WO2007103252A2 - Novel crystalline forms of antidiabetic compounds - Google Patents
Novel crystalline forms of antidiabetic compounds Download PDFInfo
- Publication number
- WO2007103252A2 WO2007103252A2 PCT/US2007/005493 US2007005493W WO2007103252A2 WO 2007103252 A2 WO2007103252 A2 WO 2007103252A2 US 2007005493 W US2007005493 W US 2007005493W WO 2007103252 A2 WO2007103252 A2 WO 2007103252A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- crystalline anhydrous
- free acid
- crystalline
- benzenesulfonate salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- ATDYDMPYGUIGMP-ZDUSSCGKSA-N CCCc1nc(Oc(cc2)ccc2Cl)ccc1-c1n[o]c(cc2Cl)c1cc2O[C@@H](C)C(O)=O Chemical compound CCCc1nc(Oc(cc2)ccc2Cl)ccc1-c1n[o]c(cc2Cl)c1cc2O[C@@H](C)C(O)=O ATDYDMPYGUIGMP-ZDUSSCGKSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
Definitions
- the present invention relates to novel crystalline forms and salt forms of compounds that are useful as pharmaceutically active ingredients for the treatment of type 2 diabetes and other diseases that are modulated by PPAR gamma agonists, including hyperglycemia, obesity, dyslipidemia, and the metabolic condition.
- the invention also relates to a process for making the compounds, crystalline forms, and salts.
- Type 2 diabetes remains a serious medical problem. There is an ongoing need for new treatments that are more effective and that have fewer side effects.
- PPAR gamma agonists including the two marketed products rosiglitazone and pioglitazone, are important medications for the treatment of type 2 diabetes.
- Treatment of a patient with PPAR gamma agonists improves insulin sensitivity, but the treatment is often accompanied by side effects, such as weight gain and edema.
- Selective PPAR gamma partial agonists also known as selective PPAR gamma modulators (SPPARM's or SPPARgM's) are effective in reducing serum glucose with reduced weight gain and/or edema.
- the present invention is concerned with novel crystal forms, salts, and crystal forms of the salts of a compound that is an active PPAR gamma partial agonist, and methods of making the compound, salts and crystal forms.
- the compound was originally disclosed as a solid in US Provisional Application No. 60/658,661, now WO2006/096564, but the solid did not have the crystal form disclosed herein.
- the crystalline forms disclosed herein are novel and well characterized, and have advantages over the solid forms disclosed in WO2006/096564 that make them useful in preparing pharmaceutical formulations, such as ease of purification, ease of processing, and thermodynamic stability with respect to other forms of the compound.
- the anhydrous free acid crystalline form is non-hygroscopic, and exhibits good bioavailability in animals, even though it has low water solubility at neutral pH.
- the invention also concerns pharmaceutical compositions comprising the novel crystalline polymorphs; processes for the preparation of these polymorphic forms and their pharmaceutical compositions; and methods for using them for the treatment of type 2 diabetes, hyperglycemia, obesity, dyslipidemia, and the metabolic condition.
- FIG. 1 is a characteristic X-ray diffraction pattern of the crystalline anhydrous free acid form.
- FIG. 2 is a carbon-13 cross-polarization magic-angle spinning (CPMAS) nuclear magnetic resonance (NMR) spectrum of the crystalline anhydrous free acid form.
- CPMAS cross-polarization magic-angle spinning
- FIG. 3 is a typical DSC curve of the crystalline anhydrous free acid form.
- FIG. 4 is a typical thermogravimetric (TG) curve of the crystalline anhydrous free acid form.
- FIG. 5 is a characteristic X-ray diffraction pattern of the crystalline anhydrous benzenesulfonate
- FIG. 6 is a typical DSC curve of the crystalline anhydrous besylate salt.
- this invention provides a novel crystalline anhydrous polymorphic form of the free acid of (2S)-2-( ⁇ 6-chloro-3-[6-(4-chlorophenoxy)-2-propylpyridin-3-yl]-l,2- benzisoxazol-5-yl ⁇ oxy)propanoic acid (Compound I):
- This compound was first disclosed as Example 14 in WO 2006/096564.
- the compound that was isolated using the synthetic methodology in the above-mentioned PCT patent application does not have the crystal form that is disclosed herein. Improvements in the process for making the compound led to the discovery of a crystalline free acid anhydrate of Compound I 1 which is described and characterized herein.
- the invention also provides a benzenesulfonate (besylate) salt of Compound I, and more specifically, an anhydrous crystalline benzenesulfonic acid (besylate) salt of Compound I.
- the besylate salt and specifically the anhydrous crystalline besylate salt, has advantageous properties compared with the non-crystalline free acid and amorphous sodium salts of Compound I that were originally made.
- the two crystalline compounds (crystalline free acid anhydrate and anhydrous crystalline besylate salt) are readily used in the preparation of pharmaceutical compositions.
- the benzenesulfonic acid (besylate) salt of Compound I is also a new composition of matter. This is generally referred to herein as the benzenesulfonic acid (besylate) salt of Compound I, but it can also be written as a chemical compound having Formula Ia:
- compositions, drug substances, formulations, and pharmaceutical uses that are described herein for the crystalline anhydrous besylate salt are also representative of compositions, drug substances, formulations, and pharmaceutical uses of the besylate salt in general.
- a further embodiment of the present invention provides a drug substance that comprises the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of Compound I in a detectable amount.
- drug substance is meant the active pharmaceutical ingredient (API).
- API active pharmaceutical ingredient
- the amount of crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt in the drug substance can be quantified by the use of physical methods such as X-ray powder diffraction (XRPD), solid-state fluorine-19 magic-angle spinning (MAS) nuclear magnetic resonance spectroscopy, solid-state carbon-13 cross-polarization magic-angle spinning (CPMAS) nuclear magnetic resonance spectroscopy, solid state Fourier-transform infrared spectroscopy, and Raman spectroscopy.
- XRPD X-ray powder diffraction
- MAS solid-state fluorine-19 magic-angle spinning
- CPMAS cross-polarization magic-angle spinning
- a sub-class of this embodiment about 5% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance. In a second sub-class of this embodiment, about 10% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance. In a third sub-class of this embodiment, about 25% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance.
- a fourth sub-class of this embodiment about 50% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance. In a fifth sub-class of this embodiment, about 75% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance. In a sixth sub-class of this embodiment, substantially all of the Compound I drug substance is the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt, i.e., the Compound I drug substance is the substantially phase pure crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt.
- Another aspect of the present invention provides a method for the treatment or control of clinical conditions for which a PPAR gamma agonist is indicated, which method comprises administering to a patient in need of such treatment or control a therapeutically effective amount of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of Compound I or a pharmaceutical composition containing a therapeutically effective amount of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of Compound I.
- Such clinical conditions include Type 2 diabetes, hyperglycemia, obesity, dyslipidemia, and metabolic syndrome.
- a "patient” is a mammal, including a human. A patient is most often a human patient.
- the present invention also provides for the use of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of the present invention in the manufacture of a medicament for the treatment or control in a patient of one or more clinical conditions for which a PPAR gamma agonist is indicated.
- the clinical condition is Type 2 diabetes.
- Another aspect of the present invention provides the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt for use in the treatment or control in a patient of one or more clinical conditions for which a PPAR gamma agonist is indicated.
- the clinical condition is Type 2 diabetes.
- the present invention also provides pharmaceutical compositions comprising the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt, in association with one or more pharmaceutically acceptable carriers or excipients.
- the pharmaceutical composition comprises the active pharmaceutical ingredient (API) in admixture with pharmaceutically acceptable excipients wherein the API comprises a detectable amount of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of the present invention.
- the pharmaceutical composition comprises the API in admixture with pharmaceutically acceptable excipients wherein the API comprises about 5% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of the present invention.
- the API in such compositions comprises about 10% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt. In a subclass of this embodiment, the API in such compositions comprises about 25% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt. In a sub-class of this embodiment, the API in such compositions comprises about 50% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt.
- the API in such compositions comprises about 75% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt.
- substantially all of the API is crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of Compound I, i.e., the API is substantially phase pure Compound I in the crystalline free acid anhydrate form or substantially phase pure Compound I in the form of a crystalline anhydrous benzenesulfonate salt.
- compositions in accordance with the invention are suitably in unit dosage forms such as tablets, pills, capsules, powders, granules, sterile solutions or suspensions, metered aerosol or liquid sprays, drops, ampoules, auto-injector devices or suppositories.
- the compositions are intended for oral, parenteral, intranasal, sublingual, or rectal administration, or for administration by inhalation or insufflation.
- Formulation of the compositions according to the invention can conveniently be effected by methods known in the art, for example, as described in Remington's Pharmaceutical Sciences. 17* ed., 1995.
- the dosage regimen is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the route of administration; and the renal and hepatic function of the patient.
- An ordinarily skilled physician, veterinarian, or clinician can readily determine and prescribe the effective amount of the drug required to prevent, counter or arrest the progress of the condition or to treat or control the condition.
- Oral administration is the preferred method of administering the crystal forms and salt forms of Compound I described herein.
- the drug can be administered 1-2 times per day, with once daily being preferred.
- the daily dosage for an adult human patient is generally 1-25 mg, and preferably 2-10 mg administered once daily.
- the Compound I crystalline free acid anhydrate and the crystalline anhydrous benzenesulfonate salt described herein in detail can form the API, and are typically administered in admixture with suitable pharmaceutical diluents, excipients or carriers (collectively referred to herein as 'carrier' materials) suitably selected with respect to the intended form of administration, that is, oral tablets, capsules, elixirs, syrups and the like, and consistent with conventional pharmaceutical practices.
- the active pharmaceutical ingredient can be combined with an oral, non-toxic, pharmaceutically acceptable, inert carrier such as lactose, starch, sucrose, glucose, methyl cellulose, microcrystalline cellulose, magnesium stearate, calcium sulfate, mannitol, sorbitol and the like;
- an oral, non-toxic, pharmaceutically acceptable, inert carrier such as lactose, starch, sucrose, glucose, methyl cellulose, microcrystalline cellulose, magnesium stearate, calcium sulfate, mannitol, sorbitol and the like
- the oral API can be combined with any oral, non-toxic, pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like.
- suitable binders, lubricants, disintegrating agents and coloring agents can also be incorporated into the mixture.
- Suitable binders include starch, gelatin, some natural sugars, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like.
- Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, and the like.
- Disintegrants include, without limitation, starch, methyl cellulose, croscarmellose sodium, agar, bentonite, xanthan gum and the like.
- Surfactants such as sodium lauryl sulfate, can also be included in the formulations.
- the reaction mixture was allowed to cool to RT and then poured into a biphasic mixture containing IPAc (220 mL) and aq. K 2 CO 3 (20.7 g in 117.3 g water) at RT with stirring.
- the organic layer was separated, washed with sat. NaHCO 3 (80 mL), and then water (80 mL).
- the isolated IPAc solution was subjected to a solvent switch to DMF (80 mL) in vacuo.
- the reaction was then quenched with 10% citric acid solution (120 mL) and diluted with MTBE (120 mL). The mixture was stirred over 15 min. The organic layer was cut and was washed with 10% NaCl solution (120 mL). The organic layer (188 mL) was concentrated to 90 mL (1/2 volume), and 90 mL of MeOH was then added. The volume was again reduced to 90 mL by vacuum distillation. This was repeated 2 additional times to complete the solvent switch to MeOH. The final volume was about 90 mL.
- nicotinic acid 5 (7200 g, 24.68 MoI), which was then dissolved in 17 L of trifluoroacetic anhydride (TFAA). l,4-Dimethoxy-2-chlorobenzene (6337 mL, 44.42 MoI) was added, followed by slow addition of triflic acid (4426 mL, 2 equivalents), while maintaining the temperature at ⁇ 40 0 C. A reflux condenser was attached, and the reaction was heated to 42 0 C and stirred overnight. The reaction was assayed, showing a 70 % conversion by mass of 5 to 7.
- TFAA trifluoroacetic anhydride
- the reaction was cooled to ambient temperature with an ice bath, and was then quenched into a 100 L extractor at 0 0 C onto 30 L (6 molar equivalents) of 5 N KOH and 25 L (3.5 volumes) of toluene, maintaining the temperature at ⁇ 50 0 C for 1 hour.
- the 100 L flask was rinsed into the extractor with 2 x 2 L of toluene and 2 x 2 L of 5N KOH.
- the phases were separated at room temperature, and the organic phase was washed with 18 L of IN HCl.
- the organic solution was transferred back into the rinsed 100 L vessel and was treated with Darco G-60 (3.6 kg, 50 wt %).
- the mixture of solution and carbon was heated at 35 0 C for 30 min.
- the charcoal mixture was then filtered through a pad of solka floe, rinsed with 8 L of toluene and vacuum transferred through a 5 uM poly cap, into a visually clean 100 L round bottom flask, with a mark at the 16 L level.
- the 100 L flask was attached to a batch concentrator and distilled down to the 16 L mark at 35 0 C. At this point the batch was seeded with 1O g of seed crystals of 7 obtained from an earlier batch, and heptane addition began.
- the slurry was filtered, and the flask was rinsed with 9 L of a mixture of 95 % heptane / 5 % toluene. The cake was then slurry washed with 9L of 95 % heptane / 5 % toluene, and then 18 L heptane.
- the product 7 was dried on the frit under a stream of N 2 at ambient temperature.
- the batch was cooled to room temperature, filtered, and sequentially washed with 50 mL HOAc (displacement wash), 50 mL HOAc (slurry wash) and 5% MeOH in water (3x 50 mL, slurry washes).
- the isolated product was dried at r.t. under vacuum over the weekend.
- the dry powder product was then suspended in 5% MeOH in water (100 mL) for 4 hours and filtered.
- the filter cake was washed with 50 mL of water and dried under vacuum to give the final product as the free base.
- n-Propanol 60 L was fed and distilled at 35-40 0 C, 28-30 m Hg, while maintaining a constant volume of 20 L
- the final batch KF was 860 ppm water
- the resulting solution was heated on a steam pot to 93-97 0 C
- the reaction was monitored for isome ⁇ zation conversion After 6 hours, the batch was allowed to cool to ambient temperature. 200 mL of the batch was sampled for seed formation.
- 50 mL of water was added, and then 1 g of seed was added, and the batch was aged to form a seed bed.
- the remaining 250 mL of water was added to complete the crystallization
- 5 L of water was added, followed by the seed slurry.
- the mixture was aged, giving a thick slurry.
- the remaining 25 L of water was added over 1 hour.
- the slurry was heated to 50 0 C and cooled to ambient temperature.
- the solid was isolated by filtration. The cake was washed with 2:1 water/n-propanol (8L 3 8L, 12L, 12L), water (8L), then hexanes (12L, 8L). The solid was d ⁇ ed on the filter under a nitrogen tent The E-oxime was obtained as an orange solid.
- Step 8 Benzisoxazole formation
- the filtrate was fed into a 100 L round bottom flask equipped with mechanical stirrer, thermocouple, and nitrogen inlet, and was attached to a batch concentrator.
- the batch was fed and distilled at 35-40 0 C, 16-20 in. Hg, maintaining the batch volume at 20 - 25 L.
- EtOAc (40 L) was then fed and distilled at 35-40 0 C, 20-23 in. Hg at a constant volume of 15-20 L.
- the solid product was isolated by filtration. The cake was washed with EtOAc (16L), then with MeCN (24 L), and was dried on the filter under a nitrogen tent. The benzisoxazole tosylate was obtained as a pale yellow solid.
- the batch was then filtered through a 20 uM poly cap filter into a Buchi rotary evaporator, yielding the product as an oil containing residual ethyl acetate (3 wt%) and 700 PPM water.
- the batch was transferred to a container and was stored in a cold room until it was used.
- the product had an ee of 98.2 %.
- the mixture was transferred to a 180 L cyclindrical vessel, and an additional 30L MTBE and 30 L cold water were added. The layers were cut and the aqueous layer was back extracted with 25 L MTBE. The combined organic layers were washed with 18 L 2% NaHCO 3 . The final organic layer was fed with concurrent distillation into a 100 L RBF and solvent switched to acetonitrile. The batch was kept at 25-30 0 C to prevent crystallization.
- the batch volume was adjusted to 45 L with acetonitile, and 36 L water was added slowly (product crystallizes after 4 L water is added). After overnight aging, the batch was filtered, and the cake was washed with 10 L 1/1 MeCN/water. Solid methyl ester 5-13 on the funnel was dried with suction under nitrogen flow for 4 days.
- the methyl ester S-XS (2.3 kg) was dissolved in 12.5 L MeCN and mixed with 10 L IN NaOH. The solution was aged for 2-3 hrs at ambient temperature.
- the batch was filtered, and the cake was washed with 10 L of 4:1 methylcyclohexane/toluene, then 2 x 10 L of methylcyclohexane. It was dried on the filter pot under vacuum and nitrogen flow overnight, and was then transferred to a vacuum oven and dried with nitrogen flow overnight.
- the crystals isolated using the method described above are the preferred anhydrous crystalline free acid crystals.
- the crystals are anhydrous. They have very low water solubility at native pH, are stable with respect to retaining their crystal form, are chemically stable, and are non-hygroscopic. For example, they gain about 0.2 wt% when placed in an environment with up to 95% RH. Their melting point is 113-114 0 C. They have a small needle morphology and a high surface area without milling or grinding. They exhibit good bioavailability in laboratory studies in dogs and rats.
- the preferred crystalline anhydrate is obtained on crystallization from MTBE/hexanes or toluene/methylcyclohexane. Crystallization from toluene/methylcyclohexane is used in the synthetic procedure described above.
- the benzenesulfonate salt of the compound having Formula I as described herein is crystalline and non-hygroscopic.
- the benzenesulfonate salt is chemically stable, remaining unchanged after 8 months at 40 0 C and 75% RH.
- the benzenesulfonate salt has properties that make it suitable in pharmaceutical formulations.
- the salt has been made by the following procedure.
- a solution of benzenesulfonic acid (1.58 g, 10 mmol) in 10 ml acetonitrile was added to a solution of Compound I (4.87 g, 10 mmol) in 50 ml acetonitrile at 50 0 C.
- the reaction mixture was seeded at 40 0 C with crystals of the Compound I benzenesulfonate salt from earlier batches, yielding a crystalline product.
- the same crystalline product can also be obtained without seeds if none are available.
- the mixture was cooled to room temperature and then was stirred for 2.5 hours. It was cooled to 0-5 0 C and stirred for an additional 30 min. The solid was collected by filtration, and the cake was washed with 10 ml acetonitrile. The solid was dried on the funnel with suction, yielding 6.4 g (99% yield).
- a toluenesulfonate (tosylate) salt of Compound I has also been prepared.
- the tosylate salt also can be prepared as a crystalline anhydrous material.
- the crystalline anhydrous tosylate salt of Compound I was prepared by the following method from the methyl ester of Compound I. MeCN (110 kg) was charged to a reactor. The methyl ester of Compound I (e.g. from step 9 of Example 1; 29.9 kg; 59.6 moles) was charged to the reactor, followed by a MeOH flush of the charge valve. 135 kg of 1.0N NaOH ( ⁇ 131 moles) was added, followed by a water flush at 15-25 0 C. The solution was aged for 2-3 hours at 15-25 0 C and then assayed for completion of the reaction.
- the batch was filtered via a 0.6 micron filter and concentrated to 200-220 L at ⁇ 40 0 C and reduced pressure.
- the solvent was switched at constant volume to EtOAc at ⁇ 40 0 C and reduced pressure ( ⁇ 125 to 252 mmHg).
- the water concentration by Karl Fischer titration was 72.3 ⁇ g/ml, the product concentration was 135.5 g/L, and the acetonitrile content was 0.36 v/v%.
- the batch was collected in drums.
- a charge of 60 kg EtOAc was added to the crystallizer through a 0.6 micron filter.
- a seed slurry (about 12.9 kg containing about 1 kg of media-milled tosylate seed in about 10 L ethyl acetate) was added to the reactor followed by about 10 kg of a pre-f ⁇ ltered EtOAc wash. The seed slurry was recycled from the bottom of the reactor through the outlet and back in through the inlet. Then, the batch of Compound I in EtOAc and the solution of p-toluenesulfonic acid (p-TSA) in EtOAc solution were charged simultaneously into the reactor over a period of about 8 hours.
- p-TSA p-toluenesulfonic acid
- the charge rates for the concentrated batch and p-TSA/EtOAc solution were 0.3 kg/L and 0.4 kg/L respectively.
- the temperature was maintained at 15 to 25 0 C. After crystallization the batch was aged at 15 to 25 0 C for 2 hours.
- Seeds for the crystallization step above are saved from earlier batches of Compound I tosylate.
- the same crystalline product can also be obtained without seed crystals if none are available.
- the batch was filtered and the cake was washed with a total of ⁇ 240 kg ethyl acetate.
- the batch was dried under vacuum at 40 0 C, yielding about 35.8 kg of the desired tosylate salt, for a yield of 90.5% for the salt preparation.
- the dried batch was delumped prior to further use.
- the crystalline free acid anhydrate and the crystalline anhydrous benzenesulfonate salt of Compound I are formulated as either dry filled capsules or compressed tablets in doses that generally will range from 1 mg to 25 mg of API as the free acid (non-salt). Typically, the doses will be in the range of 2-10 mg.
- a typical capsule or tablet formulation contains the crystalline free acid anhydrate or the crystalline anhydrous benzenesulfonate salt, microcrystalline cellulose (Avicel), lactose monohydrate, croscarmellose sodium, sodium lauryl sulfate, and magnesium stearate.
- the capsule formulations are transferred to a capsule made of gelatin, titanium dioxide, and ferric oxide. Tablet formulations are coated with a functional film coat containing lactose, hypromellose, triacetin, titanium dioxide, and ferric oxide.
- the capsule shell and tablet film coating are opaque to protect the active compound from light.
- the formulations are manufactured by first blending the excipients, then compressing the mixture into ribbons by roller compaction, and then milling the ribbons into granules. The granules are then lubricated and either filled into capsules or compressed into tablets. If tablets are selected, a film coat is applied to the compressed tablets. Exemplary fill formulations that provide a 5 mg or 10 mg dose of Compound I (free acid) in a standard gelatin capsule are shown below. The components are combined, compressed and milled as described above, and then the amount of milled formulation that contains the 5 mg or 10 mg dose of Compound I is transferred to each capsule.
- CHARACTERIZATION OF THE CRYSTALLINE FREE ACID ANHYDRATE X-ray powder diffraction studies are widely used to characterize molecular structures, crystallinity, and polymorphism.
- the X-ray powder diffraction patterns of the crystalline anhydrous free acid form of Compound I were generated on a Philips Analytical X'Pert PRO X-ray Diffraction System with PW3040/60 console.
- a PW3373/00 ceramic Cu LEF X-ray tube K-Alpha radiation was used as the source.
- Silicon powder (NIST reference standard 640C) was mixed in the sample and was used as a reference for d-spacing assignment.
- FIG. 1 shows the X-ray diffraction pattern for the crystalline free acid anhydrate.
- the crystalline free acid anhydrate exhibited characteristic reflections corresponding to d-spacings of 17.13, 5.11, and 4.82 angstroms.
- the crystalline free acid anhydrate was further characterized by reflections corresponding to d-spacings of 11.63, 7.88 and 7.42 angstroms.
- the crystalline free acid anhydrate was even further characterized by reflections corresponding to d-spacings of 10.27, 4.64 and 4.01 angstroms.
- the crystalline free acid anhydrate of Compound I was further characterized by solid-state carbon-13 nuclear magnetic resonance (NMR) spectra.
- the solid-state carbon- 13 NMR spectra were obtained on a Bruker DSX 500WB NMR system using a Bruker 4 mm H/X/Y CPMAS probe.
- the carbon-13 NMR spectra utilized proton/carbon- 13 cross-polarization magic-angle spinning with variable-amplitude cross polarization, total sideband suppression, and SPESfAL decoupling at 100kHz.
- the samples were spun at 10.0 kHz, and a total of 10k scans were collected with a recycle delay of 5 seconds. A line broadening of 10 Hz was applied to the spectra before FT was performed.
- Chemical shifts are reported on the TMS scale using the carbonyl carbon of glycine (176.03 p.p.m.) as a secondary reference.
- Figure 2 shows the solid-state carbon-13 CPMAS NMR spectrum for the crystalline free acid anhydrate.
- the crystalline free acid anhydrate exhibited characteristic signals with chemical shift values of 118.7, 17.8, 149.3, and 76.4 p.p.m. Further characteristic of the crystalline free acid anhydrate are the signals with chemical shift values of 115.4, 19.6, 162.7, and 76.0 p.p.m.
- the crystalline free acid anhydrate is even further characterized by signals with chemical shift values of 13.6, 113.3, 173.1, and 38.1 p.p.m.
- DSC data for the crystalline free acid anhydrate were acquired using TA Instruments DSC 2910 or equivalent instrumentation. Between 1 and 6 mg sample was weighed into an open pan. This pan was then placed at the sample position in the calorimeter cell. An empty pan was placed at the reference position. The calorimeter cell was closed and a flow of nitrogen was passed through the cell. The heating program was set to heat the sample at a heating rate of 10 °C/min to a temperature of approximately 200 0 C. The heating program was started. When the run was completed, the data were analyzed using the DSC analysis program contained in the system software. The melting endotherm was integrated between baseline temperature points that are above and below the temperature range over which the endotherm was observed. The data reported are the onset temperature, peak temperature and enthalpy.
- FIG. 3 shows the differential calorimetry scan for the crystalline free acid anhydrate.
- the crystalline free acid anhydrate exhibited an endotherm due to melting and decomposition with an onset temperature of 109.4 0 C, a peak temperature of 113.6°C, and an enthalpy change of 56.8 J/g.
- Thermogravimetric (TG) data were acquired using a Perkin Elmer model TGA 7 or equivalent instrumentation. Experiments were performed under a flow of nitrogen and using a heating rate of 10 °C/min to a maximum temperature of approximately 250 °C. After automatically taring the balance, 1 to 10 mg of sample was added to the platinum pan, the furnace was raised, and the heating program started. Weight/temperature data were collected automatically by the instrument. Analysis of the results was carried out by selecting the Delta Y function within the instrument software and choosing the temperatures between which the weight loss was to be calculated. Weight losses are reported up to the onset of decomposition/evaporation.
- FIG. 4 shows a characteristic thermogravimetric analysis (TGA) curve for the crystalline free acid anhydrate. TGA indicated a weight loss less than 0.1% from ambient temperature to about 109 0 C. CHARACTERIZATION OF THE CRYSTALLINE ANHYDROUS
- the X-ray powder diffraction patterns of the crystalline anhydrous benzenesulfonate salt were generated on a Philips Analytical XTert PRO X-ray Diffraction System with PW3040/60 console.
- a PW3373/00 ceramic Cu LEF X-ray tube K-Alpha radiation was used as the source.
- FIG. 5 shows the X-ray diffraction pattern of the crystalline anhydrous benzenesulfonate salt.
- the crystalline anhydrous benzenesulfonate salt exhibited characteristic reflections corresponding to d- spacings of 13.36, 8.38, and 6.86 angstroms.
- the crystalline anhydrous benzenesulfonate salt was further characterized by reflections corresponding to d-spacings of 9.85, 6.23 and 5.66 angstroms.
- the crystalline anhydrous benzenesulfonate salt was even further characterized by reflections corresponding to d-spacings of 7.23, 6.04 and 5.28 angstroms.
- DSC data of the crystalline anhydrous benzenesulfonate salt were acquired using TA Instruments DSC 2910 or equivalent instrumentation. Between 1 and 5 mg of sample was weighed into an open pan. The lid was placed lightly to cover the sample. The covered pan was then placed at the sample position in the calorimeter cell. An empty pan with lid was placed at the reference position. The calorimeter cell was closed and a flow of nitrogen was passed through the cell. The heating program was set to heat the sample at a heating rate of 10 0 C /min to a temperature of approximately 250 °C. The heating program was then started. When the run was completed, the data were analyzed using the DSC analysis program contained in the system software. The melting endotherm was integrated between baseline temperature points that are above and below the temperature range over which the endotherm was observed. The data reported are the onset temperature, peak temperature and enthalpy.
- FIG. 6 shows the differential calorimetry scan for the crystalline anhydrous benzenesulfonate salt.
- the crystalline anhydrous benzenesulfonate salt exhibited a single endotherm due to melting with an onset temperature of 206.6 0 C, a peak temperature of 208.1 0 C, and an enthalpy change of 95.4 J/g.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- General Chemical & Material Sciences (AREA)
- Emergency Medicine (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Endocrinology (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
A novel crystalline anhydrate of the free acid and a crystalline anhydrous besylate salt of a selective PPAR gamma partial agonist which has a fused bicyclic aromatic group attached to an oxypropanoic acid moiety are stable and non-hygroscopic. The compounds are suitable for preparing pharmaceutical formulations for the treatment of type 2 diabetes, hyperglycemia, obesity, and dyslipidemia.
Description
TITLE OF THE INVENTION
NOVEL CRYSTALLINE FORMS OF ANTIDIABETIC COMPOUNDS
FIELD OF THE INVENTION The present invention relates to novel crystalline forms and salt forms of compounds that are useful as pharmaceutically active ingredients for the treatment of type 2 diabetes and other diseases that are modulated by PPAR gamma agonists, including hyperglycemia, obesity, dyslipidemia, and the metabolic condition. The invention also relates to a process for making the compounds, crystalline forms, and salts.
BACKGROUND OF THE INVENTION
Type 2 diabetes remains a serious medical problem. There is an ongoing need for new treatments that are more effective and that have fewer side effects. PPAR gamma agonists, including the two marketed products rosiglitazone and pioglitazone, are important medications for the treatment of type 2 diabetes. Treatment of a patient with PPAR gamma agonists improves insulin sensitivity, but the treatment is often accompanied by side effects, such as weight gain and edema. Selective PPAR gamma partial agonists, also known as selective PPAR gamma modulators (SPPARM's or SPPARgM's), are effective in reducing serum glucose with reduced weight gain and/or edema.
SUMMARY OF THE INVENTION
The present invention is concerned with novel crystal forms, salts, and crystal forms of the salts of a compound that is an active PPAR gamma partial agonist, and methods of making the compound, salts and crystal forms. The compound was originally disclosed as a solid in US Provisional Application No. 60/658,661, now WO2006/096564, but the solid did not have the crystal form disclosed herein. The crystalline forms disclosed herein are novel and well characterized, and have advantages over the solid forms disclosed in WO2006/096564 that make them useful in preparing pharmaceutical formulations, such as ease of purification, ease of processing, and thermodynamic stability with respect to other forms of the compound. The anhydrous free acid crystalline form is non-hygroscopic, and exhibits good bioavailability in animals, even though it has low water solubility at neutral pH. The invention also concerns pharmaceutical compositions comprising the novel crystalline polymorphs; processes for the preparation of these polymorphic forms and their pharmaceutical compositions; and methods for using them for the treatment of type 2 diabetes, hyperglycemia, obesity, dyslipidemia, and the metabolic condition.
BRIEF DESCRIPTION OF THE FIGURES
FIG. 1 is a characteristic X-ray diffraction pattern of the crystalline anhydrous free acid form.
FIG. 2 is a carbon-13 cross-polarization magic-angle spinning (CPMAS) nuclear magnetic resonance (NMR) spectrum of the crystalline anhydrous free acid form.
FIG. 3 is a typical DSC curve of the crystalline anhydrous free acid form.
FIG. 4 is a typical thermogravimetric (TG) curve of the crystalline anhydrous free acid form.
FIG. 5 is a characteristic X-ray diffraction pattern of the crystalline anhydrous benzenesulfonate
(besylate) salt.
FIG. 6 is a typical DSC curve of the crystalline anhydrous besylate salt.
DETAILED DESCRIPTION OF THE INVENTION
In one embodiment, this invention provides a novel crystalline anhydrous polymorphic form of the free acid of (2S)-2-({6-chloro-3-[6-(4-chlorophenoxy)-2-propylpyridin-3-yl]-l,2- benzisoxazol-5-yl}oxy)propanoic acid (Compound I):

I
This compound was first disclosed as Example 14 in WO 2006/096564. The compound that was isolated using the synthetic methodology in the above-mentioned PCT patent application does not have the crystal form that is disclosed herein. Improvements in the process for making the compound led to the discovery of a crystalline free acid anhydrate of Compound I1 which is described and characterized herein. The invention also provides a benzenesulfonate (besylate) salt of Compound I, and more specifically, an anhydrous crystalline benzenesulfonic acid (besylate) salt of Compound I. The besylate salt, and specifically the anhydrous crystalline besylate salt, has advantageous properties compared with the non-crystalline free acid and amorphous sodium salts of Compound I that were originally made. The two crystalline compounds (crystalline free acid anhydrate and anhydrous crystalline besylate salt) are readily used in the preparation of pharmaceutical compositions.
The benzenesulfonic acid (besylate) salt of Compound I is also a new composition of matter. This is generally referred to herein as the benzenesulfonic acid (besylate) salt of Compound I, but it can also be written as a chemical compound having Formula Ia:

The compositions, drug substances, formulations, and pharmaceutical uses that are described herein for the crystalline anhydrous besylate salt are also representative of compositions, drug substances, formulations, and pharmaceutical uses of the besylate salt in general.
A further embodiment of the present invention provides a drug substance that comprises the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of Compound I in a detectable amount. By "drug substance" is meant the active pharmaceutical ingredient (API). The amount of crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt in the drug substance can be quantified by the use of physical methods such as X-ray powder diffraction (XRPD), solid-state fluorine-19 magic-angle spinning (MAS) nuclear magnetic resonance spectroscopy, solid-state carbon-13 cross-polarization magic-angle spinning (CPMAS) nuclear magnetic resonance spectroscopy, solid state Fourier-transform infrared spectroscopy, and Raman spectroscopy. In a sub-class of this embodiment, about 5% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance. In a second sub-class of this embodiment, about 10% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance. In a third sub-class of this embodiment, about 25% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance. In a fourth sub-class of this embodiment, about 50% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance. In a fifth sub-class of this embodiment, about 75% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt is present in the drug substance. In a sixth sub-class of this embodiment, substantially all of the Compound I drug substance is the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt, i.e., the Compound I drug substance is the substantially phase pure crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt.
Another aspect of the present invention provides a method for the treatment or control of clinical conditions for which a PPAR gamma agonist is indicated, which method comprises administering to a patient in need of such treatment or control a therapeutically effective amount of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of Compound I or a pharmaceutical composition containing a therapeutically effective amount of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of Compound I. Such clinical conditions include Type 2
diabetes, hyperglycemia, obesity, dyslipidemia, and metabolic syndrome. A "patient" is a mammal, including a human. A patient is most often a human patient.
The present invention also provides for the use of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of the present invention in the manufacture of a medicament for the treatment or control in a patient of one or more clinical conditions for which a PPAR gamma agonist is indicated. In one embodiment, the clinical condition is Type 2 diabetes.
Another aspect of the present invention provides the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt for use in the treatment or control in a patient of one or more clinical conditions for which a PPAR gamma agonist is indicated. In one embodiment of this aspect the clinical condition is Type 2 diabetes.
The present invention also provides pharmaceutical compositions comprising the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt, in association with one or more pharmaceutically acceptable carriers or excipients. In one embodiment the pharmaceutical composition comprises the active pharmaceutical ingredient (API) in admixture with pharmaceutically acceptable excipients wherein the API comprises a detectable amount of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of the present invention. In a sub-class of this embodiment the pharmaceutical composition comprises the API in admixture with pharmaceutically acceptable excipients wherein the API comprises about 5% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of the present invention. In a sub- class of this second embodiment, the API in such compositions comprises about 10% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt. In a subclass of this embodiment, the API in such compositions comprises about 25% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt. In a sub-class of this embodiment, the API in such compositions comprises about 50% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt. In a sub-class of this embodiment, the API in such compositions comprises about 75% to about 100% by weight of the crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt. In a sub-class of this embodiment, substantially all of the API is crystalline free acid anhydrate or crystalline anhydrous benzenesulfonate salt of Compound I, i.e., the API is substantially phase pure Compound I in the crystalline free acid anhydrate form or substantially phase pure Compound I in the form of a crystalline anhydrous benzenesulfonate salt.
The compositions in accordance with the invention are suitably in unit dosage forms such as tablets, pills, capsules, powders, granules, sterile solutions or suspensions, metered aerosol or liquid sprays, drops, ampoules, auto-injector devices or suppositories. The compositions are intended for oral, parenteral, intranasal, sublingual, or rectal administration, or for administration by inhalation or insufflation. Formulation of the compositions according to the invention can conveniently be effected by
methods known in the art, for example, as described in Remington's Pharmaceutical Sciences. 17* ed., 1995.
The dosage regimen is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the route of administration; and the renal and hepatic function of the patient. An ordinarily skilled physician, veterinarian, or clinician can readily determine and prescribe the effective amount of the drug required to prevent, counter or arrest the progress of the condition or to treat or control the condition.
Oral administration is the preferred method of administering the crystal forms and salt forms of Compound I described herein. The drug can be administered 1-2 times per day, with once daily being preferred. The daily dosage for an adult human patient is generally 1-25 mg, and preferably 2-10 mg administered once daily.
In the methods of the present invention, the Compound I crystalline free acid anhydrate and the crystalline anhydrous benzenesulfonate salt described herein in detail can form the API, and are typically administered in admixture with suitable pharmaceutical diluents, excipients or carriers (collectively referred to herein as 'carrier' materials) suitably selected with respect to the intended form of administration, that is, oral tablets, capsules, elixirs, syrups and the like, and consistent with conventional pharmaceutical practices.
For instance, for oral administration in the form of a tablet or capsule, the active pharmaceutical ingredient can be combined with an oral, non-toxic, pharmaceutically acceptable, inert carrier such as lactose, starch, sucrose, glucose, methyl cellulose, microcrystalline cellulose, magnesium stearate, calcium sulfate, mannitol, sorbitol and the like; for oral administration in liquid form, the oral API can be combined with any oral, non-toxic, pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like. Moreover, when desired or necessary, suitable binders, lubricants, disintegrating agents and coloring agents can also be incorporated into the mixture. Suitable binders include starch, gelatin, some natural sugars, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like. Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, and the like. Disintegrants include, without limitation, starch, methyl cellulose, croscarmellose sodium, agar, bentonite, xanthan gum and the like. Surfactants, such as sodium lauryl sulfate, can also be included in the formulations.
The following non-limiting Examples are intended to illustrate the present invention and should not be construed as being limitations on the scope or spirit of the instant invention.
EXAMPLE 1 Synthesis of (2SV2-(f6-chloro-3-r6-C4-chlorophenoxyV2-propylpyridin-3-vn-lπ2-ben2isoxazol-5- yUoxy)propanoic acid (Compound I)
Compound I is made by the multi-step process shown in Schemes 1 and 2 below. The process is described in detail in the description after the schemes. Compound I is (S)-14 in the schemes and description below.
Synthesis of Hy droxybenzisoxazole Intermediate 10
Cl

10
Synthesis of Chiral Acid 14
TsCI, Et3N, DABCO
HO CO2Me
TsO CO2Me (R)-11 12

Steps 1 and 2. Esterifϊcation and aryl ether formation

The reaction mixture was allowed to cool to RT and then poured into a biphasic mixture containing IPAc (220 mL) and aq. K2CO3 (20.7 g in 117.3 g water) at RT with stirring. The organic layer was separated, washed with sat. NaHCO3 (80 mL), and then water (80 mL). The isolated IPAc solution was subjected to a solvent switch to DMF (80 mL) in vacuo.
A solution of 4-chloroρhenol (12.2 g, 0.095 mol) in 36.6 mL of DMF was added at room temperature to the above solution (19.6 g of ester 2, 0.095 mol), followed by addition of triethylamine (17.3 mL, 0.124 mol) at 20-22 0C over 15 min. Solid DABCO (1.6 g, 14.2 mmol) was added to the resulting solution in one portion. A temperature increase of ~3 0C was observed. A water bath was used to maintain the reaction temperature. The reaction was stirred at 22-24 0C for 4-5 h while monitoring by LC until all of the 4-chlorophenol was consumed, resulting in a light slurry. AcOH (2.72 mL, 47.5 mmol) and EPA (57.5 mL) were added to the light slurry, followed by cold water (30 mL) to maintain the internal temperature at 20-25 0C. When the water was added, a clear solution first formed, and then a
slurry of product formed. After stirring at RT for 0.5 h, additional water (86 mL) was added over 0.5 h. After the slurry was stirred at RT for 1-2 h, it was filtered. The filter cake was washed with mixed solvents (60 mL OfIPAiH2O = 1:1). The isolated solid was dried in a vacuum-oven at 50 0C for 8 h to provide the product as white cotton-like solid.
Step 3. Propylation

To a solution of methyl 2-chloro-6-(4-chlorophenoxy)nicotinate (12.53 g, 42.03 mmol) and NiCl2dppe (11 I.mg, 0.5 mol%) in THF (63 mL) was added n-PrMgCl (2.0 M in diethyl ether, 22.5 mL, 45.0 mmol) over Vi h. The reaction was aged at 25 0C to 28 0C for 15 minutes.
The reaction was then quenched with 10% citric acid solution (120 mL) and diluted with MTBE (120 mL). The mixture was stirred over 15 min. The organic layer was cut and was washed with 10% NaCl solution (120 mL). The organic layer (188 mL) was concentrated to 90 mL (1/2 volume), and 90 mL of MeOH was then added. The volume was again reduced to 90 mL by vacuum distillation. This was repeated 2 additional times to complete the solvent switch to MeOH. The final volume was about 90 mL.
Step 4. Methyl Ester Hydrolysis

To the solution of 4 from above was added 5N NaOH (13 mL, 65 mmol). The mixture was heated to 68 0C for 2.5 h. LC assay showed the reaction was complete. The reaction can also be run at 50 0C, in which case it is typically complete in 4 h. Water (90 mL) was then added to the solution at 68 0C, followed by 36 mL of 20% citric acid. The product crystallized from the solution. Water (90 mL) was then added. The slurry was stirred for 2h and was then filtered. The white cake was washed with 150 mL of water/MeOH (2:1) and was dried in an oven at 620C overnight.
Step 5. Friedel-Crafts Acylation

To a 100 L round bottom vessel was charged nicotinic acid 5 (7200 g, 24.68 MoI), which was then dissolved in 17 L of trifluoroacetic anhydride (TFAA). l,4-Dimethoxy-2-chlorobenzene (6337 mL, 44.42 MoI) was added, followed by slow addition of triflic acid (4426 mL, 2 equivalents), while maintaining the temperature at <40 0C. A reflux condenser was attached, and the reaction was heated to 42 0C and stirred overnight. The reaction was assayed, showing a 70 % conversion by mass of 5 to 7.
An additional triflic acid charge (440 mL, 0.20 equivalents) was made, and a distillation setup was substituted for the reflux condenser. The batch was heated to 55 0C, and ~9 L of TFAA was distilled into an ice cooled 22 L RBF. The batch was aged at 55 0C for 4 hours. At this point the reaction had reached completion.
The reaction was cooled to ambient temperature with an ice bath, and was then quenched into a 100 L extractor at 0 0C onto 30 L (6 molar equivalents) of 5 N KOH and 25 L (3.5 volumes) of toluene, maintaining the temperature at <50 0C for 1 hour. The 100 L flask was rinsed into the extractor with 2 x 2 L of toluene and 2 x 2 L of 5N KOH. The phases were separated at room temperature, and the organic phase was washed with 18 L of IN HCl.
The organic solution was transferred back into the rinsed 100 L vessel and was treated with Darco G-60 (3.6 kg, 50 wt %). The mixture of solution and carbon was heated at 35 0C for 30 min. The charcoal mixture was then filtered through a pad of solka floe, rinsed with 8 L of toluene and vacuum transferred through a 5 uM poly cap, into a visually clean 100 L round bottom flask, with a mark at the 16 L level. The 100 L flask was attached to a batch concentrator and distilled down to the 16 L mark at 35 0C. At this point the batch was seeded with 1O g of seed crystals of 7 obtained from an earlier batch, and heptane addition began. After 20 L of heptane had been added the slurry grew thick. The batch was heated to 55 0C, and an additional 4 L of heptane was added bringing the total batch volume to the 40 L mark. The slurry was aged at 55 0C for 15 minutes with rapid stirring. At this point a constant volume distillation with the addition of heptane was begun, and the batch temperature was cooled and then was maintained between 30 and 35 0C. A total of 80 L of heptane (including the original 24 L) was added to the batch. The solvent composition was checked by 1H NMR, and was found to contain 94 mole % heptane. The slurry was then heated to 65 0C and allowed to slowly cool to room temperature overnight.
The slurry was filtered, and the flask was rinsed with 9 L of a mixture of 95 % heptane / 5 % toluene. The cake was then slurry washed with 9L of 95 % heptane / 5 % toluene, and then 18 L heptane. The product 7 was dried on the frit under a stream of N2 at ambient temperature.
Step 6. Demethylation of 7 to 8

Into a visually clean 200 mL two-neckRBF was charged 1 1.1 g of solid 93.5 wt% dimethoxyketone 7 (25 mmol), 18.75 g sodium iodide (125 mmol), HBr (48% aqueous, 50 mL, 0.5 mol), and HOAc (50 mL, 5x vol). The slurry was heated to 100 0C (dial -in temp.) in 0.5 hours, and the internal temperature gradually stabilized at 95-95.5 0C.
The slurry turned dark brown within two hours after the reaction temperature reached 90 0C. Further heating for one hour gradually generated bright yellow crystals, and the precipitate became thicker with time. The reaction was stirred at 95-95.5 0C (Internal T) for 24 hours.
The batch was cooled to room temperature, filtered, and sequentially washed with 50 mL HOAc (displacement wash), 50 mL HOAc (slurry wash) and 5% MeOH in water (3x 50 mL, slurry washes). The isolated product was dried at r.t. under vacuum over the weekend.
The dry powder product was then suspended in 5% MeOH in water (100 mL) for 4 hours and filtered. The filter cake was washed with 50 mL of water and dried under vacuum to give the final product as the free base.
Step 7. Oxime Formation and Isomerization
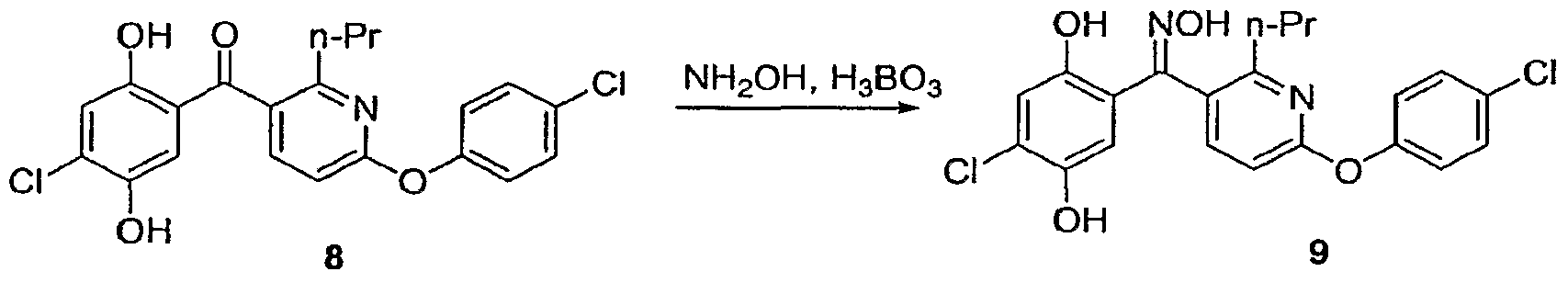
To a 100 L, 4-neck round bottom flask, with mechanical stirrer, reflux condenser, thermocouple and nitrogen/vacuum line, was charged n-propanol (24 L), dihydroquinone ketone (7.598 kg, 89% purity, 6.762 assay kg, 12.38 mol), and boric acid (808 g, 13.07 mol). Hydroxylamine (2.3 L, 37.60 mol) was then poured into the flask. The reaction was heated to reflux (90-92 0C) for 60 minutes.
The reaction was cooled to 30 0C and transferred into a 180-L extractor containing 35 L of water. 15 L of water and 50 L of MTBE were added to the extractor and the mixture was vigorously stirred and allowed to settle The bottom aqueous layer was cut. The organic layer was washed with 50 L of 20 wt% NaCl (aq), and then with 18 L of 20 wt% NaCl (aq) The organic layer was agitated with 3 kg of sodium sulfate and 1 kg of DARCO G-60 and filtered through a bed of Solkailok. The cake bed was πnsed with 15 L of MTBE The filtrate was concentrated to approximately 20 L at 35-40 0C, 20-25 in Hg. n-Propanol (60 L) was fed and distilled at 35-40 0C, 28-30 m Hg, while maintaining a constant volume of 20 L The final batch KF was 860 ppm water The resulting solution was heated on a steam pot to 93-97 0C The reaction was monitored for isomeπzation conversion After 6 hours, the batch was allowed to cool to ambient temperature. 200 mL of the batch was sampled for seed formation. To the stirring solution, 50 mL of water was added, and then 1 g of seed was added, and the batch was aged to form a seed bed. The remaining 250 mL of water was added to complete the crystallization To the batch, 5 L of water was added, followed by the seed slurry. The mixture was aged, giving a thick slurry. The remaining 25 L of water was added over 1 hour. The slurry was heated to 50 0C and cooled to ambient temperature.
The solid was isolated by filtration. The cake was washed with 2:1 water/n-propanol (8L3 8L, 12L, 12L), water (8L), then hexanes (12L, 8L). The solid was dπed on the filter under a nitrogen tent The E-oxime was obtained as an orange solid.
Step 8 Benzisoxazole formation

The reaction was then warmed to 25 0C. MeOH (1 3 L) was added, and the solution was aged for 1 hour
To the reaction, 35 L of MTBE, 20 L of water, and 2 5 L of 85% phosphoπc acid were added with vigorous stirring. After settling, the bottom aqueous layer was cut The organic layer was washed with water (20 L), 0.5 M Na2CO3 (2 x 20 L), IM H3PO4 (20 L), then 10 wt% KH2PO4 (4L).
The batch was stirred with 1 kg of DARCO G-60 for 1.5 hours. The mixture was filtered through Solkaflok and the bed was washed with 14 L of MTBE.
The filtrate was fed into a 100 L round bottom flask equipped with mechanical stirrer, thermocouple, and nitrogen inlet, and was attached to a batch concentrator. The batch was fed and distilled at 35-40 0C, 16-20 in. Hg, maintaining the batch volume at 20 - 25 L. EtOAc (40 L) was then fed and distilled at 35-40 0C, 20-23 in. Hg at a constant volume of 15-20 L.
To a 100 L cylindrical vessel with heating coils were charged EtOAc (20 L) and TsOH/H2O (2.304 kg, 12.11 mol), and the mixture was heated to 35-45 0C to dissolve. The acid solution was fed into the isoxazole batch with further distilling, maintaining a constant volume of 25 L. An additional 20 L of EtOAc was distilled to azeotropically dry the mixture. A slurry began to form, and it continued to thicken on addition and concentration. The final KF was 400 ppm water. The batch was heated to 60 0C and allowed to slowly cool to ambient temperature overnight.
The solid product was isolated by filtration. The cake was washed with EtOAc (16L), then with MeCN (24 L), and was dried on the filter under a nitrogen tent. The benzisoxazole tosylate was obtained as a pale yellow solid.
Step 9A. Lactate Tosylate Formation OMe

To a 50 L RBF was added 1.50 kg R-methyl lactate, which was then dissolved in EtOAc
(7.5 L) with 3.02 kg tosyl chloride. The batch was cooled with ice to 6 0C. A mild endotherm was noted on mixing.
'DABCO (242 g) and triethylamine (3.01 L) were separately dissolved in the 7.5 L of EtOAc. The solution was charged to a 50 L vessel, maintaining the temperature below 25 0C. The reaction was aged 2 h at room temperature. A mild to moderate delayed exotherm was seen. A white slurry formed during the addition.
To a 50 L extractor 4 L of water and 3 L of EtOAc were added with stirring. Water (3.5 L) was added to the reaction vessel, and the biphasic solution was transferred to the extractor. The vessel was then rinsed with 4.5 L EtOAc. To the stirred extraction was added 7.5 L of 2 N HCl, bringing the total extraction volume to 40 L. The extraction was aged 10 min and phase separated. The organic was washed with 7.5 L of water and then 15 L of 4 % NaHCO3 (aq). The organic solution was then transferred to clean plastic carboys, and dried over Na2SO4 (5 kg) in the carboys.
The batch was then filtered through a 20 uM poly cap filter into a Buchi rotary evaporator, yielding the product as an oil containing residual ethyl acetate (3 wt%) and 700 PPM water.
The batch was transferred to a container and was stored in a cold room until it was used. The product had an ee of 98.2 %.
Step 9. Methyl Lactate Attachment

To a 100 L RBF was added benzisoxazole tosylate 10 (5.7kg, 10 moles), then K2CO3 powder (5.7 kg, 42 moles), and then 25 L DMSO. A slight exotherm was noted. The reaction was stirred for 10 min, and the mixture was degassed and placed under N2. The slurry was cooled to <30 0C, and the lactate tosylate 12 (2.8 kg, 11 moles) was added. The mixture was stirred for 1-A hrs until HPLC showed >98% conversion. To the reaction was added 20 L MTBE and 30 L cold water. The cold water was added to moderate the slight exotherm on quenching. The layers were agitated for 10 min.
The mixture was transferred to a 180 L cyclindrical vessel, and an additional 30L MTBE and 30 L cold water were added. The layers were cut and the aqueous layer was back extracted with 25 L MTBE. The combined organic layers were washed with 18 L 2% NaHCO3. The final organic layer was fed with concurrent distillation into a 100 L RBF and solvent switched to acetonitrile. The batch was kept at 25-30 0C to prevent crystallization.
The batch volume was adjusted to 45 L with acetonitile, and 36 L water was added slowly (product crystallizes after 4 L water is added). After overnight aging, the batch was filtered, and the cake was washed with 10 L 1/1 MeCN/water. Solid methyl ester 5-13 on the funnel was dried with suction under nitrogen flow for 4 days.
Step 10. Hydrolysis and Final Crystallization
In a 50 L cyclindrical vessel, the methyl ester S-XS (2.3 kg) was dissolved in 12.5 L MeCN and mixed with 10 L IN NaOH. The solution was aged for 2-3 hrs at ambient temperature.
Toluene (25 L) was added, followed by cone. HCl to bring the pH to 2-3 (0.85 L). The resulting layers were separated. The organic layer was washed with 15 L brine and dried with Na2SO4 and 0.7 kg Ecorsorb C-933. The slurry was filtered and the cake was washed with 10 L toluene. In a IOOL RBF, the filtrate was batch concentrated to 15 L.
The batch volume was then adjusted to 18 L (8 L toluene/kg product). The batch was heated to 50 0C, and 56 L of methylcyclohexane was added at 50 0C. The batch was seeded with crystals from earlier batches after 18 L of methylcyclohexane was added. The batch was cooled slowly to ambient temparature (about 10 min per degree) to yield crystalline product 5-14. The batch became thick at around 39 0C. The batch was cooled further to ambient temperature over 4-8 hrs. It was aged a total of 16 hrs.
The batch was filtered, and the cake was washed with 10 L of 4:1 methylcyclohexane/toluene, then 2 x 10 L of methylcyclohexane. It was dried on the filter pot under vacuum and nitrogen flow overnight, and was then transferred to a vacuum oven and dried with nitrogen flow overnight.
CRYSTALLINE FREE ACID ANHYDRATE
The crystals isolated using the method described above are the preferred anhydrous crystalline free acid crystals. The crystals are anhydrous. They have very low water solubility at native pH, are stable with respect to retaining their crystal form, are chemically stable, and are non-hygroscopic. For example, they gain about 0.2 wt% when placed in an environment with up to 95% RH. Their melting point is 113-114 0C. They have a small needle morphology and a high surface area without milling or grinding. They exhibit good bioavailability in laboratory studies in dogs and rats. The preferred crystalline anhydrate is obtained on crystallization from MTBE/hexanes or toluene/methylcyclohexane. Crystallization from toluene/methylcyclohexane is used in the synthetic procedure described above.
CRYSTALLINE BENZENESULFONATE SALT
The benzenesulfonate salt of the compound having Formula I as described herein is crystalline and non-hygroscopic. The benzenesulfonate salt is chemically stable, remaining unchanged after 8 months at 40 0C and 75% RH. The benzenesulfonate salt has properties that make it suitable in pharmaceutical formulations. The salt has been made by the following procedure.
A solution of benzenesulfonic acid (1.58 g, 10 mmol) in 10 ml acetonitrile was added to a solution of Compound I (4.87 g, 10 mmol) in 50 ml acetonitrile at 50 0C. The reaction mixture was seeded at 40 0C with crystals of the Compound I benzenesulfonate salt from earlier batches, yielding a crystalline product. The same crystalline product can also be obtained without seeds if none are available.
The mixture was cooled to room temperature and then was stirred for 2.5 hours. It was cooled to 0-5 0C and stirred for an additional 30 min. The solid was collected by filtration, and the cake was washed with 10 ml acetonitrile. The solid was dried on the funnel with suction, yielding 6.4 g (99% yield).
CRYSTALLINE ANHYDROUS TOSYLATE SALT
A toluenesulfonate (tosylate) salt of Compound I has also been prepared. The tosylate salt also can be prepared as a crystalline anhydrous material. The crystalline anhydrous tosylate salt of Compound I was prepared by the following method from the methyl ester of Compound I. MeCN (110 kg) was charged to a reactor. The methyl ester of Compound I (e.g. from step 9 of Example 1; 29.9 kg; 59.6 moles) was charged to the reactor, followed by a MeOH flush of the charge valve. 135 kg of 1.0N NaOH (~131 moles) was added, followed by a water flush at 15-25 0C. The solution was aged for 2-3 hours at 15-25 0C and then assayed for completion of the reaction.
Concentrated 5N HCl (26.7 kg) was added using a pump to adjust the pH to 2-3. The solution was extracted with 295 kg ethyl acetate. The organic layer was separated from the aqueous layer and washed with 215 kg of 18% brine solution.
The batch was filtered via a 0.6 micron filter and concentrated to 200-220 L at <40 0C and reduced pressure. The solvent was switched at constant volume to EtOAc at <40 0C and reduced pressure (~125 to 252 mmHg). The water concentration by Karl Fischer titration was 72.3 μg/ml, the product concentration was 135.5 g/L, and the acetonitrile content was 0.36 v/v%. The batch was collected in drums.
A solution of p-toluenesulfonic acid monohydrate (12 kgs; 62 moles) in ethyl acetate (135 kgs) was prepared and was also collected in drums.
A charge of 60 kg EtOAc was added to the crystallizer through a 0.6 micron filter. A seed slurry (about 12.9 kg containing about 1 kg of media-milled tosylate seed in about 10 L ethyl acetate) was added to the reactor followed by about 10 kg of a pre-fϊltered EtOAc wash. The seed slurry was recycled from the bottom of the reactor through the outlet and back in through the inlet. Then, the batch of Compound I in EtOAc and the solution of p-toluenesulfonic acid (p-TSA) in EtOAc solution were charged simultaneously into the reactor over a period of about 8 hours. The charge rates for the concentrated batch and p-TSA/EtOAc solution were 0.3 kg/L and 0.4 kg/L respectively. The temperature was maintained at 15 to 25 0C. After crystallization the batch was aged at 15 to 25 0C for 2 hours.
Seeds for the crystallization step above are saved from earlier batches of Compound I tosylate. The same crystalline product can also be obtained without seed crystals if none are available.
The batch was filtered and the cake was washed with a total of ~240 kg ethyl acetate. The batch was dried under vacuum at 40 0C, yielding about 35.8 kg of the desired tosylate salt, for a yield of 90.5% for the salt preparation. The dried batch was delumped prior to further use.
DOSAGE FORM
The crystalline free acid anhydrate and the crystalline anhydrous benzenesulfonate salt of Compound I are formulated as either dry filled capsules or compressed tablets in doses that generally will range from 1 mg to 25 mg of API as the free acid (non-salt). Typically, the doses will be in the range of 2-10 mg. A typical capsule or tablet formulation contains the crystalline free acid anhydrate or the crystalline anhydrous benzenesulfonate salt, microcrystalline cellulose (Avicel), lactose
monohydrate, croscarmellose sodium, sodium lauryl sulfate, and magnesium stearate. The capsule formulations are transferred to a capsule made of gelatin, titanium dioxide, and ferric oxide. Tablet formulations are coated with a functional film coat containing lactose, hypromellose, triacetin, titanium dioxide, and ferric oxide. The capsule shell and tablet film coating are opaque to protect the active compound from light.
The formulations are manufactured by first blending the excipients, then compressing the mixture into ribbons by roller compaction, and then milling the ribbons into granules. The granules are then lubricated and either filled into capsules or compressed into tablets. If tablets are selected, a film coat is applied to the compressed tablets. Exemplary fill formulations that provide a 5 mg or 10 mg dose of Compound I (free acid) in a standard gelatin capsule are shown below. The components are combined, compressed and milled as described above, and then the amount of milled formulation that contains the 5 mg or 10 mg dose of Compound I is transferred to each capsule.
Components 5 me Dose 10 mε Dose
Compound I (free acid weight) 5 mg 10 mg Microcrystalline cellulose (Avicel) 44.5 mg 42 mg Lactose monohydrate (Diluent) 44.5 mg 42 mg Croscarmellose sodium (Disintegrant) 3 mg 3 mg Sodium lauryl sulfate (surfactant) 2 mg 2 τng Magnesium stearate (lubricant) 1 mg 1 mg
CHARACTERIZATION OF THE CRYSTALLINE FREE ACID ANHYDRATE X-ray powder diffraction studies are widely used to characterize molecular structures, crystallinity, and polymorphism. The X-ray powder diffraction patterns of the crystalline anhydrous free acid form of Compound I were generated on a Philips Analytical X'Pert PRO X-ray Diffraction System with PW3040/60 console. A PW3373/00 ceramic Cu LEF X-ray tube K-Alpha radiation was used as the source. Silicon powder (NIST reference standard 640C) was mixed in the sample and was used as a reference for d-spacing assignment.
FIG. 1 shows the X-ray diffraction pattern for the crystalline free acid anhydrate. The crystalline free acid anhydrate exhibited characteristic reflections corresponding to d-spacings of 17.13, 5.11, and 4.82 angstroms. The crystalline free acid anhydrate was further characterized by reflections corresponding to d-spacings of 11.63, 7.88 and 7.42 angstroms. The crystalline free acid anhydrate was even further characterized by reflections corresponding to d-spacings of 10.27, 4.64 and 4.01 angstroms.
In addition to the X-ray powder diffraction patterns described above, the crystalline free acid anhydrate of Compound I was further characterized by solid-state carbon-13 nuclear magnetic resonance
(NMR) spectra. The solid-state carbon- 13 NMR spectra were obtained on a Bruker DSX 500WB NMR system using a Bruker 4 mm H/X/Y CPMAS probe. The carbon-13 NMR spectra utilized proton/carbon- 13 cross-polarization magic-angle spinning with variable-amplitude cross polarization, total sideband suppression, and SPESfAL decoupling at 100kHz. The samples were spun at 10.0 kHz, and a total of 10k scans were collected with a recycle delay of 5 seconds. A line broadening of 10 Hz was applied to the spectra before FT was performed. Chemical shifts are reported on the TMS scale using the carbonyl carbon of glycine (176.03 p.p.m.) as a secondary reference.
Figure 2 shows the solid-state carbon-13 CPMAS NMR spectrum for the crystalline free acid anhydrate. The crystalline free acid anhydrate exhibited characteristic signals with chemical shift values of 118.7, 17.8, 149.3, and 76.4 p.p.m. Further characteristic of the crystalline free acid anhydrate are the signals with chemical shift values of 115.4, 19.6, 162.7, and 76.0 p.p.m. The crystalline free acid anhydrate is even further characterized by signals with chemical shift values of 13.6, 113.3, 173.1, and 38.1 p.p.m.
DSC data for the crystalline free acid anhydrate were acquired using TA Instruments DSC 2910 or equivalent instrumentation. Between 1 and 6 mg sample was weighed into an open pan. This pan was then placed at the sample position in the calorimeter cell. An empty pan was placed at the reference position. The calorimeter cell was closed and a flow of nitrogen was passed through the cell. The heating program was set to heat the sample at a heating rate of 10 °C/min to a temperature of approximately 2000C. The heating program was started. When the run was completed, the data were analyzed using the DSC analysis program contained in the system software. The melting endotherm was integrated between baseline temperature points that are above and below the temperature range over which the endotherm was observed. The data reported are the onset temperature, peak temperature and enthalpy.
FIG. 3 shows the differential calorimetry scan for the crystalline free acid anhydrate. The crystalline free acid anhydrate exhibited an endotherm due to melting and decomposition with an onset temperature of 109.40C, a peak temperature of 113.6°C, and an enthalpy change of 56.8 J/g.
Thermogravimetric (TG) data were acquired using a Perkin Elmer model TGA 7 or equivalent instrumentation. Experiments were performed under a flow of nitrogen and using a heating rate of 10 °C/min to a maximum temperature of approximately 250 °C. After automatically taring the balance, 1 to 10 mg of sample was added to the platinum pan, the furnace was raised, and the heating program started. Weight/temperature data were collected automatically by the instrument. Analysis of the results was carried out by selecting the Delta Y function within the instrument software and choosing the temperatures between which the weight loss was to be calculated. Weight losses are reported up to the onset of decomposition/evaporation. FIG. 4 shows a characteristic thermogravimetric analysis (TGA) curve for the crystalline free acid anhydrate. TGA indicated a weight loss less than 0.1% from ambient temperature to about 109 0C.
CHARACTERIZATION OF THE CRYSTALLINE ANHYDROUS
BENZENESULFONATE SALT
The X-ray powder diffraction patterns of the crystalline anhydrous benzenesulfonate salt were generated on a Philips Analytical XTert PRO X-ray Diffraction System with PW3040/60 console. A PW3373/00 ceramic Cu LEF X-ray tube K-Alpha radiation was used as the source.
FIG. 5 shows the X-ray diffraction pattern of the crystalline anhydrous benzenesulfonate salt. The crystalline anhydrous benzenesulfonate salt exhibited characteristic reflections corresponding to d- spacings of 13.36, 8.38, and 6.86 angstroms. The crystalline anhydrous benzenesulfonate salt was further characterized by reflections corresponding to d-spacings of 9.85, 6.23 and 5.66 angstroms. The crystalline anhydrous benzenesulfonate salt was even further characterized by reflections corresponding to d-spacings of 7.23, 6.04 and 5.28 angstroms.
DSC data of the crystalline anhydrous benzenesulfonate salt were acquired using TA Instruments DSC 2910 or equivalent instrumentation. Between 1 and 5 mg of sample was weighed into an open pan. The lid was placed lightly to cover the sample. The covered pan was then placed at the sample position in the calorimeter cell. An empty pan with lid was placed at the reference position. The calorimeter cell was closed and a flow of nitrogen was passed through the cell. The heating program was set to heat the sample at a heating rate of 10 0C /min to a temperature of approximately 250 °C. The heating program was then started. When the run was completed, the data were analyzed using the DSC analysis program contained in the system software. The melting endotherm was integrated between baseline temperature points that are above and below the temperature range over which the endotherm was observed. The data reported are the onset temperature, peak temperature and enthalpy.
FIG. 6 shows the differential calorimetry scan for the crystalline anhydrous benzenesulfonate salt. The crystalline anhydrous benzenesulfonate salt exhibited a single endotherm due to melting with an onset temperature of 206.60C, a peak temperature of 208.10C, and an enthalpy change of 95.4 J/g.
Claims
1. The compound (2S)-2-({6-chloro-3-[6-(4-chlorophenoxy)-2-propylpyridin-3-yl]-l,2- benzisoxazol-5-yl}oxy)propanoic acid having formula I:

characterized as being a crystalline anhydrous free acid or a crystalline anhydrous benzenesulfonate salt.
2. The compound of Claim 1 characterized as being a crystalline anhydrous free acid.
3. The compound of Claim 1 characterized as being a crystalline anhydrous free acid having one or more spectral characteristics selected from the X-ray powder diffraction pattern, solid-state carbon-13 CPMAS nuclear magnetic resonance spectrum, a differential scanning calorimetric (DSC) curve, and a thermogravimetric analyis (TGA) curve.
4. The compound of Claim 1 characterized as being a crystalline anhydrous free acid having X-ray powder diffraction peaks corresponding to d-spacings of 17.13, 5.11, and 4.82 angstroms.
5. The compound of Claim 1 characterized as being a crystalline anhydrous free acid having X-ray powder diffraction peaks corresponding to d-spacings of 11.63, 7.88 and 7.42 angstroms.
6. The compound of Claim 1 characterized as being a crystalline anhydrous free acid having X-ray powder diffraction peaks corresponding to d-spacings of 10.27, 4.64 and 4.01 angstroms.
7. The compound of Claim 1 characterized as being a crystalline anhydrous free acid having peaks in the solid-state carbon-13 CPMAS nuclear magnetic resonance spectrum having chemical shift values of 118.7, 17.8, 149.3, and 76.4 p.p.m.
8. The compound of Claim 1 characterized as being a crystalline anhydrous free acid having peaks in the solid-state carbon-13 CPMAS nuclear magnetic resonance spectrum having chemical shift values of 115.4, 19.6, 162.7, and 76.0 p.p.m.
9. The compound of Claim 1 characterized as being a crystalline anhydrous free acid having peaks in the solid-state carbon-13 CPMAS nuclear magnetic resonance spectrum having chemical shift values of 13.6, 113.3, 173.1, and 38.1 p.p.m.
10. The compound of Claim 1 characterized as being a crystalline anhydrous free acid having an endotherm in the differential calorimetry scan with an onset temperature of 109.40C and a peak temperature of 113.60C.
1 1. The compound of Claim 1 characterized as being a crystalline anhydrous benzenesulfonate salt.
12. The compound of Claim 1 characterized as being a crystalline anhydrous benzenesulfonate salt having one oτ more spectral characteristics selected from the X-ray powder diffraction pattern and a differential scanning calorimetric (DSC) curve.
13. The compound of Claim 1 characterized as being a crystalline anhydrous benzenesulfonate salt having X-ray powder diffraction peaks corresponding to d-spacings of 13.36, 8.38, and 6.86 angstroms.
14. The compound of Claim 1 characterized as being a crystalline anhydrous benzenesulfonate salt having X-ray powder diffraction peaks corresponding to d-spacings of 9.85, 6.23 and 5.66 angstroms.
15. The compound of Claim 1 characterized as being a crystalline anhydrous benzenesulfonate salt having X-ray powder diffraction peaks corresponding to d-spacings of 7.23, 6.04 and 5.28 angstroms.
16. The compound of Claim 1 characterized as being a crystalline anhydrous benzenesulfonate salt having an endotherm in the differential calorimetry scan with an onset temperature of 206.60C and a peak temperature of 208.10C.
17. A method of synthesizing the crystalline anhydrous free acid form of the compound of Claim 1, comprising the crystallization of the free acid from a solution comprising the free acid, toluene and methylcyclohexane.
18. A pharmaceutical composition comprising a therapeutically effective amount of the crystalline anhydrous free acid form of the compound of Claim 1 in association with one or more pharmaceutically acceptable carriers or excipients.
19. A pharmaceutical composition comprising a therapeutically effective amount of the crystalline anhydrous benzenesulfonate salt of the compound of Claim 1 in association with one or more pharmaceutically acceptable carriers or excipients.
20. The benzenesulfonic acid salt of (2S)-2-({6-chloro-3-[6-(4-chlorophenoxy)-2- propylpyridm-3-yl]-l,2-benzisoxazol-5-yl}oxy)propanoic acid having formula Ia:

21. A method of treating Type 2 diabetes comprising the administration to a patient in need of such treatment a therapeutically effective amount of the crystalline anhydrous free acid form of the compound of Claim 1.
22. A method of treating Type 2 diabetes comprising the administration to a patient in need of such treatment a therapeutically effective amount of the crystalline anhydrous benzenesulfonate salt of the compound of Claim 1.
23. The use of the crystalline anhydrous free acid form of the compound of Claim 1 in the manufacture of a medicament for the treatment of Type 2 diabetes in a human or mammalian patient.
24. The use of the crystalline anhydrous benzenesulfonate salt of the compound of
Claim 1 in the manufacture of a medicament for the treatment of Type 2 diabetes in a human or mammalian patient.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07752209A EP1993543A2 (en) | 2006-03-03 | 2007-03-02 | Novel crystalline forms of antidiabetic compounds |
| US12/224,527 US20090264473A1 (en) | 2006-03-03 | 2007-03-02 | Novel Crystalline Forms of Antidiabetic Compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US77899806P | 2006-03-03 | 2006-03-03 | |
| US60/778,998 | 2006-03-03 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007103252A2 true WO2007103252A2 (en) | 2007-09-13 |
| WO2007103252A3 WO2007103252A3 (en) | 2008-03-06 |
Family
ID=38475439
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/005493 Ceased WO2007103252A2 (en) | 2006-03-03 | 2007-03-02 | Novel crystalline forms of antidiabetic compounds |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20090264473A1 (en) |
| EP (1) | EP1993543A2 (en) |
| WO (1) | WO2007103252A2 (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| DE102010015123A1 (en) | 2010-04-16 | 2011-10-20 | Sanofi-Aventis Deutschland Gmbh | New benzylamidic diphenylazetidinone compounds, useful for treating lipid disorders, hyperlipidemia, atherosclerotic manifestations or insulin resistance, and for reducing serum cholesterol levels |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013068486A1 (en) | 2011-11-08 | 2013-05-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the diagnosis and treatment of male infertility |
| US10485236B2 (en) | 2015-08-14 | 2019-11-26 | Bayer Cropscience Aktiengesellschaft | Triazole derivatives, intermediates thereof and their use as fungicides |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008109334A1 (en) * | 2007-03-02 | 2008-09-12 | Merck & Co., Inc. | Novel crystalline salt form of an antidiabetic compound |
| CN116253678A (en) * | 2021-12-10 | 2023-06-13 | 都创(重庆)药业有限公司 | A method for rapid preparation of 2,6-dichloropyridine-3-carboxylic acid based on microchannel continuous flow technology |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7834036B2 (en) * | 2005-03-04 | 2010-11-16 | Merck Sharp & Dohme Corp | Fused-aromatic compounds having anti-diabetic activity |
-
2007
- 2007-03-02 EP EP07752209A patent/EP1993543A2/en not_active Withdrawn
- 2007-03-02 US US12/224,527 patent/US20090264473A1/en not_active Abandoned
- 2007-03-02 WO PCT/US2007/005493 patent/WO2007103252A2/en not_active Ceased
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| DE102010015123A1 (en) | 2010-04-16 | 2011-10-20 | Sanofi-Aventis Deutschland Gmbh | New benzylamidic diphenylazetidinone compounds, useful for treating lipid disorders, hyperlipidemia, atherosclerotic manifestations or insulin resistance, and for reducing serum cholesterol levels |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013068486A1 (en) | 2011-11-08 | 2013-05-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the diagnosis and treatment of male infertility |
| US10485236B2 (en) | 2015-08-14 | 2019-11-26 | Bayer Cropscience Aktiengesellschaft | Triazole derivatives, intermediates thereof and their use as fungicides |
Also Published As
| Publication number | Publication date |
|---|---|
| US20090264473A1 (en) | 2009-10-22 |
| WO2007103252A3 (en) | 2008-03-06 |
| EP1993543A2 (en) | 2008-11-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20090264473A1 (en) | Novel Crystalline Forms of Antidiabetic Compounds | |
| US8258161B2 (en) | Crystalline salt form of an antidiabetic compound | |
| EP2468750A1 (en) | Polymorphic forms of asenapine maleate and processes for their preparation | |
| US20250243223A1 (en) | Solid state forms of ixazomib citrate | |
| US20090076272A1 (en) | Polymorphs of eszopiclone malate | |
| US20220281879A1 (en) | Salt and crystal forms of an activin receptor-like kinase inhibitor | |
| JPH04364179A (en) | Sulfur-containing heterocyclic compound | |
| AU2005207925B2 (en) | Novel crystalline forms of an inhibitor of 11-beta-hydroxysteroid dehydrogenase Type 1 | |
| US7932249B2 (en) | Olanzapine pamoate dihydrate | |
| CN111094241B (en) | Crystal form of oxopyridine amide derivative and preparation method thereof | |
| US20230271967A1 (en) | Solid state forms of fezolinetant and salts thereof | |
| JP2014501753A (en) | Polymorph of 3-chloro-4 [(2R) -2- (4-chlorophenyl) -4-[(1R) -1- (4-cyanophenyl) ethyl] -1-piperazinyl] -benzonitrile, said polymorph Containing the pharmaceutical composition and methods of use, and methods of preparation thereof | |
| US8618112B2 (en) | Crystalline forms of an inhibitor of 11-β-hydroxysteroid dehydrogenase type 1 | |
| KR20260012243A (en) | Preparation of (S)-6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide in enantiomerically enriched form by separation process | |
| WO2026042788A1 (en) | Heterocyclic derivative | |
| CN121226356A (en) | Parmolate crystal forms of genomelene or its deuterated derivatives, their preparation methods and uses | |
| HK40025351B (en) | Crystal form of oxopicolinamide derivative and preparation method therefor | |
| US20100056583A1 (en) | Polymorphic forms of rosiglitazone hydrobromide and processes for their preparation | |
| HK40025351A (en) | Crystal form of oxopicolinamide derivative and preparation method therefor | |
| WO2013181384A1 (en) | Solid state forms of aleglitazar sodium | |
| EP2109613A2 (en) | Polymorphs of eszopiclone malate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 12224527 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007752209 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |