WO2006070878A1 - Carboxylic acid derivative or salt thereof - Google Patents
Carboxylic acid derivative or salt thereof Download PDFInfo
- Publication number
- WO2006070878A1 WO2006070878A1 PCT/JP2005/024096 JP2005024096W WO2006070878A1 WO 2006070878 A1 WO2006070878 A1 WO 2006070878A1 JP 2005024096 W JP2005024096 W JP 2005024096W WO 2006070878 A1 WO2006070878 A1 WO 2006070878A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- methyl
- ring
- added
- tert
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/30—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by doubly bound oxygen or sulfur atoms or by two oxygen or sulfur atoms singly bound to the same carbon atom
- C07D211/32—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by doubly bound oxygen or sulfur atoms or by two oxygen or sulfur atoms singly bound to the same carbon atom by oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/39—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by esterified hydroxy groups
- C07C205/42—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by esterified hydroxy groups having nitro groups or esterified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C205/43—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by esterified hydroxy groups having nitro groups or esterified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton to carbon atoms of the same non-condensed six-membered aromatic ring or to carbon atoms of six-membered aromatic rings being part of the same condensed ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/44—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by —CHO groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/40—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having the nitrogen atom of the carboxamide group bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/11—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms containing cyano groups and singly-bound oxygen atoms bound to the same saturated acyclic carbon skeleton
- C07C255/13—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms containing cyano groups and singly-bound oxygen atoms bound to the same saturated acyclic carbon skeleton containing cyano groups and etherified hydroxy groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C257/00—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines
- C07C257/10—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines
- C07C257/18—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines having carbon atoms of amidino groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C257/00—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines
- C07C257/10—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines
- C07C257/20—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines having nitrogen atoms of amidino groups acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C259/00—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups
- C07C259/12—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. N-hydroxyamidines
- C07C259/18—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. N-hydroxyamidines having carbon atoms of hydroxamidine groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/62—Compounds containing any of the groups, X being a hetero atom, Y being any atom, e.g. N-acylcarbamates
- C07C271/64—Y being a hydrogen or a carbon atom, e.g. benzoylcarbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C307/00—Amides of sulfuric acids, i.e. compounds having singly-bound oxygen atoms of sulfate groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C307/04—Diamides of sulfuric acids
- C07C307/06—Diamides of sulfuric acids having nitrogen atoms of the sulfamide groups bound to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C307/00—Amides of sulfuric acids, i.e. compounds having singly-bound oxygen atoms of sulfate groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C307/04—Diamides of sulfuric acids
- C07C307/10—Diamides of sulfuric acids having nitrogen atoms of the sulfamide groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/02—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C311/08—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/30—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/37—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/26—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C317/32—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/31—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton
- C07C323/32—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton having at least one of the nitrogen atoms bound to an acyclic carbon atom of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/52—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/12—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/18—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D207/22—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/24—Oxygen or sulfur atoms
- C07D207/26—2-Pyrrolidones
- C07D207/263—2-Pyrrolidones with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to other ring carbon atoms
- C07D207/267—2-Pyrrolidones with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to other ring carbon atoms with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/42—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/28—Radicals substituted by singly-bound oxygen or sulphur atoms
- C07D213/30—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/84—Nitriles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/12—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with radicals, substituted by hetero atoms, attached to carbon atoms of the nitrogen-containing ring
- C07D217/18—Aralkyl radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/24—Benzimidazoles; Hydrogenated benzimidazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D235/30—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/06—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having one or two double bonds between ring members or between ring members and non-ring members
- C07D241/08—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having one or two double bonds between ring members or between ring members and non-ring members with oxygen atoms directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D241/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with nitrogen atoms directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D243/00—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms
- C07D243/06—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4
- C07D243/08—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4 not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/32—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/30—1,4-Oxazines; Hydrogenated 1,4-oxazines not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/10—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/08—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/12—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/38—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/52—Radicals substituted by nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to a novel carboxylic acid derivative or a salt thereof useful as a pharmaceutical, particularly an active blood coagulation factor VII inhibitor, and its use as a pharmaceutical.
- the compound of the present invention is useful as an active blood coagulation factor VII inhibitor.
- thromboembolic diseases such as myocardial infarction, cerebral thrombosis, and peripheral arterial thrombosis have increased year by year due to the westernization of lifestyle and the aging of the population.
- Anticoagulation therapy along with fibrinolysis and antiplatelet therapy, plays a part in medical treatment in the treatment and prevention of thrombosis (Non-patent Document 1).
- Non-patent Document 1 In particular, in the prevention of thrombosis, safety that can withstand long-term administration and the development of reliable and appropriate anticoagulant activity are essential.
- Lufarin potassium is widely used around the world as the only oral anticoagulant, but it is difficult to control its anticoagulant ability due to its characteristic mechanism.
- Non-patent Documents 2 and 3 it is a drug that is very difficult to use clinically, and the appearance of an anticoagulant that is more useful and easy to use is desired.
- the blood coagulation reaction is an extrinsic pathway and an intrinsic pathway force, and thrombin is generated by any pathway.
- Thrombin is not only responsible for the conversion of fibrinogen to fibrin, which is the final stage of coagulation, but is also deeply involved in platelet activation and aggregation (Non-patent Document 4), and its inhibitor is a target for drug discovery. As long as it has been at the center of anticoagulant research. However, there are still problems with safety, such as the occurrence of force S and bleeding (Non-patent Document 5).
- Active ⁇ blood coagulation factor VII is an enzyme located at the start of the extrinsic clotting pathway, and its enzymatic activity increases markedly by binding to tissue factor (TF), a protein cofactor. Tissue factor is known to be upregulated in various thrombotic diseases, suggesting involvement of the extrinsic coagulation pathway in thrombotic diseases (Non-patent Document 6).
- Activated blood coagulation Selective inhibitors of factor VII may be superior anticoagulants with less bleeding side effects by not inhibiting endogenous coagulation.
- Patent Document 1 discloses that a novel amidino derivative represented by the following general formula has an action of inhibiting activated blood coagulation factor VII, including generalized intravascular coagulation syndrome and coronary artery thrombosis. It is described that it is useful as a preventive or therapeutic agent.
- the E 2 ring and the E 4 ring are directly bonded to each other, and the B and C rings are bonded via the linker (L), so that the structure is different from that of the present compound. .
- Patent Document 2 describes a wide range of compounds represented by the following general formulas as active ⁇ blood coagulation factor VII and active ⁇ blood coagulation factor X inhibitors. However, the compounds described as examples are limited to compounds having a biaryl structure in which X is a bond, and the compounds of the present invention having a linker are not disclosed.
- E 1 and L represent carbocycle, heterocycle, etc.
- X represents a bond, unsubstituted or substituted C-methylene, etc.
- Patent Document 3 describes the following as inhibitors of active blood coagulation factor VII, activated blood coagulation factor IX, activated blood coagulation factor X, activated blood coagulation factor XI and the like.
- General Compounds of the formula are described. However, these compounds differ in structure from the compounds of the present invention in that there is no ring or linker corresponding to the B ring of the compounds of the present invention.
- Patent Document 4 describes a compound represented by the following general formula as a serine protease inhibitor. However, there is no description that these compounds selectively inhibit active blood coagulation factor VII. Further, these compounds differ in structure from the compounds of the present invention in that there is no ring corresponding to the B ring of the compounds of the present invention or no linker exists.
- Patent Document 5 describes a wide variety of compounds represented by the following general formulas as inhibitors of active blood coagulation factor VII, active blood coagulation factor IX and the like.
- the example compounds having a carboxylic acid moiety which is a feature of the present invention, have vias in which Z Q is a bond. It is limited to compounds having a reel structure, and no compound having a linker is disclosed.
- Patent Document 6 describes that a phenyldaricin derivative represented by the following general formula has an inhibitory effect on activated blood coagulation factor VII.
- these compounds differ in structure from the present invention compounds in which the J part is not an amino acid, in that the site that connects the two rings is an amino acid.
- Patent Document 7 describes that (thio) urea derivatives having the following general formula have an activity of inhibiting blood coagulation factor VII.
- the structure is different from that of the compound of the present invention in that there is no ring corresponding to the ring B of the compound of the present invention.
- Patent Document 8 describes that a compound represented by the following general formula has an inhibitory action on platelet aggregation. However, there is no description that it has an activity of inhibiting blood coagulation factor VII. In addition, these compounds have a pyrrolidine ring, but the compounds of the present invention differ in structure from the compounds of the present invention in that the C ring does not contain a pyrrolidine ring.
- Patent Document 9 describes that a compound represented by the following general formula has an activity of inhibiting active blood coagulation factor X. However, there is no description that it has an activity of inhibiting blood coagulation factor VII. Further, these compounds are limited to the substituent force S on Q 1 corresponding to the A ring of the compound of the present invention, and there is no substituent corresponding to R 1 of the compound of the present invention. In this respect, the structure is different from the compound of the present invention.
- Patent Document 10 a wide range of compounds represented by the following general formulas are activated blood coagulation.
- R 4 is hydrogen, alkyl, halo, haloalkyl, nitro-nitro, -OR 5 , -C (0) OR 5 , -N (R 5 ) R 6 , -C (0) N (R 5 ) R 6 or -R 8 — N (R 5 ) R 6 , R 8 represents a straight-chain or branched alkylene, alkylidene, or alkylidyne chain (for the symbols in other formulas, see the publication of Patent Document 10).
- Non-Patent Document 1 "General Clinical", 1989, 41st page, p.2141-2145
- Non-Patent Document 2 “The New England Journal of Medicine”, (USA), 1991, No. 324, No. 26, p.1865- 1875
- Non-Patent Document 3 "The Journal of CI inical Pharmacology” (USA), 1992, 32nd, p.196-209
- Non-Patent Document 4 Osamu Matsuo, “t-PA and pro-UK”, Interdisciplinary Planning, 1986, Blood Coagulation, p.5-40
- Non-Patent Document 5 “Biomedica Biochimica Acta” (Germany), 1985, No. 44, P.1201-1210
- Non-Patent Document 6 “Thrombosis and Haemostasis” (Germany), 1997, 78th, No. 1, p.759-764
- Patent Document 1 International Publication No. 99/41231 Pamphlet
- Patent Document 2 Pamphlet of International Publication No. 02/34711
- Patent Document 3 International Publication No. 02/37937 Pamphlet
- Patent Document 4 International Publication No. 02/42273 Pamphlet
- Patent Document 5 Pamphlet of International Publication No. 01/87854
- Patent Document 6 International Publication No. 00/35858 Pamphlet
- Patent Document 7 European Patent Application Publication No. 1162194
- Patent Document 8 European Patent Publication No. 0528369
- Patent Document 9 International Publication No. 00/39118 Pamphlet
- Patent Document 10 International Publication No. 99/32477 pamphlet
- the active ⁇ blood coagulation factor VII selective inhibitor can be expected to be more efficient than the thrombin inhibitor and selectively inhibit the coagulation system in anticoagulation therapy.
- an active ⁇ blood coagulation factor VII selective inhibitor is expected as a drug with fewer side effects. Therefore, the creation of an activated blood coagulation factor VII selective inhibitor having a chemical structure different from that of the compound described in the above-mentioned patent document and having a superior effect is eagerly desired.
- the present invention relates to a carboxylic acid derivative represented by the formula (I) or a pharmaceutically acceptable salt thereof.
- Ring A Aaryl ring or heteroaryl ring.
- Ring B benzene ring, naphthalene ring, monocyclic to bicyclic heteroaryl ring.
- Ring C a cycloalkyl ring, an aryl ring or a hetero ring.
- n, and p each independently an integer from 0 to 3. However, if m, n or p is 2 or more, R 2 , R 3 or R 4 are the same or different from each other! /
- R °°, -NR ° C ( 0) _halogeno lower alkyl, cycloalkyl, aryl or hetero ring group.
- the lower alkyl, cycloalkyl, aryl or hetero ring group in R 2 and R 3 may be substituted.
- each of two R 2 or R 3 may be in the form of -0-lower alkylene-0-.
- R Q and R QQ each independently -H or lower alkyl.
- the lower alkyl, lower alkenyl, cycloalkyl, aryl, and heterocyclic groups in R 4 may each be substituted.
- R 6 , R 6a and R 6b each independently -H, or an optionally substituted lower alkyl, lower alkenyl, cycloalkyl, aryl or heterocyclic group.
- R 5 -OR 0 , -NR ° R. . Or -N (R °) -lower alkylene-0R. . .
- Y Single bond, each substituted, may be lower alkylene or lower alkylene. However, when C ring is a hetero ring, pyrrolidine is excluded. The same applies below. ]
- the present invention also relates to a pharmaceutical composition
- a pharmaceutical composition comprising a carboxylic acid derivative represented by the above formula (I) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, in particular, activated.
- Diseases caused by blood coagulation factor VII inhibition, blood coagulation inhibitors, or thrombus or embolism especially ischemic heart disease, restenosis after angioplasty, cerebral thrombosis, transient ischemic attack, peripheral Arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular coagulation syndrome (DIC), thrombus formation after heart valve replacement, coagulation or inflammation of perfused blood during extracorporeal circulation, arteries
- the present invention also relates to a pharmaceutical composition that is a prophylactic or therapeutic agent for sclerosis and cancer.
- composition comprising the compound of the general formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier;
- composition according to (1) which is a preventive or therapeutic agent for diseases caused by thrombus or embolism;
- composition according to (1) which is a preventive or therapeutic agent for syndrome (DIC), thrombus formation after heart valve replacement, blood coagulation or inflammation during arterial circulation, arteriosclerosis, and cancer;
- Ischemic heart disease restenosis after angioplasty, cerebral thrombosis, transient ischemic attack, peripheral arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular coagulation Claims (1) for the manufacture of drugs for the prevention or treatment of syndrome (DIC), thrombus formation after heart valve replacement, coagulation or inflammation of perfused blood during extracorporeal circulation, arteriosclerosis, cancer Use of the described compounds or pharmaceutically acceptable salts thereof;
- a method for preventing or treating a disease caused by thrombus or embolism comprising administering to a patient a therapeutically effective amount of a compound of the general formula (I) or a salt thereof;
- (10) Ischemic heart disease, restenosis after angioplasty, cerebral thrombosis, transient cerebral imagination, including administering to a patient a therapeutically effective amount of a compound of general formula (I) or a salt thereof Blood attack, peripheral arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular coagulation syndrome (DIC), thrombus formation after heart valve replacement, blood coagulation of perfused blood during extracorporeal circulation Or prevention or treatment of inflammation, arteriosclerosis, cancer
- lower means 1 to 6 carbon atoms (hereinafter referred to as “C” unless otherwise specified).
- Alkyl alkylene
- alkylene alkylene
- alkylene may each be linear or branched.
- lower alkyl means C alkyl. Specifically, methyl,
- Examples include til, normal propyl, isopropyl, normal butyl, isobutyl, sec-butyl, tert-butyl, normal pentyl, normal hexyl and the like.
- C til, normal propyl, isopropyl, normal butyl, isobutyl, sec-butyl, tert-butyl, normal pentyl, normal hexyl and the like.
- “Lower alkellule” is a C alkell having one or more double bonds at any position.
- “Lower alkylene” means a divalent group formed by removing one hydrogen at any position of the “lower alkyl”. Specifically,-(CH)-,-CH (CH)-,-C (CH)-,-CH CH (C
- Cycloalkyl means a non-aromatic hydrocarbon ring of C, and a bridged ring spiro ring.
- Specific examples include cyclopropinole, cyclobutinole, cyclopentinole, cyclohexenole, cycloheptinole, cyclooctyl, cyclohexenyl, cyclooctanegel, adamantyl and norbol.
- Preferred is C cycloalkyl, more preferred is cyclobuty
- Halogen means a halogen atom. Specific examples include fluoro, black mouth, bromo and iodine. Preferred are fluoro, black mouth and bromo, and more preferred are fluoro and black mouth.
- halogeno lower alkyl means one or more arbitrary hydrogen atom forces of the “lower alkyl” which are the same or different from each other and substituted with the “halogen”. Specific examples include trifluoromethyl, pentafluoroethyl and the like. Preferred is trifluoromethyl.
- aryl means a monocyclic to tricyclic C aromatic hydrocarbon ring, specifically
- Examples include phenyl and naphthyl, and is preferably phenyl.
- the cycloalkyl ring may be condensed.
- indanyl or tetrahydronaphthyl may be formed.
- “Monocyclic heteroreel” means a group power consisting of N, O, and S forces.
- the ring atom S or N may be oxidized to form an oxide or dioxide.
- Specific examples include pyridyl, pyridazyl, pyrimidinyl, pyrazinyl, furyl, chael, pyrrolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, thiadiazolyl, imidazolyl, triazolyl, tetrazolyl and the like.
- Pyridyl and birazinyl are preferable, and pyridyl is more preferable.
- Heteroaryl refers to the above-mentioned “monocyclic heteroreel", the monocyclic heteroreel or “bicyclic heteroreel” in which the monocyclic heteroreel is fused with a benzene ring, and
- the bicyclic heteroaryl is a monocyclic heteroaryl or a tricyclic heteroaryl fused with a benzene ring.
- the ring atom S or N may be oxidized to form a dioxide.
- bicyclic heteroaryl examples include benzofuranyl, benzochel, benzoxazolyl, benzoimidazolyl, indolyl, benzothiazolyl, quinolinyl, quinazolyl, quinoxalinyl, cinnolyl and the like. Indolyl and benzimidazolyl are preferred.
- Specific examples of the tricyclic heteroaryl include carbazolyl and fluorenyl.
- Specific examples of the hetero reel include the groups described in the above-mentioned “monocyclic hetero reel”, “bicyclic hetero reel”, and “tricyclic hetero reel”. Pyridyl, pyradyl, indolyl and benzimidazolyl are preferable, and pyridyl is more preferable.
- Heterocycle refers to a 3- to 8-membered monocyclic heterocycle having 1 to 4 heteroatoms in which a group force consisting of N, O and S is also selected.
- the ring atom N or S may be oxidized to form an oxydoxide or a bridged ring or a spiro ring.
- Substituents which may be substituted in R 2 and R 3 and are allowed by “lower alkyl” are preferably groups selected from the following group P 1 .
- Q Group 1 lower alkyl, halogeno lower alkyl, halogen, oxo, -OR. And -0-halogeno lower alkyl.
- R 4, R 6, R 6a and R may be substituted in 6b "lower alkyl” and "lower Aruke - Le" As preferred substituents allowed in, include groups selected from the following P 2 group Be
- R 6A and R 6B which may be substituted, respectively, in R 6A and R 6B "cycloalkyl", as preferred substituents allowed in " ⁇ Re window” and “heterocyclic group” is a group selected from the Q 2 group Can be mentioned.
- the A ring is preferably a benzene ring, a pyridine ring or a benzoimidazole ring, more preferably a benzene ring or a pyridine ring, and still more preferably a benzene ring.
- B ring is preferably a monocyclic 6-membered aromatic ring, and more preferably a benzene ring or a pyridine ring.
- the C ring is preferably a benzene ring, piperidine ring, piperazine ring or morpholine ring, and more preferably a benzene ring or piperidine ring.
- R 2 is preferably halogen.
- R 3 is preferably lower alkyl, -0-lower alkyl, or halogen, and more preferably methyl, -0-methyl, black mouth, or fluoro.
- R 4 is preferably a heterocyclic group, -0-lower alkyl, -lower alkylene-OH, -0- Cycloalkyl or —O-lower alkylene- (substituted with lower alkyl !, may! /, Heterocycle), more preferably —0-lower alkyl, —lower alkylene-OH, or —0— Lower alkylene- (which may be substituted with lower alkyl, or a heterocycle), and more preferably is 0-ethyl, -0-isopropyl or -lower alkylene-OH.
- R 5 is preferably -0H or -0-lower alkyl, more preferably -0H or -0-ethyl.
- L is preferably * -NHCH- (where * represents a bond to the B ring).
- X is preferably a single bond or -0-, more preferably -0-.
- Y is preferably lower alkylene, more preferably methylene or ethylene.
- B 2 , B 3 , B 4 each independently CH, CR 3 , N or N (0). However, among B 4 , CR 3 is 3 or less, and each CR 3 may be the same or different from each other. The same applies below. ]
- the group force which also has power The compound or its pharmaceutically acceptable salt as described in (1) selected. Further, other preferred compounds in the compound of the present invention represented by the general formula (I) are shown below.
- [B 5 , B, B 7 and B are each independently CH, CR or N. However, among B 5 , B, B 7 and B, CR 3a is 3 or less, and each CR 3a may be the same or different from each other.
- R QQ cycloalkyl, aryl or heterocyclic group.
- the cycloalkyl, aryl, and heterocyclic groups in R 2a and R 3a may be substituted with lower alkyl, halogeno lower alkyl, halogen, oxo, -OR °, or 0-0-norogeno lower alkyl. .
- each of two R 2a or R 3a may be in the form of -0-lower alkylene-0-.
- R 6e lower alkyl in R 1 ⁇ 2 is substituted with a group selected also P a group force and may, cycloalkyl, be the heterocyclic group Ariru and to be substituted with a group selected respectively Q a group force Good.
- R 6c , R 6d and R 6e each independently —H, lower alkyl, cycloalkyl, aryl or hetero ring group.
- R 6e , R 6d and R 6e is a cycloalkyl, aryl and hetero ring group which may be substituted with a group selected from group P a and each is selected from group Q a May be substituted with a group.
- cycloalkyl, aryl or heterocycle 0
- R °° cycloalkyl, aryl or heterocycle.
- the cycloalkyl, aryl and hetero ring group in the P a group may be substituted with a group selected from the Q a group.
- R 5a —OH or —0-lower alkyl.
- Y a lower alkylene substituted with halogen or —OR °. ]
- the compound of the present invention may have various stereoisomers such as geometric isomers, tautomers and optical isomers.
- the present invention includes mixtures thereof and isolated ones.
- the compound of the present invention may form an acid addition salt. It may also form a salt with a base.
- strong salts include mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, Fumaric acid, maleic acid, lactic acid, malic acid, tartaric acid, citrate, trifluoroacetic acid, methanesulfonic acid, ethanesulfonic acid and other organic acids, acid addition salts with aspartic acid, glutamic acid and other acidic amino acids, sodium, Examples thereof include inorganic bases such as potassium, magnesium, calcium, and aluminum, organic bases such as methylamine, ethylamine, and ethanolamine, salts with basic amino acids such as lysine and oltin, and ammonium salts.
- the present invention includes hydrates of the compounds of the present invention, various pharmaceutically acceptable solvates, crystal polymorphs, and the like. It should be understood that the present invention is not limited to the compounds described in the examples below, and all of the carboxylic acid derivatives represented by the general formula (I) or pharmaceutically acceptable salts thereof. Is included.
- the compounds of the present invention also include all compounds that are metabolized in vivo and converted to the compounds having general formula (I) or salts thereof, so-called prodrugs. Examples of the group that forms a prodrug of the compound of the present invention include those described in Prog. Med.
- a prodrug of the compound of the present invention a prodrug having a hydroxyl group and an ester group that undergoes metabolism in a living body and becomes a carboxylic acid derivative represented by the general formula (I) can be considered.
- a prodrug having a hydroxyl group and an ester group that undergoes metabolism in a living body and becomes a carboxylic acid derivative represented by the general formula (I) can be considered.
- the compound of the present invention and a pharmaceutically acceptable salt thereof can be produced by applying various known synthetic methods using characteristics based on the basic skeleton or the type of substituent.
- a typical production method is illustrated below.
- it is effective in terms of production technology to replace the functional group with a suitable protecting group at the raw material or intermediate stage, that is, a group that can be easily converted to the functional group. There is a case. Thereafter, the protecting group can be removed as necessary to obtain the desired compound.
- functional groups include a hydroxyl group, a carboxyl group, an amino group, and the like, and examples of protective groups thereof include those described in “Protective Groups in Organic Synthesis (third) by Greene and Wuts”. edition) ”, and these may be appropriately used depending on the reaction conditions.
- R 8 , R 9 , R 1Q , R U , R 12 and X 1 are R 1 R 2 , R 3 , A substituent that can be converted to R 5 and X.
- J 2 and J 3 are substituents that can be converted into a bond J through a process that can be usually employed by those skilled in the art, and specifically include methyl, hydroxymethyl, halogenomethyl, and the like. Group, formyl group, alkoxymethyl group, acetoxymethyl group, amino group, alkoxycarboamino group, nitro group, carboxyl group, alkoxycarbo group and the like.
- L 1 and L 2 mean a substituent that can be converted to a bond L through a process that can be usually employed by those skilled in the art, and specifically include a methyl group, a hydroxymethyl group, and a halogenomethyl group.
- R 8 , R 9 , R 1Q , R u , R 12 and X 1 are each It may be the same as R 5 and X. The same shall apply hereinafter. ) [0048] Process A
- any step that can be usually employed by those skilled in the art for example, halogenation reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction, alkylation reaction, reductive alkyl-rich reaction, etc. It can be implemented by combining with.
- This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
- the halogenation reaction can be a halogenation reaction that can be usually used by those skilled in the art.
- the method described in “Chemical Experiment Course (4th edition)” edited by The Chemical Society of Japan, 19 ⁇ (1992) (Maruzen) can be applied.
- N-promosuccinimide or the like can be used as a halogenating agent in the presence of 2,2′-azobisisobutyryl-tolyl or peroxybenzoyl.
- Reactions include aromatic hydrocarbons such as benzene, toluene, xylene, esters such as ethyl acetate, ethers such as dimethyl ether, tetrahydrofuran (THF), 1,4-dioxane, dichloromethane, 1,2-dichloroethane, Halogenated hydrocarbons such as black mouth form, alcohols such as methanol and ethanol, ⁇ , ⁇ -dimethylformamide (DMF), ⁇ , ⁇ -dimethylacetamide (DMA), ⁇ -methylpyrrolidone ( ⁇ ), dimethyl sulfoxide
- the reaction can be carried out in a solvent inert to the reaction such as (DMSO), acetonitrile, pyridine, water, etc. under cooling to heating under reflux.
- an acid-acid reaction that can be usually used by those skilled in the art can be used.
- the method described in “Chemical Experiment Course (4th edition)” edited by The Chemical Society of Japan, 23 ⁇ (1992) (Maruzen) can be applied.
- a reduction reaction which can be usually used by those skilled in the art can be adopted.
- a reducing agent such as lithium aluminum hydride and sodium borohydride
- Room temperature to Caro in solvent It can be carried out under heat reflux.
- a hydrolysis reaction which can be usually used by those skilled in the art can be used.
- sulfuric acid, hydrochloric acid, hydrobromic acid in a solvent inert to the reaction such as the above aromatic hydrocarbons, ethers, halogenated hydrocarbons, alcohols, DMF, DMA, NMP, DMSO, pyridine, etc.
- an acid such as mineral acid such as formic acid and acetic acid
- a base such as lithium hydroxide, sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate, cesium carbonate or ammonia.
- the reaction can be carried out under cooling to heating under reflux.
- the hydrogenolysis reaction such as debenzylation
- a hydrogenolysis reaction which can be usually used by those skilled in the art can be used.
- a catalyst use is made of noradium carbon, Raney nickel, platinum, etc., and the above-mentioned aromatic hydrocarbons, esters, ethers, halogenated hydrocarbons, DMF.
- the reaction can be carried out in a solvent inert to the reaction, such as DMA, NMP, ethyl acetate, acetonitrile, and acetic acid, from room temperature to under reflux.
- an acid preferably hydrochloric acid, acetic acid, etc.
- amidy salt which can be usually used by those skilled in the art can be adopted.
- methods using acid chloride, carbodiimidazole (CDI), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (WSC 'HCl), dicyclohexylcarbodiimide, diphenylphosphoryl A method using a condensing agent such as ruazide and jetylphosphyl cyanide, and a method using a mixed acid anhydride using isobutyl chloroformate, ethyl ethyl formate, and the like are preferable.
- reaction is carried out in a solvent inert to the reaction such as the above-mentioned halogenated hydrocarbons, aromatic hydrocarbons, ethers, DMF, DMA, NMP, DMSO or the like, under cooling or under reflux with heating.
- a solvent inert such as the above-mentioned halogenated hydrocarbons, aromatic hydrocarbons, ethers, DMF, DMA, NMP, DMSO or the like, under cooling or under reflux with heating.
- a base preferably triethylamine, di-sodium pyrethylamine, N-methylmorpholine, pyridine, 4- (N, N-dimethylamino) pyridine, etc.
- alkyl groups such as a hydroxyl group and an amino group
- alkyl groups generally used by those skilled in the art can be employed.
- solvent or inactive solvents such as aromatic hydrocarbons, esters, ethers, halogenated hydrocarbons, DMF, DMA, NMP, DMSO, and acetonitrile, or alcohols
- the reaction can be carried out in a solvent such as a mixture at room temperature or under reflux.
- organic bases triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4- (N, N-dimethylamino) pyridine, etc. are preferably used
- metal salt bases potassium carbonate
- reductive alkyl group a reductive alkyl group commonly used by those skilled in the art can be adopted.
- solvents inert to the reaction such as halogenated hydrocarbons, aromatic hydrocarbons, esters, ethers, alcohols, acetic acid, sodium borohydride, sodium triacetoxyborohydride, etc. It is preferable to use a reducing agent under cooling and at room temperature or under reflux with heating.
- the reaction in the presence of an acid such as a mineral acid such as sulfuric acid, hydrochloric acid or hydrobromic acid, or an organic acid such as formic acid or acetic acid.
- an acid such as a mineral acid such as sulfuric acid, hydrochloric acid or hydrobromic acid, or an organic acid such as formic acid or acetic acid.
- the reductive alkyl group uses, for example, noradium carbon, Raney nickel, platinum or the like as a catalyst, and the above-mentioned aromatic hydrocarbons, esters, ethers, halogenated substances in a hydrogen atmosphere of normal pressure to pressure.
- the reaction can be carried out in a solvent inert to the reaction, such as hydrocarbons, DMF, DMA, NMP, acetonitrile, and acetic acid, at room temperature to under reflux.
- an acid preferably hydrochloric acid, acetic acid, etc.
- This step is a step that can be usually employed by those skilled in the art, for example, halogenation reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction,
- the reaction can be carried out by arbitrarily combining a rukylation reaction, a reductive alkyl reaction or the like. This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
- Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
- any step that can be usually employed by those skilled in the art for example, halogenation reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction, alkylation reaction, reductive alkylation reaction, etc. It can implement by combining.
- This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
- Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
- the compounds of the present invention can also be produced by the following typical production methods.
- any step that can be usually employed by those skilled in the art for example, halogenation reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction, alkylation reaction, reductive alkyl-rich reaction, etc. It can be implemented by combining with.
- This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
- Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
- This step is a step usually employed by those skilled in the art, such as halogen.
- the reaction can be carried out by arbitrarily combining oxidization reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction, alkylation reaction, reductive alkyl reaction, and the like. This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
- Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
- R 9 , R 1Q , R U , R 12 or X 1 are each R 8 , R 9 , R 1Q , R U , R 12 or X 1 as required if different from R 5 and X
- This step is a step that can be usually employed by those skilled in the art, such as halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, reductive alkylation, etc. It can implement by combining arbitrarily.
- This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
- Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
- R 8 , R 9 , R 1Q , R U , R 12 or X 1 is R 8 , R 9 , R 1Q , R U , R 12 or X 1 as R 1 R 2 , if different from R 4 , R 5 and X, respectively, as required
- Process C or process F converted to R 5 and X is process
- the raw material used for the production of the compound of the present invention can be produced, for example, by using the method described in the Reference Examples described below, a known method, a method obvious to those skilled in the art, or a modification thereof. it can.
- the compound of the present invention thus produced can be isolated and purified by a known method such as extraction, precipitation, fractional chromatography, fractional crystallization, recrystallization and the like.
- the salt of this invention compound can be guide
- optical isomer when the compound of the present invention has an asymmetric carbon, an optical isomer exists. These optical isomers can be used for fractional crystallization, column chromatography, etc. It can be divided by conventional methods. Moreover, it can manufacture by using a suitable optically active raw material.
- a pharmaceutical composition containing, as an active ingredient, one or more of the compounds of the present invention represented by the general formula (I) or a pharmaceutically acceptable salt thereof, Prepared into tablets, powders, fine granules, granules, capsules, pills, solutions, injections, suppositories, ointments, patches, etc. using carriers and excipients and other additives, and orally Or administered parenterally.
- the clinical dose of the compound of the present invention to humans is appropriately determined in consideration of the patient's symptoms, body weight, age, sex, etc., but it is usually 0.1 to 500 mg orally per day for adults and 0 to parenterally.
- 01 ⁇ : LOOmg which is administered once or in several divided doses. Since the dosage varies depending on various conditions, the dosage may be less than the above dosage range.
- the solid composition for oral administration according to the present invention tablets, powders, granules and the like are used.
- one or more active substance forces at least one inert diluent such as lactose, mannitol, glucose, hydroxypropyl cellulose, microcrystalline cellulose, starch, poly It is mixed with bull pyrrolidone and magnesium aluminate metasilicate.
- the composition may contain an additive other than the inert diluent, for example, a lubricant, a disintegrant, a stabilizer, a solubilizer, or a solubilizing agent according to a conventional method.
- tablets or pills may be coated with sugar coating such as sucrose, gelatin, hydroxypropylcellulose, hydroxypropylmethylcellulose phthalate, etc., or gastric soluble sputum may be coated with an enteric film!
- Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, elixirs and the like, commonly used inert diluents, Examples include purified water and ethyl alcohol.
- This composition may contain solubilizers, solubilizers, wetting agents, suspending agents, sweeteners, flavors, fragrances, preservatives in addition to the inert diluent. Good.
- Injections for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions.
- aqueous solution and suspension diluent include distilled water for injection and physiological saline.
- diluents for non-aqueous solutions and suspensions include Examples include propylene glycol, polyethylene glycol, vegetable oils such as olive oil, alcohols such as ethyl alcohol, and polysorbate 80 (a pharmacopeia name).
- Such compositions may further contain additives such as isotonic agents, preservatives, wetting agents, emulsifiers, dispersants, stabilizers, solubilizers or solubilizers.
- These are sterilized by, for example, filtration through a bacteria-retaining filter, blending with a bactericide or irradiation.
- These can be prepared by preparing a sterile solid composition and dissolving it in sterile water or a sterile solvent for injection before use.
- Me methyl, Et: ethyl, cPr: cyclopropyl, iPr: isopropyl, nBu: normal butyl, iBu: isobutyl, tBu: tert-butyl, cBu: cyclobutyl, cHex: cyclohexyl, Ac: acetyl, Ph: phenyl, Bn: benzyl, Bo tert-butoxycarbonyl, TFA: trifluoroacetic acid, Ms: methanesulfonyl, null: unsubstituted, bond: bond.
- the obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as an elution solvent to obtain 1.08 g of 5-cyan-3-methoxysalicylaldehyde as a colorless solid.
- the obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as the elution solvent, and 3.9 g of 4-chloro-2- (hydroxymethyl) -6-odophenol was obtained. Obtained as a colorless oil (EP: 285). 841 mg of the resulting oil was dissolved in 6 mL of ⁇ , ⁇ -dimethylformamide, 496 mg of benzaldehyde dimethylacetal and 10 mg of p-toluenesulfonic acid were added at room temperature, and then 1 at 100 ° C. Stir for hours.
- the flask was charged with 22.6 mL of concentrated hydrochloric acid, 2.7 g of zinc chloride and 4.1 g of paraformaldehyde and bubbled with saline and hydrogen until dissolved.
- a solution of 15 g of 3-ethoxysalicylaldehyde in toluene (68 mL) was added thereto, and hydrogen chloride was blown in at room temperature for 2 hours with vigorous stirring.
- To the reaction mixture 45 mL of toluene was added and poured into about 150 g of ice. After stirring at room temperature for 20 minutes, the insoluble material was filtered off through Celite. After washing with toluene, the filtrate was separated.
- the organic layer was washed twice with water and then dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure to obtain a red-black solid.
- 29.6 g of sodium acetate was added at room temperature, and the mixture was stirred at the same temperature for 1.5 hours.
- About 300 mL of water was added to the reaction mixture, and the mixture was extracted with ethyl acetate (about 300 mL).
- the organic layer was washed twice with water, twice with saturated aqueous sodium hydrogen carbonate, and saturated brine, and dried over anhydrous magnesium sulfate.
- the obtained crude product was dissolved in 2 mL of ⁇ , ⁇ -dimethylformamide, 112 mg of potassium carbonate and 169 mg of tert-butyl bromoacetate were added, and the mixture was stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- the suspension (8.5 mL) was stirred at 70 ° C. for 14 hours under an argon atmosphere.
- the reaction mixture was allowed to cool, filtered through celite, and washed with ethyl acetate.
- the filtrate was washed with water and saturated brine, and dried over anhydrous magnesium sulfate.
- Ethyl (2-formyl-6-isopropoxy-4-butylphenoxy) acetate was added dropwise to a solution of AD-MIX- ⁇ 7.7 g in tert-butyl alcohol (15 mL) at 0 ° C. The mixture was warmed to room temperature and stirred for 12 hours. After adding 865 mg of sodium thiosulfate and stirring for 1 hour, 15 mL of water was added and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate and concentrated under reduced pressure.
- Tetrahydrofuran (5 mL) solution of tert-butyl (4-acetoxymethyl-2-formyl-6-hydroxyphenoxy) acetate 0.32 4 g, 1-tert-butoxy-2-propanol 0.145 g, triphenylphosphine 0.288 g
- 0.48 mL of a toluene solution (2.30 M) of jetillazodicarboxylate was added at room temperature and stirred at the same temperature for 1.5 hours.
- reaction mixture was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as an elution solvent, and tert-butyl [4-acetoxymethyl-2- (2-tert-butoxy-1-methyl) was obtained. Ethoxy) -6-formylphenoxy] ase A tarte of 0.085 g was obtained.
- NMR (CDC1): 1.20 (9H, s), 1.36 (3H, d, J 6.5 Hz), 1.49 (9
- the obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 3) as an elution solvent to obtain 11.24 g of a yellow oily substance.
- 11.24 g of the obtained yellow oily substance was dissolved in 300 mL of ethanol, added with 4.384 g of potassium carbonate, and stirred at room temperature for 5.5 hours. Potassium carbonate was removed by filtration, and the filtrate was concentrated under reduced pressure.
- the residue was diluted with ethyl acetate and washed successively with 0.5N hydrochloric acid, water and saturated brine.
- the reaction mixture was washed successively with water, diluted hydrochloric acid three times, saturated aqueous sodium hydrogen carbonate, water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1:10) as an elution solvent to obtain 10.985 g of a yellow oily substance. 100 mg of the obtained yellow oily substance was dissolved in ethanol (2 mL), 156 mg of potassium carbonate was added, and the mixture was stirred at room temperature for 2 hours and allowed to stand for 13 hours. Potassium carbonate was removed by filtration, and the filtrate was concentrated under reduced pressure.
- the reaction mixture was diluted with ethyl acetate, poured into 1N hydrochloric acid and extracted. Organic layer with water and saturated saline After washing, it was dried over anhydrous magnesium sulfate.
- the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 2) as the elution solvent, and tert-butyl 3- (4-acetoxymethyl-2- 148 mg of hydroxymethyl-6-isopropoxyphenol) propanoate was obtained as a colorless oil.
- the obtained colorless solid 500 mg and phenylboronic acid 317 mg were dissolved in ⁇ , ⁇ -dimethylformamide 10 mL, and potassium phosphate 552 mg and tetrakis (triphosphine) palladium 150 mg were dissolved at room temperature. The mixture was stirred at 100 ° C for 1.5 hours. Ethyl acetate and water were added to the reaction solution and extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- the obtained residue was dissolved in 100 mL of ⁇ , dimethylformamide, and 4.15 g of imidazole and 9.18 g of tert-butyldimethylchlorosilane were sequentially stirred at room temperature for 17 hours.
- the reaction solution was diluted with water and extracted with ethyl acetate.
- the organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (8: 1) as an elution solvent.
- the obtained yellow oil was dissolved in 100 mL of ethanol, an ethanol suspension of Raney nickel (3 mL) was added, and the mixture was stirred at room temperature for 17 hours under a hydrogen atmosphere of 1 atm.
- the reaction solution was filtered, and the filtrate was concentrated under reduced pressure.
- the obtained orange oil was dissolved in 50 mL of 1,2-dichloroethane, and 1.24 g of acetic acid and 4.30 g of 37% formaldehyde aqueous solution were prepared at room temperature, and then 3.30 g of sodium triacetoxyhydrogen hydride was brought to room temperature. And stirred for 1 hour.
- Nickel chloride hexahydrate A solution of 607 mg of methanol (50 mL) in sodium borohydride (300 mg) stirred at room temperature for 30 minutes was mixed with tert-butyl (2-formyl-6-methoxy-4- To a solution of 1.59 g of nitrophenoxy) acetate in methanol (30 mL) was added 193 mg of sodium borohydride under ice cooling, and the mixture was stirred for 5 minutes. To the reaction solution, 950 mg of sodium borohydride was added and stirred at room temperature for 1 hour. The reaction mixture was filtered through celite and concentrated under reduced pressure.
- tert-Butyl [4-amino-2- (hydroxymethyl) -6-methoxyphenoxy] acetate 480 mg in ⁇ , ⁇ -dimethylformamide (1 mL) solution, imidazole 230 mg and tert-butyldimethylchlorosilane 281 mg Were sequentially stirred at room temperature for 20 minutes. The reaction mixture was diluted with jetyl ether-ethyl acetate (1: 1), and the organic layer was washed successively with 5% aqueous citrate and then with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- the obtained residue was dissolved in 0.5 mL of pyridine, 0.08 mL of acetic anhydride was added at room temperature, and the mixture was stirred for 10 minutes.
- the reaction solution was concentrated under reduced pressure.
- Ammonium 555 mg was added and stirred at room temperature for 30 minutes.
- the reaction mixture was diluted with ethyl acetate, washed successively with saturated brine, aqueous ammonia and then saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- Jetyl ether was added to the residue, and the resulting precipitate was collected by filtration, and the filtrate was dried under reduced pressure to obtain 4.72 g of a pale yellow solid.
- the same procedure as in Reference Example 14 was performed to obtain tert-butyl ⁇ 4-[(dimethylamino) methyl] -2-ethoxy-6-formylphenoxy ⁇ acetate 1.12 g was obtained as a pale yellow solid.
- the obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (9: 1) as an elution solvent to obtain a colorless oil.
- the obtained colorless oil was dissolved in a mixed solvent of methylene chloride and ethanol (1: 1, 40 mL), and ozone gas was blown into the solution at ⁇ 78 ° C. for 1.5 hours, followed by stirring. After adding 355 mg of thiourea to the reaction solution at room temperature and stirring for 2 hours, insoluble matters were filtered off.
- the obtained red solid (2.00 g) was dissolved in 2-butanone (20 mL), potassium carbonate (4.88 g) and methyl iodide (1.95 g) were added at room temperature, and the mixture was heated to reflux for 2 hours. Add ethyl acetate and water to the reaction solution. And extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as an eluent to obtain 1.02 g of a yellow solid.
- the obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as an elution solvent to obtain 475 mg of a yellow foam. Dissolve the resulting yellow foam in 10 mL of ethanol and add Raney A water suspension (3 g) of Kell was added, and the mixture was stirred at room temperature for 30 minutes under a hydrogen atmosphere of 1 atm. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure to obtain 365 mg of tert-butyl [ ⁇ 4-[(2-amino-5-chlorobenzyl) amino] phenol ⁇ (imino) methyl] force rubamate. It was. FP: 375
- the organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain a crude product as a pale yellow solid.
- the obtained pale yellow solid 0.37 g, ammonium chloride 0.23 g, 1-hydroxybenzotriazole hydrate 0.16 g, (3-dimethylaminopropyl) ethylcarbodiimide monohydrochloride 0.20 g and ⁇ ,
- a solution of 0.55 g of) -diisopropylethylamine in ⁇ , N-dimethylformamide (1.5 mL) was stirred at room temperature for 13 hours. Water was added to the reaction solution and extracted with ethyl acetate.
- tert-Butyl [ ⁇ 4- [(5-Chromium-2-2-nitrobenzene) amino] phenol ⁇ (imino) methyl] strength rubamate 5.00 g of Raney nickel in a solution of 5.00 g of 1,4-dioxane (100 mL) 1.0 g of the water suspension was added and stirred for 2 days at room temperature in a hydrogen atmosphere of 1 atm. The reaction mixture was filtered, and the filtrate was concentrated under reduced pressure to give 4.43 g of tert-butyl [ ⁇ 4-[(2-amino-5-chlorobenzoyl) amino] phenol ⁇ (imino) methyl] force rubamate. Obtained.
- N- (4-Chanophenol) -5-methyl-2--trobensamide 14.3 g, hydroxylamine hydrochloride 4.9 g, ⁇ , ⁇ -diisopropylethylamine 14 mL and ethanol 100 mL Stir at ° C overnight.
- the reaction mixture was concentrated under reduced pressure, water was added to the resulting residue, and the precipitate was collected by filtration to obtain 12.5 g of a yellow solid.
- the obtained yellow solid was dissolved in tetrahydrofuran (50 mL) and ethanol (50 mL), 10% palladium-carbon (4.23 g) was added, and the mixture was stirred overnight at room temperature under a hydrogen atmosphere of 1 atm.
- the obtained colorless oil was dissolved in 20 mL of ⁇ , ⁇ -dimethylformamide, and 0.29 g of 4-cyanobenzoic acid, 0.40 g of 1-hydroxybenzotriazole and 1-ethyl-3- (3-dimethylaminopropyl) carpositimide 17 hours of monohydrochloride 0.57 g at room temperature Stir.
- the reaction solution was concentrated under reduced pressure, and the resulting residue was dissolved in ethyl acetate and washed successively with water and saturated brine.
- the organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure.
- the solvent was distilled off under reduced pressure.
- the obtained residue was suspended in 35 mL of acetic acid, 1.60 g of sodium acetate was added, and the mixture was heated to reflux for 16 hours. After naturally cooling to room temperature, the solvent was distilled off under reduced pressure. Water was added to the residue and extracted with ethyl acetate. The organic layer was washed sequentially with water and saturated Japanese brine and dried over anhydrous magnesium sulfate.
- Example 1 In the same manner as in Reference Examples 1 to 87, Reference Example compounds 88 to 230 shown in Tables 4 to 24 below were produced using the corresponding raw materials. Tables 4 to 24 show the structures and physicochemical data of the reference compounds. [0158] Example 1
- tert-Butyl ⁇ 2-Ethoxy-6-[[hydroxyimino) methyl] phenoxy ⁇ acetate 50 mg of 10% palladium carbon powder is added to an ethanol solution (17 mL) of 500 mg at room temperature under a hydrogen atmosphere of 3 atm. Stir for hours. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure. Dissolve the resulting colorless oil in 15 mL of ⁇ , ⁇ -dimethylformamide, and add tert-butyl [ ⁇ 4- [(5-ciano-2-fluorobenzoyl) amino] phenol ⁇ (imino) methyl.
- tert-butyl ⁇ 2-[( ⁇ 2-[( ⁇ 4-[[(tert-butoxycarbonyl) amino] (imino) methyl] phenol ⁇ amino was prepared in the same manner as in Reference Example 61. ) Carboxyl] -4-chlorophenol ⁇ amino) methyl] -6-hydroxyphenoxy ⁇ acetate was obtained as a yellow solid.
- reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by ODS column chromatography using ethanol / water (1: 2) as the elution solvent, and ethyl [ 2 — ( ⁇ [ 2 — ( ⁇ 4 — [amino ( Imino) methyl] Benzoiru ⁇ Amino) - 4 - chloro port Hue - Le] Amino) methyl) -6-ethoxy-phenoxyethanol] Asetato 0.70 g as a yellow solid.
- the obtained residue was purified by silica gel column chromatography using methanol / ethyl acetate-hexane (1: 4: 4) as an elution solvent, and tert-butyl [2-[( ⁇ 2-[( ⁇ 4-[[ (tert-butoxycarbonyl) amino] (imino) methyl] phenyl ⁇ amino) carbonyl] -4-chloro-amino ⁇ methyl) methyl] -4-chloro-6- (4-methyl-2-o Xisopiperazine-1-yl) phenoxy] acetate 27 mg was obtained as a colorless oil.
- the obtained residue was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as a solvent, and tert-butyl ⁇ 2-[( ⁇ 2-[( ⁇ 6- [amino (imino) methyl] pyridine- 3-yl ⁇ amino) carbonyl] -4-chlorophenyl ⁇ amino) methyl] -6-ethoxyphenoxy ⁇ acetoformate 16 mg (Example 8) and tert-butyl ⁇ 2-[( ⁇ 2 -[( ⁇ 6- [Amino (imino) methyl] pyridine-3-yl ⁇ amino) carbol] phenyl ⁇ amino) methyl] -6-ethoxyphenoxy ⁇ acetate formate 5 mg (Example) 9) was obtained.
- the resulting residue was purified by silica gel column chromatography using chloroform / methanol (20: 1) as the elution solvent, and te rt-butyl ⁇ 2-[( ⁇ 2-[( ⁇ 4- [amino (imino 1.16 g of) methyl] phenyl ⁇ amino) carbonyl] -4-chlorophenol-amino ⁇ methyl] phenoxy ⁇ acetate was obtained as a yellow foam.
- the reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by aminopropyl silica gel column chromatography using ethyl acetate / n-hexane (7: 1) as an elution solvent, and ethyl 3- ⁇ 2-[( ⁇ 2-[( ⁇ 4- [Amino (hydroxyimino) methyl] phenol ⁇ amino) carbol] -4-chlorophine ⁇ amino) methyl] piperidine-1-yl ⁇ propanoate 28 mg was obtained as a pale yellow oil.
- tert-Butyl ⁇ 2-[( ⁇ 2-[( ⁇ 4-[[(tert-Butoxycarbonyl) amino] (imino) methyl] phenyl ⁇ amino) carbol] -4-chlorophine ⁇ Amino) methyl] -6-hydroxyphenoxy ⁇ acetate 250 mg was dissolved in tetrahydrofuran (5 mL), and 2- (dimethylamino) ethanol (53 mg), triphenylphosphine (157 mg) and diisopropylazodicarboxylate (0.15 mL) were dissolved at room temperature. The mixture was stirred overnight.
- the reaction solution was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (7: 3) as an elution solvent.
- the obtained compound was charged with 4 mL of methylene chloride and 4 mL of trifluoroacetic acid at room temperature and stirred for 4 hours.
- 250 mg was dissolved in 5 mL of tetrahydrofuran, and 3- (methyloxetane-3-yl) was dissolved at room temperature.
- Methanol 61 mg, triphenylphosphine 157 mg, and diisopropylazodicarboxylate 0.15 mL were added and stirred overnight.
- reaction solution was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (7: 3) as an elution solvent.
- the resulting compound was charged with 4 mL of methylene chloride and 4 mL of trifluoroacetic acid at room temperature and stirred for 4 hours.
- Trifluoroacetic acid (4 mL) was stirred at room temperature for 2 hours in 300 mg of methylene chloride solution (4 mL). The reaction solution was concentrated under reduced pressure. The obtained residue was dissolved in 20 mL of ethyl acetate, a solution of hydrogen chloride in ethyl acetate (4 N, 1 mL) was added at room temperature, and the mixture was stirred for 30 minutes.
- Example 17 Ethyl 4- ⁇ 2- [( ⁇ 2- [( ⁇ 4- [[(tert-Butoxycarbonyl) amino] (imino) methyl] phenyl ⁇ amino) carbol] -4 ⁇ Amino) methyl] piperidine-1-yl ⁇ Pytolate
- methylene chloride 1.5 mL
- trifluoroacetic acid 1.5 mL
- the reaction solution was concentrated under reduced pressure.
- the obtained residue was dissolved in ethanol 0.80 mL, 1N aqueous sodium hydroxide solution 0.78 mL was added, and the mixture was stirred for 9 hr.
- the reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the solvent, and 4- ⁇ 2-[( ⁇ 2-[( ⁇ 4- [amino ( Imino) methyl] phenyl ⁇ amino) carbol] -4-chlorophenyl ⁇ amino) methyl] piperidine-1-yl ⁇ petitic acid formate 72 mg was obtained as a pale yellow oil.
- Trifluoroacetic acid (0.35 mL) was added to 3 mg of methylene chloride solution (0.35 mL) at room temperature and stirred for 4 hours.
- the reaction mixture was concentrated under reduced pressure and purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the solvent, and 3 [( ⁇ 2 [( ⁇ 4 [Amino (imino) methyl] phenol ⁇ amino)
- 15 mg of (carbol-4-phenyl) amino) methyl] piperidine-1-yl ⁇ propanoic acid formate was obtained as a pale yellow solid.
- the obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 9) as an elution solvent to obtain 124 mg of a colorless foam.
- the obtained colorless foam was used as a raw material, and the same operation as in Example 15 was performed.
- tert-butyl ⁇ 4-amino- 2- [( ⁇ 2- [( ⁇ 4- [[(tert-butoxycarbonyl) amino] (imino) methyl] phenyl ⁇ amino) carbol] -4-chloro Mouthphenol ⁇ amino) methyl] -6-isopropoxyphenoxyacetate 100 mg, pyridine 13 ⁇ L, dichloromethane 1.5 mL and tetrahydrofuran 0.5 mL in a mixture (N-tert-butoxycarbol) 35 mg of sulfamyl was added and stirred overnight at room temperature. To the reaction solution was added a saturated aqueous solution of ammonium chloride and extracted with ethyl acetate.
- the organic layer was washed with water and then saturated Japanese brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography using chloroform / methanol (99: 1) as an elution solvent.
- To the obtained residue were added 3 mL of a 1,4-dioxane solution (4 M) of hydrochloric acid and hydrogen and 0.030 mL of water at room temperature, and the mixture was stirred overnight at the same temperature.
- the obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (3: 1) as an elution solvent.
- ethanol 1.0 mL
- 2N aqueous sodium hydroxide solution 0.04 mL
- the reaction solution was concentrated under reduced pressure.
- the obtained crude product was dissolved in 5 mL of 1,4-dioxane, 0.03 mL of a 1,4-dioxane solution (4 M) in hydrochloric acid and hydrogen was added at room temperature, and the mixture was stirred for 4 hours.
- the reaction mixture was concentrated under reduced pressure, and 5 mL of 1N aqueous sodium hydroxide solution was added.
- the resulting reaction solution was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the elution solvent, and 3- [2-[( ⁇ 2-[( ⁇ 2- [amino (imino) methyl] pyrimidine- 5-yl ⁇ amino) carbol] -4-methylphenyl ⁇ amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenyl] propanoic acid formate 5 mg was obtained.
- tert-butyl [ ⁇ 4-[(2-amino-5-methylbenzoyl) amino] phenol ⁇ (imino) methyl] carbamate 100 mg and sodium triacetoxyborohydride 173 mg in acetic acid (3.3 mL) 4 _ ⁇ [tert-Butyl (dimethyl) silyl] oxy ⁇ -2_formyl-6_isopropoxyphenoxy) acetate 107 mg acetic acid (2.7 mL) solution at room temperature and stirred for 14 hours under the same conditions did. The solvent was distilled off under reduced pressure, and the resulting residue was diluted with water and extracted with ethyl acetate.
- the organic layer was washed successively with saturated aqueous sodium hydrogen carbonate twice, water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 3) as an elution solvent to obtain 94 mg of a yellow solid.
- 94 mg of the obtained yellow solid was dissolved in 3 mL of tetrahydrofuran, and 0.04 mL of tetrahydrofuran solution (1 M) of n-tetraptylammonium fluoride was stored at room temperature, and 1.5 hours under the same conditions. Stir.
- the reaction solution was purified by silica gel column chromatography using ethyl acetate / n-hexane (2: 3) as an elution solvent to obtain 47 mg of a yellow oily substance.
- 1.6 mL of trifluoroacetic acid was added to a solution of 104 mg of the yellow oil obtained by the above method in 1,2-dichloroethane (5 mL). After stirring at room temperature for 2 hours, the mixture was concentrated under reduced pressure. The residue obtained was added to 7 mL of acetonitrile. After dissolution, 0.59 mL of 1N aqueous sodium hydroxide solution was added. After stirring at room temperature for 1 hour, 1.2 mL of 1N aqueous sodium hydroxide solution was added.
- tert-Butyl [ ⁇ 4- [(2-amino-5-methylbenzoyl) amino] phenol ⁇ (imino) methyl] carbamate In a suspension of 71 mg acetic acid (0.5 mL), 123 mg sodium triacetoxyborohydride was added. The mixture was further stirred at room temperature for 5 minutes. Next, a solution of tert-butyl [4-acetoxymethyl-2- (2-tert-butoxy-1-methylethoxy) -6-formyl] phenoxyacetate in acetic acid (1 mL) was added, and the mixture was stirred at room temperature for 1.5 hours. did. The reaction mixture was poured into saturated aqueous sodium hydrogen carbonate and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (10: 1) as an elution solvent. 4N Hydrochloric acid (10 mL) was added to the obtained residue, and the mixture was stirred at room temperature for 5 hr.
- the organic layer was washed successively with water, 5% aqueous sodium bicarbonate and then saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography using ethyl acetate / n_hexane (3: 1) as an elution solvent to obtain a colorless foam.
- the obtained compound was dissolved in 4 mL of methylene chloride, 4 mL of trifluoroacetic acid was added, and the mixture was stirred at room temperature for 3 hours.
- reaction solution was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the elution solvent, and ethyl 3- [2-[( ⁇ 2-[( ⁇ 4- [amino (imino) methyl] phenol ⁇ Amino) carbol] -4-methylphenol ⁇ amino) methyl] -4_ (hydroxymethyl) _6_isopropoxyphenol] propanoate formate 16 mg was obtained as a yellow foam.
- tert-Butyl [ ⁇ 4- [(2-amino-5-methylpyridine-3-carbol) amino] phenol ⁇ (imino) methyl] force rubamate 0.10 g and sodium triacetoxyborohydride 0.17 g acetic acid (1.0 mL) solution was charged with 0.10 g of tert-butyl 3- (4-acetoxymethyl-2-formyl-6-isopropoxyphenol) propanoate at room temperature. After stirring for 30 minutes, 0.25 g of sodium triacetoxyborohydride and 0.13 g of tert-butyl 3- (4-acetoxymethyl-2-formyl-6-isopropoxyphenyl) propanoate were obtained at room temperature.
- the reaction solution was diluted with ethyl acetate, poured into saturated aqueous sodium bicarbonate and extracted.
- the organic layer was washed with water and saturated brine, and dried over anhydrous magnesium sulfate.
- the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (3: 1) as an elution solvent to obtain 78 mg of a pale yellow oil.
- 2 mL of trifluoroacetic acid was added at room temperature and stirred for 3 hours.
- reaction solution was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as a solvent, and 3- (2- ⁇ [(2- ⁇ 4- [amino (imino) methyl] phenol ⁇ amino) carbo- L] -4-methylphenylamino ⁇ methyl-4- (2-hydroxyethylamino) -6-isopropoxyphenol) propanoic acid formate 22 mg was obtained as a yellow solid.
- Example 38 2-Amino-N- ⁇ 4- [Amino (hydroxyimino) methyl] phenol ⁇ -5-chlorobensamide Add 277 mg of sodium triacetoxyborohydride to a suspension of 133 mg of acetic acid (2 mL). After stirring at room temperature for 5 minutes, 129 mg of ethyl (2-formyl-4-hydroxymethyl _6_isopropoxyphenoxy) acetate was added, and the mixture was stirred at the same temperature for 1.5 hours. The reaction solution was poured into saturated aqueous sodium hydrogen carbonate and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated.
- the obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (1:10) as an elution solvent to obtain 180 mg of a pale yellow solid.
- 180 mg of the obtained pale yellow solid was dissolved in ethyl acetate, and 0.135 mL of an ethyl acetate solution (4 M) of hydrochloric acid and hydrogen salt was obtained at 0 ° C.
- the resulting solid was collected by filtration and dried under reduced pressure to give ethyl [2-[( ⁇ 2-[( ⁇ 4- [amino (hydroxyimino) methyl] phenol ⁇ amino) carbol] -4-chloro].
- Mouthphenol ⁇ amino) methyl] -4- (hydroxymethyl) _6_isopropoxyphenoxy] acetate hydrochloride was obtained as a pale yellow solid.
- the obtained residue was purified by silica gel column chromatography using ethyl acetate-hexane (3: 1) as an elution solvent.
- the obtained compound was dissolved in 8 mL of ethanol, and 2 mL of 2N sodium hydroxide aqueous solution was added at room temperature, followed by stirring for 2.5 hours.
- 2.3 mL of 2N hydrochloric acid was added at 0 ° C, and the solvent was concentrated under reduced pressure. Water was added to the residue, and the resulting precipitate was collected by filtration.
- the residue was washed successively with water and n-hexane and then dried under reduced pressure.
- the organic layer was washed successively with saturated aqueous sodium hydrogen carbonate twice, water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography using ethyl acetate-hexane (2: 3) as an elution solvent to obtain 1.1 g of a yellow solid.
- 1.1 g of the obtained yellow solid was dissolved in 33 mL of tetrahydrofuran, and 0.47 mL of tetrahydrofuran solution (1 M) of tetra n-butylammonium fluoride was prepared at room temperature and stirred for 1.5 hours under the same conditions. .
- a tetrahydrofuran solution (1 M) of tetrahydrofuran-tetra-n-butyl ammonium (0.235 mL) was added at room temperature, and the mixture was stirred for 3 hours under the same conditions.
- 0.141 mL of a tetrahydrofuran solution (1 M) of tetraptyl ammonium fluoride was added at room temperature, and the mixture was stirred for 2 hours under the same conditions.
- reaction solution was purified by silica gel column chromatography using ethyl acetate / n-hexane (2: 3) as an elution solvent, and ethyl ⁇ 2-[( ⁇ 2-[( ⁇ 4- [amino (hydroxyimino 616 mg of) methyl] phenyl ⁇ amino) carbol] -4-methylphenol ⁇ amino) methyl] -4-hydroxy-6-isopropoxyphenoxy ⁇ acetate was obtained as a yellow solid.
- the residue obtained by distilling off the solvent under reduced pressure was dissolved in 15 mL of ethanol, and 268 mg of potassium carbonate was added at room temperature, followed by stirring for 3 hours.
- the reaction mixture was poured into 2N hydrochloric acid, and neutralized with saturated aqueous sodium hydrogen carbonate.
- the precipitate was collected by filtration to obtain 162 mg of a colorless solid.
- the resulting colorless solid 132 mg was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as a solvent.
- the elution fraction was concentrated under reduced pressure to about 20 mL, saturated aqueous sodium hydrogen carbonate was added, and the precipitated solid was collected by filtration.
- the residue obtained by distilling off the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 2) as an elution solvent to obtain 26 mg of a pale yellow oily substance.
- 2 mL of trifluoroacetic acid was added at room temperature, followed by stirring for 3.5 hours.
- the solvent was distilled off under reduced pressure, methylene chloride was added to the resulting residue, and the mixture was distilled off again under reduced pressure to obtain a pale yellow solid.
- the obtained pale yellow solid was dissolved in 5 mL of ethanol, and 53 mg of potassium carbonate was stirred at room temperature for 1 hour and at 40 ° C. for 1 hour.
- Potassium carbonate (53 mg) was added and stirred at 60 ° C for 30 minutes.
- a phosphate buffer solution of PH7.4 was added, and 1N hydrochloric acid was added dropwise to adjust the pH to 6.
- Example 43 tert-Butyl [ ⁇ 4- [(2-amino-5-chlorobenzobenzo) amino] phenol Kimino) methyl] rubamate 250 mg of sodium triacetoxyborohydride in a solution of 229 mg of acetic acid (6 mL) After stirring at room temperature and stirring for 10 minutes, 200 mg of methyl 4- (2_tert-butoxy_2_oxoethoxy) -3-ethoxy-5-formylbenzoate was added at room temperature and 3 Stir for hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with water, 5% aqueous sodium bicarbonate, and then saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (2: 1) as an elution solvent.
- the obtained residue was dissolved in 3 mL of methanol, 0.3 mL of 1N aqueous sodium hydroxide solution was added under ice cooling, and the mixture was stirred at room temperature for 2 hours. 0.6 mL of 1N aqueous solution of sodium hydroxide and sodium chloride was stirred at room temperature for 2 hours.
- the obtained residue was purified by silica gel column chromatography using ethyl acetate as an elution solvent, and ethyl 3- [2-[( ⁇ 3-[( ⁇ 4-[[(ethoxycarbonyl) amino] (imino) methyl [Fuel ⁇ amino) carbol] pyridine-2-yl ⁇ amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenyl] propanoate 19 mg was obtained as a pale yellow solid.
- the resulting residue was purified by aminopropyl silica gel column chromatography using ethyl acetate-hexane (7: 1) as an elution solvent. 0.07 mL of an ethyl acetate solution (4 M) of sodium chloride and hydrogen was added to an ethyl acetate solution (1.0 mL) of the obtained compound, and the mixture was stirred for 30 minutes.
- reaction solution was purified by ODS column chromatography using water / acetonitrile as an elution solvent, and 3- [2-[( ⁇ 2-[( ⁇ 4-[[(ethoxycarbonyl) amino] (imino) methyl] phenyl. L-amino) force] -4-methylphenol ⁇ amino) methyl] -4_ (hydroxymethyl) _6_isopropoxyphenyl] propanoic acid 42 mg was obtained as a yellow solid.
- N- ⁇ 4- [Amino (hydroxyimino) methyl] phenol ⁇ -2-amino-5-methylbenzamide and tert-butyl 3_ (4-acetoxymethyl-2_formyl-6_isopropoxyphenyl) propanoate was used as a raw material to give a yellow solid by the same operation as in Example 54 described later.
- 1N aqueous sodium hydroxide solution 5.5 mL was added, and the mixture was stirred at room temperature for 1 hour.
- the reaction solution was purified by ODS column chromatography using water / acetonitrile as an elution solvent, and sodium 3- [2-[( ⁇ 2-[( ⁇ 4- [amino (hydroxyimino) methyl] fale was obtained.
- a yellow solid was obtained in the same manner as in Example 7, using tert-butyl [ ⁇ 4-[(2-amino-5-chlorobenzobenzo) amino] phenol Kimino) methyl] rubamate as a raw material. .
- ⁇ 2-[( ⁇ 2-[( ⁇ 4- [amino] imino] methyl ⁇ amino) carbol] -4 was prepared in the same manner as in Example 15.
- -Chlorophenyl ⁇ amino) methyl] -4-[(dimethylamino) methyl] -6-ethoxyphenoxy ⁇ acetic acid hydrochloride was obtained as a yellow solid.
- Example compounds 61 to 654 shown in Tables 25 to 107 to be described later were produced using the corresponding raw materials.
- Tables 25 to 107 show the structures and physicochemical data of the example compounds.
- Tables 108 to 118 below show structures of other compounds of the present invention. These can be easily produced by using the production methods described above, the methods described in the examples, methods obvious to those skilled in the art, or variations thereof.
- Test Method 1 Enzyme inhibition measurement test by synthetic substrate method
- Table 1 shows the activity of the representative compounds of the present invention. Ex represents the example number (the same applies hereinafter) 0
- the compound of Example 16 is active against blood coagulation factor X and its inhibitory activity IC is 1
- Thrombin inhibitory activity 50 ⁇ L of reaction buffer (pH 7.4) in 96-well microplate, compound solution 10 ⁇ 2 mM synthetic substrate S-2238 (Chromogenix) 20 ⁇ L, 0.2 U / Add 20 ⁇ L of mL of human thrombin (Sigma), react for 1 hour at room temperature, and measure the change in absorbance at 405 nm.
- the compound of Example 16 has an inhibitory activity IC of over 100 ⁇ IC against thrombin.
- the compound (I) of the present invention has a strong activity ⁇ blood coagulation factor VII inhibitory activity, and is also active selectively against the activity ⁇ blood coagulation factor X and thrombin. It was confirmed that the blood coagulation factor VII was inhibited.
- Test method Ex vivo clotting time measurement test using guinea pigs To male Hartley guinea pigs (5-7 weeks old, Japan SLC), a test compound dissolved in a solvent containing 1 volume of dimethylacetamide and 1 volume of 5% glucose was administered intravenously under anesthesia with jetyl ether. (1 mg / kg) After 2 minutes, 0.5 mL of blood was collected from the jugular vein with 1/10 volume of 3.13% sodium citrate, and plasma was separated by centrifugation at 3000 rpm for 10 minutes. Using this plasma, the extrinsic clotting time (PT) was measured according to the following method a).
- Guinea pig plasma 50 / z L was warmed at 37 ° C for 1 minute, and 100 L of moth moth recombin plastin was added to measure the clotting time.
- an automatic blood coagulation measuring device ST4 Roche Diagno Status
- the clotting time of guinea pig plasma without the test compound was used as a control, and the activity of the test compound was shown as a relative value when this control was 1.
- the compound of the present invention was found to prolong the extrinsic clotting time by intravenous administration.
- Table 2 below shows the action of prolonging the extrinsic clotting time of the representative compounds of the present invention.
- the compound (I) of the present invention includes a compound that is metabolized in vivo and exhibits an active blood coagulation factor VII inhibitory activity.
- R 1 is —C ( ⁇ NH) NH and / or —C (0) R 5 is a carboxylic acid in vivo.
- Test Method 3 There are compounds that have been converted to certain compounds and show active activity, blood coagulation factor VII inhibitory activity. The usefulness of such compounds is shown below in Test Method 3, and Test Methods 1 and Z or Test This can be confirmed by combining the tests of Method 2.
- Rats Male; Sprague-Dawley (SD-IGS), 7 weeks old (purchased at 6 weeks of age), Nippon Chiyers River, food condition: free intake of water and food) were fasted from the evening before administration .
- the compound was suspended in 0.5% MC to a concentration of 10 mg / 5 mL and administered orally once into the stomach using a sonde.
- the compound was dissolved in ⁇ , ⁇ -dimethylacetamide to a concentration of 1 mg / 0.2 mL, diluted with the same amount of 5% glucose (Otsuka Pharmaceutical Factory), and then rat (male; Sprague-Dawley (SD_IGS), 7-week-old (purchased at 6-week-old), Nippon Chisuriba Co., Ltd., feeding conditions: free intake of water and food) Single intravenous administration via tail vein.
- samples were collected at approximately 200 / zL with a syringe that had been parinized from the jugular vein at 15, 30 minutes, 1, 2, 4, and 8 hours after administration, and immediately stored under ice cooling.
- intravenous administration about 200 ⁇ L of blood was collected from a jugular vein-treated syringe at 2, 10, 30 minutes, 1, 2, and 4 hours after administration, and immediately stored under ice cooling.
- the blood was centrifuged at 10,000 rpm for 1 minute (room temperature) to collect the plasma.
- the prepared sample was stored at about 4 ° C until measurement.
- a plasma sample at each point was dispensed into a 25 ⁇ L tube, and 100 ⁇ L of an internal standard solution dissolved in acetonitrile and 875 ⁇ L of acetonitrile were added. Thereafter, the mixture was stirred with a mixer, centrifuged at 3300 rpm for 20 minutes, and 5 ⁇ L of the supernatant was injected into HPLC.
- a calibration curve solution was prepared by dissolving the measurement compound in 50% aqueous acetonitrile and serially diluting with acetonitrile to 10, 50, 100, 500, 1000, 5000, 10000, 50000 ng / mL.
Abstract
It is intended to provide a compound which has an anticoagulant effect based on the inhibition of the activated blood coagulation factor VII and, therefore, is useful as a blood coagulation inhibitor or a preventive/remedy for diseases caused by thrombus or embolus. The above-described substance is a carboxylic acid derivative characterized by having three cyclic moieties or a pharmaceutically acceptable salt thereof that has a potent activity of inhibiting the activated blood coagulation factor VII. Because of showing an excellent anticoagulant effect, the above-described carboxylic acid derivative is usable as a preventive/remedy for diseases caused by thrombus or embolus.
Description
明 細 書 Specification
カルボン酸誘導体またはその塩 技術分野 Carboxylic acid derivatives or salts thereof
[0001] 本発明は、医薬、特に活性ィヒ血液凝固第 VII因子阻害剤として有用な新規なカル ボン酸誘導体又はその塩、及びその医薬としての利用に関する。殊に、本発明化合 物は、活性ィ匕血液凝固第 VII因子阻害剤として有用である。 [0001] The present invention relates to a novel carboxylic acid derivative or a salt thereof useful as a pharmaceutical, particularly an active blood coagulation factor VII inhibitor, and its use as a pharmaceutical. In particular, the compound of the present invention is useful as an active blood coagulation factor VII inhibitor.
背景技術 Background art
[0002] 近年、生活習慣の欧米化、人口の高齢化などに伴い、心筋梗塞、脳血栓症、末梢 動脈血栓症をはじめとする血栓塞栓性疾患は年々増加し、その治療の社会的重要 性は益々高まっている。抗凝固療法は、線溶療法及び抗血小板療法とともに血栓症 の治療及び予防における内科的治療法の一端を担っている(非特許文献 1)。特に、 血栓症の予防においては長期投与に耐えうる安全性と、確実且つ適切な抗凝固活 性の発現が必須となる。ヮルフアリンカリウムは、唯一の経口抗凝固剤として世界中で 繁用されているが、その作用機序に基づく特性力も抗凝固能のコントロールが難しく [0002] In recent years, thromboembolic diseases such as myocardial infarction, cerebral thrombosis, and peripheral arterial thrombosis have increased year by year due to the westernization of lifestyle and the aging of the population. Increasingly. Anticoagulation therapy, along with fibrinolysis and antiplatelet therapy, plays a part in medical treatment in the treatment and prevention of thrombosis (Non-patent Document 1). In particular, in the prevention of thrombosis, safety that can withstand long-term administration and the development of reliable and appropriate anticoagulant activity are essential.ヮ Lufarin potassium is widely used around the world as the only oral anticoagulant, but it is difficult to control its anticoagulant ability due to its characteristic mechanism.
(非特許文献 2及び 3)、臨床的には非常に使用しづらい薬剤であり、より有用で使い やす 、抗凝固剤の登場が望まれて 、る。 (Non-patent Documents 2 and 3), it is a drug that is very difficult to use clinically, and the appearance of an anticoagulant that is more useful and easy to use is desired.
[0003] 血液凝固反応は外因系経路と内因系経路力 なり、トロンビンはいずれの経路によ つても生成する。トロンビンは、凝固の最終段階であるフイブリノ一ゲンのフイブリンへ の転ィ匕を司るばかりか、血小板の活性化及び凝集にも深く関与し (非特許文献 4)、 その阻害剤は創薬のターゲットとして長い間抗凝固剤研究の中心にあった。しかしな 力 Sら出血の惹起など、安全性面で依然として問題がある (非特許文献 5)。 [0003] The blood coagulation reaction is an extrinsic pathway and an intrinsic pathway force, and thrombin is generated by any pathway. Thrombin is not only responsible for the conversion of fibrinogen to fibrin, which is the final stage of coagulation, but is also deeply involved in platelet activation and aggregation (Non-patent Document 4), and its inhibitor is a target for drug discovery. As long as it has been at the center of anticoagulant research. However, there are still problems with safety, such as the occurrence of force S and bleeding (Non-patent Document 5).
活性ィ匕血液凝固第 VII因子は外因系凝固経路の開始点に位置する酵素であり、蛋 白性補因子である組織因子 (TF)と結合することによりその酵素活性は著しく上昇す る。組織因子は様々な血栓性疾患で発現が亢進することが知られており、外因系凝 固経路の血栓性疾患への関与が示唆されて!ヽる (非特許文献 6)。活性化血液凝固 第 VII因子の選択的阻害剤は、内因系凝固を阻害しないことにより、より出血性の副 作用が少な 、優れた抗凝固薬となる可能性がある。
[0004] 一方、特許文献 1には、下記一般式で示される新規なアミジノ誘導体が、活性化血 液凝固第 VII因子を阻害する作用を有し、汎発性血管内凝固症候群、冠動脈血栓 症等の予防又は治療剤として有用であることが記載されている。し力しながら E2環と E 4環が直接結合して 、る点で、リンカ一 (L)を介して B環と C環が結合して 、る本発明 化合物とは構造を異にする。 Active 匕 blood coagulation factor VII is an enzyme located at the start of the extrinsic clotting pathway, and its enzymatic activity increases markedly by binding to tissue factor (TF), a protein cofactor. Tissue factor is known to be upregulated in various thrombotic diseases, suggesting involvement of the extrinsic coagulation pathway in thrombotic diseases (Non-patent Document 6). Activated blood coagulation Selective inhibitors of factor VII may be superior anticoagulants with less bleeding side effects by not inhibiting endogenous coagulation. [0004] On the other hand, Patent Document 1 discloses that a novel amidino derivative represented by the following general formula has an action of inhibiting activated blood coagulation factor VII, including generalized intravascular coagulation syndrome and coronary artery thrombosis. It is described that it is useful as a preventive or therapeutic agent. However, the E 2 ring and the E 4 ring are directly bonded to each other, and the B and C rings are bonded via the linker (L), so that the structure is different from that of the present compound. .
[0005] [化 1] [0005] [Chemical 1]

(式中の記号については、特許文献 1の公報参照。 ) (Refer to the publication of Patent Document 1 for symbols in the formula.)
[0006] また、特許文献 2には、活性ィ匕血液凝固第 VII因子及び活性ィ匕血液凝固第 X因子 阻害剤として、広範な下記一般式で示される化合物が記載されている。しかしながら 、実施例として記載されて 、る化合物は Xが結合であるビアリール構造を有する化合 物に限られており、リンカ一を有する本発明化合物については開示されていない。 [0006] Patent Document 2 describes a wide range of compounds represented by the following general formulas as active 活性 blood coagulation factor VII and active 匕 blood coagulation factor X inhibitors. However, the compounds described as examples are limited to compounds having a biaryl structure in which X is a bond, and the compounds of the present invention having a linker are not disclosed.
[0007] [化 2] [0007] [Chemical 2]

(E1, Lは炭素環、ヘテロ環等を示す。 Xは結合、無置換または置換 C メチレン等を (E 1 and L represent carbocycle, heterocycle, etc. X represents a bond, unsubstituted or substituted C-methylene, etc.
1-4 1-4
示す。他の式中の記号については、特許文献 2の公報参照。 ) Show. For the symbols in other formulas, see Patent Document 2. )
[0008] 更に、特許文献 3には、活性ィ匕血液凝固第 VII因子、活性化血液凝固第 IX因子、 活性化血液凝固第 X因子、活性化血液凝固第 XI因子等の阻害剤として、下記一般
式で示される化合物が記載されている。し力しながら、それらの化合物は本発明化合 物の B環に相当する環またはリンカ一が存在しない点で本発明化合物とは構造を異 にする。 [0008] Furthermore, Patent Document 3 describes the following as inhibitors of active blood coagulation factor VII, activated blood coagulation factor IX, activated blood coagulation factor X, activated blood coagulation factor XI and the like. General Compounds of the formula are described. However, these compounds differ in structure from the compounds of the present invention in that there is no ring or linker corresponding to the B ring of the compounds of the present invention.
[化 3] [Chemical 3]

(式中の記号については、特許文献 3の公報参照。 ) (Refer to Patent Document 3 for symbols in the formula.)
[0010] 更に、特許文献 4にはセリンプロテアーゼ阻害剤として、下記一般式で示される化 合物が記載されている。しかしながら、それらの化合物が活性ィ匕血液凝固第 VII因子 を選択的に阻害するとの記載はない。また、それらの化合物は本発明化合物の B環 に相当する環が存在しない点またはリンカ一が存在しない点で本発明化合物とは構 造を異にする。 Furthermore, Patent Document 4 describes a compound represented by the following general formula as a serine protease inhibitor. However, there is no description that these compounds selectively inhibit active blood coagulation factor VII. Further, these compounds differ in structure from the compounds of the present invention in that there is no ring corresponding to the B ring of the compounds of the present invention or no linker exists.
[0011] [化 4] [0011] [Chemical 4]

(式中の記号については、特許文献 4の公報参照。 ) (For the symbols in the formula, refer to the publication of Patent Document 4.)
更に特許文献 5では、活性ィ匕血液凝固第 VII因子、活性ィ匕血液凝固第 IX因子等 の阻害剤として、広範な下記一般式で示される化合物が記載されている。し力しなが ら、本発明の特徴であるカルボン酸部を有する実施例化合物は ZQが結合であるビア
リール構造を有する化合物に限られており、リンカ一を有する化合物については開示 されていない。 Furthermore, Patent Document 5 describes a wide variety of compounds represented by the following general formulas as inhibitors of active blood coagulation factor VII, active blood coagulation factor IX and the like. However, the example compounds having a carboxylic acid moiety, which is a feature of the present invention, have vias in which Z Q is a bond. It is limited to compounds having a reel structure, and no compound having a linker is disclosed.
[0013] [化 5] [0013] [Chemical 5]
B ΨB Ψ

(R2は Z°-Qを、 Z°は結合等を、 Qはフエ-ル、ヘテロ環等を示す。他の式中の記号に ついては、特許文献 5の公報参照。 ) (R 2 represents Z ° -Q, Z ° represents a bond, Q represents a file, a heterocycle, etc. For symbols in other formulas, refer to Patent Document 5)
[0014] 更に特許文献 6では、下記一般式で示されるフエニルダリシン誘導体が、活性化血 液凝固第 VII因子阻害作用を有することが記載されている。し力しながら、それらの 化合物は 2つの環を結合する部位がアミノ酸である点で、 J部がアミノ酸ではな ヽ本発 明化合物とは構造を異にする。 [0014] Further, Patent Document 6 describes that a phenyldaricin derivative represented by the following general formula has an inhibitory effect on activated blood coagulation factor VII. However, these compounds differ in structure from the present invention compounds in which the J part is not an amino acid, in that the site that connects the two rings is an amino acid.
[0015] [化 6] [0015] [Chemical 6]

(式中の記号については、特許文献 6の公報参照。 ) (For the symbols in the formula, refer to Patent Document 6)
[0016] 更に特許文献 7では、下記一般式を有する (チォ)ゥレア誘導体が活性ィ匕血液凝固 第 VII因子阻害作用を有することが記載されている。し力しながら、本発明化合物の B環に相当する環がない点等で、本発明化合物とは構造を異にする。 [0016] Further, Patent Document 7 describes that (thio) urea derivatives having the following general formula have an activity of inhibiting blood coagulation factor VII. However, the structure is different from that of the compound of the present invention in that there is no ring corresponding to the ring B of the compound of the present invention.
[0017] [化 7]
 [0017] [Chemical 7]
[0017] [Chemical 7]
(式中の記号については、特許文献 7の公報参照。 ) (Refer to the publication of Patent Document 7 for symbols in the formula.)
[0018] 更に特許文献 8では、下記一般式で示される化合物が血小板凝集抑制作用を有 することが記載されている。しカゝしながら、活性ィ匕血液凝固第 VII因子阻害作用を有 するとの記載はない。また、それらの化合物はピロリジン環を有するが、本発明化合 物は C環にピロリジン環を含まない点で、本発明化合物とは構造を異にする。 [0018] Further, Patent Document 8 describes that a compound represented by the following general formula has an inhibitory action on platelet aggregation. However, there is no description that it has an activity of inhibiting blood coagulation factor VII. In addition, these compounds have a pyrrolidine ring, but the compounds of the present invention differ in structure from the compounds of the present invention in that the C ring does not contain a pyrrolidine ring.
[0019] [化 8] [0019] [Chemical 8]

(Uはメチレンまたはカルボ-ルを示す。他の式中の記号については、特許文献 8の 公報参照。 ) (U represents methylene or carbo- yl. For symbols in other formulas, see Patent Document 8)
[0020] 更に特許文献 9では、下記一般式で示される化合物が活性ィ匕血液凝固第 X因子阻 害作用を有することが記載されている。しカゝしながら、活性ィ匕血液凝固第 VII因子阻 害作用を有するとの記載はない。また、それらの化合物は本発明化合物の A環に相 当する Q1上の置換基力 Sメチル、フルォロまたはクロ口に限定されており、本発明化合 物の R1に相当する置換基がない点で本発明化合物とは構造を異にする。 [0020] Further, Patent Document 9 describes that a compound represented by the following general formula has an activity of inhibiting active blood coagulation factor X. However, there is no description that it has an activity of inhibiting blood coagulation factor VII. Further, these compounds are limited to the substituent force S on Q 1 corresponding to the A ring of the compound of the present invention, and there is no substituent corresponding to R 1 of the compound of the present invention. In this respect, the structure is different from the compound of the present invention.
[0021] [化 9]
 [0021] [Chemical 9]
[0021] [Chemical 9]
(Q1は 5位カ チル、フルォロ若しくはクロ口で置換されていてもよい 2-ピリジ-ル、 6位 がメチル、フルォロ若しくはクロ口で置換されていてもよい 3-ピリジ-ル、または 6位が
メチル、フルォロ若しくはクロ口で置換されていてもよいピリダジ -ルを示す。他の式 中の記号については、特許文献 9の公報参照。 ) (Q 1 is 2-pyridyl optionally substituted with 5-position chloro, fluoro or chloro, 6-position 3-pyridyl optionally substituted with methyl, fluoro or chloro, or 6 Rank Pyridazyl optionally substituted with methyl, fluoro, or black mouth is shown. For the symbols in the other formulas, see Patent Document 9. )
[0022] 更に特許文献 10では、下記一般式で示される広範な化合物が活性化血液凝固第[0022] Further, in Patent Document 10, a wide range of compounds represented by the following general formulas are activated blood coagulation.
X因子阻害作用を有することが記載されている。し力しながら、活性化血液凝固第 VIIt is described to have a factor X inhibitory action. Activated blood coagulation VI
I因子阻害作用を有するとの記載はない。 There is no description that it has a factor I inhibitory action.
[0023] [化 10] [0023] [Chemical 10]

(R4は水素、アルキル、ハロ、ハロアルキル、シァ入ニトロ、 -OR5, - C(0)OR5、 -N(R5 )R6、 - C(0)N(R5)R6または- R8— N(R5)R6を、 R8は直鎖または分枝のアルキレン、アルキ リデンまたはアルキリジン鎖を示す。他の式中の記号については、特許文献 10の公 報参照。 ) (R 4 is hydrogen, alkyl, halo, haloalkyl, nitro-nitro, -OR 5 , -C (0) OR 5 , -N (R 5 ) R 6 , -C (0) N (R 5 ) R 6 or -R 8 — N (R 5 ) R 6 , R 8 represents a straight-chain or branched alkylene, alkylidene, or alkylidyne chain (for the symbols in other formulas, see the publication of Patent Document 10).
[0024] 非特許文献 1:「総合臨床」、 1989年、第 41卷、 p.2141-2145 [0024] Non-Patent Document 1: "General Clinical", 1989, 41st page, p.2141-2145
非特許文献 2:「ザ' -ュ一'イングランド ·ジャーナル ·ォブ ·メデイシン(The New Engla nd Journal of Medicine)] , (米国)、 1991年、第 324卷、第 26号、 p.1865- 1875 非特許文献 3 :「ザ'ジャーナル'ォブ 'タリ-カル 'ファーマコロジー(The Journal of CI inical Pharmacology)」、(米国)、 1992年、第 32卷、 p.196- 209 Non-Patent Document 2: “The New England Journal of Medicine”, (USA), 1991, No. 324, No. 26, p.1865- 1875 Non-Patent Document 3: "The Journal of CI inical Pharmacology" (USA), 1992, 32nd, p.196-209
非特許文献 4 :松尾 理編、「t-PAと pro-UK」、学際企画、 1986年、血液凝固、 p.5-40 非特許文献 5:「ビオメディ力 ·ビォヒミカ ·ァクタ(Biomedica Biochimica Acta)」 (独国)、 1985年、第 44卷、 P.1201- 1210 Non-Patent Document 4: Osamu Matsuo, “t-PA and pro-UK”, Interdisciplinary Planning, 1986, Blood Coagulation, p.5-40 Non-Patent Document 5: “Biomedica Biochimica Acta” (Germany), 1985, No. 44, P.1201-1210
非特許文献 6 :「スロンボシス 'アンド'へモスタシス(Thrombosis and Haemostasis)」、( 独国)、 1997年、第 78卷、第 1号、 p.759-764 Non-Patent Document 6: “Thrombosis and Haemostasis” (Germany), 1997, 78th, No. 1, p.759-764
特許文献 1:国際公開第 99/41231号パンフレット Patent Document 1: International Publication No. 99/41231 Pamphlet
特許文献 2 :国際公開第 02/34711号パンフレット Patent Document 2: Pamphlet of International Publication No. 02/34711
特許文献 3:国際公開第 02/37937号パンフレット Patent Document 3: International Publication No. 02/37937 Pamphlet
特許文献 4 :国際公開第 02/42273号パンフレット Patent Document 4: International Publication No. 02/42273 Pamphlet
特許文献 5:国際公開第 01/87854号パンフレット
特許文献 6:国際公開第 00/35858号パンフレット Patent Document 5: Pamphlet of International Publication No. 01/87854 Patent Document 6: International Publication No. 00/35858 Pamphlet
特許文献 7:欧州特許出願公開第 1162194号明細書 Patent Document 7: European Patent Application Publication No. 1162194
特許文献 8 :欧州特許公開第 0528369号明細書 Patent Document 8: European Patent Publication No. 0528369
特許文献 9 :国際公開第 00/39118号パンフレット Patent Document 9: International Publication No. 00/39118 Pamphlet
特許文献 10:国際公開第 99/32477号パンフレット Patent Document 10: International Publication No. 99/32477 pamphlet
発明の開示 Disclosure of the invention
[0025] 上述の通り、活性ィ匕血液凝固第 VII因子選択的阻害剤は、抗凝固療法において、 トロンビン阻害剤よりも効率的で且つ、選択的な凝固系の阻害を期待できる。また、 活性ィ匕血液凝固第 VII因子選択的阻害剤は、より副作用の少ない薬剤として期待さ れる。従って、上記の特許文献に記載された化合物とは化学構造が異なり、更に優 れた効果を有する、活性化血液凝固第 VII因子選択的阻害剤の創製が切望されて いる。 [0025] As described above, the active 匕 blood coagulation factor VII selective inhibitor can be expected to be more efficient than the thrombin inhibitor and selectively inhibit the coagulation system in anticoagulation therapy. In addition, an active 匕 blood coagulation factor VII selective inhibitor is expected as a drug with fewer side effects. Therefore, the creation of an activated blood coagulation factor VII selective inhibitor having a chemical structure different from that of the compound described in the above-mentioned patent document and having a superior effect is eagerly desired.
[0026] 本発明者等は、下記一般式 (I)で示される誘導体又はその塩が、優れた活性化血 液凝固第 VII因子選択的阻害作用を有することを見出し本発明を完成した。 [0026] The present inventors have found that a derivative represented by the following general formula (I) or a salt thereof has an excellent activated blood coagulation factor VII selective inhibitory action and completed the present invention.
すなわち本発明は、式 (I)で示されるカルボン酸誘導体またはその製薬学的に許 容される塩に関する。 That is, the present invention relates to a carboxylic acid derivative represented by the formula (I) or a pharmaceutically acceptable salt thereof.
[化 11] [Chemical 11]

[式中の記号は、以下の意味を示す。 [The symbols in the formula have the following meanings.
A環:ァリール環またはへテロアリール環。 Ring A: Aaryl ring or heteroaryl ring.
B環:ベンゼン環、ナフタレン環または単環乃至 2環式へテロァリール環。 Ring B: benzene ring, naphthalene ring, monocyclic to bicyclic heteroaryl ring.
C環:シクロアルキル環、ァリール環またはへテロ環。 Ring C: a cycloalkyl ring, an aryl ring or a hetero ring.
m、 n及び p :それぞれ独立して 0乃至 3の整数。ただし m、 nまたは pが 2以上の場合、そ
れぞれ R2、 R3または R4は同一または互いに異なって!/、てもよ!/、。 m, n, and p: each independently an integer from 0 to 3. However, if m, n or p is 2 or more, R 2 , R 3 or R 4 are the same or different from each other! /
R1 :— NH、 -CH NH、— C(=0)NH、— C(=NH)NH、— C(=N0H)NH、— C(=NH)NH— CO -R 1 :-NH, -CH NH,-C (= 0) NH,-C (= NH) NH,-C (= N0H) NH,-C (= NH) NH- CO-
2 2 2 2 2 2 22 2 2 2 2 2 2
(置換されて 、てもよ 、低級アルキル)、または 5-ォキソ -2,5-ジヒドロ- 1,2,4-ォキサジ ァゾール -3-ィル。 (Substituted or lower alkyl), or 5-oxo-2,5-dihydro-1,2,4-oxadiazol-3-yl.
R2及び R3 :それぞれ独立して、低級アルキル、ハロゲノ低級アルキル、ハロゲン、ォキ ソ、 - CN、 -NO、 -OR°, -0-ハロゲノ低級アルキル、 - NR°R°°、 - SR°、 - S(=0)R°、 - S(=0 R 2 and R 3 : each independently lower alkyl, halogeno lower alkyl, halogen, oxo, -CN, -NO, -OR °, -0-halogeno lower alkyl, -NR ° R °°, -SR °,-S (= 0) R °,-S (= 0
2 2
) R0、 - S(=0) NR°R°°、 - NR°S(=0) R°°、 - C(=0)R°、 -CO R°、 - C(=0)NR°R°°、 - NR°C(=0)) R 0 ,-S (= 0) NR ° R °°,-NR ° S (= 0) R °°,-C (= 0) R °, -CO R °,-C (= 0) NR ° R °°,-NR ° C (= 0)
2 2 2 2 2 2 2 2
R°°、 -NR°C(=0)_ハロゲノ低級アルキル、シクロアルキル、ァリールまたはへテロ環基 。ただし、 R2及び R3における低級アルキル、シクロアルキル、ァリールまたはへテロ環 基は置換されていてもよい。 R °°, -NR ° C (= 0) _halogeno lower alkyl, cycloalkyl, aryl or hetero ring group. However, the lower alkyl, cycloalkyl, aryl or hetero ring group in R 2 and R 3 may be substituted.
あるいは、それぞれ 2つの R2または R3がー体となって- 0-低級アルキレン- 0-を形 成していてもよい。 Alternatively, each of two R 2 or R 3 may be in the form of -0-lower alkylene-0-.
RQ及び RQQ:それぞれ独立して、 -Hまたは低級アルキル。 R Q and R QQ : each independently -H or lower alkyl.
R4 :それぞれ独立して、低級アルキル、低級アルケニル、シクロアルキル、ァリール、 ヘテロ環基、ハロゲン、ォキソ、 - CN、 -NO、 - OR6、 - NR6R6a、 - SR6、 - S(=0)R6、 - S(=0) R 4 : each independently lower alkyl, lower alkenyl, cycloalkyl, aryl, heterocyclic group, halogen, oxo, -CN, -NO, -OR 6 , -NR 6 R 6a , -SR 6 , -S ( = 0) R 6 ,-S (= 0)
2 2
R6、 - S(=0) NR6R6a、 - NR6S(=0) R6 - NR¾(=0) - NR6aR6b、 - NR (=0) - NR6aC(=0)ORR 6 ,-S (= 0) NR 6 R 6a ,-NR 6 S (= 0) R 6 -NR¾ (= 0)-NR 6a R 6b ,-NR (= 0)-NR 6a C (= 0) OR
2 2 2 2 2 2 2 2 2 2
6b、 - C(=0)R6、 -CO R6、 - C(=0)NR6R6a、 - C(=0)N(R6)-低級アルキレン- NR6aR6b、 - NH- 6b, - C (= 0) R 6, -CO R 6, - C (= 0) NR 6 R 6a, - C (= 0) N (R 6) - lower alkylene - NR 6a R 6b, - NH-
2 2
C(=N— CN)NH、— C(=0)— N=C(NH )、— C(=0)— N=C(R6)— NH、— C(=0)NR6— S(=〇)— NR C (= N— CN) NH, — C (= 0) — N = C (NH), — C (= 0) — N = C (R 6 ) — NH, — C (= 0) NR 6 — S (= 〇) — NR
2 2 2 2 2 2 2 2 2 2
6aR6b、— C(=0)NR6— S(=〇) R6a、— OC(=〇)R6、— OC(=0)OR6、— OC(=0)NR6R6a、— NR6C(= 6a R 6b , — C (= 0) NR 6 — S (= 〇) R 6a , — OC (= 〇) R 6 , — OC (= 0) OR 6 , — OC (= 0) NR 6 R 6a , — NR 6 C (=
2 2
0)R6a、 - NR6C(=0)OR6aまたは- NR6C(=0)NR6aR6b。ただし、 R4における低級アルキル、 低級アルケニル、シクロアルキル、ァリール及びへテロ環基はそれぞれ置換されてい てもよい。 0) R 6a , -NR 6 C (= 0) OR 6a or -NR 6 C (= 0) NR 6a R 6b . However, the lower alkyl, lower alkenyl, cycloalkyl, aryl, and heterocyclic groups in R 4 may each be substituted.
R6、 R6a及び R6b :それぞれ独立して、 - H、またはそれぞれ置換されていてもよい低級ァ ルキル、低級アルケニル、シクロアルキル、ァリール若しくはヘテロ環基。 R 6 , R 6a and R 6b : each independently -H, or an optionally substituted lower alkyl, lower alkenyl, cycloalkyl, aryl or heterocyclic group.
R5: -OR0, - NR°R。。または- N(R°)-低級アルキレン- 0R。。。 R 5 : -OR 0 , -NR ° R. . Or -N (R °) -lower alkylene-0R. . .
J : *- NR°C(=0)-、 *- C(=0)NR°-、 - NR°C(=0)NR°-、 *- NR。-低級アルキレン-、または * J: * -NR ° C (= 0)-, * -C (= 0) NR °-, -NR ° C (= 0) NR °-, * -NR. -Lower alkylene- or *
-低級アルキレン- NR°C(=0)-。ただし、 *は A環への結合を示す。 -Lower alkylene-NR ° C (= 0)-. However, * indicates a bond to the A ring.
L : *-NR°-低級アルキレン-、 *-NR°-低級ァルケ-レン-、 -低級アルキレン-または-
低級ァルケ-レン-。ただし、 *は B環への結合を示す。 L: * -NR ° -lower alkylene-, * -NR ° -lower alkylene-, -lower alkylene- or- Lower alkylene. However, * indicates a bond to the B ring.
X:単結合、 - 0-、 - NR7-、 - S -、 - C(=0)-、 - S(=0)-、 - S(=0) -、 *-低級アルキレン- O-X: a single bond, - 0-, - NR 7 - , - S -, - C (= 0) -, - S (= 0) -, - S (= 0) -, * - lower alkylene - O-
2 2
または *-低級アルキレン- NR7-。ただし、 *は C環への結合を示す。 Or * - lower alkylene - NR 7 -. * Indicates a bond to the C ring.
R7: - R°、 - C(=0)-低級アルキルまたは- C(=0)-ハロゲノ低級アルキル。 R 7 : -R °, -C (= 0) -lower alkyl or -C (= 0) -halogeno lower alkyl.
Y:単結合、それぞれ置換されて 、てもよ 、低級アルキレン若しくは低級ァルケ-レン ただし、 C環がヘテロ環である場合、ピロリジンを除く。以下同様。 ] Y: Single bond, each substituted, may be lower alkylene or lower alkylene. However, when C ring is a hetero ring, pyrrolidine is excluded. The same applies below. ]
また、本発明は、前記式 (I)で示されるカルボン酸誘導体またはその製薬学的に許 容される塩と、製薬学的に許容される担体とからなる医薬組成物、殊に、活性化血液 凝固第 VII因子阻害、血液凝固抑制剤、又は血栓若しくは塞栓によって引き起こされ る疾病、殊に、虚血性心疾患、血管形成術後の再狭窄、脳血栓症、一過性脳虚血 発作、末梢動脈閉塞症、間歇性跛行、深部静脈血栓症、肺塞栓症、播種性血管内 凝固症候群 (DIC)、心臓弁置換術後の血栓形成、血液体外循環時における灌流血 液の凝固若しくは炎症、動脈硬化症、癌の予防または治療薬である医薬組成物にも 関する。 The present invention also relates to a pharmaceutical composition comprising a carboxylic acid derivative represented by the above formula (I) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, in particular, activated. Diseases caused by blood coagulation factor VII inhibition, blood coagulation inhibitors, or thrombus or embolism, especially ischemic heart disease, restenosis after angioplasty, cerebral thrombosis, transient ischemic attack, peripheral Arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular coagulation syndrome (DIC), thrombus formation after heart valve replacement, coagulation or inflammation of perfused blood during extracorporeal circulation, arteries The present invention also relates to a pharmaceutical composition that is a prophylactic or therapeutic agent for sclerosis and cancer.
即ち、(1)一般式 (I)記載の化合物またはその製薬学的に許容される塩と、製薬学 的に許容される担体とからなる医薬組成物; That is, (1) a pharmaceutical composition comprising the compound of the general formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier;
(2)活性ィ匕血液凝固第 VII因子阻害剤である(1)記載の医薬組成物; (2) The pharmaceutical composition according to (1), which is an active 匕 blood coagulation factor VII inhibitor;
(3)血液凝固抑制剤である(1)記載の医薬組成物; (3) The pharmaceutical composition according to (1), which is a blood coagulation inhibitor;
(4)血栓若しくは塞栓によって引き起こされる疾病の予防または治療薬である(1)記 載の医薬組成物; (4) The pharmaceutical composition according to (1), which is a preventive or therapeutic agent for diseases caused by thrombus or embolism;
(5)虚血性心疾患、血管形成術後の再狭窄、脳血栓症、一過性脳虚血発作、末梢 動脈閉塞症、間歇性跛行、深部静脈血栓症、肺塞栓症、播種性血管内凝固症候群 (DIC)、心臓弁置換術後の血栓形成、血液体外循環時における灌流血液の凝固若 しくは炎症、動脈硬化症、癌の予防または治療薬である(1)記載の医薬組成物; (5) Ischemic heart disease, restenosis after angioplasty, cerebral thrombosis, transient ischemic attack, peripheral arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular coagulation The pharmaceutical composition according to (1), which is a preventive or therapeutic agent for syndrome (DIC), thrombus formation after heart valve replacement, blood coagulation or inflammation during arterial circulation, arteriosclerosis, and cancer;
(6)活性化血液凝固第 VII因子阻害剤、血液凝固抑制剤の製造のための、請求の範 囲(1)記載の化合物またはその製薬学的に許容される塩の使用; (6) Use of a compound according to claim (1) or a pharmaceutically acceptable salt thereof for the manufacture of an activated blood coagulation factor VII inhibitor or blood coagulation inhibitor;
(7)血栓若しくは塞栓によって引き起こされる疾病の予防または治療薬の製造のた
めの、請求の範囲(1)記載の化合物またはその製薬学的に許容される塩の使用;(7) For the manufacture of preventive or therapeutic drugs for diseases caused by thrombi or emboli Use of a compound according to claim (1) or a pharmaceutically acceptable salt thereof;
(8)虚血性心疾患、血管形成術後の再狭窄、脳血栓症、一過性脳虚血発作、末梢 動脈閉塞症、間歇性跛行、深部静脈血栓症、肺塞栓症、播種性血管内凝固症候群 (DIC)、心臓弁置換術後の血栓形成、血液体外循環時における灌流血液の凝固若 しくは炎症、動脈硬化症、癌の予防または治療薬の製造のための、請求の範囲(1) 記載の化合物またはその製薬学的に許容される塩の使用; (8) Ischemic heart disease, restenosis after angioplasty, cerebral thrombosis, transient ischemic attack, peripheral arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular coagulation Claims (1) for the manufacture of drugs for the prevention or treatment of syndrome (DIC), thrombus formation after heart valve replacement, coagulation or inflammation of perfused blood during extracorporeal circulation, arteriosclerosis, cancer Use of the described compounds or pharmaceutically acceptable salts thereof;
(9)一般式 (I)記載の化合物またはその塩の治療有効量を患者に投与することを含 む、血栓若しくは塞栓によって引き起こされる疾病の予防または治療方法; (9) A method for preventing or treating a disease caused by thrombus or embolism, comprising administering to a patient a therapeutically effective amount of a compound of the general formula (I) or a salt thereof;
(10)—般式 (I)記載の化合物またはその塩の治療有効量を患者に投与することを含 む、虚血性心疾患、血管形成術後の再狭窄、脳血栓症、一過性脳虚血発作、末梢 動脈閉塞症、間歇性跛行、深部静脈血栓症、肺塞栓症、播種性血管内凝固症候群 (DIC)、心臓弁置換術後の血栓形成、血液体外循環時における灌流血液の凝固若 しくは炎症、動脈硬化症、癌の予防または治療方法 (10) —Ischemic heart disease, restenosis after angioplasty, cerebral thrombosis, transient cerebral imagination, including administering to a patient a therapeutically effective amount of a compound of general formula (I) or a salt thereof Blood attack, peripheral arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular coagulation syndrome (DIC), thrombus formation after heart valve replacement, blood coagulation of perfused blood during extracorporeal circulation Or prevention or treatment of inflammation, arteriosclerosis, cancer
に関する。 About.
発明を実施するための最良の形態 BEST MODE FOR CARRYING OUT THE INVENTION
[0028] 本発明をさらに詳細に説明すると以下の通りである。 [0028] The present invention is described in further detail as follows.
本明細書中、「低級」なる語は、特に断らない限り炭素数 1乃至 6個(以下、 C と表 In this specification, the term “lower” means 1 to 6 carbon atoms (hereinafter referred to as “C” unless otherwise specified).
1-6 記することがある)の炭化水素鎖を意味する。「アルキル」、「ァルケ-ル」、「アルキレ ン」、「ァルケ-レン」はそれぞれ直鎖または分枝状であってもよ 、。 1-6) hydrocarbon chain. “Alkyl”, “alkylene”, “alkylene” and “alkylene” may each be linear or branched.
従って、「低級アルキル」とは、 C のアルキルを意味する。具体的には、メチル、ェ Thus, “lower alkyl” means C alkyl. Specifically, methyl,
1-6 1-6
チル、ノルマルプロピル、イソプロピル、ノルマルブチル、イソブチル、 sec-ブチル、 te rt-ブチル、ノルマルペンチル、ノルマルへキシル等が挙げられる。好ましくは C ァ Examples include til, normal propyl, isopropyl, normal butyl, isobutyl, sec-butyl, tert-butyl, normal pentyl, normal hexyl and the like. Preferably C
1-4 ノレキノレのメチノレ、ェチル、ノルマルプロピル、イソプロピル、ノノレマノレブチノレ、イソブチ ル、 sec-ブチル、 tert-ブチルであり、より好ましくはメチル、ェチル、ノルマルプロピル 、イソプロピノレである。 1-4 Norequinole methinole, ethyl, normal propyl, isopropyl, nonolemanole bubutenore, isobutyl, sec-butyl, tert-butyl, more preferably methyl, ethyl, normal propyl, isopropino.
[0029] 「低級ァルケ-ル」とは、任意の位置に 1個以上の二重結合を有する C のァルケ- [0029] "Lower alkellule" is a C alkell having one or more double bonds at any position.
2-6 ルを意味する。具体的には、ビュル、 1-プロペン- 1-ィル、ァリル、イソプロべ-ル、 3 -ブテン- 1-ィル、 1,3 -ブタジェ-ル等が挙げられる。好ましくはじ ァルケ-ルのビ
-ル、 1-プロペン- 1-ィル、ァリル、イソプロべ-ルであり、より好ましくはビュル、ァリ ルである。 2-6. Specific examples include bulle, 1-propen-1-yl, aryl, iso-probe, 3-butene-1-yl, 1,3-butane gel, and the like. Preferably -L, 1-propene-1-yl, aryl, and isopropyl, more preferably bull and aryl.
[0030] 「低級アルキレン」とは、前記「低級アルキル」の任意の位置の水素を 1個除去して なる 2価基を意味する。具体的には- (CH ) -、 - CH(CH ) -、 - C(CH ) -、 - CH CH(C “Lower alkylene” means a divalent group formed by removing one hydrogen at any position of the “lower alkyl”. Specifically,-(CH)-,-CH (CH)-,-C (CH)-,-CH CH (C
2 1-6 3 3 2 2 2 1-6 3 3 2 2
1"0-、 -0"[ 0"[ ) -等が挙げられる。好ましくはじ のアルキレンの- (CH ) -、 - CH(1 "0-, -0" [0 "[)-, etc., preferably-(CH)-, -CH (
3 2 3 2 1-3 2 1-33 2 3 2 1-3 2 1-3
CH ) -、 - C(CH ) -、 - CH CH(CH ) -であり、より好ましくは- (CH ) -である。 CH 2 —, —C (CH 2) 2 —, —CH 2 CH (CH 2) 2 —, and more preferably — (CH 2) 2 —.
3 3 2 2 3 2 1-3 3 3 2 2 3 2 1-3
[0031] 「低級ァルケ-レン」とは、前記「低級アルケニル」の任意の位置の水素を 1個除去 してなる 2価基を意味する。具体的には、 - CH=CH -、 -CH=C(CH ) -、 - C(CH )=C(CH [0031] "Lower alkene" means a divalent group formed by removing one hydrogen at any position of the "lower alkenyl". Specifically, -CH = CH-, -CH = C (CH)-, -C (CH) = C (CH
3 3 3 3
)―、— CH=CH— CH―、—CH=CH—CH=CH—等が挙げられる。好ましくは C ァルケ-レ) —, —CH═CH—CH—, —CH═CH—CH═CH— and the like. Preferably C
3 2 2-3 ンであり、より好ましくはビ-レンである。 3 2 2-3 and more preferably beylene.
[0032] 「シクロアルキル」とは、 C の非芳香族の炭化水素環を意味し、架橋環ゃスピロ環 [0032] "Cycloalkyl" means a non-aromatic hydrocarbon ring of C, and a bridged ring spiro ring.
3-10 3-10
を形成していてもよぐまた部分的に不飽和結合を有していてもよい。具体的には、シ クロプロピノレ、シクロブチノレ、シクロペンチノレ、シクロへキシノレ、シクロへプチノレ、シクロ ォクチル、シクロへキセニル、シクロオクタンジェ -ル、ァダマンチル及びノルボル- ル等が挙げられる。好ましくは C のシクロアルキルであり、より好ましくはシクロブチ May be formed or may have a partially unsaturated bond. Specific examples include cyclopropinole, cyclobutinole, cyclopentinole, cyclohexenole, cycloheptinole, cyclooctyl, cyclohexenyl, cyclooctanegel, adamantyl and norbol. Preferred is C cycloalkyl, more preferred is cyclobuty
3-6 3-6
ル、シクロペンチル若しくはシクロへキシルである。 , Cyclopentyl or cyclohexyl.
[0033] 「ハロゲン」とは、ハロゲン原子を意味する。具体的にはフルォロ、クロ口、ブロモ、ョ ードが挙げられる。好ましくはフルォロ、クロ口、ブロモであり、より好ましくはフルォロ、 クロ口である。 “Halogen” means a halogen atom. Specific examples include fluoro, black mouth, bromo and iodine. Preferred are fluoro, black mouth and bromo, and more preferred are fluoro and black mouth.
[0034] 「ハロゲノ低級アルキル」とは、前記「低級アルキル」の 1個以上の任意の水素原子 力 同一または互いに異なって前記「ハロゲン」で置換された基を意味する。具体的 には、トリフルォロメチル、ペンタフルォロェチル等が挙げられる。好ましくは、トリフル ォロメチルである。 The “halogeno lower alkyl” means one or more arbitrary hydrogen atom forces of the “lower alkyl” which are the same or different from each other and substituted with the “halogen”. Specific examples include trifluoromethyl, pentafluoroethyl and the like. Preferred is trifluoromethyl.
[0035] 「ァリール」とは、単環乃至 3環式の C の芳香族の炭化水素環を意味し、具体的に [0035] The "aryl" means a monocyclic to tricyclic C aromatic hydrocarbon ring, specifically
6-14 6-14
は例えば、フエニル、ナフチル等が挙げられ、好ましくはフエニルである。また、 C の Examples include phenyl and naphthyl, and is preferably phenyl. C's
5-8 シクロアルキル環が縮環していてもよぐ例えば、インダニル、テトラヒドロナフチルを 形成していてもよい。 5-8 The cycloalkyl ring may be condensed. For example, indanyl or tetrahydronaphthyl may be formed.
[0036] 「単環式へテロァリール」とは、 N、 O及び S力 なる群力 選択される 1種以上のへ
テロ原子を 1乃至 4個有する 5乃至 6員の単環式芳香族へテロ環を意味する。環原子 である Sまたは Nが酸ィ匕されォキシドゃジォキシドを形成してもよい。具体的には、ピリ ジル、ピリダジ -ル、ピリミジニル、ピラジ -ル、フリル、チェ-ル、ピロリル、ォキサゾリ ル、イソキサゾリル、ォキサジァゾリル、チアゾリル、チアジアゾリル、イミダゾリル、トリ ァゾリル、テトラゾリル等が挙げられる。好ましくは、ピリジル、ビラジニルであり、より好 ましくはピリジルである。 [0036] "Monocyclic heteroreel" means a group power consisting of N, O, and S forces. A 5- to 6-membered monocyclic aromatic heterocycle having 1 to 4 tera atoms. The ring atom S or N may be oxidized to form an oxide or dioxide. Specific examples include pyridyl, pyridazyl, pyrimidinyl, pyrazinyl, furyl, chael, pyrrolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, thiadiazolyl, imidazolyl, triazolyl, tetrazolyl and the like. Pyridyl and birazinyl are preferable, and pyridyl is more preferable.
[0037] 「ヘテロァリール」とは、前記「単環式へテロァリール」、該単環式へテロァリール同 士または該単環式へテロァリールがベンゼン環と縮環した「2環式へテロァリール」、 及び、該 2環式へテロァリールが該単環式へテロァリールまたはベンゼン環と縮環し た 3環式へテロァリールを意味する。環原子である Sまたは Nが酸ィ匕されォキシドゃジ ォキシドを形成してもよい。「2環式へテロァリール」として具体的には、ベンゾフラ二 ル、ベンゾチェ-ル、ベンゾォキサゾリル、ベンゾイミダゾリル、インドリル、ベンゾチア ゾリル、キノリニル、キナゾリ-ル、キノキサリニル、シンノリ-ル等が挙げられる。好ま しくはインドリル、ベンゾイミダゾリルである。また、 3環式へテロァリールとして具体的 にはカルバゾリル、フルォレニルが挙げられる。ヘテロァリールとして具体的には前記 「単環式へテロァリール」、 「2環式へテロァリール」及び「3環式へテロァリール」で記 載の基が挙げられる。好ましくは、ピリジル、ピラジュル、インドリル、ベンゾイミダゾリ ルであり、より好ましくはピリジルである。 [0037] "Heteroaryl" refers to the above-mentioned "monocyclic heteroreel", the monocyclic heteroreel or "bicyclic heteroreel" in which the monocyclic heteroreel is fused with a benzene ring, and The bicyclic heteroaryl is a monocyclic heteroaryl or a tricyclic heteroaryl fused with a benzene ring. The ring atom S or N may be oxidized to form a dioxide. Specific examples of the “bicyclic heteroaryl” include benzofuranyl, benzochel, benzoxazolyl, benzoimidazolyl, indolyl, benzothiazolyl, quinolinyl, quinazolyl, quinoxalinyl, cinnolyl and the like. Indolyl and benzimidazolyl are preferred. Specific examples of the tricyclic heteroaryl include carbazolyl and fluorenyl. Specific examples of the hetero reel include the groups described in the above-mentioned “monocyclic hetero reel”, “bicyclic hetero reel”, and “tricyclic hetero reel”. Pyridyl, pyradyl, indolyl and benzimidazolyl are preferable, and pyridyl is more preferable.
[0038] 「ヘテロ環」とは、 N、 O及び Sからなる群力も選択される 1種以上のへテロ原子を 1乃 至 4個有する 3乃至 8員の単環式へテロ環、該単環式へテロ環同士並びに該単環式 ヘテロ環がベンゼン環またはシクロアルキル環と縮環した 2環式へテロ環、及び、該 2 環式へテロ環が該単環式へテロ環、ベンゼン環またはシクロアルキル環と縮環した 3 環式へテロ環を意味し、飽和、一部不飽和、及び、芳香族のへテロ環が含まれる。ま た環原子である Nまたは Sが酸ィ匕され、ォキシドゃジォキシドを形成して 、てもよく、 架橋環ゃスピロ環を形成していてもよい。具体的には、前記「単環式へテロァリール」 または「ヘテロァリール」で記載の基、ジヒドロピリジル、ジヒドロピロリル、ジヒドロォキ サゾリル、ジヒドロチアゾリル、ジヒドロイミダゾリル、ピペリジル、モルホリニル、チォモ ルホリニル、ピぺラジュル、ビラゾリジニル、イミダゾリジニル、ピロリジニル、ォキサゾリ
ジニル、チアゾリジニル、ホモピぺラジュル、テトラヒドロフラニル、テトラヒドロビラニル 、テトラヒドロピリミジニル、クロマニル、ジォキソラニル、ホモモルホリニル、ジヒドロイン ドリル等が挙げられる。好ましくは、ピリジル、ピラジュル、ベンゾイミダゾリル、ピロリジ ル、ピペリジル、モルホリニル、インドリル、ジヒドロインドリルであり、より好ましくは、ピ ジジノレ、ピぺジジノレ、モノレホジ-ノレである Q [0038] "Heterocycle" refers to a 3- to 8-membered monocyclic heterocycle having 1 to 4 heteroatoms in which a group force consisting of N, O and S is also selected. A bicyclic heterocycle in which the cyclic heterocycles and the monocyclic heterocycle are condensed with a benzene ring or a cycloalkyl ring, and the bicyclic heterocycle is the monocyclic heterocycle, benzene This means a tricyclic heterocycle condensed with a ring or a cycloalkyl ring, and includes saturated, partially unsaturated, and aromatic heterocycles. The ring atom N or S may be oxidized to form an oxydoxide or a bridged ring or a spiro ring. Specifically, the group described in the above-mentioned “monocyclic heteroaryl” or “heteroaryl”, dihydropyridyl, dihydropyrrolyl, dihydroxazolyl, dihydrothiazolyl, dihydroimidazolyl, piperidyl, morpholinyl, thiomorpholinyl, piperazil , Virazolidinyl, imidazolidinyl, pyrrolidinyl, oxazoly Examples thereof include dinyl, thiazolidinyl, homopiperadil, tetrahydrofuranyl, tetrahydrobiranyl, tetrahydropyrimidinyl, chromanyl, dioxolanyl, homomorpholinyl, dihydroindolyl and the like. Preferably, pyridyl, Pirajuru, benzimidazolyl, pyrrolidine Le, piperidyl, morpholinyl, indolyl, a dihydroindolyl, more preferably, pin Jijinore, piperidines Jijinore, Monorehoji - a Honoré Q
[0039] 「置換されていてもよい」とは、「置換されていない」あるいは「同一または異なる 1乃 至 5個の置換基で置換された」ことを意味する。 [0039] The "optionally substituted" means "unsubstituted" or "substituted with 1 to 5 substituents which are the same or different".
[0040] 本明細書にぉ 、て、「置換されて 、てもよ 、」の語の許容される置換基としては、そ れぞれの基の置換基として、当該技術分野で通常用いられる置換基であれば 、ず れでもよい。 In the present specification, as the permissible substituent of the word “substituted or may be used”, it is commonly used in the art as a substituent of each group. Any substituent may be used.
R1における「- C(=NH)NH- CO -(置換されて 、てもよ 、低級アルキル)」で許容される Accepted by "-C (= NH) NH-CO- (substituted, optionally lower alkyl)" in R 1
2 2
低級アルキル上の置換基として好ましくは、 - oc(=o)-低級アルキルが挙げられる。 Preferred examples of the substituent on the lower alkyl include -oc (= o) -lower alkyl.
R2及び R3における置換されて 、てもよ 、「低級アルキル」で許容される置換基として 好ましくは、下記 P1群より選択される基が挙げられる。 Substituents which may be substituted in R 2 and R 3 and are allowed by “lower alkyl” are preferably groups selected from the following group P 1 .
P1群:ハロゲン、 -OR0, - NR°R°°、 -CO R。及び- C(=0)NR°R。。。 P 1 group: halogen, -OR 0 , -NR ° R °°, -CO R. And -C (= 0) NR ° R. . .
2 2
R2及び R3におけるそれぞれ置換されていてもよい「シクロアルキル」、「ァリール」及 び「ヘテロ環基」で許容される置換基として好ましくは、下記 Q1群より選択される基が 挙げられる。 Which may be substituted, respectively, in R 2 and R 3 "cycloalkyl", as preferred substituents allowed in "Ariru"及beauty "heterocyclic group" includes groups selected from the following Q 1 group .
Q1群:低級アルキル、ハロゲノ低級アルキル、ハロゲン、ォキソ、 -OR。及び- 0-ハロゲ ノ低級アルキル。 Q Group 1 : lower alkyl, halogeno lower alkyl, halogen, oxo, -OR. And -0-halogeno lower alkyl.
R4、 R6、 R6a及び R6bにおける置換されていてもよい「低級アルキル」及び「低級ァルケ -ル」で許容される置換基として好ましくは、下記 P2群より選択される基が挙げられる R 4, R 6, R 6a and R may be substituted in 6b "lower alkyl" and "lower Aruke - Le" As preferred substituents allowed in, include groups selected from the following P 2 group Be
P2群:ノヽロゲン、 - CN、 -OR°, -0-ハロゲノ低級アルキル、 - OC(=0)R°、 -OC(=0)NR°R 00、 -ォキソ、 - SR°、 - S(=0)R°、 - S(=0) R°、 - S(=0) NR°R°°、 - NR°S(=0) R°°、 - NHS(=0) P 2 group: neurogen, -CN, -OR °, -0-halogeno lower alkyl, -OC (= 0) R °, -OC (= 0) NR ° R 0 0 , -oxo, -SR °,- S (= 0) R °,-S (= 0) R °,-S (= 0) NR ° R °°,-NR ° S (= 0) R °°,-NHS (= 0)
2 2 2 2 2 2 2 2
NR。R°°、 - N (低級アルキル) - S(=0) NR°R°°、 - NR°R°°、 - NH-低級アルキレン- NR°R00、 NR. R °°, -N (lower alkyl) -S (= 0) NR ° R °°, -NR ° R °°, -NH-lower alkylene-NR ° R 00 ,
2 2
- N (低級アルキル) -低級アルキレン - NR°R°°、 - C(=0)R°、 -CO R°、 - C(=0)NR°R°°、 - C -N (lower alkyl)-Lower alkylene-NR ° R °°,-C (= 0) R °, -CO R °,-C (= 0) NR ° R °°,-C
2 2
(=〇)N(R°)-ヘテロ環基、 - C(=〇)N(R°)-低級アルキレン- OR°、 -C(=〇)NH-低級アルキ
レン - NR°R°°、 - C(=0)N (低級アルキル)-低級アルキレン - NR°R°°、 - C(=0)N(R°)-低級 アルキレン-シクロアルキル、 - C(=0)_ヘテロ環基、 - N(R°)C(=0)R°°、シクロアルキル、 ァリール及びへテロ環基。ただし、 P2群におけるシクロアルキル、ァリール及びへテロ 環基は、 Q2群力 選択される基で置換されて 、てもよ 、。 (= 〇) N (R °) -heterocyclic group, -C (= 〇) N (R °) -lower alkylene-OR °, -C (= 〇) NH-lower alkyl Ren-NR ° R °°,-C (= 0) N (lower alkyl) -lower alkylene-NR ° R °°,-C (= 0) N (R °) -lower alkylene-cycloalkyl,-C ( = 0) _heterocyclic group, -N (R °) C (= 0) R °°, cycloalkyl, aryl and heterocyclic group. However, cycloalkyl of P 2 group, Ariru and hetero ring group is substituted with a group selected Q 2 groups force, even.
Q2群:低級アルキル、ハロゲノ低級アルキル、ハロゲン、ォキソ、 -OR0, - 0-ハロゲノ 低級アルキル、 - NR°R°°、 - C(=0)R°、 -CO 2 R°、 - NR°S(=0) 2 R°°、 - C(=0)NR°R°°、 -NR°C(Q 2 group: lower alkyl, halogeno lower alkyl, halogen, oxo, -OR 0 ,-0-halogeno lower alkyl, -NR ° R °°, -C (= 0) R °, -CO 2 R °, -NR ° S (= 0) 2 R °°, -C (= 0) NR ° R °°, -NR ° C (
=0)R。。、低級アルキレン- NR。R。。、低級アルキレン- C〇2R。及び-低級アルキレン- C(== 0) R. . , Lower alkylene-NR. R. . , Lower alkylene - C_〇 2 R. And -lower alkylene-C (=
O)NRV0。 O) NRV 0 .
R6A及び R6Bにおけるそれぞれ置換されていてもよい「シクロアルキル」、「ァリ ール」及び「ヘテロ環基」で許容される置換基として好ましくは、上記 Q2群より選択さ れる基が挙げられる。 Which may be substituted, respectively, in R 6A and R 6B "cycloalkyl", as preferred substituents allowed in "§ Re window" and "heterocyclic group" is a group selected from the Q 2 group Can be mentioned.
Yにおける置換されて 、てもよ ヽ「低級アルキレン」、「低級ァルケ-レン」、で許容さ れる置換基として好ましくは、ハロゲン及び- ORQより選択される基が挙げられる。 一般式 (I)に示される本発明化合物における好ましい態様を以下に示す。 Substituents which may be substituted in Y but are allowed by “lower alkylene” and “lower alkylene” are preferably groups selected from halogen and —OR Q. Preferred embodiments of the compound of the present invention represented by the general formula (I) are shown below.
A環として好ましくはベンゼン環、ピリジン環またはべンゾイミダゾール環であり、より 好ましくはベンゼン環またはピリジン環であり、さらにより好ましくはベンゼン環である The A ring is preferably a benzene ring, a pyridine ring or a benzoimidazole ring, more preferably a benzene ring or a pyridine ring, and still more preferably a benzene ring.
B環として好ましくは、単環 6員芳香環であり、より好ましくは、ベンゼン環またはピリ ジン環である。 B ring is preferably a monocyclic 6-membered aromatic ring, and more preferably a benzene ring or a pyridine ring.
C環として好ましくはベンゼン環、ピぺリジン環、ピぺラジン環またはモルホリン環で あり、より好ましくはベンゼン環またはピぺリジン環である。 The C ring is preferably a benzene ring, piperidine ring, piperazine ring or morpholine ring, and more preferably a benzene ring or piperidine ring.
R1として好ましくは、 -CH NH、— C(=0)NH、— C(=NH)NH、— C(=NOH)NHまたは— R 1 is preferably -CH NH, -C (= 0) NH, -C (= NH) NH, -C (= NOH) NH or-
2 2 2 2 2 C 2 2 2 2 2 C
(=NH)NH- CO -低級アルキルであり、より好ましくは- C(=NH)NH、 - C(=NOH)NHま (= NH) NH-CO-lower alkyl, more preferably -C (= NH) NH, -C (= NOH) NH
2 2 2 たは- C(=NH)NH- CO -低級アルキルである。 2 2 2 or —C (═NH) NH—CO 2 -lower alkyl.
2 2
R2として好ましくはハロゲンである。 R 2 is preferably halogen.
R3として好ましくは、低級アルキル、 -0-低級アルキルまたはハロゲンであり、より好 ましくはメチル、 - 0-メチル、クロ口またはフルォロである。 R 3 is preferably lower alkyl, -0-lower alkyl, or halogen, and more preferably methyl, -0-methyl, black mouth, or fluoro.
R4として好ましくは、ヘテロ環基、 -0-低級アルキル、—低級アルキレン- OH、 -0-
シクロアルキルまたは- O-低級アルキレン- (低級アルキルで置換されて!、てもよ!/、へ テロ環)であり、より好ましくは- 0-低級アルキル、—低級アルキレン- OH、または- 0- 低級アルキレン- (低級アルキルで置換されて 、てもよ 、ヘテロ環)であり、さらにより 好ましくは- 0-ェチル、 -0-イソプロピルまたは—低級アルキレン- OHである。 R 4 is preferably a heterocyclic group, -0-lower alkyl, -lower alkylene-OH, -0- Cycloalkyl or —O-lower alkylene- (substituted with lower alkyl !, may! /, Heterocycle), more preferably —0-lower alkyl, —lower alkylene-OH, or —0— Lower alkylene- (which may be substituted with lower alkyl, or a heterocycle), and more preferably is 0-ethyl, -0-isopropyl or -lower alkylene-OH.
R5として好ましくは、 -0Hまたは- 0-低級アルキルであり、より好ましくは- 0Hまたは- 0-ェチルである。 R 5 is preferably -0H or -0-lower alkyl, more preferably -0H or -0-ethyl.
Jとして好ましくは、 *-NHC(=0)_ (ただし *は A環への結合を表す)である。 J is preferably * -NHC (= 0) _ (where * represents a bond to the A ring).
Lとして好ましくは、 *- NHCH -(ただし *は B環への結合を表す)である。 L is preferably * -NHCH- (where * represents a bond to the B ring).
2 2
Xとして好ましくは、単結合または- 0-であり、より好ましくは- 0-である。 X is preferably a single bond or -0-, more preferably -0-.
Yとして好ましくは、低級アルキレンであり、より好ましくはメチレンまたはエチレンで ある。 Y is preferably lower alkylene, more preferably methylene or ethylene.
また、一般式 (I)に示される本発明化合物における別の好ましいィ匕合物を以下に示 す。 Further, another preferred compound in the compound of the present invention represented by the general formula (I) is shown below.
(1)Jが *-NHC(=0)-または *-NH-低級アルキレン- (ただし、 *は A環への結合を表す )であり、 Lが *-NH-低級アルキレン- (ただし、 *は B環への結合を表す)である一般式 (I)記載の化合物。 (1) J is * -NHC (= 0)-or * -NH-lower alkylene- (where * represents a bond to the A ring) and L is * -NH-lower alkylene- (where * Represents a bond to ring B), and the compound according to general formula (I).
(2)下記一般式 (I 1)で示される(1)記載の化合物。 (2) The compound according to (1), which is represented by the following general formula (I 1).
[化 12] [Chemical 12]

[Ba環:ベンゼン環または 5乃至 6員単環式へテロァリール環。ただし、 Ba環に隣接する 窒素原子及び J1は、 Ba環を構成する隣接する 2つの炭素原子にそれぞれ結合する。 J1 :- C(=0)-または- CH -。以下同様。 ] [B a ring: Teroariru ring to a benzene ring or a 5 or 6-membered monocyclic. However, the nitrogen atom and J 1 adjacent to B a ring, each bonded to adjacent two carbon atoms constituting the B a ring. J 1 : -C (= 0)-or -CH-. The same applies below. ]
2 2
(3)下記一般式 (I 2)で示される(2)記載の化合物。
[化 13] (3) The compound according to (2), which is represented by the following general formula (I 2). [Chemical 13]

(4) J1が- C(=0)-である(3)記載の化合物。 (4) The compound according to (3), wherein J 1 is —C (= 0) —.
(5) !?1が- C(=NH)NH、 - C(=NOH)NH、 - C(=NH)NH- CO -(置換されていてもよい低 (5)!? 1 is -C (= NH) NH, -C (= NOH) NH, -C (= NH) NH-CO- (optionally low
2 2 2 2 2 2
級アルキル)である (4)記載の化合物。 (4) The compound according to (4).
(6) Yがハロゲンまたは- OR°で置換されて 、てもよ 、低級アルキレンである(5)記載 の化合物。 (6) The compound according to (5), wherein Y is substituted with halogen or —OR °, and is lower alkylene.
(7) Xが単結合または- 0-である(6)記載の化合物。 (7) The compound according to (6), wherein X is a single bond or -0-.
(8) A環がベンゼン環またはピリジン環である(6)記載の化合物。 (8) The compound according to (6), wherein the A ring is a benzene ring or a pyridine ring.
(9) B環がベンゼン環またはピリジン環である(6)記載の化合物。 (9) The compound according to (6), wherein the B ring is a benzene ring or a pyridine ring.
(10) C環がベンゼン環、ピぺリジン環、ピぺラジン環またはモルホリン環である(6)記 載の化合物。 (10) The compound according to (6), wherein the C ring is a benzene ring, piperidine ring, piperazine ring or morpholine ring.
(11) {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル} ァミノ)メチル ]-4- [(アミノスルホ -ル)ァミノ] -6-イソプロポキシフエノキシ }ァセチックァ シッド、 (11) {2- [({2- [({4- [Amino (imino) methyl] phenol} amino) carbol] -4-chlorophine} amino) methyl] -4- [ (Aminosulfoyl) amino] -6-isopropoxyphenoxy} acetic acid,
ェチル {2- [({2- [({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ 口フエ-ル}ァミノ)メチル ]-4- [(アミノスルホ -ル)ァミノ] -6-イソプロポキシフエノキシ }ァ セタート、 Ethyl {2- [({2- [({4- [Amino (hydroxyimino) methyl] phenol} amino) carbol] -4-chlorophenol} amino) methyl] -4- [ (Aminosulfol) amino] -6-isopropoxyphenoxy} acetate,
[2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-フルオロフェ-ル }アミ
ノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ]ァセチックアシッド、 {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-メトキシフエ-ル}アミ ノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ }ァセチックアシッド、 [2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-メチルフエ-ル}ァミノ) メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ]ァセチックアシッド、 ェチル [2- {[(2- {[(4- {[(エトキシカルボニル)ァミノ] (ィミノ)メチル }フエニル)ァミノ]カルボ -ル }-4-フルオロフェ -ル)ァミノ]メチル }- 4- (ヒドロキシメチル) -6-イソプロポキシフエノ キシ]ァセタート、 [2- [({2- [({4- [Amino (imino) methyl] phenol} amino) carbol] -4-fluorophenyl} amino ) Methyl] -4- (hydroxymethyl) -6-isopropoxyphenoxy] acetic acid, {2-[({2-[({4- [amino (imino) methyl] phenol} amino ) Carbon] -4-methoxyphenyl} amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenoxy} acetic acid, [2-[({2-[({4 -[Amino (methyl) phenol} amino) carbol] -4-methylphenol} amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenoxy] acetic acid, Ethyl [2- {[(2- {[(4- {[(Ethoxycarbonyl) amino] (imino) methyl} phenyl) amino] carbol} -4-fluorophenyl) amino] methyl}-4- ( Hydroxymethyl) -6-isopropoxyphenoxy] acetate,
ェチル [2- {[(2- {[(4- {[(エトキシカルボニル)ァミノ] (ィミノ)メチル }フエニル)ァミノ]カルボ -ル }-4-メトキシフエ-ル)ァミノ]メチル }-4- (ヒドロキシメチル) -6-イソプロポキシフエノ キシ]ァセタート、 Ethyl [2- {[(2- {[(4- {[(Ethoxycarbonyl) amino] (imino) methyl} phenyl) amino] carbol} -4-methoxyphenyl) amino] methyl} -4- ( Hydroxymethyl) -6-isopropoxyphenoxy] acetate,
ェチル [2- {[(2- {[(4- {[(エトキシカルボニル)ァミノ] (ィミノ)メチル }フエニル)ァミノ]カルボ -ル }-4-メチルフエ-ル)ァミノ]メチル }-4- (ヒドロキシメチル) -6-イソプロポキシフエノキ シ]ァセタート、 Ethyl [2- {[(2- {[(4- {[(Ethoxycarbonyl) amino] (imino) methyl} phenyl) amino] carbol} -4-methylphenol) amino] methyl} -4- ( Hydroxymethyl) -6-isopropoxyphenoxy] acetate,
[2- [({2- [({6- [ァミノ (ィミノ)メチル]ピリジン- 3-ィル }ァミノ)カルボ二ル]- 4-メチルフエ二 ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ]ァセチックァシッ ド、及び [2-[({2-[({6- [Amino (imino) methyl] pyridine-3-yl} amino) carboyl] -4-methylphenyl} amino) methyl] -4- (hydroxymethyl ) -6-Isopropoxyphenoxy] acetic acid, and
ェチル [2- {[(2- {[(6- {[(エトキシカルボニル)ァミノ] (ィミノ)メチル }ピリジン- 3-ィル)ァミノ] カルボ-ル}-4-メチルフエ-ル)ァミノ]メチル }-4- (ヒドロキシメチル) -6-イソプロポキシ フエノキシ]ァセタート Ethyl [2- {[(2- {[(6- {[(Ethoxycarbonyl) amino] (imino) methyl} pyridine-3-yl) amino] carbol} -4-methylphenol) amino] methyl } -4- (Hydroxymethyl) -6-isopropoxyphenoxy] acetate
力もなる群力 選択される(1)記載の化合物またはその製薬学的に許容される塩。 また、一般式 (I)に示される本発明化合物におけるさらに別の好ましいィ匕合物を以 下に示す。 The group force which also has power The compound or its pharmaceutically acceptable salt as described in (1) selected. Further, other preferred compounds in the compound of the present invention represented by the general formula (I) are shown below.
(12)下記式 (I 3)で示される式 (I)記載の化合物。 (12) A compound of the formula (I) represented by the following formula (I 3).
[化 14]
[Chemical 14]

[B5、 B、 B7及び B :それぞれ独立して、 CH、 CR または N。ただし、 B5、 B、 B7及び B のうち、 CR3aは 3個以下であり、また、それぞれの CR3aは同一または互いに異なってい てもよい。 [B 5 , B, B 7 and B are each independently CH, CR or N. However, among B 5 , B, B 7 and B, CR 3a is 3 or less, and each CR 3a may be the same or different from each other.
Rla: -NH、 -CH NH、— C(=0)NH、— C(=NH)NH、— C(=NOH)NHまたは— C(=NH)NH—R la : -NH, -CH NH,-C (= 0) NH,-C (= NH) NH,-C (= NOH) NH or-C (= NH) NH-
2 2 2 2 2 2 2 2 2 2 2 2
CO -低級アルキル。 CO 2 -lower alkyl.
2 2
R2a及び R3a:それぞれ独立して、低級アルキル、ハロゲノ低級アルキル、ハロゲン、ォ キソ、 - CN、 -NO、 -OR°, -0-ハロゲノ低級アルキル、 - NR°R°°、 - SR°、 - S(=0)R°、 - S(= R 2a and R 3a : each independently lower alkyl, halogeno lower alkyl, halogen, oxo, -CN, -NO, -OR °, -0-halogeno lower alkyl, -NR ° R °°, -SR ° ,-S (= 0) R °,-S (=
2 2
O) R。、— S(=0) NR。R。。、— NR。S(=0) R。。、— C(=0)R。、—CO R。、— C(=0)NR。R。。、— NR。C(= O) R. , — S (= 0) NR. R. . , — NR. S (= 0) R. . , — C (= 0) R. , —CO R. , — C (= 0) NR. R. . , — NR. C (=
2 2 2 2 2 2 2 2
0)RQQ、シクロアルキル、ァリールまたはへテロ環基。ただし、 R2a及び R3aにおけるシク 口アルキル、ァリール及びへテロ環基は、低級アルキル、ハロゲノ低級アルキル、ハロ ゲン、ォキソ、 -OR°または- 0-ノヽロゲノ低級アルキルで置換されていてもよい。 0) R QQ , cycloalkyl, aryl or heterocyclic group. However, the cycloalkyl, aryl, and heterocyclic groups in R 2a and R 3a may be substituted with lower alkyl, halogeno lower alkyl, halogen, oxo, -OR °, or 0-0-norogeno lower alkyl. .
あるいは、それぞれ 2つの R2aまたは R3aがー体となって- 0-低級アルキレン- 0-を形 成していてもよい。 Alternatively, each of two R 2a or R 3a may be in the form of -0-lower alkylene-0-.
R½:それぞれ独立して、低級アルキル、シクロアルキル、ァリール、ヘテロ環基、ハロ ゲン、ォキソ、 - CNゝ -NO、 - 0R6c、 - NR6cR6d、 - SR6c、 - S(=0)R6c、 - S(=0) R6c、 - S(=0) N R ½: each independently, lower alkyl, cycloalkyl, Ariru, heterocyclic group, halo gen, Okiso, - CNゝ-NO, - 0R 6c, - NR 6c R 6d, - SR 6c, - S (= 0 ) R 6c ,-S (= 0) R 6c ,-S (= 0) N
2 2 2 2 2 2
R6cR6d、 -NR6cS(=0) R6d、 - C(=0)R6c、 -CO Re\ - C(=0)NR6 6d、 - 0C(=0)R6c、 - 0C(=0 R 6c R 6d , -NR 6c S (= 0) R 6d ,-C (= 0) R 6c , -CO R e \-C (= 0) NR 6 6d ,-0C (= 0) R 6c ,- 0C (= 0
2 2 twenty two
)0R6c、 - OC(=0)NR6cR6d、 - NR6cC(=0)R6d、 - NR6cC(=0)OR6dまたは- NR6cC(=0)NR6dR6e ただし、 R½における低級アルキルは Pa群力も選択される基で置換されて 、てもよく、 シクロアルキル、ァリール及びへテロ環基はそれぞれ Qa群力 選択される基で置換さ れていてもよい。
R6c、 R6d及び R6e:それぞれ独立して、 - H、低級アルキル、シクロアルキル、ァリールま たはへテロ環基。ただし、 R6e、 R6d及び R6eにおける低級アルキルは Pa群カゝら選択され る基で置換されていてもよぐシクロアルキル、ァリール及びへテロ環基はそれぞれ Qa 群から選択される基で置換されて 、てもよ 、。 ) 0R 6c , -OC (= 0) NR 6c R 6d , -NR 6c C (= 0) R 6d , -NR 6c C (= 0) OR 6d or -NR 6c C (= 0) NR 6d R 6e lower alkyl in R ½ is substituted with a group selected also P a group force and may, cycloalkyl, be the heterocyclic group Ariru and to be substituted with a group selected respectively Q a group force Good. R 6c , R 6d and R 6e : each independently —H, lower alkyl, cycloalkyl, aryl or hetero ring group. However, the lower alkyl in R 6e , R 6d and R 6e is a cycloalkyl, aryl and hetero ring group which may be substituted with a group selected from group P a and each is selected from group Q a May be substituted with a group.
pa群:ノヽロゲン、 - CN、 -OR°, -O-ハロゲノ低級アルキル、ォキソ、 - SR°、 - S(=0)R°、 - S (=0) R°、 -S(=0) NR°R°°、 -NR°S(=0) R°°、 - NR°R°°、 -CO R°、 - C(=0)NR°R°°、 -N(R°)C( p a group: neurogen, -CN, -OR °, -O-halogeno lower alkyl, oxo, -SR °, -S (= 0) R °, -S (= 0) R °, -S (= 0 ) NR ° R °°, -NR ° S (= 0) R °°, -NR ° R °°, -CO R °, -C (= 0) NR ° R °°, -N (R °) C (
2 2 2 2 2 2 2 2
=0)R°°、シクロアルキル、ァリールまたはへテロ環。ただし、 Pa群におけるシクロアルキ ル、ァリール及びへテロ環基は、 Qa群カゝら選択される基で置換されていてもよい。 = 0) R °°, cycloalkyl, aryl or heterocycle. However, the cycloalkyl, aryl and hetero ring group in the P a group may be substituted with a group selected from the Q a group.
Qa群:低級アルキル、ハロゲノ低級アルキル、ハロゲン、ォキソ、 -OR0, - 0-ハロゲノ 低級アルキル、 NR。R。。、 -CO R。、— NR。S(=0) R。。、— C(=0)NR。R。。、— NR。C(=0)R。。、低 Q a group: lower alkyl, halogeno-lower alkyl, halogen, Okiso, -OR 0, - 0- halogeno-lower alkyl, NR. R. . -CO R. , — NR. S (= 0) R. . , — C (= 0) NR. R. . , — NR. C (= 0) R. . Low
2 2 twenty two
級アルキレン- NR°R°°、低級アルキレン- CO R°、 -低級アルキレン- C(=0)NR°R°°。 Grade alkylene-NR ° R °°, lower alkylene-CO R °, -lower alkylene-C (= 0) NR ° R °°.
2 2
R5a: -OHまたは- 0-低級アルキル。 R 5a : —OH or —0-lower alkyl.
Ya:ハロゲンまたは- OR°で置換されて 、てもよ 、低級アルキレン。 ] Y a : lower alkylene substituted with halogen or —OR °. ]
また、本発明の化合物には、幾何異性体、互変異性体、光学異性体などの各種の 立体異性体が存在することがある。本発明はそれらの混合物や単離されたものが含 まれる。 The compound of the present invention may have various stereoisomers such as geometric isomers, tautomers and optical isomers. The present invention includes mixtures thereof and isolated ones.
本発明化合物は、酸付加塩を形成する場合がある。また、塩基との塩を形成する場 合もある。力かる塩としては、具体的には、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、 硝酸、リン酸等の鉱酸、ギ酸、酢酸、プロピオン酸、シユウ酸、マロン酸、コハク酸、フ マル酸、マイレン酸、乳酸、リンゴ酸、酒石酸、クェン酸、トリフルォロ酢酸、メタンスル ホン酸、エタンスルホン酸等の有機酸、ァスパラギン酸、グルタミン酸などの酸性アミ ノ酸との酸付加塩、ナトリウム、カリウム、マグネシウム、カルシウム、アルミニウムなど 無機塩基、メチルァミン、ェチルァミン、エタノールァミンなどの有機塩基、リジン、ォ ル-チンなどの塩基性アミノ酸との塩やアンモ-ゥム塩等が挙げられる。 The compound of the present invention may form an acid addition salt. It may also form a salt with a base. Specific examples of strong salts include mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, Fumaric acid, maleic acid, lactic acid, malic acid, tartaric acid, citrate, trifluoroacetic acid, methanesulfonic acid, ethanesulfonic acid and other organic acids, acid addition salts with aspartic acid, glutamic acid and other acidic amino acids, sodium, Examples thereof include inorganic bases such as potassium, magnesium, calcium, and aluminum, organic bases such as methylamine, ethylamine, and ethanolamine, salts with basic amino acids such as lysine and oltin, and ammonium salts.
更に本発明には、本発明化合物の水和物、製薬学的に許容可能な各種溶媒和物 や結晶多形等も含まれる。なお、当然のことながら、本発明は後記の実施例に記載さ れた化合物に限定されるものではなぐ一般式 (I)で示されるカルボン酸誘導体又は その製薬学的に許容される塩の全てを包含するものである。
また、本発明化合物には、生体内において代謝されて前記一般式 (I)を有するィ匕 合物またはその塩に変換される化合物、いわゆるプロドラッグもすベて含まれる。本 発明化合物のプロドラッグを形成する基としては、 Prog. Med. 5 : 2157— 2161 (19 85)に記載されている基や、広川書店 1990年刊「医薬品の開発」第 7卷分子設計 1 63〜198頁に記載されている基が挙げられる。特に本発明化合物のプロドラッグとし ては、水酸基及びエステル基を有するプロドラッグが生体内で代謝を受け、一般式 (I )で示されるカルボン酸誘導体となるプロドラッグが考えられる力 そのようなプロドラッ グも本発明に含まれるものである。 Furthermore, the present invention includes hydrates of the compounds of the present invention, various pharmaceutically acceptable solvates, crystal polymorphs, and the like. It should be understood that the present invention is not limited to the compounds described in the examples below, and all of the carboxylic acid derivatives represented by the general formula (I) or pharmaceutically acceptable salts thereof. Is included. The compounds of the present invention also include all compounds that are metabolized in vivo and converted to the compounds having general formula (I) or salts thereof, so-called prodrugs. Examples of the group that forms a prodrug of the compound of the present invention include those described in Prog. Med. 5: 2157-2161 (1985), Hirokawa Shoten, 1990, “Drug Development”, 7th Molecular Design 1 63 And the groups described on page 198. In particular, as a prodrug of the compound of the present invention, a prodrug having a hydroxyl group and an ester group that undergoes metabolism in a living body and becomes a carboxylic acid derivative represented by the general formula (I) can be considered. Are also included in the present invention.
[0045] (製造法) [0045] (Production method)
本発明化合物及びその製薬学的に許容される塩は、その基本骨格あるいは置換 基の種類に基づく特徴を利用し、種々の公知の合成法を適用して製造することがで きる。以下に代表的な製造法を例示する。なお、官能基の種類によっては、当該官 能基を原料乃至中間体の段階で適当な保護基、即ち、容易に当該官能基に転化可 能な基に置き換えておくことが製造技術上効果的な場合がある。しかるのち、必要に 応じて保護基を除去し、所望の化合物を得ることができる。このような官能基としては 例えば水酸基やカルボキシル基、アミノ基等を挙げることができ、それらの保護基とし ては例えばグリーン(Greene)及びウッツ(Wuts)著、「Protective Groups in Organic S ynthesis (third edition)」に記載の保護基を挙げることができ、これらを反応条件に応 じて適宜用いればよい。 The compound of the present invention and a pharmaceutically acceptable salt thereof can be produced by applying various known synthetic methods using characteristics based on the basic skeleton or the type of substituent. A typical production method is illustrated below. Depending on the type of functional group, it is effective in terms of production technology to replace the functional group with a suitable protecting group at the raw material or intermediate stage, that is, a group that can be easily converted to the functional group. There is a case. Thereafter, the protecting group can be removed as necessary to obtain the desired compound. Examples of such functional groups include a hydroxyl group, a carboxyl group, an amino group, and the like, and examples of protective groups thereof include those described in “Protective Groups in Organic Synthesis (third) by Greene and Wuts”. edition) ”, and these may be appropriately used depending on the reaction conditions.
[0046] (製造法) [0046] (Production method)
以下に本発明化合物の代表的な製造法を説明する。 Hereinafter, representative production methods of the compound of the present invention will be described.
[0047] [化 15]
[0047] [Chemical 15]



( I ) (I)
(式中、 R8、 R9、 R1Q、 RU、 R12及び X1は、必要に応じ当業者が通常採用し得る工程を 経てそれぞれ R1 R2、 R3、
 R5及び Xへ変換可能な置換基を意味する。式中、 J2及 び J3は必要に応じ当業者が通常採用し得る工程を経て結合 Jへと変換可能な置換基 を意味し、具体的には、メチル基、ヒドロキシメチル基、ハロゲノメチル基、ホルミル基 、アルコキシメチル基、ァセトキシメチル基、アミノ基、アルコキシカルボ-ルァミノ基、 ニトロ基、カルボキシル基、アルコキシカルボ-ル基等が挙げられる。式中、 L1及び L 2は必要に応じ当業者が通常採用し得る工程を経て結合 Lへと変換可能な置換基を 意味し、具体的には、メチル基、ヒドロキシメチル基、ハロゲノメチル基、ホルミル基、 アルコキシメチル基、ァセトキシメチル基、アミノ基、アルコキシカルボ-ルァミノ基、 ニトロ基、カルボキシル基、アルコキシカルボ-ル基等が挙げられる。なお、 R8、 R9、 R1Q、 Ru、 R12及び X1は、各々
R5及び Xへ変換可能な置換基を意味する。式中、 J2及 び J3は必要に応じ当業者が通常採用し得る工程を経て結合 Jへと変換可能な置換基 を意味し、具体的には、メチル基、ヒドロキシメチル基、ハロゲノメチル基、ホルミル基 、アルコキシメチル基、ァセトキシメチル基、アミノ基、アルコキシカルボ-ルァミノ基、 ニトロ基、カルボキシル基、アルコキシカルボ-ル基等が挙げられる。式中、 L1及び L 2は必要に応じ当業者が通常採用し得る工程を経て結合 Lへと変換可能な置換基を 意味し、具体的には、メチル基、ヒドロキシメチル基、ハロゲノメチル基、ホルミル基、 アルコキシメチル基、ァセトキシメチル基、アミノ基、アルコキシカルボ-ルァミノ基、 ニトロ基、カルボキシル基、アルコキシカルボ-ル基等が挙げられる。なお、 R8、 R9、 R1Q、 Ru、 R12及び X1は、各々
 R5及び Xと同一であっても良い。以下 同様。)
[0048] 工程 A ( Where R 8 , R 9 , R 1Q , R U , R 12 and X 1 are R 1 R 2 , R 3 , A substituent that can be converted to R 5 and X. In the formula, J 2 and J 3 are substituents that can be converted into a bond J through a process that can be usually employed by those skilled in the art, and specifically include methyl, hydroxymethyl, halogenomethyl, and the like. Group, formyl group, alkoxymethyl group, acetoxymethyl group, amino group, alkoxycarboamino group, nitro group, carboxyl group, alkoxycarbo group and the like. In the formula, L 1 and L 2 mean a substituent that can be converted to a bond L through a process that can be usually employed by those skilled in the art, and specifically include a methyl group, a hydroxymethyl group, and a halogenomethyl group. A formyl group, an alkoxymethyl group, an acetoxymethyl group, an amino group, an alkoxycarboamino group, a nitro group, a carboxyl group, an alkoxycarbo group, and the like. R 8 , R 9 , R 1Q , R u , R 12 and X 1 are each It may be the same as R 5 and X. The same shall apply hereinafter. ) [0048] Process A
R5及び Xと同一であっても良い。以下 同様。)
[0048] 工程 A ( Where R 8 , R 9 , R 1Q , R U , R 12 and X 1 are R 1 R 2 , R 3 , A substituent that can be converted to R 5 and X. In the formula, J 2 and J 3 are substituents that can be converted into a bond J through a process that can be usually employed by those skilled in the art, and specifically include methyl, hydroxymethyl, halogenomethyl, and the like. Group, formyl group, alkoxymethyl group, acetoxymethyl group, amino group, alkoxycarboamino group, nitro group, carboxyl group, alkoxycarbo group and the like. In the formula, L 1 and L 2 mean a substituent that can be converted to a bond L through a process that can be usually employed by those skilled in the art, and specifically include a methyl group, a hydroxymethyl group, and a halogenomethyl group. A formyl group, an alkoxymethyl group, an acetoxymethyl group, an amino group, an alkoxycarboamino group, a nitro group, a carboxyl group, an alkoxycarbo group, and the like. R 8 , R 9 , R 1Q , R u , R 12 and X 1 are each It may be the same as R 5 and X. The same shall apply hereinafter. ) [0048] Process A
化合物(II)と化合物(III)の組み合わせ力 なる二つの化合物から、化合物(IV)を 合成する工程である。本工程は、当業者が通常採用し得る工程、例えば、ハロゲン 化反応、酸化反応、還元反応、加水分解反応、加水素分解反応、アミド化反応、ァ ルキル化反応、還元的アルキルィヒ反応等を任意に組み合わせることにより、実施す ることができる。本工程は一段階の反応とは限らず、多段階の反応により構成される 場合もある。 This is a step of synthesizing compound (IV) from two compounds having the combined power of compound (II) and compound (III). In this step, any step that can be usually employed by those skilled in the art, for example, halogenation reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction, alkylation reaction, reductive alkyl-rich reaction, etc. It can be implemented by combining with. This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
[0049] A- 1ハロゲン化 [0049] A-1 Halogenation
ノ、ロゲン化反応は当業者が通常用いうるハロゲン化反応を用いることができる。例 えば日本化学会編「実験化学講座 (第 4版)」 19卷 (1992年) (丸善)等に記載の方法が 適用できる。例えば、 2, 2'—ァゾビスイソブチ口-トリルあるいは過酸ィ匕べンゾィル存 在下、ハロゲン化剤として N—プロモスクシンイミド等を用いることができる。反応はべ ンゼン、トルエン、キシレン等の芳香族炭化水素類、酢酸ェチル等のエステル類、ジ ェチルエーテル、テトラヒドロフラン(THF)、 1,4-ジォキサン等のエーテル類、ジクロロ メタン、 1,2-ジクロロエタン、クロ口ホルム等のハロゲン化炭化水素類、メタノール、ェ タノール等のアルコール類、 Ν,Ν-ジメチルホルムアミド(DMF)、 Ν,Ν-ジメチルァセト アミド(DMA)、 Ν-メチルピロリドン(ΝΜΡ)、ジメチルスルホキシド(DMSO)、ァセトニトリ ル、ピリジン、水等の反応に不活性な溶媒中、冷却下乃至加熱還流下に行うことがで きる。 The halogenation reaction can be a halogenation reaction that can be usually used by those skilled in the art. For example, the method described in “Chemical Experiment Course (4th edition)” edited by The Chemical Society of Japan, 19 卷 (1992) (Maruzen) can be applied. For example, N-promosuccinimide or the like can be used as a halogenating agent in the presence of 2,2′-azobisisobutyryl-tolyl or peroxybenzoyl. Reactions include aromatic hydrocarbons such as benzene, toluene, xylene, esters such as ethyl acetate, ethers such as dimethyl ether, tetrahydrofuran (THF), 1,4-dioxane, dichloromethane, 1,2-dichloroethane, Halogenated hydrocarbons such as black mouth form, alcohols such as methanol and ethanol, Ν, Ν-dimethylformamide (DMF), Ν, Ν-dimethylacetamide (DMA), Ν-methylpyrrolidone (ΝΜΡ), dimethyl sulfoxide The reaction can be carried out in a solvent inert to the reaction such as (DMSO), acetonitrile, pyridine, water, etc. under cooling to heating under reflux.
[0050] A— 2 :酸化 [0050] A—2: oxidation
アルコール、アルデヒド等の酸ィ匕反応は当業者が通常用いうる酸ィ匕反応を用いるこ とができる。例えば日本化学会編「実験化学講座 (第 4版)」 23卷 (1992年) (丸善)等に 記載の方法が適用できる。 For the acid-acid reaction of alcohol, aldehyde, etc., an acid-acid reaction that can be usually used by those skilled in the art can be used. For example, the method described in “Chemical Experiment Course (4th edition)” edited by The Chemical Society of Japan, 23 卷 (1992) (Maruzen) can be applied.
[0051] A— 3 :還元 [0051] A-3: Reduction
エステル、アミド、二トリル等の還元反応は当業者が通常用いうる還元反応を採用 することができる。例えば、水素化アルミニウムリチウム、水素化ホウ素ナトリウム等の 還元剤を用いて、前述の芳香族炭化水素類、エーテル類、ハロゲンィ匕炭化水素類、 DMF、 DMA, NMP、 DMSO、ァセトニトリル、アルコール類等の溶媒中、室温乃至カロ
熱還流下に行うことができる。 For the reduction reaction of ester, amide, nitrile and the like, a reduction reaction which can be usually used by those skilled in the art can be adopted. For example, by using a reducing agent such as lithium aluminum hydride and sodium borohydride, the above aromatic hydrocarbons, ethers, halogenated hydrocarbons, DMF, DMA, NMP, DMSO, acetonitrile, alcohols, etc. Room temperature to Caro in solvent It can be carried out under heat reflux.
[0052] A-4 :加水分解 [0052] A-4: Hydrolysis
エステル、アミド、二トリル等の加水分解反応は当業者が通常用いうる加水分解反 応を用いることができる。例えば、前述の芳香族炭化水素類、エーテル類、ハロゲン 化炭化水素類、アルコール類、 DMF、 DMA, NMP、 DMSO、ピリジン等の反応に不活 性な溶媒中、硫酸、塩酸、臭化水素酸等の鉱酸、ギ酸、酢酸等の有機酸等の酸存 在下;又は水酸化リチウム、水酸化ナトリウム、水酸ィ匕カリウム、炭酸カリウム、炭酸ナ トリウム、炭酸セシウム若しくはアンモニア等の塩基存在下、冷却下乃至加熱還流下 に行うことができる。 For the hydrolysis reaction of ester, amide, nitrile and the like, a hydrolysis reaction which can be usually used by those skilled in the art can be used. For example, sulfuric acid, hydrochloric acid, hydrobromic acid in a solvent inert to the reaction such as the above aromatic hydrocarbons, ethers, halogenated hydrocarbons, alcohols, DMF, DMA, NMP, DMSO, pyridine, etc. In the presence of an acid such as mineral acid such as formic acid and acetic acid; or in the presence of a base such as lithium hydroxide, sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate, cesium carbonate or ammonia. The reaction can be carried out under cooling to heating under reflux.
[0053] A-5 :加水素分解 [0053] A-5: Hydrogenolysis
脱べンジル等の加水素分解反応は当業者が通常用いうる加水素分解反応を用い ることができる。例えば、触媒として、ノ ラジウム 炭素、ラネーニッケル、白金等を用 い、常圧乃至加圧の水素雰囲気下、前述の芳香族炭化水素類、エステル類、エー テル類、ハロゲン化炭化水素類、 DMF、 DMA, NMP、酢酸ェチル、ァセトニトリル、酢 酸等反応に不活性な溶媒中、室温乃至加熱還流下に行うことができる。また、化合 物によっては酸 (好ましくは、塩酸、酢酸等)の存在下に反応させることが、反応を円 滑に進行させる上で有利な場合がある。 As the hydrogenolysis reaction such as debenzylation, a hydrogenolysis reaction which can be usually used by those skilled in the art can be used. For example, as a catalyst, use is made of noradium carbon, Raney nickel, platinum, etc., and the above-mentioned aromatic hydrocarbons, esters, ethers, halogenated hydrocarbons, DMF, The reaction can be carried out in a solvent inert to the reaction, such as DMA, NMP, ethyl acetate, acetonitrile, and acetic acid, from room temperature to under reflux. Depending on the compound, it may be advantageous to allow the reaction to proceed smoothly in the presence of an acid (preferably hydrochloric acid, acetic acid, etc.).
[0054] A— 6 :アミドィ匕 [0054] A—6: Amido
アミドィ匕反応は当業者が通常用いうるアミドィ匕を採用することができる。特に、酸クロ リドを用いる方法、カルボ-ルジイミダゾール(CDI)、 1-ェチル -3-(3-ジメチルァミノ プロピル)カルボジイミドー塩酸塩 (WSC 'HCl)、ジシクロへキシルカルボジイミド、ジフ ェ-ルホスフォリルアジド、ジェチルホスフオリルシア-ド等の縮合剤を使用する方法 、クロロギ酸イソブチル、クロ口ギ酸ェチル等を用いて混合酸無水物を経由する方法 が好適である。反応は前述のハロゲンィ匕炭化水素類、芳香族炭化水素類、エーテル 類、 DMF、 DMA, NMP、 DMSO等の反応に不活性な溶媒中、冷却下乃至加熱還流 下に行われる。反応の種類によっては、塩基 (好ましくは、トリェチルァミン、ジィソプ 口ピルェチルァミン、 N-メチルモルホリン、ピリジン、 4-(N,N-ジメチルァミノ)ピリジン等 )の存在下に反応させることが、反応を円滑に進行させる上で有利な場合がある。
[0055] A-7 :アルキル化 For the amidy reaction, amidy salt which can be usually used by those skilled in the art can be adopted. In particular, methods using acid chloride, carbodiimidazole (CDI), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (WSC 'HCl), dicyclohexylcarbodiimide, diphenylphosphoryl A method using a condensing agent such as ruazide and jetylphosphyl cyanide, and a method using a mixed acid anhydride using isobutyl chloroformate, ethyl ethyl formate, and the like are preferable. The reaction is carried out in a solvent inert to the reaction such as the above-mentioned halogenated hydrocarbons, aromatic hydrocarbons, ethers, DMF, DMA, NMP, DMSO or the like, under cooling or under reflux with heating. Depending on the type of reaction, the reaction can be carried out smoothly by reacting in the presence of a base (preferably triethylamine, di-sodium pyrethylamine, N-methylmorpholine, pyridine, 4- (N, N-dimethylamino) pyridine, etc.). In some cases, it may be advantageous. [0055] A-7: Alkylation
水酸基、アミノ基等のアルキルィ匕は当業者が通常用いうるアルキルィ匕を採用するこ とができる。例えば、無溶媒下、若しくは前述の芳香族炭化水素類、エステル類、ェ 一テル類、ハロゲン化炭化水素類、 DMF、 DMA, NMP、 DMSO、ァセトニトリル等の反 応に不活性な溶媒、あるいはアルコール類等の溶媒中、室温乃至加熱還流下に行う ことができる。化合物によっては、有機塩基(トリエチルァミン、ジイソプロピルェチル ァミン、 N-メチルモルホリン、ピリジン、 4-(N,N-ジメチルァミノ)ピリジン等が好適に用 いられる)、又は金属塩塩基 (炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水酸ィ匕 カリウム、水素化ナトリウム、 tert-ブトキシカリウム等が好適に用いられる)の存在下に 行うことが、反応を円滑に進行させる上で有利な場合がある。 As alkyl groups such as a hydroxyl group and an amino group, alkyl groups generally used by those skilled in the art can be employed. For example, in the absence of solvent or inactive solvents such as aromatic hydrocarbons, esters, ethers, halogenated hydrocarbons, DMF, DMA, NMP, DMSO, and acetonitrile, or alcohols The reaction can be carried out in a solvent such as a mixture at room temperature or under reflux. Depending on the compound, organic bases (triethylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 4- (N, N-dimethylamino) pyridine, etc. are preferably used) or metal salt bases (potassium carbonate, In the presence of cesium carbonate, sodium hydroxide, potassium hydroxide, sodium hydride, potassium tert-butoxy and the like, it may be advantageous for the reaction to proceed smoothly.
[0056] A-8 :還元的アルキルィ匕 [0056] A-8: Reductive alkyl
還元的アルキルィ匕は当業者が通常用いうる還元的アルキルィ匕を採用することがで きる。例えば日本化学会編「実験化学講座 (第 4版)」 20卷 (1992年) (丸善)等に記載の 方法が挙げられる。通常、前述のハロゲンィ匕炭化水素類、芳香族炭化水素類、エス テル類、エーテル類、アルコール類、酢酸等の反応に不活性な溶媒中、水素化ホウ 素ナトリウム、トリァセトキシ水素化ホウ素ナトリウム等の還元剤を用い冷却下、室温下 乃至加熱還流下に行うのが好適である。化合物によっては硫酸、塩酸、臭化水素酸 等の鉱酸、ギ酸、酢酸等の有機酸等の酸存在下反応を行うことが有利な場合がある 。また、還元的アルキルィ匕は、例えば触媒として、ノ ラジウム 炭素、ラネーニッケル 、白金等を用い、常圧乃至加圧の水素雰囲気下、前述の芳香族炭化水素類、エス テル類、エーテル類、ハロゲン化炭化水素類、 DMF、 DMA, NMP、ァセトニトリル、酢 酸等反応に不活性な溶媒中、室温乃至加熱還流下に行うことができる。化合物によ つては酸 (好ましくは、塩酸、酢酸等)の存在下に反応させることが、反応を円滑に進 行させる上で有利な場合がある。 As the reductive alkyl group, a reductive alkyl group commonly used by those skilled in the art can be adopted. For example, the method described in “Chemical Experiment Course (4th edition)” 20th edition (1992) (Maruzen) edited by the Chemical Society of Japan can be cited. Usually, in the above-mentioned solvents inert to the reaction such as halogenated hydrocarbons, aromatic hydrocarbons, esters, ethers, alcohols, acetic acid, sodium borohydride, sodium triacetoxyborohydride, etc. It is preferable to use a reducing agent under cooling and at room temperature or under reflux with heating. Depending on the compound, it may be advantageous to carry out the reaction in the presence of an acid such as a mineral acid such as sulfuric acid, hydrochloric acid or hydrobromic acid, or an organic acid such as formic acid or acetic acid. In addition, the reductive alkyl group uses, for example, noradium carbon, Raney nickel, platinum or the like as a catalyst, and the above-mentioned aromatic hydrocarbons, esters, ethers, halogenated substances in a hydrogen atmosphere of normal pressure to pressure. The reaction can be carried out in a solvent inert to the reaction, such as hydrocarbons, DMF, DMA, NMP, acetonitrile, and acetic acid, at room temperature to under reflux. For some compounds, the reaction in the presence of an acid (preferably hydrochloric acid, acetic acid, etc.) may be advantageous in order to facilitate the reaction.
[0057] 工程 B [0057] Process B
化合物(IV)と化合物 (V)の組み合わせ力 なる二つの化合物から、化合物 (VI)を 合成する工程である。本工程は、当業者が通常採用し得る工程、例えば、ハロゲン 化反応、酸化反応、還元反応、加水分解反応、加水素分解反応、アミド化反応、ァ
ルキル化反応、還元的アルキルィヒ反応等を任意に組み合わせることにより、実施す ることができる。本工程は一段階の反応とは限らず、多段階の反応により構成される 場合もある。 This is a step of synthesizing compound (VI) from two compounds having the combined power of compound (IV) and compound (V). This step is a step that can be usually employed by those skilled in the art, for example, halogenation reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction, The reaction can be carried out by arbitrarily combining a rukylation reaction, a reductive alkyl reaction or the like. This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
ハロゲン化、酸化、還元、加水分解、加水素分解、アミド化、アルキル化、還元的ァ ルキルイ匕はそれぞれ工程 Aの A—l〜8に準じて行うことができる。 Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
[0058] 工程 C [0058] Process C
化合物 (VI)において R8、 R9、 R1Q、 RU、
 R5 及び Xと異なる場合に、必要に応じて R8、 R9、 R1Q、 Ru、 R12或
R5 及び Xと異なる場合に、必要に応じて R8、 R9、 R1Q、 Ru、 R12或
 In compound (VI), R 8 , R 9 , R 1Q , R U , If different from R 5 and X, R 8 , R 9 , R 1Q , R u , R 12 or as required
In compound (VI), R 8 , R 9 , R 1Q , R U , If different from R 5 and X, R 8 , R 9 , R 1Q , R u , R 12 or as required
R2、 R5及び Xへと変換し、本発明化合物 (I)を合成する工程である。 本工程 は、当業者が通常採用し得る工程、例えば、ハロゲン化反応、酸化反応、還元反応、 加水分解反応、加水素分解反応、アミド化反応、アルキル化反応、還元的アルキル 化反応等を任意に組み合わせることにより実施することができる。本工程は一段階の 反応とは限らず、多段階の反応により構成される場合もある。 This is a step of converting to R 2 , R 5 and X to synthesize the compound (I) of the present invention. In this step, any step that can be usually employed by those skilled in the art, for example, halogenation reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction, alkylation reaction, reductive alkylation reaction, etc. It can implement by combining. This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
ハロゲン化、酸化、還元、加水分解、加水素分解、アミド化、アルキル化、還元的ァ ルキルイ匕はそれぞれ工程 Aの A—l〜8に準じて行うことができる。 Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
[0059] また本発明化合物は、以下の代表的な製造法にて製造することもできる。 [0059] The compounds of the present invention can also be produced by the following typical production methods.
[0060] [化 16]
[0060] [Chemical 16]



[0061] 工程 D [0061] Process D
化合物(III)と化合物 (V)の組み合わせ力 なる二つの化合物から、化合物 (VII)を 合成する工程である。本工程は、当業者が通常採用し得る工程、例えば、ハロゲン 化反応、酸化反応、還元反応、加水分解反応、加水素分解反応、アミド化反応、ァ ルキル化反応、還元的アルキルィヒ反応等を任意に組み合わせることにより、実施す ることができる。本工程は一段階の反応とは限らず、多段階の反応により構成される 場合もある。 This is a step of synthesizing compound (VII) from two compounds having the combined ability of compound (III) and compound (V). In this step, any step that can be usually employed by those skilled in the art, for example, halogenation reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction, alkylation reaction, reductive alkyl-rich reaction, etc. It can be implemented by combining with. This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
ハロゲン化、酸化、還元、加水分解、加水素分解、アミド化、アルキル化、還元的ァ ルキルイ匕はそれぞれ工程 Aの A—l〜8に準じて行うことができる。 Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
[0062] 工程 E [0062] Process E
化合物 (VII)と化合物(II)の組み合わせ力 なる二つの化合物から、化合物 (VIII) を合成する工程である。本工程は、当業者が通常採用し得る工程、例えば、ハロゲン
化反応、酸化反応、還元反応、加水分解反応、加水素分解反応、アミド化反応、ァ ルキル化反応、還元的アルキルィヒ反応等を任意に組み合わせることにより、実施す ることができる。本工程は一段階の反応とは限らず、多段階の反応により構成される 場合もある。 This is a step of synthesizing compound (VIII) from two compounds having the combined ability of compound (VII) and compound (II). This step is a step usually employed by those skilled in the art, such as halogen. The reaction can be carried out by arbitrarily combining oxidization reaction, oxidation reaction, reduction reaction, hydrolysis reaction, hydrogenolysis reaction, amidation reaction, alkylation reaction, reductive alkyl reaction, and the like. This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
ハロゲン化、酸化、還元、加水分解、加水素分解、アミド化、アルキル化、還元的ァ ルキルイ匕はそれぞれ工程 Aの A—l〜8に準じて行うことができる。 Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
[0063] 工程 F [0063] Process F
化合物 (VIII)において 、 R9、 R1Q、 RU、 R12或いは X1が各々
 R5及 び Xと異なる場合に、必要に応じて R8、 R9、 R1Q、 RU、 R12或いは X1を各々
R5及 び Xと異なる場合に、必要に応じて R8、 R9、 R1Q、 RU、 R12或いは X1を各々
 In compound (VIII), R 9 , R 1Q , R U , R 12 or X 1 are each R 8 , R 9 , R 1Q , R U , R 12 or X 1 as required if different from R 5 and X
In compound (VIII), R 9 , R 1Q , R U , R 12 or X 1 are each R 8 , R 9 , R 1Q , R U , R 12 or X 1 as required if different from R 5 and X
R5及び Xへと変換し、本発明化合物 (I)を合成する工程である。 This is a step of converting to R 5 and X to synthesize the compound (I) of the present invention.
本工程は、当業者が通常採用し得る工程、例えば、ハロゲン化反応、酸化反応、還 元反応、加水分解反応、加水素分解反応、アミド化反応、アルキル化反応、還元的 アルキルィ匕反応等を任意に組み合わせることにより実施することができる。本工程は 一段階の反応とは限らず、多段階の反応により構成される場合もある。 This step is a step that can be usually employed by those skilled in the art, such as halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, reductive alkylation, etc. It can implement by combining arbitrarily. This process is not limited to a one-step reaction and may be composed of a multi-step reaction.
ハロゲン化、酸化、還元、加水分解、加水素分解、アミド化、アルキル化、還元的ァ ルキルイ匕はそれぞれ工程 Aの A—l〜8に準じて行うことができる。 Halogenation, oxidation, reduction, hydrolysis, hydrogenolysis, amidation, alkylation, and reductive alkylation can be carried out according to steps A to 8 in Step A, respectively.
[0064] また本発明化合物 (I)を製造するにあたり、 R8、 R9、 R1Q、 RU、 R12或いは X1がそれぞ
 R4、 R5及び Xと異なる場合に、必要に応じて R8、 R9、 R1Q、 RU、 R12或い は X1をそれぞれ R1 R2、
R4、 R5及び Xと異なる場合に、必要に応じて R8、 R9、 R1Q、 RU、 R12或い は X1をそれぞれ R1 R2、
 R5及び Xへと変換する工程 C或いは工程 Fは、工程[0064] In the production of the compound (I) of the present invention, R 8 , R 9 , R 1Q , R U , R 12 or X 1 is R 8 , R 9 , R 1Q , R U , R 12 or X 1 as R 1 R 2 , if different from R 4 , R 5 and X, respectively, as required Process C or process F converted to R 5 and X is process
R5及び Xへと変換する工程 C或いは工程 Fは、工程[0064] In the production of the compound (I) of the present invention, R 8 , R 9 , R 1Q , R U , R 12 or X 1 is R 8 , R 9 , R 1Q , R U , R 12 or X 1 as R 1 R 2 , if different from R 4 , R 5 and X, respectively, as required Process C or process F converted to R 5 and X is process
A, B、 D或いは Eの工程において実施しても良い。 You may implement in the process of A, B, D, or E.
[0065] 本発明化合物の製造に使用する原料は、例えば、後述の参考例に記載の方法、 公知の方法若しくは当業者にとって自明の方法、またはそれらの変法を用いることに より製造することができる。 [0065] The raw material used for the production of the compound of the present invention can be produced, for example, by using the method described in the Reference Examples described below, a known method, a method obvious to those skilled in the art, or a modification thereof. it can.
[0066] この様にして製造された本発明化合物は、公知の方法、例えば、抽出、沈澱、分画 クロマトグラフィー、分別結晶化、再結晶等により単離、精製することができる。また、 本発明化合物の塩は、通常の造塩反応により所望の塩に導くことができる。 [0066] The compound of the present invention thus produced can be isolated and purified by a known method such as extraction, precipitation, fractional chromatography, fractional crystallization, recrystallization and the like. Moreover, the salt of this invention compound can be guide | induced to a desired salt by normal salt-forming reaction.
また、本発明化合物が不斉炭素を有する場合には光学異性体が存在する。これら の光学異性体は適切な塩と再結晶する分別結晶化やカラムクロマトグラフィー等の
常法により分割することができる。また、適当な光学活性な原料を用いることにより製 造することができる。 Further, when the compound of the present invention has an asymmetric carbon, an optical isomer exists. These optical isomers can be used for fractional crystallization, column chromatography, etc. It can be divided by conventional methods. Moreover, it can manufacture by using a suitable optically active raw material.
[0067] 一般式 (I)で示される本発明化合物やその製薬学的に許容される塩の 1種又は 2 種以上を有効成分として含有する医薬組成物は、通常用いられている製剤用の担体 ゃ賦形剤、その他の添加剤を用いて、錠剤、散剤、細粒剤、顆粒剤、カプセル剤、丸 剤、液剤、注射剤、坐剤、軟膏、貼付剤等に調製され、経口的又は非経口的に投与 される。 [0067] A pharmaceutical composition containing, as an active ingredient, one or more of the compounds of the present invention represented by the general formula (I) or a pharmaceutically acceptable salt thereof, Prepared into tablets, powders, fine granules, granules, capsules, pills, solutions, injections, suppositories, ointments, patches, etc. using carriers and excipients and other additives, and orally Or administered parenterally.
本発明化合物のヒトに対する臨床投与量は適用される患者の症状、体重、年齢や 性別等を考慮して適宜決定されるが、通常成人 1日当たり経口で 0. l〜500mg、非 経口で 0. 01〜: LOOmgであり、これを 1回あるいは数回に分けて投与する。投与量は 種々の条件で変動するので、上記投与量範囲より少な!、量で十分な場合もある。 本発明による経口投与のための固体組成物としては、錠剤、散剤、顆粒剤等が用 いられる。このような固体組成物においては、一つ又はそれ以上の活性物質力 少な くとも一つの不活性な希釈剤、例えば乳糖、マン-トール、ブドウ糖、ヒドロキシプロピ ルセルロース、微結晶セルロース、デンプン、ポリビュルピロリドン、メタケイ酸アルミン 酸マグネシウムと混合される。組成物は、常法に従って、不活性な希釈剤以外の添 加剤、例えば滑沢剤、崩壊剤、安定化剤、可溶化剤又は溶解補助剤を含有していて もよい。錠剤又は丸剤は必要によりショ糖、ゼラチン、ヒドロキシプロピルセルロース、 ヒドロキシプロピルメチルセルロースフタレートなどの糖衣、又は胃溶性ある ヽは腸溶 性物質のフィルムで被膜してもよ!/、。 The clinical dose of the compound of the present invention to humans is appropriately determined in consideration of the patient's symptoms, body weight, age, sex, etc., but it is usually 0.1 to 500 mg orally per day for adults and 0 to parenterally. 01 ~: LOOmg, which is administered once or in several divided doses. Since the dosage varies depending on various conditions, the dosage may be less than the above dosage range. As the solid composition for oral administration according to the present invention, tablets, powders, granules and the like are used. In such a solid composition, one or more active substance forces at least one inert diluent such as lactose, mannitol, glucose, hydroxypropyl cellulose, microcrystalline cellulose, starch, poly It is mixed with bull pyrrolidone and magnesium aluminate metasilicate. The composition may contain an additive other than the inert diluent, for example, a lubricant, a disintegrant, a stabilizer, a solubilizer, or a solubilizing agent according to a conventional method. If necessary, tablets or pills may be coated with sugar coating such as sucrose, gelatin, hydroxypropylcellulose, hydroxypropylmethylcellulose phthalate, etc., or gastric soluble sputum may be coated with an enteric film!
[0068] 経口投与のための液体組成物は、薬剤的に許容される乳濁剤、溶液剤、懸濁剤、 シロップ剤、エリキシル剤等を含み、一般的に用いられる不活性な希釈剤、例えば精 製水、エチルアルコールを含む。この組成物は不活性な希釈剤以外に可溶化剤、溶 解補助剤、湿潤剤、懸濁剤のような補助剤、甘味剤、風味剤、芳香剤、防腐剤を含 有していてもよい。 [0068] Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, elixirs and the like, commonly used inert diluents, Examples include purified water and ethyl alcohol. This composition may contain solubilizers, solubilizers, wetting agents, suspending agents, sweeteners, flavors, fragrances, preservatives in addition to the inert diluent. Good.
非経口投与のための注射剤としては、無菌の水性又は非水性の溶液剤、懸濁剤、 乳濁剤を包含する。水性の溶液剤、懸濁剤の希釈剤としては、例えば注射剤用蒸留 水及び生理食塩水が含まれる。非水溶性の溶液剤、懸濁剤の希釈剤としては、例え
ばプロピレングリコール、ポリエチレングリコール、ォリーブ油のような植物油、ェチル アルコールのようなアルコール類、ポリソルベート 80 (局方名)等がある。このような組 成物は、更に等張化剤、防腐剤、湿潤剤、乳化剤、分散剤、安定化剤、可溶化剤又 は溶解補助剤のような添加剤を含んでもょ ヽ。これらは例えばバクテリア保留フィルタ 一を通す濾過、殺菌剤の配合又は照射によって無菌化される。また、これらは無菌の 固体組成物を製造し、使用前に無菌水又は無菌の注射用溶媒に溶解して使用する ことちでさる。 Injections for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions. Examples of the aqueous solution and suspension diluent include distilled water for injection and physiological saline. Examples of diluents for non-aqueous solutions and suspensions include Examples include propylene glycol, polyethylene glycol, vegetable oils such as olive oil, alcohols such as ethyl alcohol, and polysorbate 80 (a pharmacopeia name). Such compositions may further contain additives such as isotonic agents, preservatives, wetting agents, emulsifiers, dispersants, stabilizers, solubilizers or solubilizers. These are sterilized by, for example, filtration through a bacteria-retaining filter, blending with a bactericide or irradiation. These can be prepared by preparing a sterile solid composition and dissolving it in sterile water or a sterile solvent for injection before use.
実施例 Example
以下、本発明化合物の製造例を挙げ、本発明化合物の製造方法を具体的に説明 するが本発明はこれらの実施例により何ら制限されるものではない。なお、本発明化 合物の原料化合物には新規な化合物も含まれており、これらの化合物の製造方法を 参考例として説明する。 Hereinafter, production examples of the compound of the present invention will be given and the production method of the compound of the present invention will be specifically described. However, the present invention is not limited to these examples. Note that the raw material compounds of the compounds of the present invention include novel compounds, and the production methods of these compounds will be described as reference examples.
なお、参考例、実施例中の記号は以下の意味を示す (以下同様)。 The symbols in Reference Examples and Examples have the following meanings (the same applies hereinafter).
Rf:参考例番号、 Ex:実施例番号、 No :化合物番号、 Structure:構造式、 DATA:物 理学的データ。 NMR( δ (ppm)) ^H-NMRにおけるピークの δ (ppm)、 sep:septet、 FP: FAB-MS (Pos) (特に断らない限り [M+H]+)、 FN:FAB— MS(Neg) ([M— H]―)、 EP:ESI— MS (Pos) (特に断らない限り [M+H]+)、 EN:ESI- MS(Neg) ([M- H]つ、 AN:API- MS(Neg) ([M - H]—)、 GC:GC-MS ([M 。 Sal:塩 (特に、塩との表示が無い場合は、フリー体であるこ とを示し、また、例えば HC1と記載されている場合、その化合物が塩酸塩であることを 示す。)。 Rf: Reference example number, Ex: Example number, No: Compound number, Structure: Structural formula, DATA: Physical data. NMR (δ (ppm)) ^ H-NMR peak δ (ppm), sep: septe, FP: FAB-MS (Pos) ([M + H] + unless otherwise noted), FN: FAB— MS ( Neg) ([M—H] —), EP: ESI—MS (Pos) ([M + H] + unless otherwise noted), EN: ESI-MS (Neg) ([M-H], AN: API-MS (Neg) ([M-H] —), GC: GC-MS ([M. Sal: Salt (especially, when there is no indication of salt, it indicates that it is a free form. If it is described as HC1, it indicates that the compound is hydrochloride.)
Me:メチル、 Et :ェチル、 cPr:シクロプロピル、 iPr:イソプロピル、 nBu:ノルマルブチ ル、 iBu:イソブチル、 tBu:tert-ブチル、 cBu:シクロブチル、 cHex:シクロへキシル、 Ac :ァセチル、 Ph:フエニル、 Bn:ベンジル、 Bo tert-ブトキシカルボニル、 TFA:トリフル ォロ酢酸、 Ms:メタンスルホニル、 null:無置換、 bond:結合。 Syn:製造方法(その数字 の番号を実施例番号として有する実施例化合物と同様にして、対応する原料を用い て製造したことを示す。 ) o RSyn:製造方法 (その数字の番号を参考例番号として有す る参考例化合物と同様にして、対応する原料を用いて製造したことを示す。 ) o置換 基の前の数字は、置換位置を示し、複数個ある場合は複数個の置換を意味する。例
えば 4, 5 ,6-triOMeは 4, 5 ,6位それぞれに- OMe基を有することを示す。 Me: methyl, Et: ethyl, cPr: cyclopropyl, iPr: isopropyl, nBu: normal butyl, iBu: isobutyl, tBu: tert-butyl, cBu: cyclobutyl, cHex: cyclohexyl, Ac: acetyl, Ph: phenyl, Bn: benzyl, Bo tert-butoxycarbonyl, TFA: trifluoroacetic acid, Ms: methanesulfonyl, null: unsubstituted, bond: bond. Syn: Manufacturing method (Indicates that the product was prepared using the corresponding raw material in the same manner as the Example compound having the numerical number as the Example number.) O RSyn: Manufacturing method (The numerical number is the reference example number. In the same manner as in the reference example compound, the corresponding starting material is used.) O Substituent The number before the substituent indicates the substitution position, and when there are multiple, it means multiple substitution. To do. Example For example, 4,5,6-triOMe indicates that each of the 4,5,6 positions has an -OMe group.
[0070] 参考例 1 [0070] Reference Example 1
2-ピぺリジン- 2-ィルエタノール 3.9 g、ブロモ酢酸 tert-ブチル 6.4 g及びァセトニトリ ル 6.4 gの混合物に室温にて炭酸カリウム 5.0 gを加え 48時間攪拌した。反応液に水 を加え、酢酸ェチルにて抽出した。有機層を水次いで飽和食塩水で順次洗浄し、無 水硫酸マグネシウムで乾燥後減圧下濃縮し、得られた残渣をクロ口ホルム/メタノール (40 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し、 tert-プチ ル [2-(2-ヒドロキシェチル)ピぺリジン- 1-ィル]ァセタート 6.6 gを黄色油状物として得 た。 EP: 244 To a mixture of 3.9 g of 2-piperidin-2-ylethanol, tert-butyl bromoacetate and 6.4 g of acetonitrile, 5.0 g of potassium carbonate was added at room temperature and stirred for 48 hours. Water was added to the reaction solution and extracted with ethyl acetate. The organic layer is washed successively with water and then with saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The resulting residue is purified by silica gel column chromatography using chloroform / methanol (40: 1) as an elution solvent. Purification gave 6.6 g of tert-butyl [2- (2-hydroxyethyl) piperidin-1-yl] acetate as a yellow oil. EP: 244
[0071] 参考例 2 [0071] Reference Example 2
ピぺリジン- 2-ィルメタノール 192 mg、アクリル酸 tert-ブチル 179 mg及びメタノール 1.0 mLの混合物を室温にて 3時間攪拌した。反応液を減圧下濃縮し、得られた残渣 を n-へキサン/酢酸ェチル (3: 2)を溶出溶媒とするシリカゲルカラムクロマトグラフィー により精製し、 tert-ブチル 3-[2- (ヒドロキシメチル)ピぺリジン- 1-ィル]プロパノアート 1 99 mgを無色油状物として得た。 NMR (CDC1 ) : 1.28-1.48 (11H, m), 1.50—1.73 (4H, A mixture of 192 mg of piperidine-2-ylmethanol, 179 mg of tert-butyl acrylate and 1.0 mL of methanol was stirred at room temperature for 3 hours. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (3: 2) as an elution solvent, and tert-butyl 3- [2- (hydroxymethyl) Piperidine-1-yl] propanoate 1 99 mg was obtained as a colorless oil. NMR (CDC1): 1.28-1.48 (11H, m), 1.50—1.73 (4H,
3 Three
m), 2.14-2.23 (IH, m), 2.27-2.56 (4H, m), 2.71-2.93 (IH, br), 2.94—3.01 (IH, m), 3 .08-3.17 (IH, m), 3.42 (IH, dd, J = 3.8, 11.3 Hz), 3.83 (IH, dd, J = 3.8, 11.3 Hz) [0072] 参考例 3 m), 2.14-2.23 (IH, m), 2.27-2.56 (4H, m), 2.71-2.93 (IH, br), 2.94—3.01 (IH, m), 3.08-3.17 (IH, m), 3.42 (IH, dd, J = 3.8, 11.3 Hz), 3.83 (IH, dd, J = 3.8, 11.3 Hz) [0072] Reference Example 3
塩化ォキサリル 0.125 mLの塩化メチレン(1.3 mL)溶液に- 78 °Cにてジメチルスル ホキシド 0.166 mLの塩化メチレン(0.33 mL)溶液を滴カ卩し 15分間攪拌した。この混 合物に tert-ブチル 3-[2- (ヒドロキシメチル)ピぺリジン- 1-ィル]プロパノアート 0.12 gの 塩化メチレン(0.51 mL)溶液を- 78 °Cにて滴加し 15分間攪拌した。この混合物にトリ ェチルァミン 0.60 mLを- 78°Cにて滴カ卩し、室温にて 30分間攪拌した。反応液に水を 加え、塩化メチレンで抽出した。有機層を水及び飽和食塩水で順次洗い、無水硫酸 マグネシウムで乾燥後減圧下濃縮し、 tert-ブチル 3-(2-ホルミルピぺリジン- 1-ィル) プロパノアート 0.135 gを赤色油状物として得た。 To a solution of 0.125 mL of oxalyl chloride in methylene chloride (1.3 mL) was added dropwise a solution of 0.166 mL of dimethyl sulfoxide in methylene chloride (0.33 mL) at -78 ° C and stirred for 15 minutes. To this mixture was added dropwise a solution of 0.12 g of tert-butyl 3- [2- (hydroxymethyl) piperidin-1-yl] propanoate in methylene chloride (0.51 mL) at -78 ° C and stirred for 15 minutes. did. To this mixture, 0.60 mL of triethylamine was added dropwise at -78 ° C and stirred at room temperature for 30 minutes. Water was added to the reaction solution and extracted with methylene chloride. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to give 0.135 g of tert-butyl 3- (2-formylpiperidine-1-yl) propanoate as a red oil. .
[0073] 参考例 4 [0073] Reference Example 4
4-シァノ -2-メトキシフヱノール 7.21 gのトリフルォロ酢酸(50 mL)溶液に氷冷下へキ
サメチレンテトラミン 13.6 gを加え、 100 °Cにて 2時間攪拌した。氷冷下反応液に 50% 硫酸 24 mLと水 160 mLを順次カ卩えた。反応液を酢酸ェチルで抽出し、抽出液を水で 四回洗浄した。有機層を 1規定水酸ィ匕ナトリウム水溶液にて中和し、無水硫酸マグネ シゥムで乾燥して減圧下濃縮した。得られた残渣を n-へキサン/酢酸ェチル(4: 1)を 溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し、 5-シァノ -3-メトキシ サリチルアルデヒド 1.08 gを無色固体として得た。 4-Ciano-2-methoxyphenol Into a solution of 7.21 g of trifluoroacetic acid (50 mL) under ice-cooling. 13.6 g of samethylenetetramine was added and stirred at 100 ° C. for 2 hours. Under ice-cooling, 24 mL of 50% sulfuric acid and 160 mL of water were sequentially added. The reaction solution was extracted with ethyl acetate, and the extract was washed four times with water. The organic layer was neutralized with 1N aqueous sodium hydroxide, dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as an elution solvent to obtain 1.08 g of 5-cyan-3-methoxysalicylaldehyde as a colorless solid.
[0074] 参考例 5 [0074] Reference Example 5
4-クロ口- 2- (ヒドロキシメチル)フエノール 2.3 gのメタノール (40 mL)溶液に、炭酸ナ トリウム 7.6 gと一塩化ヨウ素 2.3 gを室温にて加え 4時間攪拌した。反応液を減圧下濃 縮した。残渣にクロ口ホルムをカ卩え、水、 2規定塩酸次いで 10%チォ硫酸ナトリウム水 溶液で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥して減圧下濃縮した。 得られた残渣を n-へキサン/酢酸ェチル (4: 1)を溶出溶媒とするシリカゲルカラムク 口マトグラフィ一により精製し、 4-クロ口- 2- (ヒドロキシメチル) -6-ョードフエノール 3.9 g を無色油状物として得た (EP:285)。得られた油状物 841 mgを Ν,Ν-ジメチルホルムアミ ド 6 mLに溶解し、ベンズアルデヒドジメチルァセタール 496 mgと p-トルエンスルホン 酸 10 mgを室温にて加えた後、 100 °Cにて 1時間攪拌した。反応液を減圧下濃縮し、 得られた残渣を n-へキサン/酢酸ェチル (4: 1)を溶出溶媒とするシリカゲルカラムク 口マトグラフィ一により精製し、 6-クロ口- 8-ョード -2-フエ-ル- 4H- 1,3-ベンゾジォキシ ン 578 mgを無色固体として得た。 EP: 372 [M]+ To a solution of 4-chloro-2,2- (hydroxymethyl) phenol 2.3 g in methanol (40 mL), 7.6 g sodium carbonate and 2.3 g iodine monochloride were added at room temperature and stirred for 4 hours. The reaction solution was concentrated under reduced pressure. The residue was filled with black mouth form and washed successively with water, 2N hydrochloric acid and then 10% aqueous sodium thiosulfate solution. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as the elution solvent, and 3.9 g of 4-chloro-2- (hydroxymethyl) -6-odophenol was obtained. Obtained as a colorless oil (EP: 285). 841 mg of the resulting oil was dissolved in 6 mL of Ν, Ν-dimethylformamide, 496 mg of benzaldehyde dimethylacetal and 10 mg of p-toluenesulfonic acid were added at room temperature, and then 1 at 100 ° C. Stir for hours. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as an elution solvent, and 6-chloro-8-odo-2 578 mg of -phenol-4H-1,3-benzodioxin was obtained as a colorless solid. EP: 372 [M] +
[0075] 参考例 6 [0075] Reference Example 6
6-クロ口- 8-ョード -2-フエニル- 4H- 1,3-ベンゾジォキシン 228 mg、 4-メチルピペラジ ン- 2-オン 114 mg、リン酸カリウム 259 mg、ヨウ化銅 38 mg、 1,2- cis-シクロへキサンジ ァミン 228 mg及び 1,4-ジォキサン 1 mLの混合物を還流下 24時間攪拌した。反応液 を減圧下濃縮し、得られた残渣をクロ口ホルム/メタノール(30 : 1)を溶出溶媒とするシ リカゲルカラムクロマトグラフィーにより精製し、 1- (6-クロ口- 2-フエ-ル- 4H- 1,3-ベン ゾジォキシン- 8-ィル) -4-メチルビペラジン- 2-オン 192 mgを得た。得られた残渣をテ トラヒドロフラン 3 mLとメタノール 1 mLに溶解し、 0.5規定塩酸 3 mLを加え還流下 30 分間攪拌した。反応液を飽和重曹水で中和し、酢酸ェチルで抽出した。有機層を無
水硫酸マグネシウムで乾燥して減圧下濃縮した。残渣をクロ口ホルム/メタノール(10 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製して 1-[5-クロ口- 2- ヒドロキシ- 3- (ヒドロキシメチル)フエ-ル] -4-メチルビペラジン- 2-オン 63 mgを無色油 状物として得た。 FP: 271 6-Black mouth-8-Lodo-2-phenyl-4H- 1,3-benzodioxin 228 mg, 4-methylpiperazin-2-one 114 mg, Potassium phosphate 259 mg, Copper iodide 38 mg, 1,2- A mixture of cis-cyclohexanediamine 228 mg and 1,4-dioxane 1 mL was stirred under reflux for 24 hours. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using chloroform-form / methanol (30: 1) as the elution solvent, and 1- (6-chromato-2-phenyl) was obtained. -4H- 1,3-Benzozoxin-8-yl) -4-methylbiperazin-2-one 192 mg was obtained. The obtained residue was dissolved in tetrahydrofuran (3 mL) and methanol (1 mL), 0.5N hydrochloric acid (3 mL) was added, and the mixture was stirred under reflux for 30 min. The reaction solution was neutralized with saturated aqueous sodium hydrogen carbonate, and extracted with ethyl acetate. No organic layer The extract was dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using black mouth form / methanol (10: 1) as an elution solvent to give 1- [5-black mouth-2-hydroxy-3- (hydroxymethyl) phenol] -4- Methylbiperazin-2-one (63 mg) was obtained as a colorless oil. FP: 271
[0076] 参考例 7 [0076] Reference Example 7
3-エトキシ- 2-ヒドロキシベンズアルデヒド 2.5gの酢酸(15 mL)溶液に硝酸 1.6mLを 室温にて加え、 2時間攪拌した。反応液に水を加え、生じた析出物を濾取した。濾物 を水で洗浄した後、減圧下乾燥して 3-エトキシ -2-ヒドロキシ- 5-ニトロべンズアルデヒ ド 2.3 gを得た。 To a solution of 2.5 g of 3-ethoxy-2-hydroxybenzaldehyde in acetic acid (15 mL), 1.6 mL of nitric acid was added at room temperature and stirred for 2 hours. Water was added to the reaction solution, and the resulting precipitate was collected by filtration. The filtrate was washed with water and then dried under reduced pressure to obtain 2.3 g of 3-ethoxy-2-hydroxy-5-nitrobenzaldehyde.
[0077] 参考例 8 [0077] Reference Example 8
フラスコに濃塩酸 22.6 mL、塩化亜鉛 2.7 gおよびパラホルムアルデヒド 4.1 gを入れ 、溶解するまで塩ィ匕水素を吹き込んだ。そこへ 3—エトキシサリチルアルデヒド 15 gの トルエン(68 mL)溶液を加え、激しく撹拌しながら、室温にて塩化水素を 2時間吹き 込んだ。反応混合物にトルエン 45 mLを加え、氷約 150 gに注ぎ込んだ。室温下 20分 間撹拌後、不溶物をセライト濾去した。トルエンで洗浄後、濾液を分離した。有機層 を水で 2回洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下に留去して、赤 黒色固体を得た。得られた固体 18.0 gの酢酸 (170 mL)懸濁液に、室温にて酢酸ナトリ ゥム 29.6 gを加え、同温で 1.5時間撹拌した。反応混合物に水約 300mLを加え、酢酸 ェチル(約 300mL)で抽出した。有機層を、水で 2回、飽和重曹水で 2回、および飽和 食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下に留去し、得ら れた残渣を酢酸ェチル /n-へキサン(1:3)を溶出溶媒とするシリカゲルカラムクロマト グラフィ一にて精製して、 3-エトキシ -5-ホルミル- 4-ヒドロキシベンジルァセタート 5.9 0 gを黄色固体として得た。 The flask was charged with 22.6 mL of concentrated hydrochloric acid, 2.7 g of zinc chloride and 4.1 g of paraformaldehyde and bubbled with saline and hydrogen until dissolved. A solution of 15 g of 3-ethoxysalicylaldehyde in toluene (68 mL) was added thereto, and hydrogen chloride was blown in at room temperature for 2 hours with vigorous stirring. To the reaction mixture, 45 mL of toluene was added and poured into about 150 g of ice. After stirring at room temperature for 20 minutes, the insoluble material was filtered off through Celite. After washing with toluene, the filtrate was separated. The organic layer was washed twice with water and then dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to obtain a red-black solid. To a suspension of the obtained solid 18.0 g in acetic acid (170 mL), 29.6 g of sodium acetate was added at room temperature, and the mixture was stirred at the same temperature for 1.5 hours. About 300 mL of water was added to the reaction mixture, and the mixture was extracted with ethyl acetate (about 300 mL). The organic layer was washed twice with water, twice with saturated aqueous sodium hydrogen carbonate, and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 3) as an elution solvent to obtain 3-ethoxy-5-formyl. -5.90 g of 4-hydroxybenzylacetate was obtained as a yellow solid.
[0078] 参考例 9 [0078] Reference Example 9
37%ホルムアルデヒド水溶液 12.4 mLと 50%ジメチルァミン水溶液 17.5 mLをエタ ノール 70 mLに溶解し、 2-ヒドロキシ -3-イソプロポキシベンズアルデヒド 20gのェタノ ール(30 mL)溶液を室温にて添加し、 13時間加熱還流した。放冷後溶媒を減圧留 去した。残渣をトルエンに懸濁し、減圧下濃縮した。得られた残渣をジェチルエーテ
ルにて洗浄することで、 5- [(ジメチルァミノ)メチル ]-2-ヒドロキシ -3-イソプロポキシベ ンズアルデヒド 23.4gを黄色固体として得た。 EP: 238 Dissolve 12.4 mL of 37% aqueous formaldehyde and 17.5 mL of 50% aqueous dimethylamine in 70 mL of ethanol, add 20 g of ethanol (30 mL) of 2-hydroxy-3-isopropoxybenzaldehyde at room temperature for 13 hours. Heated to reflux. After standing to cool, the solvent was distilled off under reduced pressure. The residue was suspended in toluene and concentrated under reduced pressure. The residue obtained By washing with water, 23.4 g of 5-[(dimethylamino) methyl] -2-hydroxy-3-isopropoxybenzaldehyde was obtained as a yellow solid. EP: 238
[0079] 参考例 10 [0079] Reference Example 10
5- [(ジメチルァミノ)メチル ]-2-ヒドロキシ -3-イソプロポキシベンズアルデヒド 23.17g をメタノール 175 mLとアセトン 115 mLの混合溶媒に溶解し、ョードメタン 6.4 mLをカロ え、室温にて 22時間攪拌した。溶媒を減圧留去した。残渣をトルエンに懸濁し、減圧 下濃縮した。固体性残渣を酢酸 223 mLに懸濁し、酢酸ナトリウム 9.6 gを加え、 16時 間加熱還流した。放冷後溶媒を減圧留去し、残渣に水を加え、酢酸ェチルで抽出し た。有機層を水で 3回、及び飽和食塩水で順次洗浄後、無水硫酸マグネシウムで乾 燥し、減圧下濃縮した。得られた残渣を酢酸ェチル -へキサン(1 : 3)を溶出溶媒と するシリカゲルカラムクロマトグラフィーで精製し、 3-ホルミル- 4-ヒドロキシ -5-イソプロ ポキシベンジルァセタート 15.3 gを淡褐色固体として得た。 23.17 g of 5-[(dimethylamino) methyl] -2-hydroxy-3-isopropoxybenzaldehyde was dissolved in a mixed solvent of 175 mL of methanol and 115 mL of acetone, and 6.4 mL of iodine methane was added thereto and stirred at room temperature for 22 hours. The solvent was removed under reduced pressure. The residue was suspended in toluene and concentrated under reduced pressure. The solid residue was suspended in 223 mL of acetic acid, 9.6 g of sodium acetate was added, and the mixture was heated to reflux for 16 hours. After allowing to cool, the solvent was distilled off under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water three times and with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using ethyl acetate-hexane (1: 3) as the elution solvent, and 15.3 g of 3-formyl-4-hydroxy-5-isopropoxybenzylacetate was obtained as a light brown solid. Got as.
[0080] 参考例 11 [0080] Reference Example 11
5-ホルミル- 4-ヒドロキシ -3-メトキシベンジルァセタート 7.25 gの塩化メチレン(70 m L)溶液をメタノール-氷浴につけ、三臭化ホウ素の塩化メチレン溶液 (1.0 M) 97 mL を滴下漏斗よりゆっくり加えた。混合物を同条件下 1時間撹拌した。反応混合物を氷 に注ぎ、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄後、無水硫酸マグネシ ゥムで乾燥した。溶媒を減圧下に留去して、粗成績体を得た。得られた成績体の酢 酸(60 mL)懸濁液に室温撹拌下、酢酸ナトリウム 7.96 gを加え、同温で 1時間撹拌し た。減圧下溶媒を留去した後、反応混合物に水を加え、酢酸ェチルで抽出した。有 機層を、水で 2回、および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。 溶媒を減圧下に留去し、残渣を酢酸ェチル /n-へキサン(1 : 2)を溶出溶媒とするシリ 力ゲルカラムクロマトグラフィーで精製して、 3,4-ジヒドロキシ- 5-ホルミルべンジルァ セタート 5.75 gを得た。 NMR (CDC1 ): 2.10 (3H, s), 5.05 (2H, s), 5.65 (1H, br s), 7. 5-Formyl-4-hydroxy-3-methoxybenzylacetate 7.25 g of methylene chloride (70 mL) was placed in a methanol-ice bath, and boron tribromide in methylene chloride (1.0 M) (97 mL) was added to the dropping funnel. Added more slowly. The mixture was stirred for 1 hour under the same conditions. The reaction mixture was poured onto ice and extracted with ethyl acetate. The organic layer was washed with saturated brine and then dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to obtain a crude product. To a suspension of the resulting product in acetic acid (60 mL), 7.96 g of sodium acetate was added with stirring at room temperature, and the mixture was stirred at the same temperature for 1 hour. After evaporating the solvent under reduced pressure, water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed twice with water and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 2) as an elution solvent to obtain 3,4-dihydroxy-5-formylbenzilya. 5.75 g of setart was obtained. NMR (CDC1): 2.10 (3H, s), 5.05 (2H, s), 5.65 (1H, br s), 7.
3 Three
17 (1H, d, J = 2.0 Hz), 7.20 (1H, d, J = 2.0 Hz), 9.89 (1H, s), 11.12 (1H, s) 17 (1H, d, J = 2.0 Hz), 7.20 (1H, d, J = 2.0 Hz), 9.89 (1H, s), 11.12 (1H, s)
[0081] 参考例 12 [0081] Reference Example 12
メタノール 氷浴中、 2-ヒドロキシ -3-イソプロポキシベンズアルデヒド 2.0 gの Ν,Ν-ジ メチルホルムアミド (22 mL)溶液に撹拌下 1-ョードピロリジン- 2,5-ジオン 2.62 gを加
え、同条件下 17時間撹拌した。反応混合物に水を加え、酢酸ェチルで 2回抽出した。 有機層を合わせ、水で 3回および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾 燥した。溶媒を減圧下に留去し、得られた残渣を n-へキサン/酢酸ェチル (92:8)を 溶出溶媒とするシリカゲルカラムクロマトグラフィーにて精製して、 2-ヒドロキシ -5-ョー ド -3-イソプロポキシベンズアルデヒド 582 mgを赤色油状物として得た。 In a methanol ice bath, add 2-hydroxy-3-isopropoxybenzaldehyde (2.0 g) in Ν, Ν-dimethylformamide (22 mL) with stirring and add 2.62 g of 1-yodopyrrolidine-2,5-dione. The mixture was stirred for 17 hours under the same conditions. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The organic layers were combined, washed 3 times with water and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (92: 8) as an elution solvent to give 2-hydroxy-5-iodo- 582 mg of 3-isopropoxybenzaldehyde was obtained as a red oil.
[0082] 参考例 13 [0082] Reference Example 13
4-ヒドロキシ -3-イソプロポキシベンズアルデヒド 31.6 gをクロロメチルメチルエーテル 178 mLに溶解し、室温にて四塩化スズ 100 gを添加した。室温にて 4.5時間攪拌後、 反応液を氷水に注いだ。析出した結晶を濾取し、 1規定塩酸及び水で順次洗浄して 乾燥した。得られた固体を酢酸 300mLに溶解し、酢酸ナトリウム 15.824 gを加え、室温 にて 2時間攪拌した。溶媒を減圧留去し、残渣に水と酢酸ェチルを加え、不溶物をセ ライト濾過した。濾液を分液し、有機層を水で 3回、及び飽和食塩水で順次洗浄後、 無水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られた残渣を酢酸ェチル /n-へ キサン(1 : 3)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し、 5-ホルミ ル- 2-ヒドロキシ -3-イソプロポキシベンジルァセタート 13.525 gを淡黄色油状物として 得た。 EP: 253 31.6 g of 4-hydroxy-3-isopropoxybenzaldehyde was dissolved in 178 mL of chloromethyl methyl ether, and 100 g of tin tetrachloride was added at room temperature. After stirring at room temperature for 4.5 hours, the reaction solution was poured into ice water. The precipitated crystals were collected by filtration, washed successively with 1N hydrochloric acid and water and dried. The obtained solid was dissolved in 300 mL of acetic acid, 15.824 g of sodium acetate was added, and the mixture was stirred at room temperature for 2 hours. The solvent was distilled off under reduced pressure, water and ethyl acetate were added to the residue, and the insoluble material was filtered through Celite. The filtrate was separated, and the organic layer was washed three times with water and then with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 3) as an elution solvent, and 13.525 g of 5-form-2-hydroxy-3-isopropoxybenzylacetate was added. Obtained as a pale yellow oil. EP: 253
[0083] 参考例 14 [0083] Reference Example 14
サリチルアルデヒド 6.40 gとブロモ酢酸 tert-ブチル 10.2 gの Ν,Ν-ジメチルホルムァ ミド(52 mL)溶液に炭酸カリウム 10.8 gを室温で加え 60 °Cにて 22時間攪拌した。反 応液を減圧下濃縮し得られた残渣を酢酸ェチルに溶解し、水及び飽和食塩水で順 次洗浄した。有機層を無水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られた残 渣を酢酸ェチル -へキサン(1 : 6)を溶出溶媒とするシリカゲルカラムクロマトグラフ ィ一で精製し tert-ブチル(2-ホルミルフエノキシ)ァセタート 4.23 gを無色固体として得 た。 To a solution of 6.40 g of salicylaldehyde and 10.2 g of tert-butyl bromoacetate in Ν, ジ メ チ ル -dimethylformamide (52 mL) was added 10.8 g of potassium carbonate at room temperature, and the mixture was stirred at 60 ° C for 22 hours. The reaction solution was concentrated under reduced pressure, and the resulting residue was dissolved in ethyl acetate and washed successively with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using ethyl acetate-hexane (1: 6) as an elution solvent to obtain 4.23 g of tert-butyl (2-formylphenoxy) acetate as a colorless solid. .
[0084] 参考例 15 [0084] Reference Example 15
水素化アルミニウムリチウム 76 mgのテトラヒドロフラン(2 mL)懸濁液に、(3—ェトキ シ- 5-ホルミル- 4-ヒドロキシ)フエ-ルァセチックアシッド 224 mgのテトラヒドロフラン(8 mL)溶液を室温で加え、 50°Cで 6時間攪拌した。反応液を冷却後 1規定塩酸 60 mL
に注ぎ、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシ ゥムで乾燥後減圧下溶媒を留去し粗成績体を得た。得られた粗成績体を Ν,Ν-ジメチ ルホルムアミド 2 mL〖こ溶解し、炭酸カリウム 112mg、ブロモ酢酸 tert-ブチル 169 mgを 加え、室温で終夜攪拌した。反応液に水を加え、酢酸ェチルで抽出した。有機層を 水、飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥後減圧下溶媒を留去し た。残渣を n-へキサン/酢酸ェチル (85:15)を溶出溶媒とするシリカゲルカラムクロマ トグラフィ一で精製し、 tert-ブチル [2-エトキシ -4-(2-ヒドロキシェチル) -6- (ヒドロキ シメチル)フエノキシ]ァセタート 63 mgを得た。 To a suspension of 76 mg of lithium aluminum hydride in tetrahydrofuran (2 mL), a solution of 224 mg of (3-ethoxy-5-formyl-4-hydroxy) phenolic acid in tetrahydrofuran (8 mL) was added at room temperature. The mixture was stirred at 50 ° C for 6 hours. 1N hydrochloric acid 60 mL after cooling the reaction solution And extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure to obtain a crude product. The obtained crude product was dissolved in 2 mL of Ν, Ν-dimethylformamide, 112 mg of potassium carbonate and 169 mg of tert-butyl bromoacetate were added, and the mixture was stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (85:15) as an elution solvent, and tert-butyl [2-ethoxy-4- (2-hydroxyethyl) -6- (hydroxy 63 mg of (methyl) phenoxy] acetate was obtained.
[0085] 参考例 16 [0085] Reference Example 16
tert-ブチル 2-ホルミル- 4-ョード -6-イソプロポキシフエノキシァセタート 796 mg、トリ ブチルビ-ルスズ 781 mg、トリス (ジベンジリデンアセトン)ジパラジウム 174 mgおよびト リブチルホスフィン 58 mgのトルエン懸濁液(8.5 mL)をアルゴン雰囲気下、 70°Cで 14 時間撹拌した。反応混合物を放冷後、セライト濾過し、酢酸ェチルで洗浄した。濾液 を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下 に留去し、残渣を n_へキサン/酢酸ェチル (85:15)を溶出溶媒とするシリカゲルカラ ムクロマトグラフィーで精製後、得られた油状物を塩化メチレンを溶出溶媒とするシリ 力ゲルカラムクロマトグラフィーで精製して、 tert-ブチル 2-ホルミル- 6-イソプロポキシ -4-ビュルフエノキシァセタート 236 mgを淡黄色油状物として得た。 tert-butyl 2-formyl-4-iodo-6-isopropoxyphenoxyacetate 796 mg, tributyl beryltin 781 mg, tris (dibenzylideneacetone) dipalladium 174 mg and tributylphosphine 58 mg in toluene The suspension (8.5 mL) was stirred at 70 ° C. for 14 hours under an argon atmosphere. The reaction mixture was allowed to cool, filtered through celite, and washed with ethyl acetate. The filtrate was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography using n_hexane / ethyl acetate (85:15) as the eluting solvent, and the resulting oil was eluted with methylene chloride. Purification by silica gel gel chromatography gave 236 mg of tert-butyl 2-formyl-6-isopropoxy-4-butfenoxyacetate as a pale yellow oil.
[0086] 参考例 17 [0086] Reference Example 17
氷冷撹拌下 tert-ブチル 2-ホルミル- 6-イソプロポキシ -4-ビュルフエノキシァセター ト 0.26 gのテトラヒドロフラン(2.6 mL)溶液にボラン-テトラヒドロフラン錯体テトラヒドロ フラン溶液 (1 M) 1.62 mLを加え、同条件下で 5分、室温で 1時間撹拌した。反応混 合物を氷浴中で冷却し、撹拌下水 2.6 mLを加えた。同条件下 5分間撹拌後、 2規定 水酸化ナトリウム水溶液 5.2 mLおよび 30 %過酸化水素水 2.6 mLを順次カ卩えた。混合 物を同条件下で 5分、室温で 3.5時間撹拌した。反応混合物に水を加え、酢酸ェチル で 2回抽出した。有機層を合わせ、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾 燥した。溶媒を減圧下に留去した。残渣を n-へキサン/酢酸ェチル(1:4)を溶出溶媒 とするシリカゲルカラムクロマトグラフィーで精製して、 tert-ブチル 4-(2-ヒドロキシェ
チル) -2- (ヒドロキシメチル) -6-イソプロポキシフエノキシァセタート 142 mgを淡黄色油 状物として得た。 NMR (CDC1 ) : 1.36 (6H, d, J = 6.0 Hz), 1.39 (1H, t, J = 6.3 Hz), 1 To a solution of 0.26 g of tetrahydrofuran (2.6 mL) in tert-butyl 2-formyl-6-isopropoxy-4-butylphenoxyacetate with ice cooling and stirring, add 1.62 mL of borane-tetrahydrofuran complex tetrahydrofuran solution (1 M), The mixture was stirred for 5 minutes under the same conditions and for 1 hour at room temperature. The reaction mixture was cooled in an ice bath and 2.6 mL of water was added with stirring. After stirring for 5 minutes under the same conditions, 5.2 mL of 2N aqueous sodium hydroxide solution and 2.6 mL of 30% hydrogen peroxide solution were sequentially added. The mixture was stirred under the same conditions for 5 minutes and at room temperature for 3.5 hours. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The organic layers were combined, washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (1: 4) as an elution solvent, and tert-butyl 4- (2-hydroxyl 142 mg of (til) -2- (hydroxymethyl) -6-isopropoxyphenoxyacetate was obtained as a pale yellow oil. NMR (CDC1): 1.36 (6H, d, J = 6.0 Hz), 1.39 (1H, t, J = 6.3 Hz), 1
3 Three
.48 (9H, s), 2.79 (2H, t, J = 6.3 Hz), 3.83 (2H, q, J = 6.3 Hz), 4.39 (1H, t, J = 7.0 Hz), 4.53 (1H, sep, J = 6.0 Hz), 4.66 (2H, d, J = 7.0 Hz), 4.68 (2H, s), 6.74 (2H, s) [0087] 参考例 18 .48 (9H, s), 2.79 (2H, t, J = 6.3 Hz), 3.83 (2H, q, J = 6.3 Hz), 4.39 (1H, t, J = 7.0 Hz), 4.53 (1H, sep, J = 6.0 Hz), 4.66 (2H, d, J = 7.0 Hz), 4.68 (2H, s), 6.74 (2H, s) [0087] Reference Example 18
AD-MIX- α 7.7 gの tert-ブチルアルコール(15 mL)溶液に 0°Cでェチル(2-ホルミ ル -6-イソプロポキシ -4-ビュルフエノキシ)ァセタートを滴下した。室温に昇温後、 12 時間攪拌した。チォ硫酸ナトリウム 865 mgを加え 1時間攪拌後、水 15 mLを加え酢酸 ェチルにて抽出した。有機層を硫酸マグネシウムで乾燥し、減圧下濃縮した。得られ た残渣を酢酸ェチル /n-へキサン(1: 1)を溶出溶媒とするシリカゲルカラムクロマトグ ラフィ一で精製してェチル {4- [(1S)-1, 2-ジヒドロキシェチル] -2-ホルミル- 6-イソプロ ポキシフエノキシ }ァセタート 1.25 gを無色油状物質として得た。 FP: 327 Ethyl (2-formyl-6-isopropoxy-4-butylphenoxy) acetate was added dropwise to a solution of AD-MIX-α 7.7 g in tert-butyl alcohol (15 mL) at 0 ° C. The mixture was warmed to room temperature and stirred for 12 hours. After adding 865 mg of sodium thiosulfate and stirring for 1 hour, 15 mL of water was added and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 1) as an elution solvent to obtain ethyl {4-[(1S) -1,2-dihydroxyethyl] -2 1.25 g of -formyl-6-isopropoxyphenoxy} acetate was obtained as a colorless oil. FP: 327
[0088] 参考例 19 [0088] Reference Example 19
tert-ブチル(2-エトキシ- 6-ホルミル- 4-ョードフエノキシ)ァセタート 426 mg、 2- (メチ ルァミノ)メタノール 413 mg、ヨウ化銅 209 mg、酢酸セシウム 528 mg及びジメチルスル ホキシド 1 mLの混合物をアルゴン雰囲気下 90 °Cにて 15時間攪拌した。反応液に水 を加え酢酸ェチルで抽出した。抽出液を水、飽和塩ィ匕アンモ-ゥム水そして飽和食 塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥して減圧下濃縮した。残渣 をクロ口ホルム/メタノール (97 : 3)を溶出溶媒とするシリカゲルカラムクロマトグラフィ 一により精製して tert-ブチル {2-エトキシ- 6-ホルミル- 4- [(2-ヒドロキシェチル) (メチ ル)ァミノ]フエノキシ }ァセタート 110 mgを黄色泡状物として得た。 FP: 354 A mixture of tert-butyl (2-ethoxy-6-formyl-4-iodophenoxy) acetate 426 mg, 2- (methylamino) methanol 413 mg, copper iodide 209 mg, cesium acetate 528 mg, and dimethyl sulfoxide 1 mL The mixture was stirred at 90 ° C for 15 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water, saturated saline water and saturated saline. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using chloroform / methanol (97: 3) as an elution solvent and purified by tert-butyl {2-ethoxy-6-formyl-4-[(2-hydroxyethyl) (methyl) ) Amino] phenoxy} acetate 110 mg was obtained as a yellow foam. FP: 354
[0089] 参考例 20 [0089] Reference Example 20
tert-ブチル(4-ァセトキシメチル- 2-ホルミル- 6-ヒドロキシフエノキシ)ァセタート 0.32 4 g、 1- tert-ブトキシ- 2-プロパノール 0.145 g、トリフエ-ルホスフィン 0.288 gのテトラヒ ドロフラン(5 mL)溶液にジェチルァゾジカルボキシラートのトルエン溶液(2.30 M) 0 .48 mLを室温でカ卩え、同温で 1.5時間攪拌した。反応混合物を n-へキサン/酢酸ェチ ル(4:1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し、 tert-ブチル [ 4-ァセトキシメチル- 2-(2-tert-ブトキシ -1-メチルエトキシ) -6-ホルミルフエノキシ]ァセ
タート 0.085 gを得た。 NMR (CDC1 ): 1.20 (9H, s), 1.36 (3H, d, J = 6.5 Hz), 1.49 (9 Tetrahydrofuran (5 mL) solution of tert-butyl (4-acetoxymethyl-2-formyl-6-hydroxyphenoxy) acetate 0.32 4 g, 1-tert-butoxy-2-propanol 0.145 g, triphenylphosphine 0.288 g Into this, 0.48 mL of a toluene solution (2.30 M) of jetillazodicarboxylate was added at room temperature and stirred at the same temperature for 1.5 hours. The reaction mixture was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as an elution solvent, and tert-butyl [4-acetoxymethyl-2- (2-tert-butoxy-1-methyl) was obtained. Ethoxy) -6-formylphenoxy] ase A tarte of 0.085 g was obtained. NMR (CDC1): 1.20 (9H, s), 1.36 (3H, d, J = 6.5 Hz), 1.49 (9
3 Three
H, s), 2.10 (3H, s), 3.47 (IH, dd, J = 9.5, 4.0 Hz), 3.58 (IH, dd, J = 9.5, 6.5 Hz), 4. 45 - 4.55 (IH, m), 4.79 (IH, d, J = 16.0 Hz), 4.83 (IH, d, J = 16.0 Hz), 5.04 (2H, s ), 7.21 (IH, d, J = 2.0 Hz), 7.43 (IH, d, J = 2.0 Hz), 10.65 (IH, s) H, s), 2.10 (3H, s), 3.47 (IH, dd, J = 9.5, 4.0 Hz), 3.58 (IH, dd, J = 9.5, 6.5 Hz), 4. 45-4.55 (IH, m) , 4.79 (IH, d, J = 16.0 Hz), 4.83 (IH, d, J = 16.0 Hz), 5.04 (2H, s), 7.21 (IH, d, J = 2.0 Hz), 7.43 (IH, d, J = 2.0 Hz), 10.65 (IH, s)
[0090] 参考例 21 [0090] Reference Example 21
ェチル(2-ァセトキシメチル- 4-ホルミル- 6-イソプロポキシフエノキシ)ァセタート 12 g を 1,2-ジクロロェタン 200mLに溶解し、 3-クロ口過安息香酸 12.241 gを 0°Cにて少し ずつ添加した。室温にて 3時間攪拌後、析出した固体を濾去した。濾液を飽和重曹 水で 2回、水及び飽和食塩水で順次洗浄後、無水硫酸マグネシウムで乾燥し、減圧 下濃縮した。得られた残渣を酢酸ェチル /n-へキサン(1 : 3)を溶出溶媒とするシリカ ゲルカラムクロマトグラフィーで精製し黄色油状物 11.24 gを得た。得られた黄色油状 物 11.24 gをエタノール 300mLに溶解し、炭酸カリウム 4.384 gを添カ卩して、室温にて 5.5時間攪拌した。炭酸カリウムを濾去し、濾液を減圧下濃縮した。残渣を酢酸ェチ ルで希釈し、 0.5規定塩酸、水及び飽和食塩水で順次洗浄した。無水硫酸マグネシ ゥムで乾燥し、減圧下濃縮することで、ェチル(2-ァセトキシメチル -4-ヒドロキシ -6-ィ ソプロボキシフエノキシ)ァセタート 10.563 gを淡褐色油状物として得た。 EN : 325 [0091] 参考例 22 Ethyl (2-acetoxymethyl-4-formyl-6-isopropoxyphenoxy) acetate (12 g) was dissolved in 1,2-dichloroethane (200 mL) and 3-chloroperbenzoic acid (12.241 g) was added little by little at 0 ° C. did. After stirring at room temperature for 3 hours, the precipitated solid was removed by filtration. The filtrate was washed successively with saturated aqueous sodium hydrogen carbonate twice, water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 3) as an elution solvent to obtain 11.24 g of a yellow oily substance. 11.24 g of the obtained yellow oily substance was dissolved in 300 mL of ethanol, added with 4.384 g of potassium carbonate, and stirred at room temperature for 5.5 hours. Potassium carbonate was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was diluted with ethyl acetate and washed successively with 0.5N hydrochloric acid, water and saturated brine. The extract was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain 10.563 g of ethyl (2-acetoxymethyl-4-hydroxy-6-isopropoxyphenoxy) acetate as a light brown oil. EN: 325 [0091] Reference Example 22
ェチル(2-ァセトキシメチル- 4-ヒドロキシ -6-イソプロポキシフエノキシ)ァセタート 9.5 63 gとピリジン 4.74 mLを 1,2-ジクロロェタン (lOOmL)に溶解し、 tert-ブチルジメチ ルクロロシラン 4.638 gを添カ卩した。室温で 8時間攪拌後、 tert-ブチルジメチルクロ口 シラン 2,208 gとトリェチルァミン 8.2 mLを添加し、室温で 12時間攪拌した。反応液 を水、希塩酸で 3回、飽和重曹水、水及び飽和食塩水で順次洗浄後、無水硫酸マグ ネシゥムで乾燥し、減圧下濃縮した。得られた残渣を酢酸ェチル /n-へキサン(1: 10) を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し黄色油状物 10.985 gを 得た。得られた黄色油状物 100 mgをエタノール(2mL)に溶解し、炭酸カリウム 156 mgを添加して、室温にて 2時間攪拌後、 13時間放置した。炭酸カリウムを濾去し、濾 液を減圧下濃縮した。残渣を酢酸ェチルで希釈し、 0.5規定塩酸、水及び飽和食塩 水で順次洗浄した。無水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られた残
渣を酢酸ェチル /n-へキサン(1 : 3)、次いでクロ口ホルム/メタノール(20 : 1)を溶出溶 媒とするシリカゲルカラムクロマトグラフィーで精製しェチル [4-{[tert-ブチル (ジメチ ル)シリル]ォキシ }- 2-ヒドロキシメチル -6-イソプロポキシフエノキシ]ァセタート 26 mgを 無色油状物として得た。 NMR (CDC1 ) : 0.18 (6H, s), 0.97 (9H, s), 1.30 (3H, t, J = 7. Ethyl (2-acetoxymethyl-4-hydroxy-6-isopropoxyphenoxy) acetate 9.5 63 g and pyridine 4.74 mL are dissolved in 1,2-dichloroethane (lOOmL), and tert-butyldimethylchlorosilane 4.638 g is added. did. After stirring at room temperature for 8 hours, 2,208 g of tert-butyldimethylchlorosilane and 8.2 mL of triethylamine were added, and the mixture was stirred at room temperature for 12 hours. The reaction mixture was washed successively with water, diluted hydrochloric acid three times, saturated aqueous sodium hydrogen carbonate, water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1:10) as an elution solvent to obtain 10.985 g of a yellow oily substance. 100 mg of the obtained yellow oily substance was dissolved in ethanol (2 mL), 156 mg of potassium carbonate was added, and the mixture was stirred at room temperature for 2 hours and allowed to stand for 13 hours. Potassium carbonate was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was diluted with ethyl acetate and washed successively with 0.5N hydrochloric acid, water and saturated brine. The extract was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. Obtained residue The residue was purified by silica gel column chromatography using ethyl acetate / n- hexane (1: 3) and chloroform / methanol (20: 1) as an elution solvent, and purified with ethyl 4-[[tert-butyl (dimethyl). 26 mg of (l) silyl] oxy} -2-hydroxymethyl-6-isopropoxyphenoxy] acetate was obtained as a colorless oil. NMR (CDC1): 0.18 (6H, s), 0.97 (9H, s), 1.30 (3H, t, J = 7.
3 Three
0 Hz), 1.34 (6H, d, J = 7.0 Hz), 4.24 (2H, q, J = 7.0 Hz), 4.47 (IH, sep, J = 7.0 Hz) , 4.63 (2H, s), 4.72 (2H, s), 6.34 (2H, d, J = 2.0 Hz) 0 Hz), 1.34 (6H, d, J = 7.0 Hz), 4.24 (2H, q, J = 7.0 Hz), 4.47 (IH, sep, J = 7.0 Hz), 4.63 (2H, s), 4.72 (2H , s), 6.34 (2H, d, J = 2.0 Hz)
[0092] 参考例 23 [0092] Reference Example 23
3-ホルミル- 4-ヒドロキシ -5-イソプロポキシベンジルァセタート 12.4 gとピリジン 8.0 mLのジクロロメタン (100 mL)溶液にトリフルォロメタンスルホン酸無水物 9.1 mLを氷 冷下加え、室温で 30分間攪拌した。反応液を減圧下濃縮し、得られた残渣に飽和塩 化アンモ-ゥム水溶液をカ卩ぇ酢酸ェチルで抽出した。有機層を水および飽和食塩水 で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥して減圧下濃縮した。得ら れた残渣を酢酸ェチル /n—へキサン(40:60)を溶出溶媒とするシリカゲルカラムクロ マトグラフィ一で精製し、 3-ホルミル- 5-イソプロポキシ -4-{ [(トリフルォロメチル)スルホ -ル]ォキシ }ベンジルァセタート 10.4gを黄色油状物として得た。 Add 9.1 mL of trifluoromethanesulfonic anhydride to a solution of 12.4 g of 3-formyl-4-hydroxy-5-isopropoxybenzylacetate and 8.0 mL of pyridine in dichloromethane (100 mL) under ice-cooling, and stir at room temperature for 30 minutes did. The reaction mixture was concentrated under reduced pressure, and a saturated aqueous ammonium chloride solution was extracted from the resulting residue with ketyl acetate. The organic layer was washed successively with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (40:60) as the elution solvent, and 3-formyl-5-isopropoxy-4-{[(trifluoromethyl ) Sulfo-l] oxy} benzylate 10.4 g was obtained as a yellow oil.
[0093] 参考例 24 [0093] Reference Example 24
3-ホルミル- 5-イソプロポキシ -4-{ [(トリフルォロメチル)スルホ -ル]ォキシ }ベンジル ァセタート 300 mg、アクリル酸 tert-ブチル 2.24 mL、 1,3-ビス (ジフエ-ルホスフイノ)プ 口パン 64 mg、酢酸パラジウム 18 mg、 Ν,Ν-ジイソプロピルェチルァミン 0.204 mLの Ν, N-ジメチルホルムアミド混液 (2 mL)を、封管中マイクロ波照射下 150 °Cにて 10分間 撹拌した。反応液に水を加え酢酸ェチルで抽出し、得られた有機層を水および飽和 食塩水で洗浄後、無水硫酸マグネシウムで乾燥させた。溶媒を減圧留去し得た残渣 を酢酸ェチル -へキサン(1:5)を溶出溶媒とするシリカゲルカラムクロマトグラフィー にて精製し、 tert-ブチル(E)-3-(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキシフ ェ -ル)アタリラート 188 mgを黄色油状物として得た。 NMR (CDC1 ) : 1.40 (6H, d, J = 6 3-Formyl-5-isopropoxy-4-{[(trifluoromethyl) sulfol] oxy} benzyl acetate 300 mg, tert-butyl acrylate 2.24 mL, 1,3-bis (diphenylphosphino) Pan 64 mg, palladium acetate 18 mg, Ν, Ν-diisopropylethylamine 0.204 mL Ν, N-dimethylformamide mixed solution (2 mL) was stirred for 10 minutes at 150 ° C under microwave irradiation in a sealed tube . Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by distilling off the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate-hexane (1: 5) as an elution solvent, and tert-butyl (E) -3- (4-acetoxymethyl-2- 188 mg of formyl-6-isopropoxyphenyl) attalylate was obtained as a yellow oil. NMR (CDC1): 1.40 (6H, d, J = 6
3 Three
.0 Hz), 1.54 (9H, s), 2.14 (3H, s), 4.64 (IH, sep, J = 6.0 Hz), 5.12 (2H, s), 6.14 (IH , d, J = 16.1 Hz), 7.11 (IH, d, J = 1.4 Hz), 7.48 (IH, d, J = 1.4 Hz), 8.03 (IH, d, J = 16.1 Hz), 10.21 (IH, s)
[0094] 参考例 25 .0 Hz), 1.54 (9H, s), 2.14 (3H, s), 4.64 (IH, sep, J = 6.0 Hz), 5.12 (2H, s), 6.14 (IH, d, J = 16.1 Hz), 7.11 (IH, d, J = 1.4 Hz), 7.48 (IH, d, J = 1.4 Hz), 8.03 (IH, d, J = 16.1 Hz), 10.21 (IH, s) [0094] Reference Example 25
トリフルォロメタンスルホン酸 2-ホルミル- 6-イソプロポキシ -4-ニトロフエニルエステ ル 3.65 gのトルエン(30 mL)溶液に臭化テトラブチルアンモ -ゥム 3.95 gを加え、 110 °Cで 2.5時間撹拌した。臭化テトラプチルアンモ-ゥムをさらに 1.0 g加え、 110 °Cで 1. 5時間撹拌した。溶媒を減圧留去し得た残渣を酢酸ェチル /n-へキサン(1:10)を溶 出溶媒とするシリカゲルカラムクロマトグラフィーにて精製し、 2-ブロモ -3-イソプロボ キシ- 5-二トロべンズアルデヒド 2.47 gを淡黄色固体として得た。 NMR (CDC1 ) : 1.48 ( 2-Formyl-6-isopropoxy-4-nitrophenyl trifluoromethanesulfonate Add 3.95 g of tetrabutylammonium bromide to a solution of 3.65 g of toluene (30 mL) and heat at 110 ° C for 2.5 hours. Stir. An additional 1.0 g of tetraptyl ammonium bromide was added, and the mixture was stirred at 110 ° C. for 1.5 hours. The residue obtained by distilling off the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1:10) as an elution solvent, and 2-bromo-3-isopropoxy-5-nitro 2.47 g of benzaldehyde was obtained as a pale yellow solid. NMR (CDC1): 1.48 (
3 Three
6H, d, J = 6.0 Hz), 4.76 (1H, sep, J = 6.0 Hz), 7.89 (1H, d, J = 1.6 Hz), 8.32 (1H, J = 1.6 Hz), 10.46 (1H, s) 6H, d, J = 6.0 Hz), 4.76 (1H, sep, J = 6.0 Hz), 7.89 (1H, d, J = 1.6 Hz), 8.32 (1H, J = 1.6 Hz), 10.46 (1H, s)
[0095] 参考例 26 [0095] Reference Example 26
2-ブロモ -3-イソプロポキシ -5--トロべンズアルデヒド 2.47 g、アクリル酸 tert-ブチ ル 1.56 mL、酢酸パラジウム 96 mg、臭化テトラブチルアンモ -ゥム 276 mg、炭酸力リウ ム 3.55 gの Ν,Ν-ジメチルホルムアミド混液 (20 mL)を 60 °Cで 2時間撹拌した。反応液 に水を加え酢酸ェチルで抽出し、得られた有機層を水および飽和食塩水で洗浄後、 無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去し得られた残渣を酢酸ェチル /n-へキサン(1:5)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにて精製し、 t ert-ブチル(E)-3-(2-ホルミル- 6-イソプロポキシ -4--トロフエ-ル)アタリラート 2.47 g を淡黄色固体として得た。 NMR (CDC1 ) : 1.46 (6H, d, J = 6.0 Hz), 1.55 (9H, s), 4.76 2-Bromo-3-isopropoxy-5-trobensaldehyde 2.47 g, tert-butyl acrylate 1.56 mL, palladium acetate 96 mg, tetrabutylammonium bromide 276 mg, carbonated power 3.55 g The Ν, Ν-dimethylformamide mixture (20 mL) was stirred at 60 ° C for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 5) as an elution solvent, and tert-butyl (E) -3- (2 There was obtained 2.47 g of -formyl-6-isopropoxy-4-trophenol) attalylate as a pale yellow solid. NMR (CDC1): 1.46 (6H, d, J = 6.0 Hz), 1.55 (9H, s), 4.76
3 Three
(1H, sep, J = 6.0 Hz), 6.23 (1H, d, J = 16.1 Hz), 7.90 (1H, d, J = 2.3 Hz), 7.98 (1H , d, J = 16.1 Hz), 8.33 (1H, d, J = 2.3 Hz), 10.24 (1H, s) (1H, sep, J = 6.0 Hz), 6.23 (1H, d, J = 16.1 Hz), 7.90 (1H, d, J = 2.3 Hz), 7.98 (1H, d, J = 16.1 Hz), 8.33 (1H , d, J = 2.3 Hz), 10.24 (1H, s)
[0096] 参考例 27 [0096] Reference Example 27
tert-ブチル(E)-3-(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキシフエ-ル)ァク リラート 234 mg、メタノール 3 mLおよびテトラヒドロフラン 3 mLの混合物に、氷冷下臭 化ニッケル 14 mgおよび水素化ホウ素ナトリウム 24 mgを順次カ卩え、室温にて 30分間 撹拌した。反応液を再度氷冷し、臭化ニッケル 28 mgおよび水素化ホウ素ナトリウム 48 mgをカ卩え、室温にて 30分間撹拌した。反応液を再度氷冷し、臭化ニッケル 28 mgお よび水素化ホウ素ナトリウム 48 mgをさらに加え、室温にて 30分間撹拌した。反応液を 酢酸ェチルで希釈後 1規定塩酸に注ぎ抽出した。有機層を水および飽和食塩水で
洗浄後、無水硫酸マグネシウムで乾燥させた。溶媒を減圧留去して得た残渣を酢酸 ェチル /n-へキサン(1:2)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにて精 製し、 tert-ブチル 3-(4-ァセトキシメチル- 2-ヒドロキシメチル -6-イソプロポキシフエ- ル)プロパノアート 148 mgを無色油状物として得た。 Add tert-butyl (E) -3- (4-acetoxymethyl-2-formyl-6-isopropoxyphenyl) acrylate 234 mg, methanol 3 mL and tetrahydrofuran 3 mL to a mixture of nickel odorant under ice-cooling. 14 mg and 24 mg of sodium borohydride were sequentially added and stirred at room temperature for 30 minutes. The reaction mixture was ice-cooled again, and 28 mg of nickel bromide and 48 mg of sodium borohydride were added and stirred at room temperature for 30 minutes. The reaction mixture was ice-cooled again, 28 mg nickel bromide and 48 mg sodium borohydride were further added, and the mixture was stirred at room temperature for 30 min. The reaction mixture was diluted with ethyl acetate, poured into 1N hydrochloric acid and extracted. Organic layer with water and saturated saline After washing, it was dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 2) as the elution solvent, and tert-butyl 3- (4-acetoxymethyl-2- 148 mg of hydroxymethyl-6-isopropoxyphenol) propanoate was obtained as a colorless oil.
[0097] 参考例 28 [0097] Reference Example 28
tert-ブチル 3-(4-ァミノ- 2-ヒドロキシメチル -6-イソプロポキシフエニル)プロパノア ート 272 mgと tert-ブチルジメチルシリルォキシァセトアルデヒド 0.167 mLのテトラヒドロ フラン(10 mL)溶液に、室温でトリァセトキシ水素化ホウ素ナトリウム 373 mgをカ卩え、 2. 5時間撹拌した。反応液に水を加え酢酸ェチルで抽出し、得られた有機層を水およ び飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥させた。溶媒を減圧留去して 得た残渣を酢酸ェチル /n-へキサン(1:10)を溶出溶媒とするシリカゲルカラムクロマ トグラフィ一にて精製し、 tert-ブチル 3-{4-[2-(tert-ブチルジメチルシリルォキシ)ェ チルァミノ] -2-ヒドロキシメチル -6-イソプロポキシフエ-ル}プロパノアート 411 mgを淡 黄色油状物として得た。 NMR (CDC1 ) : 0.07 (6H, s), 0.91 (9H, s), 1.33 (6H, d, J = 6. To a solution of tert-butyl 3- (4-amino-2-hydroxymethyl-6-isopropoxyphenyl) propanoate 272 mg and tert-butyldimethylsilyloxyacetaldehyde 0.167 mL in tetrahydrofuran (10 mL), At room temperature, 373 mg of sodium triacetoxyborohydride was added and stirred for 2.5 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1:10) as an elution solvent, and tert-butyl 3- {4- [2- ( tert-butyldimethylsilyloxy) ethylamino] -2-hydroxymethyl-6-isopropoxyphenol} propanoate 411 mg was obtained as a pale yellow oil. NMR (CDC1): 0.07 (6H, s), 0.91 (9H, s), 1.33 (6H, d, J = 6.
3 Three
0 Hz), 1.40 (9H, s), 2.52 (2H, t, J = 7.4 Hz), 2.58 - 2.72 (1H, br), 2.84 (1H, t, J = 7.4 Hz), 3.20 (2H, t, J = 5.4 Hz), 3.81 (2H, t, J = 5.4 Hz), 4.51 (1H, sep, J = 6.0 H z), 4.61 (2H, s), 6.13 (1H, d, J = 2.2 Hz), 6.27 (1H, d, J = 2.2 Hz) 0 Hz), 1.40 (9H, s), 2.52 (2H, t, J = 7.4 Hz), 2.58-2.72 (1H, br), 2.84 (1H, t, J = 7.4 Hz), 3.20 (2H, t, J = 5.4 Hz), 3.81 (2H, t, J = 5.4 Hz), 4.51 (1H, sep, J = 6.0 H z), 4.61 (2H, s), 6.13 (1H, d, J = 2.2 Hz), 6.27 (1H, d, J = 2.2 Hz)
[0098] 参考例 29 [0098] Reference Example 29
tert-ブチル 3-{4-[2-(tert-ブチルジメチルシリルォキシ)ェチルァミノ] -2-ヒドロキシ メチル -6-イソプロポキシフエ二ル}プロパノアート 461 mg、ジ -tert-ブチルジカーボナ ート 646 mgの Ν,Ν-ジメチルホルムアミド(10 mL)混液に、室温で Ν,Ν-ジイソプロピル ェチルァミン 0.515 mLをカ卩ぇ終夜撹拌した。反応液に水を加え酢酸ェチルで抽出し 、得られた有機層を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥さ せた。溶媒を減圧留去して得た残渣を酢酸ェチル /n-へキサン(1:3)を溶出溶媒と するシリカゲルカラムクロマトグラフィーにて精製し、 tert-ブチル 3-(4_{tert-ブトキシ カルボ-ル -[2-(tert-ブチルジメチルシリルォキシ)ェチル]アミノ}-2-ヒドロキシメチル -6-イソプロポキシフエ-ル)プロパノアート 492 mgを淡黄色油状物として得た。 NMR ( CDC1 ) : 0.04 (6H, s), 0.87 (9H, s), 1.34 (6H, d, J = 6.0 Hz), 1.40 (9H, s), 1.44 (9H,
br s), 2.50-2.57 (3H, m), 2.91 (2H, t, J = 7.5 Hz), 3.67-3.77 (4H, m), 4.52 (IH, sep , J = 6.0 Hz), 4.66 (2H, d, J = 6.0 Hz), 6.71 (IH, d, J = 2.0 Hz), 6.85 (IH, d, J = 2. 0 Hz) tert-butyl 3- {4- [2- (tert-butyldimethylsilyloxy) ethylamino] -2-hydroxymethyl-6-isopropoxyphenyl} propanoate 461 mg, di-tert-butyl dicarbonate 646 mg To a mixture of Ν, Ν-dimethylformamide (10 mL), 0.515 mL of Ν, Ν-diisopropylethylamine was stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by distilling off the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 3) as an elution solvent, and tert-butyl 3- (4_ {tert-butoxycarbo- 492 mg of ru- [2- (tert-butyldimethylsilyloxy) ethyl] amino} -2-hydroxymethyl-6-isopropoxyphenyl) propanoate was obtained as a pale yellow oil. NMR (CDC1): 0.04 (6H, s), 0.87 (9H, s), 1.34 (6H, d, J = 6.0 Hz), 1.40 (9H, s), 1.44 (9H, br s), 2.50-2.57 (3H, m), 2.91 (2H, t, J = 7.5 Hz), 3.67-3.77 (4H, m), 4.52 (IH, sep, J = 6.0 Hz), 4.66 (2H, d, J = 6.0 Hz), 6.71 (IH, d, J = 2.0 Hz), 6.85 (IH, d, J = 2.0 Hz)
[0099] 参考例 30 [0099] Reference Example 30
tert-ブチル 3-(4-{tert-ブトキシカルボ-ル -[2-(tert-ブチルジメチルシリルォキシ) ェチル]アミノ}-2-ヒドロキシメチル -6-イソプロポキシフエ-ル)プロパノアートを原料と し、参考例 3と同様の操作を行い tert-ブチル 3-(4-{tert-ブトキシカルボ-ル _[2-(ter t-ブチルジメチルシリルォキシ)ェチル]アミノ}-2-ホルミル- 6-イソプロポキシフエニル) プロパノアートを得た。 NMR (CDC1 ) : 0.03 (6H, s), 0.85 (9H, s), 1.36 (6H, d, J = 6.0 tert-Butyl 3- (4- {tert-Butoxycarbol- [2- (tert-Butyldimethylsilyloxy) ethyl] amino} -2-hydroxymethyl-6-isopropoxyphenyl) propanoate as raw material Tert-butyl 3- (4- {tert-butoxycarbol_ [2- (ter-butyldimethylsilyloxy) ethyl] amino} -2-formyl-6 -Isopropoxyphenyl) Propanoate was obtained. NMR (CDC1): 0.03 (6H, s), 0.85 (9H, s), 1.36 (6H, d, J = 6.0
3 Three
Hz), 1.43 (9H, s), 1.44 (9H, br s), 2.43-2.49 (2H, m), 3.25-3.30 (2H, m), 3.71-3.7 9 (4H, m), 4.55 (IH, sep, J = 6.0 Hz), 7.03 (IH, d, J = 2.1 Hz), 7.34 (IH, d, J = 2.1 Hz), 10.28 (IH, s) Hz), 1.43 (9H, s), 1.44 (9H, br s), 2.43-2.49 (2H, m), 3.25-3.30 (2H, m), 3.71-3.7 9 (4H, m), 4.55 (IH, sep, J = 6.0 Hz), 7.03 (IH, d, J = 2.1 Hz), 7.34 (IH, d, J = 2.1 Hz), 10.28 (IH, s)
[0100] 参考例 31 [0100] Reference Example 31
tert-ブチル 3-(4-{tert-ブトキシカルボ-ル -[2-(tert-ブチルジメチルシリルォキシ) ェチル]アミノ}-2-ホルミル- 6-イソプロポキシフエ-ル)プロパノアート 80 mgを塩化水 素のエタノール溶液 (15wt%)に溶解し、 60°Cにて 4.5時間撹拌した。反応液を飽和重 曹水に注ぎ、酢酸ェチルで抽出した。有機層を水および飽和食塩水で洗浄後、無水 硫酸マグネシウムで乾燥した。溶媒を減圧留去して得た残渣を酢酸ェチル /n-へキ サン(1:1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにて精製し、ェチル 3 -[2-ホルミル- 4-(2-ヒドロキシェチルァミノ) -6-イソプロポキシフエ-ル]プロパノアート 26 mgを黄色油状物として得た。 NMR (CDC1 ) : 1.23 (3H, t, J = 7.1 Hz), 1.35 (6H, d, tert-Butyl 3- (4- {tert-Butoxycarbol- [2- (tert-butyldimethylsilyloxy) ethyl] amino} -2-formyl-6-isopropoxyphenyl) propanoate 80 mg This was dissolved in an ethanol solution of hydrogen (15 wt%) and stirred at 60 ° C for 4.5 hours. The reaction mixture was poured into saturated aqueous sodium hydrogen carbonate and extracted with ethyl acetate. The organic layer was washed with water and saturated brine, and then dried over anhydrous magnesium sulfate. The residue obtained by distilling off the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 1) as an elution solvent, and ethyl 3- [2-formyl-4- (2 -Hydroxyethylamino) -6-isopropoxyphenol] propanoate 26 mg was obtained as a yellow oil. NMR (CDC1): 1.23 (3H, t, J = 7.1 Hz), 1.35 (6H, d,
3 Three
J = 6.0 Hz), 2.53 (2H, t, J = 7.8 Hz), 3.24 (2H, t, J = 7.8 Hz), 3.33 (2H, t, J = 5.2 Hz), 3.86 (2H, t, J = 5.2 Hz), 4.11 (2H, q, J = 7.1 Hz), 4.52 (IH, sep, J = 6.0 Hz), 6 .42 (IH, d, J = 2.4 Hz), 6.70 (IH, d, J = 2.4 Hz), 10.28 (IH, s) J = 6.0 Hz), 2.53 (2H, t, J = 7.8 Hz), 3.24 (2H, t, J = 7.8 Hz), 3.33 (2H, t, J = 5.2 Hz), 3.86 (2H, t, J = 5.2 Hz), 4.11 (2H, q, J = 7.1 Hz), 4.52 (IH, sep, J = 6.0 Hz), 6.42 (IH, d, J = 2.4 Hz), 6.70 (IH, d, J = 2.4 Hz), 10.28 (IH, s)
[0101] 参考例 32 [0101] Reference Example 32
3-ホルミル- 5-イソプロポキシ -4-{ [(トリフルォロメタン)スルホ -ル]ォキシ }ベンジル ァセタート 1.00 g、ビス (ジベンジリデンアセトン)パラジウム 0.075 g、硼弗化トリ- tert-ブ チルホスホ-ゥム 0.0379 g、 Ν,Ν-ジイソプロピルェチルァミン 0.506 g、(1- tert-ブトキ
シビュルォキシ) -tert-ブチルジメチルシラン 5.99 gとフッ化亜鉛 0.135 gの Ν,Ν-ジメ チルホルムアミド(10 mL)溶液を 60 °Cで 1時間攪拌した。反応液を冷却後、水を加え 、ジェチルエーテルで抽出した。有機層を水及び飽和食塩水で順次洗浄した後、無 水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られた残渣を n-へキサン/酢酸 ェチル (5 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し、 tert- ブチル(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキシフエ-ル)ァセタート 1.18 g を無色油状物として得た。 NMR (CDC1 ) : 1.35 (6H, d, J = 6.0 Hz), 1.44 (9H, s), 2.1 3-formyl-5-isopropoxy-4-{[(trifluoromethane) sulfol] oxy} benzyl acetate 1.00 g, bis (dibenzylideneacetone) palladium 0.075 g, borofluorinated tri-tert-butylphosphor 0.0379 g, Ν, Ν-Diisopropylethylamine 0.506 g, (1-tert-butoxy A solution of 99, Ν-dimethylformamide (10 mL) of 5.99 g of cybroxy) -tert-butyldimethylsilane and 0.135 g of zinc fluoride was stirred at 60 ° C for 1 hour. The reaction mixture was cooled, water was added, and the mixture was extracted with jetyl ether. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (5: 1) as the eluting solvent, and tert-butyl (4-acetoxymethyl-2-formyl-6-isopropoxyphenyl) 1.18 g of acetate was obtained as a colorless oil. NMR (CDC1): 1.35 (6H, d, J = 6.0 Hz), 1.44 (9H, s), 2.1
3 Three
2 (3H, s), 4.07 (2H, s), 4.59 (1H, sep, J = 6.0 Hz), 5.13 (2H, s), 7.12 (2H, d, J = 1. 2 (3H, s), 4.07 (2H, s), 4.59 (1H, sep, J = 6.0 Hz), 5.13 (2H, s), 7.12 (2H, d, J = 1.
3 Hz), 7.41 (1H, d, J = 1.3 Hz), 10.12 (1H, s) 3 Hz), 7.41 (1H, d, J = 1.3 Hz), 10.12 (1H, s)
[0102] 参考例 33 [0102] Reference Example 33
tert-ブチル(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキシフエニル)ァセタート 150 mgにトリフルォロ酢酸 3 mLを室温にて加え、 2.5時間撹拌した。トリフルォロ酢酸 を減圧留去して得た残渣を酢酸ェチル /n-へキサン(1:2)を溶出溶媒とするシリカゲ ルカラムクロマトグラフィーにて精製し、黄色固体 73 mgを得た。得られた黄色固体 73 mgと炭酸カリウム 41 mgの Ν,Ν-ジメチルホルムアミド(2 mL)混液に室温にてョードエ タン 0.04 mLを加え、 1.5時間撹拌した。反応液を酢酸ェチルで希釈し、 1規定塩酸に 注ぎ抽出した。有機層を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾 燥した。溶媒を減圧下留去し、褐色油状物を得た。得られた褐色油状物のエタノー ル(3 mL)溶液に室温にて炭酸カリウム 69 mgを加え、 1.5時間撹拌した。反応液を酢 酸ェチルで希釈し、 1規定塩酸に注ぎ抽出した。有機層を水および飽和食塩水で洗 浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し得た残渣を酢酸ェチ ル /n-へキサン(1:3)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにて精製し 、ェチル(2-ホルミル- 4-ヒドロキシメチル -6-イソプロポキシフエ-ル)ァセタート 39 mg を淡黄色油状物として得た。 NMR (CDC1 ) : 1.26 (3H, t, J = 7.1 Hz), 1.34 (6H, d, J = To 150 mg of tert-butyl (4-acetoxymethyl-2-formyl-6-isopropoxyphenyl) acetate, 3 mL of trifluoroacetic acid was added at room temperature and stirred for 2.5 hours. The residue obtained by distilling off trifluoroacetic acid under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 2) as an elution solvent to obtain 73 mg of a yellow solid. To a mixed solution of 73 mg of the obtained yellow solid and 41 mg of potassium carbonate in Ν, mg-dimethylformamide (2 mL) was added 0.04 mL of odoethane at room temperature, and the mixture was stirred for 1.5 hours. The reaction solution was diluted with ethyl acetate, poured into 1N hydrochloric acid and extracted. The organic layer was washed with water and saturated brine, and then dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure to give a brown oil. To a solution of the obtained brown oil in ethanol (3 mL), 69 mg of potassium carbonate was added at room temperature and stirred for 1.5 hours. The reaction mixture was diluted with ethyl acetate and poured into 1N hydrochloric acid for extraction. The organic layer was washed with water and saturated brine, and then dried over anhydrous magnesium sulfate. The residue obtained by distilling off the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 3) as an elution solvent, and ethyl (2-formyl-4-hydroxymethyl- 39 mg of 6-isopropoxyphenol) acetate was obtained as a pale yellow oil. NMR (CDC1): 1.26 (3H, t, J = 7.1 Hz), 1.34 (6H, d, J =
3 Three
6.1 Hz), 4.12-4.18 (4H, m), 4.61 (1H, sep, J = 6.1 Hz), 4.77 (2H, s), 7.19 (1H, s), 7.39 (1H, s), 10.11 (1H, s) 6.1 Hz), 4.12-4.18 (4H, m), 4.61 (1H, sep, J = 6.1 Hz), 4.77 (2H, s), 7.19 (1H, s), 7.39 (1H, s), 10.11 (1H, s)
[0103] 参考例 34 [0103] Reference Example 34
2,3-ジヒドロキシベンズアルデヒド 23.17 gとブロモ酢酸 tert-ブチル 32.8 gの Ν,Ν-ジ
メチルホルムアミド(170 mL)溶液に炭酸水素カリウム 16.8 gを室温にてカ卩ぇ 16時間 攪拌した。反応液に水を加え酢酸ェチルで抽出し、水及び飽和食塩水で順次洗浄 した。有機層を無水硫酸マグネシウムで乾燥後減圧下濃縮した。得られた残渣を 2- プロパノール /n-へキサンから結晶化させ tert-ブチル(2-ホルミル- 6-ヒドロキシフエノ キシ)ァセタート 19.2 gを無色固体として得た。 FP: 253 2,3-dihydroxybenzaldehyde 23.17 g and tert-butyl bromoacetate 32.8 g In a methylformamide (170 mL) solution, 16.8 g of potassium hydrogen carbonate was stirred at room temperature for 16 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate and washed successively with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was crystallized from 2-propanol / n- hexane to obtain 19.2 g of tert-butyl (2-formyl-6-hydroxyphenoxy) acetate as a colorless solid. FP: 253
[0104] 参考例 35 [0104] Reference Example 35
3-エトキシサリチルアルデヒド 5.23 gを 1規定水酸ィ匕ナトリウム水溶液 32mLに溶解し 、 β -プロピオラタトン 2.27 gを加熱還流下加え、 1時間攪拌した。反応液に氷冷下濃 塩酸を加え酸性とした後クロ口ホルムで抽出した。有機層を重曹水で抽出し、水層に 濃塩酸を加え酸性とした。析出物を濾取し、濾物を減圧下乾燥して 3-(2-エトキシ -6- ホルミルフエノキシ)プロパノイツクアシッド 5.29 gを無色固体として得た。 EP: 260 [M+ Na]+ 5.23 g of 3-ethoxysalicylaldehyde was dissolved in 32 mL of 1N sodium hydroxide aqueous solution, 2.27 g of β-propiolatatone was added with heating under reflux, and the mixture was stirred for 1 hour. The reaction mixture was acidified with concentrated hydrochloric acid under ice-cooling, and extracted with black mouth form. The organic layer was extracted with aqueous sodium bicarbonate, and the aqueous layer was acidified with concentrated hydrochloric acid. The precipitate was collected by filtration, and the filtrate was dried under reduced pressure to obtain 5.29 g of 3- (2-ethoxy-6-formylphenoxy) propanoic acid as a colorless solid. EP: 260 [M + Na] +
[0105] 参考例 36 [0105] Reference Example 36
tert-ブチル(2-ホルミル- 6-ヒドロキシフエノキシ)ァセタート 2.06 gを塩化メチレン 40 mLに溶解し、室温にてピリジン 3.3 mL次いでトリフルォロメタンスルホン酸無水物 2.7 mLを加え、 30分間攪拌した。反応液に水を加え、塩化メチレンで抽出した。有機層 を無水硫酸マグネシウムで乾燥し減圧下濃縮した。得られた残渣を n-へキサン/酢酸 ェチル (9 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し無色 固体 1.89 gを得た。得られた無色固体 500 mgとフエ-ルボロン酸 317 mgを Ν,Ν-ジメチ ルホルムアミド 10 mLに溶解し、室温にてリン酸カリウム 552 mgとテトラキス (トリフエ- ルホスフィン)パラジウム 150 mgをカ卩え、 100 °Cにて 1.5時間攪拌した。反応液に酢酸 ェチル及び水をカ卩え、酢酸ェチルで抽出した。有機層を水及び飽和食塩水で順次 洗浄した後、無水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られた残渣を n-へ キサン/酢酸ェチル (9 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより 精製し、 tert-ブチル [(3-ホルミルビフエ-ル -2-ィル)ォキシ]ァセタートを 406 mg得た 。 FP: 313 Dissolve 2.06 g of tert-butyl (2-formyl-6-hydroxyphenoxy) acetate in 40 mL of methylene chloride, add 3.3 mL of pyridine and 2.7 mL of trifluoromethanesulfonic anhydride at room temperature, and stir for 30 minutes . Water was added to the reaction mixture, and the mixture was extracted with methylene chloride. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (9: 1) as an elution solvent to obtain 1.89 g of a colorless solid. The obtained colorless solid 500 mg and phenylboronic acid 317 mg were dissolved in Ν, Ν-dimethylformamide 10 mL, and potassium phosphate 552 mg and tetrakis (triphosphine) palladium 150 mg were dissolved at room temperature. The mixture was stirred at 100 ° C for 1.5 hours. Ethyl acetate and water were added to the reaction solution and extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (9: 1) as an elution solvent, and tert-butyl [(3-formylbiphenyl-2-yl) oxy] acetate was purified. Obtained 406 mg. FP: 313
[0106] 参考例 37 [0106] Reference Example 37
2- (ブロモメチル)ベンジルァセタート 0.67 gのァセトニトリル (28 mL)溶液に、サル
コシン tert-ブチルエステル一塩酸塩 0.50 gと炭酸カリウム 0.76 gを室温にて加え 12 時間攪拌した。反応液を濾別し、濾液を減圧下濃縮した。残渣を酢酸ェチルで希釈 し、水次いで飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥して減 圧下濃縮し、無色油状物 0.83 gを得た。得られた無色油状物 0.35 gをメタノール 11 m Lに溶解し、 1規定水酸化ナトリウム水溶液 1.2 mLを氷冷下加え 30分間攪拌した。 1 規定塩酸 1.2 mLを氷冷下加え、減圧下濃縮した。得られた残渣を酢酸ェチルで希 釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥して減圧下濃 縮し、得られた残渣を酢酸ェチル /n-へキサン(1 : 3)を溶出溶媒とするシリカゲル力 ラムクロマトグラフィーで精製し、 tert-ブチル {[2- (ヒドロキシメチル)ベンジル] (メチル) アミノ}ァセタート 0.24 gを無色油状物として得た。 FP: 266 2- (Bromomethyl) benzylacetate 0.67 g of acetonitrile (28 mL) Cosin tert-butyl ester monohydrochloride (0.50 g) and potassium carbonate (0.76 g) were added at room temperature and stirred for 12 hours. The reaction solution was filtered off, and the filtrate was concentrated under reduced pressure. The residue was diluted with ethyl acetate and washed with water and then saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain 0.83 g of a colorless oil. 0.35 g of the obtained colorless oil was dissolved in 11 mL of methanol, and 1.2 mL of 1N aqueous sodium hydroxide solution was added under ice-cooling and stirred for 30 minutes. 1 N hydrochloric acid (1.2 mL) was added under ice-cooling, and the mixture was concentrated under reduced pressure. The obtained residue was diluted with ethyl acetate and washed with saturated brine. The organic layer is dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The resulting residue is purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 3) as the eluting solvent, and tert-butyl 0.24 g of {[2- (hydroxymethyl) benzyl] (methyl) amino} acetate was obtained as a colorless oil. FP: 266
参考例 38 Reference Example 38
2- (ブロモメチル)ベンジルァセタート 3.98 gのァセトニトリル (80 mL)溶液に、グリシ ン tert-ブチルエステル一塩酸塩 2.73 gと炭酸カリウム 4.53 gを氷冷下カ卩え、室温に て 12時間攪拌した。反応液を濾別し、濾液を減圧下濃縮した。残渣を酢酸ェチル /n —へキサン(1 : 3)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し無色 油状物 3.94 gを得た。得られた無色油状物 2.00 gを 1,4-ジォキサン 20 mLに溶解し、 ジ -tert-ブチルジカルボナート 1.49 gを室温にてカ卩ぇ 12時間攪拌した。反応液を減 圧下濃縮した。得られた残渣を酢酸ェチルで希釈し、 10%クェン酸水溶液次いで飽 和重曹水で洗浄した。有機層を無水硫酸マグネシウムで乾燥して減圧下濃縮した。 得られた残渣を酢酸ェチル /n-へキサン(1 :4)を溶出溶媒とするシリカゲルカラムク 口マトグラフィ一で精製し無色油状物 1.03 gを得た。得られた無色油状物 0.51 gをメタ ノール 13 mLに溶解し、 1規定水酸化ナトリウム水溶液 1.4 mLを氷冷下加え 30分間 攪拌した。 1規定塩酸 1.4 mLを氷冷下加え、減圧下濃縮した。得られた残渣を酢酸 ェチルで希釈し、飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥し て減圧下濃縮した。得られた残渣を酢酸ェチル -へキサン(1 : 3)を溶出溶媒とする シリカゲルカラムクロマトグラフィーで精製し、 tert-ブチル {(tert-ブトキシカルボ-ル) [ 2- (ヒドロキシメチル)ベンジル]アミノ}ァセタート 0.36 gを無色油状物として得た。 FP: 3 52
[0108] 参考例 39 2- (Bromomethyl) benzylacetate In a solution of 3.98 g of acetonitrile (80 mL), add 2.73 g of glycine tert-butyl ester monohydrochloride and 4.53 g of potassium carbonate under ice-cooling, and stir at room temperature for 12 hours. did. The reaction solution was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using ethyl acetate / n-hexane ( 1: 3 ) as an elution solvent to obtain 3.94 g of a colorless oil. 2.00 g of the obtained colorless oil was dissolved in 20 mL of 1,4-dioxane, and 1.49 g of di-tert-butyl dicarbonate was stirred at room temperature for 12 hours. The reaction solution was concentrated under reduced pressure. The obtained residue was diluted with ethyl acetate and washed with a 10% aqueous citrate solution and then with a saturated aqueous sodium bicarbonate solution. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 4) as an elution solvent to obtain 1.03 g of a colorless oil. 0.51 g of the obtained colorless oil was dissolved in 13 mL of methanol, and 1.4 mL of a 1N aqueous sodium hydroxide solution was added under ice-cooling and stirred for 30 minutes. 1N Hydrochloric acid (1.4 mL) was added under ice-cooling, and the mixture was concentrated under reduced pressure. The obtained residue was diluted with ethyl acetate and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using ethyl acetate-hexane (1: 3) as an elution solvent, and tert-butyl {(tert-butoxycarbol) [2- (hydroxymethyl) benzyl] amino } 0.36 g of acetate was obtained as a colorless oil. FP: 3 52 [0108] Reference Example 39
5-フルォロサリチル酸 1.77 gのテトラヒドロフラン(9.0 mL)溶液に氷冷下ボランーテ トラヒドロフラン錯体のテトラヒドロフラン溶液 (1.0 M) 22 mLを加え室温にて 19時間攪 拌した。反応液に水 3.0 mLと 1規定塩酸 3.0 mLを氷冷下にて順次カ卩え、室温にて 2 時間攪拌した。反応液を酢酸ェチルで抽出した。有機層を無水硫酸マグネシウムで 乾燥した後減圧下濃縮し、 4-フルォロ- 2- (ヒドロキシメチル)フエノール 1.60 gを褐色 油状物として得た。 NMR (DMSO-d ) : 4.45 (2H, s), 6.70-6.75 (1H, m), 6.81-6.87 (1 To a solution of 1.77 g of 5-fluorosalicylic acid in tetrahydrofuran (9.0 mL) was added 22 mL of a tetrahydrofuran solution (1.0 M) of a borane-tetrahydrofuran complex under ice-cooling, and the mixture was stirred at room temperature for 19 hours. Water (3.0 mL) and 1N hydrochloric acid (3.0 mL) were successively added to the reaction solution under ice-cooling, and the mixture was stirred at room temperature for 2 hours. The reaction solution was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain 1.60 g of 4-fluoro-2- (hydroxymethyl) phenol as a brown oil. NMR (DMSO-d): 4.45 (2H, s), 6.70-6.75 (1H, m), 6.81-6.87 (1
6 6
H, m), 7.03-7.07 (1H, m), 9.33 (1H, br s) H, m), 7.03-7.07 (1H, m), 9.33 (1H, br s)
[0109] 参考例 40 [0109] Reference Example 40
4-フルォロ- 2- (ヒドロキシメチル)フエノール 1.42 gを原料とし、参考例 14と同様の操 作を行い、 tert-ブチル [4-フルォロ- 2- (ヒドロキシメチル)フエノキシ]ァセタート 2.37 g を無色油状物として得た。 FP: 257 Using 1.42 g of 4-fluoro-2- (hydroxymethyl) phenol as the starting material, the same procedure as in Reference Example 14 was carried out, and 2.37 g of tert-butyl [4-fluoro-2- (hydroxymethyl) phenoxy] acetate was a colorless oil. Obtained as a thing. FP: 257
[0110] 参考例 41 [0110] Reference Example 41
3-ニトロサリチルアルデヒド 3.43 gのメタノール(100 mL)溶液に氷冷下水素化ホウ 素ナトリウム 1.55 gを加え 30分間攪拌し、その後室温にて 17時間攪拌した。反応液 を減圧下濃縮して得られた残渣を酢酸ェチルで希釈し、 1規定塩酸次!ヽで飽和食塩 水で洗浄した。有機層を無水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られた 残渣を Ν,Ν-ジメチルホルムアミド 100 mLに溶解し、イミダゾール 4.15 gと tert-ブチル ジメチルクロロシラン 9.18 gを室温にて順次カ卩ぇ 17時間攪拌した。反応液を水で希釈 し、酢酸ェチルで抽出した。有機層を水、飽和食塩水で順次洗浄した後、無水硫酸 マグネシウムで乾燥し減圧下濃縮した。得られた残渣を n-へキサン/酢酸ェチル (8 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製した。得られた黄色 油状物をエタノール 100 mLに溶解し、ラネーニッケルのエタノール懸濁物(3 mL)を 加え、 1気圧の水素雰囲気下室温にて 17時間攪拌した。反応液を濾過し、濾液を 減圧下濃縮した。得られた橙色油状物を 1,2-ジクロロェタン 50 mLに溶解し、酢酸 1.2 4 gと 37%ホルムアルデヒド水溶液 4.30 gを室温にてカ卩えた後、トリァセトキシ水素化ホ ゥ素ナトリウム 3.30 gを室温にて加え 1時間攪拌した。反応液を 10%重曹水で洗浄し、 無水硫酸マグネシウムで乾燥して減圧下濃縮した。得られた無色油状物を 1 ,4-ジォ
キサン 45 mLに溶解し、 1規定塩酸 45 mLをカ卩ぇ室温にて 2時間攪拌した。得られた 褐色油状物を原料とし、参考例 14と同様の操作を行い tert-ブチル [2- (ジメチルアミ ノ) -6- (ヒドロキシメチル)フエノキシ]ァセタート 1.68 gを黄色油状物として得た。 FP: 28 2 To a solution of 3-nitrosalicylaldehyde 3.43 g in methanol (100 mL) was added 1.55 g of sodium borohydride under ice-cooling, and the mixture was stirred for 30 minutes, and then stirred at room temperature for 17 hours. The residue obtained by concentrating the reaction solution under reduced pressure was diluted with ethyl acetate and washed with 1N hydrochloric acid and then with saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was dissolved in 100 mL of Ν, dimethylformamide, and 4.15 g of imidazole and 9.18 g of tert-butyldimethylchlorosilane were sequentially stirred at room temperature for 17 hours. The reaction solution was diluted with water and extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (8: 1) as an elution solvent. The obtained yellow oil was dissolved in 100 mL of ethanol, an ethanol suspension of Raney nickel (3 mL) was added, and the mixture was stirred at room temperature for 17 hours under a hydrogen atmosphere of 1 atm. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure. The obtained orange oil was dissolved in 50 mL of 1,2-dichloroethane, and 1.24 g of acetic acid and 4.30 g of 37% formaldehyde aqueous solution were prepared at room temperature, and then 3.30 g of sodium triacetoxyhydrogen hydride was brought to room temperature. And stirred for 1 hour. The reaction mixture was washed with 10% aqueous sodium bicarbonate, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The colorless oil obtained was converted to 1,4-diazo The product was dissolved in 45 mL of hexane, and 45 mL of 1N hydrochloric acid was stirred at room temperature for 2 hours. The obtained brown oil was used as a raw material, and the same operation as in Reference Example 14 was carried out to obtain 1.68 g of tert-butyl [2- (dimethylamino) -6- (hydroxymethyl) phenoxy] acetate as a yellow oil. FP: 28 2
[0111] 参考例 42 [0111] Reference Example 42
tert-ブチル [4-フルォロ- 2- (ヒドロキシメチル)フエノキシ]ァセタート 2.37 gをクロロホ ルム 50 mLに溶解し、二酸ィ匕マンガン 8.09 gをカ卩ぇ 60 °Cにて 24時間攪拌した。反応 液を濾過し、濾液を減圧下濃縮して tert-ブチル(4-フルォロ- 2-ホルミルフエノキシ) ァセタート 2.02 gを無色固体として得た。 2.37 g of tert-butyl [4-fluoro-2- (hydroxymethyl) phenoxy] acetate was dissolved in 50 mL of chloroform, and 8.09 g of manganese dioxide was stirred at 60 ° C. for 24 hours. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure to obtain 2.02 g of tert-butyl (4-fluoro-2-formylphenoxy) acetate as a colorless solid.
[0112] 参考例 43 [0112] Reference Example 43
tert-ブチル 2-ホルミル- 6-イソプロポキシ -4-ビュルフエノキシァセタートを原料とし 、参考例 17と同様の操作、次いで参考例 42と同様の操作を行い tert-ブチル [2-ホ ルミル- 4-(2-ヒドロキシェチル)- 6-イソプロポキシフエノキシ]ァセタートを得た。 FP: 3 11 Using tert-butyl 2-formyl-6-isopropoxy-4-buluphenoxyacetate as the starting material, the same procedure as in Reference Example 17 was performed, followed by the same procedure as in Reference Example 42. -4- (2-hydroxyethyl) -6-isopropoxyphenoxy] acetate was obtained. FP: 3 11
[0113] 参考例 44 [0113] Reference Example 44
3- (メチルスルファ -ル)サリチルアルデヒド 3.26 gのトルエン(150 mL)溶液にェチレ ングリコール 2.41 gと P-トルエンスルホン酸 370 mgをカ卩ぇ還流下 28時間攪拌した。反 応液を水、飽和食塩水で順次洗浄した後、無水硫酸マグネシウムで乾燥して減圧下 濃縮した。得られた残渣を原料として参考例 14と同様の操作を行い、 tert-ブチル [2 -(1,3-ジォキソラン- 2-ィル) -6- (メチルスルファ -ル)フエノキシ]ァセタート 2.09 gを黄 色油状物として得た。 EP: 349 [M+Na]+ Ethylene glycol (2.41 g) and P-toluenesulfonic acid (370 mg) were stirred in a solution of 3- (methylsulfuryl) salicylaldehyde (3.26 g) in toluene (150 mL) for 28 hours under reflux. The reaction solution was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. Using the obtained residue as a raw material, the same operation as in Reference Example 14 was carried out, and 2.09 g of tert-butyl [2- (1,3-dioxolan-2-yl) -6- (methylsulfuryl) phenoxy] acetate was Obtained as a colored oil. EP: 349 [M + Na] +
[0114] 参考例 45 [0114] Reference Example 45
tert-ブチル [2- (1,3-ジォキソラン- 2-ィル) -6- (メチルスルファ -ル)フエノキシ]ァセタ ート 1.15 gのクロ口ホルム(25 mL)溶液にォキソン(登録商標) 6.48 gとアルミナ 3 gを 室温にて加え、還流下 5時間攪拌した。反応液を濾過し、濾液を減圧下濃縮して tert -ブチル [2-(1, 3-ジォキソラン- 2-ィル) -6- (メチルスルホ -ル)フエノキシ]ァセタート 1. 16 gを黄色固体として得た。 EP: 381 [M+Na]+ tert-Butyl [2- (1,3-Dioxolan-2-yl) -6- (methylsulfuryl) phenoxy] acetate 1.48 g of OXON® 6.48 g in a 25 ml solution And 3 g of alumina were added at room temperature and stirred for 5 hours under reflux. The reaction mixture was filtered, and the filtrate was concentrated under reduced pressure to give tert-butyl [2- (1,3-dioxolan-2-yl) -6- (methylsulfol) phenoxy] acetate 1.16 g as a yellow solid. Obtained. EP: 381 [M + Na] +
[0115] 参考例 46
tert-ブチル [2- (1,3-ジォキソラン- 2-ィル) -6- (メチルスルファ -ル)フエノキシ]ァセタ ート 567 mgを 80%酢酸水 10 mLに溶解し還流下 20時間攪拌した。反応液を減圧下濃 縮し、 [2-ホルミル- 6- (メチルスルファ -ル)フエノキシ]ァセチックアシッド 259 mgを黄 色固体として得た。 [0115] Reference Example 46 567 mg of tert-butyl [2- (1,3-dioxolan-2-yl) -6- (methylsulfuryl) phenoxy] acetate was dissolved in 10 mL of 80% aqueous acetic acid and stirred for 20 hours under reflux. The reaction solution was concentrated under reduced pressure to obtain 259 mg of [2-formyl-6- (methylsulfuryl) phenoxy] acetic acid as a yellow solid.
[0116] 参考例 47 [0116] Reference Example 47
tert-ブチル(2-ホルミル- 6-ヒドロキシフエノキシ)ァセタート 250 mgをテトラヒドロフラ ン 5 mLに溶解し、室温にて 1-ジメチルァミノ- 2-プロパノール 204 mg、トリフエ-ルホス フィン 312 mg及びジイソプロピルァゾジカルボキシラート 0.23 mLをカ卩ぇ終夜攪拌した 。反応液を減圧下濃縮し、得られた残渣を n_へキサン/酢酸ェチル(1: 1)、その後、ク ロロホルム/メタノール (9 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーによ り精製し、 tert-ブチル {2- [2- (ジメチルァミノ)プロポキシ ]-6-ホルミルフエノキシ }ァセタ ート 80 mgを無色固体として得た。 FP: 338 Dissolve 250 mg of tert-butyl (2-formyl-6-hydroxyphenoxy) acetate in 5 mL of tetrahydrofuran and dissolve at room temperature with 1-dimethylamino-2-propanol 204 mg, triphenylphosphine 312 mg and diisopropylazo Dicarboxylate 0.23 mL was stirred overnight. The reaction solution was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n_hexane / ethyl acetate (1: 1) and then chloroform / methanol (9: 1) as an elution solvent. As a result, 80 mg of tert-butyl {2- [2- (dimethylamino) propoxy] -6-formylphenoxy} acetate was obtained as a colorless solid. FP: 338
[0117] 参考例 48 [0117] Reference Example 48
塩化ニッケル六水和物 607 mgのメタノール(50 mL)溶液に水素化ホウ素ナトリウム 300 mgをカ卩ぇ室温にて 30分攪拌した溶液に、 tert-ブチル(2-ホルミル- 6-メトキシ -4- ニトロフエノキシ)ァセタート 1.59 gのメタノール (30 mL)溶液に氷冷下水素化ホウ素 ナトリウム 193 mgを加え 5分間攪拌した。反応液に水素化ホウ素ナトリウム 950 mgを 加え、室温にて 1時間攪拌した。反応液をセライト濾過後、減圧下濃縮した。残渣を 酢酸ェチルで希釈し、飽和塩ィ匕アンモニア水次いで飽和食塩水で順次洗浄した。有 機層を無水硫酸マグネシウムで乾燥し、減圧下濃縮し tert-ブチル [4-ァミノ- 2- (ヒド 口キシメチル) -6-メトキシフエノキシ]ァセタート 1.36 gを淡黄色固体として得た。 EP: 2 84 Nickel chloride hexahydrate A solution of 607 mg of methanol (50 mL) in sodium borohydride (300 mg) stirred at room temperature for 30 minutes was mixed with tert-butyl (2-formyl-6-methoxy-4- To a solution of 1.59 g of nitrophenoxy) acetate in methanol (30 mL) was added 193 mg of sodium borohydride under ice cooling, and the mixture was stirred for 5 minutes. To the reaction solution, 950 mg of sodium borohydride was added and stirred at room temperature for 1 hour. The reaction mixture was filtered through celite and concentrated under reduced pressure. The residue was diluted with ethyl acetate and washed successively with saturated saline and aqueous ammonia and then saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain 1.36 g of tert-butyl [4-amino-2- (hydroxymethyl) -6-methoxyphenoxy] acetate as a pale yellow solid. EP: 2 84
[0118] 参考例 49 [0118] Reference Example 49
tert-ブチル [4-ァミノ- 2- (ヒドロキシメチル) -6-メトキシフエノキシ]ァセタート 1.36 gの エタノール(5 mL)溶液に 1H-ベンゾトリアゾール -1-メタノール 714 mgを室温にて加 え 2時間攪拌した。反応液に室温にて水素化ホウ素ナトリウムを 455 mgカ卩ぇ 30分間 攪拌した。反応液を減圧下濃縮した。得られた残渣を酢酸ェチルで希釈し、飽和塩 化アンモニゥム水、飽和重曹水次いで飽和食塩水で順次洗浄した。有機層を無水
硫酸マグネシウムで乾燥し、減圧下濃縮後、残渣をクロ口ホルム/メタノール(97 : 3)を 溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製して tert-ブチル [2- (ヒドロ キシメチル) -6-メトキシ- 4- (メチルァミノ)フエノキシ]ァセタート 1.12 gを黄色固体として 得た。 EP: 298 tert-Butyl [4-amino-2- (hydroxymethyl) -6-methoxyphenoxy] acetate Add 714 mg of 1H-benzotriazole-1-methanol at room temperature to a solution of 1.36 g of ethanol (5 mL) 2 Stir for hours. The reaction solution was stirred at room temperature for 30 minutes with 455 mg of sodium borohydride. The reaction solution was concentrated under reduced pressure. The obtained residue was diluted with ethyl acetate and washed successively with saturated aqueous ammonium chloride, saturated aqueous sodium bicarbonate and then saturated brine. Organic layer anhydrous After drying over magnesium sulfate and concentrating under reduced pressure, the residue was purified by silica gel column chromatography using chloroform-form / methanol (97: 3) as an elution solvent, and tert-butyl [2- (hydroxymethyl) -6-methoxy -1.12 g of 4- (methylamino) phenoxy] acetate was obtained as a yellow solid. EP: 298
[0119] 参考例 50 [0119] Reference Example 50
tert-ブチル [2- (ヒドロキシメチル) -6-メトキシ- 4- (メチルァミノ)フエノキシ]ァセタート 450 mgの 2-プロパノール(5 mL)溶液にジ -tert-ブチルジカルボナート 330 mgを室 温にて加え 6時間攪拌した。反応液を減圧下濃縮し、得られた残渣を原料として参 考例 42と同様の操作を行 、tert-ブチル {4-[(tert-ブトキシカルボ-ル) (メチル)ァミノ] -2-ホルミル- 6-メトキシフエノキシ }ァセタート 560 mgを無色固体として得た。 EP: 418 [ M+Na]+ tert-Butyl [2- (hydroxymethyl) -6-methoxy-4- (methylamino) phenoxy] acetate 450 mg of 2-propanol (5 mL) solution with 330 mg of di-tert-butyl dicarbonate at room temperature The mixture was stirred for 6 hours. The reaction mixture was concentrated under reduced pressure, and the obtained residue was used as a starting material. The same procedure as in Reference Example 42 was carried out to obtain tert-butyl {4-[(tert-butoxycarbol) (methyl) amino] -2-formyl. 560 mg of 6-methoxyphenoxy} acetate was obtained as a colorless solid. EP: 418 [M + Na] +
[0120] 参考例 51 [0120] Reference Example 51
tert-ブチル [2- (ヒドロキシメチル) -6-メトキシ- 4- (メチルァミノ)フエノキシ]ァセタート 670 mgのァセトニトリル (5 mL)溶液に、 37%ホルムアルデヒド水溶液 0.7 mLとトリァセ トキシ水素化ホウ素ナトリウム 958 mgを室温にてカ卩ぇ 5分間攪拌した。反応液を 10% 重曹水で洗浄し、無水硫酸マグネシウムで乾燥して減圧下濃縮した。得られた残渣 を原料として参考例 42と同様の操作を行 、得られた残渣を n-へキサン/酢酸ェチル (3 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製して tert-ブチル [4 - (ジメチルァミノ) -2-ホルミル- 6-メトキシフエノキシ]ァセタート 117 mgを黄色固体とし て得た。 EP: 310 tert-Butyl [2- (hydroxymethyl) -6-methoxy-4- (methylamino) phenoxy] acetate A solution of 670 mg of acetonitrile (5 mL), 37% formaldehyde aqueous solution 0.7 mL and sodium triacetoxyborohydride 958 mg Stir at room temperature for 5 minutes. The reaction mixture was washed with 10% aqueous sodium bicarbonate, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was used as a starting material and the same procedure as in Reference Example 42 was performed. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (3: 1) as an elution solvent, and tert-butyl 117 mg of [4- (dimethylamino) -2-formyl-6-methoxyphenoxy] acetate was obtained as a yellow solid. EP: 310
[0121] 参考例 52 [0121] Reference Example 52
tert-ブチル [4-ァミノ- 2- (ヒドロキシメチル) -6-メトキシフエノキシ]ァセタート 480 mg の Ν,Ν-ジメチルホルムアミド(1 mL)溶液に、イミダゾール 230 mgと tert-ブチルジメチ ルクロロシラン 281 mgを室温にて順次カ卩ぇ 20分間攪拌した。反応液をジェチルエー テル-酢酸ェチル (1 : 1)で希釈し、有機層を 5%クェン酸水次いで飽和食塩水で順次 洗浄した後、無水硫酸マグネシウムで乾燥し減圧下濃縮した。得られた残渣をピリジ ン 0.5 mLに溶解し、無水酢酸 0.08 mLを室温にて加え 10分間攪拌した。反応液を減 圧下濃縮した。得られた残渣をテトラヒドロフラン 3 mLに溶解し、フッ化テトラ n-ブチル
アンモ-ゥム 555 mgをカ卩え、室温にて 30分間攪拌した。反応液を酢酸ェチルで希釈 し、飽和塩ィ匕アンモニア水次いで飽和食塩水で順次洗浄した後、無水硫酸マグネシ ゥムで乾燥し減圧下濃縮した。得られた無色固体を原料として参考例 42と同様の操 作を行い、 tert-ブチル [4- (ァセチルァミノ) -2-ホルミル- 6-メトキシフエノキシ]ァセタ ート 300 mgを黄色固体として得た。 EP: 346 [M+Na]+ tert-Butyl [4-amino-2- (hydroxymethyl) -6-methoxyphenoxy] acetate 480 mg in Ν, Ν-dimethylformamide (1 mL) solution, imidazole 230 mg and tert-butyldimethylchlorosilane 281 mg Were sequentially stirred at room temperature for 20 minutes. The reaction mixture was diluted with jetyl ether-ethyl acetate (1: 1), and the organic layer was washed successively with 5% aqueous citrate and then with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was dissolved in 0.5 mL of pyridine, 0.08 mL of acetic anhydride was added at room temperature, and the mixture was stirred for 10 minutes. The reaction solution was concentrated under reduced pressure. Dissolve the resulting residue in 3 mL of tetrahydrofuran, and add tetra n-butyl fluoride. Ammonium 555 mg was added and stirred at room temperature for 30 minutes. The reaction mixture was diluted with ethyl acetate, washed successively with saturated brine, aqueous ammonia and then saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained colorless solid was used as a starting material and the same procedure as in Reference Example 42 was carried out to obtain 300 mg of tert-butyl [4- (acetylylamino) -2-formyl-6-methoxyphenoxy] acetate as a yellow solid. It was. EP: 346 [M + Na] +
[0122] 参考例 53 [0122] Reference Example 53
3-エトキシサリチルアルデヒド 3.50 gのエタノール (15 mL)溶液に、ジメチルァミンの テトラヒドロフラン溶液(2 M) 15.8 mLと 37%ホルムアルデヒド水溶液 2.63 mLを室温に て加え、 21時間還流下攪拌した。ジメチルァミンのテトラヒドロフラン溶液 (2 M) 5.3 m Lと 37%ホルムアルデヒド水溶液 0.5 mLを室温にてカ卩え、 6時間還流下攪拌した。反 応液を減圧下濃縮した。残渣にジェチルエーテルを加えて生じた析出物を濾取し、 濾物を減圧下乾燥し、淡黄色固体 4.72gを得た。得られた淡黄色固体 3.00 gを原料と し、参考例 14と同様の操作を行い、 tert-ブチル {4- [(ジメチルァミノ)メチル ]-2-ェトキ シ- 6-ホルミルフエノキシ }ァセタート 1.12 gを淡黄色固体として得た。 To a solution of 3-ethoxysalicylaldehyde 3.50 g in ethanol (15 mL) were added 15.8 mL of a dimethylamine tetrahydrofuran solution (2 M) and 2.63 mL of 37% formaldehyde aqueous solution at room temperature, and the mixture was stirred under reflux for 21 hours. Dimethylamine in tetrahydrofuran (2 M) (5.3 mL) and 37% formaldehyde aqueous solution (0.5 mL) were placed at room temperature and stirred under reflux for 6 hours. The reaction solution was concentrated under reduced pressure. Jetyl ether was added to the residue, and the resulting precipitate was collected by filtration, and the filtrate was dried under reduced pressure to obtain 4.72 g of a pale yellow solid. Using 3.00 g of the obtained pale yellow solid as a starting material, the same procedure as in Reference Example 14 was performed to obtain tert-butyl {4-[(dimethylamino) methyl] -2-ethoxy-6-formylphenoxy} acetate 1.12 g was obtained as a pale yellow solid.
[0123] 参考例 54 [0123] Reference Example 54
氷冷撹拌下、 tert-ブチル 2-エトキシ- 6-ホルミル- 4-ヒドロキシメチルフエノキシァセ タート 80 mgおよびトリェチルァミン 0.043 mLの塩化メチレン(1.0 mL)溶液にメタンス ルホニルクロリド 0.024 mLをカ卩え、同条件下 1.3時間撹拌した。反応混合物に 10%塩 化アンモ-ゥム水溶液をカ卩え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄 し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下に留去して、無色油状物を得た 。室温撹拌下、水素化ナトリウム 11 mgのテトラヒドロフラン懸濁液(0.40 mL)にジ -ter t-ブチルイミノカルボキシレート 62 mgをカ卩え、同温で 15分撹拌した。そこへ、先に得 られた油状物のテトラヒドロフラン(0.60 mL)溶液を加え、同温で 1.5時間撹拌した。 反応混合物に水を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、無 水硫酸マグネシウムで乾燥した。溶媒を減圧下に留去し、残渣を n-へキサン/酢酸ェ チル(8:2)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製して、 tert-ブ チル 4- [(ジ -tert-ブトキシカルボニル)アミノメチル]- 2-エトキシ- 6-ホルミルフエノキシ ァセタート 83 mgを無色油状物として得た。 EP: 532 [M+Na]+
[0124] 参考例 55 Under ice-cooling, add 0.024 mL of methanesulfonyl chloride to a solution of 80 mg of tert-butyl 2-ethoxy-6-formyl-4-hydroxymethylphenoxyacetate and 0.043 mL of triethylamine in 1.04 mL of methylene chloride. The mixture was stirred for 1.3 hours. A 10% aqueous ammonium chloride solution was added to the reaction mixture and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to obtain a colorless oil. Under stirring at room temperature, 62 mg of di-ter t-butyliminocarboxylate was added to a tetrahydrofuran suspension (0.40 mL) of 11 mg of sodium hydride and stirred at the same temperature for 15 minutes. Thereto was added a solution of the oil obtained earlier in tetrahydrofuran (0.60 mL), and the mixture was stirred at the same temperature for 1.5 hours. Water was added to the reaction mixture and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (8: 2) as an elution solvent, and tert-butyl 4-[(di-tert- 83 mg of butoxycarbonyl) aminomethyl] -2-ethoxy-6-formylphenoxyacetate was obtained as a colorless oil. EP: 532 [M + Na] + [0124] Reference Example 55
2-ブロモベンズアルデヒド 6.47 g、塩化テトラ n-ブチルアンモ -ゥム 2.92 g、炭酸力 リウム 12.1 g、酢酸パラジウム 393 mg及びアクリル酸 tert-ブチル 22.4 gの Ν,Ν-ジメチ ルホルムアミド(5 mL)溶液を 100 °Cにて終夜攪拌した。反応液に酢酸ェチル及び水 を加え、酢酸ェチルで抽出した。有機層を水及び飽和食塩水で順次洗浄した後、無 水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られた残渣を n-へキサン/酢酸ェ チル(9 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し、無色油 状物を得た。得られた無色油状物をメチレンクロライド及びエタノールの混合溶媒(1 : 1、 40 mL)に溶解し、この溶液に- 78 °Cにてオゾンガスを 1.5時間吹き込み撹拌した 。室温にて反応液にチォ尿素 355 mgを加え 2時間撹拌した後、不溶物を濾別した。 濾液を減圧下濃縮し、得られた残渣を n-へキサン/酢酸ェチル(9 : 1)を溶出溶媒とす るシリカゲルカラムクロマトグラフィーにより精製し、 tert-ブチル 3-(2—ホルミルフエ- ル)プロパノアート 1.30 gを無色油状物として得た。 NMR (DMSO-d ) : 1.34 (9H, s), 2. A solution of 2-bromobenzaldehyde 6.47 g, tetra n-butylammonium chloride 2.92 g, potassium carbonate 12.1 g, palladium acetate 393 mg and tert-butyl acrylate 22.4 g in Ν, Ν-dimethylformamide (5 mL) The mixture was stirred at 100 ° C overnight. Ethyl acetate and water were added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (9: 1) as an elution solvent to obtain a colorless oil. The obtained colorless oil was dissolved in a mixed solvent of methylene chloride and ethanol (1: 1, 40 mL), and ozone gas was blown into the solution at −78 ° C. for 1.5 hours, followed by stirring. After adding 355 mg of thiourea to the reaction solution at room temperature and stirring for 2 hours, insoluble matters were filtered off. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (9: 1) as an elution solvent, and tert-butyl 3- (2-formylphenol) was obtained. Propanoate 1.30 g was obtained as a colorless oil. NMR (DMSO-d): 1.34 (9H, s), 2.
6 6
51(2H, t, J = 7.5 Hz), 3.23(2H, t, J = 7.5 Hz), 7.39(1H, d, J = 7.5 Hz), 7.42-7.50 (1 H, m), 7.55-7.64 (1H, m), 7.86 (1H, dd, J = 1.4, 7.5 Hz), 10.22 (1H, s) 51 (2H, t, J = 7.5 Hz), 3.23 (2H, t, J = 7.5 Hz), 7.39 (1H, d, J = 7.5 Hz), 7.42-7.50 (1 H, m), 7.55-7.64 ( 1H, m), 7.86 (1H, dd, J = 1.4, 7.5 Hz), 10.22 (1H, s)
[0125] 参考例 56 [0125] Reference Example 56
3-メトキシ- 2-二トロべンズアルデヒド 15.0 g、エタンジオール 9.3 mL、 p—トルエンス ルホン酸 315 mgのベンゼン(600 mL)溶液を脱水条件下で 2日間加熱還流した。反 応液を室温に冷却後、反応液に重曹水を加えた後、酢酸ェチルで抽出した。有機層 を飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、減圧下濃縮して灰色 油状物 18.6 gを得た。得られた灰色油状物 10.0 gをメタノール 200 mLに溶解し、 10% パラジウム炭素粉末 1 gを加え、 1気圧の水素雰囲気下室温にて 2時間攪拌した。反 応液を濾過し、濾液を減圧下濃縮して灰色油状物を得た。得られた灰色油状物を塩 化メチレン 100 mLに溶解し、氷冷下ピリジン 4.3 mL及び無水トリフルォロ酢酸 6.3 mL を順次加え、 1時間撹拌した。反応液に水を加えた後、クロ口ホルムで抽出した。有 機層を無水硫酸マグネシウムで乾燥し、減圧下濃縮して赤色固体 12 gを得た。得ら れた赤色固体 2.00 gを 2-ブタノン 20 mLに溶解し、室温にて炭酸カリウム 4.78 g及びョ ゥ化メチル 1.95 gを加えた後、 2時間加熱還流した。反応液に酢酸ェチル及び水を加
え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄した後、無水硫酸マグネシ ゥムで乾燥し、減圧下濃縮した。得られた残渣を n-へキサン/酢酸ェチル (4: 1)を溶 出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し、黄色固体 1.02 g得た。 得られた黄色固体 970 mgをエタノール 10 mLに溶解し、室温にてナトリウムエトキシド 325 mgを加え、 80 °Cにて終夜撹拌した。反応液を減圧下濃縮した後、残渣に酢酸 ェチル及び水をカ卩え、酢酸ェチルで抽出した。有機層を無水硫酸マグネシウムで乾 燥し、減圧下濃縮して茶色固体 668 mgを得た。得られた茶色固体 300 mg、ブロモ酢 酸 tert-ブチル 308 mg、炭酸カリウム 297 mg及びテトラプチルアンモ -ゥム硫酸水素 塩 24 mgを 2-ブタノン及び水の混合溶媒 (1 : 1, 10 mL)に混合し終夜加熱還流した。 反応液を酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄した後、無水硫酸マグ ネシゥムで乾燥し、減圧下濃縮した。得られた残渣を n-へキサン/酢酸ェチル (9 : 1) を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し、黄色油状物 417 m g得た。得られた黄色油状物 410 mgにテトラヒドロフラン 10 mL及び 1規定塩酸 10 mL を加え、終夜撹拌した。反応液を酢酸ェチルで希釈し、 1規定水酸化ナトリウム水溶 液 8 mLをカ卩えた。反応液に重曹水をカ卩えてアルカリ性とした後、反応液を酢酸ェチ ルで抽出した。有機層を飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し 、減圧下濃縮して tert-ブチル [(2—ホルミル- 6—メトキシフエ-ル) (メチル)ァミノ]ァセ タート 330 mgを黄色油状物として得た。 FP: 280 A solution of 3-methoxy-2-nitrobenzenealdehyde (15.0 g), ethanediol (9.3 mL) and p-toluenesulfonic acid (315 mg) in benzene (600 mL) was heated to reflux for 2 days under dehydrating conditions. The reaction solution was cooled to room temperature, sodium bicarbonate water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to give 18.6 g of a gray oil. 10.0 g of the obtained gray oily substance was dissolved in 200 mL of methanol, 1 g of 10% palladium carbon powder was added, and the mixture was stirred at room temperature for 2 hours under a hydrogen atmosphere of 1 atm. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure to obtain a gray oil. The obtained gray oil was dissolved in 100 mL of methylene chloride, and 4.3 mL of pyridine and 6.3 mL of trifluoroacetic anhydride were sequentially added under ice-cooling, followed by stirring for 1 hour. Water was added to the reaction solution, followed by extraction with black mouth form. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain 12 g of a red solid. The obtained red solid (2.00 g) was dissolved in 2-butanone (20 mL), potassium carbonate (4.88 g) and methyl iodide (1.95 g) were added at room temperature, and the mixture was heated to reflux for 2 hours. Add ethyl acetate and water to the reaction solution. And extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as an eluent to obtain 1.02 g of a yellow solid. 970 mg of the obtained yellow solid was dissolved in 10 mL of ethanol, 325 mg of sodium ethoxide was added at room temperature, and the mixture was stirred at 80 ° C. overnight. After the reaction solution was concentrated under reduced pressure, ethyl acetate and water were added to the residue, and the residue was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain 668 mg of a brown solid. 300 mg of the obtained brown solid, tert-butyl bromoacetate 308 mg, potassium carbonate 297 mg, and tetraptyl ammonium hydrogen sulfate 24 mg were mixed with 2-butanone and water (1: 1, 10 mL) And heated to reflux overnight. The reaction solution was extracted with ethyl acetate, and the organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (9: 1) as an elution solvent to obtain 417 mg of a yellow oily substance. To 410 mg of the obtained yellow oil, 10 mL of tetrahydrofuran and 10 mL of 1N hydrochloric acid were added and stirred overnight. The reaction solution was diluted with ethyl acetate, and 8 mL of 1N aqueous sodium hydroxide solution was added. After adding sodium bicarbonate water to the reaction solution to make it alkaline, the reaction solution was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to give 330 mg of tert-butyl [(2-formyl-6-methoxyphenyl) (methyl) amino] acetate in yellow. Obtained as an oil. FP: 280
[0126] 参考例 57 [0126] Reference Example 57
2- (1,3-ジォキソラン- 2-ィル)ァ-リン 1.68 gとピリジン 1.61 gのジクロロェタン(50 mL )溶液に無水トリフルォロ酢酸 3.21 gを氷冷下加え 40分間攪拌した。反応液を 5%重 曹水、 5%クェン酸水溶液次いで飽和食塩水で順次洗浄し、無水硫酸マグネシウムで 乾燥後減圧下濃縮して無色油状物 2.52 gを得た。得られた無色油状物 1.26 g、炭酸 カリウム 1.33 g、ヨウ化メチル 2.05 g及び 2-ブタノン 50 mLの混合物を 60 °Cにて 6時間 撹拌した。反応液を水次いで飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾 燥後減圧下濃縮し、 N- [2- (1,3-ジォキソラン- 2-ィル)フエ-ル]- 2,2,2-トリフルォロ- N -メチルァセトアミド 1.32 gを得た。 FP: 276 To a solution of 1.68 g of 2- (1,3-dioxolan-2-yl) aline and 1.61 g of pyridine in dichloroethane (50 mL) was added 3.21 g of trifluoroacetic anhydride under ice cooling, and the mixture was stirred for 40 minutes. The reaction mixture was washed successively with 5% aqueous sodium bicarbonate, 5% aqueous citrate solution and saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give 2.52 g of a colorless oil. A mixture of the obtained colorless oily substance 1.26 g, potassium carbonate 1.33 g, methyl iodide 2.05 g and 2-butanone 50 mL was stirred at 60 ° C. for 6 hours. The reaction mixture was washed successively with water and then with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to give N- [2- (1,3-dioxolan-2-yl) phenol] -2,2 Thus, 1.32 g of 2-trifluoro-N-methylacetamide was obtained. FP: 276
[0127] 参考例 58
N- [2- (1,3-ジォキソラン- 2-ィル)フエ-ル]- 2,2,2-トリフルォロ- N-メチルァセトアミド 1.31 g、炭酸カリウム 3.30 g、水 5.0 mL及びメタノール 50 mLの混合物を 60 °Cにて 9 時間撹拌した。反応液に水を加え、酢酸ェチルで抽出した。有機層を水次いで飽和 食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥後減圧下濃縮し、黄色油状物 0. 63 gを得た。得られた黄色油状物 0.62 g、 50%ポリマー型ダリオキシル酸ェチルのトル ェン溶液 5.65 g及び酢酸 17 mLの混合物にトリァセトキシ水素化ホウ素ナトリウム 1.06 gを室温にて加え 14時間撹拌した。反応液に水を加え、酢酸ェチルで抽出した。有 機層を水次 、で飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥して減圧 下濃縮した。得られた残渣を酢酸ェチル -へキサン(1 : 3)を溶出溶媒とするシリカ ゲルカラムクロマトグラフィーで精製しェチル [(2-ホルミルフエ-ル) (メチル)ァミノ]ァ セタート 0.23 gを黄色油状物として得た。 FP: 222 [0127] Reference Example 58 N- [2- (1,3-Dioxolan-2-yl) phenol] -2,2,2-trifluoro-N-methylacetamide 1.31 g, potassium carbonate 3.30 g, water 5.0 mL and methanol 50 The mixture of mL was stirred at 60 ° C for 9 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water and then with saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give 0.63 g of a yellow oil. To a mixture of 0.62 g of the obtained yellow oil, 5.65 g of a 50% polymer-type ethyl dalioxylate toluene solution and 17 mL of acetic acid, 1.06 g of sodium triacetoxyborohydride was added at room temperature and stirred for 14 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using ethyl acetate-hexane (1: 3) as an elution solvent, and 0.23 g of ethyl [(2-formylphenol) (methyl) amino] acetate was obtained as a yellow oil. Got as. FP: 222
[0128] 参考例 59 [0128] Reference Example 59
4-ァミノべンズアミジン二塩酸塩 37.9 gを 1,4-ジォキサン 180 mLに懸濁し、 4規定 水酸化ナトリウム水溶液 364 mLを氷冷下加え 30分間攪拌した。ジ -tert-ブチルジカ ルボナート 39.8 g及び 4規定水酸化ナトリウム水溶液 46 mLを氷冷下順次加え 7時 間攪拌した。反応液を酢酸ェチルで抽出し、飽和食塩水で洗浄した。有機層を無水 硫酸マグネシウムで乾燥して減圧下濃縮した。析出物を濾取し、 2-プロパノール 100 mLに懸濁させ室温にて 30分間攪拌した。析出物を濾取し、濾物を減圧下乾燥して t ert-ブチル [(4-ァミノフエ-ル) (ィミノ)メチル]力ルバマート 38.0 gを無色固体として得 た。 FP: 236 4-Aminobenzamidine dihydrochloride (37.9 g) was suspended in 1,4-dioxane (180 mL), 4N aqueous sodium hydroxide solution (364 mL) was added with ice cooling, and the mixture was stirred for 30 min. Di-tert-butyl dicarbonate 39.8 g and 4N aqueous sodium hydroxide solution 46 mL were sequentially added under ice-cooling and stirred for 7 hours. The reaction mixture was extracted with ethyl acetate and washed with saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The precipitate was collected by filtration, suspended in 100 mL of 2-propanol, and stirred at room temperature for 30 minutes. The precipitate was collected by filtration, and the filtrate was dried under reduced pressure to obtain 38.0 g of tert-butyl [(4-aminophenol) (imino) methyl] force rubamate as a colorless solid. FP: 236
[0129] 参考例 60 [0129] Reference Example 60
5-クロ口- 2--トロべンズアルデヒド 394 mgと tert-ブチル [(4-ァミノフエ-ル) (イミノ)メ チル]力ルバマート 500 mgの酢酸(10 mL)溶液にトリァセトキシ水素化ホウ素ナトリウ ム 901 mgを室温にて加え 2時間攪拌した。反応液を減圧下留去した後、残渣に酢酸 ェチル及び水を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄した後、 無水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られた残渣を n-へキサン/酢酸 ェチル (4: 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し、黄色 泡状物 475 mgを得た。得られた黄色泡状物をエタノール 10 mLに溶解し、ラネー-ッ
ケルの水懸濁物(3 g)を加え、 1気圧の水素雰囲気下室温にて 30分攪拌した。反応 液を濾過し、濾液を減圧下濃縮して tert-ブチル [{4-[(2-ァミノ- 5-クロ口ベンジル)アミ ノ]フエ-ル }(ィミノ)メチル]力ルバマート 365 mgを得た。 FP: 375 5-black mouth 2-Trobensaldehyde 394 mg and tert-butyl [(4-aminophenol) (imino) methyl] strength rubamate 500 mg acetic acid (10 mL) in acetic acid (10 mL) solution with sodium triacetoxyborohydride 901 mg was added at room temperature and stirred for 2 hours. After the reaction solution was distilled off under reduced pressure, ethyl acetate and water were added to the residue, followed by extraction with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (4: 1) as an elution solvent to obtain 475 mg of a yellow foam. Dissolve the resulting yellow foam in 10 mL of ethanol and add Raney A water suspension (3 g) of Kell was added, and the mixture was stirred at room temperature for 30 minutes under a hydrogen atmosphere of 1 atm. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure to obtain 365 mg of tert-butyl [{4-[(2-amino-5-chlorobenzyl) amino] phenol} (imino) methyl] force rubamate. It was. FP: 375
[0130] 参考例 61 [0130] Reference Example 61
tert-ブチル(2-ホルミルフエノキシ)ァセタート 1.47 gと 5-クロ口アントラ-ル酸 1.07 g の酢酸(6.0 mL)溶液にトリァセトキシ水素化ホウ素ナトリウム 2.64 gを室温でカ卩ぇ 1.5 時間攪拌した。反応液に水を加え生じた析出物を濾取し、濾物を減圧下乾燥して 2- [2- (2-tert-ブトキシ -2-ォキソエトキシ)ベンジル]アミノ}-5-クロ口安息香酸 2.23 gを黄 色固体として得た。 To a solution of 1.47 g of tert-butyl (2-formylphenoxy) acetate and 1.07 g of 5-chloroanthralic acid in acetic acid (6.0 mL) was stirred 2.64 g of sodium triacetoxyborohydride at room temperature for 1.5 hours. . Water was added to the reaction mixture, and the resulting precipitate was collected by filtration, and the filtrate was dried under reduced pressure to give 2- [2- (2-tert-butoxy-2-oxoethoxy) benzyl] amino} -5-chlorobenzoic acid. 2.23 g was obtained as a yellow solid.
[0131] 参考例 62 [0131] Reference Example 62
4-ァミノべンズアミジン二塩酸塩 49.5 gと 1,4-ジォキサン 240 mLの混合物に、氷冷 下 2規定水酸化ナトリウム水溶液 240 mLをカ卩ぇ 10分間攪拌した。ェチルクロ口ホル マート 27.1 gを氷冷下加え 30分間攪拌した。反応液を酢酸ェチルで抽出し、抽出液 を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、減圧下濃縮した。得られた残 渣をシリカゲルカラムクロマトグラフィーにて精製し、ェチル [(4-ァミノフエニル) (ィミノ) メチル]力ルバマート 19.0 gを無色固体として得た。 EP: 208 To a mixture of 49.5 g of 4-aminobenzamidine dihydrochloride and 240 mL of 1,4-dioxane, 240 mL of 2N aqueous sodium hydroxide solution was stirred for 10 minutes under ice cooling. Ethyl black mouth formate 27.1 g was added under ice cooling and stirred for 30 minutes. The reaction solution was extracted with ethyl acetate, and the extract was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to obtain 19.0 g of ethyl [(4-aminophenyl) (imino) methyl] force rubamate as a colorless solid. EP: 208
[0132] 参考例 63 [0132] Reference Example 63
(4-ヒドロキシ- 3-二トロフエ-ル)ァセチックアシッド 2.31 gに塩化水素のエタノール 溶液 (25.1wt%) 23 mLを室温にて加え、同温度にて 3時間攪拌した。反応液を減圧 下濃縮し、ェチル(4-ヒドロキシ -3--トロフエニル)ァセタート 2.61 gを黄色油状物とし て得た。 NMR (CDC1 ) : 1.27 (3H, t, J = 7.3 Hz), 3.61 (2H, s), 4.17 (2H, q, J = 7.3 To 2.31 g of (4-hydroxy-3-nitrophenyl) acetic acid, 23 mL of an ethanol solution of hydrogen chloride (25.1 wt%) was added at room temperature, and the mixture was stirred at the same temperature for 3 hours. The reaction mixture was concentrated under reduced pressure to obtain 2.61 g of ethyl (4-hydroxy-3-trophenyl) acetate as a yellow oil. NMR (CDC1): 1.27 (3H, t, J = 7.3 Hz), 3.61 (2H, s), 4.17 (2H, q, J = 7.3
3 Three
Hz), 7.14 (1H, d, J = 8.8 Hz), 7.53 (1H, dd, J = 8.8, 2.2 Hz), 8.03 (1H, d, J = 2.2 H z), 10.54 (1H, s) Hz), 7.14 (1H, d, J = 8.8 Hz), 7.53 (1H, dd, J = 8.8, 2.2 Hz), 8.03 (1H, d, J = 2.2 H z), 10.54 (1H, s)
[0133] 参考例 64 [0133] Reference Example 64
氷冷撹拌下、ェチル(4-ヒドロキシ- 3-ニトロフエ-ル)ァセタート 2.61 g及びピリジン 1 .83 gの塩化メチレン(25 mL)溶液にトリフルォロメタンスルホン酸無水物 4.91 gをカロ え、同条件下 1時間撹拌した。反応液を減圧下濃縮し、残渣に水を加え、酢酸ェチル で抽出した。有機層を飽和重曹水、 1規定塩酸及び飽和食塩水で洗浄し、無水硫酸
マグネシウムで乾燥後減圧下濃縮した。得られた残渣を n-へキサン/酢酸ェチル (75 : 25)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにて精製し、ェチル(3-二ト 口- 4-{[(トリフルォロメチル)スルホ -ル]ォキシ }フエ-ル)ァセタート 4.10 gを黄色油状 物として得た。 NMR(CDC1 ) : 1.29 (3H, t, J = 7.1 Hz), 3.74 (2H, s), 4.21 (2H, q, J = Under ice-cooling, add 4.91 g of trifluoromethanesulfonic anhydride to a solution of 2.61 g of ethyl (4-hydroxy-3-nitrophenol) acetate and 1.83 g of pyridine in methylene chloride (25 mL) under the same conditions. Stirred for 1 hour. The reaction mixture was concentrated under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated aqueous sodium bicarbonate, 1N hydrochloric acid and saturated brine, and anhydrous sulfuric acid The extract was dried over magnesium and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (75:25) as an elution solvent, and ethyl (3-nitro-4-{[(trifluoromethyl) sulfoyl 4.10 g of (l-oxy) phenol) acetate was obtained as a yellow oil. NMR (CDC1): 1.29 (3H, t, J = 7.1 Hz), 3.74 (2H, s), 4.21 (2H, q, J =
3 Three
7.1 Hz), 7.42 (IH, d, J = 8.5 Hz), 7.68 (IH, dd, J = 8.5, 2.3 Hz), 8.12 (IH, d, J = 2. 3 Hz) 7.1 Hz), 7.42 (IH, d, J = 8.5 Hz), 7.68 (IH, dd, J = 8.5, 2.3 Hz), 8.12 (IH, d, J = 2.3 Hz)
[0134] 参考例 65 [0134] Reference Example 65
ェチル(3--トロ- 4- {[(トリフルォロメチル)スルホ -ル]ォキシ }フエ-ル)ァセタート 2.5 0 g、トリブチルビ-ルスズ 3.33 g、ビス (トリフエ-ルホスフィン)パラジウムジクロリド 490 mg及び塩化リチウム 1.48 gのテトラヒドロフラン(15 mL)溶液を室温にて 14時間撹拌 した。反応液を減圧下濃縮し、残渣に酢酸ェチルを加え、セライト濾過した。濾液を 水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後減圧下濃縮した。得 られた残渣を n-へキサン/酢酸ェチル (3 : 1)を溶出溶媒とするシリカゲルカラムクロマ トグラフィ一にて精製し、ェチル(3-二トロ- 4-ビュルフエ-ル)ァセタート 1.32 gを黄色 油状物として得た。 NMR(CDC1 ) : 1.27 (3H, t, J = 7.1 Hz), 3.68 (2H, s), 4.18 (2H, q, Ethyl (3-tro-4-{[(trifluoromethyl) sulfoyl] oxy} phenol) acetate 2.50 g, tributylbutyltin 3.33 g, bis (triphenylphosphine) palladium dichloride 490 mg and A solution of lithium chloride (1.48 g) in tetrahydrofuran (15 mL) was stirred at room temperature for 14 hours. The reaction mixture was concentrated under reduced pressure, ethyl acetate was added to the residue, and the mixture was filtered through celite. The filtrate was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (3: 1) as an elution solvent, and 1.32 g of ethyl (3-nitro-4-butylphenol) acetate was yellow. Obtained as an oil. NMR (CDC1): 1.27 (3H, t, J = 7.1 Hz), 3.68 (2H, s), 4.18 (2H, q,
3 Three
J = 7.1 Hz), 5.48 (IH, d, J = 11.0 Hz), 5.74 (IH, d, J = 17.4 Hz), 7.16 (IH, dd, J = 17.4, 11.0 Hz), 7.51 (IH, dd, J = 8.1, 1.7 Hz), 7.59 (IH, d, J = 8.1 Hz), 7.87 (IH, d , J = 1.7 Hz) J = 7.1 Hz), 5.48 (IH, d, J = 11.0 Hz), 5.74 (IH, d, J = 17.4 Hz), 7.16 (IH, dd, J = 17.4, 11.0 Hz), 7.51 (IH, dd, J = 8.1, 1.7 Hz), 7.59 (IH, d, J = 8.1 Hz), 7.87 (IH, d, J = 1.7 Hz)
[0135] 参考例 66 [0135] Reference Example 66
ェチル(3--トロ- 4-ビュルフエ-ル)ァセタート 20.0 mgの塩化メチレン(1 mL)溶液 に 78°Cでオゾンガスを 5分間通じた。反応液に 78°Cで窒素を通じ過剰のオゾン ガスを留去した後、同条件下ジメチルスルフイド 31 Lを加えた。攪拌下室温まで昇 温し、室温にて 1時間攪拌した。反応液を減圧下濃縮し、得られた残渣を n_へキサン /酢酸ェチル (6 :4)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにて精製し、 ェチル(4-ホルミル- 3-ニトロフエ-ル)ァセタート 15.8 mgを淡黄色油状物として得た。 NMR(CDC1 ) : 1.28 (3H, t, J = 7.1 Hz), 3.78 (2H, s), 4.20 (2H, q, J = 7.1 Hz), 7.71 ( Ethyl (3-tro-4-butylphenol) acetate 20.0 mg of methylene chloride (1 mL) was passed through ozone gas at 78 ° C for 5 minutes. Excess ozone gas was removed by passing nitrogen through the reaction solution at 78 ° C, and then 31 L of dimethylsulfide was added under the same conditions. The mixture was warmed to room temperature with stirring and stirred at room temperature for 1 hour. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n_hexane / ethyl acetate (6: 4) as an elution solvent, and ethyl (4-formyl-3-nitrophenyl) was purified. ) 15.8 mg of acetate was obtained as a pale yellow oil. NMR (CDC1): 1.28 (3H, t, J = 7.1 Hz), 3.78 (2H, s), 4.20 (2H, q, J = 7.1 Hz), 7.71 (
3 Three
IH, dd, J = 7.9, 1.6 Hz), 7.94 (IH, d, J = 7.9 Hz), 8.06 (IH, d, J = 1.6 Hz), 10.42 ( IH, br s)
[0136] 参考例 67 IH, dd, J = 7.9, 1.6 Hz), 7.94 (IH, d, J = 7.9 Hz), 8.06 (IH, d, J = 1.6 Hz), 10.42 (IH, br s) [0136] Reference Example 67
ェチル(4-ホルミル- 3--トロフエ-ル)ァセタート 0.56 g、 2-メチル -2-ブテン 0.51 g、 ァセトニトリル 1 mL、水 2 mLおよび tert-ブチルアルコール 5 mLの混合物にリン酸二 水素ナトリウム 0.23 g及び亜塩素酸ナトリウム 0.66 gを加え、室温にて 1時間撹拌した。 反応液に水を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫 酸マグネシウムで乾燥後減圧下濃縮し、 4-エトキシカルボニルメチル -2--トロ安息 香酸 0.63 gを無色固体として得た。 NMR (DMSO-d ): 1.20 (3H, t, J = 7.1 Hz), 3.90 Ethyl (4-formyl-3-trophenyl) acetate 0.56 g, 2-methyl-2-butene 0.51 g, acetonitrile 1 mL, water 2 mL and tert-butyl alcohol 5 mL in a mixture of sodium dihydrogen phosphate 0.23 g and 0.66 g of sodium chlorite were added, and the mixture was stirred at room temperature for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give 0.63 g of 4-ethoxycarbonylmethyl-2-trobenzoic acid as a colorless solid. NMR (DMSO-d): 1.20 (3H, t, J = 7.1 Hz), 3.90
6 6
(2H, s), 4.11 (2H, q, J = 7.1 Hz), 7.69 (1H, dd, J = 7.9, 1.3 Hz), 7.83 (1H, d, J = 7. 9 Hz), 7.91 (1H, d, J = 1.3 Hz), 13.80 (1H, br s) (2H, s), 4.11 (2H, q, J = 7.1 Hz), 7.69 (1H, dd, J = 7.9, 1.3 Hz), 7.83 (1H, d, J = 7.9 Hz), 7.91 (1H, d, J = 1.3 Hz), 13.80 (1H, br s)
[0137] 参考例 68 [0137] Reference Example 68
2- {[2- (2- tert-ブトキシ- 2-ォキソエトキシ)ベンジル]アミノ}- 5-クロ口安息香酸 300 mg 、 tert-ブチル(4-ァミノベンジル)力ルバマート 168 mgの Ν,Ν-ジメチルホルムアミド(6. 0 mL)溶液に 1-ヒドロキシベンゾトリァゾールー水和物 112 mg及び 1-ェチル -3-(3-ジ メチルァミノプロピル)カルポジイミド一塩酸塩 158 mgを室温にて順次カ卩ぇ終夜攪拌 した。反応液に酢酸ェチル及び水を加え、酢酸ェチルで抽出した。有機層を水及び 飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥した後減圧下濃縮した。得 られた残渣を n-へキサン/酢酸ェチル (7: 3)を溶出溶媒とするシリカゲルカラムクロマト グラフィ一にて精製し tert-ブチル(2-{[(2-{[(4-{[(tert-ブトキシカルボ-ル)ァミノ]メチ ル}フエ-ル)ァミノ]カルボ-ル}-4-クロ口フエ-ル)ァミノ]メチル }-6-エトキシフエノキシ) ァセタート 320 mgを無色固体として得た。 FP: 640 2-{[2- (2-tert-Butoxy-2-oxoethoxy) benzyl] amino} -5-chlorobenzoic acid 300 mg, tert-butyl (4-aminobenzyl) force rubamate 168 mg Ν, Ν-dimethylformamide (6.0 mL) Into the solution, add 112 mg of 1-hydroxybenzotriazole hydrate and 158 mg of 1-ethyl-3- (3-dimethylaminopropyl) carpositimide monohydrochloride sequentially at room temperature. Stir overnight. Ethyl acetate and water were added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (7: 3) as the elution solvent and purified by tert-butyl (2-{[(2-{[(4-{[( tert-Butoxycarbol) amino] methylene} phenol) amino] carbol-4-chlorophenol) amino] methyl} -6-ethoxyphenoxy) Acetate 320 mg as colorless solid Obtained. FP: 640
[0138] 参考例 69 [0138] Reference Example 69
5-クロ口- 2--トロ安息香酸 15.6 g、 Ν,Ν-ジメチルホルムアミド 3滴及び 1,2-ジクロロ ェタン(150 mL)の混合物に氷冷下塩化ォキサリル 13.3 mLを加え、室温にて 3時間 攪拌した。反応液を減圧下濃縮して得られた残渣をァセトニトリル 150 mLに溶解し、 t ert-ブチル [(4-ァミノフエ-ル) (ィミノ)メチル]力ルバマート 18.2 gとピリジン 18.3 gのァ セトニトリル(150 mL)溶液に氷冷下滴加し、室温にて 18時間攪拌した。反応液が 20 0 mL程度になるまで減圧下溶媒を留去した後、室温にて 0.5規定水酸ィ匕ナトリウム水 溶液 500 mLに注カ卩し 2時間攪拌した。析出物を濾取し、濾物をメタノール 100 mLに
懸濁させた。室温にて 1時間攪拌した後に析出物を濾取し、濾物を減圧下乾燥して t ert-ブチル [{4- [(5-クロ口- 2--トロべンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]力ルバマ ート 24.6 gを得た。 Add 13.3 mL of oxalyl chloride under ice-cooling to a mixture of 15.6 g of 2-chlorobenzo-2-trobenzoic acid, 3 drops of Ν, Ν-dimethylformamide and 1,2-dichloroethane (150 mL) at room temperature. Stir for hours. The residue obtained by concentrating the reaction solution under reduced pressure was dissolved in 150 mL of acetonitrile, and 18.2 g of tert-butyl [(4-aminophenol) (imino) methyl] strength rubamate and 18.3 g of acetonitrile (150 The solution was added dropwise to the solution under ice-cooling and stirred at room temperature for 18 hours. After distilling off the solvent under reduced pressure until the reaction volume reached about 200 mL, the mixture was poured into 500 mL of 0.5N aqueous sodium hydroxide solution at room temperature and stirred for 2 hours. The precipitate was collected by filtration, and the filtrate was added to 100 mL of methanol. Suspended. After stirring at room temperature for 1 hour, the precipitate is collected by filtration, and the filtrate is dried under reduced pressure to give t-tert-butyl [{4- [(5-chloro-2-2-trobenzyl) amino] phenol. } (Imino) methyl] strength rubamate 24.6 g was obtained.
[0139] 参考例 70 [0139] Reference Example 70
5—クロ口— N— (6—シァノピリジン— 3—ィル)—2—-トロべンズアミド 4.0 gのメタノール (40 m L)懸濁液にナトリウムメトキシド 71mgを室温でカ卩えて 12時間攪拌した。室温にてナトリ ゥムメトキシド 713mgを加え 48時間攪拌した。室温にてナトリウムメトキシド 356mgをカロ え 12時間攪拌した。塩ィ匕アンモ-ゥム 706 mgを加え 80°Cに昇温し 3時間攪拌した。室 温に冷却後、溶媒を減圧下濃縮し 1,4-ジォキサン 40 mLに溶解した後、 1規定水酸 化ナトリウム水溶液 13.3 mL,ジ -tert-ブチルジカルボナート 2.9 gを室温でカ卩ぇ 12時 間攪拌した。水 50 mLを加え酢酸ェチルで抽出し、有機層を無水硫酸マグネシウム にて乾燥し減圧濃縮した。得られた残渣を n-へキサン/酢酸ェチル (1 : 1)を溶出溶 媒とするシリカゲルカラムクロマトグラフィーにより精製し tert-ブチル [{5- [(5-クロ口- 2- ニトロべンゾィル)ァミノ]ピリジン- 2-ィル }(ィミノ)メチル]力ルバマート 2.2 gを淡黄色固 体として得た。 5—Black mouth— N— (6-Cyanopyridine-3-yl) —2—-Trobensamide In a suspension of 4.0 g of methanol (40 mL), 71 mg of sodium methoxide was added at room temperature and stirred for 12 hours. did. At room temperature, 713 mg of sodium methoxide was added and stirred for 48 hours. At room temperature, 356 mg of sodium methoxide was added and stirred for 12 hours. 706 mg of salt ammonium was added and the temperature was raised to 80 ° C and stirred for 3 hours. After cooling to room temperature, the solvent was concentrated under reduced pressure and dissolved in 40 mL of 1,4-dioxane, then 13.3 mL of 1N aqueous sodium hydroxide solution and 2.9 g of di-tert-butyl dicarbonate were stirred at room temperature. Stir for 12 hours. 50 mL of water was added and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (1: 1) as an elution solvent, and purified by tert-butyl [{5- [(5-chloro-2-nitronitrosyl)]. Amino] pyridin-2-yl} (imino) methyl] force rubamate 2.2 g was obtained as a pale yellow solid.
[0140] 参考例 71 [0140] Reference Example 71
室温攪拌下、ェチル [4- (4- [[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ- ルカルバモイル)- 3--トロフエ-ル]ァセタート 0.40 gのエタノール (2 mL)溶液に 1規 定水酸ィ匕ナトリウム水溶液 0.85 mLを加え 15分間攪拌した。反応液に 1規定塩酸 0.8 5 mLをカ卩えて中和し、反応液を減圧下濃縮した。残渣に水を加え、酢酸ェチルで抽 出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後減圧下濃縮 し、淡黄色固体の粗生成物を得た。得られた淡黄色固体 0.37 g、塩化アンモ -ゥム 0 .23 g、 1-ヒドロキシベンゾトリァゾールー水和物 0.16 g、(3-ジメチルァミノプロピル)ェ チルカルボジイミド 一塩酸塩 0.20 g及び Ν,Ν-ジイソプロピルェチルァミン 0.55 gの Ν, N-ジメチルホルムアミド(1.5 mL)溶液を室温にて 13時間攪拌した。反応液に水を加 え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウム で乾燥後減圧下濃縮した。得られた残渣をメタノール/酢酸ェチル (5 : 95)を溶出溶 媒とするシリカゲルカラムクロマトグラフィーにて精製し、 tert-ブチル [(4-{[4-(2-ァミノ
-2-ォキソェチル )-2- -トロべンゾィル]アミノ}フエ-ル) (ィミノ)メチル]力ルバマート 0.22 gを得た。 EN:442 While stirring at room temperature, ethyl [4- (4- [[(tert-butoxycarbonyl) amino] (imino) methyl] phenylcarbamoyl) -3--3-trifluoro] acetate was added to a solution of 0.40 g of ethanol (2 mL). 0.85 mL of a standard aqueous sodium hydroxide solution was added, and the mixture was stirred for 15 minutes. The reaction mixture was neutralized with 1N hydrochloric acid (0.85 mL), and the reaction mixture was concentrated under reduced pressure. Water was added to the residue and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain a crude product as a pale yellow solid. The obtained pale yellow solid 0.37 g, ammonium chloride 0.23 g, 1-hydroxybenzotriazole hydrate 0.16 g, (3-dimethylaminopropyl) ethylcarbodiimide monohydrochloride 0.20 g and Ν , A solution of 0.55 g of) -diisopropylethylamine in Ν, N-dimethylformamide (1.5 mL) was stirred at room temperature for 13 hours. Water was added to the reaction solution and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using methanol / ethyl acetate (5:95) as an elution solvent, and tert-butyl [(4-{[4- (2-amino) was obtained. -2-Oxoethyl) -2-trobenzoyl] amino} phenol) (imino) methyl] power rubamate 0.22 g was obtained. EN: 442
[0141] 参考例 72 [0141] Reference Example 72
5-クロ口- N- (4-シァノフエ-ル)- 2--トロべンズアミド 1.58 g、塩酸ヒドロキシルァミン 400 mg、トリェチルァミン 583 mg及びエタノール 50 mLの混合物を 60 °Cにて終夜攪 拌した。反応液を減圧下濃縮し、得られた残渣に酢酸ェチル及び水を加えた後、酢 酸ェチルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、減圧下濃縮した。 得られた残渣をクロ口ホルムで洗浄して N-{4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル} -5-クロ口- 2-ニトロべンズアミド 1.43 gを黄色固体として得た。 5-Chloro-N- (4-Chanophenol) -2-Trobensamide 1.58 g, hydroxylamine hydrochloride 400 mg, triethylamine 583 mg and ethanol 50 mL were stirred overnight at 60 ° C. . The reaction solution was concentrated under reduced pressure, and ethyl acetate and water were added to the resulting residue, followed by extraction with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was washed with black mouth form to obtain 1.43 g of N- {4- [amino (hydroxyimino) methyl] phenol} -5-black mouth-2-nitrobenzamide as a yellow solid.
[0142] 参考例 73 [0142] Reference example 73
tert-ブチル(イミノ {4- [(5-メチル -2--トロべンゾィル)ァミノ]フエ-ル}メチル)力ルバ マート 3.08 g、塩化メチレン 15 mLおよびトリフルォロ酢酸 15 mLの混合物を室温にて 15時間攪拌した。反応液を減圧下濃縮した後、酢酸ェチルを加えて得られた析出物 を濾取し N- {4- [ァミノ (ィミノ)メチル]フエ-ル}- 5-メチル -2--トロべンズアミドトリフル ォロ酢酸塩 2.63 gを淡黄色固体として得た。 EP: 299 tert-Butyl (imino {4-[(5-methyl-2-trobenzoyl) amino] phenyl} methyl) rubamate 3.08 g, methylene chloride 15 mL and trifluoroacetic acid 15 mL mixture at room temperature Stir for 15 hours. The reaction mixture was concentrated under reduced pressure, and the precipitate obtained by adding ethyl acetate was collected by filtration. N- {4- [Amino (imino) methyl] phenol} -5-methyl-2-trobens 2.63 g of amide trifluoroacetate was obtained as a pale yellow solid. EP: 299
[0143] 参考例 74 [0143] Reference Example 74
N- {4- [ァミノ (ィミノ)メチル]フエ-ル}- 5-メチル -2--トロべンズアミドトリフルォロ酢酸 塩 1.23 gおよび 1,4-ジォキサン 30 mLの混合物に、室温にて 1規定水酸化ナトリウム 水溶液 3.0 mLを加え、 30分間攪拌した。反応液にジェチルジカルボナート 484 mgと 1規定水酸化ナトリウム水溶液 3.0 mLを室温にて加え、 2時間攪拌した。反応液に炭 酸カリウム 484 mgおよびェチルクロ口ホルマート 323 mgを室温にて加え、 2時間攪拌 した。反応液に炭酸カリウム 484 mgおよびェチルクロ口ホルマート 323 mgを室温にて 加え、 14時間攪拌した。反応液に炭酸カリウム 484 mgおよびェチルクロ口ホルマート 323 mgを室温にて加え、 4時間攪拌した。反応液に水を加え、酢酸ェチルで抽出し た。有機層を飽和食塩水にて洗浄し、無水硫酸ナトリウムにて乾燥後、減圧下濃縮し た。残渣をジェチルエーテルにて洗浄し、ェチル(ィミノ {4- [(5-メチル -2-二トロべンゾ ィル)ァミノ]フエ-ル}メチル)力ルバマート 970 mgを淡黄色固体として得た。 EP: 371 [0144] 参考例 75
N- {4- [ァミノ (ィミノ)メチル]フエ-ル}- 5-メチル -2--トロべンズアミドトリフルォロ酢酸 塩 1.28 g、テトラヒドロフラン 30 mLおよび水 6.0 mLの混合物に室温にて炭酸カリウム 2 .57 gを加え、 15分間攪拌した。反応液に室温にて n-へキシルクロ口ホルマート 511 m gを加え 2時間攪拌した。反応液に室温にて炭酸カリウム 428 mgを加え 2時間攪拌した 。反応液に室温にて炭酸カリウム 428 mgおよび n-へキシルクロ口ホルマート 511 mg をカロえ 2時間攪拌した。反応液に室温にて炭酸カリウム 428 mgおよび n-へキシルクロ 口ホルマート 511 mgをカ卩ぇ 14時間攪拌した。反応液に室温にて炭酸カリウム 428 mg および n-へキシルクロ口ホルマート 511 mgをカ卩ぇ 4時間攪拌した。反応液に水を加え 、酢酸ェチルで抽出した。有機層を飽和食塩水にて洗浄し、無水硫酸ナトリウムにて 乾燥後、減圧下濃縮した。残渣をジェチルエーテルにて洗浄し、 n-へキシル(イミノ{ 4- [(5-メチル -2-二トロべンゾィル)ァミノ]フエ-ル}メチル)力ルバマート 1.25 gを無色固 体として得た。 N- {4- [Amino (imino) methyl] phenol} -5-methyl-2--trobensamide trifluoroacetate 1.23 g and 1,4-dioxane 30 mL at room temperature 1 N aqueous sodium hydroxide solution (3.0 mL) was added, and the mixture was stirred for 30 min. To the reaction solution, 484 mg of jetyl dicarbonate and 3.0 mL of 1N aqueous sodium hydroxide solution were added at room temperature, and the mixture was stirred for 2 hours. To the reaction solution, 484 mg of potassium carbonate and 323 mg of ethyl chloroformate were added at room temperature, and the mixture was stirred for 2 hours. To the reaction solution, 484 mg of potassium carbonate and 323 mg of ethyl acetate were added at room temperature and stirred for 14 hours. To the reaction solution, 484 mg of potassium carbonate and 323 mg of ethyl chloride formate were added at room temperature and stirred for 4 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was washed with jetyl ether to obtain 970 mg of ethyl (imino {4-[(5-methyl-2-nitrobenzene) amino] phenol} methyl) strength rubamate as a pale yellow solid. It was. EP: 371 [0144] Reference Example 75 N- {4- [Amino (imino) methyl] phenol} -5-methyl-2--trobensamide trifluoroacetate 1.28 g, tetrahydrofuran 30 mL and water 6.0 mL 2.57 g of potassium was added and stirred for 15 minutes. To the reaction solution, 511 mg of n-hexylchloroform formate was added at room temperature and stirred for 2 hours. To the reaction solution, 428 mg of potassium carbonate was added at room temperature and stirred for 2 hours. To the reaction solution, 428 mg of potassium carbonate and 511 mg of n-hexylchloroform formate were added at room temperature and stirred for 2 hours. To the reaction solution, 428 mg of potassium carbonate and 511 mg of n-hexylchloroformate were stirred at room temperature for 14 hours. To the reaction solution, 428 mg of potassium carbonate and 511 mg of n-hexylchloroform formate were stirred at room temperature for 4 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was washed with jetyl ether to obtain 1.25 g of n-hexyl (imino {4-[(5-methyl-2-nitrobenzene) amino] phenol} methyl) strength rubamate as a colorless solid. It was.
[0145] 参考例 76 [0145] Reference Example 76
tert-ブチル(2-エトキシ- 6-ホルミルフエノキシ)ァセタート 1.00 g、塩酸ヒドロキシル ァミン 260 mg及び 2-プロパノール 18 mLの混合物に酢酸カリウム 368 mgの水(3.8 mL )溶液を室温にて加え、 20時間攪拌した。反応液に水を加え、酢酸ェチルで抽出し た。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後減圧下濃縮して tert-ブチル {2-エトキシ -6- [(ヒドロキシィミノ)メチル]フエノキシ }ァセタート 1.04 gを無 色固体として得た。 FP: 296 To a mixture of tert-butyl (2-ethoxy-6-formylphenoxy) acetate 1.00 g, hydroxylamine hydrochloride 260 mg and 2-propanol 18 mL, a solution of potassium acetate 368 mg in water (3.8 mL) was added at room temperature. Stir for 20 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to give 1.04 g of tert-butyl {2-ethoxy-6-[(hydroxyimino) methyl] phenoxy} acetate as a colorless solid. It was. FP: 296
[0146] 参考例 77 [0146] Reference Example 77
tert-ブチル [{4- [(5-クロ口- 2-二トロべンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]力ルバ マート 5.00 gの 1,4-ジォキサン(100 mL)溶液にラネーニッケルの水懸濁物 1.0 gをカロ え、 1気圧の水素雰囲気下室温にて 2 日間攪拌した。反応液を濾過し、濾液を減圧 下濃縮して tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル }(ィミノ)メチ ル]力ルバマート 4.43 gを得た。 tert-Butyl [{4- [(5-Chromium-2-2-nitrobenzene) amino] phenol} (imino) methyl] strength rubamate 5.00 g of Raney nickel in a solution of 5.00 g of 1,4-dioxane (100 mL) 1.0 g of the water suspension was added and stirred for 2 days at room temperature in a hydrogen atmosphere of 1 atm. The reaction mixture was filtered, and the filtrate was concentrated under reduced pressure to give 4.43 g of tert-butyl [{4-[(2-amino-5-chlorobenzoyl) amino] phenol} (imino) methyl] force rubamate. Obtained.
[0147] 参考例 78 [0147] Reference Example 78
N- (4-シァノフエ-ル)- 5-メチル -2--トロべンズアミド 14.3 g、塩酸ヒドロキシルァミン 4.9 g、 Ν,Ν-ジイソプロピルェチルァミン 14 mL及びエタノール 100 mLの混合物を 60
°cにて終夜攪拌した。反応液を減圧下濃縮し、得られた残渣に水を加え析出物を濾 取し黄色固体 12.5 gを得た。得られた黄色固体をテトラヒドロフラン 50 mLとエタノール 50 mLに溶解し、 10%パラジウム-炭素 4.23 gを加えて、 1気圧の水素雰囲気下室温に て終夜攪拌した。反応液を濾過し、濾液を減圧濃縮し 2-ァミノ- N-{4- [ァミノ (ヒドロキ シィミノ)メチル]フエ-ルト 5-メチルベンズアミド 11 gを淡黄色固体として得た。 NMR ( DMSO-d ) : 2.22 (3H, s), 5.76 (2H, brs), 6.10 (2H, brs), 6.68 (1H, d, J = 8.3 Hz), 7. N- (4-Chanophenol) -5-methyl-2--trobensamide 14.3 g, hydroxylamine hydrochloride 4.9 g, Ν, Ν-diisopropylethylamine 14 mL and ethanol 100 mL Stir at ° C overnight. The reaction mixture was concentrated under reduced pressure, water was added to the resulting residue, and the precipitate was collected by filtration to obtain 12.5 g of a yellow solid. The obtained yellow solid was dissolved in tetrahydrofuran (50 mL) and ethanol (50 mL), 10% palladium-carbon (4.23 g) was added, and the mixture was stirred overnight at room temperature under a hydrogen atmosphere of 1 atm. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure to obtain 11 g of 2-amino-N- {4- [amino (hydroxyimino) methyl] felt 5-methylbenzamide as a pale yellow solid. NMR (DMSO-d): 2.22 (3H, s), 5.76 (2H, brs), 6.10 (2H, brs), 6.68 (1H, d, J = 8.3 Hz), 7.
6 6
04 (1H, dd, J = 1.5, 8.3 Hz), 7.44 (1H, J = 1.5 Hz), 7.64 (2H, d, J = 8.9 Hz), 7.72 ( 2H, d, J = 8.9 Hz), 9.45 (1H, brs), 10.05 (1H, brs) 04 (1H, dd, J = 1.5, 8.3 Hz), 7.44 (1H, J = 1.5 Hz), 7.64 (2H, d, J = 8.9 Hz), 7.72 (2H, d, J = 8.9 Hz), 9.45 ( 1H, brs), 10.05 (1H, brs)
[0148] 参考例 79 [0148] Reference Example 79
tert-ブチル [(4-ァミノフエ-ル) (イミノ)メチル]力ルバマート 706 mgのテトラヒドロフラ ン(30 mL)溶液に 0 °Cにて塩化ァリルマグネシウムのテトラヒドロフラン溶液(2 M) 3 mLを加え 0.5時間攪拌した。反応液に 5-ブロモイサトイン酸無水物 363 mgを室温に て加え、 4時間攪拌した。反応液を減圧下濃縮して得られた残渣に水を加え、酢酸 ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後 減圧下濃縮した。得られた残渣を酢酸ェチル -へキサン (2: 1)を溶出溶媒とするシリ 力ゲルカラムクロマトグラフィーで精製し tert-ブチル [{4-[(2-ァミノ- 5-ブロモベンゾィ ル)ァミノ]フエ-ル Kィミノ)メチル]力ルバマート 80 mgを黄色固体として得た。 FP: 434 [0149] 参考例 80 tert-Butyl [(4-aminophenol) (imino) methyl] strength rubamate To a solution of 706 mg of tetrahydrofuran (30 mL) at 0 ° C was added 3 mL of a solution of gallium magnesium chloride in tetrahydrofuran (2 M) 0.5 Stir for hours. To the reaction solution, 363 mg of 5-bromoisatoic anhydride was added at room temperature and stirred for 4 hours. The reaction mixture was concentrated under reduced pressure, water was added to the resulting residue, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel chromatography using ethyl acetate-hexane (2: 1) as an elution solvent, and purified by tert-butyl [{4-[(2-amino-5-bromobenzoyl) amino] phenol. -LeKimino) methyl] rubamate 80 mg was obtained as a yellow solid. FP: 434 [0149] Reference Example 80
tert-ブチル(2-エトキシ- 6-ホルミルフエノキシ)ァセタートと 4-クロ口- 2-ニトロァニリ ンを原料とし、参考例 61と同様の操作を行い tert-ブチル(2-{[(4-クロ口- 2-ニトロフエ -ル)ァミノ]メチル }- 6-エトキシフエノキシ)ァセタートを得た。 FP: 437 Using tert-butyl (2-ethoxy-6-formylphenoxy) acetate and 4-chloro-2-nitroaniline as raw materials, the same procedure as in Reference Example 61 was performed, and tert-butyl (2-{[(4- Chromium-2-nitrophenol) amino] methyl} -6-ethoxyphenoxy) acetate was obtained. FP: 437
[0150] 参考例 81 [0150] Reference Example 81
tert-ブチル (2- {[(4-クロ口- 2--トロフエ-ル)ァミノ]メチル }- 6-エトキシフエノキシ)ァ セタート 0.98 gのエタノール (20 mL)溶液にラネーニッケルのエタノール懸濁物 2.0 m Lを加え、 1気圧の水素雰囲気下室温にて 3時間攪拌した。反応液を濾過し、濾液を 減圧下濃縮した。得られた無色油状物を Ν,Ν-ジメチルホルムアミド 20 mLに溶解し、 4 -シァノ安息香酸 0.29 g、 1-ヒドロキシベンゾトリアゾール 0.40 g及び 1-ェチル -3-(3- ジメチルァミノプロピル)カルポジイミド一塩酸塩 0.57 gを室温にて順次カ卩ぇ 17時間
攪拌した。反応液を減圧下濃縮し、得られた残渣を酢酸ェチルに溶解し、水及び飽 和食塩水で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥し減圧下濃縮したtert-Butyl (2- {[(4-Chromium-2 -trophenyl) amino] methyl} -6-ethoxyphenoxy) acetate Ethanol suspension of Raney nickel in 0.98 g ethanol (20 mL) 2.0 mL of the product was added and the mixture was stirred at room temperature for 3 hours under a hydrogen atmosphere of 1 atm. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure. The obtained colorless oil was dissolved in 20 mL of Ν, Ν-dimethylformamide, and 0.29 g of 4-cyanobenzoic acid, 0.40 g of 1-hydroxybenzotriazole and 1-ethyl-3- (3-dimethylaminopropyl) carpositimide 17 hours of monohydrochloride 0.57 g at room temperature Stir. The reaction solution was concentrated under reduced pressure, and the resulting residue was dissolved in ethyl acetate and washed successively with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure.
。得られた残渣をクロ口ホルム/メタノール(20 : 1)を溶出溶媒とするシリカゲルカラム クロマトグラフィーにて精製し tert-ブチル {2- [({4-クロ口- 2- [(4-シァノベンゾィル)アミ ノ]フエ-ル}ァミノ)メチル ]-6-エトキシフエノキシ }ァセタート 0.73 gを無色固体として得 た。 FP: 536 . The resulting residue was purified by silica gel column chromatography using chloroform-form / methanol (20: 1) as an elution solvent and purified by tert-butyl {2-[({4-cloguchi-2-[(4-cyanobenzoyl) Amino] phenol} amino) methyl] -6-ethoxyphenoxy} acetate 0.73 g was obtained as a colorless solid. FP: 536
[0151] 参考例 82 [0151] Reference Example 82
ェチル(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキシフエノキシ)ァセタート 6.76 7 gのエタノール (60 ml)溶液にアルゴン雰囲気下、炭酸カリウム 1.659 gを加え、 1. 5 時間攪拌した。不溶物を濾別し減圧下濃縮後、酢酸ェチルで希釈し、水を加え、分 液操作を行った。有機層を飽和食塩水で洗浄後無水硫酸ナトリウムで乾燥し、減圧 下濃縮した。得られた残渣を n-へキサン/酢酸ェチル (1:1)を溶出溶媒とするシリカ ゲルカラムクロマトグラフィーにより精製し、無色固体としてェチル [2-ホルミル- 4- (ヒ ドロキシメチル) -6-イソプロポキシフエノキシ]ァセタート 4.836 gを得た。 FP: 297 Ethyl (4-acetoxymethyl-2-formyl-6-isopropoxyphenoxy) acetate 6.76 To a solution of 7 g of ethanol (60 ml) was added 1.659 g of potassium carbonate under an argon atmosphere, and the mixture was stirred for 1.5 hours. Insoluble material was filtered off, concentrated under reduced pressure, diluted with ethyl acetate, water was added, and liquid separation was performed. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (1: 1) as an elution solvent, and ethyl [2-formyl-4- (hydroxymethyl) -6-iso Propoxyphenoxy] acetate 4.836 g was obtained. FP: 297
[0152] 参考例 83 [0152] Reference Example 83
2-ベンジルォキシ- 3-ヒドロキシベンズアルデヒド 8.92 gと炭酸セシウム 16.55 gの N, N-ジメチルホルムアミド(80 mL)懸濁液に室温にてメタンスルホン酸 2-tert-ブトキシ -1-メチルェチル 10.68 gを加え、 60°Cにて 20時間攪拌した。反応液に水を加え、酢 酸ェチルで抽出した。有機層を水および飽和食塩水で順次洗浄した。有機層を無水 硫酸マグネシウムで乾燥して減圧下濃縮した。得られた残渣を酢酸ェチル 一へキ サン(30 : 70)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し 2-ベンジ ルォキシ- 3-(2-tert-ブトキシ -1-メチルエトキシ)ベンズアルデヒドを 10.66g得た。 NM R (CDC1 ) : 1.18 (9H, s), 1.39 (3H, d, J = 6.3 Hz), 3.45—3.50 (IH, m), 3.58—3.64 (IH To a suspension of 8.92 g of 2-benzyloxy-3-hydroxybenzaldehyde and 16.55 g of cesium carbonate in N, N-dimethylformamide (80 mL) at room temperature was added 10.68 g of 2-tert-butoxy-1-methylethyl methanesulfonate. The mixture was stirred at 60 ° C for 20 hours. Water was added to the reaction solution and extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate monohexan (30:70) as an elution solvent, and 2-benzyloxy-3- (2-tert-butoxy-1-methylethoxy) benzaldehyde was purified by 10.66. g got. NM R (CDC1): 1.18 (9H, s), 1.39 (3H, d, J = 6.3 Hz), 3.45—3.50 (IH, m), 3.58—3.64 (IH
3 Three
, m), 4.53-4.61 (IH, m), 5.19—5.27 (2H, m), 7.10 (IH, td, J = 8.0, 0.7 Hz), 7.26 (1 H, dd, J = 8.0, 1.5 Hz), 7.30-7.44 (6H, m), 10.22 (IH, d, J = 0.7 Hz) , m), 4.53-4.61 (IH, m), 5.19—5.27 (2H, m), 7.10 (IH, td, J = 8.0, 0.7 Hz), 7.26 (1 H, dd, J = 8.0, 1.5 Hz) , 7.30-7.44 (6H, m), 10.22 (IH, d, J = 0.7 Hz)
[0153] 参考例 84 [0153] Reference Example 84
2-ベンジルォキシ- 3-(2-tert-ブトキシ -1-メチルエトキシ)ベンズアルデヒド 10.66g、 トルエン 100 mLおよびジェチルエーテル 30 mLの混合物に室温にてマグネシウムブ
ロミドジェチルエーテル錯体 8.84 gを加え、 90°Cにて 6時間攪拌した後終夜で自然 冷却した。反応液に室温でマグネシウムブロミドジェチルエーテル錯体 4.02 gを加え 、 90°Cにて 4時間攪拌した後、終夜で自然冷却した。反応液に 0°Cで 1規定塩酸 100 mLを加え、 10分間攪拌した。反応液に飽和食塩水を加え、有機層を抽出した。有機 層に 1規定水酸ィ匕ナトリウム水溶液 40 mLを加え水層を抽出し、再度 1規定水酸化ナ トリウム水溶液 20 mLを力卩ぇ水層を抽出した。合わせた水層に 1規定塩酸 70 mLをカロ え酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで 乾燥した。溶媒を減圧下留去し、得られた残渣を酢酸ェチル 一へキサン(1 : 1)を 溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し 3-(2-tert-ブトキシ -1-メ チルェトキシ)- 2-ヒドロキシベンズアルデヒドを 4.18 g得た。 NMR (CDC1 ) : 1.25 (9H, s 2-Benzyloxy-3- (2-tert-butoxy-1-methylethoxy) benzaldehyde 10.66 g, toluene 100 mL, and jetyl ether 30 mL were mixed with magnesium Romidjetyl ether complex (8.84 g) was added, and the mixture was stirred at 90 ° C for 6 hours, and then naturally cooled overnight. To the reaction solution was added 4.02 g of magnesium bromide jetyl ether complex at room temperature, and the mixture was stirred at 90 ° C. for 4 hours, and then naturally cooled overnight. 100 mL of 1N hydrochloric acid was added to the reaction solution at 0 ° C, and the mixture was stirred for 10 minutes. Saturated saline was added to the reaction solution, and the organic layer was extracted. To the organic layer, 40 mL of 1N aqueous sodium hydroxide solution was added to extract the aqueous layer, and 20 mL of 1N aqueous sodium hydroxide solution was extracted again to extract the aqueous layer. The combined aqueous layer was extracted with 70 mL of 1N hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography using ethyl acetate monohexane (1: 1) as an elution solvent, and 3- (2-tert-butoxy-1-methylethyl)- 4.18 g of 2-hydroxybenzaldehyde was obtained. NMR (CDC1): 1.25 (9H, s
3 Three
), 1.37 (3H, d, J = 6.3 Hz), 3.46(1H, dd, J = 9.5, 4.3 Hz), 3.61 (1H, dd, J = 9.5, 7. 3 Hz), 4.17- 4.25 (1H, m), 6.88 (1H, t, J = 8.0 Hz), 7.24 (1H, dd, J = 8.0, 1.5 Hz), 7.36 (1H, dd, J = 8.0, 1.5 Hz), 10.17 (1H, s), 10.25 (1H, br s) ), 1.37 (3H, d, J = 6.3 Hz), 3.46 (1H, dd, J = 9.5, 4.3 Hz), 3.61 (1H, dd, J = 9.5, 7.3 Hz), 4.17- 4.25 (1H, m), 6.88 (1H, t, J = 8.0 Hz), 7.24 (1H, dd, J = 8.0, 1.5 Hz), 7.36 (1H, dd, J = 8.0, 1.5 Hz), 10.17 (1H, s), 10.25 (1H, br s)
参考例 85 Reference Example 85
37%ホルムアルデヒド水溶液 1.82 mLと 50%ジメチルァミン水溶液 2.56 mLをェタノ ール 9 mLに溶解し、 3-(2-tert-ブトキシ -1-メチルエトキシ) - 2-ヒドロキシベンズアルデ ヒド 4.10 gのエタノール (6 mL)溶液を室温で滴加した後、 14時間加熱還流した。溶 媒を減圧下留去して得られた残渣にトルエンを加え、再度減圧下濃縮した。得られた 化合物、メタノール 30 mLおよびアセトン 15 mLの混合物にョードメタン 1.06 mLを加え 、室温で 3日間攪拌した。溶媒を減圧下留去した。得られた残渣を酢酸 35 mLに懸濁 させ、酢酸ナトリウム 1.60 gを加えて 16時間加熱還流した。室温まで自然冷却した後、 溶媒を減圧下留去した。残渣に水を加え、酢酸ェチルで抽出した。有機層を水、飽 和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥した。減圧下濃縮し、得られ た残渣を酢酸ェチル 一へキサン(8:2)を溶出溶媒とするシリカゲルカラムクロマトグ ラフィ一で精製し 3-(2-ァセトキシ -1-メチルエトキシ) -5-ホルミル- 4-ヒドロキシベンジ ルァセタートを 2.23 g得た。 NMR (CDC1 ) : 1.39 (3H, d, J = 6.3 Hz), 2.06 (3H, s), 2. Dissolve 1.82 mL of 37% aqueous formaldehyde solution and 2.56 mL of 50% aqueous dimethylamine solution in 9 mL of ethanol, and add 4- (2-tert-butoxy-1-methylethoxy) -2-hydroxybenzaldehyde 4.10 g of ethanol (6 mL) The solution was added dropwise at room temperature and then heated to reflux for 14 hours. Toluene was added to the residue obtained by evaporating the solvent under reduced pressure, and the mixture was concentrated again under reduced pressure. 1.06 mL of odomethane was added to a mixture of the obtained compound, methanol (30 mL) and acetone (15 mL), and the mixture was stirred at room temperature for 3 days. The solvent was distilled off under reduced pressure. The obtained residue was suspended in 35 mL of acetic acid, 1.60 g of sodium acetate was added, and the mixture was heated to reflux for 16 hours. After naturally cooling to room temperature, the solvent was distilled off under reduced pressure. Water was added to the residue and extracted with ethyl acetate. The organic layer was washed sequentially with water and saturated Japanese brine and dried over anhydrous magnesium sulfate. Concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using ethyl acetate monohexane (8: 2) as the elution solvent, and 3- (2-acetoxy-1-methylethoxy) -5-formyl- There were obtained 2.23 g of 4-hydroxybenzyl sulfate. NMR (CDC1): 1.39 (3H, d, J = 6.3 Hz), 2.06 (3H, s), 2.
3 Three
10 (3H, s), 4.21-4.31 (2H, m), 4.60—4.68 (1H, m), 5.05 (2H, s), 7.24 (1H, d, J = 2.0 Hz), 7.27 (1H, d, J = 2.0 Hz), 9.92 (1H, s), 10.99 (1H, br s)
[0155] 参考例 86 10 (3H, s), 4.21-4.31 (2H, m), 4.60-4.68 (1H, m), 5.05 (2H, s), 7.24 (1H, d, J = 2.0 Hz), 7.27 (1H, d, J = 2.0 Hz), 9.92 (1H, s), 10.99 (1H, br s) [0155] Reference Example 86
3-(2-ァセトキシ -1-メチルエトキシ) -5-ホルミル- 4-ヒドロキシベンジルァセタート 1.6 3 gとピリジン 0.85 mLのジクロロメタン (15 mL)溶液にトリフルォロメタンスルホン酸無 水物 1.06 mLを水冷下加え、室温で 1.5時間攪拌した。溶媒を減圧下留去し、得られ た残渣に飽和塩ィ匕アンモ-ゥム水溶液を加え酢酸ェチルで抽出した。有機層を水お よび飽和食塩水で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥して減圧下 濃縮した。得られた残渣を酢酸ェチル 一へキサン(1 : 1)を溶出溶媒とするシリカゲ ルカラムクロマトグラフィーで精製し、 3- (2-ァセトキシ- 1-メチルエトキシ) -5-ホルミル- 4- {[(トリフルォロメチル)スルホ -ル]ォキシ }ベンジルァセタートを 2.03 g得た。 NMR ( CDC1 ) : 1.44(3H, d, J = 6.3 Hz), 2.07 (3H, s), 2.15 (3H, s), 4.20-4.34 (2H, m), 4.75 3- (2-acetoxy-1-methylethoxy) -5-formyl-4-hydroxybenzylacetate 1.63 g and pyridine 0.85 mL in dichloromethane (15 mL) solution with trifluoromethanesulfonic acid anhydrous 1.06 mL The mixture was added with water cooling and stirred at room temperature for 1.5 hours. The solvent was distilled off under reduced pressure, a saturated aqueous solution of ammonium chloride was added to the resulting residue, and the mixture was extracted with ethyl acetate. The organic layer was washed sequentially with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate-hexane (1: 1) as an elution solvent, and 3- (2-acetoxy-1-methylethoxy) -5-formyl-4-{[ 2.03 g of (trifluoromethyl) sulfoyl] oxy} benzylacetate was obtained. NMR (CDC1): 1.44 (3H, d, J = 6.3 Hz), 2.07 (3H, s), 2.15 (3H, s), 4.20-4.34 (2H, m), 4.75
3 Three
-4.84 (1H, m), 5.14 (2H, s), 7.36 (1H, d, J = 2.0 Hz), 7.51 (1H, d, J = 2.0 Hz), 10.2 3 (1H, s) -4.84 (1H, m), 5.14 (2H, s), 7.36 (1H, d, J = 2.0 Hz), 7.51 (1H, d, J = 2.0 Hz), 10.2 3 (1H, s)
[0156] 参考例 87 [0156] Reference Example 87
3-(2-ァセトキシ -1-メチルエトキシ) -5-ホルミル- 4-{[ (トリフルォロメチル)スルホ -ル] ォキシ }ベンジルァセタート 2.03 g、アクリル酸 tert-ブチル 13.1 mL、 1,3-ビス (ジフエ -ルホスフイノ)プロパン 379 mg、酢酸パラジウム 103 mg、 Ν,Ν-ジイソプロピルェチル ァミン 1.2 mLの Ν,Ν-ジメチルホルムアミド(12mL)溶液を、封管中マイクロ波照射下 1 50°Cで 10分間攪拌した。反応液に水を加え、酢酸ェチルで抽出した。有機層を水お よび飽和食塩水で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥して減圧下 濃縮した。得られた残渣を酢酸ェチル 一へキサン(33 : 67)を溶出溶媒とするシリカ ゲルカラムクロマトグラフィーで精製し、 tert-ブチル(E)-3-[4-ァセトキシメチル -2-(2- ァセトキシ -1-メチルエトキシ) -6-ホルミルフエ-ル]アタリラートを 1.03 g得た。 NMR (C DC1 ) : 1.40 (3H, d, J = 6.3 Hz), 1.54 (9H, s), 2.07 (3H, s), 2.14 (3H, s), 4.12—4.31 ( 3- (2-acetoxy-1-methylethoxy) -5-formyl-4-{[(trifluoromethyl) sulfol] oxy} benzylacetate 2.03 g, tert-butyl acrylate 13.1 mL, 1, 3-Bis (diphenol-phosphino) propane 379 mg, palladium acetate 103 mg, Ν, Ν-diisopropylethylamine 1.2 mL of Ν, Ν-dimethylformamide (12 mL) in a sealed tube under microwave irradiation at 150 ° Stir at C for 10 min. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed sequentially with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using ethyl acetate / hexane (33:67) as an elution solvent, and tert-butyl (E) -3- [4-acetoxymethyl-2- (2-acetoxy- 1.03 g of 1-methylethoxy) -6-formylphenol] talylate was obtained. NMR (C DC1): 1.40 (3H, d, J = 6.3 Hz), 1.54 (9H, s), 2.07 (3H, s), 2.14 (3H, s), 4.12—4.31 (
3 Three
2H, m), 4.66-4.73 (1H, m), 5.13 (2H, s), 6.16 (1H, d, J = 16.1 Hz), 7.19 (1H, d, J = 1.2 Hz), 7.52 (1H, d, J = 1.2 Hz), 8.01 (1H, d, J = 16.1 Hz), 10.22 (1H, s) 2H, m), 4.66-4.73 (1H, m), 5.13 (2H, s), 6.16 (1H, d, J = 16.1 Hz), 7.19 (1H, d, J = 1.2 Hz), 7.52 (1H, d , J = 1.2 Hz), 8.01 (1H, d, J = 16.1 Hz), 10.22 (1H, s)
[0157] 上記参考例 1〜87の方法と同様にして、後記表 4〜24に示す参考例化合物 88〜 230をそれぞれ対応する原料を使用して製造した。表 4〜24に参考例化合物の構 造及び物理化学的データを示す。
[0158] 実施例 1 [0157] In the same manner as in Reference Examples 1 to 87, Reference Example compounds 88 to 230 shown in Tables 4 to 24 below were produced using the corresponding raw materials. Tables 4 to 24 show the structures and physicochemical data of the reference compounds. [0158] Example 1
tert-ブチル {2-エトキシ -6- [(ヒドロキシィミノ)メチル]フエノキシ }ァセタート 500 mgの エタノール溶液 (17 mL)に 10%パラジウム炭素粉末 50 mgを加え 3気圧の水素雰囲気 下室温にて 24時間攪拌した。反応液を濾過し、濾液を減圧下濃縮した。得られた無 色油状物を Ν,Ν-ジメチルホルムアミド 15 mLに溶解し、 tert-ブチル [{4- [(5-シァノ - 2- フルォ口べンゾィル)ァミノ]フエ-ル} (ィミノ)メチル]力ルバマート 0.37 gと炭酸カリウム 4 14 mgを室温にて加え、 80 °Cにて 12時間攪拌した。反応液を減圧下濃縮した。残渣 を酢酸ェチルで希釈し、水次いで飽和食塩水で洗浄した。有機層を無水硫酸マグネ シゥムで乾燥し減圧下濃縮した後、得られた残渣をクロ口ホルム/メタノール(100 : 1) を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し tert-ブチル {2_[({2_[({4 -[[(tert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-シァ ノフエ-ル}ァミノ)メチル ]-6-エトキシフエノキシ }ァセタート 0.26 gを無色固体として得 た。 tert-Butyl {2-Ethoxy-6-[[hydroxyimino) methyl] phenoxy} acetate 50 mg of 10% palladium carbon powder is added to an ethanol solution (17 mL) of 500 mg at room temperature under a hydrogen atmosphere of 3 atm. Stir for hours. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure. Dissolve the resulting colorless oil in 15 mL of Ν, Ν-dimethylformamide, and add tert-butyl [{4- [(5-ciano-2-fluorobenzoyl) amino] phenol} (imino) methyl. ] Powerful rubamate 0.37 g and potassium carbonate 4 14 mg were added at room temperature, and the mixture was stirred at 80 ° C for 12 hours. The reaction solution was concentrated under reduced pressure. The residue was diluted with ethyl acetate and washed with water and then saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using chloroform / methanol (100: 1) as an elution solvent, and tert-butyl {2 _ [( {2 _ [({4-[[(tert-Butoxycarbol) amino] (imino) methyl] phenol} amino) carbol] -4-cyanophen} amino) methyl] -6-ethoxy 0.26 g of phenoxy} acetate was obtained as a colorless solid.
[0159] 実施例 2 [0159] Example 2
tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル Kィミノ)メチル]力ルバ マートと tert-ブチル(2-ホルミル- 6-ヒドロキシフエノキシ)ァセタートを原料として、参 考例 61と同様の操作により tert-ブチル {2-[({2- [({4-[[(tert-ブトキシカルボニル)アミ ノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6-ヒドロ キシフエノキシ }ァセタートを黄色固体として得た。 tert-Butyl [{4- [(2-amino-5-chlorobenzoyl) amino] phenol Kimino) methyl] rubamate and tert-butyl (2-formyl-6-hydroxyphenoxy) acetate As a starting material, tert-butyl {2-[({2-[({4-[[(tert-butoxycarbonyl) amino] (imino) methyl] phenol} amino was prepared in the same manner as in Reference Example 61. ) Carboxyl] -4-chlorophenol} amino) methyl] -6-hydroxyphenoxy} acetate was obtained as a yellow solid.
[0160] 実施例 3 [0160] Example 3
tert-ブチル {2-[({2-[({4-[[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル} ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6-ヒドロキシフエノキシ }ァセタート を原料として参考例 47と同様の操作を行い、 tert-ブチル {2- [({2- [({4-[[(tert-ブトキ シカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}アミ ノ)メチル ]-6-[(1-メチルピロリジン- 2-ィル)メトキシ]フエノキシ }ァセタートを黄色固体と して得た。 tert-Butyl {2-[({2-[({4-[[(tert-Butoxycarbonyl) amino] (imino) methyl] phenyl} amino) carbol] -4-chlorophine} The same procedure as in Reference Example 47 was performed using amino) methyl] -6-hydroxyphenoxy} acetate as a raw material, and tert-butyl {2-[({2-[({4-[[(tert-butoxycarboxy- ) Amino] (imino) methyl] phenol} amino) carbol] -4-chloro-phenol} amino) methyl] -6-[(1-methylpyrrolidine-2-yl) methoxy] The phenoxy} acetate was obtained as a yellow solid.
[0161] 実施例 4 [0161] Example 4
tert-ブチル {2-[({2-[({4-[[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル}
ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6-メトキシ- 4--トロフエノキシ }ァ セタートを原料として参考例 77と同様の操作を行 ヽ tert-ブチル {4-ァミノ- 2- [({2- [({4 -[[(tert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ 口フエ-ル}ァミノ)メチル ]-6-メトキシフエノキシ }ァセタートを黄色固体として得た。 tert-butyl {2-[({2-[({4-[[(tert-butoxycarbonyl) amino] (imino) methyl] phenyl} [Amino) Carbol]-4-chlorophenol} amino) methyl] -6-methoxy-4-trophenoxy} acetate and the same operation as in Reference Example 77 tert-butyl {4- Amino-2-[({2-[({4-[[(tert-Butoxycarbole) amino] (imino) methyl] phenol} Amino) carbole] -4-chlorophine} Amino) methyl] -6-methoxyphenoxy} acetate was obtained as a yellow solid.
[0162] 実施例 5 [0162] Example 5
tert-ブチル {2- [({4-クロ口- 2- [(4-シァノベンゾィル)ァミノ]フエ-ル}ァミノ)メチル ]-6 tert-Butyl {2- [({4-Black-Two-2-[(4-Cyanobenzoyl) amino] phenol} amino) methyl] -6
-エトキシフエノキシ }ァセタート 0.72 gのエタノール溶液 (7 mL)を- 78°Cに冷却し塩化 水素ガスを 15分間通導した。反応液を室温にて 18時間攪拌した後、減圧下濃縮し た。得られた残渣をエタノール 30 mLに溶解し、酢酸アンモ-ゥム 0.52 gを室温にて 加え 24時間攪拌した後、 60°Cにて 4時間攪拌した。反応液を減圧下濃縮し、得られ た残渣をエタノール/水(1 : 2)を溶出溶媒とする ODSカラムクロマトグラフィーで精製 し、ェチル [2— ({[2— ({4— [ァミノ (ィミノ)メチル]ベンゾィル }ァミノ )—4—クロ口フエ-ル]ァミノ) メチル )-6-エトキシフエノキシ]ァセタート 0.70 gを黄色固体として得た。 -Ethoxyphenoxy} acetate An ethanol solution (7 mL) of 0.72 g was cooled to -78 ° C, and hydrogen chloride gas was introduced for 15 minutes. The reaction solution was stirred at room temperature for 18 hours and then concentrated under reduced pressure. The obtained residue was dissolved in 30 mL of ethanol, 0.52 g of ammonium acetate was added at room temperature and stirred for 24 hours, and then stirred at 60 ° C for 4 hours. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by ODS column chromatography using ethanol / water (1: 2) as the elution solvent, and ethyl [ 2 — ({[ 2 — ({ 4 — [amino ( Imino) methyl] Benzoiru} Amino) - 4 - chloro port Hue - Le] Amino) methyl) -6-ethoxy-phenoxyethanol] Asetato 0.70 g as a yellow solid.
[0163] 実施例 6 [0163] Example 6
tert-ブチル [4-クロ口- 2- (ヒドロキシメチル) -6-(4-メチル -2-ォキソピペラジン- 1-ィ ル)フエノキシ]ァセタート 28 mgを、ジクロロェタン 3 mUこ溶解し、デス一マーチンパー ョージナンのジクロロメタン溶液 (15%) 1 mLを室温にてカ卩え、 1時間撹拌した。減圧下 濃縮し、得られた残渣に tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ- ル }(ィミノ)メチル]力ルバマート 40 mg、酢酸 1 mL及びトリァセトキシ水素化ホウ素ナトリ ゥム 42 mgを室温にて順次加え 1時間攪拌した。反応液に水を加え酢酸ェチルで抽 出した。有機層を水、 5%重曹水次いで飽和食塩水で順次洗浄し、無水硫酸マグネシ ゥムで乾燥後減圧下濃縮した。得られた残渣をメタノール/酢酸ェチル -へキサン( 1 :4:4)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し、 tert-ブチル [2 -[({2-[({4-[[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル}ァミノ)カルボニル ]-4-クロ口フエ-ル}ァミノ)メチル ]-4-クロ口- 6-(4-メチル -2-ォキソピペラジン- 1-ィル) フエノキシ]ァセタート 27 mgを無色油状物として得た。 Dissolve 28 mg of tert-butyl [4-chromo-2- (hydroxymethyl) -6- (4-methyl-2-oxopiperazine-1-yl) phenoxy] acetate in 3 mU of dichloroethane, 1 mL of a solution of martin peroxide in dichloromethane (15%) was placed at room temperature and stirred for 1 hour. Concentrate under reduced pressure, and add tert-butyl [{4- [(2-amino-5-chlorobenzoyl) amino] phenol} (imino) methyl] strength rubamate 40 mg, acetic acid 1 mL and 42 mg of sodium triacetoxyborohydride was sequentially added at room temperature and stirred for 1 hour. Water was added to the reaction solution and extracted with ethyl acetate. The organic layer was washed successively with water, 5% aqueous sodium bicarbonate and then saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using methanol / ethyl acetate-hexane (1: 4: 4) as an elution solvent, and tert-butyl [2-[({2-[({4-[[ (tert-butoxycarbonyl) amino] (imino) methyl] phenyl} amino) carbonyl] -4-chloro-amino} methyl) methyl] -4-chloro-6- (4-methyl-2-o Xisopiperazine-1-yl) phenoxy] acetate 27 mg was obtained as a colorless oil.
[0164] 実施例 7 [0164] Example 7
2-ァミノ- N- {4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}- 5-クロ口べンズアミド 0.65 gと
ェチル(2-エトキシ- 6-ホルミルフエノキシ)ァセタート 0.54 gの酢酸(10 mL)溶液にトリ ァセトキシ水素化ホウ素ナトリウム 0.90 gを室温にてカ卩ぇ 30分間攪拌した。反応液に 水を加え酢酸ェチルで抽出した。有機層を水、 5%重曹水次いで飽和食塩水で順次 洗浄し、無水硫酸マグネシウムで乾燥後減圧下濃縮した。得られた残渣を酢酸ェチ ル /n-へキサン(3 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製しェ チル {2-[({2-[({4- [ァミノ (ヒドロキシィミノ)メチル]フエ二ル}ァミノ)カルボ二ル]- 4-クロ口 フエ-ル}ァミノ)メチル ]-6-エトキシフエノキシ }ァセタート 467 mgを黄色固体として得 た。 2-Amino- N- {4- [Amino (hydroxyimino) methyl] phenol} -5-Black mouth Bensamide 0.65 g Ethyl (2-ethoxy-6-formylphenoxy) acetate 0.54 g of acetic acid (10 mL) solution was stirred with sodium triacetoxyborohydride (0.90 g) at room temperature for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water, 5% aqueous sodium bicarbonate and then saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (3: 1) as an elution solvent. Ethyl {2-[({2-[({4- [amino (hydroxy 467 mg of (imino) methyl] phenyl} amino) carbonyl] -4-chloro (phenyl) amino) methyl] -6-ethoxyphenoxy} acetate was obtained as a yellow solid.
[0165] 実施例 8及び実施例 9 [0165] Example 8 and Example 9
tert-ブチル {2- [({2- [({6- [ァミノ (ヒドロキシィミノ)メチル]ピリジン- 3-ィル }ァミノ)カル ボ-ル ]-4-クロ口フエ-ル}ァミノ)メチル ]-6-エトキシフエノキシ }ァセタート 57 mgの酢 酸溶液 (1 mL)に 10%パラジウム炭素粉末 20 mgを加え 4気圧の水素雰囲気下室温 にて 18時間攪拌した。反応液を濾過し、濾液を減圧下濃縮した。得られた残渣を 0.1 %ギ酸水/ァセトニトリルを溶媒とする ODSカラムクロマトグラフィーにて精製し、 tert-ブ チル {2-[({2-[({6- [ァミノ (ィミノ)メチル]ピリジン- 3-ィル }ァミノ)カルボニル] -4-クロロフ ェ-ル }ァミノ)メチル ]-6-エトキシフエノキシ }ァセタートギ酸塩 16 mg (実施例 8)と tert- ブチル {2-[({2-[({6- [ァミノ (ィミノ)メチル]ピリジン- 3-ィル }ァミノ)カルボ-ル]フエ二ル} ァミノ)メチル ]-6-エトキシフエノキシ }ァセタートギ酸塩 5 mg (実施例 9)を得た。 tert-Butyl {2- [({2- [({6- [Amino (hydroxyimino) methyl] pyridine-3-yl} amino) carbol] -4-chloro-phenyl} amino) Methyl] -6-ethoxyphenoxy} acetate 20 mg of 10% palladium carbon powder was added to 57 mg of an acetic acid solution (1 mL), and the mixture was stirred at room temperature for 18 hours in a hydrogen atmosphere of 4 atm. The reaction solution was filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as a solvent, and tert-butyl {2-[({2-[({6- [amino (imino) methyl] pyridine- 3-yl} amino) carbonyl] -4-chlorophenyl} amino) methyl] -6-ethoxyphenoxy} acetoformate 16 mg (Example 8) and tert-butyl {2-[({2 -[({6- [Amino (imino) methyl] pyridine-3-yl} amino) carbol] phenyl} amino) methyl] -6-ethoxyphenoxy} acetate formate 5 mg (Example) 9) was obtained.
[0166] 実施例 10 [0166] Example 10
2- {[2- (2- tert-ブトキシ- 2-ォキソエトキシ)ベンジル]アミノ}- 5-クロ口安息香酸 3.00 g 、 4-ァミノべンズアミジン二塩酸塩 4.78 gの Ν,Ν-ジメチルホルムアミド溶液(30 mL) にピリジン 30 mLをカ卩えた後、 Ν,Ν'-ジシクロへキシルカルボジイミド 1.74 gを加え、室 温にて終夜攪拌した。反応液を濾過し、減圧下濃縮した。得られた残渣をクロ口ホル ム /メタノール (20 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにて精製し te rt-ブチル {2-[({2-[({4- [ァミノ (ィミノ)メチル]フエ二ル}ァミノ)カルボ二ル]- 4-クロ口フエ -ル }ァミノ)メチル]フエノキシ }ァセタート 1.16 gを黄色泡状物として得た。 2-{[2- (2-tert-butoxy-2-oxoethoxy) benzyl] amino} -5-chlorobenzoic acid 3.00 g, 4-aminobenzamidine dihydrochloride 4.78 g in Ν, Ν-dimethylformamide solution ( After 30 mL of pyridine was added to 30 mL), 1.74 g of Ν, Ν'-dicyclohexylcarbodiimide was added and stirred overnight at room temperature. The reaction solution was filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using chloroform / methanol (20: 1) as the elution solvent, and te rt-butyl {2-[({2-[({4- [amino (imino 1.16 g of) methyl] phenyl} amino) carbonyl] -4-chlorophenol-amino} methyl] phenoxy} acetate was obtained as a yellow foam.
[0167] 実施例 11 [0167] Example 11
tert-ブチル 3-{2-[({2-[({4- [ァミノ (ヒドロキシィミノ)メチル]フエ二ル}ァミノ)カルボニル
]-4-クロ口フエ-ル}ァミノ)メチル]ピぺリジン- 1-ィル }プロパノアート 40mgに塩化水素 ガスのエタノール溶液 (21重量%、 1.0 mL)を加え、室温にて 3時間攪拌した。反応 液を減圧下濃縮し、得られた残渣を酢酸ェチル /n-へキサン(7 : 1)を溶出溶媒とする ァミノプロピルィ匕シリカゲルカラムクロマトグラフィーにて精製し、ェチル 3-{2-[({2-[({4 -[ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メ チル]ピぺリジン- 1-ィル }プロパノアート 28 mgを淡黄色油状物として得た。 tert-Butyl 3- {2-[({2-[({4- [Amino (hydroxyimino) methyl] phenyl} amino) carbonyl ] -4-Chlorophenyl} amino) methyl] piperidine-1-yl} propanoate 40 mg of hydrogen chloride gas in ethanol (21 wt%, 1.0 mL) was added and stirred at room temperature for 3 hours. . The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by aminopropyl silica gel column chromatography using ethyl acetate / n-hexane (7: 1) as an elution solvent, and ethyl 3- {2-[({ 2-[({4- [Amino (hydroxyimino) methyl] phenol} amino) carbol] -4-chlorophine} amino) methyl] piperidine-1-yl} propanoate 28 mg was obtained as a pale yellow oil.
[0168] 実施例 12 [0168] Example 12
tert-ブチル {2-[({2-[({4-[[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル} ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル]フエノキシ }ァセタート 500 mgの塩 化メチレン溶液 (10 mL)にトリフルォロ酢酸 10 mLを室温でカ卩ぇ 3時間攪拌した。反 応液を減圧下濃縮した。得られた残渣に酢酸ェチルを加え、析出物を濾取し、濾物 を減圧下乾燥して {2-[({2-[({4- [ァミノ (ィミノ)メチル]フエ二ル}ァミノ)カルボニル] -4-ク ロロフエ-ル}ァミノ)メチル]フエノキシ }ァセチックアシッドトリフルォロ酢酸塩 254 mgを 淡黄色固体として得た。 tert-Butyl {2-[({2-[({4-[[(tert-Butoxycarbonyl) amino] (imino) methyl] phenyl} amino) carbol] -4-chlorophine} Amino) methyl] phenoxy} acetate 500 mL of methylene chloride solution (10 mL) was stirred with 10 mL of trifluoroacetic acid at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure. Ethyl acetate was added to the resulting residue, the precipitate was collected by filtration, and the filtrate was dried under reduced pressure to give {2-[({2-[({4- [amino (imino) methyl] phenyl} amino}. 254 mg)] carbonyl] -4-chlorophenol} amino) methyl] phenoxy} acetic acid trifluoroacetate was obtained as a pale yellow solid.
[0169] 実施例 13 [0169] Example 13
tert-ブチル {2-[({2-[({4-[[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル} ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6-ヒドロキシフエノキシ }ァセタート 250 mgをテトラヒドロフラン 5 mLに溶解し、室温にて 2- (ジメチルァミノ)エタノール 53 m g、トリフエニルホスフィン 157 mg及びジイソプロピルァゾジカルボキシラート 0.15mLを 加え終夜攪拌した。反応液を減圧下濃縮し、得られた残渣を n-へキサン/酢酸ェチ ル(7 : 3)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製した。得られ た化合物に塩化メチレン 4 mL及びトリフルォロ酢酸 4 mLを室温にてカ卩え、 4時間撹 拌した。反応液を減圧下濃縮した後、酢酸ェチル 10 mL及び塩ィ匕水素の酢酸ェチル 溶液 (4 N, 1 mL)を加え、生じた固体を濾別乾燥し、 {2- [({2- [({4- [ァミノ (ィミノ)メチル] フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6- [2- (ジメチルァミノ)ェ トキシ]フエノキシ }ァセチックアシッド塩酸塩 99 mgを淡黄色固体として得た。 tert-Butyl {2-[({2-[({4-[[(tert-Butoxycarbonyl) amino] (imino) methyl] phenyl} amino) carbol] -4-chlorophine} Amino) methyl] -6-hydroxyphenoxy} acetate (250 mg) was dissolved in tetrahydrofuran (5 mL), and 2- (dimethylamino) ethanol (53 mg), triphenylphosphine (157 mg) and diisopropylazodicarboxylate (0.15 mL) were dissolved at room temperature. The mixture was stirred overnight. The reaction solution was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (7: 3) as an elution solvent. The obtained compound was charged with 4 mL of methylene chloride and 4 mL of trifluoroacetic acid at room temperature and stirred for 4 hours. After concentrating the reaction solution under reduced pressure, 10 mL of ethyl acetate and an ethyl acetate solution of sodium hydrogen carbonate (4 N, 1 mL) were added, and the resulting solid was filtered and dried, and {2- [({2- [ ({4- [Amino (methyl) phenyl) amino) carbol]-4-chlorophenol} amino) methyl] -6- [2- (dimethylamino) ethoxy] phenoxy} case 99 mg of tic acid hydrochloride was obtained as a pale yellow solid.
[0170] 実施例 14 [0170] Example 14
tert-ブチル {2-[({2-[({4-[[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル}
ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6-ヒドロキシフエノキシ }ァセタート 250 mgをテトラヒドロフラン 5 mLに溶解し、室温にて 3- (メチルォキセタン- 3-ィル)メタ ノール 61 mg、トリフエニルホスフィン 157 mg及びジイソプロピルァゾジカルボキシラー ト 0.15mLを加え終夜攪拌した。反応液を減圧下濃縮し、得られた残渣を n-へキサン/ 酢酸ェチル (7 : 3)を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し た。得られた化合物に塩化メチレン 4 mL及びトリフルォロ酢酸 4 mLを室温にてカロえ、 4時間撹拌した。反応液を減圧下留去した後、酢酸ェチル 10 mLを加え、生じた固体 を濾別乾燥し、 {2-[({2-[({4- [ァミノ (ィミノ)メチル]フエ二ル}ァミノ)カルボニル] -4-クロ口 フエ-ル}ァミノ)メチル ]-6-[(3-メチルォキセタン- 3-ィル)メトキシ]フエノキシ }ァセチッ クアシッドトリフルォロ酢酸塩 66 mgを淡黄色固体として得た。 tert-butyl {2-[({2-[({4-[[(tert-butoxycarbonyl) amino] (imino) methyl] phenyl} Amino) carbol] -4-chlorophenol} amino) methyl] -6-hydroxyphenoxy} acetate 250 mg was dissolved in 5 mL of tetrahydrofuran, and 3- (methyloxetane-3-yl) was dissolved at room temperature. ) Methanol 61 mg, triphenylphosphine 157 mg, and diisopropylazodicarboxylate 0.15 mL were added and stirred overnight. The reaction solution was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (7: 3) as an elution solvent. The resulting compound was charged with 4 mL of methylene chloride and 4 mL of trifluoroacetic acid at room temperature and stirred for 4 hours. After evaporating the reaction solution under reduced pressure, 10 mL of ethyl acetate was added, and the resulting solid was separated by filtration and dried, and {2-[({2-[({4- [amino (imino) methyl] phenyl} Amino) carbonyl] -4-chloro-phenyl} amino) methyl] -6-[(3-methyloxetane-3-yl) methoxy] phenoxy} acetic acid trifluoroacetate 66 mg as a pale yellow solid Obtained.
[0171] 実施例 15 [0171] Example 15
tert-ブチル(2- {[(2- {[(4- {[(tert-ブトキシカルボニル)ァミノ]メチル }フエニル)ァミノ]力 ルポ-ル}-4-クロ口フエ-ル)ァミノ]メチル }-6-エトキシフエノキシ)ァセタート 300 mgの 塩化メチレン溶液 (4 mL)にトリフルォロ酢酸(4 mL)を室温でカ卩ぇ 2時間攪拌した。 反応液を減圧下濃縮した。得られた残渣を酢酸ェチル 20 mLに溶解し、塩化水素の 酢酸ェチル溶液 (4 N, 1 mL)を室温で加え 30分間攪拌した。析出物を濾取し、濾物 を減圧下乾燥して [2- ({[2- ({[4- (アミノメチル)フエニル]アミノ}カルボ二ル)- 4-クロ口フエ -ル]アミノ}メチル )-6-エトキシフエノキシ]ァセチックアシッド塩酸塩 216 mgを淡黄色 固体として得た。 tert-Butyl (2- {[(2- {[(4- {[(tert-Butoxycarbonyl) amino] methyl} phenyl) amino] force Lolol-4--4-phenyl) amino) methyl} -6-Ethoxyphenoxy) acetate Trifluoroacetic acid (4 mL) was stirred at room temperature for 2 hours in 300 mg of methylene chloride solution (4 mL). The reaction solution was concentrated under reduced pressure. The obtained residue was dissolved in 20 mL of ethyl acetate, a solution of hydrogen chloride in ethyl acetate (4 N, 1 mL) was added at room temperature, and the mixture was stirred for 30 minutes. The precipitate was collected by filtration, and the filtrate was dried under reduced pressure to give [2-({[2- ({[4- (aminomethyl) phenyl] amino} carbonyl) -4-chlorophenyl] -amino] amino. } 216 mg of methyl) -6-ethoxyphenoxy] acetic acid hydrochloride was obtained as a pale yellow solid.
[0172] 実施例 16 [0172] Example 16
ェチル [(3- {[(2- {[(2-ァミノ- 1H-ベンズイミダゾール- 5-ィル)ァミノ]カルボ-ル}- 4-ク ロロフエ-ル)ァミノ]メチル }ビフエ-ル -2-ィル)ォキシ]ァセタート 200 mgをエタノール 5 mLに溶解し、室温にて 1規定水酸化ナトリウム水溶液 0.71 mLを加え 3時間攪拌し た。室温にて 1規定塩酸 0.71 mLを加え攪拌した後、生成した沈殿を濾取し水で洗 浄した。得られた無色固体を減圧下乾燥し、 [(3-{[(2-{[(2-ァミノ- 1H-ベンズイミダゾ ール -5-ィル)ァミノ]カルボ-ル}-4-クロ口フエ-ル)ァミノ]メチル }ビフエ-ル -2-ィル)ォ キシ]ァセテイクアシッドを 148 mg得た。 Ethyl [(3- {[(2- {[(2-Amino-1H-benzimidazol-5-yl) amino] carbol} -4-chlorophenol) amino] methyl} biphenyl -2 -Ill) oxy] acetate (200 mg) was dissolved in ethanol (5 mL), 1N aqueous sodium hydroxide solution (0.71 mL) was added at room temperature, and the mixture was stirred for 3 hr. After adding 0.71 mL of 1N hydrochloric acid at room temperature and stirring, the formed precipitate was collected by filtration and washed with water. The obtained colorless solid was dried under reduced pressure, and [(3-{[(2-{[(2-Amino-1H-benzimidazol-5-yl) amino] carbol} -4-black mouth was obtained. 148 mg of vinyl) amino] methyl} biphenyl-2-yl) oxy] acetic acid was obtained.
[0173] 実施例 17
ェチル 4- {2- [({2- [({4- [[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル}アミ ノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル]ピぺリジン- 1-ィル }プチラート 185 mgの 塩化メチレン(1.5 mL)溶液にトリフルォロ酢酸 1.5 mLを室温にてカロえ 5時間攪拌し た。反応液を減圧下濃縮した。得られた残渣をエタノール 0.80 mLに溶解し、 1規定 水酸化ナトリウム水溶液 0.78 mLを加え 9時間撹拌した。反応液を減圧下濃縮し、得 られた残渣を 0.1%ギ酸水/ァセトニトリルを溶媒とする ODSカラムクロマトグラフィーに て精製し、 4- {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロロフ ェニル }ァミノ)メチル]ピぺリジン- 1-ィル }プチリックアシッドギ酸塩 72 mgを淡黄色油 状物として得た。 [0173] Example 17 Ethyl 4- {2- [({2- [({4- [[(tert-Butoxycarbonyl) amino] (imino) methyl] phenyl} amino) carbol] -4 } Amino) methyl] piperidine-1-yl} Pytolate To a solution of 185 mg of methylene chloride (1.5 mL) was added 1.5 mL of trifluoroacetic acid at room temperature and stirred for 5 hours. The reaction solution was concentrated under reduced pressure. The obtained residue was dissolved in ethanol 0.80 mL, 1N aqueous sodium hydroxide solution 0.78 mL was added, and the mixture was stirred for 9 hr. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the solvent, and 4- {2-[({2-[({4- [amino ( Imino) methyl] phenyl} amino) carbol] -4-chlorophenyl} amino) methyl] piperidine-1-yl} petitic acid formate 72 mg was obtained as a pale yellow oil.
[0174] 実施例 18 [0174] Example 18
tert-ブチル 3- {2- [({2- [({4- [[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエニル tert-butyl 3- {2- [({2- [({4- [[(tert-butoxycarbonyl) amino] (imino) methyl] phenyl
}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル]ピぺリジン- 1-ィル }プロパノアート 4} Amino) Carbol]-4-Black Mole} Amino) Methyl] piperidine-1-yl} Propanoate 4
3 mgの塩化メチレン溶液 (0.35 mL)にトリフルォロ酢酸(0.35 mL)を室温にて加え 4 時間攪拌した。反応液を減圧下濃縮し、 0.1%ギ酸水/ァセトニトリルを溶媒とする ODS カラムクロマトグラフィーにて精製し、 3 [({2 [({4 [ァミノ (ィミノ)メチル]フエ-ル}アミ ノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル]ピぺリジン- 1-ィル }プロパノイツクァシ ッドギ酸塩 15 mgを淡黄色固体として得た。 Trifluoroacetic acid (0.35 mL) was added to 3 mg of methylene chloride solution (0.35 mL) at room temperature and stirred for 4 hours. The reaction mixture was concentrated under reduced pressure and purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the solvent, and 3 [({ 2 [({ 4 [Amino (imino) methyl] phenol} amino) Thus, 15 mg of (carbol-4-phenyl) amino) methyl] piperidine-1-yl} propanoic acid formate was obtained as a pale yellow solid.
[0175] 実施例 19 [0175] Example 19
tert-ブチル(2-ァセチルフエノキシ)ァセタート 14.0 g、(2-ァミノ- 5-クロ口フエ-ル)メ タノール 8.82 g、酢酸 30 mL及び石油エーテル 90 mLの混合物に室温にてボラン-ピ リジン錯体 7.0 mLをカ卩え、室温にて 2時間攪拌した。反応液に 5規定塩酸 34 mLを室 温にて加え 1時間攪拌した。反応液に重曹水を加え酢酸ェチルで抽出し、有機層を 飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後減圧下濃縮した。得られた残 渣を酢酸ェチル -へキサン(1 : 9)を溶出溶媒とするシリカゲルカラムクロマトグラフ ィ一で精製し、無色油状物 14.5 gを得た。得られた無色油状物 500 mgをアセトン 200 mLに溶解し、無水硫酸マグネシウム 1.08 gと 1%過マンガン酸カリウム水溶液 80 mL を順次加え、 80 °Cにおいて終夜撹拌した。反応液を減圧留去した後、酢酸ェチルで 抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後減圧下濃
縮した。得られた残渣を酢酸ェチル /n-へキサン(1 : 9)を溶出溶媒とするシリカゲル カラムクロマトグラフィーで精製し、無色泡状物 124 mgを得た。得られた無色泡状物 1 24 mgと tert-ブチル(4-ァミノベンジル)力ルバマートを原料とし、参考例 68と同様の 操作を行い無色泡状物 73 mgを得た。得られた無色泡状物を原料とし、実施例 15と 同様の操作を行い [2- (1- {[2- ({[4- (アミノメチル)フエ-ル]アミノ}カルボ-ル)- 4-クロ口 フエ-ル]アミノ}ェチル)フエノキシ]ァセチックアシッド塩酸塩 43 mgを無色固体として 得た。 tert-Butyl (2-acetyl-phenoxy) acetate 14.0 g, (2-amino-5-phenyl) methanol 8.82 g, acetic acid 30 mL and petroleum ether 90 mL 7.0 mL of pyridine complex was added and stirred at room temperature for 2 hours. To the reaction solution, 34 mL of 5N hydrochloric acid was added at room temperature and stirred for 1 hour. Sodium bicarbonate water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography using ethyl acetate-hexane (1: 9) as an elution solvent to obtain 14.5 g of a colorless oil. 500 mg of the obtained colorless oil was dissolved in 200 mL of acetone, 1.08 g of anhydrous magnesium sulfate and 80 mL of 1% aqueous potassium permanganate solution were sequentially added, and the mixture was stirred at 80 ° C. overnight. The reaction solution was evaporated under reduced pressure, and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. Shrinked. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 9) as an elution solvent to obtain 124 mg of a colorless foam. Using the obtained colorless foam 1 24 mg and tert-butyl (4-aminobenzyl) strength rubamate as raw materials, the same operation as in Reference Example 68 was carried out to obtain 73 mg of a colorless foam. The obtained colorless foam was used as a raw material, and the same operation as in Example 15 was performed. [2- (1-{[2-({[4- (aminomethyl) phenol] amino} carbol)- 43 mg of 4-chloro (phenyl) amino} ethyl) phenoxy] acetic acid hydrochloride was obtained as a colorless solid.
[0176] 実施例 20 [0176] Example 20
tert-ブチル {2- [({2- [({6- [ァミノ (ィミノ)メチル]ピリジン- 3-ィル }ァミノ)カルボ-ル]- 4- クロ口フエ-ル}ァミノ)メチル ]-6-エトキシフエノキシ }ァセタートギ酸塩 16 mgを塩化メ チレン(0.75 mL)に溶解し、トリフルォロ酢酸 0.75 mLを室温にてカ卩ぇ終夜攪拌した。 反応液を減圧下濃縮し、 {2-[({2-[({6- [ァミノ (ィミノ)メチル]ピリジン- 3-ィル }ァミノ)カル ボニル ]-4-クロ口フエ二ル}ァミノ)メチル ]-6-エトキシフエノキシ }ァセチックアシッドトリ フルォロ酢酸塩 13 mgを淡黄色固体として得た。 tert-Butyl {2- [({2- [({6- [Amino (imino) methyl] pyridine-3-yl} amino) carbol] -4- 4-chlorophenol} amino) methyl]- 16 mg of 6-ethoxyphenoxy} acetate was dissolved in methyl chloride (0.75 mL), and 0.75 mL of trifluoroacetic acid was stirred overnight at room temperature. The reaction solution was concentrated under reduced pressure, and {2-[({2-[({6- [amino (imino) methyl] pyridine-3-yl} amino) carbonyl] -4-chlorophenyl} amino ) Methyl] -6-ethoxyphenoxy} acetic acid trifluoroacetate 13 mg was obtained as a pale yellow solid.
[0177] 実施例 21 [0177] Example 21
tert-ブチル {2-[({2-[({4-[[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル} ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6-シァノフエノキシ }ァセタート 425 mg及び塩化ニッケル六水和物 319 mgのエタノール(10 mL)溶液に室温にて水素化 ホウ素ナトリウム 254 mgを加え、室温にて 1時間撹拌した。反応液に酢酸ェチル及び 1規定塩酸を加えた後、重曹水を加え、セライト濾過した。濾液を酢酸ェチルにて抽 出し、有機層を飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、減圧下 濃縮した。得られた残渣をクロ口ホルム/メタノール(20 : 1)を溶出溶媒とするシリカゲ ルカラムクロマトグラフィーにより精製した。得られた残渣に塩化メチレン 4 mL及びトリ フルォロ酢酸 4 mLを加え、室温にて 4時間撹拌した。反応液を減圧下濃縮した後、 酢酸ェチル 10 mLを加え、生じた固体を濾別乾燥して [2-[({2-[({4- [ァミノ (ィミノ)メチ ル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6- (アミノメチル)フエノ キシ]ァセチックアシッドトリフルォロ酢酸塩 68 mgを黄色固体として得た。 tert-Butyl {2-[({2-[({4-[[(tert-Butoxycarbonyl) amino] (imino) methyl] phenyl} amino) carbol] -4-chlorophine} To a solution of (amino) methyl] -6-cyanophenoxy} acetate (425 mg) and nickel chloride hexahydrate (319 mg) in ethanol (10 mL) was added sodium borohydride (254 mg) at room temperature, and the mixture was stirred at room temperature for 1 hour. Ethyl acetate and 1N hydrochloric acid were added to the reaction solution, sodium bicarbonate water was added, and the mixture was filtered through celite. The filtrate was extracted with ethyl acetate, and the organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using chloroform / methanol (20: 1) as an elution solvent. Methylene chloride (4 mL) and trifluoroacetic acid (4 mL) were added to the resulting residue, and the mixture was stirred at room temperature for 4 hours. After concentrating the reaction solution under reduced pressure, 10 mL of ethyl acetate was added, and the resulting solid was separated by filtration and dried to give [2-[({2-[({4- [amino] methyl] phenol}. Amino) carbol] -4-chlorophenol} amino) methyl] -6- (aminomethyl) phenoxy] acetic acid trifluoroacetate 68 mg was obtained as a yellow solid.
[0178] 実施例 22
tert-ブチル {2- [2- (ベンジルォキシ)カルボ-ルメトキシ]- 6- [({2- [({4- [[(tert-ブトキ シカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}アミ ノ)メチル]フエノキシ }ァセタート 492mg、テトラヒドロフラン 2 mL、エタノール 2 mLおよび クロ口ベンゼン 2 mLの混合物に 10%パラジウム炭素粉末 30mgをカ卩え、水素雰囲気下 2時間攪拌した。触媒をセライトを通して濾別し、濾液を濃縮した。得られた残渣を酢 酸ェチルで粉末ィ匕し [3- [({2- [({4- [[(tert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]フエ -ル }ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-2- (2- tert-ブトキシ- 2-ォキソ エトキシ)フエノキシ]ァセチックアシッド 390mgを得た。 [0178] Example 22 tert-Butyl {2- [2- (Benzyloxy) carbolmethoxy] -6-[({2-[({4-[[(tert-Butoxycarbonyl) amino] (imino) methyl] phenol} Amino) Carbol] -4-chloro-phenyl} amino) methyl] phenoxy} acetate 492 mg, tetrahydrofuran 2 mL, ethanol 2 mL and chloroform benzene 2 mL were mixed with 10 mg palladium carbon powder 30 mg. The mixture was stirred for 2 hours in a hydrogen atmosphere. The catalyst was filtered off through celite and the filtrate was concentrated. The resulting residue was pulverized with ethyl acetate and [3- [({2- [({4- [[(tert-butoxycarbol) amino] (imino) methyl] phenol} amino) carbo). -Lu] -4-chlorophenol} amino) methyl] -2- (2-tert-butoxy-2-oxoethoxy) phenoxy] acetic acid 390 mg was obtained.
[0179] 実施例 23 [0179] Example 23
tert-ブチル {4-ァミノ- 2- [({2- [({4- [[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フ ェ-ル }ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6-イソプロポキシフエノキ シ}ァセタート 100 mg、ピリジン 13 μ L、ジクロロメタン 1.5 mLおよびテトラヒドロフラン 0. 5 mLの混合物に塩化(N-tert-ブトキシカルボ-ル)スルファミル 35 mgをカ卩え、室温で 終夜攪拌した。反応液に飽和塩ィ匕アンモ-ゥム水溶液を加え、酢酸ェチルで抽出し た。有機層を水および飽和食塩水で順次洗浄した。有機層を無水硫酸マグネシウム で乾燥して減圧下濃縮した。得られた残渣を酢酸ェチル /n—へキサン (1 : 2)を溶出 溶媒とするシリカゲルカラムクロマトグラフィーで精製し、 tert-ブチル [2-[({2-[({4-[[(t ert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ -ル }ァミノ)メチル ]-4-({[(tert-ブトキシカルボ-ル)ァミノ]スルホ-ル }ァミノ) -6-イソプ ロボキシフエノキシ]ァセタート 71 mgを黄色固体として得た。 tert-butyl {4-amino- 2- [({2- [({4- [[(tert-butoxycarbonyl) amino] (imino) methyl] phenyl} amino) carbol] -4-chloro Mouthphenol} amino) methyl] -6-isopropoxyphenoxyacetate 100 mg, pyridine 13 μL, dichloromethane 1.5 mL and tetrahydrofuran 0.5 mL in a mixture (N-tert-butoxycarbol) 35 mg of sulfamyl was added and stirred overnight at room temperature. To the reaction solution was added a saturated aqueous solution of ammonium chloride and extracted with ethyl acetate. The organic layer was washed successively with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography eluting with ethyl acetate / n-hexane (1: 2) as the solvent, and tert-butyl [2-[({2-[({4-[[(t ert-butoxycarbol) amino] (imino) methyl] phenol} amino) carbol] -4-chlorophenol -amino} methyl] -4-({[(tert-butoxycarbole) ) Amino] sulfol} amino) -6-isopropoxyphenoxy] acetate 71 mg was obtained as a yellow solid.
[0180] 実施例 24 [0180] Example 24
メチル 3-[({4-[[(tert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ -ル] -4- {[2- (2- tert-ブトキシ- 2-ォキソエトキシ) -3-エトキシベンジル]アミノ}ベンゾァ ート 50mg、水 0.3 mLおよび 1,4-ジォキサン 0.7 mLの混合物に室温にて水酸化リチウ ム一水和物 (19mg)をカ卩え、 2日間攪拌した。反応液を 0.1%ギ酸水/ァセトニトリルを溶 出溶媒とする ODSカラムクロマトグラフィーにより精製し、 3-[({4- [ァミノ (ィミノ)メチル]フ ェ-ル }ァミノ)カルボ-ル] -4-{[2- (カルボキシェトキシ)- 3-エトキシベンジル]アミノ}安 息香酸ギ酸塩を 18mg得た。
[0181] 実施例 25 Methyl 3-[({4-[[(tert-butoxycarbol) amino] (imino) methyl] phenol} amino) carbol] -4- {[2- (2- tert-butoxy-2 -Oxoethoxy) -3-ethoxybenzyl] amino} benzoate 50 mg, water 0.3 mL and 1,4-dioxane 0.7 mL were mixed with lithium hydroxide monohydrate (19 mg) at room temperature. Stir for days. The reaction solution was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the elution solvent, and 3-[({4- [amino (imino) methyl] phenyl} amino) carbol] -4 18 mg of-{[2- (carboxyethoxy) -3-ethoxybenzyl] amino} benzoic acid formate was obtained. [0181] Example 25
tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]力ルバ マート 79mgとトリァセトキシ水素化ホウ素ナトリウム 200 mgの酢酸(1 mL)溶液に、 tert -ブチル {2-エトキシ- 6-ホルミル- 4-[(2-ヒドロキシェチル) (メチル)ァミノ]フエノキシ }ァ セタート 110 mgの酢酸(1 mL)溶液を室温にてカ卩えて同温度で 30分間攪拌した。反 応液を減圧下濃縮した後に水を加え、酢酸ェチルで抽出した。有機層を水次いで飽 和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、減圧下濃縮した。得られた残 渣をクロ口ホルム/メタノール (99 : 1)を溶出溶媒とするシリカゲルカラムクロマトグラフ ィ一により精製した。得られた残渣に塩ィ匕水素の 1,4-ジォキサン溶液 (4 M)3 mLおよ び水 0.030 mLを室温にて加え、同温度にて終夜攪拌した。反応液を減圧下濃縮して {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ) メチル ]-6-エトキシ -4- [(2-ヒドロキシェチル) (メチル)ァミノ]フエノキシ }ァセチックァシ ッド 塩酸塩 35 mgを黄色固体として得た。 tert-Butyl [{4- [(2-amino-5-chlorobenzobenzoyl) amino] phenol} (imino) methyl] strength rubamate 79 mg and sodium triacetoxyborohydride 200 mg in acetic acid (1 mL) Tert-Butyl {2-Ethoxy-6-formyl-4-[(2-hydroxyethyl) (methyl) amino] phenoxy} acetate 110 mg of acetic acid (1 mL) was added at room temperature. Stir at temperature for 30 minutes. The reaction solution was concentrated under reduced pressure, water was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and then saturated Japanese brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using chloroform / methanol (99: 1) as an elution solvent. To the obtained residue were added 3 mL of a 1,4-dioxane solution (4 M) of hydrochloric acid and hydrogen and 0.030 mL of water at room temperature, and the mixture was stirred overnight at the same temperature. The reaction solution was concentrated under reduced pressure to obtain {2- [({2- [({4- [amino (imino) methyl] phenol} amino) carbol] -4-chlorophenol} amino) methyl ] -6-Ethoxy-4-[(2-hydroxyethyl) (methyl) amino] phenoxy} acetic acid hydrochloride 35 mg was obtained as a yellow solid.
[0182] 実施例 26 [0182] Example 26
tert-ブチル [{4- [(2-ァミノ- 5-メトキシベンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]カル バマート 107 mgの酢酸(2.00 mL)溶液にトリァセトキシ水素化ホウ素ナトリウム 118 m gおよびェチル(4-ァセトキシメチル- 2_ホルミル- 6_イソプロポキシフエノキシ)ァセタ ート 99 mgの酢酸(2.00 mL)溶液を加え、室温にて 2時間攪拌した。反応液に水を加 え酢酸ェチルで抽出し、有機層を飽和重曹水、水および飽和食塩水で洗浄し、無水 硫酸ナトリウムで乾燥後減圧下濃縮した。得られた残渣をジクロロメタン 2.0 mLに溶 解し、室温にてトリフルォロ酢酸 2.00 mLを加え 1時間攪拌した。減圧下濃縮し、得ら れた残渣を酢酸ェチルおよびジェチルエーテルを用いて洗浄し黄色固体を得た。得 られたィ匕合物に室温にてァセトニトリル 3.2 mLおよび 1規定水酸ィ匕ナトリウム水溶液 0.81 mLをカ卩ぇ 4時間攪拌した。室温にて 1規定水酸化ナトリウム水溶液 1.62 mLを 加え、 2時間攪拌した。反応液を減圧下濃縮し、 1規定塩酸 2.43 mLを加えた。再度 反応液を減圧下濃縮し、得られた残渣を水/ァセトニトリル(1:5)を溶出溶媒とする 0 DSカラムクロマトグラフィーにて精製し、 {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}アミ ノ)カルボ-ル] -4-メトキシフエ-ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロボ
キシフエノキシ }ァセチックアシッド 46 mgを黄色固体として得た。 tert-Butyl [{4- [(2-amino-5-methoxybenzoyl) amino] phenol} (imino) methyl] carbamate 107 mg acetic acid (2.00 mL) in sodium triacetoxyborohydride 118 mg A solution of 99 mg of ethyl (4-acetoxymethyl-2-formyl-6-isopropoxyphenoxy) acetate in acetic acid (2.00 mL) was added, and the mixture was stirred at room temperature for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated aqueous sodium hydrogen carbonate, water and saturated brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained residue was dissolved in 2.0 mL of dichloromethane, 2.00 mL of trifluoroacetic acid was added at room temperature, and the mixture was stirred for 1 hour. Concentration was carried out under reduced pressure, and the resulting residue was washed with ethyl acetate and jetyl ether to obtain a yellow solid. To the obtained compound, 3.2 mL of acetonitrile and 0.81 mL of 1N sodium hydroxide aqueous solution were stirred at room temperature for 4 hours. At room temperature, 1.6N mL of 1N aqueous sodium hydroxide solution was added and stirred for 2 hours. The reaction mixture was concentrated under reduced pressure, and 2.43 mL of 1N hydrochloric acid was added. The reaction solution was again concentrated under reduced pressure, and the resulting residue was purified by 0 DS column chromatography using water / acetonitrile (1: 5) as an elution solvent, and {2- [({2- [({4- [Amino (imino) methyl] phenol} amino) carbol] -4-methoxyphenol} amino) methyl] -4- (hydroxymethyl) -6-isoprobo 46 mg of xyphenoxy} acetic acid was obtained as a yellow solid.
[0183] 実施例 27 [0183] Example 27
tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル Kィミノ)メチル]力ルバ マート 295 mgと tert-ブチル {[4- (1R)- 1,2-ジヒドロキシェチル] -2-エトキシ- 6-ホルミル フエノキシ }ァセタート 258 mgの酢酸(5 mL)溶液にトリァセトキシ水素化ホウ素ナトリウ ム 321 mgを室温にて加え 1時間攪拌した。反応液に水を加え酢酸ェチルで抽出した 。有機層を水、 5%重曹水次いで飽和食塩水で順次洗浄し、無水硫酸マグネシウムで 乾燥後減圧下濃縮した。得られた残渣を酢酸ェチル /n-へキサン(3 : 1)を溶出溶媒 とするシリカゲルカラムクロマトグラフィーで精製し淡黄色泡状物を得た。得られたィ匕 合物に 4規定塩酸 6 mLを加え、室温にて 7時間撹拌した。反応液に 1規定水酸化ナト リウム水溶液 24 mLを加え、生じた固体を濾別乾燥し、 [2-[({2-[({4- [ァミノ (ィミノ)メチ ル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-4- (1R)- 1,2-ジヒドロ キシェチル) -6-エトキシフエノキシ]ァセチックアシッド 100 mgを黄色固体として得た。 tert-Butyl [{4- [(2-Amino-5-clobenbenzoyl) amino] phenol Kimino) methyl] rubamate 295 mg and tert-butyl {[4- (1R)-1,2 -Dihydroxyethyl] -2-ethoxy-6-formylphenoxy} acetate To a solution of 258 mg of acetic acid (5 mL) was added 321 mg of sodium triacetoxyborohydride at room temperature, and the mixture was stirred for 1 hour. Water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water, 5% aqueous sodium bicarbonate and then saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (3: 1) as an elution solvent to obtain a pale yellow foam. To the obtained compound, 6 mL of 4N hydrochloric acid was added, and the mixture was stirred at room temperature for 7 hours. 24 mL of 1N aqueous sodium hydroxide solution was added to the reaction solution, and the resulting solid was filtered, dried, and [2-[({2-[({4- [amino (imino) methyl] phenol} amino]. ) Carbol] -4-chlorophenol} amino) methyl] -4- (1R) -1,2-dihydrochichetyl) -6-ethoxyphenoxy] acetic acid 100 mg as a yellow solid Obtained.
[0184] 実施例 28 [0184] Example 28
ェチル [{4-[(2-ァミノ- 5-メチルピリジン- 3-カルボ-ル)ァミノ]フエ-ル }(ィミノ)メチル ]力ルバマート 0.138 gとトリァセトキシ水素化ホウ素ナトリウム 0.17 gの酢酸(1.5 mL) 溶液に、ェチル 3-(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキシフエ-ル)プロパ ノアート 0.14 gを加え、室温にて 4時間攪拌した。反応液に飽和重曹水をカ卩ぇ中和し 、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで 乾燥後減圧下濃縮した。得られた残渣を酢酸ェチル /n-へキサン(3 : 1)を溶出溶媒 とするシリカゲルカラムクロマトグラフィーにより精製した。得られたィ匕合物のエタノー ル(1.0 mL)溶液に、 2規定水酸化ナトリウム水溶液 0.04 mLを室温にてカ卩え、 15分 間撹拌した。反応液に 2規定塩酸水溶液 0.04 mLを加え、反応液を減圧下濃縮した 。得られた残渣を酢酸ェチル: n-へキサン(3 : 1)を溶出溶媒とするシリカゲルカラムク 口マトグラフィ一により精製し、ェチル 3- [2- [({3- [({4- [[(エトキシカルボ-ル)ァミノ] (ィ ミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -5-メチルピリジン- 2-ィル }ァミノ)メチル ]-4- (ヒ ドロキシメチル) -6-イソプロポキシフエ-ル]プロパノアート 28 mgを黄色固体として得
[0185] 実施例 29 Ethyl [{4-[(2-Amino-5-methylpyridine-3-carbol) amino] phenol} (imino) methyl] rrubamate 0.138 g and sodium triacetoxyborohydride 0.17 g acetic acid (1.5 mL ) 0.14 g of ethyl 3- (4-acetoxymethyl-2-formyl-6-isopropoxyphenyl) propanoate was added to the solution, and the mixture was stirred at room temperature for 4 hours. The reaction solution was neutralized with saturated aqueous sodium bicarbonate and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (3: 1) as an elution solvent. To a solution of the resulting compound in ethanol (1.0 mL) was added 2N aqueous sodium hydroxide solution (0.04 mL) at room temperature, and the mixture was stirred for 15 minutes. To the reaction solution was added 2N aqueous hydrochloric acid 0.04 mL, and the reaction solution was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate: n-hexane (3: 1) as an elution solvent, and ethyl 3- [2-[({3-[({4-[[ (Ethoxycarbol) amino] (imino) methyl] phenol} amino) carbol] -5-methylpyridine-2-yl} amino) methyl] -4- (hydroxymethyl) -6-iso Propoxyphenyl] propanoate 28 mg as a yellow solid [0185] Example 29
2-ァミノ- N- {2- [ァミノ (ィミノ)メチル]ピリミジン- 5-ィルト 5-メチルベンズアミド塩酸塩 100 mgの酢酸 (5 mL)溶液に tert-ブチル 3-(4-ァセトキシメチル- 2-ホルミル- 6-イソ プロポキシフエ-ル)プロパノアート 130 mgおよびトリァセトキシ水素化ホウ素ナトリウム 207 mgを室温で加えて同温度で 3時間攪拌した。反応液を減圧下濃縮し、水を加え て析出した粗結晶を 0.1%ギ酸水/ァセトニトリルを溶出溶媒とする ODSカラムクロマトグ ラフィ一にて精製し粗生成物を得た。得られた粗生成物を 1,4-ジォキサン 5 mLに溶 解し室温で塩ィ匕水素の 1,4-ジォキサン溶液 (4 M) 0.03 mLを加えて 4時間攪拌した 。反応液を減圧下濃縮し 1規定水酸化ナトリウム水溶液 5 mLを加えた。得られた反応 液を 0.1%ギ酸水/ァセトニトリルを溶出溶媒とする ODSカラムクロマトグラフィーにて精 製し 3- [2- [({2- [({2- [ァミノ (ィミノ)メチル]ピリミジン- 5-ィル }ァミノ)カルボ-ル]- 4-メチ ルフエ-ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエ-ル]プロパノィ ックアシッドギ酸塩 5 mgを得た。 2-Amino- N- {2- [Amino (imino) methyl] pyrimidine-5-ylto 5-methylbenzamide hydrochloride in 100 mg of acetic acid (5 mL) in tert-butyl 3- (4-acetoxymethyl-2-formyl -6-Isopropoxyphenol) propanoate 130 mg and sodium triacetoxyborohydride 207 mg were added at room temperature and stirred at the same temperature for 3 hours. The reaction solution was concentrated under reduced pressure, and water was added to precipitate the crude crystal, which was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as an elution solvent to obtain a crude product. The obtained crude product was dissolved in 5 mL of 1,4-dioxane, 0.03 mL of a 1,4-dioxane solution (4 M) in hydrochloric acid and hydrogen was added at room temperature, and the mixture was stirred for 4 hours. The reaction mixture was concentrated under reduced pressure, and 5 mL of 1N aqueous sodium hydroxide solution was added. The resulting reaction solution was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the elution solvent, and 3- [2-[({2-[({2- [amino (imino) methyl] pyrimidine- 5-yl} amino) carbol] -4-methylphenyl} amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenyl] propanoic acid formate 5 mg was obtained.
[0186] 実施例 30 [0186] Example 30
tert-ブチル [{4- [(2-ァミノ- 5-メチルベンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]カル バマート 100 mgとトリァセトキシ水素化ホウ素ナトリウム 173 mgの酢酸(3.3 mL)溶液 にェチル(4_{[tert-ブチル (ジメチル)シリル]ォキシ }- 2_ホルミル- 6_イソプロポキシフエ ノキシ)ァセタート 107 mgの酢酸 (2.7 mL)溶液を室温にてカ卩え、同条件にて 14時間 攪拌した。減圧下溶媒を留去し、得られた残渣を水で希釈して酢酸ェチルで抽出し た。有機層を飽和重曹水で 2回、水及び飽和食塩水で順次洗浄後、無水硫酸マグネ シゥムで乾燥し、減圧下濃縮した。得られた残渣を酢酸ェチル /n-へキサン(1 : 3)を 溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し、黄色固体 94 mgを得た。 得られた黄色固体 94 mgをテトラヒドロフラン 3 mLに溶解し、フッ化 n-テトラプチルアン モ-ゥムのテトラヒドロフラン溶液 (1 M) 0.04 mLを室温にてカ卩え、同条件にて 1.5時 間攪拌した。反応液を酢酸ェチル /n-へキサン(2 : 3)を溶出溶媒とするシリカゲル力 ラムクロマトグラフィーで精製し、黄色油状物 47 mgを得た。上記の手法にて得られた 黄色油状物 104 mgの 1,2-ジクロロエタン (5 mL)溶液にトリフルォロ酢酸 1.6 mLをカロ えた。室温にて 2時間攪拌後、減圧下濃縮した。得られた残渣をァセトニトリル 7 mLに
溶解し、 1規定水酸化ナトリウム水溶液 0.59 mLを加えた。室温にて 1時間攪拌後、 1 規定水酸化ナトリウム水溶液 1.2 mLを加えた。 4時間攪拌後、 1規定塩酸 1.59 mLを 加え、減圧下濃縮した。得られた残渣を 0.1%ギ酸水/ァセトニトリルを溶出溶媒とする ODSカラムクロマトグラフィーにて精製し {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}アミ ノ)カルボ-ル] -4-メチルフエ-ル}ァミノ)メチル ]-4-ヒドロキシ -6-イソプロポキシフエノ キシ }ァセチックアシッドギ酸塩 21 mgを黄色固体として得た。 tert-butyl [{4-[(2-amino-5-methylbenzoyl) amino] phenol} (imino) methyl] carbamate 100 mg and sodium triacetoxyborohydride 173 mg in acetic acid (3.3 mL) 4 _ {[tert-Butyl (dimethyl) silyl] oxy} -2_formyl-6_isopropoxyphenoxy) acetate 107 mg acetic acid (2.7 mL) solution at room temperature and stirred for 14 hours under the same conditions did. The solvent was distilled off under reduced pressure, and the resulting residue was diluted with water and extracted with ethyl acetate. The organic layer was washed successively with saturated aqueous sodium hydrogen carbonate twice, water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 3) as an elution solvent to obtain 94 mg of a yellow solid. 94 mg of the obtained yellow solid was dissolved in 3 mL of tetrahydrofuran, and 0.04 mL of tetrahydrofuran solution (1 M) of n-tetraptylammonium fluoride was stored at room temperature, and 1.5 hours under the same conditions. Stir. The reaction solution was purified by silica gel column chromatography using ethyl acetate / n-hexane (2: 3) as an elution solvent to obtain 47 mg of a yellow oily substance. 1.6 mL of trifluoroacetic acid was added to a solution of 104 mg of the yellow oil obtained by the above method in 1,2-dichloroethane (5 mL). After stirring at room temperature for 2 hours, the mixture was concentrated under reduced pressure. The residue obtained was added to 7 mL of acetonitrile. After dissolution, 0.59 mL of 1N aqueous sodium hydroxide solution was added. After stirring at room temperature for 1 hour, 1.2 mL of 1N aqueous sodium hydroxide solution was added. After stirring for 4 hours, 1.59 mL of 1N hydrochloric acid was added, and the mixture was concentrated under reduced pressure. The resulting residue was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the elution solvent, and was purified by {2- [({2- [({4- [amino (imino) methyl] phenol} amino}. ) Carbol] -4-methylphenol} amino) methyl] -4-hydroxy-6-isopropoxyphenoxy} acetic acid formate 21 mg was obtained as a yellow solid.
[0187] 実施例 31 [0187] Example 31
tert-ブチル [{4- [(2-ァミノ- 5-メチルベンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]カル バマート 71 mgの酢酸 (0.5 mL)懸濁液にトリァセトキシ水素化ホウ素ナトリウム 123 mg を加え、室温で 5分攪拌した。ついで、 tert-ブチル [4-ァセトキシメチル- 2-(2-tert-ブ トキシ -1-メチルエトキシ) -6-ホルミル]フエノキシァセタートの酢酸(1 mL)溶液を加え 、室温で 1.5時間攪拌した。反応液を飽和重曹水に注ぎ、酢酸ェチルで抽出した。有 機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を留去した。得 られた残渣を n-へキサン/酢酸ェチル (3 : 2)を溶出溶媒とするシリカゲルカラムクロマ トグラフィ一により精製し、 tert-ブチル [4-ァセトキシメチル- 2- [({2- [({4- [[(tert-ブトキ シカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -4-メチルフエ-ル}アミ ノ)メチル ]- 6-(2-tert-ブトキシ -1-メチルエトキシ)フエノキシ]ァセタート 65 mgを得た (E P:791)o得られた tert-ブチル [4-ァセトキシメチル -2-{[2-(4- [(tert-ブトキシカルボ- ル)ァミノ] (ィミノ)メチル)フエ-ル]アミノ}カルボ-ル -4-メチルフエ-ルァミノ]メチル ]-6 -(2-tert-ブトキシ -1-メチルエトキシ)フエノキシ]ァセタート 63 mgを塩化メチレン 0.5 m Lに溶解し、トリフルォロ酢酸 0.5 mLを加え、室温で 4時間攪拌した。溶媒を減圧下留 去した後、残渣をエタノール 2 mLに溶解し、 0°Cで 2規定水酸化ナトリウム水溶液 0.4 mLを加え 1時間攪拌した。同温で 2規定塩酸 0.4 mLを加え溶媒を減圧下留去した後 、残渣を 0.1%ギ酸水/ァセトニトリルを溶出溶媒とする ODSカラムクロマトグラフィーに より精製し、 [2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ二ル}ァミノ)カルボ二ル]- 4-メチルフ ェ-ル }ァミノ)メチル ]-4- (ヒドロキシメチル) -6-(2-ヒドロキシ -1-メチルエトキシ)フエノキ シ]ァセチックアシッドギ酸塩 29 mgを淡黄色固体として得た。 tert-Butyl [{4- [(2-amino-5-methylbenzoyl) amino] phenol} (imino) methyl] carbamate In a suspension of 71 mg acetic acid (0.5 mL), 123 mg sodium triacetoxyborohydride was added. The mixture was further stirred at room temperature for 5 minutes. Next, a solution of tert-butyl [4-acetoxymethyl-2- (2-tert-butoxy-1-methylethoxy) -6-formyl] phenoxyacetate in acetic acid (1 mL) was added, and the mixture was stirred at room temperature for 1.5 hours. did. The reaction mixture was poured into saturated aqueous sodium hydrogen carbonate and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated. The resulting residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (3: 2) as the elution solvent, and tert-butyl [4-acetoxymethyl-2-[({2-[({4 -[[(tert-Butoxycarbonyl) amino] (imino) methyl] phenol} amino) carbol] -4-methylphenol} amino) methyl] -6- (2-tert-butoxy- 65 mg of 1-methylethoxy) phenoxy] acetate was obtained (EP: 791) o tert-butyl [4-acetoxymethyl-2-{[2- (4-[(tert-butoxycarbol) amino]] obtained (Imino) methyl) phenol] amino} carbol-4-methylphenolamino] methyl] -6- (2-tert-butoxy-1-methylethoxy) phenoxy] acetate 63 mg in methylene chloride 0.5 mL After dissolution, 0.5 mL of trifluoroacetic acid was added, and the mixture was stirred at room temperature for 4 hours. After the solvent was distilled off under reduced pressure, the residue was dissolved in 2 mL of ethanol, and 0.4 mL of 2N aqueous sodium hydroxide solution was added at 0 ° C and stirred for 1 hour. After adding 0.4 mL of 2N hydrochloric acid at the same temperature and distilling off the solvent under reduced pressure, the residue was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the elution solvent, [2- [({2- [ ({4- [Amino (imino) methyl] phenyl} amino) carbonyl] -4-methylphenyl} amino) methyl] -4- (hydroxymethyl) -6- (2-hydroxy-1- Methylethoxy) phenoxy] acetic acid formate 29 mg was obtained as a pale yellow solid.
[0188] 実施例 32
tert-ブチル [{4- [(2-ァミノ- 5-メチルベンゾィル)ァミノ]フエ-ル Kィミノ)メチル]力ルバ マート 463 mgの酢酸 (10 mL)溶液にェチル [2-ホルミル- 4- (ヒドロキシメチル)フエノ キシ]ァセタート 300 mgおよびトリァセトキシ水素化ホウ素ナトリウム 800 mgを室温で加 えて同温度で 3時間攪拌した。反応液を減圧下濃縮し、酢酸ェチル及び飽和重曹水 を加えて酢酸ェチルにて抽出した。有機層を飽和食塩水で洗浄した後、無水硫酸ナ トリウムで乾燥し、減圧下濃縮した。得られた残渣を n-へキサン/酢酸ェチル (10 : 1) を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製した。得られた残渣に 4規定塩酸(10 mL)を加えて室温にて 5時間攪拌した。 1規定水酸化ナトリウム水溶 液を pH 7.0まで加えて析出した粗結晶を 0.1%ギ酸水/ァセトニトリルを溶出溶媒とする ODSカラムクロマトグラフィーにて精製し [2-[({2-[({4- [ァミノ (ィミノ)メチル]フエ二ル}アミ ノ)カルボ-ル] -4-メチルフエ-ル}ァミノ)メチル ]-4- (ヒドロキシメチル)フエノキシ]ァセ チックアシッドギ酸塩 50 mgを黄色固体として得た。 [0188] Example 32 tert-Butyl [{4- [(2-Amino-5-methylbenzoyl) amino] phenol Kimino) methyl] rubamate Ethyl [2-formyl-4- (hydroxy) in a solution of 463 mg of acetic acid (10 mL) Methyl) phenoxy] acetate (300 mg) and sodium triacetoxyborohydride (800 mg) were added at room temperature, and the mixture was stirred at the same temperature for 3 hours. The reaction mixture was concentrated under reduced pressure, ethyl acetate and saturated aqueous sodium hydrogen carbonate were added, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (10: 1) as an elution solvent. 4N Hydrochloric acid (10 mL) was added to the obtained residue, and the mixture was stirred at room temperature for 5 hr. 1N aqueous sodium hydroxide solution was added to pH 7.0 and the precipitated crude crystals were purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the elution solvent [2-[({2-[({4- [Amino (imino) methyl] phenyl} amino) carbol] -4-methylphenol} amino) methyl] -4- (hydroxymethyl) phenoxy] acetic acid formate 50 mg as a yellow solid Obtained.
実施例 33 Example 33
tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル Kィミノ)メチル]力ルバ マート 538 mgと tert-ブチル [(2-ホルミル- 6-メトキシフエ-ル) (トリフルォロアセチル)ァ ミノ]ァセタート 500 mgの酢酸(10 mL)溶液にトリァセトキシ水素化ホウ素ナトリウム 880 mgを室温にて加え 1時間攪拌した。反応液に水を加え酢酸ェチルで抽出した。有機 層を水、 5%重曹水次いで飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥 後減圧下濃縮した。得られた残渣を酢酸ェチル /n_へキサン(3 : 1)を溶出溶媒とす るシリカゲルカラムクロマトグラフィーで精製し無色泡状物を得た。得られたィ匕合物を 塩化メチレン 4 mLに溶解し、トリフルォロ酢酸 4 mLを加え、室温にて 3時間撹拌した。 反応液を減圧留去した後、残渣を酢酸ェチル 20 mLに溶解させ、塩化水素の酢酸ェ チル溶液 (4 M) 1 mLを加え、生じた固体を濾別乾燥した。得られた固体をエタノー ル 5 mLに溶解し、ナトリウムエトキシド 61 mgを加え終夜加熱還流した。反応液を減圧 留去し、得られた残渣を水/ァセトニトリルを溶出溶媒とする ODSカラムクロマトグラフ ィ一にて精製し、({2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ 口フエ-ル}ァミノ)メチル ]-6-メトキシフエ-ル}ァミノ)ァセチックアシッド 18 mgを黄色 固体として得た。
[0190] 実施例 34 tert-butyl [{4- [(2-amino-5-chlorobenzoyl) amino] phenol Kimino) methyl] rubamate 538 mg and tert-butyl [(2-formyl-6-methoxyphenol) ) (Trifluoroacetyl) amino] acetate To a solution of 500 mg of acetic acid (10 mL) was added 880 mg of sodium triacetoxyborohydride at room temperature, and the mixture was stirred for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed successively with water, 5% aqueous sodium bicarbonate and then saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate / n_hexane (3: 1) as an elution solvent to obtain a colorless foam. The obtained compound was dissolved in 4 mL of methylene chloride, 4 mL of trifluoroacetic acid was added, and the mixture was stirred at room temperature for 3 hours. After evaporating the reaction solution under reduced pressure, the residue was dissolved in 20 mL of ethyl acetate, 1 mL of an ethyl acetate solution of hydrogen chloride (4 M) was added, and the resulting solid was separated by filtration and dried. The obtained solid was dissolved in 5 mL of ethanol, 61 mg of sodium ethoxide was added, and the mixture was heated to reflux overnight. The reaction solution was distilled off under reduced pressure, and the resulting residue was purified by ODS column chromatography using water / acetonitrile as an elution solvent, and ({2- [({2- [({4- [amino (imino ) Methyl] phenol} amino) carbol] -4-chlorophenol} amino) methyl] -6-methoxyphenol} amino) acetic acid 18 mg was obtained as a yellow solid. [0190] Example 34
2-ァミノ- N- {4- [ァミノ (ィミノ)メチル]フエ-ル}- 5-メチルベンズアミド 36 mg及びェチ ル 3-[2-ホルミル- 4- (ヒドロキシメチル) -6-イソプロポキシフエ-ル]プロパノアート 78 m gの酢酸(10 mL)溶液にトリァセトキシ水素化ホウ素ナトリウム 47 mgを室温にてカロえ 2 時間攪拌した。反応液を 0.1%ギ酸水/ァセトニトリルを溶出溶媒とする ODSカラムクロ マトグラフィ一にて精製し、ェチル 3- [2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ) カルボ-ル] -4-メチルフエ-ル}ァミノ)メチル ]-4_ (ヒドロキシメチル) _6_イソプロポキシ フエ-ル]プロパノアートギ酸塩 16 mgを黄色泡状物として得た。 2-Amino- N- {4- [Amino (imino) methyl] phenol} -5-methylbenzamide 36 mg and ethyl 3- [2-formyl-4- (hydroxymethyl) -6-isopropoxyphene -Lu] propanoate To a solution of 78 mg of acetic acid (10 mL) was added sodium triacetoxyborohydride 47 mg and stirred at room temperature for 2 hours. The reaction solution was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the elution solvent, and ethyl 3- [2-[({2-[({4- [amino (imino) methyl] phenol} Amino) carbol] -4-methylphenol} amino) methyl] -4_ (hydroxymethyl) _6_isopropoxyphenol] propanoate formate 16 mg was obtained as a yellow foam.
[0191] 実施例 35 [0191] Example 35
tert-ブチル [{4- [(2-ァミノ- 5-メチルピリジン- 3-カルボ-ル)ァミノ]フエ-ル }(ィミノ)メ チル]力ルバマート 0.10 gとトリァセトキシ水素化ホウ素ナトリウム 0.17 gの酢酸(1.0 mL )溶液に tert-ブチル 3-(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキシフエ-ル) プロパノアート 0.10 gを室温にてカ卩えた。 30分間攪拌した後、トリァセトキシ水素化ホ ゥ素ナトリウム 0.25 gと tert-ブチル 3-(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキ シフエニル)プロパノアート 0.13 gを室温にてカ卩えた。 30分間攪拌した後、トリァセトキ シ水素化ホウ素ナトリウム 0.25 gと tert-ブチル 3-(4-ァセトキシメチル- 2-ホルミル- 6- イソプロポキシフエ-ル)プロパノアート 0.13 gを室温にて加えた。反応液に飽和重曹 水を加え中和し、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸 マグネシウムで乾燥後減圧下濃縮した。得られた残渣を酢酸ェチル /n-へキサン(3 : 1)を溶出溶媒とするアミノプロピルィ匕シリカゲルカラムクロマトグラフィーにより精製し た。得られた化合物の塩化メチレン(1.0 mL)溶液にトリフルォロ酢酸 1.0 mLを室温 にて加え 1.5時間攪拌した。反応液を減圧下濃縮した。残渣にジェチルエーテルを 加え、析出物を濾別後、減圧乾燥し、黄色固体を得た。得られた化合物のエタノール tert-Butyl [{4- [(2-amino-5-methylpyridine-3-carbol) amino] phenol} (imino) methyl] force rubamate 0.10 g and sodium triacetoxyborohydride 0.17 g acetic acid (1.0 mL) solution was charged with 0.10 g of tert-butyl 3- (4-acetoxymethyl-2-formyl-6-isopropoxyphenol) propanoate at room temperature. After stirring for 30 minutes, 0.25 g of sodium triacetoxyborohydride and 0.13 g of tert-butyl 3- (4-acetoxymethyl-2-formyl-6-isopropoxyphenyl) propanoate were obtained at room temperature. After stirring for 30 minutes, 0.25 g of sodium triacetoxyborohydride and 0.13 g of tert-butyl 3- (4-acetoxymethyl-2-formyl-6-isopropoxyphenyl) propanoate were added at room temperature. The reaction mixture was neutralized with saturated aqueous sodium hydrogen carbonate, and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was purified by aminopropyl silica gel column chromatography using ethyl acetate / n-hexane (3: 1) as an elution solvent. To a methylene chloride (1.0 mL) solution of the obtained compound was added 1.0 mL of trifluoroacetic acid at room temperature, and the mixture was stirred for 1.5 hours. The reaction solution was concentrated under reduced pressure. Jetyl ether was added to the residue, and the precipitate was filtered off and dried under reduced pressure to give a yellow solid. Ethanol of the obtained compound
(1.0 mL)溶液に、 2規定水酸化ナトリウム水溶液 0.44 mLを室温にてカ卩え、 30分間 撹拌した。反応液に 2規定塩酸 0.44 mLを加え、反応液を減圧下濃縮した。得られた 残渣に水を加え、析出物を濾別後、減圧乾燥し、 3-[2-[({3-[({4- [ァミノ (ィミノ)メチル] フエ-ル}ァミノ)カルボ-ル] -5-メチルピリジン- 2-ィル }ァミノ)メチル ]-4- (ヒドロキシメチ ル) -6-イソプロポキシフエ-ル]プロパノイツクアシッド塩酸塩 11 mgを淡黄色固体とし
て得た。 To the (1.0 mL) solution, 0.44 mL of 2N aqueous sodium hydroxide solution was added at room temperature and stirred for 30 minutes. To the reaction solution was added 0.44 mL of 2N hydrochloric acid, and the reaction solution was concentrated under reduced pressure. Water was added to the obtained residue, and the precipitate was filtered off and dried under reduced pressure. 3- [2-[({3-[({4- [Amino (imino) methyl] phenol} amino) carbo- L] -5-methylpyridine-2-yl} amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenyl] propanoic acid hydrochloride 11 mg as a pale yellow solid I got it.
[0192] 実施例 36 [0192] Example 36
tert-ブチル [{4- [(2-ァミノ- 5-メチルベンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]カル ノ マート 50 mg、 tert-ブチノレ 3- (4- {tert-ブトキシカルボ-ル- [2- (tert-ブチルジメチ ルシリルォキシ)ェチル]アミノ}-2-ホルミル- 6-イソプロポキシフエニル)プロパノアート 7 7 mgの酢酸(3 mL)混液に室温にてトリァセトキシ水素化ホウ素ナトリウム 58 mgをカロ え、 1時間撹拌した。反応液を酢酸ェチルで希釈し、飽和重曹水に注ぎ抽出した。有 機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥させた。溶媒を 減圧留去し得た残渣を酢酸ェチル /n-へキサン(3 : 1)を溶出溶媒とするシリカゲル力 ラムクロマトグラフィーにより精製し、淡黄色油状物 78 mgを得た。得られた淡黄色油 状物 78 mgに室温にてトリフルォロ酢酸 2 mLを加え、 3時間撹拌した。反応液を減圧 下濃縮して得た残渣をァセトニトリル 2 mLに溶解し、フッ化テトラプチルアンモ -ゥム のテトラヒドロフラン溶液 (1 M) 0.1 mLをカ卩え、室温で 10分間撹拌した。反応液を 0.1 %ギ酸水/ァセトニトリルを溶媒とする ODSカラムクロマトグラフィーにて精製し、 3-(2-{[( 2-{4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -4-メチルフエ-ルァミノ }メチル- 4-(2-ヒドロキシェチルァミノ) -6-イソプロポキシフエ-ル)プロパノイツクアシッド ギ酸 塩 22mgを黄色固体として得た。 tert-Butyl [{4- [(2-amino-5-methylbenzoyl) amino] phenol} (imino) methyl] carnomat 50 mg, tert-butinole 3- (4- {tert-butoxycarbol- [2- (tert-Butyldimethylsilyloxy) ethyl] amino} -2-formyl-6-isopropoxyphenyl) propanoate 7 Caroylate 7 mg sodium triacetoxyborohydride at room temperature in a mixture of 7 mg acetic acid (3 mL). Stir for 1 hour. The reaction solution was diluted with ethyl acetate, poured into saturated aqueous sodium bicarbonate and extracted. The organic layer was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (3: 1) as an elution solvent to obtain 78 mg of a pale yellow oil. To 78 mg of the obtained pale yellow oily substance, 2 mL of trifluoroacetic acid was added at room temperature and stirred for 3 hours. The residue obtained by concentrating the reaction solution under reduced pressure was dissolved in 2 mL of acetonitrile, and 0.1 mL of a tetrahydrofuran solution (1 M) of tetraptylammonium fluoride was prepared and stirred at room temperature for 10 minutes. The reaction solution was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as a solvent, and 3- (2-{[(2- {4- [amino (imino) methyl] phenol} amino) carbo- L] -4-methylphenylamino} methyl-4- (2-hydroxyethylamino) -6-isopropoxyphenol) propanoic acid formate 22 mg was obtained as a yellow solid.
[0193] 実施例 37 [0193] Example 37
[4- (ァセトキシメチル)- 2- [({3- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] ピリジン- 2-ィル }ァミノ)メチル ]-6-エトキシフエノキシ]ァセチックアシッドトリフルォロ 酢酸塩 43 mg、 1規定水酸化ナトリウム水溶液 0.197 mL、メタノール 0.25 mLおよびテト ラヒドロフラン 0.20 mLの混合物を室温にて 45分間撹拌した。反応混合物に 1規定塩 酸 0.20 mLを加え、さらに水を加えた。混合物を減圧下濃縮し、残渣に塩化水素の 1, 4-ジォキサン溶液 (4 M) 0.10 mLおよび水をカ卩えた。析出物をろ取し、水および酢酸 ェチルで順次洗浄後、減圧下乾燥して、 [2- [({3- [({4- [ァミノ (ィミノ)メチル]フエ二ル}ァ ミノ)カルボ-ル]ピリジン- 2-ィル }ァミノ)メチル ]-6-エトキシ- 4- (ヒドロキシメチル)フエノ キシ]ァセチックアシッド塩酸塩 11 mgを無色固体として得た。 [4- (Acetoxymethyl)-2- [({3- [({4- [Amino (imino) methyl] phenol} amino) carbol] pyridine-2-yl} amino) methyl] -6- Ethoxyphenoxy] acetic acid trifluoroacetate (43 mg), 1N aqueous sodium hydroxide solution (0.197 mL), methanol (0.25 mL) and tetrahydrofuran (0.20 mL) were stirred at room temperature for 45 minutes. To the reaction mixture was added 0.20 mL of 1N hydrochloric acid, and water was further added. The mixture was concentrated under reduced pressure, and 0.10 mL of hydrogen chloride in 1,4-dioxane (4 M) and water were added to the residue. The precipitate was collected by filtration, washed successively with water and ethyl acetate, and then dried under reduced pressure to give [2-[({3- [({4- [amino (imino) methyl] phenyl} amino) carbo). 11 mg of -l] pyridine-2-yl} amino) methyl] -6-ethoxy-4- (hydroxymethyl) phenoxy] acetic acid hydrochloride was obtained as a colorless solid.
[0194] 実施例 38
2-ァミノ- N- {4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}- 5-クロ口べンズアミド 133 mg の酢酸(2 mL)懸濁液にトリァセトキシ水素化ホウ素ナトリウム 277 mgをカ卩ぇ室温で 5 分攪拌した後、ェチル(2-ホルミル- 4_ヒドロキシメチル _6_イソプロポキシフエノキシ) ァセタート 129 mgを加え、同温で 1.5時間攪拌した。反応液を飽和重曹水に注ぎ、酢 酸ェチルで抽出した。有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥 し、溶媒を留去した。得られた残渣を n-へキサン/酢酸ェチル(1:10)を溶出溶媒とす るシリカゲルカラムクロマトグラフィーにより精製し淡黄色固体 180 mgを得た。得られ た淡黄色固体 180 mgを酢酸ェチルに溶解し、 0°Cにて塩ィ匕水素の酢酸ェチル溶液 ( 4 M) 0.135 mLをカ卩えた。生じた固体を濾取した後、減圧乾燥してェチル [2- [({2- [({4 -[ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メ チル] -4- (ヒドロキシメチル) _6_イソプロポキシフエノキシ]ァセタート塩酸塩 153 mgを 淡黄色固体として得た。 [0194] Example 38 2-Amino-N- {4- [Amino (hydroxyimino) methyl] phenol} -5-chlorobensamide Add 277 mg of sodium triacetoxyborohydride to a suspension of 133 mg of acetic acid (2 mL). After stirring at room temperature for 5 minutes, 129 mg of ethyl (2-formyl-4-hydroxymethyl _6_isopropoxyphenoxy) acetate was added, and the mixture was stirred at the same temperature for 1.5 hours. The reaction solution was poured into saturated aqueous sodium hydrogen carbonate and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (1:10) as an elution solvent to obtain 180 mg of a pale yellow solid. 180 mg of the obtained pale yellow solid was dissolved in ethyl acetate, and 0.135 mL of an ethyl acetate solution (4 M) of hydrochloric acid and hydrogen salt was obtained at 0 ° C. The resulting solid was collected by filtration and dried under reduced pressure to give ethyl [2-[({2-[({4- [amino (hydroxyimino) methyl] phenol} amino) carbol] -4-chloro]. Mouthphenol} amino) methyl] -4- (hydroxymethyl) _6_isopropoxyphenoxy] acetate hydrochloride was obtained as a pale yellow solid.
実施例 39 Example 39
2-ァミノ- N- {4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ベンズアミド 504 mgの酢酸 (7 mL)懸濁液にトリァセトキシ水素化ホウ素ナトリウム 1.1 gを加えた。反応液にェチル(4 -ァセトキシメチル- 2_ホルミル- 6_イソプロポキシフエノキシ)ァセタート 600 mgを加え、 室温で 1.5時間攪拌した。反応液を酢酸ェチルで希釈し、飽和重曹水を加え、有機 層を抽出した。有機層を水および飽和食塩水で順次洗浄した。有機層を無水硫酸マ グネシゥムで乾燥し、減圧下濃縮した。得られた残渣を酢酸ェチル -へキサン (3 : 1) を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製した。得られたィ匕合 物をエタノール 8 mLに溶解し、室温にて 2規定水酸ィ匕ナトリウム水溶液 2.3 mLを加え 、 2.5時間攪拌した。反応液に 0°Cで 2規定塩酸 2.3 mLを加え溶媒を減圧下濃縮した 。残渣に水を加え、生じた析出物を濾取した。濾物を水、 n-へキサンで順次洗浄した 後、減圧下乾燥した。得られたィ匕合物に室温にて塩ィ匕水素のメタノール溶液 (30wt%) 2-Amino-N- {4- [Amino (hydroxyimino) methyl] phenol} benzamide To a suspension of 504 mg of acetic acid (7 mL) was added 1.1 g of sodium triacetoxyborohydride. Ethyl (4-acetoxymethyl-2-formyl-6-isopropoxyphenoxy) acetate (600 mg) was added to the reaction mixture, and the mixture was stirred at room temperature for 1.5 hours. The reaction solution was diluted with ethyl acetate, saturated aqueous sodium hydrogen carbonate was added, and the organic layer was extracted. The organic layer was washed successively with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate-hexane (3: 1) as an elution solvent. The obtained compound was dissolved in 8 mL of ethanol, and 2 mL of 2N sodium hydroxide aqueous solution was added at room temperature, followed by stirring for 2.5 hours. To the reaction solution, 2.3 mL of 2N hydrochloric acid was added at 0 ° C, and the solvent was concentrated under reduced pressure. Water was added to the residue, and the resulting precipitate was collected by filtration. The residue was washed successively with water and n-hexane and then dried under reduced pressure. Methanol solution of salt and hydrogen at room temperature (30wt%)
4 mLを加え、 1時間攪拌した。反応液を減圧下濃縮し、得られた残渣に酢酸ェチル を加えた後、飽和重曹水を加え、有機層を抽出した。有機層を水および飽和食塩水 で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥し、減圧下濃縮した。得られ た残渣をメタノール/酢酸ェチル (5 : 95)を溶出溶媒とするアミノプロピルィ匕シリカゲル
カラムクロマトグラフィーにより精製した。得られた化合物に o°cで塩化水素の酢酸ェ チル溶液 (4 M) 1.5 mLを加え、 30分間攪拌した。生じた析出物を濾取し、濾物を酢 酸ェチルで洗净した後、減圧下乾燥してメチル [2- ({2-[({4- [ァミノ (ヒドロキシィミノ)メ チル]フエ-ル}ァミノ)カルボ-ル] -4-メチルフエ-ルァミノ }メチル )-4-ヒドロキシメチル -6-イソプロポキシフエノキシ]ァセタート 塩酸塩 153 mgを得た。 4 mL was added and stirred for 1 hour. The reaction mixture was concentrated under reduced pressure, ethyl acetate was added to the resulting residue, saturated aqueous sodium hydrogen carbonate was added, and the organic layer was extracted. The organic layer was washed successively with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. Aminopropyl silica gel using methanol / ethyl acetate (5:95) as an elution solvent Purified by column chromatography. To the obtained compound, 1.5 mL of an ethyl acetate solution of hydrogen chloride (4 M) was added at o ° C., and the mixture was stirred for 30 minutes. The resulting precipitate was collected by filtration, washed with ethyl acetate, and then dried under reduced pressure to give methyl [2-({2-[({4- [amino (hydroxyimino) methyl] phenol]. -L} amino) carbol] -4-methylphenolamino} methyl) -4-hydroxymethyl-6-isopropoxyphenoxy] acetate hydrochloride 153 mg was obtained.
[0196] 実施例 40 [0196] Example 40
2-ァミノ- N- {4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}- 5-メチルベンズアミド 900 mg とトリァセトキシ水素化ホウ素ナトリウム 2.013 gの酢酸(30 mL)溶液にェチル(4-{[ter t-ブチル (ジメチル)シリル]ォキシ }_2_ホルミル- 6_イソプロポキシフエノキシ)ァセタート 1.255 gの酢酸 (24 mL)溶液を室温にて加え、同条件にて 6.5時間攪拌した。減圧下 溶媒を留去し、得られた残渣を水で希釈して酢酸ェチルで抽出した。有機層を飽和 重曹水で 2回、水及び飽和食塩水で順次洗浄後、無水硫酸マグネシウムで乾燥し、 減圧下濃縮した。得られた残渣を酢酸ェチル -へキサン(2 : 3)を溶出溶媒とするシ リカゲルカラムクロマトグラフィーで精製し、黄色固体 1.1 gを得た。得られた黄色固体 1.1 gをテトラヒドロフラン 33 mLに溶解し、フッ化テトラ n-ブチルアンモ-ゥムのテトラヒ ドロフラン溶液 (1 M) 0.47 mLを室温にてカ卩え、同条件にて 1.5時間攪拌した。フツイ匕 テトラ n-ブチルアンモ-ゥムのテトラヒドロフラン溶液(1 M) 0.235 mLを室温にて加え 、同条件にて 3時間攪拌した。フッ化テトラプチルアンモ-ゥムのテトラヒドロフラン溶 液 (1 M) 0.141 mLを室温にて加え、同条件にて 2時間攪拌した。反応液を酢酸ェチ ル /n-へキサン(2 : 3)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製し、 ェチル {2-[({2-[({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -4-メチ ルフエ-ル}ァミノ)メチル ]-4-ヒドロキシ -6-イソプロポキシフエノキシ }ァセタート 616 m gを黄色固体として得た。 2-Amino- N- {4- [Amino (hydroxyimino) methyl] phenol}-5-methylbenzamide 900 mg and sodium triacetoxyborohydride 2.013 g in acetic acid (30 mL) in ethyl acetate (4- { [ter t-Butyl (dimethyl) silyl] oxy} _2_formyl-6_isopropoxyphenoxy) acetate 1.255 g of acetic acid (24 mL) was added at room temperature, and the mixture was stirred for 6.5 hours under the same conditions. The solvent was distilled off under reduced pressure, and the resulting residue was diluted with water and extracted with ethyl acetate. The organic layer was washed successively with saturated aqueous sodium hydrogen carbonate twice, water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate-hexane (2: 3) as an elution solvent to obtain 1.1 g of a yellow solid. 1.1 g of the obtained yellow solid was dissolved in 33 mL of tetrahydrofuran, and 0.47 mL of tetrahydrofuran solution (1 M) of tetra n-butylammonium fluoride was prepared at room temperature and stirred for 1.5 hours under the same conditions. . A tetrahydrofuran solution (1 M) of tetrahydrofuran-tetra-n-butyl ammonium (0.235 mL) was added at room temperature, and the mixture was stirred for 3 hours under the same conditions. 0.141 mL of a tetrahydrofuran solution (1 M) of tetraptyl ammonium fluoride was added at room temperature, and the mixture was stirred for 2 hours under the same conditions. The reaction solution was purified by silica gel column chromatography using ethyl acetate / n-hexane (2: 3) as an elution solvent, and ethyl {2-[({2-[({4- [amino (hydroxyimino 616 mg of) methyl] phenyl} amino) carbol] -4-methylphenol} amino) methyl] -4-hydroxy-6-isopropoxyphenoxy} acetate was obtained as a yellow solid.
[0197] 実施例 41 [0197] Example 41
2-ァミノ- N- {6- [ァミノ (ヒドロキシィミノ)メチル]ピリジン- 3-ィル }べンズアミド 300 mg、 ェチル(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキシフエノキシ)ァセタート 374 m gの酢酸(5 mL)混液に室温にてトリァセトキシ水素化ホウ素ナトリウム 469 mgを加え、 1.5時間撹拌した。溶媒を減圧留去し得た残渣に飽和重曹水および酢酸ェチルを加
え抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥 させた。溶媒を減圧留去し得た残渣をエタノール 15 mLに溶解し、室温にて炭酸カリ ゥム 268 mgを加え 3時間撹拌した。反応液を 2規定塩酸に注いだ後、飽和重曹水を 加え中和した。析出物を濾取し無色固体 162 mgを得た。得られた無色固体 132 mgを 0.1%ギ酸水/ァセトニトリルを溶媒とする ODSカラムクロマトグラフィーにて精製した。溶 出画分を 20 mL程度まで減圧濃縮した後飽和重曹水を加え、析出した固体を濾取し ェチル [2- ({2-[({6- [ァミノ (ヒドロキシィミノ)メチル]ピリジン- 3-ィル }ァミノ)カルボニル] フエ-ルァミノ }メチル )-4-ヒドロキシメチル -6-イソプロポキシフエノキシ]ァセタート 35 mgを無色固体として得た。 2-Amino- N- {6- [Amino (hydroxyimino) methyl] pyridine-3-yl} benzamide 300 mg, ethyl (4-acetoxymethyl-2-formyl-6-isopropoxyphenoxy) acetate 374 To a mixed solution of mg acetic acid (5 mL), 469 mg of sodium triacetoxyborohydride was added at room temperature and stirred for 1.5 hours. Saturated aqueous sodium bicarbonate and ethyl acetate were added to the residue obtained by distilling off the solvent under reduced pressure. Extracted. The organic layer was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by distilling off the solvent under reduced pressure was dissolved in 15 mL of ethanol, and 268 mg of potassium carbonate was added at room temperature, followed by stirring for 3 hours. The reaction mixture was poured into 2N hydrochloric acid, and neutralized with saturated aqueous sodium hydrogen carbonate. The precipitate was collected by filtration to obtain 162 mg of a colorless solid. The resulting colorless solid 132 mg was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as a solvent. The elution fraction was concentrated under reduced pressure to about 20 mL, saturated aqueous sodium hydrogen carbonate was added, and the precipitated solid was collected by filtration. Ethyl [2- ({2-[({6- [Amino (hydroxyimino) methyl] pyridine- 35 mg of 3-yl} amino) carbonyl] phenolamino} methyl) -4-hydroxymethyl-6-isopropoxyphenoxy] acetate was obtained as a colorless solid.
[0198] 実施例 42 [0198] Example 42
ェチル [{4- [(2-ァミノ- 5-メチルベンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]力ルバマー ト 30 mg、 tert-ブチル(4-ァセトキシメチル- 2-ホルミル- 6-イソプロポキシフエ-ル)ァ セタート 31 mgの酢酸(2 mL)混液に室温でトリァセトキシ水素化ホウ素ナトリウム 37 m gを加え、 1.5時間撹拌した。反応液を酢酸ェチルで希釈し、飽和重曹水に注ぎ抽出 した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥させた。 溶媒を減圧留去し得た残渣を酢酸ェチル /n-へキサン(1 : 2)を溶出溶媒とするシリカ ゲルカラムクロマトグラフィーにより精製し、淡黄色油状物 26 mgを得た。得られた淡 黄色油状物 26 mgに室温にてトリフルォロ酢酸 2 mLを加え、 3.5時間撹拌した。溶媒 を減圧留去し得た残渣に塩化メチレンを加え再度減圧留去し、淡黄色固体を得た。 得られた淡黄色固体をエタノール 5 mLに溶解し、炭酸カリウム 53 mgをカ卩ぇ室温で 1 時間、 40 °Cで 1時間撹拌した。炭酸カリウム 53 mgをカ卩え、 60 °Cで 30分間撹拌した。 溶媒を減圧留去し得た残渣に PH7.4のリン酸緩衝液をカロえ、 1規定塩酸を滴加し pH6 とした。得られた混合物にジメチルスルホキシドをカロえ析出物を溶解し、水/ァセトニト リルを溶媒とする ODSカラムクロマトグラフィーにて精製し、 [2- [({2- [({4- [[(エトキシカ ルポ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -4-メチルフエ-ル}ァミノ)メ チル] -4- (ヒドロキシメチル) _6_イソプロポキシフエ-ル]ァセチックアシッド 9 mgを黄色 固体として得た。 Ethyl [{4- [(2-amino-5-methylbenzoyl) amino] phenol} (imino) methyl] strength rubamate 30 mg, tert-butyl (4-acetoxymethyl-2-formyl-6-isopropoxyphenol- Lu) acetate 31 mg of acetic acid (2 mL) was mixed with 37 mg of sodium triacetoxyborohydride at room temperature and stirred for 1.5 hours. The reaction solution was diluted with ethyl acetate, poured into saturated aqueous sodium bicarbonate and extracted. The organic layer was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by distilling off the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 2) as an elution solvent to obtain 26 mg of a pale yellow oily substance. To 26 mg of the obtained pale yellow oil, 2 mL of trifluoroacetic acid was added at room temperature, followed by stirring for 3.5 hours. The solvent was distilled off under reduced pressure, methylene chloride was added to the resulting residue, and the mixture was distilled off again under reduced pressure to obtain a pale yellow solid. The obtained pale yellow solid was dissolved in 5 mL of ethanol, and 53 mg of potassium carbonate was stirred at room temperature for 1 hour and at 40 ° C. for 1 hour. Potassium carbonate (53 mg) was added and stirred at 60 ° C for 30 minutes. To the residue obtained by distilling off the solvent under reduced pressure, a phosphate buffer solution of PH7.4 was added, and 1N hydrochloric acid was added dropwise to adjust the pH to 6. The resulting mixture was dissolved in dimethyl sulfoxide, and the precipitate was dissolved and purified by ODS column chromatography using water / acetononitrile as the solvent, and [2-[({2-[({4-[[(ethoxy Lolol) amino] (imino) methyl] phenol} amino) carbol] -4-methylphenyl} amino) methyl] -4- (hydroxymethyl) _6_isopropoxyphenyl] ase 9 mg of tic acid was obtained as a yellow solid.
[0199] 実施例 43
tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル Kィミノ)メチル]力ルバ マート 229mgの酢酸(6 mL)溶液にトリァセトキシ水素化ホウ素ナトリウム 250 mgを室 温にてカ卩えて同温度で 10分間攪拌した後、メチル 4-(2_tert-ブトキシ _2_ォキソエト キシ) -3-エトキシ -5-ホルミルべンゾアート 200 mgを室温でカ卩えて同温度で 3時間攪 拌した。反応液に水を加え、酢酸ェチルで抽出した。有機層を水、 5%重曹水次いで 飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、減圧下濃縮した。得られた 残渣を n-へキサン/酢酸ェチル(2 : 1)を溶出溶媒とするシリカゲルカラムクロマトダラ フィ一により精製した。得られた残渣をメタノール 3 mLに溶解し、 1規定水酸化ナトリ ゥム水溶液 0.3 mLを氷冷下加え室温にて 2時間攪拌した。 1規定水酸ィ匕ナトリウム水 溶液 0.6 mLを室温にて力卩ぇ同温度にて 2時間攪拌した。 1規定塩酸 0.9 mLを室温に て加え減圧下濃縮した。得られた残渣と 1,4-ジォキサン 1 mLの混合物に、塩化水素 の 1,4-ジォキサン溶液 (4 M) 10 mLを室温にて加え、同温度にて 4時間攪拌した。反 応液を減圧下濃縮した。得られた残渣とメタノール 10 mLおよび水 10 mLの混合物に 、室温にて 1規定水酸化ナトリウム水溶液 3 mLをくわえ、同温度にて 3.5時間攪拌した 。 1規定塩酸を加えて反応液を pH3とし、得られた析出物を濾取した。濾物を水で洗 浄後、減圧下乾燥して 3-[({2-[({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -4 -クロ口フエ二ル}ァミノ)メチル ]-4- (カルボキシメトキシ)- 5-エトキシ安息香酸塩酸塩 2 18 mgを黄色固体として得た。 [0199] Example 43 tert-Butyl [{4- [(2-amino-5-chlorobenzobenzo) amino] phenol Kimino) methyl] rubamate 250 mg of sodium triacetoxyborohydride in a solution of 229 mg of acetic acid (6 mL) After stirring at room temperature and stirring for 10 minutes, 200 mg of methyl 4- (2_tert-butoxy_2_oxoethoxy) -3-ethoxy-5-formylbenzoate was added at room temperature and 3 Stir for hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water, 5% aqueous sodium bicarbonate, and then saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using n-hexane / ethyl acetate (2: 1) as an elution solvent. The obtained residue was dissolved in 3 mL of methanol, 0.3 mL of 1N aqueous sodium hydroxide solution was added under ice cooling, and the mixture was stirred at room temperature for 2 hours. 0.6 mL of 1N aqueous solution of sodium hydroxide and sodium chloride was stirred at room temperature for 2 hours. 0.9 mL of 1N hydrochloric acid was added at room temperature and concentrated under reduced pressure. To a mixture of the obtained residue and 1 mL of 1,4-dioxane, 10 mL of a 1,4-dioxane solution (4 M) of hydrogen chloride was added at room temperature, and the mixture was stirred at the same temperature for 4 hours. The reaction solution was concentrated under reduced pressure. To a mixture of the obtained residue, 10 mL of methanol and 10 mL of water, 3 mL of 1N aqueous sodium hydroxide solution was added at room temperature, and the mixture was stirred at the same temperature for 3.5 hours. 1N Hydrochloric acid was added to adjust the reaction mixture to pH 3, and the resulting precipitate was collected by filtration. The filtrate is washed with water and then dried under reduced pressure. 3-[({2-[({4- [Amino (imino) methyl] phenol} amino) carbol] -4- Ru} amino) methyl] -4- (carboxymethoxy) -5-ethoxybenzoic acid hydrochloride 2 18 mg was obtained as a yellow solid.
実施例 44 Example 44
[2-[({2-[({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-フルォロ フエ-ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ]ァセチック アシッド 370 mgに、塩化水素のエタノール溶液 (27wt%) 3 mLを室温でカ卩えて 20分攪 拌した。反応液を酢酸ェチル 10 mLで希釈し、飽和重曹水(5 mL)を加えて酢酸ェチ ルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、得られた残渣を酢酸ェチ ル /n-へキサン(1: 1)を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製しェ チル [2-[({2-[({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-フル オロフェ-ル }ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ]ァセタ ート 90 mgを黄色固体として得た。
[0201] 実施例 45 [2-[({2-[({4- [Amino (hydroxyimino) methyl] phenyl} amino) carbol] -4-fluorophenyl} amino) methyl] -4- (hydroxymethyl ) -6-Isopropoxyphenoxy] acetic acid 370 mg, 3 mL of an ethanol solution of hydrogen chloride (27 wt%) was added at room temperature and stirred for 20 minutes. The reaction mixture was diluted with 10 mL of ethyl acetate, saturated aqueous sodium hydrogen carbonate (5 mL) was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, and the resulting residue was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 1) as an elution solvent. Ethyl [2-[({2 -[({4- [Amino (hydroxyimino) methyl] phenol} amino) carbol] -4-fluorophenol} amino) methyl] -4- (hydroxymethyl) -6-isopropoxyv 90 mg of enoxy] acetate was obtained as a yellow solid. [0201] Example 45
3-[2-[({3-[({4- [[(エトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル}ァミノ)カルボ二 ル]ピリジン- 2-ィル }ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエ-ル]プ ロパノイツクアシッド 30 mgと炭酸カリウム 14 mgの Ν,Ν-ジメチルホルムアミド (1.0 mL) 溶液にョードエタン 12 mgを室温にてカ卩ぇ 14時間攪拌した。反応液に水を加え、酢 酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥 後減圧下濃縮した。得られた残渣を酢酸ェチルを溶出溶媒とするシリカゲルカラムク 口マトグラフィ一で精製し、ェチル 3- [2- [({3- [({4- [[(エトキシカルボニル)ァミノ] (ィミノ) メチル]フエ-ル}ァミノ)カルボ-ル]ピリジン- 2-ィル }ァミノ)メチル ]-4- (ヒドロキシメチル )-6-イソプロポキシフエ-ル]プロパノアート 19 mgを淡黄色固体として得た。 3- [2-[({3-[({4- [[(Ethoxycarbonyl) amino] (imino) methyl] phenyl} amino) carboyl] pyridine-2-yl} amino) methyl]- 4- (Hydroxymethyl) -6-isopropoxyphenyl] propanoic acid 30 mg and potassium carbonate 14 mg in Ν, ホ ル -dimethylformamide (1.0 mL) in a solution of 12 mg of odoethane at room temperature Stir for 14 hours. Water was added to the reaction solution and extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate as an elution solvent, and ethyl 3- [2-[({3-[({4-[[(ethoxycarbonyl) amino] (imino) methyl [Fuel} amino) carbol] pyridine-2-yl} amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenyl] propanoate 19 mg was obtained as a pale yellow solid.
[0202] 実施例 46 [0202] Example 46
[2-[({2-[({6- [ァミノ (ヒドロキシィミノ)メチル]ピリジン- 3-ィル }ァミノ)カルボ-ル] -4-メ チルフエ-ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ]ァセチ ックアシッド 0.20 gに塩化水素のエタノール溶液 (25wt%) 3.7 mLを加え、室温にて 2 時間攪拌した。反応液に飽和重曹水を加え、酢酸ェチルで抽出した。有機層を飽和 食塩水で洗浄し、無水硫酸マグネシウムで乾燥後減圧下濃縮した。得られた残渣を 酢酸ェチル -へキサン(7 : 1)を溶出溶媒とするアミノプロピルィ匕シリカゲルカラムク 口マトグラフィ一にて精製した。得られたィ匕合物の酢酸ェチル (1.0 mL)溶液に、塩ィ匕 水素の酢酸ェチル溶液 (4 M) 0.07 mLを氷冷にて加え、 30分間撹拌した。析出した 沈殿物を濾取し、ェチル [2-[({2-[({6- [ァミノ (ヒドロキシィミノ)メチル]ピリジン- 3-ィル } ァミノ)カルボ-ル] -4-メチルフエ-ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロ ポキシフエノキシ]ァセタート塩酸塩 0.12 gを黄色固体として得た。 [2-[({2-[({6- [Amino (hydroxyimino) methyl] pyridine-3-yl} amino) carbol] -4-methylphenol} amino) methyl] -4- To 0.20 g of (hydroxymethyl) -6-isopropoxyphenoxy] acetic acid was added 3.7 mL of an ethanol solution of hydrogen chloride (25 wt%), and the mixture was stirred at room temperature for 2 hours. Saturated aqueous sodium hydrogen carbonate was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The resulting residue was purified by aminopropyl silica gel column chromatography using ethyl acetate-hexane (7: 1) as an elution solvent. 0.07 mL of an ethyl acetate solution (4 M) of sodium chloride and hydrogen was added to an ethyl acetate solution (1.0 mL) of the obtained compound, and the mixture was stirred for 30 minutes. The deposited precipitate was collected by filtration, and ethyl [2-[({2-[({6- [amino (hydroxyimino) methyl] pyridine-3-yl} amino) carbol] -4-methylphenol- L} amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenoxy] acetate hydrochloride 0.12 g was obtained as a yellow solid.
[0203] 実施例 47 [0203] Example 47
3- [2- [({3- [({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 5-メチル ピリジン- 2-ィル }ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエ-ル]プロパ ノイツクアシッド塩酸塩 44 mgと炭酸カリウム 11 mgの Ν,Ν-ジメチルホルムアミド (0.5 m L)溶液にョードエタン 12 mgを室温にて加え 15時間攪拌した。反応液に炭酸力リウ ム 6 mgとョードエタン 6mgを加え、室温にて 1時間攪拌した。反応液に水を加え、酢酸
ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後 減圧下濃縮した。得られた残渣を酢酸ェチルを溶出溶媒とするシリカゲルカラムクロ マトグラフィ一で精製した。得られた化合物の酢酸ェチル (1.0 mL)溶液に、塩ィ匕水 素の酢酸ェチル溶液 (4 M) 0.03 mLを氷冷にて加え、 1時間撹拌した。析出した沈 殿物を濾取し、ェチル 3-[2-[({3-[({4- [ァミノ (ヒドロキシィミノ)メチル]フエ二ル}ァミノ)力 ルポ-ル] -5-メチルピリジン- 2-ィル }ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロボ キシフエ-ル]プロパノアート塩酸塩 0.02 gを淡黄色固体として得た。 3- [2- [({3- [({4- [Amino (hydroxyimino) methyl] phenol} amino) carbol] -5-methylpyridine-2-yl} amino) methyl]- 4- (Hydroxymethyl) -6-isopropoxyphenyl] propanoic acid hydrochloride 44 mg and potassium carbonate 11 mg in Ν, Ν-dimethylformamide (0.5 mL) solution at room temperature was added 12 mg Stir for 15 hours. To the reaction solution, 6 mg of sodium carbonate and 6 mg of odoethane were added and stirred at room temperature for 1 hour. Add water to the reaction mixture and add acetic acid. Extracted with ethyl. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography using ethyl acetate as an elution solvent. To a solution of the obtained compound in ethyl acetate (1.0 mL) was added 0.03 mL of a solution of sodium chloride in ethyl acetate (4 M) under ice cooling, and the mixture was stirred for 1 hour. The deposited precipitate was collected by filtration, and ethyl 3- [2-[({3-[({4- [amino (hydroxyimino) methyl] phenyl} amino) force] -l-methyl 0.02 g of pyridine-2-yl} amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenyl] propanoate hydrochloride was obtained as a pale yellow solid.
[0204] 実施例 48 [0204] Example 48
ェチル {4-ァセトキシメチル- 2- [({2- [({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}アミ ノ)カルボ-ル] -4-メチルフエ-ル}ァミノ)メチル ]-6-イソプロポキシフエノキシ }ァセター ト 200 mgのエタノール (3.3 mL)溶液に 2- (メチルァミノ)エタノール 0.21 mLを加え、 7 5°Cで終夜攪拌した。反応液を減圧下濃縮し、得られた残渣を 0.1%ギ酸水/ァセトニト リルを溶出溶媒とする ODSカラムクロマトグラフィーにて精製し、 N-{4- [ァミノ (ヒドロキ シィミノ)メチル]フエ-ルト 2- {[2- {2- [(2-ヒドロキシェチル) (メチル)ァミノ]- 2-ォキソエト キシ }-5- (ヒドロキシメチル) -3-イソプロポキシベンジル]アミノ}-5-メチルベンズアミド 1 04 mgを淡黄色固体として得た。 Ethyl {4-Acetoxymethyl-2-[({2-[({4- [Amino (hydroxyimino) methyl] phenol} amino) carbol] -4-methylphenyl} amino) methyl]- To a solution of 6-isopropoxyphenoxy} acetate 200 mg in ethanol (3.3 mL) was added 0.21 mL of 2- (methylamino) ethanol and stirred at 75 ° C. overnight. The reaction solution was concentrated under reduced pressure, and the resulting residue was purified by ODS column chromatography using 0.1% aqueous formic acid / acetonitrile as the eluting solvent, and N- {4- [amino (hydroxyimino) methyl] felt. 2- {[2- {2- [(2-Hydroxyethyl) (methyl) amino]-2-oxoethoxy} -5- (hydroxymethyl) -3-isopropoxybenzyl] amino} -5-methylbenzamide 1 04 mg was obtained as a pale yellow solid.
[0205] 実施例 49 [0205] Example 49
tert-ブチル 3-[2-[({2-[({4- [[(エトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル}アミ ノ)カルボ-ル] -4-メチルフエ-ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキ シフエ-ル]プロパノアートの塩化メチレン(1.4 mL)溶液にトリフルォロ酢酸 1.4 mLを 加え、室温にて 4時間撹拌した。反応液を減圧留去した後、飽和重曹水を加え中和 した。反応液を水/ァセトニトリルを溶出溶媒とする ODSカラムクロマトグラフィーにて 精製し、 3- [2- [({2- [({4- [[(エトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル}ァミノ)力 ルポ-ル] -4-メチルフエ-ル}ァミノ)メチル ]-4_ (ヒドロキシメチル) _6_イソプロポキシフ ェ -ル]プロパノイツクアシッド 42 mgを黄色固体として得た。 tert-Butyl 3- [2-[({2-[({4-[[(Ethoxycarbonyl) amino] (imino) methyl] phenyl} amino) carbol] -4-methylphenol} amino ) Methyl] -4- (hydroxymethyl) -6-isopropoxyphenyl] propanoate in methylene chloride (1.4 mL) was added with 1.4 mL of trifluoroacetic acid and stirred at room temperature for 4 hours. After the reaction solution was distilled off under reduced pressure, saturated sodium bicarbonate water was added for neutralization. The reaction solution was purified by ODS column chromatography using water / acetonitrile as an elution solvent, and 3- [2-[({2-[({4-[[(ethoxycarbonyl) amino] (imino) methyl] phenyl. L-amino) force] -4-methylphenol} amino) methyl] -4_ (hydroxymethyl) _6_isopropoxyphenyl] propanoic acid 42 mg was obtained as a yellow solid.
[0206] 実施例 50 [0206] Example 50
tert-ブチル [4-ァミノ- 2- ({4-クロ口- 2- [({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァ ミノ)カルボ-ル]フエ-ルァミノ }メチル )-6-イソプロポキシフエノキシ]ァセタート 201 mg
、 tert-ブトキシカルボニルアミノアセチックアシッド 88 mgの Ν,Ν-ジメチルホルムアミド (5 mL)混液に室温にて 1-ェチル -3-(3-ジメチルァミノプロピル)カルボジイミド一塩 酸塩 97 mgをカ卩ぇ終夜撹拌した。反応液に飽和重曹水および酢酸ェチルを加え抽出 した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥させた。 溶媒を減圧留去し得た残渣を酢酸ェチル /n-へキサン(1: 1)を溶出溶媒とするシリカ ゲルカラムクロマトグラフィーにより精製し、淡黄色固体 80 mgを得た。得られた淡黄 色固体 10 mgの酢酸ェチル(1 mL)溶液に室温にて塩化水素の 1,4-ジォキサン溶液 (4 M) 1 mLを加え、生じた懸濁液を終夜撹拌した。析出物を濾取し、酢酸ェチルで 洗浄後減圧乾燥して [2-[({2-[({4- [ァミノ (ヒドロキシィミノ)メチル]フエ二ル}ァミノ)カルボ -ル] -4-クロ口フエ-ル}ァミノ)メチル ]-4- (グリシルァミノ) -6-イソプロポキシフエノキシ] ァセチックアシッド塩酸塩 6 mgを淡黄色粉末として得た。 tert-Butyl [4-Amino-2- ({4-Black Mouth 2- [({4- [Amino (hydroxyimino) methyl] phenol} amino) carbol] phenolamino} methyl) -6-Isopropoxyphenoxy] acetate 201 mg Tert-butoxycarbonylaminoacetic acid 88 mg of Ν, Ν-dimethylformamide (5 mL) was mixed with 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide monohydrochloride 97 mg at room temperature. Stir overnight. Saturated aqueous sodium hydrogen carbonate and ethyl acetate were added to the reaction solution for extraction. The organic layer was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The residue obtained by distilling off the solvent under reduced pressure was purified by silica gel column chromatography using ethyl acetate / n-hexane (1: 1) as an elution solvent to obtain 80 mg of a pale yellow solid. To a solution of the obtained pale yellow solid 10 mg in ethyl acetate (1 mL) was added 1 mL of a 1,4-dioxane solution (4 M) of hydrogen chloride at room temperature, and the resulting suspension was stirred overnight. The precipitate was collected by filtration, washed with ethyl acetate and dried under reduced pressure. [2-[({2-[({4- [Amino (hydroxyimino) methyl] phenyl} amino) carbol] -4 -Black mouth phenol} amino) methyl] -4- (glycylamino) -6-isopropoxyphenoxy] Acetic acid hydrochloride 6 mg was obtained as a pale yellow powder.
実施例 51 Example 51
tert-ブチル [{4- [(2-ァミノ- 5-メチルベンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]カル バマートとェチル 3- (4- {[N- (tert-ブトキシカルボ-ル)グリシル]アミノ}- 2-ホルミル- 6- イソプロポキシフエ-ル)プロパノアートを原料として、実施例 7と同様の操作によりェ チル 3- [4- (2- tert-ブトキシカルボ-ルアミノアセチルァミノ)- 2- ({2- [({4- [ァミノ (ヒドロ キシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -4-メチルフエ-ルァミノ }メチル )-6-イソ プロポキシフエ-ル]プロパノアートを無色固体として得た(EP:705)。ェチル 3-[4-(2- tert-ブトキシカルボ-ルアミノアセチルァミノ) -2-({2-[({4- [ァミノ (ヒドロキシィミノ)メチ ル]フエ-ル}ァミノ)カルボ-ル] -4-メチルフエ-ルァミノ }メチル )-6-イソプロポキシフエ -ル]プロパノアート 50 mg、水 0.3 mLおよびァセトニトリル 1 mLの混合物に室温にて 2 規定水酸化ナトリウム水溶液 72 μ Lを加え 4.5時間攪拌した。反応液に室温にて 2規 定塩酸 72 μ Lを加え、溶媒を減圧下濃縮した。残渣に水を加え、生じた析出物を粉 末状にした後、濾取した。濾物を水で洗浄し、減圧下乾燥した。得られた化合物の塩 ィ匕メチレン(0.6 mL)溶液にトリフルォロ酢酸 0.6 mLを室温にてカロえ 1.5時間攪拌した 。反応液を減圧下濃縮した。残渣にジェチルエーテルを加え、生じた析出物を粉末 状にした後、濾取した。濾物をジェチルエーテルで洗浄し、減圧下乾燥して 3-[4-(2- ァミノ-ァセチルァミノ)- 2- ({2- [({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カル
ボ-ル ]-4-メチルフエ-ルァミノ }メチル )-6-イソプロポキシフエ-ル]プロパノイツクァ シッドトリフルォロ酢酸塩 48 mgを得た。 tert-Butyl [{4- [(2-amino-5-methylbenzoyl) amino] phenol} (imino) methyl] carbamate and ethyl 3- (4- {[N- (tert-butoxycarbol) glycyl ] Amino} -2-formyl-6-isopropoxyphenyl) propanoate as a starting material and the same procedure as in Example 7 to produce ethyl 3- [4- (2-tert-butoxycarbolaminoacetylamino) -2- ({2- [({4- [Amino (hydroxyxymino) methyl] phenol} amino) carbol] -4-methylphenylamino} methyl) -6-isopropoxyphenyl] propanoate Obtained as a colorless solid (EP: 705). Ethyl 3- [4- (2-tert-Butoxycarbolaminoacetylamino) -2-({2-[({4- [Amino (hydroxyimino) methyl] phenol} amino) carbo- L] -4-Methylphenol-methyl})-6-isopropoxyphenol] propanoate 50 mg, water 0.3 mL, and acetonitrile 1 mL at room temperature with 2 N aqueous sodium hydroxide solution 72 μL added for 4.5 hours Stir. To the reaction solution, 72 μL of 2N hydrochloric acid was added at room temperature, and the solvent was concentrated under reduced pressure. Water was added to the residue, and the resulting precipitate was powdered and collected by filtration. The residue was washed with water and dried under reduced pressure. To a solution of the obtained compound in methylene chloride (0.6 mL), 0.6 mL of trifluoroacetic acid was added at room temperature and stirred for 1.5 hours. The reaction solution was concentrated under reduced pressure. Jetyl ether was added to the residue, and the resulting precipitate was powdered and collected by filtration. The filtrate was washed with jetyl ether and dried under reduced pressure to give 3- [4- (2-amino-acetylamino) -2-({2-[({4- [amino (hydroxyimino) methyl] phenol- Le} amino) Cal Bol] -4-methylphenolamino} methyl) -6-isopropoxyphenyl] propanoic acid trifluoroacetate 48 mg was obtained.
[0208] 実施例 52 [0208] Example 52
N-{4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}-2-ァミノ- 5-メチルベンズアミドとェチ ル 3-[4- (ァセトキシァセチルァミノ) -2-ホルミル- 6-イソプロポキシフエ-ル]プロパノ アートを原料として、実施例 7と同様の操作によりェチル 3-[4-(2-ァセトキシァセチル ァミノ)- 2- ({2- [({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-メチル フエ-ルァミノ }メチル )-6-イソプロポキシフエ-ル]プロパノアートを得た (EP:648)。得ら れたェチル 3-[4-(2-ァセトキシァセチルァミノ)- 2-({2-[({4- [ァミノ (ヒドロキシィミノ)メチ ル]フエ-ル}ァミノ)カルボ-ル] -4-メチルフエ-ルァミノ }メチル )-6-イソプロポキシフエ -ル]プロパノアート 100 mgのエタノール (1.5 mL)懸濁液にナトリウムエトキシド 21 mg のエタノール (0.5 mL)溶液を加え、室温で 4.5時間攪拌した。反応液にテトラヒドロフラ ン 2 mLをカ卩え、室温で 3時間攪拌した。反応液に室温にて 2規定塩酸 155 μ Lを加え 、溶媒を減圧下濃縮した。残渣に酢酸ェチル /テトラヒドロフラン/エタノールを加えた 後、飽和重曹水を加え、有機層を抽出した。有機層を水で洗浄し、無水硫酸マグネ シゥムで乾燥した後、減圧下濃縮した。残渣にジェチルエーテルを加え、生じた析出 物を粉末状にした後、濾取した。濾物をジェチルエーテルで洗浄し、減圧下乾燥し てェチル 3- [4- (2-ヒドロキシァセチルァミノ)- 2- ({2- [({4- [ァミノ (ヒドロキシィミノ)メチル ]フエ-ル}ァミノ)カルボ-ル] -4-メチルフエ-ルァミノ }メチル )-6-イソプロポキシフエ- ル]プロパノアート 77mgを得た。 N- {4- [Amino (hydroxyimino) methyl] phenol} -2-amino-5-methylbenzamide and ethyl 3- [4- (acetoxyacetylamino) -2-formyl-6 -Isopropoxyphenyl] propanoate was used as a starting material in the same manner as in Example 7 to perform ethyl 3- [4- (2-acetoxycetylamino) -2- ({2- [({4- [amino (Hydroxyimino) methyl] phenol} amino) carbol] -4-methyl phenolamino} methyl) -6-isopropoxyphenyl] propanoate was obtained (EP: 648). The resulting ethyl 3- [4- (2-acetoxyacetylylamino) -2-({2-[({4- [amino (hydroxyimino) methyl] phenyl} amino) carbo- L] -4-methylphenol-methyl) -6-isopropoxyphenol-propanoate To a suspension of 100 mg ethanol (1.5 mL), add a solution of sodium ethoxide 21 mg in ethanol (0.5 mL) at room temperature. Stir for 4.5 hours. To the reaction solution, 2 mL of tetrahydrofuran was added and stirred at room temperature for 3 hours. To the reaction solution, 155 μL of 2N hydrochloric acid was added at room temperature, and the solvent was concentrated under reduced pressure. Ethyl acetate / tetrahydrofuran / ethanol was added to the residue, saturated aqueous sodium hydrogen carbonate was added, and the organic layer was extracted. The organic layer was washed with water, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. Jetyl ether was added to the residue, and the resulting precipitate was powdered and collected by filtration. The filtrate was washed with jetyl ether, dried under reduced pressure, and ethyl 3- [4- (2-hydroxyacetylamino) -2-({2-[({4- [amino (hydroxyimino) methyl]. [Fuel} amino) Carbon] -4-methylphenolamino} methyl) -6-isopropoxyphenol] propanoate 77 mg was obtained.
[0209] 実施例 53 [0209] Example 53
N- {4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}- 2-ァミノ- 5-メチルベンズアミドと tert- ブチル 3_(4-ァセトキシメチル- 2_ホルミル- 6_イソプロポキシフエ-ル)プロパノアート を原料として、後述の実施例 54と同様の操作により黄色固体を得た。得られた黄色 固体 317 mgの水(3.2 mL)溶液に 1規定水酸ィ匕ナトリウム水溶液 5.5 mLをカ卩え、室温 にて 1時間撹拌した。反応液を水/ァセトニトリルを溶出溶媒とする ODSカラムクロマト グラフィーを用いて精製し、ナトリウム 3- [2- [({2- [({4- [ァミノ (ヒドロキシィミノ)メチル]フ ェ-ル }ァミノ)カルボ-ル] -4-メチルフエ-ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-ィ
ソプロポキシフエニル]プロパノアート 176 mgを得た。 N- {4- [Amino (hydroxyimino) methyl] phenol}-2-amino-5-methylbenzamide and tert-butyl 3_ (4-acetoxymethyl-2_formyl-6_isopropoxyphenyl) propanoate Was used as a raw material to give a yellow solid by the same operation as in Example 54 described later. To a solution of the obtained yellow solid (317 mg) in water (3.2 mL), 1N aqueous sodium hydroxide solution (5.5 mL) was added, and the mixture was stirred at room temperature for 1 hour. The reaction solution was purified by ODS column chromatography using water / acetonitrile as an elution solvent, and sodium 3- [2-[({2-[({4- [amino (hydroxyimino) methyl] fale was obtained. } Amino) carbo] -4-methylphenol} amino) methyl] -4- (hydroxymethyl) -6- Sopropoxyphenyl] propanoate 176 mg was obtained.
[0210] 実施例 54 [0210] Example 54
tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]力ルバ マートと tert-ブチル(2-ホルミル- 6-ヒドロキシフエノキシ)ァセタートを原料として、実 施例 7と同様の操作により tert-ブチル {2- [({2- [({4-[[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6-エトキシ フエノキシ }ァセタートを黄色固体として得た(FP: 653)。得られた tert-ブチル {2-[({2-[ ({4-[[(tert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-ク ロロフエ-ル}ァミノ)メチル ]-6-エトキシフエノキシ }ァセタートを原料として、実施例 12 と同様の操作により {2-[({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -4-ク ロロフエ-ル}ァミノ)メチル ]-6-エトキシフエノキシ }ァセチックアシッドトリフルォロ酢酸 塩を淡黄色固体として得た。 tert-Butyl [{4- [(2-amino-5-chlorobenzoyl) amino] phenol} (imino) methyl] rubamate and tert-butyl (2-formyl-6-hydroxyphenoxy) Using cetate as a raw material, tert-butyl {2-[({2-[({4-[[(tert-butoxycarbonyl) amino] (imino) methyl] phenol} amino ) Carboyl] -4-chlorophenol} amino) methyl] -6-ethoxyphenoxy} acetate was obtained as a yellow solid (FP: 653). The resulting tert-butyl {2-[({2- [({4-[[(tert-butoxycarbol) amino] (imino) methyl] phenol} amino) carbol] -4-alkyl [2-[({2-[({4- [Amino (imino) methyl] phenol] phenol by the same procedure as in Example 12 using chlorophenol} amino) methyl] -6-ethoxyphenoxy} acetate as a raw material. -Le} amino) carbol] -4-chlorophenol} amino) methyl] -6-ethoxyphenoxy} acetic acid trifluoroacetate was obtained as a pale yellow solid.
[0211] 実施例 55 [0211] Example 55
tert-ブチル [{4- [(2-ァミノ- 5-メチルベンゾィル)ァミノ]フエ-ル }(ィミノ)メチル]力ルバ マートと tert-ブチル(2-ホルミル- 6-ヒドロキシフエノキシ)ァセタートを原料として、実 施例 7と同様の操作により黄色固体として得た。得られた黄色固体を原料として、実 施例 18と同様の操作により {2-[({2-[({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ- ル] -4-メチルフエ-ル}ァミノ)メチル ]-6-ヒドロキシフエノキシ }ァセチックアシッドギ酸 塩を淡黄色固体として得た。 tert-Butyl [{4- [(2-Amino-5-methylbenzoyl) amino] phenol} (imino) methyl] rubamate and tert-butyl (2-formyl-6-hydroxyphenoxy) acetate As a result, a yellow solid was obtained in the same manner as in Example 7. Using the obtained yellow solid as a raw material, {2-[({2-[({4- [Amino (imino) methyl] phenol} amino) carbol] -4 was prepared in the same manner as in Example 18. -Methylphenol} amino) methyl] -6-hydroxyphenoxy} acetic acid formate was obtained as a pale yellow solid.
[0212] 実施例 56 [0212] Example 56
[3- [({2- [({4- [[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチル]フエ二ル}ァミノ)カルボ -ル] -4-クロ口フエ-ル}ァミノ)メチル ]-2- (2- tert-ブトキシ- 2-ォキソエトキシ)フエノキ シ]ァセチックアシッドを原料として、実施例 10と同様の操作により tert-ブチル {2-[({ 2- [({4- [[(tert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4 -クロ口フエ-ル}ァミノ)メチル ]-6-[2- (メチルァミノ) -2-ォキソエトキシ]フエノキシ }ァセ タートを淡黄色油状物として得た(EP:696)。得た。得られた tert-ブチル {2- [({2- [({4-[ [(tert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口 フエ-ル}ァミノ)メチル ]-6- [2- (メチルァミノ)- 2-ォキソエトキシ]フエノキシ }ァセタートを
原料として、実施例 18と同様の操作により {2-[({2-[({4- [ァミノ (ィミノ)メチル]フエ二ル} ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-6-[2- (メチルァミノ) -2-ォキソエト キシ]フエノキシ }ァセチックアシッドギ酸塩を淡黄色固体として得た。 [3- [({2- [({4- [[(tert-Butoxycarbonyl) amino] (imino) methyl] phenyl} amino) carbol] -4-chlorophine} amino) methyl ] -2- (2- tert-Butoxy-2-oxoethoxy) phenoxy] acetic acid as a starting material, tert-butyl {2-[({2- [({4- [[(tert-Butoxycarbol) amino] (imino) methyl] phenol} amino) carbol]-4 -chlorophenol} amino) methyl] -6- [2- (methylamino)- 2-Oxoethoxy] phenoxy} acetate was obtained as a pale yellow oil (EP: 696). Obtained. The resulting tert-butyl {2- [({2- [({4- [[(tert-butoxycarbol) amino] (imino) methyl] phenol} amino) carbol] -4-chloro Oral phenol} amino) methyl] -6- [2- (methylamino) -2-oxoethoxy] phenoxy} acetate As a raw material, {2-[({2-[({4- [Amino (imino) methyl] phenyl} amino) carbol] -4-chlorophole} was prepared in the same manner as in Example 18. Amino) methyl] -6- [2- (methylamino) -2-oxoethoxy] phenoxy} acetic acid formate was obtained as a pale yellow solid.
[0213] 実施例 57 [0213] Example 57
tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル Kィミノ)メチル]力ルバ マートを原料として、実施例 7と同様の操作により黄色固体を得た。得られた黄色固 体を原料として、実施例 15と同様の操作により {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ- ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル ]-4- [(ジメチルァミノ)メチル ]-6- エトキシフエノキシ }ァセチックアシッド塩酸塩を黄色固体として得た。 A yellow solid was obtained in the same manner as in Example 7, using tert-butyl [{4-[(2-amino-5-chlorobenzobenzo) amino] phenol Kimino) methyl] rubamate as a raw material. . Using the obtained yellow solid as a raw material, {2-[({2-[({4- [amino] imino] methyl} amino) carbol] -4 was prepared in the same manner as in Example 15. -Chlorophenyl} amino) methyl] -4-[(dimethylamino) methyl] -6-ethoxyphenoxy} acetic acid hydrochloride was obtained as a yellow solid.
[0214] 実施例 58 [0214] Example 58
tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル Kィミノ)メチル]力ルバ マートとェチル(4-ァセトキシメチル- 2-ホルミル- 6-プロポキシフエノキシ)ァセタートを 原料として、実施例 7と同様の操作によりェチル {4-ァセトキシメチル -2-[({2- [({4-[[(t ert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -4-メチルフ ェ-ル }ァミノ)メチル ]-6-プロポキシフエノキシ }ァセタートを得た(EP: 691)。得られた ェチル {4-ァセトキシメチル- 2- [({2- [({4- [[(tert-ブトキシカルボニル)ァミノ] (ィミノ)メチ ル]フエ-ル}ァミノ)カルボ-ル] -4-メチルフエ-ル}ァミノ)メチル ]-6-プロポキシフエノ キシ }ァセタート 90 mgとトリフルォロ酢酸 1 mLの混合物を室温にて 30分間攪拌した。 反応液を減圧下濃縮して得られた残渣をエタノール 2mLに希釈し、氷冷下 1規定水 酸ィ匕ナトリウム水溶液 0.20 mLを加えた。室温にて 1時間攪拌した後減圧下有機溶媒 を溜去し、 0.1%トリフルォロ酢酸水/ァセトニトリルを溶出溶媒とする ODSクロマトグラフ ィ一にて精製して、 {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル] -4-メ チルフエ-ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-プロポキシフエノキシ }ァセチック アシッドトリフルォロ酢酸塩 83 mgを無色固体として得た。 tert-Butyl [{4- [(2-amino-5-chlorobenzobenzo) amino] phenol Kimino) methyl] rubamate and ethyl (4-acetoxymethyl-2-formyl-6-propoxyphenoxy ) Acetate as a starting material, the same procedure as in Example 7 was followed by the same procedure as in Example 7. Ethyl {4-acetoxymethyl-2-[({2-[({4-[[(tert-butoxycarbol) amino] (imino) methyl] Phenol} amino) carbol] -4-methylphenyl} amino) methyl] -6-propoxyphenoxy} acetate was obtained (EP: 691). The resulting ethyl {4-acetoxymethyl-2-[({2-[({4-[[(tert-butoxycarbonyl) amino] (imino) methyl] phenol} amino) carbol] -4- A mixture of 90 mg of methylphenol} amino) methyl] -6-propoxyphenoxy} acetate and 1 mL of trifluoroacetic acid was stirred at room temperature for 30 minutes. The residue obtained by concentrating the reaction solution under reduced pressure was diluted with 2 mL of ethanol, and 0.20 mL of 1N sodium hydroxide aqueous solution was added under ice cooling. After stirring at room temperature for 1 hour, the organic solvent was distilled off under reduced pressure, and purified by ODS chromatography using 0.1% aqueous trifluoroacetic acid / acetonitrile as the elution solvent, {2- [({2- [( {4- [amino] phenyl} amino) carbol] -4-methylphenyl} amino) methyl] -4- (hydroxymethyl) -6-propoxyphenoxy} acetic acid triflu The chloroacetate 83 mg was obtained as a colorless solid.
[0215] 実施例 59 [0215] Example 59
tert-ブチル [{4- [(2-ァミノ- 5-クロ口べンゾィル)ァミノ]フエ-ル Kィミノ)メチル]力ルバ マートとェチル N-(2-ホルミルフエ-ル)- N-メチルダリシナートを原料として、実施例 2 8と同様の操作により N- {2- [({2- [({4- [[(tert-ブトキシカルボ-ル)ァミノ] (ィミノ)メチル]
フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル]フエ- }-N-メチルダリシ ンを得た(FP: 566)。得られた N-{2-[({2-[({4-[[(tert-ブトキシカルボニル)ァミノ] (ィミノ) メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ)メチル]フエ- }-N-メチ ルグリシン 75 mg、 0.2規定塩酸 1 mLおよびテトラヒドロフラン 0.5 mLの混合物を 50°C にて 3時間撹拌した。 1規定水酸ィ匕ナトリウム水溶液にて中和し減圧下濃縮して得ら れた残渣を 0.1%ギ酸水/ァセトニトリルを溶出溶媒とする ODSクロマトグラフィーにて精 製して、 [{2-[({2-[({4- [ァミノ (ィミノ)メチル]フエ二ル}ァミノ)カルボニル] -4-クロ口フエ- ル}ァミノ)メチル]フエ-ル }(メチル)ァミノ]ァセチックアシッドギ酸塩 12 mgを黄色固体 として得た。 tert-Butyl [{4- [(2-amino-5-chlorobenzene) amino] phenol Kimino) methyl] rubamate and ethyl N- (2-formylphenol)-N-methyldarlici N- {2- [({2- [({4- [[(tert-butoxycarbol) amino] (imino) methyl] Phenyl} amino) carbol] -4-chloro-phenyl} amino) methyl] phenol} -N-methyldaricin was obtained (FP: 566). The obtained N- {2-[({2-[({4-[[(tert-butoxycarbonyl) amino] (imino) methyl] phenol} amino) carbol] -4-chlorophine- A mixture of 75 mg of ru} amino) methyl] phenol-N-methylglycine, 1 mL of 0.2N hydrochloric acid and 0.5 mL of tetrahydrofuran was stirred at 50 ° C. for 3 hours. The residue obtained by neutralizing with 1N aqueous sodium hydroxide and concentrating under reduced pressure was purified by ODS chromatography using 0.1% aqueous formic acid / acetonitrile as the elution solvent, and [{2- [ ({2-[({4- [Amino (imino) methyl] phenyl} amino) carbonyl] -4-chloroform} amino) methyl] phenol} (methyl) amino] acetic acid Formate 12 mg was obtained as a yellow solid.
[0216] 実施例 60 [0216] Example 60
3-{[2_(2_エトキシ- 2_ォキソエトキシ) -5- (ヒドロキシメチル) _3_イソプロポキシベンジ ル]アミノ}-2-ナフトイックアシッドを原料として実施例 10と同様の操作により得た黄色 固体を原料として、実施例 24と同様の操作により {2-[({3-[({4- [ァミノ (ィミノ)メチル]フ ェ-ル }ァミノ)カルボ-ル]- 2-ナフチル }ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロ ポキシフエノキシ }ァセチックアシッドギ酸塩を黄色固体として得た。 3-{[2_ (2_Ethoxy-2_oxoethoxy) -5- (hydroxymethyl) _3_isopropoxybenzyl] amino} -2-naphthoic acid was used as a starting material to give a yellow color obtained in the same manner as in Example 10. {2-[({3-[({4- [Amino (imino) methyl] phenyl} amino) carbol]-2-naphthyl} amino in the same manner as in Example 24, using solid as a raw material. ) Methyl] -4- (hydroxymethyl) -6-isopropoxyphenoxy} acetic acid formate was obtained as a yellow solid.
[0217] 上記実施例 1〜60の方法と同様にして、後記表 25〜107に示す実施例化合物 61 〜654をそれぞれ対応する原料を使用して製造した。表 25〜107に実施例化合物 の構造及び物理化学的データを示す。 [0217] In the same manner as in the above Examples 1 to 60, Example compounds 61 to 654 shown in Tables 25 to 107 to be described later were produced using the corresponding raw materials. Tables 25 to 107 show the structures and physicochemical data of the example compounds.
[0218] また、後記表 108〜118に本発明の別の化合物の構造を示す。これらは、上記の 製造法や実施例記載の方法若しくは当業者にとって自明である方法、またはこれら の変法を用いることによって容易に製造することができる。 [0218] Further, Tables 108 to 118 below show structures of other compounds of the present invention. These can be easily produced by using the production methods described above, the methods described in the examples, methods obvious to those skilled in the art, or variations thereof.
[0219] 本発明の化合物の優れた活性ィ匕血液凝固第 VII因子阻害活性は、以下に示す試 験方法により確認された。 [0219] The excellent activity of the compound of the present invention, ie, blood coagulation factor VII inhibitory activity, was confirmed by the test method shown below.
[0220] 試験方法 1.合成基質法による酵素阻害測定試験 [0220] Test Method 1. Enzyme inhibition measurement test by synthetic substrate method
(1)活性ィ匕血液凝固第 VII因子/ TF複合体阻害活性 : 96穴マイクロプレートに反応 緩衝液(pH 7.4) 50 μ L、化合物溶液 10 L、 2 mMの合成基質 S- 2288 (Chromogeni X社) 20 μ L、ヒト TF(American Diagnostics社)とヒト活性化血液凝固第 VII因子 (登録 商標 NovoSeven、 Novo Nordisk社)の混合液(各 30 ηΜ) 20 μ Lを加え、 1時間室温で
反応させ、 405 nmの吸光度変化を吸光マイクロプレートリーダー(商品名: SPECTRA max340PC384、 Molecular Devices社)で測定し、 IC を算出した。 (1) Activity 阻 害 Blood coagulation factor VII / TF complex inhibitory activity: Reaction to 96-well microplate Buffer (pH 7.4) 50 μL, Compound solution 10 L, 2 mM synthetic substrate S-2288 (Chromogeni X 20 μL, add 20 μL of a mixture of human TF (American Diagnostics) and human activated blood coagulation factor VII (registered trademark NovoSeven, Novo Nordisk) (30 η 各 each) for 1 hour at room temperature After the reaction, the change in absorbance at 405 nm was measured with an absorbance microplate reader (trade name: SPECTRA max340PC384, Molecular Devices), and IC was calculated.
50 50
本発明の代表的化合物の活性ィ匕血液凝固第 VII因子阻害活性を下記表 1に示す 。尚、 Exは実施例番号を示す (以下同様。 )0 Table 1 below shows the activity of the representative compounds of the present invention. Ex represents the example number (the same applies hereinafter) 0
[0221] [表 1] [0221] [Table 1]

[0222] (2)活性ィ匕血液凝固第 X因子阻害活性: 96穴マイクロプレートに反応緩衝液 (ρΗ 7.4 ) 50 μ 化合物溶液 10 μ 2 mMの合成基質 S- 2222 (Chromogenix社) 20 μ L、 0. 02U/mLのヒト活性化血液凝固第 X因子 (Enzyme Research Laboratories社) 20 μ Lを 加え、 1時間室温で反応させ、 405應の吸光度変化を測定した。 [0222] (2) Activity 匕 Blood coagulation factor X inhibitory activity: Reaction buffer (ρΗ 7.4) in a 96-well microplate 50 μ Compound solution 10 μ 2 mM synthetic substrate S-2222 (Chromogenix) 20 μ L Then, 20 μL of 0.02 U / mL human activated blood coagulation factor X (Enzyme Research Laboratories) was added and allowed to react at room temperature for 1 hour, and the change in absorbance at 405 was measured.
実施例 16の化合物は活性ィ匕血液凝固第 X因子に対して、その阻害活性 IC は 1 The compound of Example 16 is active against blood coagulation factor X and its inhibitory activity IC is 1
50 50
00 M以上の数値を示した。 A numerical value of 00 M or more was shown.
[0223] (3)トロンビン阻害活性: 96穴マイクロプレートに反応緩衝液 (pH 7.4) 50 μ L、化合 物溶液 10 μ 2 mMの合成基質 S- 2238 (Chromogenix社) 20 μ L、 0.2U/mLのヒトト ロンビン (Sigma社) 20 μ Lを加え、 1時間室温で反応させ、 405 nmの吸光度変化を測 し 7こ。 [0223] (3) Thrombin inhibitory activity: 50 μL of reaction buffer (pH 7.4) in 96-well microplate, compound solution 10 μ2 mM synthetic substrate S-2238 (Chromogenix) 20 μL, 0.2 U / Add 20 μL of mL of human thrombin (Sigma), react for 1 hour at room temperature, and measure the change in absorbance at 405 nm.
実施例 16の化合物はトロンビンに対して、その阻害活性 IC は 100 μ Μ以上の The compound of Example 16 has an inhibitory activity IC of over 100 μ IC against thrombin.
50 50
数値を示した。 Numerical values are shown.
[0224] 以上の結果から、本発明化合物 (I)は強力な活性ィ匕血液凝固第 VII因子阻害活性 を有すること、また、活性ィ匕血液凝固第 X因子、トロンビンに対して選択的に活性ィ匕 血液凝固第 VII因子を阻害することが確認できた。 [0224] From the above results, the compound (I) of the present invention has a strong activity 匕 blood coagulation factor VII inhibitory activity, and is also active selectively against the activity 匕 blood coagulation factor X and thrombin. It was confirmed that the blood coagulation factor VII was inhibited.
[0225] 試験方法 2.モルモットを用いた ex vivoでの凝固時間測定試験
雄性ハートレー系モルモット (5- 7週齢、 日本 SLC社)に対し、ジメチルァセトアミド 1容 、5%グルコース 1容を混合した溶媒に溶解した試験化合物をジェチルエーテル麻酔 下で静脈内投与し (1 mg/kg)、 2分後に頸静脈より 3.13%クェン酸ナトリウム 1/10容にて 0.5 mL採血し、 3000 rpm 10分の遠心処理により血漿を分離した。この血漿を用いて 以下 a)の方法に従い外因系凝固時間 (PT)の測定を行った。 [0225] Test method 2.Ex vivo clotting time measurement test using guinea pigs To male Hartley guinea pigs (5-7 weeks old, Japan SLC), a test compound dissolved in a solvent containing 1 volume of dimethylacetamide and 1 volume of 5% glucose was administered intravenously under anesthesia with jetyl ether. (1 mg / kg) After 2 minutes, 0.5 mL of blood was collected from the jugular vein with 1/10 volume of 3.13% sodium citrate, and plasma was separated by centrifugation at 3000 rpm for 10 minutes. Using this plasma, the extrinsic clotting time (PT) was measured according to the following method a).
a)外因系凝固時間 (PT) a) Extrinsic clotting time (PT)
モルモット血漿 50 /z Lを 37°Cにて 1分間加温し、ヒーモスアイエル'リコンビプラスチ ン 100 Lを添加し凝固時間の測定を行った。凝固時間の測定には血液凝固自動 測定装置 ST4 (ロシュ ·ダイァグノステイタス社)を使用した。試験化合物非投与のモル モット血漿の凝固時間を対照とし、この対照を 1としたときの相対値で試験化合物の 活性を示した。 Guinea pig plasma 50 / z L was warmed at 37 ° C for 1 minute, and 100 L of moth moth recombin plastin was added to measure the clotting time. For the measurement of the coagulation time, an automatic blood coagulation measuring device ST4 (Roche Diagno Status) was used. The clotting time of guinea pig plasma without the test compound was used as a control, and the activity of the test compound was shown as a relative value when this control was 1.
外因系凝固時間 (PT)の測定の結果、本発明化合物は静脈内投与において外因系 凝固時間の延長作用が認められた。 As a result of measuring the extrinsic clotting time (PT), the compound of the present invention was found to prolong the extrinsic clotting time by intravenous administration.
本発明の代表的化合物の外因系凝固時間の延長作用を下記表 2に示す。 Table 2 below shows the action of prolonging the extrinsic clotting time of the representative compounds of the present invention.
[0226] [表 2] [0226] [Table 2]

[0227] 以上の結果から、本発明化合物 (I)は外因系凝固時間の延長作用を示すことから、 血液凝固抑制剤として有効であることが確認できた。 [0227] From the above results, it was confirmed that the compound (I) of the present invention was effective as a blood coagulation inhibitor since it exhibited an action of extending the extrinsic clotting time.
[0228] 本発明化合物 (I)には、生体内において代謝されて活性ィ匕血液凝固第 VII因子阻 害活性を示す化合物も含まれる。例えば、 R1カ C(=NOH)NH、 - C(=NH)NH- CO - ( [0228] The compound (I) of the present invention includes a compound that is metabolized in vivo and exhibits an active blood coagulation factor VII inhibitory activity. For example, R 1 C (= NOH) NH,-C (= NH) NH- CO-(
2 2 置換されていてもよい低級アルキル)若しくは 5-ォキソ -2,5-ジヒドロ- 1,2,4-ォキサジ ァゾール -3-ィルであり、及び Zまたは、 -C(0)R5がエステルまたはアミドである化合 物には、生体内で R1が- C(=NH)NHであり、及び/または、 - C(0)R5がカルボン酸で 2 2 optionally substituted lower alkyl) or 5-oxo-2,5-dihydro-1,2,4-oxadiazol-3-yl, and Z or -C (0) R 5 is For compounds that are esters or amides, R 1 is —C (═NH) NH and / or —C (0) R 5 is a carboxylic acid in vivo.
2 2
ある化合物に変換され活性ィ匕血液凝固第 VII因子阻害活性を示すィ匕合物がある。こ のような化合物の有用性は下記試験方法 3、並びに、試験方法 1及び Zまたは試験
方法 2の試験を組み合わせることによって確認することができる。 There are compounds that have been converted to certain compounds and show active activity, blood coagulation factor VII inhibitory activity. The usefulness of such compounds is shown below in Test Method 3, and Test Methods 1 and Z or Test This can be confirmed by combining the tests of Method 2.
[0229] 試験方法 3. [0229] Test method 3.
a)経口投与 a) Oral administration
ラット (雄性; Sprague- Dawley (SD- IGS)、 7週齢 (6週齢で購入)、日本チヤ一ルスリ バー社、摂餌条件:水、餌共に自由摂取)は投与前日の夕方より絶食した。化合物は 10 mg/5 mLの濃度になるように 0.5%MCで懸濁させて、ゾンデを用いて胃内部へを単 回経口投与した。 Rats (male; Sprague-Dawley (SD-IGS), 7 weeks old (purchased at 6 weeks of age), Nippon Chiyers River, food condition: free intake of water and food) were fasted from the evening before administration . The compound was suspended in 0.5% MC to a concentration of 10 mg / 5 mL and administered orally once into the stomach using a sonde.
[0230] b)静脈内投与 [0230] b) Intravenous administration
化合物は 1 mg/0.2 mLの濃度になるように Ν,Ν-ジメチルァセトアミドで溶解させ、さ らに同量の 5%グルコース(大塚製薬工場)で希釈後、ラット(雄性; Sprague- Dawley (S D_IGS)、 7週齢 (6週齢で購入)、日本チヤ一ルスリバ一社、摂餌条件:水、餌共に自 由摂取)尾静脈より単回静脈内投与した。 The compound was dissolved in Ν, Ν-dimethylacetamide to a concentration of 1 mg / 0.2 mL, diluted with the same amount of 5% glucose (Otsuka Pharmaceutical Factory), and then rat (male; Sprague-Dawley (SD_IGS), 7-week-old (purchased at 6-week-old), Nippon Chisuriba Co., Ltd., feeding conditions: free intake of water and food) Single intravenous administration via tail vein.
[0231] c)試料採取方法 [0231] c) Sampling method
経口投与後の試料採取は、投与後 15、 30分、 1、 2、 4、 8時間に頸静脈よりへパリン 処理したシリンジで約 200 /z L採血し、直ちに氷冷下に保存した。静脈内投与後の場 合は、投与後 2、 10、 30分、 1、 2、 4時間に頸静脈よりへノリン処理したシリンジで約 20 0 μ L採血し、直ちに氷冷下に保存した。血液は 10000 rpmで 1分間(室温)遠心し血 漿を分取した。調製した試料は測定まで約 4°Cで保存した。 After oral administration, samples were collected at approximately 200 / zL with a syringe that had been parinized from the jugular vein at 15, 30 minutes, 1, 2, 4, and 8 hours after administration, and immediately stored under ice cooling. In the case of intravenous administration, about 200 μL of blood was collected from a jugular vein-treated syringe at 2, 10, 30 minutes, 1, 2, and 4 hours after administration, and immediately stored under ice cooling. The blood was centrifuged at 10,000 rpm for 1 minute (room temperature) to collect the plasma. The prepared sample was stored at about 4 ° C until measurement.
[0232] d)定量分析 [0232] d) Quantitative analysis
各ポイントの血漿サンプルを 25 μ Lチューブに分注し、ァセトニトリルに溶解した内 部標準物質溶液を 100 μ Lおよびァセトニトリルを 875 μ L加えた。その後、ミキサー で攪拌し、 3300 rpmで 20分間遠心分離後、上清 5 μ Lを HPLCに注入した。検量線は 、測定化合物を 50%ァセトニトリル水溶液に溶解し、ァセトニトリルで 10、 50、 100、 500 、 1000、 5000、 10000、 50000 ng/mLに段階希釈した検量線溶液を用意した。続いて ブランク血漿を 25 μ Lチューブに分注し、先に用意した各濃度の検量線溶液を 25 μ L、ァセトニトリルに溶解した内部標準物質溶液 100 μ Lおよびァセトニトリルを 850 μ L加えた。その後、ミキサーで攪拌し、 3300 rpmで 20分間遠心分離後、上清 5 μ Lを Η PLCに注入した。定量分析には、 LC- MS/MSを用いた。
HPLC : HP1100(Agilent)カラム: Cadenza CD- C18, 3 ^ m, 50*4.6 mm (Imtakt) カラム温度: 40°C A plasma sample at each point was dispensed into a 25 μL tube, and 100 μL of an internal standard solution dissolved in acetonitrile and 875 μL of acetonitrile were added. Thereafter, the mixture was stirred with a mixer, centrifuged at 3300 rpm for 20 minutes, and 5 μL of the supernatant was injected into HPLC. For the calibration curve, a calibration curve solution was prepared by dissolving the measurement compound in 50% aqueous acetonitrile and serially diluting with acetonitrile to 10, 50, 100, 500, 1000, 5000, 10000, 50000 ng / mL. Subsequently, blank plasma was dispensed into a 25 μL tube, and 25 μL of the calibration curve solution prepared at each concentration, 100 μL of the internal standard solution dissolved in acetonitrile, and 850 μL of acetonitrile were added. Thereafter, the mixture was stirred with a mixer, centrifuged at 3300 rpm for 20 minutes, and 5 μL of the supernatant was injected into the ΗPLC. LC-MS / MS was used for quantitative analysis. HPLC: HP1100 (Agilent) Column: Cadenza CD-C18, 3 ^ m, 50 * 4.6 mm (Imtakt) Column temperature: 40 ° C
移動相: 10 mM酢酸アンモ-ゥム水溶液 (pH4.5) ;:ァセトニトリル; B Mobile phase: 10 mM ammonium acetate aqueous solution (pH 4.5);: acetonitrile; B
結果は、表 3に示す。 The results are shown in Table 3.
[表 3] [Table 3]

MS/MS: API-4000(MDS/Sciex) MS / MS: API-4000 (MDS / Sciex)
[0234] [表 4]

 [0234] [Table 4]
[0234] [Table 4]
[0235] [表 5]
[0235] [Table 5]

Rf RSyn R4a R4 DATA Rf RSyn R 4a R 4 DATA
NMR (DMS0-d6) : 3.92 (3H, s), 7.65NMR (DMS0-d 6 ): 3.92 (3H, s), 7.65
4 4 NC - - OMe (2H, s), 10.27 (IH, s), 11.31 (IH, br 4 4 NC--OMe (2H, s), 10.27 (IH, s), 11.31 (IH, br
o o s) o o s)
丽 R (DMS0-d6) : 1.36 (3H, t, J = 7.0 Hz), 3.53 (2H, s), 4.08 (2H, q, J =丽 R (DMS0-d 6 ): 1.36 (3H, t, J = 7.0 Hz), 3.53 (2H, s), 4.08 (2H, q, J =
89 4 H02CCH2- -OEt 7.0Hz), 7.12 (IH, d, J = 1.5 Hz), 89 4 H0 2 CCH 2 --OEt 7.0Hz), 7.12 (IH, d, J = 1.5 Hz),
7.15 (IH, d, J = 1.5 Hz), 10.02 (IH, br s), 10.23 (IH, s) 7.15 (IH, d, J = 1.5 Hz), 10.02 (IH, br s), 10.23 (IH, s)
NMR (CDC13) : 1.55 (3H, t, J = 7.0 Hz), 4.24 (2H, q, J = 7.0 Hz), 7.93 (IH,NMR (CDC1 3 ): 1.55 (3H, t, J = 7.0 Hz), 4.24 (2H, q, J = 7.0 Hz), 7.93 (IH,
7 7 02N- -OEt 7 7 0 2 N- -OEt
d, J = 2.4 Hz), 8.22 (IH, d, J = 2.4 Hz), 10.00 (IH, s), 11.66 (IH, s) 丽 R (DMS0-d6) : 1.34 (6H, d, J = 6.0 Hz), 4.85 (IH, sep, J = 6.0 Hz), 7.95d, J = 2.4 Hz), 8.22 (IH, d, J = 2.4 Hz), 10.00 (IH, s), 11.66 (IH, s) 丽 R (DMS0-d 6 ): 1.34 (6H, d, J = 6.0 Hz), 4.85 (IH, sep, J = 6.0 Hz), 7.95
90 7 02N- -OiPr (1H, d, J = 2.7 Hz), 8.10 (IH, d, J = 90 7 0 2 N- -OiPr (1H, d, J = 2.7 Hz), 8.10 (IH, d, J =
2.7 Hz), 10.33 (1H, s), 11.20-11.50 (1H, br) 2.7 Hz), 10.33 (1H, s), 11.20-11.50 (1H, br)
91 7 02N- -OiBu EN: 238 91 7 0 2 N- -OiBu EN: 238
92 7 02N- Me FN: 180 92 7 0 2 N-Me FN: 180
NMR (CDC13) : 1.49 (3H, t, J = 7.0 Hz), 2.10 (3H, s), 4.15 (2H, q, J =NMR (CDC1 3 ): 1.49 (3H, t, J = 7.0 Hz), 2.10 (3H, s), 4.15 (2H, q, J =
8 8 -OEt 7.0 Hz), 5.06 (2H, s), 7.11 (IH, d, J 8 8 -OEt 7.0 Hz), 5.06 (2H, s), 7.11 (IH, d, J
= 1.7 Hz), 7.20 (IH, d, J= 1.7 Hz), 9.92 (IH, s), 11.06 (1H, br s) = 1.7 Hz), 7.20 (IH, d, J = 1.7 Hz), 9.92 (IH, s), 11.06 (1H, br s)
NMR (CDC") : 2.11 (3H, s), 3.94 (3H, s), 5.07 (2H , s), 7.12 (IH, d, J =NMR (CDC "): 2.11 (3H, s), 3.94 (3H, s), 5.07 (2H, s), 7.12 (IH, d, J =
93 8 AcOCH2- -OMe 93 8 AcOCH 2 --OMe
1.6 Hz), 7.21 (IH, d, J = 1.6 Hz), 9.92 (1H, s), 11.16 (IH, s) 1.6 Hz), 7.21 (IH, d, J = 1.6 Hz), 9.92 (1H, s), 11.16 (IH, s)
10 10 AcOCH2- -OiPr EN: 251 10 10 AcOCH 2 --OiPr EN: 251
NMR (CDCI3) : 1.39 (3H, d, J = 6.3 Hz), 2.06 (3H, s), 2.10 (3H, s), 4.21-4.31 (2H, m), 4.60-4.68 (IH, m) , NMR (CDCI3): 1.39 (3H, d, J = 6.3 Hz), 2.06 (3H, s), 2.10 (3H, s), 4.21-4.31 (2H, m), 4.60-4.68 (IH, m),
94 10 94 10
Me 5.05 (2H, s), 7.24 (IH, d, J = 2.0 Me 5.05 (2H, s), 7.24 (IH, d, J = 2.0
Hz), 7.27 (IH, d, J = 2.0 Hz), 9.92 (1H, s), 10.99 (1H, br s) Hz), 7.27 (IH, d, J = 2.0 Hz), 9.92 (1H, s), 10.99 (1H, br s)
删 R (CDCI3) : 1.39 (6H, d, J = 6.0 Hz), 4.57 (IH, sep, J = 6.0 Hz), 7.35 删 R (CDCI3): 1.39 (6H, d, J = 6.0 Hz), 4.57 (IH, sep, J = 6.0 Hz), 7.35
12 12 I -OiPr 12 12 I -OiPr
(IH, d, J = 1.9 Hz), 7.49 (IH, d, J = 1.9 Hz), 9.84 (IH, s), 10.85 (IH, s) (IH, d, J = 1.9 Hz), 7.49 (IH, d, J = 1.9 Hz), 9.84 (IH, s), 10.85 (IH, s)
95 12 Br -OiPr EP: 260 6]
 95 12 Br -OiPr EP: 260 6]
95 12 Br -OiPr EP: 260 6]
Rf RSyn R4a R4 R5 DATA Rf RSyn R 4a R 4 R 5 DATA
14 14 -OtBu FP: 237 14 14 -OtBu FP: 237
96 14 0,N- -OMe -OtBu GC: 311 96 14 0, N- -OMe -OtBu GC: 311
NMR (CDC13) : 1.45 (9H, s), 1.5 (3H, t, J = 7.0 Hz), 4.22 (2H, tNMR (CDC1 3 ): 1.45 (9H, s), 1.5 (3H, t, J = 7.0 Hz), 4.22 (2H, t
97 14 0,N- -OEt -OtBu J = 7.0 Hz), 4.93 (2H, s), 7.9 97 14 0, N- -OEt -OtBu J = 7.0 Hz), 4.93 (2H, s), 7.9
(1H, d, J = 2.5 Hz), 8.35 (1H, d J = 2.5 Hz), 10.61 (1H, s) (1H, d, J = 2.5 Hz), 8.35 (1H, d J = 2.5 Hz), 10.61 (1H, s)
NMR (CDCI3) : 1.44 (9H, s), 1.46 (6H, d, J = 6.0 Hz), 4.72 (1H,NMR (CDCI3): 1.44 (9H, s), 1.46 (6H, d, J = 6.0 Hz), 4.72 (1H,
98 14 0,N- - OiPr -OtBu sep, J = 6.0 Hz), 7.93 (1H, d, J 98 14 0, N--OiPr -OtBu sep, J = 6.0 Hz), 7.93 (1H, d, J
二 2.5 Hz), 8.33 (1H, d, J = 2.5 Hz), 10.60 (1H, s) 2 2.5 Hz), 8.33 (1H, d, J = 2.5 Hz), 10.60 (1H, s)
99 14 0,N- - OiBu -OtBu EP: 354 99 14 0, N--OiBu -OtBu EP: 354
00 14 Ο,Ν- Me -OtBu FN: 294 00 14 Ο, Ν- Me -OtBu FN: 294
01 14 -OEt -OtBu FP: 281 01 14 -OEt -OtBu FP: 281
02 14 -OEt -OEt FP: 253 02 14 -OEt -OEt FP: 253
匪 R (CDC13): 1.51 (9H, s), 2.11匪 R (CDC1 3 ): 1.51 (9H, s), 2.11
(3H, s), 4.57 (2H, s), 5.07 (2H, 03 14 AcOCH,- -OH -OtBu s), 7.24 (1H, d, J = 2.0 Hz), (3H, s), 4.57 (2H, s), 5.07 (2H, 03 14 AcOCH,--OH -OtBus), 7.24 (1H, d, J = 2.0 Hz),
7.27 (1H, d, J = 2.0 Hz), 9.00 7.27 (1H, d, J = 2.0 Hz), 9.00
(1H, s), 10.11 (1H, s)(1H, s), 10.11 (1H, s)
04 14 AcOCH,- -OEt -OtBu EP: 375 [M+Na] + 04 14 AcOCH,--OEt -OtBu EP: 375 [M + Na] +
NMR (CDC : 1.28 (3H, t, J = 6.9 NMR (CDC: 1.28 (3H, t, J = 6.9
Hz), 1.48 (3H, t, 〗 = 6.9 Hz),Hz), 1.48 (3H, t,〗 = 6.9 Hz),
2.11 (3H, s), 4.11 (2H, q, J =2.11 (3H, s), 4.11 (2H, q, J =
6.9 Hz), 4.22 (2H, q, J = 6.9 05 14 AcOCH, - -OEt -OEt 6.9 Hz), 4.22 (2H, q, J = 6.9 05 14 AcOCH,--OEt -OEt
Hz), 4.86 (2H, s), 5.05 (2H, s), Hz), 4.86 (2H, s), 5.05 (2H, s),
7.11 (1H, d, J = 1.9 Hz), 7.437.11 (1H, d, J = 1.9 Hz), 7.43
(1H, d, J = 1.9 Hz), 10.62 (1H, br s)(1H, d, J = 1.9 Hz), 10.62 (1H, br s)
06 14 AcOCH,- -OiPr -OtBu EP: 367 06 14 AcOCH,--OiPr -OtBu EP: 367
07 14 AcOCH,- -OiPr 一 OEt FP 339 07 14 AcOCH,--OiPr OEt FP 339
08 14 -OiPr -OtBu EP 443 [M+Na] 08 14 -OiPr -OtBu EP 443 [M + Na]
09 14 Br -OiPr -OEt FP: 345 09 14 Br -OiPr -OEt FP: 345
NMR (DMS0-d6") : 1.39 (9H, si, 3.92 (3H, s), 4.89 (2H, s), 7.68 (1H, 10 14 NC- -OMe -OtBu d, J = 2.0 Hz), 7.84 (1H, d, J = NMR (DMS0-d 6 "): 1.39 (9H, si, 3.92 (3H, s), 4.89 (2H, s), 7.68 (1H, 10 14 NC- -OMe -OtBud, J = 2.0 Hz), 7.84 (1H, d, J =
2.0 Hz), 10.45 (1H, s) 7]
16 16 H2C=CH- -OiPr -OtBu EP: 343 [M+Na] *2.0 Hz), 10.45 (1H, s) 7] 16 16 H 2 C = CH- -OiPr -OtBu EP: 343 [M + Na] *
1 11 16 H2C=CH- -OiPr -OEt FP: 2931 11 16 H 2 C = CH- -OiPr -OEt FP: 293
42 42 F H -OtBu GC: 25442 42 F H -OtBu GC: 254
112 42 H -NMe2 -OtBu FP: 280112 42 H -NMe 2 -OtBu FP: 280
113 42 . -OEt -OtBu EP: 347 [M+Na] ÷113 42. -OEt -OtBu EP: 347 [M + Na] ÷
114 42 H0- (CH2) 2- -OEt -OEt EP: 297114 42 H0- (CH 2 ) 2 --OEt -OEt EP: 297
115 42 tBuMe2SiO- -OiPr -OEt EP: 397115 42 tBuMe 2 SiO- -OiPr -OEt EP: 397
46 46 H -SMe -OH EN: 22546 46 H -SMe -OH EN: 225
116 46 H -S02Me -OH EN: 257116 46 H -S0 2 Me -OH EN: 257
53 53 Me2NCH2- -OEt -OtBu EP: 33853 53 Me 2 NCH 2 --OEt -OtBu EP: 338
117 53 -OiPr -OtBu EP: 352 117 53 -OiPr -OtBu EP: 352
[0238] [表 8] [0238] [Table 8]

[0239] [表 9]
[0239] [Table 9]

Rf RSyn R4a R4b R5 DATA Rf RSyn R 4a R 4b R 5 DATA
120 14 CI -OtBu FP: 385 120 14 CI -OtBu FP: 385
zN zN
15 15 HO-(C¾)2- -OEt -OtBu EP: 349 15 15 HO- (C¾) 2 --OEt -OtBu EP: 349
NMR (CDC13) : 1.30 (3H, t, J = 7.0 Hz), 1.45 (3H, t, J = 7.0 Hz), 2.79 (2H, t, J =NMR (CDC1 3 ): 1.30 (3H, t, J = 7.0 Hz), 1.45 (3H, t, J = 7.0 Hz), 2.79 (2H, t, J =
6.5 Hz), 3.80-3.88 (2H, m), 3.97 (IH, t, J = 7.0 Hz),6.5 Hz), 3.80-3.88 (2H, m), 3.97 (IH, t, J = 7.0 Hz),
121 15 HO- (CH2) 2 - -OEt -OEt 4.06 (2H, q, J = 7.0 Hz), 121 15 HO- (CH 2 ) 2 --OEt -OEt 4.06 (2H, q, J = 7.0 Hz),
4.25 (2H, q, J = 7.0 Hz), 4.69 (2H, d, J = 7.0 Hz), 4.25 (2H, q, J = 7.0 Hz), 4.69 (2H, d, J = 7.0 Hz),
4.80 (2H, s), 6.73 (IH, d, J = 2.0 Hz), 6.76 (IH, d, J = 2.0 Hz) 4.80 (2H, s), 6.73 (IH, d, J = 2.0 Hz), 6.76 (IH, d, J = 2.0 Hz)
[0240] [表 10] [0240] [Table 10]


[0241] [表 11]
 [0241] [Table 11]
[0241] [Table 11]


m [swo]m [swo]


w\ o]
 w \ o]
w \ o]
960丽 SOOZdf/ェ:) d 66 8.80.0/900Z OAV
mm [9 wo] 960 丽 SOOZdf / e :) d 66 8.80.0 / 900Z OAV mm [9 wo]


960丽 SOOZdf/ェ:) d 001· 8.80.0/900Z OAV
m [ so] 960 丽 SOOZdf / e :) d 001 8.80.0 / 900Z OAV m [so]


960丽 SOOZdf/ェ:) d !•01· 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d! • 01 · 8.80.0 / 900Z OAV
m ¾N- H ■ - 11 m m ¾N- H ■-11 m
: ZHN- H H a 161: Z HN- HH a 161
: N3 ZHM- H ί hi 061: N3 Z HM- H ί hi 061
Ml : άά ZHM- ά 11 681 Ml : άά Z HM- ά 11 681
: Ν3 zON- H 69 881 m : 3 zon- ί H 69 181 m : dd zON- i 69 981 ooc : άά zON- ά 69 581: Ν3 z ON- H 69 881 m: 3 z on- ί H 69 181 m: dd z ON- i 69 981 ooc: άά z ON- H 69 691
08Z - Μ zOM- H 69 n\08Z-Μ z OM- H 69 n \
(s 'HI) ει ·π '(ZH 6(s 'HI) ει · π' (ZH 6
, = f 'PP 'HI) '(in Ή9) 98 -i - zOM- H 69 m , = f 'PP' HI) '(in Ή9) 98 -i- z OM- H 69 m
(ω Ήΐ) 69 ·ί - 29-i: (9P-OSM) MN (ω Ήΐ) 69 · ί-29-i: ( 9 P-OSM) MN
892 : <ia zON - H H 69 892: <ia z ON-HH 69
H 69 181 丽 ¾ u人 S3 H 69 181 ¾ ¾ u people S3

960丽 SOOZdf/ェ:) d 301- 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 301- 8.80.0 / 900Z OAV

Rf RSyn R3a R3t R DATA Rf RSyn R 3a R 3t R DATA
72 72 CI H -N02 EP: 335 72 72 CI H -N0 2 EP: 335
NMR (DMS0-d6) : 6.27 (2H, brs), 6.79 (1H, dd, J = 4.2, 9.2 Hz), 7.11 一 7.19 (1H, m), 7.52 (1H, dd, J = 3.0,NMR (DMS0-d 6 ): 6.27 (2H, brs), 6.79 (1H, dd, J = 4.2, 9.2 Hz), 7.11 one 7.19 (1H, m), 7.52 (1H, dd, J = 3.0,
193 72 F H -N02 193 72 FH -N0 2
10.0 Hz), 7.80 (2H, d, J = 8.8 Hz), 7.92 (2H, d, J = 8.8 Hz), 10.37 (1H, s) 10.0 Hz), 7.80 (2H, d, J = 8.8 Hz), 7.92 (2H, d, J = 8.8 Hz), 10.37 (1H, s)
194 72 Me F -N02 EP: 333 194 72 Me F -N0 2 EP: 333
195 72 F F -NH2 FP: 307 195 72 FF -NH 2 FP: 307
196 72 H F -NH2 EP: 289 196 72 HF -NH 2 EP: 289
197 72 H H -NHZ FP: 551 197 72 HH -NH Z FP: 551
198 72 - OMe H -NH2 EP: 301 198 72-OMe H -NH 2 EP: 301
199 77 CI H -NH2 EP: 305 199 77 CI H -NH 2 EP: 305
200 77 F H -NH2 FP: 289 200 77 FH -NH 2 FP: 289
201 77 F Me - NH2 FP: 303 201 77 F Me-NH 2 FP: 303
[0249] [表 19] [0249] [Table 19]


[0250] [表 20]
[0250] [Table 20]
692 : dH H 69 692: dH H 69
■ dH 69 on ■ dH 69 on
(s 'HI) 'll '(ZH (s' HI) 'll' (ZH
9 = f 'P ΉΙ) 06 "8 '(ZH 9 'ZH 0 "6 = f 'PP 'HI) 9 = f 'P ΉΙ) 06 "8' (ZH 9 'ZH 0" 6 = f' PP 'HI)
ID 69 602 K' '(ZH 0·6 = ί 'P 'HI) "8 '(ω 'Μ) 01 "8-S0"8 ID 69 602 K '' (ZH 0 · 6 = ί 'P' HI) "8 '(ω' Μ) 01" 8-S0 "8
'(ZH 0 'ZH 0·6 = ί 'PP 'HI) 16 : (9P- OSW) 漏 '(ZH 0' ZH 0 · 6 = ί 'PP' HI) 16: ( 9 P-OSW) leakage
viva viva



960丽 SOOZdf/ェ:) d 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 8.80.0 / 900Z OAV

Rf RSyn R3 R DATA Rf RSyn R 3 R DATA
NMR (DMSO— d6) : 5.82 (2H, br s), 7.85-7.92 (2H, ra), 8.04 (IH, d, J = 2.5 Hz), 8.11 (1H,NMR (DMSO— d 6 ): 5.82 (2H, br s), 7.85-7.92 (2H, ra), 8.04 (IH, d, J = 2.5 Hz), 8.11 (1H,
212 72 CI - N02 dd, J = 8.5 Hz, 2.5 Hz), 8.22 (IH, d, J = 8.5 212 72 CI-N0 2 dd, J = 8.5 Hz, 2.5 Hz), 8.22 (IH, d, J = 8.5
Hz), 8.80 (IH, d, J = 2.5 Hz), 9.87 (1H, s), 11.07 (1H, s) Hz), 8.80 (IH, d, J = 2.5 Hz), 9.87 (1H, s), 11.07 (1H, s)
213 72 Me -N02 EP 316 213 72 Me -N0 2 EP 316
214 72 H -N02 EP 302 214 72 H -N0 2 EP 302
215 77 CI - N¾ EP 306 215 77 CI-N¾ EP 306
216 77 Me -NH2 EP 286 216 77 Me -NH 2 EP 286
217 77 H -NH2 EP 272 217 77 H -NH 2 EP 272
[0253] [表 23]

 [0253] [Table 23]
[0253] [Table 23]
[0254] [表 24]
[0254] [Table 24]


960丽 SOOZdf/ェ:) d 901· 8.80.0/900Z OAV
 960 丽 SOOZdf / e :) d 901 · 8.80.0 / 900Z OAV
960 丽 SOOZdf / e :) d 901 · 8.80.0 / 900Z OAV
960丽 SOOZdf/ェ:) d LOV 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d LOV 8.80.0 / 900Z OAV

960丽 SOOZdf/ェ:) d 801· 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 801 · 8.80.0 / 900Z OAV



960丽 SOOZdf/ェ:) d 0 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 0 8.80.0 / 900Z OAV


[0260] [表 30]

 [0260] [Table 30]
[0260] [Table 30]
[0261] [表 31]
 [0261] [Table 31]
[0261] [Table 31]


[0263] [表 33] [0263] [Table 33]

[0264] [表 34]
[0264] [Table 34]

Ex Syn - (R3)„ DATA(Sal)Ex Syn-(R 3 ) „DATA (Sal)
54 54 4- CI FP 497 Sal TFA 18 54 4-F FP 481 Sal TFA 19 54 4-OMe FP 493 Sal TFA 20 54 nul l FP 463 Sal TFA 21 54 4-CN FP 488 Sal TFA 22 54 5- CF3 FP 531 Sal TFA 23 54 5- CI FP 497 Sal TFA 24 54 4, 5-diOMe FP 523 Sal TFA 25 54 4- Br FP 541 , 543 Sal: TFA 26 54 4, 5, 6-t r iOMe FP 553; Sal TFA 27 54 3- CI FP 497; Sal TFA 28 54 4-CF3 FP 531; Sal TFA 29 54 4-Me FP 477; Sal TFA 30 54 5-F FP 481; Sal TFA 31 54 4, 5-diF FP 499; Sal TFA 32 54 4-C02Me EP 521; Sal: TFA54 54 4- CI FP 497 Sal TFA 18 54 4-F FP 481 Sal TFA 19 54 4-OMe FP 493 Sal TFA 20 54 nul l FP 463 Sal TFA 21 54 4-CN FP 488 Sal TFA 22 54 5- CF 3 FP 531 Sal TFA 23 54 5- CI FP 497 Sal TFA 24 54 4, 5-diOMe FP 523 Sal TFA 25 54 4- Br FP 541, 543 Sal: TFA 26 54 4, 5, 6-tr iOMe FP 553; Sal TFA 27 54 3- CI FP 497; Sal TFA 28 54 4-CF 3 FP 531; Sal TFA 29 54 4-Me FP 477; Sal TFA 30 54 5-F FP 481; Sal TFA 31 54 4, 5-diF FP 499; Sal TFA 32 54 4-C0 2 Me EP 521; Sal: TFA
24 24 4-C02H EP 507; Sal HCOOH 33 54 4-Et EP 491; Sal TFA 34 54 4- (CH2) 2NH2 EP 506; Sal TFA 35 54 5-C¾C (0) NH2 EP 520; Sal TFA ]
24 24 4-C0 2 H EP 507; Sal HCOOH 33 54 4-Et EP 491; Sal TFA 34 54 4- (CH 2 ) 2 NH 2 EP 506; Sal TFA 35 54 5-C¾C (0) NH 2 EP 520 Sal TFA]





960丽 SOOZdf/ェ:) d 8 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 8 8.80.0 / 900Z OAV


[0270] [表 40]

 [0270] [Table 40]
[0270] [Table 40]
[0271] [表 41]
[0271] [Table 41]



960丽 SOOZdf/ェ:) d 1-31. 8.80.0/900Z OAV
 960 丽 SOOZdf / e :) d 1-31.8.80.0 / 900Z OAV
960 丽 SOOZdf / e :) d 1-31.8.80.0 / 900Z OAV
[0274] [表 44]

 [0274] [Table 44]
[0274] [Table 44]
[0275] [表 45]
[0275] [Table 45]

Ex Syn R4 DATA(Sal) Ex Syn R 4 DATA (Sal)
236 54 へ EP 609 ; Sal : TFA 236 54 to EP 609; Sal: TFA
57 57 Me2NCH2- EP 554 ; Sal : HC157 57 Me 2 NCH 2 -EP 554; Sal: HC1
237 57 Qへ EP 596 ; Sal : HC1 α 237 57 Q Go to EP 596; Sal: HC1 α
238 54 iPrN(Me) CH2- EP 582 ; Sal : TFA 238 54 iPrN (Me) CH 2 -EP 582; Sal: TFA
239 57 Me - N、 ) FP 623 ; Sal : HC1 239 57 Me-N,) FP 623; Sal: HC1
240 57 EP 580 ; Sal : HC1
 240 57 EP 580; Sal: HC1
240 57 EP 580; Sal: HC1
241 57 Me2N- (CH2) 2-N (Me) -C¾- EP 611 ; Sal : HC1241 57 Me 2 N- (CH 2 ) 2 -N (Me) -C¾- EP 611; Sal: HC1
242 27 H0C¾CH(0H)- EP 557 242 27 H0C¾CH (0H)-EP 557
25 25 H0- (CH2) 2-N (Me) - EP 570 ; Sal : HC125 25 H0- (CH 2 ) 2 -N (Me)-EP 570; Sal: HC1
243 25 N\ EP 596 ; Sal : HC1243 25 N \ EP 596; Sal: HC1
244 25 Me2N- (CH2) 2- N(Me) - EP 597 ; Sal : HC1 244 25 Me 2 N- (CH 2 ) 2 -N (Me)-EP 597; Sal: HC1
Me、N Me, N
245 25 EP 595 ; Sal : HC1 245 25 EP 595; Sal: HC1
246 25 N\ EP 582 ; Sal : HC1 246 25 N \ EP 582; Sal: HC1
247 25 FP 582 ; Sal : HC1 247 25 FP 582; Sal: HC1
248 25 N FP 563 ; Sal : HC1 、 248 25 N FP 563; Sal: HC1,
43 43 H02C- FP 541 ; Sal : HC143 43 H0 2 C- FP 541 ; Sal : HC1
249 26 H0- EP 513 249 26 H0- EP 513
250 57 Me02C- FP 555 ; Sal : HC1250 57 Me0 2 C- FP 555; Sal: HC1
251 54 CH2=CH- FP 523 ; Sal : TFA 6]
251 54 CH 2 = CH- FP 523; Sal: TFA 6]

t, J = 7.0 Hz), 4.02 (2H, q, J = 7.0 Hz), 4.48 (2H, d, J = 5.9 Hz), 4.59 (2H, s), 6.64 (1H, d, J = 2.4 Hz), 6.76 (1H, d, J = 9.0 Hz), 6.83 (1H, d, J = 2.4 Hz), 6.98 (2H, s), 7.33 t, J = 7.0 Hz), 4.02 (2H, q, J = 7.0 Hz), 4.48 (2H, d, J = 5.9 Hz), 4.59 (2H, s), 6.64 (1H, d, J = 2.4 Hz) , 6.76 (1H, d, J = 9.0 Hz), 6.83 (1H, d, J = 2.4 Hz), 6.98 (2H, s), 7.33
275 12 H,NS (0),NH- (1H, dd, J = 9.0, 2.5 Hz), 7.79-7.85 (3H, m), 7.85 (1H, t, I = 5.9 Hz), 7.93 (2H, d, J = 8.9 Hz), 8.88 (2H, br s), 9.20 (2H, br s), 9.24 (1H, s),275 12 H, NS (0), NH- (1H, dd, J = 9.0, 2.5 Hz), 7.79-7.85 (3H, m), 7.85 (1H, t, I = 5.9 Hz), 7.93 (2H, d , J = 8.9 Hz), 8.88 (2H, br s), 9.20 (2H, br s), 9.24 (1H, s),
10.53 (1H, s), 12.78 (1H, br s) Sal: TFA 10.53 (1H, s), 12.78 (1H, br s) Sal: TFA
276 12 MeNHS (0) ,NH- EP 605 Sal TFA 276 12 MeNHS (0), NH- EP 605 Sal TFA
277 12 MsNH- EP 590 Sal TFA 277 12 MsNH- EP 590 Sal TFA
278 55 MeNHC (0)- EP 554 Sal HCOOH 278 55 MeNHC (0)-EP 554 Sal HCOOH
279 55 H2NC (0)- EP 540 Sal HCOOH 279 55 H 2 NC (0)-EP 540 Sal HCOOH
280 55 H2NS (0) 2NHC (0)- EP 619 Sal HCOOH 280 55 H 2 NS (0) 2 NHC (0)-EP 619 Sal HCOOH
281 12 H2NC (O)NH- EP 555 Sal TFA 281 12 H 2 NC (O) NH- EP 555 Sal TFA
282 54 H2NS (0) 2N(Me)- EP 605 Sal TFA 282 54 H 2 NS (0) 2 N (Me)-EP 605 Sal TFA
283 54 H2NC (0) OCH2 EP 570 Sal TFA 283 54 H 2 NC (0) OCH 2 EP 570 Sal TFA
284 55 H2NS (0) 2NHCH2- EP 605 Sal HCOOH 284 55 H 2 NS (0) 2 NHCH 2 -EP 605 Sal HCOOH
285 54 EP: 579; Sal: TFA 285 54 EP: 579; Sal: TFA
to to
[0278] [表 48] [0278] [Table 48]


[0279] [表 49]
 [0279] [Table 49]
[0279] [Table 49]
Ex Syn R4 DATA (Sal) Ex Syn R 4 DATA (Sal)
289 29 FP : 551 ; Sal: HCOOH 289 29 FP: 551; Sal: HCOOH
OH OH
290 29 FP : 551 ; Sal: HCOOH 290 29 FP: 551; Sal: HCOOH
30 30 H0_ FP : 507 ; Sal : HCOOH 30 30 H0_ FP: 507; Sal: HCOOH
TQ TQ
291 31 FP 558 ; Sal: HCOOH 291 31 FP 558; Sal: HCOOH
292 31 MeS (0) JHC0- FP 612; Sal: HCOOH 292 31 MeS (0) JHC0- FP 612; Sal: HCOOH
EP : 585; NMR: (DMSO - ) ; 1.29 (6H, d, J = 6.0 Hz) , 2.24 (3H, s), 4.43 (2H, br s), 4.49 - .56 (IH, m), 4.58 (2H, s), 6.65 (IH, d, J 二 2.5 Hz) , 6.68 (1H, d, J = 8.6 Hz), 6.83 (IH, d, J = 2.5Hz), 6.98 (2H, br s), 7.15 (IH, EP: 585; NMR: (DMSO-); 1.29 (6H, d, J = 6.0 Hz), 2.24 (3H, s), 4.43 (2H, br s), 4.49-.56 (IH, m), 4.58 ( 2H, s), 6.65 (IH, d, J 2 2.5 Hz), 6.68 (1H, d, J = 8.6 Hz), 6.83 (IH, d, J = 2.5 Hz), 6.98 (2H, br s), 7.15 (IH,
293 12 H2NS(0)2NH- dd, J = 8.6, 1.1 Hz), 7.56 (IH, d, J = 1.1 Hz), 7.47-7.68 (IH, br), 7.81 (2H, d, J = 8.7 Hz), 7.94 (2H, d, J = 8.7 Hz), 8.79 (2H, br s), 9.19 (2H, br s), 9.20 (IH, s), 10.42 (IH, s), 12.76 (IH, br s) ; Sal: TFA 293 12 H 2 NS (0) 2 NH- dd, J = 8.6, 1.1 Hz), 7.56 (IH, d, J = 1.1 Hz), 7.47-7.68 (IH, br), 7.81 (2H, d, J = 8.7 Hz), 7.94 (2H, d, J = 8.7 Hz), 8.79 (2H, br s), 9.19 (2H, br s), 9.20 (IH, s), 10.42 (IH, s), 12.76 (IH, br s); Sal: TFA
294 54 AcOCH,- EP 563; Sal: TFA 294 54 AcOCH,-EP 563; Sal: TFA
EP : 521 ; MR (DMSO- d6) : 1.28 (6H, d, J = 6.0 Hz), 2.23 (3H, s), 4.34 (2H, d, J = 2.8 Hz), 4.43-4.62 (5H, m), 5.02-5.11 (IH, m), 6.71 (1H, d, J = 8.5 Hz), 6.79 (1H, d, J = 1.6EP: 521; MR (DMSO- d 6): 1.28 (6H, d, J = 6.0 Hz), 2.23 (3H, s), 4.34 (2H, d, J = 2.8 Hz), 4.43-4.62 (5H, m ), 5.02-5.11 (IH, m), 6.71 (1H, d, J = 8.5 Hz), 6.79 (1H, d, J = 1.6
295 26 HOCH, 295 26 HOCH,
Hz), 6.87 (IH, d, J = 1.6 Hz), 7.13 (IH, dd, J= 8.5, 1.9 Hz), 7.45-7.69 (2H, m), 7.78 (2H, d, J = 8.5 Hz), 7.93 (2H, d, J = 8.5 Hz), 8.88-9.16 (2H, br), 9.35-10.60 (2H, m) Hz), 6.87 (IH, d, J = 1.6 Hz), 7.13 (IH, dd, J = 8.5, 1.9 Hz), 7.45-7.69 (2H, m), 7.78 (2H, d, J = 8.5 Hz), 7.93 (2H, d, J = 8.5 Hz), 8.88-9.16 (2H, br), 9.35-10.60 (2H, m)
EP : 535 ; NMR (DMSO- ) : 1.27 (6H, d, J = 6.0 Hz), 2.23 (3H, s), 2.58 (2H, t, J = 7.3 Hz), 3.47-3.54 (2H, m) , 4.42-4.63 (6H, m), 6.67 (IH, d, J = 1.6 Hz), 6.71 (1H, d, J = 8.5 EP: 535; NMR (DMSO-): 1.27 (6H, d, J = 6.0 Hz), 2.23 (3H, s), 2.58 (2H, t, J = 7.3 Hz), 3.47-3.54 (2H, m), 4.42-4.63 (6H, m), 6.67 (IH, d, J = 1.6 Hz), 6.71 (1H, d, J = 8.5
296 26 HO(CH2). 296 26 HO (CH 2 ).
Hz), 6.78 (1H, d, J = 1.6 Hz), 7.13 (1H, dd, J = 8.5,1-9 Hz), 7.48-7.58 (2H, m), 7.79 (2H, d, J = 8.5 Hz), 7.93 (2H, d, J = 8.5 Hz), 8.85- 10.00 (3H, m), 10.40-10.50 (IH, br) Hz), 6.78 (1H, d, J = 1.6 Hz), 7.13 (1H, dd, J = 8.5, 1-9 Hz), 7.48-7.58 (2H, m), 7.79 (2H, d, J = 8.5 Hz) ), 7.93 (2H, d, J = 8.5 Hz), 8.85-10.00 (3H, m), 10.40-10.50 (IH, br)
297 54 H,NC (0) - EP 534; Sal: TFA 0]
 1]
297 54 H, NC (0)-EP 534; Sal: TFA 0] 1]
1]
297 54 H, NC (0)-EP 534; Sal: TFA 0] 1]


960丽 SOOZdf/ェ:) d 831- 8.80.0/900Z OAV
[SS挲] [S820] 960 丽 SOOZdf / e :) d 831- 8.80.0 / 900Z OAV [SS 挲] [S820]


960丽 SOOZdf/ェ:) d 631- 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 631- 8.80.0 / 900Z OAV



Ex Syn R a R4b DATA (Sal) Ex Syn R a R 4b DATA (Sal)
329 54 CI EP: 599 ; Sal: TFA
 329 54 CI EP: 599; Sal: TFA
329 54 CI EP: 599; Sal: TFA
330 25 F -C¾OH EP: 599 ; Sal: HC1 330 25 F -C¾OH EP: 599; Sal: HC1
331 55 Br -OMe EP: 561 ; Sal: HC02H331 55 Br -OMe EP: 561; Sal: HC0 2 H
332 55 CI CI EP: 521 ; Sal: HC02H332 55 CI CI EP: 521; Sal: HC0 2 H
333 55 CI Br EP: 565 ; Sal: HC02H 333 55 CI Br EP: 565; Sal: HC0 2 H
N'Me N ' Me
334 25 CI EP: 585 ; Sal : HC 1 334 25 CI EP: 585; Sal: HC 1
335 54 F EP: 569 ; Sal : TFA NH 335 54 F EP: 569; Sal: TFA NH
336 54 F EP: 555 ; Sal : TFA 336 54 F EP: 555; Sal: TFA
337 54 F EP: 597 ; Sal • TFA 337 54 F EP: 597; Sal • TFA
338 25 F -CH(0H) iPr FP : 543 ; Sal: HC1 5]
338 25 F-CH (0H) iPr FP: 543; Sal: HC1 5]

Ex Syn R4 DATA(Sal) Ex Syn R 4 DATA (Sal)
EP: 561; NMR (DMSO-d6) 2.23 (3H, s), 4.41-4.43 (4H, m), 6.73 (IH, d, J = 9.4 Hz), 6.91 (IH, d, J = 2.4 Hz), 6.98 (ΙΗ' d, J = 2.4 Hz), 7.35 (1H, dd, J = 9.4, 2.4EP: 561; NMR (DMSO-d 6 ) 2.23 (3H, s), 4.41-4.43 (4H, m), 6.73 (IH, d, J = 9.4 Hz), 6.91 (IH, d, J = 2.4 Hz) , 6.98 (ΙΗ 'd, J = 2.4 Hz), 7.35 (1H, dd, J = 9.4, 2.4
339 12 Me Hz), 7.82 (2H, d, J = 2.4 Hz), 7.83 (2H, d, J = 8.8 339 12 Me Hz), 7.82 (2H, d, J = 2.4 Hz), 7.83 (2H, d, J = 8.8
Hz), 7.93 (2H, d, J = 8.8 Hz), 8.91 (2H, s), 9.20 (2H, s), 9.26 (IH, s), 10.54 (IH, s), 12.89 (IH, s) ; Sal: TFA Hz), 7.93 (2H, d, J = 8.8 Hz), 8.91 (2H, s), 9.20 (2H, s), 9.26 (IH, s), 10.54 (IH, s), 12.89 (IH, s); Sal: TFA
340 12 -OnPr EP: 605 Sal: TFA 340 12 -OnPr EP: 605 Sal: TFA
EP: 619; ; NMR (DMSO-d6) : 1.01 (6H, d, J = 6.6 Hz), 2.00-2.12 (IH, m), 3.74 (2H, d, J = 6.6 Hz), 4.49 (2H, d, J = 5. 7 Hz), 4.61 (2H , s), 6.62 (1H, d, J = 1.9 Hz), 6.75 (1H, d, J = 9.5 Hz), 6.83 (1H, d, J = 1.9 Hz),EP: 619; NMR (DMSO-d 6 ): 1.01 (6H, d, J = 6.6 Hz), 2.00-2.12 (IH, m), 3.74 (2H, d, J = 6.6 Hz), 4.49 (2H, d, J = 5.7 Hz), 4.61 (2H, s), 6.62 (1H, d, J = 1.9 Hz), 6.75 (1H, d, J = 9.5 Hz), 6.83 (1H, d, J = 1.9 Hz),
341 12 -OiBu 341 12 -OiBu
7.01 (2H, s), 7.34 (1H, dd, J = 9.5, 1.9 Hz), 7.77-7.84 (3H, m), 7.86 (IH, t, J = 5.7 Hz), 7.93 (2H, d, J = 8.8 Hz), 8.82 (2H, s), 9.20 (2H, s), 9.25 (IH, s), 10.55 (IH, s), 12.70-13.00 (IH br) ; Sal: TFA 7.01 (2H, s), 7.34 (1H, dd, J = 9.5, 1.9 Hz), 7.77-7.84 (3H, m), 7.86 (IH, t, J = 5.7 Hz), 7.93 (2H, d, J = 8.8 Hz), 8.82 (2H, s), 9.20 (2H, s), 9.25 (IH, s), 10.55 (IH, s), 12.70-13.00 (IH br); Sal: TFA
[0286] [表 56] [0286] [Table 56]


[0287] [表 57]
[0287] [Table 57]

Ex Syn R3 -X-Y- DATA (Sal) Ex Syn R 3 -XY- DATA (Sal)
346 55 CI -OCH2- EP 453 Sal HC02H346 55 CI -OCH 2 -EP 453 Sal HC0 2 H
347 55 Me -OCH2- EP 433 Sal HC02H347 55 Me -OCH 2 -EP 433 Sal HC0 2 H
348 55 CI -OCH(Me)- EP 467 Sal HC02H348 55 CI -OCH (Me)-EP 467 Sal HC0 2 H
349 55 CI -OCH(Me)- EP 447 Sal HC02H349 55 CI -OCH (Me)-EP 447 Sal HC0 2 H
350 55 CI - OCH (Me) C¾- EP 481 Sal HC02H 58] 350 55 CI-OCH (Me) C¾- EP 481 Sal HC0 2 H 58]

Ex Syn R3 - X- Y - DATA(Sal)Ex Syn R 3 -X- Y-DATA (Sal)
351 54 CI -0-(CH2)2- FP: 467 ; Sal TFA351 54 CI -0- (CH 2 ) 2 -FP: 467; Sal TFA
352 54 CI - SCH2- EP: 469 ; Sal TFA352 54 CI-SCH 2 -EP: 469; Sal TFA
353 54 CI — (CH2)2 - FP: 451 ; Sal TFA353 54 CI — (CH 2 ) 2 -FP: 451; Sal TFA
59 59 CI -N(Me)-CH2- EP: 466 ; Sal HC02H59 59 CI -N (Me) -CH 2 -EP: 466; Sal HC0 2 H
354 54 CI bond FP: 423 ; Sal TFA354 54 CI bond FP: 423; Sal TFA
355 55 CI - OCH (Me) - EP: 467 ; Sal HC02H355 55 CI-OCH (Me)-EP: 467; Sal HC0 2 H
356 55 Me -OCH (Me) - EP: 447 ; Sal HC02H356 55 Me -OCH (Me)-EP: 447; Sal HC0 2 H
357 55 Me -SCH2- EP: 449 ; Sal HC02H357 55 Me -SCH 2 -EP: 449; Sal HC0 2 H
358 55 Me - - EP: 431 ; Sal HC02H358 55 Me--EP: 431; Sal HC0 2 H
359 55 CI -CH2N(Me) CH2- EP: 480 ; Sal HC02H359 55 CI -CH 2 N (Me) CH 2 -EP: 480; Sal HC0 2 H
360 55 CI -CH2NHCH2- EP: 466 ; Sal HC02H360 55 CI -CH 2 NHCH 2 -EP: 466; Sal HC0 2 H
361 55 Me -CH2N(Me) CH2- EP: 460 Sal: HC02H361 55 Me -CH 2 N (Me) CH 2 -EP: 460 Sal: HC0 2 H
362 55 Me -CHZNHCH2- EP: 446 Sal: HC02H
[0289] [表 59] 362 55 Me -CH Z NHCH 2 -EP: 446 Sal: HC0 2 H [0289] [Table 59]


[0290] [表 60] [0290] [Table 60]

Ex Syn R3 - X - Y - 醒 (Sal)Ex Syn R 3 -X-Y-Awakening (Sal)
370 54 CI — 0— (CH2)2— FP: 511 ; Sal: TFA370 54 CI — 0— (CH 2 ) 2 — FP: 511; Sal: TFA
371 26 CI FP: 533371 26 CI FP: 533
372 55 CI - OCH(Me) - EP: 511 ; Sal: HC02H372 55 CI-OCH (Me)-EP: 511; Sal: HC0 2 H
373 55 Me -OCH(Me)- EP: 491 ; Sal: HC02H373 55 Me -OCH (Me)-EP: 491; Sal: HC0 2 H
374 55 CI -0CH(Me)CH2- EP: 525 ; Sal: HC02H374 55 CI -0CH (Me) CH 2 -EP: 525; Sal: HC0 2 H
375 55 CI -OCH(iPr)- EP: 539 ; Sal: HC0ZH375 55 CI -OCH (iPr)-EP: 539; Sal: HC0 Z H
376 55 CI -OCH(nBu)- EP: 553 ; Sal: HC02H376 55 CI -OCH (nBu)-EP: 553; Sal: HC0 2 H
377 55 Me -OCH(Me) CH2- EP: 505 ; Sal: HC02H377 55 Me -OCH (Me) CH 2 -EP: 505; Sal: HC0 2 H
378 55 Me -OCH(iPr)- EP: 519 ; Sal: HC02H378 55 Me -OCH (iPr)-EP: 519; Sal: HC0 2 H
379 55 Me -OCH(nBu)- EP: 533 ; Sal: HC02H
IDH IBS I6 : da -¾) - S6S379 55 Me -OCH (nBu)-EP: 533; Sal: HC0 2 H IDH IBS I6: da -¾)-S6S
I3H : <ia _¾>- I3- S£I3H: <ia _¾>-I3- S £
I3H IBS 60S -ZH3- C6S I3H IBS 60S- Z H3- C6S
I3H goe άί -ZH3-I3H goe άί- Z H3-
- (3皿: ))- 0_ I6S -(3 dishes:))-0_ I6S
I¾S di - ε(¾))- 0_ IS I¾S di- ε (¾))-0_ IS
I^S S£S di - (¾))-。- ε 68ε I ^ S S £ S di-(¾))-. -ε 68ε
I¾S 6SS da - z(¾3) - I3-t ιε 88C I¾S 6SS da- z (¾3)-I3-t ιε 88C
I^S S£9 ii - z(¾)) - 3ΚΪ -9 ' ιε I ^ SS £ 9 ii- z (¾))-3ΚΪ -9 'ιε
HZ03H i 9 da -z(¾3)- η η H Z 03H i 9 da- z (¾3)-η η
IBS id -z(¾))_ ■- 1 π 98ε IBS id- z (¾)) _ ■-1 π 98ε
HzODH i3 」(¾))- IC S8C H z ODH i3 "(¾))-IC S8C
SOS -z(¾3)- Iinu m SOS- z (¾3)-Iinu m
tfODH : I¾S : (ZH 8·8 = ί 'P tfODH: I¾S: (ZH 8 · 8 = ί 'P
¾) 68 'i '(ZH 8·8 = r 'p 'm) 8i 'L ¾) 68 'i' (ZH 8/8 = r 'p' m) 8i 'L
'(s ΉΙ) iS'i '(ZH S- 8 = r 'p ΉΙ) '(s ΉΙ) iS'i' (ZH S-8 = r 'p ΉΙ)
81 -i '(s 'HI) 98 '9 '(s ΉΙ) '(ZH 81 -i '(s' HI) 98' 9 '(s ΉΙ)' (ZH
5'8 = ί 'P ΉΙ) 89 '9 '(III ΉΙ) S9 - z(¾3)- ιε C8S 5'8 = ί 'P ΉΙ) 89' 9 '(III ΉΙ) S9- z (¾3)-ιε C8S
- ss '(s ¾) 6ε '(s n -f -ss '(s ¾) 6ε' (s n -f
'(in 'HZ) £8 Ί - IVl '(HI 'H '(in' HZ) £ 8 Ί-IVl '(HI' H
一 '(s ) SZ 0'9 = r '(S) SZ 0'9 = r
'p Ή9) u : (9p- osra) 翻 : 613 id 'p Ή9) u: ( 9 p-osra) translation: 613 id
(IBS)丽 -A-X- Χ3 (IBS) 丽 -A-X- Χ3

[29¾ [2620] [29¾ [2620]


[19挲] [Ϊ620] [19 挲] [Ϊ620]
960丽 SOOZdf/ェ:) d 981- 8.80.0/900Z OAV
[0293] [表 63] 960 丽 SOOZdf / e :) d 981- 8.80.0 / 900Z OAV [0293] [Table 63]

Ex Syn R3 R4 DATA (Sal)Ex Syn R 3 R 4 DATA (Sal)
396 12 H2NCH2C (O)NH- EP: 561; Sal: TFA396 12 H 2 NCH 2 C (O) NH- EP: 561; Sal: TFA
397 35 HOCH,C(0)NH- EP 562; Sal HC1397 35 HOCH, C (0) NH- EP 562; Sal HC1
398 56 H2NC(0)- EP 532; Sal: HCOOH 398 56 H 2 NC (0)-EP 532; Sal: HCOOH
EP: 548; NMR (DMSO-d6) : 1.26 (6H, d, J = 6.0 Hz), 2.25 (3H, s), 2.29-2.34 (2H, ra), 2.63- 2.69 (2H, m), 3.01 (2H, t, J = 6.0Hz), 3.13-3.19 (1H, m), 3.49 (2H, t, J = 6.0 Hz), 4.20 (2H, br s), 4.50 (1H, sep, J =EP: 548; NMR (DMSO-d 6 ): 1.26 (6H, d, J = 6.0 Hz), 2.25 (3H, s), 2.29-2.34 (2H, ra), 2.63- 2.69 (2H, m), 3.01 (2H, t, J = 6.0Hz), 3.13-3.19 (1H, m), 3.49 (2H, t, J = 6.0 Hz), 4.20 (2H, br s), 4.50 (1H, sep, J =
Me 6.0 5.24-5.36 (IH, br), Me 6.0 5.24-5.36 (IH, br),
6.13 d, J = 1.8 Hz), 6.17 6.13 d, J = 1.8 Hz), 6.17
36 36 H0-(CH2) ,-NH-36 36 H0- (CH 2 ), -NH-
(1H, d J = 1.8 Hz), 6.70 (IH, d, J = 8.5 Hz), 7.19 (1H, dd,(1H, d J = 1.8 Hz), 6.70 (IH, d, J = 8.5 Hz), 7.19 (1H, dd,
J = 8.5, 1.7 Hz), 7.32-7.39J = 8.5, 1.7 Hz), 7.32-7.39
(IH, br), 7.56-7.58 (1H, m),(IH, br), 7.56-7.58 (1H, m),
7.79 (2H, d, J = 8.8 Hz), 7.907.79 (2H, d, J = 8.8 Hz), 7.90
(2H, d, J = 8.8 Hz), 8.37 (IH, s), 8.65-9.20 (2H, br), 10.41(2H, d, J = 8.8 Hz), 8.37 (IH, s), 8.65-9.20 (2H, br), 10.41
(IH, br s), 10.55-11.00 (2H, br) ; Sal: HCOOH (IH, br s), 10.55-11.00 (2H, br); Sal: HCOOH
399 54 H,NCH2C(0)NH- EP 565; Sal: TFA399 54 H, NCH 2 C (0) NH-EP 565; Sal: TFA
400 26 HOCH,C(0)NH- EP 566 400 26 HOCH, C (0) NH- EP 566
401 24 HOCH,C(0)NH- EP 582; Sal: HCOOH 401 24 HOCH, C (0) NH- EP 582; Sal: HCOOH
CI CI
402 12 H2NC¾C(0)NH- EP 581; Sal: TFA402 12 H 2 NC¾C (0) NH- EP 581; Sal: TFA
403 49 Me HOCH,CH(OH)CH,NH- EP 578 403 49 Me HOCH, CH (OH) CH, NH- EP 578
[0294] [表 64]
[0294] [Table 64]





[0299] [表 69]

 [0299] [Table 69]
[0299] [Table 69]
[0300] [表 70]
[0300] [Table 70]

[0301] [表 71]
[0301] [Table 71]

Ex Syn R3 R4a R4b DATA (Sal)Ex Syn R 3 R 4a R 4b DATA (Sal)
455 54 H H - OEt FP 464 ; Sal: TFA455 54 H H-OEt FP 464; Sal: TFA
456 54 CI H -OEt FP 498 ; Sal: TFA456 54 CI H -OEt FP 498; Sal: TFA
457 27 H HOCH2CH (OH) - -OEt EP 524 457 27 H HOCH 2 CH (OH)--OEt EP 524
458 54 H AcOCH2- - OEt EP 536 Sal TFA458 54 H AcOCH 2 --OEt EP 536 Sal TFA
37 37 H HOC¾ - - OEt EP 494 Sal HC137 37 H HOC¾--OEt EP 494 Sal HC1
459 55 H H02CCH2- -OEt EP 522 Sal HCOOH459 55 H H0 2 CCH 2 --OEt EP 522 Sal HCOOH
460 55 H ¾NC (0) CH2- - OEt EP 521 Sal HCOOH460 55 H ¾NC (0) CH 2 --OEt EP 521 Sal HCOOH
461 35 H HOCH2- -OiPr EP 508 Sal HC1461 35 H HOCH 2 --OiPr EP 508 Sal HC1
462 26 CI HOCHr -OiPr EP 542 462 26 CI HOCH r -OiPr EP 542
463 31 Me HOCH2- -OiPr EP 522 ; Sal: HCOOH 72]
463 31 Me HOCH 2 --OiPr EP 522; Sal: HCOOH 72]

Ex Syn R3 DATA (Sal) Ex Syn R 3 DATA (Sal)
464 35 Me -CH, EP: 506 ; Sal: HC1 464 35 Me -CH, EP: 506; Sal: HC1
465 35 -CH, EP 492 ; Sal: HCl 465 35 -CH, EP 492; Sal: HCl
466 26 CI -CH, - EP 526 466 26 CI -CH,-EP 526
EP: 520 ; NMR (DMS0-d6) : 1.28 (6H, d, J = 5.9 Hz), 2.23 (3H, s) , 2.34-2.42 (2H, m), 2.75-2.85 (2H, m), 4.40 (2H, s), 4.55- 4.65 (3H, m), 6.83 (1H, s), 6.87 (1H, s),EP: 520; NMR (DMS0-d 6 ): 1.28 (6H, d, J = 5.9 Hz), 2.23 (3H, s), 2.34-2.42 (2H, m), 2.75-2.85 (2H, m), 4.40 (2H, s), 4.55- 4.65 (3H, m), 6.83 (1H, s), 6.87 (1H, s),
35 35 Me ■(CH,), 35 35 Me (CH,),
7.88 (2H, d, J = 8.9 Hz), 7.93 (2H, d, J = 8.9 Hz), 7.99-8.21 (3H, m), 8.98 (2H, s), 9.26 (2H, s), 10.68 (1H, br s), 11.95 (1H, br s) ; Sal: HCl 7.88 (2H, d, J = 8.9 Hz), 7.93 (2H, d, J = 8.9 Hz), 7.99-8.21 (3H, m), 8.98 (2H, s), 9.26 (2H, s), 10.68 (1H , br s), 11.95 (1H, br s); Sal: HCl
EP ·· 506 ; NMR (DMS0_d6) : 1.28 (6H, d, J = 6.0 Hz), 2.39 (2H, t, J = 7.5 Hz), 2.82 (2H, t, J = 7.5 Hz), 4.40 (2H, s), 4.55- 4.70 (3H, m), 6.71 (1H, dd, J = 7.5, 4.5 EP ··· 506, NMR (DMS0_d6): 1.28 (6H, d, J = 6.0 Hz), 2.39 (2H, t, J = 7.5 Hz), 2.82 (2H, t, J = 7.5 Hz), 4.40 (2H, s), 4.55- 4.70 (3H, m), 6.71 (1H, dd, J = 7.5, 4.5
467 35 (CHZ); Hz), 6.84 (1H, s), 6.86 (1H, s), 7.81 (2H, d, J = 9.0 Hz), 7.91 (2H, d, J = 9.0 Hz), 8.10-8.20 (2H, m), 8.26 (1H, d, J = 4.5 Hz), 8.79 (2H, s). 9.20 (2H, s), 10.56(1H, s), 11.80-12.10 OH, br) ; Sal: HCl467 35 (CH Z ) ; Hz), 6.84 (1H, s), 6.86 (1H, s), 7.81 (2H, d, J = 9.0 Hz), 7.91 (2H, d, J = 9.0 Hz), 8.10- 8.20 (2H, m), 8.26 (1H, d, J = 4.5 Hz), 8.79 (2H, s). 9.20 (2H, s), 10.56 (1H, s), 11.80-12.10 OH, br); Sal: HCl
468 26 CI -(C¾)2- EP: 540 468 26 CI-(C¾) 2 -EP: 540
[0303] [表 73] [0303] [Table 73]


[0304] [表 74]
[0304] [Table 74]
NH2 CI
 NH 2 CI
NH 2 CI
[0305] [表 75]
 [0305] [Table 75]
[0305] [Table 75]

[0306] [表 76]
[0306] [Table 76]

[0307] [表 77]

 [0307] [Table 77]
[0307] [Table 77]
[0308] [表 78]
[0308] [Table 78]


[08挲] [οτεο] [08 挲] [οτεο]


960丽 SOOZdf/ェ:) d 8 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 8 8.80.0 / 900Z OAV

[0311] [表 81]
[0311] [Table 81]


[£s [ετεο] [£ s [ετεο]

960丽 SOOZdf/ェ:) d 1-91. 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 1-91.8.80.0 / 900Z OAV
m m


960丽 SOOZdf/ェ:) d 391- 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 391- 8.80.0 / 900Z OAV
[S8挲] [STSO] [S8 挲] [STSO]


960丽 SOOZdfAlOd 8ん 80ム0/900 OM
 86]
960 丽 SOOZdfAlOd 8 80m 0/900 OM 86]
86]
960 丽 SOOZdfAlOd 8 80m 0/900 OM 86]

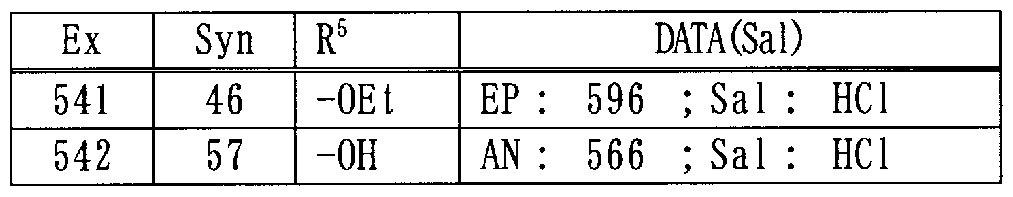


Ex Syn 3 R5 DATA (Sal)Ex Syn 3 R 5 DATA (Sal)
548 7 -OEt 548 7 -OEt
-OMe -OMe
549 16 -OH EP: 551 549 16 -OH EP: 551
550 45 -OEt EP: 567 550 45 -OEt EP: 567
F F
551 53 - ONa EP: 539 551 53-ONa EP: 539
[0319] [表 89] [0319] [Table 89]


[0320] [表 90] [0320] [Table 90]


[0321] [表 91]
/v: O 96030sooifcl£AV 891· [0321] [Table 91] / v: O 96030sooifcl £ AV 891 ·
〔〕稷

 [] 稷
[] 稷
I3H: I¾S: 99S : J3 HO- 98 I3H: I¾S: 99S: J3 HO-98
13 13
130- SIS 130- SIS
d3 HO- 9Z d3 HO-9Z
(ZH 9 = ί 'P 'HI) ' ( (ZH 9 = ί 'P' HI) '(
9'S = f ') 'HI) 00 '8 '(s Ήΐ) 6i"i '(ZH 9'S = f ')' HI) 00 '8' (s Ήΐ) 6i "i '(ZH
'9 -i = f 'PP Ήΐ) Sl'i '(ZH S'8 = Γ ' 'HZ) '9 -i = f' PP Ήΐ) Sl'i '(ZH S'8 = Γ' 'HZ)
19 Ί '(ZH S—8 = ί 'P ΈΖ) S9'l '(s 'HI) 06 '9 19 Ί '(ZH S—8 = ί' P ΈΖ) S9'l '(s' HI) 06' 9
'(s 'HI) C8"9 '(ZH 9 'f '9 = f 'P 'HI) 6S -9 '(s' HI) C8 "9' (ZH 9 'f' 9 = f 'P' HI) 6S -9
130- 9^ US 130-9 9 US
'(s Jq Έ 28 -t '(ZH S '9 = ί 'P Έ2) Zff '(s '(s Jq Έ 28 -t' (ZH S '9 = ί' P Έ2) Zff '(s
Ή2) 19 '(in 'HI) 19^- 9^ '(^H O'i = ί 't Ή2) 19 '(in' HI) 19 ^-9 ^ '(^ H O'i = ί' t
Ή2) LQ'f '(ZH 3Ί = f Ί '¾) 6 '(ΖΗΠ = Ή2) LQ'f '(ZH 3Ί = f Ί' ¾) 6 '(ΖΗΠ =
r ) '(ZH 0-9 - r 'p Ή9) ε·ι '(ΖΗ r) '(ZH 0-9-r' p Ή9) ε · ι '(ΖΗ
0·い J: '\ 'HQ Ο に: lDQD) N J 0S5 _: d3 0 · J: '\' HQ Ο: lDQD) N J 0S5 _: d3
I3H: I¾S: 9SS : J3 HO- 013 I3H: I¾S: 9SS: J3 HO-013
I OH : S : (s Jq ΉΙ) 80 'II '(s iq 'HI) I OH: S: (s Jq ΉΙ) 80 'II' (s iq 'HI)
Z9-01 '(J Ή2) 09-6-09 '8 '( 'Η2) ΟΓ8—00 ·8 Z9-01 '(J Ή2) 09-6-09' 8 '(' Η2) ΟΓ8-00 8
'(ΖΗ 6·8 = Γ 'Ρ 'ΗΖ) 06 Ί ' (Ζ 6 ·8 = Γ 'Ρ '(ΖΗ 6 ・ 8 = Γ' Ρ 'ΗΖ) 06 Ί' ( Ζ 6 · 8 = Γ 'Ρ
ΈΖ) U'i '(s Ήΐ) 88 ·9 '(s 'ΗΙ) '(ω Ήε) 3W ΈΖ) U'i '(s Ήΐ) 88 9' (s 'ΗΙ)' (ω Ήε) 3W
130- i 130- i
'(s ΉΖ) '(ΖΗ 0 = ί 'ΕΖ) '(s ΉΖ)' (ΖΗ 0 = ί 'ΕΖ)
66■£ 'Cm 98 —8 '(m 'ΗΖ) it — 66 ■ £ 'Cm 98 —8' (m 'ΗΖ) it —
'(s Ή2) '(ΖΗ 0·9 = ί 'Ρ Ή9) 2Ζ '(ΖΗ '(s Ή2)' (ΖΗ 0 9 = ί 'Ρ Ή9) 2Ζ' (ΖΗ
O'i = ί Ί Ήδ) Π Ί : (9P-0SKQ) : ·' i3 O'i = ί Ί Ήδ) Π Ί: ( 9 P-0SKQ): · 'i3
(IBS)丽 HAS (IBS) 丽 HAS

960丽 SOOZdf/ェ:) d 691- 8.80.0/900Z OAV
[96挲] [92S0]960 丽 SOOZdf / e :) d 691- 8.80.0 / 900Z OAV [96 挲] [92S0]




960丽 SOOZdf/ェ:) d 091· 8.80.0/900Z OAV
 7]
7]
 960 丽 SOOZdf / e :) d 091 8.80.0 / 900Z OAV 7]
960 丽 SOOZdf / e :) d 091 8.80.0 / 900Z OAV 7]
Ex Syn R3 R5 DATA (Sal) Ex Syn R 3 R 5 DATA (Sal)
596 45 -OMe FP: 607 596 45 -OMe FP: 607
EP: 621 ; NMR (DMS0-d6) : 1.18 (3H, t, J = 7.0 Hz), 1.23 (3H, t, J = 7.1 Hz), 1.28 (6H, d, J = 5.9 Hz), 2.23 (3H, s), 4.07 (2H, q, J = 7.0 Hz), 4.12 (2H, q, J = 7.1 Hz), 4.36 (2H, d, J = 5.7 Hz), 4.45 (2H, d, J = 5.9 Hz), 4.55 - 4.62EP: 621; NMR (DMS0-d 6 ): 1.18 (3H, t, J = 7.0 Hz), 1.23 (3H, t, J = 7.1 Hz), 1.28 (6H, d, J = 5.9 Hz), 2.23 ( 3H, s), 4.07 (2H, q, J = 7.0 Hz), 4.12 (2H, q, J = 7.1 Hz), 4.36 (2H, d, J = 5.7 Hz), 4.45 (2H, d, J = 5.9 Hz), 4.55-4.62
597 -OEt (IH, in), 4.69 (2H, s), 5.09 (1H, t, J = 5.7 597 -OEt (IH, in), 4.69 (2H, s), 5.09 (1H, t, J = 5.7
Me Hz), 6.67 (IH, d, J = 8.6 Hz), 6.82 (IH, s), Me Hz), 6.67 (IH, d, J = 8.6 Hz), 6.82 (IH, s),
6.90 (IH, s), 7.13 (1H, d, J = 8.6 Hz), 7.51 ― 7.58 (2H, m), 7.82 (2H, d, J = 8.8 Hz), 7.99 (2H, d, J = 8.8 Hz), 9.07 (2H, brs), 10.30 (IH, s) 6.90 (IH, s), 7.13 (1H, d, J = 8.6 Hz), 7.51 ― 7.58 (2H, m), 7.82 (2H, d, J = 8.8 Hz), 7.99 (2H, d, J = 8.8 Hz) ), 9.07 (2H, brs), 10.30 (IH, s)
598 45 -OnPr FP: 635 598 45 -OnPr FP: 635
599 45 -OiPr FP: 635 599 45 -OiPr FP: 635
600 16 -OH FP: 593 600 16 -OH FP: 593
FP: 625; NMR (DMSO— ) : 1.17 (3H, t, J = 7.2 Hz), 1.23 (3H, t, J = 7.1 Hz), 1.28 (6H, d, J = 6.1 Hz), 3.99 - 4.17 (4H, m), 4.36 (2H, d, J = 5.7 Hz), 4.46 (2H, d, J = 5.9 Hz), 4.54 - 4.65 (1H, m), 4.70 (2H, s), 5.11 (IH, t, J = 5.7 FP: 625; NMR (DMSO—): 1.17 (3H, t, J = 7.2 Hz), 1.23 (3H, t, J = 7.1 Hz), 1.28 (6H, d, J = 6.1 Hz), 3.99-4.17 ( 4H, m), 4.36 (2H, d, J = 5.7 Hz), 4.46 (2H, d, J = 5.9 Hz), 4.54-4.65 (1H, m), 4.70 (2H, s), 5.11 (IH, t , J = 5.7
601 -OEt 601 -OEt
Hz), 6.74 (IH, dd, J = 4.7, 9.3 Hz), 6.82 (IH, d, J = 1.4 Hz), 6.91 (1H, d, J = 1.4 Hz), 7.21 (IH, dt, J = 2.9, 9.3 Hz), 7.56-7.66 (2H, m), 7.82 (2H, d, J = 8.8 Hz), 7.99 (2H, d, J = 8.8 Hz), 10.35 (IH, s) Hz), 6.74 (IH, dd, J = 4.7, 9.3 Hz), 6.82 (IH, d, J = 1.4 Hz), 6.91 (1H, d, J = 1.4 Hz), 7.21 (IH, dt, J = 2.9 , 9.3 Hz), 7.56-7.66 (2H, m), 7.82 (2H, d, J = 8.8 Hz), 7.99 (2H, d, J = 8.8 Hz), 10.35 (IH, s)
602 16 -OH FP: 597 602 16 -OH FP: 597
FP: 607; NMR (DMSO— d6) : 1.18 (3H, t, J = 7.1 Hz), 1.23 (3H, t, J = 7.1 Hz), 1.28 (6H, d, J = 6.0 Hz), 3.99 - 4.16 (4H, ra), 4.36 (2H, d, J = 5.8 Hz), 4.47 (2H, d, J = 5.8 Hz), 4.55-4.63 (IH, m), 4.70 (2H, s), 5.09 (IH, t, J = 5.8FP: 607; NMR (DMSO—d 6 ): 1.18 (3H, t, J = 7.1 Hz), 1.23 (3H, t, J = 7.1 Hz), 1.28 (6H, d, J = 6.0 Hz), 3.99- 4.16 (4H, ra), 4.36 (2H, d, J = 5.8 Hz), 4.47 (2H, d, J = 5.8 Hz), 4.55-4.63 (IH, m), 4.70 (2H, s), 5.09 (IH , t, J = 5.8
603 -OEt Hz), 6.64 (IH, t, J = 7.8 Hz), 6.75 (1H, d, J = 603 -OEt Hz), 6.64 (IH, t, J = 7.8 Hz), 6.75 (1H, d, J =
7.8 Hz), 6.83 (1H, d, J = 1.5 Hz), 6.91 (IH, d, J = 1.5 Hz), 7.30 (IH, dt, J = 1.4, 7.8 Hz), 7.71 (IH, dd, J = 1.4, 7.8 Hz), 7.77 (1H, t, J = 5.8 Hz), 7.82 (2H, d, J = 8.9 Hz), 7.98 (2H, d, J = 8.9 Hz), 10.32 (IH, s) 7.8 Hz), 6.83 (1H, d, J = 1.5 Hz), 6.91 (IH, d, J = 1.5 Hz), 7.30 (IH, dt, J = 1.4, 7.8 Hz), 7.71 (IH, dd, J = 1.4, 7.8 Hz), 7.77 (1H, t, J = 5.8 Hz), 7.82 (2H, d, J = 8.9 Hz), 7.98 (2H, d, J = 8.9 Hz), 10.32 (IH, s)
604 16 -OH FP: 579 604 16 -OH FP: 579
605 -OEt FP: 641 605 -OEt FP: 641
CI CI
606 16 -OH EP: 613; Sal: HC1 98]
[οο ΐ挲] [οεεο] 606 16 -OH EP: 613; Sal: HC1 98] [οο ΐ 挲] [οεεο]


[66挲] [62S0] [66 挲] [62S0]
H003H : IBS : 609 : J3 HO- 91 809 H003H: IBS: 609: J3 HO- 91 809
(s 'HI) S'OI '(ΖΗ 6·8 = ί 'Ρ ' Ζ) 66 ·丄 '(ΖΗ (s' HI) S'OI '(ΖΗ 6 ・ 8 = ί' Ρ 'Ζ) 66 · 丄' (ΖΗ
6·8 = ί 'Ρ 'Η Ζ8Ί '("I ' Ζ) 0Π - fZ 'L '(¾ 6 · 8 = ί 'Ρ' Η Ζ8Ί '("I' Ζ) 0Π-fZ 'L' (¾
6 '6'Ζ = Γ 'ΡΡ 'ΗΙ) 66 '9 '(s 'ΗΙ) 68 "9 '(¾ 6 '6'Ζ = Γ' ΡΡ 'ΗΙ) 66' 9 '(s' ΗΙ) 68 '9' (¾
ΈΙ) Ζ8'9 '(ΖΗ Γ6 = ί 'Ρ Έΐ) U '9 '( 1 '9 ΈΙ) Ζ8'9 '(ΖΗ Γ6 = ί' Ρ Έΐ) U '9' (1 '9
= ί Ί Ήΐ) 60 '9 '(s 'RZ) 89· '(ω 'ΗΙ) 29^ 3W0- = ί Ί Ήΐ) 60 '9' (s' RZ) 89 · '(ω' ΗΙ) 29 ^ 3W0-
130- 109 130- 109
- SS' '(ΖΗ 8'S = f 'Ρ ' t '(ZH i'S = f -SS '' (ΖΗ 8'S = f 'Ρ' t '(ZH i'S = f
'p 'm) 9c- '(in 'Η ) si · - ΐϋ '(s ε) trc 'p' m) 9c- '(in' Η) si ·-ΐϋ '(s ε) trc
'(ΖΗ 0.9 = ί 'Ρ 'Η9) 8Π '( Γ1 = ί Ί Ήε) '(ΖΗ 0.9 = ί' Ρ 'Η9) 8' '(Γ1 = ί Ί Ήε)
εΖΊ '(ΖΗ I Ί = ί Ί Ή8) 8ΐ Ί : (9Ρ - OSWCQ N εΖΊ '(ΖΗ I Ί = ί Ί Ή8) 8 ΐ : ( 9 Ρ-OSWCQ N
iS9 : iS9:
960丽 SOOZdf/ェ:) d 891- 8.80.0/900Z OAV
960 丽 SOOZdf / e :) d 891- 8.80.0 / 900Z OAV

表table

Ex Syn R a R4b R5 DATA(Sal)Ex Syn R a R 4b R 5 DATA (Sal)
629 38 -OEt FP: 635 629 38 -OEt FP: 635
HOCH2CH2- -OiPr ; Sal: HC1HOCH 2 CH 2 --OiPr; Sal: HC1
630 16 -OH 630 16 -OH
631 7 -OEt 631 7 -OEt
HOCH2- -OiBu HOCH 2 --OiBu
632 16 -OH FP: 607
[0332] [表 102] 632 16 -OH FP: 607 [0332] [Table 102]

[0333] [表 103]
[0333] [Table 103]

Ex Syn R1 -X- Y R3 R5 DATA (Sal) Ex Syn R 1 -X- YR 3 R 5 DATA (Sal)
EP EP
NH 535NH 535
635 31 CH Me -OH 635 31 CH Me -OH
U Sal U Sal
-CH2 - : HCOOH-CH 2- : HCOOH
636 31 H2N EP 522 636 31 H 2 N EP 522
N H -OH N H -OH
Sal: HCOOH Sal: HCOOH
637 45 - OEt EP 637 637 45-OEt EP 637
Et。2 NH -0- CH Me Et. 2 NH -0- CH Me
638 26 -OH EP 609 638 26 -OH EP 609
639 45 u人 -OEt EP 635 639 45 u people -OEt EP 635
HNへ - CH2- CH Me EP 607To HN-CH 2 -CH Me EP 607
640 35 -OH 640 35 -OH
Sal: HC1 Sal: HC1
641 45 -o- -OEt EP 581 641 45 -o- -OEt EP 581
CH Me CH Me
642 28 H0 N -OH EP 553642 28 H0 N -OH EP 553
643 45 -OEt EP 579 643 45 -OEt EP 579
H2NH 2 N
4 35 人 -CH CH Me EP 551 4 35 people -CH CH Me EP 551
64 - OH 64-OH
Sal: HC1 04]
Sal: HC1 04]
[soi挲] [esso] [soi 挲] [esso]

960丽 SOOZdf/ェ:) d 19 V 8.80.0/900Z OAV
 960 丽 SOOZdf / e :) d 19 V 8.80.0 / 900Z OAV
960 丽 SOOZdf / e :) d 19 V 8.80.0 / 900Z OAV
Ex Syn R3 -X-Y-C (0)R5 DATA(Sal) Ex Syn R 3 -XYC (0) R 5 DATA (Sal)
648 45 -0CH,C0,Et EP: 608 648 45 -0CH, C0, Et EP: 608
649 28 -0C¾C02H EP: 580 649 28 -0C¾C0 2 H EP: 580
EP: 606; NMR (DMSO- d6) : 1.10 (3H, t, J = 7.0 Hz), 1.22 (3H, t, J = 7.0 Hz), 1.28 (6H, t, J = 6.1 Hz), 2.42-2.48 (2H, m), 2.80-2.87 (2H, m), 3.98 (2H, q, J = 7.0 Hz), 4.06 (2H, q, J = 7.0 Hz), 4.41 (2H, d, J = 5.8 Hz), 4.56-4.66 (3H, m) , 5.09EP: 606; NMR (DMSO- d 6 ): 1.10 (3H, t, J = 7.0 Hz), 1.22 (3H, t, J = 7.0 Hz), 1.28 (6H, t, J = 6.1 Hz), 2.42- 2.48 (2H, m), 2.80-2.87 (2H, m), 3.98 (2H, q, J = 7.0 Hz), 4.06 (2H, q, J = 7.0 Hz), 4.41 (2H, d, J = 5.8 Hz ), 4.56-4.66 (3H, m), 5.09
45 45 -(CH,)2-C0,Et 45 45-(CH,) 2 -C0, Et
(1H, t, J = 5.8 Hz), 6.70 (1H, dd, J = 7.7, 4.7 Hz), 6.86 (1H, s), 6.87 (1H, s), 7.79 (2H, d, J = 8.9 Hz), 7.98 (2H, d, J = 8.9 Hz), 8.13-8.19 (2H, m) , 8.25 (1H, dd, J = 4.7, 1.6 Hz), 8.80-9.30 (2H, br), 10.41 (1H, br s) (1H, t, J = 5.8 Hz), 6.70 (1H, dd, J = 7.7, 4.7 Hz), 6.86 (1H, s), 6.87 (1H, s), 7.79 (2H, d, J = 8.9 Hz) , 7.98 (2H, d, J = 8.9 Hz), 8.13-8.19 (2H, m), 8.25 (1H, dd, J = 4.7, 1.6 Hz), 8.80-9.30 (2H, br), 10.41 (1H, br s)
650 28 -(CH,),-C0,H EP: 578 650 28-(CH,),-C0, H EP: 578
EP: 620; NMR (DMS0-d6) : 1.10 (3H, t, J = 7.1 Hz), 1.22 (3H, t, J = 7.1 Hz), 1.27 (6H, d, J = 6.0 Hz), 2.22 (3H, s), 2.40- 2.47 (2H, ffl), 2.75-2.95 (2H, m) , 3.98 (2H, q, J = 7.1 Hz), 4.06 (2H, q, J = 7.1 Hz),EP: 620; NMR (DMS0-d 6 ): 1.10 (3H, t, J = 7.1 Hz), 1.22 (3H, t, J = 7.1 Hz), 1.27 (6H, d, J = 6.0 Hz), 2.22 ( 3H, s), 2.40- 2.47 (2H, ffl), 2.75-2.95 (2H, m), 3.98 (2H, q, J = 7.1 Hz), 4.06 (2H, q, J = 7.1 Hz),
28 28 - (CH,) ,-C0,Et 4.40 (2H, d, J = 5.8 Hz), 4.55-4.65 (3H, 28 28-(CH,), -C0, Et 4.40 (2H, d, J = 5.8 Hz), 4.55-4.65 (3H,
Me Me
m), 5.11 (1H, t, J = 5.8 Hz), 6.85 (1H, s), 6.86 (1H, s), 7.79 (2H, d, J = 8.9 Hz), 7.90-8.05 (4H, m) , 8.10 (1H, d, J = 1.8 Hz), 8.75-9.35 (2H, m), 10.40 (1H, br s) m), 5.11 (1H, t, J = 5.8 Hz), 6.85 (1H, s), 6.86 (1H, s), 7.79 (2H, d, J = 8.9 Hz), 7.90-8.05 (4H, m), 8.10 (1H, d, J = 1.8 Hz), 8.75-9.35 (2H, m), 10.40 (1H, br s)
651 28 - (CH2) 2- C02H EP: 592 651 28-(CH 2 ) 2 -C0 2 H EP: 592
[0336] [表 106] [0336] [Table 106]

[0337] [表 107]

 [0337] [Table 107]
[0337] [Table 107]
[0338] [表 108]

 [0338] [Table 108]
[0338] [Table 108]
[0339] [表 109]
[0339] [Table 109]






No R1 R3 R4 -X-Y-No R 1 R 3 R 4 -XY-
NH NH
81 81
H2N人 Me MeS(0)2NHC(0)- -OCH2- H 2 N people Me MeS (0) 2 NHC ( 0) - -OCH 2 -
H0 N OH H H0 N OH H
82 Me 一 (CH2)2 -82 Me i (CH 2 ) 2-
H2N H 2 N
83 B02C、NH OH 83 B0 2 C, NH OH
-OMe AcO^\ -OCH2--OMe AcO ^ \ -OCH 2-
84 HN人 Me HO- - OCH2- 3]
84 HN people Me HO- - OCH 2 - 3]





[Lii [ εο] [Lii [εο]



/v: O 96030sooifcl£/-0AV s- _■ / v: O 96030sooifcl £ / -0AV s- _ ■


本発明化合物は、活性ィ匕血液凝固第 VII因子を選択的に阻害し、強力な抗凝固作 用を有する。従って、血液凝固抑制剤又は血栓若しくは塞栓によって引きおこされる 疾病の予防 ·治療剤として有用である。 The compound of the present invention selectively inhibits active blood coagulation factor VII and has a potent anticoagulant action. Therefore, it is useful as a blood coagulation inhibitor or a preventive / therapeutic agent for diseases caused by thrombus or embolism.
適応する上記疾病として脳梗塞、脳血栓、脳塞栓、一過性脳虚血発作 (TIA)、くも 膜下出血 (血管れん縮)等の脳血管障害における疾病、急性及び慢性心筋梗塞、不 安定狭心症、冠動脈血栓溶解等の虚血性心疾患における疾病、肺梗塞、肺塞栓等 の肺血管障害における疾病、更に末梢動脈閉塞症、深部静脈血栓症、播種性血管 内凝固症候群(DIC : disseminated intravascular coagulation)、汎発性血管内凝固症 候群、冠動脈バイパス術、経皮的冠動脈形成術(PTCA : Percutaneous transluminal coronary angioplasty)等の血管形成術後における再閉塞及び再狭窄、冠動脈内血 溶解療法 (PTCR: Percutaneous transluminal coronary recanalization)後における 再閉塞及び再狭窄、心臓弁置換術後の血栓形成、血液体外循環時における灌流血 液の凝固若しくは炎症、等の各種血管障害における疾病、間歇性跛行、動脈硬化症 、癌が挙げられる。
Applicable diseases are cerebral vascular disorders such as cerebral infarction, cerebral thrombosis, cerebral embolism, transient cerebral ischemic attack (TIA), subarachnoid hemorrhage (vascular spasm), acute and chronic myocardial infarction, unstable narrow Diseases in ischemic heart diseases such as cardiomyopathy and coronary thrombolysis, diseases in pulmonary vascular disorders such as pulmonary infarction and pulmonary embolism, peripheral artery occlusion, deep vein thrombosis, disseminated intravascular coagulation syndrome (DIC) coagulation), generalized intravascular coagulation syndrome, coronary artery bypass surgery, percutaneous transluminal coronary angioplasty (PTCA) after angioplasty and other reocclusions and restenosis, intracoronary hemolysis therapy ( PTCR: in various vascular disorders such as reocclusion and restenosis after percutaneous transluminal coronary recanalization), thrombus formation after heart valve replacement, coagulation or inflammation of perfused blood during extracorporeal circulation Disease, intermittent claudication, atherosclerosis, and cancer.
Claims
請求の範囲 The scope of the claims
式 (I)で示されるカルボン酸誘導体またはその製薬学的に許容される塩。 A carboxylic acid derivative represented by the formula (I) or a pharmaceutically acceptable salt thereof.
[化 1] [Chemical 1]

[式中の記号は、以下の意味を示す。 [The symbols in the formula have the following meanings.
A環:ァリール環またはへテロアリール環。 Ring A: Aaryl ring or heteroaryl ring.
B環:ベンゼン環、ナフタレン環または単環乃至 2環式へテロァリール環。 Ring B: benzene ring, naphthalene ring, monocyclic to bicyclic heteroaryl ring.
C環:シクロアルキル環、ァリール環またはへテロ環。 Ring C: a cycloalkyl ring, an aryl ring or a hetero ring.
m、 n及び p :それぞれ独立して 0乃至 3の整数。ただし m、 nまたは pが 2以上の場合、そ れぞれ R2、 R3または R4は同一または互いに異なって!/、てもよ!/、。 m, n, and p: each independently an integer from 0 to 3. However, when m, n or p is 2 or more, R 2 , R 3 or R 4 are the same or different from each other!
R1 :— NH、 -CH NH、— C(=0)NH、— C(=NH)NH、— C(=NOH)NH、— C(=NH)NH— CO -R 1 :-NH, -CH NH,-C (= 0) NH,-C (= NH) NH,-C (= NOH) NH,-C (= NH) NH- CO-
2 2 2 2 2 2 22 2 2 2 2 2 2
(置換されて 、てもよ 、低級アルキル)、または 5-ォキソ -2,5-ジヒドロ- 1,2,4-ォキサジ ァゾール -3-ィル。 (Substituted or lower alkyl), or 5-oxo-2,5-dihydro-1,2,4-oxadiazol-3-yl.
R2及び R3 :それぞれ独立して、低級アルキル、ハロゲノ低級アルキル、ハロゲン、ォキ ソ、 - CN、 -NO、 -OR°, -0-ハロゲノ低級アルキル、 - NR。R。。、 - SR。、 - S(=0)R。、 - S(=0 R 2 and R 3 are each independently lower alkyl, halogeno lower alkyl, halogen, oxo, —CN, —NO, —OR °, —0-halogeno lower alkyl, —NR. R. . ,-SR. ,-S (= 0) R. ,-S (= 0
2 2
) R0、 - S(=0) NR°R°°、 - NR°S(=0) R°°、 - C(=0)R°、 -CO R°、 - C(=0)NR°R°°、 - NR°C(=0)) R 0 ,-S (= 0) NR ° R °°,-NR ° S (= 0) R °°,-C (= 0) R °, -CO R °,-C (= 0) NR ° R °°,-NR ° C (= 0)
2 2 2 2 2 2 2 2
R°°、 -NR°C(=0)_ハロゲノ低級アルキル、シクロアルキル、ァリールまたはへテロ環基 。ただし、 R2及び R3における低級アルキル、シクロアルキル、ァリールまたはへテロ環 基は置換されていてもよい。 R °°, -NR ° C (= 0) _halogeno lower alkyl, cycloalkyl, aryl or hetero ring group. However, the lower alkyl, cycloalkyl, aryl or hetero ring group in R 2 and R 3 may be substituted.
あるいは、それぞれ 2つの R2または R3がー体となって- 0-低級アルキレン- 0-を形 成していてもよい。 Alternatively, each of two R 2 or R 3 may be in the form of -0-lower alkylene-0-.
RQ及び RQQ:それぞれ独立して、 -Hまたは低級アルキル。 R Q and R QQ : each independently -H or lower alkyl.
R4:それぞれ独立して、低級アルキル、低級アルケニル、シクロアルキル、ァリール、
ヘテロ環基、ハロゲン、ォキソ、 - CN、 -NO、 -OR6, - NR6R6a、 - SR6、 - S(=0)R6、 - S(=0) R 4 : each independently lower alkyl, lower alkenyl, cycloalkyl, aryl, A heterocyclic group, halogen, Okiso, - CN, -NO, -OR 6 , - NR 6 R 6a, - SR 6, - S (= 0) R 6, - S (= 0)
2 2
R6、 - S(=0) NR6R6a、 - NR6S(=0) R6a、 - NR (=0) - NR6aR6b、 - NR (=0) - NR6aC(=0)ORR 6 ,-S (= 0) NR 6 R 6a ,-NR 6 S (= 0) R 6a ,-NR (= 0)-NR 6a R 6b ,-NR (= 0)-NR 6a C (= 0 ) OR
2 2 2 2 2 2 2 2 2 2
6b、 - C(=0)R6、 -CO R6、 - C(=0)NR6R6a、 - C(=0)N(R6)-低級アルキレン- NR6aR6b、 - NH- 6b, - C (= 0) R 6, -CO R 6, - C (= 0) NR 6 R 6a, - C (= 0) N (R 6) - lower alkylene - NR 6a R 6b, - NH-
2 2
C(=N-CN)NH、 -C(=0)-N=C(NH )、 - C(=0)- N=C(R6)- NH、 - C(=0)NR6- S(=0) - NR C (= N-CN) NH, -C (= 0) -N = C (NH),-C (= 0)-N = C (R 6 )-NH,-C (= 0) NR 6 -S (= 0)-NR
2 2 2 2 2 2 2 2 2 2
6aR6b、 -C(=0)NR6-S(=0) R6a、— OC(=0)R6、— OC(=0)OR6、— OC(=0)NR6R6a、— NR6C(= 6a R 6b , -C (= 0) NR 6 -S (= 0) R 6a , — OC (= 0) R 6 , — OC (= 0) OR 6 , — OC (= 0) NR 6 R 6a , — NR 6 C (=
2 2
0)R6a、 - NR6C(=0)OR6aまたは- NR6C(=0)NR6aR6b。ただし、 R4における低級アルキル、 低級アルケニル、シクロアルキル、ァリール及びへテロ環基はそれぞれ置換されてい てもよい。 0) R 6a , -NR 6 C (= 0) OR 6a or -NR 6 C (= 0) NR 6a R 6b . However, the lower alkyl, lower alkenyl, cycloalkyl, aryl, and heterocyclic groups in R 4 may each be substituted.
R6a及び R6b :それぞれ独立して、 - H、またはそれぞれ置換されていてもよい低級ァ ルキル、低級アルケニル、シクロアルキル、ァリール若しくはヘテロ環基。 R 6a and R 6b are each independently —H, or an optionally substituted lower alkyl, lower alkenyl, cycloalkyl, aryl or heterocyclic group.
R5: -OR0, - NR。R。。または- N(R°)-低級アルキレン- OR。。。 R 5 : -OR 0 , -NR. R. . Or -N (R °) -lower alkylene-OR. . .
J : *- NR°C(=0)-、 *- C(=0)NR°-、 - NR°C(=0)NR°-、 *- NR。-低級アルキレン-、または * -低級アルキレン- NR°C(=0)-。ただし、 *は A環への結合を示す。 J: * -NR ° C (= 0)-, * -C (= 0) NR °-, -NR ° C (= 0) NR °-, * -NR. -Lower alkylene- or * -Lower alkylene-NR ° C (= 0)-. However, * indicates a bond to the A ring.
L : *-NR°-低級アルキレン-、 *-NR°-低級ァルケ-レン-、 -低級アルキレン-または- 低級ァルケ-レン-。ただし、 *は B環への結合を示す。 L: * -NR ° -lower alkylene-, * -NR ° -lower alkylene-, -lower alkylene- or -lower alkylene-. However, * indicates a bond to the B ring.
X:単結合、 - 0-、 - NR7-、 - S -、 - C(=0)-、 - S(=0)-、 - S(=0) -、 *-低級アルキレン- O-X: a single bond, - 0-, - NR 7 - , - S -, - C (= 0) -, - S (= 0) -, - S (= 0) -, * - lower alkylene - O-
2 2
または *-低級アルキレン- NR7-。ただし、 *は C環への結合を示す。 Or * - lower alkylene - NR 7 -. * Indicates a bond to the C ring.
R7: - R°、 - C(=0)-低級アルキルまたは- C(=0)-ハロゲノ低級アルキル。 R 7 : -R °, -C (= 0) -lower alkyl or -C (= 0) -halogeno lower alkyl.
Y:単結合、それぞれ置換されて 、てもよ 、低級アルキレン若しくは低級ァルケ-レン ただし、 C環がヘテロ環である場合、ピロリジンを除く。 ] Y: Single bond, each substituted, may be lower alkylene or lower alkylene. However, when C ring is a hetero ring, pyrrolidine is excluded. ]
[2] Jが *- NHC(=0)-または *- NH-低級アルキレン- (ただし、 *は A環への結合を表す) であり、 Lが *-NH-低級アルキレン- (ただし、 *は B環への結合を表す)である請求の 範囲 1記載の化合物。 [2] J is *-NHC (= 0)-or *-NH-lower alkylene- (where * represents a bond to the A ring) and L is * -NH-lower alkylene- (where * The compound according to claim 1, wherein represents a bond to the B ring.
[3] 下記一般式 (I 1)で示される請求の範囲 2記載の化合物。 [3] The compound according to claim 2, represented by the following general formula (I 1):
[化 2]
 [Chemical 2]
[Chemical 2]
[式中の A環、 C環、 、 R R R R5、 m、 n、 p、 X及び Yは請求の範囲 1記載の意味 を示す。他の記号は以下の意味を示す。 [In the formula, A ring, C ring, RRRR 5 , m, n, p, X and Y have the meanings described in claim 1.] Other symbols have the following meanings.
BA環:ベンゼン環または 5乃至 6員単環式へテロァリール環。ただし、 BA環に隣接する 窒素原子及び J1は、 BA環を構成する隣接する 2つの炭素原子にそれぞれ結合する。 J1 :- C(=0)-または- CH -。 ] B ring A : benzene ring or 5- to 6-membered monocyclic heteroaryl ring. However, the nitrogen atom adjacent to the B A ring and J 1 are each bonded to the two adjacent carbon atoms constituting the B A ring. J 1 : -C (= 0)-or -CH-. ]
2 2
下記一般式 (I 2)で示される請求の範囲 3記載の化合物。 The compound according to claim 3, which is represented by the following general formula (I 2).
[化 3] [Chemical 3]

[式中の A環、 C環、 、 R2、
 R5、 m、 p、 X及び Yは請求の範囲 1記載の意味を 示し、 J1は請求の範囲 3記載の意味を示す。[In the formula, A ring, C ring, R 2 , R 5 , m, p, X and Y have the meaning described in claim 1 , and J 1 has the meaning described in claim 3.
R5、 m、 p、 X及び Yは請求の範囲 1記載の意味を 示し、 J1は請求の範囲 3記載の意味を示す。[In the formula, A ring, C ring, R 2 , R 5 , m, p, X and Y have the meaning described in claim 1 , and J 1 has the meaning described in claim 3.

アルキル)である請求の範囲 5記載の化合物。
[7] Yがハロゲンまたは- OR°で置換されて!、てもよ!/、低級アルキレンである請求の範囲6. The compound according to claim 5, which is alkyl). [7] Y is substituted by halogen or -OR ° !, may! /, Lower alkylene
6記載の化合物。 6. The compound according to 6.
[8] Xが単結合または- 0-である請求の範囲 7記載の化合物。 [8] The compound according to claim 7, wherein X is a single bond or -0-.
[9] A環がベンゼン環またはピリジン環である請求の範囲 7記載の化合物。 [9] The compound according to claim 7, wherein the A ring is a benzene ring or a pyridine ring.
[10] B環がベンゼン環またはピリジン環である請求の範囲 7記載の化合物。 [10] The compound according to claim 7, wherein the B ring is a benzene ring or a pyridine ring.
[11] C環がベンゼン環、ピぺリジン環、ピぺラジン環またはモルホリン環である請求の範 囲 7記載の化合物。 [11] The compound according to claim 7, wherein the C ring is a benzene ring, piperidine ring, piperazine ring or morpholine ring.
[12] {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ口フエ-ル}ァミノ) メチル ]-4- [(アミノスルホ -ル)ァミノ] -6-イソプロポキシフエノキシ }ァセチックアシッド ェチル {2- [({2- [({4- [ァミノ (ヒドロキシィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-クロ 口フエ-ル}ァミノ)メチル ]-4- [(アミノスルホ -ル)ァミノ] -6-イソプロポキシフエノキシ }ァ セタート、 [12] {2- [({2- [({4- [Amino (imino) methyl] phenol} amino) carbol] -4-chlorophenol} amino) Methyl] -4- [ (Aminosulfol) amino] -6-isopropoxyphenoxy} acetic acid ethyl {2- [({2- [({4- [amino (hydroxyimino) methyl] phenol} amino) carbo -L] -4-chlorophenyl} amino) methyl] -4-[(aminosulfol) amino] -6-isopropoxyphenoxy} phosphate,
[2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-フルオロフェ-ル }アミ ノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ]ァセチックアシッド、 {2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-メトキシフエ-ル}アミ ノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ }ァセチックアシッド、 [2- [({2- [({4- [ァミノ (ィミノ)メチル]フエ-ル}ァミノ)カルボ-ル]- 4-メチルフエ-ル}ァミノ) メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ]ァセチックアシッド、 ェチル [2- {[(2- {[(4- {[(エトキシカルボニル)ァミノ] (ィミノ)メチル }フエニル)ァミノ]カルボ -ル }-4-フルオロフェ -ル)ァミノ]メチル }- 4- (ヒドロキシメチル) -6-イソプロポキシフエノ キシ]ァセタート、 [2-[({2-[({4- [Amino (imino) methyl] phenol} amino) carbol] -4-fluorophenyl} amino) methyl] -4- (hydroxymethyl)- 6-Isopropoxyphenoxy] acetic acid, {2- [({2- [({4- [Amino (imino) methyl] phenol} amino) carbol] -4-methoxyphenol} Amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenoxy} acetic acid, [2- [({2- [({4- [amino (imino) methyl] phenol} Amino) carbol] -4-methylphenyl} amino) methyl] -4- (hydroxymethyl) -6-isopropoxyphenoxy] acetic acid, ethyl [2- {[(2- {[( 4- {[(Ethoxycarbonyl) amino] (imino) methyl} phenyl) amino] carbol} -4-fluorophenyl) amino] methyl}-4- (hydroxymethyl) -6-isopropoxyphenoxy] Acetate,
ェチル [2- {[(2- {[(4- {[(エトキシカルボニル)ァミノ] (ィミノ)メチル }フエニル)ァミノ]カルボ -ル }-4-メトキシフエ-ル)ァミノ]メチル }-4- (ヒドロキシメチル) -6-イソプロポキシフエノ キシ]ァセタート、 Ethyl [2- {[(2- {[(4- {[(Ethoxycarbonyl) amino] (imino) methyl} phenyl) amino] carbol} -4-methoxyphenyl) amino] methyl} -4- ( Hydroxymethyl) -6-isopropoxyphenoxy] acetate,
ェチル [2- {[(2- {[(4- {[(エトキシカルボニル)ァミノ] (ィミノ)メチル }フエニル)ァミノ]カルボ -ル }-4-メチルフエ-ル)ァミノ]メチル }-4- (ヒドロキシメチル) -6-イソプロポキシフエノキ シ]ァセタート、
[2- [({2- [({6- [ァミノ (ィミノ)メチル]ピリジン- 3-ィル }ァミノ)カルボ二ル]- 4-メチルフエ二 ル}ァミノ)メチル ]-4- (ヒドロキシメチル) -6-イソプロポキシフエノキシ]ァセチックァシッ ド、及び Ethyl [2- {[(2- {[(4- {[(Ethoxycarbonyl) amino] (imino) methyl} phenyl) amino] carbol} -4-methylphenol) amino] methyl} -4- ( Hydroxymethyl) -6-isopropoxyphenoxy] acetate, [2-[({2-[({6- [Amino (imino) methyl] pyridine-3-yl} amino) carboyl] -4-methylphenyl} amino) methyl] -4- (hydroxymethyl ) -6-Isopropoxyphenoxy] acetic acid, and
ェチル [2- {[(2- {[(6- {[(エトキシカルボニル)ァミノ] (ィミノ)メチル }ピリジン- 3-ィル)ァミノ] カルボ-ル}-4-メチルフエ-ル)ァミノ]メチル }-4- (ヒドロキシメチル) -6-イソプロポキシ フエノキシ]ァセタート Ethyl [2- {[(2- {[(6- {[(Ethoxycarbonyl) amino] (imino) methyl} pyridine-3-yl) amino] carbol} -4-methylphenol) amino] methyl } -4- (Hydroxymethyl) -6-isopropoxyphenoxy] acetate
力 なる群力 選択される請求の範囲 1記載の化合物またはその製薬学的に許容さ れる塩。 Power of group force The compound according to claim 1 or a pharmaceutically acceptable salt thereof selected.
下記式 (I 3)で示される請求項 1記載の化合物。 The compound according to claim 1, which is represented by the following formula (I 3):
[化 4] [Chemical 4]

[式中の A環、 C環、 m、 p、 X及び Yは請求の範囲 1記載の意味を示し、 J1は請求の範 囲 3記載の意味を示す。 [In the formula, A ring, C ring, m, p, X and Y have the meanings described in claim 1 , and J 1 has the meaning described in claim 3.
Β5、
 Β7及び Β8:それぞれ独立して、 CH、 CR3Aまたは N。ただし、 B5、 B7及び B8の うち、 CR3Aは 3個以下であり、また、それぞれの CR3Aは同一または互いに異なっていて ちょい。 Β 5 , Β 7 and Β 8 : each independently CH, CR 3A or N. However, among B 5 , B 7 and B 8 , CR 3A is 3 or less, and each CR 3A may be the same or different from each other.
Β7及び Β8:それぞれ独立して、 CH、 CR3Aまたは N。ただし、 B5、 B7及び B8の うち、 CR3Aは 3個以下であり、また、それぞれの CR3Aは同一または互いに異なっていて ちょい。 Β 5 , Β 7 and Β 8 : each independently CH, CR 3A or N. However, among B 5 , B 7 and B 8 , CR 3A is 3 or less, and each CR 3A may be the same or different from each other.
RLA : -NH、 -CH NH、— C(=0)NH、— C(=NH)NH、— C(=NOH)NHまたは— C(=NH)NH—R LA : -NH, -CH NH,-C (= 0) NH,-C (= NH) NH,-C (= NOH) NH or-C (= NH) NH-
2 2 2 2 2 2 2 2 2 2 2 2
CO -低級アルキル。 CO 2 -lower alkyl.
2 2
R2A及び R3A :それぞれ独立して、低級アルキル、ハロゲノ低級アルキル、ハロゲン、ォ キソ、 - CN、 -NO、 -OR°, -0-ハロゲノ低級アルキル、 - NR°R°°、 - SR°、 - S(=0)R°、 - S(= R 2A and R 3A : each independently lower alkyl, halogeno lower alkyl, halogen, oxo, -CN, -NO, -OR °, -0-halogeno lower alkyl, -NR ° R °°, -SR ° ,-S (= 0) R °,-S (=
2 2
O) R。、 - S(=0) NR。R。。、 - NR。S(=0) R。。、 - C(=0)R。、 -CO R。、 - C(=0)NR。R。。、 - NR。C(= O) R. ,-S (= 0) NR. R. . ,-NR. S (= 0) R. . ,-C (= 0) R. -CO R. ,-C (= 0) NR. R. . ,-NR. C (=
2 2 2 2 2 2 2 2
0)RQQ、シクロアルキル、ァリールまたはへテロ環基。ただし、 R2A及び R3Aにおけるシク 口アルキル、ァリール及びへテロ環基は、低級アルキル、ハロゲノ低級アルキル、ハロ
ゲン、ォキソ、 -OR°または- 0-ノヽロゲノ低級アルキルで置換されていてもよい。 0) R QQ , cycloalkyl, aryl or heterocyclic group. However, the cycloalkyl, aryl, and heterocyclic groups in R 2A and R 3A are lower alkyl, halogeno lower alkyl, halo. Optionally substituted by gen, oxo, -OR ° or -0-nologeno lower alkyl.
あるいは、それぞれ 2つの R2aまたは R3aがー体となって- 0-低級アルキレン- 0-を形 成していてもよい。 Alternatively, each of two R 2a or R 3a may be in the form of -0-lower alkylene-0-.
R½:それぞれ独立して、低級アルキル、シクロアルキル、ァリール、ヘテロ環基、ハロ ゲン、ォキソ、 - CNゝ -NO 、 - OR6c、 - NR6cR6d、 - SR6c、 - S(=0)R6c、 - S(=0) R6c、 - S(=0) N R ½: each independently, lower alkyl, cycloalkyl, Ariru, heterocyclic group, halo gen, Okiso, - CNゝ-NO, - OR 6c, - NR 6c R 6d, - SR 6c, - S (= 0 ) R 6c ,-S (= 0) R 6c ,-S (= 0) N
2 2 2 2 2 2
R6cR6d、 -NR6cS(=0) R6d、 - C(=0)R6c、 -CO Re\ - C(=0)NR6 6d、 - OC(=0)R6c、 - OC(=0 R 6c R 6d , -NR 6c S (= 0) R 6d ,-C (= 0) R 6c , -CO R e \-C (= 0) NR 6 6d ,-OC (= 0) R 6c ,- OC (= 0
2 2 twenty two
)OR6c、 - OC(=0)NR6cR6d、 - NR6cC(=0)R6d、 - NR6cC(=0)OR6dまたは- NR6cC(=0)NR6dR6e ただし、 R½における低級アルキルは Pa群力も選択される基で置換されて 、てもよく、 シクロアルキル、ァリール及びへテロ環基はそれぞれ Qa群力 選択される基で置換さ れていてもよい。 ) OR 6c , -OC (= 0) NR 6c R 6d , -NR 6c C (= 0) R 6d , -NR 6c C (= 0) OR 6d or -NR 6c C (= 0) NR 6d R 6e lower alkyl in R ½ is substituted with a group selected also P a group force and may, cycloalkyl, be the heterocyclic group Ariru and to be substituted with a group selected respectively Q a group force Good.
R6c、 R6d及び R6e:それぞれ独立して、 - H、低級アルキル、シクロアルキル、ァリールま たはへテロ環基。ただし、 R6e、 R6d及び R6eにおける低級アルキルは Pa群カゝら選択され る基で置換されていてもよぐシクロアルキル、ァリール及びへテロ環基はそれぞれ Qa 群から選択される基で置換されて 、てもよ 、。 R 6c , R 6d and R 6e : each independently —H, lower alkyl, cycloalkyl, aryl or hetero ring group. However, the lower alkyl in R 6e , R 6d and R 6e is a cycloalkyl, aryl and hetero ring group which may be substituted with a group selected from group P a and each is selected from group Q a May be substituted with a group.
pa群:ノヽロゲン、 - CN、 -OR°, -0-ハロゲノ低級アルキル、ォキソ、 - SR°、 - S(=0)R°、 - S (=0) R°、 - S(=0) NR°R°°、 - NR°S(=0) R°°、 - NR°R°°、 -CO R°、 - C(=0)NR°R°°、 -N(R°)C(p a group: neurogen, -CN, -OR °, -0-halogeno lower alkyl, oxo, -SR °, -S (= 0) R °, -S (= 0) R °, -S (= 0 ) NR ° R °°, -NR ° S (= 0) R °°, -NR ° R °°, -CO R °, -C (= 0) NR ° R °°, -N (R °) C (
2 2 2 2 2 2 2 2
=0)R°°、シクロアルキル、ァリールまたはへテロ環。ただし、 Pa群におけるシクロアルキ ル、ァリール及びへテロ環基は、 Qa群カゝら選択される基で置換されていてもよい。 Qa群:低級アルキル、ハロゲノ低級アルキル、ハロゲン、ォキソ、 -OR0, - 0-ハロゲノ 低級アルキル、 - NR°R°°、 -CO R°、 - NR°S(=0) R°°、 - C(=0)NR°R°°、 - NR°C(=0)R°°、低 = 0) R °°, cycloalkyl, aryl or heterocycle. However, the cycloalkyl, aryl and hetero ring group in the P a group may be substituted with a group selected from the Q a group. Q a group: lower alkyl, halogeno lower alkyl, halogen, oxo, -OR 0 , -0-halogeno lower alkyl, -NR ° R °°, -CO R °, -NR ° S (= 0) R °°, -C (= 0) NR ° R °°,-NR ° C (= 0) R °°, low
2 2 twenty two
級アルキレン- NR°R°°、低級アルキレン- CO R°、 -低級アルキレン- C(=0)NR°R°°。 Grade alkylene-NR ° R °°, lower alkylene-CO R °, -lower alkylene-C (= 0) NR ° R °°.
2 2
R5a: -OHまたは- 0-低級アルキル。 R 5a : —OH or —0-lower alkyl.
Ya:ハロゲンまたは- 0R°で置換されて 、てもよ 、低級アルキレン。 ] Y a : lower alkylene substituted with halogen or −0 °. ]
[14] 請求の範囲 1記載の化合物またはその製薬学的に許容される塩と、製薬学的に許 容される担体とからなる医薬組成物。 [14] A pharmaceutical composition comprising the compound according to claim 1 or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
[15] 活性ィ匕血液凝固第 VII因子阻害剤である請求の範囲 14記載の医薬組成物。 15. The pharmaceutical composition according to claim 14, which is an active 匕 blood coagulation factor VII inhibitor.
[16] 血液凝固抑制剤である請求の範囲 14記載の医薬組成物。
[17] 血栓若しくは塞栓によって引き起こされる疾病の予防または治療薬である請求の範 囲 14記載の医薬組成物。 [16] The pharmaceutical composition according to claim 14, which is a blood coagulation inhibitor. [17] The pharmaceutical composition according to claim 14, which is a prophylactic or therapeutic agent for a disease caused by thrombus or embolism.
[18] 虚血性心疾患、血管形成術後の再狭窄、脳血栓症、一過性脳虚血発作、末梢動 脈閉塞症、間歇性跛行、深部静脈血栓症、肺塞栓症、播種性血管内凝固症候群 (D IC)、心臓弁置換術後の血栓形成、血液体外循環時における灌流血液の凝固若しく は炎症、動脈硬化症、癌の予防または治療薬である請求の範囲 14記載の医薬組成 物。 [18] Ischemic heart disease, restenosis after angioplasty, cerebral thrombosis, transient ischemic attack, peripheral arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular 15. The pharmaceutical composition according to claim 14, which is a prophylactic or therapeutic agent for coagulation syndrome (D IC), thrombus formation after heart valve replacement, or coagulation or inflammation of perfused blood during blood extracorporeal circulation, arteriosclerosis, or cancer. object.
[19] 活性ィ匕血液凝固第 VII因子阻害剤、血液凝固抑制剤の製造のための、請求の範囲 1記載の化合物またはその製薬学的に許容される塩の使用。 [19] Use of the compound according to claim 1 or a pharmaceutically acceptable salt thereof for the production of an active blood coagulation factor VII inhibitor and a blood coagulation inhibitor.
[20] 血栓若しくは塞栓によって引き起こされる疾病の予防または治療薬の製造のため の、請求の範囲 1記載の化合物またはその製薬学的に許容される塩の使用。 [20] Use of the compound according to claim 1 or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for preventing or treating a disease caused by thrombus or embolism.
[21] 虚血性心疾患、血管形成術後の再狭窄、脳血栓症、一過性脳虚血発作、末梢動 脈閉塞症、間歇性跛行、深部静脈血栓症、肺塞栓症、播種性血管内凝固症候群 (D IC)、心臓弁置換術後の血栓形成、血液体外循環時における灌流血液の凝固若しく は炎症、動脈硬化症、癌の予防または治療薬の製造のための、請求の範囲 1記載の 化合物またはその製薬学的に許容される塩の使用。 [21] Ischemic heart disease, restenosis after angioplasty, cerebral thrombosis, transient ischemic attack, peripheral arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular Coagulation syndrome (D IC), thrombus formation after heart valve replacement, coagulation of perfused blood during extracorporeal circulation or inflammation, arteriosclerosis, production of preventive or therapeutic drugs for cancer 1 Use of the described compounds or pharmaceutically acceptable salts thereof.
[22] 請求の範囲 1記載の化合物またはその塩の治療有効量を患者に投与することを含 む、血栓若しくは塞栓によって引き起こされる疾病の予防または治療方法。 [22] A method for preventing or treating a disease caused by thrombus or embolism, comprising administering to a patient a therapeutically effective amount of the compound according to claim 1 or a salt thereof.
[23] 請求の範囲 1記載の化合物またはその塩の治療有効量を患者に投与することを含 む、虚血性心疾患、血管形成術後の再狭窄、脳血栓症、一過性脳虚血発作、末梢 動脈閉塞症、間歇性跛行、深部静脈血栓症、肺塞栓症、播種性血管内凝固症候群 (DIC)、心臓弁置換術後の血栓形成、血液体外循環時における灌流血液の凝固若 しくは炎症、動脈硬化症、癌の予防または治療方法。
[23] Ischemic heart disease, restenosis after angioplasty, cerebral thrombosis, transient ischemic attack, comprising administering to a patient a therapeutically effective amount of the compound according to claim 1 or a salt thereof Peripheral arterial occlusion, intermittent claudication, deep vein thrombosis, pulmonary embolism, disseminated intravascular coagulation syndrome (DIC), thrombus formation after heart valve replacement, coagulation of perfused blood during extracorporeal circulation Methods for preventing or treating inflammation, arteriosclerosis and cancer.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006550852A JPWO2006070878A1 (en) | 2004-12-28 | 2005-12-28 | Carboxylic acid derivative or salt thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004-380131 | 2004-12-28 | ||
| JP2004380131 | 2004-12-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006070878A1 true WO2006070878A1 (en) | 2006-07-06 |
Family
ID=36614989
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2005/024096 WO2006070878A1 (en) | 2004-12-28 | 2005-12-28 | Carboxylic acid derivative or salt thereof |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2006070878A1 (en) |
| WO (1) | WO2006070878A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010088000A2 (en) * | 2009-02-02 | 2010-08-05 | Angion Biomedica Corp. | Antifibrotic compounds and uses thereof |
| WO2011118672A1 (en) * | 2010-03-25 | 2011-09-29 | アステラス製薬株式会社 | Plasma kallikrein inhibitor |
| US20110263556A1 (en) * | 2009-10-23 | 2011-10-27 | Boehringer Ingelheim International Gmbh | New Compounds |
| JP2013511482A (en) * | 2009-11-18 | 2013-04-04 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Method for producing dabigatran etexilate |
| JP2013515026A (en) * | 2009-12-22 | 2013-05-02 | エフ.ホフマン−ラ ロシュ アーゲー | Substituted benzamide derivatives |
| JP2013521278A (en) * | 2010-03-03 | 2013-06-10 | ゲンナディエヴィッヒ トヴビン,ドミトリー | Factor Xa urethane, urea, amidine, and related inhibitors |
| US9925174B2 (en) | 2002-03-07 | 2018-03-27 | Boehringer Ingelheim International Gmbh | Administration form for the oral application of 3-[(2-{[4-(hexyloxycarbonyl-amino-imino-methyl)-phenylamino]-methyl}-1-methyl-1 H-benzimidazol acid ethyl ester and the salts thereof |
| JP2019500380A (en) * | 2015-12-28 | 2019-01-10 | セルジーン クオンティセル リサーチ,インク. | Histone demethylase inhibitor |
| US10508107B2 (en) | 2016-03-17 | 2019-12-17 | Hoffmann-La Roche Inc. | Morpholine derivative |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08188563A (en) * | 1993-12-27 | 1996-07-23 | Eisai Co Ltd | Anthranilic acid derivative |
| JP2001523256A (en) * | 1997-05-01 | 2001-11-20 | イーライ・リリー・アンド・カンパニー | Antithrombotic substance |
| JP2002533454A (en) * | 1998-12-23 | 2002-10-08 | イーライ・リリー・アンド・カンパニー | Aromatic amides |
| WO2003040101A1 (en) * | 2001-11-08 | 2003-05-15 | Novartis Ag | Anthranilic acid amides and pharmaceutical use thereof |
| JP2003520853A (en) * | 2000-01-27 | 2003-07-08 | ノバルティス アクチエンゲゼルシャフト | 2-amino-nicotinamide derivatives |
| WO2004013102A1 (en) * | 2002-07-31 | 2004-02-12 | Schering Aktiengesellschaft | Vegfr-2 and vegfr-3 inhibitory anthranylamidopyridines |
| JP2004522689A (en) * | 1998-12-23 | 2004-07-29 | イーライ・リリー・アンド・カンパニー | Antithrombotic amides |
| JP2004528379A (en) * | 2001-05-08 | 2004-09-16 | シエーリング アクチエンゲゼルシャフト | Selective anthranilamidopyridine amides as VEGFR-2 and VEGFR-3 inhibitors |
| JP2004315395A (en) * | 2003-04-14 | 2004-11-11 | Yamanouchi Pharmaceut Co Ltd | New benzoic acid derivative or its salt |
| JP2004323504A (en) * | 2002-08-30 | 2004-11-18 | Japan Tobacco Inc | Dibenzylamine compound and its pharmaceutical use |
-
2005
- 2005-12-28 WO PCT/JP2005/024096 patent/WO2006070878A1/en active Application Filing
- 2005-12-28 JP JP2006550852A patent/JPWO2006070878A1/en not_active Withdrawn
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08188563A (en) * | 1993-12-27 | 1996-07-23 | Eisai Co Ltd | Anthranilic acid derivative |
| JP2001523256A (en) * | 1997-05-01 | 2001-11-20 | イーライ・リリー・アンド・カンパニー | Antithrombotic substance |
| JP2002533454A (en) * | 1998-12-23 | 2002-10-08 | イーライ・リリー・アンド・カンパニー | Aromatic amides |
| JP2004522689A (en) * | 1998-12-23 | 2004-07-29 | イーライ・リリー・アンド・カンパニー | Antithrombotic amides |
| JP2003520853A (en) * | 2000-01-27 | 2003-07-08 | ノバルティス アクチエンゲゼルシャフト | 2-amino-nicotinamide derivatives |
| JP2004528379A (en) * | 2001-05-08 | 2004-09-16 | シエーリング アクチエンゲゼルシャフト | Selective anthranilamidopyridine amides as VEGFR-2 and VEGFR-3 inhibitors |
| WO2003040101A1 (en) * | 2001-11-08 | 2003-05-15 | Novartis Ag | Anthranilic acid amides and pharmaceutical use thereof |
| WO2004013102A1 (en) * | 2002-07-31 | 2004-02-12 | Schering Aktiengesellschaft | Vegfr-2 and vegfr-3 inhibitory anthranylamidopyridines |
| JP2004323504A (en) * | 2002-08-30 | 2004-11-18 | Japan Tobacco Inc | Dibenzylamine compound and its pharmaceutical use |
| JP2004315395A (en) * | 2003-04-14 | 2004-11-11 | Yamanouchi Pharmaceut Co Ltd | New benzoic acid derivative or its salt |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9925174B2 (en) | 2002-03-07 | 2018-03-27 | Boehringer Ingelheim International Gmbh | Administration form for the oral application of 3-[(2-{[4-(hexyloxycarbonyl-amino-imino-methyl)-phenylamino]-methyl}-1-methyl-1 H-benzimidazol acid ethyl ester and the salts thereof |
| WO2010088000A3 (en) * | 2009-02-02 | 2011-04-07 | Angion Biomedica Corp. | Antifibrotic compounds and uses thereof |
| WO2010088000A2 (en) * | 2009-02-02 | 2010-08-05 | Angion Biomedica Corp. | Antifibrotic compounds and uses thereof |
| US8951999B2 (en) * | 2009-10-23 | 2015-02-10 | Orexo Ab | Compounds |
| US20110263556A1 (en) * | 2009-10-23 | 2011-10-27 | Boehringer Ingelheim International Gmbh | New Compounds |
| JP2013511482A (en) * | 2009-11-18 | 2013-04-04 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Method for producing dabigatran etexilate |
| KR101802132B1 (en) * | 2009-11-18 | 2017-11-28 | 베링거 인겔하임 인터내셔날 게엠베하 | Method for producing dabigatran etexilate |
| JP2013515026A (en) * | 2009-12-22 | 2013-05-02 | エフ.ホフマン−ラ ロシュ アーゲー | Substituted benzamide derivatives |
| JP2016041740A (en) * | 2009-12-22 | 2016-03-31 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | Substituted benzamide derivatives |
| US9452980B2 (en) | 2009-12-22 | 2016-09-27 | Hoffmann-La Roche Inc. | Substituted benzamides |
| JP2013521278A (en) * | 2010-03-03 | 2013-06-10 | ゲンナディエヴィッヒ トヴビン,ドミトリー | Factor Xa urethane, urea, amidine, and related inhibitors |
| US9708265B2 (en) | 2010-03-03 | 2017-07-18 | Dmitry Gennadievich Tovbin | Urethanes, ureas, amidines and related inhibitors of factor Xa |
| WO2011118672A1 (en) * | 2010-03-25 | 2011-09-29 | アステラス製薬株式会社 | Plasma kallikrein inhibitor |
| JP2019500380A (en) * | 2015-12-28 | 2019-01-10 | セルジーン クオンティセル リサーチ,インク. | Histone demethylase inhibitor |
| US11001568B2 (en) | 2015-12-28 | 2021-05-11 | Celgene Quanticel Research, Inc. | Histone demethylase inhibitors |
| JP2021121599A (en) * | 2015-12-28 | 2021-08-26 | セルジーン クオンティセル リサーチ,インク. | Histone demethylase inhibitors |
| US11731954B2 (en) | 2015-12-28 | 2023-08-22 | Celgene Quanticel Research, Inc. | Histone demethylase inhibitors |
| US10508107B2 (en) | 2016-03-17 | 2019-12-17 | Hoffmann-La Roche Inc. | Morpholine derivative |
| US11312711B2 (en) | 2016-03-17 | 2022-04-26 | Hoffmann-La Roche Inc. | Morpholine derivative |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2006070878A1 (en) | 2008-06-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006070878A1 (en) | Carboxylic acid derivative or salt thereof | |
| TWI828008B (en) | Glp-1r modulating compounds | |
| JP5795630B2 (en) | Cyclopropyldicarboxamide and analogs exhibiting anticancer and antiproliferative activity | |
| JP4894517B2 (en) | 2-Phenylpyridine derivative | |
| JP6476205B2 (en) | Heterocyclic sulfones as RORγ modifiers | |
| US20100179137A1 (en) | Pyridone compound | |
| WO2011118672A1 (en) | Plasma kallikrein inhibitor | |
| JP2019502709A (en) | Inhibitors of PD-1 / PD-L1 protein / protein interaction | |
| JP2010501630A (en) | Pyrimidine compounds for treating GPR119 related disorders | |
| SK61399A3 (en) | HETEROCYCLE DERIVATIVES WHICH INHIBIT FACTOR Xa | |
| JP7301861B2 (en) | Inhibitor of TRPC6 | |
| AU2002238855B2 (en) | N-phenylarylsulfonamide compound, drug containing the compound as active ingredient, intermediate for the compound, and processes for producing the same | |
| WO2008029217A2 (en) | Dipeptidyl peptidase iv inhibitors | |
| JP7102388B2 (en) | Monocyclic heteroaryl substituted compound | |
| JPH03255080A (en) | Benzene compound | |
| WO2005030706A1 (en) | Amide type carboxamide derivative | |
| WO2015162452A1 (en) | Substituted pyrazole compounds as cb1 receptor antagonists and uses thereof | |
| JP5249484B2 (en) | Pharmaceutical composition | |
| WO2019144811A1 (en) | Tetrahydroisoquinoline derivative and preparation method therefor and use thereof | |
| JPH0841039A (en) | Substituted fluoxane | |
| JP2006219481A (en) | Method for producing acylaminothiazole derivative | |
| JP2007084437A (en) | Aminoalkylpyrazole derivative | |
| WO2022122035A1 (en) | Coagulation factor xia inhibitor, preparation method therefor and use thereof | |
| JP7427665B2 (en) | 6-Hydroxy-8-oxatricyclo[3.2.1.02,4]octane-2-carboxamide derivatives for inducing cartilage formation for treating joint injuries | |
| JP2004315395A (en) | New benzoic acid derivative or its salt |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006550852 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 05822689 Country of ref document: EP Kind code of ref document: A1 |