WO2007042815A1 - Compounds containing more than one human neutrophil elastase inhibiting moiety for use in the treatment of respiratory diseases - Google Patents
Compounds containing more than one human neutrophil elastase inhibiting moiety for use in the treatment of respiratory diseases Download PDFInfo
- Publication number
- WO2007042815A1 WO2007042815A1 PCT/GB2006/003791 GB2006003791W WO2007042815A1 WO 2007042815 A1 WO2007042815 A1 WO 2007042815A1 GB 2006003791 W GB2006003791 W GB 2006003791W WO 2007042815 A1 WO2007042815 A1 WO 2007042815A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- crc
- group
- compound according
- heteroaryl
- Prior art date
Links
- IZIQJRCONFJKDC-UHFFFAOYSA-N CC(C)(C)OC(N1CCN(CCN(CC2)CCN2C(OC(C)(C)C)=O)CC1)=O Chemical compound CC(C)(C)OC(N1CCN(CCN(CC2)CCN2C(OC(C)(C)C)=O)CC1)=O IZIQJRCONFJKDC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug, i.e. a dimer, oligomer or polymer of pharmacologically or therapeutically active compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/20—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D239/22—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
Definitions
- This invention relates to heterocyclic compounds and their use in therapy. Background to the invention
- Human neutrophil elastase is a 32 kDa serine proteinase found in the azurophilic granules of neutrophils. It has a role in the degradation of a wide range of extracellular matrix proteins, including fibronectin, laminin, proteoglycans, Type III and Type IV collagens as well as elastin (Bieth, G. In Regulation of Matrix accumulation, Mecham, R. P. (Eds), Academic Press, NY 1 USA 1986, 217-306). HNE has long been considered to
- HNE has been implicated in the upregulation of IL-8 gene expression and also induces IL-8 release from the epithelial cells of the lung.
- HNE ulcerative colitis induced by tobacco smoke exposure
- small molecule inhibitors and protein inhibitors of HNE inhibit the inflammatory response and the development of emphysema (Wright, J. L. et al. Am. J. Respir. Crit. Care Med.2002, 166, 954-960; Churg, A. et al. Am. J. Respir. Crit. Care Med. 2003, 168, 199-207).
- HNE may play a role both in matrix destruction and in amplifying inflammatory responses
- HNE chronic obstructive pulmonary disease
- COPD chronic obstructive pulmonary disease
- CF cystic fibrosis
- ARDS acute respiratory distress syndrome
- pulmonary emphysema pulmonary emphysema
- pneumonia pulmonary fibrosis
- COPD chronic bronchitis
- emphysema emphysema
- small- airway disease Generally all three will exist to varying extents in patients presenting with
- COPD bronchoalveolar leakage
- Multimeric ligands consist of multiple binding domains which are tethered together through a suitable scaffold. Hence individual binding domains are linked together into a single molecule, increasing the probability that the multimerwill bind simultaneously with multiple active sites resulting in high-affinity interactions (Handl, H. L. etal. Expert Opin. Ther. Targets 2004, 8, 565-586; Han, Y. F. et al., Bioorg. Med. Chem. Letts. 1999, 7, 2569-2575). Also, multiple binding interactions with relatively high off-rates can combine to yield an overall low off-rate for the multimeric ligand.
- a molecule consisting of a suitable linker and ligands may be expected to show advantage over the monomeric ligands alone in terms of potency and/or duration of action.
- Multimeric compounds are unlikely to be orally bioavailable (as predicted by Lipinski's "Rule of 5") which may be advantageous where an inhaled route of administration to the lungs is targeted, since even after inhaled administration, a large proportion of drug is likely to enter the Gl tract.
- Such compounds may be expected to show reduced systemic exposure after inhalation administration and hence an improved toxicity profile over orally administered therapies.
- a first aspect of the invention is a compound of formula (I) or formula (IV):
- each M is independently a group of Formula (II); each L is independently a linker group; t is 2 to 20;
- G is aryl, heteroaryl, alkyl, cycloalkyl, nitrogen, a dendrimer or a group of any of formulae (V) to (VII): Ar' 0* Ar Ar-Ar NT JN
- Ar is aryl or heteroaryl; and u is 2-20; or a pharmaceutically acceptable salt, solvate, N-oxide or prodrug thereof.
- each and any of the sites on the Ar groups may be the site or sites of attachment.
- each nitrogen atom may be substituted with between 1 to 3 attachments. When the nitrogen has 3 attachments it is to be understood that the nitrogen is quatemised and bears a positive charge.
- Compounds of the invention may be useful in the treatment or prevention of diseases in which HNE is implicated, for example chronic obstructive pulmonary disease (COPD), chronic bronchitis, lung fibrosis, pneumonia, acute respiratory distress syndrome (ARDS), pulmonary emphysema, smoking-induced emphysema, cystic fibrosis, asthma, rhinitis, psoriasis, dermatitis, Chrohn's disease, ulcerative colitis, and irritable bowel disease.
- COPD chronic obstructive pulmonary disease
- COPD chronic obstructive pulmonary disease
- ARDS acute respiratory distress syndrome
- pulmonary emphysema smoking-induced emphysema
- cystic fibrosis asthma
- rhinitis psoriasis
- dermatitis dermatitis
- Chrohn's disease Crohn's disease
- ulcerative colitis ulcerative colitis
- Another aspect of the invention is a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the invention and a pharmaceutically acceptable carrier or excipient.
- Another aspect of the invention is the use of a compound of the invention for the manufacture of a medicament for the treatment or prevention of a disease or condition in which HNE is implicated.
- Acyl means a -CO-alkyl group in which the alkyl group is as described herein.
- acyl groups include -COCH 3 and -COCH(CH 3 ) 2 .
- acylamino means a -NR-acyl group in which R and acyl are as described herein.
- exemplary acylamino groups include -NHCOCH 3 and -N(CH 3 )COCH 3 .
- Alkoxy and “alkyloxy” means an -O-alkyl group in which alkyl is as described below.
- exemplary alkoxy groups include methoxy (-OCH 3 ) and ethoxy (-OC 2 H 5 ).
- Alkoxycarbonyl means a -COO-alkyl group in which alkyl is as defined below.
- exemplary alkoxycarbonyl groups include methoxycarbonyl and ethoxycarbonyl.
- Alkyl as a group or part of a group refers to a straight or branched chain saturated hydrocarbon group having from 1 to 12, preferably 1 to 6, carbon atoms, in the chain.
- exemplary alkyl groups include methyl, ethyl, 1 -propyl and 2-propyl.
- alkenyl as a group or part of a group refers to a straight or branched chain hydrocarbon group having from 1 to 12, preferably 1 to 6, carbon atoms and one carbon- carbon double bond in the chain.
- alkenyl groups include ethenyl, 1 -propenyl, and 2-propenyl.
- Alkylamino means a -NH-alkyl group in which alkyl is as defined above.
- exemplary alkylamino groups include methylamino and ethylamino.
- Alkylene means an -alkyl- group in which alkyl is as defined previously.
- exemplary alkylene groups include -CH 2 -, -(CH 2 ) 2 - and -C(CH 3 )HCH 2 -.
- Alkenylene means an -alkenyl- group in which alkenyl is as defined previously.
- Alkylsulfinyl means a -SO-alkyl group in which alkyl is as defined above.
- exemplary alkylsulfinyl groups include methylsulfinyl and ethylsulfinyl.
- Alkylsulfonyl means a -SO 2 -alkyl group in which alkyl is as defined above.
- exemplary alkylsulfonyl groups include methylsulfonyl and ethylsulfonyl.
- Alkylthio means a -S-alkyl group in which alkyl is as defined above.
- exemplary alkylthio groups include methylthio and ethylthio.
- aminoacyl means a -CO-NRR group in which R is as herein described.
- exemplary aminoacyl groups include -CONH 2 and -CONHCH 3 .
- Aminoalkyl means an alkyl-NH 2 group in which alkyl is as previously described.
- Exemplary aminoalkyl groups include -CH 2 NH 2 .
- aminosulfonyl means a -SO 2 -NRR group in which R is as herein described.
- exemplary aminosulfonyl groups include -SO 2 NH 2 and -SO 2 NHCH 3 .
- Aryl as a group or part of a group denotes an optionally substituted monocyclic or multicyclic aromatic carbocyclic.moiety of from 6 to 14 carbon atoms, preferably from 6 to 10 carbon atoms, such as phenyl or naphthyl.
- the aryl group may be substituted by one or more substituent groups.
- Arylalkyl means an aryl-alkyl- group in which the aryl and alkyl moieties are as previously described. Preferred arylalkyl groups contain a C 1 4 alkyl moiety. Exemplary arylalkyl groups include benzyl, phenethyl and naphthlenemethyl.
- Arylalkyloxy means an aryl-alkyloxy- group in which the aryl and alkyloxy moieties are as previously described. Preferred arylalkyloxy groups contain a C 1 4 alkyl moiety. Exemplary arylalkyl groups include benzyloxy.
- Aryl-fused-cycloalkyl means a monocyclic aryl ring, such as phenyl, fused to a cycloalkyl group, in which the aryl and cycloalkyl are as described herein.
- Exemplary aryl- fused-cycloalkyl groups include tetrahydronaphthyl and indanyl.
- the aryl and cycloalkyl rings may each be substituted by one or more substituent groups.
- the aryl-fused- cycloalkyl group may be attached to the remainder of the compound by any available carbon atom.
- Aryl-fused-heterocycloalkyl means a monocyclic aryl ring, such as phenyl, fused to a heterocycloalkyl group, in which the aryl and heterocycloalkyl are as described herein.
- exemplary aryl-fused-heterocycloalkyl groups include tetrahydroquinolinyl, indolinyl, benzodioxinyl, benxodioxolyl, dihydrobenzofuranyl and isoindolonyl.
- the aryl and heterocycloalkyl rings may each be substituted by one or more substituent groups.
- the aryl-fused-heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
- aryloxy means an -O-aryl group in which aryl is described above.
- exemplary aryloxy groups include phenoxy.
- Cyclic amine means an optionally substituted 3 to 8 membered monocyclic cycloalkyl ring system where one of the ring carbon atoms is replaced by nitrogen, and which may optionally contain an additional heteroatom selected from O, S or NR (where
- cyclic amines include pyrrolidine, piperidine, morpholine, piperazine and ⁇ /-methylpiperazine.
- the cyclic amine group may be substituted by one or more substituent groups.
- Cycloalkyl means an optionally substituted saturated monocyclic or bicyclic ring system of from 3 to 12 carbon atoms, preferably from 3 to 8 carbon atoms, and more preferably from 3 to 6 carbon atoms.
- Exemplary monocyclic cycloalkyl rings include cyclopropyl, cyclopentyl, cyclohexyl and cycloheptyl.
- the cycloalkyl group may be substituted by one or more substituent groups.
- Cycloalkylalkyl means a cycloalkyl-alkyl- group in which the cycloalkyl and alkyl moieties are as previously described.
- Exemplary monocyclic cycloalkylalkyl groups include cyclopropylmethyl, cyclopentylmethyl, cyclohexylmethyl and cycloheptylmethyl.
- Dialkylamino means a -N(alkyl)2 group in which alkyl is as defined above.
- dialkylamino groups include dimethylamino and diethylamino.
- Halo or halogen means fluoro, chloro, bromo, or iodo. Preferred are fluoro or chloro.
- Haloalkoxy means an -O-alkyl group in which the alkyl is substituted by one or more halogen atoms.
- exemplary haloalkyl groups include trifluoromethoxy and difluoromethoxy.
- Haloalkyl means an alkyl group which is substituted by one or more halo atoms.
- Exemplary haloalkyl groups include trifluoromethyl.
- Heteroaryl as a group or part of a group denotes an optionally substituted aromatic monocyclic or multicyclic organic moiety of from 5 to 14 ring atoms, preferably from 5 to 10 ring atoms, in which one or more of the ring atoms is/are element(s) other than carbon, for example nitrogen, oxygen or sulfur.
- Examples of such groups include benzimidazolyl, benzoxazolyl, benzothiazolyl, benzofuranyl, benzothienyl, furyl, imidazolyl, indolyl, indolizinyl, isoxazolyl, isoquinolinyl, isothiazolyl, oxazolyl, oxadiazolyl, pyrazinyl, pyridazinyl, pyrazolyl, pyridyl, pyrimidinyl, pyrrolyl, quinazolinyl, quinolinyl, tetrazolyl, 1 ,3,4-thiadiazolyl, thiazolyl, thienyl and triazolyl groups.
- the heteroaryl group may be substituted by one or more substituent groups.
- the heteroaryl group may be attached to the remainder of the compound of the invention by any available carbon or nitrogen atom.
- Heteroarylalkyl means a heteroaryl-alkyl- group in which the heteroaryl and alkyl moieties are as previously described. Preferred heteroarylalkyl groups contain a lower alkyl moiety. Exemplary heteroarylalkyl groups include pyridylmethyl.
- Heteroarylalkyloxy means a heteroaryl-alkyloxy- group in which the heteroaryl and alkyloxy moieties are as previously described. Preferred heteroarylalkyloxy groups contain a lower alkyl moiety. Exemplary heteroarylalkyloxy groups include pyridylmethyloxy. "Heteroaryloxy” means a heteroaryloxy- group in which the heteroaryl is as previously described. Exemplary heteroaryioxy groups include pyridyloxy.
- Heteroaryl-fused-cycloalkyl means a monocyclic heteroaryl group, such as pyridyl or furanyl, fused to a cycloalkyl group, in which heteroaryl and cycloalkyl are as previously described.
- Exemplary heteroaryl-fused-cycloalkyl groups include tetrahydroquinolinyl and tetrahydrobenzofuranyl.
- the heteroaryl and cycloalkyl rings may each be substituted by one or more substituent groups.
- the heteroaryl-fused-cycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
- Heteroaryl-fused-heterocycloalkyl means a monocyclic heteroaryl group, such as pyridyl or furanyl, fused to a heterocycloalkyl group, in which heteroaryl and heterocycloalkyl are as previously described.
- Exemplary heteroaryl-fused- heterocycloalkyl groups include dihydrodioxinopyridinyl, dihydropyrrolopyridinyl, dihydrofuranopyridinyl and dioxolopyridinyl.
- the heteroaryl and heterocycioalkyl rings may each be substituted by one or more substituents groups.
- heteroaryl-fused- heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
- Heterocycloalkyl means: (i) an optionally substituted cycloalkyl group of from 4 to 8 ring members which contains one or more heteroatoms selected from O, S or NR; (ii) a cycloalkyl group of from 4 to 8 ring members which contains CONR and CONRCO (examples of such groups include succinimidyl and 2-oxopyrrolidinyl).
- the heterocycloalkyl group may be substituted by one or more substituent groups.
- the heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
- Heterocycloalkylalkyl means a heterocycloalkyl-alkyl- group in which the heterocycloalkyl and alkyl moieties are as previously described.
- “Lower alkyl” as a group means unless otherwise specified, an aliphatic hydrocarbon group which may be straight or branched having 1 to 4 carbon atoms in the chain, i.e. methyl, ethyl, propyl (propyl or /s ⁇ -propyl) or butyl (butyl, /so-butyl or fert-butyl).
- Sulfonyl means a -SO 2 -alkyl group in which alkyl is as described herein.
- Exemplary sulfonyl groups include methanesulfonyl.
- “Sulfonylamino” means a -NR-sulfonyl group in which R and sulfonyl are as described herein.
- Exemplary sulfonylamino groups include -NHSO 2 CI-I 3 .
- R means alkyl, aryl, or heteroaryl as described herein.
- “Pharmaceutically acceptable salt” means a physiologically or toxicologically tolerable salt and include, when appropriate, pharmaceutically acceptable base addition salts and pharmaceutically acceptable acid addition salts.
- pharmaceutically acceptable base addition salts that may be formed include sodium, potassium, calcium, magnesium and ammonium salts, or salts with organic amines, such as, diethylamine, ⁇ /-methyl-glucamine, diethanolamine or amino acids (e.g.
- a compound of the invention contains a basic group, such as an amino group
- pharmaceutically acceptable acid addition salts that may be formed include hydrochlorides, hydrobromides, phosphates, acetates, citrates, lactates, tartrates, malonates, methanesulphonates and the like.
- Prodrug refers to a compound which is convertible in vivo by metabolic means (e.g. by hydrolysis, reduction or oxidation) to a compound of the invention.
- metabolic means e.g. by hydrolysis, reduction or oxidation
- an ester prodrug of a compound of the invention containing a hydroxy group may be convertible by hydrolysis in vivoto the parent molecule.
- Suitable esters of compounds of the invention containing a hydroxy group are for example acetates, citrates, lactates, tartrates, malonates, oxalates, salicylates, propionates, succinates, fumarates, maleates, methylene-bis- ⁇ -hydroxynaphthoates, gentisates, isothionates, di-p-toluoyltartrates, methanesulphonates, ethanesulphonates, benzenesulphonates, p-toluenesulphonates, cyclohexylsulphamates and quinates.
- ester prodrug of a compound of the invention containing a carboxy group may be convertible by hydrolysis in vivo to the parent molecule.
- ester prodrugs are those described by F. J. Leinweber, Drug Metab. Res., 1987, 18, 379.
- “Saturated” pertains to compounds and/or groups which do not have any carbon- carbon double bonds or carbon-carbon triple bonds.
- cyclic groups referred to above namely, aryl, heteroaryl, cycloalkyl, aryl- fused-cycloalkyl, heteroaryl-fused-cycloalkyl, heterocycloalkyl, aryl-fused- heterocycloalkyl, heteroaryl-fused-heterocycloalkyl and cyclic amine may be substituted by one or more substituent groups.
- Suitable optional substituent groups include acyl (e.g. -COCH 3 ), alkoxy (e.g., -OCH 3 ), alkoxycarbonyl (e.g. -COOCH 3 ), alkyiamino (e.g. -
- alkylsulfinyl e.g. -SOCH 3
- alkylsulfonyl e.g. -SO 2 CH 3
- alkylthio e.g. -SCH 3
- - NH 2 aminoacyl (e.g. -CON(CH 3 ) 2 ), aminoalkyl (e.g. -CH 2 NH 2 ), arylalkyl (e.g. -CH 2 Ph or -CH 2 -CH 2 -Ph), cyano, dialkylamino (e.g. -N(CH 3 ) 2 ), halo, haloalkoxy (e.g.
- haloalkyl e.g. -CF 3
- alkyl e.g. -CH 3 or -CH 2 CH 3
- -OH, -CHO, -NO 2 aryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heteroaryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heterocycloalkyl, aminoacyl (e.g. -CONH 2 , -CONHCH 3 ), aminosulfonyl (e.g.
- acylamino e.g. -NHCOCH 3
- sulfonylamino e.g. -NHSO 2 CH 3
- heteroarylalkyl cyclic amine (e.g. morpholine), aryloxy, heteroaryloxy, arylalkyloxy (e.g. benzyloxy) and heteroarylalkyloxy.
- Alkylene or alkenylene groups may be optionally substituted. Suitable optional substituent groups include alkoxy (e.g., -OCH 3 ), alkyiamino (e.g. -NHCH 3 ), alkylsulfinyl
- alkylsulfonyl e.g. -SOXHJ, alkylthio (e.g. -SCHJ, -NH 2 , aminoalkyl (e.g. - CH 2 NH 2 ), arylalkyl (e.g. -CH 2 Ph or -CH 2 -CH 2 -Ph), cyano, dialkylamino (e.g. -N(CH 3 J 2 ), halo, haloalkoxy (e.g. -OCF, or -OCHFJ, haloalkyl (e.g. -CFJ, alkyl (e.g. -CH, or - CH 2 CH 3 ), -OH, -CHO, and -NO 2 .
- -SOCHJ alkylsulfonyl
- alkylthio e.g. -SCHJ
- -NH 2 aminoalkyl (e.g. - CH 2 NH 2 )
- Compounds of the invention may exist in one or more geometrical, optical, enantiomeric, diastereomeric and tautomeric forms, including but not limited to cis- and frans-forms, E- and Z-forms, R-, S- and meso-forms, keto-, and enol-forms. Unless otherwise stated a reference to a particular compound includes all such isomeric forms, including racemic and other mixtures thereof. Where appropriate such isomers can be separated from their mixtures by the application or adaptation of known methods (e.g. chromatographic techniques and recrystallisation techniques). Where appropriate such isomers may be prepared by the application of adaptation of known methods (e.g. asymmetric synthesis).
- Each M may independently be a group of Formula (II)
- A is aryl or heteroaryl;
- R 1 , R 2 and R 3 are independently each hydrogen, halogen, nitro, cyano, C r C 6 - alkyl, hydroxy or C r C 6 -alkoxy, wherein d-C 6 -alkyl and CrC 6 -alkoxy can be further substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy and C r C 4 -alkoxy;
- R 4 is hydrogen, CrCe-alkyl, formyl, aminocarbonyl, mono- or di-C r C 4 - alkylaminocarbonyl, C 3 -C 8 -cycloalkylcarbonyl, CrC 6 -alkylcarbonyl, CrC ⁇ -alkoxycarbonyl, N-(CrC 4 -alkylsulfonyl)-aminocarbonyl, N-(Ci-C 4 -alkylsulfonyl)-N-(C r C 4 -alkyl)- aminocarbonyl, heteroaryl, heterocycloalkyl, heteroarylcarbonyl or heterocycloalkylcarbonyl; wherein CrC 6 -alkyl, mono- and di-CrC ⁇ alkylaminocarbonyl, CrCe-alkylcarbonyl, C r C 6 -alkoxycarbonyl, heteroaryl and heterocycloalkyl can be substituted with one to three identical
- R 4 represents a group of Formula (VIII)
- R 4A is hydrogen or CrCe-alkyl, and n is 1 or 2;
- R 5 is CrC 4 -alkyl, which can be substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy, CrC 6 -alkoxy, C 1 -C 6 - alkenoxy, CrC 6 -alkylthio, amino, mono- and di- C r C 6 -alkylamino, arylamino, hydroxycarbonyl, C r C 6 -alkoxycarbonyl and the radical -0-(C 1 -C 4 -BIkVl)-O-(C 1 -C 4 -alkyl); or R 5 is amino;
- R 6 is halogen, nitro, cyano, CrC 6 -alkyl, hydroxy or CrC 6 -alkoxy, wherein
- CrCe-alkyl and C r C 6 -alkoxy can be further substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy and C 1 -C 4 - alkoxy; and
- Y 1 , Y 2 , Y 3 , Y 4 and Y 5 are independently each CH or N, with the proviso that the ring in which they are comprised contains no more than 2 N atoms.
- A is a phenyl ring.
- R 1 is hydrogen.
- R 2 is methyl. In a further embodiment, R 2 is -CN. In a preferred embodiment, R 3 is hydrogen. In another preferred embodiment, R 4 is alkoxycarbonyl.
- R 5 is methyl.
- R 6 is trifluoromethyl.
- Y 1 , Y 2 , Y 3 , Y 4 and Y 5 are each CH.
- L may be a group of formula (III): -L a -Z-(W a -Z) m -W b -Z-L ⁇ (III)
- each L a is independently , -S(O) W -, -, -C(O)O-, -C(O)-, -C(O)NR 7 -, -C(S)-, -C(S)O-, -C(O)S-, -C(S)NR 7 - or a covalent bond;
- each Z is independently optionally substituted alkylene, optionally substituted cycloalkylene, optionally substituted alkenylene, optionally substituted alkynylene, optionally substituted cycloalkenylene, aryi, heteroaryl, heterocycloalkyl or a covalent bond;
- R 7 and R 8 are independently hydrogen, optionally substituted alkyl, optionally substituted cycloalkyl, optionally substituted alkenyl, optionally substituted cycloalkenyl, optionally substituted alkynyl, aryl, heteroaryl or heterocycloalkyl; and each W is independently 0,1 or 2.
- the group -L a -Z-(W a -Z) m -W b -Z-L a - represents an alkylene group.
- W a and W b are each -0-, -NR 7 , or a covalent bond.
- a compound of the invention is of formula (IV).
- a compound of the invention is as defined in Table 1 , or in Examples 1 to 15.
- each M is the same or different and is a group of formula (II) as defined herein.
- the arrow denotes the point of attachment of M to the linker L.
- L is a group of Formula (III) as defined herein.
- the linkers, L, used in this invention can be selected so as to allow multivalent binding of ligands to binding sites on human neutrophil elastase, whether such sites are located at the substrate binding site and/or at other binding sites on the same enzyme molecule, or on binding sites of more than one enzyme molecule via all combinations of available binding sites.
- G may be a group of any of formulae (V) to (VII) or a dendrimer.
- groups of formulae (V) to (VII) include, but are not limited to phenoxyphenyl, biphenyl, bipyridyl, ethylenediamino, propylenediamino and the like. It is to be understood that the number of possible attachment points is dictated by the valency of the groups present, so that for example, biphenyl can contain up to 10 possible attachments (5 on each phenyl ring), and ethylenediamine can possess up to 4 possible attachments (2 on each terminal amine).
- An example of a dendrimer suitable for use in the invention is:
- Preferred compounds of the invention include:
- the therapeutic utility of the present compounds is pertinent to any disease that is known to be at least partially mediated by the action of human neutrophil elastase.
- the present compounds may be beneficial in the treatment of chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), acute respiratory distress syndrome (ARDS), pulmonary emphysema, pneumonia and lung fibrosis.
- COPD chronic obstructive pulmonary disease
- CF cystic fibrosis
- ARDS acute respiratory distress syndrome
- pulmonary emphysema pulmonary emphysema
- pneumonia and lung fibrosis lung fibrosis.
- the present invention is also concerned with pharmaceutical formulations comprising, as an active ingredient, a compound of the invention.
- Other compounds may be combined with compounds of this invention for the prevention and treatment of inflammatory diseases of the lung.
- the present invention is also concerned with pharmaceutical compositions for preventing and treating inflammatory diseases of the lung comprising a therapeutically effective amount of a compound of the invention and one or more other therapeutic agents.
- Suitable therapeutic agents for a combination therapy with compounds of the invention include: (1 ) a corticosteroid, for example fluticasone or budesonide; (2) a ⁇ 2- adrenoreceptor agonist, for example salmeterol or formeterol; (3) a leukotriene modulator, for example montelukast or pranlukast; (4) anticholinergic agents, for example selective muscarinic-3 (M3) receptor antagonists such as tiotropium bromide; (5) phosphodiesterase-IV (PDE-IV) inhibitors, for example roflumilast or cilomilast; (6) an antitussive agent, such as codeine or dextramorphan; and (7) a non-steroidal antiinflammatory agent (NSAID), for example ibuprofen or ketoprofen.
- a corticosteroid for example fluticasone or budesonide
- a ⁇ 2- adrenoreceptor agonist for
- the weight ratio of the first and second active ingredients may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used.
- the magnitude of prophylactic or therapeutic dose of a compound of the invention will, of course, vary with the nature of the severity of the condition to be treated and with the particular compound and its route of administration. It will also vary according to the age, weight and response of the individual patient. In general, the daily dose range will lie within the range of from about 0.001 mg to about 100 mg per kg body weight of a mammal, preferably 0.01 mg to about 50 mg per kg, and most preferably 0.1 to 10 mg per kg, in single or divided doses. On the other hand, it may be necessary to use dosages outside these limits in some cases.
- compositions which comprise a compound of the invention and a pharmaceutically acceptable carrier.
- composition is intended to encompass a product comprising the active ingredient(s), and the inert ingredient(s) (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the invention, additional active ingredient(s), and pharmaceutically acceptable excipients.
- compositions of the present invention comprise a compound of the invention as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
- any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dosage of a compound of the present invention.
- the active compound may be administered by any convenient, suitable or effective route. Suitable routes of administration are known to those skilled in the art, and include oral, intravenous, rectal, parenteral, topical, ocular, nasal, buccal and pulmonary. Delivery by inhalation is preferred.
- Compositions suitable for administration by inhalation are known, and may include carriers and/or diluents that are known for use in such compositions.
- the composition may contain 0.01-99% by weight of active compound.
- a unit dose comprises the active compound in an amount of 1 ⁇ g to 10 mg.
- the most suitable dosage level may be determined by any suitable method known to one skilled in the art. It will be understood, however, that the specific amount for any particular patient will depend upon a variety of factors, including the activity of the specific compound that is used, the age, body weight, diet, general health and sex of the patient, time of administration, the route of administration, the rate of excretion, the use of any other drugs, and the severity of the disease undergoing treatment.
- the active compound is preferably in the form of microparticles. They may be prepared by a variety of techniques, including spray-drying, freeze-drying and micronisation.
- a composition of the invention may be prepared as a suspension for delivery from a nebuliser or as an aerosol in a liquid propellant, for example for use in a pressurised metered dose inhaler (PMDI).
- PMDI pressurised metered dose inhaler
- Propellants suitable for use in a PMDI are known to the skilled person, and include CFC-12, HFA-134a, HFA- 227, HCFC-22 (CCI 2 F 2 ) and HFA-152 (CH 2 F 2 ) and isobutane.
- a composition of the invention is in dry powder form, for delivery using a dry powder inhaler (DPI).
- DPI dry powder inhaler
- Microparticles for delivery by administration may be formulated with excipients that aid delivery and release.
- microparticles may be formulated with large carrier particles that aid flow from the DPI into the lung.
- Suitable earner particles are known, and include lactose particles; they may have a mass median aerodynamic diameter of greater than 90 ⁇ m.
- a preferred composition is: Compound of the invention 24 mg / canister Lecithin, NF Liq. Cone. 1.2 mg / canister
- compositions of the invention may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which present compounds are useful. Such other drugs may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with a compound of the invention.
- a pharmaceutical composition containing such other drugs in addition to the compound of the invention is preferred.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the invention.
- the agents of the invention may be administered in inhaled form. Aerosol generation can be carried out using, for example, pressure-driven jet atomizers or ultrasonic atomizers, preferably using propellant-driven metered aerosols or propellant- free administration of micronized active compounds from, for example, inhalation capsules or other "dry powder" delivery systems.
- the active compounds may be dosed as described depending on the inhaler system used.
- the administration forms may additionally contain excipients, such as, for example, propellants (e.g. Frigen in the case of metered aerosols), surface-active substances, emulsifiers, stabilizers, preservatives, flavorings, fillers (e.g. lactose in the case of powder inhalers) or, if appropriate, further active compounds.
- the compounds of the invention of the present invention can be prepared according to the procedures of the following schemes and examples, using appropriate materials, and are further exemplified by the following specific examples. Moreover, by utilising the procedures described with the disclosure contained herein, one of ordinary skill in the art can readily prepare additional compounds of the present invention claimed herein.
- the compounds illustrated in the examples are not, however, to be construed as forming the only genus that is considered as the invention.
- the examples further illustrate details for the preparation of the compounds of the present invention. Those skilled in the art will readily understand that known variations of the conditions and processes of the following preparative procedures can be used to prepare these compounds.
- the compounds of the invention may be isolated in the form of their pharmaceutically acceptable salts, such as those described previously herein above.
- the free acid form corresponding to isolated salts can be generated by neutralisation with a suitable acid such as acetic acid and hydrochloric acid and extraction of the liberated free acid into an organic solvent followed by evaporation.
- a suitable acid such as acetic acid and hydrochloric acid
- the free acid form isolated in this manner can be further converted into another pharmaceutically acceptable salt by dissolution in an organic solvent followed by addition of the appropriate base and subsequent evaporation, precipitation, or crystallisation.
- Compounds of formula 1-1 wherein A, Y 1 , Y 2 , Y 3 , Y 4 , Y 5 , R 1 , R 2 , R 3 , R 4 , R 5 and R 6 are as defined above, may be prepared from compounds of formula IX-, wherein X is a suitable leaving group such as halogen or a sulfonate ester, by reaction with a suitably substituted diamine.
- the reaction may be conducted in the presence of a base, such as sodium bicarbonate in a solvent such as acetonitrile. Heating the reaction in a microwave reactor may be beneficial.
- Compounds of formula I-2 may be prepared from compounds of formula IX-4 by reaction with a suitably substituted coupling partner of formula IX-3. The reaction takes place in the presence or absence of base. Suitable bases include NaH and potassium carbonate in a solvents such as DMF.
- Compounds of formula IX-4 may be prepared from compounds of IX-3 in a similar manner to that used to prepare compounds of formula 1-2 from compounds of formula IX-4.
- Compounds of formula IX-3 may be prepared from compounds of formula IX-5 by reaction with a suitable bis-electrophile, such as an ⁇ , ⁇ - dihalide or disulfonate.
- a suitable bis-electrophile such as an ⁇ , ⁇ - dihalide or disulfonate.
- the reaction may be conducted in dlute aqueous sodium hydroxide in the presence of a phase-transfer catalyst such as a trialkylammonium halide.
- a phase-transfer catalyst such as a trialkylammonium halide.
- Phenyl hexyl column (250 x 21.20 mm Luna column with 5 ⁇ m particle size), eluting with a gradient of A: water + 0.1 % TFA; B: acetonitrile + 0.1 % TFA at a flow rate of 5 ml/min with UV detection at 254 nm.
- HPLC system 2 C18-reverse-phase column (100 x 22.5 mm i.d Genesis column with 7 ⁇ m particle size), eluting with a gradient of A: water + 0.1 % HCO 2 H; B: acetonitrile + 0.1 % HCO 2 H at a flow rate of 5 ml/min and gradient of 1 %/min increasing in B. UV detection at 230 nm.
- HPLC system 3
- Phenyl hexyl column (250 x 21.20 mm Luna column with 5 ⁇ m particle size), eluting with a gradient of A: water + 0.1 % HCO 2 H; B: acetonitrile + 0.1 % HCO 2 H at a flow rate of 5 ml/min with UV detection at 254 nm.
- HPLC system 4
- Amylose tris(3,5-dimethylphenylcarbamate) (250 x 20 mm CHIRALPAK IA column with 5 ⁇ m particle size), eluting with an isocratic mixture of ethanol (10%) in n-heptane at a flow rate of 15 ml/min with UV detection at 230 nm.
- LC/MS Systems
- Micromass Platform LCT with a C18-reverse-phase column (100 x 3.0 mm Higgins Clipeus with 5 ⁇ m particle size), elution with A: water + 0.1% formic acid; B: acetonitrile + 0.1% formic acid.
- Gradient Gradient - Time flow ml/min %A %B
- Intermediate 3 was prepared by a similar procedure from intermediate 1 (500 mg, 1.1 mmol), 1 ,5-diiodopentane (1.1 g, 0.5 ml, 3.3 mmol), 1 M aqueous sodium hydroxide (3 ml, 3 mmol), tetra-n-butylammonium iodide (45 mg, 0.12 mmol) and water (3 ml).
- Boc-Piperazine 0.5 g, 2.69 mmol
- 1 ,2-dibromoethane (253 mg, 1.34 mmol)
- NaHCO 3 564 mg, 6.72 mmol
- acetonitrile 20 ml
- the solvent was removed and the residue was dissolved in ethyl acetate (80 ml).
- the organic solution was washed with water (50 ml) and saturated brine (20 ml), dried (Na 2 SO 4 ) and evaporated to give intermediate 7 as a white solid.
- Example 2 was prepared by a similar procedure from intermediate 2 (77 mg) and N-methyl-2,2 ' -diaminodiethylamine. The crude solution was diluted with water and purified by HPLC (System 1).
- Example 3 was prepared by a similar procedure from intermediate 2 (77 mg) and N,N ' -Bis(2-hydroxyethyl)ethylenediamine. The crude solution was diluted with water and purified by HPLC (System 1 ).
- Example 4 was prepared by a similar procedure from intermediate 3 (108 mg) and piperazine. The crude solution was diluted with water and purified by HPLC (System 1).
- Example 5 was prepared by a similar procedure from intermediate 3 (108 mg) and N-methyl-2,2 ' -diaminodiethylamine. The crude solution was diluted with water and purified by HPLC (System 1).
- Example 6 was isolated from the same reaction as example 5.
- Example 7 was prepared by a similar procedure from intermediate 3 (96 mg) and 4,9-dioxa-1 ,12-dodecanediamine. Purification was achieved using HPLC (System 3). Yield: 16 mg (16%) LC-MS (Method 1): Rt 9.55 min, m/z 1231.34 [MH] +
- Example 9 was prepared by a similar procedure from intermediate 3 (69 mg) and N,N ' -bis(2-hydroxyethyl)ethylenediamine. Purification was effected by HPLC (System 1). Yield: 19 mg (28%) LC-MS (Method 1): Rt 10.84/11.34 min, m/z 1175.34 [MH] +
- Example 10 was prepared by a similar procedure from intermediate 5 (100 mg) and N,N ' -bis(2-hydroxyethyl)ethylenediamine. Purification was effected by HPLC (System 2).
- Example 11 was prepared by a similar procedure from intermediate 5 (100 mg) and N-(2-aminoethyl)-N-methylethanediamine. Purification was effected by HPLC
- Example 14 was made from Intermediate 9 in an analogous manner to Example 5. Purification was achieved using HPLC (System 2).
- Example 15 was obtained as a by-product in the synthesis of Intermediate 9.
- Fluorescent peptide substrate Assays were performed in 96-well plates at a total assay volume of 100 ⁇ l. The final concentration of the enzyme (human leukocyte elastase, Sigma E8140) was 0.00036 units/well. A peptide substrate (MeO-Suc-Ala-Ala-Pro-ValAMC, Calbiochem #324745) was used, at the final concentration of 100 ⁇ M. The final concentration of DMSO was 1 % in the assay buffer (0.05 M Tris.HCI, pH 7.5, 0.1 M NaCI; 0.1 M CaCI 2 ; 0.0005% brij-35).
- the enzymatic reaction was started by adding the enzyme.
- the enzymatic reaction was performed at RT and after 30 mins stopped by adding 50 ⁇ l soybean trypsin inhibitor (Sigma T-9003) at a final concentration of 50 ⁇ g/well.
- Fluorescence was read on the FLEXstation (Molecular Devices) using 380 nm excitation and 460 nm emission filters.
- the potency of the compounds was determined from a concentration series of 10 concentrations in range from 1000 nM to 0.051 nM. The results are means of two independent experiments, each performed in duplicate. Using Fluorescently labelled elastin
- Assays were performed in 96-well plate at a total assay volume of 100 ⁇ l.
- the final concentration of the enzyme human leukocyte elastase, Sigma E8140
- Fluorescently labelled, solubilised elastin from bovine neck ligament (Molecular Probes, E-12056) was used at the final concentration of 15 ⁇ g/ml.
- the final concentration of DMSO was 2.5% in the assay buffer (0.1 M Tris-HCL, pH 8.0, containing 0.2 mM sodium azide).
- the enzymatic reaction was started by adding the enzyme.
- the enzymatic reaction was performed at RT and read after 120 minutes. Fluorescence was read on the FLEXstation (Molecular Devices) using 485 nm excitation and 530 nm emission filters.
- the potency of the compounds was determined from a concentration series of 10 concentrations in range from 25000 nM to 1 nM. The results are means of two independent experiments, each performed in duplicate.
- the compounds tested were shown to have IC 50 values for HNE in the range 1- 100O nM.
- a common, generic substrate was used for all proteases: fluorescently labelled casein (Molecular Probes, E-6639), at the final concentration of 20 ⁇ g/ml (Cathepsin G and Chymotrypsin), 10 ⁇ g/ml (Plasmin and Thrombin) or 5 ⁇ g/ml (Proteinase 3 and Trypsin).
- the final concentration of the substrate was close to the respective K m values as determined for this substrate.
- the final concentration of DMSO was 5% in the assay buffer (0.05 M Tris.HCl, pH 7.5, 0.1 M NaCI; 0.1 M CaCI 2 ; 0.0005% brij-35). The enzymatic reaction was started by adding the enzyme.
- the enzymatic reaction was performed at RT for 60 min. Fluorescence was read on the FLEXstation (Molecular Devices) using 589 nm excitation and 617 nm emission filters. The potency of the compounds was determined from a concentration series of 8 concentrations in range from 500 ⁇ M to 0.2 ⁇ M. The results are means of two independent experiments, each performed in duplicate.
- Membrane Bound Elastase Blood was collected from healthy human volunteers. PMNs were isolated by density centrifugation on ficol and red blood cells lysed hypotonically Cells were fixed with paraformaldehyde / gluteraldehyde and washed by centrifugation. Compounds were made up in HBSS containing and incubated for 5 minutes at 37°C with cells. Fluorogenic AAPV substrate (Calbiochem #324745) was added to each well to make 100 ⁇ l final volume and the plate read using a Spectramax Gemini Ex 380 nm Em 460 for 30 min at 37°C. Intracellular Elastase (controlled with lysed cell elastase)
- PMNs were isolated as described previously. PMNs were added to 96-well polypropylene plates and DMSO or compound added to each well to give 150 ⁇ l volume. The plate was incubated at 37 0 C for 30 min. Cells were washed by centrifugation and lysed with HBSS containing 0.04% triton. Cell debris was pelleted and the supernatant transferred to a fresh pate, with compounds or DMSO. Fluorogenic AAPV substrate was added to all wells and the plate was read using a Spectramax Gemini Ex 380 nm Em 460 for 30 min at 37°C. Neutrophil Released Elastase Activity Assay (Human, Mouse, Guinea Pig) Generation of Released Neutrophil Elastase, from Guinea Pigs
- Guinea pigs were treated with an LPS aerosol. Animals were left for 4 hours, euthanized and the lungs lavaged to recover PMN. Bronchoalveolar lavage fluid (BAL) was spun at 400 g for 10minut.es and the cells resuspended in HBSS. 10 ⁇ M cytocholasin B was added to the cell suspension and incubated at 37°C for 5 minutes after which 1 ⁇ M f MLP was added for a further 5 minutes. Cells were centrifuged at 400 g for 10 minutes. 'Elastase rich supernatant' was transferred to a fresh tube. Generation of Released Neutrophil Elastase, from Mice
- mice were anaesthetised and treated with LPS i.n. Animals were left for 4 hours, euthanized and the lungs lavaged to recover PMN.
- Bronchoalveolar lavage fluid (BAL) was centrifuged at 400 g for 10 minutes and the cells resuspended in 1ml of HBSS. 10 ⁇ M cytocholasin B was added to the cell suspension and incubated at 37°C for 5 minutes after which 1 ⁇ M fMLP was added for a further 5 minutes. Cells were centrifuged at 400 g for 10 minutes. 'Elastase rich supernatant' was transferred to a fresh tube. Generation of Human Released Neutrophil Elastase, from Humans
- HNE induced lung haemorrhage in the rat Instillation of human neutrophil elastase (HNE) into rat lung causes acute lung damage. The extent of this injury can be assessed by measuring lung haemorrhage.
- mice Male Sprague Dawley rats (175-220 g) were obtained from Harlan UK Ltd., full barrier- bred and certified free from specified micro-organisms on receipt. Animals were weighed and randomly assigned to treatment groups (7-12 animals per group). The vehicle used was 1 % DMSO/Saline. Inhibitors were dissolved in 1 % DMSO before the addition of 0.9% saline.
- the BALF was centrifuged at 1000 r.p.m. for 10 minutes in a centrifuge cooled to between 4 and 10 0 C. The supernatant was discarded and the cell pellet resuspended in 1 ml 0.1 % CETAB/PBS to lyse the cells. Cell lysates were frozen until spectrophotometric analysis for blood content could be made. Standards were prepared by making solutions of whole rat blood in 0.1% CETAB/PBS.
- a standard curve was constructed by measuring the OD (at 415 nm) of different concentrations of blood in 0.1% CETAB/PBS (30, 10, 7, 3, 1 , 0.3, 0.1 ⁇ l/ml).
- the compounds were shown to have desirable HNE inhibitory activity.
Abstract
A compound of formula (I) (M)-(L)-(M) or formula (IV) [(M)-(L)t]-G: wherein each M is independently an inhibitor of HNE; each L is independently a linker group; t is 2 to 20; G is aryl, heteroaryl, alkyl, cycloalkyl, nitrogen, a dendrimer or a group of any of formulae (V) to (VII): wherein Ar is aryl or heteroaryl; and u is 2 to 20; or a pharmaceutically acceptable salt, solvate, N-oxide or prodrug thereof.
Description
COMPOUNDS CONTAINING MORE THAN ONE HUMAN NEUTROPHIL ELASTASE INHIBITING MOIETY FOR USE IN THE TREATMENT OF RESPIRATORY DISEASES
Field of the Invention
This invention relates to heterocyclic compounds and their use in therapy. Background to the invention
5 Human neutrophil elastase is a 32 kDa serine proteinase found in the azurophilic granules of neutrophils. It has a role in the degradation of a wide range of extracellular matrix proteins, including fibronectin, laminin, proteoglycans, Type III and Type IV collagens as well as elastin (Bieth, G. In Regulation of Matrix accumulation, Mecham, R. P. (Eds), Academic Press, NY1 USA 1986, 217-306). HNE has long been considered to
10 play an important role in homeostasis through repair and disposal of damaged tissues via degradation of the tissue structural proteins. It is also relevant in the defence against bacterial invasion by means of degradation of the bacterial body. In addition to its effects on matrix tissues, HNE has been implicated in the upregulation of IL-8 gene expression and also induces IL-8 release from the epithelial cells of the lung. In animal models of
15 Chronic Obstructive Pulmonary Disease induced by tobacco smoke exposure both small molecule inhibitors and protein inhibitors of HNE inhibit the inflammatory response and the development of emphysema (Wright, J. L. et al. Am. J. Respir. Crit. Care Med.2002, 166, 954-960; Churg, A. et al. Am. J. Respir. Crit. Care Med. 2003, 168, 199-207). Thus, HNE may play a role both in matrix destruction and in amplifying inflammatory responses
20 in chronic respiratory diseases where neutrophil influx is a characteristic feature. Indeed, HNE is believed to play a role in several pulmonary diseases, including chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), acute respiratory distress syndrome (ARDS), pulmonary emphysema, pneumonia and lung fibrosis. It is also implicated in several cardiovascular diseases in which tissue remodelling is involved, for
25 example, in heart failure and the generation of ischaemic tissue injury following acute myocardial infarction.
COPD is an umbrella term encompassing three different pathological conditions, all of which contribute to limitation of airflow: chronic bronchitis, emphysema and small- airway disease. Generally all three will exist to varying extents in patients presenting with
30 COPD, and all three may be due to neutrophil-mediated inflammation, as supported by the increased number of neutrophils observed in bronchoalveolar leakage (BAL) fluids of COPD patients (Thompson, A. B.; Daughton, D.; et al. Am. Rev. Respir. Dis. 1989, 140, 1527-1537). The major pathogenic determinant in COPD has long been considered to be the protease-anti-protease balance (also known as the 'eiastase:anti-elastase
35 hypothesis'), in which an imbalance of HNE and endogenous antiproteases such as α1 - antitrypsin (O1 -AT), Secretory leukocyte protease inhibitor (SLPI) and pre-elafin leads to
the various inflammatory disorders of COPD. Individuals that have a genetic deficiency of the protease inhibitor α1 -antitrypsin develop emphysema that increases in severity over time (Laurrell, C. B.; Erikkson, S Scand. J. Clin. Invest. 1963 75, 132-140). An excess of HNE is therefore destructive, leading to the breakdown of pulmonary morphology with loss of elasticity and destruction of alveolar attachments of airways in the lung (emphysema) whilst simultaneously increasing microvascular permeability and mucus hypersecretion (chronic bronchitis).
Multimeric ligands consist of multiple binding domains which are tethered together through a suitable scaffold. Hence individual binding domains are linked together into a single molecule, increasing the probability that the multimerwill bind simultaneously with multiple active sites resulting in high-affinity interactions (Handl, H. L. etal. Expert Opin. Ther. Targets 2004, 8, 565-586; Han, Y. F. et al., Bioorg. Med. Chem. Letts. 1999, 7, 2569-2575). Also, multiple binding interactions with relatively high off-rates can combine to yield an overall low off-rate for the multimeric ligand. Thus, a molecule consisting of a suitable linker and ligands may be expected to show advantage over the monomeric ligands alone in terms of potency and/or duration of action. Multimeric compounds are unlikely to be orally bioavailable (as predicted by Lipinski's "Rule of 5") which may be advantageous where an inhaled route of administration to the lungs is targeted, since even after inhaled administration, a large proportion of drug is likely to enter the Gl tract. Thus such compounds may be expected to show reduced systemic exposure after inhalation administration and hence an improved toxicity profile over orally administered therapies. Summary of the Invention
A first aspect of the invention is a compound of formula (I) or formula (IV):
(M)-(L)-(M) (I)
[(M)-(L)JrG (IV) wherein each M is independently a group of Formula (II); each L is independently a linker group; t is 2 to 20;
G is aryl, heteroaryl, alkyl, cycloalkyl, nitrogen, a dendrimer or a group of any of formulae (V) to (VII):
Ar'0* Ar Ar-Ar NT JN
U
(V) (Vl) (VII)
wherein Ar is aryl or heteroaryl; and u is 2-20; or a pharmaceutically acceptable salt, solvate, N-oxide or prodrug thereof. In the structures of Formulae (V) or (Vl) each and any of the sites on the Ar groups may be the site or sites of attachment. In the structure of formula (VII) each nitrogen atom may be substituted with between 1 to 3 attachments. When the nitrogen has 3 attachments it is to be understood that the nitrogen is quatemised and bears a positive charge.
Compounds of the invention may be useful in the treatment or prevention of diseases in which HNE is implicated, for example chronic obstructive pulmonary disease (COPD), chronic bronchitis, lung fibrosis, pneumonia, acute respiratory distress syndrome (ARDS), pulmonary emphysema, smoking-induced emphysema, cystic fibrosis, asthma, rhinitis, psoriasis, dermatitis, Chrohn's disease, ulcerative colitis, and irritable bowel disease.
Another aspect of the invention is a pharmaceutical composition comprising a compound of the invention and a pharmaceutically acceptable carrier or excipient.
Another aspect of the invention is the use of a compound of the invention for the manufacture of a medicament for the treatment or prevention of a disease or condition in which HNE is implicated. Description of Preferred Embodiments "Acyl" means a -CO-alkyl group in which the alkyl group is as described herein.
Exemplary acyl groups include -COCH3 and -COCH(CH3)2.
"Acylamino" means a -NR-acyl group in which R and acyl are as described herein. Exemplary acylamino groups include -NHCOCH3 and -N(CH3)COCH3.
"Alkoxy" and "alkyloxy" means an -O-alkyl group in which alkyl is as described below. Exemplary alkoxy groups include methoxy (-OCH3) and ethoxy (-OC2H5).
"Alkoxycarbonyl" means a -COO-alkyl group in which alkyl is as defined below. Exemplary alkoxycarbonyl groups include methoxycarbonyl and ethoxycarbonyl.
"Alkyl" as a group or part of a group refers to a straight or branched chain saturated hydrocarbon group having from 1 to 12, preferably 1 to 6, carbon atoms, in the
chain. Exemplary alkyl groups include methyl, ethyl, 1 -propyl and 2-propyl.
"Alkenyl" as a group or part of a group refers to a straight or branched chain hydrocarbon group having from 1 to 12, preferably 1 to 6, carbon atoms and one carbon- carbon double bond in the chain. Exemplary alkenyl groups include ethenyl, 1 -propenyl, and 2-propenyl.
"Alkylamino" means a -NH-alkyl group in which alkyl is as defined above. Exemplary alkylamino groups include methylamino and ethylamino.
"Alkylene means an -alkyl- group in which alkyl is as defined previously. Exemplary alkylene groups include -CH2-, -(CH2)2- and -C(CH3)HCH2-. "Alkenylene" means an -alkenyl- group in which alkenyl is as defined previously.
Exemplary alkenylene groups include -CH=CH-, -CH=CHCH2-, and -CH2CH=CH-.
"Alkylsulfinyl" means a -SO-alkyl group in which alkyl is as defined above. Exemplary alkylsulfinyl groups include methylsulfinyl and ethylsulfinyl.
"Alkylsulfonyl" means a -SO2-alkyl group in which alkyl is as defined above. Exemplary alkylsulfonyl groups include methylsulfonyl and ethylsulfonyl.
"Alkylthio" means a -S-alkyl group in which alkyl is as defined above. Exemplary alkylthio groups include methylthio and ethylthio.
"Aminoacyl" means a -CO-NRR group in which R is as herein described. Exemplary aminoacyl groups include -CONH2 and -CONHCH3. "Aminoalkyl" means an alkyl-NH2 group in which alkyl is as previously described.
Exemplary aminoalkyl groups include -CH2NH2.
"Aminosulfonyl" means a -SO2-NRR group in which R is as herein described. Exemplary aminosulfonyl groups include -SO2NH2 and -SO2NHCH3.
. "Aryl" as a group or part of a group denotes an optionally substituted monocyclic or multicyclic aromatic carbocyclic.moiety of from 6 to 14 carbon atoms, preferably from 6 to 10 carbon atoms, such as phenyl or naphthyl. The aryl group may be substituted by one or more substituent groups.
"Arylalkyl" means an aryl-alkyl- group in which the aryl and alkyl moieties are as previously described. Preferred arylalkyl groups contain a C1 4 alkyl moiety. Exemplary arylalkyl groups include benzyl, phenethyl and naphthlenemethyl.
"Arylalkyloxy" means an aryl-alkyloxy- group in which the aryl and alkyloxy moieties are as previously described. Preferred arylalkyloxy groups contain a C1 4 alkyl moiety. Exemplary arylalkyl groups include benzyloxy.
"Aryl-fused-cycloalkyl" means a monocyclic aryl ring, such as phenyl, fused to a cycloalkyl group, in which the aryl and cycloalkyl are as described herein. Exemplary aryl- fused-cycloalkyl groups include tetrahydronaphthyl and indanyl. The aryl and cycloalkyl
rings may each be substituted by one or more substituent groups. The aryl-fused- cycloalkyl group may be attached to the remainder of the compound by any available carbon atom.
"Aryl-fused-heterocycloalkyl" means a monocyclic aryl ring, such as phenyl, fused to a heterocycloalkyl group, in which the aryl and heterocycloalkyl are as described herein. Exemplary aryl-fused-heterocycloalkyl groups include tetrahydroquinolinyl, indolinyl, benzodioxinyl, benxodioxolyl, dihydrobenzofuranyl and isoindolonyl. The aryl and heterocycloalkyl rings may each be substituted by one or more substituent groups.
The aryl-fused-heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
"Aryioxy" means an -O-aryl group in which aryl is described above. Exemplary aryloxy groups include phenoxy.
"Cyclic amine" means an optionally substituted 3 to 8 membered monocyclic cycloalkyl ring system where one of the ring carbon atoms is replaced by nitrogen, and which may optionally contain an additional heteroatom selected from O, S or NR (where
R is as described herein). Exemplary cyclic amines include pyrrolidine, piperidine, morpholine, piperazine and Λ/-methylpiperazine. The cyclic amine group may be substituted by one or more substituent groups.
"Cycloalkyl" means an optionally substituted saturated monocyclic or bicyclic ring system of from 3 to 12 carbon atoms, preferably from 3 to 8 carbon atoms, and more preferably from 3 to 6 carbon atoms. Exemplary monocyclic cycloalkyl rings include cyclopropyl, cyclopentyl, cyclohexyl and cycloheptyl. The cycloalkyl group may be substituted by one or more substituent groups.
"Cycloalkylalkyl" means a cycloalkyl-alkyl- group in which the cycloalkyl and alkyl moieties are as previously described. Exemplary monocyclic cycloalkylalkyl groups include cyclopropylmethyl, cyclopentylmethyl, cyclohexylmethyl and cycloheptylmethyl.
"Dendrimer" means a multifunctional core group with a branching group attached to each functional site. Each branching site can be attached to another branching molecule and this process may be repeated multiple times. "Dialkylamino" means a -N(alkyl)2 group in which alkyl is as defined above.
Exemplary dialkylamino groups include dimethylamino and diethylamino.
"Halo" or "halogen" means fluoro, chloro, bromo, or iodo. Preferred are fluoro or chloro.
"Haloalkoxy" means an -O-alkyl group in which the alkyl is substituted by one or more halogen atoms. Exemplary haloalkyl groups include trifluoromethoxy and difluoromethoxy.
"Haloalkyl" means an alkyl group which is substituted by one or more halo atoms. Exemplary haloalkyl groups include trifluoromethyl.
"Heteroaryl" as a group or part of a group denotes an optionally substituted aromatic monocyclic or multicyclic organic moiety of from 5 to 14 ring atoms, preferably from 5 to 10 ring atoms, in which one or more of the ring atoms is/are element(s) other than carbon, for example nitrogen, oxygen or sulfur. Examples of such groups include benzimidazolyl, benzoxazolyl, benzothiazolyl, benzofuranyl, benzothienyl, furyl, imidazolyl, indolyl, indolizinyl, isoxazolyl, isoquinolinyl, isothiazolyl, oxazolyl, oxadiazolyl, pyrazinyl, pyridazinyl, pyrazolyl, pyridyl, pyrimidinyl, pyrrolyl, quinazolinyl, quinolinyl, tetrazolyl, 1 ,3,4-thiadiazolyl, thiazolyl, thienyl and triazolyl groups. The heteroaryl group may be substituted by one or more substituent groups. The heteroaryl group may be attached to the remainder of the compound of the invention by any available carbon or nitrogen atom.
"Heteroarylalkyl" means a heteroaryl-alkyl- group in which the heteroaryl and alkyl moieties are as previously described. Preferred heteroarylalkyl groups contain a lower alkyl moiety. Exemplary heteroarylalkyl groups include pyridylmethyl.
"Heteroarylalkyloxy" means a heteroaryl-alkyloxy- group in which the heteroaryl and alkyloxy moieties are as previously described. Preferred heteroarylalkyloxy groups contain a lower alkyl moiety. Exemplary heteroarylalkyloxy groups include pyridylmethyloxy. "Heteroaryloxy" means a heteroaryloxy- group in which the heteroaryl is as previously described. Exemplary heteroaryioxy groups include pyridyloxy.
"Heteroaryl-fused-cycloalkyl" means a monocyclic heteroaryl group, such as pyridyl or furanyl, fused to a cycloalkyl group, in which heteroaryl and cycloalkyl are as previously described. Exemplary heteroaryl-fused-cycloalkyl groups include tetrahydroquinolinyl and tetrahydrobenzofuranyl. The heteroaryl and cycloalkyl rings may each be substituted by one or more substituent groups. The heteroaryl-fused-cycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
"Heteroaryl-fused-heterocycloalkyl" means a monocyclic heteroaryl group, such as pyridyl or furanyl, fused to a heterocycloalkyl group, in which heteroaryl and heterocycloalkyl are as previously described. Exemplary heteroaryl-fused- heterocycloalkyl groups include dihydrodioxinopyridinyl, dihydropyrrolopyridinyl, dihydrofuranopyridinyl and dioxolopyridinyl. The heteroaryl and heterocycioalkyl rings may each be substituted by one or more substituents groups. The heteroaryl-fused- heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
"Heterocycloalkyl" means: (i) an optionally substituted cycloalkyl group of from 4 to 8 ring members which contains one or more heteroatoms selected from O, S or NR; (ii) a cycloalkyl group of from 4 to 8 ring members which contains CONR and CONRCO (examples of such groups include succinimidyl and 2-oxopyrrolidinyl). The heterocycloalkyl group may be substituted by one or more substituent groups. The heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
"Heterocycloalkylalkyl" means a heterocycloalkyl-alkyl- group in which the heterocycloalkyl and alkyl moieties are as previously described. "Lower alkyl" as a group means unless otherwise specified, an aliphatic hydrocarbon group which may be straight or branched having 1 to 4 carbon atoms in the chain, i.e. methyl, ethyl, propyl (propyl or /sσ-propyl) or butyl (butyl, /so-butyl or fert-butyl).
"Sulfonyl" means a -SO2-alkyl group in which alkyl is as described herein.
Exemplary sulfonyl groups include methanesulfonyl. "Sulfonylamino" means a -NR-sulfonyl group in which R and sulfonyl are as described herein. Exemplary sulfonylamino groups include -NHSO2CI-I3. R means alkyl, aryl, or heteroaryl as described herein.
"Pharmaceutically acceptable salt" means a physiologically or toxicologically tolerable salt and include, when appropriate, pharmaceutically acceptable base addition salts and pharmaceutically acceptable acid addition salts. For example (i) where a compound of the invention contains one or more acidic groups, for example carboxy groups, pharmaceutically acceptable base addition salts that may be formed include sodium, potassium, calcium, magnesium and ammonium salts, or salts with organic amines, such as, diethylamine, Λ/-methyl-glucamine, diethanolamine or amino acids (e.g. lysine) and the like; (ii) where a compound of the invention contains a basic group, such as an amino group, pharmaceutically acceptable acid addition salts that may be formed include hydrochlorides, hydrobromides, phosphates, acetates, citrates, lactates, tartrates, malonates, methanesulphonates and the like.
It will be understood that, as used herein, references to the compounds of the invention are meant to also include the pharmaceutically acceptable salts.
"Prodrug" refers to a compound which is convertible in vivo by metabolic means (e.g. by hydrolysis, reduction or oxidation) to a compound of the invention. For example an ester prodrug of a compound of the invention containing a hydroxy group may be convertible by hydrolysis in vivoto the parent molecule. Suitable esters of compounds of the invention containing a hydroxy group, are for example acetates, citrates, lactates, tartrates, malonates, oxalates, salicylates, propionates, succinates, fumarates, maleates,
methylene-bis-β-hydroxynaphthoates, gentisates, isothionates, di-p-toluoyltartrates, methanesulphonates, ethanesulphonates, benzenesulphonates, p-toluenesulphonates, cyclohexylsulphamates and quinates. As another example an ester prodrug of a compound of the invention containing a carboxy group may be convertible by hydrolysis in vivo to the parent molecule. Examples of ester prodrugs are those described by F. J. Leinweber, Drug Metab. Res., 1987, 18, 379.
It will be understood that, as used in herein, references to the compounds of the invention are meant to also include the prodrug forms.
"Saturated" pertains to compounds and/or groups which do not have any carbon- carbon double bonds or carbon-carbon triple bonds.
The cyclic groups referred to above, namely, aryl, heteroaryl, cycloalkyl, aryl- fused-cycloalkyl, heteroaryl-fused-cycloalkyl, heterocycloalkyl, aryl-fused- heterocycloalkyl, heteroaryl-fused-heterocycloalkyl and cyclic amine may be substituted by one or more substituent groups. Suitable optional substituent groups include acyl (e.g. -COCH3), alkoxy (e.g., -OCH3), alkoxycarbonyl (e.g. -COOCH3), alkyiamino (e.g. -
NHCH3), alkylsulfinyl (e.g. -SOCH3), alkylsulfonyl (e.g. -SO2CH3), alkylthio (e.g. -SCH3), - NH2, aminoacyl (e.g. -CON(CH3)2), aminoalkyl (e.g. -CH2NH2), arylalkyl (e.g. -CH2Ph or -CH2-CH2-Ph), cyano, dialkylamino (e.g. -N(CH3)2), halo, haloalkoxy (e.g. -OCF3 or -OCHF2), haloalkyl (e.g. -CF3), alkyl (e.g. -CH3 or -CH2CH3), -OH, -CHO, -NO2, aryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heteroaryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heterocycloalkyl, aminoacyl (e.g. -CONH2, -CONHCH3), aminosulfonyl (e.g. -SO2NH2, - SO2NHCH3), acylamino (e.g. -NHCOCH3), sulfonylamino (e.g. -NHSO2CH3), heteroarylalkyl, cyclic amine (e.g. morpholine), aryloxy, heteroaryloxy, arylalkyloxy (e.g. benzyloxy) and heteroarylalkyloxy.
Alkylene or alkenylene groups may be optionally substituted. Suitable optional substituent groups include alkoxy (e.g., -OCH3), alkyiamino (e.g. -NHCH3), alkylsulfinyl
(e.g. -SOCHJ, alkylsulfonyl (e.g. -SOXHJ, alkylthio (e.g. -SCHJ, -NH2, aminoalkyl (e.g. - CH2NH2), arylalkyl (e.g. -CH2Ph or -CH2-CH2-Ph), cyano, dialkylamino (e.g. -N(CH3J2), halo, haloalkoxy (e.g. -OCF, or -OCHFJ, haloalkyl (e.g. -CFJ, alkyl (e.g. -CH, or - CH2CH3), -OH, -CHO, and -NO2.
Compounds of the invention may exist in one or more geometrical, optical, enantiomeric, diastereomeric and tautomeric forms, including but not limited to cis- and frans-forms, E- and Z-forms, R-, S- and meso-forms, keto-, and enol-forms. Unless
otherwise stated a reference to a particular compound includes all such isomeric forms, including racemic and other mixtures thereof. Where appropriate such isomers can be separated from their mixtures by the application or adaptation of known methods (e.g. chromatographic techniques and recrystallisation techniques). Where appropriate such isomers may be prepared by the application of adaptation of known methods (e.g. asymmetric synthesis).
Certain compound and combinations of substituents are preferred; in particular see the subclaims.
Each M may independently be a group of Formula (II)

(H)
wherein
A is aryl or heteroaryl; R1, R2 and R3 are independently each hydrogen, halogen, nitro, cyano, CrC6- alkyl, hydroxy or CrC6-alkoxy, wherein d-C6-alkyl and CrC6-alkoxy can be further substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy and CrC4-alkoxy;
R4 is hydrogen, CrCe-alkyl, formyl, aminocarbonyl, mono- or di-CrC4- alkylaminocarbonyl, C3-C8-cycloalkylcarbonyl, CrC6-alkylcarbonyl, CrCδ-alkoxycarbonyl, N-(CrC4-alkylsulfonyl)-aminocarbonyl, N-(Ci-C4-alkylsulfonyl)-N-(CrC4-alkyl)- aminocarbonyl, heteroaryl, heterocycloalkyl, heteroarylcarbonyl or heterocycloalkylcarbonyl; wherein CrC6-alkyl, mono- and di-CrC^alkylaminocarbonyl, CrCe-alkylcarbonyl, CrC6-alkoxycarbonyl, heteroaryl and heterocycloalkyl can be substituted with one to three identical or different radicals selected from the group consisting of aryl, heteroaryl, hydroxyl, CrC4-alkoxy, hydroxycarbonyl, CrC6- alkoxycarbonyl, aminocarbonyl, mono and di-CrC4-alkylaminocarbonyl, amino, mono- and di-CrC4-alkylamino, CrCValkylcarbonylamino, M-(C1 -C6)-alkyl)-silyl, cyano, N-
(mono- and di-CrC4-alkylamino-CrC4-alkyl)-aminocarbonyl, N-(C1 -C4-alkoxy-Ci -C4- alkyl)-aminocarbonyl and halogen; or
R4 represents a group of Formula (VIII)

R4A is hydrogen or CrCe-alkyl, and n is 1 or 2; R5 is CrC4-alkyl, which can be substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy, CrC6-alkoxy, C1-C6- alkenoxy, CrC6-alkylthio, amino, mono- and di- CrC6-alkylamino, arylamino, hydroxycarbonyl, CrC6-alkoxycarbonyl and the radical -0-(C1 -C4-BIkVl)-O-(C1 -C4-alkyl); or R5 is amino; R6 is halogen, nitro, cyano, CrC6-alkyl, hydroxy or CrC6-alkoxy, wherein
CrCe-alkyl and CrC6-alkoxy can be further substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy and C1-C4- alkoxy; and
Y1, Y2, Y3, Y4 and Y5 are independently each CH or N, with the proviso that the ring in which they are comprised contains no more than 2 N atoms. In a preferred embodiment, A is a phenyl ring. In one embodiment, R1 is hydrogen.
In another embodiment, R2 is methyl. In a further embodiment, R2 is -CN. In a preferred embodiment, R3 is hydrogen. In another preferred embodiment, R4 is alkoxycarbonyl.
In a further preferred embodiment, R5 is methyl. In yet another preferred embodiment, R6 is trifluoromethyl. In yet another preferred embodiment, Y1, Y2, Y3, Y4 and Y5 are each CH. L may be a group of formula (III): -La-Z-(Wa-Z)m-Wb-Z-LΛ (III)
wherein m is O to 20;
each La is independently , -S(O)W-, -, -C(O)O-, -C(O)-, -C(O)NR7-, -C(S)-, -C(S)O-, -C(O)S-, -C(S)NR7- or a covalent bond; each Z is independently optionally substituted alkylene, optionally substituted cycloalkylene, optionally substituted alkenylene, optionally substituted alkynylene, optionally substituted cycloalkenylene, aryi, heteroaryl, heterocycloalkyl or a covalent bond;
Wa and Wb are independently -0-, -C(O)-, -OC(O)-, -C(O)O-, -NR7-, S(O)n-, - C(O)NR7-, -NR7C(O)-, NR7C(O)NR7-, NR7C(S)NR7-, -C(=NR7)NR7-, -OC(O)NR7-, - NR7C(O)O-, -N=C(La)NR7-, -P(O)(OR7)O-, -OP(O)(OR7)-, S(O)nCR7R8-, -S(O)nNR7-, - NR7S(O)n-, -S-S- or a covalent bond,
R7 and R8 are independently hydrogen, optionally substituted alkyl, optionally substituted cycloalkyl, optionally substituted alkenyl, optionally substituted cycloalkenyl, optionally substituted alkynyl, aryl, heteroaryl or heterocycloalkyl; and each W is independently 0,1 or 2. In a preferred embodiment, the group -La-Z-(Wa-Z)m-Wb-Z-La- represents an alkylene group.
In another preferred embodiment, Wa and Wb are each -0-, -NR7, or a covalent bond.
In a further preferred embodiment, a compound of the invention is of formula (IV). In yet another preferred embodiment, a compound of the invention is as defined in Table 1 , or in Examples 1 to 15.
In a preferred embodiment, each M is the same or different and is a group of formula (II) as defined herein. In formula (II), the arrow denotes the point of attachment of M to the linker L. Preferably, L is a group of Formula (III) as defined herein. The linkers, L, used in this invention can be selected so as to allow multivalent binding of ligands to binding sites on human neutrophil elastase, whether such sites are located at the substrate binding site and/or at other binding sites on the same enzyme molecule, or on binding sites of more than one enzyme molecule via all combinations of available binding sites. G may be a group of any of formulae (V) to (VII) or a dendrimer. Examples of groups of formulae (V) to (VII) include, but are not limited to phenoxyphenyl, biphenyl, bipyridyl, ethylenediamino, propylenediamino and the like. It is to be understood that the number of possible attachment points is dictated by the valency of the groups present, so that for example, biphenyl can contain up to 10 possible attachments (5 on each phenyl ring), and ethylenediamine can possess up to 4 possible attachments (2 on each terminal amine). An example of a dendrimer suitable for use in the invention is:

Preferred compounds of the invention include:





The therapeutic utility of the present compounds is pertinent to any disease that is known to be at least partially mediated by the action of human neutrophil elastase. For example, the present compounds may be beneficial in the treatment of chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), acute respiratory distress syndrome (ARDS), pulmonary emphysema, pneumonia and lung fibrosis.
The present invention is also concerned with pharmaceutical formulations comprising, as an active ingredient, a compound of the invention. Other compounds may be combined with compounds of this invention for the prevention and treatment of inflammatory diseases of the lung. Thus the present invention is also concerned with
pharmaceutical compositions for preventing and treating inflammatory diseases of the lung comprising a therapeutically effective amount of a compound of the invention and one or more other therapeutic agents.
Suitable therapeutic agents for a combination therapy with compounds of the invention include: (1 ) a corticosteroid, for example fluticasone or budesonide; (2) a β2- adrenoreceptor agonist, for example salmeterol or formeterol; (3) a leukotriene modulator, for example montelukast or pranlukast; (4) anticholinergic agents, for example selective muscarinic-3 (M3) receptor antagonists such as tiotropium bromide; (5) phosphodiesterase-IV (PDE-IV) inhibitors, for example roflumilast or cilomilast; (6) an antitussive agent, such as codeine or dextramorphan; and (7) a non-steroidal antiinflammatory agent (NSAID), for example ibuprofen or ketoprofen.
The weight ratio of the first and second active ingredients may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. The magnitude of prophylactic or therapeutic dose of a compound of the invention will, of course, vary with the nature of the severity of the condition to be treated and with the particular compound and its route of administration. It will also vary according to the age, weight and response of the individual patient. In general, the daily dose range will lie within the range of from about 0.001 mg to about 100 mg per kg body weight of a mammal, preferably 0.01 mg to about 50 mg per kg, and most preferably 0.1 to 10 mg per kg, in single or divided doses. On the other hand, it may be necessary to use dosages outside these limits in some cases.
Another aspect of the present invention provides pharmaceutical compositions which comprise a compound of the invention and a pharmaceutically acceptable carrier. The term "composition", as in pharmaceutical composition, is intended to encompass a product comprising the active ingredient(s), and the inert ingredient(s) (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. Accordingly, the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the invention, additional active ingredient(s), and pharmaceutically acceptable excipients.
The pharmaceutical compositions of the present invention comprise a compound of the invention as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic
ingredients. The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dosage of a compound of the present invention. In therapeutic use, the active compound may be administered by any convenient, suitable or effective route. Suitable routes of administration are known to those skilled in the art, and include oral, intravenous, rectal, parenteral, topical, ocular, nasal, buccal and pulmonary. Delivery by inhalation is preferred. Compositions suitable for administration by inhalation are known, and may include carriers and/or diluents that are known for use in such compositions. The composition may contain 0.01-99% by weight of active compound. Preferably, a unit dose comprises the active compound in an amount of 1 μg to 10 mg.
The most suitable dosage level may be determined by any suitable method known to one skilled in the art. It will be understood, however, that the specific amount for any particular patient will depend upon a variety of factors, including the activity of the specific compound that is used, the age, body weight, diet, general health and sex of the patient, time of administration, the route of administration, the rate of excretion, the use of any other drugs, and the severity of the disease undergoing treatment. For delivery by inhalation, the active compound is preferably in the form of microparticles. They may be prepared by a variety of techniques, including spray-drying, freeze-drying and micronisation.
By way of example, a composition of the invention may be prepared as a suspension for delivery from a nebuliser or as an aerosol in a liquid propellant, for example for use in a pressurised metered dose inhaler (PMDI). Propellants suitable for use in a PMDI are known to the skilled person, and include CFC-12, HFA-134a, HFA- 227, HCFC-22 (CCI2F2) and HFA-152 (CH2F2) and isobutane.
In a preferred embodiment of the invention, a composition of the invention is in dry powder form, for delivery using a dry powder inhaler (DPI). Many types of DPI are known.
Microparticles for delivery by administration may be formulated with excipients that aid delivery and release. For example, in a dry powder formulation, microparticles may be formulated with large carrier particles that aid flow from the DPI into the lung. Suitable earner particles are known, and include lactose particles; they may have a mass median aerodynamic diameter of greater than 90 μm.
In the case of an aerosol-based formulation, a preferred composition is:
Compound of the invention 24 mg / canister Lecithin, NF Liq. Cone. 1.2 mg / canister
Trichlorofluoromethane, NF 4.025 g / canister Dichlorodifluoromethane, NF 12.15 g / canister. Compounds of the invention may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which present compounds are useful. Such other drugs may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with a compound of the invention. When a compound of the invention is used contemporaneously with one or more other drugs, a pharmaceutical composition containing such other drugs in addition to the compound of the invention is preferred. Accordingly, the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the invention. The agents of the invention may be administered in inhaled form. Aerosol generation can be carried out using, for example, pressure-driven jet atomizers or ultrasonic atomizers, preferably using propellant-driven metered aerosols or propellant- free administration of micronized active compounds from, for example, inhalation capsules or other "dry powder" delivery systems. The active compounds may be dosed as described depending on the inhaler system used. In addition to the active compounds, the administration forms may additionally contain excipients, such as, for example, propellants (e.g. Frigen in the case of metered aerosols), surface-active substances, emulsifiers, stabilizers, preservatives, flavorings, fillers (e.g. lactose in the case of powder inhalers) or, if appropriate, further active compounds.
For the purposes of inhalation, a large number of systems are available with which aerosols of optimum particle size can be generated and administered, using an inhalation technique which is appropriate for the patient. In addition to the use of adaptors (spacers, expanders) and pear-shaped containers (e.g. Nebulator®, Volumatic®), and automatic devices emitting a puffer spray (Autohaler®), for metered aerosols, in particular in the case of powder inhalers, a number of technical solutions are available (e.g. Diskhaler®, Rotadisk®, Turbohaler® or the inhalers for example as described EP-A-0505321). Methods of Synthesis The compounds of the invention of the present invention can be prepared according to the procedures of the following schemes and examples, using appropriate
materials, and are further exemplified by the following specific examples. Moreover, by utilising the procedures described with the disclosure contained herein, one of ordinary skill in the art can readily prepare additional compounds of the present invention claimed herein. The compounds illustrated in the examples are not, however, to be construed as forming the only genus that is considered as the invention. The examples further illustrate details for the preparation of the compounds of the present invention. Those skilled in the art will readily understand that known variations of the conditions and processes of the following preparative procedures can be used to prepare these compounds. The compounds of the invention may be isolated in the form of their pharmaceutically acceptable salts, such as those described previously herein above. The free acid form corresponding to isolated salts can be generated by neutralisation with a suitable acid such as acetic acid and hydrochloric acid and extraction of the liberated free acid into an organic solvent followed by evaporation. The free acid form isolated in this manner can be further converted into another pharmaceutically acceptable salt by dissolution in an organic solvent followed by addition of the appropriate base and subsequent evaporation, precipitation, or crystallisation.
It may be necessary to protect reactive functional groups (e.g. hydroxy, amino, thio orcarboxy) in intermediates used in the preparation of compounds of the invention to avoid their unwanted participation in a reaction leading to the formation of the compounds. Conventional protecting groups, for example those described by T. W. Greene and P. G. M. Wuts in "Protective groups in organic chemistry" John Wiley and Sons, 1999, may be used.
Compounds of the invention may be prepared according to the routes illustrated in Scheme 1.

1-2
Scheme 1
Compounds of formula 1-1 wherein A, Y1, Y2, Y3, Y4, Y5, R1, R2, R3, R4, R5 and R6 are as defined above, may be prepared from compounds of formula IX-, wherein X is a suitable leaving group such as halogen or a sulfonate ester, by reaction with a suitably substituted diamine. The reaction may be conducted in the presence of a base, such as sodium bicarbonate in a solvent such as acetonitrile. Heating the reaction in a microwave reactor may be beneficial.
Compounds of formula I-2 may be prepared from compounds of formula IX-4 by reaction with a suitably substituted coupling partner of formula IX-3. The reaction takes place in the presence or absence of base. Suitable bases include NaH and potassium carbonate in a solvents such as DMF.
Compounds of formula IX-4 may be prepared from compounds of IX-3 in a similar manner to that used to prepare compounds of formula 1-2 from compounds of formula IX-4.
Compounds of formula IX-3 may be prepared from compounds of formula IX-5 by reaction with a suitable bis-electrophile, such as an α, ω- dihalide or disulfonate. The reaction may be conducted in dlute aqueous sodium hydroxide in the presence of a phase-transfer catalyst such as a trialkylammonium halide. Compounds of formula IX-5 are known in the art (WO04024701 ).
The following Examples illustrate the invention. It will be understood that the processes detailed below are solely for the purpose of illustrating the invention and should not be construed as limiting. A process utilising similar or analogous reagents and/or conditions known to one skilled in the art may also be used to obtain a compound
of the invention.
General Experimental Details
All reactions were carried out under an atmosphere of nitrogen unless specified otherwise. Microwave irradiation was carried out using a Biotage EmrysTM Optimizer. All solvents and commercial reagents were used as received.
Where products were purified by column chromatography, lsolute Si Il SPE cartridges were used. Compounds containing a basic centre(s), which were purified by HPLC, were obtained as the TFA or formate salts. Preparative HPLC conditions: HPLC system 1
Phenyl hexyl column (250 x 21.20 mm Luna column with 5 μm particle size), eluting with a gradient of A: water + 0.1 % TFA; B: acetonitrile + 0.1 % TFA at a flow rate of 5 ml/min with UV detection at 254 nm. HPLC system 2 C18-reverse-phase column (100 x 22.5 mm i.d Genesis column with 7 μm particle size), eluting with a gradient of A: water + 0.1 % HCO2H; B: acetonitrile + 0.1 % HCO2H at a flow rate of 5 ml/min and gradient of 1 %/min increasing in B. UV detection at 230 nm. HPLC system 3
Phenyl hexyl column (250 x 21.20 mm Luna column with 5 μm particle size), eluting with a gradient of A: water + 0.1 % HCO2H; B: acetonitrile + 0.1 % HCO2H at a flow rate of 5 ml/min with UV detection at 254 nm. HPLC system 4
Amylose tris(3,5-dimethylphenylcarbamate) (250 x 20 mm CHIRALPAK IA column with 5 μm particle size), eluting with an isocratic mixture of ethanol (10%) in n-heptane at a flow rate of 15 ml/min with UV detection at 230 nm. LC/MS Systems
The Liquid Chromatography Mass Spectroscopy (LC/MS) systems used: LC-MS method 1 (long run)
Micromass Platform LCT with a C18-reverse-phase column (100 x 3.0 mm Higgins Clipeus with 5 μm particle size), elution with A: water + 0.1% formic acid; B: acetonitrile + 0.1% formic acid. Gradient:
Gradient - Time flow ml/min %A %B
0.00 1.0 95 5
1.00 1.0 95 5
15.00 1.0 5 95
20.00 1.0 5 95
22.00 1.0 95 5
25.00 1.0 95 5
Detection - MS, ELS, UV (100 μl split to MS with in-line UV detector)
MS ionisation method - Electrospray (positive ion) LC-MS method 2 (open-access 1)
Micromass Platform LCT with a C18-reverse-phase column (30 x 4.6 mm Phenomenex Luna 3 μm particle size), elution with A: water + 0.1 % formic acid; B: acetonitrile + 0.1 % formic acid. Gradient:
Gradient - Time flow ml/min %A %B 0.00 2.0 95 5
0.50 2.0 95 5
4.50 2.0 5 95
5.50 2.0 5 95
6.00 2.0 95 5 Detection - MS, ELS, UV (100 μl split to MS with in-line UV detector)
MS ionisation method - Electrospray (positive and negative ion) LC-MS method 3 (open-access 2)
Waters Micromass ZQ with a C18-reverse-phase column (30 x 4.6 mm Phenomenex Luna 3 μm particle size), elution with A: water + 0.1 % formic acid; B: acetonitrile + 0.1 % formic acid. Gradient:
Gradient - Time flow ml/min %A %B
0.00 2.0 95 5
0.50 2.0 95 5
4.50 2.0 5 95 5.50 2.0 5 95
6.00 2.0 95 5
Detection - MS, ELS, UV (100 μl split to MS with in-line UV detector)
MS ionisation method - Electrospray (positive and negative ion)
Abbreviations used in the experimental section:
DCM = dichloromethane
CHCI3 = chloroform
EtOAc = ethyl acetate
HCI = hydrochloric acid
THF = tetrahydrofuran
RT = room temperature
DCE = 1 ,1-dichloroethane
DMF = N,N-dirnethylformamide
Rt = retention time
Intermediate 1 (IX-5: A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C)

A solution of 4-cyanobenzaldehyde (1.875 g, 14.3 mmol) and 3- trifluoromethylphenylthiourea (1.5 g, 6.82 mmol) in THF (40 ml) was added to polyphosphoric acid (3.4 g). Ethyl acetoacetate (1.75 ml, 13.75 mmol) was added and the mixture mechanically stirred and heated to reflux for 2.5 h. The reaction was cooled and diluted with 1 M HCI (100 ml). The resulting solution was extracted with EtOAc and the combined EtOAc fractions were reduced to give 4.6 g of a thick orange oil. Purification was achieved via column chromatography (loaded onto diatomaceous earth) using 100% CHCI3 as eiuent, followed by 100% cyclohexane, followed by 30% EtOAc in cyclohexane to remove the product as a yellow gum.
Yield: 3.1O g (83%)
LC-MS (Method 3): Rt 3.87 min, m/z 446 [MH+]
Intermediate 2 (IX-3: A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, X = I, n = 10)

Intermediate 1 (300 mg, 0.67 mmol), 1 ,10-diiododecane (600 mg, 1.5 mmol), 1 M aqueous sodium hydroxide (1.5 ml, 1.5 mmol), tetra-n-butylamonium iodide (27 mg, 0.072 mmol) and water (2 ml) were charged into a 5 ml Biotage microwave vial. The mixture was then irradiated at 1400C for 5 min. The resulting orange solution was extracted with EtOAc and the combined EtOAc fractions were reduced to afford an orange gum. Purification was achieved via column chromatography (loaded onto diatomaceous earth) using 0-14% step gradient of EtOAc in cyclohexane (2% steps) as eluent, followed by 100% cyclohexane, followed by 30% EtOAc in cyclohexane to afford the product as a colorless gum.
Yield: 272 mg (57%)
LC-MS (Method 3): Rt 5.57 min, m/z 711 [MH+]
Intermediate 3 (IX-3: A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = WIe, R6 = CF3, Y1-Y5 = C, X = I, n = 5)

Intermediate 3 was prepared by a similar procedure from intermediate 1 (500 mg, 1.1 mmol), 1 ,5-diiodopentane (1.1 g, 0.5 ml, 3.3 mmol), 1 M aqueous sodium hydroxide (3 ml, 3 mmol), tetra-n-butylammonium iodide (45 mg, 0.12 mmol) and water (3 ml). Purification was achieved via column chromatography (loaded onto diatomaceous earth) using 0-14% step gradient of EtOAc in cyclohexane (2% steps ) as eluent, followed by
100% cyclohexane, followed by 30% EtOAc in cyclohexane to afford the product as a colorless gum.
Yield: 324 mg (46%)
LC-MS (Method 3): Rt 4.79 min, m/z 642 [MH+]
Intermediate 4 (IX-3: A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, X = Br, n = 5)

Intermediate 1 (1 g, 2.2 mmol), 1 ,5-dibromopentane (1.2 ml), 1 M aqueous sodium hydroxide (10 ml), tetra-n-butylammonium iodide (90 mg, 0.24 mmol) and DCM (10 ml ml) were stirred together at RT for 18 h. The organic layer was separated, dried (Na2SO4) and evaporated. Purification was achieved via SPE cartridge chromatography using a 5-20% step gradient of EtOAc in cyclohexane. Intermediate 4 was obtained as an orange gum.
Yield: 734 mg (56%)
LC-MS (Method 2): Rt 4.83 min, m/z 596 [MH+]
Intermediate 5 (IX-3: A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, X = Br, n = 6)

Intermediate 5 was prepared in a similar manner from Intermediate 1 (1 g) and 1 ,6-dibromohexane. Purification was achieved via SPE cartridge chromatography using a 5-20% step gradient of EtOAc in cyclohexane.
Yield: 861 mg (64%)
LC-MS (Method 3): Rt 4.72 min, m/z 610 [MH+]
Intermediate 6 (IX-4: A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1 -Y5 = C, n = 5)

Imidazole (8 mg, 0.118 mmol) was dissolved in DMF (1 ml) and the solution was cooled to -78°C. Sodium hydride (60%, 4.4 mg, 0.129 mmol) was added and the reaction was stirred at -200C for 1 h. Intermediate 4 (70 mg, 0.118 mmol) in DMF (1 ml) was added and stirring was continued at 700C for 18 h. The DMF was evaporated and the residue partitioned between EtOAc and water. The organic layer was separated and reduced. Purification was achieved by HPLC (System 2).
Yield: 37 mg (53%)
LC-MS (Method 3): Rt 2.62 min, m/z 582 [MH+] Intermediate 7

Boc-Piperazine (0.5 g, 2.69 mmol), 1 ,2-dibromoethane (253 mg, 1.34 mmol) and NaHCO3 (564 mg, 6.72 mmol) in acetonitrile (20 ml) were heated at 900C for 17h. After cooling to RT, the solvent was removed and the residue was dissolved in ethyl acetate (80 ml). The organic solution was washed with water (50 ml) and saturated brine (20 ml), dried (Na2SO4) and evaporated to give intermediate 7 as a white solid.
Yield: 426 mg (80%)
LC-MS (Method 2): Rt 0.34/2.05 min, m/z 399 [MH+]
Intermediate 8 (IX-5: A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C)
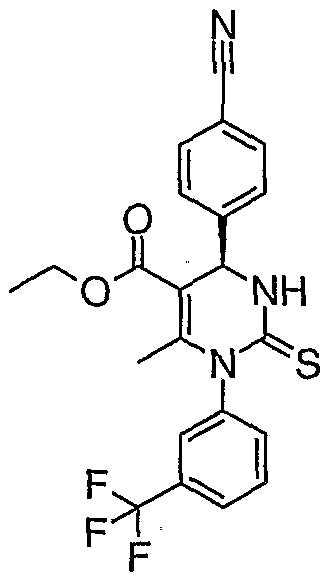
Intermediate 1 was separated into its constituent enantiomers using HPLC (System 4). Intermediate 8 was the second enantiomer to elute from the column. Intermediate 9 (IX-3: A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, X = I, n = 5)

Intermediate 9 was made from Intermediate 8 (638 mg) in an analogous manner to Intermediate 3.
Yield: 683 mg (75%) LC-MS (Method 2): Rt 9.45 min, m/z 1144.24 [MH]+
Example 1 (1-1 : A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1 -Y5 = C, -N(R)XN(R)- = piperazin-1 ,4-diyI, n = 10)

A solution of intermediate 2 (77 mg, 0.108 mmol) and piperazine (3 mg, 0.035 mmoi) in acetonitrile (1.3 ml) was added to sodium hydrogencarbonate (30 mg) in a 2 ml
Biotage microwave vial. The mixture was irradiated at 1000C for 25 min. The crude solution was diluted with water and purified by HPLC (System 1).
Yield: 49.4 mg (95%)
LC-MS (Method 1): Rt 13.69 min, m/z 1253.43 [MH]+
Example 2 (1-1 : A = Ph5 R1 = CN5 R2 = H, R3 = H5 R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, X = -(CHa)2NCH3(CHz)2-, R = H, n = 10)
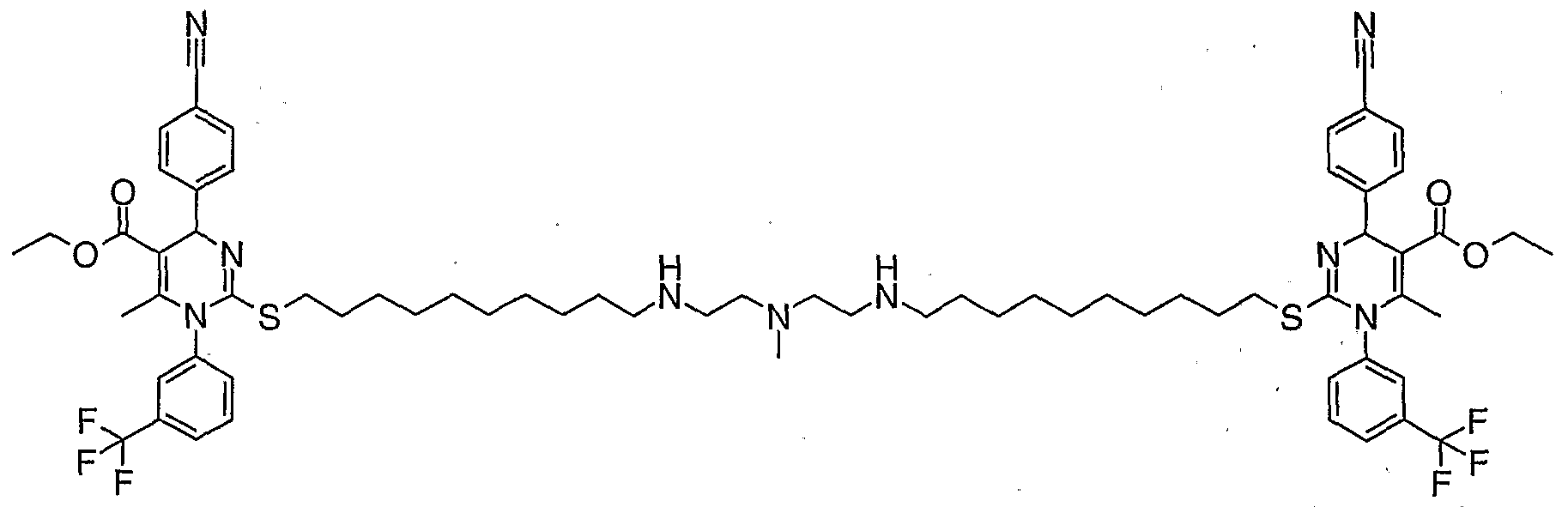
Example 2 was prepared by a similar procedure from intermediate 2 (77 mg) and N-methyl-2,2'-diaminodiethylamine. The crude solution was diluted with water and purified by HPLC (System 1).
Yield: 36.8 mg (68%)
LC-MS (Method 1): Rt 10.86 min, m/z 1284.47 [MH]+
Example 3 (1-1 : A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et5 R5 = Me, R6 = CF3, Y1 -Y5 = C, X = -(CH2)2-, R = -(CHz)2OH, n = 10)

Example 3 was prepared by a similar procedure from intermediate 2 (77 mg) and N,N'-Bis(2-hydroxyethyl)ethylenediamine. The crude solution was diluted with water and purified by HPLC (System 1 ).
Yield: 5.6 mg (10%)
LC-MS (Method 1): Rt 13.16 min, m/z 1315.47 [MH]+
Example 4 (1-1 : A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, -N(R)XN(R)- = piperazin-1,4-diyl, n = 5)

Example 4 was prepared by a similar procedure from intermediate 3 (108 mg) and piperazine. The crude solution was diluted with water and purified by HPLC (System 1).
Yield: 56.8 mg (73%)
LC-MS (Method 1): Rt 11.31 min, m/z 1113.29 [MH]+
Example 5 (M : A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, X = -(CH2)2NCH3(CH2)2-, R = H, n = 5)
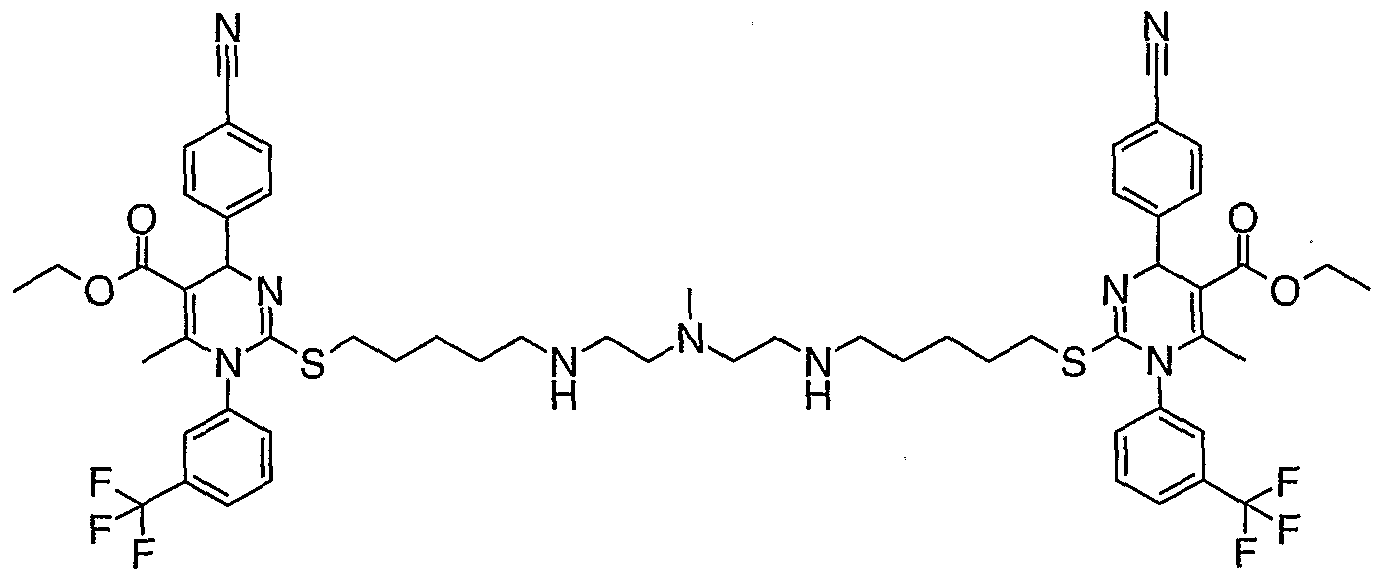
Example 5 was prepared by a similar procedure from intermediate 3 (108 mg) and N-methyl-2,2'-diaminodiethylamine. The crude solution was diluted with water and purified by HPLC (System 1).
Yield: 31.4 mg (39%)
LC-MS (Method 1): Rt 9.44 min, m/z 1144.33 [MH]+
Also isolated from this reaction was;
Example 6

Example 6 was isolated from the same reaction as example 5.
Yield: 13.6 mg (17%)
LC-MS (Method 1): Rt 9.52 min, m/z 1144.34 [MH]+
Example 7 (1-1 : A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = WIe5 R6 = CF3, Y1 -Y5 : C, X = -(CH2)3O(CH2)4θ(CH2)3-5 R = H, n = 5)

Example 7 was prepared by a similar procedure from intermediate 3 (96 mg) and 4,9-dioxa-1 ,12-dodecanediamine. Purification was achieved using HPLC (System 3). Yield: 16 mg (16%) LC-MS (Method 1): Rt 9.55 min, m/z 1231.34 [MH]+
Example 8 (1-1 : A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 - CF3, Y1 -Y5 = C, X = -(CHa)10-, R = H, n = 5)

Intermediate 4 (100 mg, 0.17 mmol), 1 ,10-diaminodecane (14.6 mg, 0.085 mmol) and cesium carbonate (111 mg, 0.34 mmol) were stirred in DMF (2 ml) containing water (9.18 μl, 0.51 mmol). After 60 h the mixture was evaporated and purified by HPLC (System 2)
Yield: 4 mg (4%)
LC-MS (Method 1): Rt 9.72 min, m/z 1199.41 [MH]+
Example 9 (1-1 : A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1 -Y5 = C, X = -(CH2)2-, R = -(CH2J2OH, n = 5)

Example 9 was prepared by a similar procedure from intermediate 3 (69 mg) and N,N'-bis(2-hydroxyethyl)ethylenediamine. Purification was effected by HPLC (System 1). Yield: 19 mg (28%) LC-MS (Method 1): Rt 10.84/11.34 min, m/z 1175.34 [MH]+
Example 10 (1-1 : A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, X = -(CHa)2-, R = -(CH2)2OH, n = 6)

Example 10 was prepared by a similar procedure from intermediate 5 (100 mg) and N,N'-bis(2-hydroxyethyl)ethylenediamine. Purification was effected by HPLC (System 2).
Yield: 9 mg (8%)
LC-MS (Method 1 ): Rt 10.94 min, m/z 1203.14 [MH]+
Example 11 (1-1 : A = Ph, R1 = CN, R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, X = -(CHa)2NCH3(CHa)2-, R = H, n = 6)

Example 11 was prepared by a similar procedure from intermediate 5 (100 mg) and N-(2-aminoethyl)-N-methylethanediamine. Purification was effected by HPLC
(System 2).
Yield: 27 mg (22%)
LC-MS (Method 1): Rt 9.67 min, m/z 1172.27 [MH]+
Example 12 (1-1 : A = Ph, R1 = CN5 R2 = H, R3 = H, R4 = CO2Et5 R5 = Me5 R6 = CF3, Y1 -Y5 = C, -N(R)XN(R)- = piperazine-(CH2)2-piperazinyl, n = 5)

Intermediate 7 (29.9 mg, 0.075 mmol) was treated with 25% TFA in DCM (4 ml). After 30 min the solution was evaporated to dryness. The residue was dissolved in acetonitrile (1 ml) and sodium hydrogen carbonate (63 mg, 0.75 mmol) and Intermediate 3 (96 mg, 0.15 mmol) were added. The mixture was heated at 1500C for 35 min in the microwave. The reaction mixture was purified by HPLC (System 2 then System 3).
Yield: 20 mg (20%)
LC-MS (Method 1 ): Rt 9.17 min, m/z 1225.39 [MH]+
Example 13 (I-2: A = Ph5 R1 = CN5 R2 = H5 R3 = H5 R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C)

Intermediate 6 (37.5 mg, 0.0645 mmol) and intermediate 4 (38 mg, 0.0645 mmol) were dissolved in DMF (2 ml). The solution was stirred at 75°C for 18 h and then at 85°C for a further 48 h. The DMF was removed and the residue was purified by HPLC (System
2).
Yield: 20 mg (28%)
LC-MS (Method 1): Rt 11.85 min, m/z 1095.26 [M]+
Example 14 (1-1 : A = Ph, R1 = CN5 R2 = H, R3 = H, R4 = CO2Et, R5 = Me, R6 = CF3, Y1-Y5 = C, X = -(CHa)2NCH3(CHa)2-, R = H, n = 5)

Example 14 was made from Intermediate 9 in an analogous manner to Example 5. Purification was achieved using HPLC (System 2).
LC-MS (Method 1 ): Rt 9.45 min, m/z 1144.24 [MH]+ Example 15

Example 15 was obtained as a by-product in the synthesis of Intermediate 9. LC-MS (Method 1 ): Rt 16.49 min, m/z 959.20 [MH]+
Example 6: elastase inhibition assays
Various compounds of the invention were tested for their inhibitory activity towards HNE. Fluorescent peptide substrate Assays were performed in 96-well plates at a total assay volume of 100μl. The final concentration of the enzyme (human leukocyte elastase, Sigma E8140) was 0.00036 units/well. A peptide substrate (MeO-Suc-Ala-Ala-Pro-ValAMC, Calbiochem #324745) was used, at the final concentration of 100μM. The final concentration of DMSO was 1 % in the assay buffer (0.05 M Tris.HCI, pH 7.5, 0.1 M NaCI; 0.1 M CaCI2; 0.0005% brij-35).
The enzymatic reaction was started by adding the enzyme. The enzymatic reaction was performed at RT and after 30 mins stopped by adding 50 μl soybean trypsin
inhibitor (Sigma T-9003) at a final concentration of 50 μg/well. Fluorescence was read on the FLEXstation (Molecular Devices) using 380 nm excitation and 460 nm emission filters. The potency of the compounds was determined from a concentration series of 10 concentrations in range from 1000 nM to 0.051 nM. The results are means of two independent experiments, each performed in duplicate. Using Fluorescently labelled elastin
Assays were performed in 96-well plate at a total assay volume of 100 μl. The final concentration of the enzyme (human leukocyte elastase, Sigma E8140) was 0.002 units/well. Fluorescently labelled, solubilised elastin from bovine neck ligament (Molecular Probes, E-12056) was used at the final concentration of 15 μg/ml. The final concentration of DMSO was 2.5% in the assay buffer (0.1 M Tris-HCL, pH 8.0, containing 0.2 mM sodium azide).
The enzymatic reaction was started by adding the enzyme. The enzymatic reaction was performed at RT and read after 120 minutes. Fluorescence was read on the FLEXstation (Molecular Devices) using 485 nm excitation and 530 nm emission filters. The potency of the compounds was determined from a concentration series of 10 concentrations in range from 25000 nM to 1 nM. The results are means of two independent experiments, each performed in duplicate.
The compounds tested were shown to have IC50 values for HNE in the range 1- 100O nM.
Elastase Selectivity Assays
Selectivity for elastase inhibition was determined by testing the compounds against a panel of 6 proteases: plasmin, thrombin, cathepsin G, proteinase 3, trypsin, chymotrypsin (all sourced from Sigma, cat. No. P1867, T1063, C4428, P0615, T6424, C8949 respectively). Assays were performed in 96-well plate at a total assay volume of 100 μl. A common, generic substrate was used for all proteases: fluorescently labelled casein (Molecular Probes, E-6639), at the final concentration of 20 μg/ml (Cathepsin G and Chymotrypsin), 10μg/ml (Plasmin and Thrombin) or 5 μg/ml (Proteinase 3 and Trypsin). The final concentration of the substrate was close to the respective Km values as determined for this substrate. The final concentration of DMSO was 5% in the assay buffer (0.05 M Tris.HCl, pH 7.5, 0.1 M NaCI; 0.1 M CaCI2; 0.0005% brij-35). The enzymatic reaction was started by adding the enzyme. The enzymatic reaction was performed at RT for 60 min. Fluorescence was read on the FLEXstation (Molecular Devices) using 589 nm excitation and 617 nm emission filters. The potency of the compounds was determined from a concentration series of 8 concentrations in range from 500 μM to 0.2 μM. The results are means of two independent experiments, each
performed in duplicate.
The compounds tested showed selectivities for a range of proteases fromi to 300 fold. Membrane Bound Elastase Blood was collected from healthy human volunteers. PMNs were isolated by density centrifugation on ficol and red blood cells lysed hypotonically Cells were fixed with paraformaldehyde / gluteraldehyde and washed by centrifugation. Compounds were made up in HBSS containing and incubated for 5 minutes at 37°C with cells. Fluorogenic AAPV substrate (Calbiochem #324745) was added to each well to make 100 μl final volume and the plate read using a Spectramax Gemini Ex 380 nm Em 460 for 30 min at 37°C. Intracellular Elastase (controlled with lysed cell elastase)
PMNs were isolated as described previously. PMNs were added to 96-well polypropylene plates and DMSO or compound added to each well to give 150 μl volume. The plate was incubated at 370C for 30 min. Cells were washed by centrifugation and lysed with HBSS containing 0.04% triton. Cell debris was pelleted and the supernatant transferred to a fresh pate, with compounds or DMSO. Fluorogenic AAPV substrate was added to all wells and the plate was read using a Spectramax Gemini Ex 380 nm Em 460 for 30 min at 37°C. Neutrophil Released Elastase Activity Assay (Human, Mouse, Guinea Pig) Generation of Released Neutrophil Elastase, from Guinea Pigs
Guinea pigs were treated with an LPS aerosol. Animals were left for 4 hours, euthanized and the lungs lavaged to recover PMN. Bronchoalveolar lavage fluid (BAL) was spun at 400 g for 10minut.es and the cells resuspended in HBSS. 10 μM cytocholasin B was added to the cell suspension and incubated at 37°C for 5 minutes after which 1 μM f MLP was added for a further 5 minutes. Cells were centrifuged at 400 g for 10 minutes. 'Elastase rich supernatant' was transferred to a fresh tube. Generation of Released Neutrophil Elastase, from Mice
Mice were anaesthetised and treated with LPS i.n. Animals were left for 4 hours, euthanized and the lungs lavaged to recover PMN. Bronchoalveolar lavage fluid (BAL) was centrifuged at 400 g for 10 minutes and the cells resuspended in 1ml of HBSS. 10μM cytocholasin B was added to the cell suspension and incubated at 37°C for 5 minutes after which 1 μM fMLP was added for a further 5 minutes. Cells were centrifuged at 400 g for 10 minutes. 'Elastase rich supernatant' was transferred to a fresh tube. Generation of Human Released Neutrophil Elastase, from Humans
Human PMN were isolated as described previously. 10 μM cytocholasin B was
added to the cell suspension and incubated at 370C for 5 minutes after which 1 μM fMLP was added for a further 5 minutes. Cells were centrifuged at 400 g for 10 minutes. Εlastase rich supernatant' was transferred to a fresh tube.
To a clear bottomed 96-well plate compounds were added and incubated for 5 minutes at 37°C with 'elastase rich' supernatant. Fluorogenic AAPV substrate was added to all wells and the plate read using a Spectramax Gemini Ex 380 nm Em 460 for 30 minutes at 37°C. For comparison, an activity matched control of human elastase was also run. HNE induced lung haemorrhage in the rat Instillation of human neutrophil elastase (HNE) into rat lung causes acute lung damage. The extent of this injury can be assessed by measuring lung haemorrhage. Male Sprague Dawley rats (175-220 g) were obtained from Harlan UK Ltd., full barrier- bred and certified free from specified micro-organisms on receipt. Animals were weighed and randomly assigned to treatment groups (7-12 animals per group). The vehicle used was 1 % DMSO/Saline. Inhibitors were dissolved in 1 % DMSO before the addition of 0.9% saline.
Animals in each study used to determine the efficacy of the elastase inhibitors delivered locally to the lung by a variety of routes. Rats were anaesthetised with the inhaled anaesthetic lsoflurane (4%) when the dose was given from 30 minutes to 6 h prior to human neutrophil elastase (HNE) administration or terminally anaesthetised with hypnorm:hypnovel:water (1.5:1 :2 at 2.7 mi/kg) when the predose was given at less than 30 minutes prior to HNE administration and dosed either intratracheally (i.t.) by transoral administration using a Penn Century microsprayer or intranasally (i.n.) by dropping the fluid on to the nares. Animals either received vehicle or compound at a dose volume of 0.5 ml/kg.
Animals that had been allowed to recover after dosing were terminally anaesthetised with hypnorm:hypnovel:water (1.5:1 :2 at 2.7 ml/kg). Once sufficiently anaesthetised, HNE (600 units/ml) or sterile saline was administered by transoral tracheal instillation at a volume of 100 μl using a Penn Century microsprayer. Animals were kept warm in a temperature controlled box and given top up doses of anaesthetic as required to ensure continuous anaesthesia until termination.
Animals were sacrificed (0.5 ml to 1 ml sodium pentobarbitone) one hour post HNE challenge. The trachea was exposed and a small incision made between two tracheal rings allowing a cannula (10 gauge, O. D. 2-10mm, Portex Ltd.) to be inserted approximately 2 cm into the trachea towards the lung. This was secured into place with a cotton ligature. The lungs were then lavaged (BAL) three times with fresh 4 ml aliquots
of heparinised (10 units/ml) phosphate buffered saline (PBS). The resultant BALF was kept on ice until it was centrifuged.
The BALF was centrifuged at 1000 r.p.m. for 10 minutes in a centrifuge cooled to between 4 and 100C. The supernatant was discarded and the cell pellet resuspended in 1 ml 0.1 % CETAB/PBS to lyse the cells. Cell lysates were frozen until spectrophotometric analysis for blood content could be made. Standards were prepared by making solutions of whole rat blood in 0.1% CETAB/PBS.
Once defrosted 100 μl of each lysed cell suspension was placed into a separate well of a 96 well flat bottomed plate. All samples were tested in duplicate and 100 μl 0.1 % CETAB/PBS was included on the plate as a blank. The OD of the contents of each well was measured at 415 nm using a spectramax 250 (Molecular devices).
A standard curve was constructed by measuring the OD (at 415 nm) of different concentrations of blood in 0.1% CETAB/PBS (30, 10, 7, 3, 1 , 0.3, 0.1 μl/ml).
The amount of blood in each experimental sample was calculated by comparison to the standard curve. Data were then analysed as below:
1 ) The mean OD for duplicates was calculated
2) The value for the blank was subtracted from the value for all other samples
3) Data were assessed to evaluate the normality of distribution.
The compounds were shown to have desirable HNE inhibitory activity.
Claims
1. A compound of formula (I) or formula (IV):
(M)-(L)-(M) (I)
[(M)-(L)]rG (IV) wherein each M is independently an inhibitor of HNE; each L is independently a linker group; t is 2 to 20;
G is aryl, heteroaryl, alkyl, cycloalkyl, nitrogen, a dendrimer or a group of any of formulae (V) to (VII):
U
(V) (Vl) (VII) wherein
Ar is aryl or heteroaryl; and u is 2 to 20; or a pharmaceutically acceptable salt, solvate, N-oxide or prodrug thereof.
2. A compound according to claim 1 , where each M is independently a group of Formula (II)

(H)
wherein A is aryl or heteroaryl;
R1, R2 and R3 are independently each hydrogen, halogen, nitro, cyano, Ci-C6- alkyl, hydroxy or CrC6-alkoxy, wherein Ci-C6-alkyl and CrC6-alkoxy can be further substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy and CrC4-alkoxy;
R4 is hydrogen, CrC6-alkyl, formyl, aminocarbonyl, mono- or di-d-C4- alkylaminocarbonyl, C3-C8-cycloalkylcarbonyl, d-C6-alkylcarbonyl, CrC6-alkoxycarbonyl,
N^d-d-alkylsulfonyO-aminocarbonyl, N-(Ci-C4-alkylsulfonyl)-N-(CrC4-alkyl)- aminocarbonyl, heteroaryl, heterocycloalkyl, heteroarylcarbonyl or heterocycloalkylcarbonyl; wherein CrC6-alkyl, mono- and di-d-Cralkylaminocarbonyl,
CrC6-alkylcarbonyl, CrC6-alkoxycarbonyl, heteroaryl and heterocycloalkyl can be substituted with one to three identical or different radicals selected from the group consisting of aryl, heteroaryl, hydroxyl, CrC4-alkoxy, hydroxycarbonyl, CrC6- alkoxycarbonyl, aminocarbonyl, mono and di-d-Cralkylaminocarbonyl, amino, mono- and di-CrC4-alkylamino, CrC4-alkylcarbonylamino, tri-(CrC6)-alkyl)-silyl, cyano, N-
(mono- and di-CrC4-alkylamino-CrC4-alkyl)-aminocarbonyl, N-(Ci-C4-alkoxy-CrC4- alkyl)-aminocarbonyl and halogen; or
R4 represents a group of Formula (VIII)

R4A is hydrogen or d-C6-alkyl, and n is 1 or 2;
R5 is Ci-C4-alkyl, which can be substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy, d-Cβ-alkoxy, CrC6- alkenoxy, d-C6-alkylthio, amino, mono- and di- CrC6-alkylamino, arylamino, hydroxycarbonyl, d-C6-alkoxycarbonyl and the radical -O-(CrC4-alkyl)-O-(CrC4-alkyl); or R5 is amino;
R6 is halogen, nitro, cyano, CrC6-alkyl, hydroxy or CrC6-alkoxy, wherein d-C6-alkyl and d-C6-alkoxy can be further substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy and CrC4- alkoxy; and
Y1, Y2, Y3, Y4 and Y5 are independently each CH or N, with the proviso that the ring in which they are comprised contains no more than 2 N atoms.
3. A compound according to claim 2, wherein each A is phenyl.
4. A compound according to claim 2 or claim 3, wherein R1 is hydrogen.
5. A compound according to any of claims 2 to 4, wherein each R2 is independently methyl or -CN.
6. A compound according to any of claims 2 to 5, wherein each R3 is hydrogen.
7. A compound according to any of claims 2 to 6, wherein each R4 is alkoxycarbonyl.
8. A compound according to any of claims 2 to 7, wherein each R5 is methyl.
9. A compound according to any of claims 2 to 8, wherein each R6 is trifluoromethyl.
10. A compound according to any of claims 2 to 9, wherein Y1 , Y2, Y3, Y4 and Y5 are each CH.
11. A compound according to any preceding claim, wherein L is a group of formula (III):
-La-Z-(Wa-Z)m-Wb-Z-La- (III)
wherein m is 0 to 20; each La is independently , -S(OV, -, -C(O)O-, -C(O)-, -C(O)NR7-, -C(S)-, -C(S)O-, -C(O)S-, -C(S)NR7- or a covalent bond; each Z is independently optionally substituted alkylene, optionally substituted cycloalkylene, optionally substituted alkenylene, optionally substituted alkynylene, optionally substituted cycloalkenylene, aryl, heteroaryl, heterocycloalkyl or a covalent bond;
Wa and Wb are independently -0-, -C(O)-, -OC(O)-, -C(O)O-, -NR7-, S(O)n-, - C(O)NR7-, -NR7C(O)-, NR7C(O)NR7-, NR7C(S)NR7-, -C(=NR7)NR7-, -OC(O)NR7-, - NR7C(O)O-, -N=C(La)NR7-, -P(O)(OR7)O-, -OP(O)(OR7)-, S(O)nCR7R8-, -S(O)nNR7-, - NR7S(O)n-, -S-S- or a covalent bond,
R7 and R8 are independently hydrogen, optionally substituted alkyl, optionally substituted cycloalkyl, optionally substituted alkenyl, optionally substituted cycloalkenyl, optionally substituted alkynyl, aryl, heteroaryl or heterocycloalkyl; and each w is independently 0,1 or 2.
12. A compound according to claim 11 wherein -La-Z-(Wa-Z)m-Wb-Z-lΛ represents an alkylene group.
13. A compound according to claim 11 , wherein Wa and Wb are each -0-, -NR7-, or a covalent bond.
14. A compound according to any preceding claim, which is of formula (IV).
15. A compound according to claim 1 , as defined in Table 1 or in Examples 1 to 15.
16. A compound according to any preceding claim, for use in therapy.
17. A pharmaceutical composition comprising a compound of any of claims 1 to 15 and a pharmaceutically acceptable carrier or excipient.
18. Use of a compound of any of claims 1 to 19, for the manufacture of a medicament for use in the treatment of prevention of a disease or condition in which HNE is implicated.
19. Use according to claim 18, wherein the disease or condition is chronic obstructive pulmonary disease (COPD), chronic bronchitis, lung fibrosis, pneumonia, acute respiratory distress syndrome (ARDS), pulmonary emphysema, smoking-induced emphysema or cystic fibrosis.
20. Use according to claim 18, wherein the disease or condition is asthma, rhinitis, psoriasis, dermatitis, (atopic and non-atopic), Crohn's disease, ulcerative colitis or irritable bowel disease.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0520743.6A GB0520743D0 (en) | 2005-10-12 | 2005-10-12 | Compounds and their use |
| GB0520743.6 | 2005-10-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007042815A1 true WO2007042815A1 (en) | 2007-04-19 |
Family
ID=35451608
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2006/003791 WO2007042815A1 (en) | 2005-10-12 | 2006-10-12 | Compounds containing more than one human neutrophil elastase inhibiting moiety for use in the treatment of respiratory diseases |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB0520743D0 (en) |
| WO (1) | WO2007042815A1 (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007129060A1 (en) * | 2006-05-04 | 2007-11-15 | Argenta Discovery Limited | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors |
| WO2008135537A1 (en) * | 2007-05-03 | 2008-11-13 | Argenta Discovery Limited | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors |
| WO2009060203A1 (en) * | 2007-11-07 | 2009-05-14 | Argenta Discovery Limited | 3,4,6,7-tetrahydro-1 h-pyrrolo[3,4-d]pyrimidine-2,5-diones and their therapeutic use |
| DE102007061766A1 (en) | 2007-12-20 | 2009-06-25 | Bayer Healthcare Ag | New 4-(4-cyano-2-thioaryl)-dihydro-pyrimidinone compounds are human neutrophil elastase inhibitor, useful for the treatment or prevention of e.g. pulmonary arterial hypertonia, acute lung injury and diseases of the cardiovascular system |
| DE102008022521A1 (en) | 2008-05-07 | 2009-11-12 | Bayer Schering Pharma Aktiengesellschaft | 1,4-Diaryl-pyrimidopyridazine-2,5-diones and their use |
| DE102008052013A1 (en) | 2008-10-17 | 2010-04-22 | Bayer Schering Pharma Aktiengesellschaft | New 4-(4-cyano-2-thioaryl)dihydropyrimidinone compounds are neutrophil elastase inhibitors useful to treat or prevent e.g. pulmonary arterial hypertension, chronic obstructive pulmonary disease, acute lung injury, or cystic fibrosis |
| DE102009016553A1 (en) | 2009-04-06 | 2010-10-07 | Bayer Schering Pharma Aktiengesellschaft | Sulfonamide- and sulfoximine-substituted diaryldihydropyrimidinones and their use |
| DE102010030187A1 (en) | 2010-06-16 | 2011-12-22 | Bayer Schering Pharma Aktiengesellschaft | New 4-cyano-2-sulfonylphenyl-pyrazolyl-substituted pyridinones and pyrazinones compounds are human neutrophil elastase inhibitors, useful to treat and prevent e.g. pulmonary arterial hypertension and chronic obstructive pulmonary disease |
| US8198288B2 (en) | 2006-05-04 | 2012-06-12 | Pulmagen Therapeutics (Inflammation) Limited | Tetrahydropyrrolopyrimidinediones and their use in therapy |
| US8288402B2 (en) | 2007-12-20 | 2012-10-16 | Bayer Intellectual Property Gmbh | 4-(4-cyano-2-thioaryl)dihydropyrimidinones and their use |
| CN102858774A (en) * | 2010-03-12 | 2013-01-02 | 奇斯药制品公司 | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as copd |
| CN102858773A (en) * | 2010-03-12 | 2013-01-02 | 奇斯药制品公司 | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as copd |
| US10316001B2 (en) | 2015-03-18 | 2019-06-11 | Ph Pharma Co., Ltd. | Method for producing (4S)-4-[4-cyano-2-(methylsulfonyl)phenyl]-3,6-dimethyl-2-oxo-1-[3-(trifluoromethyl)phenyl]-1,2,3,4-tetrohydro pyrimidine-5-carbonitrile |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004024701A1 (en) * | 2002-09-10 | 2004-03-25 | Bayer Healthcare Ag | Heterocyclic derivatives |
| WO2006082412A2 (en) * | 2005-02-03 | 2006-08-10 | Argenta Discovery Limited | Multimers of pyrimidinone derivatives and their use as human neutrophil elastase inhibitors |
-
2005
- 2005-10-12 GB GBGB0520743.6A patent/GB0520743D0/en not_active Ceased
-
2006
- 2006-10-12 WO PCT/GB2006/003791 patent/WO2007042815A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004024701A1 (en) * | 2002-09-10 | 2004-03-25 | Bayer Healthcare Ag | Heterocyclic derivatives |
| WO2006082412A2 (en) * | 2005-02-03 | 2006-08-10 | Argenta Discovery Limited | Multimers of pyrimidinone derivatives and their use as human neutrophil elastase inhibitors |
Non-Patent Citations (2)
| Title |
|---|
| HAN YI FAN ET AL: "Dual-site binding of bivalent 4-aminopyridine- and 4-aminoquinoline-based AChE inhibitors: contribution of the hydrophobic alkylene tether to monomer and dimer affinities", BIOORGANIC & MEDICINAL CHEMISTRY, ELSEVIER SCIENCE LTD, GB, vol. 7, no. 11, 1999, pages 2569 - 2575, XP002365936, ISSN: 0968-0896 * |
| HANDL H L ET AL: "HITTING MULTIPLE TARGETS WITH MULTIMERIC LIGANDS", EXPERT OPINION ON THERAPEUTIC TARGETS, ASHLEY PUBLICATIONS, LONDON, GB, vol. 8, no. 6, 2004, pages 565 - 586, XP009069513, ISSN: 1472-8222 * |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8198288B2 (en) | 2006-05-04 | 2012-06-12 | Pulmagen Therapeutics (Inflammation) Limited | Tetrahydropyrrolopyrimidinediones and their use in therapy |
| WO2007129060A1 (en) * | 2006-05-04 | 2007-11-15 | Argenta Discovery Limited | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors |
| US8957082B2 (en) | 2006-05-04 | 2015-02-17 | Chiesi Farmaceutici S.P.A | Tetrahydropyrrolopyrimidinediones and their use in therapy |
| EA018061B1 (en) * | 2006-05-04 | 2013-05-30 | Пульмаджен Терапьютикс (Инфлэмэйшн) Лимитед | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors |
| WO2008135537A1 (en) * | 2007-05-03 | 2008-11-13 | Argenta Discovery Limited | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors |
| WO2009013444A1 (en) * | 2007-07-25 | 2009-01-29 | Argenta Discovery Limited | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors |
| WO2009060203A1 (en) * | 2007-11-07 | 2009-05-14 | Argenta Discovery Limited | 3,4,6,7-tetrahydro-1 h-pyrrolo[3,4-d]pyrimidine-2,5-diones and their therapeutic use |
| DE102007061766A1 (en) | 2007-12-20 | 2009-06-25 | Bayer Healthcare Ag | New 4-(4-cyano-2-thioaryl)-dihydro-pyrimidinone compounds are human neutrophil elastase inhibitor, useful for the treatment or prevention of e.g. pulmonary arterial hypertonia, acute lung injury and diseases of the cardiovascular system |
| US9174997B2 (en) | 2007-12-20 | 2015-11-03 | Bayer Intellectual Property Gmbh | 4-(4-cyano-2-thioaryl)dihydropyrimidinones and use thereof |
| US8288402B2 (en) | 2007-12-20 | 2012-10-16 | Bayer Intellectual Property Gmbh | 4-(4-cyano-2-thioaryl)dihydropyrimidinones and their use |
| DE102008022521A1 (en) | 2008-05-07 | 2009-11-12 | Bayer Schering Pharma Aktiengesellschaft | 1,4-Diaryl-pyrimidopyridazine-2,5-diones and their use |
| US8580800B2 (en) | 2008-05-07 | 2013-11-12 | Bayer Intellectual Property Gmbh | 1,4-diaryl-pyrimidopyridazine-2,5-diones and their use |
| DE102008052013A1 (en) | 2008-10-17 | 2010-04-22 | Bayer Schering Pharma Aktiengesellschaft | New 4-(4-cyano-2-thioaryl)dihydropyrimidinone compounds are neutrophil elastase inhibitors useful to treat or prevent e.g. pulmonary arterial hypertension, chronic obstructive pulmonary disease, acute lung injury, or cystic fibrosis |
| WO2010115548A1 (en) | 2009-04-06 | 2010-10-14 | Bayer Schering Pharma Aktiengesellschaft | Sulfonic amide and sulfoximine-substituted diaryl-dihydropyrimidinones and usage thereof |
| US8691817B2 (en) | 2009-04-06 | 2014-04-08 | Bayer Intellectual Property Gmbh | Sulfonic amide and sulfoximine-substituted diaryl-dihydropyrimidinones and usage thereof |
| DE102009016553A1 (en) | 2009-04-06 | 2010-10-07 | Bayer Schering Pharma Aktiengesellschaft | Sulfonamide- and sulfoximine-substituted diaryldihydropyrimidinones and their use |
| CN102858774A (en) * | 2010-03-12 | 2013-01-02 | 奇斯药制品公司 | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as copd |
| CN102858773A (en) * | 2010-03-12 | 2013-01-02 | 奇斯药制品公司 | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as copd |
| CN102858773B (en) * | 2010-03-12 | 2015-05-06 | 奇斯药制品公司 | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as COPD |
| CN102858774B (en) * | 2010-03-12 | 2015-05-06 | 奇斯药制品公司 | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as COPD |
| DE102010030187A1 (en) | 2010-06-16 | 2011-12-22 | Bayer Schering Pharma Aktiengesellschaft | New 4-cyano-2-sulfonylphenyl-pyrazolyl-substituted pyridinones and pyrazinones compounds are human neutrophil elastase inhibitors, useful to treat and prevent e.g. pulmonary arterial hypertension and chronic obstructive pulmonary disease |
| US10316001B2 (en) | 2015-03-18 | 2019-06-11 | Ph Pharma Co., Ltd. | Method for producing (4S)-4-[4-cyano-2-(methylsulfonyl)phenyl]-3,6-dimethyl-2-oxo-1-[3-(trifluoromethyl)phenyl]-1,2,3,4-tetrohydro pyrimidine-5-carbonitrile |
| US10676443B2 (en) | 2015-03-18 | 2020-06-09 | Ph Pharma Co., Ltd. | Method for producing (4S)-4-[4-cyano-2-(methylsulfonyl)phenyl]-3,6-dimethyl-2-oxo-1-[3-(trifluoromethyl)phenyl]-1,2,3,4-tetrahydro pyrimidine-5-carbonitrile |
Also Published As
| Publication number | Publication date |
|---|---|
| GB0520743D0 (en) | 2005-11-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007042815A1 (en) | Compounds containing more than one human neutrophil elastase inhibiting moiety for use in the treatment of respiratory diseases | |
| EP1856059B1 (en) | Multimers of pyrimidinone derivatives and their use as human neutrophil elastase inhibitors | |
| US20090105268A1 (en) | Dihydropyrimidone Multimers and Their Use as Human Neutrophil, Elastase Inhibitors | |
| US8957082B2 (en) | Tetrahydropyrrolopyrimidinediones and their use in therapy | |
| US9156844B2 (en) | Pyrimidine derivatives and their use in the treatment of respiratory diseases such a COPD | |
| US9120802B2 (en) | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as COPD | |
| US8198288B2 (en) | Tetrahydropyrrolopyrimidinediones and their use in therapy | |
| WO2009013444A1 (en) | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors | |
| WO2009060158A1 (en) | 4- (4-cyanophenyl) -1- (3-trifluoromethylphenyl) -3,4, 6, 7-tetrahydro-1h-pyrrolo [3, 4- d] pyrimidine-2, 5-dione derivatives and their use as human neutrophil elastase inhibitors | |
| WO2009037413A1 (en) | Dimers of 5- [ (4-cyanophenyl) sulfinyl] -6-methyl-2-oxo-1- [3- (trifluoromethyl)phenyl] -1,2-dihydropyridine-3-carboxamide as inhibitors of human neutrophil elastase for treating respiratory diseases | |
| WO2009060206A1 (en) | 3,4,6,7-tetrahydro-1h-pyrrolo[3,4-d]pyrimidine-2,5-diones and their therapeutic use | |
| EP2391629B1 (en) | Dimeric pyrrolopyrimidinedione and its use in therapy of respiratory diseases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06794739 Country of ref document: EP Kind code of ref document: A1 |