GENERATION OF ONCOLYTIC ADENOVIRUSES AND USES THEREOF
FIELD OF THE INVENTION
The invention described herein relates to a method for generation of oncolytic adenoviruses having an increased potency/increased therapeutic index for use in the treatment of solid tumors.
BACKGROUND OF THE INVENTION
Successful selection of clinically effective oncolytic agents depends on the use of cells or tissues that accurately model target tumors, while being routinely reproducible in the laboratory. Recent work has provided evidence that three dimensional associations of extracellular matrix nanofibrils and the cellular architecture they induce are critical to development of in vitro systems that sufficiently mimic the physiological patterns of cell adhesion, cytoskeletal organization, signal transduction and gene expression, morphogenesis and differentiation in cultures of both normal and transformed cells. A number of analytical methods, including Gene Expression Profiling (GEP), proteomics and analyses of cellular function have demonstrated that 3D cultures are generally better tumor models than are monolayer cultures (Birgersdotter et al. (2005) Semin Cancer Bb. 15:405-412; Nelson and Bissell (2005) Semin Cancer Biol 15:342-352).
SUMMARY OF THE INVENTION
The present invention provides a method for the isolation of oncolytic adenoviruses useful for /iral-based therapy of solid or haemotologic tumors, wherein the isolated adenoviruses display an enhanced potency as compared with a reference virus or viruses.
In one embodiment, the method comprises the steps of
(a) pooling a group of adenoviruses, wherein the adenoviruses are selected from the group insisting of the adenoviral serotypes B, C, D1 E and F;
(b) passaging the pooled adenoviral mixture from step (a) on an actively growing culture of umor cells;
(c) harvesting the supernatant from step (b);
(d) infecting a quiescent culture of tumor cells with the supernatant harvested in step (c);
(e) harvesting the cell culture supernatant from step (d) prior to any sign of CPE;
(f) infecting a quiescent culture of tumor cells with the supernatant harvested in step (e);
(g) harvesting the cell culture supernatant from step (f) prior to any sign of CPE; and (h) isolating a virus from the supernatant of step (g) by plaque purification, rherein the tumor cells in steps (b), (d) and (f) are all grown on, or in, an extracellular matrix.
In a preferred embodiment, the extracellular matrix is the reconstituted basement membrane nown as MATRIGEL™.
In another embodiment of the invention, the pooled adenoviral mixture further comprises CoIoAdI (SEQ ID N0:3).
In another embodiment of the invention, a portion of the pool of step (a) is mutagenized prior to passaging.
In another embodiment of the invention, the passaging of step (b) is performed twice before the first harvesting of supernatant.
The present invention provides for the isolation of oncolytic adenoviruses that are specifically targeted to tumor cells derived from colon, ovarian, lung, prostate, breast or pancreas.
In one embodiment of the invention, oncolytic adenoviruses isolated by this method target ovarian tumor cells. Two isolated oncolytic adenoviruses of the invention that target ovarian tumor cells are OvAdI (SEQ ID NO:1) and OvAd2 (SEQ ID NO:2).
In another embodiment of the invention, oncolytic adenoviruses isolated by this method target cancer progenitor cells. One isolated oncolytic adenovirus of the invention that targets ovarian cancer progenitor cells, otherwise known as ovarian cancer stem cells, is OvAdI (SEQ ID NO:1).
The present invention further encompasses conservatively modified variants of the oncolytic adenoviruses of the invention, where said variant shows equal or greater potency, when compared with the oncolytic adenovirus of which it is a variant.
In another embodiment of the invention, oncolytic adenoviruses of the invention have been modified to produce adenoviral vectors which are replication deficient. In a preferred embodiment, an oncolytic adenovirus of the invention has been rendered replication deficient through deletion of one or more regions of the adenoviral genome, or parts thereof, which encode proteins involved in adenoviral replication, e.g. the E1 , E2 or E4 regions. Particularly preferred is deletion of the E1 or E2 regions.
In another embodiment of the invention, the oncolytic adenoviruses of the invention are modified to increase their potency by reducing the size of the adenoviral genome. A preferred modification is deletion of the E3 region or parts thereof. Such a modification can be made either on the original adenovirus of the invention or on a replication deficient derivative thereof.
In another embodiment, the oncolytic adenoviruses of the invention are modified to increase tumor specificity. A preferred modification is production of an adenovirus with a "delta 24" deletion.
The present invention further provides methods for use of the oncolytic adenoviruses of the invention for therapeutic purposes. In one embodiment, the oncolytic adenoviruses of the invention can be used to inhibit the growth of tumor cells, in vitro or in vivo, by infecting the tumor cells with the oncolytic adenovirus.
In a preferred embodiment, the OvAdI (SEQ ID NO:1) or OvAd2 (SEQ ID NO:2) adenoviruses are useful for inhibiting the growth of ovarian tumor cells (i.e. for the treatment of ovarian cancer in a patient). In a particularly preferred embodiment, the OvAdI (SEQ ID NO:1) oncolytic adenovirus is useful for inhibiting the growth of ovarian tumor cells, particularly chemotherapy-resistant ovarian tumor cells.
In another embodiment, an oncolytic adenovirus of the invention further comprises a heterologous gene whose expression serves to attenuate adenoviral replication, such that any therapeutic dose of the virus can be eliminated in vivo when desired. In a preferred embodiment, expression of this infection-attenuating gene can be regulated, for example through use of the let-on" system of gene expression regulation. In another preferred embodiment, the gene is thymidine kinase, the expression of which leads to the death of infected cells upon administration of gancyclovir, leading to attenuation of viral infection.
In another embodiment, an oncolytic adenovirus of the invention further comprises a heterologous gene, wherein the heterologous gene is expressed within a cell infected with the adenovirus and encodes a therapeutic protein or factor. In a preferred embodiment, the therapeutic protein is selected from the group consisting of cytokines and chemokines, antibodies, known inducers of cell death, pro-drug converting enzymes and immunoregulatory proteins and peptides. Therapeutic factors can be, but are not limited to, small RNAs ( e.g. shRNA, miRNA) and aptamers.
BRIEF DESCRIPTION OF THE FIGURES
Figure 1. Potency of bioselected pools. SM10 (— ■ — ) and SP10 (— • — ) viral pools, bioselected on MATRIGEL™ and monolayers, respectively, both show an enhanced potency on SKOV3 cells compared to the potencies of viruses in the starting pool: Ad3 (— O — ); Ad5 (— □ — ); Ad11 p (— Δ— ); and Ad35 (— 0— ■). MTS assays were read at 7 days post infection.
Figurβ 2. Therapeutic index of bioselected pools. Fig 2A. The therapeutic index of the adenoviral pool selected on MATRIGEL™ (SM10), is determined by comparing its potency (ICso) on HUVEC endothelial cells (— • — ) and SKOV3 platinum-resistant ovarian tumor cells (— O — ). Fig 2B. The therapeutic index of the adenoviral pool selected on monolayers (SP10), is determined by comparing its potency (IC50) on HUVEC endothelial cells {— • — ) and SKOV3 platinum-resistant ovarian tumor
cells ( — O — ). MTS assays were read at 7 days post infection.
Figure 3: Potency of adenoviruses OvAdI and OvAd2. Individual adenoviruses, OvAdI and OvAd2, were isolated from the SM10 (MATRIGEL™-selected) and SP10 (monolayer-selected) pools,
respectively. The potency of the isolates OvAdI (— O — ) and OvAd2 (— □ — ) on SKOV3 cells was
determined using the MTS assay and compared with the potency of the parental adenoviruses CoIoAdI
( — Δ — ) and Ad3 ( — O — ) and the known oncolytic adenovirus, Onyx-015 ( — ■ — ). Results are
shown at 7 days post infection.
Figure 4: Therapeutic index of adenoviruses OvAdI and OvAd2. Individual adenoviruses, OvAdI and OvAd2, were isolated from the SM10 (MATRIGEL™-selected) and SP10 (monolayer-selected) pools, respectively. The potency of each isolate was determined on SKOV3 and HUVEC cells. The symbols are as follows: OvAdI on HUVEC (— • — ); OvAdI on SKOV3 (—▼—); OvAd2 on HUVEC
( — O — ); and OvAd2 on SKOV3 (— Δ— ). Therapeutic index is determined by comparing adenoviral potency (ICs0) on SKOV3 platinum-resistant ovarian tumor cells and on HUVEC primary normal endothelial cells. Results are shown at 7 days post infection. Both OvAdI and OvAd2 are about 100- fold more potent on SKOV3 cells than on HUVEC cells.
Figure 5. Relationship of OvAdI and OvAd2 sequences to CoIoAdI and Ad3. DNA sequence analysis of OvAdI (SEQ IO N0:1)and OvAd2 (SEQ ID N0:2) revealed that both viruses are chimeras of the sequences CoIoAdI (SEQ ID N0:3) and Ad3 (SEQ ID N0:4). The region between 10,000bp and 13,060bp on the OvAdI sequence is an area of non-homology between OvAdI and OvAd2.
Figure 6. In vivo efficacy of OvAdI and 0vAd2 in an intraperitoneal (IP) model. Fig 6A. SKOV3 tumor cells were seeded to the peritoneal cavity of 25 mice on day O. Mice were divided into 5 groups, and one of four kinds of virus OvAdI (— • — ), OvAd2 (— ■ —). ONYX-015 ( — x — ) and
CoIoAdI ( —A—) or the vehicle control ( — + —). was injected ip on days 3, 5, and 7 post tumor seeding. Mice were weighed every 4 to 6 days. Fig 6B. On day 41 , mice were euthanized, the tumors dissected out and weighed (n=5).
Figure 7: From Lieber and Strauss; potency of OvAdI against ovarian cancer progenitor, or stem, cells from a Stage 4 ovarian cancer patient.
DETAILED DESCRIPTION OF THE INVENTION
All publications, including patents and patent applications, mentioned in this specification are herein incorporated by reference to the same extent as if each individual publication was specifically and individually indicated to be incorporated by reference in its entirety.
Definitions
Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. Generally, the nomenclature used herein and the laboratory procedures described below are those well known and commonly employed in the art.
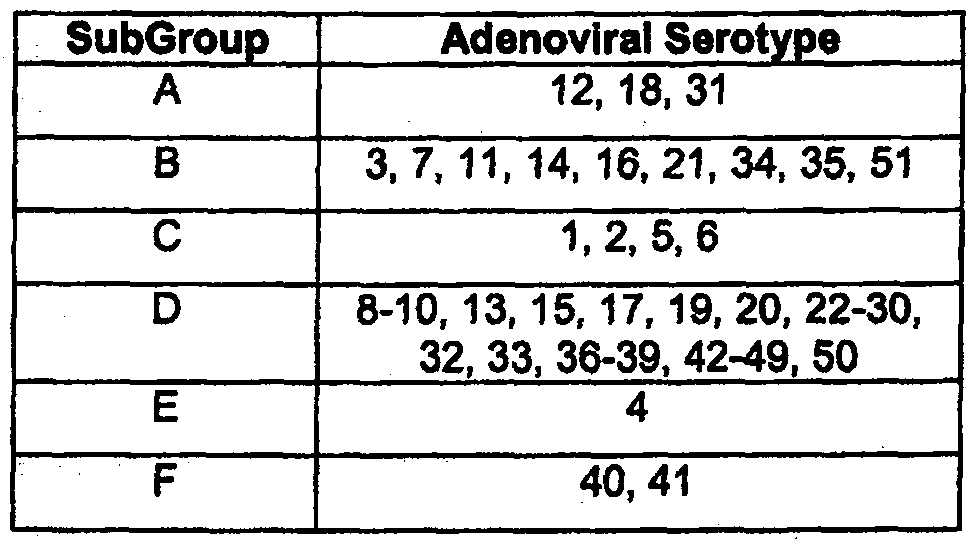
As used herein, the term "adenovirus", "serotype" or "adenoviral serotype" refers to any of the 51 human adenoviral serotypes currently known, or isolated in the future. See, for example, Strauss, "Adenovirus infections in humans," in The Adenoviruses, Ginsberg, ed., Plenum Press, New York, NY, pp.451-596 (1984). These serotypes are classified in the subgroups A-F (see, Shenk, "Adenoviridae: The Viruses and Their Replication," in Fields Virology, Vol.2, Fourth Edition, Knipe, ea., Lippincott Williams & Wilkins, pp. 2265-2267 (2001), as shown in Table 1.
Table 1
As used herein, "chimeric adenovirus" refers to an adenovirus whose nucleic acid sequence is comprised of the nucleic acid sequences of at least two of the adenoviral genomes included within the initial adenoviral pool on which selection is performed.
As used herein, the term "homologous recombination" refers to two nucleic acid molecules, each having homologous sequences, where the two nucleic acid molecules cross over or undergo recombination in the region of homology.
As used herein, the term "potency" refers to the lytic potential of a virus and represents its ability to replicate, lyse, and spread. For the purposes of the instant invention, potency is the IC5O of any given adenovirus on a given cell line.
As used herein, the term "oncolytic virus" refers to a virus that preferentially kills cancer cells as compared with normal cells.
As used herein, the term "therapeutic index" or "therapeutic window" refers to a number indicating the oncolytic selectivity of a given adenovirus and is determined by dividing the potency of an adenovirus in a normal (i.e. non-cancerous) cell line by the potency of the adenovirus in a chosen cancer cell line.
As used herein, the term "modified" refers to a molecule with a nucleotide or amino acid sequence differing from a naturally-occurring, e.g. a wild-type nucleotide or amino acid sequence or from the nucleotide sequence or amino acid sequence of an adenovirus generated by the methods of the invention. A modified molecule can retain the function or activity of a wild-type molecule, i.e. a modified adenovirus may retain its oncolytic activity. Modifications include mutations to nucleic acids, encompassing deletions, insertions and substitutions, as described below. Polynucleotides and polypeptides having such mutations can be isolated or generated using methods well known in the art.
As used herein, "mutation" with reference to a polynucleotide or polypeptide, refers to a naturally-occurring, synthetic, recombinant, or chemical change or difference to the primary, secondary, or tertiary structure of a polynucleotide or polypeptide, as compared to a reference polynucleotide or polypeptide, respectively (e.g., as compared to a wild-type polynucleotide or polypeptide).
As used herein, "deletion" is defined as a change in either polynucleotide or amino acid sequences in which one or more nucleotides or amino acid residues, respectively, are absent.
As used herein, "insertion" or "addition" is that change in a polynucleotide or amino acid sequence which has resulted in the addition of one or more nucleotides or amino acid residues, respectively, as compared to the naturally occurring polynucleotide or amino acid sequence.
As used herein, "substitution" results from the replacement of one or more nucleotides or amino acids by different nucleotides or amino acids, respectively.
As used herein, the term "adenoviral derivative" refers to an adenovirus of the invention that has been modified such that an addition, deletion or substitution has been made to or in the viral genome, such that the resulting adenoviral derivative exhibits a potency and/or therapeutic index equal to or greater than that of the parent adenovirus, or in some other way is more therapeutically useful (i.e. less immunogenic, improved clearance profile). For example, a derivative of an adenovirus of the invention may have a deletion in one of the early genes of the viral genome, including, but not limited to, the E1 A or E2B region of the viral genome.
A used herein, "conservatively modified variants" applies to modifications in both amino acid and nucleic acid sequences of the adenoviruses of the invention. With respect to particular nucleic acid sequences, conservatively modified variants refers to those nucleic acids which encode identical or essentially identical amino acid sequences, or where the nucleic acid does not encode an amino acid sequence, to essentially identical sequences. Because of the degeneracy of the genetic code, a large number of functionally identical nucleic acids encode any given protein. For instance, the codons GCA, GCC, GCG and GCU all encode the amino acid alanine. Thus, at every position where an alanine is specified by a codon, the codon can be altered to any of the corresponding codons described without altering the encoded polypeptide. Such nucleic acid variations are "silent variations," which are one species of conservatively modified variations. Every nucleic acid sequence herein that encodes a polypeptide also describes every possible silent variation of the nucleic acid. One of skill will recognize that each codon in a nucleic acid (except AUG, which is ordinarily the only codon for methionine, and TGG, which is ordinarily the only codon for tryptophan) can be modified to yield a functionally identical molecule. Accordingly, each silent variation of a nucleic acid that encodes a polypeptide is implicit in each described sequence.
As to amino acid sequences, one of skill will recognize that individual substitutions, deletions or additions to a nucleic acid, peptide, polypeptide, or protein sequence that alters, adds or deletes a single amino acid or a small percentage of amino acids in the encoded sequence is a "conservatively modified variant" where the alteration results in the substitution of an amino acid with a chemically similar amino acid. Conservative substitution tables providing functionally similar amino acids are well known in the art. Such conservatively modified variants are in addition to and do not exclude polymorphic variants, interspecies homologs, and alleles of the invention.
The following eight groups each contain amino acids that are conservative substitutions for one another:
1) Alanine (A)1 Glycine (G);
2) Aspartic acid (D), Glutamic acid (E);
3) Asparagine (N), Glutamine (Q);
4) Arginine (R), Lysine (K);
5) lsoleucine (I), Leucine (L), Methionine (M), Valine (V);
7 088415
-8-
6) Phenylalanine (F)1 Tyrosine (Y), Tryptophan (W);
7) Serine (S), Threonine (T); and
8) Cysteine (C), Methionine (M)
Nonconservative substitutions include, but are not limited to aspartic acid (D) being replaced with glycine (G); asparagine (N) being replaced with lysine (K); or alanine (A) being replaced with arginine (R). See, e.g., Creighton, Proteins (1984).
It will be appreciated that polypeptides often contain amino acids other than the 20 amino acids commonly referred to as the 20 naturally occurring amino acids, and that many amino acids, including the terminal amino acids, may be modified in a given polypeptide, either by natural processes such as glycosylation and other post-translational modifications, or by chemical modification techniques which are well known in the art. Even the common modifications that occur naturally in polypeptides are too numerous to list exhaustively here, but they are well described in basic texts and in more detailed monographs, as well as in a voluminous research literature, and they are well known to those of skill in the art. Among the known modifications which may be present in polypeptides of the present invention are, to name an illustrative few, acetylation, acylation, ADP-ribosylation, amidation, covalent attachment of flavin, covalent attachment of a heme moiety, covalent attachment of a polynucleotide or polynucleotide derivative, covalent attachment of a lipid or lipid derivative, covalent attachment of phosphotidylinositol, cross-linking, cyclization, disulfide bond formation, demethylation, formation of covalent cross-links, formation of cystine, formation of pyroglutamate, formylation, gamma- carboxylation, glycation, glycosylation, GPI anchor formation, hydroxylation, iodination, methylation, myristoylation, oxidation, proteolytic processing, phosphorylation, prenylation, racemization, selenoylation, sulfation, transfer-RNA mediated addition of amino acids to proteins such as arginylation, and ubiquitination.
Such modifications are well known to those of skill and have been described in great detail in the scientific literature. Several particularly common modifications, glycosylation, lipid attachment, sulfation, gamma-carboxylation of glutamic acid residues, hydroxylation and ADP-ribosylation, for instance, are described in most basic texts, such as, for instance, I. E. Creighton, Proteins-Structure and Molecular Properties, 2nd Ed., W.H. Freeman and Company, New York, I993. Many detailed reviews are available on this subject, such as, for example, those provided by Wold, F., in Posttranslational Covalent Modification of Proteins, B. C. Johnson, Ed., Academic Press, New York, pp 1-12, 1983; Seifter et al., Meth. Enzymol. 182: 626-646, 1990 and Rattan et al., Protein Synthesis: Posttranslational Modifications and Aging, Ann. N.Y. Acad. Sci. 663: 48-62, 1992.
It will be appreciated, as is well known and as noted above, that polypeptides are not always entirety linear. For instance, polypeptides may be branched as a result of ubiquitination, and they may be circular, with or without branching, generally as a result of posttranslational events, including natural
processing events and events brought about by human manipulation which do not occur naturally. Circular, branched and branched circular polypeptides may be synthesized by non-translational natural processes and by entirely synthetic methods, as well.
Modifications can occur anywhere in a polypeptide, including the peptide backbone, the amino acid side-chains and the amino or carboxyl termini. In fact, blockage of the amino or carboxyl group in a polypeptide, or both, by a covalent modification, is common in naturally occurring and synthetic polypeptides and such modifications may be present in polypeptides of the present invention, as well. For instance, the amino terminal residue of polypeptides made in E coli, prior to proteolytic processing, almost invariably will be N-formylmethionine.
The modifications that occur in a polypeptide often will be a function of how it is made. For polypeptides made by expressing a cloned gene in a host, for instance, the nature and extent of the modifications in large part will be determined by the host cell posttranslational modification capacity and the modification signals present in the polypeptide amino acid sequence. For instance, as is well known, glycosylation often does not occur in bacterial hosts such as E. coli. Accordingly, when glycosylate is desired, a polypeptide should be expressed in a glycosylating host, generally a eukaryotic cell. Insect cells often carry out the same posttranslational glycosylates as mammalian cells and, for this reason, insect cell expression systems have been developed to efficiently express mammalian proteins having native patterns of glycosylation, inter alia. Similar considerations apply to other modifications.
It will be appreciated that the same type of modification may be present to the same or varying degree at several sites in a given polypeptide. Also, a given polypeptide may contain many types of modifications.
As used herein, the following terms are used to describe the sequence relationships between two or more polynucleotide or amino acid sequences: "reference sequence", "comparison window", "sequence identity", "percentage of sequence identity", "substantial identity", "similarity", and "homologous". A "reference sequence" is a defined sequence used as a basis for a sequence comparison; a reference sequence may be a subset of a larger sequence, for example, as a segment of a full-length cDNA or gene sequence given in a sequence listing or may comprise a complete cDNA or gene sequence. Generally, a reference sequence is at least 18 nucleotides or 6 amino acids in length, frequently at least 24 nucleotides or 8 amino acids in length, and often at least 48 nucleotides or 16 amino acids in length. Since two polynucleotides or amino acid sequences may each (1) comprise a sequence (i.e., a portion of the complete polynucleotide or amino acid sequence) that is similar between the two molecules, and (2) may further comprise a sequence that is divergent between the two polynucleotides or amino acid sequences, sequence comparisons between two (or more) molecules are typically performed by comparing sequences of the two molecules over a "comparison window" to
identlfy and compare local regions of sequence similarity. A "comparison window", as used herein, refers to a conceptual segment of at least 18 contiguous nucleotide positions or 6 amino acids wherein a polynucleotide sequence or amino acid sequence may be compared to a reference sequence of at least 18 contiguous nucleotides or 6 amino acid sequences and wherein the portion of the polynucleotide sequence in the comparison window may comprise additions, deletions, substitutions, and the like (i.e., gaps) of 20 percent or less as compared to the reference sequence (which does not comprise additions or deletions) for optimal alignment of the two sequences. Optimal alignment of sequences for aligning a comparison window may be conducted, for example, by the local homology algorithm of Smith and Waterman, Adv. Appl. Math. 2:482 (1981), by the homology alignment algorithm of Needleman and Wunsch, J. MoI. Biol. 48:443 (1970), by the search for similarity method of Pearson and Lipman, Proc. Natl. Acad. ScL ( U.S.A .) 85:2444 (1988), by computerized implementations of these algorithms (GAP, BESTFIT, FASTA1 and TFASTA in the Wisconsin Genetics Software Package Release 7.0, (Genetics Computer Group, 575 Science Dr., Madison, Wis.), VectorNTI from Informatix, Geneworks, or MacVector software packages), or by inspection, and the best alignment (i. e., resulting in the highest percentage of homology over the comparison window) generated by the various methods is selected.
As used herein, the term "sequence identity" means that two polynucleotide or amino acid sequences are identical (i.e., on a nucleotide-by-nucleotide or residue-by-residue basis) over the comparison window. The term "percentage of sequence identity" is calculated by comparing two optimally aligned sequences over the window of comparison, determining the number of positions at which the identical nucleic acid base (e.g., A, T, C, G, U1 or I) or residue occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the comparison window (i.e., the window size), and multiplying the result by 100 to yield the percentage of sequence identity. The terms "substantial identity" as used herein denotes a characteristic of a polynucleotide or amino acid sequence, wherein the polynucleotide or amino acid comprises a sequence that has at least 85 percent sequence identity, preferably at least 90 to 95 percent sequence identity, more usually at least 99 percent sequence identity as compared to a reference sequence over a comparison window of at least 18 nucleotide (6 amino acid) positions, frequently over a window of at least 24-48 nucleotide (8-16 amino acid) positions, wherein the percentage of sequence identity is calculated by comparing the reference sequence to the sequence which may include deletions or additions which total 20 percent or less of the reference sequence over the comparison window. The reference sequence may be a subset of a larger sequence. The term "similarity", when used to describe a polypeptide, is determined by comparing the amino acid sequence and the conserved amino acid substitutes of one polypeptide to the sequence of a second polypeptide. The term "homologous", when used to describe a polynucleotide, indicates that two polynucleotides, or designated sequences thereof, when optimally aligned and compared, are identical, with appropriate nucleotide insertions or deletions, in at least 70% of the nucleotides, usually from about 75% to 99%, and more preferably at least about 98 to 99% of the nucleotides.
As used herein, "homologous", when used to describe a polynucleotide, indicates that two polynucleotides, or designated sequences thereof, when optimally aligned and compared, are identical, with appropriate nucleotide insertions or deletions, in at least 70% of the nucleotides, usually from about 75% to 99%, and more preferably at least about 98 to 99% of the nucleotides.
As used herein, "polymerase chain reaction" or "PCR" refers to a procedure wherein specific pieces of DNA are amplified as described in U.S. Pat. No. 4,683,195. Generally, sequence information from the ends of the polypeptide fragment of interest or beyond needs to be available, such that oligonucleotide primers can be designed; these primers will point towards one another, and will be identical or similar in sequence to opposite strands of the template to be amplified. The 5' terminal nucleotides of the two primers will coincide with the ends of the amplified material. PCR can be used to amplify specific DNA sequences from total genomic DNA, cDNA transcribed from total cellular RNA, piasmid sequences, etc. (See generally MuIHs et al., Cold Spring Harbor Symp. Quant. Biol., 51: 263, 1987; Erlich, ed., PCR Technology, Stockton Press, NY, 1989).
As used herein, "stringency" typically occurs in a range from about Tn, (melting temperature)-50C (5° below the Tn, of the probe) to about 2O0C to 250C below Tn,. As will be understood by those of skill in the art, a stringent hybridization can be used to identify or detect identical polynucleotide sequences or to identify or detect similar or related polynucleotide sequences. As herein used, the term "stringent conditions" means hybridization will occur only if there is at least 95% and preferably at least 97% identity between the sequences.
As used herein, "hybridization" as used herein, shall include "any process by which a polynucleotide strand joins with a complementary strand through base pairing" (Coombs, J., Dictionary of Biotechnology, Stockton Press, New York, N.Y., 1994).
Adenoviruses of the Invention
The present invention provides a novel method that takes advantage of the biodiversity of adenoviral serotypes for the isolation of oncolytic adenoviruses that demonstrate an enhanced potency and/or increased selectivity toward tumors cells derived from tumor types including, but not limited to, colon, ovary, lung, prostate, breast and pancreas.
Isolation of Oncolytic Adenoviruses
The oncolytic adenoviruses of the invention are produced by directed evolution, a method that uniquely combines mutagenesis, serotype biodiversity, and stringent selection conditions to isolate an adenovirus with desired properties, such as enhanced potency or cell type specificity. Directed evolution for adenoviruses targeting particular tissues can be performed using tumor cells derived from
the tissue of choice. Preferred tumor cell lines useful in the directed evolution process include, but are not limited to, those derived from breast, colon, pancreas, lung, prostate and ovary. Examples of solid tumor cell lines useful for the "directed evolution" passaging of the adenoviral mixture include, but are not limited to, MDA231, HT29 and PC-3 cells (for targeting tumors derived from breast, colon or pancreas, respectively). Particularly preferred for directed evolution of adenoviruses showing an enhanced potency in ovarian tissues include, but are not limited to, the tumor cell lines SKOV3, OVCAR3 and CaOV3. In a preferred embodiment, SKOV3 tumor cells are used for selection of oncolytic adenoviruses that show efficacy in ovarian tumors which have become resistant to chemotherapy. Any other tumor cell line which is representative of a target tissue of interest, available through such sources as the ATCC, can be used in isolating adenoviruses of the invention.
Alternatively, directed evolution of the adenoviruses of the invention can be done directly on freshly excised human primary or metastatic tumor cells, including tumor cells from haematological malignancies.
In the present invention, adenoviral selection is performed using as the starting material an adenoviral mixture that includes serotypes representative of the adenoviral subgroups B, C, D, E and F. Group A adenoviruses are not included in the mixture as they are associated with tumor formation in rodents. In a preferred embodiment of the invention, the mixture also includes the chimeric adenovirus CoIoAdI (see U. S. Patent Application Serial No. 11/136,912).
The pooled adenoviral mixture is passaged once, more preferably at least twice, on a subconfluent culture of tumor cells or on cells grown on in vitro 3D associations of extracellular matrix nanofibrils including, but not limited to, collagen and basement membrane matrix (MATRIGEL™, Becton Dickinson). It has been shown that the cellular architecture induced by such matrices are critical to the reproduction of physiological patterns of cell adhesion, cytoskeletal organization, signal transduction and gene expression, morphogenesis, and differentiation in cultures of both normal and transformed cells and that such extracts induce a cellular architecture important for the creation of model systems that more closely mimic the in vivo characteristics of abnormal cells (Schindler et al. J Cell Biochem Biophys (2006) 45:215-227; Birgersdotter et al. (2005) Semin. Cancer Bio. 15:405-412; Boyd et al. (2002) J Gene Λfeo* 4:567-576).
Initial passage of adenovirus is performed at a particle per cell ratio high enough to encourage recombination between serotypes, but not so high as to produce premature cell death. A preferred particle per cell ratio is between approximately 100-500 particles per cell, and is easily determined by one skilled in the art. As used herein, a "subconfluent culture" of cells refers to a culture in which the cells are actively growing. For cells grown as a monolayer, a subconfluent culture would be one in which approximately 50% to 80%, preferably 75%, of the area available for cell growth is covered with cells.
For cells grown on a biological matrix material, a "subconfluent culture" would be one in which the cells do not confluβntly cover the matrix material.
Adenoviruses produced during these initial passages are used to infect quiescent tumor cells at a particle to cell ratio low enough to permit the infection of a cell by no more than one adenovirus and the supernatant for the subsequent passage harvested prior to visible cytopathic effect (CPE, see Fields Virology, Vol. 2, Fourth Edition, Knipe, ea., Lippincott Williams & Wilkins, pp. 135-136) to increase selection of highly potent viruses . After up to 20 passages under these conditions, the supernatant from the final passage, is again harvested prior to visible CPE and is then concentrated by techniques well known to those skilled in the art. One method for attaining quiescent cells, i.e. ones in which active cell growth has stopped, in a monolayer culture is to allow the culture to grow for 3 days following confluence, where confluence means that the entire area available for cell growth is occupied (covered with cells). For cultures grown on a 3D matrix, confluence is dependent on the cell type and can easily be determined by one of skill in the art. Suspension cultures can be grown to densities characterized by the absence of active cell growth.
The serotype profile of the concentrated supernatant harvested from the extracellular matrix material, which contains the bioselected adenoviral pool, can be examined by determining the retention times present within the harvested viral pool using an anion exchange column, where different adenoviral serotypes are known to have characteristic retention times (Blanche et al. (2000) Gene Therapy 7:1055-1062). Adenoviruses of the invention can be isolated from the concentrated supernatant by dilution and plaque purification, or other techniques well know in the art, and grown for further characterization. Techniques well known in the art are used to determine the sequences of isolated oncolytic adenoviruses (see Example 5). Examples of oncolytic adenoviruses of the invention with selectivity for ovarian tumor cells derived by this method are OvAdI (SEQ ID NO:1 ) and OvAd2 (SEQ ID NO:2), which were bioselected using MATRIGEL ™ or a monolayer culture, respectively, during the selection process.
Adenoviruses of the invention have an enhanced potency/therapeutic index as compared with the adenoviral serotypes from which they are derived. Figures 1 and 2 demonstrate the potency and therapeutic index, respectively, of the viral pools derived on the MATRIGEL™ 3D extracellular cell matrix and on monolayer cells, while Figure 4 shows similar data for individual adenoviral isolates from each pool: OvAdI (from the MATRIGEL™ pool) and OvAd2 (from the monolayer pool).
Adenoviral Derivatives
The invention also encompasses an oncolytic adenovirus of the invention that is modified to provide other therapeutically useful oncolytic adenoviruses. Modifications include, but are not limited to, those described below.
One modification is production of derivatives of the oncolytic adenoviruses of the invention substantially lacking the ability to bind p53, as a result of a mutation in the adenoviral gene that encodes the E1B-55K protein. Such viruses generally have some, or all, of the E1B-55K region deleted. U.S. Patent No. 6,080,578 describes, among other things, Ad5 mutants that have deletions in the region of the E1B-55K protein that is responsible for binding p53 (see also U.S. Patent No. 5,677,178). Another preferred modification to the oncolytic adenoviruses of the instant invention are mutations in the E1 A region, as described in U.S. Patent Nos. 5,801 ,029 and 5,972,706. These types of modifications provide derivatives of the oncolytic adenoviruses of the invention with greater selectivity for tumor cells.
Another example of a modification encompassed by the invention is modification of an oncolytic adenovirus such that it exhibits an enhanced degree of tissue specificity due to placement of viral replication under the control of a tissue specific promoter as described in U.S. Patent No. 5,998,205. Replication of an oncolytic adenovirus of the invention can also be put under the control of an E2F responsive element as described in U.S. Patent Application No. 09/714,409. This modification affords a viral replication control mechanism based on the presence of E2F, resulting in enhanced tumor tissue specificity, and is distinct from the control realized by a tissue specific promoter. In both of these embodiments, the tissue specific promoter and the E2F responsive element are operably linked to an adenoviral gene that is essential for the replication of the adenovirus.
Another modification encompassed by the invention is use of an oncolytic adenovirus of the invention, e.g. OvAdI (SEQ ID NO:1) or OvAd2 (SEQ ID NO:2), as the backbone for production of novel replication-deficient adenoviral vectors. As described in Lai et al. ((2002) DNA Cell Bio. 21:895-913), adenoviral vectors which are replication deficient can be used to deliver and express therapeutic genes. Adenoviral vectors of the invention can be modified to produce replication deficient derivatives by deletion of the E1, E2 or E4 regions of the viral genome. Particularly preferred is deletion of the E1 or E2 regions. Such modified adenoviral vectors are easily produced using techniques well known to those skilled in the art (see lmperiale and Kochanek (2004) Cυrr. Top. Microbiol. Immunol. 273:335- 357; Vogels et al. (2003) J. Virol. 77:8263-8271).
Similarly, other modifications can be made to the oncolytic adenoviruses of the invention to increase their potency by reducing the size of the viral genome by deletion of one or more regions or parts thereof. Also encompassed within the invention is modification of the adenoviruses of the invention through deletion of a section of the adenoviral genome which binds to the cellular Rb protein, commonly known as a "delta 24" deletion. Such deletions can result in adenoviruses with enhanced tumor specificity (Fueyo et al. (2000) Oncogene 19:2-12).
Another modification encompassed by the invention is deletion of the E3 region of the oncolytic adenoviruses of the invention to increase their potency by a mechanism distinct from reduction of genome size.
Methods for the construction of modified adenoviruses are generally known in the art. See, Mittal, S. K. (1993) Virus Res. 28:67-90 and Hermiston, T. et al. (1999) Methods in Molecular Medicine: Adenovirus Methods and Protocols, W.S.M. Wold, ed, Humana Press. Standard techniques are used for recombinant nucleic acid methods, polynucleotide synthesis, and microbial culture and transformation (e.g., electroporation, lipofection). Generally, enzymatic reactions and purification steps are performed according to the manufacturer's specifications. The techniques and procedures are generally performed according to conventional methods in the art and various general references (see generally, Sambrook et al., Molecular Cloning: A Laboratory Manual, 2nd edition (1989) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.) which are provided throughout this document. The nomenclature used herein and the laboratory procedures in analytical chemistry, organic synthetic chemistry, and pharmaceutical formulation described below are those well known and commonly employed in the art.
Determination of Therapeutic Potential
Oncolytic adenoviruses of the invention, or variants or derivatives thereof, can be evaluated for their therapeutic utility by examination of their lytic potential in tumor cells derived from tissues of interest as therapeutic targets. Tumor cell lines useful for testing adenoviruses of the invention include, but are not limited to, colon cell lines, including but not limited to, DLD-1, HCT116, HT29, LS1034 and SW48 cell lines; prostate cell lines, including but not limited to, DU145 and PC-3 cell lines; pancreatic cell lines, including but not limited to, the Panc-1 cell line; breast tumor cell lines, including but not limited to, the MDA231 cell line; and ovarian cell lines, including but not limited to, the OVCAR-3, CaOV3, BG1 , ES-2 and IGROV and SKOV3 cell lines. Any other tumor cell lines, available through sources such as the American Type Culture Collection, which are representative of a tissue target of interest, can be used in identifying and evaluating adenoviruses of the invention for use in the treatment of neoplasia of that tissue type. Alternatively, evaluation of the oncolytic adenoviruses of the invention can also be performed using matched human primary tumor and normal explants, e.g. through quantitation of viral burst (U.S. Patent Application No. 11/136,912) or reporter gene expression (Lam et al. (2003) Cancer Gene Therapy 10:377-387; Grill et al. (2003) MoI. Therapy 6:609-614). See Example 8.
The cytolytic activity of adenoviruses of the invention can be determined in representative tumor cell lines and the data converted to a measurement of potency (i.e. IC50). A preferred method for determining cytolytic activity is an MTS assay (see Example 4).
The therapeutic index of an adenovirus of the invention in a particular tumor cell can be calculated by comparison of the potency of the given adenovirus in the tumor cell with the potency of that same adenovirus in a matched normal cell. Preferred non-cancerous cells are SAEC cells (Cambrex/Clonetics, Inc., Walkersville, MD), which are epithelial in origin, and HUVEC cells (VEC
Technologies, Rennselaer, NY), which are endothelial in origin (see Figures 2 and 4). These two cell types represent normal cells from which organs and vasculature, respectively, are derived, and are representative of likely sites of toxicity during adenoviral therapy, depending on the mode of delivery of the adenovirus. However, practice of the invention is not limited to the use of these cells, and other non-cancerous cells (e.g. B cells, T ceils, macrophages, monocytes, fibroblasts) may also be used.
The oncolytic adenoviruses of the invention can be further evaluated for their ability to target neoplastic cell growth (i.e. cancer) by their capacity to reduce tumorigenesis or neoplastic cell burden in nude mice harboring a transplant of neoplastic cells, as compared to untreated mice harboring an equivalent neoplastic cell burden (see Example 6, Figures 6A and B).
Therapeutic Utility
The present invention provides for the use of the oncolytic adenoviruses of the invention for the inhibition of tumor cell growth, as well as for the use of adenoviral vectors derived from these adenoviruses to deliver therapeutic proteins useful in the treatment of neoplasia.
Pharmaceutical Compositions and Administration
The present invention also relates to pharmaceutical compositions which comprise the chimeric/oncolytic adenoviruses of the invention, including variants and derivatives thereof, formulated for therapeutic administration to a patient. For therapeutic use, a sterile composition containing a pharmacologically effective dosage of adenovirus is administered to a human patient or veterinary non- human patient for treatment, for example, of a neoplastic condition. Generally, the composition will comprise about 1011 or more adenovirus particles in an aqueous suspension. A pharmaceutically acceptable carrier or excipient is often employed in such sterile compositions. A variety of aqueous solutions can be used, e.g. water, buffered water, 0.4% saline, 0.3% -glycine and the like. These solutions are sterile and generally free of particulate matter other than the desired adenoviral vector. The compositions may contain pharmaceutically acceptable auxiliary substances as required to approximate physiological conditions such as pH adjusting and buffering agents, toxicity adjusting agents and the like, e.g. sodium acetate, sodium chloride, potassium chloride, calcium chloride, sodium lactate, etc. Excipients which enhance infection of cells by adenovirus may be included, (see U.S. Patent No. 6,392,069).
Adenoviruses of the invention may also be delivered to neoplastic cells by liposome or immunoliposome delivery; such delivery may be selectively targeted to neoplastic cells on the basis of a cell surface property present on the neoplastic cell population (e.g., the presence of a cell surface protein which binds an immunoglobulin in an immunoliposome). Typically, an aqueous suspension containing the virions are encapsulated in liposomes or immunoliposomes. For example, a suspension of adenovirus virions can be encapsulated in micelles to form immunoliposomes by conventional
methods (U.S. Patent No. 5,043,164, U.S. Patent No.4,957,735, U.S. Patent No.4,925,661; Connor and Huang, (1985) J. Cell Biol. 101: 581; Lasic D.D. (1992) Nature 355: 279; Novel Drug Delivery (eds. Prescott and Nimmo, Wiley, New York, 1989); Reddy et al. (1992) J. Immunol. 148:1585). lmmunoliposomes comprising an antibody that binds specifically to a cancer cell antigen (e.g., CALLA, CEΞA) present on the cancer cells of the individual may be used to target virions to those cells (Fisher (2001) Gene Therapy 8:341-348).
To further increase the efficacy of the adenoviruses of the invention, they may be modified to exhibit enhanced tropism for particular tumor cell types. For example, as shown in PCT/US98/04964, a protein on the exterior coat of an adenovirus may be modified to display a chemical agent, preferably a polypeptide, that binds to a receptor present on tumor cells to a greater degree than normal cells. (See also, U. S. Patent Nos. 5,770,442 and 5,712,136). The polypeptide can be an antibody, and preferably is a single chain antibody.
Adenoviral Therapy
The adenoviruses of the invention, or pharmaceutical compositions thereof, can be administered for therapeutic treatment of neoplastic disease or cancer. In therapeutic applications, compositions are administered to a patient already affected by the particular neoplastic disease, in an amount sufficient to cure or at least partially arrest the condition and its complications. An amount adequate to accomplish this is defined as a "therapeutically effective dose" or "efficacious dose". Amounts effective for this use will depend upon the severity of the condition, the general state of the patient, and the route of administration.
For example, but not by way of limitation, a human patient or non-human mammal having a solid or haemotologic neoplastic disease, (e.g. pancreatic, colon, ovarian, lung, or breast carcinoma, leukemia or multiple myeloma) may be treated by administering a therapeutically effective dosage of an appropriate oncolytic adenovirus of the invention, i.e. one which has been shown to have an improved therapeutic index for that tissue type. A preferred oncolytic adenovirus for the treatment of ovarian cancer would be the adenovirus OvAd2 (SEQ ID NO: 2). A particularly preferred oncolytic adenovirus for the treatment of ovarian cancer would be OvAdI(SEQ ID NO:1).
Suspensions of infectious adenovirus particles may be delivered to neoplastic tissue by various routes, including intravenous, intraperitoneal, intramuscular, intratumoral, subdermal, and topical. An adenovirus suspension containing about 103 to 1012or more virion particles per ml may be administered by infusion (e.g., into the peritoneal cavity for treating ovarian cancer).
Adenoviral therapy using the adenoviruses of the instant invention may be combined with other antineoplastic protocols, such as conventional chemotherapy or x-ray therapy to treat a particular cancer.
Adβnoviral therapy using the adenoviruses of the instant invention as adenoviral vectors may also be combined with other genes known to be useful in viral based therapy. See U. S. Patent No. 5,648,478. In such cases, the chimeric/oncolytic adenovirus further comprises a heterologous gene that encodes a therapeutic protein, incorporated within the viral genome, such that the heterologous gene is expressed within an infected cell. A therapeutic protein, as used herein, refers to a protein that would be expected to provide some therapeutic benefit when expressed in a given cell.
In one embodiment, the heterologous gene can be the thymidine kinase (TK) gene, which is useful as a pro-drug converting enzyme (Freeman, S.M. (2000J Adv Exp Med Biol 465:411-422). TK can also be used as a marker or reporter for tracking the efficiency of viral infection ( Sangro et al. (2002) MoI. Imaging Biol. 4:27-33).
In one embodiment, the heterologous gene is a pro-drug activator gene, such as cytosine deaminase (CD) (See, U.S. Patent Nos. 5,631,236; 5,358,866; and 5,677,178). In other embodiments, the heterologous gene is a known inducer of cell-death, e.g apoptin or adenoviral death protein (ADP), or a fusion protein, e.g. fusogenic membrane glycoprotien (Danen-Van Oorschot et al. (1997) Proc. Nat. Acad. Sc/. 94:5843-5847; Tollefson et al.(1996) J. Virol. 70:2296-2306; Fu et al. (2003) MoI. Therapy 7; 48-754, 2003; Ahmed et al. (2003) Gene Therapy 10:1663-1671; Galanis et al. (2001) Human Gene Therapy 12(7): 811-821).
Further examples of heterologous genes, or fragments thereof, include those that encode immunomodulatory proteins, such as cytokines or chemokines. Examples include interieukin 2, U.S. Patent Nos. 4,738,927 or 5,641,665; interieukin 7, U. S. Patent Nos. 4,965,195 or 5,328,988; and interieukin 12, U. S. Patent No. 5,457,038; tumor necrosis factor alpha, U. S. Patent Nos. 4,677,063 or 5,773,582; interferon gamma, U.S. Patent Nos. 4,727,138 or 4,762,791; or GM CSF, U.S. Patent Nos. 5,393,870 or 5,391 ,485, Mackensen et al. (1997) Cytokine Growth Factor Rev. 8:119-128). Additional immunomodulatory proteins include macrophage inflammatory proteins, including MIP- 3. Monocyte chemotatic protein (MCP-3 alpha) may also be used; a preferred embodiment of a heterologous gene is a chimeric gene consisting of a gene that encodes a protein that traverses cell membranes, for example, VP22 or TAT, fused to a gene that encodes a protein that is preferably toxic to cancer but not normal cells.
Another example of a heterologous gene is an antibody or antibody fragment. Preferred antibodies are targeted against epidermal growth factor (EGF) or tissue factor (TF) (Jiang et al. (2006) Clin Cancer Res 12:6179-6185; Kasuya et al. (2005) MoI Ther 11 :237-244)
The oncolytic adenoviruses of the invention can also be used as vectors to deliver genes encoding therapeutically useful RNA molecules, i.e. siRNA (Dorsett and Tuschl (2004) Nature Rev Drug Disc 3:318-329), shRNA, or miRNA or aptamers.
In some cases, genes can be incorporated into an oncolytic adenovirus of the invention to further enhance the ability of the oncolytic virus to eradicate the tumor, although not having any direct impact on the tumor itself - these include genes encoding proteins that compromise MHC class I presentation (Hewitt et al. (2003^ Immunology 110: 163-169), block complement, inhibit IFNs and IFN- induced mechanisms, chemokines and cytokines, NK cell based killing (Orange et al., (2002) Nature Immunol. 3: 1006-1012; Mireille et al. (2002) immunogenetics 54: 527-542; Alcami (2003) Nature Rev. Immunol. 3: 36-50; down regulate the immune response (e.g. IL-10, TGF- Beta, Khong and Restifo (2002) Nature Immunol. 3: 999-1005; 2002) and metalloproteases which can breakdown the extracelluar matrix and enhance spread of the virus within the tumor (Bosman and Stamenkovic (2003) J. Pathol. 2000: 423-428; Visse and Nagase (2003) Circulation Res. 92: 827-839).
Kits
The invention further relates to pharmaceutical packs and kits comprising one or more containers filled with one or more of the ingredients of the aforementioned compositions of the invention. Associated with such container(s) can be a notice in the form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals or biological products, reflecting approval by the agency of the manufacture, use or sale of the product for human administration.
The present invention is further described by the following examples, which are illustrative of specific embodiments of the invention, and various uses thereof. These exemplifications, which illustrating certain specific aspects of the invention, do not portray the limitations or circumscribe the scope of the disclosed invention.
Unless otherwise indicated, the practice of the present invention employs conventional techniques of cell culture, molecular biology, microbiology, recombinant DNA manipulation, immunology science, which are within the skill of the art. Such techniques are explained fully in the literature. See, e.g. Cell Biology: a Laboratory Handbook: J. Celis (Ed).Academic Press. N.Y. (1996); Graham, Fl. and Prevec, L. Adenovirus-based expression vectors and recombinant vaccines. In: Vaccines: New Approaches to Immunological Problems. R.W. Ellis (ed) Butterworth. Pp 363-390; Grahan and Prevec Manipulation of adenovirus vectors. In: Methods in Molecular Biology, Vol. 7: Gene Transfer and Expression Techniques. EJ. Murray and J.M. Walker (eds) Humana Press Inc., Clifton, NJ. pp 109- 128, 1991; Sambrook et al. (1989), Molecular Cloning, A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press; Sambrook et al. (1989), and Ausubel et al. (1995), Short Protocols in Molecular Biology, John Wiley and Sons.
EXAMPLES
Methods
Standard techniques are used for recombinant nucleic acid methods, polynucleotide synthesis, and microbial culture and transformation (e.g., eiectroporation, lipofection). Generally, enzymatic reactions and purification steps are performed according to the manufacturer's specifications. The techniques and procedures are generally performed according to conventional methods in the art and various general references (see generally, Sambrook et al., Molecular Cloning: A Laboratory Manual, 2nd. edition (1989) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.) which are provided throughout this document. The nomenclature used herein and the laboratory procedures in analytical chemistry, organic synthetic chemistry, and pharmaceutical formulation and delivery, and treatment of patients. Methods for the construction of adenoviral mutants are generally known in the art. See, Mittal, S. K. Virus Res.,1993, vol: 28, pages 67-90; and Hermiston, T. et al., Methods in Molecular Medicine: Adenovirus Methods and Protocols, W.S.M. Wold, ed., Humana Press, 1999. The adenovirus 5 genome is registered as Genbank 10 accession #M73260, and the virus is available from the American Type Culture Collection, Rockville, Maryland, U. S. A., under accession number VR-5.
Viruses and Cell Lines
The human Ad serotypes Ad3 (GB strain), Ad4 (RI-67 strain), Ad5 (Adenoid 75 strain), Ad9 (Hicks strain), Ad16 (Ch. 79 strain); and the SKOV3, OVCAR3, CaOV3, IGROV, BG1, ES-2, PC-3, and HT-29 cell lines used were all purchased from the ATCC. The chimeric adenovirus CoIoAdI is described in U.S. Patent Application Serial No. 11/136,912. Ad35, Ad11 p (Slobitski strain), and Ad40 were kind gifts from Dr. William S.M. Wold at St. Louis University. Other cells used were MDA-231mt1 (a cell line derivative isolated by Dr. Deb Zajchowski, Berlex Laboratories, from a rapidly growing subcutaneously implanted xenograft of MDA-231 cells) and Panel -set (derived by Dr. Sandra Biroc, Berlex Laboratories, from a rapidly growing subcutaneously implanted xenograft of Panel cells), HUVEC (Vec Technologies, Rensselaer, NY)1 and SAEC (Clonetics, Walkersville, MD).
Example 1 • Viral Purification and Quantitation
Viral stocks were propagated on either 293 cells or SKOV3 cells, purified on CsCI gradients, and titered (viral particles per ml, vp/ml, is the unit used throughout this report) by spectroscopy (Tollefson, A., Hermiston, T.W., and Wold, W.S.M.; "Preparation and Titration of CsCI-banded Adenovirus Stock" in Adenovirus Methods and Protocols. Humana Press, 1999, pp 1-10, W.S.M. Wold, Ed )and by aπion-exchange (AIEX) chromatography (Kuhn et al. (2006) Gene Therapy ). Most Ad serotypes have distinct, characteristic retention profiles when analyzed by the AIEX method used, and viral purification is not necessary to determine an accurate titer by AIEX, allowing accurate quantification of crude lysates as described (ibid.). Therefore AIEX chromatography was used to verify the
spectroscopic titer of pure viral stocks, to determine the titer of crude viral lysates, and to partially characterize the serotype-relatedness of all viral stocks.
The method used to quantitate viral particles is as described in Kuhn et al. (2006) Gene Therapy published on-line. In brief, a 1.25 ml column was packed with Q Sepharose XL Media (Pharmacia). HPLC separation was performed on an Agilent HP 1100 HPLC using the following conditions: Buffer A = 20 mM TrisHCI, pH 7.5; Buffer B = 1.0 M NaCI in Buffer A; flow rate of 1 ml per minute. After column equilibration for not less than 30 minutes in Buffer A, approximately 109-1011 viral particles of sample were loaded onto the column in 10-100 ul volume, followed by 4 column volumes of Buffer A. A linear gradient extending over 16 column volumes and ending in 100% Buffer B was applied.
The column effluent was monitored at A260 and A280 nm, peak areas calculated, and the 260 to 280 nm ratio determined. Viral peaks were identified as those narrow, sharp peaks having a A260/A280 ratio close to 1.33. A virus standard was included with each sample series. The number of viral particles per ml of the standard had been determined using the method of Lehmberg et al. (1999) J. Chrom. B, 732:411-423}. In the viral concentration range used, the A260 nm peak area of each sample is directly proportional to the number of viral particles in the sample. The number of viral particles per ml in each test sample was calculated by multiplying the known number of viral particles per ml in the standard by the ratio of the A260 nm viral peak area of the sample to the A260 nm viral peak area of the standard.
The column was regenerated after each sample gradient by washing with at least two column volumes of 0.1-0.5 N NaOH followed by two column volumes of 100% Buffer A, 3 column volumes of 100% Buffer B, and then 4 column volumes of 100% Buffer A.
Example 2 - Directed Evolution
Viral serotypes representing subgroup Ads B-F, namely Ad3, Ad4, Ad5, Ad9, Ad11p, Ad16, Ad35, Ad40, and the chimeric virus CoIoAdI (U. S. Patent Application Serial No. 11/136,912)) were assembled into the starting viral pool. A portion of the starting pool, containing 1012 viral particles of each viral type, was subjected to random mutagenesis by nitrous acid (Williams et al. (1971) J Gen Virol 11 :95-101; Kfessig, D.F. (1977) J. Virol. 21:1243-1246). The reaction was stopped by neutralization of the nitrous acid after 2.5-3 logs of kill. A 356 bp region of the 19 K protein was amplified from each of 10 viral isolates from a parallel Ad5 mutagenized stock and sequenced. Sequencing showed that one in ten isolates carried a point mutation. Extrapolation from this result indicates that approximately 10 mutations were introduced on average per viral genome. The pool of mutagenized virus serotypes was called the mutagenized pool. Another portion of the starting pool,
containing 109 viral particles of each viral type, was then added to the mutagenized pool, resulting in a combined pool containing approximately equal numbers of mutagenized and non-mutagenized viral particles of each viral type. This combined pool was used to infect, at a multiplicity of infection (MOI)=IO, subconfluent monolayers of SKOV3, HT-29, and OVCAR3 cells. These infection conditions were chosen to invite recombination between all viral types present in the combined (mutagenized and non-mutagenized) viral pool. Viral lysates were harvested from these infected cultures at 24 and 48 hours post infection (hpi), then mixed together to produce the "recombined pool." A fresh aliquot of the combined pool was then added to this recombined pool to generate the viral pool used for directed evolution. The titer of this pool was determined by anion exchange chromatography as described above.
Directed Evolution on monolayer cultures: The biodiverse viral pool was passaged once on a sub-confluent culture of SKOV3 cells at MOI=IO, a high particle-per-cell ratio again used to invite recombination between all viruses present in the biodiverse pool. The titer of the viral lysate supernatant from this round of high viral particle-per-cell infection of subconfluent SKOV3 monolayer cells was determined by AIEX chromatography, then used in a 10-fold dilution series, starting at an MOI=0.1 , to infect a series of over-confluent SKOV3 cultures grown in 6-well plates. To achieve over- confluency, SKOV3 cells were seeded at split ratios that allowed that cell line to reach confluency between 24 and 40 hours post seeding, and the cells were allowed to grow a total of 72 hours post seeding prior to infection. This cell density was 150,000 cells per cm2. This high cell density and prolonged growth was used to maximize confluency at time of infection, with the goal of mimicking growth conditions in human solid tumors. Cell culture supernatant was harvested from the well infected with the most concentrated innocula in the 10-fold dilution series that did not show any sign of CPE at day 3 or 4 post-infection. Each harvest served as the starting material for the next passage of the virus. This process was repeated until the viral pool achieved 10 passages.
Directed evolution on MATRIGEL™ cultures: Directed evolution using SKOV3 cells grown on growth factor reduced MATRIGEL™ (Becton Dickinson Labware, Bedford, MA) coated tissue cultures plates as the target cell culture was done generally as described for directed evolution on monolayer cultures, with the exceptions that the culture dishes or wells were coated, following manufacturer's directions, with MATRIGEL™ at 15OuI/ cm2 prior to seeding the cells. SKOV3 cells were seeded onto MATRIGEL™-coated plates at about 150,000 cells per cm2, a density that generated obvious three-dimensional growth patterns in these cells by 24 hours post seeding (hps). The cells were infected 24-36 hours post seeding, i.e. soon after the three-dimensional growth patterns induced by MATRIGEL™ had become apparent in the cultures. One passage at MOI=IO was done to invite recombination between all the viral genomes in the pool before starting selective passaging. Selective passaging on MATRIGEL™ was done, as described for monolayer directed evolution, starting at an MOI of less than one viral particle per cell (to avoid complementation between genotypes), followed by three 10-fold serial dilutions. In this way, 10-fold serial dilutions (starting at an MOI=O.1) of the previous
selective passage supernatant were used to infect a series of SK0V3 cultures grown on MATRIGEL™. At each passage, culture supernatant was harvested from the culture infected with the most concentrated inoculates in the 10-fold dilution series that did not show any sign of CPE at day 3 or 4 post-infection. A total of 10 passages were performed before individual viruses were isolated and characterized from each selected pool.
The selected pools were analyzed by ion exchange chromatography, which demonstrated that the viral pool selected on SKOV3 monolayers was composed of Ad-3 related viruses, while the pool derived on MATRIGEL™ contains both Ad3 and Ad11 p/35-related viruses.
Example 3 - Isolation and Characterization of Selected Viruses
Individual viruses were isolated from each selected pool by two rounds of plaque purification on SKOV3 cells using standard methods (Tollefson, A., Hermiston, T.W., and Wold, W.S.M.; "Preparation and Titration of CsCI-banded Adenovirus Stock" in Adenovirus Methods and Protocols. Humana Press, 1999, pp 1-10, W.S.M. Wold, Ed). In brief, dilutions of the supernatant harvested from the 10th passage on SKOV3 cells grown either as monolayers, or on top of MATRIGEL™, were used to infect SKOV3 cells in a standard plaque assay. Individual plaques were harvested, and the same plaque assay method was used to generate a second round of individual plaques from these harvests. Plaques from the second round of plaque purification were deemed pure, infected cultures of A549 cells were prepared using these purified plaques, and the oncolytic potency of these culture lysates determined by MTS assay as described.
Example 4 - Cytolytic assay
Viral lytic capacity was measured using a modification of the MTT assay (Yan et al. (2003) J Virol 77:2640-2650). Briefly, the MTS assay (Promega, CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay) was used in place of the MTT assay because conversion of MTS by cells into aqueous, soluble formazan reduces time and eliminates the use of a volatile organic solvent associated with the MTT assay.
To perform the MTS assay, cells were seeded at a density determined for each tumor cell line to generate a confluent monolayer within 24 hours. These densely seeded cells were allowed to grow for 2 additional days prior to exposure to the test virus(es). Viral lysates or stocks to be assayed for potency by MTS assay were titered by anion exchange (AIEX) chromatography as described above. Infections of both tumor and primary normal cells were carried out in quadruplicate, using serial three fold dilutions of the viruses starting at a particle per cell ratio of 100 and ending at a particle per cell ratio of 0.005. Infected cells were incubated at 37° C and the MTS assay was performed at the time points indicated for the individual primary cells or tumor cell lines. Mock-infected cells served as negative
controls and established the 100% survival point for the given assay. Each data point in any given MTS assay was assessed in quadruplicate, and IC50 values were derived from dose response curves with R2V value of 0.9 or greater. Each MTS assay was repeated at least twice, with consistent results.
When performed on MATRIGEL™-grown cell cultures, the MTS assay method was modified as follows. 96-well plates were coated with Growth Factor Reduced MATRIGEL™ (Becton-Dickinson) at 0.15 ml/cm2. Cells were seeded on top of the MATRIGEL™ at densities that generated 3-dimensional growth within 24 hours post seeding. For SKOV3 cells, the seeding density was 150,000 cells/cm2. For HUVEC cells, the seeding density was 100,000 cells/cm2. Viruses were added to cells at 24 hours post seeding, using 3-fold serial dilutions of the viruses starting at a particle per cell ratio of 1 ,000 and ending at a particle per cell ratio of 0.05.
Example 5 - DNA sequencing
Sequencing of the isolated adenoviruses OvAdI (SEQ IO NO:1) and OvAd2 (SEQ ID NO:2) was accomplished as follows. A shotgun library was prepared using sheared purified template DNA. The sheared DNA was size-selected for the range 2-4 kb before insertion into the pUC18 vector. Library construction was done by the double-adapter method described in Anderson et al. (1996) Anal. Biochβm. 236:107-113. DNA cycle sequencing was performed on the PCR products using Big Dye Terminator v3.1 chemistry in conjunction with primers provided to SeqWright (M13 forward primer, 5■-GTAAAACGACGGCCAGT-31 (SEQ ID NO:5); M13 reverse primer, δ'-CAGGAAACAGCTATGAC (SEQ ID NO:6)). Sequence delineation and base-calling was performed using automated fluorescent DNA sequencers, ABI model 373OxI. All data including the final contig assembly was evaluated using a Phred20 scoring criteria. Sequence assembly and editing was carried out using Sequencher 4.5 Software (GeneCodes, Inc.). Sequence information was analyzed using the Vector NTI program (Informatix).
The sequences of OvAdI (SEQ ID NO:1 ) and OvAd2 (SEQ ID NO:2) were compared to the DNA sequence of CoIoAdI (SEQ ID NO:3; see U.S. Patent Application Serial No. 11/136,912) and to the DNA sequences of each of the other serotypes included in the starting viral pool. These analyses showed that OvAdI and OvAd2 are chimeras of CoIoAdI (including the CoIoAdI chimeric E2B region) and serotype Ad3 (right end) (SEQ ID NO:4). See Figure 5.
Example 6 - In Vivo Efficacy of Adenoviruses
Efficacy of OvAdI and OvAd2 in reducing tumor burden was shown in SKOV3 intraperitoneal tumour burden studies comparing anti cancer efficacy of Ad5, Onyx-015, OvAdL OvAd2, CoIoAdI and PBS. The studies were done using MF1 nude mice (5 per group first run; 7 per group on the repeat experiment). SKOV3 cells were administered into the peritoneal cavity of each mouse, 5x10e6 cells per
mouse, on day 0. The viruses were administered intraperitoneal^, 5x10e10 virus particles or vehicle administered on days 3, 5, and 7 in 0.5 ml PBS. There were no signs of acute toxicity (breathing difficulties, hunch, death, starry coat) observed following administration of any vector. Extensive peritoneal organ adhesions were observed in mice treated with either Ad5 or ONYX-015 viruses. These adhesions can be fatal. None of the non Ad5-based viruses (OvAdI , OvAd2, CoIoAdI1 or Ad11p) showed any signs of adhesions. Blood samples were taken 1 hour following the final vector administration; no vectors were detected in the blood stream, indicating that there is no peritoneal leakage of these viruses. All mice were euthanized and tumor burden measured after 18 days (the day on which the tumor burden in control mice became excessive). In this model the OvAdI virus was the most effective in reducing tumor burden, followed in efficacy by OvAd2 and the parental virus CoIoAdI and Ad 11 p. Ad5 was effective at reducing tumor burden but also caused extensive adhesions. Onyx- 015 was ineffective. (See Figures 6A and 6B).
This study was repeated, except that SK0V3-luciferase cells were used and the mice were not euthanized until they acquired excessive tumor burdens. Tumor burden was followed during the study using whole-mouse imaging after luciferin injection. The imaging results were consistent with the tumor burdens measured in the in vivo SKOV3 study described above. Further, survival of mice in the various treatment groups showed that mice in the Onyx-015 group died within the first 21 days while OvAdI treated mice were still alive after 45 days.
Example 7 - Ex vivo Efficacy of Adenoviruses
Ovarian ascites tumor cell samples removed during surgery were placed in culture media and infected with equal numbers of OvAdI, OvAd2, CoIoAdI, Ad3 or Onyx-015 adenoviral particles. Cell viability is measured after 5 days using MTS assay (described under cytolytic assay).
Example 8 - Activity of OvAdI on Primary Human Pluripotent Ovarian Cancer Progenitor
Cells
Ovarian tumor and matched normal tissue samples are removed during surgery, cultured separately and infected with equal numbers of OvAdI , OvAd2, Ad5, or Onyx-015 adenoviral particles. After 2-6 days the culture supernatant and the cells are harvested, ONA purified from each sample, and the number of viral genomes measured in each sample by Taqman assay using genome-specific probes.