 15 or N; each Re and each R 7 are independently selected from H, halogen, -CN, Ci-C 6 alkyl, Ci-Cehaloalkyl, -OCi-C 6 alkyl, -OCi-Cehaloalkyl, -Q 2 , -C(O)OR 8 , -C(O)N(Rg) 2 , -NR 8 C(O)R 8 , -N(Rg) 2 , -S(O) 2 R 8 , - NR 8 C(O)Q 2 and -OR 8 ; each R 8 is independently selected from H and Ci-C ⁇ alkyl; each R 15 is independently selected from H, halogen, -CN, Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, -OCi_C 6 alkyl, -OCi-Cehaloalkyl, -Q 2 , -C(O)OR 8 , -C(O)N(
- heteroatoms independently selected from N, O and S, wherein:
- X 6 is C(R 9 ) 2 , NR 9 , O or S; X 7 is N or CR 9 ; X 8 is CR 9 , NR 9 , O or S; X 9 is N or CR 9 ; Xio is S or O; Xn is CR 9 or N; each R 9 is independently H or Ci-C ⁇ alkyl; and wherein the C 2 -C 6 alkenylene, C 2 -Coalkynylene, arylenes and heterocycloalkylenes of L 2 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, CN, -OC 1 -
- cyclopropyl L 3 Q 2 , -(CR 10 Rn) n Q 2 , wherein: the phenyl, aryls, heteroaryls, heterocycloalkyls and C 3 -C 8 cycloalkyls of Q 1 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, halogen, -CN, -Q 2 , -OCCRIORIOMORIO, -OCCRIORIOMD , -0(CR 10 RII)I- 4 RIO, -OCi-Cealkyl, -OCi-C ⁇ haloalkyl, -C(O)OR 14 , -N(R 14 ) 2 , -C(O)N(R 14 ) 2 , - NR 14 C(O)R 14 , -NR 14 C(O)Q 2 , -S(O) 2 R 14 , -SR 14 and -
- X 2 is O or NRg
- X 3 is S or N.
- L 1 is a bond, -C(O)NH-
- L 2 is a a bond, -O-,
- L 2 is a bond, -O-, -C(O)-,
- L 2 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl and halogen.
- L 1 is a C 2 -C 6 alkynylene
- Q 1 is phenyl, pyridyl, C 2 -
- phenyl, pyridyl, C 2 - Coheterocycloalkyl and C 3 -C 6 cycloalkyl of Q 1 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, -CN, -Q 2 , -OC 1- C ⁇ alkyl, -OCi-Cehaloalkyl, -C(O)ORi 4 , -C(O)N(Rw) 2 , -S(O) 2 Ri 4 , halogen, - NRi 4 C(O)Ri 4 ,
- Ri is H, Ci-C ⁇ alkyl, or - (CH 2 XC(O)OR 8 ;
- R 2 is H, d-C 6 alkyl, halogen, -CN, -C(O)ORi or -C(O)NRiRi;
- R 3 and R 4 are independently selected from Ci-C ⁇ alkyl and halogen;
- R 5 is H, Ci-C ⁇ alkyl or C 2 - Coheterocycloalkyl;
- each R 6 and each R 7 are independently selected from H, Ci-C ⁇ alkyl and halogen;
- Rg is H or Ci-C ⁇ alkyl;
- each R 9 is independently H or Ci-C ⁇ alkyl;
- each Rio and each Rn are independently selected from H, halogen and Ci-C ⁇ alkyl, or Ri 0 and Rn are independently Ci-C 3 alkyl and together
- R 1 is H or - (CH 2 )C(O)OR 8 ;
- R 2 is H, halogen, -C(O)NR 8 R 8 or CN ;
- R 3 is halo;
- R 4 is halo;
- R 5 is H, C 1 - C ⁇ alkyl or C 2 -C 6 heterocycloalkyl;
- R 6 is H or halogen;
- R 7 is H or halogen;
- R 8 is H or C 1 - C 4 alkyl;
- R 9 is H or CrC 4 alkyl;
- R 10 is H, F or CrC 4 alkyl;
- R 11 is H, F or CrC 4 alkyl; or
- R 10 and R 11 are independently Ci-C 3 alkyl and together with the carbon to which they are attached form a C 3 cycloalkyl;
- R 12 is H or CrC 4 alkyl;
- R 13 is H,
- R 1 is H or -CH 2 C(O)OH
- R 2 is H, Br, -C(O)NH 2 or CN, R 3 is F, R 4 is Cl, R 5 is H or O — ' , R 6 is H or Br, R 7 is
- R 8 is H or Qalkyl
- R 9 is H or Qalkyl
- R 11 is OH, F, C ⁇ alkyl or phenyl substituted with 1 or 2 halogen groups
- n is 0 or 1 and m is 0.
- each R 1 is independently selected from H and Ci-C ⁇ alkyl
- R 2 is H, halogen, -CN, -C(O)OR 1 or -C(O)NR 1 R 1
- R 3 is Ci-C ⁇ haloalkyl or halogen
- R 4 is H, CrC ⁇ haloalkyl or halogen
- L 1 is a bond, , ⁇ . X H 3 , X2 A ⁇ T ⁇ or X ⁇ T? " ;
- X 1 is CR 15 or N
- X 2 is O, S or NR 8 ;
- X 3 is S, O or NR 8 ;
- X 4 is CR 15 or N
- X 5 is CR 15 or N; each Re and each R 7 are independently selected from H, halogen, C 1 - C 6 alkyl, Ci-C ⁇ haloalkyl, and -C(O)OR 8 ; each R 8 is independently selected from H and Ci-C ⁇ alkyl; each R 15 is independently selected from H, halogen and -C(O)OR 8 ; L 2 is a bond, C 2 -C 6 alkynylene, phenylene, -0-, -C(O)-, -C(O)NR 8 -, -NR 8 C(O)-, -
- X 6 is C(R 9 ) 2 , NR 9 , O or S;
- X 7 is N or CR 9 ;
- X 8 is CR 9 , NR 9 , O or S;
- X 9 is N or CR 9 ;
- each R 9 is independently E or Ci-C 6 alkyl; and wherein the C 2 -Coalkynylene and phenylene of L 2 are each optionally substituted with 1-4 substituents independently selected from C 1-
- cyclopropyl -(CR 1 ORn) n Q 2 , wherein: the phenyl, heteroaryl, C 2 -C 6 heterocycloalkyl and C 3 -Cgcycloalkyl groups of Q 1 are optionally substituted with 1-4 substituents independently selected from Ci-C ⁇ alkyl, Ci-C ⁇ haloalkyl, halogen, -CN, -Q 2 , -O(CR 10 Rn)i- 4 OR 10 , -O(CR 10 Rn)i- 4 D , - 0(CR 10 Rn) 1 ⁇ R 10 , -OCi-C ⁇ alkyl, -OCi-C ⁇ haloalkyl, -C(O)OR 14 , -N(Rw) 2 , -C(O)N(Rw) 2 , - NRi 4 C(O)Ri 4 , -NRi 4 C(O)Q 2 , - S(O) 2
- Xi 3 is -(CH 2 ),- or -O(CH 2 ) P -;
- R 12 is H, Ci-Cealkyl or -C(O)OR 14 ; each R 14 is independently selected from H and C 1 -C ⁇ alkyl; n is O, 1, 2 or 3; p is O, 1, 2 or 3; q is 1, 2 or 3; s is 0 or 1 and provided that when the combination of L 1 , L 2 and Q 1 is, L 1 is pyridyl and L 2 is C 2 alkynylene and Q 1 is phenyl, pyridyl, thiazolyl, cyclohexyl or cyclopropyl, then R 2 is -CN, -C(O)OR 1 or -C(O)NR 1 R 1
- the compounds of Formula (I) and Formula (Ia), pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, and pharmaceutical compositions provided herein include all suitable isotopic variations of such compounds, and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, and pharmaceutical compositions.
- An isotopic variation of a compound provided herein or a pharmaceutically acceptable salt thereof is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature.
- isotopes that may be incorporated into the compounds provided herein and pharmaceutically acceptable salts thereof include but are not limited to isotopes of hydrogen, carbon, nitrogen and oxygen such as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 17 O, 18 0, 35 S, 18 F, 36 Cl and 123 I.
- Certain isotopic variations of the compounds provided herein and pharmaceutically acceptable salts thereof, for example, those in which a radioactive isotope such as 3 H or 14 C is incorporated, are useful in drug and/or substrate tissue distribution studies.
- 3 H and 14 C isotopes may be used for their ease of preparation and detectability.
- substitution with isotopes such as H may afford certain therapeutic advantages resulting from greater metabolic stability, such as increased in vivo half-life or reduced dosage requirements.
- isotopic variations of the compounds, and pharmaceutically acceptable salts, solvates, N- oxides, prodrugs and isomers thereof, and pharmaceutical compositions provided herein are prepared by conventional procedures using appropriate isotopic variations of suitable reagents.
- the compounds of Formula (I) and Formula (Ia) described herein are prepared as a pharmaceutically acceptable acid addition salt by reacting the free base form of the compound of Formula (I) and Formula (Ia) with a pharmaceutically acceptable organic acid or inorganic acid.
- a pharmaceutically acceptable base addition salt of compounds of Formula (I) and Formula (Ia) described herein is prepared by reacting the free acid form of the compound of Formula (I) and Formula (Ia) with a pharmaceutically acceptable organic base or inorganic base.
- the salt forms of the compounds of Formula (I) and Formula (Ia) described herein are prepared using salts of the starting materials or intermediates.
- the compounds of Formula (I) and Formula (Ia) described herein are in the form of other salts including, but not limited to, oxalates and trifluoroacetates.
- hemisalts of acids and bases are formed, for example, hemisulphate and hemicalcium salts.
- Such pharmaceutically acceptable acid addition salts of compounds of Formula (I) and Formula (Ia) include, but are not limited to, a hydrobromide, hydrochloride, hydroiodide, sulfate, bisulphate, nitrate, phosphate, succinate, maleate, formate, acetate, adipate, besylatye, bicarbonate/carbonate, propionate, fumarate, citrate, tartrate, lactate, benzoate, salicylate, glutamate, aspartate, p-toluenesulfonate, benzenesulfonate, methanesulfonate, ethanesulfonate, naphthalenesulfonate (e.g.
- 2-naphthalenesulfonate hexanoate salt, bisulphate/sulphate, borate, camsylate, cyclamate, edisylate, esylate, gluceptate, gluconate, glucuronate, pyruvate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, orotate, oxalate, oxaloacetate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, pyroglutamate, saccharate, stearate, tannate, tosylate, trifluoroacetate and xinofoate salts.
- organic acid or inorganic acids used to form certain pharmaceutically acceptable acid addition salts of compounds of Formula (I) and Formula (Ia) include, but are not limited to, hydrobromic, hydrochloric, sulfuric, nitric, phosphoric, succinic, maleic, formic, acetic, propionic, fumaric, citric, tartaric, lactic, benzoic, salicylic, glutamic, aspartic, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic, or hexanoic acid.
- Such pharmaceutically acceptable base addition salt of a compound of Formula (I) and Formula (Ia) include, but are not limited to, aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
- the free acid or free base forms of the compounds of Formula (I) and Formula (Ia) described herein are prepared from the corresponding base addition salt or acid addition salt from, respectively.
- a compound Formula (I) and Formula (Ia) in an acid addition salt form is converted to the corresponding free base by treating with a suitable base (by way of example only, an ammonium hydroxide solution, a sodium hydroxide, and the like).
- a compound of Formula (I) and Formula (Ia) in a base addition salt form is converted to the corresponding free acid by treating with a suitable acid (by way of example only, hydrochloric acid).
- the compounds of Formula (I) and Formula (Ia) described herein in unoxidized form are prepared from N-oxides of compounds Formula (I) and Formula (Ia) by treating with a reducing agent (by way of example only, sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like) in a suitable inert organic solvent (by way of example only, acetonitrile, ethanol, aqueous dioxane, or the like) at 0 to 8O 0 C.
- a reducing agent by way of example only, sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like
- a suitable inert organic solvent by way of example only, acetonitrile, ethanol, aqueous dioxane, or the like
- prodrug derivatives of compounds Formula (I) and Formula (Ia) described herein are prepared using methods known to those of ordinary skill in the art (e.g., for further details see Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985).
- appropriate prodrugs are prepared by reacting a non-derivatized compound of Formula (I) and Formula (Ia) with a suitable carbamylating agent (by way of example only, 1,1-acyloxyalkylcarbanochloridate, para- nitrophenyl carbonate, or the like).
- the compounds of Formula (I) and Formula (Ia) described herein are prepared or formed, as solvates (e.g., hydrates).
- hydrates of compounds of Formula (I) and Formula (Ia) are prepared by recrystallization from an aqueous/organic solvent mixture, using organic solvents such as dioxin, tetrahydrofuran or methanol.
- the compounds of Formula (I) and Formula (Ia) described herein are prepared as their individual stereoisomers.
- the compounds of Formula (I) and Formula (Ia) described herein are prepared as their individual stereoisomers by reacting a racemic mixture of the compound with an optically active resolving agent to form a pair of diastereoisomeric compounds, separating the diastereomers and recovering the optically pure enantiomers.
- resolution of enantiomers is carried out using covalent diastereomeric derivatives of the compounds of Formula (I) and Formula (Ia), or by using dissociable complexes (e.g., crystalline diastereomeric salts).
- Diastereomers have distinct physical properties (e.g., melting points, boiling points, solubility, reactivity, etc.) and are readily separated by taking advantage of these dissimilarities.
- the diastereomers are separated by chromatography, or by separation/resolution techniques based upon differences in solubility.
- the optically pure enantiomer is then recovered, along with the resolving agent, by any practical means that would not result in racemization.
- a more detailed description of the techniques applicable to the resolution of stereoisomers of compounds from their racemic mixture can be found in Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions," John Wiley And Sons, Inc., 1981.
- Compounds of Formula (I) and Formula (Ia) are made by processes described herein and as illustrated in the Examples.
- compounds of Formula (I) and Formula (Ia) are made by:
- Non-limiting examples of synthetic schemes used to make compounds of Formula (I) and Formula (Ia) described herein are illustrated in reaction schemes (I)-(IX), wherein n, Q 1 , L 1 , L 2 , X 1 ' R 1 , R 2 , R 3 , R 4 R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , R 11 and R 12 are as defined herein.
- Scheme (I) illustrates the use of Sonogashira coupling in the synthesis of certain compounds of Formula (I).
- a Q 1 group having a terminal alkyne reacts with an L 1 group having a halogen group attached.
- L 1 groups are aryl or vinyl halides.
- Reaction scheme (II) illustrates the synthesis of certain embodiments of compounds of Formula (I) by Sonogashira coupling of a halo-substituted pyridines or pyrimidines with a Q 1 group having a terminal alkyne.
- Scheme (III) illustrates the use of Suzuki coupling in the synthesis of certain compounds of Formula (I).
- a Q 1 group having a terminal boronic acid undergoes palladium catalyzed cross-coupling with an L 1 group (aryl or heteroaryl moiety) having a halogen group attached.
- Reaction scheme (IV) illustrates the synthesis of certain embodiments of compounds of Formula (I) by Suzuki coupling of a halo-substituted pyridines or pyrimidines with a Q 1 group having a terminal boronic acid.
- Scheme (V) illustrates the the synthesis of certain compounds of Formula (I) wherein imidazole formation is followed by Sonogashira coupling.
- Reaction scheme (VI) illustrates the use of Suzuki coupling in the synthesis of certain compounds of Formula (I).
- a Q 1 group linked to an L 2 group undergoes palladium catalyzed cross-coupling with an L 1 group (aryl or heteroaryl moiety) having a halogen group attached and a terminal boronic acid on the L 2 .
- Reaction scheme (VII) illustrates the synthesis of certain embodiments of compounds of Formula (I) by Suzuki coupling of a halo-substituted pyridines or pyrimidines with phenyl boronic acid having an attached Q 1 group.
- Reaction scheme (VIII) illustrates the the synthesis of certain compounds of Formula (I) wherein an oxadiazole moiety is formed as an L 2 group between an L 1 group and a Q 1 group.
- the Q 1 is substituted with a carboxylic acid moiety and the L 1 is substituted with a hydroxy imidamide moiety.
- Reaction scheme (IX) illustrates the synthesis of certain embodiments of compounds of Formula (I) wherein an oxadiazole moiety is formed as an L 2 group between an L 1 group and a Q 1 group.
- the Q 1 is substituted with a carboxylic acid moiety and the L 1 is a pyridine or pyrimidine substituted with a hydroxy imidamide moiety.
- Reaction scheme (X) illustrates the the synthesis of certain compounds of Formula (I) wherein an thiazole moiety or oxazole moiety is formed as an L 2 group between an L 1 group and a Q 1 group.
- the Q 1 is substituted with an aminopropanone moiety and the L 1 is substituted with a carboxylic acid moiety.
- Reaction scheme (XII) illustrates the use of Suzuki coupling in the synthesis of certain compounds of Formula (I).
- a Q 1 -L 2 -L 1 group undergoes palladium catalyzed cross-coupling with an imidazole moiety having a halogen group attached and a terminal boronic acid on the L 1 .
- Reaction scheme (XIII) illustrates the synthesis of certain embodiments of compounds of Formula (I) by Suzuki coupling of Q 1 -L 2 -L 1 group with an imidazole moiety having a halogen group attached, wherein the L 1 group is a pyridine or pyrimidine with a terminal boronic acid having an attached Q 1 group.
- Non-steroidal anti-inflammatory therapies and aspirin-related drugs block the production of all prostaglandins through inhibiting the ability of cyclooxygenases to convert arachidonic acid into prostaglandin H 2 (PGH 2 ).
- PGH 2 can subsequently be metabolized into a spectrum of lipid mediators including PGE 2 , PGD 2 and PGF 2 ⁇ as well as thromboxane and prostacyclin (PGI 2 ) through the action of a number of different synthases. All of these products activate G protein-coupled receptors; the phenotypes resulting from deletion of these receptors has considerably enhanced the understanding of prostanoid biology.
- Non-steroidal anti-inflammatory drugs which include both traditional NSAIDs (tNSAIDs) and selective inhibitors of COX-2, relieve pain and inflammation by suppressing the COX function of prostaglandin H synthases (PGHS; COX-I or COX-2) and the consequent formation of PGE 2 and prostacyclin (PGI 2 ).
- NSAIDs traditional NSAIDs
- PGHS prostaglandin H synthases
- PGE 2 and prostacyclin PGE 2 and prostacyclin
- the tNSAIDs gastrointestinal (GI) intolerance is attributed to inhibition of COX-I derived protective PGE 2 and PGI 2 by gastroduodenal epithelium and platelet COX-1-derived TxA 2
- the first three selective COX-2 inhibitors approved by the FDA, celecoxib, rofecoxib, and valdecoxib showed clinical superiority in terms of efficacy in rheumatoid arthritis patients and lack of a drug-induced ulceration compared to a tNSAID.
- randomized clinical trials of these structurally distinct COX-2 inhibitors also indicate that such compounds elevate the risk of myocardial infarction and stroke.
- a combination of pharmacological, genetic and neutralizing antibody approaches demonstrates the importance of PGE 2 in inflammation.
- disruption of PGE 2 dependent signaling in animal models can be as effective as treatment with tNSAIDs or COX-2 inhibitors.
- prostaglandin E synthases the enzymes that catalyze the nonoxidative rearrangmenet of the COX product PGH 2 into PGE 2 , have been isolated and identified to date including microsomal prostaglandin E synthase (mPGES)-l, mPGES-2, GSTM2-2, GSTM3-3, and cPGES/p23.
- mPGES-1 has emerged as an important modulator of inflammation in-vivo. Not only does this enzyme occupy the terminal position in the PGE2-synthesis cascade, but it also preferentially couples with COX-2.
- constitutive mPGES-1 expression in tissues such as stomach and kidney appear to contribute to small amounts of basal PGE2 production.
- LPS lipopolysaccharide
- mPGES-1 null mice show impaired delayed-type hypersensitivity responses, as well as some evidence for impaired formation of inflammatory granulation tissue and angiogenesis.
- the role of mPGES-1 in the central nervous system regulation of fever and nociception in inflammatory states is also well described. Animal pyresis models show transient up-regulation of mPGES-1 mRNA in brain endothelial cells after systemic injection with endotoxin or IL-I. Additionally, peripheral inflammation from carageenan-injected paws was associated with increased mPGES-1 expression and PGE2 levels in the central nervous system.
- mice with adjuvant-induced arthritis displayed sustained induction of mPGES-1 by immunohistochemistry in the brain vasculature and the paraventricular nucleus of the hypothalamus.
- mPGES-1 null mice did not generate fevers in response to peripheral LPS injection but did become febrile after intracerebroventricular PGE2 injection.
- PGE2 derived from mPGES-1 in the central nervous system as a critical mediator in inflammation-induced pyresis.
- mPGES-1 null mice exhibit diminished writhing in response to intraperitoneal injections of acetic acid (especially after LPS priming), similar to NSAID-treated wild-type mice. There was no difference between null and wild-type mice in withdrawal latency upon thermal stimuli, a model of noninflammatory pain. However, in a model of neuropathic pain involving hyperalgesia following transection of an L5 (fifth lumbar) spinal nerve, mPGES-1 null mice had higher withdrawal thresholds and latency upon both mechanical and thermal stimuli compared with wild-type mice.
- mPGES-1 null mice do not display increased thrombogenicity. Decreased expression or pharmacological inhibiiton of COX2 results in increased thrombogenicity in several animal models. These effects are likely mediated by decreased COX-2-dependent prostacyclin (PGI 2 ) formation. In mPGES-1 null mice, prostacyclin (PGI 2 ) formation levels are actually increased while PGE2 levels are decreased. The result is a lack of effect on thrombogenicity and protection from the formation of atherosclerosis on an apoE null background.
- Microsomal prostaglandin E synthase- 1 (mPGES-1) is a novel target for pain (inflammatory, dental, osteoarthritic and neuropathic), inflammation, rheumatoid arthritis (RA), coronary artery disease and fever.
- Selective inhibitors of mPGES-1 have the potential to be efficacious in reducing inflammation, fever and pain while potentially avoiding the side effects associated with COX-2 inhibitors or traditional NSAIDs.
- targeting mPGES-1 may provide a novel strategy which provides similar efficacy without increasing risk of myocardial infarction and stroke.
- the compounds provided herein are inhibitors of prostaglandin E synthase (PGES) activity, and in certain embodiments are inhibitors of microsomal prostaglandin E synthase- 1 (mPGES-1) activity.
- the compounds provided herein are inhibitors of prostaglandin E synthases (PGES), and in certain embodiments are inhibitors of microsomal prostaglandin E synthase- 1 (mPGES-1).
- the compounds provided herein reduce the formation of the specific arachidonic acid metabolite PGE 2 without reducing the formation of other COX generated arachidonic acid metabolites, and do not give rise to the associated side-effects mentioned herein.
- compounds provided herein are inhibitors of microsomal prostaglandin E synthase- 1 (mPGES-1) activity, wherein they prevent the action of mPGES- 1 or a complex of which the mPGES-1 enzyme forms a part, and/or elicit an mPGES-1 modulating effect.
- mPGES-1 microsomal prostaglandin E synthase- 1
- Compounds provided herein are useful in the treatment of a disease or disorder in which inhibition of PGES activity, prevents, inhibits or ameliorates the pathology and/or symptomatology of such diseases or disorders.
- the compounds provided herein are useful in the treatment of a disease or disorder in which inhibition of mPGES-1 activity, prevents, inhibits or ameliorates the pathology and/or symptomatology of such diseases or disorders.
- Compounds of Formula (I) and Formula (Ia) provided herein are inhibitors of microsomal prostaglandin E synthase- 1 (mPGES-1) activity, and as such are useful for treating diseases or disorders in which mPGES-1 contribute to the pathology and/or symptomatology of the disease. Also, such compounds of Formula (I) and Formula (Ia) are useful for the prevention or treatment of mPGES-1 mediated diseases or conditions. In addition, such compounds of Formula (I) and Formula (Ia) are used in the preparation of medicaments for the treatment of diseases or disorders in which mPGES-1 contribute to the pathology and/or symptomatology of the disease.
- mPGES-1 microsomal prostaglandin E synthase- 1
- Such compounds of Formula (I) and Formula (Ia) are useful in a method of medical treatment, wherein the method of medical treatment is for treating a mPGES-1 mediated diseases or conditions.
- mPGES-1 mediated diseases or conditions and the diseases or disorders in which mPGES-1 contribute to the pathology and/or symptomatology of the disease include pain, including but not limited to, inflammatory pain, dental pain, postdental procedure pain, osteoarthritic pain, nociceptive pain, neuropathic pain muscoskeletal pain, low back pain, headaches, sinus headaches, migraines, post-surgical pain, surgical pain, burn injury and pain associated with bacterial, fungal or viral illnesses (by way of example only, influenza, the common cold, a viral infections (such as, by way of example only, influenza, common cold, herpes zoster, hepatitis C and AIDS), a bacterial infection or a fungal infection) and periodontal disease; inflammatory diseases, inflammation, arthritis, rheumatoid arthritis (RA
- provided herein are methods for preventing or treating inflammation, methods for preventing or treating inflammatory diseases, and methods for preventing or treating pain.
- the compounds of Formula (I) and Formula (Ia) provided herein are useful for treating or preventing various diseases with an inflammatory component, including, but not limited to, rheumatoid arthritis, osteoarthritis and asthma.
- methods for preventing or treating any of the diseases or disorders described herein in a subject in need thereof of such treatment comprises administering to the subject a therapeutically effective amount of a compound of Formula (I) or Formula (Ia) or a pharmaceutically acceptable salt thereof.
- such methods described herein involve administering to the subject in need thereof a pharmaceutical composition that contains one or more compounds of Formula (I) or Formula (Ia), or a pharmaceutically acceptable salt thereof.
- kits for treatment of a disease or disorder which is associated with, and/or wherein the activity of prostaglandin E synthase is implicated are provided herein.
- Such methods include administration of a therapeutically effective amount of a compound or composition provided herein to a subject in need thereof.
- Compounds and compositions provided herein are useful in both the therapeutic and/or prophylactic treatment of the disease and/or disorders provided herein.
- the methods provided herein are either prophylactic or therapeutic treatments of mPGES-1 related disorders and/or mPGES-1 related diseases.
- Such methods involve administering to a subject in need thereof a therapeutically effective amount of one or more compounds of Formula (I) or Formula (Ia), wherein such compounds are inhibitors of mPGES-1. Also, such methods involve administering to a subject in need thereof a pharmaceutical composition that contains a therapeutically effective amount of one or more compounds of Formula (I) or Formula (Ia), wherein such compounds are inhibitors of mPGES-1.
- Such prophylactic or therapeutic treatment methods are used to treat any of the mPGES-1 mediated diseases, disorders or conditions provided herein.
- Animal subjects include both domestic animals and livestock, raised either as pets or for commercial purposes. Examples include, but are not limited to, dogs, cats, cattle, horses, sheep, hogs, and goats.
- the compounds and composition provided herein are useful in the treatment of inflammation. Accordingly, compounds and composition provided herein are useful in the treatment of pain, asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, inflammatory bowel disease, irritable bowel syndrome, inflammatory pain, fever, migraine, headache, low back pain, fibromyalgia, myofascial disorders, viral infections (such as, by way of example only, influenza, common cold, herpes zoster, hepatitis C and AIDS), bacterial infections, fungal infections, dysmenorrhea, burns, surgical or dental procedures, malignancies (such as, by way of example only, breast cancer, colon cancer, and prostate cancer), hyperprostaglandin E syndrome, classic Bartter syndrome, atherosclerosis, gout, arthritis, osteoarthritis, juvenile arthritis, rheumatoid arthritis, rheumatic fever, ankylosing spondylitis, Hodgkin's disease, systemic
- the compounds and composition provided herein are useful in the treatment of diseases or disorders associated with bone loss in a subject.
- diseases and/or disorders include, but are not limited to, osteoporosis, osteoarthritis, Paget's disease and/or periodontal diseases.
- Compounds and composition provided herein are useful in the reduction of bone loss, increasing bone mineral density, and/or the reduction in incidence and/or healing of fractures.
- the methods of treatment are used to treat any disease and/or disorder provided herein including, but not limited to, those associated with inflammation, pain and/or bone loss.
- such methods of treatment are also useful in the reduction of bone loss, increasing bone mineral density, and/or the reduction in incidence and/or healing of fractures.
- compositions which comprise at least one compound provided herein, including at least one compound of Formulas (I) or Formula (Ia), pharmaceutically acceptable salts and/or solvates thereof, and one or more pharmaceutically acceptable carriers, diluents, adjuvant or excipients.
- such compounds and compositions are administered singly or in combination with one or more additional therapeutic agents.
- the method of administration of such compounds and compositions include, but are not limited to, oral administration, rectal administration, parenteral, intravenous administration, intravitreal administration, subcutaneous administration, intramuscular administration, inhalation, intranasal administration, dermal administration, topical administration, ophthalmic administration or buccal administration, tracheal administration, bronchial administration, sublingual administration or otic administration.
- such compounds of Formula (I) or Formula (Ia) are inhibitors of prostaglandin E synthases, including microsomal prostaglandin E synthase- 1 (mPGES-1), formulated in an amount sufficient to prevent, inhibit or ameliorate the pathology and/or symptomatology of diseases or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity.
- the therapeutically effective amount will vary depending on, among others, the disease indicated, the severity of the disease, the age and relative health of the subject, the potency of the compound administered, the mode of administration and the treatment desired. .
- the daily dosage of a compound of Formula (I) or Formula (Ia), satisfactory results are indicated to be obtained systemically at daily dosages of from about 0.03 to 150 mg/kg per body weight. In certain embodiments, the daily dosage of a compound of Formula (I) or Formula (Ia), satisfactory results are indicated to be obtained systemically at daily dosages of from about 0.03 to 2.5 mg/kg per body weight. In certain embodiments, the daily dosage of a compound of Formula (I) or Formula (Ia), administered by inhalation, is in the range from 0.05 micrograms per kilogram body weight ( ⁇ g/kg) to 100 micrograms per kilogram body weight ( ⁇ g/kg).
- the daily dosage of a compound of Formula (I) or Formula (Ia), administered orally is in the range from 0.01 micrograms per kilogram body weight ( ⁇ g/kg) to 100 milligrams per kilogram body weight (mg/kg).
- An indicated daily dosage in the larger mammal, e.g. humans is in the range from about 0.5mg to about lOOmg of a compound of Formula (I) or Formula (Ia), conveniently administered, e.g. in divided doses up to four times a day or in controlled release form.
- unit dosage forms for oral administration comprise from about 1 to 50 mg of a compound of Formula (I) or Formula (Ia).
- oral, pulmonary and topical dosages range from between about 0.01 mg/kg of body weight per day (mg/kg/day) to about 100 mg/kg/day. In certain embodiments, dosages range from between about 0.01 to about 10 mg/kg/day, and in other embodiments, dosages range from between about 0.1 to about 5.0 mg/kg/day.
- the compositions contain between about 0.01 mg to about 500 mg. In other embodiments, between about 1 mg to about 100 mg, of the active ingredient.
- the doses will range from about 0.001 to about 10 mg/kg/hour during constant rate infusion.
- the physician or the skilled person, will be able to determine the actual dosage which will be most suitable for an individual patient, which is likely to vary with the route of administration, the type and severity of the condition that is to be treated, as well as the species, age, weight, sex, renal function, hepatic function and response of the particular patient to be treated.
- the above-mentioned dosages are exemplary of the average case; there can, of course, be individual instances where higher or lower dosage ranges are merited, and such are within the scope of this invention.
- Compounds provided herein are administered alone, or are administered by way of known pharmaceutical formulations, including tablets, capsules or elixirs for oral administration, suppositories for rectal administration, sterile solutions or suspensions for parenteral or intramuscular administration, lotions, gels, ointments or creams for topical administration, and the like.
- compositions which comprise at least one compound of Formula (I) or Formula (Ia), or pharmaceutically acceptable salts and/or solvates thereof.
- processes include admixing a compound of the Formula (I) or Formula (Ia), and pharmaceutically acceptable salts and solvates thereof, with one or more pharmaceutically acceptable carriers, diluents or excipients.
- the pharmaceutical compositions comprising a compound of Formula (I) or Formula (Ia) in free form or in a pharmaceutically acceptable salt or solvate form, in association with at least one pharmaceutically acceptable carrier, diluent or excipient are manufactured by mixing, granulating and/or coating methods.
- compositions optionally contain excipients, such as adjuvants, preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers.
- excipients such as adjuvants, preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers.
- such compositions are sterilized.
- pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 1% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 5% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 10% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 20% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 30% by weight.
- pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 40% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 50% by weight. That is, the ratio of active ingredient to the other components (by way of example, the addition of adjuvant, diluent and carrier) of the pharmaceutical composition is at least 1:99, 5:95, 10:90, 20:80, 30:70, 40:60 or at least 50:50 by weight.
- the pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered orally as discrete dosage forms, wherein such dosage forms include, but are not limited to, capsules, gelatin capsules, caplets, tablets, chewable tablets, powders, granules, syrups, flavored syrups, solutions or suspensions in aqueous or non-aqueous liquids, edible foams or whips, and oil-in-water liquid emulsions or water- in-oil liquid emulsions.
- the capsules, gelatin capsules, caplets, tablets, chewable tablets, powders or granules, used for the oral administration of at least one compound of Formula (I) or Formula (Ia) are prepared by admixing at least one compound of Formula (I) or Formula (Ia) (active ingredient) together with at least one excipient using conventional pharmaceutical compounding techniques.
- excipients used in oral dosage forms described herein include, but are not limited to, binders, fillers, disintegrants, lubricants, absorbents, colorants, flavors, preservatives and sweeteners.
- Non-limiting examples of such binders include, but are not limited to, corn starch, potato starch, starch paste, pre- gelatinized starch, or other starches, sugars, gelatin, natural and synthetic gums such as acacia, sodium alginate, alginic acid, other alginates, tragacanth, guar gum, cellulose and its derivatives (by way of example only, ethyl cellulose, cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethylcellulose, methyl cellulose, hydroxypropyl methylcellulose and microcrystalline cellulose), magnesium aluminum silicate, polyvinyl pyrrolidone and combinations thereof.
- acacia sodium alginate, alginic acid, other alginates, tragacanth, guar gum
- cellulose and its derivatives by way of example only, ethyl cellulose, cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethylcellulose, methyl cellulose, hydroxypropyl methylcellulose and microcrystalline
- Non-limiting examples of such fillers include, but are not limited to, talc, calcium carbonate (e.g., granules or powder), microcrystalline cellulose, powdered cellulose, dextrates, kaolin, mannitol, silicic acid, sorbitol, starch, pre- gelatinized starch, and mixtures thereof.
- the binder or filler in pharmaceutical compositions provided herein are present in from about 50 to about 99 weight percent of the pharmaceutical composition or dosage form.
- Non-limiting examples of such disintegrants include, but are not limited to, agar- agar, alginic acid, sodium alginate, calcium carbonate, sodium carbonate, microcrystalline cellulose, croscarmellose sodium, crospovidone, polacrilin potassium, sodium starch glycolate, potato or tapioca starch, pre-gelatinized starch, other starches, clays, other algins, other celluloses, gums, and combinations thereof.
- the amount of disintegrant used in the pharmaceutical compositions provided herein is from about 0.5 to about 15 weight percent of disintegrant, while in other embodiments the amount is from about 1 to about 5 weight percent of disintegrant.
- Non-limiting examples of such lubricants include, but are not limited to, sodium stearate, calcium stearate, magnesium stearate, stearic acid, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, sodium lauryl sulfate, talc, hydrogenated vegetable oil (by way of example only, peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil, corn oil, and soybean oil), zinc stearate, sodium oleate, ethyl oleate, ethyl laureate, agar, silica, a syloid silica gel (AEROSIL 200, manufactured by W.R.
- AEROSIL 200 AEROSIL 200, manufactured by W.R.
- the amount of lubricants used in the pharmaceutical compositions provided herein is in an amount of less than about 1 weight percent of the pharmaceutical compositions or dosage forms.
- Non-limiting examples of such diluents include, but are not limited to, lactose, dextrose, sucrose, mannitol, sorbitol, cellulose, glycine or combinations thereof.
- tablets and capsules are prepared by uniformly admixing at least one compound of Formula (I) or Formula (Ia) (active ingredients) with liquid carriers, finely divided solid carriers, or both, and then shaping the product into the desired presentation if necessary.
- tablets are prepared by compression.
- tablets are prepared by molding.
- At least one compound of Formula (I) or Formula (Ia) is orally administered as a controlled release dosage form.
- dosage forms are used to provide slow or controlled-release of one or more compounds of Formula (I) or Formula (Ia). Controlled release is obtained using, for example, hydroxypropylmethyl cellulose, other polymer matrices, gels, permeable membranes, osmotic systems, multilayer coatings, microparticles, liposomes, microspheres, or a combination thereof.
- controlled-release dosage forms are used to extend activity of the compound of Formula (I) or Formula (Ia), reduce dosage frequency, and increase patient compliance.
- Administration of compounds of Formula (I) or Formula (Ia) as oral fluids such as solution, syrups and elixirs are prepared in unit dosage forms such that a given quantity of solution, syrups or elixirs contains a predetermined amount of a compound of Formula (I) or Formula (Ia).
- Syrups are prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle.
- Suspensions are formulated by dispersing the compound in a non-toxic vehicle.
- Non-limiting examples of excipients used in as oral fluids for oral administration include, but are not limited to, solubilizers, emulsifiers, flavoring agents, preservatives, and coloring agents.
- solubilizers and emulsifiers include, but are not limited to, water, glycols, oils, alcohols, ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers.
- Non-limiting examples of preservatives include, but are not limited to, sodium benzoate.
- Non-limiting examples of flavoring agents include, but are not limited to, peppermint oil or natural sweeteners or saccharin or other artificial sweeteners. Parenteral Dosage Forms
- compositions containing at least one compound of Formula (I) or Formula (Ia) are administered parenterally by various routes including, but not limited to, subcutaneous, intravenous (including bolus injection), intramuscular, and intraarterial.
- parenteral dosage forms are administered in the form of sterile or sterilizable injectable solutions, suspensions, dry and/or lyophylized products ready to be dissolved or suspended in a pharmaceutically acceptable vehicle for injection (reconstitutable powders) and emulsions.
- Vehicles used in such dosage forms include, but are not limited to, Water for Injection USP; aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride Injection, and Lactated Ringer's Injection; water-miscible vehicles such as, but not limited to, ethyl alcohol, polyethylene glycol, and polypropylene glycol; and non-aqueous vehicles such as, but not limited to, corn oil, cottonseed oil, peanut oil, sesame oil, ethyl oleate, isopropyl myristate, and benzyl benzoate.
- Water for Injection USP Water for Injection USP
- aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride Injection, and Lactated Ringer's Injection
- compositions containing at least one compound of Formula (I) or Formula (Ia) are administered transdemally.
- transdermal dosage forms include "reservoir type” or “matrix type” patches, which are applied to the skin and worn for a specific period of time to permit the penetration of a desired amount of a compound of Formula (I) or Formula (Ia).
- transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound to the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin.
- matrix transdermal formulations are used.
- Formulations for transdermal delivery of a compound of Formula (I) or Formula (Ia) include an effective amount of a compound of Formula (I) or Formula (Ia), a carrier and an optional diluent.
- a carrier includes, but is not limited to, absorbable pharmacologically acceptable solvents to assist passage through the skin of the host, such as water, acetone, ethanol, ethylene glycol, propylene glycol, butane- 1,3-diol, isopropyl myristate, isopropyl palmitate, mineral oil, and combinations thereof.
- such transdermal delivery systems include penetration enhancers to assist in delivering one or more compounds of Formula (I) or Formula (Ia) to the tissue.
- Such penetration enhancers include, but are not limited to, acetone; various alcohols such as ethanol, oleyl, and tetrahydrofuryl; alkyl sulfoxides such as dimethyl sulfoxide; dimethyl acetamide; dimethyl formamide; polyethylene glycol; pyrrolidones such as polyvinylpyrrolidone; Kollidon grades (Povidone, Polyvidone); urea; and various water- soluble or insoluble sugar esters such as Tween 80 (polysorbate 80) and Span 60 (sorbitan monostearate).
- the pH of such a transdermal pharmaceutical composition or dosage form, or of the tissue to which the pharmaceutical composition or dosage form is applied is adjusted to improve delivery of one or more compounds of Formula (I) or Formula (Ia).
- the polarity of a solvent carrier, its ionic strength, or tonicity are adjusted to improve delivery.
- compounds such as stearates are added to advantageously alter the hydrophilicity or lipophilicity of one or more compounds of Formula (I) or Formula (Ia) so as to improve delivery.
- such stearates serve as a lipid vehicle for the formulation, as an emulsifying agent or surfactant, and as a delivery-enhancing or penetration-enhancing agent.
- different salts, hydrates or solvates of the compounds of Formula (I) or Formula (Ia) are used to further adjust the properties of the resulting composition.
- At least one compound of Formula (I) or Formula (Ia) is administered by topical application of pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia) in the form of lotions, gels, ointments solutions, emulsions, suspensions or creams.
- suitable formulations for topical application to the skin are aqueous solutions, ointments, creams or gels, while formulations for ophthalmic administration are aqueous solutions.
- Such formulations optionally contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
- Such topical formulations include at least one carrier, and optionally at least one diluent.
- Such carriers and diluents include, but are not limited to, water, acetone, ethanol, ethylene glycol, propylene glycol, butane- 1,3-diol, isopropyl myristate, isopropyl palmitate, mineral oil, and combinations thereof.
- such topical formulations include penetration enhancers to assist in delivering one or more compounds of Formula (I) or Formula (Ia) to the tissue.
- penetration enhancers include, but are not limited to, acetone; various alcohols such as ethanol, oleyl, and tetrahydrofuryl; alkyl sulfoxides such as dimethyl sulfoxide; dimethyl acetamide; dimethyl formamide; polyethylene glycol; pyrrolidones such as polyvinylpyrrolidone; Kollidon grades (Povidone, Polyvidone); urea; and various water- soluble or insoluble sugar esters such as Tween 80 (polysorbate 80) and Span 60 (sorbitan monostearate).
- compositions containing at least one compound of Formula (I) or Formula (Ia) are administered by inhalation.
- Dosage forms for inhaled administration are formulated as aerosols or dry powders.
- Aerosol formulations for inhalation administration comprise a solution or fine suspension of at least one compound of Formula (I) or Formula (Ia) in a pharmaceutically acceptable aqueous or non-aqueous solvent.
- such pharmaceutical compositions optionally comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, and optionally a performance modifier such as L-leucine or another amino acid, and/or metals salts of stearic acid such as magnesium or calcium stearate.
- compounds of Formula (I) or Formula (Ia) are be administered directly to the lung by inhalation using a Metered Dose Inhaler (“MDI”), which utilizes canisters that contain a suitable low boiling propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas, or a Dry Powder Inhaler (DPI) device which uses a burst of gas to create a cloud of dry powder inside a container, which is then be inhaled by the patient.
- MDI Metered Dose Inhaler
- DPI Dry Powder Inhaler
- capsules and cartridges of gelatin for use in an inhaler or insufflator are formulated containing a powder mixture of a compound of Formula (I) or Formula (Ia) and a powder base such as lactose or starch.
- compounds of Formula (I) or Formula (Ia) are delivered to the lung using a liquid spray device, wherein such devices use extremely small nozzle holes to aerosolize liquid drug formulations that can then be directly inhaled into the lung.
- compounds of Formula (I) or Formula (Ia) are delivered to the lung using a nebulizer device, wherein a nebulizers creates an aerosols of liquid drug formulations by using ultrasonic energy to form fine particles that can be readily inhaled.
- compounds of Formula (I) or Formula (Ia) are delivered to the lung using an electrohydrodynamic ("EHD") aerosol device wherein such EHD aerosol devices use electrical energy to aerosolize liquid drug solutions or suspensions.
- EHD electrohydrodynamic
- the pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), or pharmaceutically acceptable salts and solvates thereof, described herein also contain one or more absorption enhancers.
- such absorption enhancers include, but are not limited to, sodium glycocholate, sodium caprate, N-lauryl- ⁇ -D-maltopyranoside, EDTA, and mixed micelles.
- pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered nasally.
- the dosage forms for nasal administration are formulated as aerosols, solutions, drops, gels or dry powders.
- pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered rectally in the form of suppositories, enemas, ointment, creams rectal foams or rectal gels.
- such suppositories are prepared from fatty emulsions or suspensions, cocoa butter or other glycerides.
- compositions containing at least one compound of Formula (I) or Formula (Ia) are administered opthamically as eye drops.
- Such formulations are aqueous solutions that optionally contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
- compositions containing at least one compound of Formula (I) or Formula (Ia) are administered otically as ear drops.
- Such formulations are aqueous solutions that optionally contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
- compositions containing at least one compound of Formula (I) or Formula (Ia) are formulated as a depot preparation.
- Such long acting formulations are administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection.
- such formulations include polymeric or hydrophobic materials (for example, as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- a compound of Formula (I) or Formula (Ia) described herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing such compounds of Formula (I) or Formula (Ia), is administered to a patient who has not previously undergone or is not currently undergoing treatment with another therapeutic agent.
- one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), is administered alone (without an additional therapeutic agent) for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
- mPGES-1 microsomal prostaglandin E synthase- 1
- one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), is administered in combination with one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
- mPGES-1 microsomal prostaglandin E synthase- 1
- one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), is formulated in combination with one or more additional therapeutic agents and administered for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
- mPGES-1 microsomal prostaglandin E synthase- 1
- one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), is administered sequentially with one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
- mPGES-1 microsomal prostaglandin E synthase- 1
- the combination treatments provided herein include administration of one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), prior to administration of one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
- mPGES-1 microsomal prostaglandin E synthase- 1
- the combination treatments provided herein include administration of one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), subsequent to administration of one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
- mPGES-1 microsomal prostaglandin E synthase- 1
- the combination treatments provided herein include administration of one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), concurrently with one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
- mPGES-1 microsomal prostaglandin E synthase- 1
- the combination treatments provided herein include administration of one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), formulated with one or more additional therapeutic agents, for the treatment of one or more of diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
- compounds of Formula (I) or Formula (Ia) provided herein are administered in therapeutically effective amounts in combination with other therapies, such as radiation therapy, bone marrow transplantation or hormone therapy.
- the compounds of Formula (I) or Formula (Ia) provided herein, or a pharmaceutically acceptable salts, N-oxides, isomers or solvates thereof, and the additional therapeutics agent(s) act additively.
- the compounds of Formula (I) or Formula (Ia) provided herein, or a pharmaceutically acceptable salts, N-oxides, isomers or solvates thereof, and the additional therapeutics agent(s) act synergistically.
- the additional therapeutic agents used in combination with at least one compound of Formula (I) or Formula (Ia) described herein, or a pharmaceutically acceptable salt or solvate thereof include, but are not limited to, anti-inflammatory agents, therapeutic agents for osteoporosis, anti-tumor therapeutic agents, chemotherapeutic agents, antineoplastic agents and other anticancer agents.
- the additional therapeutic agents used in combination with a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia) described herein, or a pharmaceutically acceptable salt or solvate thereof include, but are not limited to, anti-inflammatory agents, therapeutic agents for osteoporosis, anti-tumor therapeutic agents, chemotherapeutic agents, antineoplastic agents and other anticancer agents.
- anti-inflammatory agents used in such combinations described herein include, but are not limited to, non-steroidal anti-inflammatory drugs such as salicylic acid, acetylsalicylic acid, methyl salicylate, diflunisal, salsalate, olsalazine, sulfasalazine, acetaminophen, indomethacin, sulindac, etodolac, mefenamic acid, meclofenamate sodium, tolmetin, ketorolac, dichlofenac, ibuprofen, naproxen, naproxen sodium, fenoprofen, ketoprofen, flurbiprofen, oxaprozin, piroxicam, meloxicam, ampiroxicam, droxicam, pivoxicam, tenoxicam, nabumetome, phenylbutazone, oxyphenbutazone, antipyrine, aminopyrine, apazone and nimes
- chemotherapeutic agents used in such combinations described herein include, but are not limited to, anthracyclines, alkylating agents (e.g., mitomycin C), alkyl sulfonates, aziridines, ethylenimines, methylmelamines, nitrogen mustards, nitrosoureas, antibiotics, antimetabolites, folic acid analogs (e.g., dihydrofolate reductase inhibitors such as methotrexate), purine analogs, pyrimidine analogs, enzymes, podophyllotoxins, platinum- containing agents, interferons, and interleukins.
- alkylating agents e.g., mitomycin C
- alkyl sulfonates e.g., aziridines, ethylenimines, methylmelamines, nitrogen mustards, nitrosoureas, antibiotics, antimetabolites, folic acid analogs (e.g., dihydrofolate reductas
- chemotherapeutic agents which may be used in the compositions and methods of the invention include, but are not limited to, busulfan, improsulfan, piposulfan, benzodepa, carboquone, meturedepa, uredepa, altretamine, triethylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide, trimethylolomelamine, chlorambucil, chlornaphazine, cyclophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard, carmustine, chlorozotocin, fotemustine, lomustine, nimustine, ranimustine, dacarbazine, mannomustine, mitobronitol, mitolactol, pipobroman, aclacinomycin
- the therapeutic agents for treating osteoporosis used in such combinations described herein include, but are not limited to, alfacalcidol, calcitriol, elcatonin, calcitonin salmon, estriol, ipriflavone, pamidronate disodium, alendronate sodium hydrate, reminderonate disodium and the like.
- such combination therapies includes the combination of one or more compounds of Formula (I) or Formula (Ia) provided herein with other therapeutic agents that are useful in the treatment of inflammation, including, but not limited to, NSAIDs and coxibs.
- kits that include one or more containers containing a compound of Formula (I) or Formula (Ia) useful for the treatment or prevention of a disease or disorder associated with the activity of mPGES-1.
- such pharmaceutical packs or kits include one or more containers containing a compound of Formula (I) or Formula (Ia) useful for the treatment or prevention of a disease or disorder associated with the activity of mPGES-1 and one or more containers containing an additional therapeutic agent.
- such pharmaceutical packs or kits optionally include instructions for its administration of a compound of Formula (I).
- the compound of Formula (I) is in free form or in pharmaceutically acceptable salt or N-oxide form.
- Glacial acetic acid (12 mL) was added dropwise to a 50 mL reaction flask charged with commercially available 5-acetyl-2-bromo-pyrimidine (R-4) (12.9 mmol). The solution was cooled in an ice bath to 0 0 C. A 48% solution of HBr (10 mL) was then added slowly, followed by dropwise addition of bromine (12.9 mmol). The reaction mixture was stirred at room temperature for 12 hours. The pale orange solution was quenched by slow addition of 10% aq.
- R-7 70 0 C, 12h R-9 rt, 2h R-IO
- EX-3 Ethyl 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethynyl) pyridin-3-yl)-lH- imidazole-5-carboxylate (EX-3) (0.078 mmol) was dissolved in a T ⁇ F:water (1 : 1) mixture (4 rnL), LiOH (0.31 mmol) was added and the reaction mixture was stirred at room temperature for 12 hours.
- N-(5-Bromo-2-(methylamino)phenyl)-2-(2-chloro-6-fluorophenyl)-lH- imidazole-4-carboxamide (1-34) was converted to 5-bromo-2-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)-l -methyl- lH-benzo [J] imidazole (EX-96) using similar procedure as for the synthesis of 5-bromo-2-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-lH- imidazo[4,5-b]pyridine (EX-95).
- Ethyl 5-(2-(2-chloro-6-fluorophenyl)- l-(ethoxycarbonyl)- lH-imidazol-4-yl)-2- (phenylethynyl)isonicotinate (1-37) was prepared by employing the same Sonogashira condition as described for the synthesis of 1-2.
- Membrane preparations of tagless full length human mPGESl protein were generated from baculovirus mediated expression in insect cells.
- Membrane preparations containing human mPGESl were incubated with potential antagonist compounds for 30 minutes prior to addition of substrate.
- the substrate prostaglandin H 2 (PGH 2 ; 20 nM final concentrations) was added in acetone at 4 0 C and the reaction was incubated for 5 minutes.
- the reaction was quenched with SnCl 2 in HCl.
- the amount of PGE 2 generated was detected by using a competitive HTRF PGE 2 assay kit (CisBio Bioassays, Bedford MA, USA) and determined by comparing values to a standard curve of PGE 2 .
- A549 human adenocarcinoma cells were pre-treated with test or positive control compounds for 30 minutes. Cells were then stimulated with ILl ⁇ (20 ng/mL) for 24 hours. An aliquot of cell media was transferred to a fresh assay plate and the level of PGE 2 generated was detected by using a competitive HTRF PGE 2 assay kit (CisBio Bioassays, Bedford MA, USA) and determined by comparing values to a standard curve of PGE 2 . IC 50 values are calculated based on PGE 2 levels by using a 4-parameter fit curve fitting model.
- A549 human adenocarcinoma cells were pre-treated with test or positive control compounds for 30 minutes prior to stimulation with ILl ⁇ (20 ng/mL) for 24 hours.
- the quantity of PGF 2a accumulated in the media was determined by transferring an aliquot of media to a fresh plate.
- PGF 2a was detected with the PGF 2a EIA Kit (Cayman Chemical, Ann Arbor, Michigan; Catalog # 516011) and absolute levels are determined by comparison to a standard curve of PGF 2a .
- Various compounds of Formula (I) in free form or in pharmaceutically acceptable salt form exhibit pharmacological properties, for example, as indicated by the in vitro tests described in this application.
- the IC 50 value in those experiments is given as that concentration of the test compound in question that provoke a response halfway between the baseline and maximum responses.
- compounds of Formula (I) have IC 50 values from 1 nM to 200 ⁇ M.
- compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 100 ⁇ M.
- compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 50 ⁇ M.
- compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 25 ⁇ M.
- compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 20 ⁇ M. In other examples, compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 15 ⁇ M. In other examples, compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 10 ⁇ M. In other examples, compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 5 ⁇ M. In other examples, compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 2 ⁇ M. In other examples, compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 1 ⁇ M.
- compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 0.8 ⁇ M. In other examples, compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 0.6 ⁇ M. In other examples, compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 0.4 ⁇ M. In other examples, compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 0.2 ⁇ M. In other examples, compounds of Formula (I) have IC 50 values from 0.001 ⁇ M to 0.1 ⁇ M.
- compounds of Formula (Ia) have IC 50 values from 1 nM to 200 ⁇ M. In some examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 100 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 50 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 25 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 20 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 15 ⁇ M.
- compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 10 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 5 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 2 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 1 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 0.8 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 0.6 ⁇ M.
- compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 0.4 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 0.2 ⁇ M. In other examples, compounds of Formula (Ia) have IC 50 values from 0.001 ⁇ M to 0.1 ⁇ M.
- the IC 50 for mPGES-1 inhibition by certain compounds of Formula (Ia) are also listed in Table 1 above.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Provided herein are compounds, and pharmaceutical compositions comprising such compounds, wherein the compounds are inhibitors of mPGES-1 activity. Also provided herein are methods of using such compounds and composition to treat or prevent diseases or disorders associated with the activity of mPGES-1.
Description
COMPOUNDS AND COMPOSITIONS AS MICROSOMAL PROSTAGLANDIN E SYNTHASE-I INHIBITORS
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. provisional application serial number 61/173,795, filed April 29, 2009; which is incorporated herein by reference its entirety.
FIELD OF THE INVENTION
[0002] Described herein are compounds, pharmaceutical compositions comprising such compounds, and methods of using such compounds to treat or prevent diseases or conditions associated with the activity of microsomal prostaglandin E synthase-1 (mPGES-1).
BACKGROUND OF THE INVENTION
[0003] Inflammation is a common cause of pain, with inflammatory pain resulting from a variety of causes, such as infection, surgery or other trauma. Other inflammatory diseases include asthma, chronic obstructive pulmonary disease (COPD), inflammatory bowel disease, rheumatoid arthritis, osteoarthritis, rhinitis, conjunctivitis and dermatitis. Moreover, several diseases including malignancies and cardioavascular diseases are known to have inflammatory components adding to the symptomatology of the patients. [0004] The cyclooxygenase (COX) is the enzyme that mediates biosynthesis of prostaglandins (PGs) and thromboxane (TXA2) from arachidonic acid. The cyclooxygenase (COX) enzyme activity originates from two distinct and independently regulated isozymes: COX-I which is constitutively expressed in many cells and tissues and catalyzes the formation of prostaglandins that subserve housekeeping functions, such as the maintenance of gastrointestinal (GI) integrity; and COX-2 which is induced by pro-inflammatory stimuli, such as cytokines, during an inflammatory response and is the dominant source of prostaglandins which mediate pain and inflammation. These enzymes metabolize arachidonic acid to the unstable intermediate prostaglandin H2 (PGH2). PGH2 is further metabolized to other prostaglandins including PGE2, PGF2CC, PGD2, prostacyclin and thromboxane A2. These arachidonic acid metabolites are known to have pronounced physiological and pathophysiological activity including pro-inflammatory effects.
[0005] Prostaglandin H2 (PGH2) is transformed to PGE2 by prostaglandin E synthases (PGES). Three prostaglandin E synthases (PGES)are known: two microsomal prostaglandin E synthases (mPGES-1 and mPGES-2), and one cytosolic prostaglandin E synthase (cPGES). Microsomal prostaglandin E synthase-1 (mPGES-1) belongs to the membrane- associated proteins in the eicosanoid and glutathione metabolism (MAPEG) family. Other members of this family include the microsomal glutathione S-transferases (MGSTl, MGST2 and MGST3).
[0006] PGE2 in particular is a strong pro-inflammatory mediator, and is known to induce fever and pain. PGE2 is also involved in arthritis and inflammation. The inhibition of PGE2 formation by inhibiting the COX enzymes underlies the effectiveness of a variety of antiinflammatory drugs. Thus, numerous drugs, including non-steroidal anti-inflammatory drugs (NSAIDs) and coxibs (selective COX-2 inhibitors), have been developed for inhibiting the formation of PGE2. Such drugs act predominantly by inhibition of COX-I and/or COX-2, thereby reducing the formation of PGE2. Unfortunately, the inhibition of COX-I and/or COX-2 also results in the reduction of the formation of all metabolites of arachidonic acid, some of which are known to have beneficial properties. Traditional non-steroidal antiinflammatory drugs (tNS AIDs) inhibit both COX-I and COX-2. However, a consequence of such non-selective inhibition has been adverse gastrointestinal effects, including both direct and indirect irritation of the gastrointestinal tract, and affects on platelet and renal function. [0007] The coxibs, selective inhibitors of COXS-2, were designed to inhibit the major enzymatic source of the prostaglandins which mediate pain and inflammation, while sparing PGHS-I -derived prostaglandins which contribute dominantly to gastric cytoprotection. However, while the selective inhibition of COX-2 by coxibs reduces such gastrointestinal side-effects, cardiovascular problems arise.
[0008] All of the coxibs depress substantially the level of prostacyclin (PGI2), leaving platelet COX-I -derived thromboxane A2 (TxA2) level unaffected. PGI2, the dominant product of arachidonic acid in macrovascular endothelial cells, is formed by prostacyclin synthase (PGIS) action on prostaglandin endoperoxide intermediates, which are produced catalytically by COX-2. PGI2 exhibits properties of potential relevance to atheroprotection by inhibiting platelet aggregation, vascular smooth muscle contraction and proliferation,
leukocyte-endothelial cell interactions and cholesteryl ester hydrolase. PGI2 also activates reverse cholesterol transport. Indirect evidence suggests that PGI2 protects against oxidant- induced tissue injury. Deletion of the PGI2 receptor (IP) or suppression of PGI2 biosynthesis augments cardiac injury caused by schemia/reperfusion or the anthracycline, doxarubacin. PGI2 also limits the cardiovascular effects of thromboxane A2 (TxA2), the major PGHS-I product of platelets. The cardiovascular effects of TxA2 include: platelet aggregation, elevation of blood pressure and acceleration of atherogenesis.
[0009] Inhibition of the transformation of PGH2 to the pro-inflammatory mediator PGE2 might be expected to reduce the inflammatory response, in the absence of a corresponding reduction of the formation of other, beneficial arachidonic acid metabolites, and thereby alleviate the undesirable side-effects mentioned above. Thus, agents that are capable of inhibiting the action of in PGES, in particular inhibitors of microsomal prostaglandin E synthase-1 (mPGES-1), and thus reducing the formation of the specific arachidonic acid metabolite PGE2, are likely to be of benefit in the treatment of inflammation and pain.
SUMMARY OF THE INVENTION
[00010] Provided herein are compounds and pharmaceutical compositions thereof, which are useful inhibitors of microsomal prostaglandin E synthase-1 (mPGES-1). Provided herein are compounds and pharmaceutical compositions thereof, which are useful inhibitors of microsomal prostaglandin E synthase-1 (mPGES-1). Also provided herein are methods of using such compound and pharmaceutical compositions for treating diseases or disorders associated with mPGES-1 activity. Further provided herein are methods of using such compound and pharmaceutical compositions for treating diseases or disorders in which mPGES-1 activity contributes to the pathology and/or symptomolgy of the disease or disorder.
[00011] In one aspect provided herein such compounds, and the pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, have a structure according to Formula (I):
 wherein, each R1 is independently selected from H, CrCβalkyl, or -(CH2XC(O)ORg, ;
wherein, each R1 is independently selected from H, CrCβalkyl, or -(CH2XC(O)ORg, ;
R2 is H, Ci-Cealkyl, aryl, halogen, -CN, -C(O)OR1 or -C(O)NR1R1;
R3 is -CN, CrCealkylA-Cόhaloalkyl or halogen;
R4 is H, -CN, Ci-Cealkyl , CrCehaloalkyl or halogen;
R5 is H, halogen, CrCβalkyl, CrCβalkoxy or 4-6 membered heterocycloalkyl containing
1-3 heteroatoms independently selected from N, O and S; L1 is a bond, C2-C6alkenylene, C2-Coalkynylene, -C(O)NRs-,

X1 is CR15 or N;
X2 is O, S or NR8;
X3 is S, O or NR8;
X4 is CR15 or N;
X5 is CR15 or N; each Re and each R7 are independently selected from H, halogen, -CN, CrCealkyl, Ci.Cehaloalkyl, -OQ-Cealkyl, -OCi.Cehaloalkyl, -Q2, -C(O)OR8, -C(O)N(Rg)2, -NR8C(O)R8, -N(Rg)2, -S(O)2R8, - NR8C(O)Q2 and -OR8; each R8 is independently selected from H and CrCόalkyl;
each R45 is independently selected from H, halogen, -CN, CrCβalkyl, Ci-Cβhaloalkyl, -OCi.Cβalkyl, -OCi.Cβhaloalkyl, -Q2, -C(O)OR8, -C(O)N(Rg)2, -NR8C(O)R8, -N(Rg)2, -S(O)2R8, - NR8C(O)Q2 and -OR8; L2 is a bond, -0-, -C(O)-, -C(O)NR8-, -NR8C(O)-, -NR8C(O)NR8-,
-(CR9Rg)nNR8C(O)NR8-, -C2-C6alkenylene, C2-C6alkynylene, phenylene, Cioarylene, C14arylene, 4-6 membered heterocycloalkylene containing 1-3
heteroatoms independently selected from N, O and S,

 wherein:
wherein:
X6 is C(R9)2, NR9, O or S; X7 is N or CR9; X8 is CR9, NR9, O or S; X9 is N or CR9; X10 is S or O; X11 is CR9 or N; each R9 is independently H or Ci-Cβalkyl; and wherein the C2-C6alkenylene, C2-Coalkynylene, arylenes and heterocycloalkylenes of L2 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, CN, -OC1- Cβalkyl, halogen and OH; Q1 is phenyl, Cioaryl, C14aryl, 5-6 membered heteroaryl containing 1-2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S, C3-C8cycloalkyl, C4- C8cycloalkyl optionally substituted with =CH2 cyclohexyl substituted with 2 Qalkyl which together with the carbon to which they are attached form a
cyclopropyl, L3Q2, -(CRi0RiI)nQ2,


Q2 is phenyl, Cioaryl, Ci4aryl, 5-6 membered heteroaryl containing 1- 2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S or C3-C8cycloalkyl, each of which is optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, -CN, - N(Rw)2, -OCi_C6alkyl, halogen and -OH;
L3 is C1-C βalkylene, C2-C βalkenylene or C2-C βalkynylene;
X12 is NR12, S or O;
X13 is (CH2)q or O(CH2)P;
R12 is H, Ci-Cealkyl or -C(O)OR14;
R13 is H, -C(O)OR14 or Ci-ealkyl, each R14 is independently selected from H or C1-C βalkyl; n is O, 1, 2 or 3; m is O, 1, 2, 3 or 4; p is O, 1, 2 or 3; q is 1, 2 or 3; s is 0 or 1 ; t is O, 1, 2, 3 or 4; provided that when L1 and L2 are each a bond, then Q1 is phenyl, Cioaryl, C14aryl, 5-6 membered heteroaryl containing 1-2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S, C3-C8cycloalkyl, C4-C8cycloalkyl optionally substituted with =CH2 cyclohexyl substituted with 2 Cialkyl which together with the carbon to which they are attached form a cyclopropyl,
L3Q2, -(CR10Rn)nQ2,
 , , or
, , or


[00012] Also provided herein are certain embodiments of such compounds of Formula (I), or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, wherein such embodiments are compounds having the structure of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof:

(Ia) wherein, each R1 is independently selected from H and Ci-Cβalkyl; R2 is H, halogen, -CN, -C(O)OR1 or -C(O)NR1R1; R3 is Ci-Cόhaloalkyl or halogen; R4 is H, CrCόhaloalkyl or halogen;

X1 is CR15 or N;
X2 is O, S or NR8;
X3 is S, O or NR8;
X4 is CR15 or N;
X5 is CR15 or N; each Re and each R7 are independently selected from H, halogen,
Ci-Cβalkyl, Ci.Cβhaloalkyl, and -C(O)OR8; each R8 is independently selected from H and CrCβalkyl; each R1S is independently selected from H, halogen and -
C(O)OR8; L2 is a bond, C2-C6alkynylene, phenylene, -0-, -C(O)-, -C(O)NR8-, -
NR8C(O)-, -NR8C(O)NR8-,


X6 is C(R9)2, NR9, O or S; X7 is N or CR9; X8 is CR9, NR9, O or S; X9 is N or CR9;
 X11 is CR9 or N; each R9 is independently H or CrCόalkyl; and wherein the C2-C6alkynylene and phenylene of L2 are each optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, CN, -OCi-Cβalkyl, halogen and OH;
X11 is CR9 or N; each R9 is independently H or CrCόalkyl; and wherein the C2-C6alkynylene and phenylene of L2 are each optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, CN, -OCi-Cβalkyl, halogen and OH;
Q1 is phenyl, 5-6 membered heteroaryl containing 1-2 N heteroatoms, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently
selected from N, O and S, Cs-Cscycloalkyl, C4-C8cycloalkyl optionally substituted with =CH2 cyclohexyl substituted with 2 Cialkyl which together with the carbon to which they are attached

Q2 is phenyl, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S or C3- Cgcycloalkyl, each of which is optionally substituted with 1-4 substituents independently selected from halogen and C1- C6haloalkyl;
X12 is NR12, S or O;
Xi3 is -(CH2),- or -O(CH2)P-;
R12 is H, Ci-Cealkyl or -C(O)OR14; each R14 is independently selected from H and C1-C βalkyl; n is O, 1, 2 or 3; p is O, 1, 2 or 3; q is 1, 2 or 3; s is 0 or 1, provided that when the combination of L1, L2 and Q1 is, L1 is pyridyl and L2 is
C2alkynylene and Q1 is phenyl, pyridyl, thiazolyl, cyclohexyl or cyclopropyl, then R2 is -CN, -C(O)OR1 or -C(O)NR1R1
[00013] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of
isomers thereof, L1 is a bond,


[00014] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of
isomers thereof L2 is a bond, -O-, -C(O)-, C2alkynylene, phenylene,


[00015] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of
isomers thereof, Q1 is phenyl, pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl optionally substituted with =CH2, cyclopentyl optionally substituted with =CH2, cyclohexyl optionally substituted with =CH2, cyclohexyl substituted with 2 Cialkyl which together with the carbon to which they
are attached form a Cscycloalkyl, -(CR1ORn)nQ2,


, or R6 , where the phenyl, pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl of Q1 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -0(CRioRn)i- 4OR10, -0(CRioR11)1-4D , -0(CRiOR11)WR1O, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORi4, N(Rw)2, -C(O)N(RM)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, -S(O)2Ri4, -SRi4 and -ORi4. [00016] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of
isomers thereof, Li is
 or ancj Q1 js phenyl optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -0(CRioRn)i-4θRio, -O(CRi0Rn)wD , -0(CRi0Rn)i-4Rio, -OCi_C6alkyl, -OCi-
Cβhaloalkyl, -C(O)OR14, -N(Rw)2, -C(O)N(R14)2, - NR14C(O)R145 -NR14C(O)Q2, - S(O)2R14, -SR14 and -OR14.
or ancj Q1 js phenyl optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -0(CRioRn)i-4θRio, -O(CRi0Rn)wD , -0(CRi0Rn)i-4Rio, -OCi_C6alkyl, -OCi-
Cβhaloalkyl, -C(O)OR14, -N(Rw)2, -C(O)N(R14)2, - NR14C(O)R145 -NR14C(O)Q2, - S(O)2R14, -SR14 and -OR14.
[00017] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of
isomers thereof, L1 is
 , L2 is C2alkynylene and Q1 is phenyl optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, C1- Cβhaloalkyl, halogen, -CN, -Q2, -O(CRi0R11)1-4ORi0, -O(CRi0R11)1-4D , -0(CRi0R1 i)i-4Rio, - OCi-Cβalkyl, -OCi-Cehaloalkyl, -C(O)ORi4, -N(Rw)2, -C(O)N(Rw)2, - NRi4C(O)Rw, - NRwC(O)Q2, -S(O)2Rw, -SRw and -ORi4.
, L2 is C2alkynylene and Q1 is phenyl optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, C1- Cβhaloalkyl, halogen, -CN, -Q2, -O(CRi0R11)1-4ORi0, -O(CRi0R11)1-4D , -0(CRi0R1 i)i-4Rio, - OCi-Cβalkyl, -OCi-Cehaloalkyl, -C(O)ORi4, -N(Rw)2, -C(O)N(Rw)2, - NRi4C(O)Rw, - NRwC(O)Q2, -S(O)2Rw, -SRw and -ORi4.
[00018] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of
isomers thereof, Li is
 or , L2 is a bond and Qi is phenyl optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -0(CRi0Ri i)i-4ORio, -O(CRi0Rn)i_4D , -O(CRi0Rn)i_4Ri0, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORw, -N(Rw)2, -C(O)N(Rw)2, - NRWC(O)RW5 -NRWC(O)Q2, - S(O)2Rw, -SRw and -ORw.
or , L2 is a bond and Qi is phenyl optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -0(CRi0Ri i)i-4ORio, -O(CRi0Rn)i_4D , -O(CRi0Rn)i_4Ri0, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORw, -N(Rw)2, -C(O)N(Rw)2, - NRWC(O)RW5 -NRWC(O)Q2, - S(O)2Rw, -SRw and -ORw.
[00019] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of
isomers thereof, Li is
 or , L2 is a bond and Qi is pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl optionally substituted with =CH2, cyclopentyl optionally substituted with =CH2, cyclohexyl optionally substituted with =CH2, cyclohexyl substituted with 2
dalkyl which together with the carbon to which they are attached form a Cscycloalkyl, -
or , L2 is a bond and Qi is pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl optionally substituted with =CH2, cyclopentyl optionally substituted with =CH2, cyclohexyl optionally substituted with =CH2, cyclohexyl substituted with 2
dalkyl which together with the carbon to which they are attached form a Cscycloalkyl, -

[00020] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, Li is a bond , L2 is a bond and Qi is phenyl, pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl optionally substituted with =CH2, cyclopentyl optionally substituted with =CH2, cyclohexyl optionally substituted with =CH2, cyclohexyl substituted with 2 Cialkyl which together with the carbon to which they are attached form a Cscycloalkyl, -(CR1ORn)nQ2,


[00021] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, each R1 is independently selected from H, methyl and ethyl. [00022] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, R2 is H, Br, -CN, -C(O)OR1 or -C(O)NR1R1; provided that when the combination of L1, L2 and Q1 is, L1 is pyridyl and L2 is C2alkynylene and Q1 is phenyl, pyridyl, thiazolyl, cyclohexyl or cyclopropyl, then R2 is -CN, -C(O)OR1 or -C(O)NR1R1. [00023] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, R3 is -CF3, Cl or F.
[00024] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, R4 is H, -CF3, Cl or F.
[00025] In certain embodiments of such compounds of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, Q2 is phenyl, 5 optionally substituted with 1-4 halogen groups. [00026] Certain embodiments of such compounds of Formula (Ia) are selected from: 2- (2- chloro-6-fluorophenyl)-5-[6-(2-phenylethynyl)pyridin-3-yl]-lH-imidazole-4-carbonitrile; 2- (2-chloro-6-fluorophenyl)-5-[6-(2-phenylethynyl)pyridin-3-yl]-lH-imidazole-4- carboxamide; ethyl 2-(2-chloro-6-fluorophenyl)-4-[6-(2-phenylethynyl)pyridin-3-yl]- IH- imidazole-5-carboxylate; 2-(2-chloro-6-fluorophenyl)-5-[6-(2-phenylethynyl)pyridin-3-yl]- lH-imidazole-4-carboxylic acid; 2-(2-chloro-6-fluorophenyl)-N-methyl-5-[6-(2- phenylethynyl)pyridin-3-yl]-lH-imidazole-4-carboxamide; 5-[5-bromo-2-(2-chloro-6- fluorophenyl)-lH-imidazol-4-yl]-2-(2-phenylethynyl)pyrimidine; 5-[4-bromo-2-(2-chloro-6- fluorophenyl)- lH-imidazol-5-yl]-2-phenylpyrimidine; 2-{ 5-[2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl]pyrimidin-2-yl}-lH-indole; 2-(2-chloro-6-fluorophenyl)-4-[5-(2- phenylethynyl)thiophen-2-yl]- lH-imidazole; 5-[5-bromo-2-(2-chloro-6-fluorophenyl)- IH- imidazol-4-yl]-2-(4-tert-butylphenyl)pyridine; 2-{5-[2-(2-chloro-6-fluorophenyl)-lH- imidazol-5-yl]pyridin-2-yl}-lH-indole; 5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH- imidazol-5-yl]-2-[4-(2-methylpropyl)phenyl]pyridine; 5-[4-bromo-2-(2-chloro-6- fluorophenyl)-lH-imidazol-5-yl]-2-[3-(trifluoromethyl)phenyl]pyridine; 4-{5-[4-bromo-2- (2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridin-2-yl}benzonitrile; 3-{5-[4-bromo-2-(2- chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridin-2-yl}benzonitrile; 5-[4-bromo-2-(2- chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[4-(trifluoromethoxy)phenyl]pyridine; 5-[4- bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[3-
(trifluoromethoxy)phenyl]pyridine; tert-butyl 5-{ 5-[4-bromo-2-(2-chloro-6-fluorophenyl)- lH-imidazol-5-yl]pyridin-2-yl}-lH-indole-l-carboxylate; 5-{5-[2-(2-chloro-6- fluorophenyl)- lH-imidazol-5-yl]pyridin-2-yl}- 1-methyl- lH-indole; 2-(l-benzothiophen-5- yl)-5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridine; 5-[2-(2-chloro-6- fluorophenyl)-lH-imidazol-5-yl]-2-[4-(trifluoromethyl)phenyl]pyridine; 5-[2-(2-chloro-6- fluorophenyl)- lH-imidazol-5-yl]-2-(2,3-dihydro- l-benzofuran-5-yl)pyridine; 5-[2-(2-chloro- 6-fluorophenyl)-lH-imidazol-5-yl]-2-(3,4-dihydro-2H-l,5-benzodioxepin-7-yl)pyridine; 5-
[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-phenyl-l,2,4-oxadiazol-3-yl)pyridine; 5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-phenyl-l,2,4-oxadiazol-3- yl)pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-cyclohexyl- 1,2,4- oxadiazol-3-yl)pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5-(propan-2- yl)-l,2,4-oxadiazol-3-yl]pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5- (2-methylpropyl)-l,2,4-oxadiazol-3-yl]pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH- imidazol-5-yl]-2-[5-(4,4-difluorocyclohexyl)-l,2,4-oxadiazol-3-yl]pyridine; 5-[2-(2-chloro- 6-fluorophenyl)-lH-imidazol-5-yl]-2-{5-[l-(4-fluorophenyl)cyclopropyl]-l,2,4-oxadiazol-3- yljpyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-cyclopentyl- 1,2,4- oxadiazol-3-yl)pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5-(pentan-3- yl)-l,2,4-oxadiazol-3-yl]pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-{5- [2-(3-fluorophenyl)ethyl]-l,2,4-oxadiazol-3-yl}pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH- imidazol-5-yl]-2-[5-(2-phenylethyl)-l,2,4-oxadiazol-3-yl]pyridine; 5-[2-(2-chloro-6- fluorophenyl)-lH-imidazol-5-yl]-2-(5-{spiro[2.5]octan-6-yl}-l,2,4-oxadiazol-3-yl)pyridine; 2-(5-benzyl-l,2,4-oxadiazol-3-yl)-5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5- yl]pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-phenyl-l,3-oxazol-2- yl)pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-phenyl-l,3-thiazol-2- yl)pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5-(4-methoxyphenyl)-l,3- thiazol-2-yl]pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-phenoxypyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(4-methoxyphenoxy)pyridine; 2-(2- chloro-6-fluorophenyl)-5-(2-methoxypyrimidin-5-yl)-lH-imidazole-4-carbonitrile; 2-(2- chloro-6-fluorophenyl)-5-(2-phenoxypyrimidin-5-yl)-lH-imidazole-4-carbonitrile; 2-(2- chloro-6-fluorophenyl)-5-[2-(l-cyclopropylethoxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-(2-cyclobutoxypyrimidin-5-yl)-lH-imidazole-4- carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(l-phenylethoxy)pyrimidin-5-yl]-lH- imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-{2-[(l-methoxypropan-2- yl)oxy]pyrimidin-5-yl}-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2- (oxan-4-yloxy)pyrimidin-5-yl]-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5- [2-(propan-2-yloxy)pyrimidin-5-yl]-lH-imidazole-4-carbonitrile; 5-[2- (benzyloxy)pyrimidin-5-yl]-2-(2-chloro-6-fluorophenyl)-lH-imidazole-4-carbonitrile; 2-(2-
chloro-6-fluorophenyl)-5-[2-(propan-2-yloxy)pyrimidin-5-yl]-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(cyclopentyloxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(cyclohexyloxy)pyrimidin-5-yl]-lH- imidazole-4-carbonitrile; 5-[2-(butan-2-yloxy)pyrimidin-5-yl]-2-(2-chloro-6-fluorophenyl)- lH-imidazole-4-carbonitrile; 5-[2-(benzyloxy)pyrimidin-5-yl]-2-[2-fluoro-6- (trifluoromethyl)phenyl]-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-{2- [(1,1,1, 3,3, 3-hexafluoropropan-2-yl)oxy]pyrimidin-5-yl}-lH-imidazole-4-carbonitrile; 2- (2,6-dichlorophenyl)-5-[2-(propan-2-yloxy)pyrimidin-5-yl]-lH-imidazole-4-carbonitrile; 2- [2-fluoro-6-(trifluoromethyl)phenyl]-4-[2-(propan-2-yloxy)pyrimidin-5-yl]-lH-imidazole-5- carbonitrile; 5-{2-[(4-tert-butylcyclohexyl)oxy]pyrimidin-5-yl}-2-(2-chloro-6-fluorophenyl)- lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-(2-{ [(lS,2R,5R)-2-methyl-5- (propan-2-yl)cyclohexyl]oxy}pyrimidin-5-yl)-lH-imidazole-4-carbonitrile; 2-(2-chloro-6- fluorophenyl)-5-{2-[(4,4-dimethylcyclohexyl)oxy]pyrimidin-5-yl}-lH-imidazole-4- carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-(2-{ [(2S)-l,l,l-trifluoropropan-2- yl]oxy}pyrimidin-5-yl)-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-(2- { [(2R)-l,l,l-trifluoropropan-2-yl]oxy}pyrimidin-5-yl)-lH-imidazole-4-carbonitrile; 2-(2- chloro-6-fluorophenyl)-5-[2-(2,2,4,4-tetrafluorocyclobutoxy)pyrimidin-5-yl]-lH-imidazole- 4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(2,3-dihydro-lH-inden-2-yloxy)pyrimidin- 5-yl]-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-(2-{ [4- (trifluoromethyl)cyclohexyl]oxy}pyrimidin-5-yl)-lH-imidazole-4-carbonitrile; 2-(2-chloro- 6-fluorophenyl)-4-{2-[(l,l,l-trifluoropropan-2-yl)oxy]pyrimidin-5-yl}-lH-imidazole-5- carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(pentan-3-yloxy)pyrimidin-5-yl]-lH- imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-(2-ethoxypyrimidin-5-yl)-lH- imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(4-methoxyphenoxy)pyrimidin- 5-yl]-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(3- methoxyphenoxy)pyrimidin-5-yl]-lH-imidazole-4-carbonitrile; 5-[2-(benzyloxy)pyrimidin- 5-yl]-2-[2-chloro-6-(trifluoromethyl)phenyl]-lH-imidazole-4-carbonitrile; 2-(2-chloro-6- fluorophenyl)-4-[2-(methylsulfanyl)pyrimidin-5-yl]-lH-imidazole-5-carbonitrile; 2-(2- chloro-6-fluorophenyl)-5-(2-methanesulfonylpyrimidin-5-yl)-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(propan-2-ylsulfanyl)pyrimidin-5-yl]-lH-imidazole-4-
carbonitrile; 5-[2-(butan-2-ylsulfanyl)pyrimidin-5-yl]-2-(2-chloro-6-fluorophenyl)-lH- imidazole-4-carbonitrile; 5-[2-(tert-butylsulfanyl)pyrimidin-5-yl]-2-(2-chloro-6- fluorophenyl)-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-4-(5-phenyl-l- benzofuran-2-yl)-lH-imidazole; 2-(2-chloro-6-fTuorophenyl)-4-[5-(2-phenylethynyl)-l- benzofuran-2-yl] - lH-imidazole; 5- { 2- [2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl] - 1 - benzofuran-5-yl}-3-(propan-2-yl)-l,2,4-oxadiazole; 2-(2-chloro-6-fluorophenyl)-4-[5-(4- phenylphenyl)- l-benzofuran-2-yl]- lH-imidazole; 2-(2-chloro-6-fTuorophenyl)-4-[5-(3,5- dimethylphenyl)-l-benzofuran-2-yl]-lH-imidazole; 2-(2-chloro-6-fTuorophenyl)-4-{5-[4-(2- methylpropyl)phenyl]- l-benzofuran-2-yl}- lH-imidazole; 4-[5-(4-tert-butylphenyl)- 1- benzofuran-2-yl]-2-(2-chloro-6-fluorophenyl)-lH-imidazole; 5-bromo-2-(2-chloro-6- fluorophenyl)-4-[5-(2-phenylethynyl)- l-benzofuran-2-yl]- lH-imidazole; 5-bromo-4-[3- bromo-5-(2-phenylethynyl)-l-benzofuran-2-yl]-2-(2-chloro-6-fluorophenyl)-lH-imidazole; methyl 4-{2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5-yl}benzoate; methyl 2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-6-carboxylate; 5-{2- [2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5-yl}-3-methyl- 1,2,4- oxadiazole; 5-{2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5-yl}-3- cyclopropyl-l,2,4-oxadiazole; 3-(5-{2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l- benzofuran-5-yl}-l,2,4-oxadiazol-3-yl)pyridine; 4-(5-{2-[2-(2-chloro-6-fluorophenyl)-lH- imidazol-4-yl]-l-benzofuran-5-yl}-l,2,4-oxadiazol-3-yl)pyridine; ethyl 4-{2-[2-(2- fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5-yl}benzoate; 5-bromo-2-[2-(2-chloro-6- fluorophenyl)-lH-imidazol-4-yl]-lH-l,3-benzodiazole; 4-{5-bromo-lH-imidazo[4,5- b]pyridin-2-yl}-2-(2-chloro-6-fluorophenyl)-lH-imidazole; 5-bromo-2-[2-(2-chloro-6- fluorophenyl)-lH-imidazol-4-yl]-l-methyl-lH-l,3-benzodiazole; ethyl 5-[2-(2-chloro-6- fluorophenyl)- lH-imidazol-4-yl]-2-(2-phenylethynyl)pyridine-4-carboxylate; methyl 5-[2- (2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-2-(2-phenylethynyl)pyridine-4-carboxylate; 2- (2-chloro-6-fluorophenyl)-5-(2-phenylethynyl)-lH-imidazole-4-carbonitrile; 2-(2-chloro-6- fluorophenyl)-5- { 2-[4-(propan-2-yl)phenyl]ethynyl }- lH-imidazole-4-carbonitrile; 2-(2- chloro-6-fluorophenyl)-5-[2-(4-phenylphenyl)ethynyl]-lH-imidazole-4-carbonitrile; 4- chloro-N-(3-{2-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5- yl]ethynyl}phenyl)benzamide; 2-(2-chloro-6-fluorophenyl)-5-{2-[4-
(trifluoromethoxy)phenyl]ethynyl}-lH-imidazole-4-carbonitrile; 2-(2-chloro-6- fluorophenyl)-5-{2-[3-(trifluoromethyl)phenyl]ethynyl}-lH-imidazole-4-carbonitrile; 2- (2- chloro-6-fluorophenyl)-5-[2-(4-methoxyphenyl)ethynyl]-lH-imidazole-4-carbonitrile; 2- (2- chloro-6-fluorophenyl)-5-[2-(4-methoxy-2-methylphenyl)ethynyl]-lH-imidazole-4- carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(4-ethoxyphenyl)ethynyl]-lH-imidazole-4- carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(3,5-dimethoxyphenyl)ethynyl]-lH- imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(l-hydroxycyclohexyl)ethynyl]- lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(4-cyanophenyl)ethynyl]-lH- imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-{2-[4-(piperidin-l- yl)phenyl]ethynyl}-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(pyridin- 3-yl)ethynyl]-lH-imidazole-4-carbonitrile; 5-[2-(3-aminophenyl)ethynyl]-2-(2-chloro-6- fluorophenyl)- lH-imidazole-4-carbonitrile; 5-[4-bromo-2-(2-chloro-6-fluorophenyl)- IH- imidazol-5-yl]-3-phenyl-l,2,4-oxadiazole; 5-[5-bromo-2-(2-chloro-6-fluorophenyl)-lH- imidazol-4-yl]-3-(4-fluorophenyl)-l,2,4-oxadiazole; 2-(2-chloro-6-fluorophenyl)-4-{6-[3- (propan-2-yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-lH-imidazole-5-carbonitrile; 2-(2-chloro- 6-fluorophenyl)-5-[6-(3-cyclopropyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl]-lH-imidazole-4- carbonitrile; 5-{6-[3-(butan-2-yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-2-(2-chloro-6- fluorophenyl)-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-{6-[3-(oxan-4- yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-lH-imidazole-4-carbonitrile; 2-(2-chloro-6- fluorophenyl)-4-[6-(3-cyclopentyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl]-lH-imidazole-5- carbonitrile; tert-butyl 3-(5-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5- yl]pyridin-2-yl}-l,2,4-oxadiazol-3-yl)azetidine-l-carboxylate; 2-(2-chloro-6-fluorophenyl)- 5-{6-[3-(3,3-difluorocyclobutyl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-lH-imidazole-4- carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[6-(3-cyclobutyl-l,2,4-oxadiazol-5-yl)pyridin-3- yl]-lH-imidazole-4-carbonitrile; 5-{6-[3-(butan-2-yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-2- (2-chloro-6-fluorophenyl)-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-{6- [3-(4,4-difluorocyclohexyl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-lH-imidazole-4-carbonitrile; 5-[6-(3-tert-butyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl]-2-(2-chloro-6-fluorophenyl)-lH- imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-{6-[3-(3-methylidenecyclobutyl)- l,2,4-oxadiazol-5-yl]pyridin-3-yl}-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-
fluorophenyl)-4-{6-[3-(3-methylphenyl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-lH-imidazole-5- carbonitrile; 2-(2-chloro-6-fluorophenyl)-4-{6-[3-(4-methylphenyl)-l,2,4-oxadiazol-5- yl]pyridin-3-yl}-lH-imidazole-5-carbonitrile; 2-[2-fluoro-6-(trifluoromethyl)phenyl]-5-{6- [3-(propan-2-yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-lH-imidazole-4-carbonitrile; 2-(2,6- dichlorophenyl)-5-{6-[3-(propan-2-yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-lH-imidazole-4- carbonitrile; 2-(2-chloro-6-fluorophenyl)-4-[6-(3-phenyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl]- lH-imidazole-5-carbonitrile; 2-(2-chloro-6-fluorophenyl)-4-{6-[3-(4-fluorophenyl)-l,2,4- oxadiazol-5-yl]pyridin-3-yl}-lH-imidazole-5-carbonitrile; 2-(2-chloro-6-fluorophenyl)-4-[6- (5-phenyl-l,3,4-oxadiazol-2-yl)pyridin-3-yl]-lH-imidazole-5-carbonitrile; 5-[2-(2-chloro-6- fluorophenyl)-5-cyano-lH-imidazol-4-yl]-N-(4-fluorophenyl)pyridine-2-carboxamide; 5-[2- (2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]-N-(3-fluorophenyl)pyridine-2- carboxamide; N-(2-amino-5-fluorophenyl)-5-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH- imidazol-4-yl]pyridine-2-carboxamide; 2-(2-chloro-6-fluorophenyl)-4-(6-{ [2-(3- fluoropheny^pyrrolidin-l-y^carbonylJpyridin-S-y^-lH-imidazole-S-carbonitrile; 5-[2-(2- chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]-N-phenylpyridine-2-carboxamide; 5-[2- (2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(4-fluorophenyl)pyridine-2-carboxamide; 5- [2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(3-fluorophenyl)pyridine-2-carboxamide; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(2-fluorophenyl)pyridine-2- carboxamide; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(3- methylphenyl)pyridine-2-carboxamide; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]- N-(4-methylphenyl)pyridine-2-carboxamide; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5- yl]-N-phenylpyridine-2-carboxamide; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N- (3-cyanophenyl)pyridine-2-carboxamide; 5-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH- imidazol-4-yl]-N-(3-fluorophenyl)-N-methylpyridine-2-carboxamide; 2-(2-chloro-6- fluorophenyl)-4-(6-{ [(3S)-3-phenylmorpholin-4-yl]carbonyl}pyridin-3-yl)-lH-imidazole-5- carbonitrile; N-{5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridin-2-yl}benzamide; N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-4- fluorobenzamide; N- { 5-[2-(2-chloro-6-fluorophenyl)-4-cyano- lH-imidazol-5-yl]pyridin-2- yl}-3-(trifluoromethyl)benzamide; N- { 5-[2-(2-chloro-6-fluorophenyl)-4-cyano- lH-imidazol- 5-yl]pyridin-2-yl}-2-(trifluoromethyl)benzamide; N-{5-[2-(2-chloro-6-fluorophenyl)-4-
cyano-lH-imidazol-5-yl]pyridin-2-yl}-4-methylbenzamide; N-{5-[2-(2-chloro-6- fluorophenyl)-4-cyano- lH-imidazol-5-yl]pyridin-2-yl}-4-ethylbenzamide; N- { 5-[2-(2- chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-4- (trifluoromethyl)benzamide; N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5- yl]pyridin-2-yl}-3-fluorobenzamide; N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH- imidazol-5-yl]pyridin-2-yl}-3,4-difluorobenzamide; N-{5-[2-(2-chloro-6-fluorophenyl)-4- cyano-lH-imidazol-5-yl]pyridin-2-yl}-2-fluorobenzamide; N-{5-[2-(2-chloro-6- fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}benzamide; N-{5-[2-(2-chloro-6- fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3-methoxybenzamide; N-{5-[2-(2- chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-2,4-difluorobenzamide; N- {5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-4- (dimethylamino)benzamide; N-{ 5-[2-(2-chloro-6-fluorophenyl)-4-cyano- lH-imidazol-5- yl]pyridin-2-yl}-3-cyanobenzamide; N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH- imidazol-5-yl]pyridin-2-yl}-3-(trifluoromethoxy)benzamide; N-{5-[2-(2-chloro-6- fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3-(trifluoromethoxy)benzamide; 1- {5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3-(4- fluorophenyl)urea; N-{4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]phenyl}-4- fluorobenzamide; N-{4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]phenyl}-3- fluorobenzamide; N-{4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]phenyl}-4- (trifluoromethyl)benzamide; N-{4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4- yl]phenyl}benzamide; 4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]-N-(3- fluorophenyl)benzamide; methyl 2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l- benzofuran-5-carboxylate; 5-(5-bromo-l-benzofuran-2-yl)-2-(2-chloro-6-fluorophenyl)-lH- imidazole, and 5-bromo-2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l,3-benzoxazole. [00027] Another aspect provided herein are pharmaceutical compositions comprising a therapeutically effective amount of a compound of Formula (Ia) and a pharmaceutically acceptable carrier.
[00028] Another aspect provided herein is a method for treating a disease or disorder associated with mPGES-1 activity, wherein the method comprises administering to a subject in need thereof a therapeutically effective amount of a compound of Formula (Ia).
[00029] In certain embodimenst of such a method, the diseases or disorder is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease. In certain embodimenst of such a method the compound of Formula (Ia) is an inhibitor of mPGES-1.
[00030] Another aspect provided herein is the use of a compound of Formula (I) in the manufacture of a medicament for treating a disease or disorder associated with mPGES-1 activity, wherein the disease or disorder is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease.
[00031] Another aspect provided herein is a medicament for treating a disease or disorder associated with mPGES-1 activity, wherein the medicament comprises a therapeutically effective amount of a compound of Formula (Ia). In certain embodiments of such medicaments, the disease or disorder associated with mPGES-1 activity is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease. [00032] Another aspect provided herein is a compound of Formula (Ia) for use in a method of medical treatment, wherein the method of medical treatment is for treating a disease or disorder associated with mPGES-1 activity, wherein the disease or disorder is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease.
DETAILED DESCRIPTION OF THE INVENTION
[00033] Provided herein are compounds and composition that inhibit the activity of microsomal prostaglandin E synthase- 1 (mPGES-1) and are, therefore useful in the treatment of mPGES-1-associated diseases such as pain, including but not limited to, inflammatory pain, dental pain, postdental procedure pain, osteoarthritic pain, nociceptive pain, neuropathic pain muscoskeletal pain, low back pain, headaches, sinus headaches, migraines, post-surgical pain, surgical pain, burn injury and pain associated with bacterial, fungal or viral illnesses (by way of example only, influenza, the common cold, a viral infections (such as, by way of example only, influenza, common cold, herpes zoster, hepatitis C and AIDS), a bacterial infection or a fungal infection) and periodontal disease; inflammatory diseases, inflammation, arthritis, rheumatoid arthritis (RA), juvenile arthritis, inflammatory arthritis, arthritis, atherosis, tendonitis, bursitis, gouty arthritis, polymyalgia rheumatica, fibermyalgia, fibromyalgia, neuropathy, systemic lupus erythematosus, soft tissue injury, myalgia,
neuralgia, neuritis, osteoarthritis, primary dysmenorrheal, secondary dysmenorrheal, coronary artery disease, a myofascial disorder, atherosclerosis, fever, rheumatic fever, angiogenesis, granulation, cancer, tumorigenesis, gastrointestinal cancer, breast cancer, colorectal, colon cancer, lung and prostate cancer gastrointestinal homeostasis, Crohn's disease, ulcerative colitis, inflammatory bowel disease, irritable bowel syndrome, reproduction disorders, renal homeostasis, edema, Ductus Arteriosus Closure, Alzheimer's, a malignancy, ankylosing spondylitis, systemic lupus erythematosus, vasculitis, pancreatitis, nephritis, opthlamic conditions, conjunctivitis, iritis, scleritis, uveitis, wound healing, dermatitis, eczema, psoriasis, stroke, diabetes, a neurodegenerative disorder, a neurologic disorder, myofascial disorders, dysmenorrhea, hyperprostaglandin E syndrome, classic Bartter syndrome, Hodgkin's disease, multiple sclerosis, an immunological disorder, an autoimmune disease, osteoporosis, asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, an allergic disorder, rhinitis, an ulcer, a cardiovascular disorder, coronary heart disease, sarcoidosis, ischemia/reperfusion injury and any other disease with an inflammatory component. Definitions
[00034] The term "alkene" or "alkenyl", as used herein, refers to a partially unsaturated branched or straight chain hydrocarbon having at least one carbon-carbon double bond. Atoms oriented about the double bond are in either the cis (Z) or trans (E) conformation. As used herein, the terms "C2-C4alkenyl", "C2-C5alkenyl", "C2-C6alkenyl", "C2-C7alkenyl", and "C2-C8alkenyl" refer to an alkenyl group containing at least 2, and at most 4, 5, 6, 7 or 8 carbon atoms, respectively. Non-limiting examples of alkenyl groups, as used herein, include ethenyl, propenyl, allyl (2-propenyl), butenyl, pentenyl, hexenyl, heptenyl, octenyl, nonenyl, decenyl and the like. In certain embodiments an alkene or alkenyl group is optionally substituted.
[00035] The term "alkenylene," as used herein, refers to a saturated branched or straight chain divalent hydrocarbon radical derived from an alkenyl group. In certain embodiments an alkenylene group is optionally substituted. As used herein, the terms "C2-C3 alkenylene", "C2-C4alkenylene", "C2-C5alkenylene", "C2-C6alkenylene", "C2-C7alkenylene" and "C2- Cgalkenylene" refer to an alkenylene group containing at least 2, and at most 3, 4, 5, 6, 7 or 8
carbon atoms respectively. Non-limiting examples of alkenylene groups as used herein include, ethenlene, n-propenylene, isopropenylene, n-butenylene, isobutenylene, sec- butenylene, t-butenylene, n-pentenylene, isopentenylene, hexenylene and the like. [00036] The term "alkyl," as used herein, refers to a saturated branched or straight chain hydrocarbon. An alkyl group can be optionally substituted. As used herein, the terms "C1- C3alkyl", "Ci-C4alkyl", "Ci-C5alkyl", "Ci-C6alkyl", "Ci-C7alkyl" and "Ci-C8alkyl" refer to an alkyl group containing at least 1, and at most 3, 4, 5, 6, 7 or 8 carbon atoms, respectively. Non-limiting examples of alkyl groups as used herein include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec -butyl, t-butyl, n-pentyl, isopentyl, hexyl, heptyl, octyl, nonyl, decyl and the like. In certain embodiments an alkyl group is optionally substituted. [00037] The term "alkylene," as used herein, refers to a saturated branched or straight chain divalent hydrocarbon radical derived from an alkyl group. In certain embodiments an alkylene group is optionally substituted. As used herein, the terms "CrCsalkylene", "C1- C4alkylene", "Ci-C5alkylene", "Ci-C6alkylene", "Ci-C7alkylene" and "CrQalkylene" refer to an alkylene group containing at least 1, and at most 3, 4, 5, 6, 7 or 8 carbon atoms respectively. Non-limiting examples of alkylene groups as used herein include, methylene, ethylene, n-propylene, isopropylene, n-butylene, isobutylene, sec-butylene, t-butylene, n- pentylene, isopentylene, hexylene and the like.
[00038] The term "alkynyl", as used herein, refers to a partially unsaturated branched or straight chain hydrocarbon radical having at least one carbon-carbon triple bond. As used herein, the terms "C2-C4alkynyl", "C2-C5alkynyl", "C2-C6alkynyl", "C2-C7alkynyl", and "C2- Cgalkynyl" refer to an alkynyl group containing at least 2, and at most 4, 5, 6, 7 or 8 carbon atoms, respectively. Non-limiting examples of alkynyl groups, as used herein, include ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, octynyl, nonynyl, decynyl and the like. In certain embodiments an alkynyl group is optionally substituted. [00039] The term "alkynylene," as used herein, refers to a saturated branched or straight chain divalent hydrocarbon radical derived from an alkynyl group. In certain embodiments an alkynylene group is optionally substituted. As used herein, the terms "C2-C3alkynylene", "C2-C4alkynylene", "C2-C5alkynylene", "C2-C6alkynylene", "C2-C7alkynylene" and "C2- Cgalkynylene" refer to an alkynylene group containing at least 2, and at most 3, 4, 5, 6, 7 or
8 carbon atoms respectively. Non-limiting examples of alkenylene groups as used herein include, ethynlene, propynylene, butynylene, pentynylene, hexynylene and the like. [00040] The term "alkoxy," as used herein, refers to the group -ORa, where Ra is an alkyl group as defined herein. An alkoxy group can be optionally substituted. As used herein, the terms "Ci-C3alkoxy", "C1-C4alkoxy", "Ci-C5alkoxy", "Ci-C6alkoxy", "Ci-C7alkoxy" and "Ci-Cgalkoxy" refer to an alkoxy group wherein the alkyl moiety contains at least 1, and at most 3, 4, 5, 6, 7 or 8, carbon atoms. Non-limiting examples of alkoxy groups, as used herein, include methoxy, ethoxy, n-propoxy, isopropoxy, n-butyloxy, t-butyloxy, pentyloxy, hexyloxy, heptyloxy, octyloxy, nonyloxy, decyloxy and the like.
[00041] The term "aryl," as used herein, refers to monocyclic, fused bicyclic, and fused tricyclic ring systems having a total of six to fourteen ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains 3 to 7 ring members. In certain embodiments an aryl group is optionally substituted. Non-limiting examples of aryl groups, as used herein, include phenyl, naphthyl, fluorenyl, indenyl, azulenyl, anthracenyl and the like.
[00042] The term "arylene," as used means a divalent radical derived from an aryl group. In certain embodiments an arylene group is optionally substituted. [00043] The term "cyano," as used herein, refers to a -CN group.
[00044] The term "cycloalkyl," as used herein, refers to a saturated or partially unsaturated, monocyclic, fused bicyclic, fused tricyclic or bridged polycyclic ring assembly. As used herein, the terms "C3-C5cycloalkyl", "C3-C6cycloalkyl", " C3 -C7cyclo alkyl", "C3- Cgcycloalkyl, "Q-Cgcycloalkyl and "Q-Ciocycloalkyl refer to a cycloalkyl group wherein the saturated or partially unsaturated, monocyclic, fused bicyclic or bridged polycyclic ring assembly contain at least 3, and at most 5, 6, 7, 8, 9 or 10, carbon atoms. In certain embodiments a cycloalkyl group is optionally substituted. Non-limiting examples of cycloalkyl groups, as used herein, include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cyclopentenyl, cyclohexenyl, decahydronaphthalenyl, 2,3,4,5,6,7-hexahydro-lH-indenyl and the like. [00045] The term "cycloalkylene," as used means a divalent radical derived from a cycloalkyl group. In certain embodiments a cycloalkylene group is optionally substituted.
[00046] The term "halogen," as used herein, refers to fluorine (F), chlorine (Cl), bromine (Br), or iodine (I).
[00047] The term "halo," as used herein, refers to the halogen radicals: fluoro (-F), chloro (- Cl), bromo (-Br), and iodo (-1).
[00048] The terms "haloalkyl" or "halo-substituted alkyl," as used herein, refers to an alkyl group as defined herein, substituted with one or more halogen groups, wherein the halogen groups are the same or different. A haloalkyl group can be optionally substituted. Non- limiting examples of such branched or straight chained haloalkyl groups, as used herein, include methyl, ethyl, propyl, isopropyl, isobutyl and n-butyl substituted with one or more halogen groups, wherein the halogen groups are the same or different, including, but not limited to, trifluoromethyl, pentafluoroethyl, and the like.
[00049] The term "halo-substituted alkenyl", as used herein, refers to an alkenyl group as defined above, substituted with at least one halo group or combinations thereof. Non- limiting examples of such branched or straight chained haloalkenyl groups, as used herein, include ethenyl, propenyl, allyl (2-propenyl), butenyl, pentenyl, hexenyl, heptenyl, octenyl, nonenyl, decenyl and the like substituted with one or more halo groups or combinations thereof.
[00050] The term "halo-substituted alkynyl", as used herein, refers to an alkynyl group as defined above, substituted with at least one halo group or combinations thereof. Non- limiting examples of such branched or straight chained haloalkynyl groups, as used herein, include ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, octynyl, nonynyl, decynyl and the like substituted with one or more halo groups or combinations thereof. [00051] The term "haloalkoxy" or "halo-substituted -alkoxy," as used herein, refers to an alkoxy group as defined herein, substituted with one or more halogen groups, wherein the halogen groups are the same or different. A haloalkoxy group can be optionally substituted. Non-limiting examples of such branched or straight chained haloalkynyl groups, as used herein, include methoxy, ethoxy, n-propoxy, isopropoxy, n-butyloxy, t-butyloxy, pentyloxy, hexyloxy, heptyloxy, octyloxy, nonyloxy, decyloxy and the like, substituted with one or more halogen groups, wherein the halogen groups are the same or different.
[00052] The term "heteroalkyl," as used herein, refers to an alkyl group as defined herein wherein one or more carbon atoms are independently replaced by one or more of oxygen, sulfur, nitrogen, or combinations thereof.
[00053] The term "heteroaryl," as used herein, refers to monocyclic, fused bicyclic, and fused tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic, at least one ring in the system contains one or more heteroatoms selected from nitrogen, oxygen and sulfur, and wherein each ring in a multi-ring system contains 3 to 7 ring members. A heteroaryl group may contain one or more substituents. In certain embodiments a heteroaryl group is optionally substituted. Non- limiting examples of heteroaryl groups, as used herein, include benzofuranyl, benzofurazanyl, benzoxazolyl, benzopyranyl, benzthiazolyl, benzothienyl, benzazepinyl, benzimidazolyl, benzothiopyranyl, benzo[l,3]dioxole, benzo[b]furyl, benzo[b]thienyl, cinnolinyl, furazanyl, furyl, furopyridinyl, imidazolyl, indolyl, indolizinyl, indolin-2-one, indazolyl, isoindolyl, isoquinolinyl, isoxazolyl, isothiazolyl, 1,8-naphthyridinyl, oxazolyl, oxaindolyl, oxadiazolyl, pyrazolyl, pyrrolyl, phthalazinyl, pteridinyl, purinyl, pyridyl, pyridazinyl, pyrazinyl, pyrimidinyl, quinoxalinyl, quinolinyl, quinazolinyl, 4H-quinolizinyl, thiazolyl, thiadiazolyl, thienyl, triazinyl,triazolyl and tetrazolyl.
[00054] The term "heterocycloalkyl," as used herein, refers to a cycloalkyl, as defined herein, wherein one or more of the ring carbons are replaced by a moiety selected from -O-, - N=, -NR-, -C(O)-, -S-, -S(O) - or -S(O)2-, wherein R is hydrogen, C1-C4alkyl or a nitrogen protecting group, with the proviso that the ring of said group does not contain two adjacent O or S atoms. In certain embodiments a heterocycloalkyl group is optionally substituted. Non-limiting examples of heterocycloalkyl groups, as used herein, include morpholino, pyrrolidinyl, pyrrolidinyl-2-one, piperazinyl, piperidinyl, piperidinylone, l,4-dioxa-8-aza- spiro[4.5]dec-8-yl, 2H-pyrrolyl, 2-pyrrolinyl, 3-pyrrolinyl, 1,3-dioxolanyl, 2-imidazolinyl, imidazolidinyl, 2-pyrazolinyl, pyrazolidinyl, 1,4-dioxanyl, 1,4-dithianyl, thiomorpholinyl, azepanyl, hexahydro-l,4-diazepinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, thioxanyl, azetidinyl, oxetanyl, thietanyl, oxepanyl, thiepanyl, 1,2,3,6-tetrahydropyridinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1,3-dioxolanyl, dithianyl, dithiolanyl, dihydropyranyl, dihydrothienyl,
dihydrofuranyl, imidazolinyl, imidazolidinyl, 3-azabicyclo[3.1.0]hexanyl, and 3- azabicyclo[4.1.0]heptanyl.
[00055] The terai "heteroatom," as used herein, refers to one or more of oxygen, sulfur, nitrogen, phosphorus, or silicon.
[00056] The term "heterocycloalkylene," as used herein, refers to a divalent radical derived from a heterocycloalkyl group. In certain embodiments a heterocycloalkylene group is optionally substituted.
[00057] The term "hydroxyl," as used herein, refers to the group -OH. [00058] The term "hydroxyalkyl" or hydroxyl-substituted-alkyl," as used herein, refers to an alkyl group as defined herein substituted with one or more hydroxyl group. Non-limiting examples of branched or straight chained "C1-CO hydroxyalkyl groups as used herein include methyl, ethyl, propyl, isopropyl, isobutyl and n-butyl groups substituted with one or more hydroxyl groups.
[00059] The term "optionally substituted," as used herein, means that the referenced group may or may not be substituted with one or more additional group(s) individually and independently selected from alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocycloalkyl, hydroxyl, alkoxy, mercaptyl, cyano, halo, carbonyl, thiocarbonyl, isocyanato, thiocyanato, isothiocyanato, nitro, perhaloalkyl, perfluoroalkyl, and amino, including mono- and di- substituted amino groups, and the protected derivatives thereof. Non-limiting examples of optional substituents include, halogen, -CN, =0, -OR, -C(O)R, - C(O)OR, -OC(O)R, -OC(O)OR, -C(O)NHR, -C(O)NR2, -OC(O)NHR, -OC(O)NR2, -SR-, - S(O)R, -S(O)2R, -NHR, -N(R)2, -NHC(O)R, -NRC(O)R, -NHC(O)OR, -NRC(O)OR, S(O)2NHR, -S(O)2N(R)2, -NHS(O)2, -NRS(O)2, -NHS(O)2R, -NRS(O)2R, Ci-C8alkyl, C1- Cgalkoxy, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, halo-substituted CrCgalkyl, halo- substituted CrCgalkoxy, where each R is independently selected from H, halogen, C1- Cgalkyl, CrCgalkoxy, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, halo-substituted C1- Cgalkyl, and halo-substituted CrCgalkoxy. The placement and number of such substituent groups is done in accordance with the well-understood valence limitations of each group, for example =0 is a suitable substituent for an alkyl group but not for an aryl group.
[00060] The term "solvate," as used herein, refers to a complex of variable stoichiometry formed by a solute (by way of example, a compound of Formula (I), or a salt thereof, as described herein) and a solvent. Non-limiting examples of a solvent are water, acetone, methanol, ethanol and acetic acid.
[00061] The term "acceptable" with respect to a formulation, composition or ingredient, as used herein, means having no persistent detrimental effect on the general health of the subject being treated.
[00062] The term "administration" or "administering" of the subject compound means providing a compound of Formula (I), a pharmaceutically acceptable salt, a pharmaceutically acceptable solvate, or prodrug thereof to a subject in need of treatment.
[00063] The term "agent" or "test agent," as used herein, includes any substance, molecule, element, compound, entity, or a combination thereof. It includes, but is not limited to a protein, a polypeptide, a small organic molecule, a polysaccharide, a polynucleotide, and the like. It can be a natural product, a synthetic compound, or a chemical compound, or a combination of two or more substances. Unless otherwise specified, the terms "agent",
"substance", and "compound" can be used interchangeably.
[00064] The term "carrier," as used herein, refers to chemical compounds or agents that facilitate the incorporation of a compound described herein into cells or tissues.
[00065] The terms "co-administration" or "combined administration" or the like as used herein are meant to encompass administration of the selected therapeutic agents to a single patient, and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time.
[00066] The term "diluent," as used herein, refers to chemical compounds that are used to dilute a compound described herein prior to delivery. Diluents can also be used to stabilize compounds described herein.
[00067] The terms "effective amount" or "therapeutically effective amount," as used herein, refer to a sufficient amount of a compound described herein being administered which will relieve to some extent one or more of the symptoms of the disease or condition being treated.
The result can be reduction and/or alleviation of the signs, symptoms, or causes of a disease, or any other desired alteration of a biological system. For example, an "effective amount" for
therapeutic uses is the amount of the composition comprising a compound as disclosed herein required to provide a clinically significant decrease in disease symptoms. An appropriate "effective" amount in any individual case may be determined using techniques, such as a dose escalation study.
[00068] The terms "enhance" or "enhancing," as used herein, means to increase or prolong either in potency or duration a desired effect. Thus, in regard to enhancing the effect of therapeutic agents, the term "enhancing" refers to the ability to increase or prolong, either in potency or duration, the effect of other therapeutic agents on a system. An "enhancing- effective amount," as used herein, refers to an amount adequate to enhance the effect of another therapeutic agent in a desired system.
[00069] The term "iatrogenic," as used herein, means a condition, disorder, or disease created or worsened by medical or surgical therapy.
[00070] The term "inflammation," as used herein, includes any condition characterized by a localized or a systemic protective response, which may be elicited by physical trauma, infection, chronic diseases, such as those mentioned herein, and/or chemical and/or physiological reactions to external stimuli (for example, as part of an allergic response). Any such response, which may serve to destroy, dilute or sequester both the injurious agent and the injured tissue, may be manifest by, for example, heat, swelling, pain, redness, dilation of blood vessels and/or increased blood flow, invasion of the affected area by white blood cells, loss of function and/or any other symptoms known to be associated with inflammatory conditions. The term "inflammation" also includes any inflammatory disease, disorder or condition per se, any condition that has an inflammatory component associated with it, and/or any condition characterized by inflammation as a symptom, including inter alia acute, chronic, ulcerative, specific, allergic and necrotic inflammation, and other forms of inflammation known to those skilled in the art. The term also includes inflammatory pain, pain generally and/or fever.
[00071] The term "inflammatory disorders," as used herein, refers to those diseases or conditions that are characterized by one or more of the signs of pain (dolor, from the generation of noxious substances and the stimulation of nerves), heat (calor, from vasodilatation), redness (rubor, from vasodilatation and increased blood flow), swelling
(tumor, from excessive inflow or restricted outflow of fluid), and loss of function (functio laesa, which may be partial or complete, temporary or permanent). Inflammation takes many forms and includes, but is not limited to, inflammation that is one or more of the following: acute, adhesive, atrophic, catarrhal, chronic, cirrhotic, diffuse, disseminated, exudative, fibrinous, fibrosing, focal, granulomatous, hyperplastic, hypertrophic, interstitial, metastatic, necrotic, obliterative, parenchymatous, plastic, productive, proliferous, pseudomembranous, purulent, sclerosing, seroplastic, serous, simple, specific, subacute, suppurative, toxic, traumatic, and/or ulcerative. Inflammatory disorders further include, without being limited to those affecting the blood vessels (polyarteritis, temporarl arteritis); joints (arthritis: crystalline, osteo-, psoriatic, reactive, rheumatoid, Reiter's); gastrointestinal tract; skin (dermatitis); or multiple organs and tissues (systemic lupus erythematosus). [00072] The terms "inhibiting" or "inhibition," as used herein in the context of modulation of enzymatic activities, inhibition relates to reversible suppression or reduction of an enzymatic activity including competitive, uncompetitive, and noncompetitive inhibition. This can be experimentally distinguished by the effects of the inhibitor on the reaction kinetics of the enzyme, which may be analyzed in terms of the basic Michaelis- Menten rate equation. Competitive inhibition occurs when the inhibitor can combine with the free enzyme in such a way that it competes with the normal substrate for binding at the active site. A competitive inhibitor reacts reversibly with the enzyme to form an enzyme- inhibitor complex [EI], analogous to the enzyme-substrate complex.
[00073] The term "pharmaceutically acceptable," as used herein, refers a material, such as a carrier or diluent, which does not abrogate the biological activity or properties of the compounds described herein. Such materials are administered to an individual without causing undesirable biological effects or interacting in a deleterious manner with any of the components of the composition in which it is contained.
[00074] The term "pharmaceutically acceptable salt," as used herein, refers to a formulation of a compound that does not cause significant irritation to an organism to which it is administered and does not abrogate the biological activity and properties of the compounds described herein.
[00075] The term "prevent" or "prevention," as used herein, refers to a complete inhibition of development of primary or secondary tumors or any secondary effects of disease.
[00076] The terms "combination" or "pharmaceutical combination," as used herein mean a product that results from the mixing or combining of more than one active ingredient and includes both fixed and non-fixed combinations of the active ingredients. The term "fixed combination" means that the active ingredients, by way of example, a compound of Formula (I) and an additional therapeutic agent, are both administered to a patient simultaneously in the form of a single entity or dosage. The term "non-fixed combination" means that the active ingredients, by way of example, a compound of Formula (I) and an additional therapeutic agent, are both administered to a patient as separate entities either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the 2 compounds in the body of the patient. The latter also applies to cocktail therapy, e.g. the administration of 3 or more active ingredients.
[00077] The terms "composition" or "pharmaceutical composition," as used herein, refers to a mixture of at least one compound of Formula (I) described herein with other chemical components, such as carriers, stabilizers, diluents, dispersing agents, suspending agents, thickening agents, and/or excipients.
[00078] The term "prodrug," as used herein, refers to an agent that is converted into the parent drug in vivo. A non-limiting example of a prodrug of the compounds described herein is a compound described herein administered as an ester which is then metabolically hydrolyzed to a carboxylic acid, the active entity, once inside the cell. A further example of a prodrug is a short peptide bonded to an acid group where the peptide is metabolized to reveal the active moiety.
[00079] The term "subject" or "patient", as used herein, encompasses mammals and non- mammals. Examples of mammals include, but are not limited to, humans, chimpanzees, apes, monkeys, cattle, horses, sheep, goats, swine; rabbits, dogs, cats, rats, mice, guinea pigs, and the like. Examples of non-mammals include, but are not limited to, birds, fish and the like.
[00080] The term "therapeutically effective amount," as used herein, refers to any amount of a compound which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder. The term also includes within its scope amounts effective to enhance normal physiological function.
[00081] The terms "treat," "treating" or "treatment," as used herein, refers to methods of alleviating, abating or ameliorating a disease or condition symptoms, preventing additional symptoms, ameliorating or preventing or delaying the underlying metabolic causes of symptoms, inhibiting the disease or condition, arresting the development of the disease or condition, relieving the disease or condition, causing regression of the disease or condition, relieving a condition caused by the disease or condition, or stopping the symptoms of the disease or condition either prophylactically (prevent or delay the onset of the disease, or to prevent the manifestation of clinical or subclinical symptoms thereof) and/or therapeutically. [00082] The compound names provided herein were obtained using ChemDraw Ultra 10.0 (CambridgeSoft®) or JChem version 5.2.2 (ChemAxon). [00083] Other objects, features and advantages of the methods, compositions and combinations described herein will become apparent from the following detailed description. It should be understood, however, that the detailed description and the specific examples, while indicating specific embodiments, are given by way of illustration only.
Description of Embodiments
[00084] Provided herein are compounds, pharmaceutically acceptable salts, solvates, N- oxides, prodrugs and isomers thereof, and pharmaceutical compositions, which are inhibitors of prostaglandin E synthases, including microsomal prostaglandin E synthase-1 (mPGES-1), mPGES-2, GSTM2-2, GSTM3-3, and cPGES/p23. In particular, such compounds, pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, and such pharmaceutical compositions provided herein are inhibitors of microsomal prostaglandin E synthase-1 (mPGES-1).
[00085] Further provided herein are compounds, pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, pharmaceutical compositions and methods
for the treatment and/or prevention of diseases or conditions/disorders, associated with prostaglandin E synthases activity. In certain embodiments, are compounds, pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, pharmaceutical compositions and methods for the treatment and/or prevention of diseases or conditions/disorders, associated with microsomal prostaglandin E synthase- 1 (mPGES-1). [00086] The prostaglandin E synthases inhibitors provided herein are compounds having the structure of Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof:

R2 is H, Ci-Cealkyl, aryl, halogen, -CN, -C(O)OR1 or -C(O)NR1R1;
R3 is -CN, Ci-CealkylA-Cehaloalkyl or halogen;
R4 is H, -CN, Ci-Cealkyl , d-Cehaloalkyl or halogen;
R5 is H, halogen, Ci-Cβalkyl, Ci-Cβalkoxy or 4-6 membered heterocycloalkyl containing
1-3 heteroatoms independently selected from N, O and S; L1 is a bond, C2-C6alkenylene, C2-Coalkynylene, -C(O)NRs-,

X1 is CR15 or N; X2 is O, S or NR8;
X3 is S, O or NR8;
X4 is CR15 or N;
X5 is CR15 or N; each Re and each R7 are independently selected from H, halogen, -CN, Ci-C6alkyl, Ci-Cehaloalkyl, -OCi-C6alkyl, -OCi-Cehaloalkyl, -Q2, -C(O)OR8, -C(O)N(Rg)2, -NR8C(O)R8, -N(Rg)2, -S(O)2R8, - NR8C(O)Q2 and -OR8; each R8 is independently selected from H and Ci-Cόalkyl; each R15 is independently selected from H, halogen, -CN, Ci-Cβalkyl, Ci-Cβhaloalkyl, -OCi_C6alkyl, -OCi-Cehaloalkyl, -Q2, -C(O)OR8, -C(O)N(Rg)2, -NR8C(O)R8, -N(Rg)2, -S(O)2R8, - NR8C(O)Q2 and -OR8; L2 is a bond, -0-, -C(O)-, -C(O)NR8-, -NR8C(O)-, -NR8C(O)NR8-,
-(CR9Rg)nNR8C(O)NR8-, -C2-C6alkenylene, C2-C6alkynylene, phenylene, Cioarylene, C14arylene, 4-6 membered heterocycloalkylene containing 1-3
heteroatoms independently selected from N, O and S,

 wherein:
wherein:
X6 is C(R9)2, NR9, O or S; X7 is N or CR9; X8 is CR9, NR9, O or S; X9 is N or CR9; Xio is S or O; Xn is CR9 or N; each R9 is independently H or Ci-Cβalkyl; and wherein the C2-C6alkenylene, C2-Coalkynylene, arylenes and heterocycloalkylenes of L2 are optionally substituted with 1-4
substituents independently selected from Ci-Cβalkyl, CN, -OC1-
Cβalkyl, halogen and OH; Q1 is phenyl, Cioaryl, C14aryl, 5-6 membered heteroaryl containing 1-2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S, Cs-Cscycloalkyl, C4- Cgcycloalkyl optionally substituted with =CH2 cyclohexyl substituted with 2 Qalkyl which together with the carbon to which they are attached form a
cyclopropyl, L3Q2, -(CR10Rn)nQ2,

 wherein: the phenyl, aryls, heteroaryls, heterocycloalkyls and C3-C8cycloalkyls of Q1 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -OCCRIORIOMORIO, -OCCRIORIOMD , -0(CR10RII)I-4RIO, -OCi-Cealkyl, -OCi-Cβhaloalkyl, -C(O)OR14, -N(R14)2, -C(O)N(R14)2, - NR14C(O)R14, -NR14C(O)Q2, -S(O)2R14, -SR14 and -OR14; and wherein: each R1O and each R11 are independently selected from H, C1- 6haloalkyl, halogen and C1-6alkyl, or R10 and R11 are independently Ci^alkyl and together with the carbon to which they are attached form a C3-C7cycloalkyl;
Q2 is phenyl, Cioaryl, C14aryl, 5-6 membered heteroaryl containing 1- 2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S or C3-Cgcycloalkyl, each of which is optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, -CN, - N(R14)2, -OCi-Cβalkyl, halogen and -OH; L3 is C1-C 6alkylene, C2-C 6alkenylene or C2-C 6alkynylene; X12 is NR12, S or O; Xi3 is (CH2)q or O(CH2)P; R12 is H, Ci-Cealkyl or -C(O)OR14; R13 is H, -C(O)OR14 or d-ealkyl, each R14 is independently selected from H or C1-C 6alkyl; n is O, 1, 2 or 3; m is O, 1, 2, 3 or 4; p is O, 1, 2 or 3; q is 1, 2 or 3; s is 0 or 1 ; t is O, 1, 2, 3 or 4; providing that when L1 and L2 are each a bond, then Q1 is phenyl, Qoaryl, C14aryl, 5-6 membered heteroaryl containing 1-2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S, C3-C8cycloalkyl, C4-C8cycloalkyl optionally substituted with =CH2 cyclohexyl substituted with 2 Qalkyl which together with the carbon to which they are attached form a cyclopropyl,
wherein: the phenyl, aryls, heteroaryls, heterocycloalkyls and C3-C8cycloalkyls of Q1 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -OCCRIORIOMORIO, -OCCRIORIOMD , -0(CR10RII)I-4RIO, -OCi-Cealkyl, -OCi-Cβhaloalkyl, -C(O)OR14, -N(R14)2, -C(O)N(R14)2, - NR14C(O)R14, -NR14C(O)Q2, -S(O)2R14, -SR14 and -OR14; and wherein: each R1O and each R11 are independently selected from H, C1- 6haloalkyl, halogen and C1-6alkyl, or R10 and R11 are independently Ci^alkyl and together with the carbon to which they are attached form a C3-C7cycloalkyl;
Q2 is phenyl, Cioaryl, C14aryl, 5-6 membered heteroaryl containing 1- 2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S or C3-Cgcycloalkyl, each of which is optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, -CN, - N(R14)2, -OCi-Cβalkyl, halogen and -OH; L3 is C1-C 6alkylene, C2-C 6alkenylene or C2-C 6alkynylene; X12 is NR12, S or O; Xi3 is (CH2)q or O(CH2)P; R12 is H, Ci-Cealkyl or -C(O)OR14; R13 is H, -C(O)OR14 or d-ealkyl, each R14 is independently selected from H or C1-C 6alkyl; n is O, 1, 2 or 3; m is O, 1, 2, 3 or 4; p is O, 1, 2 or 3; q is 1, 2 or 3; s is 0 or 1 ; t is O, 1, 2, 3 or 4; providing that when L1 and L2 are each a bond, then Q1 is phenyl, Qoaryl, C14aryl, 5-6 membered heteroaryl containing 1-2 heteroatoms indpendently selected from N, O and S, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S, C3-C8cycloalkyl, C4-C8cycloalkyl optionally substituted with =CH2 cyclohexyl substituted with 2 Qalkyl which together with the carbon to which they are attached form a cyclopropyl,
L3Q2, -(CR10Rn)nQ2,
 , , or
, , or
 wherein: the phenyl, aryls, heteroaryls, heterocycloalkyls and Cs-Cscycloalkyls of Q1 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -OCCRIORIOMORIO, -OCCRIORIOMD , -0(CR10RII)I-4RIO, -OCi-Cealkyl, -OCi-Cβhaloalkyl, -C(O)OR14, -N(R14)2, -C(O)N(R14)2, - NR14C(O)R14, -NR14C(O)Q2, -S(O)2R14, -SR14 and -OR14, and provided that when the combination of L1, L2 and Q1 is, L1 is pyridyl and L2 is C2alkynylene and Q1 is phenyl, pyridyl, thiazolyl, cyclohexyl or cyclopropyl, then R2 is -CN, -C(O)OR1 or -C(O)NR1R1
wherein: the phenyl, aryls, heteroaryls, heterocycloalkyls and Cs-Cscycloalkyls of Q1 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -OCCRIORIOMORIO, -OCCRIORIOMD , -0(CR10RII)I-4RIO, -OCi-Cealkyl, -OCi-Cβhaloalkyl, -C(O)OR14, -N(R14)2, -C(O)N(R14)2, - NR14C(O)R14, -NR14C(O)Q2, -S(O)2R14, -SR14 and -OR14, and provided that when the combination of L1, L2 and Q1 is, L1 is pyridyl and L2 is C2alkynylene and Q1 is phenyl, pyridyl, thiazolyl, cyclohexyl or cyclopropyl, then R2 is -CN, -C(O)OR1 or -C(O)NR1R1
[00087] In certain embodiments of such compounds of Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, L1
is a bond, C2-C6alkenylene, C2-Coalkynylene, -C(O)NRs-,


[00088] In certain embodiments of such compounds Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, X2 is O or NRg, and X3 is S or N.
[00089] In certain embodiments of such compounds Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, L1 is a bond, -C(O)NH-,

[00090] In certain embodiments of such compounds Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, L2 is a a bond, -O-,
-C(O)-, -C(O)NR8-, -NR8C(O)-, -NR8C(O)NR8-, -C2-C6alkenylene, -
(CR9Rg)nNR8C(O)NR8-, C2-C6alkynylene, phenylene,

 , where X6 is C(R9)2 or O; X7 is N; X8 is O or S; X9 is N or
, where X6 is C(R9)2 or O; X7 is N; X8 is O or S; X9 is N or
CR9; X1O is S or O; X11 is CR9; R9 is H or Cialkyl; and wherein the C2-C6alkenylene, C2- Cβalkynylene and phenylene of L2 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, CN, -OCi-Cβalkyl, halogen and OH. [00091] In certain embodiments of such compounds Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, L2 is a bond, -O-, -C(O)-,
C2alkenylene, C2alkynylene, phenylene, piperidinylene,

 , where R9 is H or Cialkyl, and the alkenylene, phenylene, piperidinylene of
, where R9 is H or Cialkyl, and the alkenylene, phenylene, piperidinylene of
L2 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl and halogen.
[00092] In certain embodiments of such compounds Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, L1 is a C2-C6alkynylene
or pyridylene, and L2 is

[00093] In certain embodiments of such compounds Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, Q1 is phenyl, pyridyl, C2-
Cβheterocycloalkyl, C3-C6cycloalkyl, L3Q2, -(CR10Rn)nQ2,


[00094] In certain embodiments of such compounds Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, Ri is H, Ci-Cβalkyl, or - (CH2XC(O)OR8; R2 is H, d-C6alkyl, halogen, -CN, -C(O)ORi or -C(O)NRiRi; R3 and R4 are independently selected from Ci-Cβalkyl and halogen; R5 is H, Ci-Cβalkyl or C2- Coheterocycloalkyl; each R6 and each R7 are independently selected from H, Ci-Cβalkyl and halogen; Rg is H or Ci-Cβalkyl; each R9 is independently H or Ci-Cβalkyl; each Rio and each Rn are independently selected from H, halogen and Ci-C βalkyl, or Ri0 and Rn are independently Ci-C3alkyl and together with the carbon to which they are attached form a C3- Qcycloalkyl; Ri2 is H or CrC 6alkyl; Ri3 is H, -C(O)ORi4 or CrC 6alkyl; R14 is H or Q-C βalkyl; L3 is Ci-C βalkylene; Q2 is phenyl substituted with 1, 2, 3 or 4 halogen groups; n is O, 1 or 2; m is O, lor 2; p is 0 or 1; q is 1, and t is 0, 1 or 2.
[00095] In certain embodiments of such compounds Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, R1 is H or - (CH2)C(O)OR8; R2 is H, halogen, -C(O)NR8R8 or CN ; R3 is halo; R4 is halo; R5 is H, C1- Cβalkyl or C2-C6heterocycloalkyl; R6 is H or halogen; R7 is H or halogen; R8 is H or C1- C4alkyl; R9 is H or CrC4alkyl; R10 is H, F or CrC4alkyl; R11 is H, F or CrC4alkyl; or R10 and R11 are independently Ci-C3alkyl and together with the carbon to which they are attached form a C3cycloalkyl; R12 is H or CrC4alkyl; R13 is H, -C(O)OR14 or CrC4alkyl; R14 is H or Ci-C4alkyl; L3 is Ci-C4alkylene, and Q2 is phenyl substituted with 1 or 2 halogen groups.
[00096] In certain embodiments of such compounds Formula (I), and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, R1 is H or -CH2C(O)OH,
R2 is H, Br, -C(O)NH2 or CN, R3 is F, R4 is Cl, R5 is H or O — ' , R6 is H or Br, R7 is
H or Br, R8 is H or Qalkyl, R9 is H or Qalkyl, R11 is OH, F, C^alkyl or phenyl substituted with 1 or 2 halogen groups, n is 0 or 1 and m is 0.
[00097] Also provided herein are certain embodiments of such compounds of Formula (I), or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof, wherein such embodiments are compounds having the structure of Formula (Ia) or pharmaceutically acceptable salts, pharmaceutically acceptable solvates (e.g. hydrates), the N-oxide derivatives, prodrug derivatives, protected derivatives, individual isomers and mixture of isomers thereof:

L1 is a bond,
 , Λ. XH3 , X2A ~T~ or X< T?" ; wherein:
, Λ. XH3 , X2A ~T~ or X< T?" ; wherein:
X1 is CR15 or N;
X2 is O, S or NR8;
X3 is S, O or NR8;
X4 is CR15 or N;
X5 is CR15 or N; each Re and each R7 are independently selected from H, halogen, C1- C6alkyl, Ci-Cβhaloalkyl, and -C(O)OR8; each R8 is independently selected from H and Ci-Cβalkyl; each R15 is independently selected from H, halogen and -C(O)OR8; L2 is a bond, C2-C6alkynylene, phenylene, -0-, -C(O)-, -C(O)NR8-, -NR8C(O)-, -

X6 is C(R9 )2, NR9, O or S;
X7 is N or CR9;
X8 is CR9, NR9, O or S;
X9 is N or CR9;
Xio i 5 S or O;
Xn i 5 CR9 or N; each R9 is independently E or Ci-C6alkyl; and wherein the C2-Coalkynylene and phenylene of L2 are each optionally
substituted with 1-4 substituents independently selected from C1-
C6alkyl, CN, -OCi_C6alkyl, halogen and OH;
Q1 is phenyl, 5-6 membered heteroaryl containing 1-2 N heteroatoms, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S, C3-C8cycloalkyl, C4-C8cycloalkyl optionally substituted with =CH2 cyclohexyl substituted with 2 Cialkyl which together with the carbon to which
they are attached form a cyclopropyl, -(CR1ORn)nQ2,

 wherein: the phenyl, heteroaryl, C2-C6heterocycloalkyl and C3-Cgcycloalkyl groups of Q1 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -O(CR10Rn)i-4OR10, -O(CR10Rn)i-4D , - 0(CR10Rn)1^R10, -OCi-Cβalkyl, -OCi-Cβhaloalkyl, -C(O)OR14, -N(Rw)2, -C(O)N(Rw)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, - S(O)2Ri4, -SRi4 and -0Ri4; each Rio and each Rn are independently selected from H, , Ci-βhaloalkyl and C1- 6alkyl; Q2 is phenyl, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S or C3-Cgcycloalkyl, each of which is optionally substituted with 1-4 substituents independently selected from C1- Cβalkyl and halogen groups;
X12 is NR12, S or O;
wherein: the phenyl, heteroaryl, C2-C6heterocycloalkyl and C3-Cgcycloalkyl groups of Q1 are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -O(CR10Rn)i-4OR10, -O(CR10Rn)i-4D , - 0(CR10Rn)1^R10, -OCi-Cβalkyl, -OCi-Cβhaloalkyl, -C(O)OR14, -N(Rw)2, -C(O)N(Rw)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, - S(O)2Ri4, -SRi4 and -0Ri4; each Rio and each Rn are independently selected from H, , Ci-βhaloalkyl and C1- 6alkyl; Q2 is phenyl, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S or C3-Cgcycloalkyl, each of which is optionally substituted with 1-4 substituents independently selected from C1- Cβalkyl and halogen groups;
X12 is NR12, S or O;
Xi3 is -(CH2),- or -O(CH2)P-;
R12 is H, Ci-Cealkyl or -C(O)OR14; each R14 is independently selected from H and C1-C βalkyl; n is O, 1, 2 or 3; p is O, 1, 2 or 3; q is 1, 2 or 3; s is 0 or 1 and provided that when the combination of L1, L2 and Q1 is, L1 is pyridyl and L2 is C2alkynylene and Q1 is phenyl, pyridyl, thiazolyl, cyclohexyl or cyclopropyl, then R2 is -CN, -C(O)OR1 or -C(O)NR1R1
[00098] In certain embodiments, the compounds of Formula (I) and Formula (Ia), pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, and pharmaceutical compositions provided herein include all suitable isotopic variations of such compounds, and pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, and pharmaceutical compositions. An isotopic variation of a compound provided herein or a pharmaceutically acceptable salt thereof is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature. Examples of isotopes that may be incorporated into the compounds provided herein and pharmaceutically acceptable salts thereof include but are not limited to isotopes of hydrogen, carbon, nitrogen and oxygen such as 2H, 3H, 11C, 13C, 14C, 15N, 17O, 180, 35S, 18F, 36Cl and 123I. Certain isotopic variations of the compounds provided herein and pharmaceutically acceptable salts thereof, for example, those in which a radioactive isotope such as 3H or 14C is incorporated, are useful in drug and/or substrate tissue distribution studies. In particular examples, 3H and 14C isotopes may be used for their ease of preparation and detectability. In other examples, substitution with isotopes such as H may afford certain therapeutic advantages resulting from greater metabolic stability, such as increased in vivo half-life or reduced dosage requirements. Isotopic variations of the compounds, and pharmaceutically acceptable salts, solvates, N- oxides, prodrugs and isomers thereof, and pharmaceutical compositions provided herein are
prepared by conventional procedures using appropriate isotopic variations of suitable reagents.
Processes for Making Compounds of Formula (I) and Formula (Ia)
[00099] General procedures for preparing compounds of Formula (I) and Formula (Ia) are described in the Examples, infra. In the reactions described, reactive functional groups, for example hydroxyl, amino, imino, thio or carboxy groups, where these are desired in the final product, may be protected to avoid their unwanted participation in the reactions. A detailed description of the techniques applicable to the creation of protecting groups and their removal can be found in T. W. Greene, "Protecting Groups in Organic Chemistry," 3rd edition, John Wiley and Sons, Inc., 1999.
[000100] In certain embodiments, the compounds of Formula (I) and Formula (Ia) described herein are prepared as a pharmaceutically acceptable acid addition salt by reacting the free base form of the compound of Formula (I) and Formula (Ia) with a pharmaceutically acceptable organic acid or inorganic acid. In other embodiments, a pharmaceutically acceptable base addition salt of compounds of Formula (I) and Formula (Ia) described herein is prepared by reacting the free acid form of the compound of Formula (I) and Formula (Ia) with a pharmaceutically acceptable organic base or inorganic base. Alternatively, the salt forms of the compounds of Formula (I) and Formula (Ia) described herein are prepared using salts of the starting materials or intermediates. In certain embodiments, the compounds of Formula (I) and Formula (Ia) described herein are in the form of other salts including, but not limited to, oxalates and trifluoroacetates. In certain embodiments, hemisalts of acids and bases are formed, for example, hemisulphate and hemicalcium salts. [000101] Such pharmaceutically acceptable acid addition salts of compounds of Formula (I) and Formula (Ia) include, but are not limited to, a hydrobromide, hydrochloride, hydroiodide, sulfate, bisulphate, nitrate, phosphate, succinate, maleate, formate, acetate, adipate, besylatye, bicarbonate/carbonate, propionate, fumarate, citrate, tartrate, lactate, benzoate, salicylate, glutamate, aspartate, p-toluenesulfonate, benzenesulfonate, methanesulfonate, ethanesulfonate, naphthalenesulfonate (e.g. 2-naphthalenesulfonate), hexanoate salt, bisulphate/sulphate, borate, camsylate, cyclamate, edisylate, esylate, gluceptate, gluconate, glucuronate, pyruvate, hexafluorophosphate, hibenzate,
hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, orotate, oxalate, oxaloacetate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, pyroglutamate, saccharate, stearate, tannate, tosylate, trifluoroacetate and xinofoate salts.
[000102] The organic acid or inorganic acids used to form certain pharmaceutically acceptable acid addition salts of compounds of Formula (I) and Formula (Ia) include, but are not limited to, hydrobromic, hydrochloric, sulfuric, nitric, phosphoric, succinic, maleic, formic, acetic, propionic, fumaric, citric, tartaric, lactic, benzoic, salicylic, glutamic, aspartic, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic, or hexanoic acid.
[000103] Such pharmaceutically acceptable base addition salt of a compound of Formula (I) and Formula (Ia) include, but are not limited to, aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
[000104] In certain embodiments, the free acid or free base forms of the compounds of Formula (I) and Formula (Ia) described herein are prepared from the corresponding base addition salt or acid addition salt from, respectively. For example a compound Formula (I) and Formula (Ia) in an acid addition salt form is converted to the corresponding free base by treating with a suitable base (by way of example only, an ammonium hydroxide solution, a sodium hydroxide, and the like). For example, a compound of Formula (I) and Formula (Ia) in a base addition salt form is converted to the corresponding free acid by treating with a suitable acid (by way of example only, hydrochloric acid).
[000105] In certain embodiments, the compounds of Formula (I) and Formula (Ia) described herein in unoxidized form are prepared from N-oxides of compounds Formula (I) and Formula (Ia) by treating with a reducing agent (by way of example only, sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like) in a suitable inert organic solvent (by way of example only, acetonitrile, ethanol, aqueous dioxane, or the like) at 0 to 8O0C.
[000106] In certain embodiments, prodrug derivatives of compounds Formula (I) and Formula (Ia) described herein are prepared using methods known to those of ordinary skill in the art (e.g., for further details see Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985). For example, appropriate prodrugs are prepared by reacting a non-derivatized compound of Formula (I) and Formula (Ia) with a suitable carbamylating agent (by way of example only, 1,1-acyloxyalkylcarbanochloridate, para- nitrophenyl carbonate, or the like).
[000107] In certain embodiments, the compounds of Formula (I) and Formula (Ia) described herein are prepared or formed, as solvates (e.g., hydrates). In certain embodiments, hydrates of compounds of Formula (I) and Formula (Ia) are prepared by recrystallization from an aqueous/organic solvent mixture, using organic solvents such as dioxin, tetrahydrofuran or methanol.
[000108] In certain embodiments, the compounds of Formula (I) and Formula (Ia) described herein are prepared as their individual stereoisomers. In other embodiments, the compounds of Formula (I) and Formula (Ia) described herein are prepared as their individual stereoisomers by reacting a racemic mixture of the compound with an optically active resolving agent to form a pair of diastereoisomeric compounds, separating the diastereomers and recovering the optically pure enantiomers. In certain embodiments, resolution of enantiomers is carried out using covalent diastereomeric derivatives of the compounds of Formula (I) and Formula (Ia), or by using dissociable complexes (e.g., crystalline diastereomeric salts). Diastereomers have distinct physical properties (e.g., melting points, boiling points, solubility, reactivity, etc.) and are readily separated by taking advantage of these dissimilarities. In certain embodiments, the diastereomers are separated by chromatography, or by separation/resolution techniques based upon differences in solubility. The optically pure enantiomer is then recovered, along with the resolving agent, by any practical means that would not result in racemization. A more detailed description of the techniques applicable to the resolution of stereoisomers of compounds from their racemic mixture can be found in Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions," John Wiley And Sons, Inc., 1981.
[000109] Compounds of Formula (I) and Formula (Ia) are made by processes described herein and as illustrated in the Examples. In certain embodiments, compounds of Formula (I) and Formula (Ia) are made by:
(a) optionally converting a compound of the invention into a pharmaceutically acceptable salt;
(c) optionally converting a salt form of a compound of the invention to a non-salt form;
(d) optionally converting an unoxidized form of a compound of the invention into a pharmaceutically acceptable N-oxide;
(e) optionally converting an N-oxide form of a compound of the invention to its unoxidized form;
(f) optionally resolving an individual isomer of a compound of the invention from a mixture of isomers;
(g) optionally converting a non-derivatized compound of the invention into a pharmaceutically acceptable prodrug derivative; and
(h) optionally converting a prodrug derivative of a compound of the invention to its non-derivatized form.
[000110] Non-limiting examples of synthetic schemes used to make compounds of Formula (I) and Formula (Ia) described herein are illustrated in reaction schemes (I)-(IX), wherein n, Q1, L1, L2, X1' R1, R2 , R3, R4 R5, R6, R7, R8, R9, R10, R11 and R12 are as defined herein.
[000111] Scheme (I) illustrates the use of Sonogashira coupling in the synthesis of certain compounds of Formula (I). In such reactions a Q1 group having a terminal alkyne reacts with an L1 group having a halogen group attached. Such L1 groups are aryl or vinyl halides.
Reaction Scheme (I)

Reaction Scheme (II)

Y = Cl or Br
[000113] Scheme (III) illustrates the use of Suzuki coupling in the synthesis of certain compounds of Formula (I). In such reactions a Q1 group having a terminal boronic acid undergoes palladium catalyzed cross-coupling with an L1 group (aryl or heteroaryl moiety) having a halogen group attached.
Reaction Scheme (III)

[000114] Reaction scheme (IV) illustrates the synthesis of certain embodiments of compounds of Formula (I) by Suzuki coupling of a halo-substituted pyridines or pyrimidines with a Q1 group having a terminal boronic acid.
Reaction Scheme (IV)

[000115] Scheme (V) illustrates the the synthesis of certain compounds of Formula (I) wherein imidazole formation is followed by Sonogashira coupling.
Reaction Scheme (V)
CuI


[000116] Reaction scheme (VI) illustrates the use of Suzuki coupling in the synthesis of certain compounds of Formula (I). In such reactions a Q1 group linked to an L2 group undergoes palladium catalyzed cross-coupling with an L1 group (aryl or heteroaryl moiety) having a halogen group attached and a terminal boronic acid on the L2.
Reaction Scheme (VI)
,B(OH)2
Q — 4

Y = Cl or Br
[000117] Reaction scheme (VII) illustrates the synthesis of certain embodiments of compounds of Formula (I) by Suzuki coupling of a halo-substituted pyridines or pyrimidines with phenyl boronic acid having an attached Q1 group.
Reaction Scheme (VII)

[000118] Reaction scheme (VIII) illustrates the the synthesis of certain compounds of Formula (I) wherein an oxadiazole moiety is formed as an L2 group between an L1 group and a Q1 group. In such reactions the Q1 is substituted with a carboxylic acid moiety and the L1 is substituted with a hydroxy imidamide moiety.
Reaction Scheme (VIII)

[000119] Reaction scheme (IX) illustrates the synthesis of certain embodiments of compounds of Formula (I) wherein an oxadiazole moiety is formed as an L2 group between an L1 group and a Q1 group. In such reactions the Q1 is substituted with a carboxylic acid moiety and the L1 is a pyridine or pyrimidine substituted with a hydroxy imidamide moiety.
Reaction Scheme (IX)

[000120] Reaction scheme (X) illustrates the the synthesis of certain compounds of Formula (I) wherein an thiazole moiety or oxazole moiety is formed as an L2 group between an L1 group and a Q1 group. In such reactions the Q1 is substituted with an aminopropanone moiety and the L1 is substituted with a carboxylic acid moiety.
Reaction Scheme (X)

Reaction Scheme (XII)
H2O
 0C
0C

[000122] Reaction scheme (XIII) illustrates the synthesis of certain embodiments of compounds of Formula (I) by Suzuki coupling of Q1-L2-L1 group with an imidazole moiety having a halogen group attached, wherein the L1 group is a pyridine or pyrimidine with a terminal boronic acid having an attached Q1 group.
Reaction Scheme (XIII)

[000123] Preferred compounds of Formula I are detailed in the Examples and Table I, infra.
[000124] One of skill in the art will appreciate that the above transformations are only representative of methods for preparation of the compounds of the present invention, and that other methods can similarly be used.
Pharmacology and Utility
[000125] Non-steroidal anti-inflammatory therapies and aspirin-related drugs block the production of all prostaglandins through inhibiting the ability of cyclooxygenases to convert arachidonic acid into prostaglandin H2 (PGH2). PGH2 can subsequently be metabolized into a spectrum of lipid mediators including PGE2, PGD2 and PGF2α as well as thromboxane and prostacyclin (PGI2) through the action of a number of different synthases. All of these
products activate G protein-coupled receptors; the phenotypes resulting from deletion of these receptors has considerably enhanced the understanding of prostanoid biology. [000126] Non-steroidal anti-inflammatory drugs (NSAIDs), which include both traditional NSAIDs (tNSAIDs) and selective inhibitors of COX-2, relieve pain and inflammation by suppressing the COX function of prostaglandin H synthases (PGHS; COX-I or COX-2) and the consequent formation of PGE2 and prostacyclin (PGI2). Thus selective inhibitors of COX-2 were developed that suppressed COX-2 mediated inflammation and fever and minimized the frequent complication of tNSAIDs, gastrointestinal (GI) intolerance. The tNSAIDs gastrointestinal (GI) intolerance is attributed to inhibition of COX-I derived protective PGE2 and PGI2 by gastroduodenal epithelium and platelet COX-1-derived TxA2 [000127] The first three selective COX-2 inhibitors approved by the FDA, celecoxib, rofecoxib, and valdecoxib showed clinical superiority in terms of efficacy in rheumatoid arthritis patients and lack of a drug-induced ulceration compared to a tNSAID. However randomized clinical trials of these structurally distinct COX-2 inhibitors also indicate that such compounds elevate the risk of myocardial infarction and stroke. These findings are compatible with the substantial body of preclinical evidence that demonstrates suppression of COX-2-dependent prostacyclin (PGI2) formation, can both augment the response to thrombotic and hypertensive stimuli and initiate and accelerate atherogenesis. These clinical findings have resulted in the worldwide withdrawal of rofecoxib, the withdrawal of valdecoxib from the US, Australian, and European markets, and the substantial decline in the number of prescriptions and "black-box" labeling for celecoxib.
[000128] A combination of pharmacological, genetic and neutralizing antibody approaches demonstrates the importance of PGE2 in inflammation. In many respects, disruption of PGE2 dependent signaling in animal models can be as effective as treatment with tNSAIDs or COX-2 inhibitors. Several prostaglandin E synthases, the enzymes that catalyze the nonoxidative rearrangmenet of the COX product PGH2 into PGE2, have been isolated and identified to date including microsomal prostaglandin E synthase (mPGES)-l, mPGES-2, GSTM2-2, GSTM3-3, and cPGES/p23.
[000129] mPGES-1 has emerged as an important modulator of inflammation in-vivo. Not only does this enzyme occupy the terminal position in the PGE2-synthesis cascade, but it
also preferentially couples with COX-2. In wild-type rats and mice, constitutive mPGES-1 expression in tissues such as stomach and kidney appear to contribute to small amounts of basal PGE2 production. However, upon injection with lipopolysaccharide (LPS), these animals show up-regulation of mPGES-1 in most tissues and markedly increased PGE2 production. In rat paws of acute (carrageenan-induced) and chronic (adjuvant-induced) arthritis models, significant up-regulation of mPGES-1 RNA and protein expression was observed, compared with other PGE synthases, and was associated with elevated PGE2 levels. Consistent with these findings, increased mPGES-1 expression is detected in synovial tissue of patients with rheumatoid arthritis. mPGES-1 expression is down-regulated in-vitro by classical anti-inflammatory drugs such as dexamethasone. These findings suggest that mPGESl provides a target for the treatment of inflammatory disease and pain associated with inflammatory states.
[000130] Insight into the role of mPGES-1 in inflammatory disease has been facilitated by the development of mPGES-1 null mice by two different groups: one on a C57BL/6 x 129/SvJ background, the other on a DBAl/lacJ background. Both strains of mice exhibit normal longevity and fertility and are phenotypically indistinguishable from their wild-type litter-mates. In mPGES-1 knockout mice injected with LPS, PGE2 synthase activity was only minimally increased in tissues. In peritoneal macrophages derived from LPS -stimulated mPGES-1 knockout mice, minimal elevation in PGE2 production, but no difference in TNF- α, IL-6, or IL- 12 levels was observed. Together, these findings establish mPGES-1 as a critical source of inducible PGE2 synthetic activity in-vivo.
[000131] The importance of mPGES-1 in inflammatory arthritis was further demonstrated by a study of collagen-induced arthritis in mPGES-1 null mice. These mice exhibited reduced incidence and severity of disease compared with wild-type controls, both clinically and histopathologically. This difference was not associated with alterations in IL-6 production by peritoneal macrophages or significant differences in circulating IgG2a anticollagen antibodies. In collagen-antibody-induced arthritis, mPGES-1 null mice had a similar incidence but a lesser severity of arthritis than wild-type mice, as well as a 50% reduction in paw levels of PGE2. In other models of inflammation, mPGES-1 null mice
show impaired delayed-type hypersensitivity responses, as well as some evidence for impaired formation of inflammatory granulation tissue and angiogenesis. [000132] The role of mPGES-1 in the central nervous system regulation of fever and nociception in inflammatory states is also well described. Animal pyresis models show transient up-regulation of mPGES-1 mRNA in brain endothelial cells after systemic injection with endotoxin or IL-I. Additionally, peripheral inflammation from carageenan-injected paws was associated with increased mPGES-1 expression and PGE2 levels in the central nervous system. In a chronic inflammation model, rats with adjuvant-induced arthritis displayed sustained induction of mPGES-1 by immunohistochemistry in the brain vasculature and the paraventricular nucleus of the hypothalamus. mPGES-1 null mice did not generate fevers in response to peripheral LPS injection but did become febrile after intracerebroventricular PGE2 injection. Together, these findings highlight PGE2 derived from mPGES-1 in the central nervous system as a critical mediator in inflammation-induced pyresis.
[000133] Studies also describe a role for mPGES-1 in pain. mPGES-1 null mice exhibit diminished writhing in response to intraperitoneal injections of acetic acid (especially after LPS priming), similar to NSAID-treated wild-type mice. There was no difference between null and wild-type mice in withdrawal latency upon thermal stimuli, a model of noninflammatory pain. However, in a model of neuropathic pain involving hyperalgesia following transection of an L5 (fifth lumbar) spinal nerve, mPGES-1 null mice had higher withdrawal thresholds and latency upon both mechanical and thermal stimuli compared with wild-type mice.
[000134] Importantly, mPGES-1 null mice do not display increased thrombogenicity. Decreased expression or pharmacological inhibiiton of COX2 results in increased thrombogenicity in several animal models. These effects are likely mediated by decreased COX-2-dependent prostacyclin (PGI2) formation. In mPGES-1 null mice, prostacyclin (PGI2) formation levels are actually increased while PGE2 levels are decreased. The result is a lack of effect on thrombogenicity and protection from the formation of atherosclerosis on an apoE null background.
[000135] Microsomal prostaglandin E synthase- 1 (mPGES-1) is a novel target for pain (inflammatory, dental, osteoarthritic and neuropathic), inflammation, rheumatoid arthritis (RA), coronary artery disease and fever. Selective inhibitors of mPGES-1 have the potential to be efficacious in reducing inflammation, fever and pain while potentially avoiding the side effects associated with COX-2 inhibitors or traditional NSAIDs. In light of the cardiovascular risk posed by selective COX-2 inhibitors, targeting mPGES-1 may provide a novel strategy which provides similar efficacy without increasing risk of myocardial infarction and stroke.
[000136] The biology and pharmacology of mPGES-1 is discussed in Pharmacol. Rev., 2007, 59, 207-224 and in Curr. Opin. Invest. Drugs, 2007, 8, 411-415. In addition, a review of mPGES-1 in drug development is presented in J. Med. Chem., 2008, 57, 4059-4067. The review presents that inhibition of mPGES-1 is effective in providing symptomatic relief in models of inflammation, pain and fever, and suggests that mPGES-1 inhibition may be a viable therapeutic approach for treating inflammatory diseases, pain and arthritis. [000137] The compounds provided herein are inhibitors of prostaglandin E synthase (PGES) activity, and in certain embodiments are inhibitors of microsomal prostaglandin E synthase- 1 (mPGES-1) activity. In certain embodiments, the compounds provided herein are inhibitors of prostaglandin E synthases (PGES), and in certain embodiments are inhibitors of microsomal prostaglandin E synthase- 1 (mPGES-1). In other embodiments, the compounds provided herein reduce the formation of the specific arachidonic acid metabolite PGE2 without reducing the formation of other COX generated arachidonic acid metabolites, and do not give rise to the associated side-effects mentioned herein.
[000138] In certain embodiments, compounds provided herein are inhibitors of microsomal prostaglandin E synthase- 1 (mPGES-1) activity, wherein they prevent the action of mPGES- 1 or a complex of which the mPGES-1 enzyme forms a part, and/or elicit an mPGES-1 modulating effect.
[000139] Compounds provided herein are useful in the treatment of a disease or disorder in which inhibition of PGES activity, prevents, inhibits or ameliorates the pathology and/or symptomatology of such diseases or disorders. In certain embodiments, the compounds provided herein are useful in the treatment of a disease or disorder in which inhibition of
mPGES-1 activity, prevents, inhibits or ameliorates the pathology and/or symptomatology of such diseases or disorders.
[000140] Compounds of Formula (I) and Formula (Ia) provided herein are inhibitors of microsomal prostaglandin E synthase- 1 (mPGES-1) activity, and as such are useful for treating diseases or disorders in which mPGES-1 contribute to the pathology and/or symptomatology of the disease. Also, such compounds of Formula (I) and Formula (Ia) are useful for the prevention or treatment of mPGES-1 mediated diseases or conditions. In addition, such compounds of Formula (I) and Formula (Ia) are used in the preparation of medicaments for the treatment of diseases or disorders in which mPGES-1 contribute to the pathology and/or symptomatology of the disease. Also, such compounds of Formula (I) and Formula (Ia) are useful in a method of medical treatment, wherein the method of medical treatment is for treating a mPGES-1 mediated diseases or conditions. Such mPGES-1 mediated diseases or conditions, and the diseases or disorders in which mPGES-1 contribute to the pathology and/or symptomatology of the disease include pain, including but not limited to, inflammatory pain, dental pain, postdental procedure pain, osteoarthritic pain, nociceptive pain, neuropathic pain muscoskeletal pain, low back pain, headaches, sinus headaches, migraines, post-surgical pain, surgical pain, burn injury and pain associated with bacterial, fungal or viral illnesses (by way of example only, influenza, the common cold, a viral infections (such as, by way of example only, influenza, common cold, herpes zoster, hepatitis C and AIDS), a bacterial infection or a fungal infection) and periodontal disease; inflammatory diseases, inflammation, arthritis, rheumatoid arthritis (RA), juvenile arthritis, inflammatory arthritis, arthritis, atherosis, tendonitis, bursitis, gouty arthritis, polymyalgia rheumatica, fibermyalgia, fibromyalgia, neuropathy, systemic lupus erythematosus, soft tissue injury, myalgia, neuralgia, neuritis, osteoarthritis, primary dysmenorrhea!, secondary dysmenorrhea!, coronary artery disease, a myofascial disorder, atherosclerosis, fever, rheumatic fever, angiogenesis, granulation, cancer, tumorigenesis, gastrointestinal cancer, breast cancer, colorectal, colon cancer, lung and prostate cancer gastrointestinal homeostasis, Crohn's disease, ulcerative colitis, inflammatory bowel disease, irritable bowel syndrome, reproduction disorders, renal homeostasis, edema, Ductus Arteriosus Closure, Alzheimer's, a malignancy, ankylosing spondylitis, systemic lupus erythematosus, vasculitis, pancreatitis,
nephritis, opthlamic conditions, conjunctivitis, iritis, scleritis, uveitis, wound healing, dermatitis, eczema, psoriasis, stroke, diabetes, a neurodegenerative disorder, a neurologic disorder, myofascial disorders, dysmenorrhea, hyperprostaglandin E syndrome, classic Bartter syndrome, Hodgkin's disease, multiple sclerosis, an immunological disorder, an autoimmune disease, osteoporosis, asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, an allergic disorder, rhinitis, an ulcer, a cardiovascular disorder, coronary heart disease, sarcoidosis, ischemia/reperfusion injury and any other disease with an inflammatory component. Accordingly, provided herein are methods for preventing or treating inflammation, methods for preventing or treating inflammatory diseases, and methods for preventing or treating pain. In addition, the compounds of Formula (I) and Formula (Ia) provided herein are useful for treating or preventing various diseases with an inflammatory component, including, but not limited to, rheumatoid arthritis, osteoarthritis and asthma.
[000141] In accordance with the foregoing, further provided herein are methods for preventing or treating any of the diseases or disorders described herein in a subject in need thereof of such treatment, which method comprises administering to the subject a therapeutically effective amount of a compound of Formula (I) or Formula (Ia) or a pharmaceutically acceptable salt thereof. In certain embodiments, such methods described herein involve administering to the subject in need thereof a pharmaceutical composition that contains one or more compounds of Formula (I) or Formula (Ia), or a pharmaceutically acceptable salt thereof.
[000142] Also, provided herein are methods of treatment of a disease or disorder which is associated with, and/or wherein the activity of prostaglandin E synthase is implicated. In certain embodiments, are methods of treatment of a disease or disorder which is associated with, and/or wherein the activity of mPGES-1 is implicated. Such diseases or disorder are provided herein. Such methods include administration of a therapeutically effective amount of a compound or composition provided herein to a subject in need thereof. [000143] Compounds and compositions provided herein are useful in both the therapeutic and/or prophylactic treatment of the disease and/or disorders provided herein.
[000144] The methods provided herein are either prophylactic or therapeutic treatments of mPGES-1 related disorders and/or mPGES-1 related diseases. Such methods involve administering to a subject in need thereof a therapeutically effective amount of one or more compounds of Formula (I) or Formula (Ia), wherein such compounds are inhibitors of mPGES-1. Also, such methods involve administering to a subject in need thereof a pharmaceutical composition that contains a therapeutically effective amount of one or more compounds of Formula (I) or Formula (Ia), wherein such compounds are inhibitors of mPGES-1. Such prophylactic or therapeutic treatment methods are used to treat any of the mPGES-1 mediated diseases, disorders or conditions provided herein.
[000145] The methods of treatment provide herein are effective for both human and animal subjects. Animal subjects include both domestic animals and livestock, raised either as pets or for commercial purposes. Examples include, but are not limited to, dogs, cats, cattle, horses, sheep, hogs, and goats.
[000146] In certain embodiments, the compounds and composition provided herein are useful in the treatment of inflammation. Accordingly, compounds and composition provided herein are useful in the treatment of pain, asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, inflammatory bowel disease, irritable bowel syndrome, inflammatory pain, fever, migraine, headache, low back pain, fibromyalgia, myofascial disorders, viral infections (such as, by way of example only, influenza, common cold, herpes zoster, hepatitis C and AIDS), bacterial infections, fungal infections, dysmenorrhea, burns, surgical or dental procedures, malignancies (such as, by way of example only, breast cancer, colon cancer, and prostate cancer), hyperprostaglandin E syndrome, classic Bartter syndrome, atherosclerosis, gout, arthritis, osteoarthritis, juvenile arthritis, rheumatoid arthritis, rheumatic fever, ankylosing spondylitis, Hodgkin's disease, systemic lupus erythematosus, vasculitis, pancreatitis, nephritis, bursitis, conjunctivitis, iritis, scleritis, uveitis, wound healing, dermatitis, eczema, psoriasis, stroke, diabetes mellitus, neurodegenerative disorders such as Alzheimer's disease and multiple sclerosis, autoimmune diseases, allergic disorders, rhinitis, ulcers, coronary heart disease, sarcoidosis and any other disease with an inflammatory component.
[000147] In certain embodiments, the compounds and composition provided herein are useful in the treatment of diseases or disorders associated with bone loss in a subject. Such diseases and/or disorders include, but are not limited to, osteoporosis, osteoarthritis, Paget's disease and/or periodontal diseases. Compounds and composition provided herein are useful in the reduction of bone loss, increasing bone mineral density, and/or the reduction in incidence and/or healing of fractures.
[000148] The methods of treatment provide herein are used to treat any disease and/or disorder provided herein including, but not limited to, those associated with inflammation, pain and/or bone loss. In certain embodiments, such methods of treatment are also useful in the reduction of bone loss, increasing bone mineral density, and/or the reduction in incidence and/or healing of fractures.
Administration and Pharmaceutical Compositions
[000149] For the therapeutic uses of compounds of Formula (I) or Formula (Ia), or pharmaceutically acceptable salts, solvates, N-oxides, prodrugs and isomers thereof, such compounds are administered in therapeutically effective amounts either alone or as part of a pharmaceutical composition. Accordingly, provided herein are pharmaceutical compositions, which comprise at least one compound provided herein, including at least one compound of Formulas (I) or Formula (Ia), pharmaceutically acceptable salts and/or solvates thereof, and one or more pharmaceutically acceptable carriers, diluents, adjuvant or excipients. In addition, such compounds and compositions are administered singly or in combination with one or more additional therapeutic agents. The method of administration of such compounds and compositions include, but are not limited to, oral administration, rectal administration, parenteral, intravenous administration, intravitreal administration, subcutaneous administration, intramuscular administration, inhalation, intranasal administration, dermal administration, topical administration, ophthalmic administration or buccal administration, tracheal administration, bronchial administration, sublingual administration or otic administration.
[000150] In certain embodiments such compounds of Formula (I) or Formula (Ia) are inhibitors of prostaglandin E synthases, including microsomal prostaglandin E synthase- 1 (mPGES-1), formulated in an amount sufficient to prevent, inhibit or ameliorate the
pathology and/or symptomatology of diseases or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity. [000151] The therapeutically effective amount will vary depending on, among others, the disease indicated, the severity of the disease, the age and relative health of the subject, the potency of the compound administered, the mode of administration and the treatment desired. . In certain embodiments, the daily dosage of a compound of Formula (I) or Formula (Ia), satisfactory results are indicated to be obtained systemically at daily dosages of from about 0.03 to 150 mg/kg per body weight. In certain embodiments, the daily dosage of a compound of Formula (I) or Formula (Ia), satisfactory results are indicated to be obtained systemically at daily dosages of from about 0.03 to 2.5 mg/kg per body weight. In certain embodiments, the daily dosage of a compound of Formula (I) or Formula (Ia), administered by inhalation, is in the range from 0.05 micrograms per kilogram body weight (μg/kg) to 100 micrograms per kilogram body weight (μg/kg). In other embodiments, the daily dosage of a compound of Formula (I) or Formula (Ia), administered orally, is in the range from 0.01 micrograms per kilogram body weight (μg/kg) to 100 milligrams per kilogram body weight (mg/kg). An indicated daily dosage in the larger mammal, e.g. humans, is in the range from about 0.5mg to about lOOmg of a compound of Formula (I) or Formula (Ia), conveniently administered, e.g. in divided doses up to four times a day or in controlled release form. In certain embodiment, unit dosage forms for oral administration comprise from about 1 to 50 mg of a compound of Formula (I) or Formula (Ia).
[000152] In certain embodiments, oral, pulmonary and topical dosages range from between about 0.01 mg/kg of body weight per day (mg/kg/day) to about 100 mg/kg/day. In certain embodiments, dosages range from between about 0.01 to about 10 mg/kg/day, and in other embodiments, dosages range from between about 0.1 to about 5.0 mg/kg/day. By way of example only, in certain embodiments of oral administration, the compositions contain between about 0.01 mg to about 500 mg. In other embodiments, between about 1 mg to about 100 mg, of the active ingredient. By way of example only, in certain embodiments of intravenous administration, the doses will range from about 0.001 to about 10 mg/kg/hour during constant rate infusion.
[000153] In any event, the physician, or the skilled person, will be able to determine the actual dosage which will be most suitable for an individual patient, which is likely to vary with the route of administration, the type and severity of the condition that is to be treated, as well as the species, age, weight, sex, renal function, hepatic function and response of the particular patient to be treated. The above-mentioned dosages are exemplary of the average case; there can, of course, be individual instances where higher or lower dosage ranges are merited, and such are within the scope of this invention.
[000154] Compounds provided herein are administered alone, or are administered by way of known pharmaceutical formulations, including tablets, capsules or elixirs for oral administration, suppositories for rectal administration, sterile solutions or suspensions for parenteral or intramuscular administration, lotions, gels, ointments or creams for topical administration, and the like.
[000155] Other aspects provided herein are processes for the preparation of pharmaceutical composition which comprise at least one compound of Formula (I) or Formula (Ia), or pharmaceutically acceptable salts and/or solvates thereof. In certain embodiments, such processes include admixing a compound of the Formula (I) or Formula (Ia), and pharmaceutically acceptable salts and solvates thereof, with one or more pharmaceutically acceptable carriers, diluents or excipients. In certain embodiments, the pharmaceutical compositions comprising a compound of Formula (I) or Formula (Ia) in free form or in a pharmaceutically acceptable salt or solvate form, in association with at least one pharmaceutically acceptable carrier, diluent or excipient are manufactured by mixing, granulating and/or coating methods. In other embodiments, such compositions optionally contain excipients, such as adjuvants, preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In other embodiments, such compositions are sterilized.
[000156] In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 1% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 5% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical
compositions) provided herein include those in which the active ingredient is present in at least 10% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 20% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 30% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 40% by weight. In certain embodiments, pharmaceutical formulations (pharmaceutical compositions) provided herein include those in which the active ingredient is present in at least 50% by weight. That is, the ratio of active ingredient to the other components (by way of example, the addition of adjuvant, diluent and carrier) of the pharmaceutical composition is at least 1:99, 5:95, 10:90, 20:80, 30:70, 40:60 or at least 50:50 by weight. Oral Dosage Forms
[000157] In certain embodiments, the pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered orally as discrete dosage forms, wherein such dosage forms include, but are not limited to, capsules, gelatin capsules, caplets, tablets, chewable tablets, powders, granules, syrups, flavored syrups, solutions or suspensions in aqueous or non-aqueous liquids, edible foams or whips, and oil-in-water liquid emulsions or water- in-oil liquid emulsions.
[000158] The capsules, gelatin capsules, caplets, tablets, chewable tablets, powders or granules, used for the oral administration of at least one compound of Formula (I) or Formula (Ia) are prepared by admixing at least one compound of Formula (I) or Formula (Ia) (active ingredient) together with at least one excipient using conventional pharmaceutical compounding techniques. Non-limiting examples of excipients used in oral dosage forms described herein include, but are not limited to, binders, fillers, disintegrants, lubricants, absorbents, colorants, flavors, preservatives and sweeteners.
[000159] Non-limiting examples of such binders include, but are not limited to, corn starch, potato starch, starch paste, pre- gelatinized starch, or other starches, sugars, gelatin, natural and synthetic gums such as acacia, sodium alginate, alginic acid, other alginates, tragacanth, guar gum, cellulose and its derivatives (by way of example only, ethyl cellulose,
cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethylcellulose, methyl cellulose, hydroxypropyl methylcellulose and microcrystalline cellulose), magnesium aluminum silicate, polyvinyl pyrrolidone and combinations thereof. [000160] Non-limiting examples of such fillers include, but are not limited to, talc, calcium carbonate (e.g., granules or powder), microcrystalline cellulose, powdered cellulose, dextrates, kaolin, mannitol, silicic acid, sorbitol, starch, pre- gelatinized starch, and mixtures thereof. In certain embodiments, the binder or filler in pharmaceutical compositions provided herein are present in from about 50 to about 99 weight percent of the pharmaceutical composition or dosage form.
[000161] Non-limiting examples of such disintegrants include, but are not limited to, agar- agar, alginic acid, sodium alginate, calcium carbonate, sodium carbonate, microcrystalline cellulose, croscarmellose sodium, crospovidone, polacrilin potassium, sodium starch glycolate, potato or tapioca starch, pre-gelatinized starch, other starches, clays, other algins, other celluloses, gums, and combinations thereof. In certain embodiments, the amount of disintegrant used in the pharmaceutical compositions provided herein is from about 0.5 to about 15 weight percent of disintegrant, while in other embodiments the amount is from about 1 to about 5 weight percent of disintegrant.
[000162] Non-limiting examples of such lubricants include, but are not limited to, sodium stearate, calcium stearate, magnesium stearate, stearic acid, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, sodium lauryl sulfate, talc, hydrogenated vegetable oil (by way of example only, peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil, corn oil, and soybean oil), zinc stearate, sodium oleate, ethyl oleate, ethyl laureate, agar, silica, a syloid silica gel (AEROSIL 200, manufactured by W.R. Grace Co. of Baltimore, Md.), a coagulated aerosol of synthetic silica (marketed by Degussa Co. of Piano, Tex.), CAB-O-SIL (a pyrogenic silicon dioxide product sold by Cabot Co. of Boston, Mass.) and combinations thereof. In certain embodiments, the amount of lubricants used in the pharmaceutical compositions provided herein is in an amount of less than about 1 weight percent of the pharmaceutical compositions or dosage forms.
[000163] Non-limiting examples of such diluents include, but are not limited to, lactose, dextrose, sucrose, mannitol, sorbitol, cellulose, glycine or combinations thereof.
[000164] In certain embodiments, tablets and capsules are prepared by uniformly admixing at least one compound of Formula (I) or Formula (Ia) (active ingredients) with liquid carriers, finely divided solid carriers, or both, and then shaping the product into the desired presentation if necessary. In certain embodiments, tablets are prepared by compression. In other embodiments, tablets are prepared by molding.
[000165] In certain embodiments, at least one compound of Formula (I) or Formula (Ia) is orally administered as a controlled release dosage form. Such dosage forms are used to provide slow or controlled-release of one or more compounds of Formula (I) or Formula (Ia). Controlled release is obtained using, for example, hydroxypropylmethyl cellulose, other polymer matrices, gels, permeable membranes, osmotic systems, multilayer coatings, microparticles, liposomes, microspheres, or a combination thereof. In certain embodiments, controlled-release dosage forms are used to extend activity of the compound of Formula (I) or Formula (Ia), reduce dosage frequency, and increase patient compliance. [000166] Administration of compounds of Formula (I) or Formula (Ia) as oral fluids such as solution, syrups and elixirs are prepared in unit dosage forms such that a given quantity of solution, syrups or elixirs contains a predetermined amount of a compound of Formula (I) or Formula (Ia). Syrups are prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle. Suspensions are formulated by dispersing the compound in a non-toxic vehicle. Non-limiting examples of excipients used in as oral fluids for oral administration include, but are not limited to, solubilizers, emulsifiers, flavoring agents, preservatives, and coloring agents. Non-limiting examples of solubilizers and emulsifiers include, but are not limited to, water, glycols, oils, alcohols, ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers. Non-limiting examples of preservatives include, but are not limited to, sodium benzoate. Non-limiting examples of flavoring agents include, but are not limited to, peppermint oil or natural sweeteners or saccharin or other artificial sweeteners. Parenteral Dosage Forms
[000167] In certain embodiments pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered parenterally by various routes
including, but not limited to, subcutaneous, intravenous (including bolus injection), intramuscular, and intraarterial.
[000168] Such parenteral dosage forms are administered in the form of sterile or sterilizable injectable solutions, suspensions, dry and/or lyophylized products ready to be dissolved or suspended in a pharmaceutically acceptable vehicle for injection (reconstitutable powders) and emulsions. Vehicles used in such dosage forms include, but are not limited to, Water for Injection USP; aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride Injection, and Lactated Ringer's Injection; water-miscible vehicles such as, but not limited to, ethyl alcohol, polyethylene glycol, and polypropylene glycol; and non-aqueous vehicles such as, but not limited to, corn oil, cottonseed oil, peanut oil, sesame oil, ethyl oleate, isopropyl myristate, and benzyl benzoate. Transdermal Dosage Forms
[000169] In certain embodiments pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered transdemally. Such transdermal dosage forms include "reservoir type" or "matrix type" patches, which are applied to the skin and worn for a specific period of time to permit the penetration of a desired amount of a compound of Formula (I) or Formula (Ia). By way of example only, such transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound to the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin. In other embodiments, matrix transdermal formulations are used.
[000170] Formulations for transdermal delivery of a compound of Formula (I) or Formula (Ia) include an effective amount of a compound of Formula (I) or Formula (Ia), a carrier and an optional diluent. A carrier includes, but is not limited to, absorbable pharmacologically acceptable solvents to assist passage through the skin of the host, such as water, acetone, ethanol, ethylene glycol, propylene glycol, butane- 1,3-diol, isopropyl myristate, isopropyl palmitate, mineral oil, and combinations thereof.
[000171] In certain embodiments, such transdermal delivery systems include penetration enhancers to assist in delivering one or more compounds of Formula (I) or Formula (Ia) to the tissue. Such penetration enhancers include, but are not limited to, acetone; various alcohols such as ethanol, oleyl, and tetrahydrofuryl; alkyl sulfoxides such as dimethyl sulfoxide; dimethyl acetamide; dimethyl formamide; polyethylene glycol; pyrrolidones such as polyvinylpyrrolidone; Kollidon grades (Povidone, Polyvidone); urea; and various water- soluble or insoluble sugar esters such as Tween 80 (polysorbate 80) and Span 60 (sorbitan monostearate).
[000172] In other embodiments, the pH of such a transdermal pharmaceutical composition or dosage form, or of the tissue to which the pharmaceutical composition or dosage form is applied, is adjusted to improve delivery of one or more compounds of Formula (I) or Formula (Ia). In other embodiments, the polarity of a solvent carrier, its ionic strength, or tonicity are adjusted to improve delivery. In other embodiments, compounds such as stearates are added to advantageously alter the hydrophilicity or lipophilicity of one or more compounds of Formula (I) or Formula (Ia) so as to improve delivery. In certain embodiments, such stearates serve as a lipid vehicle for the formulation, as an emulsifying agent or surfactant, and as a delivery-enhancing or penetration-enhancing agent. In other embodiments, different salts, hydrates or solvates of the compounds of Formula (I) or Formula (Ia) are used to further adjust the properties of the resulting composition. Topical Dosage Forms
[000173] In certain embodiments at least one compound of Formula (I) or Formula (Ia) is administered by topical application of pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia) in the form of lotions, gels, ointments solutions, emulsions, suspensions or creams. Suitable formulations for topical application to the skin are aqueous solutions, ointments, creams or gels, while formulations for ophthalmic administration are aqueous solutions. Such formulations optionally contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
[000174] Such topical formulations include at least one carrier, and optionally at least one diluent. Such carriers and diluents include, but are not limited to, water, acetone, ethanol,
ethylene glycol, propylene glycol, butane- 1,3-diol, isopropyl myristate, isopropyl palmitate, mineral oil, and combinations thereof.
[000175] In certain embodiments, such topical formulations include penetration enhancers to assist in delivering one or more compounds of Formula (I) or Formula (Ia) to the tissue. Such penetration enhancers include, but are not limited to, acetone; various alcohols such as ethanol, oleyl, and tetrahydrofuryl; alkyl sulfoxides such as dimethyl sulfoxide; dimethyl acetamide; dimethyl formamide; polyethylene glycol; pyrrolidones such as polyvinylpyrrolidone; Kollidon grades (Povidone, Polyvidone); urea; and various water- soluble or insoluble sugar esters such as Tween 80 (polysorbate 80) and Span 60 (sorbitan monostearate).
[000176] In certain embodiments pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered by inhalation. Dosage forms for inhaled administration are formulated as aerosols or dry powders. Aerosol formulations for inhalation administration comprise a solution or fine suspension of at least one compound of Formula (I) or Formula (Ia) in a pharmaceutically acceptable aqueous or non-aqueous solvent. In addition, such pharmaceutical compositions optionally comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, and optionally a performance modifier such as L-leucine or another amino acid, and/or metals salts of stearic acid such as magnesium or calcium stearate.
[000177] In certain embodiments, compounds of Formula (I) or Formula (Ia) are be administered directly to the lung by inhalation using a Metered Dose Inhaler ("MDI"), which utilizes canisters that contain a suitable low boiling propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas, or a Dry Powder Inhaler (DPI) device which uses a burst of gas to create a cloud of dry powder inside a container, which is then be inhaled by the patient. In certain embodiments, capsules and cartridges of gelatin for use in an inhaler or insufflator are formulated containing a powder mixture of a compound of Formula (I) or Formula (Ia) and a powder base such as lactose or starch. In certain embodiments, compounds of Formula (I) or Formula (Ia) are delivered to the lung using a liquid spray device, wherein such devices use extremely small nozzle holes to aerosolize liquid drug formulations that can then
be directly inhaled into the lung. In other embodiments, compounds of Formula (I) or Formula (Ia) are delivered to the lung using a nebulizer device, wherein a nebulizers creates an aerosols of liquid drug formulations by using ultrasonic energy to form fine particles that can be readily inhaled. In other embodiments, compounds of Formula (I) or Formula (Ia) are delivered to the lung using an electrohydrodynamic ("EHD") aerosol device wherein such EHD aerosol devices use electrical energy to aerosolize liquid drug solutions or suspensions. [000178] In certain embodiments, the pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), or pharmaceutically acceptable salts and solvates thereof, described herein, also contain one or more absorption enhancers. In certain embodiments, such absorption enhancers include, but are not limited to, sodium glycocholate, sodium caprate, N-lauryl-β-D-maltopyranoside, EDTA, and mixed micelles. [000179] In certain embodiments pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered nasally. The dosage forms for nasal administration are formulated as aerosols, solutions, drops, gels or dry powders. [000180] In certain embodiments pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered rectally in the form of suppositories, enemas, ointment, creams rectal foams or rectal gels. In certain embodiments such suppositories are prepared from fatty emulsions or suspensions, cocoa butter or other glycerides.
[000181] In certain embodiments pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered opthamically as eye drops. Such formulations are aqueous solutions that optionally contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
[000182] In certain embodiments pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are administered otically as ear drops. Such formulations are aqueous solutions that optionally contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
[000183] In certain embodiments pharmaceutical compositions containing at least one compound of Formula (I) or Formula (Ia) are formulated as a depot preparation. Such long acting formulations are administered by implantation (for example subcutaneously or
intramuscularly) or by intramuscular injection. In certain embodiments, such formulations include polymeric or hydrophobic materials (for example, as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
[000184] In other embodiments, a compound of Formula (I) or Formula (Ia) described herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing such compounds of Formula (I) or Formula (Ia), is administered to a patient who has not previously undergone or is not currently undergoing treatment with another therapeutic agent. Combination Treatment
[000185] In certain embodiments, one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), is administered alone (without an additional therapeutic agent) for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein. [000186] In other embodiments, one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), is administered in combination with one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
[000187] In other embodiments, one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), is formulated in combination with one or more additional therapeutic agents and administered for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
[000188] In other embodiments, one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), is administered sequentially with one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein.
[000189] In other embodiments, the combination treatments provided herein include administration of one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), prior to administration of one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein. [000190] In other embodiments, the combination treatments provided herein include administration of one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), subsequent to administration of one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein. [000191] In certain embodiments, the combination treatments provided herein include administration of one or more compounds of Formulas (I) provided herein, or a pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), concurrently with one or more additional therapeutic agents, for the treatment of one or more diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein. [000192] In certain embodiments, the combination treatments provided herein include administration of one or more compounds of Formulas (I) provided herein, or a
pharmaceutically acceptable salt, N-oxide, isomer or solvate thereof, or a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia), formulated with one or more additional therapeutic agents, for the treatment of one or more of diseases and/or disorders associated with prostaglandin E synthase activity, including microsomal prostaglandin E synthase- 1 (mPGES-1) activity, described herein. [000193] In certain embodiments, compounds of Formula (I) or Formula (Ia) provided herein are administered in therapeutically effective amounts in combination with other therapies, such as radiation therapy, bone marrow transplantation or hormone therapy. [000194] In certain embodiments of the combination therapies described herein, the compounds of Formula (I) or Formula (Ia) provided herein, or a pharmaceutically acceptable salts, N-oxides, isomers or solvates thereof, and the additional therapeutics agent(s) act additively. In other embodiments of the combination therapies described herein, the compounds of Formula (I) or Formula (Ia) provided herein, or a pharmaceutically acceptable salts, N-oxides, isomers or solvates thereof, and the additional therapeutics agent(s) act synergistically.
[000195] The additional therapeutic agents used in combination with at least one compound of Formula (I) or Formula (Ia) described herein, or a pharmaceutically acceptable salt or solvate thereof, include, but are not limited to, anti-inflammatory agents, therapeutic agents for osteoporosis, anti-tumor therapeutic agents, chemotherapeutic agents, antineoplastic agents and other anticancer agents.
[000196] The additional therapeutic agents used in combination with a pharmaceutical composition containing at least one compound of Formula (I) or Formula (Ia) described herein, or a pharmaceutically acceptable salt or solvate thereof, include, but are not limited to, anti-inflammatory agents, therapeutic agents for osteoporosis, anti-tumor therapeutic agents, chemotherapeutic agents, antineoplastic agents and other anticancer agents. [000197] The anti-inflammatory agents used in such combinations described herein include, but are not limited to, non-steroidal anti-inflammatory drugs such as salicylic acid, acetylsalicylic acid, methyl salicylate, diflunisal, salsalate, olsalazine, sulfasalazine, acetaminophen, indomethacin, sulindac, etodolac, mefenamic acid, meclofenamate sodium, tolmetin, ketorolac, dichlofenac, ibuprofen, naproxen, naproxen sodium, fenoprofen,
ketoprofen, flurbiprofen, oxaprozin, piroxicam, meloxicam, ampiroxicam, droxicam, pivoxicam, tenoxicam, nabumetome, phenylbutazone, oxyphenbutazone, antipyrine, aminopyrine, apazone and nimesulide, leukotriene antagonists including, but not limited to, zileuton, aurothioglucose, gold sodium thiomalate and auranofin, steroids including, but not limited to, alclometasone diproprionate, amcinonide, beclomethasone dipropionate, betametasone, betamethasone benzoate, betamethasone diproprionate, betamethasone sodium phosphate, betamethasone valerate, clobetasol proprionate, clocortolone pivalate, hydrocortisone, hydrocortisone derivatives, desonide, desoximatasone, dexamethasone, flunisolide, flucoxinolide, flurandrenolide, halcinocide, medrysone, methylprednisolone, methprednisolone acetate, methylprednisolone sodium succinate, mometasone furoate, paramethasone acetate, prednisolone, prednisolone acetate, prednisolone sodium phosphate, prednisolone tebuatate, prednisone, triamcinolone, triamcinolone acetonide, triamcinolone diacetate, and triamcinolone hexacetonide and other anti-inflammatory agents including, but not limited to, methotrexate, colchicine, allopurinol, probenecid, thalidomide or a derivative thereof, 5-aminosalicylic acid, retinoid, dithranol or calcipotriol, sulfinpyrazone and benzbromarone.
[000198] The chemotherapeutic agents used in such combinations described herein include, but are not limited to, anthracyclines, alkylating agents (e.g., mitomycin C), alkyl sulfonates, aziridines, ethylenimines, methylmelamines, nitrogen mustards, nitrosoureas, antibiotics, antimetabolites, folic acid analogs (e.g., dihydrofolate reductase inhibitors such as methotrexate), purine analogs, pyrimidine analogs, enzymes, podophyllotoxins, platinum- containing agents, interferons, and interleukins. Particular examples of known chemotherapeutic agents which may be used in the compositions and methods of the invention include, but are not limited to, busulfan, improsulfan, piposulfan, benzodepa, carboquone, meturedepa, uredepa, altretamine, triethylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide, trimethylolomelamine, chlorambucil, chlornaphazine, cyclophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard, carmustine, chlorozotocin, fotemustine, lomustine, nimustine, ranimustine, dacarbazine, mannomustine, mitobronitol,
mitolactol, pipobroman, aclacinomycins, actinomycin F(I), anthramycin, azaserine, bleomycin, cactinomycin, carubicin, carzinophilin, chromomycin, dactinomycin, daunorubicin, daunomycin, 6-diazo-5-oxo-l-norleucine, doxorubicin, epirubicin, mitomycin C, mycophenolic acid, nogalamycin, olivomycin, peplomycin, plicamycin, porfiromycin, puromycin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin, denopterin, methotrexate, pteropterin, trimetrexate, fludarabine, 6-mercaptopurine, thiamiprine, thioguanine, ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine, fluorouracil, tegafur, L-asparaginase, pulmozyme, aceglatone, aldophosphamide glycoside, aminolevulinic acid, amsacrine, bestrabucil, bisantrene, carboplatin, cisplatin, defofamide, demecolcine, diaziquone, elfornithine, elliptinium acetate, etoglucid, etoposide, flutamide, gallium nitrate, hydroxyurea, interferon- alpha, interferon-beta, interferon-gamma, interleukin-2, lentinan, lonidamine, mitoguazone, mitoxantrone, mopidamol, nitracrine, pentostatin, phenamet, pirarubicin, podophyllinic acid, 2-ethylhydrazide, procarbazine, razoxane, sizofiran, spirogermanium, paclitaxel, tamoxifen, teniposide, tenuazonic acid, triaziquone, 2,2',2"- trichlorotriethylamine, urethane, vinblastine, vincristine, and vindesine. [000199] The therapeutic agents for treating osteoporosis used in such combinations described herein include, but are not limited to, alfacalcidol, calcitriol, elcatonin, calcitonin salmon, estriol, ipriflavone, pamidronate disodium, alendronate sodium hydrate, incadronate disodium and the like.
[000200] In certain embodiments, such combination therapies includes the combination of one or more compounds of Formula (I) or Formula (Ia) provided herein with other therapeutic agents that are useful in the treatment of inflammation, including, but not limited to, NSAIDs and coxibs.
[000201] Where the compounds of Formula (I) or Formula (Ia) are administered in conjunction with other therapies, dosages of the co-administered compounds will of course vary depending on the type of co-drug employed, on the specific drug employed, on the condition being treated and so forth. Kits
[000202] Also provided herein are pharmaceutical packs or kits that include one or more containers containing a compound of Formula (I) or Formula (Ia) useful for the treatment or prevention of a disease or disorder associated with the activity of mPGES-1. In other embodiments, such pharmaceutical packs or kits include one or more containers containing a compound of Formula (I) or Formula (Ia) useful for the treatment or prevention of a disease or disorder associated with the activity of mPGES-1 and one or more containers containing an additional therapeutic agent. In certain embodiments, such pharmaceutical packs or kits optionally include instructions for its administration of a compound of Formula (I). In certain embodiments of such kits, the compound of Formula (I) is in free form or in pharmaceutically acceptable salt or N-oxide form.
Examples
[000203] The present invention is further exemplified, but not limited, by the following examples that illustrate the preparation of compounds of Formula (Ia) according to the invention.
Preparation of intermediates
Synthesis of 2-bromo-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl)pyridine (1-1)

LiN(SiMe3)2 R-I TΗF rt, 2h

[000204] A solution of 2-chloro-6-fluorobenzonitrile (R-2) (19.3 mmol) in THF (1 mL) was added drop wise to a 25 mL dry reaction flask charged with commercially available IN lithium bis(trimethylsilyl)-amide (R-I) in THF (21.2 mmol) and the reaction mixture was then stirred at room temperature for 2 hours. The reaction mixture was cooled in an ice bath to 0 0C, and 4N HCl in 1,4-dioxane (38.6 mmol) was carefully added followed by addition
of isopropanol (IPA) (15 niL). The mixture was stirred for 15 minutes and then was kept at 0 0C for 12 hours. The precipitated product was collected by filtration, washed with ether, and dried to yield 2-chloro-6-fluorobenzimidamide hydrochloride (R- 3) as an orange solid. 1H NMR (400 MHz, d6-OMSO) δ 9.69 (bs, 2H), 9.51 (bs, 2H), 7.69 (m, IH), 7.51 (m, IH), 7.43 (m, IH). MS (jn/z) (M+l)+: 173.1.
[000205] Glacial acetic acid (12 mL) was added dropwise to a 50 mL reaction flask charged with commercially available 5-acetyl-2-bromo-pyrimidine (R-4) (12.9 mmol). The solution was cooled in an ice bath to 0 0C. A 48% solution of HBr (10 mL) was then added slowly, followed by dropwise addition of bromine (12.9 mmol). The reaction mixture was stirred at room temperature for 12 hours. The pale orange solution was quenched by slow addition of 10% aq. Na2CO3 and the precipitate formed was collected by filtration, washed with water, and dried under high vacuum to yield 2-bromo-l-(6-bromopyridin-3-yl) ethanone (R-5) as a yellow solid which was used without further purification. 1H NMR (400MHz, CDC13) δ 8.88 (m, IH), 8.05 (m, IH), 7.59 (m, IH), 4.32 (s, 2H). MS (jn/z) (M+l)+: 279.1.
[000206] A solution of 2-chloro-6-fluorobenzimidamide hydrochloride (R- 3) (7.4 mmol) and K2CO3 (29.6 mmol) in THF (13 mL) and H2O (3 mL) was heated to 70 0C in a seal tube, then 2-bromo-l-(6-bromopyridin-3-yl) ethanone (R-5) (6.7 mmol) in THF (2 mL) was added in 5 portions over 5 hours. The reaction mixture was quenched with water and extracted with EtOAc (3 x 50 mL). The combined organic layers were washed with water, brine, dried over Na2SO4 and concentrated. The crude residue was purified by flash column chromatography (DCM : MeCN = 9 : 1) to afford 2-bromo-5-[2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl] pyridine (1-6) as a pale yellow solid. 1H NMR (400 MHz, CD3CN) δ 10.71(bs, IH), 8.83 (d, J = 2.0 Hz, IH), 8.07 (dd, J = 2.0 and 4.0 Hz, IH), 7.75 (m, IH), 7.58 (d, J = 8.0 Hz, IH), 7.55 (m, IH), 7.45 (m, IH), 7.28 (m, IH). MS (m/z) (M+l)+: 351.0 and 353.0.
Synthesis of 5-(5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-2-
(phenvlethvnvl)pvridine (1-3)

[000207] To a solution of 2-bromo-5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl] pyridine (1-1) (2.84 mmol), phenyl acetylene (R-6) (3.12 mmol) and triethylamine (TEA) (8.5 mmol) in MeCN (15 mL) was added copper iodide (I) (0.03 mmol) and Pd(PPh3)4 (0.6 mmol). The reaction mixture was degassed for 10 minutes, sealed and heated in a microwave oven at 200 0C for 10 minutes. The mixture was cooled to room temperature, diluted with DCM (20 mL), filtered through celite. The combined organic layers were concentrated under vacuum to give a yellow oil residue. Purification by silica chromatography (hexane : EtOAc = 1 : 1) gave 5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol- 4-yl)-2-(phenylethynyl)pyridine (1-2) as brownish solid. 1H NMR (400MHz, J4-CH3OH) δ 9.01 (s, IH), 8.24 (m, IH), 7.90 (s, IH), 7.67 (d, J = 8.0 Hz, 1 H), 7.62 (m, 2H), 7.56, (m, IH), 7.43 (m, 5H), 7.29 (t, J = 8.0 Hz, IH). MS (m/z) (M+l)+: 374.1. [000208] NBS (2.3 mmol) was added to a solution of 5-(2-(2-chloro-6-fluorophenyl)-lH- imidazol-4-yl)-2-(phenylethynyl)pyridine (1-2) (2.3 mmol) in CCl4 (20 mL). The reaction mixture was stirred at room temperature until complete conversion (approx. 30 minutes), then quenched with water (30 mL) and extracted with DCM (3 x 40 mL). The combined organic layers were washed with brine (2 x 50 mL), dried over Na2SO4 and concentrated under vacuum. The residue was purified by flash chromatography (hexane : EtOAc = 3 : 1) to yield 5-(5-bromo-2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)-2- (phenylethynyl)pyridine (1-3) in a 3:1 mixture of two tautomers. 1H NMR (400 MHz, d6- DMSO) δ 13.58 (s, 1 H), 9.02 (s, IH), 8.24 (d, J = 8.7 Hz, 1 H), 7.80 (d, J= 8.0 Hz, IH), 7.49-7.65 (m, 7 H). MS (m/z) (M+l)+: 452.0 and 454.1.
Synthesis of 2-chloro-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)pyrimidine (I-4a) and 2-bromo-5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)pyrimidine (I-4b)

R-7 70 0C, 12h R-9 rt, 2h R-IO

[000209] Pd Cl2(PPh3)2 (0.65 mmol) was added to a solution of 5-bromo-2- chloropyrimidine (R-7) (12.9 mmol) and tributyl(l-ethoxy vinyl) stannane (R-8) (14.2 mmol) in THF (25 niL). The flask was evacuated and backfilled with nitrogen twice and then heated at 70 0C for 12 hours. The reaction mixture was quenched with 2M KF (30 mL) and extracted with EtOAc (3 x 30 mL). The combined organic layers were washed with 2M KF (1 x 20 mL), brine, dried over Na2SO4, and concentrated under vacuum to afford 2-chloro-5- (1-ethoxy vinyl )pyrimidine (R-9) as a yellow residue which was used in the next step without purification.
[000210] The above 2-chloro-5-(l-ethoxyvinyl)pyrimidine (R-9) crude (12.9 mmol) was diluted with MeOH (30 mL) and IM HCl (10 mL) added. The resulting yellow solution was stirred at room temperature for 2 hours, and then the organic solvent was removed under reduced pressure. The residue was diluted with water (50 mL), the pH neutralized with IN NaOH and extracted with EtOAc (3 x 50 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under vacuum. The crude was purified by silica chromatography (hexane : DCM = 8 : 2) to yield l-(2-chloropyrimidin-5-yl)ethanone (R-IO). 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 2H), 2.67 (s, 3H). MS (m/z) (M+l)+: 157.1. [000211] Pyridine tribromide (1.4 mmol) was added over 1 hour to a solution of l-(2- chloropyrimidin-5-yl)ethanone (R-10) (1.4 mmol) in AcOH (10 mL). After the addition was complete the reaction mixture was stirred at room temperature until complete conversion (approx. 6 hours). The flask was cooled in an ice-bath and 5N NaOH was slowly added to adjust to pH 6. The aqueous layer was extracted with EtOAc (3 x 20 mL). The combined
organic layers were washed with 2N NaOH (2 x 20 rnL) and brine, dried over Na2SO4 and concentrated under vacuum to afford a reddish residue containing a 2: 1 mixture of 2-chloro- l-(2-chloropyrimidin-5-yl)ethanone (R-IIa) and 2-bromo-l-(2-bromopyrimidin-5- yl)ethanone (R-IIb), which was used in the next step without further purification. MS (m/z) (M+l)+: 236.1 for (R-Ha); 278.0 and 280.1 for (R-Hb).
[000212] Using the same conditions as described for the synthsis of (1-1) the above mixture of 2-chloro-l-(2-chloropyrimidin-5-yl)ethanone (R-Ha) and 2-bromo-l-(2-bromopyrimidin- 5-yl)ethanone (R-Hb) was converted to 2-halo-5-(2-(2-chloro-6-fluorophenyl)-lH- imidazol-4-yl)pyrimidine mixture (I-4a and I-4b). The mixture was purified by preparative ΗPLC (MeCN gradient 10-90%) to afford a 2:1 mixture of 2-chloro-5-(2-(2-chloro-6- fluorophenyl)-lH-imidazol-4-yl)pyrimidine (I-4a) and 2-bromo-5-(2-(2-chloro-6- fluorophenyl)-lH-imidazol-4-yl)pyrimidine (I-4b). MS (m/z) (M+l)+: 310.1 for (I-4a); 352.0 and 354.1 for (I-4b).
Synthesis of 5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)picolinic acid (1-6)

[000213] To a solution of 2-bromo-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)pyridine (1-1) (1.13 mmol) in DMF (10 mL) was added Zn(CN)2 (1.46 mmol) and Pd(PPh3)4 (0.34 mmol). The reaction flask was evacuated and backfilled with nitrogen twice and then heated at 80 0C for 12 hours. The reaction mixture was quenched with water and extracted with EtOAc (3 x 40 mL). The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated under reduced pressure. Purification on silica chromatography (hexane : EtOAc = 1 : 1) gave 5-(2-(2-chloro-6-fluorophenyl)-lH- imidazol-4-yl)picolinonitrile (1-5) as a light brown solid. MS (m/z) (M+l)+: 299.1. [000214] KOΗ (2.31 mmol) was added to a suspension of 5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)picolinonitrile (1-5) (0.57 mmol) in MeOH (2 mL) and H2O (2 mL). The reaction was heated at 80 0C for 12 hours. After this time the reaction mixture was then cooled in an ice-bath and neutralized with IN HCl to pH 7. The resulting suspension was
extracted with DCM (4 x 50 rnL). The combined organic layers were washed once with brine, dried over Na2SO4 and concentrated under reduced pressure to afford 5-(2-(2-chloro- 6-fluorophenyl)-lH-imidazol-4-yl)picolinic acid (1-6) as a light brown solid which was used without further purification. 1H NMR (400MHz, J4-CH3OH) δ 9.11 (s, IH), 8.38 (d, J = 8.0 Hz, IH), 8.21 (d, J= 8.0 Hz, IH), 7.97 (s, IH), 7.57 (m, IH), 7.47 (m,lH), 7.29 (m, IH). MS (m/z) (M+l)+: 318.1.
Synthesis of 5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)-N-hvdroxypicolinimidamide
OzZ.

[000215] A solution of 5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)picolinonitrile (I- 5) (0.184 mmol) and hydroxylamine hydrochloride (0.20 mmol) in EtOH (2 rnL) was heated at 70 0C for 12 hours. The solvent was removed under reduced pressure to afford 5-(2-(2- chloro-6-fluorophenyl)-lH-imidazol-4-yl)-N-hydroxypicolinimidamide (1-7), which was used without further purification. MS (m/z) (M+l)+: 332.0.
Synthesis of 1-fe/t-butyl 5-ethyl 4-(6-bromopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)-lH- imidazole-l,5-dicarboxylate (I-9a) or 1-tert-butyl 4-ethyl 5-(6-bromopyridin-3-yl)-2-(2- chloro-6-fluorophenyl)- lH-imidazole- 1 ,4-dicarboxvlate (I-9b)

1-1 1-8 2 regioisomers

I-9a I -9b
[000216] To a solution of 2-bromo-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)pyridine (1-1) (1.41 mmol) in dry TΗF (16 niL) Boc-anhydride (2.84 mmol) and K2CO3 (2.84 mmol) were added sequentially. The reaction mixture was refluxed for 12 hours. The solvent was removed under vacuum, the residue diluted with water (20 mL) and the organic layer was extracted with EtOAc (3 x 20 mL). The organic portions were combined, dried over Na2SO4 and purified by flash chromatography (DCM : MeOH = 9.5 : 0.5) to yield tert- butyl 4-(6-bromopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)- lH-imidazole- 1 -carboxylate and tert-butyl 5-(6-bromopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)- lH-imidazole- 1 -carboxylate 19 in a 1 : 1 mixture of regioisomers, which were used in the next step. MS (m/z) (M+l)+ : 452.2 and 454.0.
[000217] A solution of the regioisomeric mixture (1-8) (1.14 mmol) in dry TΗF (20 mL) was cooled to -78 0C. rø-BuLi (1.42 mmol) was slowly added over 40 minutes. After the addition was complete the reaction mixture was stirred at -78 0C for 30 minutes. Ethyl chloroformate (2.85 mmol) was then added dropwise over a period of 10 minutes. The reaction mixture was stirred at -78 0C for additional 8 hours then quenched by addition of ice-cold water (5 mL). The reaction mixture was warmed to room temperature and the organic layer was extracted with EtOAc (3 x 40 mL). The combined organic layers were dried over Na2SO4 and the crude was purified by flash chromatography (DCM : MeOH = 9 : 1) to yield l-tert-buty\ 5-ethyl 4-(6-bromopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)-lH- imidazole-l,5-dicarboxylate (I-9a) or 1-tert-butyl 4-ethyl 5-(6-bromopyridin-3-yl)-2-(2- chloro-6-fluorophenyl)-lΗ-imidazole-l,4-dicarboxylate (I-9b). 1H NMR (400 MHz, d4- MeOD) δ 8.71 (d, J = 2.0 Hz, IH), 8.07 (dd, J = 8.0 and 2.0 Hz, IH), 7.62 (d, J = 8.0 Hz, IH), 7.53 (m, IH), 7.37 (m, IH), 7.27 (m, IH), 4.27 (q, J = 8.0 Hz, 2H), 1.25 (s, 9H), 1.22 (t, J= 8.0 Hz, 3H). MS (m/z) (M+l)+ : 524.2 and 526.0.
Synthesis of 5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazole-4-carbonitrile (1-10)

[000218] 3,3-Dibromo-l,l,l-trifluoracetone (R-12) (37 mmol ) was added to a solution of NaOAc (70 mmol) in H2O (15 mL) and the mixture was heated for 30 minutes atlOO 0C. After cooling to room temperature, 2-chloro-6-fluoro benzaldehyde (R-13) (30 mmol) in MeOH (75mL) was added followed by cone. NH4OH (18 mL). The reaction mixture was allowed to stir for 12 hours and then the MeOH was removed under vacuum. The precipitate was collected by filtration, washed with water and dried under vacuum to yield 2-(2-chloro- 6-fluorophenyl)-4-(trifluoromethyl)-lH-imidazole (R-14). MS (m/z) (M+l)+: 265.1. [000219] A mixture of 2-(2-chloro-6-fluorophenyl)-4-(trifluoromethyl)- lH-imidazole (R-
14) (1.4 mmol) and 5% NH4OH (200 mL) was heated to 60 0C. The reaction progress was monitored by TLC. The reaction was cooled to room temperature and carefully neutralized with glacial acetic acid to precipitate 2-(2-chloro-6-fluorophenyl)-lH-imidazole-4- carbonitrile (R-15). MS (m/z) (M+l)+: 300.2.
[000220] To a solution of the 2-(2-chloro-6-fluorophenyl)-lH-imidazole-4-carbonitrile (R-
15) (5.5 mmol) in DMF (50 mL) was added NBS (5.5 mmol). The resulting mixture was stirred for 2 hours at room temperature and then quenched with water (200 mL). The resulting precipitate was filtered and dried to yield 5-bromo-2-(2-chloro-6-fluorophenyl)-lH- imidazole-4-carbonitrile (1-10) as a white solid. 1H NMR (400MHz, J6-DMSO) δ 7.66 (m, IH), 7.55 (d, J = 8.0 Hz, IH), 7.47 (t, J = 8.0 Hz, IH). MS (m/z) (M+l)+: 299.0 and 301.4.
Synthesis of 5-bromo-2-(2-6-dichloro)-lH-imidazole-4-carbonitrile (1-11)

[000221] 3,3-Dibromo-l,l,l-trifluoracetone (R-12) (14.3 mmol ) was added to a solution of NaOAc (28.6 mmol) in H2O (10 mL) and the mixture was heated for 30 minutes atlOO 0C. After cooling to room temperature, 2,6-dichloro benzaldehyde (R-16) (11.4 mmol) in MeOH (30 mL) was added followed by cone. NH4OH (7 mL). The reaction mixture was allowed to stir for 12 hours and then the MeOH was removed under vacuum. The precipitate was collected by filtration, washed with water and dried under vacuum to yield 2-(2,6- dichlorophenyl)-4-(trifluoromethyl)-lH-imidazole (R-17). MS (m/z) (M+l)+: 281.0 [000222] A mixture of 2-(2,6-dichlorophenyl)-4-(trifluoromethyl)- lH-imidazole (R-17) (3.5 mmol) and 5% NH4OH (75 mL) was heated to 60 0C. The reaction progress was monitored by TLC. The reaction was cooled to room temperature and carefully neutralized with glacial acetic acid to precipitate 2-(2,6-dichlorophenyl)-lH-imidazole-4-carbonitrile (R- 18). MS (m/z) (M+l)+: 238.0
[000223] To a solution of the 2-(2,6-dichlorophenyl)- lH-imidazole-4-carbonitrile (R-18) (2.0 mmol) in DMF (10 mL) was added NBS (2.5 mmol). The resulting mixture was stirred for 2 hours at room temperature and then quenched with water (100 mL). The resulting precipitate was filtered and dried to yield 5-bromo-2-(2,6-dichlorophenyl)-lH-imidazole-4- carbonitrile (Ml) 1H NMR (400MHz, J6-DMSO) δ 7.67 (m, 3H). MS (m/z) (M+l)+: 316.0
Synthesis of 5-bromo-2-(2,6-difluoro)-lH-imidazole-4-carbonitrile (1-12)

[000224] 3,3-Dibromo-l,l,l-trifluoracetone (R-12) (37 mmol ) is added to a solution of NaOAc (70 mmol) in H2O (15 mL) and the mixture is heated for 30 minutes atlOO 0C. After cooling to room temperature, 2,6-difluoro benzaldehyde (R-19) (30 mmol) in MeOH (75mL) is added followed by cone. NH4OH (18 mL). The reaction mixture is allowed to stir for 12 hours and then the MeOH is removed under vacuum. The precipitate is collected by filtration, washed with water and dried under vacuum to yield 2-(2,6-difluorophenyl)-4- (trifluoromethyl)- lH-imidazole (R- 20).
[000225] A mixture of 2-(2,6-difluorophenyl)-4-(trifluoromethyl)- lH-imidazole (R-20) (1.4 mmol) and 5% NH4OH (200 mL) is heated to 60 0C. The reaction progress is monitored by TLC. The reaction is cooled to room temperature and carefully neutralized with glacial acetic acid to precipitate 2-(2,6-difluorophenyl)-lH-imidazole-4-carbonitrile (R-21). [000226] To a solution of the 2-(2,6-difluorophenyl)-7H-imidazole-4-carbonitrile (R-21) (5.5 mmol) in DMF (50 mL) is added NBS (5.5 mmol). The resulting mixture is stirred for 2 hours at room temperature and then quenched with water (200 mL). The resulting precipitate is filtered and dried to yield 5-bromo-2-(2,6-difluorophenyl)-7H-imidazole-4- carbonitrile (1-12).
Synthesis of 4-bromo-2-(2-chloro-6-(trifluoromethyl)phenyl)- lΗ-imidazole-5-carbonitrile
(1-13)

[000227] 3,3-Dibromo-l,l,l-trifluoracetone (R-12) (12.0 mmol ) was added to a solution of NaOAc (24.0 mmol) in H2O (6.0 mL) and the mixture was heated for 30 minutes atlOO 0C. After cooling to room temperature, 2-chloro-6-(trifluoromethyl)benzaldehyde (R- 22) (9.6 mmol) in MeOH (40 mL) was added followed by cone. NH4OH (14 mL). The reaction mixture was allowed to stir for 12 hours and then the MeOH was removed under vacuum. The precipitate was collected by filtration, washed with water and dried under vacuum to yield 2-(2-chloro-6-(trifluoromethyl)phenyl)-5-(trifluoromethyl)-lH-imidazole (R-23). MS (m/z) (M+ 1)+: 315.0
[000228] A mixture of 2-(2-chloro-6-(trifluoromethyl)phenyl)-5-(trifluoromethyl)- IH- imidazole (R-23) (2.9 mmol) and 5% NH4OH (60 mL) was heated to 60 0C. The reaction progress was monitored by TLC. The reaction was cooled to room temperature and carefully neutralized with glacial acetic acid to precipitate 2-(2-chloro-6- (trifluoromethyl)phenyl)-lH-imidazole-5-carbonitrile (R-24). MS (m/z) (M+l)+: 272.0 [000229] To a solution of the 2-(2-chloro-6-(trifluoromethyl)phenyl)- lH-imidazole-5- carbonitrile (R-24) 1.8 mmol) in DMF (20 mL) was added NBS (1.95 mmol). The resulting mixture was stirred for 2 hours at room temperature and then quenched with water (10349.90 mL). The resulting precipitate was filtered and dried to yield 4-bromo-2-(2-chloro-6- (trifluoromethyl)phenyl)-lH-imidazole-5-carbonitrile (1-13) ) 1H NMR (400MHz, d6- DMSO) δ 8.17 (s, IH), 8.04(d, IH, J = 8.0 Hz), 7.97(m, IH). ). MS (m/z) (M+l)+:349.9
Synthesis of 4-bromo-2-(2-fluoro-6-(trifluoromethvl)phenvl)-
lH-imidazole-5-carbonitrile (1-14)

[000230] 3,3-Dibromo-l,l,l-trifluoracetone (R-12) (5.6 mmol ) was added to a solution of NaOAc (11.1 mmol) in H2O (3.0 mL) and the mixture was heated for 30 minutes atlOO °C. After cooling to room temperature, 2-fluoro-6-(trifluoromethyl)benzaldehyde (R-25) (4.45 mmol) in MeOH (20 mL) was added followed by cone. NH4OH (7 mL). The reaction mixture was allowed to stir for 12 hours and then the MeOH was removed under vacuum. The precipitate was collected by filtration, washed with water and dried under vacuum to yield 2-(2-fluoro-6-(trifluoromethyl)phenyl)-5-(trifluoromethyl)-lH-imidazole (R-26). MS (m/z) (M+ 1)+: 299
[000231] A mixture of 2-(2-fluoro-6-(trifluoromethyl)phenyl)-5-(trifluoromethyl)- IH- imidazole (R-26) (3.4 mmol) and 5% NH4OH (60 mL) was heated to 60 0C. The reaction progress was monitored by TLC. The reaction was cooled to room temperature and carefully neutralized with glacial acetic acid to precipitate 2-(2-fluoro-6- (trifluoromethyl)phenyl)-lH-imidazole-5-carbonitrile (R-27). MS (m/z) (M+l)+: 256.0 [000232] To a solution of the 2-(2-fluoro-6-(trifluoromethyl)phenyl)-lH-imidazole-5- carbonitrile (R-27) (6.3 mmol) in DMF (50 mL) was added NBS (7.5 mmol). The resulting mixture was stirred for 2 hours at room temperature and then quenched with water (200 mL). The resulting precipitate was filtered and dried to yield 4-bromo-2-(2-fluoro-6- (trifluoromethyl)phenyl)-lH-imidazole-5-carbonitrile (1-14) 1H NMR (400MHz, J6-DMSO) δ 7.89 (m, 3H). MS (m/z) (M+l)+: 339.9
Synthesis of 2-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl)benzofuran-
5-carboxylic acid (1-15)

[000233] Chloroacetone (7.94 mmol) was added dropwise to a solution of methyl 3- formyl-4-hydroxybenzoate (R-28) (7.22 mmol) and anhydrous K2CO3 (7.22 mmol) in MeCN. After the addition was complete, the reaction mixture was heated at 80 0C for 5 hours. Subsequently the reaction was cooled to room temperature and concentrated. The precipitate formed was collected by filtration and washed with small amounts of MeCN to afford methyl 2-acetylbenzofuran-5-carboxylate (R-29) as a yellow solid. 1HNMR (400 MHz, J6-DMSO) δ 8.49 (m, IH), 8.11 (m, IH), 8.01 (s, IH), 7.85 (s, IH), 3.90 (s, 3H), 2.59 (s, 3H). MS (m/z) (M+l)+: 219.0.
[000234] A solution of methyl 2-acetylbenzofuran-5-carboxylate (R-29) (21.2 mmol) in glacial acetic acid (100 mL) was heated to 55 0C. Pyridinium tribromide (22.3 mmol) was added in several portions during a period of 30 minutes. After the addition was complete, the reaction mixture was stirred at 55 0C for 2 hours. After cooling to room temperature, the acetic acid was evaporated and the pH was adjusted to pH 6 with saturated aq NaHCO3. The precipitate was filtered, washed with water and dried to give methyl 2- (2- bromoacetyl)benzofuran-5-carboxylate (R30) as a mixture containing 5 % of methyl 2-(2, 2'-dibromoacetyl)benzofuran-5-carboxylate as by-produc. 1HNMR (400 MHz, J6-DMSO) δ 8.53 (m, IH), 8.16 (m, 2H), 7.88 (m, IH), 4.87 (s, 2H), 3.91 (s, 3H). MS (m/z) (M+l)+: 296.0 and 298.0.
[000235] A solution of 2-chloro-6-fluorobenzamidine hydrochloride (R- 3) (1.52 mmol) and K2CO3 (3.54 mmol) in THF (25 mL) and water (5 mL) was heated to 70 0C. A solution of methyl 2-(2-bromoacetyl)benzofuran-5-carboxylate (R-30) (1.01 mmol) in THF (20 mL)
was then added in five portions over a period of 5 hours. After the addition was complete, the reaction mixture was stirred at 70 0C for 1 hour and then cooled to room temperature. The reaction mixture was extracted with EtOAc (3 x 30 mL). The combined organic phases were washed with water, brine and dried over MgSO4. After filtration and concentration, the residue was purified by preparative HPLC (MeCN gradient 10-90%) to give of methyl 2- (2- (2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)benzofuran-5-carboxylate (R-31). MS (m/z) (M+l)+: 371.
[000236] Methyl 2-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)benzofuran-5- carboxylate (R-31) was dissolved in MeOH (2 mL) and then IN NaOH (4 mL) was added. After stirring at room temperature for 4 hours, the pΗ of the solution was adjusted to pΗ 6 with IN HCl. The resulting suspension was extracted with EtOAc. The extracts were combined, washed with brine and dried over MgSO4. Filtration and concentration gave 2-(2- (2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)benzofuran-5-carboxylic acid (1-15) as a yellow solid. 1HNMR (400 MHz, J4-CH3OH) δ 8.25 (m, IH), 7.94 (m IH), 7.91 (s, IH), 7.55 (m, 2H), 7.43 (m, IH), 7.27 (m, IH), 7.17 (s, IH). MS (m/z) (M+l)+: 357.
Synthesis of 4-(5-bromobenzofuran-2-yl)-2-(2-chloro-6-fluorophenyl)- IH- imidazole (1-16)

[000237] Bromine (0.44 mmol) was added dropwise to a suspension of l-(5- bromobenzofuran-2-yl)ethanone (R-32) (0.42 mmol) in glacial acetic acid (0.84 mL). After the addition was complete, the reaction mixture was stirred at room temperature for 12 hours. The acetic acid was evaporated and the residue was purified by silica chromatography (hexanes : EtOAc = 9 : 1) to afford 2-bromo-l-(5-bromobenzofuran-2- yl)ethanone (R-33). 1H NMR (400Mz, dά-DMSO) δ 8.13 (m, IH), 8.03 (m, IH), 7.74 (m, 2H). MS (m/z) (M+l)+: 318.0.
[000238] To a solution of 2-chloro-6-fluorobenzamidine hydrochloride (R-3) (0.19 mmol) and K2CO3 (0.70 mmol) in THF (5 mL) and water (1 mL) at 70 0C was added 2-bromo-l-(5- bromobenzofuran-2-yl)ethanone (R-33) (0.16 mmol) in THF (3 mL) in five portions over a 5
hour period. After the addition was complete, the reaction mixture was stirred at 70 0C for an additional hour and then cooled to room temperature. The resulting mixture was extracted by EtOAc (3 x 20 mL). The organic phases were combined, washed with water and brine, dried over MgSO4 and concentrated. The resulting residue was purified by preparative HPLC (MeCN gradient 10-90%) to give 4-(5-bromobenzofuran-2-yl)-2-(2- chloro-6-fluorophenyl)-lH-imidazole (1-16). MS (m/z) (M+l)+: 390.0 and 392.0.
Synthesis of 2-(2-chloro-6-fluorophenyl)-lH-imidazole-4-carboxyric acid (1-17)

R-14 1-17
[000239] A solution of 2-(2-chloro-6-fluorophenyl)-5-(trifluoromethyl)- lH-imidazole (R- 14) (0.01 mol) and 2N NaOH (25 mL) was stirred at 90 0C for 1 hour. The reaction mixture was brought to pΗ 6 using 2N HCl, filtered and the filtrate extracted with EtOAc (5 x 100 mL). The combined organic layers were dried over MgSO4 and concentrated to give the 2- (2-chloro-6-fluorophenyl)-lH-imidazole-4-carboxylic acid (1-17) as a yellow solid. MS (m/z) (M+ 1)+: 241.2.
Preparation certain exemplary compounds
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethvnyl)pyridin-3-yl)- lH-imidazole-5- carbonitrile (EX-I)

1-3 EX-I
[000240] To a solution of 5-(5-bromo-2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)-2- (phenylethynyl)pyridine (1-3) (1.34 mmol) in DMF (15 mL) was added Zn(CN)2 (1.54 mmol) and Pd(PPh3)4 (0.4 mmol). The flask was evacuated and backfilled with nitrogen three times and heated at 80 0C for 12 hours. The reaction was diluted with EtOAc (50 mL)
and filtered through celite. The filtrate was washed with brine (2 x 20 rnL), dried over Na2SO4, and concentrated under vacuum. The brown residue was purified by silica chromatography (DCM : MeCN = 9 : 1) to afford 2-(2-chloro-6-fluorophenyl)-4-(6- (phenylethynyl)pyridin-3-yl)-lH-imidazole-5-carbonitrile (EX-I). 1H NMR (400MHz, d4- CH3OH) δ 9.07 (s, IH), 8.36 (dd, J = 4.0 and 8.0 Hz, IH), 7.82 (d, J = 8.0 Hz, IH), 7.65 (m, 3H), 7.49 (m, 4H), 7.32 (t, J= 8.0 Hz, IH). MS (m/z) (M+l)+: 399.1.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethvnyl)pyridin-3-yl)- lH-imidazole-5- carboxamide (EX-2)

EX l EX-2
[000241] A solution of 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethynyl)pyridin-3-yl)- IH- imidazole-5-carbonitrile (EX-I) (0.025 mmol) in AcOH (0.25 mL) and 12N ΗCl (0.5 mL) was stirred at room temperature for 12 hours. The reaction mixture was diluted with water (0.25 mL) and 3N NaOH added until pΗ 7. The mixture was extracted with DCM and the organic layer concentrated under reduced pressure. Purification by preparative TLC (DCM : MeOH = 97 : 3) gave 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethynyl)pyridin-3-yl)-lH- imidazole-5-carboxamide (EX-2) as an off white solid. 1H NMR (400MHz, J4-CH3OH) δ 8.66 (s, IH), 8.22 (dd, J = 4.0 and 8.0 Hz, IH), 7.63 (d, J = 8.0 Hz, IH), 7.54 (m, 3H), 7.36 (m, 4H), 7.19 (t, J = 8.0 Hz, IH). MS (jn/z) (M+l)+: 417.1.
Synthesis of ethyl 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethvnyl)pyridin-3-yl)-lH- imidazole-5-carboxvlate (EX-3)

I-9a or I-9-b 1-18 1-19

EX-3
[000242] To a solution of 1-tert-butyl 5-ethyl 4-(6-bromopyridin-3-yl)-2-(2-chloro-6- fluorophenyl)-lH-imidazole-l,5-dicarboxylate (I-9a) (2.84 mmol) or 1-tert-butyl 4-ethyl 5- (6-bromopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)- lΗ-imidazole- 1 ,4-dicarboxylate (I-9b) (2.84 mmol), phenyl acetylene (R-6) (3.12 mmol) and triethylamine (TEA) (8.5 mmol) in MeCN (15 mL) was added copper iodide (I) (0.03 mmol) and Pd(PPh3)4 (0.6 mmol). The reaction mixture was degassed for 10 minutes, sealed and heated in a microwave oven at 200 0C for 10 minutes. The mixture was cooled to room temperature, diluted with DCM (20 mL), filtered through celite. The combined organic layers were concentrated under vacuum to give a yellow oil residue. Purification by silica chromatography (hexane : EtOAc = 1 : 1) gave 1-tert-butyl 5-ethyl 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethynyl)pyridin-3-yl)-lH- imidazole-l,5-dicarboxylate (1-18) or (1-19). MS (m/z) (M+l)+: 546.1. [000243] l-7ert-butyl 5-ethyl 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethynyl)pyridin-3- yl)- IH- imidazole- 1,5-dicarboxylate (1-18) or (1-19) (0.21 mmol) was dissolved in a TFA : DCM (1 : 1) mixture (5 mL) and stirred for 30 minutes at room temperature. The solvent was removed and the residue was purified via preparative ΗPLC (MeCN gradient 20-90%) to yield ethyl 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethynyl)pyridin-3-yl)-lH-imidazole- 5-carboxylate (EX-3) as a TFA salt. 1H NMR (400 MHz, Acetone-d6) δ 9.2 (brs, IH), 8.43 (s, IH), 7.62 (m, IH), 7.51-7.54 (m, 4H), 7.36 (m, 4H), 7.24 (m, IH), 4.30 (q, J = 8.2 Hz, 2H), 1.29 (t, J = 8.2 Hz, 3H). MS (m/z) (M+l)+: 446.2.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethynyl)pyridin-3-yl)- lH-imidazole
-5-carboxylic acid (EX-4)

EX-3 EX-4
[000244] Ethyl 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethynyl) pyridin-3-yl)-lH- imidazole-5-carboxylate (EX-3) (0.078 mmol) was dissolved in a TΗF:water (1 : 1) mixture (4 rnL), LiOH (0.31 mmol) was added and the reaction mixture was stirred at room temperature for 12 hours. After this time the solvent was removed under vacuum and the residue was purified by preparative HPLC (MeCM gradient 10-70%) to afford 2-(2-chloro- 6-fluorophenyl)-4-(6-(phenylethynyl)pyridin-3-yl)-lH-imidazole-5-carboxylic acid (EX-4). 1H NMR (400MHz, J4-CH3OH) δ 9.08 (s, IH), 8.36 (s, IH), 7.71 (d, J = 8.0 Hz, IH), 7.64 (m, 3H), 7.45 (m, 4H), 7.28 (m, IH). MS (m/z) (M+l)+: 417.2.
Synthesis of 2-(2-chloro-6-fluorophenyl)-N-methyl-4-(6-(phenylethvnyl)pyridin-3-yl)- IH- imidazole -5-carboxamide (EX-5)

EX-4 EX 5
[000245] A solution of 2-(2-chloro-6-fluorophenyl)-4-(6-(phenylethynyl)pyridin-3-yl)- IH- imidazole-5-carboxylic acid (EX-4) (0.023 mmol), ΗATU (0.029 mmol), DIEA (0.057) and methyl amine hydrochloride (0.057 mmol) in DCM (2.5 mL) was stirred at room temperature for 12 hours. After this time the reaction mixture was purified by preparative ΗPLC (MeCN gradient 10-90%) to yield 2-(2-chloro-6-fluorophenyl)-N-methyl-4-(6-
(phenylethynyl)pyridin-3-yl)-lH-imidazole-5-carboxamide (EX-5). MS (m/z) (M+l)+: 431.2.
Synthesis of 5-(5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-2- (phenvlethynyl)pvrimidine (EX-6)
[000246] 2-Chloro-5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)pyrimidine (1-20) was prepared from I-4a/I-4b mixture using the same Sonogashira condition as described for 1-2. The reaction crude was purified by preparative ΗPLC (MeCN gradient 10-70%) to afford the title compound (1-20) as a yellow solid. MS (m/z) (M+ 1)+: 375.1. [000247] 5-(5-Bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-2- (phenylethynyl)pyrimidine (EX-6) was prepared from (1-20) by bromination with NBS using same procedure as described for 1-3. The crude reaction mixture was purified by preparative TLC (DCM : MeCN : MeOH = 8 : 1.5 : 0.5) to afford 5-(5-bromo-2-(2-chloro-6- fluorophenyl)-lH-imidazol-4-yl)-2(phenylethynyl)pyrimidine (EX-6) as a pale yellow solid. 1H NMR (400MHz, d6-OMSO) δ 9.01 (s, 2H), 7.48 (m, 2H), 7.28 (m, 6H). MS (m/z) (M+l)+: 452.0 and 454.1.
Synthesis of 5-(5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-
2-phenylpyrimidine (EX-7)

[000248] To a solution of the 2-chloro-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)pyrimidine (I-4a) and 2-bromo-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)pyrimidine (I-4b) mixture (0.054 mmol) in toluene (1.5 niL) was added phenyl boronic
acid (R-34) (0.065 mmol), 2M aq K2CO3 (O.l lmmol), and Pd(PPh3)4 (0.0005 mmol). The vial was evacuated and backfilled with nitrogen twice and heated at 80 0C for 4 hours. The reaction was then diluted with a saturated solution of NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude material was purified by prepapative TLC (hexane : EtOAc = 1 : 1) to afford 5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-2-phenylpyrimidine (1-21). MS (m/z) (M+ 1)+: 351.1.
[000249] A solution of 5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)-2- phenylpyrimidine (1-21) was brominated with NBS using same procedure as described for the synthesis of 1-3. The crude reaction mixture was purified by preparative TLC (hexane : EtOAc = 1 : 1) to afford 5-(5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-2- phenylpyrimidine (EX-7) as pale yellow solid. MS (m/z) (M+l)+: 428.0 and 430.1.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(5-(phenylethvnyl)thiophen-2-yl)
-lH-imidazole (EX-9)

[000250] To a solution of amidine hydrochloride (R-3) (0.48 mmol) and K2CO3 (1.92 mmol) in THF (3 mL) and H2O (1 mL), bromo thiophene (R-35) (0.48 mmol) was added as described above for the synthesis of 1-1. The reaction was stirred at 70 0C for 12 hours. The reaction mixture was quenched with water and extracted with EtOAc (3 x 50 mL). The organic layer was washed with water and brine, dried over Na2SO4 and concentrated. The crude residue was purified by preparative HPLC (MeCN gradient 10-70%) to afford 4-(5- bromothiophen-2-yl)-2-(2-chloro-6-fluorophenyl)-lH-imidazole (1-22) as a pale yellow solid. 1H NMR (400 MHz, J4-CH3OH) δ 7.53 (m, 2H), 7.43 (m, IH), 7.26 (t, J= 8.0 Hz, IH), 7.11 (d, J = 4.0 Hz, IH), 7.05 (d, J = 4.0 Hz, IH). MS (m/z) (M+l)+ : 356.0 and 358.0. [000251] To a solution of 4-(5-bromothiophen-2-yl)-2-(2-chloro-6-fluorophenyl)-lH- imidazole (1-22) (0.099 mmol), phenyl acetylene (R-6) (0.12 mmol) and triethylamine (0.5 mmol) in MeCN (1 mL) was added copper iodide (I) (0.01 mmol) and Pd(PPh3)4 (0.02 mmol). The reaction mixture was degassed for 10 minutes and heated in a microwave oven
at 200 0C for 10 minutes. The mixture was then cooled to room temperature, diluted with DCM (10 mL) filtered through celite and the cake washed with additional 30 mL DCM. The combined organic layers were concentrated under vaccum to give a yellow oil residue which was purified by preparative TLC (hexane : EtOAc = 7 : 3) to yield 2-(2-chloro-6- fluorophenyl)-4-(5-(phenylethynyl)thiophen-2-yl)-lH-imidazole (EX-9). 1H NMR (400MHz, J4-CH3OH) δ 7.69 (s, IH), 7.54 (m, 1 H), 7.41 (m, 3H), 7.30 (m, 3H), 7.26 (m, 2H), 7.20 (d, J = 4.0 Hz, IH). MS (m/z) (M+l)+: 379.1.
Synthesis of 5-(4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl)-2-(4-fe/t- butvlphenvPpvridine (EX-10)

[000252] A suspension of 2-bromo-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)pyridine (1-1) (0.071 mmol), 4-tert-butylphenylboronic acid (R-36) (0.093 mmol), K2CO3 (0.14 mmol), and Pd(PPh3)4 (0.007 mmol) in IPA (0.8 mL) and H2O (0.2 mL) was heated in an oil bath at 80 0C for 12 hours. The reaction mixure was then diluted with isopropanol (1 mL) and filtered through a short celite pad. The filtrate was purified by preparative HPLC (MeCN gradient 10-60%) to afford 2-(4-?ert-butylphenyl)-5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)pyridine (1-23). MS (m/z) (M+l)+: 406.1.
[000253] To a solution of 2-(4-?er?-butylphenyl)-5-(2-(2-chloro-6-fluorophenyl)- IH- imidazol-4-yl)pyridine (1-23) (0.037 mmol) in CCl4 (1 mL) was added NBS (0.038 mmol). The reaction mixture was stirred at room temperature for 30 minutes, the solvent was then removed and the residue purified by preparative ΗPLC (MeCN gradient 20-80%) to afford 5-(4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl)-2-(4-?ert-butylphenyl)pyridine (EX-10) as TFA salt. 1H NMR (400MHz, J4-CH3OH) δ 9.08 (s, IH), 8.43 (dd, J = 4.0 and 8.0 Hz, IH), 8.07 (d, J = 8.0 Hz, 1 H), 7.97 (m, 2H), 7.61, (m, IH), 7.47 (d, J= 8.0 Hz, IH), 7.31 (t, J = 8.0 Hz, IH), 1.39 (s, 9H). MS (m/z) (M+l)+: 484.0 and 486.1.
Synthesis of 2-(5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)pyridin-2-yl)- lH-indole (EX-Il)

[000254] A suspension of 2-bromo-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)pyridine (1-1) (0.071 mmol), l-(ter^butoxycarbonyl)-lH-indol-2-ylboronic acid (R- 37) (0.093 mmol), K2CO3 (0.14 mmol) and Pd(PPh3)4 (0.007 mmol) in IPA (0.8 mL) and H2O (0.2 mL) was heated in an oil bath at 80 0C for 12 hours. The reaction mixure was diluted with isopropanol (1 mL) and filtered through a short celite pad. The filtrate was purified by preparative HPLC (MeCN gradient 10-60%) to afford tert-bυtyl 2-(5-(2-(2-chloro-6- fluorophenyl)-lH-imidazol-4-yl)pyridin-2-yl)-lH-indole-l-carboxylate (1-24). MS (m/z) (M+l)+: 489.2.
[000255] 7e/t-butyl 2-(5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)pyridin-2-yl)- IH- indole-1-carboxylate (1-24) (0.025 mmol) was stirred with TFA (2 mL) at room temperature for 10 minutes. Purification by preparative TLC (hexane : EtOAc = 1:1) afforded 2-(5-(2-(2- chloro-6-fluorophenyl)-lH-imidazol-4-yl)pyridin-2-yl)-lH-indole (EX-Il). 1U NMR (400MHz, J4-CH3OH) δ 11.69 (s, IH), 9.08 (s, IH), 8.25 (bs, IH), 8.09 (m, 2H), 7.56 (m, 5H), 7.17 (m, 3H). MS (m/z) (M+l)+: 389.1.
Synthesis of 3-(5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl)pyridin-2-yl)-5-phenyl-
1,2,4-oxadiazole (EX-24)

[000256] To a solution of 5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-5-yl)-N- hydroxypicolinimidamide (1-7) (0.18 mmol), ΗATU (0.22 mmol) and benzoic acid (R- 38) (0.18 mmol) in DMF (1.5 mL) was added DIEA (0.55 mmol) and the resulting mixture was heated at 80 0C for 12 hours. After this time the reaction mixture was cooled in an ice bath and H2O was added dropwise. The precipitate formed was collected by filtration and
purified by preparative TLC (hexane : EtOAc = 1 : 3) to yield 3-(5-(2-(2-chloro-6- fluorophenyl)-lH-imidazol-5-yl)pyridin-2-yl)-5-phenyl-l,2,4-oxadiazole (EX-24). 1H NMR (400MHz, J4-CH3OH) δ 9.10 (s, IH), 8.32 (m, IH), 8.18 (m, 3H) 7.89 (m, IH), 7.60 (m, IH), 7.57 (m, 3H), 7.46 (m, IH), 7.35 (m, IH), 7.20 (t, J = 8.0 Hz, IH). MS (m/z) (M+l)+: 418.1.
Synthesis of 3-(5-(4-bromo-2-(2-chloro-6-fluorophenyl)- lH-imidazol-5- yl)pyridin-2-yl)-5- phenyl-l,2,4-oxadiazole (EX-25)

[000257] To a solution of 3-(5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-5-yl)pyridin-2- yl)-5-phenyl-l,2,4-oxadiazole (EX-24) (0.047 mmol) in CCl4 (1 niL) was added NBS (0.047 mmol) and the reaction mixture was stirred at room temperature for 30 minutes. The solvent was evaporated and the residue purified by preparative TLC (hexane : EtOAc = 1 : 1) to afford 3-(5-(4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl)pyridin-2-yl)-5-phenyl- 1,2,4-oxadiazole (EX-25). 1H NMR (400MΗz, J4-CH3OH) δ 9.08 (s, IH), 8.39 (d, J = 8.0 Hz, IH), 8.31 (d, J= 8.0 Hz, IH), 8.18 (m, 2H), 7.61 (m, IH), 7.56 (m, 2H), 7.48 (m, IH), 7.39 (m, IH), 7.21 (m, IH). MS (m/z) (M+l)+: 495.0 and 497.1.
Synthesis of 2-(5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-5-yl)pyridin-2-yl)-
5-phenvloxazole (EX-37)

[000258] A solution of 5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl)picolinic acid (I- 6) (0.12 mmol), ΗATU (0.23 mmol) and 2-amino-l-phenylethanone hydrochloride (R-39) (0.13 mmol) in DMF (0.5 mL) and DIEA (0.50 mmol) was stirred at room temperature for 12 hours. The mixture was cooled in an ice bath and H2O was added dropwise. The precipitate formed was collected by filtration and dried under high vacuum to yield 5-(2-(2-
chloro-6-fluorophenyl)- lH-imidazol-5-yl)-N-(2-oxo-2-phenylethyl)picolinamide (1-25). MS (m/z) (M+l)+:435.1.
[000259] 5-(2-(2-Chloro-6-fluorophenyl)-lH-imidazol-5-yl)-N-(2-oxo-2- phenylethyl)picolinamide (1-25) (0.03 mmol) was dissolved in concentrated H2SO4 (1 rnL) and heated at 50 0C for 12 hours. The reaction mixture was diluted with MeCN and purified by preparative HPLC (MeCN gradient 10-60%) to afford 2-(5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-5-yl)pyridin-2-yl)-5-phenyloxazole (EX-37). 1H NMR (400MΗz, J4-CH3OH) δ 9.17 (s, IH), 8.42 (dd, J = 4.0 and 8.0 Hz, IH), 8.34 (d, J= 8.0 Hz, IH), 8.21 (s, IH), 7.93 (m, 2H), 7.77 (s, IH), 7.69 (m, 1H),7.54 (m, 3H), 7.44 (m, 2H). MS (jn/z) (M+l)+: 417.1.
Synthesis of 2-(5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-5-yl)pyridin-2-yl)-
5-phenylthiazole (EX-38)

[000260] 5-(2-(2-Chloro-6-fluorophenyl)-lH-imidazol-5-yl)-N-(2-oxo-2- phenylethyl)picolinamide (1-25) (0.23 mmol) and Lawesson's reagent (0.43 mmol) in toluene (2 ml) were heated at 100 0C for 12 hours. The reaction mixture was then directly purified by ΗPLC (MeCN gradient 10-80%) to afford 2-(5-(2-(2-chloro-6-fluorophenyl)-lH- imidazol-5-yl)pyridin-2-yl)-5-phenylthiazole (EX-38). 1U NMR (400MΗz, J4-CH3OH) δ 9.06 (s, IH), 8.30 (m, 2H), 8.24 (s, IH), 8.20 (s, IH), 7.76 (m, 3H), 7.59 (m, IH), 7.48 (m, 4H). MS (m/z) (M+l)+: 433.2.
Synthesis of 5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl)-2-phenoxypyridine (EX-40)

[000261] A suspension of 2-bromo-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)pyridine (1-1) (0.78 mmol), K2CO3 (1.06 mmol) and SEM-Cl (0.85 mmol) in acetone was heated at 80 0C for 12 hours. The solvent was removed under reduced pressure and the residue dissolved in water and extracted with EtOAc (3 x 40 rnL). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated. The crude material was purified by silica chromatography (hexane : EtOAc = 2 : 1) to afford (1-26) as a single regioisomer by LCMS. MS (m/z) (M+l)+: 482.0 and 484.2.
[000262] Phenol (R-40) (0.04 mmol) and NaH (0.06 mmol) in DMSO (1 niL) were stirred at room temperature for 1 hour. After this time (1-26) (0.04 mmol) in DMSO (1 mL) was added and the reaction mixture heated at 100 0C for 12 hours. The reaction mixture was purified by preparative HPLC (MeCN gradient 30-90%) to yield the phenoxypyridine (1-27). MS (jn/z) (M+ 1)+: 497.2.
[000263] In a small vial, the phenoxypyridine (1-27) (0.025 mmol), TBAF (0.20 mmol) and THF (1 mL) were stirred at 70 0C for 12 hours. The reaction mixture was then directly purified by preparative HPLC (MeCN gradient 10-70%) to afford 5-(2-(2-chloro-6- fluorophenyl)-lH-imidazol-4-yl)-2-phenoxypyridine (EX-40). 1H NMR (400MHz, d4- MeOH) δ 8.56 (s, IH), 8.22 (dd, J = 4.0 and 8.0 Hz, IH), 8.03 (s, IH), 7.73 (m, IH), 7.59 (d, J = 8.0 Hz, IH), 7.48 (m, 3H), 7.29 (m, IH), 7.16 (m, 2H), 7.11 (d, J = 8.0 Hz, IH). MS (m/z) (M+l)+: 366.1.
Synthessis of 2-(2-chloro-6-fluorophenyl)-5-(2-methoxypyrimidin-5-yl)- lH-imidazole-4-carbonitrile (EX-42)

[000264] To a solution of 2-methoxypyrimidin-5-yl-5-boronic acid (R-41) (0.13 mmol) in 1 : 1: 1.5 = toluene : ethanol : water solution (0.9 rnL) was added 5-bromo-2-(2-chloro-6- fluorophenyl)-lH-imidazole-4-carbonitrile (1-10) (0.083 mmol) and Na2CO3 (0.25 mmol). The reaction mixture was degassed for 5 minutes with nitrogen and Pd(PPh3)4 (0.083 mmol) was added. The reaction was heated in a microwave oven at 150 0C for 10 minutes. Upon cooling the reaction was partitioned with EtOAc and water. The organic layer was washed with brine, dried over MgSO4, filtered and reduced to dryness. The crude product was purified by flash column chromatography (EtOAc : hexanes = 2 : 1) to afford 2-(2-chloro-6- fluorophenyl)-5-(2-methoxypyrimidin-5-yl)-lH-imidazole-4-carbonitrile (EX-42) as a glassy white solid. 1H NMR (400MΗz, J6-DMSO) δ 9.02 (2, IH), 7.68 (m, IH), 7.58 (d, J = 8.0 Hz, IH), 7.50 (t, J = 8.0 Hz, IH), 4.02 (s, 3H). MS (m/z) (M+l)+: 330.5.
Synthesis of 2-(2-chloro-6-fluorophenyl)-5-(2-phenoxypyrimidin-5-yl)- lH-imidazole-4-carbonitrile (EX-43)

[000265] A solution of 5-bromo-2-phenoxypyrimidine (1-28) (0.3 mmol) and hexamethylditin (0.3 mmol) in DME (3 mL) was degassed with nitrogen for 5 minutes. Pd(PPh3 )4 (0.015 mmol) was added and the reaction was heated at 80 0C for 12 hours. The reaction was subsequently cooled to room temperature and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered and reduced to dryness. The crude product was purified by flash column chromatography (hexanes : EtOAc = 9 : 1) to afford 5-(trimethylstannyl)-2-phenoxypyrimidine (1-29) as a clear oil. MS (m/z) (M+l)+: 335.0, 337.0.
[000266] 5-(Trimethylstannyl)-2-phenoxypyrimidine (1-29) (0.16 mmol) and 5-bromo-2- (2-chloro-6-fluorophenyl)-lH-imidazole-4-carbonitrile (1-10) (0.083 mmol) were dissolved in DME (2 mL) and degassed for 5 minutes with nitrogen. Pd(PPh3)4 (0.0082 mmol) was added and the reaction was heated at 85 0C for 12 hours. The reaction was cooled to room temperature and partitioned with EtOAc and water. The organic layer was washed with brine, dried over MgSO4, filtered and reduced to dryness. The crude product was purified by flash column chromatography (hexanes : EtOAc = 3 : 1) to afford 2-(2-chloro-6- fluorophenyl)-5-(2-phenoxypyrimidin-5-yl)-lH-imidazole-4-carbonitrile (EX-43) as white glassy solid. 1U NMR (400MΗz, d4-MeOO) δ 8.95 (s, 2H), 7.50 (m, IH), 7.37 (m, 3H), 7.21 (m, 2H), 7.14 (m, 2H). MS (m/z) (M+l)+: 392.5.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(2-(l-cyclopropylethoxy)pyrimidin-5-yl)-lH- imidazole-5-carbonitrile (EX-44)

[000267] To a solution of 2-(2-chloro-6-fluorophenyl)-4-(2-methoxypyrimidin-5-yl)-lH- imidazole-5-carbonitrile (EX-42) (0.42 mmol) in anhydrous MeCN (6 mL) TMSiI (0.93 mmol) was added dropwise. The reaction mixture was stirred at 60 0C until complete conversion (15 minutes) and then the solvent was removed under nitrogen to afford 2- (2- chloro-6-fluorophenyl)-4-(2-hydroxypyrimidin-5-yl)-lH-imidazole-5-carbonitrile (1-30) as a brownish solid residue that was used in the next step without further purification. MS (m/z) (M+l)+: 316.1.
[000268] A sealed tube charged with crude 2-(2-chloro-6-fluorophenyl)-4-(2- hydroxypyrimidin-5-yl)-lH-imidazole-5-carbonitrile (1-30) (0.3 mmol) and phosphoryl trichloride (1 mL) was heated at 11O0C for 40 minutes. The reaction mixture was poured
onto ice and the pH adjusted to 7 with 5N NaOH. The mixture was extract with EtOAc (3 x 20 rnL). The combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated. The crude material was purified by silica chromatography (hexane : EtOAc = 1:1) to afford 2-(2-chloro-6-fluorophenyl)-4-(2-chloropyrimidin-5-yl)- IH- imidazole-5-carbonitrile (1-31). MS (m/z) (M+ 1)+: 335.2.
[000269] 1-Cyclopropylethanol (R-42) (0.1 mmol) was dissolved in DME (1 mL) and NaH (0.2 mmol) was added. The mixture was stirred at room temperature for 30 minutes. Subsequently a solution of 2-(2-chloro-6-fluorophenyl)-4-(2-chloropyrimidin-5-yl)-lH- imidazole-5-carbonitrile (1-31) (0.05 mmol) in DME (1 mL) was added and the reaction mixture was stirred at 8O0C for 12 hours. The solvent was removed under vacuum and the crude was directly purified by preparative ΗPLC (MeCN gradient 30-90%) to yield 2-(2- chloro-6-fluorophenyl)-4-(2-(l-cyclopropylethoxy)pyrimidin-5-yl)-lH-imidazole-5- carbonitrile (EX-U) as a white solid. 1U NMR (400MΗz, d4-MeOO) δ 8.89 (s, 2H), 7.48 (m, IH), 7.38 (m, IH), 7.22 (m, IH), 4.67 (m, IH), 1.37 (d, J= 6.5 Hz, 3H), 1.12 (m, IH), 0.49 (m, 2H), 0.40 (m, IH), 0.27 (m, IH). MS (m/z) (M+l)+: 384.1.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(2-(methylthio)pyrimidin-5-yl)- lH-imidazole-5- carbonitrile (EX-73)

[000270] A microwave vial charged with 5-bromo-2-(2-chloro-6-fluorophenyl)-lH- imidazole-4-carbonitrile (1-10) (2.6 mmol), 2-(methylthio)pyrimidin-5-ylboronic acid (R-32) (3.2 mmol), Na2CO3 (5.2 mmol), Pd(PPh3)4 (0.13 mmol), IPA (15 mL) and water (3 mL) was irradiated in a microwave oven at 150 0C for 15 minutes. The reaction mixture was diluted with a 10% aq NH4Cl solution (50 mL) and the organic phase extracted with EtOAc (3 x 30 mL). The combined organic layer was washed with additional 10% NH4Cl and brine, dried over Na2SO4 and concentrated. The crude material was purification by silica chromatography (hexane : EtOAc = 7 : 3) to afford 2-(2-chloro-6-fluorophenyl)-4-(2- (methylthio)pyrimidin-5-yl)-lH-imidazole-5-carbonitrile (EX-73) as a white solid. 1H NMR
(400 MHz, d6-OMSO) δ 9.03 (s, 2H), 7.68 (m, IH), 7.55 (m, IH), 7.49 (m, IH), 2.60 (s, 3H). MS (m/z) (M+l)+: 346.2.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(2-(methylsulfonyl)pyrimidin-5-yl)-lH- imidazole-5-carbonitrile (EX-74)

[000271] A suspension of 2-(2-chloro-6-fluorophenyl)-4-(2-(methylthio)pyrimidin-5-yl)- lH-imidazole-5-carbonitrile (EX-73) (0.7 mmol) in DCM (5 niL) was cooled to 0 0C and 70% MCPBA (1.4 mmol) was added. The reaction was stirred at room temerature for 12 hours, at which point a white solid formed. The solid was separated by filtration and further purified by preparative ΗPLC (MeCN gradient 20-70%) to afford 2-(2-chloro-6- fluorophenyl)-4-(2-(methylsulfonyl)pyrimidin-5-yl)-lH-imidazole-5-carbonitrile (EX-74). 1H NMR (400MHz, d6-OMSO) δ 9.44 (s, 2H), 7.68 (m, IH), 7.54 (m, IH), 7.53(m, IH), 3.48 (s, 3H), 1.36 (d, J = 6.5 Hz, 6H). MS (m/z) (M+l)+: 378.1.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(2-(isopropylthio)pyrimidin-5-yl)-lH-imidazole-
5-carbonitrile (EX-21)

[000272] Propane-2-thiol (R-33) (0.23 mmol) in DMSO (1 mL) was treated with NaH (0.3 mmol) and stirred at room temperature for 20 minutes. 2-(2-Chloro-6-fluorophenyl)-4-(2- (methylsulfonyl)pyrimidin-5-yl)-lH-imidazole-5-carbonitrile (EX-74) dissolved in DMSO (1 mL) was then added and the reaction was stirred at 80 0C for 30 minutes. The reaction mixture was quenched with water (5 mL) and extracted with EtOAc (3 x 15 mL). The combined organic layer was washed with brine, dried over Na2SO4 and concentrated. Filtration through a short silica plug (hexane : EtOAc = 7 : 3) gave 2-(2-chloro-6-
fluorophenyl)-4-(2-(isopropylthio)pyrimidin-5-yl)-lH-imidazole-5-carbonitrile (EX-75) as a white solid. 1H NMR (400MΗz, d6-OMSO) δ 8.9 (s, 2H), 7.61 (m, IH), 7.51 (m, IH), 7.42 (m, IH), 3.90 (hep, J = 6.5 Hz, IH), 1.36 (d, J = 6.5 Hz, 6 H). MS (m/z) (M+l)+: 374.1.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(5-phenylbenzofuran-2- yl)- IH- imidazole (EX-78)

1-16 1.4 Dioxane. 85 0C. EX-78
12h
[000273] A solution of 4-(5-bromobenzofuran-2-yl)-2-(2-chloro-6-fluorophenyl)-lH- imidazole (1-16) (0.038 mmol), phenylboronic acid (R-34) (0.058 mmol) and 2M Na2CO3 (1.5 niL) in 1,4-dioxane (1 niL) was purged with nitrogen. [1,1' Bis(diphenylphosphino)- ferrocene]diclororopalladium(II) (0.002 mmol) was added and the reaction was purged with nitrogen. After heating at 85 0C forl2 hours, the reaction mixture was cooled to room temperature, filtered and concentrated. Purification of the residue by preparative ΗPLC (MeCN gradient 10-90%) gave 2-(2-chloro-6-fluorophenyl)-4-(5-phenylbenzofuran-2-yl)- lH-imidazole (EX-78) as a white solid. 1HNMR(400 MHz, J4-CH3OH) δ 7.93 (s, IH), 7.85 (m, IH), 7.65 (m, 3H), 7.60 (m, 2H), 7.51 (m, IH), 7.46 (m, 2H), 7.35 (m, 2H), 7.21 (s, IH). MS (m/z) (M+l)+: 389.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(5-phenyethynyllbenzofuran-2-yl)- lH-imidazole (EX-79)
CuI h,


1 16 EX-79
[000274] A solution of 4-(5-bromobenzofuran-2-yl)-2-(2-chloro-6-fluorophenyl)-lH- imidazole (1-16) (0.38 mmol) and phenyl acetylene (R-6) (0.46 mmol) in DMF (5 mL) was purged with nitrogen. Copper iodide (I) (0.038 mmol), bis(triphenylphosphine) dichloropalladium (0.019 mmol) and triethylamine (1 mL) were added. The reaction
mixture was degassed with nitrogen and heated at 100 0C under nitrogen atmosphere for 8 hours. After cooling to room temperature, the suspension was filtered and concentrated. The residue was purified by preparative HPLC (MeCN gradient 10-90%) to give 2- (2- chloro-6-fluorophenyl)-4-(5-phenyethynylbenzofuran-2-yl)- IH- imidazole (EX-79) as a yellow solid. 1HNMR (400 MHz, J4-CH3OH) δ 8.06 (s, IH), 7.84 (m, IH), 7.69 (m, IH), 7.54 (m, 5H), 7.39 (m, 4H), 7.24 (m, IH). MS (m/z) (M+l)+: 413.
Synthesis of 5-(2-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)benzofuran-5-yl)-3- isopropyl- 1 ,2,4-oxadiazole (EX-80)

[000275] To a solution of 2-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)benzofuran-5- carboxylic acid (1-15) (0.034 mmol) and ΗOBt (0.040 mmol) in anhydrous MeCN was added EDC (0.040 mmol) followed by (Z)-N'-hydroxyisobutyrimidamide (R-34) (0.040 mmol). The reaction mixture was stirred at room temperature for 2 hours and then heated to 80 0C for 8 hours. After cooling, the mixture was filtered and purified by preparative ΗPLC (MeCN gradient 10-90%) to afford 5-(2-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)benzofuran-5-yl)-3-isopropyl-l,2,4-oxadiazole (EX-80) as a white solid. 1HNMR (400 MHz, J4-,CH3OH) δ 8.32 (m, IH), 8.00 (m, IH), 7.81 (s, IH), 7.63 (m, IH), 7.51 (m, IH), 7.40 (m, IH), 7.24 (m, IH), 7.15 (s, IH), 3.05 (m, IH), 1.31 (m, 6H). MS (m/z) (M+l)+: 423.
Synthesis of 5-bromo-2-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)- lH-benzoMimidazole (EX-94)

[000276] A solution of 4-bromobenzene-l,2-diamine (R-35) (0.16 mmol), 2-(2-chloro-6- fluorophenyl)-lH-imidazole-4-carboxylic acid (1-17) (0.13 mmol), O-(benzotriazol-l-yl)-
AWN',N'-tetramethyluronium tetrafluoroborate (TBTU) (0.17 mmol) and TEA (0.33 mmol) in DMF (5 niL) was stirred at room temperature for 16 hours. Purification by preparative HPLC (MeCN gradient 10-90%) afforded iV-(2-amino-5-bromophenyl)-2-(2-cMoro-6- fluorophenyl)-lH-imidazole-4-carboxamide (1-32) as a white solid. 1H NMR (400 MHz, d4 -CH3OH), δ 8.05 (s, IH), 7.62 (m, 2H), 7.49 (m, IH), 7.33 (m, IH), 6.84 (m, IH). MS (m/z) (M+l)+: 408.0 and 410.0.
[000277] To N-(2-amino-5-bromophenyl)-2-(2-chloro-6-fluorophenyl)- lH-imidazole-4- carboxamide (1-32) was added PPA until dissolved. After heating at 150 0C for 2 hours, the solution was cooled to 0 0C and slowly neutralized with K2CO3 to pΗ 7. The mixture was extracted with EtOAc (3 x 40 mL) and the extracts combined, concentrated and purified by ΗPLC to give 5-bromo-2-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-lH- benzo [d\ imidazole (EX-94) as a white solid. MS (m/z) (M+l)+: 390.0 and 392.0.
Synthesis of 5-bromo-2-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)- lH-imidazor4,5-blpvridine (EX-95)

[000278] To a suspension of 2,6-dibromo-3-nitropyridine (R-36) (14.2 mmol) in EtOH was added TEA (28.4 mmol) followed by dropwise addition of 4-methoxybenzylamine (R-37) (14.2 mmol). After the addition was complete, the solution was stirred at room temperature for 12 hours. The reaction mixture was concentrated and the residue was washed with EtOAc to afford 6-bromo-N-(4-methoxybenzyl)-3-nitropyridin-2-amine (R-38) as a yellow solid. 1U NMR(400 MHz, d6-OMSO) δ 8.29 (m, IH), 7.33 (m, 2H), 6.90 (m, 3H), 4.63 (m, 2H), 3.73 (s, 3H). MS (m/z) (M+l)+: 338.0 and 340.0.
[000279] To a solution of 6-bromo-N-(4-methoxybenzyl)-3-nitropyridin-2-amine (R-38) (10.4 mmol) in EtOH was added tin (II) chloride dihydrate (52.0 mmol), and the mixture heated at 50 0C for 2 hours. After cooling to room temperature, 10% aq NaHCO3 was added to the reaction mixture and a white precipitate formed. The precipitate was filtrated and washed with EtOH. The filtrate was concentrated and filtered through a silica gel pad. The resulting solution was concentrated to give 6-bromo-iV-(4-methoxybenzyl)pyridine-2,3- diamine (R-39). MS (m/z) (M+l)+: 308.0 and 310.0.
[000280] A solution of 6-bromo-N-(4-methoxybenzyl)pyridine-2,3-diamine (R-39) in TFA was heated at 85 0C for 2 hours. The solution was concentrated, neutralized with a 10% aq NaHCO3 and extracted with EtOAc. The extracts were combined and concentrated to give 6-bromopyridine-2,3-diamine (R-40) as a syrup. 1H NMR(400 MHz, d6-OMSO) δ 6.74 (m, IH), 6.54 (m, IH). MS (m/z) (M+l)+: 187.0 and 189.0.
[000281] A solution of 6-bromopyridine-2,3-diamine (R-40) (0.267 mmol), 2-(2-chloro-6- fluorophenyl)-lH-imidazole-4-carboxylic acid (1-17) (0.223 mmol), O-(benzotriazol-l-yl)- AWN',N'-tetramethyluronium tetrafluoroborate (TBTU) (0.290 mmol) and TEA (0.558 mmol) in anhydrous DMF (5 mL) was stirred at room temperature for 24 hours. The reaction mixture was filtered, concentrated and purified by preparative ΗPLC (MeCN gradient 10-90%) to afford N-(2-amino-6-bromopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)- lH-imidazole-4-carboxamide (1-33) as a light yellow solid. 1H NMR (400 MHz, d4- CH3OH) δ 8.05 (s, IH), 7.63 (m, 2H), 7.49 (m, IH), 7.33 (m, IH), 6.84 (m, IH). MS (m/z) (M+l)+: 409.0 and 411.0.
[000282] To N-(2-amino-6-bromopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)- lH-imidazole- 4-carboxamide (1-33) was added PPA until dissolved. The solution was heated at 150 0C for 4 hours. The reaction mixture was cooled to 0 0C, and then slowly neutralized with K2CO3 to pΗ 7. The resulting mixture was extracted with EtOAc (3 x 30 mL). The extracts were combined, concentrated and purified with preparative ΗPLC (MeCN gradient 10-90%) to give 5-bromo-2-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)- lH-imidazo[4,5-b]pyridine (EX-95) as a white solid. 1H NMR(400 MHz, J4-CH3OH) δ 8.07 (s, IH), 7.81 (m, IH), 7.46 (m, IH), 7.41 (m, IH), 7.36 (m, IH), 7.21 (m, IH). MS (m/z) (M+l)+: 391.0 and 393.0.
Synthesis of 5-bromo-2-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-l-methyl-lH-
benzoMimidazole (EX-96)

[000283] To 4-bromo-l-fluoro-2-nitrobenzene (R-41) (03.27 mmol) was added a 2M methylamine solution in methanol. The solution was stirred at room temperature for 2 hours and then concentrated to give 4-bromo-./V-methyl-2-nitroaniline (R-42) which was used in the next step without further purification.
[000284] To a solution of 4-bromo-N-methyl-2-nitroaniline (R-42) dissolved in EtOH (20 mL) was added tin (II) choloride dihyrate (13.1 mmol) and the resulting suspension was heated at 55 0C for 2 hours. The reaction mixture was cooled and 10% aq NaHCO3 was added. The precipitate was removed by filtration. The filtrate was concentrated and purified by preparative HPLC (MeCN gradient 10-90%) to give 4-bromo-N1-methylbenzene-l,2- diamine (R-43). MS (m/z) (M+l)+: 200.0 and 202.0.
[000285] 4-Bromo-N1-methylbenzene- 1 ,2-diamine (R-43) was converted to iV-(5-bromo-2- (methylamino)phenyl)-2-(2-chloro-6-fluorophenyl)-lH-imidazole-4-carboxamide (1-34) using a similar procedure as for the synthesis of N-(2-amino-6-bromopyridin-3-yl)-2-(2- chloro-6-fluorophenyl)-lH-imidazole-4-carboxamide (1-33). MS (m/z) (M+l)+: 422.0 and 424.0.
[000286] N-(5-Bromo-2-(methylamino)phenyl)-2-(2-chloro-6-fluorophenyl)-lH- imidazole-4-carboxamide (1-34) was converted to 5-bromo-2-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)-l -methyl- lH-benzo [J] imidazole (EX-96) using similar procedure as for the synthesis of 5-bromo-2-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-lH- imidazo[4,5-b]pyridine (EX-95). 1U NMR(400 MHz, J4-CH3OH) δ 8.03 (s, IH), 7.83 (m, IH), 7.69 (m, IH), 7.61 (m, IH), 7.48 (m, IH), 7.38 (m, IH), 7.22 (m, IH), 4.21 (s, 3H). MS (m/z) (M+l)+: 404.0 and 406.0.
Synthesis of ethyl 5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-vl)-
2-(phenylethvnyl)isonicotinate (EX-97)

[000287] To a solution of 2-bromo-5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)pyridine (1-1) (1.9 mmol) in TΗF (20 ML) and cooled to -780C w-BuLi (2.11 mmol) was slowly added over 15 minuntes. After 30 minutes, ethyl chloroformate (1.9 mmol) was added drop wise over 15 minutes and the reaction mixture was stirred for 1 hour. After this time 10% aq NH4Cl (5 mL) was slowly added and the mixture was warmed to room temperature. The organic phase was extracted with EtOAc (3 x 50 mL) to afford ethyl 4-(6- bromopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)-lH-imidazole-l-carboxylate (1-35) as a pale yellow solid which was used without further purification. MS (m/z) (M+l)+: 423.0 and 425.1.
[000288] To a solution of ethyl 4-(6-bromopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)- IH- imidazole-1-carboxylate (1-35) (0.6 mmol) in TΗF (20 mL) at -780C w-BuLi (4.0 mmol) was added slowly over 50 minutes and stirred for 1 hour. Ethylchloroformate (2.4 mmol) was then added to the cold mixture and stirred for 30 minutes. After this time the reaction was quenched with 10 % aq NH4Cl (5 mL) and warmed to room temperature. The organic phase was extracted with EtOAc (3 x 50 mL), washed with brine, dried over Na2SO4 and concentrated. The crude residue was purified by silica chromatography (hexane : EtOAc = 1 : 1) to afford ethyl 2-bromo-5-(2-(2-chloro-6-fluorophenyl)-l-(ethoxycarbonyl)-lH- imidazol-4-yl)isonicotinate (1-36) containing 10% of starting material (1-35). MS (m/z) (M+l)+: 497.1 and 499.0.
[000289] Ethyl 5-(2-(2-chloro-6-fluorophenyl)- l-(ethoxycarbonyl)- lH-imidazol-4-yl)-2- (phenylethynyl)isonicotinate (1-37) was prepared by employing the same Sonogashira condition as described for the synthesis of 1-2. 1H NMR (400MHz, J4-CH3OH) δ 9.00 (s, IH), 8.24 (s, IH), 7.84 (s, IH), 7.64 (s, IH), 7.64 (m, 2H), 7.52 (m, IH), 7.45 (m, 5H), 7.24 (m, IH), 4.35 (m, 4H), 1.29 (t, J = 8.0 Hz, 3H), 1.21 (t, J = 8.0 Hz, 3H). MS (m/z) (M+l)+: 446.1.
[000290] In a small vial a suspension of ethyl 5-(2-(2-chloro-6-fluorophenyl)-l- (ethoxycarbonyl)-lH-imidazol-4-yl)-2-(phenylethynyl)isonicotinate (1-37) (0.03 mmol) in water (1 mL) was treated with concentrated H2SO4 (1 mL). The mixture was heated at 100 0C for 10 minutes and then cooled to room temperature, diluted with water (1 mL) and neutralized with 6N NaOH. The mixture was concentrated and the crude purified by preparative HPLC (MeCN gradient 20-70%) to yield ethyl 5-(2-(2-chloro-6-fluorophenyl)- lH-imidazol-4-yl)-2-(phenylethynyl)isonicotinate (EX-97) as a yellow solid. MS (m/z) (M+l)+: 446.1.
Synthesis of methyl 5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-2- (phenylethvnvl)isonicotinate (EX-98)

[000291] In a small vial a suspension of ethyl 5-(2-(2-chloro-6-fluorophenyl)-l- (ethoxycarbonyl)-lH-imidazol-4-yl)-2-(phenylethynyl)isonicotinate (1-37) (0.04 mmol) in MeOH (1 mL) was treated with 6N NaOH (0.24 mmol) and stirred at room temperature until complete conversion (30 minutes). The mixture was neutralized with 6N HCl (0.024 mmol) and the suspension was concentrated to yield a residue that was purified by preparative ΗPLC (MeCN gradient 20-70%) to afford 5-(2-(2-chloro-6-fluorophenyl)-lH-imidazol-4- yl)-2-(phenylethynyl)-isonicotinic acid (1-38) as a yellow solid. 1H NMR (400MHz, d4- CH3OH) δ 8.90 (s, IH), 7.74 (s, IH), 7.68 (s, IH), 7.53 (m, IH), 7.46 (m, IH), 7.34 (m, 5H), 7.19 (m, IH). MS (m/z) (M+l)+: 418.2.
Ill
[000292] 5-(2-(2-Chloro-6-fluorophenyl)-lH-imidazol-4-yl)-2-(phenylethynyl)- isonicotinic acid (1-38) (0.025 mmol) was dissolved in MeOH (1.5 niL) and IM TMSCH2N2 ether solution was added dropwise. The bright yellow solution was stirred at room temperature for 30 minutes then solvent was removed under vacuum and the crude residue purified by preparative HPLC (MeCN gradient 20-90%) to yield (EX-98)as a yellow solid. 1H NMR (400MHz, J4-CH3OH) δ 8.97 (s, IH), 7.82 (s, IH), 7.64 (s, IH), 7.63 (m, IH), 7.54 (m, IH), 7.45 (m, 5H), 7.27 (m, IH), 3.89 (s, 3H). MS (m/z) (M+l)+: 432.2.
Synthesis of 2-(2-chloro-6-fruorophenyl)-4-(phenylethvnyl)- lH-imidazole-5-carbonitrile
(EX-99)

[000293] To a solution of 5-bromo-2-(2-chloro-6-fluorophenyl)- lH-imidazole-4- carbonitrile (1-10) (0.06 mmol) in DMF (1 mL) was added TEA (0.040 mL). The mixture was degassed with nitrogen for 10 minutes and then was added phenylacetylene (R-6) (0.26 mmol), Copper Iodide (I) (0.006 mmol) and Pd(PPh3) (0.006 mmol). The reaction vessel was sealed and heat in a microwave oven for 10 minutes at 150 0C. Upon cooling the reaction was filtered and purified by preparative ΗPLC (MeCN gradient 10-90%) to afford 2-(2-chloro-6-fluorophenyl)-4-(phenylethynyl)-lH-imidazole-5-carbonitrile (EX-99) as a white solid. 1H NMR (400MΗz, d6-OMSO) δ 7.70-7.64 (m, 3H), 7.57-7.46 (m, 5H). MS (jn/z) (M+ 1)+: 322.1.
Synthesis of 5-(5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-3- phenyl-l,2,4-oxadiazole (EX-114)

[000295] To a solution of 5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazole-4- carboxylic acid (1-39) (0.03 mmol) in DMF (0.5 mL) was added ΗATU (0.03 mmol) and DIEA (0.011 mL). The reaction was stirred at room temperature for 10 minutes then N- hydroxybenzimidamide (R-44) (0.03 mmol) was added and the reaction was heated to 100 0C for 8 hours. Upon cooling the reaction mixture was partitioned with water and EtOAc. The organic layer was separated, washed with brine, dried over MgSO4, filtered and reduced to dryness. The crude material was purified by preparative TLC (hexane : EtOAc = 6 : 1) to afford 5-(5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)-3-phenyl- 1,2,4- oxadiazole (EX-114) as a white solid. 1U NMR (400MΗz, CD3OD) δ 8.16 (dd, J = 8.0, 2.0 Hz, 2H), 7.54 (m, 4H), 7.41 (bd, J = 8.4 Hz, IH), 7.25 (dt, J = 8.4, 1.0 Hz, IH). MS (jn/z) (M+l)+: 418.0 and 420.9.
Synthesis of 2-(2-chloro-6-fluorophenyl)-5-(6-(3-isopropyl-l,2,4-oxadiazol-5-yl)pyridin-3- vP- lH-imidazole-4-carbonitrile (EX-116)

[000296] To a solution of 5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazole-4- carbonitrile (1-10) (2.5 mmol) and methyl 5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)picolinate (R-45) (1.67 mmol) in a 2 : 1 : 1 toluene : ethanol : water solution was added Pd(PPh3)4 (0.17 mmol) and K2CO3 (3.34 mmol). The reaction mixture was degassed with nitrogen, and then heated at 100 0C for 16 hours. After cooling to room temperature the
crude product was purified by preparative HPLC (MeCN gradient 5 -70%) to yield 5-(2-(2- chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl)picolinic acid (1-40) as a white solid. MS (m/z) (M+ 1)+: 343.1.
[000297] To a solution of S-^-^-chloro-β-fluorophenylM-cyano-lH-imidazol-S- yl)picolinic acid (1-40) (0.29 mmol) in DMF (5 rnL) was added iV-hydroxybenzotriazole (0.44 mmol) followed by l-ethyl-3-(3'-dimethylaminopropyl)carbodiimide (0.44 mmol) and (Z)-N'-hydroxyisobutyrimidamide (R- 34) (0.44 mmol). The resulting solution was stirred at room temperature for 2 hours and then heated to 85 0C for 4 hours. After cooling to room temperature the reaction mixture was purified by preparative ΗPLC (MeCN gradient 10- 90%). 2-(2-Chloro-6-fluorophenyl)-5-(6-(3-isopropyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl)- lH-imidazole-4-carbonitrile (EX-116) was obtained as a white solid. MS (m/z) (M+l)+: 409.1.
Synthesis of 2-(2-chloro-6-fluorophenyl)-4-(6-(5-phenyl-l,3,4-oxadiazol-2-yl)pyridin-3-yl)- lH-imidazole-5-carbonitrile (EX-134)

[000298] To a suspension of 5-(2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4- yl)picolinic acid (1-40) (0.058 mmol), benzohydrazide (R-46) (0.064 mmol) and polymer supported triphenylphosphine (0.17 mmol) in MeCN (5 mL) was added trichloroacetonitrle (0.12, 8.4 uL). The resulting suspension was heated at 150 0C in a microwave oven for 30 minutes. The mixture was then cooled to room temperature and the polymer removed by filtration. The filtrate was concentrated and purified by preparative ΗPLC (MeCN gradient 10- 90%) to give 2-(2-Chloro-6-fluorophenyl)-4-(6-(5-phenyl-l,3,4-oxadiazol-2-yl)pyridin- 3-yl)-lH-imidazole-5-carbonitrile (EX-134). MS (m/z) (M+l)+: 433.1.
Synthesis of 5-(2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl)-N-(4- fluorophenvl)picolinamide (EX-135)

[000299] To a solution of 5-(2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4- yl)picolinic acid (1-40) (0.029 mmol) in DMF (1 niL) was added ΗATU (0.058 mmol) and aniline (R-47) (0.035 mmol). The resulting solution was stirred at room temperature for 6 hours. After purification by preparative ΗPLC (MeCN gradient 10 - 90%) 5-(2-(2-chloro-6- fluorophenyl)-5-cyano- lΗ-imidazol-4-yl)-N-(4-fluorophenyl)picolinamide (EX-135) was obtained as a white solid. MS (m/z) (M+l)+: 436.1.
Synthesis of N-(5-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl)pyridine-2-yl)benzamide

[000300] A sealed tube charged with 2-bromo-5-(2-(2-chloro-6-fluorophenyl)- IH- imidazol-4-yl)pyridine (1-1) (0.28 mmol), benzamide (R-48) (0.42 mmol), Xantphos (0.084 mmol), Pd2(dba)3 (0.028 mmol) and Cs2CO3 (0.56 mmol) in 1,4-dioxane (3.0 ml) was evacuated under vacuum and purged with nitrogen. The resulting mixture was heated at 100 0C for 12 hours. After this time, the solvent was removed and the residue was dissolved in DMSO. Purification by preparative ΗPLC (MeCN gradient 10-90%) gave N-(5-2-(2-chloro- 6-fluorophenyl)-lH-imidazol-4-yl)pyridine-2-yl)benzamide (EX-149) as a white solid. MS (m/z) (M+l)+: 393.0.
Synthesis of N-(5-2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl)pyridine-
2-vl)benzamide (EX-150)

[000301] To a sealed tube charged with 4-bromo-2-(2-chloro-6-fluorophenyl)-lH- imidazole-5-carbonitrile (1-10) (1.67 mmol), methyl 5-(4,4,5,5-tetramethyl- 1,3,2- dioxaborolan-2-yl)pyridine-2-amine (R-49) (3.35 mmol) and Pd(PPh3)4 (0.17 mmol) was added a 2 : 1 : 1 = toluene : ethanol : water mixture (9 mL) followed by Na2CO3 (3.34 mmol). The reaction mixture was purged with nitrogen and heated at 100 0C for 16 hours. The reaction mixture was then cooled to room temperature and the solvent was removed. The residue was dissolved in DMSO and purification by preparative ΗPLC (MeCN gradient 10-90%) gave 4-(6-aminopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)-lH-imidazole-5- carbonitrile (1-41) as a white solid. MS (m/z) (M+l)+: 314.
[000302] 4-Fluorobenzoyl chloride (R-50) (0.048 mmol) was added to a solution of 4-(6- aminopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)- lH-imidazole-5-carbonitrile (1-41) (0.032 mmol) in pyridine (2 mL) and the resulting mixture was stirred for 1 hour at room temperature. The solvent was removed and purification by preparative ΗPLC (MeCN gradient 10-90%) gave N-(5-(2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4- yl)pyridin-2-yl)-4-fluorobenzamide (EX-150) as a white solid. 1H NMR (400 MHz, dβ- DMSO) δ 10.48 (s, IH), 8.01 (m, 2H), 7.92 (d, 2H), 7.77 (d, 2H), 7.61 (m, IH), 7.51 (d, IH), 7.42 (t, IH), 7.34 (m, 2H). MS (m/z) (M+l)+: 436.
Synthesis of l-(5-(2-(2-chloro-6-fluorophenyl)-5-cyano- lH-imidazol-4-yl)pyridin-2-yl)-3-
(4-fluorophenyl)urea (EX- 166)

1-41
[000303] 4-Fluorophenylisocyanate (R-51) (0.048 mmol) was added to a solution of 4-(6- aminopyridin-3-yl)-2-(2-chloro-6-fluorophenyl)- lH-imidazole-5-carbonitrile (1-41) (0.032 mmol) in TΗF and the resulting mixture was heated at 70 0C for 2 hours. The solvent was removed and purification by preparative ΗPLC (MeCM gradient 10-90%) gave l-(5-(2- chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl)3-(4-fluorophenyl)urea (EX-166) as a white solid. 1H NMR (400 MHz, J6-DMSO) δ 10.1 (s, IH), 9.69 (s, IH), 8.76 (s, IH), 8.15 (m, IH), 7.81 (m, IH), 7.67 (m, IH), 7.52 (m, 4H), 7.17 (t, J = 8.8 Hz, 2H). MS (m/z) (M+l)+: 451.1.
Synthesis of N-(4-(2-(2-chloro-6-fluorophenyl)-5-cyano- lH-imidazol-4-yl)phenyl)-4- fluorobenzamide (EX-167)

[000304] To A solution of 4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazole-5- carbonitrile (1-10) (0.33 mmol) in a 2 : 1 : 1 toluene : ethanol : water solution (3.5 mL) was added 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)aniline (R-52) (0.67 mmol), Pd(PPh3)4 (0.017 mmol) and K2CO3 (0.67 mmol). The resulting suspension was bubbled with nitrogen and then heated at 100 0C for 12 hours. After cooled down to room temperature, the reaction mixture was extracted with EtOAc (3x 30 mL). The organics were combined and washed with water and brine, then dried over MgSO4. After filtration and concentration, the crude product was purified by preparative ΗPLC (MeCN gradient 10 to 90 %) to yield 4- (4-
aminophenyl)-2-(2-chloro-6-fluorophenyl)-lH-imidazole-5-carbonitrile (1-42) (41% yields). MS (m/z) (M+l)+: 313.0
[000305] A solution of 4-(4-aminophenyl)-2-(2-chloro-6-fluorophenyl)-lH-imidazole-5- carbonitrile (1-42) (0.032 mmol) in DCM(I niL) was added TEA(0.064 mmol), followed by 4-fluorobenzoyl chloride (R-50) (0.032 mmol) dropwise. The resulting reaction mixture was stirred at room temperature for 2 hours. After that the reaction mixture was concentrated, dissolved in DMSO and purified by preparative ΗPLC (MeCN gradient 10 to 70 %) to gave N-(4-(2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl)phenyl)-4-fluorobenzamide (EX-167) as a white solid. 1U NMR (400MΗz, d4- CH3OH) δ 10.48 (s, IH), 8.01 (m, 2H), 7.92 (m, 2H), 7.77 (m, 2H), 7.61 (m, IH), 7.51 (m, IH), 7.42 (m, IH), 7.36 (m, 2H). MS (m/z) (M+ 1)+: 435.0
Synthesis of 4-(2-(2-Chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl)-N-(3- fluorophenvDbenzamide (Ex-168)

[000306] To A solution of 4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazole-5- carbonitrile (1-10) (1.0 mmol) in a 2 : 1 : 1 toluene : ethanol : water solution (3.5 mL) was added methyl 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzoate (R-53) (1.45 mmol), Pd(PPh3)4 (0.017 mmol) and K2CO3 (2.0 mmol). The resulting suspension was bubbled with nitrogen and then heated at 100 0C for 12 hours. After cooled down to room temperature, the reaction mixture was extracted with EtOAc (3x 30 mL). The organics were combined and washed with water and brine, then dried over MgSO4. Purification by preparative ΗPLC (MeCN gradient 10 to 90 %) gave 4-(2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4- yl)benzoic acid (1-43) as white solid (20% yield). MS (m/z) (M+l)+: 342.0. [000307] To a solution of 4-(2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4- yl)benzoic acid (1-43) (0.03 mmol) in DMF (1 mL) was added ΗATU (0.06 mmol) and aniline (R-54) (0.04 mmol). The resulting solution was stirred at room temperature for 6
hours. After purification by preparative HPLC (MeCN gradient 10 - 90%) 4-(2-(2-chloro-6- fluorophenyl)-5-cyano- lH-imidazol-4-yl)-N-(3-fluorophenyl)benzamide (EX-171) was obtained as a white solid. 1H NMR (400MΗz, d6-OMSO) δ 10.6 (s, 1H),8.15 (m, 2H), 8.01 (m, 2H), 7.78 (dt, J = 12.0 , 2.4 Hz, IH), 7.70 (m, IH), 7.60 (m, 2H), 4.50 (m, IH), 7.41 (m, IH), 6.96 (m, IH). MS (m/z) (M+l)+: 435.0.
[000308] By repeating the procedures described in the above examples, using appropriate starting materials, the following compounds of Formula I, as identified in Table 1 were obtained.
Table 1





























Assays
Biochemical assay for human mPGES-1
[000309] Membrane preparations of tagless full length human mPGESl protein were generated from baculovirus mediated expression in insect cells. Membrane preparations containing human mPGESl were incubated with potential antagonist compounds for 30 minutes prior to addition of substrate. The substrate prostaglandin H2 (PGH2; 20 nM final concentrations) was added in acetone at 4 0C and the reaction was incubated for 5 minutes. The reaction was quenched with SnCl2 in HCl. The amount of PGE2 generated was detected by using a competitive HTRF PGE2 assay kit (CisBio Bioassays, Bedford MA, USA) and determined by comparing values to a standard curve of PGE2.
Cellular assay for human mPGES-1
[000310] A549 human adenocarcinoma cells were pre-treated with test or positive control compounds for 30 minutes. Cells were then stimulated with ILlβ (20 ng/mL) for 24 hours. An aliquot of cell media was transferred to a fresh assay plate and the level of PGE2 generated was detected by using a competitive HTRF PGE2 assay kit (CisBio Bioassays, Bedford MA, USA) and determined by comparing values to a standard curve of PGE2. IC50 values are calculated based on PGE2 levels by using a 4-parameter fit curve fitting model.
Cellular selectivity assay
[000311] A549 human adenocarcinoma cells were pre-treated with test or positive control compounds for 30 minutes prior to stimulation with ILl β (20 ng/mL) for 24 hours. The quantity of PGF2a accumulated in the media was determined by transferring an aliquot of media to a fresh plate. PGF2a was detected with the PGF2a EIA Kit (Cayman Chemical, Ann Arbor, Michigan; Catalog # 516011) and absolute levels are determined by comparison to a standard curve of PGF2a.
Biochemical assay for human COX2
[000312] The ability of compounds to inhibit COX-2 was determined using the COX Activity Assay from Cayman Chemical (Ann Arbor, Michigan, Catalog # 760151). Briefly, the assay was performed in a 384 well plate using recombinant COX-2 enzyme. The test compounds were added to the COX-2 enzyme before addition of substrate. The reaction was initiated by addition of arachadonic acid (final cone of 100 μM) and allowed to proceed for 10 minutes at 37 0C. Activity is determined by using the colorimetric substrate N,N,N',N'- tetramethyl-p-phenylenediamine (TMPD) at 590 nM.
Certain Assay Results
[000313] Various compounds of Formula (I) in free form or in pharmaceutically acceptable salt form, exhibit pharmacological properties, for example, as indicated by the in vitro tests described in this application. The IC50 value in those experiments is given as that concentration of the test compound in question that provoke a response halfway between the baseline and maximum responses. In certain examples compounds of Formula (I) have IC50 values from 1 nM to 200 μM. In some examples, compounds of Formula (I) have IC50 values from 0.001 μM to 100 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 50 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 25 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 20 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 15 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 10 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 5 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 2 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 1 μM. In other examples, compounds of Formula (I) have IC50
values from 0.001 μM to 0.8 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 0.6 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 0.4 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 0.2 μM. In other examples, compounds of Formula (I) have IC50 values from 0.001 μM to 0.1 μM.
[000314] In certain examples compounds of Formula (Ia) have IC50 values from 1 nM to 200 μM. In some examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 100 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 50 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 25 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 20 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 15 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 10 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 5 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 2 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 1 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 0.8 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 0.6 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 0.4 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 0.2 μM. In other examples, compounds of Formula (Ia) have IC50 values from 0.001 μM to 0.1 μM. By way of example only, the IC50 for mPGES-1 inhibition by certain compounds of Formula (Ia) are also listed in Table 1 above.
[000315] It is understood that the examples and embodiments described herein are for illustrative purposes only and that various modifications or changes in light thereof will be suggested to persons skilled in the art and are to be included within the spirit and purview of this application and scope of the appended claims.
Claims
1. A compound of Formula (Ia):

(Ia) wherein, each Ri is independently selected from H and Ci-Cβalkyl;
R2 is H, halogen, -CN, -C(O)ORi or -C(O)NRiRi; R3 is Ci-Cόhaloalkyl or halogen; R4 is H, Ci-Cόhaloalkyl or halogen;

X2 is O, S or NR8;
X3 is S, O or NR8;  each Re and each R7 are independently selected from H, halogen,
each Re and each R7 are independently selected from H, halogen,
Ci-C6alkyl, d_C6haloalkyl, and -C(O)OR8; each R8 is independently selected from H and Ci-Cβalkyl; each Ri5 is independently selected from H, halogen and -
C(O)OR8; L2 is a bond, C2-C6alkynylene, phenylene, -0-, -C(O)-, -C(O)NR8-, - NR8C(O)-, -NR8C(O)NR8-, 
 wherein:
wherein:
X6 is C(Rg)2, NR9, O or S; X7 is N or CR9; X8 is CR9, NR9, O or S; X9 is N or CR9; Xio is S or O; Xn is CR9 or N; each R9 is independently H or Ci-Cόalkyl; and wherein the d-Cβalkynylene and phenylene of L2 are each optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, CN, -OCi-Cβalkyl, halogen and OH;
Qi is phenyl, 5-6 membered heteroaryl containing 1-2 N heteroatoms, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S, C3-C8cycloalkyl, C4-C8cycloalkyl optionally substituted with =CH2 cyclohexyl substituted with 2 Cialkyl which together with the carbon to which they are attached
form a cyclopropyl, -(CRiORn)nQ2, 

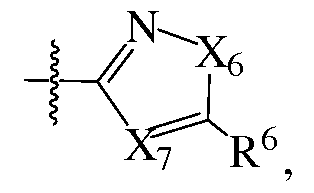 wherein: the phenyl, heteroaryl, C2-C6heterocycloalkyl and C3-C8cycloalkyl groups of Qi are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -0(CRiORn)1-4ORi0, -O(CRi0Rπ)i-4D , - 0(CRIORII)I-4RIO, -OCi.Cβalkyl, -OCi_C6haloalkyl, -C(O)ORi4, -N(Ri4)2, -C(O)N(Ri4)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, - S(O)2Ri4, -SRi4 and -0Ri4; each Rio and each Rn are independently selected from H, , C1- όhaloalkyl and Ci_6alkyl;
wherein: the phenyl, heteroaryl, C2-C6heterocycloalkyl and C3-C8cycloalkyl groups of Qi are optionally substituted with 1-4 substituents independently selected from Ci-Cβalkyl, Ci-Cβhaloalkyl, halogen, -CN, -Q2, -0(CRiORn)1-4ORi0, -O(CRi0Rπ)i-4D , - 0(CRIORII)I-4RIO, -OCi.Cβalkyl, -OCi_C6haloalkyl, -C(O)ORi4, -N(Ri4)2, -C(O)N(Ri4)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, - S(O)2Ri4, -SRi4 and -0Ri4; each Rio and each Rn are independently selected from H, , C1- όhaloalkyl and Ci_6alkyl;
Q2 is phenyl, 4-6 membered heterocycloalkyl containing 1-3 heteroatoms independently selected from N, O and S or C3- Cgcycloalkyl, each of which is optionally substituted with 1-4 substituents independently selected from halogen and C1- C6haloalkyl; 
Xi3 is -(CH2),- or -O(CH2)P-;
Ri2 is H, Ci-C ealkyl or -C(O)ORi4; each Ri4 is independently selected from H and C1-C βalkyl; n is O, 1, 2 or 3; p is O, 1, 2 or 3; q is 1, 2 or 3; s is O or 1 ;
provided that when the combination of L1, L2 and Qi is, Li is pyridyl and L2 is C2alkynylene and Qi is phenyl, pyridyl, thiazolyl, cyclohexyl or cyclopropyl, then R2 is -CN, -C(O)ORi or -C(O)NRiRi, or pharmaceutically acceptable salts, pharmaceutically acceptable solvates, N-oxides, prodrugs and isomers thereof. The compound of claim 1, wherein Li is a bond, 

The compound of claim 1 or claim 2, wherein L2 is a bond, -O-, -C(O)-,
C2alkynylene, phenylene,  or
or

The compound of any of claims 1 to 3, wherein Qi is phenyl, pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl optionally substituted with =CH2, cyclopentyl optionally substituted with =CH2, cyclohexyl optionally substituted with =CH2, cyclohexyl substituted with 2 Cialkyl which together with the carbon to which they are attached
form a C3cycloalkyl, -(CRi0RiI)nQ2, 


5. The compound of claim 1, wherein Li is  or and Qi is phenyl optionally substituted with 1-4 substituents independently selected from C1- C6alkyl, Ci_C6haloalkyl, halogen, -CN, -Q2, -O(CRi0Rii)i-4ORi0, -0(CR10Rn)1-4D , - 0(CR10Rn)1-4Ri0, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORi4, -N(R14)2, - C(O)N(R14)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, -S(O)2Ri4, -SRi4 and -0Ri4.
or and Qi is phenyl optionally substituted with 1-4 substituents independently selected from C1- C6alkyl, Ci_C6haloalkyl, halogen, -CN, -Q2, -O(CRi0Rii)i-4ORi0, -0(CR10Rn)1-4D , - 0(CR10Rn)1-4Ri0, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORi4, -N(R14)2, - C(O)N(R14)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, -S(O)2Ri4, -SRi4 and -0Ri4.
6. The compound of claim 1, wherein Li is  or , L2 is C2alkynylene and Qi is phenyl substituted with 1-4 substituents independently selected from d_C6alkyl, d_C6haloalkyl, halogen, -CN, -Q2, -O(CRi0Rπ)i-4ORi0, - 0(CR10Rn)1-4D , -0(CR10Rn)1-4R10, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORi4, - N(RH)2, -C(O)N(R14)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, -S(O)2Ri4, -SRi4 and -OR14.
or , L2 is C2alkynylene and Qi is phenyl substituted with 1-4 substituents independently selected from d_C6alkyl, d_C6haloalkyl, halogen, -CN, -Q2, -O(CRi0Rπ)i-4ORi0, - 0(CR10Rn)1-4D , -0(CR10Rn)1-4R10, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORi4, - N(RH)2, -C(O)N(R14)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, -S(O)2Ri4, -SRi4 and -OR14.
7. The compound of claim 1, wherein Li is  or , L2 is a bond and Qi is phenyl optionally substituted with 1-4 substituents independently selected from Ci_C6alkyl, d_C6haloalkyl, halogen, -CN, -Q2, -0(CR10Rn)1-4OR10, - 0(CR10Rn)1-4D 5 -O(CR10Rn)1-4R10, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORi4, - N(RH)2, -C(O)N(R14)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, -S(O)2Ri4, -SRi4 and -OR14.
or , L2 is a bond and Qi is phenyl optionally substituted with 1-4 substituents independently selected from Ci_C6alkyl, d_C6haloalkyl, halogen, -CN, -Q2, -0(CR10Rn)1-4OR10, - 0(CR10Rn)1-4D 5 -O(CR10Rn)1-4R10, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORi4, - N(RH)2, -C(O)N(R14)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, -S(O)2Ri4, -SRi4 and -OR14.
8. The compound of claim 1, wherein Li is  or , L2 is a bond and Qi is pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl optionally substituted with =CH2, cyclopentyl optionally substituted with =CH2, cyclohexyl optionally substituted with =CH2, cyclohexyl substituted with 2 Cialkyl which together with the carbon to which
or , L2 is a bond and Qi is pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl optionally substituted with =CH2, cyclopentyl optionally substituted with =CH2, cyclohexyl optionally substituted with =CH2, cyclohexyl substituted with 2 Cialkyl which together with the carbon to which
they are attached form a C3cycloalkyl, -(CRioRn)nQ2, 

are attached form a C3cycloalkyl, -(CRioRn)nQ2, 

 where the phenyl, pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl of Qi are optionally substituted with 1-4 substituents independently selected from C1- C6alkyl, d_C6haloalkyl, halogen, -CN, -Q2, -0(CR10Rn)1-4OR10, -0(CR10Rn)1-4D , -0(CR10Rn)1-4R10, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORi4, -N(R14)2, -C(O)N(Ru)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, -S(O)2Ri4, -SRi4 and -0Ri4.
where the phenyl, pyridyl, pyrimidinyl, morpholinyl, piperidinyl, azetidinyl, pyrrolidinyl, tetrahydro-2H-pyranyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl of Qi are optionally substituted with 1-4 substituents independently selected from C1- C6alkyl, d_C6haloalkyl, halogen, -CN, -Q2, -0(CR10Rn)1-4OR10, -0(CR10Rn)1-4D , -0(CR10Rn)1-4R10, -OCi_C6alkyl, -OCi_C6haloalkyl, -C(O)ORi4, -N(R14)2, -C(O)N(Ru)2, - NRi4C(O)Ri4, -NRi4C(O)Q2, -S(O)2Ri4, -SRi4 and -0Ri4.
10. The compound of any of claims 1-9, wherein each Ri is independently selected from H, methyl and ethyl.
11. The compound of any of claims 1-10, wherein R2 is H, Br, -CN, -C(O)ORi or - C(O)NRiRi; provided that when the combination of L1, L2 and Qi is, Li is pyridyl and L2 is C2alkynylene and Qi is phenyl, pyridyl, thiazolyl, cyclohexyl or cyclopropyl, then R2 is -CN, -C(O)ORi or -C(O)NRiRi.
12. The compound of any of claims 1-11, wherein R3 is -CF3, Cl or F.
13. The compound of any of claims 1-12, wherein R4 is H, -CF3, Cl or F.
14. The compound of any of claims 1-13, wherein Q2 is phenyl optionally substituted with 1-4 halogen groups.
15. The compound of claim 1 selected from:
2-(2-chloro-6-fluorophenyl)-5-[6-(2-phenylethynyl)pyridin-3-yl]-lH-imidazole-4- carbonitrile;
2- (2-chloro-6-fluorophenyl)-5 - [6- (2-phenylethynyl)pyridin-3 -yl] - 1 H-imidazole-4- carboxamide; ethyl 2-(2-chloro-6-fluorophenyl)-4-[6-(2-phenylethynyl)pyridin-3-yl]-lH-imidazole-
5-carboxylate;
2- (2-chloro-6-fluorophenyl)-5 - [6- (2-phenylethynyl)pyridin-3 -yl] - 1 H-imidazole-4- carboxylic acid;
2-(2-chloro-6-fluorophenyl)-N-methyl-5-[6-(2-phenylethynyl)pyridin-3-yl]-lH- imidazole-4-carboxamide;
5-[5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-2-(2- phenylethynyl)pyrimidine;
5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-phenylpyrimidine;
2-{5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]pyrimidin-2-yl}-lH-indole;
2-(2-chloro-6-fluorophenyl)-4-[5-(2-phenylethynyl)thiophen-2-yl]-lH-imidazole;
5-[5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-2-(4-tert- butylphenyl)pyridine;
2-{5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridin-2-yl}-lH-indole;
5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[4-(2- methylpropyl)phenyl]pyridine;
5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[3-
(trifluoromethyl)phenyl]pyridine;
4-{5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridin-2- yljbenzonitrile;
3-{5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridin-2- yljbenzonitrile;
5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[4-
(trifluoromethoxy)phenyl]pyridine;
5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[3-
(trifluoromethoxy)phenyl]pyridine; tert-butyl 5-{5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridin-2- yl } - 1 H-indole- 1 -carboxylate;
5-{5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridin-2-yl}-l-methyl-lH- indole;
2-(l-benzothiophen-5-yl)-5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5- yl]pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[4-
(trifluoromethyl)phenyl]pyridine; 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(2,3-dihydro-l-benzofuran-5- yl)pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(3,4-dihydro-2H-l,5- benzodioxepin-7-yl)pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-phenyl-l,2,4-oxadiazol-3- yl)pyridine;
5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-phenyl-l,2,4- oxadiazol-3-yl)pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-cyclohexyl-l,2,4-oxadiazol-
3-yl)pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5-(propan-2-yl)-l,2,4- oxadiazol-3-yl]pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5-(2-methylpropyl)-l,2,4- oxadiazol-3-yl]pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5-(4,4-difluorocyclohexyl)-
1 ,2,4-oxadiazol-3-yl]pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-{5-[l-(4- fluorophenyl)cyclopropyl]-l,2,4-oxadiazol-3-yl}pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-cyclopentyl-l,2,4-oxadiazol-
3-yl)pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5-(pentan-3-yl)-l,2,4- oxadiazol-3-yl]pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-{5-[2-(3-fluorophenyl)ethyl]- l,2,4-oxadiazol-3-yl}pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5-(2-phenylethyl)-l,2,4- oxadiazol-3-yl]pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-{spiro[2.5]octan-6-yl}-l,2,4- oxadiazol-3-yl)pyridine;
2-(5-benzyl-l,2,4-oxadiazol-3-yl)-5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5- yl]pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(5-phenyl-l,3-oxazol-2- yl)pyridine;
S-tl-Cl-chloro-ό-fluoropheny^-lH-imidazol-S-yll-l-CS-phenyl-l^-thiazol-l- yl)pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-[5-(4-methoxyphenyl)-l,3- thiazol-2-yl]pyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-phenoxypyridine;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-2-(4-methoxyphenoxy)pyridine;
2-(2-chloro-6-fluorophenyl)-5-(2-methoxypyrimidin-5-yl)-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-(2-phenoxypyrimidin-5-yl)-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(l-cyclopropylethoxy)pyrimidin-5-yl]-lH- imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-(2-cyclobutoxypyrimidin-5-yl)-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(l-phenylethoxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-{2-[(l-methoxypropan-2-yl)oxy]pyrimidin-5-yl}-lH- imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(oxan-4-yloxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(propan-2-yloxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile;
5-[2-(benzyloxy)pyrimidin-5-yl]-2-(2-chloro-6-fluorophenyl)-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(propan-2-yloxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(cyclopentyloxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(cyclohexyloxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile; 5-[2-(butan-2-yloxy)pyrimidin-5-yl]-2-(2-chloro-6-fluorophenyl)-lH-imidazole-4- carbonitrile;
5-[2-(benzyloxy)pyrimidin-5-yl]-2-[2-fluoro-6-(trifluoromethyl)phenyl]-lH- imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-{2-[(l,l,l,3,3,3-hexafluoropropan-2-yl)oxy]pyrimidin-
5-yl}-lH-imidazole-4-carbonitrile;
2-(2,6-dichlorophenyl)-5-[2-(propan-2-yloxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile;
2-[2-fluoro-6-(trifluoromethyl)phenyl]-4-[2-(propan-2-yloxy)pyrimidin-5-yl]-lH- imidazole-5-carbonitrile;
5-{2-[(4-tert-butylcyclohexyl)oxy]pyrimidin-5-yl}-2-(2-chloro-6-fluorophenyl)-lH- imidazole-4-carbonitrile;
2-(2-cmoro-6-fluorophenyl)-5-(2-{ [(lS,2R,5R)-2-methyl-5-(propan-2- yl)cyclohexyl]oxy}pyrimidin-5-yl)-lH-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-{2-[(4,4-dimethylcyclohexyl)oxy]pyrimidin-5-yl}-lH- imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-(2-{ [(2S)-l,l,l-trifluoropropan-2-yl]oxy}pyrimidin-5- yl)- 1 H-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-(2- { [(2R)- 1,1,1 -trifluoropropan-2-yl] oxy }pyrimidin-5- yl)- 1 H-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(2,2,4,4-tetrafluorocyclobutoxy)pyrimidin-5-yl]- lH-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(2,3-dihydro-lH-inden-2-yloxy)pyrimidin-5-yl]- lH-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-(2-{ [4-(trifluoromethyl)cyclohexyl]oxy}pyrimidin-5- yl)-l H-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-4-{2-[(l,l,l-trifluoropropan-2-yl)oxy]pyrimidin-5-yl}- lH-imidazole-5-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(pentan-3-yloxy)pyrimidin-5-yl]-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-(2-ethoxypyrimidin-5-yl)-lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-[2-(4-methoxyphenoxy)pyrimidin-5-yl]-lH-imidazole-
4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(3-methoxyphenoxy)pyrimidin-5-yl]-lH-imidazole-
4-carbonitrile;
5-[2-(benzyloxy)pyrimidin-5-yl]-2-[2-chloro-6-(trifluoromethyl)phenyl]-lH- imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-4-[2-(methylsulfanyl)pyrimidin-5-yl]-lH-imidazole-5- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-(2-methanesulfonylpyrimidin-5-yl)-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(propan-2-ylsulfanyl)pyrimidin-5-yl]-lH- imidazole-4-carbonitrile;
5-[2-(butan-2-ylsulfanyl)pyrimidin-5-yl]-2-(2-chloro-6-fluorophenyl)-lH-imidazole-
4-carbonitrile;
5-[2-(tert-butylsulfanyl)pyrimidin-5-yl]-2-(2-chloro-6-fluorophenyl)-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-4-(5-phenyl-l-benzofuran-2-yl)-lH-imidazole;
2-(2-chloro-6-fluorophenyl)-4-[5-(2-phenylethynyl)-l-benzofuran-2-yl]-lH- imidazole;
5-{2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5-yl}-3-(propan-
2-yl)- 1 ,2,4-oxadiazole;
2-(2-chloro-6-fluorophenyl)-4-[5-(4-phenylphenyl)-l-benzofuran-2-yl]-lH- imidazole;
2-(2-chloro-6-fluorophenyl)-4-[5-(3,5-dimethylphenyl)-l-benzofuran-2-yl]-lH- imidazole;
2-(2-chloro-6-fluorophenyl)-4-{5-[4-(2-methylpropyl)phenyl]-l-benzofuran-2-yl}- lH-imidazole;
4-[5-(4-tert-butylphenyl)-l-benzofuran-2-yl]-2-(2-chloro-6-fluorophenyl)-lH- imidazole;
5-bromo-2-(2-chloro-6-fluorophenyl)-4-[5-(2-phenylethynyl)-l-benzofuran-2-yl]-lH- imidazole; 5-bromo-4-[3-bromo-5-(2-phenylethynyl)-l-benzofuran-2-yl]-2-(2-chloro-6- fluorophenyl)- lH-imidazole; methyl 4-{2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5- yljbenzoate; methyl 2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-6- carboxylate;
5-{2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5-yl}-3-methyl-
1,2,4-oxadiazole;
5-{2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5-yl}-3- cyclopropyl- 1 ,2,4-oxadiazole;
3-(5-{2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5-yl}-l,2,4- oxadiazol-3-yl)pyridine;
4- (5- { 2- [2- (2-chloro-6-fluorophenyl)- 1 H-imidazol-4-yl] - 1 -benzofuran-5 -yl } - 1 ,2,4- oxadiazol-3-yl)pyridine; ethyl 4- { 2- [2- (2-fluorophenyl) - 1 H-imidazol-4- yl] - 1 -benzofuran- 5 - yl } benzoate ;
5-bromo-2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-lH-l,3-benzodiazole;
4-{5-bromo-lH-imidazo[4,5-b]pyridin-2-yl}-2-(2-chloro-6-fluorophenyl)-lH- imidazole;
5-bromo-2-[2-(2-chloro-6-fluorophenyl)- 1 H-imidazol-4-yl]- 1 -methyl- IH- 1,3- benzodiazole; ethyl 5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-2-(2-phenylethynyl)pyridine-
4-carboxylate; methyl 5-[2-(2-chloro-6-fluorophenyl)- 1 H-imidazol-4-yl] -2- (2- phenylethynyl)pyridine-4-carboxylate;
2-(2-chloro-6-fluorophenyl)-5-(2-phenylethynyl)-lH-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-{2-[4-(propan-2-yl)phenyl]ethynyl}-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(4-phenylphenyl)ethynyl]-lH-imidazole-4- carbonitrile;
4-chloro-N-(3-{2-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5- yl] ethynyl } phenyl)benzamide ; 2-(2-chloro-6-fluorophenyl)-5- { 2- [4-(trifluoromethoxy)phenyl]ethynyl } - 1 H- imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-{2-[3-(trifluoromethyl)phenyl]ethynyl}-lH-imidazole-
4-carbonitrile;
2- (2-chloro-6-fluorophenyl)-5 - [2- (4-methoxyphenyl)ethynyl] - 1 H-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(4-methoxy-2-methylphenyl)ethynyl]-lH- imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(4-ethoxyphenyl)ethynyl]-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(3,5-dimethoxyphenyl)ethynyl]-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(l-hydroxycyclohexyl)ethynyl]-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(4-cyanophenyl)ethynyl]-lH-imidazole-4- carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-{2-[4-(piperidin-l-yl)phenyl]ethynyl}-lH-imidazole-
4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[2-(pyridin-3-yl)ethynyl]-lH-imidazole-4-carbonitrile;
5-[2-(3-aminophenyl)ethynyl]-2-(2-chloro-6-fluorophenyl)-lH-imidazole-4- carbonitrile;
5-[4-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-3-phenyl- 1,2,4- oxadiazole;
5-[5-bromo-2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-3-(4-fluorophenyl)- 1,2,4- oxadiazole;
2-(2-chloro-6-fluorophenyl)-4-{6-[3-(propan-2-yl)-l,2,4-oxadiazol-5-yl]pyridin-3- yl } - lH-imidazole-5-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[6-(3-cyclopropyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl]- lH-imidazole-4-carbonitrile;
5-{6-[3-(butan-2-yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-2-(2-chloro-6-fluorophenyl)- lH-imidazole-4-carbonitrile; 2-(2-chloro-6-fluorophenyl)-5-{6-[3-(oxan-4-yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}- lH-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-4-[6-(3-cyclopentyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl]- lH-imidazole-5-carbonitrile; tert-butyl 3-(5-{ 5-[2-(2-chloro-6-fluorophenyl)-4-cyano- lH-imidazol-5-yl]pyridin-2- yl }- 1 ,2,4-oxadiazol-3-yl)azetidine- 1 -carboxylate;
2-(2-chloro-6-fluorophenyl)-5-{6-[3-(3,3-difluorocyclobutyl)-l,2,4-oxadiazol-5- yl]pyridin-3-yl}-lH-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-[6-(3-cyclobutyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl]- lH-imidazole-4-carbonitrile;
5-{6-[3-(butan-2-yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-2-(2-chloro-6-fluorophenyl)- lH-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-{6-[3-(4,4-difluorocyclohexyl)-l,2,4-oxadiazol-5- yl]pyridin-3-yl}-lH-imidazole-4-carbonitrile;
5-[6-(3-tert-butyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl]-2-(2-chloro-6-fluorophenyl)-lH- imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-5-{6-[3-(3-methylidenecyclobutyl)-l,2,4-oxadiazol-5- yl]pyridin-3-yl}-lH-imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-4-{6-[3-(3-methylphenyl)-l,2,4-oxadiazol-5-yl]pyridin-
3-yl}-lH-imidazole-5-carbonitrile;
2-(2-chloro-6-fluorophenyl)-4-{6-[3-(4-methylphenyl)-l,2,4-oxadiazol-5-yl]pyridin-
3-yl}-lH-imidazole-5-carbonitrile;
2-[2-fluoro-6-(trifluoromethyl)phenyl]-5-{6-[3-(propan-2-yl)-l,2,4-oxadiazol-5- yl]pyridin-3-yl}-lH-imidazole-4-carbonitrile;
2-(2,6-dichlorophenyl)-5-{6-[3-(propan-2-yl)-l,2,4-oxadiazol-5-yl]pyridin-3-yl}-lH- imidazole-4-carbonitrile;
2-(2-chloro-6-fluorophenyl)-4-[6-(3-phenyl-l,2,4-oxadiazol-5-yl)pyridin-3-yl]-lH- imidazole-5-carbonitrile;
2-(2-chloro-6-fluorophenyl)-4-{6-[3-(4-fluorophenyl)-l,2,4-oxadiazol-5-yl]pyridin-3- yl } - lH-imidazole-5-carbonitrile;
2-(2-chloro-6-fluorophenyl)-4-[6-(5-phenyl-l,3,4-oxadiazol-2-yl)pyridin-3-yl]-lH- imidazole- 5 -carbonitrile ;
5-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]-N-(4- fluorophenyl)pyridine-2-carboxamide;
5-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]-N-(3- fluorophenyl)pyridine-2-carboxamide;
N-(2-amino-5-fluorophenyl)-5-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4- yl]pyridine-2-carboxamide;
2-(2-chloro-6-fluorophenyl)-4-(6-{ [2-(3-fluorophenyl)pyrrolidin-l- yl]carbonyl}pyridin-3-yl)-lH-imidazole-5-carbonitrile;
5-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]-N-phenylpyridine-2- carboxamide;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(4-fluorophenyl)pyridine-2- carboxamide;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(3-fluorophenyl)pyridine-2- carboxamide;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(2-fluorophenyl)pyridine-2- carboxamide;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(3-methylphenyl)pyridine-2- carboxamide;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(4-methylphenyl)pyridine-2- carboxamide;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-phenylpyridine-2-carboxamide;
5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]-N-(3-cyanophenyl)pyridine-2- carboxamide;
5-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]-N-(3-fluorophenyl)-N- methylpyridine-2-carboxamide;
2-(2-chloro-6-fluorophenyl)-4-(6-{ [(3S)-3-phenylmorpholin-4-yl]carbonyl}pyridin-3- yl)-lH-imidazole-5-carbonitrile;
N-{5-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-5-yl]pyridin-2-yl}benzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-4- fluorobenzamide ; N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3-
(trifluoromethyl)benzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-2-
(trifluoromethyl)benzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-4- methylbenzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-4- ethylbenzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-4-
(trifluoromethyl)benzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3- fluorobenzamide ;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3,4- difluorobenzamide ;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-2- fluorobenzamide ;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2- yljbenzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3- methoxybenzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-2,4- difluorobenzamide ;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-4-
(dimethylamino)benzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3- cyanobenzamide ;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3-
(trifluoromethoxy)benzamide;
N-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3-
(trifluoromethoxy)benzamide; l-{5-[2-(2-chloro-6-fluorophenyl)-4-cyano-lH-imidazol-5-yl]pyridin-2-yl}-3-(4- fluorophenyl)urea;
N-{4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]phenyl}-4- fluorobenzamide ;
N-{4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]phenyl}-3- fluorobenzamide ;
N-{4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]phenyl}-4-
(trifluoromethyl)benzamide;
N-{4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]phenyl}benzamide;
4-[2-(2-chloro-6-fluorophenyl)-5-cyano-lH-imidazol-4-yl]-N-(3- fluorophenyl)benzamide; methyl 2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l-benzofuran-5- carboxylate;
5-(5-bromo-l-benzofuran-2-yl)-2-(2-chloro-6-fluorophenyl)-lH-imidazole, and
5-bromo-2-[2-(2-chloro-6-fluorophenyl)-lH-imidazol-4-yl]-l,3-benzoxazole.
16. A pharmaceutical composition comprising a therapeutically effective amount of a compound of any one of claims 1-15 and a pharmaceutically acceptable carrier.
17. A method for treating a disease or disorder associated with mPGES-1 activity, wherein the method comprises administering to a subject in need thereof a therapeutically effective amount of a compound of any of claims 1-15.
18. The method of claim 17, wherein the diseases or disorder is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease.
19. The use of a compound of Formula (Ia) of claim 1 in the manufacture of a medicament for treating a disease or disorder associated with mPGES-1 activity, wherein the disease or disorder is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease.
20. A medicament for treating a disease or disorder associated with mPGES-1 activity, wherein the medicament comprises a therapeutically effective amount of a compound of any of claims 1-15.
21. The medicament of claim 20, wherein disease or disorder associated with mPGES-1 activity is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease.
2. A compound for use in a method of medical treatment, wherein the method of medical treatment is for treating a disease or disorder associated with mPGES-1 activity, wherein the disease or disorder is selected from pain, a cardiovascular disease, a neurodegenerative diseases and an inflammatory disease, and wherein the compound is a compound of Formula (Ia) of claim 1.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17379509P | 2009-04-29 | 2009-04-29 | |
| US61/173,795 | 2009-04-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2010127152A2 true WO2010127152A2 (en) | 2010-11-04 |
| WO2010127152A3 WO2010127152A3 (en) | 2011-09-29 |
Family
ID=42272690
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/033022 WO2010127152A2 (en) | 2009-04-29 | 2010-04-29 | Compounds and compositions as microsomal prostaglandin e synthase-1 inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2010127152A2 (en) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012087771A1 (en) * | 2010-12-21 | 2012-06-28 | Eli Lilly And Company | Novel imidazole-2-benzamide compounds useful for the treatment of osteoarthritis |
| EP2521451A1 (en) * | 2010-01-07 | 2012-11-14 | Selexagen Therapeutics, Inc. | Hedgehog inhibitors |
| WO2012161965A1 (en) * | 2011-05-26 | 2012-11-29 | Eli Lilly And Company | Novel imidazole derivatives useful for the treatment of arthritis |
| EP2552208A1 (en) * | 2010-03-31 | 2013-02-06 | Glaxo Group Limited | Imidazolyl-imidazoles as kinase inhibitors |
| WO2013170774A1 (en) * | 2012-05-16 | 2013-11-21 | 上海医药集团股份有限公司 | Acetylene derivative having antineoplastic activity |
| WO2013186089A2 (en) | 2012-06-14 | 2013-12-19 | Basf Se | Pesticidal methods using substituted 3-pyridyl thiazole compounds and derivatives for combating animal pests |
| JP2015503505A (en) * | 2011-12-23 | 2015-02-02 | ミレニアム ファーマシューティカルズ, インコーポレイテッドMillennium Pharmaceuticals, Inc. | Heteroaryl and uses thereof |
| US8957066B2 (en) | 2011-02-28 | 2015-02-17 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US9056874B2 (en) | 2012-05-04 | 2015-06-16 | Novartis Ag | Complement pathway modulators and uses thereof |
| US9265734B2 (en) | 2008-09-03 | 2016-02-23 | Biomarin Pharmaceutical Inc. | Compositions including 6-aminohexanoic acid derivatives as HDAC inhibitors |
| CN105899504A (en) * | 2014-01-21 | 2016-08-24 | 豪夫迈·罗氏有限公司 | Imidazoles for the treatment and prophylaxis of respiratory syncytial virus infection |
| US9475806B2 (en) | 2013-03-14 | 2016-10-25 | Novartis Ag | Complement factor B inhibitors and uses there of |
| US9540395B2 (en) | 2011-02-28 | 2017-01-10 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US9676728B2 (en) | 2013-10-30 | 2017-06-13 | Novartis Ag | 2-benzyl-benzimidazole complement factor B inhibitors and uses thereof |
| US10029988B2 (en) | 2013-03-15 | 2018-07-24 | Biomarin Pharmaceutical Inc. | HDAC inhibitors |
| US10059723B2 (en) | 2011-02-28 | 2018-08-28 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| WO2019101826A1 (en) | 2017-11-22 | 2019-05-31 | Khondrion Ip B.V. | Compounds as mpges-1 inhibitors |
| US11465987B2 (en) | 2010-03-01 | 2022-10-11 | Oncternal Therapeutics, Inc. | Compounds for treatment of cancer |
| WO2024041555A1 (en) * | 2022-08-24 | 2024-02-29 | Insilico Medicine Ip Limited | Methods of manufacturing kinase inhibitors |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2005254782A1 (en) * | 2004-06-18 | 2005-12-29 | Biolipox Ab | Indoles useful in the treatment of inflammation |
| US7442716B2 (en) * | 2004-12-17 | 2008-10-28 | Merck Frosst Canada Ltd. | 2-(phenyl or heterocyclic)-1H-phenantrho[9,10-d]imidazoles as mPGES-1 inhibitors |
| WO2008084218A1 (en) * | 2007-01-12 | 2008-07-17 | Boehringer Ingelheim International Gmbh | Benzazole derivatives for the treatment of inflammations |
| GB0709031D0 (en) * | 2007-05-10 | 2007-06-20 | Sareum Ltd | Pharmaceutical compounds |
-
2010
- 2010-04-29 WO PCT/US2010/033022 patent/WO2010127152A2/en active Application Filing
Non-Patent Citations (4)
| Title |
|---|
| CURR. OPIN. INVEST. DRUGS, vol. 8, 2007, pages 411 - 415 |
| J. MED. CHEM., vol. 51, 2008, pages 4059 - 4067 |
| PHARMACOL. REV., vol. 59, 2007, pages 207 - 224 |
| SAULNIER ET AL., BIOORGANIC AND MEDICINAL CHEMISTRY LETTERS, vol. 4, 1985 |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9796664B2 (en) | 2008-09-03 | 2017-10-24 | Biomarin Pharmaceutical Inc. | Compositions including 6-aminohexanoic acid derivatives as HDAC inhibitors |
| US9265734B2 (en) | 2008-09-03 | 2016-02-23 | Biomarin Pharmaceutical Inc. | Compositions including 6-aminohexanoic acid derivatives as HDAC inhibitors |
| US9174949B2 (en) | 2010-01-07 | 2015-11-03 | Selexagen Therapeutics, Inc. | Hedgehog inhibitors |
| EP2521451A1 (en) * | 2010-01-07 | 2012-11-14 | Selexagen Therapeutics, Inc. | Hedgehog inhibitors |
| EP2521451A4 (en) * | 2010-01-07 | 2013-08-21 | Selexagen Therapeutics Inc | Hedgehog inhibitors |
| US11465987B2 (en) | 2010-03-01 | 2022-10-11 | Oncternal Therapeutics, Inc. | Compounds for treatment of cancer |
| EP2552208A4 (en) * | 2010-03-31 | 2014-07-09 | Glaxo Group Ltd | Imidazolyl-imidazoles as kinase inhibitors |
| EP2552208A1 (en) * | 2010-03-31 | 2013-02-06 | Glaxo Group Limited | Imidazolyl-imidazoles as kinase inhibitors |
| US8252831B2 (en) | 2010-12-21 | 2012-08-28 | Eli Lilly And Company | Imidazole-2-benzamide compounds useful for the treatment of osteoarthritis |
| WO2012087771A1 (en) * | 2010-12-21 | 2012-06-28 | Eli Lilly And Company | Novel imidazole-2-benzamide compounds useful for the treatment of osteoarthritis |
| US8957066B2 (en) | 2011-02-28 | 2015-02-17 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US10059723B2 (en) | 2011-02-28 | 2018-08-28 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US9908899B2 (en) | 2011-02-28 | 2018-03-06 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US10301323B2 (en) | 2011-02-28 | 2019-05-28 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US10526346B2 (en) | 2011-02-28 | 2020-01-07 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US10280182B2 (en) | 2011-02-28 | 2019-05-07 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US10981933B2 (en) | 2011-02-28 | 2021-04-20 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US9512143B2 (en) | 2011-02-28 | 2016-12-06 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| US9540395B2 (en) | 2011-02-28 | 2017-01-10 | Biomarin Pharmaceutical Inc. | Histone deacetylase inhibitors |
| WO2012161965A1 (en) * | 2011-05-26 | 2012-11-29 | Eli Lilly And Company | Novel imidazole derivatives useful for the treatment of arthritis |
| US8648200B2 (en) | 2011-05-26 | 2014-02-11 | Eli Lilly And Company | Imidazole derivatives useful for the treatment of arthritis |
| CN103562200A (en) * | 2011-05-26 | 2014-02-05 | 伊莱利利公司 | Novel imidazole derivatives useful for the treatment of arthritis |
| JP2015503505A (en) * | 2011-12-23 | 2015-02-02 | ミレニアム ファーマシューティカルズ, インコーポレイテッドMillennium Pharmaceuticals, Inc. | Heteroaryl and uses thereof |
| US9056874B2 (en) | 2012-05-04 | 2015-06-16 | Novartis Ag | Complement pathway modulators and uses thereof |
| CN103421005A (en) * | 2012-05-16 | 2013-12-04 | 上海医药集团股份有限公司 | Acetylene derivative capable of resisting activity of tumor |
| WO2013170774A1 (en) * | 2012-05-16 | 2013-11-21 | 上海医药集团股份有限公司 | Acetylene derivative having antineoplastic activity |
| WO2013186089A2 (en) | 2012-06-14 | 2013-12-19 | Basf Se | Pesticidal methods using substituted 3-pyridyl thiazole compounds and derivatives for combating animal pests |
| US9475806B2 (en) | 2013-03-14 | 2016-10-25 | Novartis Ag | Complement factor B inhibitors and uses there of |
| US10029988B2 (en) | 2013-03-15 | 2018-07-24 | Biomarin Pharmaceutical Inc. | HDAC inhibitors |
| US10428028B2 (en) | 2013-03-15 | 2019-10-01 | Biomarin Pharmaceutical Inc. | HDAC inhibitors |
| US9676728B2 (en) | 2013-10-30 | 2017-06-13 | Novartis Ag | 2-benzyl-benzimidazole complement factor B inhibitors and uses thereof |
| US9790213B2 (en) * | 2014-01-21 | 2017-10-17 | Hoffmann-La Roche Inc. | Imidazoles for the treatment and prophylaxis of respiratory syncytial virus infection |
| JP2017503821A (en) * | 2014-01-21 | 2017-02-02 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | Imidazole for treatment and prevention of respiratory syncytial virus infection |
| US20160326148A1 (en) * | 2014-01-21 | 2016-11-10 | Hoffmann-La Roche Inc. | Novel Imidazoles for the Treatment and Prophylaxis of Respiratory Syncytial Virus Infection |
| CN105899504A (en) * | 2014-01-21 | 2016-08-24 | 豪夫迈·罗氏有限公司 | Imidazoles for the treatment and prophylaxis of respiratory syncytial virus infection |
| WO2019101826A1 (en) | 2017-11-22 | 2019-05-31 | Khondrion Ip B.V. | Compounds as mpges-1 inhibitors |
| US11672787B2 (en) | 2017-11-22 | 2023-06-13 | Khondrion Ip B.V. | Compounds as mPGES-1 inhibitors |
| WO2024041555A1 (en) * | 2022-08-24 | 2024-02-29 | Insilico Medicine Ip Limited | Methods of manufacturing kinase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010127152A3 (en) | 2011-09-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2010127152A2 (en) | Compounds and compositions as microsomal prostaglandin e synthase-1 inhibitors | |
| EP2549875B1 (en) | Soluble guanylate cyclase activators | |
| EP1753743B1 (en) | Pyridazinone derivatives, methods for producing them and their use as pharmaceuticals | |
| EP1943219B1 (en) | 5-lipoxygenase-activating protein (flap) inhibitors | |
| AU2008247672B2 (en) | 5-lipoxygenase-activating protein (FLAP) inhibitors | |
| EA028626B1 (en) | Tnf-alpha modulating benzimidazoles | |
| BR112019000716A2 (en) | cyclin dependent kinase inhibitors (cdk7) | |
| AU2014298017B2 (en) | Heterobicycloaryl RORC2 inhibitors and methods of use thereof | |
| WO2012082947A1 (en) | Compounds and compositions as tgr5 agonists | |
| WO2007056228A2 (en) | 5-lipoxygenase-activating protein (flap) inhibitors | |
| JP2012246302A (en) | Indole derivative as crac modulator | |
| CA2693214A1 (en) | Beta carboline derivatives as antidiabetic compounds | |
| AU2002341921A1 (en) | Heteroaryl substituted tetrazole modulators of metabotropic glutamate receptor-5 | |
| EP1434773A2 (en) | Heteroaryl substituted tetrazole modulators of metabotropic glutamate receptor-5 | |
| JP2012528165A (en) | Benzoxazepines as inhibitors of PI3K / mTOR and methods for their use and production | |
| CA2503715A1 (en) | Kinase inhibitors | |
| EP2763672A1 (en) | 1,3-substituted azetidine pde10 inhibitors | |
| AU2005243493A1 (en) | Novel pyridazinone derivatives as inhibitors of CDK2 | |
| WO2013116182A1 (en) | Heterocyclic compounds as inhibitors of leukotriene production | |
| WO2015143653A1 (en) | TrkA KINASE INHIBITORS,COMPOSITIONS AND METHODS THEREOF | |
| WO2014150114A1 (en) | Substituted pyridizinone derivatives as pde10 inhibitors | |
| EP4267564A1 (en) | Indole derivatives useful in treating conditions associated with cgas | |
| KR20230026288A (en) | Sos1 inhibitors and use thereof | |
| OA18716A (en) | Heterobicycloaryl RORC2 inhibitors and methods of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10717391 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10717391 Country of ref document: EP Kind code of ref document: A2 |