WO2010045505A1 - Boron-containing small molecules as anti-protozoal agents - Google Patents
Boron-containing small molecules as anti-protozoal agents Download PDFInfo
- Publication number
- WO2010045505A1 WO2010045505A1 PCT/US2009/060917 US2009060917W WO2010045505A1 WO 2010045505 A1 WO2010045505 A1 WO 2010045505A1 US 2009060917 W US2009060917 W US 2009060917W WO 2010045505 A1 WO2010045505 A1 WO 2010045505A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- exemplary embodiment
- compound
- alkyl
- substituted
- group
- Prior art date
Links
- 0 *B(c1c2)OCc1ccc2OCc1c(*)c(*)c(*)c(*)c1* Chemical compound *B(c1c2)OCc1ccc2OCc1c(*)c(*)c(*)c(*)c1* 0.000 description 5
- AGEZXYOZHKGVCM-UHFFFAOYSA-N BrCc1ccccc1 Chemical compound BrCc1ccccc1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- DPXQAZQQGXDNPC-UHFFFAOYSA-N C=[Br]c1cc(C2(c3ccccc3)OCCO2)ccc1C1OCCO1 Chemical compound C=[Br]c1cc(C2(c3ccccc3)OCCO2)ccc1C1OCCO1 DPXQAZQQGXDNPC-UHFFFAOYSA-N 0.000 description 1
- XGNYZUAZHLLLFC-UHFFFAOYSA-N COCOCc(c(Br)c1)ccc1S(c1cc(Cl)ccc1)(=O)=O Chemical compound COCOCc(c(Br)c1)ccc1S(c1cc(Cl)ccc1)(=O)=O XGNYZUAZHLLLFC-UHFFFAOYSA-N 0.000 description 1
- KXDYMGOOAZKIBG-UHFFFAOYSA-N Cc1cccc(S(Nc2ccc(COB3O)c3c2)(=O)=O)n1 Chemical compound Cc1cccc(S(Nc2ccc(COB3O)c3c2)(=O)=O)n1 KXDYMGOOAZKIBG-UHFFFAOYSA-N 0.000 description 1
- USCKQKQTFCBECF-UHFFFAOYSA-N Nc1ccc(COB2O)c2c1 Chemical compound Nc1ccc(COB2O)c2c1 USCKQKQTFCBECF-UHFFFAOYSA-N 0.000 description 1
- BCSPNFQGTHFFBY-UHFFFAOYSA-N OB(c1c(C=O)ccc(C(c2ccccc2)=O)c1)O Chemical compound OB(c1c(C=O)ccc(C(c2ccccc2)=O)c1)O BCSPNFQGTHFFBY-UHFFFAOYSA-N 0.000 description 1
- BGWLQHAPVCGARL-UHFFFAOYSA-N OB(c1c2)OCc1ccc2NS(c1cc2ccccc2cc1)(=O)=O Chemical compound OB(c1c2)OCc1ccc2NS(c1cc2ccccc2cc1)(=O)=O BGWLQHAPVCGARL-UHFFFAOYSA-N 0.000 description 1
- VVRHWLMUVDGIJJ-UHFFFAOYSA-N OB(c1c2)OCc1ccc2O Chemical compound OB(c1c2)OCc1ccc2O VVRHWLMUVDGIJJ-UHFFFAOYSA-N 0.000 description 1
- HLFDUGGPWTUHOZ-UHFFFAOYSA-N OB(c1c2)OCc1ccc2Sc1cc(Cl)ccc1 Chemical compound OB(c1c2)OCc1ccc2Sc1cc(Cl)ccc1 HLFDUGGPWTUHOZ-UHFFFAOYSA-N 0.000 description 1
- VXDGEAYKTHEYPI-UHFFFAOYSA-N OB1OCc(cc2)c1cc2C(O)=O Chemical compound OB1OCc(cc2)c1cc2C(O)=O VXDGEAYKTHEYPI-UHFFFAOYSA-N 0.000 description 1
- LRBPMXBSPAREEW-UHFFFAOYSA-N OB1OCc(cc2)c1cc2NCc1ccccc1 Chemical compound OB1OCc(cc2)c1cc2NCc1ccccc1 LRBPMXBSPAREEW-UHFFFAOYSA-N 0.000 description 1
- OKUPWNMQXIWLLF-UHFFFAOYSA-N OB1OCc(cc2)c1cc2S(c(cccc1)c1Cl)=O Chemical compound OB1OCc(cc2)c1cc2S(c(cccc1)c1Cl)=O OKUPWNMQXIWLLF-UHFFFAOYSA-N 0.000 description 1
- HUSTVPXUPTTWOG-UHFFFAOYSA-N OB1OCc(cc2)c1cc2S(c1cc(Cl)ccc1)(=O)=O Chemical compound OB1OCc(cc2)c1cc2S(c1cc(Cl)ccc1)(=O)=O HUSTVPXUPTTWOG-UHFFFAOYSA-N 0.000 description 1
- MKIGPENLEQKNLT-UHFFFAOYSA-N OB1OCc2c1cc(C(c1ccccc1)O)cc2 Chemical compound OB1OCc2c1cc(C(c1ccccc1)O)cc2 MKIGPENLEQKNLT-UHFFFAOYSA-N 0.000 description 1
- PWBFUKYLAOVVJB-UHFFFAOYSA-N OB1OCc2c1cc(Cc1ccccc1)cc2 Chemical compound OB1OCc2c1cc(Cc1ccccc1)cc2 PWBFUKYLAOVVJB-UHFFFAOYSA-N 0.000 description 1
- JZQMKSZUNPDPSJ-UHFFFAOYSA-N ONc(c(C(F)(F)F)c1)ccc1[Zn] Chemical compound ONc(c(C(F)(F)F)c1)ccc1[Zn] JZQMKSZUNPDPSJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic System
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- oxaboroles useful as antimicrobials have been described previously, such as in U.S. Pat. Pubs. US20060234981 and US20070155699.
- an oxaborole has the following structure and substituent numbering system:
- This invention provides, among other things, novel compounds useful for treating protozoa infections, pharmaceutical compositions containing such compounds, as well as combinations of these compounds with at least one additional therapeutically effective agent.
- FIG. 1 provides biological data for compounds of the invention.
- an active agent includes a single active agent as well as two or more different active agents in combination. It is to be understood that present teaching is not limited to the specific dosage forms, carriers, or the like, disclosed herein and as such may vary.
- aq. is aqueous; AcOH is acetic acid; ACTBr is cetyltrimethylammonium bromide; B 2 pin 2 - bis(pinacolato)diboron; Boc is tert-butoxy carbonyl; Boc 2 O-di-te/t-butyl dicarbonate; BzOOH-benzoyl peroxide; Cs 2 COs is cesium carbonate; DABCO is 1,4- diazabicyclo[2.2.2]octane; DCM is dichloromethane or methylene chloride; DIAD is diisopropyl azodicarboxylate; DIEA is diisopropylethylamine; NJSf- Diisopropylethylamine is DIPEA; DMAP is 4-(dimethylamino)pyridine; DME is 1 ,2- dimethoxyethane; DMF is N,N-dimethylformamide; DM
- EtOAc is ethyl acetate
- EtOH is ethanol
- Et 2 O is diethyl ether
- m-CPB A-3 chloroperoxybenzoic acid; equiv-equivalent
- h is hours
- HATU is O-(7- azabenzotriazol-l-yl)- ⁇ , ⁇ , ⁇ ', ⁇ '-tetramethyluronium hexafluorophosphate
- HCl is hydrochloric acid
- HPLC high pressure liquid chromatography
- ISCO Companion is automated flash chromatography equipment with fraction analysis by UV absorption available from Presearch
- KOAc is potassium acetate
- K 2 C ⁇ 3 is potassium carbonate
- LiAlH 4 or LAH is lithium aluminum hydride
- LDA is lithium diisopropylamide
- LHMDS is lithium bis(
- Compound of the invention refers to the compounds discussed herein, salts (e.g. pharmaceutically acceptable salts), prodrugs, solvates and hydrates of these compounds.
- Combination of the invention refers to the compounds and antiprotozoals discussed herein as well as acids, bases, salt forms (such as pharmaceutically acceptable salts), prodrugs, solvates and hydrates of these compounds and antiprotozoals.
- Boon containing compounds refers to the compounds of the invention that contain boron as part of their chemical formula.
- MIC or minimum inhibitory concentration
- MIC is the point where the compound stops more than 50% of cell growth, preferably 60% of cell growth, preferably 70% of cell growth, preferably 80% of cell growth, preferably 90% of cell growth, relative to an untreated control.
- substituent groups are specified by their conventional chemical formulae, written from left to right, they equally encompass the chemically identical substituents, which would result from writing the structure from right to left, e.g., -CH 2 O- is intended to also recite -OCH 2 -.
- poly as used herein means at least 2.
- a polyvalent metal ion is a metal ion having a valency of at least 2.
- Moiety refers to a radical of a molecule that is attached to the remainder of the molecule.
- v/wvr whether utilized as a bond or displayed perpendicular to a bond, indicates the point at which the displayed moiety is attached to the remainder of the molecule.
- alkyl by itself or as part of another substituent, means, unless otherwise stated, a straight or branched chain, or cyclic hydrocarbon radical, or combination thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals, having the number of carbon atoms designated (i.e. Ci-Cio means one to ten carbons).
- the term “alkyl” means a straight or branched chain, or combinations thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals.
- saturated hydrocarbon radicals include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, cyclohexyl, (cyclohexyl)methyl, cyclopropylmethyl, homologs and isomers of, for example, n- pentyl, n-hexyl, n-heptyl, n-octyl, and the like.
- An unsaturated alkyl group is one having one or more double bonds or triple bonds.
- unsaturated alkyl groups include, but are not limited to, vinyl, 2-propenyl, crotyl, 2-isopentenyl, 2- (butadienyl), 2,4-pentadienyl, 3-(l,4-pentadienyl), ethynyl, 1- and 3-propynyl, 3- butynyl, and the higher homologs and isomers.
- alkylene by itself or as part of another substituent means a divalent radical derived from an alkane, as exemplified, but not limited, by - CH2CH2CH2CH2-, and further includes those groups described below as
- alkylene typically, an alkyl (or alkylene) group will have from 1 to 24 carbon atoms, with those groups having 10 or fewer carbon atoms being preferred in the present invention.
- a “lower alkyl” or “lower alkylene” is a shorter chain alkyl or alkylene group, generally having eight or fewer carbon atoms.
- alkoxy alkylamino
- alkylthio or thioalkoxy
- heteroalkyl by itself or in combination with another term, means, unless otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom.
- the term “heteroalkyl,” by itself or in combination with another term means a stable straight or branched chain, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom.
- the heteroatoms can be selected from the group consisting of B, O, N and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized.
- the heteroatom(s) B, O, N and S may be placed at any interior position of the heteroalkyl group or at the position at which the alkyl group is attached to the remainder of the molecule.
- heteroalkylene by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified, but not limited by, -CH 2 -CH 2 -S-CH 2 -CH 2 - and -CH 2 -S-CH 2 -CH 2 -NH-CH 2 -.
- heteroatoms can also occupy either or both of the chain termini (e.g., alkyleneoxy, alkylenedioxy, alkyleneamino, alkylenediamino, and the like).
- cycloalkyl examples include, but are not limited to, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like.
- heterocycloalkyl examples include, but are not limited to, 1 -(1,2,5,6- tetrahydropyridyl), 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 4-morpholinyl, 3- morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl, tetrahydrothien-3-yl, 1 -piperazinyl, 2-piperazinyl, and the like.
- halo or halogen
- haloalkyl by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom.
- terms such as “haloalkyl,” are meant to include monohaloalkyl and polyhaloalkyl.
- halo(Ci-C 4 )alkyl is mean to include, but not be limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
- aryl means, unless otherwise stated, a polyunsaturated, aromatic, substituent that can be a single ring or multiple rings (preferably from 1 to 3 rings), which are fused together or linked covalently.
- heteroaryl refers to aryl groups (or rings) that contain from one to four heteroatoms.
- the heteroatom is selected from B, N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- a heteroaryl group can be attached to the remainder of the molecule through a heteroatom.
- Non- limiting examples of aryl and heteroaryl groups include phenyl, 1-naphthyl, 2-naphthyl, 4-biphenyl, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3- pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 2-phenyl-4- oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2- pyrimidyl, 4-pyrimidyl, 5-benzothiazolyl, purinyl, 2-benzimidazolyl, 5-indolyl, 1-
- aryl when used in combination with other terms (e.g. , aryloxy, arylthioxy, arylalkyl) includes both aryl and heteroaryl rings as defined above.
- arylalkyl is meant to include those radicals in which an aryl group is attached to an alkyl group (e.g.
- an oxygen atom e.g., phenoxymethyl, 2- pyridyloxymethyl, 3-(l-naphthyloxy)propyl, and the like.
- -NR""-C(NR'R") NR'", -S(O)R', -S(O) 2 R', -S(O) 2 NR 5 R", -NR"S0 2 R', -CN, -NO 2 , -N 3 , -CH(Ph) 2 , fluoro(Ci-C 4 )alkoxy, and fluoro(Ci-C 4 )alkyl, in a number ranging from zero to (2m'+l), where m' is the total number of carbon atoms in such radical.
- R', R", R'", R"" and R'" each preferably independently refer to hydrogen, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl, e.g. , aryl substituted with 1-3 halogens, substituted or unsubstituted alkyl, alkoxy or thioalkoxy groups, or arylalkyl groups.
- each of the R groups is independently selected as are each R', R", R'", R"" and R'"" groups when more than one of these groups is present.
- R' and R" When R' and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6-, or 7-membered ring.
- -NR'R is meant to include, but not be limited to, 1-pyrrolidinyl and 4-morpholinyl.
- alkyl is meant to include groups including carbon atoms bound to groups other than hydrogen groups, such as haloalkyl (e.g., -CF 3 and -CH 2 CF 3 ) and acyl (e.g., - C(O)CH 3 , -C(O)CF 3 , -C(O)CH 2 OCH 3 , and the like).
- haloalkyl e.g., -CF 3 and -CH 2 CF 3
- acyl e.g., - C(O)CH 3 , -C(O)CF 3 , -C(O)CH 2 OCH 3 , and the like.
- substituents for the aryl and heteroaryl groups are generically referred to as "aryl group substituents.”
- Two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)-(CRR') q -U-, wherein T and U are independently -NR-, -O-, -CRR'- or a single bond, and q is an integer of from 0 to 3.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula - A-(CH 2 ) r -B-, wherein A and B are independently -CRR'-, -O-, -NR-, -S-, -S(O)-, -S(O) 2 -, -S(O) 2 NR'- or a single bond, and r is an integer of from 1 to 4.
- One of the single bonds of the new ring so formed may optionally be replaced with a double bond.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CRR') s -X-(CR"R'")d-, where s and d are independently integers of from 0 to 3, and X is -O-, -NR'-, -S-, -S(O)-, -S(O) 2 -, or -S(O) 2 NR'-.
- the substituents R, R', R" and R'" are preferably independently selected from hydrogen or substituted or unsubstituted (Ci-C 6 )alkyl.
- Ring means a substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl.
- a ring includes fused ring moieties. The number of atoms in a ring is typically defined by the number of members in the ring. For example, a "5- to 7-membered ring" means there are 5 to 7 atoms in the encircling arrangement. Unless otherwise specified, the ring optionally includes a heteroatom.
- the term “5- to 7-membered ring” includes, for example phenyl, pyridinyl and piperidinyl.
- the term “ring” further includes a ring system comprising more than one "ring”, wherein each "ring” is independently defined as above.
- heteroatom includes atoms other than carbon (C) and hydrogen (H). Examples include oxygen (O), nitrogen (N) sulfur (S), silicon (Si), germanium (Ge), aluminum (Al) and boron (B).
- leaving group means a functional group or atom which can be displaced by another functional group or atom in a substitution reaction, such as a nucleophilic substitution reaction.
- representative leaving groups include triflate, chloro, bromo and iodo groups; sulfonic ester groups, such as mesylate, tosylate, brosylate, nosylate and the like; and acyloxy groups, such as acetoxy, trifluoroacetoxy and the like.
- R is a general abbreviation that represents a substituent group that is selected from substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted cycloalkyl and substituted or unsubstituted heterocycloalkyl groups.
- effective amount of a drug, formulation, or permeant is meant a sufficient amount of an active agent to provide the desired local or systemic effect.
- Topicically effective refers to the amount of drug needed to effect the desired therapeutic result.
- Topicically effective refers to a material that, when applied to the skin, nail, hair, claw or hoof produces a desired pharmacological result either locally at the place of application or systemically as a result of transdermal passage of an active ingredient in the material.
- Cosmetically effective refers to a material that, when applied to the skin, nail, hair, claw or hoof, produces a desired cosmetic result locally at the place of application of an active ingredient in the material.
- pharmaceutically acceptable salt is meant to include a salt of a compound of the invention which are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the compounds described herein.
- base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent.
- pharmaceutically acceptable base addition salts include sodium, potassium, calcium, ammonium, organic amino, or magnesium salt, or a similar salt.
- acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent.
- Examples of pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, maleic, malonic, benzoic, succinic, suberic, fumaric, lactic, mandelic, phthalic, benzenesulfonic, p-tolylsulfonic, citric, tartaric, methanesulfonic, and the like.
- inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and
- salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galactunoric acids and the like (see, for example, Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)).
- Certain specific compounds of the invention contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts.
- the neutral forms of the compounds are preferably regenerated by contacting the salt with a base or acid and isolating the parent compounds in the conventional manner.
- the parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents.
- the present invention provides compounds which are in a prodrug form.
- Prodrugs of the compounds described herein readily undergo chemical changes under physiological conditions to provide the compounds of the invention. Additionally, prodrugs can be converted to the compounds of the invention by chemical or biochemical methods in an ex vivo environment.
- Certain compounds of the invention can exist in unsolvated forms as well as solvated forms, including hydrated forms. In general, the solvated forms are equivalent to unsolvated forms and are encompassed within the scope of the present invention. Certain compounds of the invention may exist in multiple crystalline or amorphous forms.
- Certain compounds of the invention possess asymmetric carbon atoms (optical centers) or double bonds; the racemates, diastereomers, geometric isomers and individual isomers are encompassed within the scope of the present invention.
- the graphic representations of racemic, ambiscalemic and scalemic or enantiomerically pure compounds used herein are taken from Maehr, J. Chem. Ed. 1985, 62: 114-120. Solid and broken wedges are used to denote the absolute configuration of a stereocenter unless otherwise noted.
- the compounds described herein contain olefmic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers. Likewise, all tautomeric forms are included.
- Compounds of the invention can exist in particular geometric or stereoisomeric forms.
- the invention contemplates all such compounds, including cis- and trans -isomers, (-)- and (+)-enantiomers, (R)- and (S)-enantiomers, diastereomers, (D)-isomers, (L)-isomers, the racemic mixtures thereof, and other mixtures thereof, such as enantiomerically or diastereomerically enriched mixtures, as falling within the scope of the invention.
- Additional asymmetric carbon atoms can be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention.
- Optically active (R)- and (5)-isomers and d and / isomers can be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. If, for instance, a particular enantiomer of a compound of the present invention is desired, it can be prepared by asymmetric synthesis, or by derivatization with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers.
- diastereomeric salts can be formed with an appropriate optically active acid or base, followed by resolution of the diastereomers thus formed by fractional crystallization or chromatographic means known in the art, and subsequent recovery of the pure enantiomers.
- separation of enantiomers and diastereomers is frequently accomplished using chromatography employing chiral, stationary phases, optionally in combination with chemical derivatization (e.g., formation of carbamates from amines).
- the compounds of the invention may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds.
- the compounds may be radiolabeled with radioactive isotopes, such as for example tritium ( 3 H), iodine-125 ( 125 I) or carbon-14 ( 14 C). All isotopic variations of the compounds of the invention, whether radioactive or not, are intended to be encompassed within the scope of the present invention.
- pharmaceutically acceptable carrier or “pharmaceutically acceptable vehicle” refers to any formulation or carrier medium that provides the appropriate delivery of an effective amount of an active agent as defined herein, does not interfere with the effectiveness of the biological activity of the active agent, and that is sufficiently non-toxic to the host or patient.
- Representative carriers include water, oils, both vegetable and mineral, cream bases, lotion bases, ointment bases and the like. These bases include suspending agents, thickeners, penetration enhancers, and the like. Their formulation is well known to those in the art of cosmetics and topical pharmaceuticals. Additional information concerning carriers can be found in Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005) which is incorporated herein by reference.
- “Pharmaceutically acceptable topical carrier” and equivalent terms refer to pharmaceutically acceptable carriers, as described herein above, suitable for topical application.
- An inactive liquid or cream vehicle capable of suspending or dissolving the active agent(s), and having the properties of being nontoxic and non-inflammatory when applied to the skin, nail, hair, claw or hoof is an example of a pharmaceutically- acceptable topical carrier. This term is specifically intended to encompass carrier materials approved for use in topical cosmetics as well.
- compositions refers to preservatives, antioxidants, fragrances, emulsif ⁇ ers, dyes and excipients known or used in the field of drug formulation and that do not unduly interfere with the effectiveness of the biological activity of the active agent, and that is sufficiently non-toxic to the host or patient.
- Additives for topical formulations are well-known in the art, and may be added to the topical composition, as long as they are pharmaceutically acceptable and not deleterious to the epithelial cells or their function. Further, they should not cause deterioration in the stability of the composition.
- inert fillers for example, inert fillers, anti- irritants, tackif ⁇ ers, excipients, fragrances, opacif ⁇ ers, antioxidants, gelling agents, stabilizers, surfactant, emollients, coloring agents, preservatives, buffering agents, other permeation enhancers, and other conventional components of topical or transdermal delivery formulations as are known in the art.
- the terms “enhancement,” “penetration enhancement” or “permeation enhancement” relate to an increase in the permeability of the skin, nail, hair, claw or hoof to a drug, so as to increase the rate at which the drug permeates through the skin, nail, hair, claw or hoof.
- the enhanced permeation effected through the use of such enhancers can be observed, for example, by measuring the rate of diffusion of the drug through animal skin, nail, hair, claw or hoof using a diffusion cell apparatus.
- a diffusion cell is described by Merritt et al. Diffusion Apparatus for Skin Penetration, J of Controlled Release, 1 (1984) pp. 161-162.
- the term “permeation enhancer” or “penetration enhancer” intends an agent or a mixture of agents, which, alone or in combination, act to increase the permeability of the skin, nail, hair or hoof to a drug.
- excipients is conventionally known to mean carriers, diluents and/or vehicles used in formulating drug compositions effective for the desired use.
- Topical administration refers to the application of a pharmaceutical agent to the external surface of the skin, nail, hair, claw or hoof, such that the agent crosses the external surface of the skin, nail, hair, claw or hoof and enters the underlying tissues.
- Topical administration includes application of the composition to intact skin, nail, hair, claw or hoof, or to a broken, raw or open wound of skin, nail, hair, claw or hoof.
- Topical administration of a pharmaceutical agent can result in a limited distribution of the agent to the skin and surrounding tissues or, when the agent is removed from the treatment area by the bloodstream, can result in systemic distribution of the agent.
- Transdermal delivery refers to the diffusion of an agent across the barrier of the skin, nail, hair, claw or hoof resulting from topical administration or other application of a composition.
- stratum corneum acts as a barrier and few pharmaceutical agents are able to penetrate intact skin.
- the epidermis and dermis are permeable to many solutes and absorption of drugs therefore occurs more readily through skin, nail, hair, claw or hoof that is abraded or otherwise stripped of the stratum corneum to expose the epidermis.
- Transdermal delivery includes injection or other delivery through any portion of the skin, nail, hair, claw or hoof or mucous membrane and absorption or permeation through the remaining portion.
- Absorption through intact skin, nail, hair, claw or hoof can be enhanced by placing the active agent in an appropriate pharmaceutically acceptable vehicle before application to the skin, nail, hair, claw or hoof.
- Passive topical administration may consist of applying the active agent directly to the treatment site in combination with emollients or penetration enhancers.
- transdermal delivery is intended to include delivery by permeation through or past the integument, i.e. skin, nail, hair, claw or hoof.
- an "effective amount” of one active of the combination is the amount of that active that is effective to provide the desired effect when used in combination with the other active of the combination.
- the amount that is “effective” will vary from subject to subject, depending on the age and general condition of the individual, the particular active agent or agents, and the appropriate “effective” amount in any individual case may be determined by one of ordinary skill in the art using routine experimentation.
- phrases "active ingredient”, “therapeutic agent”, “active”, or “active agent” mean a chemical entity which can be effective in treating a targeted disorder, disease or condition.
- pharmaceutically acceptable means moieties or compounds that are, within the scope of medical judgment, suitable for use in humans without causing undesirable biological effects such as undue toxicity, irritation, allergic response, and the like, for example.
- oral dosage form means any pharmaceutical composition administered to a subject via the oral cavity.
- exemplary oral dosage forms include tablets, capsules, films, powders, sachets, granules, solutions, solids, suspensions or as more than one distinct unit (e.g., granules, tablets, and/or capsules containing different actives) packaged together for co-administration, and other formulations known in the art.
- An oral dosage form can be one, two, three, four, five or six units. When the oral dosage form has multiple units, all of the units are contained within a single package, (e.g. a bottle or other form of packaging such as a blister pack). When the oral dosage form is a single unit, it may or may not be in a single package.
- the oral dosage form is one, two or three units. In a particularly preferred embodiment, the oral dosage form is one unit.
- the dosage form includes a compound of the invention in one capsule. This is a single unit. In some embodiments, the dosage form includes a compound of the invention as part of a therapeutically effective dosage of a cream or ointment. This is also a single unit. In some embodiments, the dosage form includes a compound of the invention and another active ingredient contained within one capsule, or as part of a therapeutically effective dosage of a cream or ointment. This is a single unit, whether or not the interior of the capsule includes multiple discrete granules of the active ingredient.
- the dosage form includes a compound of the invention in one capsule, and the active ingredient in a second capsule.
- This is a two unit dosage form, such as two capsules or tablets, and so such units are contained in a single package.
- the term 'unit' refers to the object which is administered to the animal, not to the interior components of the object.
- prodrug is a derivative of a parent drug molecule that exerts its pharmacological effect only after chemical and/or enzymatic conversion to its active form in vivo.
- Prodrugs include those designed to circumvent problems associated with delivery of the parent drug. This may be due to poor physicochemical properties, such as poor chemical stability or low aqueous solubility, and may also be due to poor pharmacokinetic properties, such as poor bioavailability or poor half- life. Thus, certain advantages of prodrugs may include improved chemical stability, absorption, and/or PK properties of the parent carboxylic acids.
- Prodrugs may also be used to make drugs more "patient friendly,” by minimizing the frequency (e.g., once daily) or route of dosing (e.g., oral), or to improve the taste or odor if given orally, or to minimize pain if given parenterally .
- the prodrugs are chemically more stable than the active drug, thereby improving formulation and delivery of the parent drug, compared to the drug alone.
- Prodrugs for carboxylic acid analogs of the invention may include a variety of esters.
- the pharmaceutical compositions of the invention include a carboxylic acid ester.
- the prodrug is suitable for treatment /prevention of those diseases and conditions that require the drug molecule to cross the blood brain barrier.
- the prodrug enters the brain, where it is converted into the active form of the drug molecule.
- a prodrug is used to enable an active drug molecule to reach the inside of the eye after topical application of the prodrug to the eye.
- a prodrug can be converted to its parent compound by chemical or biochemical methods in an ex vivo environment. For example, a prodrug can be slowly converted to its parent compound when placed in a transdermal patch reservoir with a suitable enzyme or chemical reagent.
- Antibiotic is a compound which can kill or inhibit the growth of bacteria.
- the term antibiotic is broad enough to encompass acids, bases, salt forms (such as pharmaceutically acceptable salts), prodrugs, solvates and hydrates of the antibiotic compound.
- Antiprotozoal or antiprotozoa is a compound which can kill or inhibit the growth of protozoa.
- the term antiprotozoal or antiprotozoa is broad enough to encompass acids, bases, salt forms (such as pharmaceutically acceptable salts), prodrugs, solvates and hydrates of the antiprotozoal or antiprotozoa compound.
- microbial infection or "infection by a microorganism” refers to any infection of a host by an infectious agent including, but not limited to, viruses, bacteria, mycobacteria, fungus and parasites (see, e.g., Harrison's Principles of Internal Medicine, pp. 93-98 (Wilson et al., eds., 12th ed. 1991); Williams et al, J. of Medicinal Chem. 42:1481-1485 (1999), herein each incorporated by reference in their entirety).
- Biological medium refers to both in vitro and in vivo biological milieus. Exemplary in vitro "biological media” include, but are not limited to, cell culture, tissue culture, homogenates, plasma and blood. In vivo applications are generally performed in mammals, preferably humans.
- Inhibiting and blocking are used interchangeably herein to refer to the partial or full blockade of an enzyme, such as a beta-lactamase or a leucyl t-RNA synthetase.
- leaving group means a functional group or atom which can be displaced by another functional group or atom in a substitution reaction, such as a nucleophilic substitution reaction.
- representative leaving groups include triflate, chloro, bromo and iodo groups; sulfonic ester groups, such as mesylate, tosylate, brosylate, nosylate and the like; and acyloxy groups, such as acetoxy, trifluoroacetoxy and the like.
- amino-protecting group means a protecting group suitable for preventing undesired reactions at an amino nitrogen.
- Representative amino-protecting groups include, but are not limited to, formyl; acyl groups, for example alkanoyl groups, such as acetyl, trichloroacetyl or trifluoroacetyl; alkoxycarbonyl groups, such as tert-butoxycarbonyl (Boc); arylmethoxycarbonyl groups, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethoxycarbonyl (Fmoc); arylmethyl groups, such as benzyl (Bn), trityl (Tr), and l,l-di-(4'-methoxyphenyl)methyl; silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
- hydroxy-protecting group means a protecting group suitable for preventing undesired reactions at a hydroxy group.
- Representative hydroxy- protecting groups include, but are not limited to, alkyl groups, such as methyl, ethyl, and tert-butyl; acyl groups, for example alkanoyl groups, such as acetyl; arylmethyl groups, such as benzyl (Bn), p-methoxybenzyl (PMB), 9-fluorenylmethyl (Fm), and diphenylmethyl (benzhydryl, DPM); silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
- alkyl groups such as methyl, ethyl, and tert-butyl
- acyl groups for example alkanoyl groups, such as acetyl
- arylmethyl groups such as benzyl (Bn), p-methoxy
- Dative bonds are usually weaker than covalent bonds.
- a boron is covalently bonded to at least one oxygen, sulfur or nitrogen, and is at the same time datively bonded to an oxygen, sulfur or nitrogen, respectively, the dative bond and covalent bond between the boron and the two identical heteroatoms can interconvert or be in the form of a resonance hybrid.
- the electrons in such bonds may be partially or fully delocalized.
- Embodiments of the present invention also encompass compounds that are poly- or multi-valent species, including, for example, species such as dimers, trimers, tetramers and higher homo logs of the compounds of use in the invention or reactive analogues thereof.
- Salt counterion refers to positively charged ions that associate with a compound of the invention when the boron is fully negatively or partially negatively charged.
- salt counterions include H + , H 3 O + , ammonium, potassium, calcium, magnesium and sodium.
- the compounds comprising a boron bonded to a carbon and three heteroatoms can optionally contain a fully negatively charged boron or partially negatively charged boron, due to the nature of the dative bond between the boron and one of the oxygens. Due to the negative charge, a positively charged counterion may associate with this compound, thus forming a salt.
- positively charged counterions include H + , H 3 O + , calcium, sodium, ammonium and potassium. The salts of these compounds are implicitly contained in descriptions of these compounds.
- the present invention provides novel boron compounds.
- novel compounds, as well as pharmaceutical compositions containing such compounds or combinations of these compounds with at least one additional therapeutically effective agent, can be used for, among other things, treating protozoal infections.
- the invention provides a compound of the invention.
- the invention is a compound described herein.
- the invention is a compound according to a formula described herein.
- the compound of the invention has the following structure:
- n is an integer from 0 to 5
- each X is an independently selected halogen
- R* is selected from the group consisting of H, a negative charge and a positively charged counterion
- L is selected from the group consisting of -S-, -S(O)-, -SO 2 -, -O-, - C(O)-, -C(OH)-, " -CH 2 O- ' , " -CH 2 NH- ' , " -C(O)NH- ' , " -NHC(O)- ' , " -NHC(O)O- ' , "-SO 2 NH-" and -CH 2 -, wherein " indicates a covalent linkage between L and the phenyl ring, and wherein ' indicates a covalent linkage between L and the boron- containing moiety, or a salt thereof.
- n is 0 and L is selected from the group consisting of -S(O)-, -SO 2 -, -C(O)-, -C(OH)-, "-CH 2 O-', "-NHC(O)O-” and -CH 2 -, wherein " indicates a covalent linkage between L and the phenyl ring, and wherein ' indicates a covalent linkage between L and the boron-containing moiety.
- n is O and L is "-NHC(O)-', wherein " indicates a covalent linkage between L and the phenyl ring, and wherein ' indicates a covalent linkage between L and the boron-containing moiety.
- R* is H.
- n is 1 and L is selected from the group consisting of -S-, -S(O)- and -SO 2 -, and there is a proviso that the compound is not In an exemplary embodiment, there is a proviso that the compound is not
- n is 2 and L is selected from the group consisting of -S-, -S(O)- and -SO 2 -.
- n is 2
- X is Cl and L is selected from the group consisting of -S-, -S(O)- and -SO 2 -.
- n is 2
- X is F and L is selected from the group consisting of -S-, -S(O)- and -SO 2 -.
- each X is F or Cl, wherein at least one X is Cl, and L is selected from the group consisting of -S-, -S(O)- and -SO 2 -.
- n is 3 and L is selected from the group consisting of -S-, -S(O)- and -SO 2 -. In an exemplary embodiment, n is 4 and L is selected from the group consisting of -S-, -S(O)- and -SO 2 -. In an exemplary embodiment, n is 5 and L is selected from the group consisting of -S-, -S(O)- and - SO 2 -.
- the compound has a structure according to the following formula:
- R* is selected from the group consisting of H, a negative charge and a positively charged counterion
- L is selected from the group consisting of -S-, -S(O)-, -SO 2 -, -O-, -C(O)-, -C(OH)-, ' -CH 2 O- ⁇ " -CH 2 NH- ' , " -C(O)NH- ' , " -NHC(O)- ' , " - NHC(O)O-", "-SO 2 NH-" and -CH 2 -, wherein " indicates a covalent linkage between L and the phenyl ring, and wherein ' indicates a covalent linkage between L and the boron-containing moiety, and R 2 , R 3 , R 4 , R 5 and R 6 are members selected from the following table, or a salt thereof.
- L is -S-, and R 2 , R 3 , R 4 , R 5 and R 6 are members selected from the following table. In an exemplary embodiment, L is -S(O)-, and R 2 , R 3 , R 4 , R 5 and R 6 are members selected from the following table. In an exemplary embodiment, L is -SO 2 -, and R 2 , R 3 , R 4 , R 5 and R 6 are according to the entries in the following table, or a salt thereof.
- the compound of the invention has the following structure:
- L is selected from the group consisting of -S-, -S(O)-, -SO 2 -, -O-, -C(O)-, -C(OH)-, " -CH 2 O- ' , " -CH 2 NH- ' , " -C(O)NH- ' , " -NHC(O)- ' , " -NHC(O)O- ' , " - SO 2 NH-” and -CH 2 -, wherein " indicates a covalent linkage between L and the phenyl ring, and wherein ' indicates a covalent linkage between L and the boron- containing moiety, or a salt thereof.
- L is "
- the compound has the following structure:
- the compound has the proviso that the compound is not
- the compound has structure which is
- the compound has structure which is
- the compound of the invention is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- R 2 is unsubstituted linear alkylene, and R 3 is halosubstituted aryl.
- R 2 is unsubstituted linear alkylene, and R 3 is monohaloaryl.
- R 2 is unsubstituted linear alkylene, and R 3 is A- haloaryl.
- R 2 is unsubstituted linear alkylene, and R 3 is 3-haloaryl.
- R 2 is unsubstituted linear alkylene, and R 3 is 2-haloaryl.
- R 2 is methylene, and R 3 is 4-haloaryl.
- R 2 is methylene, and R 3 is 3-haloaryl.
- R 2 is methylene, and R 3 is 2-haloaryl.
- the compound has a structure according to the following formula:
- R* is selected from the group consisting of H, a negative charge and a positively charged counterion
- R 2 , R 3 , R 4 , R 5 and R 6 are members selected from the following table, or a salt thereof.
- the compound has a structure which is , or a salt thereof.
- the compound has a structure which is , or a salt thereof.
- the compound of the invention has a structure which is
- the compound of the invention has a structure which is .
- the compound of the invention has a structure which is or salts thereof.
- the compound of the compound of the invention has a structure which is or salts thereof.
- the compound of the invention is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- R 27 is selected from the group consisting of unsubstituted indolyl, unsubstituted benzothiooxazolyl, and halosubstituted or unsubstituted pyrimidinyl.
- the compound of the invention is wherein R 27 is unsubstituted pyrimidinyl. In an exemplary embodiment, the compound is
- the compound of the invention is wherein R 27 is halosubstituted pyrimidinyl. In an exemplary embodiment, R 27 is monohalosubstituted pyrimidinyl. In an exemplary embodiment, R 27 is chlorosubstituted pyrimidinyl. In an exemplary embodiment, the compound is
- R , 28 is selected from the group consisting of F, Cl, Br and I.
- the compound is , wherein R , 28 is selected from the group consisting of F, Cl, Br and I.
- R , 28 is selected from the group consisting of F, Cl, Br and I.
- R , 28 is F.
- the compound is
- the compound of the invention is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- R , 27 is unsubstituted indolyl.
- the compound is
- the compound of the invention is wherein R , 27 is unsubstituted indolyl. In an exemplary embodiment, the compound is
- the invention provides a compound which is
- Z is selected from the group consisting of phenyl, pyridinyl and pyrazolyl
- b is 1 or 2 or 3 and each R a is independently selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl.
- Z is phenyl, and b and R a are as described herein.
- Z is pyridinyl, and b and R a are as described herein.
- Z is pyrazolyl, and b and R a are as described herein.
- the compound has a structure according to the following formula:
- b is 1 or 2 or 3 or 4 or 5, and each R a is independently selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl.
- b is 1.
- b is 2.
- b is 3.
- b is 4.
- b is 5.
- b is 1, R a is fluoro.
- b is 2, and at least one R a is fluoro.
- b is 3, and at least one R a is fluoro.
- b is 4, and at least one R a is fluoro.
- b is 5, and at least one R a is fluoro.
- b is 1, R a is chloro.
- b is 2, and at least one R a is chloro.
- b is 3, and at least one R a is chloro.
- b is 4, and at least one R a is chloro.
- b is 5, and at least one R a is chloro.
- b is 1, R a is bromo.
- b is 2, and at least one R a is bromo.
- b is 3, and at least one R a is bromo.
- b is 4, and at least one R a is bromo. In an exemplary embodiment, b is 5, and at least one R a is bromo. In an exemplary embodiment, b is 1, R a is iodo. In an exemplary embodiment, b is 2, and at least one R a is iodo. In an exemplary embodiment, b is 3, and at least one R a is iodo. In an exemplary embodiment, b is 4, and at least one R a is iodo. In an exemplary embodiment, b is 5, and at least one R a is iodo. In an exemplary embodiment, b is 1, R a is methyl.
- b is 2, and at least one R a is methyl. In an exemplary embodiment, b is 3, and at least one R a is methyl. In an exemplary embodiment, b is 4, and at least one R a is methyl. In an exemplary embodiment, b is 5, and at least one R a is methyl. In an exemplary embodiment, b is 1, R a is ethyl. In an exemplary embodiment, b is 2, and at least one R a is ethyl. In an exemplary embodiment, b is 3, and at least one R a is ethyl. In an exemplary embodiment, b is 4, and at least one R a is ethyl.
- b is 5, and at least one R a is ethyl. In an exemplary embodiment, b is 1, R a is an unsubstitued C 3 alkyl. In an exemplary embodiment, b is 2, and at least one R a is an unsubstitued C 3 alkyl. In an exemplary embodiment, b is 3, and at least one R a is an unsubstitued C 3 alkyl. In an exemplary embodiment, b is 4, and at least one R a is an unsubstitued C 3 alkyl. In an exemplary embodiment, b is 5, and at least one R a is an unsubstitued C 3 alkyl.
- b is 1, R a is an unsubstitued C 4 alkyl. In an exemplary embodiment, b is 2, and at least one R a is an unsubstitued C 4 alkyl. In an exemplary embodiment, b is 3, and at least one R a is an unsubstitued C 4 alkyl. In an exemplary embodiment, b is 4, and at least one R a is an unsubstitued C 4 alkyl. In an exemplary embodiment, b is 5, and at least one R a is an unsubstitued C 4 alkyl. In an exemplary embodiment, b is 1, R a is an unsubstitued C 5 alkyl. In an exemplary embodiment, b is 2, and at least one R a is an unsubstitued C 5 alkyl. In an exemplary embodiment, b is
- R a is an unsubstitued C 5 alkyl.
- b is
- R a is an unsubstitued C 5 alkyl.
- b is
- R a is an unsubstitued C 5 alkyl.
- b is 1, R a is an unsubstitued C 6 alkyl.
- b is 2, and at least one R a is an unsubstitued C 6 alkyl.
- b is 3, and at least one R a is an unsubstitued C 6 alkyl.
- b is 4, and at least one R a is an unsubstitued C 6 alkyl.
- b is 5, and at least one R a is an unsubstitued C 6 alkyl.
- b is 1 or 2, and at least one R a is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl.
- b is 1 and R a is halo-substituted C 1 - C 6 alkyl.
- b is 2, and at least one R a is halo-substituted Ci-C 6 alkyl.
- b is 3, and at least one R a is halo- substituted Ci-C 6 alkyl.
- b is 4, and at least one R a is halo-substituted Ci-C 6 alkyl.
- b is 5, and at least one R a is halo-substituted Ci-C 6 alkyl.
- b is 1 and R a is Ci-C 6 alkyl, substituted with one halogen.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with one halogen.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with one halogen.
- b is 1 and R a is Ci-C 6 alkyl, substituted with two halogens.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 3, and at least one R a is Ci-C 6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 3, and at least one R a is Ci-C 6 alkyl, substituted with three halogens.
- b is 1 and R a is Ci-C 6 alkyl, substituted with one fluorine.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with one fluorine.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with one fluorine.
- b is 1 and R a is Ci-C 6 alkyl, substituted with two fluorines.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with two fluorines.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 3, and at least one R a is Ci-C 6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with one chlorine.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 3, and at least one R a is Ci-C 6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 3, and at least one R a is Ci-C 6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with three chlorines.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 3, and at least one R a is Ci-C 6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 1 or 2 or 3 and R a is Ci-C 6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, b is 1 or 2 or 3 and R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one chlorine. In an exemplary embodiment, b is 1 or 2 or 3 and R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one bromine.
- b is 1 or 2 or 3 and R a is Ci-C 6 alkyl, substituted with at least one chlorine and at least one bromine.
- b is 1 or 2 or 3 and R a is fluoro-substituted Ci-C 6 alkyl.
- R a is trifluoro- substituted Ci-C 6 alkyl.
- the compound has a structure which is selected from the group consisting of: wherein R a is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl.
- R a is fluoro.
- R a is chloro.
- R a is bromo.
- R a is iodo.
- R a is methyl.
- R a is ethyl.
- R a is an unsubstitued C 3 alkyl.
- R a is an unsubstitued C 4 alkyl. In an exemplary embodiment, R a is an unsubstitued C 5 alkyl. In an exemplary embodiment, R a is an unsubstitued C 6 alkyl. In an exemplary embodiment, R a is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, R a is halo-substituted Ci-C 6 alkyl. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with one halogen.
- R a is Ci-C 6 alkyl, substituted with two halogens. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with three halogens. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with four halogens. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with one fluorine. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with two fluorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with three fluorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with four fluorines.
- R a is Ci-C 6 alkyl, substituted with one chlorine. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with two chlorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with three chlorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with four chlorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one chlorine.
- R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one bromine. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with at least one chlorine and at least one bromine. In an exemplary embodiment, R a is fluoro-substituted Ci-C 6 alkyl. In an exemplary embodiment, R a is trifluoro-substituted Ci-C 6 alkyl.
- the compound has a structure which is: wherein b is 1 or 2 or 3 or 4, B is pyridinyl, R a is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl.
- b is 1.
- b is 2.
- b is 3.
- b is 4.
- b is 1, R a is fluoro.
- b is 2, and at least one R a is fluoro.
- b is 3, and at least one R a is fluoro.
- b is 4, and at least one R a is fluoro.
- b is 1, R a is chloro.
- b is 2, and at least one R a is chloro.
- b is 3, and at least one R a is chloro.
- b is 4, and at least one R a is chloro.
- b is 1, R a is bromo.
- b is 2, and at least one R a is bromo.
- b is 3, and at least one R a is bromo.
- b is 4, and at least one R a is bromo.
- b is 1, R a is iodo. In an exemplary embodiment, b is 2, and at least one R a is iodo. In an exemplary embodiment, b is 3, and at least one R a is iodo. In an exemplary embodiment, b is 4, and at least one R a is iodo. In an exemplary embodiment, b is 1, R a is methyl. In an exemplary embodiment, b is 2, and at least one R a is methyl. In an exemplary embodiment, b is 3, and at least one R a is methyl. In an exemplary embodiment, b is 4, and at least one R a is methyl. In an exemplary embodiment, b is 1, R a is ethyl.
- b is 2, and at least one R a is ethyl. In an exemplary embodiment, b is 3, and at least one R a is ethyl. In an exemplary embodiment, b is 4, and at least one R a is ethyl. In an exemplary embodiment, b is 1, R a is an unsubstitued C 3 alkyl. In an exemplary embodiment, b is 2, and at least one R a is an unsubstitued C 3 alkyl. In an exemplary embodiment, b is
- R a is an unsubstitued C 3 alkyl.
- b is
- R a is an unsubstitued C 3 alkyl.
- b is 1, R a is an unsubstitued C 4 alkyl.
- b is 2, and at least one R a is an unsubstitued C 4 alkyl.
- b is 3, and at least one R a is an unsubstitued C 4 alkyl.
- b is 4, and at least one R a is an unsubstitued C 4 alkyl.
- b is 1, R a is an unsubstitued C 5 alkyl.
- b is 2, and at least one R a is an unsubstitued C 5 alkyl. In an exemplary embodiment, b is 3, and at least one R a is an unsubstitued C 5 alkyl. In an exemplary embodiment, b is 4, and at least one R a is an unsubstitued C5 alkyl. In an exemplary embodiment, b is 1, R a is an unsubstitued C 6 alkyl. In an exemplary embodiment, b is 2, and at least one R a is an unsubstitued C 6 alkyl. In an exemplary embodiment, b is 3, and at least one R a is an unsubstitued C 6 alkyl.
- b is 4, and at least one R a is an unsubstitued C 6 alkyl. In an exemplary embodiment, b is 1 or 2, and at least one R a is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, b is 1 and R a is halo-substituted Ci-C 6 alkyl. In an exemplary embodiment, b is 2, and at least one R a is halo-substituted Ci-C 6 alkyl. In an exemplary embodiment, b is 3, and at least one R a is halo-substituted Ci-C 6 alkyl.
- b is 4, and at least one R a is halo-substituted Ci-C 6 alkyl.
- b is 1 and R a is Ci-C 6 alkyl, substituted with one halogen.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with one halogen.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with one halogen.
- b is 4, and at least one R a is Ci-C 6 alkyl, substituted with one halogen.
- b is 1 and R a is Ci-C 6 alkyl, substituted with two halogens.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with two halogens.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with two halogens.
- b is 4, and at least one R a is Ci-C 6 alkyl, substituted with two halogens.
- b is 1 and R a is Ci-C 6 alkyl, substituted with three halogens.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 3, and at least one R a is Ci-C 6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 4, and at least one R a is Ci-C 6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with one fluorine.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with one fluorine.
- b is 4, and at least one R a is Ci-C 6 alkyl, substituted with one fluorine.
- b is 1 and R a is Ci-C 6 alkyl, substituted with two fluorines.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with two fluorines.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with two fluorines.
- b is 4, and at least one R a is Ci-C 6 alkyl, substituted with two fluorines.
- b is 1 and R a is Ci-C 6 alkyl, substituted with three fluorines.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with three fluorines.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with three fluorines.
- b is 4, and at least one R a is Ci-C 6 alkyl, substituted with three fluorines.
- b is 1 and R a is Ci-C 6 alkyl, substituted with one chlorine.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with one chlorine.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with one chlorine.
- b is 4, and at least one R a is Ci-C 6 alkyl, substituted with one chlorine.
- b is 1 and R a is Ci-C 6 alkyl, substituted with two chlorines.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with two chlorines.
- b is 3, and at least one R a is Ci-C 6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 4, and at least one R a is Ci-C 6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 3, and at least one R a is Ci-C 6 alkyl, substituted with three chlorines.
- b is 4, and at least one R a is Ci-C 6 alkyl, substituted with three chlorines.
- b is 1 or 2 or 3 or 4 and R a is C 1 - C 6 alkyl, substituted with a combination of two different halogens.
- b is 1 or 2 or 3 or 4 and R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one chlorine.
- b is 1 or 2 or 3 or 4 and R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one bromine.
- b is 1 or 2 or 3 or 4 and R a is Ci-C 6 alkyl, substituted with at least one chlorine and at least one bromine.
- b is 1 or 2 or 3 or 4 and R a is fluoro-substituted Ci-C 6 alkyl.
- R a is trifluoro-substituted Ci-C 6 alkyl.
- the compound has a structure which is selected from the group consisting of: wherein R a is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl.
- R a is fluoro.
- R a is chloro.
- R a is bromo.
- R a is iodo.
- R a is methyl.
- R a is ethyl.
- R a is an unsubstitued C 3 alkyl.
- R a is an unsubstitued C 4 alkyl. In an exemplary embodiment, R a is an unsubstitued C 5 alkyl. In an exemplary embodiment, R a is an unsubstitued C 6 alkyl. In an exemplary embodiment, R a is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, R a is halo-substituted Ci-C 6 alkyl. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with one halogen.
- R a is Ci-C 6 alkyl, substituted with two halogens. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with three halogens. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with four halogens. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with one fluorine. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with two fluorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with three fluorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with four fluorines.
- R a is Ci-C 6 alkyl, substituted with one chlorine. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with two chlorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with three chlorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with four chlorines. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one chlorine.
- R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one bromine. In an exemplary embodiment, R a is Ci-C 6 alkyl, substituted with at least one chlorine and at least one bromine. In an exemplary embodiment, R a is fluoro-substituted Ci-C 6 alkyl. In an exemplary embodiment, R a is trifluoro-substituted Ci-C 6 alkyl.
- the compound has a structure according to the following formula:
- R 2 , R 3 , R 4 , R 5 and R 6 are according to the entries in the following table, or a salt thereof.
- the compound has a structure according to the following formula:
- R 3 , R 4 , R 5 and R 6 are according to the entries in the following table, or a salt thereof.
- the compound has a structure which is: wherein b is 1 or 2, C is pyrazolyl, R a is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl.
- b is 1.
- b is 2.
- R a is fluoro.
- b is 2, and at least one R a is fluoro.
- b is 1, R a is chloro.
- b is 2, and at least one R a is chloro.
- b is 1, R a is bromo.
- b is 2, and at least one R a is bromo. In an exemplary embodiment, b is 1, R a is iodo. In an exemplary embodiment, b is 2, and at least one R a is iodo. In an exemplary embodiment, b is 1, R a is methyl. In an exemplary embodiment, b is 2, and at least one R a is methyl. In an exemplary embodiment, b is
- R a is ethyl. In an exemplary embodiment, b is 2, and at least one R a is ethyl. In an exemplary embodiment, b is 1, R a is an unsubstitued C 3 alkyl. In an exemplary embodiment, b is 2, and at least one R a is an unsubstitued C 3 alkyl. In an exemplary embodiment, b is 1, R a is an unsubstitued C 4 alkyl. In an exemplary embodiment, b is
- R a is an unsubstitued C 4 alkyl.
- b is 1, R a is an unsubstitued Cs alkyl.
- b is 2, and at least one R a is an unsubstitued C 5 alkyl.
- b is 1, R a is an unsubstitued C 6 alkyl.
- b is 2, and at least one R a is an unsubstitued C 6 alkyl.
- b is 1 or 2, and at least one R a is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl.
- b is 1 and R a is halo-substituted Ci-C 6 alkyl.
- b is 2, and at least one R a is halo-substituted Ci-C 6 alkyl.
- b is 1 and R a is Ci-C 6 alkyl, substituted with one halogen.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with one halogen.
- b is 1 and R a is Ci-C 6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with one fluorine.
- b is 2, and at least one R a is Ci-C 6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with three fluorines.
- b is 1 and R a is Ci-C 6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 1 and R a is Ci-C 6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 2, and at least one R a is Ci-C 6 alkyl, substituted with three chlorines.
- b is 1 or 2 and R a is Ci-C 6 alkyl, substituted with a combination of two different halogens.
- b is 1 or 2 and R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one chlorine.
- b is 1 or 2 and R a is Ci-C 6 alkyl, substituted with at least one fluorine and at least one bromine.
- b is 1 or 2 and R a is Ci-C 6 alkyl, substituted with at least one chlorine and at least one bromine.
- b is 1 or 2 and R a is fluoro-substituted Ci-C 6 alkyl.
- R a is trifluoro-substituted Ci-C 6 alkyl.
- the compound has a structure according to the following formula: wherein R 2 , R 3 and R 4 are according to the entries in the following table, or a salt thereof.
- the compound has a structure which is selected from the group consisting of
- the cytotoxicity on murine L929 IC50 of a compound of the invention is a member selected from about 1 ⁇ M to 20 ⁇ M. In an exemplary embodiment, the cytotoxicity on murine L929 IC50 of a compound of the invention is a member selected from about 10 ⁇ M to 15 ⁇ M.
- the selectivity index (SI) of a compound of the invention is between about 50-150. In an exemplary embodiment, the selectivity index (SI) of 6-(4-chlorophenylsulfinyl)benzo[c][l,2]oxaborol-l(3H)-ol is greater than about 75-100.
- the in vitro metabolism T 1/2 (Mouse/human liver microsomes) of a compound of the invention is a member selected from about 300 minutes to 400 minutes. In an exemplary embodiment, the in vitro metabolism T 1/2 (Mouse/human liver microsomes) of a compound of the invention is a member selected from about 340 minutes to 360 minutes. In an exemplary embodiment, the in vitro metabolism T 1/2 (Mouse/human liver microsomes) of 6-(4-chlorophenyl sulfmyl)benzo[c][l,2]oxaborol-l(3H)-ol is greater than 350 minutes. [0103] In an exemplary embodiment, the in vitro metabolism T 1/2 (Mouse S9) of a compound of the invention is a member selected from about 100 minutes to 300 minutes.
- a compound of the invention essentially does not inhibit a cytochrome P450 enzyme. In an exemplary embodiment, a compound of the invention does not inhibit a cytochrome P450 enzyme. In an exemplary embodiment, 6-(4-chlorophenylsulf ⁇ nyl) benzo[c][l,2]oxaborol-l(3H)-ol essentially does not inhibit a cytochrome P450 enzyme. In an exemplary embodiment, 6-(4- chlorophenylsulfinyl)benzo[c][l,2]oxaborol-l(3H)-ol does not inhibit a cytochrome P450 enzyme. In an exemplary embodiment, the cytochrome P450 enzyme is a member selected from CP1A2, 2C9, 2D6 and 3A4. In an exemplary embodiment, the cytochrome P450 enzyme is CYP2C19.
- a compound of the invention is essentially not a substrate for the P-gp transporter. In an exemplary embodiment, a compound of the invention is not a substrate for the P-gp transporter. [0106] In an exemplary embodiment, the invention provides a compound described herein, or a salt, hydrate or solvate thereof, or a combination thereof. In an exemplary embodiment, the invention provides a compound described herein, or a salt, hydrate or solvate thereof. In an exemplary embodiment, the invention provides a compound described herein, or a salt thereof. In an exemplary embodiment, the salt is a pharmaceutically acceptable salt. In an exemplary embodiment, the invention provides a compound described herein, or a hydrate thereof.
- the invention provides a compound described herein, or a solvate thereof.
- the invention provides a compound described herein, or a prodrug thereof.
- the invention provides a salt of a compound described herein.
- the invention provides a pharmaceutically acceptable salt of a compound described herein.
- the invention provides a hydrate of a compound described herein.
- the invention provides a solvate of a compound described herein.
- the invention provides a prodrug of a compound described herein.
- alkyl is linear alkyl. In another exemplary embodiment, alkyl is branched alkyl.
- heteroalkyl is linear heteroalkyl. In another exemplary embodiment, heteroalkyl is branched heteroalkyl.
- HLb Compositions involving stereoisomers
- the term "chiral”, “enantiomerically enriched” or “diastereomerically enriched” refers to a composition having an enantiomeric excess (ee) or a diastereomeric excess (de) of greater than about 50%, preferably greater than about 70% and more preferably greater than about 90%. In general, higher than about 90% enantiomeric or diastereomeric excess is particularly preferred, e.g., those compositions with greater than about 95%, greater than about 97% and greater than about 99% ee or de.
- first compound and a second compound are present in a composition, and the first compound is a non-superimposable mirror image of the second compound, and the first compound is present in the composition in a greater amount than the second compound, then the first compound is referred to herein as being present in "enantiomeric excess".
- e e ⁇ ( COnC - ° f Z - C ° nC - ° f A x 100 yconc. of z + cone, of y) wherein z is a first compound in a composition, y is a second compound in the composition, and the first compound is a non-superimposable mirror image of the second compound.
- enantiomeric excess is related to the older term “optical purity” in that both are measures of the same phenomenon.
- the value of ee will be a number from 0 to 100, zero being racemic and 100 being enantiomerically pure.
- a composition which in the past might have been called 98% optically pure is now more precisely characterized by 96% ee.
- a 90% ee reflects the presence of 95% of one enantiomer and 5% of the other(s) in the material in question.
- _ cone, of major diastereomer - cone, of ram or diastereomer (s) ⁇ yconc. of major diastereomer + cone, of ram or diastereomer is) J wherein the major diastereomer is a first compound in a composition, and the minor diastereomer(s) is at least one additional compound in the composition, and the major diastereomer and minor diastereomer(s) are stereoisomers, but not mirror images, of one another.
- the value of de will likewise be a number from 0 to 100, zero being an equal mixture of a first diastereomer and the remaining diastereomer(s), and 100 being 100% of a single diastereomer and zero% of the other(s) - i.e. diastereomerically pure.
- 90% de reflects the presence of 95% of one diastereomer and 5% of the other diastereomer(s) in the material in question.
- the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has at least one stereocenter, and at least one stereoisomer of the first compound of the invention.
- the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has at least one stereocenter, and a second compound of the invention, wherein the first compound of the invention is a stereoisomer of the second compound of the invention.
- the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has at least one stereocenter, and only one stereoisomer of the first compound of the invention.
- the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has only one stereocenter, and an enantiomer of the first compound of the invention.
- the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has two stereocenters, and an enantiomer of the first compound of the invention.
- the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has two stereocenters, and at least one diasteromer of the first compound of the invention.
- the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has two stereocenters, and only one diasteromer of the first compound of the invention.
- the first compound of the invention can be present in an enantiomeric excess of at least about 80%, or at least about 90%, or at least about 92% or at least about 95%.
- the first compound of the invention can be present in an enantiomeric excess of at least about 96%, at least about 97%, at least about 98%, at least about 99% or at least about 99.5%.
- the first compound of the invention has at least one stereocenter and is enantiomerically pure (enantiomeric excess is about 100%).
- the first compound of the invention can be present in a diastereomeric excess of at least about 80%, or at least about 90%, or at least about 92% or at least about 95%. In situations where the first compound of the invention and at least one diastereomer of the first compound of the invention are present in a composition, the first compound of the invention can be present in a diastereomeric excess of at least about 96%, at least about 97%, at least about 98%, at least about 99% or at least about 99.5%. In another embodiment, the first compound of the invention has at least two stereocenters and is diastereomerically pure (diastereomeric excess is about 100%).
- Enantiomeric or diastereomeric excess can be determined relative to exactly one other stereoisomer, or can be determined relative to the sum of at least two other stereoisomers. In an exemplary embodiment, enantiomeric or diastereomeric excess is determined relative to all other detectable stereoisomers, which are present in the mixture. Stereoisomers are detectable if a concentration of such stereoisomer in the analyzed mixture can be determined using common analytical methods, such as chiral HPLC.
- composition that is "substantially free" of a compound means that the composition contains less than about 20% by weight, or less than about 15% by weight, or less than about 10% by weight, or less than about 5% by weight, or less than about 3% by weight, or less than about 2% by weight, or less than about 1% by weight of the compound.
- the term "substantially free of the (or its) enantiomer” means that a composition contains a significantly greater proportion of a first compound of the invention than a second compound of the invention, wherein the first compound is a non-superimposable mirror image of the second compound.
- the term “substantially free of the enantiomer” means that the composition is made up of at least about 90% by weight of a first compound of the invention, and about 10% by weight or less of a second compound of the invention, wherein the first compound is a non-superimposable mirror image of the second compound.
- the term "substantially free of the (R) enantiomer” means that the composition is made up of at least about 90% by weight of a first compound of the invention which has only one stereocenter and the stereocenter is in an (S) configuration, and about 10% by weight or less of a second compound of the invention, wherein the second compound is the enantiomer of the first compound.
- the term “substantially free of the enantiomer” means that the composition is made up of at least about 95% by weight of a first compound of the invention, and about 5% by weight or less of a second compound of the invention, wherein the first compound is a non- superimposable mirror image of the second compound.
- the term "substantially free of the (R) enantiomer” means that the composition is made up of at least about 95% by weight of a first compound of the invention which has only one stereocenter and the stereocenter is in an (S) configuration, and about 5% by weight or less of a second compound of the invention, wherein the second compound is the enantiomer of the first compound.
- the term “substantially free of the enantiomer” means that the composition is made up of at least about 98% by weight of a first compound of the invention, and about 2% by weight or less of a second compound of the invention, wherein the first compound is a non-superimposable mirror image of the second compound.
- the term "substantially free of the (R) enantiomer” means that the composition is made up of at least about 98% by weight of a first compound of the invention which has only one stereocenter and the stereocenter is in an (S) configuration, and about 2% by weight or less of a second compound of the invention, wherein the second compound is the enantiomer of the first compound.
- the term “substantially free of the enantiomer” means that the composition is made up of at least about 99% by weight of a first compound of the invention, and about 1% by weight or less of a second compound of the invention, wherein the first compound is a non- superimposable mirror image of the second compound.
- the term "substantially free of the (R) enantiomer” means that the composition is made up of at least about 99% by weight of a first compound of the invention which has only one stereocenter and the stereocenter is in an (S) configuration, and about 1% by weight or less of a second compound of the invention, wherein the second compound is the enantiomer of the first compound.
- the compounds of the invention may also be used in combination with additional therapeutic agents.
- the invention thus provides, in a further aspect, a combination comprising a compound described herein or a pharmaceutically acceptable salt thereof together with at least one additional therapeutic agent.
- the additional therapeutic agent is a compound of the invention.
- the additional therapeutic agent includes a boron atom.
- the additional therapeutic agent does not contain a boron atom.
- the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art. It will be appreciated that the amount of a compound of the invention required for use in treatment will vary with the nature of the condition being treated and the age and the condition of the patient and will be ultimately at the discretion of the attendant physician or veterinarian.
- the additional therapeutic agent is Berenil.
- the additional therapeutic agent is an antiprotozoa.
- the additional therapeutic agent is a member selected from
- the additional therapeutic agent is pentamidine.
- the additional therapeutic agent is suramin.
- the additional therapeutic agent is Eflornithine.
- the additional therapeutic agent is Melarsoprol. In an exemplary embodiment, the additional therapeutic agent is Nifurtimox. In an exemplary embodiment, the additional therapeutic agent is an antiparasitic. In an exemplary embodiment, the additional therapeutic agent is a member selected from Amitraz, Avermectin, Carbadox, Diethylcarbamazine, Dimetridazole, Diminazene, Ivermectin, Macro filaricide, Malathion, Mitaban, Organophosphate, Oxamniquine, Permethrin, Praziquantel, Pyrantel pamoate, Selamectin, Sodium stibogluconate and Thiabendazole.
- the additional therapeutic agent is a member selected from antimony, meglumine antimoniate, sodium stibogluconate, amphotericin, miltefosine and paromomycin.
- the compounds of the invention, or pharmaceutical formulations thereof may also be used in combination with other therapeutic agents, for example immune therapies [e.g.
- interferon such as interferon alfa-2a (ROFERONd)-A; Hoffmann-La Roche), interferon alpha-2b (INTRONd)-A; Schering-Plough), interferon alfacon-1 (INFERGEN®; Intermune), peginterferon alpha-2b (PEGINTRONTM; Schering- Plough) or peginterferon alpha-2a (PEGAS YSd); Hoffmann-La Roche)]
- therapeutic vaccines such as interferon alfa-2a (ROFERONd)-A; Hoffmann-La Roche), interferon alpha-2b (INTRONd)-A; Schering-Plough), interferon alfacon-1 (INFERGEN®; Intermune), peginterferon alpha-2b (PEGINTRONTM; Schering- Plough) or peginterferon alpha-2a (PEGAS YSd); Hoffmann-La Roche)]
- therapeutic vaccines such as interferon alfa-2a (ROFERONd
- compositions according to the invention may also be used in combination with gene replacement therapy.
- the individual components of such combinations may be administered either simultaneously or sequentially in a unit dosage form.
- the unit dosage form may be a single or multiple unit dosage forms.
- the invention provides a combination in a single unit dosage form.
- An example of a single unit dosage form is a capsule wherein both the compound of the invention and the additional therapeutic agent are contained within the same capsule.
- the invention provides a combination in a two unit dosage form.
- An example of a two unit dosage form is a first capsule which contains the compound of the invention and a second capsule which contains the additional therapeutic agent.
- the term 'single unit' or 'two unit' or 'multiple unit' refers to the object which the patient ingests, not to the interior components of the object. Appropriate doses of known therapeutic agents will be readily appreciated by those skilled in the art.
- an exemplary embodiment of the invention is a pharmaceutical formulation comprising a) a compound of the invention; b) an additional therapeutic agent and c) a pharmaceutically acceptable excipient.
- the pharmaceutical formulation is a unit dosage form.
- the pharmaceutical formulation is a single unit dosage form.
- the pharmaceutical formulation is a two unit dosage form.
- the pharmaceutical formulation is a two unit dosage form comprising a first unit dosage form and a second unit dosage form, wherein the first unit dosage form includes a) a compound of the invention and b) a first pharmaceutically acceptable excipient; and the second unit dosage form includes c) an additional therapeutic agent and d) a second pharmaceutically acceptable excipient.
- Thioether, sulfoxide and sulfone derivatives such as B, C and D can be prepared by the following reactions.
- Thioethers such as B can be obtained by subjecting bromide A to boronylation conditions, such as treatment with n-butyl lithium and triisopropyl borate followed by addition of acid.
- Sulfoxides such as C can be obtained by subjecting B to oxidation conditions, such as sodium periodate or one equivalent of mCPBA.
- Sulfones such as D can be obtained by subjecting B to oxidation conditions such as sodium periodate over extended reaction time or two equivalents of mCPBA.
- Carbamates such as F and G can be prepared by reacting compound E with corresponding isocyanate RNCO or ArNCO in the presence of base such as triethylamine.
- Ethers such as H and I can be obtained by reacting compound E under basic conditions with alkylbromide RBr or arylbromide ArBr.
- Benzylethers such as K can be obtained by reacting compound E under basic conditions with substituted benzyl bromide J.
- Carbinol derivatives such as M can be obtained by sujecting ketone L to reducing conditions such as sodium borohydride.
- Ketone derivatives such as N can be obtained by subjecting alcohol M to oxidation conditions such as PCC.
- Amide derivatives such as P can be prepared from compound O and corresponding anilines by standard peptide coupling conditions as shown below.
- Amide derivatives such as S can be prepared from compound Q and corresponding acyl chlorides by standard peptide coupling conditions such as shown below.
- Benzylamine derivatives such as T can be obtained by reacting compound Q with corresponding benzyl bromides under basic conditions such as shown below.
- Sulfonamide derivatives such as U can be prepared from compound Q and corresponding sulfonyl chlorides under basic conditions such as shown below.
- Compounds such as W can be prepared by subjecting protected amines such as V to acidic conditions such as shown below.
- aldehyde X can be first subjected to reducing conditions such as sodium borohydride, then subjected to acidic conditions such as shown below.
- unsubstituted phenyl or unsubstituted pyridinyl sulfonyl chloride (1-1.2 equiv) and a base (such as NMM, K2CO3, or pyridine 3-4 equiv) can be added sequentially to a solution of the amine in MeCN (20 mL/g) at rt. After completion (typical duration O/N) the volatiles can be removed in vacuo. H 2 O can be added to the residue and the mixture adjusted to ⁇ pH 6 with dilute HCl.
- the aqueous layer can be then extracted with an organic solvent (such as EtOAc), and the combined organic fractions can be dried with a desiccant, such as Na 2 SO 4 or MgSO 4 , filtered, and concentrated in vacuo.
- an organic solvent such as EtOAc
- a desiccant such as Na 2 SO 4 or MgSO 4
- the product can be typically purified by either recrystallization from H 2 O, trituration with CH 2 Cl 2 or EtOAc, or flash chromatography.
- the compounds of the present invention exhibit potency against microorganisms, such as protozoa, and therefore have the potential to kill and/or inhibit the growth of microorganisms.
- the invention provides a method of killing and/or inhibiting the growth of a microorganism, said method comprising: contacting said microorganism with an effective amount of a compound of the invention, thereby killing and/or inhibiting the growth of the microorganism.
- the microorganism is a protozoa.
- the microorganism is a kinetoplastid.
- the protozoa is a Trypanosoma.
- the Trypanosoma is selected from the group consisting of T. avium, T. boissoni, T. brucei, T. carassii, T. cruzi, T.
- the protozoa is a Trypanosoma brucei. In another exemplary embodiment, the protozoa is Trypanosoma brucei brucei.
- the protozoa is Trypanosoma brucei rhodesiense. In another exemplary embodiment, the protozoa is Trypanosoma brucei gambiense. In another exemplary embodiment, the protozoa is a member of the genus Leishmania. In another exemplary embodiment, the protozoa is a member of Leishmania Viannia. In an exemplary embodiment, the protozoa is selected from the group consisting of L. donovani, L. infantum, L. chagasi; L. mexicana, L. amazonensis, L. venezuelensis, L. tropica, L. major, L. aethiopica, L.
- the protozoa is L. donovani. In an exemplary embodiment, the protozoa is L. infantum. In another exemplary embodiment, the protozoa is a member of the genus Plasmodium. In another exemplary embodiment, the protozoa is selected from the group consisting of Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium vivax, Plasmodium malariae and Plasmodium knowlesi.
- the protozoa is selected from the group consisting of Plasmodium vivax, Plasmodium ovale, Plasmodium vivax and Plasmodium malariae.
- the protozoa is Plasmodium falciparum.
- the protozoa is transmitted to the animal described herein by a mosquito infected with the protozoa. In another exemplary embodiment, wherein the protozoa is transmitted to the animal described herein by an Anopheles mosquito containing the protozoa.
- the compound is selected from the group consisting of 6-(2-chlorophenyl sulf ⁇ nyl)benzo[c][l,2]oxaborol-l(3H)-ol, 6-(3- chlorophenylsulfmyl) benzo[c][l,2] oxaborol-l(3H)-ol and 6-(4-chlorophenylsulfmyl) benzo[c][l,2] oxaborol-l(3H)-ol, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof.
- the compound is 6-(4- chlorophenylsulfmyl) benzo[c][l,2]oxaborol-l(3H)-ol, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof.
- the compound is 6-(4-chlorophenylsulfmyl)benzo[c][l,2] oxaborol-l(3H)-ol.
- the compound is described herein, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof.
- the invention provides a compound described herein, or a salt, hydrate or solvate thereof.
- the invention provides a compound described herein, or a prodrug thereof.
- the invention provides a compound described herein, or a salt thereof.
- the compound of the invention is a compound described herein, or a pharmaceutically acceptable salt thereof.
- the compound is described by a formula listed herein, or a pharmaceutically acceptable salt thereof.
- the compound is part of a pharmaceutical formulation described herein.
- the contacting occurs under conditions which permit entry of the compound into the organism. Such conditions are known to one skilled in the art and specific conditions are set forth in the Examples appended hereto.
- the microorganism is inside, or on the surface of an animal.
- the animal is selected from the group consisting of human, cattle, deer, reindeer, goat, honey bee, pig, sheep, horse, cow, bull, dog, guinea pig, gerbil, rabbit, cat, camel, yak, elephant, ostrich, otter, chicken, duck, goose, guinea fowl, pigeon, swan, and turkey.
- the animal is a human.
- the microorganism is killed or its growth is inhibited through oral administration of the compound of the invention.
- the microorganism is killed or its growth is inhibited through intravenous administration of the compound of the invention. In an exemplary embodiment, the microorganism is killed or its growth is inhibited through topical administration of the compound of the invention. In an exemplary embodiment, the microorganism is killed or its growth is inhibited through intraperitoneal administration of the compound of the invention. In an exemplary embodiment, the compound is administered in a topically effective amount. In an exemplary embodiment, the compound is administered in a cosmetically effective amount. In an exemplary embodiment, the pharmaceutical formulation is administered in an orally effective amount. V.
- the compounds of the present invention exhibit potency against microorganisms, such as protozoa, and therefore have the potential to achieve therapeutic efficacy in the animals described herein.
- the invention provides a method of treating and/or preventing a disease. The method includes administering to the animal a therapeutically effective amount of the compound of the invention, sufficient to treat and/or prevent the disease.
- the animal is not otherwise is need of treatment with the compound of the invention.
- the compound of the invention can be used in human or veterinary medical therapy, particularly in the treatment or prophylaxis of protozoa-associated disease.
- the compound of the invention can be used in human or veterinary medical therapy, particularly in the treatment or prophylaxis of kinetoplastid-associated disease.
- the disease is associated with a Trypanosoma.
- the Trypanosoma is selected from the group consisting of T. avium, T. boissoni, T. brucei, T. carassii, T. cruzi, T. congolense, T equinum, T equiperdum, T evansi, T hosei, T levisi, T melophagium, T parroti, T percae, T rangeli, T rotatorium, T.
- the disease is associated with a Trypanosoma brucei. In an exemplary embodiment, the disease is associated with Trypanosoma brucei brucei. In an exemplary embodiment, the disease is associated with Trypanosoma brucei rhodesiense. In an exemplary embodiment, the disease is associated with Trypanosoma brucei gambiense. In an exemplary embodiment, the disease is a typanosomiasis. In an exemplary embodiment, the disease is a human typanosomiasis.
- the disease is an animal typanosomiasis.
- the disease is selected from the group consisting of nagana, surra, mal de caderas, murrina de caderas, dourine, cachexial fevers, Gambian horse sickness, baleri, kaodzera, tahaga, galziekte or galzietzke and peste-boba.
- the disease is selected from the group consisting of Chagas disease (or Human American trypanosomiasis), nagana, surra, Covering sickness (or dourine) and sleeping sickness (or African sleeping sickness or Human African trypanosomiasis).
- the disease is Chagas disease.
- the disease is sleeping sickness (or African sleeping sickness). In an exemplary embodiment, the disease is acute phase sleeping sickness. In an exemplary embodiment, the disease is chronic phase sleeping sickness. In an exemplary embodiment, the disease is an acute phase of a typanosomiasis. In an exemplary embodiment, the disease is a chronic phase of a typanosomiasis. In an exemplary embodiment, the disease is the non-CNS form of a typanosomiasis. In an exemplary embodiment, the disease is the CNS form of a typanosomiasis. In an exemplary embodiment, the disease is the non-CNS form of sleeping sickness. In an exemplary embodiment, the disease is the CNS form of sleeping sickness. In an exemplary embodiment, the disease is the CNS form of sleeping sickness.
- the disease is early stage Human African trypanosomiasis. In an exemplary embodiment, the disease is late stage Human African trypanosomiasis. In another exemplary embodiment, the disease is associated with a member of the genus Leishmania. In another exemplary embodiment, the disease is associated with a member of Leishmania Viannia. In an exemplary embodiment, the disease is associated with a member selected from the group consisting of L. donovani, L. infantum, L. chagasi; L. mexicana, L. amazonensis, L. venezuelensis, L. tropica, L. major, L. aethiopica, L. (V.) braziliensis, L.
- the disease is associated with L. donovani. In an exemplary embodiment, the disease is associated with L. infantum. In an exemplary embodiment, the disease is leshmaniasis. In an exemplary embodiment, the disease is a member selected from visceral leshmaniasis and/or cutaneous leshmaniasis. In an exemplary embodiment, the disease is diffuse cutaneous leshmaniasis and/or mucocutaneous leshmaniasis. In another exemplary embodiment, the disease is associated with a member of the genus Plasmodium.
- the disease is associated with a member selected from the group consisting of Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium vivax, Plasmodium malariae and Plasmodium knowlesi.
- the disease is associated with a member selected from the group consisting of Plasmodium vivax, Plasmodium ovale, Plasmodium vivax and Plasmodium malariae.
- the disease is associated with Plasmodium falciparum.
- the disease is transmitted to the animal described herein by a mosquito infected with the protozoa.
- the disease is transmitted to the animal described herein by an Anopheles mosquito containing the protozoa.
- the disease is malaria.
- the disease is cerebral malaria.
- the disease is chronic malaria.
- the compound is selected from the group consisting of 6-(2-chlorophenylsulfmyl)benzo[c][l,2] oxaborol-l(3H)-ol, 6-(3- chlorophenylsulfmyl)benzo[c] [ 1 ,2]oxaborol- 1 (3H)-ol and 6-(4-chlorophenylsulfmyl) benzo[c][l,2] oxaborol-l(3H)-ol, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof.
- the compound is 6-(4- chlorophenylsulfinyl)benzo[c][l,2]oxaborol-l(3H)-ol, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof.
- the compound is 6-(4-chlorophenylsulfmyl)benzo[c][l,2] oxaborol-l(3H)-ol.
- the compound is described herein, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof.
- the invention provides a compound described herein, or a salt, hydrate or solvate thereof.
- the invention provides a compound described herein, or a prodrug thereof.
- the invention provides a compound described herein, or a salt thereof.
- the compound of the invention is a compound described herein, or a pharmaceutically acceptable salt thereof.
- the compound is described by a formula listed herein, or a pharmaceutically acceptable salt thereof.
- the compound is part of a pharmaceutical formulation described herein.
- the contacting occurs under conditions which permit entry of the compound into the organism. Such conditions are known to one skilled in the art and specific conditions are set forth in the Examples appended hereto.
- the animal is selected from the group consisting of human, cattle, deer, reindeer, goat, honey bee, pig, sheep, horse, cow, bull, dog, guinea pig, gerbil, rabbit, cat, camel, yak, elephant, ostrich, otter, chicken, duck, goose, guinea fowl, pigeon, swan, and turkey.
- the animal is a human.
- the animal is a mouse.
- the animal is selected from the group consisting of a human, cattle, goat, pig, sheep, horse, cow, bull, dog, guinea pig, gerbil, rabbit, cat, chicken and turkey.
- the disease is treated through oral administration of the compound of the invention.
- the disease is treated through intravenous administration of the compound of the invention.
- the disease is treated through topical administration of the compound of the invention.
- the disease is treated through intraperitoneal administration of the compound of the invention.
- the compound is administered in a topically effective amount.
- the compound is administered in a cosmetically effective amount.
- the pharmaceutical formulation is administered in an orally effective amount.
- the disease is associated with an infection by a microorganism described herein. In an exemplary embodiment, the disease is associated with an infection by a protozoa described herein.
- the invention is a pharmaceutical formulation which includes: (a) a pharmaceutically acceptable excipient; and (b) a compound of the invention.
- the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound according to a formula described herein.
- the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound described herein, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof.
- the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound described herein, or a salt, hydrate or solvate thereof, or a combination thereof.
- the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound described herein, or a salt, hydrate or solvate thereof.
- the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a salt of a compound described herein.
- the salt is a pharmaceutically acceptable salt.
- the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a prodrug of a compound described herein.
- the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound described herein.
- the pharmaceutical formulation is a unit dosage form. In an exemplary embodiment, the pharmaceutical formulation is a single unit dosage form.
- the invention is a pharmaceutical formulation which includes: (a) a pharmaceutically acceptable excipient; and (b) a compound which is
- the invention is a pharmaceutical formulation which includes: (a) a pharmaceutically acceptable excipient; and (b) a compound which is
- the pharmaceutical formulations of the invention can take a variety of forms adapted to the chosen route of administration. Those skilled in the art will recognize various synthetic methodologies that may be employed to prepare non-toxic pharmaceutical formulations incorporating the compounds described herein. Those skilled in the art will recognize a wide variety of non-toxic pharmaceutically acceptable solvents that may be used to prepare solvates of the compounds of the invention, such as water, ethanol, propylene glycol, mineral oil, vegetable oil and dimethylsulfoxide (DMSO).
- DMSO dimethylsulfoxide
- the pharmaceutical formulation of the invention may be administered orally, topically, intraperitoneally, parenterally, by inhalation or spray or rectally in unit dosage forms containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles. It is further understood that the best method of administration may be a combination of methods. Oral administration in the form of a pill, capsule, elixir, syrup, lozenge, troche, or the like is particularly preferred.
- parenteral as used herein includes subcutaneous injections, intradermal, intravascular (e.g., intravenous), intramuscular, spinal, intrathecal injection or like injection or infusion techniques.
- the pharmaceutical formulation is administered orally.
- the pharmaceutical formulation is administered intravenously.
- the pharmaceutical formulation is administered in a topically effective dose.
- the pharmaceutical formulation is administered in a cosmetically effective dose.
- the pharmaceutical formulation is administered in an orally effective dose.
- the pharmaceutical formulations containing compounds of the invention are preferably in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, or syrups or elixirs.
- compositions intended for oral use may be prepared according to any method known in the art for the manufacture of pharmaceutical formulations, and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets may contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients that are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia; and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate may be employed.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- an oil medium for example peanut oil, liquid paraffin or olive oil.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; and dispersing or wetting agents, which may be a naturally-occurring phosphatide, for example, lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbito
- the aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
- preservatives for example ethyl, or n-propyl p-hydroxybenzoate
- coloring agents for example ethyl, or n-propyl p-hydroxybenzoate
- flavoring agents for example ethyl, or n-propyl p-hydroxybenzoate
- sweetening agents such as sucrose or saccharin.
- Oily suspensions may be formulated by suspending the active ingredients in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol.
- Sweetening agents such as those set forth above, and flavoring agents may be added to provide palatable oral preparations. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present.
- Pharmaceutical formulations of the invention may also be in the form of oil-in-water emulsions and water-in-oil emulsions.
- the oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these.
- Suitable emulsifying agents may be naturally-occurring gums, for example gum acacia or gum tragacanth; naturally-occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol; anhydrides, for example sorbitan monooleate; and condensation products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monooleate.
- the emulsions may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, and flavoring and coloring agents.
- the pharmaceutical formulations may be in the form of a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents, which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
- Suitable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- composition of the invention may also be administered in the form of suppositories, e.g., for rectal administration of the drug.
- suppositories e.g., for rectal administration of the drug.
- These compositions can be prepared by mixing the drug with a suitable non-irritating excipient that is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- suitable non-irritating excipient that is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- Such materials are cocoa butter and polyethylene glycols.
- compositions can be administered parenterally in a sterile medium.
- the drug depending on the vehicle and concentration used, can either be suspended or dissolved in the vehicle.
- adjuvants such as local anesthetics, preservatives and buffering agents can be dissolved in the vehicle.
- the composition containing the therapeutic compound may be added to the animal's feed or drinking water. Also, it will be convenient to formulate animal feed and drinking water products so that the animal takes in an appropriate quantity of the compound in its diet. It will further be convenient to present the compound in a composition as a premix for addition to the feed or drinking water. The composition can also added as a food or drink supplement for humans.
- the amount of active ingredient that may be combined with the carrier materials to produce a unit dosage form will vary depending upon the condition being treated and the particular mode of administration. Unit dosage forms will generally contain between from about 1 mg to about 500 mg of an active ingredient.
- Frequency of dosage may also vary depending on the compound used and the particular disease treated. However, for treatment of most disorders, a dosage regimen of 4 times daily or less is preferred. It will be understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration and rate of excretion, drug combination and the severity of the particular disease undergoing therapy.
- the unit dosage form contains from about 1 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 1 mg to about 500 mg of an active ingredient. In an exemplary embodiment, the unit dosage form contains from about 100 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 200 mg to about 500 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 500 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 1 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 10 mg to about 100 mg of a compound of the invention.
- the unit dosage form contains from about 50 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 25 mg to about 75 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 40 mg to about 60 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 75 mg to about 200 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 1 mg to about 5 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 10 mg to about 25 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 50 mg to about 350 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 200 mg to about 400 mg of a compound of the invention.
- the daily dosage contains from about 1 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the the daily dosage contains from about 1 mg to about 500 mg of an active ingredient. In an exemplary embodiment, the daily dosage contains from about 100 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 200 mg to about 500 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 500 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 1 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 10 mg to about 100 mg of a compound of the invention.
- the daily dosage contains from about 50 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 75 mg to about 200 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 1 mg to about 5 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 10 mg to about 25 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 50 mg to about 350 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 200 mg to about 400 mg of a compound of the invention.
- Preferred compounds of the invention will have desirable pharmacological properties that include, but are not limited to, oral bioavailability, low toxicity, low serum protein binding and desirable in vitro and in vivo half- lives. Penetration of the blood brain barrier for compounds used to treat CNS disorders is necessary, while low brain levels of compounds used to treat peripheral disorders are often preferred.
- Assays may be used to predict these desirable pharmacological properties. Assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco-2 cell monolayers. Toxicity to cultured hepatocyctes may be used to predict compound toxicity. Penetration of the blood brain barrier of a compound in humans may be predicted from the brain levels of laboratory animals that receive the compound intravenously.
- Serum protein binding may be predicted from albumin binding assays. Such assays are described in a review by Oravcova, et al. (Journal of Chromatography B (1996) volume 677, pages 1-27).
- Compound half- life is inversely proportional to the frequency of dosage of a compound.
- In vitro half- lives of compounds may be predicted from assays of microsomal half-life as described by Kuhnz and Gieschen (Drug Metabolism and Disposition, (1998) volume 26, pages 1120-1127).
- compositions required for use in treatment will vary not only with the particular compound selected but also with the route of administration, the nature of the condition being treated and the age and condition of the patient and will ultimately be at the discretion of the attendant physician or clinician.
- Preferred compounds for use in the pharmeceutical formulations described herein will have certain pharmacological properties. Such properties include, but are not limited to, low toxicity, low serum protein binding and desirable in vitro and in vivo half-lives. Assays may be used to predict these desirable pharmacological properties. Assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco-2 cell monolayers. Serum protein binding may be predicted from albumin binding assays. Such assays are described in a review by Oravcova et al. (1996, J. Chromat. B677: 1-27). Compound half-life is inversely proportional to the frequency of dosage of a compound.
- Toxicity and therapeutic efficacy of such compounds can be determined by Standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50 (the dose lethal to 50% of the population) and the ED50 (the dose therapeutically effective in 50% of the population).
- the dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio between LD50 and ED50.
- Compounds that exhibit high therapeutic indices are preferred.
- the data obtained from these cell culture assays and animal studies can be used in formulating a range of dosage for use in humans.
- the dosage of such compounds lies preferably within a range of circulating concentrations that include the ED50 with little or no toxicity.
- the dosage can vary within this range depending upon the unit dosage form employed and the route of administration utilized.
- the exact formulation, route of administration and dosage can be chosen by the individual physician in view of the patient's condition. (See, e.g. Fingl et al., 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1, p. 1). VI. b) Administration
- the therapeutically effective dose can be estimated initially from cell culture assays, as disclosed herein.
- a dose can be formulated in animal models to achieve a circulating concentration range that includes the EC 50 (effective dose for 50% increase) as determined in cell culture, i.e., the concentration of the test compound which achieves a half-maximal inhibition of protozoa cell growth.
- EC 50 effective dose for 50% increase
- concentration of the test compound which achieves a half-maximal inhibition of protozoa cell growth Such information can be used to more accurately determine useful doses in humans.
- the compounds prepared by the methods, and from the intermediates, described herein will be administered in a therapeutically or cosmetically effective amount by any of the accepted modes of administration for agents that serve similar utilities. It will be understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration, and rate of excretion, drug combination, the severity of the particular disease undergoing therapy and the judgment of the prescribing physician.
- the drug can be administered from once or twice a day, or up to 3 or 4 times a day.
- Dosage amount and interval can be adjusted individually to provide plasma levels of the active moiety that are sufficient to maintain protozoa cell growth inhibitory effects.
- Usual patient dosages for systemic administration range from 0.1 to 1000 mg/day, preferably, 1-500 mg/day, more preferably 10 - 200 mg/day, even more preferably 100 - 200 mg/day. Stated in terms of patient body surface areas, usual dosages range from 50-91 mg/m 2 /day.
- the amount of the compound in a formulation can vary within the full range employed by those skilled in the art.
- the formulation will contain, on a weight percent (wt%) basis, from about 0.01-10 wt% of the drug based on the total formulation, with the balance being one or more suitable pharmaceutical excipients.
- the compound is present at a level of about 0.1-3.0 wt%, more preferably, about 1.0 wt%.
- the invention provides a compound having a structure which is
- R* is selected from the group consisting of H, a negative charge and a positively charged counterion
- L is selected from the group consisting of -S-, -S(O)-, - SO 2 -
- R 2 , R 3 , R 4 , R 5 and R 6 are independently selected from H and halogen with the proviso that when R 4 is Cl, at least one of R 2 or R 3 or R 5 or R 6 is halogen, or a salt thereof.
- two members selected from R 2 , R 3 , R 4 , R 5 and R 6 are each an independently selected halogen; and the remaining members of R 2 , R 3 , R 4 , R 5 and R 6 are H.
- the compound is [0190] In an exemplary embodiment, according to any of the above paragraphs, the compound is selected from the group consisting of
- the invention provides a combination comprising the compound according to any of the above paragraphs, together with at least one other therapeutically active agent.
- the invention provides a pharmaceutical composition comprising the compound according to any of the above paragraphs, and a pharmaceutically acceptable excipient.
- the pharmaceutical formulation is a unit dosage form.
- the salt of the compound according to any of the above paragraphs is a pharmaceutically acceptable salt.
- the invention provides a method of killing and/or preventing the growth of a protozoa, comprising: contacting the protozoa with an effective amount of the compound according to any of the above paragraphs, thereby killing and/or preventing the growth of the protozoa.
- the protozoa is Trypanosoma.
- the protozoa is Trypanosoma brucei.
- the Trypanosoma brucei is a member selected from Trypanosoma brucei brucei, Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense.
- the invention provides a method of treating and/or preventing a disease in an animal, comprising: administering to the animal a therapeutically effective amount of the compound according to any of the above paragraphs, thereby treating and/or preventing the disease.
- the disease is sleeping sickness.
- the animal is a human.
- DCM/CH 2 C1 2 dichloromethane
- DIEA diisopropylethylamine
- DMAP 4-dimethylaminopyridine
- DMF NJV- dimethylformamide
- EtOAc ethyl acetate
- EDCl l-ethyl-3-(3'- dimethylaminopropyl)carbodiimide
- ELS evaporative light scattering
- HATU 2- (lH-7-azabenzotriazol-l-yl)-l,l,3,3-tetramethyl uranium
- HOBt N- hydroxybenzotriazole
- HCO 2 H formic acid
- MeOH methanol
- TEA triethylamine
- THF tetrahydrofuran.
- LCMS spectra were obtained using a ThermoFinnigan AQA MS ESI instrument utilizing a Phenomenex Aqua 5 micron C 18 125 A 50 x 4.60 mm column.
- the spray setting for the MS probe was at 350 ⁇ L/min with a cone voltage at 25 mV and a probe temperature at 450 0 C.
- the spectra were recorded using ELS and UV (254 nm) detection.
- LCMS spectra were obtained using an Agilent 1200SL HPLC equipped with a 6130 mass spectrometer operating with electrospray ionization.
- the eluent was 0-100% EtOAc in heptane or 0-10% MeOH in CH 2 Cl 2 as a linear gradient over the length of the run (14-20 minutes). Peaks were detected by variable wavelength UV absorption (200-360 nm). The resulting fractions were analyzed, combined as appropriate, and evaporated under reduced pressure to provide purified material.
- HPLC purification was performed using a 50 mm Varian Dynamax HPLC 21.4 mm Microsorb Guard-8 C 18 column, Dyonex Chromeleon operating system coupled with a Varian Prostar 320 UV-vis detector (254 nm) and a Sedex55 ELS detector. Conditions: Solvent A: H 2 O/1% acetonitrile/0.1% HCO 2 H; Solvent B: MeOH. The appropriate solvent gradient for purification was determined based on the results of analytical HPLC experiments. The resulting fractions were analyzed, combined as appropriate, and evaporated under reduced pressure to provide purified material.
- Mass spectra were recorded on a Waters MS consisting of an Alliance 2795 (LC) and Waters Micromass ZQ detector at 120 0 C.
- the mass spectrometer was equipped with an electrospray ion source (ESI) operated in a positive or negative mode.
- Elemental Analysis for C, H and N composition was performed using a Costech Instrument Elemental Combustion System ECS4010 with a helium flow of 100 mL/min (14 psi), oxygen 20 mL/min (10 psi), air 25 psi and purge of 50 mL/min. The reported analyses are an average of two runs.
- HPLC analyses were performed on a Water 600 Controller system with a Waters 717 Plus Autosampler and a Waters 2996 Photodiode Array Detector. The column used was an ACE C 18 , 5 ⁇ m, 4.6 x 150 mm.
- a linear gradient was applied, starting at 95% A (A: 0.1% H 3 PO 4 in water) and ending at 90% B (B: MeCN) over 6 min and then maintained at 90% B until the 10 min mark.
- the column was then re- equilibrated over 3 min to 95:5 with a total run time of 20 min.
- the column temperature was at rt with the flow rate of 1.0 mL/min.
- the Diode Array Detector was scanned from 200-400 nm. For high purity samples requiring baseline subtraction, a linear gradient was applied, starting at 99% A (A: 0.1% H 3 PO 4 in water) and ending at 90% B (B: MeCN) over 15 min.
- the column was then re- equilibrated over 3 min to 99% A with a total run time of 23 min.
- the column temperature was at rt with the flow rate of 1.0 mL/min.
- the Diode Array Detector was scanned from 200-400 nm. A blank MeOH sample was run immediately prior to the sample of which purity was to be determined: this was then subtracted to obtain the baseline subtracted chromatogram.
- TLC Thin layer chromatography
- Alugram ® Silica gel 60 F 254
- UV was typically used to visualize the spots. Additional visualization methods were also employed in some cases.
- the TLC plate was developed with iodine (generated by adding approximately 1 g of I 2 to 10 g silica gel and thoroughly mixing), vanillin (generated by dissolving about 1 g vanillin in 100 mL 10% H 2 SO 4 ), potassium permanganate (generated by dissolving 1.5 g KMnO 4 and 10 g K 2 CO 3 in 1.25 mL NaOH and 200 mL H 2 O), ninhydrin (available commercially from Aldrich), or Magic Stain (generated by thoroughly mixing 25 g (NH 4 ) 6 Mo 7 O 24 «4H 2 O, 5 g (NH 4 ) 2 Ce(IV)(NO 3 ) 6 in 450 mL H 2 O and 50 mL cone H 2 SO 4 )
- Flash chromatography was preformed using typically 40-63 ⁇ m (230-400 mesh) silica gel from Silicycle following analogous techniques to those disclosed by Still et al.
- Typical solvents used for flash chromatography or thin layer chromatography (TLC) were mixtures of CHCl 3 /MeOH, CH 2 Cl 2 MeOH, EtOAc/MeOH and hexane/EtOAc.
- Reverse phase flash chromatography were performed on a Biotage ® using a Biotage C 18 cartridges and a H 2 O/MeOH gradient (typically eluting from 5% MeOH/H 2 O to 90% MeOH/H 2 O).
- Preparative chromatography was performed on either a Waters Prep LC 4000 System using a Waters 2487 Diode Array or on a Waters LC Module 1 plus.
- the column used were either a Waters x Terra Prep C 18 , 5 ⁇ m, 30 x 100 mm, Phenomenex Luna C 18 , 5 ⁇ m, 21.6 x 250 mm, or a Phenomenex Gemini C 18 , 5 ⁇ m, 100 x 30 mm.
- Narrow gradients with MeCNZH 2 O water containing either 0.1% TFA, 0.1% AcOH, 0.1% HCO 2 H or 0.1% NH 4 OAc) were used to elute the compound at a flow rate of approximately 20 mL/min and a total run time between 20-30 min.
- This compound can be produced by the same method as H109 by substituting 3,4-dichlorobenzenethiol with 2,3-dichlorobenzenethiol.
- This compound can be producd by the same method as H109 by substituting 3,4-dichlorobenzenethiol with 2,5-dichlorobenzenethiol.
- This compound can be produced by the same method as H109 by substituting 3,4-dichlorobenzenethiol with 2,6-dichlorobenzenethiol. 6-(3 ⁇ 4 '-dichlorophenyl)sulfinyl-l,5-dihydro-l-hydroxy-2,l-benzoxaborole (Hl 14)
- This compound can be produced by the same method as Hl 14 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,3- dichlorobenzenethiol.
- This compound can be produced by the same method as Hl 14 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,5- dichlorobenzenethiol.
- This compound can be produced by the same method as Hl 14 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,6- dichlorobenzenethiol.
- This compound can be produced by the same method as Hl 19 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,3- dichlorobenzenethiol.
- This compound can be produced by the same method as Hl 19 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,5- dichlorobenzenethiol.
- This compound can be produced by the same method as Hl 19 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,6- dichlorobenzenethiol. 6-(phenylthio)benzo / cj [l,2]oxaborol-l (3H)-ol (H124)
- H142 can be synthesized using the following procedure:
- H157 was prepared using a procedure similar to that of H156 with 3- chlorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H158 was prepared using a procedure similar to that of H156 with 4- chlorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H159 was prepared using a procedure similar to that of H156 with 2- fluorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H160 was prepared using a procedure similar to that of H156 with 3- fluorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- 4-Fluoro-N-(l-hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)- benzenesulfonamide (H161)
- H161 was prepared using a procedure similar to that of H156 with 4- fluorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H162 was prepared using a procedure similar to that of H156 with 3,4- dichlorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H163 was prepared using a procedure similar to that of H156 with 3,5- dichlorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H166 was prepared using a procedure similar to that of H156 with 3- methylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H167 was prepared using a procedure similar to that of H156 with 4- methylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H168 was prepared using a procedure similar to that of H156 with 4- isopropylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H169 was prepared using a procedure similar to that of H156 with 4-t- butylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H170 was prepared using a procedure similar to that of H156 with 3- trifluoromethylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- H172 was prepared using a procedure similar to H171 with 6- methylpyridine-2-sulfonyl chloride replacing 4-(trifluoromethyl)benzenesulfonyl chloride.
- H173 was prepared using a procedure similar to H171 with 5- methylpyridine-2-sulfonyl chloride replacing 4-(trifluoromethyl)benzenesulfonyl chloride.
- H174 was prepared using a procedure similar to H171 with 4- methylpyridine-2-sulfonyl chloride replacing 4-(trifluoromethyl)benzenesulfonyl chloride.
- H175 was prepared using a procedure similar to H171 with 1-ethyl-lH- pyrazole-4-sulfonyl chloride replacing 4-(trifluoromethyl)benzenesulfonyl chloride. Purification: recrystallization from H 2 O. H175 was isolated as light yellow needles: yield 0.401 g (56%).
- H176 was prepared using a procedure similar to H171 with 1-methyl-lH- imidazole-4-sulfonyl chloride replacing 4-(trifluoromethyl)benzenesulfonyl chloride.
- H177 was prepared using a procedure similar to that of H156 with 2- naphthalenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride.
- Trypanosoma brucei brucei High-Throughput Screening Assay Procedure [0316] Experiments were conducted with the bloodstream- form trypanosome T. brucei brucei S427 strain and the T. brucei brucei STIB 795 strain. Parasites were cultured in T-25 vented cap flasks and kept in humidified incubators at 37°C and 5% CO 2 . The parasite culture media was complete HMI- 9 medium (c.f. Hirumi, Journal of Parasitology, 40:75, 985-989 (1989)) containing 10% FBS, 10% Serum Plus medium and penicillin/streptomycin. To ensure log growth phase, trypanosomes were sub-cultured at appropriate dilutions every 2-3 days.
- Rat skeletal myoblasts (L-6 cells) were seeded in 96-well microtitre plates at 2 x 10 3 cells/well in 100 ⁇ L RPMI 1640 medium with 10% FBS and 2 mM L- glutamine. After 24 h the medium was removed and replaced by 100 ⁇ l per well containing 5 x 10 3 trypomastigote forms of T. cruzi Tulahuen strain C2C4 containing the ⁇ -galactosidase (Lac Z) gene (Buckner, et al., Antimicrobial Agents and Chemotherapy, 40: 2592-2597 (1996)).
- Lac Z ⁇ -galactosidase
- the medium was removed from the wells and replaced by 100 ⁇ l fresh medium with or without a serial drug dilution of seven 3-fold dilution steps covering a range from 90 to 0.123 ⁇ g/ml.
- the plates were inspected under an inverted microscope to assure growth of the controls and sterility.
- the substrate (50 ⁇ l) chlorophenol red- ⁇ -D- galactopyranoside (CPRG, Roche Diagnostics Ltd) in 0.25% Nonidet P-40/PBS was added to all wells and a color reaction developed within 2-6 h. which was read photometrically at 540 nm. Data were transferred into the graphic programme Softmax Pro (Molecular Devices), which calculated IC50 values.
- Compound solutions/dilutions Compound stock solutions were prepared in 100% DMSO at 20 mM or mg/ml for natural products, drug mixtures and if the molecular weight was not known. The compounds were serially pre-diluted (2-fold or 4-fold) in DMSO followed by a further (intermediate) dilution in demineralized water to assure a final in-test DMSO concentration of ⁇ 1%.
- Parasite burdens were assessed after adding the substrate CPRG (chlorophenolred ⁇ -D-galactopyranoside): 50 ⁇ l/well of a stock solution containing 15.2 mg CPRG + 250 ⁇ l Nonidet in 100 ml PBS. The change in color was measured spectrophotometrically at 540 nm after 4 hours incubation at 37 0 C. The results were expressed as % reduction in parasite burdens compared to control wells and an IC50 (50% inhibitory concentration) was calculated.
- substrate CPRG chlorophenolred ⁇ -D-galactopyranoside
- Nifurtimox or benznidazole were included as the reference drugs.
- the test compound was classified as inactive when the IC50 was higher than 30 ⁇ M. When the IC50 was between 30 and 5 ⁇ M, the compound was regarded as being moderate active. When the IC50 was lower than 5 ⁇ M, the compound was classified as highly active on the condition that it also demonstrated selective action (absence of cytotoxicity). A final recommendation for activity was given after confirmatory evaluation in a secondary screening.
- T. b. gambiense strains were isolated from sleeping sickness patients as described, and were subsequently propagated in mice at Swiss Tropical Institute.
- T. b. gambiense 4OR, 108R were isolated by Pati Pyana in Mbuji Mayi (D. R Congo) in 2005 and then propagated in STI (Swiss Tropical Institute, Basel, Switzerland) in different mice in winter 2006.
- o 4OR is a relapse 6 months after a 10 days melarsoprol treatment.
- o 108R is a relapse 8 months after a 10 days melarsoprol treatment
- T. b. gambiense DAL 1402 was obtained from the cryobank of the "Project de mecanics Cliniques Sur Ia Trypanosomiase," in Daloa. It was isolated inl990 from a human patient in Cote d'lrium. • T. b. gambiense ITMAP 141267 was isolated in Bandundu/Lac Mai-Ndombe,
- T. b. gambiense Drani was isolated from a patient in Kenya; West.Nile, 1995; original stabilate ID: UTRO 210396 A.
- Trypanosoma brucei rhodesiense STIB 900 Screening Assay Procedure [0330] The Trypanosoma brucei rhodesiense STIB900 strain was isolated in 1982 from a human patient in Africa and after several mouse passages cloned and adapted to axenic culture conditions (Baltz, et al., EMBO Journal 4: 1273-1277 (1985); Thuita, et al., Acta Tropica 108:6-10 (2008)).
- Amastigotes of L. donovani strain MHOM/ET/67/L82 were grown in axenic culture at 37 0 C in SM medium (Cunningham, I. J Protozol. 24:325-329 (1977)) at pH 5.4 supplemented with 10% heat-inactivated fetal bovine serum under an atmosphere of 5% CO 2 in air.
- SM medium Cunningham, I. J Protozol. 24:325-329 (1977)
- fetal bovine serum under an atmosphere of 5% CO 2 in air.
- One hundred microlitres of culture medium with 10 5 amastigotes from axenic culture with or without a serial drug dilution were seeded in 96-well microtitre plates.
- MHOM/MA(BE)/67 and L.donovani MHOM/ET/67/L82 were used.
- the strains were maintained in the Golden Hamster (Mesocricetus auratus). Amastigotes were collected from the spleen of an infected donor hamster using three centrifugation purification steps (300 rpm, keeping the supernatant, 2200 rpm, keeping the supernatants and 3500 rpm, keeping the pellet) and spleen parasite burdens were assessed using the Stauber technique (Stauber LA., Exp Parasitol. 18: 1-11 (1966)).
- Drug sensitivity assays were performed in 96-well microtiter plates, each well contained 10 ⁇ l of the compound dilutions together with 190 ⁇ l of macrophage/parasite inoculum (3 x 10 3 cells + 4.5 x 10 5 parasites/well).
- the inoculum was prepared in RPMI- 1640 medium, supplemented with 200 mM L-glutamine, 16.5 mM NaHCOs, and 5% heat-inactivated fetal calf serum. Parasite multiplication was compared to untreated-infected controls (100% growth) and uninfected controls (0% growth).
- a test compound was classified as inactive when the IC50 was higher than 30 ⁇ M. When the IC50 was between 30 and 10 ⁇ M, the compound was regarded as moderately active. If the IC 50 is lower than 10 ⁇ M, the compound was classified as highly active on the condition that it also demonstrates selective action (absence of cytotoxicity against primary peritoneal macrophages). A final recommendation for activity was given after confirmatory evaluation in a secondary screening. [0337] Secondary screen.
- Leishmania infantum MHOM/MA(BE)/67 and L.donovani MHOM/ET/67/L82 strains were used and the ICso-values were determined using an extended dose range (2-fold compound dilutions).
- Pentostam®, miltefosine, fungizone and PX-6518 were included as reference drugs. Advanced selectivity evaluations were performed against a panel of unrelated organisms (bacteria, yeasts, fungi and other protozoan parasites).
- a solution can be formed including a compound described herein, 40 mM Mops, pH 7.5, 0.8 mM EGTA, 15 mM magnesium acetate, 0.2 mg/ml bovine serum albumin (BSA) and 100000 c.p.m. of [ 3 H]cAMP, in a final volume of 250 ⁇ l at 30 0 C.
- the assay reaction can be started with the addition of the trypanosoma PDE (the amount will be in the linear hydrolysis vs. time zone as determined in a preliminary experiment) to this mixture.
- trypanosoma PDE source is a cell lysate or protein homogenate
- an inhibitor of endogenous PDEs such as, but not limited to, IBMX (3-isobutylmethylxanthine)
- IBMX 3-isobutylmethylxanthine
- the reaction can be terminated by boiling the mixture for 2 minutes and the resulting AMP can be converted to adenosine by the addition of 10 mg/ml snake venom nucleotidase and further incubation at 37 0 C for 10 minutes. Unhydrolyzed cAMP can be bound to AG1-X2 resin, and the remaining [ 3 H]Adenosine in the aqueous phase can be quantitated by scintillation counting.
- Trypanosoma brucei PDEA TbrPDEA, formerly known as TbPDEl
- TbPDEl Trypanosoma brucei PDEA
- TbPD E2C Trypanosoma brucei PDEBl
- Zoraghi et al., Proc. Natl. Acad. Sci. U.S.A. 99: 4343-4348 (2002).
- Trypanosoma brucei PDEB2 (TbrPDEB2, formerly known as TbPDE2B) can be obtained through the methods described in Rascon, et al., Proc. Natl. Acad. Sci. U.S.A. 99: 4714-4719 (2002). Trypanosoma brucei PDE2A (TbPDE2A) can be obtained through the methods described in Zoraghi, et al., J. Biol. Chem, 276: 11559-11566 (2001).
Abstract
This invention provides, among other things, novel compounds useful for treating protozoal infections, pharmaceutical compositions containing such compounds, as well as combinations of these compounds with at least one additional therapeutically effective agent.
Description
BORON-CONTAINING SMALL MOLECULES AS ANTI-PROTOZOAL
AGENTS
CROSS-REFERENCES TO RELATED APPLICATIONS [0001] This application claims the benefit of U.S. Provisional Pat. App. No.
61/105,759, filed October 15, 2008, U.S. Provisional Pat. App. No. 61/105,763, filed October 15, 2008, U.S. Provisional Pat. App. No. 61/110,907, filed November 3, 2008, U.S. Provisional Pat. App. No. 61/119,956, filed December 4, 2008, and U.S. Provisional Pat. App. No. 61/148,241, filed January 29, 2009, each of which is incorporated by reference in its entirety for all purposes.
BACKGROUND OF THE INVENTION
[0002] The global rise of protozoa resistant to antimicrobials in general, poses a major threat. Deployment of massive quantities of antimicrobial agents into the ecosphere during the past 60 years has introduced a powerful selective pressure for the emergence and spread of antimicrobial-resistant pathogens. Thus, there is a need to discover new broad spectrum antimicrobials, such as antiprotozoals, useful in combating microorganisms, especially those with multidrug-resistance.
[0003] Boron-containing molecules, such as oxaboroles, useful as antimicrobials have been described previously, such as in U.S. Pat. Pubs. US20060234981 and US20070155699. Generally speaking, an oxaborole has the following structure and substituent numbering system:

It has now been discovered that certain classes of oxaboroles which are surprisingly effective antiprotozoals. This, and other uses of these oxaboroles are described herein.
SUMMARY OF THE INVENTION
[0004] This invention provides, among other things, novel compounds useful for treating protozoa infections, pharmaceutical compositions containing such
compounds, as well as combinations of these compounds with at least one additional therapeutically effective agent.
BRIEF DESCRIPTION OF THE DRAWING [0005] FIG. 1 provides biological data for compounds of the invention.
DETAILED DESCRIPTION OF THE INVENTION
/. Definitions and Abbreviations
[0006] As used herein, the singular forms "a," "an", and "the" include plural references unless the context clearly dictates otherwise. For example, reference to "an active agent" includes a single active agent as well as two or more different active agents in combination. It is to be understood that present teaching is not limited to the specific dosage forms, carriers, or the like, disclosed herein and as such may vary.
[0007] The abbreviations used herein generally have their conventional meaning within the chemical and biological arts.
[0008] The following abbreviations have been used: aq. is aqueous; AcOH is acetic acid; ACTBr is cetyltrimethylammonium bromide; B2pin2- bis(pinacolato)diboron; Boc is tert-butoxy carbonyl; Boc2O-di-te/t-butyl dicarbonate; BzOOH-benzoyl peroxide; Cs2COs is cesium carbonate; DABCO is 1,4- diazabicyclo[2.2.2]octane; DCM is dichloromethane or methylene chloride; DIAD is diisopropyl azodicarboxylate; DIEA is diisopropylethylamine; NJSf- Diisopropylethylamine is DIPEA; DMAP is 4-(dimethylamino)pyridine; DME is 1 ,2- dimethoxyethane; DMF is N,N-dimethylformamide; DMSO is dimethylsulfoxide; eq. is equivalent; EtOAc is ethyl acetate; EtOH is ethanol; Et2O is diethyl ether; EDCI-N- (3 -dimethylaminopropy^-N-ethylcarbodiimide hydrochloride; m-CPB A-3 - chloroperoxybenzoic acid; equiv-equivalent; h is hours; HATU is O-(7- azabenzotriazol-l-yl)-Ν,Ν,Ν',Ν'-tetramethyluronium hexafluorophosphate; HCl is hydrochloric acid; HPLC is high pressure liquid chromatography; ISCO Companion is automated flash chromatography equipment with fraction analysis by UV absorption available from Presearch; KOAc is potassium acetate; K2Cθ3 is potassium carbonate; LiAlH4 or LAH is lithium aluminum hydride; LDA is lithium diisopropylamide; LHMDS is lithium bis(trimethylsilyl) amide; KHMDS is potassium
bis(trimethylsilyl) amide; LiOH is lithium hydroxide; m-CPBA is 3- chloroperoxybenzoic acid; MeCN is acetonitrile; MeOH is methanol; MgSO4 is magnesium sulfate; mins or min is minutes; Mp or MP is melting point; NaCNBH3 is sodium cyanoborohydride; NaOH is sodium hydroxide; Na2SO4 is sodium sulfate; NH4Cl is ammonium chloride; N2 is nitrogen; NMM-JV-methylmorpholine; n-BuLi is n-butyllithium; overnight is O/N; PdCl2(pddf) is l,l'-Bis(diphenylphosphino) ferrocene] dichloro palladium(II); PrOH is 1-propanol; iPrOH is 2-propanol; POCl3 is phosphorus chloride oxide; RT or rt is room temperature; TFA is trifluoroacetic acid; Tf2O is trifluoromethanesulfonic anhydride; THF is tetrahydrofuran; THP- tetrahydopyranyl; TMSI is trimethylsilyl iodide; H2O is water; Ac-acetyl; PTSA- /?αra-toluene sulfonic acid; Pyr.-Pyridine; Cbz-benzyloxycarbonyl; PMB-/?- methoxybenzyl; DHP-dihydropyran; CSA-camphor sulfonic acid; CTAB- cetyltrimethylammonium bromide; sat. -saturated; Cy-cyclohexyl; Ph-phenyl; Ar-aryl.
[0009] "Compound of the invention," as used herein refers to the compounds discussed herein, salts (e.g. pharmaceutically acceptable salts), prodrugs, solvates and hydrates of these compounds.
[0010] "Combination of the invention," as used herein refers to the compounds and antiprotozoals discussed herein as well as acids, bases, salt forms (such as pharmaceutically acceptable salts), prodrugs, solvates and hydrates of these compounds and antiprotozoals.
[0011] "Boron containing compounds", as used herein, refers to the compounds of the invention that contain boron as part of their chemical formula.
[0012] MIC, or minimum inhibitory concentration, is the point where the compound stops more than 50% of cell growth, preferably 60% of cell growth, preferably 70% of cell growth, preferably 80% of cell growth, preferably 90% of cell growth, relative to an untreated control.
[0013] Where substituent groups are specified by their conventional chemical formulae, written from left to right, they equally encompass the chemically identical substituents, which would result from writing the structure from right to left, e.g., -CH2O- is intended to also recite -OCH2-.
[0014] The term "poly" as used herein means at least 2. For example, a polyvalent metal ion is a metal ion having a valency of at least 2.
[0015] "Moiety" refers to a radical of a molecule that is attached to the remainder of the molecule. [0016] The symbol v/wvr ; whether utilized as a bond or displayed perpendicular to a bond, indicates the point at which the displayed moiety is attached to the remainder of the molecule.
[0017] The term "alkyl," by itself or as part of another substituent, means, unless otherwise stated, a straight or branched chain, or cyclic hydrocarbon radical, or combination thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals, having the number of carbon atoms designated (i.e. Ci-Cio means one to ten carbons). In some embodiments, the term "alkyl" means a straight or branched chain, or combinations thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals. Examples of saturated hydrocarbon radicals include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, cyclohexyl, (cyclohexyl)methyl, cyclopropylmethyl, homologs and isomers of, for example, n- pentyl, n-hexyl, n-heptyl, n-octyl, and the like. An unsaturated alkyl group is one having one or more double bonds or triple bonds. Examples of unsaturated alkyl groups include, but are not limited to, vinyl, 2-propenyl, crotyl, 2-isopentenyl, 2- (butadienyl), 2,4-pentadienyl, 3-(l,4-pentadienyl), ethynyl, 1- and 3-propynyl, 3- butynyl, and the higher homologs and isomers.
[0018] The term "alkylene" by itself or as part of another substituent means a divalent radical derived from an alkane, as exemplified, but not limited, by - CH2CH2CH2CH2-, and further includes those groups described below as
"heteroalkylene." Typically, an alkyl (or alkylene) group will have from 1 to 24 carbon atoms, with those groups having 10 or fewer carbon atoms being preferred in the present invention. A "lower alkyl" or "lower alkylene" is a shorter chain alkyl or alkylene group, generally having eight or fewer carbon atoms. [0019] The terms "alkoxy," "alkylamino" and "alkylthio" (or thioalkoxy) are used in their conventional sense, and refer to those alkyl groups attached to the remainder of the molecule via an oxygen atom, an amino group, or a sulfur atom, respectively.
[0020] The term "heteroalkyl," by itself or in combination with another term, means, unless otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom. In some embodiments, the term "heteroalkyl," by itself or in combination with another term, means a stable straight or branched chain, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom. In an exemplary embodiment, the heteroatoms can be selected from the group consisting of B, O, N and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized. The heteroatom(s) B, O, N and S may be placed at any interior position of the heteroalkyl group or at the position at which the alkyl group is attached to the remainder of the molecule. Examples include, but are not limited to, -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CHs)-CH3, -CH2-S-CH2-CH3, -CH2-CH25-S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3, and -CH=CH-N(CH3)-CH3. Up to two heteroatoms may be consecutive, such as, for example, -CH2-NH-OCH3. Similarly, the term "heteroalkylene" by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified, but not limited by, -CH2-CH2-S-CH2-CH2- and -CH2-S-CH2-CH2-NH-CH2-. For heteroalkylene groups, heteroatoms can also occupy either or both of the chain termini (e.g., alkyleneoxy, alkylenedioxy, alkyleneamino, alkylenediamino, and the like). Still further, for alkylene and heteroalkylene linking groups, no orientation of the linking group is implied by the direction in which the formula of the linking group is written. For example, the formula -C(O)2R'- represents both -C(O)2R'- and - R5C(O)2-. [0021] The terms "cycloalkyl" and "heterocycloalkyl", by themselves or in combination with other terms, represent, unless otherwise stated, cyclic versions of "alkyl" and "heteroalkyl", respectively. Additionally, for heterocycloalkyl, a heteroatom can occupy the position at which the heterocycle is attached to the remainder of the molecule. Examples of cycloalkyl include, but are not limited to, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like. Examples of heterocycloalkyl include, but are not limited to, 1 -(1,2,5,6- tetrahydropyridyl), 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 4-morpholinyl, 3-
morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl, tetrahydrothien-3-yl, 1 -piperazinyl, 2-piperazinyl, and the like.
[0022] The terms "halo" or "halogen," by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as "haloalkyl," are meant to include monohaloalkyl and polyhaloalkyl. For example, the term "halo(Ci-C4)alkyl" is mean to include, but not be limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
[0023] The term "aryl" means, unless otherwise stated, a polyunsaturated, aromatic, substituent that can be a single ring or multiple rings (preferably from 1 to 3 rings), which are fused together or linked covalently. The term "heteroaryl" refers to aryl groups (or rings) that contain from one to four heteroatoms. In an exemplary embodiment, the heteroatom is selected from B, N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized. A heteroaryl group can be attached to the remainder of the molecule through a heteroatom. Non- limiting examples of aryl and heteroaryl groups include phenyl, 1-naphthyl, 2-naphthyl, 4-biphenyl, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3- pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 2-phenyl-4- oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2- pyrimidyl, 4-pyrimidyl, 5-benzothiazolyl, purinyl, 2-benzimidazolyl, 5-indolyl, 1- isoquinolyl, 5-isoquinolyl, 2-quinoxalinyl, 5-quinoxalinyl, 3-quinolyl, 6-quinolyl, dioxaborolane, dioxaborinane and dioxaborepane. Substituents for each of the above noted aryl and heteroaryl ring systems are selected from the group of acceptable substituents described below.
[0024] For brevity, the term "aryl" when used in combination with other terms (e.g. , aryloxy, arylthioxy, arylalkyl) includes both aryl and heteroaryl rings as defined above. Thus, the term "arylalkyl" is meant to include those radicals in which an aryl group is attached to an alkyl group (e.g. , benzyl, phenethyl, pyridylmethyl and the like) including those alkyl groups in which a carbon atom (e.g., a methylene group) has been replaced by, for example, an oxygen atom (e.g., phenoxymethyl, 2- pyridyloxymethyl, 3-(l-naphthyloxy)propyl, and the like).
[0025] Each of the above terms (e.g., "alkyl," "heteroalkyl," "aryl" and "heteroaryl") are meant to include both substituted and unsubstituted forms of the indicated radical. Preferred substituents for each type of radical are provided below.
[0026] Substituents for the alkyl and heteroalkyl radicals (including those groups often referred to as alkylene, alkenyl, heteroalkylene, heteroalkenyl, alkynyl, cycloalkyl, heterocycloalkyl, cycloalkenyl, and heterocycloalkenyl) are generically referred to as "alkyl group substituents," and they can be one or more of a variety of groups selected from, but not limited to: -R', -OR', =0, =NR', =N-0R', -NR'R", - SR', -halogen, -SiR'R"R'", -OC(O)R', -C(O)R', -CO2R', -CONR'R", -0C(0)NR'R", -NR"C(0)R', -NR'-C(0)NR"R"', -NR"C(0)2R', -NR'""-C(NR'R"R'")=NR"",
-NR""-C(NR'R")=NR'", -S(O)R', -S(O)2R', -S(O)2NR5R", -NR"S02R', -CN, -NO2, -N3, -CH(Ph)2, fluoro(Ci-C4)alkoxy, and fluoro(Ci-C4)alkyl, in a number ranging from zero to (2m'+l), where m' is the total number of carbon atoms in such radical. R', R", R'", R"" and R'"" each preferably independently refer to hydrogen, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl, e.g. , aryl substituted with 1-3 halogens, substituted or unsubstituted alkyl, alkoxy or thioalkoxy groups, or arylalkyl groups. When a compound of the invention includes more than one R group, for example, each of the R groups is independently selected as are each R', R", R'", R"" and R'"" groups when more than one of these groups is present. When R' and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6-, or 7-membered ring. For example, -NR'R" is meant to include, but not be limited to, 1-pyrrolidinyl and 4-morpholinyl. From the above discussion of substituents, one of skill in the art will understand that the term "alkyl" is meant to include groups including carbon atoms bound to groups other than hydrogen groups, such as haloalkyl (e.g., -CF3 and -CH2CF3) and acyl (e.g., - C(O)CH3, -C(O)CF3, -C(O)CH2OCH3, and the like).
[0027] Similar to the substituents described for the alkyl radical, substituents for the aryl and heteroaryl groups are generically referred to as "aryl group substituents." The substituents are selected from, for example: -R', -OR', =0, =NR', =N-0R', -NR'R", -SR', -halogen, -SiR'R"R'", -OC(O)R', -C(O)R', -CO2R', -CONR'R", -0C(0)NR'R", -NR"C(O)R', -NR'-C(0)NR"R"', -NR"C(0)2R', -NR'""-C(NR'R"R'")=NR"", -NR""-C(NR'R")=NR'", -S(O)R', -S(O)2R', -S(O)2NR5R", -NR"S02R', -CN, -NO2, -N3, -CH(Ph)2, fiuoro(Ci-C4)alkoxy, and
fluoro(Ci-C4)alkyl, in a number ranging from zero to the total number of open valences on the aromatic ring system; and where R', R", R'", R"" and R'"" are preferably independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl and substituted or unsubstituted heteroaryl. When a compound of the invention includes more than one R group, for example, each of the R groups is independently selected as are each R', R", R'", R"" and R'"" groups when more than one of these groups is present.
[0028] Two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)-(CRR')q-U-, wherein T and U are independently -NR-, -O-, -CRR'- or a single bond, and q is an integer of from 0 to 3. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula - A-(CH2)r-B-, wherein A and B are independently -CRR'-, -O-, -NR-, -S-, -S(O)-, -S(O)2-, -S(O)2NR'- or a single bond, and r is an integer of from 1 to 4. One of the single bonds of the new ring so formed may optionally be replaced with a double bond. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CRR')s-X-(CR"R'")d-, where s and d are independently integers of from 0 to 3, and X is -O-, -NR'-, -S-, -S(O)-, -S(O)2-, or -S(O)2NR'-. The substituents R, R', R" and R'" are preferably independently selected from hydrogen or substituted or unsubstituted (Ci-C6)alkyl.
[0029] "Ring" as used herein, means a substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl. A ring includes fused ring moieties. The number of atoms in a ring is typically defined by the number of members in the ring. For example, a "5- to 7-membered ring" means there are 5 to 7 atoms in the encircling arrangement. Unless otherwise specified, the ring optionally includes a heteroatom. Thus, the term "5- to 7-membered ring" includes, for example phenyl, pyridinyl and piperidinyl. The term "5- to 7-membered heterocycloalkyl ring", on the other hand, would include pyridinyl and piperidinyl, but not phenyl. The term "ring" further includes a ring system comprising more than one "ring", wherein each "ring" is independently defined as above.
[0030] As used herein, the term "heteroatom" includes atoms other than carbon (C) and hydrogen (H). Examples include oxygen (O), nitrogen (N) sulfur (S), silicon (Si), germanium (Ge), aluminum (Al) and boron (B).
[0031] The term "leaving group" means a functional group or atom which can be displaced by another functional group or atom in a substitution reaction, such as a nucleophilic substitution reaction. By way of example, representative leaving groups include triflate, chloro, bromo and iodo groups; sulfonic ester groups, such as mesylate, tosylate, brosylate, nosylate and the like; and acyloxy groups, such as acetoxy, trifluoroacetoxy and the like. [0032] The symbol "R" is a general abbreviation that represents a substituent group that is selected from substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted cycloalkyl and substituted or unsubstituted heterocycloalkyl groups. [0033] By "effective" amount of a drug, formulation, or permeant is meant a sufficient amount of an active agent to provide the desired local or systemic effect. A "Topically effective," "Cosmetically effective," "pharmaceutically effective," or "therapeutically effective" amount refers to the amount of drug needed to effect the desired therapeutic result. [0034] "Topically effective" refers to a material that, when applied to the skin, nail, hair, claw or hoof produces a desired pharmacological result either locally at the place of application or systemically as a result of transdermal passage of an active ingredient in the material.
[0035] "Cosmetically effective" refers to a material that, when applied to the skin, nail, hair, claw or hoof, produces a desired cosmetic result locally at the place of application of an active ingredient in the material.
[0036] The term "pharmaceutically acceptable salt" is meant to include a salt of a compound of the invention which are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the compounds described herein. When compounds of the invention contain relatively acidic functionalities, base addition salts can be obtained by contacting the neutral form of such compounds with
a sufficient amount of the desired base, either neat or in a suitable inert solvent. Examples of pharmaceutically acceptable base addition salts include sodium, potassium, calcium, ammonium, organic amino, or magnesium salt, or a similar salt. When compounds of the invention contain relatively basic functionalities, acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent. Examples of pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, maleic, malonic, benzoic, succinic, suberic, fumaric, lactic, mandelic, phthalic, benzenesulfonic, p-tolylsulfonic, citric, tartaric, methanesulfonic, and the like. Also included are salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galactunoric acids and the like (see, for example, Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)). Certain specific compounds of the invention contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts. [0037] The neutral forms of the compounds are preferably regenerated by contacting the salt with a base or acid and isolating the parent compounds in the conventional manner. The parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents.
[0038] In addition to salt forms, the present invention provides compounds which are in a prodrug form. Prodrugs of the compounds described herein readily undergo chemical changes under physiological conditions to provide the compounds of the invention. Additionally, prodrugs can be converted to the compounds of the invention by chemical or biochemical methods in an ex vivo environment.
[0039] Certain compounds of the invention can exist in unsolvated forms as well as solvated forms, including hydrated forms. In general, the solvated forms are equivalent to unsolvated forms and are encompassed within the scope of the present
invention. Certain compounds of the invention may exist in multiple crystalline or amorphous forms.
[0040] Certain compounds of the invention possess asymmetric carbon atoms (optical centers) or double bonds; the racemates, diastereomers, geometric isomers and individual isomers are encompassed within the scope of the present invention. The graphic representations of racemic, ambiscalemic and scalemic or enantiomerically pure compounds used herein are taken from Maehr, J. Chem. Ed. 1985, 62: 114-120. Solid and broken wedges are used to denote the absolute configuration of a stereocenter unless otherwise noted. When the compounds described herein contain olefmic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers. Likewise, all tautomeric forms are included.
[0041] Compounds of the invention can exist in particular geometric or stereoisomeric forms. The invention contemplates all such compounds, including cis- and trans -isomers, (-)- and (+)-enantiomers, (R)- and (S)-enantiomers, diastereomers, (D)-isomers, (L)-isomers, the racemic mixtures thereof, and other mixtures thereof, such as enantiomerically or diastereomerically enriched mixtures, as falling within the scope of the invention. Additional asymmetric carbon atoms can be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention.
[0042] Optically active (R)- and (5)-isomers and d and / isomers can be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. If, for instance, a particular enantiomer of a compound of the present invention is desired, it can be prepared by asymmetric synthesis, or by derivatization with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers. Alternatively, where the molecule contains a basic functional group, such as an amino group, or an acidic functional group, such as a carboxyl group, diastereomeric salts can be formed with an appropriate optically active acid or base, followed by resolution of the diastereomers thus formed by fractional crystallization or chromatographic means known in the art, and subsequent recovery of the pure enantiomers. In addition, separation of enantiomers and diastereomers is frequently accomplished using
chromatography employing chiral, stationary phases, optionally in combination with chemical derivatization (e.g., formation of carbamates from amines).
[0043] The compounds of the invention may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. For example, the compounds may be radiolabeled with radioactive isotopes, such as for example tritium (3H), iodine-125 (125I) or carbon-14 (14C). All isotopic variations of the compounds of the invention, whether radioactive or not, are intended to be encompassed within the scope of the present invention.
[0044] The term "pharmaceutically acceptable carrier" or "pharmaceutically acceptable vehicle" refers to any formulation or carrier medium that provides the appropriate delivery of an effective amount of an active agent as defined herein, does not interfere with the effectiveness of the biological activity of the active agent, and that is sufficiently non-toxic to the host or patient. Representative carriers include water, oils, both vegetable and mineral, cream bases, lotion bases, ointment bases and the like. These bases include suspending agents, thickeners, penetration enhancers, and the like. Their formulation is well known to those in the art of cosmetics and topical pharmaceuticals. Additional information concerning carriers can be found in Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005) which is incorporated herein by reference. [0045] "Pharmaceutically acceptable topical carrier" and equivalent terms refer to pharmaceutically acceptable carriers, as described herein above, suitable for topical application. An inactive liquid or cream vehicle capable of suspending or dissolving the active agent(s), and having the properties of being nontoxic and non-inflammatory when applied to the skin, nail, hair, claw or hoof is an example of a pharmaceutically- acceptable topical carrier. This term is specifically intended to encompass carrier materials approved for use in topical cosmetics as well.
[0046] The term "pharmaceutically acceptable additive" refers to preservatives, antioxidants, fragrances, emulsifϊers, dyes and excipients known or used in the field of drug formulation and that do not unduly interfere with the effectiveness of the biological activity of the active agent, and that is sufficiently non-toxic to the host or patient. Additives for topical formulations are well-known in the art, and may be added to the topical composition, as long as they are pharmaceutically acceptable and
not deleterious to the epithelial cells or their function. Further, they should not cause deterioration in the stability of the composition. For example, inert fillers, anti- irritants, tackifϊers, excipients, fragrances, opacifϊers, antioxidants, gelling agents, stabilizers, surfactant, emollients, coloring agents, preservatives, buffering agents, other permeation enhancers, and other conventional components of topical or transdermal delivery formulations as are known in the art.
[0047] The terms "enhancement," "penetration enhancement" or "permeation enhancement" relate to an increase in the permeability of the skin, nail, hair, claw or hoof to a drug, so as to increase the rate at which the drug permeates through the skin, nail, hair, claw or hoof. The enhanced permeation effected through the use of such enhancers can be observed, for example, by measuring the rate of diffusion of the drug through animal skin, nail, hair, claw or hoof using a diffusion cell apparatus. A diffusion cell is described by Merritt et al. Diffusion Apparatus for Skin Penetration, J of Controlled Release, 1 (1984) pp. 161-162. The term "permeation enhancer" or "penetration enhancer" intends an agent or a mixture of agents, which, alone or in combination, act to increase the permeability of the skin, nail, hair or hoof to a drug.
[0048] The term "excipients" is conventionally known to mean carriers, diluents and/or vehicles used in formulating drug compositions effective for the desired use.
[0049] The term "topical administration" refers to the application of a pharmaceutical agent to the external surface of the skin, nail, hair, claw or hoof, such that the agent crosses the external surface of the skin, nail, hair, claw or hoof and enters the underlying tissues. Topical administration includes application of the composition to intact skin, nail, hair, claw or hoof, or to a broken, raw or open wound of skin, nail, hair, claw or hoof. Topical administration of a pharmaceutical agent can result in a limited distribution of the agent to the skin and surrounding tissues or, when the agent is removed from the treatment area by the bloodstream, can result in systemic distribution of the agent.
[0050] The term "transdermal delivery" refers to the diffusion of an agent across the barrier of the skin, nail, hair, claw or hoof resulting from topical administration or other application of a composition. The stratum corneum acts as a barrier and few pharmaceutical agents are able to penetrate intact skin. In contrast, the epidermis and dermis are permeable to many solutes and absorption of drugs therefore occurs more
readily through skin, nail, hair, claw or hoof that is abraded or otherwise stripped of the stratum corneum to expose the epidermis. Transdermal delivery includes injection or other delivery through any portion of the skin, nail, hair, claw or hoof or mucous membrane and absorption or permeation through the remaining portion. Absorption through intact skin, nail, hair, claw or hoof can be enhanced by placing the active agent in an appropriate pharmaceutically acceptable vehicle before application to the skin, nail, hair, claw or hoof. Passive topical administration may consist of applying the active agent directly to the treatment site in combination with emollients or penetration enhancers. As used herein, transdermal delivery is intended to include delivery by permeation through or past the integument, i.e. skin, nail, hair, claw or hoof.
[0051] The terms "effective amount" or a "therapeutically effective amount" of a drug or pharmacologically active agent refers to a nontoxic but sufficient amount of the drug or agent to provide the desired effect. In the oral dosage forms of the present disclosure, an "effective amount" of one active of the combination is the amount of that active that is effective to provide the desired effect when used in combination with the other active of the combination. The amount that is "effective" will vary from subject to subject, depending on the age and general condition of the individual, the particular active agent or agents, and the appropriate "effective" amount in any individual case may be determined by one of ordinary skill in the art using routine experimentation.
[0052] The phrases "active ingredient", "therapeutic agent", "active", or "active agent" mean a chemical entity which can be effective in treating a targeted disorder, disease or condition. [0053] The phrase "pharmaceutically acceptable" means moieties or compounds that are, within the scope of medical judgment, suitable for use in humans without causing undesirable biological effects such as undue toxicity, irritation, allergic response, and the like, for example.
[0054] The phrase "oral dosage form" means any pharmaceutical composition administered to a subject via the oral cavity. Exemplary oral dosage forms include tablets, capsules, films, powders, sachets, granules, solutions, solids, suspensions or as more than one distinct unit (e.g., granules, tablets, and/or capsules containing different
actives) packaged together for co-administration, and other formulations known in the art. An oral dosage form can be one, two, three, four, five or six units. When the oral dosage form has multiple units, all of the units are contained within a single package, (e.g. a bottle or other form of packaging such as a blister pack). When the oral dosage form is a single unit, it may or may not be in a single package. In a preferred embodiment, the oral dosage form is one, two or three units. In a particularly preferred embodiment, the oral dosage form is one unit.
[0055] The phrase "unit", as used herein, refers to the number of discrete objects to be administered which comprise the dosage form. In some embodiments, the dosage form includes a compound of the invention in one capsule. This is a single unit. In some embodiments, the dosage form includes a compound of the invention as part of a therapeutically effective dosage of a cream or ointment. This is also a single unit. In some embodiments, the dosage form includes a compound of the invention and another active ingredient contained within one capsule, or as part of a therapeutically effective dosage of a cream or ointment. This is a single unit, whether or not the interior of the capsule includes multiple discrete granules of the active ingredient. In some embodiments, the dosage form includes a compound of the invention in one capsule, and the active ingredient in a second capsule. This is a two unit dosage form, such as two capsules or tablets, and so such units are contained in a single package. Thus the term 'unit' refers to the object which is administered to the animal, not to the interior components of the object.
[0056] The term, "prodrug", as defined herein, is a derivative of a parent drug molecule that exerts its pharmacological effect only after chemical and/or enzymatic conversion to its active form in vivo. Prodrugs include those designed to circumvent problems associated with delivery of the parent drug. This may be due to poor physicochemical properties, such as poor chemical stability or low aqueous solubility, and may also be due to poor pharmacokinetic properties, such as poor bioavailability or poor half- life. Thus, certain advantages of prodrugs may include improved chemical stability, absorption, and/or PK properties of the parent carboxylic acids. Prodrugs may also be used to make drugs more "patient friendly," by minimizing the frequency (e.g., once daily) or route of dosing (e.g., oral), or to improve the taste or odor if given orally, or to minimize pain if given parenterally .
[0057] In some embodiments, the prodrugs are chemically more stable than the active drug, thereby improving formulation and delivery of the parent drug, compared to the drug alone.
[0058] Prodrugs for carboxylic acid analogs of the invention may include a variety of esters. In an exemplary embodiment, the pharmaceutical compositions of the invention include a carboxylic acid ester. In an exemplary embodiment, the prodrug is suitable for treatment /prevention of those diseases and conditions that require the drug molecule to cross the blood brain barrier. In an exemplary embodiment, the prodrug enters the brain, where it is converted into the active form of the drug molecule. In one embodiment, a prodrug is used to enable an active drug molecule to reach the inside of the eye after topical application of the prodrug to the eye. Additionally, a prodrug can be converted to its parent compound by chemical or biochemical methods in an ex vivo environment. For example, a prodrug can be slowly converted to its parent compound when placed in a transdermal patch reservoir with a suitable enzyme or chemical reagent.
[0059] "Antibiotic", as used herein, is a compound which can kill or inhibit the growth of bacteria. The term antibiotic is broad enough to encompass acids, bases, salt forms (such as pharmaceutically acceptable salts), prodrugs, solvates and hydrates of the antibiotic compound. [0060] "Antiprotozoal" or "antiprotozoa", as used herein, is a compound which can kill or inhibit the growth of protozoa. The term antiprotozoal or antiprotozoa is broad enough to encompass acids, bases, salt forms (such as pharmaceutically acceptable salts), prodrugs, solvates and hydrates of the antiprotozoal or antiprotozoa compound.
[0061] The term "microbial infection" or "infection by a microorganism" refers to any infection of a host by an infectious agent including, but not limited to, viruses, bacteria, mycobacteria, fungus and parasites (see, e.g., Harrison's Principles of Internal Medicine, pp. 93-98 (Wilson et al., eds., 12th ed. 1991); Williams et al, J. of Medicinal Chem. 42:1481-1485 (1999), herein each incorporated by reference in their entirety). [0062] "Biological medium," as used herein refers to both in vitro and in vivo biological milieus. Exemplary in vitro "biological media" include, but are not limited
to, cell culture, tissue culture, homogenates, plasma and blood. In vivo applications are generally performed in mammals, preferably humans.
[0063] "Inhibiting" and "blocking," are used interchangeably herein to refer to the partial or full blockade of an enzyme, such as a beta-lactamase or a leucyl t-RNA synthetase.
[0064] The term "leaving group" means a functional group or atom which can be displaced by another functional group or atom in a substitution reaction, such as a nucleophilic substitution reaction. By way of example, representative leaving groups include triflate, chloro, bromo and iodo groups; sulfonic ester groups, such as mesylate, tosylate, brosylate, nosylate and the like; and acyloxy groups, such as acetoxy, trifluoroacetoxy and the like.
[0065] The term "amino-protecting group" means a protecting group suitable for preventing undesired reactions at an amino nitrogen. Representative amino-protecting groups include, but are not limited to, formyl; acyl groups, for example alkanoyl groups, such as acetyl, trichloroacetyl or trifluoroacetyl; alkoxycarbonyl groups, such as tert-butoxycarbonyl (Boc); arylmethoxycarbonyl groups, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethoxycarbonyl (Fmoc); arylmethyl groups, such as benzyl (Bn), trityl (Tr), and l,l-di-(4'-methoxyphenyl)methyl; silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like. [0066] The term "hydroxy-protecting group" means a protecting group suitable for preventing undesired reactions at a hydroxy group. Representative hydroxy- protecting groups include, but are not limited to, alkyl groups, such as methyl, ethyl, and tert-butyl; acyl groups, for example alkanoyl groups, such as acetyl; arylmethyl groups, such as benzyl (Bn), p-methoxybenzyl (PMB), 9-fluorenylmethyl (Fm), and diphenylmethyl (benzhydryl, DPM); silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
[0067] Boron is able to form dative bonds with oxygen, sulfur or nitrogen under some circumstances in this invention. Dative bonds are usually weaker than covalent bonds. In situations where a boron is covalently bonded to at least one oxygen, sulfur or nitrogen, and is at the same time datively bonded to an oxygen, sulfur or nitrogen, respectively, the dative bond and covalent bond between the boron and the two identical heteroatoms can interconvert or be in the form of a resonance hybrid. There
is potential uncertainty surrounding the exact nature and extent of electron sharing in these situations. Generally, in boron compounds comprising both covalent and coordinate covalent (dative) bonds, the electrons in such bonds may be partially or fully delocalized. [0068] Embodiments of the present invention also encompass compounds that are poly- or multi-valent species, including, for example, species such as dimers, trimers, tetramers and higher homo logs of the compounds of use in the invention or reactive analogues thereof.
[0069] "Salt counterion", as used herein, refers to positively charged ions that associate with a compound of the invention when the boron is fully negatively or partially negatively charged. Examples of salt counterions include H+, H3O+, ammonium, potassium, calcium, magnesium and sodium.
[0070] The compounds comprising a boron bonded to a carbon and three heteroatoms (such as three oxygens described in this section) can optionally contain a fully negatively charged boron or partially negatively charged boron, due to the nature of the dative bond between the boron and one of the oxygens. Due to the negative charge, a positively charged counterion may associate with this compound, thus forming a salt. Examples of positively charged counterions include H+, H3O+, calcium, sodium, ammonium and potassium. The salts of these compounds are implicitly contained in descriptions of these compounds.
//. Introduction
[0071] The present invention provides novel boron compounds. The novel compounds, as well as pharmaceutical compositions containing such compounds or combinations of these compounds with at least one additional therapeutically effective agent, can be used for, among other things, treating protozoal infections.
///. The Compounds III. a) Cyclic Boronic Esters
[0072] In one aspect, the invention provides a compound of the invention. In an exemplary embodiment, the invention is a compound described herein. In an exemplary embodiment, the invention is a compound according to a formula described herein.
[0073] In an exemplary embodiment, the compound of the invention has the following structure:

[0074] In an exemplary embodiment, n is 0 and L is selected from the group consisting of -S(O)-, -SO2-, -C(O)-, -C(OH)-, "-CH2O-', "-NHC(O)O-" and -CH2-, wherein " indicates a covalent linkage between L and the phenyl ring, and wherein ' indicates a covalent linkage between L and the boron-containing moiety. In an exemplary embodiment, n is O and L is "-NHC(O)-', wherein " indicates a covalent linkage between L and the phenyl ring, and wherein ' indicates a covalent linkage between L and the boron-containing moiety. In another exemplary embodiment, R* is H.
[0075] In an exemplary embodiment, n is 1 and L is selected from the group consisting of -S-, -S(O)- and -SO2-, and there is a proviso that the compound is not
 In an exemplary embodiment, there is a proviso that the compound is not
In an exemplary embodiment, there is a proviso that the compound is not

[0076] In an exemplary embodiment, n is 2 and L is selected from the group consisting of -S-, -S(O)- and -SO2-. In an exemplary embodiment, n is 2, X is Cl and L is selected from the group consisting of -S-, -S(O)- and -SO2-. In an exemplary embodiment, n is 2, X is F and L is selected from the group consisting of -S-, -S(O)-
and -SO2-. In an exemplary embodiment, n is 2, each X is F or Cl, wherein at least one X is Cl, and L is selected from the group consisting of -S-, -S(O)- and -SO2-.
[0077] In an exemplary embodiment, n is 3 and L is selected from the group consisting of -S-, -S(O)- and -SO2-. In an exemplary embodiment, n is 4 and L is selected from the group consisting of -S-, -S(O)- and -SO2-. In an exemplary embodiment, n is 5 and L is selected from the group consisting of -S-, -S(O)- and - SO2-.
[0078] In an exemplary embodiment, the compound has a structure according to the following formula:





[0079] In an exemplary embodiment, the compound of the invention has the following structure:

[0080] In an exemplary emboidment, the compound has the the following structure:

In an exemplary embodiment, the compound has the proviso that the compound is not

In an exemplary embodiment, the compound has structure which is

In an exemplary embodiment, the compound has structure which is

[0081] In an exemplary embodiment, the compound of the invention is

[0082] In an exemplary embodiment, the compound has a structure according to the following formula:



[0083] In an exemplary embodiment, the compound has a structure which is
 , or a salt thereof. In an exemplary embodiment, the compound
, or a salt thereof. In an exemplary embodiment, the compound
has a structure which is
 or a salt thereof.
or a salt thereof.
[0084] In an exemplary embodiment, the compound of the invention has a structure which is

invention has a structure which is
 . In an exemplary embodiment, the compound of the invention has a structure which is
. In an exemplary embodiment, the compound of the invention has a structure which is
 or salts thereof. In an exemplary embodiment, the compound of the
or salts thereof. In an exemplary embodiment, the compound of the
invention has a structure which is

[0085] In an exemplary embodiment, the compound of the invention is

[0086] In an exemplary embodiment, the compound of the invention is
 wherein R27 is unsubstituted pyrimidinyl. In an exemplary embodiment, the compound is
wherein R27 is unsubstituted pyrimidinyl. In an exemplary embodiment, the compound is

[0087] In an exemplary embodiment, the compound of the invention is
 wherein R27 is halosubstituted pyrimidinyl. In an exemplary embodiment, R27 is monohalosubstituted pyrimidinyl. In an exemplary embodiment, R27 is chlorosubstituted pyrimidinyl. In an exemplary embodiment, the compound is
wherein R27 is halosubstituted pyrimidinyl. In an exemplary embodiment, R27 is monohalosubstituted pyrimidinyl. In an exemplary embodiment, R27 is chlorosubstituted pyrimidinyl. In an exemplary embodiment, the compound is




[0088] In an exemplary embodiment, the compound of the invention is


[0089] In an exemplary embodiment, the compound of the invention is
 wherein R , 27 is unsubstituted indolyl. In an exemplary embodiment, the compound is
wherein R , 27 is unsubstituted indolyl. In an exemplary embodiment, the compound is

[0090] In another aspect, the invention provides a compound which is


3, and at least one Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, b is
4, and at least one Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, b is
5, and at least one Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 3, and at least one Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 4, and at least one Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 5, and at least one Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 1 or 2, and at least one Ra is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, b is 1 and Ra is halo-substituted C1- C6 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, b is 3, and at least one Ra is halo- substituted Ci-C6 alkyl. In an exemplary embodiment, b is 4, and at least one Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, b is 5, and at least one Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary
embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 1 or 2 or 3 and Ra is Ci-C6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, b is 1 or 2 or 3 and Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one chlorine. In an exemplary embodiment, b is 1 or 2 or 3 and Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one bromine. In an exemplary embodiment, b is 1 or 2 or 3 and Ra is Ci-C6 alkyl, substituted with at least one chlorine and at least one bromine. In an exemplary embodiment, b is 1 or 2 or 3 and Ra is fluoro-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is trifluoro- substituted Ci-C6 alkyl.
[0092] In an exemplary embodiment, the compound has a structure which is selected from the group consisting of:
 wherein Ra is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl. In an exemplary embodiment, Ra is fluoro. In an exemplary embodiment, Ra is chloro. In an exemplary embodiment, Ra is bromo. In an exemplary embodiment, Ra is iodo. In an exemplary embodiment, Ra is methyl. In an exemplary embodiment, Ra is ethyl. In an exemplary embodiment, Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, Ra is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one chlorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one bromine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one chlorine and at least one bromine. In an exemplary embodiment, Ra is fluoro-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is trifluoro-substituted Ci-C6 alkyl.
wherein Ra is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl. In an exemplary embodiment, Ra is fluoro. In an exemplary embodiment, Ra is chloro. In an exemplary embodiment, Ra is bromo. In an exemplary embodiment, Ra is iodo. In an exemplary embodiment, Ra is methyl. In an exemplary embodiment, Ra is ethyl. In an exemplary embodiment, Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, Ra is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one chlorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one bromine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one chlorine and at least one bromine. In an exemplary embodiment, Ra is fluoro-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is trifluoro-substituted Ci-C6 alkyl.
[0093] In an exemplary embodiment, the compound has a structure which is:
 wherein b is 1 or 2 or 3 or 4, B is pyridinyl, Ra is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl. In an exemplary embodiment, b is 1. In an exemplary embodiment, b is 2. In an exemplary embodiment, b is 3. In an exemplary embodiment, b is 4. In an exemplary embodiment, b is 1, Ra is fluoro. In an exemplary embodiment, b is 2, and at least one Ra is fluoro. In an exemplary embodiment, b is 3, and at least one Ra is fluoro. In an exemplary embodiment, b is 4, and at least one Ra is fluoro. In an exemplary embodiment, b is 1, Ra is chloro. In an exemplary embodiment, b is 2, and at least one Ra is chloro. In an exemplary embodiment, b is 3, and at least one Ra is chloro. In an exemplary embodiment, b is 4, and at least one Ra is chloro. In an exemplary embodiment, b is 1, Ra is bromo. In an exemplary embodiment, b is 2, and at least one Ra is bromo. In an exemplary embodiment, b is 3, and at least one Ra is bromo. In an exemplary embodiment, b is 4, and at least one Ra is bromo. In an exemplary embodiment, b is 1, Ra is iodo. In an exemplary embodiment, b is 2, and at least one Ra is iodo. In an exemplary embodiment, b is 3, and at least one Ra is iodo. In an exemplary embodiment, b is 4, and at least one Ra is iodo. In an exemplary embodiment, b is 1, Ra is methyl. In an exemplary embodiment, b is 2, and at least one Ra is methyl. In an exemplary embodiment, b is 3, and at least one Ra is methyl. In an exemplary embodiment, b is 4, and at least one Ra is methyl. In an exemplary embodiment, b is 1, Ra is ethyl. In an exemplary embodiment, b is 2, and at least one Ra is ethyl. In an exemplary embodiment, b is 3, and at least one Ra is ethyl. In an exemplary embodiment, b is 4, and at least one Ra is ethyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, b is
wherein b is 1 or 2 or 3 or 4, B is pyridinyl, Ra is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl. In an exemplary embodiment, b is 1. In an exemplary embodiment, b is 2. In an exemplary embodiment, b is 3. In an exemplary embodiment, b is 4. In an exemplary embodiment, b is 1, Ra is fluoro. In an exemplary embodiment, b is 2, and at least one Ra is fluoro. In an exemplary embodiment, b is 3, and at least one Ra is fluoro. In an exemplary embodiment, b is 4, and at least one Ra is fluoro. In an exemplary embodiment, b is 1, Ra is chloro. In an exemplary embodiment, b is 2, and at least one Ra is chloro. In an exemplary embodiment, b is 3, and at least one Ra is chloro. In an exemplary embodiment, b is 4, and at least one Ra is chloro. In an exemplary embodiment, b is 1, Ra is bromo. In an exemplary embodiment, b is 2, and at least one Ra is bromo. In an exemplary embodiment, b is 3, and at least one Ra is bromo. In an exemplary embodiment, b is 4, and at least one Ra is bromo. In an exemplary embodiment, b is 1, Ra is iodo. In an exemplary embodiment, b is 2, and at least one Ra is iodo. In an exemplary embodiment, b is 3, and at least one Ra is iodo. In an exemplary embodiment, b is 4, and at least one Ra is iodo. In an exemplary embodiment, b is 1, Ra is methyl. In an exemplary embodiment, b is 2, and at least one Ra is methyl. In an exemplary embodiment, b is 3, and at least one Ra is methyl. In an exemplary embodiment, b is 4, and at least one Ra is methyl. In an exemplary embodiment, b is 1, Ra is ethyl. In an exemplary embodiment, b is 2, and at least one Ra is ethyl. In an exemplary embodiment, b is 3, and at least one Ra is ethyl. In an exemplary embodiment, b is 4, and at least one Ra is ethyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, b is
3, and at least one Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, b is
4, and at least one Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, b is 3, and at least one Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, b is 4, and at least one Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is an
unsubstitued C5 alkyl. In an exemplary embodiment, b is 3, and at least one Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, b is 4, and at least one Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 3, and at least one Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 4, and at least one Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 1 or 2, and at least one Ra is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, b is 1 and Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, b is 3, and at least one Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, b is 4, and at least one Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, b is 4, and at least one Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 4, and at least one Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 4, and at least one Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 4, and at least one Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 3,
and at least one Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 4, and at least one Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 4, and at least one Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 4, and at least one Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 4, and at least one Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 3, and at least one Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 4, and at least one Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 1 or 2 or 3 or 4 and Ra is C1- C6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, b is 1 or 2 or 3 or 4 and Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one chlorine. In an exemplary embodiment, b is 1 or 2 or 3 or 4 and Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one bromine. In an exemplary embodiment, b is 1 or 2 or 3 or 4 and Ra is Ci-C6 alkyl, substituted with at least one chlorine and at least one bromine. In an exemplary embodiment, b is 1 or 2 or 3 or 4 and Ra is fluoro-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is trifluoro-substituted Ci-C6 alkyl.
[0094] In an exemplary embodiment, the compound has a structure which is selected from the group consisting of:
 wherein Ra is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl. In an exemplary embodiment, Ra is fluoro. In an exemplary embodiment, Ra is chloro. In an exemplary embodiment, Ra is bromo. In an exemplary embodiment, Ra is iodo. In an exemplary embodiment, Ra is methyl. In an exemplary embodiment, Ra is ethyl. In an exemplary embodiment, Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, Ra is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one chlorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one bromine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one chlorine and at least one
bromine. In an exemplary embodiment, Ra is fluoro-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is trifluoro-substituted Ci-C6 alkyl.
wherein Ra is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl. In an exemplary embodiment, Ra is fluoro. In an exemplary embodiment, Ra is chloro. In an exemplary embodiment, Ra is bromo. In an exemplary embodiment, Ra is iodo. In an exemplary embodiment, Ra is methyl. In an exemplary embodiment, Ra is ethyl. In an exemplary embodiment, Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, Ra is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four fluorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with four chlorines. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one chlorine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one bromine. In an exemplary embodiment, Ra is Ci-C6 alkyl, substituted with at least one chlorine and at least one
bromine. In an exemplary embodiment, Ra is fluoro-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is trifluoro-substituted Ci-C6 alkyl.
[0095] In an exemplary embodiment, the compound has a structure according to the following formula:





[0096] In an exemplary embodiment, the compound has a structure according to the following formula:





[0097] In an exemplary embodiment, the compound has a structure which is:
 wherein b is 1 or 2, C is pyrazolyl, Ra is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl. In an exemplary embodiment, b is 1. In an exemplary embodiment, b is 2. In an exemplary embodiment, b is 1, Ra is fluoro. In an exemplary embodiment, b is 2, and at least one Ra is fluoro. In an exemplary embodiment, b is 1, Ra is chloro. In an exemplary embodiment, b is 2, and at least one Ra is chloro. In an exemplary embodiment, b is 1, Ra is bromo. In an exemplary embodiment, b is 2, and at least one Ra is bromo. In an exemplary embodiment, b is 1, Ra is iodo. In an exemplary embodiment, b is 2, and at least one Ra is iodo. In an exemplary embodiment, b is 1, Ra is methyl. In an exemplary embodiment, b is 2, and at least one Ra is methyl. In an exemplary embodiment, b is
wherein b is 1 or 2, C is pyrazolyl, Ra is selected from the group consisting of halogen, unsubstituted alkyl and halosubstituted alkyl. In an exemplary embodiment, b is 1. In an exemplary embodiment, b is 2. In an exemplary embodiment, b is 1, Ra is fluoro. In an exemplary embodiment, b is 2, and at least one Ra is fluoro. In an exemplary embodiment, b is 1, Ra is chloro. In an exemplary embodiment, b is 2, and at least one Ra is chloro. In an exemplary embodiment, b is 1, Ra is bromo. In an exemplary embodiment, b is 2, and at least one Ra is bromo. In an exemplary embodiment, b is 1, Ra is iodo. In an exemplary embodiment, b is 2, and at least one Ra is iodo. In an exemplary embodiment, b is 1, Ra is methyl. In an exemplary embodiment, b is 2, and at least one Ra is methyl. In an exemplary embodiment, b is
1, Ra is ethyl. In an exemplary embodiment, b is 2, and at least one Ra is ethyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is an unsubstitued C3 alkyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, b is
2, and at least one Ra is an unsubstitued C4 alkyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued Cs alkyl. In an exemplary embodiment, b is 2, and at least one Ra is an unsubstitued C5 alkyl. In an exemplary embodiment, b is 1, Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 2, and at least one Ra is an unsubstitued C6 alkyl. In an exemplary embodiment, b is 1 or 2, and at least one Ra is selected from the group consisting of methyl, ethyl, propyl, butyl, pentyl and hexyl. In an exemplary embodiment, b is 1 and Ra is halo-substituted Ci-C6 alkyl. In an
exemplary embodiment, b is 2, and at least one Ra is halo-substituted Ci-C6 alkyl. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with one halogen. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with two halogens. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with three halogens. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with one fluorine. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with two fluorines. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with three fluorines. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with one chlorine. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with two chlorines. In an exemplary embodiment, b is 1 and Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 2, and at least one Ra is Ci-C6 alkyl, substituted with three chlorines. In an exemplary embodiment, b is 1 or 2 and Ra is Ci-C6 alkyl, substituted with a combination of two different halogens. In an exemplary embodiment, b is 1 or 2 and Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one chlorine. In an exemplary embodiment, b is 1 or 2 and Ra is Ci-C6 alkyl, substituted with at least one fluorine and at least one bromine. In an exemplary embodiment, b is 1 or 2 and Ra is Ci-C6 alkyl, substituted with at least one chlorine and at least one bromine. In an exemplary embodiment, b is 1 or 2 and Ra is fluoro-substituted Ci-C6 alkyl. In an exemplary embodiment, Ra is trifluoro-substituted Ci-C6 alkyl.
[0098] In an exemplary embodiment, the compound has a structure according to the following formula:
 wherein R2, R3 and R4 are according to the entries in the following table, or a salt thereof.
wherein R2, R3 and R4 are according to the entries in the following table, or a salt thereof.
K K K
906 H H H
907 Cl H H
908 H Cl H
909 H H Cl
910 Cl Cl H
911 Cl H Cl
912 H Cl Cl
913 Cl CpHpH PHl Cl
914 F H H
915 H H
916 H H F
917 F F H
918 F H PH PH PH PH PH PH PH PH
919 H
920 F F
921 Cl F H
922 F Cl H
923 Cl H
924 F H Cl
925 H Cl
926 H Cl
927 Cl F
928 F Cl
929 Cl Cl F
930 Cl F Cl
931 F Cl
932 F Cl
933 Cl F F
934 F Cl Cl
935 -CF3 H H
936 H -CF3 H
937 H H -CF3
938 -CH3 H H
939 H -CH3 H
940 H H -CH3
941 -CH2CH3 H H
942 H -CH2CH3 H
943 H H -CH2CH3
944 -CH(CH3)2 H H
945 H -CH(CH3)2 H
946 H H -CH(CH3),
947 -C(CH3), H H
948 H -C(CHs)3 H 949 H H -C(CHs)3
[0099] In an exemplary embodiment, the compound has a structure which is selected from the group consisting of

[0101] In an exemplary embodiment, the selectivity index (SI) of a compound of the invention is between about 50-150. In an exemplary embodiment, the selectivity index (SI) of 6-(4-chlorophenylsulfinyl)benzo[c][l,2]oxaborol-l(3H)-ol is greater than about 75-100.
[0102] In an exemplary embodiment, the in vitro metabolism T 1/2 (Mouse/human liver microsomes) of a compound of the invention is a member selected from about 300 minutes to 400 minutes. In an exemplary embodiment, the in vitro metabolism T 1/2 (Mouse/human liver microsomes) of a compound of the invention is a member selected from about 340 minutes to 360 minutes. In an exemplary embodiment, the in vitro metabolism T 1/2 (Mouse/human liver microsomes) of 6-(4-chlorophenyl sulfmyl)benzo[c][l,2]oxaborol-l(3H)-ol is greater than 350 minutes. [0103] In an exemplary embodiment, the in vitro metabolism T 1/2 (Mouse S9) of a compound of the invention is a member selected from about 100 minutes to 300 minutes.
[0104] In an exemplary embodiment, a compound of the invention essentially does not inhibit a cytochrome P450 enzyme. In an exemplary embodiment, a compound of the invention does not inhibit a cytochrome P450 enzyme. In an exemplary
embodiment, 6-(4-chlorophenylsulfϊnyl) benzo[c][l,2]oxaborol-l(3H)-ol essentially does not inhibit a cytochrome P450 enzyme. In an exemplary embodiment, 6-(4- chlorophenylsulfinyl)benzo[c][l,2]oxaborol-l(3H)-ol does not inhibit a cytochrome P450 enzyme. In an exemplary embodiment, the cytochrome P450 enzyme is a member selected from CP1A2, 2C9, 2D6 and 3A4. In an exemplary embodiment, the cytochrome P450 enzyme is CYP2C19.
[0105] In an exemplary embodiment, a compound of the invention is essentially not a substrate for the P-gp transporter. In an exemplary embodiment, a compound of the invention is not a substrate for the P-gp transporter. [0106] In an exemplary embodiment, the invention provides a compound described herein, or a salt, hydrate or solvate thereof, or a combination thereof. In an exemplary embodiment, the invention provides a compound described herein, or a salt, hydrate or solvate thereof. In an exemplary embodiment, the invention provides a compound described herein, or a salt thereof. In an exemplary embodiment, the salt is a pharmaceutically acceptable salt. In an exemplary embodiment, the invention provides a compound described herein, or a hydrate thereof. In an exemplary embodiment, the invention provides a compound described herein, or a solvate thereof. In an exemplary embodiment, the invention provides a compound described herein, or a prodrug thereof. In an exemplary embodiment, the invention provides a salt of a compound described herein. In an exemplary embodiment, the invention provides a pharmaceutically acceptable salt of a compound described herein. In an exemplary embodiment, the invention provides a hydrate of a compound described herein. In an exemplary embodiment, the invention provides a solvate of a compound described herein. In an exemplary embodiment, the invention provides a prodrug of a compound described herein.
[0107] In an exemplary embodiment, alkyl is linear alkyl. In another exemplary embodiment, alkyl is branched alkyl.
[0108] In an exemplary embodiment, heteroalkyl is linear heteroalkyl. In another exemplary embodiment, heteroalkyl is branched heteroalkyl. HLb) Compositions involving stereoisomers
[0109] As used herein, the term "chiral", "enantiomerically enriched" or "diastereomerically enriched" refers to a composition having an enantiomeric excess
(ee) or a diastereomeric excess (de) of greater than about 50%, preferably greater than about 70% and more preferably greater than about 90%. In general, higher than about 90% enantiomeric or diastereomeric excess is particularly preferred, e.g., those compositions with greater than about 95%, greater than about 97% and greater than about 99% ee or de.
[0110] When a first compound and a second compound are present in a composition, and the first compound is a non-superimposable mirror image of the second compound, and the first compound is present in the composition in a greater amount than the second compound, then the first compound is referred to herein as being present in "enantiomeric excess".
[0111] The term "enantiomeric excess" of a compound z, as used herein, is defined as: eeχ = ( COnC- °f Z - C°nC- °f A x 100 yconc. of z + cone, of y) wherein z is a first compound in a composition, y is a second compound in the composition, and the first compound is a non-superimposable mirror image of the second compound.
[0112] The term "enantiomeric excess" is related to the older term "optical purity" in that both are measures of the same phenomenon. The value of ee will be a number from 0 to 100, zero being racemic and 100 being enantiomerically pure. A composition which in the past might have been called 98% optically pure is now more precisely characterized by 96% ee. A 90% ee reflects the presence of 95% of one enantiomer and 5% of the other(s) in the material in question.
[0113] When a first compound and at least one additional compound are present in a composition, and the first compound and each of the additional compounds are stereoisomers, but not mirror images, of one another, and the first compound is present in the composition in a greater amount than each of the additional compounds, then the first compound is referred to herein as being present in "diastereomeric excess".
[0114] When dealing with mixtures of diastereomers, the term "diastereomeric excess" or "de" is defined analagously to enantiomeric excess. Thus:
_ ( cone, of major diastereomer - cone, of ram or diastereomer (s) Λ yconc. of major diastereomer + cone, of ram or diastereomer is) J wherein the major diastereomer is a first compound in a composition, and the minor diastereomer(s) is at least one additional compound in the composition, and the major diastereomer and minor diastereomer(s) are stereoisomers, but not mirror images, of one another.
[0115] The value of de will likewise be a number from 0 to 100, zero being an equal mixture of a first diastereomer and the remaining diastereomer(s), and 100 being 100% of a single diastereomer and zero% of the other(s) - i.e. diastereomerically pure. Thus, 90% de reflects the presence of 95% of one diastereomer and 5% of the other diastereomer(s) in the material in question.
[0116] Hence, in one embodiment, the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has at least one stereocenter, and at least one stereoisomer of the first compound of the invention. In another embodiment, the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has at least one stereocenter, and a second compound of the invention, wherein the first compound of the invention is a stereoisomer of the second compound of the invention. In another embodiment, the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has at least one stereocenter, and only one stereoisomer of the first compound of the invention.
[0117] In another embodiment, the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has only one stereocenter, and an enantiomer of the first compound of the invention. In another embodiment, the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has two stereocenters, and an enantiomer of the first compound of the invention. In another embodiment, the invention provides a composition including a first compound of the invention, wherein the first compound of the invention has two stereocenters, and at least one diasteromer of the first compound of the invention. In another embodiment, the invention provides a composition including a first compound of the invention,
wherein the first compound of the invention has two stereocenters, and only one diasteromer of the first compound of the invention.
[0118] In situations where the first compound of the invention and its enantiomer are present in a composition, the first compound of the invention can be present in an enantiomeric excess of at least about 80%, or at least about 90%, or at least about 92% or at least about 95%. In another embodiment, where the first compound of the invention and its enantiomer are present in a composition, the first compound of the invention can be present in an enantiomeric excess of at least about 96%, at least about 97%, at least about 98%, at least about 99% or at least about 99.5%. In another embodiment, the first compound of the invention has at least one stereocenter and is enantiomerically pure (enantiomeric excess is about 100%).
[0119] In situations where the first compound of the invention and at least one diastereomer of the first compound of the invention are present in a composition, the first compound of the invention can be present in a diastereomeric excess of at least about 80%, or at least about 90%, or at least about 92% or at least about 95%. In situations where the first compound of the invention and at least one diastereomer of the first compound of the invention are present in a composition, the first compound of the invention can be present in a diastereomeric excess of at least about 96%, at least about 97%, at least about 98%, at least about 99% or at least about 99.5%. In another embodiment, the first compound of the invention has at least two stereocenters and is diastereomerically pure (diastereomeric excess is about 100%).
[0120] Enantiomeric or diastereomeric excess can be determined relative to exactly one other stereoisomer, or can be determined relative to the sum of at least two other stereoisomers. In an exemplary embodiment, enantiomeric or diastereomeric excess is determined relative to all other detectable stereoisomers, which are present in the mixture. Stereoisomers are detectable if a concentration of such stereoisomer in the analyzed mixture can be determined using common analytical methods, such as chiral HPLC.
[0121] As used herein, and unless otherwise indicated, a composition that is "substantially free" of a compound means that the composition contains less than about 20% by weight, or less than about 15% by weight, or less than about 10% by
weight, or less than about 5% by weight, or less than about 3% by weight, or less than about 2% by weight, or less than about 1% by weight of the compound.
[0122] As used herein, the term "substantially free of the (or its) enantiomer" means that a composition contains a significantly greater proportion of a first compound of the invention than a second compound of the invention, wherein the first compound is a non-superimposable mirror image of the second compound. In one embodiment of the invention, the term "substantially free of the enantiomer" means that the composition is made up of at least about 90% by weight of a first compound of the invention, and about 10% by weight or less of a second compound of the invention, wherein the first compound is a non-superimposable mirror image of the second compound. In one embodiment of the invention, the term "substantially free of the (R) enantiomer" means that the composition is made up of at least about 90% by weight of a first compound of the invention which has only one stereocenter and the stereocenter is in an (S) configuration, and about 10% by weight or less of a second compound of the invention, wherein the second compound is the enantiomer of the first compound. In one embodiment of the invention, the term "substantially free of the enantiomer" means that the composition is made up of at least about 95% by weight of a first compound of the invention, and about 5% by weight or less of a second compound of the invention, wherein the first compound is a non- superimposable mirror image of the second compound. In one embodiment of the invention, the term "substantially free of the (R) enantiomer" means that the composition is made up of at least about 95% by weight of a first compound of the invention which has only one stereocenter and the stereocenter is in an (S) configuration, and about 5% by weight or less of a second compound of the invention, wherein the second compound is the enantiomer of the first compound. In one embodiment of the invention, the term "substantially free of the enantiomer" means that the composition is made up of at least about 98% by weight of a first compound of the invention, and about 2% by weight or less of a second compound of the invention, wherein the first compound is a non-superimposable mirror image of the second compound. In one embodiment of the invention, the term "substantially free of the (R) enantiomer" means that the composition is made up of at least about 98% by weight of a first compound of the invention which has only one stereocenter and the stereocenter is in an (S) configuration, and about 2% by weight or less of a
second compound of the invention, wherein the second compound is the enantiomer of the first compound. In one embodiment of the invention, the term "substantially free of the enantiomer" means that the composition is made up of at least about 99% by weight of a first compound of the invention, and about 1% by weight or less of a second compound of the invention, wherein the first compound is a non- superimposable mirror image of the second compound. In one embodiment of the invention, the term "substantially free of the (R) enantiomer" means that the composition is made up of at least about 99% by weight of a first compound of the invention which has only one stereocenter and the stereocenter is in an (S) configuration, and about 1% by weight or less of a second compound of the invention, wherein the second compound is the enantiomer of the first compound..
HLb) Combinations comprising additional therapeutic agents
[0123] The compounds of the invention may also be used in combination with additional therapeutic agents. The invention thus provides, in a further aspect, a combination comprising a compound described herein or a pharmaceutically acceptable salt thereof together with at least one additional therapeutic agent. In an exemplary embodiment, the additional therapeutic agent is a compound of the invention. In an exemplary embodiment, the additional therapeutic agent includes a boron atom. In an exemplary embodiment, the additional therapeutic agent does not contain a boron atom.
[0124] When a compound of the invention is used in combination with a second therapeutic agent active against the same disease state, the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art. It will be appreciated that the amount of a compound of the invention required for use in treatment will vary with the nature of the condition being treated and the age and the condition of the patient and will be ultimately at the discretion of the attendant physician or veterinarian. In an exemplary embodiment, the additional therapeutic agent is Berenil. In an exemplary embodiment, the additional therapeutic agent is an antiprotozoa. In an exemplary embodiment, the additional therapeutic agent is a member selected from
Benznidazole, Buparvaquone, Carbarsone, Clioquinol, Disulfiram, Eflornithine, Emetine, Etofamide, Furazolidone, Meglumine antimoniate, Melarsoprol, Metronidazole, Miltefosine, Nifurtimox, Nimorazole, Nitazoxanide, Ornidazole,
Paromomycin sulfate, Pentamidine, Pyrimethamine, Secnidazole and Tinidazole. In an exemplary embodiment, the additional therapeutic agent is pentamidine. In an exemplary embodiment, the additional therapeutic agent is suramin. In an exemplary embodiment, the additional therapeutic agent is Eflornithine. In an exemplary embodiment, the additional therapeutic agent is Melarsoprol. In an exemplary embodiment, the additional therapeutic agent is Nifurtimox. In an exemplary embodiment, the additional therapeutic agent is an antiparasitic. In an exemplary embodiment, the additional therapeutic agent is a member selected from Amitraz, Avermectin, Carbadox, Diethylcarbamazine, Dimetridazole, Diminazene, Ivermectin, Macro filaricide, Malathion, Mitaban, Organophosphate, Oxamniquine, Permethrin, Praziquantel, Pyrantel pamoate, Selamectin, Sodium stibogluconate and Thiabendazole. In an exemplary embodiment, the additional therapeutic agent is a member selected from antimony, meglumine antimoniate, sodium stibogluconate, amphotericin, miltefosine and paromomycin. [0125] The compounds of the invention, or pharmaceutical formulations thereof may also be used in combination with other therapeutic agents, for example immune therapies [e.g. interferon, such as interferon alfa-2a (ROFERONd)-A; Hoffmann-La Roche), interferon alpha-2b (INTRONd)-A; Schering-Plough), interferon alfacon-1 (INFERGEN®; Intermune), peginterferon alpha-2b (PEGINTRON™; Schering- Plough) or peginterferon alpha-2a (PEGAS YSd); Hoffmann-La Roche)], therapeutic vaccines, antifibrotic agents, anti-inflammatory agents [such as corticosteroids or NSAIDs], bronchodilators [such as beta-2 adrenergic agonists and xanthines (e.g. theophylline)], mucolytic agents, anti-muscarinics, anti-leukotrienes, inhibitors of cell adhesion [e.g. ICAM antagonists], anti-oxidants [e.g. N-acetylcysteine], cytokine agonists, cytokine antagonists, lung surfactants and/or antimicrobial. The compositions according to the invention may also be used in combination with gene replacement therapy.
[0126] The individual components of such combinations may be administered either simultaneously or sequentially in a unit dosage form. The unit dosage form may be a single or multiple unit dosage forms. In an exemplary embodiment, the invention provides a combination in a single unit dosage form. An example of a single unit dosage form is a capsule wherein both the compound of the invention and the additional therapeutic agent are contained within the same capsule. In an
exemplary embodiment, the invention provides a combination in a two unit dosage form. An example of a two unit dosage form is a first capsule which contains the compound of the invention and a second capsule which contains the additional therapeutic agent. Thus the term 'single unit' or 'two unit' or 'multiple unit' refers to the object which the patient ingests, not to the interior components of the object. Appropriate doses of known therapeutic agents will be readily appreciated by those skilled in the art.
[0127] The combinations referred to herein may conveniently be presented for use in the form of a pharmaceutical formulation. Thus, an exemplary embodiment of the invention is a pharmaceutical formulation comprising a) a compound of the invention; b) an additional therapeutic agent and c) a pharmaceutically acceptable excipient. In an exemplary embodiment, the pharmaceutical formulation is a unit dosage form. In an exemplary embodiment, the pharmaceutical formulation is a single unit dosage form. In an exemplary embodiment, the pharmaceutical formulation is a two unit dosage form. In an exemplary embodiment, the pharmaceutical formulation is a two unit dosage form comprising a first unit dosage form and a second unit dosage form, wherein the first unit dosage form includes a) a compound of the invention and b) a first pharmaceutically acceptable excipient; and the second unit dosage form includes c) an additional therapeutic agent and d) a second pharmaceutically acceptable excipient.
[0128] It is to be understood that the present invention covers all combinations of aspects and/or embodiments, as well as suitable, convenient and preferred groups described herein.
HLc) Preparation of Boron-Containing Compounds [0129] Compounds of use in the present invention can be prepared using commercially available starting materials or known intermediates. Compounds of use in the present invention can be prepared using synthetic methods known in the art or described herein.
[0130] The following general procedures were used as indicated in generating the examples and can be applied, using the knowledge of one of skill in the art, to other appropriate compounds to obtain additional analogues.
[0131] Thioether, sulfoxide and sulfone derivatives such as B, C and D can be prepared by the following reactions. Thioethers such as B can be obtained by subjecting bromide A to boronylation conditions, such as treatment with n-butyl lithium and triisopropyl borate followed by addition of acid. Sulfoxides such as C can be obtained by subjecting B to oxidation conditions, such as sodium periodate or one equivalent of mCPBA. Sulfones such as D can be obtained by subjecting B to oxidation conditions such as sodium periodate over extended reaction time or two equivalents of mCPBA.

or tO RT

[0132] Carbamates such as F and G can be prepared by reacting compound E with corresponding isocyanate RNCO or ArNCO in the presence of base such as triethylamine.

[0133] Ethers such as H and I can be obtained by reacting compound E under basic conditions with alkylbromide RBr or arylbromide ArBr.

H I
[0134] Benzylethers such as K can be obtained by reacting compound E under basic conditions with substituted benzyl bromide J.

[0135] Carbinol derivatives such as M can be obtained by sujecting ketone L to reducing conditions such as sodium borohydride. Ketone derivatives such as N can be obtained by subjecting alcohol M to oxidation conditions such as PCC.

M
[0136] Amide derivatives such as P can be prepared from compound O and corresponding anilines by standard peptide coupling conditions as shown below.

O
[0137] Amide derivatives such as S can be prepared from compound Q and corresponding acyl chlorides by standard peptide coupling conditions such as shown below.

[0138] Benzylamine derivatives such as T can be obtained by reacting compound Q with corresponding benzyl bromides under basic conditions such as shown below.


[0140] Compounds such as W can be prepared by subjecting protected amines such as V to acidic conditions such as shown below.

V W
[0141] To make derivatives with a methylene linkage group such as Y, aldehyde X can be first subjected to reducing conditions such as sodium borohydride, then subjected to acidic conditions such as shown below.

X Y
General Procedure: Synthesis of Amino 3H-benzofc] fl,2]oxaborol-l-ols

[0142] Reference: JACS 1960, 82, 2172. Benzoxaboroles can be mixed with concentrated nitric acid at -40 0C. The mixture can be stirred for 30min then can be added to ice water to precipitate a solid that can be collected by filtration. This crude material can be recrystallized, such as from water, to produce the appropriately substituted 6-nitro 3H-benzo[c][l,2]oxaborol-l-ol. The nitro group can be reduced by dissolving in EtOH and combining with Raney-Ni. This mixture can be subjected to 1.6atm of hydrogen with agitation in a Parr apparatus for 16hrs. Nickel catalyst can be removed via filtration and the solvent can be removed under reduced pressure. The resulting residues can be purified by recrystallization from 25% EtOH.
General Procedure: Sulfonylation of Amino 3H-benzofcJ [l,2]oxaborol-l-ols

[0143] Through subjecting it to sulfonylation conditions, compound 1 * can be converted to compound 2*.
[0144] In some applications of this general procedure, unsubstituted phenyl or unsubstituted pyridinyl sulfonyl chloride (1-1.2 equiv) and a base (such as NMM, K2CO3, or pyridine 3-4 equiv) can be added sequentially to a solution of the amine in MeCN (20 mL/g) at rt. After completion (typical duration O/N) the volatiles can be removed in vacuo. H2O can be added to the residue and the mixture adjusted to ~ pH 6 with dilute HCl. The aqueous layer can be then extracted with an organic solvent (such as EtOAc), and the combined organic fractions can be dried with a desiccant, such as Na2SO4 or MgSO4, filtered, and concentrated in vacuo. The product can be typically purified by either recrystallization from H2O, trituration with CH2Cl2 or EtOAc, or flash chromatography.
[0145] Compounds described herein can be converted into hydrates and solvates by methods similar to those described herein.
IV. Methods of Inhibiting Microorganism Growth or Killing Microorganisms
[0146] The compounds of the present invention exhibit potency against microorganisms, such as protozoa, and therefore have the potential to kill and/or inhibit the growth of microorganisms.
[0147] In a further aspect, the invention provides a method of killing and/or inhibiting the growth of a microorganism, said method comprising: contacting said microorganism with an effective amount of a compound of the invention, thereby killing and/or inhibiting the growth of the microorganism. In an exemplary embodiment, the microorganism is a protozoa. In an exemplary embodiment, the microorganism is a kinetoplastid. In another exemplary embodiment, the protozoa is a Trypanosoma. In an exemplary embodiment, the Trypanosoma is selected from the
group consisting of T. avium, T. boissoni, T. brucei, T. carassii, T. cruzi, T. congolense, T. equinum, T. equiperdum, T. evansi, T. hosei, T levisi, T. melophagium, T parroti, T. percae, T. rangeli, T. rotatorium, T. rugosae, T sergenti, T. simiae, T sinipercae, T. suis, T. theileri, T. triglae and T. vivax. In another exemplary embodiment, the protozoa is a Trypanosoma brucei. In another exemplary embodiment, the protozoa is Trypanosoma brucei brucei. In another exemplary embodiment, the protozoa is Trypanosoma brucei rhodesiense. In another exemplary embodiment, the protozoa is Trypanosoma brucei gambiense. In another exemplary embodiment, the protozoa is a member of the genus Leishmania. In another exemplary embodiment, the protozoa is a member of Leishmania Viannia. In an exemplary embodiment, the protozoa is selected from the group consisting of L. donovani, L. infantum, L. chagasi; L. mexicana, L. amazonensis, L. venezuelensis, L. tropica, L. major, L. aethiopica, L. (V.) braziliensis, L. (V.) guyanensis, L. (V.) panamensis, and L. (V.) peruviana. In an exemplary embodiment, the protozoa is L. donovani. In an exemplary embodiment, the protozoa is L. infantum. In another exemplary embodiment, the protozoa is a member of the genus Plasmodium. In another exemplary embodiment, the protozoa is selected from the group consisting of Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium vivax, Plasmodium malariae and Plasmodium knowlesi. In another exemplary embodiment, the protozoa is selected from the group consisting of Plasmodium vivax, Plasmodium ovale, Plasmodium vivax and Plasmodium malariae. In another exemplary embodiment, the protozoa is Plasmodium falciparum. In another exemplary embodiment, the protozoa is transmitted to the animal described herein by a mosquito infected with the protozoa. In another exemplary embodiment, wherein the protozoa is transmitted to the animal described herein by an Anopheles mosquito containing the protozoa. In an exemplary embodiment, the compound is selected from the group consisting of 6-(2-chlorophenyl sulfϊnyl)benzo[c][l,2]oxaborol-l(3H)-ol, 6-(3- chlorophenylsulfmyl) benzo[c][l,2] oxaborol-l(3H)-ol and 6-(4-chlorophenylsulfmyl) benzo[c][l,2] oxaborol-l(3H)-ol, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof. In an exemplary embodiment, the compound is 6-(4- chlorophenylsulfmyl) benzo[c][l,2]oxaborol-l(3H)-ol, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof. In an exemplary embodiment, the compound is 6-(4-chlorophenylsulfmyl)benzo[c][l,2] oxaborol-l(3H)-ol. In an exemplary embodiment, the compound is described herein, or a salt, prodrug, hydrate
or solvate thereof, or a combination thereof. In an exemplary embodiment, the invention provides a compound described herein, or a salt, hydrate or solvate thereof. In an exemplary embodiment, the invention provides a compound described herein, or a prodrug thereof. In an exemplary embodiment, the invention provides a compound described herein, or a salt thereof. In another exemplary embodiment, the compound of the invention is a compound described herein, or a pharmaceutically acceptable salt thereof. In another exemplary embodiment, the compound is described by a formula listed herein, or a pharmaceutically acceptable salt thereof. In an exemplary embodiment, the compound is part of a pharmaceutical formulation described herein. In another exemplary embodiment, the contacting occurs under conditions which permit entry of the compound into the organism. Such conditions are known to one skilled in the art and specific conditions are set forth in the Examples appended hereto.
[0148] In another aspect, the microorganism is inside, or on the surface of an animal. In an exemplary embodiment, the animal is selected from the group consisting of human, cattle, deer, reindeer, goat, honey bee, pig, sheep, horse, cow, bull, dog, guinea pig, gerbil, rabbit, cat, camel, yak, elephant, ostrich, otter, chicken, duck, goose, guinea fowl, pigeon, swan, and turkey. In another exemplary embodiment, the animal is a human. [0149] In an exemplary embodiment, the microorganism is killed or its growth is inhibited through oral administration of the compound of the invention. In an exemplary embodiment, the microorganism is killed or its growth is inhibited through intravenous administration of the compound of the invention. In an exemplary embodiment, the microorganism is killed or its growth is inhibited through topical administration of the compound of the invention. In an exemplary embodiment, the microorganism is killed or its growth is inhibited through intraperitoneal administration of the compound of the invention. In an exemplary embodiment, the compound is administered in a topically effective amount. In an exemplary embodiment, the compound is administered in a cosmetically effective amount. In an exemplary embodiment, the pharmaceutical formulation is administered in an orally effective amount.
V. Methods of Treating and/or Preventing Disease [0150] The compounds of the present invention exhibit potency against microorganisms, such as protozoa, and therefore have the potential to achieve therapeutic efficacy in the animals described herein. [0151] In another aspect, the invention provides a method of treating and/or preventing a disease. The method includes administering to the animal a therapeutically effective amount of the compound of the invention, sufficient to treat and/or prevent the disease. In an exemplary embodiment, the animal is not otherwise is need of treatment with the compound of the invention. In an exemplary embodiment, the compound of the invention can be used in human or veterinary medical therapy, particularly in the treatment or prophylaxis of protozoa-associated disease. In an exemplary embodiment, the compound of the invention can be used in human or veterinary medical therapy, particularly in the treatment or prophylaxis of kinetoplastid-associated disease. In an exemplary embodiment, the disease is associated with a Trypanosoma. In an exemplary embodiment, the Trypanosoma is selected from the group consisting of T. avium, T. boissoni, T. brucei, T. carassii, T. cruzi, T. congolense, T equinum, T equiperdum, T evansi, T hosei, T levisi, T melophagium, T parroti, T percae, T rangeli, T rotatorium, T. rugosae, T sergenti, T simiae, T sinipercae, T suis, T theileri, T triglae and T. vivax. In an exemplary embodiment, the disease is associated with a Trypanosoma brucei. In an exemplary embodiment, the disease is associated with Trypanosoma brucei brucei. In an exemplary embodiment, the disease is associated with Trypanosoma brucei rhodesiense. In an exemplary embodiment, the disease is associated with Trypanosoma brucei gambiense. In an exemplary embodiment, the disease is a typanosomiasis. In an exemplary embodiment, the disease is a human typanosomiasis. In an exemplary embodiment, the disease is an animal typanosomiasis. In an exemplary embodiment, the disease is selected from the group consisting of nagana, surra, mal de caderas, murrina de caderas, dourine, cachexial fevers, Gambian horse sickness, baleri, kaodzera, tahaga, galziekte or galzietzke and peste-boba. In an exemplary embodiment, the disease is selected from the group consisting of Chagas disease (or Human American trypanosomiasis), nagana, surra, Covering sickness (or dourine) and sleeping sickness (or African sleeping sickness or Human African trypanosomiasis). In an exemplary embodiment, the disease is
Chagas disease. In an exemplary embodiment, the disease is sleeping sickness (or African sleeping sickness). In an exemplary embodiment, the disease is acute phase sleeping sickness. In an exemplary embodiment, the disease is chronic phase sleeping sickness. In an exemplary embodiment, the disease is an acute phase of a typanosomiasis. In an exemplary embodiment, the disease is a chronic phase of a typanosomiasis. In an exemplary embodiment, the disease is the non-CNS form of a typanosomiasis. In an exemplary embodiment, the disease is the CNS form of a typanosomiasis. In an exemplary embodiment, the disease is the non-CNS form of sleeping sickness. In an exemplary embodiment, the disease is the CNS form of sleeping sickness. In an exemplary embodiment, the disease is early stage Human African trypanosomiasis. In an exemplary embodiment, the disease is late stage Human African trypanosomiasis. In another exemplary embodiment, the disease is associated with a member of the genus Leishmania. In another exemplary embodiment, the disease is associated with a member of Leishmania Viannia. In an exemplary embodiment, the disease is associated with a member selected from the group consisting of L. donovani, L. infantum, L. chagasi; L. mexicana, L. amazonensis, L. venezuelensis, L. tropica, L. major, L. aethiopica, L. (V.) braziliensis, L. (V.) guyanensis, L. (V.) panamensis, and L. (V.) peruviana. In an exemplary embodiment, the disease is associated with L. donovani. In an exemplary embodiment, the disease is associated with L. infantum. In an exemplary embodiment, the disease is leshmaniasis. In an exemplary embodiment, the disease is a member selected from visceral leshmaniasis and/or cutaneous leshmaniasis. In an exemplary embodiment, the disease is diffuse cutaneous leshmaniasis and/or mucocutaneous leshmaniasis. In another exemplary embodiment, the disease is associated with a member of the genus Plasmodium. In another exemplary embodiment, the disease is associated with a member selected from the group consisting of Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium vivax, Plasmodium malariae and Plasmodium knowlesi. In another exemplary embodiment, the disease is associated with a member selected from the group consisting of Plasmodium vivax, Plasmodium ovale, Plasmodium vivax and Plasmodium malariae. In another exemplary embodiment, the disease is associated with Plasmodium falciparum. In another exemplary embodiment, the disease is transmitted to the animal described herein by a mosquito infected with the protozoa. In another exemplary embodiment, the disease is transmitted to the animal described
herein by an Anopheles mosquito containing the protozoa. In another exemplary embodiment, the disease is malaria. In another exemplary embodiment, the disease is cerebral malaria. In another exemplary embodiment, the disease is chronic malaria. In an exemplary embodiment, the compound is selected from the group consisting of 6-(2-chlorophenylsulfmyl)benzo[c][l,2] oxaborol-l(3H)-ol, 6-(3- chlorophenylsulfmyl)benzo[c] [ 1 ,2]oxaborol- 1 (3H)-ol and 6-(4-chlorophenylsulfmyl) benzo[c][l,2] oxaborol-l(3H)-ol, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof. In an exemplary embodiment, the compound is 6-(4- chlorophenylsulfinyl)benzo[c][l,2]oxaborol-l(3H)-ol, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof. In an exemplary embodiment, the compound is 6-(4-chlorophenylsulfmyl)benzo[c][l,2] oxaborol-l(3H)-ol. In an exemplary embodiment, the compound is described herein, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof. In an exemplary embodiment, the invention provides a compound described herein, or a salt, hydrate or solvate thereof. In an exemplary embodiment, the invention provides a compound described herein, or a prodrug thereof. In an exemplary embodiment, the invention provides a compound described herein, or a salt thereof. In another exemplary embodiment, the compound of the invention is a compound described herein, or a pharmaceutically acceptable salt thereof. In another exemplary embodiment, the compound is described by a formula listed herein, or a pharmaceutically acceptable salt thereof. In an exemplary embodiment, the compound is part of a pharmaceutical formulation described herein. In another exemplary embodiment, the contacting occurs under conditions which permit entry of the compound into the organism. Such conditions are known to one skilled in the art and specific conditions are set forth in the Examples appended hereto.
[0152] In another exemplary embodiment, the animal is selected from the group consisting of human, cattle, deer, reindeer, goat, honey bee, pig, sheep, horse, cow, bull, dog, guinea pig, gerbil, rabbit, cat, camel, yak, elephant, ostrich, otter, chicken, duck, goose, guinea fowl, pigeon, swan, and turkey. In another exemplary embodiment, the animal is a human. In another exemplary embodiment, the animal is a mouse. In another exemplary embodiment, the animal is selected from the group consisting of a human, cattle, goat, pig, sheep, horse, cow, bull, dog, guinea pig, gerbil, rabbit, cat, chicken and turkey.
[0153] In an exemplary embodiment, the disease is treated through oral administration of the compound of the invention. In an exemplary embodiment, the disease is treated through intravenous administration of the compound of the invention. In an exemplary embodiment, the disease is treated through topical administration of the compound of the invention. In an exemplary embodiment, the disease is treated through intraperitoneal administration of the compound of the invention. In an exemplary embodiment, the compound is administered in a topically effective amount. In an exemplary embodiment, the compound is administered in a cosmetically effective amount. In an exemplary embodiment, the pharmaceutical formulation is administered in an orally effective amount.
[0154] In an exemplary embodiment, the disease is associated with an infection by a microorganism described herein. In an exemplary embodiment, the disease is associated with an infection by a protozoa described herein.
VI. Pharmaceutical Formulations [0155] In another aspect, the invention is a pharmaceutical formulation which includes: (a) a pharmaceutically acceptable excipient; and (b) a compound of the invention. In another aspect, the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound according to a formula described herein. In another aspect, the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound described herein, or a salt, prodrug, hydrate or solvate thereof, or a combination thereof. In another aspect, the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound described herein, or a salt, hydrate or solvate thereof, or a combination thereof. In another aspect, the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound described herein, or a salt, hydrate or solvate thereof. In another aspect, the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a salt of a compound described herein. In an exemplary embodiment, the salt is a pharmaceutically acceptable salt. In another aspect, the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a prodrug of a compound described herein. In another aspect, the pharmaceutical formulation includes: (a) a pharmaceutically acceptable excipient; and (b) a compound described herein. In an
exemplary embodiment, the pharmaceutical formulation is a unit dosage form. In an exemplary embodiment, the pharmaceutical formulation is a single unit dosage form.
[0156] In an exemplary embodiment, the invention is a pharmaceutical formulation which includes: (a) a pharmaceutically acceptable excipient; and (b) a compound which is

[0157] In an exemplary embodiment, the invention is a pharmaceutical formulation which includes: (a) a pharmaceutically acceptable excipient; and (b) a compound which is

[0158] The pharmaceutical formulations of the invention can take a variety of forms adapted to the chosen route of administration. Those skilled in the art will recognize various synthetic methodologies that may be employed to prepare non-toxic pharmaceutical formulations incorporating the compounds described herein. Those skilled in the art will recognize a wide variety of non-toxic pharmaceutically acceptable solvents that may be used to prepare solvates of the compounds of the invention, such as water, ethanol, propylene glycol, mineral oil, vegetable oil and dimethylsulfoxide (DMSO).
[0159] The pharmaceutical formulation of the invention may be administered orally, topically, intraperitoneally, parenterally, by inhalation or spray or rectally in unit dosage forms containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles. It is further understood that the best method of administration may be a combination of methods. Oral administration in the form of a pill, capsule, elixir, syrup, lozenge, troche, or the like is particularly preferred. The term parenteral as used herein includes subcutaneous injections, intradermal, intravascular (e.g., intravenous), intramuscular, spinal, intrathecal injection or like injection or infusion techniques. In an exemplary embodiment, the pharmaceutical formulation is administered orally. In an exemplary embodiment, the pharmaceutical formulation is administered intravenously. In an exemplary embodiment, the pharmaceutical formulation is administered in a topically effective dose. In an
exemplary embodiment, the pharmaceutical formulation is administered in a cosmetically effective dose. In an exemplary embodiment, the pharmaceutical formulation is administered in an orally effective dose.
[0160] The pharmaceutical formulations containing compounds of the invention are preferably in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, or syrups or elixirs.
[0161] Compositions intended for oral use may be prepared according to any method known in the art for the manufacture of pharmaceutical formulations, and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets may contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients that are suitable for the manufacture of tablets. These excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia; and lubricating agents, for example magnesium stearate, stearic acid or talc. The tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate may be employed.
[0162] Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin or olive oil.
[0163] Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions. Such excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; and dispersing or wetting agents, which may be a
naturally-occurring phosphatide, for example, lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate. The aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
[0164] Oily suspensions may be formulated by suspending the active ingredients in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin. The oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set forth above, and flavoring agents may be added to provide palatable oral preparations. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
[0165] Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present. [0166] Pharmaceutical formulations of the invention may also be in the form of oil-in-water emulsions and water-in-oil emulsions. The oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these. Suitable emulsifying agents may be naturally-occurring gums, for example gum acacia or gum tragacanth; naturally-occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol; anhydrides, for example sorbitan monooleate; and condensation products of
the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monooleate. The emulsions may also contain sweetening and flavoring agents.
[0167] Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, and flavoring and coloring agents. The pharmaceutical formulations may be in the form of a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents, which have been mentioned above. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, for example as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid find use in the preparation of injectables.
[0168] The composition of the invention may also be administered in the form of suppositories, e.g., for rectal administration of the drug. These compositions can be prepared by mixing the drug with a suitable non-irritating excipient that is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug. Such materials are cocoa butter and polyethylene glycols.
[0169] Alternatively, the compositions can be administered parenterally in a sterile medium. The drug, depending on the vehicle and concentration used, can either be suspended or dissolved in the vehicle. Advantageously, adjuvants such as local anesthetics, preservatives and buffering agents can be dissolved in the vehicle.
[0170] For administration to non-human animals, the composition containing the therapeutic compound may be added to the animal's feed or drinking water. Also, it will be convenient to formulate animal feed and drinking water products so that the animal takes in an appropriate quantity of the compound in its diet. It will further be convenient to present the compound in a composition as a premix for addition to the
feed or drinking water. The composition can also added as a food or drink supplement for humans.
[0171] Dosage levels of the order of from about 5 mg to about 250 mg per kilogram of body weight per day and more preferably from about 25 mg to about 150 mg per kilogram of body weight per day, are useful in the treatment of the above- indicated conditions. The amount of active ingredient that may be combined with the carrier materials to produce a unit dosage form will vary depending upon the condition being treated and the particular mode of administration. Unit dosage forms will generally contain between from about 1 mg to about 500 mg of an active ingredient.
[0172] Frequency of dosage may also vary depending on the compound used and the particular disease treated. However, for treatment of most disorders, a dosage regimen of 4 times daily or less is preferred. It will be understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration and rate of excretion, drug combination and the severity of the particular disease undergoing therapy.
[0173] In an exemplary embodiment, the unit dosage form contains from about 1 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 1 mg to about 500 mg of an active ingredient. In an exemplary embodiment, the unit dosage form contains from about 100 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 200 mg to about 500 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 500 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 1 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 10 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 50 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 25 mg to about 75 mg of a compound of the
invention. In an exemplary embodiment, the unit dosage form contains from about 40 mg to about 60 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 75 mg to about 200 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 1 mg to about 5 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 10 mg to about 25 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 50 mg to about 350 mg of a compound of the invention. In an exemplary embodiment, the unit dosage form contains from about 200 mg to about 400 mg of a compound of the invention.
[0174] In an exemplary embodiment, the daily dosage contains from about 1 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the the daily dosage contains from about 1 mg to about 500 mg of an active ingredient. In an exemplary embodiment, the daily dosage contains from about 100 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 200 mg to about 500 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 500 mg to about 800 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 1 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 10 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 50 mg to about 100 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 75 mg to about 200 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 1 mg to about 5 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 10 mg to about 25 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 50 mg to about 350 mg of a compound of the invention. In an exemplary embodiment, the daily dosage contains from about 200 mg to about 400 mg of a compound of the invention.
[0175] Preferred compounds of the invention will have desirable pharmacological properties that include, but are not limited to, oral bioavailability, low toxicity, low serum protein binding and desirable in vitro and in vivo half- lives. Penetration of the
blood brain barrier for compounds used to treat CNS disorders is necessary, while low brain levels of compounds used to treat peripheral disorders are often preferred.
[0176] Assays may be used to predict these desirable pharmacological properties. Assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco-2 cell monolayers. Toxicity to cultured hepatocyctes may be used to predict compound toxicity. Penetration of the blood brain barrier of a compound in humans may be predicted from the brain levels of laboratory animals that receive the compound intravenously.
[0177] Serum protein binding may be predicted from albumin binding assays. Such assays are described in a review by Oravcova, et al. (Journal of Chromatography B (1996) volume 677, pages 1-27).
[0178] Compound half- life is inversely proportional to the frequency of dosage of a compound. In vitro half- lives of compounds may be predicted from assays of microsomal half-life as described by Kuhnz and Gieschen (Drug Metabolism and Disposition, (1998) volume 26, pages 1120-1127).
[0179] The amount of the composition required for use in treatment will vary not only with the particular compound selected but also with the route of administration, the nature of the condition being treated and the age and condition of the patient and will ultimately be at the discretion of the attendant physician or clinician. VI. a) Testins
[0180] Preferred compounds for use in the pharmeceutical formulations described herein will have certain pharmacological properties. Such properties include, but are not limited to, low toxicity, low serum protein binding and desirable in vitro and in vivo half-lives. Assays may be used to predict these desirable pharmacological properties. Assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco-2 cell monolayers. Serum protein binding may be predicted from albumin binding assays. Such assays are described in a review by Oravcova et al. (1996, J. Chromat. B677: 1-27). Compound half-life is inversely proportional to the frequency of dosage of a compound. In vitro half- lives of compounds may be predicted from assays of microsomal half- life as described by Kuhnz and Gieschen (Drug Metabolism and Disposition, (1998) volume 26, pages 1120-1127).
[0181] Toxicity and therapeutic efficacy of such compounds can be determined by Standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50 (the dose lethal to 50% of the population) and the ED50 (the dose therapeutically effective in 50% of the population). The dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio between LD50 and ED50. Compounds that exhibit high therapeutic indices are preferred. The data obtained from these cell culture assays and animal studies can be used in formulating a range of dosage for use in humans. The dosage of such compounds lies preferably within a range of circulating concentrations that include the ED50 with little or no toxicity. The dosage can vary within this range depending upon the unit dosage form employed and the route of administration utilized. The exact formulation, route of administration and dosage can be chosen by the individual physician in view of the patient's condition. (See, e.g. Fingl et al., 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1, p. 1). VI. b) Administration
[0182] For any compound used in the method of the invention, the therapeutically effective dose can be estimated initially from cell culture assays, as disclosed herein. For example, a dose can be formulated in animal models to achieve a circulating concentration range that includes the EC50 (effective dose for 50% increase) as determined in cell culture, i.e., the concentration of the test compound which achieves a half-maximal inhibition of protozoa cell growth. Such information can be used to more accurately determine useful doses in humans.
[0183] In general, the compounds prepared by the methods, and from the intermediates, described herein will be administered in a therapeutically or cosmetically effective amount by any of the accepted modes of administration for agents that serve similar utilities. It will be understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration, and rate of excretion, drug combination, the severity of the particular disease undergoing therapy and the judgment of the prescribing physician. The drug can be administered from once or twice a day, or up to 3 or 4 times a day.
[0184] Dosage amount and interval can be adjusted individually to provide plasma levels of the active moiety that are sufficient to maintain protozoa cell growth inhibitory effects. Usual patient dosages for systemic administration range from 0.1 to 1000 mg/day, preferably, 1-500 mg/day, more preferably 10 - 200 mg/day, even more preferably 100 - 200 mg/day. Stated in terms of patient body surface areas, usual dosages range from 50-91 mg/m2/day.
[0185] The amount of the compound in a formulation can vary within the full range employed by those skilled in the art. Typically, the formulation will contain, on a weight percent (wt%) basis, from about 0.01-10 wt% of the drug based on the total formulation, with the balance being one or more suitable pharmaceutical excipients. Preferably, the compound is present at a level of about 0.1-3.0 wt%, more preferably, about 1.0 wt%.
[0186] Exemplary embodiments are summarized herein below.
[0187] In an exemplary embodiment, the invention provides a compound having a structure which is

[0188] In an exemplary embodiment, according to the above paragraph, two members selected from R2, R3, R4, R5 and R6 are each an independently selected halogen; and the remaining members of R2, R3, R4, R5 and R6 are H. [0189] In an exemplary embodiment, according to any of the above paragraphs, the compound is
 [0190] In an exemplary embodiment, according to any of the above paragraphs, the compound is selected from the group consisting of
[0190] In an exemplary embodiment, according to any of the above paragraphs, the compound is selected from the group consisting of

[0192] In an exemplary embodiment, the invention provides a pharmaceutical composition comprising the compound according to any of the above paragraphs, and a pharmaceutically acceptable excipient.
[0193] In an exemplary embodiment, according to any of the above paragraphs, the pharmaceutical formulation is a unit dosage form.
[0194] In an exemplary embodiment, according to any of the above paragraphs, the salt of the compound according to any of the above paragraphs is a pharmaceutically acceptable salt.
[0195] In an exemplary embodiment, the invention provides a method of killing and/or preventing the growth of a protozoa, comprising: contacting the protozoa with an effective amount of the compound according to any of the above paragraphs, thereby killing and/or preventing the growth of the protozoa. [0196] In an exemplary embodiment, according to any of the above paragraphs, the protozoa is Trypanosoma.
[0197] In an exemplary embodiment, according to any of the above paragraphs, the protozoa is Trypanosoma brucei.
[0198] In an exemplary embodiment, according to any of the above paragraphs, the Trypanosoma brucei is a member selected from Trypanosoma brucei brucei, Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense.
[0199] In an exemplary embodiment, the invention provides a method of treating and/or preventing a disease in an animal, comprising: administering to the animal a therapeutically effective amount of the compound according to any of the above paragraphs, thereby treating and/or preventing the disease. [0200] In an exemplary embodiment, according to any of the above paragraphs, the disease is sleeping sickness.
[0201] In an exemplary embodiment, according to any of the above paragraphs, the animal is a human.
[0202] The invention is further illustrated by the Examples that follow. The Examples are not intended to define or limit the scope of the invention.
EXAMPLES
[0203] The following Examples illustrate the synthesis of representative compounds used in the present invention and the following Reference Examples illustrate the synthesis of intermediates in their preparation. These examples are not intended, nor are they to be construed, as limiting the scope of the invention. It will be clear that the invention may be practiced otherwise than as particularly described herein. Numerous modifications and variations of the present invention are possible in view of the teachings herein and, therefore, are within the scope of the invention.
[0204] Abbreviations: DCM/CH2C12: dichloromethane; DIEA: diisopropylethylamine; DMAP: 4-dimethylaminopyridine; DMF: NJV- dimethylformamide; EtOAc: ethyl acetate; EDCl: l-ethyl-3-(3'- dimethylaminopropyl)carbodiimide; ELS: evaporative light scattering; HATU: 2- (lH-7-azabenzotriazol-l-yl)-l,l,3,3-tetramethyl uranium; HOBt: N- hydroxybenzotriazole; HCO2H: formic acid; MeOH: methanol; TEA: triethylamine; THF: tetrahydrofuran.
[0205] All temperatures are given in degrees Centigrade. Room temperature means 20 to 250C. Reagents were purchased from commercial sources or prepared following standard literature procedures. Unless otherwise noted, reactions were carried out under a positive pressure of nitrogen. Reaction vessels were sealed with either rubber septa or Teflon screw caps. Nitrogen was introduced through Tygon
tubing, fitted with a large bore syringe needle. Concentration under vacuum refers to the removal of solvent on a Bϋchi Rotary Evaporator.
[0206] Analytical HPLC was performed using a Supelco discovery C18 15 cm x 4.6 mm / 5 μm column coupled with an Agilent 1050 series VWD UV detector at 210 nm. Conditions: Solvent A: H2O/1% acetonitrile/0.1% HCO2H; Solvent B: methanol.
[0207] Proton magnetic resonance (1H NMR) spectra were recorded on a Varian INOVA NMR spectrometer [400 MHz (1H) or 500 MHz (1H)]. AU spectra were determined in the solvents indicated. Although chemical shifts are reported in ppm downfield of tetramethylsilane, they are referenced to the residual proton peak of the respective solvent peak for 1H NMR. Interproton coupling constants are reported in Hertz (Hz).
[0208] LCMS spectra were obtained using a ThermoFinnigan AQA MS ESI instrument utilizing a Phenomenex Aqua 5 micron C18 125 A 50 x 4.60 mm column. The spray setting for the MS probe was at 350 μL/min with a cone voltage at 25 mV and a probe temperature at 450 0C. The spectra were recorded using ELS and UV (254 nm) detection. Alternatively, LCMS spectra were obtained using an Agilent 1200SL HPLC equipped with a 6130 mass spectrometer operating with electrospray ionization. [0209] Silica gel chromatography was carried out on either a Teledyne ISCO CombiFlash Companion or Companion Rf Flash Chromatography System with a variable flow rate from 5-100 mL/min. The columns used were Teledyne ISCO RediSep Disposable Flash Columns (4, 12, 40, 80, or 120 g prepacked silica gel), which were run with a maximum capacity of 1 g crude sample per 1O g silica gel. Samples were preloaded on Celite in Analogix Sample Loading Cartridges with frits (I/in, I/out). The eluent was 0-100% EtOAc in heptane or 0-10% MeOH in CH2Cl2 as a linear gradient over the length of the run (14-20 minutes). Peaks were detected by variable wavelength UV absorption (200-360 nm). The resulting fractions were analyzed, combined as appropriate, and evaporated under reduced pressure to provide purified material.
[0210] HPLC purification was performed using a 50 mm Varian Dynamax HPLC 21.4 mm Microsorb Guard-8 C18 column, Dyonex Chromeleon operating system
coupled with a Varian Prostar 320 UV-vis detector (254 nm) and a Sedex55 ELS detector. Conditions: Solvent A: H2O/1% acetonitrile/0.1% HCO2H; Solvent B: MeOH. The appropriate solvent gradient for purification was determined based on the results of analytical HPLC experiments. The resulting fractions were analyzed, combined as appropriate, and evaporated under reduced pressure to provide purified material.
[0211] The following experimental sections illustrate procedures for the preparation of intermediates and methods for the preparation of products according to this invention. It should be evident to those skilled in the art that appropriate substitution of both the materials and methods disclosed herein will produce the examples illustrated below and those encompassed by the scope of the invention.
[0212] All solvents used were commercially available and were used without further purification. Reactions were typically run using anhydrous solvents under an inert atmosphere of N2. [0213] 1H, 13C, and 19F NMR spectra were recorded at 400 MHz for proton, 100 MHz for carbon- 13, and 376 MHz for fluorine- 19 on a Varian 300 MercuryPlus station with an Oxford AS400 Spectrometer equipped with a Varian 400 ATB PFG probe. All deuterated solvents typically contained 0.03% to 0.05% v/v tetramethylsilane, which was used as the reference signal (set at δ 0.00 for both 1H and 13C).
[0214] Compounds are named using ChemDraw 7.0 or their catalogue name if commercially available.
[0215] Mass spectra were recorded on a Waters MS consisting of an Alliance 2795 (LC) and Waters Micromass ZQ detector at 120 0C. The mass spectrometer was equipped with an electrospray ion source (ESI) operated in a positive or negative mode. The mass spectrometer was scanned between m/z = 100-1000 with a scan time of 0.3 s.
[0216] Elemental Analysis for C, H and N composition was performed using a Costech Instrument Elemental Combustion System ECS4010 with a helium flow of 100 mL/min (14 psi), oxygen 20 mL/min (10 psi), air 25 psi and purge of 50 mL/min. The reported analyses are an average of two runs.
[0217] HPLC analyses were performed on a Water 600 Controller system with a Waters 717 Plus Autosampler and a Waters 2996 Photodiode Array Detector. The column used was an ACE C18, 5 μm, 4.6 x 150 mm. A linear gradient was applied, starting at 95% A (A: 0.1% H3PO4 in water) and ending at 90% B (B: MeCN) over 6 min and then maintained at 90% B until the 10 min mark. The column was then re- equilibrated over 3 min to 95:5 with a total run time of 20 min. The column temperature was at rt with the flow rate of 1.0 mL/min. The Diode Array Detector was scanned from 200-400 nm. For high purity samples requiring baseline subtraction, a linear gradient was applied, starting at 99% A (A: 0.1% H3PO4 in water) and ending at 90% B (B: MeCN) over 15 min. The column was then re- equilibrated over 3 min to 99% A with a total run time of 23 min. The column temperature was at rt with the flow rate of 1.0 mL/min. The Diode Array Detector was scanned from 200-400 nm. A blank MeOH sample was run immediately prior to the sample of which purity was to be determined: this was then subtracted to obtain the baseline subtracted chromatogram.
[0218] Thin layer chromatography (TLC) was performed on Alugram® (Silica gel 60 F254) from Mancherey-Nagel and UV was typically used to visualize the spots. Additional visualization methods were also employed in some cases. In these cases the TLC plate was developed with iodine (generated by adding approximately 1 g of I2 to 10 g silica gel and thoroughly mixing), vanillin (generated by dissolving about 1 g vanillin in 100 mL 10% H2SO4), potassium permanganate (generated by dissolving 1.5 g KMnO4 and 10 g K2CO3 in 1.25 mL NaOH and 200 mL H2O), ninhydrin (available commercially from Aldrich), or Magic Stain (generated by thoroughly mixing 25 g (NH4)6Mo7O24«4H2O, 5 g (NH4)2Ce(IV)(NO3)6 in 450 mL H2O and 50 mL cone H2SO4) to visualize the compound. Flash chromatography was preformed using typically 40-63 μm (230-400 mesh) silica gel from Silicycle following analogous techniques to those disclosed by Still et al. Typical solvents used for flash chromatography or thin layer chromatography (TLC) were mixtures of CHCl3/MeOH, CH2Cl2MeOH, EtOAc/MeOH and hexane/EtOAc. Reverse phase flash chromatography were performed on a Biotage® using a Biotage C18 cartridges and a H2O/MeOH gradient (typically eluting from 5% MeOH/H2O to 90% MeOH/H2O).
[0219] Preparative chromatography was performed on either a Waters Prep LC 4000 System using a Waters 2487 Diode Array or on a Waters LC Module 1 plus.
The column used were either a Waters x Terra Prep C18, 5 μm, 30 x 100 mm, Phenomenex Luna C18, 5 μm, 21.6 x 250 mm, or a Phenomenex Gemini C18, 5 μm, 100 x 30 mm. Narrow gradients with MeCNZH2O (water containing either 0.1% TFA, 0.1% AcOH, 0.1% HCO2H or 0.1% NH4OAc) were used to elute the compound at a flow rate of approximately 20 mL/min and a total run time between 20-30 min.
[0220] Starting materials used were either available from commercial sources or prepared according to literature procedures and had experimental data in accordance with those reported. 6-aminobenzo[c][l,2]oxaborol-l(3H)-ol, for example, can be synthesized according to the methods described in U.S. Pat. Pubs. US20060234981 and US20070155699.
EXAMPLE 1
6-N-phenylformamide-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (\\99)

[0222] To a mixture of the carboxylic acid intermediate (175mg, 0.98mmol) and aniline (108μL, 1.18mmol, 1.2eq) in DCM (8mL) were added EDCI (377mg, 1.97mmol, 2.0eq) and DMAP (5mg, 0.04mmol, 0.04eq). The mixture was stirred at room temperature for 60 hours before evaporation. The residue was dissolved in ethyl acetate, and washed with IM HCl and brine. The residue after evaporation was purified by column chromatography over silica gel to give the title compound (128mg, 51.6% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.29 (s, IH), 9.34 (s, IH), 8.30 (d, J = 0.8 Hz, IH), 8.03 (dd, J = 8 & 1.6 Hz, IH), 7.77 (dd, J = 8 & 0.8 Hz, 2H), 7.54 (d, J = 8 Hz, IH), 7.34 (t, J = 8 Hz, IH), 7.08 (t, J = 8 Hz, IH) and 5.06 (s, 2H) ppm.
Alternate synthesis of l-hvdroxy-N-phenyl-l,3-dihvdrobenzo[cl[l,21oxaborole-6- carboxamide -78°C;

 cone HCI,
cone HCI,
85 reflux, O/N
rt, O/N


Synthesis of3-bromo-4-(hydroxymethyl)benzonitrile (B)

A B
[0223] A solution of 3-bromo-4-formylbenzonitrile A (1.0 g, 4.8 mmol) in CH3OH
(30 niL) was cooled to 00C. NaBH4 (180 mg, 4.8 mmol) was added portionwise. The mixture was allowed to warm to room temperature and stirr at room temperature for 1 h. The mixture was qunched with IN HCl and concentrated under vacuum. The residue was extracted with ethyl acetate (25 mL*3). The combined organic layers were washed with brine (20 mL), dried (Na2SO4) and concentrated under vacuum to give a white solid of the desired compound B (1.Og, 990Zo)-1H NMR (300 MHz, CDCl3): δ 7.82 (s, IH), 7.49-7.71 (m, 2H), 4.75 (s, 2H). LC-MS: 212 (M + I)+.
Synthesis of l-hydroxy-l,3-dihydrobenzo[c] [l,2]oxaborole-6-carbonitrile (C)

[0224] A solution of compound B (211 mg, 1.0 mmol) and triisopropyl borate (282 mg, 1.5 mmol) in anhydrous THF (10 mL) at N2 atmosphere was cooled to -78°C. n- BuLi (0.9 mL, 2.25 mmol) was added dropwise at -78°C. Then the mixture was allowed to warm to room temperature and stirr at room temperature for 1 h. The mixture was qunched with IN HCl and extracted with ethyl acetate (25 mL*3). The combined organic layers were washed with brine (20 mL), dried (Na2SO4) and concentrated under vacuum. The residue was purified by column chromatography (eluting with CH3OH and EtOAc =1 :1) on silica gel to give the desired compound C
as a yellow solid (80 mg, 50 %).1H NMR (300 MHz, CDCl3): δ 9.50 (s, IH), 8.08 (s, IH), 7.83-7.92 (m, IH), 7.61-7.66 (m, IH), 5.06 (s, 2H). LC-MS: 160 (M + I)+.
Synthesis of l-hydroxy-l,3-dihydrobenzo[c] [l,2]oxaborole-6-carboxylic acid (D)


DBU (62 mg, 0.40 mmol) in dry DMF (1 mL). The reaction mixture is stirred at room temperature overnight and diluted with EtOAc (100 mL) and is washed with aqueous NaOAc buffer (30 mL*2), 5% NaHCO3 (30 mL) and brine (50 mL). The organic layer is dried over Na2SO4 and is concentrated. The residue is purified by prep-HPLC to give the title compound.
6-(4-chloronhenylsuffinyl)benzorcIfl,2Ioxaborol-l(3H)-ol (HlOO)
[0227] The title compound was synthesized as shown in the scheme.

Synthesis of2-bromo-4-(4-chlorophenylthio)benzaldehyde (B) [0228] A mixture of 4-chlorophenylthiol (10g, 69.15mmol), 2-bromo-4- fluorobenzaldehyde (14.04g, 69.15mmol) and K2CO3 (19g) in dry DMF (15OmL) was heated to 1000C and stirred for 4h. The mixture was filtered and the filtrate was evaporated to remove the solvent. The residue was dissolved in Et2O and filtered to remove insoluble solid. Again, the filtrate was evaporated to give a brown liquid that became solid after being pumped on vacuum (22.17g, yield 97.9%). 1HNMR (300Hz, DMSO-de): δ 10.08 (s, IH), 7.72 (d, IH), 7.57 (s, 4H), 7.43 (s, IH) and 7.21 (d, IH) ppm.
Synthesis of (2-bromo-4-(4-chlorophenylthio)phenyl)methanol (C) [0229] 2-Bromo-4-(4-chlorophenylthio)benzaldehyde (22.1g, 67.45 mmol) in methanol (30OmL) was reduced with NaBH4 (3.1 g) at O0C for 20 min after the completion OfNaBH4 addition. A normal work-up and purification by silica gel column chromatography (hexane:EtOAc=5;l, v/v) provided the desired alcohol (19.8g, yield 89%) as a white solid. 1HNMR (300Hz, DMSO-d6): δ 7.54-7.50 (m, 2H), 7.43 (d, 2H), 7.37 (dd, IH), 7.32 (d, 2H), 5.49 (t, IH) and 4.48 (d, 2H) ppm. Synthesis of(3-bromo-4-((methoxymethoxy)methyl)phenyl)(4-chlorophenyl)sulfane
(D)
[0230] To a solution of (2-bromo-4-(4-chlorophenylthio)phenyl)methanol (20 g,
61.3 mmol) and N,N-diisopropyl-N-ethyl amine (184 mmol) in DCM (200 mL) was added dropwise methoxymethyl chloride (MOM-Cl, 14.7 g, 184 mmol) at O0C under N2. The mixture was stirred at r.t. for 2 days. The mixture was washed with water and
concentrated under vacuum to give a crude residue, which was purified by column to afford the desired product as a white solid (16 g, yield 70%).
Synthesis of 2-bromo-4-(4-chlorophenylsulfinyl)-l- ((methoxymethoxy)methyl)benzene (E) [0231] To a solution of (3-bromo-4-((methoxymethoxy)methyl)phenyl)(4- chlorophenyl)sulfane (16 g, 43 mmol) in DCM (100 mL) was added NaIO4 (10.1 g, 47.3 mmol) at O0C under N2. The mixture was stirred at r.t. for 1 hour. The mixture was washed with water and the organic layer was concentrated under vacuum to give the crude residue that was purified by column chromatography to afford the desired product as a white solid (10 g, yield 64%). 1HNMR (300 MHz, CDCl3) δ 7.81 (d, J = 1.5 Hz, 1 H), 7.60-7.55 (m, 4 H), 7.44 (d, J= 8.7 Hz, 2 H), 4.75 (s, 2 H), 4.63 (s, 2 H), 4.00 (s, 3 H) ppm.
Synthesis of 6-(4-chlorophenylsulfinyl)benzofcl fl,2]oxaborol-l(3H)-ol (F) from compound E [0232] To a solution of 2-bromo-4-(4-chlorophenylsulfinyl)-l-
((methoxymethoxy)methyl)- benzene (10 g, 37.5 mmol) in dioxane (100 mL) was added 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(l,3,2-dioxaborolane) (11.4 g, 45 mmol), Pd(dppf)2Cl2 (765 mg, 2.5 mol%), and KOAc (11 g, 112.5 mmol) at r.t. under N2. The mixture was stirred at 7O0C for 10 hours. The mixture was filtered and the filtrate was extracted with EtOAc. The combined organic layers were concentrated in vacuo to give the crude residue, which was purified by column chromatography to provide 2- (5-(4-chlorophenylsulfinyl)-2-((methoxymethoxy)methyl)phenyl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane as a white solid (10 g, yield 70 %). 1HNMR (300 MHz, CDCl3) δ 8.05 (d, J= 1.8 Hz, 1 H), 7.70-7.56 (m, 4 H), 7.31 (d, J= 8.4 Hz, 2 H), 4.84 (s, 2 H), 4.72 (s, 2 H), 3.38 (s, 3 H), 1.33 (s, 12 H) ppm.
[0233] This pinacol-boron intermediate (10 g, 22.3 mmol) and 6N HCl (200 mL) were stirred at rt overnight. The precipitate was filtered and the solid was dried to give the desired title compound as a white power (6 g, 89.5%). 1HNMR (300 MHz, DMSO-d6) δ 9.40 (s, 1 H), 8.08 (s, 1 H), 7.80 (dd, J= 7.8, 1.2 Hz, 1 H), 7.72 (d, 2H), 7.61 (d, 2H), 7.56 (d, IH) and 5.01 (s, 2 H) ppm. The data are consistent with the NMR of the compound generated with another method described above.
6-(3-chlorophenylsulfinyl)benzo[c][l,2]oxaborol-l(3H)-ol (HlOl)

28
[0234] Compound 28 (1.45mmol) was dissolved in 10% (v/v) H2O in MeOH
(3OmL). To this solution was added sodium periodate (3.2g, 14.5mmol, lO.Oeq). The reaction mixture was stirred for 12 hours then treated with IM HCl (10ml). After extraction with ethyl acetate, the organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After rotary evaporation the residue was purified by crystallization to give the title compound (275mg, 65% yield). 1H NMR (300 MHz, DMSO-de): δ 9.39 (s, IH), 8.11 (s, IH), 7.85 (dd, J = 8.1 & 1.8 Hz, IH), 7.78 (s, IH), 7.69-7.63 (m, IH), 7.59-7.56 (m, 3H) and 5.01 (s, 2H) ppm. Mp 152-153°C.
6-(2-chlorophenylsulfinyl)benzo[c][l,2]oxaborol-l(3H)-ol (H102)

[0235] This compound was produced by the same method as HlOl by substituting 4-chlorophenylthiol with 2-chlorophenylthiol. 1H NMR (300 MHz, DMSO-d6): δ 9.41 (s, IH), 8.09 (d, J = 1.2 Hz, IH), 8.01-7.97 (m, IH), 7.83 (dd, J = 8.1 & 1.8 Hz, IH), 7.71-7.65 (m, IH), 7.61-7.51 (m, 3H) and 5.02 (s, 2H) ppm. MS: found: 315 (M
+ Na)+, 347 (M + Na + MeOH)+. Mp 174-175°C.
6-(4'-chlorophenylsulfanyl)-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H103)

[0236] The synthesis of the title compound has been described previously in U.S. Pat. App. Nos. 11/357,687; 11/505,591 and 11/676,120.
6-(3'-chlorophenylsulfanyl)-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H104)

2-bromo-4-(3-chlorophenylsulfanyl)benzaldehvde (25)
[0237] Compound 24 (7.39mmol) was dissolved in DMF (25mL) and cooled to 00C with ice bath. To this solution under nitrogen were added in sequence potassium carbonate (2.04g, 14.78mmol, 2.0eq) and 3-chlorobenzenethiol (0.835ml, 7.39mmol, 1.Oeq). The reaction mixture was stirred for 0.5 hour then treated with cooled water (50ml). After extraction with ethyl acetate, the organic layer was washed with water and brine, and dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by column chromatography over silica gel to give compound 25 (1.92g, 80% yield). 1H NMR (400 MHz, CDCl3): δ 10.25 (s, IH), 7.78 (d, J = 8 Hz, IH) and 7.52- 7.12 (m, 6H) ppm.

25 26
2-bromo-4-(3 '-chlorophenylthiojbenzyl alcohol (26)
[0238] Compound 25 (6.08mmol) was dissolved in MeOH (25mL) and cooled to 00C with ice bath. To this solution was added NaBH4 (459.6mg, 12.16mmol, 2. Oeq). The reaction mixture was stirred for 0.5 hour then treated with saturated NaHCO3. After evaporation, the residue was extracted with ethyl acetate, washed with water and brine, and dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by column chromatography over silica gel to give compound 26 (1.8g, 90% yield). 1H NMR (400 MHz, DMSO-d6): δ 7.60-7.23 (m, 7H), 5.53 (t, J = 5.2 Hz, IH) and 4.51 (d, J = 5.2 Hz, 2H) ppm.

(3-bromo-4-((methoxymethoxy)methyl)phenyl) (3-chlorophenyl)sulfane (27)
[0239] Compound 26 (5.94mmol) was dissolved in anhydrous CH2Cl2 (25mL). To this solution under nitrogen were added in sequence N,N-diisopropylethylamine
(3.67ml, 20.79mmol, 3.5eq) and chloromethyl methyl ether (0.96mL, 13.07mmol, 2.2eq). The reaction mixture was stirred for 14 hours then treated with water (10ml). After extraction with dichloromethane, the organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After rotary evaporation, the residue was purified by column chromatography over silica gel to give compound 27 (1.79g, 80% yield). 1H NMR (400 MHz, CDCl3): δ 7.56 (d, J = 1.6 Hz, IH), 7.46 (d, J = 8.0 Hz, IH), 7.32-7.17 (m, 5H), 4.77 (s, 2H), 4.64 (s, 2H) and 3.43 (s, 3H) ppm.
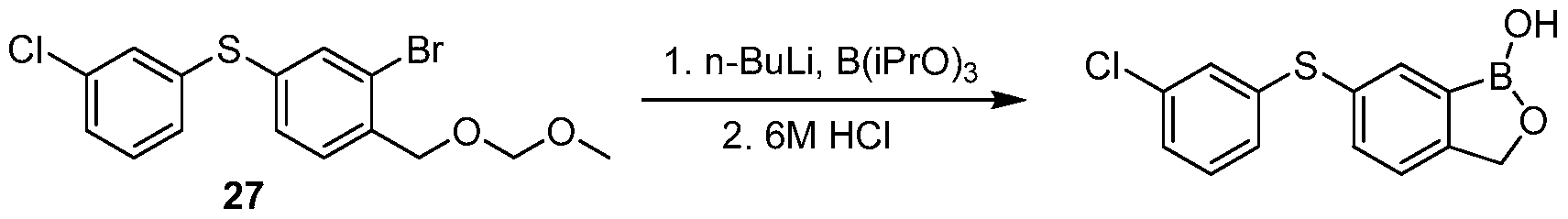
6- (3 '-chlorophenylsulfanyl)-l,3-dihvdro-l-hvdroxy-2, 1-benzoxaborole [0240] Compound 27 (1.Og, 2.67mmol) was dissolved in anhydrous THF (2OmL) and cooled to -800C. To this solution under nitrogen was added dropwise 1.6M n- BuLi (1.84mL, 2.94mmol, l.leq) over 20 minutes. After stirring for another 20 minutes at -800C, B(iPrO)3 (0.68mL, 2.94mmol, l.leq) was added dropwise over 8 minutes. The reaction mixture was allowed to warm to room temperature gradually and stirred overnight at room temperature. After 6M HCl (1OmL) was added and stirred for 2 hours, the mixture was evaporated and extracted with ethyl acetate (25mLx5) and dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by column chromatography over silica gel to give the title compound (380mg, 51.5% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.30 (s, IH), 7.81 (d, J = 1.6 Hz, IH), 7.58 (dd, J = 8 & 1.6 Hz, IH), 7.5 (d, J = 8 Hz, IH), 7.45-7.15 (m, 4H) and 5.02 (s, 2H) ppm.
6-(2'-chlorophenylsulfanyl)-l,3-dihydro-l-hydroxy-2, 1-benzoxaborole (H105)

[0241] This compound was produced by the same method as H104 by substituting 3-chlorobenzenethiol with 2-chlorobenzenethiol. 1H NMR (300 MHz, DMSO-d6): δ 9.30 (s, IH), 7.80 (d, J = 0.9 Hz, IH), 7.50-7.59 (m, 3H), 7.25 (m, 2H), 6.91 (m, IH) and 5.03 (s, 2H) ppm. MS: found: 275 (M - H)". Mp 116-118°C.
6-(4'-chlorophenylsulfanyl)-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H106)

[0242] The synthesis of the title compound has been described previously in U.S. Pat. App. Nos. 11/357,687; 11/505,591 and 11/676,120.
6-(3'-chloro)phenylsulfonyl-l,3-dihydro-l-hydwxy-2,l-benzoxabowle (H107)

MCPBA, DCM


27 30 l-(3 '-bromo-4 '-((methoxymethoxy)methyl)phenylsulfonyl)-3-chlorobenzene (30)
[0243] Compound 27 (4.78mmol) was dissolved in CH2Cl2 (45mL) and cooled to 00C with ice bath. To this solution was added meta-chloroperoxybenzoic acid (2.48g, 14.36mmol, 3.0eq). The reaction mixture was stirred for 18 hours then treated with 1.25M NaOH (20ml). After evaporation the residue was extracted with ethyl acetate, washed with water and brine, and dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by column chromatography over silica gel to give compound 30 (1.5g, 77% yield). 1H NMR (300 MHz, DMSO-d6): δ 8.24 (d, J = 1.8 Hz, IH), 8.11-7.97 (m, 6H), 4.72 (s, 2H), 4.60 (s, 2H) and 3.29 (s, 3H) ppm.

6- (3 '-chloro)phenylsulfonyl-l,3-dihvdro-l-hvdroxy-2, 1-benzoxaborole [0244] Compound 30 (l.Og, 2.46mmol) was dissolved in anhydrous THF (16mL) and cooled to -800C. To this solution under nitrogen was added dropwise 1.6M n- BuLi (1.69mL, 2.71mmol, l.leq) over 20 minutes. After stirring for 20 minutes at - 800C, B(iPrO)3 (0.62mL, 2.71mmol, l.leq) was added dropwise over 8 minutes. The reaction mixture was allowed to warm to room temperature gradually and stirred overnight at room temperature. After 6M HCl (1OmL) was added and stirred for 2 hours, the mixture was evaporated, extracted with ethyl acetate (25mLx5) and dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by
crystallization to give the title compound (123mg, 16.2% yield). 1H NMR (300 MHz, DMSO-de): δ 9.48 (s, IH), 8.35-7.63 (m, 7H), and 5.06 (s, 2H) ppm.
6-(2'-chlorophenylsulfanyl)-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H108)

[0245] This compound was produced by the same method as H104 by substituting 3-chlorobenzenethiol with 2-chlorobenzenethiol. 1H NMR (300 MHz, DMSO-d6): δ 9.50 (s, IH), 8.35-8.30 (m, 2H), 8.00 (dd, J = 8.1 & 1.8 Hz, IH), 7.78-7.62 (m, 4H) and 5.08 (s, 2H) ppm. MS: found: 331 (M + Na)+, 363 (M + Na + MeOH)+. Mp 130- 132°C.
6-(3',4'-dichlorophenyl)sulfenyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H109)

24 32
2-bromo-4-(3 'A '-dichlorophenylsulfanvDbenzaldehvde (32)
[0246] Compound 24 (14.78mmol) was dissolved in DMF (7OmL) and cooled to 00C with ice bath. To this solution under nitrogen were added in sequence potassium carbonate (4.08g, 29.55mmol, 2.0eq) and 3,4-dichlorobenzenethiol (1.88ml, 14.78mmol, l.Oeq). The reaction mixture was stirred for 0.5 hour then treated with cooled water (100ml). After extraction with ethyl acetate the organic layer was washed with water and brine, and dried over anhydrous Na2SO4. The residue was evaporated to give compound 32 (4.49g, 84% yield).

32 33
(2-bromo-4-(3 'A '-dichlorophenylsulfanvDbenzyl alcohol (33)
[0247] Compound 32 (12.46mmol) was dissolved in MeOH (5OmL) and cooled to 00C with ice bath. To this solution was added NaBH4 (942mg, 24.93mmol, 2.0eq). The reaction mixture was stirred for 0.5 hour then treated with saturated NaHCOs. After evaporation the residue was extracted with ethyl acetate, washed with water and
brine, and dried over anhydrous Na2SO4. The residue was evaporated to give compound 33 (4.3g, 95% yield).
MOMCI, DCM


33 34
(3-bromo-4-((methoxymethoxy)methyl)phenyl) (3, 4-dichlorophenyl)sulfane (34)
Compound 33 (11.84mmol) was dissolved in anhydrous CH2Cl2 (5OmL). To this solution under nitrogen were added in sequence N, N-Diisopropylethylamine (7.32ml, 41.45mmol, 3.5eq) and chloromethyl methyl ether (1.9ImL, 26.05mmol, 2.2eq). The reaction mixture was stirred for 14 hours then treated with water (15ml). After extraction with dichloromethane, the organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After rotary evaporation the residue was purified by column chromatography over silica gel to give compound 34 (4.04g, 83% yield). 1H NMR (400 MHz, DMSO-d6): δ 7.66-7.58 (m, 3H), 7.53 (d, J = 8.4 Hz, IH), 7.43 (dd, J = 8.4 & 2 Hz, IH), 7.28-7.25 (m, IH), 4.71 (s, 2H), 4.55 (s, 2H) and 3.31 (s, 3H) ppm

34
6-(3 ',4 '-dichlorophenyl)sulfenyl-l,3-dihvdro-l-hvdroxy-2,l-benzoxaborole [0248] Compound 34 (2.39g, 5.86mmol) was dissolved in anhydrous THF (25mL) and cooled to -800C. To this solution under nitrogen was added dropwise 1.6M n-BuLi (4.03mL, 6.45mmol, 1.1 eq) over 20 minutes. After stirred for another 20 minutes at -800C, B(iPrO)3 (1.48mL, 6.45mmol, l.leq) was added dropwise over 8 minutes. The reaction mixture was allowed to warm to room temperature gradually and stirred overnight at room temperature. After 6M HCl (2OmL) was added and stirred overnight, the mixture was evaporated and extracted with ethyl acetate (25mLx5) and dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by column chromatography over silica gel to give the title compound (607.5mg, 33.3% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.28 (s, IH), 7.81 (s, IH), 7.60-7.45 (m, 4H), 7.15 (dd, J = 8.4 & 2 Hz, IH) and 5.03 (s, 2H) ppm.
6-(2 \3 '-dichlorophenyl)sulfenyl-l,5-dihydro-l-hydroxy-2,l-benzoxaborole (Hl 10)

[0249] This compound can be produced by the same method as H109 by substituting 3,4-dichlorobenzenethiol with 2,3-dichlorobenzenethiol.
6-(2 ',4 '-dichlowphenyl)sulfenyl-l,3-dihydro-l-hydwxy-2,l-benzoxabowle (Hill)

[0250] This compound was produced by the same method as H109 by substituting 3,4-dichlorobenzenethiol with 2,4-dichlorobenzenethiol. 1H NMR (400 MHz, DMSO-de): δ 9.30 (s, IH), 7.79 (s, IH), 7.70 (d, J = 2.0 Hz, IH), 7.54 (m, 2H), 7.33 (dd, J = 8.4 & 2.0 Hz, IH), 6.86 (d, J = 8.4 Hz, IH) and 5.02 (s, 2H) ppm. MS: found: 309 (M +I)+. Mp 125-127°C.
6-(2 ',5'-dichlorophenyl)sulfenyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (Hl 12)

[0251] This compound can be producd by the same method as H109 by substituting 3,4-dichlorobenzenethiol with 2,5-dichlorobenzenethiol.
6-(2 ',6'-dichlowphenyl)sulfenyl-l,3-dihydro-l-hydwxy-2,l-benzoxabowle (Hl 13)

[0252] This compound can be produced by the same method as H109 by substituting 3,4-dichlorobenzenethiol with 2,6-dichlorobenzenethiol.
6-(3 \4 '-dichlorophenyl)sulfinyl-l,5-dihydro-l-hydroxy-2,l-benzoxaborole (Hl 14)

35
6-C3 ',4 '-dichlorophenyl)sulfinyl-l,3-dihvdro-l-hvdroxy-2,l-benzoxaborole (36) [0253] Compound 35 (0.64mmol) was dissolved in 10% (v/v) H2O in MeOH (15mL). To this solution was added sodium periodate (688mg, 3.21mmol, 5.0eq). The reaction mixture was stirred for 12 hours then evaporated and extracted with ethyl acetate. The organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After rotary evaporation the residue was purified by crystallization to give the title compound (112.4mg, 53.5% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.39 (s, IH), 8.11 (s, IH), 7.99 (d, J = 2 Hz, IH), 7.88-7.81 (m, 2H), 7.69-7.60 (m, 2H) and 5.02 (s, 2H) ppm.
6-(2 \3 '-dichlorophenyl)sulfinyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (Hl 15)

[0254] This compound can be produced by the same method as Hl 14 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,3- dichlorobenzenethiol.
6-(2',4'-dichlorophenyl)sulfinyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H116)

[0255] This compound was produced by the same method as Hl 14 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,4- dichlorobenzenethiol. 1H NMR (300 MHz, DMSO-d6): δ 9.41 (s, IH), 8.07 (d, J = 1.5 Hz, IH), 7.97 (m, IH), 7.83 (dd, J = 8.1 & 1.8 Hz, IH), 7.78 (m, 2H), 7.58 (d, J = 8.1 Hz, IH) and 5.03 (s, 2H) ppm. MS: found: 327 (M - I)". Mp 155-157°C.
6-(2',5'-dichlorophenyl)sulfinyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H117)

[0256] This compound can be produced by the same method as Hl 14 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,5- dichlorobenzenethiol.
6-(2 ',6'-dichlowphenyl)sulfinyl-l,3-dihydw-l-hydwxy-2,l-benzoxabowle (Hl 18)

[0257] This compound can be produced by the same method as Hl 14 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,6- dichlorobenzenethiol.
6-(3',4'-dichlorophenyl)sulfonyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H119)

35
6-C3 \4 '-dichlorophenvDsulfonyl- 1 ,3-dihvdro-l -hvdroxy-2 , 1-benzoxaborole [0258] Compound 35 (0.83mmol) was dissolved in 10% (v/v) H2O in MeOH (2OmL). To this solution was added sodium periodate (894mg, 4.17mmol, 6.5eq). The mixture was stirred for 2 days at 65°C then evaporated and extracted with ethyl acetate, the organic layer was washed with water and brine, and dried over anhydrous Na2SO4. After rotary evaporation, the residue was purified by crystallization to give the title compound (86mg, 30% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.49 (s, IH), 8.35 (s, IH), 8.20 (s, IH), 8.12 (dd, J = 8.4 & 2 Hz, IH), 7.90 (d, J = 1.2 Hz, 2H), 7.68 (d, J = 8.4 Hz, IH), and 5.06 (s, 2H) ppm.
6-(2 ',3 '-dichlorophenyl)sulfonyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H120)

[0259] This compound can be produced by the same method as Hl 19 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,3- dichlorobenzenethiol.
6-(2',4'-dichlowphenyl)sulfonyl-l,3-dihydw-l-hydwxy-2,l-benzoxabowle (H121)

[0260] This compound was produced by the same method as Hl 19 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,4- dichlorobenzenethiol. 1H NMR (300 MHz, DMSO-d6): δ 9.49 (s, IH), 8.31 (d, J = 8.7 Hz, 2H), 8.00 (dd, J = 8.4 & 2.1 Hz, IH), 7.87 (d, J = 1.8 Hz, 1H),7.8O (dd, J = 8.7 & 2.1 Hz, IH), 7.67 (d, J = 8.4 Hz, IH) and 5.09 (s, 2H) ppm. Mp 169-17FC.
6-(2 ',5'-dichlowphenyl)sulfonyl-l,3-dihydw-l-hydwxy-2,l-benzoxabowle (H122)

[0261] This compound can be produced by the same method as Hl 19 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,5- dichlorobenzenethiol.
6-(2',6'-dichlorophenyl)sulfonyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H123)

[0262] This compound can be produced by the same method as Hl 19 by substituting 3,4-dichlorobenzenethiol used in making compound 35 with 2,6- dichlorobenzenethiol.
6-(phenylthio)benzo / cj [l,2]oxaborol-l (3H)-ol (H124)

[0263] The synthesis of the title compound has been described previously in U.S. Pat. App. Nos. 11/357,687; 11/505,591 and 11/676,120.
6-(phenoxy)benzo[c][l,2]oxaborol-l(3H)-ol (H125)

[0264] The synthesis of the title compound has been described previously in U.S. Pat. App. Nos. 11/357,687; 11/505,591 and 11/676,120.
N-(l-hydroxy-l,3-dihydrobenzo[c][l,2]oxaborol-6-yl)-benzamide (H126)

[0265] The synthesis of the title compound has been described previously in U.S.
Pat. App. Nos. 11/357,687; 11/505,591 and 11/676,120. Alternatively, the title compound can be synthesized according to the methods described above by using benzoyl chloride in place of 2-(trifluoromethyl)benzoyl chloride. N-(l-hydroxy-l,3-dihydrobenzo[c][l,2]oxaborol-6-yl)-benzamine (H127)

[0266] The synthesis of the title compound has been described previously in U.S. Pat. App. Nos. 11/357,687; 11/505,591 and 11/676,120.
6-(phenylsulfonamido)benzo[c][l,2]oxaborol-l(3H)-ol (H128)

[0267] The synthesis of the title compound has been described previously in U.S. Pat. App. Nos. 11/357,687; 11/505,591 and 11/676,120.
6-(phenylsulfinyl)benzo[c][l,2]oxaborol-l(3H)-ol (H129)

[0268] The title compound was synthesized according to the methods above by using phenylthiol in place of 4-chlorophenylthiol.
6-(phenylsulfonyl)benzo[c][l,2]oxaborol-lβH)-ol (H130)

[0269] The title compound was synthesized according to the methods described above by using phenylthiol in place of 4-chlorophenylthiol.
[6-(l-phenyl-l-hydroxylmethyl)]-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole (H 131 )

(2-bromo-4-benzoyl)toluene (15)
[0270] To a mixture OfAlCl3 (1.46g, 1 l.Ommol, 1.1 eq) in benzene (16mL) was added a solution of compound 2 (2.33g, lO.Ommol) in benzene (8mL) dropwise at room temperature. This mixture was stirred for 2 hours at 500C. The mixture was washed with 3M HCl (2OmL) and saturated brine (2OmL), and dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by column chromatography over silica gel to give compound 15 (2.7g, 98% yield). 1H NMR (300 MHz, CDCl3): δ 7.98 (d, J = 1.8 Hz, IH), 7.78 (d, J = 6.9 Hz, 2H), 7.65 (dd, J = 7.8 & 1.5 Hz, IH), 7.59 (d, J = 7.5 Hz, IH), 7.49 (t, J = 7.5 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H) and 2.49 (s, 3H) ppm.

(2-bromomethyl-5-benzoyl)bromobenzene (16)
[0271] To a solution of compound 15 (2.7g, 9.8mmol) in CCl4 (5OmL) was added NBS (1.75g, 9.8mmol, l.Oeq) and Bz2O2 (0.12g, 0.5mmol, 0.05eq). This mixture was heated to reflux and stirred overnight. The residue after rotary evaporation was
purified by column chromatography over silica gel to give compound 16 (1.7 Ig, 49.3% yield). 1H NMR (300 MHz, CDCl3): δ 8.01 (d, J = 1.5 Hz, IH), 7.79 (d, J = 7.8 Hz, 2H), 7.71 (dd, J = 8.1& 1.5 Hz, IH) and 4.64 (s, 2H) ppm.

(2-bromo-4- benzovDbenzyl acetate (17)
[0272] To a solution of compound 16 (1.7 Ig, 4.83mmol) in DMF (3OmL) was added sodium acetate (1.98g, 24.15mmol, 5.0eq). This mixture was heated to 600C and stirred overnight. The mixture was poured into ice-water (5Og). The precipitate was filtered, washed with water and dried under vacuum to give compound 17 (1.63g, 100% yield).

(2-bromo-4-benzoyl)benzyl alcohol (18)
[0273] To a solution of compound 17 (1.63g, 4.9mmol) in methanol (25mL) was added 15% aq NaOH (5mL). This mixture was heated to reflux and stirred for 1 hour. After methanol was evaporated the mixture was extracted with ethyl acetate, dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by crystallization to give compound 18 (1.5Og, 100% yield).

(2-bromo-4-benzoyl)benzaldehvde (19)
[0274] To a solution of compound 18 (1.5Og, 5.15mmol) in DCM (3OmL) was added PCC (2.22g, 10.3mmol, 2.0eq) and celite (2.5g). This mixture was stirred overnight at room temperature. Then the mixture was filtered and the residue after rotary evaporation was purified by crystallization to give compound 19 (1.32g, 88.7% yield).
glycol,TsOH toluene


[2-(3-bromo-4-(l ,3-dioxolan-2-yl)phenyl)-2-phenyl] -1 ,3-dioxolane (20) [0275] To a solution of compound 19 (1.32g, 4.57mmol) in toluene (5OmL) was added ethylene glycol (2.83g, 45.70mmol, lO.Oeq) and p-toluenesulfonate monohydrate (69mg, 0.36mmol, 0.08eq). This mixture was heated to reflux and stirred for 96 hours. The mixture was washed with saturated NaHCO3 , water and brine, dried over anhydrous Na2SO4. The solvent was evaporated to give compound 20 (1.5 Ig, 98% yield).

(2-formyl-5-benzoyl)phenylboronic acid (21)
[0276] Compound 20 (0.5Og, 1.23mmol) was dissolved in anhydrous THF (1OmL) and cooled to -800C. To this solution under nitrogen was added dropwise 1.6M n-BuLi (0.88mL, 1.41mmol, 1.15eq) over 15 minutes. After stirred for another 20 minutes at -800C, B(IPrO)3 (0.33mL, 1.41mmol, 1.15eq) was added dropwise over 10 minutes. The reaction mixture was allowed to warm to room temperature gradually and stirred overnight at room temperature. After 6M HCl (6mL) was added and stirred for 2 hours, the mixture was evaporated and extracted with ethyl acetate (25mLx5) and dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by column chromatography over silica gel to give compound 21 (0.3 Ig, 88% yield).

[6-(l -phenyl- 1 -hvdroxylmethyl)] -1 ,3-dihvdro-l -hydroxy-2, 1 -benzoxaborole [0277] Compound 21 (0.75g, 2.95mmol) was dissolved in THF (8mL) and water (0.5mL). To this solution under stirring was added NaBH4 (217mg, 5.90mmol, 2.0eq) at room temperature. After stirred for 3 hours, 3M HCl (1OmL) was added to quench the reaction, and the mixture was evaporated and extracted with ethyl acetate
(30mLx3), dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by crystallization to give the title compound (0.35g, 50% yield). 1H NMR (300 MHz, DMSO-de): δ 9.1 (s, IH), 7.71 (s, IH), 7.46 (dd, J = 8.1 & 1.8 Hz, IH), 7.31 (m, 5H), 7.18 (d, J = 7.5 Hz, IH), 5.89 (d, J = 3.9 Hz, IH), 5.72 (d, J = 3.6 Hz, IH) and 4.91 (s, 2H) ppm.
6-Benzoyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H132)

[0278] To a solution of compound 22 (0.2g, 0.83mmol) in 15mL DCM was added PCC (0.45g, 2.08mmol, 2.5eq) and celite (0.45g). This mixture was stirred for 3 hours at room temperature before filtration. The residue after rotary evaporation was purified by crystallization to give the title compound (0.15g, 76% yield). 1H NMR (400 MHz, DMSO-de): δ 9.35 (s, IH), 8.12 (s, IH), 7.87 (dd, J = 8 & 1.6 Hz, IH), 7.69 (m, 3H), 7.57 (m, 3H) and 5.08 (s, 2H) ppm.
6-Benzyloxy-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H133)

24 38
2-bromo-4-fluorobenzyl alcohol (38)
[0279] Compound 24 (49mmol) was dissolved in MeOH (10OmL) and cooled to 00C with ice bath. To this solution was added NaBH4 (3.7g, 98mmol, 2.0eq). The reaction mixture was stirred for 1 hour then treated with saturated NaHCO3. After evaporation, the residue was extracted with ethyl acetate and the organic layer was washed with water and brine. The residue after rotary evaporation was purified by column chromatography over silica gel to give compound 38 (9.04g, 90% yield).

[3-fIuoro-6-(((tetrahvdro-2H-pyran-2-yl)oxy)methyl)]bromobenzene (39) [0280] Compound 38 (4.88mmol) and 3, 4-dihydro-2H-pyran (24.4mmol, 5.0eq) was dissolved in DCM (2OmL). To this solution were added in sequence pyridinium p-toluenesulfonate (0.5mmol, O.leq). The reaction mixture was stirred overnight at room temperature then treated with saturated NaHCO3. After extraction with ethyl acetate, the organic layer was washed with water and brine. The residue after rotary evaporation was purified by column chromatography over silica gel to give compound 39 (1.2g, 85.1% yield).

[3-benzyloxy-6-(((tetrahvdro-2H-pyran-2-yl)oxy)methyl)lbromobenzene(40) [0281] Compound 39 (lmmol) was dissolved in DMF (2OmL) and cooled to 00C with ice bath. To this solution under nitrogen were added in sequence sodium hydride (353mg, 8.8mmol, 2.5eq) and benzyl alcohol (0.76g, 7.06mmol, 2.0eq). The reaction mixture was stirred for 1 hour at 1000C then treated with cold water (30ml). After extraction with ethyl acetate, the organic layer was washed with water and brine. The residue after rotary evaporation was purified by column chromatography over silica gel to give compound 40 (0.86g, 64.8% yield).
[0282] 1H NMR (300 MHz, CDCl3): δ 7.39 (m, 6H), 7.20 (dd, J = 3.2 Hz, IH), 6.92 (dd, J = 11.2 & 3.2 Hz, IH), 5.04 (s, 2H), 4.79 (s, 0.5H), 4.75 (s, 1.5H), 4.52 (s, IH), 3.93 (m, IH), 3.55 (m, IH) and 1.70 (m, 6H) ppm.

[0283] Compound 40 (0.86g, 2.28mmol) was dissolved in anhydrous THF (20OmL) and cooled to -800C. To this solution under nitrogen was added dropwise 1.6M n-BuLi (1.64mL, 2.62mmol, 1.15eq) over 20 minutes. After the mixture was
stirred for another 20 minutes at -800C, B(iPrO)3 (0.6ImL, 2.62mmol, 1.15eq) was added dropwise over 8 minutes. The mixture was allowed to warm to room temperature gradually and stirred overnight at room temperature. After 6M HCl (6mL) was added and stirred for 2 hours, the mixture was evaporated and extracted with ethyl acetate (25mLx5) and dried over anhydrous Na2SO4. The residue after rotary evaporation was purified by crystallization to give the title compound (0.36g, 66% yield). 1H NMR (300 MHz, DMSO-d6): δ 9.12 (s, IH), 7.38 (m, 7H), 7.11 (dd, J = 7.2 & 3.2 Hz, IH), 5.10 (s, 2H) and 4.90 (s, 2H) ppm.
6-Hydroxyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H134)

[0284] H134 (13mmol) was dissolved in MeOH (30OmL). To this solution under nitrogen was added 10% Pd/C (200mg). The reaction mixture was vacuumed and backfilled hydrogen for 3 times, then stirred overnight at room temperature. After filtration and rotary evaporation, the residue was purified by recrystallization to give the title compound (1.98mg, 98% yield). 1H NMR (300 MHz, DMSO-d6): δ 9.29 (s, IH), 9.04 (s, IH), 7.18 (d, J = 8.4 Hz, 2H), 6.87 (dd, J = 8.1 & 2.4 Hz, IH) and 4.86 (s, 2H) ppm. Mp 133-135°C. l,3-dihydro-l-hydroxy-2,l-benzoxaborol-6-ylN-phenylcarbamate (H135)


[0286] 6-Hydroxyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (0.667mmol) was dissolved in toluene (1OmL) and cooled to 00C with ice bath. To this solution under nitrogen were added in sequence triethylamine (0.28ml, 2mmol, 3.0eq) and isocyanatocyclohexane (0.852mL, 6.67mmol, lO.Oeq). The reaction mixture was stirred for 2 days at room temperature then treated with IM HCl (10ml). The white solid was filtrated then purified by column chromatography over silica gel and recrystallization to give the title compound (24mg, 13% yield). 1H NMR (300 MHz, Acetone-de): δ 8.09 (s, IH), 7.42-7.37 (m, 2H), 7.19 (dd, J = 8.4 & 2.1 Hz, IH), 6.65 (m, IH), 5.00 (s, 2H), 3.46 (m, IH) and 1.98-1.15 (m, 10H) ppm. Mp 198-2000C.
6-(2'-Fluoro)benzyloxy-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (β\31)

[0287] 6-Hydroxyl-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (lmmol) was dissolved in DMF (1OmL) and cooled to 00C with ice bath. To this solution under nitrogen were added in sequence sodium hydride (160mg, 4mmol, 4.0eq) and 1- (chloromethyl)-2-fluorobenzene (0.485mL, 4mmol, 4.0eq). The reaction mixture was stirred for 2 hours then treated with IM HCl (10ml). After extraction with ethyl acetate, the organic layer was washed with water and brine. The residue after rotary evaporation was purified by column chromatography over silica gel to give the title compound (143.6mg, 55.7% yield). 1H NMR (300 MHz, DMSO-d6): δ 9.13 (s, IH), 7.58-7.12 (m, 7H), 5.16(s, 2H) and 4.92 (s, 2H) ppm. Mp 129-131°C.
6-(4'-Fluoro)benzyloxy-l,3-dihydro-l-hydroxy-2,l-benzoxaborole (H138)

6-(2-Nitrobenzyloxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole (H 139)

[0289] Compound 42 (200 mg, 1.33 mmol) was dissolved in DMF (9.0 mL) and cooled to 0 0C with ice bath. To this solution under nitrogen were added in sequence NaH (60% in mineral oil, 133 mg, 3.33 mmol) and l-(bromomethyl)-2-nitrobenzene (432 mg, 2.00 mmol). The reaction mixture was stirred for 1 h then treated with 1.0 M HCl (10.0 mL). After extraction with ethyl acetate, the organic phase was washed with water and saturated brine. After rotary evaporation, the residue was purified by column chromatography over silica gel to give the title compound (153 mg, 40% yield). 1H NMR (300 MHz, DMSO-d6): δ 9.17 (s, IH), 8.15 (d, J = 7.2 Hz, IH), 7.83- 7.76 (m, 2H), 7.66-7.59 (m, IH), 7.37-7.30 (m, 2H), 7.15 (dd, J = 8.1 & 2.4 Hz, IH), 5.48 (s, 2H) and 4.92 (s, 2H) ppm; Mp: 135-138 0C.
6-(Benzyl)benzo[c][l,2]oxaborol-l(3H)-ol (H140)

[0290] Compound 22 (125mg, 0.5mmol) and NaBH4 (190mg, 5mmol) was slowly added in portions with vigorous stirring to trifluoroacetic acid (2OmL) at O0C under nitrogen. After 15 min, aqueous NaHCO3 was slowly added and the solution was extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, and evaporated under vacuum to a solid residue. The crude product was recrystallized from hexane and ethyl acetae to give 105mg of the product (90%).
6-Formyl-l-hydroxy-l, 3-dihydro-2, 1-benzoxaborole (H 141 )

[0291] To a solution of compound 121 (24.Og, 62.3mmol) in THF (25OmL) under nitrogen at -78°C was added dropwise n-BuLi solution (1.6M in hexane, 42.9mL, 68.6mmol, 1. leq) over 30 minutes. After the mixture was stirred at -78°C for another 20 minutes, triisopropyl borate (15.7mL, 68.6mmol, l.leq) was added over 10 minutes. The mixture was allowed to warm to room temperature gradually and stirred overnight before quenched with 6M HCl (10OmL). The mixture was stirred for another 6 hours at room temperature. All of solvents were evaporated under reduced pressure to give crude product 122 which was used to oxidative reaction without purification.
[0292] To a mixture of crude product 122 in CH2Cl2 (25OmL) were added PCC (26.8g, 124.6mmol, 2.0eq) and Celite (40.2g). The mixture was stirred overnight at room temperature. The reaction mixture was filtered through celite and silica gel pad and the filtrate was washed with IM HCl and IM NaOH. The aqueous phase was acidified by concentrated HCl to pH 2 and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous Na2SO4, and evaporated to give crude product (4.7Og) which was purified by column chromatography and recrystallization to give the title compound (1.6Og, 15.8% yield). 1H NMR (300 MHz, DMSO-de): δ 10.06 (s, IH), 9.44 (s, IH), 8.28 (s, IH), 8.01 (m, IH), 7.63 (m, IH) and 5.09 (s, 2H) ppm. Mp 133-135°C.
7-Formyl-l-hydroxy-l, 3-dihydro-2, 1-benzoxaborole (H 142)
[0293] H142 can be synthesized using the following procedure:
PCC, DCM

2-Chloro-N-(l-hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)- benzenesulfonamide (Hl 56)

[0294] A 40 mL scintillation vial was charged with 5-amino-2- hydroxymethylphenylboronic acid hydrochloride (100 mg, 0.54 mmol, 1 eq) in dry DCM (10 mL). Pyridine (55 μl, 0.67 mmol, 1.2 eq) was then added followed by 2- chlorobenzenesulfonylchloride (92 μl, 0.67 mmol, 1.2 eq). The mixture was allowed to stir at room temperature overnight. Aqueous hydrochloric acid (1 M, 3 mL) was added and the resulting mixture was extracted twice with DCM (5 mL). The combined organic phases were dried over sodium sulfate, and the material was concentrated under reduced pressure. The residue was purified by silica gel chromatography to furnish the title compound as a clear oil. LCMS (m/z) 324 [M+H]; 1H NMR (400 MHz, DMSO-d6) δ ppm 4.86 (s, 2 H) 7.17 - 7.29 (m, 2 H) 7.45 - 7.53 (m, 2 H) 7.54 - 7.67 (m, 2 H) 7.92 - 8.04 (m, 1 H) 9.21 (s, 1 H) 10.55 (s, 1 H).
3-Chloro-N-(l-hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)- benzenesulfonamide (Hl 57)

[0295] H157 was prepared using a procedure similar to that of H156 with 3- chlorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (m/z) 324 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 4.89 (s, 2 H) 7.14 - 7.21
(m, 1 H) 7.26 - 7.33 (m, 1 H) 7.49 (dd, J= 1.8, 0.4 Hz, 1 H) 7.53 - 7.62 (m, 1 H) 7.62 7.74 (m, 2 H) 7.73 - 7.78 (m, 1 H) 9.23 (s, 1 H) 10.36 (s, 1 H).
4-Chloro-N-(l-hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)- benzenesulfonamide (H158)

[0296] H158 was prepared using a procedure similar to that of H156 with 4- chlorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (mix) 324 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 4.88 (s, 2 H) 7.16 (dd, J=8.1, 2.0 Hz, 1 H) 7.27 (d, J=8.2 Hz, 1 H) 7.47 (dd, J=I.9, 0.5 Hz, 1 H) 7.58 - 7.65 (m, 2 H) 7.67 - 7.76 (m, 2 H) 9.21 (s, 1 H) 10.31 (s, 1 H).
2-Fluoro-N-(l-hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)- benzenesulfonamide (H159)

[0297] H159 was prepared using a procedure similar to that of H156 with 2- fluorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (m/z) 308 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 4.87 (s, 2 H) 7.17 - 7.24 (m, 1 H) 7.23 - 7.30 (m, 1 H) 7.33 (td, J=7.6, 1.0 Hz, 1 H) 7.40 (dd, J=9.9, 9.1 Hz, 1 H) 7.49 (d, J=I.8 Hz, 1 H) 7.66 (dddd, J=7.9, 5.3, 5.1, 2.5 Hz, 1 H) 7.79 (td, J=7.6, 1.8 Hz, 1 H) 9.21 (s, 1 H) 10.50 (s, 1 H).
3-Fluow-N-(l-hydwxy-l,3-dihydro-benzofcJfl,2Joxaborol-6-yl)- benzenesulfonamide (H160)

[0298] H160 was prepared using a procedure similar to that of H156 with 3- fluorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (m/z) 308 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 4.89 (s, 2 H) 7.18 (dd, J=8.2, 2.1 Hz, 1 H) 7.29 (d, J=8.2 Hz, 1 H) 7.40 - 7.69 (m, 5 H) 9.22 (s, 1 H) 10.35 (s, I H).
4-Fluoro-N-(l-hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)- benzenesulfonamide (H161)

[0299] H161 was prepared using a procedure similar to that of H156 with 4- fluorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (mix) 308 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 4.88 (s, 2 H) 7.16 (dd, J=8.1, 2.1 Hz, 1 H) 7.27 (d, J=8.2 Hz, 1 H) 7.38 (t, J=8.9 Hz, 2 H) 7.48 (d, J=I.8 Hz, 1 H) 7.78 (dd, J=9.0, 5.1 Hz, 2 H) 9.22 (s, 1 H) 10.25 (s, 1 H).
3,4-Dichloro-N-(l-hydroxy-l,3-dihydw-benzofcJfl,2Joxabowl-6-yl)- benzenesulfonamide (H162)
[0300] H162 was prepared using a procedure similar to that of H156 with 3,4- dichlorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (m/z) 358 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 4.90 (s, 2 H) 7.17 (dd, J=8.2, 2.1 Hz, 1 H) 7.31 (d, J=8.2 Hz, 1 H) 7.48 (d, J=2.1 Hz, 1 H) 7.63 (dd, J=8.4, 2.1 Hz, 1 H) 7.83 (d, J=8.4 Hz, 1 H) 7.92 (d, J=2.1 Hz, 1 H) 9.24 (s, 1 H) 10.41 (s, 1 H).
3,5-Dichloro-N-(l-hydroxy-l,3-dihydro-benzofcJfl,2Joxaborol-6-yl)- benzenesulfonamide (H163)

[0301] H163 was prepared using a procedure similar to that of H156 with 3,5- dichlorobenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (m/z) 379 [M+Na]; 1H NMR (400 MHz, DMSO- d6) δ ppm 4.91 (s, 2 H) 7.19 (dd, J=8.2, 2.1 Hz, 1 H) 7.33 (d, J=8.2 Hz, 1 H) 7.49 (d, J=2.1 Hz, 1 H) 7.69 (d, J=2.0 Hz, 2 H) 7.95 (t, J=I.9 Hz, 1 H) 9.25 (s, 1 H) 10.46 (s, 1 H).
2,4-Dichloro-N-(l-hydroxy-l,3-dihydro-benzo[c] [l,2]oxaborol-6-yl)- benzenesulfonamide (H164)

[0302] General Procedure 1 : 6-amino-3H-benzo[c][l,2]oxaborol-l-ol (300 mg, 2.01 mmol), acetonitrile (10 mL), 2,4-dichloro-benzenesulfonyl chloride (594 mg, 2.42 mmol), ,N-methyl morpholine (815 mg, 8.06 mmol). Column purification using 5% MeOH in DCM followed by preparative ΗPLC purification with 0.1% formic acid/water and acetonitrile provided 132 mg (0.36 mmol, 18%) of the title compound as a white solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 9.21 (s, 1 H), 7.95 (d, J=8.6 Hz, 1 H), 7.83 (d, J=2.2 Hz, 1 H), 7.56 (dd, J=8.6, 2.2 Hz, 1 H), 7.44 (d, J=I.9 Hz, 1 H), 7.24 (d, J=8.3 Hz, 1 H), 7.17 (dd, J=8.3, 1.9 Hz, 1 H), 4.85 (s, 2 H); MS (ESI) m/z = 357 (M-I, negative); HPLC purity: 97.86 % (MaxPlot 200 - 400 nm), 98.28% (220 nm); Anal. Calcd for C13H10BCI2NO4S: C 43.61%; H 2.82%; N 3.91%. Found: C 43.59%; H 2.92%; N 4.03%.
3,4-Difluoro-N-(l-hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)- benzenesulfonamide (H165)

[0303] General Procedure 1 : 6-amino-3H-benzo[c][l,2]oxaborol-l-ol (250 mg, 1.7 mmol), 3,4-difluorobenzenesulfonyl chloride (0.26 mL, 2.0 mmol), Si-pyridine (4.7 g, 6.8 mmol), and MeCN (20 mL) at rt O/N. The mixture was filtered through a pad of Celite® and the filtrate was concentrated in vacuo at 40 0C. The residue was recrystallization from MeCNZH2O to give the title compound as a white solid; yield: 233 mg (42%). mp 153-155 0C; 1H NMR (400 MHz, DMSO-J6) δ (ppm): 10.34 (s, IH), 9.23 (s, IH), 7.78-7.74 (m, IH), 7.65-7.56 (m, 2H), 7.45 (s, IH), 7.29-7.27 (m, IH), 7.17-7.15 (m, IH), 4.87 (s, 2H); 19F NMR (376 MHz, DMSO-J6) δ (ppm): - 131.3 (s), -135.4 (s); MS (ESI) m/z = 324 (M-I, negative); HPLC purity: 92.90% (MaxPlot 200-400 nm), 94.20% (220 nm).
N-(l-Hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)-3-methyl- benzenesulfonamide (Hl 66)

[0304] H166 was prepared using a procedure similar to that of H156 with 3- methylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (mix) 304 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 2.33 (s, 3 H) 4.87 (s, 2 H) 7.10 - 7.20 (m, 1 H) 7.22 - 7.29 (m, 1 H) 7.40 (d, J=5.3 Hz, 2 H) 7.48 (d, J=2.0 Hz, 1 H) 7.52 (td, J=4.5, 1.4 Hz, 1 H) 7.58 (s, 1 H) 9.21 (s, 1 H) 10.21 (s, 1 H).
N-(l-Hydroxy-l,3-dihydw-benzofcJfl,2Joxabowl-6-yl)-4-methyl- benzenesulfonamide (Hl 67)

[0305] H167 was prepared using a procedure similar to that of H156 with 4- methylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (mix) 304 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 2.33 (s, 3 H) 4.88 (s, 2 H) 7.10 - 7.21 (m, 1 H) 7.22 - 7.28 (m, 1 H) 7.33 (d, J=7.8 Hz, 2 H) 7.49 (d, J=2.0 Hz, 1 H) 7.62 (d, J=8.4 Hz, 2 H) 9.21 (s, 1 H) 10.17 (s, 1 H).
N-(l-Hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)-4-isopropyl- benzenesulfonamide (H168)

[0306] H168 was prepared using a procedure similar to that of H156 with 4- isopropylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (mix) 332 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 1.17 (d, J=7.0 Hz, 6 H) 2.92 (q, J=13.7, 6.9 Hz, 1 H) 4.88 (s, 2 H) 7.15 - 7.21 (m, 1 H) 7.23 - 7.29 (m, 1 H) 7.41 (d, J=8.4 Hz, 2 H) 7.50 (d, J=I.8 Hz, 1 H) 7.64 - 7.69 (m, 2 H) 9.21 (s, 1 H) 10.22 (s, 1 H).
4-tert-Butyl-N-(l-hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)- benzenesulfonamide (H169)

[0307] H169 was prepared using a procedure similar to that of H156 with 4-t- butylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (m/z) 346 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 1.26 (s, 9 H) 4.88 (s, 2 H) 7.17 - 7.23 (m, 1 H) 7.24 - 7.29 (m, 1 H) 7.52 (d, J=2.0 Hz, 1 H) 7.56 (d, J=8.6 Hz, 2 H) 7.69 (d, J=8.6 Hz, 2 H) 9.21 (s, 1 H) 10.25 (s, 1 H).
N-(l-Hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl)-3-trifluoromethyl- benzenesulfonamide (Hl 70)

[0308] H170 was prepared using a procedure similar to that of H156 with 3- trifluoromethylbenzenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (m/z) 358 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 4.88 (s, 2 H) 7.15 (dd, J=8.1, 2.0 Hz, 1 H) 7.28 (d, J=8.4 Hz, 1 H) 7.47 (d, J=I.6 Hz, 1 H) 7.79 (t, J=8.0 Hz, 1 H) 7.96 (s, 1 H) 7.99 (br. s., 2 H) 9.21 (s, 1 H) 10.41 (br. s., 1 H).
N-(l-Hydroxy-l,3-dihydro-benzofcJfl,2Joxabowl-6-yl)-4-trifluowmethyl- benzenesulfonamide (H171)

[0309] A mixture of 6-amino-3H-benzo[c][l,2]oxaborol-l-ol (250 mg, 1.7 mmol), 4-(trifluoromethyl)benzenesulfonyl chloride (488 mg, 2.0 mmol), NMM (0.74 mL, 6.8 mmol) and MeCN (5 mL) was stirred overnight at room temperature, then was concentrated in vacuo, water (5 mL) andl M HCl (5 drops) were added. EtOAc (15 mL) was added and the mixture was stirred until a clear biphasic solution was observed. The aqueous layer was loaded onto an Isolute ΗM-N column and left to stand for 10 min. The organic layer was then eluted through the column. The column was then further washed with EtOAc (20 mL). The organic fractions were
concentrated in vacuo and the residue was dissolved in MeOH and loaded onto a pre- column (silica, 12 g). Purification by flash chromatography (20-100% EtOAc/hexane) gave a white solid; yield: 187 mg (31%). Recrystallization from MeCN/H2O gave a white solid that was further purified by prep HPLC (0.1% TFA (aq)/MeCN. The major fraction was isolated, concentrated in vacuo at 40 0C, and then lyophilized to give H171 as a white solid (17 mg). 1H NMR (400 MHz, DMSO- d6) δ (ppm): 10.48 (bs, IH), 9.23 (s, IH), 7.96-7.91 (m, 4H), 7.49-7.48 (m, IH), 7.28- 7.27 (m, IH), 7.17-7.15 (m, IH), 4.88 (s, 2H); 19F NMR (376 MHz, DMSO-J6) δ (ppm): -62.1 (s); MS (ESI) m/z = 356 (M-I, negative); HPLC purity: 99.78% (MaxPlot 200-400 nm), 99.74% (220 nm).
N-(l-hydroxy-l,3-dihydrobenzo[c][l,2]oxaborol-6-yl)-6-methylpyridine-2- sulfonamide (H172)

[0310] H172 was prepared using a procedure similar to H171 with 6- methylpyridine-2-sulfonyl chloride replacing 4-(trifluoromethyl)benzenesulfonyl chloride. 1H NMR (300 MHz, DMSO-J6) δ (ppm): 10.46 (s, IH), 9.19 (s, IH), 7.87- 7.84 (m, IH), 7.70-7.68 (d, IH), 7.49-7.44 (m, 2H), 7.22-7.21 (m, 2H), 4.84 (s, 2H), 2.50 (s, 3H); MS (ESI): m/z = 305.0 (M+l, positive); HPLC purity: 100% (254 nm), 90.3% (220 nm). N-(l-hydwxy-l,3-dihydrobenzofcJfl,2Joxaborol-6-yl)-5-methylpyridine-2- sulfonamide (H173)

[0311] H173 was prepared using a procedure similar to H171 with 5- methylpyridine-2-sulfonyl chloride replacing 4-(trifluoromethyl)benzenesulfonyl chloride. 1H NMR (300 MHz, DMSO-J6) δ (ppm): 8.99 (s, IH), 8.40 (s, IH), 7.68- 7.73 (m, 3H), 7.30 (s, IH), 7.05 (s, IH), 4.78 (s, 2H), 2.28 (s, 3H); MS (ESI): m/z = 305 (M+l, positive); HPLC purity: 93.73% (254 nm), 93.84% (220 nm).
N-(l-hydroxy-l,3-dihydrobenzo[c][l,2]oxaborol-6-yl)-4-methylpyridine-2- sulfonamide (H174)

[0312] H174 was prepared using a procedure similar to H171 with 4- methylpyridine-2-sulfonyl chloride replacing 4-(trifluoromethyl)benzenesulfonyl chloride. 1H NMR (300 MHz, DMSO-J6) δ (ppm): 10.49 (s, IH), 9.21 (s, IH), 8.51- 8.53 (d, IH), 7.78 (s, IH), 7.48 (s, IH), 7.41-7.43 (d, IH), 7.21 (s, 2H), 4.84 (s, 2H), 1.96 (s, IH); MS (ESI): m/z = 305 (M+ 1, positive); HPLC purity: 99.22% (254 nm), 99.36% (220 nm). l-Ethyl-lH-pyrazole-4-sulfonic acid (l-hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol- 6-yl)-amide (H175)

[0313] H175 was prepared using a procedure similar to H171 with 1-ethyl-lH- pyrazole-4-sulfonyl chloride replacing 4-(trifluoromethyl)benzenesulfonyl chloride. Purification: recrystallization from H2O. H175 was isolated as light yellow needles: yield 0.401 g (56%). mp 162-164 0C; 1H NMR (400 MHz, DMSO-J6) δ (ppm): 10.06 (s, IH), 9.25 (s, IH), 8.19 (s, IH), 7.67 (d, J= 0.8 Hz, IH), 7.51 (d, J= 2.0 Hz, IH), 7.30 (d, J= 8.2 Hz, IH), 7.20 (dd, J= 8.2, 2.0 Hz, IH), 4.91 (s, 2H), 4.11 (q, J= 7.2 Hz, 2H), 1.30 (t, J= 7.2 Hz, 3H); MS (ESI): m/z = 306 (M-I, negative); HPLC purity: 98.10% (MaxPlot 200-400 nm), 98.02% (220 nm); Anal. Calcd for Ci2Hi4BN3O4S: C 46.93%; H 4.59%; N 13.68%. Found: C 46.99%; H 4.66%; N 13.61%.
N-(l-hydwxy-l,3-dihydrobenzofcJfl,2Joxabowl-6-yl)-l-methyl-lH-imidazole-4- sulfonamide (H176)

Naphthalene-2-sulfonic acid (l-hydroxy-l,3-dihydro-benzo[c] [1,2] oxaborol-6-yl)- amide (H177)

[0315] H177 was prepared using a procedure similar to that of H156 with 2- naphthalenesulfonylchloride replacing 2-chlorobenzenesulfonylchloride. LCMS (m/z) 340 [M+H]; 1H NMR (400 MHz, DMSO- d6) δ ppm 4.83 (s, 2 H) 7.11 - 7.27 (m, 2 H) 7.51 (s, 1 H) 7.66 (ddd, J=I 1.9, 8.1, 1.2 Hz, 2 H) 7.76 (dd, J=8.6, 2.0 Hz, 1 H) 7.99 (d, J=8.0 Hz, 1 H) 8.09 (t, J=9.2 Hz, 2 H) 8.41 (d, J=I.6 Hz, 1 H) 9.17 (s, 1 H) 10.34 (s, 1 H).
EXAMPLE 2
Trypanosoma brucei brucei High-Throughput Screening Assay Procedure [0316] Experiments were conducted with the bloodstream- form trypanosome T. brucei brucei S427 strain and the T. brucei brucei STIB 795 strain. Parasites were cultured in T-25 vented cap flasks and kept in humidified incubators at 37°C and 5% CO2. The parasite culture media was complete HMI- 9 medium (c.f. Hirumi, Journal of Parasitology, 40:75, 985-989 (1989)) containing 10% FBS, 10% Serum Plus medium and penicillin/streptomycin. To ensure log growth phase, trypanosomes were sub-cultured at appropriate dilutions every 2-3 days.
In Vitro Drug Sensitivity Assays
[0317] Log phase cultures were diluted 1 : 10 in HMI-9 and 10 uL was counted using hemocytometer to determine parasite concentration. Parasites were diluted to 2 x 105 /mL in HMI-9 to generate a 2-fold working concentration for assay. Compounds to be tested were serially diluted in DMSO and 0.5 uL added to 49.5 uL HMI-9 in triplicate 96-well plates using a Biomek NX liquid handler. Parasites from the diluted stock were added to each well (50 uL) using a Multidrop 384 dispenser to give a final concentration of 1.0xl05/ml parasites in 0.4% for DMSO. Trypanosomes were incubated with compounds for 72 hrs at 37°C with 5% CO2. Resazurin (20 uL of 12.5
mg/ml stock) from Sigma- Aldrich was added to each well and plates were incubated for an additional 2-4 hrs. Assay plates were read using an En Vision plate reader at an excitation wavelength of 544 nm and emission of 590 nm. Triplicate data points were averaged to generate sigmoidal dose response curve and determine IC50 values using XLfit curve fitting software from IDBS (Guildford, UK).
[0318] Results from the S427 strain and the STIB 795 strain are provided in FIG.l.
EXAMPLE 3
Trypanosoma cruzi C2C4 Screening Assay Procedure
[0319] Rat skeletal myoblasts (L-6 cells) were seeded in 96-well microtitre plates at 2 x 103 cells/well in 100 μL RPMI 1640 medium with 10% FBS and 2 mM L- glutamine. After 24 h the medium was removed and replaced by 100 μl per well containing 5 x 103 trypomastigote forms of T. cruzi Tulahuen strain C2C4 containing the β-galactosidase (Lac Z) gene (Buckner, et al., Antimicrobial Agents and Chemotherapy, 40: 2592-2597 (1996)). After 48 h the medium was removed from the wells and replaced by 100 μl fresh medium with or without a serial drug dilution of seven 3-fold dilution steps covering a range from 90 to 0.123 μg/ml. After 96 h of incubation the plates were inspected under an inverted microscope to assure growth of the controls and sterility. The substrate (50 μl) chlorophenol red-β-D- galactopyranoside (CPRG, Roche Diagnostics Ltd) in 0.25% Nonidet P-40/PBS was added to all wells and a color reaction developed within 2-6 h. which was read photometrically at 540 nm. Data were transferred into the graphic programme Softmax Pro (Molecular Devices), which calculated IC50 values.
[0320] Results from this assay are provided in FIG.l.
EXAMPLE 4 Trypanosoma cruzi CL2 Screening Assay Procedure
[0321] Parasite and cell cultures. Trypanosoma cruzi, Tulahuen CL2, β galactosidase strain (nifurtimox-sensitive) was used (Buckner et al., Antimicrob Agents Chemother. 40: 2592-2597 (1996)). The strain was maintained in MRC-5SV2 (human lung fibroblast) cells. A SV-40 transformed cell line was available to obtain unlimited subcultivation characteristics in MEM medium, supplemented with 200
niM. L-glutamine, 16.5 niM NaHCOs, and 5% inactivated fetal calf serum. All cultures and assays were conducted at 37°C with 5% CO2.
[0322] Compound solutions/dilutions. Compound stock solutions were prepared in 100% DMSO at 20 mM or mg/ml for natural products, drug mixtures and if the molecular weight was not known. The compounds were serially pre-diluted (2-fold or 4-fold) in DMSO followed by a further (intermediate) dilution in demineralized water to assure a final in-test DMSO concentration of <1%.
[0323] Drug sensitivity assays. Assays were performed in sterile 96-well micro titer plates, each well containing 10 μl of the watery compound dilutions together with 190 μl of MRC-5 cell/parasite inoculum (4 x 103 cells/well + 4 x 104 parasites/well). Parasite growth was compared to untreated-infected controls (100% growth) and noninfected controls (0% growth) after 7 days incubation at 37°C and 5% CO2. Parasite burdens were assessed after adding the substrate CPRG (chlorophenolred β-D-galactopyranoside): 50 μl/well of a stock solution containing 15.2 mg CPRG + 250 μl Nonidet in 100 ml PBS. The change in color was measured spectrophotometrically at 540 nm after 4 hours incubation at 37 0C. The results were expressed as % reduction in parasite burdens compared to control wells and an IC50 (50% inhibitory concentration) was calculated.
[0324] Primary screen. Trypanosoma cruzi β galactosidase strain was used. Compounds were tested at 5 concentrations (64 - 16 - 4 -1 and 0.25 μM or ug/ml).
Nifurtimox or benznidazole were included as the reference drugs. The test compound was classified as inactive when the IC50 was higher than 30 μM. When the IC50 was between 30 and 5 μM, the compound was regarded as being moderate active. When the IC50 was lower than 5 μM, the compound was classified as highly active on the condition that it also demonstrated selective action (absence of cytotoxicity). A final recommendation for activity was given after confirmatory evaluation in a secondary screening.
[0325] Secondary screen. Trypanosoma cruzi β galactosidase strain was used and IC50-values were determined using an extended dose range (2-fold compound dilutions). Nifurtimox or benznidazole was included as reference drugs. Advanced selectivity evaluation was performed against a panel of unrelated organisms (bacteria, yeasts, fungi and other protozoan parasites).
[0326] Results from this assay are provided in FIG.l.
EXAMPLE 5
Trypansoma brucei satnbiense Screening Assay Procedure
[0327] The following T. b. gambiense strains were isolated from sleeping sickness patients as described, and were subsequently propagated in mice at Swiss Tropical Institute.
• T. b. gambiense 4OR, 108R were isolated by Pati Pyana in Mbuji Mayi (D. R Congo) in 2005 and then propagated in STI (Swiss Tropical Institute, Basel, Switzerland) in different mice in winter 2006. o 4OR is a relapse 6 months after a 10 days melarsoprol treatment. o 108R is a relapse 8 months after a 10 days melarsoprol treatment
• T. b. gambiense DAL 1402 was obtained from the cryobank of the "Project de Recherches Cliniques Sur Ia Trypanosomiase," in Daloa. It was isolated inl990 from a human patient in Cote d'lvoire. • T. b. gambiense ITMAP 141267 was isolated in Bandundu/Lac Mai-Ndombe,
DRC, 1960
• T. b. gambiense Drani was isolated from a patient in Uganda; West.Nile, 1995; original stabilate ID: UTRO 210396 A.
[0328] In vitro culture and assay procedure: 50 μl HMI-9 medium (Hirumi et al, J. Parasitology, 75: 985-989 (1989)) supplemented 10% heat inactivated fetal calf serum and 5% heat inactivated human serum was added to each well of a 96-well microtiter plate. Serial drug dilutions of seven 3 -fold dilution steps covering a range from 90 to 0.123 μg/ml were prepared. Then 2x105 bloodstream forms of T. b. gambiense in 50 μl medium was added to each well and the plate incubated at 37 0C under a 5 % CO2 atmosphere for 72 h. 10 μl Alamar Blue (resazurin, 12.5 mg in 100 ml double-distilled water) was then added to each well and incubation continued for a further 2-4 h (Raz et al, Acta Trop 68: 139-47 (1997)). Then the plates were read with a Spectramax Gemini XS microplate fluorometer (Molecular Devices Cooperation, Sunnyvale, CA, USA) using an excitation wave length of 536 nm and an emission wave length of 588 nm. Data were analyzed using the microplate reader software Softmax Pro (Molecular Devices Cooperation, Sunnyvale, CA, USA).
[0329] Results from the 108R, 4OR, DAL 1402, DRANI, and ITMAP strains are provided in FI G.I.
EXAMPLE 6
Trypanosoma brucei rhodesiense STIB 900 Screening Assay Procedure [0330] The Trypanosoma brucei rhodesiense STIB900 strain was isolated in 1982 from a human patient in Tanzania and after several mouse passages cloned and adapted to axenic culture conditions (Baltz, et al., EMBO Journal 4: 1273-1277 (1985); Thuita, et al., Acta Tropica 108:6-10 (2008)). Minimum Essential Medium (50 μl) supplemented with 25 mM HEPES, lg/L additional glucose, 1% MEM non- essential amino acids (10Ox), 0.2 mM 2-mercaptoethanol, ImM Na-pyruvate and 15% heat inactivated horse serum was added to each well of a 96-well microtiter plate. Serial drug dilutions of seven 3-fold dilution steps covering a range from 90 to 0.123 μg/ml were prepared. Then 104 bloodstream forms of T. b. rhodesiense STIB 900 in 50 μl was added to each well and the plate incubated at 37 0C under a 5 % CO2 atmosphere for 72 h. 10 μl Alamar Blue (resazurin, 12.5 mg in 100 ml double- distilled water) was then added to each well and incubation continued for a further 2- 4 h (Raz, et al., Acta Trop 68:139-47 (1997)). Then the plates were read with a Spectramax Gemini XS microplate fluorometer (Molecular Devices Cooperation, Sunnyvale, CA, USA) using an excitation wave length of 536 nm and an emission wave length of 588 nm. Data were analyzed using the microplate reader software Softmax Pro (Molecular Devices Cooperation, Sunnyvale, CA, USA).
[0331] Results from this assay are provided in FIG.l.
EXAMPLE 7
Leishmania donovani Axenic Amastigote Screening Assay Procedure [0332] Amastigotes of L. donovani strain MHOM/ET/67/L82 were grown in axenic culture at 370C in SM medium (Cunningham, I. J Protozol. 24:325-329 (1977)) at pH 5.4 supplemented with 10% heat-inactivated fetal bovine serum under an atmosphere of 5% CO2 in air. One hundred microlitres of culture medium with 105 amastigotes from axenic culture with or without a serial drug dilution were seeded in 96-well microtitre plates. Serial drug dilutions of seven 3-fold dilution steps covering a range from 90 to 0.123 μg/ml were prepared. After 72 h of incubation the plates
were inspected under an inverted microscope to assure growth of the controls and sterile conditions. 10 μl of Alamar Blue (12.5 mg resazurin dissolved in 100 ml distilled water) (Mikus and Steverdig, Parasitology International 48:265-269 (2000)) were then added to each well and the plates incubated for another 2 h. Then the plates were read with a Spectramax Gemini XS microplate fluorometer (Molecular Devices Cooperation, Sunnyvale, CA, USA) using an excitation wave length of 536 nm and an emission wave length of 588 nm. Data were analyzed using the software Softmax Pro (Molecular Devices Cooperation, Sunnyvale, CA, USA). Decrease of fluorescence (= inhibition) was expressed as percentage of the fluorescence of control cultures and plotted against the drug concentrations. From the sigmoidal inhibition curves the IC50 values were calculated.
EXAMPLE 8
Leishmania donovani Macrophage and Leishmania infantum Macrophage Screening Assay Procedure [0333] Parasite and cell cultures. Two Leishmania species (L.infantum
MHOM/MA(BE)/67 and L.donovani MHOM/ET/67/L82) were used. The strains were maintained in the Golden Hamster (Mesocricetus auratus). Amastigotes were collected from the spleen of an infected donor hamster using three centrifugation purification steps (300 rpm, keeping the supernatant, 2200 rpm, keeping the supernatants and 3500 rpm, keeping the pellet) and spleen parasite burdens were assessed using the Stauber technique (Stauber LA., Exp Parasitol. 18: 1-11 (1966)). Primary peritoneal mouse macrophages were used as host cell and were collected 2 days after peritoneal stimulation with a 2% potato starch suspension. All cultures and assays were conducted at 37°C under an atmosphere of 5% CO2. [0334] Compound solutions/dilutions. Compound stock solutions were prepared in 100% DMSO at 20 mM or mg/ml. Concentrations were standard and expressed in molar concentrations, except for natural products, drug mixtures and if the molecular weight was not known. The compounds were serially pre-diluted (2-fold or 4-fold) in DMSO followed by a further (intermediate) dilution in demineralized water to assure a final in-test DMSO concentration of <1%.
[0335] Drug sensitivity assays. Assays were performed in 96-well microtiter plates, each well contained 10 μl of the compound dilutions together with 190 μl of
macrophage/parasite inoculum (3 x 103 cells + 4.5 x 105 parasites/well). The inoculum was prepared in RPMI- 1640 medium, supplemented with 200 mM L-glutamine, 16.5 mM NaHCOs, and 5% heat-inactivated fetal calf serum. Parasite multiplication was compared to untreated-infected controls (100% growth) and uninfected controls (0% growth). After 5 days incubation, parasite burdens (mean number of amastigotes/macrophage) were microscopically assessed after staining the cells with a 10% Giemsa solution. The results were expressed as % reduction in parasite burden compared to untreated control wells and an IC50 (50% inhibitory concentration) was calculated. [0336] Primary screen. Leishmania infantum MHOM/MA(BE)/67 strain was used. The compounds were tested at 5 concentrations (64 - 16 - 4 - 1 and 0.25 μM or μg/ml). Sodium-stibogluconate (IC50 = 8.7 + 2.1 μM) and miltefosin (IC50 = 4.3 + 1.1 μM) were included as the reference drugs. A test compound was classified as inactive when the IC50 was higher than 30 μM. When the IC50 was between 30 and 10 μM, the compound was regarded as moderately active. If the IC50 is lower than 10 μM, the compound was classified as highly active on the condition that it also demonstrates selective action (absence of cytotoxicity against primary peritoneal macrophages). A final recommendation for activity was given after confirmatory evaluation in a secondary screening. [0337] Secondary screen. Leishmania infantum MHOM/MA(BE)/67 and L.donovani MHOM/ET/67/L82 strains were used and the ICso-values were determined using an extended dose range (2-fold compound dilutions). Pentostam®, miltefosine, fungizone and PX-6518 were included as reference drugs. Advanced selectivity evaluations were performed against a panel of unrelated organisms (bacteria, yeasts, fungi and other protozoan parasites).
[0338] Results from this assay are provided in FIG.l.
EXAMPLE 9
Plasmodium falciparum Screening Assay Procedure
[0339] In vitro activity against erythrocytic stages of P. falciparum was determined using a H-hypoxanthine incorporation assay (Desjardins et al.,
Antimicrobial Agents and Chemotherapy 16:710-718 (1979), Matiel and Pink.
Plasmodium falciparum malaria parasite cultures and their use in immunology. In I. Lefkovits and B. Pernis (ed.), Immunological Methods. Academic Press, San Diego(1990)), using the chloroquine and pyrimethamine resistant Kl strain that originate from Thailand (Thaitong et al. Transactions of the Royal Society of Tropical Medicine and Hygiene 77:228-231 (1983)) and the standard drug chloroquine (Sigma C6628). Compounds were dissolved in DMSO at 10 mg/ml and added to parasite cultures incubated in RPMI 1640 medium without hypoxanthine, supplemented with HEPES (5.94 g/1), NaHCO3 (2.1 g/1), neomycin (100 U/ml), AlbumaxR (5 g/1) and washed human red cells A+ at 2.5% haematocrit (0.3% parasitaemia). Serial drug dilutions of seven 2-fold dilution steps covering a range from 5 to 0.156 μg/ml were prepared. The 96-well plates were incubated in a humidified atmosphere at 37 C; 4% CO2, 3% O2, 93% N2. After 48 h 50 μl of 3H-hypoxanthine (=0.5 μCi) was added to each well of the plate. The plates were incubated for a further 24 h under the same conditions. The plates were then harvested with a Betaplate™ cell harvester (Wallac, Zurich, Switzerland), and the red blood cells transferred onto a glass fibre filter then washed with distilled water. The dried filters were inserted into a plastic foil with 10 ml of scintillation fluid, and counted in a Betaplate™ liquid scintillation counter (Wallac, Zurich, Switzerland). IC50 values were calculated from sigmoidal inhibition curves using Microsoft Excel. [0340] Results from this assay are provided in FIG.l.
EXAMPLE 10
In vitro Trypanosoma PDE assays
[0341] A solution can be formed including a compound described herein, 40 mM Mops, pH 7.5, 0.8 mM EGTA, 15 mM magnesium acetate, 0.2 mg/ml bovine serum albumin (BSA) and 100000 c.p.m. of [3H]cAMP, in a final volume of 250 μl at 300C. The assay reaction can be started with the addition of the trypanosoma PDE (the amount will be in the linear hydrolysis vs. time zone as determined in a preliminary experiment) to this mixture. If the trypanosoma PDE source is a cell lysate or protein homogenate, then an inhibitor of endogenous PDEs, such as, but not limited to, IBMX (3-isobutylmethylxanthine), may be added. The reaction can be terminated by boiling the mixture for 2 minutes and the resulting AMP can be converted to adenosine by the addition of 10 mg/ml snake venom nucleotidase and further incubation at 37 0C for 10
minutes. Unhydrolyzed cAMP can be bound to AG1-X2 resin, and the remaining [3H]Adenosine in the aqueous phase can be quantitated by scintillation counting. Compounds described herein can be tested at concentrations varying from 0.001 to 100 μM for IC50 determination. Trypanosoma brucei PDEA (TbrPDEA, formerly known as TbPDEl) can be obtained through the methods described in Kunz, et al., Eur. J. Biochem. 271: 637-647 (2004). Trypanosoma brucei PDEBl (TbrPDEBl, formerly known as TbPD E2C) can be obtained through the methods described in Zoraghi, et al., Proc. Natl. Acad. Sci. U.S.A. 99: 4343-4348 (2002). Trypanosoma brucei PDEB2 (TbrPDEB2, formerly known as TbPDE2B) can be obtained through the methods described in Rascon, et al., Proc. Natl. Acad. Sci. U.S.A. 99: 4714-4719 (2002). Trypanosoma brucei PDE2A (TbPDE2A) can be obtained through the methods described in Zoraghi, et al., J. Biol. Chem, 276: 11559-11566 (2001).
[0342] It is understood that the examples and embodiments described herein are for illustrative purposes only and that various modifications or changes in light thereof will be suggested to persons skilled in the art and are to be included within the spirit and purview of this application and scope of the appended claims. All publications, patents, and patent applications cited herein are hereby incorporated by reference in their entirety for all purposes.
Claims
1. A compound having a structure according to the following formula:

R* is selected from the group consisting of H, a negative charge and a positively charged counterion,
L is selected from the group consisting of -S-, -S(O)-, -SO2-, and R2, R3, R4, R5 and R6 are independently selected from H and halogen with the proviso that when R4 is Cl, at least one of R2 or R3 or R5 or R6 is halogen, or a salt thereof.
2. The compound according to claim 1, wherein two members sseelleecctteed from R2, R3, R4, R5 and R6 are each an independently selected halogen; and the remaining members of R2, R3, R4, R5 and R6 are H.
3. The compound according to claim 1, wherein the compound is 
4. The compound according to claim 1, wherein the compound is selected from the group consisting of

5. A combination comprising the compound of claim 1, together with at least one other therapeutically active agent.
6. A pharmaceutical formulation comprising: a) the compound of claim 1; and b) a pharmaceutically acceptable excipient.
7. The pharmaceutical formulation of claim 6, wherein the pharmaceutical formulation is a unit dosage form.
8. The pharmaceutical formulation of claim 6, wherein the salt of said compound of a preceding claim is a pharmaceutically acceptable salt.
9. A method of killing and/or preventing the growth of a protozoa, comprising: contacting the protozoa with an effective amount of the compound of claim 1, thereby killing and/or preventing the growth of the protozoa.
10. The method of claim 9, wherein the compound is according to claim 1.
11. The method of claim 9, wherein the protozoa is Trypanosoma.
12. The method of claim 9, wherein the protozoa is Trypanosoma brucei.
13. The method of claim 12, wherein the Trypanosoma brucei is selected from the group consisting of Trypanosoma brucei brucei, Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense.
14. A method of treating and/or preventing a disease in an animal, comprising: administering to the animal a therapeutically effective amount of the compound of claim 1, thereby treating and/or preventing the disease.
15. The method of claim 14, wherein the compound is according to claim 1.
16. The method of claim 14, wherein the disease is sleeping sickness.
17. The method of claim 14, wherein the animal is a human.
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10576308P | 2008-10-15 | 2008-10-15 | |
| US10575908P | 2008-10-15 | 2008-10-15 | |
| US61/105,763 | 2008-10-15 | ||
| US61/105,759 | 2008-10-15 | ||
| US11090708P | 2008-11-03 | 2008-11-03 | |
| US61/110,907 | 2008-11-03 | ||
| US11995608P | 2008-12-04 | 2008-12-04 | |
| US61/119,956 | 2008-12-04 | ||
| US14824109P | 2009-01-29 | 2009-01-29 | |
| US61/148,241 | 2009-01-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010045505A1 true WO2010045505A1 (en) | 2010-04-22 |
Family
ID=42106904
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/060917 WO2010045505A1 (en) | 2008-10-15 | 2009-10-15 | Boron-containing small molecules as anti-protozoal agents |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2010045505A1 (en) |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011019618A1 (en) * | 2009-08-14 | 2011-02-17 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as antiprotozoal agents |
| WO2011019612A1 (en) * | 2009-08-14 | 2011-02-17 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as antiprotozoal agents |
| WO2011019616A1 (en) * | 2009-08-14 | 2011-02-17 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as antiprotozoal agents |
| WO2011116348A1 (en) * | 2010-03-19 | 2011-09-22 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-protozoal agent |
| US8039451B2 (en) | 2005-02-16 | 2011-10-18 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8039450B2 (en) | 2008-03-06 | 2011-10-18 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US8106031B2 (en) | 2006-05-02 | 2012-01-31 | Anacor Pharmaceuticals, Inc. | Hydrolytically-resistant boron-containing therapeutics and methods of use |
| US8115026B2 (en) | 2005-02-16 | 2012-02-14 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8168614B2 (en) | 2006-02-16 | 2012-05-01 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US8343944B2 (en) | 2009-07-28 | 2013-01-01 | Anacor Pharmaceuticals, Inc. | Trisubstituted boron-containing molecules |
| US8461134B2 (en) | 2009-11-11 | 2013-06-11 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8461364B2 (en) | 2008-12-17 | 2013-06-11 | Glaxosmithkline Llc | Polymorphs of (S)-3-aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[C][1,2]oxaborol-1-OL |
| US8461336B2 (en) | 2008-09-04 | 2013-06-11 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8470803B2 (en) | 2008-09-04 | 2013-06-25 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8703742B2 (en) | 2010-09-07 | 2014-04-22 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8716478B2 (en) | 2010-01-27 | 2014-05-06 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9138002B2 (en) | 2013-01-30 | 2015-09-22 | Agrofresh Inc. | Compounds and compositions |
| WO2016001834A1 (en) | 2014-07-01 | 2016-01-07 | Daiichi Sankyo Company, Limited | Tricyclic benzoxaboroles as antibacterial agents |
| US9346834B2 (en) | 2009-10-20 | 2016-05-24 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as antiprotozoal agents |
| US9426996B2 (en) | 2013-01-30 | 2016-08-30 | Agrofresh Inc. | Use of benzoxaboroles as volatile antimicrobial agents on meats, plants, or plant parts |
| US9493489B2 (en) | 2008-10-15 | 2016-11-15 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-protozoal agents |
| US9585396B2 (en) | 2013-01-30 | 2017-03-07 | Agrofresh Inc. | Volatile applications against pathogens |
| US10070649B2 (en) | 2013-01-30 | 2018-09-11 | Agrofresh Inc. | Volatile applications against pathogens |
| KR20190005982A (en) * | 2016-05-12 | 2019-01-16 | 아나코르 파마슈티칼스 인코포레이티드 | Oxaborol ester and its use |
| WO2020252679A1 (en) * | 2019-06-19 | 2020-12-24 | Shanghai Jiao Tong University | A new type of benzoxaborole-based anti-pneumococcal compounds targeting leucyl-trna synthetase |
| US10961261B2 (en) | 2017-03-01 | 2021-03-30 | Anacor Pharmaceuticals, Inc. | Oxaborole analogs and uses thereof |
| US10966429B2 (en) | 2016-03-07 | 2021-04-06 | Agrofresh Inc. | Synergistic methods of using benzoxaborole compounds and preservative gases as an antimicrobial for crops |
| US11039617B2 (en) | 2013-01-30 | 2021-06-22 | Agrofresh Inc. | Large scale methods of uniformly coating packaging surfaces with a volatile antimicrobial to preserve food freshness |
| JP2022514015A (en) * | 2018-12-18 | 2022-02-09 | エランコ・ティアゲゾンタイト・アーゲー | Bicyclic derivative |
| WO2022043936A1 (en) | 2020-08-31 | 2022-03-03 | Pfizer Inc. | Methods of protecting rna |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6521619B2 (en) * | 2000-06-29 | 2003-02-18 | Icos Corporation | Aryl phenylcyclopropyl sulfide derivatives and their use as cell adhesion inhibiting anti-inflammatory and immune suppressive agents |
| US20040077601A1 (en) * | 2002-07-09 | 2004-04-22 | Point Therapeutics, Inc. | Methods and compositions relating to isoleucine boroproline compounds |
| US20070286822A1 (en) * | 2006-06-12 | 2007-12-13 | Anacor Pharmaceuticals Inc. | Compounds for the Treatment of Periodontal Disease |
| US20070293457A1 (en) * | 2006-02-16 | 2007-12-20 | Baker Stephen J | Boron-containing small molecules as anti-inflammatory agents |
-
2009
- 2009-10-15 WO PCT/US2009/060917 patent/WO2010045505A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6521619B2 (en) * | 2000-06-29 | 2003-02-18 | Icos Corporation | Aryl phenylcyclopropyl sulfide derivatives and their use as cell adhesion inhibiting anti-inflammatory and immune suppressive agents |
| US20040077601A1 (en) * | 2002-07-09 | 2004-04-22 | Point Therapeutics, Inc. | Methods and compositions relating to isoleucine boroproline compounds |
| US20070293457A1 (en) * | 2006-02-16 | 2007-12-20 | Baker Stephen J | Boron-containing small molecules as anti-inflammatory agents |
| US20070286822A1 (en) * | 2006-06-12 | 2007-12-13 | Anacor Pharmaceuticals Inc. | Compounds for the Treatment of Periodontal Disease |
Cited By (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8440642B2 (en) | 2005-02-16 | 2013-05-14 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9549938B2 (en) | 2005-02-16 | 2017-01-24 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9572823B2 (en) | 2005-02-16 | 2017-02-21 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9566289B2 (en) | 2005-02-16 | 2017-02-14 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8039451B2 (en) | 2005-02-16 | 2011-10-18 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8889656B2 (en) | 2005-02-16 | 2014-11-18 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9353133B2 (en) | 2005-02-16 | 2016-05-31 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8115026B2 (en) | 2005-02-16 | 2012-02-14 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8722917B2 (en) | 2005-02-16 | 2014-05-13 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9566290B2 (en) | 2005-02-16 | 2017-02-14 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9029353B2 (en) | 2006-02-16 | 2015-05-12 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US8501712B2 (en) | 2006-02-16 | 2013-08-06 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US8168614B2 (en) | 2006-02-16 | 2012-05-01 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US9682092B2 (en) | 2006-02-16 | 2017-06-20 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US8106031B2 (en) | 2006-05-02 | 2012-01-31 | Anacor Pharmaceuticals, Inc. | Hydrolytically-resistant boron-containing therapeutics and methods of use |
| US8039450B2 (en) | 2008-03-06 | 2011-10-18 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US8461135B2 (en) | 2008-03-06 | 2013-06-11 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US9416146B2 (en) | 2008-03-06 | 2016-08-16 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US9012431B2 (en) | 2008-03-06 | 2015-04-21 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US8470803B2 (en) | 2008-09-04 | 2013-06-25 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8461336B2 (en) | 2008-09-04 | 2013-06-11 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9493489B2 (en) | 2008-10-15 | 2016-11-15 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-protozoal agents |
| US8461364B2 (en) | 2008-12-17 | 2013-06-11 | Glaxosmithkline Llc | Polymorphs of (S)-3-aminomethyl-7-(3-hydroxy-propoxy)-3H-benzo[C][1,2]oxaborol-1-OL |
| US8343944B2 (en) | 2009-07-28 | 2013-01-01 | Anacor Pharmaceuticals, Inc. | Trisubstituted boron-containing molecules |
| US9440994B2 (en) | 2009-08-14 | 2016-09-13 | Anacor Pharmaceuticals, Inc. | Boron containing small molecules as antiprotozoal agents |
| US10301329B2 (en) | 2009-08-14 | 2019-05-28 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as antiprotozoal agents |
| WO2011019612A1 (en) * | 2009-08-14 | 2011-02-17 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as antiprotozoal agents |
| WO2011019616A1 (en) * | 2009-08-14 | 2011-02-17 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as antiprotozoal agents |
| WO2011019618A1 (en) * | 2009-08-14 | 2011-02-17 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as antiprotozoal agents |
| US9346834B2 (en) | 2009-10-20 | 2016-05-24 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as antiprotozoal agents |
| US8461134B2 (en) | 2009-11-11 | 2013-06-11 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9145429B2 (en) | 2010-01-27 | 2015-09-29 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8716478B2 (en) | 2010-01-27 | 2014-05-06 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US9499570B2 (en) | 2010-01-27 | 2016-11-22 | Anacor Pharmaceuticals, Inc. | Boron containing small molecules |
| US8623911B2 (en) | 2010-03-19 | 2014-01-07 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-protozoal agent |
| WO2011116348A1 (en) * | 2010-03-19 | 2011-09-22 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-protozoal agent |
| US9751898B2 (en) | 2010-09-07 | 2017-09-05 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8703742B2 (en) | 2010-09-07 | 2014-04-22 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US11008345B2 (en) | 2010-09-07 | 2021-05-18 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US10070649B2 (en) | 2013-01-30 | 2018-09-11 | Agrofresh Inc. | Volatile applications against pathogens |
| US9138002B2 (en) | 2013-01-30 | 2015-09-22 | Agrofresh Inc. | Compounds and compositions |
| US11917997B2 (en) | 2013-01-30 | 2024-03-05 | Agrofresh Inc. | Volatile applications against pathogens |
| US11771089B2 (en) | 2013-01-30 | 2023-10-03 | Agrofresh Inc. | Large-scale methods of uniformly coating packaging surfaces with a volatile antimicrobial to preserve food freshness |
| US11202448B2 (en) | 2013-01-30 | 2021-12-21 | Agrofresh Inc. | Volatile applications against pathogens |
| US11039617B2 (en) | 2013-01-30 | 2021-06-22 | Agrofresh Inc. | Large scale methods of uniformly coating packaging surfaces with a volatile antimicrobial to preserve food freshness |
| US10765117B2 (en) | 2013-01-30 | 2020-09-08 | Agrofresh Inc. | Volatile applications against pathogens |
| US9585396B2 (en) | 2013-01-30 | 2017-03-07 | Agrofresh Inc. | Volatile applications against pathogens |
| US9426996B2 (en) | 2013-01-30 | 2016-08-30 | Agrofresh Inc. | Use of benzoxaboroles as volatile antimicrobial agents on meats, plants, or plant parts |
| WO2016001834A1 (en) | 2014-07-01 | 2016-01-07 | Daiichi Sankyo Company, Limited | Tricyclic benzoxaboroles as antibacterial agents |
| US10966429B2 (en) | 2016-03-07 | 2021-04-06 | Agrofresh Inc. | Synergistic methods of using benzoxaborole compounds and preservative gases as an antimicrobial for crops |
| US10882872B2 (en) | 2016-05-12 | 2021-01-05 | Anacor Pharmaceuticals, Inc. | Oxaborole esters and uses thereof |
| KR102243775B1 (en) | 2016-05-12 | 2021-04-22 | 아나코르 파마슈티칼스 인코포레이티드 | Oxaborole ester and uses thereof |
| US10562921B2 (en) | 2016-05-12 | 2020-02-18 | Anacor Pharmaceuticals, Inc. | Oxaborole esters and uses thereof |
| JP2019520326A (en) * | 2016-05-12 | 2019-07-18 | アナコール・ファーマシューティカルズ・インコーポレーテッド | Oxaborole ester and use thereof |
| US11578087B2 (en) | 2016-05-12 | 2023-02-14 | Anacor Pharmaceuticals, Inc. | Oxaborole esters and uses thereof |
| JP2019081770A (en) * | 2016-05-12 | 2019-05-30 | アナコール・ファーマシューティカルズ・インコーポレーテッド | Oxaborole ester and use thereof |
| KR20190005982A (en) * | 2016-05-12 | 2019-01-16 | 아나코르 파마슈티칼스 인코포레이티드 | Oxaborol ester and its use |
| US10961261B2 (en) | 2017-03-01 | 2021-03-30 | Anacor Pharmaceuticals, Inc. | Oxaborole analogs and uses thereof |
| US11691992B2 (en) | 2017-03-01 | 2023-07-04 | Anacor Pharmaceuticals, Inc. | Oxaborole analogs and uses thereof |
| JP2022514015A (en) * | 2018-12-18 | 2022-02-09 | エランコ・ティアゲゾンタイト・アーゲー | Bicyclic derivative |
| JP7324846B2 (en) | 2018-12-18 | 2023-08-10 | エランコ・ティアゲゾンタイト・アーゲー | bicyclic derivative |
| WO2020252679A1 (en) * | 2019-06-19 | 2020-12-24 | Shanghai Jiao Tong University | A new type of benzoxaborole-based anti-pneumococcal compounds targeting leucyl-trna synthetase |
| WO2022043936A1 (en) | 2020-08-31 | 2022-03-03 | Pfizer Inc. | Methods of protecting rna |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2010045505A1 (en) | Boron-containing small molecules as anti-protozoal agents | |
| US9493489B2 (en) | Boron-containing small molecules as anti-protozoal agents | |
| US9346834B2 (en) | Boron-containing small molecules as antiprotozoal agents | |
| US11161858B2 (en) | Boron-containing small molecules as antiprotozoal agents | |
| US10301329B2 (en) | Boron-containing small molecules as antiprotozoal agents | |
| WO2011019612A1 (en) | Boron-containing small molecules as antiprotozoal agents | |
| WO2011022337A1 (en) | Boron-containing small molecules as antiprotozoal agents | |
| US8623911B2 (en) | Boron-containing small molecules as anti-protozoal agent | |
| AU2009221793B2 (en) | Boron-containing small molecules as anti-inflammatory agents | |
| WO2011019616A1 (en) | Boron-containing small molecules as antiprotozoal agents | |
| WO2009111676A2 (en) | Boron-containing small molecules as anti-inflammatory agents | |
| WO2011094450A1 (en) | Boron-containing small molecules | |
| WO2011063293A1 (en) | Boron-containing small molecules as antihelminth agents | |
| WO2011060199A1 (en) | Boron-containing small molecules | |
| IL185080A (en) | Boron-containing small molecules, pharmaceutical formulations comprising them and use thereof in the preparation of medicaments for treating infections | |
| US20180273553A1 (en) | Boron-containing small molecules as antiprotozoal agents | |
| OA17452A (en) | Boron-containing small molecules as antiprotozoal agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09821289 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09821289 Country of ref document: EP Kind code of ref document: A1 |