WO2009068531A2 - Organic compounds - Google Patents
Organic compounds Download PDFInfo
- Publication number
- WO2009068531A2 WO2009068531A2 PCT/EP2008/066159 EP2008066159W WO2009068531A2 WO 2009068531 A2 WO2009068531 A2 WO 2009068531A2 EP 2008066159 W EP2008066159 W EP 2008066159W WO 2009068531 A2 WO2009068531 A2 WO 2009068531A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- pharmaceutically acceptable
- dpp
- compounds
- nvp
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/26—Acyclic or carbocyclic radicals, substituted by hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B41—PRINTING; LINING MACHINES; TYPEWRITERS; STAMPS
- B41J—TYPEWRITERS; SELECTIVE PRINTING MECHANISMS, i.e. MECHANISMS PRINTING OTHERWISE THAN FROM A FORME; CORRECTION OF TYPOGRAPHICAL ERRORS
- B41J2/00—Typewriters or selective printing mechanisms characterised by the printing or marking process for which they are designed
- B41J2/005—Typewriters or selective printing mechanisms characterised by the printing or marking process for which they are designed characterised by bringing liquid or particles selectively into contact with a printing material
- B41J2/01—Ink jet
- B41J2/17—Ink jet characterised by ink handling
- B41J2/175—Ink supply systems ; Circuit parts therefor
- B41J2/17593—Supplying ink in a solid state
Definitions
- the present invention provides new dipeptidyl peptidase-IV (DPP-IV) inhibitors which are effective in treating conditions mediated by DPP-IV. It was discovered that DPP-IV is responsible for inactivating glucagon-like peptide-1 (GLP-1 ). Since GLP-1 is a major stimulator of pancreatic insulin secretion and has direct beneficial effects on glucose disposal, DPP-IV inhibition appears to represent an attractive approach for treating conditions such as non-insulin-dependent diabetes mellitus (NIDDM).
- NIDDM non-insulin-dependent diabetes mellitus
- the instant invention relates to a compound of formulae (I A), (I B), (X A), (X B), (Y A) or (Y B) (compounds of the invention)
- R' represents
- OH and R" represents hydrogen, hydroxy, CrC 7 alkoxy, CrC 8 -alkanoyloxy, or R 5 R 4 N-CO- O-, where R 4 and R 5 independently are CrC 7 alkyl or phenyl which is unsubstituted or substituted by a substitutent selected from d-C 7 alkyl, d-C 7 alkoxy, halogen and trifluoromethyl and where R 4 additionally is hydrogen; or R 4 and R 5 together represent C 3 -C 6 alkylene; in free form or in form of a pharmaceutically acceptable acid addition salt.
- the invention also concerns a compound of formulae (I A), (I B), (X A), (X B), (Y A) or (Y B) as hereinabove described, wherein R" represents hydrogen.
- the compounds of formulae (I A), (I B), (X A), (X B), (Y A) or (Y B) as hereinabove described are in a substantially pure form.
- the compounds of the invention can exist in free form or in acid addition salt form.
- Pharmaceutically acceptable (i.e., non-toxic, physiologically acceptable) salts are preferred, although other salts are also useful, e.g., in isolating or purifying the compounds of this invention.
- the preferred acid addition salts are the hydrochlorides, salts of methanesulfonic, sulfuric, phosphoric, citric, lactic and acetic acid may also be utilized.
- the compounds of the invention may exist in the form of optically active isomers or diastereoisomers and can be separated and recovered by conventional techniques, such as chromatography.
- alkyl refers to straight or branched chain hydrocarbon groups having 1 to 10 carbon atoms, preferably 1 to 7 carbon atoms, most preferably 1 to 5 carbon atoms.
- exemplary alkyl groups include methyl, ethyl, propyl, isopropyl, n-butyl, t-butyl, isobutyl, pentyl, hexyl and the like.
- alkanoyl refers to alkyl-C(O)-.
- substituted adamantyl refers to adamantyl, i.e. 1- or 2-adamantyl, substituted by one or more, for example two, substitutents selected from alkyl, -OR 1 or - NR 2 R 3 ; where R-i, R 2 and R 3 are independently hydrogen, alkyl, (d-C 8 -alkanoyl), carbamyl, or -CO-NR 4 R 5 ; where R 4 and R 5 are independently alkyl, unsubstituted or substituted aryl and where one of R 4 and R 5 additionally is hydrogen or R 4 and R 5 together represent C 2 - C 7 alkylene;.
- aryl preferably represents phenyl.
- Substituted phenyl preferably is phenyl substituted by one or more, e.g. two, substitutents selected from e.g. alkyl, alkoxy, halogen and trifluoromethyl.
- alkoxy refers to alkyl-O-.
- halogen refers to fluorine, chlorine, bromine and iodine.
- alkylene refers to a straight chain bridge of 2 to 7 carbon atoms, preferably of 3 to 6 carbon atoms, most preferably 5 carbon atoms.
- substantially pure is understood in the context of the present invention to mean substantially free of biological material such as found in the blood, especially less than 10%, preferably less than 1 %, and most preferably free of such biological material.
- the compounds of the invention may be prepared e.g. by a process which comprises coupling a reactive (2-cyanopyrrolidino)carbonylmethylene compound with an appropriate substituted amine; more particularly, for the preparation of the compounds of formula I, it comprises reacting a compound of formula Il
- Y is a reactive group (preferably a halogen such as bromine, chlorine or iodine) with a compound of formula III
- the R' moiety can be attached to the chemical structure by any of the method well known by the person skilled in the art or by the process described herein after for the structures of the chemical formula IA or IB.
- the O-glucuronide of vildagliptin have been prepared as described herein after.
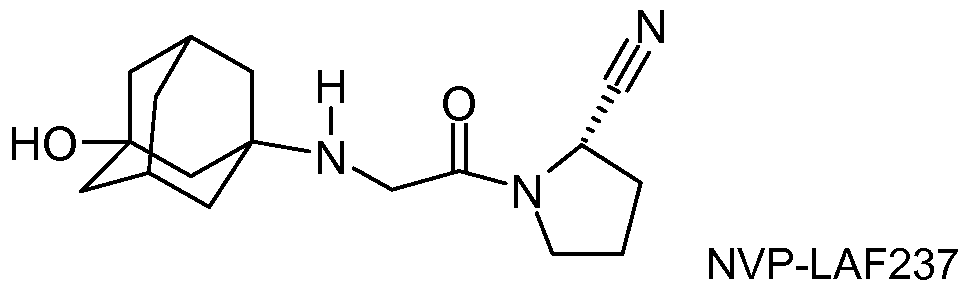
- Vildagliptin (NVP-LAF237-NX or Galvus®) is a new oral hypoglycemic (anti-diabetic) drug of the dipeptidyl peptidase-4 (DPP-4) inhibitor class currently undergoing regulatory review by the U. S, FDA.
- DPP-4 dipeptidyl peptidase-4
- Figure AA the O-glucuronide of vildagliptin
- Figure AA remains as active as vildagliptin.
- the O-glucuronide of vildagliptin in addition to be equally potent to vildagliptin at inhibiting the DPP-4 enzyme, is less potent at inhibiting DPP-8 or DPP-9 enzymes.
- the O-glucuronide of vildagliptin can provide further pharmacokinetic or pharmacological advantages e.g. less side effects, better bioavailability.
- Figure AA the O-glucuronide of vildagliptin ( Figure AA) is in a substantially pure form.
- the process of the invention may be effected in conventional manner.
- the compound of formula Il is reacted with 1 to 3 equivalents, preferably 3 equivalents of a primary amine of formula III.
- the reaction is conveniently conducted in the presence of an inert, organic solvent, such as methylene chloride or a cyclic ether such as tetrahydrofuran.
- the temperature preferably is of from about 0° to about 35 0 C, preferably between about 0° and about 25 0 C.
- the compounds of the invention may be isolated from the reaction mixture and purified in conventional manner, e.g. by chromatography.
- the starting materials may also be prepared in conventional manner.
- the compounds of formula Il may be prepared by the following two-step reaction scheme:
- Step 1 involves the reaction of the pyrrolidine of formula IV with a slight molar excess of a haloacetylhalide such as bromoacetylbromide or chloroacetylchloride and a base such as potassium carbonate or triethylamine.
- a haloacetylhalide such as bromoacetylbromide or chloroacetylchloride
- a base such as potassium carbonate or triethylamine.
- the reaction conveniently is conducted in the presence of an inert, organic solvent, such as tetrahydrofuran or a chlorinated, aliphatic hydrocarbon such as methylene chloride, at a temperature of from about 0° to about 25 0 C, preferably at a temperature between about 0° and about 15 0 C.
- Step 2 concerns the dehydration of the compound of formula V, prepared in Step 1 , with 1 to 2 equivalents of trifluoroacetic anhydride (TFAA).
- TFAA trifluoroacetic anhydride
- the dehydration preferably is conducted in the presence of an inert, organic solvent such as tetrahydrofuran or a chlorinated, aliphatic hydrocarbon such as methylene chloride, at a temperature of from about 0° to about 25 0 C, preferably at a temperature between about 0° and about 15 0 C.
- an inert, organic solvent such as tetrahydrofuran or a chlorinated, aliphatic hydrocarbon such as methylene chloride
- a compound used as starting material is known or may be prepared from known compounds in known manner or analogously to known methods or analogously to methods described in the Example.
- the primary amine compounds of formula III are known and may be prepared by procedures documented in the literature, for example, Khim. -Farm. Zh. (1986), 20(7), 810 -15.
- Compounds of the invention having basic groups can be converted into acid addition salts, especially pharmaceutically acceptable acid addition salts.
- acid addition salts are formed, for example, with inorganic acids, such as mineral acids, for example sulfuric acid, a phosphoric or hydrohalic acid, or with organic carboxylic acids.
- inorganic acids such as mineral acids, for example sulfuric acid, a phosphoric or hydrohalic acid, or with organic carboxylic acids.
- the compounds, including their salts, can also be obtained in the form of their hydrates, or include other solvents used for their crystallization.
- the instant invention also includes pharmaceutical compositions, for example, useful in inhibiting DPP-IV, comprising a pharmaceutically acceptable carrier or diluent and a therapeutically effective amount of a compound of formula I, or a pharmaceutically acceptable acid addition salt thereof.
- the instant invention provides a method of inhibiting DPP-IV comprising administering to a mammal in need of such treatment a therapeutically effective amount of a compound of the invention, or a pharmaceutically acceptable acid addition salt thereof.
- the instant invention provides a method of treating conditions mediated by DPP-IV inhibition comprising administering to a mammal in need of such treatment a therapeutically effective amount of a compound of the invention above, or a pharmaceutically acceptable acid addition salt thereof.
- the present invention also relates to the use of a compound according to the instant invention or a pharmaceutically acceptable salt thereof e.g. for the manufacture of a medicament for the prevention or treatment of diseases or conditions associated with elevated levels of DPP-IV.
- a compound according to the instant invention or a pharmaceutically acceptable salt thereof e.g. for the manufacture of a medicament for the prevention or treatment of diseases or conditions associated with elevated levels of DPP-IV.
- all of the compounds of formulae (I A), (I B), (X A), (X B), (Y A) or (Y B), and their corresponding pharmaceutically acceptable acid addition salts are useful in inhibiting DPP-IV.
- the ability of the compounds of the invention, and their corresponding pharmaceutically acceptable acid addition salts, to inhibit DPP-IV may be demonstrated employing the Caco-2 DPP-IV Assay which measures the ability of test compounds to inhibit DPP-IV activity from human colonic carcinoma cell extracts.
- the human colonic carcinoma cell line Caco-2 was obtained from the American Type Culture Collection (ATCC HTB 37). Differentiation of the cells to induce DPP-IV expression was accomplished as described by Reisher, et al. in an article entitled "Increased expression of intestinal cell line Caco-2" in Proc. Natl. Acad. Sci., Vol. 90, pgs. 5757-5761 (1993).
- Cell extract is prepared from cells solubilized in 1 OmM Tris HCI, 0.15 M NaCI, 0.04 t.i.u. aprotinin, 0.5% nonidet-P40, pH 8.0, which is centrifuged at 35,000 g for 30 min. at 4 0 C. to remove cell debris.
- the assay is conducted by adding 20 ⁇ g solubilized Caco-2 protein, diluted to a final volume of 125 ⁇ l in assay buffer (25 mM Tris HCI pH 7.4, 14OmM NaCI, 10 mM KCI, 1 % bovine serum albumin) to microtiter plate wells. After a 60 min. incubation at room temperature, the reaction is initiated by adding 25 ⁇ l of 1 mM substrate (H-Alanine-Proline-pNA; pNA is p-nitroaniline). The reaction is carried out at room temperature for 10 minutes after which time a 19 ⁇ l volume of 25% glacial acetic acid is added to stop the reaction.
- assay buffer 25 mM Tris HCI pH 7.4, 14OmM NaCI, 10 mM KCI, 1 % bovine serum albumin
- Test compounds are typically added as 30 ⁇ l additions and the assay buffer volume is reduced to 95 ⁇ l.
- a standard curve of free p-nitroaniline is generated using 0-500 ⁇ M solutions of free pNA in assay buffer. The curve generated is linear and is used for interpolation of substrate consumption (catalytic activity in nmoles substrate cleaved /min). The endpoint is determined by measuring absorbance at 405 nm in a Molecular Devices UV Max microtiter plate reader.
- the potency of the test compounds as DPP-IV inhibitors is calculated from 8-point, dose-response curves using a 4-parameter logistic function.
- the ability of the compounds of the invention, and their corresponding pharmaceutically acceptable acid addition salts, to inhibit DPP-IV may also be demonstrated by measuring the effects of test compounds on DPP-IV activity in human and rat plasma employing a modified version of the assay described by Kubota, et al. in an article entitled "Involvement of dipeptidylpeptidase IV in an in vivo immune response" in Clin. Exp. Immunol., Vol. 89, pgs. 192-197 (1992).
- fluorescence is measured using a CytoFluor 2350 fluorimeter (Excitation 380 nm Emission 460nm; sensitivity setting 4). Test compounds are typically added as 2 ⁇ l additions and the assay buffer volume is reduced to 13 ⁇ l.
- a fluorescence-concentration curve of free AMC is generated using 0-50 ⁇ M solutions of AMC in assay buffer. The curve generated is linear and is used for interpolation of substrate consumption (catalytic activity in nmoles substrate cleaved/min).
- the potency of the test compounds as DPP-IV inhibitors expressed as IC 5O , is calculated from 8-point, dose-response curves using a 4 parameter logistic function.
- the compounds of the invention are useful in treating conditions mediated by DPP-IV inhibition.
- the compounds disclosed herein are useful in the treatment of conditions such as non-insulin-dependent diabetes mellitus, arthritis, obesity, allograft transplantation and calcitonin-osteoporosis.
- glucagon-like peptides such as GLP-1 and GLP-2
- DPP-IV inhibition DPP-IV inhibition
- the compounds disclosed herein are useful for example, to produce a sedative or anxiolytic effect, or to attenuate post-surgical catabolic changes and hormonal responses to stress, or to reduce mortality and morbidity after myocardial infarction, or in the treatment of conditions related to the above effects which may be mediated by GLP-1 and/or GLP-2 levels.
- the compounds of the invention, and their corresponding pharmaceutically acceptable acid addition salts improve early insulin response to an oral glucose challenge and, therefore, are useful in treating non-insulin-dependent diabetes mellitus.
- the ability of the compounds of the invention, and their corresponding pharmaceutically acceptable acid addition salts, to improve early insulin response to an oral glucose challenge may be measured in insulin resistant rats according to the following method:
- RIA radioimmunoassay
- the RIA has a lower limit of detection of 0.5 ⁇ ll/mL with intra- and inter-assay variations of less than 5%. Data are expressed as % increase of the mean of the control animals.
- each of the compounds tested amplified the early insulin response which led to an improvement in glucose tolerance in the insulin resistant test animals. The following results were obtained:
- the precise dosage of the compounds of the invention, and their corresponding pharmaceutically acceptable acid addition salts, to be employed for treating conditions mediated by DPP-IV inhibition depends upon several factors, including the host, the nature and the severity of the condition being treated, the mode of administration and the particular compound employed. However, in general, conditions mediated by DPP-IV inhibition are effectively treated when a compound of the invention, or a corresponding pharmaceutically acceptable acid addition salt, is administered enterally, e.g., orally, or parenterally, e.g., intravenously, preferably orally, at a daily dosage of 0.002-5, preferably 0.02-2.5 mg/kg body weight or, for most larger primates, a daily dosage of 0.1-250, preferably 1-100 mg.
- a typical oral dosage unit is 0.01-0.75 mg/kg, one to three times a day. Usually, a small dose is administered initially and the dosage is gradually increased until the optimal dosage for the host under treatment is determined. The upper limit of dosage is that imposed by side effects and can be determined by trial for the host being treated.
- the compounds of the invention may be combined with one or more pharmaceutically acceptable carriers and, optionally, one or more other conventional pharmaceutical adjuvants and administered enterally, e.g., orally, in the form of tablets, capsules, caplets, etc. or parenterally, e.g., intravenously, in the form of sterile injectable solutions or suspensions.
- enteral and parenteral compositions may be prepared by conventional means. Examples of suitable formulations comprising the compounds of the invention are described in the patent applications WO 2005/067976 or WO 2006/078593.
- the invention also concerns a pharmaceutical composition
- a pharmaceutical composition comprising a compound according to any of claims 1 to 4 or herein described compounds, in free form or in pharmaceutically acceptable acid addition salt form, together with at least one pharmaceutically acceptable carrier or diluent.
- the pharmaceutical compositions according to the invention are those suitable for enteral, such as oral or rectal; transdermal and parenteral administration to mammals, including man, for the treatment of conditions mediated by DPP4 activity. Such conditions include impaired glucose tolerance, Type 2 diabetes and obesity.
- the pharmacologically active compounds of the invention may be employed in the manufacture of pharmaceutical compositions comprising an effective amount thereof in conjunction or admixture with excipients or carriers suitable for either enteral or parenteral application.
- Injectable compositions are preferably aqueous isotonic solutions or suspensions, and suppositories are advantageously prepared from fatty emulsions or suspensions.
- compositions may be sterilized and/or contain adjuvants, such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In addition, they may also contain other therapeutically valuable substances.
- adjuvants such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers.
- Said compositions are prepared according to conventional mixing, granulating or coating methods, respectively, and contain about 0.1-75%, preferably about 1-50%, of the active ingredient.
- transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound of the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin.
- the present invention provides pharmaceutical compositions as described above for the treatment of conditions mediated by dipeptidyl peptidase-IV inhibition, preferably, impaired glucose tolerance, Type 2 diabetes and obesity.
- the present invention also provides the use of a pharmaceutical compositions as described above for the treatment of conditions mediated by dipeptidyl peptidase-IV inhibition, preferably, impaired glucose tolerance, Type 2 diabetes and obesity, wherein the compound of the invention (as defined in any of claims 1 to 4) is administered in combination with another therapeutic agent, e.g. one or two additional agent, as hereinafter described.
- a pharmaceutical compositions as described above for the treatment of conditions mediated by dipeptidyl peptidase-IV inhibition, preferably, impaired glucose tolerance, Type 2 diabetes and obesity, wherein the compound of the invention (as defined in any of claims 1 to 4) is administered in combination with another therapeutic agent, e.g. one or two additional agent, as hereinafter described.
- compositions may contain a therapeutically effective amount of a compound of the invention as defined herein (e.g. in claims 1 to 4), either alone or in a combination with another therapeutic agent, e.g., each at an effective therapeutic dose as reported in the art.
- Such therapeutic agents include: a) antidiabetic agents, such as insulin, insulin derivatives and mimetics; insulin secretagogues such as the sulfonylureas, e.g., Glipizide, glyburide and Amaryl; insulinotropic sulfonylurea receptor ligands such as meglitinides, e.g., nateglinide and repaglinide; protein tyrosine phosphatase-1 B (PTP-1 B) inhibitors such as PTP-1 12; GSK3 (glycogen synthase kinase-3) inhibitors such as SB-517955, SB-4195052, SB-216763, NN-57-05441 and NN-57- 05445; RXR ligands such as GW-0791 and AGN-194204; sodium-dependent glucose cotransporter inhibitors such as T-1095; glycogen phosphorylase A inhibitors such as BAY R3401 ; biguan
- GLP-1 glycopeptide 1
- GLP-1 analogs such as Exendin-4 and GLP-1 mimetics
- DPPIV dipeptidyl peptidase IV
- vildagliptin b) hypolipidemic agents such as 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase inhibitors, e.g., lovastatin, pitavastatin, simvastatin, pravastatin, cerivastatin, mevastatin, velostatin, fluvastatin, dalvastatin, atorvastatin, rosuvastatin and rivastatin; squalene synthase inhibitors; FXR (famesoid X receptor) and LXR (liver X receptor) ligands; cholestyramine; fibrates; nicotinic acid bile acid binding resins such as cholestyramine; fibrates;
- anti-hypertensive agents e.g., loop diuretics such as ethacrynic acid, furosemide and torsemide; angiotensin converting enzyme (ACE) inhibitors such as benazepril, captopril, enalapril, fosinopril, lisinopril, moexipril, perinodopril, quinapril, ramipril and trandolapril; inhibitors of the Na-K-ATPase membrane pump such as digoxin; neutralendopeptidase (NEP) inhibitors; ACE/NEP inhibitors such as omapatrilat, sampatrilat and fasidotril; angiotensin Il antagonists such as candesartan, eprosartan, irbesartan, losartan, telmisartan and valsartan, in particular val
- ACE angiotensin converting
- agonists of peroxisome proliferator-activator receptors such as fenofibrate, pioglitazone, rosiglitazone, tesaglitazar, BMS-298585, L-796449, the compounds specifically described in the patent application WO 2004/103995 i.e. compounds of examples 1 to 35 or compounds specifically listed in claim 21 , or the compounds specifically described in the patent application WO 03/043985 i.e.
- compositions comprising; i) a compound according to any of claims 1 to 4, and ii) at least one compound selected from a) antidiabetic agents, b) hypolipidemic agents, c) anti-obesity agents, d) anti-hypertensive agents, e) agonists of peroxisome proliferator-activator receptors, ii) one or more pharmaceutically acceptable carriers.
- a compound of the present invention may be administered either simultaneously, before or after the other active ingredient, either separately by the same or different route of administration or together in the same pharmaceutical formulation.
- compositions comprising a therapeutically effective amount of a compound of the invention in combination with a therapeutically effective amount of another therapeutic agent, preferably selected from antidiabetics, hypolipidemic agents, anti-obesity agents or anti-hypertensive agents, most preferably from antidiabetics or hypolipidemic agents as described above.
- another therapeutic agent preferably selected from antidiabetics, hypolipidemic agents, anti-obesity agents or anti-hypertensive agents, most preferably from antidiabetics or hypolipidemic agents as described above.
- the present invention further relates to pharmaceutical compositions as described above for use as a medicament.
- the present invention further relates to use of pharmaceutical compositions or combinations as described above for the preparation of a medicament for the treatment of conditions mediated by dipeptidyl peptidase-IV inhibition, preferably, impaired glucose tolerance, Type 2 diabetes and obesity.
- the compounds of the invention may be formulated into enteral and parenteral pharmaceutical compositions containing an amount of the active substance that is effective for treating conditions mediated by DPP-IV inhibition, such compositions in unit dosage form and such compositions comprising a pharmaceutically acceptable carrier.
- the compounds of the invention may be administered in enantiomerically pure form (e.g., ee >98%, preferably >99%) or together with the R enantiomer, e.g., in racemic form.
- enantiomerically pure form e.g., ee >98%, preferably >99%
- R enantiomer e.g., in racemic form.
- the above dosage ranges are based on the compounds of the invention (excluding the amount of the R enantiomer).
- the solution is placed in an ice-water bath and allowed to stir for 30 minutes. Approximately 55Og of 89% pure KOH (8,74 mol) is then added in small portions over 45 minutes. During this addition, the reaction is exothermic; reaching 80 0 C and producing copious amounts of brown NO 2 gas. By the end of the addition, the reaction is thick with white solids (both product and salts).
- the resulting white paste is then poured onto a buchner funnel/celite pad and washed with 1.2 L of CH 2 CI 2 . The CH 2 CI 2 layer is then extracted from the water layer and dried over Na2SO4. The solution is then filtered and concentrated (rotovap/pump) to provide 1-aminoadamantane-3-ol as a white solid.
- reaction is then magnetically stirred for 1 hour at room temperature and the resulting clear yellow/orange solution is concentrated via rotovap.
- the excess trifluoroacetic anhydride is removed by adding ethyl acetate to the concentrated oil and reconcentrating via rotovap. This removing operation is performed three times.
- the resulting oil is partitioned between ethyl acetate and water.
- the product is then extracted into the ethyl acetate and the aqueous layer is then washed twice with ethyl acetate.
- the combined organic layers are then washed successively with water and brine dried over magnesium sulfate, filtered and concentrated to obtain 1-chloroacetyl-2- cyanopyrrolidine as a yellow solid.
- NVP-BQS867 the O-qlucuronide of vildaqliptin
- NVP- BRU563 the labeled O-qlucuronide of vildaqliptin
- rat liver homogenate Two portions of 15 g and one of 1 1 g of frozen rat liver were defrosted and cut into small pieces. After addition of 0.5 volume equivalent of ice cold 0.9 % NaCI solution to each portion of liver and mixing in a Dispomix blender, the tissue was homogenized in a "Potter S" Tissue Homogenizer (Braun Biotech Inc., Melsungen, Germany) under cooling in ice water by moving the teflon pistil 3 times up and down at 100 % stirrer speed.
- NVP-LAF237 (vildagliptin) on 340 mg scale were: NVP-LAF237 5 mM, UDP-glucuronate (Yamasa Co., Tokyo, JP) 20 mM, MgCI 2 20 mM, HEPES, pH 8.5 160 mM, rat liver homogenate 20 % v/v, total volume 224 ml, incubation for 5 h at 37 0 C and further 14 h at 30 0 C.
- the reactions were performed in 50 ml Nunc tubes each filled with 20 ml of reaction mixture, which were shaken at 170 rpm on a microbiological lab shaker with 5 cm shaking radius.
- the preparative reaction was stopped by addition of 224 ml of acetonitrile and mixing for 10 min at 20 0 C. After centrifugation for 15 min at 8000 rpm (rotor JA-10), the pellet was suspended in 50 ml of 50 % v/v acetonitrile in deionized water and centrifuged again. The combined supematants were concentrated to 50 ml under reduced pressure at 25 0 C. The turbid concentrate was centrifuged at 5000 rpm, and after filtering the supernatant through fiberglass it was subjected to preparative HPLC.
- the biotransformation conditions were the same as for NVP-LAF237 on 340 mg scale except that the concentration of UDP-glucuronate was 80 mM, of rat liver homogenate 30 % v/v, the filling volume of the Nunc tubes was 16.5 ml, the total volume 33 ml, the reaction time 4 h and the shaking speed 200 rpm.
- the reaction was stopped by addition of 16.5 ml of acetonitrile to each tube, incubation for 15 min on ice and subsequent mixing. After centrifugation for 15 min at 17,000 rpm (rotor JA- 10), the sediment was resuspended in 20 ml of 50 % v/v acetonitrile in deionized water and centrifuged again. The combined supematants were concentrated to 10 ml under reduced pressure at 30 0 C, again diluted to 25 ml final volume and centrifuged for final clarification. The pellet was resuspended with 5 ml of deionized water, and after centrifugation, the latter two supematants were combined and subjected to preparative HPLC.
- the supernatant was analyzed by analytical RP 18 -HPLC-DAD (HPLC-DAD 1 100 series from Agilent Technologies, Basel, Switzerland; two connected columns of the type Chromolith Performance RP-18e 100 x 4.6 mm with a Chromolith Guard Cartridge RP-18e 5 x 4.6 mm (Merck); flow 2 ml / min; mobile phase A: 3 mM aqueous H 3 PO 4 , mobile phase B: acetonitrile; gradient from 3 % to 15 % B in 5 min; DAD detection at 200 - 400 nm). Conversion was based on the UV-absorption peak areas at 205 nm of the glucuronide and NVP-LAF237.
- the preparative liquid chromatography-mass spectrometry system consisted of a Waters Autopurification system (Waters Corp. Milford, MA, USA) with 2525 pump, 2767 sample manager, 2996 photodiode detector, 515 make-up pump, ZQ2000 mass spectrometer and MassLynx 4.0 and FractionLynx 4.0 software.
- the column effluent was split, a very small part was mixed in-line with make-up liquid 2-propanol/H 2 O/HCOOH 400:100:1 and introduced into the ion source of the mass spectrometer; the rest entered the DAD detector recording from 200 - 600 nm, and subsequently the sample manager for fraction collection.
- the mass spectrometer was equipped with ESCi interface used in the positive ESI mode. A capillary voltage of 3 kV and a cone voltage of 30 V was applied; mass range 250 - 550 Da.
- the target fraction was collected time-based from 5.3 - 6.7 min. Target fractions from 12 HPLC runs were combined, the organic solvent was removed under reduced pressure at 30 0 C, and the remaining solution freeze-dried at -80 0 C and 0.2 mbar yielding 15 mg as almost colorless lyophilizate.
- the mass spectrometer was equipped with ESCi interface used in the positive ESI mode. A capillary voltage of 3 kV and a cone voltage of 30 V was applied; mass range 100 - 1000 Da.
- the target fraction was collected time-based from 7.0 - 9.0 min. Target fractions from 12 HPLC runs were combined, the organic solvent was removed under reduced pressure at 30 0C, and the remaining solution freeze-dried at -80 0 C and 0.2 mbar yielding 63 mg as almost colorless lyophilizate.
- the liquid chromatograph consisted of a Waters UPLC Acquity (Waters) equipped with a Waters Acquity 2996 PDA detector.
- a TSQ Quantum AM mass spectrometer (Thermo, San Jose, CA, USA) equipped with electrospray interface in the positive mode was used. It was operated with Xcalibur software version 2.0. A sheath gas setting of 25 units and auxiliary gas of 5 units was used and a spray voltage of 3 kV applied. The heated metal capillary was maintained at 300 0 C; mass range 200 to 800 Da. MS/MS parameters: collision gas 1.5 mTorr argon; collision energy 25 V.
- NVP-BQS867-NX-2 15 mg was obtained by preparative HPLC-MS from 5 ml of aqueous culture extract obtained from biotransformation. From the whole preparative reaction with 340 mg of NVP-LAF237, four batches of NVP- BQS867-NX (NX-1 to NX-4) were isolated with a total amount of 295 mg of O-glucuronide. 3. Structure elucidation
- NVP-BQS867-NX-2 shows a [M + H] + of 480.1 suggesting a molecular weight of 479. This is consistent with a glucuronide of NVP-LAF237.
- the MS/MS product ion spectrum shows the loss of glucuronide (m/z 304) as well as glucuronic acid (m/z 286).
- the structure of NVP-BQS867-NX-2 ( Figure 3-1 ) was elucidated unambiguously on the basis of NMR spectroscopy.
- the major differences in the NMR spectra of NVP-BQS867 compared with NVP-LAF237 are mainly the resonances obtained from the glucuronide moiety.
- NVP-BRU563-NX-3 biosynthesized from [U-pyrrolidin,cyano- 13 C 5 ,pyrrolidin- 15 N] labeled NVP-LAF237, showed identical spectral properties to NVP-BQS867-NX with the expected exceptions for the stable label:
- the MH + of 486.2 is 6 Da higher than that of NVP- BQS867-NX and in the MS/MS product ion spectrum some fragments are also shifted.
- Another preferred compound is :
- the compound of Figure BB is in a substantially pure form.
- This O-glucuronide compound ( Figure BB) can be obtained by adapting the herein described process.
- the process to obtain the starting compound from wherein R' is -OH is described in the patent application WO 01/068603, or WO 05/095339.
- a salt can be the HCL salt .
- HCI as hydrochloride. All HCI salts of final products are prepared by passing HCI gas through a 0.1 Molar solution of the free base in tetrahydrofuran until solution is clearly acidic followed by removal of the solvent (rotovap/pump).
- the amino-adamantane starting materials are known in the literatrue or can be prepared as follows:
- 3-Methoxy-1-adamantylamine can be prepared as follows:
- the filtrate is then concentrated and purified on silica gel employing a SIMS/Biotage apparatus and 19% methanol and 1 % ammonium hydroxide in methylene chloride as eluent to yield 3-methoxy-1-adamantylamine as an opaque oil.
- the procedure for the synthesis of 3-[[(phenylamino)carbonyl1oxy1 -1-aminoadamantane is essentially the procedure of 3-[[(tertbutylamino)carbonyl]oxy]-1-aminoadamantane except in the second step where an equivalent of phenyl isocyanate replaces the tert-butylisocyanate, 1 ,2-dichloroethane is used as solvent instead of methylene chloride and the reaction is stirred at 5O 0 C for 18 hours.
- the final amine intermediate is provided as a clear oil.
- the procedure to make 2-aminoadamantane-5-ol is the same as in Example 1 except that the starting material is 2-aminoadamantane instead of 1-aminoadamantane.
- the procedure for the synthesis of the nucleophile 3-acetoxy-1 -aminoadamantane is essentially the procedure of 3-[[(tertbutylamino)carbonyl]oxy]-1 -aminoadamantane except for a standard acylation of 1-benzylcarbamoyladamantane-3-ol using 1.2 eq of acetyl chloride,
- Tablets each containing 50 mg of the active ingredient, NVP-BQS867, can be prepared as follows:
- composition for 10,000 tablets
- the active ingredient is mixed with the lactose and 292 g of potato starch, and the mixture is moistened using an alcoholic solution of the gelatin and granulated by means of a sieve. After drying, the remainder of the potato starch, the talc, the magnesium stearate and the highly disperse silica are admixed and the mixture is compressed to give tablets of weight 145.0 mg each and active ingredient content 50.0 mg which, if desired, can be provided with breaking notches for finer adjustment of the dose.
- FAP fibroblast activation protein alpha
- Fl measurements using a Rh1 10 dye were done with a Safire2 reader (TECAN, Maennedorf, Switzerland).
- the Safire2 is a monochomator-based instrument and wavelengths of 485 nm and 535 nm were taken for fluorescence excitation and emission acquisition, respectively. The bandwidths were set to 20 nm in both the excitation and the emission path.
- the fluorophores in each well were excited by three flashes per measurement.
- the assays were performed at room temperature in 384-well plates. All final assay volumes were 30 ⁇ l. Test compounds were dissolved in 90 % (v/v) DMSO/water and diluted in water (containing 0.05 % (w/v) CHAPS) to 3-times the desired assay concentration. For each assay, 10 ⁇ l water/CHAPS ( ⁇ test compound) were added per well, followed by 10 ⁇ l protease solution (diluted with assay buffer; for final assay concentration cf. ⁇ Assay conditions). After 1 hour of incubation at room temperature, the reaction was started by addition of 10 ⁇ l substrate solution (for final concentrations cf. ⁇ Assay conditions).
- the eleven final compound concentrations were either 0.9 nM, 3 nM, 9 nM, 30 nM, 90 nM, 300 nM, 900 nM, 3 ⁇ M, 9 ⁇ M, 30 ⁇ M and 90 ⁇ M or 3 nM, 10 nM, 30 nM, 10O nM, 30O nM, 1 ⁇ M, 3 ⁇ M, 10 ⁇ M, 30 ⁇ M, 100 ⁇ M and 300 ⁇ M.
- the IC 50 value was calculated from the plot of percentage of inhibition vs. inhibitor concentration using non-linear regression analysis software (XLfit, Vers. 4.0; ID Business Solution Ltd., Guildford, Surrey, UK).
- DPP-2 dipeptidyl peptidase 2 (dipeptidyl peptidase 2) enzyme: human DPP-2 covering amino acid 30-492; expressed in and purified from insect cells (baculovirus expression system) substrate: Nle-Pro-AMC, purchased from Biosyntan (www.biosyntan.de), product number
- DPP-IV dipeptidyl peptidase IV enzyme: human DPP-IV covering amino acid 39-766; expressed in and purified from insect cells (baculovirus expression system) substrate: Gly-Pro-AMC, purchased from Bachem (www.bachem.com), catalogue number I-
- DPP-8 (dipeptidyl-peptidase 8) enzyme: human DPP-8 covering amino acid 1-882; expressed in and purified from insect cells (baculovirus expression system) substrate: Gly-Pro-AMC, purchased from Bachem (www.bachem.com), catalogue number I-
- DPP-9 dipeptidyl-peptidase 9 enzyme: human DPP-9 covering amino acid 1-863; expressed in and purified from Pichia pastoris substrate: Gly-Pro-AMC, purchased from Bachem (www.bachem.com), catalogue number I-
- FAP fibroblast activation protein, alpha
- enzyme human FAP ⁇ covering amino acid 27-760 excluding the cytoplasmic and transmebrane domains; expressed in and purified from insect cells (baculovirus expression system) substrate: Z-Gly-Pro-AMC, purchased from Bachem (www.bachem.com), catalogue number
Abstract
Description













Claims




Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/744,510 US8252751B2 (en) | 2007-11-30 | 2008-11-25 | Organic compounds |
| PL08853345T PL2247602T3 (en) | 2007-11-30 | 2008-11-25 | Adamantyl o-glucuronide derivatives as inhibitors of dipeptidyl peptidase iv for the treatment of diabetes |
| RU2010126056/04A RU2489439C2 (en) | 2007-11-30 | 2008-11-25 | Organic compounds |
| ES08853345T ES2418440T3 (en) | 2007-11-30 | 2008-11-25 | Adamantil O-glucuronide derivatives as dipeptidyl peptidase IV inhibitors for the treatment of diabetes |
| CN2008801181558A CN101918423B (en) | 2007-11-30 | 2008-11-25 | Dipeptidyl peptidase-IV inhibitor |
| JP2010535355A JP5401465B2 (en) | 2007-11-30 | 2008-11-25 | Organic compounds |
| MX2010005914A MX2010005914A (en) | 2007-11-30 | 2008-11-25 | Organic compounds. |
| CA2704493A CA2704493A1 (en) | 2007-11-30 | 2008-11-25 | Organic compounds |
| EP08853345A EP2247602B1 (en) | 2007-11-30 | 2008-11-25 | Adamantyl o-glucuronide derivatives as inhibitors of dipeptidyl peptidase iv for the treatment of diabetes |
| AU2008328845A AU2008328845C1 (en) | 2007-11-30 | 2008-11-25 | Organic compounds |
| US13/560,421 US8569250B2 (en) | 2007-11-30 | 2012-07-27 | Organic compounds |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US484607P | 2007-11-30 | 2007-11-30 | |
| US61/004,846 | 2007-11-30 | ||
| US60/004,846 | 2007-11-30 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/744,510 A-371-Of-International US8252751B2 (en) | 2007-11-30 | 2008-11-25 | Organic compounds |
| US13/560,421 Continuation US8569250B2 (en) | 2007-11-30 | 2012-07-27 | Organic compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009068531A2 true WO2009068531A2 (en) | 2009-06-04 |
| WO2009068531A3 WO2009068531A3 (en) | 2010-09-10 |
Family
ID=40679052
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/066159 WO2009068531A2 (en) | 2007-11-30 | 2008-11-25 | Organic compounds |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US8252751B2 (en) |
| EP (1) | EP2247602B1 (en) |
| JP (1) | JP5401465B2 (en) |
| KR (1) | KR20100092461A (en) |
| CN (1) | CN101918423B (en) |
| AU (1) | AU2008328845C1 (en) |
| CA (1) | CA2704493A1 (en) |
| ES (1) | ES2418440T3 (en) |
| MX (1) | MX2010005914A (en) |
| PL (1) | PL2247602T3 (en) |
| PT (1) | PT2247602E (en) |
| RU (2) | RU2489439C2 (en) |
| WO (1) | WO2009068531A2 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101798270A (en) * | 2010-02-25 | 2010-08-11 | 东华大学 | Method for preparing 3-amino-1-adamantane alcohol |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| US8796468B2 (en) | 2009-12-22 | 2014-08-05 | Shionogi & Co., Ltd. | Adamantanamine derivative |
| WO2013179300A3 (en) * | 2012-05-04 | 2014-08-28 | Megafine Pharma (P) Ltd. | A process for the preparation of vildagliptin and its intermediate thereof |
| US8901316B2 (en) | 2010-08-09 | 2014-12-02 | Shionogi & Co., Ltd. | Process for preparing aminoadamantyl carbamate derivatives |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| WO2023233024A1 (en) * | 2022-06-03 | 2023-12-07 | Universiteit Antwerpen | Dpp9 binding compounds |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5929555B2 (en) * | 2012-06-28 | 2016-06-08 | 三菱瓦斯化学株式会社 | Method for producing hydroxyadamantanecarboxylic acid compound |
| CN103804267B (en) * | 2014-02-21 | 2016-06-08 | 张家港威胜生物医药有限公司 | A kind of synthesis technique of vildagliptin |
| CN105439873A (en) * | 2014-08-20 | 2016-03-30 | 天津药物研究院 | 3-hydroxy-1-amantadine preparation method |
| CN104744334A (en) * | 2015-03-25 | 2015-07-01 | 合肥创新医药技术有限公司 | Preparation method for vildagliptin |
| CN107311907A (en) * | 2017-07-29 | 2017-11-03 | 合肥创新医药技术有限公司 | A kind of preparation method of vildagliptin isomer impurities |
| CN111138334A (en) * | 2020-01-13 | 2020-05-12 | 天津民祥生物医药股份有限公司 | Preparation method of vildagliptin |
| JP7349475B2 (en) | 2021-06-22 | 2023-09-22 | シャープ株式会社 | Relay control circuit and power supply circuit |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000034241A1 (en) * | 1998-12-10 | 2000-06-15 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2005012249A2 (en) * | 2003-08-01 | 2005-02-10 | Bristol-Myers Squibb Company | Adamantyglycine- based inhibitors of dipeptidyl peptidase iv for the treatment of diabetes |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6683056B2 (en) * | 2000-03-30 | 2004-01-27 | Bristol-Myers Squibb Company | O-aryl glucoside SGLT2 inhibitors and method |
| US6555519B2 (en) * | 2000-03-30 | 2003-04-29 | Bristol-Myers Squibb Company | O-glucosylated benzamide SGLT2 inhibitors and method |
| JP5064237B2 (en) * | 2005-12-09 | 2012-10-31 | Meiji Seikaファルマ株式会社 | Lincomycin derivatives and antibacterial agents containing the same as active ingredients |
-
2008
- 2008-11-25 RU RU2010126056/04A patent/RU2489439C2/en not_active IP Right Cessation
- 2008-11-25 CA CA2704493A patent/CA2704493A1/en not_active Abandoned
- 2008-11-25 AU AU2008328845A patent/AU2008328845C1/en not_active Ceased
- 2008-11-25 MX MX2010005914A patent/MX2010005914A/en active IP Right Grant
- 2008-11-25 US US12/744,510 patent/US8252751B2/en not_active Expired - Fee Related
- 2008-11-25 CN CN2008801181558A patent/CN101918423B/en not_active Expired - Fee Related
- 2008-11-25 PT PT88533450T patent/PT2247602E/en unknown
- 2008-11-25 KR KR1020107011725A patent/KR20100092461A/en not_active Application Discontinuation
- 2008-11-25 PL PL08853345T patent/PL2247602T3/en unknown
- 2008-11-25 EP EP08853345A patent/EP2247602B1/en not_active Not-in-force
- 2008-11-25 WO PCT/EP2008/066159 patent/WO2009068531A2/en active Application Filing
- 2008-11-25 JP JP2010535355A patent/JP5401465B2/en not_active Expired - Fee Related
- 2008-11-25 ES ES08853345T patent/ES2418440T3/en active Active
-
2012
- 2012-07-27 US US13/560,421 patent/US8569250B2/en not_active Expired - Fee Related
-
2013
- 2013-04-24 RU RU2013119129/04A patent/RU2013119129A/en not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000034241A1 (en) * | 1998-12-10 | 2000-06-15 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO2005012249A2 (en) * | 2003-08-01 | 2005-02-10 | Bristol-Myers Squibb Company | Adamantyglycine- based inhibitors of dipeptidyl peptidase iv for the treatment of diabetes |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8796468B2 (en) | 2009-12-22 | 2014-08-05 | Shionogi & Co., Ltd. | Adamantanamine derivative |
| CN101798270A (en) * | 2010-02-25 | 2010-08-11 | 东华大学 | Method for preparing 3-amino-1-adamantane alcohol |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| US8901316B2 (en) | 2010-08-09 | 2014-12-02 | Shionogi & Co., Ltd. | Process for preparing aminoadamantyl carbamate derivatives |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013179300A3 (en) * | 2012-05-04 | 2014-08-28 | Megafine Pharma (P) Ltd. | A process for the preparation of vildagliptin and its intermediate thereof |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| WO2023233024A1 (en) * | 2022-06-03 | 2023-12-07 | Universiteit Antwerpen | Dpp9 binding compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2008328845C1 (en) | 2012-11-08 |
| RU2010126056A (en) | 2012-01-10 |
| RU2489439C2 (en) | 2013-08-10 |
| JP2011504903A (en) | 2011-02-17 |
| RU2013119129A (en) | 2014-10-27 |
| EP2247602B1 (en) | 2013-04-03 |
| PL2247602T3 (en) | 2013-08-30 |
| US20120295860A1 (en) | 2012-11-22 |
| KR20100092461A (en) | 2010-08-20 |
| JP5401465B2 (en) | 2014-01-29 |
| US8569250B2 (en) | 2013-10-29 |
| MX2010005914A (en) | 2010-12-14 |
| US8252751B2 (en) | 2012-08-28 |
| EP2247602A2 (en) | 2010-11-10 |
| ES2418440T3 (en) | 2013-08-13 |
| AU2008328845B2 (en) | 2012-04-19 |
| CA2704493A1 (en) | 2009-06-04 |
| CN101918423B (en) | 2013-05-15 |
| WO2009068531A3 (en) | 2010-09-10 |
| US20100256080A1 (en) | 2010-10-07 |
| AU2008328845A1 (en) | 2009-06-04 |
| CN101918423A (en) | 2010-12-15 |
| PT2247602E (en) | 2013-07-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2008328845B2 (en) | Organic compounds | |
| EP1638963B1 (en) | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors | |
| EP1137635B1 (en) | N-substituted 2-cyanopyrrolidines | |
| EP2402319B1 (en) | DGAT Inhibitors | |
| US7538135B2 (en) | Benzenesulfonylamino compounds and pharmaceutical compositions containing these compounds | |
| KR20210069000A (en) | Glp-1 receptor agonist | |
| CN101326180A (en) | Piperazine derivative renin inhibitors | |
| AU2004293177B2 (en) | 4-phenylpiperidine derivatives as renin inhibitors | |
| EP1664002A1 (en) | Organic compounds | |
| EP2527338A1 (en) | N5-(2-ethoxyethyl)-n3-(2-pyridinyl) -3,5-piperidinedicarboxamide derivatives for use as renin inhibitors | |
| CN101466668A (en) | Pyrrolidine compounds as renin inhibitors | |
| AU2004272285B2 (en) | Organic compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880118155.8 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008328845 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2848/DELNP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008853345 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2704493 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2008328845 Country of ref document: AU Date of ref document: 20081125 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12744510 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 20107011725 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/005914 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010535355 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010126056 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0819767 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100531 |