WO2008009897A1 - Process for preparing pregabalin and its opposite enantiomer - Google Patents
Process for preparing pregabalin and its opposite enantiomer Download PDFInfo
- Publication number
- WO2008009897A1 WO2008009897A1 PCT/GB2007/002607 GB2007002607W WO2008009897A1 WO 2008009897 A1 WO2008009897 A1 WO 2008009897A1 GB 2007002607 W GB2007002607 W GB 2007002607W WO 2008009897 A1 WO2008009897 A1 WO 2008009897A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- pregabalin
- iii
- pharmaceutically
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)CC(C*=[N+][O-])CC(ON)=O Chemical compound CC(C)CC(C*=[N+][O-])CC(ON)=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/02—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C229/04—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C229/06—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one amino and one carboxyl group bound to the carbon skeleton
- C07C229/08—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one amino and one carboxyl group bound to the carbon skeleton the nitrogen atom of the amino group being further bound to hydrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C265/00—Derivatives of isocyanic acid
- C07C265/02—Derivatives of isocyanic acid having isocyanate groups bound to acyclic carbon atoms
- C07C265/04—Derivatives of isocyanic acid having isocyanate groups bound to acyclic carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/08—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides with the hydroxy or O-metal group of organic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/34—Esters of acyclic saturated polycarboxylic acids having an esterified carboxyl group bound to an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/32—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Definitions
- the invention relates to the preparation of pregabalin and its opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, and to the intermediates used for their preparation.
- Pregabalin is approved for the treatment of epilepsy, neuropathic pain and Generalised Anxiety Disorder (GAD).
- GAD Generalised Anxiety Disorder
- Pregabalin acts by binding to the ⁇ 2 ⁇ subunit of the voltage-dependent calcium channel in the central nervous system.
- EP828704 describes a synthetic route for the preparation of pregabalin.
- the synthetic route described in EP828704 includes a key step which is the classical resolution of racemic 3- (carbamoylmethy ⁇ -S-methylhexanoic acid by the use of a chiral amine in ethanol/chloroform to give (i?)-3-(carbamoylmethyl)-5-methylhexanoic acid in 35% yield (max. theoretical yield is 50%) This is followed by a Hofmann rearrangement and hydrolysis with a strong acid to give pregabalin.
- this route provides (S)-3-(aminomethyl)-5-methylhexanoic acid (pregabalin) in a high enantiomeric purity, it suffers from several drawbacks that can not be avoided:
- the present invention represents a better and more efficient method of preparing either enantiomer of 3-(aminomethyl)-5-methylhexanoic acid, but preferably the (S) enantiomer, avoiding the above mentioned problems, giving the final product in a good yield with good chemical purity and with excellent enantiomeric purity.
- EP828704 describes that 4-isobutyl-dihydro-3H-pyran-2,6-dione is opened but, however, the racemate is formed that requires classical resolution to obtain the desired enantiomerically enriched intermediate.
- 4-isobutyl-dihydro-3H-pyran-2,6-dione can be enantioselectively opened by enatioselective alcoholysis using a chiral amine base [preferred chiral amine bases are quinine, quinidine, hydroquinine, cinchonine, cinchonidine, epiquinidine, epicinchonidine, epicinchonine or epiquinine] and with the use of an allyl alcohol to give the desired enantiomerically enriched monoester.
- reaction could be further improved to create further enantiomeric excess of the desired enantiomer, and improve the total yield, by lowering the reaction temperature and/or by using 0.5-1.1 mol of the chiral amine base per mol [preferably at 0.9-1.0 mol per mol or ideally at about lmol per mol] of 4-isobutyl-dihydro-3H-pyran-2,6-dione.
- the chiral amine base after the reaction is completed, can also be easily separated and recycled (yield over 90%).
- the product of this reaction can be then isolated as a salt with a base [preferred bases are 1 -adamantylamine, dicyclohexylamine, tert-butylamine or (S)- or (i?)- ⁇ -phenylethylamine], or optionally it can be isolated after a base/acid work-up and then selectively precipitated as a salt with the base to further improve enantiomer purity.
- bases are 1 -adamantylamine, dicyclohexylamine, tert-butylamine or (S)- or (i?)- ⁇ -phenylethylamine
- the salt formed has high chemical and enantiomeric purity and there is no need for further recrystallization.
- A represents the allyl group
- B represents the chiral amine base
- A represents the allyl group
- R 4 represents phenyl or C 1 -C 5 branched or straight alkyl chain, in an inert solvent and in the presence of a base, or f) ii) by activation of the carboxy group of the compound of Formula (III) and subsequent reaction with alkali metal azides or trialkylazides to obtain acid azides, which are then converted to the corresponding rearranged isocyanates,
- A represents the allyl group
- A' is an allyl group that may be the same or different from A ;
- the final product of the reaction may be recrystallised from an inert solvent in order to improve the purity of the product or to change the solid state characteristics of the product, such as morphology or physical form.
- intermediates (VI) and (VII) are not isolated during the reaction.
- intermediate (IV) is isolated during the reaction.
- step b) and/ or step g) the allyl alcohol is one of Formula (E)
- each Ri, R 2 and R 3 can be identical or different and are selected from H, Ci-C 5 branched or straight alkyl chain, phenyl [optionally substituted by halogen, cyano, trifluoromethoxy, nitro, trifluoromethyl or by Ci-C 5 branched or straight alkyl chain], or R 2 may also represents a 5- to 7- membered aromatic heterocyclic group.
- Preferred 5- to 7-membered aromatic heterocyclic groups are selected from, pyridyl, thienyl, furyl, oxazolyl or imidazolyl.
- Preferred allyl alcohols of Formula (E) are cinnamyl alcohol, allyl alcohol, crotyl alcohol, 3- penten-2-ol, 2-methyl-2-propen-l-ol, l,5-hexadien-3-ol, 2-hexen-l-ol, 2-methyl-3-phenyl-2- propen-1-ol, 2-hexen-l-ol, 2-penten-l-ol, 4-nitrocinnamyl alcohol, 3-buten-2-ol, l-hexen-3-ol, 1- octen-3-ol or l-penten-3-ol.
- preferred chiral amine bases for enantioselective alcoholysis are cinchona alkaloids, especially: quinine, quinidine, hydroquinine, cinchonine, cinchonidine, epiquinidine, epicinchonidine, epicinchonine or epiquinine.
- the chiral amine is added in 0.5-1.1 mol per mol of Formula (I) [ideally at 0.9-1.0 mol per mol, ideally about 1 mol per mol] and/or is preferably present as an enantiomerically pure form.
- the process is performed in an inert solvent selected from toluene, xylene, tetrahydrofuran, dioxane, methylene chloride, acetonitrile, dimethylformamide, diisopropyl ether or methyl tert-butyl ether, or a mixture of any thereof.
- an inert solvent selected from toluene, xylene, tetrahydrofuran, dioxane, methylene chloride, acetonitrile, dimethylformamide, diisopropyl ether or methyl tert-butyl ether, or a mixture of any thereof.
- the reaction is conducted at a temperature from -70 to 3O 0 C.
- preferred bases D for precipitation of compound of Formula (III), are 1 - adamantylamine, dicyclohexylamine, tert-butylamine, (S)- or ( ⁇ )- ⁇ -phenylethylamine.
- the compounds of Formula (III) may first be reacted with a simple base, such as potassium carbonate, sodium carbonate or sodium-hydrogen carbonate, then by washing of the water layer containing the salt compound of Formula (III) with an organic solvent, liberating the compound of Formula (III) by dilute acid.
- the compound of formula (III) can then be reacted with a base D as described above
- step f) compounds of Formula (VII) are obtained from compounds of Formula (III) via a Curtius rearrangement, see for Curtius rearrangement ,for example, E.F. Scriven, K. Turnbull, Chem. Rev., 1988, 88, 297.
- Suitable azides of Formula (V) for the Curtius rearrangement are phosphoric acid ester-azide, such as phosphoric acid diphenyl ester-azide or phosphoric acid diethyl ester-azide.
- Curtius rearrangement is in general carried out in suitable inert solvents such as toluene, benzene, xylene, dioxane or tetrahydrofuran.
- Suitable bases for Curtius rearrangement are organic amines selected from N-ethylmorpholine, N-methylmorpholine, pyridine or triethylamine preferably in an amount of lmol to 3mol per mol of compounds of Formula (III). Also, it is possible first to convert carboxylic acid to corresponding derivatives by activating reagents, such as C 1 -C 4 alkyl chloroformates in presence of amine, thionyl chloride or phosphorus pentachloride and then prepare carboxylic acid azides by reaction with alkali metal azides or trialkylsilyl azide.
- reagents such as C 1 -C 4 alkyl chloroformates
- the acid azides can be transformed to corresponding isocyanates, which can be optionally isolated and reacted with an allyl alcohol, preferably of Formula (E), to give compounds of Formula (VII).
- an allyl alcohol preferably of Formula (E)
- the Curtius rearrangement reaction is conducted at a temperature of from 20 to HO 0 C.
- step h) the splitting off of urethane and ester function of the compounds of Formula (VII) is conducted with a catalytic amount of palladium catalyst and/or a phosphine and a nucleophilic auxiliary in an inert solvent.
- the inert solvent in this reaction is selected from ethanol, tetrahydrofuran, 2-propanol, n-propanol or dioxane, or a mixture of any thereof.
- the reaction is conducted at a temperature of from 10 to 9O 0 C.
- Suitable nucleophilic auxiliaries for the splitting off of urethane and ester function are selected from triethylamine, dimedone, tributyltin hydride, ammonium formate, morpholine or pyrrolidine, preferably in an amount of 1 mol to 4 mol per mol of compounds of Formula (VII).
- Suitable palladium catalysts are selected from, Pd(PPh 3 ) 4 , Pd(OAc) 2 , PdCl 2 , Pd(dba) 2 or (Pd 2 (dba) 3 ), preferably in an amount of 0.0005 mol to 0.2 mol per mol of compounds of Formula (VII).
- Suitable phosphines are selected from, triphenylphosphine, triisopropylphosphine or tri-o- tolylphosphine, preferably in an amount of 0.002 mol to 0.8 mol per mol of compounds of Formula (VII).
- enantiomerically enriched we mean that the enantiomeric excess (ee) is greater than 60%, greater than 65% or greater than 70%.
- enantiomerically pure we mean that the enantiomeric excess (ee) is greater than 90%, ideally greater than 99% and ideally enantiomerically pure means greater than 99.5%.
- Diacid 50 g, 266 mmol was placed in propionic anhydride (130 ml) and the reaction mixture was heated at 140-145 °C during 6 hours.
- Propionic acid and propionic anhydride were distilled in vacuo (30-60 °C/15 mm Hg).
- Product was collected at 100-105 °C/0.5 mm Hg as yellowish oil. Yield 41 g (91 %), GC purity: >98% (HP-I, 25 m, 70 kPa, gradient 70-240 0 C, 15 °C/min).
- reaction mixture was washed with a solution of 1 g NaNO 2 and 1,5. g NaHCO 3 in H 2 O (10OmL), than with H 2 O (10OmL) and evaporated under reduced pressure to yield 25.8g of crude oily (S)- Cinnamyl 3-((cinnamyloxycarbonylamino)methyl)-5-methylhexanoate which was used without further purification in the next step.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The invention relates to the preparation of pregabalin and its opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, and to the intermediates used for their preparation.
Description
PROCESS FOR PREPARING PREGABALIN AND ITS OPPOSITE ENANTIOMER
The invention relates to the preparation of pregabalin and its opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, and to the intermediates used for their preparation.
(S)-3-(aminomethyl)-5-methylhexanoic acid (pregabalin) is approved for the treatment of epilepsy, neuropathic pain and Generalised Anxiety Disorder (GAD). Pregabalin acts by binding to the α2δ subunit of the voltage-dependent calcium channel in the central nervous system.
Background of the invention
EP828704 describes a synthetic route for the preparation of pregabalin. The synthetic route described in EP828704 includes a key step which is the classical resolution of racemic 3- (carbamoylmethy^-S-methylhexanoic acid by the use of a chiral amine in ethanol/chloroform to give (i?)-3-(carbamoylmethyl)-5-methylhexanoic acid in 35% yield (max. theoretical yield is 50%) This is followed by a Hofmann rearrangement and hydrolysis with a strong acid to give pregabalin. Although this route provides (S)-3-(aminomethyl)-5-methylhexanoic acid (pregabalin) in a high enantiomeric purity, it suffers from several drawbacks that can not be avoided:
-to increase low overall yield and productivity, unwanted enantiomer should be recovered by hydrolysis to γ-isobutylglutaric acid,
-potentially toxic solvent (chloroform) must be used for crystallization of (R)-a- phenylethylamine salt of (/?)-3-(carbamoylmethyl)-5-methylhexanoic acid, -Hofmann rearrangement could be problematic on scale-up because it may cause a very exothermic reaction and be difficult to control,
-strong acid hydrolysis in the last step may cause low yield and formation of impurities that could be problematic to remove from the final product.
The present invention represents a better and more efficient method of preparing either enantiomer of 3-(aminomethyl)-5-methylhexanoic acid, but preferably the (S) enantiomer, avoiding the above mentioned problems, giving the final product in a good yield with good chemical purity and with excellent enantiomeric purity.
Summary of the invention
Compounds 3-isobutylglutaric acid and 4-isobutyl-dihydro-3H-pyran-2,6-dione (3-isobutylglutaric acid anhydride) are known from J. Chem. Soc, 1923, 123, 3131; preparation of 4-isobutyl- dihydro-3H-pyran-2,6-dione is also described in patents EP828704 and US5,629,447 using acetyl- chloride under reflux for 16 hours. However, acetyl -chloride is a corrosive chemical and can cause serious damage to the equipment. We have found that by using 3-isobutylglutaric acid and propionic acid anhydride, 4-isobutyl-dihydro-3H-pyran-2,6-dione can be obtained in high yield and in high chemical purity after only 6 hours of reaction.
EP828704 describes that 4-isobutyl-dihydro-3H-pyran-2,6-dione is opened but, however, the racemate is formed that requires classical resolution to obtain the desired enantiomerically enriched intermediate. Surprisingly, we have found that 4-isobutyl-dihydro-3H-pyran-2,6-dione can be enantioselectively opened by enatioselective alcoholysis using a chiral amine base [preferred chiral amine bases are quinine, quinidine, hydroquinine, cinchonine, cinchonidine, epiquinidine, epicinchonidine, epicinchonine or epiquinine] and with the use of an allyl alcohol to give the desired enantiomerically enriched monoester. After further investigations it was also surprisingly found that the reaction could be further improved to create further enantiomeric excess of the desired enantiomer, and improve the total yield, by lowering the reaction temperature and/or by using 0.5-1.1 mol of the chiral amine base per mol [preferably at 0.9-1.0 mol per mol or ideally at about lmol per mol] of 4-isobutyl-dihydro-3H-pyran-2,6-dione. The chiral amine base, after the reaction is completed, can also be easily separated and recycled (yield over 90%). The product of this reaction can be then isolated as a salt with a base [preferred bases are 1 -adamantylamine, dicyclohexylamine, tert-butylamine or (S)- or (i?)-α-phenylethylamine], or optionally it can be isolated after a base/acid work-up and then selectively precipitated as a salt with the base to further improve enantiomer purity. Usually, the salt formed has high chemical and enantiomeric purity and there is no need for further recrystallization.
We present as a feature of the invention a process for the preparation of pregabalin or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, the process comprising the steps of:
a) optionally forming of anhydride of Formula I

I by reacting 3-isobutylglutaric acid with propionic anhydride, preferably at elevated temperature, ideally at a temperature of from 110 to 160 0C;
b) enantioselective alcoholysis of the anhydride of Formula (I) by the use of a chiral amine base and an allyl alcohol, in an inert solvent, to form an enantiomerically enriched compound of Formula (II), in either of its enatiomeric forms, but where the (R) form is preferred,

wherein A represents the allyl group, and B represents the chiral amine base,
c) converting the compound of Formula (II) into a compound of Formula (III), in either of its enatiomeric forms, but where the (R) form is preferred, by the removal of the base B, by the addition of an acid;

d) reacting a compound of Formula (III) with a base D, in an inert solvent, to obtain an enantiomerically pure salt of a compound of Formula (IV) in either of its enatiomeric forms, but where the (R) form is preferred,
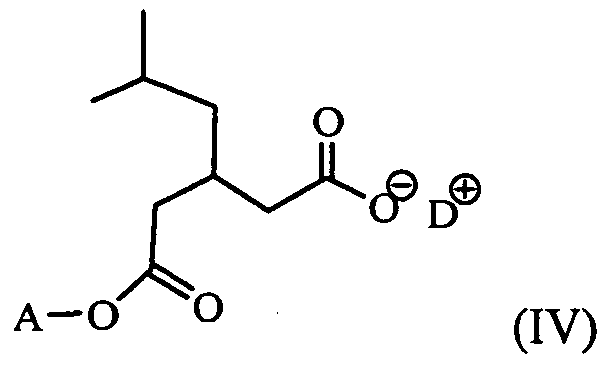 wherein A represents the allyl group and D is the base;
wherein A represents the allyl group and D is the base;
e) reacting the compound of Formula (IV) with a dilute acid to obtain an enantiomerically pure compound of Formula (III) in either of its enatiomeric forms, but where the (R) form is preferred;
f) i) reacting the compound of Formula (III), with an azide of Formula (V), in a Curtius rearrangement reaction,
(R4O)2-P(O)-N3 (V)
wherein R4 represents phenyl or C1-C5 branched or straight alkyl chain, in an inert solvent and in the presence of a base, or f) ii) by activation of the carboxy group of the compound of Formula (III) and subsequent reaction with alkali metal azides or trialkylazides to obtain acid azides, which are then converted to the corresponding rearranged isocyanates,
to form an enantiomerically pure compound of Formula (VI), in either of its enatiomeric forms, but where the (S) form is preferred
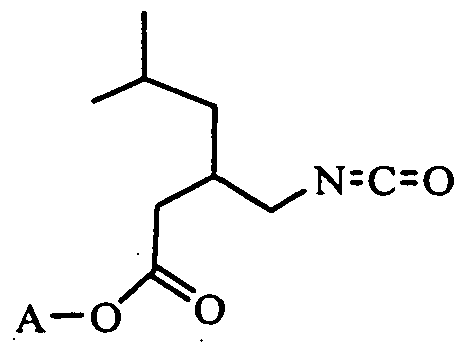
(VI)
wherein A represents the allyl group;
g) reacting a enantiomerically pure compound of Formula (VI), in an inert solvent, preferably at elevated temperature, with an allyl alcohol, to provide an enantiomerically pure compound of Formula (VII) in either of its enatiomeric forms, but where the (S) form is preferred

wherein A' is an allyl group that may be the same or different from A ;
h) splitting off of the urethane and ester functions of the compound of Formula (VII) to provide pregabalin, or its opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof.
Optionally the final product of the reaction may be recrystallised from an inert solvent in order to improve the purity of the product or to change the solid state characteristics of the product, such as morphology or physical form.
Preferably intermediates (VI) and (VII) are not isolated during the reaction. Preferably intermediate (IV) is isolated during the reaction.
Each process step a), b), c), d), e), f)i), f)ii), g) and h) are separately described herein as independent process steps for the purpose of supporting claims to such steps.
Preferably in step b) and/ or step g) the allyl alcohol is one of Formula (E)

wherein each Ri, R2 and R3 can be identical or different and are selected from H, Ci-C5 branched or straight alkyl chain, phenyl [optionally substituted by halogen, cyano, trifluoromethoxy, nitro, trifluoromethyl or by Ci-C5 branched or straight alkyl chain], or R2 may also represents a 5- to 7- membered aromatic heterocyclic group. Preferred 5- to 7-membered aromatic heterocyclic groups are selected from, pyridyl, thienyl, furyl, oxazolyl or imidazolyl.
Preferred allyl alcohols of Formula (E) are cinnamyl alcohol, allyl alcohol, crotyl alcohol, 3- penten-2-ol, 2-methyl-2-propen-l-ol, l,5-hexadien-3-ol, 2-hexen-l-ol, 2-methyl-3-phenyl-2- propen-1-ol, 2-hexen-l-ol, 2-penten-l-ol, 4-nitrocinnamyl alcohol, 3-buten-2-ol, l-hexen-3-ol, 1- octen-3-ol or l-penten-3-ol.
In step b) preferred chiral amine bases for enantioselective alcoholysis are cinchona alkaloids, especially: quinine, quinidine, hydroquinine, cinchonine, cinchonidine, epiquinidine, epicinchonidine, epicinchonine or epiquinine. Preferably the chiral amine is added in 0.5-1.1 mol per mol of Formula (I) [ideally at 0.9-1.0 mol per mol, ideally about 1 mol per mol] and/or is preferably present as an enantiomerically pure form. Preferably the process is performed in an inert solvent selected from toluene, xylene, tetrahydrofuran, dioxane, methylene chloride, acetonitrile, dimethylformamide, diisopropyl ether or methyl tert-butyl ether, or a mixture of any thereof. Preferably the reaction is conducted at a temperature from -70 to 3O0C.
In step d) preferred bases D, for precipitation of compound of Formula (III), are 1 - adamantylamine, dicyclohexylamine, tert-butylamine, (S)- or (Λ)-α-phenylethylamine. In addition alternatively the compounds of Formula (III) may first be reacted with a simple base, such as potassium carbonate, sodium carbonate or sodium-hydrogen carbonate, then by washing of the water layer containing the salt compound of Formula (III) with an organic solvent, liberating the compound of Formula (III) by dilute acid. The compound of formula (III) can then be reacted with a base D as described above
Preferably in step f) compounds of Formula (VII) are obtained from compounds of Formula (III) via a Curtius rearrangement, see for Curtius rearrangement ,for example, E.F. Scriven, K. Turnbull, Chem. Rev., 1988, 88, 297. Suitable azides of Formula (V) for the Curtius rearrangement are phosphoric acid ester-azide, such as phosphoric acid diphenyl ester-azide or phosphoric acid diethyl ester-azide. Curtius rearrangement is in general carried out in suitable inert solvents such as toluene, benzene, xylene, dioxane or tetrahydrofuran. Suitable bases for Curtius rearrangement are organic amines selected from N-ethylmorpholine, N-methylmorpholine, pyridine or triethylamine preferably in an amount of lmol to 3mol per mol of compounds of Formula (III). Also, it is possible first to convert carboxylic acid to corresponding derivatives by activating reagents, such as C1-C4 alkyl chloroformates in presence of amine, thionyl chloride or phosphorus pentachloride and then prepare carboxylic acid azides by reaction with alkali metal azides or trialkylsilyl azide. The acid azides can be transformed to corresponding isocyanates, which can be optionally isolated
and reacted with an allyl alcohol, preferably of Formula (E), to give compounds of Formula (VII). Preferably the Curtius rearrangement reaction is conducted at a temperature of from 20 to HO0C.
Preferably in step h) the splitting off of urethane and ester function of the compounds of Formula (VII) is conducted with a catalytic amount of palladium catalyst and/or a phosphine and a nucleophilic auxiliary in an inert solvent. Preferably the inert solvent in this reaction is selected from ethanol, tetrahydrofuran, 2-propanol, n-propanol or dioxane, or a mixture of any thereof. Preferably the reaction is conducted at a temperature of from 10 to 9O0C. Preferably the splitting off of urethane and ester functions in compounds of Formula (VII) takes place in a single step; product can crystallize out of the reaction mixture and can be isolated by filtration. Suitable nucleophilic auxiliaries for the splitting off of urethane and ester function are selected from triethylamine, dimedone, tributyltin hydride, ammonium formate, morpholine or pyrrolidine, preferably in an amount of 1 mol to 4 mol per mol of compounds of Formula (VII). Suitable palladium catalysts are selected from, Pd(PPh3)4, Pd(OAc)2, PdCl2, Pd(dba)2 or (Pd2(dba)3), preferably in an amount of 0.0005 mol to 0.2 mol per mol of compounds of Formula (VII). Suitable phosphines are selected from, triphenylphosphine, triisopropylphosphine or tri-o- tolylphosphine, preferably in an amount of 0.002 mol to 0.8 mol per mol of compounds of Formula (VII). These very mild and neutral hydrolysis conditions allow for the formation of either enantiomer of 3-(aminomethyl)-5-methylhexanoic acid in high chemical and enantiomeric purity.
Compounds or intermediates mentioned in the present method can be found or isolated in the form of hydrates or solvates, which considered as falling within the scope of invention. The process according to invention represents an efficient and highly enantioselective route to target compounds.
By the use of the term enantiomerically enriched we mean that the enantiomeric excess (ee) is greater than 60%, greater than 65% or greater than 70%.
By use of the term enantiomerically pure we mean that the enantiomeric excess (ee) is greater than 90%, ideally greater than 99% and ideally enantiomerically pure means greater than 99.5%.
Preferred Scheme of synthesis
Propionic anhydride, Δ


Cinnamyl alcohol, quinine, toluene, -35 0C

Example 1
4-Isobutyl-dihydro-3H-pyran-2 ,6-dione (I)
Diacid (50 g, 266 mmol) was placed in propionic anhydride (130 ml) and the reaction mixture was heated at 140-145 °C during 6 hours. Propionic acid and propionic anhydride were distilled in vacuo (30-60 °C/15 mm Hg). Product was collected at 100-105 °C/0.5 mm Hg as yellowish oil. Yield 41 g (91 %), GC purity: >98% (HP-I, 25 m, 70 kPa, gradient 70-240 0C, 15 °C/min). 1H NMR (CDCl3), δ/ppm: 0.91 (d, 6H, J=6.6 Hz), 1.27 (t, 2H, J=7.1 Hz), 1.61-1.75 (m, IH), 2.12- 2.50 (m, 3H), 2.85 (dd, 2H, J1= 17 Hz, J2=4.1 Hz). 13C NMR (CDCl3), δ/ppm:_21.86, 24.23, 25.89, 35.43, 43.08, 166.48.
Example 2 (i?)-3-(2-(Cinnamyloxy)-2-oxoethyl)-5-methylhexanoic acid, (III) Procedure A
Quinine (28.7g, 88 mmol) was suspended in toluene (380 mL). Cinnamyl alcohol (15.5 g, 115 mmol) was added and the reaction mixture was cooled to -35 0C. The solution of 3- isobutylglutaric anhydride (15.0 g, 88 mmol) in toluene (10 mL) was added during 15 min and the reaction mixture was stirred at -35 °C for 24 hours. Toluene solution was washed with 5% HCl (250 mL) and evaporated. Oily residue was dissolved in 2-PrOH (300 mL), warmed to 45 0C, and the solution of 1-adamantylamine (12.0 g, 79 mmol) in MTBE (100 mL) was added. The mixture was stirred at 25 0C for 6 hours, filtered, washed with 2-PrOH (100 mL) and dried under reduced pressure to yield 28.1 g of 1-adamantylamine salt of (/?)-3-(2-(cinnamyloxy)-2-oxoethyl)-5- methylhexanoic acid. The salt was suspended in toluene (15OmL) and stirred with 3% HCl (100 mL) until a clear solution was obtained. Aqueous acidic solution was separated and organic layer was washed once again with 3% HCl (3OmL). Evaporation of toluene afforded 18.1 g (69%) of monoester as viscous yellowish oil. HPLC analysis on Chiralpak AS column, hexane/EtOH/TFA=95/5/0.1 revealed 91.2 % ee. 1H NMR (CDCl3), δ/ppm: 0.87 (d, 6H, J=6.5 Hz), 1.21-1.27 (m, 2H), 1.56-1.70 (m, IH), 2.38-2.48 (m, 5H), 4.73 (dd, 2H, J/=6.5 Hz, J2=1.2 Hz), 6.27 (dt, IH J/=15.8 Hz, J2=6.5 Hz), 6.65 (d, IH, J=I 5.8 HZ), 7.22-7.40 (m, 2H).
13C NMR (CDCl3), δ/ppm: 22.34, 25.07, 29.64, 38.30, 38.48, 43.26, 64.92, 122.95, 126.50, 127.96, 128.49, 134.18, 136.07, 172.26, 178.64.
Example 3
(i?)-3-(2-(Cinnamyloxy)-2-oxoethyl)-5-methylhexanoic acid, (III) Procedure B
Quinine (28.7g, 88mmol) was suspended in toluene (38OmL). Cinnamyl alcohol (15.5 g, 115mmol) was added and the reaction mixture was cooled to -35 0C. The solution of 3- isobutylglutaric anhydride (15.0 g, 88 mmol) in toluene (1OmL) was added during 15 min and the reaction mixture was stirred at -35 0C for 24 hours. Toluene solution was washed with 5% HCl (250 + 5OmL), and than extracted with 2% K2CO3 solution (1000 + 25OmL). Aqueous extracts were washed with EtOAc (3x10OmL), acidified to pH 1 with cone. HCl and extracted with diisopropylether (150 + 5OmL). Combined extracts were warmed to 35 0C and (S)-α- phenylethylamine (9.7 g, 80 mmol) was added, followed by seed crystals (10 mg). Mixture was stirred for 4 hours at 25 °C and filtered to obtain 24.8 g of (5)-α-phenylethylamine salt of (/?)-3-(2- (cinnamyloxy)-2-oxoethyl)-5-methylhexanoic acid. The salt was suspended in toluene (15OmL) and stirred with 3% HCl (10OmL) until clear solution was obtained. The aqueous acidic solution was separated and organic layer was washed once again with 3% HCl (30 mL). Evaporation of toluene afforded 17.3 g (66%) of monoester as viscous yellowish oil. HPLC analysis on Chiralpak AS column, hexane/EtOH/TFA=95/5/0.1 revealed 90.7 % ee.
Example 4
(5)-3-(2-(Cinnamyloxy)-2-oxoethyl)-5-methylhexanoic acid (III) - (opposite enantiomer to Example 3)
Quinidine (17.2g, 52.8mmol) was suspended in toluene (23OmL). Cinnamyl alcohol (9.3 g, 69mmol) was added and the reaction mixture was cooled to -35 0C. The solution of 3- isobutylglutaric anhydride (9.Og, 52.8mmol) in toluene (6mL) was added during 15 min and the reaction mixture was stirred at -38 °C for 24 hours. Working up is carried out analogously to the preparation of the compound from Example 3. Combined extracts were warmed to 35 0C and (S)- α-phenylethylamine (5.8 g, 48mmol) was added, followed by seed crystals (10 mg). The mixture was stirred for 4 hours at 25°C and filtered to obtain 14.6 g of (5)-α-phenylethylamine salt of (S)- 3-(2-(cinnamyloxy)-2-oxoethyl)-5-methylhexanoic acid. Salt was suspended in toluene (90 mL) and stirred with 3% HCl (600 mL) until clear solution was obtained. Aqueous acidic solution was separated and organic layer was washed once again with 3% HCl (2OmL). Evaporation of toluene
afforded 10.2 g (65%) of monoester as viscous yellowish oil. HPLC analysis on Chiralpak AS column, hexane/EtOH/TFA=95/5/0.1 revealed 91.4 % ee.
1H NMR (CDCl3), δ/ppm: 0.87 (d, 6H, J=6.5 Hz), 1.21-1.27 (m, 2H), 1.56-1.70 (m, IH), 2.38-2.48 (m, 5H), 4.73 (dd, 2H, Jy=6.5 Hz, J2=1.2 Hz), 6.27 (dt, IH J/=15.8 Hz, J2=6.5 Hz), 6.65 (d, IH, J=15.8 HZ), 7.22-7.40 (m, 2H).
13C NMR (CDCl3), δ/ppm: 22.34, 25.07, 29.64, 38.30, 38.48, 43.26, 64.92, 122.95, 126.50, 127.96, 128.49, 134.18, 136.07, 172.26, 178.64.
Example 5 (S)-Cinnamyl 3-((cinnamyloxycarbonylamino)methyl)-5-methylhexanoate (VII)
To the dry toluene solution of (/?)-3-(2-(cinnamyloxy)-2-oxoethyl)-5-methylhexanoic acid (18.1 g, 60mmol in 10OmL), triethylamine (8.36ml, 60mmol) and diphenylphosphoryl azide (14.25ml, 65mmol) were added at 20 0C. The reaction mixture was stirred for 30 min and was slowly added to toluene (80 mL) in separated flask at 90 0C. When nitrogen evolution cased (30-45 min), cinnamyl alcohol (9.65 g, 72 mmol) was added and the mixture was refluxed overnight. The reaction mixture was washed with a solution of 1 g NaNO2 and 1,5. g NaHCO3 in H2O (10OmL), than with H2O (10OmL) and evaporated under reduced pressure to yield 25.8g of crude oily (S)- Cinnamyl 3-((cinnamyloxycarbonylamino)methyl)-5-methylhexanoate which was used without further purification in the next step.
1H NMR (CDCl3), δ/ppm: 0.88 (d, 3H, J=6.4 Hz), 0.90 (d, 3H, J=6.4 Hz), 1.09-1.25 (m, 2H), 1.63- 1.71 (m, IH), 2.15-2.38 (m, 3H), 3.05-3.18 (m, IH), 3.25-3.33 (m, IH), 4.68-4.75 (m, 4H), 4.96 (br.s, IH), 6.22-6.32 (m, 2H), 6.62 (d, 1H, J=15.8 Hz), 6.65 (d, 1H, J=15.8 Hz), 7.15-7.50 (m, 10H) 13C NMR (CDCl3), δ/ppm: 22.53, 25.05, 33.51, 37.30, 41.36, 44.61, 65.01, 65.29, 122.89, 123.83, 126.45, 126.48, 127.80, 127.96, 128.43, 128.47, 133.42, 134.25, 136.01, 136.20, 156.35, 172.77.
Example 6
(<S)-3-(aminomethyl)-5-methylhexanoic acid (pregabalin)
The solution of crude carbamate (25.8g) in abs. EtOH (12OmL) was refluxed for 10 min, and than cooled to 70 0C. Morpholine (21 ml, 237 mmol) was added followed by triphenylphosphine (202 mg, 0.77 mmol) and Pd(OAc)2 (27 mg, 0.12 mmol). The reaction mixture was refluxed for 3 hours, then slowly cooled to room temperature. After 5 hours the product was collected by
filtration and dried at reduced pressure to give 5.7 g (61%) of crude product. Crystallization from aqueous 2-PrOH afforded pure amino acid (the derivative of aminoacid was prepared with Marfey's reagent and analysed on Nucleosil 100-5 RP 18, 0.05 M aqueous triethylamine (adjusted to pH 3 with phosphoric acid)-acetonitrile, 50:50, revealed 99.8% ee). Mp 190 0C (decomp.), [α]D +12 (c=1.0, H2O).
1H NMR (D2O), δ/ppm: 0.87 (d, 3H, J=6.5Hz), 0.89 (d, 3H, J=6.5Hz), 1.21 (t, 2H, J=7.0 Hz), 1.58-1.72 (m, IH), 2.07-2.35 (m, 3H), 2.90-3.05 (m, 2H). 13C NMR (D2O), δ/ppm: 21.64, 22.12, 24.51, 41.82, 40.72, 40.88, 43.82, 181.31.
Claims
1. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, the process comprising the steps of:
a) enantioselective alcoholysis of the anhydride of Formula (I)

(I) by the use of a chiral amine base and an allyl alcohol , in an inert solvent to form an enantiomerically enriched compound of Formula (II), in either of its enatiomeric forms, but where the (R) form is preferred,

wherein A represents the allyl group, and B represents the chiral amine base;
b) converting the compound of Formula (II) into a compound of Formula (III), in either of its enatiomeric forms, but where the (R) form is preferred, by the removal of the base B, by the addition of an acid;


d) reacting the compound of Formula (IV) with a dilute acid to obtain an enantiomerically pure compound of Formula (III) in either of its enatiomeric forms, but where the (R) form is preferred;
e) i) reacting the compound of Formula (III) with an azide of Formula (V), in a Curtius rearrangement reaction,
(RiO)2-P(O)-N3 (V)
wherein R4 represents phenyl or Ci -C5 branched or straight alkyl chain, in an inert solvent and in the presence of a base, or e) ii) by activation of the carboxy group of the compound of Formula (III) and subsequent reaction with alkali metal azides or trialkylazides to obtain acid azides, which are then converted to the corresponding rearranged isocyanates,
to form an enantiomerically pure compound of Formula (VI), in either of its enatiomeric forms, but where the (S) form is preferred,

(VI)
wherein A represents the allyl group; f) reacting a enantiomerically pure compound of Formula (VI) with an allyl alcohol, in an inert solvent, to provide an enantiomerically pure compound of Formula (VII) in either of its enatiomeric forms, but where the (S) form is preferred,

wherein A' is an allyl group that may be the same or different from A;
g) splitting off of the urethane and ester functions of the compound of Formula (VII) with a catalytic amount of palladium catalyst to provide pregabalin, or its opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof.
2. A process as claimed in claim 1 wherein the chiral amine base used in step a) is selected from quinine, quinidine, hydroquinine, cinchonine, cinchonidine, epiquinidine, epicinchonidine, epicinchonine or epiquinine.
3. A process as claimed in claim 1 or 2 wherein the allyl alcohol in step a) and step f), may be the same or different, and each is a compound of Formula (E)

4. A process as claimed in any claim from 1 to 3 wherein the base in step c) is selected from 1- adamantylamine, dicyclohexylamine, ter/-butylamine, (S)- or (Λ)-α-phenylethylamine.
5. A process of as claimed in claim 1 wherein, the anhydride of Formula (I)

(D is formed by reacting 3-isobutylglutaric acid with propionic anhydride.
6. A process for the preparation of a compound of Formula (I)

(I) by reacting 3-isobutylglutaric acid with propionic anhydride.
7. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof as claimed in claim 1 wherein, the process starts from an intermediate of Formula (III).
8. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof as claimed in claim 1 wherein, the process starts from an intermediate of Formula (FV).
9. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof as claimed in claim 1 wherein, the process starts from an intermediate of Formula (VI).
10. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof as claimed in claim 1 wherein, the process starts from an intermediate of Formula (VII).
11. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, the process comprising the use of the intermediate of Formula (II)

wherein A is an allyl group and B is a chiral amine base selected from quinine, quinidine, hydroquinine, cinchonine, cinchonidine, epiquinidine, epicinchonidine, epicinchonine or epiquinine.
12. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, the process comprising the use of an intermediate of Formula (III)

wherein A is an allyl group.
13. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, the process comprising the use of an intermediate of Formula (IV) 
wherein A is an allyl group and D is a base selected from 1-adamantylamine, dicyclohexylamine, ferf-butylamine, (S)- or (Λ)-α-phenylethylamine.
14. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, the process comprising the use of an intermediate of Formula (VI)

(VI) wherein A is a allyl group.
15. A process for the preparation of pregabalin, or the opposite enantiomer, or a pharmaceutically-acceptable salt of either thereof, the process comprising use of an intermediate of Formula (VII)

16. A process as claimed in claim 11, 12 or 13 wherein, the compound of Formula (II), (III) or (IV), respectively, is in the (R) form.
17. A process as claimed in claim 14 or 15 wherein, the compound of Formula (VI) or (VII), respectively, is in the (S) form.
18. A compound of Formula (II), as defined in claim 11.
19. A compound of Formula (III), as defined in claim 12.
20. A compound of Formula (IV), as defined in claim 13.
21. A compound of Formula (VI), as defined in claim 14.
22. A compound of Formula (VII), as defined in claim 15.
23. A compound of Formula (VII), as defined in claim 16.
24. A compound of Formula (II), (III) or (IV), as defined in claims 11, 12 and 13 respectively, which is in the (R) form.
25. A compound of Formula (VI) or (VII), as defined in claim 14 and 15 respectively, which is in the (S) form.
26. A compound as claimed in claim 24 or 25 which is enatiomerically enriched.
27. A compound as claimed in claim 26 wherein the compound is enatiomerically pure.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EA200970126A EA200970126A1 (en) | 2006-07-15 | 2007-07-11 | METHOD OF OBTAINING PREGABALIN AND ITS ENANTIOMER |
| EP07766194A EP2046728A1 (en) | 2006-07-15 | 2007-07-11 | Process for preparing pregabalin and its opposite enantiomer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0614133A GB0614133D0 (en) | 2006-07-15 | 2006-07-15 | Process for preparing a pharmaceutical compound |
| GB0614133.7 | 2006-07-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008009897A1 true WO2008009897A1 (en) | 2008-01-24 |
Family
ID=36955756
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2007/002607 Ceased WO2008009897A1 (en) | 2006-07-15 | 2007-07-11 | Process for preparing pregabalin and its opposite enantiomer |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP2046728A1 (en) |
| EA (1) | EA200970126A1 (en) |
| GB (1) | GB0614133D0 (en) |
| WO (1) | WO2008009897A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7417165B2 (en) | 2005-04-06 | 2008-08-26 | Teva Pharmaceutical Industries Ltd. | Crystalline forms of pregabalin |
| US7446220B2 (en) | 2005-09-19 | 2008-11-04 | Teva Pharmaceutical Industries Ltd. | Asymmetric synthesis of (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid |
| US7462737B2 (en) | 2005-05-10 | 2008-12-09 | Teva Pharmaceutical Industries Ltd. | Pregabalin free of isobutylglutaric acid and a process for preparation thereof |
| US7462738B2 (en) | 2006-05-24 | 2008-12-09 | Teva Pharmaceutical Industries Ltd. | Processes for the preparation of R-(+)-3-(carbamoyl methyl)-5-methylhexanoic acid and salts thereof |
| US7488846B2 (en) | 2005-04-11 | 2009-02-10 | Teva Pharmaceuical Industries Ltd. | Pregabalin free of lactam and a process for preparation thereof |
| US7619112B2 (en) | 2005-05-10 | 2009-11-17 | Teva Pharmaceutical Industries Ltd. | Optical resolution of 3-carbamoylmethyl-5-methyl hexanoic acid |
| US7763749B2 (en) | 2005-05-10 | 2010-07-27 | Teva Pharmaceutical Industries Ltd. | Method for the preparation of Pregabalin and salts thereof |
| CN101333165B (en) * | 2008-08-05 | 2011-05-04 | 浙江大学 | Method for synthesizing monomethyl glutarate |
| US8063244B2 (en) * | 2009-05-07 | 2011-11-22 | Dipharma Francis S.R.L. | Process for the synthesis of pregabalin |
| US8097754B2 (en) | 2007-03-22 | 2012-01-17 | Teva Pharmaceutical Industries Ltd. | Synthesis of (S)-(+)-3-(aminomethyl)-5-methyl hexanoic acid |
| WO2012059797A1 (en) * | 2010-11-04 | 2012-05-10 | Lupin Limited | Process for synthesis of (s) - pregabalin |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996038405A1 (en) * | 1995-06-02 | 1996-12-05 | Warner-Lambert Company | Methods of making (s)-3-(aminomethyl)-5-methylhexanoic acid |
-
2006
- 2006-07-15 GB GB0614133A patent/GB0614133D0/en not_active Ceased
-
2007
- 2007-07-11 WO PCT/GB2007/002607 patent/WO2008009897A1/en not_active Ceased
- 2007-07-11 EA EA200970126A patent/EA200970126A1/en unknown
- 2007-07-11 EP EP07766194A patent/EP2046728A1/en not_active Withdrawn
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996038405A1 (en) * | 1995-06-02 | 1996-12-05 | Warner-Lambert Company | Methods of making (s)-3-(aminomethyl)-5-methylhexanoic acid |
Non-Patent Citations (2)
| Title |
|---|
| HAMERSAK ET AL: "An efficient synthesis of (S)-3-aminomethyl-5-methylhexanoic acid (Pregabalin) via quinine-mediated desymmetrization of cyclic anhydride", 4 July 2007, TETRAHEDRON: ASYMMETRY, PERGAMON, OXFORD, GB, PAGE(S) 1481-1485, ISSN: 0957-4166, XP022157447 * |
| MARVIN S. HOEKSTRA ET AL.: "Chemical Development of CI-1008, an Enatiomerically Pure Anticonvulsant", ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 1, 1997, Washington D.C., pages 26 - 38, XP002454624 * |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7417165B2 (en) | 2005-04-06 | 2008-08-26 | Teva Pharmaceutical Industries Ltd. | Crystalline forms of pregabalin |
| US7488846B2 (en) | 2005-04-11 | 2009-02-10 | Teva Pharmaceuical Industries Ltd. | Pregabalin free of lactam and a process for preparation thereof |
| US7763749B2 (en) | 2005-05-10 | 2010-07-27 | Teva Pharmaceutical Industries Ltd. | Method for the preparation of Pregabalin and salts thereof |
| US7462737B2 (en) | 2005-05-10 | 2008-12-09 | Teva Pharmaceutical Industries Ltd. | Pregabalin free of isobutylglutaric acid and a process for preparation thereof |
| US7678938B2 (en) | 2005-05-10 | 2010-03-16 | Teva Pharmaceutical Industries Ltd. | Optical resolution of 3-carbamoylmethyl-5-methyl hexanoic acid |
| US7619112B2 (en) | 2005-05-10 | 2009-11-17 | Teva Pharmaceutical Industries Ltd. | Optical resolution of 3-carbamoylmethyl-5-methyl hexanoic acid |
| US7470812B2 (en) | 2005-09-19 | 2008-12-30 | Teva Pharmaceutical Industries Ltd. | Chiral 3-carbamoylmethyl-5-methyl hexanoic acids, key intermediates for the synthesis of (S)-Pregabalin |
| US7960583B2 (en) | 2005-09-19 | 2011-06-14 | Teva Pharmaceutical Industries Ltd. | Asymmetric synthesis of (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid |
| US7586005B2 (en) | 2005-09-19 | 2009-09-08 | Teva Pharmaceutical Industries Ltd. | Asymmetric synthesis of (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid |
| US7465826B2 (en) | 2005-09-19 | 2008-12-16 | Teva Pharmaceutical Industries Ltd. | Chiral 3-carbamoylmethyl-5-methyl hexanoic acids, key intermediates for the synthesis of (S)-pregabalin |
| US8212071B2 (en) | 2005-09-19 | 2012-07-03 | Teva Pharmaceutical Industries Ltd. | Asymmetric synthesis of (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid |
| US7687656B2 (en) | 2005-09-19 | 2010-03-30 | Teva Pharmaceutical Industries Ltd. | Asymmetric synthesis of (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid |
| US7446220B2 (en) | 2005-09-19 | 2008-11-04 | Teva Pharmaceutical Industries Ltd. | Asymmetric synthesis of (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid |
| US7851651B2 (en) | 2005-09-19 | 2010-12-14 | Teva Pharmaceutical Industries Ltd. | Asymmetric synthesis of (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid |
| US7923575B2 (en) | 2005-09-19 | 2011-04-12 | Teva Pharmaceutical Industries Limited | Asymmetric synthesis of (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid |
| US7973196B2 (en) | 2005-09-19 | 2011-07-05 | Teva Pharmaceutical Industries Ltd. | Asymmetric synthesis of (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid |
| US7563923B2 (en) | 2005-09-19 | 2009-07-21 | Teva Pharmaceutical Industries Ltd. | Chiral 3-carbamoylmethyl-5-methyl hexanoic acids, key intermediates for the synthesis of (S)-Pregabalin |
| US7462738B2 (en) | 2006-05-24 | 2008-12-09 | Teva Pharmaceutical Industries Ltd. | Processes for the preparation of R-(+)-3-(carbamoyl methyl)-5-methylhexanoic acid and salts thereof |
| US8097754B2 (en) | 2007-03-22 | 2012-01-17 | Teva Pharmaceutical Industries Ltd. | Synthesis of (S)-(+)-3-(aminomethyl)-5-methyl hexanoic acid |
| CN101333165B (en) * | 2008-08-05 | 2011-05-04 | 浙江大学 | Method for synthesizing monomethyl glutarate |
| US8063244B2 (en) * | 2009-05-07 | 2011-11-22 | Dipharma Francis S.R.L. | Process for the synthesis of pregabalin |
| WO2012059797A1 (en) * | 2010-11-04 | 2012-05-10 | Lupin Limited | Process for synthesis of (s) - pregabalin |
Also Published As
| Publication number | Publication date |
|---|---|
| EA200970126A1 (en) | 2009-06-30 |
| GB0614133D0 (en) | 2006-08-23 |
| EP2046728A1 (en) | 2009-04-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008009897A1 (en) | Process for preparing pregabalin and its opposite enantiomer | |
| EP0830338B1 (en) | Method of making 3-(aminomethyl)-5-methylhexanoic acid | |
| US6891059B2 (en) | Asymmetric synthesis of pregabalin | |
| EP0828704B1 (en) | Methods of making (s)-3-(aminomethyl)-5-methylhexanoic acid | |
| WO2008062460A2 (en) | Crystalline forms of pregabalin | |
| KR20080027880A (en) | R-(+)-3- (carbamoylmethyl) -5-methylhexanoic acid and salts thereof | |
| WO2014068333A2 (en) | New process | |
| JP2002508345A (en) | Asymmetric hydrogenation | |
| Resnick et al. | A practical synthesis of 3-ethyl-l-norvaline | |
| EP2050738B1 (en) | Novel imidazolidinone derivative, method of producing the same and method of producing optically active amino acid | |
| EP1489066B1 (en) | Process for production of optically active carboxylic acid | |
| AU700091C (en) | Method of making (S)-3-(aminomethyl)-5-methylhexanoic acid | |
| JP3845884B2 (en) | Process for producing N- [1- (2,4-dichlorophenyl) ethyl] -2-cyano-3,3-dimethylbutanamide | |
| JPH1059947A (en) | Process for producing chiral, non-racemic (4-aryl-2,5-dioxoimidazolidin-1-yl) acetic acids | |
| KR20110086167A (en) | Enantioselective Synthesis of γ-Amino-α, β-unsaturated Carboxylic Acid Derivatives | |
| WO2014012832A1 (en) | Process for the preparation of 2-(3-n,n-diisopropylamino-1-phenylpropyl)-4-hydroxymethyl-phenol and its derivatives | |
| EP3154930A1 (en) | Method for the preparation of beta-substituted gamma-amino carboxylic acids | |
| KR19990046899A (en) | Method for preparing pure optical isomer of 1.1'-bi-2-naphthol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07766194 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007766194 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200970126 Country of ref document: EA |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |