WO2009082152A2 - Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient - Google Patents
Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient Download PDFInfo
- Publication number
- WO2009082152A2 WO2009082152A2 PCT/KR2008/007585 KR2008007585W WO2009082152A2 WO 2009082152 A2 WO2009082152 A2 WO 2009082152A2 KR 2008007585 W KR2008007585 W KR 2008007585W WO 2009082152 A2 WO2009082152 A2 WO 2009082152A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dihydro
- thiazol
- indol
- ethyl
- cyclopentylamino
- Prior art date
Links
- 108010021582 Glucokinase Proteins 0.000 title claims abstract description 51
- 102000030595 Glucokinase Human genes 0.000 title claims abstract description 47
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 16
- 239000004480 active ingredient Substances 0.000 title claims abstract description 13
- 239000012190 activator Substances 0.000 title description 14
- 150000001875 compounds Chemical class 0.000 claims abstract description 754
- 239000000203 mixture Substances 0.000 claims description 179
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 155
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Natural products OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 152
- -1 C)-C6-alkylcarbonyl Chemical group 0.000 claims description 115
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N EtOH Substances CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 83
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 76
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 62
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical group C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 54
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 38
- 229910052739 hydrogen Inorganic materials 0.000 claims description 34
- 239000001257 hydrogen Substances 0.000 claims description 34
- 125000000623 heterocyclic group Chemical group 0.000 claims description 27
- 125000005842 heteroatom Chemical group 0.000 claims description 26
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Substances C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 25
- 150000003839 salts Chemical class 0.000 claims description 23
- 150000002431 hydrogen Chemical group 0.000 claims description 22
- 206010012601 diabetes mellitus Diseases 0.000 claims description 21
- 125000001072 heteroaryl group Chemical group 0.000 claims description 21
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 20
- 125000003118 aryl group Chemical group 0.000 claims description 20
- 229910052760 oxygen Inorganic materials 0.000 claims description 18
- 229910052736 halogen Inorganic materials 0.000 claims description 15
- 150000002367 halogens Chemical group 0.000 claims description 15
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 claims description 14
- 201000010099 disease Diseases 0.000 claims description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 14
- 229910052717 sulfur Inorganic materials 0.000 claims description 14
- 125000000217 alkyl group Chemical group 0.000 claims description 13
- 125000001424 substituent group Chemical group 0.000 claims description 13
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical group C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 12
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 10
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Chemical group COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 10
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 10
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 10
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 claims description 9
- 239000003937 drug carrier Substances 0.000 claims description 9
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 9
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 8
- 230000002265 prevention Effects 0.000 claims description 8
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 7
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 125000003545 alkoxy group Chemical group 0.000 claims description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 6
- MUOMJAMAPIPYOZ-OAHLLOKOSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-(methylsulfonylmethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@@H](CC(O)=O)N=3)=CC2=CC(CS(=O)(=O)C)=CC=1NC1CCCC1 MUOMJAMAPIPYOZ-OAHLLOKOSA-N 0.000 claims description 5
- 125000000041 C6-C10 aryl group Chemical class 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 5
- NTXZLLLQCAOISR-QGZVFWFLSA-N n-cyclopentyl-2-[(4r)-4-(pyrrolidin-1-ylmethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C([C@H](N=1)CN2CCCC2)SC=1C(NC1=2)=CC1=CC=CC=2NC1CCCC1 NTXZLLLQCAOISR-QGZVFWFLSA-N 0.000 claims description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 5
- 125000006714 (C3-C10) heterocyclyl group Chemical group 0.000 claims description 4
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 4
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 4
- VUCVBMBIOHUZAT-CQSZACIVSA-N 2-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound OCC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)=N1 VUCVBMBIOHUZAT-CQSZACIVSA-N 0.000 claims description 4
- LHNFZHRTDUVHDB-CQSZACIVSA-N 2-[(4r)-2-[5-methyl-7-[(4-oxocyclohexyl)amino]-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@@H](CC(O)=O)N=3)=CC2=CC(C)=CC=1NC1CCC(=O)CC1 LHNFZHRTDUVHDB-CQSZACIVSA-N 0.000 claims description 4
- NEULLOCJPYHOSQ-CQSZACIVSA-N 2-[(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n-methylacetamide Chemical compound CNC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 NEULLOCJPYHOSQ-CQSZACIVSA-N 0.000 claims description 4
- WSSGVFFLIVEOAE-CYBMUJFWSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 WSSGVFFLIVEOAE-CYBMUJFWSA-N 0.000 claims description 4
- DSXPECSRXYNDES-QGZVFWFLSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-methoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-morpholin-4-ylethanone Chemical compound C=12NC(C=3SC[C@@H](CC(=O)N4CCOCC4)N=3)=CC2=CC(OC)=CC=1NC1CCCC1 DSXPECSRXYNDES-QGZVFWFLSA-N 0.000 claims description 4
- FSZRYQNVPUUJJA-CQSZACIVSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@@H](CC(O)=O)N=3)=CC2=CC(C)=CC=1NC1CCCC1 FSZRYQNVPUUJJA-CQSZACIVSA-N 0.000 claims description 4
- FMDXMBNYFQQBQP-MRXNPFEDSA-N 2-[(4r)-4-[2-(dimethylamino)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-5-fluoro-n-(oxan-4-yl)-1h-indol-7-amine Chemical compound CN(C)CC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(F)C=C3C=2)=N1 FMDXMBNYFQQBQP-MRXNPFEDSA-N 0.000 claims description 4
- FVDYXUXRKWHFOH-LJQANCHMSA-N 2-hydroxy-1-[4-[2-[(4r)-2-[5-methoxy-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-1-yl]ethanone Chemical compound C=12NC(C=3SC[C@@H](CCN4CCN(CC4)C(=O)CO)N=3)=CC2=CC(OC)=CC=1NC1CCOCC1 FVDYXUXRKWHFOH-LJQANCHMSA-N 0.000 claims description 4
- SUGSSXONAVGVTF-QGZVFWFLSA-N 3-[2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]-5-methylimidazole-4-carboxylic acid Chemical compound OC(=O)C1=C(C)N=CN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 SUGSSXONAVGVTF-QGZVFWFLSA-N 0.000 claims description 4
- VGTAGLXGWXFORQ-QGZVFWFLSA-N 4-[2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-2-one Chemical compound C=12NC(C=3SC[C@@H](CCN4CC(=O)NCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 VGTAGLXGWXFORQ-QGZVFWFLSA-N 0.000 claims description 4
- MNCAODPCJHHLAD-QFIPXVFZSA-N 4-[2-[(4s)-2-[7-(oxan-4-ylmethylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-2-one Chemical compound C1CNC(=O)CN1CC[C@@H]1N=C(C=2NC3=C(NCC4CCOCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)SC1 MNCAODPCJHHLAD-QFIPXVFZSA-N 0.000 claims description 4
- WFLYOFAEOXUSMW-QGZVFWFLSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-(2-pyrazol-1-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4N=CC=C4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 WFLYOFAEOXUSMW-QGZVFWFLSA-N 0.000 claims description 4
- 230000004913 activation Effects 0.000 claims description 4
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 4
- 230000009849 deactivation Effects 0.000 claims description 4
- QJYDRFXMFDGZCU-OAHLLOKOSA-N ethyl 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound CCOC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 QJYDRFXMFDGZCU-OAHLLOKOSA-N 0.000 claims description 4
- HXGLHGOHWUIOII-OAHLLOKOSA-N ethyl 2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound CCOC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 HXGLHGOHWUIOII-OAHLLOKOSA-N 0.000 claims description 4
- RGASNXICKYMZGF-MRXNPFEDSA-N ethyl 3-[(4r)-2-[5-bromo-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound CCOC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Br)C=C3C=2)=N1 RGASNXICKYMZGF-MRXNPFEDSA-N 0.000 claims description 4
- 230000002218 hypoglycaemic effect Effects 0.000 claims description 4
- 150000002475 indoles Chemical class 0.000 claims description 4
- HNJNNAPYEODGCI-CQSZACIVSA-N methyl 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 HNJNNAPYEODGCI-CQSZACIVSA-N 0.000 claims description 4
- ADYYTORVODGGNU-OAHLLOKOSA-N methyl 2-[(4r)-2-[7-(cyclopentylamino)-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(C)C=C3C=2)=N1 ADYYTORVODGGNU-OAHLLOKOSA-N 0.000 claims description 4
- 238000002156 mixing Methods 0.000 claims description 4
- BCOYMDFZVCEFOH-MRXNPFEDSA-N n-cyclopentyl-2-[(4r)-4-[2-(dimethylamino)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-5-fluoro-1h-indol-7-amine Chemical compound CN(C)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 BCOYMDFZVCEFOH-MRXNPFEDSA-N 0.000 claims description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 4
- NDAGUOLXHHPDOP-XLIONFOSSA-N (2s)-1-[2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 NDAGUOLXHHPDOP-XLIONFOSSA-N 0.000 claims description 3
- BEUNTKQGUNXHAO-CYBMUJFWSA-N 2-[(4r)-2-[5-bromo-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Br)C=C3C=2)=N1 BEUNTKQGUNXHAO-CYBMUJFWSA-N 0.000 claims description 3
- RZPDCVNGOIJGSY-CQSZACIVSA-N 2-[(4r)-2-[5-bromo-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound OCC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Br)C=C3C=2)=N1 RZPDCVNGOIJGSY-CQSZACIVSA-N 0.000 claims description 3
- PTMZTYWDDQIPQO-MRTLOADZSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-[3-(dimethylamino)pyrrolidin-1-yl]ethanone Chemical compound C1C(N(C)C)CCN1C(=O)C[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 PTMZTYWDDQIPQO-MRTLOADZSA-N 0.000 claims description 3
- NYINCTOKYLFKMS-OAHLLOKOSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n,n-dimethylacetamide Chemical compound CN(C)C(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 NYINCTOKYLFKMS-OAHLLOKOSA-N 0.000 claims description 3
- DIUQOMSDTBYHJE-HXUWFJFHSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n-(3-morpholin-4-ylpropyl)acetamide Chemical compound C=12NC(C=3SC[C@@H](CC(=O)NCCCN4CCOCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 DIUQOMSDTBYHJE-HXUWFJFHSA-N 0.000 claims description 3
- TYTKCGWYMJNGMW-CYBMUJFWSA-N 2-[(4r)-2-[5-fluoro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(F)C=C3C=2)=N1 TYTKCGWYMJNGMW-CYBMUJFWSA-N 0.000 claims description 3
- WCZBLXULYUFRIF-CQSZACIVSA-N 2-[(4r)-2-[5-fluoro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound OCC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(F)C=C3C=2)=N1 WCZBLXULYUFRIF-CQSZACIVSA-N 0.000 claims description 3
- UIJKUPNCTDZVOP-CYBMUJFWSA-N 2-[(4r)-2-[5-methoxy-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@@H](CC(O)=O)N=3)=CC2=CC(OC)=CC=1NC1CCOCC1 UIJKUPNCTDZVOP-CYBMUJFWSA-N 0.000 claims description 3
- BUKKUUIIFNRZSS-CYBMUJFWSA-N 2-[(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 BUKKUUIIFNRZSS-CYBMUJFWSA-N 0.000 claims description 3
- XCPPPJFWGAQTON-CQSZACIVSA-N 2-[(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound OCC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 XCPPPJFWGAQTON-CQSZACIVSA-N 0.000 claims description 3
- MQXNNVVCPHVDTF-LJQANCHMSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-(phenoxymethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(COC=4C=CC=CC=4)C=C3C=2)=N1 MQXNNVVCPHVDTF-LJQANCHMSA-N 0.000 claims description 3
- UMRWBXZGGKOEQL-CQSZACIVSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-ethoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@@H](CC(O)=O)N=3)=CC2=CC(OCC)=CC=1NC1CCCC1 UMRWBXZGGKOEQL-CQSZACIVSA-N 0.000 claims description 3
- BXORQYBJKSLIHU-CYBMUJFWSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-methoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@@H](CC(O)=O)N=3)=CC2=CC(OC)=CC=1NC1CCCC1 BXORQYBJKSLIHU-CYBMUJFWSA-N 0.000 claims description 3
- PLTKYFWJWHDBLW-OAHLLOKOSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-propoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@@H](CC(O)=O)N=3)=CC2=CC(OCCC)=CC=1NC1CCCC1 PLTKYFWJWHDBLW-OAHLLOKOSA-N 0.000 claims description 3
- NNMSAXVTDPQBQV-MRXNPFEDSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-pyridin-3-yloxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC=4C=NC=CC=4)C=C3C=2)=N1 NNMSAXVTDPQBQV-MRXNPFEDSA-N 0.000 claims description 3
- PRHPRWULEUZEID-CYBMUJFWSA-N 2-[(4r)-2-[7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=CC=C3C=2)=N1 PRHPRWULEUZEID-CYBMUJFWSA-N 0.000 claims description 3
- BNEJVZQAPCGPTC-QGZVFWFLSA-N 2-[(4r)-2-[7-(oxan-4-ylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)=N1 BNEJVZQAPCGPTC-QGZVFWFLSA-N 0.000 claims description 3
- LRZWTWOPTYMKTF-FUHWJXTLSA-N 2-[(4r)-4-[2-[(3s)-3-aminopyrrolidin-1-yl]ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-5-chloro-n-(oxan-4-yl)-1h-indol-7-amine Chemical compound C1[C@@H](N)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)SC1 LRZWTWOPTYMKTF-FUHWJXTLSA-N 0.000 claims description 3
- UPTMPOOMRXKLLP-CQSZACIVSA-N 3-[(4r)-2-[5-bromo-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound OC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Br)C=C3C=2)=N1 UPTMPOOMRXKLLP-CQSZACIVSA-N 0.000 claims description 3
- YXVLLHIDVWSDOL-LJQANCHMSA-N 3-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-(4-methylpiperazin-1-yl)propan-1-one Chemical compound C1CN(C)CCN1C(=O)CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 YXVLLHIDVWSDOL-LJQANCHMSA-N 0.000 claims description 3
- NUVGUEZXELGIPI-CQSZACIVSA-N 3-[(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound OC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 NUVGUEZXELGIPI-CQSZACIVSA-N 0.000 claims description 3
- HPJVLKJBOSASQY-CYBMUJFWSA-N 3-[(4r)-2-[7-(cyclopentylamino)-5-(trifluoromethoxy)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound OC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC(F)(F)F)C=C3C=2)=N1 HPJVLKJBOSASQY-CYBMUJFWSA-N 0.000 claims description 3
- ZKATTYKCDHWBGG-CQSZACIVSA-N 3-[(4r)-2-[7-(cyclopentylamino)-5-methoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound C=12NC(C=3SC[C@@H](CCC(O)=O)N=3)=CC2=CC(OC)=CC=1NC1CCCC1 ZKATTYKCDHWBGG-CQSZACIVSA-N 0.000 claims description 3
- HJLGOFIEZJNOOU-GOSISDBHSA-N 3-[(4r)-2-[7-(cyclopentylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound OC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)=N1 HJLGOFIEZJNOOU-GOSISDBHSA-N 0.000 claims description 3
- ZSSFSRMUUPQLBO-GOSISDBHSA-N 4-[2-[(4r)-2-(7-phenoxy-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]ethyl]morpholine Chemical compound C([C@H]1N=C(SC1)C=1NC2=C(OC=3C=CC=CC=3)C=CC=C2C=1)CN1CCOCC1 ZSSFSRMUUPQLBO-GOSISDBHSA-N 0.000 claims description 3
- YGWCETVEGUAPDJ-QGZVFWFLSA-N 4-[2-[(4r)-2-[5-fluoro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-2-one Chemical compound C=12NC(C=3SC[C@@H](CCN4CC(=O)NCC4)N=3)=CC2=CC(F)=CC=1NC1CCOCC1 YGWCETVEGUAPDJ-QGZVFWFLSA-N 0.000 claims description 3
- FYFHQRVBGCRNNP-QGZVFWFLSA-N 4-[2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-2-one Chemical compound C=12NC(C=3SC[C@@H](CCN4CC(=O)NCC4)N=3)=CC2=CC(F)=CC=1NC1CCCC1 FYFHQRVBGCRNNP-QGZVFWFLSA-N 0.000 claims description 3
- ZNGQSKARSZOHBW-GOSISDBHSA-N 5-chloro-n-(oxan-4-yl)-2-[(4r)-4-(2-piperazin-1-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCNCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCOCC1 ZNGQSKARSZOHBW-GOSISDBHSA-N 0.000 claims description 3
- QMKMPFYGUYBFKG-HXUWFJFHSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-[2-(4-ethylsulfonylpiperazin-1-yl)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C1CN(S(=O)(=O)CC)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 QMKMPFYGUYBFKG-HXUWFJFHSA-N 0.000 claims description 3
- LZENYYYULVBFQH-LJQANCHMSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-[2-(4-methylpiperazin-1-yl)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C1CN(C)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 LZENYYYULVBFQH-LJQANCHMSA-N 0.000 claims description 3
- UJACHDBABNNGKV-MRXNPFEDSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-[2-(dimethylamino)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound CN(C)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 UJACHDBABNNGKV-MRXNPFEDSA-N 0.000 claims description 3
- XIYOXQGSJBSWRZ-OAHLLOKOSA-N 5-fluoro-2-[(4r)-4-(2-methylsulfonylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-n-(oxan-4-yl)-1h-indol-7-amine Chemical compound CS(=O)(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(F)C=C3C=2)=N1 XIYOXQGSJBSWRZ-OAHLLOKOSA-N 0.000 claims description 3
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 3
- 206010020772 Hypertension Diseases 0.000 claims description 3
- 208000008589 Obesity Diseases 0.000 claims description 3
- 208000001647 Renal Insufficiency Diseases 0.000 claims description 3
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 3
- UTKQTLRDJKXHQO-CYBMUJFWSA-N [(4r)-2-[5-bromo-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Br)C=C3C=2)=N1 UTKQTLRDJKXHQO-CYBMUJFWSA-N 0.000 claims description 3
- CTBQMRMUYUVENX-CYBMUJFWSA-N [(4r)-2-[5-bromo-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Br)C=C3C=2)=N1 CTBQMRMUYUVENX-CYBMUJFWSA-N 0.000 claims description 3
- CKNJKLBMTYFMCG-CYBMUJFWSA-N [(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)=N1 CKNJKLBMTYFMCG-CYBMUJFWSA-N 0.000 claims description 3
- MFBYUZUPPRPRDI-CYBMUJFWSA-N [(4r)-2-[5-chloro-7-(thian-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCSCC4)C=C(Cl)C=C3C=2)=N1 MFBYUZUPPRPRDI-CYBMUJFWSA-N 0.000 claims description 3
- LMTJGOWQFJMETO-CYBMUJFWSA-N [(4r)-2-[5-fluoro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(F)C=C3C=2)=N1 LMTJGOWQFJMETO-CYBMUJFWSA-N 0.000 claims description 3
- VZJZPUUQQUNBNB-GOSISDBHSA-N [(4r)-2-[7-(cyclopentylamino)-5-(1h-pyrrol-3-ylmethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(CC4=CNC=C4)C=C3C=2)=N1 VZJZPUUQQUNBNB-GOSISDBHSA-N 0.000 claims description 3
- DFZQFOPSTZLGBN-OAHLLOKOSA-N [(4r)-2-[7-(cyclopentylamino)-5-(methylsulfonylmethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound C=12NC(C=3SC[C@@H](CO)N=3)=CC2=CC(CS(=O)(=O)C)=CC=1NC1CCCC1 DFZQFOPSTZLGBN-OAHLLOKOSA-N 0.000 claims description 3
- WRYMUDJKFPBFIG-CYBMUJFWSA-N [(4r)-2-[7-(cyclopentylamino)-5-methoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound C=12NC(C=3SC[C@@H](CO)N=3)=CC2=CC(OC)=CC=1NC1CCCC1 WRYMUDJKFPBFIG-CYBMUJFWSA-N 0.000 claims description 3
- DMFUQWDGRCACKE-MRXNPFEDSA-N [(4r)-2-[7-(cyclopentylamino)-5-pyridin-3-yloxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC=4C=NC=CC=4)C=C3C=2)=N1 DMFUQWDGRCACKE-MRXNPFEDSA-N 0.000 claims description 3
- QXMQZZPKJFWNEU-CYBMUJFWSA-N [(4r)-2-[7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=CC=C3C=2)=N1 QXMQZZPKJFWNEU-CYBMUJFWSA-N 0.000 claims description 3
- XISAEFPRGFMGNX-PIJUOVFKSA-N [(4r)-2-[7-(oxolan-3-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4COCC4)C=CC=C3C=2)=N1 XISAEFPRGFMGNX-PIJUOVFKSA-N 0.000 claims description 3
- ZZXRBKDKZTYYRY-ZGTCLIOFSA-N [(4r)-2-[7-[(1-methylsulfonylpyrrolidin-3-yl)amino]-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound C1N(S(=O)(=O)C)CCC1NC1=CC=CC2=C1NC(C=1SC[C@@H](CO)N=1)=C2 ZZXRBKDKZTYYRY-ZGTCLIOFSA-N 0.000 claims description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 3
- OCFAHPDQRHJFGA-OAHLLOKOSA-N ethyl 2-[(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound CCOC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 OCFAHPDQRHJFGA-OAHLLOKOSA-N 0.000 claims description 3
- DMDDLNXTVJZAKS-MRXNPFEDSA-N ethyl 2-[[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methoxy]acetate Chemical compound CCOC(=O)COC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 DMDDLNXTVJZAKS-MRXNPFEDSA-N 0.000 claims description 3
- DVBWZUPJRKSPKH-MRXNPFEDSA-N ethyl 3-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound CCOC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 DVBWZUPJRKSPKH-MRXNPFEDSA-N 0.000 claims description 3
- PAAZDMQPBVIYMF-OAHLLOKOSA-N ethyl 3-[(4r)-2-[7-(cyclopentylamino)-5-(trifluoromethoxy)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound CCOC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC(F)(F)F)C=C3C=2)=N1 PAAZDMQPBVIYMF-OAHLLOKOSA-N 0.000 claims description 3
- RTLMTHJCAPGORR-QGZVFWFLSA-N ethyl 3-[(4r)-2-[7-(cyclopentylamino)-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound CCOC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(C)C=C3C=2)=N1 RTLMTHJCAPGORR-QGZVFWFLSA-N 0.000 claims description 3
- AUWZXVQIVPVJCT-LJQANCHMSA-N ethyl 3-[2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]-5-methylimidazole-4-carboxylate Chemical compound CCOC(=O)C1=C(C)N=CN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 AUWZXVQIVPVJCT-LJQANCHMSA-N 0.000 claims description 3
- 201000006370 kidney failure Diseases 0.000 claims description 3
- ITIBEUMDYKNRLW-CQSZACIVSA-N methyl 2-[(4r)-2-[5-methoxy-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(OC)C=C3C=2)=N1 ITIBEUMDYKNRLW-CQSZACIVSA-N 0.000 claims description 3
- PDLQARYLJBVLSM-CQSZACIVSA-N methyl 2-[(4r)-2-[7-(cyclopentylamino)-5-methoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC)C=C3C=2)=N1 PDLQARYLJBVLSM-CQSZACIVSA-N 0.000 claims description 3
- XZMBJFHTFJXFPN-QGZVFWFLSA-N methyl 2-[(4r)-2-[7-(oxan-4-ylamino)-5-pyridin-3-yloxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(OC=4C=NC=CC=4)C=C3C=2)=N1 XZMBJFHTFJXFPN-QGZVFWFLSA-N 0.000 claims description 3
- OEOLQZQKZZYEMA-VQTJNVASSA-N n-[(3s)-1-[2-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]pyrrolidin-3-yl]acetamide Chemical compound C1[C@@H](NC(=O)C)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)SC1 OEOLQZQKZZYEMA-VQTJNVASSA-N 0.000 claims description 3
- WTQOBEPDBMQZGD-OAHLLOKOSA-N n-cyclopentyl-2-[(4r)-4-(2-methoxyethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound COCC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 WTQOBEPDBMQZGD-OAHLLOKOSA-N 0.000 claims description 3
- CEMMHWUGNHZBQQ-LJQANCHMSA-N n-cyclopentyl-2-[(4r)-4-(2-piperidin-1-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C([C@H]1N=C(SC1)C=1NC2=C(NC3CCCC3)C=CC=C2C=1)CN1CCCCC1 CEMMHWUGNHZBQQ-LJQANCHMSA-N 0.000 claims description 3
- VGPMTLJNNYOWED-MRXNPFEDSA-N n-cyclopentyl-2-[(4r)-4-[2-(dimethylamino)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound CN(C)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 VGPMTLJNNYOWED-MRXNPFEDSA-N 0.000 claims description 3
- VZMORDAZACZUHU-CQSZACIVSA-N n-methyl-2-[(4r)-2-[7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetamide Chemical compound CNC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=CC=C3C=2)=N1 VZMORDAZACZUHU-CQSZACIVSA-N 0.000 claims description 3
- 150000002825 nitriles Chemical class 0.000 claims description 3
- 235000020824 obesity Nutrition 0.000 claims description 3
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 claims description 3
- 229940080818 propionamide Drugs 0.000 claims description 3
- WNTQGLURLUEVTM-GOSISDBHSA-N (4r)-2-(7-phenoxy-1h-indol-2-yl)-4-(2-pyrrolidin-1-ylethyl)-4,5-dihydro-1,3-thiazole Chemical compound C([C@H]1N=C(SC1)C=1NC2=C(OC=3C=CC=CC=3)C=CC=C2C=1)CN1CCCC1 WNTQGLURLUEVTM-GOSISDBHSA-N 0.000 claims description 2
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims description 2
- NVSFJTBVHCYHKT-LJQANCHMSA-N 1-(4-acetylpiperazin-1-yl)-2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanone Chemical compound C1CN(C(=O)C)CCN1C(=O)C[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)SC1 NVSFJTBVHCYHKT-LJQANCHMSA-N 0.000 claims description 2
- OGNTVOHERFTPHV-QGZVFWFLSA-N 1-[2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]pyrrolidin-2-one Chemical compound C=12NC(C=3SC[C@@H](CCN4C(CCC4)=O)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 OGNTVOHERFTPHV-QGZVFWFLSA-N 0.000 claims description 2
- VXOQSLZAKKUYJW-JOCHJYFZSA-N 1-[4-[2-[(4r)-2-[7-(cyclopentylamino)-5-(methylsulfonylmethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-1-yl]ethanone Chemical compound C1CN(C(=O)C)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(CS(C)(=O)=O)C=C3C=2)SC1 VXOQSLZAKKUYJW-JOCHJYFZSA-N 0.000 claims description 2
- BOPWVEFNKJHQRB-LJQANCHMSA-N 1-[4-[2-[(4r)-2-[7-(cyclopentylamino)-5-methoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-1-yl]-2-hydroxyethanone Chemical compound C=12NC(C=3SC[C@@H](CCN4CCN(CC4)C(=O)CO)N=3)=CC2=CC(OC)=CC=1NC1CCCC1 BOPWVEFNKJHQRB-LJQANCHMSA-N 0.000 claims description 2
- KDISMIMTGUMORD-UHFFFAOYSA-N 1-acetylpiperidine Chemical compound CC(=O)N1CCCCC1 KDISMIMTGUMORD-UHFFFAOYSA-N 0.000 claims description 2
- GMQPIGJXSZAXSJ-UHFFFAOYSA-N 1-methylsulfonylpyrrolidine Chemical compound CS(=O)(=O)N1CCCC1 GMQPIGJXSZAXSJ-UHFFFAOYSA-N 0.000 claims description 2
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Chemical compound C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 claims description 2
- HGDLYINUPQPJEP-CYBMUJFWSA-N 2-[(4r)-2-(7-phenoxy-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(OC=4C=CC=CC=4)C=CC=C3C=2)=N1 HGDLYINUPQPJEP-CYBMUJFWSA-N 0.000 claims description 2
- ZUWKEEMNYNQXOK-TZHYSIJRSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-(3-hydroxypyrrolidin-1-yl)ethanone Chemical compound C1C(O)CCN1C(=O)C[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 ZUWKEEMNYNQXOK-TZHYSIJRSA-N 0.000 claims description 2
- HSKBFJODQMFOIH-GOSISDBHSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-(4-methylpiperazin-1-yl)ethanone Chemical compound C1CN(C)CCN1C(=O)C[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 HSKBFJODQMFOIH-GOSISDBHSA-N 0.000 claims description 2
- PKZUNHKAAWULMY-GOSISDBHSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-piperidin-1-ylethanone Chemical compound C=12NC(C=3SC[C@@H](CC(=O)N4CCCCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 PKZUNHKAAWULMY-GOSISDBHSA-N 0.000 claims description 2
- AKJCQAIKURYVRF-CYBMUJFWSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 AKJCQAIKURYVRF-CYBMUJFWSA-N 0.000 claims description 2
- OOKSGEKSARLCJO-QGZVFWFLSA-N 2-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-morpholin-4-ylethanone Chemical compound C=12NC(C=3SC[C@@H](CC(=O)N4CCOCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCOCC1 OOKSGEKSARLCJO-QGZVFWFLSA-N 0.000 claims description 2
- WISKYOCMGBKSTL-CYBMUJFWSA-N 2-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)=N1 WISKYOCMGBKSTL-CYBMUJFWSA-N 0.000 claims description 2
- IMPVOZJUDYSWHF-CQSZACIVSA-N 2-[(4r)-2-[5-methyl-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@@H](CC(O)=O)N=3)=CC2=CC(C)=CC=1NC1CCOCC1 IMPVOZJUDYSWHF-CQSZACIVSA-N 0.000 claims description 2
- AEDZMPVAESRWEP-QGZVFWFLSA-N 2-[(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-morpholin-4-ylethanone Chemical compound C([C@H](N=1)CC(=O)N2CCOCC2)SC=1C(NC1=2)=CC1=CC=CC=2NC1CCCC1 AEDZMPVAESRWEP-QGZVFWFLSA-N 0.000 claims description 2
- JXKGDIRTJVYORJ-OAHLLOKOSA-N 2-[(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n-ethylacetamide Chemical compound CCNC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 JXKGDIRTJVYORJ-OAHLLOKOSA-N 0.000 claims description 2
- XZRCKUSTRUUVJF-MRXNPFEDSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-(methylsulfonylmethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound C=12NC(C=3SC[C@@H](CCO)N=3)=CC2=CC(CS(=O)(=O)C)=CC=1NC1CCCC1 XZRCKUSTRUUVJF-MRXNPFEDSA-N 0.000 claims description 2
- DVFHISWYLNRPHY-GOSISDBHSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-(4-methylpiperazin-1-yl)ethanone Chemical compound C1CN(C)CCN1C(=O)C[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)SC1 DVFHISWYLNRPHY-GOSISDBHSA-N 0.000 claims description 2
- LYBAMZQSMJNDIY-LJQANCHMSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n-(2-morpholin-4-ylethyl)acetamide Chemical compound C=12NC(C=3SC[C@@H](CC(=O)NCCN4CCOCC4)N=3)=CC2=CC(F)=CC=1NC1CCCC1 LYBAMZQSMJNDIY-LJQANCHMSA-N 0.000 claims description 2
- ICJKSONIYVQJLC-CQSZACIVSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound OCC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 ICJKSONIYVQJLC-CQSZACIVSA-N 0.000 claims description 2
- JPURYDFXLFVCIJ-QGZVFWFLSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)=N1 JPURYDFXLFVCIJ-QGZVFWFLSA-N 0.000 claims description 2
- UAWNTRFHTIPPOZ-CQSZACIVSA-N 2-[(4r)-2-[7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound OCC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=CC=C3C=2)=N1 UAWNTRFHTIPPOZ-CQSZACIVSA-N 0.000 claims description 2
- JRPUXGVBCFOJNE-MRXNPFEDSA-N 2-[(4r)-2-[7-(oxan-4-ylamino)-5-pyridin-3-yloxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(OC=4C=NC=CC=4)C=C3C=2)=N1 JRPUXGVBCFOJNE-MRXNPFEDSA-N 0.000 claims description 2
- RUUFMHOJJBHODO-CQSZACIVSA-N 2-[(4r)-4-(2-aminoethyl)-4,5-dihydro-1,3-thiazol-2-yl]-n-cyclopentyl-5-fluoro-1h-indol-7-amine Chemical compound NCC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 RUUFMHOJJBHODO-CQSZACIVSA-N 0.000 claims description 2
- JKMDCHQVOUVOPL-OAHLLOKOSA-N 2-[(4r)-4-(2-methylsulfonylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-n-(oxan-4-yl)-1h-indol-7-amine Chemical compound CS(=O)(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=CC=C3C=2)=N1 JKMDCHQVOUVOPL-OAHLLOKOSA-N 0.000 claims description 2
- AKJCQAIKURYVRF-ZDUSSCGKSA-N 2-[(4s)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 AKJCQAIKURYVRF-ZDUSSCGKSA-N 0.000 claims description 2
- WISKYOCMGBKSTL-ZDUSSCGKSA-N 2-[(4s)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)=N1 WISKYOCMGBKSTL-ZDUSSCGKSA-N 0.000 claims description 2
- FBPKPRZFVXTWQN-ABLWVSNPSA-N 2-[(4s)-2-[5-methyl-7-(oxolan-3-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@H](CC(O)=O)N=3)=CC2=CC(C)=CC=1NC1CCOC1 FBPKPRZFVXTWQN-ABLWVSNPSA-N 0.000 claims description 2
- OXOLNQUKGFLRSA-ZDUSSCGKSA-N 2-[(4s)-2-[7-(cyclobutylamino)-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@H](CC(O)=O)N=3)=CC2=CC(C)=CC=1NC1CCC1 OXOLNQUKGFLRSA-ZDUSSCGKSA-N 0.000 claims description 2
- BUKKUUIIFNRZSS-ZDUSSCGKSA-N 2-[(4s)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 BUKKUUIIFNRZSS-ZDUSSCGKSA-N 0.000 claims description 2
- JPURYDFXLFVCIJ-KRWDZBQOSA-N 2-[(4s)-2-[7-(cyclopentylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)=N1 JPURYDFXLFVCIJ-KRWDZBQOSA-N 0.000 claims description 2
- KLSLWDLSIWCTDG-ZDUSSCGKSA-N 2-[(4s)-2-[7-(cyclopropylmethylamino)-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@H](CC(O)=O)N=3)=CC2=CC(C)=CC=1NCC1CC1 KLSLWDLSIWCTDG-ZDUSSCGKSA-N 0.000 claims description 2
- PRHPRWULEUZEID-ZDUSSCGKSA-N 2-[(4s)-2-[7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@H]1CSC(C=2NC3=C(NC4CCOCC4)C=CC=C3C=2)=N1 PRHPRWULEUZEID-ZDUSSCGKSA-N 0.000 claims description 2
- BNEJVZQAPCGPTC-KRWDZBQOSA-N 2-[(4s)-2-[7-(oxan-4-ylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)=N1 BNEJVZQAPCGPTC-KRWDZBQOSA-N 0.000 claims description 2
- RSILHZIDSLVYOE-AWEZNQCLSA-N 2-[(4s)-2-[7-[(4,4-difluorocyclohexyl)amino]-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@H](CC(O)=O)N=3)=CC2=CC(C)=CC=1NC1CCC(F)(F)CC1 RSILHZIDSLVYOE-AWEZNQCLSA-N 0.000 claims description 2
- ZKEAARRZKOMPLD-LJAQVGFWSA-N 2-[(4s)-4-[2-(4-benzylpiperazin-1-yl)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-n-cyclopentyl-5-phenoxy-1h-indol-7-amine Chemical compound C([C@@H]1N=C(SC1)C=1NC2=C(NC3CCCC3)C=C(OC=3C=CC=CC=3)C=C2C=1)CN(CC1)CCN1CC1=CC=CC=C1 ZKEAARRZKOMPLD-LJAQVGFWSA-N 0.000 claims description 2
- GFRKCUZZPPNUQE-AEFFLSMTSA-N 2-[(4s)-4-[2-[(3r)-3-aminopyrrolidin-1-yl]ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-5-chloro-n-cyclopentyl-1h-indol-7-amine Chemical compound C1[C@H](N)CCN1CC[C@@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 GFRKCUZZPPNUQE-AEFFLSMTSA-N 0.000 claims description 2
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 claims description 2
- GUFDLJIHPZVQAL-OAHLLOKOSA-N 3-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propan-1-ol Chemical compound OCCC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 GUFDLJIHPZVQAL-OAHLLOKOSA-N 0.000 claims description 2
- NLNTWGBEWGJHRY-CQSZACIVSA-N 3-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound OC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 NLNTWGBEWGJHRY-CQSZACIVSA-N 0.000 claims description 2
- ZEYLFBUGYITYNZ-CQSZACIVSA-N 3-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound OC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)=N1 ZEYLFBUGYITYNZ-CQSZACIVSA-N 0.000 claims description 2
- UYDANJMZZNMWDI-OAHLLOKOSA-N 3-[(4r)-2-[7-(cyclopentylamino)-5-ethoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound C=12NC(C=3SC[C@@H](CCC(O)=O)N=3)=CC2=CC(OCC)=CC=1NC1CCCC1 UYDANJMZZNMWDI-OAHLLOKOSA-N 0.000 claims description 2
- IJKBLVUAVNAXLP-CQSZACIVSA-N 3-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound OC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 IJKBLVUAVNAXLP-CQSZACIVSA-N 0.000 claims description 2
- KOBDFYZKXUWLRZ-OAHLLOKOSA-N 3-[(4r)-2-[7-(cyclopentylamino)-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoic acid Chemical compound C=12NC(C=3SC[C@@H](CCC(O)=O)N=3)=CC2=CC(C)=CC=1NC1CCCC1 KOBDFYZKXUWLRZ-OAHLLOKOSA-N 0.000 claims description 2
- CGHUTNQOIIDFJE-NRFANRHFSA-N 4-[2-[(4s)-2-[7-(cyclopentylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-2-one Chemical compound C1CNC(=O)CN1CC[C@@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)SC1 CGHUTNQOIIDFJE-NRFANRHFSA-N 0.000 claims description 2
- QDWGBWJTJLBSIT-OAQYLSRUSA-N 5-chloro-n-(oxan-4-yl)-2-[(4r)-4-[2-(4-pyrimidin-2-ylpiperazin-1-yl)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCN(CC4)C=4N=CC=CN=4)N=3)=CC2=CC(Cl)=CC=1NC1CCOCC1 QDWGBWJTJLBSIT-OAQYLSRUSA-N 0.000 claims description 2
- XTXQOUDFSHKZTA-OAHLLOKOSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-(2-methylsulfonylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound CS(=O)(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 XTXQOUDFSHKZTA-OAHLLOKOSA-N 0.000 claims description 2
- CHAFDQLKRURXLK-LJQANCHMSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-(2-piperidin-1-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCCCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 CHAFDQLKRURXLK-LJQANCHMSA-N 0.000 claims description 2
- MOSKTDWRZQHWIR-VQTJNVASSA-N 5-chloro-n-cyclopentyl-2-[(4s)-4-[2-[(3r)-3-(dimethylamino)pyrrolidin-1-yl]ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C1[C@H](N(C)C)CCN1CC[C@@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 MOSKTDWRZQHWIR-VQTJNVASSA-N 0.000 claims description 2
- HMHXVZNOKBRDIC-GOSISDBHSA-N 5-fluoro-n-(oxan-4-yl)-2-[(4r)-4-(2-pyrrolidin-1-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCCC4)N=3)=CC2=CC(F)=CC=1NC1CCOCC1 HMHXVZNOKBRDIC-GOSISDBHSA-N 0.000 claims description 2
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 claims description 2
- YPWFISCTZQNZAU-UHFFFAOYSA-N Thiane Chemical compound C1CCSCC1 YPWFISCTZQNZAU-UHFFFAOYSA-N 0.000 claims description 2
- QYQHMTBXLBCVSW-LEWJYISDSA-N [(2r)-1-[2-[(4s)-2-[5-methyl-7-(oxan-4-ylmethylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]pyrrolidin-2-yl]methanol Chemical compound C=12NC(C=3SC[C@H](CCN4[C@H](CCC4)CO)N=3)=CC2=CC(C)=CC=1NCC1CCOCC1 QYQHMTBXLBCVSW-LEWJYISDSA-N 0.000 claims description 2
- OBPQXHIXHPEZKD-GOSISDBHSA-N [(4r)-2-[5-(morpholin-4-ylmethyl)-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(CN4CCOCC4)C=C3C=2)=N1 OBPQXHIXHPEZKD-GOSISDBHSA-N 0.000 claims description 2
- UKGQVVKDULGYLP-CYBMUJFWSA-N [(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 UKGQVVKDULGYLP-CYBMUJFWSA-N 0.000 claims description 2
- KZRVHYKRYDOGCF-CQSZACIVSA-N [(4r)-2-[7-(cyclopentylamino)-5-(hydroxymethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(CO)C=C3C=2)=N1 KZRVHYKRYDOGCF-CQSZACIVSA-N 0.000 claims description 2
- HPDNCJPIRIPRBE-CYBMUJFWSA-N [(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 HPDNCJPIRIPRBE-CYBMUJFWSA-N 0.000 claims description 2
- MVSFJSJYGUKSPE-MRXNPFEDSA-N [(4r)-2-[7-(oxan-4-ylamino)-5-pyridin-3-yloxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(OC=4C=NC=CC=4)C=C3C=2)=N1 MVSFJSJYGUKSPE-MRXNPFEDSA-N 0.000 claims description 2
- ONXCLZXWCPPOSF-SUHMBNCMSA-N [4-[2-[(4s)-2-[7-(cyclopentylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-1-yl]-(oxolan-2-yl)methanone Chemical compound C1CN(CC[C@@H]2N=C(SC2)C=2NC3=C(NC4CCCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)CCN1C(=O)C1CCCO1 ONXCLZXWCPPOSF-SUHMBNCMSA-N 0.000 claims description 2
- WCVXIPQTPMZXOO-OAHLLOKOSA-N ethyl 2-[(4r)-2-[7-(cyclopentylamino)-5-methoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound CCOC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC)C=C3C=2)=N1 WCVXIPQTPMZXOO-OAHLLOKOSA-N 0.000 claims description 2
- XNDKIXLBTURDKG-QGZVFWFLSA-N ethyl 3-[(4r)-2-[7-(cyclopentylamino)-5-ethoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound CCOC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OCC)C=C3C=2)=N1 XNDKIXLBTURDKG-QGZVFWFLSA-N 0.000 claims description 2
- YFLJBPSBONFFGA-MRXNPFEDSA-N ethyl 3-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound CCOC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 YFLJBPSBONFFGA-MRXNPFEDSA-N 0.000 claims description 2
- 125000001153 fluoro group Chemical group F* 0.000 claims description 2
- IFMHHDLVPNMFDM-CQSZACIVSA-N methyl 2-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)=N1 IFMHHDLVPNMFDM-CQSZACIVSA-N 0.000 claims description 2
- BHHWDQNDDXFSBY-HXUWFJFHSA-N methyl 2-[(4r)-2-[7-(cyclopentylamino)-5-(phenoxymethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(COC=4C=CC=CC=4)C=C3C=2)=N1 BHHWDQNDDXFSBY-HXUWFJFHSA-N 0.000 claims description 2
- MDEHTEXRKNQTKB-LJQANCHMSA-N methyl 2-[(4r)-2-[7-(cyclopentylamino)-5-(pyrrolidin-1-ylmethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(CN4CCCC4)C=C3C=2)=N1 MDEHTEXRKNQTKB-LJQANCHMSA-N 0.000 claims description 2
- BXJGTPXAZFNJOI-QGZVFWFLSA-N methyl 2-[(4r)-2-[7-(cyclopentylamino)-5-pyridin-3-yloxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC=4C=NC=CC=4)C=C3C=2)=N1 BXJGTPXAZFNJOI-QGZVFWFLSA-N 0.000 claims description 2
- ADYYTORVODGGNU-HNNXBMFYSA-N methyl 2-[(4s)-2-[7-(cyclopentylamino)-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(C)C=C3C=2)=N1 ADYYTORVODGGNU-HNNXBMFYSA-N 0.000 claims description 2
- IMAKHNTVDGLIRY-UHFFFAOYSA-N methyl prop-2-ynoate Chemical compound COC(=O)C#C IMAKHNTVDGLIRY-UHFFFAOYSA-N 0.000 claims description 2
- XGXUTMSHYCOHAW-MRXNPFEDSA-N n,n-dimethyl-2-[(4r)-2-(7-phenoxy-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]ethanamine Chemical compound CN(C)CC[C@@H]1CSC(C=2NC3=C(OC=4C=CC=CC=4)C=CC=C3C=2)=N1 XGXUTMSHYCOHAW-MRXNPFEDSA-N 0.000 claims description 2
- MZVCOZSKRPPBPU-SFHVURJKSA-N n-(4,4-difluorocyclohexyl)-2-[(4s)-4-(2-morpholin-4-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C1CC(F)(F)CCC1NC1=CC=CC2=C1NC(C=1SC[C@H](CCN3CCOCC3)N=1)=C2 MZVCOZSKRPPBPU-SFHVURJKSA-N 0.000 claims description 2
- GESREBYZPQEXFY-QGZVFWFLSA-N n-cyclopentyl-2-[(4r)-4-(morpholin-4-ylmethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C([C@H](N=1)CN2CCOCC2)SC=1C(NC1=2)=CC1=CC=CC=2NC1CCCC1 GESREBYZPQEXFY-QGZVFWFLSA-N 0.000 claims description 2
- OEYSACMTSFPEIU-GOSISDBHSA-N n-cyclopentyl-2-[(4r)-4-[(3-cyclopentyl-1,2,4-oxadiazol-5-yl)methyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C([C@H](N=1)CC=2ON=C(N=2)C2CCCC2)SC=1C(NC1=2)=CC1=CC=CC=2NC1CCCC1 OEYSACMTSFPEIU-GOSISDBHSA-N 0.000 claims description 2
- GFQWEJAHDKYMRD-OAHLLOKOSA-N n-cyclopentyl-5-fluoro-2-[(4r)-4-(2-methylsulfonylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound CS(=O)(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 GFQWEJAHDKYMRD-OAHLLOKOSA-N 0.000 claims description 2
- YFUXYZCFYRUGEN-QFIPXVFZSA-N n-cyclopentyl-5-phenoxy-2-[(4s)-4-(2-piperazin-1-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C([C@@H]1N=C(SC1)C=1NC2=C(NC3CCCC3)C=C(OC=3C=CC=CC=3)C=C2C=1)CN1CCNCC1 YFUXYZCFYRUGEN-QFIPXVFZSA-N 0.000 claims description 2
- HARQVKPQRZPZTO-QFIPXVFZSA-N n-cyclopentyl-5-phenoxy-2-[(4s)-4-(2-pyrrolidin-1-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C([C@@H]1N=C(SC1)C=1NC2=C(NC3CCCC3)C=C(OC=3C=CC=CC=3)C=C2C=1)CN1CCCC1 HARQVKPQRZPZTO-QFIPXVFZSA-N 0.000 claims description 2
- AHQLZUZQHIPPLI-SANMLTNESA-N n-cyclopentyl-5-phenoxy-2-[(4s)-4-[2-(4-pyridin-2-ylpiperazin-1-yl)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C([C@@H]1N=C(SC1)C=1NC2=C(NC3CCCC3)C=C(OC=3C=CC=CC=3)C=C2C=1)CN(CC1)CCN1C1=CC=CC=N1 AHQLZUZQHIPPLI-SANMLTNESA-N 0.000 claims description 2
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 claims description 2
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 claims 1
- WHQLFLQXEGWMQJ-QGZVFWFLSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-1-morpholin-4-ylethanone Chemical compound C=12NC(C=3SC[C@@H](CC(=O)N4CCOCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 WHQLFLQXEGWMQJ-QGZVFWFLSA-N 0.000 claims 1
- MXNPEJZZTUYYOH-LJQANCHMSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n-(2-morpholin-4-ylethyl)acetamide Chemical compound C=12NC(C=3SC[C@@H](CC(=O)NCCN4CCOCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 MXNPEJZZTUYYOH-LJQANCHMSA-N 0.000 claims 1
- XRQOWZOBMHKRIQ-CQSZACIVSA-N 2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n-methylacetamide Chemical compound CNC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)=N1 XRQOWZOBMHKRIQ-CQSZACIVSA-N 0.000 claims 1
- YGQUQSWUCQFWGS-QGZVFWFLSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-(4-methylsulfonylphenoxy)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C1=CC(S(=O)(=O)C)=CC=C1OC1=CC(NC2CCCC2)=C(NC(=C2)C=3SC[C@@H](CC(O)=O)N=3)C2=C1 YGQUQSWUCQFWGS-QGZVFWFLSA-N 0.000 claims 1
- IMPVOZJUDYSWHF-AWEZNQCLSA-N 2-[(4s)-2-[5-methyl-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C=12NC(C=3SC[C@H](CC(O)=O)N=3)=CC2=CC(C)=CC=1NC1CCOCC1 IMPVOZJUDYSWHF-AWEZNQCLSA-N 0.000 claims 1
- BVSHIKSZLVZATA-OAHLLOKOSA-N 3-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propan-1-ol Chemical compound OCCC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)=N1 BVSHIKSZLVZATA-OAHLLOKOSA-N 0.000 claims 1
- VBPWELMGFRRLGW-GOSISDBHSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-(2-piperazin-1-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCNCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 VBPWELMGFRRLGW-GOSISDBHSA-N 0.000 claims 1
- 230000001272 neurogenic effect Effects 0.000 claims 1
- 125000004495 thiazol-4-yl group Chemical group S1C=NC(=C1)* 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 10
- 230000001747 exhibiting effect Effects 0.000 abstract description 2
- 238000002360 preparation method Methods 0.000 description 373
- 230000015572 biosynthetic process Effects 0.000 description 259
- 238000003786 synthesis reaction Methods 0.000 description 259
- 238000006243 chemical reaction Methods 0.000 description 251
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 238
- 238000000034 method Methods 0.000 description 236
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 225
- 238000005160 1H NMR spectroscopy Methods 0.000 description 212
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 139
- 239000000243 solution Substances 0.000 description 136
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 96
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 90
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 85
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 82
- 235000019439 ethyl acetate Nutrition 0.000 description 80
- 230000002829 reductive effect Effects 0.000 description 79
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 76
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 75
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 66
- 238000004440 column chromatography Methods 0.000 description 64
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 62
- 239000000706 filtrate Substances 0.000 description 58
- 239000002585 base Substances 0.000 description 55
- 239000002253 acid Substances 0.000 description 52
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 40
- 229960004756 ethanol Drugs 0.000 description 38
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 38
- 229960000583 acetic acid Drugs 0.000 description 35
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 34
- 230000035484 reaction time Effects 0.000 description 33
- 229960004132 diethyl ether Drugs 0.000 description 28
- 239000012442 inert solvent Substances 0.000 description 27
- 239000007787 solid Substances 0.000 description 26
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 25
- 230000008569 process Effects 0.000 description 24
- 101150041968 CDC13 gene Proteins 0.000 description 23
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 23
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 21
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 19
- 150000002148 esters Chemical class 0.000 description 19
- 239000008103 glucose Substances 0.000 description 19
- 229910000027 potassium carbonate Inorganic materials 0.000 description 19
- PKDPUENCROCRCH-UHFFFAOYSA-N 1-piperazin-1-ylethanone Chemical compound CC(=O)N1CCNCC1 PKDPUENCROCRCH-UHFFFAOYSA-N 0.000 description 18
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 18
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 16
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 16
- 150000004702 methyl esters Chemical class 0.000 description 16
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Substances CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 15
- 239000003795 chemical substances by application Substances 0.000 description 15
- 229940095574 propionic acid Drugs 0.000 description 15
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 14
- 239000008280 blood Substances 0.000 description 14
- 210000004369 blood Anatomy 0.000 description 14
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 14
- 150000002170 ethers Chemical class 0.000 description 14
- 229940052303 ethers for general anesthesia Drugs 0.000 description 14
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 13
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 13
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 13
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 13
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 13
- 239000002904 solvent Substances 0.000 description 13
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 238000005859 coupling reaction Methods 0.000 description 12
- 229910052751 metal Inorganic materials 0.000 description 12
- 239000002184 metal Substances 0.000 description 12
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 11
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 11
- 125000005233 alkylalcohol group Chemical group 0.000 description 11
- 125000003277 amino group Chemical group 0.000 description 11
- 229920006395 saturated elastomer Polymers 0.000 description 11
- 235000011054 acetic acid Nutrition 0.000 description 10
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical group [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 10
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 239000007822 coupling agent Substances 0.000 description 9
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium;hydroxide;hydrate Chemical compound [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 9
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 9
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 9
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 9
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 8
- APWRZPQBPCAXFP-UHFFFAOYSA-N 1-(1-oxo-2H-isoquinolin-5-yl)-5-(trifluoromethyl)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrazole-4-carboxamide Chemical compound O=C1NC=CC2=C(C=CC=C12)N1N=CC(=C1C(F)(F)F)C(=O)NC1=CC(=NC=C1)C(F)(F)F APWRZPQBPCAXFP-UHFFFAOYSA-N 0.000 description 8
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 8
- 239000012448 Lithium borohydride Substances 0.000 description 8
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 8
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 8
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 125000003158 alcohol group Chemical group 0.000 description 8
- 235000019270 ammonium chloride Nutrition 0.000 description 8
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 8
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 8
- 239000001632 sodium acetate Substances 0.000 description 8
- 235000017281 sodium acetate Nutrition 0.000 description 8
- 229910000104 sodium hydride Inorganic materials 0.000 description 8
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 8
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 8
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 7
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 7
- 239000003638 chemical reducing agent Substances 0.000 description 7
- 150000001989 diazonium salts Chemical class 0.000 description 7
- 238000002474 experimental method Methods 0.000 description 7
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 7
- 150000007530 organic bases Chemical class 0.000 description 7
- 239000012312 sodium hydride Substances 0.000 description 7
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 7
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 6
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 6
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 6
- 239000012346 acetyl chloride Substances 0.000 description 6
- 239000003377 acid catalyst Substances 0.000 description 6
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 6
- 229960002433 cysteine Drugs 0.000 description 6
- 125000004494 ethyl ester group Chemical group 0.000 description 6
- 210000004185 liver Anatomy 0.000 description 6
- JMJRYTGVHCAYCT-UHFFFAOYSA-N oxan-4-one Chemical compound O=C1CCOCC1 JMJRYTGVHCAYCT-UHFFFAOYSA-N 0.000 description 6
- 229920000137 polyphosphoric acid Polymers 0.000 description 6
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 6
- PTDVPWWJRCOIIO-UHFFFAOYSA-N (4-methoxyphenyl)methanethiol Chemical compound COC1=CC=C(CS)C=C1 PTDVPWWJRCOIIO-UHFFFAOYSA-N 0.000 description 5
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical group COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 5
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 5
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 5
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 5
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 5
- XIWBSOUNZWSFKU-UHFFFAOYSA-N ethyl piperidine-3-carboxylate Chemical compound CCOC(=O)C1CCCNC1 XIWBSOUNZWSFKU-UHFFFAOYSA-N 0.000 description 5
- 230000004153 glucose metabolism Effects 0.000 description 5
- 238000006460 hydrolysis reaction Methods 0.000 description 5
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 5
- 229910000000 metal hydroxide Inorganic materials 0.000 description 5
- 150000004692 metal hydroxides Chemical class 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 238000006722 reduction reaction Methods 0.000 description 5
- 238000007363 ring formation reaction Methods 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 239000012279 sodium borohydride Substances 0.000 description 5
- 229910000033 sodium borohydride Inorganic materials 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 4
- 0 C*(CNCC1)CS1(=O)=O Chemical compound C*(CNCC1)CS1(=O)=O 0.000 description 4
- NBSCHQHZLSJFNQ-GASJEMHNSA-N D-Glucose 6-phosphate Chemical compound OC1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H](O)[C@H]1O NBSCHQHZLSJFNQ-GASJEMHNSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- VFRROHXSMXFLSN-UHFFFAOYSA-N Glc6P Natural products OP(=O)(O)OCC(O)C(O)C(O)C(O)C=O VFRROHXSMXFLSN-UHFFFAOYSA-N 0.000 description 4
- 102000004877 Insulin Human genes 0.000 description 4
- 108090001061 Insulin Proteins 0.000 description 4
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 150000001348 alkyl chlorides Chemical class 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 239000007853 buffer solution Substances 0.000 description 4
- 229910000024 caesium carbonate Inorganic materials 0.000 description 4
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 4
- 235000018417 cysteine Nutrition 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 230000002255 enzymatic effect Effects 0.000 description 4
- 125000004185 ester group Chemical group 0.000 description 4
- NNZMPPYNNJODFX-UHFFFAOYSA-N ethyl 7-nitro-5-pyridin-3-yloxy-1h-indole-2-carboxylate Chemical compound C=1C([N+]([O-])=O)=C2NC(C(=O)OCC)=CC2=CC=1OC1=CC=CN=C1 NNZMPPYNNJODFX-UHFFFAOYSA-N 0.000 description 4
- 230000014101 glucose homeostasis Effects 0.000 description 4
- 210000003494 hepatocyte Anatomy 0.000 description 4
- OAKJQQAXSVQMHS-UHFFFAOYSA-N hydrazine group Chemical group NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 4
- 229940125396 insulin Drugs 0.000 description 4
- 230000003914 insulin secretion Effects 0.000 description 4
- 238000012423 maintenance Methods 0.000 description 4
- 238000001819 mass spectrum Methods 0.000 description 4
- 229940098779 methanesulfonic acid Drugs 0.000 description 4
- KDHSUTRVEOAJBH-UHFFFAOYSA-N methyl 5-bromo-7-nitro-1h-indole-2-carboxylate Chemical compound BrC1=CC([N+]([O-])=O)=C2NC(C(=O)OC)=CC2=C1 KDHSUTRVEOAJBH-UHFFFAOYSA-N 0.000 description 4
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 239000011368 organic material Substances 0.000 description 4
- IWELDVXSEVIIGI-UHFFFAOYSA-N piperazin-2-one Chemical compound O=C1CNCCN1 IWELDVXSEVIIGI-UHFFFAOYSA-N 0.000 description 4
- VWDWKYIASSYTQR-UHFFFAOYSA-N sodium nitrate Chemical compound [Na+].[O-][N+]([O-])=O VWDWKYIASSYTQR-UHFFFAOYSA-N 0.000 description 4
- LYPGDCWPTHTUDO-UHFFFAOYSA-M sodium;methanesulfinate Chemical compound [Na+].CS([O-])=O LYPGDCWPTHTUDO-UHFFFAOYSA-M 0.000 description 4
- 239000011877 solvent mixture Substances 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- NDOVLWQBFFJETK-UHFFFAOYSA-N 1,4-thiazinane 1,1-dioxide Chemical compound O=S1(=O)CCNCC1 NDOVLWQBFFJETK-UHFFFAOYSA-N 0.000 description 3
- YLPRHFLWIYEAGT-UHFFFAOYSA-N 4-(4-methylsulfonylphenoxy)aniline Chemical compound C1=CC(S(=O)(=O)C)=CC=C1OC1=CC=C(N)C=C1 YLPRHFLWIYEAGT-UHFFFAOYSA-N 0.000 description 3
- ZSLIXJKSPVCNHZ-UHFFFAOYSA-N 4-pyridin-3-yloxyaniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CN=C1 ZSLIXJKSPVCNHZ-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 102100040880 Glucokinase regulatory protein Human genes 0.000 description 3
- 101710148430 Glucokinase regulatory protein Proteins 0.000 description 3
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 3
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 3
- 238000002835 absorbance Methods 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 3
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000012954 diazonium Substances 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 230000010030 glucose lowering effect Effects 0.000 description 3
- 230000004190 glucose uptake Effects 0.000 description 3
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- OWGXPBRLKFGUQB-UHFFFAOYSA-N methyl 7-nitro-1h-indole-2-carboxylate Chemical compound C1=CC([N+]([O-])=O)=C2NC(C(=O)OC)=CC2=C1 OWGXPBRLKFGUQB-UHFFFAOYSA-N 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 229910052759 nickel Inorganic materials 0.000 description 3
- 229910017604 nitric acid Inorganic materials 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- CLDWGXZGFUNWKB-UHFFFAOYSA-M silver;benzoate Chemical compound [Ag+].[O-]C(=O)C1=CC=CC=C1 CLDWGXZGFUNWKB-UHFFFAOYSA-M 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000006277 sulfonation reaction Methods 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 3
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- 238000013293 zucker diabetic fatty rat Methods 0.000 description 3
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 2
- XJRIDJAGAYGJCK-UHFFFAOYSA-N (1-acetyl-5-bromoindol-3-yl) acetate Chemical compound C1=C(Br)C=C2C(OC(=O)C)=CN(C(C)=O)C2=C1 XJRIDJAGAYGJCK-UHFFFAOYSA-N 0.000 description 2
- PQPZSPJVMUCVAQ-JTQLQIEISA-N (2r)-2-azaniumyl-3-[(4-methoxyphenyl)methylsulfanyl]propanoate Chemical compound COC1=CC=C(CSC[C@H](N)C(O)=O)C=C1 PQPZSPJVMUCVAQ-JTQLQIEISA-N 0.000 description 2
- HMLXAWGHSCTGQJ-UHFFFAOYSA-N (4-chloro-2-nitrophenyl)hydrazine Chemical compound NNC1=CC=C(Cl)C=C1[N+]([O-])=O HMLXAWGHSCTGQJ-UHFFFAOYSA-N 0.000 description 2
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 2
- PYADZVVITOSLGZ-LJQANCHMSA-N 1-[4-[2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-1-yl]-2-hydroxyethanone Chemical compound C1CN(C(=O)CO)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)SC1 PYADZVVITOSLGZ-LJQANCHMSA-N 0.000 description 2
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- WBOWIYTVTTUXMY-CQSZACIVSA-N 2-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n-methylacetamide Chemical compound CNC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)=N1 WBOWIYTVTTUXMY-CQSZACIVSA-N 0.000 description 2
- VKGVHIJUKWDPSY-OAHLLOKOSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl methanesulfonate Chemical compound CS(=O)(=O)OCC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 VKGVHIJUKWDPSY-OAHLLOKOSA-N 0.000 description 2
- IYFOLDCJEYIXGP-CQSZACIVSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-methoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n-methylacetamide Chemical compound CNC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC)C=C3C=2)=N1 IYFOLDCJEYIXGP-CQSZACIVSA-N 0.000 description 2
- GGVDLEGJMVIZNZ-CQSZACIVSA-N 2-[[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methoxy]acetic acid Chemical compound OC(=O)COC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)=N1 GGVDLEGJMVIZNZ-CQSZACIVSA-N 0.000 description 2
- TXASRMXSWLNSLD-LHIURRSHSA-N 2-[[2-[(4r)-4-(hydroxymethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-yl]amino]propanoic acid Chemical compound N1C=2C(NC(C)C(O)=O)=CC=CC=2C=C1C1=N[C@H](CO)CS1 TXASRMXSWLNSLD-LHIURRSHSA-N 0.000 description 2
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 2
- DLURHXYXQYMPLT-UHFFFAOYSA-N 2-nitro-p-toluidine Chemical compound CC1=CC=C(N)C([N+]([O-])=O)=C1 DLURHXYXQYMPLT-UHFFFAOYSA-N 0.000 description 2
- SRVXSISGYBMIHR-UHFFFAOYSA-N 3-[3-[3-(2-amino-2-oxoethyl)phenyl]-5-chlorophenyl]-3-(5-methyl-1,3-thiazol-2-yl)propanoic acid Chemical compound S1C(C)=CN=C1C(CC(O)=O)C1=CC(Cl)=CC(C=2C=C(CC(N)=O)C=CC=2)=C1 SRVXSISGYBMIHR-UHFFFAOYSA-N 0.000 description 2
- GRFNBEZIAWKNCO-UHFFFAOYSA-N 3-pyridinol Chemical compound OC1=CC=CN=C1 GRFNBEZIAWKNCO-UHFFFAOYSA-N 0.000 description 2
- MSHFRERJPWKJFX-UHFFFAOYSA-N 4-Methoxybenzyl alcohol Chemical compound COC1=CC=C(CO)C=C1 MSHFRERJPWKJFX-UHFFFAOYSA-N 0.000 description 2
- ISFYBUAVOZFROB-UHFFFAOYSA-N 4-ethoxy-2-nitroaniline Chemical compound CCOC1=CC=C(N)C([N+]([O-])=O)=C1 ISFYBUAVOZFROB-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- VXJVICSCLUFGGG-UHFFFAOYSA-N 5-(hydroxymethyl)-7-nitro-1h-indole-2-carboxylic acid Chemical compound OCC1=CC([N+]([O-])=O)=C2NC(C(O)=O)=CC2=C1 VXJVICSCLUFGGG-UHFFFAOYSA-N 0.000 description 2
- MCQQBEZMEXVVEL-GOSISDBHSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-(2-morpholin-4-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCOCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 MCQQBEZMEXVVEL-GOSISDBHSA-N 0.000 description 2
- IFYNIBBCCGUGRG-LJQANCHMSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-(3-morpholin-4-ylpropyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCCN4CCOCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 IFYNIBBCCGUGRG-LJQANCHMSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 2
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- 238000011740 C57BL/6 mouse Methods 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 208000000454 Congenital Hyperinsulinism Diseases 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 102000005548 Hexokinase Human genes 0.000 description 2
- 108700040460 Hexokinases Proteins 0.000 description 2
- 101001133924 Homo sapiens Gamma-glutamyl phosphate reductase Proteins 0.000 description 2
- 101000905392 Homo sapiens Glycerol kinase Proteins 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 206010022489 Insulin Resistance Diseases 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 235000013878 L-cysteine Nutrition 0.000 description 2
- 239000004201 L-cysteine Substances 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- 208000035180 MODY Diseases 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 2
- PMDCZENCAXMSOU-UHFFFAOYSA-N N-ethylacetamide Chemical compound CCNC(C)=O PMDCZENCAXMSOU-UHFFFAOYSA-N 0.000 description 2
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- FMGILQSRTUVOEB-LJQANCHMSA-N [(2r)-2-[[5-(hydroxymethyl)-7-nitro-1h-indole-2-carbonyl]amino]-3-[(4-methoxyphenyl)methylsulfanyl]propyl] 2,2-dimethylpropanoate Chemical compound C1=CC(OC)=CC=C1CSC[C@@H](COC(=O)C(C)(C)C)NC(=O)C1=CC2=CC(CO)=CC([N+]([O-])=O)=C2N1 FMGILQSRTUVOEB-LJQANCHMSA-N 0.000 description 2
- CDGGFTHRMTXKGS-LLVKDONJSA-N [(4r)-2-(7-amino-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]methyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OC[C@@H]1CSC(C=2NC3=C(N)C=CC=C3C=2)=N1 CDGGFTHRMTXKGS-LLVKDONJSA-N 0.000 description 2
- BJCCDIDCRGSUIK-MRXNPFEDSA-N [(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 BJCCDIDCRGSUIK-MRXNPFEDSA-N 0.000 description 2
- CGHKLPGZPWDMPO-CQSZACIVSA-N [(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methyl methanesulfonate Chemical compound CS(=O)(=O)OC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 CGHKLPGZPWDMPO-CQSZACIVSA-N 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 125000005907 alkyl ester group Chemical group 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 125000005605 benzo group Chemical group 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000012153 distilled water Substances 0.000 description 2
- SLUXJQCYWLQAAY-UHFFFAOYSA-N ethyl 2-[(4-fluoro-2-nitrophenyl)hydrazinylidene]propanoate Chemical compound CCOC(=O)C(C)=NNC1=CC=C(F)C=C1[N+]([O-])=O SLUXJQCYWLQAAY-UHFFFAOYSA-N 0.000 description 2
- 229940035423 ethyl ether Drugs 0.000 description 2
- 229940124828 glucokinase activator Drugs 0.000 description 2
- 230000009229 glucose formation Effects 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 230000002140 halogenating effect Effects 0.000 description 2
- 238000005658 halogenation reaction Methods 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- 230000003345 hyperglycaemic effect Effects 0.000 description 2
- 201000001421 hyperglycemia Diseases 0.000 description 2
- 208000033066 hyperinsulinemic hypoglycemia Diseases 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- OHZZTXYKLXZFSZ-UHFFFAOYSA-I manganese(3+) 5,10,15-tris(1-methylpyridin-1-ium-4-yl)-20-(1-methylpyridin-4-ylidene)porphyrin-22-ide pentachloride Chemical compound [Cl-].[Cl-].[Cl-].[Cl-].[Cl-].[Mn+3].C1=CN(C)C=CC1=C1C(C=C2)=NC2=C(C=2C=C[N+](C)=CC=2)C([N-]2)=CC=C2C(C=2C=C[N+](C)=CC=2)=C(C=C2)N=C2C(C=2C=C[N+](C)=CC=2)=C2N=C1C=C2 OHZZTXYKLXZFSZ-UHFFFAOYSA-I 0.000 description 2
- 201000006950 maturity-onset diabetes of the young Diseases 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- WBMKFAZWUVPTFD-NSHDSACASA-N methyl (2r)-2-amino-3-[(4-methoxyphenyl)methylsulfanyl]propanoate Chemical compound COC(=O)[C@@H](N)CSCC1=CC=C(OC)C=C1 WBMKFAZWUVPTFD-NSHDSACASA-N 0.000 description 2
- OYEFYPPGPGHHNX-AWEZNQCLSA-N methyl (2r)-3-[(4-methoxyphenyl)methylsulfanyl]-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound CC(C)(C)OC(=O)N[C@H](C(=O)OC)CSCC1=CC=C(OC)C=C1 OYEFYPPGPGHHNX-AWEZNQCLSA-N 0.000 description 2
- GAPPVHRJJGEYEL-UHFFFAOYSA-N methyl 2-[(4-chloro-2-nitrophenyl)hydrazinylidene]propanoate Chemical compound COC(=O)C(C)=NNC1=CC=C(Cl)C=C1[N+]([O-])=O GAPPVHRJJGEYEL-UHFFFAOYSA-N 0.000 description 2
- JHQJSOFNLZAANJ-SECBINFHSA-N methyl 2-[(4r)-2-(7-amino-5-bromo-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(N)C=C(Br)C=C3C=2)=N1 JHQJSOFNLZAANJ-SECBINFHSA-N 0.000 description 2
- QJIGYWXDKVZPHN-LLVKDONJSA-N methyl 2-[(4r)-2-[5-(methylsulfonylmethyl)-7-nitro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C([N+]([O-])=O)C=C(CS(C)(=O)=O)C=C3C=2)=N1 QJIGYWXDKVZPHN-LLVKDONJSA-N 0.000 description 2
- SSYXWSITBDEGIS-QGZVFWFLSA-N methyl 2-[(4r)-2-[7-(1,4-dioxaspiro[4.5]decan-8-ylamino)-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCC5(CC4)OCCO5)C=C(C)C=C3C=2)=N1 SSYXWSITBDEGIS-QGZVFWFLSA-N 0.000 description 2
- FDDMAVQFVJWNDE-CYBMUJFWSA-N methyl 2-[(4r)-2-[7-amino-5-(4-methylsulfonylphenoxy)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(N)C=C(OC=4C=CC(=CC=4)S(C)(=O)=O)C=C3C=2)=N1 FDDMAVQFVJWNDE-CYBMUJFWSA-N 0.000 description 2
- FGYZPCMEXFXZIP-UHFFFAOYSA-N methyl 5-(4-methylsulfonylphenoxy)-7-nitro-1h-indole-2-carboxylate Chemical compound C=1C([N+]([O-])=O)=C2NC(C(=O)OC)=CC2=CC=1OC1=CC=C(S(C)(=O)=O)C=C1 FGYZPCMEXFXZIP-UHFFFAOYSA-N 0.000 description 2
- VEZJPSMVGQWPNA-UHFFFAOYSA-N methyl 5-methoxy-7-nitro-1h-indole-2-carboxylate Chemical compound COC1=CC([N+]([O-])=O)=C2NC(C(=O)OC)=CC2=C1 VEZJPSMVGQWPNA-UHFFFAOYSA-N 0.000 description 2
- LPVNQMGIHZNRRW-UHFFFAOYSA-N methyl 5-methyl-7-nitro-1h-indole-2-carboxylate Chemical compound CC1=CC([N+]([O-])=O)=C2NC(C(=O)OC)=CC2=C1 LPVNQMGIHZNRRW-UHFFFAOYSA-N 0.000 description 2
- CWKLZLBVOJRSOM-UHFFFAOYSA-N methyl pyruvate Chemical compound COC(=O)C(C)=O CWKLZLBVOJRSOM-UHFFFAOYSA-N 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- FJABAFACLDVRTR-UHFFFAOYSA-N n'-hydroxycyclopentanecarboximidamide Chemical compound ON=C(N)C1CCCC1 FJABAFACLDVRTR-UHFFFAOYSA-N 0.000 description 2
- AVAWMINJNRAQFS-UHFFFAOYSA-N n,n-dimethylpyrrolidin-3-amine Chemical compound CN(C)C1CCNC1 AVAWMINJNRAQFS-UHFFFAOYSA-N 0.000 description 2
- RWIVICVCHVMHMU-UHFFFAOYSA-N n-aminoethylmorpholine Chemical compound NCCN1CCOCC1 RWIVICVCHVMHMU-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- DVAIJVUXWCQJHE-CQSZACIVSA-N n-cyclopentyl-2-[(4r)-4-(2-iodoethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound ICC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 DVAIJVUXWCQJHE-CQSZACIVSA-N 0.000 description 2
- VHFQKAVXEJVKAW-GOSISDBHSA-N n-cyclopentyl-2-[(4r)-4-(2-morpholin-4-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C([C@H]1N=C(SC1)C=1NC2=C(NC3CCCC3)C=CC=C2C=1)CN1CCOCC1 VHFQKAVXEJVKAW-GOSISDBHSA-N 0.000 description 2
- UMAQPNRREQCZMN-OAHLLOKOSA-N n-cyclopentyl-2-[(4r)-4-[(dimethylamino)methyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound CN(C)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 UMAQPNRREQCZMN-OAHLLOKOSA-N 0.000 description 2
- XMTXXITZYLHBSU-HXUWFJFHSA-N n-cyclopentyl-5-(methylsulfonylmethyl)-2-[(4r)-4-(2-morpholin-4-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCOCC4)N=3)=CC2=CC(CS(=O)(=O)C)=CC=1NC1CCCC1 XMTXXITZYLHBSU-HXUWFJFHSA-N 0.000 description 2
- GKLSPAFTTDTBCT-GOSISDBHSA-N n-cyclopentyl-5-fluoro-2-[(4r)-4-(2-morpholin-4-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCOCC4)N=3)=CC2=CC(F)=CC=1NC1CCCC1 GKLSPAFTTDTBCT-GOSISDBHSA-N 0.000 description 2
- WEEVLNYXKDIXOJ-GOSISDBHSA-N n-cyclopentyl-5-fluoro-2-[(4r)-4-(2-pyrrolidin-1-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCCC4)N=3)=CC2=CC(F)=CC=1NC1CCCC1 WEEVLNYXKDIXOJ-GOSISDBHSA-N 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 2
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 238000005932 reductive alkylation reaction Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- 239000004317 sodium nitrate Substances 0.000 description 2
- 235000010344 sodium nitrate Nutrition 0.000 description 2
- RPACBEVZENYWOL-XFULWGLBSA-M sodium;(2r)-2-[6-(4-chlorophenoxy)hexyl]oxirane-2-carboxylate Chemical compound [Na+].C=1C=C(Cl)C=CC=1OCCCCCC[C@]1(C(=O)[O-])CO1 RPACBEVZENYWOL-XFULWGLBSA-M 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 150000003871 sulfonates Chemical class 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- MHXXDNMSJBMXNI-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3,3,3-trifluoropropanoate Chemical compound FC(F)(F)CC(=O)ON1C(=O)CCC1=O MHXXDNMSJBMXNI-UHFFFAOYSA-N 0.000 description 1
- VRTXRNJMNFVTOM-ZDUSSCGKSA-N (2r)-3-[(4-methoxyphenyl)methylsulfanyl]-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound COC1=CC=C(CSC[C@H](NC(=O)OC(C)(C)C)C(O)=O)C=C1 VRTXRNJMNFVTOM-ZDUSSCGKSA-N 0.000 description 1
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 1
- OSFPMHMPAGDBQO-JTQLQIEISA-N (3s)-3-amino-3-methoxycarbonyl-4-[(2-methylpropan-2-yl)oxy]-4-oxobutanoic acid Chemical compound COC(=O)[C@@](N)(CC(O)=O)C(=O)OC(C)(C)C OSFPMHMPAGDBQO-JTQLQIEISA-N 0.000 description 1
- LFBBZAYPJSLJCR-UHFFFAOYSA-N (4-chloro-2-nitrophenyl)hydrazine;hydrochloride Chemical compound Cl.NNC1=CC=C(Cl)C=C1[N+]([O-])=O LFBBZAYPJSLJCR-UHFFFAOYSA-N 0.000 description 1
- FYSTYLZVGRXXPD-CQSZACIVSA-N (4r)-4-(2-iodoethyl)-2-(7-phenoxy-1h-indol-2-yl)-4,5-dihydro-1,3-thiazole Chemical compound ICC[C@@H]1CSC(C=2NC3=C(OC=4C=CC=CC=4)C=CC=C3C=2)=N1 FYSTYLZVGRXXPD-CQSZACIVSA-N 0.000 description 1
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 1
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 1
- FNQJDLTXOVEEFB-UHFFFAOYSA-N 1,2,3-benzothiadiazole Chemical compound C1=CC=C2SN=NC2=C1 FNQJDLTXOVEEFB-UHFFFAOYSA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- KTZQTRPPVKQPFO-UHFFFAOYSA-N 1,2-benzoxazole Chemical compound C1=CC=C2C=NOC2=C1 KTZQTRPPVKQPFO-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
- KPZGRMZPZLOPBS-UHFFFAOYSA-N 1,3-dichloro-2,2-bis(chloromethyl)propane Chemical compound ClCC(CCl)(CCl)CCl KPZGRMZPZLOPBS-UHFFFAOYSA-N 0.000 description 1
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 1
- VKRKCBWIVLSRBJ-UHFFFAOYSA-N 1,4-dioxaspiro[4.5]decan-8-one Chemical compound C1CC(=O)CCC21OCCO2 VKRKCBWIVLSRBJ-UHFFFAOYSA-N 0.000 description 1
- BIFVGOGQSLKDOT-GOSISDBHSA-N 1-[2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperidin-4-ol Chemical compound C1CC(O)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 BIFVGOGQSLKDOT-GOSISDBHSA-N 0.000 description 1
- JNIKLRXJPRJRTR-XCWJXAQQSA-N 1-[2-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperidine-3-carboxylic acid Chemical compound C1C(C(=O)O)CCCN1CC[C@H]1N=C(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)SC1 JNIKLRXJPRJRTR-XCWJXAQQSA-N 0.000 description 1
- ZPSJMPVFKKBEBQ-LJQANCHMSA-N 1-[4-[2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-1-yl]-2-hydroxyethanone Chemical compound C1CN(C(=O)CO)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 ZPSJMPVFKKBEBQ-LJQANCHMSA-N 0.000 description 1
- DBVHRGVYDHSNKS-HXUWFJFHSA-N 1-[4-[2-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-1-yl]ethanone Chemical compound C1CN(C(=O)C)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 DBVHRGVYDHSNKS-HXUWFJFHSA-N 0.000 description 1
- RBHIYEZXBCEOJZ-HXUWFJFHSA-N 1-[4-[2-[(4r)-2-[7-(cyclopentylamino)-5-fluoro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-1-yl]ethanone Chemical compound C1CN(C(=O)C)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(F)C=C3C=2)SC1 RBHIYEZXBCEOJZ-HXUWFJFHSA-N 0.000 description 1
- DTZRMWGEDPQBNU-OAQYLSRUSA-N 1-[4-[3-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propyl]piperazin-1-yl]ethanone Chemical compound C1CN(C(=O)C)CCN1CCC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 DTZRMWGEDPQBNU-OAQYLSRUSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- BIYGAOBOLDXNHM-UHFFFAOYSA-N 1-ethylsulfonylpiperazine Chemical compound CCS(=O)(=O)N1CCNCC1 BIYGAOBOLDXNHM-UHFFFAOYSA-N 0.000 description 1
- WFQDTOYDVUWQMS-UHFFFAOYSA-N 1-fluoro-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(F)C=C1 WFQDTOYDVUWQMS-UHFFFAOYSA-N 0.000 description 1
- XVXPNHFFFHVWIH-UHFFFAOYSA-N 1-o-tert-butyl 2-o-methyl 5-(bromomethyl)-7-nitroindole-1,2-dicarboxylate Chemical compound BrCC1=CC([N+]([O-])=O)=C2N(C(=O)OC(C)(C)C)C(C(=O)OC)=CC2=C1 XVXPNHFFFHVWIH-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- SYOANZBNGDEJFH-UHFFFAOYSA-N 2,5-dihydro-1h-triazole Chemical compound C1NNN=C1 SYOANZBNGDEJFH-UHFFFAOYSA-N 0.000 description 1
- RAGPEAXCMHCJQG-PNESFZTOSA-N 2-[(4R)-2-(7-amino-1H-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetic acid methyl 2-[(4R)-2-(7-amino-1H-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound NC=1C=CC=C2C=C(NC12)C=1SC[C@H](N1)CC(=O)O.COC(C[C@H]1N=C(SC1)C=1NC2=C(C=CC=C2C1)N)=O RAGPEAXCMHCJQG-PNESFZTOSA-N 0.000 description 1
- LHRRNEAONOYJOG-GOSISDBHSA-N 2-[(4R)-2-[7-(cyclopentylamino)-5-(pyrrolidin-1-ylmethyl)-1H-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C1(CCCC1)NC=1C=C(C=C2C=C(NC12)C=1SC[C@H](N1)CC(=O)O)CN1CCCC1 LHRRNEAONOYJOG-GOSISDBHSA-N 0.000 description 1
- UJWAHCPIKWRHAV-CQSZACIVSA-N 2-[(4r)-2-(7-phenoxy-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound OCC[C@@H]1CSC(C=2NC3=C(OC=4C=CC=CC=4)C=CC=C3C=2)=N1 UJWAHCPIKWRHAV-CQSZACIVSA-N 0.000 description 1
- SAACNHGJKIZYBW-CQSZACIVSA-N 2-[(4r)-2-[5-bromo-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound OCC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(Br)C=C3C=2)=N1 SAACNHGJKIZYBW-CQSZACIVSA-N 0.000 description 1
- PEDFTBRDESRHPX-CYBMUJFWSA-N 2-[(4r)-2-[5-bromo-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound OC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(Br)C=C3C=2)=N1 PEDFTBRDESRHPX-CYBMUJFWSA-N 0.000 description 1
- XTWKDFQUGXULDZ-OAHLLOKOSA-N 2-[(4r)-2-[5-fluoro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl methanesulfonate Chemical compound CS(=O)(=O)OCC[C@@H]1CSC(C=2NC3=C(NC4CCOCC4)C=C(F)C=C3C=2)=N1 XTWKDFQUGXULDZ-OAHLLOKOSA-N 0.000 description 1
- RFMHTUNCHZAFBB-CQSZACIVSA-N 2-[(4r)-2-[5-methoxy-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound C=12NC(C=3SC[C@@H](CCO)N=3)=CC2=CC(OC)=CC=1NC1CCOCC1 RFMHTUNCHZAFBB-CQSZACIVSA-N 0.000 description 1
- QDRDBVLIGZXKAF-CQSZACIVSA-N 2-[(4r)-2-[7-(cyclopentylamino)-5-methoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethanol Chemical compound C=12NC(C=3SC[C@@H](CCO)N=3)=CC2=CC(OC)=CC=1NC1CCCC1 QDRDBVLIGZXKAF-CQSZACIVSA-N 0.000 description 1
- ZFMWFMHBONZYKP-GOSISDBHSA-N 2-[(4r)-4-[2-(1,1-dioxo-1,4-thiazinan-4-yl)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-5-fluoro-n-(oxan-4-yl)-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCS(=O)(=O)CC4)N=3)=CC2=CC(F)=CC=1NC1CCOCC1 ZFMWFMHBONZYKP-GOSISDBHSA-N 0.000 description 1
- GSNDDAHVVNMOLV-INIZCTEOSA-N 2-[(4s)-2-[7-[(1-acetylpiperidin-4-yl)amino]-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C1CN(C(=O)C)CCC1NC1=CC(C)=CC2=C1NC(C=1SC[C@H](CC(O)=O)N=1)=C2 GSNDDAHVVNMOLV-INIZCTEOSA-N 0.000 description 1
- GBDMCQFOXLWVGC-LOACHALJSA-N 2-[(4s)-2-[7-[(1-acetylpyrrolidin-3-yl)amino]-5-methyl-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetic acid Chemical compound C1N(C(=O)C)CCC1NC1=CC(C)=CC2=C1NC(C=1SC[C@H](CC(O)=O)N=1)=C2 GBDMCQFOXLWVGC-LOACHALJSA-N 0.000 description 1
- MGDKCUWISFMFPR-QHCPKHFHSA-N 2-[(4s)-4-(2-morpholin-4-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-n-(oxan-4-ylmethyl)-5-phenoxy-1h-indol-7-amine Chemical compound C([C@@H]1N=C(SC1)C=1NC2=C(NCC3CCOCC3)C=C(OC=3C=CC=CC=3)C=C2C=1)CN1CCOCC1 MGDKCUWISFMFPR-QHCPKHFHSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- VUMCVANUWQZNGD-UHFFFAOYSA-N 2-nitro-4-propoxyaniline Chemical compound CCCOC1=CC=C(N)C([N+]([O-])=O)=C1 VUMCVANUWQZNGD-UHFFFAOYSA-N 0.000 description 1
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical compound C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 1
- BXRQEJKKEDDSBF-UHFFFAOYSA-N 3-(4-nitrophenoxy)pyridine Chemical compound C1=CC([N+](=O)[O-])=CC=C1OC1=CC=CN=C1 BXRQEJKKEDDSBF-UHFFFAOYSA-N 0.000 description 1
- IPDWOBMNIBXCEU-UHFFFAOYSA-N 3-(4-nitrophenoxy)pyridine 4-pyridin-3-yloxyaniline Chemical compound N1=CC(=CC=C1)OC1=CC=C(C=C1)[N+](=O)[O-].N1=CC(=CC=C1)OC1=CC=C(C=C1)N IPDWOBMNIBXCEU-UHFFFAOYSA-N 0.000 description 1
- XLIOBMAAHXWNOR-HXUWFJFHSA-N 3-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]-n-(2-morpholin-4-ylethyl)propanamide Chemical compound C=12NC(C=3SC[C@@H](CCC(=O)NCCN4CCOCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 XLIOBMAAHXWNOR-HXUWFJFHSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- UIKUBYKUYUSRSM-UHFFFAOYSA-N 3-morpholinopropylamine Chemical compound NCCCN1CCOCC1 UIKUBYKUYUSRSM-UHFFFAOYSA-N 0.000 description 1
- HRASFKASEOUOPD-UHFFFAOYSA-N 3-oxopyrrolidine-1-carboxylic acid Chemical compound OC(=O)N1CCC(=O)C1 HRASFKASEOUOPD-UHFFFAOYSA-N 0.000 description 1
- MRLGCTNJRREZHZ-UHFFFAOYSA-N 3-phenoxybenzaldehyde Chemical compound O=CC1=CC=CC(OC=2C=CC=CC=2)=C1 MRLGCTNJRREZHZ-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- QASBCTGZKABPKX-UHFFFAOYSA-N 4-(methylsulfanyl)phenol Chemical compound CSC1=CC=C(O)C=C1 QASBCTGZKABPKX-UHFFFAOYSA-N 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- ZCWBZRBJSPWUPG-UHFFFAOYSA-N 4-bromo-2-nitroaniline Chemical compound NC1=CC=C(Br)C=C1[N+]([O-])=O ZCWBZRBJSPWUPG-UHFFFAOYSA-N 0.000 description 1
- PBGKNXWGYQPUJK-UHFFFAOYSA-N 4-chloro-2-nitroaniline Chemical compound NC1=CC=C(Cl)C=C1[N+]([O-])=O PBGKNXWGYQPUJK-UHFFFAOYSA-N 0.000 description 1
- IMPPGHMHELILKG-UHFFFAOYSA-N 4-ethoxyaniline Chemical compound CCOC1=CC=C(N)C=C1 IMPPGHMHELILKG-UHFFFAOYSA-N 0.000 description 1
- DENDDENDRRUXHZ-UHFFFAOYSA-N 4-ethoxyaniline;4-ethoxy-2-nitroaniline Chemical compound CCOC1=CC=C(N)C=C1.CCOC1=CC=C(N)C([N+]([O-])=O)=C1 DENDDENDRRUXHZ-UHFFFAOYSA-N 0.000 description 1
- PUGDHSSOXPHLPT-UHFFFAOYSA-N 4-fluoro-2-nitroaniline Chemical compound NC1=CC=C(F)C=C1[N+]([O-])=O PUGDHSSOXPHLPT-UHFFFAOYSA-N 0.000 description 1
- QFMJFXFXQAFGBO-UHFFFAOYSA-N 4-methoxy-2-nitroaniline Chemical compound COC1=CC=C(N)C([N+]([O-])=O)=C1 QFMJFXFXQAFGBO-UHFFFAOYSA-N 0.000 description 1
- WOYZXEVUWXQVNV-UHFFFAOYSA-N 4-phenoxyaniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC=C1 WOYZXEVUWXQVNV-UHFFFAOYSA-N 0.000 description 1
- HLBMLMPGXJXCJU-UHFFFAOYSA-N 5-(4-methylsulfonylphenoxy)-7-nitro-1h-indole-2-carboxylic acid Chemical compound C1=CC(S(=O)(=O)C)=CC=C1OC1=CC([N+]([O-])=O)=C(NC(=C2)C(O)=O)C2=C1 HLBMLMPGXJXCJU-UHFFFAOYSA-N 0.000 description 1
- LHZQZRKAUYIMLK-UHFFFAOYSA-N 5-(acetyloxymethyl)-7-nitroindole-1,2-dicarboxylic acid Chemical compound CC(=O)OCC1=CC([N+]([O-])=O)=C2N(C(O)=O)C(C(O)=O)=CC2=C1 LHZQZRKAUYIMLK-UHFFFAOYSA-N 0.000 description 1
- IRPVABHDSJVBNZ-RTHVDDQRSA-N 5-[1-(cyclopropylmethyl)-5-[(1R,5S)-3-(oxetan-3-yl)-3-azabicyclo[3.1.0]hexan-6-yl]pyrazol-3-yl]-3-(trifluoromethyl)pyridin-2-amine Chemical compound C1=C(C(F)(F)F)C(N)=NC=C1C1=NN(CC2CC2)C(C2[C@@H]3CN(C[C@@H]32)C2COC2)=C1 IRPVABHDSJVBNZ-RTHVDDQRSA-N 0.000 description 1
- OITGMXKMPMPOGW-UHFFFAOYSA-N 5-chloro-7-nitro-1h-indole-2-carboxylic acid Chemical compound ClC1=CC([N+]([O-])=O)=C2NC(C(=O)O)=CC2=C1 OITGMXKMPMPOGW-UHFFFAOYSA-N 0.000 description 1
- CPLCNLFLUCOWNC-OAQYLSRUSA-N 5-chloro-n-(oxan-4-yl)-2-[(4r)-4-[2-(4-pyrazin-2-ylpiperazin-1-yl)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCN(CC4)C=4N=CC=NC=4)N=3)=CC2=CC(Cl)=CC=1NC1CCOCC1 CPLCNLFLUCOWNC-OAQYLSRUSA-N 0.000 description 1
- NRMIOTIHCYMNRO-CQSZACIVSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-(2-iodoethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCI)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 NRMIOTIHCYMNRO-CQSZACIVSA-N 0.000 description 1
- MOSKTDWRZQHWIR-FIWHBWSRSA-N 5-chloro-n-cyclopentyl-2-[(4r)-4-[2-[3-(dimethylamino)pyrrolidin-1-yl]ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C1C(N(C)C)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(Cl)C=C3C=2)SC1 MOSKTDWRZQHWIR-FIWHBWSRSA-N 0.000 description 1
- WLVUAOONUBXNRT-UHFFFAOYSA-N 5-ethoxy-7-nitro-1H-indole-2-carboxylic acid Chemical compound CCOC1=CC([N+]([O-])=O)=C2NC(C(O)=O)=CC2=C1 WLVUAOONUBXNRT-UHFFFAOYSA-N 0.000 description 1
- QOHCYYZLRMSMHX-CQSZACIVSA-N 5-fluoro-2-[(4r)-4-(2-iodoethyl)-4,5-dihydro-1,3-thiazol-2-yl]-n-(oxan-4-yl)-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCI)N=3)=CC2=CC(F)=CC=1NC1CCOCC1 QOHCYYZLRMSMHX-CQSZACIVSA-N 0.000 description 1
- WLGKXXCARXACND-GOSISDBHSA-N 5-fluoro-2-[(4r)-4-(2-morpholin-4-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-n-(oxan-4-yl)-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCOCC4)N=3)=CC2=CC(F)=CC=1NC1CCOCC1 WLGKXXCARXACND-GOSISDBHSA-N 0.000 description 1
- VCIWSABEZMBHJL-UHFFFAOYSA-N 5-fluoro-7-nitro-1h-indole-2-carboxylic acid Chemical compound FC1=CC([N+]([O-])=O)=C2NC(C(=O)O)=CC2=C1 VCIWSABEZMBHJL-UHFFFAOYSA-N 0.000 description 1
- BNZBLYSWCZRLBL-IBGZPJMESA-N 5-methyl-2-[(4s)-4-(2-morpholin-4-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-n-(oxan-4-yl)-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@H](CCN4CCOCC4)N=3)=CC2=CC(C)=CC=1NC1CCOCC1 BNZBLYSWCZRLBL-IBGZPJMESA-N 0.000 description 1
- LOQDTQDUEYLYQM-UHFFFAOYSA-N 5-methyl-7-nitroindole-1,2-dicarboxylic acid Chemical compound CC1=CC([N+]([O-])=O)=C2N(C(O)=O)C(C(O)=O)=CC2=C1 LOQDTQDUEYLYQM-UHFFFAOYSA-N 0.000 description 1
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 1
- 102100031126 6-phosphogluconolactonase Human genes 0.000 description 1
- 108010029731 6-phosphogluconolactonase Proteins 0.000 description 1
- HYSYLJJPLMYRCN-UHFFFAOYSA-N 7-nitro-5-phenoxy-1H-indole-2-carboxylic acid Chemical compound [N+](=O)([O-])C=1C=C(C=C2C=C(NC=12)C(=O)O)OC1=CC=CC=C1 HYSYLJJPLMYRCN-UHFFFAOYSA-N 0.000 description 1
- BIUCOFQROHIAEO-UHFFFAOYSA-N 7-nitroindole-2-carboxylic acid Chemical compound C1=CC([N+]([O-])=O)=C2NC(C(=O)O)=CC2=C1 BIUCOFQROHIAEO-UHFFFAOYSA-N 0.000 description 1
- ZSCJSFSYTPYDQT-UHFFFAOYSA-N 7-phenoxy-1h-indole-2-carboxylic acid Chemical compound C=12NC(C(=O)O)=CC2=CC=CC=1OC1=CC=CC=C1 ZSCJSFSYTPYDQT-UHFFFAOYSA-N 0.000 description 1
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- OEULFNLWJZJBDT-UHFFFAOYSA-N C1=C=CNN=C1 Chemical compound C1=C=CNN=C1 OEULFNLWJZJBDT-UHFFFAOYSA-N 0.000 description 1
- TUFPKZVGLUFPSO-OEMAIJDKSA-N CC(C)(C)C(OC[C@H]1N=C(c2cc(cccc3NC(CC4)CN4S(C)(=O)=O)c3[nH]2)SC1)=O Chemical compound CC(C)(C)C(OC[C@H]1N=C(c2cc(cccc3NC(CC4)CN4S(C)(=O)=O)c3[nH]2)SC1)=O TUFPKZVGLUFPSO-OEMAIJDKSA-N 0.000 description 1
- SLUXJQCYWLQAAY-NTUHNPAUSA-N CCOC(/C(/C)=N/Nc(c([N+]([O-])=O)c1)ccc1F)=O Chemical compound CCOC(/C(/C)=N/Nc(c([N+]([O-])=O)c1)ccc1F)=O SLUXJQCYWLQAAY-NTUHNPAUSA-N 0.000 description 1
- VBIXZCPHFVFLNY-UHFFFAOYSA-N CCOC(C1CN(CCC2N=C(c3cc(cc(cc4NC5CCOCC5)Cl)c4[nH]3)SC2)CCC1)=O Chemical compound CCOC(C1CN(CCC2N=C(c3cc(cc(cc4NC5CCOCC5)Cl)c4[nH]3)SC2)CCC1)=O VBIXZCPHFVFLNY-UHFFFAOYSA-N 0.000 description 1
- ZQHUPTRYZRCHPI-UHFFFAOYSA-N CCOC(CCC(CSCc(cc1)ccc1OC)N)=O Chemical compound CCOC(CCC(CSCc(cc1)ccc1OC)N)=O ZQHUPTRYZRCHPI-UHFFFAOYSA-N 0.000 description 1
- NYINCTOKYLFKMS-UHFFFAOYSA-N CN(C)C(CC1N=C(c2cc(cc(cc3NC4CCCC4)Cl)c3[nH]2)SC1)=O Chemical compound CN(C)C(CC1N=C(c2cc(cc(cc3NC4CCCC4)Cl)c3[nH]2)SC1)=O NYINCTOKYLFKMS-UHFFFAOYSA-N 0.000 description 1
- MOSKTDWRZQHWIR-UHFFFAOYSA-N CN(C)C1CN(CCC2N=C(c3cc(cc(cc4NC5CCCC5)Cl)c4[nH]3)SC2)CC1 Chemical compound CN(C)C1CN(CCC2N=C(c3cc(cc(cc4NC5CCCC5)Cl)c4[nH]3)SC2)CC1 MOSKTDWRZQHWIR-UHFFFAOYSA-N 0.000 description 1
- SOYJBGSYZULRAD-UHFFFAOYSA-N COC(CC(CSCc(cc1)ccc1OC)NC(c1cc(cc(CO)cc2[N+]([O-])=O)c2[nH]1)=O)=O Chemical compound COC(CC(CSCc(cc1)ccc1OC)NC(c1cc(cc(CO)cc2[N+]([O-])=O)c2[nH]1)=O)=O SOYJBGSYZULRAD-UHFFFAOYSA-N 0.000 description 1
- BHQWZLMEJCOUDV-UHFFFAOYSA-N COC(CC1N=C(c2cc(cccc3N)c3[nH]2)SC1)=O Chemical compound COC(CC1N=C(c2cc(cccc3N)c3[nH]2)SC1)=O BHQWZLMEJCOUDV-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- GSXOAOHZAIYLCY-UHFFFAOYSA-N D-F6P Natural products OCC(=O)C(O)C(O)C(O)COP(O)(O)=O GSXOAOHZAIYLCY-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 241001198387 Escherichia coli BL21(DE3) Species 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 208000002705 Glucose Intolerance Diseases 0.000 description 1
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 1
- 108010018962 Glucosephosphate Dehydrogenase Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 208000013016 Hypoglycemia Diseases 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- HCUARRIEZVDMPT-UHFFFAOYSA-N Indole-2-carboxylic acid Chemical class C1=CC=C2NC(C(=O)O)=CC2=C1 HCUARRIEZVDMPT-UHFFFAOYSA-N 0.000 description 1
- 238000006147 Japp-Klingemann synthesis reaction Methods 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229910010084 LiAlH4 Inorganic materials 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241001024304 Mino Species 0.000 description 1
- ZRKWMRDKSOPRRS-UHFFFAOYSA-N N-Methyl-N-nitrosourea Chemical compound O=NN(C)C(N)=O ZRKWMRDKSOPRRS-UHFFFAOYSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 229910020889 NaBH3 Inorganic materials 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- VGTAGLXGWXFORQ-UHFFFAOYSA-N O=C1NCCN(CCC2N=C(c3cc(cc(cc4NC5CCCC5)Cl)c4[nH]3)SC2)C1 Chemical compound O=C1NCCN(CCC2N=C(c3cc(cc(cc4NC5CCCC5)Cl)c4[nH]3)SC2)C1 VGTAGLXGWXFORQ-UHFFFAOYSA-N 0.000 description 1
- BEUNTKQGUNXHAO-UHFFFAOYSA-N OC(CC1N=C(c2cc(cc(cc3NC4CCCC4)Br)c3[nH]2)SC1)=O Chemical compound OC(CC1N=C(c2cc(cc(cc3NC4CCCC4)Br)c3[nH]2)SC1)=O BEUNTKQGUNXHAO-UHFFFAOYSA-N 0.000 description 1
- JPURYDFXLFVCIJ-UHFFFAOYSA-N OC(CC1N=C(c2cc(cc(cc3NC4CCCC4)Oc4ccccc4)c3[nH]2)SC1)=O Chemical compound OC(CC1N=C(c2cc(cc(cc3NC4CCCC4)Oc4ccccc4)c3[nH]2)SC1)=O JPURYDFXLFVCIJ-UHFFFAOYSA-N 0.000 description 1
- JRPUXGVBCFOJNE-UHFFFAOYSA-N OC(CC1N=C(c2cc(cc(cc3NC4CCOCC4)Oc4cnccc4)c3[nH]2)SC1)=O Chemical compound OC(CC1N=C(c2cc(cc(cc3NC4CCOCC4)Oc4cnccc4)c3[nH]2)SC1)=O JRPUXGVBCFOJNE-UHFFFAOYSA-N 0.000 description 1
- CKNJKLBMTYFMCG-UHFFFAOYSA-N OCC1N=C(c2cc(cc(cc3NC4CCOCC4)Cl)c3[nH]2)SC1 Chemical compound OCC1N=C(c2cc(cc(cc3NC4CCOCC4)Cl)c3[nH]2)SC1 CKNJKLBMTYFMCG-UHFFFAOYSA-N 0.000 description 1
- VUCVBMBIOHUZAT-UHFFFAOYSA-N OCCC1N=C(c2cc(cc(cc3NC4CCOCC4)Cl)c3[nH]2)SC1 Chemical compound OCCC1N=C(c2cc(cc(cc3NC4CCOCC4)Cl)c3[nH]2)SC1 VUCVBMBIOHUZAT-UHFFFAOYSA-N 0.000 description 1
- BVSHIKSZLVZATA-UHFFFAOYSA-N OCCCC1N=C(c2cc(cc(cc3NC4CCOCC4)Cl)c3[nH]2)SC1 Chemical compound OCCCC1N=C(c2cc(cc(cc3NC4CCOCC4)Cl)c3[nH]2)SC1 BVSHIKSZLVZATA-UHFFFAOYSA-N 0.000 description 1
- IDRGFNPZDVBSSE-UHFFFAOYSA-N OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F Chemical compound OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F IDRGFNPZDVBSSE-UHFFFAOYSA-N 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108010016731 PPAR gamma Proteins 0.000 description 1
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 description 1
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 description 1
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- FOIXSVOLVBLSDH-UHFFFAOYSA-N Silver ion Chemical compound [Ag+] FOIXSVOLVBLSDH-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- UGWNOJVBOYHGTH-CYBMUJFWSA-N [(2r)-2-amino-3-[(4-methoxyphenyl)methylsulfanyl]propyl] 2,2-dimethylpropanoate Chemical compound COC1=CC=C(CSC[C@H](N)COC(=O)C(C)(C)C)C=C1 UGWNOJVBOYHGTH-CYBMUJFWSA-N 0.000 description 1
- ABPAXYKXSJNMTO-MRXNPFEDSA-N [(2r)-3-[(4-methoxyphenyl)methylsulfanyl]-2-[(2-methylpropan-2-yl)oxycarbonylamino]propyl] 2,2-dimethylpropanoate Chemical compound COC1=CC=C(CSC[C@@H](COC(=O)C(C)(C)C)NC(=O)OC(C)(C)C)C=C1 ABPAXYKXSJNMTO-MRXNPFEDSA-N 0.000 description 1
- FIWQRJPNXLKGFC-ZWKOTPCHSA-N [(3s)-1-[2-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]pyrrolidin-3-yl]carbamic acid Chemical compound C1[C@@H](NC(=O)O)CCN1CC[C@H]1N=C(C=2NC3=C(NC4CCOCC4)C=C(Cl)C=C3C=2)SC1 FIWQRJPNXLKGFC-ZWKOTPCHSA-N 0.000 description 1
- ZYMIZTZVSCVCJB-LLVKDONJSA-N [(4r)-2-(7-nitro-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]methyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OC[C@@H]1CSC(C=2NC3=C([N+]([O-])=O)C=CC=C3C=2)=N1 ZYMIZTZVSCVCJB-LLVKDONJSA-N 0.000 description 1
- ZISUTZNUSHYYQY-QGZVFWFLSA-N [(4r)-2-[7-(cyclopentylamino)-5-(imidazol-1-ylmethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methanol Chemical compound OC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(CN4C=NC=C4)C=C3C=2)=N1 ZISUTZNUSHYYQY-QGZVFWFLSA-N 0.000 description 1
- FXLDVICYDMVXEH-OAHLLOKOSA-N [(4r)-2-[7-amino-5-(2,2-dimethylpropanoyloxymethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]methyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OC[C@@H]1CSC(C=2NC3=C(N)C=C(COC(=O)C(C)(C)C)C=C3C=2)=N1 FXLDVICYDMVXEH-OAHLLOKOSA-N 0.000 description 1
- MQVPKHZEWBQLTE-OAQYLSRUSA-N [4-[2-[(4r)-2-[5-chloro-7-(oxan-4-ylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]piperazin-1-yl]-(furan-2-yl)methanone Chemical compound C=12NC(C=3SC[C@@H](CCN4CCN(CC4)C(=O)C=4OC=CC=4)N=3)=CC2=CC(Cl)=CC=1NC1CCOCC1 MQVPKHZEWBQLTE-OAQYLSRUSA-N 0.000 description 1
- VLSOAXRVHARBEQ-UHFFFAOYSA-N [4-fluoro-2-(hydroxymethyl)phenyl]methanol Chemical compound OCC1=CC=C(F)C=C1CO VLSOAXRVHARBEQ-UHFFFAOYSA-N 0.000 description 1
- GQSDJPCQSDHEHH-UHFFFAOYSA-N [O-][N+](c1cc(Oc2cnccc2)cc2c1[nH]c(C(O)=O)c2)=O Chemical compound [O-][N+](c1cc(Oc2cnccc2)cc2c1[nH]c(C(O)=O)c2)=O GQSDJPCQSDHEHH-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 210000001789 adipocyte Anatomy 0.000 description 1
- 210000000577 adipose tissue Anatomy 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 239000003888 alpha glucosidase inhibitor Substances 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 229940054051 antipsychotic indole derivative Drugs 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- BGWGXPAPYGQALX-ARQDHWQXSA-N beta-D-fructofuranose 6-phosphate Chemical compound OC[C@@]1(O)O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O BGWGXPAPYGQALX-ARQDHWQXSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000005997 bromomethyl group Chemical group 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- 150000001734 carboxylic acid salts Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- BTJPVWMQGCSFJY-GFCCVEGCSA-N cyclohexyl (4r)-5-hydroxy-4-[(2-methylpropan-2-yl)oxycarbonylamino]pentanoate Chemical compound CC(C)(C)OC(=O)N[C@@H](CO)CCC(=O)OC1CCCCC1 BTJPVWMQGCSFJY-GFCCVEGCSA-N 0.000 description 1
- UUIHRIIYISQCLV-CQSZACIVSA-N cyclohexyl 3-[(4r)-2-(7-amino-5-chloro-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound C([C@H]1N=C(SC1)C1=CC=2C=C(Cl)C=C(C=2N1)N)CC(=O)OC1CCCCC1 UUIHRIIYISQCLV-CQSZACIVSA-N 0.000 description 1
- CDHKBQIKUUKULZ-LJQANCHMSA-N cyclohexyl 3-[(4r)-2-[5-chloro-7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound C=12NC(C=3SC[C@@H](CCC(=O)OC4CCCCC4)N=3)=CC2=CC(Cl)=CC=1NC1CCCC1 CDHKBQIKUUKULZ-LJQANCHMSA-N 0.000 description 1
- GCFAUZGWPDYAJN-UHFFFAOYSA-N cyclohexyl 3-phenylprop-2-enoate Chemical compound C=1C=CC=CC=1C=CC(=O)OC1CCCCC1 GCFAUZGWPDYAJN-UHFFFAOYSA-N 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- XWBDWHCCBGMXKG-UHFFFAOYSA-N ethanamine;hydron;chloride Chemical compound Cl.CCN XWBDWHCCBGMXKG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- HUUPGSZROXPMLA-UTONKHPSSA-N ethyl (3r)-3-amino-4-[(4-methoxyphenyl)methylsulfanyl]butanoate;hydrochloride Chemical compound Cl.CCOC(=O)C[C@@H](N)CSCC1=CC=C(OC)C=C1 HUUPGSZROXPMLA-UTONKHPSSA-N 0.000 description 1
- BHMHVCMFKFFOIE-SNVBAGLBSA-N ethyl (4r)-4-[(2-methylpropan-2-yl)oxycarbonylamino]-5-methylsulfonyloxypentanoate Chemical compound CCOC(=O)CC[C@H](COS(C)(=O)=O)NC(=O)OC(C)(C)C BHMHVCMFKFFOIE-SNVBAGLBSA-N 0.000 description 1
- AZLUOJLQQQJXEL-MRXNPFEDSA-N ethyl (4r)-5-[(4-methoxyphenyl)methylsulfanyl]-4-[(2-methylpropan-2-yl)oxycarbonylamino]pentanoate Chemical compound CCOC(=O)CC[C@@H](NC(=O)OC(C)(C)C)CSCC1=CC=C(OC)C=C1 AZLUOJLQQQJXEL-MRXNPFEDSA-N 0.000 description 1
- YJJLBIIKYNSDRO-SECBINFHSA-N ethyl (4r)-5-hydroxy-4-[(2-methylpropan-2-yl)oxycarbonylamino]pentanoate Chemical compound CCOC(=O)CC[C@H](CO)NC(=O)OC(C)(C)C YJJLBIIKYNSDRO-SECBINFHSA-N 0.000 description 1
- FNENWZWNOPCZGK-UHFFFAOYSA-N ethyl 2-methyl-3-oxobutanoate Chemical compound CCOC(=O)C(C)C(C)=O FNENWZWNOPCZGK-UHFFFAOYSA-N 0.000 description 1
- CKJVHZBWCCVCDT-LLVKDONJSA-N ethyl 3-[(4r)-2-(7-amino-5-chloro-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound CCOC(=O)CC[C@@H]1CSC(C=2NC3=C(N)C=C(Cl)C=C3C=2)=N1 CKJVHZBWCCVCDT-LLVKDONJSA-N 0.000 description 1
- SLUMRZSTONIYOP-MRXNPFEDSA-N ethyl 3-[(4r)-2-[7-(cyclopentylamino)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound CCOC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=CC=C3C=2)=N1 SLUMRZSTONIYOP-MRXNPFEDSA-N 0.000 description 1
- MNNRZDWBZQDJJX-HXUWFJFHSA-N ethyl 3-[(4r)-2-[7-(cyclopentylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]propanoate Chemical compound CCOC(=O)CC[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)=N1 MNNRZDWBZQDJJX-HXUWFJFHSA-N 0.000 description 1
- OXZLIZKJTGLEPV-UHFFFAOYSA-N ethyl 5-fluoro-7-nitro-1h-indole-2-carboxylate Chemical compound FC1=CC([N+]([O-])=O)=C2NC(C(=O)OCC)=CC2=C1 OXZLIZKJTGLEPV-UHFFFAOYSA-N 0.000 description 1
- VLDUBDZWWNLZCU-UHFFFAOYSA-N ethyl 5-methyl-1h-imidazole-4-carboxylate Chemical compound CCOC(=O)C=1NC=NC=1C VLDUBDZWWNLZCU-UHFFFAOYSA-N 0.000 description 1
- KKGGTNMEIMZVQJ-UHFFFAOYSA-N ethyl 5-methyl-7-nitro-1h-indole-2-carboxylate Chemical compound CC1=CC([N+]([O-])=O)=C2NC(C(=O)OCC)=CC2=C1 KKGGTNMEIMZVQJ-UHFFFAOYSA-N 0.000 description 1
- OTRIOPGZNFTKLU-UHFFFAOYSA-N ethyl 7-nitro-5-propoxy-1h-indole-2-carboxylate Chemical compound CCCOC1=CC([N+]([O-])=O)=C2NC(C(=O)OCC)=CC2=C1 OTRIOPGZNFTKLU-UHFFFAOYSA-N 0.000 description 1
- MVEAAGBEUOMFRX-UHFFFAOYSA-N ethyl acetate;hydrochloride Chemical compound Cl.CCOC(C)=O MVEAAGBEUOMFRX-UHFFFAOYSA-N 0.000 description 1
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- SADPINFEWFPMEA-UHFFFAOYSA-N furan-2-yl(piperazin-1-yl)methanone Chemical compound C=1C=COC=1C(=O)N1CCNCC1 SADPINFEWFPMEA-UHFFFAOYSA-N 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 102000010705 glucose-6-phosphate dehydrogenase activity proteins Human genes 0.000 description 1
- 108040005050 glucose-6-phosphate dehydrogenase activity proteins Proteins 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- LRDFRRGEGBBSRN-UHFFFAOYSA-N isobutyronitrile Chemical compound CC(C)C#N LRDFRRGEGBBSRN-UHFFFAOYSA-N 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- ZKLLSNQJRLJIGT-UYFOZJQFSA-N keto-D-fructose 1-phosphate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C(=O)COP(O)(O)=O ZKLLSNQJRLJIGT-UYFOZJQFSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000004777 loss-of-function mutation Effects 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- SLBPQEIOHNQCBH-MRXNPFEDSA-N methyl (3r)-3-[(5-chloro-7-nitro-1h-indole-2-carbonyl)amino]-4-[(4-methoxyphenyl)methylsulfanyl]butanoate Chemical compound C([C@@H](CC(=O)OC)NC(=O)C=1NC2=C([N+]([O-])=O)C=C(Cl)C=C2C=1)SCC1=CC=C(OC)C=C1 SLBPQEIOHNQCBH-MRXNPFEDSA-N 0.000 description 1
- JIQILOIUMXNRRO-VZYDHVRKSA-N methyl (3r)-3-amino-4-tritylsulfanylbutanoate;hydrochloride Chemical compound Cl.C=1C=CC=CC=1C(C=1C=CC=CC=1)(SC[C@H](N)CC(=O)OC)C1=CC=CC=C1 JIQILOIUMXNRRO-VZYDHVRKSA-N 0.000 description 1
- YDJJENVBJVMVTO-CQSZACIVSA-N methyl (3r)-4-[(4-methoxyphenyl)methylsulfanyl]-3-[(2-methylpropan-2-yl)oxycarbonylamino]butanoate Chemical compound CC(C)(C)OC(=O)N[C@H](CC(=O)OC)CSCC1=CC=C(OC)C=C1 YDJJENVBJVMVTO-CQSZACIVSA-N 0.000 description 1
- LZBHGEKCKNEDEH-SECBINFHSA-N methyl 2-[(4r)-2-(5-chloro-7-nitro-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C([N+]([O-])=O)C=C(Cl)C=C3C=2)=N1 LZBHGEKCKNEDEH-SECBINFHSA-N 0.000 description 1
- MKSATFSMURXYCA-SECBINFHSA-N methyl 2-[(4r)-2-(7-amino-5-chloro-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(N)C=C(Cl)C=C3C=2)=N1 MKSATFSMURXYCA-SECBINFHSA-N 0.000 description 1
- NKPVKNVFXNIJBJ-SECBINFHSA-N methyl 2-[(4r)-2-(7-amino-5-fluoro-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(N)C=C(F)C=C3C=2)=N1 NKPVKNVFXNIJBJ-SECBINFHSA-N 0.000 description 1
- KWMZOADDJCAHSA-SECBINFHSA-N methyl 2-[(4r)-2-(7-amino-5-methoxy-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(N)C=C(OC)C=C3C=2)=N1 KWMZOADDJCAHSA-SECBINFHSA-N 0.000 description 1
- ZBVDAISNRCVMHY-SNVBAGLBSA-N methyl 2-[(4r)-2-(7-amino-5-methyl-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(N)C=C(C)C=C3C=2)=N1 ZBVDAISNRCVMHY-SNVBAGLBSA-N 0.000 description 1
- UPNIUYHNEJCBGM-CYBMUJFWSA-N methyl 2-[(4r)-2-(7-amino-5-phenoxy-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(N)C=C(OC=4C=CC=CC=4)C=C3C=2)=N1 UPNIUYHNEJCBGM-CYBMUJFWSA-N 0.000 description 1
- YIVZZOFOXJQRKA-CQSZACIVSA-N methyl 2-[(4r)-2-(7-phenoxy-1h-indol-2-yl)-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(OC=4C=CC=CC=4)C=CC=C3C=2)=N1 YIVZZOFOXJQRKA-CQSZACIVSA-N 0.000 description 1
- HCTSTQKCUZEREN-SNVBAGLBSA-N methyl 2-[(4r)-2-[5-(chloromethyl)-7-nitro-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C([N+]([O-])=O)C=C(CCl)C=C3C=2)=N1 HCTSTQKCUZEREN-SNVBAGLBSA-N 0.000 description 1
- NUPBLPLITSAREG-MRXNPFEDSA-N methyl 2-[(4r)-2-[7-(cyclopentylamino)-5-(methylsulfonylmethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C(NC4CCCC4)C=C(CS(C)(=O)=O)C=C3C=2)=N1 NUPBLPLITSAREG-MRXNPFEDSA-N 0.000 description 1
- JXMIFSJSDBFTTC-CQSZACIVSA-N methyl 2-[(4r)-2-[7-nitro-5-(pyrrolidin-1-ylmethyl)-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]acetate Chemical compound COC(=O)C[C@@H]1CSC(C=2NC3=C([N+]([O-])=O)C=C(CN4CCCC4)C=C3C=2)=N1 JXMIFSJSDBFTTC-CQSZACIVSA-N 0.000 description 1
- RXYCUKSOYJWGPE-UHFFFAOYSA-N methyl 2-azidoacetate Chemical compound COC(=O)CN=[N+]=[N-] RXYCUKSOYJWGPE-UHFFFAOYSA-N 0.000 description 1
- SKWNZLRKRWZFFF-UHFFFAOYSA-N methyl 5-(acetyloxymethyl)-7-nitro-1h-indole-2-carboxylate Chemical compound CC(=O)OCC1=CC([N+]([O-])=O)=C2NC(C(=O)OC)=CC2=C1 SKWNZLRKRWZFFF-UHFFFAOYSA-N 0.000 description 1
- GRSWIYZNMCDFPS-UHFFFAOYSA-N methyl 5-ethoxy-7-nitro-1h-indole-2-carboxylate Chemical compound CCOC1=CC([N+]([O-])=O)=C2NC(C(=O)OC)=CC2=C1 GRSWIYZNMCDFPS-UHFFFAOYSA-N 0.000 description 1
- VPAHIKCVBUGHIH-UHFFFAOYSA-N methyl 7-nitro-5-(trifluoromethoxy)-1h-indole-2-carboxylate Chemical compound FC(F)(F)OC1=CC([N+]([O-])=O)=C2NC(C(=O)OC)=CC2=C1 VPAHIKCVBUGHIH-UHFFFAOYSA-N 0.000 description 1
- NFMZHWMNCKZBAI-UHFFFAOYSA-N methyl 7-nitro-5-phenoxy-1h-indole-2-carboxylate Chemical compound C=1C([N+]([O-])=O)=C2NC(C(=O)OC)=CC2=CC=1OC1=CC=CC=C1 NFMZHWMNCKZBAI-UHFFFAOYSA-N 0.000 description 1
- LIUUACIJFNCIOW-UHFFFAOYSA-N methyl 7-phenoxy-1h-indole-2-carboxylate Chemical compound C=12NC(C(=O)OC)=CC2=CC=CC=1OC1=CC=CC=C1 LIUUACIJFNCIOW-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- HQEIPVHJHZTMDP-UHFFFAOYSA-N methyl pyrrolidin-1-ium-2-carboxylate;chloride Chemical compound Cl.COC(=O)C1CCCN1 HQEIPVHJHZTMDP-UHFFFAOYSA-N 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- WDKQNCOJYGKYPL-UHFFFAOYSA-N n'-hydroxypiperidine-1-carboximidamide Chemical compound ON=C(N)N1CCCCC1 WDKQNCOJYGKYPL-UHFFFAOYSA-N 0.000 description 1
- FXULRBQINTUJDB-NRFANRHFSA-N n,5-dimethyl-2-[(4s)-4-(2-morpholin-4-ylethyl)-4,5-dihydro-1,3-thiazol-2-yl]-n-(oxan-4-ylmethyl)-1h-indol-7-amine Chemical compound C=1C(C)=CC=2C=C(C=3SC[C@H](CCN4CCOCC4)N=3)NC=2C=1N(C)CC1CCOCC1 FXULRBQINTUJDB-NRFANRHFSA-N 0.000 description 1
- XSEOKZXDDVGRFR-RPWUZVMVSA-N n-[(3r)-1-[2-[(4s)-2-[7-(cyclopentylamino)-5-phenoxy-1h-indol-2-yl]-4,5-dihydro-1,3-thiazol-4-yl]ethyl]pyrrolidin-3-yl]acetamide Chemical compound C1[C@H](NC(=O)C)CCN1CC[C@@H]1N=C(C=2NC3=C(NC4CCCC4)C=C(OC=4C=CC=CC=4)C=C3C=2)SC1 XSEOKZXDDVGRFR-RPWUZVMVSA-N 0.000 description 1
- QUPNQKXYVIUPOZ-CQSZACIVSA-N n-cyclopentyl-2-[(4r)-4-(2-iodoethyl)-4,5-dihydro-1,3-thiazol-2-yl]-5-methoxy-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCI)N=3)=CC2=CC(OC)=CC=1NC1CCCC1 QUPNQKXYVIUPOZ-CQSZACIVSA-N 0.000 description 1
- YPRRLYUKLKZPLD-GOSISDBHSA-N n-cyclopentyl-2-[(4r)-4-[2-(1,1-dioxo-1,4-thiazinan-4-yl)ethyl]-4,5-dihydro-1,3-thiazol-2-yl]-5-fluoro-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCN4CCS(=O)(=O)CC4)N=3)=CC2=CC(F)=CC=1NC1CCCC1 YPRRLYUKLKZPLD-GOSISDBHSA-N 0.000 description 1
- PBYRXDJUXQSZDM-CQSZACIVSA-N n-cyclopentyl-5-fluoro-2-[(4r)-4-(2-iodoethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-amine Chemical compound C=12NC(C=3SC[C@@H](CCI)N=3)=CC2=CC(F)=CC=1NC1CCCC1 PBYRXDJUXQSZDM-CQSZACIVSA-N 0.000 description 1
- VBEGHXKAFSLLGE-UHFFFAOYSA-N n-phenylnitramide Chemical compound [O-][N+](=O)NC1=CC=CC=C1 VBEGHXKAFSLLGE-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 208000029140 neonatal diabetes Diseases 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- JLPJFSCQKHRSQR-UHFFFAOYSA-N oxolan-3-one Chemical compound O=C1CCOC1 JLPJFSCQKHRSQR-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 150000003003 phosphines Chemical class 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 230000000865 phosphorylative effect Effects 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- HDOWRFHMPULYOA-UHFFFAOYSA-N piperidin-4-ol Chemical compound OC1CCNCC1 HDOWRFHMPULYOA-UHFFFAOYSA-N 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 229940074439 potassium sodium tartrate Drugs 0.000 description 1
- IHUSQSJUVFOHIQ-ZDUSSCGKSA-N propan-2-yl (3s)-3-amino-4-[(4-methoxyphenyl)methylsulfanyl]butanoate Chemical compound COC1=CC=C(CSC[C@@H](N)CC(=O)OC(C)C)C=C1 IHUSQSJUVFOHIQ-ZDUSSCGKSA-N 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- FKRCODPIKNYEAC-UHFFFAOYSA-N propionic acid ethyl ester Natural products CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- USPWKWBDZOARPV-UHFFFAOYSA-N pyrazolidine Chemical compound C1CNNC1 USPWKWBDZOARPV-UHFFFAOYSA-N 0.000 description 1
- DNXIASIHZYFFRO-UHFFFAOYSA-N pyrazoline Chemical compound C1CN=NC1 DNXIASIHZYFFRO-UHFFFAOYSA-N 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- JHHZLHWJQPUNKB-UHFFFAOYSA-N pyrrolidin-3-ol Chemical compound OC1CCNC1 JHHZLHWJQPUNKB-UHFFFAOYSA-N 0.000 description 1
- ZVJHJDDKYZXRJI-UHFFFAOYSA-N pyrroline Natural products C1CC=NC1 ZVJHJDDKYZXRJI-UHFFFAOYSA-N 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 238000011808 rodent model Methods 0.000 description 1
- 102220015875 rs6734111 Human genes 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 235000019553 satiation Nutrition 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- KZJPVUDYAMEDRM-UHFFFAOYSA-M silver;2,2,2-trifluoroacetate Chemical compound [Ag+].[O-]C(=O)C(F)(F)F KZJPVUDYAMEDRM-UHFFFAOYSA-M 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- KFNRRLINWBIAKW-QRWMCTBCSA-N tert-butyl 3-[[2-[(4r)-4-(2,2-dimethylpropanoyloxymethyl)-4,5-dihydro-1,3-thiazol-2-yl]-1h-indol-7-yl]amino]pyrrolidine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC1NC1=CC=CC2=C1NC(C=1SC[C@@H](COC(=O)C(C)(C)C)N=1)=C2 KFNRRLINWBIAKW-QRWMCTBCSA-N 0.000 description 1
- PFTUSRWQSBWUAG-AWEZNQCLSA-N tert-butyl N-[(2R)-4-diazo-1-[(4-methoxyphenyl)methylsulfanyl]-3-oxobutan-2-yl]carbamate Chemical compound COc1ccc(CSC[C@H](NC(=O)OC(C)(C)C)C(=O)C=[N+]=[N-])cc1 PFTUSRWQSBWUAG-AWEZNQCLSA-N 0.000 description 1
- ZZGLZZVBFFBGDK-CYBMUJFWSA-N tert-butyl n-[(2r)-1-hydroxy-3-[(4-methoxyphenyl)methylsulfanyl]propan-2-yl]carbamate Chemical compound COC1=CC=C(CSC[C@@H](CO)NC(=O)OC(C)(C)C)C=C1 ZZGLZZVBFFBGDK-CYBMUJFWSA-N 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 description 1
- OVRJVKCZJCNSOW-UHFFFAOYSA-N thian-4-one Chemical compound O=C1CCSCC1 OVRJVKCZJCNSOW-UHFFFAOYSA-N 0.000 description 1
- 150000001467 thiazolidinediones Chemical class 0.000 description 1
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- JABYJIQOLGWMQW-UHFFFAOYSA-N undec-4-ene Chemical compound CCCCCCC=CCCC JABYJIQOLGWMQW-UHFFFAOYSA-N 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to new compounds exhibiting excellent activity for glucokinase (glucokinase activators, GKAs), and pharmaceutical compositions comprising the same as an active ingredient.
- GKAs glucokinase activators
- Diabetes affects harmful influences on human health, causing various complications. Diabetes may be classified into type 1 diabetes where insulin is not excreted due to the destruction of pancreatic cells, and type 2 diabetes where insulin is not produced due to the other conditions or the body does not response to insulin. The type 2 diabetes occupies 90% or more of the total patients suffered from diabetes. Typical complications accompanied with diabetes include hyperlipidemia, hypertension, retinosis, renal failure, etc. (Zimmer P., et al., Nature, 2001, 414, 782).
- sulfonyl ureas facilitating insulin secretion in the pancreatic cells
- biguanides suppressing glucose production in the liver
- ⁇ - glucosidase inhibitors suppressing glucose uptake in the bowels
- PPAR Y peroxisome proliferator-activated receptor gamma
- these agents show some side effects, such as weight gain, according to the respective mechanisms of action (Moller D.E., Nature, 2001, 414, 821).
- PPAR Y peroxisome proliferator-activated receptor gamma
- glucose is accurately controlled within a safe and narrow physiological range by means of various endocrine glucostatic systems. If such glucostatic systems do not work, glucose intolerance occurs first, which is gradually grown to the type 2 diabetes. Dysfunction of such control mechanism is resulted from (i) decrease of secretion of insulin from the pancreatic cells, (ii) increases of insulin resistance in the liver, cells of adipose tissue, and cells of skeletal muscle, and (iii) excess production of blood glucose by the liver.
- glucokinase that belongs to hexokinase IV series are involved in the first step of glucose metabolism to directly control the glucose content in the blood, whereby it plays an important role in the maintenance of glucose homeostasis in the body.
- the glucokinase in the pancreatic cells can determine the thresholds of glucose- stimulated insulin release (GSIR) by acting as a glucose sensor.
- GSIR glucose- stimulated insulin release
- the glucokinase decreases blood glucose by phosphorylating glucose into glucose-6-phosphate consuming ATP, and keeping glucose-6-phosphate in the cells (Meglasson M.D. and Matschinsky F.M., Diabetes Metab Rev, 1986, 2, 163).
- the glucokinase in hepatocytes has the feature of being short- term controlled by glucokinase regulatory protein.
- Glucokinase regulatory protein forms a 1:1 complex with glucokinase, and acts as a "competitive inhibitor" against glucose to confine the inactivated glucokinase within the nucleus and to protect and stabilize it from other proteins such as decomposition enzymes, etc. It has been reported that fructose-6-phosphate further stabilizes glukinase regulatory protein, whereas fructose- 1 -phosphate separates glucokinase from glucokinase regulatory protein and transfers it from nucleus to cytoplasm to keep its activated state (Van Schaftingen E., Eur J Biochem, 1989, 179). The glucokinase in hepatocytes appropriately controls the glucose metabolism in the liver. That is, glucose uptake and production are effectively controlled under the satiation or fast state (Agius L., et al., J Biol Chem, 1996, 271, 30479).
- glucokinase activates the two functions of (i) direct control of blood glucose in the liver, and (ii) facilitation of insulin secretion within the physiological range after detection of glucose concentration in the pancreas, and thus, plays a very important role in the maintenance of glucose homeostasis.
- glucokinase is a key regulator in the maintenance of glucose homeostasis. Rats lacking the glucokinase gene function in pancreatic beta cells show a significant hyperglycemic symptom, and rats lacking the glucokinase gene function in hepatocytes show depressed glucose uptake and hyperglycemic symptom. On the other hand, when the glucokinase gene is over expressed in hepatocytes of normal rats, amelioration effect of glucose tolerance is shown (Rossetti L., et al., Am J Physiol, 1997, 273, E743). And, the over expression of glucokinase in diabetic rats induces amelioration of glucose tolerance and blood glucose lowering effect under the fast state (Desai UJ., et al., Am J Diabetes, 2001, 50, 2285).
- glucokinase gene mutants have been clinically reported for humans.
- patients of PNDM (permanent neonatal diabetes) and PHHI (persistent hyperinsulinemia hypoglycemia of infancy) showed serious hypoglycemia due to the glucokinase activation based on the gain-of-function mutation (Matsinsky F.M., et al., Frontiers in Daibetes, 2004, 16, chapter 4-7).
- glucokinase-associated diseases suggest that glucokinase plays an important role in the maintenance of glucose homeostasis in the body, which leaves a clue to develop a drug for enhancing the glucokinase activity.
- glucokinase activators facilitate pancreatic beta cell division to improve the glucose metabolism by maintaining the pancreatic cell mass.
- the present inventors extensively studied glucokinase activators, and as a result have confirmed that the indole compounds of formula (1) are effective as glucokinase activators. Thus, they completed the present invention that relates to glucokinase activators based on indole structure.
- the object of the present invention is to provide glucokinase activators of the indole compounds of formula (1). It is also another object of the present invention to provide a composition for the prevention or treatment of diseases caused by the decline of glucokinase activity, which comprises said compounds as an active ingredient.
- the present invention provides the compounds of the following formula (1): [Formula 1] in which
- X represents O or NH
- n denotes a number of 0 to 3
- Y represents a direct bond, -(CH 2 ) P O-, -(CH 2 ),-, or -(CH 2 ) q SO 2 -
- p denots a number of 0 to 2
- q denotes a number of 1 to 3
- Rl represents hydrogen, -(CR4R5) P -A-R6 or ⁇ (CR4R5) q -R6, p and q are as defined above, R4 and R5 independently of one another represent hydrogen or Ci-C 5 -alkyl,
- A represents 6-12 membered aryl or optionally oxo-containing C 3 -C 8 - cycloalkyl, or represents 3-10 membered heterocyclyl or heteroaryl each of which has 1 to 3 hetero atoms selected from O, S, and N,
- R6 represents hydrogen, hydroxy, halogen, nitro, d-C ⁇ -alkylcarbonyl, C 1 - C 6 -alkylsulfonyl, Q-C ⁇ -alkoxycarbonyl or carboxy,
- R2 represents hydrogen, nitro, halogen, Ci-C 6 -alkyl or trifluoromethyl, represents 5-12 membered heteroaryl or heterocyclyl each of which has 1 to 3 hetero atoms selected from N and O, or represents optionally Ci-C 6 -alkylsulfonyl-substituted 6-12 membered aryl, R3 represents R7-X-B-X'-,
- B represents a direct bond, or represents 3-10 membered heterocyclyl or heteroaryl each of which optionally contains oxo, is optionally fused, and has 1 to 4 hetero atoms selected from N, O and S
- X and X' independently of one another represent a direct bond, or are selected from the group consisting of -CO-, -(CH 2 ),-, -NR4C(O)-, -NR4-, -OC(O)-, -O-, - (CH 2 ) P C(O)-, -(CH 2 ) P O-, -(CH 2 ) P NR4-, -C(0)NR4- and -S(OV, wherein p and q are as defined above, r denotes a number of O to 2, and R4 represents hydrogen or Ci-C 5 -alkyl, R7 represents hydrogen, hydroxy, Ci-C 6 -alkyl, Cj-C 6 -alkoxy, halogeno-Ci-
- C 6 -alkyl or C 3 -C 6 -cycloalkyl represents 6-12 membered aryl, or represents 4-8 membered heteroaryl or heterocyclyl each of which has 1 to 4 hetero atoms selected from N and O, where alkyl, alkoxy, aryl, cycloalkyl, heterocyclyl and heteroaryl may be optionally substituted, and the substituents are one or more selected from the group consisting of hydroxy, halogen, nitrile, amino, d-C ⁇ -alkylamino, di(Ci-C 6 -alkyl)amino,
- Ci-C ⁇ -alkyl Ci-C ⁇ -alkyl, halogeno-Q-C ⁇ -alkyl, Ci-C 6 -alkylsulfonyl, aryl-Q-C ⁇ -alkoxy and oxo, pharmaceutically acceptable salts or isomers thereof.
- alkyl' means an aliphatic hydrocarbon radical.
- Alkyl may be saturated alkyl that does not comprise alkenyl or alkynyl moiety, or unsaturated alkyl that comprises at least one alkenyl or alkynyl moiety.
- Alkenyl means a group containing at least one carbon-carbon double bond
- alkynyl means a group containing at least one carbon-carbon triple bond.
- Alkyl may be branched or straight-chain when used alone or in a composite form such as alkoxy.
- Alkyl group may have 1 to 20 carbon atoms unless otherwise defined. Alkyl group may be a medium sized alkyl having 1 to 10 carbon atoms. Otherwise, alkyl group may be a lower alkyl having 1 to 6 carbon atoms. Typical examples thereof include, but not limited to, methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, t-butyl, pentyl, hexyl, ethenyl, propenyl, butenyl, etc.
- Ci-C 4 -alkyl has 1 to 4 carbon atoms in the alkyl chain, and is selected from the group consisting of methyl, ethyl, propyl, isopropyl, n-butyl, iso-butyl, sec-butyl and t-butyl.
- alkoxy means an alkyloxy having 1 to 10 carbon atoms unless otherwise defined.
- 'cycloalkyl' means a saturated aliphatic 3-10 membered cycle unless otherwise defined. Typical examples thereof include, but not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, etc.
- aryl' includes at least one ring having covalent ⁇ electron system, for example, monocyclic or fused polycyclic (i.e., cycles that share the adjacent carbon atom pairs) group.
- aryl means an aromatic 4-10 membered, preferably 6-10 membered, monocyclic or multicyclic ring including phenyl, naphthyl, etc., unless otherwise defined.
- heteroaryl means an aromatic 3-10 membered, preferably 4-8 membered, more preferably 5-6 membered cycle that has 1 to 3 hetero atoms selected from N, O and S, and may be fused with benzo or C 3 -C 8 cycloalkyl, unless otherwise defined.
- the monocyclic heteroaryl includes, but not limited to, thiazole, oxazole, thiophene, furan, pyrrole, imidazole, isoxazole, isothiazole, pyrazole, triazole, triazine, thiadiazole, tetrazole, oxadiazole, pyridine, pyridazine, pyrimidine, pyrazine and the like.
- the bicyclic heteroaryl includes, but not limited to, indole, indoline, benzothiophene, benzofuran, benzimidazole, benzoxazole, benzisoxazole, benzthiazole, benzthiadiazole, benztriazole, quinoline, isoquinoline, purine, puropyridine and the like.
- heterocycle means a 3-10 membered, preferably 4-8 membered, more preferably 5-6 membered cycle that has 1 to 3 hetero atoms selected from N, O and S, may be fused with benzo or C 3 -C 8 cycloalkyl, and is saturated or contains 1 or 2 double bonds, unless otherwise defined.
- the heterocycle includes, but not limited to, pyrroline, pyrrolidine, imidazoline, imidazolidine, pyrazoline, pyrazolidine, pyran, piperidine, morpholine, thiomorpholine, piperazine, hydrofuran and the like.
- Preferred compounds among the compounds of formula (1) above are those wherein
- X represents O or NH
- n denotes a number of 0 to 3
- Y represents a direct bond, -(CH 2 ) P O, -(CH 2 ) q -, or -(CH 2 ) q SO 2 -, p denotes a number of 0 to 2, q denotes a number of 1 to 3,
- Rl represents -(CR4R5) P -A-R6 or -(CR4R5) q -R6, p and q are as defined above,
- R4 and R5 independently of one another represent hydrogen or Ci-C 5 -alkyl
- A represents 6—12 membered aryl or optionally oxo-containing C 3 -C 7 - cycloalkyl, or represents 4-8 membered heterocyclyl or heteroaryl each of which has 1 to 3 hetero atoms selected from O, S, and N,
- R6 represents hydrogen, hydroxy, halogen, nitro, Ci-C 6 -alkylcarbonyl, C 1 - C ⁇ -alkylsulfonyl, Ci-C ⁇ -alkoxycarbonyl or carboxy,
- R2 represents hydrogen, halogen, Q-C ⁇ -alkyl or trifluoromethyl, represents 5-8 membered heteroaryl or heterocyclyl each of which has 1 to 3 hetero atoms selected from N and O, or represents optionally Ci-C 6 -alkylsulfonyl-substituted 6-10 membered aryl,
- R3 represents R7-X-B-X'-
- B represents a direct bond, or represents 4-10 membered heterocyclyl or heteroaryl each of which optionally contains oxo, is optionally fused, and has 1 to 4 hetero atoms selected from N, O and S,
- X and X' independently of one another represent a direct bond, or are selected from the group consisting of -CO-, -(CH 2 ) q -, -NR4C(O)-, -NR4-, -OC(O)-, -O-, - (CH 2 ) P C(O)-, -C(0)NR4- and -S(O) r , wherein p and q are as defined above, r denotes a number of 0 to 2, and R4 represents hydrogen or Q-Cs-alkyl, R7 represents hydrogen, hydroxy, Ci-C 6 -alkyl, halogeno-Ci-C 6 -alkyl or C 3 -
- C 6 -cycloalkyl represents 6-12 membered aryl, or represents 4-8 membered heteroaryl or heterocyclyl each of which has 1 to 4 hetero atoms selected from N and O, where alkyl, alkoxy, aryl, cycloalkyl, heterocyclyl and heteroaryl may be optionally substituted, and the substituents are one or more selected from the group consisting of hydroxy, halogen, nitrile, amino, Ci-C 6 -alkylamino, di(C 1 -C 6 -alkyl)amino, Ci-C ⁇ -alkyl, halogeno-C ! -C 6 -alkyl, Ci-C 6 -alkylsulfonyl, aryl-C ! -C 6 -alkoxy and oxo.
- the substituent Y more preferably represents a direct bond, -O-, -(CH 2 )O-, -(CH 2 )- or -(CH 2 )SO 2 -.
- Rl more preferably represents -(CH 2 ) P -A-R6 or -(CR4R5) q -R6, wherein p denotes a number of O to 2, q denotes a number of 1 to 3, R4 and R5 independently of one another represent hydrogen or d-C 5 -alkyl, A represents 6-12 membered aryl or optionally oxo-containing C 3 -C 6 -cycloalkyl or represents 5 ⁇ 6 membered heterocyclyl which has 1 to 2 hetero atoms selected from O, S, and N, and R6 represents hydrogen, halogen, nitro, Ci-C ⁇ -alkylcarbonyl, Ci-C 6 -alkylsulfonyl, Ci-C 6 - alkoxycarbonyl or carboxy.
- Rl is selected from the group consisting of cyclopropyl, cyclobutyl, cyclopentyl, difluorocyclohexyl, tetrahydrofuran, tetrahydropyran, (tetrahydropyran-4-yl)methyl, tetrahydrothiopyran, 4-oxo-cyclohexyl, (l-methanesulfonyl)pyrrolidine, (l-acetyl)piperidine, 4-nitrophenyl and methylpropiolate.
- the substituent R2 more preferably represents hydrogen, halogen, Ci-C 3 -alkyl or trifluoromethyl, represents 5 ⁇ 6 membered heteroaryl or heterocyclyl each of which has 1 to 3 hetero atoms selected from N and O, or represents optionally methanesulfonyl-substituted 6 ⁇ 10 membered aryl.
- R2 is selected from the group consisting of hydrogen, fluoro, chloro, bromo, methyl, ethyl, propyl, phenyl, methanesulfonylphenyl, pyridine, morpholine, 1,2-imidazole, 1,3-imidazole, pyrrolidine and pyrrole.
- the substituent B more preferably represents a direct bond, represents pyrazole, imidazole or oxadiazole each of which is optionally substituted by Q-C ⁇ -alkyl, or represents 5 ⁇ 9 membered heterocyclyl which optionally contains oxo, is optionally fused, and has 1 to 4 hetero atoms selected from N, S and O.
- B represents a direct bond, or may be a structure selected from the following formulae (i) to (xi)
- the substituent X' more preferably represents a direct bond, or is selected from the group consisting of -CO-, - NR4CO-, -SO 2 - and -0-.
- the substituent X more preferably represents a direct bond, or is selected from the group consisting of -C(0)NR4-, -NR4-, -OC(O)-, -NR4C(0)-, -(CH 2 )C(O)-, - S(O) 2 - and -C(O)-.
- X represents a direct bond, or is selected from the group consisting of -C(O)NH-, -C(O)N(Me)-, -NH-, -N(Me)-, -OC(O)-, - N(Me)C(O)-, -(CH 2 )C(O)-, -S(O) 2 - and -C(O)-.
- the substituent R7 more preferably represents hydrogen, hydroxy, Ci-C ⁇ -alkyl, halogeno-Ci-C 6 -alkyl or C 4 -C 6 -cycloalkyl, represents optionally halogen-substituted 6—10 membered aryl, or represents 5-6 membered heteroaryl or heterocyclyl each of which has 1 to 4 hetero atoms selected from N and O.
- R7 is selected from the group consisting of hydrogen, hydroxy, methyl, trifluoromethyl, ethyl, t-butyl, cyclohexyl, pyrrolidine, phenyl, 2-fluorophenyl, piperidine, pyridine, 1,3-pyrazine, 1,4- pyrazine, furan, trifluoromethyl, 1,2,3,4-tetrazole and tetrahydrofuran.
- Typical compounds among the compounds of formula (1) are those selected from the following:
- the compounds of formula (1) according to the present invention can also form a pharmaceutically acceptable salt.
- a pharmaceutically acceptable salt includes non-toxic acid addition salt containing pharmaceutically acceptable anion, for example, a salt with inorganic acids such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, hydrobromic acid, hydriodic acid, etc.; a salt with organic acids such as tartaric acid, formic acid, citric acid, acetic acid, trichloroacetic acid, trifluoroacetic acid, gluconic acid, benzoic acid, lactic acid, fumaric acid, maleic acid, salicylic acid, etc.; or a salt with sulfonic acids such as methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, etc.
- inorganic acids such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid,
- the pharmaceutically acceptable carboxylic acid salt includes a salt with alkali metals or alkaline earth metals such as lithium, sodium, potassium, calcium, magnesium, etc.; a salt with amino acids such as lysine, arginine, guanidine, etc.; or an organic salt with dicyclohexylamine, N-methyl-D-glucamine, tris(hydroxymethyl)methylamine, diethanolamine, choline, triethylamine, etc.
- the compounds of formula (I) of the present invention may be converted to their salts according to any of the conventional methods.
- the compounds of formula (1) of the present invention may have an asymmetric carbon center(s) in the structure, and so may exist in the form of R or S isomer, racemate, mixture of diastereomers, or individual diastereomer, etc. All the isomers are also covered by the present invention.
- the present invention also provides processes for preparing the compounds of formula (1).
- the processes for preparing the compounds of formula (1) are illustrated by exemplary reaction schemes for the purpose of better understanding.
- a skilled artisan in the field to which the present invention pertains may prepare the compounds of formula (1) via various routes according to their structures, and such processes should be construed to fall under the scope of the present invention.
- the compounds of formula (1) may be prepared by optionally combining various synthetic methods which are described in the present specification or disclosed in the prior arts.
- the processes for preparing the compounds of formula (1) cover even such processes, and are not limited to those explained below.
- the compounds of formula (1) can be prepared according to the following
- Reaction Scheme (1) by reducing the nitro group of Compound (2) to give an amine Compound (3), and introducing Rl substituent to the resulting amine group.
- the compounds of formula (1) can be prepared according to the following
- Compound (5) can be prepared according to the following Reaction Schemes (8) and (9).
- Compound (7) can be prepared according to the following Reaction Scheme (10), and
- Compound (20) can be prepared according to the following Reaction Scheme (11).
- a represents Fe, Zn, Pd/C, etc.
- Rl, R2, and R3 are as defined in formula (1)
- R8 represents Y-R2, wherein Y and R2 are as defined in formula (1).
- Compound (2) can be prepared according to the following Reaction Schemes (2) to (9).
- Compound (3) can be prepared by reducing the Compound (2).
- the reduction reaction may be carried out using an acid catalyst and metal, or using a metal catalyst in the presence of hydrogen gas.
- the acid that can be used in the reduction reaction using an acid catalyst and metal includes, for example, inorganic acids such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, etc., organic carboxylic acids such as acetic acid, trifluoroacetic acid, etc., aminates such as ammonium chloride, preferably hydrochloric acid, acetic acid, ammonium chloride, etc.
- the acid is typically used in the amount of 0.01 ⁇ 10 eq., preferably 0.1 - 5 eq., with respect to 1 eq. of the Compound (2).
- the metal that can be used includes, for example, iron, zinc, lithium, sodium, tin (usually, tin chloride), etc., particularly preferably iron, zinc, tin chloride, etc.
- the metal is typically used in the amount of 1 ⁇ 20 eq., preferably 1 - 10 eq., with respect to 1 eq. of the Compound (2).
- the reaction of metal in the presence of an acid catalyst may be carried out in an inert solvent.
- alkyl alcohols such as methanol, ethanol, etc.
- ethers such as tetrahydrofuran, diethylether, etc.
- alkyl esters such as ethyl acetate, etc., preferably methanol, ethanol, tetrahydrofuran, ethyl acetate, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C , preferably 25 ⁇ 120 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- the metal catalyst that can be used in the reduction reaction using a metal catalyst in the presence of hydrogen gas includes palladium, nickel, platinum, ruthenium, rhodium, etc., particularly preferably palladium, nickel, etc.
- the metal catalyst is typically used in the amount of 0.001 ⁇ 2 eq., preferably 0.01 ⁇ 1 eq., with respect to 1 eq. of the Compound (2).
- the hydrogen gas pressure is typically in the range of 1 ⁇ 10 atm, preferably 1 ⁇ 3 atm.
- the reaction may be carried out in an inert solvent, for example, alkyl alcohols such as methanol, ethanol, etc., ethers such as tetrahydrofuran, diethylether, etc., alkyl acetates such as methyl acetate, ethyl acetate, etc., preferably methanol, ethanol, ethyl acetate, etc.
- the reaction temperature using the metal catalyst is typically in the range of -10 ⁇ 200 " C, preferably 25 ⁇ 50 ° C, and the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (4) can be prepared via a reductive alkylation reaction of the Compound (3).
- the reductive alkylation on the amine group of the Compound (3) may be carried out with a ketone using a reducing agent, and if necessary, using an acid catalyst.
- the ketone is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 3 eq., with respect to 1 eq. of the Compound (3).
- the reducing agent that can be used includes sodium borohydride, sodium cyanoborohydride, sodium triacetoxyborohydride, etc.
- the reducing agent is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 3 eq., with respect to 1 eq. of the Compound (3).
- the acid catalyst that can be used includes, for example, inorganic acids such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, etc., organic carboxylic acids such as acetic acid, trifluoroacetic acid, etc., aminates such as ammonium chloride, particularly preferably hydrochloric acid, acetic acid, etc.
- the acid is typically used in the amount of 0.1 ⁇ 10 eq., preferably 1 ⁇ 5 eq., with respect to 1 eq. of the Compound (3).
- the reaction may be carried out in an inert solvent selected, for example, from ethers such as tetrahydrofuran, diethylether, etc., chloroalkanes such as dichloromethane, chloroform, dichloroethane, etc., preferably dichloroethane, chloroform, etc.
- the reaction temperature is typically in the range of - 10 ⁇ 100 ° C , preferably -10 - 50 "C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- the Compound (1) or (2) of the present invention can be prepared according to the processes that are specifically exemplified in the following Reaction Schemes (2) to (9).
- a represents a metal hydroxide (for example, NaOH, LiOH)
- b represents a coupling agent (for example, EDC, CDI, BOP-Cl) and Compound (7)
- c represents PCl 5 or Tf 2 O and Ph 3 PO
- d represents a coupling agent (for example, EDC, CDI, BOP-Cl) and
- R8 is as defined in the Reaction Scheme (1), R9 represents Ci-C 6 -alkyl,
- RlO represents NO 2 or Rl-X, wherein X and Rl are as defined in formula
- RI l represents p-MeOBn or Ph 3 C
- R3' and R3" independently of one another represent R7-X-B-, wherein R7, X and B are as defined in formula (1).
- Compound (5) can be prepared according to Reaction Schemes (8) and (9).
- Compound (6) can be prepared via hydrolysis reaction of the Compound (5) using a base.
- the base that can be used includes lithium hydroxide, sodium hydroxide, potassium hydroxide, etc.
- the base is typically used in the amount of 1 ⁇ 10 eq., preferably 1 - 5 eq., with respect to 1 eq. of the Compound (5).
- the hydrolysis reaction may be carried out in an inert solvent selected, for example, from water, alkyl alcohols such as methanol, ethanol, etc., ethers such as tetrahydrofuran, diethylether, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably 25 ⁇
- reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (8) can be prepared via a coupling reaction of the carboxylic acid of
- the known coupling agent that can be used in the coupling reaction includes, but not limited to, carboimides such as dicyclohexylcarbodiimide (DCC), l-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC), l,r-dicarbonyldiimidazole (CDI), etc.
- DCC dicyclohexylcarbodiimide
- EDC l-(3-dimethylaminopropyl)-3-ethylcarbodiimide
- CDI l,r-dicarbonyldiimidazole
- the coupling agent is typically used in the amount of 1 ⁇ 10 eq., preferably 1 - 3 eq., with respect to 1 eq. of the Compound (6).
- the amount of HOBT or HOAT used is typically in the range of 1 - 10 eq., preferably 1 ⁇ 3 eq., with respect to 1 eq. of the Compound (6).
- the base that can be used includes organic bases such as triethylamine, diisopropylethylamine.
- the base is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 3 eq., with respect to 1 eq. of the Compound (7).
- the coupling reaction may be carried out in an inert solvent selected from tetrahydrofuran, diethylether, N,N-dimethylformamide, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 "C, preferably 25 ⁇ 120 ° C, and the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (9) can be prepared by cyclizing the Compound (8) as described in Journal of Organic Chemistry, 68(24), 2003, 9506-9509, Tetrahedron, 55(34), 1999, 10271-10282, etc.
- PCI 5 is typically used in the amount of 1 - 10 eq., preferably 1 - 3 eq., with respect to 1 eq. of the Compound (8).
- the reaction temperature is typically in the range of -10 - 50 ° C , preferably 0 - 25 ° C, and the reaction time is typically in the range of 10 min - 60 h, preferably 10 min - 12 h.
- Rl 1 is triphenylmethyl (Ph 3 C) group
- the cyclization reaction is carried out in dichloromethane solvent using trifluoromethanesulfonic-anhydride (Tf 2 O) and triphenylphosphineoxide (Ph 3 PO), which are typically used in the amount of 1 - 10 eq., preferably 1 - 3 eq., with respect to 1 eq. of the Compound (8).
- the reaction temperature is typically in the range of -10 ⁇ 50 ° C , preferably 0 - 25 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (11) is an amine compound that is commercially available.
- Compound (12) can be prepared via a coupling reaction of the carboxylic acid of the Compound (10) with the Compound (11) according to the preparing process of the Compound (8).
- a represents a reducing agent (for example, NaBH 4 , LiAlH 4 ), b represents I 2 or MsCl, etc., c represents a base and the Compound (11), d represents a base and Compound (15), R8 is as defined in Reaction Scheme (1),
- R9 represents d-C 6 -alkyl
- RlO represents NO 2 or Rl-X, wherein X and Rl are as defined in formula
- R12 represents Ci -C ⁇ -alkoxy, cyano or 5-6 membered heteroaryl
- R' and R" are as defined in the Reaction Scheme (2), and W represents a leaving group, for example, halides such as chloride, bromide, iodide, etc., or sulfonates such as methane sulfonate, p-toluene sulfonate, etc.
- Compound (13) can be prepared by converting the ester group of Compound (9) to an alcohol group.
- the reducing agent that can be used to reduce the ester group to the alcohol group includes, for example, sodium borohydride, lithium borohydride, borane, lithium aluminum hydride, diisobutyl aluminum hydride (DIBAL-H), etc.
- the reducing agent is typically used in the amount of 1 ⁇ 10 eq., preferably 1 - 3 eq., with respect to 1 eq. of the Compound (9).
- the reaction may be carried out in an inert solvent selected, for example, from alcohols such as methanol, ethanol, etc., ethers such as tetrahydrofuran, diethylether, etc., preferably tetrahydrofuran, diethylether, etc.
- the reaction temperature is typically in the range of -78 ⁇ 100 °C, preferably -78 - 50 ° C, and the reaction time is typically in the range of 10 min — 60 h, preferably 10 min - 12 h.
- Compound (14) can be prepared by converting the alcohol group of the Compound (13) to a leaving group W.
- the leaving group W can be introduced via halogenation or sulfonation reaction.
- the halogenation reaction may be carried out using a halogenating agent selected from iodine, bromine, N-iodosuccimide (NIS), N- bromosuccimide (NBS), carbon tetrachloride (CCl 4 ), carbon tetrabromide (CBr 4 ), etc. in the presence of a base such as imidazole, dimethylaminopyridine (DMAP), etc.
- phosphines such as triphenylphosphine (Ph 3 P), tributylphosphine (Bu 3 P), etc.
- Each of the halogenating agent, base and phosphine is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 3 eq., with respect to 1 eq. of the Compound (13).
- This reaction may be carried out in an inert solvent selected, for example, from ethers such as tetrahydrofuran, diethylether, etc. and dichloromethane, chloroform, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C , preferably 0 - 50 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- the sulfonation reaction may be carried out using a sulfonating agent selected from methanesulfonyl chloride, p-toluenesulfonyl chloride, etc. in the presence of an organic base such as pyridine, triethylamine, etc.
- a sulfonating agent selected from methanesulfonyl chloride, p-toluenesulfonyl chloride, etc.
- an organic base such as pyridine, triethylamine, etc.
- Each of the sulfonating agent and base is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 5 eq., with respect to 1 eq. of the Compound (13).
- This reaction may be carried out in an inert solvent selected, for example, from ethers such as tetrahydrofuran, diethylether, etc., chloroalkanes such as dichloromethane, dichloroethane, chloroform, etc., preferably dichloromethane, dichloroethane, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably 0 ⁇ 50 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (16) can be prepared by a coupling reaction of the Compound (11) with the Compound (14) using a base.
- a base for example, inorganic bases such as sodium carbonate, potassium carbonate, cesium carbonate, etc., organic bases such as triethylamine, diisopropylethylamine, l,8-diazabicyclo[5,4,0]undeca-7-ene (DBU), etc. can be mentioned.
- the base is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 5 eq., with respect to 1 eq. of the Compound (14).
- This reaction may be carried out in an inert solvent selected, for example, from ethers such as tetrahydrofuran, diethylether, etc., alkyl nitriles such as acetonitrile, propionitrile, etc., amides such as N,N-dimethylformamide, etc., preferably tetrahydrofuran, acetonitrile, N,N- dimethylformamide, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably 25 ⁇ 120 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (17) can be prepared via a coupling reaction of the Compound (14) with the Compound (15) according to the preparing process of the Compound (16).
- a represents a coupling agent (for example, EDC, CDI, BOP-Cl) and
- R8 is as defined in the Reaction Scheme (1), RlO represents NO 2 or Rl-X, wherein X and Rl are as defined in formula
- Rl 3 represents C 3 -C 6 -cyc loalkyl or piperidinyl.
- Compound (18) can be prepared according to a method known in Heterocycles, 60(10), 2087, 2003 or Bioorganic & Medicinal Chemistry Letters, 11(24), 3164, 2001.
- Compound (19) can be prepared via a coupling reaction of the Compound (10) with the Compound (18).
- the coupling agent dicyclohexylcarbodiimide (DCC), 1- (3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC), l,l'-dicarbonyldiimidazole (CDI), etc. can be used, but not limited thereto.
- the coupling agent is typically used in the amount of 1 ⁇ 10 eq., preferably 1 - 3 eq., with respect to 1 eq. of the Compound (10).
- This reaction may be carried out in an inert solvent selected from tetrahydrofuran, diethylether, N,N-dimethylformamide, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably 25 ⁇ 120 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- a represents a coupling agent (for example, EDC, CDI, BOP-Cl)
- b represents PCl 5 or Tf 2 O and Ph 3 PO
- c represents a metal hydroxide (for example, NaOH, LiOH)
- d represents I 2 or MsCl, etc.
- e represents a base and the Compound (11), R8 is as defined in the Reaction Scheme (1),
- RlO represents NO 2 or Rl-X, wherein X and Rl are as defined in formula
- Rl 1 represents p-MeOBn or Ph 3 C
- R14 represents Ci-C 6 -alkyl
- R' and R" are as defined in the Reaction Scheme (2), and
- W represents a leaving group, for example, halides such as chloride, bromide, iodide, etc., or sulfonates such as methane sulfonate, p-toluene sulfonate, etc.
- Compound (21) can be prepared using the Compounds (6) and (20) according to the preparing process of the Compound (8) in the Reaction Scheme (2).
- Compound (22) can be prepared using the Compound (21) according to the preparing process of the Compound (9) in the Reaction Scheme (2).
- Compound (23) can be prepared using the Compound (22) according to the preparing process of the Compound (6) in the Reaction Scheme (2).
- Compound (24) can be prepared using the Compound (23) according to the preparing process of the Compound (14) in the Reaction Scheme (3).
- Compound (25) can be prepared using the Compound (24) according to the preparing process of the Compound (16) in the Reaction Scheme (3).
- a represents di-t-butyloxy-dicarbonyl (BoC 2 O), a base (for example,
- b represents a brominating agent (for example, N-bromosuccinimide
- NBS sodium acetate
- d represents an acid (for example, hydrochloric acid, trifluoroacetic acid)
- e represents a metal hydroxide (for example, NaOH, LiOH)
- f represents a coupling agent (for example, EDC, CDI, BOP-Cl) and
- R2 is as defined in formula (1)
- R9 represents Ci-C 6 -alkyl
- Rl 1 represents p-MeOBn
- R15 represents CrC ⁇ -alkoxycarbonyl or Ci-C ⁇ -alkylcarbonyloxy.
- Compound (27) can be prepared by protecting the amine group of the
- Boc 2 O used in the protection reaction of amine group is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 3 eq., with respect to 1 eq. of the Compound (26).
- the base is typically used in the amount of 1 ⁇ 10 eq., preferably 1 - 3 eq., with respect to 1 eq. of the Compound (26).
- a catalyst may be used for facilitating the reaction.
- the catalyst used is dimethylaminopyridine (DMAP), and typically used in the amount of
- This reaction may be carried out in an inert solvent selected from tetrahydrofuran, diethylether, N,N-dimethylformamide, dichloromethane, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C , preferably 25 ⁇ 120 " C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- the brominating agent used in the bromomethylation reaction includes N- bromosuccinimide (NBS) and l,3-dibromo-5,5-dimethylhydantoin, and is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 3 eq., with respect to 1 eq. of the Compound
- a catalyst may be used for facilitating the reaction.
- the catalyst used is 2,2'- azidobis(2-methylpropionitrile) (AIBN) or benzoyl peroxide, and typically used in the amount of 0.001 ⁇ 2 eq., preferably 0.01 ⁇ 0.3 eq., with respect to 1 eq. of the
- This reaction may be carried out in an inert solvent selected from benzene, toluene, carbon tetrachloride, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably 25 - 120 " C, and the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (28) can be prepared by reacting sodium acetate (NaOAc) with the Compound (27).
- Sodium acetate is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 5 eq., with respect to 1 eq. of the Compound (27).
- This reaction may be carried out in an inert solvent, for example, selected from ethers such as tetrahydrofuran, diethylether, etc., alkyl nitriles such as acetonitrile, propionitrile, etc., amides such as N,N-dimethylformamide, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably 25 ⁇ 120 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (29) can be prepared by removing the BOC group using an acid, and hydrolysis reaction using a base, in the order.
- the acid used in the removal of BOC group is hydrochloric acid, trifiuoroacetic acid, etc.
- the acid is typically used in the amount of 1 ⁇ 10 eq., preferably 2 - 5 eq., with respect to 1 eq. of the Compound (28).
- This reaction may be carried out in an inert solvent, for example, selected from ethers such as tetrahydrofuran, diethylether, dioxane, etc., alkyl alcohols such as methanol, ethanol, etc., chloroalkanes such as dichloromethane, chloroform, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C , preferably 25 ⁇ 120 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- the base used in the hydrolysis reaction includes lithium hydroxide, sodium hydroxide, potassium hydroxide, etc.
- the base is typically used in the amount of 2 ⁇ 20 eq., preferably 2 - 10 eq., with respect to 1 eq. of the Compound (28).
- This hydrolysis reaction may be carried out in an inert solvent, for example, selected from alkyl alcohols such as methanol, ethanol, etc., ethers such as tetrahydrofuran, diethylether, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably 25 ⁇ 120 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (30) can be prepared according to Reaction Schemes (10) and (11).
- Compound (31) can be prepared via a coupling reaction of the Compound (29) with the Compound (30) according to the preparing process of the Compound (8) in the Reaction Scheme (2).
- Compound (32) can be prepared by reacting PClswith the Compound (31). hi this reaction of using PCl 5 , cyclization and chlorination of the alcohol group occur simultaneously.
- PCl 5 is typically used in the amount of 1 ⁇ 10 eq., preferably 1 - 3 eq., with respect to 1 eq. of the Compound (31).
- This reaction may be carried out in a solvent selected from dichloromethane, chloroform, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C , preferably 0 ⁇ 50 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (33) is commercially available.
- Compound (34) can be prepared via a coupling reaction of the Compound (32) with the Compound (33) according to the preparing process of the Compound (16).
- a represents an acylating agent [for example, Rl 1 -CO-Cl, (Rl 1-CO) 2 O]
- b represents PCl 5
- c represents a metal hydroxide (for example, NaOH, LiOH)
- Rl 1 represents p-MeOBn
- Rl 4 represents Ci-C 6 -alkyl
- Rl 5 represents C ! -C 6 -alkoxycarbonyl or Ci-C ⁇ -alkylcarbonyloxy.
- Compound (35) can be prepared by protecting the alcohol group of the Compound (31) with an acyl group, and cyclizing using PCl 5 .
- the protection reaction of the alcohol group is carried out using a base and an acylating agent.
- the base used includes organic bases such as triethylamine, diisopropylethylamine, pyridine, etc.
- the base is typically used in the amount of 1 - 10 eq., preferably 1 - 5 eq., with respect to 1 eq. of the Compound (31).
- the acylating agent is typically used in the amount of 1 ⁇ 10 eq., preferably 1 - 3 eq., with respect to 1 eq. of the Compound (31).
- This reaction may be carried out in a solvent selected from dichloromethane, chloroform, dichloroethane, etc.
- the reaction temperature is typically in the range of - 10 - 200 ° C , preferably 0 - 50 ° C, and the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- the cyclization reaction uses PCl 5 .
- PCl 5 is typically used in the amount of 1 - 10 eq., preferably 2 - 5 eq., with respect to 1 eq. of the Compound (31).
- This reaction may be carried out in a solvent selected from dichloromethane, chloroform, etc.
- the reaction temperature is typically in the range of -10 - 200 ° C, preferably 0 - 50 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min - 12 h.
- Compound (36) can be prepared via a deprotection reaction of the hydroxyl group of the Compound (35) using a base.
- the base used in the deprotection reaction includes lithium hydroxide, sodium hydroxide, potassium hydroxide, etc.
- the base is typically used in the amount of 1 - 10 eq., preferably 1 - 5 eq., with respect to 1 eq. of the Compound (35).
- This reaction may be carried out in an inert solvent, for example, selected from water, alkyl alcohols such as methanol, ethanol, etc., ethers such as tetrahydrofuran, diethylether, etc.
- the reaction temperature is typically in the range of -10 - 200 ° C , preferably 25 - 120 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- a represents sodium nitrite (NaNO 2 ); tin chloride (SnCl 2 ), b represents a ketone compound (39), a base (for example, NaOAc), c represents an acid (for example, polyphosphoric acid PPA), d represents NaNO 2 , e represents Compound (42), a base (for example, NaOH),
- R8 is as defined in the Reaction Scheme (1), and
- R9 and RlO are as defined in the Reaction Scheme (2).
- Compound (37) is commercially available, or can be prepared by a method known in Heterocycles, 68(11), 2285-99, 2006, or Bioorganic & Medicinal Chemistry Letters, 14(19), 4903-4906, 2004.
- Compound (38) is commercially available, or can be prepared by converting the amine group of the Compound (37) to hydrazine group according to a method known in Journal of the American Chemical Society, 198(48), 15374-75, 2006.
- the hydrazine Compound (38) can be prepared by reacting the amine group of the Compound (37) with NaNO 2 in the presence of hydrochloric acid to give a diazonium salt (41), which is not separated and reduced by using SnCl 2 .
- NaNO 2 is typically used in the amount of 1 ⁇ 10 eq., preferably 2 - 5 eq., with respect to 1 eq. of the Compound (37).
- SnCl 2 is typically used in the amount of 1 ⁇ 10 eq., preferably 2 ⁇
- reaction temperature is in the range of -10 ⁇ 50 ° C
- reaction time is typically in the range of 10 min ⁇ 6O h, preferably 10 min ⁇ 6 h.
- Compound (39) is commercially available.
- Hydrazone Compound (40) can be prepared via a coupling reaction of the Compound (38) with the ketone Compound (39).
- a base is not used when the Compound (38) is in neutral form, but should be used when the Compound (38) is in the form of an acid salt to make the neutral form.
- the base for example, metal hydroxides such as sodium hydroxide, lithium hydroxide, etc., metal carbonates such as sodium bicarbonate, potassium carbonate, etc., metal acetates such as sodium acetate, etc., organic bases such as triethylamine, pyridine, etc., preferably sodium acetate, sodium bicarbonate, etc. can be used.
- the base is typically used in the amount of 1 ⁇ 5 eq., preferably 1 - 2 eq., with respect to 1 eq. of the Compound (38).
- This reaction may be carried out in an inert solvent selected from tetrahydrofuran, methanol, ethanol, etc.
- the reaction temperature is in the range of -10 ⁇ 100 "C, and the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- the Compound (40) can also be prepared by reacting the diazonium salt (41) with the Compound (42) in the presence of a base according to Japp-Klingemann rearrangement method described in Organic Process Research & Development, 2, 1988,
- Hydrochloric acid is used in the preparation of the diazonium salt (41) typically in the amount of 1 ⁇ 10 eq., preferably 2 ⁇ 4 eq., with respect to 1 eq. of the
- the base used in the reaction of the Compounds (41) and (42) is sodium hydroxide, which is typically used in the amount of 1 ⁇ 20 eq., preferably 1 ⁇ 10 eq., with respect to 1 eq. of the Compound (42).
- 80% aqueous ethanol solution is used as the solvent, and the reaction temperature is in the range of -10 ⁇ 50 ° C .
- the reaction time is typically in the range of 10 mim ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (5) can be prepared using an acid catalyst and the Compound (40).
- the acid used in the synthesis is polyphosphoric acid, hydrochloric acid, p- toluenesulfonic acid, sulfuric acid, acetic acid, etc., preferably polyphosphoric acid.
- Polyphosphoric acid can be used alone, or as a mixture with aromatic hydrocarbons such as benzene, toluene, etc.
- the reaction temperature is in the range of 25 - 150 ° C, and the reaction time is typically in the range of 5 mim ⁇ 60 h, preferably 5 min - 12 h.
- a represents a sodium alkoxide (for example, sodium methoxide)
- b represents heat
- Rl is as defined in formula (1)
- R8 is as defined in the Reaction Scheme (1), and R9 represents Ci-C 6 -alkyl.
- Compound (43) is commercially available.
- Compound (44) can be prepared by a method known in Journal of Medicinal Chemistry, 31(11), 2145, 1988.
- Compound (45) is commercially available, or can be prepared by a method known in WO 2007040289, WO200601079 or Organic Letters 9(3), 397-400, 2007.
- the Compound (45) can be prepared via a coupling reaction of the
- the base used is sodium methoxide, sodium ethoxide, etc.
- the base is typically used in the amount of 1 ⁇ 10 eq., preferably 1 ⁇ 3 eq., with respect to 1 eq. of the Compound (43).
- This reaction may be carried out in an inert solvent, for example, selected from alkyl alcohols such as methanol, ethanol, etc., ethers such as tetrahydrofuran, diethylether, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably -10 - 25 ° C
- the reaction time is typically in the range of 10 min - 60 h, preferably 10 min - 12 h.
- Compound (46) can be prepared by cyclizing the Compound (45).
- the cyclization reaction may be carried out by dissolving the Compound (45) in an inert solvent, and heating the solution.
- the inert solvent that can be used includes tetrahydrofuran, benzene, toluene, etc.
- the reaction temperature is typically in the range of 25 ⁇ 200 ° C , preferably 50 ⁇ 120 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- a represents p-methoxybenzylchloride (PMBCl) or triphenylmethyl- chloride (TrCl), a base (for example, NaOH), b represents di-t-butyloxy-dicarbonyl (BoC 2 O), a base (for example,
- c represents an alkylchloroformate (for example, EtOCOCl), a base (for example, N-methylmorpholine), d represents diazomethane (CH 2 N 2 ), a base (for example, KOH), e represents a silver ion (for example, silver benzoate), f represents an acid, g represents MsCl, Et 3 N, h represents p-methoxybenzylthiol (PMBSH), NaH, R9 represents Ci-C 6 -alkyl, and Rl 1 represents p-MeOBn or Ph 3 C.
- alkylchloroformate for example, EtOCOCl
- a base for example, N-methylmorpholine
- d represents diazomethane (CH 2 N 2 )
- a base for example, KOH
- e represents a silver ion (for example, silver benzoate)
- f represents an acid
- g represents MsCl
- Et 3 N
- h represents
- Compound (47) can be prepared by protecting the thiol group of cysteine using p-methoxybenzyl chloride (PMBCl) or triphenylmethyl chloride (TrCl) in the presence of a base.
- PMBCl p-methoxybenzyl chloride
- TrCl triphenylmethyl chloride
- PMBCl or TrCl used in the protection reaction of the thiol group is typically used in the amount of 1 ⁇ 5 eq., preferably 1 - 2 eq., with respect to 1 eq. of cysteine.
- the base used is sodium hydroxide, potassium carbonate, etc., and is typically used in the amount of 1 ⁇ 5 eq, preferably 1 ⁇ 2 eq., with respect to 1 eq. of cysteine.
- This reaction may be carried out in an inert solvent selected from tetrahydrofuran, methanol, ethanol, water, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C , preferably 0 ⁇ 50 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (48) can be prepared by protecting the amine group of the Compound (47) using BOC group.
- Boc 2 O used in the protection reaction of the amine group is typically used in the amount of 1 ⁇ 5 eq., preferably 1 ⁇ 2 eq., with respect to 1 eq. of cysteine.
- the base used is selected, for example, from hydroxides such as sodium hydroxide, lithium hydroxide, etc., carbonates such as sodium carbonate, sodium bicarbonate, potassium carbonate, cesium carbonate, etc., organic bases such as diisopropylethylamine, triethylamine, etc., preferably potassium carbonate, triethylamine, etc.
- This reaction may be carried out in an inert solvent selected from tetrahydrofuran, methanol, ethanol, water, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably 0 ⁇ 50 " C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- Compound (49) can be prepared by a method known in Helvetica Chimica Acta, 87, 2004, 3131-3159.
- an Ag ion for example, silver trifluoroacetate (CF 3 CO 2 Ag), silver benzoate, etc.
- an alkyl alcohol for example, methanol, ethanol, etc.
- the reaction for removing the BOC group can be made using an acid.
- the acid used includes hydrochloric acid, trifluoroacetic acid, etc.
- the acid is typically used in the amount of 1 - 10 eq., preferably 2 - 5 eq., with respect to 1 eq. of the Compound (48).
- This reaction may be carried out in an inert solvent, for example, selected from ethers such as tetrahydrofuran, diethylether, dioxane, etc., alkyl alcohols such as methanol, ethanol, etc., chloroalkanes such as dichloromethane, chloroform, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C, preferably 25 ⁇ 120 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min - 12 h.
- Compound (50) can be prepared from glutamic acid or aspartic acid by a method known in Synlett, 15, 2005, 2397-2399, Journal of Organic Chemistry, 66(5), 2001, 1919-1923, etc.
- Compound (51) can be prepared by sulfonating the Compound (50).
- the sulfonation reaction may be carried out using methanesulfonyl chloride in the presence of an organic base such as pyridine, triethylamine, etc.
- the sulfonylating agent and base each are used in the amount of 1 ⁇ 10 eq., preferably 1 - 5 eq., with respect to 1 eq. of the Compound (50).
- This reaction may be carried out in an inert solvent selected from dichloromethane, dichloroethane, etc.
- the reaction temperature is typically in the range of -10 - 200 ° C , preferably 0 - 50 ° C, and the reaction time is typically in the range of 10 min - 60 h, preferably 10 min - 12 h.
- the Compound (7) can be prepared by reacting p-methoxybenzylthiol
- PMBSH PMBSH with the Compound (51) in the presence of a base, and removing the BOC group using an acid.
- the base used is sodium hydride, potassium carbonate, cesium carbonate, etc., preferably sodium hydride.
- the base is typically used in the amount of 1 - 10 eq., preferably 2 - 5 eq., with respect to 1 eq. of the Compound (51).
- p- Methoxybenzylthiol (PMBSH) is typically used in the amount of 1 ⁇ 10 eq., preferably 2 ⁇ 5 eq., with respect to 1 eq. of the Compound (51).
- This reaction may be carried out in an inert solvent selected from tetrahydrofuran, dimethylformamide, N- methylpyrrolidinone, etc.
- the reaction temperature is typically in the range of -10 ⁇ 200 ° C , preferably 25 ⁇ 100 ° C
- the reaction time is typically in the range of 10 min ⁇ 60 h, preferably 10 min ⁇ 12 h.
- the reaction for removing BOC group may be carried out in the same manner as the removal of BOC group explained in the preparing process of the Compound (49).
- a represents an alkyl alcohol (for example, methanol, ethanol), acetyl chloride or thionyl chloride, b represents di-t-butyloxy-dicarbonyl (BoC 2 O), a base (for example,
- c represents a reducing agent (for example, NaBH 4 )
- d represents an alkylcarbonyl chloride (for example, t-butylcarbonyl chloride ( 1 BuCOCl))
- a base for example, Et 3 N
- e represents an acid
- R9 represents Ci-C 6 -alkyl
- Rl 1 represents p-MeOBn or Ph 3 C
- R14 represents Ci-C 6 -alkyl
- Compound (52) can be prepared by esterifying the carboxylic group of the Compound (47), and protecting the amine group with BOC group.
- the esterification reaction may be carried out using acetyl chloride or thionyl chloride in an alkyl alcohol solvent.
- Acetyl chloride or thionyl chloride is used in the amount of 1 ⁇ 10 eq., preferably 1 - 5 eq., with respect to 1 eq. of the Compound (47).
- the reaction temperature is typically in the range of 25 - 200 ° C, preferably 25 - 100 ° C
- the reaction time is typically in the range of 10 min - 60 h, preferably 10 min - 12 h.
- the protection reaction of the amine group may be carried out in the same manner as the preparing process of the Compound (48).
- the Compound (20) can be prepared from the starting Compound (52) via reduction of the ester group, protection of the alcohol group, and removal of BOC, in the order.
- the reduction reaction of the ester group may be carried out by reacting with an alkylchloro formate (for example, ethylchloroformate, isobutylchloroformate) in tetrahydrofuran solvent of room temperature in the presence of 1 ⁇ 5 eq. of a base (for example, triethylamine, diisopropylethylamine, N-methylmorpholine, etc.) to give an anhydride, which is then reacted with 1 - 5 eq. of lithium borohydride or sodium borohydride in aqueous tetrahydrofuran solution of 0 ⁇ 25 "C for 10 min — 12 h.
- an alkylchloro formate for example, ethylchloroformate, isobutylchloroformate
- the protection reaction of the alcohol group may be carried out by reacting with an alkylcarbonylchloride, for example, t-BuCOCl, in dichloromethane solvent of 0 ⁇ 25 ° C in the presence of 1 ⁇ 5 eq. of a base selected from triethylamine, pyridine, etc. for 10 min ⁇ 12 h.
- an alkylcarbonylchloride for example, t-BuCOCl
- the reaction for removing BOC group may be carried out by dissolving the reactant in an inert solvent selected from tetrahydrofuran, dioxane, ethyl acetate, dichloromethane, etc. and reacting with 1 ⁇ 10 eq. of hydrochloric acid or trifluoroacetic acid at 0 ⁇ 50 ° C for lO min - 12 h.
- an inert solvent selected from tetrahydrofuran, dioxane, ethyl acetate, dichloromethane, etc.
- the compounds whose preparation methods are not specifically explained in the present specification are known per se, or can be prepared from a known compound according to a known process or a similar process thereto.
- the compounds of formula (1) obtained by the above processes may be separated or purified from the reaction product by various methods such as recrystallization, ionophoresis, silica gel column chromatography, ion exchange chromatography, etc.
- the compounds according to the present invention may be obtained by various processes, and such processes for preparing the compounds of formula (1) should be construed to fall under the scope of the present invention.
- the present invention further provides a pharmaceutical composition for the activation of glucokinase, which comprises the compounds of formula (1), pharmaceutically acceptable salts or isomers thereof as an active ingredient together with pharmaceutically acceptable carriers.
- Diseases which are caused by the deactivation of glucokinase, and can be prevented or treated by the pharmaceutical composition of the present invention include, but not limited to, diabetes, complications of diabetes, obesity, etc.
- the pharmaceutical composition of the present invention can be used for the prevention or treatment of type 1 diabetes or type 2 diabetes, and is particularly preferable for type 2 diabetes.
- the complications of diabetes that can be prevented or treated by the pharmaceutical composition of the present invention include, but not limited to, hyperlipidemia, hypertension, retinosis, renal failure, etc.
- the present invention further provides a hypoglycemic composition which comprises the compounds of formula (1), pharmaceutically acceptable salts or isomers thereof as an active ingredient together with pharmaceutically acceptable carriers.
- the present invention further provides a process for preparing a pharmaceutical composition for the activation of glucokinase, more specifically, for the prevention or treatment of diabetes, complications of diabetes, or obesity, which comprises the step of mixing the compounds of formula (1), pharmaceutically acceptable salts or isomers thereof as an active ingredient together with pharmaceutically acceptable carriers.
- compositions may comprise pharmaceutically acceptable carriers, diluents, excipients, or their combinations, if needed, together with the compounds of the present invention.
- Pharmaceutical composition facilitates the administration of the compound into a living organism. There exist a number of techniques to administer the compound, and they include, but not limited to, oral, injectable, aerosol, parenteral and topical administration.
- carrier means a substance which facilitates the incorporation of the compound into the cells or tissues.
- DMSO dimethylsulfoxide
- carrier is a typical carrier which is used to facilitate the introduction of various organic compounds into the cells or tissues of living organisms.
- diluent is defined as a substance that is diluted in water which dissolves the compound, as well as stabilizes the biologically active form of the subject compound.
- the salts dissolved in buffer solution are utilized as diluents in the art.
- buffer solution is phosphate buffered saline which mimics the salt form of human solution. Buffer diluents rarely alter the biological activities of the compound, as the buffer salts can control the pH of solution at a low concentration.
- pharmaceutically acceptable means the property that does not impair the biological activities and physical properties of the compound.
- the compounds of the present invention can be formulated as various pharmaceutical dosage forms according to the purpose.
- the active ingredient specifically, the compounds of formula (1), pharmaceutically acceptable salts or isomers thereof are mixed together with various pharmaceutically acceptable carriers which can be selected according to the formulation to be prepared.
- the pharmaceutical composition of the present invention can be formulated as injectable preparation, oral preparation, etc., according to the purpose.
- the compounds of the present invention can be formulated by the methods known in the art, which utilize pharmaceutical carriers and excipients known in the art, and be incorporated into the containers of unit dose form or multi-dose form.
- the form of the preparation can be solutions, suspensions or emulsions in oily or aqueous media, and may contain typical dispersing agents, suspending agents or stabilizers. Further, for example, it can be a form of dry powder which is intended to be reconstructed by dissolving in sterile, pyrogen-free water prior to use.
- the compounds of the present invention also can be formulated into suppository forms utilizing typical suppository bases such as cocoa butter or other glycerides.
- solid dosage forms for oral administration capsules, tablets, pills, powder and granule can be prepared, and capsules and tablets are especially useful.
- tablets and pills are prepared as enteric coated forms.
- Solid dosage forms can be prepared by mixing the compounds of the present invention together with carriers, for example, one or more inert diluents such as sucrose, lactose, starch, etc., lubricants such as magnesium stearate, disintegrant, binder, etc.
- the compounds of the present invention or the pharmaceutical compositions containing the same can also be administered in combination with other active agents, for example, other agents for treating diabetes.
- the dosage of the compounds of formula (1) depends on the prescription of a physician, taking into account such factors as body weight or age of a patient, specific nature of the disease, and severity of the disease, etc. However, dosage needed for the treatment of an adult is typically from about 1 to 500 mg per day, depending on the intensity and frequency of the administration. When administered to an adult via intramuscular or intravenous routes, total dosage typically from about 5 to 300 mg per day will be sufficient when separately administered in a single dosage, but for some patients a higher daily dosage may be desirable.
- the present invention further provides a method for the prevention or treatment of diseases which are caused by the deactivation of glucokinase, using effective amount of the compounds of formula (1), pharmaceutically acceptable salts or isomers thereof as an active ingredient.
- the present invention further provides a process for preparing a pharmaceutical composition for the prevention or treatment of diseases which are caused by the deactivation of glucokinase, which comprises the step of mixing the compounds of formula (1), pharmaceutically acceptable salts or isomers thereof as an active ingredient together with pharmaceutically acceptable carriers.
- treatment means the interrupting or delaying the progress of the disease when applied to the subject showing the onset of disease symptoms
- prevention means the interrupting or delaying the sign of the onset of disease when applied to the subject that does not show, but is at risk of, the onset of disease symptoms.
- M means molar concentration
- N means normal concentration
- the resulting acetamide compound was dissolved in dichloromethane (200ml), and fuming nitric acid (13ml, 0.29 mol) was added in drops thereto at 0 ° C .
- the mixture was stirred for 1 h at 0 " C ⁇ room temperature.
- Saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with ethyl acetate. The extract was washed with saturated sodium chloride solution, and dried over anhydrous magnesium sulfate.
- the resulting nitrate compound was dissolved in methanol (100ml) and tetrahydrofuran (100ml), and 6N sodium hydride was added in drops thereto. The mixture was stirred for 6 h at room temperature. After completion of the reaction, the reaction solution was neutralized to about pH 7 using 6N hydrochloric acid solution, and extracted with ethyl acetate. The extract was washed with saturated sodium chloride solution, and dried over anhydrous magnesium sulfate to give the title compound (44g, Yield 83%).
- the compound thus obtained was dissolved using water (100ml), tetrahydrofuran (100ml) and methanol (100ml). Iron powder (103g, 1.84 mol) and ammonium chloride (99g, 1.84 mol) were added thereto, and the mixture was stirred using a mechanical stirrer for 3 h at 80 "C . After completion of the reaction, the reaction solution was filtered through a celite, washed with methanol, and concentrated.
- Step 2 nylann ⁇ no-3-(4 yl)-prop ⁇ on ⁇ c
- the compound (30.7g, 127.3mmol) prepared in Step 1 was dissolved in tetrahydrofuran (150ml) and water (150ml). Potassium carbonate (26.4g, 190mmol) and di-t-butyloxy-dicarbonyl (27.7g, 127.3mmol) were added thereto, and the mixture was stirred for 2 h at room temperature. After completion of the reaction, the reaction solution was distilled under reduced pressure to remove tetrahydrofuran. The residue was cooled to 0 ° C , and acidified to pH 3 using 3N aqueous hydrochloric acid solution.
- the compound prepared in Step 3 was dissolved in methanol (1000ml), silver benzoate (7.1g, 31.1mmol) was added thereto, and the mixture was sonicated for 1 h.
- the compound prepared in Step 1 was dissolved in tetrahydrofuran (200ml) and water (200ml). Triethylamine (87ml, 621.6mmol) was added thereto, and di-t- butyloxy-dicarbonyl (43.Og, 196.8mmol) dissolved in tetrahydrofuran (100ml) was added in drops thereto while stirring. The mixture was stirred for 8 h at room temperature. After completion of the reaction, water was added to the reaction solution, which was then extracted with ethyl acetate.
- Triethylamine (58ml, 414.4mmol) and trimethylacetyl chloride (28ml, 227.9mmol) were added thereto, and the mixture was stirred for 6 h at 0 ° C . After completion of the reaction, water was added, and the reaction solution was extracted with ethyl acetate.
- the compound (900mg, 2.7mmol) prepared in Preparation 19 was dissolved in 1 ,2-dichloroethane (100ml). Tetrahydro-4H-pyran-4-one (0.8ml, 8.13mmol), sodium triacetoxyborohydride (1.72g, 8.13mmol) and acetic acid (0.47ml, 8.13mmol) were added thereto, and the mixture was stirred for 48 h at room temperature. After completion of the reaction, the reaction solution was diluted with dichloromethane, washed with saturated sodium bicarbonate solution, dried over anhydrous magnesium sulfate, and filtered.
- the compound prepared in Step 1 was dissolved in methanol (32ml), tetrahydrofuran (32ml) and water (16ml). IN sodium hydroxide (7ml) was added thereto, and the mixture was stirred for 4 h at room temperature. After completion of the reaction, the reaction solution was distilled under reduced pressure, extracted with dichloromethane, washed with saturated sodium chloride solution, dried over anhydrous magnesium sulfate, and filtered. The filtrate was distilled under reduced pressure, and purified by column chromatography to give the title compound (700mg, Yield 78%).
- the compound (460mg, 0.9mmol) prepared in Step 1 was dissolved in methanol (50ml), and 4N hydrochloric acid solution (0.8ml, 2.7mmol) was added thereto. The mixture was stirred for 8 h at room temperature, distilled under reduced pressure, and purified by column chromatography.
- Example 8 Synthesis of ⁇ (R)-2-[5-chloro-7-(tetrahydro-pyran-4-ylamino)- lH-indol-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -methanol ⁇ (R)-2-[5-Chloro-7-(tetrahydro-pyra n-4-ylamino)-1 H-indol-2-yl]-4,5-dih ydro-thiazol-4-yl ⁇ -methanol
- Example 14 Synthesis of ⁇ (R)-2-[5-(pyridin-3-yloxy)-7-(tetrahydro-pyran- 4-ylamino)-lH-indol-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -methanoI H-indol-2-
- Example 17 Synthesis of cyclopentyl-[2-((R)-4-dimethylaminomethyl-4,5- dihydro-thiazol-2-yl)-lH-indol-7-yl]-amine Cyclopentyl-[2-((R)-4-dimethylamino methyl-4,5-dihydro-thiazol-2-yl)-1H
- the compound (24.Og, lOOmmol) prepared in Preparation 7 was dissolved in dichloromethane (500ml). Triethylamine (84ml, ⁇ Olmmol) and 4- (dimethylamino)pyridine (600mg, 5mmol) were added, and di-t-butyloxy-dicarbonyl (43. Ig, 200mmol) dissolved in dichloromethane (100ml) was added in drops thereto. The mixture was stirred for 8 h at room temperature. After completion of the reaction, water was added.
- Preparation 29 Synthesis of 2,2-dimethyl-propionic acid (R)-2-(5- chIoromethyl-7-nitro-lH-indol-2-yI)-4,5-dihydro-thiazol-4-ylmethyl ester acid (R)-2-( e
- the compound (1.Og, 1.9mmol) prepared in Preparation 28 was dissolved in dichloromethane (30ml). Phosphorus pentachloride (0.8g, 3.9mmol) was added thereto, and the mixture was stirred for 6 h at room temperature. After completion of the reaction, saturated sodium bicarbonate solution was added. The mixture was extracted with ethyl acetate, dried over anhydrous magnesium sulfate, and filtered. The filtrate was distilled under reduced pressure to give the title compound (0.7g, Yield 90%).
- Step 1 2,2-Dimethyl-propionic acid (R)-2-( 7-nitro-5-pyrazol-1 -ylmethyl-1 H-ind ol-2-yl)-4,5-dihydro-thiazol-4-ylme thyl ester
- Step 1 The compound (300mg, 0.68mmol) prepared in Step 1 was reacted according to the same procedures as Step 4 of Preparation 19, Preparation 20 and Example 1 in the order to give the title compound (42mg, Yield 24%).
- Example 25 Synthesis of [7-cyclopentylamino-2-((R)-4-hydroxymethyl-4,5- dihydro-thiazol-2-yl)-l H-indoI-5-yl] -methanol [7-Cyclopentylam ⁇ no-2-((R)-4-hydrox ymethyl-4,5-d ⁇ hydro-th ⁇ azol-2-yl)-1
- Step 2 The compound (15. Ig, 60.8mmol) prepared in Step 1 was reacted according to the same procedures as Preparation 23 and Preparation 24 to give the title compound (6.3g, Yield 24%).
- Preparation 33 Synthesis of S-chloro- ⁇ -nitro-lH-indole- ⁇ -carboxylic acid -7-n ⁇ tro-1 H- ⁇ ndole-2-carbox
- the compound (15.Og, 59.1mmol) prepared in Preparation 5 was dissolved in tetrahydrofuran (300ml) and methanol (100ml).
- Lithium hydroxide monohydrate (7.43g, 177mmol) was dissolved in water (100ml), and added to the reaction solution, which was then stirred for 3 h at room temperature. After completion of the reaction, the reaction solution was distilled under reduced pressure to remove tetrahydrofuran and methanol. The residue was neutralized to about pH 6 using 3N hydrochloric acid solution. The resulting solid was filtered and dried to give the title compound (13. Ig, Yield 92%).
- Step 2 ⁇ H ⁇ --i ⁇ n ⁇ iduoul ⁇ --2. yl]-acet ⁇ es cf t ⁇ e r r
- dichloromethane 200ml
- Phosphorus pentachloride 17. Ig, 82mmol
- the reaction solution was concentrated, and diethylether (200ml) was added.
- Example 26 The compound (1.5g, 3.83mmol) prepared in Example 26 was dissolved in tetrahydrofuran (100ml) and methanol (50ml). Lithium hydroxide monohydrate (640mg, 15.3mmol) was dissolved in water (50ml) and added to the reaction solution, which was then stirred for 4 h at room temperature. After completion of the reaction, the reaction solution was distilled under reduced pressure to remove tetrahydrofuran and methanol. IN hydrochloric acid solution was added to the residue, and the mixture was extracted with ethyl acetate, washed with saturated sodium chloride solution, dried over anhydrous magnesium sulfate, and filtered. The filtrate was distilled under reduced pressure and separated by column chromatography to give the title compound (13. Ig, Yield 92%).
- Example 28 Synthesis of [(R)-2-(5-chloro-7-cyclopentylamino-lH-indol-2- yl)-4,5-dihydro-thiazol-4-yl] -acetic acid ethyl ester
- the compound (l.Og, 3.1mmol) prepared in Preparation 34 was dissolved in 1,2- dichloroethane (100ml). Tetrahydro-4H-pyran-4-one (0.57ml, 6.18mmol), sodium triacetoxyborohydride (1.31g, 6.18mmol) and acetic acid (0.18ml, 3.09mmol) were added thereto, and the mixture was stirred for 24 h at room temperature. After completion of the reaction, the reaction solution was diluted with dichloromethane, washed with saturated sodium bicarbonate solution, dried over anhydrous magnesium sulfate, and filtered. The filtrate was distilled under reduced pressure, and purified by column chromatography to give the title compound (0.5g, Yield 40%).
- Example 30 The compound (400mg, l.Ommol) prepared in Example 30 was reacted according to the same procedure as Example 27 to give the title compound (360mg, Yield 92%).
- Example 30 The compound (2.5g, 6.12mmol) prepared in Example 30 was reacted according to the same procedure as Example 29 to give the title compound (2.19g, 5.76mmol, Yield 94%).
- Example 34 Synthesis of 2-[(R)-2-(5-bromo-7-cyclopentylamino-lH-indol- 2-yl)-4,5-dihydro-l,3-thiazol-4-yl]-ethanol 2-[(R)-2-(5-Bromo-7-cyclopentylamin o-1 H-indol-2-yl)-4,5-dihydro-thiazo l-4-yl]-ethanol
- Example 35 Synthesis of ⁇ (R)-2-[5-bromo-7-(tetrahydro-pyran-4-ylamino)- lH-indoI-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -acetic acid ⁇ (R)-2-[5-Bromo-7-(tetrahydro-pyran -4-ylamino)-1 H-indol-2-yl]-4,5-dihy dro-thiazol-4-yl ⁇ -acetic acid
- Example 37 The compound (80mg, 0.22mmol) prepared in Example 37 was dissolved in ethanol (2ml). Acetyl chloride (0.1ml) was added thereto, and the mixture was stirred for 8 h at room temperature. After completion of the reaction, the reaction solution was diluted with ethyl acetate, washed with saturated sodium bicarbonate solution, dried over anhydrous magnesium sulfate, and filtered. The filtrate was distilled under reduced pressure, and purified by column chromatography to give the title compound (50mg, Yield 58%).
- Example 40 Synthesis of ⁇ (R)-2-[5-fluoro-7-(tetrahydro-pyran-4-ylamino)- lH-indol-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -acetic acid ⁇ (R)-2-[5-Fluoro-7-(tetrahydro-pyra n-4-ylamino)-1 H-indol-2-yl]-4,5-dih ydro-thiazol-4-yl ⁇ -acetic acid
- Step 2 The compound prepared in Step 1 was reacted according to the same procedure as Example 27 to give the title compound (5.44g, 2 steps, Yield 91%).
- Example 43 Synthesis of [(R)-2-(7-cyclopentylamino-lH-indol-2-yl)-4,5- dihydro-thiazol-4-yl]-acetic acid ethyl ester [(R)-2-(7-Cyclopentylamino-1 H-indol -2-yl)-4,5-dihydro-thiazol-4-yl]-ac etic acid ethyl ester
- Example 42 The compound (500mg, 1.46mmol) prepared in Example 42 was reacted according to the same procedure as Example 38 to give the title compound (420mg, Yield 78%).
- Example 48 Synthesis of [(R)-2-(7-cyclopentylamino-5-methoxy-lH-indol- 2-yl)-4,5-dihydro-thiazol-4-yl] -acetic acid [(R)-2-(7-Cyclopentylam ⁇ no-5-methox y-1 H- ⁇ ndol-2-yl)-4,5-d ⁇ hydro-th ⁇ azo l-4-yl]-acet ⁇ c acid
- Example 47 The compound (300mg, 0.78mmol) prepared in Example 47 was reacted according to the same procedure as Example 27 to give the title compound (240mg, Yield 82%).
- Example 48 The compound (200mg, 0.54mmol) prepared in Example 48 was reacted according to the same procedure as Example 38 to give the title compound (124mg, Yield 57%).
- Example 50 Synthesis of ⁇ (R)-2-[5-methoxy-7-(tetrahydro-pyran-4- ylamino)-lH-indol-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -acetic acid methyl ester
- Example 51 Synthesis of ⁇ (R)-2-[5-methoxy-7-(tetrahydro-pyran-4- ylamino)-lH-indol-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -acetic acid
- Example 52 Synthesis of [(R)-2-(7-cyclopentylamino-5-ethoxy-lH-indol-2- yl)-4,5-dihydro-thiazol-4-yl] -acetic acid [(R)-2-(7-Cyclopentylam ⁇ no-5-ethoxy -1H- ⁇ ndol-2-yl)-4,5-d ⁇ hydro-th ⁇ azol
- Example 55 Synthesis of ⁇ (R)-2-[5-phenoxy-7-(tetrahydro-pyran-4- ylamino)-lH-indol-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -acetic acid
- Example 56 Synthesis of ⁇ (R)-2-[7-cycIopentylamino-5-(pyridin-3-yloxy)- lH-indol-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -acetic acid methyl ester ⁇ (R)-2-[7-Cyclopentylam ⁇ no-5-(pyr ⁇ d ⁇ n-3-yloxy)-1 H- ⁇ ndol-2-yl]-4,5-d ⁇ hy dro-th ⁇ azol-4-yl ⁇ -acet ⁇ c acid methy I ester
- Example 56 The compound (35mg, 0.08mmol) prepared in Example 56 was reacted according to the same procedure as Example 27 to give the title compound (15mg, Yield 44%).
- Example 58 Synthesis of ⁇ (R)-2-[5-(pyridin-3-yloxy)-7-(tetrahydro-pyran- 4-ylamino)-lH-indol-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -acetic acid methyl ester ⁇ (R)-2-[5-(Py ⁇ d ⁇ n-3-yloxy)-7-(tetr ahydro-pyran-4-ylam ⁇ no)-1 H- ⁇ ndol-2- yl]-4,5-d ⁇ hydro-th ⁇ azol-4-yl ⁇ -acet ⁇ c acid methyl ester
- Example 58 The compound (25mg, 0.05mmol) prepared in Example 58 was reacted according to the same procedure as Example 27 to give the title compound (15mg, Yield 58%).
- Example 60 The compound (12mg, 0.03mmol) prepared in Example 60 was reacted according to the same procedure as Example 27 to give the title compound (5mg, Yield 43%).
- Example 62 Synthesis of ⁇ (R)-2-[5-methyI-7-(tetrahydro-pyran-4-ylamino)- 1 H-indol-2-yl] -4,5-dihydro-thiazol-4-yl ⁇ -acetic acid
- Example 64 Synthesis of ⁇ (R)-2-[7-cycIopentylamino-5-(4-methanesulfonyl- phenoxy)-lH-indol-2-yl]-4,5-dihydro-thiazol-4-yl ⁇ -acetic acid
- Example 65 The compound (l lmg, 0.02mmol) prepared in Example 65 was reacted according to the same procedure as Example 27 to give the title compound (5mg, Yield 45%).
- Example 68 The compound (29mg, 0.07mmol) prepared in Example 68 was reacted according to the same procedure as Example 27 to give the title compound (19mg, Yield 67%).
- Example 68 The compound (720mg, l. ⁇ Ommol) prepared in Example 68 was dissolved in tetrahydrofuran (20ml). 2 M lithium borohydride tetrahydrofuran solution (1.6ml, 3.2mmol) was added thereto, and the mixture was stirred for 3 h at room temperature.
- Example 70 The compound (178mg, 0.42mmol) prepared in Example 70 was dissolved in tetrahydrofuran (10ml). Iodine (161mg, 0.63mmol), triphenylphosphine (166mg,
- Example 72 Synthesis of l-(4- ⁇ 2-[(R)-2-(7-cyclopentylamino-5- methanesulfonylmethyl-lH-indol-2-yl)-4,5-dihydro-thiazol-4-yl]-ethyl ⁇ -piperazin-l- yl)-ethanone 1-(4- ⁇ 2-[(R)-2-(7-Cyclopentylamino- 5-methanesulfonylmethyl-1 H-indol-2- yl)-4,5-dihydro-thiazol-4-yl]-ethyl ⁇ -piperazin-1-yl)-ethanone
- Example 27 The compound (50mg, 0.13mmol) prepared in Example 27 was dissolved in
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Ophthalmology & Optometry (AREA)
- Emergency Medicine (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Child & Adolescent Psychology (AREA)
- Urology & Nephrology (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
Abstract
Description


































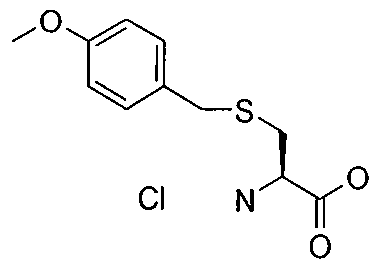









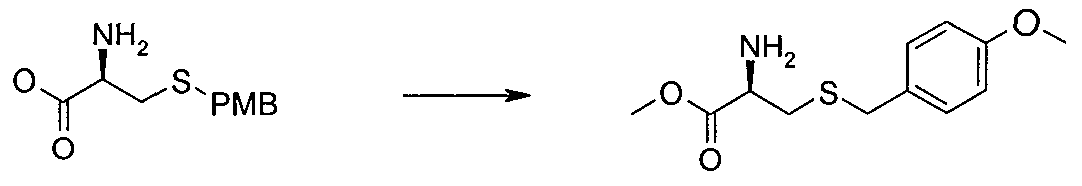




















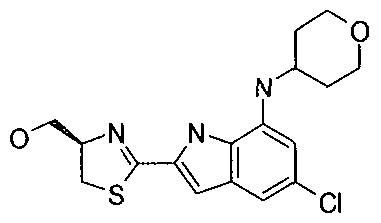
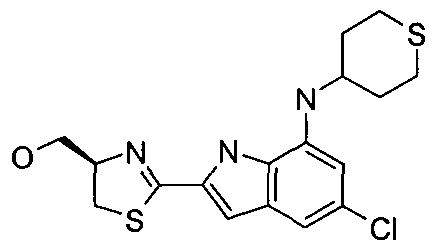












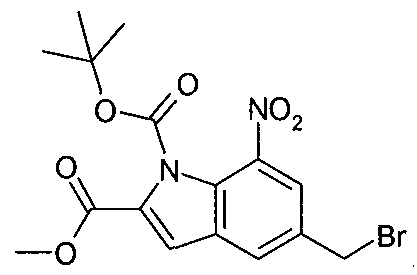


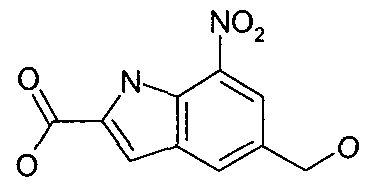
































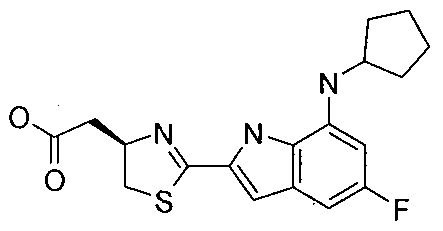





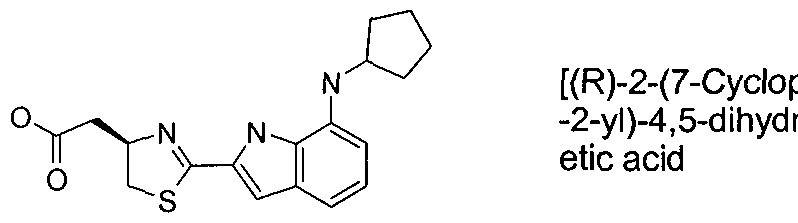


























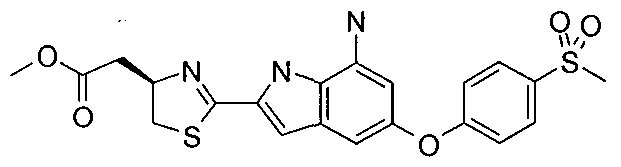




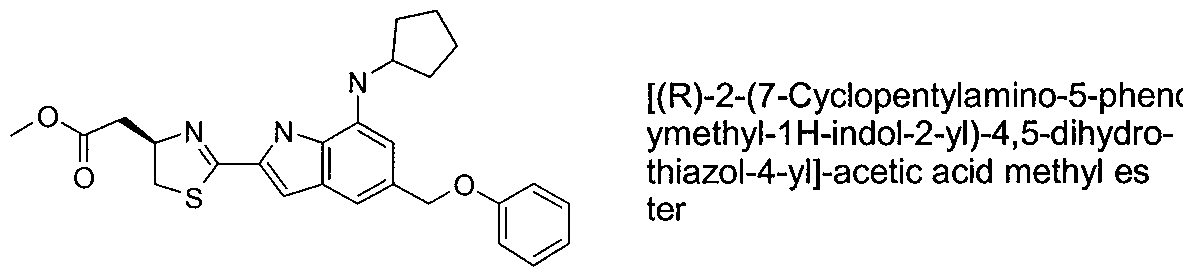


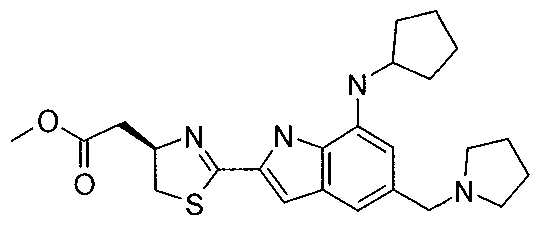





























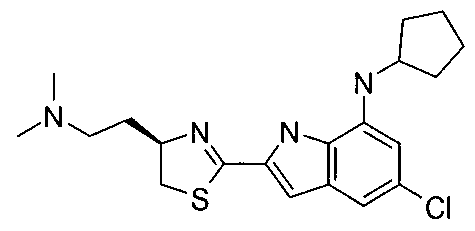




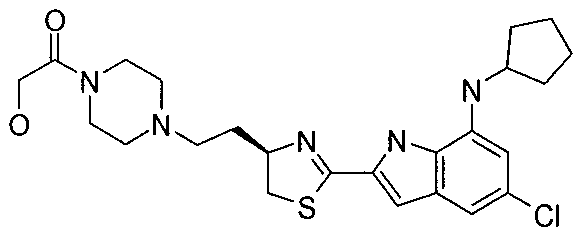















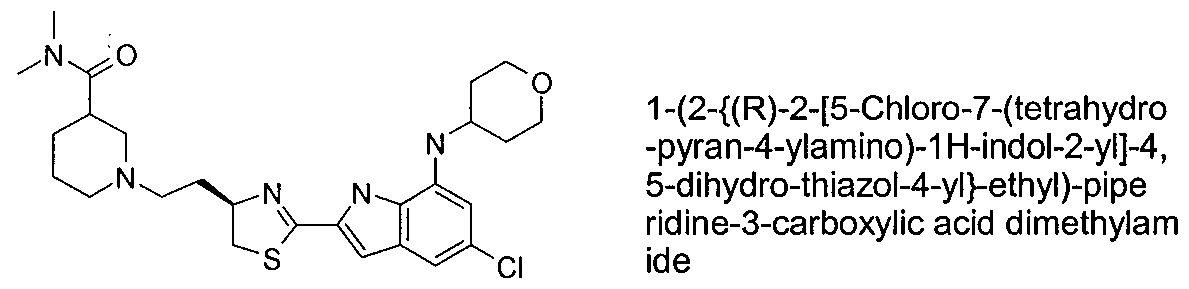



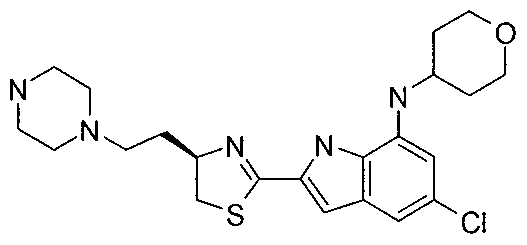
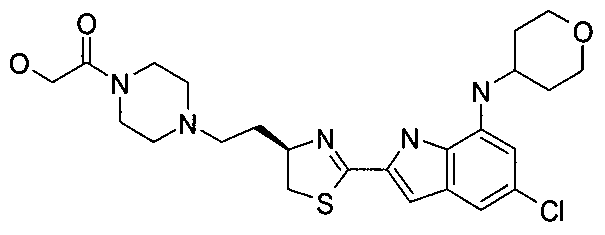


































































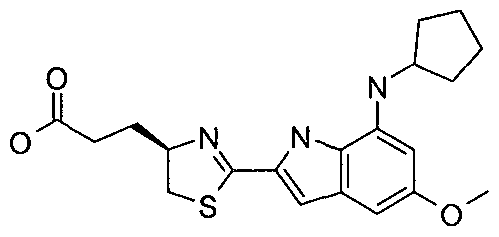




























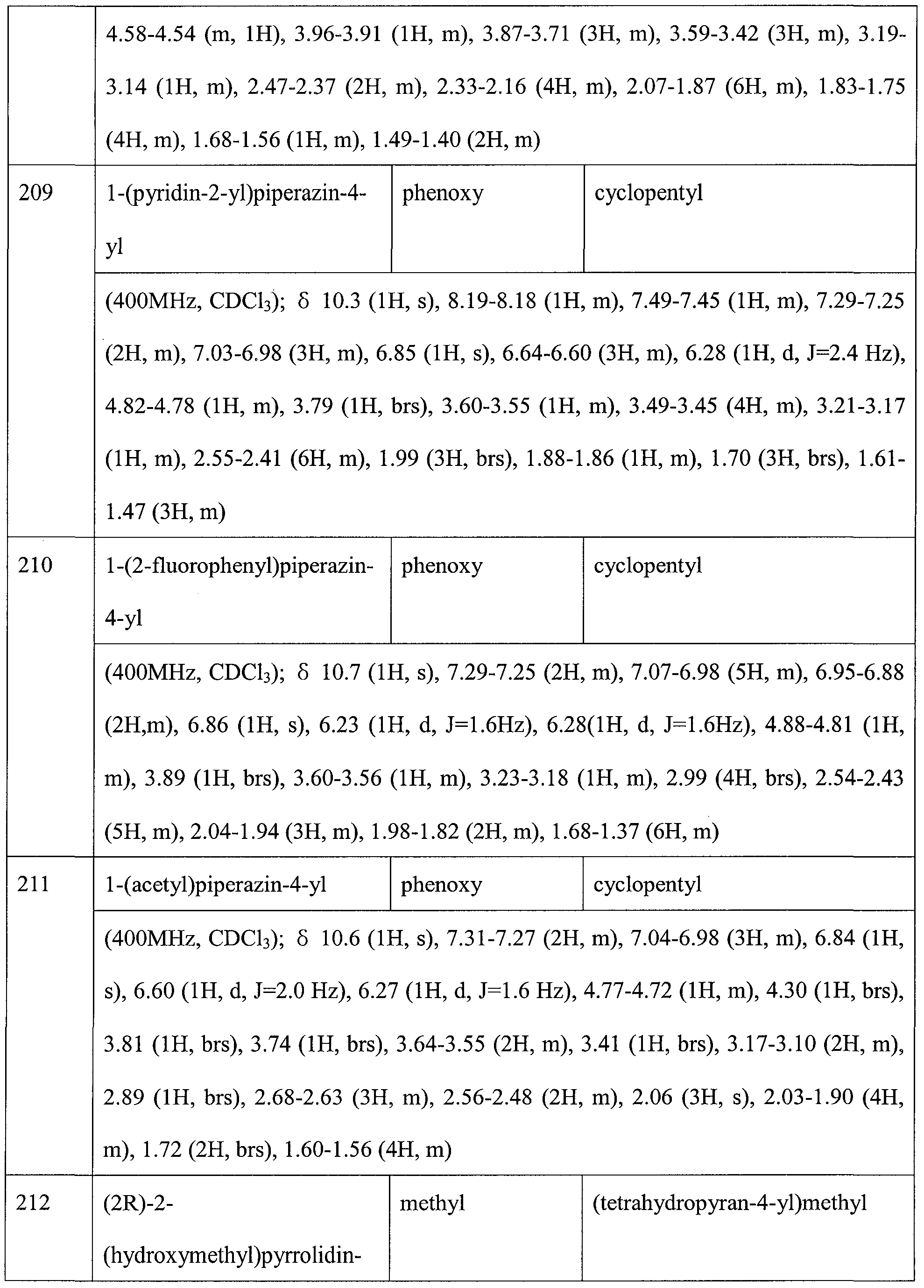







Claims




Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010539310A JP5364103B2 (en) | 2007-12-20 | 2008-12-22 | Glucokinase activator and pharmaceutical composition containing it as an active ingredient |
| US12/742,991 US20100267708A1 (en) | 2007-12-20 | 2008-12-22 | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| CN2008801208137A CN101896483B (en) | 2007-12-20 | 2008-12-22 | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| EP08864050A EP2220081A4 (en) | 2007-12-20 | 2008-12-22 | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| BRPI0819655-9A BRPI0819655B1 (en) | 2007-12-20 | 2008-12-22 | COMPOUND AND PHARMACEUTICAL COMPOSITION |
| US13/329,831 US8309586B2 (en) | 2007-12-20 | 2011-12-19 | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20070134705 | 2007-12-20 | ||
| KR10-2007-0134705 | 2007-12-20 |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/742,991 A-371-Of-International US20100267708A1 (en) | 2007-12-20 | 2008-12-22 | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| US13/329,831 Division US8309586B2 (en) | 2007-12-20 | 2011-12-19 | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| US13/329,831 Continuation US8309586B2 (en) | 2007-12-20 | 2011-12-19 | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009082152A2 true WO2009082152A2 (en) | 2009-07-02 |
| WO2009082152A3 WO2009082152A3 (en) | 2009-09-24 |
Family
ID=40801688
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2008/007585 WO2009082152A2 (en) | 2007-12-20 | 2008-12-22 | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US20100267708A1 (en) |
| EP (1) | EP2220081A4 (en) |
| JP (1) | JP5364103B2 (en) |
| KR (1) | KR101133772B1 (en) |
| CN (1) | CN101896483B (en) |
| RU (1) | RU2450001C2 (en) |
| WO (1) | WO2009082152A2 (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2230238A2 (en) * | 2008-01-04 | 2010-09-22 | LG Life Sciences Ltd. | Indole and indazole derivatives having a cell-, tissue- and organ-preserving effect |
| JP2010536845A (en) * | 2007-08-17 | 2010-12-02 | エルジー・ライフ・サイエンシーズ・リミテッド | Indole compounds as cell necrosis inhibitors |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011123572A1 (en) | 2010-03-31 | 2011-10-06 | The Scripps Research Institute | Reprogramming cells |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| EP2493469A2 (en) * | 2009-10-26 | 2012-09-05 | LG Life Sciences Ltd | Pharmaceutical composition comprising indole compound |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8673942B2 (en) | 2008-04-10 | 2014-03-18 | Takeda Pharmaceutical Company Limited | Fused ring compounds and use thereof |
| EP3107919A4 (en) * | 2014-02-20 | 2017-11-22 | University of Florida Research Foundation | Macrocyclic therapeutic agents, methods of manufacture, and methods of treatment |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101186502B1 (en) * | 2011-02-16 | 2012-09-27 | 한국생산기술연구원 | Composition of acid catalysts for preparation of 5-chloromethyl-2-furfural from algal galactans and preparing method of 5-chloromethyl-2-furfural from algal galactans under biphasic condition using thereof |
| WO2014099578A1 (en) * | 2012-12-17 | 2014-06-26 | Merck Sharp & Dohme Corp. | Novel glucokinase activator compounds, compositions containing such compounds, and methods of treatment |
| KR101941004B1 (en) * | 2013-03-25 | 2019-01-23 | 주식회사 엘지화학 | A pharmaceutical composition for suppressing immune response via promoting differentiation and proliferation of regulatory T cell |
| GB201714777D0 (en) | 2017-09-14 | 2017-11-01 | Univ London Queen Mary | Agent |
| CN109134325A (en) * | 2018-09-14 | 2019-01-04 | 成都市科隆化学品有限公司 | A kind of S-(trityl)-L-cysteine preparation method |
| EP3971185B1 (en) * | 2019-06-19 | 2024-04-17 | LG Chem, Ltd. | Method for preparing indole or indazole compound |
| CN111554969A (en) * | 2020-06-19 | 2020-08-18 | 中节能万润股份有限公司 | Lithium ion battery electrolyte additive containing sulfonic acid phosphonium salt, preparation method and application thereof |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1510207A1 (en) | 2002-06-05 | 2005-03-02 | Institute of Medicinal Molecular Design, Inc. | Therapeutic drug for diabetes |
| WO2005049019A1 (en) | 2003-11-24 | 2005-06-02 | Novo Nordisk A/S | N-heteroaryl indole carboxamides and analogues thereof, for use as glucokinase activators in the treatment of diabetes |
| WO2006049304A1 (en) | 2004-11-02 | 2006-05-11 | Banyu Pharmaceutical Co., Ltd | Aryloxy-substituted benzimidazole derivatives |
| WO2006112549A1 (en) | 2005-04-20 | 2006-10-26 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| WO2007007910A1 (en) | 2005-07-13 | 2007-01-18 | Banyu Pharmaceutical Co., Ltd. | Heterocycle-substituted benzimidazole derivative |
| WO2007028135A2 (en) | 2005-09-01 | 2007-03-08 | Takeda Pharmaceutical Company Limited | Imidazopyridine compounds |
| WO2007031739A1 (en) | 2005-09-16 | 2007-03-22 | Astrazeneca Ab | Heterobicyclic compounds as glucokinase activators |
| WO2007037534A1 (en) | 2005-09-30 | 2007-04-05 | Banyu Pharmaceutical Co., Ltd. | 2-heteroaryl-substituted indole derivative |
| WO2007041365A2 (en) | 2005-09-30 | 2007-04-12 | Novartis Ag | 3-cyclyl-2- (4-sulfamo yl-phenyl) -n-cyclyl-propionamide derivatives useful in the treatment of impaired glucose tolerance and diabetes |
| WO2007043638A1 (en) | 2005-10-14 | 2007-04-19 | Astellas Pharma Inc. | Condensed heterocyclic compound |
| US20070099930A1 (en) | 2005-11-01 | 2007-05-03 | Joseph Dudash | Substituted Dihydroisoindolones As Allosteric Modulators of Glucokinase |
| WO2007053345A1 (en) | 2005-11-01 | 2007-05-10 | Array Biopharma Inc. | Glucokinase activators |
| WO2007051847A1 (en) | 2005-11-03 | 2007-05-10 | Prosidion Ltd | Tricyclo substituted amides as glucokinase modulators |
| WO2009025477A1 (en) | 2007-08-17 | 2009-02-26 | Lg Life Sciences Ltd. | Indole compounds as an inhibitor of cellular necrosis |
| EP2230238A2 (en) | 2008-01-04 | 2010-09-22 | LG Life Sciences Ltd. | Indole and indazole derivatives having a cell-, tissue- and organ-preserving effect |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2508524A3 (en) * | 2006-10-19 | 2012-10-24 | Takeda Pharmaceutical Company Limited | Indole compound |
-
2008
- 2008-12-22 EP EP08864050A patent/EP2220081A4/en not_active Withdrawn
- 2008-12-22 RU RU2010130095/04A patent/RU2450001C2/en active
- 2008-12-22 CN CN2008801208137A patent/CN101896483B/en active Active
- 2008-12-22 US US12/742,991 patent/US20100267708A1/en not_active Abandoned
- 2008-12-22 WO PCT/KR2008/007585 patent/WO2009082152A2/en active Application Filing
- 2008-12-22 KR KR1020080131504A patent/KR101133772B1/en active IP Right Grant
- 2008-12-22 JP JP2010539310A patent/JP5364103B2/en active Active
-
2011
- 2011-12-19 US US13/329,831 patent/US8309586B2/en active Active
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1510207A1 (en) | 2002-06-05 | 2005-03-02 | Institute of Medicinal Molecular Design, Inc. | Therapeutic drug for diabetes |
| WO2005049019A1 (en) | 2003-11-24 | 2005-06-02 | Novo Nordisk A/S | N-heteroaryl indole carboxamides and analogues thereof, for use as glucokinase activators in the treatment of diabetes |
| WO2006049304A1 (en) | 2004-11-02 | 2006-05-11 | Banyu Pharmaceutical Co., Ltd | Aryloxy-substituted benzimidazole derivatives |
| WO2006112549A1 (en) | 2005-04-20 | 2006-10-26 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| WO2007007910A1 (en) | 2005-07-13 | 2007-01-18 | Banyu Pharmaceutical Co., Ltd. | Heterocycle-substituted benzimidazole derivative |
| WO2007028135A2 (en) | 2005-09-01 | 2007-03-08 | Takeda Pharmaceutical Company Limited | Imidazopyridine compounds |
| WO2007031739A1 (en) | 2005-09-16 | 2007-03-22 | Astrazeneca Ab | Heterobicyclic compounds as glucokinase activators |
| WO2007037534A1 (en) | 2005-09-30 | 2007-04-05 | Banyu Pharmaceutical Co., Ltd. | 2-heteroaryl-substituted indole derivative |
| WO2007041365A2 (en) | 2005-09-30 | 2007-04-12 | Novartis Ag | 3-cyclyl-2- (4-sulfamo yl-phenyl) -n-cyclyl-propionamide derivatives useful in the treatment of impaired glucose tolerance and diabetes |
| WO2007043638A1 (en) | 2005-10-14 | 2007-04-19 | Astellas Pharma Inc. | Condensed heterocyclic compound |
| US20070099930A1 (en) | 2005-11-01 | 2007-05-03 | Joseph Dudash | Substituted Dihydroisoindolones As Allosteric Modulators of Glucokinase |
| WO2007053345A1 (en) | 2005-11-01 | 2007-05-10 | Array Biopharma Inc. | Glucokinase activators |
| WO2007051847A1 (en) | 2005-11-03 | 2007-05-10 | Prosidion Ltd | Tricyclo substituted amides as glucokinase modulators |
| WO2009025477A1 (en) | 2007-08-17 | 2009-02-26 | Lg Life Sciences Ltd. | Indole compounds as an inhibitor of cellular necrosis |
| EP2230238A2 (en) | 2008-01-04 | 2010-09-22 | LG Life Sciences Ltd. | Indole and indazole derivatives having a cell-, tissue- and organ-preserving effect |
Non-Patent Citations (1)
| Title |
|---|
| ZIMMER P. ET AL., NATURE, vol. 414, 2001, pages 782 |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010536845A (en) * | 2007-08-17 | 2010-12-02 | エルジー・ライフ・サイエンシーズ・リミテッド | Indole compounds as cell necrosis inhibitors |
| EP2230238A4 (en) * | 2008-01-04 | 2012-11-07 | Lg Life Sciences Ltd | Indole and indazole derivatives having a cell-, tissue- and organ-preserving effect |
| EP2230238A2 (en) * | 2008-01-04 | 2010-09-22 | LG Life Sciences Ltd. | Indole and indazole derivatives having a cell-, tissue- and organ-preserving effect |
| US8673942B2 (en) | 2008-04-10 | 2014-03-18 | Takeda Pharmaceutical Company Limited | Fused ring compounds and use thereof |
| US10322135B2 (en) | 2009-10-26 | 2019-06-18 | Lg Chem, Ltd. | Pharmaceutical composition comprising indole compound |
| EP2493469A4 (en) * | 2009-10-26 | 2013-05-29 | Lg Life Sciences Ltd | Pharmaceutical composition comprising indole compound |
| JP2013508452A (en) * | 2009-10-26 | 2013-03-07 | エルジー・ライフ・サイエンシーズ・リミテッド | Pharmaceutical composition comprising an indole compound |
| EP2493469A2 (en) * | 2009-10-26 | 2012-09-05 | LG Life Sciences Ltd | Pharmaceutical composition comprising indole compound |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| EP3199623A1 (en) | 2010-03-31 | 2017-08-02 | The Scripps Research Institute | Reprogramming cells |
| WO2011123572A1 (en) | 2010-03-31 | 2011-10-06 | The Scripps Research Institute | Reprogramming cells |
| EP3936608A1 (en) | 2010-03-31 | 2022-01-12 | The Scripps Research Institute | Reprogramming cells |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| EP3107919A4 (en) * | 2014-02-20 | 2017-11-22 | University of Florida Research Foundation | Macrocyclic therapeutic agents, methods of manufacture, and methods of treatment |
| US11718645B2 (en) | 2014-02-20 | 2023-08-08 | University Of Florida Research Foundation, Incorporated | Macrocyclic therapeutic agents, methods of manufacture, and methods of treatment |
Also Published As
| Publication number | Publication date |
|---|---|
| KR101133772B1 (en) | 2012-04-24 |
| EP2220081A2 (en) | 2010-08-25 |
| WO2009082152A3 (en) | 2009-09-24 |
| KR20090067125A (en) | 2009-06-24 |
| RU2450001C2 (en) | 2012-05-10 |
| US20120088760A1 (en) | 2012-04-12 |
| CN101896483B (en) | 2013-11-20 |
| US8309586B2 (en) | 2012-11-13 |
| US20100267708A1 (en) | 2010-10-21 |
| JP5364103B2 (en) | 2013-12-11 |
| BRPI0819655A2 (en) | 2019-03-19 |
| EP2220081A4 (en) | 2011-08-24 |
| CN101896483A (en) | 2010-11-24 |
| RU2010130095A (en) | 2012-01-27 |
| JP2011507833A (en) | 2011-03-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009082152A2 (en) | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient | |
| KR101098583B1 (en) | Indole and indazole compounds having the effect of the cell, tissue and organ protection | |
| EP2178869B1 (en) | Indole compounds as an inhibitor of cellular necrosis | |
| KR100681724B1 (en) | Tyrosine kinase inhibitors and pharmaceutical compositions comprising the same | |
| KR102151288B1 (en) | Orexin receptor antagonists which are [ortho bi (hetero )aryl]-[2-(meta bi (hetero )aryl)-pyrrolidin-1-yl]-methanone derivatives | |
| JPH10511687A (en) | Substituted N- (indole-2-carbonyl) -glycinamides and derivatives as glycogen phosphorylase inhibitors | |
| EA007968B1 (en) | N-substituted pyrrolidin derivatives as dipeptidyl peptidase iv inhibitors | |
| ZA200600821B (en) | 3,5 Disubstituted indazole compounds, pharmaceutical compositions, and methods for mediating or inhibiting cell proliferation | |
| WO2012017020A1 (en) | N-((6-amino-pyridin-3-yl)methyl)-heteroaryl-carboxamides as inhibitors of plasma kallikrein | |
| AU2018361364A1 (en) | Novel, highly active amino-thiazole substituted indole-2-carboxamides active against the hepatitis B virus (HBV) | |
| WO2007063928A1 (en) | Novel noncyclic amine carboxamide derivative and salt thereof | |
| KR20210153051A (en) | macrocyclic compounds | |
| KR20130087283A (en) | Composition containing indole and indazole derivatives for inhibition of cancer metastasis | |
| CN107949562A (en) | The positive allosteric modulators of muscarinic M2 acceptors | |
| WO2024026484A2 (en) | Cdk2 inhibitors and methods of using the same | |
| KR20080065674A (en) | Novel indole-containing beta agonists, method for producing them and their use as drugs | |
| US20230014137A1 (en) | Compound having lysophosphatidic acid receptor agonist activity and pharmaceutical use thereof | |
| US20120028931A1 (en) | Heterocyclic m-glu5 antagonists | |
| BRPI0819655B1 (en) | COMPOUND AND PHARMACEUTICAL COMPOSITION | |
| KR20220052934A (en) | Substituted 1,3-phenyl heteroaryl derivatives and their use in the treatment of diseases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880120813.7 Country of ref document: CN |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08864050 Country of ref document: EP Kind code of ref document: A2 |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2008864050 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12742991 Country of ref document: US Ref document number: 3742/CHENP/2010 Country of ref document: IN Ref document number: MX/A/2010/006777 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010539310 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010130095 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0819655 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100531 |