WO2006078752A2 - Inhibitors of dna methyltransferase - Google Patents
Inhibitors of dna methyltransferase Download PDFInfo
- Publication number
- WO2006078752A2 WO2006078752A2 PCT/US2006/001791 US2006001791W WO2006078752A2 WO 2006078752 A2 WO2006078752 A2 WO 2006078752A2 US 2006001791 W US2006001791 W US 2006001791W WO 2006078752 A2 WO2006078752 A2 WO 2006078752A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- aryl
- optionally substituted
- heteroaryl
- mmol
- Prior art date
Links
- 0 CC=C1N(C*(*)c2*c(*)**(*(C3OC(C*)C(*)C3O)C=C)c2C=C)C1 Chemical compound CC=C1N(C*(*)c2*c(*)**(*(C3OC(C*)C(*)C3O)C=C)c2C=C)C1 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
Definitions
- This invention relates to inhibition of the DNA methyltransferase isoforms DNMT1 and DNMT3b2. More particularly, the invention relates to compounds and methods for the inhibition of DNMT1 and DNMT3b2.
- DNA methyltransferase 1 (DNMT1) protein is a major contributor to DNA methyltransferase activity in human cells and is required to maintain methylation patterns in differentiated cells (M. F. Robert, S. Morin, N. Beaulieu, F. Gauthier, I.C. Chute, A. Barsalou, A.R. MacLeod, Nat. Genet. 33 (2003) 61-65).
- the de novo DNA methyltransferases DNMT3A and DNMT3B establish DNA methylation during early embryogenesis (M. Okano, S. Xie, E. Li, Nat. Genet, 1998, 19, 219-220; M. Okano, D.W. Bell, DA Haber, E.
- SAH can bind to DNA methyltransferases and inhibit their catalytic reaction and is an important molecule in the regulation of biological transmethylation (T. Deguchi and J. Barchas, J. Biol. Chem., 1971 , 246, 3175; A. Oliva, P. Galletti, and V. Zappia, Eurp. J. Biochem., 1980, 104, 595; P.M. Ueland and J. Saebo, Biochemistry, 1979, 18, 4130). [0005] Inhibitors of DNMT1 and DNMT3B are expected to have value as anti-cancer agents (M. Szyf, Fontiers in Bioscience 2001 , 6, 599-609; M.F. Robert, S. Morin, N.
- SAH is a known nonselective inhibitor of human DNA methyltransferase and many other methyltransferases.
- a number of simplified and less rigid analogues of SAH have been reported. Such compounds, however, were devoid of activity against human DNA methyltransferase (M. Botta, R. Saladino, G. Pedraly-Noy, and R. Nicoletti, Med. Chem. Res., 1994, 4, 323).
- SAM and SAH as inhibitors are not good drugs and they are unstable in plasma due to hydrolases and ribonucleases, they have poor absorption due to the Zwitterionic nature, are rapidly excreted, and have a short half life.
- SAH is an endogenous inhibitor of numerous methyltransferases and as such is non-selective, making it undesirable as a drug.
- Some more stable nitrogen anlogues of SAM and SAH have been reported. Chi-Deu Chang and J. K. Coward, J. Med.
- Sinefungin a natural product, is a nitrogen analogue of SAH and has been reported to inhibit human DNA methyltransferase. It is also a non-selective inhibitor with potential for toxicity (C. Barbes, J. Sanchez, M.J. Yebra, M.Robert-Gero, and C. Hardisson, FEMS Microbiology Letters, 1990, 69, 239).
- DNA methyltransferase inhibitors are known to be DNA binding agents, however, and thus may inhibit the DNMTs indirectly.
- This invention provides compounds and methods for the inhibition of human C-5
- the compounds of the invention are inhibitors of the DNA methyltransferase isoforms DNMT1 and DNMT3b2. Accordingly, the invention provides new inhibitors of DNMT1 and DNMT3b2. [0016] In a first aspect, the invention provides compounds of formula (I) (and their pharmaceutically acceptable salts) that are useful as inhibitors of DNMT1 and/or DNMT3b2 and therefore are useful for studying the role of DNA methyltransferase in biological processes.
- compositions comprising a compound that is an inhibitor of DNMT1 and/or DNMT3b2, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, excipient, or diluent.
- the invention provides a method of inhibiting DNMT1 and/or DNMT3b2 enzymes in a cell, comprising contacting a cell in which inhibition of DNMT1 or DNMT3b2 is desired with a compound of the invention.
- Figure 1 displays the full length DNMT1 (Swissprot accession number P26358 (SEQ ID NO.1)).
- Figure 2 displays the DNMT3 splice variant 2 of DNMT3b2 (Swissprot accession number Q9UBC3-2 (SEQ ID NO.2))
- the invention provides compounds and methods for inhibiting DNMT1 and DNMT3b2.
- inhibitor of DNMT1 and DNMT3b2 is used to identify a compound having a structure as defined herein, which is capable of interacting with DNMT1 , DNMT3b2 or both DNMT1 and DNMT3b2 and inhibiting the activity of DNMT1 , DNMT3b2, or both DNMT1 and DNMT3b2. In some preferred embodiments, such reduction of activity is at least about 50%, more preferably at least about 75%, and still more preferably at least about 90%.
- chemical moieties are defined and referred to throughout primarily as univalent chemical moieties (e.g., alkyl, aryl, etc.).
- alkyl generally refers to a monovalent radical (e.g. CH 3 -CH 2 -)
- a bivalent linking moiety can be "alkyl,” in which case those skilled in the art will understand the alkyl to be a divalent radical (e.g., -CH 2 - CH 2 -), which is equivalent to the term "alkylene.”
- aryl refers to the corresponding divalent moiety, arylene.
- All atoms are understood to have their normal number of valences for bond formation ⁇ i.e., 4 for carbon, 3 for N, 2 for O, and 2, 4, or 6 for S, depending on the oxidation state of the S).
- a moiety may be defined, for example, as (A) 3 -B-, wherein a is 0 or 1. In such instances, when a is 0 the moiety is B- and when a is 1 the moiety is A-B-. Also, a number of moieties disclosed herein exist in multiple tautomeric forms, all of which are intended to be encompassed by any given tautomeric structure.
- hydrocarbyl refers to a straight, branched, or cyclic alkyl, alkenyl, or alkynyl, each as defined herein.
- a “C 0 " hydrocarbyl is used to refer to a covalent bond.
- C 0 -C 3 -hydrocarbyF' includes a covalent bond, methyl, ethyl, ethenyl, ethynyl, propyl, propenyl, propynyl, and cyclopropyl.
- alkyl refers to straight and branched chain aliphatic groups having from 1 to 12 carbon atoms, preferably 1-8 carbon atoms, and more preferably 1-6 carbon atoms, which is optionally substituted with one, two or three substituents.
- Preferred alkyl groups include, without limitation, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert- butyl, pentyl, and hexyl.
- a "C 0 " alkyl (as in "C 0 -C 3- alkyr') is a covalent bond (like "C 0 " hydrocarbyl).
- alkenyl as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon double bonds, having from 2 to 12 carbon atoms, preferably 2-8 carbon atoms, and more preferably 2-6 carbon atoms, which is optionally substituted with one, two or three substituents.
- Preferred alkenyl groups include, without limitation, ethenyl, propenyl, butenyl, pentenyl, and hexenyl.
- alkynyl as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon triple bonds, having from 2 to 12 carbon atoms, preferably 2-8 carbon atoms, and more preferably 2-6 carbon atoms, which is optionally substituted with one, two or three substituents.
- Preferred alkynyl groups include, without limitation, ethynyl, propynyl, butynyl, pentynyl, and hexynyl.
- alkylene is an alkyl, alkenyl, or alkynyl group, as defined hereinabove, that is positioned between and serves to connect two other chemical groups.
- Preferred alkylene groups include, without limitation, methylene, ethylene, propylene, and butylene.
- Preferred alkenylene groups include, without limitation, ethenylene, propenylene, and butenylene.
- Preferred alkynylene groups include, without limitation, ethynylene, propynylene, and butynylene.
- cycloalkyl as employed herein includes saturated and partially unsaturated cyclic hydrocarbon groups having 3 to 12 carbons, preferably 3 to 8 carbons, and more preferably 3 to 6 carbons, wherein the cycloalkyl group additionally is optionally substituted.
- Preferred cycloalkyl groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, and cyclooctyl.
- heteroalkyl refers to an alkyl group, as defined hereinabove, wherein one or more carbon atoms in the chain are replaced by a heteroatom selected from the group consisting of O, S, NH, N-alkyl, SO, SO 2 , SO 2 NH, or NHSO 2 .
- An "aryl” group is a C 6 -Ci 4 aromatic moiety comprising one to three aromatic rings, which is optionally substituted.
- the aryl group is a C 6 -Ci 0 aryl group.
- Preferred aryl groups include, without limitation, phenyl, naphthyl, anthracenyl, and fluorenyl.
- An “aralkyl” or “arylalkyl” group comprises an aryl group covalently linked to an alkyl group, either of which may independently be optionally substituted or unsubstituted.
- the aralkyl group is (C 1 - C ⁇ aIk(C 6 -C 10 )aryl, including, without limitation, benzyl, phenethyl, and naphthylmethyl.
- a "heterocyclyl” or “heterocyclic” group is a ring structure having from about 3 to about 12 atoms, wherein one or more atoms are selected from the group consisting of N, O, S, SO, and SO 2 .
- the heterocyclic group is optionally substituted on carbon at one or more positions.
- heterocyclic group is also independently optionally substituted on nitrogen with alkyl, aryl, aralkyl, alkylcarbonyl, alkylsulfonyl, arylcarbonyl, arylsulfonyl, alkoxycarbonyl, or aralkoxycarbonyl.
- Preferred heterocyclic groups include, without limitation, epoxy, aziridinyl, tetrahydrofuranyl, pyrrolidinyl, piperidinyl, piperazinyl, thiazolidinyl, oxazolidinyl, oxazolidinonyl, and morpholino.
- the heterocyclic group is fused to an aryl, heteroaryl, or cycloalkyl group.
- fused heterocyles include, without limitation, tetrahydroquinoline and dihydrobenzofuran.
- tetrahydroquinoline and dihydrobenzofuran.
- compounds where an annular O or S atom is adjacent to another O or S atom are also included.
- heteroaryl refers to groups having 5 to 14 ring atoms, preferably 5, 6, 9, or 10 ring atoms; having 6, 10, or 14 ⁇ -electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to three heteroatoms per ring selected from the group consisting of N, O, and S.
- heteroaryl is also meant to encompass monocyclic and bicyclic groups.
- a heteroaryl group may be pyrimidinyl, pyridinyl, benzimidazolyl, thienyl, benzothiazolyl, benzofuranyl and indolinyl.
- Preferred heteroalkyl groups comprise a C 1 -C 6 alkyl group and a heteroaryl group having 5, 6, 9, or 10 ring atoms.
- Specifically excluded from the scope of this term are compounds having adjacent annular O and/or S atoms.
- heteroaralkyl groups examples include pyridylmethyl, pyridylethyl, pyrrolylmethyl, pyrrolylethyl, imidazolylmethyl, imida ⁇ olylethyl, thiazolylmethyl, and thiazolylethyl.
- pyridylmethyl pyridylethyl
- pyrrolylmethyl pyrrolylethyl
- imidazolylmethyl imida ⁇ olylethyl
- thiazolylmethyl examples of preferred heteroaralkyl groups.
- C n -C n heterocyclyl or “C n -C m “ heteroaryl means a heterocyclyl or heteroaryl having from “n” to "m” annular atoms, where "n” and “m” are integers.
- a C 5 -C 6 -heterocyclyl is a 5- or 6- membered ring having at least one heteroatom, and includes pyrrolidinyl (C 5 ) and piperidinyl (C 6 );
- C 6 -hetoaryl includes, for example, pyridyl and pyrimidyl.
- arylene is an aryl, heteroaryl, or heterocyclyl group, as defined hereinabove, that is positioned between and serves to connect two other chemical groups.
- Preferred heterocyclyls and heteroaryls include, but are not limited to, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, pyridotriazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H.6H-1 ,5,2- dithiazinyl, dihydrofuro[2,3-b]tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, 1 H-indazolyl, indolenyl, ind
- a moiety e.g., cycloalkyl, hydrocarbyl, aryl, heteroaryl, heterocyclic, urea, etc.
- a moiety e.g., cycloalkyl, hydrocarbyl, aryl, heteroaryl, heterocyclic, urea, etc.
- the group optionally has from one to four, preferably from one to three, more preferably one or two, non- hydrogen substituents.
- Suitable substituents include, without limitation, halo, hydroxy, oxo (e.g., an annular -CH- substituted with oxo is -C(O)-) nitro, halohydrocarbyl, hydrocarbyl, aryl, aralkyl, alkoxy, aryloxy, amino, acylamino, alkylcarbamoyl, arylcarbamoyl, aminoalkyl, acyl, carboxy, hydroxyalkyl, alkanesulfonyl, arenesulfonyl, alkanesulfonamido, arenesulfonamido, aralkylsulfonamido, alkylcarbonyl, acyloxy, cyano, and ureido groups.
- Preferred substituents, which are themselves not further substituted are:
- R 30 and R 31 are each independently hydrogen, cyano, oxo, carboxamido, amidino, Ci-C 8 hydroxyalkyl, C 1 -C 3 alkylaryl, aryl-C r C 3 alkyl, C 1 -C 8 alkyl, C r C 8 alkenyl, C 1 -C 8 alkoxy, Ci-C 8 alkoxycarbonyl, aryloxycarbonyl, 3IyI-C 1 -C 3 alkoxycarbonyl, C 2 -C 8 acyl, Ci-C 8 alkylsulfonyl, arylalkylsulfonyl, arylsulfonyl, aroyl, aryl, cycloalkyl, heterocyclyl, or heteroaryl, wherein
- halohydrocarbyl is a hydrocarbyl moiety in which from one to all hydrogens have been replaced with one or more halo.
- halogen refers to chlorine, bromine, fluorine, or iodine.
- acyl refers to an alkylcarbonyl or arylcarbonyl substituent.
- acylamino refers to an amide group attached at the nitrogen atom (Ae., R-CO-NH-).
- carbamoyl refers to an amide group attached at the carbonyl carbon atom (i.e., NH 2 -CO-). The nitrogen atom of an acylamino or carbamoyl substituent is additionally substituted.
- sulfonamido refers to a sulfonamide substituent attached by either the sulfur or the nitrogen atom.
- amino is meant to include NH 2 , alkylamino, arylamino, and cyclic amino groups.
- ureido refers to a substituted or unsubstituted urea moiety.
- radical means a chemical moiety comprising one or more unpaired electrons.
- a moiety that is substituted is one in which one or more hydrogens have been independently replaced with another chemical substituent.
- substituted phenyls include 2-flurophenyl, 3,4-dichlorophenyl, 3-chloro-4-fluoro-phenyl, 2-fluoro- 3-propylphenyl.
- substituted n-octyls include 2,4-dimethyI-5- ethyl-octyl and 3-cyclopentyl-octyl. Included within this definition are methylenes (-CH 2 -) substituted with oxygen to form carbonyl -CO-).
- an "unsubstituted" moiety as defined above e.g., unsubstituted cycloalkyl, unsubstituted heteroaryl, etc. means that moiety as defined above that does not have any of the optional substituents for which the definition of the moiety (above) otherwise provides.
- an "aryl” includes phenyl and phenyl substituted with a halo
- "unsubstituted aryl” does not include phenyl substituted with a halo.
- the invention comprises compounds of formula (I), that are inhibitors DNMT1 and DNMT3b2:
- R 3 and R 4 independently represent H, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -C r C 6 -alkyl-cycloalkyl, -Ci-C 6 -alkyl- heterocyclyl, -C r C 6 -alkyl-aryl, -CrC 6 -alkyl-heteroaryl, -d-Cealkoxy-aryl or - (CH 2 ) 1-6 -T, wherein C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, cycloalkyl, heterocyclyl, aryl and heteroaryl, at each occurrence, are optionally substituted; or R 3 and R 4 taken together with the nitrogen to which they are attached form a C 5 -C 9 heterocyclyl
- R 2 is H, halo, CF 3 , C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 alkoxy, -NH-C 1 -C 6 alkyl, or -S- C 1 -C 6 alkyl, wherein C 1 -C 6 alkyl, C 2 -C 6 alkenyl, and C 2 -C 6 alkynyl are each optionally substituted;
- a and B independently are F, Cl, -OH, H 1 -NHR, or -OR;
- R at each occurrence is independently benzyl or C 1 -C 4 alkyl, wherein benzyl and C 1 -C 4 alkyl are optionally substituted;
- W is CH, N, CR, or C-halogen;
- X is CH, N, C-C r C 6 -alkyl, or C-halogen;
- D is CH, or N;
- Z is -L-C(H)(NH 2 )-COOR 7 , -L-NR 19 R 20 , or heterocyclyl, wherein heterocyclyl is optionally substituted;
- L is a bond or is -(CR 17 Ri 8 )I -6 -;
- each R 17 and R 18 independently is H or C r C 6 -alkyl, wherein Ci-C 6 -alkyl is optionally substituted;
- C-rC 6 -alkyl and heteroaryl are optionally substituted; and R 7 is H or Ci-C 6 -alkyl.
- R 2 is H, halo, C 1 -C 6 alkyl, C 2 -C 6 alkenyl or -S- Ci-C 6 alkyl, wherein C 1 -C 6 alkyl and C 2 -C 6 alkenyl, at each occurrence, are optionally substituted;
- R 3 and R 4 independently represent H, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -C r C 6 -alkyl-cycloalkyl, -C-i-Ce-alkyl-heterocyclyl, -C r C 6 -a!ky!-aryl, -C 1 -C 6 - alkyl-heteroaryl, -C r C 6 alkoxy-aryl or -(CH 2 )i
- Ri 4 is C 1 -C 6 alkyl, aryl or heteroaryl and R 15 is aryl, wherein C 1 -C 6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted;
- W and X are independently CH or N;
- R 16 is H, C 1 -C 6 alkyl, -C-pCe-alkyl-aryl, -CrCe-alkyl-heteroaryl, or -C 2 -C 6 alkenyl-aryl, wherein C 1 -C 6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted;
- Z is -L-C(H)(NH 2 )-COOR 7 , -L-NR 19 R 20 , or heterocyclyl, wherein heterocyclyl is optionally substituted;
- L is a bond or is -(CR 17 Ris)i-6-;
- R 7 is H or CrCe-alkyl.
- H-A and pharmaceutically acceptable salts and complexes thereof, wherein A is H, halogen, or OH;
- R 2 is H, halo, Ci-C 6 alkyl, C 2 -C 6 alkenyl or -S- C 1 -C 6 alkyl, wherein Ci-C 6 alkyl and C 2 -C 6 alkenyl, at each occurrence, are optionally substituted;
- R 3 and R 4 independently represent H, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -C-i-Ce-alkyl-cycloalkyl, -Ci-Ce-alkyl-heterocyclyl, -Ci-C 6 -alkyl-aryl, -C 1 -C 6 - alkyl-heteroaryl, -Ci-C 6 alkoxy-aryl or -(CH 2 )i
- R 14 is C 1 -C 6 alkyl, aryl or heteroaryl and R 15 is aryl, wherein C 1 -C 6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted
- R 7 is H or Ci-C 6 -alky ⁇
- Y is S, O, or -N(R 16 )-
- R 16 is H, C 1 -C 6 alkyl, -C r C 6 -alkyl-aryl, -CrCe-alkyl-heteroaryl, Or -C 2 -C 6 alkenyl-aryl, wherein C 1 -C 6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted
- R 7 is H or Ci-C 6 -alky
- L 1 is -(CR 17 Ri 8 )i-6-;
- R 17 and R 18 independently are H or C r C 6 -alkyl, wherein C r C 6 -alkyl is optionally substituted.
- R 7 is H.
- L 1 is -CH 2 CH 2 -.
- R 2 is H, halogen, C 1 -C 3 alkyl, -S-C 1 -C 2 alkyl, or C 2 -C 3 alkenyl.
- R 2 is H or halogen.
- Y is S.
- R 3 and R 4 are independently H, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -CrCe-alkyl-cycloalkyl, -CrC 6 -alkyl-aryl, -CpCe-alkyl-heteroaryl, -CrCealkoxy-aryl or
- R 4 are both H.
- R 3 is H and R 4 is C 1 -C 6 alkyl optionally substituted with 1, 2, or 3 groups independently selected from
- both R 3 and R 4 are C 1 -C 6 alkyl, wherein Ci-C 6 alkyl is independently optionally substituted with 1 , 2, or 3 groups independently selected from OH, CO 2 H, NH 2 , N(C 1 -C 3 alkyl) 2 , C 1 -C 3 alkoxy, and phenyl.
- R 3 is -
- C- ⁇ -C 3 -all ⁇ yl-aryl and R 4 is C 1 -C 6 alkyl, wherein C 1 -C 6 alkyl is optionally substituted with 1 , 2, or 3 groups independently selected from OH, CO 2 H 1 NH 2 , N(C 1 -C 3 alkyl) 2 , C 1 -C 3 alkoxy, and phenyl.
- R 3 is H and R 4 is C 3 -C 8 -cycloalkyl.
- cycloalkyl is cyclopropyl, cyclohexyl, or cyclooctanyl.
- R 3 is H and R 4 is aryl, wherein aryl is optionally substituted.
- aryl is phenyl, naphthyl, or fluorenyl.
- aryl is unsubstituted or is substituted with 1 , 2, or 3 groups independently selected from OCH 3 , NH 2 , NHCH 3 , NO 2 , C 1 -C 3 alkyl, halogen, CF 3 , CN, OH, NH 2 SO 2 -, and phenyl.
- R 3 is H and R 4 is heterocyclyl.
- heterocyclyl is pyrrolidinonyl.
- R 3 is H and R 4 is -C r C 6 -alkyl-heteroaryl, wherein heteroaryl and C 1 -C 6 alkyl are optionally substituted.
- heteroaryl is imidazolyl, indolyl, thiophenyl, pyridinyl, or dihydroindenyl.
- heteroaryl is unsubstituted or is substituted with 1, 2, or 3 groups independently selected from methoxy and phenyl-CrC 3 -alkoxy-.
- R 3 is H and R 4 is -C r C 6 -alkyl-aryl, wherein aryl and C 1 -C 6 alkyl are optionally substituted.
- aryl is phenyl, naphthyl, or fluorenyl.
- aryl is unsubstituted or is substituted with 1 , 2, or 3 groups independently selected from OCH 3 , NH 2 , NHCH 3 , NO 2 , C 1 -C 3 alkyl, halogen, CF 3 ,
- aryl is phenyl, optionally substituted with 1 , 2, or 3 groups independently selected from OCH 3 , NH 2 ,
- T is selected from -NH-CO-phenyl, NH-SO 2 -naphthyl, -S-CH 2 -phenyl, -NH-CO-methyl, and -NH-CO- furanyl.
- R 3 and R 4 are selected from -NH-CO-phenyl, NH-SO 2 -naphthyl, -S-CH 2 -phenyl, -NH-CO-methyl, and -NH-CO- furanyl.
- R 4 taken together with the nitrogen to which they are attached form a C 5 -C 9 heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted.
- R 4 taken together with the nitrogen to which they are attached form a C 4 -C 8 heterocyclyl ring, wherein said ring is optionally substituted.
- R 3 and R 4 taken together with the nitrogen to which they are attached form a pyrrolidinyl, azetidinyl, or piperidinyl ring.
- Y is -N(R 16 )-.
- R 16 is
- R 16 is
- CrC 6 -alkyl is unsubstituted or is substituted with NO 2 .
- R 16 is aryl, wherein aryl is optionally substituted.
- R 16 is -
- Ri 6 is -
- aryl is phenyl, wherein phenyl is unsubstituted or is substituted with 1 , 2, or 3 groups independently selected from NO 2 , Ci-C 3 -alkoxy, CN, or CF 3 .
- R 16 is -
- heteroaryl is pyridinyl.
- R 3 and R 4 are both H.
- R 3 is H, and R 4 is CrC 6 -alkyl.
- L 2 is a bond or is -CH 2 -;
- R 2 is H or halogen;
- R 3 is H, C 1 -C 6 alkyl, or -Ci-C 6 -alkyl-aryl, wherein C 1 -C 6 alkyl and aryl, at each occurrence, are optionally substituted;
- R 4 is H or Ci-C 6 alkyl, wherein C 1 -C 6 alkyl is optionally substituted;
- R 8 is H, -CO 2 H, or CO 2 CH 3 ;
- R 9 is absent, H or C 1 -C 6 alkyl, wherein C 1 -C 6 alkyl is optionally substituted; W is N or CH; Y is S or O; and
- Q is N, CH or O, provided that when Q is O, R 9 is absent.
- R 3 and R 4 are both H.
- R 3 is - C-i-Ce-alkyl-aryl and R 4 is H, wherein aryl is optionally substituted.
- aryl is phenyl.
- phenyl is unsubstituted or is substituted with phenyl.
- R 2 is H, or halogen
- R 3 and R 4 independently represent H, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, cycloalkyl, aryl or (-Ci-C 6 -alkyl)-aryl, wherein C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, cycloalkyl, aryl and (-Ci-C 6 -alkyl)-aryl are each optionally substituted; or R 3 and R 4 taken together with the nitrogen to which they are attached form a C 5 -C 9 - heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted;
- L 3 is a bond or is -(CRi 7 Ri8)i-e-;
- Ri 7 and R 18 at each occurrence are independently H or Ci-C 6 -alkyl, wherein C 1 -
- C 6 -alkyl is optionally substituted;
- L 3 is - CHRi 7 CHR 18 -, wherein R 17 and Ri 8 independently are H, or CrC 6 alkyl, and where C 1 -C 6 alkyl at each occurrence is optionally substituted.
- C 1 -C 6 alkyl is unsubstituted or is substituted with NH 2 .
- L 3 is - CH 2 CH 2 CH 2 -.
- R 19 is C 1 -C 6 alkyl, wherein C 1 -C 6 alkyl is optionally substituted.
- C 1 -C 6 alkyl is unsubstituted or is substituted with NH 2 .
- R 19 is heteroaryl.
- heteroaryl is pyrimidin-2(1 H)-one.
- pyrimidin-2(1H)-one is unsubstituted or is substituted with amine.
- the invention provides a composition comprising a compound according to any one of paragraphs [0047]-[0096] or as depicted in any of the examples and tables herein together with a pharmaceutically acceptable excipient, diluent, or carrier.
- a pharmaceutically acceptable excipient diluent, or carrier.
- Compounds of the invention may be formulated by any method well known in the art.
- pharmaceutically acceptable means a non-toxic material that is compatible with a biological system in a cell, cell culture, or tissue sample and that does not interfere with the effectiveness of the biological activity of the active ingredient(s).
- compositions according to the invention may contain, in addition to the inhibitor, diluents, fillers, salts, buffers, stabilizers, solubilizers, and other materials well known in the art.
- diluents fillers, salts, buffers, stabilizers, solubilizers, and other materials well known in the art.
- the preparation of pharmaceutically acceptable formulations is described in, e.g., Remington's Pharmaceutical Sciences, 18th Edition, ed. A. Gennaro, Mack Publishing Co., Easton, PA, 1990.
- pharmaceutically acceptable salts refers to salts that retain the desired biological activity of the above-identified compounds and exhibit minimal or no undesired toxicological effects.
- salts include, but are not limited to acid addition salts formed with inorganic acids (for example, hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric add, and the like), and salts formed with organic acids such as acetic acid, oxalic acid, tartaric acid, succinic acid, malic acid, ascorbic acid, benzoic acid, tannic acid, pamoic acid, alginic acid, polyglutamic acid, naphthalenesulfonic acid, naphthalenedisulfonic acid, and polygalacturonic acid.
- inorganic acids for example, hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric add, and the like
- organic acids such as acetic acid, oxalic acid, tartaric acid, succinic acid, malic acid, ascorbic acid, benzoic acid, tannic acid, pamoic acid, alginic acid, polyglutamic acid
- the compounds can also be administered as pharmaceutically acceptable quaternary salts known by those skilled in the art, which specifically include the quaternary ammonium salt of the formula -NR + Z-, wherein R is hydrogen, alkyl, or benzyl, and Z is a counterion, including chloride, bromide, iodide, -O-alkyl, toluenesulfonate, methylsulfonate, sulfonate, phosphate, or carboxylate (such as benzoate, succinate, acetate, glycolate, maleate, malate, citrate, tartrate, ascorbate, benzoate, cinnamoate, mandeloate, benzyloate, and diphenylacetate).
- R is hydrogen, alkyl, or benzyl
- Z is a counterion, including chloride, bromide, iodide, -O-alkyl, toluenesulfonate, methylsulf
- salt is also meant to encompass complexes, such as with an alkaline metal or an alkaline earth metal.
- the active compound is included in the pharmaceutically acceptable carrier or diluent in an amount sufficient to deliver to an enzyme-inhibiting effective amount without causing serious toxic effects to the cell.
- the third aspect of the invention provides a method of inhibiting DNMT1 and/or DNMT3b2 enzymes, the method comprising contacting the enzyme(s) with a compound according to any one of paragraphs [0047]-[0097], or as depicted in any of the tables herein, or with a composition according to paragraph [0098]-[0102].
- l ⁇ h ⁇ bitiori of DNMT1 ahd/or DNMT3b2 " enzymes can be in a cell or a multicellular organism.
- the method according to this aspect of the invention comprises administering to the organism an effective DNMT1- and/or DNMT3b2-inhibiting amount of a compound according to any one of paragraphs [0047]-[0097] or as depicted in any of the tables herein, or a composition according to paragraph [0098]-[0102].
- the organism is a mammal, more preferably a primate, most preferably a human.
- Preferred compounds according to the invention include those described in the examples below. Compounds were named using Chemdraw Ultra version 6.0.2 or version 8.0.3, which are available through Cambridgesoft.com, 100 Cambridge Park Drive, Cambridge, MA 02140, Namepro version 5.09, which is available from ACD labs, 90 Adelaide Street West, Toronto, Ontario, M5H, 3V9, Canada, or were derived therefrom.
- the compounds of the invention can be prepared according to the reaction schemes or the examples illustrated below utilizing methods known to one of ordinary skill in the art. These schemes serve to exemplify some procedures that can be used to make the compounds of the invention. One skilled in the art will recognize that other general synthetic procedures may be used.
- the compounds of the invention can be prepared from starting components that are commercially available. Any kind of substitutions can be made to the starting components to obtain the compounds of the invention according to procedures that are well known to those skilled in the art.
- Step 3 methyl ⁇ -ftert-butoxycarbonyla ' minoy ⁇ -C ⁇ SaS ⁇ S.eR ⁇ aR ⁇ .Z-dimethyl-e-r ⁇ - (phenethylamino)-9H-purin-9-yl)-tetrahvdrofurof3,4-di ⁇ ,31dioxol-4-v ⁇ methylthio')butanoate 5a
- NaH (30 mg 60% mineral oil suspension, 0.72 mmol) was added to a solution of ((3aR,4R,6R,6aR)-2,2-dimethyl-6-(6-(phenethylamino)-9H-purin-9-yl)-tetrahydrofuro[3,4- d][1,3]dioxol-4-yl)methanol 3a (149 mg, 0.36 mmol) in THF (4 ml_) at O 0 C and stirred for 15 min.
- pTsCl (76 mg, 0.4 mmol) was then added and the reaction mixture was allowed to stir for 1 hour at O 0 C. It was diluted in EtOAc (15 ml_) and washed sequentially with water (10 mL) and NaCI sat solution (10 mL). The organic layer was dried with Na 2 SO 4 , filtered and concentrated in vacuo. The crude product was dissolved in dry MeOH and treated with a solution prepared by mixing tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate 4 (114 mg, 0.52 mmol) and NaOMe (1.05 mL 0,5M solution, 0.52 mmol) for 15 min at room temperature.
- Step 4 2-amino-4-f((2S,3S,4R.5R)-3,4-dihvdroxy-5-(6-(phenethylaminoV9H-purin-9-yl)- tetrahvdrofuran-2-yl)methylthio)butanoic acid 6a.
- Examples 2-9, compounds 5b-5i, Table 2, were prepared in a manner similar to example 1 , scheme 1a, utilizing the appropriate amine.
- Step 1 methyl 4-(((3aS,4S,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2.2-dimethyl-tetrahvdrofurol ' 3.4- d1f1 ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 8
- NaH 310 mg 60% mineral oil suspension, 7.74 mmol
- ((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyl-tetrahydrofuro[3,4-d][1 ,3]dioxol-4- yOmethanol 7 (1.19 g, 3.87 mmol) in THF (4 ml_) at O 0 C and stirred for 15 min.
- pTsCI (810 mg, 4.25 mmol) was then added and the reaction mixture was allowed to stir for 1 hour at O 0 C. It was diluted in EtOAc (15 mL) and washed sequentially with water (10 ml_) and NaCI sat solution (10 mL).
- Step 2 methyl 4-(((3aS,4S.6R.6aR)-6-(6-(4H-t2,4-triazol-4-yl)-9H-purin-9-yl)-2,2-dimethyl- tetrahydrofuro[3,4-di ⁇ ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 1
- the dihydrochloride salt 9 (398 mg, 1.85, 5 equiv.) was added to a solution of 8 (134 mg, 0.37 mmol) in dry pyridine (4 mL) and the reaction mixture was refluxed for 16h.
- Step 3 methyl 2-(tert-butoxycarbonylamino)-4-(((3aS,4S.6R,6aRV2,2-dimethyl-6-(6-(3- morpholinopropylamino)-9H-purin-9-yl)-tetrahvdrofuror3,4-d1[1 ,31dioxol-4- yl)methylthio)butanoate 11a
- Step 4 2-amino-4-(((2S.3S.4R,5R)-3,4-dihvdroxy-5-(6-(3-morpholinopropylamino)-9H-purin-9- yl)-tetrahvdrofuran-2-yl)methylthio)butanoic acid 12a.
- Step 1 methyl 2-(tert-butoxycarbonylamino)-4-(((3aS,4S.6R,6aR)-6-methoxy-2.2-dimethyl- tetrahvdrofurof3,4-di ⁇ ,31dioxol-4-yl)methylthio)butanoate 14
- Step 2 methyl 2-(((9H-fluoren-9-vnmethoxy ⁇ carbonylaminoV4-(((3aS,4S,6R,6aRV6- methoxy-2,2-dimethyl-tetrahvdrofurof3,4-cn ⁇ ,31dioxol-4-yl)methylthio)butanoate 15
- a solution of 14 (11.0 g, 25.3 mmol) in dry 1 ,2-dichloroethane (50 ml) was placed under a nitrogen atmosphere and cooled to O 0 C.
- TMSOTf (4.6 ml, 25.4 mmol) was then added and the reaction was stirred for 1.5 hours at O 0 C.
- Acetyl chloride (8.9 ml, 0.125 mol) was added slowly to this solution via syringe over a period of one hour and the reaction mixture was stirred overnight as it warmed to room temperature. The reaction was then quenched slowly with saturated NaHCO 3 .
- Step 5 2-amino-4-(((2S,3S,4R.5R)-5-(6-(2-(dimethylamino)ethylaminoV9H-purin-9-v ⁇ -3,4- dihvdroxy-tetrahvdrofuran-2-yl)methylthio)butanoic acid 19a.
- Step 1 Tert-butyl 2-(1-(dimethylamino)naphthalene-5-su]fonamido ⁇ ethylcarbamate 20
- Step 3 2-amino-4-f((2S,3S,4R,5R)-5-(6-(2-(5-(dimethylamino)naphthalene-1- sulfonamido)ethylamino)-9H-purin-9-v ⁇ -3.4-dihvdroxy-tetrahvdrofuran-2- yl)methylthio)butanoic acid 19d.
- Examples 26-42, compounds 24c-24s, Table 3, were prepared according to scheme 2, utilizing either method A or method B.
- Step 1 (2R,3R,4S,5R)-2-(6-(2-(biphenyl-4-yl)ethylamino)-2-chloro-9H-purin-9-yl)-5- (hydroxymethyl)-tetrahydrofuran-3,4-diol 26
- Step 2 ((3aR,4R,6R.6aRV6-(6-(2-(biphenyl-4-vnethylamino)-2-chloro-9H-purin-9-yl)-2,2- dimethyl-tetrahvdrofuror3,4-d1H ,31dioxol-4-v0methanol 27
- Step 3 ((3aR.4R,6R.6aR)-6-(6-(2-(biphenyl-4-yl)ethylamino)-2-chloro-9H-purin-9-yl)-2,2- dimethyl-tetrahvdrofurof3,4-diri ,31dioxol-4-yl)methyl 4-methylbenzenesulfonate 28
- Step 4 Methyl 4-(((3aS.4S.6R,6aR)-6-(6-(2-( ' biphenyl-4-vnethylamino)-2-chloro-9H-purin-9-vn- 2.2-dimethyl-tetrahvdrofurof3,4-dlf1.31dioxol-4-vnmethylthio)-2-(tert- butoxycarbonylamino)butanoate 29
- Step 1 (2S.3S.4R.5RV2-((3-(((9H-fluoren-9-yl)methoxy)carbonylamino)-4-methoxy-4- oxobutylth) ⁇ )methyl)-5-(6-(2-(tert-butoxycarbonylamino)ethylamino)-2-chloro-9H-purin-9-v ⁇ - tetrahydrofuran-3,4-diyl diacetate 31
- Step 2 (2S,3S.4R,5RV2-((3-(((9H-fluoren-9-yl)methoxy)carbonylamino)-4-methoxy-4- oxobutylthio)methyl)-5-(6-(2-arninoethylamino)-2-chloro-9H-pur ' m-9-v ⁇ -tetrahvdrofuran- 3,4-diyl diacetate 32
- Step 3 2-amino-4-(((2S,3S.4R.5RV5-(2-chloro-6-(2-( r 4-cvanobenzamido)ethylamino)-9H-Durin- 9-yl)-3,4-dihvdroxy-tetrahvdrofuran-2-yl)methylthio)butanoic acid 33a [0143] A solution of 32 (76mg, 0.075 mmol), triethylamine (62.6 ⁇ l, 0.45 mmol) and 4- cyanobenzoyl chloride (18.5 mg, 0.112 mmol) in 0.75 -1.0 ml THF was stirred for 16 hours at room temperature.
- Examples 51-61 compounds 33b-33l, Table 4 were prepared from compound 32 and the appropriate acid chloride, scheme 3, as described for example 50, step 3. Table 4
- the title compound 35a was prepared in 35% yield (610 mg) using the general procedure and employing 3.92 g of di-ferf-butyl dicarbonate instead of the indicated amount and the product was obtained as white solid after recrystalized from hexanes.
- Methane sulfonyl chloride (103 mg, 0.9 mmol) was added to a solution of di-tert-butyl 2-hydroxypropane-1,3-diyldicarbamate 35a (174.2 mg, 0.6 mmol) in 1.0 ml pyridine. The reaction was mixed and left to stand for 30 minutes. Methylene chloride (10 ml) was then added and the reaction was washed with H 2 O (3 x 10 ml). The organic phase was dried with MgSO 4 , filtered and evaporated to give the crude mesylate.
- Examples 63-66, Table 5, were prepared in a manner similar to example 62, scheme 3, using compounds 35b-e in place of 35a The compounds were isolated as the formate salts after prep-HPLC purification in 20.6%; 33.8%; 18.9; and 47% yields respectively.
- Step 1 (2S,4S)-methyl 4-(acetylthio)-1-methylpyrrolidine-2-carbo ⁇ ylate 44
- Mesyl chloride (170 ⁇ L, 251 mg, 2.25 mmol) was added to a solution of (2S.4R)- methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43 (239 mg, 1.5 mmol, prepared according to the procedure of R.L. Elliott et al., Synthesis, 1995, 772) in pyridine (3 mL) and stirred at room temperature overnight.
- the reaction mixture was diluted in DCM (20 mL) and washed with water (15 mL).
- Step 2 (2S.4SVmethyl 4-(((3aS,4S.6R.6aRV6-(6-amino-9H-Purin-9-vn-2.2-dimethyl- tetrahvdrofuro[3,4-di ⁇ ,31dioxol-4-yl)methylthio)-1 -methylpyrrolidine-2-carboxylate 46
- a solution of thioacetate 44 (95 mg, 0.43 mmol) in MeOH (2mL) was treated with 0.5 M solution of NaOMe (876 ⁇ l_).
- Step 3 (2S.4SV4-(((2S.3S.4R.5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihvdroxy-tetrahvdrofuran-2- yl)methylthio)-1 -methylpyrrolidine-2-carboxylic 47
- Example 72 compound 48a, was prepared in a manner similar to example 71, were step 1 , was conducted according to the procedure in J. Org. Chem. 1196, 61 , 2226, replacing
- Example 73, compound 48b, was prepared in a manner similar to example 71, scheme 7. Step 1 was carried out according to the procedure for example 72 replacing (2S.4R)- methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43, with (2S,4R)-1-tert-butyl 2-methyl 4- hydroxypiperidine-1 ,2-dicarboxylate.
- Example 74 compound 48c, was prepared in a manner similar to example 71. Step
- Example 75 compound 48d, was prepared in a manner similar to example 71, replacing (2S,4R)-methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43, with (R)-tert-butyl 2-
- Example 76 compound 48e, was prepared in a manner similar to example 71, replacing (2S,4R)-methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43, with (SJ-tert-butyl 2-
- Step 1 (2S,4S)-1-tert-butyl 2-methyl 4-(((3aS,4S.6R,6aR)-6-(6-(4H-1 ,2,4-triazol-4-vn-9H-purin-
- Step 2 (2S,4S)-1-tert-butyl 2-methyl 4-( «3aS,4S,6R,6aRV2,2-dimethyl-6-(6-(3- phenylpropylamino)-9H-purin-9-yl)-tetrahvdrofuror3,4-dlf1 ,31dioxol-4-yl)methylthio)pyrrolidine-
- Step 3 (2S ⁇ S)-4-(((2S,3S.4R,5R)-3.4-dihvdroxy-5-(6-(3-phenylpropylaminoV9H-purin-9-vO- tetrahvdrofuran-2-v ⁇ methylthio)pyrrolidine-2-carboxylic acid 52a
- Example 78, compound 52b was prepared according to the method shown in scheme 2a, example 43, replacing 2-(biphenyl-4-yl)ethanamine with 3-phenylpropan-1 -amine in stepi, and utilizing (2S,4S)-1-tert-butyl 2-methyl 4-(acetylthio)pyrrolidine-1,2-dicarboxylate in place of tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate in step 4.
- Example 79, 52c was prepared as shown in scheme 1a, example 1, in step 3 replacing tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate with (2S,4S)-1 -tert-butyl 2-methyl 4-
- Example 80, 52d was prepared according to scheme 2a, example 43, step 4 reacting 28 with (2S,4S)-1 -tert-butyl 2-methyl 4-(acetylthio)pyrrolidine-1 ,2-dicarboxylate in place of tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate.
- Example 81 , 52e was prepared as shown in scheme 1a, example 1, in step 3 replacing tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate with (S)-tert-butyl 3-
- Example 82, 52f, was prepared accordinglo " scheme 2a ⁇ example 43, " step 4 reacting "
- Example 83, 52g was prepared according to the method shown in scheme 2a, example 43, replacing 2-(biphenyl-4-yl)ethanamine with 3-phenylpropan-1 -amine in stepi , and utilizing (S)-tert-butyl 3-(acetylthio)pyrrolidine-1-carboxylate in place of tert-butyl 2-oxo- tetrahydrothiophen-3-ylcarbamate in step 4.
- Step 1 9- ⁇ 3aR,4R,6R.6aR)-6-((tert-butyldimethylsilyloxy)methyl)-2,2-dimethyl- tetrahvdrofurof3,4-d1f1,3ldioxol-4-yl)-6-chloro-2-iodo-9H-purine 55a
- Step 2 9-((3aR,4R,6R,6aR)-6-((tert-butyldimethylsilyloxy ' )methylV2,2-dimethyl- tetrahvdrofurof3,4-d1f1 ,31dioxol-4-yl)-2-iodo-9H-purin-6-ami ⁇ e 56a
- Step 3 ( r (3aR.4R,6R,6aR)-6-(6-amino-2-iodo-9H-purin-9-yl)-2.2-dimethyl-tetrahvdrofuror3,4- d1[1 ,31dioxol-4-yl)methanol 57a
- Step 4 methyl 4-(((3aS,4S,6R,6aR)-6-(6-amino-2-iodo-9H-purin-9-yl)-2,2-dimethyl- tetrahvdrofurof3,4-din ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 58a
- Step 5 2-amino-4-(((2S,3S,4R,5R)-5-(6-amino-2-iodo-9H-purin-9-yl)-3.4-dihvdroxy- tetrahvdrofuran-2-v0methylthio)butanoic acid 59a
- Step 1 methyl 4-(((3aS.4S.6R.6aR)-6-(6-amino-2-methyl-9H-purin-9-yl)-2,2-dimethyl- tetrahvdrofuror3,4-diri ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 60
- Step 2 2-Amino-4-(((2S.3S,4R,5R)-5-(6-amino-2-methyl-9H-purin-9-v ⁇ -3,4-dihvdroxy- tetrahydrofuran-2-yl)methylthio)butanoic acid 61
- Stepi methyl 4-(((3aS,4S,6R,6aR)-6-(6-amino-2-chloro-9H-purin-9-ylV2,2-dimethyl- tetrahvdrofuror3,4-diri ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 64
- Step 2 2-amino-4-(((2S.3S.4R,5R)-5-(6-amino-2-chloro-9H-purin-9-vn-3,4-dihvdroxy- tetrahvdrofuran-2-vDrnethylthio)butanoic acid 65
- Step 1 (2S.4R)-methyl 4-(((2S,3S.4R.5R)-5-(6-amino-2-chloro-9H-purin-9-vO-3,4-dihvdroxy- tetrahvdrofuran-2-yl)methylthio)pyrrolidine-2-carboxylate 67a
- Step2 (2S.4R)-4- «(2S,3S ⁇ R,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-3.4-dihvdroxy- tetrahvdrofuran-2-yl)methylthio)pyrrolidine-2-carboxylic acid 68a
- Stepi (R)-tert-butyl 3-(((3aS,4S.6R,6aR)-6-(6-amino-2-chloro-9H-purin-9-ylV2.2-dimethyl- tetrahvdrofuror3,4-dlf 1 ,3ldioxol-4-vDmethylthio)pyrrolidine-1 -carboxylate 67b
- Compound 67b was prepared in 33% yield as a clear syrup following the procedure detailed in step 4 for 58a, example 84, scheme 9, replacing 57a with (S)-tert-butyl 3- (acetylthio)pyrrolidine-i -carboxylate 66b.
- Step 1 benzyl (S)-3-(((3aR,4R,6R,6aR)-4-(6-amino-9H-purin-9-v ⁇ -tetrahvdro-2.2- dimethylfuro[3,4-cnf 1 ,3ldioxol-6-vDmethylamino)-1 -(tert-butoxycarbonyl)propylcarbamate 71 [0194] To a solution of (S)-tert-butyl 2-(benzyloxycarbonylamino)-4-oxobutanoate 69 (4.0 ml, 1.41 mmol, prepared according to I. Weits et al, J. Org. Chem.
- Step 3 (S)-4-(N-(2-nitrocinnamv ⁇ -N-(((2R.3S.4R,5RV5-(6-amino-9H-purin-9-yl)-tetrahvdro-3.4- dihvdroxyfuran-2-v0methv0amino)-2-aminobutanoic acid (74a)
- Example 94 compound 74d, Table 8, was prepared according to the procedure described for example 91 , in step 2 using 72d, 4-nitrobutanal (previously reported by R. Kimura et al. Bull. Chem. Soc. Jpn., 2002, 75(11), 2517-2526, however it was prepared in 94% yield by DIBAL-H reduction of methyl 4-nitrobutanoate in DCM at -78 0 C) in place of aldehyde 72a.
- Step 1 (S)-tert-butyl 4-((((3aR,4R,6R.6aRV6-(6-amino-9H-purin-9-vn-2.2-dimethyl- tetrahvdrofuror3,4-cn ⁇ ,31dioxol-4-vnmethyl)(methv ⁇ amino)-2-
- Step 2 benzyl (S)-1-(tert-butoxycarbonv ⁇ -3-(N-(((3aR.4R.6R.6aRV4-(6-(ethvIaminoV9H-purin-)
- Step 3 (S)-4-(N-(((2R,3S.4R,5R)-5-(6-(ethylamino)-9H-purin-9-vn-tetrahvdro-3.4- dihydroxyfuran-2-yl)methyl)-N-methylamino)-2-aminobutanoic acid 77
- P26358 (SEQ ID NO.1) was cloned into the BamHt/Sall site of the pBlueBac4.5 vector
- Hi-5 cells grown in suspension and maintained in serum-free medium (Sf900 Il supplemented with gentamycin) were infected with the abovementioned viruses at multiplicity of infection (MOI) varying from 1 to 3 during 84 hours at 27 0 C with agitation at 120 rpm on a rotary shaker. Infected cells were harvested by centrifugation at 398g for 15 min. after which a nuclear and cytosolic fractionation was performed. Fractions were frozen at -8O 0 C until purifications were performed.
- MOI multiplicity of infection
- DTT 0.5% NP-40, 25% glycerol, 1 ⁇ g/ml pepstatin, 2 ⁇ g/ml Aprotinin and leupeptin, 50 ⁇ g/ml
- Buffer B (2OmM Tris pH 8.0, 1.5mM MgCI 2 , 5mM KCI, 5mM DTT, 0.5% NP-40, 25% glycerol,
- Buffer B was 10%.
- DNMT-1 enriched fractions were pooled according to SDS-PAGE analysis (coomassie staining) Final DNMT-1 protein preparations concentration were about 7mg/ml and purity above 95%.
- DNMT3b2 was purified using only the nuclear extract as starting material.
- the NaCI concentration of the latter was adjusted to 0.2M by diluting with Buffer D (5OmM NaPO 4 pH 7.8, 10% glycerol and 1mM EDTA) supplemented with the protease inhibitors described in Buffer A followed by centrifugation at 3000Og for 10 min..
- the supernatant was loaded onto a Hitrap SPsepharose column (Amersham Biosciences) equilibrated with Buffer D with 0.05M NaCI. Column was washed with 8 CV of equilibration buffer and proteins were eluted with an 8 CV linear gradient of NaCI (from 0.05 to 1 M) in Buffer D.
- DNMT3b2 containing fractions were pooled based on SDS-PAGE analysis. Selected fractions from this elution had conductivity varying from 15.1 to 28.2mS/cm. Finally, this SPsepharose eluate underwent buffer exchange, using PD-10 column (Amersham Biosciences) against Buffer D + 0.3M NaCI. Typical DNMT3b2 enzyme preparation had concentration of about 2.5mg/ml and approximately 70% purity. [0211] Purified DNMT-1 and DNMT3b2 protein stocks were aliquoted and frozen at -8O 0 C prior to use in enzymatic assay. DNA Methyltransferase Assay Reagents : -Enzyme: Cloned human DNA Methyltransferase (DNMT1 and DNMT3b2)
- MYG 167 ATC GCA TCG ATC GCG ATT CGC GCA TCG GCG ATC MYG 166: GAT XCG XGA TGX GXG AAT XGX GAT XGA TGX GAT (X: 5-methylcytosine) -Substrate2: "" “ " “ ⁇ ' " " "
- the mixture is pre-incubated at 37 0 C for 10min and then the SAM mix is added (2.25uL of 4OuM of S- Adenosyl-L-Methionine to make 3uM final, 1.65uL of 10X Buffer [5OmM Tris-HCI pH7.6, 5%Glycerol, 1mM EDTA, 100ug/mL BSA, 1 mM DTT]).
- the final reaction is in 3OuL.
- the reaction is incubated at 37°C for 15min and then transferred onto DEAE filtermat (Wallac cat# 1450-522) using Tomtec cell harvester.
- the DEAE filtermat is washed with cold 2OmM NH4HCO3. When the filter is dry, it is covered with MeltiLex TM scintillant (Wallac cat#1450- 441 ). Counting is done in a Wallac beta-counter.
- DNMT3b2 assay follows identical protocol, at enzyme concentration of 188nM. [0214] The activities of a number of compounds according to the invention measured by various assays are displayed in the table below.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Hematology (AREA)
- Medicinal Chemistry (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
The invention relates to the inhibition of DNA methyltransferase isoforms DNMT1 and DNMT3b2. The invention provides compounds and methods for inhibiting DNMT1 and DNMT3b2.
Description
INHIBITORS OF DNA METHYLTRANSFERASE
BACKGROUND OF THE INVENTION Field of the Invention
[0001] This invention relates to inhibition of the DNA methyltransferase isoforms DNMT1 and DNMT3b2. More particularly, the invention relates to compounds and methods for the inhibition of DNMT1 and DNMT3b2.
Summary of the Related Art
[0002] Changes in human DNA methylation patterns are commonly found in human tumors and are implicated in development and maintenance of human cancer (A. Bird, Genes Dev., 2002, 16, 6-21; P. Siedlecki, R.G. Boy, S. Comagic, R. Schirrmacher, M. Wiessler, P. Zielenkiewicz, S. Suhai, and F. Lyko, Biochemical and Biophysical Research communications, 2003, 306, 558). DNA hypermethylation in cancer cells results in alteration of gene expression patterns and most notably the loss of expression of tumor suppressor genes (K. D. Robertson, A.P. Baylin, Nat. Rev. Genet., 2000, 1, 11-19; PA Jones, S.B. Baylin, Trends Genet, 2000, 16, 168-174).
[0003] DNA methyltransferase 1 (DNMT1) protein is a major contributor to DNA methyltransferase activity in human cells and is required to maintain methylation patterns in differentiated cells (M. F. Robert, S. Morin, N. Beaulieu, F. Gauthier, I.C. Chute, A. Barsalou, A.R. MacLeod, Nat. Genet. 33 (2003) 61-65). The de novo DNA methyltransferases DNMT3A and DNMT3B establish DNA methylation during early embryogenesis (M. Okano, S. Xie, E. Li, Nat. Genet, 1998, 19, 219-220; M. Okano, D.W. Bell, DA Haber, E. Li, Cell, 1999, 99, 247- 257, T.M. Geiman, UT. Sankpal, A.K. Robertson, Y. Chen, M. Mazumdar, J.T. Heale, JA. Schmeising, W. Kim, K. Yokomori, Y. Zhao, and K.D. Robertson, Nucleic Acids Research, 2004, 32, 2716-2729).
[0004] Regulation of methylation of human DNA requires the activity of several DNA methyltransferases. The DNA is covaiently modified by the cofactor (L)-S-Adenosyl-L- methionine (SAM) at carbon-5 of cytosine residues. This biological methylating agent is converted to its demethylated metabolite (L)-S-Adenosyl-L-homocysteine (L-SAH). In tissue the SAM and SAH levels are equivalent (F. Salvatore, R. Utili, V. Zappia, and S.K. Shapiro, Anal. Biochem. 1971, 41, 16; J. Hoffman, Anal. Biochem, 1975, 68, 522). SAH can bind to DNA methyltransferases and inhibit their catalytic reaction and is an important molecule in the regulation of biological transmethylation (T. Deguchi and J. Barchas, J. Biol. Chem., 1971 , 246, 3175; A. Oliva, P. Galletti, and V. Zappia, Eurp. J. Biochem., 1980, 104, 595; P.M. Ueland and J. Saebo, Biochemistry, 1979, 18, 4130).
[0005] Inhibitors of DNMT1 and DNMT3B are expected to have value as anti-cancer agents (M. Szyf, Fontiers in Bioscience 2001 , 6, 599-609; M.F. Robert, S. Morin, N. Beaulieu, F. Gauthier, !.C. Chute, A. Barsalou, A.R. MacLeod, Nat. Genet. 2003, 33, 61-65; D. S. Schrump et al, abstract 442, 16th EORTC-AACR Symposium on Molecular Targets and Cancer Therapeutics, 28 September-1 October, 2004, Geneva, Switzerland).

[0006] Borchardt et al. described the synthesis and biological activity of a number of SAH analogues having structural modifications in the amino acid, sugar, or base portions. However, these analogues were synthesized in an effort to develop inhibitors for O- or N- methyltransferases, specifically catechol O-methyltransferase (COMT), hydroxyindole O- methyltransferase (HIMT), indoleethylamine N-methyltransferase (INMT), phenylethanolamine N-methyltransferase (PNMT), and histamine N-methyltransferase (HMT), but not human DNA methyltransferase (DNMT) (RT. Borchardt and Y.S. Wu, J. Med. Chem., 1974, 17, 862; RT. Borchardt, J.A. Huber, and Y.S. Wu, J. Med. Chem., 1974, 17, 868; RT. Borchardt and Y.S. Wu, J. Med. Chem., 1975, 18, 300; RT. Borchardt and Y.S. Wu, J. Med. Chem., 1976, 19, 197; RT. Borchardt, Biochem. Pharmacol., 1975, 24, 1542; RT. Borchardt, J.A. Huber, and Y.S. Wu, J. Med. Chem., 1976, 19, 1094). Some N-6-substituted SAH analogues have been tested against protein arginine methyltransferases (Q. Lin et al, J. Am. Chem. Soc. 2001 , 123, 11608- 11613).
[0007] SAH is a known nonselective inhibitor of human DNA methyltransferase and many other methyltransferases. A number of simplified and less rigid analogues of SAH have been reported. Such compounds, however, were devoid of activity against human DNA methyltransferase (M. Botta, R. Saladino, G. Pedraly-Noy, and R. Nicoletti, Med. Chem. Res., 1994, 4, 323).
[0008] The 2'- and 3'-hydroxy groups of the sugar portion and the 6-amino group of the adenine moiety of SAH were found to be important structural requirements for inhibition of DNA methyltransferases. T. Borchardt, Y.S. Wu, and J.A. Huber, J. Med. Chem., 1976, 19, 1104; M. D. Houston, B. Matuszewska, and RT. Borchardt, J. Med. Chem. 1985, 28, 478; P.A. Crooks, MJ. tribe, and R.J. Pinney, J. Pharm. Pharmacol, 1984, 36, 85.
[0009] SAM and SAH as inhibitors (in vivo) are not good drugs and they are unstable in plasma due to hydrolases and ribonucleases, they have poor absorption due to the Zwitterionic nature, are rapidly excreted, and have a short half life. A.A. Minnick and G. L. Kenyon, J. Org. Chem., 1988, 53, 4952. In addition, SAH is an endogenous inhibitor of numerous methyltransferases and as such is non-selective, making it undesirable as a drug. [0010] Some more stable nitrogen anlogues of SAM and SAH have been reported. Chi-Deu Chang and J. K. Coward, J. Med. Chem., 1976, 19, 684; AA Minnick and G. Kenyon, J. Org. Chem., 1988, 53, 4952; M. Thompson, a. Makhalfia, D.P. Hornby, and G. M. Blackburn, J. Org. Chem. 1999, 64, 7467 When these analogues were evaluated as inhibitors of catechol O- methyltransferase and tRNA methylases, however, they were found to have poor activity. [0011] Sinefungin, a natural product, is a nitrogen analogue of SAH and has been reported to inhibit human DNA methyltransferase. It is also a non-selective inhibitor with potential for toxicity (C. Barbes, J. Sanchez, M.J. Yebra, M.Robert-Gero, and C. Hardisson, FEMS Microbiology Letters, 1990, 69, 239).

[0012] In addition, inhibition of DNMT1 with 5-azacytidine or related compounds has been reported. Such inhibitors, however, are incorporated into the DNA of the target cell and, thus, suffer from high toxicity and low specificity (D.V. Santi, A. Norment, CE. Garrett, Proc. Natl.
Acad. Sci. USA 81(1984) 6993-6997).
[0013] More recently indolyl-2-phenyl bisamidines have been reported as a new class of
DNA methyltransferase inhibitors. These are known to be DNA binding agents, however, and thus may inhibit the DNMTs indirectly. S. W. Goldstein, B.L. Mylari, J. R. Perez, and E.A. Glazer,
US 6,699,862 B1, Mar 2, 2004.
[0014] This invention provides compounds and methods for the inhibition of human C-5
(cytosine) DNA methyltransferases DNMT1 and DNMT3b2 enzymes.
BRIEF SUMMARY OF THE INVENTION
[0015] The compounds of the invention are inhibitors of the DNA methyltransferase isoforms DNMT1 and DNMT3b2. Accordingly, the invention provides new inhibitors of DNMT1 and DNMT3b2.
[0016] In a first aspect, the invention provides compounds of formula (I) (and their pharmaceutically acceptable salts) that are useful as inhibitors of DNMT1 and/or DNMT3b2 and therefore are useful for studying the role of DNA methyltransferase in biological processes. [0017] In a second aspect, the invention provides compositions comprising a compound that is an inhibitor of DNMT1 and/or DNMT3b2, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, excipient, or diluent.
[0018] In a third aspect, the invention provides a method of inhibiting DNMT1 and/or DNMT3b2 enzymes in a cell, comprising contacting a cell in which inhibition of DNMT1 or DNMT3b2 is desired with a compound of the invention.
[0019] The foregoing merely summarizes certain aspects of the invention and is not intended to be limiting in nature. These aspects and other aspects and embodiments are described more fully below.
BRIEF DESCRIPTION OF THE DRAWINGS
[0020] Figure 1 displays the full length DNMT1 (Swissprot accession number P26358 (SEQ ID NO.1)).
[0021] Figure 2 displays the DNMT3 splice variant 2 of DNMT3b2 (Swissprot accession number Q9UBC3-2 (SEQ ID NO.2))
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS [0022] The invention provides compounds and methods for inhibiting DNMT1 and DNMT3b2.
[0023] The patent and scientific literature referred to herein establishes knowledge that is available to those with skill in the art. The issued patents, applications, and references that are cited herein are hereby incorporated by reference to the same extent as if each was specifically and individually indicated to be incorporated by reference. In the case of inconsistencies, the present disclosure will prevail.
[0024] For purposes of the present invention, the following definitions will be used (unless expressly stated otherwise):
[0025] The term "inhibitor of DNMT1 and DNMT3b2" is used to identify a compound having a structure as defined herein, which is capable of interacting with DNMT1 , DNMT3b2 or both DNMT1 and DNMT3b2 and inhibiting the activity of DNMT1 , DNMT3b2, or both DNMT1 and DNMT3b2. In some preferred embodiments, such reduction of activity is at least about 50%, more preferably at least about 75%, and still more preferably at least about 90%.
[0026] For simplicity, chemical moieties are defined and referred to throughout primarily as univalent chemical moieties (e.g., alkyl, aryl, etc.). Nevertheless, such terms are also used to convey corresponding multivalent moieties under the appropriate structural circumstances clear to those skilled in the art. For example, while an "alkyl" moiety generally refers to a monovalent radical (e.g. CH3-CH2-), in certain circumstances a bivalent linking moiety can be "alkyl," in which case those skilled in the art will understand the alkyl to be a divalent radical (e.g., -CH2- CH2-), which is equivalent to the term "alkylene." (Similarly, in circumstances in which a divalent moiety is required and is stated as being "aryl," those skilled in the art will understand that the term "aryl" refers to the corresponding divalent moiety, arylene.) All atoms are understood to have their normal number of valences for bond formation {i.e., 4 for carbon, 3 for N, 2 for O, and 2, 4, or 6 for S, depending on the oxidation state of the S). On occasion a moiety may be defined, for example, as (A)3-B-, wherein a is 0 or 1. In such instances, when a is 0 the moiety is B- and when a is 1 the moiety is A-B-. Also, a number of moieties disclosed herein exist in multiple tautomeric forms, all of which are intended to be encompassed by any given tautomeric structure.
[0027] The term "hydrocarbyl" refers to a straight, branched, or cyclic alkyl, alkenyl, or alkynyl, each as defined herein. A "C0" hydrocarbyl is used to refer to a covalent bond. Thus, "C0-C3-hydrocarbyF' includes a covalent bond, methyl, ethyl, ethenyl, ethynyl, propyl, propenyl, propynyl, and cyclopropyl.
[0028] The term "alkyl" as employed herein refers to straight and branched chain aliphatic groups having from 1 to 12 carbon atoms, preferably 1-8 carbon atoms, and more preferably 1-6 carbon atoms, which is optionally substituted with one, two or three substituents. Preferred alkyl groups include, without limitation, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert- butyl, pentyl, and hexyl. A "C0" alkyl (as in "C0-C3-alkyr') is a covalent bond (like "C0" hydrocarbyl).
[0029] The term "alkenyl" as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon double bonds, having from 2 to 12 carbon atoms, preferably 2-8 carbon atoms, and more preferably 2-6 carbon atoms, which is optionally substituted with one, two or three substituents. Preferred alkenyl groups include, without limitation, ethenyl, propenyl, butenyl, pentenyl, and hexenyl.
[0030] The term "alkynyl" as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon triple bonds, having from 2 to 12 carbon atoms, preferably 2-8 carbon atoms, and more preferably 2-6 carbon atoms, which is optionally
substituted with one, two or three substituents. Preferred alkynyl groups include, without limitation, ethynyl, propynyl, butynyl, pentynyl, and hexynyl.
[0031] An "alkylene," "alkenylene," or "alkynylene" group is an alkyl, alkenyl, or alkynyl group, as defined hereinabove, that is positioned between and serves to connect two other chemical groups. Preferred alkylene groups include, without limitation, methylene, ethylene, propylene, and butylene. Preferred alkenylene groups include, without limitation, ethenylene, propenylene, and butenylene. Preferred alkynylene groups include, without limitation, ethynylene, propynylene, and butynylene.
[0032] The term "cycloalkyl" as employed herein includes saturated and partially unsaturated cyclic hydrocarbon groups having 3 to 12 carbons, preferably 3 to 8 carbons, and more preferably 3 to 6 carbons, wherein the cycloalkyl group additionally is optionally substituted. Preferred cycloalkyl groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, and cyclooctyl. [0033] The term "heteroalkyl" refers to an alkyl group, as defined hereinabove, wherein one or more carbon atoms in the chain are replaced by a heteroatom selected from the group consisting of O, S, NH, N-alkyl, SO, SO2, SO2NH, or NHSO2.
[0034] An "aryl" group is a C6-Ci4 aromatic moiety comprising one to three aromatic rings, which is optionally substituted. Preferably, the aryl group is a C6-Ci0 aryl group. Preferred aryl groups include, without limitation, phenyl, naphthyl, anthracenyl, and fluorenyl. An "aralkyl" or "arylalkyl" group comprises an aryl group covalently linked to an alkyl group, either of which may independently be optionally substituted or unsubstituted. Preferably, the aralkyl group is (C1- C^aIk(C6-C10)aryl, including, without limitation, benzyl, phenethyl, and naphthylmethyl. [0035] A "heterocyclyl" or "heterocyclic" group is a ring structure having from about 3 to about 12 atoms, wherein one or more atoms are selected from the group consisting of N, O, S, SO, and SO2. The heterocyclic group is optionally substituted on carbon at one or more positions. The heterocyclic group is also independently optionally substituted on nitrogen with alkyl, aryl, aralkyl, alkylcarbonyl, alkylsulfonyl, arylcarbonyl, arylsulfonyl, alkoxycarbonyl, or aralkoxycarbonyl. Preferred heterocyclic groups include, without limitation, epoxy, aziridinyl, tetrahydrofuranyl, pyrrolidinyl, piperidinyl, piperazinyl, thiazolidinyl, oxazolidinyl, oxazolidinonyl, and morpholino. In certain preferred embodiments, the heterocyclic group is fused to an aryl, heteroaryl, or cycloalkyl group. Examples of such fused heterocyles include, without limitation, tetrahydroquinoline and dihydrobenzofuran. Specifically excluded from the scope of this term are compounds where an annular O or S atom is adjacent to another O or S atom.
[0036] As used herein, the term "heteroaryl" refers to groups having 5 to 14 ring atoms, preferably 5, 6, 9, or 10 ring atoms; having 6, 10, or 14 π-electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to three heteroatoms per ring selected from the group consisting of N, O, and S. The term "heteroaryl" is also meant to encompass monocyclic and bicyclic groups. For example, a heteroaryl group may be pyrimidinyl, pyridinyl, benzimidazolyl, thienyl, benzothiazolyl, benzofuranyl and indolinyl. A "heteroaralkyl" or "heteroarylalkyl" group comprises a heteroaryl group covalently linked to an alkyl group, either of which is independently optionally substituted or unsubstituted. Preferred heteroalkyl groups comprise a C1-C6 alkyl group and a heteroaryl group having 5, 6, 9, or 10 ring atoms. Specifically excluded from the scope of this term are compounds having adjacent annular O and/or S atoms. Examples of preferred heteroaralkyl groups include pyridylmethyl, pyridylethyl, pyrrolylmethyl, pyrrolylethyl, imidazolylmethyl, imida∑olylethyl, thiazolylmethyl, and thiazolylethyl. Specifically excluded from the scope of this term are compounds having adjacent annular O and/or S atoms.
[0037] For simplicity, reference to a "Cn-Cn," heterocyclyl or "Cn-Cm" heteroaryl means a heterocyclyl or heteroaryl having from "n" to "m" annular atoms, where "n" and "m" are integers. Thus, for example, a C5-C6-heterocyclyl is a 5- or 6- membered ring having at least one heteroatom, and includes pyrrolidinyl (C5) and piperidinyl (C6); C6-hetoaryl includes, for example, pyridyl and pyrimidyl.
[0038] An "arylene," "heteroarylene," or "heterocyclylene" group is an aryl, heteroaryl, or heterocyclyl group, as defined hereinabove, that is positioned between and serves to connect two other chemical groups.
[0039] Preferred heterocyclyls and heteroaryls include, but are not limited to, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, pyridotriazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H.6H-1 ,5,2- dithiazinyl, dihydrofuro[2,3-b]tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, 1 H-indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, 3H-indolyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isoxazolyl, methylenedioxyphenyl, morpholinyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1 ,2,3- oxadiazolyl, 1 ,2,4-oxadiazolyl, 1 ,2,5-oxadiazolyl, 1 ,3,4-oxadiazolyl, oxazolidinyl, oxazolyl, oxazolidinyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, piperidonyl, 4-piperidonyl, piperonyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl,
pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, 2H-pyrrolyl, pyrrolyl, quinazolinyl, quinolinyl, 4-H-quinolizinyI, quinoxalinyl, quinuclidinyl, tetrahydrofuranyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, tetrazolyl, 6H-1 ,2,5- thiadiazinyl, 1 ,2,3-thiadiazolyl, 1 ,2,4-thiadiazolyl, 1,2,5-thiadiazolyl, 1 ,3,4-thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl, 1 ,2,3-triazolyl, 1 ,2,4-triazolyl, 1 ,3,4-triazolyl, and xanthenyl. [0040] As employed herein, when a moiety (e.g., cycloalkyl, hydrocarbyl, aryl, heteroaryl, heterocyclic, urea, etc.) is described as "optionally substituted" it is meant that the group optionally has from one to four, preferably from one to three, more preferably one or two, non- hydrogen substituents. Suitable substituents include, without limitation, halo, hydroxy, oxo (e.g., an annular -CH- substituted with oxo is -C(O)-) nitro, halohydrocarbyl, hydrocarbyl, aryl, aralkyl, alkoxy, aryloxy, amino, acylamino, alkylcarbamoyl, arylcarbamoyl, aminoalkyl, acyl, carboxy, hydroxyalkyl, alkanesulfonyl, arenesulfonyl, alkanesulfonamido, arenesulfonamido, aralkylsulfonamido, alkylcarbonyl, acyloxy, cyano, and ureido groups. Preferred substituents, which are themselves not further substituted (unless expressly stated otherwise) are:
(a) halo, hydroxy, cyano, oxo, carboxy, formyl, nitro, amino, amidino, guanidino,
(b) C1-C5 alkyl or alkenyl or arylalkyl imino, carbamoyl, azido, carboxamido, mercapto, hydroxy, hydroxyalkyl, alkylaryl, arylalkyl, C1-C8 alkyl, C1-C8 alkenyl, C1-C8 alkoxy, C1-C8 alkoxycarbonyl, aryloxycarbonyl, C2-C8 acyl, C2-C8 acylamino, C1-C6 alkylthio, arylalkylthio, arylthio, C1-C8 alkylsulfinyl, arylalkylsulfinyl, arylsulfinyl, C1-C8 alkylsulfonyl, arylalkylsulfonyl, arylsulfonyl, C0-C6 W.alkyl carbamoyl, C2-C15 N,N- dialkylcarbamoyl, C3-C7 cycloalkyl, aroyl, aryloxy, arylalkyl ether, aryl, aryl fused to a cycloalkyl or heterocycle or another aryl ring, C3-C7 heterocycle, C5-Ci4 heteroaryl, or any of these rings fused or spiro-fused to a cycloalkyl, heterocyclyl, or aryl, wherein each of the foregoing is further optionally substituted with one more moieties listed in (a), above; and
(c) -(CH2)s-NR30R31, wherein s is from 0 (in which case the nitrogen is directly bonded to the moiety that is substituted) to 6, and R30 and R31 are each independently hydrogen, cyano, oxo, carboxamido, amidino, Ci-C8 hydroxyalkyl, C1-C3 alkylaryl, aryl-CrC3 alkyl, C1-C8 alkyl, CrC8 alkenyl, C1-C8 alkoxy, Ci-C8 alkoxycarbonyl, aryloxycarbonyl, 3IyI-C1-C3 alkoxycarbonyl, C2-C8 acyl, Ci-C8 alkylsulfonyl, arylalkylsulfonyl, arylsulfonyl, aroyl, aryl, cycloalkyl, heterocyclyl, or heteroaryl, wherein each of the foregoing is further optionally substituted with one more moieties listed in (a), above; or
R30 and R31 taken together with the N to which they are attached form a heterocyclyl or heteroaryl, each of which is optionally substituted with from 1 to 3 substituents from (a), above.
[0041] A "halohydrocarbyl" is a hydrocarbyl moiety in which from one to all hydrogens have been replaced with one or more halo.
[0042] The term "halogen" or "halo" as employed herein refers to chlorine, bromine, fluorine, or iodine. As herein employed, the term "acyl" refers to an alkylcarbonyl or arylcarbonyl substituent. The term "acylamino" refers to an amide group attached at the nitrogen atom (Ae., R-CO-NH-). The term "carbamoyl" refers to an amide group attached at the carbonyl carbon atom (i.e., NH2-CO-). The nitrogen atom of an acylamino or carbamoyl substituent is additionally substituted. The term "sulfonamido" refers to a sulfonamide substituent attached by either the sulfur or the nitrogen atom. The term "amino" is meant to include NH2, alkylamino, arylamino, and cyclic amino groups. The term "ureido" as employed herein refers to a substituted or unsubstituted urea moiety.
[0043] The term "radical" as used herein means a chemical moiety comprising one or more unpaired electrons.
[0044] A moiety that is substituted is one in which one or more hydrogens have been independently replaced with another chemical substituent. As a non-limiting example, substituted phenyls include 2-flurophenyl, 3,4-dichlorophenyl, 3-chloro-4-fluoro-phenyl, 2-fluoro- 3-propylphenyl. As another non-limiting example, substituted n-octyls include 2,4-dimethyI-5- ethyl-octyl and 3-cyclopentyl-octyl. Included within this definition are methylenes (-CH2-) substituted with oxygen to form carbonyl -CO-).
[0045] An "unsubstituted" moiety as defined above (e.g., unsubstituted cycloalkyl, unsubstituted heteroaryl, etc.) means that moiety as defined above that does not have any of the optional substituents for which the definition of the moiety (above) otherwise provides. Thus, for example, while an "aryl" includes phenyl and phenyl substituted with a halo, "unsubstituted aryl" does not include phenyl substituted with a halo.
[0046] Throughout the specification, preferred embodiments of one or more chemical substituents are identified. Also preferred are combinations of preferred embodiments. For example, paragraph [0052] describes preferred embodiments of L1 in the compounds of formula (N-A) and paragraph [0053] describes preferred embodiments of R2 in the compounds of formula (H-A). Thus, also contemplated as within the scope of the invention are compounds of formula (A) in which L1 and R2 are as described in paragraph [0052] and Ri is as described in paragraph [0053]. Furthermore, compounds excluded from any one particular genus of
compounds (e.g., through a proviso clause) are intended to be excluded from the scope of the invention entirely, including from other disclosed genera, unless expressly stated to the contrary.
Compounds
[0047] In the first aspect, the invention comprises compounds of formula (I), that are inhibitors DNMT1 and DNMT3b2:

R3 and R4 independently represent H, C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -CrC6-alkyl-cycloalkyl, -Ci-C6-alkyl- heterocyclyl, -CrC6-alkyl-aryl, -CrC6-alkyl-heteroaryl, -d-Cealkoxy-aryl or - (CH2)1-6-T, wherein C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, cycloalkyl, heterocyclyl, aryl and heteroaryl, at each occurrence, are optionally substituted; or R3 and R4 taken together with the nitrogen to which they are attached form a C5-C9 heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted; T is NH-C(=O)-R14, -NH-SO2-R15, or -S-(CH2)1-3-R14, R14 is Ci-C6 alkyl, aryl or heteroaryl and R15 is aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted;
R2 is H, halo, CF3, C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C1-C6 alkoxy, -NH-C1-C6 alkyl, or -S- C1-C6 alkyl, wherein C1-C6 alkyl, C2-C6 alkenyl, and C2-C6 alkynyl are each optionally substituted; A and B independently are F, Cl, -OH, H1 -NHR, or -OR;
R at each occurrence is independently benzyl or C1-C4 alkyl, wherein benzyl and C1-C4 alkyl are optionally substituted; W is CH, N, CR, or C-halogen; X is CH, N, C-CrC6-alkyl, or C-halogen; D is CH, or N; Y is -S-, -O-, N(R16)-, -CH=CH-, -S-CH2-, -0-CH2-,
where R16 is H, d-C6 alkyl, -CrCe-alkyl-aryl, -CrCe-alkyl-heteroaryl, or -C2-C6 alkenyl- aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted;
Z is -L-C(H)(NH2)-COOR7, -L-NR19R20, or heterocyclyl, wherein heterocyclyl is optionally substituted; L is a bond or is -(CR17Ri8)I-6-; each R17 and R18 independently is H or CrC6-alkyl, wherein Ci-C6-alkyl is optionally substituted; R19 and R20 independently are H, Ci-C6-alkyl, heteroaryl, or H2N-C(=NH)-CH2-, wherein
C-rC6-alkyl and heteroaryl are optionally substituted; and R7 is H or Ci-C6-alkyl.
[0048] In a preferred embodiment of the compounds according to paragraph [0047], the compounds are represented by formula (II)

(II) and pharmaceutically acceptable salts and complexes thereof, wherein A is H, halogen, or OH; R2 is H, halo, C1-C6 alkyl, C2-C6 alkenyl or -S- Ci-C6 alkyl, wherein C1-C6 alkyl and C2-C6 alkenyl, at each occurrence, are optionally substituted; R3 and R4 independently represent H, C1-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -CrC6-alkyl-cycloalkyl, -C-i-Ce-alkyl-heterocyclyl, -CrC6-a!ky!-aryl, -C1-C6- alkyl-heteroaryl, -CrC6alkoxy-aryl or -(CH2)i-6-T, wherein Ci-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl and heteroaryl, at each occurrence, are optionally substituted; or R3 and R4 taken together with the nitrogen to which they are attached form a C5-C9 heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted;
T is NH-C(=O)-Ri4, -NH-SO2-Ri5, or -S-(CH2)i-3-Ri4,
Ri4 is C1-C6 alkyl, aryl or heteroaryl and R15 is aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted; W and X are independently CH or N;
 R16 is H, C1-C6 alkyl, -C-pCe-alkyl-aryl, -CrCe-alkyl-heteroaryl, or -C2-C6 alkenyl-aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted;
R16 is H, C1-C6 alkyl, -C-pCe-alkyl-aryl, -CrCe-alkyl-heteroaryl, or -C2-C6 alkenyl-aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted;
Z is -L-C(H)(NH2)-COOR7, -L-NR19R20, or heterocyclyl, wherein heterocyclyl is optionally substituted; L is a bond or is -(CR17Ris)i-6-;
Ri7 and R18 independently are H or Ci-C6-alkyl, wherein CrC6-alkyl is optionally substituted; Rig and R20 independently are H, CrC6-alkyl, heteroaryl, or H2N-C(=NH)-CH2-, wherein
C-i-Cβ-alkyl and heteroaryl are optionally substituted; and R7 is H or CrCe-alkyl.
[0049] In a preferred embodiment of the compounds according to paragraph [0048], the compounds are of formula H-A:

(H-A) and pharmaceutically acceptable salts and complexes thereof, wherein A is H, halogen, or OH; R2 is H, halo, Ci-C6 alkyl, C2-C6 alkenyl or -S- C1-C6 alkyl, wherein Ci-C6 alkyl and C2-C6 alkenyl, at each occurrence, are optionally substituted; R3 and R4 independently represent H, C1-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -C-i-Ce-alkyl-cycloalkyl, -Ci-Ce-alkyl-heterocyclyl, -Ci-C6-alkyl-aryl, -C1-C6- alkyl-heteroaryl, -Ci-C6alkoxy-aryl or -(CH2)i-6-T, wherein C1-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl and heteroaryl, at each occurrence, are optionally substituted; or R3 and R4 taken together with the nitrogen to which they are attached form a C5-C9 heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted;
T is NH-C(=O)-Ri4, -NH-SO2-Ri5, or -S-(CH2)1-3-R14, R14 is C1-C6 alkyl, aryl or heteroaryl and R15 is aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted; R7 is H or Ci-C6-alkyϊ, Y is S, O, or -N(R16)-,
R16 is H, C1-C6 alkyl, -CrC6-alkyl-aryl, -CrCe-alkyl-heteroaryl, Or -C2-C6 alkenyl-aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted; and
L1 is -(CR17Ri8)i-6-; and
R17 and R18 independently are H or CrC6-alkyl, wherein CrC6-alkyl is optionally substituted.
[0050] In a preferred embodiment of the compounds according to paragraph [0049], A is
OH.
[0051] In a preferred embodiment of the compounds according to paragraph [0049]-[0050],
R7 is H.
[0052] In a preferred embodiment of the compounds according to paragraphs [0049]-[0051],
L1 is -CH2CH2-.
[0053] In a preferred embodiment of the compounds according to paragraphs [0049]-[0052],
R2 is H, halogen, C1-C3 alkyl, -S-C1-C2 alkyl, or C2-C3 alkenyl.
[0054] In a preferred embodiment of the compounds according to paragraphs [0049]-[0053],
R2 is H or halogen.
[0055] In a preferred embodiment of the compounds according to paragraphs [0049]-[0054],
Y is S.
[0056] In a preferred embodiment of the compounds according to paragraphs [0049]-[0055],
R3 and R4 are independently H, C1-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -CrCe-alkyl-cycloalkyl, -CrC6-alkyl-aryl, -CpCe-alkyl-heteroaryl, -CrCealkoxy-aryl or
-(CH2)1-6-T, wherein C1-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl and heteroaryl, at each occurrence, are optionally substituted, or R3 and R4 taken together with the nitrogen to which they are attached form a C5-C9 heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted.
[0057] In a preferred embodiment of the compounds according to paragraph [0056], R3 and
R4 are both H.
[0058] In a preferred embodiment of the compounds according to paragraph [0056], R3 is H and R4 is C1-C6 alkyl optionally substituted with 1, 2, or 3 groups independently selected from
OH, CO2H, NH2, N(C1-C3 alkyl)2, C1-C3 alkoxy, and phenyl.
[0059] In a preferred embodiment of the compounds according to paragraph [0056], both R3 and R4 are C1-C6 alkyl, wherein Ci-C6 alkyl is independently optionally substituted with 1 , 2, or 3 groups independently selected from OH, CO2H, NH2, N(C1-C3 alkyl)2, C1-C3 alkoxy, and phenyl.
[0060] In a preferred embodiment of the compounds according to paragraph [0056], R3 is -
C-ι-C3-all<yl-aryl and R4 is C1-C6 alkyl, wherein C1-C6 alkyl is optionally substituted with 1 , 2, or 3 groups independently selected from OH, CO2H1 NH2, N(C1-C3 alkyl)2, C1-C3 alkoxy, and phenyl.
[0061] In a preferred embodiment of the compounds according to paragraph [0056], R3 is H and R4 is C3-C8-cycloalkyl. Preferably, cycloalkyl is cyclopropyl, cyclohexyl, or cyclooctanyl.
[0062] In a preferred embodiment of the compounds according to paragraph [0056], R3 is H and R4 is aryl, wherein aryl is optionally substituted. Preferably aryl is phenyl, naphthyl, or fluorenyl. Preferably, aryl is unsubstituted or is substituted with 1 , 2, or 3 groups independently selected from OCH3, NH2, NHCH3, NO2, C1-C3 alkyl, halogen, CF3, CN, OH, NH2SO2-, and phenyl.
[0063] In a preferred embodiment of the compounds according to paragraph [0056], R3 is H and R4 is heterocyclyl. Preferably, heterocyclyl is pyrrolidinonyl.
[0064] In a preferred embodiment of the compounds according to paragraph [0056], R3 is H and R4 is -CrC6-alkyl-heteroaryl, wherein heteroaryl and C1-C6 alkyl are optionally substituted.
Preferably, heteroaryl is imidazolyl, indolyl, thiophenyl, pyridinyl, or dihydroindenyl. Preferably, heteroaryl is unsubstituted or is substituted with 1, 2, or 3 groups independently selected from methoxy and phenyl-CrC3-alkoxy-.
[0065] In a preferred embodiment of the compounds according to paragraph [0056], R3 is H and R4 is -CrC6-alkyl-aryl, wherein aryl and C1-C6 alkyl are optionally substituted. Preferably, aryl is phenyl, naphthyl, or fluorenyl. Preferably, aryl is unsubstituted or is substituted with 1 , 2, or 3 groups independently selected from OCH3, NH2, NHCH3, NO2, C1-C3 alkyl, halogen, CF3,
CN, OH, NH2SO2-, or phenyl.
[0066] In a preferred embodiment of the compounds according to paragraph [0065], aryl is phenyl, optionally substituted with 1 , 2, or 3 groups independently selected from OCH3, NH2,
NHCH3, NO2, C1-C3 alkyl, halogen, CF3, CN, OH, NH2SO2-, or phenyl.
[0067] In a preferred embodiment of the compounds according to paragraph [0056], R3 is H and R4 is -(CH2)i-6-T, wherein T is NH-C(=O)-R-|4, -NH-SO2-R15, or -S-(CH2)i-3-Ri4, Ru is C1-C6 alkyl, aryl or heteroaryl and R15 is aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted.
[0068] In a preferred embodiment of the compounds according to paragraph [0067], T is selected from -NH-CO-phenyl, NH-SO2-naphthyl, -S-CH2-phenyl, -NH-CO-methyl, and -NH-CO- furanyl.
[0069] In a preferred embodiment of the compounds according to paragraph [0056], R3 and
R4 taken together with the nitrogen to which they are attached form a C5-C9 heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted.
[0070] In a preferred embodiment of the compounds according to paragraph [0069], R3 and
R4 taken together with the nitrogen to which they are attached form a C4-C8 heterocyclyl ring, wherein said ring is optionally substituted.
[0071] In a preferred embodiment of the compounds according to paragraph [0069]-[0070],
R3 and R4 taken together with the nitrogen to which they are attached form a pyrrolidinyl, azetidinyl, or piperidinyl ring.
[0072] In a preferred embodiment of the compounds according to paragraphs [0049]-[0054],
Y is -N(R16)-.
[0073] In a preferred embodiment of the compounds according to paragraph [0072], R16 is
H.
[0074] In a preferred embodiment of the compounds according to paragraph [0072], R16 is
CrC6-alkyl. Preferably, CrC6-alkyl is unsubstituted or is substituted with NO2.
[0075] In a preferred embodiment of the compounds according to paragraph [0072], R16 is aryl, wherein aryl is optionally substituted.
[0076] In a preferred embodiment of the compounds according to paragraph [0072], R16 is -
CrC6-alkyl-aryl, wherein CrC6-alkyl and aryl are optionally substituted.
[0077] In a preferred embodiment of the compounds according to paragraph [0072], Ri6 is -
C2-CQ alkenyl-aryl, wherein aryl is optionally substituted.
[0078] In a preferred embodiment of the compounds according to paragraphs [0075]-[0077], aryl is phenyl, wherein phenyl is unsubstituted or is substituted with 1 , 2, or 3 groups independently selected from NO2, Ci-C3-alkoxy, CN, or CF3.
[0079] In a preferred embodiment of the compounds according to paragraph [0072], R16 is -
C-pCe-alkyl-heteroaryl, wherein CrC6-alkyl and heteroaryl are optionally substituted.
[0080] In a preferred embodiment of the compounds according to paragraph [0079], heteroaryl is pyridinyl.
[0081] In a preferred embodiment of the compounds according to paragraphs [0072]-[0080],
R3 and R4 are both H.
[0082] In a preferred embodiment of the compounds according to paragraphs [0072]-[0080],
R3 is H, and R4 is CrC6-alkyl.
[0083] In a preferred embodiment of the compounds according to paragraphs [0049]-[0054],
Y is oxygen.
[0084] In a preferred embodiment of the compounds according to paragraph [0048], the compounds are of formula H-B:

(H-B) and pharmaceutically acceptable salts and complexes thereof, wherein m is 0 or 1 ; n is 1 or 2;
L2 is a bond or is -CH2-; R2 is H or halogen; R3 is H, C1-C6 alkyl, or -Ci-C6-alkyl-aryl, wherein C1-C6 alkyl and aryl, at each occurrence, are optionally substituted;
R4 is H or Ci-C6 alkyl, wherein C1-C6 alkyl is optionally substituted; R8 is H, -CO2H, or CO2CH3;
R9 is absent, H or C1-C6 alkyl, wherein C1-C6 alkyl is optionally substituted; W is N or CH; Y is S or O; and
Q is N, CH or O, provided that when Q is O, R9 is absent.
[0085] In a preferred embodiment of the compounds according to paragraph [0084], R3 and R4 are both H.
[0086] In a preferred embodiment of the compounds according to paragraph [0084], R3 is - C-i-Ce-alkyl-aryl and R4 is H, wherein aryl is optionally substituted.
[0087] In a preferred embodiment of the compounds according to paragraph [0086], aryl is phenyl. Preferably, phenyl is unsubstituted or is substituted with phenyl. [0088] In a preferred embodiment of the compounds according to paragraphs [0084]-[0087],

[0089] In a preferred embodiment of the compounds according to paragraph [0048], the compounds are of formula H-C:

H-C and pharmaceutically acceptable salts and complexes thereof, wherein R2 is H, or halogen; R3 and R4 independently represent H, C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, cycloalkyl, aryl or (-Ci-C6-alkyl)-aryl, wherein C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, cycloalkyl, aryl and (-Ci-C6-alkyl)-aryl are each optionally substituted; or R3 and R4 taken together with the nitrogen to which they are attached form a C5-C9- heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted; L3 is a bond or is -(CRi7Ri8)i-e-;
Ri7 and R18 at each occurrence are independently H or Ci-C6-alkyl, wherein C1-
C6-alkyl is optionally substituted; R19 is H, C-i-Ce-alkyl, heteroaryl, or H2N-C(=NH)-CH2-, wherein Ci-C6-alkyl and heteroaryl are optionally substituted.
[0090] In a preferred embodiment of the compounds according to paragraph [0089], L3 is - CHRi7CHR18-, wherein R17 and Ri8 independently are H, or CrC6 alkyl, and where C1-C6 alkyl at each occurrence is optionally substituted. Preferably, C1-C6 alkyl is unsubstituted or is substituted with NH2.
[0091] In a preferred embodiment of the compounds according to paragraph [0089], L3 is - CH2CH2CH2-.
[0092] In a preferred embodiment of the compounds according to paragraphs [0089]-[0091],
[0093] In a preferred embodiment of the compounds according to paragraphs [0089]-[0091], R19 is C1-C6 alkyl, wherein C1-C6 alkyl is optionally substituted. Preferably, C1-C6 alkyl is unsubstituted or is substituted with NH2.
[0094] In a preferred embodiment of the compounds according to paragraphs [0089]-[0091], R19 is H2N-C(=NH)-CH2-.
[0095] In a preferred embodiment of the compounds according to paragraphs [0089]-[0091], R19 is heteroaryl.
[0096] In a preferred embodiment of the compounds according to paragraph [0095], heteroaryl is pyrimidin-2(1 H)-one. Preferably, pyrimidin-2(1H)-one is unsubstituted or is substituted with amine.
[0097] In a preferred embodiment, the compounds listed in Table 1 are excluded from the compounds of paragraphs [0047]-[0096]:



[0098] In a second aspect, the invention provides a composition comprising a compound according to any one of paragraphs [0047]-[0096] or as depicted in any of the examples and tables herein together with a pharmaceutically acceptable excipient, diluent, or carrier. [0099] Compounds of the invention may be formulated by any method well known in the art. [0100] As used herein, the term "pharmaceutically acceptable" means a non-toxic material that is compatible with a biological system in a cell, cell culture, or tissue sample and that does not interfere with the effectiveness of the biological activity of the active ingredient(s). Thus, compositions according to the invention may contain, in addition to the inhibitor, diluents, fillers, salts, buffers, stabilizers, solubilizers, and other materials well known in the art. The preparation of pharmaceutically acceptable formulations is described in, e.g., Remington's Pharmaceutical Sciences, 18th Edition, ed. A. Gennaro, Mack Publishing Co., Easton, PA, 1990. [0101] As used herein, the term pharmaceutically acceptable salts refers to salts that retain the desired biological activity of the above-identified compounds and exhibit minimal or no undesired toxicological effects. Examples of such salts include, but are not limited to acid addition salts formed with inorganic acids (for example, hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric add, and the like), and salts formed with organic acids such as acetic acid, oxalic acid, tartaric acid, succinic acid, malic acid, ascorbic acid, benzoic acid, tannic acid, pamoic acid, alginic acid, polyglutamic acid, naphthalenesulfonic acid, naphthalenedisulfonic acid, and polygalacturonic acid. The compounds can also be administered as pharmaceutically acceptable quaternary salts known by those skilled in the art, which specifically include the quaternary ammonium salt of the formula -NR + Z-, wherein R is hydrogen, alkyl, or benzyl, and Z is a counterion, including chloride, bromide, iodide, -O-alkyl, toluenesulfonate, methylsulfonate, sulfonate, phosphate, or carboxylate (such as benzoate, succinate, acetate, glycolate, maleate, malate, citrate, tartrate, ascorbate, benzoate, cinnamoate, mandeloate, benzyloate, and diphenylacetate). As used herein, the term "salt" is also meant to encompass complexes, such as with an alkaline metal or an alkaline earth metal. [0102] The active compound is included in the pharmaceutically acceptable carrier or diluent in an amount sufficient to deliver to an enzyme-inhibiting effective amount without causing serious toxic effects to the cell.
Methods of Inhibiting DNMT1 and/or DNMT3b2 Enzymes
[0103] The third aspect of the invention provides a method of inhibiting DNMT1 and/or DNMT3b2 enzymes, the method comprising contacting the enzyme(s) with a compound according to any one of paragraphs [0047]-[0097], or as depicted in any of the tables herein, or
with a composition according to paragraph [0098]-[0102]. lήhϊbitiori of DNMT1 ahd/or DNMT3b2" enzymes can be in a cell or a multicellular organism. If in a multicellular organism, the method according to this aspect of the invention comprises administering to the organism an effective DNMT1- and/or DNMT3b2-inhibiting amount of a compound according to any one of paragraphs [0047]-[0097] or as depicted in any of the tables herein, or a composition according to paragraph [0098]-[0102]. Preferably the organism is a mammal, more preferably a primate, most preferably a human.
[0104] Preferred compounds according to the invention include those described in the examples below. Compounds were named using Chemdraw Ultra version 6.0.2 or version 8.0.3, which are available through Cambridgesoft.com, 100 Cambridge Park Drive, Cambridge, MA 02140, Namepro version 5.09, which is available from ACD labs, 90 Adelaide Street West, Toronto, Ontario, M5H, 3V9, Canada, or were derived therefrom.
Synthetic Schemes and Experimental Procedures
[0105] The compounds of the invention can be prepared according to the reaction schemes or the examples illustrated below utilizing methods known to one of ordinary skill in the art. These schemes serve to exemplify some procedures that can be used to make the compounds of the invention. One skilled in the art will recognize that other general synthetic procedures may be used. The compounds of the invention can be prepared from starting components that are commercially available. Any kind of substitutions can be made to the starting components to obtain the compounds of the invention according to procedures that are well known to those skilled in the art.
[0106] Compounds belonging to this class were prepared according to scheme 1a as detailed for example 1.
Scheme 1a

HO2C HO" ΛH N-N

Example 1, Comp 6a, R= CH2CH2-Ph 5a
Example 1
2-AMINO-4-(((2S,3S,4R,5R)-3,4-DIHYDROXY-5-(6-(PHENETHYLAMINO)-9H-PURIN-9-YL)- TETRAHYDROFURAN-Z-YL)METHYLTHIO)BUTANOIC ACID 6a.
Step1: (2R,3S,4R,5R)-2-(hvdroxymethvπ-5-(6-(phenethylaminoV9H-purin-9-vπ-tetrahvdrofuran- 3.4-diol 2a
[0107] A solution of 2a (220 mg, 0.48 mmol, prepared according to the method of E.A. Veliz, et al., Tet. Lett. 2000, 41 , 1695) and phenylethyl amine (182 μL, 175 mg, 1.45 mmol) in DME (5 mL) was stirred for 4 hours at room temperature. The reaction mixture was then diluted in EtOAc (15 mL) and washed with NaCI sat solution (15 mL), dried with Na2SO4, filtered and concentrated in vacuo. The crude product was stirred in saturated methanolic solution of ammonia at room temperature for overnight. The mixture was concentrated in vacuo and purified by flash chromatography using 5% MeOH in DCM as the eluent giving the title compound 2a in 94% yield as white solid (168 mg). MS: calc 371; found 372 (MH+). Step 2: ((3aR.4R,6R,6aR)-2,2-dimethyl-6-(6-(phenethylamino)-9H-purin-9-yl)-tetrahvdrofuror3,4- diπ,31dioxol-4-yl)methanol 3a
[0108] A solution of (2R,3S,4R,5R)-2-(hydroxymethyl)-5-(6-(phenethylamino)-9H-purin-9-yl)- tetrahydrofuran-3,4-diol 2a (168 mg, 0.45 mmol), 2,2-dimethoxypropane (300 μL, 250 mg, 2.4 mmol) and pTsOH (100 mg, 0.52 mmol) in acetone (5 mL) was stirred at room temperature for 3 hrs. The reaction was quenched by the addition of Et3N (200 μL) and concentrated in vacuo. The crude product was purified by flash chromatography using the gradient eluent from 70- 100% EtOAc in hexanes giving the title compound 3a in 81% yield (149 mg) white solid MS: calc 411 ; found 412 (MH+).
Step 3: methyl Σ-ftert-butoxycarbonyla'minoy^-CαSaS^S.eR^aR^.Z-dimethyl-e-rø- (phenethylamino)-9H-purin-9-yl)-tetrahvdrofurof3,4-diπ ,31dioxol-4-vπmethylthio')butanoate 5a [0109] NaH (30 mg 60% mineral oil suspension, 0.72 mmol) was added to a solution of ((3aR,4R,6R,6aR)-2,2-dimethyl-6-(6-(phenethylamino)-9H-purin-9-yl)-tetrahydrofuro[3,4- d][1,3]dioxol-4-yl)methanol 3a (149 mg, 0.36 mmol) in THF (4 ml_) at O0C and stirred for 15 min. pTsCl (76 mg, 0.4 mmol) was then added and the reaction mixture was allowed to stir for 1 hour at O0C. It was diluted in EtOAc (15 ml_) and washed sequentially with water (10 mL) and NaCI sat solution (10 mL). The organic layer was dried with Na2SO4, filtered and concentrated in vacuo. The crude product was dissolved in dry MeOH and treated with a solution prepared by mixing tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate 4 (114 mg, 0.52 mmol) and NaOMe (1.05 mL 0,5M solution, 0.52 mmol) for 15 min at room temperature. The resultant reaction mixture was heated at reflux for 4 hours. It was allowed to cool to room temperature and concentrated in vacuo. The mixture was diluted in EtOAc (10 mL) and washed with water (5 mL) and sat'd NaCI solution (5 mL). The organic layer was dried with Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography using the gradient from 50- 70% EtOAc in hexanes as the eluent giving the title compound 5a in 43% yield (97 mg) white solid, MS: calc 642; found 643 (MH+).
Step 4: 2-amino-4-f((2S,3S,4R.5R)-3,4-dihvdroxy-5-(6-(phenethylaminoV9H-purin-9-yl)- tetrahvdrofuran-2-yl)methylthio)butanoic acid 6a.
[0110] A solution of methyl 2-(tert-butoxycarbonyIamino)-4-(((3aS,4S,6R,6aR)-2,2-dimethyl- 6-(6-(phenethylamino)-9H-purin-9-yl)-tetrahydrofuro[3,4-d][1 ,3]dioxol-4-yl)methylthio)butanoate 5a (97 mg, 0.15 mmol) and KOH (40 mg, 0.71 mmol) in 1:1 mixture of THF and water (3 mL) was stirred at room temperature for 2 hours. The reaction mixture was then concentrated in vacuo and diluted in water (5 mL). It was washed with Et2O (5 mL) and acidified to pH=2 by addition of citric acid. The aqueous phase was then extracted with EtOAc (3x5 mL) and the combined organic layer was dried with Na2SO4, filtered and concentrated in vacuo. [0111] The crude product above was treated with 2:1 mixture of TFA/water (3 mL) containing trace amount of BHT. The reaction mixture was stirred at room temperature for 45 min and then it was treated with 5 drops of water. It was allowed to stir for another 1 hour before it was concentrated in vacuo. The title compound 6a was obtained in 40% yield (29 mg) after purification by flash chromatography using the gradient from 30% MeOH/DCM to 60:30:10 CHCI3/MeOH/NH4OH. 1H NMR (DMSO-d6)) 58.26 (s, 1 H), 8.16 (s, 1 H), 7.79 (br.s, 1H), 7.0-7.2 (m, 5H), 5.80 (d, 1H, J=5.6 Hz), 4.64 (m, 1H), 4.08 (m, 1 H), 3.94 (m, 1 H), 3.61 (br.s, 2H)1 2H
assumed under H2θ"at 3.33, 2.83 (t, 2R;J=7.5 HZ)"; 2J1 (mTi H), 2.54 (mr2H),' 1.9OχmT1H)7
1.73 (m, 1 H). MS: calc 488; found 489 (MH+).
[0112] Examples 2-9, compounds 5b-5i, Table 2, were prepared in a manner similar to example 1 , scheme 1a, utilizing the appropriate amine.
[0113] Alternatively, compounds belonging to this class were prepared as illustrated in scheme 1b, example 10, compound 12a.
Scheme 1b

Example 10
2-amino-4-(((2S,3S,4R,5R)-3,4-dihydroxy-5-(6-(3-morpholinopropylamino)-9H-purin-9-yI)- tetrahydrofuran-2-yl)methylthio)butanoic acid 12a.
Step 1 : methyl 4-(((3aS,4S,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2.2-dimethyl-tetrahvdrofurol'3.4- d1f1 ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 8 [0114] NaH (310 mg 60% mineral oil suspension, 7.74 mmol) was added to a solution of ((3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyl-tetrahydrofuro[3,4-d][1 ,3]dioxol-4- yOmethanol 7 (1.19 g, 3.87 mmol) in THF (4 ml_) at O0C and stirred for 15 min. pTsCI (810 mg, 4.25 mmol) was then added and the reaction mixture was allowed to stir for 1 hour at O0C. It was diluted in EtOAc (15 mL) and washed sequentially with water (10 ml_) and NaCI sat solution (10 mL). The organic layer was dried with Na2SO4, filtered and concentrated in vacuo, dissolved in MeOH and added to a pre-formed solution of thiolactone 4 (1.26 g, 5.81 mmol, 1.5 equiv.) in dry MeOH (5 mL) was treated with 0.5 M solution of NaOMe (11.6 mL, 5.81 mmol, 1.5 equiv.) at
room temperature and stirred for 15 min- The mixture was refluxed for 2 hours, and then it was cooled down and concentrated. The residue was diluted in EtOAc (20 ml_), washed with water (20 mL) and sat'd NaCI solution (20 mL), dried with Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography using 5% MeOH in DCM as the eluent to afford the title compound 8 in 59% yield as white solid (1.27 g) MS: calc 538; found 539 (MH+)
Step 2: methyl 4-(((3aS,4S.6R.6aR)-6-(6-(4H-t2,4-triazol-4-yl)-9H-purin-9-yl)-2,2-dimethyl- tetrahydrofuro[3,4-diπ ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 1 Q [0115] The dihydrochloride salt 9 (398 mg, 1.85, 5 equiv.) was added to a solution of 8 (134 mg, 0.37 mmol) in dry pyridine (4 mL) and the reaction mixture was refluxed for 16h. It was then cooled to room temperature and concentrated in under reduced pressure. Crude material was purified by flash chromatography using EtOAc as the eluent to afford the title compound 10 in 55% yield (80 mg). MS: calc 590; found 591 (MH+)
Step 3: methyl 2-(tert-butoxycarbonylamino)-4-(((3aS,4S.6R,6aRV2,2-dimethyl-6-(6-(3- morpholinopropylamino)-9H-purin-9-yl)-tetrahvdrofuror3,4-d1[1 ,31dioxol-4- yl)methylthio)butanoate 11a
[0116] A solution of 10 (45 mg, 0.085 mmol, 1 equiv.) and 3-morpholinopropan-1 -amine (37 μL, 36 mg, 0.25 mmol, 3 equiv.) in dry DMF (7 mL) was stirred at room temperature for 2 days.
The reaction mixture was concentrated, diluted in EtOAc (20 mL) and washed with sat'd NaCI solution (10 mL). Organic phase was dried with Na2SO4, filtered and concentrated. The crude material was purified by flash chromatography using EtOAc as the eluent to give the title compound 11a in 30% yield (15 mg). MS: calc 665; found 666 (MH+)
Step 4: 2-amino-4-(((2S.3S.4R,5R)-3,4-dihvdroxy-5-(6-(3-morpholinopropylamino)-9H-purin-9- yl)-tetrahvdrofuran-2-yl)methylthio)butanoic acid 12a.
[0117] The deprotection was carried out similar to example 1.scheme 1 a, step 4 described earlier to give the title compound as white solid. 1H NMR (D2O) 58.24 (s, 1 H), 8.17 (s, 1 H), 6.01
(d, 1 H, J=5.3 Hz), 4.38 (m, 1H), 4.29 (m, 1H), 3.78 (m, 6H), 3.58 (m, 2H), 2.9-3.1 (m, 2H)1 2.5-
2.8 (m, 8H), 1.8-2.2 (m, 4H)
MS: calc 511 ; found 512 (MH+).
[0118] Examples 11-19, compounds 12b-12j, Table 2, were prepared similar to example 10, scheme 1b, by reacting compound 10 with the appropriate commercial amine.
Table 2




Scheme 1c
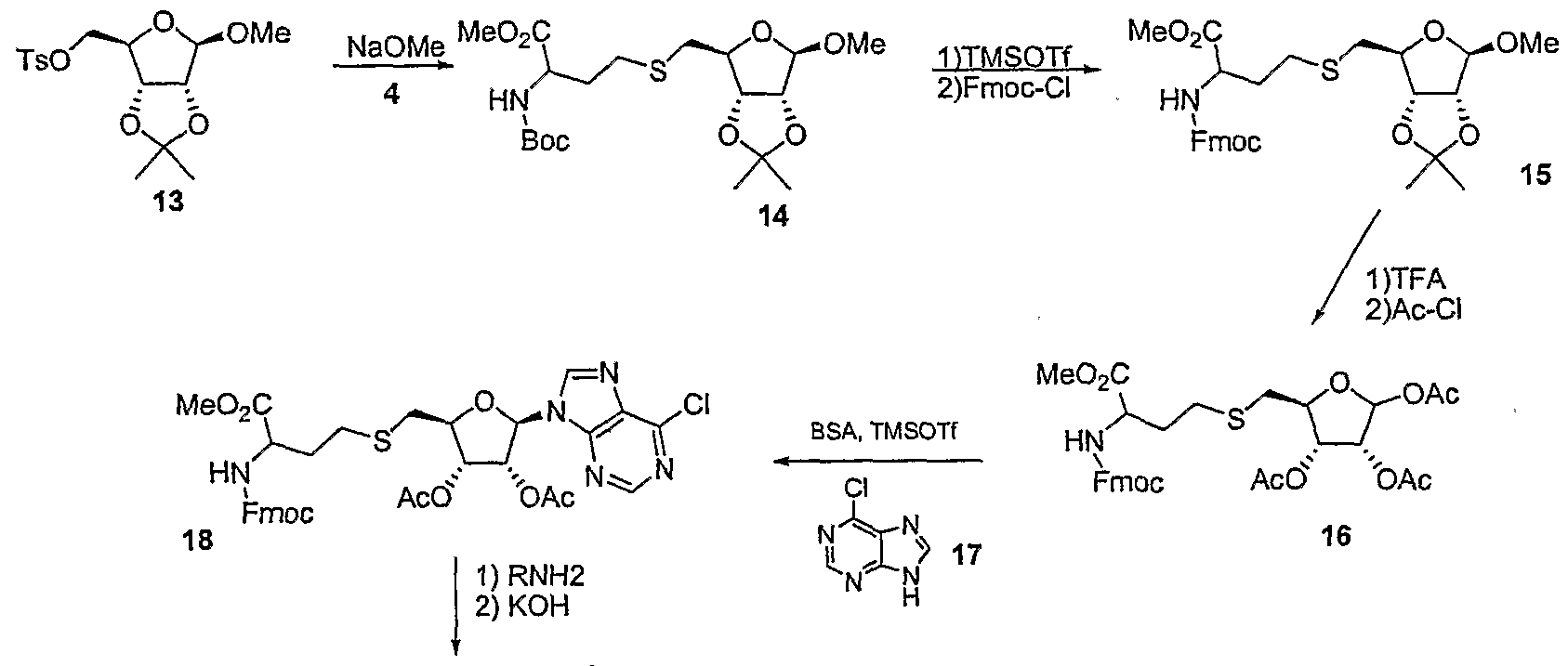

Example 20
2-amino-4-(((2S,3S,4R,5R)-5-(6-(2-(dimethylamino)ethylamino)-9H-purin-9-yl)-3,4- dihydroxy-tetrahydrofuran-2-yi)methylthio)butanoic acid 19a.
Step 1 : methyl 2-(tert-butoxycarbonylamino)-4-(((3aS,4S.6R,6aR)-6-methoxy-2.2-dimethyl- tetrahvdrofurof3,4-diπ ,31dioxol-4-yl)methylthio)butanoate 14
[0119] A solution of /V-terf-butoxycarbonyl-DL-homocysteine thiolactone 4 (9.1 g, 41.9 mmol) in NaOMe (0.5 M in methanol, 84 ml, 42 mmol) was stirred under a nitrogen atmosphere
for 10 minutes. Methyl 2>3-0-isopropylidene-5-0-p-tolylsulfonyl-β-D-ribofuranoside 13 (10.0 g, 27.9 mmol) was then added and the mixture was reflux for 3 hours. After the reaction had cooled to room temperature, the solvent was evaporated and the crude material was added to ethyl acetate (200 ml). The ethyl acetate solution was washed with saturated NaHCO3 (2 x 100 ml), 5% HCI (2 x 100 ml), and brine (100 ml). The ethyl acetate solution was then dried with MgSO4, filtered and evaporated to give the crude product. The title compound 14 was obtained in 83% yield (10.1 g) after purification by flash chromatography using 25% ethyl acetate and 75% hexanes. MS:calc 435.53; found 458 (M+Na+)
Step 2: methyl 2-(((9H-fluoren-9-vnmethoxy^carbonylaminoV4-(((3aS,4S,6R,6aRV6- methoxy-2,2-dimethyl-tetrahvdrofurof3,4-cnπ ,31dioxol-4-yl)methylthio)butanoate 15 [0120] A solution of 14 (11.0 g, 25.3 mmol) in dry 1 ,2-dichloroethane (50 ml) was placed under a nitrogen atmosphere and cooled to O0C. TMSOTf (4.6 ml, 25.4 mmol) was then added and the reaction was stirred for 1.5 hours at O0C. The reaction was then quenched with saturated NaHCO3 (IOO ml) and 9-fluorenylmethyl chloroformate (6.5 g, 25.2 mmol) was added slowly as a gas evolved. The bi-phasic mixture was stirred vigoursouly for 5 hours and then diluted with CH2CI2 (50 ml). The organic layer was separated, washed with saturated NaHCO3 (2 x 50 ml), 10% HCI (2 x 50 ml), and brine (50 ml). The organic layer was then dried with MgSO4, filtered and evaporated and crude material was purified by flash chromatography using 30% ethyl acetate and 70% hexanes to give 15 in 85.8% yield (12.1 g) as a clear oil. MS: calc 557.66; found 580 (M+Na+)
Step 3: (3R,4S,5S)-5-((3-(((9H-fluoren-9-yl)methoxy)carbonylamino)-4-methoxy-4- oxobutylthio)methyl)-tetrahvdrofuran-2,3,4-triyl triacetate 16
[0121] A solution of 15 (10 g, 17.9 mmol) in trifluoroacetic acid (20 ml) and H2O (20 ml) was stirred at room temperature for 5 hours. H2O (100 ml) was then added slowly and a white solid precipitated from the solution. After this mixture had stirred for 0.5 hours, the precipitate was filtered and dried under high vacuum. The precipitate was added to a solution of dry CH2CI2 (100 ml) and dry pyridine (20 ml) under a nitrogen atmosphere and cooled to O0C. Acetyl chloride (8.9 ml, 0.125 mol) was added slowly to this solution via syringe over a period of one hour and the reaction mixture was stirred overnight as it warmed to room temperature. The reaction was then quenched slowly with saturated NaHCO3. The quenched reaction was stirred for an additional 0.5 hours and then the organic phase was separated, washed with saturated NaHCO3 (2 x 100 ml) and brine (100 ml), dried with MgSO4, filtered and evaporated and the crude material was purified by flash chromatography using 30% ethyl acetate and 70% hexanes
to give the title compound 16 in 77.2% yield~(8J'g) as a mixture of ά ana1 β isomers (1:1.7 molar ratio respectively by 1H NMR). MS: calc 629.67; found 652 (M+Na+) Step 4: (2S.3S,4R,5R)-2-((3-(((9H-fluoren-9-yl)methoxy)carbonylamino)-4-methoxy-4- oxobutylthio)methyl)-5-(6-chloro-9H-purin-9-yl)-tetrahvdrofuran-3,4-diyl diacetate 18 [0122] A solution of 6-chloropurine 17 (368 mg, 2.38 mmol) and N1O- bistrimethylsilylacetomide (588 μl, 2.38 mmol) in dry acetonitrile (10 ml) was stirred for ΛA hour at room temperature under an N2 atmosphere. 16 (1.0 g, 1.59 mmol), in dry acetonitrile (10 ml), was then added followed by TMSOTf (287 μl, 1.59 mmol) and the mixture was refluxed for Vz hour. After the reaction had cooled to room temperature, it was poured into saturated NaHCO3 (100ml) and extracted into ethyl acetate. The organic phase was then washed with brine (50ml), dried with Na2SO4, filtered and evaporated and the crude material was purified by flash chromatography using 95% CH2CI2 and 5% acetone to give the title compound 18 in 78.2% yield (900 mg).
Step 5: 2-amino-4-(((2S,3S,4R.5R)-5-(6-(2-(dimethylamino)ethylaminoV9H-purin-9-vπ-3,4- dihvdroxy-tetrahvdrofuran-2-yl)methylthio)butanoic acid 19a.
[0123] A solution of 18 (75 mg, 0.104 mmol) and Λ/,Λ/-dimethylethylenediamine (36.5 mg, 0.414mmol) in 3 ml of ethanol was refluxed for 2 days. The solvent was then evaporated and THF (0.5 ml) was added followed by 1 N KOH 1 (.5 ml). After stirring for 3 hours, the solvent was evaporated and the product was purified by flash chromatography using 60% CHCI3, 30% methanol and 10% NH4OH. The title compound 19a was obtained in 31.7% yield after trituration with CH3CN (15 mg). 1H NMR (DMSO-Cf6ZD2O) δ ppm: 8.32 (d, 1 H, J=2.4 Hz), 8.20 (s, 1 H), 5.86 (d, 1H, J=5.2 Hz), 4.72 (m, 1 H), 4.12-4.14 (m, 1 H), 4.00 (m, 1H), 3.59 (bs, 2H), 3.16-3.23 (m, 1H), 2.75-2.92 (m, 2H), 2.54-2.61 (m, 4H), 2.28 (s, 6H), 1.91 (m, 1 H)11.76 (m, 1 H). MS calc 455, found 456 (MH+)
Example 21
2-amino-4-(((2S,3S,4R,5R)-3,4-dihydroxy-5-(6-(2-(2-(methylamino)benzarnido)ethylamino)- 9H-purin-9-yl)-tetrahydrofuran-2-yl)methyIthio)butanoic acid 19b. [0124] The title compound was prepared similar to example 20, in step 5 replacing N,N'- dimethylethylenediamine with Λ/-(2-aminoethyl)-2~(methylamino)benzamide (prepared according to the method of Fassa, A. A.; Refat, H. M.; Zaki, M. E.;A.; Monir, E. ; Synth. Commun. 2001 , 31, 3537-3545). Compound 19b was obtained after flash chromatography using 60% CHCI3, 30% methanol and 10% NH4OH in 56.6 % yield (33 mg). 1H NMR (DMSO- Cl6ID2O) δ ppm:1.80 (m, 1H), 1.97 (m, 1 H), 2.59 (t, 2H, J=7.6Hz), 2.72-2.93 (m, 2H), 2.72 (s, 3H), 3.28 (m, 1 H), 2H assumed under D2O, 3.62 (m, 2H), 3.99 (m, 1H), 4.10-4.16 (m, 1H), 4.71
(dd, 1H,~J=5:2; 5.2Hz)75786 (an H; J=5.6HZ); 6.50 (nrr, 1H), 6:57 (d, 1HτJ=7.6Hz), 7.23 (m, 1 H), 7.46 (d, 1 H1 J=7.6Hz), 8.21 (s, 1H), 8.33 (s, 1 H), 8.38 (bs,1H). MS calc 560, found 561 (MH+).
Example 22
2-amino-4-(((2S,3S,4R,5R)-5-(6-(2-(2-aminobenzamido)ethylamino)-9H-purin-9-yl)-3,4- dihydroxy-tetrahydrofuran-2-yl)methylthio)butanoic acid 19c.
[0125] The title compound was prepared similar to example 20, in step 5 replacing N,N'- dimethylethylenediamine with 2-amino-N-(2-aminoethyl)-benzamide. Compound 19c was obtained in 67.6% yield (37 mg) after flash chromatography using 60% CHCI3, 30% methanol and 10% NH4OH followed by recrystallisation from water and acetonitrile. 1H NMR (DMSO- CVD2O) δ ppm: 8.32 (s, 1H), 8.30 (bs, 1H), 8.21 (s, 1 H), 7.41 (d, 1H, J=7.2Hz), 7.09 (m, 1H), 6.64 (dd, 1 H, J=1.2, 8.4Hz), 6.46 (m, 1H), 5.86 (d, 1H, J=6.0Hz), 4.71 (dd, 1 H, J=5.2, 5.6Hz), 4.10-4.17 (m, 1H), 4.00 (m, 1H), 3.62 (m, 2H), 3.45 (m, 2H), 3.27 (m, 1H), 2.75-2.95 (m, 2H), 2.59 (t, 2H, J=7.6Hz), 1.97 (m, 1H), 1.81(m, 1 H). MS 546, found 547 (MH+).
Example 23
2-amino-4-(((2S,3S,4R,5R)-5-(6-(2-(5-(dimethylamino)naphthalene-1- suIfonamido)ethylamino)-9H-purin-9-yl)-3,4-dihydroxy-tetrahydrofuran-2- yl)methylthio)butanoic acid 19d.
Step 1 : Tert-butyl 2-(1-(dimethylamino)naphthalene-5-su]fonamido^ethylcarbamate 20
[0126] A solution of tert-butyl-Λ/-(2-aminoethyl)carbamate (1.0 g, 6.24 mmol) and dansyl chloride (1.6 g, 5.93 mmol) was added to a mixture of acetonitrile (20 ml) and saturated NaHCO3 (IO ml) according to the method of Corrie, John E. T. (J. Chem. Soc. Perkin. Trans. I, 1994, 20, 2975-2982). After the reaction was stirred for 30 minutes, ethyl acetate (100ml) was then added and the organic phase was washed with saturated NaHCO3 (2 x 50 ml) and brine (50 ml). The organic phase was then dried with MgSO4, filtered and evaporated to give a crude oil which was purified by flash chromatography using 30% ethyl acetate and 70% hexanes to give the title compound 20 in 84.9% yield (1.98g). Step 2: 5-dimethylamino-naphthalene-1 -sulfonic acid (2-amino-ethvO-amide 21
[0127] The protected amine 20 (1.0g, 2.54 mmol) was added to a solution of CH2CI2 (5 ml) and TFA (3 ml). The solution was stirred for 1 hour and then the solvent was evaporated. The crude material was then dissolved in water (30 ml) and neutralized with NaHCO3. The aqueous phase was then extracted with ethyl acetate (4 x 50 ml) and the organic phases where combined, washed with brine (50 ml), dried with Na2SO4, filtered and evaporated. Precipitation
from "methylene chloride using hexanes gave" the title" compound"21 in 47% yield (350 mg)~as~ - white solid.
Step 3: 2-amino-4-f((2S,3S,4R,5R)-5-(6-(2-(5-(dimethylamino)naphthalene-1- sulfonamido)ethylamino)-9H-purin-9-vπ-3.4-dihvdroxy-tetrahvdrofuran-2- yl)methylthio)butanoic acid 19d.
[0128] Amine 21 (91.5 mg, 0.312 mmol) was used in place of N1N'- dimethylethylenediamine and reacted with compound 18 (75 mg, 0.104 mmol) as described in example 20, step 5. After the reaction was refluxed overnight, 1.0 ml of ethanol was added before the reaction was refluxed for an additional 24 hours. The title compound 19d was obtained in 46.5 % yield (32 mg ) after flash chromatography using 60% CHCI3, 30% methanol and 10% NH4OH followed by recrystallization from CH3CN. 1H NMR (DMSO-Qf6ZD2O) δ ppm: 8.37 (d, 1 H, J=8.4Hz), 8.30 (d, 1H, J=1.6Hz), 8.18 (d, 1H, J=8.8Hz), 8.11 (s, 1 H), 8.05 (m, 1 H), 7.53 (m, 1H), 7.46 (m, 1H), 7.15 (d, 1H, J=7.2Hz), 5.85 (d, 1H, J=6.0Hz), 4.70 (dd, 1H, J=5.2, 5.6Hz), 4.10-4.16 (m, 1H), 4.00 (m, 1H), 2H assumed under D2O, 3.26 (m, 1 H), 2.87-2.98 (m, 4H), 2.78 (s, 6H), 2.59 (t, 2H, J=7.6Hz), 1.98 (m, 1H),1.81 (m, 1H). MS calc 660, found 661 (MH+).
[0129] Compounds belonging to this class, were prepared as illustrated in scheme 2, and detailed for example 24 (method A) and example 25 (method B) starting from the common intermediate 23
Scheme 2
BSA, TMSOTf


Example 24, comp 24a R1=H,
Example 25, comp 24b R1=H,

Synthesis of the common intermediate (2S,3S,4R,5R)-2-((3-(((9H-fluoren-9- ylJmethoxyJcarbonylaminoJ^-methoxy^-oxobutylthioJmethyO-δ^.e-dichloro-ΘH-purin-θ- yl)-tetrahydrofuran-3,4-diyl diacetate 23
[0130] " A solution of 2,6-dichloropύrihe 22 (164 mg", 0.868 mmol)~and N,O- bistrimethylsilylacetomide (392 μl, 1.59 mmol) in dry CH3CN (10 ml) was stirred under a nitrogen atmosphere for 0.5 hours. 15 (500 mg, 0.79 mmol), in dry CH3CN (3 ml), and TMSOTf (144 μl, 0.794 mmol) were then added and the reaction was refluxed for 0.5 hours. After the reaction had cooled to room temperature, it was poured into 80 ml of saturated NaHCO3 and extraced with ethyl acetate (2 x 80 ml). The organic layers were combined, washed with saturated NaHCO3 (80 ml) and brine (80 ml_), dried with Na2SO4, filtrated and evaporated. Purification of the crude product by flash chromatography using 95% CH2CI2 and 5% acetone gave the title compound 23 in 81.8% yield (490 mg). 1H NMR (DMSO-d6) δ ppm: 8.93 (d, 1H, J=6.0Hz), 7.86 (d, 2H, J = 7.6), 7.76 (d, 1H, J=8.0 Hz), 7.67 (d, 2H, J = 7.2 Hz), 7.39 (dd, 2H, J=7.2, 7.6 Hz), 7.30 (dd, 2H, J = 7.2, 7.6 Hz), 6.27 (m, 1H), 5.94 (m, 1H), 5.55 (m, 1H), 4.35 (m, 1H), 4.29 (m, 2H), 4.21 (m, 1 H), 4.10 (m, 1 H), 3.59 (s, 3H), 3.03 (m, 2H), 2.56 (m, 2H), 2.12 (s, 3H), 2.02 (s, 3H),1.89 (m, 2H). MS calc 757.14 (100%), found 780 (M+Na+).
Example 24
4-(((2S,3S,4R,5R)-5-(6-(3-(1H-imidazol-1-yl)propylamino)-2-chloro-9H-purin-9-yl)- 3,4-dihydroxy-tetrahydrofuran-2-yl)methylthio)-2-aminobutanoic acid 24a.
Method A
[0131] A solution of 23 (100 mg, 0.132 mmol) and 3-(1H-imidazol-1-yl)propan-1 -amine (50 mg, 0.4 mmoi) in dry 1,2-dichloroethane (3 ml) was stirred overnight at room temperature. The solvent was evaporated and THF (0.5 ml) was added followed by 1.0 M KOH (1.3 ml, 1.3 mmol). The solution was stirred for 3 to 5 hours and the solvent was removed to give the crude product which was purified by flash chromatograp using 70% CHCI3, 25% methanol and 5% NH4OH followed by recrystallized from a mixture of water and acetonitrile giving the title compound 24a in 75% yield as white solid (39 mg). 1H NMR (DMSO-Qf6ZD2O) δ ppm: 8.40 (s, 1H), 7.69 (s, 1 H), 7.23 (s, 1 H), 6.88 (s, 1H), 5.82 (d, 1 H, J = 5.6 Hz), 4.64 (m, 1H), 4.12 (m, 1H), 4.03 (m, 3H), 3.38 (m, 2H) and 3.80, 3.27 (m, 1 H), 2.87 (m, 1H), 2.79 (m, 1 H), 2.58 (m, 2H), 2.00 (m, 3H), 1.81 (m, 1H). MS calc 526, found 527 (MH+).
Example 25
4-(((2S,3S,4RJ5R)-5-(6-(9H-fluoren-9-ylamino)-2-chloro-9H-purϊn-9-yl)-3s4-dihydroxy- tetrahydrofuran-2-yl)methylthio)-2-aminobutanoic acid 24b
Method B
[0132] A solution of 23 (75 mg, 0.0989 mmol), triethylamine (83 μl, 6 eq) and 9- aminofluorene hydrochloride (64.6 mg, 0.3 mmol) in a mixture of 1 ,2-dichloroethane (0.75 ml) and ethanol (0.75 ml) was heated to 5O0C -600C overnight. The reaction was then cooled to
room temperature and the solvent was evaporated. THF (0.5 ml) was added followed by 1.0 M KOH (1.5 ml). The solution was stirred for 3 hours and the solvent was removed to give the crude product which was purified by flash chromatography using 60% CHCI3, 30% methanol and 10% NH4OH followed by trituration with CH3CN to give the title compound 24b in 57.7% yield(25 mg). 1H NMR (DMSO-CZ6ZD2O, 400 MHz): δ ppm: 8.38 (s, 1 H), 7.84 (d, 2H, J=7.2Hz), 7.25-7.48 (m, 4H), 7.25-7.31 (m, 2H), 6.50 (m, 1H), 5.84 (d, 1H, J=6.0 Hz), 4.65 (m, 1H), 4.10- 4.16 (m, 1H), 4.05 (m, 1H), 3.22-3.37 (m, 1 H), 2.77-2.96 (m, 2H), 2.62 (t, 2H, J=7.6 Hz), 1.99 (m, 1 H), 1.79 (m, 1 H). MS calc 582, found 583 (MH+).
[0133] Examples 26-42, compounds 24c-24s, Table 3, were prepared according to scheme 2, utilizing either method A or method B.
[0134] Alternatively, compounds belonging to this class were prepared according to scheme 2a, as detailed for example 43.
Scheme 2a
CI
 NaoMe
NaoMe


Example 43, comp 30a, 29 28

Example 43
Z-Amino-^^ZS.SS^R.SRJ-S^β^^biphenyM-yOethylaminoJ-a-chloro-ΘH-purin-θ- yl)-3,4-dihydroxy-tetrahydrofuran-2-yl)methylthio)butanoic acid 30a
Step 1 : (2R,3R,4S,5R)-2-(6-(2-(biphenyl-4-yl)ethylamino)-2-chloro-9H-purin-9-yl)-5- (hydroxymethyl)-tetrahydrofuran-3,4-diol 26
[0135] A solution of dichloride 25 (863 mg, 1.36 mmol, prepared according to the method of Andrzejewska, Mariola; Kaminski, Jaroslaw; Kazimierczuk, Zygmunt Nucleosides Nucleotides 21 , 1 , 2002, 73-78) and biphenylethyl amine (803 mg, 4.08 mmol) in DME (8 mL) was stirred for 4 hours at room temperature. The reaction mixture was then diluted in EtOAc (15 mL) and washed with brine (15 mL), dried with Na2SO4, filtered and concentrated in vacuo. The crude
product was stirred in saturated methanόlic sόlution of ammonia at 7O0C for overnight. The mixture was concentrated in vacuo and purified by flash chromatography using 10% MeOH in DCM to give the title compound in 67% yield (412 mg). MS: calc 481; found 482 (MH+)
Step 2: ((3aR,4R,6R.6aRV6-(6-(2-(biphenyl-4-vnethylamino)-2-chloro-9H-purin-9-yl)-2,2- dimethyl-tetrahvdrofuror3,4-d1H ,31dioxol-4-v0methanol 27
[0136] A solution of 26 (412 mg, 0.85 mmol), 2,2-dimethoxypropane (530 μl_, 446 mg, 4.28 mmol) and pTsOH (180 mg, 0.94 mmol) in acetone (5 ml_) was stirred at room temperature for 2 hrs. The reaction was quenched by the addition of Et3N (200 μL) and concentrated in vacuo. The crude product was purified by flash chromatography using the gradient eluent from 75% to 100% EtOAc in hexanes giving the title compound 27 in 66% yield (292 mg). MS: calc 521; found 522 (MH+)
Step 3: ((3aR.4R,6R.6aR)-6-(6-(2-(biphenyl-4-yl)ethylamino)-2-chloro-9H-purin-9-yl)-2,2- dimethyl-tetrahvdrofurof3,4-diri ,31dioxol-4-yl)methyl 4-methylbenzenesulfonate 28
[0137] NaH (13 mg 60% mineral oil suspension, 0.32 mmol) was added to a solution of 27
(83 mg, 0.16 mmol) in THF (2 mL) at O0C and stirred for 15 min. pTsCI (33 mg, 0.17 mmol) was then added and the reaction mixture was allowed to stir for 1 hour at O0C. It was diluted in
EtOAc (5 mL) and washed sequentially with water (5 mL) and brine (5 mL). The organic layer was dried with Na2SO4, filtered and concentrated in vacuo giving the title compound 28 quantitatively (107 mg)
MS: calc 675; found 676 (MH+)
Step 4: Methyl 4-(((3aS.4S.6R,6aR)-6-(6-(2-('biphenyl-4-vnethylamino)-2-chloro-9H-purin-9-vn- 2.2-dimethyl-tetrahvdrofurof3,4-dlf1.31dioxol-4-vnmethylthio)-2-(tert- butoxycarbonylamino)butanoate 29
[0138] Crude 28 (107 mg, 0.16 mmol) was dissolved in dry MeOH (2 mL) and treated with a solution prepared by mixing Boc thiolactone 4 (51 mg, 0.24 mmol) and NaOMe (480 μL 0,5M solution, 0.24 mmol) for 15 min at room temperature. The resultant reaction mixture was heated at reflux for 4 hours. It was allowed to cool to room temperature and concentrated in vacuo. The residue was diluted in EtOAc (5 mL) and washed with water (5 mL) and brine (5 mL). The organic layer was dried with Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography eluting with 40% -50% EtOAc in hexanes to give the title compound 29 in 94% yield (87 mg). MS: calc 752; found 753 (MH+)
Step 5: 2-Amino-4-(((28,3S,4R.5RV5-(6-(2-(biphenyl-4-vnethylamino)-2-chloro-9H- - Durin-9-ylV3,4-dihvdroxy-tetrahvdrofuran-2-yl)methylthio)butanoic acid 30a
[0139] A solution of 29 (87 mg, 0.11 mmol) and KOH (40 mg, 0.71 mmol) in 1:1 mixture of THF and water (3 ml_) was stirred at room temperature for 2 hours. The reaction mixture was then concentrated in vacuo and diluted in water (5 mL). It was washed with Et2O (5 ml_) and acidified to pH=2 by addition of citric acid. The aqueous phase was then extracted with EtOAc (3x5 mL) and the combined organic layer was dried with Na2SO4, filtered and concentrated in vacuo. The obtained crude product was treated with 2:1 mixture of TFA/water (3 mL) containing trace amount of BHT. The reaction mixture was stirred at room temperature for 45 min and then it was treated with 5 drops of water. It was allowed to stir for another 1 hour before it was concentrated in vacuo. The crude product was purified by flash chromatography using the gradient from 30% MeOH/DCM to 60:30:10 CHCI3ZIvIeOHZNH4OH to give the title compound 30a in 32% yield (22 mg). 1H NMR (DMSO-d6) δ ppιηD8.45 (m, 1H), 8.35 (s, 1 H), 7.55 (m, 4H), 7.2-7.4 (m, 5H), 5.79 (d, 1H, J=5.9 Hz)1 4.61 (m, 1H), 3.99-4.11 (m, 3H), 3.65 (m, 2H), 2.93 (m, 3H), 2.80 (m, 1H), 2.60 (m, 1 H), 2.47 (m, 1H), 1.97 (m, 1H), 1.80 (m, 1H) MS: calc 598; found 599 (MH+)
[0140] Compounds belonging to this series, presented in Table 3, were prepared either following Scheme 2, examples 26-42, compounds 24c-s or scheme 2a, example 44-49, compounds 30b-g.
Table 3






Scheme 3

TFA

Example 50, comp 33a, R= -\-\ />— CN
Example 50
2-amino-4-(((2S,3S,4R,5R)-5-(2-chloro-6-(2-(4-cyanobenzamido)ethylamino)-9H-purin-9- yl)-3,4-dihydroxy-tetrahydrofuran-2-yl)methylthio)butanoJc acid 33a
Step 1 : (2S.3S.4R.5RV2-((3-(((9H-fluoren-9-yl)methoxy)carbonylamino)-4-methoxy-4- oxobutylth)θ)methyl)-5-(6-(2-(tert-butoxycarbonylamino)ethylamino)-2-chloro-9H-purin-9-vπ- tetrahydrofuran-3,4-diyl diacetate 31
[0141] A solution of 23 (500mg, 0.659 mmol), /V-boc-ethylenediamine (211 mg, 1.32 mmol) and triethylamine (275 ml, 1.98 mmol) in ethanol (5 ml) and 1 ,2-dichloroethane (5 ml) was stirred overnight at room temperature. The reaction was then diluted with CH2CI2 (50 ml) and
washed with 5% HCI(aq) (2 x 25 ml), saturated NaHCO3 (25 ml) and brine (25'ml). The organic phase was then dried with Na2SO4, filtered and evaporated to give the crude product. The crude product was purified by flash chromatography using 80% CH2CI2 and 20% acetone to give the title compound 31 in 74.8 % yield (435 mg) as white solid. MS: calc. 882.4, found 882.3 (MH+).
Step 2: (2S,3S.4R,5RV2-((3-(((9H-fluoren-9-yl)methoxy)carbonylamino)-4-methoxy-4- oxobutylthio)methyl)-5-(6-(2-arninoethylamino)-2-chloro-9H-pur'm-9-vπ-tetrahvdrofuran- 3,4-diyl diacetate 32
[0142] To a solution of 31 (300 mg, 0.340 mmol) in 1.0 ml CH2CI2 was added trifluoroacetic acid (1.0 ml). The reaction was stirred for 1 hour and the solvent was evaporated. The residue was re-dissolved in methylene chloride and the solvent was removed (twice) to give the title compound 32 in a quantitative yield as the di-trifluoroacetic acid salt.
Step 3: 2-amino-4-(((2S,3S.4R.5RV5-(2-chloro-6-(2-(r4-cvanobenzamido)ethylamino)-9H-Durin- 9-yl)-3,4-dihvdroxy-tetrahvdrofuran-2-yl)methylthio)butanoic acid 33a [0143] A solution of 32 (76mg, 0.075 mmol), triethylamine (62.6 μl, 0.45 mmol) and 4- cyanobenzoyl chloride (18.5 mg, 0.112 mmol) in 0.75 -1.0 ml THF was stirred for 16 hours at room temperature. 1.5 ml of 1 N KOH was then added and the reaction was stirred for 3 hours. The reaction was then quenched with formic acid (100μl) and the solvent was evaporated. The crude product was purified by flash chromatography using 65% CHCI3, 30% methanol and 5% NH4OH then triturated with CH3CN. The title compound 33a was obtained in 15% yield as white solid (10 mg). 1H NMR (DMSO-GVD2O): δ 1.83 (m, 1H), 1.97 (m, 1 H), 2.60 (t, 2H, J=7.6Hz), 2.77-289 (m, 2H), 3.27 (m, 1H), 2H presumed under D2O, 3.65 (m, 2H), 4.01 (m, 1H), 4.08-4.13 (m, 1 H), 4.62 (dd, 1 H, J=4.8, 5.6Hz), 5.79 (d, 1H, J=5.6Hz), 7.89-7.95 (m, 4H), 8.35 (s, 1 H). MS: calc 590, found 591 ((MH+).
[0144] Examples 51-61 compounds 33b-33l, Table 4, were prepared from compound 32 and the appropriate acid chloride, scheme 3, as described for example 50, step 3.
Table 4





Example 66, comp 36e, R1= H, R2= i~\
NH2
General procedure for the synthesis of the BOC-protected amino alcohols 35a-e
[0145] A solution of the appropriate amino alcohol (6.0 mmol), di-terf-butyl dicarbonate (1.96 g, 9.0 mmol) and NaHCO3 (2.5 g, 30.0 mmol) in 10 ml of dioxane and 10 ml H2O was stirred overnight at room temperature. Ethyl acetate (50 ml) was then added and the organic phase was washed with saturated NaHCO3 (2 x 50 ml), 10% HCI (2 x 50 ml), and brine (50 ml). The organic phase was then dried with MgSO4, filtered and evaporated. The product was purified as indicated.
Di-tert-butyl 2-hydroxypropane-1,3-diyldicarbamate 35a
[0146] The title compound 35a was prepared in 35% yield (610 mg) using the general procedure and employing 3.92 g of di-ferf-butyl dicarbonate instead of the indicated amount and the product was obtained as white solid after recrystalized from hexanes.
(R)-Tert-butyl 2-hydroxypropylcarbamate 35b
[0147] The title compound 35b was obtained in 69.5% yield (730 mg) as white solid after flash chromatography using 50% hexanes and 50% ethyl acetate.
Tert-butyl 2-hydroxyethyl(methyl)carbamate 35c
[0148] The title compound 35c was obtained in 32.4% yield (340 mg) as white solid after flash chromatography using 50% hexanes and 50% ethyl acetate.
Tert-butyl 2-tert-butyloxycarbonyl-amidoethyl(2-hydroxyethyl)carbamate 35d
[0149] The title compound 35d was prepared using the general procedure with the following modification. 3.92g of di-fe/t-butyl dicarbonate was used instead of the indicated amount.
Purification by flash chromatography using 50% hexanes and 50% ethyl acetate gave the desired in 18.6% yield (340 mg) as white solid.
(R)-Tert-butyl 1 -hydroxypropan-2-ylcarbamate 35e
[0150] The title compound 35e was obtained in 72.4% yield (760 mg) as white solid after purification by flash chromatography using 50% hexanes and 50% ethyl acetate.
Example 62
(2R,3R,4S,5S)-2-(6-amino-9H-purin-9-yl)-5-((1,3-diaminopropan-2-ylthio)methyl)- tetrahydrofuran-3,4-dioI 36a
[0151] Methane sulfonyl chloride (103 mg, 0.9 mmol) was added to a solution of di-tert-butyl 2-hydroxypropane-1,3-diyldicarbamate 35a (174.2 mg, 0.6 mmol) in 1.0 ml pyridine. The reaction was mixed and left to stand for 30 minutes. Methylene chloride (10 ml) was then added and the reaction was washed with H2O (3 x 10 ml). The organic phase was dried with MgSO4, filtered and evaporated to give the crude mesylate.
[0152] A solution of 34 (73 mg, 0.200 mmol, Baddiley and Jamieson, J. Chem. Soc. 1955, 1085; Guillerm et al. J. Med Chem. 2001 , 44, 2743Pignot et al. Eur. J. Org. Chem 2000, 549) and NaOMe (400 μl, 0.20 mmol, 0.5 M in methanol) in DMF (1.0 ml) was stirred for 10 minutes under an N2 atmosphere. Then, the mesylate of 35a from above, dissolved in 1.OmI DMF, was added and the reaction was stirred overnight at room temperature under an N2 atmosphere. The reaction was diluted with ethyl acetate (10 ml) and washed with H2O (3 x 10 ml). The organic phase was evaporated and 1.0 ml of 50% TFA in dichloromethane was added. After 1 h the solvent was evaporated and the title compound 36a was purified by prep- HPLC (compound could be isolated as di-TFA salt). 1H NMR (DMSO-Cf6) δ (ppm): 8.32 (s, 1H), 8.13 (s, 1H), 7.23 (s, 2H), 5.88 (d, 1 H, J=5.6H-z), 4.73 (dd, 1H, J=4.8, 5.2Hz), 4.15 (dd, 1 H, J=4.0, 4.4Hz), 4.05 (m, 1H), 3.09 (m, 3H), 2.87-3.03 (m, 4H). MS: calc 355.4; found 356 (MH)+. [0153] Examples 63-66, Table 5, were prepared in a manner similar to example 62, scheme 3, using compounds 35b-e in place of 35a The compounds were isolated as the formate salts after prep-HPLC purification in 20.6%; 33.8%; 18.9; and 47% yields respectively.
Table 5

Ex. Cpd R/ Structure Name Characterization Scheme
(2R,3R,4S,5S) 1H NMR (DMSO-Cf6) δ (ppm): 8.33
-2-(6-amino- (s, 1H), 8.29 (s, 1H), 8.12 (s, 1 H),
9H-purin-9-yl)- 7.28 (s, 2H), 5.87 (d, 1H, J=5.6Hz),
5-(((S)-I- 5.87 (d, 1H, J=5.6Hz), 4.73 (dd,
63 36b aminopropan- 1 H, J=5.2, 5.6Hz), 4.14 (m, 1 H),
 2- 4.00 (m, 1 H), 3.17 (m, 1 H), 2.84- ylthio)methyl)- 2.98 (m, 2H), 2.57-2.70 (m, tetrahydrofuran 2H), 1.11 (d, 3H, J=6.4Hz). MS calc -3,4-diol 340.4, found 341 (MH)+
2- 4.00 (m, 1 H), 3.17 (m, 1 H), 2.84- ylthio)methyl)- 2.98 (m, 2H), 2.57-2.70 (m, tetrahydrofuran 2H), 1.11 (d, 3H, J=6.4Hz). MS calc -3,4-diol 340.4, found 341 (MH)+

Scheme 5

Example 68, comp 39
Example 67
1-(2-(((2SJ3S>4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxy-tetrahydrofuran-2- yl)methylthio)ethyl)guanidine 38
[0154] A solution of (2R,3R,4S,5S)-2-(6-amino-9H-purin-9-yl)-5-((2-aminoethylthio)methyl)- tetrahydrofuran-3,4-diol 37 (65 mg, 0.2 mmol, Jamieson, GA, J. Org. Chem. 1963, 28, 2397- 2400 ), 7fy-pyrazole-1-carboxamidine hydrochloride (32 mg, 0.22 mmol) and DIPEA (77 μl, 0.44 mmol) in dry DMF (1.0 ml) was stirred at room temperature for 4 hours. The solvent was then evaporated and the product was purified by preparative HPLC to give the title compound 38 as the formate salt in 48.8% yield (36 mg). 1H NMR (DMSO-Cy6ZD2O) δ (ppm): 8.45 (s, 1 H), 8.34 (s, 1H)1 8.29 (s, 1H), 8.11 (s, 1H), 5.84 (d, 1H, J=6.0 Hz), 4.69 (dd, 1 H, J=5.6, 5.6 Hz ), 4.11 (m, 1H), 3.99 (m, 1H), 3.23 (m, 2H), 2.82-2.94 (m, 2H), 2.61 (m, 2H). MS calc 368.4; found 369 (MH)+.
Example 68 e^a-C^aSjaS^RjSRJ-S^β-amino-ΘH-purin-θ-yO-S^-dihydroxy-tetrahydrofuran^- yl)methylthio)ethylamino)pyrimidin-2(1 H)-one 39
[0155] A solid mixture of 34 (100 mg, 0.306 mmol) and 4-methylthiouraciI (14.5 mg, 0.102mmol) was heated at 15O0C for 15 minutes according to the method of Delia, T. J. et al (J. Org. Chem. 1965, 30, 2766-2768). After the reaction had cooled to room temperature, the product was purified by preparative HPLC to give the title compound 39 in 28% yield (12 mg). 1H NMR (DMSO-Cf6ZD2O) δ (ppm): 8.46 (s, 1 H), 8.28 (s, 1 H), 7.69 (d, 1 H, J=7.6Hz), 5.91 (d, 1 H, J=7.6Hz), 5.89 (d, 1H, J=6.0Hz), 4.68 (dd, 1H, J=5.2, 5.6Hz), 4.12 (m, 1H), 4.03 (m, 1H), 2H assumed under D2O, 2.93 (m, 2H), 2.71 (m, 2H). MS: calc 420.45; found 421 (MH)+
Example 69
4-amino-5-(2-(((2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxy-tetrahydrofuran-2- yl)methylthio)ethylamino)pyrimidin-2(1 H)-one 40
[0156] A solid mixture of 34 (100 mg, 0.306 mmol) and 5-bromocytosine (58 mg, 0.305 mmol) was heated at 15O0C for 4 hours. After the reaction had cooled to room temperature, the product was purified by preparative HPLC to give the title compound 40 as the formate salt in 8.4% yield (11 mg). 1H NMR (DMSO-CZ6ZD2O) δ (ppm): 8.31 (s, 1H), 8.15 (s, 1 H), 8.11 (s, 1 H), 6.62 (s, 1H), 5.85 (d, 1H, J=5.6Hz), 4.70 (dd, 1H, J=5.2, 5.6Hz), 4.13 (m, 1H), 4.00 (m, 1H), 2.90 (m, 4H), 2.65 (m, 2H). MS calc 435.46; found 436 (MH)+.
Scheme 6

Example 70
1-(3-(((2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxy-tetrahydrofuran-2- yl)methylthio)propyl)guanidine 42
[0157] The title compound 42, was prepared in 57.5% yield (44 mg) as the formate salt according to the procedure described in example 67 starting from (2R,3R,4S,5S)-2-(6-amino- 9H-purin-9-yl)-5-((3-aminopropylthio)methyl)-tetrahydrofuran-3,4-diol 41 (68 mg, 0.2 mmol, prepared according to the method of Borchardt, R. T., and Wu, Y. S. J. Med. Chem. 1974, 17, 862-7). 1H NMR (CD3OD) δ (ppm): 8.43 (s, 1H), 8.29 (s, 1 H), 8.19 (s, 1 H), 5.99 (d, 1 H, J=4.8 Hz), 4.81 (dd, 1H, J=4.8, 5.2Hz), 4.34 (dd, 1 H, J=4.8, 5.2 Hz), 4.20 (m, 1H), 3.23 (t, 2H, J=6.8 Hz)1 2.97 (m, 2H), 2.63 (t, 2H, J=6.8Hz),1.82 (m, 2H). MS calc 382.4; found 383 (MH)+
Scheme 7
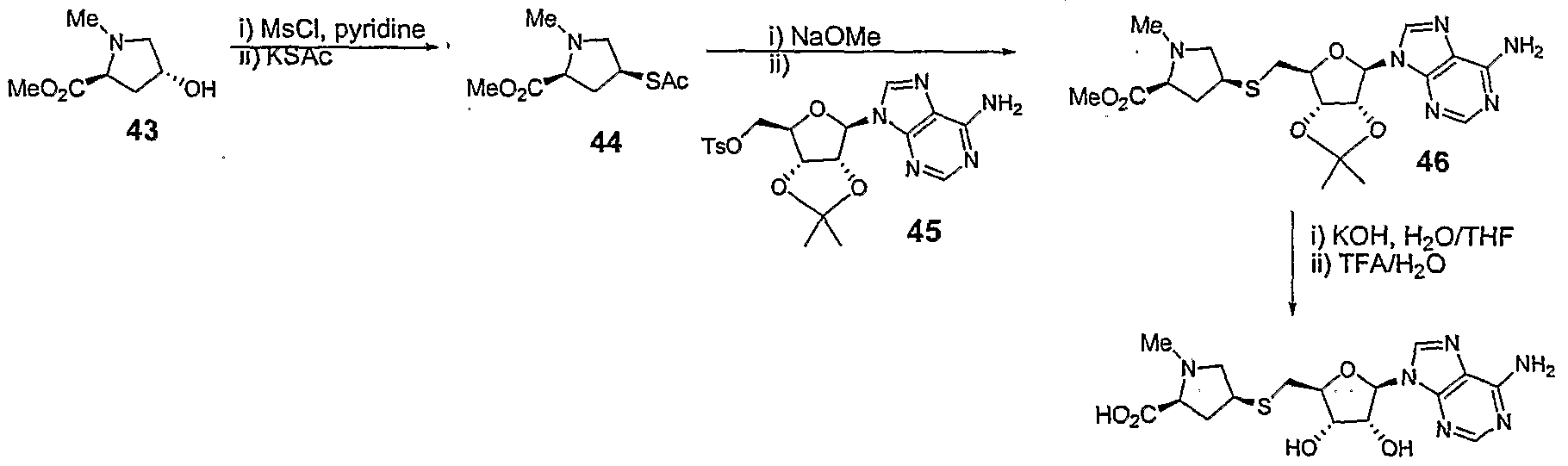
Example 71, comp 47
EXAMPLE 71
(2S,4S)-4-(((2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxy-tetrahydrofuran-2- yl)methylthio)-1-methylpyrrolidine-2-carboxylic acid 47
Step 1 : (2S,4S)-methyl 4-(acetylthio)-1-methylpyrrolidine-2-carboχylate 44 [0158] Mesyl chloride (170 μL, 251 mg, 2.25 mmol) was added to a solution of (2S.4R)- methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43 (239 mg, 1.5 mmol, prepared according to the procedure of R.L. Elliott et al., Synthesis, 1995, 772) in pyridine (3 mL) and stirred at room temperature overnight. The reaction mixture was diluted in DCM (20 mL) and washed with
water (15 mL). The'organic phase was separated, dried with Na" 2SO4, filtered and concentrated in vacuo. Thecrude mesylate was diluted in DMF and treated with KSAc (800 mg, 7.5 mmol). The reaction mixture was stirred for 2 hours at 8O0C and then it was concentrated in vacuo. The title compound 44 was obtained in 29% yield (95 mg) after flash chromatography using 30% EtOAC in hexane. MS: calc 217; found 240 (M+Na+)
Step 2: (2S.4SVmethyl 4-(((3aS,4S.6R.6aRV6-(6-amino-9H-Purin-9-vn-2.2-dimethyl- tetrahvdrofuro[3,4-diπ ,31dioxol-4-yl)methylthio)-1 -methylpyrrolidine-2-carboxylate 46 [0159] A solution of thioacetate 44 (95 mg, 0.43 mmol) in MeOH (2mL) was treated with 0.5 M solution of NaOMe (876 μl_). The reaction mixture was stirred for 15 minutes at room temperature and then it was transferred to a solution of ((3aR,4R,6R,6aR)-6-(6-amino-9H-purin- 9-yl)-2,2-dimethyl-tetrahydrofuro[3,4-d][1 ,3]dioxol-4-yl)methyl 4-methylbenzenesulfonate 45 (184 mg, 0.4 mmol, Thompson et al., J. Org. Chem. 1999, 64,(20), 7467-73) in MeOH (2 mL). The combined mixture was further refluxed for 2 hours. After it was allowed to cool down, it was concentrated in vacuo. The crude material was diluted in EtOAc (10 mL) and water. The layers were separated and the organic phase was washed with brine, dried with Na2SO4, filtered and concentrated in vacuo. The title compound 46 was obtained in 24% yield (45 mg) after flash chromatography using 5% MeOH in DCM. MS: calc 464; found 465 (MH+)
Step 3: (2S.4SV4-(((2S.3S.4R.5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihvdroxy-tetrahvdrofuran-2- yl)methylthio)-1 -methylpyrrolidine-2-carboxylic 47
[0160] A solution of 46 (24 mg, 0.05 mmol) and KOH (40 mg, 0.71 mmol) in 1:1 mixture of THF and water (3 mL) was stirred at room temperature for 2 hours. The reaction mixture was then concentrated in vacuo and diluted in water (5 mL). It was washed with Et2O (5 mL) and acidified to pH=2 by addition of citric acid. The aqueous phase was then extracted with EtOAc (3x5 mL) and the combined organic layer was dried with Na2SO4, filtered and concentrated in vacuo. The residue was treated with 2:1 mixture of TFA/water (3 mL) containing trace amount of BHT. The reaction mixture was stirred at room temperature for 45 min and then it was treated with 5 drops of water. It was allowed to stir for another 1 hour before it was concentrated in vacuo. The title compound 47 was obtained in 44% yield (9 mg) after flash chromatography eluting with 30% MeOH/DCM to 60:30:10 CHCI3/MeOH/NH4OH. 1H NMR (D2O) tfpprη; 8.12 (s, 1H), 8.02 (s, 1H), 5.86 (d, 1 H, J=4.9 Hz), 4.70 (t, 1H, J=5.1 Hz), 4.26 (t, 1H, J=5.2 Hz), 4.17 (dd, 1 H, J=4.9 Hz, J=11.2 Hz), 3.72 (dd, 1H, J=6.9 Hz, J=9.5 Hz), 3.54 (m, 1 H), 3.39 (dd, 1 H, J=4.0
Hz, J=11.6 Hz), 3.13 (dd, 1H, J=6.7 Hz, J=12.1 Hz), 2.92 (dd, 1H, J=4.5 Hz, J=14.3 Hz), 2.83
(dd, J=6.2 Hz, J=14.3 Hz), 2.69 (s, 3H), 1.92 (m, 1H)
MS: calc 410; found 411 (MH+)
[0161] Examples 72-76, compounds 48a-48e, Table 6, were prepared in a manner similar to example 71 , scheme 7 with the following modifications
[0162] Example 72, compound 48a, was prepared in a manner similar to example 71, were step 1 , was conducted according to the procedure in J. Org. Chem. 1196, 61 , 2226, replacing
(2S,4R)-methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43, with (2S,4S)-1-tert-butyl 2- methyl 4-hydroxypiperidine-1 ,2-dicarboxylate.
[0163] Example 73, compound 48b, was prepared in a manner similar to example 71, scheme 7. Step 1 was carried out according to the procedure for example 72 replacing (2S.4R)- methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43, with (2S,4R)-1-tert-butyl 2-methyl 4- hydroxypiperidine-1 ,2-dicarboxylate.
[0164] Example 74, compound 48c, was prepared in a manner similar to example 71. Step
1 was conducted according to the procedure in J. Med, Chem. 2001 , 44, 94, replacing (2S.4R)- methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43, with tert-butyl 3-hydroxyazetidine-1- carboxylate.
[0165] Example 75, compound 48d, was prepared in a manner similar to example 71, replacing (2S,4R)-methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43, with (R)-tert-butyl 2-
(hydroxymethyl)pyrrolidine-i-carboxylate.
[0166] Example 76, compound 48e, was prepared in a manner similar to example 71, replacing (2S,4R)-methyl 4-hydroxy-1-methylpyrrolidine-2-carboxylate 43, with (SJ-tert-butyl 2-
(hydroxymethyl)pyrrolidine-i-carboxylate.
Table 6


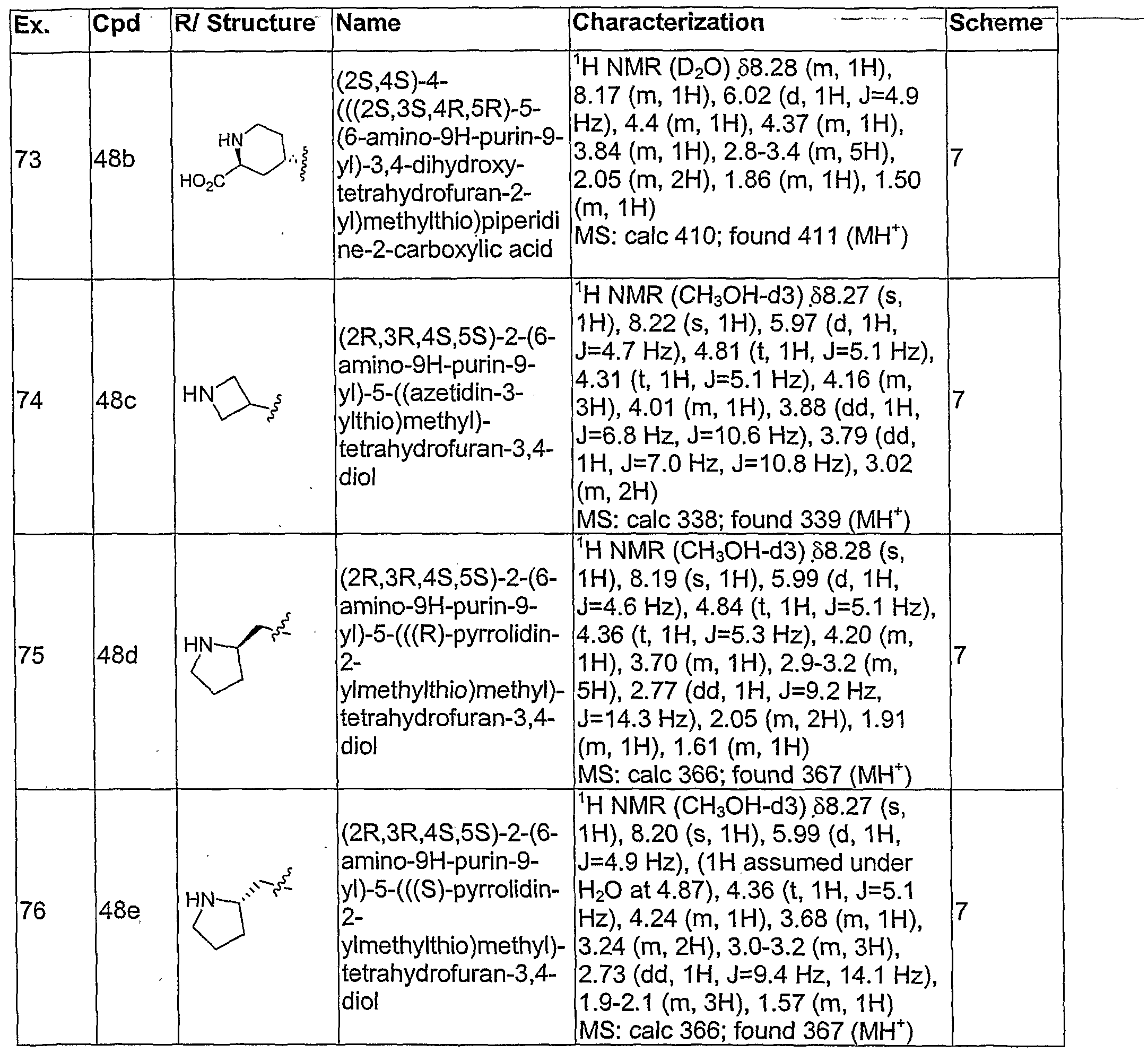

Example 77
(2S,4S)-4-(((2S,3S,4R,5R)-3,4-cIihyclroxy-5-(6-(3-phenylpropylammo)-9H-purin-9-yl)- tetrahydrofuran-2-yl)methylthio)pyrrolidine-2-carboxylic acid 52a
Step 1 : (2S,4S)-1-tert-butyl 2-methyl 4-(((3aS,4S.6R,6aR)-6-(6-(4H-1 ,2,4-triazol-4-vn-9H-purin-
9-yl)-2,2-dimethyl-tetrahvdrofuror3,4-d1f1 ,3ldioxol-4-yl)methylthio)pyrrolidine-1 ,2-dicarboxylate
50
[0167] Azine dihydrochloride (603 mg, 2.82 mmol) was added to a solution of (2S,4S)-1-tert- butyl 2-methyl 4-(((3aS,4S,6R,6aR)-6-(6-annino-9H-purin-9-yl)-2,2-dimethyl-tetrahydrofuro[3,4- d][1 ,3]dioxol-4-yI)methy!thio)pyrroIidine-1 ,2-dicarboxylate 49 (310 mg, 0.56 mmol, prepared as described by J.Q. Guo et al, Bioorg Med. Chem. Let, 1993, 3 (2), 147-152) in dry pyridine (3 ml_) and the reaction mixture was refluxed overnight. It was then cooled to room temperature and concentrated in vacuo. The crude product was purified by flash chromatography using
EtOAc giving the title compound 50 in 32% yield (108 mg).
MS: calc 602; found 603 (MH+)
Step 2: (2S,4S)-1-tert-butyl 2-methyl 4-(«3aS,4S,6R,6aRV2,2-dimethyl-6-(6-(3- phenylpropylamino)-9H-purin-9-yl)-tetrahvdrofuror3,4-dlf1 ,31dioxol-4-yl)methylthio)pyrrolidine-
1 ,2-dicarboxylate 51 a
[0168] A solution of 50 (45 mg, 0.085 mmol, 1 equiv.) and 3-phenylpropan-1 -amine (77 μL,
72 mg, 0.54 mmol) in dry DMF (1 mL) was stirred at room temperature for 2 days. The reaction mixture was concentrated, diluted in EtOAc (10 mL) and washed with brine (10 mL). The organic phase was dried with Na2SO4, filtered and concentrated. The crude product was purified
by flash chromatography using 60% EtOΑc in hexanes giving the title~compound 51a1ιτ37%~ " yield (44 mg).
MS: calc 668; found 669 (MH+)
Step 3: (2SΛS)-4-(((2S,3S.4R,5R)-3.4-dihvdroxy-5-(6-(3-phenylpropylaminoV9H-purin-9-vO- tetrahvdrofuran-2-vπmethylthio)pyrrolidine-2-carboxylic acid 52a
[0169] A solution of 51a (44 mg, 0.06 mmol) and KOH (40 mg, 0.71 mmol) in 1:1 mixture of
THF and water (3 mL) was stirred at room temperature for 3 hours. The reaction mixture was then concentrated in vacuo and diluted in water (5 mL). It was washed with Et2O (5 mL) and acidified to pH=2 by addition of citric acid. The aqueous phase was then extracted with EtOAc
(3x5 mL) and the combined organic layer was dried with Na2SO4, filtered and concentrated in vacuo. The crude product was treated with 2:1 mixture of DCM/TFA (3 mL) containing trace amount of BHT. The reaction mixture was stirred at room temperature for 45 min and then it was treated with 5 drops of water. It was allowed to stir for another 1 hour before it was concentrated in vacuo. The crude product was purified by flash chromatography using the gradient from 30% MeOH/DCM to 60:30:10 CHCI3/MeOH/NH4OH to give the title compound
52a in 47% yield (14 mg). 1H NMR (DMSO-d6) .88.32 (s, 1H), 8.19 (s, 2H), 7.89 (br.s, 1 H), 7.1-
7.3 (m, 5H), 5.87 (d, 1H, J=5.5 Hz), 4.70 (m, 1H), 4.13 (m, 1H), 4.02 (m, 1H), 3.60 (m, 2H), 2.8-
3.0 (m, 5H), 2.63 (m, 2H), 2.47 (m, 1 H), 1.91 (m, 2H), 1.70 (m, 2H).
MS: calc 514; found 515 (MH+).
[0170] Examples 78-83, compounds 52b-52g, Table 7, were prepared according to the descriptions below:
[0171] Example 78, compound 52b, was prepared according to the method shown in scheme 2a, example 43, replacing 2-(biphenyl-4-yl)ethanamine with 3-phenylpropan-1 -amine in stepi, and utilizing (2S,4S)-1-tert-butyl 2-methyl 4-(acetylthio)pyrrolidine-1,2-dicarboxylate in place of tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate in step 4.
[0172] Example 79, 52c, was prepared as shown in scheme 1a, example 1, in step 3 replacing tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate with (2S,4S)-1 -tert-butyl 2-methyl 4-
(acetylthio)pyrrolidine-1 ,2-dicarboxylate.
[0173] Example 80, 52d, was prepared according to scheme 2a, example 43, step 4 reacting 28 with (2S,4S)-1 -tert-butyl 2-methyl 4-(acetylthio)pyrrolidine-1 ,2-dicarboxylate in place of tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate.
[0174] Example 81 , 52e, was prepared as shown in scheme 1a, example 1, in step 3 replacing tert-butyl 2-oxo-tetrahydrothiophen-3-ylcarbamate with (S)-tert-butyl 3-
(acetylthio)pyrrolidine-i-carboxylate.
[0175] Example 82, 52f, was prepared accordinglo" scheme 2a~ example 43," step 4 reacting"
28 with (S)-tert-butyl 3-(acetylthio)pyrrolidine-1-carboxylate in place of tert-butyl 2-oxo- tetrahydrothiophen-3-ylcarbamate.
[0176] Example 83, 52g, was prepared according to the method shown in scheme 2a, example 43, replacing 2-(biphenyl-4-yl)ethanamine with 3-phenylpropan-1 -amine in stepi , and utilizing (S)-tert-butyl 3-(acetylthio)pyrrolidine-1-carboxylate in place of tert-butyl 2-oxo- tetrahydrothiophen-3-ylcarbamate in step 4.
Table 7



Scheme 9

Example 84, comp 59a, X=I Example 85, comp 59b, X=Br Example 86, comp 59c, X=F

Synthesis of the common intermediate, compound 54 used for Examples 84-86
9-((3aR,4R,6RI6aR)-6-((tert-butyldimethylsilyloxy)methyl)-2J2-dimethyl- tetrahydrofuro[3)4-d][1 ,3]dioxol-4-yl)-6-chloro-2-(tributylstannyl)-9H-purine 54
[0177] The reaction is a modification of the procedures reported procedures (K. Kato et al Tet Let 1995, 36, 6507-10; and K. Kato et al, J. Org. Chem 1997, 62, 6833-41 ). n-BuLi (2.5 M in hexanes, 15 mL) was added to a solution of 2, 2, 6, 6-tetramethylpiperidine (141.26 ml_, 0.837 mmol) in THF (76 mL) at 0 0C and the reaction mixture stirred 30 min. The solution was cooled down to -78 0C and a solution of 53 (3.37 g, 7.64 mmol, prepared according to the method of Camp, David; Li, Ying; McCluskey, Adam; Moni, Roger W.; Quinn, Ronald J; Biorg. Med. Chem. Leff.1998, 8 (6), 695-698) in THF (50 mL) was added drop wise maintaining the temperature
bellow -70 0C. The mixture stirred 30 mih at"-78°C. Bu3SnCI (10.3~ml_, 38.2 mmol) was added and the reaction mixture stirred 1 h at -78 °C. The mixture was quenched with NH4CI (aq. sat. solution), warmed up at room temperature and concentrated under reduced pressure. The residue dissolved in EtOAc, washed with water and brine, dried (Na2SO4) and concentrated. The residue was purified by flash chromatography (EtOAc/Hex 1/5) to give the title compoundin 82% yield as clear syrup (4.59 g). 1H-NMR CDCI3 δ (ppm): 7.90 (s, 1H), 6.15 (d, J=2.3 Hz, 1 H), 5.59 (br, 1H), 5.0 (dd, J=2.9 Hz, J=6.3 Hz, 1H), 4.36 (m, 1H), 3.82 (dd, J=4.5 Hz, J=11.2 Hz, 1H)1 3.72 (dd, J=5.9 Hz, J=11.2 Hz, 1H), 1.61 (s, 9H), 1.41 (s, 3H), 1.37 (m, 6H), 1.27 (m, 6H)1 0.91 (t, J=7.2 Hz, 1H), 0.85 (s, 9H), -0.016 (s, 3H) -0.024 (s, 3H). MS calc 730.27, found 730.5 (MH+).
Example 84
2-amino-4-(((2S,3S,4R,5R)-5-(6-amino-2-iodo-9H-purin-9-yl)-3,4-dihydroxy- tetrahydrofuran-2-yl)methylthio)butanoic acid 59a
Step 1 : 9-α3aR,4R,6R.6aR)-6-((tert-butyldimethylsilyloxy)methyl)-2,2-dimethyl- tetrahvdrofurof3,4-d1f1,3ldioxol-4-yl)-6-chloro-2-iodo-9H-purine 55a
[0178] Iodine was added in one portion to a solution of 54 (1.5 gr. 2.04 mmol) in THF (20 mL), and the solution was stirred for 1 h at room temperature. The crude was diluted with EtOAc, washed with aqueous Na3S8O3, brine, dried (Na2SO4) and concentrated. The crude was purified by flash chromatography (EtOAc/Hex 1/5) giving the title compound 55a in 95% yield (1.1 g) as a pale yellow solid.
1H-NMR: CDCI3 δ (ppm) 8.27 (s, 1 H), 6.16 (d, J=2.2 Hz, 1 H), 5.09 (dd, J=2.2, J=5.7 Hz, 1 H), 4.91 (dd, J=2.4 Hz, J=5.7 Hz, 1H), 4.41 (m, 1H), 3.89 (dd, J=3.1 Hz, J=11.4 Hz, 1H), 3.79 (dd, J=3.5 Hz, J=3.8 Hz, 1H), 1.63 (s, 3H), 1.40 (s, 3H), 0.88 (s, 9H), 0.07 (s, 3H). MS: calc 566.06 (100%), 568.06 (38%); found 567.1 (100%), 569.1 (38%) (MH+) Step 2: 9-((3aR,4R,6R,6aR)-6-((tert-butyldimethylsilyloxy')methylV2,2-dimethyl- tetrahvdrofurof3,4-d1f1 ,31dioxol-4-yl)-2-iodo-9H-purin-6-amiπe 56a
[0179] Ammonia gas was bubbled at 0 0C in a suspension of 55a (1.1 gr, 1.94 mmol) in i- PrOH. The mixture was stirred over night in a sealed tube at 100 0C. The mixture was cooled down to 0 0C, the sealed tube opened and the system slowly warmed up to room temperature. The solvent was removed under reduced pressure and the residue purified by flash chromatography EtOAc/Hex 1:1 giving the title compound 56a in 79% yield (871 mg) as white solid. 1H-NMR: CDCI3 δ (ppm) 7.92 (s, 1H), 6.12 (d, J=2.2 Hz, 1 H), 6.06 (s, 2H), 5.19 (dd, J=2.2 Hz, J=6.2 Hz, 1 H), 4.96 (dd, J=3.1 Hz, J=4.9 Hz, 1 H), 4.36 (m, 1 H), 3.89 (dd, J=3.9 Hz,
J=11.4 Hz, 1H), 3.80 (dd, J=4.8 Hz7 J=11.4 Hz; 1H), 1.63 (s; 3H), 1.40(s, 3H), 0.88 (s, 9H), 0.05
(s, 3H). MS: calc 547.46; found 548.3 (MH+)
Step 3: (r(3aR.4R,6R,6aR)-6-(6-amino-2-iodo-9H-purin-9-yl)-2.2-dimethyl-tetrahvdrofuror3,4- d1[1 ,31dioxol-4-yl)methanol 57a
[0180] TBAF was added to a solution of 56a (250 mg, 0.46 mmol) in THF at -250C. The solution was allowed to warm up to room temperature. The crude was concentrated, and the residue purified by flash chromatography (MeOH/DCM 20:1) giving the title compound 57a in
80% yield (158.6 mg) as light yellow solid. 1H-NMR : CDCI3 δ (ppm): 7.75 (s, 1H), 6.77 (br, 1H),
5.82 (d, J=4.7 Hz1 1H), 5.2 (dd, J=4.7 Hz, J=5.9 Hz, 1H), 5.09 (dd, J=1 Hz, J=5.9 Hz, 1H), 4.51
(m, 1H), 3.98 (dd, J=1.6 Hz, J=12.7 Hz, 1H), 3.83 (dd, J=2.3 Hz, J=12.7 Hz, 1H), 1.64 (s, 3H),
1.38 (s, 3H).
MS: calc 433.2; found 434.0 (MH+)
Step 4: methyl 4-(((3aS,4S,6R,6aR)-6-(6-amino-2-iodo-9H-purin-9-yl)-2,2-dimethyl- tetrahvdrofurof3,4-din ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 58a
[0181] Sodium hydride (60% in mineral oil, 28 mg, 0.62 mmol) was added in one portion to a solution of 57a (150 mg, 0.346 mmol) in THF at 0 0C and the mixture stirred 30 min. TsCI (73 mg, 0.38 mmol) was added in one portion, the system warmed up to room temperature and stirred 1 h. The mixture was poured into EtOAc, washed and water, and the organic layer was separated, dried (Na2SO4) and concentrated and used without further purification. A mixture of tert-butyl 2-oxo~tetrahydrothiophen-3-ylcarbamate 4 (112 mg, 0.519 mmol) and sodium methoxide (0.5 N in MeOH, 1 mL) was stirredifor 10 min at room temperature, then it was added to the crude tosylate in MeOH (3.5 ml) and the resulting solution was refluxed for 1 h.
The crude material was concentrated under reduced pressure; the residue dissolved in EtOAc, washed with water, dried (Na2SO4) and concentrated. The residue was purified by flash chromatography (pure EtOAc) giving the title compound 58a in 76% yield (175 mg) as clear oil.
1H-NMR: CDCI3 δ (ppm) 7.96 (s, 1H), 6.05 (d, H=1.7 Hz, 1H), 6.1 (d, J=6.1 Hz, 1H), 5.94 (dd,
J=1.7 Hz, J=6.1 Hz, 1H), 5.06 (dd, J=3.3 Hz, J=6.6 Hz, 1H), 4.37 (m, 2H), 2.95 (ddd, J=1.5 Hz,
J=7.4 Hz, J=13.5 Hz1 1 H), 2.86 (ddd, J=1.5 Hz, J=6.3 Hz, J=13.5 Hz, 1 H), 6.23 (m, 2H), 1.64 (s,
3H), 1.45 (s, 9H), 1.43 (s, 3H).
MS: calc 664.5; found 665.2 (MH+)
Step 5: 2-amino-4-(((2S,3S,4R,5R)-5-(6-amino-2-iodo-9H-purin-9-yl)-3.4-dihvdroxy- tetrahvdrofuran-2-v0methylthio)butanoic acid 59a
[0182] A mixture of 58a (175.1 mg, 0.263 mmol), KOH ( 29.5 mg, 0.53 mmol), H2O (1.75 mL) and THF (1.75 mL) was stirred for 1.5 h at room temperature (The reaction mixture turned
homogeneous afterwards)! The THF was removed under reduced pressure; water was added (10 mL) and the solution extracted with Et2O. The aqueous solution was acidified (pH ~ 3) by addition of 1 N HCI; the suspension extracted with DCM (2X), the combined organic extracts dried (Na2SO4) and concentrated. The residue was dissolved in DCM (2 mL), TFA (2 mL) was added at room temperature and the mixture stirred 1 h. H2O (1 mL) was added and the mixture was stirred for one more hour. The solvent was removed under reduced pressure, the residue dissolved in water and lyophilized. The crude material was purified by preparative HPLC (MeOH 5% to 20% in water, in 35 min, C-18 reverse phase column) giving the title compound 59a in 7% yield as white solid (10 mg). 1H-NMR: DMSO-d6 δ (ppm) 8.31 (s, 1 H), 7.73 (br, 2H), 5.80 (d, J=5.7 Hz, 1H), 5.64 (br, 1H), 4.69 (m, 1H), 4.14 (m, 1H), 4.10 (m, 1H), 4.03 (m, 1H), 3.2-3.8 (br, 2H), 2.93 (m, 1H)1 2.80 (m, 1 H), 2.70 ( m, 2H), 2.14 (m, 1 H), 1.9 (m, 1 H). MS: calc 510.5; found 511.0 (MH+)
Example 85
2-amino-4-(((2S,3S,4R,5R)-5-(6-amino-2-bromo-9H-purin-9-y[)-3,4-dihydroxy- tetrahydrofuran-2-yl)methylthio)butanoic acid 59b
[0183] The title compound 59b,was prepared in 5.4% yield as light brown solid in a manner similar to example 84, scheme 9, replacing I2 with NBS in step 1. 1H-NMR: DMSO-d6 δ (ppm) 8.38 (s, 1H), 7.86 (br, 2H), 5.81 (d, J=4.8 Hz, 1H), 4.63 (m, 1H), 4.10 (m, 1H), 4.03 (m, 1H), 3.2 (br, 3H), 2.9 (m, 1H), 2.82 (m, 1 H), 2.63 (m, 1 H), 2.24 (m, 1H), 1.86 (m, 1H). MS: calc 464 (100%), 462 (97%); found 463.1 (100%), 465.1 (97%) (MH+)
Example 86
2-amino-4-(((2S,3S,4R,5R)-5-(6-amino-2-fluoro-9H-purin-9-yI)-3,4-dihydroxy- tetrahydrofuran-2-yl)methylthio)butanoic acid 59c
[0184] The title compound 59c, was prepared in a manner similar to example 84, scheme 9, with the following modifications to step 1. XeF2 (524 mg, 2.04 mmol) was added in one portion to a solution of 54 (500 mg, 0.68 mmol), 2,6-di-tert-butyl-4-methylpyridine (140 mg, 0.68 mmol) and AgTfO (542 mg, 2.04 mmol) in DCM and the mixture stirred 15 min at room temperature. The reaction mixture was poured over NaHCO3 (aq. sat. solution) and extracted with DCM (2X). The combined organic layers were filtered through Celite™ and concentrated. The residue was dissolved in DCM, filtered again and concentrated. The crude was purified by flash chromatography (EtOAc/Hex 1:5) giving 55c in 48% yield (150 mg) as a yellow syrup. MS: calc 458.16 (100%), 460.15 (35%); found 459.3 (100%) 461.3 (34%) (MH+)
[0185] All other steps are the same as described for example 84. The tilte compound 59cr was obtained in 15% yield as white solid. 1H-NMR: DMSO-d6 δ (ppm) 8.32 (s, 1H), 8.12 (s, 1H), 7.80 (br, 2H), 6.56 (s, 1H), 5.75 (d, J=4.8 Hz, 1H), 5.72 (br, 1 H), 4.68 (m, 1H)1 4.52 (m, 1H), 4.20 (m, 2H), 4.05 (m, 1H), 3.92 (m, 1H), 2.80 (br, 2H), 2.70 (m, 2H), 2.00 (m, 1H), 1.80 (m, 1 H). MS: calc 402.11 ; found 403.4 (MH+)
Scheme 9a

7 gQ Example 87, comp 61
Example 87
2-amino-4-(((2S,3S,4R,5R)-5-(6-amino-2-methyl-9H-purin-9-yl)-3,4-dihydroxy-tetrahydrofuran-2- yl)methylthio)butanoic acid 61
Step 1 : methyl 4-(((3aS.4S.6R.6aR)-6-(6-amino-2-methyl-9H-purin-9-yl)-2,2-dimethyl- tetrahvdrofuror3,4-diri ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 60
[0186] The title compound 60 was prepared following the procedure detailed for example
84, scheme 9, steps 4, replacing 57a with ((3aR,4R,6R,6aR)-6-(6-amino-2-methyI-9H-purin-9- yl)-2,2-dimethyl-tetrahydrofuro[3,4-d][1 ,3]dioxol-4-yl)methanol 7 (51 mg, 0.16mmol, prepared according to the procedure of Yamazaki et al, J. Org. Chem. 1968, 33, 2583). MS calc 552.64; found 553.2 (MH+)
Step 2: 2-Amino-4-(((2S.3S,4R,5R)-5-(6-amino-2-methyl-9H-purin-9-vπ-3,4-dihvdroxy- tetrahydrofuran-2-yl)methylthio)butanoic acid 61
[0187] The title compound 61, was obtained as white solid in 81 % yield (30 mg) utilizing the procedures detailed in scheme 9 for example 84, in step 5, replacing 58a, with 60. 1H NMR
(CD3OD): ppm: 8.32(s, 1H), 6.06(d, 1H, J=4.8, 1-H), 4.80(m, 1H), 4.68 (s, 1H), 4.40(m, 1H),
4.30 (m, 1H), 3.74 (m, 1H), 3.08 (m, 2H), 2.80(m, 2H), 2.58(s, 3H, Me), 2.40(m, 1H), 2.10(m,
1H).
MS: calc 398.44; found 399.4(MH+)
Scheme 10

Example 89, Comp 68a R=COOH Example 90, comp 68b R=H
Example 88
2-amino-4-(((2S,3S14R,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-3,4-dihydroxy-tetrahyclrofuran-2- yl)methylthio)butanoic acid 65
Stepi : methyl 4-(((3aS,4S,6R,6aR)-6-(6-amino-2-chloro-9H-purin-9-ylV2,2-dimethyl- tetrahvdrofuror3,4-diri ,31dioxol-4-yl)methylthio)-2-(tert-butoxycarbonylamino)butanoate 64
[0188] Compound 64 was prepared in quantitative yield as a clear syrup following the procedure detailed in step 4 for compound 58a, example 84, scheme 9, replacing 57a with 62
(prepared according to the method of Andrzejewska, Mariola; Kaminski, Jaroslaw;
Kazimierczuk, Zygmunt Nucleosides Nucleotides 2002, 21 (1 ), 73-78).
MS: calc 572.18 (100%, 574.18 (37%); found 573.3 (100%), 575.2 (41%) (MH+)
Step 2: 2-amino-4-(((2S.3S.4R,5R)-5-(6-amino-2-chloro-9H-purin-9-vn-3,4-dihvdroxy- tetrahvdrofuran-2-vDrnethylthio)butanoic acid 65
I
[0189] The title compound 65 was prepared in 14% yield as white solid following the procedure detailed in step 5 for compound 59a, example 84, scheme 9, and replacing 58a with 64. 1H-NMR: DMSO-d6 δ (ppm) 8.4 (s, 1 H), 7.86 (br, 2H), 5.82 (d, J=6.2 Hz, 1 H), 4.63 (m, 1H), 4.14 (m, 1H), 4.10 (m, 1H), 3.2-3.8 (br, 3H), 2.9 (m, 1H), 2.80 (m, 1H), 2.62 (m, 2H), 2.26 (m, 1 H)1 1.86 (m, 1H). MS: calc 418.08 (100%), 420.08 (37%); found 419.2 (100%), 421.1 (38%) (MH+)
Example 89 -
(2S,4R)-4-(((2S,3S,4R,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-3,4-dihydroxy- tetrahydrofuran^-yOmethylthioJpyrrolidine-S-carboxylic acid 68a
Step 1 : (2S.4R)-methyl 4-(((2S,3S.4R.5R)-5-(6-amino-2-chloro-9H-purin-9-vO-3,4-dihvdroxy- tetrahvdrofuran-2-yl)methylthio)pyrrolidine-2-carboxylate 67a
[0190] Compound 67a was prepared in 47% yield as a clear syrup following the procedure detailed in step 4 for compound 58a, example 84, scheme 9, replacing 57a with (2S,4S)-1-tert- butyl 2-methyl 4-(acetylthio)pyrrolidine-1 ,2-dicarboxylate 66a.
Step2: (2S.4R)-4-«(2S,3SΛR,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-3.4-dihvdroxy- tetrahvdrofuran-2-yl)methylthio)pyrrolidine-2-carboxylic acid 68a
[0191] The title compound 68a,was prepared in 36% yield as white solid following the procedure detailed in step 5 for compound 59a, example 84, scheme 9, replacing 58a with 67a.
1H NMR (D2O) δ (ppm) 7.88 (s, 1 H), 5.61 (d, 1H, J=4.5 Hz, 1H), 4.46 (dd, J=4.5 Hz, J=5 Hz,
1 H), 4.14 (dd, J=5 Hz, J=5.3 Hz, 1 H), 4.05 (m, 2H), 3.45 (m, 2H), 3.16 (m, 1 H), 2.88 (dd, J=14
Hz, J=5.1 Hz, 1 H), 2.78 (dd, J=14 Hz, J=6.1 Hz, 1H), 2.57 (m, 1H).
MS: calc 430.08 (100%), 432.08 (37%); found 431.3 (100%), 433.3 (17%) (MH+)
Example 90
(2R,3R,4S,5S)-2-(6-amino-2-chloro-9H-purin-9-yl)-5-(((R)-pyrrolidin-3-ylthio)methyl)- tetrahydrofuran-3,4-diol 68b
Stepi : (R)-tert-butyl 3-(((3aS,4S.6R,6aR)-6-(6-amino-2-chloro-9H-purin-9-ylV2.2-dimethyl- tetrahvdrofuror3,4-dlf 1 ,3ldioxol-4-vDmethylthio)pyrrolidine-1 -carboxylate 67b [0192] Compound 67b was prepared in 33% yield as a clear syrup following the procedure detailed in step 4 for 58a, example 84, scheme 9, replacing 57a with (S)-tert-butyl 3- (acetylthio)pyrrolidine-i -carboxylate 66b.
MS: CaIc 526.18 (100%), 528.17 (37%); found 565.2 (100%), 567.2 (45%) (MH+) Step 2: (2R.3R.4S.5S)-2-(6-amino-2-chloro-9H-purin-9-vπ-5-(((R)-pyrrolidin-3-ylthio)methvπ- tetrahvdrofuran-3,4-diol 68b
[0193] The title compound 68bwas prepared in 12% yield as white solid following the procedure detailed in step 5 for compound 59a, example 84, scheme 9 and replacing 58a with 67b. 1H NMR (D2O) δ (ppm) 8.26 (br, 1 H), 8.08 (s, 1 H), 5.8 (d, 1 H, J=4.7 Hz, 1 H), 4.7 (dd, J=4.7 Hz, J=5.1 Hz, 1 H), 4.27 (dd, J=5.1 Hz, J=5.1 Hz, 1 H), 4.16 (m, 1 H), 3.48 (m, 1 H), 3.33 (m, 1 H), 3.33 (m, 2H), 3.21 (m, 1 H), 3.02 (dd, J=4.9 Hz, J=12.1 Hz, 1 H), 2.26 (dd, J=4.5 Hz, J=14.1 Hz, 1H), 2.89 (dd, J=7 Hz, J=14.1 Hz, 1 H), 2.20 (m, 1H), 1.82 (m, 1 H) MS: calc 386.09 (100%), 388.09 (37%); found 387.2 (100%), 389.1 (40%) (MH+)
"ScHefneiT

Example 91
(S)-4-(N-(2-nitrocinnamyl)-N-(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yI)-tetrahydro-3,4- dihydroxyfuran-2-yl)methyl)amino)-2-aminobutanoic acid 74a
Step 1 : benzyl (S)-3-(((3aR,4R,6R,6aR)-4-(6-amino-9H-purin-9-vπ-tetrahvdro-2.2- dimethylfuro[3,4-cnf 1 ,3ldioxol-6-vDmethylamino)-1 -(tert-butoxycarbonyl)propylcarbamate 71 [0194] To a solution of (S)-tert-butyl 2-(benzyloxycarbonylamino)-4-oxobutanoate 69 (4.0 ml, 1.41 mmol, prepared according to I. Weits et al, J. Org. Chem. 1997, 62, 2527-2534) in dichloroethane (7.5 ml) at room temperature was added to 9-((3aR,4R,6R,6aR)-6- (aminomethyl)-2,2-dimethyI-tetrahydrofuro[3,4-d][1 ,3]dioxol-4-yl)-9H-purin-6-amine 70 (538.0 mg, 1.76 mmol, prepared according to A. M. Reeve et al, Tetrahedron 1998, 54, 15959-15974). The mixture was heated slightly until it became homogeneous and then was allowed to cool to room temperature. Sodium triacetoxyborohydride (418.0 mg, 1.97 mmol) was added portion wise and the mixture was stirred for 2 h. The reaction was quenched with aqueous sodium bicarbonate and then the aqueous layer was extracted repeatedly with dichloromethane. The organic washings were dried over sodium sulfate, filtered and concentrated under reduced pressure. Column chromatography using a solvent gradient of ethyl acetate to 10%MeOH/EtOAc yielded the title compound 71 as white solid (538 mg, 64%). 1H NMR: (300 MHz, DMSO-Cf6) δ (ppm): 8.39 (1 H, s), 8.22 (1 H, s), 7.70 (1 H, d, J=7.7), 7.40-7.35 (7H, m), 6.12 (1 H1 d, J=3.3), 5.50 (1H, dd, J=6.2, 3.5), 5.08 (2H, s), 5.02 (1 H, dd, J=6.3, 2.5), 4.25 (1 H, m),
"4~1 (T(IHTmX 278O"(1H,~m), 2.69"(1H1Tn)T 2762-2:57 (2H7m)72:08 (1H1 m)T 1.84 (1H, m), 1:73
(1H, m), 1.59 (3H, s), 1.43 (9H, s), 1.36 (3H, s). MS calc: 597.66 observed.: 598.2 Step 2: benzyl (S)-1-(tert-butoxycarbonvn-3-(N-(2-nitrocinnamvIVN-(((3aR.4R.6R.6aRV4-(6- amino-9H-purin-9-ylHetrahvdro-2,2-dimethylfuror3,4-d1H,31dioxol-6- Vl)methv0amino)propylcarbamate 73a
[0195] To a solution of amine 71 (300.0 mg, 0.502 mmol) in dichloroethane (4 ml) at room temperature was added (E)-3-(2-nitrophenyl)acrylaldehyde 72a (156.0 mg, 0.88 mmol). The mixture was allowed to stir at rtfor 15 min. then sodium triacetoxyborohydride (150.0 mg, 0.703 mmol) was added and the mixture was stirred for 3 h. The reaction was quenched with aqueous sodium bicarbonate, allowed to stir vigorously for 30 min. and then the aqueous layer was extracted repeatedly with dichloromethane. The organic extracts were dried over sodium sulfate, filtered and concentrated under reduced pressure. Column chromatography using a solvent gradient of ethyl acetate to 5%MeOH/EtOAc yielded the title compound 73a as white solid (376 mg, 98%). 1H NMR: (300 MHz, DMSO-c/6) δ (ppm): 8.37 (1 H, s), 8.15 (1H, s), 7.97 (1 H, d, J=8.0), 7.76-7.67 (2H, m), 7.63 (1H, d, J=7.7), 7.54 (1H, m), 7.39-7.34 (7H, m), 6.83 (1 H, d, J=16.0), 6.34 (1 H, m), 6.21 (1 H, d, J=2.2), 5.54 (1H, dd, J=6.2, 2.3), 5.09-5.05 (2H, m), 4.92 (1H, d, J=12.6), 4.34 (1H, m), 4.12 (1H, m), 3.39 (1H, m), 3.23 (1H, dd, J=14.7, 7.9), 2.64-2.60 (2H, m), 2.50 (1H, m). MS calc/. 758.8 observed.: 759.3
Step 3: (S)-4-(N-(2-nitrocinnamvπ-N-(((2R.3S.4R,5RV5-(6-amino-9H-purin-9-yl)-tetrahvdro-3.4- dihvdroxyfuran-2-v0methv0amino)-2-aminobutanoic acid (74a)
[0196] To a solution of EtSH (0.44 ml, 5.94 mmol)) and BF3 OEt2 (0.25 ml, 1.98 mmol) in dichloromethane (1.5 ml) at O0C was added a solution of 73a (150 mg, 0.20 mmol) in dichloromethane (0.5 ml) and then a cap was placed tightly on top. After 30 min. the mixture was allowed to warm to rt, the cap removed and mixture vented and then the cap was placed tightly on top again. After 18 h. the mixture was concentrated, re-dissolved in acetone/H2O and concentrated again. The crude was purified by column chromatography (CHCI3ZMeOHZNH4OH, 6:3:1) and then Dowex 50WX8-200 resin to afford the title compound 74a as an off-white solid in 36% yield (38 mg). 1H NMR: (300 MHz, DMSO-Cf6) δ (ppm): 8.22 (1 H, s), 8.13 (1 H, s), 7.97 (1 H, d, J=8.2), 7.65 (1H, m), 7.57-7.49 (2H, m), 7.01 (1 H, d, J=15.9), 6.25 (1H, m), 6.05 (1 H1 d, J=3.8), 4.76 (1 H, m), 4.48 (1H, t, J=5.6), 4.40 (1 H, m), 3.74 (1H, dd, J=9.4, 2.9), 3.57 (1H1 dd, J=15.7, 5.5), 3.34 (1 H, m), 3.15 (1 H, dd, J=13.3, 9.8), 3.03 (1 H, m), 2.94-2.85 (2H, m), 2.21 (1 H, m), 2.00 (1H1 m).
[0197] "" " Examples 92", compound 74b, Table~8, was" prepared" according to the procedure — ■ described for example 91, in step 2 using 72b,3-(pyridin-3-yl)propanal (prepared according to the procedure of M. Stocks et al, Tet. Let.1995, 36, 6555-8) in place of aldehyde 72a. [0198] Example 93, compound 74c, Table 8, was prepared according to the procedure described for example 91 , in step 2 using 72c, 3-(2-nitrophenyl)propanal (previously reported by Harmon et al. J. Org. Chem. 1969, 34, 3684; however, it was prepared according to the method in JOC, 1992, 57(11), 3218-3225) in place of aldehyde 72a. [0199] Example 94, compound 74d, Table 8, was prepared according to the procedure described for example 91 , in step 2 using 72d, 4-nitrobutanal (previously reported by R. Kimura et al. Bull. Chem. Soc. Jpn., 2002, 75(11), 2517-2526, however it was prepared in 94% yield by DIBAL-H reduction of methyl 4-nitrobutanoate in DCM at -78 0C) in place of aldehyde 72a. [0200] Examples 95-102, compounds 74e-74l, Table 8, were prepared according to the method described for example 91 , utilizing commercially available aldehydes 72e-72l.
Table 8




Scheme 12

(SJ-^N^tCZR.SS^R.SRJ-S-tβ-CethylaminoJ-ΘH-purin-θ-ylJ-tetrahydro-S^-dihydroxyfuran-a- yl)methyl)-N-methylamino)-2-aminobutanoic acid 77
Step 1 : (S)-tert-butyl 4-((((3aR,4R,6R.6aRV6-(6-amino-9H-purin-9-vn-2.2-dimethyl- tetrahvdrofuror3,4-cnπ ,31dioxol-4-vnmethyl)(methvπamino)-2-
(benzyloxycarbonylamino)butanoate 75
[0201] To a solution of amine 71, scheme 11 (0.75 g, 1.25 mmol) in DCE (dichloroethane) at rt was added formaldehyde (37% in H2O, 0.14 ml, 1.9 mmol). To this emulsion was added sodium cyanoborohydride (0.37, 1.75 mmol) and the resulting mixture was stirred 1 h. When reaction was complete, solvent was removed in vacuo and the crude residue was purified by column chromatography to afford the title compound 75 in 50% yield as a solid. MS calc:
611.69, observed: 612.5.
Step 2: benzyl (S)-1-(tert-butoxycarbonvπ-3-(N-(((3aR.4R.6R.6aRV4-(6-(ethvIaminoV9H-purin-
9-yl)-tetrahvdro-2.2-dimethylfuroF3,4-diπ,31dioxol-6-yl)methyl)-N-methylamino)propylcarbamate
76
[0202] Acetaldehyde (0.05 ml, 0.90 mmol), Bu2SnCI2 (12.0 mg, 0.039 mmol) was added to a solution of 75 (250.0 mg, 0.409 mmol) in THF (0.75 ml) was added and then PhSiH3 (0.10 ml,
0.82 mmol) quickly and the reaction flask sealed tightly. After 18 h. the reaction mixture was concentrated and the crude was purified by column chromatography (2%MeOH/EtOAc) to afford the title compound 76 in 34% yield as an off-white solid (89 mg). MS calc: 639.7; found: 640.5.
Step 3: (S)-4-(N-(((2R,3S.4R,5R)-5-(6-(ethylamino)-9H-purin-9-vn-tetrahvdro-3.4- dihydroxyfuran-2-yl)methyl)-N-methylamino)-2-aminobutanoic acid 77
[0203] Following the procedure for compounds described for example 91 , the title compound 77 was obtained as a white solid in 56% yield (9 mg). 1H NMR: (400 MHz, CD3OD)
£(ppm): 8.26 (s, 1 H), 8.22 (s, 1H), 5.98 (d, J=4.70, 1H), 4.74 (t, J=5.09, 1H), 4.26 (m, 1 H), 4.21
(t, J=5.28, 1H), 3.63 (dd, J=9.0, 3.13, 2H), 2.93 (dd, J=13.11, 9.59, 1H), 2.82-2.65 (m, 3-4H),
2.08 (m, 1H), 1.92 (m, 1H), 1.32 (t, J=7.24, 3H), MS calc: 409.44; found: 410.2
ASSAY EXAMPLES Assay Example 1
Inhibition of DNMT1 and DNMT3b2 Activity [0204] The following protocols were used to assay the compounds of the invention.
Human DNA methyltransferases (hDRMTsTcloning, expression and purification^ ~~ "
Construction Of Bacuioviruses
[0205] A 5.0-kb cDNA corresponding to the full length DNMT1 (Swissprot accession number
P26358 (SEQ ID NO.1)) was cloned into the BamHt/Sall site of the pBlueBac4.5 vector
(Invitrogen) Also, a 2.7-kb cDNA encoding the DNMT3 splice variant 2 of DNMT3b2 (Swissprot accession number Q9UBC3-2 (SEQ ID NO.2)) was cloned into the BamHI/Xbal site of the abovementioned vector. These constucts were used to generate recombinant bacuioviruses using the Bac-N-Blue™ DNA according to the manucfacturer's instructions (Invitrogen).
Protein Expression
[0206] Full length hDNMTs proteins were expressed in Hi-5 cells {Trichoplusia Ni) upon infection with recombinant bacuioviruses constructs. Briefly, Hi-5 cells grown in suspension and maintained in serum-free medium (Sf900 Il supplemented with gentamycin) were infected with the abovementioned viruses at multiplicity of infection (MOI) varying from 1 to 3 during 84 hours at 270C with agitation at 120 rpm on a rotary shaker. Infected cells were harvested by centrifugation at 398g for 15 min. after which a nuclear and cytosolic fractionation was performed. Fractions were frozen at -8O0C until purifications were performed.
Nuclear Extraction
[0207] After harvesting of Hi-5 cells infected with the DNMT-1 recombinant baculovirus, cell pellets were gently resuspended in Buffer A (1OmM Tris pH 8.0, 1.5mM MgCI2, 5mM KCI, 5mM
DTT, 0.5% NP-40, 25% glycerol, 1μg/ml pepstatin, 2μg/ml Aprotinin and leupeptin, 50μg/ml
PMSF, 50μg/ml TLCK and 10μM E64) and left on ice for 10 min.. Nuclei were pelleted down at
50Og and supernatant (cytoplasmic fraction) kept on ice. Nuclei pellets were resuspended in
Buffer B (2OmM Tris pH 8.0, 1.5mM MgCI2, 5mM KCI, 5mM DTT, 0.5% NP-40, 25% glycerol,
0.2mM EDTA, 40OmM NaCI, 1μg/ml pepstatin, 2μg/ml Aprotinin and leupeptin, 50μg/ml PMSF,
50μg/ml TLCK and 10μM E64) and left on ice for 45 min. followed by a centrifugation at 3000Og for 30 min. The supernatant was recovered as the nuclear fraction.
[0208] The nuclear extraction of Hi-5 cells infected with the DNMT3b2 recombinant baculovirus construct was performed as described above with the following modifications in buffers composition: Buffer A did not have glycerol and DTT. Also, glycerol concentration in
Buffer B was 10%.
Purification
[0209] All the purification steps described below were performed at 40C. For DNMT-1 purification, both cytoplasmic and nuclear fractions were pooled together and NaCI concentration was adjusted to 0.1 M by diluting with Buffer C (2OmM Tris pH 7.4, 10% sucrose
"ahcfϊmM EDTAJ This material was centfifϋged for' 10 rtiin. at"30000g~arfd the~sOpematant was — loaded onto a QsepharoseFF column (Amersham Biosciences) equilibrated with Buffer C + 0.1 M NaCI. Following a ten column volume (CV) wash with equilibration buffer, bound proteins were eluted with a 10 CV salt linear gradient spanning from 0.1 to 1 M NaCI in Buffer C. DNMT-1 containing fractions were pooled together. Typically, the conductivity of selected fractions ranked between 13.5 and 18.6mS/cm. This Qsepharose eluate was diluted 4-fold with Buffer C and centrifuged as described above. The supernatant was applied to a Hitrap Heparin column (Amersham Biosciences) equilibrated with Buffer C + 0.1 M NaCI. Column was washed with 10 CV of equilibration buffer and proteins were eluted with a 10 CV salt linear gradient (0.1 to 1.5M NaCI in buffer C) The DNMT-1 enriched fractions were pooled according to SDS-PAGE analysis (coomassie staining) Final DNMT-1 protein preparations concentration were about 7mg/ml and purity above 95%.
[0210] DNMT3b2 was purified using only the nuclear extract as starting material. The NaCI concentration of the latter was adjusted to 0.2M by diluting with Buffer D (5OmM NaPO4 pH 7.8, 10% glycerol and 1mM EDTA) supplemented with the protease inhibitors described in Buffer A followed by centrifugation at 3000Og for 10 min.. The supernatant was loaded onto a Hitrap SPsepharose column (Amersham Biosciences) equilibrated with Buffer D with 0.05M NaCI. Column was washed with 8 CV of equilibration buffer and proteins were eluted with an 8 CV linear gradient of NaCI (from 0.05 to 1 M) in Buffer D. DNMT3b2 containing fractions were pooled based on SDS-PAGE analysis. Selected fractions from this elution had conductivity varying from 15.1 to 28.2mS/cm. Finally, this SPsepharose eluate underwent buffer exchange, using PD-10 column (Amersham Biosciences) against Buffer D + 0.3M NaCI. Typical DNMT3b2 enzyme preparation had concentration of about 2.5mg/ml and approximately 70% purity. [0211] Purified DNMT-1 and DNMT3b2 protein stocks were aliquoted and frozen at -8O0C prior to use in enzymatic assay. DNA Methyltransferase Assay Reagents : -Enzyme: Cloned human DNA Methyltransferase (DNMT1 and DNMT3b2)
Expressed in insect cells and purified at 98% -Substratel:
Oligonucleotides
MYG 167: ATC GCA TCG ATC GCG ATT CGC GCA TCG GCG ATC MYG 166: GAT XCG XGA TGX GXG AAT XGX GAT XGA TGX GAT (X: 5-methylcytosine)
-Substrate2:"" " " ~ ' " "
S-adenosyl-L-methionine -10X buffer:
0.5M Tris, pH 7.6, 50% Glycerol, 1OmM EDTA, 1mg/ml BSA, 1OmM DTT DNMT1 Assay:
Protocol for screening at 15 μM and 45 μM inhibitor concentrations. Assay format: 96-well
[0212] One μL of 450 μM Inhibitor: (15 μM final)or add 1 μL of 1.35mM Inhibitor: (45 μM final) is added to the Enzyme Mix (0.03 μL of 25uM of DNA Methyltransferasel to make 25nM final, 0.2 μL of 39OuM of Hemi-methylated oligo to make 2.6uM final, 1.65 μL of 10X Buffer [5OmM Tris-HCI pH7.6, 5%Glycerol, 1mM EDTA, 100ug/mL BSA, 1mM DTT]. The mixture is pre-incubated at 370C for 10min and then the SAM mix is added (2.25uL of 4OuM of S- Adenosyl-L-Methionine to make 3uM final, 1.65uL of 10X Buffer [5OmM Tris-HCI pH7.6, 5%Glycerol, 1mM EDTA, 100ug/mL BSA, 1 mM DTT]). The final reaction is in 3OuL. The reaction is incubated at 37°C for 15min and then transferred onto DEAE filtermat (Wallac cat# 1450-522) using Tomtec cell harvester. The DEAE filtermat is washed with cold 2OmM NH4HCO3. When the filter is dry, it is covered with MeltiLex TM scintillant (Wallac cat#1450- 441 ). Counting is done in a Wallac beta-counter.
[0213] DNMT3b2 assay follows identical protocol, at enzyme concentration of 188nM. [0214] The activities of a number of compounds according to the invention measured by various assays are displayed in the table below.
Enzymatic DNMT1 and DNMT3b2 data




Claims
1. A compound of formula (I):

R3 and R4 independently represent H, C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -CrCβ-alkyl-cycloalkyl, -CrC6-alkyl- heterocyclyl, -CrCe-alkyl-aryl, -Ci-Ce-alkyl-heteroaryl, -CrCealkoxy-aryl or - (CH2)I-6-T, wherein Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, cycloalkyl, heterocyclyl, aryl and heteroaryl, at each occurrence, are optionally substituted; or R3 and R4 taken together with the nitrogen to which they are attached form a C5-Cg heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted; T is NH-C(=O)-R14, -NH-SO2-R15, or -S-(CH2)1-3-R14, Ri4 is C1-C6 alkyl, aryl or heteroaryl and R15 is aryl, wherein Ci-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted;
R2 is H, halo, CF3, C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C1-C6 alkoxy, -NH-C1-C6 alkyl, or -S- C1-C6 alkyl, wherein Ci-C6 alkyl, C2-C6 alkenyl, and C2-C6 alkynyl are each optionally substituted; A and B independently are F, Cl1 -OH, H, -NHR, or -OR;
R at each occurrence is independently benzyl or Ci-C4 alkyl, wherein benzyl and Ci-C4 alkyl are optionally substituted; W is CH, N, CR, or C-halogen; X is CH, N, C-Ci-C6-alkyl, or C-halogen; D is CH, or N; Y is -S-, -O-, N(R16)-, -CH=CH-, -S-CH2-, -0-CH2-, where Ri6 is H, Ci-C6 alkyl, -Ci-C6-alkyl-aryl, -Ci-C6-alkyl-heteroaryl, Or -C2-C6 alkenyl- aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted; 'Z iS--IT- -C(H)(NHa)-COORTr -Ir-NR19R2OrOr heterocyclylrwherein^heterocyclyl 1 is optionally ■ substituted;
L is a bond or is -(CR17Ri8)i-6-; each R17 and R18 independently is H or CrC6-alkyl, wherein CrCe-alkyl is optionally substituted; Rig and R20 independently are H, CrC6-alkyl, heteroaryl, or H2N-C(=NH)-CH2-, wwrherein
CrC6-alkyl and heteroaryl are optionally substituted; and R7 is H or CrCe-alkyl, wherein compounds selected from the group consisting of:
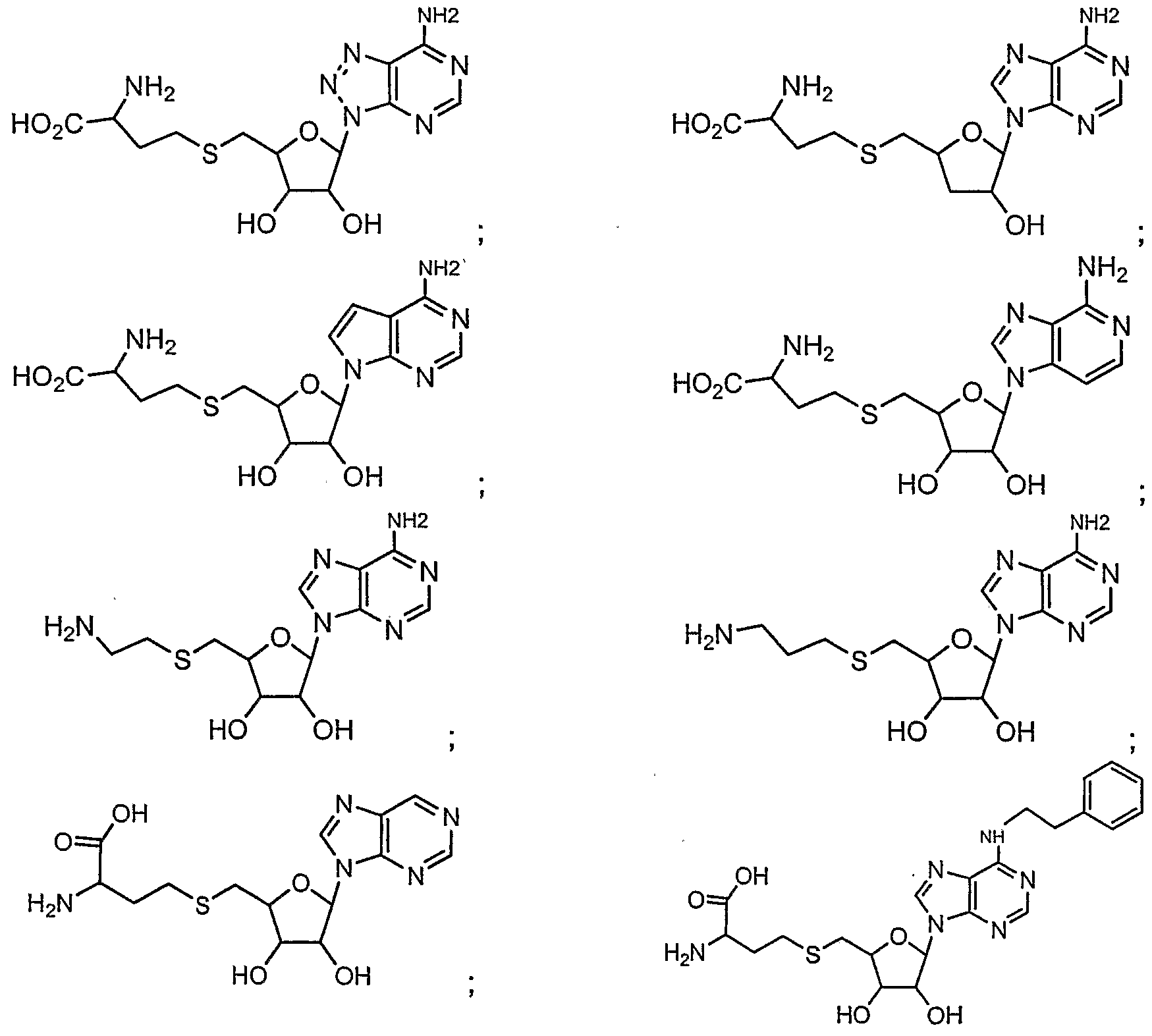




2. A compound according to claim 1 , represented by formula (II)

R3 and R4 independently represent H, C1-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -CrCe-alkyl-cycloalkyl, -CrC6-alkyl-heterocyclyl, -Ci-C6-alkyl-aryl, -C1-C6- alkyl-heteroaryl, -C-i-C6alkoxy-aryl or -(CH2)i-6-T, wherein C1-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl and heteroaryl, at each occurrence, are optionally substituted; or
R3 and R4 taken together with the nitrogen to which they are attached form a C5-C9 heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted; T is NH-C(=O)-R14, -NH-SO2-R15, or -S-(CH2)1-3-R14, R14 is C1-C6 alkyl, aryl or heteroaryl and Ri5 is aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted; W and X are independently CH or N; 
Ri6 is H, C1-C6 alkyl, -CrC6-alkyl-aryl, -CrCe-alkyl-heteroaryl, or -C2-C6 alkenyl-aryl, wherein C1-C6 alkyl, aryl, and heteroaryl, at each occurrence, are optionally substituted; Z is -L-C(H)(NH2)-COOR7, -L-NIR19R2O, or heterbcyclyl, wherein heterocyclyl is optionally substituted; L is a bond or is -(CR17R18)1-6-;
R17 and Rt8 independently are H or CrCe-alkyl, wherein CrC6-aikyl is optionally substituted; and R19 and R20 independently are H, C^Ce-alkyl, heteroaryl, or H2N-C(=NH)-CH2-, wherein
CrCe-alkyl and heteroaryl are optionally substituted R7 is H or d-Ce-alkyl.
3. A compound according to claim 2, of the formula H-A:

R17 and Ri8 independently are H or CrC6-alkyl, wherein CrC6-alkyl is optionally substituted.
4. A compound according to claim 3, wherein A is OH.
5. A compound according to claim 3, wherein R7 is H.
6. A compound according to claim 3, wherein L1 is -CH2CH2-.
7. A compound according to claim 3, wherein R2 is H1 halogen, C1-C3 alkyl, -S-C1-C2 alkyl, or C2-C3 alkenyl.
8. A compound according to claim 3, wherein Y is S.
9. A compound according to claim 3, wherein R3 and R4 are independently H, C1-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -CrC6-alkyl-cycloalkyl, -C^Ce-alkyl-aryl, -C-i-Ce-alkyl-heteroaryl, -CrC6alkoxy-aryl or -(CH2)i-6-T, wherein C1-C6 alkyl, C2-C6 alkenyl, cycloalkyl, heterocyclyl, aryl and heteroaryl, at each occurrence, are optionally substituted, or R3 and R4 taken together with the nitrogen to which they are attached form a C5-C9 heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted.
10. A compound according to claim 2, of formula H-B: 
(H-B) or a pharmaceutically acceptable salt or complex thereof, wherein m is 0 or 1 ; n is 1 or 2;
L2 is a bond or is -CH2-; R2 is H or halogen; R3 is H, C1-C6 alkyl, or -Ci-C6-alkyl-aryl, wherein Ci-C6 alkyl and aryl, at each occurrence, are optionally substituted;
R4 is H or CrC6 alkyl, wherein C1-C6 alkyl is optionally substituted; R8 is H, -CO2H, or CO2CH3;
R9 is absent, H or C1-C6 alkyl, wherein C1-C6 alkyl is optionally substituted; W is N or CH; Y is S or O; and Q is N, CH or O, provided that when Q is O, R9 is absent.
11. A compound according to claim 10 wherein R3 and R4 are both H.
12. A compound according to claim 10 wherein R3 is -Ci-C6-alkyl-aryl and R4 is H, wherein aryl is optionally substituted.
13. A compound according to claim 2, of the formula U-C:

H-C or a pharmaceutically acceptable salt or complex thereof, wherein R2 is H or halo; R3 and R4 independently represent H, C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, cycloalkyl, aryl - or (-CrCe-alkyO-aryl, wherein C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, cycloalkyl, aryl and (-CrC6-alkyl)-aryl are each optionally substituted; or R3 and R4 taken together with the nitrogen to which they are attached form a C5-C9- heterocyclyl ring or a heteroaryl ring, wherein said ring is optionally substituted; L3 is a bond or is -(CR17R1B)1-6-;
R17 and R18 at each occurrence are independently H or C-pCe-alkyl, wherein C1-
C6-alkyl is optionally substituted;
R19 is H, d-Ce-alkyl, heteroaryl, or H2N-C(=NH)-CH2-, wherein CτC6-alkyl and heteroaryl are optionally substituted.
14. A compound according to claim 13 wherein L3 is -CHR17CHRi8-, R17 and Ri8 independently are H, or Ci-C6 alkyl, and C1-C6 alkyl at each occurrence is optionally substituted.
15. A compound according to claim 14 wherein R17 and Ri8 independently are H, or C1-C6 alkyl, and Ci-C6 alkyl is unsubstituted or is substituted with NH2.
16. A compound according to claim 13 wherein L3 is -CH2CH2CH2-.
17. A method of inhibiting DNMT1 and/or DNMT3b2 enzymes in a cell, comprising contacting a cell in which inhibition of DNMT1 or DNMT3b2 is desired with a compound according to claim 1.
18. A composition comprising a compound according to claim 1 , or salt or complex thereof, together with a pharmaceutically acceptable excipient, diluent, or carrier.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06718809A EP1844062A2 (en) | 2005-01-21 | 2006-01-18 | Inhibitors of dna methyltransferase |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US64615605P | 2005-01-21 | 2005-01-21 | |
| US60/646,156 | 2005-01-21 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006078752A2 true WO2006078752A2 (en) | 2006-07-27 |
| WO2006078752A3 WO2006078752A3 (en) | 2007-02-15 |
Family
ID=36692836
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/001791 WO2006078752A2 (en) | 2005-01-21 | 2006-01-18 | Inhibitors of dna methyltransferase |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP1844062A2 (en) |
| WO (1) | WO2006078752A2 (en) |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8093264B2 (en) | 2005-05-20 | 2012-01-10 | Methylgene Inc. | Fused heterocycles as inhibitors of VEGF receptor and HGF receptor signaling |
| JP2012509351A (en) * | 2008-11-20 | 2012-04-19 | プレジデント アンド フェローズ オブ ハーバード カレッジ | Fluorination of organic compounds |
| WO2013062945A1 (en) * | 2011-10-24 | 2013-05-02 | Glaxosmithkline Intellectual Property Development Limited | Chemical compounds |
| US8580762B2 (en) | 2010-12-03 | 2013-11-12 | Epizyme, Inc. | Substituted purine and 7-deazapurine compounds |
| US8722877B2 (en) | 2010-12-03 | 2014-05-13 | Epizyme, Inc. | 7-deazapurine modulators of histone methyltransferase, and methods of use thereof |
| JP2014530910A (en) * | 2011-10-24 | 2014-11-20 | グラクソスミスクライン、インテレクチュアル、プロパティー、ナンバー2、リミテッドGlaxosmithkline Intellectual Property No.2 Limited | New compounds |
| WO2015040169A1 (en) * | 2013-09-19 | 2015-03-26 | Pierre Fabre Medicament | Quinazoline derivatives and their use as dna methyltransferase inhibitors |
| WO2014039860A3 (en) * | 2012-09-07 | 2015-07-16 | University Of Louisville Research Foundation, Inc. | Compositions and methods for modulating dnmt1 inhibitor activity |
| US9150516B2 (en) | 2011-04-12 | 2015-10-06 | President And Fellows Of Harvard College | Fluorination of organic compounds |
| US9175032B2 (en) | 2013-03-15 | 2015-11-03 | Epizyme, Inc. | Methods of synthesizing substituted purine compounds |
| US9273083B2 (en) | 2012-09-26 | 2016-03-01 | President And Fellows Of Harvard College | Nickel fluorinating complexes and uses thereof |
| EP3207932A1 (en) | 2016-02-19 | 2017-08-23 | Universität Stuttgart | Dna methyltransferase inhibitors for rett syndrome therapy |
| WO2017216727A1 (en) | 2016-06-13 | 2017-12-21 | Glaxosmithkline Intellectual Property Development Limited | Substituted pyridines as inhibitors of dnmt1 |
| US10112968B2 (en) | 2012-08-10 | 2018-10-30 | Epizyme, Inc. | Inhibitors of protein methyltransferase DOT1L and methods of use thereof |
| WO2020013712A1 (en) * | 2018-07-09 | 2020-01-16 | Victoria Link Limited | Inhibitors of dnmt1 as anticancer therapeutic agents |
| US10653711B2 (en) | 2015-08-26 | 2020-05-19 | Janssen Pharmaceutica Nv | 6-6 bicyclic aromatic ring substituted nucleoside analogues for use as PRMT5 inhibitors |
| US10759764B2 (en) | 2013-10-18 | 2020-09-01 | President And Fellows Of Harvard College | Fluorination of organic compounds |
| US10881680B2 (en) | 2012-09-06 | 2021-01-05 | Epizyme, Inc. | Method of treating leukemia |
| US10898504B2 (en) | 2016-03-10 | 2021-01-26 | Janssen Pharmaceutica Nv | Substituted nucleoside analogues for use as PRMT5 inhibitors |
| US11059850B2 (en) | 2017-12-08 | 2021-07-13 | Janssen Pharmaceutica Nv | Spirobicyclic analogues |
| US11098062B2 (en) | 2016-10-03 | 2021-08-24 | Janssen Pharmaceutica Nv | Monocyclic and bicyclic ring system substituted carbanucleoside analogues for use as PRMT5 inhibitors |
| US11279970B2 (en) | 2017-02-27 | 2022-03-22 | Janssen Pharmaceutica Nv | Use of biomarkers in identifying cancer patients that will be responsive to treatment with a PRMT5 inhibitor |
| US11571437B2 (en) | 2019-06-06 | 2023-02-07 | Janssen Pharmaceutica Nv | Methods of treating cancer using PRMT5 inhibitors |
| CN116284188A (en) * | 2023-02-07 | 2023-06-23 | 南开大学 | Double-substrate inhibitor of DNA methyltransferase DNMT1 and application thereof |
| WO2024156288A1 (en) * | 2023-01-29 | 2024-08-02 | Shanghai Kygent Pharmaceutical Co., Ltd | Dnmt1 inhibitors, pharmaceutical compositions, and therapeutic applications |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011018435A1 (en) | 2009-08-10 | 2011-02-17 | Institut Curie | Method for predicting the sensitivity of a tumor to an epigenetic treatment |
-
2006
- 2006-01-18 WO PCT/US2006/001791 patent/WO2006078752A2/en active Application Filing
- 2006-01-18 EP EP06718809A patent/EP1844062A2/en not_active Withdrawn
Non-Patent Citations (8)
| Title |
|---|
| BORCHARDT, R. T. ET AL: "Potential inhibitors of S-adenosylmethionine-dependent methyltransferases . 4. Further modifications of the amino acid and base portions of S-adenosyl-L-homocysteine" JOURNAL OF MEDICINAL CHEMISTRY , 19(9), 1094-9 CODEN: JMCMAR; ISSN: 0022-2623, 1976, XP002412379 * |
| BORCHARDT, RONALD T. ET AL: "Potential inhibitors of S-adenosylmethionine-dependent methyltransferases . 2. Modification of the base portion of S-adenosylhomocysteine" JOURNAL OF MEDICINAL CHEMISTRY , 17(8), 868-73 CODEN: JMCMAR; ISSN: 0022-2623, 1974, XP002412378 * |
| BRUECKNER B ET AL: "DNA methyltransferase inhibitors: old and new drugs for an epigenetic cancer therapy" TRENDS IN PHARMACOLOGICAL SCIENCES, ELSEVIER, AMSTERDAM, NL, vol. 25, no. 11, November 2004 (2004-11), pages 551-554, XP004604038 ISSN: 0165-6147 * |
| COHEN, HELEN M. ET AL: "Determinants of cofactor binding to DNA methyltransferases : insights from a systematic series of structural variants of S-adenosylhomocysteine" ORGANIC & BIOMOLECULAR CHEMISTRY , 3(1), 152-161 CODEN: OBCRAK; ISSN: 1477-0520, 2005, XP002412376 * |
| COWARD, JAMES K. ET AL: "Analogs of S-adenosylhomocysteine as potential inhibitors of biological transmethylation. Specificity of the S-adenosylhomocysteine binding site" JOURNAL OF MEDICINAL CHEMISTRY , 16(5), 460-3 CODEN: JMCMAR; ISSN: 0022-2623, 1973, XP002412377 * |
| LIN, QING ET AL: "Design of allele-specific protein methyltransferase inhibitors" JOURNAL OF THE AMERICAN CHEMICAL SOCIETY , 123(47), 11608-11613 CODEN: JACSAT; ISSN: 0002-7863, 2001, XP002959577 * |
| MINNICK, ALBERT A. ET AL: "General synthetic approach to stable nitrogen analogs of S-adenosylmethionine" JOURNAL OF ORGANIC CHEMISTRY , 53(21), 4952-61 CODEN: JOCEAH; ISSN: 0022-3263, 1988, XP002412380 * |
| SERAFINOWSKI, PAWEL ET AL: "Synthesis and antiviral activity of some new S-adenosyl-L-homocysteine derivatives" JOURNAL OF MEDICINAL CHEMISTRY , 35(24), 4576-83 CODEN: JMCMAR; ISSN: 0022-2623, 1992, XP002955749 * |
Cited By (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8329726B2 (en) | 2005-05-20 | 2012-12-11 | Methylgene Inc. | Inhibitors of VEGF receptor and HGF receptor signaling |
| US8093264B2 (en) | 2005-05-20 | 2012-01-10 | Methylgene Inc. | Fused heterocycles as inhibitors of VEGF receptor and HGF receptor signaling |
| JP2012509351A (en) * | 2008-11-20 | 2012-04-19 | プレジデント アンド フェローズ オブ ハーバード カレッジ | Fluorination of organic compounds |
| US9024093B2 (en) | 2008-11-20 | 2015-05-05 | President And Fellows Of Harvard College | Fluorination of organic compounds |
| US8580762B2 (en) | 2010-12-03 | 2013-11-12 | Epizyme, Inc. | Substituted purine and 7-deazapurine compounds |
| US8722877B2 (en) | 2010-12-03 | 2014-05-13 | Epizyme, Inc. | 7-deazapurine modulators of histone methyltransferase, and methods of use thereof |
| US9096634B2 (en) | 2010-12-03 | 2015-08-04 | Epizyme, Inc. | Substituted purine and 7-deazapurine compounds |
| US9145438B2 (en) | 2010-12-03 | 2015-09-29 | Epizyme, Inc. | 7-deazapurine modulators of histone methyltransferase, and methods of use thereof |
| US9150516B2 (en) | 2011-04-12 | 2015-10-06 | President And Fellows Of Harvard College | Fluorination of organic compounds |
| WO2013062945A1 (en) * | 2011-10-24 | 2013-05-02 | Glaxosmithkline Intellectual Property Development Limited | Chemical compounds |
| JP2014530910A (en) * | 2011-10-24 | 2014-11-20 | グラクソスミスクライン、インテレクチュアル、プロパティー、ナンバー2、リミテッドGlaxosmithkline Intellectual Property No.2 Limited | New compounds |
| US9174984B2 (en) | 2011-10-24 | 2015-11-03 | Glaxosmithkline Intellectual Property (No.2) Limited | Chemical compounds |
| US10112968B2 (en) | 2012-08-10 | 2018-10-30 | Epizyme, Inc. | Inhibitors of protein methyltransferase DOT1L and methods of use thereof |
| US10881680B2 (en) | 2012-09-06 | 2021-01-05 | Epizyme, Inc. | Method of treating leukemia |
| US11633420B2 (en) | 2012-09-06 | 2023-04-25 | Epizyme, Inc. | Method of treating leukemia |
| US9737493B2 (en) | 2012-09-07 | 2017-08-22 | University Of Louisville Research Foundation, Inc. | Compositions and methods for modulating DNMT1 inhibitor activity |
| WO2014039860A3 (en) * | 2012-09-07 | 2015-07-16 | University Of Louisville Research Foundation, Inc. | Compositions and methods for modulating dnmt1 inhibitor activity |
| US9273083B2 (en) | 2012-09-26 | 2016-03-01 | President And Fellows Of Harvard College | Nickel fluorinating complexes and uses thereof |
| US9701707B2 (en) | 2013-03-15 | 2017-07-11 | Epizyme, Inc. | Methods of synthesizing substituted purine compounds |
| US9175032B2 (en) | 2013-03-15 | 2015-11-03 | Epizyme, Inc. | Methods of synthesizing substituted purine compounds |
| US9738679B2 (en) | 2013-03-15 | 2017-08-22 | Epizyme, Inc. | Methods of synthesizing substituted purine compounds |
| US11572383B2 (en) | 2013-03-15 | 2023-02-07 | Epizyme, Inc. | Methods of synthesizing substituted purine compounds |
| US11753433B2 (en) | 2013-03-15 | 2023-09-12 | Epizyme, Inc. | Methods of synthesizing substituted purine compounds |
| US10968247B2 (en) | 2013-03-15 | 2021-04-06 | Epizyme, Inc. | Methods of synthesizing substituted purine compounds |
| WO2015040169A1 (en) * | 2013-09-19 | 2015-03-26 | Pierre Fabre Medicament | Quinazoline derivatives and their use as dna methyltransferase inhibitors |
| US9951044B2 (en) | 2013-09-19 | 2018-04-24 | Pierre Fabre Medicament | Quinazoline derivatives and their use as DNA methyltransferase inhibitors |
| CN105555781A (en) * | 2013-09-19 | 2016-05-04 | 皮埃尔法布雷医药公司 | Quinazoline derivatives and their use as DNA methyltransferase inhibitors |
| US10759764B2 (en) | 2013-10-18 | 2020-09-01 | President And Fellows Of Harvard College | Fluorination of organic compounds |
| US11318157B2 (en) | 2015-08-26 | 2022-05-03 | Janssen Pharmaceutica Nv | 6-6 bicyclic aromatic ring substituted nucleoside analogues for use as PRMT5 inhibitors |
| US10653711B2 (en) | 2015-08-26 | 2020-05-19 | Janssen Pharmaceutica Nv | 6-6 bicyclic aromatic ring substituted nucleoside analogues for use as PRMT5 inhibitors |
| US11883367B2 (en) | 2015-08-26 | 2024-01-30 | Janssen Pharmaceutica Nv | 6-6 bicyclic aromatic ring substituted nucleoside analogues for use as PRMT5 inhibitors |
| EP3207932A1 (en) | 2016-02-19 | 2017-08-23 | Universität Stuttgart | Dna methyltransferase inhibitors for rett syndrome therapy |
| US10898504B2 (en) | 2016-03-10 | 2021-01-26 | Janssen Pharmaceutica Nv | Substituted nucleoside analogues for use as PRMT5 inhibitors |
| WO2017216726A1 (en) | 2016-06-13 | 2017-12-21 | Glaxosmithkline Intellectual Property Development Limited | Substituted pyridines as inhibitors of dnmt1 |
| WO2017216727A1 (en) | 2016-06-13 | 2017-12-21 | Glaxosmithkline Intellectual Property Development Limited | Substituted pyridines as inhibitors of dnmt1 |
| US11098062B2 (en) | 2016-10-03 | 2021-08-24 | Janssen Pharmaceutica Nv | Monocyclic and bicyclic ring system substituted carbanucleoside analogues for use as PRMT5 inhibitors |
| US11993614B2 (en) | 2016-10-03 | 2024-05-28 | Janssen Pharmaceutica Nv | Monocyclic and bicyclic ring system substituted carbanucleoside analogues for use as PRMT5 inhibitors |
| US11279970B2 (en) | 2017-02-27 | 2022-03-22 | Janssen Pharmaceutica Nv | Use of biomarkers in identifying cancer patients that will be responsive to treatment with a PRMT5 inhibitor |
| US11999993B2 (en) | 2017-02-27 | 2024-06-04 | Janssen Pharmaceutica Nv | Use of biomarkers in identifying cancer patients that will be responsive to treatment with a PRMT5 inhibitor |
| US11059850B2 (en) | 2017-12-08 | 2021-07-13 | Janssen Pharmaceutica Nv | Spirobicyclic analogues |
| US11702441B2 (en) | 2017-12-08 | 2023-07-18 | Janssen Pharmaceutica Nv | Spirobicyclic analogues |
| US11518779B2 (en) | 2018-07-09 | 2022-12-06 | Victoria Link Limited | Inhibitors of DNMT1 as anticancer therapeutic agents |
| WO2020013712A1 (en) * | 2018-07-09 | 2020-01-16 | Victoria Link Limited | Inhibitors of dnmt1 as anticancer therapeutic agents |
| US11571437B2 (en) | 2019-06-06 | 2023-02-07 | Janssen Pharmaceutica Nv | Methods of treating cancer using PRMT5 inhibitors |
| WO2024156288A1 (en) * | 2023-01-29 | 2024-08-02 | Shanghai Kygent Pharmaceutical Co., Ltd | Dnmt1 inhibitors, pharmaceutical compositions, and therapeutic applications |
| CN116284188A (en) * | 2023-02-07 | 2023-06-23 | 南开大学 | Double-substrate inhibitor of DNA methyltransferase DNMT1 and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1844062A2 (en) | 2007-10-17 |
| WO2006078752A3 (en) | 2007-02-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006078752A2 (en) | Inhibitors of dna methyltransferase | |
| US20080132525A1 (en) | Inhibitors of DNA Methyltransferase | |
| US5736549A (en) | Hypoxanthine and guanine compounds | |
| Mansuri et al. | Preparation of 1-(2, 3-dideoxy-. beta.-D-glycero-pent-2-enofuranosyl) thymine (d4T) and 2', 3'-dideoxyadenosine (ddA): general methods for the synthesis of 2', 3'-olefinic and 2', 3'-dideoxy nucleoside analogs active against HIV | |
| FI93217B (en) | Process for the preparation of esters and amides of therapeutically active nucleoside derivatives | |
| MXPA97002488A (en) | Compounds of purine and guanine as inhibitors of | |
| US20030144501A1 (en) | Conformationally constrained L-nucleosides | |
| MXPA92005623A (en) | Pyrimidine nucleoside derivatives having anti-tumor activity, their preparation and use. | |
| WO2011005595A2 (en) | 2-5a analogs and their methods of use | |
| EP2227481A2 (en) | Antiviral nucleoside compounds | |
| EP1827460A1 (en) | Nucleosides with antiviral and anticancer activity | |
| KR20190077606A (en) | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs | |
| WO1994018215A1 (en) | Adenosine kinase inhibitors comprising lyxofuranosyl derivatives | |
| DK148745B (en) | RIBOFURANOSYL DERIVATIVES USED AS INTERMEDIATES IN THE PREPARATION OF 5'-DEOXY-5-FLUORCYTIDINE OR 5'-DEOXY-5-FLUORURIDINE OR PHYSIOLOGICALLY TOLERABLE ACID ADDITIONAL SALTS | |
| JP2022531899A (en) | Modified cyclic dinucleoside compound as a STING modulator | |
| WO2003053989A1 (en) | Masked phosphate containing nucleoside derivatives and their use as antivirals | |
| EP1198458B1 (en) | Ceramide analogs, process for their preparation and their use as antitumor agents | |
| Zhang et al. | Synthesis of neplanocin F analogues as potential antiviral agents | |
| CA2290364A1 (en) | Method of producing tiazofurin and other c-nucleosides | |
| US20040122223A1 (en) | Non-natural nucleotides and dinucleotides | |
| EP1968971B1 (en) | Dioxolane derivates for the treatment of cancer | |
| Silamkoti et al. | Synthesis and biological activity of 2-fluoro adenine and 6-methyl purine nucleoside analogs as prodrugs for suicide gene therapy of cancer | |
| AU626600B2 (en) | Nucleoside derivatives | |
| JPH05148261A (en) | Substituted 2',3'-dideoxy-5-trifluoromethyl- uridines | |
| Bhattacharya et al. | Studies on the synthesis of furo [3, 2-d] pyrimidine C-nucleosides: new inosine analogues with antiprotozoan activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006718809 Country of ref document: EP |