WO2007041630A1 - Deuterated inhibitors of gastric h+, k+-atpase with enhanced therapeutic properties - Google Patents
Deuterated inhibitors of gastric h+, k+-atpase with enhanced therapeutic properties Download PDFInfo
- Publication number
- WO2007041630A1 WO2007041630A1 PCT/US2006/038819 US2006038819W WO2007041630A1 WO 2007041630 A1 WO2007041630 A1 WO 2007041630A1 US 2006038819 W US2006038819 W US 2006038819W WO 2007041630 A1 WO2007041630 A1 WO 2007041630A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- enantiomer
- compound
- deuterium
- mixture
- formula
- Prior art date
Links
- 0 *C(*)(c1c(*)c(*)c(*)c(*)n1)S(c1nc(c(*)c(*)c(*)c2*)c2[n]1)=O Chemical compound *C(*)(c1c(*)c(*)c(*)c(*)n1)S(c1nc(c(*)c(*)c(*)c2*)c2[n]1)=O 0.000 description 32
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention is directed to inhibitors of the gastric H + , K + - ATPase and pharmaceutically acceptable salts and prodrugs thereof, the chemical synthesis thereof, and the medical use of such compounds for the treatment and/or management of duodenal ulcers, heartburn, acid reflux, other conditions mediated by gastric acid secretion and/or psoriasis.
- the human body expresses various enzymes (e.g. the cytochrome P 450 enzymes or CYPs, esterases, proteases, reductases, dehydrogenases, and the like) that react with the chemicals and nutrients to produce novel intermediates or metabolites.
- enzymes e.g. the cytochrome P 450 enzymes or CYPs, esterases, proteases, reductases, dehydrogenases, and the like
- Some of the most common metabolic reactions of pharmaceutical compounds involve the oxidation of a carbon-hydrogen (C-H) bond to either a carbon-oxygen (C-O) or carbon-carbon (C-C) ⁇ -bond.
- the resultant metabolites may be stable or unstable under physiological conditions, and can have substantially different pharmacokinetic, pharmacodynamic, acute and long-term toxicity profiles relative to the parent compounds. For most drugs, such oxidations are generally rapid and ultimately lead to administration of multiple or high daily doses. There is therefore an obvious and immediate need for improvements of such drugs.
- Chemical kinetics is the study of reaction rates.
- the activation energy E aot in chemistry is the energy that must be supplied to a system in order to initiate a particular chemical process. In other words, this is the minimum energy required for a specific chemical reaction to take place. A reaction will occur between two properly oriented molecules if they possess a minimum requisite energy. During the approach, the outer shell electrons of each molecule will induce repulsion.
- RT is the average amount of thermal energy that molecules possess at a certain temperature T, where R is the molar gas constant, k is the rate constant for the reaction and A (the frequency factor) is a constant specific to each reaction that depends on the probability that the molecules will collide with the correct orientation.
- the transition state in a reaction is a short lived state (on the order of 10 ⁇ 14 sec) along the reaction pathway during which the original bonds have stretched to their limit.
- the activation energy E act for a reaction is the energy required to reach the transition state of that reaction. Reactions that involve multiple steps will necessarily have a number of transition states, and in these instances, the activation energy for the reaction is equal to the energy difference between the reactants and the most unstable transition state. Once the transition state is reached, the molecules can either revert, thus reforming the original reactants, or the new bonds form giving rise to the products. This dichotomy is possible because both pathways, forward and reverse, result in the release of energy.
- a catalyst facilitates a reaction process by lowering the activation energy leading to a transition state.
- Enzymes are examples of biological catalysts that reduce the energy necessary to achieve a particular transition state.
- a carbon-hydrogen bond is by nature a covalent chemical bond. Such a bond forms when two atoms of similar electronegativity share some of their valence electrons, thereby creating a force that holds the atoms together. This force or bond strength can be quantified and is expressed in units of energy, and as such, covalent bonds between various atoms can be classified according to how much energy must be applied to the bond in order to break the bond or separate the two atoms.
- the bond strength is directly proportional to the absolute value of the ground-state vibrational energy of the bond.
- This vibrational energy which is also known as the zero-point vibrational energy, depends on the mass of the atoms that form the bond.
- the absolute value of the zero-point vibrational energy increases as the mass of one or both of the atoms making the bond increases. Since deuterium (D) is two-fold more massive than hydrogen (H), it follows that a C-D bond is stronger than the corresponding C-H bond.
- Compounds with C-D bonds are frequently indefinitely stable in H 2 O, and have been widely used for isotopic studies. If a C-H bond is broken during a rate-determining step in a chemical reaction (i.e.
- DKIE deuterium Kinetic Isotope Effect
- 1 no isotope effect
- very large numbers such as 50 or more, meaning that the reaction can be fifty, or more, times slower when deuterium is substituted for hydrogen.
- High DKIE values may be due in part to a phenomenon known as tunneling, which is a consequence of the uncertainty principle. Tunneling is ascribed to the small size of a hydrogen atom, and occurs because transition states involving a proton can sometimes form in the absence of the required activation energy. A deuterium is larger and statistically has a much lower probability of undergoing this phenomenon. Substitution of tritium for hydrogen results in yet a stronger bond than deuterium and gives numerically larger isotope effects.
- deuterium is a stable and non-radioactive isotope of hydrogen. It was the first isotope to be separated from its element in pure form and is twice as massive as hydrogen, and makes up about 0.02% of the total mass of hydrogen (in this usage meaning all isotopes) on earth.
- deuterium oxide D 2 O or "heavy water"
- D 2 O looks and tastes like H 2 O but it has different physical properties. It boils at 101.41 0 C and freezes at 3.79 °C. Its heat capacity, heat of fusion, heat of vaporization, and entropy are all higher than H 2 O. It is also more viscous and is not as powerful a solvent as H 2 O.
- Tritium (T) is a radioactive isotope of hydrogen, used in research, fusion reactors, neutron generators and radiopharmaceuticals. Mixing tritium with a phosphor provides a continuous light source, a technique that is commonly used in wristwatches, compasses, rifle sights and exit signs. It was discovered by Rutherford, Oliphant and Harteck in 1934 and is produced naturally in the upper atmosphere when cosmic rays react with H 2 molecules. Tritium is a hydrogen atom that has 2 neutrons in the nucleus and has an atomic weight close to 3. It occurs naturally in the environment in very low concentrations, most commonly found as T 2 O, a colorless and odorless liquid.
- PK pharmacokinetics
- PD pharmacodynamics
- toxicity profiles have been demonstrated previously with some classes of drugs.
- DKIE was used to decrease the hepatotoxicity of halothane by presumably limiting the production of reactive species such as trifluoroacetyl chloride.
- this method may not be applicable to all drug classes.
- deuterium incorporation can lead to metabolic switching which may even give rise to an oxidative intermediate with a faster off-rate from an activating Phase I enzyme (e.g. cytochrome P 450 3A4).
- Omeprazole (PRILOSEC ® ) is an inhibitor of the gastric H + , K + -ATPase.
- This class of drugs includes, among others, esomeprazole, lansoprazole, pantoprazole, rabeprazole, leminoprazole, ilaprazole, nepaprazole, saviprazole and tenatoprazole.
- the mechanism of action of these drugs has been extensively studied, and it is postulated that they react transiently with a critical cysteine in the gastric H + , K + -ATPase ("gastric H + pump").
- Omeprazole has been shown to degrade to inactive and less active metabolites as part of its metabolic clearance from systemic circulation. This degradation is so rapid that the producer of omeprazole (AstraZeneca) undertook a full development/clinical program to develop and market its successor (i.e. esomeprazole (NEXIUM ® )), a homochiral analog of omeprazole with a very similar pharmacodynamic, pharmacokinetic and toxicological profile. The only noticeable difference resides in the fact that the half-life of esomeprazole in human plasma is 20% higher than that of omeprazole. Clinical advantages of esomeprazole versus omeprazole are unclear.
- Esomeprazole is extensively metabolized in the liver by the cytochrome P 450 (CYP) enzyme system.
- the metabolites of esomeprazole lack antisecretory activity.
- the major part of esomeprazole 's metabolism is dependent upon the CYP2C19 isoenzyme which forms the hydroxy- and desmethyl metabolites.
- the remaining amount is dependent on CYP3A4 which forms the sulphone metabolite.
- CYP2C19 isoenzyme exhibits polymorphism in the metabolism of esomeprazole, since some 3% of Caucasians and 15-20% of Asians lack CYP2C19 and are termed poor metabolizers.
- AUC Area Under the Curve
- R 1 , R 4 , R 9 and R 10 are independently selected from the group consisting of hydrogen, and deuterium;
- R 2 , R 3 , R 6 , and R 8 are independently selected from the group consisting of hydrogen, deuterium, C 1 -C 6 alkyl, and C 1 -C 6 alkyloxy;
- R. 5 is selected from the group consisting Of-CH 2 -, -CHD- and -CD 2 -;
- R 7 is selected from the group consisting of hydrogen, deuterium, -NO 2 , Ci-C 6 alkyl, and Cj-C 6 alkyloxy; provided that compounds of Formula 1 contain at least one deuterium atom, and provided that deuterium enrichment in compounds of Formula 1 is at least about 1%.
- compositions comprising a compound according to Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)-enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, with a pharmaceutically acceptable carrier.
- the carbon-hydrogen bonds of these gastric H + , K + -ATPase modulators contain a naturally occurring distribution of hydrogen isotopes, namely 1 H or protium (about 99.9844%), 2 H or deuterium (about 0.0156%), and 3 H or tritium (in the range between about 0.5 and 67 tritium atoms per 10 18 protium atoms).
- Increased levels of deuterium incorporation produce a detectable Kinetic Isotope Effect (KIE) that could affect the pharmacokinetic, pharmacologic and/or toxicologic parameters of these gastric H + , K + - ATPase modulators relative to compounds having naturally occurring levels of deuterium.
- KIE Kinetic Isotope Effect
- aspects of the present invention disclosed herein describe a novel approach to designing and synthesizing new analogs of these gastric H + , K + -ATPaSe modulators through chemical modifications and derivations of the carbon-hydrogen bonds of the modulators and/or of the chemical precursors used to synthesize said modulators.
- Suitable modifications of certain carbon-hydrogen bonds into carbon-deuterium bonds may generate novel gastric H + , K + - ATPase modulators with unexpected and non-obvious improvements of pharmacological, pharmacokinetic and toxicological properties in comparison to the non-isotopically enriched gastric H + , K + -ATPaSe modulators.
- This invention relies on the judicious and successful application of chemical kinetics to drug design. Deuterium incorporation levels in the compounds of the invention are significantly higher than the naturally-occurring levels and are sufficient to induce at least one substantial improvement as described herein.
- the deuterated analogs of this invention have the potential to uniquely maintain the beneficial aspects of the non-isotopically enriched drugs while substantially increasing the half-life (T 1 Z 2 ), lowering the maximum plasma concentration (C max ) of the minimum efficacious dose (MED), lowering the efficacious dose and thus decreasing the non-mechanism-related toxicity, and/or lowering the probability of drug-drug interactions.
- These drugs also have strong potential to reduce the cost-of-goods (COG) owing to the ready availability of inexpensive sources of deuterated reagents combined with previously mentioned potential for lowering the therapeutic dose.
- R 1 , R 4 , Rg and Rio are independently selected from the group consisting of hydrogen, and deuterium;
- R 2 , R 3 , R 6 , and R 8 are independently selected from the group consisting of hydrogen, deuterium, C 1 -C 6 alkyl, and C 1 -C 6 alkyloxy;
- R 5 is selected from the group consisting Of-CH 2 -, -CHD- and -CD 2 -;
- R 7 is selected from the group consisting of hydrogen, deuterium, -NO 2 , Ci-C 6 alkyl, Ci-C 6 alkyloxy, and C 1 -C 6 alkoxy(Ci-C 6 )alkyloxy; provided that compounds of Formula 1 contain at least one deuterium atom, and provided that deuterium enrichment in compounds of Formula 1 is at least about 1%.
- Compounds of this invention have the potential to uniquely maintain the beneficial aspects of non-isotopically enriched gastric H + , K + -ATPase modulators while substantially altering the half-life (T 1/2 ), lowering the maximum plasma concentration (C ma ⁇ ) of the minimum efficacious dose (MED), lowering the efficacious dose and thus decreasing non-mechanism-related toxicities, and/or lowering the probability of drug-drug interactions.
- These drugs also have potential to reduce the cost-of-goods (COG) due to a potential for lowering the therapeutic dose when compared to the non-isotopically enriched gastric H , K + -ATPase modulators.
- COG cost-of-goods
- Agents in the present invention will expose patients to a maximum of 0.000005% D 2 O (can also be expressed as 0.00001% DHO). This quantity is a small fraction of the naturally occurring background levels of D 2 O (or DHO) in circulation. Even this minute exposure would require complete metabolism of every drug-incorporated C-D bond, and yet this C-D metabolism is precisely the phenomenon that can be eliminated or diminished through creative incorporation. Recall the levels of D 2 O shown to cause toxicity in animals (vide supra). The safety factor is thus extraordinary for this approach.
- Deuterium enrichment refers to the percentage of incorporation of deuterium at a given site on the molecule instead of a hydrogen atom. For example, deuterium enrichment of 1% means that in 1% of molecules in a given sample a particular site is occupied by deuterium. Because the naturally occurring distribution of deuterium is about 0.0156%, deuterium enrichment in compounds synthesized using non-enriched starting materials is about 0.0156%. In some embodiments, the deuterium enrichment in the compounds of the present invention is greater than 10%. In other embodiments, the deuterium enrichment in the compounds of the present invention is greater than 20%. In further embodiments, the deuterium enrichment in the compounds of the present invention is greater than 50%. In some embodiments, the deuterium enrichment in the compounds of the present invention is greater than 70%. In some embodiments, the deuterium enrichment in the compounds of the present invention is greater than 90%.
- Isotopic enrichment refers to the percentage of incorporation of a less prevalent isotope of an element at a given site on the molecule instead of the more prevalent isotope of the element.
- Non-isotopically enriched refers to a molecule in which the percentage of the various isotopes is substantially the same as the naturally occurring percentages.
- the compound of Formula 1 contains about 60% or more by weight of the (-)-enantiomer of the compound and about 40% or less by weight of (+)-enantiomer of the compound. In some embodiments, the compound of Formula 1 contains about 70% or more by weight of the (-)-enantiomer of the compound and about 30% or less by weight of (+)-enantiomer of the compound. In some embodiments, the compound of Formula 1 contains about 80% or more by weight of the (-)-enantiomer of the compound and about 20% or less by weight of (+)-enantiomer of the compound.
- the compound of Formula 1 contains about 90% or more by weight of the (-)-enantiomer of the compound and about 10% or less by weight of the (+)-enantiomer of the compound. In some embodiments, the compound of Formula 1 contains about 95% or more by weight of the (-)-enantiomer of the compound and about 5% or less by weight of (+)-enantiomer of the compound. In some embodiments, the compound of Formula 1 contains about 99% or more by weight of the (-)-enantiomer of the compound and about 1% or less by weight of (+)- enantiomer of the compound.
- the compound of Formula 1 contains about 60% or more by weight of the (+)-enantiomer of the compound and about 40% or less by weight of (-)-enantiomer of the compound. In some embodiments, the compound of Formula 1 contains about 70% or more by weight of the (+)-enantiomer of the compound and about 30% or less by weight of (-)-enantiomer of the compound. In some embodiments, the compound of Formula 1 contains about 80% or more by weight of the (+)-enantiomer of the compound and about 20% or less by weight of (-)-enantiomer of the compound.
- the compound of Formula 1 contains about 90% or more by weight of the (+)- enantiomer of the compound and about 10% or less by weight of the (-)-enantiomer of the compound. In some embodiments, the compound of Formula 1 contains about 95% or more by weight of the (+)-enantiomer of the compound and about 5% or less by weight of (-)-enantiomer of the compound. In some embodiments, the compound of Formula 1 contains about 99% or more by weight of the (+)-enantiomer of the compound and about 1% or less by weight of (-)-enantiomer of the compound.
- the alkyl is selected from the group consisting of methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, and tert-butyl.
- the alkyloxy is selected from the group consisting of methoxy, ethoxy, n- propoxy, iso-propoxy, n-butoxy, sec-butoxy, iso-butoxy, and tert-butoxy.
- R 1 is hydrogen. In other embodiments, R 2 is hydrogen. In some embodiments, R 3 is hydrogen. In other embodiments, R 4 is hydrogen. In still other embodiments, R 6 is hydrogen. In yet other embodiments, R 7 is hydrogen. In yet other embodiments, R 8 is hydrogen. In still other embodiments, R 9 is hydrogen. In still other embodiments, R 10 is hydrogen.
- Rj is deuterium.
- R 2 is deuterium.
- R 3 is deuterium.
- R 4 is deuterium.
- R 6 is deuterium.
- R 7 is deuterium.
- R 8 is deuterium.
- R 9 is deuterium.
- R 1O is deuterium.
- R 2 is a Ci -6 alkyloxy, wherein any one or more of the hydrogen atoms on the alkoxy group can be substituted by deuterium.
- R 2 is selected from the group consisting of -OCH 3 , -OCD 3 , -OCHF 2 , and - OCDF 2 . In other embodiments, R 2 is -OCD 3 or -OCDF 2 .
- R 5 is -CH 2 -, -CHD- or -CD 2 -;
- R 6 is a C 1-6 alkyloxy, wherein any one or more of the hydrogen atoms on the alkoxy group can be substituted by deuterium. In some of these embodiments, R 6 is -OCH 3 , or -OCD 3 ;
- R 6 is a Ci -6 alkyl, wherein any one or more of the hydrogen atoms on the alkyl group can be substituted by deuterium. In some of these embodiments, R 6 is -CH 3 , or -CD 3 ;
- R 7 is a Ci -6 alkyloxy, wherein any one or more of the hydrogen atoms on the alkoxy group can be substituted by deuterium.
- R 7 is selected from the group consisting of -OCH 3 , -OCD 3 , -OCH 2 CF 3 , - OCD 2 CF 3 , and -O(CH 2 ) 3 OCH 3 wherein any one or more of the hydrogen atoms in - O(CH 2 ) 3 OCH 3 can be substituted by deuterium.
- R 8 is a C 1-6 alkyl wherein any one or more of the hydrogen atoms on the alkyl group can be substituted by deuterium. In some of these embodiments, R 8 is -CH 3 , or -CD 3 ;
- R 8 is a C 1-6 alkyloxy wherein any one or more of the hydrogen atoms on the alkoxy group can be substituted by deuterium.
- Ri is not hydrogen. In other embodiments, R 2 is not hydrogen. In some embodiments, R 3 is not hydrogen. In other embodiments, R 4 is not hydrogen. In still other embodiments, R 6 is not hydrogen. In yet other embodiments, R 7 is not hydrogen. In yet other embodiments, R 8 is not hydrogen. In still other embodiments, R 9 is not hydrogen. In still other embodiments, Ri 0 is not hydrogen.
- Ri is not deuterium.
- R 2 is not deuterium.
- R 3 is not deuterium.
- R 4 is not deuterium.
- R 6 is not deuterium.
- R 7 is not deuterium.
- R 8 is not deuterium.
- R 9 is not deuterium.
- Rio is not deuterium.
- R 2 is not a Ci -6 alkyloxy. In some of these embodiments, R 2 is not -OCH 3 , -OCD 3 , -OCHF 2 , or -OCDF 2 . In other embodiments, R 2 is not -OCD 3 or -OCDF 2 .
- R 5 is not -CH 2 , -CHD or -CD 2 ;
- R 6 is not a C 1-6 alkyloxy. In some of these embodiments, R 6 is not -OCH 3 , or -OCD 3 ;
- R 6 is not a C 1-6 alkyl. In some of these embodiments, R 6 Is not -CH 3 , or -CD 3 ;
- R 7 is not a Ci -6 alkyloxy. In some of these embodiments, R 7 is not -OCH 3 , -OCD 3 , -OCH 2 CF 3 , -OCD 2 CF 3 , or -O(CH 2 ) 3 OCH 3 wherein any of the hydrogen atoms in -O(CH 2 ) 3 OCH 3 can be substituted by a deuterium.
- R 8 is not a C 1-6 alkyl. In some of these embodiments, R 8 is not -CH 3 , or -CD 3 ;
- R 8 is not a Ci -6 alkyloxy.
- compositions comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or excipient, or a combination thereof, for enteral, intravenous infusion, oral, parenteral, topical or ocular administration.
- compositions comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or excipient, or a combination thereof, for the treatment of conditions involving the gastric H + , K + -ATPase, duodenal ulcers, other conditions mediated by gastric acid secretion, or a combination thereof, for the treatment of conditions involving the gastric H
- methods of modulating the activity of the gastric H + , K + -ATPase with one or more of the compounds or compositions of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)-enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)- enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof.
- a mammalian subject particularly a human, suspected of having, or being prone to a disease or condition involving the gastric H + , K + -ATPase, gastric ulcers, duodenal ulcers, esophageal ulcers, other conditions mediated by gastric acid secretion, or psoriasis.
- the administering step in the above methods comprises administering the compound of the invention in some composition, i.e., a single tablet, pill, or capsule, or a single solution for intravenous injection, or a single drinkable solution, or a single dragee formulation or patch, wherein the amount administered is about 0.5 milligram to 80 milligram total daily dose.
- a gastric H + , K + -ATPase inhibitor comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or
- the inter-individual variation in plasma levels of the compounds of the invention, or metabolites thereof is decreased by greater than about 5%, as compared to the non-isotopically enriched compounds. In other embodiments, the inter-individual variation in plasma levels of the compounds of the invention, or metabolites thereof, is decreased by greater than about 10%, as compared to the non-isotopically enriched compounds. In other embodiments, the inter-individual variation in plasma levels of the compounds of the invention, or metabolites thereof, is decreased by greater than about 20%, as compared to the non-isotopically enriched compounds.
- the inter- individual variation in plasma levels of the compounds of the invention, or metabolites thereof is decreased by greater than about 30%, as compared to the non-isotopically enriched compounds. In other embodiments, the inter-individual variation in plasma levels of the compounds of the invention, or metabolites thereof, is decreased by greater than about 40%, as compared to the non-isotopically enriched compounds. In other embodiments, the inter- individual variation in plasma levels of the compounds of the invention, or metabolites thereof, is decreased by greater than about 50%, as compared to the non-isotopically enriched compounds. Plasma levels of the compounds of the invention, or metabolites thereof, are measured by the methods of Li et al Rapid Communications in Mass Spectrometry 2005, 19(14), 1943-1950, which is hereby incorporated by reference in its entirety.
- a gastric H + , K + -ATPase inhibitor comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or
- the average plasma levels of the compounds of the invention are increased by greater than about 5%, as compared to the non-isotopically enriched compounds. In other embodiments, the average plasma levels of the compounds of the invention are increased by greater than about 10%, as compared to the non-isotopically enriched compounds. In other embodiments, the average plasma levels of the compounds of the invention are increased by greater than about 20%, as compared to the non-isotopically enriched compounds. In other embodiments, the average plasma levels of the compounds of the invention are increased by greater than about 30%, as compared to the non-isotopically enriched compounds.
- the average plasma levels of the compounds of the invention are increased by greater than about 40%, as compared to the non-isotopically enriched compounds. In other embodiments, the average plasma levels of the compounds of the invention are increased by greater than about 50%, as compared to the non-isotopically enriched compounds.
- the average plasma levels of a metabolite of the compounds of the invention are decreased by greater than about 5%, as compared to the non- isotopically enriched compounds. In other embodiments, the average plasma levels of a metabolite of the compounds of the invention are decreased by greater than about 10%, as compared to the non-isotopically enriched compounds. In other embodiments, the average plasma levels of a metabolite of the compounds of the invention are decreased by greater than about 20%, as compared to the non-isotopically enriched compounds. In other embodiments, the average plasma levels of a metabolite of the compounds of the invention are decreased by greater than about 30%, as compared to the non-isotopically enriched compounds.
- the average plasma levels of a metabolite of the compounds of the invention are decreased by greater than about 40%, as compared to the non-isotopically enriched compounds. In other embodiments, the average plasma levels of a metabolite of the compounds of the invention are decreased by greater than about 50%, as compared to the non-isotopically enriched compounds.
- Plasma levels of the compounds of the invention, or metabolites thereof, are measured by the methods of Li et al Rapid Communications in Mass Spectrometry 2005, 19(14), 1943-1950.
- methods for treatment of gastric acid related diseases by inhibition of gastric acid secretion comprising administering to a mammalian subject in need of treatment a therapeutically effective amount of a gastric H + , K + -ATPase inhibitor comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (H-)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of
- a gastric H + , K + -ATPase inhibitor comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or
- cytochrome P 450 isoforms in mammalian subjects include CYPlAl, CYP1A2, CYPlBl, CYP2A6, CYP2A13, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2D6, CYP2E1, CYP2G1, CYP2J2, CYP2R1, CYP2S1, CYP3A4, CYP3A5, CYP3A5P1, CYP3A5P2, CYP3A7, CYP4A11, CYP4B1, CYP4F2, CYP4F3, CYP4F8, CYP4F11, CYP4F12, CYP4X1, CYP4Z1, CYP5A1, CYP7A1, CYP7B1, CYP8A1, CYP8B1, CYPIlAl, CYPIl,
- the decrease in inhibition of the cytochrome P 450 isoform by compounds of the invention is greater than about 5%, as compared to the non- isotopically enriched compounds. In other embodiments, the decrease in inhibition of the cytochrome P 450 isoform by compounds of the invention is greater than about 10%, as compared to the non-isotopically enriched compounds. In other embodiments, the decrease in inhibition of the cytochrome P 450 isoform by compounds of the invention is greater than about 20%, as compared to the non-isotopically enriched compounds.
- the decrease in inhibition of the cytochrome P 45 o isoform by compounds of the invention is greater than about 30%, as compared to the non-isotopically enriched compounds. In other embodiments, the decrease in inhibition of the cytochrome P 450 isoform by compounds of the invention is greater than about 40%, as compared to the non-isotopically enriched compounds. In other embodiments, the decrease in inhibition of the cytochrome P 450 isoform by compounds of the invention is greater than about 50%, as compared to the non- isotopically enriched compounds.
- a gastric H + , K + -ATPase inhibitor comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or
- a gastric H + , K + -ATPase inhibitor comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or
- oral multiple unit tablet pharmaceutical compositions comprising two components A and B, wherein component A comprises one or more antibacterial agent with similar or different activities, such as for example amoxicillin, clarithromycin, metronidazole, and the like, and a combination thereof, and component B comprises at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)-enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, d
- effervescent dosage forms comprising two components A and B, wherein, component A is one or more effervescent excipients, and component B is made up of a mixture comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)-enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula I 5 a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or excipient, or a combination thereof, a beta
- oral multiple unit tablet pharmaceutical compositions comprising three components A, B and C, wherein component A comprises at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or excipient, or a combination thereof, in the form of pellets covered with an enteric coating polymer layer with mechanical properties such that the acid resistance of the enteric
- a bacterial infection caused or mediated by Helicobacter pylori comprising simultaneously, separately or sequentially administering to a mammalian subject in need, an effective amount of an Nitric Oxide releasing Non Steroidal Anti-inflammatory Drug (NSAE)) and at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)-enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (H-)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically
- NSAE Nitric Oxide releasing Non Steroidal Anti-inflammatory Drug
- extended release pharmaceutical dosage forms comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (H-)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or excipient, or a combination thereof, a hydrophilic or hydrophobic matrix, a water- soluble separating layer, an enteric coating layer, and optionally one or more pharmaceutically acceptable excipients.
- enteric coated pharmaceutical dosage forms comprising at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)- enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or excipient, or a combination thereof, a disruptable semi-permeable membrane and one or more swellable substances, wherein the dosage form has an instant inhibitor-releasing 6 038819
- the part and at least one delayed inhibitor-releasing part is capable of giving a discontinuous release of the compound in the form of at least two consecutive pulses separated in time from 0.1 up to 24 hours.
- stable pharmaceutical dosage forms for oral administration to mammalian subjects which comprises at least one of the compounds of Formula 1, a single enantiomer of a compound of Formula 1, a mixture of the (+)-enantiomer and the (-)-enantiomer, a mixture of about 90% or more by weight of the (-)-enantiomer and about 10% or less by weight of the (+)-enantiomer, a mixture of about 90% or more by weight of the (+)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer of a compound of Formula 1, a mixture of diastereomers, or a pharmaceutically acceptable salt, solvate, or prodrug thereof, in a pharmaceutically acceptable vehicle, carrier, diluent, or excipient, or a combination thereof, and optionally one or more pharmaceutical adjuvants, enclosed in an intermediate reactive layer comprising a gastric juice-resistant polymeric layered material partially neutralized
- the present invention is intended to include all isotopes of all atoms occurring in the present compounds.
- Isotopes include those atoms having the same atomic number but different mass numbers.
- isotopes of hydrogen include deuterium (D) and tritium (T).
- isotopes of carbon include 13 C and 14 C.
- Isotopes of sulfur include 32 S, 33 S, 34 S, and 36 S.
- Isotopes of nitrogen include 14 N and 15 N.
- Isotopes of oxygen include 16 0, 17 O, and 18 O.
- Isotopic hydrogen can be introduced into organic molecules by synthetic techniques that employ deuterated reagents whereby incorporation rates are pre-determined and/or by exchange techniques wherein incorporation rates are determined by equilibrium conditions and maybe highly variable depending on the reaction conditions. Synthetic techniques, where tritium or deuterium is directly and specifically inserted by tritiated or deuterated reagents of known isotopic content, may yield high tritium or deuterium abundance, but can be limited by the chemistry required. In addition, the molecule being labeled may be changed, depending upon the severity of the synthetic reaction employed.
- substituent is a group that may be substituted with one or more group(s) individually and independently selected from the group consisting of hydrogen, deuterium, alkyl, cycloalkyl, aryl, heteroaryl, heterocyclic, hydroxy, alkoxy, aryloxy, mercapto, alkylthio, arylthio, cyano, halo, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, C-amido, N-amido, S-sulfonamido, N-sulfonamido, C-carboxy, O-carboxy, isocyanato, thiocyanato, isothiocyanato, nitro, silyl, trihalomethanesulfonyl, and amino,
- the compounds according to this invention may occur as any reasonable tautomer as recognized by one skilled in the art or a mixture of such tautomers.
- tautomer or “tautomerism” refers to one of two or more structural isomers that exist in equilibrium and are readily converted from one isomeric form to another. Examples include keto-enol tautomers, such as acetone/propen-2-ol and the like, ring-chain tautomers, such as glucose/ 2,3,4,5,6-pentahydroxy-hexanal and the like.
- the compounds described herein may have one or more tautomers and therefore include various isomers. AU such isomeric forms of these compounds are expressly included in the present invention.
- the following example of tautomerism is provided for reference:
- the compounds according to this invention may contain one or more asymmetric atoms and can thus occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures or individual diastereomers.
- stereoisomer refers to a chemical compound having the same molecular weight, chemical composition, and constitution as another, but with the atoms grouped differently. That is, certain identical chemical moieties are at different orientations in space and, therefore, when pure, have the ability to rotate the plane of polarized light. However, some pure stereoisomers may have an optical rotation that is so slight that it is undetectable with present instrumentation.
- the compounds described herein may have one or more asymmetrical atoms and therefore include various stereoisomers. All such isomeric forms of these compounds are expressly included in the present invention.
- Each stereogenic carbon or sulfur may be of R or S configuration. Although the specific compounds exemplified in this application may be depicted in a particular configuration, compounds having the opposite stereochemistry at any given chiral center or mixtures thereof are also envisioned. When chiral centers are found in the derivatives of this invention, it is to be understood that this invention encompasses all possible stereoisomers.
- optically pure compound or “optically pure isomer” refers to a single stereoisomer of a chiral compound regardless of the configuration of the said compound.
- substantially homogeneous refers to collections of molecules wherein at least about 80%, preferably at least about 90% and more preferably at least about 95% of the molecules are a single compound or a single stereoisomer thereof, or to collections of molecules wherein at least about 80%, preferably at least about 90% and more preferably at least about 95% of the molecules are fully substituted (e.g., deuterated) at the positions stated.
- attachment signifies a stable covalent bond, certain preferred points of attachment being apparent to those skilled in the art.
- the term "effective amount" of a compound refers a sufficient amount of the compound that provides a desired effect but with no- or acceptable- toxicity. This amount may vary from subject to subject, depending on the species, age, and physical condition of the subject, the severity of the disease that is being treated, the particular compound used, its mode of administration, and the like. A suitable effective amount may be determined by one of ordinary skill in the art.
- pharmaceutically acceptable refers to a compound, additive or composition that is not biologically or otherwise undesirable.
- the additive or composition may be administered to a subject along with a compound of the invention without causing any undesirable biological effects or interacting in an undesirable manner with any of the other components of the pharmaceutical composition in which it is contained.
- salts includes hydrochloric salt, hydrobromic salt, hydroiodic salt, hydrofluoric salt, sulfuric salt, citric salt, maleic salt, acetic salt, lactic salt, nicotinic salt, succinic salt, oxalic salt, phosphoric salt, malonic salt, salicylic salt, phenylacetic salt, stearic salt, pyridine salt, ammonium salt, piperazine salt, diethylamine salt, nicotinamide salt, formic salt, urea salt, sodium salt, potassium salt, calcium salt, magnesium salt, zinc salt, lithium salt, cinnamic salt, methylamino salt, methanesulfonic salt, picric salt, tartaric salt, triethylamino salt, dimethylamino salt, tris(hydroxymethyl)aminomethane salt and the like. Additional pharmaceutically acceptable salts are known to those of skill in the art.
- the terms “elicit”, “eliciting,” “modulator”, “modulate”, “modulating”, “regulator”, “regulate” or “regulating” the activity refer to a compound that can act as an inhibitor, or an antagonist of a particular enzyme or receptor, such as for example the gastric H + , K + -ATPase and the like.
- drug refers to a compound or compounds and pharmaceutically acceptable compositions thereof that are administered to mammalian subjects as prophylactic or remedy in the treatment of a disease or medical condition.
- Such compounds may be administered to the subject via oral formulation, inhalation, ocular application, transdermal formulation or by injection.
- the term "subject” refers to an animal, preferably a mammal, and most preferably a human, who is the object of treatment, observation or experiment.
- the mammal may be selected from the group consisting of mice, rats, hamsters, gerbils, rabbits, guinea pigs, dogs, cats, sheep, goats, cows, horses, giraffes, platypuses, primates, such as monkeys, chimpanzees, and apes, and humans.
- the term "therapeutically effective amount” is used to indicate an amount of an active compound, or pharmaceutical agent, that elicits the biological or medicinal response indicated. This response may occur in a tissue, system (animal including human) that is being sought by a researcher, veterinarian, medical doctor or other clinician.
- treatment does not necessarily mean total loss of nociception. Any alleviation of any undesired signs or symptoms of a disease, such as inhibition of the gastric H + , K + -ATPase, duodenal ulcers and other conditions mediated by gastric acid secretion, or a subset of these conditions, to any extent can be considered treatment or therapy.
- treatment may include acts that may worsen the patient's overall feeling of well-being or appearance.
- Lewis acid refers to a molecule that can accept an unshared pair of electrons and as such would be obvious to one of ordinary skill and knowledge in the art.
- the definition of "Lewis acid” includes but is not limited to: boron trifluoride, boron trifluoride etherate, boron trifluoride tetrahydrofuran complex, boron trifluoride tert-butyl- methyl ether complex, boron trifluoride dibutyl ether complex, boron trifluoride dihydrate, boron trifluoride di-acetic acid complex, boron trifluoride dimethyl sulfide complex, boron trichloride, boron trichloride dimethyl sulfide complex, boron tribromide, boron tribromide dimethyl sulfide complex, boron triiodide, triimethoxyborane, triethoxyborane, trimethylalumin
- Lewis acids may have optically pure ligands attached to the electron acceptor atom, as set forth in Corey, E. J. Angewandte Chemie, International Edition (2002), 41(10), 1650-1667; Aspinall, H. C. Chemical Reviews (Washington, DC, United States) (2002), 102(6), 1807-1850; Groger, H. Chemistry-A European Journal (2001), 7(24), 5246-5251; Davies, H. M. L. Chemtracts (2001), 14(11), 642-645; Wan, Y. Chemtracts (2001), 14(11), 610-615; Kim, Y. H. Accounts of Chemical Research (2001), 34(12), 955-962; Seebach, D.
- Such Lewis acids may be used by one of ordinary skill and knowledge in the art to produce optically pure compounds from achiral starting materials.
- acylating agent refers to a molecule that can transfer an alkylcarbonyl, substituted alkylcarbonyl or aryl carbonyl group to another molecule.
- the definition of "acylating agent” includes but is not limited to ethyl acetate, vinyl acetate, vinyl propionate, vinyl butyrate, isopropenyl acetate, 1-ethoxyvinyl acetate, trichloroethyl butyrate, trifluoroethyl butyrate, trifluoroethyl laureate, S-ethyl thiooctanoate, biacetyl monooxime acetate, acetic anhydride, acetyl chloride, succinic anhydride, diketene, diallyl carbonate, carbonic acid but-3-enyl ester cyanomethyl ester, amino acid and the like.
- nucleophile refers to a negatively charged or neutral molecule that has an unshared pair of electrons and as such would be obvious to one of ordinary skill and knowledge in the art.
- the definition of “nucleophile” includes but is not limited to: water, alkylhydroxy, alkoxy anion, arylhydroxy, aryloxy anion, alkylthiol, alkylthio anion, arylthiol, arylthio anion, ammonia, alkylamine, arylamine, alkylamine anion, arylamine anion, hydrazine, alkyl hydrazine, arylhydrazine, alkylcarbonyl hydrazine, arylcarbonyl hydrazine, hydrazine anion, alkyl hydrazine anion, arylhydrazine anion, alkylcarbonyl hydrazine anion, arylhydrazine anion, alkylcarbonyl hydrazin
- electrophilic reagent refers to a positively charged or neutral molecule that has an open valence shell or an attraction for an electron- rich reactant and as such would be obvious to one of ordinary skill and knowledge in the art.
- electrotrophile includes but is not limited to: hydronium, acylium, Lewis acids, such as for example, Boron trifluoride and the like, halogens, such as for example Br 2 and the like, carbocations, such as for example tert-butyl cation and the like, diazomethane, trimethylsilyldiazomethane, alkyl halides, such as for example methyl iodide, trideuteromethyl iodide (CD 3 I), benzyl bromide and the like, alkyl triflates, such as for example methyl triflate and the like, alkyl sulfonates, such as for example ethyl toluenesulfonate, butyl methanesulfonate, dimethylsulfate, hexadeuterodimethylsulfate ((CD 3 ) 2 SO 4 ) and the like, acyl halides, such as for example acet
- LG refers to any atom (or group of atoms) that is stable in its anion or neutral form after it has been displaced by a nucleophile and as such would be obvious to one of ordinary skill and knowledge in the art.
- the definition of “leaving group” includes but is not limited to: water, methanol, ethanol, chloride, bromide, iodide, methanesulfonate, tolylsulfonate, trifluoromethanesulfonate, acetate, trichloroacetate, benzoate and the like.
- oxidant refers to any reagent that will increase the oxidation state of an atom, such as for example, hydrogen, carbon, nitrogen, sulfur, phosphorus and the like in the starting material by either adding an oxygen to this atom or removing an electron from this atom and as such would be obvious to one of ordinary skill and knowledge in the art.
- the definition of "oxidant” includes but is not limited to: osmium tetroxide, ruthenium tetroxide, ruthenium trichloride, potassium permanganate, meta-chloroperbenzoic acid, hydrogen peroxide, dimethyl dioxirane and the like.
- metal ligand refers to a molecule that has an unshared pair of electrons and can coordinate to a metal atom and as such would be obvious to one of ordinary skill and knowledge in the art.
- metal ligand includes but is not limited to: water, alkoxy anion, alkylthio anion, ammonia, trialkylamine, triarylamine, trialkylphosphine, triarylphosphine, cyanide, azide and the like.
- reducing reagent refers to any reagent that will decrease the oxidation state of an atom in the starting material by either adding a hydrogen to this atom, or adding an electron to this atom, or by removing an oxygen from this atom and as such would be obvious to one of ordinary skill and knowledge in the art.
- reducing reagent includes but is not limited to: borane-dimethyl sulfide complex, 9- borabicyclo[3.3.1.]nonane (9-BBN), catechol borane, lithium borohydride, lithium borodeuteride, sodium borohydride, sodium borodeuteride, sodium borohydride-methanol complex, potassium borohydride, sodium hydroxyborohydride, lithium triethylborohydride, lithium n-butylborohydride, sodium cyanoborohydride, sodium cyanoborodeuteride, calcium (II) borohydride, lithium aluminum hydride, lithium aluminum deuteride, diisobutylAluminum hydride, n-butyl-diisobutylaluminum hydride, Sodium bis- methoxyethoxyAluminum hydride, triethoxysilane, diethoxymethylsilane, lithium hydride, lithium, sodium, hydrogen Ni/B, and the like.
- Certain acidic and Lewis acidic reagents enhance the activity of reducing reagents.
- acidic reagents include: acetic acid, methanesulfonic acid, hydrochloric acid, and the like.
- Lewis acidic reagents include: trimethoxyborane, triethoxyborane, aluminum trichloride, lithium chloride, vanadium trichloride, dicyclopentadienyl titanium dichloride, cesium fluoride, potassium fluoride, zinc (II) chloride, zinc (II) bromide, zinc (II) iodide, and the like.
- Coupled reagent refers to any reagent that will activate the carbonyl of a carboxylic acid and facilitate the formation of an ester or amide bond.
- the definition of “coupling reagent” includes but is not limited to: acetyl chloride, ethyl chloroformate, dicyclohexylcarbodiimide (DCC), diisopropyl carbodiiimide (DIC), 1-ethyl- 3-(3-dimethylaminopropyl) carbodiimide (EDCI), N-hydroxybenzotriazole (HOBT), N- hydroxysuccinimide (HOSu), 4-nitrophenol, pentafluorophenol, 2-(lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU), O-benzotriazole-N,N,N'N'- tetramethyluronium hexafluorophosphate
- removable protecting group or “protecting group” refers to any group which when bound to a functionality, such as the oxygen atom of a hydroxyl or carboxyl group or the Nitrogen atom of an amino group, prevents reactions from occurring at these functional groups and which protecting group can be removed by conventional chemical or enzymatic steps to reestablish the functional group.
- the particular removable protecting group employed is not critical.
- hydroxyl protecting group includes but is not limited to: a) Methyl, tert-butyl, allyl, propargyl, p-chlorophenyl, p-methoxyphenyl, p- nitrophenyl, 2,4-dinitrophenyl, 2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenyl, methoxymethyl, methylthiomethyl, (phenyldimethylsilyl)methoxymethyl, benzyloxymethyl, p-methoxy-benzyloxymethyl, p-nitrobenzyloxymethyl, o-nitrobenzyloxymethyl, (4- methoxyphenoxy)methyl, guaiacolmethyl, tert-butoxymethyl, 4-pentenyloxymethyl, tert- butyldimethylsiloxymethyl, thexyldimethylsiloxymethyl, tert-butyldiphenylsiloxymethyl, 2- methoxymethyl, tert-
- amino protecting group includes but is not limited to: a) 2-methylthioethyl, 2-methylsulfonylethyl, 2-(p-toluenesulfonyl)ethyl, [2-(l,3- dithianyl)]methyl, 4-methylthiophenyl, 2,4-dimethylthiophenyl, 2-phosphonioethyl, 1- methyl- l-(triphenylphosphonio)ethyl, l,l-dimethyl-2-cyanoethyl, 2-dansylethyl, 2-(4- nitrophenyl)ethyl, 4- ⁇ henylacetoxybenzyl, 4-azidobenzyl, 4-azidomethoxybenzyl, m-chloro- p-acyloxybenzyl, p-(dihydroxyboryl)benzyl, 5-benzisoxazolylmethyl, 2-(trifluoromethyl)
- N-2-nitro-4-methoxybenzenesulfenyl N- triphenylmethylsulfenyl, N-l-(2,2,2-trifluoro-l,l-diphenyl)ethylsulfenyl, N-3-nitro-2- pyridinesulfenyl, N-p-toluenesulfonyl, N-benzenesulfonyl, N-2,3,6-trimethyl-4- methoxybenzenesulfonyl, N-2,4,6-trimethoxybenzene-sulfonyl, N-2,6-dimethyl-4- methoxybenzenesulfonyl, N-pentamethylbenzenesulfonyl, N-2,3,5.6-tetramethyl-4- methoxybenzenesulfonyl and the like; b) -C(O)OR 20 , where R 20 is selected from the group consisting of alkyl, substituted alkyl,
- carboxyl protecting groups are given in Greene and Wutts, above.
- the definition of "thiol protecting group” includes but is not limited to: a) Alkyl, benzyl, 4-methoxybenzyl, .
- amino acid refers to any of the naturally occurring amino acids, as well as synthetic analogs and derivatives thereof.
- Alpha-Amino acids comprise a carbon atom to which is bonded an amino group, a carboxy group, a hydrogen atom, and a distinctive group referred to as a "side chain".
- side chains of naturally occurring amino acids include, for example, hydrogen (e.g., as in glycine), alkyl (e.g., as in alanine, valine, leucine, isoleucine, proline), substituted alkyl (e.g., as in threonine, serine, methionine, cysteine, aspartic acid, asparagine, glutamic acid, glutamine, arginine, and lysine), arylalkyl (e.g., as in phenylalanine), substituted arylalkyl (e.g., as in tyrosine), heteroarylalkyl (e.g., as in tryptophan, histidine) and the like.
- hydrogen e.g., as in glycine
- alkyl e.g., as in alanine, valine, leucine, isoleucine, proline
- substituted alkyl e.g., as in thre
- amino acid can also include beta-, gamma-, delta-, omega- amino acids, and the like.
- Unnatural amino acids are also known in the art, as set forth in, Natchus, M. G. Organic Synthesis: Theory and Applications (2001), 5, 89-196; Ager, D. J. Current Opinion in Drug Discovery & Development (2001), 4(6), 800; Reginato, G. Recent Research Developments in Organic Chemistry (2000), 4(Pt. 1), 351-359; Dougherty, D. A. Current Opinion in Chemical Biology (2000), 4(6), 645-652; Lesley, S. A.
- Stereoisomers e.g., D-amino acids
- unnatural amino acids such as alpha, alpha-disubstituted amino acids
- unconventional amino acids include: 4-hydroxyproline, 3-methylhistidine, 5-hydroxylysine, and other similar amino acids and imino acids (e.g., 4-hydroxyproline).
- N-protected amino acid refers to any amino acid which has a protecting group bound to the nitrogen of the amino functionality. This protecting group prevents reactions from occurring at the amino functional group and can be removed by conventional chemical or enzymatic steps to reestablish the amino functional group.
- O-protected amino acid refers to any amino acid which has a protecting group bound to the oxygen of the carboxyl functionality. This protecting group prevents reactions from occurring at the carboxyl functional group and can be removed by conventional chemical or enzymatic steps to reestablish the carboxyl functional group.
- the particular protecting group employed is not critical.
- prodrug refers to an agent that is converted into the parent drug in vivo. Prodrugs are often useful because, in some situations, they may be easier to administer than the parent drug. They may, for instance, be bioavailable by oral administration whereas the parent drug is not. The prodrug may also have improved solubility in pharmaceutical compositions over the parent drug. A prodrug may be converted into the parent drug by various mechanisms, including enzymatic processes and metabolic hydrolysis. See Harper, “Drag Latentiation” in Jucker, ed. Progress in Drug Research 4:221- 294 (1962); Morozowich et al., "Application of Physical Organic Principles to Prodrug Design” in E. B. Roche ed.
- references to reagents ordinarily containing hydrogens, hydrides, or protons may include partially or fully deuterated versions (containing deuterium, deuteride, or deuteronhim) as required to affect transformation to the improved drug substances outlined herein.
- halogen includes fluorine, chlorine, bromine, and iodine.
- alkyl and substituted alkyl are interchangeable and include substituted, optionally substituted and unsubstituted Q -do straight chain saturated aliphatic hydrocarbon groups, substituted, optionally substituted and unsubstituted C 2 -Ci 0 straight chain unsaturated aliphatic hydrocarbon groups, substituted, optionally substituted and unsubstituted C 2 -C 10 branched saturated aliphatic hydrocarbon groups, substituted and unsubstituted C 2 -C 10 branched unsaturated aliphatic hydrocarbon groups, substituted, optionally substituted and unsubstituted C 3 -C 8 cyclic saturated aliphatic hydrocarbon groups, substituted, optionally substituted and unsubstituted Cs-C 8 cyclic unsaturated aliphatic hydrocarbon groups having the specified number of carbon atoms.
- alkyl shall include but is not limited to: methyl (Me), trideuteromethyl (-CD 3 ), ethyl (Et), propyl (Pr), butyl (Bu), pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, ethenyl, propenyl, butenyl, penentyl, hexenyl, heptenyl, octenyl, nonenyl, decenyl, undecenyl, isopropyl (i-Pr), isobutyl (i-Bu), tert-butyl (t-Bu), sec-butyl (s-Bu), isopentyl, neopentyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooc
- alkyloxy (e.g. methoxy, ethoxy, propyloxy, allyloxy, cyclohexyloxy) represents a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms attached through an oxygen bridge.
- alkyloxyalkyl represents an alkyloxy group attached through an alkyl or substituted alkyl group as defined above having the indicated number of carbon atoms.
- alkyloxycarbonyl e.g. methoxycarbonyl, ethoxycarbonyl, tert- butoxycarbonyl, allyloxycarbonyl
- alkyloxycarbonyl represents a substituted or unsubstituted alkyloxy group as defined above having the indicated number of carbon atoms attached through a carbonyl bridge.
- alkylthio (e.g. methylthio, ethylthio, propylthio, cyclohexenylthio and the like) represents a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms attached through a sulfur bridge.
- alkylthioalkyl represents an alkylthio group attached through an alkyl or substituted alkyl group as defined above having the indicated number of carbon atoms.
- alkylamino (e.g. methylamino, diethylamino, butylamino, N- propyl-N-hexylamino, (2-cyclopentyl) ⁇ ropylamino, hexenylamino, and the like) represents one or two substituted or unsubstituted alkyl groups as defined above having the indicated number of carbon atoms attached through an amine bridge.
- the substituted or unsubstituted alkyl groups maybe taken together with the nitrogen to which they are attached forming a saturated cyclic or unsaturated cyclic system containing 3 to 10 carbon atoms with at least one substituent as defined above.
- alkylaminoalkyl represents an alkylamino group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- alkylhydrazino (e.g. methylhydrazino, diethylhydrazino, butylhydrazino, (2-cyclopentyl)propylhydrazino, cyclohexanehydrazino, and the like) represents one or two substituted or unsubstituted alkyl groups as defined above having the indicated number of carbon atoms attached through a nitrogen atom of a hydrazine bridge.
- the substituted or unsubstituted alkyl groups maybe taken together with the nitrogen to which they are attached forming a saturated cyclic or unsaturated cyclic system containing 3 to 10 carbon atoms with at least one substituent as defined above.
- alkylhydrazinoalkyl represents an alkylhydrazino group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- alkylcarbonyl (e.g. cyclooctylcarbonyl, pentylcarbonyl, 3- hexenylcarbonyl and the like) represents a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms attached through a carbonyl group.
- alkylcarbonylalkyl represents an alkylcarbonyl group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- alkylcarboxy (e.g. heptylcarboxy, cyclopropylcarboxy, 3- pentenylcarboxy and the like) represents an alkylcarbonyl group as defined above wherein the carbonyl is in turn attached through an oxygen.
- alkylcarboxyalkyl represents an alkylcarboxy group attached through an alkyl group as defined above having the indicated number of carbon atoms.
- alkylcarbonylamino (e.g. hexylcarbonylamino, cyclopentylcarbonyl-aminomethyl, methylcarbonylaminophenyl and the like) represents an alkylcarbonyl group as defined above wherein the carbonyl is in turn attached through the nitrogen atom of an amino group.
- the nitrogen group may itself be substituted with a substituted or unsubstituted alkyl or aryl group.
- alkylcarbonylaminoalkyl represents an alkylcarbonylamino group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- alkylcarbonylhydrazino (e.g. ethylcarbonylhydrazino, tert- butylcarbonylhydrazino and the like) represents an alkylcarbonyl group as defined above wherein the carbonyl is in turn attached through the nitrogen atom of a hydrazino group.
- aryl represents an unsubstituted, mono-, or polysubstituted monocyclic, polycyclic, biaryl aromatic groups covalently attached at any ring position capable of forming a stable covalent bond, certain preferred points of attachment being apparent to those skilled in the art (e.g., 3-phenyl, 4-naphthyl and the like).
- the aryl substituents are independently selected from the group consisting of hydrogen, deuterium, halogen, -OH, -SH, -CN, -NO 2 , trihalomethyl, hydroxypyronyl, C 1-10 alkyl, arylC o-1 oalkyl, Co- alkyloxyCo-ioalkyl, arylCcioalkyloxyCo-ioalkyl, Co -1 oalkylthioC 0-1 oalkyl, arylCo-iQalkylthioCo-ioalkyl, C 0-10 alkylaminoCQ -1 Qalkyl, arylCc-toalkylaminoCo-ioalkyl, N-aryl- N-Co-ioalkylaminoCo-ioalkyl, Cj.toalkylcarbonylCo-ioalkyl, arylCo-ioalkylcarbonylC 0-1 oalkyl, d
- R 21 , R 22 and R 23 are independently selected from the group consisting of hydrogen, deuterium, alkyl, aryl or R 22 and R 23 are taken together with the nitrogen to which they are attached forming a saturated cyclic or unsaturated cyclic system containing 3 to 8 carbon atoms with at least one substituent as defined above.
- aryl includes but is not limited to phenyl, pentadeuterophenyl, biphenyl, naphthyl, dihydronaphthyl, tetrahydronaphthyl, indenyl, indanyl, azulenyl, anthryl, phenanthryl, fluorenyl, pyrenyl and the like.
- arylalkyl e.g. (4-hydroxyphenyl)ethyl, (2- aminonaphthyl)hexenyl and the like
- arylalkyl represents an aryl group as defined above attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- arylcarbonyl e.g. 2-thiophenylcarbonyl, 3- methoxyanthrylcarbonyl and the like
- arylcarbonyl represents an aryl group as defined above attached through a carbonyl group.
- arylalkylcarbonyl e.g. (2,3-dimethoxyphenyl)propylcarbonyl, (2-chloronaphthyl)pentenyl-carbonyl and the like
- arylalkylcarbonyl represents an arylalkyl group as defined above wherein the alkyl group is in turn attached through a carbonyl.
- aryloxy (e.g. phenoxy, naphthoxy, 3-methylphenoxy, and the like) represents an aryl or substituted aryl group as defined above having the indicated number of carbon atoms attached through an oxygen bridge.
- aryloxyalkyl represents an aryloxy group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- aryloxycarbonyl e.g. phenoxycarbonyl, naphthoxycarbonyl
- aryloxycarbonyl represents a substituted or unsubstituted aryloxy group as defined above having the indicated number of carbon atoms attached through a carbonyl bridge.
- arylthio (e.g. phenylthio, naphthylthio, 3-bromophenylthio, and the like) represents an aryl or substituted aryl group as defined above having the indicated number of carbon atoms attached through a sulfur bridge.
- arylthioalkyl represents an arylthio group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- arylamino e.g. phenylamino, diphenylamino, naphthylamino, N- ⁇ henyl-N-naphthylamino, o-methylphenylamino, p-methoxyphenylamino, and the like
- arylaminoalkyl represents an arylamino group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- arylalkylamino represents an aryl group attached through an alkylamino group as defined above having the indicated number of carbon atoms.
- N-aryl-N-alkylamino e.g. N-phenyl-N-methylamino, N-naphthyl- N-butylamino, and the like
- arylhydrazino (e.g. phenylhydrazino, naphthylhydrazino, 4- methoxyphenylhydrazino, and the like) represents one or two aryl groups as defined above having the indicated number of carbon atoms attached through a hydrazine bridge.
- arylhydrazinoalkyl represents an arylhydrazino group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- arylalkylhydrazino represents an aryl group attached through an alkylhydrazino group as defined above having the indicated number of carbon atoms.
- N-aryl-N- alkylhydrazino e.g. N-phenyl-N-methylhydrazino, N-naphthyl-N-butylhydrazino, and the like
- N-aryl-N- alkylhydrazino represents one aryl and one a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms independently attached through an amine atom of a hydrazine bridge.
- arylcarboxy e.g. phenylcarboxy, naphthylcarboxy, 3- fluorophenylcarboxy and the like
- arylcarboxyalkyl represents an arylcarboxy group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- arylcarbonylamino (e.g. phenylcarbonylamino, naphthylcarbonylamino, 2-methylphenylcarbonylamino and the like) represents an arylcarbonyl group as defined above wherein the carbonyl is in turn attached through the nitrogen atom of an amino group.
- the nitrogen group may itself be substituted with a substituted or unsubstituted alkyl or aryl group.
- arylcarbonylaminoalkyl represents an arylcarbonylamino group attached through a substituted or unsubstituted alkyl group as defined above having the indicated number of carbon atoms.
- the Nitrogen group may itself be substituted with a substituted or unsubstituted alkyl or aryl group.
- arylcarbonylhydrazino e.g. phenylcarbonylhydrazino, naphthylcarbonylhydrazino, and the like
- arylcarbonylhydrazino represents an arylcarbonyl group as defined above wherein the carbonyl is in turn attached through the Nitrogen atom of a hydrazino group.
- heteroaryl refers to a monovalent unsaturated group having a single ring or multiple condensed rings, from 1 to 13 carbon atoms and from 1 to 10 hetero atoms selected from the group consisting of nitrogen, sulfur, and oxygen, within the ring.
- heteroaryl groups in this invention can be optionally substituted with 1 to 10 substituents selected from the group consisting of: hydrogen, deuterium, halogen, -OH, -SH, -CN, -NO 2 , trihalomethyl, hydroxypyronyl, Ci-ioalkyl, arylCo- loalkyl, C 0-1 oalkyloxyC 0-10 alkyl, arylCo. ⁇ alkyloxyCo.ioalkyl, C 0-1 oalkylthioCo-ioalkyl, arylCo- i 0 alkylthioC 0-1 oalkyl, Co-ioalkylaminoCo-ioalkyl, arylCo-ioalkylaminoCo-ioalkyl, N-aryl-N-Co- l oalkylaminoCo-ioalkyl, Ci-walkylcarbonylC d oalkyl, arylCo-ioalky
- heteroaryl includes but is not limited to thienyl, benzothienyl, isobenzothienyl, 2,3-dihydrobenzothienyl, furyl, pyranyl, benzofuranyl, isobenzofuranyl, 2,3-dihydrobenzofuranyl, pyrrolyl, pyrrolyl-2,5-dione, 3-pyrrolinyl, indolyl, isoindolyl, 3H-indolyl, indolinyl, indolizinyl, indazolyl, phthalimidyl (or isoindoly-1,3- dione), imidazolyl, 2H-imidazolinyl, benzimidazolyl, deuterobenzimidazolyl, dideuterobenzimidazolyl, trideuterobenzimidazolyl, tetradeuterobenzimidazolyl, pyridy
- saturated heterocyclic represents an unsubstituted, mono-, and polysubstituted monocyclic, polycyclic saturated heterocyclic group covalently attached at any ring position capable of forming a stable covalent bond, certain preferred points of attachment being apparent to those skilled in the art (e.g., 1-piperidinyl, 4-piperazinyl, DBU, and the like).
- the saturated heterocyclic substituents are independently selected from the group consisting of halo, -OH, -SH 5 -CN, -NO 2 , trihalomethyl, hydroxypyronyl, C 1 . l oalkyl, arylCo-i O alkyl, Co-ioalkyloxyCo-ioalkyl, arylCo-ioalkyloxyCo-ioalkyl, Co.i 0 alkylthioCo- ioalkyl, arylCo-ioalkylthioCo-ioalkyl, CQ-ioalkylaminoCo.ioalkyl, arylCo -10 alkylaminoC 0-10 alkyl, N-aryl-N-Co-ioalkylaminoCo-ioalkyl, Ci-ioalkylcarbonylCo- ⁇ alkyl, arylC 0-10 alkylcarbonylC 0- t oalkyl, Ci-io
- R 21 , R 22 and R 23 are independently selected from the group consisting of hydrogen, deuterium, alkyl, aryl, or R 22 and R 23 are taken together with the nitrogen to which they are attached forming a saturated cyclic or unsaturated cyclic system containing 3 to 8 carbon atoms with at least one substituent as defined above.
- saturated heterocyclic includes but is not limited to pyrrolidinyl, pyrazolidinyl, piperidinyl, 1,4-dioxanyl, morpholinyl, 1,4-dithienyl, thiomorpholinyl, piperazinyl, quinuclidinyl, and the like.
- alpha-beta-unsaturated carbonyl refers to a molecule that has a carbonyl group directly attached to a double or triple bonded carbon and which would be obvious to one of ordinary skill and knowledge in the art.
- the definition of alpha-beta- unsaturated carbonyl includes but is not limited to acrolein, methyl vinyl ketone, and the like.
- acetal refers to a molecule that contains a carbon atom C 1 that is directly attached to a hydrogen atom (H 1 ), a substituted carbon atom (C 2 ) and two oxygen atoms (Oi and O 2 ). These oxygen atoms are in turn attached to other substituted carbon atoms (C 3 and C 4 ), which would be obvious to one of ordinary skill and knowledge in the art.
- the definition of acetal includes but is not limited to 1,1-dimethoxypropane, 1,1-bis- allyloxybutane and the like.
- cyclic acetal refers to an acetal as defined above where C 3 and C 4 , together with the oxygen atoms to which they are attached, combine thru an alkyl bridge to form a 5- to 10-membered ring, which would be obvious to one of ordinary skill and knowledge in the art.
- the definition of cyclic acetal includes but is not limited to 2-methyl- [l,3]dioxolane, 2-ethyl-[l,3]dioxane, 2-phenyl-[l,3]dioxane, 2-phenyl-hexahydro- pyrano[3,2-d][l,3]dioxine and the like.
- ketal refers to a molecule that contains a carbon atom Ci that is directly attached to two substituted carbon atom (C 2 and C 3 ) and two oxygen atoms (Oi and O 2 ). These oxygen atoms are in turn attached to other substituted carbon atoms (C 4 and C 5 ), which would be obvious to one of ordinary skill and knowledge in the art.
- the definition of acetal includes but is not limited to 2,2-dimethoxy-butane, 3,3-diethoxy-pentane and the like.
- cyclic ketal refers to a ketal as defined above where C 4 and C 5 , together with the oxygen atoms to which they are attached, combine thru an alkyl bridge to form a 5- to 10-membered ring, which would be obvious to one of ordinary skill and knowledge in the art.
- the definition of cyclic acetal includes but is not limited to 2,2,4,5- tetramethyl ⁇ [l,3]dioxolane, 2,2-diethyl-[l,3]dioxe ⁇ ane, 2,2-dimethyl-hexahydro-pyrano[3,2- d][l,3]dioxine and the like.
- An "acetyl” group refers to a -C( ⁇ O)CH 3 , group.
- a "cyano” group refers to a -CN group.
- An “isocyanato” group refers to a -NCO group. .
- a “thiocyanato” group refers to a -CNS group.
- An “isothiocyanato” group refers to a -NCS group.
- perhaloalkyl refers to an alkyl group where all of the hydrogen atoms are replaced by halogen atoms.
- composition refers to a mixture of a compound disclosed herein with other chemical components, such as diluents or carriers.
- the pharmaceutical composition facilitates administration of the compound to an organism. Multiple techniques of administering a compound exist in the art including, but not limited to, oral, injection, aerosol, parenteral, and topical administration.
- Pharmaceutical compositions can also be obtained by reacting compounds with inorganic or organic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like.
- carrier defines a chemical compound that facilitates the incorporation of a compound into cells or tissues.
- DMSO dimethyl sulfoxide
- carrier facilitates the uptake of many organic compounds into the cells or tissues of an organism.
- the term "diluent” defines a solution, typically one that is aqueous or partially aqueous, that dissolves chemical compounds of interest and may stabilize the biologically active form of the compound. Salts dissolved in buffered solutions are utilized as diluents in the art.
- One commonly used buffered solution is phosphate buffered saline because it mimics the salt conditions of human blood. Since buffer salts can control the pH of a solution at low concentrations, a buffered diluent rarely modifies the biological activity of a compound.
- gastric related diseases defines a medical or physiological condition that includes, but is not limited to, peptic ulcer, duodenal ulcer, stomach ulcer, gastric ulcer, chronic gastritis, gastroesophageal reflux disease, GERD, heartburn, acid reflux, reflux esophagitis, indigestion, non-ulcer dyspepsia, functional dyspepsia, dyspepsia caused by structural or biochemical disease, biliary tract disease, gastroparesis, pancreatitis, carbohydrate malabsorption, including but not limited to lactose, sorbitol, fructose, and mannitol malabsorption, infiltrative diseases of the stomach, Crohn's disease, sarcoidosis, ischemic bowel disease, inflammatory bowel disease, diverticulitis, Helicobacter infections, Helicobacter pylori infections, intestinal parasites including but not limited to giardia species and strongyloides species, abdominal cancer,
- the present invention provides a process for preparing a compound of formula 3 wherein R 11 and R 14 are independently selected from the group consisting of hydrogen and deuterium; Rj 2 and R 13 are independently selected from the group consisting of -CH 3 , -CDH 2 , -CD 2 H, and -CD 3 .
- Such a process can be performed, for example, by contacting compound of formula 2 with deuterium oxide under conditions suitable to form a compound of formula 3, as set forth below:
- Compound of formula 2 may be prepared by known processes.
- Compound 2 is typically contacted with deuterium oxide in the presence of a catalyst.
- Catalysts contemplated for use in the practice of this particular invention process are typically sodium carbonate, potassium carbonate, DBU and the like.
- Solvents contemplated for use in the practice of this particular invention process are typically polar solvents, such as for example, 1,4-dioxane, acetone, acetonitrile, dimethyl formamide, dimethyl acetamide, N- methylpyrrolidine, dimethyl sulfoxide and the like, or any suitable mixtures thereof.
- the process is typically carried out at a temperature in the range of about 0 0 C up to about 500 0 C, for 0.01 to 240 hours, at a pH in the range of about 1 up to about 14, at a pressure in the range of about 1 mBar up to about 350 Bar.
- Compound 2 is typically contacted with deuterium oxide in the presence of a catalyst, with or without another solvent.
- Catalysts contemplated for use in the practice of this particular invention process are typically sodium carbonate, potassium carbonate, DBU and the like.
- Solvents contemplated for use in the practice of this particular invention process are typically polar solvents, such as for example, 1,4-dioxane, acetone, acetonitrile, dimethyl formamide, dimethyl acetamide, N- methylpyrrolidine, dimethyl sulfoxide and the like, or any suitable mixtures thereof.
- the process is typically carried out in the presence of focused microwave radiation using a quartz reactor at a pressure in the range of about 1 Bar to about 25 Bar, a power setting in the range of about 1 W per liter of solvent to about 900 W per liter of solvent, at a temperature in the range of about O 0 C up to about 500 0 C, for 0.01 to 5 hours, at a pH in the range of about 1 up to about 14.
- the present invention provides a process for preparing a compound of formula 5 wherein R 14 is hydrogen or deuterium, R 12 , R 13 and R 15 are independently selected from the group consisting of -CH 3 , -CDH 2 , -CD 2 H, and -CD 3 .
- Such a process can be performed, for example, by contacting compound of formula 4 with deuterium oxide under conditions suitable to form a compound of formula 5, as set forth below:
- Compound of formula 4 may be prepared by known processes. Compound 4 is typically contacted with deuterium oxide in the presence of a catalyst. Catalysts contemplated for use in the practice of this particular invention process are typically sodium carbonate, potassium carbonate, DBU and the like. Solvents contemplated for use in the practice of this particular invention process are typically polar solvents, such as for example, 1,4-dioxane, acetone, acetonitrile, dimethyl formamide, dimethyl acetamide, N- methylpyrrolidine, dimethyl sulfoxide and the like, or any suitable mixtures thereof.
- the process is typically carried out at a temperature in the range of about 0 0 C up to about 500 0 C, for 0.01 to 240 hours, at a pH in the range of about 1 up to about 14, and at a pressure in the range of about 1 mBar up to about 350 Bar.
- Compound 4 is typically contacted with Deuterium oxide in the presence of a catalyst, with or without another solvent.
- Catalysts contemplated for use in the practice of this particular invention process are typically Sodium carbonate, Potassium carbonate, DBU and the like.
- Solvents contemplated for use in the practice of this particular invention process are typically polar solvents, such as for example, 1,4-dioxane, acetone, acetonitrile, dimethyl formamide, dimethyl acetamide, N- methylpyrrolidine, dimethyl sulfoxide and the like, or any suitable mixtures thereof.
- the process is typically carried out in the presence of focused microwave radiation using a quartz reactor at a pressure in the range of about 1 Bar to about 25 Bar, a power setting in the range of about 1 W per liter of solvent to about 900 W per liter of solvent, at a temperature in the range of about O 0 C up to about 500 0 C, for 0.01 to 5 hours, at a pH in the range of about 1 up to about 14.
- Certain pharmaceutically acceptable salts of the invention are prepared by treating the novel compounds of the invention with an appropriate amount of pharmaceutically acceptable base.
- Representative pharmaceutically acceptable bases are ammonium hydroxide, sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide, magnesium hydroxide, ferrous hydroxide, zinc hydroxide, copper hydroxide, Aluminum hydroxide, ferric hydroxide, isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine, 2-dimethylaminoethanol, 2- diethylaminoethanol, lysine, arginine, histidine, and the like.
- the reaction is conducted in water or D 2 O, alone or in combination with an inert, water-miscible organic solvent, at a temperature of from about 0 °C to about 100 °C, preferably at room temperature.
- the molar ratio of compounds of structural Formula 1 to base used is chosen to provide the ratio desired for any particular salts.
- compounds of Formula 1 can be treated with approximately one equivalent of the pharmaceutically acceptable base to yield a neutral salt.
- Calcium salts approximately one-half a molar equivalent of base is used to yield a neutral salt, while for Aluminum salts, approximately one-third a molar equivalent of base will be used.
- the compounds of the invention may be conveniently formulated into pharmaceutical compositions composed of one or more of the compounds together with a pharmaceutically acceptable carrier as described in Remington's Pharmaceutical Sciences, latest edition, by E. W. Martin (Mack Publ. Co., Easton Pa.).
- the compounds of the invention may be administered orally, parenterally (e.g., intravenously), by intramuscular injection, by intraperitoneal injection, topically, transdermally, or the like, although oral or topical administration is typically preferred.
- parenterally e.g., intravenously
- intramuscular injection by intraperitoneal injection, topically, transdermally, or the like, although oral or topical administration is typically preferred.
- the amount of active compound administered will, of course, be dependent on the subject being treated, the subject's weight, the manner of administration and the judgment of the prescribing physician.
- the dosage will be in the range of about 1 microgram per kilogram per day to 100 milligram per kilogram per day.
- the pharmaceutical compositions may be in the form of solid, semi-solid or liquid dosage forms, such as, for example, tablets, suppositories, pills, capsules, powders, liquids, suspensions, lotions, creams, gels and the like, preferably in unit dosage form suitable for single administration of a precise dosage.
- the compositions will include, as noted above, an effective amount of the selected drug in combination with a pharmaceutically acceptable carrier and, in addition, may include other medicinal agents, pharmaceutical agents, carriers, adjuvants, diluents and the like.
- conventional non-toxic solid carriers include, for example, pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharin, talc, cellulose, glucose, sucrose, magnesium carbonate, and the like.
- Liquid pharmaceutically administrable-compositions can, for example, be prepared by dissolving, dispersing, etc., an active compound as described herein and optional pharmaceutical adjuvants in an excipient, such as, for example, water, saline, aqueous dextrose, glycerol, ethanol, and the like, to thereby form a solution or suspension.
- the pharmaceutical composition to be administered may also contain minor amounts of nontoxic auxiliary substances such as wetting or emulsifying agents, pH buffering agents and the like, for example, sodium acetate, sorbitan monolaurate, triethanolamine sodium acetate, triethanolamine oleate, etc.
- auxiliary substances such as wetting or emulsifying agents, pH buffering agents and the like, for example, sodium acetate, sorbitan monolaurate, triethanolamine sodium acetate, triethanolamine oleate, etc.
- fine powders or granules may contain diluting, dispersing, and/or surface active agents, and may be presented in water or in a syrup, in capsules or sachets in the dry state, or in a non-aqueous solution or suspension wherein suspending agents may be included, in tablets wherein binders and lubricants may be included, or in a suspension in water or a syrup. Wherever required, flavoring, preserving, suspending, thickening, or emulsifying agents may also be included. Tablets and granules are preferred oral administration forms, and these may be coated.
- Parenteral administration if used, is generally characterized by injection.
- Injectables can be prepared in conventional forms, either as liquid solutions or suspensions, solid forms suitable for solution or suspension in liquid prior to injection, as emulsions, or as sustained release delivery system.
- Systemic administration can also be by transmucosal or transdermal means.
- penetrants appropriate to the barrier to be permeated are used in the formulation.
- penetrants are generally known in the art, and include, for example, for transmucosal administration, bile salts and fusidic acid derivatives.
- detergents can be used to facilitate permeation.
- Transmucosal administration can be through nasal sprays, for example, or using suppositories.
- the agents are formulated into ointments, creams, salves, powders and gels.
- the transdermal delivery agent can be DMSO.
- Transdermal delivery systems can include, such as for example, patches.
- compositions containing the compounds of the invention as an active ingredient can take the form of tablets, capsules, powders, suspensions, solutions, emulsions as well as salves and creams, and can be used for parenteral (intravenous, intradermal, intramuscular, intrathecal etc.) injections, infiltration, topical application, central injection at spinal cord, oral, rectal, intravaginal and intranasal administering or for local application.
- parenteral intravenous, intradermal, intramuscular, intrathecal etc.
- Such compositions can be prepared by combining the active ingredient(s) with pharmaceutically acceptable excipients normally used for this purpose.
- excipients can comprise aqueous and non-aqueous solvents, stabilizers, suspension agents, dispersing agents, moisturizers and the like, and will be known to the skilled person in the pharmaceutical field.
- the composition may further contain likewise suitable additives such as for instance polyethylene glycols and, if necessary, colorants, fragrances and the like.
- the pharmaceutical compositions will preferably contain at least 0.1 volume % by weight of the active ingredient.
- the actual concentration will depend on the human subject and the chosen administering route. In general this concentration will lie between 0.1 and 100% for the above applications and indications.
- the dose of the active ingredient to be administered can further vary between 1 microgram and 100 milligram per kilogram body weight per day, preferably between 1 microgram and 50 milligram per kilogram body weight per day, and most preferably between 1 microgram and 20 milligram per kilogram body weight per day. Also, all of the specific dosages which lie between the upper and lower dosages stated above are contemplated in the present invention.
- the desired dose is preferably presented in the form of one, two, three, four, five, six or more sub-doses that are administered at appropriate intervals per day.
- the dose or sub-doses can be administered in the form of dosage units containing for instance from 0.5 to 1500 milligram, preferably from 1 to 100 milligram and most preferably from 1 to 40 milligram active constituent per dosage unit, and if the condition of the patient requires the dose can, by way of alternative, be administered as a continuous infusion.
- Me refers to methyl (CH 3 -)
- Et refers to ethyl (CH 3 CH 2 -)
- i-Pr refers to isopropyl ((CH 3 ) 2 CH 2 -)
- t-Bu or tert-butyl refers to tertiary butyl ((CH 3 ) 3 CH-)
- Ph refers to phenyl
- Bn refers to benzyl (PhCH 2 -)
- Bz refers to benzoyl (PhCO- )
- MOM refers to methoxymethyl
- Ac refers to acetyl
- TMS refers to trimethylsilyl
- TBS refers to tert-butyldimethylsilyl
- Ms refers to methanesulfonyl (CH 3 SO 2 -)
- Ts refers to p- toluenesulfonyl (p-CH 3 PhSO 2
- HMQC proton detected heteronuclear multiplet-quantum coherence
- HMBC heteronuclear multiple-bond connectivity
- s refers to singlet
- br s refers to broad singlet
- d refers to doublet
- br d refers to broad doublet
- t refers to triplet
- q refers to quartet
- dd refers to double doublet
- m refers to multiple!
- ppm refers to parts per million
- IR infrared spectrometry
- MS mass spectrometry
- HRMS high resolution mass spectrometry
- EI electron impact
- FAB fast atom bombardment
- CI refers to chemical ion
- a dry heavy-walled teflon screw cap glass tube equipped with a magnetic stirrer was charged with 2,3,5-trimethyl-4-nitropyridine-l-oxide (5 g, 27.5 mmol), K 2 CO 3 (3.8 g, 27.5 mmol) and D 2 O (30 mL) under nitrogen.
- the apparatus was sealed and the mixture was placed in an oil bath at 15O 0 C for 2 hours.
- the reaction was cooled to ambient temperature, NaCl (1Og) and brine (50 mL) were added and the mixture was extracted with ethyl acetate (5 x 50 mL).
- the organic layer was dried over Na 2 SO 4 .
- a solution of magnesium methoxide was prepared by adding magnesium turnings (1.31 g, 0.054 mol) and dichloromethane (5 mL) to methanol (150 niL) and stirring under nitrogen atmosphere for 2-3 hours, at 40-45°C.
- the solution was cooled to 5-1O 0 C and added to a stirred mixture of esomeprazole (42.0 g, 0.121 mol) and methanol (150.0 mL), and stirring was maintained for 3 hours. Water (2.0 mL) was added and stirring was continued for 1 hour and the solution was filtered.
- Human liver microsomal stability assays were conducted at 1 mg per niL protein concentration with an NADPH-generating system (1.3 mM NADPH, 3.3 mM glucose 6-phosphate and 0.4 U per mL glucose 6-phosphate dehydrogenase) and 3.3 mM MgCl 2 .
- Test compounds were added as acetonitrile solutions (final assay concentration of acetonitrile should be ⁇ 1%) and incubated at 37 0 C with shaking. Aliquots (150 ⁇ L) were removed at 0, 15, 30, 60, and 120 minutes, and ice cold acetonitrile (300 ⁇ L) was added to stop the reactions. Samples were centrifuged at 4000 RPM for 5 minutes to precipitate all proteins.
- Example 69 In vitro metabolism using human cytochrome Pa ⁇ n enzymes
- the cytochrome P 450 enzymes are expressed from the corresponding human cDNA using a baculo virus expression system (BD Biosciences).
- a 0.25 milliliter reaction mixture containing 0.8 milligrams per milliliter protein, 1.3 millimolar NADP + , 3.3 millimolar glucose-6-phosphate, 0.4 U/mL glucose-6-phosphate dehydrogenase, 3.3 millimolar magnesium chloride and 0.2 millimolar of a compound of Formula 1, the corresponding non-isotopically enriched compound or standard or control in 100 millimolar potassium phosphate (pH 7.4) is incubated at 37°C for 20 min. After incubation, the reaction is stopped by the addition of an appropiate solvent (e.g.
- Example 70 In vitro inhibition of dog kidnev H + ZK + -ATPaSe activity
- Example 71 In vitro inhibition of pig stomach gastric vesicle H + /K + -ATPase activity
- the medium of pH 6.0 is adjusted with a HEPES-Tris buffer to pH 7.4.
- the enzyme reaction is started by the addition of Tris-ATP.
- the total reaction volume is 1 milliliter, containing 20 micrograms of vesicular protein, 4 millimolar MgCl 2 , 10 millimolar KCl, 20 micrograms Nigericin, 2 millimolar Tris-ATP, 10 millimolar Hepes, and additionally 2 millimolar Pipes for the preincubation medium at pH 6.0.
- the reaction is stopped by the addition of 10 microliters of 50% trichloroacetic acid.
- the denatured protein is spun down, and the P 1 content is determined as described (Le Bel, 1978).
- the hydrolysis of ATP should not exceed 15%.
- Inhibition is calculated as percent inhibition against maximal stimulation, and IC5 0 is calculated by probit analysis.
- the gastric mucosa in the corpus part is scraped off and minced with a pair of scissors. Mucosa pieces are incubated in a collagenase-containing medium (1 milligrams per milliliter) for 30-45 min at 37 0 C.
- the medium composition (in millimolar) is as follows: 100.0 NaCl, 5.0 KCl, 0.5 NaH 2 PO 4 , 1.0 Na 2 HPO 4 , 1.0 CaCl 2 , 1.5 MgCl 2 , 20.0 NaHCO 3 , 20.0 HEPES, 2 milligrams per milliliter glucose, and 1 milligrams per milliliter rabbit albumin.
- the pH is adjusted to 7.4 with 1 M Tris.
- the glands are filtered through a nylon mesh to remove coarse fragments and rinsed three times with incubation medium.
- the glands are diluted to a final concentration of 2-4 mg dry weight/milliliter.
- the ability of gastric glands to form acid is measured based on aminopyrine (AP) accumulation (Berglindh, 1976).
- Samples of 1.0- milliliter gland suspension are equilibrated in 1.0 milliliter of medium containing 0.1 microcurie per milliliter 14 C-AP at 37 0 C in a shaking water bath together with compounds of Formula 1 or the corresponding non-isotopically enriched compounds or standards or controls.
- dbcAMP is added, followed by a 45-min incubation period.
- the glands are then separated from the medium by brief centrifugation, and aliquots of supernatant and the digested gland pellet are used for measurements in a liquid scintillation counter.
- the AP accumulation is calculated as the ratio between AP in intraglandular water and AP in the incubation medium (Sack, 1982). All determinations are made in triplicate.
- IC 50 is calculated by probit analysis where 0% corresponds to basal and 100% to maximal stimulated AP ratio.
- White New Zealander Rabbit fundic glands are obtained by high-pressure perfusion of the circulation of the stomach and subsequent collagenase treatment of pieces of fundic mucosa. After the glands have been washed several times, they are placed in 20- milliliter vials with dibutyryl cyclic AMP (1 millimolar) and the test compound (3 x 10 " to 10 "4 molar) in the presence of [ l4 C]-aminopyrine (0.125 micromolar) and are incubated at 37 0 C. The incubate is agitated (150 oscillations per minute) for 30 minutes and the reaction stopped by centrifugation (10 seconds at 20,000 g).
- the ability of the glands to maintain a pH gradient to the medium (pH 7.4) on stimulation with dibutyryl cyclic AMP is measured by means of the concentration ratio of [ C]-aminopyrine between glands and medium as described in Berglindh et al. Acta Physiol. Scand. 1976, 96, 150-169.
- the concentration of purified Jack bean Urease used (18 micrograms per milliliter, 1.28 U/mL) gave the same Urease activity as the bacterial suspension.
- Compounds of Formula 1 or the corresponding non-isotopically enriched compounds or standards or controls are dissolved in MeOH or DMSO and when necessary sonicated for some minutes. Aliquots are added to the test solutions to final concentrations of 1, 10, and 100 micromolar (with the exception of Flurofamide where the concentrations used are 1, 10, and 100 nanomolar), and the organic solvent component amounted to 51%. The samples are incubated for 30 min at 37 0 C in a water bath with gentle shaking.
- the reaction is started by adding 1 part 200 millimolar urea solution to 1 part test solution and stopped 10 min later by adding 25 parts reagent A (10 gram of phenol and 50 milligrams of Na 2 Fe(CN) 5 NO dissolved in 1 liter of water) and 25 parts reagent B (5 gram of NaOH and 8.4 milliliter of NaOCl (Sigma- Aldrich) dissolved in 1 liter of water).
- reagent A 10 gram of phenol and 50 milligrams of Na 2 Fe(CN) 5 NO dissolved in 1 liter of water
- reagent B 5 gram of NaOH and 8.4 milliliter of NaOCl (Sigma- Aldrich) dissolved in 1 liter of water.
- the samples are incubated for a further 15 min to allow color development, after which 200 microliters aliquots are transferred to 96-well microliter plates.
- the absorbance at 650 nm is determined at ambient temperature using (NH 3 ) 2 SO 4 as standard.
- SPF mice are challenged with bacteria three times during a 6-day period, and 3 weeks after inoculation animals are treated orally according to different regimens for 4 weeks.
- Six different regimens are selected as follows: an uninfected no treatment control, an infected no treatment group to check for spontaneous elimination of the infection, an infected group receiving vehicle only, a triple therapy group used as a positive eradication control, and, finally, the three groups to be studied, using a compound of Formula 1, the corresponding non-isotopically enriched compound or standard or control and a therapeutic dose Flurofamide.
- Methocel vehicle (0.1 milliliter) adjusted to pH 6 with citric acid is used and given twice daily. Compounds are given either dissolved or suspended in the vehicle, and the amounts stated are per mouse, mean body weight of 30 gram, and day. Stock solutions or suspensions are stored frozen.
- Triple therapy is made up by 0.185 milligrams of bismuth, 0.675 milligrams of metronidazole, and 1.500 milligrams of tetracycline and is administered once daily for 2 weeks followed by bismuth alone once daily for another 2 weeks. In this group of animals, vehicle alone is administered at the second daily dosing occasion.
- Compounds of Formula 1, the corresponding non-isotopically enriched compounds or standards or controls (125 micromole per kg) or Flurofamide (230 micromole per kg) are each dosed twice daily for 4 weeks. Animals are sacrificed 24 h or 5 weeks after cessation of the treatment to measure suppression and eradication, respectively. The assessment is done by checking mouse stomach specimens for Urease activity, and the rate of both suppression and eradication for each regimen is expressed as the number of Urease positive animals divided by the number of animals checked x 100% as described in Dick- Hegedus et al Scand. J . Gastroenterol. 1991, 26, 909-915 Hazell et ⁇ Am. J. Gastroenterol. 1987, 82, 292-296, both of which are incorporated by reference in their entireties.
- the stomach is perfused continuously with warm (37 0 C) saline at a rate of 1 milliliter per minute.
- the perfusate is collected at 15-minute periods and its acid concentration measured by electrotitration against 100 millimolar NaOH to an endpoint of pH 7, and acid output (micromolar of H + /15 minute) is calculated.
- histamine (10 milligrams per kilogram per hour) is administered after a basal period of 45 minutes by iv infusion into the jugular vein, and observation is continued until acid output reaches a stable plateau (Herling, 1986).
- Compounds of Formula 1 or the corresponding non-isotopically enriched compounds or standards or controls are administered iv (25% DMSO, 1 milliliter per rat). Maximal inhibition is calculated as percent change versus pre-dose value and presented as mean +/- SEM.
- Gastric acid secretion is induced with an iv infusion of 0.05 milligrams per kilogram per h of histamine, which produces a maximal stimulation.
- Gastric juice is collected from the pouch at 30-min intervals, and acidity is measured by titration against 100 millimolar NaOH to an endpoint of pH 7, and acid output (millimole H + /30 minute) is calculated.
- Compounds of Formula 1 or the corresponding non-isotopically enriched compounds or standards or controls are administered at doses of 0.3 milligrams per kilogram iv or 1 milligrams per kilogram id at a volume of 20 milliliter per dog as soon as acid secretion stabilizes.
- mice Female Wistar rats are treated orally for 10 weeks with 30 milligrams per kilogram per day of compounds of Formula 1 or the corresponding non-isotopically enriched compounds or standards or controls. At days 1 to 3, rats receive said compound by intraperitoneal (ip) administration, to cause gastric acid inhibition and therefore to reduce the acidic degradation of subsequent orally administered test compounds to 10 weeks. Said compounds are suspended in potato starch mucilage (20 milligrams per milliliter) and administered at a volume of 2 milliliter per kilogram. A control group is also included in the experiment. Blood samples are collected retroorbitally during anesthesia. Serum Gastrin levels (picogram per milliliter) are determined by using a commercially available RIA kit and presented as means +/- SEM. Significant differences (p ⁇ 0.05) are calculated by Students t- test.
- a mixture containing an equal amount of a compound of Formula 1 and the corresponding non-isotopically enriched compound or standard or control are administered to the subjects either orally or by intravenous infusion.
- Blood specimens are 6 038819
- Serum is decanted immediately, and stored at -1O 0 C. Serum concentrations of said compound and the corresponding non-isotopically enriched compound or standard or control are analyzed by HPLC/MS/MS.
- Omeprazole analogues VI. The reaction of Omeprazole in the absence of 2- mercaptoethanol" Browne, Synthesis and Applications of ' Isotopically Labelled Compounds, Proceedings of the
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description







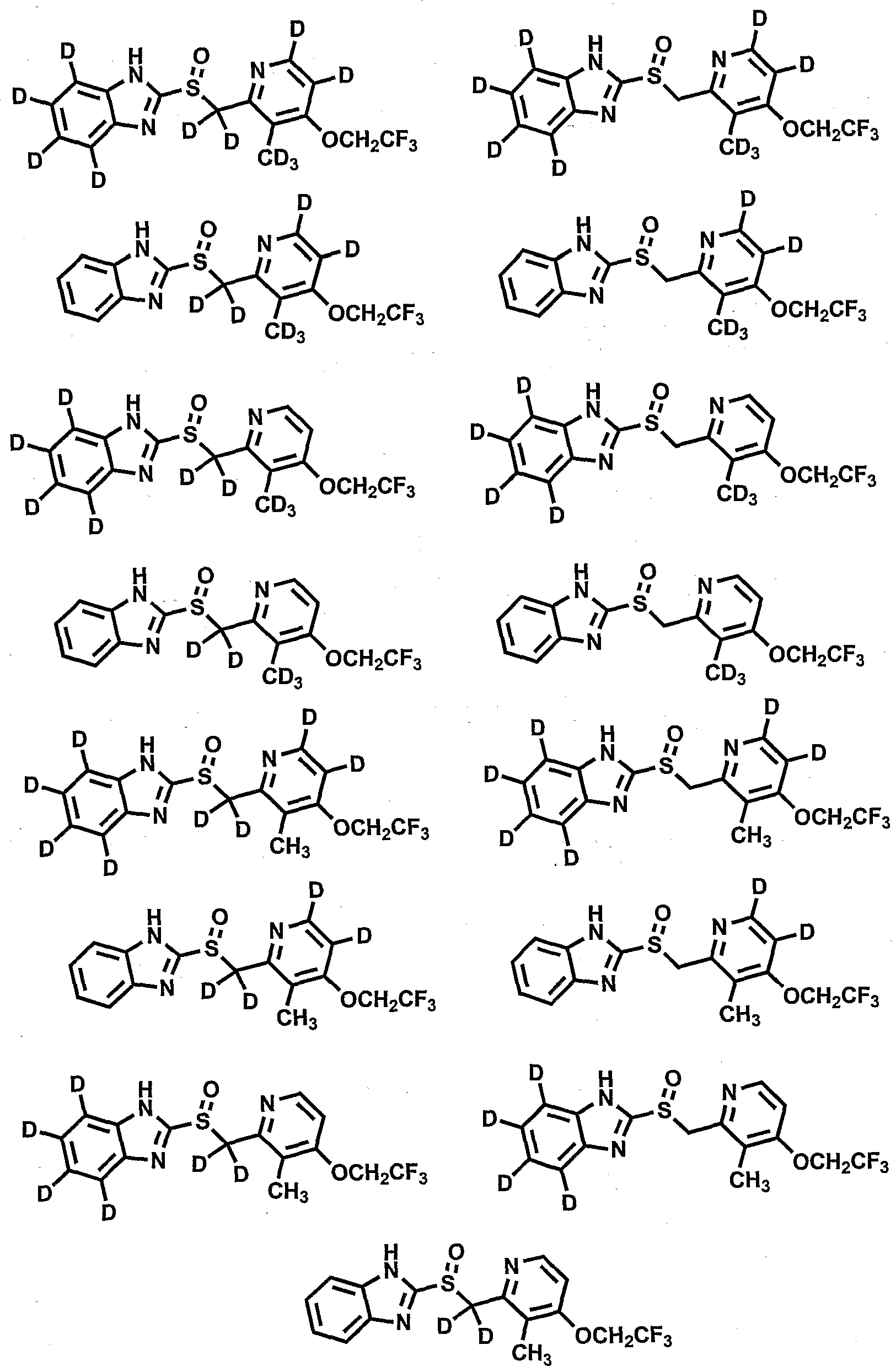






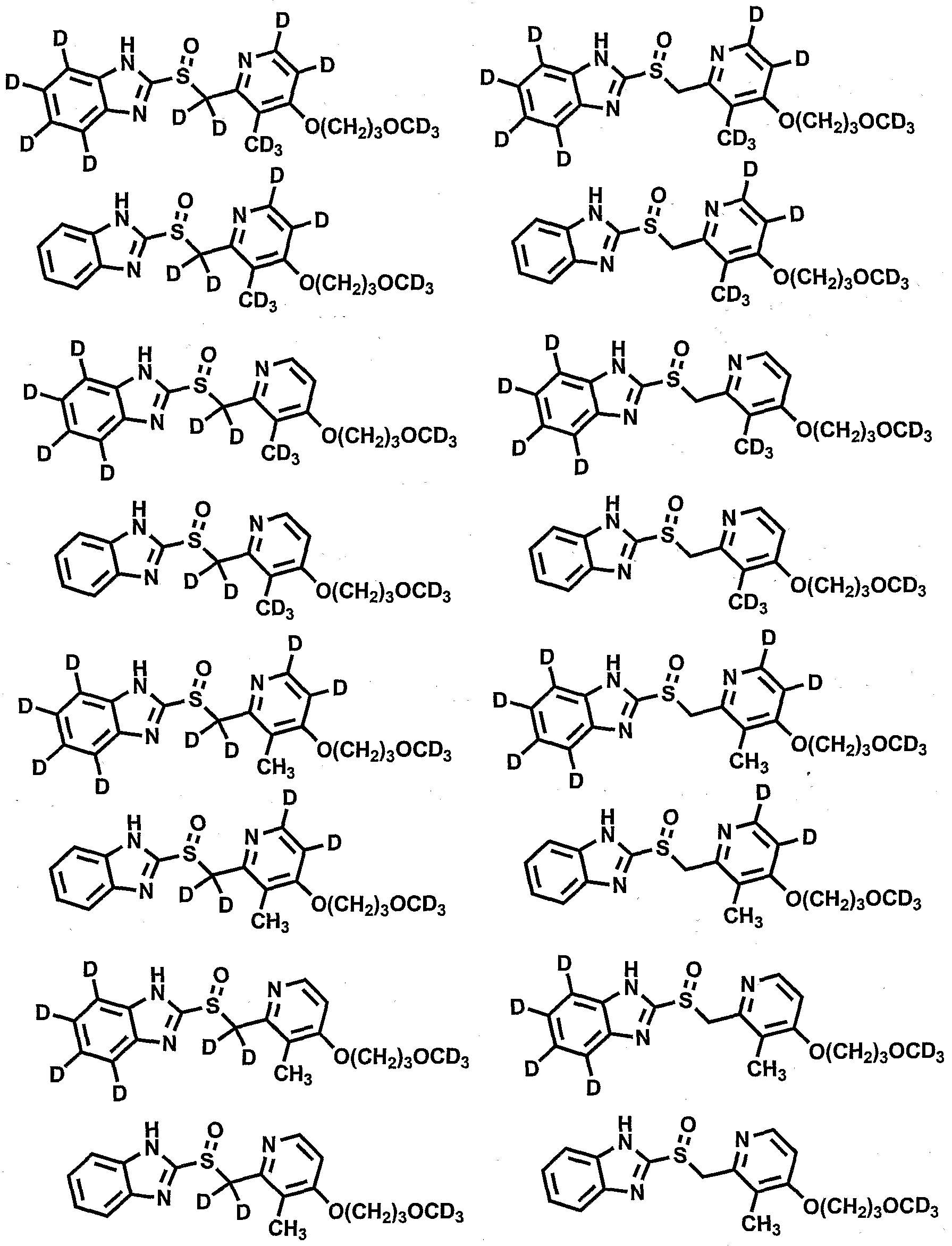












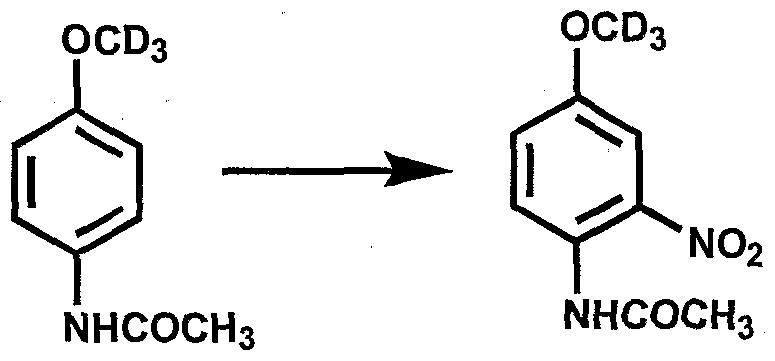

























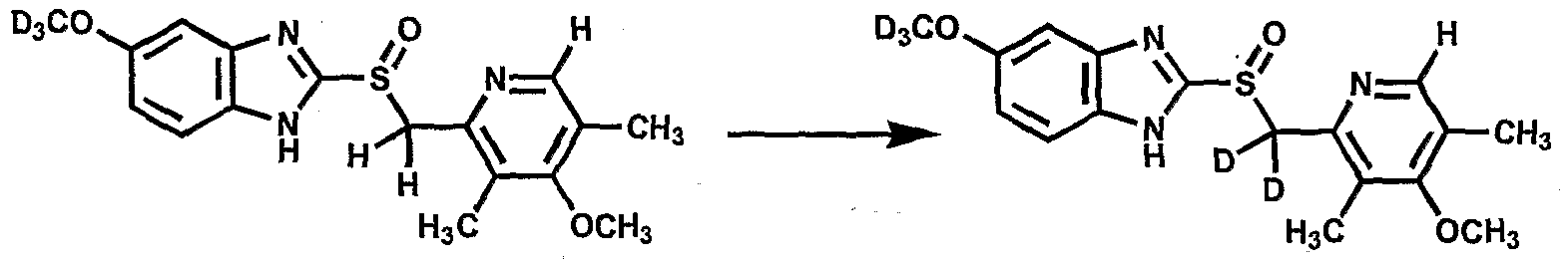





















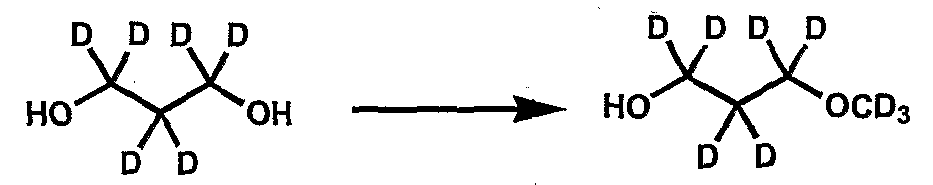

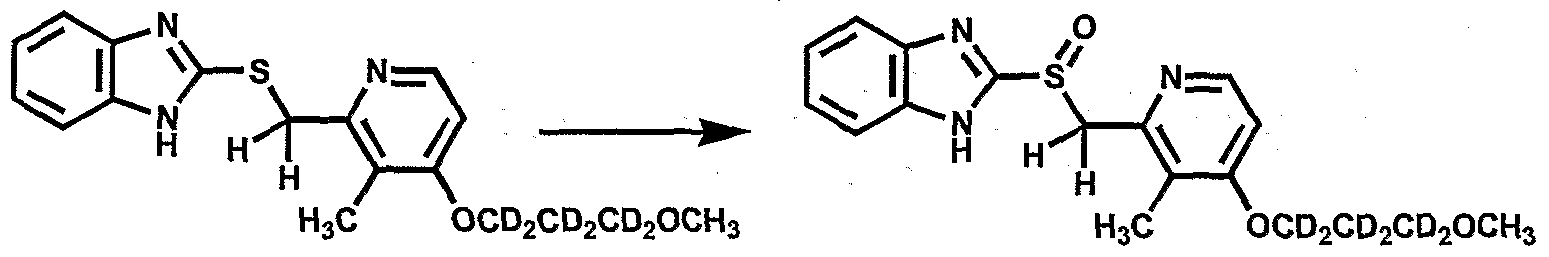















Claims







































Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2006800426099A CN101309917B (en) | 2005-10-06 | 2006-10-04 | Inhibitors of the gastric h+, k+-atpase with enhanced therapeutic properties |
| AU2006299424A AU2006299424A1 (en) | 2005-10-06 | 2006-10-04 | Deuterated inhibitors of gastric H+, K+-ATPase with enhanced therapeutic properties |
| BRPI0617987-8A BRPI0617987A2 (en) | 2005-10-06 | 2006-10-04 | composition, pharmaceutical composition, effervescent dosage form, multiple unit oral oral pharmaceutical composition, extended release pharmaceutical dosage form, enteric coated pharmaceutical dosage form, stable pharmaceutical dosage form for oral administration to mammalian subjects, method for treating diseases acid-related diseases by inhibiting gastric acid secretion, method for treating a bacterial infection caused or caused by helicobacter pylori, process for preparing a compound of formula 3, process for preparing a compound of formula 5, use of a compound of formula 1 for the preparation of a medicament for treating gastric acid-related diseases by inhibiting gastric acid secretion, use of a compound of formula 1 for the preparation of a medicament for treating a bacterial infection caused or caused by helicobacter pyl or use of a compound of formula 1 for the preparation of a medicament for treating gastric acid-related diseases by inhibiting gastric acid secretion |
| CA002624179A CA2624179A1 (en) | 2005-10-06 | 2006-10-04 | Deuterated inhibitors of gastric h+, k+-atpase with enhanced therapeutic properties |
| JP2008534658A JP2009511481A (en) | 2005-10-06 | 2006-10-04 | Gastric H +, K + -ATPase deuteration inhibitors with enhanced therapeutic properties |
| EP06816227A EP1934201A1 (en) | 2005-10-06 | 2006-10-04 | Deuterated inhibitors of gastric h+, k+-atpase with enhanced therapeutic properties |
| IL190331A IL190331A0 (en) | 2005-10-06 | 2008-03-20 | Deuterated inhibitors of the gastric h+, k+-atpase with enhanced therapeutic properties |
| US12/413,365 US20090215831A1 (en) | 2005-10-06 | 2009-03-27 | Inhibitors of the gastric h+, k+-atpase with enhanced therapeutic properties |
| US12/413,323 US20090209592A1 (en) | 2005-10-06 | 2009-03-27 | Inhibitors of the gastric h+, k+-atpase with enhanced therapeutic properties |
| HK09103859.5A HK1124336A1 (en) | 2005-10-06 | 2009-04-27 | Deuterated inhibitors of gastric h+, k+-atpase with enhanced therapeutic properties |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US72416005P | 2005-10-06 | 2005-10-06 | |
| US60/724,160 | 2005-10-06 | ||
| US74131605P | 2005-12-01 | 2005-12-01 | |
| US60/741,316 | 2005-12-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007041630A1 true WO2007041630A1 (en) | 2007-04-12 |
Family
ID=37603090
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/038819 WO2007041630A1 (en) | 2005-10-06 | 2006-10-04 | Deuterated inhibitors of gastric h+, k+-atpase with enhanced therapeutic properties |
Country Status (10)
| Country | Link |
|---|---|
| US (3) | US7598273B2 (en) |
| EP (1) | EP1934201A1 (en) |
| JP (1) | JP2009511481A (en) |
| CN (1) | CN101309917B (en) |
| AU (1) | AU2006299424A1 (en) |
| BR (1) | BRPI0617987A2 (en) |
| CA (1) | CA2624179A1 (en) |
| HK (1) | HK1124336A1 (en) |
| IL (1) | IL190331A0 (en) |
| WO (1) | WO2007041630A1 (en) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008127640A2 (en) * | 2007-04-11 | 2008-10-23 | Auspex Pharmaceuticals, Inc. | Substituted benzimidazoles |
| US7598273B2 (en) | 2005-10-06 | 2009-10-06 | Auspex Pharmaceuticals, Inc | Inhibitors of the gastric H+, K+-ATPase with enhanced therapeutic properties |
| US7601737B2 (en) | 2005-07-26 | 2009-10-13 | Nycomed Gmbh | Isotopically substituted proton pump inhibitors |
| CN101492452B (en) * | 2008-01-25 | 2010-11-17 | 山东轩竹医药科技有限公司 | Sulfhydryl benzimidazole derivative containing dioxepane-pyridine |
| CN101492464B (en) * | 2008-01-25 | 2011-01-12 | 山东轩竹医药科技有限公司 | Sulfhydryl imidazopyridine derivative containing dioxepane-pyridine |
| WO2010120797A3 (en) * | 2009-04-13 | 2011-03-31 | Auspex Pharmaceuticals, Inc. | Methods of reduction of interpatient variability |
| JP2011516426A (en) * | 2008-03-28 | 2011-05-26 | コンサート ファーマシューティカルズ インコーポレイテッド | Quinazoline derivatives and methods of treatment |
| JP2012501328A (en) * | 2008-09-02 | 2012-01-19 | アクチミス ファーマシューティカルズ インコーポレーテッド | Isotopically enriched pyrimidin-5-yl acetic acid derivatives as CRTH2 antagonists |
| JP2012502038A (en) * | 2008-09-03 | 2012-01-26 | テバ ファーマシューティカル インダストリーズ リミティド | 2-oxo-1,2-dihydro-quinoline modulator of immune function |
| JP2013519708A (en) * | 2010-02-19 | 2013-05-30 | ノバルティス アーゲー | Deuterated pyrrolopyrimidine compounds as CDK4 / 6 inhibitors |
| CN103204819A (en) * | 2013-04-15 | 2013-07-17 | 公安部物证鉴定中心 | Deuterated diazepam and preparation method thereof |
| WO2014039421A1 (en) * | 2012-09-04 | 2014-03-13 | Celgene Corporation | Isotopologues of 3-(5-amino-2-methyl-4-oxoquinazolin-3(4h)-yl) piperidine-2-6-dione and methods of preparation thereof |
| US9035050B2 (en) | 2008-02-29 | 2015-05-19 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
| US9061060B2 (en) | 2008-07-15 | 2015-06-23 | Theracos Inc. | Deuterated benzylbenzene derivatives and methods of use |
| US9074233B2 (en) | 2010-09-01 | 2015-07-07 | Concert Pharmaceuticals, Inc. | Process for preparing an enantiomerically enriched, deuterated secondary alcohol from a corresponding ketone without reducing deuterium incorporation |
| US9085788B2 (en) | 2010-09-01 | 2015-07-21 | Concert Pharmaceuticals, Inc. | Process for preparing an enantiomerically enriched, deuterated secondary alcohol from a corresponding ketone without reducing deuterium incorporation |
| US9260432B2 (en) | 2009-09-02 | 2016-02-16 | Concert Pharmaceuticals, Inc. | Substituted derivatives of bicyclic [4.3.0] heteroaryl compounds |
| US9328113B2 (en) | 2012-04-13 | 2016-05-03 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
| US9492457B2 (en) | 2009-02-27 | 2016-11-15 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
Families Citing this family (275)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7683177B2 (en) * | 2003-06-10 | 2010-03-23 | Teva Pharmaceutical Industries Ltd | Process for preparing 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles and novel chlorinated derivatives of pantoprazole |
| US7863274B2 (en) * | 2005-07-29 | 2011-01-04 | Concert Pharmaceuticals Inc. | Deuterium enriched analogues of tadalafil as PDE5 inhibitors |
| CA2616383C (en) * | 2005-07-29 | 2015-06-09 | Concert Pharmaceuticals Inc. | Novel benzo[d][1,3]-dioxol derivatives |
| CA2646229A1 (en) * | 2006-03-16 | 2007-09-27 | Vertex Pharmaceuticals Incorporated | Deuterated hepatitis c protease inhibitors |
| US20070276042A1 (en) * | 2006-05-26 | 2007-11-29 | Auspex Pharmaceuticals, Inc. | Preparation and utility of substituted carboxylic acid compounds |
| US20080051422A1 (en) * | 2006-08-22 | 2008-02-28 | Concert Pharmaceuticals Inc. | 4-Aminoquinazoline derivatives and methods of use thereof |
| US20110053964A1 (en) * | 2006-08-22 | 2011-03-03 | Roger Tung | 4-aminoquinazoline derivatives and methods of use thereof |
| CA2664940A1 (en) * | 2006-10-20 | 2008-06-12 | Concert Pharmaceuticals Inc. | 3-(dihydro-1h-pyrazolo [4,3-d] pyrimidin-5-yl)-4-propoxybenzenesulfonamide derivatives and methods of use |
| US20080280927A1 (en) * | 2006-10-20 | 2008-11-13 | Concert Pharmaceuticals Inc. | 3-(dihydro-1h-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxybenzenesulfonamide derivatives and methods of use |
| US20090197899A1 (en) * | 2006-10-20 | 2009-08-06 | Concert Pharmaceuticals Inc. | 3-(Dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxybenzenesulfonamide Derivatives and Methods of Use |
| US20090270425A1 (en) * | 2006-10-20 | 2009-10-29 | Concert Pharmaceuticals Inc. | 3-(dihydro-1h-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxybenzenesulfonamide derivatives and methods of use |
| WO2008066620A2 (en) * | 2006-10-20 | 2008-06-05 | Concert Pharmaceuticals Inc. | Dibenzothiazepine derivatives |
| JP5302200B2 (en) * | 2006-10-23 | 2013-10-02 | コンサート ファーマシューティカルズ インコーポレイテッド | Oxazolidinone derivatives and methods of use |
| US8796267B2 (en) | 2006-10-23 | 2014-08-05 | Concert Pharmaceuticals, Inc. | Oxazolidinone derivatives and methods of use |
| US7932235B2 (en) * | 2006-11-17 | 2011-04-26 | Concert Pharmaceuticals, Inc. | Triazolyl tropane derivatives |
| WO2008076949A2 (en) * | 2006-12-15 | 2008-06-26 | Concert Pharmaceuticals Inc. | Quinazoline derivatives and methods of treatment |
| US20100260674A1 (en) * | 2006-12-15 | 2010-10-14 | Concert Pharmaceuticals, Inc. | Quinazoline derivatives and methods of treatment |
| US20090325976A1 (en) * | 2006-12-21 | 2009-12-31 | Concert Pharmaceuticals Inc. | Prostacyclin derivatives |
| US8071596B2 (en) * | 2007-01-12 | 2011-12-06 | Concert Pharmaceuticals, Inc. | Endothelin receptor antagonists |
| US8080549B2 (en) * | 2007-01-12 | 2011-12-20 | Concert Pharmaceuticals, Inc. | Endothelin receptor antagonists |
| US20080262006A1 (en) * | 2007-02-02 | 2008-10-23 | Harbeson Scott L | Selective endothelin type-a antagonists |
| WO2008106125A2 (en) * | 2007-02-26 | 2008-09-04 | Concert Pharmaceuticals, Inc. | Deuterated derivatives of silodosin as alpha la-adrenoceptor antagonists |
| US20100076010A1 (en) * | 2007-02-26 | 2010-03-25 | Concert Pharmaceuticals, Inc. | Alpha 1a-adrenoceptor antagonists |
| WO2008109175A1 (en) * | 2007-03-07 | 2008-09-12 | Concert Pharmaceuticals, Inc. | Deuterated piperazine derivatives as anti-anginal compounds |
| CA2681628C (en) | 2007-03-16 | 2016-10-18 | Concert Pharmaceuticals, Inc. | Inhibitors of cholesterol ester transfer protein |
| WO2008130863A2 (en) * | 2007-04-11 | 2008-10-30 | Auspex Pharmaceuticals, Inc. | Substituted benzimidazoles |
| US8198305B2 (en) * | 2007-04-13 | 2012-06-12 | Concert Pharmaceuticals Inc. | 1,2-benzisoxazol-3-yl compounds |
| ES2401914T3 (en) | 2007-04-25 | 2013-04-25 | Concert Pharmaceuticals Inc. | Cilostazol analogues |
| US8349817B2 (en) * | 2007-04-25 | 2013-01-08 | Concert Pharmaceuticals, Inc. | Analogues of cilostazol |
| PL2522667T3 (en) * | 2007-05-01 | 2015-01-30 | Concert Pharmaceuticals Inc | Morphinan compounds |
| CA2685924A1 (en) * | 2007-05-01 | 2008-11-13 | Concert Pharmaceuticals Inc. | Naphthyl(ethyl)acetamides |
| DK2792662T3 (en) | 2007-05-01 | 2016-07-25 | Concert Pharmaceuticals Inc | MORPHINANE COMPOUNDS |
| WO2008141021A1 (en) * | 2007-05-08 | 2008-11-20 | Concert Pharmaceuticals, Inc. | Deuterated derivatives of tetrahydrotriazolopyrazine compounds and their use as dpp-iv inhibitors |
| US20080306151A1 (en) * | 2007-06-11 | 2008-12-11 | Protia, Llc | Deuterium-enriched fenofibrate |
| WO2008156632A1 (en) * | 2007-06-12 | 2008-12-24 | Concert Pharmaceuticals, Inc. | Azapeptide derivatives |
| CA2690379A1 (en) * | 2007-06-13 | 2008-12-24 | Auspex Pharmaceuticals, Inc. | Substituted piperazines |
| US20090005366A1 (en) * | 2007-06-19 | 2009-01-01 | Protia, Llc | Deuterium-enriched olanzapine |
| US20080318920A1 (en) * | 2007-06-19 | 2008-12-25 | Protia, Llc | Deuterium-enriched ezetimibe |
| US7915309B2 (en) * | 2007-06-20 | 2011-03-29 | Protia, Llc | Deuterium-enriched oseltamivir |
| US20080319007A1 (en) * | 2007-06-21 | 2008-12-25 | Protia, Llc | Deuterium-enriched montelukast |
| CA2702317A1 (en) * | 2007-07-09 | 2009-01-15 | Concert Pharmaceuticals, Inc. | Novel pyrimidinecarboxamide derivatives |
| EP2190841B1 (en) * | 2007-08-14 | 2013-05-15 | Concert Pharmaceuticals Inc. | Substituted oxazolidinone derivatives |
| US20090062561A1 (en) * | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched latanoprost |
| US20090062299A1 (en) * | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched doxazosin |
| US20090062385A1 (en) * | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched fesoterodine |
| US20090062393A1 (en) * | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched pregabalin |
| US20090062374A1 (en) * | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched lasofoxifene |
| US20090062305A1 (en) * | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched cetirizine |
| US20090062399A1 (en) * | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched sertraline |
| US20090062283A1 (en) * | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched linezolid |
| US20090062316A1 (en) * | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched pelitrexol |
| US20090069410A1 (en) * | 2007-09-09 | 2009-03-12 | Protia, Llc | Deuterium-enriched paclitaxel |
| US20090069413A1 (en) * | 2007-09-09 | 2009-03-12 | Protia, Llc | Deuterium-enriched olopatadine |
| US20090131485A1 (en) * | 2007-09-10 | 2009-05-21 | Concert Pharmaceuticals, Inc. | Deuterated pirfenidone |
| US20090069339A1 (en) * | 2007-09-11 | 2009-03-12 | Protia, Llc | Deuterium-enriched ciprofloxacin |
| US20090069431A1 (en) * | 2007-09-12 | 2009-03-12 | Protia, Llc | Deuterium-enriched milnacipran |
| WO2009035662A1 (en) * | 2007-09-12 | 2009-03-19 | Concert Pharmaceuticals, Inc. | Deuterated 4 -oxoquinoline derivatives for the treatment of hiv infection |
| US20090076165A1 (en) * | 2007-09-12 | 2009-03-19 | Protia, Llc | Deuterium-enriched memantine |
| US8288414B2 (en) * | 2007-09-12 | 2012-10-16 | Deuteria Pharmaceuticals, Inc. | Deuterium-enriched lenalidomide |
| EP2200997B9 (en) | 2007-09-13 | 2016-01-13 | Concert Pharmaceuticals Inc. | Synthesis of deuterated benzodioxoles |
| US20090076112A1 (en) * | 2007-09-13 | 2009-03-19 | Protia, Llc | Deuterium-enriched eltrombopag |
| US20090076118A1 (en) * | 2007-09-13 | 2009-03-19 | Protia, Llc | Deuterium-enriched saxagliptin |
| US7919516B2 (en) * | 2007-09-13 | 2011-04-05 | Protia, Llc | Deuterium-enriched ondansetron |
| US20090076161A1 (en) * | 2007-09-13 | 2009-03-19 | Protia, Llc | Deuterium-enriched bupropion |
| US20090076116A1 (en) * | 2007-09-13 | 2009-03-19 | Protia, Llc | Deuterium-enriched carvediolo |
| US20090075965A1 (en) * | 2007-09-13 | 2009-03-19 | Protia, Llc | Deuterium-enriched amoxicilin |
| US20090076010A1 (en) * | 2007-09-13 | 2009-03-19 | Protia, Llc | Deuterium-enriched lamotrigine |
| US20090075991A1 (en) * | 2007-09-14 | 2009-03-19 | Protia, Llc | Deuterium-enriched efavirenz |
| US20090076148A1 (en) * | 2007-09-14 | 2009-03-19 | Protia, Llc | Deuterium-enriched pravastatin |
| US20090076038A1 (en) * | 2007-09-14 | 2009-03-19 | Protia, Llc | Deuterium-enriched entecavir |
| US20090076034A1 (en) * | 2007-09-14 | 2009-03-19 | Protia, Llc | Deuterium-enriched bms-690514 |
| US20090076039A1 (en) * | 2007-09-14 | 2009-03-19 | Protia, Llc | Deuterium-enriched valacyclovir |
| US20090076102A1 (en) * | 2007-09-14 | 2009-03-19 | Protia, Llc | Deuterium-enriched muraglitazar |
| US20090075994A1 (en) * | 2007-09-14 | 2009-03-19 | Protia, Llc | Deuterium-enriched radafaxine |
| US20090076028A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched itraconazole |
| US20090076000A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched mycophenolate mofetil |
| US20090076164A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched tapentadol |
| US20090076128A1 (en) * | 2007-09-15 | 2009-03-19 | Protia Llc | Deuterium-enriched topiramate |
| US20090076163A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched dapoxetine |
| US20090076138A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched darunavir |
| US20090076018A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched ranolazine |
| US20090076157A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched senicapoc |
| US20090076040A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched valganciclovir |
| US20090075944A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched ibandronate |
| US20090076264A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched rivaroxaban |
| US20090076017A1 (en) * | 2007-09-15 | 2009-03-19 | Protia, Llc | Deuterium-enriched trabectedin |
| US20090076144A1 (en) * | 2007-09-17 | 2009-03-19 | Protia, Llc | Deuterium-enriched bazedoxifene |
| US20090076027A1 (en) * | 2007-09-17 | 2009-03-19 | Protia, Llc | Deuterium-enriched lurasidone |
| US20090076126A1 (en) * | 2007-09-17 | 2009-03-19 | Protia, Llc | Deuterium-enriched enalapril |
| US20090118238A1 (en) * | 2007-09-17 | 2009-05-07 | Protia, Llc | Deuterium-enriched alendronate |
| US20090076154A1 (en) * | 2007-09-17 | 2009-03-19 | Protia, Llc | Deuterium-enriched vorinostat |
| US20090076117A1 (en) * | 2007-09-17 | 2009-03-19 | Protia, Llc | Deuterium-enriched laropiprant |
| US20090076058A1 (en) * | 2007-09-17 | 2009-03-19 | Protia, Llc | Deuterium-enriched finasteride |
| US20090076167A1 (en) * | 2007-09-17 | 2009-03-19 | Protia, Llc | Deuterium-enriched tramiprosate |
| US20090076011A1 (en) * | 2007-09-19 | 2009-03-19 | Protia, Llc | Deuterium-enriched tirapazamine |
| US20090075997A1 (en) * | 2007-09-19 | 2009-03-19 | Protia, Llc | Deuterium-enriched satavaptan |
| US20090076127A1 (en) * | 2007-09-19 | 2009-03-19 | Protia, Llc | Deuterium-enriched larotaxel |
| US20090076159A1 (en) * | 2007-09-19 | 2009-03-19 | Protia, Llc | Deuterium-enriched eplivanserin |
| US20090082454A1 (en) * | 2007-09-25 | 2009-03-26 | Protia, Llc | Deuterium-enriched disufenton |
| US20090082334A1 (en) * | 2007-09-25 | 2009-03-26 | Protia, Llc | Deuterium-enriched quetiapine |
| US20090082462A1 (en) * | 2007-09-25 | 2009-03-26 | Protia, Llc | Deuterium-enriched armodafinil |
| US20090082364A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched levocedtirizine |
| US20090082335A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched oxcarbazepine |
| US20090082459A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched guanfacine |
| US20090082452A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched lumiracoxib |
| US20090082363A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched posaconazole |
| US20090082436A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched rivastigmine |
| US20090082387A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched nvp-bez234 |
| US8722710B2 (en) | 2007-09-26 | 2014-05-13 | Deuterx, Llc | Deuterium-enriched pioglitazone |
| US20090082471A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched fingolimod |
| US20090082442A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched lubiprostone |
| US20090143363A1 (en) * | 2007-10-15 | 2009-06-04 | Concert Pharmaceuticals, Inc. | Deuterated lorcaserin |
| US8410124B2 (en) * | 2007-10-18 | 2013-04-02 | Concert Pharmaceuticals Inc. | Deuterated etravirine |
| WO2009054964A1 (en) * | 2007-10-22 | 2009-04-30 | Concert Pharmaceuticals, Inc. | Alpha-aminoamide derivatives |
| US20090131363A1 (en) * | 2007-10-26 | 2009-05-21 | Harbeson Scott L | Deuterated darunavir |
| US8592487B2 (en) * | 2007-10-26 | 2013-11-26 | Concert Pharmaceuticals, Inc. | Deuterated darunavir |
| US20090175824A1 (en) * | 2007-11-20 | 2009-07-09 | Craig Masse | Peptides for the treatment of HCV infections |
| EP2222166B9 (en) * | 2007-12-10 | 2012-11-07 | Concert Pharmaceuticals Inc. | Heterocyclic kinase inhibitors |
| US8084464B2 (en) * | 2007-12-18 | 2011-12-27 | Concert Pharmaceuticals, Inc. | Tetrahydroisoquinoline derivatives |
| WO2009094216A1 (en) * | 2008-01-22 | 2009-07-30 | Concert Pharmaceuticals Inc. | Derivatives of gefitinib |
| WO2009094210A1 (en) * | 2008-01-22 | 2009-07-30 | Concert Pharmaceuticals Inc. | Vandetanib derivatives |
| WO2009105604A1 (en) * | 2008-02-20 | 2009-08-27 | Auspex Pharmaceuticals, Inc. | Substituted triazolopyridines |
| EP2268604B1 (en) * | 2008-03-14 | 2017-02-15 | Retrotope, Inc. | Therapies for cancer using isotopically substituted lysine |
| EP2276774B1 (en) * | 2008-03-14 | 2016-08-17 | Retrotope, Inc. | Therapeutic substances that modulate genome methylation |
| WO2009126844A2 (en) * | 2008-04-09 | 2009-10-15 | Concert Pharmaceuticals Inc. | Derivatives of 3-(2-hydroxy-5-methyphenyl)-n,n-diisopropyl-3-phenylpropylamine and methods of use thereof |
| US8367674B2 (en) * | 2008-04-17 | 2013-02-05 | Concert Pharmaceuticals, Inc. | Piperazine derivatives |
| US20110129549A1 (en) * | 2008-04-17 | 2011-06-02 | Liu Julie F | Tricyclic benzo[5,6]cyclohepta[1,2-b]pyridine derivatives and uses thereof |
| US20100120786A1 (en) * | 2008-04-17 | 2010-05-13 | Concert Pharmaceuticals, Inc. | Piperazine Derivatives |
| WO2009146310A1 (en) * | 2008-05-28 | 2009-12-03 | Concert Pharmaceuticals Inc. | Deuterated tizanidine |
| US8354557B2 (en) * | 2008-06-17 | 2013-01-15 | Concert Pharmaceuticals, Inc. | Synthesis of deuterated morpholine derivatives |
| WO2010011868A2 (en) * | 2008-07-23 | 2010-01-28 | Auspex Pharmaceuticals | Pyridine sulfonamide modulators of endothelin-a-receptor |
| WO2010019557A1 (en) * | 2008-08-12 | 2010-02-18 | Concert Pharmaceuticals Inc. | N-phenyl-2-pyrimidineamine derivatives |
| ES2451542T3 (en) | 2008-08-29 | 2014-03-27 | Concert Pharmaceuticals Inc. | Substituted triazolo-pyridazine derivatives |
| US20100105755A1 (en) * | 2008-09-12 | 2010-04-29 | Auspex Pharmaceuticals, Inc. | Substituted benzamide modulators of dopamine receptor |
| US8704001B2 (en) * | 2008-09-16 | 2014-04-22 | Concert Pharmaceuticals, Inc. | Deuterated 2-amino-3-hydroxypropanoic acid derivatives |
| WO2010033270A1 (en) * | 2008-09-17 | 2010-03-25 | Auspex Pharmaceuticals, Inc. | Dibenzothiazepine modulators of dopamine, alpha adrenergic, and serotonin receptors |
| ES2750825T3 (en) * | 2008-09-19 | 2020-03-27 | Concert Pharmaceuticals Inc | Deuterated morphinan compounds |
| US20100087455A1 (en) * | 2008-10-06 | 2010-04-08 | Auspex Pharmaceuticals, Inc. | Substituted xanthine compounds |
| AU2009305563C1 (en) | 2008-10-17 | 2015-09-03 | Signature Therapeutics, Inc. | Pharmaceutical compositions with attenuated release of phenolic opioids |
| US20100099701A1 (en) * | 2008-10-20 | 2010-04-22 | Auspex Pharmaceuticals, Inc. | Isoquinolinone modulators of 5-ht3 receptors |
| US9045453B2 (en) | 2008-11-14 | 2015-06-02 | Concert Pharmaceuticals, Inc. | Substituted dioxopiperidinyl phthalimide derivatives |
| US20100137215A1 (en) * | 2008-11-25 | 2010-06-03 | Concert Pharmaceuticals Inc. | Novel tetrahydro-1h-pyrido[4,3-b]indoles |
| US20110313004A1 (en) * | 2008-12-04 | 2011-12-22 | Concert Pharmaceuticals, Inc. | Deuterated pyridinones |
| US20100143354A1 (en) * | 2008-12-04 | 2010-06-10 | Auspex Pharmaceuticals, Inc. | Triazine dna modifiers |
| US20100144657A1 (en) * | 2008-12-09 | 2010-06-10 | Auspex Pharmaceuticals, Inc. | PHENYLPIPERIDINE MODULATORS OF mu-OPIOID RECEPTORS |
| WO2010075086A2 (en) * | 2008-12-15 | 2010-07-01 | Auspex Pharmaceuticals, Inc. | Pyrrolidinone inhibitors of pde-4 |
| ES2575689T3 (en) * | 2008-12-23 | 2016-06-30 | Aquilus Pharmaceuticals, Inc | Compounds and methods for the treatment of pain and other diseases |
| EP2396312A1 (en) * | 2009-02-11 | 2011-12-21 | Celgene Corporation | Isotopologues of lenalidomide |
| US20110053961A1 (en) * | 2009-02-27 | 2011-03-03 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
| US8563554B2 (en) | 2009-03-17 | 2013-10-22 | Concert Pharmaceuticals, Inc. | Deuterated pyrazinoisoquinoline compounds |
| US8778998B2 (en) * | 2009-04-10 | 2014-07-15 | Auspex Pharmaceuticals, Inc. | Biphenyl-3-carboxylic acid modulators of beta-3-adrenoreceptor |
| WO2010123999A2 (en) * | 2009-04-21 | 2010-10-28 | Auspex Pharmaceuticals, Inc. | 1-methylpyrazole modulators of substance p, calcitonin gene-related peptide, adrenergic receptor, and/or 5-ht receptor |
| NZ620074A (en) | 2009-04-22 | 2015-09-25 | Axikin Pharmaceuticals Inc | 2,5-disubstituted arylsulfonamide ccr3 antagonists |
| CA2759251C (en) | 2009-04-23 | 2017-01-03 | Concert Pharmaceuticals, Inc. | 4-hydroxybutyric acid analogs |
| US20100305173A1 (en) * | 2009-04-30 | 2010-12-02 | Concert Pharmaceuticals, Inc. | Hydroxyethylamino sulfonamide derivatives |
| JP2012526810A (en) | 2009-05-13 | 2012-11-01 | イントラ−セルラー・セラピーズ・インコーポレイテッド | Organic compounds |
| US8410082B2 (en) | 2009-05-22 | 2013-04-02 | Concert Pharmaceuticals, Inc. | Fluorinated diaryl urea derivatives |
| WO2010144499A2 (en) * | 2009-06-09 | 2010-12-16 | Medolution Limited | Urea derivatives as kinase inhibitors |
| JP5852565B2 (en) | 2009-06-18 | 2016-02-03 | コンサート ファーマシューティカルズ インコーポレイテッド | Deuterated isoindoline-1,3-dione derivatives as PDE4 and TNF-α inhibitors |
| WO2011005520A1 (en) | 2009-06-23 | 2011-01-13 | Concert Pharmaceuticals, Inc. | Deuterium-modified triazolo-pyridazine derivatives as gaba-a receptor modulators |
| WO2011009075A2 (en) * | 2009-07-17 | 2011-01-20 | Rigel Pharmaceuticals, Inc. | Deuterated 2, 4-pyrimidinediamine compounds and prodrugs thereof and their uses |
| EP2456311A4 (en) * | 2009-07-24 | 2013-01-23 | Concert Pharmaceuticals Inc | Substituted imidazotriazines |
| US8575348B2 (en) * | 2009-07-28 | 2013-11-05 | Auspex Pharmaceuticals, Inc | Quinolone inhibitors of lipoprotein-associated phospholipase A2 |
| US9512104B2 (en) | 2009-07-28 | 2016-12-06 | Auspex Pharmaceuticals, Inc. | Quinolone inhibitors of lipoprotein-associated phospholipase A2 |
| US8637524B2 (en) * | 2009-07-28 | 2014-01-28 | Auspex Pharmaceuticals, Inc | Pyrimidinone inhibitors of lipoprotein-associated phospholipase A2 |
| US8618116B2 (en) * | 2009-08-03 | 2013-12-31 | Daljit Singh Dhanoa | Deuterium-enriched pyrimidine compounds and derivatives |
| EP2464355A4 (en) * | 2009-08-14 | 2013-01-02 | Concert Pharmaceuticals Inc | Substituted triazolophthalazine derivatives |
| US8658236B2 (en) | 2009-08-21 | 2014-02-25 | Deuteria Beverages, Llc | Alcoholic compositions having a lowered risk of acetaldehydemia |
| EP2473052A4 (en) * | 2009-09-02 | 2013-03-20 | Concert Pharmaceuticals Inc | Substituted xanthine derivatives |
| US9493477B2 (en) | 2009-09-08 | 2016-11-15 | Signature Therapeutics, Inc. | Compositions comprising enzyme-cleavable ketone-modified opioid prodrugs and optional inhibitors thereof |
| WO2011041584A2 (en) | 2009-09-30 | 2011-04-07 | President And Fellows Of Harvard College | Methods for modulation of autophagy through the modulation of autophagy-enhancing gene products |
| US8278460B2 (en) * | 2009-10-15 | 2012-10-02 | Concert Pharmaceuticals, Inc. | Substituted benzimidazoles |
| CA2778907A1 (en) * | 2009-10-28 | 2011-05-12 | Anadys Pharmaceuticals, Inc. | Deuterated 5,6-dihydro-1h-pyridin-2-one compounds |
| WO2011060112A1 (en) | 2009-11-12 | 2011-05-19 | Osi Pharmaceuticals, Inc. | Deuterated tyrosine kinase inhibitors |
| WO2011063001A1 (en) * | 2009-11-18 | 2011-05-26 | Concert Pharmaceuticals, Inc. | Niacin prodrugs and deuterated versions thereof |
| BR112012012085A2 (en) * | 2009-11-21 | 2016-05-17 | Hoffmann La Roche | heterocyclic antiviral compounds |
| US20110178180A1 (en) * | 2010-01-18 | 2011-07-21 | Kurt Nielsen | Deuterium-enriched colchicine, thiocolchicine, and derivatives thereof; methods of preparation; and use thereof |
| EP2539347A2 (en) * | 2010-02-24 | 2013-01-02 | Auspex Pharmaceuticals, Inc. | Trimethoxyphenyl inhibitors of tyrosine kinase |
| EP2542075A4 (en) * | 2010-03-01 | 2014-11-05 | Concert Pharmaceuticals Inc | Fluorouracil derivatives |
| US8513434B2 (en) * | 2010-03-02 | 2013-08-20 | Concert Pharmaceuticals Inc. | Tetrahydronaphthalene derivatives |
| US8575361B2 (en) | 2010-03-02 | 2013-11-05 | Concert Pharmaceuticals Inc. | Tetrahydronaphthalene derivatives |
| US8575221B2 (en) | 2010-03-17 | 2013-11-05 | Concert Pharmaceuticals, Inc. | Derivatives of dimethylcurcumin |
| WO2011133149A1 (en) * | 2010-04-21 | 2011-10-27 | Pharmacofore, Inc. | Compositions comprising enzyme-cleavable opioid prodrugs and inhibitors thereof |
| EP2560486B1 (en) | 2010-04-21 | 2018-11-21 | Signature Therapeutics, Inc. | Compositions comprising enzyme-cleavable amphetamine prodrugs and inhibitors thereof |
| US9238020B2 (en) | 2010-04-21 | 2016-01-19 | Signature Therapeutics, Inc. | Compositions comprising enzyme-cleavable phenol-modified tapentadol prodrug |
| US20110262355A1 (en) | 2010-04-21 | 2011-10-27 | Jenkins Thomas E | Compositions comprising enzyme-cleavable opioid prodrugs and inhibitors thereof |
| EP2560490A1 (en) * | 2010-04-21 | 2013-02-27 | Signature Therapeutics, Inc. | Compositions comprising enzyme-cleavable prodrugs of active agents and inhibitors thereof |
| US9107922B2 (en) | 2010-07-16 | 2015-08-18 | Concert Pharmaceuticals, Inc. | Pyrimidinecarboxamide derivatives |
| CN103249697B (en) * | 2010-09-03 | 2016-06-15 | 梯瓦制药国际有限责任公司 | It is useful as fixed (pridopidine) deuterated analogue of Puli many of dopaminergic stabilizer |
| WO2012096886A1 (en) | 2011-01-11 | 2012-07-19 | Signature Therapeutics, Inc. | Compositions comprising enzyme-cleavable oxycodone prodrug |
| ES2584634T3 (en) | 2011-01-11 | 2016-09-28 | Signature Therapeutics, Inc. | Compositions comprising an enzymatically cleavable oxycodone prodrug |
| WO2012099915A1 (en) * | 2011-01-19 | 2012-07-26 | Hongwen Zhu | Thiazolidine derivatives and their therapeutic use |
| ES2804540T3 (en) | 2011-02-14 | 2021-02-08 | Concert Pharmaceuticals Inc | Deuterated analogs of 4-hydroxybutyric acid |
| US9145390B2 (en) | 2011-03-03 | 2015-09-29 | Concert Pharmaceuticals, Inc. | Derivatives of pyrazole-substituted amino-heteroaryl compounds |
| WO2012122028A2 (en) | 2011-03-04 | 2012-09-13 | Concert Pharmaceuticals Inc. | Pyrazinoisoquinoline compounds |
| WO2012122420A2 (en) | 2011-03-09 | 2012-09-13 | Pharmacofore, Inc. | Opioid prodrugs with heterocyclic linkers |
| AU2012225337B2 (en) | 2011-03-09 | 2016-04-28 | Signature Therapeutics, Inc. | Active agent prodrugs with heterocyclic linkers |
| US9090585B2 (en) | 2011-03-28 | 2015-07-28 | Deuterx, Llc | 2,6-dioxo-3-deutero-piperdin-3-yl-isoindoline compounds |
| US20140213553A1 (en) * | 2011-05-03 | 2014-07-31 | Concert Pharmaceuticals Inc. | Carbamoylpyridone derivatives |
| JP6092849B2 (en) | 2011-05-04 | 2017-03-08 | バランス セラピューティックス, インコーポレイテッドBalance Therapeutics, Inc. | Pentylenetetrazole derivatives |
| HUE047354T2 (en) | 2011-05-18 | 2020-04-28 | Vertex Pharmaceuticals Europe Ltd | Deuterated derivatives of ivacaftor |
| CA2860489C (en) * | 2012-02-08 | 2019-11-12 | Merck Patent Gmbh | Deuterated thiazolidinone analogues as agonists for follicle stimulating hormone receptor |
| WO2013159026A1 (en) | 2012-04-20 | 2013-10-24 | Concert Pharmaceuticals, Inc. | Deuterated rigosertib |
| US20150197525A1 (en) * | 2012-06-15 | 2015-07-16 | Concert Pharmaceuticals, Inc. | Deuterated derivatives of ruxolitinib |
| WO2014008417A1 (en) | 2012-07-04 | 2014-01-09 | Concert Pharmaceuticals, Inc. | Deuterated vercirnon |
| ES2694202T3 (en) | 2012-07-12 | 2018-12-19 | Concert Pharmaceuticals, Inc. | Deuterated Idebenone |
| EA201590296A1 (en) | 2012-07-30 | 2015-07-30 | Консерт Фармасьютикалс Инк. | DEUTERATED IBRUTINIB |
| MX361499B (en) | 2012-08-17 | 2018-12-06 | Concert Pharmaceuticals Inc | Deuterated baricitinib. |
| EP2888222A1 (en) * | 2012-08-22 | 2015-07-01 | Concert Pharmaceuticals Inc. | Deuterated 4-hydroxybutyric acid analogs |
| EP2892884A1 (en) * | 2012-09-07 | 2015-07-15 | Axikin Pharmaceuticals, Inc. | Isotopically enriched arylsulfonamide ccr3 antagonists |
| EP2922838B1 (en) | 2012-10-22 | 2018-03-14 | Concert Pharmaceuticals Inc. | Solid forms of {s-3-(4-amino-1-oxo-isoindolin-2-yl)(piperidine-3,4,4,5,5-d5)-2,6-dione} . |
| WO2014110189A1 (en) | 2013-01-09 | 2014-07-17 | Concert Pharmaceuticals Inc. | Deuterated momelotinib |
| US9540340B2 (en) | 2013-01-14 | 2017-01-10 | Deuterx, Llc | 3-(5-substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| WO2014121036A1 (en) * | 2013-02-01 | 2014-08-07 | Deuterx, Llc | 5-deutero-2,4-thiazolidinedione and 5-deutero-2,4-oxazolidinedione derivatives and compositions comprising and methods of using the same |
| WO2014127331A1 (en) | 2013-02-17 | 2014-08-21 | Intra-Cellular Therapies, Inc. | Novel uses |
| CA2941560A1 (en) | 2013-03-14 | 2014-09-25 | Deuterx, Llc | 3-(substituted-4-oxo-quinazolin-3(4h)-yl)-3-deutero-piperidine-2,6-dione derivatives |
| WO2014152843A1 (en) | 2013-03-14 | 2014-09-25 | Deuterx, Llc | Deuterium-enriched 2,4-thiazolidinediones and methods of treatment |
| EP2968268B1 (en) | 2013-03-15 | 2020-07-29 | Concert Pharmaceuticals Inc. | Inhibitors of the enzyme udp-glucose: n-acyl-sphingosine glucosyltransferase |
| US9676790B2 (en) | 2013-08-30 | 2017-06-13 | Concert Pharmaceuticals, Inc. | Substituted thienotriazolodiazapines |
| WO2015085004A1 (en) | 2013-12-03 | 2015-06-11 | Intra-Cellular Therapies, Inc. | Novel methods |
| EP3077397B1 (en) * | 2013-12-06 | 2019-09-18 | Vertex Pharmaceuticals Inc. | 2-amino-6-fluoro-n-[5-fluoro-pyridin-3-yl]pyrazolo[1,5-a]pyrimidin-3-carboxamide compound useful as atr kinase inhibitor, its preparation, different solid forms and radiolabelled derivatives thereof |
| WO2015095713A1 (en) | 2013-12-20 | 2015-06-25 | Deuterx, Llc | Methods of treating neurological and other disorders using enantiopure deuterium-enriched bupropion |
| ES2831326T3 (en) | 2014-01-15 | 2021-06-08 | Poxel Sa | Methods to treat neurological, metabolic, and other disorders using deuterium-enriched enantiopure pioglitazone |
| EP3099299A4 (en) * | 2014-01-27 | 2017-10-04 | Auspex Pharmaceuticals, Inc. | Benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| MX2016010213A (en) | 2014-02-07 | 2017-04-13 | Auspex Pharmaceuticals Inc | Novel pharmaceutical formulations. |
| US9694017B2 (en) | 2014-02-10 | 2017-07-04 | Concert Pharmaceuticals, Inc. | Substituted triazolobenzodiazepines |
| MX2021014508A (en) | 2014-04-04 | 2023-05-18 | Intra Cellular Therapies Inc | Organic compounds. |
| ES2732442T3 (en) * | 2014-06-20 | 2019-11-22 | Intra Cellular Therapies Inc | Organic compounds |
| WO2016022893A1 (en) | 2014-08-07 | 2016-02-11 | Intra-Cellular Therapies, Inc. | Organic compounds |
| JP2017527614A (en) * | 2014-09-02 | 2017-09-21 | ブピンダー シン | Deuterated or non-deuterated molecules and pharmaceutical formulations |
| WO2016069630A1 (en) | 2014-10-27 | 2016-05-06 | Concert Pharmaceuticals, Inc. | Pyrimidine phosphonic acid esters bearing at least one deuterium atom |
| CA2966452A1 (en) * | 2014-12-17 | 2016-06-23 | Samuel David Brown | Substituted bicyclic compounds as bromodomain inhibitors |
| US20180022710A1 (en) | 2015-01-29 | 2018-01-25 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10196384B2 (en) | 2015-03-31 | 2019-02-05 | Vertex Pharmaceuticals (Europe) Limited | Deuterated CFTR modulators |
| SI3277681T1 (en) * | 2015-04-02 | 2019-09-30 | Merck Patent Gmbh | Imidazolonylquinolines and their use as atm kinase inhibitors |
| WO2016176335A1 (en) * | 2015-04-27 | 2016-11-03 | Concert Pharmaceuticals, Inc. | Deuterated otx-015 |
| CN107698620A (en) * | 2015-06-23 | 2018-02-16 | 江苏天士力帝益药业有限公司 | A kind of deuterated thieno piperidine derivative, preparation method and applications |
| AU2015399642B2 (en) * | 2015-06-26 | 2020-04-09 | Chongqing Dikang Erle Pharma Co. Ltd. | Novel phosphodiesterase type-5 inhibitor and application thereof |
| US9809603B1 (en) | 2015-08-18 | 2017-11-07 | Deuterx, Llc | Deuterium-enriched isoindolinonyl-piperidinonyl conjugates and oxoquinazolin-3(4H)-yl-piperidinonyl conjugates and methods of treating medical disorders using same |
| EP3352758A4 (en) | 2015-09-25 | 2018-08-01 | Vertex Pharmaceuticals (Europe) Limited | Deuterated cftr potentiators |
| US9688659B2 (en) * | 2015-10-21 | 2017-06-27 | NeuForm Pharmaceuticals, Inc. | Deuterated compounds for treating hematologic malignancies, and compositions and methods thereof |
| PT3377476T (en) * | 2015-11-16 | 2022-12-29 | Ptc Therapeutics Inc | Hydrogen isotope-enriched analogues of 1,2,4-oxadiazole benzoic acid compounds, compositions and uses thereof |
| WO2017087795A1 (en) | 2015-11-19 | 2017-05-26 | Concert Pharmaceuticals, Inc. | Deuterated epi-743 |
| PT3838274T (en) | 2016-01-26 | 2023-12-18 | Intra Cellular Therapies Inc | Pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxaline derivative for use in the treatment of cns disorders |
| CN108541255B (en) * | 2016-02-02 | 2019-06-14 | 深圳市塔吉瑞生物医药有限公司 | A kind of steroid compound and the composition comprising the compound and application thereof |
| US10442761B2 (en) | 2016-02-16 | 2019-10-15 | Concert Pharmaceuticals, Inc. | Deuterated GFT-505 |
| CN113754661A (en) | 2016-03-25 | 2021-12-07 | 细胞内治疗公司 | Organic compounds |
| US10682354B2 (en) | 2016-03-28 | 2020-06-16 | Intra-Cellular Therapies, Inc. | Compositions and methods |
| US11331316B2 (en) | 2016-10-12 | 2022-05-17 | Intra-Cellular Therapies, Inc. | Amorphous solid dispersions |
| US10961245B2 (en) | 2016-12-29 | 2021-03-30 | Intra-Cellular Therapies, Inc. | Substituted heterocycle fused gamma-carbolines for treatment of central nervous system disorders |
| JP6987868B2 (en) | 2016-12-29 | 2022-01-05 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | Organic compounds |
| RU2767410C2 (en) | 2017-03-24 | 2022-03-17 | Интра-Селлулар Терапиз, Инк. | New compositions and methods |
| KR102714561B1 (en) * | 2017-05-16 | 2024-10-10 | 버텍스 파마슈티칼스 인코포레이티드 | Deuterated pyridone amides and their prodrugs as modulators of sodium channels |
| CN109498811A (en) * | 2017-09-15 | 2019-03-22 | 江苏吉贝尔药业股份有限公司 | A kind of compound preparation containing potassium ion competitive sour retarding agent and non-steroidal anti-inflammatory drugs |
| MX2020005336A (en) | 2017-11-22 | 2020-08-13 | Concert Pharmaceuticals Inc | Deuterated analogs of d-serine and uses thereof. |
| WO2019109021A1 (en) | 2017-12-01 | 2019-06-06 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| CN111362949B (en) * | 2017-12-22 | 2021-12-21 | 深圳市塔吉瑞生物医药有限公司 | Substituted pyrazolo [1,5-a ] pyrimidine compound, and pharmaceutical composition and application thereof |
| WO2019144885A1 (en) | 2018-01-23 | 2019-08-01 | 深圳市塔吉瑞生物医药有限公司 | Substituted pyrazolo[1,5-a]pyrimidine macrocyclic compound |
| US11839614B2 (en) | 2018-01-31 | 2023-12-12 | Intra-Cellular Therapies, Inc. | Methods for treating or mitigating cardiotoxicity characterized by inhibition of adenosine A2 signaling and/or adenosine A2 receptor expression |
| JP2021517902A (en) * | 2018-04-16 | 2021-07-29 | 深▲チェン▼市塔吉瑞生物医薬有限公司Shenzhen TargetRx, Inc. | Substituted pyrorotriazine compounds and their pharmaceutical compositions and their use |
| EP3810134A4 (en) * | 2018-06-19 | 2022-03-30 | Celecor Therapeutics, Inc. | Deuterated ruc-4 |
| EP3816162A4 (en) * | 2018-07-12 | 2021-08-11 | Shenzhen TargetRx, Inc. | Diarylpyrazole compound, composition comprising same, and use thereof |
| CN112584837A (en) | 2018-08-31 | 2021-03-30 | 细胞内治疗公司 | New method |
| EP3843739A4 (en) | 2018-08-31 | 2022-06-01 | Intra-Cellular Therapies, Inc. | Novel methods |
| WO2020063676A1 (en) * | 2018-09-26 | 2020-04-02 | 江苏恒瑞医药股份有限公司 | Ligand-drug conjugate of exatecan analogue, preparation method therefor and application thereof |
| US11851409B2 (en) * | 2019-01-25 | 2023-12-26 | Qingdao Jião Pharmaceutical Technology Co., Ltd | Deuterated benzylaminopyrimidinedione derivatives and use thereof |
| US12065395B2 (en) | 2019-04-03 | 2024-08-20 | Sun Pharmaceutical Industries, Inc. | Processes for the preparation of deuterated D-serine |
| WO2020227491A1 (en) * | 2019-05-07 | 2020-11-12 | The Trustees Of The University Of Pennsylvania | Deuterated triazolopyrimidines |
| CN114072150A (en) | 2019-07-07 | 2022-02-18 | 细胞内治疗公司 | New method |
| CN110194773B (en) * | 2019-07-26 | 2019-11-22 | 上海美迪西生物医药股份有限公司 | Pyrrolo-pyrazole analog derivative, preparation method and its application in medicine |
| US11319313B2 (en) | 2020-06-30 | 2022-05-03 | Poxel Sa | Crystalline forms of deuterium-enriched pioglitazone |
| US11767317B1 (en) | 2020-06-30 | 2023-09-26 | Poxel Sa | Methods of synthesizing enantiopure deuterium-enriched pioglitazone |
| US20230150965A1 (en) * | 2021-11-05 | 2023-05-18 | Terran Biosciences, Inc. | Isotopically enriched n-methyl-1,3-benzodioxolylbutanamine (mbdb) and stereoisomers thereof |
| WO2023092044A2 (en) | 2021-11-17 | 2023-05-25 | Terran Biosciences, Inc. | Phenethylamine compounds salts, polymorphic forms and methods of use thereof |
| CN116726011A (en) * | 2022-03-04 | 2023-09-12 | 丽珠医药集团股份有限公司 | Application of ilaprazole in regulating digestive tract microecology |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0005129A1 (en) * | 1978-04-14 | 1979-10-31 | Aktiebolaget Hässle | Substituted pyridylsulfinylbenzimidazoles having gastric acid secretion properties, pharmaceutical preparations containing same, and intermediates for their preparation |
Family Cites Families (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL75400A (en) | 1984-06-16 | 1988-10-31 | Byk Gulden Lomberg Chem Fab | Dialkoxypyridine methyl(sulfinyl or sulfonyl)benzimidazoles,processes for the preparation thereof and pharmaceutical compositions containing the same |
| JPS6150978A (en) | 1984-08-16 | 1986-03-13 | Takeda Chem Ind Ltd | Pyridine derivative and preparation thereof |
| CA1327010C (en) | 1986-02-13 | 1994-02-15 | Tadashi Makino | Stabilized solid pharmaceutical composition containing antiulcer benzimidazole compound and its production |
| US5433959A (en) | 1986-02-13 | 1995-07-18 | Takeda Chemical Industries, Ltd. | Stabilized pharmaceutical composition |
| US6749864B2 (en) | 1986-02-13 | 2004-06-15 | Takeda Chemical Industries, Ltd. | Stabilized pharmaceutical composition |
| US5026560A (en) | 1987-01-29 | 1991-06-25 | Takeda Chemical Industries, Ltd. | Spherical granules having core and their production |
| DK0382489T3 (en) | 1989-02-10 | 1995-01-16 | Takeda Chemical Industries Ltd | Monoclonal Anti-Human Papillomavirus Antibody, Hybridoma Cell Producing This, and Method of Preparation thereof |
| YU48263B (en) | 1991-06-17 | 1997-09-30 | Byk Gulden Lomberg Chemische Fabrik Gmbh. | PROCEDURE FOR OBTAINING PANTOPRAZOLE PHARMACEUTICAL PRODUCT |
| US5464632C1 (en) | 1991-07-22 | 2001-02-20 | Prographarm Lab | Rapidly disintegratable multiparticular tablet |
| US5877192A (en) | 1993-05-28 | 1999-03-02 | Astra Aktiebolag | Method for the treatment of gastric acid-related diseases and production of medication using (-) enantiomer of omeprazole |
| SE9301830D0 (en) | 1993-05-28 | 1993-05-28 | Ab Astra | NEW COMPOUNDS |
| US6875872B1 (en) | 1993-05-28 | 2005-04-05 | Astrazeneca | Compounds |
| SE9302395D0 (en) | 1993-07-09 | 1993-07-09 | Ab Astra | NEW PHARMACEUTICAL FORMULATION |
| SE9302396D0 (en) | 1993-07-09 | 1993-07-09 | Ab Astra | A NOVEL COMPOUND FORM |
| US6334997B1 (en) | 1994-03-25 | 2002-01-01 | Isotechnika, Inc. | Method of using deuterated calcium channel blockers |
| US6221335B1 (en) * | 1994-03-25 | 2001-04-24 | Isotechnika, Inc. | Method of using deuterated calcium channel blockers |
| CN1087725C (en) * | 1994-03-25 | 2002-07-17 | 同位素技术有限公司 | Enhancement of the effect of drugs by deuteration |
| US6645988B2 (en) | 1996-01-04 | 2003-11-11 | Curators Of The University Of Missouri | Substituted benzimidazole dosage forms and method of using same |
| US6699885B2 (en) | 1996-01-04 | 2004-03-02 | The Curators Of The University Of Missouri | Substituted benzimidazole dosage forms and methods of using same |
| US5840737A (en) | 1996-01-04 | 1998-11-24 | The Curators Of The University Of Missouri | Omeprazole solution and method for using same |
| US6489346B1 (en) | 1996-01-04 | 2002-12-03 | The Curators Of The University Of Missouri | Substituted benzimidazole dosage forms and method of using same |
| SE510650C2 (en) | 1997-05-30 | 1999-06-14 | Astra Ab | New association |
| US6884429B2 (en) * | 1997-09-05 | 2005-04-26 | Isotechnika International Inc. | Medical devices incorporating deuterated rapamycin for controlled delivery thereof |
| DE69832984T2 (en) * | 1997-10-08 | 2006-09-21 | Isotechnika, Inc., Edmonton | DEUTERATED AND UNINTERRATED CYCLOSPORIN ANALOGUES AND THEIR USE AS IMMUNOMODULATING AGENTS |
| ES2559766T3 (en) | 1998-05-18 | 2016-02-15 | Takeda Pharmaceutical Company Limited | Disintegrable tablets in the mouth |
| DE69942777D1 (en) | 1998-07-28 | 2010-10-28 | Takeda Pharmaceutical | Easily disintegrating solid preparation |
| US6191148B1 (en) | 1998-08-11 | 2001-02-20 | Merck & Co., Inc. | Omerazole process and compositions thereof |
| US6166213A (en) | 1998-08-11 | 2000-12-26 | Merck & Co., Inc. | Omeprazole process and compositions thereof |
| SE9803772D0 (en) | 1998-11-05 | 1998-11-05 | Astra Ab | Pharmaceutical formulation |
| UA72748C2 (en) | 1998-11-10 | 2005-04-15 | Astrazeneca Ab | A novel crystalline form of omeprazole |
| US6440710B1 (en) * | 1998-12-10 | 2002-08-27 | The Scripps Research Institute | Antibody-catalyzed deuteration, tritiation, dedeuteration or detritiation of carbonyl compounds |
| NO309305B1 (en) * | 1999-02-19 | 2001-01-15 | Norsk Hydro As | Use of benzaldehyde derivatives in the manufacture of pharmaceutical preparations for the prevention and / or treatment of cancer, as well as certain new benzaldehyde derivatives |
| TWI275587B (en) | 1999-06-17 | 2007-03-11 | Takeda Chemical Industries Ltd | A crystal of (R)-2-[[[3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridyl]methyl]sulfinyl]-1H-benzimidazole |
| PT1104760E (en) * | 1999-12-03 | 2003-06-30 | Pfizer Prod Inc | SULFAMOYL-HETEROARILPIRAZOLE COMPOUNDS AS ANALGESIC AND ANTI-INFLAMMATORY AGENTS |
| EP1134290A3 (en) | 2000-03-14 | 2004-01-02 | Pfizer Products Inc. | Pharmacophore models for the identification of the CYP2D6 inhibitory potency of selective serotonin reuptake inhibitors |
| WO2002015908A1 (en) | 2000-08-18 | 2002-02-28 | Takeda Chemical Industries, Ltd. | Injections |
| MY137726A (en) | 2000-11-22 | 2009-03-31 | Nycomed Gmbh | Freeze-dried pantoprazole preparation and pantoprazole injection |
| EP1337525B8 (en) | 2000-12-01 | 2011-10-05 | Takeda Pharmaceutical Company Limited | Process for the crystallization of (r)- or (s)-lansoprazole |
| DE10123129A1 (en) * | 2001-05-02 | 2002-11-14 | Berolina Drug Dev Ab Svedala | Deuterated 3-piperidinopropiophenones and medicinal products containing these compounds |
| DE10162121A1 (en) * | 2001-12-12 | 2003-06-18 | Berolina Drug Dev Ab Svedala | Deuterated substituted pyrazolyl-benzenesulfonamides and drugs containing these compounds |
| TW200413273A (en) * | 2002-11-15 | 2004-08-01 | Wako Pure Chem Ind Ltd | Heavy hydrogenation method of heterocyclic rings |
| EA016814B1 (en) | 2005-07-26 | 2012-07-30 | Никомед Гмбх | Isotopically substituted proton pump inhibitors |
| AR054583A1 (en) * | 2005-07-26 | 2007-06-27 | Altana Pharma Ag | PANTOPRAZOL ISOTOPICALLY REPLACED |
| US7601737B2 (en) | 2005-07-26 | 2009-10-13 | Nycomed Gmbh | Isotopically substituted proton pump inhibitors |
| US20080033011A1 (en) | 2005-07-29 | 2008-02-07 | Concert Pharmaceuticals Inc. | Novel benzo[d][1,3]-dioxol derivatives |
| AU2006283876A1 (en) | 2005-08-22 | 2007-03-01 | Nycomed Gmbh | Isotopically substituted benzimidazole derivatives |
| EP1934215A1 (en) | 2005-09-22 | 2008-06-25 | Nycomed GmbH | Isotopically substituted imidazopyridine derivatives for the treatment of gastrointestinal disorders |
| US20100076087A1 (en) * | 2005-10-06 | 2010-03-25 | Auspex Pharmaceuticals, Inc. | Methods of reduction of interpatient variability |
| CA2624179A1 (en) | 2005-10-06 | 2007-04-12 | Auspex Pharmaceuticals, Inc. | Deuterated inhibitors of gastric h+, k+-atpase with enhanced therapeutic properties |
| US7750168B2 (en) * | 2006-02-10 | 2010-07-06 | Sigma-Aldrich Co. | Stabilized deuteroborane-tetrahydrofuran complex |
| CA2654450A1 (en) | 2006-06-05 | 2007-12-13 | Auspex Pharmaceuticals, Inc. | Preparation and utility of substituted erythromycin analogs |
| EP2081939B1 (en) | 2006-09-21 | 2011-06-15 | RaQualia Pharma Inc | Benzimidazole derivatives as selective acid pump inhibitors |
| WO2008114123A1 (en) | 2007-03-21 | 2008-09-25 | Raqualia Pharma Inc. | Spiro benzimidazole derivatives as acid pump inhibitors |
| WO2008127640A2 (en) | 2007-04-11 | 2008-10-23 | Auspex Pharmaceuticals, Inc. | Substituted benzimidazoles |
| WO2008130863A2 (en) | 2007-04-11 | 2008-10-30 | Auspex Pharmaceuticals, Inc. | Substituted benzimidazoles |
| KR20100105802A (en) | 2007-04-19 | 2010-09-29 | 콘서트 파마슈티컬즈, 인크. | Deuterated morpholinyl compounds |
| US20090076089A1 (en) * | 2007-09-17 | 2009-03-19 | Protia, Llc | Deuterium-enriched pantoprazole |
| US20090258897A1 (en) * | 2008-04-11 | 2009-10-15 | Auspex Pharmaceuticals, Inc. | Substituted benzimidazoles |
-
2006
- 2006-10-04 CA CA002624179A patent/CA2624179A1/en not_active Abandoned
- 2006-10-04 JP JP2008534658A patent/JP2009511481A/en active Pending
- 2006-10-04 BR BRPI0617987-8A patent/BRPI0617987A2/en not_active IP Right Cessation
- 2006-10-04 EP EP06816227A patent/EP1934201A1/en not_active Withdrawn
- 2006-10-04 US US11/544,407 patent/US7598273B2/en active Active
- 2006-10-04 WO PCT/US2006/038819 patent/WO2007041630A1/en active Application Filing
- 2006-10-04 AU AU2006299424A patent/AU2006299424A1/en not_active Abandoned
- 2006-10-04 CN CN2006800426099A patent/CN101309917B/en active Active
-
2008
- 2008-03-20 IL IL190331A patent/IL190331A0/en unknown
-
2009
- 2009-03-27 US US12/413,323 patent/US20090209592A1/en not_active Abandoned
- 2009-03-27 US US12/413,365 patent/US20090215831A1/en not_active Abandoned
- 2009-04-27 HK HK09103859.5A patent/HK1124336A1/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0005129A1 (en) * | 1978-04-14 | 1979-10-31 | Aktiebolaget Hässle | Substituted pyridylsulfinylbenzimidazoles having gastric acid secretion properties, pharmaceutical preparations containing same, and intermediates for their preparation |
Non-Patent Citations (1)
| Title |
|---|
| TONINI M ET AL: "Novel therapeutic strategies in acid-related disorders", EXPERT OPINION ON THERAPEUTIC PATENTS, ASHLEY PUBLICATIONS, GB, vol. 13, no. 5, 5 May 2003 (2003-05-05), pages 639 - 649, XP002269191, ISSN: 1354-3776 * |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7601737B2 (en) | 2005-07-26 | 2009-10-13 | Nycomed Gmbh | Isotopically substituted proton pump inhibitors |
| US7598273B2 (en) | 2005-10-06 | 2009-10-06 | Auspex Pharmaceuticals, Inc | Inhibitors of the gastric H+, K+-ATPase with enhanced therapeutic properties |
| WO2008127640A2 (en) * | 2007-04-11 | 2008-10-23 | Auspex Pharmaceuticals, Inc. | Substituted benzimidazoles |
| WO2008127640A3 (en) * | 2007-04-11 | 2008-12-04 | Auspex Pharmaceuticals Inc | Substituted benzimidazoles |
| CN101492452B (en) * | 2008-01-25 | 2010-11-17 | 山东轩竹医药科技有限公司 | Sulfhydryl benzimidazole derivative containing dioxepane-pyridine |
| CN101492464B (en) * | 2008-01-25 | 2011-01-12 | 山东轩竹医药科技有限公司 | Sulfhydryl imidazopyridine derivative containing dioxepane-pyridine |
| US9035050B2 (en) | 2008-02-29 | 2015-05-19 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
| US9493463B2 (en) | 2008-02-29 | 2016-11-15 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
| JP2011516426A (en) * | 2008-03-28 | 2011-05-26 | コンサート ファーマシューティカルズ インコーポレイテッド | Quinazoline derivatives and methods of treatment |
| US9061060B2 (en) | 2008-07-15 | 2015-06-23 | Theracos Inc. | Deuterated benzylbenzene derivatives and methods of use |
| JP2012501328A (en) * | 2008-09-02 | 2012-01-19 | アクチミス ファーマシューティカルズ インコーポレーテッド | Isotopically enriched pyrimidin-5-yl acetic acid derivatives as CRTH2 antagonists |
| JP2015007060A (en) * | 2008-09-03 | 2015-01-15 | テバ ファーマシューティカル インダストリーズ リミティド | 2-oxo-1,2-dihydro-quinoline modulators of immune function |
| JP2016183158A (en) * | 2008-09-03 | 2016-10-20 | テバ ファーマシューティカル インダストリーズ リミティド | 2-oxo-1,2-dihydro-quinoline modulators of immune function |
| JP2012502038A (en) * | 2008-09-03 | 2012-01-26 | テバ ファーマシューティカル インダストリーズ リミティド | 2-oxo-1,2-dihydro-quinoline modulator of immune function |
| US9918987B2 (en) | 2009-02-27 | 2018-03-20 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
| US10335413B2 (en) | 2009-02-27 | 2019-07-02 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
| US9492457B2 (en) | 2009-02-27 | 2016-11-15 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
| WO2010120797A3 (en) * | 2009-04-13 | 2011-03-31 | Auspex Pharmaceuticals, Inc. | Methods of reduction of interpatient variability |
| US9260432B2 (en) | 2009-09-02 | 2016-02-16 | Concert Pharmaceuticals, Inc. | Substituted derivatives of bicyclic [4.3.0] heteroaryl compounds |
| JP2013519708A (en) * | 2010-02-19 | 2013-05-30 | ノバルティス アーゲー | Deuterated pyrrolopyrimidine compounds as CDK4 / 6 inhibitors |
| US9085788B2 (en) | 2010-09-01 | 2015-07-21 | Concert Pharmaceuticals, Inc. | Process for preparing an enantiomerically enriched, deuterated secondary alcohol from a corresponding ketone without reducing deuterium incorporation |
| US9074233B2 (en) | 2010-09-01 | 2015-07-07 | Concert Pharmaceuticals, Inc. | Process for preparing an enantiomerically enriched, deuterated secondary alcohol from a corresponding ketone without reducing deuterium incorporation |
| US9328113B2 (en) | 2012-04-13 | 2016-05-03 | Concert Pharmaceuticals, Inc. | Substituted xanthine derivatives |
| US9682952B2 (en) | 2012-09-04 | 2017-06-20 | Celgene Corporation | Isotopologues of 3-(5-amino-2-methyl-4-oxoquinazolin-3(4H)-yl) piperidine-2-6-dione and methods of preparation thereof |
| WO2014039421A1 (en) * | 2012-09-04 | 2014-03-13 | Celgene Corporation | Isotopologues of 3-(5-amino-2-methyl-4-oxoquinazolin-3(4h)-yl) piperidine-2-6-dione and methods of preparation thereof |
| CN103204819B (en) * | 2013-04-15 | 2015-03-11 | 公安部物证鉴定中心 | Deuterated diazepam and preparation method thereof |
| CN103204819A (en) * | 2013-04-15 | 2013-07-17 | 公安部物证鉴定中心 | Deuterated diazepam and preparation method thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1934201A1 (en) | 2008-06-25 |
| AU2006299424A1 (en) | 2007-04-12 |
| IL190331A0 (en) | 2009-09-22 |
| CA2624179A1 (en) | 2007-04-12 |
| CN101309917A (en) | 2008-11-19 |
| US20090215831A1 (en) | 2009-08-27 |
| BRPI0617987A2 (en) | 2011-08-16 |
| JP2009511481A (en) | 2009-03-19 |
| US7598273B2 (en) | 2009-10-06 |
| US20090209592A1 (en) | 2009-08-20 |
| US20070082929A1 (en) | 2007-04-12 |
| HK1124336A1 (en) | 2009-07-10 |
| CN101309917B (en) | 2013-09-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7598273B2 (en) | Inhibitors of the gastric H+, K+-ATPase with enhanced therapeutic properties | |
| US9422225B2 (en) | Substituted phenethylamines with serotoninergic and/or norepinephrinergic activity | |
| JP2009517480A5 (en) | ||
| US20110130424A1 (en) | Substituted phenylpiperidines with serotoninergic activity and enhanced therapeutic properties | |
| US20110136914A1 (en) | Substituted aryloxypropylamines with serotoninergic and/or norepinephrinergic activity | |
| US20090312435A1 (en) | Substituted phenethylamines with serotoninergic and/or norepinephrinergic activity | |
| US20100137244A1 (en) | Preparation and utility of hmg-coa reductase inhibitors | |
| MX2008004622A (en) | Deuterated inhibitors of gastric h+, k+-atpase with enhanced therapeutic properties |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680042609.9 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006299424 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 566700 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1145/KOLNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006816227 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 190331 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2624179 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/004622 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2008534658 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2006299424 Country of ref document: AU Date of ref document: 20061004 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0617987 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080404 |