WO2010055028A2 - Organic compounds - Google Patents
Organic compounds Download PDFInfo
- Publication number
- WO2010055028A2 WO2010055028A2 PCT/EP2009/064891 EP2009064891W WO2010055028A2 WO 2010055028 A2 WO2010055028 A2 WO 2010055028A2 EP 2009064891 W EP2009064891 W EP 2009064891W WO 2010055028 A2 WO2010055028 A2 WO 2010055028A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- salt
- crystalline
- fty720
- peaks
- weak
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C215/00—Compounds containing amino and hydroxy groups bound to the same carbon skeleton
- C07C215/02—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C215/22—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being unsaturated
- C07C215/28—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being unsaturated and containing six-membered aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/02—Local antiseptics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- the present invention relates to crystalline forms and hydrates of the compound FTY720 hydrochloride, and to the use thereof.
- the present invention is based in part on a discovery that the FTY720 hydrochloride exhibits polymorphism.
- FTY720 hydrochloride exists in a particular crystalline form (hereinafter Form I) at room temperature. Crystalline Form I undergoes a change to an alternative crystalline form (Form II) at a transition temperature of approximately 40 °C. Moreover, crystalline Form Il undergoes a transition to a third crystalline form (Form II! at a temperature of approximately 66 "C. At a temperature of approximately 107 °C, FTY720 hydrochloride forms a phase with lower crystalline order.
- Crystalline Form I of FYT720 hydrochloride is characterised by an X-ray powder diffraction pattern having peaks at least two, preferably at least four, and more preferably all, of the following 2-theta values: 3.6, 7.1 , 10.7, 12.5, 15.4 and 20.6 degrees 2-theta.
- the peaks at said 2-theta values may have the following relative intensities: 3.6 (strong), 7.1 (weak), 10.7 (weak), 12.5 (weak), 15.4 (medium) and 20.6 (medium).
- this crystalline form is characterised by an X-ray powder diffraction pattern having peaks at least two, preferably at least four, and more preferably all, of the following 2-theta values: 3.55. 7.12, 10.71 , 12.48, 15.42 and 20.59 degrees 2-theta.
- the peaks at said 2-theta values may have the following relative intensities: 3.55 (strong), 7.12 (weak), 10.71 (weak), 12.48 (weak). 15.42 (medium) and 20.59 (medium).
- this crystalline form is characterised by an X-ray powder diffraction pattern corresponding substantially to that shown in Fig. 1.
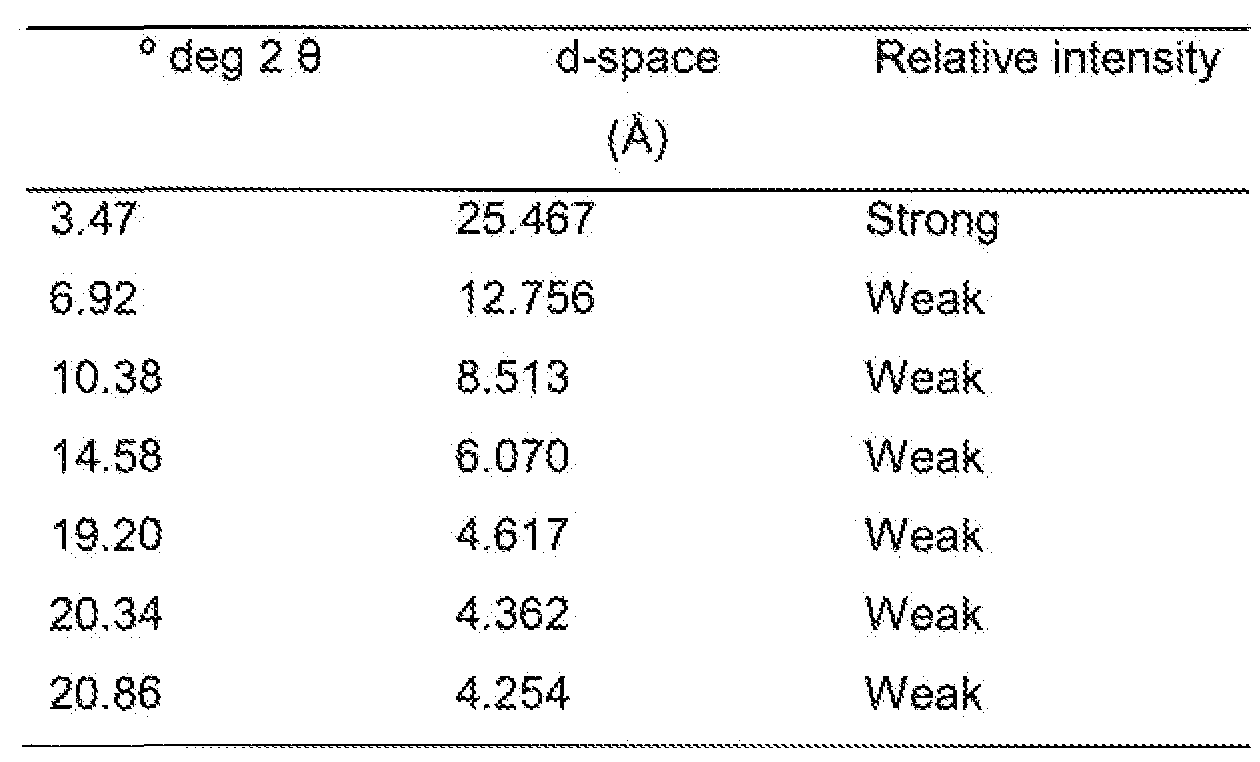
- Crystalline (Form II) of FTY720 hydrochloride is characterised by an X-ray powder diffraction pattern having peaks at at least two, preferably at least four, and more preferably all, of the following 2-theta values: 3.5, 6.9, 10.4, 14.6, 19.2. 20.3 and 20.9 degrees 2-theta.
- the peaks at said 2-theta values may have the following relative intensities: 3.5 (strong), 6.9 (weak), 10.4 (weak), 14.6 (weak), 19.2 (weak), 20.3 (weak) and 20.9 (weak).
- this crystalline form is characterised by an X-ray powder diffraction pattern having peaks at least two, preferably at least four, and more preferably all, of the following 2-theta values: 3.47, 6.92, 10.38, 14.58, 19.20, 20.34 and 20.86 degrees 2-theta.
- the peaks at said 2-theta values may have the following relative intensities: 3.47 (strong), 6.92 (weak), 10.38 (weak), 14.58 (weak), 19.20 (weak), 20.34 (weak) and 20.86 (weak).
- this crystalline form is characterised by an X-ray powder diffraction pattern corresponding substantially to that shown in Fig. 2.
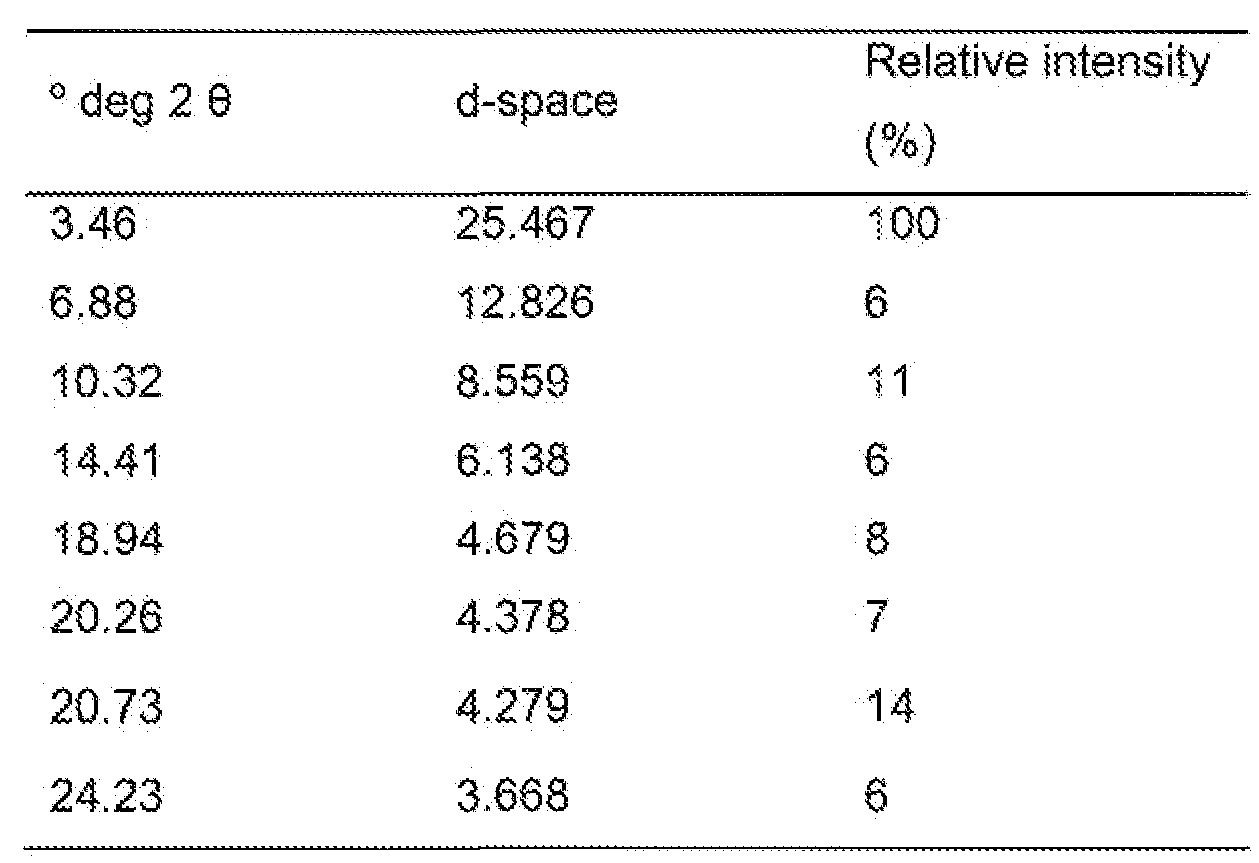
- Crystalline form (Form III) of FTY720 hydrochloride is characterised by an X-ray powder diffraction pattern having peaks at at least two, preferably at least four, and more preferably all, of the following 2-theta values: 3.5, 6.9, 10.3, 14.4, 18.9, 20.3, 20.7 and 24.2 degrees 2- theta.
- the peaks at said 2-theta values may have the following relative intensities: 3.5 (strong), 6.9 (weak), 10.3 (weak), 14.4 (weak), 18.9 (weak), 20.3 (weak), 20.7 (weak) and 24.2 (weak).
- this crystalline form is characterised by an X-ray powder diffraction pattern having peaks at least two, preferably at least four, and more preferably all, of the following 2-theia values. 3.46, 6.88, 10.32, 14.41 , 18.94, 20.26, 20.73 and 24.23 degrees 2- theta.
- the peaks at said 2-theta values may have the following relative intensities: 3.46 (strong), 6.88 (weak), 10.32 (weak), 14.41 (weak), 18.94 (weak), 20.26 (weak), 20.73 (weak) and 24.23 (weak).
- this crystalline form is characterised by an X-ray powder diffraction pattern corresponding substantially to that shown in Fig. 3.
- the invention provides a process for the production of crystalline Form I of FTY720 hydrochloride, which comprises cooling crystalline Form Il or Form III of FTY720 hydrochloride to a temperature of less than 40 °C.
- the process comprises cooling to a temperature of 30 °C or less, more preferably 20 °C or less, more preferably still 10 °C or less, e.g. still 8 °C or less, e.g. 2 to 8 °C, in order to ensure conversion to crystalline Form I.
- FTY720 hydrochloride may exist substantially in the form of a hydrate.
- the hydrate is characterised by an X-ray powder diffraction pattern having at least two, preferably at least four and more preferably all, peaks at about 2.9, 17.2, 30.6, 28.2. 24.4, 8.6 and 25.9 degrees 2-theta.
- the peaks at said 2-theta values may have the following relative intensities: 2.9 (strong). 17.2 (medium), 30.6 (weak), 28.2 (weak), 24.4 (weak), 8.6 (weak) and 25.9 (weak).
- a hydrate of FTY720 hydrochloride characterised by an X-ray powder diffraction pattern corresponding substantially to that shown in Fig. 4,
- FTY720 hydrochloride may be obtained according to the procedures given in the Examples herein.
- interconversion between the various polymorphic forms of FYT720 hydrochloride may be achieved by heating or cooling FTY720 hydrochloride in accordance with the procedures described in the Examples.
- the various crystalline salt forms of the invention may have one or more desirable properties compared with the free base form of FTY720.
- crystalline salts of the invention may be more stable and of better quality than the free base, in particular during storage and distribution.
- the salts may have a high degree of dissociation in water and thus substantially improved water solubility.
- the salts may also be advantageous in that they show no measurable water absorption or loss.
- Crystalline forms may be characterized by the major peaks of an X-ray powder diffraction spectrum, as illustrated in the Examples herein. Crystalline forms may also differ with respect to their thermodynamic stability, in their physical parameters, such as the absorption pattern in an infrared spectroscopy (IR) or phase transition signals in differential scanning calorimetry (DSC).
- IR infrared spectroscopy
- DSC differential scanning calorimetry
- the various crystalline salt forms of the present invention are in substantially pure crystalline form.
- substantially pure includes reference to crystalline forms of, or greater than, 90%, more preferably 95%, more preferably 96%, more preferably 97%, more preferably 98%, more preferably 99% polymorphic purity as determined, for example, by X-ray powder diffraction, Raman spectroscopy or IR spectroscopy.
- a pharmaceutical formulation of the invention preferably contains 0.01 to 20% by weight of the salt, more preferably 0.1 to 10%, e.g 0.5 to 5% by weight, based on the total weight of the formulation.
- the pharmaceutical formulation may be a solid pharmaceutical composition in a form suitable for oral administration, e.g. a tablet or capsule.
- the composition may be manufactured in a conventional manner, e.g. by mixing a salt of the invention with a pharmaceutically acceptable carrier or diluent.
- the formulation is a solid pharmaceutical composition comprising a salt of the invention and a sugar alcohol.
- compositions of this type are disclosed in WO 2004/089341, the contents of which are incorporated herein by reference.
- the solid compositions disclosed in this publication are particularly well suited to the oral administration of the salts of the present invention.
- the compositions provide a convenient meari5 of systemic administration of the compounds, do not suffer from the disadvantages of liquid formulations for injection or oral use, and have good physicochemical and storage properties.
- the compositions of the present invention may show a high level of uniformity in the distribution of the compound throughout the composition, as well as high stability. The compositions may therefore be manufactured on high speed automated equipment, and thus do not require hand encapsulation.
- the sugar alcohol may act as a diluent, carrier, filler or bulking agent, and may suitably be mannitol, maltitol, inositol, xylitol or lactitol, preferably a substantially non-hygroscopic sugar alcohol, e.g. mannitol (D-mannitol).
- a single sugar alcohol may be used, or a mixture of two or more sugar alcohols, e.g a mixture of mannitol and xylitol, e.g. in a ratio of 1 :1 to 4.1.
- the sugar alcohol is prepared from a spray-dried composition, e.g. mannitol composition, having a high specific surface area.
- a spray-dried composition e.g. mannitol composition
- the use of this type of mannitol composition may assist in promoting uniform distribution of the compound throughout the mannitol in the compositon.
- a higher surface area may be achieved by providing a sugar alcohol, e.g. mannitol, preparation consisting of particles having a smaller mean size and/or a rougher surface on each particle.
- a spray- dried sugar alcohol, e.g. mannitol e.g. with a mean particle size of 300 ⁇ m or less, has also been found to improve compressibility and hardness of tablets formed from the composition.
- the single point surface area of the sugar alcohol preparation is 1 to 7 m 2 /g, e g. 2 to 6 m 2 /g or 3 to 5 m z /g.
- the mannitol preparation may suitably have a mean particle size of 100 to 300 ⁇ m, e.g 150 to 250 ⁇ m and a bulk density of 0.4 to 0.6 g/mL, e.g. 0.45 to 0.55 g/mL
- a suitable high surface area mannitol is Part ⁇ ck M200, available commercially from E. Merck.
- the composition preferably contains 75 to 99.99% by weight of the sugar alcohol, more preferably 85 to 99.9%, e.g. 90 to 99.5% by weight, based on the total weight of the composition.
- the composition preferably further comprises a lubricant.
- Suitable lubricants include stearic acid, magnesium stearate. calcium stearate, zinc stearate. glyceryl palmitostearate, sodium stearyl fumarate, canola oil, hydrogenated vegetable oil such as hydrogenated castor oil (e.g C ⁇ tina® or lubriwax® 101 ), mineral oil, sodium lauryl sulfate, magnesium oxide, colloidal silicon dioxide, silicone fluid, polyethylene glycol, polyvinyl alcohol, sodium benzoate, talc, poloxamer. or a mixture of any of the above.
- the lubricant comprises magnesium stearate, hydrogenated castor oil or mineral oil. Colloidal silicon dioxide and polyethylene glycol are less preferred as the lubricant.
- the composition preferably contains 0.01 to 5% by weight of a lubricant, more preferably 1 to 3% by weight, e.g. about 2% by weight, based on the total weight of the composition.
- the composition may comprise one or more further excipients such as carriers, binders or diluents.
- the composition may comprise microcrystalline cellulose (e.g. Avicel®), methylcellulose, hydroxypropylcellulose, hydroxypropylmethylceilulose, starch (e.g corn starch) or dicalcium phosphate, preferably in an amount of from 0.1 to 90% by weight, e.g. 1 to 30% by weight, based on the total weight of the composition.
- a binder e.g microcrystalline cellulose, methylcellulose, hydroxypropyl cellulose, hydroxypropylmethyl cellulose is used, it is preferably incl ⁇ deo in an amount of 1 to 8 %, e.g.
- a binder increases the granule strength of the formulation, which is particularly important for fine granulations.
- Micro- crystalline cellulose and methylcellulose are particularly preferred where a high tablet hardness and/or longer disintegration time is required. Hydroxypropyl cellulose is preferred where faster distintegralion is required.
- xylitol may also be added as an additional binder, for example in addition to microcrystalline cellulose, e.g. in an amount up to 20% by weight of the sugar alcohol, e.g. xylitol.
- the composition further comprises a stabiliser, preferably glycine HCI or sodium bicarbonate.
- the stabiliser may be present in an amount of e.g. 0.1 to 30%. preferably 1 to 20% by weight.
- the composition may be in the form of a powder, granule or pellets or a unit dosage form, for example as a tablet or capsule.
- the compositions of the present invention are well- adapted for encapsulation into an orally administrable capsule shell, particularly a hard gelatin shell.
- the compositions may be compacted into tablets.
- the tablets may optionally be coated, for instance with talc or a polysaccharide (e.g. cellulose) or hydroxypropylmethyl- cellulose coating.
- each unit dosage may, for example, contain from about 0.5 to about 10 mg of a salt of the invention.
- compositions of the invention may show good stability characteristics as indicated by standard stability trials, for example having a shelf life stability of up to one, two or three years, and even longer. Stability characteristics may be determined, e.g. by measuring decomposition products by HPLC analysis after storage for particular times, at particular temperatures, e.g. 20, 40 or 60 °C.
- compositions of the present invention may be produced by standard processes, for instance by conventional mixing, granulating, sugar-coating, dissolving or lyophilizing processes. Procedures which may be used are known in the art, e.g. those described In L. Lachman et al. The Theory and Practice of Industrial Pharmacy, 3rd Ed, 1986, H Sucker et al, Pharmazeutician Technologie, Thieme, 1991 , Hagers Handbuch der pharmazeutica fürtechnik, 4th Ed. (Springer Veriag, 1971 ) and Remington's Pharmaceutical Sciences. 13th Ed., (Mack Publ., Co., 1970) or later editions.
- the pharmaceutical composition is produced by a process comprising:
- a preparation having a good level of content and blend uniformity i.e. a substantially uniform distribution of the salt throughout the composition
- the salt may optionally be micronized, and/or prescreened, e.g. with a 400 to 500 ⁇ m mesh screen, before step (a) in order to remove lumps.
- the mixing step (a) may suitably comprise blending the salt and the sugar alcohol, e.g. mannitol in any suitable blender or mixer for e.g. 100 to 400 revolutions.
- the process may be carried out by dry mixing the components.
- the milling step (b) may suitably comprise passing the mixture obtained in (a) through a screen, which preferably has a mesh size of 400 to 500 ⁇ m.
- Process step (a) may comprise the step of mixing the total amount of the salt at first with a low amount of sugar alcohol, e.g. from 5 to 25% by weight of the total weight of sugar alcohol, in order to form a pre-mix. Subsequently the remaining amount of sugar alcohol is added to the pre-mix.
- Step (a) may also comprise the step of adding a binder solution, e.g. methylcellulose and/or xytitol, e.g. an aqueous solution, to the mixture. Alternatively the binder is added to the mix dry and water is added in the granulation step.
- a binder solution e.g. methylcellulose and/or xytitol, e.g. an aqueous solution
- the milled mixture obtained in (b) may optionally be blended once more before mixing with the lubricant.
- the lubricant e.g. magnesium stearate
- the salt is preferably first dry-mixed with the desired sugar alcohol, e.g. mannitol, and the obtained sugar alcohol/salt mixture is then dry-mixed with a binder such as hydroxypropyl cellulose or hydroxypropylmethyl cellulose. Water is then added and the mixture granulated, e.g. using an automated granulator. The granulation is then dried and milled.
- a binder such as hydroxypropyl cellulose or hydroxypropylmethyl cellulose.
- an additional amount of binder may be added in step (c) to the mixture obtained in (b).
- the process may comprise a further step of tabletting or encapsulating the mixture obtained in (c), e.g. into a hard gelatin capsule using an automated encapsulation device.
- the capsules may be coloured or marked so as to impart an individual appearance and to make them instantly recognizable.
- the use of dyes can serve to enhance the appearance as well as to identify the capsules.
- Dyes suitable for use in pharmacy typically include carotinoids, iron oxides, and chlorophyll.
- the capsules of the invention are marked using a code.
- Salts and polymorphs of the invention may be useful in: a) treatment and prevention of organ or tissue transplant rejection, for example for the treatment of the recipients of heart, lung, combined heart-lung, liver, kidney, pancreatic, skin or corneal transplants, and the prevention of graft-versus-host disease, such as sometimes occurs following bone marrow transplantation; particularly in the treatment of acute or chronic allo- and xenograft rejection or in the transplantation of insulin producing cells, e.g. pancreatic islet cells; and b) treatment and prevention of autoimmune disease or of inflammatory conditions, e.g.
- rheumatoid arthritis systemic lupus erythematosus, hashimoto ' s thyroidis, multiple sclerosis, myasthenia gravis, diabetes type I or Il and the disorders associated therewith, vasculitis, pernicious anemia, Sjoegren syndrome, uveitis, psoriasis, Graves ophthalmopathy, alopecia areata and others, allergic diseases, e g. allergic asthma, atopic dermatitis, allergic rhinitis/conjunctivitis, allergic contact dermatitis, inflammatory diseases optionally with underlying aberrant reactions, e.g.
- inflammatory bowel disease Crohn's disease or ulcerative colitis
- intrinsic asthma inflammatory lung injury, inflammatory liver injury, inflammatory glomerular injury, atherosclerosis, osteoarthritis, irritant contact dermatitis and further eczematous dermatitises, seborrhoeic dermatitis, cutaneous manifestations of immunologically-mediated disorders, inflammatory eye disease, keratoconjunctivitis, myocarditis or hepatitis.
- the required dosage will of course vary depending on the mode of administration, the particular condition to be treated and the effect desired. In general, satisfactory results are indicated to be obtained at daily dosages of from about 0.1 to about 100 mg/kg body weight.
- An indicated daily dosage in the larger mammal, e.g. humans, is in the range of from about 0.5 mg to 2000 mg, conveniently administered, for example, in divided doses up to four times a day or in retard form.
- the salts may be administered by any appropriate route, e.g. orally, for example in the form of a tablet or capsule, topically or parenterally. for example intravenously.
- Pharmaceutical compositions comprising a salt of the invention in association with at least one pharmaceutically acceptable carrier or diluent may be manufactured in conventional manner by mixing with a pharmaceutically acceptable carrier or diluent.
- Unit dosage forms for oral administration contain, for example, from about 0.1 mg to about 500 mg of active substance.
- the salts may be administered as the sole active ingredient or together with other drugs in immunomodulating regimens or other anti-inflammatory agents e.g. for the treatment or prevention of allograft acute or chronic rejection or inflammatory or autoimmune disorders.
- they may be used in combination with calcineurin inhibitors, e.g. cyclosporin A, cyclosporin G, FK-506, ABT-281 , ASM 981 ; an mTOR inhibitor, e.g.
- rapamycin 40-O-(2- hydroxy)ethyl-rapamycin, CCI779, ABT578 or AP23573 elc; corticosteroids; cyclophosphamide; azathioprene; methotrexate; another S1P receptor agonist, e.g. FTY 720 or an analogue thereof; leflunomide or analogs thereof; mizoribine; mycophenolic acid; mycophenolate mofetil; 15-deoxyspergualine or analogs thereof; immunosuppressive monoclonal antibodies, e.g., monoclonal antibodies to leukocyte receptors, e.g.. MHC, CD2, CD3, CD4, CD 11a/CD18. CD7, CD25.
- S1P receptor agonist e.g. FTY 720 or an analogue thereof
- leflunomide or analogs thereof mizoribine
- mycophenolic acid mycophenolate mofetil
- mAbs or low molecular weight inhibitors including LFA-1 antagonists, Selectin antagonists and VLA-4 antagonists.
- dosages of the co-administered immunomodulating or anti-inflammatory agent will of course vary depending on the type of co-drug employed, on the condition to be treated and so forth.
- the present invention thus provides:
- a method of treating or preventing organ or tissue transplant rejection comprising administering to a subject a therapeutically effective amount of a crystalline salt of the invention.
- a method of treating or preventing an autoimmune disease or inflammatory condition comprising administering to a subject a therapeutically effective amount of a crystalline salt of the invention.
- a crystalline salt of the invention for use as a pharmaceutical.
- a pharmaceutical composition comprising a crystalline salt of the invention and a pharmaceutically acceptable diluent or carrier.
- a pharmaceutical combination comprising (a) a crystalline salt of the invention and (b) a second drug substance, said second drug substance being suitable for the prevention or treatment of a condition described above.
- a method as defined above comprising co-administration, e.g. concomitantly or in sequence, of (a) a crystalline salt of the invention and ⁇ b) a second drug substance, said second drug substance being suitable for the prevention or treatment of a condition described above.
- Micronized Compound A e g. 2-amino-2-[2- ⁇ 4-octylphenyl)ethyl]propane-1 ,3-diol hydrochloride salt (FTY720), is screened and 116.7 g of the screened compound is mixed with 9683.3 g of a microcrystalline cellulose agent. The mixture is then milled in a Frewitt MGI device (Key International Inc. USA) using a 30 mesh screen. Magnesium stearate is screened using a 20 mesh screen and 200 g of the screened compound blended with the FTY720 mixture to produce a product composition.
- FTY720 2-amino-2-[2- ⁇ 4-octylphenyl)ethyl]propane-1 ,3-diol hydrochloride salt
- the product composition is then compacted on a tablet press using a 7 mm die to form 120 mg tablets, each containing:
- Compound A 1 e.g. FTY720 * 1.4 mg Microcrystalline cellulose, e.g. Avicel PH 102 116.2 mg Magnesium stearate 2.4 mg
- example 2 the process of example 1 is repeated except that the magnesium stearate is replaced by Cutina® (hydrogenated castor oil).
- Cutina® hydrogenated castor oil
- Compound A 1 e.g. FTY720, and Microcrystal ⁇ ne cellulose, e.g. Avicel PH 102 are each screened separately using an 18 mesh screen.
- 1.9 g screened FTY720 is mixed with 40 g screened microcrystalline cellulose agent for 120 revolutions in a blender at 32 rpm. The FTY720 mixture is then screened through a 35 mesh screen.
- the screened FTY720 mixture is added to a granulator along with a further 340.1 g Microcrystalline cellulose, e.g. Avicel PH 102 and 12 g hydroxypropylcellulose. The mixture is mixed for 3 minutes. Water is then added at a rate of 100 ml/minute and the mixture granulated for 2 minutes. The granulation is transferred into a tray dryer and dried at 50°C for 150 minutes.
- Microcrystalline cellulose e.g. Avicel PH 102 and 12 g hydroxypropylcellulose.
- Water is then added at a rate of 100 ml/minute and the mixture granulated for 2 minutes.
- the granulation is transferred into a tray dryer and dried at 50°C for 150 minutes.
- the mixture is then milled in a Frewitt MGI device using a 35 mesh screen.
- Magnesium stearate is screened and 6 g of the screened compound is blended for 90 revolutions at 32 rpm with the FTY720 mixture to produce a product composition showing a substantially uniform distribution of the S1P receptor agonist throughout the Microcrystalline cellulose, e.g. Avicel PH 102 in the blend.
- the product composition is then filled into size 3 hard gelatin shells on an H & K 400 encapsulation device. 120 mg of the product composition is added to each capsule. Therefore each capsule contains:
- example 3 the process of example 3 is repeated except that the magnesium stearate is replaced by Cutina® (hydrogenated castor oil).
- Cutina® hydrogenated castor oil
- Micronized Compound A e.g. FTY720
- FTY720 is screened using a 400 ⁇ m (40 mesh) screen.
- 58.35 g of the screened compound is mixed with 4841.65 g Microcrystalline cellulose, e.g. Avicel PH 102 in a 25L Bohle bin blender for 240 blending revolutions.
- the mixture is then milled in a Frewitt MGI device using a 425 ⁇ m mesh screen, and the milled mixture is blended once more.
- Magnesium stearate is screened and 100 g of the screened compound is blended with the FTY720 mixture to produce a product composition showing a substantially uniform distribution of the S1P receptor agonist throughout the blend.
- the product composition is then filled into size 3 hard gelatin shells on an H & K 400 encapsulation device. 120 mg of the product composition is added to each capsule. Therefore each capsule contains:
- capsules are manufactured using the components and in the amounts described in Example 6a. but the FTY720 is first mixed with 14 mg mannitol (before screening). This mixture is then screened as described above. The screened mixture is then blended with the remaining mannitol and the magnesium stearate is added, followed by additional blending and filling into capsules.
- capsules are prepared as described in example 6, except that each capsule contains each component in the following amounts:
- capsules are prepared as described in examples 6 to 8, except that the magnesium stearate is replaced in each case by Cutina® (hydrogenated castor oil).
- Cutina® hydrogenated castor oil
- Capsules containing the following ingredients are prepared, by weighing each component and mixing in a mortar, ⁇ hen filling into capsules:
- DSC Differential scanning calorimetry
- DSC heating curves showed three characteristic transitions at approximately 40 °C, 66 °C and 107 °C.
- the first endothermic peak at 40 *C is followed by a smalt exothermic peak which hints to melting of Form I followed by recrystallization into Form II.
- the second transition between Form Il and Form III is a solid-solid transition.
- a third transition was observed at 107 °C. Above 107 °C, the X-ray powder pattern almost disappeared and only a single strong peak at 2.9° remained, suggesting formation of a phase with lower crystalline order above this temperature.
- Thermomicroscopy showed birefringence above 107 "C which disappeared only at ca. 230 °C, which is below the onset of decomposition at ca.
- Variable temperature XRPD was then performed in order to investigate the nature of the different transitions seen in DSC.
- the heating rate was 10 K/min and the stage time was 5min for each experiment.
- X-ray powder diagrams were recorded between 2° and 35* (2 theta) with Cu Ka radiation using a Scintag X1 diffraction system.
- Temperature variable and humidity variable XRPD was performed using the Scintag XDS 2000 system equipped with a temperature and humidity control unit.
- the XRPD diagram of FTY720 hydrochloride Form Il is:
- the XRPD diagram of FTY720 hydrochloride Form Il is:
- a water desorptio ⁇ isotherm recorded at 25 *C showed between 90% and 60% relative humidity (r.h.) a nearly constant water content of 5.2 to 5.9%. This suggests the formation of a hydrate (theoretical water content of a monohydrate is 4.98%).
- the water sorption isotherm recorded at 40 °C showed a first significant water uptake already at 80% r.h., whereas the isotherm recorded at 25 °C showed the first uptake at 90% r.h..
- Samples of Form I stored for 1 month at 60°C and 75%r.h. and 1 month at 80°C and 75%r.h. show conversion to the hydrate form with water contents of 10.2 to 10.6%, which is close to the calculated water content of 9.48% for two moles of water).
- the XRPD diagram of the hvdrate is
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- Emergency Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Communicable Diseases (AREA)
- Transplantation (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description









Claims
Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PL09748350T PL2356090T3 (en) | 2008-11-11 | 2009-11-10 | Crystalline forms of fingolimod hcl |
| KR1020117010560A KR101393994B1 (en) | 2008-11-11 | 2009-11-10 | Crystalline forms of fingolimod hcl |
| CA2743232A CA2743232C (en) | 2008-11-11 | 2009-11-10 | Crystalline forms of fingolimod hcl |
| MX2011004962A MX2011004962A (en) | 2008-11-11 | 2009-11-10 | Crystalline forms of fingolimod hcl. |
| AU2009315736A AU2009315736B2 (en) | 2008-11-11 | 2009-11-10 | Crystalline forms of fingolimod HCL |
| BRPI0921826A BRPI0921826A2 (en) | 2008-11-11 | 2009-11-10 | organic compounds |
| JP2011535130A JP2012508216A (en) | 2008-11-11 | 2009-11-10 | Organic compounds |
| ES09748350.7T ES2643161T3 (en) | 2008-11-11 | 2009-11-10 | Hing crystalline forms of fingolimod |
| US13/128,825 US8530522B2 (en) | 2008-11-11 | 2009-11-10 | Organic compounds |
| KR1020137024001A KR20130109254A (en) | 2008-11-11 | 2009-11-10 | Crystalline forms of fingolimod hcl |
| EP09748350.7A EP2356090B1 (en) | 2008-11-11 | 2009-11-10 | Crystalline forms of fingolimod hcl |
| CN200980144998XA CN102209705A (en) | 2008-11-11 | 2009-11-10 | Crystalline form of fingolimod hydrochloride |
| RU2011123371/04A RU2549899C2 (en) | 2008-11-11 | 2009-11-10 | Crystalline forms of fingolimod hydrochloride |
| US13/964,817 US20140051766A1 (en) | 2008-11-11 | 2013-08-12 | Organic Compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08168865 | 2008-11-11 | ||
| EP08168865.7 | 2008-11-11 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/128,825 A-371-Of-International US8530522B2 (en) | 2008-11-11 | 2009-11-10 | Organic compounds |
| US13/964,817 Continuation US20140051766A1 (en) | 2008-11-11 | 2013-08-12 | Organic Compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2010055028A2 true WO2010055028A2 (en) | 2010-05-20 |
| WO2010055028A3 WO2010055028A3 (en) | 2010-07-08 |
Family
ID=40521487
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/064891 Ceased WO2010055028A2 (en) | 2008-11-11 | 2009-11-10 | Organic compounds |
Country Status (14)
| Country | Link |
|---|---|
| US (2) | US8530522B2 (en) |
| EP (1) | EP2356090B1 (en) |
| JP (4) | JP2012508216A (en) |
| KR (2) | KR101393994B1 (en) |
| CN (3) | CN104788325A (en) |
| AU (1) | AU2009315736B2 (en) |
| BR (1) | BRPI0921826A2 (en) |
| CA (1) | CA2743232C (en) |
| ES (1) | ES2643161T3 (en) |
| MX (1) | MX2011004962A (en) |
| PL (1) | PL2356090T3 (en) |
| PT (1) | PT2356090T (en) |
| RU (2) | RU2549899C2 (en) |
| WO (1) | WO2010055028A2 (en) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012041358A1 (en) | 2010-10-01 | 2012-04-05 | Synthon B.V. | Process for making fingolimod hydrochloride crystals |
| WO2012071524A1 (en) | 2010-11-24 | 2012-05-31 | Ratiopharm Gmbh | Arylsulfonate salts of fingolimod and processes for preparation thereof |
| WO2012089238A1 (en) | 2010-12-28 | 2012-07-05 | Synthon Bv | Process for making fingolimod hydrochloride crystals |
| WO2012097330A3 (en) * | 2011-01-14 | 2012-11-01 | University Of Washington | Compositions and methods for treating degenerative muscle conditions |
| WO2012160187A1 (en) * | 2011-05-26 | 2012-11-29 | Jado Technologies Gmbh | Amino- or ammonium-containing sulfonic acid, phosphonic acid and carboxylic acid derivatives and their medical use |
| WO2013019872A1 (en) | 2011-08-01 | 2013-02-07 | Teva Pharmaceutical Industries Ltd. | Process for preparing pharmaceutical compositions comprising fingolimod |
| US8735627B2 (en) | 2010-10-28 | 2014-05-27 | Mapi Pharma Ltd. | Intermediate compounds and process for the preparation of fingolimod |
| WO2014136047A2 (en) | 2013-03-05 | 2014-09-12 | Biocon Limited | A process for the preparation of 2-amino-1,3-propane diol compounds and salts thereof |
| WO2015015254A1 (en) * | 2013-07-29 | 2015-02-05 | Aizant Drug Research Solutions Pvt Ltd | Pharmaceutical compositions of fingolimod |
| EP2646409A4 (en) * | 2010-11-25 | 2015-12-16 | Shilpa Medicare Ltd | FINGOLIMODE POLYMORPHS AND METHODS THEREOF |
| WO2016042493A1 (en) * | 2014-09-19 | 2016-03-24 | Aizant Drug Research Pvt. Ltd | Pharmaceutical compositions of fingolimod |
| EP2945927A4 (en) * | 2013-01-17 | 2017-01-18 | Shilpa Medicare Limited | Process for preparation of fingolimod and its salts |
| US9573886B2 (en) | 2011-05-26 | 2017-02-21 | Glycoregimmune, Inc. | Hydroxy-substituted amino and ammonium derivatives and their medical use |
| RU2627691C1 (en) * | 2016-07-06 | 2017-08-10 | Олег Ростиславович Михайлов | Crystalline h-modification of 2-amino-2-(2-(4-octylphenyl) ethyl)propane-1,3-diole hydrochloride, method for its production and pharmaceutical composition based thereon |
| US9751834B2 (en) | 2011-05-26 | 2017-09-05 | Gri Bio, Inc. | Oxygenated amino- or ammonium-containing sulfonic acid, phosphonic acid and carboxylic acid derivatives and their medical use |
| US9925138B2 (en) | 2015-01-20 | 2018-03-27 | Handa Pharmaceuticals, Llc | Stable solid fingolimod dosage forms |
| WO2020043325A1 (en) | 2018-08-31 | 2020-03-05 | Pharmathen S.A. | Pharmaceutical composition comprising an immunomodulatory agent and method for the preparation thereof |
| WO2020245775A1 (en) * | 2019-06-05 | 2020-12-10 | Biocon Limited | Crystalline forms of 2-amino-2-(2-(4-octylphenyl) ethyl) propane-1,3-diol |
| WO2021084068A1 (en) | 2019-10-31 | 2021-05-06 | Idorsia Pharmaceuticals Ltd | Combination of a cxcr7 antagonist with an s1p1 receptor modulator |
| WO2022253077A1 (en) | 2021-05-31 | 2022-12-08 | 上海博志研新药物技术有限公司 | Pharmaceutical salt of fingolimod, preparation method therefor, pharmaceutical composition containing same and use thereof |
| EP4212156A1 (en) | 2022-01-13 | 2023-07-19 | Abivax | Combination of 8-chloro-n-(4-(trifluoromethoxy)phenyl)quinolin-2-amine and its derivatives with a s1p receptor modulator |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2702034A4 (en) | 2011-04-29 | 2015-03-25 | Reddys Lab Ltd Dr | Preparation of fingolimod and its salts |
| WO2013091704A1 (en) * | 2011-12-22 | 2013-06-27 | Synthon Bv | Pharmaceutical composition comprising fingolimod |
| CN102887829B (en) * | 2012-09-05 | 2014-07-02 | 中国科学院上海药物研究所 | Method for preparing fingolimod mucate and crystals thereof and application of fingolimod mucate and crystals thereof |
| CN106860440A (en) * | 2017-01-16 | 2017-06-20 | 南京医科大学第二附属医院 | Application of the fingolimod hydrochloride in systemic lupus erythematosus encephalopathic medicine is prepared |
| CN106667981B (en) * | 2017-01-16 | 2019-05-14 | 南京医科大学第二附属医院 | Fingolimod hydrochloride is preparing the application in medicine physical property liver injury medicament |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5604229A (en) | 1992-10-21 | 1997-02-18 | Yoshitomi Pharmaceutical Industries, Ltd. | 2-amino-1,3-propanediol compound and immunosuppressant |
| US6476004B1 (en) * | 1996-07-18 | 2002-11-05 | Mitsubishi Pharma Corporation | Pharmaceutical composition |
| JP4079505B2 (en) * | 1998-04-28 | 2008-04-23 | 田辺三菱製薬株式会社 | Novel process for the preparation of 2-amino-2- [2- (4-octylphenyl) ethyl] propane-1,3-diol |
| EP1457478B1 (en) * | 1998-11-11 | 2017-07-19 | Novartis AG | Production of 2-amino-2-[2-(4-alkyl-phenyl)ethyl]propane-1,3-diols |
| PT1031347E (en) * | 1999-01-27 | 2002-09-30 | Idea Ag | TRANSNATIONAL TRANSMISSION / IMMUNIZATION WITH MOST ADAPTABLE VEHICLES |
| CN1230421C (en) * | 1999-02-10 | 2005-12-07 | 三菱制药株式会社 | Amide compounds and their medicinal uses |
| CN1144779C (en) * | 1999-03-11 | 2004-04-07 | 杭州中美华东制药有限公司 | Process for the preparation of 2-[2-(4-alkylphenyl)-ethyl]-2-amino-propanediol and intermediates produced therein |
| ES2322442T3 (en) * | 2001-06-08 | 2009-06-22 | Novartis Ag | TREATMENT OR PROFILAXIS OF THE REJECTION OF INSULIN CELL PRODUCTION CELLS. |
| US20030144712A1 (en) * | 2001-12-20 | 2003-07-31 | Jackson Streeter, M.D. | Methods for overcoming organ transplant rejection |
| PL372103A1 (en) * | 2002-05-16 | 2005-07-11 | Novartis Ag | Use of edg receptor binding agents in cancer |
| GB0217152D0 (en) * | 2002-07-24 | 2002-09-04 | Novartis Ag | Organic compounds |
| CA2521325C (en) | 2003-04-08 | 2010-09-14 | Novartis Ag | Solid pharmaceutical compositions comprising a s1p receptor agonist and a sugar alcohol |
| CN1212308C (en) * | 2003-07-24 | 2005-07-27 | 漆又毛 | Process for preparing amino-2-(2-(4-otylphenyl) ethyl)-1, 3-propylene glycol hydrochloride |
| CN1241903C (en) * | 2003-10-14 | 2006-02-15 | 马启明 | Method for preparing 2-para octylphenyl ehtyl-2-amino propanediol |
| TWI418350B (en) * | 2005-06-24 | 2013-12-11 | Sankyo Co | Use of pharmaceutical compositions comprising ppar modulator |
| RU2008116578A (en) * | 2005-09-30 | 2009-11-10 | Новартис АГ (CH) | APPLICATION OF DPP-IV INHIBITORS FOR TREATMENT OF AUTOIMMUNE DISEASES AND TRANSPLANT rejection |
| CN1310869C (en) * | 2005-11-22 | 2007-04-18 | 江苏吴中苏药医药开发有限责任公司 | 2-amido-2-[2-(4-alkylphenyl)ethyl]-1,3-methyl glycol preparation method |
| CN100548968C (en) * | 2006-03-01 | 2009-10-14 | 徐州师范大学 | A kind of method for preparing 2-to octyl group styroyl-2-amino-propanediol hydrochloride |
| ES2414205T3 (en) * | 2006-06-02 | 2013-07-18 | The Ohio State University Research Foundation | Therapeutic agents for the treatment of mantle cell lymphoma |
| KR20090057399A (en) * | 2006-09-26 | 2009-06-05 | 노파르티스 아게 | Pharmaceutical composition containing S1P regulator |
| KR20110020928A (en) * | 2008-06-20 | 2011-03-03 | 노파르티스 아게 | Pediatric compositions for treating multiple sclerosis |
| EP2621889B1 (en) | 2010-10-01 | 2019-07-17 | Synthon BV | Process for making fingolimod hydrochloride crystals |
-
2009
- 2009-11-10 CA CA2743232A patent/CA2743232C/en active Active
- 2009-11-10 US US13/128,825 patent/US8530522B2/en not_active Expired - Fee Related
- 2009-11-10 WO PCT/EP2009/064891 patent/WO2010055028A2/en not_active Ceased
- 2009-11-10 CN CN201510141353.8A patent/CN104788325A/en active Pending
- 2009-11-10 PL PL09748350T patent/PL2356090T3/en unknown
- 2009-11-10 CN CN201710377640.8A patent/CN107233336A/en active Pending
- 2009-11-10 EP EP09748350.7A patent/EP2356090B1/en active Active
- 2009-11-10 RU RU2011123371/04A patent/RU2549899C2/en active
- 2009-11-10 AU AU2009315736A patent/AU2009315736B2/en not_active Ceased
- 2009-11-10 JP JP2011535130A patent/JP2012508216A/en not_active Withdrawn
- 2009-11-10 RU RU2015111229/04A patent/RU2015111229A/en not_active Application Discontinuation
- 2009-11-10 CN CN200980144998XA patent/CN102209705A/en active Pending
- 2009-11-10 KR KR1020117010560A patent/KR101393994B1/en not_active Expired - Fee Related
- 2009-11-10 KR KR1020137024001A patent/KR20130109254A/en not_active Withdrawn
- 2009-11-10 MX MX2011004962A patent/MX2011004962A/en active IP Right Grant
- 2009-11-10 BR BRPI0921826A patent/BRPI0921826A2/en not_active Application Discontinuation
- 2009-11-10 ES ES09748350.7T patent/ES2643161T3/en active Active
- 2009-11-10 PT PT97483507T patent/PT2356090T/en unknown
-
2013
- 2013-08-12 US US13/964,817 patent/US20140051766A1/en not_active Abandoned
-
2014
- 2014-02-06 JP JP2014020950A patent/JP2014139179A/en not_active Withdrawn
-
2015
- 2015-09-03 JP JP2015173474A patent/JP2016028056A/en not_active Withdrawn
-
2017
- 2017-09-07 JP JP2017172276A patent/JP2018035160A/en active Pending
Cited By (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012041358A1 (en) | 2010-10-01 | 2012-04-05 | Synthon B.V. | Process for making fingolimod hydrochloride crystals |
| EP2621889B1 (en) | 2010-10-01 | 2019-07-17 | Synthon BV | Process for making fingolimod hydrochloride crystals |
| CN103228617A (en) * | 2010-10-01 | 2013-07-31 | 斯索恩有限公司 | Process for making fingolimod hydrochloride crystals |
| US8735627B2 (en) | 2010-10-28 | 2014-05-27 | Mapi Pharma Ltd. | Intermediate compounds and process for the preparation of fingolimod |
| WO2012071524A1 (en) | 2010-11-24 | 2012-05-31 | Ratiopharm Gmbh | Arylsulfonate salts of fingolimod and processes for preparation thereof |
| EP2646409A4 (en) * | 2010-11-25 | 2015-12-16 | Shilpa Medicare Ltd | FINGOLIMODE POLYMORPHS AND METHODS THEREOF |
| US9266816B2 (en) | 2010-11-25 | 2016-02-23 | Shilpa Medicare Limited | Fingolimod polymorphs and their processes |
| WO2012089238A1 (en) | 2010-12-28 | 2012-07-05 | Synthon Bv | Process for making fingolimod hydrochloride crystals |
| EP2658840B1 (en) | 2010-12-28 | 2019-07-03 | Synthon BV | Process for making fingolimod hydrochloride crystals |
| WO2012097330A3 (en) * | 2011-01-14 | 2012-11-01 | University Of Washington | Compositions and methods for treating degenerative muscle conditions |
| US10829506B2 (en) | 2011-05-26 | 2020-11-10 | Gri Bio, Inc. | Amino- or ammonium-containing sulfonic acid, phosphonic acid and carboxylic acid derivatives and their medical use |
| US10952977B2 (en) | 2011-05-26 | 2021-03-23 | Gri Bio, Inc. | Hydroxy-substituted amino and ammonium derivatives and their medical use |
| US12528775B2 (en) | 2011-05-26 | 2026-01-20 | Gri Bio, Inc. | Oxygenated and amino- or ammonium-containing phosphonic acid derivatives and their medical use |
| US11564895B2 (en) | 2011-05-26 | 2023-01-31 | Gri Bio, Inc. | Hydroxy-substituted amino and ammonium derivatives and their medical use |
| US11453642B2 (en) | 2011-05-26 | 2022-09-27 | Gri Bio, Inc. | Oxygenated amino- or ammonium-containing sulfonic acid, phosphonic acid and carboxylic acid derivatives and their medical use |
| US10143668B2 (en) | 2011-05-26 | 2018-12-04 | Gri Bio, Inc. | Hydroxy-substituted amino and ammonium derivatives and their medical use |
| US9573886B2 (en) | 2011-05-26 | 2017-02-21 | Glycoregimmune, Inc. | Hydroxy-substituted amino and ammonium derivatives and their medical use |
| US10815195B2 (en) | 2011-05-26 | 2020-10-27 | Gri Bio, Inc. | Oxygenated amino- or ammonium-containing sulfonic acid, phosphonic acid and carboxylic acid derivatives and their medical use |
| WO2012160187A1 (en) * | 2011-05-26 | 2012-11-29 | Jado Technologies Gmbh | Amino- or ammonium-containing sulfonic acid, phosphonic acid and carboxylic acid derivatives and their medical use |
| US9850265B2 (en) | 2011-05-26 | 2017-12-26 | Gri Bio, Inc. | Amino- or ammonium-containing sulfonic acid, phosphonic acid and carboxylic acid derivatives and their medical use |
| US9751834B2 (en) | 2011-05-26 | 2017-09-05 | Gri Bio, Inc. | Oxygenated amino- or ammonium-containing sulfonic acid, phosphonic acid and carboxylic acid derivatives and their medical use |
| US9186333B2 (en) | 2011-08-01 | 2015-11-17 | Teva Pharmaceutical Industries Ltd. | Process for preparing pharmaceutical compositions of fingolimod |
| WO2013019872A1 (en) | 2011-08-01 | 2013-02-07 | Teva Pharmaceutical Industries Ltd. | Process for preparing pharmaceutical compositions comprising fingolimod |
| US9732030B2 (en) | 2013-01-17 | 2017-08-15 | Shilpa Medicare Limited | Process for the preparation of fingolimod and its salts |
| EP2945927A4 (en) * | 2013-01-17 | 2017-01-18 | Shilpa Medicare Limited | Process for preparation of fingolimod and its salts |
| WO2014136047A2 (en) | 2013-03-05 | 2014-09-12 | Biocon Limited | A process for the preparation of 2-amino-1,3-propane diol compounds and salts thereof |
| AU2014224237B2 (en) * | 2013-03-05 | 2017-07-20 | Biocon Limited | A process for the preparation of 2-amino-1,3-propane diol compounds and salts thereof |
| WO2014136047A3 (en) * | 2013-03-05 | 2015-04-02 | Biocon Limited | A process for the preparation of 2-amino-1,3-propane diol compounds and salts thereof |
| RU2663833C2 (en) * | 2013-03-05 | 2018-08-10 | Биокон Лимитед | Process for preparation of 2-amino-1,3-propanediol compounds and salts thereof |
| EP3027174B1 (en) | 2013-07-29 | 2019-07-24 | Aizant Drug Research Solutions Private Limited | Pharmaceutical compositions of fingolimod |
| WO2015015254A1 (en) * | 2013-07-29 | 2015-02-05 | Aizant Drug Research Solutions Pvt Ltd | Pharmaceutical compositions of fingolimod |
| WO2016042493A1 (en) * | 2014-09-19 | 2016-03-24 | Aizant Drug Research Pvt. Ltd | Pharmaceutical compositions of fingolimod |
| US9925138B2 (en) | 2015-01-20 | 2018-03-27 | Handa Pharmaceuticals, Llc | Stable solid fingolimod dosage forms |
| RU2627691C1 (en) * | 2016-07-06 | 2017-08-10 | Олег Ростиславович Михайлов | Crystalline h-modification of 2-amino-2-(2-(4-octylphenyl) ethyl)propane-1,3-diole hydrochloride, method for its production and pharmaceutical composition based thereon |
| WO2020043325A1 (en) | 2018-08-31 | 2020-03-05 | Pharmathen S.A. | Pharmaceutical composition comprising an immunomodulatory agent and method for the preparation thereof |
| WO2020245775A1 (en) * | 2019-06-05 | 2020-12-10 | Biocon Limited | Crystalline forms of 2-amino-2-(2-(4-octylphenyl) ethyl) propane-1,3-diol |
| WO2021084068A1 (en) | 2019-10-31 | 2021-05-06 | Idorsia Pharmaceuticals Ltd | Combination of a cxcr7 antagonist with an s1p1 receptor modulator |
| WO2022253077A1 (en) | 2021-05-31 | 2022-12-08 | 上海博志研新药物技术有限公司 | Pharmaceutical salt of fingolimod, preparation method therefor, pharmaceutical composition containing same and use thereof |
| EP4212156A1 (en) | 2022-01-13 | 2023-07-19 | Abivax | Combination of 8-chloro-n-(4-(trifluoromethoxy)phenyl)quinolin-2-amine and its derivatives with a s1p receptor modulator |
| WO2023135207A1 (en) | 2022-01-13 | 2023-07-20 | Abivax | Combination of 8-chloro-n-(4-(trifluoromethoxy)phenyl)quinolin-2-amine and its derivatives with a s1p receptor modulator |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010055028A3 (en) | 2010-07-08 |
| EP2356090A2 (en) | 2011-08-17 |
| ES2643161T3 (en) | 2017-11-21 |
| AU2009315736A1 (en) | 2010-05-20 |
| PL2356090T3 (en) | 2017-12-29 |
| US20110229501A1 (en) | 2011-09-22 |
| JP2014139179A (en) | 2014-07-31 |
| JP2016028056A (en) | 2016-02-25 |
| RU2549899C2 (en) | 2015-05-10 |
| KR101393994B1 (en) | 2014-05-14 |
| PT2356090T (en) | 2017-10-16 |
| AU2009315736B2 (en) | 2013-08-29 |
| RU2011123371A (en) | 2012-12-20 |
| JP2012508216A (en) | 2012-04-05 |
| CA2743232C (en) | 2015-12-29 |
| JP2018035160A (en) | 2018-03-08 |
| CN104788325A (en) | 2015-07-22 |
| BRPI0921826A2 (en) | 2016-01-12 |
| KR20130109254A (en) | 2013-10-07 |
| CN107233336A (en) | 2017-10-10 |
| US8530522B2 (en) | 2013-09-10 |
| CN102209705A (en) | 2011-10-05 |
| MX2011004962A (en) | 2011-05-30 |
| RU2015111229A3 (en) | 2018-11-08 |
| US20140051766A1 (en) | 2014-02-20 |
| CA2743232A1 (en) | 2010-05-20 |
| RU2015111229A (en) | 2015-11-20 |
| EP2356090B1 (en) | 2017-07-05 |
| KR20110069156A (en) | 2011-06-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2356090B1 (en) | Crystalline forms of fingolimod hcl | |
| US8680146B2 (en) | Organic compounds | |
| SG181466A1 (en) | Formulations, salts and polymorphs of transnorsertraline and uses thereof | |
| AU2013100533A4 (en) | Crystalline forms of fingolimod HCL | |
| AU2013203470A1 (en) | Salts of fingolimod |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980144998.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09748350 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3125/DELNP/2011 Country of ref document: IN |
|
| REEP | Request for entry into the european phase |
Ref document number: 2009748350 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009315736 Country of ref document: AU Ref document number: 2009748350 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20117010560 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011535130 Country of ref document: JP Ref document number: 2743232 Country of ref document: CA Ref document number: MX/A/2011/004962 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13128825 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2009315736 Country of ref document: AU Date of ref document: 20091110 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011123371 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0921826 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110510 |