WO2010050628A2 - Foamed rubber composition for shoe sole and outersole - Google Patents
Foamed rubber composition for shoe sole and outersole Download PDFInfo
- Publication number
- WO2010050628A2 WO2010050628A2 PCT/JP2010/051833 JP2010051833W WO2010050628A2 WO 2010050628 A2 WO2010050628 A2 WO 2010050628A2 JP 2010051833 W JP2010051833 W JP 2010051833W WO 2010050628 A2 WO2010050628 A2 WO 2010050628A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- weight
- parts
- rubber
- foaming agent
- shoe sole
- Prior art date
Links
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
-
- A—HUMAN NECESSITIES
- A43—FOOTWEAR
- A43B—CHARACTERISTIC FEATURES OF FOOTWEAR; PARTS OF FOOTWEAR
- A43B13/00—Soles; Sole-and-heel integral units
- A43B13/02—Soles; Sole-and-heel integral units characterised by the material
- A43B13/04—Plastics, rubber or vulcanised fibre
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0061—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof characterized by the use of several polymeric components
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/06—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a chemical blowing agent
- C08J9/10—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a chemical blowing agent developing nitrogen, the blowing agent being a compound containing a nitrogen-to-nitrogen bond
- C08J9/102—Azo-compounds
- C08J9/103—Azodicarbonamide
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/32—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof from compositions containing microballoons, e.g. syntactic foams
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L21/00—Compositions of unspecified rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2203/00—Foams characterized by the expanding agent
- C08J2203/22—Expandable microspheres, e.g. Expancel®
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2321/00—Characterised by the use of unspecified rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2409/00—Characterised by the use of homopolymers or copolymers of conjugated diene hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/22—Expanded, porous or hollow particles
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L7/00—Compositions of natural rubber
Definitions
- the present invention is a rubber foam composition for a shoe sole, which is lightweight and has an appropriate hardness, and excellent in tensile strength, tear strength, impact absorbability, in-mold fluidity, and dimensional stability after foaming, and further its rubber
- the present invention relates to an outsole using the composition.
- Patent Document 1 In order to achieve low hardness and light weight, a chemical foaming agent (Patent Document 1) and a physical foaming agent (Patent Document 2) are used, and low hardness and light weight are achieved by mixing bubbles in the product.
- Patent Document 2 a chemical foaming agent
- Patent Document 2 a physical foaming agent
- problems such as destruction of the foam due to abnormal foaming, deterioration of the surface of the foamed product, and unstable product dimensions.
- thermoplastic resin having a glass transition temperature (Tg) lower than that of conventional 1,2-polybutadiene and vinyl cis polybutadiene (VCR) are used together and crosslinked with an organic oxide to increase the thickness accuracy.
- Tg glass transition temperature
- VCR vinyl cis polybutadiene
- excellent, moderate hardness, excellent smoothness, and further improved workability Patent Document 3
- 1,2-polybutadiene, polyethylene-based polymer, ethylene-vinyl acetate polymer resin is blended with natural rubber (isoprene) as a third component, which is rich in rubber elasticity, light weight and excellent hardness, Improves impact resistance and tear strength.
- the present invention relates to a foam rubber composition for a shoe sole having a tensile strength, a tear strength, and an impact absorption property with a light weight and an appropriate hardness by using a pyrolytic foaming agent and a thermal expansion foaming agent in combination.
- the outsole used is provided.
- the flowability in the mold because the temperature dependency during pressurization is low, the viscosity is low and there is no air bubble inclusion.
- air voids the product is free from chipping and becomes a molded product.
- a foam rubber composition for a shoe sole which is free from defects and excellent in dimensional stability after foaming.
- a vinyl cis-butadiene rubber (VCR) and a thermoplastic resin (C) are combined with a foaming agent comprising (D1) a thermal expansion foaming agent and (D2) a thermal decomposition foaming agent, and (A) + (B) +
- a shoe characterized in that (D1) is 0.5 to 3.0 parts by weight and (D2) is 0.5 to 7.0 parts by weight with respect to 100 parts by weight of the total rubber component comprising (C).
- the present invention relates to a foam rubber composition for a bottom.
- the present invention also relates to an outsole characterized by using the foam rubber composition for a shoe sole.
- the foam rubber composition for shoe soles and the outsole using the same obtained in the present invention are lightweight and have an appropriate hardness, and have tensile strength, tear strength, and impact absorption.
- the fluidity in the mold is improved (because the temperature dependency at the time of pressurization is low, the viscosity decrease is small and the air void is also improved). Therefore, it is possible to provide a foam rubber composition for a shoe sole that is free from defects in the product, has no defects in the molded product, and is excellent in dimensional stability after foaming.
- FIG. 4 shows the relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Examples 1 to 4.
- FIG. FIG. 6 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Examples 5 to 8.
- FIG. FIG. 5 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Comparative Examples 1 to 5.
- FIG. FIG. 4 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Comparative Examples 6 to 10.
- FIG. 4 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Examples 9 to 12.
- FIG. FIG. 6 shows the relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Examples 13 to 15.
- FIG. FIG. 6 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Comparative Examples 11 to 14.
- FIG. It is a relationship between the foaming pressure in a reactor in the initial stage of vulcanization (heating and pressurization) in Comparative Example 15 and time.
- the in-reactor foaming pressure from the start to the end of vulcanization in Examples 9-12.
- FIG. 6 shows the relationship between the in-reactor foaming pressure and time at the initial stage of vulcanization (heating and pressurization) in Examples 16 to 18 and Comparative Example 16.
- Component (A) As vulcanizable rubber of (A), natural rubber (NR), isoprene rubber (IR), butadiene rubber (BR), styrene-butadiene rubber (SBR), acrylonitrile-butadiene rubber (NBR) ), Butyl rubber (IIR), acrylonitrile-chloroprene rubber, acrylonitrile-isoprene rubber, styrene-chloroprene rubber, styrene-isoprene rubber and other diene rubbers, ethylene-propylene rubber, ethylene-butene rubber, ethylene-vinyl acetate copolymer (EVA) And ethylene- ⁇ -olefin copolymer rubbers such as ethylene-propylene-diene rubber (EPDM). These may be used alone or in combination.
- NR natural rubber
- IR isoprene rubber
- BR butadiene rubber
- SBR styrene-buta
- (A-2) As the styrene-butadiene rubber, it is particularly preferable to use a water-added styrene-butadiene rubber. Further, as the (a-2) styrene-butadiene rubber, it is particularly preferable to use a styrene-butadiene rubber (S-SBR) obtained by solution polymerization. Use of SBR other than S-SBR is not preferable because it causes problems such as deterioration of shrinkage and deterioration of wear resistance (or use of S-SBR exhibits excellent characteristics in terms of shrinkage and wear resistance). Preferred to use).
- S-SBR styrene-butadiene rubber
- the blending amount of (A) is 2 to 30 parts by weight, preferably 5 to 25 parts by weight, particularly preferably 7 to 17 parts by weight of vulcanizable rubber in 100 parts by weight of all rubber components.
- properties necessary for roll processability sheet skin shape appearance
- various physical properties of footwear tensile strength, elongation, tear strength
- the blending amount is less than 2 parts by weight, problems such as roughness of the sheet and workability occur, and conversely if it exceeds 30 parts by weight, problems such as an increase in specific gravity and deterioration of shrinkage occur.
- the blending amount of (a-1) natural rubber (NR) is: It is preferable that 2 to 30 parts by weight, preferably 5 to 20 parts by weight, particularly preferably 7 to 15 parts by weight are blended in 100 parts by weight of the total rubber component.
- properties necessary for roll processability sheet skin shape appearance
- various physical properties of footwear tensile strength, elongation, tear strength
- the amount of the solution-polymerized styrene butadiene rubber (S-SBR) is 2 to 30 parts by weight, preferably 5 to 25 parts by weight, particularly preferably 8 to 20 parts by weight based on 100 parts by weight of the total rubber. What mix
- blends a part is preferable. When blended, properties necessary for roll processability (sheet skin shape appearance, viscosity adjustment) can be imparted.
- the blending ratio (a-1) / (a-2) is preferably 0.07 to 15 More preferably, it is 0.2 to 4, particularly preferably 0.35 to 1.88. Use within the above-mentioned ratio range is preferable because it exhibits excellent properties in terms of shrinkage and wear resistance.
- VCR vinyl cis-butadiene rubber
- SPB 2-polybutadiene resin
- the boiling n-hexane insoluble matter mentioned here means the part recovered as an insoluble matter when the VCR is refluxed in boiling n-hexane, and the boiling n-hexane soluble matter means that the VCR is refluxed in boiling n-hexane. It is the part that dissolves when The boiling n-hexane insoluble matter has a reduced viscosity (135 ° C, concentration 0.20 g / dl orthodichlorobenzene solution) measured with an orthodichlorobenzene solution of 0.5 to 4, preferably 0.8 to 3 is there. When the reduced viscosity of boiling n-hexane insolubles is less than 0.5, the die swell of the formulation is not improved sufficiently.
- the boiling n-hexane insoluble matter is SPB having a melting point of preferably 170 ° C. or higher, particularly preferably 190 to 220 ° C. If the melting point of SPB is less than 170 ° C., problems such as deterioration in workability and mechanical strength (such as tensile stress and tensile strength) occur due to the decrease in SPB reinforcement.
- the weight average molecular weight of the boiling n-hexane soluble part is preferably in the range of 300,000 to 800,000, and if it is less than 300,000, the durability and impact resilience of the vulcanizate are lowered, which is not preferred.
- Exceeding 800,000 is not preferable because the Mooney viscosity of the compound becomes too high and processing becomes difficult.
- VCR production methods are disclosed in, for example, JP-B-49-17666, JP-B-49-17667, JP-B-61-57858, JP-B-62-171, JP-B-63-36324, The methods described in JP-A-2-37927, JP-B-2-38081, JP-B-3-63566 and the like can be used.
- the manufacturing method of the VCR used in the present invention is not limited to these methods.
- the blending amount of the VCR of the component (B) is preferably 40 to 80 parts by weight, and more preferably 50 to 70 parts by weight with respect to 100 parts by weight of all rubber components.
- various physical properties of the footwear tensile strength, elongation, tear strength, abrasion resistance
- the blending amount is less than 40 parts by weight, the problem of deterioration of wear resistance occurs, and conversely if it exceeds 80 parts by weight, problems such as an increase in specific gravity and an increase in hardness occur.
- thermoplastic resin examples include syndiotactic 1,2-polybutadiene resin (SPB), polyethylene and its maleic anhydride graft polymer, polyisobutylene, ethylene vinyl acetate copolymer, Ethylene acrylate, ethylene acrylic acid copolymer, polypropylene and its maleic anhydride graft polymer, chlorinated polypropylene, 4-methylpentene-1 resin, polystyrene, ABS resin, AS resin, acrylic resin, methacrylic resin, vinyl chloride resin, Examples include vinylidene chloride resin, polyamide resin, and polycarbonate. These thermoplastic resins (C) can be used alone or in combination of two or more.
- SPB syndiotactic 1,2-polybutadiene resin
- polyisobutylene polyisobutylene
- ethylene vinyl acetate copolymer Ethylene acrylate
- ethylene acrylic acid copolymer polypropylene and its maleic anhydride graft polymer
- the thermoplastic resin is preferably a polyolefin resin.
- polyolefin resins include polyethylene (PE), particularly low density PE (LDPE), medium density PE (MDPE), high density PE (HDPE), and linear low density PE (LLDPE), ethylene-propylene elastomer copolymer.
- EPM ethylene-propylene-diene rubber
- EVA ethylene-vinyl ester copolymers
- EVA ethylene-vinyl acetate copolymer
- EVA ethylene-acrylate copolymers
- ethylene- ⁇ -olefin thermoplastic copolymers as well as propylene, butene -1, and copolymers such as pentene-1.
- These polyolefin resins can be used alone or in combination of two or more.
- thermoplastic resin examples include 1,2-polybutadiene resins having a melting point of 70 ° C. to 110 ° C., and ethylene-vinyl acetate copolymer (EVA).
- EVA ethylene-vinyl acetate copolymer
- the blending amount of the thermoplastic resin (C) is preferably 10 to 35 parts by weight, more preferably 15 to 30 parts by weight, and more preferably 18 to 28 parts by weight based on 100 parts by weight of the total rubber component. It is particularly preferred. When blending within this range, various physical properties (gas barrier properties, dimensional stability) of footwear can be imparted. On the other hand, if the blending amount is less than 10 parts by weight, problems such as an increase in specific gravity and dimensional stability (shrinkage) due to a decrease in gas barrier properties occur, and conversely, if it exceeds 35 parts by weight, a problem of an increase in hardness will occur. Absent.
- the total rubber component means (A) + (B) + (C).
- the thermal expansion type foaming agent is not particularly limited as long as it is a foaming agent that expands by heat, but it is preferable to use thermally expandable microcapsules.
- a heat-expandable microcapsule is an encapsulated liquid or gas that expands when heated in a synthetic resin capsule, and is encapsulated by heat of kneading and melting by a screw or the like during extrusion molding or injection molding.
- the microcapsules that form the outer shell are expanded by the expansion of the liquid or gas. However, depending on the temperature conditions during molding, a microcapsule that melts and completes without bursting is used.
- a copolymer having acrylonitrile as one of monomer components is used, and other monomer components that may be copolymerized with acrylonitrile include, for example, acrylic acid, methacrylic acid, acrylate ester, methacrylic ester, and the like. Examples thereof include, but are not limited to, acid esters, styrene, vinyl acetate, and vinylidene chloride.
- the liquid or gas encapsulated in the microcapsule is one that expands as a gas at a temperature below the softening point of the microcapsule.
- Low boiling point liquids such as methane halides such as heptane, petroleum ether, methyl chloride and methylene chloride, chlorofluorocarbons such as CCl 3 F and CCl 2 F 2 , and tetraalkylsilanes such as tetramethylsilane and trimethylethylsilane
- a compound such as AIBN that is thermally decomposed by heating and becomes gaseous can be mentioned.
- the amount and size of the bubbles are determined by the number (mixing amount) and the particle size of the thermally expandable microcapsules after expansion, and the molding is performed from these. Since the specific gravity of the body is determined, by predicting the number of unexpanded thermally expandable microcapsules (mixed amount) to be mixed with the thermoplastic resin raw material and the particle size of the thermally expandable microcapsules after expansion by the heating amount during molding The foaming state can be controlled, and only the gas does not escape to the outside as compared with the case where the conventional gas is directly expanded, and the target specific gravity can be easily achieved.
- a foam can be formed in a state where the thermally expandable microcapsule is expanded and contained in the thermoplastic resin by extrusion molding or injection molding, and each bubble is stable in a state of being encapsulated in the microcapsule.
- extrusion molding even when the pressure is released by being extruded from a die, the gas does not escape directly to the outside of the molded product, and a foam having a good surface appearance can be obtained.
- thermoly expandable microcapsules for example, EXPANCEL 920DU40, EXPANCEL 920DU80, EXPANCEL009DU80, EXPANCEL920DU40 master batch products 920MB40 and 920MB50, Matsumoto Yushi Pharmaceutical Matsumoto Microsphere F-D, etc.
- the particle size of the thermally expandable microcapsule is preferably 8 to 30 ⁇ m, more preferably 8 to 17 ⁇ m, and even more preferably 10 to 16 ⁇ m. In the said range, the effect of low specific gravity and the smoothness of the surface skin of a molded article can be expected.
- the compounding amount of the thermal expansion foaming agent is 0.5 to 3.0 parts by weight, preferably 0.75 to 3.0 parts by weight, more preferably 0.80 to 2. It is 5 parts by weight, more preferably 0.85 to 2.5 parts by weight, still more preferably 0.90 to 2.0 parts by weight, and particularly preferably 1.2 to 2.0 parts by weight.
- air removal, appearance of molded products (surface state smoothness and dimensional stability) with increasing foaming pressure in the initial stage of vulcanization, and various physical properties (tensile strength, elongation at break and tear strength) associated with lower specific gravity. Can be suppressed.
- the blending amount is less than 0.5 parts by weight, there is a problem that the effect of lowering the specific gravity is reduced.
- the blending amount is more than 3.0 parts by weight, the surface roughness of the molded product (smoothness) is reduced. Problems arise.
- D2 Pyrolytic foaming agent (chemical foaming agent)
- thermally decomposable foaming agent examples include organic foaming agents such as azocicarbonamide complex (ADCA), azobisisobutyronitrile (AIBN), dinitrosopentamethylenetetramine (DPT), N, N′-dimethyl.
- ADCA azocicarbonamide complex
- AIBN azobisisobutyronitrile
- DPT dinitrosopentamethylenetetramine
- N, N′-dimethyl N, N′-dimethyl.
- BSH benzenesulfonyl hydrazide
- TSH p-toluenesulfonyl hydrazide
- OBSH 4,4'-oxybis (benzenesulfonyl hydrazide)
- 3,3'-disulfone hydrazide diphenyl Sulfone toluene disulfonyl hydrazine, p-toluene disulfonyl hydrazide, p-toluene sulfonyl semi strength rubazide, diethyl azodicarboxylate and the like can be used.
- an inorganic foaming agent can also be used as the thermally decomposable foaming agent.
- sodium bicarbonate or ammonium bicarbonate can be used.
- thermal decomposable foaming agents may be used alone or in combination.
- An organic foaming agent having a decomposition temperature of about 195 to 210 ° C. and a generated gas amount of 190 to 240 (ml / g) is preferable for use as a thermally decomposable foaming agent.
- azodicarbonamide complex ADCA
- OBSH 4,4′-oxybis (benzenesulfonylhydrazide)
- the foaming effect with the combined thermal expansion foaming agent (D1) can be maximized, and the air accompanying the increase in foaming pressure at the initial stage of vulcanization can be completely removed.
- the compounding amount of the thermally decomposable foaming agent is 0.5 to 7.0 parts by weight, preferably 0.75 to 7.0 parts by weight, more preferably 0.8 to 7. 0 parts by weight, more preferably 0.85 to 7.0 parts by weight, still more preferably 0.85 to 6.0 parts by weight, and particularly preferably 1.0 to 4.5 parts by weight.
- the blending amount is less than 0.5 parts by weight, there is a problem that the effect of reducing the specific gravity and the hardness is reduced. Conversely, if the blending amount is more than 7.0 parts by weight, the dimensional stability is reduced due to high foaming. Problem arises.
- Examples of the rubber reinforcing agent (E) blended in the rubber composition according to the present invention include various types of carbon black, white carbon, silica, activated calcium carbonate, and ultrafine magnesium silicate. . Of these, silica and / or carbon black are preferred. Above all, silica having an average primary particle size of 5 to 100 nm, such as silicic anhydride by a dry method, hydrous silicic acid by a wet method, and synthetic silicate, and a particle size of 90 nm or less and a dibutyl phthalate (DBP) oil supply amount of 70 ml / 100 g or more.
- Carbon black is preferred. Examples of carbon black include furnace black, channel black, thermal black, and the like. 110, 212, N242, S315, N330, N550, N660, N765 and the like.
- Silica can be either wet silica or dry silica, and can be used in combination. Further, silica can be used alone. Moreover, it is preferable that the silica to be used has a silica purity of 97% or more. Further, pulverized natural quartz having a silica purity of 97% or more can be used. Furthermore, particles obtained by hydrolyzing an organosilicon compound such as silane are also preferably used. Silica having a purity of less than 97% does not have a sufficient rubber reinforcing effect, so that a rubber composition blended with such silica does not have sufficient wear resistance.
- the average particle diameter of the silica particles is preferably in the range of 0.1 to 50 ⁇ m, more preferably 1 to 50 ⁇ m, and particularly preferably 5 to 50 ⁇ m.
- a silica particle size can be obtained, for example, by a method of pulverizing a completely melted quartz glass so as to have an appropriate particle size.
- the average particle size is smaller than 0.1 ⁇ m, it is difficult to mix in the rubber, causing problems such as loss and poor dispersion, and conversely if larger than 50 ⁇ m, the reinforcing effect is low, which is not preferable.
- the amount of the rubber reinforcing agent used is 10 to 30 parts by weight, preferably 12 to 25 parts by weight, more preferably 15 to 20 parts by weight based on 100 parts by weight of the total rubber components. Within the above-mentioned range, a sufficient reinforcing effect can be obtained, and an effect of improving wear resistance can be obtained. On the other hand, if the amount is less than 10 parts by weight, the reinforcing effect is low, causing problems of deterioration in various physical properties (mechanical strength and wear resistance). Conversely, if the amount is more than 30 parts by weight, the workability is lowered due to the increase in viscosity. This is not preferable because it causes problems.
- (F) Vulcanization accelerator examples include aldehydes, ammonia, amines, guanidines, thioureas, thiazoles, thiurams, dithiocarbamates, xanthates and the like.
- TMTD tetramethylthiuram disulfide
- OBS N-oxydiethylene-2-benzothiazolylsulfenamide
- CBS N-cyclohexyl-2-benzothiazylsulfenamide
- MBTS dibenzo Thiazyl sulfide
- ZnBDC zinc di-n-butyldithiocarbide
- ZnMDC zinc dimethyldithiocarbide
- MBTS dibenzothiazyl sulfide
- DAG diortolyl guanidine
- the amount of the vulcanization accelerator is 0.5 to 1.5 parts by weight, preferably 0.7 to 1.3 parts by weight, more preferably 0.8 to 1.2 parts by weight based on 100 parts by weight of the total rubber component. Part. Within the above range, a sufficient vulcanization effect can be obtained. On the other hand, if the amount is less than 0.5 parts by weight, problems such as a delay in vulcanization time and various physical properties due to a decrease in crosslink density are caused. This is not preferable.
- (G) Vulcanizing agent As the vulcanizing agent used in the present invention, sulfur, a compound that generates sulfur by heating, an organic peroxide, a metal oxide such as magnesium oxide, a polyfunctional monomer, a silanol compound, and the like Is mentioned.
- the compound that generates sulfur by heating include tetramethyl thiuram disulfide and tetraethyl thiuram disulfide. Among these, sulfur is particularly preferable.
- the amount of the vulcanizing agent is 0.3 to 2.2 parts by weight, preferably 0.5 to 1.7 parts by weight, more preferably 1.0 to 1.5 parts by weight, based on 100 parts by weight of all rubber components. It is. Within the above range, a sufficient vulcanization effect can be obtained. On the other hand, if the amount is less than 0.3 parts by weight, problems such as a delay in vulcanization time and a decrease in physical properties due to a decrease in crosslink density are caused. This is not preferable.
- Anti-aging agent examples include amine / ketone-based, imidazole-based, amine-based, phenol-based, sulfur-based and phosphorus-based anti-aging agents.
- H2 Vulcanization aids As vulcanization aids, known vulcanization aids such as aldehydes, ammonia, amines, guanidines, thioureas, thiazoles, thiurams, dithiocarbamates, and xanthates Etc. are used.
- H3 Scorch inhibitor (retarder)
- organic acids nitroso compounds, N-cyclohexylthiophthalimide, sulfonamide derivatives and the like are used.
- H4 Process oil may be any of aromatic, naphthenic, and paraffinic.
- (Processing method) (A) Vulcanizable rubber other than (A) (B) Boiling n-hexane insoluble content: 1 to 25% by weight, boiling n-hexane soluble content: 99 to 75% by weight vinyl cisbutadiene
- the rubber (VCR) and (C) thermoplastic resin are used in combination with a foaming agent comprising (D1) a thermal expansion type foaming agent and (D2) a thermal decomposition type foaming agent.
- the rubber reinforcing agent is an open type such as a roll. It can be obtained by kneading using a kneader such as a closed kneader such as a kneader or Banbury mixer, and can be vulcanized after molding and applied to various rubber products.
- Another main point of the present invention is the adjustment of the pressure in the reactor in the vulcanization process.
- the foaming pressure in the reactor at the initial stage of vulcanization (after heating and pressurization 30 seconds) to 2000 kPa or more, preferably 2200 kPa, air voids are not generated, and there is no chipping in the product. It is possible to provide a method for producing a rubber foam composition for a shoe sole that is excellent in dimensional stability after foaming.
- the foaming pressure in the reactor in the initial stage of vulcanization (after heating and pressurization 30 seconds) is less than 2000 kP, sufficient deaeration cannot be performed in the initial stage of vulcanization, causing air voids. Therefore, the foaming pressure in the reactor in the initial stage of vulcanization (after 30 seconds of heating and pressurization) is a major factor for improving the product.
- the foaming pressure in the reactor is larger than 2000 kPa, and the roles of the two types of foaming agents at the initial stage of vulcanization and the final stage of vulcanization are greatly affected.
- the foaming effect of (D1) thermal expansion type foaming agent affects the rapid increase of the foaming pressure in the reactor.
- the foaming effect of (D2) pyrolytic foaming agent is affected.
- the foaming effect by the pyrolytic foaming agent can suppress the increase in the hardness of the resin and realize weight reduction. Therefore, the combined use of (D1) and (D2) is important.
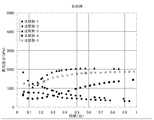
- FIGS. 1 and 2 show the relationship between the vulcanization time (especially at the initial stage of vulcanization) and the foaming pressure in the reactor of Examples 1 to 8.
- the pressure inside the reactor was higher than 2000 kPa at the time of 0.5 minutes from the start of vulcanization (start of vulcanization: 30 seconds after heating and pressurization).
- start of vulcanization start of vulcanization: 30 seconds after heating and pressurization
- FIGS. 3 to 4 where Comparative Examples 1 to 10 are shown, the pressure inside the reactor is less than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: after 30 seconds of heating and pressurization).
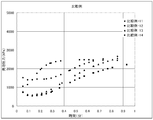
- Figures 5 to 8 show the relationship between the foaming pressure in the reactor from the start to the end of vulcanization.
- the initial pressure of vulcanization (after 30 seconds of heating and pressurization) was maintained even after the in-reactor foaming pressure exceeded 2000 kPa. It can be seen that, even at the end of vulcanization, the in-reactor foaming pressure still exceeds 1000 kPa and the pressure is maintained. It is desirable to take such a course that there is no sudden pressure decrease, and the foaming effect of the pyrolytic foaming agent (D2) is greatly influenced particularly in the late vulcanization stage.
- D2 pyrolytic foaming agent
- Table 3 shows that no air void occurred in Examples 1 to 8.
- FIGS. 9 to 10 show the relationship between the vulcanization time (particularly at the initial stage of vulcanization) and the foaming pressure in the reactor of Examples 9 to 15 in the present invention.
- the pressure in the reactor was higher than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: after 30 seconds of heating and pressurization).
- 11 to 12 show the relationship between the foaming pressure in the reactor from the start to the end of vulcanization in Comparative Examples 11 to 15.
- the reaction pressure decreases to 2000 kPa or less from the start to the end of vulcanization, there arises a problem of an increase in specific gravity and hardness accompanying a decrease in the reaction pressure.
- FIG. 17 shows the relationship between the vulcanization time (especially at the initial stage of vulcanization) and the foam pressure in the reactor of Examples 16 to 18 and Comparative Example 16 in the present invention.
- the pressure in the reactor was higher than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: 30 seconds after heating and pressurization).
- the pressure in the reactor was less than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: after 30 seconds of heating and pressurization).
- the specific gravity value at 2 mm is preferably 0.450 to 0.950 (g / cm 3 ), more preferably 0.500 to 0.800 (g / cm 3 ), and 0.550 to More preferably, it is 0.750 (g / cm 3 ).
- the specific gravity is large, there arises a problem that the hardness cannot be increased and the weight cannot be reduced.
- specific gravity is small, the problem of deterioration of various physical properties (mechanical strength, abrasion resistance, shrinkage
- the foaming pressure in the reactor is a combination of the constituent elements in the composition and the initial stage of vulcanization (after heating and pressurization 30 seconds) is 2000 kPa. It turns out that it is important to adjust so that it may become above, and to adjust so that specific gravity may become small.
- the blending ratio of all rubber components (D1) and (D2) is also important in the present invention.
- the foaming pressure in the reactor at the initial stage of vulcanization (after heating and pressurization 30 seconds) is 2000 kPa or more.
- the amount of the foaming agent is necessary, and an appropriate amount of foaming agent is required to suppress the increase in the hardness of the resin at the end of vulcanization and to realize weight reduction. From such a viewpoint, the blending ratio of (D1) of 0.5 to 3.0 parts by weight and (D2) of 0.5 to 7.0 parts by weight is the optimum condition.
- the physical properties of the obtained rubber composition were measured as follows. (1) Specific gravity (density): Measured by method A according to JIS K6268.
- Foaming pressure Using ALPHA TECHNOLOGIES RPA2000 (rotorless type, rubber processability analyzer), the time to reach 10% and 160% vulcanization degree at 160 ° C. and the foaming pressure were measured according to JIS K6300. The higher the pressure value, the better.
- Hardness Measured at room temperature using a type A durometer according to JIS K6253. Moreover, it measured at room temperature using Asker C type (sponge hardness meter) based on SR1S0101. The smaller the value, the softer and better.
- Tear strength measured in accordance with JIS K6252. The higher the value, the better it will be without tearing when the product is peeled from the mold.
- Rebound resilience measured according to JIS K6255 using a trypso type.
- Shrinkage rate Calculated from the length after 5 minutes at room temperature after molding (length after 30 minutes at room temperature). The smaller the value, the better the dimensional stability.
- Example 1 (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin in Table 1 in a blending ratio of 7/70/23 parts by weight, and rubber reinforcing agents such as silica, zinc white and stearic acid Together with a 1.7 liter closed kneader. Thereafter, the vulcanization accelerator and sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 1.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 2 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 7/65/28 parts by weight, and a vulcanized test piece was prepared in the same manner as in Example 1 below. The physical properties were evaluated. The results are shown in Table 3.
- Example 3 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 12/60/28 parts by weight, and a vulcanized test piece was prepared in the same manner as in Example 1 below. The physical properties were evaluated. The results are shown in Table 3.
- Example 4 In Table 1, (a-1) natural rubber / (a-2) water-added styrene-butadiene rubber (manufactured by Asahi Kasei Chemicals Co., Ltd.) / (B) VCR / (C) 1,2 polybutadiene resin is blended in a ratio of 7/10/65. / 18 parts by weight, vulcanized test pieces were prepared in the same manner as in Example 1, and the physical properties were evaluated. The results are shown in Table 3.
- Example 5 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator and sulfur, and 2.5 parts by weight of the thermal expansion foaming agent and 0.7 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 6 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator, sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 6.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 7 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator and sulfur, 2.5 parts by weight of the thermal expansion foaming agent, and 6.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 8 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, a vulcanization accelerator, sulfur, 2.5 parts by weight of a thermal expansion foaming agent, and 1.0 part by weight of a thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 10 (A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 5 to a blending ratio of 7/18/55/20 parts by weight, rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 11 In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 12 In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 14 In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 15 In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 11 The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used. The results are shown in Table 8.
- Example 12 The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used and the thermal decomposition foaming agent CAP-500 was used in an amount of 5.50 parts by weight. The results are shown in Table 8.
- Example 13 The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used and the thermal decomposition foaming agent CAP-500 was used in an amount of 6.50 parts by weight. The results are shown in Table 8.
- Example 14 The same procedure as in Example 9 was conducted, except that 5.00 parts by weight of the thermal expansion foaming agent EXPANCEL DU092-40 was used, and the thermal decomposition foaming agent CAP-500 was not used. The results are shown in Table 8.
- Experimental example 3 (Example 16) (A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 17 (A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 18 (A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 16 The same procedure as in Example 16 was performed except that the thermal expansion foaming agent was not used. The results are shown in Table 10.
- the foam rubber composition obtained in the present invention it is useful as a sole material in the sole field, in particular, footwear in general such as sports shoes, running shoes, and casual shoes.
Abstract
Provided is a foamed rubber composition for a shoe sole that is lightweight, has suitable hardness, and has excellent tensile strength, tearing strength, and impact absorbance, and an outersole that uses the same. The foamed rubber composition for a shoe sole uses (A) a vulcanizable rubber, (B) a vinyl cis-butadiene rubber (VCR) that is not (A) and that has a boiling n-hexane-insoluble content of 1 to 25 wt% and a boiling n-hexane-soluble content of 99 to 75 wt%, and (C) a thermoplastic resin, in combination with a foaming agent composed of (D1) a thermal expanding foaming agent and (D2) a pyrolyzing foaming agent such that there are 0.5 to 3.0 parts by weight of (D1) and 0.5 to 7.0 parts by weight of (D2) per 100 parts by weight of the total rubber components consisting of (A) + (B) + (C).
Description
本発明は軽量かつ適度な硬度で、引張強度、引裂強度、衝撃吸収性、金型内流動性、及び発泡後の寸法安定性にも優れた靴底用ゴム発泡体組成物、さらにはそのゴム組成物を用いたアウトソールに関する。
The present invention is a rubber foam composition for a shoe sole, which is lightweight and has an appropriate hardness, and excellent in tensile strength, tear strength, impact absorbability, in-mold fluidity, and dimensional stability after foaming, and further its rubber The present invention relates to an outsole using the composition.
履物分野においては、着用者の体重を支え、運動に伴う荷重や衝撃力に耐える強度と靴の履き心地、安全性を高める為の軽量化、グリップ性が求められている。その中でも近年、低硬度・軽量化と成型品の寸法安定性(低収縮率)が要求されており、これらの特性を向上させる必要がある。
In the footwear field, there is a need for strength to support the weight of the wearer, withstand the load and impact force associated with exercise, weight reduction and grip to enhance the comfort and safety of shoes. Among them, in recent years, low hardness and light weight and dimensional stability (low shrinkage) of molded products are required, and it is necessary to improve these characteristics.
低硬度・軽量化を図るには化学発泡剤(特許文献1)や物理発泡剤(特許文献2)が用いられ、製品中に気泡を混入させる事により低硬度・軽量化を達成している。しかし、軽量化を図る為に化学発泡剤を単独で大量に添加すると異常発泡による発泡体の破壊、発泡製品表面の悪化や製品寸法の不安定などの問題がある。
In order to achieve low hardness and light weight, a chemical foaming agent (Patent Document 1) and a physical foaming agent (Patent Document 2) are used, and low hardness and light weight are achieved by mixing bubbles in the product. However, when a large amount of a chemical foaming agent is added alone to reduce the weight, there are problems such as destruction of the foam due to abnormal foaming, deterioration of the surface of the foamed product, and unstable product dimensions.
上記問題を解決する為、従来の1,2-ポリブタジエンに比べガラス転移温度(Tg)が低い熱可塑性樹脂とビニル・シスポリブタジエン(VCR)を併用し有機化酸化物にて架橋し、厚み精度に優れ、適度な硬度を有し、平滑性に優れ、さらに加工性の向上を図っている(特許文献3)。また、特許文献4では1,2-ポリブタジエン及びポリエチレン系重合体、エチレン-酢酸ビニル重合体樹脂に第三成分として天然ゴム(イソプレン)を配合する事でゴム弾性に富み、軽量かつ優れた硬度、耐衝撃性、及び引裂強度の向上させている。
In order to solve the above problems, a thermoplastic resin having a glass transition temperature (Tg) lower than that of conventional 1,2-polybutadiene and vinyl cis polybutadiene (VCR) are used together and crosslinked with an organic oxide to increase the thickness accuracy. Excellent, moderate hardness, excellent smoothness, and further improved workability (Patent Document 3). In Patent Document 4, 1,2-polybutadiene, polyethylene-based polymer, ethylene-vinyl acetate polymer resin is blended with natural rubber (isoprene) as a third component, which is rich in rubber elasticity, light weight and excellent hardness, Improves impact resistance and tear strength.
本発明は、熱分解型発泡剤と熱膨張型発泡剤を併用する事により、軽量かつ適度な硬度で、引張強度、引裂強度、衝撃吸収性を有する靴底用発泡体ゴム組成物およびそれを用いたアウトソールを提供する。特に金型内流動性(加圧時の温度依存性が低い為、粘度低下が少なく気泡の抱き込みが無い。以下エアーボイドと示す。)も改良される事で製品にカケがなく成形品に不良が無く、また発泡後の寸法安定性にも優れた靴底用発泡体ゴム組成物を提供する。
The present invention relates to a foam rubber composition for a shoe sole having a tensile strength, a tear strength, and an impact absorption property with a light weight and an appropriate hardness by using a pyrolytic foaming agent and a thermal expansion foaming agent in combination. The outsole used is provided. In particular, the flowability in the mold (because the temperature dependency during pressurization is low, the viscosity is low and there is no air bubble inclusion. Hereafter referred to as air voids), the product is free from chipping and becomes a molded product. There is provided a foam rubber composition for a shoe sole which is free from defects and excellent in dimensional stability after foaming.
本発明は、(A)加硫可能なゴム、(A)以外の(B)沸騰n-ヘキサン不溶分;1~25重量%で、沸騰n-ヘキサン可溶分;99~75重量%であるビニル・シスブタジエンゴム(VCR)および(C)熱可塑性樹脂に、(D1)熱膨張型発泡剤と(D2)熱分解型発泡剤からなる発泡剤を併用し、(A)+(B)+(C)からなる全ゴム成分100重量部に対して、(D1)が0.5~3.0重量部、(D2)が0.5~7.0重量部であることを特徴とする靴底用発泡体ゴム組成物に関する。
In the present invention, (A) vulcanizable rubber, (B) other than (A) (B) boiling n-hexane insoluble content: 1 to 25% by weight, boiling n-hexane soluble content: 99 to 75% by weight A vinyl cis-butadiene rubber (VCR) and a thermoplastic resin (C) are combined with a foaming agent comprising (D1) a thermal expansion foaming agent and (D2) a thermal decomposition foaming agent, and (A) + (B) + A shoe characterized in that (D1) is 0.5 to 3.0 parts by weight and (D2) is 0.5 to 7.0 parts by weight with respect to 100 parts by weight of the total rubber component comprising (C). The present invention relates to a foam rubber composition for a bottom.
また、本発明は、前記靴底用発泡体ゴム組成物を用いたことを特徴とするアウトソールに関する。
The present invention also relates to an outsole characterized by using the foam rubber composition for a shoe sole.
本発明で得られる靴底用発泡体ゴム組成物およびそれを用いたアウトソールは、軽量かつ適度な硬度で、引張強度、引裂強度、衝撃吸収性を有する。特にその製造工程では、金型内流動性が改良される(加圧時の温度依存性が低い為、粘度低下が少なく、エアーボイドも改良されるため)。よって、製品にカケがなく成形品に不良が無く、また発泡後の寸法安定性にも優れた靴底用発泡体ゴム組成物を提供することが出来る。
The foam rubber composition for shoe soles and the outsole using the same obtained in the present invention are lightweight and have an appropriate hardness, and have tensile strength, tear strength, and impact absorption. In particular, in the manufacturing process, the fluidity in the mold is improved (because the temperature dependency at the time of pressurization is low, the viscosity decrease is small and the air void is also improved). Therefore, it is possible to provide a foam rubber composition for a shoe sole that is free from defects in the product, has no defects in the molded product, and is excellent in dimensional stability after foaming.
(A)成分の説明
(A)の加硫可能なゴムとしては、天然ゴム(NR)、イソプレンゴム(IR)、ブタジエンゴム(BR)、スチレン-ブタジエンゴム(SBR)、アクリロニトリル-ブタジエンゴム(NBR)、ブチルゴム(IIR)、アクリロニトリル-クロロプレンゴム、アクリロニトリル-イソプレンゴム、スチレン-クロロプレンゴム、スチレン-イソプレンゴムなどのジエン系ゴム、エチレン-プロピレンゴム、エチレン-ブテンゴム、エチレン-酢酸ビニル共重合体(EVA)、エチレン-プロピレン-ジエンゴム(EPDM)などのエチレン-α-オレフィン系共重合ゴムが挙げられる。これらを単独でもよいし複数組み合わせて使用してもよい。 Description of Component (A) As vulcanizable rubber of (A), natural rubber (NR), isoprene rubber (IR), butadiene rubber (BR), styrene-butadiene rubber (SBR), acrylonitrile-butadiene rubber (NBR) ), Butyl rubber (IIR), acrylonitrile-chloroprene rubber, acrylonitrile-isoprene rubber, styrene-chloroprene rubber, styrene-isoprene rubber and other diene rubbers, ethylene-propylene rubber, ethylene-butene rubber, ethylene-vinyl acetate copolymer (EVA) And ethylene-α-olefin copolymer rubbers such as ethylene-propylene-diene rubber (EPDM). These may be used alone or in combination.
(A)の加硫可能なゴムとしては、天然ゴム(NR)、イソプレンゴム(IR)、ブタジエンゴム(BR)、スチレン-ブタジエンゴム(SBR)、アクリロニトリル-ブタジエンゴム(NBR)、ブチルゴム(IIR)、アクリロニトリル-クロロプレンゴム、アクリロニトリル-イソプレンゴム、スチレン-クロロプレンゴム、スチレン-イソプレンゴムなどのジエン系ゴム、エチレン-プロピレンゴム、エチレン-ブテンゴム、エチレン-酢酸ビニル共重合体(EVA)、エチレン-プロピレン-ジエンゴム(EPDM)などのエチレン-α-オレフィン系共重合ゴムが挙げられる。これらを単独でもよいし複数組み合わせて使用してもよい。 Description of Component (A) As vulcanizable rubber of (A), natural rubber (NR), isoprene rubber (IR), butadiene rubber (BR), styrene-butadiene rubber (SBR), acrylonitrile-butadiene rubber (NBR) ), Butyl rubber (IIR), acrylonitrile-chloroprene rubber, acrylonitrile-isoprene rubber, styrene-chloroprene rubber, styrene-isoprene rubber and other diene rubbers, ethylene-propylene rubber, ethylene-butene rubber, ethylene-vinyl acetate copolymer (EVA) And ethylene-α-olefin copolymer rubbers such as ethylene-propylene-diene rubber (EPDM). These may be used alone or in combination.
好ましくは天然ゴム、イソプレンゴム、ブタジエンゴム、ブチルゴム、アクリロニトリル-ブタジエンゴム及びスチレン-ブタジエンゴムから選択されるゴム成分であり、特に好ましくは、(a-1)天然ゴム(NR)および(a-2)スチレン-ブタジエンゴム(SBR)の組合わせ、または(a-1)天然ゴムである。
Preferred are rubber components selected from natural rubber, isoprene rubber, butadiene rubber, butyl rubber, acrylonitrile-butadiene rubber and styrene-butadiene rubber, and particularly preferred are (a-1) natural rubber (NR) and (a-2 ) Styrene-butadiene rubber (SBR) combination or (a-1) natural rubber.
(a-2)スチレン-ブタジエンゴムは、水添加スチレン-ブタジエンゴムを用いる事が特に好ましい。また、(a-2)スチレン-ブタジエンゴムは、溶液重合により得られたスチレン-ブタジエンゴム(S-SBR)を用いる事が特に好ましい。S-SBR以外のSBRを用いると、収縮性の悪化や耐摩耗性の低下の問題が生ずるため好ましくない(あるいはS-SBRを用いると収縮性や耐摩耗性の点で優れた特性を示すため、使用が好ましい)。
(A-2) As the styrene-butadiene rubber, it is particularly preferable to use a water-added styrene-butadiene rubber. Further, as the (a-2) styrene-butadiene rubber, it is particularly preferable to use a styrene-butadiene rubber (S-SBR) obtained by solution polymerization. Use of SBR other than S-SBR is not preferable because it causes problems such as deterioration of shrinkage and deterioration of wear resistance (or use of S-SBR exhibits excellent characteristics in terms of shrinkage and wear resistance). Preferred to use).
(A)の配合量としては、全ゴム成分100重量部において、加硫可能なゴムを2~30重量部、好ましくは5~25重量部、特に好ましくは7~17重量部である。前記配合範囲内で配合すると、ロール加工性に必要な特性(シート肌の形状外観)や履物の諸物性(引張強度、伸び、引裂強度)を付与する事が出来る。ただし、配合量が2重量部より少ないとシート肌の荒れや加工性の問題が生じ、また逆に30重量部よりも多いと比重の上昇や収縮の悪化の問題が生ずるため好ましくない。
The blending amount of (A) is 2 to 30 parts by weight, preferably 5 to 25 parts by weight, particularly preferably 7 to 17 parts by weight of vulcanizable rubber in 100 parts by weight of all rubber components. When blended within the above blending range, properties necessary for roll processability (sheet skin shape appearance) and various physical properties of footwear (tensile strength, elongation, tear strength) can be imparted. However, if the blending amount is less than 2 parts by weight, problems such as roughness of the sheet and workability occur, and conversely if it exceeds 30 parts by weight, problems such as an increase in specific gravity and deterioration of shrinkage occur.
(A)の成分として、(a-1)天然ゴム(NR)と(a-2)スチレン-ブタジエンゴムを混合して用いる場合、(a-1)天然ゴム(NR)の配合量としては、全ゴム成分100重量部において、2~30重量部、好ましくは5~20重量部、特に好ましくは7~15重量部を配合するものが好ましい。この配合するとき、ロール加工性に必要な特性(シート肌の形状外観)や履物の諸物性(引張強度、伸び、引裂強度)を付与する事が出来る。
When (a-1) natural rubber (NR) and (a-2) styrene-butadiene rubber are mixed and used as the component (A), the blending amount of (a-1) natural rubber (NR) is: It is preferable that 2 to 30 parts by weight, preferably 5 to 20 parts by weight, particularly preferably 7 to 15 parts by weight are blended in 100 parts by weight of the total rubber component. When blended, properties necessary for roll processability (sheet skin shape appearance) and various physical properties of footwear (tensile strength, elongation, tear strength) can be imparted.
(a-2)溶液重合スチレンブタジエンゴム(S-SBR)の配合量としては、全ゴム分100重量部において、2~30重量部、好ましくは5~25重量部、特に好ましくは8~20重量部を配合するものが好ましい。この配合するとき、ロール加工性に必要な特性(シート肌の形状外観、粘度調整)を付与する事が出来る。
(A-2) The amount of the solution-polymerized styrene butadiene rubber (S-SBR) is 2 to 30 parts by weight, preferably 5 to 25 parts by weight, particularly preferably 8 to 20 parts by weight based on 100 parts by weight of the total rubber. What mix | blends a part is preferable. When blended, properties necessary for roll processability (sheet skin shape appearance, viscosity adjustment) can be imparted.
(a-1)天然ゴムの配合量と(a-2)S-SBRを混合して用いる際、配合量の比(a-1)/(a-2)は、0.07~15が良く、より好ましくは0.2~4、特に好ましくは0.35~1.88である。前述の比の範囲内では、収縮性及び耐摩耗性の点で優れた特性を示すため、使用が好ましい。
When the blending amount of (a-1) natural rubber and (a-2) S-SBR are used, the blending ratio (a-1) / (a-2) is preferably 0.07 to 15 More preferably, it is 0.2 to 4, particularly preferably 0.35 to 1.88. Use within the above-mentioned ratio range is preferable because it exhibits excellent properties in terms of shrinkage and wear resistance.
(B)成分の説明
(B)成分のビニル・シスブタジエンゴム(以下,VCRと略す)について説明する。VCRは沸騰n-ヘキサン不溶分;1~25重量%で,沸騰n-ヘキサン可溶分;99~75重量%であるビニル・シスブタジエンゴムであり,沸騰n-ヘキサン不溶分はシンジオタクチック1,2-ポリブタジエン樹脂(以下,SPBと略す)である。沸騰n-ヘキサン可溶分は高シス-1,4-ポリブタジエンであり,ミクロ構造はシス-1,4構造が90重量%以上である。ここで言う沸騰n-ヘキサン不溶分とはVCRを沸騰n-ヘキサン中に還流した時に不溶分として回収される部分をいい,沸騰n-ヘキサン可溶分とはVCRを沸騰n-ヘキサン中で還流した時に溶解する部分である。沸騰n-ヘキサン不溶分はオルトジクロロベンゼン溶液で測定した還元粘度(135℃,濃度0.20g/dlオルトジクロロベンゼン溶液)が0.5~4であり,好ましくは0.8~3の範囲である。沸騰n-ヘキサン不溶分の還元粘度が0.5より小さい時には,配合物のダイスウェルが十分改善されない。一方沸騰n-ヘキサン不溶分の還元粘度が4より大きい時には,重合時にSPBが高シス-1,4-ポリブタジエン中で凝集塊を形成するようになり分散不良を起こして加工性や耐久性が低下してこの発明の目的を達成できない。また、沸騰n-ヘキサン不溶分は融点が好ましくは170℃以上、特に好ましくは190~220℃のSPBである。SPBの融点が170℃未満であると、SPB補強性の低下により加工性や機械的強度の低下(引張応力や引張強度など)の問題が生ずるため好ましくない。また,沸騰n-ヘキサン可溶分の重量平均分子量は300,000~800,000の範囲であることが好ましく,300,000未満では加硫物の耐久性や反発弾性が低下するので好ましくない。800,000を超えると配合物ムーニー粘度が高くなりすぎて加工が困難になるので好ましくない。 (B) Explanation of Component The vinyl cis-butadiene rubber (hereinafter abbreviated as VCR) as the component (B) will be described. VCR is a boiling cis-butadiene rubber with a boiling n-hexane insoluble content of 1 to 25% by weight, a boiling n-hexane soluble content of 99 to 75% by weight, and a boiling n-hexane insoluble content is syndiotactic 1 , 2-polybutadiene resin (hereinafter abbreviated as SPB). The boiling n-hexane soluble content is high cis-1,4-polybutadiene, and the microstructure is 90% by weight or more of the cis-1,4 structure. The boiling n-hexane insoluble matter mentioned here means the part recovered as an insoluble matter when the VCR is refluxed in boiling n-hexane, and the boiling n-hexane soluble matter means that the VCR is refluxed in boiling n-hexane. It is the part that dissolves when The boiling n-hexane insoluble matter has a reduced viscosity (135 ° C, concentration 0.20 g / dl orthodichlorobenzene solution) measured with an orthodichlorobenzene solution of 0.5 to 4, preferably 0.8 to 3 is there. When the reduced viscosity of boiling n-hexane insolubles is less than 0.5, the die swell of the formulation is not improved sufficiently. On the other hand, when the reduced viscosity of boiling n-hexane insolubles is greater than 4, SPB will form agglomerates in high cis-1,4-polybutadiene during polymerization, resulting in poor dispersion and reduced workability and durability. Thus, the object of the present invention cannot be achieved. The boiling n-hexane insoluble matter is SPB having a melting point of preferably 170 ° C. or higher, particularly preferably 190 to 220 ° C. If the melting point of SPB is less than 170 ° C., problems such as deterioration in workability and mechanical strength (such as tensile stress and tensile strength) occur due to the decrease in SPB reinforcement. Further, the weight average molecular weight of the boiling n-hexane soluble part is preferably in the range of 300,000 to 800,000, and if it is less than 300,000, the durability and impact resilience of the vulcanizate are lowered, which is not preferred. Exceeding 800,000 is not preferable because the Mooney viscosity of the compound becomes too high and processing becomes difficult.
(B)成分のビニル・シスブタジエンゴム(以下,VCRと略す)について説明する。VCRは沸騰n-ヘキサン不溶分;1~25重量%で,沸騰n-ヘキサン可溶分;99~75重量%であるビニル・シスブタジエンゴムであり,沸騰n-ヘキサン不溶分はシンジオタクチック1,2-ポリブタジエン樹脂(以下,SPBと略す)である。沸騰n-ヘキサン可溶分は高シス-1,4-ポリブタジエンであり,ミクロ構造はシス-1,4構造が90重量%以上である。ここで言う沸騰n-ヘキサン不溶分とはVCRを沸騰n-ヘキサン中に還流した時に不溶分として回収される部分をいい,沸騰n-ヘキサン可溶分とはVCRを沸騰n-ヘキサン中で還流した時に溶解する部分である。沸騰n-ヘキサン不溶分はオルトジクロロベンゼン溶液で測定した還元粘度(135℃,濃度0.20g/dlオルトジクロロベンゼン溶液)が0.5~4であり,好ましくは0.8~3の範囲である。沸騰n-ヘキサン不溶分の還元粘度が0.5より小さい時には,配合物のダイスウェルが十分改善されない。一方沸騰n-ヘキサン不溶分の還元粘度が4より大きい時には,重合時にSPBが高シス-1,4-ポリブタジエン中で凝集塊を形成するようになり分散不良を起こして加工性や耐久性が低下してこの発明の目的を達成できない。また、沸騰n-ヘキサン不溶分は融点が好ましくは170℃以上、特に好ましくは190~220℃のSPBである。SPBの融点が170℃未満であると、SPB補強性の低下により加工性や機械的強度の低下(引張応力や引張強度など)の問題が生ずるため好ましくない。また,沸騰n-ヘキサン可溶分の重量平均分子量は300,000~800,000の範囲であることが好ましく,300,000未満では加硫物の耐久性や反発弾性が低下するので好ましくない。800,000を超えると配合物ムーニー粘度が高くなりすぎて加工が困難になるので好ましくない。 (B) Explanation of Component The vinyl cis-butadiene rubber (hereinafter abbreviated as VCR) as the component (B) will be described. VCR is a boiling cis-butadiene rubber with a boiling n-hexane insoluble content of 1 to 25% by weight, a boiling n-hexane soluble content of 99 to 75% by weight, and a boiling n-hexane insoluble content is syndiotactic 1 , 2-polybutadiene resin (hereinafter abbreviated as SPB). The boiling n-hexane soluble content is high cis-1,4-polybutadiene, and the microstructure is 90% by weight or more of the cis-1,4 structure. The boiling n-hexane insoluble matter mentioned here means the part recovered as an insoluble matter when the VCR is refluxed in boiling n-hexane, and the boiling n-hexane soluble matter means that the VCR is refluxed in boiling n-hexane. It is the part that dissolves when The boiling n-hexane insoluble matter has a reduced viscosity (135 ° C, concentration 0.20 g / dl orthodichlorobenzene solution) measured with an orthodichlorobenzene solution of 0.5 to 4, preferably 0.8 to 3 is there. When the reduced viscosity of boiling n-hexane insolubles is less than 0.5, the die swell of the formulation is not improved sufficiently. On the other hand, when the reduced viscosity of boiling n-hexane insolubles is greater than 4, SPB will form agglomerates in high cis-1,4-polybutadiene during polymerization, resulting in poor dispersion and reduced workability and durability. Thus, the object of the present invention cannot be achieved. The boiling n-hexane insoluble matter is SPB having a melting point of preferably 170 ° C. or higher, particularly preferably 190 to 220 ° C. If the melting point of SPB is less than 170 ° C., problems such as deterioration in workability and mechanical strength (such as tensile stress and tensile strength) occur due to the decrease in SPB reinforcement. Further, the weight average molecular weight of the boiling n-hexane soluble part is preferably in the range of 300,000 to 800,000, and if it is less than 300,000, the durability and impact resilience of the vulcanizate are lowered, which is not preferred. Exceeding 800,000 is not preferable because the Mooney viscosity of the compound becomes too high and processing becomes difficult.
上記のVCRの製造方法は例えば特公昭49-17666号公報,特公昭49-17667号公報,特公昭61-57858号公報,特公昭62-171号公報,特公昭63-36324号公報,特公平2-37927号公報,特公平2-38081号公報,特公平3-63566号公報などに記載された方法を用いることができる。但し,この発明に用いるVCRの製造方法は,これらの方法に限定されるものではない。
The above-mentioned VCR production methods are disclosed in, for example, JP-B-49-17666, JP-B-49-17667, JP-B-61-57858, JP-B-62-171, JP-B-63-36324, The methods described in JP-A-2-37927, JP-B-2-38081, JP-B-3-63566 and the like can be used. However, the manufacturing method of the VCR used in the present invention is not limited to these methods.
(B)成分のVCRの配合量としては、全ゴム成分100重量部において、40~80重量部であることが好ましく、50~70重量部であることがさらに好ましい。前記範囲内で配合すると、履物の諸物性(引張強度、伸び、引裂強度、耐摩耗性)を付与する事が出来る。ただし、配合量が40重量部より少ないと耐摩耗性の悪化の問題が生じ、また逆に80重量部よりも多いと比重の上昇や硬度の上昇の問題が生ずるため好ましくない。
The blending amount of the VCR of the component (B) is preferably 40 to 80 parts by weight, and more preferably 50 to 70 parts by weight with respect to 100 parts by weight of all rubber components. When blended within the above range, various physical properties of the footwear (tensile strength, elongation, tear strength, abrasion resistance) can be imparted. However, if the blending amount is less than 40 parts by weight, the problem of deterioration of wear resistance occurs, and conversely if it exceeds 80 parts by weight, problems such as an increase in specific gravity and an increase in hardness occur.
(C)成分の説明
(C)熱可塑性樹脂としては、例えば、シンジオタクチック1,2-ポリブタジエン樹脂(SPB)、ポリエチレン及びその無水マレイン酸グラフト重合体、ポリイソブチレン、エチレン酢ビ共重合体、エチレンアクリレート、エチレンアクリル酸共重合体、ポリプロピレン及びその無水マレイン酸グラフト重合体、塩素化ポリプロピレン、4-メチルペンテン-1樹脂、ポリスチレン、ABS樹脂、AS樹脂、アクリル樹脂、メタクリル樹脂、塩化ビニル樹脂、塩化ビニリデン樹脂、ポリアミド樹脂、ポリカーボネート等を挙げることができる。これらの熱可塑性樹脂(C)は、単独でも2種以上組み合わせて用いることもできる。 (C) Explanation of Component (C) Examples of the thermoplastic resin include syndiotactic 1,2-polybutadiene resin (SPB), polyethylene and its maleic anhydride graft polymer, polyisobutylene, ethylene vinyl acetate copolymer, Ethylene acrylate, ethylene acrylic acid copolymer, polypropylene and its maleic anhydride graft polymer, chlorinated polypropylene, 4-methylpentene-1 resin, polystyrene, ABS resin, AS resin, acrylic resin, methacrylic resin, vinyl chloride resin, Examples include vinylidene chloride resin, polyamide resin, and polycarbonate. These thermoplastic resins (C) can be used alone or in combination of two or more.
(C)熱可塑性樹脂としては、例えば、シンジオタクチック1,2-ポリブタジエン樹脂(SPB)、ポリエチレン及びその無水マレイン酸グラフト重合体、ポリイソブチレン、エチレン酢ビ共重合体、エチレンアクリレート、エチレンアクリル酸共重合体、ポリプロピレン及びその無水マレイン酸グラフト重合体、塩素化ポリプロピレン、4-メチルペンテン-1樹脂、ポリスチレン、ABS樹脂、AS樹脂、アクリル樹脂、メタクリル樹脂、塩化ビニル樹脂、塩化ビニリデン樹脂、ポリアミド樹脂、ポリカーボネート等を挙げることができる。これらの熱可塑性樹脂(C)は、単独でも2種以上組み合わせて用いることもできる。 (C) Explanation of Component (C) Examples of the thermoplastic resin include syndiotactic 1,2-polybutadiene resin (SPB), polyethylene and its maleic anhydride graft polymer, polyisobutylene, ethylene vinyl acetate copolymer, Ethylene acrylate, ethylene acrylic acid copolymer, polypropylene and its maleic anhydride graft polymer, chlorinated polypropylene, 4-methylpentene-1 resin, polystyrene, ABS resin, AS resin, acrylic resin, methacrylic resin, vinyl chloride resin, Examples include vinylidene chloride resin, polyamide resin, and polycarbonate. These thermoplastic resins (C) can be used alone or in combination of two or more.
(C)熱可塑性樹脂としては、ポリオレフィン系樹脂が好ましい。ポリオレフィン系樹脂としては、例えば、ポリエチレン(PE)、特に低密度PE(LDPE)、中密度PE(MDPE)、高密度PE(HDPE)、及び線状低密度PE(LLDPE)、エチレン-プロピレンエラストマーコポリマー(EPM)、エチレン-プロピレン-ジエンゴム(EPDM)、エチレン-酢酸ビニル共重合体(EVA)などのエチレン-ビニルエステルコポリマー、エチレン-アクリレートコポリマー、並びにエチレン-α-オレフィン熱可塑性コポリマー、さらにプロピレン、ブテン-1、及びペンテン-1などのコポリマーが挙げられる。これらのポリオレフィン系樹脂は、単独でも2種以上組み合わせて用いることもできる。
(C) The thermoplastic resin is preferably a polyolefin resin. Examples of polyolefin resins include polyethylene (PE), particularly low density PE (LDPE), medium density PE (MDPE), high density PE (HDPE), and linear low density PE (LLDPE), ethylene-propylene elastomer copolymer. (EPM), ethylene-propylene-diene rubber (EPDM), ethylene-vinyl ester copolymers such as ethylene-vinyl acetate copolymer (EVA), ethylene-acrylate copolymers, and ethylene-α-olefin thermoplastic copolymers, as well as propylene, butene -1, and copolymers such as pentene-1. These polyolefin resins can be used alone or in combination of two or more.
(C)熱可塑性樹脂として、特に好ましいものとして、融点が70℃~110℃である1,2-ポリブタジエン樹脂、及びエチレン-酢酸ビニル共重合体(EVA)が挙げられる。
(C) Particularly preferable examples of the thermoplastic resin include 1,2-polybutadiene resins having a melting point of 70 ° C. to 110 ° C., and ethylene-vinyl acetate copolymer (EVA).
(C)熱可塑性樹脂の配合量としては、全ゴム成分100重量部において、10~35重量部であることが好ましく、15~30重量部であることがさらに好ましく、18~28重量部であることが特に好ましい。この範囲内で配合をするとき、履物の諸物性(ガスバリアー性、寸法安定性)を付与する事が出来る。一方、配合量が10重量部より少ないとガスバリアー性の低下による比重上昇や寸法安定性(収縮)の問題が生じ、また逆に35重量部よりも多いと硬度の上昇の問題が生ずるため好ましくない。
The blending amount of the thermoplastic resin (C) is preferably 10 to 35 parts by weight, more preferably 15 to 30 parts by weight, and more preferably 18 to 28 parts by weight based on 100 parts by weight of the total rubber component. It is particularly preferred. When blending within this range, various physical properties (gas barrier properties, dimensional stability) of footwear can be imparted. On the other hand, if the blending amount is less than 10 parts by weight, problems such as an increase in specific gravity and dimensional stability (shrinkage) due to a decrease in gas barrier properties occur, and conversely, if it exceeds 35 parts by weight, a problem of an increase in hardness will occur. Absent.
ここに全ゴム成分とは、(A)+(B)+(C)を意味する。
Here, the total rubber component means (A) + (B) + (C).
(D1)熱膨張型発泡剤(物理発泡剤)
熱膨張型発泡剤としては、熱によって膨張する発泡剤であれば特に制限されないが、熱膨張性のマイクロカプセルを用いることが好ましい。熱膨張性マイクロカプセルとは、合成樹脂カプセルの中に、加熱することにより膨張する液体や気体を内包させたものであり、押出成形や射出成形の際のスクリューなどによる混練溶融熱で内包された液体や気体が膨張することにより外殻となるマイクロカプセルを膨張させるが、成形時の温度条件によっては溶融し、破裂することなく成形が完了するものを用いる。 (D1) Thermal expansion type foaming agent (physical foaming agent)
The thermal expansion type foaming agent is not particularly limited as long as it is a foaming agent that expands by heat, but it is preferable to use thermally expandable microcapsules. A heat-expandable microcapsule is an encapsulated liquid or gas that expands when heated in a synthetic resin capsule, and is encapsulated by heat of kneading and melting by a screw or the like during extrusion molding or injection molding. The microcapsules that form the outer shell are expanded by the expansion of the liquid or gas. However, depending on the temperature conditions during molding, a microcapsule that melts and completes without bursting is used.
熱膨張型発泡剤としては、熱によって膨張する発泡剤であれば特に制限されないが、熱膨張性のマイクロカプセルを用いることが好ましい。熱膨張性マイクロカプセルとは、合成樹脂カプセルの中に、加熱することにより膨張する液体や気体を内包させたものであり、押出成形や射出成形の際のスクリューなどによる混練溶融熱で内包された液体や気体が膨張することにより外殻となるマイクロカプセルを膨張させるが、成形時の温度条件によっては溶融し、破裂することなく成形が完了するものを用いる。 (D1) Thermal expansion type foaming agent (physical foaming agent)
The thermal expansion type foaming agent is not particularly limited as long as it is a foaming agent that expands by heat, but it is preferable to use thermally expandable microcapsules. A heat-expandable microcapsule is an encapsulated liquid or gas that expands when heated in a synthetic resin capsule, and is encapsulated by heat of kneading and melting by a screw or the like during extrusion molding or injection molding. The microcapsules that form the outer shell are expanded by the expansion of the liquid or gas. However, depending on the temperature conditions during molding, a microcapsule that melts and completes without bursting is used.
マイクロカプセルの素材としては、アクリルニトリルをモノマー成分の一つとした共重合体が用いられ、アクリルニトリルと共重合しても良い他のモノマー成分として、例えばアクリル酸、メタクリル酸、アクリル酸エステル、メタクリル酸エステル、スチレン、酢酸ビニル、及び塩化ビニリデンなどを挙げることができるが、これらに限定するものでない。
As a material of the microcapsule, a copolymer having acrylonitrile as one of monomer components is used, and other monomer components that may be copolymerized with acrylonitrile include, for example, acrylic acid, methacrylic acid, acrylate ester, methacrylic ester, and the like. Examples thereof include, but are not limited to, acid esters, styrene, vinyl acetate, and vinylidene chloride.
マイクロカプセルに内包する液体または気体としては、マイクロカプセルの軟化点以下の温度でガスになって膨張するもので、例えば、プロパン、プロピレン、ブテン、ノルマルブタン、イソブタン、イソペンタン、ネオペンタン、ノルマルペンタン、ヘキサン、ヘプタン、石油エーテル、塩化メチルやメチレンクロリドなどのメタンのハロゲン化物、CCl3FやCCl2F2などのクロロフルオロカーボン、及びテトラメチルシランやトリメチルエチルシランなどのテトラアルキルシランなどの低沸点液体のほか、加熱により熱分解してガス状になるAIBNなどの化合物を挙げることができる。
The liquid or gas encapsulated in the microcapsule is one that expands as a gas at a temperature below the softening point of the microcapsule. For example, propane, propylene, butene, normal butane, isobutane, isopentane, neopentane, normal pentane, hexane Low boiling point liquids such as methane halides such as heptane, petroleum ether, methyl chloride and methylene chloride, chlorofluorocarbons such as CCl 3 F and CCl 2 F 2 , and tetraalkylsilanes such as tetramethylsilane and trimethylethylsilane In addition, a compound such as AIBN that is thermally decomposed by heating and becomes gaseous can be mentioned.
また、押出成形や射出成形では、押出機や射出機によってホッパーから投入された原料がスクリューなどで混練されることから、熱膨張性マイクロカプセルにも混練による剪断力などが加わるとともに、ダイや金型に押出されるときに押出力や射出力が加わることから、これらの力が加わってもマイクロカプセルが壊れないものである必要があり、大きさなどが選定される。
In addition, in extrusion molding and injection molding, since the raw material introduced from the hopper by the extruder or the injection machine is kneaded by a screw or the like, shearing force due to the kneading is applied to the thermally expandable microcapsule, and the die or metal Since a pushing force and a radiant force are applied when being extruded into the mold, the microcapsule needs to be unbreakable even when these forces are applied, and the size is selected.
したがって、このような押出成形または射出成形によって成形される発泡成形体では、膨張した後の熱膨張性マイクロカプセルの数(混合量)と粒径によって気泡の量と大きさが定まり、これらから成形体の比重が定まることから、熱可塑性樹脂原料に混合する未膨張の熱膨張性マイクロカプセルの数(混合量)と成形途中の加熱量による膨張後の熱膨張性マイクロカプセルの粒径の予測によって発泡状態のコントロールが可能となり、従来のガスを直接膨張させる場合に比べてガスだけが外部に逃げることもなく、簡単に目的の比重にすることができる。
Therefore, in the foamed molded product formed by such extrusion molding or injection molding, the amount and size of the bubbles are determined by the number (mixing amount) and the particle size of the thermally expandable microcapsules after expansion, and the molding is performed from these. Since the specific gravity of the body is determined, by predicting the number of unexpanded thermally expandable microcapsules (mixed amount) to be mixed with the thermoplastic resin raw material and the particle size of the thermally expandable microcapsules after expansion by the heating amount during molding The foaming state can be controlled, and only the gas does not escape to the outside as compared with the case where the conventional gas is directly expanded, and the target specific gravity can be easily achieved.
このように押出成形または射出成形によって、熱可塑性樹脂中に熱膨張性マイクロカプセルが膨張されて包含された状態で発泡体を成形することができ、各気泡はマイクロカプセルで包まれた状態で安定しており、押出成形の場合で、ダイから押出されて圧力が開放された状態でもガスが直接成形品の外部に抜けることもなく、表面外観の良好な発泡体を得ることができる。
In this way, a foam can be formed in a state where the thermally expandable microcapsule is expanded and contained in the thermoplastic resin by extrusion molding or injection molding, and each bubble is stable in a state of being encapsulated in the microcapsule. In the case of extrusion molding, even when the pressure is released by being extruded from a die, the gas does not escape directly to the outside of the molded product, and a foam having a good surface appearance can be obtained.
また、市販の熱膨張性マイクロカプセル(たとえばEXPANCEL社製、EXPANCEL920DU40、EXPANCEL920DU80、EXPANCEL009DU80、前記EXPANCEL920DU40のマスターバッチ化品920MB40及び920MB50、及び松本油脂製薬製マツモトマイクロスフェアー F-105Dなど)を使用することができる。ただし、熱膨張性マイクロカプセルの粒径が8~30μmであることが好ましく、8~17μmであることがより好ましく、10~16μmであることがさらに好ましい。当該範囲では、低比重化と成形品の表面肌の平滑性の効果を期待できる。上記範囲より小さいマイクロカプセルを使用すると、低比重化を図る効果が少なくなるという問題が生ずる、一方、上記範囲より大きいマイクロカプセルを使用すると、成形品の表面肌の荒れ(平滑性)の問題が生ずる。
Also, commercially available heat-expandable microcapsules (for example, EXPANCEL 920DU40, EXPANCEL 920DU80, EXPANCEL009DU80, EXPANCEL920DU40 master batch products 920MB40 and 920MB50, Matsumoto Yushi Pharmaceutical Matsumoto Microsphere F-D, etc.) are used. Can do. However, the particle size of the thermally expandable microcapsule is preferably 8 to 30 μm, more preferably 8 to 17 μm, and even more preferably 10 to 16 μm. In the said range, the effect of low specific gravity and the smoothness of the surface skin of a molded article can be expected. If a microcapsule smaller than the above range is used, there is a problem that the effect of reducing the specific gravity is reduced. On the other hand, if a microcapsule larger than the above range is used, there is a problem of surface roughness (smoothness) of the molded product. Arise.
熱膨張型発泡剤の配合量としては、全ゴム成分100重量部において、0.5~3.0重量部、好ましくは0.75~3.0重量部、より好ましくは0.80~2.5重量部、さらに好ましくは0.85~2.5重量部、よりさらに好ましくは0.90~2.0重量部、特に好ましくは1.2~2.0重量部である。上記配合範囲では、加硫初期の発泡圧力上昇に伴いエアーの除去や成形品外観(表面状態の平滑性や寸法安定性)、低比重化に伴う諸物性(引張強度、破断伸びや引裂強度)の低下幅を抑える事が出来る。一方、配合量が0.5重量部より少ないと、低比重化を図る効果が少なくなるという問題が生じ、逆に3.0重量部より多いと成形品の表面肌の荒れ(平滑性)の問題が生ずる。
The compounding amount of the thermal expansion foaming agent is 0.5 to 3.0 parts by weight, preferably 0.75 to 3.0 parts by weight, more preferably 0.80 to 2. It is 5 parts by weight, more preferably 0.85 to 2.5 parts by weight, still more preferably 0.90 to 2.0 parts by weight, and particularly preferably 1.2 to 2.0 parts by weight. In the above blending range, air removal, appearance of molded products (surface state smoothness and dimensional stability) with increasing foaming pressure in the initial stage of vulcanization, and various physical properties (tensile strength, elongation at break and tear strength) associated with lower specific gravity. Can be suppressed. On the other hand, if the blending amount is less than 0.5 parts by weight, there is a problem that the effect of lowering the specific gravity is reduced. Conversely, if the blending amount is more than 3.0 parts by weight, the surface roughness of the molded product (smoothness) is reduced. Problems arise.
(D2)熱分解型発泡剤(化学発泡剤)
熱分解性発泡剤としては有機系発泡剤、例えば、アゾシカルボンアミド系複合体(ADCA)、アゾビスイソブチロニトリル(AIBN)、ジニトロソペンタメチレンテトラミン(DPT)、N、N’-ジメチル-N、N’-ジニトロソテレフタルアミド、ベンゼンスルホニルヒドラジド(BSH)、p-トルエンスルホニルヒドラジド(TSH)、4,4’-オキシビス(ベンゼンスルホニルヒドラジド)(OBSH)、3,3’-ジスルホンヒドラジドジフェニルスルホン、トルエンジスルホニルヒドラジン、p-トルエンジスルホニルヒドラジド、p-トルエンスルホニルセミ力ルバジド、ジエチルアゾジカルボキシレート等を用いることができる。 (D2) Pyrolytic foaming agent (chemical foaming agent)
Examples of the thermally decomposable foaming agent include organic foaming agents such as azocicarbonamide complex (ADCA), azobisisobutyronitrile (AIBN), dinitrosopentamethylenetetramine (DPT), N, N′-dimethyl. -N, N'-dinitrosotephthalamide, benzenesulfonyl hydrazide (BSH), p-toluenesulfonyl hydrazide (TSH), 4,4'-oxybis (benzenesulfonyl hydrazide) (OBSH), 3,3'-disulfone hydrazide diphenyl Sulfone, toluene disulfonyl hydrazine, p-toluene disulfonyl hydrazide, p-toluene sulfonyl semi strength rubazide, diethyl azodicarboxylate and the like can be used.
熱分解性発泡剤としては有機系発泡剤、例えば、アゾシカルボンアミド系複合体(ADCA)、アゾビスイソブチロニトリル(AIBN)、ジニトロソペンタメチレンテトラミン(DPT)、N、N’-ジメチル-N、N’-ジニトロソテレフタルアミド、ベンゼンスルホニルヒドラジド(BSH)、p-トルエンスルホニルヒドラジド(TSH)、4,4’-オキシビス(ベンゼンスルホニルヒドラジド)(OBSH)、3,3’-ジスルホンヒドラジドジフェニルスルホン、トルエンジスルホニルヒドラジン、p-トルエンジスルホニルヒドラジド、p-トルエンスルホニルセミ力ルバジド、ジエチルアゾジカルボキシレート等を用いることができる。 (D2) Pyrolytic foaming agent (chemical foaming agent)
Examples of the thermally decomposable foaming agent include organic foaming agents such as azocicarbonamide complex (ADCA), azobisisobutyronitrile (AIBN), dinitrosopentamethylenetetramine (DPT), N, N′-dimethyl. -N, N'-dinitrosotephthalamide, benzenesulfonyl hydrazide (BSH), p-toluenesulfonyl hydrazide (TSH), 4,4'-oxybis (benzenesulfonyl hydrazide) (OBSH), 3,3'-disulfone hydrazide diphenyl Sulfone, toluene disulfonyl hydrazine, p-toluene disulfonyl hydrazide, p-toluene sulfonyl semi strength rubazide, diethyl azodicarboxylate and the like can be used.
さらに、熱分解性発泡剤としては無機系発泡剤を用いることもできる。例えば、重炭酸ナトリウム、重炭酸アンモニウムを用いることもできる。
Furthermore, an inorganic foaming agent can also be used as the thermally decomposable foaming agent. For example, sodium bicarbonate or ammonium bicarbonate can be used.
これら熱分解性発泡剤を単独で使用しても良いし、複数組み合わせて使用しても良い。
These thermal decomposable foaming agents may be used alone or in combination.
熱分解性発泡剤として使用に好ましいのは、成形加工上、分解温度が195~210℃くらいのものであり、かつ発生ガス量が190~240(ml/g)の有機系発泡剤である。特にアゾジカルボンアミド系複合体(ADCA)または4,4’-オキシビス(ベンゼンスルホニルヒドラジド)(OBSH)が好ましい。
An organic foaming agent having a decomposition temperature of about 195 to 210 ° C. and a generated gas amount of 190 to 240 (ml / g) is preferable for use as a thermally decomposable foaming agent. In particular, azodicarbonamide complex (ADCA) or 4,4′-oxybis (benzenesulfonylhydrazide) (OBSH) is preferable.
上述した特性の熱分解性発泡剤を使用すると、併用する(D1)熱膨張型発泡剤との発泡効果が最大限に発揮でき、加硫初期の発泡圧力上昇に伴うエアーを完全に除去できるだけでなく、成形品外観(表面状態の平滑性や寸法安定性)、低比重化に伴う諸物性(引張強度、破断伸びや引裂強度)の低下幅を抑える事が出来る。
When the thermal decomposable foaming agent having the above-described characteristics is used, the foaming effect with the combined thermal expansion foaming agent (D1) can be maximized, and the air accompanying the increase in foaming pressure at the initial stage of vulcanization can be completely removed. In addition, it is possible to suppress a decrease in appearance of a molded product (smoothness and dimensional stability of the surface state) and various physical properties (tensile strength, elongation at break and tear strength) associated with a decrease in specific gravity.
熱分解性発泡剤の配合量としては、全ゴム成分100重量部において、0.5~7.0重量部、好ましくは0.75~7.0重量部、より好ましくは0.8~7.0重量部、さらに好ましくは0.85~7.0重量部、よりさらに好ましくは0.85~6.0重量部、特に好ましくは1.0~4.5重量部である。上記配合範囲では、加硫初期の発泡圧力上昇に伴いエアーの除去や成形品外観(表面状態の平滑性や寸法安定性)、低比重化に伴う諸物性(引張強度、破断伸びや引裂強度)の低下幅を抑える事が出来る。一方、配合量が0.5重量部より少ないと、低比重化と低硬度を図る効果が少なくなるという問題が生じ、逆に7.0重量部より多いと高発泡に伴う寸法安定性の低下の問題が生ずる。
The compounding amount of the thermally decomposable foaming agent is 0.5 to 7.0 parts by weight, preferably 0.75 to 7.0 parts by weight, more preferably 0.8 to 7. 0 parts by weight, more preferably 0.85 to 7.0 parts by weight, still more preferably 0.85 to 6.0 parts by weight, and particularly preferably 1.0 to 4.5 parts by weight. In the above blending range, air removal, appearance of molded products (surface state smoothness and dimensional stability) with increasing foaming pressure in the initial stage of vulcanization, and various physical properties (tensile strength, elongation at break and tear strength) associated with lower specific gravity. Can be suppressed. On the other hand, if the blending amount is less than 0.5 parts by weight, there is a problem that the effect of reducing the specific gravity and the hardness is reduced. Conversely, if the blending amount is more than 7.0 parts by weight, the dimensional stability is reduced due to high foaming. Problem arises.
(E)ゴム補強剤
本発明に係るゴム組成物に配合されるゴム補強剤(E)としては、各種のカーボンブラック、ホワイトカーボン、シリカ、活性化炭酸カルシウム、及び超微粒子珪酸マグネシウムなどが挙げられる。これらの中ではシリカ及び又はカーボンブラックが好ましい。中でも乾式法による無水ケイ酸、湿式法による含水ケイ酸、合成ケイ酸塩等の平均一次粒径5~100nmのシリカ、及び粒子径が90nm以下、ジブチルフタレート(DBP)給油量が70ml/100g以上のカーボンブラックが好ましい。カーボンブラックとしてはファーネスブラック、チャンネルブラック、サーマルブラック等が挙げられ、より具体的には、ASTMコードNo.110、212、N242、S315、N330、N550、N660、N765等が挙げられる。 (E) Rubber Reinforcing Agent Examples of the rubber reinforcing agent (E) blended in the rubber composition according to the present invention include various types of carbon black, white carbon, silica, activated calcium carbonate, and ultrafine magnesium silicate. . Of these, silica and / or carbon black are preferred. Above all, silica having an average primary particle size of 5 to 100 nm, such as silicic anhydride by a dry method, hydrous silicic acid by a wet method, and synthetic silicate, and a particle size of 90 nm or less and a dibutyl phthalate (DBP) oil supply amount of 70 ml / 100 g or more. Carbon black is preferred. Examples of carbon black include furnace black, channel black, thermal black, and the like. 110, 212, N242, S315, N330, N550, N660, N765 and the like.
本発明に係るゴム組成物に配合されるゴム補強剤(E)としては、各種のカーボンブラック、ホワイトカーボン、シリカ、活性化炭酸カルシウム、及び超微粒子珪酸マグネシウムなどが挙げられる。これらの中ではシリカ及び又はカーボンブラックが好ましい。中でも乾式法による無水ケイ酸、湿式法による含水ケイ酸、合成ケイ酸塩等の平均一次粒径5~100nmのシリカ、及び粒子径が90nm以下、ジブチルフタレート(DBP)給油量が70ml/100g以上のカーボンブラックが好ましい。カーボンブラックとしてはファーネスブラック、チャンネルブラック、サーマルブラック等が挙げられ、より具体的には、ASTMコードNo.110、212、N242、S315、N330、N550、N660、N765等が挙げられる。 (E) Rubber Reinforcing Agent Examples of the rubber reinforcing agent (E) blended in the rubber composition according to the present invention include various types of carbon black, white carbon, silica, activated calcium carbonate, and ultrafine magnesium silicate. . Of these, silica and / or carbon black are preferred. Above all, silica having an average primary particle size of 5 to 100 nm, such as silicic anhydride by a dry method, hydrous silicic acid by a wet method, and synthetic silicate, and a particle size of 90 nm or less and a dibutyl phthalate (DBP) oil supply amount of 70 ml / 100 g or more. Carbon black is preferred. Examples of carbon black include furnace black, channel black, thermal black, and the like. 110, 212, N242, S315, N330, N550, N660, N765 and the like.
シリカは、湿式シリカ及び乾式シリカのどちらでも用いる事ができ、併用も可能である。さらにシリカは単独で用いる事も出来る。また、使用するシリカは、シリカ純度が97%以上であるのが好ましい。また、シリカの純度が97%以上の天然石英を粉砕したものも用いることができる。さらに、シラン等の有機珪素化合物を加水分解して得られた粒子も好ましく用いられる。純度97%未満のシリカはゴムの補強効果が充分ではないので、このようなシリカをブレンドしたゴム組成物は耐摩耗性が充分ではない。
Silica can be either wet silica or dry silica, and can be used in combination. Further, silica can be used alone. Moreover, it is preferable that the silica to be used has a silica purity of 97% or more. Further, pulverized natural quartz having a silica purity of 97% or more can be used. Furthermore, particles obtained by hydrolyzing an organosilicon compound such as silane are also preferably used. Silica having a purity of less than 97% does not have a sufficient rubber reinforcing effect, so that a rubber composition blended with such silica does not have sufficient wear resistance.
シリカ粒子の平均粒径は、好ましくは0.1~50μm、さらに好ましくは1~50μm、特に好ましくは5~50μmの範囲である。かかるシリカ粒径は、例えば完全溶融した石英ガラスを然るべき粒径になるように粉砕するなどの方法により得る事が出来る。一方、平均粒径が0.1μmより小さいとゴム内に混入しづらく、ロスや分散不良の問題を生じ、逆に50μmより大きいと補強効果が低いという問題を生ずるため好ましくない。
The average particle diameter of the silica particles is preferably in the range of 0.1 to 50 μm, more preferably 1 to 50 μm, and particularly preferably 5 to 50 μm. Such a silica particle size can be obtained, for example, by a method of pulverizing a completely melted quartz glass so as to have an appropriate particle size. On the other hand, if the average particle size is smaller than 0.1 μm, it is difficult to mix in the rubber, causing problems such as loss and poor dispersion, and conversely if larger than 50 μm, the reinforcing effect is low, which is not preferable.
使用されるゴム補強剤の量は、全ゴム成分100重量部において、10~30重量部、好ましくは12~25重量部、より好ましくは15~20重量部である。前述の範囲内では、補強効果が充分得られ、耐摩耗性向上の効果を得る事が出来る。一方、10重量部より少ない量では、補強効果が低く、諸物性の低下(機械的強度や耐摩耗性)の問題を生じ、逆に30重量部より多いと粘度上昇に伴う加工性の低下の問題を生ずるため好ましくない。
The amount of the rubber reinforcing agent used is 10 to 30 parts by weight, preferably 12 to 25 parts by weight, more preferably 15 to 20 parts by weight based on 100 parts by weight of the total rubber components. Within the above-mentioned range, a sufficient reinforcing effect can be obtained, and an effect of improving wear resistance can be obtained. On the other hand, if the amount is less than 10 parts by weight, the reinforcing effect is low, causing problems of deterioration in various physical properties (mechanical strength and wear resistance). Conversely, if the amount is more than 30 parts by weight, the workability is lowered due to the increase in viscosity. This is not preferable because it causes problems.
(F)加硫促進剤
本発明で用いられる加硫促進剤としては、例えばアルデヒド類、アンモニア類、アミン類、グアニジン類、チオウレア類、チアゾール類、チウラム類、ジチオカーバメイト類、キサンテート類等が挙げられ、より具体的には、テトラメチルチウラムジスルフィド(TMTD)、N-オキシジエチレン-2-ベンゾチアゾリルスルフェンアミド(OBS)、N-シクロヘキシル-2-ベンゾチアジルスルフェンアミド(CBS)、ジベンゾチアジルスルフィド(MBTS)、ジンクジ-n-ブチルジチオカーバイド(ZnBDC)、ジンクジメチルジチオカーバイド(ZnMDC)等が挙げられる。この中でも特に、ジベンゾチアジルスルフィド(MBTS)、ジオルトトリルグアニジン(DOTG)が好ましい。 (F) Vulcanization accelerator Examples of the vulcanization accelerator used in the present invention include aldehydes, ammonia, amines, guanidines, thioureas, thiazoles, thiurams, dithiocarbamates, xanthates and the like. More specifically, tetramethylthiuram disulfide (TMTD), N-oxydiethylene-2-benzothiazolylsulfenamide (OBS), N-cyclohexyl-2-benzothiazylsulfenamide (CBS), dibenzo Thiazyl sulfide (MBTS), zinc di-n-butyldithiocarbide (ZnBDC), zinc dimethyldithiocarbide (ZnMDC) and the like can be mentioned. Of these, dibenzothiazyl sulfide (MBTS) and diortolyl guanidine (DOTG) are particularly preferable.
本発明で用いられる加硫促進剤としては、例えばアルデヒド類、アンモニア類、アミン類、グアニジン類、チオウレア類、チアゾール類、チウラム類、ジチオカーバメイト類、キサンテート類等が挙げられ、より具体的には、テトラメチルチウラムジスルフィド(TMTD)、N-オキシジエチレン-2-ベンゾチアゾリルスルフェンアミド(OBS)、N-シクロヘキシル-2-ベンゾチアジルスルフェンアミド(CBS)、ジベンゾチアジルスルフィド(MBTS)、ジンクジ-n-ブチルジチオカーバイド(ZnBDC)、ジンクジメチルジチオカーバイド(ZnMDC)等が挙げられる。この中でも特に、ジベンゾチアジルスルフィド(MBTS)、ジオルトトリルグアニジン(DOTG)が好ましい。 (F) Vulcanization accelerator Examples of the vulcanization accelerator used in the present invention include aldehydes, ammonia, amines, guanidines, thioureas, thiazoles, thiurams, dithiocarbamates, xanthates and the like. More specifically, tetramethylthiuram disulfide (TMTD), N-oxydiethylene-2-benzothiazolylsulfenamide (OBS), N-cyclohexyl-2-benzothiazylsulfenamide (CBS), dibenzo Thiazyl sulfide (MBTS), zinc di-n-butyldithiocarbide (ZnBDC), zinc dimethyldithiocarbide (ZnMDC) and the like can be mentioned. Of these, dibenzothiazyl sulfide (MBTS) and diortolyl guanidine (DOTG) are particularly preferable.
加硫促進剤の量としては、全ゴム成分100重量部において、0.5~1.5重量部、好ましくは0.7~1.3重量部、より好ましくは0.8~1.2重量部である。前述の範囲内では、加硫効果が充分得る事が出来る。一方、0.5重量部より少ない量では、加硫時間の遅延や架橋密度の低下による諸物性の低下の問題を生じ、逆に1.5重量部より多いと架橋先行による発泡の阻害の問題を生ずるため好ましくない。
The amount of the vulcanization accelerator is 0.5 to 1.5 parts by weight, preferably 0.7 to 1.3 parts by weight, more preferably 0.8 to 1.2 parts by weight based on 100 parts by weight of the total rubber component. Part. Within the above range, a sufficient vulcanization effect can be obtained. On the other hand, if the amount is less than 0.5 parts by weight, problems such as a delay in vulcanization time and various physical properties due to a decrease in crosslink density are caused. This is not preferable.
(G)加硫剤
また、本願発明で用いる加硫剤としては、硫黄、加熱により硫黄を生成させる化合物、有機過酸化物、酸化マグネシウム等の金属酸化物、多官能性モノマー、及びシラノール化合物等が挙げられる。加熱により硫黄を生成させる化合物として、テトラメチルチウラムジスルフィド、及びテトラエチルチウラムジスルフィド等があげられる。これらの中でも特に硫黄が好ましい。 (G) Vulcanizing agent Further, as the vulcanizing agent used in the present invention, sulfur, a compound that generates sulfur by heating, an organic peroxide, a metal oxide such as magnesium oxide, a polyfunctional monomer, a silanol compound, and the like Is mentioned. Examples of the compound that generates sulfur by heating include tetramethyl thiuram disulfide and tetraethyl thiuram disulfide. Among these, sulfur is particularly preferable.
また、本願発明で用いる加硫剤としては、硫黄、加熱により硫黄を生成させる化合物、有機過酸化物、酸化マグネシウム等の金属酸化物、多官能性モノマー、及びシラノール化合物等が挙げられる。加熱により硫黄を生成させる化合物として、テトラメチルチウラムジスルフィド、及びテトラエチルチウラムジスルフィド等があげられる。これらの中でも特に硫黄が好ましい。 (G) Vulcanizing agent Further, as the vulcanizing agent used in the present invention, sulfur, a compound that generates sulfur by heating, an organic peroxide, a metal oxide such as magnesium oxide, a polyfunctional monomer, a silanol compound, and the like Is mentioned. Examples of the compound that generates sulfur by heating include tetramethyl thiuram disulfide and tetraethyl thiuram disulfide. Among these, sulfur is particularly preferable.
加硫剤の量としては、全ゴム成分100重量部において、0.3~2.2重量部、好ましくは0.5~1.7重量部、より好ましくは1.0~1.5重量部である。前述の範囲内では、加硫効果が充分得る事が出来る。一方、0.3重量部より少ない量では、加硫時間の遅延や架橋密度の低下による諸物性の低下の問題を生じ、逆に2.2重量部より多いと架橋先行による発泡の阻害の問題を生ずるため好ましくない。
The amount of the vulcanizing agent is 0.3 to 2.2 parts by weight, preferably 0.5 to 1.7 parts by weight, more preferably 1.0 to 1.5 parts by weight, based on 100 parts by weight of all rubber components. It is. Within the above range, a sufficient vulcanization effect can be obtained. On the other hand, if the amount is less than 0.3 parts by weight, problems such as a delay in vulcanization time and a decrease in physical properties due to a decrease in crosslink density are caused. This is not preferable.
(H)その他添加剤
本発明の目的が損なわれない範囲で、所望により通常、ゴム工業界で用いられる各種薬品、例えば老化防止剤、加工助剤、プロセスオイル、スコーチ防止剤、亜鉛華、ステアリン酸などを含有させる事が出来る。 (H) Other additives Various chemicals usually used in the rubber industry, for example, anti-aging agents, processing aids, process oils, scorch inhibitors, zinc white, stearin as long as the object of the present invention is not impaired. An acid etc. can be contained.
本発明の目的が損なわれない範囲で、所望により通常、ゴム工業界で用いられる各種薬品、例えば老化防止剤、加工助剤、プロセスオイル、スコーチ防止剤、亜鉛華、ステアリン酸などを含有させる事が出来る。 (H) Other additives Various chemicals usually used in the rubber industry, for example, anti-aging agents, processing aids, process oils, scorch inhibitors, zinc white, stearin as long as the object of the present invention is not impaired. An acid etc. can be contained.
(h1)老化防止剤
老化防止剤としては、アミン・ケトン系、イミダゾール系、アミン系、フェノール系、硫黄系、及び燐系等の老化防止剤が挙げられる。 (H1) Anti-aging agent Examples of the anti-aging agent include amine / ketone-based, imidazole-based, amine-based, phenol-based, sulfur-based and phosphorus-based anti-aging agents.
老化防止剤としては、アミン・ケトン系、イミダゾール系、アミン系、フェノール系、硫黄系、及び燐系等の老化防止剤が挙げられる。 (H1) Anti-aging agent Examples of the anti-aging agent include amine / ketone-based, imidazole-based, amine-based, phenol-based, sulfur-based and phosphorus-based anti-aging agents.
(h2)加硫助剤
加硫助剤としては、公知の加硫助剤、例えばアルデヒド類、アンモニア類、アミン類、グアニジン類、チオウレア類、チアゾール類、チウラム類、ジチオカーバメイト類、及びキサンテート類等が用いられる。 (H2) Vulcanization aids As vulcanization aids, known vulcanization aids such as aldehydes, ammonia, amines, guanidines, thioureas, thiazoles, thiurams, dithiocarbamates, and xanthates Etc. are used.
加硫助剤としては、公知の加硫助剤、例えばアルデヒド類、アンモニア類、アミン類、グアニジン類、チオウレア類、チアゾール類、チウラム類、ジチオカーバメイト類、及びキサンテート類等が用いられる。 (H2) Vulcanization aids As vulcanization aids, known vulcanization aids such as aldehydes, ammonia, amines, guanidines, thioureas, thiazoles, thiurams, dithiocarbamates, and xanthates Etc. are used.
(h3)スコーチ防止剤(リターダー)
スコーチ防止剤としては、有機酸やニトロソ化合物、N-シクロヘキシルチオフタルイミド、及びスルホンアミド誘導体等が用いられる。 (H3) Scorch inhibitor (retarder)
As the scorch inhibitor, organic acids, nitroso compounds, N-cyclohexylthiophthalimide, sulfonamide derivatives and the like are used.
スコーチ防止剤としては、有機酸やニトロソ化合物、N-シクロヘキシルチオフタルイミド、及びスルホンアミド誘導体等が用いられる。 (H3) Scorch inhibitor (retarder)
As the scorch inhibitor, organic acids, nitroso compounds, N-cyclohexylthiophthalimide, sulfonamide derivatives and the like are used.
(h4)プロセスオイル
プロセスオイルはアロマティック系、ナフテン系、及びパラフィン系のいずれを用いてもよい。 (H4) Process oil Process oil may be any of aromatic, naphthenic, and paraffinic.
プロセスオイルはアロマティック系、ナフテン系、及びパラフィン系のいずれを用いてもよい。 (H4) Process oil Process oil may be any of aromatic, naphthenic, and paraffinic.
(加工方法)
(A)加硫可能なゴム、(A)以外の(B)沸騰n-ヘキサン不溶分;1~25重量%で,沸騰n-ヘキサン可溶分;99~75重量%であるビニル・シスブタジエンゴム(VCR)および(C)熱可塑性樹脂に、(D1)熱膨張型発泡剤と(D2)熱分解型発泡剤からなる発泡剤を併用かつ(E)ゴム補強剤は、ロール等の開放式混練機、バンバリーミキサー等の密閉式混練機などの混練機を用いて混練りする事によって得られ、成形加工後に加硫を行い、各種ゴム製品に適用する事が出来る。 (Processing method)
(A) Vulcanizable rubber other than (A) (B) Boiling n-hexane insoluble content: 1 to 25% by weight, boiling n-hexane soluble content: 99 to 75% by weight vinyl cisbutadiene The rubber (VCR) and (C) thermoplastic resin are used in combination with a foaming agent comprising (D1) a thermal expansion type foaming agent and (D2) a thermal decomposition type foaming agent. (E) The rubber reinforcing agent is an open type such as a roll. It can be obtained by kneading using a kneader such as a closed kneader such as a kneader or Banbury mixer, and can be vulcanized after molding and applied to various rubber products.
(A)加硫可能なゴム、(A)以外の(B)沸騰n-ヘキサン不溶分;1~25重量%で,沸騰n-ヘキサン可溶分;99~75重量%であるビニル・シスブタジエンゴム(VCR)および(C)熱可塑性樹脂に、(D1)熱膨張型発泡剤と(D2)熱分解型発泡剤からなる発泡剤を併用かつ(E)ゴム補強剤は、ロール等の開放式混練機、バンバリーミキサー等の密閉式混練機などの混練機を用いて混練りする事によって得られ、成形加工後に加硫を行い、各種ゴム製品に適用する事が出来る。 (Processing method)
(A) Vulcanizable rubber other than (A) (B) Boiling n-hexane insoluble content: 1 to 25% by weight, boiling n-hexane soluble content: 99 to 75% by weight vinyl cisbutadiene The rubber (VCR) and (C) thermoplastic resin are used in combination with a foaming agent comprising (D1) a thermal expansion type foaming agent and (D2) a thermal decomposition type foaming agent. (E) The rubber reinforcing agent is an open type such as a roll. It can be obtained by kneading using a kneader such as a closed kneader such as a kneader or Banbury mixer, and can be vulcanized after molding and applied to various rubber products.
本願発明のもう一つの主要な点は、加硫工程における反応器内圧力の調整にある。特に加硫初期(加熱及び加圧30秒後)での反応器内発泡圧力が2000kPa以上、好ましくは2200kPaになるように調整することで、エアーボイドが発生せず、製品にカケがなく成形品に不良の無い、発泡後の寸法安定性にも優れた靴底用ゴム発泡体組成物の製造方法を提供できるというものである。
Another main point of the present invention is the adjustment of the pressure in the reactor in the vulcanization process. In particular, by adjusting the foaming pressure in the reactor at the initial stage of vulcanization (after heating and pressurization 30 seconds) to 2000 kPa or more, preferably 2200 kPa, air voids are not generated, and there is no chipping in the product. It is possible to provide a method for producing a rubber foam composition for a shoe sole that is excellent in dimensional stability after foaming.
加硫初期段階(加熱及び加圧30秒後)での反応器内発泡圧力が2000kPより小さいと、加硫初期段階での十分な脱気が出来ず、エアーボイド発生の原因となる。よって、加硫初期(加熱及び加圧30秒後)での反応器内発泡圧力が、当該製品の改良の大きな要因となる。
If the foaming pressure in the reactor in the initial stage of vulcanization (after heating and pressurization 30 seconds) is less than 2000 kP, sufficient deaeration cannot be performed in the initial stage of vulcanization, causing air voids. Therefore, the foaming pressure in the reactor in the initial stage of vulcanization (after 30 seconds of heating and pressurization) is a major factor for improving the product.
ただし、単に反応器内発泡圧力が2000kPaより大きければ良いというわけではなく、加硫初期と加硫終期の2種類の発泡剤の役割が大きく影響する。
However, it is not only necessary that the foaming pressure in the reactor is larger than 2000 kPa, and the roles of the two types of foaming agents at the initial stage of vulcanization and the final stage of vulcanization are greatly affected.
加硫初期段階(加熱及び加圧30秒後)での反応器内発泡圧力の急激な上昇では、(D1)熱膨張型発泡剤の発泡効果が影響する。ここでは、加硫初期で(D1)熱膨張型発泡剤の発泡による反応器内発泡圧力が2000kPaより大きくする事で、反応器内のエアーボイドが発生しなくなり、更に成形品の熱収縮の問題も防ぐ事が出来る。ついで、加硫終期では、(D2)熱分解型発泡剤の発泡効果が影響する。この段階では、加硫が十分進んだ場合での(D2)熱分解型発泡剤による発泡効果により、樹脂の硬度上昇を抑え、軽量化を実現する事が出来る。故に、(D1)と(D2)の併用が重要である。
In the vulcanization initial stage (after heating and pressurization 30 seconds), the foaming effect of (D1) thermal expansion type foaming agent affects the rapid increase of the foaming pressure in the reactor. Here, at the initial stage of vulcanization (D1), by increasing the foaming pressure in the reactor due to foaming of the thermal expansion type foaming agent, the air void in the reactor is not generated, and there is a problem of heat shrinkage of the molded product. Can also be prevented. Next, at the end of vulcanization, the foaming effect of (D2) pyrolytic foaming agent is affected. At this stage, when the vulcanization is sufficiently advanced, (D2) the foaming effect by the pyrolytic foaming agent can suppress the increase in the hardness of the resin and realize weight reduction. Therefore, the combined use of (D1) and (D2) is important.
図1~2に実施例1~8の加硫時間(特に加硫初期)と反応器内発泡圧力の関係を示す。実施例1~8は加硫開始0.5分(加硫開始:加熱及び加圧30秒後)時点において、反応器内圧力が2000kPaより大きくなっている。一方、比較例1~10が示されている図3~4では、加硫開始0.5分(加硫開始:加熱及び加圧30秒後)時点において、反応器内圧力が2000kPaを下回っているものが多い。
FIGS. 1 and 2 show the relationship between the vulcanization time (especially at the initial stage of vulcanization) and the foaming pressure in the reactor of Examples 1 to 8. In Examples 1 to 8, the pressure inside the reactor was higher than 2000 kPa at the time of 0.5 minutes from the start of vulcanization (start of vulcanization: 30 seconds after heating and pressurization). On the other hand, in FIGS. 3 to 4 where Comparative Examples 1 to 10 are shown, the pressure inside the reactor is less than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: after 30 seconds of heating and pressurization). There are many things.
図5~8に加硫開始から終期にかけての反応器内発泡圧力の関係を示す。図5~6より、実施例1~8においては、加硫開始初期(加熱及び加圧30秒後)急激に反応器内発泡圧力が2000kPaを超えた後においても、保圧されており、特に加硫終期においても依然として反応器内発泡圧力が1000kPaを超えて、保圧されている事が分かる。こうした急激な圧力減少が無いような経過を取る事が望ましく、特に加硫後期では(D2)熱分解型発泡剤の発泡効果が大きく影響する。
Figures 5 to 8 show the relationship between the foaming pressure in the reactor from the start to the end of vulcanization. As shown in FIGS. 5 to 6, in Examples 1 to 8, the initial pressure of vulcanization (after 30 seconds of heating and pressurization) was maintained even after the in-reactor foaming pressure exceeded 2000 kPa. It can be seen that, even at the end of vulcanization, the in-reactor foaming pressure still exceeds 1000 kPa and the pressure is maintained. It is desirable to take such a course that there is no sudden pressure decrease, and the foaming effect of the pyrolytic foaming agent (D2) is greatly influenced particularly in the late vulcanization stage.
表3から、実施例1~8では、エアーボイドの発生が起こっていないことが分かる。
Table 3 shows that no air void occurred in Examples 1 to 8.
図9~10に本願発明における実施例9~15の加硫時間(特に加硫初期)と反応器内発泡圧力の関係を示す。図9~10より、実施例9~15は加硫開始0.5分(加硫開始:加熱及び加圧30秒後)時点において、反応器内圧力が2000kPaより大きくなっている。
FIGS. 9 to 10 show the relationship between the vulcanization time (particularly at the initial stage of vulcanization) and the foaming pressure in the reactor of Examples 9 to 15 in the present invention. 9 to 10, in Examples 9 to 15, the pressure in the reactor was higher than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: after 30 seconds of heating and pressurization).
図11~12に比較例11~15の加硫開始から終期にかけての反応器内発泡圧力の関係を示す。ここで加硫開始から終期にかけて反応圧力が2000kPa以下に低下すると、反応圧力の低下に伴う比重や硬度という上昇の問題が生じる。
11 to 12 show the relationship between the foaming pressure in the reactor from the start to the end of vulcanization in Comparative Examples 11 to 15. Here, when the reaction pressure decreases to 2000 kPa or less from the start to the end of vulcanization, there arises a problem of an increase in specific gravity and hardness accompanying a decrease in the reaction pressure.
表7から、実施例9~15では、エアーボイドの発生も起こっていないことが分かる。
From Table 7, it can be seen that no air voids occurred in Examples 9 to 15.
図17に本願発明における実施例16~18及び比較例16の加硫時間(特に加硫初期)と反応器内発泡圧力の関係を示す。図17より、実施例16~18は加硫開始0.5分(加硫開始:加熱及び加圧30秒後)時点において、反応器内圧力が2000kPaより大きくなっている。一方、比較例16は加硫開始0.5分(加硫開始:加熱及び加圧30秒後)時点において、反応器内圧力が2000kPaより小さくなっている。
FIG. 17 shows the relationship between the vulcanization time (especially at the initial stage of vulcanization) and the foam pressure in the reactor of Examples 16 to 18 and Comparative Example 16 in the present invention. As shown in FIG. 17, in Examples 16 to 18, the pressure in the reactor was higher than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: 30 seconds after heating and pressurization). On the other hand, in Comparative Example 16, the pressure in the reactor was less than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: after 30 seconds of heating and pressurization).
表10から、実施例16~18では、エアーボイドの発生も起こっていないことが分かる。
From Table 10, it can be seen that in Examples 16 to 18, no air void occurred.
2mmでの比重値は、0.450~0.950(g/cm3)であることが好ましく、0.500~0.800(g/cm3)であることがより好ましく、0.550~0.750(g/cm3)であることがさらに好ましい。比重が大きいと硬度の上昇や軽量化が図れないという問題が生じる。また、比重が小さいと諸物性(機械的強度、耐摩耗性、収縮性)の悪化という問題が生じる。
The specific gravity value at 2 mm is preferably 0.450 to 0.950 (g / cm 3 ), more preferably 0.500 to 0.800 (g / cm 3 ), and 0.550 to More preferably, it is 0.750 (g / cm 3 ). When the specific gravity is large, there arises a problem that the hardness cannot be increased and the weight cannot be reduced. Moreover, when specific gravity is small, the problem of deterioration of various physical properties (mechanical strength, abrasion resistance, shrinkage | contraction) will arise.
全ての比較例は、加工性やエアーボイドが改善されると軽量化、低硬度化および吸水率などが悪くなる傾向があり、逆に軽量化を目的とすると、加工性やエアーボイドが悪くなるなど、バランスが悪い事が分かる。ゆえに、全体の物性や加工性等のバランスを考慮した場合、組成物における構成要素の組み合わせであって、かつ、加硫初期(加熱及び加圧30秒後)での反応器内発泡圧力が2000kPa以上になるように調整し、かつ、比重を小さくなるように調整することが重要であることがわかる。
In all the comparative examples, when workability and air void are improved, there is a tendency for weight reduction, low hardness, water absorption, etc. to deteriorate, and conversely, for weight reduction purposes, workability and air void deteriorate. I understand that the balance is bad. Therefore, in consideration of the balance of overall physical properties and workability, the foaming pressure in the reactor is a combination of the constituent elements in the composition and the initial stage of vulcanization (after heating and pressurization 30 seconds) is 2000 kPa. It turns out that it is important to adjust so that it may become above, and to adjust so that specific gravity may become small.
(D1)と(D2)の全ゴム成分配合割合についても、本願発明において重要であり、前述したように加硫初期(加熱及び加圧30秒後)での反応器内発泡圧力が2000kPa以上になるような発泡剤の量が必要であり、かつ加硫終期での樹脂の硬度上昇を抑え、軽量化を実現するための適量の発泡剤が必要である。こうした観点から、(D1)が0.5~3.0重量部、(D2)が0.5~7.0重量部の配合割合が最適な条件である。
The blending ratio of all rubber components (D1) and (D2) is also important in the present invention. As described above, the foaming pressure in the reactor at the initial stage of vulcanization (after heating and pressurization 30 seconds) is 2000 kPa or more. The amount of the foaming agent is necessary, and an appropriate amount of foaming agent is required to suppress the increase in the hardness of the resin at the end of vulcanization and to realize weight reduction. From such a viewpoint, the blending ratio of (D1) of 0.5 to 3.0 parts by weight and (D2) of 0.5 to 7.0 parts by weight is the optimum condition.
(靴底用発泡体ゴム組成物およびアウトソール)
(A)加硫可能なゴム、(A)以外の(B)沸騰n-ヘキサン不溶分;1~25重量%で,沸騰n-ヘキサン可溶分;99~75重量%であるビニル・シスブタジエンゴム(VCR)および(C)熱可塑性樹脂からなる(A)+(B)+(C)全ゴム成分100重量部に対し、(D1)熱膨張型発泡剤0.5~3.0重量部と(D2)熱分解型発泡剤0.5~7.0重量部からなる構成にする事で、ソリッド系ゴム靴底材料(比重=1以上)と比較し、低硬度・低比重・引張強度・破断伸びが大きく、加硫後の成型品の収縮・外観及び、モールド成型性に優れ、かつそれらのバランスに優れた靴底用発泡体ゴム組成物を得る事が出来ることを見出した。 (Shoe sole foam rubber composition and outsole)
(A) Vulcanizable rubber other than (A) (B) Boiling n-hexane insoluble content: 1 to 25% by weight, boiling n-hexane soluble content: 99 to 75% by weight vinyl cisbutadiene (D1) 0.5 to 3.0 parts by weight of a thermal expansion foaming agent with respect to 100 parts by weight of the total rubber component (A) + (B) + (C) consisting of rubber (VCR) and (C) thermoplastic resin (D2) Low hardness, low specific gravity, and tensile strength compared to solid rubber sole material (specific gravity = 1 or more) -It has been found that a foam rubber composition for shoe soles having a large elongation at break, excellent shrinkage / appearance of a molded product after vulcanization, moldability, and balance thereof can be obtained.
(A)加硫可能なゴム、(A)以外の(B)沸騰n-ヘキサン不溶分;1~25重量%で,沸騰n-ヘキサン可溶分;99~75重量%であるビニル・シスブタジエンゴム(VCR)および(C)熱可塑性樹脂からなる(A)+(B)+(C)全ゴム成分100重量部に対し、(D1)熱膨張型発泡剤0.5~3.0重量部と(D2)熱分解型発泡剤0.5~7.0重量部からなる構成にする事で、ソリッド系ゴム靴底材料(比重=1以上)と比較し、低硬度・低比重・引張強度・破断伸びが大きく、加硫後の成型品の収縮・外観及び、モールド成型性に優れ、かつそれらのバランスに優れた靴底用発泡体ゴム組成物を得る事が出来ることを見出した。 (Shoe sole foam rubber composition and outsole)
(A) Vulcanizable rubber other than (A) (B) Boiling n-hexane insoluble content: 1 to 25% by weight, boiling n-hexane soluble content: 99 to 75% by weight vinyl cisbutadiene (D1) 0.5 to 3.0 parts by weight of a thermal expansion foaming agent with respect to 100 parts by weight of the total rubber component (A) + (B) + (C) consisting of rubber (VCR) and (C) thermoplastic resin (D2) Low hardness, low specific gravity, and tensile strength compared to solid rubber sole material (specific gravity = 1 or more) -It has been found that a foam rubber composition for shoe soles having a large elongation at break, excellent shrinkage / appearance of a molded product after vulcanization, moldability, and balance thereof can be obtained.
得られたゴム組成物の物性は以下のようにして測定した。
(1)比重(密度);JIS K6268に準じて、A法で測定した。 The physical properties of the obtained rubber composition were measured as follows.
(1) Specific gravity (density): Measured by method A according to JIS K6268.
(1)比重(密度);JIS K6268に準じて、A法で測定した。 The physical properties of the obtained rubber composition were measured as follows.
(1) Specific gravity (density): Measured by method A according to JIS K6268.
(2)発泡圧力;ALPHA TECHNOLOGIES社製RPA2000(ロータレス型、ゴム加工性解析装置)を用いて、JIS K6300に従って160℃における10%及び、90%加硫度に達する時間と発泡圧力を測定した。圧力の数値が高いほど良好である事を示す。
(2) Foaming pressure: Using ALPHA TECHNOLOGIES RPA2000 (rotorless type, rubber processability analyzer), the time to reach 10% and 160% vulcanization degree at 160 ° C. and the foaming pressure were measured according to JIS K6300. The higher the pressure value, the better.
(3)硬度;JIS K6253に準じて、タイプAデュロメータを、用いて室温で測定した。また、SR1S0101に準拠したAsker C型(スポンジ硬度計)を用いて室温で測定した。数値が小さいほど軟らかく良好である事を示す。
(3) Hardness: Measured at room temperature using a type A durometer according to JIS K6253. Moreover, it measured at room temperature using Asker C type (sponge hardness meter) based on SR1S0101. The smaller the value, the softer and better.
(4)引張弾性率、引張強度、破断伸び;JIS K6251に従って測定した。数値が高いほど製品の機械的特性が良好であり、数値が低いと耐摩耗性が悪化する事を示す。
(4) Tensile modulus, tensile strength, elongation at break; measured according to JIS K6251. The higher the value, the better the mechanical properties of the product, and the lower the value, the worse the wear resistance.
(5)引裂強度;JIS K6252に従い測定した。数値が高いほど金型から製品を剥がす際に引裂かれる事がなく良好である事を示す。
(5) Tear strength: measured in accordance with JIS K6252. The higher the value, the better it will be without tearing when the product is peeled from the mold.
(6)DIN摩耗;JIS K6264に従い摩耗減量を測定した。DIN Indexの数値が小さいほどDIN摩耗性能が良好な物性を示す。
(6) DIN wear; wear loss was measured according to JIS K6264. The smaller the value of DIN Index, the better the physical properties of DIN wear performance.
(7)反撥弾性率;JIS K6255に準じて、トリプソ式を用いて測定した。
(7) Rebound resilience: measured according to JIS K6255 using a trypso type.
(8)衝撃吸収性;JIS K6255に準じて、トリプソ式を用いて測定した。数値が高いほど衝撃吸収性能が良好である事を示す。
(8) Impact absorbability; measured according to JIS K6255 using a trypso type. The higher the value, the better the shock absorption performance.
(9)収縮率;成型品作成後(室温で30分経過後の長さ)、室温で5日経過後の長さより算出した。数値が小さいほど寸法安定性が良好である事を示す。
(9) Shrinkage rate: Calculated from the length after 5 minutes at room temperature after molding (length after 30 minutes at room temperature). The smaller the value, the better the dimensional stability.
(10)吸水率;加硫したゴム組成物をヤマト科学社製ウォータバスを用いて40℃、24時間浸漬し、浸漬前・後の重量変化を測定して吸水率を算出した。数値が大きいほど気泡が大きい事を示す。
(10) Water absorption rate: The vulcanized rubber composition was immersed in a water bath manufactured by Yamato Kagaku Co. at 40 ° C for 24 hours, and the weight change before and after immersion was measured to calculate the water absorption rate. Larger numbers indicate larger bubbles.
実験例1
(実施例1)
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤1.0重量部、及び熱分解型発泡剤1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 Experimental example 1
Example 1
(A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin in Table 1 in a blending ratio of 7/70/23 parts by weight, and rubber reinforcing agents such as silica, zinc white and stearic acid Together with a 1.7 liter closed kneader. Thereafter, the vulcanization accelerator and sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 1.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
(実施例1)
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤1.0重量部、及び熱分解型発泡剤1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 Experimental example 1
Example 1
(A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin in Table 1 in a blending ratio of 7/70/23 parts by weight, and rubber reinforcing agents such as silica, zinc white and stearic acid Together with a 1.7 liter closed kneader. Thereafter, the vulcanization accelerator and sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 1.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
(実施例2)
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/65/28重量部にし、以下実施例1と同様の手法により加硫試験片を作成し、物性を評価した。その結果を表3に示した。 (Example 2)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 7/65/28 parts by weight, and a vulcanized test piece was prepared in the same manner as in Example 1 below. The physical properties were evaluated. The results are shown in Table 3.
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/65/28重量部にし、以下実施例1と同様の手法により加硫試験片を作成し、物性を評価した。その結果を表3に示した。 (Example 2)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 7/65/28 parts by weight, and a vulcanized test piece was prepared in the same manner as in Example 1 below. The physical properties were evaluated. The results are shown in Table 3.
(実施例3)
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比12/60/28重量部にし、以下実施例1と同様の手法により加硫試験片を作成し、物性を評価した。その結果を表3に示した。 (Example 3)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 12/60/28 parts by weight, and a vulcanized test piece was prepared in the same manner as in Example 1 below. The physical properties were evaluated. The results are shown in Table 3.
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比12/60/28重量部にし、以下実施例1と同様の手法により加硫試験片を作成し、物性を評価した。その結果を表3に示した。 (Example 3)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 12/60/28 parts by weight, and a vulcanized test piece was prepared in the same manner as in Example 1 below. The physical properties were evaluated. The results are shown in Table 3.
(実施例4)
表1の(a-1)天然ゴム/(a-2)水添加スチレン-ブタジエンゴム(旭化成ケミカルズ社製)/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/10/65/18重量部にし、以下実施例1と同様の手法により加硫試験片を作成し、物性を評価した。その結果を表3に示した。 Example 4
In Table 1, (a-1) natural rubber / (a-2) water-added styrene-butadiene rubber (manufactured by Asahi Kasei Chemicals Co., Ltd.) / (B) VCR / (C) 1,2 polybutadiene resin is blended in a ratio of 7/10/65. / 18 parts by weight, vulcanized test pieces were prepared in the same manner as in Example 1, and the physical properties were evaluated. The results are shown in Table 3.
表1の(a-1)天然ゴム/(a-2)水添加スチレン-ブタジエンゴム(旭化成ケミカルズ社製)/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/10/65/18重量部にし、以下実施例1と同様の手法により加硫試験片を作成し、物性を評価した。その結果を表3に示した。 Example 4
In Table 1, (a-1) natural rubber / (a-2) water-added styrene-butadiene rubber (manufactured by Asahi Kasei Chemicals Co., Ltd.) / (B) VCR / (C) 1,2 polybutadiene resin is blended in a ratio of 7/10/65. / 18 parts by weight, vulcanized test pieces were prepared in the same manner as in Example 1, and the physical properties were evaluated. The results are shown in Table 3.
(実施例5)
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下実施例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤2.5重量部、及び熱分解型発泡剤0.7重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 (Example 5)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator and sulfur, and 2.5 parts by weight of the thermal expansion foaming agent and 0.7 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下実施例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤2.5重量部、及び熱分解型発泡剤0.7重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 (Example 5)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator and sulfur, and 2.5 parts by weight of the thermal expansion foaming agent and 0.7 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
(実施例6)
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下実施例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤1.0重量部、及び熱分解型発泡剤6.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 (Example 6)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator, sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 6.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下実施例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤1.0重量部、及び熱分解型発泡剤6.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 (Example 6)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator, sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 6.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
(実施例7)
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下実施例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤2.5重量部、及び熱分解型発泡剤6.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 (Example 7)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator and sulfur, 2.5 parts by weight of the thermal expansion foaming agent, and 6.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下実施例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤2.5重量部、及び熱分解型発泡剤6.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 (Example 7)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator and sulfur, 2.5 parts by weight of the thermal expansion foaming agent, and 6.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
(実施例8)
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下実施例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤2.5重量部、及び熱分解型発泡剤1.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 (Example 8)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, a vulcanization accelerator, sulfur, 2.5 parts by weight of a thermal expansion foaming agent, and 1.0 part by weight of a thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
表1の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下実施例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤2.5重量部、及び熱分解型発泡剤1.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表3に示した。 (Example 8)
In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, a vulcanization accelerator, sulfur, 2.5 parts by weight of a thermal expansion foaming agent, and 1.0 part by weight of a thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
(比較例1)
表2の(a-1)天然ゴム/(a-2)溶液重合スチレン-ブタジエンゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤を1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤と硫黄をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 1)
(A-1) natural rubber / (a-2) solution-polymerized styrene-butadiene rubber / (B) VCR / (C) 1,2 polybutadiene resin in Table 2 to a blending ratio of 7/18/55/20 parts by weight, Silica, which is a rubber reinforcing agent, and additives such as zinc white and stearic acid were kneaded using a 1.7 liter closed kneader. Thereafter, the vulcanization accelerator and sulfur were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
表2の(a-1)天然ゴム/(a-2)溶液重合スチレン-ブタジエンゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤を1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤と硫黄をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 1)
(A-1) natural rubber / (a-2) solution-polymerized styrene-butadiene rubber / (B) VCR / (C) 1,2 polybutadiene resin in Table 2 to a blending ratio of 7/18/55/20 parts by weight, Silica, which is a rubber reinforcing agent, and additives such as zinc white and stearic acid were kneaded using a 1.7 liter closed kneader. Thereafter, the vulcanization accelerator and sulfur were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
(比較例2)
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により加硫試験片を作成し、物性を評価した。その結果を表4に示した。 (Comparative Example 2)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 7/70/23 parts by weight, and a vulcanized test piece was prepared in the same manner as in Comparative Example 1 below. The physical properties were evaluated. The results are shown in Table 4.
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により加硫試験片を作成し、物性を評価した。その結果を表4に示した。 (Comparative Example 2)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 7/70/23 parts by weight, and a vulcanized test piece was prepared in the same manner as in Comparative Example 1 below. The physical properties were evaluated. The results are shown in Table 4.
(比較例3)
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤2.5重量部、及び熱分解型発泡剤0.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 3)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator and sulfur, and 2.5 parts by weight of the thermal expansion foaming agent and 0.0 part by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤2.5重量部、及び熱分解型発泡剤0.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 3)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator and sulfur, and 2.5 parts by weight of the thermal expansion foaming agent and 0.0 part by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
(比較例4)
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤0.0重量部、及び熱分解型発泡剤2.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 4)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator, sulfur, 0.0 part by weight of the thermal expansion foaming agent, and 2.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤0.0重量部、及び熱分解型発泡剤2.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 4)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator, sulfur, 0.0 part by weight of the thermal expansion foaming agent, and 2.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
(比較例5)
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤0.3重量部、及び熱分解型発泡剤1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 5)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator and sulfur, and 0.3 parts by weight of the thermal expansion foaming agent and 1.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤0.3重量部、及び熱分解型発泡剤1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 5)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator and sulfur, and 0.3 parts by weight of the thermal expansion foaming agent and 1.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
(比較例6)
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤5.0重量部、及び熱分解型発泡剤1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 6)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, vulcanization accelerator and sulfur, and 5.0 parts by weight of thermal expansion type foaming agent and 1.5 parts by weight of thermal decomposition type foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤5.0重量部、及び熱分解型発泡剤1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 6)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, vulcanization accelerator and sulfur, and 5.0 parts by weight of thermal expansion type foaming agent and 1.5 parts by weight of thermal decomposition type foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
(比較例7)
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤1.0重量部、及び熱分解型発泡剤0.3重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 7)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator and sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 0.3 part by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤1.0重量部、及び熱分解型発泡剤0.3重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 7)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator and sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 0.3 part by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
(比較例8)
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤1.0重量部、及び熱分解型発泡剤10.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 8)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, a vulcanization accelerator, sulfur, 1.0 part by weight of a thermal expansion foaming agent, and 10.0 parts by weight of a thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤1.0重量部、及び熱分解型発泡剤10.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 8)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, a vulcanization accelerator, sulfur, 1.0 part by weight of a thermal expansion foaming agent, and 10.0 parts by weight of a thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
(比較例9)
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤3.0重量部、及び熱分解型発泡剤0.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 9)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, a vulcanization accelerator, sulfur, 3.0 parts by weight of a thermal expansion foaming agent, and 0.0 part by weight of a thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤3.0重量部、及び熱分解型発泡剤0.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 9)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, a vulcanization accelerator, sulfur, 3.0 parts by weight of a thermal expansion foaming agent, and 0.0 part by weight of a thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
(比較例10)
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤0.0重量部、及び熱分解型発泡剤7.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 10)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator and sulfur, and 0.0 parts by weight of the thermal expansion type foaming agent and 7.0 parts by weight of the thermal decomposition type foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.
表2の(A)天然ゴム/(B)VCR/(C)1,2ポリブタジエン樹脂を配合比7/70/23重量部にし、以下比較例1と同様の手法により混錬した。その後、加硫促進剤と硫黄、並びに熱膨張型発泡剤0.0重量部、及び熱分解型発泡剤7.0重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表4に示した。 (Comparative Example 10)
In Table 2, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded by the same method as in Comparative Example 1. Thereafter, the vulcanization accelerator and sulfur, and 0.0 parts by weight of the thermal expansion type foaming agent and 7.0 parts by weight of the thermal decomposition type foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 4.




実験例2
(実施例9)
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 Experimental example 2
Example 9
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of vulcanization accelerator, 1.2 parts by weight of sulfur, 1.00 parts by weight of thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
(実施例9)
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 Experimental example 2
Example 9
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of vulcanization accelerator, 1.2 parts by weight of sulfur, 1.00 parts by weight of thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
(実施例10)
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.25重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 10)
(A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 5 to a blending ratio of 7/18/55/20 parts by weight, rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.25 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of a thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was carried out, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.25重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 10)
(A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 5 to a blending ratio of 7/18/55/20 parts by weight, rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.25 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of a thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was carried out, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
(実施例11)
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.50重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 Example 11
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.50 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of a thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.50重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 Example 11
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.50 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of a thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
(実施例12)
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を2.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 12)
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of vulcanization accelerator, 1.2 parts by weight of sulfur, 2.00 parts by weight of thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を2.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 12)
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of vulcanization accelerator, 1.2 parts by weight of sulfur, 2.00 parts by weight of thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
(実施例13)
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.0重量部、及び熱分解型発泡剤CAP-500 1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 13)
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.0 part by weight of a thermal expansion foaming agent EXPANCEL DU092-40, and 1.5 parts by weight of a thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.0重量部、及び熱分解型発泡剤CAP-500 1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 13)
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.0 part by weight of a thermal expansion foaming agent EXPANCEL DU092-40, and 1.5 parts by weight of a thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
(実施例14)
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.0重量部、及び熱分解型発泡剤CAP-500 6.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 14)
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of vulcanization accelerator, 1.2 parts by weight of sulfur, 1.0 part by weight of thermal expansion foaming agent EXPANCEL DU092-40, and 6.5 parts by weight of thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を1.0重量部、及び熱分解型発泡剤CAP-500 6.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 14)
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of vulcanization accelerator, 1.2 parts by weight of sulfur, 1.0 part by weight of thermal expansion foaming agent EXPANCEL DU092-40, and 6.5 parts by weight of thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
(実施例15)
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を2.5重量部、及び熱分解型発泡剤CAP-500 1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 15)
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 2.5 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-40, and 1.5 parts by weight of a thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
表5の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/55/20重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を2.5重量部、及び熱分解型発泡剤CAP-500 1.5重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表7に示した。 (Example 15)
In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 2.5 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-40, and 1.5 parts by weight of a thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 7.
(比較例11)
熱膨張型発泡剤EXPANCEL DU092-40を使用しなかった以外は、実施例9と同様にして行った。その結果を表8に示した。 (Comparative Example 11)
The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used. The results are shown in Table 8.
熱膨張型発泡剤EXPANCEL DU092-40を使用しなかった以外は、実施例9と同様にして行った。その結果を表8に示した。 (Comparative Example 11)
The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used. The results are shown in Table 8.
(比較例12)
熱膨張型発泡剤EXPANCEL DU092-40を使用せず、かつ熱分解型発泡剤CAP-500 5.50重量部にした以外は、実施例9と同様にして行った。その結果を表8に示した。 (Comparative Example 12)
The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used and the thermal decomposition foaming agent CAP-500 was used in an amount of 5.50 parts by weight. The results are shown in Table 8.
熱膨張型発泡剤EXPANCEL DU092-40を使用せず、かつ熱分解型発泡剤CAP-500 5.50重量部にした以外は、実施例9と同様にして行った。その結果を表8に示した。 (Comparative Example 12)
The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used and the thermal decomposition foaming agent CAP-500 was used in an amount of 5.50 parts by weight. The results are shown in Table 8.
(比較例13)
熱膨張型発泡剤EXPANCEL DU092-40を使用せず、かつ熱分解型発泡剤CAP-500 6.50重量部にした以外は、実施例9と同様にして行った。その結果を表8に示した。 (Comparative Example 13)
The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used and the thermal decomposition foaming agent CAP-500 was used in an amount of 6.50 parts by weight. The results are shown in Table 8.
熱膨張型発泡剤EXPANCEL DU092-40を使用せず、かつ熱分解型発泡剤CAP-500 6.50重量部にした以外は、実施例9と同様にして行った。その結果を表8に示した。 (Comparative Example 13)
The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used and the thermal decomposition foaming agent CAP-500 was used in an amount of 6.50 parts by weight. The results are shown in Table 8.
(比較例14)
熱膨張型発泡剤EXPANCEL DU092-40を5.00重量部使用し、かつ熱分解型発泡剤CAP-500を使用しなかった以外は、実施例9と同様にして行った。その結果を表8に示した。 (Comparative Example 14)
The same procedure as in Example 9 was conducted, except that 5.00 parts by weight of the thermal expansion foaming agent EXPANCEL DU092-40 was used, and the thermal decomposition foaming agent CAP-500 was not used. The results are shown in Table 8.
熱膨張型発泡剤EXPANCEL DU092-40を5.00重量部使用し、かつ熱分解型発泡剤CAP-500を使用しなかった以外は、実施例9と同様にして行った。その結果を表8に示した。 (Comparative Example 14)
The same procedure as in Example 9 was conducted, except that 5.00 parts by weight of the thermal expansion foaming agent EXPANCEL DU092-40 was used, and the thermal decomposition foaming agent CAP-500 was not used. The results are shown in Table 8.
(比較例15)
表6の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/75/0重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を2.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表8に示した。 (Comparative Example 15)
In Table 6, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended in a ratio of 7/18/75/0 parts by weight. It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of vulcanization accelerator, 1.2 parts by weight of sulfur, 2.00 parts by weight of thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 8.
表6の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/18/75/0重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに熱膨張型発泡剤EXPANCEL DU092-40を2.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表8に示した。 (Comparative Example 15)
In Table 6, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended in a ratio of 7/18/75/0 parts by weight. It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of vulcanization accelerator, 1.2 parts by weight of sulfur, 2.00 parts by weight of thermal expansion foaming agent EXPANCEL DU092-40, and 4.50 parts by weight of thermal decomposition foaming agent CAP-500 Were mixed in an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 8.




実験例3
(実施例16)
表9の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/10/65/18重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに平均粒径が10~16μmの熱膨張型発泡剤EXPANCEL DU092-40を1.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表10に示した。 Experimental example 3
(Example 16)
(A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.00 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-40 having an average particle size of 10 to 16 μm, and a thermal decomposition foaming agent 4.50 parts by weight of CAP-500 was mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 10.
(実施例16)
表9の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/10/65/18重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに平均粒径が10~16μmの熱膨張型発泡剤EXPANCEL DU092-40を1.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表10に示した。 Experimental example 3
(Example 16)
(A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.00 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-40 having an average particle size of 10 to 16 μm, and a thermal decomposition foaming agent 4.50 parts by weight of CAP-500 was mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 10.
(実施例17)
表9の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/10/65/18重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに平均粒径が18~24μmの熱膨張型発泡剤EXPANCEL DU092-80を1.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表10に示した。 (Example 17)
(A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.00 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-80 having an average particle size of 18 to 24 μm, and a thermal decomposition foaming agent 4.50 parts by weight of CAP-500 was mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 10.
表9の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/10/65/18重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに平均粒径が18~24μmの熱膨張型発泡剤EXPANCEL DU092-80を1.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表10に示した。 (Example 17)
(A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.00 parts by weight of a thermal expansion foaming agent EXPANCEL DU092-80 having an average particle size of 18 to 24 μm, and a thermal decomposition foaming agent 4.50 parts by weight of CAP-500 was mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 10.
(実施例18)
表9の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/10/65/18重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに平均粒径が18~24μmの熱膨張型発泡剤EXPANCEL DU009-80を1.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表10に示した。 (Example 18)
(A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.00 parts by weight of a thermal expansion type foaming agent EXPANCEL DU009-80 having an average particle size of 18 to 24 μm, and a thermal decomposition type foaming agent 4.50 parts by weight of CAP-500 was mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 10.
表9の(a-1)天然ゴム/(a-2)スチレン-ブタジエンゴム(SBR)/(B)VCR/(C)オレフィン系樹脂を配合比7/10/65/18重量部にし、ゴム補強剤であるシリカと亜鉛華やステアリン酸などの添加剤と共に1.7リットルの密閉式混練装置を使用して混錬した。その後、加硫促進剤0.95重量部と硫黄1.2重量部、並びに平均粒径が18~24μmの熱膨張型発泡剤EXPANCEL DU009-80を1.00重量部、及び熱分解型発泡剤CAP-500 4.50重量部をオープンロールで混合した。次いで、プレス加硫し、得られた加硫試験片により物性を評価した。その結果を表10に示した。 (Example 18)
(A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead | mixed using the 1.7-liter closed-type kneading apparatus with additives, such as silica which is a reinforcing agent, zinc white, and stearic acid. Thereafter, 0.95 parts by weight of a vulcanization accelerator, 1.2 parts by weight of sulfur, 1.00 parts by weight of a thermal expansion type foaming agent EXPANCEL DU009-80 having an average particle size of 18 to 24 μm, and a thermal decomposition type foaming agent 4.50 parts by weight of CAP-500 was mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 10.
(比較例16)
熱膨張型発泡剤を使用しなかった以外は、実施例16と同様にして行った。その結果を表10に示した。 (Comparative Example 16)
The same procedure as in Example 16 was performed except that the thermal expansion foaming agent was not used. The results are shown in Table 10.
熱膨張型発泡剤を使用しなかった以外は、実施例16と同様にして行った。その結果を表10に示した。 (Comparative Example 16)
The same procedure as in Example 16 was performed except that the thermal expansion foaming agent was not used. The results are shown in Table 10.


本発明で得られる発泡体ゴム組成物の用途としては、靴底分野、特にスポーツシューズ、ランニングシューズ、カジュアルシューズなどの履物全般の靴底材料として有用である。
As the use of the foam rubber composition obtained in the present invention, it is useful as a sole material in the sole field, in particular, footwear in general such as sports shoes, running shoes, and casual shoes.
Claims (12)
- (A)加硫可能なゴム、(A)以外の(B)沸騰n-ヘキサン不溶分;1~25重量%で、沸騰n-ヘキサン可溶分;99~75重量%であるビニル・シスブタジエンゴム(VCR)および(C)熱可塑性樹脂に、(D1)熱膨張型発泡剤と(D2)熱分解型発泡剤からなる発泡剤を併用し、(A)+(B)+(C)からなる全ゴム成分100重量部に対して、(D1)が0.5~3.0重量部、(D2)が0.5~7.0重量部であることを特徴とする靴底用発泡体ゴム組成物。 (A) Vulcanizable rubber other than (A) (B) Boiling n-hexane insoluble content: 1 to 25% by weight, boiling n-hexane soluble content; 99 to 75% by weight vinyl cisbutadiene From (A) + (B) + (C), a rubber (VCR) and (C) thermoplastic resin are combined with a foaming agent comprising (D1) a thermal expansion type foaming agent and (D2) a thermal decomposition type foaming agent. A foam for a shoe sole, wherein (D1) is 0.5 to 3.0 parts by weight and (D2) is 0.5 to 7.0 parts by weight with respect to 100 parts by weight of the total rubber component. Rubber composition.
- (A)の加硫可能なゴムが(a-1)天然ゴムおよび(a-2)スチレン-ブタジエンゴム(SBR)の組合わせ、または(a-1)天然ゴムであることを特徴とする請求項1記載の靴底用発泡体ゴム組成物。 The vulcanizable rubber (A) is a combination of (a-1) natural rubber and (a-2) styrene-butadiene rubber (SBR), or (a-1) natural rubber. Item 1. A foam rubber composition for a shoe sole according to Item 1.
- (a-2)スチレンーブタジエンゴム(SBR)が、水添加スチレン-ブタジエンゴムであることを特徴とする請求項2記載の靴底用発泡体ゴム組成物。 3. The foam rubber composition for a shoe sole according to claim 2, wherein the (a-2) styrene-butadiene rubber (SBR) is a water-added styrene-butadiene rubber.
- (a-2)スチレン-ブタジエンゴム(SBR)が、溶液重合スチレン-ブタジエンゴム(S-SBR)であることを特徴とする請求項2記載の靴底用発泡体ゴム組成物。 3. The foam rubber composition for a shoe sole according to claim 2, wherein (a-2) the styrene-butadiene rubber (SBR) is a solution-polymerized styrene-butadiene rubber (S-SBR).
- (B)沸騰n-ヘキサン不溶分の融点が170℃以上であるシンジオタクチック1,2-ポリブタジエンであることを特徴とする請求項1乃至4いずれか記載の靴底用発泡体ゴム組成物。 5. The foam rubber composition for a shoe sole according to any one of claims 1 to 4, which is syndiotactic 1,2-polybutadiene having a melting point of (B) insoluble in boiling n-hexane of 170 ° C. or higher.
- (C)の熱可塑性樹脂が、ポリオレフィン系樹脂であることを特徴とする請求項1乃至5いずれか記載の靴底用発泡体ゴム組成物。 6. The foam rubber composition for a shoe sole according to any one of claims 1 to 5, wherein the thermoplastic resin (C) is a polyolefin resin.
- 前記ポリオレフィン系樹脂が、融点が70℃~110℃である1,2-ポリブタジエン樹脂であることを特徴とする請求項6記載の靴底用発泡体ゴム組成物。 The foam rubber composition for a shoe sole according to claim 6, wherein the polyolefin resin is a 1,2-polybutadiene resin having a melting point of 70 ° C to 110 ° C.
- 前記ポリオレフィン系樹脂が、エチレン-酢酸ビニル共重合体(EVA)であることを特徴とする請求項6に記載の靴底用発泡体ゴム組成物。 The shoe rubber foam composition according to claim 6, wherein the polyolefin resin is an ethylene-vinyl acetate copolymer (EVA).
- (D1)熱膨張型発泡剤が熱膨張性マイクロカプセルであることを特徴とする請求項1乃至8いずれか記載の靴底用発泡体ゴム組成物。 (D1) The foam rubber composition for a shoe sole according to any one of claims 1 to 8, wherein the thermally expandable foaming agent is a thermally expandable microcapsule.
- 前記熱膨張性マイクロカプセルの粒径が8~30μmであることを特徴とする請求項9記載の靴底用発泡体ゴム組成物。 10. The foam rubber composition for a shoe sole according to claim 9, wherein a particle diameter of the thermally expandable microcapsule is 8 to 30 μm.
- (D2)の熱分解型発泡剤がアゾジカルボンアミド系複合発泡剤であることを特徴とする請求項1乃至10いずれか記載の靴底用発泡体ゴム組成物。 The foam rubber composition for a shoe sole according to any one of claims 1 to 10, wherein the pyrolytic foaming agent (D2) is an azodicarbonamide composite foaming agent.
- 請求項1乃至11いずれか記載の靴底用発泡体ゴム組成物を用いたことを特徴とするアウトソール。 An outsole comprising the foam rubber composition for a shoe sole according to any one of claims 1 to 11.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010535862A JP5565313B2 (en) | 2009-06-22 | 2010-02-08 | Foam rubber composition for shoe sole and outsole |
Applications Claiming Priority (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009147947 | 2009-06-22 | ||
| JP2009-147947 | 2009-06-22 | ||
| JP2009176118 | 2009-07-29 | ||
| JP2009-176118 | 2009-07-29 | ||
| JP2009229768 | 2009-10-01 | ||
| JP2009-229768 | 2009-10-01 | ||
| JP2009-257679 | 2009-11-11 | ||
| JP2009257678 | 2009-11-11 | ||
| JP2009257679 | 2009-11-11 | ||
| JP2009-258347 | 2009-11-11 | ||
| JP2009-257678 | 2009-11-11 | ||
| JP2009258347 | 2009-11-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2010050628A2 true WO2010050628A2 (en) | 2010-05-06 |
| WO2010050628A3 WO2010050628A3 (en) | 2010-06-24 |
Family
ID=42129403
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/051833 WO2010050628A2 (en) | 2009-06-22 | 2010-02-08 | Foamed rubber composition for shoe sole and outersole |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP5565313B2 (en) |
| WO (1) | WO2010050628A2 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013062685A3 (en) * | 2011-10-25 | 2013-10-17 | Exxonmobil Chemical Patents Inc. | Composition, foam, and article made therefrom |
| CN104356437A (en) * | 2014-10-30 | 2015-02-18 | 际华三五三七制鞋有限责任公司 | Calendering type leaf-yellow rubber foxing glue for liberation shoes, and preparation method and application thereof |
| CN104356438A (en) * | 2014-10-30 | 2015-02-18 | 际华三五三七制鞋有限责任公司 | Rolling type grass green foxing glue for folding-resistant liberation shoes as well as preparation method and application thereof |
| CN104371150A (en) * | 2014-10-30 | 2015-02-25 | 际华三五三七制鞋有限责任公司 | Bending-resistant calendered leaf-yellow rubber edge strip rubber for liberation shoes, and preparation method and application thereof |
| CN104371151A (en) * | 2014-10-30 | 2015-02-25 | 际华三五三七制鞋有限责任公司 | Calendered black rubber surround bar for flexing endurable liberation shoes as well as preparation method and application of calendered black rubber surround bar |
| JP2015193783A (en) * | 2014-03-20 | 2015-11-05 | 宇部興産株式会社 | Rubber composition for foam, rubber foam for shoe sole using the rubber composition, and shoe sole |
| CN105131366A (en) * | 2015-10-09 | 2015-12-09 | 际华三五三七制鞋有限责任公司 | Natural color rubber surrounding bar gum for odorless environment-friendly rolling type high-elongation shoes and preparation method and application thereof |
| WO2016014230A1 (en) | 2014-07-25 | 2016-01-28 | Exxonmobil Chemical Patents Inc. | Footwear compositions comprising propylene-based elastomers |
| EP3178340A4 (en) * | 2014-08-07 | 2017-09-20 | ASICS Corporation | Sole for shoes, and shoes |
| CN107922665A (en) * | 2015-10-19 | 2018-04-17 | 埃拉斯托米克斯株式会社 | Rubber composition and cross-linked rubber article and its manufacture method |
| CN112812383A (en) * | 2021-02-07 | 2021-05-18 | 福建美明达鞋业发展有限公司 | Sports shoe sole with good anti-skid effect and preparation method thereof |
| US11192992B2 (en) | 2016-12-29 | 2021-12-07 | Exxonmobil Chemical Patents Inc. | Thermoplastic vulcanizates for foaming applications |
| CN114085438A (en) * | 2021-11-19 | 2022-02-25 | 温州市耀阳鞋业有限公司 | Anti-slip men's shoes and preparation method thereof |
| WO2022137394A1 (en) * | 2020-12-23 | 2022-06-30 | 株式会社アシックス | Cushioning member and sole for shoe |
| DE102021214842A1 (en) | 2021-12-21 | 2023-06-22 | Continental Reifen Deutschland Gmbh | Shoe sole material, outsole and method of manufacturing the shoe sole material or outsole |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101801848B1 (en) * | 2016-12-08 | 2017-11-27 | 하관형 | Sole of Shoes |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006016518A (en) * | 2004-07-02 | 2006-01-19 | Jsr Corp | Composition for foam |
| JP2009120660A (en) * | 2007-11-13 | 2009-06-04 | Sekisui Chem Co Ltd | Thermally expandable microcapsule and foam-molded body |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5956432A (en) * | 1982-09-27 | 1984-03-31 | Toyo Rubber Chem Ind Co Ltd | Production of rubber foam |
| JPS62181342A (en) * | 1986-02-06 | 1987-08-08 | Japan Synthetic Rubber Co Ltd | Crosslinked foam |
| JP3476543B2 (en) * | 1994-06-28 | 2003-12-10 | ジーイー東芝シリコーン株式会社 | Silicone rubber sponge composition |
| JP2002088693A (en) * | 2000-09-08 | 2002-03-27 | Matsui Shikiso Chem Co Ltd | Wallpaper having 30 min or more steckigt sizing degree |
| JP4003964B2 (en) * | 2003-11-12 | 2007-11-07 | タイガースポリマー株式会社 | Method for producing fluororubber foam |
-
2010
- 2010-02-08 WO PCT/JP2010/051833 patent/WO2010050628A2/en active Application Filing
- 2010-02-08 JP JP2010535862A patent/JP5565313B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006016518A (en) * | 2004-07-02 | 2006-01-19 | Jsr Corp | Composition for foam |
| JP2009120660A (en) * | 2007-11-13 | 2009-06-04 | Sekisui Chem Co Ltd | Thermally expandable microcapsule and foam-molded body |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013062685A3 (en) * | 2011-10-25 | 2013-10-17 | Exxonmobil Chemical Patents Inc. | Composition, foam, and article made therefrom |
| JP2015193783A (en) * | 2014-03-20 | 2015-11-05 | 宇部興産株式会社 | Rubber composition for foam, rubber foam for shoe sole using the rubber composition, and shoe sole |
| WO2016014230A1 (en) | 2014-07-25 | 2016-01-28 | Exxonmobil Chemical Patents Inc. | Footwear compositions comprising propylene-based elastomers |
| EP3178340A4 (en) * | 2014-08-07 | 2017-09-20 | ASICS Corporation | Sole for shoes, and shoes |
| CN104371151A (en) * | 2014-10-30 | 2015-02-25 | 际华三五三七制鞋有限责任公司 | Calendered black rubber surround bar for flexing endurable liberation shoes as well as preparation method and application of calendered black rubber surround bar |
| CN104371150A (en) * | 2014-10-30 | 2015-02-25 | 际华三五三七制鞋有限责任公司 | Bending-resistant calendered leaf-yellow rubber edge strip rubber for liberation shoes, and preparation method and application thereof |
| CN104356438A (en) * | 2014-10-30 | 2015-02-18 | 际华三五三七制鞋有限责任公司 | Rolling type grass green foxing glue for folding-resistant liberation shoes as well as preparation method and application thereof |
| CN104356437A (en) * | 2014-10-30 | 2015-02-18 | 际华三五三七制鞋有限责任公司 | Calendering type leaf-yellow rubber foxing glue for liberation shoes, and preparation method and application thereof |
| CN105131366A (en) * | 2015-10-09 | 2015-12-09 | 际华三五三七制鞋有限责任公司 | Natural color rubber surrounding bar gum for odorless environment-friendly rolling type high-elongation shoes and preparation method and application thereof |
| CN107922665A (en) * | 2015-10-19 | 2018-04-17 | 埃拉斯托米克斯株式会社 | Rubber composition and cross-linked rubber article and its manufacture method |
| US11192992B2 (en) | 2016-12-29 | 2021-12-07 | Exxonmobil Chemical Patents Inc. | Thermoplastic vulcanizates for foaming applications |
| WO2022137394A1 (en) * | 2020-12-23 | 2022-06-30 | 株式会社アシックス | Cushioning member and sole for shoe |
| CN112812383A (en) * | 2021-02-07 | 2021-05-18 | 福建美明达鞋业发展有限公司 | Sports shoe sole with good anti-skid effect and preparation method thereof |
| CN114085438A (en) * | 2021-11-19 | 2022-02-25 | 温州市耀阳鞋业有限公司 | Anti-slip men's shoes and preparation method thereof |
| DE102021214842A1 (en) | 2021-12-21 | 2023-06-22 | Continental Reifen Deutschland Gmbh | Shoe sole material, outsole and method of manufacturing the shoe sole material or outsole |
| WO2023116989A1 (en) | 2021-12-21 | 2023-06-29 | Continental Reifen Deutschland Gmbh | Shoe sole material, outsole and method for producing the shoe sole material or the outsole |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010050628A3 (en) | 2010-06-24 |
| JP5565313B2 (en) | 2014-08-06 |
| JPWO2010050628A1 (en) | 2012-04-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5565313B2 (en) | Foam rubber composition for shoe sole and outsole | |
| JP2012213615A (en) | Shoe-sole foamed body rubber composition and outsole | |
| JP4912588B2 (en) | Open cell foam of ethylene / propylene / diene rubber | |
| Zhang et al. | Effect of carbon black content on microcellular structure and physical properties of chlorinated polyethylene rubber foams | |
| EP1872924A1 (en) | Process for producing crosslinked molded foam | |
| KR101837328B1 (en) | Compositions comprising thermoplastic vulcanizate, foamed material and articles made therfrom | |
| JP4728735B2 (en) | Olefinic thermoplastic elastomer composition and foam thereof | |
| JP2007039499A (en) | Rubber composition for tire | |
| KR102160077B1 (en) | A resin composition with hybrid blowing agents which are improved shrinkage | |
| JP5396623B2 (en) | Rubber product | |
| JP6398522B2 (en) | Rubber composition for foam and shoe sole using the same | |
| WO2017068893A1 (en) | Rubber composition, and crosslinked rubber product and method for producing same | |
| JP2004091747A (en) | Tire rubber composition and its production method | |
| WO2020059917A1 (en) | Resin composition with improved shrinkage comprising mixed blowing agent | |
| JP2001002866A (en) | Rubber composition for sound insulating sponge and vulcanized foamed item | |
| JP2006152058A (en) | Rubber composition | |
| JP2005120335A (en) | Hollow sponge and automobile sealing material | |
| JP3980001B2 (en) | Rubber composition | |
| JP2005171092A (en) | Rubber composition for tire | |
| JP2000234038A (en) | Thermosetting rubber composition for sponge and vulcanized rubber expansion molded product thereof | |
| JP3773376B2 (en) | Rubber composition for sponge and its vulcanized rubber foam molding | |
| JP2005329571A (en) | Composite molded product | |
| JPH11279290A (en) | Recycled rubber molded product and its production | |
| JP2004091746A (en) | Method for producing tire rubber composition | |
| TWI780526B (en) | High-functional foaming resin composition and manufacturing method thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10714552 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010535862 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10714552 Country of ref document: EP Kind code of ref document: A2 |