WO2008072724A1 - ピラゾール誘導体 - Google Patents
ピラゾール誘導体 Download PDFInfo
- Publication number
- WO2008072724A1 WO2008072724A1 PCT/JP2007/074092 JP2007074092W WO2008072724A1 WO 2008072724 A1 WO2008072724 A1 WO 2008072724A1 JP 2007074092 W JP2007074092 W JP 2007074092W WO 2008072724 A1 WO2008072724 A1 WO 2008072724A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pyrazole
- oxy
- phenyl
- piperidine
- pyrrolidine
- Prior art date
Links
- 150000003217 pyrazoles Chemical class 0.000 title claims abstract description 15
- 150000003839 salts Chemical class 0.000 claims abstract description 15
- 208000019116 sleep disease Diseases 0.000 claims abstract description 8
- 208000024827 Alzheimer disease Diseases 0.000 claims abstract description 7
- 208000017164 Chronobiology disease Diseases 0.000 claims abstract description 7
- 206010012289 Dementia Diseases 0.000 claims abstract description 7
- 208000030814 Eating disease Diseases 0.000 claims abstract description 7
- 208000019454 Feeding and Eating disease Diseases 0.000 claims abstract description 7
- 208000008589 Obesity Diseases 0.000 claims abstract description 7
- 206010012601 diabetes mellitus Diseases 0.000 claims abstract description 7
- 235000014632 disordered eating Nutrition 0.000 claims abstract description 7
- 201000003631 narcolepsy Diseases 0.000 claims abstract description 7
- 235000020824 obesity Nutrition 0.000 claims abstract description 7
- 201000000980 schizophrenia Diseases 0.000 claims abstract description 7
- 201000002859 sleep apnea Diseases 0.000 claims abstract description 7
- 239000004480 active ingredient Substances 0.000 claims abstract description 6
- -1 pyrrolidine 1-ylcarbonyl Chemical group 0.000 claims description 167
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 84
- 229910052757 nitrogen Inorganic materials 0.000 claims description 64
- 125000000217 alkyl group Chemical group 0.000 claims description 57
- NQRYJNQNLNOLGT-UHFFFAOYSA-N tetrahydropyridine hydrochloride Natural products C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 53
- 239000000126 substance Substances 0.000 claims description 44
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 37
- 229910052736 halogen Inorganic materials 0.000 claims description 34
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 claims description 34
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 33
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 31
- RHRLGFPTYKGJPR-UHFFFAOYSA-N 1-cyclobutylpiperidine Chemical compound C1CCC1N1CCCCC1 RHRLGFPTYKGJPR-UHFFFAOYSA-N 0.000 claims description 29
- 125000003545 alkoxy group Chemical group 0.000 claims description 29
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 28
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 28
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 26
- 150000002367 halogens Chemical group 0.000 claims description 26
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 24
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 claims description 21
- 125000000623 heterocyclic group Chemical group 0.000 claims description 19
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 19
- ZVXKYWHJBYIYNI-UHFFFAOYSA-N 1h-pyrazole-4-carboxamide Chemical compound NC(=O)C=1C=NNC=1 ZVXKYWHJBYIYNI-UHFFFAOYSA-N 0.000 claims description 18
- 239000003814 drug Substances 0.000 claims description 18
- 125000003118 aryl group Chemical group 0.000 claims description 17
- BXDZOYLPNAIDOC-UHFFFAOYSA-N N-[5-[(5-tert-butyl-1,3-oxazol-2-yl)methylsulfanyl]-1,3-thiazol-2-yl]-1-[2-[2-[2-[2-[2-[[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]amino]ethoxy]ethoxy]ethoxy]ethylamino]-2-oxoethyl]piperidine-4-carboxamide Chemical compound CC(C)(C)c1cnc(CSc2cnc(NC(=O)C3CCN(CC(=O)NCCOCCOCCOCCNc4cccc5C(=O)N(C6CCC(=O)NC6=O)C(=O)c45)CC3)s2)o1 BXDZOYLPNAIDOC-UHFFFAOYSA-N 0.000 claims description 15
- 125000001072 heteroaryl group Chemical group 0.000 claims description 15
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 14
- 125000004434 sulfur atom Chemical group 0.000 claims description 13
- 239000002253 acid Substances 0.000 claims description 12
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 12
- 229910052760 oxygen Inorganic materials 0.000 claims description 12
- 239000001301 oxygen Substances 0.000 claims description 12
- 125000004432 carbon atom Chemical group C* 0.000 claims description 11
- 125000005843 halogen group Chemical group 0.000 claims description 11
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 claims description 10
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 8
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 7
- 206010010904 Convulsion Diseases 0.000 claims description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 6
- 229910052799 carbon Inorganic materials 0.000 claims description 6
- 230000036461 convulsion Effects 0.000 claims description 6
- 206010015037 epilepsy Diseases 0.000 claims description 6
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 claims description 5
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 5
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 claims description 5
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 claims description 5
- 125000004473 dialkylaminocarbonyl group Chemical group 0.000 claims description 5
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 claims description 5
- 229940124597 therapeutic agent Drugs 0.000 claims description 5
- JUBXTBVKELTJEX-UHFFFAOYSA-N 1-cyclopropylpiperidine Chemical compound C1CC1N1CCCCC1 JUBXTBVKELTJEX-UHFFFAOYSA-N 0.000 claims description 4
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 4
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 4
- 230000006735 deficit Effects 0.000 claims description 4
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 claims description 4
- 239000003395 histamine H3 receptor antagonist Substances 0.000 claims description 4
- 208000013403 hyperactivity Diseases 0.000 claims description 4
- 125000004176 4-fluorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1F)C([H])([H])* 0.000 claims description 3
- 229940115480 Histamine H3 receptor antagonist Drugs 0.000 claims description 3
- 206010020751 Hypersensitivity Diseases 0.000 claims description 3
- 125000003342 alkenyl group Chemical group 0.000 claims description 3
- 125000004429 atom Chemical group 0.000 claims description 3
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- GXZZSIMZQGSQKH-UHFFFAOYSA-N n-(4-fluorophenyl)-1h-pyrazole-4-carboxamide Chemical compound C1=CC(F)=CC=C1NC(=O)C1=CNN=C1 GXZZSIMZQGSQKH-UHFFFAOYSA-N 0.000 claims description 3
- MEDLTPIUZUEAOV-UHFFFAOYSA-N 1-cyclobutyl-4-[4-(4-nitropyrazol-1-yl)phenoxy]piperidine Chemical compound C1=C([N+](=O)[O-])C=NN1C(C=C1)=CC=C1OC1CCN(C2CCC2)CC1 MEDLTPIUZUEAOV-UHFFFAOYSA-N 0.000 claims description 2
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical compound O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 claims description 2
- 125000004847 2-fluorobenzyl group Chemical group [H]C1=C([H])C(F)=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 2
- 125000006284 3-fluorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(F)=C1[H])C([H])([H])* 0.000 claims description 2
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 claims description 2
- UCYASVPBIJJZIG-UHFFFAOYSA-N [1-[4-(1-cyclobutylpiperidin-4-yl)oxy-2-fluorophenyl]pyrazol-4-yl]-pyrrolidin-1-ylmethanone Chemical compound C=1C=C(N2N=CC(=C2)C(=O)N2CCCC2)C(F)=CC=1OC(CC1)CCN1C1CCC1 UCYASVPBIJJZIG-UHFFFAOYSA-N 0.000 claims description 2
- PGUSDKVRWAICNY-UHFFFAOYSA-N [1-[4-(1-cyclobutylpiperidin-4-yl)oxy-3-fluorophenyl]pyrazol-4-yl]-pyrrolidin-1-ylmethanone Chemical compound FC1=CC(N2N=CC(=C2)C(=O)N2CCCC2)=CC=C1OC(CC1)CCN1C1CCC1 PGUSDKVRWAICNY-UHFFFAOYSA-N 0.000 claims description 2
- AHGAWVWRVRDWJW-UHFFFAOYSA-N [1-[4-(1-cyclobutylpiperidin-4-yl)oxyphenyl]pyrazol-4-yl]-morpholin-4-ylmethanone Chemical compound C1=NN(C=2C=CC(OC3CCN(CC3)C3CCC3)=CC=2)C=C1C(=O)N1CCOCC1 AHGAWVWRVRDWJW-UHFFFAOYSA-N 0.000 claims description 2
- TVZZMUOBQBZOOQ-UHFFFAOYSA-N [1-[4-(1-ethylpiperidin-4-yl)oxyphenyl]pyrazol-4-yl]-pyrrolidin-1-ylmethanone Chemical compound C1CN(CC)CCC1OC1=CC=C(N2N=CC(=C2)C(=O)N2CCCC2)C=C1 TVZZMUOBQBZOOQ-UHFFFAOYSA-N 0.000 claims description 2
- BKXDXDPQUDZXDI-UHFFFAOYSA-N [1-[4-[1-(3-hydroxycyclopentyl)piperidin-4-yl]oxyphenyl]pyrazol-4-yl]-pyrrolidin-1-ylmethanone Chemical compound C1C(O)CCC1N1CCC(OC=2C=CC(=CC=2)N2N=CC(=C2)C(=O)N2CCCC2)CC1 BKXDXDPQUDZXDI-UHFFFAOYSA-N 0.000 claims description 2
- RTVNFIGNQQROGA-UHFFFAOYSA-N [1-[4-[1-(3-methylbut-2-enyl)piperidin-4-yl]oxyphenyl]pyrazol-4-yl]-pyrrolidin-1-ylmethanone Chemical compound C1CN(CC=C(C)C)CCC1OC1=CC=C(N2N=CC(=C2)C(=O)N2CCCC2)C=C1 RTVNFIGNQQROGA-UHFFFAOYSA-N 0.000 claims description 2
- 230000007815 allergy Effects 0.000 claims description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 2
- VILAVOFMIJHSJA-UHFFFAOYSA-N dicarbon monoxide Chemical group [C]=C=O VILAVOFMIJHSJA-UHFFFAOYSA-N 0.000 claims description 2
- 150000004678 hydrides Chemical class 0.000 claims description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 2
- KYHLCISBAUJJDH-UHFFFAOYSA-N n-methyl-1h-pyrazole-4-carboxamide Chemical compound CNC(=O)C=1C=NNC=1 KYHLCISBAUJJDH-UHFFFAOYSA-N 0.000 claims description 2
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 claims description 2
- CRBRGXZDFQPDBI-UHFFFAOYSA-N phenol;1h-pyrazole Chemical compound C=1C=NNC=1.OC1=CC=CC=C1 CRBRGXZDFQPDBI-UHFFFAOYSA-N 0.000 claims description 2
- 230000003449 preventive effect Effects 0.000 claims description 2
- 206010039083 rhinitis Diseases 0.000 claims description 2
- KVTUSMPNLUCCQO-UHFFFAOYSA-N 3,3-difluoropyrrolidine Chemical compound FC1(F)CCNC1 KVTUSMPNLUCCQO-UHFFFAOYSA-N 0.000 claims 2
- 125000002944 cyanoaryl group Chemical group 0.000 claims 1
- SEEZDTHPQXRYPJ-UHFFFAOYSA-N pyrazol-1-yl(pyrrolidin-1-yl)methanone Chemical compound N1(N=CC=C1)C(=O)N1CCCC1 SEEZDTHPQXRYPJ-UHFFFAOYSA-N 0.000 claims 1
- 150000001875 compounds Chemical class 0.000 abstract description 230
- 102000004384 Histamine H3 receptors Human genes 0.000 abstract description 24
- 108090000981 Histamine H3 receptors Proteins 0.000 abstract description 24
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 10
- 201000010099 disease Diseases 0.000 abstract description 7
- 206010039085 Rhinitis allergic Diseases 0.000 abstract description 5
- 201000010105 allergic rhinitis Diseases 0.000 abstract description 5
- 230000003042 antagnostic effect Effects 0.000 abstract description 4
- 230000002265 prevention Effects 0.000 abstract description 3
- 208000036864 Attention deficit/hyperactivity disease Diseases 0.000 abstract description 2
- 208000015802 attention deficit-hyperactivity disease Diseases 0.000 abstract description 2
- 208000020685 sleep-wake disease Diseases 0.000 abstract description 2
- 239000008177 pharmaceutical agent Substances 0.000 abstract 2
- 201000005577 familial hyperlipidemia Diseases 0.000 abstract 1
- 238000000034 method Methods 0.000 description 79
- 238000006243 chemical reaction Methods 0.000 description 76
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 44
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 42
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 42
- 239000002904 solvent Substances 0.000 description 40
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 36
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 36
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 34
- 238000005859 coupling reaction Methods 0.000 description 31
- 238000004519 manufacturing process Methods 0.000 description 30
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 24
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 24
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 24
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 21
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 20
- 230000008569 process Effects 0.000 description 20
- 239000000243 solution Substances 0.000 description 20
- 238000012360 testing method Methods 0.000 description 20
- 230000009977 dual effect Effects 0.000 description 19
- 239000011541 reaction mixture Substances 0.000 description 19
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 19
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 229960001340 histamine Drugs 0.000 description 17
- 230000002829 reductive effect Effects 0.000 description 17
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 16
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 15
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 15
- 239000000203 mixture Substances 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 14
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 14
- 229940044551 receptor antagonist Drugs 0.000 description 14
- 239000002464 receptor antagonist Substances 0.000 description 14
- 239000007787 solid Substances 0.000 description 14
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 230000027455 binding Effects 0.000 description 12
- 230000035484 reaction time Effects 0.000 description 12
- 238000005160 1H NMR spectroscopy Methods 0.000 description 11
- 241000700159 Rattus Species 0.000 description 11
- 150000002170 ethers Chemical class 0.000 description 11
- 239000012046 mixed solvent Substances 0.000 description 11
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 11
- 235000017557 sodium bicarbonate Nutrition 0.000 description 11
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 10
- 150000001408 amides Chemical class 0.000 description 10
- 229930195733 hydrocarbon Natural products 0.000 description 10
- 150000002430 hydrocarbons Chemical class 0.000 description 10
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 9
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- 238000010898 silica gel chromatography Methods 0.000 description 9
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 8
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- 229910000027 potassium carbonate Inorganic materials 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 7
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 7
- 229910000024 caesium carbonate Inorganic materials 0.000 description 7
- 239000003054 catalyst Substances 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 150000001298 alcohols Chemical class 0.000 description 6
- 125000003277 amino group Chemical group 0.000 description 6
- 125000001153 fluoro group Chemical group F* 0.000 description 6
- 238000007429 general method Methods 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- 150000008282 halocarbons Chemical class 0.000 description 6
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 6
- 102000005962 receptors Human genes 0.000 description 6
- 108020003175 receptors Proteins 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 150000007942 carboxylates Chemical class 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 230000021824 exploration behavior Effects 0.000 description 5
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 5
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- 238000006722 reduction reaction Methods 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- XOFLBQFBSOEHOG-UUOKFMHZSA-N γS-GTP Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=S)[C@@H](O)[C@H]1O XOFLBQFBSOEHOG-UUOKFMHZSA-N 0.000 description 5
- XNQIOISZPFVUFG-RXMQYKEDSA-N (R)-alpha-methylhistamine Chemical compound C[C@@H](N)CC1=CN=CN1 XNQIOISZPFVUFG-RXMQYKEDSA-N 0.000 description 4
- IMBBXSASDSZJSX-UHFFFAOYSA-N 4-Carboxypyrazole Chemical compound OC(=O)C=1C=NNC=1 IMBBXSASDSZJSX-UHFFFAOYSA-N 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 235000011054 acetic acid Nutrition 0.000 description 4
- 238000005804 alkylation reaction Methods 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- JWHOQZUREKYPBY-UHFFFAOYSA-N rubonic acid Natural products CC1(C)CCC2(CCC3(C)C(=CCC4C5(C)CCC(=O)C(C)(C)C5CC(=O)C34C)C2C1)C(=O)O JWHOQZUREKYPBY-UHFFFAOYSA-N 0.000 description 4
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- NHVHYFAWHCJELN-UHFFFAOYSA-N benzene;n,n-dimethylformamide Chemical compound CN(C)C=O.C1=CC=CC=C1 NHVHYFAWHCJELN-UHFFFAOYSA-N 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 238000010531 catalytic reduction reaction Methods 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 3
- SHQSVMDWKBRBGB-UHFFFAOYSA-N cyclobutanone Chemical compound O=C1CCC1 SHQSVMDWKBRBGB-UHFFFAOYSA-N 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 238000005984 hydrogenation reaction Methods 0.000 description 3
- 210000004379 membrane Anatomy 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 230000009871 nonspecific binding Effects 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- PWQLFIKTGRINFF-UHFFFAOYSA-N tert-butyl 4-hydroxypiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(O)CC1 PWQLFIKTGRINFF-UHFFFAOYSA-N 0.000 description 3
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- JVZXWNIRCBRDBD-UHFFFAOYSA-N 1-(4-iodophenoxy)piperidine Chemical compound IC1=CC=C(C=C1)ON1CCCCC1 JVZXWNIRCBRDBD-UHFFFAOYSA-N 0.000 description 2
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- QQBFZTKZYQFNHA-UHFFFAOYSA-N 1-tert-butylpiperidin-4-ol Chemical compound CC(C)(C)N1CCC(O)CC1 QQBFZTKZYQFNHA-UHFFFAOYSA-N 0.000 description 2
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- CDIIZULDSLKBKV-UHFFFAOYSA-N 4-chlorobutanoyl chloride Chemical compound ClCCCC(Cl)=O CDIIZULDSLKBKV-UHFFFAOYSA-N 0.000 description 2
- KRZCOLNOCZKSDF-UHFFFAOYSA-N 4-fluoroaniline Chemical compound NC1=CC=C(F)C=C1 KRZCOLNOCZKSDF-UHFFFAOYSA-N 0.000 description 2
- XORHNJQEWQGXCN-UHFFFAOYSA-N 4-nitro-1h-pyrazole Chemical compound [O-][N+](=O)C=1C=NNC=1 XORHNJQEWQGXCN-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 2
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 239000007868 Raney catalyst Substances 0.000 description 2
- 229910000564 Raney nickel Inorganic materials 0.000 description 2
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 230000002152 alkylating effect Effects 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 230000003542 behavioural effect Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 238000006482 condensation reaction Methods 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 239000012024 dehydrating agents Substances 0.000 description 2
- 230000018044 dehydration Effects 0.000 description 2
- 238000006297 dehydration reaction Methods 0.000 description 2
- 150000004985 diamines Chemical class 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 210000004188 enterochromaffin-like cell Anatomy 0.000 description 2
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 2
- 210000001156 gastric mucosa Anatomy 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 210000003630 histaminocyte Anatomy 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 238000003402 intramolecular cyclocondensation reaction Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 229910052987 metal hydride Inorganic materials 0.000 description 2
- 150000004681 metal hydrides Chemical class 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 239000002858 neurotransmitter agent Substances 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 2
- 229910000105 potassium hydride Inorganic materials 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 2
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- XSCHRSMBECNVNS-UHFFFAOYSA-N quinoxaline Chemical compound N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000006268 reductive amination reaction Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 229910052703 rhodium Inorganic materials 0.000 description 2
- 239000010948 rhodium Substances 0.000 description 2
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000017550 sodium carbonate Nutrition 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- WKFYEWXSRFQOKX-UHFFFAOYSA-N 1,4-dioxane;toluene Chemical compound C1COCCO1.CC1=CC=CC=C1 WKFYEWXSRFQOKX-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- DFNHWMSTHVIRFR-UHFFFAOYSA-N 1-cyclobutylpiperidin-4-ol Chemical compound C1CC(O)CCN1C1CCC1 DFNHWMSTHVIRFR-UHFFFAOYSA-N 0.000 description 1
- CAYTVMPUNMZTFU-UHFFFAOYSA-N 1-cyclopropylpiperidin-4-ol Chemical compound C1CC(O)CCN1C1CC1 CAYTVMPUNMZTFU-UHFFFAOYSA-N 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- QHOINBKBMJLHPY-UHFFFAOYSA-N 2-chloroethyl formate Chemical compound ClCCOC=O QHOINBKBMJLHPY-UHFFFAOYSA-N 0.000 description 1
- 125000001340 2-chloroethyl group Chemical group [H]C([H])(Cl)C([H])([H])* 0.000 description 1
- ISPYQTSUDJAMAB-UHFFFAOYSA-N 2-chlorophenol Chemical compound OC1=CC=CC=C1Cl ISPYQTSUDJAMAB-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- CSQWLXZVUXRZFL-UHFFFAOYSA-N 3-fluoro-4-iodophenol Chemical compound OC1=CC=C(I)C(F)=C1 CSQWLXZVUXRZFL-UHFFFAOYSA-N 0.000 description 1
- YHQXBTXEYZIYOV-UHFFFAOYSA-N 3-methylbut-1-ene Chemical compound CC(C)C=C YHQXBTXEYZIYOV-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- BAJQRLZAPXASRD-UHFFFAOYSA-N 4-Nitrobiphenyl Chemical compound C1=CC([N+](=O)[O-])=CC=C1C1=CC=CC=C1 BAJQRLZAPXASRD-UHFFFAOYSA-N 0.000 description 1
- WXNZTHHGJRFXKQ-UHFFFAOYSA-N 4-chlorophenol Chemical compound OC1=CC=C(Cl)C=C1 WXNZTHHGJRFXKQ-UHFFFAOYSA-N 0.000 description 1
- VSMDINRNYYEDRN-UHFFFAOYSA-N 4-iodophenol Chemical compound OC1=CC=C(I)C=C1 VSMDINRNYYEDRN-UHFFFAOYSA-N 0.000 description 1
- SJISCEAZUHNOMD-UHFFFAOYSA-N 4-phenylcyclohexan-1-amine Chemical compound C1CC(N)CCC1C1=CC=CC=C1 SJISCEAZUHNOMD-UHFFFAOYSA-N 0.000 description 1
- CONKBQPVFMXDOV-QHCPKHFHSA-N 6-[(5S)-5-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]methyl]-2-oxo-1,3-oxazolidin-3-yl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C[C@H]1CN(C(O1)=O)C1=CC2=C(NC(O2)=O)C=C1 CONKBQPVFMXDOV-QHCPKHFHSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- UCDBLYHPJMMFMY-UHFFFAOYSA-N 8-bromoquinoline-4-carbonitrile Chemical compound C1=CN=C2C(Br)=CC=CC2=C1C#N UCDBLYHPJMMFMY-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 102000007527 Autoreceptors Human genes 0.000 description 1
- 108010071131 Autoreceptors Proteins 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- PFHPEFBTTULXTK-UHFFFAOYSA-N C(C)(C)C(C1=CC=CC=C1)C(C)C.[N+](=[N-])(C(=O)O)C(=O)O Chemical compound C(C)(C)C(C1=CC=CC=C1)C(C)C.[N+](=[N-])(C(=O)O)C(=O)O PFHPEFBTTULXTK-UHFFFAOYSA-N 0.000 description 1
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- UDHXJZHVNHGCEC-UHFFFAOYSA-N Chlorophacinone Chemical compound C1=CC(Cl)=CC=C1C(C=1C=CC=CC=1)C(=O)C1C(=O)C2=CC=CC=C2C1=O UDHXJZHVNHGCEC-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- QPLDLSVMHZLSFG-UHFFFAOYSA-N Copper oxide Chemical compound [Cu]=O QPLDLSVMHZLSFG-UHFFFAOYSA-N 0.000 description 1
- 239000005751 Copper oxide Substances 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- DSLZVSRJTYRBFB-UHFFFAOYSA-N Galactaric acid Natural products OC(=O)C(O)C(O)C(O)C(O)C(O)=O DSLZVSRJTYRBFB-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 102000000543 Histamine Receptors Human genes 0.000 description 1
- 108010002059 Histamine Receptors Proteins 0.000 description 1
- 101150056637 Hrh2 gene Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 description 1
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical class NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 1
- 238000006751 Mitsunobu reaction Methods 0.000 description 1
- FHQDWPCFSJMNCT-UHFFFAOYSA-N N(tele)-methylhistamine Chemical compound CN1C=NC(CCN)=C1 FHQDWPCFSJMNCT-UHFFFAOYSA-N 0.000 description 1
- 206010028735 Nasal congestion Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- WYNCHZVNFNFDNH-UHFFFAOYSA-N Oxazolidine Chemical compound C1COCN1 WYNCHZVNFNFDNH-UHFFFAOYSA-N 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 229920005439 Perspex® Polymers 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- NPYPAHLBTDXSSS-UHFFFAOYSA-N Potassium ion Chemical compound [K+] NPYPAHLBTDXSSS-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- FKNQFGJONOIPTF-UHFFFAOYSA-N Sodium cation Chemical compound [Na+] FKNQFGJONOIPTF-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 208000007107 Stomach Ulcer Diseases 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- PTFCDOFLOPIGGS-UHFFFAOYSA-N Zinc dication Chemical compound [Zn+2] PTFCDOFLOPIGGS-UHFFFAOYSA-N 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- BBGVRHCRDKAXBM-UHFFFAOYSA-N [1-[4-(1-cyclobutylpiperidin-4-yl)oxy-2-methylphenyl]pyrazol-4-yl]-pyrrolidin-1-ylmethanone Chemical compound C=1C=C(N2N=CC(=C2)C(=O)N2CCCC2)C(C)=CC=1OC(CC1)CCN1C1CCC1 BBGVRHCRDKAXBM-UHFFFAOYSA-N 0.000 description 1
- HPIAANYREBBWAE-UHFFFAOYSA-N [1-[4-(1-cyclopentylpiperidin-4-yl)oxyphenyl]pyrazol-4-yl]-(3,3-difluoropyrrolidin-1-yl)methanone Chemical compound C1C(F)(F)CCN1C(=O)C1=CN(C=2C=CC(OC3CCN(CC3)C3CCCC3)=CC=2)N=C1 HPIAANYREBBWAE-UHFFFAOYSA-N 0.000 description 1
- BIPLUIUVTNVMOV-UHFFFAOYSA-N [1-[4-[1-(cyclopropylmethyl)piperidin-4-yl]oxyphenyl]pyrazol-4-yl]-pyrrolidin-1-ylmethanone Chemical compound C1=NN(C=2C=CC(OC3CCN(CC4CC4)CC3)=CC=2)C=C1C(=O)N1CCCC1 BIPLUIUVTNVMOV-UHFFFAOYSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 1
- 229960004373 acetylcholine Drugs 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- SWLVFNYSXGMGBS-UHFFFAOYSA-N ammonium bromide Chemical compound [NH4+].[Br-] SWLVFNYSXGMGBS-UHFFFAOYSA-N 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 230000037007 arousal Effects 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- ZSIQJIWKELUFRJ-UHFFFAOYSA-N azepane Chemical compound C1CCCNCC1 ZSIQJIWKELUFRJ-UHFFFAOYSA-N 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- ODWXUNBKCRECNW-UHFFFAOYSA-M bromocopper(1+) Chemical compound Br[Cu+] ODWXUNBKCRECNW-UHFFFAOYSA-M 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- AOGYCOYQMAVAFD-UHFFFAOYSA-M carbonochloridate Chemical compound [O-]C(Cl)=O AOGYCOYQMAVAFD-UHFFFAOYSA-M 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 230000037411 cognitive enhancing Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 229910000431 copper oxide Inorganic materials 0.000 description 1
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- SBTSVTLGWRLWOD-UHFFFAOYSA-L copper(ii) triflate Chemical compound [Cu+2].[O-]S(=O)(=O)C(F)(F)F.[O-]S(=O)(=O)C(F)(F)F SBTSVTLGWRLWOD-UHFFFAOYSA-L 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- SSJXIUAHEKJCMH-UHFFFAOYSA-N cyclohexane-1,2-diamine Chemical compound NC1CCCCC1N SSJXIUAHEKJCMH-UHFFFAOYSA-N 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- MQYQOVYIJOLTNX-UHFFFAOYSA-N dichloromethane;n,n-dimethylformamide Chemical compound ClCCl.CN(C)C=O MQYQOVYIJOLTNX-UHFFFAOYSA-N 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- YWEUIGNSBFLMFL-UHFFFAOYSA-N diphosphonate Chemical compound O=P(=O)OP(=O)=O YWEUIGNSBFLMFL-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 230000000142 dyskinetic effect Effects 0.000 description 1
- 230000020595 eating behavior Effects 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 1
- KACZQOKEFKFNDB-UHFFFAOYSA-N ethyl 1h-pyrazole-4-carboxylate Chemical compound CCOC(=O)C=1C=NNC=1 KACZQOKEFKFNDB-UHFFFAOYSA-N 0.000 description 1
- 125000006260 ethylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 125000004175 fluorobenzyl group Chemical group 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- DSLZVSRJTYRBFB-DUHBMQHGSA-N galactaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O DSLZVSRJTYRBFB-DUHBMQHGSA-N 0.000 description 1
- 230000027119 gastric acid secretion Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 230000003370 grooming effect Effects 0.000 description 1
- 229920000591 gum Polymers 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001245 hexylamino group Chemical group [H]N([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 229940077716 histamine h2 receptor antagonists for peptic ulcer and gord Drugs 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- YIAPLDFPUUJILH-UHFFFAOYSA-N indan-1-ol Chemical compound C1=CC=C2C(O)CCC2=C1 YIAPLDFPUUJILH-UHFFFAOYSA-N 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 125000005929 isobutyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])OC(*)=O 0.000 description 1
- 125000005932 isopentyloxycarbonyl group Chemical group 0.000 description 1
- 230000000366 juvenile effect Effects 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 150000002611 lead compounds Chemical class 0.000 description 1
- 229910001416 lithium ion Inorganic materials 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229940098895 maleic acid Drugs 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000007721 medicinal effect Effects 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000005948 methanesulfonyloxy group Chemical group 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- KWKAKUADMBZCLK-UHFFFAOYSA-N methyl heptene Natural products CCCCCCC=C KWKAKUADMBZCLK-UHFFFAOYSA-N 0.000 description 1
- 125000004458 methylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])[H] 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N n-Octanol Natural products CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 210000002850 nasal mucosa Anatomy 0.000 description 1
- 125000005933 neopentyloxycarbonyl group Chemical group 0.000 description 1
- 230000003957 neurotransmitter release Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000002560 nitrile group Chemical group 0.000 description 1
- 125000005245 nitryl group Chemical group [N+](=O)([O-])* 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000002903 organophosphorus compounds Chemical class 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 210000000578 peripheral nerve Anatomy 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-M phenolate Chemical compound [O-]C1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-M 0.000 description 1
- 229940031826 phenolate Drugs 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- DLYUQMMRRRQYAE-UHFFFAOYSA-N phosphorus pentoxide Inorganic materials O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- HDOWRFHMPULYOA-UHFFFAOYSA-N piperidin-4-ol Chemical compound OC1CCNCC1 HDOWRFHMPULYOA-UHFFFAOYSA-N 0.000 description 1
- DNUTZBZXLPWRJG-UHFFFAOYSA-M piperidine-1-carboxylate Chemical compound [O-]C(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-M 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 229910001414 potassium ion Inorganic materials 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 230000003518 presynaptic effect Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000004353 pyrazol-1-yl group Chemical group [H]C1=NN(*)C([H])=C1[H] 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- JHHZLHWJQPUNKB-UHFFFAOYSA-N pyrrolidin-3-ol Chemical compound OC1CCNC1 JHHZLHWJQPUNKB-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 229910052705 radium Inorganic materials 0.000 description 1
- HCWPIIXVSYCSAN-UHFFFAOYSA-N radium atom Chemical compound [Ra] HCWPIIXVSYCSAN-UHFFFAOYSA-N 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000033764 rhythmic process Effects 0.000 description 1
- ORIHZIZPTZTNCU-YVMONPNESA-N salicylaldoxime Chemical compound O\N=C/C1=CC=CC=C1O ORIHZIZPTZTNCU-YVMONPNESA-N 0.000 description 1
- 125000005930 sec-butyloxycarbonyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000007958 sleep Effects 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 230000002889 sympathetic effect Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- QKSQWQOAUQFORH-UHFFFAOYSA-N tert-butyl n-[(2-methylpropan-2-yl)oxycarbonylimino]carbamate Chemical compound CC(C)(C)OC(=O)N=NC(=O)OC(C)(C)C QKSQWQOAUQFORH-UHFFFAOYSA-N 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- QQJLHRRUATVHED-UHFFFAOYSA-N tramazoline Chemical compound N1CCN=C1NC1=CC=CC2=C1CCCC2 QQJLHRRUATVHED-UHFFFAOYSA-N 0.000 description 1
- 229960001262 tramazoline Drugs 0.000 description 1
- HILRAMQWKMIWCB-UHFFFAOYSA-N tributyl(cyanomethyl)phosphanium Chemical compound CCCC[P+](CCCC)(CCCC)CC#N HILRAMQWKMIWCB-UHFFFAOYSA-N 0.000 description 1
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to drugs containing pyrazole derivatives having histamine H3 receptor antagonistic activity and derivatives thereof as active ingredients, particularly dementia, Alzheimer's disease, attention deficit 'hyperactivity disorder, schizophrenia, epilepsy,
- the present invention relates to a prophylactic or therapeutic agent for central convulsions, eating disorders, obesity, diabetes, hyperlipidemia, sleep disorders, narcolepsy, sleep apnea syndrome, circadian rhythm disorders, depression, or allergic rhinitis.
- Histamine is normally stored in intracellular granules in mast cells, lungs, liver, and gastric mucosa, and released to the outside by external stimuli such as antigen binding to antibody on the cell surface. Is issued. For example, when mast cells are stimulated by an antigen that has entered from the outside, histamine is released from moonlit cells and causes allergic reactions by stimulating histamine HI (HI) receptors present on blood vessels and smooth muscle. . In addition, histamine released from ECL cells (enterochromaffin-like cells) on the gastric mucosa stimulates histamine H2 (H2) receptors on mural cells and promotes gastric acid secretion. Based on these facts, HI receptor antagonists or H2 receptor antagonists have been developed as therapeutic agents for allergic diseases or gastric ulcers, and are now widely used as pharmaceuticals.
- HI receptor antagonists or H2 receptor antagonists have been developed as therapeutic agents for allergic diseases or gastric ulcers, and are now widely used as pharmaceuticals.
- histamine acts as a neurotransmitter on the third histamine receptor (histamine H3 (H3) receptor) present in the central and peripheral nerves and exhibits various physiological functions. became. This receptor was clawed in 1999, and its gene sequence and amino acid sequence were revealed. Amino acid sequence homology with S, HI receptor, and H2 receptor was as low as 22% and 21.4%, respectively. (See Non-Patent Document 1).
- H3 receptor exists in the presynaptic membrane and functions as an autoreceptor that controls histamine synthesis and release (see Non-Patent Document 2).
- H3 receptors have been shown to control the release of histamine and other neurotransmitters such as acetylcholine, serotonin, dopamine, and noradrenaline (see Non-Patent Document 3). See).
- H3 receptor antagonists are dementia, Alzheimer's disease (see Non-Patent Documents 4 and 5), lack of attention. It has the potential to be used as a therapeutic agent for dyskinetics (see Non-Patent Document 6), schizophrenia (see Non-Patent Document 7), epilepsy, and central convulsions.
- H3 receptor has been shown to be involved in eating behavior (see Non-Patent Document 8), and as an indication for H3 receptor antagonists, eating disorders, obesity, diabetes, hyperlipidemia Such metabolic diseases are also envisaged.
- H3 receptor antagonists Diseases associated with sleep disorders such as sleep disorders, narcolepsy, sleep apnea syndrome, circadian rhythm disorders, and depression are also envisaged.
- H3 receptors are present in sympathetic nerves of the nasal mucosa, and it has been reported that combined use of H3 receptor antagonists and HI receptor antagonists significantly improves nasal congestion. (See Non-Patent Document 11). This indicates that the H3 receptor antagonist may be useful for the treatment of allergic rhinitis or the like alone or in combination with the HI receptor antagonist.
- H3 receptor antagonists are summarized in a plurality of reviews (see Non-Patent Documents 12 to 15), and these can be referred to. Initially, many imidazole compounds with histamine itself as the lead compound have been reported, and concerns such as the inhibitory action of the drug metabolizing enzyme cytochrome P450 (CYP) have been shown.
- CYP drug metabolizing enzyme
- Patent Literatures 1 to 10 Recently, many literatures and patents on non-imidazole H3 receptor antagonists have been reported (see Patent Literatures 1 to 10).
- Patent Document 11 reports a histamine H3 receptor antagonist having a structure in which a benzene ring and a pyrrole ring represented by the following formula (A) are bonded.
- Patent Document 12 a histamine 3 receptor antagonist having a biphenyl structure represented by the following formula (B) is reported!
- Patent Document 13 reports a histamine 3 receptor antagonist having a structure in which a pyrimidine ring and a benzene ring represented by the following formula (C) are bonded.
- Patent Document 14 a histamine 3 receptor antagonist having a structure in which a benzene ring and a pyrazole ring represented by the following formula (D) are bonded is reported.
- Patent Document 1 WO2005 / 097751 International Publication
- Patent Document 2 WO2005 / 097778 International Publication
- Patent Document 3 WO2005 / 118547 International Publication
- Patent Document 4 WO2006 / 014136 International Publication
- Patent Document 5 WO2006 / 045416 International Publication
- Patent Document 6 WO2006 / 046131 International Publication
- Patent Document 7 WO2006 / 059778 International Publication
- Patent Document 8 ⁇ 02006/061193 International Publication
- Patent Document 9 WO2006 / 107661 International Publication
- Patent Document 10 WO2006 / 103057 International Publication
- Patent Document 11 WO2002 / 012190 International Publication
- Patent Document 12 WO2002 / 040461 International Publication
- Patent Document 13 WO2005 / 007644 International Publication
- Patent Document 14 WO2006 / 023462 International Publication
- Non-patent literature l Lovenberg T.W. et al., Molecular pharmacology, 55, 1101-1107, 1999
- Non-patent literature 2 Arrang J-M. Et al., Nature, 302, 832-837, 1983
- Non-patent literature 3 Brown RE et al., Progress in Neurobiology, 63, 637-672, 2001
- Non-patent literature 4 Huang YW. Et al., Behavioral Brain Research, 151, 287-293, 2004
- Non-patent literature 5 Komater ⁇ ⁇ ⁇ ⁇ Behavioural Brain Research, 159, 295-300, 2005
- Non-Patent Document 6 Passani ⁇ ⁇ ⁇ ⁇ ⁇ et al., Neuroscience and Biobehavioral Reviews, 24, 107—11
- Non-patent literature 7 Fox GB et al., J. Pharmacol. Exp. Ther., 313, 176-190, 2005
- Non-patent literature 8 Hancock ⁇ ⁇ ⁇ ⁇ et al., Curr. Opin. Investig. Drug, 4, 1190-1197
- Non-Patent Document 9 Huang Z_Shira, Prog. Natr. Acad. ScL, 103, 4687-4692, 2006
- Non-Patent Document 10 Babier AJ et al., Br. J. Pharmacol., 143, 649-661, 2004
- Non-Patent Document ll McLeod R ⁇ . Et al., Am. J. Rhinol., 13, 391-399, 1999
- Non-patent document 12 Schwartz JC et al., Trends in Pharmacol. ScL, 7, 24-28, 1986
- Non-patent document 13 Passani ⁇ ⁇ ⁇ , et al., Trends in Pharmacol. ScL, 25, 618-625, 2004
- Non-patent document 14 Leurs R. et al., Nature Drug Discovery, 4, 107-122, 2005
- Non-patent document 15 Leurs R. et al., Drug Discovery Today, 10, 1613-1627, 2005
- the object of the present invention is a novel pyrazole derivative, more specifically, a histamine H3 receptor antagonistic action, and disorders caused by the histamine H3 receptor, such as dementia, Alzheimer's disease, attention deficit Hyperactivity, schizophrenia, epilepsy, central convulsions, eating disorders, obesity, diabetes, hyperlipidemia, sleep disorders, narcolepsy, sleep apnea syndrome, circadian rhythm disorders, depression, or allergies It is to provide a novel pyrazole derivative useful for the prevention and treatment of diseases such as rhinitis.
- a and B differently represent a carbon atom or a nitrogen atom
- R 1 is C-C alkyl (the C-C alkyl is C-C cyclic alkyl or hydroxy
- n an integer of 0 to 2
- T represents a hydrogen atom; halogen or c-c alkyl
- R represents a group represented by the following structural formulas (I) to (VI),
- R 2 and R 3 are the same or different and each represents a hydrogen atom; C to C alkyl (the C to C alkyl
- Halogen may be substituted with C to C alkyl or hydroxyl) or mono (CH)-
- R 2 and R 3 may be bonded together with adjacent nitrogen atoms, and may contain one or more nitrogen, oxygen, or sulfur atoms in addition to the nitrogen atoms in the ring.
- Member saturated heterocycles are halogen; C to C alkyl; C to C alkoxy or hydro
- Ar is aryl (the aryl is halogen; C to C alkynole; C to C alkoxy; hydro
- heteroaryl may contain one or more other nitrogen, oxygen, or sulfur atoms, which may be substituted with a carbonyl substituted through a nitrogen atom on a 3-7 membered saturated heterocycle) or to Teroaryl (wherein the heteroaryl is halogen; C-C alkyl; C-C alkoxy; hydride
- m represents an integer of 0 to 2
- G represents CO or SO;
- R 4 is a hydrogen atom or C
- R 5 is C-C alkyl (wherein the C-C alkyl is halogen; C-C cyclic alkyl; C
- C-C may be substituted with alkoxy or hydroxyl); C-C cyclic alkyl (the C
- -C cyclic alkyl is halogen; c-c alkyl; c-c alkoxy or hydroxy
- Aryl (where the aryl is halogen; alkyl; c
- R 4 and R 5 may be bonded together with an adjacent nitrogen atom and carbonyl carbon, and may contain one or more nitrogen, oxygen, or sulfur atoms in addition to the nitrogen atom in the ring.
- 5- to 7-membered saturated heterocycle (wherein the saturated heterocycle is halogen;
- 6 Lucoxy may be substituted with hydroxyl or oxo
- R 6 is C-C alkyl; C-C cyclic alkyl; C-C alkoxy; aryl (the aryl
- heteroaryl wherein the heteroaryl is a halogen
- ⁇ C alkoxy may be substituted with hydroxyl or cyan! /,
- R 7 is C to C alkyl; C luamino; C
- heteroaryl wherein the heteroaryl is halogen; c; c
- 6 Lucoxy may be substituted with hydroxyl or cyan.
- a pharmaceutical comprising the pyrazole derivative according to any one of 1) to 3) or a pharmaceutically acceptable salt thereof as an active ingredient
- Dementia Alzheimer's disease, attention deficit / hyperactivity characterized by containing the pyrazole derivative according to any one of 1) to 3) or a pharmaceutically acceptable salt thereof as an active ingredient
- Disease schizophrenia, epilepsy, central convulsions, eating disorders, obesity, diabetes, high fat It is a preventive or therapeutic agent for blood pressure, sleep disorder, narcolepsy, sleep apnea syndrome, circadian rhythm disorder, depression, or allergic rhinitis.
- the compound of the present invention has an excellent histamine H3 receptor antagonistic action.
- the halogen is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- C to C alkyl refers to a linear or branched alkyl group having 1 to 6 carbon atoms.
- a group such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butinole, tert-butinole, n-pentinole, isopentinole, neopentinole or n-hexyl is shown.
- C to C cyclic alkyl means cyclopropyl, cyclobutyl, cyclopentyl, cyclo
- C to C alkenyl refers to a linear or branched alkenyl group having 3 to 8 carbon atoms.
- C-C alkoxy refers to a linear or branched alkoxy group having 6 to 6 carbon atoms
- a group such as methoxy, ethoxy, n propoxy, isopropoxy, n butoxy, isobutoxy, sec butoxy, tert butoxy, n pentyloxy, isopentyloxy, neopentyloxy, or n hexyloxy.
- 1-C alkylamino means a linear or branched alkyl group having 6 to 6 carbon atoms.
- a group such as hexylamino is shown.
- C to C dialkylamino refers to two linear or branched alkyls having 1 to 6 carbon atoms.
- An amino group substituted with an alkyl group for example, dimethylamino, diethylamino dipropylamino, N, N isopropylmethylamino, di-n-butylamino, diisobutylamino, N, N-sec butylethylamino, N, It represents a group such as N-tert butylmethylamino, di-n-pentylamino, N, N-isopentylmethylamino, N, N-neopentylmethylamino or di-n-hexylamino.
- the "4- to 7-membered saturated heterocyclic ring” refers to a saturated 4- to 7-membered cyclic amino group, for example, a group such as 1-azetidyl, 1-pyrrolidyl, piperidino or 1-azepanyl.
- C-C alkoxycarbonyl refers to a linear or branched alkyl group having from 6 to 6 carbon atoms.
- C-C alkylaminocarbonyl means a linear or branched alkyl having from 6 to 6 carbon atoms.
- C-C dialkylaminocarbonyl refers to a linear or branched group having 2 carbon atoms of 1 to 6 carbon atoms.
- a carbonyl group to which an amino group substituted with a branched alkyl group is bonded for example, dimethylaminocarbonyl, jetylaminocarbonyl, di-n-propylaminocarbonyl, N, N isopropylmethylamino Carbonyl, di-n-butylaminocarbonyl, diisobutylaminocarbonyl, N, N sec butylethylaminocarbonyl, N, N-tert butylmethylaminocarbonyl, di-n-pentylaminocarbonyl, N, N isopentylmethylamino
- a group such as carbonyl, N, N-neopentylmethylaminocarbonyl or di-n-hexylaminocarbonyl is shown.
- Carboninore '' means a saturated 3-7 which contains one nitrogen atom and may contain one or more heteroatoms selected from nitrogen, oxygen and sulfur atoms.
- a cyclic group for example, a group such as 1-azetidyl, 1-pyrrolidinyl, piperidino, 1-azepanyl, 1-imidazolidinyl, 1-virazolidinyl, 1-piperiduryl, 3-oxazolidinyl, morpholino or 1-thiomorpholinyl.
- Aryl refers to a phenyl group or a naphthyl group.
- Heteroaryl refers to a monocyclic or bicyclic aromatic heterocyclic group, such as pyridine, pyridazine, pyrimidine, pyrazine, quinoline, isoquinoline, quinazoline, quinoxaline, pyrrole, furan, thiophene, pyrazole, Groups such as imidazole, tale zonazore, isota xoazore, thiazo monole, isothia zonole, triazonole, indanol, benzofuran, benzothiophene, benzoimidazole, indazonore, benzoxazonole, benzothiazonole or benzotriazole Is mentioned.
- pharmaceutically acceptable salts include, for example, salts with inorganic acids such as sulfuric acid, hydrochloric acid, hydrobromic acid, phosphoric acid, nitric acid, acetic acid, oxalic acid, lactic acid, tartaric acid, fumaric acid.
- inorganic acids such as sulfuric acid, hydrochloric acid, hydrobromic acid, phosphoric acid, nitric acid, acetic acid, oxalic acid, lactic acid, tartaric acid, fumaric acid.
- Acid maleic acid, citrate, benzene sulfonic acid, methane sulfonic acid, p toluene sulfonic acid, benzoic acid, camphor sulfonic acid, ethane sulfonic acid, darcoheptonic acid, darconic acid, glutamic acid, glycolic acid, malic acid, malon Acid, mandelic acid, galactaric acid, naphthalene 2-salt with organic acid such as sulfonic acid, salt with one or more metal ions such as lithium ion, sodium ion, potassium ion, calcium ion, magnesium ion, zinc ion, aluminum ion, ammonia, arginine, lysine And salts with amines such as piperazine, choline, jetylamine, 4-phenylcyclohexylamine, 2-aminoethanol, and benzathine.
- amines such as piperazine, choline, jetylamine,
- the compounds of the present invention may also exist as various solvates. It may also be a hydrate from the viewpoint of applicability as a medicine.
- the compounds of the present invention include all enantiomers, diastereomers, equilibrium compounds, mixtures of these in arbitrary proportions, racemates and the like.
- the compounds of the present invention also include compounds in which one or more hydrogen atoms, carbon atoms, nitrogen atoms, oxygen atoms, sulfur atoms are substituted with radioactive isotopes or stable isotopes. These labeled compounds are useful in, for example, metabolic and pharmacokinetic studies, biological analysis as receptor ligands, and the like.
- the compound of the present invention can be combined with one or more pharmaceutically acceptable carriers, excipients or diluents to form a pharmaceutical preparation.
- the carriers, excipients and diluents include water, lactose, dextrose, fructose, sucrose, sorbitol, mannitol mononole, polyethylene glycolenolate, propylene glycolenolate, starch, gum, gelatin, anoreginate, calcium silicate, Included are various oils such as calcium phosphate, cellulose, water syrup, methyl cellulose, polybulurpyrrolidone, alkyl parahydroxybenzosorbate, talc, magnesium stearate, stearic acid, glycerin, sesame oil, olive oil, soybean oil, etc. .
- additives such as extenders, binders, disintegrants, pH adjusters, and solubilizers that are generally used as necessary are mixed with the above-described carriers, excipients, or diluents, so that usual preparations are mixed.
- it can be prepared as an oral or parenteral pharmaceutical such as tablets, pills, capsules, granules, powders, solutions, emulsions, suspensions, ointments, injections, skin patches, etc.
- the compound of the present invention can be orally or parenterally administered to an adult patient at a dose of 0.00 ;! to 500 mg once or several times a day.
- the dose can be increased or decreased as appropriate depending on the type of disease to be treated, the age, weight, symptoms, etc. of the patient. Desirable profiles of the compound of the present invention include excellent medicinal properties, excellent pharmacokinetics, excellent physical properties, low toxicity, etc. It can manufacture by the method of.
- the compound of the present invention can be produced by a known organic chemical method, for example, according to the method according to the following reaction formula.
- R, R′—R 7 , T, n, A, and B are as defined above.
- Xi to x 5 are the same or different and are halogen atoms such as chlorine atom, bromine atom and iodine atom, methanesulfonyloxy group, benzenesulfonyloxy group, p-toluenesulfonyloxy group, trifluoromethanesulfonyl.
- a leaving group such as an organic Suruhoniruokishi group such Okishi group
- Yi ⁇ Y 2 are the same or different and each represents a leaving group, or a hydroxyl group such as a halogen atom or an organic Suruhoniruo alkoxy group
- Z is Represents a carbon atom or an oxygen atom
- p represents an integer of 0 or 1
- R 8 and R 9 represent a hydrogen atom, an alkyl group or a cyclic alkyl group.
- R 8 and R 9 may form a cyclic alkyl ring together with the adjacent carbon atom.
- This production process is a process for producing the compound (1) of the present invention from the compound (2).
- Step la is a step for obtaining compound (4) by a coupling reaction between compound (2) and compound (3).
- compound (2) and compound (3) are known compounds or known compounds It is a compound that can be easily synthesized.
- the coupling reaction include a Mitsunobu reaction.
- an organic phosphorus compound such as triphenylphosphine or tributylphosphine is combined with an azo compound such as jetyl dicarboxylate, diisopropyl azodicarboxylate, or ditertbutyl azodicarboxylate.
- a method of carrying out in a solvent in the presence of a phosphorus ylide reagent such as cyanomethyltributylphosphorane.
- Examples of the solvent used in this reaction include ethers such as tetrahydrofuran and 1,4 dioxane; hydrocarbons such as toluene and benzene; halogenated hydrocarbons such as chloroform and dichloromethane; N, N dimethylformamide N, N dimethylacetamide, amides such as N methyl 2-pyrrolidone; dimethyl sulfoxide; acetonitrile or a mixed solvent thereof, among which tetrahydrofuran and toluene are preferable.
- the reaction temperature in this reaction is usually from 0 ° C to 120 ° C, preferably from 15 ° C to 80 ° C, and the reaction time is usually from! To 48 hours, preferably from! To 12 hours. It is.
- Step 2a is a step for obtaining the compound (1) of the present invention by condensing the compound (4) and the compound (5) by a coupling reaction.
- the compound (5) is a compound that can be easily synthesized from the power of a known compound and the power of a known compound.
- the coupling reaction can be performed by a general method of aromatizing a nitrogen atom of an azole compound in a solvent using a ligand and a catalyst in the presence of a base. For example, (Kunz et al., Synlett, 15 , 2428-2439, 2003) or a method analogous thereto.
- Examples of the catalyst used in this reaction include copper catalysts generally used in condensation reactions, such as copper (0), copper iodide (1), copper chloride (1), copper oxide (1), copper bromide. (I) tristriphenylphosphine complex, copper trifluoromethanesulfonate (I) benzene complex and the like.
- Examples of the ligand used in this reaction include ligands commonly used in condensation reactions using a copper catalyst, such as N, N'-dimethylenoethylenediamine, 1,2-cyclohexanediamine, 2-amino. Examples include pyridine, 1,10-phenanthroline, 2-hydroxybenzaldehyde oxime, and ethylene glycol.
- Examples of the base used in this reaction include potassium carbonate, potassium phosphate, potassium hydroxide, tertbutoxypotassium, cesium carbonate, sodium carbonate, sodium hydrogen carbonate, sodium acetate, sodium methoxide, tetraptyl ammonium hydroxy Etc. Of these, potassium carbonate and cesium carbonate are preferred.
- Examples of the solvent used in this reaction include alcohols such as methanol, ethanol and isopropanol; ethers such as tetrahydrofuran and 1,4 dioxane; hydrocarbons such as toluene and benzene; Halogenated hydrocarbons; N, N dimethylformamide, N, N dimethylacetamide, N amides such as 2-pyrrolidone; dimethyl sulfoxide; acetonitrile; acetone; water or a mixed solvent thereof. N, N dimethylformamide and N methyl 2-pyrrolidone are preferred.
- the reaction temperature in this reaction is usually from 0 ° C to 150 ° C, preferably from 40 ° C to 120 ° C, and the reaction time is usually from! To 48 hours, preferably from! To 12 hours. It is.
- Compound (4) can be obtained by a coupling reaction of compound (2) and compound (6).
- the compound (6) is a known compound or a compound that can be easily synthesized from a known compound.
- the coupling reaction can be carried out by a general method for performing O alkylation of phenol in the presence or absence of a base in a solvent or without a solvent. If necessary, additives such as potassium iodide and sodium bromide can be added.
- the base used in this reaction include pyridine, triethylamine, diisopropylethylamine, tert-butoxy potassium, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, sodium hydroxide, potassium hydroxide, sodium hydride and the like.
- Examples of the solvent used in this reaction include alcohols such as methanol, ethanol and isopropanol; ethers such as tetrahydrofuran and 1,4 dioxane; hydrocarbons such as toluene and benzene; halogens such as chloroform and dichloromethane. Hydrocarbons; N, N dimethylformamide, N, N dimethylacetamide, N methyl 2-pyrrolidone and other amides; Examples thereof include til sulfoxide; acetonitrile; acetone; water or a mixed solvent thereof. Of these, tetrahydrofuran, N-dimethylformamide, acetonitrile, and acetone are preferable.
- the reaction temperature in this reaction is usually from 0 ° C to 150 ° C, preferably from 15 ° C to 80 ° C, and the reaction time is usually from! To 48 hours, preferably from! To 12 hours. is there.
- This production process is a process for producing the compound (1) of the present invention from the compound (2).
- Step lb is a step for obtaining compound (8) by condensing compound (2) and known compound (7) by a coupling reaction.
- the coupling reaction can be carried out by the same method as in step la.
- Step 2b is a step for obtaining compound (9) by condensing compound (8) and compound (5) by a coupling reaction.
- the coupling reaction can be carried out by the same method as in step 2a.
- Step 4a is a step for obtaining compound (10) by deprotecting the tert-butoxycarbonyl group of compound (9).
- the reaction can be carried out by a general method of deprotecting the tert-butoxycarbonyl group. For example, the method described in ( ⁇ ⁇ W. Greene and PGM Wuts Protective Groups in Organic Synthesis) or a method analogous thereto.
- the reaction can be carried out according to the method, and includes a method of reacting in the presence or absence of a strong acid in a solvent and a method of reacting in a solvent in the presence of a base.
- Examples of the acid used in this reaction include hydrochloric acid, sulfuric acid, trifluoroacetic acid, trifluoromethanesulfonic acid, and the like.
- Examples of the base used in this reaction include sodium hydroxide and potassium hydroxide.
- Examples of the solvent used in this reaction include alcohols such as methanol, ethanol and isopropanol; ethers such as jetyl ether, tetrahydrofuran and 1,4-dioxane; hydrocarbons such as toluene and benzene; And halogenated hydrocarbons such as dichloromethane; Amides such as N, N dimethylformamide, N, N dimethylacetamide, N-methyl-2-pyrrolidone; Dimethyl sulfoxide; Acetonitrile; Acetone; Water or a mixed solvent thereof .
- the reaction temperature in this reaction is usually 0 ° C to 150 ° C, preferably 15 ° C to 40 ° C, and the reaction time is usually;!
- Step 5a is a step for condensing compound (10) and compound (11) by a coupling reaction to obtain compound (1) of the present invention.
- the compound (11) is a compound that can be easily synthesized from a known compound and a known compound.
- the coupling reaction can be carried out by a general reductive amination reaction method in which a carbonyl compound and an amine are condensed.
- the compound (8) and the compound can be used in a solvent or without a solvent.
- the method of adding a reducing agent to the mixture of (9) is mentioned.
- a catalytic reduction method by hydrogenation using a catalyst such as palladium carbon, platinum, Raney nickel, rhodium alumina or the like can be used as a reduction method.
- a catalyst such as palladium carbon, platinum, Raney nickel, rhodium alumina or the like
- the reducing agent used in this reaction include sodium borohydride, sodium triacetoxyborohydride, sodium cyanoborohydride, diborane, lithium aluminum hydride and the like.
- the acid used in this reaction include acetic acid, formic acid, hydrochloric acid and the like.
- Examples of the solvent used in this reaction include alcohols such as methanol, ethanol and isopropanol; ethers such as tetrahydrofuran and 1,4 dioxane; hydrocarbons such as toluene and benzene; acetonitrile; water or a mixed solvent thereof. Of these, ethanol, toluene, and tetrahydrofuran are preferred.
- the reaction temperature in this reaction is usually 0 ° C to 150 ° C. ° C, preferably 15 ° C to 40 ° C, and the reaction time is usually: 48 hours, preferably 12 hours.
- compound (8) can be obtained by condensing compound (2) and compound (12) by a coupling reaction.
- the compound (12) is a compound that can be easily synthesized from a known compound and a known compound. >; The reaction can be carried out by the same method as in Step 3a. (Scheme 5)
- the compound (1) of the present invention can be obtained by a coupling reaction between the compound (10) and the compound (13).
- the compound (13) is a compound that can be easily synthesized from a known compound and a known compound.
- the coupling reaction can be carried out by a general method for alkylating amines in the presence or absence of a base in a solvent or without a solvent. If necessary, additives such as potassium iodide and sodium bromide can be added.
- the base used in this reaction include pyridine, triethylamine, diisopropylethylamine, tert-butoxy potassium, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, sodium hydroxide, potassium hydroxide, sodium hydride and the like.
- Examples of the solvent used in this reaction include alcohols such as methanol, ethanol and isopropanol; ethers such as tetrahydrofuran and 1,4 dioxane; toluene and benzene.
- Halogenated hydrocarbons such as chloroform and dichloromethane; Amides such as N, N-dimethylformamide, N, N dimethylacetamide and N-methyl 2-pyrrolidone; Dimethyl sulfoxide; Acetonitrile; Acetone; water or a mixed solvent thereof may be mentioned, and among these, tetrahydrofuran, N, N dimethylformamide, and acetonitrile are preferable.
- the reaction temperature in this reaction is usually from 0 ° C to 150 ° C, preferably from 15 ° C to 80 ° C, and the reaction time is usually from! To 48 hours, preferably from! To 12 hours. is there.
- This production process is a process for producing the compounds (11) and (12) of the present invention from the compounds (8) and (14).
- Step 2c is a step for obtaining compound (15) by a coupling reaction between compound (8) and known compound (14).
- the coupling reaction can be carried out by the same method as in step 2a.
- Step 4b is a step for obtaining compound (16) by deprotecting the tert-butoxycarbonyl group of compound (15).
- the deprotection reaction can be carried out by the same method as in Step 4a.
- Step 5b is a step for obtaining the compound (1-1) of the present invention by a coupling reaction between the compound (16) and the compound (11).
- the coupling reaction can be carried out by the same method as in Step 5a. [0067] (Step 7)
- Step 7 is a step for obtaining a compound (17) by converting the ethoxycarbonyl group of the compound (11) into a carboxylic acid by hydrolysis.
- the hydrolysis reaction can be carried out by a general ester hydrolysis reaction.
- the method can be carried out according to the described method or a method analogous thereto, and includes a method of reacting in the presence or absence of a strong acid and a method of reacting in a solvent in the presence of a base.
- the reaction temperature in this reaction is usually from 0 ° C to 120 ° C, preferably from 15 ° C to 80 ° C, and the reaction time is usually from! To 48 hours, preferably from! To 12 hours. is there.
- Step 8a is a step for condensing compound (17) and compound (18) by a coupling reaction to obtain compound (12) of the present invention.
- Compound (18) is a compound that can be easily synthesized from a known compound and a known compound.
- the coupling reaction can be performed by a general method of amidation of carboxylic acid, for example, a method in which carboxylic acid is introduced into a carboxylic acid chloride such as carboxylic acid chloride or carboxylic acid bromide and then reacted with amine.
- a method of reacting a mixed acid anhydride obtained from rubonic acid and chlorocarbonate with amine a method of reacting carboxylic acid with an active ester such as 1-benzotriazolyl ester or succinimidyl ester and then reacting with amine. And a method of reacting carboxylic acid with amine in the presence of a dehydration condensing agent. These reactions can all be performed in a solvent in the presence or absence of a base.
- Examples of the dehydrating condensing agent used in this reaction include 3- (3-dimethylaminopropyl) 1 ethyl carpositimide hydrochloride, dicyclohexyl norepositimide, diphenyl phosphoryl azide, carbonyldiimidazole, 0- ( 7-azabenzotriazole 1-yl) 1, 1, 3, 3, 3-tetramethyluronium hexafluorophosphate, etc. 1-hydroxybenzotriazole, hydroxy An activator such as succinimide can be used.
- Examples of the base used in this reaction include pyridine, triethylamine, diisopropylethylamine, potassium carbonate, sodium carbonate, sodium hydrogencarbonate and the like.
- Examples of the solvent used in this reaction include ethers such as tetrahydrofuran and 1,4-dioxane; Halogenated hydrocarbons such as chloroform and dichloromethane; Amides such as N, N-dimethylformamide, N, N dimethylacetamide and N-methyl 2-pyrrolidone; Dimethyl sulfoxide; Acetonitrile; Acetone; water or a mixed solvent thereof may be mentioned, and among these, toluene, tetrahydrofuran or N, N dimethylformamide is preferable.
- the reaction temperature in this reaction is usually from 0 ° C to 120 ° C, preferably from 15 ° C to 40 ° C, and the reaction time is usually from! To 48 hours, preferably from! To 12 hours. It is.
- the compound (11) of the present invention can be obtained by a coupling reaction between the compound (16) and the compound (13).
- the coupling reaction can be carried out by the same method as in Step 6a.
- This production process is a process for producing the compounds (13) and (14) of the present invention from the compound (11).
- Step 8b is a step for obtaining compound (13) of the present invention by reacting compound (11) with ammonia.
- This reaction can be carried out in the same manner as in Step 8a.
- it can be carried out, for example, by a method of reacting carboxylic acid with aqueous ammonia in the presence of a dehydrating condensing agent.
- Step 9 is a step for obtaining the compound (1-4) of the present invention by converting the powerful ruberamoyl group of the compound (13) into a nitrile group.
- This step can be performed by a general reaction in which a cananolamoyl group is converted to a nitryl group, and can be performed, for example, by reacting in the presence of a dehydrating agent or in the absence of a solvent. If necessary, additives such as N, N dimethylformamide, sodium chloride and the like can be added.
- Examples of the dehydrating agent used in this reaction include phosphorus pentoxide, phosphorus pentachloride, phosphoryl chloride, thionyl chloride, oxalinole chloride, trifluoroacetic anhydride, trifluoromethanesulfonic anhydride, and the like.
- examples of the solvent include ethers such as tetrahydrofuran and 1,4-dioxane; hydrocarbons such as toluene and benzene; N, N dimethylformamide, N, N dimethylacetamide, N methyl Amides such as —2-pyrrolidone; acetonitrile, or a mixed solvent thereof.
- the reaction temperature in this reaction is usually from 0 ° C to 120 ° C, preferably from 15 ° C to 80 ° C, and the reaction time is usually from! To 48 hours, preferably from! To 12 hours. It is.
- This production process is a process for producing the compounds (15), (16) and (17) of the present invention from the compound (4).
- step 2d the compound (4) and the compound (19) are condensed by a coupling reaction to form the compound of the present invention.
- This reaction can be carried out by the same method as in Step 2a.
- Step 10 is a step for obtaining compound (20) by reducing the nitro group of compound (15).
- This step can be carried out by a general reduction reaction that converts a nitro group into an amino group.
- a general reduction reaction that converts a nitro group into an amino group.
- catalytic reduction reaction by hydrogenation using a catalyst such as noradium carbon, platinum, raney nickel, rhodium alumina, zinc, iron, etc.
- Reduction reaction under acidic conditions using tin or tin chloride ( ⁇ ) reduction reaction using metal hydride such as lithium aluminum hydride, and the like.
- it can be carried out, for example, by carrying out a catalytic reduction reaction by hydrogenation in a methanol solvent using a single carbon of radium as a catalyst.
- Step 1 la is a step for condensing compound (20) and compound (21) by a coupling reaction to obtain compound (16) of the present invention.
- Compound (21) is a known compound or a compound that can be easily synthesized from a known compound.
- the coupling reaction can be carried out in the same manner as in Step 8a when G is CO and Y 1 is a hydroxyl group.
- the coupling reaction can be carried out by reacting compound (20) and compound (21) in the presence or absence of a base in a solvent or without a solvent.
- a base examples include pyridine, triethylamine, diisopropylethylamine, potassium carbonate, sodium hydrogen carbonate, sodium hydroxide and the like.
- Examples of the solvent used in this reaction include ethers such as tetrahydrofuran and 1,4-dioxane; hydrocarbons such as toluene and benzene; halogenated hydrocarbons such as chloroform and dichloromethane; N, N dimethylformamide Amides such as N, N dimethylacetamide, N methyl-2-pyrrolidone, and mixed solvents thereof, among which tetrahydrofuran and toluene are preferable.
- the reaction temperature in this reaction is usually from 0 ° C to 120 ° C, preferably from 15 ° C to 80 ° C, and the reaction time is usually from! To 48 hours, preferably from! To 12 hours. is there.
- step 12 compound (16) and compound (22) are reacted to give compound (17) of the present invention. It is a process for obtaining.
- the compound (22) is a compound that can be easily synthesized from a known compound and a known compound. This reaction can be carried out by a general method for alkylating amides, for example, by reacting compound (16) and compound (22) in a solvent in the presence of a base. If necessary, additives such as tetraptyl ammonium bromide and 18 crown-6 ether can be added. Examples of the base used in this reaction include sodium hydride, potassium hydride, tert-butoxy potassium, potassium hydroxide, sodium hydroxide, sodium methoxide, n-butyl lithium and the like.
- Examples of the solvent used in this reaction include alcohols such as methanol, ethanol and isopropanol; ethers such as tetrahydrofuran and 1,4 dioxane; hydrocarbons such as toluene and benzene; N, N dimethylformamide; Amides such as N, N dimethylacetamide and N methyl-2-pyrrolidone; dimethyl sulfoxide; acetonitrile; water or a mixed solvent thereof can be mentioned, and among these, tetrahydrofuran, N 2, N dimethylformamide are preferable.
- the reaction temperature in this reaction is usually from 0 ° C to 150 ° C, preferably from 15 ° C to 100 ° C, and the reaction time is usually from! To 48 hours, preferably from 1 to 12 hours. It is.
- This production step is a step for producing the compound (17) of the present invention from the compound (20). (Scheme 10)
- Step 13 is a step for obtaining compound (23) by alkylation reaction of compound (20).
- the alkylation reaction can be carried out by a general alkylation method in which a primary amino group is converted to a secondary amino group.
- the reaction is carried out using an alkylating agent such as an alkyl halide or an alkyl methanesulfonate in the presence of a base.
- an alkylating agent such as an alkyl halide or an alkyl methanesulfonate
- a method of reduction with a metal hydride such as dehydration condensation with alcohol.
- Step 1 lb is a step for obtaining the compound (1-7) of the present invention by condensing the compound (23) and the compound (21) by a coupling reaction.
- the coupling reaction can be carried out by the same method as in Step 11a.
- This production process is a process for producing the compounds (18) and (19) of the present invention from the compound (20).
- Step 1lc is a step for condensing compound (20) and compound (24) by a coupling reaction to obtain compound (18) of the present invention.
- Compound (24) is a known compound or a compound that can be easily synthesized from a known compound.
- the coupling reaction can be carried out by the same method as in Step 11a.
- Step 14 is a step for obtaining the compound (19) of the present invention by intramolecular cyclization reaction of the compound (18).
- the intramolecular cyclization reaction can be performed by a method described in the literature (for example, Journal of Medicinal Chemistry, 2002, Vol. 45, pages 3972-3983) or a method analogous thereto.
- Examples of the base used in this reaction include sodium hydride, potassium hydride, potassium carbonate, tert-butoxy potassium, sodium hydroxide and the like.
- the solvent used in this reaction include ethers such as tetrahydrofuran and 1,4-dioxane; hydrocarbons such as toluene and benzene; N, N dimethylformamide.
- the reaction temperature in this reaction is usually from 0 ° C to 150 ° C, preferably from 15 ° C to 80 ° C, and the reaction time is usually from! To 48 hours, preferably from! To 12 hours. is there.
- Example 1 tert-Butyl 4- (4-Phodophenoxy) piperidine-1 1-carboxylate (1 ⁇ 5 g), 4 (pyrrolidine 1-ylcarbonyl) 1H-pyrazole obtained in Example 1 (1) (0. 68 g), rac -trans-N, N, monodimethylcyclohexane 1, 2 diamine (0.68 g), copper (I) iodide (0.085 g) and cesium carbonate (2.5 g) A suspension of N, N dimethylformamide (2.5 mL) was stirred at 110 ° C. for 2 hours. The reaction mixture was allowed to cool to room temperature, diluted with chloroform and diluted with celite. The filtrate was washed with water and concentrated under reduced pressure.
- tetrahydrofuran 15 mU, methanol (5 ⁇ OmU and black mouth form (lOmL)
- 4M ethyl acetate solution under ice-cooling and stirred at room temperature.
- the reaction mixture was mixed with saturated sodium hydrogen carbonate solution.
- the organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure, and the resulting residue was washed with diisopropyl ether to give the title compound (1. lg) as a colorless solid. .
- Table 1 shows compounds (Compound No. 2) to (Compound No. 5) obtained by the same method as shown in Example 1 (4).
- Example 1 4- (4) obtained in (3) ⁇ 4-[[4-((pyrrolidine-1-ylcarbonyl)]-1H-pyrazole-l-yl] phenoxy ⁇ piperidine (0.050 g), (bromomethinole) cycloprono (0.024 g) and potassium carbonate (0.040 g) in N, N dimethylenolenolemamide (0.50 mL) were stirred at 80 ° C. for 3 hours. The reaction mixture was allowed to cool to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure.
- Example 1 1 tert butyl 4 1 (4-iodophenoxy) piperidine 1 1 obtained from (1) Carboxylate (4 ⁇ Og), ethyl 1H-pyrazole 4 carboxylate (1 5g), rac-trans-N, N, monodimethylcyclohexane 1,2 diamine (0.79 g), copper iodide (I) (0.23 g) and cesium carbonate (6.8 g) N, The suspension of N dimethylformamide (6.5 mL) was stirred at 110 ° C for 3.5 hours. The reaction mixture was allowed to cool to room temperature, diluted with chloroform and diluted with celite. The filtrate was washed with water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained residue was washed with diisopropyl ether to give the title compound (2.6 g) as a pale blue solid.
- Example 3- The title compound was obtained by a method similar to the method described in (3) using cyclopentanone instead of cyclobutanone.
- Example 3 Ethyl 1 ⁇ 4 [(1-cyclobutylpiperidine 4 yl) oxy] phenolinol 1 H pyrazonole 4 carboxylate (1.4 g) obtained in (3) and lithium hydroxide (0.27 g) of ethanol (7. OmU and water (7. OmU mixed solution) was stirred for 1 hour at 90 ° C. The reaction mixture was allowed to cool to room temperature and the ethanol was distilled off under reduced pressure. The residue was neutralized with 1M aqueous hydrochloric acid, water was added, and the mixture was stirred for 30 min under ice-cooling, and the precipitated crystals were collected by filtration to give the title compound (1.2 g) as a colorless solid.
- Example 5 (1) 1 1 ⁇ 4 1 [(1 cyclobutylpiperidine 4 yl) oxy] phenyl ⁇ 1H-pyrazole 4 1 strength rubonic acid (0 ⁇ 40 g), 1 ⁇ 3 (dimethylamino ) Propylene ⁇ —3 Ethyl Carpositimide Hydrochloride (0 ⁇ 25g), 1-Hydroxybenzotriazo Monochlorohydrate (0.20g) and Pyrrolidine (0.092g) in N, N Dimethenolehonolemide (A suspension was added to 4.0 mU and stirred overnight at room temperature. To the reaction mixture was added aqueous sodium hydrogen carbonate solution and stirred for 10 minutes at room temperature. The precipitated crystals were filtered and dried.) Was washed with ethanol to give the title compound (0.28 g) as a light brown solid.
- Example 1 According to a method similar to that shown in (1), instead of tert-butyl 4-hydroxypiperidine 1-carboxylate, 1-tert-butyl-4-hydroxypiperidine (Journal of Organic Chemistry, 2005 The title compound was obtained using the method described in Y., 70, 1930–1933.
- Example 1 In the same manner as shown in (2), 1 tert-butyl-4 obtained in Example 8 (1) was used in place of tert-butyl 4-mono (piperphenoxy) piperidine 1-carboxylate. The title compound was obtained using 1- (4-iodophenoxy) piperidine.
- Example 1 1-cyclopropyl-4-hydroxypiperidine (in accordance with the method described in WO2005117865) in place of tert-butyl 4-hydroxypiperidine 1-carboxylate by the same method as shown in (1) To give the title compound.
- Example 1 1 Obtained in Example 9 1 (1) in the same manner as shown in 1 (3), instead of tert-butyl 4 4- (4-phenoxy) piperidine 1 carboxylate 1 -Cyclopropinole 4-(4-Phodophenoxy) piperidine, 4-Hydrazole (4-Fluorophenyl) amide 1H-Pyrazole 4-Holpyrazole instead of 1-Pyrazole 1H-Pyrazole To give the title compound.
- the title compound was obtained using tert butyl 4-hydroxypyrrolidine 1 carboxylate instead of 1 carboxylate.
- the title compound was obtained using tertbutyl 4-hydroxypyrrolidine 1-carboxylate instead of 1-carboxylate and cyclobutanone instead of cyclopentanone.
- the title compound was obtained using 3-methylphenol.
- Example 5 1 1 ⁇ 4 1 [(1 cyclobutylpiperidine 4 yl) oxy] phenyl ⁇ — 1H-pyrazole-4-carboxylic acid (1.00 g) obtained in (1), 1-hydroxybenzotria To a solution of sol-hydrate (0 ⁇ 535 g) and 1-ethyl-3- (3 dimethylaminopropyl) carbodiimide hydrochloride (0.673 g) in N, N dimethylformamide at room temperature Water (25%, 0.5 ml) was added and stirred at room temperature. To the reaction mixture was added saturated aqueous sodium hydrogen carbonate, and the mixture was stirred for 1 hour, and the crystals were collected by filtration. The obtained crystals were washed with water and diisopropyl ether and dried to give the title compound (0.988 g) as a colorless powder.
- Example 22 4 Chloro-N— (1 ⁇ 4 1 [(1-Cyclobutylpiperidine — 4 yl) oxy] phenyl ⁇ — 1H-pyrazole 1 4 yl) obtained in (2) Sodium hydride (0.176 g) was added to a solution of butanamide (0 ⁇ 366 g) in tetrahydrofuran (3. Oml) at room temperature, and the mixture was heated to reflux for 30 minutes in an oil bath (oil temperature 90 ° C.). The reaction mixture was ice-cooled, saturated aqueous sodium hydrogen carbonate was added, and the mixture was extracted with black mouth form, washed with water and saturated brine, dried over sodium sulfate, and concentrated under reduced pressure.
- Test Example 1 H3 receptor binding test
- Inhibition curves were obtained by reacting various concentrations of each test drug with a constant concentration (2 nM) of R () 1 ⁇ -methyl [ 3 ⁇ ] histamine under the conditions described above.
- Test Example 2 [35S] GTP- ⁇ S binding test
- the ratio of the second search action time to the first search action time was used as an indicator of social recognition.
- the compound of the present invention was orally administered to adult rats immediately after the first exploratory behavior.
- the compounds of the present invention were dissolved in 0.03N hydrochloric acid solution and examined at 0.1, 0.3 and lmg / kg.
- compound number 1 significantly reduced the second search time / first search action time at 0.3 and lmg / kg as compared to the solvent group, and showed a social cognitive enhancing action.
- the present invention has a strong binding inhibitory action on histamine H3 receptor and is caused by a disorder caused by histamine H3 receptor, such as dementia, Alzheimer's disease, attention deficit / hyperactivity disorder, schizophrenia, epilepsy, Prevention and treatment of diseases such as central convulsions, eating disorders, obesity, diabetes, hyperlipidemia, sleep disorders, narcolepsy, sleep apnea syndrome, circadian rhythm disorder, depression, or allergic rhinitis It has become possible to provide a novel pyrazole derivative that is useful in the present invention.
- a disorder caused by histamine H3 receptor such as dementia, Alzheimer's disease, attention deficit / hyperactivity disorder, schizophrenia, epilepsy, Prevention and treatment of diseases such as central convulsions, eating disorders, obesity, diabetes, hyperlipidemia, sleep disorders, narcolepsy, sleep apnea syndrome, circadian rhythm disorder, depression, or allergic rhinitis
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Immunology (AREA)
- Endocrinology (AREA)
- Child & Adolescent Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Emergency Medicine (AREA)
- Anesthesiology (AREA)
- Otolaryngology (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
下式で示されるヒスタミンH3受容体拮抗作用を有する新規なピラゾール誘導体若しくはその医薬上許容される塩、又はこれらを有効成分として含有する医薬は、認知症、アルツハイマー病、注意欠陥・多動性症、統合失調症、摂食障害、肥満、糖尿病、高脂血症、睡眠障害、ナルコレプシー、睡眠時無呼吸症候群、概日リズム障害、うつ病、又はアレルギー性鼻炎等の疾患の予防又は治療に有効である。
Description
明 細 書
ピラゾール誘導体
技術分野
[0001] 本発明は、ヒスタミン H3受容体拮抗作用を有するピラゾール誘導体及びそれら誘 導体を有効成分として含有する医薬、特に認知症、アルツハイマー病、注意欠陥 '多 動性症、統合失調症、てんかん、中枢性痙攣、摂食障害、肥満、糖尿病、高脂血症 、睡眠障害、ナルコレプシ一、睡眠時無呼吸症候群、概日リズム障害、うつ病、又は アレルギー性鼻炎の予防剤又は治療剤に関する。
背景技術
[0002] ヒスタミンは肥満細胞、肺、肝臓、及び胃粘膜などで普段は細胞内の顆粒に貯蔵さ れており、細胞表面の抗体に抗原が結合するなどの外部刺激を受けて細胞外へ放 出される。例えば、肥満細胞が外部から進入した抗原で刺激されると、ヒスタミンは月巴 満細胞から放出され、血管や平滑筋上に存在するヒスタミン HI (HI)受容体を刺激 することによりアレルギー反応を引きおこす。また、胃粘膜上の ECL細胞(enterochr omaffin-like cell)から放出されたヒスタミンは壁細胞上のヒスタミン H2 (H2)受容 体を刺激し、胃酸分泌を促進する。これらの事実に基づき、アレルギー疾患治療薬、 又は胃潰瘍治療薬として HI受容体拮抗物質、又は H2受容体拮抗物質の開発が行 われ、現在は医薬品として広く使用されている。
[0003] さらに、ヒスタミンは神経伝達物質として中枢神経及び末梢神経に存在する 3番目 のヒスタミン受容体 (ヒスタミン H3 (H3)受容体)に作用し、種々の生理機能を発揮す ることが明らかになった。この受容体は 1999年にクローユングされ、その遺伝子配列 およびアミノ酸配列が明らかになった力 S、 HI受容体及び H2受容体とのアミノ酸配列 相同性は、それぞれ 22%、及び 21. 4%と低いものであった(非特許文献 1参照)。
H3受容体はシナプス前膜に存在し、ヒスタミンの合成と遊離を制御するオートレセプ ターとして機能することが示された(非特許文献 2参照)。また、 H3受容体は、ヒスタミ ンの放出を制御するとともに、アセチルコリン、セロトニン、ドパミン、ノルアドレナリンと いった他の神経伝達物質の放出をも制御することが明らかとなった(非特許文献 3参
照)。これらの事実は、 H3受容体拮抗物質が神経における神経伝達物質の放出異 常に関連した各種疾患の治療薬になりうることを示唆している。
[0004] 合成化合物を用いた動物モデルでの検討結果は、 H3受容体拮抗物質のうち H3 受容体拮抗物質が、認知症、アルツハイマー病(非特許文献 4及び 5参照)、注意欠 陥 ·多動性症 (非特許文献 6参照)、統合失調症 (非特許文献 7参照)、てんかん、中 枢性痙攣などの治療薬として利用できる可能性を示している。
[0005] また、 H3受容体が摂食行動に関与することが示されており(非特許文献 8参照)、 H3受容体拮抗物質の適応疾患として摂食障害、肥満、糖尿病、高脂血症などの代 謝系疾患も想定される。
[0006] また、ヒスタミンは脳内の日周リズムを調節し、覚醒と睡眠のバランスを保つ役割が あることが示されており(非特許文献 9及び 10参照)、 H3受容体拮抗物質の適応疾 患として睡眠障害、ナルコレプシ一、睡眠時無呼吸症候群、概日リズム障害、うつ病 など睡眠障害に伴う疾患も想定される。
[0007] さらに、 H3受容体は鼻粘膜の交感神経に存在していることが示され、 H3受容体拮 抗物質と HI受容体拮抗物質の併用により鼻閉が顕著に改善されることが報告され ている(非特許文献 11参照)。このことは、 H3受容体拮抗物質は単独で、又は HI受 容体拮抗物質との併用でアレルギー性鼻炎等の治療に有用である可能性を示して いる。
[0008] H3受容体拮抗物質については、複数のレビューにまとめられており(非特許文献 1 2〜; 15参照)、これらを参照することができる。初期にはヒスタミン自身をリード化合物 としたイミダゾール系化合物が数多く報告された力 S、薬物代謝酵素チトクロム P450 ( CYP)の阻害作用等の懸念が示され、医薬品として開発するには至っていない。
[0009] 最近になって、非イミダゾール系の H3受容体拮抗物質の文献及び特許が数多く 報告されている(特許文献 1〜; 10参照)。
[0010] 例えば特許文献 11において、下記式 (A)で表されるベンゼン環とピロール環とが 結合した構造を有したヒスタミン H3受容体拮抗物質が報告されている。
[0011] [化 1]

[0012] また、特許文献 12において、下記式 (B)で表されるビフエ二ル構造を有したヒスタ ン Η3受容体拮抗物質が報告されて!/、る。
[0013] [化 2]

[0014] また、特許文献 13において、下記式 (C)で表されるピリミジン環とベンゼン環とが結 合した構造を有したヒスタミン Η3受容体拮抗物質が報告されている。
[0015] [化 3]

[0016] また、特許文献 14において、下記式 (D)で表されるベンゼン環とピラゾール環とが 結合した構造を有したヒスタミン Η3受容体拮抗物質が報告されている。
[0017] [化 4]

特許文献 2: WO2005/097778国際公開公報
特許文献 3:WO2005/118547国際公開公報
特許文献 4:WO2006/014136国際公開公報
特許文献 5:WO2006/045416国際公開公報
特許文献 6:WO2006/046131国際公開公報
特許文献 7: WO2006/059778国際公開公報
特許文献8:\¥02006/061193国際公開公報
特許文献 9:WO2006/107661国際公開公報
特許文献 10:WO2006/103057国際公開公報
特許文献 11 :WO2002/012190国際公開公報
特許文献 12:WO2002/040461国際公開公報
特許文献 13: WO2005/007644国際公開公報
特許文献 14:WO2006/023462国際公開公報
非特許文献 l:Lovenberg T.W.ら、 Molecular pharmacology, 55, 1101-1107, 1999 非特許文献 2:Arrang J-M.ら、 Nature, 302, 832-837, 1983
非特許文献 3: Brown R.E.ら、 Progress in Neurobiology, 63, 637-672, 2001 非特許文献 4: Huang Y-W.ら、 Behavioural Brain Research, 151, 287-293, 2004 非特許文献 5:Komater ν·Α·ら、 Behavioural Brain Research, 159, 295-300, 2005 非特許文献 6:Passani Μ·Β·ら、 Neuroscience and Biobehavioral Reviews, 24, 107—11
3, 2000
非特許文献 7:Fox G.B.ら、 J. Pharmacol. Exp. Ther., 313, 176-190, 2005 非特許文献 8: Hancock Α·Α·ら、 Curr. Opin. Investig. Drug, 4, 1190-1197 非特許文献 9: Huang Z_しら、 Prog. Natr. Acad. ScL, 103, 4687-4692, 2006 非特許文献 10:Babier A.J.ら、 Br. J. Pharmacol., 143, 649-661, 2004
非特許文献 ll:McLeod R丄.ら、 Am. J. Rhinol., 13, 391-399, 1999
非特許文献 12: Schwartz J.C.ら、 Trends in Pharmacol. ScL, 7, 24-28, 1986 非特許文献 13:Passani Μ·Β·ら、 Trends in Pharmacol. ScL, 25, 618-625, 2004
非特許文献 14 : Leurs R.ら、 Nature Drug Discovery, 4, 107-122, 2005 非特許文献 15 : Leurs R.ら、 Drug Discovery Today, 10, 1613-1627, 2005
発明の開示
発明が解決しょうとする課題
[0019] 本発明の目的は、新規なピラゾール誘導体、更に詳しくは、ヒスタミン H3受容体拮 抗作用を有し、ヒスタミン H3受容体に起因する障害、例えば、認知症、アルッハイマ 一病、注意欠陥 ·多動性症、統合失調症、てんかん、中枢性痙攣、摂食障害、肥満、 糖尿病、高脂血症、睡眠障害、ナルコレプシ一、睡眠時無呼吸症候群、概日リズム 障害、うつ病、又はアレルギー性鼻炎等の疾患の予防並びに治療に有用である新規 なピラゾール誘導体を提供することにある。
課題を解決するための手段
[0020] 本発明者らは上記目的のため鋭意研究を行った結果、ピラゾール誘導体が、ヒスタ ミン H3受容体に対して強力な拮抗作用を有することを見出し、本発明を完成した。 すなわち、本発明は、
1)式 (1 )
[0021] [化 5]

{式 (1)中、
A及び Bは、異なって炭素原子、又は窒素原子を示し、
R1は、 C〜Cアルキル(該 C〜Cアルキルは、 C〜C環状アルキル又はヒドロキシ
1 6 1 6 3 8
ルで置換されても良い); c 環状アルキル(該 c
3〜c
8 3〜c環状アルキルは、 c
8 1〜c
6 アルキル又はヒドロキシルで置換されても良い)又は c ニルを示し、
3〜cアルケ
8
nは、 0〜2の整数を示し、
Tは、水素原子;ハロゲン又は c〜cアルキルを示し、
1 6
Rは、下記構造式 (I)〜 (VI)で表される基を示し、
[化 6]

R2及び R3は、同一又は異なって、水素原子; C〜Cアルキル(該 C〜Cアルキル
1 6 1 6
は、ハロゲン; c〜c環状アルキル;ヒドロキシル; c〜cアルコキシカルボニル又は
3 8 2 7
カルボキシで置換されても良い); c〜c環状アルキル(該 c〜c環状アルキルは、
3 8 3 8
ハロゲン; C〜Cアルキル又はヒドロキシルで置換されても良い)又は一(CH ) -
1 6 2 m
Arで示される基を示すか、
又は、 R2及び R3は、隣接する窒素原子と一緒になつて互いに結合し、環中に前記窒 素原子の他に 1つ以上の窒素、酸素、又は硫黄原子を含んでもよい 4〜7員の飽和 複素環(該飽和複素環は、ハロゲン; C〜Cアルキル; C〜Cアルコキシ又はヒドロ
1 6 1 6
キシルで置換されても良い)を形成し、
Arは、ァリール(該ァリールは、ハロゲン; C〜Cアルキノレ; C〜Cアルコキシ;ヒドロ
1 6 1 6
キシル; C〜Cアルコキシカルボニル;シァノ; C〜Cアルキルアミノカルボニル; C
2 7 2 7 3
〜C ジアルキルアミノカルボニル;力ルバモイル又は環中に少なくとも 1つの窒素原
13
子を有し、他に 1つ以上の窒素、酸素、若しくは硫黄原子を含んでもよい 3〜7員の飽 和複素環に窒素原子を介して置換されたカルボニルで置換されても良い)又はへテ ロアリール(該ヘテロァリールは、ハロゲン; C〜Cアルキル; C〜Cアルコキシ;ヒド
1 6 1 6
ロキシル; c〜cアルコキシカルボニル;シァノ; c〜cアルキルアミノカルボニル;
2 7 2 7
C〜C ジアルキルアミノカルボニル;力ルバモイル又は環中に少なくとも 1つの窒素
3 13
原子を有し、他に 1つ以上の窒素、酸素、若しくは硫黄原子を含んでもよい 3〜7員の 飽和複素環に窒素原子を介して置換されたカルボニルで置換されても良い)を示し、 mは、 0〜2の整数を示し、
Gは、 CO 又は SO を示し、
R4は、水素原子又は C
1〜Cアルキルを示し、
6
R5は、 C〜Cアルキル(該 C〜Cアルキルは、ハロゲン; C〜C環状アルキル; C
1 6 1 6 3 8 1
〜cアルコキシ又はヒドロキシルで置換されても良い); C〜C環状アルキル(該 C
6 3 8 3
〜c環状アルキルは、ハロゲン; c〜cアルキル; c〜cアルコキシ又はヒドロキシ
8 1 6 1 6
ルで置換されても良い);ァリール(該ァリールは、ハロゲン;アルキル; c
1〜cアルコ 6 キシ;ヒドロキシル又はシァノで置換されても良い)又はへテロアリール(該ヘテロァリ ールは、ハロゲン;アルキル; C〜Cアルコキシ;ヒドロキシル又はシァノで置換され
1 6
ても良い)を示し、
また、 R4及び R5は、隣接する窒素原子及びカルボニル炭素と一緒になつて互いに結 合し、環中に前記窒素原子の他に 1つ以上の窒素、酸素、又は硫黄原子を含んでも よい 5〜7員の飽和複素環(該飽和複素環は、ハロゲン; C
1〜Cアルキル; C
6 1〜Cァ
6 ルコキシ;ヒドロキシル又はォキソで置換されても良い)を形成しても良く、
R6は、 C〜Cアルキル; C〜C環状アルキル; C〜Cアルコキシ;ァリール(該ァリ
1 6 3 8 1 6
ールは、ハロゲン;アルキル; c
1〜cアルコキシ;ヒドロキシル又はシァノで置換され 6
ても良い)又はへテロアリール(該ヘテロァリールは、ハロゲン; C C
1〜Cアルキル; 6 1
〜Cアルコキシ;ヒドロキシル又はシァノで置換されても良!/、)を示し、
6
R7は、 C〜Cアルキル; C ルァミノ; C
1 6 1〜Cアルコキシ;ァミノ; C
6 1〜Cアルキ
6 2〜C
12 ジアルキルアミノ; 4〜7員の飽和複素環(該飽和複素環は、ハロゲン;アルキル; C 〜Cアルコキシ;ヒドロキシル又はシァノで置換されても良い);ァリール(該ァリール
6
は、ハロゲン;アルキル; c ヒドロキシル又はシァノで置換されても良
1〜cアルコキシ;
6
い)又はへテロアリール(該ヘテロァリールは、ハロゲン; c ; c
1〜cアルキル
6 1〜cァ
6 ルコキシ;ヒドロキシル又はシァノで置換されても良!/、)を示す。 }
で表されるピラゾール誘導体、又はその医薬上許容される塩、
2)式(1)で示され、式中、 Rは構造式 (I)で表される、 1)に記載のピラゾール誘導体 、又はその医薬上許容される塩、
3)以下の群から選ばれる化合物、
1ーシクロペンチルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン、
1ーェチルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾールー 1 ィル]フエノキシ }ピペリジン、
1一(1ーメチルェチル)ー4 {4 [4一(ピロリジン一 1ーィルカルボニル)一 1H— ピラゾーノレ一 1—ィル]フエノキシ }ピペリジン、
1ーシクロへキシルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン、
1 2 メチルシクロペンチル) 4 {4 [4 (ピロリジン 1ーィルカルボニル) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1—(シクロプロピルメチル)ー4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H —ピラゾーノレ一 1—ィル]フエノキシ }ピペリジン、
1 (3—メチルー 2—ブテンー1ーィル)ー4 {4 [4 (ピロリジンー1ーィルカル ボニル) 1H—ピラゾールー 1 ィル]フエノキシ }ピペリジン、
1 (シクロブチルメチル)ー4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H— ピラゾーノレ一 1—ィル]フエノキシ }ピペリジン、
1— (シクロペンチルメチル)ー4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H —ピラゾーノレ一 1—ィル]フエノキシ }ピペリジン、
2- (4— {4— [4— (ピロリジン一 1—ィルカルボ二ル)一 1H—ピラゾール一 1—ィノレ] フエノキシ }ピペリジン 1 ィル)エタノール、
3— (4-{4-[4- (ピロリジン 1ーィルカルボニル) 1H—ピラゾールー 1 ィル] フエノキシ }ピペリジン 1 ィル)シクロペンタノール、
ェチル 1 {4 [(1 シクロブチルピペリジン 4 ィノレ)ォキシ]フエ二ノレ 1 H ーピラゾールー 4一力ルボン酸、
ェチル 1 {4 [(1 シクロペンチルビペリジン 4 ィノレ)ォキシ]フエ二ノレ 1 H ピラゾールー 4一力ルボン酸、
1ーシクロブチルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾー ルー 1 ィル]フエノキシ }ピペリジン、
1-[(1- {4一 [ (1ーシクロペンチルビペリジンー4 ィル)ォキシ]フエ二ル} -1H ーピラゾールー 4 ィル)カルボ二ノレ]ピロリジン オール、
1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ]フエニル } N シクロぺ ンチルー 1H—ピラゾールー 4 カルボキサミド、
1 {4一 [ (1ーシクロブチルピペリジンー4 ィル)ォキシ]フエ二ル} N メチノレー 1H—ピラゾールー 4 カルボキサミド、
1 {4 [ (1ーシクロブチルピぺリジンー4ーィル)ォキシ]フェニル}ーN—(2, 2, 2 トリフルォロェチル) 1H—ピラゾールー 4 カルボキサミド、
1 {4一 [ (1ーシクロブチルピペリジンー4 ィル)ォキシ]フエ二ル} -N- (シクロへ キシルメチル) 1H—ピラゾールー 4 カルボキサミド、
1ーシクロブチルー 4 {4 [4 (ピペリジン 1ーィルカルボニル) 1H—ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン、
1ーシクロブチルー 4一(4— {4一 [ (2 メチルピロリジン一 1一ィル)カルボニル]一 1 H ピラゾール一 1—イノレ}フエノキシ)ピぺリジン、
1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ]フエニル } N, N ジェ チルー 1 H ピラゾールー 4 カルボキサミド、
N— tert ブチル 1 4 1 シクロブチルピペリジン 4 ィル)ォキシ]フエ ニル } 1 H ピラゾールー 4 カルボキサミド、
1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ]フエニル } N, N ジメチ ルー 1 H ピラゾールー 4 カルボキサミド、
4- [ (1 - {4- [ (1 -シクロブチルピペリジン 4—ィル)ォキシ]フエニル }— 1 H— ピラゾールー 4 ィル)カルボニル]モルホリン、
1 - [ (1 - {4- [ (1 -シクロブチルピペリジン 4 ィル)ォキシ]フエニル } 1 H— ピラゾールー 4ーィノレ)カルボニル]ー4ーメチルピペラジン、
1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ]フエニル } N シクロへ キシルー N メチルー 1H—ピラゾールー 4 カルボキサミド、
1 {4一 [ (1ーシクロブチルピペリジンー4 ィル)ォキシ]フエ二ル} N メチノレー N- (2 メチルプロピル) 1H—ピラゾールー 4 カルボキサミド、
1ーシクロブチルー 4 4 {4 [ (3, 3—ジフルォロピロリジン 1 ィル)カルボ二 ル]― 1H—ピラゾール一 1—イノレ}フエノキシ)ピぺリジン、
1 {4 [ (1ーシクロブチルピぺリジンー4ーィル)ォキシ]フェニル}ーN—(4ーフル オロフェニル) 1H—ピラゾールー 4 カルボキサミド、
1一 { 4一 [ ( 1一シクロブチルピペリジン一 4一ィル)ォキシ]フエ二ル }一 N—(4一フル ォロベンジル) 1H—ピラゾールー 4 カルボキサミド、
4— { 4 [4— (ァゼチジン 1—ィルカルボニル) 1H—ピラゾーノレ 1—ィル]フエ ノキシ } 1ーシクロペンチルビペリジン、
4 [ (1 {4一 [ (1ーシクロペンチルビペリジンー4 ィル)ォキシ]フエ二ル} - 1H ピラゾーノレ 4ーィノレ)カルボニノレ]モルホリン、
1ーシクロぺンチルー4ー(4 {4 [ (3, 3—ジフルォロピロリジン 1 ィル)カルボ ニル] 1H—ピラゾールー 1—ィル }フエノキシ)ピぺリジン、
N -シクロペンチル 1— { 4— [ ( 1 シクロペンチルビペリジン 4 ィル)ォキシ]フ ェニル } 1 H ピラゾーノレ 4 カルボキサミド、
N シクロへキシル 1 { 4 [ ( 1 シクロペンチルビペリジン 4 ィル)ォキシ]フ ェニル } 1 H ピラゾール 4 カルボキサミド、
N ベンジル 1 { 4 ( 1 シクロペンチルビペリジン 4 ィル)ォキシ]フエ二 ノレ 1 H ピラゾーノレ 4一力ノレボキサミド、
1 { 4一 [ ( 1—シクロペンチルビペリジン一 4一ィル)ォキシ]フエ二ノレ N— (4—フ ノレォロベンジル) 1 H ピラゾーノレ 4一力ノレボキサミド、
1 { 4 [ ( 1 シクロペンチルビペリジン 4 ィル)ォキシ]フエニル } N—(3—フ ルォロベンジル) 1H—ピラゾールー 4 カルボキサミド、
1 {4 [ (1ーシクロぺンチルピぺリジンー4ーィル)ォキシ]フェニル}ーN—(2 フ ルォロベンジル) 1 H ピラゾーノレ 4 カルボキサミド、
1— { 4 ( 1 シクロペンチルビペリジン 4 ィル)ォキシ]フエニル } N フエ二 ノレ 1 H ピラゾーノレ 4 カルボキサミド、
1 { 4一 [ ( 1—シクロペンチルビペリジン一 4一ィル)ォキシ]フエ二ノレ N— (4—フ ルオロフェニル) 1 H ピラゾーノレ 4—カルボキサミド、
1 { 4 [ ( 1 シクロペンチルビペリジン 4 ィル)ォキシ]フエニル } N—(4ーメ チルフエニル) 1H—ピラゾールー 4 カルボキサミド、
1一 { 4一 [ ( 1 シクロペンチノレピぺリジン 4 ィノレ)ォキシ]フエ二ノレ } N—(4ーメ トキシフエニル) 1H—ピラゾールー 4 カルボキサミド、
1一 { 4一 [ ( 1 シクロペンチノレピぺリジン 4 ィノレ)ォキシ]フエ二ノレ } N— [4一( ピロリジン 1ーィルカルボニル)フエニル] 1H—ピラゾールー 4 カルボキサミド、
N シクロペンチル 1一 { 4一 [ ( 1 シクロペンチルビペリジン 4 ィノレ)ォキシ]フ ェニル } N メチル 1 H ピラゾールー 4 カルボキサミド、
ェチル 4 {[(1 {4 [(1 シクロペンチルビペリジン 4 ィノレ)ォキシ]フエ二 ノレ } 1H—ピラゾールー 4 ィル)カルボニル]アミノ}ブタン酸、
tert-ブチル 4 {[(1 {4 [(1 シクロペンチルビペリジン 4 ィノレ)ォキシ] フエニル] 1H—ピラゾールー 4—ィノレ]カルボ二ノレ }ァミノ]ブタン酸、
4-{[(1-{4-[(1-シクロペンチルビペリジン 4—ィル)ォキシ]フエ二ノレ }— 1 H ピラゾールー 4 ィル)カルボ二ノレ]ァミノ]ブタン酸、
1— tert ブチル 4— { 4— [4— (ピロリジン 1—ィノレカノレポ二ノレ) 1 H ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン、
1一 { 4一 [( 1ーシクロプロピルピぺリジン一 4一ィノレ)ォキシ]フエ二ノレ }一 N—(4ーフ ルオロフェニル) 1H—ピラゾールー 4 カルボキサミド、
N- (4 フルオロフェニル)ー1 (4 {[1 (1ーメチルェチノレ)ピぺリジンー4ーィ ノレ]ォキシ }フエニル) 1 H ピラゾールー 4 カルボキサミド、
1 { 4 [ ( 1—シクロペンチルピロリジン 3—ィル)ォキシ]フエ二ノレ }— 4— (ピロリ ジン 1ーィルカルボニル) 1H—ピラゾール、
1— {4— [(1—シクロブチルピロリジン— 3—ィル)ォキシ]フエ二ル}— 4— (ピロリジン 1ーィルカルボニル) 1H—ピラゾール、
1—シクロペンチノレ一 4— {3—フルオロー 4— [4— (ピロリジン一 1—ィルカルボ二ノレ) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1ーシクロブチルー 4 {3—フルオロー 4 [4 (ピロリジン 1ーィルカルボニル) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1—シクロペンチノレ一 4— {2 フルオロー 4— [4— (ピロリジン一 1—ィルカルボ二ノレ) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1ーシクロブチルー 4 { 2 フルオロー 4 [4 (ピロリジン 1ーィルカルボニル) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1—シクロペンチノレ一 4— { 3—メチノレ一 4— [4— (ピロリジン一 1—ィルカルボニル) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1ーシクロブチルー 4 { 3—メチルー 4 [4 (ピロリジン 1ーィルカルボニル) 1 H ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1 {4一 [ (1ーシクロブチルピペリジンー4 ィル)ォキシ]フエ二ル} 1H—ピラゾ 一ルー 4 カルボキサミド、
1一 { 4一 [ ( 1 シクロペンチノレピぺリジン 4 ィノレ)ォキシ]フエ二ノレ } 1 H ピラゾ 一ルー 4 カルボ二トリル、
1—シクロブチル一 4— [4— (4—ニトロ一 1H—ピラゾールー 1—ィル)フエノキシ]ピ ペリジン、
4—クロ口一 N— (1— {4— [ (1—シクロブチルピペリジン一 4—ィノレ)ォキシ]フェニル }— 1 H ピラゾール一 4—ィル)ブタンアミド、
1一(1 {4一 [ (1ーシクロペンチルビペリジンー4 ィル)ォキシ]フエ二ル} - 1H- ピラゾール一 4 ィル)ピロリジン 2 オン、
2 クロロェチノレ { 1 [4ー(1 シクロブチルピペリジン 4 ィルォキシ)フエ二ノレ ]一 1H—ピラゾールー 4ーィル }一力ルバミン酸、
3— (1 - {4- [ (1 -シクロペンチルビペリジン 4—ィル)ォキシ]フエ二ノレ }— 1 H— ピラゾール一 4 ィル) 1 , 3 ォキサゾリジン一 2 オン、
{ 1 [4一(1ーシクロブチルピペリジンー4 ィルォキシ)フエニル] 1H—ピラゾー ノレー3—イノレ}ピロリジン 1 イノレーメタノン、
4) 1)〜3)までのいずれかに記載のピラゾール誘導体、又はその医薬上許容される 塩を有効成分として含有する医薬、
5)ヒスタミン H3受容体拮抗物質である 4)の医薬、及び
6) 1)〜3)までのいずれかに記載のピラゾール誘導体、又はその医薬上許容される 塩を有効成分として含有することを特徴とする、認知症、アルツハイマー病、注意欠 陥 ·多動性症、統合失調症、てんかん、中枢性痙攣、摂食障害、肥満、糖尿病、高脂
血症、睡眠障害、ナルコレプシ一、睡眠時無呼吸症候群、概日リズム障害、うつ病、 又はアレルギー性鼻炎の予防剤又は治療剤である。
発明の効果
[0023] 本発明の化合物が、優れたヒスタミン H3受容体拮抗作用を有することを見出した。
発明を実施するための最良の形態
[0024] 本明細書で用いられる用語は、以下に定義される通りである。
[0025] 本発明におレ、て、ハロゲンとは、フッ素原子、塩素原子、臭素原子又はヨウ素原子 である。
[0026] 「C〜Cアルキル」とは、炭素数 1〜6個の直鎖状又は分枝状のアルキル基を示し
1 6
、例えば、メチル、ェチル、 n プロピル、イソプロピル、 n ブチル、イソブチル、 sec ーブチノレ、 tert ブチノレ、 n ペンチノレ、イソペンチノレ、ネオペンチノレ又は n へキ シル等の基を示す。
[0027] 「C〜C環状アルキル」とは、シクロプロピル、シクロブチル、シクロペンチル、シクロ
3 8
へキシル、シクロへプチル又はシクロォクチル基である。
[0028] 「C〜Cアルケニル」とは、炭素数 3〜8個の直鎖状又は分枝状のアルケニル基を
3 8
示し、例えば、 1—プロペン— 2 ィル、 1—ブテン— 3 ィル、 1—へキセン— 5 ィ ノレ、 1—オタテン一 7 ィル、 1— (3—メチルブテン一 2 ィル)、 1—へキセン一 2— ィル又は 1一(3, 4—ジメチルペンテン 3—ィル)等の基を示す。
[0029] 「C〜Cアルコキシ」とは、炭素数;!〜 6個の直鎖状又は分枝状のアルコキシ基を
1 6
示し、例えば、メトキシ、エトキシ、 n プロポキシ、イソプロポキシ、 n ブトキシ、イソ ブトキシ、 sec ブトキシ、 tert ブトキシ、 n ペンチルォキシ、イソペンチルォキシ、 ネオペンチルォキシ、又は n へキシルォキシ等の基を示す。
[0030] 「C
1〜Cアルキルァミノ」とは、炭素数;!〜 6個の直鎖状又は分枝状のアルキル基 6
で置換されたアミノ基を示し、例えば、メチノレアミノ、ェチルアミ入 n—プロピノレアミノ、 イソプロピルアミ入 n ブチルァミノ、イソブチルァミノ、 sec ブチルァミノ、 tert ブ チルァミノ、 n ペンチルァミノ、イソペンチルァミノ、ネオペンチルァミノ又は n へキ シルァミノ等の基を示す。
[0031] 「C〜C ジアルキルァミノ」とは、 2つの炭素数 1〜6個の直鎖状又は分枝状のァ
ルキル基で置換されたアミノ基を示し、例えば、ジメチルァミノ、ジェチルアミ入ジー n プロピルァミノ、 N, N イソプロピルメチルァミノ、ジ一 n ブチルァミノ、ジイソブ チルァミノ、 N, N— sec ブチルェチルァミノ、 N, N— tert ブチルメチルァミノ、ジ —n—ペンチルァミノ、 N, N—イソペンチルメチルァミノ、 N, N—ネオペンチルメチ ルァミノ又はジー n へキシルァミノ等の基を示す。
[0032] 「4〜7員の飽和複素環」とは、飽和された 4〜7員の環状のアミノ基を示し、例えば、 1 ァゼチジル、 1 ピロリジル、ピペリジノ又は 1ーァゼパニル等の基を示す。
[0033] 「C〜Cアルコキシカルボニル」とは、炭素数;!〜 6個の直鎖状又は分枝状のアル
2 7
コキシ基が結合したカルボ二ル基を示し、例えば、メトキシカルボニル、エトキシカノレ ボニノレ、 n—プロポキシカノレポニノレ、イソプロポキシカノレポニノレ、 n—ブトキシカノレポ二 ノレ、イソブトキシカルボニル、 sec ブトキシカルボニル、 tert ブトキシカルボニル、 n ペンチルォキシカルボニル、イソペンチルォキシカルボニル、ネオペンチルォキ シカルボニル、又は n へキシルォキシカルボニル等の基を示す。
[0034] 「C〜Cアルキルアミノカルボニル」とは、炭素数;!〜 6個の直鎖状又は分枝状のァ
2 7
ルキル基で置換されたァミノ基が結合したカルボ二ル基を示し、例えば、メチルァミノ カルボニル、ェチルァミノカルボニル、 n プロピルアミノカルボニル、イソプロピルァ ミノカノレポ二ノレ、 n ブチルァミノカルボニル、イソブチルァミノカルボニル、 sec ブ チルァミノカルボニル、 tert ブチルァミノカルボニル、 n ペンチルァミノカルボ二 ノレ、イソペンチルァミノカルボニル、ネオペンチルァミノカルボニル、又は n へキシ ルァミノカルボニル等の基を示す。
[0035] 「C〜C ジアルキルアミノカルボニル」とは、 2つの炭素数 1〜6個の直鎖状又は分
3 13
枝状のアルキル基で置換されたァミノ基が結合したカルボ二ル基を示し、例えば、ジ メチルァミノカルボニル、ジェチルァミノカルボニル、ジー n プロピルアミノカルボ二 ノレ、 N, N イソプロピルメチルァミノカルボニル、ジー n ブチルァミノカルボニル、 ジイソブチルァミノカルボニル、 N, N sec ブチルェチルァミノカルボニル、 N, N tert ブチルメチルァミノカルボニル、ジー n ペンチルァミノカルボニル、 N, N イソペンチルメチルァミノカルボニル、 N, N—ネオペンチルメチルァミノカルボ二 ル又はジー n へキシルァミノカルボニル等の基を示す。
[0036] 「環中に少なくとも 1つの窒素原子を有し、他に 1つ以上の窒素、酸素、若しくは硫 黄原子を含んでもよい 3〜7員の飽和複素環に窒素原子を介して結合したカルボ二 ノレ」とは、 1個の窒素原子を含み、更に他に窒素原子、酸素原子、硫黄原子から選択 される 1個以上のへテロ原子を含んでいても良い、飽和された 3〜7員の単環式複素 環に窒素原子を介して結合したカルボ二ル基を示し、例えば、アジリジン 1一力ノレ ボニル、ァゼチジン 1 カルボニル、ピロリジン 1 カルボニル、ピぺリジン 1 カルボニル、ァゼパン 1 カルボニル、イミダゾリジン 1 カルボニル、ビラゾリジ ン 1 カルボニル、ピぺラジン 1 カルボニル、ォキサゾリジン 1 カルボニル 、モルホリン 1 カルボニル又はチオモルホリン— 1 カルボニル等の基を示す。
[0037] 「隣接する窒素原子と一緒になつて互いに結合し、環中に前記窒素原子の他に 1 つ以上の窒素、酸素、又は硫黄原子を含んでもよい 4〜7員の飽和複素環」とは、 1 個の窒素原子を含み、更に窒素原子、酸素原子、硫黄原子から選択される 1個以上 のへテロ原子を含んでいても良い、飽和された 4〜7員の単環式複素環基を示し、例 えば、 1 ァゼチジル、 1 ピロリジニル、ピペリジノ、 1 ァゼパニル、 1 イミダゾリジ ニル、 1—ビラゾリジニル、 1ーピぺラジュル、 3—ォキサゾリジニル、モルホリノ又は 1 チオモルホリニル等の基を示す。
[0038] 「ァリール」とは、フエニル基又はナフチル基を示す。
[0039] 「ヘテロァリール」とは、単環又は双環の芳香族複素環基を示し、ピリジン、ピリダジン 、ピリミジン、ピラジン、キノリン、イソキノリン、キナゾリン、キノキサリン、ピロール、フラ ン、チ才フェン、ピラゾール、イミダゾール、才キサゾーノレ、イソ才キサゾーノレ、チアゾ 一ノレ、イソチアゾーノレ、 トリァゾーノレ、インドーノレ、ベンゾフラン、ベンゾチォフェン、ベ ンゾイミダゾーノレ、インダゾーノレ、ベンゾォキサゾーノレ、ベンゾチアゾーノレ又はべンゾ トリァゾール等の基が挙げられる。
[0040] 本発明において、医薬上許容される塩とは、例えば、硫酸、塩酸、臭化水素酸、リ ン酸、硝酸などの無機酸との塩、酢酸、シユウ酸、乳酸、酒石酸、フマル酸、マレイン 酸、クェン酸、ベンゼンスルホン酸、メタンスルホン酸、 p トルエンスルホン酸、安息 香酸、カンファースルホン酸、エタンスルホン酸、ダルコヘプトン酸、ダルコン酸、グル タミン酸、グリコール酸、リンゴ酸、マロン酸、マンデル酸、ガラクタル酸、ナフタレン一
2—スルホン酸などの有機酸との塩、リチウムイオン、ナトリウムイオン、カリウムイオン 、カルシウムイオン、マグネシウムイオン、亜鉛イオン、アルミニウムイオンなどの 1種 又は複数の金属イオンとの塩、アンモニア、アルギニン、リシン、ピぺラジン、コリン、 ジェチルァミン、 4 フエニルシクロへキシルァミン、 2 アミノエタノール、ベンザチン などのァミンとの塩が含まれる。
[0041] 本発明の化合物は、各種溶媒和物としても存在し得る。また、医薬としての適用性 の面から水和物の場合もある。
[0042] 本発明の化合物は、ェナンチォマー、ジァステレオマー、平衡化合物、これらの任 意の割合の混合物、ラセミ体等を全て含む。
[0043] 本発明の化合物には、一つ以上の水素原子、炭素原子、窒素原子、酸素原子、硫 黄原子が放射性同位元素や安定同位元素と置換された化合物も含まれる。これらの 標識化合物は、例えば代謝や薬物動態研究、受容体のリガンドとして生物学的分析 等に有用である。
[0044] 本発明の化合物は、一つ又は二つ以上の医薬的に許容される担体、賦形剤又は 希釈剤と組み合せて医薬的製剤とすることができる。上記担体、賦形剤及び希釈剤 として、例えば水、乳糖、デキストロース、フラクトース、ショ糖、ソルビトール、マンニト 一ノレ、ポリエチレングリコーノレ、プロピレングリコーノレ、デンプン、ガム、ゼラチン、ァノレ ギネート、ケィ酸カルシウム、リン酸カルシウム、セルロース、水シロップ、メチルセル口 ース、ポリビュルピロリドン、アルキルパラヒドロキシベンゾソルベート、タルク、ステアリ ン酸マグネシウム、ステアリン酸、グリセリン、ゴマ油、ォリーブ油、大豆油等の各種油 等が含まれる。
[0045] また、上記の担体、賦形剤又は希釈剤に必要に応じて一般に使用される増量剤、 結合剤、崩壊剤、 pH調整剤、溶解剤等の添加剤が混合し、常用の製剤技術によつ て錠剤、丸剤、カプセル剤、顆粒剤、粉剤、液剤、乳剤、懸濁剤、軟膏剤、注射剤、 皮膚貼付剤等の経口又は非経口用医薬として調製すること力 Sできる。本発明の化合 物は、成人患者に対して 1回の投与量として 0. 00;!〜 500mgを 1日 1回又は数回に 分けて経口又は非経口で投与することが可能である。なお、この投与量は治療対象 となる疾病の種類、患者の年齢、体重、症状等により適宜増減することが可能である
本発明の化合物における望ましいプロファイルとしては、薬効が優れていること、体 内動態が優れていること、物性が優れていること、低毒性であること、等が挙げられる 本発明の化合物は、下記の方法により製造することができる。
[0047] (本発明の化合物を製造する方法)
本発明の化合物は、公知の有機化学的手法によって製造することができ、例えば、 下記の反応式による方法に従って製造できる。
下記反応式;!〜 7において、 R、 R'-R7, T、 n、 A及び Bは前記と同義である。また、 xi〜x5は、同一又は異なって、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子 又はメタンスルホニルォキシ基、ベンゼンスルホニルォキシ基、 p—トルエンスルホニ ルォキシ基、トリフルォロメタンスルホニルォキシ基等の有機スルホニルォキシ基等の 脱離基を示し、 Yi〜Y2は、同一又は異なって、ハロゲン原子又は有機スルホニルォ キシ基等の脱離基、又は水酸基を示し、 Zは、炭素原子又は酸素原子を示し、 pは、 0又は 1の整数を示し、 R8及び R9は、水素原子、アルキル基又は環状アルキル基を 示す。また、 R8及び R9は、隣接する炭素原子と一緒に環状アルキル環を形成しても 良い。
[0048] 以下、反応式 1で示される本発明の化合物の製造方法について説明する。この製 造工程は、化合物(2)から本発明の化合物(1)を製造するための工程である。
(反応式 1)
[0049] [化 7]

(工程 la)
工程 laは、化合物(2)と化合物(3)とのカップリング反応により化合物(4)を得るため の工程である。化合物(2)及び化合物(3)は、公知化合物であるか、公知化合物か
ら容易に合成できる化合物である。該カップリング反応としてはミツノブ反応が挙げら れ、例えば、トリフエニルホスフィン、トリブチルホスフィン等の有機リン化合物とァゾジ カルボン酸ジェチル、ァゾジカルボン酸ジイソプロピル、ァゾジカルボン酸ジ tertブチ ル等のァゾ化合物とを組み合わせた試薬、或いはシァノメチルトリブチルホスホラン 等のリンイリド試薬の存在下溶媒中で行う方法が挙げられる。本反応で用いられる溶 媒としては、例えば、テトラヒドロフラン、 1 , 4 ジォキサン等のエーテル類;トルエン、 ベンゼン等の炭化水素類;クロ口ホルム、ジクロロメタン等のハロゲン化炭化水素類; N, N ジメチルホルムアミド、 N, N ジメチルァセトアミド、 N メチル 2—ピロリド ン等のアミド類;ジメチルスルホキシド;ァセトニトリル又はこれらの混合溶媒が挙げら れ、これらのうち、テトラヒドロフラン、トルエンが好ましい。本反応における反応温度 は、通常 0°C〜; 120°C、好ましくは、 15°C〜80°Cであり、反応時間は、通常;!〜 48時 間、好ましくは、;!〜 12時間である。
(工程 2a)
工程 2aは、化合物(4)と化合物(5)とをカップリング反応により縮合して本発明の化 合物(1)を得るための工程である。化合物(5)は、公知化合物である力、、公知化合物 力、ら容易に合成できる化合物である。該カップリング反応は、塩基の存在下、リガンド 及び触媒を用いて、溶媒中でァゾール化合物の窒素原子の芳香化を行う一般的な 方法により実施でき、 列えば、、(Kunzら, Synlett, 15, 2428— 2439, 2003 )に記載の方法又はそれに準じた方法に従って実施できる。本反応で用いられる触 媒としては、縮合反応に一般的に用いられる銅触媒、例えば、銅 (0)、ヨウ化銅 (1)、 塩化銅 (1)、酸化銅 (1)、臭化銅 (I)トリストリフエニルホスフィン錯体、トリフルォロメタ ンスルホン酸銅 (I)ベンゼン錯体等が挙げられる。本反応で用いられるリガンドとして は、銅触媒を用いた縮合反応に一般的に用いられるリガンド、例えば、 N, N'—ジメ チノレエチレンジァミン、 1 , 2—シクロへキサンジァミン、 2—アミノビリジン、 1 , 10—フ ェナンスロリン、 2—ヒドロキシベンズアルデヒドォキシム、エチレングリコール等が挙 げられる。本反応で用いられる塩基としては、例えば、炭酸カリウム、リン酸カリウム、 水酸化カリウム、 tert ブトキシカリウム、炭酸セシウム、炭酸ナトリウム、炭酸水素ナ トリウム、酢酸ナトリウム、ナトリウムメトキシド、テトラプチルアンモニゥムヒドロキシド等
が挙げられ、これらのうち、炭酸カリウム、炭酸セシウムが好ましい。本反応で用いら れる溶媒としては、例えば、メタノール、エタノール、イソプロパノール等のアルコール 類;テトラヒドロフラン、 1 , 4 ジォキサン等のエーテル類;トルエン、ベンゼン等の炭 化水素類;クロ口ホルム、ジクロロメタン等のハロゲン化炭化水素類; N, N ジメチル ホルムアミド、 N, N ジメチルァセトアミド、 N メチル 2—ピロリドン等のアミド類; ジメチルスルホキシド;ァセトニトリル;アセトン;水又はこれらの混合溶媒が挙げられ、 これらのうち、 N, N ジメチルホルムアミド、 N メチル 2—ピロリドンが好ましい。 本反応における反応温度は、通常 0°C〜; 150°C、好ましくは、 40°C〜; 120°Cであり、 反応時間は、通常;!〜 48時間、好ましくは、;!〜 12時間である。
(反応式 2)
[0052] [化 8]

[0053] また、化合物(4)は、化合物(2)と化合物(6)とのカップリング反応により得ることがで きる。化合物(6)は、公知化合物であるか、公知化合物から容易に合成できる化合物 である。該カップリング反応は、塩基の存在下又は非存在下、溶媒中又は無溶媒で フエノールの O アルキル化を行う一般的な方法により実施できる。また、必要に応じ て、例えば、ヨウ化カリウム、臭化ナトリウム等の添加物を加えることができる。本反応 で用いられる塩基としては、例えば、ピリジン、トリェチルァミン、ジイソプロピルェチル ァミン、 tert ブトキシカリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、水 酸化ナトリウム、水酸化カリウム、水素化ナトリウム等が挙げられる。本反応で用いられ る溶媒としては、例えば、メタノール、エタノール、イソプロパノール等のアルコール類 ;テトラヒドロフラン、 1 , 4 ジォキサン等のエーテル類;トルエン、ベンゼン等の炭化 水素類;クロ口ホルム、ジクロロメタン等のハロゲン化炭化水素類; N, N ジメチルホ ルムアミド、 N, N ジメチルァセトアミド、 N メチル 2—ピロリドン等のアミド類;ジメ
チルスルホキシド;ァセトニトリル;アセトン;水又はこれらの混合溶媒が挙げられ、これ らのうち、テトラヒドロフラン、 N—ジメチルホルムアミド、ァセトニトリル、アセトンが好ま しい。本反応における反応温度は、通常 0°C〜; 150°C、好ましくは、 15°C〜80°Cで あり、反応時間は、通常;!〜 48時間、好ましくは、;!〜 12時間である。
[0054] 以下、反応式 3で示される本発明の化合物の製造方法について説明する。この製 造工程は、化合物(2)から本発明の化合物(1)を製造するための工程である。
(反応式 3)
[0055] [化 9]

(工程 lb)
工程 lbは、化合物(2)と公知化合物(7)とをカップリング反応により縮合して化合物( 8)を得るための工程である。該カップリング反応は、工程 laと同様の方法により実施 できる。
(工程 2b)
工程 2bは、化合物(8)と化合物(5)とをカップリング反応により縮合して化合物(9)を 得るための工程である。該カップリング反応は、工程 2aと同様の方法により実施でき
(工程 4a)
工程 4aは、化合物(9)の tert—ブトキシカルボ二ル基を脱保護して化合物(10)を 得るための工程である。該反応は、一般的な tert—ブトキシカルボニル基の脱保護 の方法により実施でき、例えば、(Τ· W. Greene and P. G. M. Wuts Protecti ve Groups in Organic Synthesis)に記載されている方法又はそれに準じた方
法に従って実施でき、強酸存在下溶媒中又は無溶媒で反応させる方法、塩基存在 下溶媒中で反応させる方法等が挙げられる。本反応で用いられる酸としては、例え ば、塩酸、硫酸、トリフルォロ酢酸、トリフルォロメタンスルホン酸等が挙げられる。本 反応で用いられる塩基としては、例えば、水酸化ナトリウム、水酸化カリウム等が挙げ られる。本反応で用いられる溶媒としては、例えば、メタノール、エタノール、イソプロ パノール等のアルコール類;ジェチルエーテル、テトラヒドロフラン、 1 , 4ージォキサ ン等のエーテル類;トルエン、ベンゼン等の炭化水素類;クロ口ホルム、ジクロロメタン 等のハロゲン化炭化水素類; N, N ジメチルホルムアミド、 N, N ジメチルァセトァ ミド、 N メチル— 2—ピロリドン等のアミド類;ジメチルスルホキシド;ァセトニトリル;ァ セトン;水又はこれらの混合溶媒が挙げられる。本反応における反応温度は、通常 0 °C〜150°C、好ましくは、 15°C〜40°Cであり、反応時間は、通常;!〜 48時間、好まし くは、;!〜 12時間である。
(工程 5a)
工程 5aは、化合物(10)と化合物(11)とをカップリング反応により縮合して本発明の 化合物(1)を得るための工程である。化合物(11)は、公知化合物である力、、公知化 合物から容易に合成できる化合物である。該カップリング反応は、カルボニル化合物 とアミンを縮合させる一般的な還元的ァミノ化反応の方法によって実施でき、例えば 、酸の存在下又は非存在下、溶媒中又は無溶媒で化合物(8)と化合物(9)の混合 物に還元剤を加えて行う方法が挙げられる。また、還元方法として、例えば、パラジゥ ムー炭素、白金、ラネーニッケル、ロジウム アルミナ等の触媒を用いた水素添加に よる接触還元の方法を利用することもできる。本反応で用いられる還元剤としては、 例えば、水素化ホウ素ナトリウム、トリァセトキシ水素化ホウ素ナトリウム、シァノ水素化 ホウ素ナトリウム、ジボラン、水素化リチウムアルミニウム等が挙げられる。本反応で用 いられる酸としては、例えば、酢酸、ギ酸、塩酸等が挙げられる。本反応で用いられる 溶媒としては、例えば、メタノール、エタノール、イソプロパノール等のアルコール類; テトラヒドロフラン、 1 , 4 ジォキサン等のエーテル類;トルエン、ベンゼン等の炭化水 素類;ァセトニトリル;水又はこれらの混合溶媒が挙げられ、これらのうち、エタノール、 トルエン、テトラヒドロフランが好ましい。本反応における反応温度は、通常 0°C〜; 150
°C、好ましくは、 15°C〜40°Cであり、反応時間は、通常: 48時間、好ましくは、 12時間である。
(反応式 4)
[0058] [化 10]
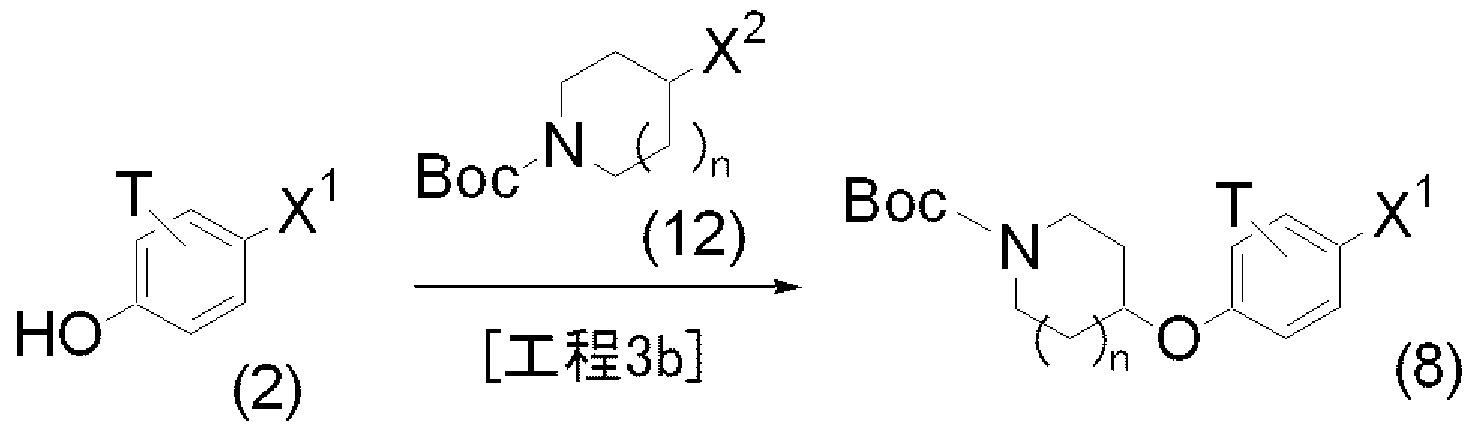
また、化合物(8)は、化合物(2)と化合物(12)とをカップリング反応により縮合して得 ること力 Sできる。化合物(12)は、公知化合物である力、、公知化合物から容易に合成 できる化合物である。該; > '反応は、工程 3aと同様の方法により実施できる。 (反応式 5)
[0060] [化 11]

[0061] また、本発明の化合物(1)は、化合物(10)と化合物(13)とのカップリング反応により 得ること力 Sできる。化合物(13)は、公知化合物である力、、公知化合物から容易に合 成できる化合物である。該カップリング反応は、塩基の存在下又は非存在下、溶媒中 又は無溶媒でァミンのアルキル化を行う一般的な方法により実施できる。また、必要 に応じて、例えば、ヨウ化カリウム、臭化ナトリウム等の添加物を加えることができる。 本反応で用いられる塩基としては、例えば、ピリジン、トリェチルァミン、ジイソプロピ ルェチルァミン、 tert ブトキシカリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナト リウム、水酸化ナトリウム、水酸化カリウム、水素化ナトリウム等が挙げられる。本反応 で用いられる溶媒としては、例えば、メタノール、エタノール、イソプロパノール等のァ ルコール類;テトラヒドロフラン、 1, 4 ジォキサン等のエーテル類;トルエン、ベンゼ
ン等の炭化水素類;クロ口ホルム、ジクロロメタン等のハロゲン化炭化水素類; N, N— ジメチルホルムアミド、 N, N ジメチルァセトアミド、 N メチル 2—ピロリドン等の アミド類;ジメチルスルホキシド;ァセトニトリル;アセトン;水又はこれらの混合溶媒が 挙げられ、これらのうち、テトラヒドロフラン、 N, N ジメチルホルムアミド、ァセトニトリ ルが好ましい。本反応における反応温度は、通常 0°C〜; 150°C、好ましくは、 15°C〜 80°Cであり、反応時間は、通常;!〜 48時間、好ましくは、;!〜 12時間である。
[0062] 以下、反応式 6で示される本発明の化合物の製造方法について説明する。この製 造工程は、化合物(8)及び化合物(14)から本発明の化合物(1 1)及び(1 2)を 製造するための工程である。
(反応式 6)
[0063] [化 12]
Boc、
[工程 5b]

[0064] (工程 2c)
工程 2cは、化合物(8)と公知化合物(14)とのカップリング反応により化合物(15)を 得るための工程である。該カップリング反応は、工程 2aと同様の方法により実施でき
[0065] (工程 4b)
工程 4bは、化合物(15)の tert ブトキシカルボ二ル基を脱保護して化合物(16)を 得るための工程である。該脱保護反応は、工程 4aと同様の方法により実施できる。
[0066] (工程 5b)
工程 5bは、化合物(16)と化合物(11)とのカップリング反応により本発明の化合物( 1— 1)を得るための工程である。該カップリング反応は、工程 5aと同様の方法により 実施できる。
[0067] (工程 7)
工程 7は、化合物(1 1)のエトキシカルボ二ル基を加水分解によりカルボン酸に変 換して化合物(17)を得るための工程である。該加水分解反応は、一般的なエステル の加水分解反応により実施することができ、例えば、(Τ· W. Greene and P. G. M. Wuts著 Protective Groups in Organic Synthesis、 3版、 John Wil ey and Sons社)に記載の方法又はそれに準じた方法に従って実施でき、強酸存 在下溶媒中又は無溶媒で反応させる方法、塩基存在下溶媒中で反応させる方法等 が挙げられる。本反応における反応温度は、通常 0°C〜; 120°C、好ましくは、 15°C〜 80°Cであり、反応時間は、通常;!〜 48時間、好ましくは、;!〜 12時間である。
[0068] (工程 8a)
工程 8aは、化合物(17)と化合物(18)とをカップリング反応により縮合して本発明の 化合物(1 2)を得るための工程である。化合物(18)は、公知化合物である力、、公 知化合物から容易に合成できる化合物である。該カップリング反応は、一般的なカル ボン酸のアミド化の方法により実施でき、例えば、カルボン酸をカルボン酸クロリドゃ カルボン酸ブロミド等のカルボン酸ノヽライドに導いた後にァミンと反応させる方法、力 ルボン酸とクロル炭酸エステル等から得られる混合酸無水物をァミンと反応させる方 法、カルボン酸を 1—ベンゾトリァゾリルエステルゃスクシンィミジルエステル等の活性 エステルに導いた後にァミンと反応させる方法、カルボン酸を脱水縮合剤存在下アミ ンと反応させる方法等が挙げられる。これらの反応は、全て塩基の存在下又は非存 在下、溶媒中で行うことができる。本反応で用いられる脱水縮合剤としては、例えば、 3—(3—ジメチルァミノプロピル) 1 ェチルカルポジイミド塩酸塩、ジシクロへキシ ノレ力ノレポジイミド、ジフエニルホスホリルアジド、カルボニルジイミダゾール、 0 - (7 - ァザべンゾトリァゾールー 1ーィル) 1 , 1 , 3, 3—テトラメチルゥロニゥム へキサフ ルォロホスフェート等が挙げられ、必要に応じて 1ーヒドロキシベンゾトリァゾール、ヒド ロキシスクシンイミド等の活性化剤を用いることができる。本反応で用いられる塩基と しては、例えば、ピリジン、トリェチルァミン、ジイソプロピルェチルァミン、炭酸カリウム 、炭酸ナトリウム、炭酸水素ナトリウム等が挙げられる。本反応で用いられる溶媒として は、例えば、テトラヒドロフラン、 1 , 4—ジォキサン等のエーテル類;トルエン、ベンゼ
ン等の炭化水素類;クロ口ホルム、ジクロロメタン等のハロゲン化炭化水素類; N, N— ジメチルホルムアミド、 N, N ジメチルァセトアミド、 N メチル 2—ピロリドン等の アミド類;ジメチルスルホキシド;ァセトニトリル;アセトン;水又はこれらの混合溶媒が 挙げられ、これらのうち、トルエン、テトラヒドロフラン又は N, N ジメチルホルムアミド が好ましい。本反応における反応温度は、通常 0°C〜; 120°C、好ましくは、 15°C〜4 0°Cであり、反応時間は、通常;!〜 48時間、好ましくは、;!〜 12時間である。
[0069] (反応式 7)
[0070] [化 13]

[0071] また、本発明の化合物(1 1)は、化合物(16)と化合物(13)とのカップリング反応に より得ること力 Sできる。該カップリング反応は、工程 6aと同様の方法により実施できる。
[0072] 以下、反応式 8で示される本発明の化合物の製造方法について説明する。この製 造工程は、化合物(1 1)から本発明の化合物(1 3)及び(1 4)を製造するため の工程である。
(反応式 8)
[0073] [化 14]

[0074] (工程 8b)
工程 8bは、化合物(1 1)をアンモニアと反応させて本発明の化合物(1 3)を得る ための工程である。該反応は、工程 8aと同様の方法により実施できる。具体例として は、例えば、カルボン酸を脱水縮合剤存在下アンモニア水と反応させる方法により実 施できる。
[0075] (工程 9)
工程 9は、化合物(1 3)の力ルバモイル基を二トリル基に変換して本発明の化合物 (1—4)を得るための工程である。本工程は、カノレバモイル基を二トリル基に変換する 一般的な反応により実施でき、例えば、脱水剤の存在下、溶媒中又は無溶媒で反応 させることにより実施できる。また、必要に応じて、例えば、 N, N ジメチルホルムアミ ド、塩化ナトリウム等の添加物を加えることができる。本反応で用いられる脱水剤とし ては、例えば、 5酸化リン、 5塩化リン、塩化ホスホリル、塩化チォニル、塩化ォキサリ ノレ、無水トリフルォロ酢酸、無水トリフルォロメタンスルホン酸等が挙げられる。本反応 で溶媒を用いる場合、溶媒としては、例えば、テトラヒドロフラン、 1 , 4ージォキサン等 のエーテル類;トルエン、ベンゼン等の炭化水素類; N, N ジメチルホルムアミド、 N , N ジメチルァセトアミド、 N メチル—2—ピロリドン等のアミド類;ァセトニトリル又 はこれらの混合溶媒が挙げられる。本反応における反応温度は、通常 0°C〜; 120°C 、好ましくは、 15°C〜80°Cであり、反応時間は、通常;!〜 48時間、好ましくは、;!〜 1 2時間である。
[0076] 以下、反応式 9で示される本発明の化合物の製造方法について説明する。この製 造工程は、化合物(4)から本発明の化合物(1 5)、(1 6)及び(1 7)を製造する ための工程である。
(反応式 9)
[0077] [化 15]

[0078] (工程 2d)
工程 2dは、化合物(4)と化合物(19)とをカップリング反応により縮合して本発明の化
合物(1 5)を得るための工程である。該反応は、工程 2aと同様の方法により実施で きる。
[0079] (工程 10)
工程 10は、化合物(1 5)のニトロ基を還元して化合物(20)を得るための工程であ る。本工程は、ニトロ基をァミノ基に変換する一般的な還元反応により実施でき、例え ば、ノ ラジウム 炭素、白金、ラネーニッケル、ロジウム アルミナ等の触媒を用いた 水素添加による接触還元反応、亜鉛、鉄、スズ又は塩化スズ (Π)を用いた酸性条件 下での還元反応、水素化リチウムアルミニウム等の金属水素化物を用いた還元反応 等が挙げられる。具体例としては、例えば、ノ ラジウム一炭素を触媒とし、メタノール 溶媒中水素添加による接触還元反応を行うことにより実施できる。
[0080] (工程 11 a)
工程 1 laは、化合物(20)と化合物(21)とをカップリング反応により縮合して本発明 の化合物(1 6)を得るための工程である。化合物(21)は、公知化合物であるか、 公知化合物から容易に合成できる化合物である。該カップリング反応は、 Gが CO、 Y 1が水酸基である場合、工程 8aと同様の方法により実施できる。
Y1がハロゲン原子である場合、該カップリング反応は、塩基の存在下又は非存在下 、溶媒中又は無溶媒で化合物(20)と化合物(21)とを反応させることにより実施でき る。本反応で用いられる塩基としては、例えば、ピリジン、トリェチルァミン、ジイソプロ ピルェチルァミン、炭酸カリウム、炭酸水素ナトリウム、水酸化ナトリウム等が挙げられ る。本反応で用いられる溶媒としては、例えば、テトラヒドロフラン、 1 , 4—ジォキサン 等のエーテル類;トルエン、ベンゼン等の炭化水素類;クロ口ホルム、ジクロロメタン等 のハロゲン化炭化水素類; N, N ジメチルホルムアミド、 N, N ジメチルァセトアミド 、 N メチルー 2—ピロリドン等のアミド類又はこれらの混合溶媒が挙げられ、これらの うち、テトラヒドロフラン、トルエンが好ましい。本反応における反応温度は、通常 0°C 〜; 120°C、好ましくは、 15°C〜80°Cであり、反応時間は、通常;!〜 48時間、好ましく は、;!〜 12時間である。
(工程 12)
工程 12は、化合物(1 6)と化合物(22)とを反応させて本発明の化合物(1 7)
を得るための工程である。化合物(22)は、公知化合物である力、、公知化合物から容 易に合成できる化合物である。該反応は、一般的なアミドのアルキル化の方法により 実施でき、例えば、塩基の存在下、溶媒中で化合物(1 6)と化合物(22)とを反応 させることにより実施できる。また、必要に応じて、例えば、テトラプチルアンモニゥム ブロミド、 18 クラウンー6 エーテル等の添加物を加えることができる。本反応で用 いられる塩基としては、例えば、水素化ナトリウム、水素化カリウム、 tert ブトキシカ リウム、水酸化カリウム、水酸化ナトリウム、ナトリウムメトキシド、 n ブチルリチウム等 が挙げられる。本反応で用いられる溶媒としては、例えば、メタノール、エタノール、ィ ソプロパノール等のアルコール類;テトラヒドロフラン、 1 , 4 ジォキサン等のエーテ ル類;トルエン、ベンゼン等の炭化水素類; N, N ジメチルホルムアミド、 N, N ジ メチルァセトアミド、 N メチルー 2—ピロリドン等のアミド類;ジメチルスルホキシド;ァ セトニトリル;水又はこれらの混合溶媒が挙げられ、これらのうち、テトラヒドロフラン、 N , N ジメチルホルムアミドが好ましい。本反応における反応温度は、通常 0°C〜; 150 °C、好ましくは、 15°C〜; 100°Cであり、反応時間は、通常;!〜 48時間、好ましくは、 1 〜; 12時間である。
[0081] 以下、反応式 10で示される本発明の化合物の製造方法について説明する。この製 造工程は、化合物(20)から本発明の化合物(1 7)を製造するための工程である。 (反応式 10)
[0082] [化 16]

(工程 13)
工程 13は、化合物(20)のアルキル化反応により化合物(23)を得るための工程であ る。該アルキル化反応は、 1級アミノ基を 2級ァミノ基へ変換する一般的なアルキル化 方法によって実施でき、例えば、塩基の存在下アルキルハライドやアルキルメタンス ルホネート等のアルキル化剤を用いて反応させる方法、アルデヒドを用いた還元的ァ ミノ化反応による方法、カルボン酸及びその誘導体を用いて酸アミドとした後にボラン
等の金属水素化物で還元する方法、アルコールとの脱水縮合による方法等が挙げら れる。
[0083] (工程 l ib)
工程 1 lbは、化合物(23)と化合物(21)とをカップリング反応により縮合して本発明 の化合物(1— 7)を得るための工程である。該カップリング反応は、工程 11aと同様の 方法により実施できる。
[0084] 以下、反応式 11で示される本発明の化合物の製造方法について説明する。この製 造工程は、化合物(20)から本発明の化合物(1 8)及び(1 9)を製造するための 工程である。
(反応式 11 )
[0085] [化 17]
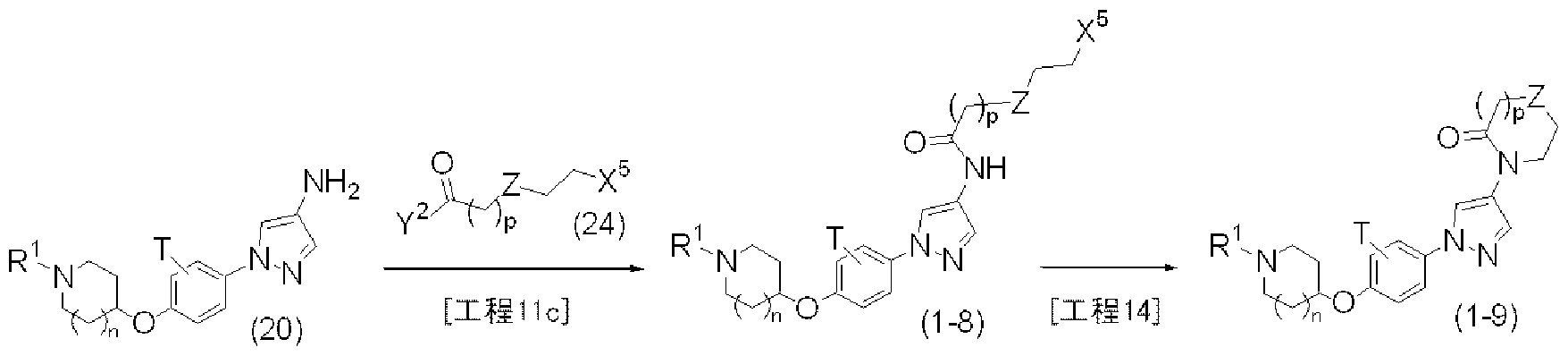
(工程 11c)
工程 1 lcは、化合物(20)と化合物(24)とをカップリング反応により縮合して本発明 の化合物(1 8)を得るための工程である。化合物(24)は、公知化合物であるか、 公知化合物から容易に合成できる化合物である。該カップリング反応は、工程 11aと 同様の方法により実施できる。
[0086] (工程 14)
工程 14は、化合物(1 8)の分子内環化反応によって本発明の化合物(1 9)を 得るための工程である。該分子内環化反応は、文献記載の方法 (例えば、 Journal of Medicinal Chemistry, 2002年、 45巻、 3972— 3983頁)又はそれに準じた 方法により実施できる。本反応で用いられる塩基としては、例えば、水素化ナトリウム 、水素化カリウム、炭酸カリウム、 tert ブトキシカリウム、水酸化ナトリウム等が挙げら れる。本反応で用いられる溶媒としては、例えば、テトラヒドロフラン、 1 , 4ージォキサ ン等のエーテル類;トルエン、ベンゼン等の炭化水素類; N, N ジメチルホルムアミ
ド、 N, N ジメチルァセトアミド、 N メチル 2—ピロリドン等のアミド類;ジメチルス ルホキシド;ァセトニトリル;アセトン又はこれらの混合溶媒が挙げられ、これらのうち、 テトラヒドロフラン、トルエンが好ましい。本反応における反応温度は、通常 0°C〜; 150 °C、好ましくは、 15°C〜80°Cであり、反応時間は、通常;!〜 48時間、好ましくは、;!〜 12時間である。
[0087] 以下に実施例及び試験例を示し、本発明を具体的に説明するが、本発明はこれら に限定されるものではない。
実施例 1
[0088] 1ーシクロペンチルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン(化合物番号 1)の製造
(1) tert ブチル 4一(4ーョードフエノキシ)ピぺリジン 1 カルボキシラートの製 造
[0089] tert ブチノレ 4 ヒドロキシピペリジン一 1 カルボキシラート(4. 3g)、 4 ョードフ ェノール(4. 8g)及びトリフエニルホスフィン(5· 8g)のテトラヒドロフラン(40mU溶液 に 40 %ァゾジカルボン酸ジイソプロピルのトルエン( 1 lmU溶液を加え、 60°Cにて 一晩攪拌した。反応混合物を室温まで放冷し、減圧下濃縮した。得られた残渣をシリ 力ゲルカラムクロマトグラフィー(展開溶媒: n へキサン:酢酸ェチル = 20: 1)にて精 製し、無色固体の表題化合物(5. 8g)を得た。
(2) tert ブチル 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾー ルー 1 ィル]フエノキシ }ピペリジン 1 カルボキシラートの製造
[0090] 実施例 1一(1)で得られた tert ブチル 4一(4ーョードフエノキシ)ピぺリジン一 1一 カルボキシラート(1 · 5g)、 4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾール (0. 68g) , rac -trans-N, N, 一ジメチルシクロへキサン一 1 , 2 ジァミン(0. 68g )、ヨウ化銅(I) (0. 085g)及び炭酸セシウム(2· 5g)の N, N ジメチルホルムアミド( 2. 5mL)懸濁液を 110°Cにて 2時間攪拌した。反応混合物を室温まで放冷し、クロ口 ホルムを加えて希釈し、セライト濾過した。濾液を水で洗浄し、減圧下濃縮した。得ら れた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:クロ口ホルム:メタノール = 9 : 1)にて精製し、酢酸ェチルで洗浄して無色固体の表題化合物(1. 5g)を得た。
(3) 4— {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾールー 1 ィル]

[0091] 実施例 1一(2)で得られた tert ブチル 4一 {4一 [4一(ピロリジン一 1ーィルカルボ ニル) 1H—ピラゾールー 1 ィル]フエノキシ }ピペリジン 1 カルボキシラート(1 . 5g)のテトラヒドロフラン(15mU、メタノール(5· OmU及びクロ口ホルム(lOmL) の混合溶液に、氷冷下 4M塩酸酢酸ェチル溶液を加え、室温にてー晚攪拌した。反 応混合物に飽和炭酸水素ナトリウム溶液を加え、クロ口ホルムで抽出した。有機層を 無水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られた残渣をジイソプロピルェ 一テルで洗浄し、無色固体の表題化合物(1. lg)を得た。
(4) 1ーシクロぺンチルー4 {4 [4ー(ピロリジン 1ーィルカルボニル) 1—ビラ ゾールー 1 ィル]フエノキシ }ピペリジン(化合物番号 1)の製造
[0092] [化 18]

実施例 1— (3)で得られた 4— {4— [4— (ピロリジン— 1—ィルカルボ二ル)— 1H— ピラゾールー 1—ィル]フエノキシ }ピペリジン(0· 30g)、シクロペンタノン(0· 089g) 及び酢酸(0· 079g)のクロロホノレム(3. OmU溶液にナトリウムトリァセトキシポロヒド リド (0. 23g)を加え、室温にて一時間攪拌した。反応混合物に飽和炭酸水素ナトリウ ム水溶液を加え、クロ口ホルムで抽出した。有機層を無水硫酸マグネシウムで乾燥し 、減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ク ロロホルム:メタノール = 8 : 1)にて精製し、ジイソプロピルエーテルで洗浄して無色固 体の表題化合物 (0. 28g)を得た。
:H NMR (600 MHz, CHLOROFORM- d) δ ppm 1.49 - 1.74 (m, 6 H), 1.81 - 2.06 ( m, 10 H), 2.29 - 2.39 (m, 2 H), 2.48 - 2.56 (m, 1 H), 2.75 - 2.86 (m, 2 H), 3.65 (t, J=6.9 Hz, 2 H), 3.74 (t, J=6.6 Hz, 2 H), 4.30 - 4.38 (m, 1 H), 6.98 (d, J=9.2 Hz, 2
H), 7.57 (d, J=9.2 Hz, 2 H), 7.95 (s, 1 H), 8.26 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 409(M+H)+
実施例 1 (4)に示した方法と同様の方法で得た化合物 (化合物番号 2)〜(化合物 番号 5)を表 1に示した。
[表 1]

[0095] 1—(シクロプロピルメチル)ー4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H ーピラゾールー 1 ィル]フエノキシ }ピペリジン(化合物番号 6 )の製造
[0096] [化 19]

[0097] 実施例 1一(3)で得られた 4一 {4一 [4一(ピロリジン一 1ーィルカルボニル)一 1H— ピラゾールー 1 ィル]フエノキシ }ピペリジン(0. 050g)、 (ブロモメチノレ)シクロプロ ノ ン(0. 024g)及び炭酸カリウム(0. 040g)の N, N ジメチノレホノレムアミド(0. 50 mL)懸濁液を 80°Cにて 3時間攪拌した。反応混合物を室温まで放冷し、水を加え、 酢酸ェチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧下濃縮した。得 られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:クロ口ホルム:メタノール = 8 : 1)にて精製し、ジイソプロピルエーテルで洗浄して無色固体の表題化合物(0. 040g)を得た。
:H NMR (600 MHz, CHLOROFORM— d) δ ppm 0.12 (d, J=3.2 Hz, 2 H), 0.54 (d, J= 7.3 Hz, 2 H), 0.89 (br. s, 1 H), 1.84 - 2.09 (m, 8 H), 2.25 - 2.45 (m, 4 H), 2.86 (br. s, 2 H), 3.66 (t, J=6.9 Hz, 2 H), 3.75 (t, J=6.9 Hz, 2 H), 4.36 (br. s, 1 H), 6.99 (d, J=9.2 Hz, 2 H), 7.58 (d, J=8.7 Hz, 2 H), 7.96 (s, 1 H), 8.28 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 395(M+H)+
実施例 2に示した方法と同様の方法で得た化合物 (化合物番号 7)〜(化合物番号 1 1)を表 2に示した。
[0098] [表 2]
 実施例 3
実施例 3
ェチル 1 {4 [(1ーシクロブチルピぺリジンー4ーィノレ)ォキシ]フェニル}ー1^1 ピラゾールー 4 カルボキシラート(化合物番号 12)の製造
(l)tert ブチル 4 {4 [4 (エトキシカルボニル) 1H—ピラゾールー 1ーィ
エノヤン cヘリンン 1一力ノレボキシラートの製造
[0100] 実施例 1一 (1)で得られた tert ブチル 4一 (4ーョードフエノキシ)ピぺリジン一 1一 カルボキシラート(4· Og)、ェチル 1H—ピラゾールー 4 カルボキシラート(1. 5g) 、 rac— trans— N, N, 一ジメチルシクロへキサン一 1 , 2 ジァミン(0. 79g)、ヨウ化 銅(I) (0. 23g)及び炭酸セシウム(6· 8g)の N, N ジメチルホルムアミド(6· 5mL) 懸濁液を 110°Cにて 3. 5時間攪拌した。反応混合物を室温まで放冷し、クロ口ホルム を加えて希釈し、セライト濾過した。濾液を水で洗浄し、無水硫酸ナトリウムで乾燥し、 減圧下濃縮した。得られた残渣をジイソプロピルエーテルで洗浄し、薄青色固体の 表題化合物 (2. 6g)を得た。
(2)ェチル 1 [4 (ピペリジンー4 ィルォキシ)フエニル] 1H—ピラゾールー 4 カルボキシラートの製造
[0101] 実施例 3—(1)で得られた tert ブチル 4 {4 [4 (エトキシカルボニル) 1H —ピラゾールー 1—ィル]フエノキシ }ピペリジン一 1—カルボキシラート(2· 6g)のクロ 口ホルム(10mU溶液に、氷冷下 4M塩酸酢酸ェチル溶液(10mUを加え、室温に て 2時間攪拌した。反応混合物にクロ口ホルムを加えて希釈し、飽和炭酸水素ナトリウ ム水溶液を加え、クロ口ホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減 圧下濃縮した。得られた残渣をジイソプロピルエーテルで洗浄し、薄青色固体の表 題化合物(1. 6g)を得た。
(3)ェチル 1一 { 4一 [ ( 1 シクロブチルピペリジン 4 - エー
H ピラゾールー 4 カルボキシラート(化合物番号 12)の製造
[0102] [化 20]

[0103] 実施例 3—(2)で得られたェチル 1 [4- -4- ェニノレ]
— 1H—ピラゾールー 4—カルボキシラート(1 · 6g) 42g)、及び
酢酸(0· 45g)のクロ口ホルム(15mU溶液にナト
g)を加え、室温にて一晩攪拌した。反応混合物を減圧下濃縮し、飽和炭酸水素ナト リゥム水溶液を加えてクロ口ホルムで抽出した。有機層を無水硫酸ナトリゥムで乾燥し 、減圧下濃縮した。得られた残渣をジイソプロピルエーテルで洗浄し、薄茶色固体の 表題化合物(1. 4g)を得た。
:H NMR (600 MHz, CHLOROFORM— d) δ ppm 1.36 (t, J=7.1 Hz, 3 H), 1.63 - 1.9 5 (m, 2 H), 1.78 - 1.93 (m, 4 H), 1.94 - 2.07 (m, 4 H), 2.16 (br.s, 2 H), 2.60 (br.s, 2 H), 2.69 - 2.78 (m, 1 H), 4.27 - 4.37 (m, 1 H), 4.32 (q, J=7.2 Hz, 2 H), 6.97 (d, J =9.2 Hz, 2 H), 7.56 (d, J=9.2 Hz, 2 H), 8.05 (s, 1 H), 8.28 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 370(M+H)+
実施例 4
[0104] ェチル 1 {4 [ (1ーシクロぺンチルピぺリジンー4ーィノレ)ォキシ]フェニル}ー1
H ピラゾールー 4 カルボキシラート(化合物番号 13)の製造
[0105] [化 21]

[0106] 実施例 3— (3)に示した方法と同様の方法により、シクロブタノンの代わりにシクロべ ンタノンを用いて表題化合物を得た。
:H NMR (600 MHz, CHLOROFORM— d) δ ppm 1.36 (t, J=7.1 Hz, 3 H), 1.35 - 1.4 5 (m, 2 H), 1.49 - 1.73 (m, 4 H), 1.80 - 1.90 (m, 4 H), 1.98 - 2.05 (m, 2 H), 2.33 ( br.s, 2 H), 2.47 - 2.56 (m, 1 H), 2.81 (br.s, 2 H), 4.28 - 4.38 (m, 1 H), 4.32 (q, J=7 .3 Hz, 2 H), 6.98 (d, J=9.2 Hz, 2 H), 7.57 (d, J=9.2 Hz, 2 H), 8.06 (s, 1 H), 8.29 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 384(M+H)+
実施例 5
[0107] 1ーシクロブチルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1ーピラゾール — 1 ィル]フエノキシ }ピペリジン (化合物番号 14)の製造
(1) 1 - {4- [ (1 -シクロブチルピペリジン 4 ィル)ォキシ]フエニル } 1 H ビラ ゾールー 4一力ルボン酸の製造
[0108] 実施例 3— (3)で得られたェチル 1 {4 [ (1ーシクロブチルピペリジン 4 ィル) ォキシ]フエ二ノレ 1 H ピラゾーノレ 4 カルボキシラート( 1. 4g)及び水酸化リチ ゥム(0. 27g)のエタノール(7. OmU及び水(7. OmU混合溶液を 90°Cにて 1時間 攪拌した。反応混合物を室温まで放冷し、減圧下エタノールを留去した。得られた残 渣に 1M塩酸水溶液を加えて中和し、水を加え、氷冷下 30分間攪拌した。析出した 結晶を濾取し、無色固体の表題化合物(1. 2g)を得た。
(2) 1ーシクロブチルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1 ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン(化合物番号 14)の製造
[0109] [化 22]

実施例 5 ( 1 )で得られた 1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ] フエ二ル} 1H—ピラゾールー 4一力ルボン酸(0· 40g)、 1 { 3 (ジメチルァミノ) プロピル }— 3 ェチルカルポジイミド塩酸塩(0· 25g)、 1—ヒドロキシベンゾトリァゾ 一ノレ水禾ロ物(0. 20g)及びピロリジン(0. 092g)を N, N ジメチノレホノレム ミド(4. 0 mUに加えて懸濁液とし、室温にて一晩攪拌した。反応混合物に炭酸水素ナトリウム 水溶液を加え、室温にて 10分間攪拌した。析出した結晶を濾過し、乾燥した。結晶 をエタノールで洗浄し、薄茶色固体の表題化合物(0. 28g)を得た。
:H NMR (600 MHz, CHLOROFORM— d) δ ppm 1.61 - 2.08 (m, 14 H), 2.10 - 2.24 (m, 2 H), 2.55 - 2.68 (m, 2 H), 2.70 - 2.78 (m, 1 H), 3.65 (t, J=6.9 Hz, 2 H), 3.73 ( t, J=6.6 Hz, 2 H), 4.29 - 4.38 (m, 3 H), 6.97 (d, J=9.2 Hz, 2 H), 7.57 (d, J=8.7 Hz, 2 H), 7.94 (s, 1 H), 8.26 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 395(M+H)
実施例 6
[0111] 1一 [ (1 {4一 [ (1ーシクロペンチルビペリジンー4 ィル)ォキシ]フエ二ル} - 1H ーピラゾールー 4 ィル)カルボニル]ピロリジン 3—オール(化合物番号 15)の製 造
[0112] [化 23]

実施例 5に示した方法と同様の方法により、ェチル 1 {4 [ (1ーシクロブチルピ ペリジン 4 ィノレ)ォキシ]フェニル } 1 H ピラゾーノレ 4 カルボキシラートの代 わりに実施例 4で得られたェチル 1 {4一 [ (1ーシクロペンチルビペリジン 4ーィ ノレ)ォキシ]フエ二ル} 1H—ピラゾールー 4 カルボキシラートを、ピロリジンの代わ りに 3
 て表題化合物を得た。
て表題化合物を得た。
1H NMR (600 MHz, CHLOROFORM- d) δ ppm 1.38 - 1.45 (m, 2 H), 1.49 - 1.64 ( m, 2 H), 1.65 - 1.74 (m, 2 H), 1.81 - 2.15 (m, 9 H), 2.34 (br. s, 2 H), 2.48 - 2.56 ( m, 1 H), 2.80 (br. s, 2 H), 3.70 - 4.00 (m, 4 H), 4.31 - 4.37 (m, 1 H), 4.55 - 4.64 ( m, 1 H), 6.98 (d, J=8.7 Hz, 2 H), 7.56 (d, J=8.7 Hz, 2 H), 7.90 - 7.99 (m, 1 H), 8.2 3 - 8.29 (m, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 425(M+H)+
実施例 5又は実施例 6に示した方法と同様の方法で得た化合物(化合物番号 16)〜 (化合物番号 48)を表 3— 1〜表 3— 7に示した。
[0113] [表 3-1]



S〕I

S〕 D¾53I 1

S〕 DI

S〕 D¾a73I 1-
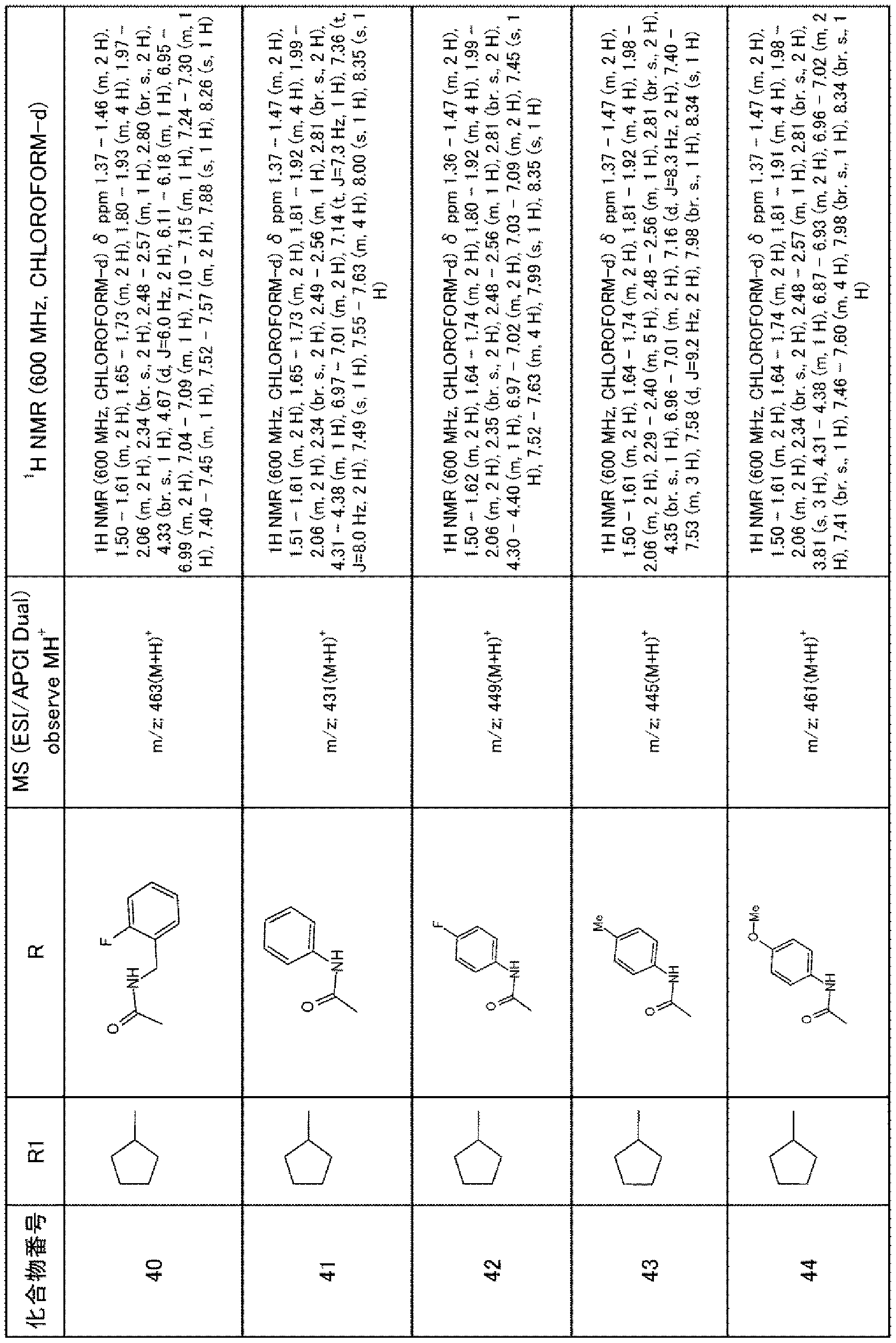
S〕 D¾83I 1
/vu O sos-oz-oozfcldAV 9
^ 〔sn ε6
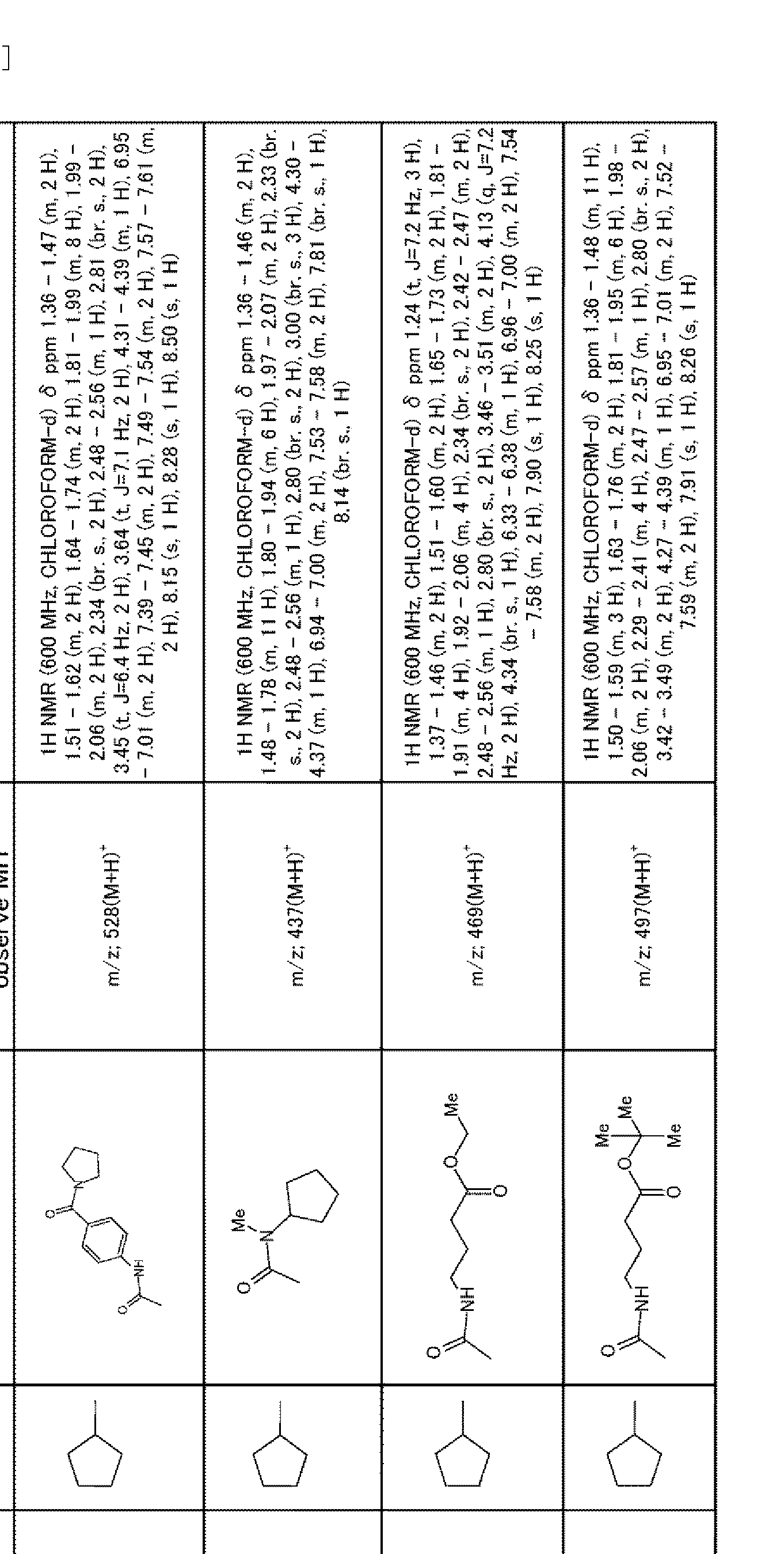
実施例 7
[0120] 4-{[(1-{4-[(1-シクロペンチルビペリジン 4—ィル)ォキシ]フエ二ノレ }— 1 H
ピラゾールー 4 ィル)カルボニル]アミノ }ブタン酸 塩酸塩 (化合物番号 49)の製 造
[0121] [化 24]

[0122] tert-ブチル 4 {[(1 {4 [(1 シクロペンチルビペリジン 4 ィノレ)ォキシ] フェニル } 1 H ピラゾールー 4 ィル)カルボニル]アミノ }ブタノエート(化合物番 号 48) (0. 26g)のクロ口ホルム溶液に、氷冷下、 4M塩酸酢酸ェチル溶液(1. 33m Uを加え、室温にて終夜攪拌した。反応混合物を減圧下濃縮して表題化合物(0. 8 8g)を得た。
1H NMR (600 MHz, DMSO-d ) δ ppm 1.44 - 1.60 (m, 2 H), 1.63 - 1.81 (m, 6 H),
1.85 - 2.07 (m, 4 H), 2.09 - 2.29 (m, 4 H), 2.93 - 3.15 (m, 2 H), 3.17 - 3.26 (m, 2 H), 3.29 - 3.57 (m, 4 H), 4.54 - 4.82 (m, 1 H), 7.05 - 7.19 (m, 2 H), 7.66 - 7.79 (m , 2 H), 8.06 (s, 1 H), 8.19 (t, J=5.5 Hz, 1 H), 8.79 (s, 1 H), 10.51 (br. s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 441(M+H)+
実施例 8
[0123] 1-tert-ブチル 4— { 4— [4— (ピロリジン 1—ィノレカノレポ二ノレ) 1 H ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン(化合物番号 50)の製造
(1) 1 -tert-ブチル 4一(4ーョードフエノキシ)ピペリジンの製造
[0124] 実施例 1一(1)に示した方法と同様の方法により、 tert ブチル 4ーヒドロキシピペリ ジン 1 カルボキシラートの代わりに 1—tert ブチルー 4ーヒドロキシピペリジン (J ournal of Organic Chemistry, 2005年, 70巻, 1930— 1933頁に記載の方 法に従って合成できる)を用いて表題化合物を得た。
(2) 1— 61ーブチルー4 {4 [4ー(ピロリジン 1ーィルカルボニル) 1H ビラ
ゾーノレ ィル]フエ 物番号 50)の製造
[0125] [化 25]
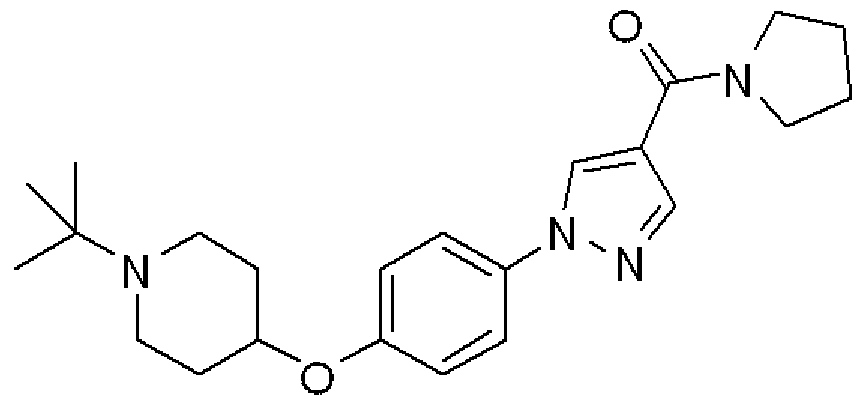
実施例 1一(2)に示した方法と同様の方法により、 tert ブチル 4一(4 ョードフエ ノキシ)ピぺリジン 1 カルボキシラートの代わりに実施例 8 (1)で得られた 1 te rtーブチルー 4一(4ーョードフエノキシ)ピペリジンを用レ、て表題化合物を得た。 1H NMR (600 MHz, DMSO-d ) δ ppm 1.11 (s, 1 H), 1.73 - 2.12 (m, 8 H), 2.46 (br
• s, 2 H), 2.88 (br.s, 2 H), 3.65 (t, J=6.9 Hz, 2 H), 3.73 (t, J=6.7 Hz, 2 H), 4.32 (br. s, 1 H), 6.97 (d, J=9.7 Hz, 2 H), 7.56 (d, J=9.2 Hz, 2 H), 7.94 (s, 1 H), 8.26 (s, 1 H )
MS (ESI/APCI Dual) (Positive) m/z; 397(M+H)+
実施例 9
[0127] 1 { 4 [ ( 1—シクロプロピルピぺリジン 4—ィノレ)ォキシ]フエ二ノレ N— (4—フ ノレオロフェニル) - 1H-ピラゾーノレ 4 カルボキサミド(化合物番号 51 )の製造
(1) 1ーシクロプロピル 4一(4ーョードフエノキシ)ピぺリジンの製造
[0128] 実施例 1一(1)に示した方法と同様の方法により、 tert ブチル 4ーヒドロキシピペリ ジン 1 カルボキシラートの代わりに 1ーシクロプロピルー4ーヒドロキシピペリジン( WO2005117865に記載の方法に従って合成できる)を用いて表題化合物を得た。
(2) 1 - {4- [ (1 -シクロプロピルピペリジン 4 ィノレ)ォキシ]フエ二ノレ N—(4 —フルオロフェニル) - 1H-ピラゾーノレ 4—カルボキサミド(化合物番号 51 )の製 造
[0129] [化 26]

[0130] 実施例 1一(3)に示した方法と同様の方法により、 tert ブチル 4一(4 ョードフエ ノキシ)ピぺリジン 1 カルボキシラートの代わりに実施例 9一(1)で得られた 1ーシ クロプロピノレ一 4— (4—ョードフエノキシ)ピぺリジンを、 4— (ピロリジン一 1—ィルカ ルポニル) 1H—ピラゾールの代わりに 1H—ピラゾールー 4一力ルボン酸 (4ーフ ルオロフェニル)アミドをそれぞれ用いて表題化合物を得た。
1H NMR (600 MHz, CHLOROFORM- d) δ ppm 0.40 - 0.49 (m, 4 H), 1.61 - 1.66 ( m, 1 H), 1.77 - 1.84 (m, 2 H), 1.95 - 2.02 (m, 2 H), 2.47 - 2.54 (m, 2 H), 2.87 - 2. 95 (m, 2 H), 4.34 - 4.40 (m, 1 H), 7.01 (d, J=8.7 Hz, 2 H), 7.07 (t, J=8.7 Hz, 2 H), 7.44 (s, 1 H), 7.55 - 7.62 (m, 4 H), 7.99 (s, 1 H), 8.36 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 421(M+H)+
実施例 10
[0131] N- (4 フルオロフェニル) 1— (4— { [1— (1ーメチルェチノレ)ピぺリジンー4ーィ ノレ]ォキシ }フエニル) - 1H-ピラゾーノレ 4 カルボキサミド(化合物番号 52)の製 造
[0132]

[0133] 実施例 1一(3)に示した方法と同様の方法により、 tert ブチル 4一(4 ョードフエ
ノキシ)ピぺリジン 1 カルボキシラートの代わりに 1 イソプロピル 4一(4ーョー ドフエノキシ)ピぺリジン (実施例 1一(1)に示した方法と同様の方法に従って合成で きる)を、 4— (ピロリジン一 1—ィルカルボニル) 1H—ピラゾールの代わりに 1H— ピラゾーノレ 4一力ルボン酸 (4 フルオロフェニル)アミドをそれぞれ用レ、て表題化 合物を得た。
IH NMR (600 MHz, CHLOROFORM— d) δ ppm 1.07 (d, J=6.4 Hz, 6 H), 1.80 - 1.8 8 (m, 2 H), 2.00 - 2.07 (m, 2 H), 2.37 - 2.46 (m, 2 H), 2.72 - 2.83 (m, 3 H), 4.31 - 4.37 (m, 1 H), 7.01 (d, J=8.7 Hz, 2 H), 7.07 (t, J=8.7 Hz, 2 H), 7.44 (s, 1 H), 7.52 - 7.65 (m, 4 H), 7.99 (s, 1 H), 8.36 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 423(M+H)+
実施例 11
[0134] 1— {4— [ (1—シクロペンチルピロリジン一 3 ィル)ォキシ]フエ二ル}— 4— (ピロリ ジン 1ーィルカルボニル) 1H—ピラゾール(化合物番号 53)の製造
[0135] [化 28]
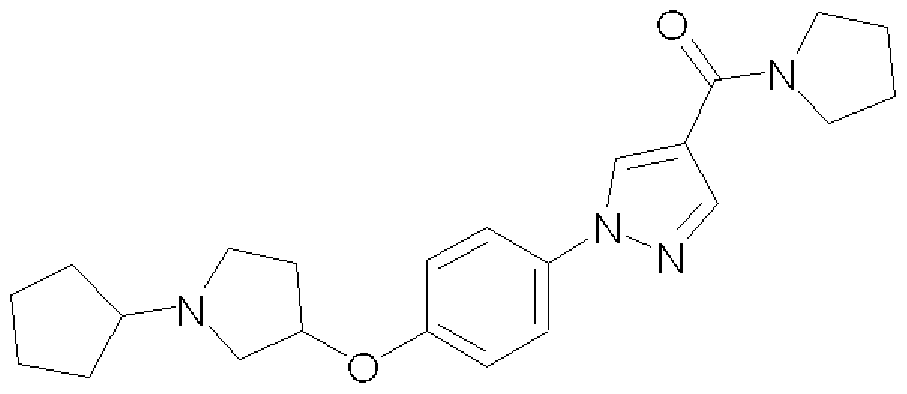
[0136] 実施例 1に示した方法と同様の方法により、 tert ブチル 4ーヒドロキシピペリジン
1 カルボキシラートの代わりに tert ブチル 4ーヒドロキシピロリジン 1 カル ボキシラートを用いて表題化合物を得た。
IH NMR (600 MHz, CHLOROFORM- d) δ ppm 1.39 - 1.61 (m, 4 H), 1.64 - 1.76 ( m, 2 H), 1.78 - 1.86 (m, 2 H), 1.89 - 2.06 (m, 5 H), 2.28 - 2.38 (m, 1 H), 2.43 - 2. 60 (m, 2 H), 2.74 - 2.87 (m, 2 H), 2.89 - 2.98 (m, 1 H), 3.65 (t, J=6.9 Hz, 2 H), 3.7 3 (t, J=6.9 Hz, 2 H), 4.80 - 4.86 (m, 1 H), 6.87 - 6.95 (m, 2 H), 7.52 - 7.60 (m, 2 H), 7.94 (s, 1 H), 8.25 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 395(M+H)
実施例 12
[0137] 1 {4 [ (1ーシクロブチルピロリジンー3 ィル)ォキシ]フエ二ル} 4ー(ピロリジン
1ーィルカルボニル) 1H—ピラゾール(化合物番号 54)の製造
[0138] [化 29]

[0139] 実施例 1に示した方法と同様の方法により、 tert ブチル 4ーヒドロキシピペリジン
1 カルボキシラートの代わりに tert ブチル 4ーヒドロキシピロリジン 1 カル ボキシラートを、シクロペンタノンの代わりにシクロブタノンをそれぞれ用いて表題化 合物を得た。
1H NMR (600 MHz, CHLOROFORM— d) δ ppm 1.65 - 1.79 (m, 2 H), 1.89 - 2.06 ( m, 9 H), 2.27 - 2.36 (m, 1 H), 2.42 - 2.50 (m, 1 H), 2.70 - 2.76 (m, 2 H), 2.81 - 2. 88 (m, 1 H), 2.92 - 2.99 (m, 1 H), 3.65 (t, J=6.9 Hz, 2 H), 3.73 (t, J=6.6 Hz, 2 H), 4.81 - 4.86 (m, 1 H), 6.89 - 6.94 (m, 2 H), 7.53 - 7.58 (m, 2 H), 7.94 (s, 1 H), 8.26
(s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 381(M+H)+
実施例 13
[0140] 1 -シクロペンチル 4— { 3 フルォロ 4— [4 (ピロリジン 1—ィルカルボ二ノレ)
1H—ピラゾールー 1 ィル]フエノキシ }ピペリジン(化合物番号 55)の製造 [0141] [化 30]

[0142] 実施例 1に示した方法と同様の方法により、 4 ョードフエノールの代わりに 3—フル オロー 4 ョードフエノールを用いて表題化合物を得た。
MS (ESI/APCI Dual) (Positive) m/z; 427(M+H)+
実施例 14
[0143] 1—シクロブチルー 4 { 3—フルオロー 4 [4 (ピロリジン 1ーィルカルボニル)
1H—ピラゾールー 1 ィル]フエノキシ }ピペリジン(化合物番号 56)の製造 [0144] [化 31]

[0145] 実施例 1に示した方法と同様の方法により、 4 ョードフエノールの代わりに 3—フル オロー 4 ョードフエノールを、シクロペンタノンの代わりに、:
用いて表題化合物を得た。
MS (ESI/APCI Dual) (Positive) m/z; 413(M+H)+
実施例 15
[0146] 1 -シクロペンチル 4— { 2 フルォロ 4— [4 (ピロリジン 1—ィルカルボ二ノレ)
1H ピラゾールー 1 ィル]フエノキシ }ピペリジン(化合物番号 57)の製造 [0147] [化 32]

[0148] 実施例 1に示した方法と同様の方法により、 4 ョードフエノールの代わりに 2 フル オロー 4 ョードフエノールを用いて表題化合物を得た。
IH NMR (600 MHz, CHLOROFORM- d) δ ppm 1.37 - 1.46 (m, 2 H), 1.49 - 1.62 ( m, 2 H), 1.64 - 1.75 (m, 2 H), 1.82 - 2.09 (m, 10 H), 2.34 (br. s, 2 H), 2.48 - 2.56 (m, 1 H), 2.81 (br. s, 2 H), 3.65 (t, J=6.9 Hz, 2 H), 3.73 (t, J=6.6 Hz, 2 H), 4.33 (br • s, 1 H), 7.06 (t, J=8.7 Hz, 1 H), 7.33 - 7.38 (m, 1 H), 7.48 (dd, J=11.7, 2.5 Hz, 1 H), 7.95 (s, 1 H), 8.27 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 427(M+H)+
実施例 16
[0149] 1ーシクロブチルー 4 { 2 フルオロー 4— [4 (ピロリジン 1ーィルカルボニル)
1H—ピラゾールー 1 ィル]フエノキシ }ピペリジン(化合物番号 58)の製造 [0150] [化 33]

[0151] 実施例 1に示した方法と同様の方法により、 4 ョードフエノールの代わりに 2 フル オロー 4 ョードフエノールを、シクロペンタノンの代わり

用いて表題化合物を得た。
IH NMR (600 MHz, CHLOROFORM— d) δ ppm 1.62
m, 12 H), 2.16 (br. s, 2 H), 2.62 (br. s, 2 H), 2.70 - 2.78 (m, 1 H), 3.65 (t, J=6.9 H z, 2 H), 3.73 (t, J=6.9 Hz, 2 H), 4.33 (br. s, 1 H), 7.06 (t, J=8.7 Hz, 1 H), 7.33 - 7. 36 (m, 1 H), 7.48 (dd, J=11.9, 2.8 Hz, 1 H), 7.95 (s, 1 H), 8.27 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 413(M+H)+
実施例 17
[0152] 1—シクロペンチノレ一 4— { 3 メチノレ一 4— [4— (ピロリジン一 1—ィルカルボニル)
1H—ピラゾールー 1 ィル]フエノキシ }ピペリジン(化合物番号 59)の製造 [0153] [化 34]

[0154] 実施例 1に示した方法と同様の方法により、 4 ョードフエノールの代わりに 4 ョード
3—メチルフエノールを用いて表題化合物を得た。
1H NMR (600 MHz, CHLOROFORM- d) δ ppm 1.35 - 1.47 (m, 2 H), 1.49 - 1.60 ( m, 2 H), 1.62 - 1.74 (m, 2 H), 1.79 - 1.97 (m, 6 H), 1.97 - 2.06 (m, 4 H), 2.15 (s, 3
H), 2.35 (br. s, 2 H), 2.47 - 2.57 (m, 1 H), 2.79 (br. s, 2 H), 3.65 (t, J=6.9 Hz, 2 H ), 3.73 (t, J=6.6 Hz, 2 H), 4.34 (br. s, 1 H), 6.75 - 6.85 (m, 2 H), 7.20 (d, J=8.3 Hz,
1 H), 7.97 (s, 2 H)
MS (ESI/APCI Dual) (Positive) m/z; 423(M+H)+
実施例 18
[0155] 1ーシクロブチルー 4 { 3—メチルー 4— [4 (ピロリジン 1ーィルカルボニル) 1
H ピラゾールー 1 ィル]フエノキシ }ピペリジン(化合物番号 60)の製造
[0156] [化 35]

[0157] 実施例 17に示した方法と同様の方法により、 4—ョードフエノールの代わりに 4—ョー ドー 3—メチルフエノールを、シクロペンタノンの代わりにシクロブタノンをそれぞれ用 いて表題化合物を得た。
1H NMR (600 MHz, CHLOROFORM- d) δ ppm 1.62 - 1.76 (m, 2 H), 1.78 - 2.09 ( m, 12 H), 2.11 - 2.24 (m, 5 H), 2.60 (br. s, 2 H), 2.69 - 2.78 (m, 1 H), 3.65 (t, J=7. 1 Hz, 2 H), 3.73 (t, J=6.9 Hz, 2 H), 4.35 (br. s, 1 H), 6.76 - 6.80 (m, 1 H), 6.81 - 6 .84 (m, 1 H), 7.16 - 7.23 (m, 1 H), 7.97 (s, 2 H)
MS (ESI/APCI Dual) (Positive) m/z; 409(M+H)+
実施例 19
[0158] 1 - {4 - [ (1 -シクロブチルピペリジン 4—ィル)ォキシ]フエニル }— 1 H ピラゾ 一ルー 4 カルボキサミド(化合物番号 61)の製造
[0159] [化 36]

実施例 5 ( 1 )で得られた 1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ] フエ二ル}— 1H—ピラゾールー 4—カルボン酸(1. 00g)、 1—ヒドロキシベンゾトリア ゾールー水和物(0· 535g)及び 1ーェチルー 3—(3 ジメチルァミノプロピル)カル ポジイミド塩酸塩(0. 673g)の N, N ジメチルホルムアミド溶液に、室温でアンモニ
ァ水(25%、 0. 5ml)を加え、室温でー晚撹拌した。反応混合物に飽和重曹水をカロ え、一時間撹拌し、結晶を濾取した。得られた結晶を水及びジイソプロピルエーテル で洗浄し、乾燥して無色粉末の表題化合物(0. 988g)を得た。
実施例 20
[0161] 1 - {4 [ (1—シクロペンチルビペリジン一 4—ィノレ)ォキシ]フエ二ル} - 1H—ピラゾ 一ルー 4 カルボ二トリル(化合物番号 62)の製造
[0162] [化 37]

[0163] 実施例 19で得られた 1一 { 4 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ]フエ 二ル}— 1H ピラゾールー 4 カルボキサミド(0. 154g)の N, N ジメチルホルム アミド溶液に、氷冷下塩化チォニル (0. 134g)を加え、氷冷下 30分撹拌し、更に室 温で 4時間撹拌した。反応混合物に氷冷下塩化チォニル (0. 825g)を加え、室温で 2時間撹拌した。反応混合物を氷冷し、水、飽和重曹水を加え、 pH8に調節して酢 酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、硫酸ナトリウムで乾燥して減圧 下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒: n へキ サン:酢酸ェチル = 1:1)にて精製して無色固体の表題化合物(0. 106g)を得た。 1H NMR (600 MHz, CHLOROFORM- d) δ ppm 1.63 - 1.75 (m, 2 H), 1.79 - 1.92 ( m, 4 H), 1.96 - 2.07 (m, 4 H), 2.16 (br. s, 2 H), 2.62 (br. s, 2 H), 2.70 - 2.77 (m, J =7.9, 7.9, 7.8, 7.6 Hz, 1 H), 4.35 (br. s, 1 H), 6.96 - 7.01 (m, 2 H), 7.51 - 7.55 (m, 2 H), 7.95 (s, 1 H), 8.18 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 323(M+H)+
実施例 21
[0164] 1—シクロブチル一 4— [4— (4—ニトロ一 1H—ピラゾール一 1—ィル)フエノキシ]ピ ペリジン (化合物番号 63)の製造
[0165] [化 38]

[0166] 1ーシクロブチルー 4一(4ーョードフエノキシ)ピぺリジン(実施例 1一(1)に示した方 法と同様の方法により、 tert ブチル 4ーヒドロキシピペリジン 1 カルボキシラー トの代わりに 1—シクロブチル— 4—ヒドロキシピペリジンを用いて合成できる) (5. 68 g)、 4 ニトロピラゾール(1 · 98g)、 (rac-trans-N, Ν'—ジメチルシクロへキサン - 1 , 2 ジァミン(0. 905g)、ヨウ化銅(0. 303g)及び炭酸セシウム(10· 9g)の N, N ジメチルホルムアミド(10ml)混合物を 130°Cにて 1. 5時間攪拌した。反応混合 物を室温に戻し、水を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し 、硫酸ナトリウムで乾燥して減圧下濃縮した。得られた残渣をシリカゲルカラムクロマト グラフィー(展開溶媒: n へキサン:酢酸ェチル = 2 : 1)にて精製し、得られた結晶を ジイソプロピルエーテルで洗浄して無色固体の表題化合物(2. 34g)を得た。
1H NMR (600 MHz, CHLOROFORM- d) δ ppm 1.63 - 1.75 (m, 2 H), 1.79 - 1.93 ( m, 4 H), 1.97 - 2.08 (m, 4 H), 2.17 (br. s, 2 H), 2.62 (br. s, 2 H), 2.70 - 2.78 (m, 1 H), 4.36 (br. s, 1 H), 6.97 - 7.03 (m, 2 H), 7.54 - 7.60 (m, 2 H), 8.22 (s, 1 H), 8.50 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 343(M+H)+
実施例 22
[0167] 4 クロ口一 N— (1— {4— [ (1—シクロブチルピペリジン一 4 ィノレ)ォキシ]フェニル }— 1Η—ピラゾール— 4 ィル)ブタンアミド(化合物番号 64)の製造
(1) 1 - {4- [ (1 -シクロペンチルビペリジン 4—ィル)ォキシ]フエ二ノレ }— 1 H ピ
ラゾール一 4 -ァミンの製造
[0168] 実施例 21で得られた 1 シクロブチルー4 [4ー(4ーニトロー1^1ーピラゾール 1 ィル)フエノキシ]ピぺリジン(1. 00g)のメタノール(20ml)溶液にパラジウム/炭素 (5%、 0. 10g)を加え、水素雰囲気下、室温にて終夜撹拌した。セライト濾過し、濾 液を減圧下濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒: 酢酸ェチル)にて精製し、淡橙色固体の表題化合物(0. 883g)を得た。
(2) 4 クロローN—(1 {4 [ (1ーシクロブチルピペリジン 4 ィノレ)ォキシ]フェ 二ル} 1H—ピラゾールー 4 ィル)ブタンアミド(化合物番号 64)の製造
[0169] [化 39]

実施例 22—(1)で得られた 1 {4 [ (1ーシクロブチルピペリジンー4 ィル)ォキ シ]フエ二ル}— 1H—ピラゾール一 4—ァミン(0· 300g)のクロロホノレム(3. 0ml)溶 液に室温でピリジン(0. 152g)及び 4 クロ口酪酸クロリド(0. 149g)を加え、室温で 30分撹拌した。反応混合物に飽和塩化アンモニゥム水溶液を加え、クロ口ホルムで 抽出し、水及び飽和食塩水で洗浄し、硫酸ナトリウムで乾燥して減圧下濃縮した。得 られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒: n へキサン:酢酸ェチ ル = 1 : 1)にて精製し、淡黄色アモルファスの表題化合物(0· 366g)を得た。
1H NMR (600 MHz, CHLOROFORM- d) δ ppm 1.63 - 1.76 (m, 2 H), 1.78 - 1.95 ( m, 4 H), 1.95 - 2.09 (m, 4 H), 2.09 - 2.24 (m, 4 H), 2.53 - 2.69 (m, 4 H), 2.69 - 2. 80 (m, 1 H), 3.61 - 3.68 (m, 2 H), 4.32 (br. s, 1 H), 6.92 - 7.00 (m, 2 H), 7.28 (s, 1 H), 7.51 - 7.58 (m, 3 H), 8.37 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 417(M+H)+
実施例 23
[0171] 1一(1 {4一 [ (1ーシクロペンチルビペリジンー4 ィル)ォキシ]フエ二ル} - 1H- ピラゾールー 4 ィル)ピロリジンー2 オン(化合物番号 65)の製造
[0172] [化 40]

[0173] 実施例 22— (2)で得られた 4 クロロー N—(1 {4一 [ (1ーシクロブチルピペリジン — 4 ィル)ォキシ]フエ二ル}— 1H—ピラゾール一 4 ィル)ブタンアミド(0· 366g) のテトラヒドロフラン(3. Oml)溶液に、室温で水素化ナトリウム(0. 176g)を加え、油 浴(油温 90°C)にて 30分間加熱還流した。反応混合物を氷冷し、飽和重曹水を加え 、クロ口ホルムで抽出し、水及び飽和食塩水で洗浄し、硫酸ナトリウムで乾燥して減圧 下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:クロロホ ルム:メタノール:アンモニア水 = 10 : 1 : 0. 1)にて精製し、得られた結晶を酢酸ェチ ルで洗浄して淡黄色固体の表題化合物(0. 082g)を得た。
1H NMR (600 MHz, CHLOROFORM- d) δ ppm 1.63 - 1.75 (m, 2 H), 1.78 - 1.93 ( m, 4 H), 1.96 - 2.08 (m, 4 H), 2.11 - 2.27 (m, 4 H), 2.54 - 2.66 (m, 4 H), 2.69 - 2. 77 (m, 1 H), 3.80 (t, J=7.1 Hz, 2 H), 4.33 (br. s, 1 H), 6.93 - 6.98 (m, 2 H), 7.54 - 7.59 (m, 2 H), 7.64 (s, 1 H), 8.43 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 381(M+H)+
実施例 24
[0174] 2 クロ口ェチル { 1 [4— (1ーシクロブチルピペリジンー4 ィルォキシ)フエニル
]一 1H—ピラゾールー 4ーィル }一力ルバミン酸(化合物番号 66)の製造
[0175] [化 41]

[0176] 実施例 22に示した方法と同様の方法により、 4 クロ口酪酸クロリドの代わりにクロ口 ギ酸 2—クロ口ェチルを用レ、て表題化合物を得た。
IH NMR (200 MHz, CHLOROFORM-d) δ ppm 1.52 - 2.23 (m, 12 H), 2.55 - 2.83 (m, 3 H), 3.75 (t, J=6.0 Hz, 2 H), 4.27 - 4.41 (m, 1 H), 4.43 (t, J=6.0 Hz, 2 H), 6.6 2 - 6.80 (m, 1 H), 6.85 - 7.00 (m, 2 H), 7.48 - 7.60 (m, 3 H), 8.09 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 419(M+H)+
実施例 25
[0177] 3- (1 - {4- [ (1 -シクロペンチルビペリジン 4—ィル)ォキシ]フエ二ノレ }— 1 H— ピラゾールー 4 ィル) 1 , 3 ォキサゾリジン 2 オン(化合物番号 67 )の製造 [0178] [化 42]

実施例 23に示した方法と同様の方法により、 4 クロロー N—(1 {4 [ (1ーシクロ ブチルビペリジン 4 ィノレ)ォキシ]フエ二ノレ 1 H ピラゾーノレ 4 ィノレ)ブタン アミドの代わりに 2 クロ口ェチル { 1 [4一(1ーシクロブチルピペリジンー4ーィル ォキシ)フエニル] 1H—ピラゾールー 4ーィル }一力ルバミン酸用いて表題化合物 を得た。
IH NMR (600 MHz, CHLOROFORM-d) δ ppm 1.45 - 2.22 (m, 12 H), 2.50 - 2.82
(m, 3 H), 3.99 (t, J=8.5 Hz, 2 H), 4.22 - 4.41 (m, 1 H), 4.55 (t, J=8.5 Hz, 2 H), 6.9 2 - 7.03 (m, 2 H), 7.56 - 7.64 (m, 3 H), 8.20 (s, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 383(M+H)+
実施例 26
[0180] { 1 [4一(1ーシクロブチルピペリジンー4 ィルォキシ)フエニル] 1H—ピラゾー ルー 3—ィル }ピロリジン 1ーィルーメタノン(化合物番号 68)の製造
[0181] [化 43]

[0182] 実施例 21に示した方法と同様の方法により、 4 ニトロピラゾールの代わりに 3— (ピ 口リジン 1ーィルカルボニル) 1H—ピラゾールを用いて表題化合物を得た。 :H NMR (600 MHz, CHLOROFORM- d) δ ppm 1.63 - 1.76 (m, 4 H), 1.79 - 2.25 ( m, 16 H), 2.56 - 2.78 (m, 3 H), 3.69 (t, J=6.9 Hz, 2 H), 4.04 (t, J=6.6 Hz, 2 H), 4.3 4 (br. s., 1 H), 6.96 - 7.00 (m, 3 H), 7.58 (d, J=9.2 Hz, 2 H), 7.81 (d, J=2.8 Hz, 1 H)
MS (ESI/APCI Dual) (Positive) m/z; 395(M+H)+
[0183] 試験例 1 : H3受容体結合試験
[0184] ヒト型 H3受容体発現 CHO— K1細胞の膜標品(ユーロスクリーン社、 ES— 392— M、タンノ ク質 15
 〃1)、 R (―)— α—メチル [3Η]ヒスタミン(アマシャム社 、 TRK— 1017、比活性 1. 74TBq/mmol、 2nM)、及び試験薬物を、室温で 1時 間反応させた。反応終了後に、反応混合物を、 0. 3%ポリエチレンィミンで処理した ガラスフィルター(GF/C)を通して吸引濾過し、ガラスフィルターを、 0· 1 %BSAと 5 mM 塩化マグネシウムを含んだ 50mM Tris— HC1洗浄液(pH7. 4)で 5回洗浄し た。洗浄後に、ガラスフィルターを乾燥し、シンチレーターを加え、フィルター上の放 射活性を液体シンチレ一ションカウンターで測定した。
[0185] ΙΟ ^ Μ R ( )一 α メチルヒスタミンの存在下で反応を実施したときの R (—)一 a メチル [3H]ヒスタミン結合量を非特異的結合度とし、全 R ( )一 a メチル [3H ]ヒスタミン結合度と非特異的結合度との差を、特異的 R (―) - a—メチル [3H]ヒスタ ミン結合度とした。一定濃度(2nM)の R ( )一 α メチル [3Η]ヒスタミンを上述の条 件下で様々な濃度の各試験薬物を反応させることにより、阻害曲線を得た。阻害曲 線から R (—) - a—メチル [ ]ヒスタミンの結合が 50%阻害される試験薬物濃度(I C
〃1)、 R (―)— α—メチル [3Η]ヒスタミン(アマシャム社 、 TRK— 1017、比活性 1. 74TBq/mmol、 2nM)、及び試験薬物を、室温で 1時 間反応させた。反応終了後に、反応混合物を、 0. 3%ポリエチレンィミンで処理した ガラスフィルター(GF/C)を通して吸引濾過し、ガラスフィルターを、 0· 1 %BSAと 5 mM 塩化マグネシウムを含んだ 50mM Tris— HC1洗浄液(pH7. 4)で 5回洗浄し た。洗浄後に、ガラスフィルターを乾燥し、シンチレーターを加え、フィルター上の放 射活性を液体シンチレ一ションカウンターで測定した。
[0185] ΙΟ ^ Μ R ( )一 α メチルヒスタミンの存在下で反応を実施したときの R (—)一 a メチル [3H]ヒスタミン結合量を非特異的結合度とし、全 R ( )一 a メチル [3H ]ヒスタミン結合度と非特異的結合度との差を、特異的 R (―) - a—メチル [3H]ヒスタ ミン結合度とした。一定濃度(2nM)の R ( )一 α メチル [3Η]ヒスタミンを上述の条 件下で様々な濃度の各試験薬物を反応させることにより、阻害曲線を得た。阻害曲 線から R (—) - a—メチル [ ]ヒスタミンの結合が 50%阻害される試験薬物濃度(I C
50 )を求めた。その結果を表 4に示した。
[0186] [表 4]
H3受容体
化合物番号 結合試験
IC50 (nM)
1 10.4
5 8.1
14 7,1
16 5,7
18 14.7
19 6.2
68 7,1
[0187] 試験例 2: [35S]GTP— γ S結合試験
ヒト型 H3受容体膜標品(タンパク質 7. 5 H g/100 1)、 30 M GDP、 100 M R (-) α メチルヒスタミン、及び試験化合物を、室温で 30分間反応させた。反 応終了後、さらに [3¾]GTP— γ— S (0. 2nM)を添加し、引き続き 30分間反応を続 けた。反応終了後に、反応混合物をガラスフィルター(GF/C)を通して吸引濾過し、 ガラスフィルターを lOOmM 塩化ナトリウム、 ImM 塩化マグネシウムを含んだ 20m M HEPES洗浄液(pH7. 4)で 3回洗浄した。洗浄後に、ガラスフィルターを乾燥し 、シンチレーターを加え、フィルター上の放射活性を液体シンチレーシヨンカウンター で測定した。
[0188] R (―)― α メチルヒスタミン非存在下で反応を実施したときの [3 ]GTP— γ— S 結合量を非特異的結合とし、 R ( )一 α メチルヒスタミン存在下得られた全結合と
の差を特異的 [ S] GTP— γ— S結合度とした。一定濃度の [d5S] GTP— γ— S (0 . 2nM)と R (—)一 α—メチルヒスタミン( 100 Μ)を上述の条件下で様々な濃度の 各試験薬物を反応させることにより、阻害曲線を得た。阻害曲線から、 [35S] GTP— γ— S結合が 50%阻害される試験薬物濃度(IC )を求めた。 その結果を表 5に示
50
した。
[表 5]
[35S]GTP- r -S
化合物番号
1 2,6
14 1.6
16 1.1
68 1.2 試験例 3 : Social Recognition試験
試験には SD系雄性ラットを用いた。成熟ラットを Perspex 製試験箱に入れ、 30分 間馴化させた。その後、幼若ラットを同じ試験箱に入れ、 5分間に成熟ラットが幼若ラ ットに対して示す探索行動時間を測定した(1回目探索行動時間)。探索行動として s niffing, groomingおよび closely followingを測定した。 5分間の探索の後、成熟 ラットおよび幼若ラットを試験箱から取り出し、それぞれホームケージに戻した。 90分 後、成熟ラットを 1回目探索行動と同じ試験箱に入れ、 30分間馴化させた。その後、 1回目と同じ幼若ラットを試験箱に入れ、 5分間に成熟ラットが幼若ラットに対して示 す探索行動時間を測定した(2回目探索行動時間)。 1回目探索行動時間に対する 2 回目探索行動時間の割合(2回目探索時間 /1回目探索行動時間)を社会性認知 の指標とした。本発明の化合物を 1回目探索行動直後に、成熟ラットに経口投与した 。本発明の化合物は 0. 03N塩酸溶液に溶解し、 0. 1 , 0. 3および lmg/kgにおい て検討した。その結果、化合物番号 1は、 0. 3および lmg/kgにおいて、溶媒群と 比較して 2回目探索時間 /1回目探索行動時間を有意に低下させ、社会認知増強 作用を示した。
産業上の利用可能性
本発明により、ヒスタミン H3受容体への強力な結合阻害作用を有し、ヒスタミン H3 受容体に起因する障害、例えば、認知症、アルツハイマー病、注意欠陥 ·多動性症、 統合失調症、てんかん、中枢性痙攣、摂食障害、肥満、糖尿病、高脂血症、睡眠障 害、ナルコレプシ一、睡眠時無呼吸症候群、概日リズム障害、うつ病、又はアレルギ 一性鼻炎等の疾患の予防並びに治療に有用である新規なピラゾール誘導体を提供 することが可能となった。
Claims
[化 1]

{式 (1)中、
A及び Bは、異なって炭素原子、又は窒素原子を示し、
R1は、 C〜Cアルキル(該 C〜Cアルキルは、 C〜C環状アルキル又はヒドロキシ
1 6 1 6 3 8
ルで置換されても良い); c

アルキル又はヒドロキシルで置換されても良い)又は c〜cアルケニルを示し、
3 8
nは、 0〜2の整数を示し、
Tは、水素原子;ハロゲン又は C〜Cアルキルを示し、
1 6
Rは、下記構造式 (I)〜 (VI)で表される基を示し、
[化 2]

R2及び R3は、同一又は異なって、水素原子; C〜Cアルキル(該 C〜Cアルキル
1 6 1 6 は、ハロゲン; c〜c環状アルキル;ヒドロキシル; c〜cアルコキシカルボニル又は
3 8 2 7
カルボキシで置換されても良い); c〜c環状アルキル(該 c〜c環状アルキルは、
3 8 3 8
ハロゲン; C〜Cアルキル又はヒドロキシルで置換されても良い)又は一(CH ) -
1 6 2 m
Arで示される基を示すか、
又は、 R2及び R3は、隣接する窒素原子と一緒になつて互いに結合し、環中に前記窒
素原子の他に 1つ以上の窒素、酸素、又は硫黄原子を含んでもよい 4〜7員の飽和 複素環(該飽和複素環は、ハロゲン; C〜Cアルキル; C〜Cアルコキシ又はヒドロ
1 6 1 6
キシルで置換されても良い)を形成し、
Arは、ァリール(該ァリールは、ハロゲン; C〜Cアルキノレ; C〜Cアルコキシ;ヒドロ
1 6 1 6
キシル; C〜Cアルコキシカルボニル;シァノ; C〜Cアルキルアミノカルボニル; C
2 7 2 7 3
〜C ジアルキルアミノカルボニル;力ルバモイル又は環中に少なくとも 1つの窒素原
13
子を有し、他に 1つ以上の窒素、酸素、若しくは硫黄原子を含んでもよい 3〜7員の飽 和複素環に窒素原子を介して置換されたカルボニルで置換されても良い)又はへテ ロアリール(該ヘテロァリールは、ハロゲン; C〜Cアルキル; C〜Cアルコキシ;ヒド
1 6 1 6
ロキシル; c〜cアルコキシカルボニル;シァノ; c〜cアルキルアミノカルボニル;
2 7 2 7
C〜C ジアルキルアミノカルボニル;力ルバモイル又は環中に少なくとも 1つの窒素
3 13
原子を有し、他に 1つ以上の窒素、酸素、若しくは硫黄原子を含んでもよい 3〜7員の 飽和複素環に窒素原子を介して置換されたカルボニルで置換されても良い)を示し、 mは、 0〜2の整数を示し、
Gは、 CO 又は SO を示し、
2
R4は、水素原子又は C〜Cアルキルを示し、
1 6
R5は、 C〜Cアルキル(該 C〜Cアルキルは、ハロゲン; C〜C環状アルキル; C
1 6 1 6 3 8 1
〜cアルコキシ又はヒドロキシルで置換されても良い); C〜C環状アルキル(該 C
6 3 8 3
〜c環状アルキルは、ハロゲン; c〜cアルキル; c〜cアルコキシ又はヒドロキシ
8 1 6 1 6
ルで置換されても良い);ァリール(該ァリールは、ハロゲン;アルキル; c〜cアルコ
1 6 キシ;ヒドロキシル又はシァノで置換されても良い)又はへテロアリール(該ヘテロァリ ールは、ハロゲン;アルキル; C〜Cアルコキシ;ヒドロキシル又はシァノで置換され
1 6
ても良い)を示し、
また、 R4及び R5は、隣接する窒素原子及びカルボニル炭素と一緒になつて互いに結 合し、環中に前記窒素原子の他に 1つ以上の窒素、酸素、又は硫黄原子を含んでも よい 5〜7員の飽和複素環(該飽和複素環は、ハロゲン; C〜Cアルキル; C〜Cァ
1 6 1 6 ルコキシ;ヒドロキシル又はォキソで置換されても良い)を形成しても良く、
R6は、 C〜Cアルキル; C〜C環状アルキル; C〜Cアルコキシ;ァリール(該ァリ
ールは、ハロゲン;アルキル; c〜cアルコキシ;ヒドロキシル又はシァノで置換され
1 6
ても良い)又はへテロアリール(該ヘテロァリールは、ハロゲン; C
1〜Cアルキル; C
6 1
〜Cアルコキシ;ヒドロキシル又はシァノで置換されても良!/、)を示し、
6
R7は、 C ァミノ; C
1〜Cアルキル; C
6 1〜Cアルコキシ;ァミノ; C キル
6 1〜Cアル
6 2〜C
12 ジアルキルアミノ; 4〜7員の飽和複素環(該飽和複素環は、ハロゲン;アルキル; C 〜Cアルコキシ;ヒドロキシル又はシァノで置換されても良い);ァリール(該ァリール
6
は、ハロゲン;アルキル; c
1〜cアルコキシ;ヒドロキシル又はシァノで置換されても良 6
い)又はへテロアリール(該ヘテロァリールは、ハロゲン; c
1〜cアルキル; c
6 1〜cァ
6 ルコキシ;ヒドロキシル又はシァノで置換されても良!/、)を示す。 }
で表されるピラゾール誘導体、又はその医薬上許容される塩。
[2] 式(1)で示され、式中、 Rは構造式 (I)で表される、請求項 1に記載のピラゾール誘導 体、又はその医薬上許容される塩。
[3] 1ーシクロペンチルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン、
1ーェチルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾールー 1 ィル]フエノキシ }ピペリジン、
1一(1ーメチルェチル)ー4 {4 [4一(ピロリジン一 1ーィルカルボニル)一 1H— ピラゾーノレ一 1—ィル]フエノキシ }ピペリジン、
1ーシクロへキシルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン、
1 2 メチルシクロペンチル) 4 {4 [4 (ピロリジン 1ーィルカルボニル) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1—(シクロプロピルメチル)ー4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H —ピラゾーノレ一 1—ィル]フエノキシ }ピペリジン、
1 (3—メチルー 2—ブテンー1ーィル)ー4 {4 [4 (ピロリジンー1ーィルカル ボニル) 1H—ピラゾールー 1 ィル]フエノキシ }ピペリジン、
1 (シクロブチルメチル)ー4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H— ピラゾーノレ一 1—ィル]フエノキシ }ピペリジン、
1— (シクロペンチルメチル)ー4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H —ピラゾーノレ一 1—ィル]フエノキシ }ピペリジン、
2- (4— {4— [4— (ピロリジン一 1—ィルカルボ二ル)一 1H—ピラゾール一 1—ィノレ] フエノキシ }ピペリジン 1 ィル)エタノール、
3— (4 - {4- [4- (ピロリジン 1ーィルカルボニル) 1H—ピラゾールー 1 ィル] フエノキシ }ピペリジン 1 ィル)シクロペンタノール、
ェチル 1 {4 [ (1 シクロブチルピペリジン 4 ィノレ)ォキシ]フエ二ノレ 1 H ーピラゾールー 4一力ルボン酸、
ェチル 1 {4 [ (1 シクロペンチルビペリジン 4 ィノレ)ォキシ]フエ二ノレ 1 H ピラゾールー 4一力ルボン酸、
1ーシクロブチルー 4 {4 [4 (ピロリジン 1ーィルカルボニル) 1H—ピラゾー ルー 1 ィル]フエノキシ }ピペリジン、
1 - [ (1 - {4一 [ (1ーシクロペンチルビペリジンー4 ィル)ォキシ]フエ二ル} - 1H ーピラゾールー 4 ィル)カルボ二ノレ]ピロリジン オール、
1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ]フエニル } N シクロぺ ンチルー 1H—ピラゾールー 4 カルボキサミド、
1 {4一 [ (1ーシクロブチルピペリジンー4 ィル)ォキシ]フエ二ル} N メチノレー 1H—ピラゾールー 4 カルボキサミド、
1 {4 [ (1ーシクロブチルピぺリジンー4ーィル)ォキシ]フェニル}ーN—(2, 2, 2 トリフルォロェチル) 1H—ピラゾールー 4 カルボキサミド、
1 {4一 [ (1ーシクロブチルピペリジンー4 ィル)ォキシ]フエ二ル} -N- (シクロへ キシルメチル) 1H—ピラゾールー 4 カルボキサミド、
1ーシクロブチルー 4 {4 [4 (ピペリジン 1ーィルカルボニル) 1H—ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン、
1ーシクロブチルー 4一(4— {4一 [ (2 メチルピロリジン一 1一ィル)カルボニル]一 1 H ピラゾール一 1—イノレ}フエノキシ)ピぺリジン、
1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ]フエニル } N, N ジェ チルー 1 H ピラゾールー 4 カルボキサミド、
N— tert一ブチル一 1一 { 4一 [ ( 1一シクロブチルピペリジン一 4一ィル)ォキシ]フエ ニル } 1 H ピラゾールー 4 カルボキサミド、
1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ]フエニル } N, N ジメチ ルー 1 H ピラゾールー 4 カルボキサミド、
4- [ (1 - {4- [ (1 -シクロブチルピペリジン 4—ィル)ォキシ]フエニル }— 1 H— ピラゾールー 4 ィル)カルボニル]モルホリン、
1 - [ (1 - {4- [ (1 -シクロブチルピペリジン 4 ィル)ォキシ]フエニル } 1 H— ピラゾールー 4ーィノレ)カルボニル]ー4ーメチルピペラジン、
1一 { 4一 [ ( 1 シクロブチルピペリジン 4 ィル)ォキシ]フエニル } N シクロへ キシルー N メチルー 1H—ピラゾールー 4 カルボキサミド、
1 {4一 [ (1ーシクロブチルピペリジンー4 ィル)ォキシ]フエ二ル} N メチノレー N- (2 メチルプロピル) 1H—ピラゾールー 4 カルボキサミド、
1ーシクロブチルー 4一(4一 {4一 [ (3, 3—ジフルォロピロリジン一 1一ィル)カルボ二 ル]― 1H—ピラゾール一 1—イノレ}フエノキシ)ピぺリジン、
1一 { 4一 [ ( 1一シクロブチルピペリジン一 4一ィル)ォキシ]フエ二ノレ }一 N—(4ーフノレ オロフェニル) 1H—ピラゾールー 4 カルボキサミド、
1一 { 4一 [ ( 1一シクロブチルピペリジン一 4一ィル)ォキシ]フエ二ノレ }一 N—(4ーフノレ ォロベンジル) 1H—ピラゾールー 4 カルボキサミド、
4— {4— [4— (ァゼチジン一 1—ィルカルボニル) 1H—ピラゾーノレ一 1—ィル]フエ ノキシ } 1ーシクロペンチルビペリジン、
4- [ (1 - {4- [ (1 -シクロペンチルビペリジン 4—ィル)ォキシ]フエ二ノレ }— 1 H ピラゾールー 4ーィノレ)カルボニル]モルホリン、
1ーシクロペンチルー 4一 (4— {4 [ (3, 3—ジフルォロピロリジン一 1一ィル)カルボ ニル]― 1H—ピラゾール一 1—イノレ}フエノキシ)ピぺリジン、
N シクロペンチル一 1一 { 4一 [ ( 1一シクロペンチルビペリジン一 4一ィノレ)ォキシ]フ ェニル } 1 H ピラゾーノレ 4 カルボキサミド、
N シクロへキシル一 1一 { 4一 [ ( 1一シクロペンチルビペリジン一 4一ィノレ)ォキシ]フ ェニル } 1 H ピラゾーノレ 4 カルボキサミド、
N -ベンジル 1 { 4— [ ( 1 シクロペンチルビペリジン 4 ィル)ォキシ]フエ二 ル} 1H—ピラゾールー 4 カルボキサミド、
1一 { 4一 [ ( 1 シクロペンチルビペリジン 4 ィル)ォキシ]フエニル } N—(4ーフ ルォロベンジル) 1 H ピラゾーノレ 4 カルボキサミド、
1— { 4— [ ( 1 シクロペンチルビペリジン一 4 -ィル)ォキシ]フエニル } N— (3—フ ルォロベンジル) 1 H ピラゾーノレ 4 カルボキサミド、
1一 { 4一 [ ( 1一シクロペンチルビペリジン一 4一ィル)ォキシ]フエニル }一 N—( 2—フ ルォロベンジル) 1 H ピラゾーノレ 4 カルボキサミド、
1— 4— [ ( 1 シクロペンチルビペリジン 4 ィル)ォキシ]フエニル } N フエ二 ルー 1H—ピラゾールー 4 カルボキサミド、
1 { 4 ( 1—シクロペンチルビペリジン 4—ィル)ォキシ]フエ二ノレ N— (4—フ ルオロフェニル) 1H ピラゾールー 4 カルボキサミド、
1 {4 [ (1ーシクロペンチルビペリジン 4 ィル)ォキシ]フエ二ル} N—(4ーメ チルフエニル) 1 H ピラゾールー 4 カルボキサミド、
1 { 4 ( 1—シクロペンチルビペリジン 4—ィル)ォキシ]フエ二ノレ N— (4—メ トキシフエニル) 1H ピラゾールー 4 カルボキサミド、
1 { 4 [ ( 1—シクロペンチルビペリジン一 4—ィル)ォキシ]フエ二ノレ } N— [4— ( ピロリジン 1ーィルカルボニル)フエニル] 1H—ピラゾールー 4 カルボキサミド、
N シクロペンチル一 1一 { 4一 [ ( 1一シクロペンチルビペリジン一 4一ィル)ォキシ]フ ェニル } N メチル 1 H ピラゾーノレ 4一力ルポキサミド、
ェチル 4 { [ (1 {4 [ (1ーシクロペンチルビペリジン 4 ィル)ォキシ]フエ二 ル} 1H—ピラゾールー 4 ィル)カルボニル]アミノ}ブタン酸、
tert ブチル 4 { [ (1 {4 [ (1ーシクロペンチルビペリジン 4 ィル)ォキシ] フエニル] - 1H-ピラゾールー 4 -ィル]カルボニル }ァミノ]ブタン酸、
4- { [ (1 - {4- [ (1 -シクロペンチルビペリジン 4 ィル)ォキシ]フエ二ノレ 1 H ピラゾールー 4 ィル)カルボ二ノレ]ァミノ]ブタン酸、
1 -tert ブチル 4 { 4 [4 (ピロリジン 1 ィルカルボニル) 1 H ピラゾ 一ルー 1 ィル]フエノキシ }ピペリジン、
1一 { 4一 [ ( 1ーシクロプロピルピぺリジン一 4一ィノレ)ォキシ]フエ二ノレ }一 N—(4ーフ ルオロフェニル) 1H—ピラゾールー 4 カルボキサミド、
N- (4 フルオロフェニル)ー1 (4 { [1 (1ーメチルェチノレ)ピぺリジンー4ーィ ノレ]ォキシ }フエニル) 1 H ピラゾールー 4 カルボキサミド、
1 { 4 [ ( 1—シクロペンチルピロリジン 3—ィル)ォキシ]フエ二ノレ }— 4— (ピロリ ジン 1ーィルカルボニル) 1H—ピラゾール、
1— {4— [ (1—シクロブチルピロリジン— 3—ィル)ォキシ]フエ二ル}— 4— (ピロリジン 1ーィルカルボニル) 1H—ピラゾール、
1—シクロペンチノレ一 4— { 3—フルオロー 4— [4— (ピロリジン一 1—ィルカルボ二ノレ) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1ーシクロブチルー 4 { 3—フルオロー 4 [4 (ピロリジン 1ーィルカルボニル) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1—シクロペンチノレ一 4— { 2 フルオロー 4— [4— (ピロリジン一 1—ィルカルボ二ノレ) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1ーシクロブチルー 4 { 2 フルオロー 4 [4 (ピロリジン 1ーィルカルボニル) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1—シクロペンチノレ一 4— { 3—メチノレ一 4— [4— (ピロリジン一 1—ィルカルボニル) - 1H—ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1ーシクロブチルー 4 { 3—メチルー 4 [4 (ピロリジン 1ーィルカルボニル) 1 H ピラゾール一 1—ィル]フエノキシ }ピペリジン、
1 {4一 [ (1ーシクロブチルピペリジンー4 ィル)ォキシ]フエ二ル} 1H—ピラゾ 一ルー 4 カルボキサミド、
1一 { 4一 [ ( 1 シクロペンチノレピぺリジン 4 ィノレ)ォキシ]フエ二ノレ } 1 H ピラゾ 一ルー 4 カルボ二トリル、
1—シクロブチル一 4— [4— (4—ニトロ一 1H—ピラゾールー 1—ィル)フエノキシ]ピ ペリジン、
4—クロ口一 N— (1— {4— [ (1—シクロブチルピペリジン一 4—ィノレ)ォキシ]フェニル }— 1 H ピラゾール一 4—ィル)ブタンアミド、
1一(1 {4一 [ (1ーシクロペンチルビペリジンー4 ィル)ォキシ]フエ二ル} - 1H- ピラゾール一 4 ィル)ピロリジン 2 オン、
2 クロロェチノレ { 1 [4ー(1 シクロブチルピペリジン 4 ィルォキシ)フエ二ノレ ]一 1H—ピラゾールー 4ーィル }一力ルバミン酸、
3- (1 - {4- [ (1 -シクロペンチルビペリジン 4—ィル)ォキシ]フエ二ノレ }— 1 H— ピラゾール一 4 ィル) 1 , 3 ォキサゾリジン一 2 オン、
{ 1 [4一(1ーシクロブチルピペリジンー4 ィルォキシ)フエニル] 1H—ピラゾー ル一 3—ィル }ピロリジン一 1—ィル一メタノン
力もなる群より選ばれる、請求項 1に記載のピラゾール誘導体、又はその医薬上許容 される塩。
[4] 請求項 1〜3までのいずれ力、 1項記載のピラゾール誘導体、又はその医薬上許容さ れる塩を有効成分として含有する医薬。
[5] ヒスタミン H3受容体拮抗物質である請求項 4の医薬。
[6] 請求項 1〜3までのいずれ力、 1項記載のピラゾール誘導体、又はその医薬上許容さ れる塩を有効成分として含有することを特徴とする、認知症、アルツハイマー病、注 意欠陥 ·多動性症、統合失調症、てんかん、中枢性痙攣、摂食障害、肥満、糖尿病、 高脂血症、睡眠障害、ナルコレプシ一、睡眠時無呼吸症候群、概日リズム障害、うつ 病、又はアレルギー性鼻炎の予防剤又は治療剤。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/519,083 US20100113776A1 (en) | 2006-12-14 | 2007-12-14 | Pyrazole derivative |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006-336506 | 2006-12-14 | ||
| JP2006336506 | 2006-12-14 | ||
| JP2007-178839 | 2007-07-06 | ||
| JP2007178839 | 2007-07-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008072724A1 true WO2008072724A1 (ja) | 2008-06-19 |
Family
ID=39511737
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/074092 WO2008072724A1 (ja) | 2006-12-14 | 2007-12-14 | ピラゾール誘導体 |
Country Status (2)
| Country | Link |
|---|---|
| US (1) | US20100113776A1 (ja) |
| WO (1) | WO2008072724A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7888354B2 (en) | 2007-11-13 | 2011-02-15 | Taisho Pharmaceutical Co., Ltd | Phenylpyrazole derivatives |
| WO2013085018A1 (ja) | 2011-12-08 | 2013-06-13 | 大正製薬株式会社 | フェニルピロール誘導体 |
| WO2013100054A1 (ja) | 2011-12-27 | 2013-07-04 | 大正製薬株式会社 | フェニルトリアゾール誘導体 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014089495A1 (en) | 2012-12-07 | 2014-06-12 | Chemocentryx, Inc. | Diazole lactams |
| WO2015084936A1 (en) * | 2013-12-04 | 2015-06-11 | The Scripps Research Institute | Novel compounds as jnk kinase inhibitors |
| JP7539231B2 (ja) | 2016-04-07 | 2024-08-23 | ケモセントリクス,インコーポレーテッド | Pd-1阻害剤又はpd-l1阻害剤と組み合わせてccr1アンタゴニストを投与することによる腫瘍負荷の低減 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004511438A (ja) * | 2000-08-08 | 2004-04-15 | オーソ−マクニール・フアーマシユーチカル・インコーポレーテツド | イミダゾールを含有しないアリールオキシピペリジン |
| WO2006023462A1 (en) * | 2004-08-23 | 2006-03-02 | Eli Lilly And Company | Histamine h3 receptor agents, preparation and therapeutic uses |
| WO2007094962A2 (en) * | 2006-02-09 | 2007-08-23 | Athersys, Inc. | Pyrazoles for the treatment of obesity and other cns disorders |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2732967B1 (fr) * | 1995-04-11 | 1997-07-04 | Sanofi Sa | 1-phenylpyrazole-3-carboxamides substitues, actifs sur la neurotensine, leur preparation, les compositions pharmaceutiques en contenant |
| US7595316B2 (en) * | 2003-06-27 | 2009-09-29 | Banyu Pharmaceutical Co., Ltd. | Heteroaryloxy nitrogenous saturated heterocyclic derivative |
| US7888354B2 (en) * | 2007-11-13 | 2011-02-15 | Taisho Pharmaceutical Co., Ltd | Phenylpyrazole derivatives |
-
2007
- 2007-12-14 US US12/519,083 patent/US20100113776A1/en not_active Abandoned
- 2007-12-14 WO PCT/JP2007/074092 patent/WO2008072724A1/ja active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004511438A (ja) * | 2000-08-08 | 2004-04-15 | オーソ−マクニール・フアーマシユーチカル・インコーポレーテツド | イミダゾールを含有しないアリールオキシピペリジン |
| WO2006023462A1 (en) * | 2004-08-23 | 2006-03-02 | Eli Lilly And Company | Histamine h3 receptor agents, preparation and therapeutic uses |
| WO2007094962A2 (en) * | 2006-02-09 | 2007-08-23 | Athersys, Inc. | Pyrazoles for the treatment of obesity and other cns disorders |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7888354B2 (en) | 2007-11-13 | 2011-02-15 | Taisho Pharmaceutical Co., Ltd | Phenylpyrazole derivatives |
| US8183387B2 (en) | 2007-11-13 | 2012-05-22 | Taisho Pharmaceutical Co., Ltd | Phenylpyrazole derivatives |
| US8193176B2 (en) | 2007-11-13 | 2012-06-05 | Taisho Pharmaceutical Co., Ltd | Phenylpyrazole derivatives |
| WO2013085018A1 (ja) | 2011-12-08 | 2013-06-13 | 大正製薬株式会社 | フェニルピロール誘導体 |
| JPWO2013085018A1 (ja) * | 2011-12-08 | 2015-04-27 | 大正製薬株式会社 | フェニルピロール誘導体 |
| US9284324B2 (en) | 2011-12-08 | 2016-03-15 | Taisho Pharmaceutical Co., Ltd | Phenylpyrrole derivative |
| AU2012349290B2 (en) * | 2011-12-08 | 2017-03-23 | Taisho Pharmaceutical Co., Ltd. | Phenylpyrrole derivative |
| WO2013100054A1 (ja) | 2011-12-27 | 2013-07-04 | 大正製薬株式会社 | フェニルトリアゾール誘導体 |
| JPWO2013100054A1 (ja) * | 2011-12-27 | 2015-05-11 | 大正製薬株式会社 | フェニルトリアゾール誘導体 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20100113776A1 (en) | 2010-05-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2005307744B2 (en) | 3-phenyl-pyrazole derivatives as modulators of the 5-HT2A serotonin receptor useful for the treatment of disorders related thereto | |
| JP5375639B2 (ja) | フェニルピラゾール誘導体 | |
| US7649002B2 (en) | (3,5-dimethylpiperidin-1yl)(4-phenylpyrrolidin-3-yl)methanone derivatives as MCR4 agonists | |
| TW200804274A (en) | Compounds comprising a lactam or a lactam derivative moiety, processes for making them, and their uses | |
| WO2008072724A1 (ja) | ピラゾール誘導体 | |
| JP2010505851A (ja) | ヒスタミン−3アンタゴニストとしてのn−置換−アザシクリルアミン | |
| JP6098892B2 (ja) | フェニルピロール誘導体 | |
| TW200901983A (en) | Azacyclylbenzamide derivatives as histamine-3 antagonists | |
| WO2008072703A1 (ja) | ビフェニルアミド誘導体 | |
| EP2346848B1 (en) | Quinazoline derivatives as nk3 receptor antagonists | |
| JP5740838B2 (ja) | フェニルピラゾール誘導体 | |
| KR101163294B1 (ko) | Nk3 수용체 길항제로서 프롤린아마이드-테트라졸 유도체 | |
| JP2015013853A (ja) | フェニルピロール誘導体を含有する医薬 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07859822 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12519083 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07859822 Country of ref document: EP Kind code of ref document: A1 |