WO2009056693A1 - Nouveaux derives de n, n'- 2, 4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk - Google Patents
Nouveaux derives de n, n'- 2, 4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk Download PDFInfo
- Publication number
- WO2009056693A1 WO2009056693A1 PCT/FR2008/001173 FR2008001173W WO2009056693A1 WO 2009056693 A1 WO2009056693 A1 WO 2009056693A1 FR 2008001173 W FR2008001173 W FR 2008001173W WO 2009056693 A1 WO2009056693 A1 WO 2009056693A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- methyl
- products
- radicals
- phenyl
- Prior art date
Links
- 0 CC(*CC1C=CC(C(C2)CC2C(C(F)(F)F)O)=CC=C1)C(CC1)CCC1C(C1C=CC(C2N=C2C2=NC(*C(CC3C)=CC=C3F)=CCN2)=CC1)=O Chemical compound CC(*CC1C=CC(C(C2)CC2C(C(F)(F)F)O)=CC=C1)C(CC1)CCC1C(C1C=CC(C2N=C2C2=NC(*C(CC3C)=CC=C3F)=CCN2)=CC1)=O 0.000 description 3
- VEVACIOMNFRTID-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1(CN1CCCC1)O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1(CN1CCCC1)O)=O VEVACIOMNFRTID-UHFFFAOYSA-N 0.000 description 1
- CTRZBSOPISRSHM-UHFFFAOYSA-O CC1(Nc(cc2)ccc2S(N(CC2)CCC2NCCS(C)=O)([OH2+])=O)N(C)CC=C(Nc(cc2)ccc2F)N1 Chemical compound CC1(Nc(cc2)ccc2S(N(CC2)CCC2NCCS(C)=O)([OH2+])=O)N(C)CC=C(Nc(cc2)ccc2F)N1 CTRZBSOPISRSHM-UHFFFAOYSA-O 0.000 description 1
- SVUQXAHOJPJPAU-UHFFFAOYSA-N CN(Cc1c[nH]nc1)C(CC1)CCN1C(c(cc1)ccc1Nc1nc(Nc(cc2)ccc2F)ccn1)=O Chemical compound CN(Cc1c[nH]nc1)C(CC1)CCN1C(c(cc1)ccc1Nc1nc(Nc(cc2)ccc2F)ccn1)=O SVUQXAHOJPJPAU-UHFFFAOYSA-N 0.000 description 1
- AATIPKPHCHPUPK-UHFFFAOYSA-N CN1C(CNCC(CCC2)CN2S(c(cc2)ccc2Nc2nccc(Nc(cc3)ccc3F)n2)(=O)=O)=CCC1 Chemical compound CN1C(CNCC(CCC2)CN2S(c(cc2)ccc2Nc2nccc(Nc(cc3)ccc3F)n2)(=O)=O)=CCC1 AATIPKPHCHPUPK-UHFFFAOYSA-N 0.000 description 1
- RAEYLUGTFJZLLH-UHFFFAOYSA-N Cc(cc(cc1)Nc2ccnc(Nc(cc3)ccc3C(N(CC3)CCC3N(C)Cc3c[s]nn3)=O)n2)c1F Chemical compound Cc(cc(cc1)Nc2ccnc(Nc(cc3)ccc3C(N(CC3)CCC3N(C)Cc3c[s]nn3)=O)n2)c1F RAEYLUGTFJZLLH-UHFFFAOYSA-N 0.000 description 1
- LRGRTYLFIHCYGK-UHFFFAOYSA-N Cc(cc(cc1)Nc2ccnc(Nc(cc3)ccc3S(N(CC3)CCC3C(c(ccnc3)c3F)O)(=O)=O)n2)c1F Chemical compound Cc(cc(cc1)Nc2ccnc(Nc(cc3)ccc3S(N(CC3)CCC3C(c(ccnc3)c3F)O)(=O)=O)n2)c1F LRGRTYLFIHCYGK-UHFFFAOYSA-N 0.000 description 1
- BBNSJAHJSUYETR-UHFFFAOYSA-N Cc(cc(cc1)Nc2ccnc(Nc(cc3)ccc3S(N(CC3)CCC3N(C)CCS=O)(O)=O)n2)c1F Chemical compound Cc(cc(cc1)Nc2ccnc(Nc(cc3)ccc3S(N(CC3)CCC3N(C)CCS=O)(O)=O)n2)c1F BBNSJAHJSUYETR-UHFFFAOYSA-N 0.000 description 1
- YCIFFBDBJFXGLW-UHFFFAOYSA-N Cc(cc(cc1)Nc2ccnc(Nc(cc3)ccc3S(N(CC3)CCC3Sc3ccncc3)O)n2)c1F Chemical compound Cc(cc(cc1)Nc2ccnc(Nc(cc3)ccc3S(N(CC3)CCC3Sc3ccncc3)O)n2)c1F YCIFFBDBJFXGLW-UHFFFAOYSA-N 0.000 description 1
- BBLUXFNVPKFBTA-UHFFFAOYSA-N NC(C(F)(F)F)O Chemical compound NC(C(F)(F)F)O BBLUXFNVPKFBTA-UHFFFAOYSA-N 0.000 description 1
- SFKAYSQAVXEORK-UHFFFAOYSA-N NC1C2CNCC1CC2 Chemical compound NC1C2CNCC1CC2 SFKAYSQAVXEORK-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to novel N, N '-2,4-dianilinopyrimidine derivatives, process for their preparation, the novel intermediates obtained, their use as medicaments, the pharmaceutical compositions containing them and the novel use of such derivatives of 2 , 4-dianilinopyrimidines.
- Patent WO200164654-A1 mentions 2, 4-di- (hetero) arylpyrimidines substituted in 5, inhibitors of CDK2 and FAK kinases, as well as other serine-threonine kinase and CDK inhibitory aminopyrimidines are presented in WO2003030909-A1.
- WO2004046118-A2 discloses 2,4-diphenylaminopyrimidine derivatives as inhibitors of cell proliferation.
- a series of 5-cyano-2-aminopyrimidines are presented as inhibitors of KDR and FGFR kinases, in WO200078731-A1, other pyrimidines as inhibitors of FAK and IGFR in WO2004080980A-1, and also ZAP-70, FAK and / or Syk tyrosine kinase in WO2003078404A1, and PLK polokinases in WO2004074244-A2, as cytostatic agents.
- the present invention thus relates to novel 2,4-dianilinopyrimidine derivatives having inhibitory effects vis-à-vis protein kinases.
- the products of the present invention can thus notably be used for the prevention or control of treatment of conditions capable of being modulated by the inhibition of the activity of protein kinases.
- protein kinases the protein kinase IKK-alpha (IKKa) and IKK-beta (IKK ⁇ ) are more particularly mentioned.
- the compounds of the present invention are kinase inhibitors, in particular IKK-alpha and IKK-beta, therefore inhibit NF-KB (nuclear factor kappa B) activity, so they can be used in the treatment of prophylaxis and inflammatory diseases, in cancer and diabetes.
- kinase inhibitors in particular IKK-alpha and IKK-beta, therefore inhibit NF-KB (nuclear factor kappa B) activity, so they can be used in the treatment of prophylaxis and inflammatory diseases, in cancer and diabetes.
- NF-kB Nuclear factor kappa B
- NF-kB belongs to a family of transcription factor complexes consisting of different combinations of Rel / NF-KB polypeptides.
- Members of this family of NF- ⁇ B-related polypeptides regulate the expression of genes involved in immune and inflammatory responses. ((Bames PJ, Karin M (1997) N Engl J Med 336, 1066-1071) and (Baeuerle PA, Baichwal VR (1997) Adv Immunol 65, 111-137)).
- NF- ⁇ B dimers are retained in inactive form in the cytoplasm by inhibitory proteins members of the IKB family (Beg et al., Genes Dev., 7: 2064-2070, 1993; Morin, Trends Genet 9: 427-43) 3), 199 '); Haskil and. al., Cell 65: 1281-1289, 1991).
- the proteins of the IKB family mask the NF-KB nuclear translocation signal.
- IKB-Kinase complex IKB-Kinase complex
- IKK IKB-Kinase complex
- IKB will be subject to ubiquitination leading to its degradation by the proteasome (26S), allowing thus the release and the translocation of NF- ⁇ B in the nucleus or it will bind to specific sequences at the level of the promoters of target genes thus inducing their transcription.
- IKK IKB-Kinase complex
- IKKa IKK1
- IKK2 IKK ⁇
- IKK2 IKK2 is the dominant kinase (Mercurio et al., Mol., Cell Biol., 19: 1526, 1999-, Zandi et al., Science, 28: 1, 3) 60, 1998; Lee and. al, Proe. Natl. Acad. Sci. USA 95:93) 19, 1998).
- NF-KB genes regulated by NF-KB encode pro-inflammatory mediators, cytokines, cell adhesion molecules and proteins of the acute phase, which in turn will induce the activation of NF- ⁇ B by autocrine or paracrine mechanisms.
- NF-KB plays a role in the growth of normal cells but also malignant cells. Proteins produced by expression of NF- ⁇ B regulated genes include cytokines, chemokines, adhesion molecules, cell growth mediators, angiogenesis. In addition, various studies have shown that NF-KB plays an essential role in neoplastic transformations. For example NF- ⁇ B may be associated with cell transformation in vitro and in vivo following overexpression, amplification, rearrangement or translocation events (Mercurio, R, and Manning, AM (1999) Oncogene, 18: 6163- 6171). In some human lymphoid tumor cells, the genes encoding the different NF-KB members are rearranged or amplified. It has been shown that NF- ⁇ B can promote cell growth by inducing transcription of cyclin D, which associated with Rb hyperphosphorylation results in the transition of G1-S phases and the inhibition of apoptosis.
- NF- ⁇ B constitutive activity of NF- ⁇ B is found following the activation of IKK2.
- NF- ⁇ B is constitutively activated in Hodgkin's disease and the inhibition of NF- ⁇ B blocks the growth of these lymphomas.
- I ⁇ B ⁇ repressor induces apoptosis of cells expressing the oncogenic H-Ras allele (Baldwin, J. Clin Invest, 107: 241 (2001), Bargou et al., J.
- NF-KB The constitutive activity of NF-KB appears to contribute to oncogenesis through the activation of several anti-apoptotic genes such as Al / Bfi-1, IEX-I, MAP, thus resulting in the suppression of the cell death pathway.Through the activation of cyclin D, NF-KB can promote the tumor cell growth The regulation of adhesion molecules and surface proteases suggests a role for NF-KB signaling in metastases.
- NF-KB is involved in the induction of chemoresistance. NF-KB is activated in response to a number of chemotherapy treatments. Inhibition of NF- ⁇ B by the use of the super-repressor form of I ⁇ B ⁇ in parallel with chemotherapy treatment has been shown to increase the efficacy of chemotherapy in xenograft models.
- R 2, R 3 and R 4 which are identical or different, are such that one represents a halogen atom or CF 3 and the other two, which are identical or different, represent a hydrogen atom or a halogen atom or an alkyl radical or an alkoxy radical optionally substituted with one or more halogen atoms;
- R5 represents a hydrogen atom or a halogen atom
- R1 and R6 represent one of the following 5 alternatives i) to v)
- R1 represents -X1-R7 with X1 represents - (CH2) m- and R7 represents a heterocycloalkyl, aryl or heteroaryl ring, all optionally substituted; and R6 represents hydrogen atom or hydroxyl radicals, - (CH2) mOH, -CO-NRaRb, -CH2-NRaRb -CO2H and -
- R1 represents -X2-R7 with X2 represents:
- R1 represents -NRc-W with W represents the hydrogen atom or an alkyl radical containing from 1 to 4 linear or branched carbon atoms from 3 carbon atoms optionally substituted with a radical chosen from -PO (OEt ) 2, -OH, -OaIk, -CF3, -CO-NR8R9 and SO2-alk; and R ⁇ represents hydrogen; it being understood that when W represents a hydrogen atom then z represents
- R1 represents -CO-N (Rc) -OR 'c and R ⁇ represents hydrogen; with n, n1 and n2, identical or different, represent an integer of 0 to 3; m represents an integer of 1 to 3;
- Rc and R 'c represent the hydrogen atom or an alkyl radical containing from 1 to 4 carbon atoms optionally substituted with one or more halogen atoms, it being understood that the halogen atoms are not not in the vicinal position of the nitrogen atom;
- NRaRb is such that either Ra and Rb, which may be identical or different, represent a hydrogen atom or an alkyl radical containing from 1 to 4 carbon atoms or a cycloalkyl radical, these alkyl and cycloalkyl radicals being optionally substituted with one or a plurality of halogen atoms, it being understood that the halogen atoms are not in the vicinal position of the nitrogen atom; a hydroxyl radical or an NH 2, NHalkyl or N (alkyl) 2 radical; either Ra and Rb form with the nitrogen atom to which they are bonded a cyclic amine which may optionally contain one or two other heteroatoms chosen from O, S, N
- NR8R9 is such that either R8 and R9, which are identical or different, are such that R8 represents a hydrogen atom or an alkyl radical containing from 1 to 4 carbon atoms or a cycloalkyl radical, these alkyl and cycloalkyl radicals being optionally substituted by one or more halogen atoms, a hydroxyl radical or an NH 2, NHalkyl or N (alkyl) 2 radical; and R 9 represents the hydrogen atom and the alkyl, cycloalkyl or heterocycloalkyl radicals themselves optionally substituted by one or more identical or different radicals chosen from halogen atoms and hydroxyl, alkoxy, NH 2, NHalkyl, N ( alkyl) 2, the alkyl radicals that represent R 9 being further optionally substituted by a phenyl, heterocycloalkyl or heteroaryl radical, themselves optionally substituted with one or more radicals selected from halogen atoms and hydroxyl, alkoxy,
- R 2 R 3, R 4, R 5, Z and the ring (N) have the meanings indicated above or below and R 1 and R6 are such that R1 represents -X1-R7 with X1 represents - (CH2) m- and R7 represents a heterocycloalkyl, aryl or heteroaryl ring, all optionally substituted; and R ⁇ represents hydrogen atom or hydroxyl radicals, - (CH2) mOH, -CO-NRaRb, -CH2-NRaRb -CO2H, and - CO2alk; with m, n and NRaRb as defined above or below and the heterocycloalkyl, aryl and heteroaryl radicals being optionally substituted with one or more radicals, which are identical or different, as defined above or below, said products of formula (I) being in all isomeric forms possible racemic, enantiomers and diastereoisomers, as well as the addition
- R7 is heterocycloalkyl, aryl, or heteroaryl, all optionally substituted, and R6 is hydrogen; with n, nl, n2, Rc and NRaRb as defined above and the heterocycloalkyl, aryl and heteroaryl radicals being optionally substituted by one or more radicals, which are identical or different, as defined above or below, said products of formula (I) being in all possible isomeric racemic, enantiomeric and diastereoisomeric forms, as well as addition salts with inorganic and organic acids of said products of formula (D.
- R1 represents -NRc-W with W represents the hydrogen atom or an alkyl radical containing from 1 to 4 linear or branched carbon atoms from 3 carbon atoms optionally substituted with a radical chosen from -PO (OEt) 2, -OH, -OaIk, -CF3, -CO-NR8R9 and SO2-alk and R ⁇ represents hydrogen, it being understood that when W represents a hydrogen atom, then z represents CO; or R1 represents -CH2-NRC-W with W represents the hydrogen atom or an alkyl radical containing from 1 to 4 linear or branched carbon atoms from 3 carbon atoms and optionally substituted with a radical chosen from -PO (OEt) 2, -OH, -OEt, -CF3, -CO- N (alk
- the ring formed can in particular be the 8-aza-bicyclo (3.2.1) octyl cycle or a ring chosen from the following: azabicyclo [ 3.3.1] nonan-3-yl, 6-azabicyclo [3.2.1] octan-3-yl, the "3-azabicyclo [3.2.1] octan-8-yl or 3-azabicyclo [3.3.1] nonan -9-yl.
- the subject of the present invention is thus the products of formula (I) as defined above in which R 2, R 3, R 4, R 5 and Z have the meanings indicated above or hereafter and the (N) cycle represents one of the rings defined below: an azetidinyl or pyrrolidinyl ring substituted in position 3 by R 1 and R 6 as defined above or hereinafter; a piperidinyl and azepinyl ring substituted in position 3 or 4 by R 1 and R 6 as defined above or hereinafter; an 8-aza bicyclo (3,2,1) octan-3-yl, 6-azabicyclo [3.2.1] octan-3-yl or 3-azabicyclo [3.2.1] octan-8-yl ring; said products of formula (I) being in all isomeric forms possible racemic, enantiomers and diastereoisomers, as well as the addition salts with the mineral and organic acids of said products of formula (I) •
- the present invention thus relates to the products
- halogen means fluorine, chlorine, bromine or iodine atoms and preferably fluorine, chlorine or bromine ;
- alkyl radical denotes a linear or branched radical containing at most 6 carbon atoms and especially the methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl or isopentyl radicals; pentyl, tert-pentyl, neopentyl, hexyl, isohexyl, sec-hexyl, tert-hexyl and their linear or branched positional isomers;
- hydroxyalkyl radical denotes the alkyl radicals indicated above substituted by one or more hydroxyl radicals
- alkoxy radical denotes a linear or branched radical containing at most 6 carbon atoms chosen, for example, from methoxy, ethoxy, propoxy, isopropoxy, linear butoxy, secondary or tertiary, pentoxy, hexoxy and heptoxy radicals, and also their positional isomers; linear or branched;
- cycloalkyl radical denotes a monocyclic or bicyclic carbocyclic radical containing from 3 to 7 ring members and in particular denotes the cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl radicals;
- aryl radical refers to unsaturated, monocyclic or fused carbocyclic ring radicals. Examples of such an aryl radical include, in particular, phenyl or naphthyl radicals; the term “heterocyclic radical” denotes a saturated (heterocycloalkyl) or partially or completely unsaturated (heteroaryl) carbocyclic radical consisting of 4 to 10 members interrupted by one to four identical or different heteroatoms chosen from oxygen, nitrogen or sulfur:
- radicals containing one to four heteroatoms chosen from N optionally oxidized, O and S optionally oxidized such as the radicals, for example thienyl radicals such as 2-thienyl, 3 thienyl, dioxidothienyl, thiazolyl (N, S), furyl (O), 2-furyl, pyrrolyl (NH, NCH 3), isothiazolyl, diazolyl, thiadiazolyl (N, N, S), 1, 3, 4-thiadiazolyl, oxazolyl, oxadiazolyl, isoxazolyl (N, O), 3-isoxazolyl, 4-isoxazolyl, imidazolyl, pyrazolyl (N, N), triazolyl groups, tetrazolyl and more particularly the oxazolyl, isoxazolyl (N, O) or pyrazolyl radicals; all
- heterocycloalkyl saturated
- heterocycloalkyl saturated
- heterocycloalkyl saturated
- heterocycloalkyl saturated
- alkylamino radical or NH (alk) radical and dialkylamino radical or N (alk) 2 denotes NH 2 amino radicals substituted respectively by one or two linear or branched alkyl radicals, which are identical or different in the case of dialkylamino, chosen from alkyl radicals as defined above and optionally substituted as indicated above or below: mention may be made, for example, of methylamino, ethylamino, propylamino or butylamino radicals, and dimethylamino, diethylamino and methylethylamino radicals.
- cycloalkylamino thus denotes an amino radical substituted in particular by a cycloalkyl radical chosen from the radicals defined above: there may thus be mentioned, for example, the cyclopropylamino, cyclobutylamino or cyclopentylamino radicals; still cyclohexylamino.
- cyclic amine denotes a monocyclic or bicyclic radical containing from 3 to 10 members in which at least one carbon atom is replaced by a nitrogen atom, this cyclic radical possibly also containing one or more other heteroatoms chosen from O, S, SO 2, N or NR 10 with R 10 as defined above:
- examples of such cyclic amines include, for example, pyrrolyl, piperidyl, morpholinyl, piperazinyl, pyrrolidinyl and azetidinyl radicals. Mention may more particularly be made of piperidinyl, morpholinyl, piperazinyl or azetidinyl radicals.
- the term patient refers to humans but also other mammals.
- Prodrug refers to a product that can be converted in vivo by metabolic mechanisms (such as hydrolysis) into a product of formula (I). For example, an ester of a product of formula (I) containing a hydroxyl group can be converted by in vivo hydrolysis to its parent molecule.
- hydroxyl group-containing esters of the formula (I) such as acetates, citrates, lactates, tartrates, malonates, oxalates, salicylates, propionates, succinates, fumarates, maleates, methylene bisulfates and the like.
- Particularly useful hydroxyl-containing products of the formula (I) can be prepared from acidic residues such as those described by Bundgaard et al. al., J. Med. Chem. , 1989, 32, page
- esters include especially
- Substituted (aminomethyl) benzoates dialkylamino-methylbenzoates in which the two alkyl groups can be bonded together or can be interrupted by an oxygen atom or by an optionally substituted nitrogen atom or an alkylated nitrogen atom or else morpholino-methyl) benzoates, eg 3- or A- (morpholinomethyl) -benzoates, and (4-alkylpiperazin-1 (yl) benzoates, eg 3- or 4- (4-alkylpiperazin-1-yl) benzoates.
- the addition salts with the inorganic or organic acids of the products of formula (I) can be, for example, the salts formed with hydrochloric, hydrobromic, hydroiodic, nitric, sulfuric, phosphoric, propionic, acetic, trifluoroacetic, formic acids, benzoic, maleic, fumaric, succinic, tartaric, citric, oxalic, glyoxylic, aspartic, ascorbic, alkylmonosulphonic acids such as, for example, methanesulfonic acid, ethanesulphonic acid, propanesulphonic acid, alkylsulphonic acids such as, for example, methanedisulfonic acid, alpha, beta-ethanedisulfonic acid, arylmonosulfonic acids such as benzenesulphonic acid and aryldisulphonic acids.
- stereoisomerism can be defined in its broad sense as the isomerism of compounds having the same developed formulas, but whose different groups are arranged differently in space, such as in particular in monosubstituted cyclohexanes whose substituent can be in axial or equatorial position.
- stereoisomerism due to the different spatial arrangements of fixed substituents, either on double bonds or on rings, often called isomerism.
- stereoisomer is used in the present application in its broadest sense and therefore relates to all of the compounds indicated above.
- the subject of the present invention is particularly the products of formula (I) as defined above or below, in which:
- R 2, R 3 and R 4, which are identical or different, are such that one represents a halogen atom or CF 3 and the other two, which are identical or different, represent a hydrogen atom or a halogen atom or an alkyl radical or an alkoxy radical optionally substituted with one or more halogen atoms;
- R5 represents a hydrogen atom or a halogen atom;
- R1 and R ⁇ represent one of the following alternatives i) to v) i) R1 represents - X1-R7 with X1 represents -CH2 and R7 represents a heterocycloalkyl, phenyl or heteroaryl ring, all optionally substituted; and R ⁇ represents a hydrogen atom or hydroxyl radicals, -CH2OH, -CO2H, -CO-NRaRb and -CO2Et;
- R1 represents -CO-N (Rc) -OR 'c and R ⁇ represents hydrogen; with n, n1 and n2, identical or different, represent an integer of 0 to 2;
- Rc and R 'c identical or different represent the hydrogen atom or an alkyl radical containing 1 to 2 carbon atoms
- NRaRb is such that either Ra and Rb, identical or different, represent the hydrogen atom or an alkyl radical containing from 1 to 4 carbon atoms optionally substituted by one or more halogen atoms, a hydroxyl radical or an NH 2 radical; , NHalkyl or N (alkyl) 2; or Ra and Rb form with the nitrogen atom to which they are bonded a morpholinyl or pyrrolidinyl radical optionally substituted with one or more identical or different radicals chosen from halogen atoms and the alkyl radicals themselves optionally substituted with one or several halogen atoms; all the heterocycloalkyl, phenyl and heteroaryl radicals being optionally substituted with one or more radicals, which may be identical or different, chosen from halogen atoms; hydroxyl radicals; cyano; NR8R9; and the alkyl, cycloalkyl, alkoxy, phenyl, heterocycloalkyl and heteroary
- NR8R9 is such that either R8 and R9, which are identical or different, are such that R8 represents a hydrogen atom, a linear or branched alkyl radical containing at most 4 carbon atoms or a cycloalkyl radical containing from 3 to 6 ring members, alkyl and cycloalkyl themselves optionally substituted by one or more halogen atoms or a hydroxyl radical; and R 9 represents the hydrogen atom or an alkyl radical optionally substituted with one or more identical or different radicals chosen from halogen atoms and hydroxyl, alkoxy, NH 2, NHalkyl, N (alkyl) 2, phenyl or heterocycloalkyl radicals; or heteroaryl themselves optionally substituted by one or more radicals selected from halogen atoms and hydroxyl radical radicals, OCH3, CH3, -CH2OH, CN, CF3, OCF3, NH2, NHaIk or N (alk) 2; or R8 and R9 form
- the subject of the present invention is particularly the products of formula (I) as defined above or below in which R2, R3, R4, R5 and Z have the meanings indicated above or below. and the ring (N) represents a piperidinyl ring substituted at the 3 or 4 position by R 1 and R 6 as defined above or below, said products of formula (I) being in all the possible isomeric forms racemic, enantiomers and diastereoisomers as well as the addition salts with the mineral and organic acids of said products of formula
- the subject of the present invention is thus the products of formula (I) as defined above in which Z, the ring (N), R 1 and R 6 have the meanings indicated above or below; R3 and R4, which are identical or different, are such that one represents a halogen atom or CF3 and the other two, which are identical or different, represent a hydrogen atom, a halogen atom or a methyl or methoxy radical, trifluoromethyl or trifluoromethoxy, and R5 represents a hydrogen atom, said products of formula (I) being in all the possible isomeric racemic, enantiomeric and diastereoisomeric forms, as well as the addition salts with the mineral and organic acids of said products of formula
- the subject of the present invention is thus the products of formula (I) as defined above in which Z, the (N) -cycle, R1 and R6 have the meanings indicated above or below and R2, R3 and R4, which are identical or different, are such that one represents a fluorine atom and the other two, which are identical or different, represent a hydrogen atom, a fluorine atom or a methyl radical; R5 represents an atom of hydrogen; said products of formula (I) being in all possible isomeric racemic, enantiomeric and diastereoisomeric forms, as well as addition salts with inorganic and organic acids of said products of formula (I).
- the subject of the present invention is therefore the products of formula (I) as defined above in which R 1, R 2, R 3, R 4, R 5 and R ⁇ and the ring (N) have the meanings indicated above or below. and Z represents SO 2, said products of formula (I) being in all possible isomeric racemic, enantiomeric and diastereoisomeric forms, as well as addition salts with inorganic and organic acids of said products of formula
- all the heterocycloalkyl, phenyl and heteroaryl radicals that R7 represents may in particular be optionally substituted with one or more radicals, which may be identical or different, chosen from halogen atoms; NR8R9 radicals; and the alkyl, cycloalkyl, alkoxy, phenyl, heterocycloalkyl and heteroaryl radicals, themselves optionally substituted by one or more identical or different radicals chosen from halogen atoms and hydroxyl, alkoxy, OCF3, CH3, -CH2OH, CN radicals; , CF3, OCF3, NH2, NHaIk, or N (alk) 2, pyrrolidinyl, piperidinyl or morpholinyl optionally substituted with one or more radicals identical or different selected from halogen atoms and alkyl radicals themselves optionally substituted by one or more halogen atoms.
- radicals which may be identical or different, chosen from halogen atoms; NR8R
- NR 8 R 9 may especially be such that either R 8 and R 9, which may be identical or different, are such that R 8 represents the hydrogen atom, a linear or branched alkyl radical containing at most 4 carbon atoms or a cycloalkyl radical containing from 3 to 6 members, alkyl and cycloalkyl themselves optionally substituted by one or more halogen atoms or a hydroxyl radical; and R 9 represents the hydrogen atom or an alkyl radical optionally substituted with one or more identical or different radicals chosen from halogen atoms and hydroxyl, alkoxy, NH 2, NHalkyl, N (alkyl) 2, phenyl or heterocycloalkyl radicals; or heteroaryl themselves optionally substituted by one or more radicals selected from halogen atoms and hydroxyl radical radicals, OCH3, CH3, -CH2OH, CN, CF3, OCF3, NH2, NHaIk or N
- R 2 R 3, R 4, R 5, Z and the ring (N) have the meanings indicated above or below and R 1 and R ⁇ are such that: either R1 is -X1-R7 with X1 is -CH2- and R6 is hydrogen or hydroxyl, CH2-OH; -CO-N (CH3) 2, -CO-NHCH3, -CO-NH- (CH2) 2- N (CH3) 2 and -CO2Et; or R1 represents -X2-R7 with X2 represents:
- R7 is selected from pyrrolidinyl, piperidinyl, piperazinyl, pyrimidinyl, morpholinyl, thiomorpholinyl, tetrahydrofuranyl, phenyl, pyridyl, thienyl, thiazolyl, dithiazolyl, pyrazolyl, pyrazinyl, furyl, imidazolyl, pyrrolyl, oxazolyl, isoxazolyl, benzodihydrofuranyl, benzoxodiazolyl, benzothiodiazolyl and benzothienyl, quinolyl, isoquinolyl; all these radicals represented by R7 being optionally substituted with one or more identical
- the subject of the present invention is particularly the products of formula (I) as defined above with the following names: - ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - [4- (methyl-oxazol-2-ylmethylamino) -piperidin-1-yl] -methanone
- the subject of the present invention is also the processes for preparing the products of formula (I) as defined above or using the methods known to those skilled in the art.
- the subject of the present invention is in particular the process for the preparation of the products of formula (I) such that defined above, characterized in that a product of formula (II) is reacted:
- products of formula (Ia) and (Ib) which may be products of formula (I) in which, respectively, z represents SO2 and z represents CO, and to obtain or of other products of formula (I), one or more of the following transformation reactions can be subjected, if desired and if necessary, in one order.
- an elimination reaction of the protective groups that the protected reactive functions can carry e) a salification reaction with an inorganic or organic acid to obtain the corresponding salt, f) a reaction of duplication racemic forms in duplicated products, said products of formula (I) thus obtained being in all possible isomeric forms racemic, enantiomers and diastereoisomers.
- the processes described above can be carried out as follows:
- the product of formula (II) is subjected to the action of the product of formula (III) as defined above in particular in an alcohol such as for example butanol, propanol, ethanol or dimethylformamide between 80 and 140 0 C, to give a product of formula (IV) as defined above.
- the product of formula (IV) thus obtained is subjected to the action of the aniline of formula (V) as defined above in particular in an alcohol such as for example butanol or dimethylformamide, in the presence or absence of a strong acid (HCl) in catalytic amount under reflux conditions to give a product of formula (VI) as defined above.
- the product of formula (VI) is subjected to the action of chlorosulfonic acid, especially first at 0 ° C. and then at ambient temperature to give a product of formula (VII) such as as defined above.
- the product of formula (VII) thus obtained is subjected to the action of an amine of formula (VIII) as defined above in particular in dichloromethane or a mixture of dichloromethane / THF or dimethylformamide at room temperature, in the presence of an organic base such as triethylamine, diisopropylethylamine or N-methyl morpholine, to give a product of formula (Ia) as defined above.
- the product of formula ( IV) as defined above is subjected to the action of the methyl ester of 4-amino benzoic acid especially in an alcohol such as butanol at a temperature of 100 to 140 ° C., to give the product of formula (IX) as defined above.
- This product of formula (IX) is saponified to its corresponding acid of formula (X) using the usual methods known to those skilled in the art such as in particular by the action of sodium hydroxide or potassium hydroxide in water.
- the products of formulas (Ia) and (Ib) as defined above can therefore constitute products of formula (I) such as as defined above, in which, respectively, z represents SO 2 and z represents CO, or may be converted into products of formula (I) by the usual methods known to those skilled in the art and for example by being subjected to one or more of reactions a) to f) indicated above.
- hydroxyl groups may be protected, for example, by alkyl radicals such as tert-butyl, trimethylsilyl, tert-butyldimethylsilyl, methoxymethyl, tetrahydropyranyl, benzyl or acetyl,
- alkyl radicals such as tert-butyl, trimethylsilyl, tert-butyldimethylsilyl, methoxymethyl, tetrahydropyranyl, benzyl or acetyl
- the amino groups may be protected, for example, by the acetyl, trityl, benzyl, tert-butoxycarbonyl, benzyloxycarbonyl or phthalimido radicals or other radicals known in the peptide chemistry: the amino functions may in particular be protected by a group such as Boc or CH 2 -phenyl and can then be released under the usual conditions known to those skilled in the art.
- the reactions to which the products of formula (I ') as defined above may be subjected, if desired or necessary, may be carried out, for example, as indicated below.
- the saponification reactions may be carried out according to the usual methods known to those skilled in the art, such as, for example, in a solvent such as methanol or ethanol, dioxane or dimethoxyethane, in the presence of sodium hydroxide or potassium hydroxide.
- the reduction or oxidation reactions may be carried out according to the usual methods known to those skilled in the art, such as, for example, in a solvent such as ethyl ether or tetrahydrofuran, in the presence of sodium borohydride or lithium aluminum hydride. ; or for example in a solvent such as acetone or tetrahydrofuran in the presence of potassium permanganate or pyridinium chlorochromate.
- Obtaining the sulphoxide function can be promoted by an equimolar mixture of the product containing an alkylthio group and the reagent such as in particular a peracid.
- Obtaining the sulphone function can be promoted by a mixture of the product containing an alkylthio group with an excess of the reagent such as in particular a peracid.
- the optional alkoxy functions, such as methoxy in particular, of the products described above may, if desired, be converted into hydroxyl function under the usual conditions known to those skilled in the art, for example by boron tribromide in a solvent such as For example, methylene chloride, with hydrobromide or pyridine hydrochloride or with hydrobromic or hydrochloric acid in water or trifluoroacetic acid under reflux.
- a solvent such as For example, methylene chloride, with hydrobromide or pyridine hydrochloride or with hydrobromic or hydrochloric acid in water or trifluoroacetic acid under reflux.
- the phthalimido group may in particular be removed by hydrazine.
- the starting products of formulas (II), (III), (V) and (VIII) can be known, can be obtained commercially or can be prepared according to the usual methods known to those skilled in the art, in particular from products for example by subjecting them to one or more reactions known to those skilled in the art such as, for example, the reactions described above in a) to f).
- the products of formula (II) which are therefore pyrimidine derivatives and the products of formulas (III) which are derivatives of aniline may be commercially available products such as for example dichloropyrimidine, trichloropyrimidine, 4-fluoroaniline, 3,4-difluoroaniline, 4-fluoro-3-chloroaniline, or aniline.
- the anilines of formula (III) can in particular be commercial anilines such as, for example, the following trihalogenated anilines: -3,4, 5-trifluoroaniline -2,3,4-trifluoroaniline -2-chloro-4, ⁇ -difluoroaniline -2,4,5-, trifluoroaniline -3-chloro-2,4-difluoroaniline -2,4 dichloro-5-fluoroaniline. 4-Trifluoromethyl-phenylamine
- the aniline of formula (V) is commercial.
- the amines of formula (VIII) can in particular be commercial amines such as, for example, the following trihalogenated anilines:
- Another subject of the present invention is, as new industrial products, compounds of formulas (VII), (IX) and (X).
- the products of formula (I) as defined above and their addition salts with acids have interesting pharmacological properties.
- the compounds of the present invention can therefore inhibit the activity of kinases, in particular IKK1 and IKK2 with an IC50 of less than 10 ⁇ M.
- the compounds of the present invention can thus inhibit the activation of NF- ⁇ B, and the production of cytokines with IC 50 of less than 10 ⁇ M.
- the compounds of the present invention can thus inhibit the proliferation of a large panel of tumor cells with IC50 values of less than 10 ⁇ M.
- the compounds of formula (I) may therefore have drug activity in particular as inhibitors of IKK1 and IKK2 and may be used in the prevention or treatment of diseases in which the inhibition of IKK1 or IKK2 is beneficial.
- diseases such as inflammatory diseases or diseases with an inflammatory component such as inflammatory arthritis including rheumatoid arthritis, spondyl osteoarthritis, Reiters syndrome, psoriatic arthritis, bone resorption diseases; multiple sclerosis, inflammatory diseases of bowel including Crohn's disease; asthma, chronic pulmonary obstruction, emphysema, rhinitis, acquired myasthenia gravis, graft disease, transplant rejection, psoriasis, dermatitis, allergic disorders, immune system diseases, cachexia, severe acute respiratory syndrome, septic shock, heart failure, myocardial infarction, atherosclerosis, reperfusion injury, AIDS, cancer and insulin resistance disorders such as diabetes , hyperglycemia, hyperinsulinemia, dyslipidemia, obesity, polycystic ovarian diseases, hypertension, cardiovascular disorders, Syndrome X, autoimmune diseases such as systemic lupus, lupus erythematosus, induced glomerulone
- the products of formula (I) according to the present invention as modulators of apoptosis may be useful in the treatment of various human diseases including aberrations in apoptosis such as cancers: such as in particular but not limited to follicular lymphomas, carcinomas with p53 mutations, hormone-dependent tumors of the breast, prostate and ovary, and precancerous lesions such as polyposis familial adenoma, viral infections (such as, but not limited to, but not limited to those caused by Herpes virus, poxvirus, Epstein - Barr virus, Sindbis virus and adenovirus), myelodysplastic syndromes, ischemic disorders associated with myocardial infarction, cerebral congestion, arrhythmia, atherosclerosis, liver disorders induced by toxins or alcohol, haematological disorders such as in particular, but not limited to, chronic anemia and aplastic anemia, degenerative diseases of the musculoskeletal system such as, but not limited to, osteoporosis, cystic fibrosis
- the compounds according to the invention have an anticancer activity and an activity in the treatment of other proliferative diseases such as psoriasis, restenosis, atherosclerosis, AIDS for example, as well as in diseases caused by proliferation.
- these compounds are useful in the prevention and treatment of leukemias, both primary and metastatic solid tumors, carcinomas and cancers, in particular: breast cancer, lung cancer, cancer of the lungs, small bowel, colon and rectal cancer, respiratory tract cancer, oropharynx and hypopharynx, esophageal cancer, liver cancer, stomach cancer, bile duct cancer, cancer of the gall bladder, pancreatic cancer, urinary tract cancers including kidney, urothelium and bladder, cancers of the female genital tract including cancer of the uterus, cervix, ovaries, chlorocarcinoma and trophoblastoma; cancers of the male genital tract including prostate cancer, seminal vesicles, testes, germ cell tumors; cancers of the endocrine glands including thyroid, pituitary, adrenal gland cancer; skin cancers including hemangiomas, melanomas, sarcomas, including Kaposi's sarcoma; tumor
- the compound (s) of formula (I) may be administered in combination with one or more anti-cancer active principle (s), in particular antitumor compounds such as alkylating agents such as alkylsulfonates ( busulfan), dacarbazine, procarbazine, nitrogen mustards (chlormethine, melphalan, chlorambucil), cyclophosphamide, ifosfamide; nitrosoureas such as carmustine, lomustine, semustine, streptozocin; antineoplastic alkaloids such as vincristine, vinblastine; taxanes such as paclitaxel or taxotere; antineoplastic antibiotics such as actinomycin; intercalators, antineoplastic antimetabolites, folate antagonists, methotrexate; inhibitors of purine synthesis; purine analogues such as mercaptopurine, ⁇ -thioguanine; inhibitors of pyrimidine synthesis, aromata
- the compounds of formula (I) may also be administered in combination with one or more other active ingredients useful in one of the pathologies indicated above, for example an anti-emetic, anti-pain, anti-inflammatory agent, anticachexia.
- the subject of the present invention is thus, as medicaments, the products of formula (I) as defined above as well as the addition salts with pharmaceutically acceptable inorganic and organic acids of said products of formula (I).
- the subject of the present invention is, in particular, as medicaments, the products of formula (I) as defined above, corresponding to the following names: - ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino ] - phenyl ⁇ - [4- (methyl-oxazol-2-ylmethylamino) -piperidin-1-yl] -methanone - ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl ⁇ - [3- (methyl-1H-pyrrole-2-ylmethyl-amino) - Piperidin-1-yl] -methanone (Racemic)
- the subject of the present invention is also the pharmaceutical compositions containing, as active principle, at least one of the products of formula (I) as defined above or a pharmaceutically acceptable salt of this product or a prodrug of this product and a pharmaceutically acceptable carrier.
- the present invention also relates to pharmaceutical compositions containing as active ingredient at least one of the products of formula (I) whose names are given above or a pharmaceutically acceptable salt of this product or a prodrug of this product and a pharmaceutically acceptable carrier.
- the present invention particularly relates to the use of the products of formula (I) as defined above or pharmaceutically acceptable salts thereof for the preparation of a medicament for the treatment or prevention of a disease by inhibition of IKK protein kinase activity.
- the present invention thus relates to the use as defined above in which the protein kinase is in a mammal.
- the subject of the present invention is therefore the use of a product of formula (I) as defined above for the preparation of a medicament for the treatment or prevention of a disease chosen from the diseases mentioned above. above.
- the subject of the present invention is in particular the use of a product of formula (I) as defined above for the preparation of a medicament for the treatment or prevention of a disease chosen from the following group: inflammatory diseases, diabetes and cancers.
- the present invention particularly relates to the use 'of a product of formula (I) as defined above for the preparation of a medicament for the treatment or prevention of inflammatory diseases.
- the present invention particularly relates to the use of a product of formula (I) as defined above for the preparation of a medicament for the treatment or prevention of diabetes.
- the present invention particularly relates to the use of a product of formula (I) as defined above for the preparation of a medicament for the treatment of cancers.
- the present invention particularly relates to the use of a product of formula (I) as defined above for the treatment of solid or liquid tumors.
- the present invention particularly relates to the use of a product of formula (I) as defined above for the treatment of cancers resistant to cytotoxic agents.
- the subject of the present invention is in particular the use of a product of formula (I) as defined above for the preparation of medicaments intended for the chemotherapy of cancers.
- the subject of the present invention is in particular the use of a product of formula (I) as defined above for the preparation of medicaments intended for cancer chemotherapy alone or in combination or in combination form as defined herein. -above.
- the present invention particularly relates to the use of a product of formula (I) as defined above as inhibitors of IKK.
- the present invention particularly relates to the products of formula (I) as defined above which constitute Examples 1 to 109 of the present invention.
- Step 1 (2-Chloro-pyrimidin-4-yl) - (4-fluoro-phenyl) -amine
- dichloropyrimidine a mixture containing 15 g of dichloropyrimidine in 200 ml of n-butanol, with stirring, 10 ml of 4- fluoroaniline and then 18 ml of di-isopropyl-ethylamine.
- the reaction mixture is stirred under reflux for 2 hours.
- the reaction medium is cooled and concentrated to dryness.
- Step 3 4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl chloride hydrochloride
- Procedure Ib 4- [4- (3,4-Difluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl chloride hydrochloride
- Stage 1 4-Chloro-N- (3,4-difluorophenyl) pyrimidin-2-amine
- the preparation of this compound is carried out according to the same process as for procedure la from the reaction of 9.21 g of dichloropyrimidine with 8 g 3,4-difluoroaniline: 10.3 g of expected product are thus obtained.
- Stage 2 N4- (3,4-Difluoro-phenyl) -N2-phenyl-pyrimidine-2,4-diamine
- the preparation of this compound is carried out according to the same process as for example 1 from the reaction of 7g (2-Chloro-pyrimidin-4-yl) - (3,4-difluoro-phenyl) amine obtained in Step 1 above with 2.72 g of aniline: 8 g of the expected product are thus obtained.
- Step 3 4- [4- (3,4-Difluorophenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl chloride hydrochloride
- the preparation of this compound is carried out according to the same procedure as in Example 1 starting from the reaction of 8 g of N-4- (3,4-difluoro-phenyl) -N-2-phenyl-pyrimidine-2,4-diamine obtained in stage 2 above with chlorosulphonic acid gives 9 g expected product.
- Step 1 (2-Chloro-pyrimidin-4-yl) - (4-fluoro-3-methylphenyl) -amine
- Step 2 N-4- (4-Fluoro-3-methyl-phenyl) -N-2-phenylpyrimidine-2,4-diamine
- Step 3 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] benzenesulfonyl chloride hydrochloride.
- Stage 1 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzoic acid methyl ester
- a mixture containing 16 g of chloropyrimidine obtained in Stage 1 of Procedure la and 10.8 g of amino 4-methyl benzoate in n-butanol is heated at 140 0 C overnight. After cooling, the precipitate is filtered. This precipitate is washed with Et2O and recrystallized from a DCM-MeOH-iPr20 mixture. 23.5 g of expected product are thus obtained.
- Step 2 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -benzoic acid
- Step 1 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzoic acid methyl ester
- Step 2 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzoic acid 2.08 g of the product obtained in Step 1, in the presence of 410 mg of sodium hydroxide in a mixture of MeOH (5 mL), water (5 mL) and dioxane (20 mL) are brought to a temperature of 40 0 C overnight. The reaction medium is concentrated to dryness and taken up in 100 ml of water. The impurities are extracted with two volumes of Et2O and the aqueous phase is then acidified to pH 6 with 1N HCl. The precipitate formed is filtered, rinsed with distilled water and suspended. in DCM and the solvent is evaporated. 1.3 g of expected acid are obtained.
- Procedure 2c 4- [4- (4-Trifluoromethyl-phenylamino) -pyrimidin-2-ylamino] -benzoic acid.
- Step 1 (2-Chloro-pyrimidin-4-yl) - (4-trifluoromethyl-phenyl) -amine
- Step 2 4- [4- (4-Trifluoromethyl-phenylamino) -pyrimidin-2-ylamino] -benzoic acid methyl ester
- Step 3 4- [4- (4-Trifluoromethyl-phenylamino) -pyrimidin-2-ylamino] -benzoic acid.
- Step 3 of Procedure 1 From 6.4 g. A mixture containing 8 g of ester obtained in Stage 2 and 2.26 g of sodium hydroxide. 4.2 g of expected product are thus obtained.
- Procedure 3 Preparation of the sulfonamide-type reaction intermediates Procedure 3a: N-4- (4-Fluoro-3-methyl-phenyl) -N-2- [4- (4-methylamino-piperidine-1-sulfonyl) -phenyl] -pyrimidine-2,4-diamine
- Step 1 (1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-yl) -methyl-carbamic acid tert-butyl ester
- Step 2 N * 4 - (4-Fluoro-3-methyl-phenyl) -N * 2 - [4- (4-methylamino-piperidine-1-sulfonyl) -phenyl] -pyrimidine-2,4-diamine
- Step 2 N-2- [4- (3-Aminomethyl-piperidine-1-sulfonyl) -phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2,4-diamine (Racemic)
- Procedure 3c N-2- [4- (3-S-Amino-pyrrolidine-1-sulfonyl) -phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2,4-diamine Step 1: ( 1- ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -pyrrolidin-3-S-yl) -carbamic acid tert-butyl ester
- Step 2 N-2- [4- (3-S-Aminopyrrolidine-1-sulfonyl) phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2,4-diamine
- Procedure 3d N-2- [4- (3-R-Amino-pyrrolidine-1-sulfonyl) -phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2,4-diamine
- Step 1 ( 1- ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -pyrrolidin-3-R-yl) -carbamic acid tert-butyl ester
- Step 1 of procedure 3a from 400 mg of sulfonyl chloride hydrochloride obtained in procedure la and 198 mg of commercial amine pyrrolidin-3-S-yl-carbamic acid tert-butyl ester, 379 mg of expected product.
- Step 1 6-Benzyl-1-oxa-6-aza-spiro [2.5] octane
- N-benzyl-4-piperidone 12.8 g of dimethyloxosulfonium methylide and 0.34 g of tetrabutylairanonium bromide in 100 mL of toluene
- a solution of 3.2 g of sodium hydroxide in 32 ml of water is added dropwise and the reaction medium is left stirring at 80 ° C. for 3 hours. After cooling, washed with water, dried over Na 2 SO 4 and concentrated to dryness. 11.7 g of epoxide are thus obtained.
- Step 2 4-Aminomethyl-1-benzyl-piperidin-4-ol
- Step 3 (1-Benzyl-4-hydroxy-piperidin-4-ylmethyl) -carbamic acid tert-butyl ester
- Step 5 4-Aminomethyl-1- ⁇ 4- [4- (4-fluoro-3-methylphenylamino) -pyrimidin-2-ylamino] benzenesulfonyl ⁇ -piperidin-4-ol
- step 1 of procedure 3a from 700 mg of sulfonyl chloride hydrochloride obtained in procedure Ic and 420 mg of piperidine obtained in stage 4, 540 mg of a compound which undergoes a decarboxylation reaction to give 150 mg of expected sulfonamide.
- step 1 of procedure 3a from 2 g of sulfonyl chloride hydrochloride obtained in procedure Ic and 1.38 g of the racemic amine pyrrolidin-3-ylmethyl-carbamic acid benzyl obtained at the stage 4, 1.8 g of a compound which undergoes a hydrogenolysis reaction to give 1.3 g of the expected sulfonamide are obtained.
- step 1 of procedure 3a from 3.5 g of sulfonyl chloride hydrochloride obtained in procedure la and 2 g of 3-N-boc-3-methylaminopiperidine, 2.65 g of a compound which undergoes a decarboxylation reaction to give 1.9 g of expected sulfonamide.
- Procedure 4a ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - (4-methylamino-piperidin-1-yl) -methanone
- Step 1 (1) ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzoyl ⁇ -piperidin-4-yl) -methyl-carbamic acid tert-butyl ester.
- Step 2 ⁇ 4- [4- (4-Fluorophenylamino) pyrimidin-2-ylamino] -phenyl ⁇ - (3-methylamino-piperidin-1-yl) -methanone
- Step 1 (1- ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzoyl ⁇ -piperidin-4-yl) -methyl-carbamic acid tert-butyl ester
- Stage 2 ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl ⁇ - (4-methylamino-piperidin-1-yl) -methanone According to the decarboxylation reaction described in stage 2 of procedure 4a, 4.3 g of the compound obtained in stage 1 make it possible to obtain 2.1 g of the expected carboxamide. .
- Procedure 4d (4-Methylamino-piperidin-1-yl) - ⁇ 4- [4- (4-trifluoromethyl-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ -methanone
- Step 1 Methyl- (1 - 4- [4- (4-Trifluoromethyl-phenylamino) -pyrimidin-2-ylamino] -benzoyl ⁇ -piperidin-4-yl) -carbamic acid tert-butyl ester
- Step 2 (4-Methylamino-piperidin-1-yl) - ⁇ 4- [4- (4-trifluoromethyl-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ -methanone
- MH + 470.9
- Melting point 225-226 0 C
- Example 1 ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - [4- (methyl) -2-
- Example 3 ⁇ 4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - ⁇ 4- [methyl- (1H-pyrazol-3-ylmethyl) -amino] -piperidin-1-yl ⁇ methanone
- Example 6 ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - [4- (methyl-oxazol-2-ylmethylamino) -piperidin-1-yl] -methanone
- Example 7 ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - [3- (methyl-oxazol-2-ylmethylamino) piperidin-1-yl] -methanone (Racemate)
- Example 8 ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl ⁇ - [3- (methyl-lH-pyrrole-2-ylmethyl-amino) • piperidin-1-yl] -methanone (Racemic)
- Example 9 ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - [3- (aminothiazol-2-yl-amino-amino) -piperidin-1-yl] -methanone (Racemic )
- Example 10 ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - ⁇ 3- [methyl- (1H-pyrazol-3-ylmethyl) -amino] -piperidin-1 yl ⁇ -methanone (Racemic)
- Example 11 N * 4 - (4-Fluoro-3-methyl-phenyl) -N * 2 * - (4- ⁇ 4 - [(2-methanesulfonyl-ethyl) -methyl-amino] -piperidine-1-sulfonyl ⁇ -phenyl) -pyrimidine-2,4-diamine
- Example 12 2- [(1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-yl) -methyl-amino] -N , N-dimethyl-acetamide
- Example 13 3 - [(1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benze-nesulfonyl ⁇ -piperidin-4-yl) methyl-amino] -N, N-dimethyl-propionamide
- Example 14 1- ⁇ 4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] -benzoyl ⁇ -3-pyridin-3-ylmethyl-piperidine-3-carboxylic acid ethyl ester (Racemic)
- Example 15 1- ⁇ 4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] -benzoyl ⁇ -3-pyridin-3-ylmethyl-piperidine-3-carboxylic acid dimethylamine (Racemic)
- Stage 1 660 mg of KOH is dissolved in 5 ml of water. 3.5 g of ester obtained in Example 14 and 50 ml of MeOH are added to this solution. After a reflux of 3 hours, the reaction medium is concentrated to dryness and taken up with acidified water at a pH of 7. The precipitate formed is filtered off.
- Example 16 1- ⁇ 4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] -benzoyl ⁇ -3-pyridin-3-ylmethyl-piperidine-3-carboxylic acid methylamide (Racemic)
- Example 17 1- ⁇ 4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] -benzoyl ⁇ -3-pyridin-3-ylmethyl-piperidine-3-carboxylic acid (2-dimethylamino-ethyl) -amide (Racemic )
- Example 18 N * 4 - (4-Fluoro-phenyl) -N * 2 - [4- (3 - ⁇ [(1-methyl-1H-pyrrol-2-ylmethyl) -amino] -methyl ⁇ -piperidine 1-sulfonyl) -phenyl] -pyrimidine-2,4-diamine (Racemic)
- Example 21 N * 4 - (4-Fluoro-phenyl) -N * 2 - (4- ⁇ 3 - [(2-methanesulfonyl-ethylamino) -methyl] -piperidine-1-sulfonyl ⁇ -phenyl) -pyrimidine -2,4-diamine (Racemic)
- Example 22 4-Dimethylaminomethyl-1- ⁇ 4- [4- (4-fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-ol
- Example 23 1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -4-imidazol-1-ylmethyl-piperidin-4-ol
- Step 1 To a solution containing 290 mg of imidazole in 5 mL of DMSO, 1.2 equivalents of sodium hydride are added. After stirring for 20 minutes at RT, 700 mg of epoxide obtained in stage 1 of procedure 3e are added and the mixture is left stirring for 18 hours at RT. It is taken up with water, extracted with DCM, dried over Na 2 SO 4 and concentrated. 540 mg of expected alcohol are obtained by trituration. Stage 2: Following a hydrogenolysis reaction described in Stage 4 of Procedure 3e, starting from 540 mg of alcohol obtained in Stage 1, 280 mg of expected piperidine is obtained. Step 3: Following the procedure described in Step 1 of Procedure 3a, from 600 mg of sulfonyl chloride hydrochloride and 280 mg of piperidine obtained in Step 2, 330 mg of expected sulfonamide is obtained.
- Example 24 N * 4 - (4-Fluoro-3-methylphenyl) -N * 2 * - (4- ⁇ 3- (2-methanesulfonyl-ethylamino) -methyl] -pyrrolidine-1-sulfonyl ⁇ -phenyl) -pyrimidine-2,4-diamine (Racemic)
- Example 25 N * 4 - (4-Fluoro-3-methyl-phenyl) -N * 2 - [4- (3 - ⁇ [(1-methyl-1H-pyrrol-2-ylmethyl) -amino] - methyl ⁇ - pyrrolidine-1-sulfonyl) -phenyl] -pyrimidine-2,4-diamine (Racemic)
- Example 26 ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - [3- (5-methyl-isoxazol-3-ylinethyl-amino) -piperidin-1-yl] -methanone (Racemic)
- Example 27 N * 4 - (4-Fluoro-phenyl) -N * 2 - (4- ⁇ 3 - [(2-methanesulfonyl-ethyl) -methyl-amino] -piperidine-1-sulfonyl ⁇ -phenyl ) -pyrimidine-2,4-diamine
- Example 28 N * 4 - (4-Fluoro-phenyl) -N * 2 - ⁇ 4- [4- (2-methanesulfonyl-ethylamino) -piperidine-1-sulfonyl] -phenyl ⁇ -pyrimidine-2,4 diamine
- Example 29 [3- (1- ⁇ 4- [4- (3,4-Difluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ piperidin-4-ylamino-methyl) -ethyl] -phosphonic acid diethyl ester
- Step 1 [2- (1-Benzylpiperidin-4-ylamino) -ethyl] -phosphonic acid diethyl ester
- Stage 3 [2- (Methyl-piperidin-4-yl-amino) -ethyl] -phosphonic acid diethyl ester
- Step 4 [3- (1- ⁇ 4- [4- (3,4-Difluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ piperidin-4-ylamino-methyl) -ethyl] -phosphonic acid diethyl ester
- Example 30 [2- (1- ⁇ 4- [4- (3,4-Difluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-ylamino) -ethyl] -phosphonic acid diethyl ester
- Step 1 ⁇ 2 - [(1-Benzyl-piperidin-4-yl) -tert-butoxycarbonyl-amino] -ethyl ⁇ -phosphonic acid diethyl ester
- CH3CN a solution of 3.4 g of piperidine obtained in Step 1 of Example 29 in CH3CN (20 mL) was added dropwise 2 g of BOC2O dissolved in CH3CN (16 mL) and stirred 18 hours at RT. It is concentrated to dryness and chromatographed (Al 2 O 3) and eluted with DCM / AcOEt (v / v; 1/1) to give 3 g of expected product.
- Step 2 [2- (tert-Butoxycarbonyl-piperidin-4-yl-amino) -ethyl] -phosphonic acid diethyl ester
- Step 3 [2- (1- ⁇ 4- [4- (3,4-Difluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-ylamino) -ethyl] -phosphonic acid diethyl ester
- 800 mg of difluorinated derivative of Procedure 2b and 880 mg of compound obtained in Stage 2 make it possible to obtain 1.3 g of an intermediate compound which undergoes a decarboxylation reaction for given 1 g of the expected compound.
- Example 31 [2- (1- ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-ylamino) -ethyl] -phosphonic acid diethyl ester
- Example 32 (1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino)] pyrimidin-2-ylamino] -benzenesulfonyl) -piperidin-4-yl) - (3-fluoro-pyridin-4-yl) -methanol
- Stage 1 (3-fluoro-pyridin-4-yl) -piperidin-4-yl-methanol
- a cold solution (-90 ° C.) of 1.82 g of 3- fluoropyridine in 50 ml of THF
- 12 ml of LDA (I.8 M) are added.
- the solution is stirred under nitrogen for 30 minutes while maintaining the same temperature.
- a solution of 2 g of 4-carboxaldehyde-piperidine-1-carboxylic acid tert-butyl ester in 22 mL of THF is added slowly keeping the temperature below -70 ° C.
- the reaction mixture is stirred at this temperature for 30 minutes. minutes.
- Step 2 (1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-yl) - (3-fluoro-pyridin-4-) yl) -methanol
- Example 35 1- (1- ⁇ 4- [4- (4-Fluoro-3-methyl-1-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-yl) -2- (3-methyl) -2- pyridin-2-yl) ethanol (racemic)
- Step 1 2- (3-Methyl-pyridin-2-yl) -1-piperidin-4-yl-ethanol
- Example 36 (1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-yl) -pyridin-4-yl-methanol (racemic )
- Step 2 Racemic (1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-yl) pyridin-4-yl-methanol
- Example 37 (1- ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzen-1-yl) -piperidin-4-yl) - (3-fluoropyridin-4-yl) -methanol (racemic)
- Example 38 1- (1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-yl) -2- (4-methyl) -2- pyridin-2-yl) ethanol (racemic)
- Step 1 2- (4-Methyl-pyridin-2-yl) -1-piperidin-4-yl-ethanol
- Step 2 1- (1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -piperidin-4-yl) -2- (3-methyl) -2- pyridin-2-yl) ethanol (racemic)
- Example 39 ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ - (piperidin-4-yl) - (3-fluoropyridin-4-yl) -methanol) -methanone (racemic)
- Example 40 [4-R- (Amino-phenyl-methyl) -piperidin-1-yl] - ⁇ 4- [4- (4-fluoro-phenylamino) -pyrimidin-2-ylamino] -phenyl ⁇ -methanone
- a solution containing 220 mg of phenylpiperidin-4-R-yl-methylamine commercial amine in 20 ml of a DCM / DMF mixture (v / v; 1/1) is added at room temperature in the order DIPEA (I.5 mL), BOP (360 mg), then, in a small portion over 30 minutes, 300 mg of acid obtained in procedure 2a. Stirred overnight. It is evaporated to dryness, carbonated water (K2CO3) is added and the mixture is extracted with AcOEt. After treatment, chromatography (SiO2) and elution with DCM / MeOH (v / v; 94/6) and recrystallization from DCM / iPr2O.
- Example 42 N * 4 - (4-Fluoro-3-methyl-phenyl) -N * 2 - ⁇ 4- [3- (pyridin-3-yloxy) -piperidine-1-sulfonyl] -phenyl ⁇ -pyrimidine Racemic -2,4-diamine
- Example 43 ⁇ 4-R- [Amino- (4-fluoro-phenyl) -methyl] -piperidin-1-yl ⁇ - ⁇ 4- [4- (4-fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl ⁇ -methanone
- Example 45 (1- ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -3-pyridin-3-ylmethyl-piperidin-3-yl) -methanol (Racemic)
- Example 46 Racemic ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) - ⁇ -trimyridin-2- ⁇ lamino] -phenyl ⁇ - (3-pyridyl-oxy-piperidin-1-yl) -methanone.
- Stage 1 To a solution containing 18.23 g of dimethyloxosulfonium in methylide and 0.485 g of tetrabutylammonium in 150 ml of toluene, a solution of 4.5 g of sodium hydroxide in 48 ml of water is added dropwise and the reaction medium is left stirring. at 80 0 C for 3 hours. After cooling, washed with water, dried over Na 2 SO 4 and concentrated to dryness. 13 gd / epoxide is thus obtained.
- Step 2 In a sealed tube, 1.5 g of the epoxide obtained in Stage 1 are heated at 80 ° C. for 4 hours in the presence of 1 g of pyrrolidine in 25 ml of ethanol. After usual treatment, 1.5 g of aminoalcohol are obtained which undergo a decarboxylation reaction to give the expected piperidine-4-methyl-pyrrolidine
- Example 48 ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyridin-2-ylamino] -phenyl ⁇ - (4-pyrrolidin-1-ylmethylpiperidin-4-ol) -methanone
- Example 79 The sulfonyl chloride of Procedure 1a, 330 mg, is suspended in 40 ml of CH 2 Cl 2. 186 mg of commercial amino alcohol (interchim BG206) are added then 0.55 ml of TEA is added and the mixture is stirred at room temperature overnight. The reaction medium is evaporated on the rotavapor and then taken up in H 2 O: 100 cm 3 and extracted with 3 ⁇ 100 cm 3 of AcOEt; the AcOEt phases are combined and evaporated with the rotavapor; Purified by prep C18 HPLC, evaporated on MeCN and freeze-dried. 152 mg of white lyophilisate are obtained.
- Example 79, 90 and 91 are separated in chiral chromatography as in Example 32, to give respectively the following enantiomers (of undefined absolute configuration): Examples 92 &93; Examples 94 &95; Examples 96 & 97.
- the rotational powers are measured using DMSO as the solvent. The concentrations are in mg / mL.
- Example 98 The compound of Example 88 (50 mg) is dissolved in 5 ml of methanol and 10 mg of sodium borohydride are added. After one hour, 3 mg NaBH4 are added and the reaction left at room temperature for 2 hr added water and then evaporated to dryness and purified by HPLC under basic conditions. 38 mg of white powder are obtained, the expected product (example 98).
- examples 99, 100, 101 are prepared by reducing the corresponding ketones.
- the ketones can be obtained according to the following synthesis scheme: stage a stage b
- This intermediate compound is itself an example of the present invention (Example 104).
- Stage b To the amide of stage a, 2.6 g in 25 ml of methylene chloride, 25 ml of trifluoroacetic acid are added under an inert atmosphere. After 3h at room temperature, the medium is evaporated to dryness and then dissolved in methanol on a Varian Mega Bond Elut SCX cartridge. After elution with pure methanol, the expected product is then eluted with a solution of 7N ammonia in methanol. This gives 1.64 g of yellow oil after evaporation to dryness.
- Stage E A 2.29g of N-BOC commercial isonipecotic acid in 40 ml methylene chloride is added several times, 1.78 g of imidazole carbonyl and the whole is stirred for 2.5 hours at room temperature. 1.072 g of N, O dimethoxy hydroxylamine hydrochloride is then added and the reaction is stirred at room temperature overnight. The medium is washed with water, then 0.01N HCl, then NaHCO3 and again with water. After drying, evaporation, the crude is purified on a silica cartridge with the eluent mixture of methylene chloride / ethyl acetate 9/1 and 8/2. 2.67 g of the expected product are obtained.
- Example 105 (1- ⁇ [4- ( ⁇ 4 - [(4-fluorophenyl) amino] pyrimidin-2-yl ⁇ amino) phenyl] sulfonyl ⁇ piperidin-4-yl) (pyridin-3-yl) methanamine
- the ketone, 290.1 mg, is dissolved in 20 ml of ethanol. 208.3 mg of commercial hydroxylamine hydrochloride are added as well as 409.7 mg of NaAcO. The resulting fine suspension is stirred at RT overnight. The reaction mixture is evaporated under reduced pressure using the rotavapor and then taken up in H 2 O: 30 ml and extracted with 3 ⁇ 20 ml of AcOEt; The AcOEt phases are combined and evaporated on the rotavapor. Purified by flash chromatography eluting the product on a cartridge of 90 g of Merck silica (15-40 microM) by a CH2Cl2 / CH3OH gradient.
- Stage 2 tert-butyl 4- [amino (pyridin-3-yl) methyl] piperidine-1-carboxylate
- EtOH a solution in 2 ml of EtOH
- 2 ml of glacial acetic acid a solution in 2 ml of water
- 171.3 mg of Zinc powder are added to the solution obtained.
- the suspension is shaken by ultrasound overnight.
- the reaction mixture was evaporated on a rotary evaporator, under reduced pressure and then taken up in methanol and the methanolic solution was deposited on a 10 g Varian SCX bonded Bond cartridge previously conditioned with MeOH.
- elution with a CH 3 OH / NH 3 (2N) solution and then evaporating in a rotavapor, under reduced pressure gives 123 mg of a white powder. corresponding to the expected product.
- Step 3 1-Piperidin-4-yl-1-pyridin-3-ylmethanamine
- the compound obtained in Step 2, 234 mg, is dissolved in 5 ml of DCM, 3 ml of CF3CO2H are added.
- the clear yellow solution obtained is stirred for 2 hours at RT and then evaporated in a rotary evaporator under reduced pressure.
- Step 4 (1 - ⁇ [4- ( ⁇ 4 - [(4-fluorophenyl) amino] pyrimidin-2-yl ⁇ amino) phenyl] sulfonyl ⁇ piperidin-4-yl) (pyridin-3-yl) methanamine
- Step 1 (2-Chloro-pyrimidin-4-yl) - (4-fluoro-phenyl) -amine
- stirring is added mL of 4-fluoroaniline followed by 18 mL of diisopropylethylamine.
- the reaction mixture is stirred under reflux for 2 hours.
- the reaction medium is cooled and concentrated to dryness.
- a solution of K 2 CO 3 was added to the residue and extracted 3 times with ethyl acetate, washed with saturated NaCl solution and dried (Na 2 SO 4).
- Step 3 4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl chloride hydrochloride
- Step 1 (2-Chloro-pyrimidin-4-yl) - (4-fluoro-3-methylphenyl) -amine
- Step 2 N-4- (4-Fluoro-3-methyl-phenyl) -N-2-phenylpyrimidine-2,4-diamine
- Example 108 1- ⁇ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -4-pyrrolidin-1-ylmethylpiperidin-4-ol
- Example 109 1- ⁇ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl ⁇ -4-pyrrolidin-1-ylmethyl-piperidin-4-ol
- the procedure is as for example 108 from 500 mg of the sulfonyl chloride hydrochloride described in procedure Ib and 359 mg of amine described in procedure 2, 210 mg of expected product is obtained.
- Example 111 Pharmaceutical Composition
- Examples 6 and 105 are taken as examples in the pharmaceutical preparations which constitute Examples 110 and 111 above, this pharmaceutical preparation can be carried out differently as indicated above and if desired with other products in examples in this application.
- IKK1 and IKK2 The compounds are tested for the inhibition of IKK1 and IKK2 using a flash-supported kinase test.
- the compounds to be tested are dissolved at 10 mM in DMSO and then diluted in kinase buffer (50 mM Tris, pH 7.4 containing 0.1 mM EGTA, 0.1 mM sodium orthovanadate and 0.1% p-mercaptoethanol).
- Serial 3-to-3 dilutions are made from this solution. 10 .mu.l of each dilution are added to the wells of a 96-well plate in duplicate. 10 ⁇ l of kinase buffer is added to the control wells which will serve as 0% inhibition and 10 ⁇ l of 0.5 mM EDTA is added to the control wells (100% inhibition). 10 ⁇ l of the IKK1 or IKK2 mixture (0.1 ⁇ g / well), 25-55 IKB-biotinylated substrate peptide and BSA (5 ⁇ g) are added to each well.
- the wells are washed twice with a solution of 50 mM Tris-EDTA pH7.5 and the radioactivity determined on a microbeta counter.
- the compounds of the invention tested in this test show an IC50 of less than 10 ⁇ M, which shows that they can be used for their therapeutic activity.
- the compounds according to the invention have been the subject of pharmacological tests for determining their anticancer activity.
- MDA-MB231 American type culture collection, Rockville, Maryland, USA, ATCC-HTB26
- MDA-A1 or MDA-ADR known as MDR-resistant multi-drug line, and described by E.Collomb et al. in Cytometry, 12 (1): 15-25, 1991
- MCF7 ATCC-HTB22
- prostate cancer DU145 (ATCC-HTB81) and PC3 (ATCC-CRL1435),
- HCT116 ATCC-CCL247
- HCT15 ATCC-CCL225
- H460 (described by Carmichael in Cancer Research 47 (4): 936-942, 1987 and issued by the National Cancer Institute, Frederick Cancer Research and Development Center, Frederick, Maryland, USA), - glioblastoma ( SF268 described by Westphal in Biochemical & Biophysical Research Communications 132 (1): 284-289, 1985 and issued by the National Cancer Institute, Frederick Cancer Research and Development Center, Frederick, Maryland, USA), Leukemia (CMLT1 described by Kuriyama et al., in Blood, 74: 1989, 1381-1387, by Soda et al in British Journal of Haematology, 59: 1985, 671-679 and by Drexler, in Leukemia Research, 18: 1994, 919-927 and issued by the company DSMZ, Mascheroder Weg Ib, 38124 Braunschweig, Germany).
- CMLT1 described by Kuriyama et al., in Blood, 74: 1989, 1381-1387, by Soda et al in British Journal of Haematology,
Abstract
L'invention concerne les produits de formule (I) : Ces produits étant sous toutes les formes isomères et les sels, à titre de médicaments notamment comme inhibiteurs de IKK.
Description
NOUVEAUX DERIVES de N,N'- 2 , 4-DIANILINOPYRIMIDINES , LEUR
PREPARATION, A TITRE DE MEDICAMENTS, COMPOSITIONS PHARMACEUTIQUES ET NOTAMMENT COMME INHIBITEURS DE IKK
La présente invention concerne de nouveaux dérivés de N,N' -2, 4-dianilinopyrimidines, leur procédé de préparation, les nouveaux intermédiaires obtenus, leur application à titre de médicaments, les compositions pharmaceutiques les renfermant et la nouvelle utilisation de tels dérivés de 2, 4-dianilinopyrimidines .
Le brevet WO200164654-A1 mentionne des 2, 4-di- (hétéro) - arylpyrimidines substituées en 5, inhibitrices des kinases CDK2 et FAK, de même d'autres aminopyrimidines inhibitrices de sérine-thréonine kinases et de CDK sont présentées dans WO2003030909-A1. Le brevet WO2004046118- A2 décrit des dérivés des 2, 4-diphénylaminopyrimidines comme inhibiteurs de la prolifération cellulaire.
Une série de 5-cyano-2-aminopyrimidines sont présentées comme inhibitrices des kinases KDR et FGFR, dans WO200078731-A1, d'autres pyrimidines comme inhibitrices de FAK et de IGFR dans WO2004080980A-1, et aussi de ZAP- 70, FAK et/ou Syk tyrosine kinase dans WO2003078404A1, et des polokinases PLK dans WO2004074244-A2, comme agents cytostatiques .
De même d'autres brevets décrivent des pyrimidines inhibitrices de la transcriptase inverse pour le traitement des infections liées à HIV (WO200185700-A2 ; WO200185699-A2 ; WO200027825A1 et WO2003094920A1) .
La présente invention a ainsi pour objet de nouveaux dérivés de 2, 4-dianilinopyrimidines dotés d'effets inhibiteurs vis-à-vis de protéines kinases.
Les produits de la présente invention peuvent ainsi notamment être utilisés pour la prévention ou le
traitement d'affections capables d'être modulées par l'inhibition de l'activité de protéines kinases. Parmi ces protéines kinases, on cite plus particulièrement la protéine kinase IKK-alpha (IKKa) et IKK-béta (IKKβ) .
Les composés de la présente invention sont des inhibiteurs de kinase en particulier de IKK-alpha et IKK-béta, par conséquent inhibent l'activité NF-KB (nuclear factor kappa B) , ainsi ils peuvent être utilisés dans le traitement de la prophilaxie et les maladies inflammatoires, dans le cancer et le diabète.
Le NF-kB (Nuclear factor kappa B) appartient à une famille de complexes de facteurs transcriptionnels constitués de différentes combinaisons de polypeptides Rel/NF-KB. Les membres de cette famille de polypeptides reliés à NF-KB régulent l'expression de gènes impliqués dans les réponses immunes et inflammatoires. ( (Bames PJ, Karin M (1997) N Engl J Med 336,1066-1071) et (Baeuerle PA, Baichwal VR (1997) Adv Immunol 65, 111-137)) . Dans les conditions basales, les dimères de NF-KB sont retenus sous forme inactive dans le cytoplasme, par des protéines inhibitrices membres de la famille IKB (Beg et. al., Gènes Dev., 7:2064-2070, 1993; Gilmore and Morin, Trends Genêt. 9:427-43)3), 199'); Haskil et. al., CeIl 65: 1281- 1289, 1991) . Les protéines de la famille IKB masquent le signal de translocation nucléaire de NF-KB. La stimulation de la cellule par différents types de ligands tels que les cytokines, le ligand anti-CD40, le lipopolysaccharide (LPS) , les oxydants, des mitogènes comme le phorbol ester, des virus ainsi que beaucoup d'autres stimulants, entraîne l'activation du complexe IKB-Kinase (IKK) qui va à son tour phosphoryler IKB au niveau des résidus serines 32 et 34. Une fois phosphorylé, IKB sera sujet à des ubiquitinations menant à sa dégradation par le protéasome (26S) , permettant
ainsi la libération et la translocation de NF-KB dans le noyau ou il va se lier à des séquences spécifiques au niveau des promoteurs de gènes cibles induisant ainsi leur transcription. Dans le complexe IKB-Kinase (IKK) , les principales kinases sont IKKl(IKKa) et IKK2 (IKKβ) qui sont capables de phosphoryler directement les différentes classes d'IKB. Dans ce complexe IKK, IKK2 est la kinase dominante (Mercurio et. al., Mol. CeIl Biol., 19:1526, 1999-, Zandi et. al., Science; 28 1: 1 3) 60, 1998; Lee et. al, Proe. Natl. Acad. Sci. USA 95:93) 19, 1998) .
Parmi les gènes régulés par NF-KB, beaucoup codent pour des médiateurs pro-inflammatoires, des cytokines, des molécules d'adhésion cellulaire, des protéines de la phase aigϋe, qui également vont à leur tour induire l'activation de NF-KB par des mécanismes autocrines ou paracrines .
L'inhibition de l'activation de NF-KB semble très importante dans le traitement des maladies inflammatoires.
En plus NF-KB, joue un rôle dans la croissance des cellules normales mais aussi des cellules malignes. Les protéines produites par l'expression de gènes régulés par NF-KB comprennent des cytokines, chemokines, molécules d'adhésion, des médiateurs de la croissance cellulaire, de l ' angiogénèse . Par ailleurs différentes études ont montré que NF-KB joue un rôle essentiel dans les transformations néoplastiques. Par exemple NF-KB peut être associé avec la transformation des cellules in vitro et in vivo suite à des événement de sur-expression, amplification, réarrangement ou translocation (Mercurio, R, and Manning, A. M. (1999) Oncogene, 18:6163-6171) . Dans certaines cellules de tumeurs lymphoïdes humaines, les gènes codant pour les différents membres NF-KB sont réarrangés ou amplifiés. Il a été montré que NF-KB peut promouvoir la croissance cellulaire en induisant la
transcription de la cycline D, qui associées à 1 ' hyperphosphorylation de Rb entraîne la transition des phases Gl à S et l'inhibition de l'apoptose.
Il a été montré que dans un nombre important de lignées de cellules tumorales, on trouve une activité constitutive de NF-KB suite à l'activation de IKK2. NF-KB est constitutivement activé dans les maladies de Hodgkin et l'inhibition de NF-KB bloque la croissance de ces lymphomes . D'autre part, l'inhibition de NF-KB par l'expression du répresseur IKBa induit l'apoptose des cellules exprimant l'allèle oncogénique H-Ras (Baldwin, J. Clin. Invest., 107:241 (2001), Bargou et al., J. Clin. Invest., 100:2961 (1997), Mayo et al., Science 178:1812 (1997) . L'activité constitutive de NF-KB semble contribuer à l'oncogénèse à travers l'activation de plusieurs gènes anti-apoptotiques tels que Al/Bfi-1, IEX-I, MAP, ce qui entraîne ainsi la suppression de la voie de mort cellulaire. A travers l'activation de la cycline D, NF-KB peut promouvoir la croissance des cellules tumorales. La régulation des molécules d'adhésion et des protéases de surface suggèrent un rôle de la signalisation NF-KB dans les métastases.
NF-KB est impliqué dans l'induction de la chimiorésistance. NF-KB est activé en réponse à un certain nombre de traitements en chimiothérapie. Il a été montré que l'inhibition de NF-KB par l'utilisation de la forme super-répresseur de IKBa en parallèle au traitement de chimiothérapie augmente l'efficacité de la chimiothérapie dans des modèles de xénogreffe.
La présente invention a pour objet les produits de formule (I) :
 dans laquelle:
dans laquelle:
R2, R3 et R4, identiques ou différents, sont tels que l'un représente un atome d'halogène ou CF3 et les deux autres, identiques ou différents, représentent un atome d'hydrogène ou un atome d'halogène ou un radical alkyle ou un radical alcoxy éventuellement substitués par un ou plusieurs atomes d'halogène;
R5 représente un atome d'hydrogène ou un atome d' halogène;
Z représente CO ou S02 ; le cycle (N) soit

Rl représente -X1-R7 avec Xl représente -(CH2)m- et R7 représente un cycle hétérocycloalkyle, aryle ou hétéroaryle, tous éventuellement substitués ; et R6 représente l'atome d'hydrogène ou les radicaux hydroxyle, -(CH2)mOH, -CO-NRaRb, -CH2-NRaRb -CO2H et -
CO2alk;
ii) Rl représente -X2-R7 avec X2 représente :
-0- ; -0-(CH2)m- ; -CH (OH) - (CH2) n-; -CO- ,--CO-NRc- ; -CO-NRc-O- ; -CH(NRaRb)- ; -C=NOH- ; -C=N-NH2- ;
- (CH2)nl-NRc- (CH2)n2- ; et R7 représente un cycle hétérocycloalkyle, aryle, ou hétéroaryle, tous éventuellement substitués ; et Rβ représente hydrogène ; iii) Rl représente -NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, -OaIk, -CF3, -CO-NR8R9 et S02-alk ; et Rβ représente hydrogène ; étant entendu que lorsque W représente un atome d'hydrogène alors z représente CO ; iv) Rl représente -CH2-NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone et éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, -OEt, -CF3, -CO- N(alk)2 et S02-alk ; et Rβ représente hydrogène ;
v) Rl représente -CO-N (Rc) -OR' c et Rβ représente hydrogène ; avec n, ni et n2, identiques ou différents, représentent un entier de 0 à 3 ; m représente un entier de 1 à 3 ;
Rc et R' c, identiques ou différents, représentent l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone éventuellement substitué par un ou plusieurs atomes d'halogène étant entendu que les atomes d'halogène ne se trouvent pas en position vicinale de l'atome d'azote; NRaRb est tel que soit Ra et Rb, identiques ou différents, représentent l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone ou un radical cycloalkyle, ces radicaux alkyle et cycloalkyle étant éventuellement substitués par un ou
plusieurs atomes d'halogène, étant entendu que les atomes d'halogène ne se trouvent pas en position vicinale de l'atome d'azote; un radical hydroxyle ou un radical NH2, NHalkyle ou N(alkyle)2; soit Ra et Rb forment avec l'atome d'azote auxquels ils sont liés une aminé cyclique pouvant éventuellement renfermer un ou deux autres hétéroatomes choisis parmi 0, S, N ou NRlO, l'aminé cyclique ainsi formée étant elle-même éventuellement substituée par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux alkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ; tous les radicaux hétérocycloalkyle, aryle et hétéroaryle étant éventuellement substitués par un ou plusieurs radicaux, identiques ou différents, choisis parmi les atomes d'halogène ; les radicaux hydroxyle ; cyano ; NR8R9 ; et les radicaux alkyle, cycloalkyle, alcoxy, phényle, hétérocycloalkyle et hétéroaryle, eux-mêmes éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, alkyle, hydroxyalkyle, alcoxyalkyle, CN, CF3, OCF3, ou NRaRb;
NR8R9 est tel que soit R8 et R9, identiques ou différents, sont tels que R8 représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone ou un radical cycloalkyle, ces radicaux alkyle et cycloalkyle étant éventuellement substitués par un ou plusieurs atomes d'halogène, un radical hydroxyle ou un radical NH2, NHalkyle ou N (alkyle) 2; et R9 représente l'atome d'hydrogène et les radicaux alkyle, cycloalkyle ou hétérocycloalkyle eux- mêmes éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, NH2, NHalkyle, N (alkyle) 2, les radicaux alkyle que représente
R9 étant de plus éventuellement substitués par un radical phényle, hétérocycloalkyle ou hétéroaryle eux-mêmes éventuellement substitués par un ou plusieurs radicaux choisis parmi les atomes d'halogène et les radicaux radicaux hydroxyle, alcoxy, alkyle, hydroxyalkyle, alcoxyalkyle, CN, CF3, OCF3, NH2, NHaIk ou N(alk)2 ; soit R8 et R9 forment avec l'atome d'azote auxquels ils sont liés une aminé cyclique pouvant éventuellement renfermer un ou deux autres hétéroatomes choisis parmi 0, S, N ou NRlO, l'aminé cyclique ainsi formée étant elle- même éventuellement substituée par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux alkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ; tous les radicaux hétérocycloalkyle et hétéroaryle ci- dessus éventuellement substitués comme indiqué ci-dessus, étant constitués de 4 à 10 chaînons et renfermant 1 à 3 hétéroatomes choisi (s) parmi 0, S, N et NRlO ; RIO représente un atome d'hydrogène ou un radical alkyle, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule
(D •
La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels R2, R3, R4, R5, Z et le cycle (N) ont les significations indiquées ci-dessus ou ci-après et Rl et R6 sont tels que Rl représente -X1-R7 avec Xl représente -(CH2)m- et R7 représente un cycle hétérocycloalkyle, aryle ou hétéroaryle, tous éventuellement substitués ; et Rβ représente l'atome d'hydrogène ou les radicaux hydroxyle, -(CH2)mOH, -CO-NRaRb, -CH2-NRaRb -CO2H, et - CO2alk;
avec m, n et NRaRb tels que définis ci-dessus ou ci-après et les radicaux hétérocycloalkyle, aryle et hétéroaryle étant éventuellement substitués par un ou plusieurs radicaux, identiques ou différents, tels que définis ci- dessus ou ci-après, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (I) .
La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels R2, R3, R4, R5, Z et le cycle (N) ont les significations indiquées ci-dessus et Rl et Rβ sont tels que Rl représente -X2-R7 avec X2 représente :
-0-,-O- (CH2)m-, -CH(OH) - (CH2)n-, -CO-, -CO-NRc-,
-CO-NRc-O-, -CH(NRaRb)-, -C=NOH-, -C=N-NH2-,
- (CH2 ) nl-NRc- (CH2 ) n2- ; et R7 représente un cycle hétérocycloalkyle, aryle, ou hétéroaryle, tous éventuellement substitués, et R6 représente hydrogène ; avec n, ni, n2, Rc et NRaRb tels que définis ci-dessus et les radicaux hétérocycloalkyle, aryle et hétéroaryle étant éventuellement substitués par un ou plusieurs radicaux, identiques ou différents, tels que définis ci- dessus ou ci-après, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (D .
La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels R2, R3, R4, R5, Z et le cycle (N) ont les significations indiquées ci-dessus ou ci-après et Rl et Rβ sont tels que :
soit Rl représente -NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, -OaIk, -CF3, -CO-NR8R9 et SO2-alk et Rβ représente hydrogène, étant entendu que lorsque W représente un atome d'hydrogène alors z représente CO; soit Rl représente -CH2-NRC-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone et éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, -OEt, -CF3, -CO- N(alk)2 et S02-alk ; et Rβ représente hydrogène ; soit Rl représente -CO-N (Rc) -OR' c et Rβ représente hydrogène ; avec Rc, R' c et NR8R9 tels que définis ci-dessus, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule
(D •
Lorsque les produits de formule (I) sont tels que R2, R3, R4, R5 et le cycle (N) ont les significations indiquées ci-dessus ou ci-après, z représente S02 et Rl et Rβ sont tels que Rl représente -NRc-W avec Rc tel que défini ci- dessus et Rβ représente hydrogène, alors la présente invention concerne notamment les produits dans lesquels W représente un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone substitué par un radical choisi parmi -PO (OEt) 2, -OH, -OaIk, -CF3,-CO-NR8R9 et SO2-alk ; lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les
acides minéraux et organiques desdits produits de formule
(D •
Lorsque le cycle (N) renferme un pont carboné constitué de 1 à 3 carbones, le cycle formé peut notamment être le cycle 8 aza bicyclo (3,2,l)oct 3yl) ou encore un cycle choisi parmi les suivants : le azabicyclo [3.3.1] nonan-3- yl, le 6-azabicyclo [3.2.1] octan-3-yl, le ' 3- azabicyclo [3.2.1] octan-8yl ou encore le 3- azabicyclo [3.3.1] nonan-9-yl. La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels R2, R3, R4, R5 et Z ont les significations indiquées ci- dessus ou ci-après et le cycle (N) représente l'un des cycles définis ci-après : - un cycle azétidinyle ou pyrrolidinyle substitué en position 3 par Rl et R6 tels que définis ci-dessus ou ci- après ; un cycle pipéridinyle et azépinyle substitués en position 3 ou 4 par Rl et R6 tels que définis ci-dessus ou ci-après; un cycle 8 aza bicyclo (3, 2, 1) octan- 3-yl, 6- azabicyclo [3.2.1] octan-3-yl ou 3-azabicyclo [3.2.1] octan- 8yl); lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (I) • La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels R2, R3, R4, R5 et Z ont les significations indiquées ci- dessus ou ci-après et le cycle (N) représente un cycle pyrrolidinyle substitué en 3 par Rl et Rβ tels que définis ci-dessus ou ci-après ou un cycle pipéridinyle substitué en position 3 ou 4 par Rl et R6 tels que définis ci-dessus ou ci-après,
lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (I) .
Dans les produits de formule (I) et dans ce qui suit, les termes indiqués ont les significations qui suivent : le terme halogène désigne les atomes de fluor, de chlore, de brome ou d'iode et de préférence de fluor, chlore ou brome ;
- le terme radical alkyle désigne un radical linéaire ou ramifié renfermant au plus 6 atomes de carbone et notamment les radicaux méthyle, éthyle, propyle, isopropyle, n-butyle, isobutyle, sec-butyle, tert-butyle, pentyle, isopentyle, sec-pentyle, tert-pentyle, néo- pentyle, hexyle, isohexyle, sec-hexyle, tert-hexyle ainsi que leurs isomères de position linéaires ou ramifiés ;
- le terme radical hydroxyalkyle désigne les radicaux alkyle indiqués ci-dessus substitués par un ou plusieurs radicaux hydroxyle ;
- le terme radical alcoxy désigne un radical linéaire ou ramifié renfermant au plus 6 atomes de carbone choisi par exemple parmi les radicaux méthoxy, éthoxy, propoxy, isopropoxy, butoxy linéaire, secondaire ou tertiaire, pentoxy, hexoxy et heptoxy ainsi que leurs isomères de position linéaires ou ramifiés ;
-le terme radical cycloalkyle désigne un radical carbocyclique monocyclique ou bicyclique renfermant de 3 a 7 chaînons et désigne notamment les radicaux cyclopropyle, cyclobutyle, cyclopentyle, cyclohexyle et cycloheptyle ;
- le terme radical aryle désigne les radicaux insaturés, monocycliques ou constitués de cycles condensés, carbocycliques . Comme exemples de tel radical aryle, on peut citer notamment les radicaux phényle ou naphtyle ;
- le terme radical hétérocyclique désigne un radical carbocylique saturé (hétérocycloalkyle) ou partiellement ou totalement insaturé (hétéroaryle) constitué de 4 à 10 chaînons interrompus par un à quatre hétéroatomes, identiques ou différents, choisis parmi les atomes d'oxygène, d'azote ou de soufre :
Parmi les radicaux hétéroaryles à 5 chaînons, on peut citer notamment les radicaux renfermant un à quatre hétéroatomes choisi (s) parmi N éventuellement oxydé, 0 et S éventuellement oxydé tels que les radicaux on peut citer les radicaux thiényle tel que 2-thiényle, 3- thiényle, dioxidothiényle, thiazolyle (N, S), furyle (0),, 2-furyle, pyrrolyle (NH, NCH3) , isothiazolyle, diazolyle, thiadiazolyle (N, N, S), 1, 3, 4-thiadiazolyle, oxazolyle, oxadiazolyle, isoxazolyle (N, 0), 3-isoxazolyle, 4- isoxazolyle, imidazolyle, pyrazolyle (N, N) , groupes triazolyle, tétrazolyle et plus particulièrement les radicaux oxazolyle, isoxazolyle (N, 0), ou pyrazolyle; tous ces cycles étant éventuellement substitués par un ou plusieurs radicaux comme défini ci-dessus ou ci-après, ces substituants se situant bien sûr aux positions chimiquement acceptables pour chacun de ces cycles ; Parmi les radicaux hétéroaryles à 6 chaînons on peut citer notamment les radicaux pyridyle tel que 2-pyridyle, 3-pyridyle et 4-pyridyle, pyridyle N oxyde, pyrimidinyle, pyridazinyle et pyrazinyle ; parmi les radicaux hétéroaryles condensés contenant au moins un hétéroatome choisi parmi le soufre, l'azote et l'oxygène, on peut citer par exemple les radicaux benzothiényle, benzofuryle, benzofurannyle, benzoxazolyle, indazolyle, indolyle, indolinyle, indolinonyle, quinolyle, isoquinolyle, azaindolyle, benzimidazolyle, benzothiazolyle, naphtyridinyle tel que
[1, 8] naphthyridinyle; imidazo (4, 5) pyridinyle ; indolizinyle ; quinazolinyle ; 2, 3-Dihydro-lH-indolyle; 2, 3-Dihydro-benzofuranyle ; 4- [ (Benzo [1, 2, 5] oxadiazolyle ;
(2, 3-Dihydro-benzofuranyle ; parmi les radicaux hétérocycles condensés, on peut citer plus particulièrement les radicaux benzothiényle, benzofuranyle, benzodihydrofuranyle, indolyle, indolinyle, indolinonyle, benzimidazolyle, benzothiazolyle, benzoxodiazolyle , benzothiodiazolyle, naphtyridinyle, indazolyle, quinolyle tel que 4- quinolyl, 5-quinolyl, isoquinolyle, azaindolyle tel que 4-azaindolyl, 3 azaindolyl, imidazo (4 , 5) pyridyle, indolizinyle, quinazolinyle .
Comme Hétérocycloalkyle (saturé) , on peut citer par exemple les radicaux oxiranyle, oxetanyle, tétrahydrofuranyle, dioxolanyle, dithiolanyle, tétrahydropyranyle, dioxanyle, aziridinyle, azétidinyle, pyrrrolidinyle, pipéridinyle, azépinyle, diazépinyle, pipérazinyle, morpholinyle, thiomorpholinyle, dioxydothiomorpholinyle, imidazolidinyle; on peut citer plus particulièrement les radicaux pyrrrolidinyle, pipéridinyle, azépinyle, pipérazinyle ou morpholinyle ; tous les radicaux cycliques étant éventuellement substitués comme indiqué ci-dessus ou ci-après. - les termes radical alkylamino ou NH(alk) et radical dialkylamino ou N(alk)2 désigne ainsi des radicaux amino NH2 substitués respectivement par un ou deux radicaux alkyles, linéaires ou ramifiés, identiques ou différents dans le cas de dialkylamino, choisis parmi les radicaux alkyles tels que définis ci-dessus et éventuellement substitué comme indiqué ci-dessus ou ci-après: on peut citer par exemple les radicaux méthylamino, éthylamino, propylamino ou butylamino, les radicaux diméthylamino, diéthylamino, méthyléthylamino. le terme radical cycloalkylamino désigne ainsi un radical amino substitué notamment par un radical cycloalkyle choisi parmi les radicaux définis ci-dessus: on peut citer ainsi par exemple les radicaux cyclopropylamino, cyclobutylamino, cyclopentylamino ou
encore cyclohexylamino .
- le terme aminé cyclique désigne un radical monocyclique ou bicyclique renfermant de 3 à 10 chaînons dans lequel au moins un atome de carbone est remplacé par un atome d'azote, ce radical cyclique pouvant renfermer également un ou plusieurs autres hétéroatomes choisi (s) parmi 0, S, S02, N ou NRlO avec RIO tel que défini ci-dessus : comme exemples de telles aminés cycliques, on peut citer par exemple les radicaux pyrrolyle, pipéridyle, morpholinyle, pipérazinyle, pyrrolidinyle, azétidinyle. On peut citer plus particulièrement les radicaux pipéridinyle, morpholinyle, pipérazinyle ou azétidinyle.
Le terme patient désigne les êtres humains mais aussi les autres mammifères. Le terme "Prodrug" désigne un produit qui peut être transformé in vivo par des mécanismes métaboliques (tel que l'hydrolyse) en un produit de formule (I) . Par exemple, un ester d'un produit de formule (I) contenant un groupe hydroxyle peut être converti par hydrolyse in vivo en sa molécule mère.
On peut citer à titre d'exemples des esters de produits de formule (I) contenant un groupe hydroxyle tels que les acétates, citrates, lactates, tartrates, malonates, oxalates, salicylates, propionates, succinates, fumarates, maléates, méthylene-bis-b-hydroxynaphthoates, gentisates, iséthionates, di-p-toluoyltartrates, méthanesulfonates, éthanesulfonates, benzenesulfonates, p-toluènesulfonates, cyclohexylsulfamates et quinates. Des esters de produits de formule (I) particulièrement utiles contenant un groupe hydroxyle peuvent être préparés à partir de restes acides tels que ceux décrits par Bundgaard et. al., J. Med. Chem. , 1989, 32, page
2503-2507: ces esters incluent notamment des
(aminométhyl) -benzoates substitués, dialkylamino- méthylbenzoates dans lesquels les deux groupements alkyle peuvent être liés ensemble ou peuvent être interrompus
par un atome d'oxygène ou par un atome d'azote éventuellement substitué soit un atome d'azote alkylé ou encore des morpholino-méthyl) benzoates, e.g. 3- ou A- (morpholinométhyl) -benzoates, et (4-alkylpiperazin-l- yl)benzoates, e.g. 3- ou 4- (4-alkylpiperazin-l- yl) benzoates .
Lorsque les produits de formule (I) comportent un radical amino salifiable par un acide il est bien entendu que ces sels d'acides font également partie de l'invention. On peut citer par exemple les sels fournis avec les acides chlorhydrique ou méthanesulfonique .
Les sels d'addition avec les acides minéraux ou organiques des produits de formule (I) peuvent être, par exemple, les sels formés avec les acides chlorhydrique, bromhydrique, iodhydrique, nitrique, sulfurique, phosphorique, propionique, acétique, trifluoroacétique, formique, benzoïque, maléique, fumarique, succinique, tartrique, citrique, oxalique, glyoxylique, aspartique, ascorbique, les acides alcoylmonosulfoniques tels que par exemple l'acide méthanesulfonique, l'acide éthanesulfonique, l'acide propanesulfonique, les acides alcoyldisulfoniques tels que par exemple l'acide méthanedisulfonique, l'acide alpha, bêta- éthanedisulfonique, les acides arylmonosulfoniques tels que l'acide benzènesulfonique et les acides aryldisulfoniques .
On peut rappeler que la stéréoisomérie peut être définie dans son sens large comme l'isomérie de composés ayant mêmes formules développées, mais dont les différents groupes sont disposés différemment dans l'espace, tels que notamment dans des cyclohexanes monosubstitués dont le substituant peut être en position axiale ou équatoriale. Cependant, il existe un autre type de stéréoisomérie, dû aux arrangements spatiaux différents de substituants fixés, soit sur des doubles liaisons, soit sur des cycles, que l'on appelle souvent isomérie
géométrique E/Z ou isomérie cis-trans ou diastéréoisomère . Le terme stéréoisomère est utilisé dans la présente demande dans son sens le plus large et concerne donc l'ensemble des composés indiqués ci-dessus. La présente invention a particulièrement pour objet les produits de formule (I) tels que définis ci-dessus ou ci- après dans lesquels:
R2, R3 et R4, identiques ou différents, sont tels que l'un représente un atome d'halogène ou CF3 et les deux autres, identiques ou différents, représentent un atome d'hydrogène ou un atome d'halogène ou un radical alkyle ou un radical alcoxy éventuellement substitués par un ou plusieurs atomes d'halogène; R5 représente un atome d'hydrogène ou un atome d'halogène;
Z représente CO ou S02 ; le cycle (N) soit

ii) Rl représente -X2-R7 avec X2 représente : -0-,-CH(OH)-, -CH(OH)-CH2-, -CO-, -CH(NRaRb)-, -C=NOH-, - C=N-NH2- et - (CH2) nl-NRc- (CH2) n2-, et R7 représente un cycle hétérocycloalkyle, phényle ou
hétéroaryle, tous éventuellement substitués, et R6 représente hydrogène ; iii) Rl représente -NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, - OEt, -CF3,-CO-NR8R9 et S02-alk ; et R6 représente hydrogène ; étant entendu que lorsque W représente un atome d'hydrogène alors z représente CO; iv) Rl représente -CH2-NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone et éventuellement substitué par un radical S02-alk ; et Rβ représente hydrogène ;
v) Rl représente -CO-N (Rc) -OR' c et Rβ représente hydrogène ; avec n, ni et n2, identiques ou différents, représentent un entier de 0 à 2;
Rc et R' c identiques ou différents représentent l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 2 atomes de carbone ;
NRaRb est tel que soit Ra et Rb, identiques ou différents, représentent l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone éventuellement substitué par un ou plusieurs atomes d'halogène, un radical hydroxyle ou un radical NH2, NHalkyle ou N (alkyle) 2; soit Ra et Rb forment avec l'atome d'azote auxquels ils sont liés un radical morpholinyle ou pyrrolidinyle éventuellement substitué par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux alkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ;
tous les radicaux hétérocycloalkyle, phényle et hétéroaryle étant éventuellement substitués par un ou plusieurs radicaux, identiques ou différents, choisis parmi les atomes d'halogène ; les radicaux hydroxyle ; cyano ; NR8R9 ; et les radicaux alkyle, cycloalkyle, alcoxy, phényle, hétérocycloalkyle et hétéroaryle, eux- mêmes éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, OCF3, CH3, -CH2OH, CN, CF3, OCF3 ou NRaRb;
NR8R9 est tel que soit R8 et R9, identiques ou différents, sont tels que R8 représente l'atome d'hydrogène, un radical alkyle linéaire ou ramifié renfermant au plus 4 atomes de carbone ou un radical cycloalkyle renfermant de 3 à 6 chaînons, alkyle et cycloalkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ou un radical hydroxyle ; et R9 représente l'atome d'hydrogène ou un radical alkyle éventuellement substitué par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, NH2, NHalkyle, N (alkyle) 2, phényle, hétérocycloalkyle ou hétéroaryle eux-mêmes éventuellement substitués par un ou plusieurs radicaux choisis parmi les atomes d'halogène et les radicaux radicaux hydroxyle, OCH3, CH3, -CH2OH, CN, CF3, OCF3, NH2, NHaIk ou N(alk)2 ; soit R8 et R9 forment avec l'atome d'azote auxquels ils sont liés une aminé cyclique choisie parmi pyrrolyle, pipéridyle, morpholinyle, pyrrolidinyle, azétidinyle et pipérazinyle éventuellement substitués par un ou plusieurs radicaux alkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ; lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule
(D -
On peut noter que la présente invention a particulièrement pour objet les produits de formule (I) tels que définis ci-dessus ou ci-après dans lesquels R2, R3, R4, R5 et Z ont les significations indiquées ci- dessus ou ci-après et le cycle (N) représente un cycle pipéridinyle substitués en position 3 ou 4 par Rl et R6 tels que définis ci-dessus ou ci-après, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule
(D • La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels Z, le cycle (N) , Rl et R6 ont les significations indiquées ci-dessus ou ci-après ; R2, R3 et R4, identiques ou différents, sont tels que l'un représente un atome d'halogène ou CF3 et les deux autres, identiques ou différents, représentent un atome d'hydrogène, un atome d'halogène ou un radical méthyle, méthoxy, trifluorométhyle ou trifluorométhoxy; et R5 représente un atome d'hydrogène; lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule
(D - La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels Z, le cycle (N) , Rl et R6 ont les significations indiquées ci-dessus ou ci-après et R2, R3 et R4, identiques ou différents, sont tels que l'un représente un atome de fluor et les deux autres, identiques ou différents, représentent un atome d'hydrogène, un atome de fluor ou un radical méthyle ; R5 représente un atome d'hydrogène;
lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (I) .
La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels Rl, R2, R3, R4, R5, Rβ et le cycle (N) ont les significations indiquées ci-dessus ou ci-après et Z représente S02, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule
(I) • La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels Rl, R2, R3, R4, R5, R6 et le cycle (N) ont les significations indiquées ci-dessus ou ci-après et Z représente CO, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule
(I) •
Dans les produits de formule (I) tels que définis ci- dessus, tous les radicaux hétérocycloalkyle, phényle et hétéroaryle que représente R7 peuvent notamment être éventuellement substitués par un ou plusieurs radicaux, identiques ou différents, choisis parmi les atomes d'halogène ; les radicaux NR8R9 ; et les radicaux alkyle, cycloalkyle, alcoxy, phényle, hétérocycloalkyle et hétéroaryle, eux-mêmes éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, OCF3, CH3, -CH2OH, CN, CF3, OCF3, NH2, NHaIk, ou N(alk)2, pyrrolidinyle, pipéridinyle ou morpholinyle éventuellement substitués par un ou plusieurs radicaux
identiques ou différents choisis parmi les atomes d'halogène et les radicaux alkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d' halogène . Dans les produits de formule (I) ' tels que définis ci- dessus, NR8R9 peut notamment être tel que soit R8 et R9, identiques ou différents, sont tels que R8 représente l'atome d'hydrogène, un radical alkyle linéaire ou ramifié renfermant au plus 4 atomes de carbone ou un radical cycloalkyle renfermant de 3 à 6 chaînons, alkyle et cycloalkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ou un radical hydroxyle; et R9 représente l'atome d'hydrogène ou un radical alkyle éventuellement substitué par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, NH2, NHalkyle, N (alkyle) 2, phényle, hétérocycloalkyle ou hétéroaryle eux-mêmes éventuellement substitués par un ou plusieurs radicaux choisis parmi les atomes d'halogène et les radicaux radicaux hydroxyle, OCH3, CH3, -CH2OH, CN, CF3, OCF3, NH2, NHaIk ou N(alk)2 ; soit R8 et R9 forment avec l'atome d'azote auxquels ils sont liés une aminé cyclique choisie parmi pyrrolyle, pipéridyle, morpholinyle, pyrrolidinyle, azétidinyle et pipérazinyle éventuellement substitués par un ou plusieurs radicaux alkyle identiques ou différents eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ; le radical NR8R9 peut également être tel que soit R8 et R9, identiques ou différents, sont tels que R8 représente l'atome d'hydrogène ou un radical alkyle renfermant au plus 3 atomes de carbone éventuellement substitué par un ou plusieurs atomes d'halogène ou un radical hydroxyle ; et R9 représente l'atome d'hydrogène ou un radical alkyle éventuellement substitué par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, NH2,
NHalkyle, N(alkyle)2, phényle, morpholinyle, pyrrolidinyle, pipéridinyle ou pyridine, ces derniers cycles étant eux-mêmes éventuellement substitués par un ou plusieurs radicaux choisis parmi les atomes d'halogène et les radicaux hydroxyle, OCH3, CH3, CF3, OCF3, NH2, NHaIk ou N(alk)2 ; soit R8 et R9 forment avec l'atome d' azote auxquels ils sont liés une aminé cyclique choisie parmi pipéridyle, morpholinyle, pyrrolidinyle et pipérazinyle éventuellement substitués par un ou plusieurs radicaux choisis parmi les atomes d'halogène et les radicaux méthyle ; le radical NR8R9 peut également représenter les valeurs définies ci-dessus pour NRaRb. La présente invention a ainsi pour objet les produits de formule (I) tels que définis ci-dessus dans lesquels R2, R3, R4, R5, Z et le cycle (N) ont les significations indiquées ci-dessus ou ci-après et Rl et Rβ sont tels que : soit Rl représente -X1-R7 avec Xl représente -CH2- et R6 représente l'atome d'hydrogène ou les radicaux hydroxyle, CH2-OH; -CO-N (CH3) 2, -CO-NHCH3, -CO-NH- (CH2) 2- N(CH3)2 et -CO2Et; soit Rl représente -X2-R7 avec X2 représente :
-0-, -CHOH-, -CH(OH) -CH2-, -CO- , -CHNH2-, -NH-CH2-, - N(CH3)-CH2- et CH2-NH-CH2-; et Rβ représente hydrogène ; et R7 est choisi parmi les radicaux pyrrolidinyle, pipéridinyle, pipérazinyle, pyrimidinyle, morpholinyle, thiomorpholinyle, tétrahydrofuranyle, phényle, pyridyle, thiényle, thiazolyle, dithiazolyle, pyrazolyle, pyrazinyle, furyle, imidazolyle, pyrrolyle, oxazolyle, isoxazolyle, benzodihydrofuranyle, benzoxodiazolyle, benzothiodiazolyle, benzothiényle, quinolyle, isoquinolyle; tous ces radicaux que représente R7 étant éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, méthyle, méthoxy, hydroxyméthyle,
alcoxyméthyle, cyano, NH2, NHaIk, et N(alk)2, -CH2-NH2, - CH2-NHalk, -CH2-N (alk) 2, phényle, morpholinyle et CH2-morpholinyle eux-mêmes éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, CH3, OCH3, -CH2OH, CN, CF3, OCF3, NH2, NHaIk ou N (alk) 2; lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule
(D •
La présente invention a particulièrement pour objet les produits de formule (I) tels que définis ci-dessus répondant aux noms suivants : - {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl}- [4- (methyl-oxazol-2-ylmethyl-amino) -piperidin-1- yl] -methanone
{ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl}- [3- (methyl-lH-pyrrole-2-ylmethyl-amino) - piperidin-1-yl] -methanone (Racémique) l-{ 4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] - benzoyl } -3-pyridin-3-ylmethyl-piperidine-3-carboxylic acid methylamide (Racémique)
N-4- (4-Fluoro-3-methyl phenyl) -N-2- (4-{ 3- [ (2- methanesulfonyl-ethylamino) -methyl] -pyrrolidine-1- sulfonyl } -phenyl) -pyrimidine-2 , 4-diamine (Racémique)
N-4- (4-Fluoro-3-methyl-phenyl) -N-2- [4- (3-{ [ (1-methyl- lH-pyrrol-2-ylmethyl ) -amino] -methyl } -pyrrolidine-l- sulfonyl ) -phenyl] -pyrimidine-2 , 4-diamine (Racémique ) - 4-pyrrolidin-l-ylmethyl-l- { 4- [ 4- ( 4-f luoro-3-methyl- phenylamino) -pyrimidin-2-ylamino] benzenesulf onyl } - piper idin-4-ol - { 4- [ 4- ( 4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -phenyl } [ 4- (methyl-pyridin-2-ylmethyl-amino) - piperidin-1-yl] -methanone ;
- { 4- [ 4- ( 4-Fluoro-3-methyl-phenylamino ) -pyrimidin-2-
ylamino] -phenyl}- [4- (methyl-pyridin-4-ylmethyl-amino) - piperidin-1-yl] -methanone;
- { 4- [ (1, 5-Dimethyl-lH-pyrazol-4-ylmethyl) -methyl-amino] - piperidin-l-yl}-{ 4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone
- { 4- [ (2-Amino-pyridin-3-ylmethyl) -methyl-amino] - piperidin-l-yl}-{4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone
- 4-{ [ (l-{4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin- 2-ylamino] -benzoyl}-piperidin-4-yl) -methyl-amino] - methyl}-l, 5-dimethyl-lH-pyrrole-2-carbonitrile
- {4- [ (2, 4-Dimethyl-thiazol-5-ylmethyl) -methyl-amino] - piperidin-1-yl} -{ 4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone - (l-{ [4- ({4- [ (4-fluorophényl) amino] pyrimidin-2- yl} amino) phenyl] sulfonyl}pipéridin-4-yl) (pyridin-3-yl) - méthanamine l-{4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - benzenesulfonyl } -4-pyrrolidin-l-ylmethyl-piperidin-4-ol - l-{4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -benzenesulfonyl } -4-pyrrolidin-l-ylmethyl- piperidin-4-ol lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (D •
La présente invention a également pour objet les procédés de préparation des produits de formule (I) tels que définis ci-dessus ou utilisant les méthodes connues de l'homme du métier.
La présente invention a notamment pour objet le procédé de préparation des produits de formule (I) tels que
définis ci-dessus caractérisé en ce que l'on fait réagir un produit de formule (II) :





voie a) z=SO2 produit de formule (VI) que l'on fait réagir avec de l'acide chlorosulfonique SO2(OH)C1 pour obtenir le produit correspondant de formule (VII) :


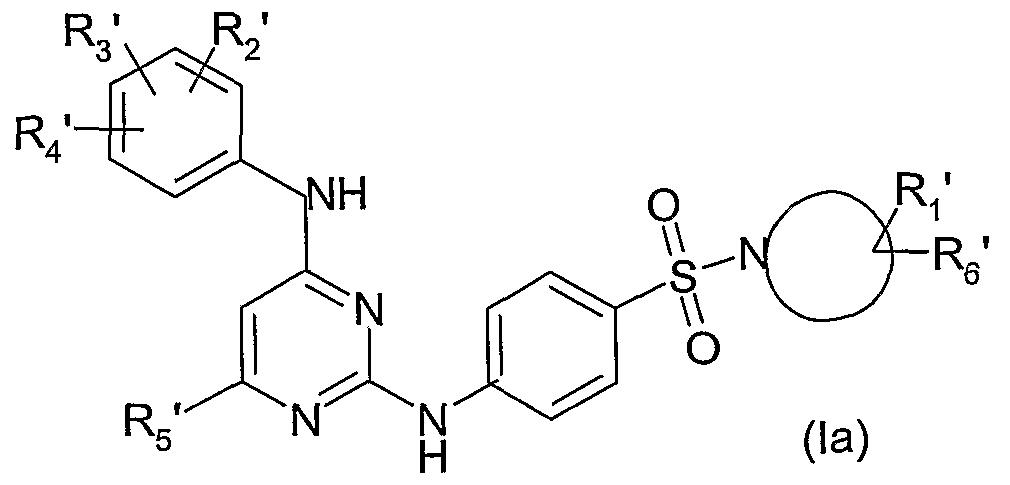


dans laquelle R2' , R3' , R4' et R5' ont les significations indiquées ci-dessus, produit de formule (X) que l'on fait réagir avec une aminé de formule (VIII) telle que définie ci-dessus :
pour obtenir un produit de formule (Ib) :

Le produit de formule (II) est soumis à l'action du produit de formule (III) telle que définie ci-dessus notamment dans un alcool tel que par exemple le butanol, le propanol, l'éthanol ou la diméthylformamide entre 80 et 140 0C, pour donner un produit de formule (IV) telle que définie ci-dessus. Le produit de formule (IV) ainsi obtenu est soumis à l'action de l'aniline de formule (V) telle que définie ci-dessus notamment dans un alcool tel que par exemple le butanol ou la diméthylformamide, en présence ou non d'un acide fort (HCl) en quantité catalytique dans les conditions de reflux pour donner un produit de formule (VI) telle que définie ci-dessus.
Selon la voie a) définie ci-dessus, le produit de formule (VI) est soumis à l'action de l'acide chlorosulfonique notamment d'abord à 00C puis à température ambiante pour donner un produit de formule (VII) telle que définie ci- dessus .
Le produit de formule (VII) ainsi obtenu est soumis à l'action d'une aminé de formule (VIII) telle que définie ci-dessus notamment dans le dichlorométhane ou un mélange dichlorométhane/THF ou la diméthylformamide à température ambiante, en présence d'une base organique telle que la triéthylamine, la diisopropyléthylamine ou la N-Méthyl morpholine, pour donner un produit de formule (Ia) telle que définie ci-dessus.) Selon la voie b) définie ci-dessus, le produit de formule (IV) tel que défini ci-dessus est soumis à l'action de l'ester méthylique de l'acide 4-amino benzoïque notamment dans un alcool tel que le butanol à une température de 100 à 1400C, pour donner le produit de formule (IX) tel que défini ci-dessus.
On saponifie ce produit de formule (IX) en son acide correspondant de formule (X) en procédant selon les méthodes usuelles connues de l'homme du métier telles que notamment par action de la soude ou de la potasse dans l'eau.
On fait réagir le produit de formule (X) ainsi obtenu avec l'aminé de formule (VIII) définie ci-dessus selon les méthodes de couplage connues de l'homme du métier telles que par exemple par un couplage peptidique en présence d'agent de couplage tel que la BOP, DCC ou TBTU dans un solvant tel que par exemple la diméthylformamide ou le dichlorométhane pour donner un produit de formule
(Ib) telle que définie ci-dessus.
Selon les valeurs de Rl', R2', R3' , R4', R5' et R6' , les produits de formules (Ia) et (Ib) telles que définies ci- dessus peuvent donc constituer des produits de formule (I) telle que définie ci-dessus, dans lesquelles respectivement z représente S02 et z représente CO, ou peuvent être transformés en produits de formule (I) par les méthodes usuelles connues de l'homme du métier et par exemple en étant soumis à une ou plusieurs des réactions a) à f) indiquées ci-dessus.
Par ailleurs, on peut noter que de telles réactions de transformation a) à f) de substituants en d'autres substituants peuvent également être effectuées sur les produits de départ ainsi que sur les intermédiaires tels que définis ci-dessus avant de poursuivre la synthèse selon les réactions indiquées dans les procédés ci- dessus. Les diverses fonctions réactives que peuvent porter certains composés des réactions définies ci-dessus peuvent, si nécessaire, être protégées : il s'agit par exemple des radicaux hydroxyle, acyle ou encore amino et monoalkylamino qui peuvent être protégés par les groupements protecteurs appropriés.
La liste suivante, non exhaustive, d'exemples de protection de fonctions réactives peut être citée :
- les groupements hydroxyle peuvent être protégés par exemple par les radicaux alkyle tels que tert-butyle, triméthylsilyle, tert-butyldiméthylsilyle, méthoxyméthyle, tétrahydropyrannyle, benzyle ou acétyle,
- les groupements amino peuvent être protégés par exemple par les radicaux acétyle, trityle, benzyle, tert- butoxycarbonyle, benzyloxycarbonyle, phtalimido ou d'autres radicaux connus dans la chimie des peptides : les fonctions aminés peuvent notamment être protégées par un groupe tel que Boc ou CH2-phényle et peut alors être libérée dans les conditions usuelles connues de l'homme du métier. Les réactions auxquelles les produits de formule (I' ) telle que définie ci-dessus peuvent être soumis, si désiré ou si nécessaire, peuvent être réalisées, par exemple, comme indiqué ci-après. Les réactions de saponification peuvent être - réalisées selon les méthodes usuelles connues de l'homme du métier, telles que par exemple dans un solvant tel que le méthanol ou l'éthanol, le dioxane ou le diméthoxyéthane, en présence de soude ou de potasse. Les réactions de réduction ou oxydation peuvent être réalisées selon les méthodes usuelles connues de l'homme du métier telles que par exemple dans un solvant tel que l'éther éthylique ou le tétrahydrofurane, en présence de borohydrure de sodium ou d'hydrure de lithium aluminium; ou par exemple dans un solvant tel que l'acétone ou le tétrahydrofurane en présence de permanganate de potassium ou de chlorochromate de pyridinium. a) Les éventuels groupements alkylthio des produits décrits ci-dessus peuvent être, si désiré, transformés en les fonctions sulfoxyde ou sulfone correspondantes dans les conditions usuelles connues de l'homme du métier telles que par exemple par les peracides comme par
exemple l'acide peracétique ou l'acide métachloroperbenzoïque ou encore par l'oxone, le périodate de sodium dans un solvant tel que par exemple le chlorure de méthylène ou le dioxanne à la température ambiante .
L'obtention de la fonction suifoxyde peut être favorisée par un mélange équimolaire du produit renfermant un groupement alkylthio et du réactif tel que notamment un peracide . L'obtention de la fonction sulfone peut être favorisée par un mélange du produit renfermant un groupement alkylthio avec un excès du réactif tel que notamment un peracide . b) Les éventuelles fonctions alcoxy telles que notamment méthoxy des produits décrits ci-dessus peuvent être, si désiré, transformées en fonction hydroxyle dans les conditions usuelles connues de l'homme du métier par exemple par du tribromure de bore dans un solvant tel que par exemple le chlorure de méthylène, par du bromhydrate ou chlorhydrate de pyridine ou encore par de l'acide bromhydrique ou chlorhydrique dans de l'eau ou de l'acide trifluoro acétique au reflux. c) Les éventuelles fonctions alcool des produits décrits ci-dessus peuvent être, si désiré, transformées en fonction aldéhyde ou cétone par oxydation dans les conditions usuelles connues de l'homme du métier telles que par exemple par action de l'oxyde de manganèse pour obtenir les aldéhydes ou par action du permanganate depotassium ou de chlorochromate de pyridinium pour accéder aux cétones pour accéder aux cétones. d) L'élimination de groupements protecteurs tels que par exemple ceux indiqués ci-dessus peut être effectuée dans les conditions usuelles connues de l'homme de métier notamment par une hydrolyse acide effectuée avec un acide tel que l'acide chlorhydrique, benzène sulfonique ou para-toluène sulfonique, formique ou trifluoroacétique ou
encore par une hydrogénation catalytique.
Le groupement phtalimido peut notamment être éliminé par 1 ' hydrazine.
On trouvera une liste de différents groupements protec- teurs utilisables par exemple dans le brevet BF 2 499 995. e) Les produits décrits ci-dessus peuvent, si désiré, faire l'objet de réactions de salification par exemple par un acide minéral ou organique selon les méthodes usuelles connues de l'homme du métier. f) Les éventuelles formes optiquement actives des produits décrits ci-dessus peuvent être préparées par dédoublement des racémiques selon les méthodes usuelles connues de l'homme du métier. Des illustrations de telles réactions définies ci-dessus sont données dans la préparation des exemples décrits ci- après .
Les produits de départ de formules (II), (III), (V) et (VIII) peuvent être connus, peuvent être obtenus commercialement ou peuvent être préparés selon les méthodes usuelles connues de l'homme du métier, notamment à partir de produits de commerciaux par exemple en les soumettant à une ou plusieurs réactions connues de l'homme du métier telles que par exemple des réactions décrites ci-dessus en a) à f) .
Les produits de formule (II) qui sont donc des dérivés de la pyrimidine et les produits de formules (III) qui sont des dérivés de l'aniline peuvent être des produits commercialisés comme par exemple la dichloropyrimidine, la trichloropyrimidine, la 4-fluoroaniline, la 3,4- difluoroaniline, la 4-fIuoro3-chloroaniline, ou 1' aniline .
Les anilines de formule (III) peuvent notamment être des anilines commerciales telles que par exemple les anilines trihalogénées suivantes :
-3,4, 5-trifluoroaniline -2,3,4 -trifluoroaniline -2-chloro- 4, β-difluoroaniline -2,4,5-, trifluoroaniline -3-chloro-2, 4-difluoroaniline -2, 4-dichloro-5-fluoroaniline. 4-trifluoromethyl-phenylamine L'aniline de formule (V) est commerciale. Les aminés de formule (VIII) peuvent notamment être des aminés commerciales telles que par exemple les anilines trihalogénées suivantes :
Dichlorhydrate de 3-carboxylate d' éthyl 3- (pyridin-2- ylméthyl) pipéridine
Dichlorhydrate de 3-carboxylate d' éthyl 3- (pyridin-3- ylméthyl) pipéridine
Dichlorhydrate de 3-carboxylate d' éthyl 3- (pyridin-4- ylméthyl) pipéridine
4-Benzyl-4-hydroxypipéridine
Dichlorhydrate de 2- (pipéridine-4-yloxy) pyrazine Dichlorhydrate de 4- (pipéridin-4-yloxy) pyridine
Dichlorhydrate de 2- (pipéridine-4-yloxy) pyrimidine
Chlorhydrate de 4-phénoxypipéridine
Dichlorhydrate de 2- (pipéridin-4-yloxy) pyridine
Dichlorhydrate de 2-pipéridin-4-ylméthylpyridine Dichlorhydrate de 4-pipéridin-4-ylméthylpyridine
Dichlorhydrate de 3-pipéridin-4-ylméthylpyridine
(R) - (4-Fluorophenyl) -l-pipéridin-4-méthanamine
(S) - (4-Fluorophenyl) -l-pipéridin-4-méthanamine
(R) -Phenyl-l-pipéridin-4-méthanamine (S) -Phenyl-l-pipéridin-4-méthanamine
(4-Fluoro-phenyl) -piperidin-4-yl-methanol
Les préparations des aminés de formule (VIII) non commerciales peuvent être réalisées selon des méthodes
connues de l'homme du métier.
On peut indiquer que pour obtenir des produits de formule (I) tels que définis ci-dessus dans lesquels le cycle (N) renferme un pont carboné constitué de 1 à 3 carbones, on peut utiliser comme produits de départ des aminés bicycliques pouvant être obtenus à partir de composés commerciaux tels que la tropinone, la pseudo-pelletrivine selon les références ci-dessous:
Tetrahedron 2002, 58, 5669-5674 J.Org.Chem. 1996, 61, 3849-3862
J.Med.Chem. 1993, 36, 3703-3720
J.Chem.Soc. Perkin Transi 1991, 1375-1381
J.Med.Chem. 1994, 37, 2831-2840
A titre d'exemples de cycle (N), on peut citer les composés suivants :
9-azabicyclo [3.3.1] nonan-3-amine

6-azabicyclo [3.2.1] octan-3-amine


3-azabicyclo [3.3.1] nonan-9-amine

Ces bicycles qui constituent des exemples de (cycle) N, étant substitués par Rl et R6 tels que définis ci-dessus et éventuellemment protégés si nécessaire, et ces bicycles étant liés à z par leur azote intracyclique .
Des exemples d'aldéhydes ou de cétones de formule (X), sont donnés dans la partie expérimentale à titre d'exemples non limitatifs.
La partie expérimentale ci-après donne des exemples non limitatifs de préparation de produits de formule (I) selon la présente invention et également des exemples de produits de départ non limitatifs utilisés dans ces préparations .
La présente invention a enfin pour objet à titre de produits industriels nouveaux, des composés de formules (VII) , (IX) et (X) .
Les produits de formule (I) tels que définis ci-dessus ainsi que leurs sels d'addition avec les acides présentent d'intéressantes propriétés pharmacologiques . Les composés de la présente invention peuvent donc inhiber l'activité des kinases, en particulier IKKl et IKK2 avec une IC50 inférieure à 10 μM.
Les composés de la présente invention peuvent ainsi inhiber l'activation de NF-KB, et la production de cytokines avec des IC50 inférieures à 10 μM.
Les composés de la présente invention peuvent ainsi inhiber la prolifération d'un large panel de cellules tumorales avec des IC50 inférieures à 10 μM. Les composés de la formule (I) peuvent donc avoir une activité de médicament en particulier comme inhibiteurs de IKKl et IKK2 et peuvent être utilisés dans la prévention ou le traitement de maladies dans lesquelles l'inhibition de IKKl ou IKK2 est bénéfique. Par exemple la prévention ou le traitement de maladies telles que les maladies inflammatoires ou maladies avec une composante inflammatoire comme par exemple l'arthrite inflammatoire y compris l'arthrite rhumatoïde, l' ostéoarthrite spondylique, le syndrome de Reiters, l'arthritis psoriatique, les maladies de résorption osseuse; la scléroses en plaques, les maladies inflammatoires de
l'intestin incluant la maladie de Crohn's; l'asthme, l'obstruction pulmonaire chronique, l'emphysème, les rhinites, la myasthénie acquise, la maladie de Graves, le rejet de greffe, le psoriasis, les dermatites, les troubles allergiques, les maladies du système immunitaire, la cachexie, le syndrome respiratoire aiguë sévère, le choc septique, l'insuffisance cardiaque, l'infarctus du myocarde, l'athérosclérose, les lésions de reperfusion, le SIDA, les cancers et les troubles caractérisés par une résistance à l'insuline tels que les diabètes, l'hyperglycémie, l' hyperinsulinémie, la dyslipidémie, l'obésité, les maladies ovariennes polycystiques, l'hypertension, les troubles cardiovasculaires, le Syndrome X, les maladies autoimmunes telles que notamment le lupus systémique, le lupus érythémateux, les glomérulonéphrites induites par des déficiences du système immunitaire, les diabètes autoimmunes insulino-dépendants, les rétinites pigmentaires, les rhinosinusites sensibles à l'aspirine. Les produits de formule (I) selon la présente invention comme modulateurs de l'apoptose, peuvent être utiles dans le traitement de différentes maladies humaines incluant des aberrations dans l'apoptose telles que des cancers: telles que notamment mais à titre non limitatif, les lymphomes folliculaires, les carcinomes avec des mutations p53, des tumeurs hormone-dépendantes du sein, de la prostate et de l'ovaire, et des lésions précancéreuses comme l'adénome familial polyposis, des infections virales (telles que notamment mais à titre non limitatif celles causées par le virus Herpès, le poxvirus, le virus d' Epstein-Barr, virus de Sindbis et l' adénovirus) , les syndromes myélodysplastiques, les désordres ischémiques associés à l'infarctus du myocarde, la congestion cérébrale, l'arythmie, l'athérosclérose, les troubles hépatiques induits par des toxines ou l'alcool, les désordres hématologiques telles que
notamment mais à titre non limitatif, l'anémie chronique et l'anémie aplasique, les maladies dégénératives du système musculosquelettal telles que notamment mais à titre non limitatif, l' ostéoporose, les fibroses cystiques, les maladies des reins et les cancers.
Il apparaît donc que les composés selon l'invention ont une activité anticancéreuse et une activité dans le traitement des autres maladies prolifératives telles que le psoriasis, la resténose, l ' arthérosclérose, le SIDA par exemple, ainsi que dans les maladies provoquées par la prolifération des cellules du muscle lisse vasculaire de l ' angiogénèse et dans la polyarthrite rhumatolde, la neuro-fibromatose, l'athérosclérose, les fibroses pulmonaires, les resténoses suivant de l' angioplastie ou de la chirurgie vasculaire, la formation de cicatrices hypertrophiques, l' angiogénèse et le choc endotoxique.
Ces médicaments trouvent leur emploi en thérapeutique, notamment dans le traitement ou la prévention des maladies causées ou exacerbées par la prolifération des cellules et en particulier des cellules tumorales.
Comme inhibiteur de la prolifération des cellules tumorales, ces composés sont utiles dans la prévention et le traitement des leucémies, des tumeurs solides à la fois primaires et métastasiques, des carcinomes et cancers, en particulier: cancer du sein, cancer du poumon, cancer de l'intestin grêle, cancer du colon et du rectum, cancer des voies respiratoires, de l' oropharynx et de l'hypopharynx, cancer de l'œsophage, cancer du foie, cancer de l'estomac, cancer des canaux biliaires, cancer de la vésicule biliaire, cancer du pancréas, cancers des voies urinaires y compris rein, urothélium et vessie, cancers du tractus génital féminin y compris le cancer de l'utérus, du col de l'utérus, des ovaires, chloriocarcinome et trophoblastome; cancers du tractus génital masculin y compris cancer de la prostate, des
vésicules séminales, des testicules, tumeurs des cellules germinales; cancers des glandes endocrines y compris cancer de la thyroïde, de l'hypophyse, des glandes surrénales ; cancers de la peau y compris hémangiomes, mélanomes, sarcomes, incluant le sarcome de Kaposi ; tumeurs du cerveau, des nerfs, des yeux, des méninges, incluant astrocytomes, gliomes, glioblastomes, rétinoblastomes, neurinomes, neuroblastomes, schwannomes, méningiomes ; tumeurs malignes hématopoïétiques ; leucémies telles que leucémie aigϋe lymphoïde, leucémie aigϋe myéloïde, leucémie myéloïde chronique, leucémie lymphoïde chronique, chloromes, plasmocytom.es, leucémies des cellules T ou B, lymphomes non hodgkiniens ou hodgkiniens, myélomes, hémopathies malignes diverses. La présente invention a notamment pour objet les combinaisons définies comme suit.
Selon la présente invention, le ou les composés de formule (I) peuvent être administrés en association avec un (ou plusieurs) principe (s) actif (s) anticancéreux, en particulier des composés antitumoraux tels que les agents alkylants tels que les alkylsulfonates (busulfan) , la dacarbazine, la procarbazine, les moutardes azotées (chlorméthine, melphalan, chlorambucil) , cyclophosphamide, ifosfamide; les nitrosourées tels que la carmustine, la lomustine, la sémustine, la streptozocine; les alcaloïdes antinéoplasiques tels que la vincristine, la vinblastine ; les taxanes tel que le paclitaxel ou le taxotère ; les antibiotiques antinéoplasiques tels que l ' actinomycine; les agents intercalants, les antimétabolites antinéoplasiques, les antagonistes des folates, le méthotrexate; les inhibiteurs de la synthèse des purines; les analogues de la purine tels que mercaptopurine, β-thioguanine; les inhibiteurs de la synthèse des pyrimidines, les inhibiteurs d'aromatase, la capécitabine, les analogues de la pyrimidine tels que fluorouracil, gemcitabine,
cytarabine et cytosine arabinoside; le bréquinar ; les inhibiteurs de topoisomérases tels que la camptothécine ou l'étoposide ; les agonistes et antagonistes hormonaux anticancéreux incluant le tamoxifène; les inhibiteurs de kinase, l' imatinib; les inhibiteurs de facteurs de croissance; les antiinflammatoires tels que le pentosane polysulfate, les corticostéroides, la prednisone, la dexamethasone; les antitopoisomérases tels que l'étoposide, les antracyclines incluant la doxorubicine, la bléomycine, la mitomycine et la méthramycine; les complexes métalliques anticancéreux, les complexes du platine, le cisplatine, le carboplatine, l ' oxaliplatine; l'interféron alpha, le triphénylthiophosphoramide, 1' altrétamine; les agents antiangiogéniques; la thalidomide; les adjuvants d'immunothérapie; les vaccins.
Selon la présente invention les composés de formule (I) peuvent également être administrés en association avec un ou plusieurs autres principes actifs utiles dans une des pathologies indiquées ci-dessus, par exemple un agent anti-émétiques, anti-douleurs, anti-inflammatoires, anticachexie .
La présente invention a ainsi pour objet à titre de médicaments, les produits de formule (I) telle que définie ci-dessus ainsi que les sels d'addition avec les acides minéraux et organiques pharmaceutiquement acceptables desdits produits de formule (I) .
La présente invention a notamment pour objet à titre de médicaments, les produits de formule (I) tels que définis ci-dessus répondant aux noms suivants : -{4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl}- [4- (methyl-oxazol-2-ylmethyl-amino) -piperidin-1- yl] -methanone - {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl}- [3- (methyl-lH-pyrrole-2-ylmethyl-amino) -
piperidin-1-yl] -methanone (Racémique)
- l-{ 4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] - benzoyl } -S-pyridin-S-ylmethyl-piperidine-S-carboxylic acid methylamide (Racémique) _ N*4*-(4-Fluoro-3-methyl phenyl) -N*2*- (4- { 3- [ (2- methanesulfonyl-ethylamino) -methyl] -pyrrolidine-1- sulfonyl} -phenyl) -pyrimidine-2, 4-diamine (Racémique)
- N*4*- (4-Fluoro-3-methyl-phenyl) -N*2*- [4- (3-{ [ (1- methyl-lH-pyrrol-2-ylmethyl) -amino] -methyl } -pyrrolidine- 1-sulfonyl) -phenyl] -pyrimidine-2, 4-diamine (Racémique)
- 4-pyrrolidin-l-ylmethyl-l-{ 4- [4- (4-fluoro-3-methyl- phenylamino) -pyrimidin-2-ylamino] benzenesulfonyl} - piperidin-4-ol
- { 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -phenyl} [4- (methyl-pyridin-2-ylmethyl-amino) - piperidin-1-yl] -methanone;
- { 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -phenyl}- [4- (methyl-pyridin-4-ylmethyl-amino) - piperidin-1-yl] -methanone; - {4- [ (l,5-Dimethyl-lH-pyrazol-4-ylmethyl) -methyl-amino] - piperidin-l-yl}-{4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl} -methanone
- {4-[ (2-Amino-pyridin-3-ylmethyl) -methyl-amino] - piperidin-1-yl } - { 4- [4- ( 4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone
- 4-{ [ (l-{4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin- 2-ylamino] -benzoyl }-piperidin-4-yl) -methyl-amino] - methyl }-l, 5-dimethyl-lH-pyrrole-2-carbonitrile
- {4- [ (2, 4-Dimethyl-thiazol-5-ylmethyl) -methyl-amino] - piperidin-l-yl}-{ 4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl} -methanone
- (l-{ [4- ( {4- [ (4-fluorophényl) amino] pyrimidin-2-
yl } amino) phényl] suifonyl }pipéridin-4-yl) (pyridin-3-yl) - méthanamine l-{ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - benzenesulfonyl } -4-pyrrolidin-l-ylmethyl-piperidin~4-ol - l-{4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -benzenesulfonyl} -4-pyrrolidin-l-ylmethyl- piperidin-4-ol ainsi que les sels d'addition avec les acides minéraux et organiques pharmaceutiquement acceptables desdits produits de formule (I) .
La présente invention a également pour objet les compositions pharmaceutiques contenant à titre de principe actif l'un au moins des produits de formule (I) tels que définis ci-dessus ou un sel pharmaceutiquement acceptable de ce produit ou un prodrug de ce produit et un support pharmaceutiquement acceptable.
La présente invention a également pour objet les compositions pharmaceutiques contenant à titre de principe actif l'un au moins des produits de formule (I) dont les noms sont donnés ci-dessus ou un sel pharmaceutiquement acceptable de ce produit ou un prodrug de ce produit et un support pharmaceutiquement acceptable .
La présente invention a particulièrement pour objet l'utilisation des produits de formule (I) tels que définis ci-dessus ou de sels pharmaceutiquement acceptables de ces produits pour la préparation d'un médicament destiné au traitement ou à la prévention d'une maladie par l'inhibition de l'activité de la protéine kinase IKK.
La présente invention a ainsi pour objet l'utilisation telle que définie ci-dessus dans laquelle la protéine kinase est dans un mammifère.
La présente invention a ainsi pour objet l'utilisation d'un produit de formule (I) tel que défini ci-dessus pour la préparation d'un médicament destiné au traitement ou à la prévention d'une maladie choisie parmi les maladies indiquées ci-dessus.
La présente invention a notamment pour objet l'utilisation d'un produit de formule (I) tel que défini ci-dessus pour la préparation d'un médicament destiné au traitement ou à la prévention d'une maladie choisie dans le groupe suivant : maladies inflammatoires, diabètes et cancers .
La présente invention a notamment pour objet l'utilisation 'd'un produit de formule (I) tel que défini ci-dessus pour la préparation d'un médicament destiné au traitement ou à la prévention de maladies inflammatoires.
La présente invention a notamment pour objet l'utilisation d'un produit de formule (I) tel que défini ci-dessus pour la préparation d'un médicament destiné au traitement ou à la prévention de diabètes . La présente invention a notamment pour objet l'utilisation d'un produit de formule (I) tel que défini ci-dessus pour la préparation d'un médicament destiné au traitement de cancers.
La présente invention a notamment pour objet l'utilisation d'un produit de formule (I) tel que défini ci-dessus destinée au traitement de tumeurs solides ou liquides .
La présente invention a notamment pour objet l'utilisation d'un produit de formule (I) tel que défini ci-dessus destinée au traitement de cancers résistant à des agents cytotoxiques .
La présente invention a notamment pour objet l'utilisation d'un produit de formule (I) tel que défini ci-dessus pour la préparation de médicaments destinés à la chimiothérapie de cancers. La présente invention a notamment pour objet l'utilisation d'un produit de formule (I) tel que défini ci-dessus pour la préparation de médicaments destinés à la chimiothérapie de cancers seul ou en en association ou sous forme de combinaison tel que défini ci-dessus. La présente invention a notamment pour objet l'utilisation d'un produit de formule (I) tel que défini ci-dessus comme inhibiteurs de IKK.
La présente invention concerne tout particulièrement les produits de formule (I) tels que définis ci-dessus qui constituent les exemples 1 à 109 de la présente invention.
Les exemples suivants illustrent l'invention sans toutefois la limiter.
Partie expérimentale: Procédure 1 : préparation des chlorhydrates de chlorure de sulfonyle.
Procédure la : Chlorhydrate de Chlorure de 4- [4- (4- Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyl
Stade 1: (2-Chloro-pyrimidin-4-yl) - (4-fluoro-phenyl) -aminé A un mélange contenant 15 g de Dichloropyrimidine dans 200 mL de n-butanol, sous agitation, on additionne 10 mL de 4-fluoroaniline puis 18 mL de di-isopropyl-ethylamine . Le mélange réactionnel est porté sous agitation, à reflux, pendant 2 heures. Le milieu réactionnel est refroidi, concentré à sec. Ajouter une solution de K2CO3 au résidu et extraire 3 fois avec de l'acétate d'éthyle, lavage avec une solution saturée de NaCl et séchage sur Na2SO4, le brut réactionnel est purifié par
chromatographie sur colonne de silice (DCM puis 30% d'acétate d' éthyle dans DCM) . Lors de la concentration 11 g de composé attendu cristallisent MH+ = 224), PF = 172-174 0C Stade 2 : N-4- (4-Fluoro-phenyl) -N-2-phenyl-pyrimidine- 2, 4-diamine
10,5 g de (2-Chloro-pyrimidin-4-yl) - (4-fluoro- phenyl) -aminé en solution dans 300 mL de n-butanol sont portés à 140 °C à reflux en présence de 4,3 mL d'aniline toute la nuit. Le milieu réactionnel est refroidi. La suspension obtenue est filtrée. Les cristaux sont repris dans l'acétate d' éthyle et lavés par une solution de 10% de K2CO3 puis par une solution saturée de NaCl. Après séchage sur Na2SO4, la phase organique est concentrée sous vide. Le brut réactionnel est purifié par chromatographie sur colonne de silice (THF/MeOH/DCM, 10/5/85) . La N-4- (4-Fluoro-phenyl) -N-2-phenyl-pyrimidine- 2,4-diamine attendue cristallise lors de la concentration et 10.5 g du produit sont obtenus par filtration. MH+ = 281, PF = 161 0C
Stade 3: Chlorhydrate de Chlorure de 4- [4- (4-Fluoro- phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyle
Dans un ballon tricol sous courant d'azote contenant l'acide chlorosulfonique à 0 0C, on additionne par petites portions 7.5 g de N-4- (4-Fluoro-phenyl) -N-2- phenyl-pyrimidine-2, 4-diamine en maintenant la température autour de 0 °C. Le milieu réactionnel est laissé à température ambiante pendant 18 h. Le mélange est versé goutte à goutte (avec précaution) sur la glace. Le précipité obtenu est filtré et lavé avec de l'eau distillée. Après dissolution du solide dans 1 L d'acétate d' éthyle, séchage sur Na2S04 et concentration sous vide, on obtient une huile blanchâtre. Cette huile précipite après dispersion dans 200 mL l'éther. 10,5 g de Chlorhydrate de Chlorure de 4- [4- (4-Fluoro-phenylamino) -
pyrimidin-2-ylamino] -benzenesulfonyle sont obtenus par filtration de la suspension éthérée. MH+ = 360, PF mal défini .
Procédure Ib : Chlorhydrate de Chlorure de 4- [4- (3,4- difluoro-phenylamino) -pyrimidin-2-ylamino] - benzenesulfonyle
Stade 1 : 4-Chloro-N- (3, 4-difluorophenyl) pyrimidin-2-amine La préparation de ce composé se fait suivant le même procédé que pour la procédure la à partir de la réaction de 9.21g de dichloropyrimidine avec 8 g de 3,4- difluoroaniline : on obtient ainsi 10.3g de produit attendu.
Stade 2 : N4- (3, 4-Difluoro-phenyl) -N2~phenyl-pyrimidine- 2, 4-diamine La préparation de ce composé se fait suivant le même procédé que pour l'exemple 1 à partir de la réaction de 7g de (2-Chloro-pyrimidin-4-yl) - (3, 4-difluoro-phenyl) - aminé obtenu au stade 1 ci-dessus avec 2.72 g d'aniline : on obtient ainsi 8 g de produit attendu. Stade 3 : Chlorhydrate de Chlorure de 4- [4- (3, 4-difluoro- phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyle La préparation de ce composé se fait suivant le même procédé que pour l'exemple 1 à partir de la réaction de 8g de N-4- (3, 4-Difluoro-phenyl) -N-2-phenyl-pyrimidine- 2,4-diamine obtenu au stade 2 ci-dessus avec l'acide chlorosulfonique : on obtient ainsi 9 g de produit attendu.
Procédure Ic : Chlorhydrate de chlorure de 4- [4- (4- Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] benzenesulfonyle
Stade 1 : (2-Chloro-pyrimidin-4-yl) - (4-fluoro-3-methyl- phenyl) -aminé
La préparation de ce composé se fait suivant le même procédé que pour l'exemple 1 à partir de la réaction de 5.3 g de 4-Fluoro-3-methyl-phenylamine avec 6.3 g de 2,4-
Dichloro-pyrimidine: On obtient 3.8 g de produit attendu (Point de fusion= 130-131 0C) ( Trituration dans l ' éther isopropylique) .
Stade 2 : N-4- (4-Fluoro-3-methyl-phenyl) -N-2-phenyl- pyrimidine-2, 4-diamine
La préparation de ce composé se fait suivant le même procédé que pour l'exemple 1 à partir de la réaction de 2.8 g de (2-Chloro-pyrimidin-4-yl) - (4-fluoro-3-methyl- phenyl) -aminé obtenu ci-dessus et de 1.2 mL d'aniline: On obtient 2.2 g de produit attendu (Point de fusion= 134-135 0C) ( Trituration dans l ' éther isopropylique) .
Stade 3 : Chlorhydrate de chlorure de 4- [4- (4-Fluoro-3- methyl-phenylamino) -pyrimidin-2-ylamino] benzenesulfonyl .
La préparation de ce composé se fait suivant le même procédé que pour l'exemple 1 à partir de la réaction 2g de N-4- (4-Fluoro-3-methyl-phenyl) -N-2-phenyl-pyrimidine- 2,4-diamine obtenu ci-dessus avec l'acide chlorosulfonique : On obtient 1.5 g de produit attendu.
Procédure 2 : préparation des dérivés pyrimidine-2- (4- amino-benzoïque acide) .
Procédure 2a : 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -benzoic acid
Stade 1 : 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -benzoic acid methyl ester Un mélange contenant 16 g de chloropyrimidine obtenu au stade 1 de la procédure la et 10.8 g de d'amino-4- benzoate de méthyle dans le n-butanol, est chauffé à 140 0C pendant une nuit. Après refroidissement, on filtre le précipité. On lave ce précipité avec de Et2O et on le recristallise dans une mélange DCM-Me0H-iPr20. On obtient ainsi 23.5 g de produit attendu. Stade 2 : 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-
ylamino] -benzoic acid
15 g de produit obtenu à stade 1, en présence de 4.5 g de soude dans un mélange de MeOH(IOO mL) , eau(100 mL) et dioxane(400 mL) sont portés à une température de 40 °C pendant une nuit. Le milieu réactionnel est concentré à sec et repris dans 100 mL d'eau. On extrait les impuretés par deux volumes de Et2O, puis on acidifie la phase acqueuse à pH 6 avec HCl I N. Le précipité formé est filtré, rincé par l'eau distillée et mis en suspension dans du DCM et le solvant est évaporé. On obtient 15 g d'acide attendu.
Procédure 2b : 4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzoic acid
Stade 1 : 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin- 2-ylamino] -benzoic acidmethyl ester
Un mélange contenant 8 g de chloropyrimidine obtenu au stade 1 de la procédure Ic et 5.1 g de d' amino-4-benzoate de méthyle, est chauffé à 140 0C pendant une nuit. Après refroidissement, on filtre le précipité. On lave ce précipité avec de Et2O et on le recristallise dans une mélange DCM-MeOH-iPr2θ. On obtient ainsi 10.5 g de produit attendu.
Stade 2 : 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin- 2-ylamino] -benzoic acid 2.08 g de produit obtenu à stade 1, en présence de 410 mg de soude dans un mélange de MeOH (5 mL) , eau (5 mL) et dioxane(20 mL) sont portés à une température de 40 0C pendant une nuit. Le milieu réactionnel est concentré à sec et repris dans 100 mL d'eau. On extrait les impuretés par deux volumes de Et2O, puis on acidifie la phase acqueuse à pH 6 avec HCl 1 N. Le précipité formé est filtré, rincé par l'eau distillée et mis en suspension
dans du DCM et le solvant est évaporé. On obtient 1.3 g d ' acide attendu .
Procédure 2c : 4- [4- (4-Trifluoromethyl-phenylamino) - pyrimidin-2-ylamino] -benzoic acid.
Stade 1 : (2-Chloro-pyrimidin-4-yl) - (4-triflu oromethyl-phenyl) -aminé
De la même manière qu'à l'exemple 1 de la procédure 2h, à partir de 15 g de dichloropyrimidine dans 200 mL de n- butanol, sous agitation, on additionne 16 g de 4- trifluoromethyl-phenylamine puis 18 mL de di-isopropyl- ethylamine. Le mélange réactionnel est porté sous agitation, à reflux, toute la nuit. Le milieu réactionnel est refroidi, concentré à sec. Ajouter une solution de K2CO3 au résidu et extraire 3 fois avec de l'acétate d'éthyle, lavage avec une solution saturée de NaCl et séchage sur Na2SO4, le brut réactionnel est purifié par chromatographie sur colonne de silice (DCM puis 2% de MeOH dans DCM) . On obtient 5 g de produit attendu.
Stade 2 : 4- [4- (4-Trifluoromethyl-phenylamino) -pyrimidin- 2-ylamino] -benzoic acid methyl ester
De même qu'au stade 2 de la procédure 1, à partir de 4.6 g Un mélange contenant 8 g de chloropyrimidine obtenu au stade 1 et de 2.6 g de d' amino-4-benzoate de méthyle, On obtient ainsi 6.4 g de produit attendu. Stade 3 : 4- [4- (4-Trifluoromethyl-phenylamino) -pyrirαidin- 2-ylamino] -benzoic acid.
De même qu'au stade 3 de la procédure 1, à partir de 6.4 g Un mélange contenant 8 g d'ester obtenu au stade 2 et de 2.26 g de de soude, On obtient ainsi 4.2 g de produit attendu.
Procédure 3 : préparation desintermédiaires réactionnels de type sulfonamide
Procédure 3a : N-4- (4-Fluoro-3-methyl-phenyl) -N-2- [4- (4- methylamino-piperidine-1-sulfonyl) -phenyl] -pyrimidine- 2, 4-diamine
Stade 1: (l-{ 4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl } -piperidin-4-yl) - methyl-carbamic acid tert-butyl ester
3 g de chlorhydrate chlorure de sulfonyle obtenu dans la procédure Ic sont traités par 1.7 g de la 4-N-Boc- méthylpipéridine dans 50 mL de DCM en présence de 2.3 ml de DIPEA pendant toute la nuit à température ambiante.
Après traitement habituel, on chromatographie sur colonne de SiO2 et élue par DCM/MeOH (97/3; v/v) . On obtient 2.75 g de produit attendu. Stade 2: N*4*- (4-Fluoro-3-methyl-phenyl) -N*2*- [4- (4- methylamino-piperidine-1-sulfonyl) -phenyl] -pyrimidine- 2, 4-diamine
Le composé obtenu au stade 1 est dissout MeOH puis traité par 35 mL Et2O/HCl 2 N pendant toute la nuit. On filtre le chlorhydrate, le redissout dans l'eau, basifie par K2CO3 solide et extrait par AcOEt. Après lavages par l'eau et séchage sur Na2So4 de la phase organique, on obtient 2.25 g d'une poudre par évaporation du solvant. MH+ = 471.3 PF = 148-150 °C RMN (IH, DMSO)
1.21-1.36 (massif, 2); 1.61 (m, 1); 1.71-1.85 (massif, 2); 2.16 (s, 3); 2.24 (d, 3); 2.27 (m, 1); 2.45 (m, 2); 3.36 (m, 2); 6.29 (d, 1); 7.12 (t, 1); 7.47 (m, 1); 7.58 (d, 2); 7.59 (m, 1) ; 7.97 (d, 2); 8.08 (d, 1); 9.43 (s, 1) ; 9.72 (s, 1) .
Procédure 3b: N-2- [4- (3-Aminomethyl-piperidine-l- sulfonyl) -phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2, 4-
diamine (Racémique)
Stade 1 : l-{4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -benzenesulfonyl } -piperidin-3-ylmethyl) carbamicacidtert-butyl ester (RacémiqueJ Suivant le mode opératoire décrit au au stade 1 de la procédure 3a, à partir de 4 g de de chlorhydrate chlorure de sulfonyle obtenu dans la procédure la et de 3.43 g de l'aminé commerciale racémique piperidin-3-ylmethyl- carbamic acid tert-butyl ester, on obtient 2.7 g de produit attendu. MH+ = 557.1
Point de fusion = 110 0C
Stade 2 : N-2- [4- (3-Aminomethyl-piperidine-l-sulfonyl) - phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2, 4- diamine (Racémique)
Suivant la réaction de décarboxylation décrite au stade 2 de la procédure 3a, à partir de 2.7 g de produit obtenu au stade 1, on obtient 2.3 g de produit attendu. MH+ = 457.1 Point de fusion = 207 0C IH RMN (DMSO) :
0.69 (m,l) ; 1.13-1.92 (massif, 7) ; 2.10 (t,l) ; 2.19- 2.43 (2m, 2) ; 3.38-3.68 (2d,2) ; 6.25 (d,l) ; 7.13 (t,2) ; 7.54 (d,2) ; 7.66 (m, 2) ; 7.93 (d,2) ; 8.10 (d, 1) ; 9.47 (s,l) ; 9.67 (s,l) .
Procédure 3c : N-2- [4- (3-S-Amino-pyrrolidine-l-sulfonyl) - phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2, 4-diamine Stade 1 : (l-{ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -benzenesulfonyl} -pyrrolidin-3-S-yl) -carbamic acid tert-butyl ester
Suivant le mode opératoire décrit au au stade 1 de la procédure 3a, à partir de 400 mg de de chlorhydrate
chlorure de sulfonyle obtenu dans la procédure la et de 198 mg de l'aminé commerciale pyrrolidin-3-S-yl-carbamic acid tert-butyl ester, on obtient 341 mg de produit attendu. MH+ = 529.2
Point de fusion = 178.2 0C
Stade 2 : N-2- [4- (3-S-Amino-pyrrolidine-lsulfonyl) - phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2, 4-diamine Suivant une réaction de décarboxylation à partir de 200 mg du composé obtenu au stade 1 dans 2.4 mL d'un mélange DCM-TFA (v/v, 1/1), on obtient 163 mg de produit attendu sous forme de sel de TFA. MH+ = 429.0 Point de fusion = 250 0C
Procédure 3d : N-2- [4- (3-R-Amino-pyrrolidine-l-sulfonyl) - phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2, 4-diamine Stade 1 : (l-{ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -benzenesulfonyl}-pyrrolidin-3-R-yl) -carbamic acid tert-butyl ester Suivant le mode opératoire décrit au au stade 1 de la procédure 3a, à partir de 400 mg de de chlorhydrate chlorure de sulfonyle obtenu dans la procédure la et de 198 mg de l'aminé commerciale pyrrolidin-3-S-yl-carbamic acid tert-butyl ester, on obtient 379 mg de produit attendu.
MH+ = 529.2
Stade 2 : N-2- [4- (3-R-Amino-pyrrolidine-lsulfonyl) - phenyl] -N-4- (4-fluoro-phenyl) -pyrimidine-2, 4-diamine Suivant la réaction de décarboxylation décrite au stade 2 de la procédure 3c, à partir de 300 mg de produit obtenu au stade 1, on obtient 410 mg de produit attendu.
MH+ = 429 . 0
Point de fusion = 250 0C
Procédure 3e : 4-Aminomethyl-l-{ 4- [4- (4-fluoro-3-methyl- phenylamino) -pyrimidin-2-ylamino] benzenesulfonyl } - piperidin-4-ol
Stade 1 : 6-Benzyl-l-oxa-6-aza-spiro [2.5] octane A une solution contenant 10 g de N-benzyl-4-piperidone, 12.8 g de dimethyloxosulfonium methylide et 0.34 g de bromure de tetrabutylairanonium dans 100 mL de toluène , on additionne goutte à goutte une solution de 3.2 g de soude dans 32 mL d'eau et on laisse le milieu réactionnel sous agitation à 80 0C pendant 3 heures. Après refroidissement, on lave à l'eau, sèche sur Na2SO4 et concentre à sec. On obtient ainsi 11.7 g d'epoxyde. Stade 2 : 4-Aminomethyl-l-benzyl-piperidin-4-ol
6 g d' epoxyde obtenu au stade 1 sont mis dans une solution de MeOH saturée en ammoniac. Et chauffé pendant 72 heures en tube scellé. On concentre sous vide et on purifie sur colonne d' alumine (gradient DCM-MeOH : v/v ; 9/1) . On obtient 5.3 g d' amino-alcool .
Stade 3 : (l-Benzyl-4-hydroxy-piperidin-4-ylmethyl) - carbamic acid tert-butyl ester
On traite 5.3 g l' amino-alcool obtenu au stade 2 en solution dans le DCM avec 5.2 g de BOC2O dissous dans DCM et on agite 15 minutes à TA. On concentre sous vide et on purifie par chromatographie sur alumine (gradient DCM- MeOH : v/v ; 98/2) . On obtient ainsi 5.2 g d' amino-alcool substitué attendu. Stade 4 : (4-Hydroxy-piperidin-4-ylmethyl) -carbamic acid tert-butyl ester
Suivant une réaction d' hydrohgenolyse, à partir de 5.1 g
d' amino-alcool au stade 2 en présence de 510 mg Pd/C 10 % dans 200 mL de methanol, on obtient 2.8 g de piperidine attendu après traitement habituel.
Stade 5 : 4-Aminomethyl-l-{ 4- [4- (4-fluoro-3-methyl- phenylamino) -pyrimidin-2-ylamino] benzenesulfonyl}- piperidin-4-ol
Suivant le mode opératoire décrit au stade 1 de la procédure 3a, à partir de 700 mg de chlorhydrate de chlorure de sulfonyl obtenu dans la procédure Ic et de 420 mg de piperidine obtenu au stade 4, on obtient 540 mg d'un composé qui subit une réaction de décarboxylation pour donner 150 mg de sulfonamide attendu. MH+ = 487.1 Point de fusion = 199 0C IH RMN (DMSO) : .
1.47-1.77 (massif, 4) ; 2.25 (s, 3) ; 2.50 (m, 2) ; 2.75 (m, 2) ; 3.42 (m, 2) ; 4.99 (m,l) ; 6.57 (d,l) ; 7.20 (t,l) ; 7.40 (m,l) ; 7.60 (m,l) ; 7.69 (d,2) ; 7.83 (d,2) ; 7.94 (Is, 3) ; 8.09 (d,l) ; 10.92-11.23 (21s, 2) Procédure 3f : N-2- [4- (3-Aminomethyl-pirrolidine-l- sulfonyl) -phenyl] -N-4- (4-Fluoro-3-methyl-phenyl) - pyrimidine-2, 4-diamine (Racémique)
Suivant le mode opératoire décrit au stade 1 de la procédure 3a, à partir de 2 g de chlorhydrate de chlorure de sulfonyle obtenu dans la procédure Ic et de 1.38 g de l'aminé racémique pyrrolidine-3-ylmethyl-carbamic acid benzyl obtenu au stade 4, on obtient 1.8 g d'un composé qui subit une réaction de d' hydrogénolyse pour donner 1.3 g de sulfonamide attendu.
Procédure 3g : N-2- [4- (3-Aminomethyl-pipéridine-l- sulfonyl) -phenyl] -N-4- (4-Fluoro-phenyl) -pyrimidine-2, 4-
diamine (Racémique)
Suivant le mode opératoire décrit au stade 1 de la procédure 3a, à partir de 3.5 g de chlorhydrate de chlorure de sulfonyle obtenu dans la procédure la et de 2 g de 3-N-boc -3-méthylaminopipéridine, on obtient 2.65 g d'un composé qui subit une réaction de décarboxylation pour donner 1.9 g de sulfonamide attendu.
MH+ = 457.2
PF = 217-218 0C Procédure 3h : N-2- [4- (4-Amino-piperidine-l-sulfonyl) - phenyl] -N-4- (3, 4-Difluoro-phenyl) -pyrimidine-2, 4- diamine (Racémique)
Suivant le mode opératoire décrit au stade 1 de la procédure 3a, à partir de 5 g de chlorhydrate de chlorure de suifonyl obtenu dans la procédure Ib et de 2.41 g de
4-N-boc -4-aminopipéridine, on obtient 2.9 g d'un composé qui subit une réaction de décarboxylation pour donner 2.9 g de sulfonamide attendu.
MH+ = 443.2
Procédure 4 : préparation des intermédiaires réactionnels de type carboxamides .
Procédure 4a : { 4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl}- (4-methylamino-piperidin-l- yl) -methanone Stade 1 : (l-{ 4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzoyl}-piperidin-4-yl) -methyl- carbamic acid tert-butyl ester.
On fait réagir à TA, toute la nuit, un mélange contenant
3 g de l'acide obtenu à la procédure 2b, 1.9 g de la 4-N- boc -4-méthylaminopipéridine en présence de 3.9 g de BOP, et de 4.5 mL de DIPEA dans 30 mL de CH2C12. On évapore à
sec, on ajoute une solution 10 % de carbonate de potassium et on extrait par l'acétate d'éthyle. Après lavage par de l'eau et séchage sur Na2SO4 de la phase organique, on filtre puis chromatographie sur une colonne de silice utilisant comme éluant du DCM/MeOH (99/1; v/v) . On obtient 3.85 g de produit attendu. Stade 2 ; { 4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } - ( 4-methylamino-piperidin-l- yl) -methanone Le produit obtenu à stade 1 est dissout dans 40 mL de
MeOH. On ajoute , à température ambiante, 40 mL de Et20 2 N et on laisse sous agitation pendant 6 heures. Après évaporation à sec, on triture le résidu dans Et2O et la filtration de la suspension génère 3.3 g de chlorhydrate de produit attendu. Le chlorhydrate est dissout dans l'eau, on le basifie par le carbonate de potassium solide. L'extraction de cette phase acqueuse se fait avec de l'acétate d'éthyle contenant un peu de THF. Après lavage et séchage, de la phase organique sur Na2SO4, on évapore à sec et on recristallise dans un mélange DCM- iPr2O pour obtenir 2.25 g de produit attendu. MH+ = 435.2 PF ≈ 195-199 0C RMN (IH, DMSO) 1.18 (m, 2); 1.80 (dl, 2); 2.23 (d, 3); 2.27 (s, 3); 2.54 (m, 1); 3.02 (t, 2); 3.66-4.34 (si, 2); 6.22 (d, 1); 7.09 (t, 1); 7.27 (d, 2); 7.47 (m, 1); 7.60 (dd, 1); 7.79 (d, 2); 8.04 (d, 1); 9.35 (s, 1); 9.40 (s, 1) .
Procédure 4b ; { 4- [ 4- ( 4-Fluoro phenylamino) pyrimidin-2-ylamino] -phenyl } - ( 3-methylamino- piperidin-1-yl ) -methanone (Racémique )
Stade 1: {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylarαino] -phenyl}- (3-methylamino-piperidin-l-yl) -methanone
(Racémique)
Suivant le mode opératoire décrit au stade 1 de la procédure 4a, à partir de 3.35 g d'acide obtenu dans la procédure 2a et de 2 g de 3-N-boc-3- méthylaminopiperidine, on obtient 3.7 g de composé attendu.
Stade 2 : { 4- [4- (4-Fluorophenylamino) pyrimidin-2- ylamino] -phenyl}- (3-methylamino-piperidin-l-yl) -methanone
(Racémique)
Suivant la réaction de décarboxylation décrite au stade 2 de la procédure 4a, 3.7 g de composé obtenu au stade 1 permettent d'obtenir 2.8 g de carboxamide attendu. MH+ = 421.1
Point de fusion = 110 0C
IH RMN (DMSO) :
1.18-2.18 (massif, 5) ; 2.28 (s, 3) ; 2.41 (m,l) ; 2.83
(m,l) ; 3.08 (m,l) ; 3.68-4.17 (2m, 2) ; 6.24 (d,l) ; 7.14 (t,2) ; 7.27 (d,2) ; 7.68 (m, 2) ; 7.77 (d,2) ; 8.04 (d,l)
; 9.17 (s,l) ; 9.24 (s,l) .
Procédure 4c : { 4~ [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl}- (4-methylamino-piperidin-l-yl) -methanone
Stade 1 : (l-{ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -benzoyl} -piperidin-4-yl) -methyl-carbamic acid tert-butyl ester
Suivant le mode opératoire décrit au stade 1 de la procédure 4a, à partir de 3.95 g d'acide obtenu dans la procédure 2a et de 2.35 g de 3-N-boc-3- méthylaminopiperidine, on obtient 4.3 g de composé attendu. MH+ = 521.3
Stade 2 ; { 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-
ylamino] -phenyl}- (4-methylamino-piperidin-l-yl) -methanone Suivant la réaction de décarboxylation décrite au stade 2 de la procédure 4a, 4.3 g de composé obtenu au stade 1 permettent d'obtenir 2.1 g de carboxamide attendu. Procédure 4d : (4-Methylamino-piperidin-l-yl) -{ 4- [4- (4- trifluoromethyl-phenylamino) -p yrimidin-2-ylamino] -phenyl } -methanone Stade 1: Methyl- (l-{4- [4- (4-trifluoromethyl- phenylamino) -pyrimidin-2-ylamino] -benzoyl} -piperidin-4- yl)-carbamic acid tert-butyl ester
Suivant le mode opératoire décrit au stade 1 de la procédure 4a, à partir de 1.5 g d'acide obtenu dans la procédure 2c et de 1.7 g de 4-N-boc-4- méthylaminopiperidine, on obtient 1.75 g de composé attendu.
Stade 2 : (4-Methylamino-piperidin-l-yl) - { 4- [4- (4- trifluoromethyl-phenylamino) -pyrimidin-2-ylamino] - phenyl } -methanone Suivant la réaction de décarboxylation décrite au stade 2 de la procédure 4a, 1.75 g de composé obtenu au stade 1 permettent d'obtenir 248 mg de carboxamide attendu. MH+ = 470.9 Point de fusion = 225-226 0C
Exemple 1 : {4- [4- (4-Fluoro-3-methyl-phenylamino) - pγrimidin-2-ylamino] -phenyl } - [4- (methyl-
[1,2,3] thiadiazol-4-γlmethyl-amino) -piperidin-1-yl] - methanone

Dans le THF(IO mL) , on mélange 370 mg d'acide de la
procédure 4a, 95 mg de 1, 2, 3-thiadiazole-4-carboxaldéhyde et on ajoute 310 mg de NaBH (OAc) 3. On agite à température ambiante pendant une nuit, ajoute le CH3OH(5 inL) et chauffe pendant une heure à 60 °C. Après évaporation des solvants, on ajoute de l'eau, basifie par quelques gouttes de soude et extrait par CH2CL2 suivi d'un traitement habituel et d'une chromâtographie sur gel de silice. On élue par CH2C12/CH3OH (98/2; v/v) . On recristallise dans CH2C12-i (Pr) 20 pour obtenir 225 mg MH+ = 533.2
PF = 183-184 0C
RMN (IH, DMSO)
1.49 (qd, 2); 1.85 (dl, 2); 2.24 (s, 3); 2.24 (s, 3);
2.67 (t, 1); 2.77-3.09 (massif, 2); 3.67-4.77 (massif, 2); 4.16 (s, 2); 6.22 (d, 1); 7.09 (t, 1); 7.30 (d, 2);
7.46 (m, 1); 7.59 (dd, 1); 7.78 (d, 2); 8.03 (d, 1); 9.02 (s, 1); 9.34 (s, 1); 9.38 (s, 1)
Exemple 2 : {4- [4- (4-Fluoro phenylamino) -pyrimidin-2- ylamino] -phenyl}-{4- [methyl- (lH-pyrazol-4-ylmethyl) - amino] -piperidin-1-yl}methanone

De la même façon qu'à l'exemple 1, à partir de 400 mg de l'acide de la procédure 4c et de lH-pyrazole-4- carboxaldéhyde (100 mg) . On obtient le produit attendu. MH+ = 501.5
PF = 140-145 0C
RMN (IH, DMSO)
1.44 (m, 2) ; 1.78 (m, 2) ; 2.14 (s, 3) ; 2.59 (m,l) ; 2.86
(Im, 2) ; 3.52( Is, 2) ; 3.65-4.71 (massif ,2) ; 6.23(d,l) ,
7.16 (t,2) ; 7.28 (d,2) ; 7.33-7.63 (massif, 2) ; 7.7 (m, 2) ; 7.81 (d,2) ; 8.04 (m, l) ; 9.39 (2s, 2) ; 12.64 (Is, 1)
Exemple 3 : {4- [4- (4-Fluorophenylamino) -pyrimidin-2- ylamino] -phenyl } - { 4- [methyl- (lH-pyrazol-3-ylmethyl) - amino] -piperidin-l-yl}-methanone

De la même façon qu'à l'exemple 1, à partir de 400 mg de l'acide de la procédure 4c et de 100 mg de lH-pyrazole-3- carboxaldehyde . On obtient le produit attendu.
MH+ = 501.3
PF ≈ 165-170 0C
RMN (IH, DMSO)
1.44 (m, 2) ; 1.78 (m,2) ; 2.14 (s, 3) ; 2.59 (m,l) ; 2.86 (Im, 2) ; 3.91( Is, 2) ; 3.65-4.71 (massif ,2) ; 6.12 (s,l)
; 6.23 (d,l) ; 7.16 (t,2) ; 7.28 (d,2) ; 7.33-7.63 (massif, 1) ; 7.7 (m, 2) ; 7.81 (d,2) ; 8.04 (m, 1) ; 9.39 (2s, 2) ; 12.64 (Is, 1)
Exemple 4 : {4- [4- (4-Fluoro phenylamino) -pyrimidin-2- ylamino] -phenyl } - { 4- [methyl- ( lH-pyrazol-4-ylmethyl) -amino] -piperidin-l-yl}methanone

De la même façon qu'à l'exemple 1, à partir de 400 mg de l'acide de la procédure 4c et de 100 mg de 3-méthyl-lH- pyrazole-5-carboxaldehyde. On obtient le produit attendu.
MH+ = 515.4 PF = 214-215 °C RMN (IH, DMSO)
1.47 (m, 2) ; 1.82 (m, 2) ; 2.19 (s, 6) ; 2.63 (m,l) ; 2.91 (t,2) ; 3.56( Is, 2) ; 4.11(lm ,2) ; 5.9(s,l) ; 6.24(d,l) ; 7.14 (t,2) ; 7.28 (d,2) ; 7.67 (m, 2) ; 7.7 (m, 2) ; 8.04 (d,l) ; 9.12(sfl) ; 9.20 (2s, 1) ; 11.97 (Is, 1)
Exemple 5 : [4- (Benzo [1,2 ,5] thiadiazol-5-ylmethyl-methyl- amino) -piperidin-1-yl] -{4- [4- (4-fluoro-phenylamino) - pyrimidin-2-ylamino] -phenyl} -methanone

De la même façon qu'à l'exemple 1, à partir de 420 mg de l'acide de la procédure 4c et de 164 mg 2,1,3- benzothiadiazole-5-carbaldehyde . On obtient 242 mg de produit attendu. MH+ = 569.1
Point de fusion = 191-192 0C
Exemple 6 : {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl}- [4- (methyl-oxazol-2-ylmethyl-amino) - piperidin-1-y1] -methanone

De la même façon qu'à l'exemple 1, à partir de 420 mg de l'acide de la procédure 4c et de 110 mg d'oxazole-2- carbaldehyde, on obtient 380 mg de produit attendu. MH+ = 501.9
Point de fusion = 175-176 0C
IH RMN (DMSO) : .
1.39 (m, 2) ; 1.79 (dl, 2) ; 2.23 (s, 3) ; 2.62 (m, 1) ;
2.88 (sl, 2) ; 3.77 (s, 2) ; 4.02 (sl, 2) ; 6.22 (d, 1) ;
7.05-7.23 (massif, 3) ; 7.23 (d, 2) ; 7.71 (m, 2) ; 7.78 (d, 2) ; 8.00-8.11 (massif, 2) ; 9.40 (S, 1) ; 9.43 (s, 1) .
Exemple 7: {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl}- [3- (methyl-oxazol-2-ylmethyl-amino) - piperidin-1-yl] -methanone (Racémique)

1.41 (m,l) ; 1.54 (m,l) ; 1.72 (m,l) ; 1.93 (m,l) ; 2.25 (Is, 3) : 2.51 (m,l) ; 2.92 (Is, 2) ; 3.49-4.75 (massif, 4) ; 6.24 (d,l) ; 7.16 (massif, 2) ; 7.27 (d,2) ; 7.71 (m,2) ; 7.79 (d,2) ; 8.04 (massif, 3) ; 9.38 (s,l) ; 9.42 (s,l)
Exemple 8 : {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl}- [3- (methyl-lH-pyrrole-2-ylmethyl-amino) • piperidin-1-yl] -methanone (Racémique)

De la même façon qu'à l'exemple 1, à partir de 400 mg de l'acide de la procédure 4b et de 100 mg de 1-methyl-lH-
pyrrole-2-carbaldehyde, on obtient 102 mg de produit attendu. MH+ = 513.9
Point de fusion = 148-151 0C IH RMN (DMSO) :
1.40 (m, 1) ; 1.60 (m, 1) ; 1.74 (m, 1) ; 1.91 (m,l) ; 2.10
(Is, 3) ; 2.54 (m,l) ; 2.59-3.14 (massif, 2) ; 3.23 (s, 3) ; 3.38-4.76 (massif, 4) ; 5.86 (Is, 2) ; 6.23 (d,l) ; 6.63
(s,l) ; 7.16 (t,2) ; 7.23 (m, 2) ; 7.72 (m, 2) ; 7.78 (d,2) ; 8.06 (d,l) ; 9.37 (s,l) ; 9.42 (s,l)
Exemple 9 : { 4- [4- (4-Fluoro-phenγlamino) -pγrimidin-2- ylamino] -phenyl } - [3- (aminothiazol-2-ylinethyl-amino) - piperidin-1-yl] -methanone (Racémique)

MH+ = 518.0 Point de fusion = 160-165 0C
IH RMN (DMSO) :
1.18-1.99 (massif, 4) ; 2.18(sl, 3) ; 2.54 (m, 1) ;
2.94(sl, 2) ; 3.86(sl, 2) ; 4.17(sl, 2) ; 6.23(d, 1) ;
7.17(t, 2) ; 7.27(d, 2) ; 7.61-7.89 (massif, 5) ; 8.06(d, 1) ; 9.00(s, 1) ; 9.41(s, 1) ; 9.45(s, 1).
Exemple 10 : {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl}-{3- [methyl- (lH-pyrazol-3-ylmethyl) - amino] -piperidin-1-yl} -methanone (Racémique)

De la même façon qu'à l'exemple 1, à partir de 340 mg de l'acide de la procédure 4b et de 120 mg de lH-pyrazole-3- carbaldehyde, on obtient 267 mg de produit attendu MH+ = 501.0
Point de fusion = 120-140 0C IH RMN (DMSO) :
1.39(m, 1) ; 1.53 (m, 1) ; 1.73(sl, 1) ; 1.95(dl,l) ; 2.16(sl, 3) ; 2.48 (m, 1) ; 2.90(Sl, 2) ; 3.61(sl, 2) ; 4.32(sl, 2) ; 6.09(sl, 1) ; 6.23(d, 1) ; 7.16(t, 2) ; 7.26(d, 2) ; 7.51 (si, 1) ; 7.71 (m, 2) ; 7.79(d, 2) ; 8.05(d, 1) ; 9.38(s, 1) ; 9.41(s, 1) .
Exemple 11: N*4*- (4-Fluoro-3-methyl-phenyl) -N*2*- (4-{4- [ (2-methanesulfonyl-ethyl) -methyl-amino] -piperidine-1- suifonyl } -phenyl) -pyrimidine-2 , 4-diamine

A une suspension de 300 mg du composé obtenu à la procédure 3a dans 18 mL d'un mélange MeOH/DMF(v/v ; 5/1), on ajoute 250 mg de TEA puis 100 mg de méthylvinylsulfone . Après agitation à TA pendant 18 heures, concentration à sec, reprise dans DCM, lavage par H2O et séchage, on concentrate à nouveau à sec. On chromatographie (Si02) et élue par DCM/MeOH (v/v ; 94/6) et recristallise dans iPr20 et on obtient 220 mg de produit attendu
MH+ = 577.1
PF = 115 0C
RMN (IH, DMSO)
1.47 (m, 2) ; 1.69 (m, 2) ; 2.12 (s, 3) ; 2.24 (massif, 5) ;
2.33 (m, 1) ; 2.76 (t,2) ; 2.93 (s, 3) ; 3.17 (t,2) ; 3.60
(Id, 2) ; 6.27 (d,l) ; 7.10 (t,l) ; 7.47 (m,l) ; 7.57 (m, 3)
; 7.97 (d,2) ; 8.07 (d,l) ; 9.41 (s,l) ; 9.69 (s,l)
Exemple 12 : 2- [ (l-{4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl}-piperidin-4-yl) - methyl-amino] -N,N-dimethyl-acetamide

On agite à température ambiante pendant une nuit dans
CH3CN(10 mL) , un mélange de 400 mg du composé obtenu à la procédure 3a, de 120 mg de 2-chloro N,N-diméthyl acétamide, de 160 mg de KI et de 260 mg de K2CO3. Après extraction de manière habituelle, le résidu est chromatographié sur silice , on élue par CH2Cl2/CH3OH(94/6; v/v) . On recristallise dans CH2C12- i (Pr) 20 pour obtenir 126 mg. MH+ = 556.2 PF = 215-218 0C RMN (IH, DMSO)
1.45 (qd, 2); 1.71 (dd, 2); 2.12 (s, 3); 2.18 (m, 2); 2.24 (s, 3); 2.38 (m, 1); 2.74 (s, 3); 2.92 (s, 3); 3.15 (s, 2); 3.59 (d, 2); 6.29 (d, 1); 7.11 (t, 1); 7.47 (m, 1); 7.58 (d, 2); 7.58 (m, 1); 7.98 (d, 2); 8.08 (d, 1) ; 9.43 (s, 1) ; 9.72 (s, 1) .
Exemple 13 : 3- [ (l-{4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benze nesulfonyl} -piperidin-4-yl) -
methyl-amino] -N,N-dimethyl-propiona_mide

A une suspension de 400 mg du composé obtenu à la procédure 3a dans 20 mL d'éthanol, on ajoute la TEA(O.35 mL) puis la N,N-diméthyl-acrylamide (0.1 mL) . On chauffe à
90 0C pendant 12 heures puis on laisse sous agitation à
TA pendant 48 heures. On évapore à sec, ajoute de
1'AcOEt. Après traitement, on chromatographie (SiO2) et élue par DCM/MeOH(v/v ; 93/7) et recristallise dans
DCM/iPr2O et on obtient 85 mg de produit attendu.
MH+ = 570.3
PF = 201 0C
IH RMN (DMSO) :
1.42 (qd, 2); 1.69 (dd, 2); 2.10 (s, 3); 2.24 (s, 3);
2.24 (d, 2); 2.29 (m, 1); 2.33 (t, 2); 2.58 (t, 2); 2.75
(s, 3); 2.91 (s, 3); 3.59 (d, 2); 6.29 (d, 1); 7.11 (t,
1); 7.47 (m, 1); 7.58 (d, 2); 7.58 (d, 1); 7.98 (d, 2) ;
8.08 (d, 1); 9.42 (si, 1); 9.70 (si, 1) .
Exemple 14 : l-{4- [4- (4-Fluorophenylamino) -pγrimidin-2- ylamino] -benzoyl} -3-pyridin-3-ylmethyl-piperidine-3- carboxylic acid ethyl ester (Racémique)

Suivant le mode opératoire décrit au stade 1 de la procédure 3a, à partir de 3 g d'acide benzolque obtenu dans la procédure 2a et de 2.25 g d'aminé commerciale, on
obtient 2.5 mg de produit attendu MH+ = 605.2
Point de fusion = 119 0C IH RMN (DMSO) : 1.12(t, 3) ; 1.26-1.85(massif, 4) ; 2.25(dl, 3) ; 2.45 (m, 2) ; 2.66-2.98 (dd, 2) ; 3.17 (dl, 1) ; 3.60(d, 1) ; 4.03 (m, 2) ; 6.29(d, 1) ; 7.12 (t, 1) ; 7.31 (m, 1) ; 7.39- 7.65 (massif, 5) ; 8.00 (d, 2) ; 8.08 (d, 1) ; 8.28 (m, 1) ; 8.43 (m, 1) ; 9.43(s, 1) ; 9.73(s, 1) .
Exemple 15 : l-{4- [4- (4-Fluorophenylamino) -pyrimidin-2- ylamino] -benzoyl} -3-pyridin-3-ylmethyl-piperidine-3- carboxylic acid dimethylam±de (Racémique)

Stade 1 : On dissout 660 mg de KOH dans 5 mL d'eau. On additionne à cette solution 3.5 g d'ester obtenu à l'exemple 14 et 50 mL de MeOH. Après un reflux de 3 heures, le milieu réactionnel est concentré à sec et repris avec de l'eau acidifiée à un pH de 7. On filtre le précipité formé. Stade 2 : L' acide (500 mg) obtenu au stade 1 est mis en réaction avec 187 mg de EDC, 138 mg de HOBT, 150 mg de chlorhydrate de dimethyl-amine et 230 mg de DIPEA. Après 18 heures de réaction, on concentre le milieu réactionnel sous vide, on reprend avec du DCM, lave à l'eau, sèche et concentre sous vide. Le brut est purifié par chromatographie sur colonne de silice et élue par un mélange DCM-MeOH (v/v, 98/2) . On obtient 290 mg de caboxamide attendu.
MH+ = 590 . 2
Point de fusion = 146 0C IH RMN (DMSO) :
1.53-1.85 (massif, 4) ; 2.56-3.28 (massif, 12) ; 6.30(d, 1) ; 7.19 (t, 2) ; 7.29 (m, 1) ; 7.50 (m, 1) ; 7.60 (d, 2) ; 7.71 (m, 2) ; 8.01(d, 2) ; 8.10 (d, 1) ; 8.32 (dl, 1) ; 8.43 (ddl, 1) ; 9.51(s, 1) ; 9.74(S, 1) .
Exemple 16 : l-{4- [4- (4-Fluorophenγlamino) -pyrimidin-2- ylamino] -benzoyl} -3-pyridin-3-ylmethyl-piperidine-3- carboxylic acid methylamide (Racémique)

Suivant le mode opératoire décrit au stade 2 de l'exemple
15, à partir de 500 mg de l'acide obtenu au stade 1 de l'exemple 15 et de 120 mg de chlorhydrate de monomethylamine, on obtient 255 mg de carboxamide attendu.
MH+ = 576.2
Point de fusion = 150 0C
IH RMN (DMSO) : 1.3-1.82 (massif, 4) ; 2.5 (s, 3) ; 2.58-3.13 (massif, 6) ;
6.25 (d,l) ; 7.13 (t,2) ; 7.26 (m,l) ; 7.40 (m,l) ; 7.54 (d*,3) ; 7.66(m,2) ; 7.96 (d,2) ; 8.04(d,l) ; 8.22(d,l) ;
8.38 (m,l) ; 9.47 (s,l) ; 9.68 (s,l)
Exemple 17 : l-{4- [4- (4-Fluorophenylamino) -pyrimidin-2- ylamino] -benzoyl}-3-pyridin-3-ylmethyl piperidine-3- carboxylicacid (2-dimethylamino-ethyl) -amide (Racémique)
Suivant le mode opératoire décrit à l'exemple 16, à partir de 500 mg de l'acide obtenu au stade 1 de l'exemple 15 et de 118 mg de N,N~dimethylamino- ethylamine, on obtient 220 mg de carboxamide attendu. MH+ = 633.4
Point de fusion = 122 0C IH RMN (DMSO) :
1.22-1.82 ( massif, 4) ; 2.08 (s, 6) ; 2.18 (t,2) ; 2.58- 3.18 ( massif, 8) ; 6.25 (d,l) ; 7.15 (t,2) ; 7.25 (m,l) . 7.52 (2m,4) ; 7.66 (m, 2) ; 7.95 (d,2) ; 8.04 ( d,l) ; 8.29 ( d,l) ; 8.37 ( d,l) ; 9.48 (s,l) ; 9.69 (s,l)
Exemple 18 : N*4*- (4-Fluoro-phenyl) -N*2*- [4- (3-{ [ (1- methyl-lH-pyrrol-2-ylmethyl) -amino] ~methyl}-piperidine-l- suifonyl) -phenyl] -pyrimidine-2 , 4-diamine (Racémique)

De la même façon qu'à l'exemple 1, à partir de 400 mg du composé de la procédure 3b et de 102 mg de 1-methyl-lH- pyrrole-2-carbaldehyde, on obtient 202 mg de produit attendu
MH+ = 550.1
Point de fusion ≈ 184.2 0C
IH RMN (DMSO) :
0.80 (m,l) ; 1.18-1.97 (massif, β) ; 1.98-2.41 ;
(massif, 3) ; 3.35-3.71 (massif, 7) ; 5.80 (d,2) ; 6.25
(d,l) ; 6.57 (t,l) ; 7.14 (t,2) ; 7.52 (d,2) ; 7.67 (m, 2) ; 7.95 (d,2) ; 8.05 (d,l) ; 9.47 (s,l) ; 9.67 (s,l)
Exemple 19 : N*4*- (4-Fluoro-phenyl) -N*2*- [4- (3-{ [ (5- methyl-isoxazol-3-ylmethyl) -amino] -methyl}-piperidine-l- sulfonyl) -phenyl] -pyrimidine-2 , 4-diamine (Racémique)

0.79 (m,l) ; 1.22-1.71 (massif, 4) ; 1.83 (t,l) ; 1.99- 2.39 (massif, 7) ;3.41 (d,l) ; 3.58 (massif, 3) ; 6.13 (s,l) ; 6.24 (d,l) ; 7.13 (t,2) ; 7.53 (d,2) ; 7.66 (m, 2) ; 7.94 (d,2) ; 8.05 (d,l) ; 9.46 (s,l) ; 9.65 (s, H
Exemple 20 : N*4*- (4-Fluoro-phenyl) -N*2*- [4- (3-
{ [ (thiophen-2-ylmethyl) -amino] -methyl}-piperidine-l- sulfonyl) -phenyl] -pyrimidine-2 , 4-diamine (Racémique)

De la même façon qu'à l'exemple 1, à partir de 500 mg du composé de la procédure 3b et de 138 mg de 1-methyl-lH- pyrrole-2-carbaldéhyde, on obtient 200 mg de produit attendu
MH+ = 553.2
Point de fusion = 98.8 0C IH RMN (DMSO) :
0.77 (m,l) ; 1.10-1.94 (massif, 5) ; 1.95-2.41 (massif, 4) ; 3.41 (d,l) ; 3.63 (d,l) ; 3.79 (s, 2) ; 6.25 (d,l) ;
6.89 (massif, 2) ; 7.13 (t,2) ; 7.31 (m,l) ; 7.53 (d,2) ; 7.66 (m, 2) ; 7.94 (d,2) ;8.04 (d,l) ; 9.47 (s,l) ; 9.67 (s,l)
Exemple 21 : N*4*- (4-Fluoro-phenyl) -N*2*- (4-{3- [ (2- methanesulfonyl-ethylamino) -methyl] -piperidine-1- sulfonyl } -phenyl) -pyrimidine-2 , 4-diamine (Racémique)

Suivant la réaction d'addition décrite à l'exemple 11, à partir de 300 mg de du composé de la procédure 3b et de 62 mg de vinyl-methylsulfone, on obtient 294 mg de produit attendu. MH+ = 563.1 Point de fusion = 184.2 0C
IH RMN (DMSO) :
0.86 (m,l) ; 1.30-1.78 (massif, 4) ; 1.94 (2m, 2) ; 2.09- 2.45 (massif, 3) ; 1.91 (m,2) ; 3 (s, 3) ; 3.19 (t,2) ; 3.47 (d,l) ; 3.6 (d,l) ; 6.30 (d,l) ; 7.21 (t,2) ; 7.58 (d,2) ; 7.72 (m, 2) ; 7.99 (d,2) ; 8.10 (d,l) ; 9.51 (s,l) ; 9.71 (s,l)
Exemple 22 : 4-Dimethylaminomethyl-l-{4- [4- (4-fluoro-3- methyl-phenylamino) -pyrimidin-2-ylamino] - benzenesulfonyl} -piperidin-4-ol

Suivant la réaction d' additon décrite à l'exemple 11, à partir de 290 mg de composé de la procédure 3e et de 82 mg de vinyl-methylsulfone, on obtient 130 mg de produit attendu. MH+ = 593.1
Point de fusion = 233 0C
IH RMN (DMSO) :
0.65 (m, 4) ; 2.24(d, 3) ; 2.55(m, 2) ; 2.93(sl, 2) ;
3.04(s, 3) ; 3.21-3.67 (massif, 6) ; 5.11(sl, 1) ; 6.31(d, 1) ; 7.11 (t, 1) ; 7.47 (m, 1) ; 7.53-7.66 (massif, 3) ;
8.01(d, 2) ; 8.08(d, 1) ; 8.75(sl, 2) ; 9.51(s, 1) ;
9.76(s, 1) .
Exemple 23 : l-{4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl}-4-imidazol-l- ylmethyl-piperidin-4-ol

Stade 1 : A une solution contenant 290 mg d'imidazole dans 5 mL de DMSO, on ajoute 1.2 équivalent d'hydrure de sodium. Après 20 minutes d'agitation à TA, on additionne 700 mg d' epoxyde obtenu au stade 1 de la procédure 3e et on laisse sous agitation pendant 18 heures à TA. On reprend avec l'eau, extrait au DCM, sèche sur Na2So4 et concentre. On obtient, par trituration 540 mg d'alcool attendu. Stade 2 : Suivant une réaction d' hydrogenolyse décrite au stade 4 de la procédure 3e, à partir de 540 mg d'alcool obtenu au stade 1, on obtient 280 mg de piperidine attendu. Stade 3 : Suivant le mode opératoire décrit au stade 1 de la procédure 3a, à partir de 600 mg de chlorhydrate de chlorure de sulfonyl et de 280 mg de piperidine obtenu au stade 2, on obtient 330 mg de sulfonamide attendu.
MH+ = 538.2
Point de fusion = 220 °C IH RMN (DMSO) : .
1.35 (d,2) ; 1.56 (m, 2) ; 2.24 (s, 3) ; 2.41 (t,2) ; 3.4 (d,2) ; 3.88 (s, 2) ; 4.66 (s,l) ; 6.28 (d,l) ; 6.84 (s,l) ; 7.06 (s,l) ; 7.10 (t,l) ; 7.46 (m,l) ; 7.49 (s,l) ; 7.52-7.61 (massif, 3) ; 7.97 (d,2) ; 8.07(d,l) ; 9.42 (s,l) ; 9.17 (s,l)
Exemple 24 : N*4*- (4~Fluoro-3-methyl phenyl) -N*2*- (4- { 3- t (2-methanesulf onyl-ethylamino) -methyl] -pyrrolidine-1- sulf onyl } -phenyl) -pyrimidine-2 , 4-diamine (Racémique)

Suivant la réaction d' additon décrite à l'exemple 11, à partir de 400 mg de du composé de la procédure 3f et de
140 mg de vinyl-methylsulfone, on obtient 260 mg de produit attendu.
MH+ = 563.2
Point de fusion = 154 0C
Exemple 25 : N*4*- (4-Fluoro-3-methyl-phenyl) -N*2*- [4- (3- { [ (l-methyl-lH-pyrrol-2-ylmethγl) -amino] -methyl}- pyrrolidine-1-sulfonyl) -phenyl] -pyrimidine-2 , 4-diamine (Racémique)

De la même façon qu'à l'exemple 1, à partir de 300 mg du composé de la procédure 3f et de 83 mg de 1-methyl-lH- pyrrole-2-carbaldéhyde, on obtient 100 mg de produit attendu
MH+ = 550.0
Point de fusion = 93 0C
IH RMN (DMSO) :
1.39 ( m, 1) ; 1.80 (m, 1) ; 2.10 (m,l) ; 2.16-2.38 ( massif, 5) ; 2.76-3.34 (massif, 4) ; 3.49(s, 2) ; 3.51(s,
3) ; 5.79 (massif, 2) ; 6.28 (d,l) ; 6,57 (s, 1) ; 7.11 (t,
1) ; 7.46 (m, 1) ; 7.58 (d, 1) ; 7.63 (d, 2) ; 7.98 (d,
2) ; 8.07 ( d, 1) ; 9.41 (s,l) ; 9.69 (s,l) .
Exemple 26 : {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl}- [3- (5-methyl-isoxazol-3-ylinethyl - amino) -piperidin-1-yl] -methanone (Racémique)

De la même façon qu'à l'exemple 1, à partir de 340 mg de l'acide de la procédure 4g et de 100 mg de 5-methyl- isoxazole-3-carbaldehyde, on obtient 360 mg de produit attendu
MH+ = 516.0
Point de fusion = 190-191 0C
IH RMN (DMSO) : 1.43 (m, 1) ; 1.56 (m, 1) ; 1.75 (m, 1) ; 1.95 (dl,l) ;
2.23(s, 3) ; 2.37(s, 3) ; 2.54 (m, 1) ; 2.94 (m, 2) ;
3.63(sl, 2) ; 3.94(sl, 1) ; 4.13(sl, 1) ; 6.06(s, 1) ;
6.24(d, 1) ; 7.12(t, 2) ; 7.26(d, 2) ; 7.66 (m, 2) ;
7.77(d, 2) ; 8.03(d, 1) ; 9.06(s, 1) ; 9.16(s, 1).
Exemple 27 : N*4*- (4-Fluoro-phenyl) -N*2*- (4-{3- [ (2- methanesulfonyl-ethyl) -methyl-amino] -piperidine-1- sulf onyl } -phenyl) -pyrimidine-2 , 4-diamine

Suivant la réaction d'additon décrite à l'exemple 11, à partir de 400 mg de du composé de la procédure 3g et de 140 mg de vinyl-methylsulfone, on obtient 360 mg de
composé attendu.
MH+ = 563.1
PF = 103 0C
RMN (IH, DMSO) 1.25 (t,6) ; 1.65 (m,2) ; 2.09 (m, 2) ; 2.14-2.37 (massif, 4) ; 2.86-3.24 (2m, 3) ; 3.74 (d,2) ; 4.02 (m, 4) 6.54 (d,l) ; 7.26 (t,2) ; 7.63 (m, 2) ; 7.68 (d,2) ;
7.81 (d,2) ; 8.09 (d,l) ; 9.33 (Is, 2) ; 10.74-11.13 (21s, 2)
Exemple 28 : N*4*- (4-Fluoro-phenyl) -N*2*-{4- [4- (2- methanesulfonyl-ethylamino) -piperidine-1-sulfonyl] - phenyl } -pyrimidine-2 , 4-diamine

Suivant la réaction d'addition décrite à l'exemple 11, à partir de 800 mg de du composé de la procédure 3h et de
190 mg de vinyl-methylsulfone, on obtient 340 mg de composé attendu.
MH+ = 549.2
PF = 157 0C RMN (IH, DMSO)
1.24 (m, 2) ; 1.76 (m,2) ; 2.38 (massif, 3) ; 2.79 (t,2) ;
2.85 (s, 3) ; 3.08(m,2) ; 3.33 (m,2) ; 6.25 (d,l) ; 7.13
(t,2) ; 7.53 (d,2) ; 7.66 (m,2) ; 7.92 (d,2) ; 8.03 (d,l)
; 9.45 (s,l) ; 9.66(s,l) .
Exemple 29 : [3- (l-{4- [4- (3,4-Difluoro-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl }piperidin-4-ylamino- methyl) -ethyl] -phosphonic acid diethyl ester

Stade 1 : [2- (l-Benzyl-piperidin-4-ylamino) -ethyl] - phosphonic acid diethyl ester
On porte à reflux dans EtOH (50 mL) , un mélange de 4- amino-1-benzyl piperidine(5 g), de (2-bromo-éthyl) - phosphonic acid diethyl ester (7 g) . Après 18 heures d'agitation à TA, on filtre le solide et concentre, on chromatographie(A1203) et élue par DCM/MeOH(v/v ; 85/15) et on obtient 6.2 g de produit attendu Stade 2 : {2- [ (l-Benzyl-piperidin-4-yl) -methyl-amino] - ethyl} -phosphonic acid diethyl ester
A un mélange de composé (2 g) obtenu au stade 1, de formaldehyde (0.6 mL, 37 % solution acqueuse) dans DCM (70 mL) , on additionne NaBH(OAC)3 (1.6 g) .Après une heure d'agitation et traitement avec une solution de Na2CO3, extraction au DCM, séchage et concentration, on obtient
1.9 g de produit attendu.
Stade 3 : [2- (Methyl-piperidin-4-yl-amino) -ethyl] - phosphonic acid diethyl ester Par une réaction d'hydrogénolyse sur 1.9 g du composé obtenu au stade 2, on obtient 1.2 g de dérivé 1-H- pipéridine.
Stade 4 : [3- (l-{ 4- [4- (3, 4-Difluoro-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl }piperidin-4~ylamino- methyl) -ethyl] -phosphonic acid diethyl ester
Suivant le mode opératoire décrit au stade 1 de l'exemple 30, 500 mg de dérivé difluoré de la procédure 2b et 420 mg de composé obtenu au stade 3 permettent d'obtenir 380 mg du compé attendu.
MH+ = 639 PF = 164 0C RMN (IH, DMSO)
1.07-1.32 (massif, 8) ; 1.52 (q, 2) ; 1.71-2.02 (massif , 4) ; 2.41 (m, 2) ; 2.64 (s, 3) ; 2.80 (d, 2) ; 3.60 (m, 1) ;
3.95 (q, 4) ; 6.28 (d, 1) ; 7.17 (t, 2) ; 7.55-7.79 (massif , 4) ; 7.95 (d, 2) ; 8.08 (d, 1) ; 9.48 (s, 1) 9.67 (s, 1) .
Exemple 30 : [2- (l-{4- [4- (3,4-Dif luoro-phenylamino) - pyrimidin-2-ylamino] -benzenesul£onyl}-piperidin-4- ylamino) -ethyl] -phosphonic acid diethyl ester

Stade 1 : {2- [ (l-Benzyl-piperidin-4-yl) -tert- butoxycarbonyl-amino] -ethyl } -phosphonic acid diethyl ester A une solution de 3.4 g de piperidine obtenu au stade 1 de l'exemple 29 dans CH3CN (20 mL) , on ajoute goutte à goutte 2 g de BOC2O dissout dans CH3CN(16 mL) et on agite 18 heures à TA. On concentre à sec et on chromatographie(A1203) et élue par DCM/AcOEt (v/v ; 1/1) et on obtient 3 g de produit attendu
Stade 2 : [2- (tert-Butoxycarbonyl-piperidin-4-yl-amino) - ethyl] -phosphonic acid diethyl ester
On porte à reflux dans EtOH (25 mL) , un mélange de composé obtenu au stade 1(3 g) et d'hydroxyde de palladium sur charbon. Après 2h30 de reflux, on filtre et on concentre pour obtenir 2.2 g de composé attendu. Stade 3 : [2- (l-{ 4- [4- (3, 4-Difluoro-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl } -piperidin-4- ylamino) -ethyl] -phosphonic acid diethyl ester
Suivant le mode opératoire décrit au stade 1 de l'exemple 2, 800 mg de dérivé difluoré de la procédure 2b et 880 mg de composé obtenu au stade 2 permettent d'obtenir 1.3 g d'un composé intermédiaire qui subit une réaction de décarboxylation pour donné 1 g du composé attendu. MH+ = 625.0 PF = 149 0C RMN (IH, DMSO)
1.25 (t,6) ; 1.65 (m, 2) ; 2.09 (m, 2) ; 2.14-2.39 (massif, 4) ; 2.91-3.21 (2m, 3) ; 3.74 (m, 2) ; 4.05 (m, 4) ; 6.53 (d,l) ; 7.35 (m,l) ; 7.44 (m,l) ; 7.71 (d,2) ; 7.87 (d,2) ;7.99 (ml) ; 8.13 (d,l) ; 9.26 (Is, 2) ; 10.75 (Is, 2)
Exemple 31 : [2- (l-{4- [4- (4-Fluoro-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl}-piperidin-4- ylamino) -ethyl] -phosphonic acid diethyl ester

Suivant le mode opératoire décrits aux stades 1 et 2 de l'exemple 32, on obtient à partir de 800 mg de dérivé fluoré de la procédure 2a et de 920 mg de l'aminé correspondant, 1 g de composé final attendu
MH+ = 607.1
PF = 195 0C
RMN (IH, DMSO) 1.25 (t,6) ; 1.65 (m, 2) ; 2.09 (m, 2) ; 2.14-2.37
(massif, 4) ; 2.86-3.24 (2m, 3) ; 3.74 (d,2) ; 4.02 (m, 4)
6.54(d,l) ; 7.26 (t,2) ; 7.63 (m, 2) ; 7.68 (d,2) ; 7.81 (d,2) ; 8.09 (d,l) ; 9.33 (Is, 2) ; 10.74-11.13 (21s, 2)
Exemple 32 : (l-{4- [4- (4-Fluoro-3-methyl-phenylamino) -
pyrimidin-2-ylamino] -benzenesulfonyl}-piperidin-4-yl) - (3- fluoro-pyridin-4-yl) -methanol

Stade 1 : (3-Fluoro-pyridin-4-yl) -piperidin-4-yl-methanol Suivant le brevet (WO/2005/059107 ), à une solution à froid (-90 0C) de 1.82 g de 3-fluoropyridine dans 50 mL de THF, on additionne 12 mL de LDA(I.8 M) . La solution est agitée sous azote pendant 30 minutes en maintenant la même tempoérature. Une solution de 2 g de 4- carboxaldehyde-piperidine-1-carboxylic acid tert-butyl ester dans 22 mL de THF est ajoutée lentement en maintenant la température en dessous de -70 0C. Le mélange réactionnel est agité à cette température pendant 30 minutes. On laisse la température remonter à -20 0C en 1 heure. 40 mL d'une solution saturée de NH4C1 sont additionnés lentement. Après décantation, la phase organique est lavée par une solution de Na2CO3 10 % , puis par une solution saturée de NaCl et séchée sur MgSO4, filtré et concentré sous vide. Après chromatographie sur silice, on ontient 1.82 g d'un intermédiaire qui subit une réaction de décarboxylation pour donner 510 mg de dérivé 1-H-pipéridine. Stade 2 : (l-{ 4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl}-piperidin-4-yl) - (3- fluoro-pyridin-4-yl) -methanol
Suivant le mode opératoire décrit au stade 1 de la procédure 3a, à partir de 600 mg de dérivé fluoré de la procédure 2c et de 407 mg de composé obtenu au stade 1, on obtient, après traitement habituel et chromatographie sur silice, DCH/MeOH (98/2; v/v) , on recristallise dans
DCM-iPr2O, 500 mg de produit attendu sous forme d'un mélange de deux isomères.
MH+ = 570.2
PF = 142 0C RMN (IH, DMSO)
1.26 (m, 1); 1.28-1.43 (massif, 2); 1.50 (massif, 1) ;
1.70 (d, 1); 2.10 (td, 2); 2.23 (s, 3); 3.62 (t, 2); 4.62 (t, 1); 5.63 (d, 1); 6.28 (d, 1); 7.10 (t, 1); 7.39-7.49 (m, 2); 7.53 (d, 2); 7.57 (dd, 1); 7.95 (d, 2); 8.07 (d, 1); 8.34 (d, 1); 8.45 (s, 1); 9.42 (s, 1); 9.69 (s, 1) .
Exemple 33 : (énantiomère 1): (l-{4- [4- (4-Fluoro-3- methyl-phenylamino) -pyrimidin-2-ylamino] - benzenesulfonyl}-piperidin-4-yl) - (3-fluoro-pyridin-4-yl) - methanol
La séparation des deux énantiomères l'exemple 32 se fait par chromatographie chirale (détection: UV 254 nm; phase stationnaire: chiralpakAD-lOμm 250x4.6mm; phase mobile: 60%EtOH-40%Heptane; débit: 1 mL/min) . Au cours de cette séparation, 99.8 mg du premier énantiomère sont obtenus. Tr = 8.47 min MH+ = 570.2
Exemple 34 : (énantiomère 2) : (l-{4- [4- (4-Fluoro-3- methyl-phenylamino) -pyrimidin-2-ylamino] - benzenesulfonyl}-piperidin-4-yl) - (3-fluoro-pyridin-4-yl) - methanol

Au cours de l'étape de chromatographie chirale décrite à l'exemple 33, 92.2 mg du deuxième énantiomère sont obtenus .
Tr = 12.26 min.
MH+ = 570.2
Exemple 35: 1- (l-{4- [4- (4-Fluoro-3-methγl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl}-piperidin-4-yl) -2- (3-methyl-pyridin-2-yl) -ethanol (racémique)

Stade 1 : 2- (3-Methyl-pyridin-2-yl) -l-piperidin-4-yl- ethanol
Suivant le mode opératoire décrit au stade 1 de l ' exemple 32, 2 g de 2, 3-dimethyl-pyridine et 2 g de 4- carboxaldehyde-piperidine-1-carboxylic acid tert-butyl ester, on obtient 650 mg de dérivé 1-H-pipéridine attendu.
Stade 2 : 1- (l-{ 4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl}-piperidin-4-yl) -2- (3-methyl-pyridin-2-yl) -ethanol (racémique) Suivant le mode opératoire décrit au stade 2 de l'exemple 32, à partir de 350 mg de dérivé fluoré de la procédure 2c et de 200 mg de pipéridine obtenu au stade 1, on obtient 200 mg de produit attendu sous forme d'un mélange de deux isomères. MH+ = 577.2 PF = 205-207 0C
IH RMN (DMSO) :
1.25 (m, 1) ; 1.30 (massif, 2) ; 1.56 (m, 1) ; 1.67 (d, 1) ; 2.16 (m, 1) ; 2.23 (s, 3) ; 2.70 (s, 3) ; 3.61 (t, 2) ; 4.32 (t, 1) ; 5.45 (d, 1) ; 6.27 (d, 2) ; 7.08 (t, 1) ; 7.27 (d, 2) ; 7.45 (m, 1) ; 7.52 (d, 2) ; 7.58 (m, 1) ; 7.96 (d, 2) ; 8.02 (d, 1) ; 8.44 (d, 2) ; 9.47 (s, 1) ; 9.62 (s, 1) .
Exemple 36 : (l-{4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl}-piperidin-4-yl) - pyridin-4-yl-methanol (racémique)

Stade 1 : Piperidin-4-yl-pyridin-4-yl-methanol
Suivant le mode opératoire décrit au stade 1 de l'exemple 32, 2 g de 3-bromopyridine et 2 g de 4-carboxaldehyde- piperidine-1-carboxylic acid tert-butyl ester, on obtient 650 mg d'un produit qui subit une hydrogénolyse à 3 bar (WO/2005/059107) pour éliminer le brome et une décarboxylation pour donner 220 mg dérivé l-JJ-pipéridine attendu. Stade 2 : (l-{ 4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl}-piperidin-4-yl) - pyridin-4-yl-methanol racémique
Suivant le mode opératoire décrit au stade 2 de l'exemple 32, à partir de 355 mg de dérivé fluoré de la procédure 2c et de 174 mg de pipéridine obtenu au stade 1, on obtient 200 mg de produit attendu sous forme d'un mélange de deux isomères. MH+ = 549.4 PF = 162 0C IH RMN (DMSO) :
1.27 (m, 1) ; 1.35 (massif, 2) ; 1.46 (m, 1) ; 1.65 (d, 1) ;
2.06 (m, 1) ; 2.23 (s, 3) ; 3.61 (t, 2) ; 4.32 (t, 1) ; 5.45
(d, 1) ; 6.27 (d, 2) ; 7.09 (t, 1) ; 7.25 (d, 2) ; 7.45 (m,
1) ; 7.52 (d, 2) ; 7.57 (m, 1) ; 7.94 (d, 2) ; 8.06 (d, 1) ;
8.46 (d, 2) ; 9.41 (s, 1) ; 9.68 (s, 1) .
Exemple 37 : (l-{4- [4- (4-Fluoro-phenγlamino) -pyrimidin-2- ylamino] -benzenest.lfonyl}-piperidin-4-yl) - (3-fluoro- pyridin-4-yl) -methanol (racémique)

Suivant le mode opératoire décrit au stade 2 de l'exemple 32, à partir de 400 mg de dérivé fluoré de la procédure 2a et de 270 mg de pipéridine obtenu au stade 1 de l'exemple 32, on obtient 115 mg de produit attendu sous forme d'un mélange de deux isomères MH+ = 553.2 PF = 140 0C RMN (IH, DMSO)
1.18-1.43 (massif, 3); 1.49 (m, 1); 1.70 (d, 1); 2.10 (t, 2); 3.62 (t, 2); 4.62 (t, 1); 5.63 (d, 1); 6.29 (d, 1) ; 7.17 (t, 2); 7.44 (t, 1); 7.55 (d, 2); 7.70 (dd, 2);.7.96 (d, 2); 8.09 (d, 1); 8.40 (d, 1); 8.46 (d, 1); 9.50 (s, 1); 9.71 (s, 1) .
Exemple 38 : 1- (l-{4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl}-piperidin-4-yl) -2- (4-methyl-pyridin-2-yl) -ethanol (racémique)
Suivant le mode opératoire décrit au stade 1 de l'exemple
32, 2 g de 2, 4-dimethyl-pyridine et 2 g de 4- carboxaldehyde-piperidine-1-carboxylic acid tert-butyl ester, on obtient 680 mg de dérivé 1-H-pipéridine attendu.
Stade 2 : 1- (l-{ 4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl }-piperidin-4-yl) -2- (3-methyl-pyridin-2-yl) -ethanol (racémique)
Suivant le mode opératoire décrit au stade 2 de l'exemple
32, à partir de 500 mg de dérivé fluoré de la procédure
2c et de 300 mg de pipéridine obtenu au stade 1, on obtient 270 mg de produit attendu sous forme d'un mélange de deux isomères.
MH+ = 563.1
PF = 103.2-104.5 0C
RMN (IH, DMSO)
IH RMN (DMSO) : 1.23 (m, 1); 1.28 (massif, 2); 1.50 (m, 1); 1.70 (d, 1) ;
2.12 (m, 1); 2.29 (s, 3); 2.68 (s, 3); 3.63 (t, 2); 4.30 (t, 1); 5.39 (d, 1); 6.20 (d, 2); 7.13 (t, 1); 7.24 (d,
2); 7.40 (m, 1); 7.58 (d, 2); 7.63 (m, 1); 7.98 (d, 2);
8.08 (d, 1); 8.40 (d, 2); 9.49 (s, 1); 9.67 (s, 1) .
Exemple 39: {4- [4- (4-Fluoro-3-methyl-phenylamino) - pyrimidin-2-γlamino] -phenyl}- (piperidin-4-yl) - (3-fluoro- pγridin-4-yl) -methanol) -methanone (racémique)

Suivant le mode opératoire décrit au stade 1 de la
procédure 4a, à partir de 320 mg d'acide obtenu dans la procédure 2c et de la pipéridine obtenu au stade 1 de l'exemple 32, on obtient 290 mg de produit attendu sous forme d'un mélange de deux isomères. MH+ = 531.2
PF = 115 0C (formation d'une mousse) RMN (IH, DMSO)
1.08-1.98 (massif, 5); 2.23(d, 3); 2.80(sl, 2); 4.70(t, 1); 5.65(d, 1); β.22(d, 1) ; 7.09(s, 1); 7.26(d, 2); 7.37-7.66(massif, 3); 7.81(d, 2); 8.04(d, 1); 8.44(d, 1); 8.50(d, 1); 9.33(s, 1); 9.38(s, 1).
Exemple 40 : [ 4-R- (Amino-phenyl-methyl) -piperidin-1-yl] - { 4- [4- (4-fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl } -methanone

Une solution contenant 220 mg d'aminé commerciale phenyl- piperidin-4-R-yl-methylamine dans 20 mL d'un mélange DCM/DMF(v/v ; 1/1), on additionne à température ambiante dans l'ordre la DIPEA(I.5 mL) , le BOP (360 mg) , puis, par petite portion en 30 minutes, 300 mg d'acide obtenu dans la procédure 2a. On agite pendant la nuit. On évapore à sec, ajoute de l'eau carbonatée (K2CO3) et on extrait par AcOEt. Après traitement, on chromatographie (SiO2) et élue par DCM/MeOH (v/v ; 94/6) et recristallise dans DCM/iPr2O. MH+ = 497.2 PF = 130-185 0C [Q]D = + 40 (c = 0.15, MeOH) RMN (IH, DMSO)
1.07-1.41 (massif, 3) ; 1.74 (m, 1) ; 1.88 (d, 2) ; 2.41 (sl, 1) ; 2.82 (m, 2) ; 3.68 (d, 1) ; 4.09 (dl, 2) ; 6.23 (d, 1) ; 7.13 (t, 2) ; 7.17-7.38 (massif , 7) ; 7.66 (m, 2) ; 7.76(d, 2) ; 8.03 (d, 1) ; 9.11 (s, 1) ; 9.20 (s, 1) .
Exemple 41 : [4-S- (Amino-phenyl-methyl) -piperidin-1-yl] - {4- [4- (4-fluoro-phenγlamino) -pyrimidin-2-ylamino] - phenyl } -methanone
N

Suivant le mode opératoire décrit à l'exemple 40, à partir de l'aminé de configuration S et de l'acide obtenu dans la procédure 2a, on obtient le produit attendu.
MH+ = 497.2
PF = 130-185 0C
[D]D = - 34 (c = 0.117, MeOH) RMN (IH, DMSO)
1.07-1.41(massif, 3) ; 1.75(m, 1) ; 1.88(d, 1) ; 2.82 (m,
2) ; 3.68(d, 1) ; 4.09(dl, 2) ; 6.23(d, 1) ; 7.13(t, 2)
; 7.17-7.38 (massif, 7) ; 7.66 (m, 2) ; 7.76 (d, 2) ;
8.03(d, 1) ; 9.11(s, 1) ; 9.20(s, 1) .
Exemple 42 : N*4*- (4-Fluoro-3-methyl-phenyl) -N*2*-{4- [3- (pyridin-3-yloxy) -piperidine-1-sulfonyl] -phenyl}- pyrimidine-2 , 4-diamine racémique

A une suspension de 800 mg de dichlorhydrate de 3- pyridyl-oxy-piperidine racémique (obtenu suivant la synthèse décrite dans J. Med. Chem. 43, 11, 2000, 2217-
2226) dans 20 mL de DCM, on ajoute la DIPEA(I mL) puis le
chlorure de sulfonyle obtenu dans la procédure Ic(I.4 g), on agite pendant une nuit. On évapore à sec, ajoute de l'eau carbonatée (K2CO3) et extrait par AcOEt. Après traitement, on chromatographie (SiO2) et élue par DCM/MeOH(v/v ; 97/3) et recristallise dans DCM/iPr2O pour obtenir le produit attendu. MH+ = 535.1 PF = 113°C RMN (IH, DMSO) 1.42- 1.96 (massif, 4) ; 2.24(s, 3) ; 2.76-3.01 (massif, 3) ; 3.24(d, 2) ; 4.61 (m, 1) ; 6.29(d, 1) ; 7.10(t, 1) ; 7.29-7.65 (massif, 6) ; 7.98 (d, 2) ; 8.07(d, 1) ; 8.19(d, 1) ; 8.30(d, 1) ; 9.42(sl, 1) ; 9.71(sl, 1) .
Exemple 43 : {4-R- [Amino- (4-fluoro-phenyl) -methyl] - piperidin-l-yl}-{4- [4- (4-fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl} -methanone

Suivant le mode opératoire décrit à l'exemple 40, à partir de l'aminé R et de l'acide obtenu dans la procédure 2a, on obtient le produit attendu.
MH+ = 515.2
PF = 184-185 0C
[D]D = + 49 (c = 0.103, MeOH)
RMN (IH, DMSO) 0.98-1.38 (massif, 3) ; 1.64 (m, 1) ; 1.87 (si, 3) ;
2.77(sl, 2) ; 3.60(d, 1) ; 3.63-4.81 (massif, 2) ; 6.22(d,
1) ; 7.03-7.20(massif, 4) ; 7.24(d, 2) ; 7.34 (m, 2) ;
7.70(m, 2) ; 7.77(d, 2) ; 8.04(d, 1) ; 9.38(3, 1) ;
9.42(s, 1) .
Exemple 44 : { 4-S- [Amino- (4-fluoro-phenyl) -methyl] - piperidin-l-yl} - { 4~ [4- (4-fluoro-phenylamino) -pyrimidin-2- ylamino] -phenyl } -methanone

0.99-2.15 (massif, 7) ; 2.72 (large multiplet, 2) ; 3.54 (multiplet,!) ; 3.61-4.97 (large signal, 2) ; 6.18 (d,l) ; 6.97-7.41 (massif, 8) ; 7.57-7.82 (massif, 4 ) ; 7.99 (d,l) ; 9.28-9.43 (2s, 2)
Exemple 45 : (l-{4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -benzenesulfonyl } -3-pyridin-3-ylmethyl-piperidin- 3-yl) -methanol (Racémique)

RMN (IH, DMSO)
1.12 ( si, 2) ; 1.45-1.90 ( 2sl, 2) ; 2.23(dl, 3) ; 2.46- 3.15(massif, 8) ; 4.81(t, 1) ; 6.29 (d,l) ; 7.12 (t, 1) ; 7.31 (m, 1) ; 7.47 (m, 1) ; 7.53-7.70 (massif, 4) ; 8.00 (d, 2) ; 8.08 (d, 1) ; 8.38-8.48 ( massif, 2) ;9.43 (s,l) ; 9.73 (S/l)
Exemple 46 : { 4- [4- (4-Fluoro-3-methyl-phenγlamino) - pγrimidin-2-γlamino] -phenyl}- (3-pγridyl-oxy-piperidin-l- yl) -methanone racémique .

Suivant le mode opératoire décrit à l'exemple 40, à partir 350 mg de la 3-pyridyl-oxy-piperidin-l-yl utilisée à l'exemple 42 et 580 mg de l'acide obtenu dans la procédure 2a, on obtient 228 mg de produit attendu. MH+ = 485.0 PF = 120 0C RMN (IH, DMSO)
1.42-2.14 (massif, 4) ; 3.37-4.23 (massif, 4) ; 4.57 (m, 1) ; 6.23(d, 1) ; 7.15(t, 2) ; 7.20-8.43 (massif, 11) ; 9.38 (s, 1) ; 9.43 (s, 1) .
Exemple 47 : 4-pyrrolidin-l-ylmethyl-l-{4- [4- (4-fluoro-3- methyl-phenylamino) -pyrimidin-2-ylamino]benzenesulfonyl} - piperidin-4-ol

Stade 1 : A une solution contenant 18.23 g de dimethyloxosulfonium dans methylide et 0.485 g de tetrabutylammonium dans 150 mL de toluène, on additionne goutte à goutte une solution de 4.5 g de soude dans 48 mL d'eau et on laisse le milieu réactionnel sous agitation à 80 0C pendant 3 heures. Après refroidissement, on lave à l'eau, sèche sur Na2SO4 et concentre à sec. On obtient ainsi 13 g d/ epoxyde Stade 2 : Dans un tube scellé, on chauffe à 80 °C pendant 4 heures 1.5 g de l ' époxyde obtenu au stade 1 en présence de 1 g de pyrrolidine dans 25 mL d'éthanol. Après traitement habituel, on obtient 1.5 g d' amino-alcool qui subissent une réaction de décarboxylation pour donner la piperidine-4-methyl-pyrrolidine attendu
Stade 3 : Suivant le mode opératoire décrit au stade 1 de la procédure 3a, à partir de 500 mg de chlorhydrate de chlorure de sulfonyl de la procédure la et de 370 mg de pipéridine obtenu au stade 2, on obtient 140 mg de sulfonamide attendu. MH+ = 526.9 Point de fusion = 148 0C
Exemple 48 : {4- [4- (4-Fluoro-3-methyl-phenylamino) - pyriiαidin-2-ylamino] -phenyl}- (4-pyrrolidin-l-ylmethyl- piperidin-4-ol) -methanone
Suivant le mode opératoire décrit à l'exemple 40, à partir 428 mg de la piperidine-4-methyl-pyrrolidine obtenu au stade 2 de l'exemple 47 et 500 mg de l'acide obtenu dans la procédure 2a, on obtient 280 mg de produit
attendu.

MH+ = 491.1
Point de fusion = 204 0C
Exemple 49 : N2-4- [ (3-{ [bis (lH-pyrazol-4- ylméthyl) amino]méthyl}pipéridin-l-yl) sulfonyl] -phenyl}
N4- (4-méthylphenyl)pyrimidine-2 , 4-diamine

De la même façon qu'à l'exemple 1, à partir de 500 mg du composé de la procédure 3b et de 138 mg de 1-methyl-lH- pyrrole-2-carbaldehyde, on obtient 150 mg de produit attendu
MH+ = 617.1
IH RMN (DMSO) : 1.18 (m,l) ; 1.24-2.88 (massif, 6) ; 2.72 (m, 2) ; 3.27-
3.54 (2d,2) ; 4.11 (t,2) ; 4.25 (d,2) ; 6.45 (d,l) ; 2.08
(t,2) ; 7.46-7.76 (massif, 6) ; 7.83 (s, 4) ; 8.05 (d,l)
Exemple 50 à 78

{4-[4-(4-Fluoro-3-methyl-phenylamino)-
50 526,29 pyrimidin-2-ylamino]-phenyl}[4-(methyl-pyridin- 2-yImethyl-amino)-piperidin-1-yl]-methanone;

{4-[4-(4-Fluoro-3-methyl-phenylamino)- pyrimidin-2-ylamino]-phenyl}-[4-(methyl- pyridin-3-ylmethyl-amino)-piperidin-1-yl]- methanone;
{4-[4-(4-Fluoro-3-methyl-phenylamino)- pyrimidin-2-ylamino]-phenyl}-[4-(methyl- pyridin-4-ylmethyl-amino)-piperidin-1-yl]- methanone;
 {4-[4-(4-Fluoro-3-methyl-phenylamino)- pyrimidin-2-ylamino]-phenyl}-{4-[methyl-(3- methyl-thiophen-2-ylmethyl)-amino]-piperidin-
{4-[4-(4-Fluoro-3-methyl-phenylamino)- pyrimidin-2-ylamino]-phenyl}-{4-[methyl-(3- methyl-thiophen-2-ylmethyl)-amino]-piperidin-
1-yl}-methanone
{4-[4-(4-Fluoro-3-methyl-phenylamino)- pyrimidin-2-ylamino]-phenyl}-{4-[methyl-(5- methyl-thiophen-2-ylmethyl)-amino]-piperidin-
1-yl}-methanone

{4-[(1 ,5-Dimethyl-1 H-pyrazol-4-ylmethyI)- ,.._ 32 methyl-amino]-piperidin-1-yl}-{4-[4-(4-fluoro-3- methyl-phenylamino)-pyrimidin-2-ylamino]- phenyl}-methanone
{4-[4-(4-Fluoro-3-methyl-phenylamino)- pyrimidin-2-ylamino]-phenyl}-{4-[methyl-(5-
529,41 methyl-3H-imidazol-4-ylmethyl)-amino]- piperidin-1 -yl}-methanone;

{4-[4-(4-Fluoro-3-methyl-phenylamino)- pyrimidin-2-ylamino]-phenyl}-{4-[methyl-(3- methyl-3H-imidazol-4-yImethyl)-amino]- piperidin-1 -yl}-methanone;

[4-(Benzo[b]thiophen-3-ylmethyl-methyl- ,-O1 ?o amino)-piperidin-1-yl]-{4-[4-(4-fluoro-3-methyl- phenylamino)-pyrimidin-2-ylamino]-phenyl}- methanone;

{4-[4-(4-Fluoro-3-methyl-phenylamino)-
59 pyrimidin-2-ylamino]-phenyl}-{4-[methyl-(2- methyl-1 H-imidazol-4-ylmethy|)-amino]- piperidin-1 -yl}-methanone;
 {4-[(2,3-Dihydro-benzofuran-5-ylmethyl)-
{4-[(2,3-Dihydro-benzofuran-5-ylmethyl)-
RΠ methyl-amino]-piperidin-1-ylH4-[4-(4-fluoro-3- methyl-phenylamino)-pyrimidin-2-ylamino]- phenyl}-methanone

R1 l}-[4-(methyl- iperidin-1-yl]-

{4-[(4,5-Dimethyl-thiophen-2-ylmethyl)-methyl- amino]-piperidin-1-yl}-{4-[4-(4-fIuoro-3-methyl- phenylamino)-pyrimidin-2-ylamino]-phenyl}- methanone

{4-[(2-Amino-pyridin-3-ylmethyl)-methyl- fi-î 4c: amino]-piperidin-1-yl}-{4-[4-(4-fluoro-3-methyl- | .4S phenylamino)-pyrimidin-2-ylamino]-phenyl}- methanone

RA

[4-(Benzo[1 ,2,5]oxadiazol-5-ylmethyl-methyl-
65 567 29 amino)-piPeridin-1-yl]-{4-[4-(4-fluoro-3-methyl- ' phenylamino)-pyrimidin-2-ylamino]-phenyl}- methanone;

{4-[(2,5-Dimethyl-2H-pyrazol-3-ylmethyl)- RR methyl-amino]-piperidin-1-yl}-{4-[4-(4-fluoro-3- bt>
 methy|-phenylamino)-pyrimidin-2-ylamino]- phenyl}-methanone
{4-[4-(4-Fiuoro-3-methyl-phenylamino)-
methy|-phenylamino)-pyrimidin-2-ylamino]- phenyl}-methanone
{4-[4-(4-Fiuoro-3-methyl-phenylamino)-
„ pyrimidin-2-yla ammiinnoo]]--pphheenny] l}-[4-(methyl- o/ quinolin-8-ylme etthhyyll--aammiinnoo))--piperidin-1-yl]- methanone

{4-[4-(4-Fluoro-3-methyl-phenylamino)-
68 Ψ 507 99 Pyrimidin-2-ylamino]-phenyl}-[4-(methyl- toΛ0 ' pyrimidin-5-ylmethyl-amino)-piperidin-1-yl]- methanone;
{4-[4-(4-Fluoro-3-methyl-phenylamino)- 69 "Ϋ 576 31 pyπmidin-2-ylamino]-phenyl}-[4-(methyl- HOΛ0 ' quinolin-7-ylmethyl-amino)-piperidin-1-yl]- methanone;

7n
70

{4-[4-(4-Fluoro-3-methyl-phenylamino)-
71 FψF ςAn 9Q pyrimidin-2-yla ammiinnoo]]--pphheennyyIl}}--{4-[methyl-(3- Λ methyl-pyridin-2 ?--yyllmmeetthhyyll))--aammiinr o]-piperidin-1 " ° y yllV}-mmeetthhaannoonnee
{4-[4-(4-Fluoro-3-methyl-phenylamino)-
79 FΨF ς7R /A Pyπmidin-2-ylamino]-phenyl}-[4-(isoquinolin-4- Xo °'4 ylmethyl-methyl-amino)-piperidin-1-yl]- methanone

4-{[(1-{4-[4-(4-Fluoro-3-methyl-phenylamino)-
7, CR7 O H pyrimidin-2-ylamino]-benzoyl}-piperidin-4-yl)- ' methyl-amino]-methyl}-1,5-dimethyl-1 H- pyrrole-2-carbonitriIe
 ino)-
ino)-
74 -[methyl-(5- peridin-
{4-[4-(4-Fluoro-3-methyl-phenylamino)-
75 24 pyrimidin-2-ylamino]-phenyl}-[4-(methyl-
/o
 ,/4 thiophen-2-ylmethyl-amino)-piperidin-1-yl]- methanone
,/4 thiophen-2-ylmethyl-amino)-piperidin-1-yl]- methanone
{4-[4-(4-Fluoro-3-methyl-phenylamino)-
76 28 Pyrimidin-2-ylamino]-phenyl}-{4-[methyl-(6- /O methyl-pyridin-2-ylmethyl)-amino]-piperidin-1- yl}-methanone
{4-[4-(4-Fluoro-3-ιmethyl-phenylamino)-
77 9Λ pyrimidin-2-ylamino]-phenyl}-[4-(methyl- thiazol-5-ylmethyl-amino)-piperidin-1-yl]- methanone
{4-[(2,4-Dimethyl-thiazol-5-ylmethyl)-methyl-
78 27 amino]-piperidin-1-yl}-{4-[4-(4-fluoro-3-methyl-
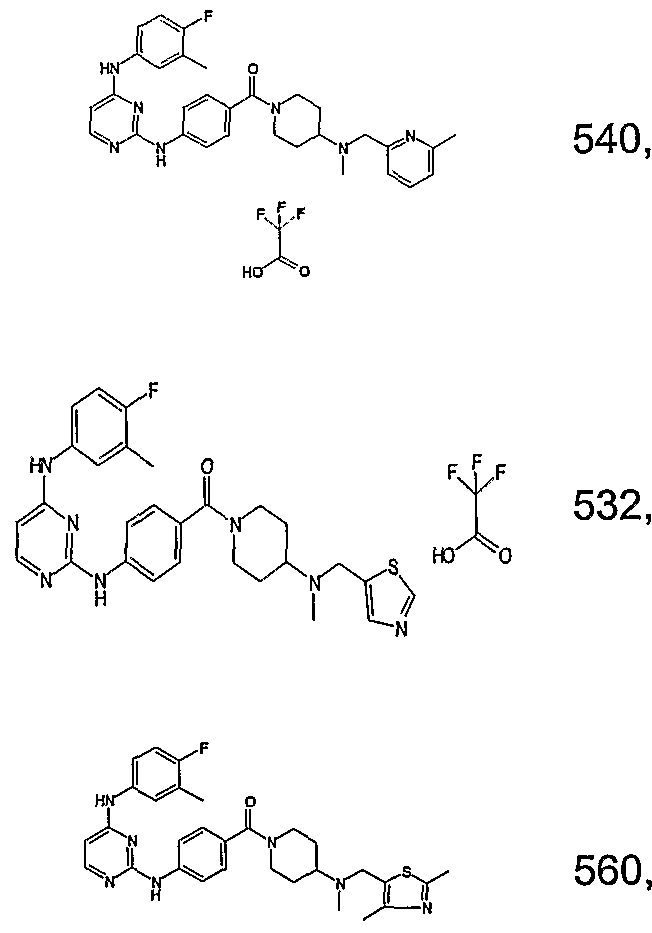 ' phenylamino)-pyrimidin-2-ylamino]-phenyl}-
' phenylamino)-pyrimidin-2-ylamino]-phenyl}-
Φ methanone
Méthodes de purifications Description de la CLHP préparative Description du matériel GILSON utilisé : 2 pompes 306 avec tête de pompe 100SC. 1 amortisseur de pulsation 806.
1 mélangeur 811C avec chambre de mélange 25 ml. 1 injecteur 231XL + racks 21 et vanne d'injection Rheodyne 7000L (boucle de 5ml en acier) . 1 module 401 avec seringue 10ml. 1 « injection valve actuator » 819 avec vanne Rheodyne
7000L utilisé en sélectionneur de colonne.
1 collecteur de fraction 215 muni de 5 racks 207 et vanne 3 voies pour collecte. 1 Détecteur 118 UV/visible. 1 boîtier 506C.
Matériel piloté par le logiciel GILSON 2.0, la collecte s'effectue en fonction de l'absorption du détecteur UV. Colonnes LC de type VP NUCLEODUR GRAVITY 100 - 10 C18 fournit par la société MACHEREY-NAGEL .
basique HCOONH4 (0, QlM) NH3aq pH9-10 Solvants utilisés :
-Eau « milli-Q » HCOONH4 0,01M NH4OH pH9-10. -Acetonitrile pour Gradient CLHP type CHROMANORM Prolabo acide (TFA 0,07%)
Solvants utilisés :
-Eau « milli-Q » à 0,07% de TFA.
-Acetonitrile à 0,07 % de TFA fournit par la société SD

Exemple 79 Le chlorure de sulfonyle de la procédure la, 330 mg, est mis en suspension dans 40 ml de CH2C12 . 186 mg d' amino- alcool commercial (interchim BG206) sont ajouté puis ajoute 0.55 ml de TEA puis laisse agiter à T amb la nuit. On évapore le milieu réactionnel au rotavapeur puis reprend par H2O : 100cm3 et extrait par 3xl00cm3 d'AcOEt ; on réunit les phases AcOEt et évapore au rotavapeur ; On purifie par CLHP prep C18 , évapore le
MeCN puis lyophilise. On obtient 152mg de lyophilisât blanc.
RMN :Comp >95% Masse : Comp >95%
Avec les différents chlorures de sulfonyle des procédures la, Ib ou Ic, en présence des aminés correspondantes, on obtient les composés suivants :



Les composés obtenus dans les exemples 79 , 90 et 91 sont séparés en chromatographie chirale comme dans l' exemple 32 , pour donner respectivement les énantiomères suivants (de configuration absolue non définie) : exemples 92 & 93 ; exemples 94 & 95 ; exemples 96 & 97 . Les pouvoirs rotatoires sont mesurés en utilisant comme solvant le DMSO . Les concentrations sont en mg/mL .
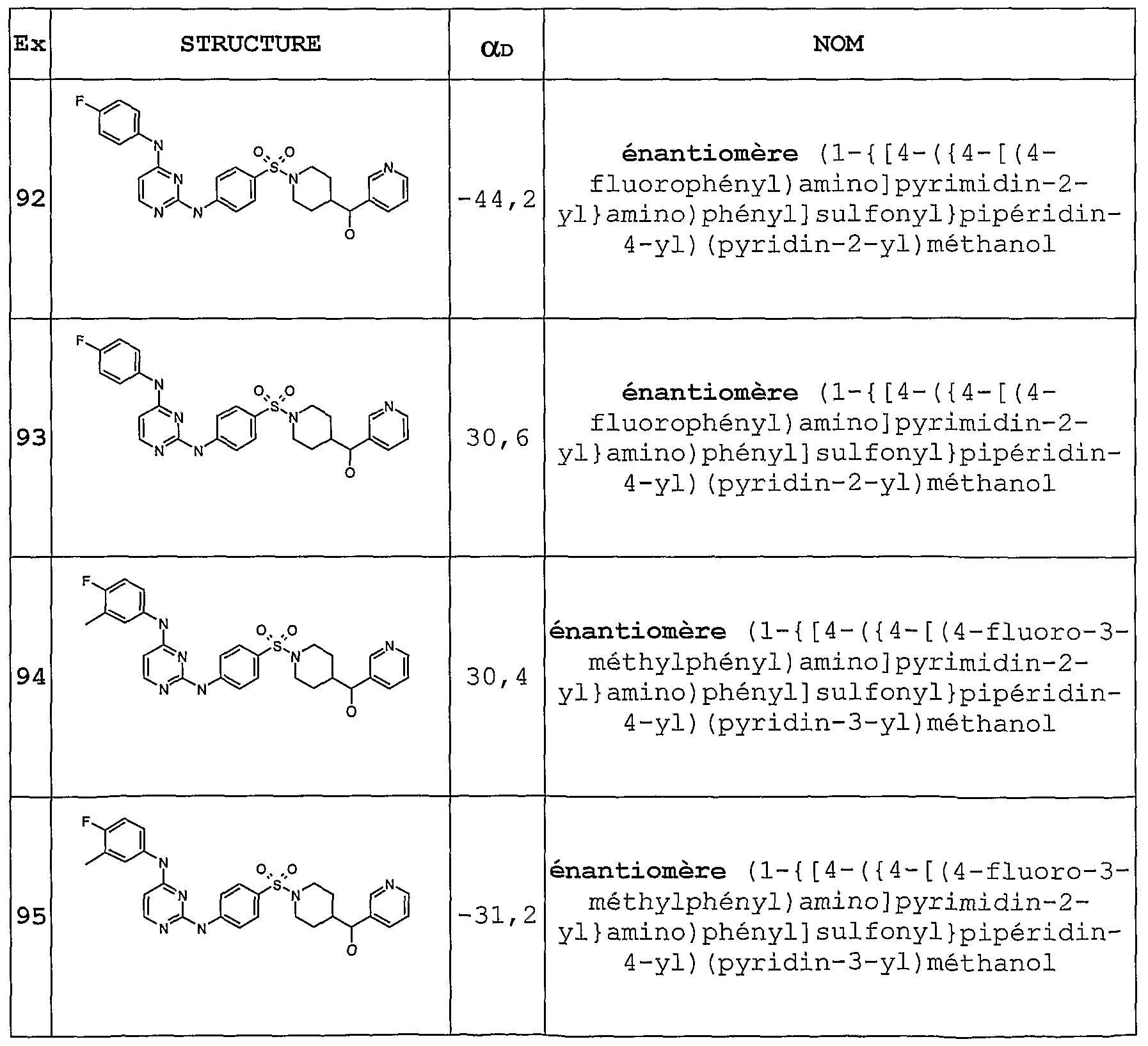

Le composé de l'exemple 88(50 mg) , est dissous dans 5 ml de Methanol et 10 mg de borohydrure de sodium sont ajoutés. Après une heure, 3 mg NaBH4 sont rajoutés et la réaction laissée à température ambiante pour 2h.0n ajoute de l'eau puis évapore à sec et purifie par CLHP en conditions basiques. On obtient 38 mg de poudre blanche, produit attendu (exemple 98) ..
De la même manière les exemples 99,100, 101 sont préparés par réduction des cétones correspondantes.

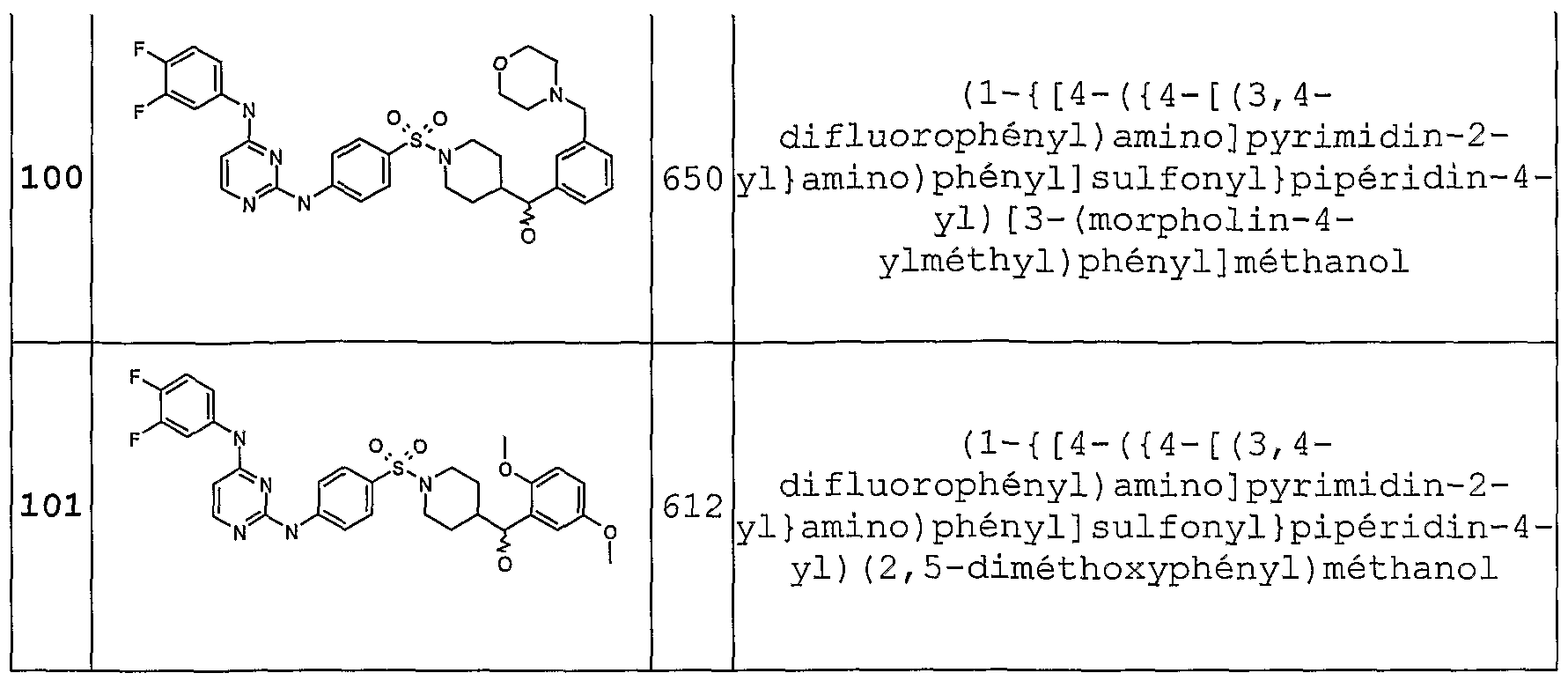
Les cétones peuvent être obtenues selon le schéma de synthèse suivant :
 stade a stade b
stade a stade b
c

Stade d
La N Methyl NMethoxy amide obtenu au stade c, 2Oing, est solubilisé dans le THF, 5 ml, puis quatre équivalents de solution commerciale de bromure de phényle magnésium sont additionnnés . Après une nuit à température ambiante, le milieu est hydrolyse avec une solution de chlorure d'ammonium et extraite par l'acétate d' éthyle puis évaporée. Après purification en CLHP conditions basiques, On obtient 10,7 mg de cétone. (M+H) (+) = 550
Stade c
A 2.06g de composé amide du stade b dans 46 ml de chlorure de méthylène et 3.3 ml de triéthylamine sont ajoutés en plusieurs fois 1.21g de chlorure de sulfonyle de la procédure Ib. Après 2h à température ambiante, le milieu réactionnel est évaporé à sec et le solide blanc obtenu est rincé deux fois avec 30 ml de chlorure de méthylène pour donner après séchage 2.08 g de solide blanc attendu. RMN : 1,53 (m, 2H) ; 1,73 (m, 2H) ; 2,31 (m, 2H) ; 2,62 (m, IH) ; 3,04 (s, 3H) ; 3,59 (s, 3H) ; 3,61 (m, 2H) ; 6,32 (d, J = 6,0 Hz, IH) ; 7,29 (m, IH) ; 7,39 (m, IH) ; 7,61 (d, J = 8,5 Hz, 2H) ; 7,99 (d, J = 8,5 Hz, 2H) ; 8,10 (m, IH) ; 8,13 (d, J = 6,0 Hz, IH) ; 9,68 (s, IH) ; 9,79 (s, IH) . (M+H) (+) = 533
Ce composé intermédiaire constitue en lui même un des exemples de la présente invention (exemple 104) .
Stade b : A l' amide du stade a, 2.6g dans 25 ml de chlorure de méthylène, sont ajoutés 25 ml d'acide trifluoroacétique sous atmosphère inerte. Après 3h à température ambiante, le milieu est évaporé à sec puis déposé en solution dans le méthanol sur une cartouche Varian Mega Bond Elut SCX. Après élution par du méthanol pur, on libère ensuite le produit attendu par élution avec une solution d'ammoniac 7N dans le méthanol. On obtient ainsi après évaporation à sec ,1.64g d'huile jaune.
Stade e : A 2.29g d'acide N-BOC isonipecotic commercial dans 40 ml
de chlorure de méthylène sont ajoutés en plusieurs fois, 1.78g de carbonyle di imidazole et le tout est agité 2.5h à température ambiante. On ajoute alors 1.072 g de chlorhydrate de N,0 dimethoxy hydroxylamine et la réaction est agitée à température ambiante pendant une nuit. Le milieu est lavé à l'eau, puis HCl 0.01N , puis NaHCO3 puis à nouveau à l'eau. Après séchage, évaporation, le brut est purifié sur cartouche de silice avec le mélange éluant chlorure de méthylène/acétate d'éthyle 9/1 puis 8/2. On obtient 2.67g du produit attendu.
Par exemples, les composés suivants sont préparés :
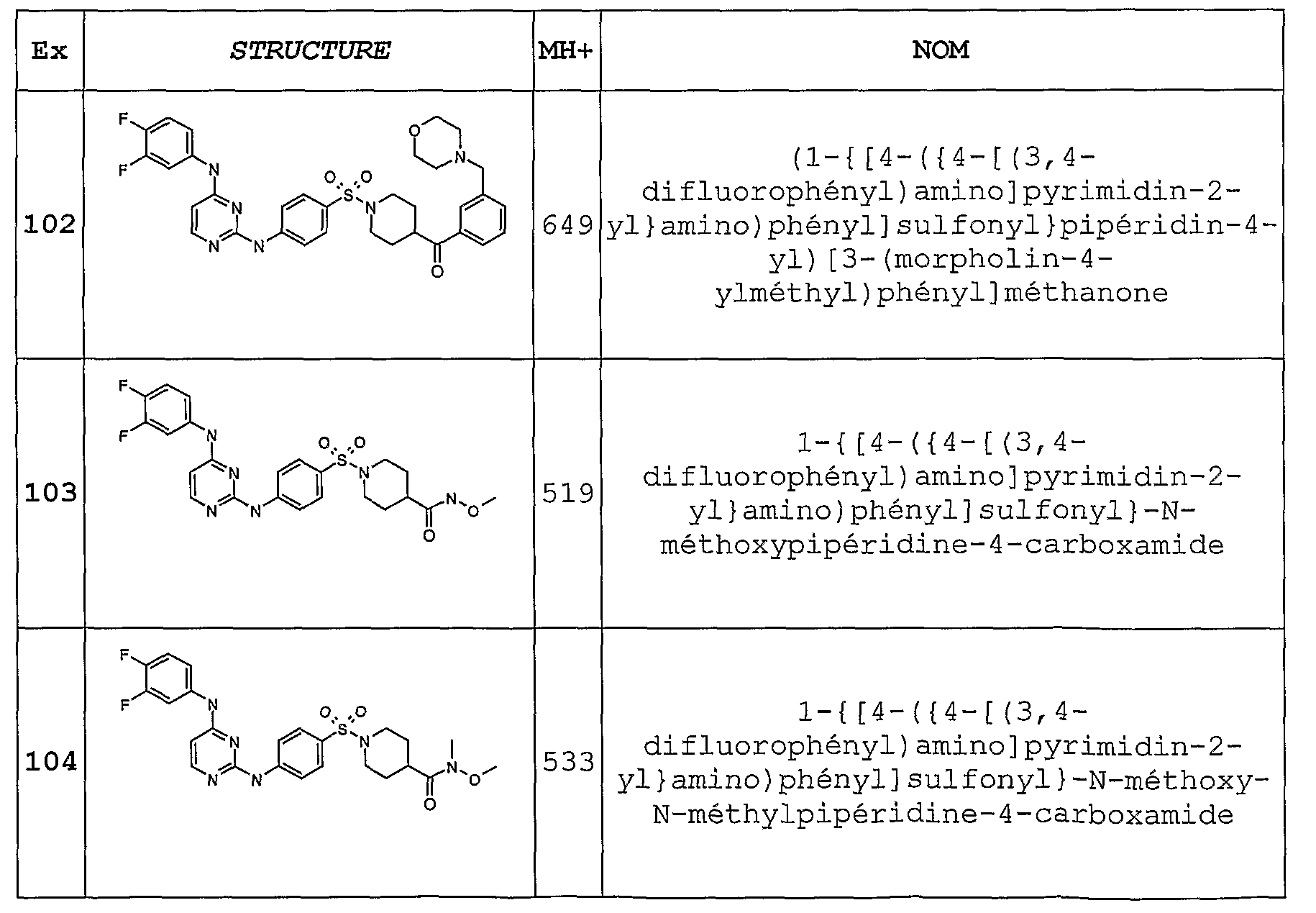
Exemple 105: (l-{ [4- ({4- [ (4-fluorophényl) amino]pyrimidin- 2-yl}amino)phényl] sulfonyl}pipéridin-4-yl) (pyridin-3-yl) - méthanamine
Stade 1 : tert-butyl 4- [ (hydroxyimino) (pyridine-3-
yl) methyl] pipéridine-1-carboxylate
La cétone, 290.1 mg, est mise en solution dans 20 ml d'éthanol. 208.3 mg de chlorhydrate d' hydroxylamine commercial sont ajoutés ainsi que 409.7 mg de NaAcO. La suspension fine obtenue est agitée à TA pendant une nuit. On évapore le mélange réactionnel au rotavapeur sous pression réduite puis reprend par H2O : 30 ml et extrait par 3 x 20 ml d'AcOEt ; On réunit les phases AcOEt et évapore au rotavapeur. On purifie par chromatographie flash en éluant le produit sur une cartouche de 90 g de silice Merck (15-40 μM) par un gradient CH2C12 / CH3OH
(98-2) en 28 mn puis (97-3) en 60 mn avec un débit de 20 ml/mn et un détection à 254 nM. Les fractions collectée, homogènes sont évaporées ensemble au rotavapeur sous pression réduite. On obtient ainsi 116 mg de poudre blanche correspondant à l'isomère Z attendu ainsi que 169 mg d'un deuxième composé correspondant à l'isomère E.
Stade 2 : tert-butyl 4- [amino (pyridin-3- yl) methyl] pipéridine-1-carboxylate A une solution dans 2 ml d'EtOH de 160 mg du dérivé oxime obtenu au stade 1 (isomère E), sont ajoutés 2 ml d'acide acétique glacial et 2 ml d'eau. A la solution obtenue sont ajoutés 171.3 mg de Zinc en poudre. La suspension est agitée par ultra-son pendant un nuit. On évapore le mélange réactionnel au rotavapeur, sous pression réduite puis reprend par du méthanol et dépose la solution méthanolique sur une cartouche Bond élut Varian SCX de 10 g préalablement conditionnée par du MeOH. Après fixation du produit, élution par une solution CH3OH/NH3 (2N) puis évapore en rotavapeur, sous pression réduite, On obtient 123 mg d'une poudre blanche
correspondant au produit attendu.
Stade 3 : l-Pipéridin-4-yl-l-pyridin-3-ylméthanamine Le composé obtenu au stade 2, 234 mg, est mis en solution dans 5 ml de DCM, 3 ml d'CF3CO2H sont ajoutés. La solution limpide jaune obtenue est agitée 2 heures à TA puis est évaporée en rotavapeur sous pression réduite. Reprend par du MeOH et dépose la solution méthanolique sur une cartouche Bond élut Varian SCX de 5 g préalablement conditionnée par du MeOH. Après fixation du produit, élue par une solution CH3OH/NH3 (2N) puis évapore en rotavapeur, sous pression réduite. On obtient 133 mg (87%) d'une huile jaune, correspondant au produit attendu.
Stade 4 : (l-{ [4- ( { 4- [ (4-fluorophényl) amino] pyrimidin-2- yl}amino) phényl] sulfonyl}pipéridin-4-yl) (pyridin-3-yl) - méthanamine
Le chlorure de sulfonyle de la procédure Ic, 273 mg et 133 mg d'aminé obtenu au stade sont mis en suspension dans 10 ml de CH2C12. Coule 447.0 μl de triéthylamine puis laisse sous agitation, à TA pendant une semaine puis évapore le solvant par rotavapeur sous pression réduite. Le produit résiduel est chromatographié sur une cartouche de 25 g de silice Merck (15-40 μM) en éluant par un gradient CH2C12/CH3OH ( 90-10) avec un débit de 30 ml/mn avec une détection à 254 nM. On obtient 113 mg d'un poudre blanche correspondant au produit attendu.
Exemples 106 et 107
Suivant le mode op-êratoire décrit au stade 4 de l'exemple 105, les produits des exemples 106 et 101 sont obtenus à partir du composé de la procédure la et
respectivement les aminés commerciales (R) -Phenyl-1- pipéridin-4-méthanamine et (S) -Phenyl-l-pipéridin-4- méthanamine.

Préparation des exemples 108 et 109 Procédure 1 : préparation des chlorhydrates de chlorure de sulfonyle.
Procédure la : Chlorhydrate de Chlorure de 4- [4- (4- Fluoro-phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyle
Stade 1 : (2-Chloro-pyrimidin-4-yl) - (4-fluoro-phenyl) -aminé A un mélange contenant 15 g de 2-4-ichloropyrimidine dans 200 πiL de n-butanol, sous agitation, on additionne 10 mL de 4-fluoroaniline puis 18 mL de di-isopropyl- ethylamine. Le mélange réactionnel est porté sous agitation, à reflux, pendant 2 heures. Le milieu réactionnel est refroidi, concentré à sec. On ajoute une solution de K2CO3 au résidu et on extrait 3 fois avec de l'acétate d'éthyle, lave avec une solution saturée de NaCl et sèche (Na2SO4 ) . Le brut réactionnel est purifié par chromatographie sur colonne de silice (DCM puis 30% d'acétate d'éthyle dans DCM) . Lors de la concentration
11 g de composé attendu cristallisent MH+ = 224), PF = 172-174 0C
Stade 2 : N-4- (4-Fluoro-phenyl) -N-2-phenyl-pyrimidine- 2, 4-diamine 10,5 g de (2-Chloro-pyrimidin-4-yl) - (4-fluoro- phenyl) -aminé en solution dans 300 mL de n-butanol sont portés à 140 0C à reflux en présence de 4,3 mL d'aniline toute la nuit. Le milieu réactionnel est refroidi. La suspension obtenue est filtrée. Les cristaux sont repris dans l'acétate d' éthyle et lavés par une solution de 10% de K2CO3 puis par une solution saturée de NaCl. Après séchage (Na2SO4) , la phase organique est concentrée sous vide. Le brut réactionnel est purifié par chromatographie sur colonne de silice (THF/MeOH/DCM, 10/5/85) . (Rdt = 10.5 g) .
MH+ = 281, PF = 161 0C
Stade 3: Chlorhydrate de Chlorure de 4- [4- (4-Fluoro- phenylamino) -pyrimidin-2-ylamino] -benzenesulfonyle
Dans un tricol, sous N2, contenant l'acide chlorosulfonique à 0 0C, on additionne par petites portions 7.5 g de N-4- (4-Fluoro-phenyl) -N-2-phenyl- pyrimidine-2, 4-diamine en maintenant la température autour de 0 0C. Le milieu réactionnel est laissé à température ambiante pendant 18 h. Le mélange est versé goutte à goutte (avec précaution) sur la glace. Le précipité obtenu est filtré et lavé avec de l'eau distillée. Après dissolution du solide dans 1 L d'acétate d' éthyle, séchage sur Na2SO4 et concentration sous vide, on obtient un huile blanchâtre. Après ajout de Et2O(100 mL) , les cristaux se forment on obtient par filtration le produit attendu (Rdt = 10,5 g) .
MH+ = 360
Procédure Ib : Chlorhydrate de chlorure de 4-[4-(4- Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] benzenesulfonyle
Stade 1 : (2-Chloro-pyrimidin-4-yl) - (4-fluoro-3-methyl- phenyl) -aminé
La préparation de ce composé se fait suivant le même procédé que pour l'exemple 1 à partir de la réaction de 5.3 g de 4-Fluoro-3-methyl-phenylamine avec 6.3 g de 2,4- Dichloro-pyrimidine : On obtient 3.8 g de produit attendu (Point de fusion= 130-131 0C) ( Trituration dans l ' éther isopropylique) .
Stade 2 : N-4- (4-Fluoro-3-methyl-phenyl) -N-2-phenyl- pyrimidine-2, 4-diamine
La préparation de ce composé se fait suivant le même procédé que pour l'exemple 1 à partir de la réaction de 2.8 g de (2-Chloro-pyrimidin-4-yl) - (4-fluoro-3-methyl- phenyl) -aminé obtenu ci-dessus et de 1.2 mL d'aniline: On obtient 2.2 g de produit attendu (PF = 134-135 0C) ( Trituration dans l ' éther isopropylique) . Stade 3 : Chlorhydrate de chlorure de 4- [4- (4-Fluoro-3- methyl-phenylamino) -pyrimidin-2-ylamino] benzenesulfonyle .
La préparation de ce composé se fait suivant le même procédé que pour l'exemple 1 à partir de 2 g de N-4~(4- Fluoro-3-methyl-phenyl) -N-2-phenyl-pyrimidine-2, 4-diamine obtenu ci-dessus et de l'acide chlorosulfonique : On obtient (Rdt = 1.5 g) .
Procédure 2 : Préparation des aminés 4-Pyrrolidin-l-ylmethyl-piperidin-4-ol
 1-Oxa-β-aza-spiro [2.5] octane-β-carboxylic acid tert-butyl ester
1-Oxa-β-aza-spiro [2.5] octane-β-carboxylic acid tert-butyl ester

A une suspension de 15 g de 4-0xo-piperidine-l-carboxylic acid tert-butyl ester dans 150 mL de toluène, on ajoute
18.22 g d' iodure de trimethylsulfoxonium et 485 mg de bromure de tetrabutylammonium. On additione goutte à goutte une solution de soude 4.5 g dans 20 mL d'eau. On laisse sous agitation pendant 3 h à 80 0C. On reprend par du toluène, on décante, lave à l'eau , sèche et concentre à sec. Après chromatographie sur colonne de silice (DCM / AcOEt : 90 / 10) , on obtient 13 g de produit attendu.
4-Hydroxy~4-pyrrolidin-l-ylmethyl-piperidine-l- carboxylic acid tert-butyl ester

2.2 g de produit obtenu à l'étape précédente sont mis en solution avec 1.46 g de pyrrolidine et 25 mL de EtOH dans un tube scellé. Le milieu réactionnel est chauffé à 75 0C pendant 18 h. Apres concentration à sec, reprise à l'eau, extraction par DCM, séchage et concentration on obtient 2.9 g de produit désiré. Dichlorhydrate de 4-Pyrrolidin-l-ylmethyl-piperidin- 4-ol
D -ans un m-élange dioxane-MeOH (50 mL) , 2.9 g de produit ci- dessus, en présence d'une solution 4 M d'HCl dans du dioxane, on agite 4 h à TA. On concentre sous vide et on triture dans l'ether isopropylique et on filtre le solide
que l'on utilise tel quel dans la réaction de couplage avec le chlorure de sulfonyle.
Exemple 108 : l-{4- [4- (4-Fluoro-phenylamino) -pyrimidin-2- ylamino] -benzenesulfonyl} -4-pyrrolidin-l-ylmethyl- piperidin-4-ol

Dans une solution contenant 500 mg de le chlorhydrate du chlorure de sulfonyle deécrit dans la procédure la et 487 mg de triéthylamine dans 20 mL d'ethanol, on ajoute 372 mg de sel de l'aminé décrit dans la procédure 2 et on laisse sous agitation pendant 3 h. Apres concentration sous vide, on reprend par le DCM et un peu de MeOH., on lave par une solution carbonatée puis par une solution saturée de NaCl. On sèche sur Mg2SO4 puis concentre sous vide. Après chromatographie sur colonne de silice (éluant : DCM / MeOH / NH4OH : 90 / 10 / 2) on obtient 140 mg de produit attendu.
Point de fusion = 148 0C, MH+ = 526.9
IH RMN (DMSO) : 1.47 - 2.10 (massif, 8) ; 2.5 (m, 2) ; 3.5(m, 2) ; 3.19 (d, 2) ; 3.43(d, 2) ; 3.56(d, 2) ; 5.17(sl, 1) 6.56 (d,l) ; 7.26 (t, 2) ; 7.56 - 7.76 (massif, 4) ; 7.82 (d, 2) ; 8.10 ( d, 1) 9.85 (si, 1) ; 11.07 (si, 1)
Exemple 109 : l- { 4- [4- (4-Fluoro~3-methyl~phenylamino) - pyrimidin-2-ylamino] -benzenesulfonyl } -4-pyrrolidin-l- ylmefchyl-piperidin-4-ol

On procède comme pour l'exemple 108 à partir de 500 mg de chlorhydrate du chlorure de sulfonyle décrit dans la procédure Ib et de 359 mg d'aminé décrit dans la procédure 2, on obtient 210 mg de produit attendu.
Point de fusion = 169 0C, MH+ = 541.1
IH RMN (DMSO) : 1.25-1.75 (massif, 8) ; 2.19 (Is, 3) ; 2.27 (Is, 2) ; 2.33-2.64 (massif, 6) ; 3.31(m,2) ; 3.97 (s,l) ; 6.25 (d,l) ; 7.07 (t,l) ; 7.41 (m,l) ; 7.48-
7.65 (massif, 3) 7.93(d,21 8.03(d,i; 9.37 (s,l) 9.65(s,l)
Exemple 110 : Composition pharmaceutique
On a préparé des comprimés répondant la formule suivante :
Produit de l ' exemple 6 0,2 g
Excipient pour un comprimé terminé à I g
(détail de l'excipient : lactose, talc, amidon, stéarate de magnésium) .
Exemple 111 : Composition pharmaceutique
On a préparé des comprimés répondant à la formule suivante :
Produit de l ' exemple 105 • 0,2 g
Excipient pour un comprimé terminé à , 1 g
(détail de l'excipient : lactose, talc, amidon, stéarate de magnésium) .
Les exemples 6 et 105 sont pris à titre d'exemples dans les préparations pharmaceutiques qui constituent les exemples 110 et 111 ci-dessus, cette préparation pharmaceutique pouvant être réalisée différemment comme indiqué ci-dessus et si désiré avec d'autres produits en exemples dans la présente demande.
Partie pharmacologique :
Protocoles d'essais biochimiques sur IKK.
I) Evaluation des composés sur IKKl et IKK2 : Les composés sont testés pour l'inhibition de IKKl et IKK2 en utilisant un test kinase sur support flash-plate. Les composés à tester sont dissous à 10 mM dans du DMSO puis dilués dans du tampon kinase (50 mM Tris, pH 7.4 contenant 0.1 mM EGTA, 0.1 mM sodium orthovanadate et 0.1% de p- mercaptoéthanol) .
Des dilutions en série de 3 en 3 sont réalisées à partir de cette solution. 10 μl de chaque dilution sont ajouté dans les puits d'une plaque 96 puits en duplicata. 10 μl de tampon kinase est ajouté dans les puits contrôles qui serviront de 0% inhibition et 10 μl de 0.5 mM EDTA est ajouté aux puits contrôles (100% d'inhibition) . 10 μl du mélange IKKl ou IKK2 (0.1 μg/puits) , peptide substrat 25-55 IKB-biotinilé et BSA (5 μg) est ajouté à chaque puit . Pour démarrer la réaction kinase, 10 μl du mélange de 10 mM magnésium acétate, 1 μM ATP froid et 0.1 μCi 33P- ATP est ajouté à chaque puit pour un volume final de 30 μl. La réaction est incubée à 30 °C pendant 90 min puis stoppée par l'ajout de 40 μl de 0.5 mM EDTA. Après agitation, 50 μl sont transférés vers une plaque flash- plate recouverte de streptavidine .
30 min après, les puits sont lavés 2 fois par une solution de 50 mM Tris-EDTA pH7.5 et la radioactivité déterminée sur un compteur microbeta.
Les composés de l'invention testés dans cet essai montrent une IC50 inférieure à 10 μM, ce qui montre qu'il peuvent être utilisés pour leur activité thérapeutique.
II) Evaluation des composés sur la viabilité et la prolifération des cellules tumorales:
Les composés selon l'invention ont fait l'objet d'essais pharmacologiques permettant de déterminer leur activité anticancéreuse .
Les composés de formule (I) selon la présente invention ont été testés in vitro sur un panel de lignées tumorales d'origine humaine provenant :
- de cancer du sein: MDA-MB231 (American Type culture collection, Rockville, Maryland, USA, ATCC-HTB26) , MDA-Al ou MDA-ADR (dite lignée multi-drug résistant MDR, et décrite par E.Collomb et al., dans Cytometry, 12(1) : 15- 25, 1991), et MCF7 (ATCC-HTB22) ,
- de cancer de la prostate: DU145 (ATCC-HTB81) et PC3 (ATCC-CRL1435) ,
- de cancer du colon: HCT116 (ATCC-CCL247) et HCT15 (ATCC-CCL225) ,
- de cancer du poumon: H460 (décrite par Carmichael dans Cancer Research 47 (4) : 936-942, 1987 et délivré par le National Cancer institute, Frederick Cancer Research and Development Center, Frederick, Maryland, USA) , - de glioblastome (SF268 décrite par Westphal dans Biochemical & Biophysical Research Communications 132 (1) : 284-289, 1985 et délivré par le National Cancer institute, Frederick Cancer Research and Development Center, Frederick, Maryland, USA) , - de leucémie (CMLTl décrite par Kuriyama et al. dans Blood, 74: 1989, 1381-1387, par Soda et al. dans British Journal of Haematology, 59: 1985, 671-679 et par Drexler, dans Leukemia Research, 18: 1994, 919-927 et délivré par la société DSMZ, Mascheroder Weg Ib, 38124 Braunschweig, Germany) .
La prolifération et la viabilité cellulaire ont été déterminées dans un test utilisant le 3-(4,5- diméthylthiazol-2-yl) -5- (3-carboxyméthoxyphényl) -2- (4- sulfophényl) -2H-tétrazolium (MTS) selon Fujishita T. et al., Oncology, 2003, 64 (4), 399-406. Dans ce test, on mesure la capacité mitochondriale des cellules vivantes à transformer le MTS en un composé coloré après 72 heures d'incubation d'un composé de formule (I) selon l'invention. Les concentrations en composé selon l'invention, qui conduisent à 50 % de perte de prolifération et de viabilité cellulaire (CI50) sont inférieure à 10 μM, selon la lignée tumorale et le composé testé. Ainsi, selon la présente invention, il apparaît que les composés de formule (I) entraînent une perte de prolifération et de viabilité des cellules tumorales avec une IC50 inférieure à 10 μM.
Claims
REVENDICATIONS
1) Produits de formule (I)

dans laquelle:
R2, R3 et R4, identiques ou différents, sont tels que l'un représente un atome d'halogène ou CF3 et les deux autres, identiques ou différents, représentent un atome d'hydrogène ou un atome d'halogène ou un radical alkyle ou un radical alcoxy éventuellement substitués par un ou plusieurs atomes d'halogène;
R5 représente un atome d'hydrogène ou un atome d' halogène;
Z représente CO ou S02 ; le cycle (N) soit

Rl représente -X1-R7 avec Xl représente -(CH2)m- et R7 représente un cycle hétérocycloalkyle, aryle ou hétéroaryle, tous éventuellement substitués ; et R6 représente l'atome d'hydrogène ou les radicaux hydroxyle, -(CH2)mOH, -CO-NRaRb, -CH2-NraRb, -CO2H, et -
C02al k;
ii) Rl représente -X2-R7 avec X2 représente : -O- ; -O-(CH2)m- ; -CH (OH) - (CH2) n-; -CO- ; -CO-NRc- ; -CO-NRc-O- ; -CH(NRaRb)- ; -C=NOH- ; -C=N-NH2- ; - (CH2)nl-NRc- (CH2)n2- ; et R7 représente un cycle hétérocycloalkyle, aryle, ou hétéroaryle, tous éventuellement substitués ; et Rβ représente hydrogène ;
iii) Rl représente -NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, -OaIk, -CF3, -CO-NR8R9 et SO2-alk ; et R6 représente hydrogène ; étant entendu que lorsque W représente un atome d' hydrogène alors z représente CO ; iv) Rl représente -CH2-NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone et éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, -OEt, -CF3, -CO- N(alk)2 et S02-alk ; et Rβ représente hydrogène ;
v) Rl représente -CO-N (Rc) -OR' c et Rβ représente hydrogène ;
avec n, ni et n2, identiques ou différents, représentent un entier de 0 à 3 ; m représente un entier de 1 à 3 •;
Rc et R' c, identiques ou différents, représentent l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone éventuellement substitué par un ou plusieurs atomes d'halogène;
NRaRb est tel que soit Ra et Rb, identiques ou différents, représentent l'atome d'hydrogène ou un
radical alkyle renfermant de 1 à 4 atomes de carbone ou un radical cycloalkyle, ces radicaux alkyle et cycloalkyle étant éventuellement substitués par un ou plusieurs atomes d'halogène, un radical hydroxyle ou un radical NH2, NHalkyle ou N (alkyle) 2; soit Ra et Rb forment avec l'atome d'azote auxquels ils sont liés une aminé cyclique pouvant éventuellement renfermer un ou deux autres hétéroatomes choisis parmi 0, S, N ou NRlO, l'aminé cyclique ainsi formée étant elle-même éventuellement substituée par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux alkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ; tous les radicaux hétérocycloalkyle, aryle et hétéroaryle étant éventuellement substitués par un ou plusieurs radicaux, identiques ou différents, choisis parmi les atomes d'halogène ; les radicaux hydroxyle ; cyano ; NR8R9 ; et les radicaux alkyle, cycloalkyle, alcoxy, phényle, hétérocycloalkyle et hétéroaryle, eux-mêmes éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, alkyle, hydroxyalkyle, alcoxyalkyle, CN, CF3, OCF3, ou NRaRb;
NR8R9 est tel que soit R8 et R9, identiques ou différents, sont tels que R8 représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone ou un radical cycloalkyle, ces radicaux alkyle et cycloalkyle étant éventuellement substitués par un ou plusieurs atomes d'halogène, un radical hydroxyle ou un radical NH2, NHalkyle ou N (alkyle) 2; et R9 représente l'atome d'hydrogène et les radicaux alkyle, cycloalkyle ou hétérocycloalkyle eux- mêmes éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les
atomes d'halogène et les radicaux hydroxyle, alcoxy, NH2, NHalkyle, N (alkyle) 2, les radicaux alkyle que représente R9 étant de plus éventuellement substitués par un radical phényle, hétérocycloalkyle ou hétéroaryle eux-mêmes éventuellement substitués par un ou plusieurs radicaux choisis parmi les atomes d'halogène et les radicaux radicaux hydroxyle, alcoxy, alkyle, hydroxyalkyle, alcoxyalkyle, CN, CF3, OCF3, NH2, NHaIk ou N(alk)2 ; soit R8 et R9 forment avec l'atome d'azote auxquels ils sont liés une aminé cyclique pouvant éventuellement renfermer un ou deux autres hétéroatomes choisis parmi 0, S, N ou NRlO, l'aminé cyclique ainsi formée étant elle- même éventuellement substituée par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux alkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ; tous les radicaux hétérocycloalkyle et hétéroaryle ci- dessus éventuellement substitués comme indiqué ci-dessus, étant constitués de 4 à 10 chaînons et renfermant 1 à 3 hétéroatomes choisi (s) parmi 0, S, N et NRlO ; RIO représente un atome d'hydrogène ou un radical alkyle, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (D -
2) Produits de formule (I) tels que définis à la revendication 1 dans lesquels R2, R3, R4 , R5, Z et le cycle (N) ont les significations indiquées à l'une quelconque des autres revendications et Rl et R6 sont tels que Rl représente -X1-R7 avec Xl représente -(CH2)m- et R7 représente un cycle hétérocycloalkyle, aryle ou hétéroaryle, tous éventuellement substitués ;
et R6 représente l'atome d'hydrogène ou les radicaux hydroxyle, -(CH2)mOH, -CO-NRaRb, ~CH2-NraRb, -CO2H, et - CO2alk; avec m,n et NRaRb tels que définis à l'une quelconque des autres revendications et les radicaux hétérocycloalkyle, aryle et hétéroaryle étant éventuellement substitués par un ou plusieurs radicaux, identiques ou différents, tels que définis à l'une quelconque des autres revendications, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule
(D .
3) Produits de formule (I) tels que définis à l'une quelconque des autres revendications dans lesquels R2, R3, R4, R5, Z et le cycle (N) ont les significations indiquées à l'une quelconque des autres revendications et Rl et R6 sont tels que Rl représente -X2-R7 avec X2 représente : -O-;-O-(CH2)m-;-CH(OH)-(CH2)n-; -CO-; -CO-NRc-; -CO-NRc-O-; -CH(NRaRb)-; -C=NOH-; -C=N-NH2-; - (CH2)nl-NRc- (CH2)n2~; et R7 représente un cycle hétérocycloalkyle, aryle, ou hétéroaryle, tous éventuellement substitués ; et Rβ représente hydrogène ; avec n, ni, n2, Rc et NRaRb tels que définis à l'une quelconque des autres revendications et les radicaux hétérocycloalkyle, aryle et hétéroaryle étant éventuellement substitués par un ou plusieurs radicaux, identiques ou différents, tels que définis à l'une quelconque des autres revendications, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les
acides minéraux et organiques desdits produits de formule (D •
4) Produits de formule (I) tels que définis ci-dessus dans lesquels R2, R3, R4, R5, Z et le cycle (N) ont les significations indiquées à l'une quelconque des autres revendications et Rl et R6 sont tels que : soit Rl représente -NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, -OaIk, -CF3, -CO-NR8R9 et SO2-alk et R6 représente hydrogène, étant entendu que lorsque W représente un atome d'hydrogène alors z représente CO; soit Rl représente -CH2-NRc-W avec W représente l'atome d/ hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone et éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, -OEt, -CF3, -CO- N(alk)2 et S02-alk ; et R6 représente hydrogène ; soit Rl représente -CO-N (Rc) -OR' c et Rβ représente hydrogène ; avec Rc, R' c et NR8R9 tels que définis à l'une quelconque des revendications, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (I) .
5) Produits de formule (I) tels que définis à l'une quelconque des autres revendications dans lesquels R2, R3, R4, R5 et Z ont les significations indiquées à l'une quelconque des autres revendications et le cycle (N) représente l'un des cycles définis ci-après :
- un cycle azétidinyle ou pyrrolidinyle substitué en position 3 par Rl et R6 tels que définis à l'une quelconque des autres revendications; un cycle pipéridinyle et azépinyle substitués en position 3 ou 4 par Rl et Rβ tels que définis à l'une quelconque des autres revendications; un cycle 8 aza bicyclo (3, 2, 1) octan- 3-yl, 6- azabicyclo [3.2.1] octan-3-yl ou 3-azabicyclo [3.2.1] octan- 8yl); lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (D • 6) Produits de formule (I) tels que définis à l'une quelconque des autres revendications dans lesquels R2, R3, R4, R5 et Z ont les significations indiquées à l'une quelconque des autres revendications et le cycle (N) représente un cycle pyrrolidinyle substitué en 3 par Rl et Rβ ou un cycle pipéridinyle substitué en position 3 ou 4 par Rl et R6, avec Rl et Rβ tels que définis à l'une quelconque des autres revendications, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (I) .
7) Produits de formule (I) tels que définis à l'une quelconque des autres revendications dans lesquels: R2, R3 et R4, identiques ou différents, sont tels que l'un représente un atome d'halogène ou CF3 et les deux autres, identiques ou différents, représentent un atome d'hydrogène ou un atome d'halogène ou un radical alkyle ou un radical alcoxy éventuellement substitués par un ou plusieurs atomes d'halogène;
R5 représente un atome d'hydrogène ou un atome d' halogène;
Z représente CO ou S02 ; le cycle (N) soit

ii) Rl représente -X2-R7 avec X2 représente :
-0-,-CH(OH)-, -CH(OH)-CH2-, -CO-, -CH(NRaRb)-, -C=NOH-, - C=N-NH2- et - (CH2) nl-NRc- (CH2) n2-,
et R7 représente un cycle hétérocycloalkyle, phényle ou hétéroaryle, tous éventuellement substitués,
et R6 représente hydrogène ; iii) Rl représente -NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone linéaire ou ramifié éventuellement substitué par un radical choisi parmi -PO (OEt) 2, -OH, - OEt, -CF3,-CO-NR8R9 et SO2-alk ; et R6 représente hydrogène ; étant entendu que lorsque W représente un atome d'hydrogène alors z représente CO;
iv) Rl représente -CH2-NRc-W avec W représente l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4
atomes de carbone linéaire ou ramifié à partir de 3 atomes de carbone et éventuellement substitué par un radical S02-alk ; et R6 représente hydrogène ; v) Rl représente -CO-N(Rc)-ORO et Rβ représente hydrogène ;
avec n, ni et n2 , identiques ou différents, représentent un entier de 0 à 2;
Rc et R' c identiques ou différents représentent l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 2 atomes de carbone ;
NRaRb est tel que soit Ra et Rb, identiques ou différents, représentent l'atome d'hydrogène ou un radical alkyle renfermant de 1 à 4 atomes de carbone éventuellement substitué par un ou plusieurs atomes d'halogène, un radical hydroxyle ou un radical NH2, NHalkyle ou N (alkyle) 2; soit Ra et Rb forment avec l'atome d'azote auxquels ils sont liés un radical morpholinyle ou pyrrolidinyle éventuellement substitué par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux alkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ; tous les radicaux hétérocycloalkyle, phényle et hétéroaryle étant éventuellement substitués par un ou plusieurs radicaux, identiques ou différents, choisis parmi les atomes d'halogène ; les radicaux hydroxyle ; cyano ; NR8R9 ; et les radicaux alkyle, cycloalkyle, alcoxy, phényle, hétérocycloalkyle et hétéroaryle, eux- mêmes éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, OCF3, CH3, -CH2OH, CN, CF3, OCF3 ou NRaRb; NR8R9 est tel que soit R8 et R9, identiques ou différents, sont tels que R8 représente l'atome d'hydrogène, un radical alkyle linéaire ou ramifié
renfermant au plus 4 atomes de carbone ou un radical cycloalkyle renfermant de 3 à 6 chaînons , alkyle et cycloalkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ou un radical hydroxyle ; et R9 représente l'atome d'hydrogène ou un radical alkyle éventuellement substitué par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, alcoxy, NH2, NHalkyle, N (alkyle) 2, phényle, hétérocycloalkyle ou hétéroaryle eux-mêmes éventuellement substitués par un ou plusieurs radicaux choisis parmi les atomes d'halogène et les radicaux radicaux hydroxyle, OCH3, CH3, -CH2OH, CN, CF3, OCF3, NH2, NHaIk ou N(alk)2 ; soit R8 et R9 forment avec l'atome d'azote auxquels ils sont liés une aminé cyclique choisie parmi pyrrolyle, pipéridyle, morpholinyle, pyrrolidinyle, azétidinyle et pipérazinyle éventuellement substitués par un ou plusieurs radicaux alkyle eux-mêmes éventuellement substitués par un ou plusieurs atomes d'halogène ; lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (I) • 8) Produits de formule (I) tels que définis à l'une quelconque des autres revendications dans lesquels Z, le cycle (N), Rl et R6 ont les significations indiquées à l'une quelconque des autres revendications; R2, R3 et R4 , identiques ou différents, sont tels que l'un représente un atome d'halogène ou CF3 et les deux autres, identiques ou différents, représentent un atome d'hydrogène, un atome d'halogène ou un radical méthyle, méthoxy, trifluorométhyle ou trifluorométhoxy; et R5 représente un atome d'hydrogène; lesdits produits de formule (I) étant sous toutes les
formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (D • 9) Produits de formule (I) tels que définis à l'une quelconque des autres revendications dans lesquels Z, le cycle (N) , Rl et R6 ont les significations indiquées à l'une quelconque des autres revendications et R2, R3 et R4, identiques ou différents, sont tels que l'un représente un atome de fluor et les deux autres, identiques ou différents, représentent un atome d'hydrogène, un atome de fluor ou un radical méthyle ;
R5 représente un atome d'hydrogène; lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (D •
10) Produits de formule (I) tels que définis ci-dessus dans lesquels Rl, R2, R3, R4, R5, R6 et le cycle (N) ont les significations indiquées ci-dessus ou ci-après et Z représente S02, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (I) .
11) Produits de formule (I) tels que définis à l'une quelconque des autres revendications dans lesquels Rl, R2, R3, R4, R5, R6 et le cycle (N) ont les significations indiquées à l'une quelconque des autres revendications et Z représente CO, lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits
produits de formule (I) .
12) Produits de formule (I) tels que définis à l'une quelconque des autres revendications dans lesquels R2, R3, R4, R5, Z et le cycle (N) ont les significations indiquées à l'une quelconque des revendications et Rl et Rβ sont tels que : soit Rl représente -X1-R7 avec Xl représente -CH2- et R6 représente l'atome d'hydrogène ou les radicaux hydroxyle, CH2-OH; -CO-N (CH3) 2, -CO-NHCH3, - CO-NH- (CH2) 2-N(CH3) 2 et -C02Et; soit Rl représente -X2-R7 avec X2 représente: -0-, -CHOH-, -CH (OH) -CH2-, -CO- , - CHNH2-, -NH-CH2-, -N(CH3)-CH2- et CH2-NH-CH2-; et R6 représente hydrogène; et R7 est choisi parmi les radicaux pyrrolidinyle, pipéridinyle, pipérazinyle, pyrimidinyle, morpholinyle, thiomorpholinyle, tetrahydrofuranyle, hexaydrofuranyle, phényle, pyridyle, thiényle, thiazolyle, dithiazolyle, pyrazolyle, pyrazinyle, furyle, imidazolyle, pyrrolyle, oxazolyle, isoxazolyle, benzodihydrofuranyle, benzoxodiazolyle, benzothiodiazolyle, benzothiényle, quinolyle, isoquinolyle; tous ces radicaux que représente R7 étant éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, méthyle, méthoxy, hydroxyméthyle, alcoxyméthyle, cyano, NH2, NHaIk, et N(alk)2, -CH2-NH2, - CH2-NHalk, -CH2-N (alk) 2, phényle, morpholinyle et CH2-morpholinyle eux-mêmes éventuellement substitués par un ou plusieurs radicaux identiques ou différents choisis parmi les atomes d'halogène et les radicaux hydroxyle, CH3, OCH3, -CH2OH, CN, CF3, 0CF3, NH2, NHaIk ou N (alk) 2; lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (I) .
13) Produits de formule (I) tels que définis à l'une quelconque des autres revendications répondant aux noms suivants :
-{ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl}- [4- (methyl-oxazol-2-ylmethyl-amino) -piperidin-1- yl] -methanone
- {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl}- [3- (methyl-lH-pyrrole-2-ylmethyl-amino) - piperidin-1-yl] -methanone (Racémique) - l-{4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] - benzoyl } ~3-pyridin-3-ylmethyl-piperidine-3-carboxylic acid methylamide (Racémique)
- N*4*-(4-Fluoro-3-methyl phenyl) -N*2*- (4-{ 3- [ (2- methanesulfonyl-ethylamino) -methyl] -pyrrolidine-1- sulfonyl} -phenyl) -pyrimidine-2, 4-diamine (Racémique)
- N*4*- (4-Fluoro-3-methyl-phenyl) -N*2*- [4- (3-{ [ (1- methyl-lH-pyrrol-2-ylmethyl) -amino] -methyl} -pyrrolidine- 1-sulfonyl) -phenyl] -pyrimidine-2, 4-diamine (Racémique)
- 4-pyrrolidin-l-ylmethyl-l-{4- [4- (4-fluoro-3-methyl- phenylamino) -pyrimidin-2-ylamino] benzenesulfonyl} - piperidin-4-ol
- {4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -phenyl} [4- (methyl-pyridin-2-ylmethyl-amino) - piperidin-1-yl] -methanone; - {4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -phenyl}- [4- (methyl-pyridin-4-ylmethyl-amino) - piperidin-1-yl] -methanone;
- {4- [ (1, 5-Dimethyl-lH-pyrazol-4-ylmethyl) -methyl-amino] - piperidin-l-yl}-{4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone
- {4-[ (2-Amino-pyridin-3-ylmethyl) -methyl-amino] - piperidin-l-yl}-{4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone
- 4-{ [ (l-{4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin- 2-ylamino] -benzoyl}-piperidin-4-yl) -methyl-amino] - methyl}-l, 5-dimethyl-lH-pyrrole-2-carbonitrile
- { 4- [ (2, 4-Dimethyl-thiazol-5-ylmethyl) -methyl-amino] - piperidin-l-yl}-{4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone
- (l-{ [4- ( { 4- [ (4-fluorophényl) amino] pyrimidin-2- yl}amino)phényl] sulfonyl}pipéridin-4-yl) (pyridin-3-yl) - méthanamine - l-{ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - benzènesuifonyl}-4-pyrrolidin-l-ylmethyl-piperidin-4-ol l-{ 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -benzenesulfonyl } -4-pyrrolidin-l-ylmethyl- piperidin-4-ol lesdits produits de formule (I) étant sous toutes les formes isomères possibles racémiques, énantiomères et diastéréoisomères, ainsi que les sels d'addition avec les acides minéraux et organiques desdits produits de formule (D • 14) Procédé de préparation des produits de formule (I) tels que définis à l'une quelconque des autres revendications caractérisé en ce que l'on fait réagir un produit de formule (II) :



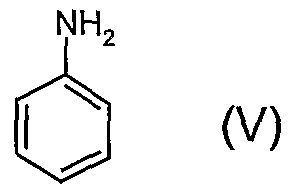

voie a) z=SO2 produit de formule (VI) que l'on fait réagir avec de l'acide chlorosulfonique SO2(OH)C1 pour obtenir le produit correspondant de formule (VII) :
 dans laquelle R2' , R3' , R4' et R5' ont les significations indiquées ci-dessus, produit de formule (VII) que l'on fait réagir avec une aminé de formule (VIII) :
dans laquelle R2' , R3' , R4' et R5' ont les significations indiquées ci-dessus, produit de formule (VII) que l'on fait réagir avec une aminé de formule (VIII) :
(VIII)
 ans uelle Rl' et R6' ont les significations indiquées à l'une quelconque des revendications ci-dessus respectivement pour Rl et R6, dans lesquelles les éventuelles fonctions réactives sont éventuellement protégées par des groupements protecteurs, pour obtenir un produit de formule (Ia) :
ans uelle Rl' et R6' ont les significations indiquées à l'une quelconque des revendications ci-dessus respectivement pour Rl et R6, dans lesquelles les éventuelles fonctions réactives sont éventuellement protégées par des groupements protecteurs, pour obtenir un produit de formule (Ia) :

voie b) Z=CO produit de formule (IV) tel que défini ci- dessus que l'on fait réagir avec l'ester méthylique de l'acide 4-amino benzoique pour obtenir le produit de formule (IX) :
 dans laquelle R2' , R3' , R4' et R5' ont les significations indiquées ci-dessus, produit de formule (IX) que l'on saponifie en son acide correspondant de formule (X) :
dans laquelle R2' , R3' , R4' et R5' ont les significations indiquées ci-dessus, produit de formule (IX) que l'on saponifie en son acide correspondant de formule (X) :


15) A titre de médicaments, les produits de formule (I) telle que définie à l'une quelconque des revendications 1 à 13 ainsi que les sels d'addition avec les acides minéraux et organiques pharmaceutiquement acceptables desdits produits de formule (I) .
16) A titre de médicaments, les produits de formule (I) telle que définie à la revendication 13 répondant aux noms suivants :
-{ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl}- [4- (naethyl-oxazol-2-ylmethyl-amino) -piperidin-1- yl] -methanone - {4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] - phenyl}- [3- (methyl-lH-pyrrole-2-ylmethyl-amino) - piperidin-1-yl] -methanone (Racémique)
- l-{4- [4- (4-Fluorophenylamino) -pyrimidin-2-ylamino] - benzoyl } -3-pyridin-3-ylmethyl-piperidine-3~carboxylic
acid methylamide (Racémi.que)
- N*4*-(4-Fluoro-3-methyl phenyl) -N*2*- (4-{ 3- [ (2- methanesulfonyl-ethylamino) -methyl] -pyrrolidine-1- sulfonyl} -phenyl) -pyrirαidlne-2, 4-diamine (Racémique) _ N*4*-(4-Fluoro-3-methyl-phenyl)-N*2*-[4-(3-{ [ (1- methyl-lH-pyrrol-2-ylmethyl) -amino] -methyl }-pyrrolidine- 1-sulfonyl) -phenyl] -pyrimidine-2, 4-diamine (Racémique)
- 4-pyrrolidin-l-ylmethyl-l-{4- [4- (4-fluoro-3-methyl- phenylamino) -pyrimidin-2-ylamino] benzenesulfonyl } - piperidin-4-ol
- { 4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -phenyl} [4- (methyl-pyridin-2-ylmethyl-amino) - piperidin-1-yl] -methanone;
- {4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin-2- ylamino] -phenyl}- [4- (methyl-pyridin-4-ylmethyl-amino) - piperidin-1-yl] -methanone;
- { 4- [ (1, 5-Dimethyl-lH-pyrazol-4-ylmethyl) -methyl-amino] - piperidin-l-yl}-{4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone - { 4- [ (2-Amino-pyridin-3-ylmethyl) -methyl-amino] - piperidin-l-yl}-{4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone
- 4-{ [ (l-{4- [4- (4-Fluoro-3-methyl-phenylamino) -pyrimidin- 2-ylamino] -benzoyl } -piperidin-4-yl) -methyl-amino] - methyl }-l, 5-dimethyl-lH-pyrrole-2-carbonitrile
- {4- [ (2, 4-Dimethyl-thiazol-5-ylmethyl) -methyl-amino] - piperidin-l-yl}-{4- [4- (4-fluoro-3-methyl-phenylamino) - pyrimidin-2-ylamino] -phenyl } -methanone
- (l-{ [4- ( {4- [ (4-fluorophényl) amino] pyrimidin-2- yl } amino) phenyl] suifonyl }pipéridin-4-yl) (pyridin-3-yl) - méthanamine l-{ 4- [4- (4-Fluoro-phenylamino) -pyrimidin-2-ylamino] -
benzenesulfonyl } -4-pyrrolidin-l-ylmethyl-piperidin-4-ol l-{ 4- [4- (4-Fluoro-3-methyl-phenylarαino) -pyrimidin-2- ylamino] -benzenesulfonyl} -4-pyrrolidin-l-ylmethyl- piperidin-4-ol ainsi que les sels d'addition avec les acides minéraux et organiques pharmaceutiquement acceptables desdits produits de formule (I) .
17) Compositions pharmaceutiques contenant à titre de principe actif l'un au moins des produits de formule (I) tels que définis à l'une quelconque des revendications 1 à 13 ou un sel pharmaceutiquement acceptable de ce produit ou un prodrug de ce produit et un support pharmaceutiquement acceptable
18) Compositions pharmaceutiques contenant à titre de principe actif l'un au moins des produits de formule (I) tels que définis à la revendication 13 ou un sel pharmaceutiquement acceptable de ce produit ou un prodrug de ce produit et un support pharmaceutiquement acceptable . 19) Utilisation des produits de formule (I) tels que définis à l'une quelconque des revendications 1 à 13) ou de sels pharmaceutiquement acceptables de ces produits pour la préparation d'un médicament destiné au traitement ou à la prévention d'une maladie par l'inhibition de l'activité de la protéine kinase IKK.
20) Utilisation telle que définie à la revendication précédente dans laquelle la protéine kinase est dans un mammifère .
21) Utilisation d'un produit de formule (I) tel que défini à l'une quelconque des revendications 1 à 13) pour la préparation d'un médicament destiné au traitement ou à la prévention d'une maladie choisie dans le groupe suivant : maladies inflammatoires, diabètes et cancers.
01173
138
22) Utilisation d'un produit de formule (I) tel que défini à l'une quelconque des revendications 1 à 13) pour la préparation d'un médicament destiné au traitement ou à la prévention de maladies inflammatoires .
23) Utilisation d'un produit de formule (I) tel que défini à l'une quelconque des revendications 1 à 13) pour la préparation d'un médicament destiné au traitement ou à la prévention de diabètes.
24) Utilisation d'un produit de formule (I) tel que défini à l'une quelconque des revendications 1 à 13) pour la préparation d' un médicament destiné au traitement de cancers .
25) Utilisation selon la revendication 20) destinée au traitement de tumeurs solides ou liquides .
26) Utilisation selon la revendication 24) ou 25) destinée au traitement de cancers résistant à des agents cytotoxiques .
27) Utilisation d'un produit de formule (I) telle que définie tel que défini à l'une quelconque des revendications 1 à 13) pour la préparation de médicaments destinés à la chimiothérapie de cancers.
28) Utilisation d'un produit de formule (I) tel que défini à l'une quelconque des revendications 1 à 13), pour la préparation de médicaments destinés à la chimiothérapie de cancers seul ou en en association.
29) Produits de formule (I) tels que définis à l'une quelconque des revendications 1 à 13) comme inhibiteurs de IKK.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0705790A FR2919869B1 (fr) | 2007-08-09 | 2007-08-09 | Nouveaux derives de n, n'-2,4-dianilinopyrimidines, leur preparation a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk |
| FR07/05790 | 2007-08-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009056693A1 true WO2009056693A1 (fr) | 2009-05-07 |
Family
ID=39198159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR2008/001173 WO2009056693A1 (fr) | 2007-08-09 | 2008-08-06 | Nouveaux derives de n, n'- 2, 4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk |
Country Status (5)
| Country | Link |
|---|---|
| AR (1) | AR068056A1 (fr) |
| FR (1) | FR2919869B1 (fr) |
| TW (1) | TW200916457A (fr) |
| UY (1) | UY31278A1 (fr) |
| WO (1) | WO2009056693A1 (fr) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (fr) | 2010-03-03 | 2011-09-09 | Sanofi | Nouveaux dérivés aromatiques de glycoside, médicaments contenants ces composés, et leur utilisation |
| WO2012004270A1 (fr) | 2010-07-05 | 2012-01-12 | Sanofi | Dérivés 1,3-propanedioxyde à substitution spirocyclique, procédé de préparation et utilisation comme médicament |
| WO2012004269A1 (fr) | 2010-07-05 | 2012-01-12 | Sanofi | Dérivés d'acide ( 2 -aryloxy -acétylamino) - phényl - propionique, procédé de production et utilisation comme médicament |
| WO2012010413A1 (fr) | 2010-07-05 | 2012-01-26 | Sanofi | Acides hydroxy-phényl-hexiniques substitués par aryloxy-alkylène, procédé de production et utilisation comme médicament |
| US10227342B2 (en) | 2014-06-19 | 2019-03-12 | Ariad Pharmaceuticals, Inc. | Heteroaryl compounds for kinase inhibition |
| CN114621206A (zh) * | 2022-03-24 | 2022-06-14 | 安徽医科大学 | 一种5-取代的嘧啶二胺类衍生物及其制备方法与应用 |
| US11827627B2 (en) | 2021-06-04 | 2023-11-28 | Vertex Pharmaceuticals Incorporated | N-(hydroxyalkyl (hetero)aryl) tetrahydrofuran carboxamides as modulators of sodium channels |
| US11834441B2 (en) | 2019-12-06 | 2023-12-05 | Vertex Pharmaceuticals Incorporated | Substituted tetrahydrofurans as modulators of sodium channels |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5607241B2 (ja) | 2010-05-21 | 2014-10-15 | ケミリア・エービー | 新規ピリミジン誘導体 |
| CA2830129C (fr) | 2011-03-24 | 2016-07-19 | Chemilia Ab | Nouveaux derives de pyrimidine |
| AU2019321526A1 (en) * | 2018-08-14 | 2021-03-25 | Mei Pharma, Inc. | Treatment of relapsed follicular lymphoma |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001064654A1 (fr) * | 2000-03-01 | 2001-09-07 | Astrazeneca Ab | Composes de pyrimidine |
| WO2006133426A2 (fr) * | 2005-06-08 | 2006-12-14 | Rigel Pharmaceuticals, Inc. | Compositions et procedes d'inhibition de la voie jak |
| WO2007006926A2 (fr) * | 2005-07-11 | 2007-01-18 | Sanofi-Aventis | Nouveaux derives de 2,4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk |
-
2007
- 2007-08-09 FR FR0705790A patent/FR2919869B1/fr not_active Expired - Fee Related
-
2008
- 2008-08-06 WO PCT/FR2008/001173 patent/WO2009056693A1/fr active Application Filing
- 2008-08-07 TW TW097130144A patent/TW200916457A/zh unknown
- 2008-08-07 AR ARP080103453A patent/AR068056A1/es unknown
- 2008-08-08 UY UY31278A patent/UY31278A1/es not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001064654A1 (fr) * | 2000-03-01 | 2001-09-07 | Astrazeneca Ab | Composes de pyrimidine |
| WO2006133426A2 (fr) * | 2005-06-08 | 2006-12-14 | Rigel Pharmaceuticals, Inc. | Compositions et procedes d'inhibition de la voie jak |
| WO2007006926A2 (fr) * | 2005-07-11 | 2007-01-18 | Sanofi-Aventis | Nouveaux derives de 2,4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (fr) | 2010-03-03 | 2011-09-09 | Sanofi | Nouveaux dérivés aromatiques de glycoside, médicaments contenants ces composés, et leur utilisation |
| WO2012004270A1 (fr) | 2010-07-05 | 2012-01-12 | Sanofi | Dérivés 1,3-propanedioxyde à substitution spirocyclique, procédé de préparation et utilisation comme médicament |
| WO2012004269A1 (fr) | 2010-07-05 | 2012-01-12 | Sanofi | Dérivés d'acide ( 2 -aryloxy -acétylamino) - phényl - propionique, procédé de production et utilisation comme médicament |
| WO2012010413A1 (fr) | 2010-07-05 | 2012-01-26 | Sanofi | Acides hydroxy-phényl-hexiniques substitués par aryloxy-alkylène, procédé de production et utilisation comme médicament |
| US10227342B2 (en) | 2014-06-19 | 2019-03-12 | Ariad Pharmaceuticals, Inc. | Heteroaryl compounds for kinase inhibition |
| US11958850B2 (en) | 2014-06-19 | 2024-04-16 | Takeda Pharmaceutical Company Limited | Heteroaryl compounds for kinase inhibition |
| US11834441B2 (en) | 2019-12-06 | 2023-12-05 | Vertex Pharmaceuticals Incorporated | Substituted tetrahydrofurans as modulators of sodium channels |
| US11919887B2 (en) | 2019-12-06 | 2024-03-05 | Vertex Pharmaceuticals Incorporated | Substituted tetrahydrofurans as modulators of sodium channels |
| US11827627B2 (en) | 2021-06-04 | 2023-11-28 | Vertex Pharmaceuticals Incorporated | N-(hydroxyalkyl (hetero)aryl) tetrahydrofuran carboxamides as modulators of sodium channels |
| CN114621206A (zh) * | 2022-03-24 | 2022-06-14 | 安徽医科大学 | 一种5-取代的嘧啶二胺类衍生物及其制备方法与应用 |
| CN114621206B (zh) * | 2022-03-24 | 2023-11-24 | 安徽医科大学 | 一种5-取代的嘧啶二胺类衍生物及其制备方法与应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| UY31278A1 (es) | 2009-03-31 |
| FR2919869B1 (fr) | 2009-09-25 |
| TW200916457A (en) | 2009-04-16 |
| FR2919869A1 (fr) | 2009-02-13 |
| AR068056A1 (es) | 2009-11-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2118092A1 (fr) | Derives de n, n' - 2, 4 -dianilino pyrimidines, leur utilisation comme inhibiteurs de ikk, leur preparation et leur compositions pharmaceutiques | |
| WO2009056693A1 (fr) | Nouveaux derives de n, n'- 2, 4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk | |
| EP2108016A1 (fr) | Derives de 2 -anilino 4 -heteroaryle pyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk | |
| WO2008099074A1 (fr) | Derives de phenyl- (4-phenyl-pyrimidin-2-yl) - amines comme inhibiteurs de ikk, leur preparation et leur compositions pharmaceutiques | |
| US11014922B2 (en) | Cyclobutane- and azetidine-containing mono and spirocyclic compounds as αV integrin inhibitors | |
| WO2007006926A2 (fr) | Nouveaux derives de 2,4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk | |
| US11028071B2 (en) | Indazole derivatives as alpha v integrin antagonists | |
| JP6948322B2 (ja) | Apj受容体のapjアゴニストとしてのヘテロアリールヒドロキシピリミジノン | |
| EP2104673A2 (fr) | Nouveaux derives de 2, 4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk | |
| JP7128811B2 (ja) | アルファvインテグリン阻害剤としてのアゾールアミドおよびアミン | |
| CA2718488A1 (fr) | Nouveaux derives de carbazole inhibiteurs d'hsp90, compositions les contenant et utilisation | |
| EP3538528A1 (fr) | Amides de pyrrole en tant qu'inhibiteurs d'intégrine alpha v | |
| WO2012176763A1 (fr) | Nouveau dérivé indazole | |
| WO2016044323A1 (fr) | Dérivés de pyrrolopyrimidine à titre d'antagonistes des récepteurs nmda nr2b | |
| FR2888239A1 (fr) | Nouveaux derives de 2,4-dianilinopyrimidines, leur preparation, a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk | |
| FR2893941A1 (fr) | Nouveaux derives de 2,4-dianilinopyridines, leur preparation a titre de medicaments, compositions pharmaceutiques et notamment comme inhibiteurs de ikk | |
| FR2955323A1 (fr) | Nouveaux derives d'indazole inhibiteurs d'hsp90, compositions les contenant et utilisation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08843930 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08843930 Country of ref document: EP Kind code of ref document: A1 |