GLYCOPEGYLATED INTERFERON ALPHA
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] The present application is related to U.S. Provisional Patent Applications 60/620,927, filed October 21, 2004; 60/609,728, filed September 13, 2004; 60/609,385, filed September 13, 2004; and 60/609,218, filed September 10, 2004; each of which are incorporated by reference in their entirety for all purposes.
BACKGROUND OF THE INVENTION
[0002] Interferons are cytokines secreted by cells (esp. white blood cells) in response to viral infections. Interferons can bind to receptors on noninfected neighboring cells and induce the cells to produce proteins that increase their resistance to viral infections. The proteins produced interfere with the transcription of the viral genetic material, as well as increase the speed and strength of the immune response through the expression of class I MHC molecules.
[0003] Interferon alpha (IFN-α) is a member of Type I interferons, which mediate the early innate immune response to viral infections. This family has been tested for both the modulation of aberrant immunological responses and as a therapy for a variety of diseases such as AIDS and hepatitis.
[0004] The use of IFN-α as a therapeutic is compromised by its limited in vivo half-life in the body. Short in vivo half- life means that therapeutic glycopeptides must be administered frequently in high dosages, which ultimately translate to higher health care costs than might be necessary if a more efficient method for making longer lasting, more effective glycoprotein therapeutics were available.
[0005] One solution to the problem of providing cost effective glycopeptide therapeutics has been to provide peptides with longer in vivo half lives. For example, glycopeptide therapeutics with improved pharmacokinetic properties have been produced by attaching synthetic polymers to the peptide backbone. An exemplary polymer that has been conjugated to peptides is poly(ethylene glycol) ("PEG"). The use of PEG to derivatize peptide therapeutics has been demonstrated to reduce the immunogenicity of the peptides. For example, U.S. Pat. No. 4,179,337 (Davis et al.) discloses non-immunogenic polypeptides such as enzymes and peptide hormones coupled to polyethylene glycol (PEG) or polypropylene glycol. In addition to reduced immunogenicity, the clearance time in
circulation is prolonged due to the increased size of the PEG-conjugate of the polypeptides in question.
[0006] The principal mode of attachment of PEG, and its derivatives, to peptides is a non¬ specific bonding through a peptide amino acid residue (see e.g., U.S. Patent No. 4,088,538 U.S. Patent No. 4,496,689, U.S. Patent No. 4,414,147, U.S. Patent No. 4,055,635, and PCT WO 87/00056). Another mode of attaching PEG to peptides is through the non-specific oxidation of glycosyl residues on a glycopeptide (see e.g., WO 94/05332).
[0007] In these non-specific methods, poly(ethyleneglycol) is added in a random, non¬ specific manner to reactive residues on a peptide backbone. Of course, random addition of PEG molecules has its drawbacks, including a lack of homogeneity of the final product, and the possibility for reduction in the biological or enzymatic activity of the peptide. Therefore, for the production of therapeutic peptides, a derivitization strategy that results in the formation of a specifically labeled, readily characterizable, essentially homogeneous product is superior. Such methods have been developed.
[0008] Specifically labeled, homogeneous peptide therapeutics can be produced in vitro through the action of enzymes. Unlike the typical non-specific methods for attaching a synthetic polymer or other label to a peptide, enzyme-based syntheses have the advantages of regioselectivity and stereoselectivity. Two principal classes of enzymes for use in the synthesis of labeled peptides are glycosyltransferases (e.g., sialyltransferases, oligosaccharyltransferases, N-acetylglucosaminyltransferases), and glycosidases. These enzymes can be used for the specific attachment of sugars which can be subsequently modified to include a therapeutic moiety. Alternatively, glycosyltransferases and modified glycosidases can be used to directly transfer modified sugars to a peptide backbone (see e.g., U.S. Patent 6,399,336, and U.S. Patent Application Publications 20030040037, 20040132640, 20040137557, 20040126838, and 20040142856, each of which are incorporated by reference herein). Methods combining both chemical and enzymatic synthetic elements are also known (see e.g., Yamamoto et al. Carbohydr. Res. 305: 415-422 (1998) and U.S. Patent Application Publication 20040137557 which is incorporated herein by reference).
[0009] Interferon α (IFN-α) is an extremely valuable therapeutic peptide. Although commercially available forms of IFN-α are in use today, these peptides can be improved by
modifications that enhance the pharmacokinetics of the resulting isolated glycoprotein product. Thus, there remains a need in the art for compositions including IFN-α with improved effectiveness and pharmacokinetics. Furthermore, to be effective for the largest number of individuals, it must be possible to produce, on an industrial scale, a composition including IFN-α with improved therapeutic pharmacokinetics that has a predictable, essentially homogeneous, structure which can be readily reproduced over and over again.
[0010] The present invention fulfills these, and other, needs.
SUMMARY OF THE INVENTION
[0011] It has now been discovered that the controlled, enzymatic modification of IFN-α peptides with one or more saccharyl unit modified with a polymeric modifying moiety, e.g., poly(ethylene glycol), affords novel IFN-α conjugates with excellent pharmacokinetic properties. These IFN-α conjugates can include a variety of IFN-α peptides, including the wild type and mutants, e.g., deletions, substitutions and additions (e.g., additional amino acids, such as those from the proteolytically cleaved leader sequence). The peptides in the conjugates are referred to as GlycoPEGylated™, or glycoPEGylated.
[0012] In an exemplary embodiment, "glycoPEGylated" IFN-α molecules of the invention are produced by the enzyme mediated formation of a conjugate between a glycosylated or non-glycosylated IFN-α peptide and an enzymatically transferable saccharyl moiety that includes a poly(ethylene glycol) moiety within its structure. The PEG moiety is attached to the saccharyl moiety directly (i.e., through a single group formed by the reaction of two reactive groups) or through a linker moiety, e.g., substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, etc. Exemplary PEG-saccharyl donor structures are set forth in FIG. 2.
[0013] Thus, in another aspect, the present invention provides a conjugate between a PEG moiety, e.g., PEG and a peptide that has an in vivo activity similar or otherwise analogous to art-recognized IFN-α. In the conjugate of the invention, the PEG moiety is covalently attached to the peptide via an intact glycosyl linking group. Exemplary intact glycosyl linking groups include sialic acid moieties and, particularly, sialic acid moieties derivatized with PEG. [0014] The polymeric modifying moiety can be attached at an amino acid residue, or at any position of a glycosyl moiety of IFN-α. Moreover, the polymeric modifying moiety can
be bound to a glycosyl residue at any position in the amino acid sequence of a wild type or mutant IFN-α peptide. '
[0015] In an exemplary embodiment, the invention provides an IFN-α peptide that is conjugated through a glycosyl linking group to a polymeric modifying moiety. Exemplary IFN-α peptide conjugates include a glycosyl linking group having a formula selected from:
[0016] In Formulae I and II, R2 is H, CH2OR7, COOR7 or OR7, in which R7 represents H, substituted or unsubstituted alkyl or substituted or unsubstituted heteroalkyl. The symbols R3, R4, R5, R6 and R6 independently represent H, substituted or unsubstituted alkyl, OR8, NHC(O)R9. The index d is 0 or 1. R8 and R9 are independently selected from H, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl or sialic acid. At least one of R3, R4, R5, R6 or R6 includes the polymeric modifying moiety e.g., PEG. In an exemplary embodiment, R6 and R6 , together with the carbon to which they are attached are components of the side chain of sialic acid. In a further exemplary embodiment, this side chain is functionalized with the polymeric modifying moiety.
[0017] In an exemplary embodiment, the polymeric moiety is bound to the glycosyl linking group, generally through a heteroatom on the glycosyl core (e.g., N, O), through a linker, L, as shown below:
(R1)w— L-
R1 is the polymeric modifying moiety and L is selected from a bond and a linking group. The index w represents an integer selected from 1-6, preferably 1-3 and more preferably 1-2. Exemplary linking groups include substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl moieties and sialic acid. An exemplary component of the linker is an acyl moiety. Another exemplary linking group is an amino acid residue (e.g., cysteine, serine, lysine, and short oligopeptides, e.g., Lys-Lys, Lys-Lys-Lys, Cys-Lys, Ser-Lys, etc.)
[0018] When L is a bond, it is formed by reaction of a reactive functional group on a precursor of R1 and a reactive functional group of complementary reactivity on a precursor of the glycosyl linking group. When L is a non-zero order linking group, L can be in place on the glycosyl moiety prior to reaction with the R1 precursor. Alternatively, the precursors of R1 and L can be incorporated into a preformed cassette that is subsequently attached to the glycosyl moiety. As set forth herein, the selection and preparation of precursors with appropriate reactive functional groups is within the ability of those skilled in the art. Moreover, coupling of the precursors proceeds by chemistry that is well understood in the art.
[0019] In an exemplary embodiment L is a linking group that is formed from an amino acid, or small peptide (e.g., 1-4 amino acid residues) providing a modified sugar in which the polymeric modifying moiety is attached through a substituted alkyl linker. Exemplary linkers include glycine, lysine, serine and cysteine. Amino acid analogs, as defined herein, are also of use as linker components. The amino acid may be modified with an additional component of a linker, e.g., alkyl, heteroalkyl, covalently attached through an acyl linkage, for example, an amide or urethane formed through an amine moiety of the amino acid residue.
[0020] In an exemplary embodiment, the glycosyl linker has a structure according to Formula I and R5 includes the polymeric modifying moiety. In another exemplary embodiment, R5 includes both the polymeric modifying moiety and a linker, L, joining the modifying moiety to the glycosyl core. L can be a linear or branched structure. Similarly, the polymeric modifying can be branched or linear.
[0021] The polymeric modifying moiety includes two or more repeating units that can be water-soluble or essentially insoluble in water. Exemplary water-soluble polymers of use in the compounds of the invention include PEG, e.g., m-PEG, PPG, e.g., m-PPG, polysialic acid, polyglutamate, polyaspartate, polylysine, polyethyeleneimine, biodegradable polymers (e.g., polylactide, polyglyceride), and functionalized PEG, e.g., terminal-functionalized PEG.
[0022] The glycosyl core of the glycosyl linking groups of use in the IFN-α conjugates of the invention is selected from both natural and unnatural furanoses and pyranoses. The unnatural saccharides optionally include an alkylated or acylated hydroxyl and/or amine moiety, e.g., ethers, esters and amide substituents on the ring. Other unnatural saccharides include an H, hydroxyl, ether, ester or amide substituent at a position on the ring at which such a substituent is not present in the natural saccharide. Alternatively, the carbohydrate is missing a substituent that would be found in the carbohydrate from which its name is derived,
e.g., deoxy sugars. Still further exemplary unnatural sugars include both oxidized (e.g., -onic and -uronic acids) and reduced (sugar alcohols) carbohydrates. The sugar moiety can be a mono-, oligo- or poly-saccharide.
[0023] Exemplary natural sugars of use as components of glycosyl linking groups in the present invention include glucose, glucosamine, galactose, galactosamine, fucose, mannose, mannosamine, xylanose, ribose, N-acetyl glucose, N-acetyl glucosamine, N-acetyl galactose, N-acetyl galactosamine, and sialic acid.
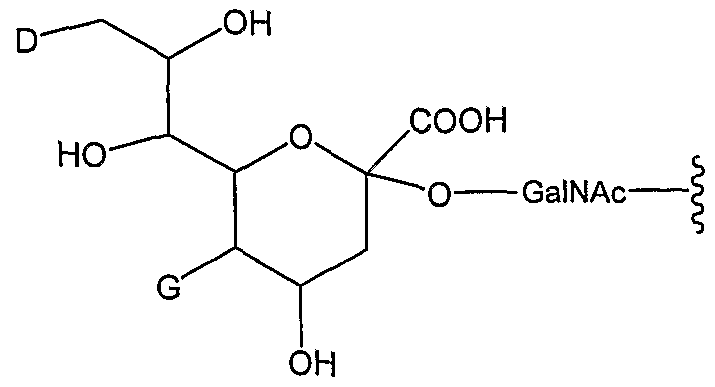
[0024] In one embodiment, the present invention provides an IFN-α conjugate including the moiety:
wherein D is a member selected from -OH and RZ-L-HN-; G is a member selected from H and R
1 -L- and -C(O)(C
1-C
6)alkyl; R
1 is a moiety including a straight-chain or branched poly(ethylene glycol) residue; and L is a linker, e.g., a bond ("zero order"), substituted or unsubstituted alkyl and substituted or unsubstituted heteroalkyl. In exemplary embodiments, when D is OH, G is R
1 -L-, and when G is -C(O)(C i-C
6)alkyl, D is R^L-NH-.
[0025] In another aspect, the invention provides an IFN-α conjugate including a glycosyl linking group having the formula:
[0026] In other embodiments, the glycosyl linking group has the formula:
[0027] In yet another embodiment, the group has the formula:
in which the index p represents and integer from 1 to 10, and c represents O or 1.
[0028] In another aspect, the invention provides a method of making a PEGylated IFN-α conjugate of the invention. The method includes: (a) contacting a substrate IFN-α peptide including a glycosyl group selected from:
-GaINAc (GaI)1. -GaI- -(Sia)c and with a PEG-sialic acid donor having the formula:
and an enzyme that transfers PEG-sialic acid from the donor onto a member selected from the GaINAc, Gal and the Sia of the glycosyl group, under conditions appropriate for the transfer. An exemplary modified sialic acid donor is CMP-sialic acid modified, through a linker moiety, with a polymer, e.g., a straight chain or branched poly(ethylene glycol) moiety. The indices c and r independently represent 0 or 1.
[0029] In another aspect, the invention provides an IFN-α conjugate including an IFN-α peptide and a glycosyl linking group attached to an amino acid residue of the IFN-α peptide. The glycosyl linking group includes a modified sialyl residue having the formula:
[0030] R
2 is H,
alkyl or substituted or unsubstituted heteroalkyl. R
3 and R are independently selected from H, substituted or unsubstituted alkyl, OR
8, NHC(O)R
9. R
8 and R
9 are independently selected from H, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl or sialic acid. L
a is a linker selected from a bond, substituted or unsubstituted alkyl and substituted or unsubstituted heteroalkyl. R
16 and R
17 are independently selected polymeric arms. X
2 and X
4 are independently selected linkage fragments joining polymeric moieties R
16 and R
17 to C; and X
5 is a non-reactive group.
[0031] In another aspect, the IFN-α conjugate of the invention includes an IFN-α peptide and a glycosyl linking group attached to an amino acid residue of the IFN-α peptide. The glycosyl linking group includes a modified sialyl residue according to Formula (III). The
IFN-α conjugate has an amino acid sequence with a threonine residue at position 106, and the threonine residue is a threonine-glycosyl linking group, and the threonine-glycosyl linking group has a structure which is a member selected from:
In a preferred embodiment, the amino acid is serine or threonine. In another exemplary embodiment, the amino acid is Thr
106.
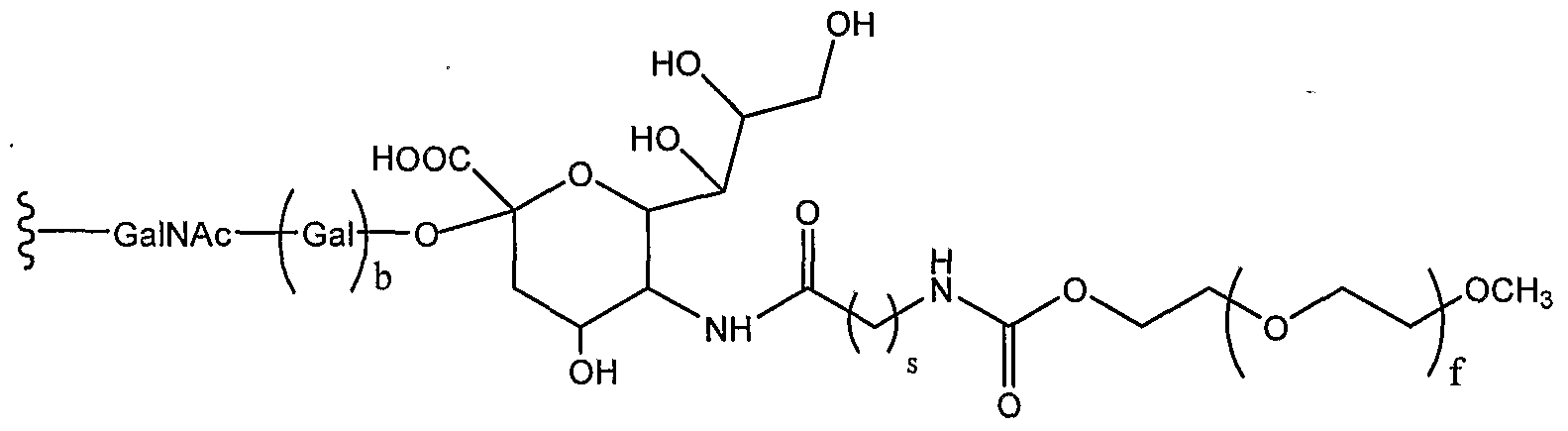
[0032] In another aspect, the invention provides an IFN-α conjugate including an IFN-α peptide and a glycosyl linking group attached to an amino acid residue of the IFN-α peptide, the glycosyl linking group including a modified sialyl residue according to Formula (IV):
in which R
2 is H, CH
2OR
7, COOR
7, COO
" or OR
7. R
7 represents H, substituted or unsubstituted alkyl or substituted or unsubstituted heteroalkyl. R
3 and R
4 are members independently selected from H, substituted or unsubstituted alkyl, OR
8, NHC(O)R
9. R
8 and R are independently selected from H, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl or sialic acid. The index s is an integer from 1 to 20. The index f is an integer from 1 to 2500. Q is a member selected from H and substituted or unsubstituted C
1-C
6 alkyl.
[0033] In another aspect, the IFN-α conjugate of the invention includes an IFN-α peptide and a glycosyl linking group attached to an amino acid residue of the IFN-α peptide. The glycosyl linking group includes a modified sialyl residue according to Formula (IV). The IFN-α conjugate has an amino acid sequence with a threonine residue at position 106, and the threonine residue is a threonine-glycosyl linking group, and the threonine-glycosyl linking group has a structure which is a member selected from:
In a preferred embodiment, the amino acid is serine or threonine. In another exemplary embodiment, the amino acid is Thr10 .
[0034] The peptide can be acquired from essentially any source, however, in one embodiment, prior to being modified as discussed above, the IFN-α peptide is expressed in a suitable host. Mammalian (e.g., CHO), bacteria (e.g., E. coli) and insect cells (e.g., Sf-9) are exemplary expression systems providing IFN-α of use in the compositions and methods set forth herein. An exmplary O-linked glycan that is glycoPEGylated is shown in FIG. 6. Exemplary glycans produced in an insect system and a mammalian system, and subsequently glycoconjugated and or remodeled and glycoconjugated to PEG are set forth in FIG. 7 and FIG. 8.
[0035] In another aspect, the invention provides a method of treating a condition in a subject in need thereof. Exemplary conditions include those characterized by compromised red blood cell production in the subject. The method includes the step of administering to the subject an amount of the polymer-modified IFN-α conjugate of the invention effective to ameliorate the condition in the subject.
[0036] In exemplary embodiments, an IFN-α conjugate of the invention may be administered to patients for the purposed of preventing infection in cancer patients undergoing certain types of radiation therapy, chemotherapy, and bone marrow transplantations, to mobilize progenitor cells for collection in peripheral blood progenitor cell transplantations, for treatment of severe chronic or relative leukopenia, irrespective of cause, and to support treatment of patients with acute myeloid leukaemia. Additionally, the
polypeptide conjugate or composition of the invention may be used for treatment of AIDS or other immunodeficiency diseases as well as bacterial infections, and Hepatisis A, B, and C.
[0037] In another aspect, the invention provides a pharmaceutical formulation including a IFN-α conjugate of the invention and a pharmaceutically acceptable carrier. [0038] In the IFN-α conjugates of the invention, essentially each of the amino acid residues to which the modifying group is bound has the same structure. For example, if one IFN-α conjugate includes a Thr linked glycosyl linking group, at least about 70%, 80%, 90%, 95%, 97%, 99%, 99.2%, 99.4%, 99.6%, or more preferably 99.8% of the IFN-α conjugates in the population will have the same glycosyl linking group covalently bound to the same Thr residue.
[0039] Other objects and advantages of the invention will be apparent to those of skill in the art from the detailed description that follows.
DESCRIPTION OF THE DRAWINGS
[0040] FIG. 1 illustrates the amino acid sequence of IFN-alpha-2a, SEQ ID NO: 1; and IFN- alpha-2b; SEQ ID NO: 2.
[0041] FIG. 2 illustrates exemplary modified sialic acid nucleotides useful in the practice of the invention. A. Structure of branched CMP-sialic acid PEG 40 kDa and 60 kDa sugar nucleotides (40 kDa: m = 441-511 ethylene oxide units, n = 441-511 ethylene oxide units, 60 kDa: m = 681-776 ethylene oxide units n = 681-776 ethylene oxide units). B. Structures of linear CMP-sialic acid PEG 20 kDa and 30 kDa sugar nucleotides (20 kDa: n = 441 -511 ethylene oxide units, 30 kDa: n = 681-776 ethylene oxide units).
[0042] FIG. 3 is a scheme showing an exemplary embodiment of the invention in which an IFN-α peptide is remodeled by enzymatically adding a GaINAc moiety at position 106 prior to adding a saccharyl moiety derivatized with PEG.
[0043] FIG. 4 is a synthetic scheme for producing an exemplary PEG-glycosyl linking group precursor (modified sugar) of use in preparing the conjugates of the invention.
[0044] FIG. 5 is a table providing exemplary sialyltransferases of use in forming the IFN-α conjugates of the invention, e.g., to glycoPEGylate peptides with a modified sialic acid.
[0045] FIG. 6A and 6B show exemplary O-linked glycan structures on IFN-α conjugates of the invention.
[0046] FIG. 7 shows an exemplary N-linked glycan structure on a mutant IFN-α conjugate of the invention expressed in insect cells (and remodeled and glycoPEGylated) in which the mutant includes one or more N-linked glycosylation sites.
[0047] FIG. 8 shows exemplary N-linked glycan structures on mutant IFN-α glycoconjugates of the invention expressed in mammalian cells (and glycoPEGylated) in which the mutant includes one or more N-linked glycosylation sites: A) exemplary glycan; B) exemplary glycan glycoPEGylated using CST-II and/or polyα2,8 sialyltransferase; C) exemplary glycan glycoPEGylated using CST-II and/or ST3Gal3.
[0048] FIG. 9 shows a MALDI Spectra of IFN-alpha-2b-GalNAc-S A-PEG (20 and 30 KDa) and IFN-alpha-2b-GalNAc-Gal-SA-PEG-40KDa.
[0049] FIG. 10 shows a site occupancy analysis of GalNAc-SA-PEG-20KDa on IFN-alpha- 2b-GalNAc-SA-PEG-20KDa. Top: Base peak chromatogram of a GIuC digest of IFN-alpha- 2b-GalNAc-SA-PEG-20KDa. Middle: Selected ion chromatogram plotting the m/z range from m/z=916 to m/z=917.5. The m/z range was selected based on the calculated m/z of peptide ACVIQGVGVTETPLMKE. Bottom: Selected ion chromatogram plotting the m/z range from m/z^lO^.S to m/z=1019.5. The m/z range was selected based on the calculated m/z of peptide ACVIQGVGVTETPLMKE plus GaINAc. Unmodified peptide and peptide- GaINAc are not found. Another peptide ACVIQGV GVTE plus GaINAc is also not found.
[0050] FIG. 11 shows a SDS-PAGE for IFN-alpha-2b-GalNAc-SA-PEG-20KDa. Lane 1 : IFN-alpha-2b; Lane 2: IFN-alpha-2b-GalNAc-SA-PEG-20KDa. Invitrogen: 4-20 % Tris- Glycine gel, EC6025BOX. Std = SeeBlue Plus2 pre-stained protein standard, LC5925. Tris- Glycine SDS running buffer, LC2675-5. Tris-Glycine SDS sample buffer, LC2676. Collodial Blue Stain, LC6025. Constant 125 V, 1 h 50 min.
[0051] FIG. 12 shows a silver stained SDS-PAGE of IFN-alpha-2b-GalNAc-S A-PEG (20 and 30 KDa) after formulation. Invitrogen: 4-20 % Tris-Glycine gel, EC6025BOX. Std. = SeeBlue Plus2 pre-stained protein standard, LC5925. Tris-Glycine SDS running buffer, LC2675-5. Tris-Glycine SDS sample buffer, LC2676. Silver stain kit, Wako, Cat 291- 50301. Constant 125 V, 1 h 50 min.
[0052] FIG. 13 shows dose-response curves of the IFN-alpha-2b reference standard, IFN- alpha-2b and the IFN-alpha-PEG variants in the anti-proliferation assay. The 2nd WHO International Standard 1999 for human IFN-alpha-2b, which is an rDNA product derived
from E. coli, was used as the reference standard. Each data point indicates the mean of three determinations.
[0053] FIG. 14 shows dose-response curves of IFN alpha-2b and IFN-alpha-2b-PEG variants in the anti-proliferation assay. Sigmoidal dose response curves for IFN-alpha-2b with the IFN-alpha-2b-PEG variants. Each data point represents the mean of 4 replicates, standard deviations at each point are represented by error bars.
[0054] FIG. 15 shows precipitate concentrations and time profiles for IFN-alpha variants injected intravenously at time zero. DPM values in acid-precipitated pellets from plasma were corrected for efficiency of acid precipitation.
[0055] FIG. 16 shows concentration and time profiles for IFN-alpha variants injected subcutaneously at time zero. DPM values in acid-precipitated pellets from plasma were converted to concentrations of IFN-alpha protein and corrected for efficiency of acid precipitation.
[0056] FIG. 17 shows phamacokinetic data for a chemoPEGylated compound vs. an IFN-α conjugate.
[0057] FIG. 18 shows phamacodynamic data against 2' -5' oligoadenylate synthetase for a chemoPEGylated compound and an IFN-α conjugate.
DETAILED DESCRIPTION OF THE INVENTION AND THE PREFERRED EMBODIMENTS Abbreviations and Definitions
[0058] IFN-α, interferon alpha; PEG, poly(ethyleneglycol); PPG, poly(propyleneglycol);
Ara, arabinosyl; Fru, fructosyl; Fuc, fucosyl; Gal, galactosyl; GaINAc, N- acetylgalactosaminyl; GIc, glucosyl; GIcNAc, N-acetylglucosaminyl; Man, mannosyl;
ManAc, mannosaminyl acetate; XyI, xylosyl; and NeuAc, sialyl (N-acetylneuraminyl); M6P, mannose-6-phosphate; Sia, sialic acid, N-acetylneuraminyl, and derivatives and analogues thereof.
[0059] Unless defined otherwise, all technical and scientific terms used herein generally have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. Generally, the nomenclature used herein and the laboratory procedures in
cell culture, molecular genetics, organic chemistry and nucleic acid chemistry and hybridization are those well known and commonly employed in the art. Standard techniques are used for nucleic acid and peptide synthesis. The techniques and procedures are generally performed according to conventional methods in the art and various general references {see generally, Sambrook et al. MOLECULAR CLONING: A LABORATORY MANUAL, 2d ed. (1989) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y., which is incorporated herein by reference), which are provided throughout this document. The nomenclature used herein and the laboratory procedures in analytical chemistry, and organic synthetic described below are those well known and commonly employed in the art. Standard techniques, or modifications thereof, are used for chemical syntheses and chemical analyses.
[0060] Oligosaccharides described herein are generally described with the name or abbreviation for the saccharide (i.e., Gal), followed by the configuration of the glycosidic bond (α or β), the ring bond (1 or 2), the ring position of the reducing saccharide involved in the bond (2, 3, 4, 6 or 8), and then the name or abbreviation of the reducing saccharide (i.e., GIcNAc). Each saccharide is preferably a pyranose. For a review of standard glycobiology nomenclature see, Essentials of Glycobiology Varki et al. eds. CSHL Press (1999).
[0061] The term "sialic acid" refers to any member of a family of nine-carbon carboxylated sugars. The most common member of the sialic acid family is N-acetyl-neuraminic acid (2- keto-5-acetamido-3,5-dideoxy-D-glycero-D-galactononulopyranos-l-onic acid (often abbreviated as Neu5Ac, NeuAc, or NANA). A second member of the family is N-glycolyl- neuraminic acid (Neu5Gc or NeuGc), in which the N-acetyl group of NeuAc is hydroxylated. A third sialic acid family member is 2-keto-3-deoxy-nonulosonic acid (KDN) (Nadano et al. (1986) J Biol. Chem. 261 : 11550-11557; Kanamori et al., J. Biol Chem. 265: 21811-21819 (1990)). Also included are 9-substituted sialic acids such as a 9-O-C1-C6 acyl-Neu5Ac like 9-O-lactyl-Neu5Ac or 9-O-acetyl-Neu5Ac, 9-deoxy-9-fluoro-Neu5Ac and 9-azido-9-deoxy- Neu5Ac. For review of the sialic acid family, see, e.g., Varki, Glycobiology 2: 25-40 (1992); Sialic Acids: Chemistry, Metabolism and Function, R. Schauer, Ed. (Springer-Verlag, New York (1992)). The synthesis and use of sialic acid compounds in a sialylation procedure is disclosed in international application WO 92/16640, published October 1, 1992.
[0062] "Peptide" refers to a polymer in which the monomers are amino acids and are joined together through amide bonds, alternatively referred to as a polypeptide. Additionally, unnatural amino acids, for example, β-alanine, phenylglycine and homoarginine are also
included. Amino acids that are not gene-encoded may also be used in the present invention. Furthermore, amino acids that have been modified to include reactive groups, glycosylation sites, polymers, therapeutic moieties, biomolecules and the like may also be used in the invention. All of the amino acids used in the present invention may be either the d - or 1 - isomer. The 1 -isomer is generally preferred. In addition, other peptidomimetics are also useful in the present invention. As used herein, "peptide" refers to both glycosylated and unglycosylated peptides. Also included are peptides that are incompletely glycosylated by a system that expresses the peptide. For a general review, see, Spatola, A. F., in Chemistry and Biochemistry of Amino Acids, Peptides and Proteins, B. Weinstein, eds., Marcel Dekker, New York, p. 267 (1983).
[0063] "Glycopeptide", as used herein, refers to peptides that are covalently attached to glycosyl residues.
[0064] The term "IFN-α peptide", refers to a family of approximately twenty peptides of approximately 18 kDa in weight. Descriptions of these different peptides are provided herein. Prototype amino acid sequences for IFN-α are known, e.g., see, SEQ ID NO:1 (FIG. 1). The present invention is not limited to these nucleotide and amino acid sequences. One of skill in the art will readily appreciate that many variants of IFN-α exist both naturally and as engineered derivatives.
[0065] IFN-α peptide conjugates of the invention include peptide sequences with additional amino acids, deletions and substitutions. These mutations can be between the carboxyl and amino termini of the wild-type sequence, as shown in PCT Application No.
PCT/US2005/000799 "O-linked Glycosylation of Peptides", filed Jan. 10, 2005. Additional amino acids, both natural and unnatural, can also be attached at the beginning or end of the amino acid sequence. The IFN-α peptide of the invention can include portions of the peptide sequence that are present before proteolytic cleavage. For example, the IFN-α peptide can include an additional cysteine residue that is a portion of the proteolytically cleaved leader sequence.
[0066] The term "chemoPEGylated IFN-α," refers to a PEGylated IFN-α peptide which has been PEGylated in the absence of an enzyme that transfers a glycosyl-PEG residue from a nucleotide sugar to the IFN-α peptide. A commercially available example of a chemoPEGylated IFN-α is PEGasys (Hoffmann-LaRoche).
[0067] The term "IFN-α conjugate," refers to species of the invention which include a glycosyl linking group which has been enzymatically attached to the IFN-α peptide. The IFN-α conjugate may be additionally or alternatively modified by further conjugation with diverse species such as therapeutic moieties, diagnostic moieties, targeting moieties and the like. These further conjugated IFN-α conjugates are also included under the term IFN-α conjugates.
[0068] As used herein, the term "modified sugar," refers to a naturally- or non-naturally- occurring carbohydrate that is enzymatically added onto an amino acid or a glycosyl residue of a peptide in a process of the invention. The modified sugar is selected from a number of enzyme substrates including, but not limited to sugar nucleotides (mono-, di-, and tri¬ phosphates), activated sugars (e.g., glycosyl halides, glycosyl mesylates) and sugars that are neither activated nor nucleotides. In some embodiments, the "modified sugar" can be covalently functionalized with a "modifying group."
[0069] As used herein, the term "modifying group" refers to a component of the IFN-α conjugate that is covalently attached to a glycosyl linking group. A modifying group can be a component of the modified sugar that is subsequently attached to the IFN-α peptide. A modifying group can also be attached directly to a sugar moiety that is already attached to the IFN-α peptide. Useful modifying groups include, but are not limited to, water-soluble polymer moieties such as PEG, water-insoluble polymer moieties, therapeutic moieties, diagnostic moieties, biomolecules, and the like. The modifying group also includes reactive functional groups, such as levulinic acid. These reactive functional groups can serve as the locus of attachment for water-soluble polymers such as PEG moieties, therapeutic moieties, diagnostic moieties, biomolecules, and the like. These reactive functional groups can also include protecting groups which can be removed at appropriate times to facilitate proper functionalization. Reactive functional groups with protecting groups are alternatively known as masked reactive functional groups. The modifying group is preferably not a naturally occurring, or an unmodified carbohydrate. The locus of functionalization with the modifying group is selected such that it does not prevent the "modified sugar" from being added enzymatically to a peptide.
[0070] The term "glycoconjugation," as used herein, refers to the enzymatically mediated conjugation of a modified sugar species to an amino acid or glycosyl residue of a polypeptide, e.g., an IFN-α peptide. A subgenus of "glycoconjugation" is "glyco-
PEGylation," in which the modifying group of the modified sugar is poly(ethylene glycol), and alkyl derivative (e.g., m-PEG) or reactive derivative (e.g., H2N-PEG, HOOC-PEG) thereof.
[0071] The term, "glycosyl linking group," as used herein, refers to a glycosyl residue to which a modifying group (e.g., water soluble polymer moiety, therapeutic moiety, diagnostic moiety, biomolecules, reactive functional moiety) is covalently attached; the glycosyl linking group joins the modifying group to the remainder of the conjugate. In the methods of the invention, the "glycosyl linking group" becomes covalently attached to a glycosylated or unglycosylated peptide, thereby linking the agent to an amino acid and/or glycosyl residue on the peptide. A "glycosyl linking group" is generally derived from a "modified sugar" by the enzymatic attachment of the "modified sugar" to an amino acid and/or glycosyl residue of the peptide. The glycosyl linking group can be a saccharide-derived structure that is degraded during formation of modifying group-modified sugar cassette (e.g., oxidation— ^Schiff base formation— ^reduction), or the glycosyl linking group may be intact. An "intact glycosyl linking group" refers to a linking group that is derived from a glycosyl moiety in which the saccharide monomer that links the modifying group and to the remainder of the conjugate is not degraded by chemical (e.g., sodium metaperiodate) or enzymatic processes (e.g., oxidase). "Intact glycosyl linking groups" of the invention may be derived from a naturally occurring oligosaccharide by addition of glycosyl unit(s) or removal of one or more glycosyl unit from a parent saccharide structure.
[0072] The term "targeting moiety," as used herein, refers to species that will selectively localize in a particular tissue or region of the body. The localization is mediated by specific recognition of molecular determinants, molecular size of the targeting agent or conjugate, ionic interactions, hydrophobic interactions and the like. Other mechanisms of targeting an agent to a particular tissue or region are known to those of skill in the art. Exemplary targeting moieties include antibodies, antibody fragments, transferrin, HS-glycoprotein, coagulation factors, serum proteins, β-glycoprotein, G-CSF, GM-CSF, M-CSF, EPO, serum proteins (e.g., Factors VII, Vila, VIII, IX, and X) and the like.
[0073] As used herein, "therapeutic moiety" means any agent useful for therapy including, but not limited to, antibiotics, anti-inflammatory agents, anti-tumor drugs, cytotoxins, and radioactive agents. "Therapeutic moiety" includes prodrugs of bioactive agents, constructs in which more than one therapeutic moiety is bound to a carrier, e.g, multivalent agents.
Therapeutic moiety also includes proteins and constructs that include proteins. Exemplary proteins include, but are not limited to, Granulocyte Colony Stimulating Factor (GCSF), Granulocyte Macrophage Colony Stimulating Factor (GMCSF), Interferon (e.g., Interferon- CC, -β, -γ), Interleukin (e.g., Interleukin II), serum proteins (e.g., Factors VII, Vila, VIII, IX, and X), Human Chorionic Gonadotropin (HCG), Follicle Stimulating Hormone (FSH) and Lutenizing Hormone (LH) and antibody fusion proteins (e.g. Tumor Necrosis Factor Receptor ((TNFR)/Fc domain fusion protein)).
[0074] The term "amino acid" refers to naturally occurring and synthetic amino acids, as well as amino acid analogs and amino acid mimetics that function in a manner similar to the naturally occurring amino acids. Naturally occurring amino acids are those encoded by the genetic code, as well as those amino acids that are later modified, e.g., hydroxyproline, γ- carboxyglutamate, and O-phosphoserine. Amino acid analogs refers to compounds that have the same basic chemical structure as a naturally occurring amino acid, i.e., an α carbon that is bound to a hydrogen, a carboxyl group, an amino group, and an R group, e.g., homoserine, norleucine, methionine sulfoxide, methionine methyl sulfonium. Such analogs have modified R groups (e.g., norleucine) or modified peptide backbones, but retain the same basic chemical structure as a naturally occurring amino acid. Amino acid mimetics refers to chemical compounds that have a structure that is different from the general chemical structure of an amino acid, but that function in a manner similar to a naturally occurring amino acid. As used herein, "amino acid," whether it is in a linker or a component of a peptide sequence refers to both the D- and L-isomer of the amino acid as well as mixtures of these two isomers.
[0075] The term "water-soluble" refers to moieties that have some detectable degree of solubility in water. Methods to detect and/or quantify water solubility are well known in the art. Exemplary water-soluble polymers include peptides, saccharides, poly(ethers), poly(amines), poly(carboxylic acids) and the like. Peptides can have mixed sequences of be composed of a single amino acid, e.g., poly(lysine). An exemplary polysaccharide is poly(sialic acid). An exemplary poly(ether) is poly(ethylene glycol). Poly (ethylene imine) is an exemplary polyamine, and poly(acrylic) acid is a representative poly(carboxylic acid).
[0076] The polymer backbone of the water-soluble polymer can be poly(ethylene glycol) (i.e. PEG). However, it should be understood that other related polymers are also suitable for use in the practice of this invention and that the use of the term PEG or poly(ethylene glycol) is intended to be inclusive and not exclusive in this respect. The term PEG includes
poly(ethylene glycol) in any of its forms, including alkoxy PEG, difunctional PEG, multiarnied PEG, forked PEG, branched PEG, pendent PEG (i.e. PEG or related polymers having one or more functional groups pendent to the polymer backbone), or PEG with degradable linkages therein.
[0077] The polymer backbone can be linear or branched. Branched polymer backbones are generally known in the art. Typically, a branched polymer has a central branch core moiety and a plurality of linear polymer chains linked to the central branch core. PEG is commonly used in branched forms that can be prepared by addition of ethylene oxide to various polyols, such as glycerol, pentaerythritol and sorbitol. The central branch moiety can also be derived from several amino acids, such as lysine. The branched poly(ethylene glycol) can be represented in general form as R(-PEG-OH)m in which R represents the core moiety, such as glycerol or pentaerythritol, and m represents the number of arms. Multi-armed PEG molecules, such as those described in U.S. Pat. No. 5,932,462, which is incorporated by reference herein in its entirety, can also be used as the polymer backbone.
[0078] Many other polymers are also suitable for the invention. Polymer backbones that are non-peptidic and water-soluble, with from 2 to about 300 termini, are particularly useful in the invention. Examples of suitable polymers include, but are not limited to, other ρoly(alkylene glycols), such as poly(propylene glycol) ("PPG"), copolymers of ethylene glycol and propylene glycol and the like, poly(oxyethylated polyol), poly(olefmic alcohol), polyvinylpyrrolidone), poly(hydroxypropylmethacrylamide), poly(α-hydroxy acid), poly(vinyl alcohol), polyphosphazene, polyoxazoline, poly(N-acryloylmorpholine), such as described in U.S. Pat. No. 5,629,384, which is incorporated by reference herein in its entirety, and copolymers, terpolymers, and mixtures thereof. Although the molecular weight of each chain of the polymer backbone can vary, it is typically in the range of from about 100 Da to about 100,000 Da, often from about 6,000 Da to about 80,000 Da.
[0079] The "area under the curve" or "AUC", as used herein in the context of administering a peptide drug to a patient, is defined as total area under the curve that describes the concentration of drug in systemic circulation in the patient as a function of time from zero to infinity.
[0080] The term "half-life" or "I1A", as used herein in the context of administering a peptide drug to a patient, is defined as the time required for plasma concentration of a drug in a patient to be reduced by one half. There may be more than one half-life associated with the
peptide drug depending on multiple clearance mechanisms, redistribution, and other mechanisms well known in the art. Usually, alpha and beta half-lives are defined such that the alpha phase is associated with redistribution, and the beta phase is associated with clearance. However, with protein drugs that are, for the most part, confined to the bloodstream, there can be at least two clearance half-lives. For some glycosylated peptides, rapid beta phase clearance may be mediated via receptors on macrophages, or endothelial cells that recognize terminal galactose, N-acetylgalactosamine, N-acetylglucosamine, mannose, or fucose. Slower beta phase clearance may occur via renal glomerular filtration for molecules with an effective radius < 2 nm (approximately 68 kD) and/or specific or non- specific uptake and metabolism in tissues. GlycoPEGylation may cap terminal sugars (e.g., galactose or N-acetylgalactosamine) and thereby block rapid alpha phase clearance via receptors that recognize these sugars. It may also confer a larger effective radius and thereby decrease the volume of distribution and tissue uptake, thereby prolonging the late beta phase. Thus, the precise impact of glycoPEGylation on alpha phase and beta phase half-lives will vary depending upon the size, state of glycosylation, and other parameters, as is well known in the art. Further explanation of "half-life" is found in Pharmaceutical Biotechnology (1997, DFA Crommelin and RD Sindelar, eds., Harwood Publishers, Amsterdam, pp 101 - 120).
[0081] The terms "large-scale" and "industrial-scale" are used interchangeably and refer to a reaction cycle that produces at least about 250 mg, preferably at least about 500 mg, and more preferably at least about 1 gram of glycoconjugate at the completion of a single reaction cycle.
[0082] As used herein, "pharmaceutically acceptable carrier" includes any material, which when combined with the conjugate retains the conjugates' activity and is non-reactive with the subject's immune systems. Examples include, but are not limited to, any of the standard pharmaceutical carriers such as a phosphate buffered saline solution, water, emulsions such as oil/water emulsion, and various types of wetting agents. Other carriers may also include sterile solutions, tablets including coated tablets and capsules. Typically such carriers contain excipients such as starch, milk, sugar, certain types of clay, gelatin, stearic acid or salts thereof, magnesium or calcium stearate, talc, vegetable fats or oils, gums, glycols, or other known excipients. Such carriers may also include flavor and color additives or other ingredients. Compositions including such carriers are formulated by well known conventional methods.
[0083] As used herein, "administering," means oral administration, administration as a suppository, topical contact, intravenous, intraperitoneal, intramuscular, intralesional, intranasal or subcutaneous administration, or the implantation of a slow-release device e.g., a mini-osmotic pump, to the subject. Adminsitration is by any route including parenteral, and transmucosal (e.g., oral, nasal, vaginal, rectal, or transdermal). Parenteral administration includes, e.g., intravenous, intramuscular, intra-arteriole, intradermal, subcutaneous, intraperitoneal, intraventricular, and intracranial. Moreover, where injection is to treat a tumor, e.g., induce apoptosis, administration may be directly to the tumor and/or into tissues surrounding the tumor. Other modes of delivery include, but are not limited to, the use of liposomal formulations, intravenous infusion, transdermal patches, etc.
[0084] The term "ameliorating" or "ameliorate" refers to any indicia of success in the treatment of a pathology or condition, including any objective or subjective parameter such as abatement, remission or diminishing of symptoms or an improvement in a patient's physical or mental well-being. Amelioration of symptoms can be based on objective or subjective parameters; including the results of a physical examination and/or a psychiatric evaluation.
[0085] The term "therapy" refers to"treating" or "treatment" of a disease or condition including preventing the disease or condition from occurring in an animal that may be predisposed to the disease but does not yet experience or exhibit symptoms of the disease (prophylactic treatment), inhibiting the disease (slowing or arresting its development), providing relief from the symptoms or side-effects of the disease (including palliative treatment), and relieving the disease (causing regression of the disease).
[0086] The term "effective amount" or "an amount effective to"or a "therapeutically effective amount" or any gramatically equivalent term means the amount that, when administered to an animal for treating a disease, is sufficient to effect treatment for that disease.
[0087] The term "isolated" refers to a material that is substantially or essentially free from components, which are used to produce the material. For IFN-α conjugates of the invention, the term "isolated" refers to material that is substantially or essentially free from components which normally accompany the material in the mixture used to prepare the IFN-α conjugates. "Isolated" and "pure" are used interchangeably. Typically, isolated IFN-α conjugates of the invention have a level of purity preferably expressed as a range. The lower end of the range of purity for the IFN-α conjugates is about 60%, about 70% or about 80% and the upper end of the range of purity is about 70%, about 80%, about 90% or more than about 90%.
[0088] When the IFN-α conjugates are more than about 90% pure, their purities are also preferably expressed as a range. The lower end of the range of purity is about 90%, about 92%, about 94%, about 96% or about 98%. The upper end of the range of purity is about 92%, about 94%, about 96%, about 98% or about 100% purity.
[0089] Purity is determined by any art-recognized method of analysis (e.g., band intensity on a silver stained gel, polyacrylamide gel electrophoresis, HPLC, or a similar means).
[0090] "Essentially each member of the population," as used herein, describes a characteristic of a population of IFN-α conjugates of the invention in which a selected percentage of the modified sugars added to an IFN-α peptide are added to multiple, identical acceptor sites on the peptide. "Essentially each member of the population" speaks to the
"homogeneity" of the sites on the IFN-α peptide conjugated to a modified sugar and refers to IFN-α conjugates of the invention, which are at least about 80%, preferably at least about 90% and more preferably at least about 95% homogenous.
[0091] "Homogeneity," refers to the structural consistency across a population of acceptor moieties to which the modified sugars are conjugated. Thus, in an IFN-α conjugate of the invention in which each modified sugar moiety is conjugated to an acceptor site having the same structure as the acceptor site to which every other modified sugar is conjugated, the IFN-α conjugate is said to be about 100% homogeneous. Homogeneity is typically expressed as a range. The lower end of the range of homogeneity for IFN-α conjugates is about 60%, about 70% or about 80% and the upper end of the range of purity is about 70%, about 80%, about 90% or more than about 90%.
[0092] When the IFN-α conjugates are more than or equal to about 90% homogeneous, their homogeneity is also preferably expressed as a range. The lower end of the range of homogeneity is about 90%, about 92%, about 94%, about 96% or about 98%. The upper end of the range of purity is about 92%, about 94%, about 96%, about 98% or about 100% homogeneity. The homogeneity of the IFN-α conjugates is typically determined by one or more methods known to those of skill in the art, e.g., liquid chromatography-mass spectrometry (LC-MS), matrix assisted laser desorption mass time of flight spectrometry (MALDITOF), capillary electrophoresis, and the like. The discussion above is equally relevant for other O-glycosylation and N-glycosylation sites.
[0093] "Substantially uniform glycoform" or a "substantially uniform glycosylation pattern," when referring to a glycopeptide species, refers to the percentage of acceptor moieties that
are glycosylated by the glycosyltransferase of interest (e.g., fucosyltransferase). For example, in the case of a α 1,2 fucosyltransferase, a substantially uniform fucosylation pattern exists if substantially all (as defined below) of the Galβl,4-GlcNAc-R and sialylated analogues thereof are fucosylated in an IFN-α conjugate of the invention. It will be understood by one of skill in the art, that the starting material may contain glycosylated acceptor moieties (e.g., fucosylated Galβl,4-GlcNAc-R moieties). Thus, the calculated percent glycosylation will include acceptor moieties that are glycosylated by the methods of the invention, as well as those acceptor moieties already glycosylated in the starting material.
[0094] The term "substantially" in the above definitions of "substantially uniform" generally means at least about 40%, at least about 70%, at least about 80%, or more preferably at least about 90%, and still more preferably at least about 95% of the acceptor moieties for a particular glycosyltransferase are glycosylated. For example, if an IFN-α conjugate includes a Ser linked glycosyl residues, at least about 70%, 80%, 90%, 95%, 97%, 99%, 99.2%, 99.4%, 99.6%, or more preferably 99.8% of the peptides in the population will have the same glycosyl residue covalently bound to the same Ser residue.
[0095] Where substituent groups are specified by their conventional chemical formulae, written from left to right, they equally encompass the chemically identical substituents, which would result from writing the structure from right to left, e.g., -CH2O- is intended to also recite -OCH2-.
[0096] The term "alkyl," by itself or as part of another substituent means, unless otherwise stated, a straight or branched chain, or cyclic hydrocarbon radical, or combination thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals, having the number of carbon atoms designated (i.e. Ci-C1O means one to ten carbons). Examples of saturated hydrocarbon radicals include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, cyclohexyl, (cyclohexyl)methyl, cyclopropylmethyl, homologs and isomers of, for example, n-pentyl, n- hexyl, n-heptyl, n-octyl, and the like. An unsaturated alkyl group is one having one or more double bonds or triple bonds. Examples of unsaturated alkyl groups include, but are not limited to, vinyl, 2-propenyl, crotyl, 2-isopentenyl, 2-(butadienyl), 2,4-pentadienyl, 3 -(1,4- pentadienyl), ethynyl, 1- and 3-propynyl, 3-butynyl, and the higher homologs and isomers. The term "alkyl," unless otherwise noted, is also meant to include those derivatives of alkyl
defined in more detail below, such as "heteroalkyl." Alkyl groups that are limited to hydrocarbon groups are termed "homoalkyl".
[0097] The term "alkylene" by itself or as part of another substituent means a divalent radical derived from an alkane, as exemplified, but not limited, by -CH2CH2CH2CH2-, and further includes those groups described below as "heteroalkylene." Typically, an alkyl (or alkylene) group will have from 1 to 24 carbon atoms, with those groups having 10 or fewer carbon atoms being preferred in the present invention. A "lower alkyl" or "lower alkylene" is a shorter chain alkyl or alkylene group, generally having eight or fewer carbon atoms.
[0098] The terms "alkoxy," "alkylamino" and "alkylthio" (or thioalkoxy) are used in their conventional sense, and refer to those alkyl groups attached to the remainder of the molecule via an oxygen atom, an amino group, or a sulfur atom, respectively.
[0099] The term "heteroalkyl," by itself or in combination with another term, means, unless otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom selected from the group consisting of O, N, Si and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quateraized. The heteroatom(s) O, N and S and Si may be placed at any interior position of the heteroalkyl group or at the position at which the alkyl group is attached to the remainder of the molecule. Examples include, but are not limited to, -CH2-CH2-O-CH3, -CH2-CH2-NH- CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH25-S(O)-CH3, -CH2-CH2-S(O)2- CH3, -CH=CH-O-CH3, -Si(CHB)3, -CH2-CH=N-OCH3, and -CH=CH-N(CH3)-CH3. Up to two heteroatoms may be consecutive, such as, for example, -CH2-NH-OCH3 and -CH2-O- Si(CH3)3. Similarly, the term "heteroalkylene" by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified, but not limited by, -CH2- CH2-S-CH2-CH2- and -CH2-S-CH2-CH2-NH-CH2-. For heteroalkylene groups, heteroatoms can also occupy either or both of the chain termini (e.g., alkyleneoxy, alkylenedioxy, alkyleneamino, alkylenediamino, and the like). Still further, for alkylene and heteroalkylene linking groups, no orientation of the linking group is implied by the direction in which the formula of the linking group is written. For example, the formula -C(O)2R'- represents both -C(O)2R'- and -R5C(O)2-.
[0100] The terms "cycloalkyl" and "heterocycloalkyl", by themselves or in combination with other terms, represent, unless otherwise stated, cyclic versions of "alkyl" and "heteroalkyl",
respectively. Additionally, for heterocycloalkyl, a heteroatom can occupy the position at which the heterocycle is attached to the remainder of the molecule. Examples of cycloalkyl include, but are not limited to, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like. Examples of heterocycloalkyl include, but are not limited to, 1 - (1,2,5,6-tetrahydropyridyl), 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 4-morpholinyl, 3- morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl, tetrahydrothien-3-yl, 1 -piperazinyl, 2-piperazinyl, and the like.
[0101] The terms "halo" or "halogen," by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as "haloalkyl," are meant to include monohaloalkyl and polyhaloalkyl. For example, the term "halo(Cl-C4)alkyl" is mean to include, but not be limited to, trifluoromethyl, 2,2,2- trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
[0102] The term "aryl" means, unless otherwise stated, a polyunsaturated, aromatic, substituent that can be a single ring or multiple rings (preferably from 1 to 3 rings), which are fused together or linked covalently. The term "heteroaryl" refers to aryl groups (or rings) that contain from one to four heteroatoms selected from N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized. A heteroaryl group can be attached to the remainder of the molecule through a heteroatom. Non-limiting examples of aryl and heteroaryl groups include phenyl, 1-naphthyl, 2-naphthyl, 4-biphenyl, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3-pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 2-phenyl-4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4- isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3- thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 5-benzothiazolyl, purinyl, 2-benzimidazolyl, 5-indolyl, 1-isoquinolyl, 5-isoquinolyl, 2-quinoxalinyl, 5-quinoxalinyl, 3- quinolyl, tetrazolyl, benzo[b]furanyl5 benzo[b]thienyl, 2,3-dihydrobenzo[l ,4]dioxin-6-yl, benzo[l,3]dioxol-5-yl and 6-quinolyl. Substituents for each of the above noted aryl and heteroaryl ring systems are selected from the group of acceptable substituents described below.
[0103] For brevity, the term "aryl" when used in combination with other terms (e.g., aryloxy, arylthioxy, arylalkyl) includes both aryl and heteroaryl rings as defined above. Thus, the term "arylalkyl" is meant to include those radicals in which an aryl group is attached to an alkyl group (e.g., benzyl, phenethyl, pyridylmethyl and the like) including those alkyl groups
in which a carbon atom (e.g., a methylene group) has been replaced by, for example, an oxygen atom (e.g., phenoxymethyl, 2-pyridyloxymethyl, 3-(l-naphthyloxy)propyl, and the like).
[0104] Each of the above terms (e.g., "alkyl," "heteroalkyl," "aryl" and "heteroaryl") is meant to include both substituted and unsubstituted forms of the indicated radical. Preferred substituents for each type of radical are provided below.
[0105] Substituents for the alkyl and heteroalkyl radicals (including those groups often referred to as alkylene, alkenyl, heteroalkylene, heteroalkenyl, alkynyl, cycloalkyl, heterocycloalkyl, cycloalkenyl, and heterocycloalkenyl) are generically referred to as "alkyl group substituents," and they can be one or more of a variety of groups selected from, but not limited to: -OR', =0, =NR\ =N-OR\ -NR'R", -SR', -halogen, -SiR'R"R"\ -OC(O)R', - C(O)R', -CO2R', -CONR'R", -OC(O)NR5R", -NR"C(0)R', -NR'-C(0)NR"R"\ - NR"C(0)2R\ -NR-C(NR'R"R'")=NR"", -NR-C(NR' R")=NR5", -S(O)R', -S(O)2R', - S(O)2NR5R", -NRSO2R', -CN and -NO2 in a number ranging from zero to (2m'+l), where m5 is the total number of carbon atoms in such radical. R', R", R'" and R"5' each preferably independently refer to hydrogen, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl, e.g., aryl substituted with 1-3 halogens, substituted or unsubstituted alkyl, alkoxy or thioalkoxy groups, or arylalkyl groups. When a compound of the invention includes more than one R group, for example, each of the R groups is independently selected as are each R', R", R5" and R"55 groups when more than one of these groups is present. When R' and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6-, or 7-membered ring. For example, -NR'R" is meant to include, but not be limited to, 1-pyrrolidinyl and 4-morpholinyl. From the above discussion of substituents, one of skill in the art will understand that the term "alkyl" is meant to include groups including carbon atoms bound to groups other than hydrogen groups, such as haloalkyl (e.g. , -CF3 and -CH2CF3) and acyl (e.g., -C(O)CH3, -C(O)CF3, -C(O)CH2OCH3, and the like).
[0106] Similar to the substituents described for the alkyl radical, substituents for the aryl and heteroaryl groups are generically referred to as "aryl group substituents." The substituents are selected from, for example: halogen, -OR', =0, =NR', =N-0R', -NR'R", -SR', -halogen, -SiR'R"R"\ -OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR5R", -NR55C(O)R5, -NR' -C(O)NR55R'", -NR55C(O)2R5, -NR-C(NR' R"R'")=NR"", -NR-C(NR' R")=NR5", - S(O)R5, -S(O)2R5, -S(O)2NR5R", -NRSO2R', -CN and -NO2, -R', -N3, -CH(Ph)2, 1TuOrO(C1-
C4)alkoxy, and fluoro(C1-C4)alkyl, in a number ranging from zero to the total number of open valences on the aromatic ring system; and where R', R", R'" and R"" are preferably independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl and substituted or unsubstituted heteroaryl. When a compound of the invention includes more than one R group, for example, each of the R groups is independently selected as are each R', R", R'" and R"" groups when more than one of these groups is present. In the schemes that follow, the symbol X represents "R" as described above.
[0107] Two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)-(CRR')q-U-, wherein T and U are independently -NR-, -O-, -CRR'- or a single bond, and q is an integer of from 0 to 3. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CH2)r-B-, wherein A and B are independently -CRR'-, -O-, -NR-, -S-, -S(O)-, -S(O)2-, -S(O)2NR'- or a single bond, and r is an integer of from 1 to 4. One of the single bonds of the new ring so formed may optionally be replaced with a double bond. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula - (CRR')s-X-(CR"R'")d-, where s and d are independently integers of from 0 to 3, and X is - O-, -NR'-, -S-, -S(O)-, -S(O)2-, or -S(O)2NR'-. The substituents R, R', R" and R'" are preferably independently selected from hydrogen or substituted or unsubstituted (Cl- C6)alkyl.
[0108] As used herein, the term "heteroatom" is meant to include oxygen (O), nitrogen (N), sulfur (S) and silicon (Si).
Introduction [0109] To improve the effectiveness of recombinant IFN-α used for therapeutic purposes, the present invention provides conjugates of glycosylated and unglycosylated IFN-α peptides with modified sugars. These modified sugars can include water-soluble polymers, e.g., PEG (m-PEG), PPG (m-PPG), etc. An IFN-α conjugate may be additionally or alternatively modified by further conjugation with diverse species such as therapeutic moieties, diagnostic moieties, targeting moieties, reactive functional moieties and the like. These further conjugated IFN-α conjugates are also included under the term "IFN-α conjugates".
[0110] The IFN-α conjugates of the invention are formed by the enzymatic attachment of a modified sugar to the glycosylated or unglycosylated IFN-α peptide. These IFN-α peptides can include the naturally occurring, completed peptides as well as variants thereof (an IFN-α peptide with additional amino acids from the proteolytically cleaved leader sequence). Glycosyl linking groups, glycosylation sites and glycosyl residues provide loci for conjugating modified sugars to the IFN-α peptide, e.g., by glycoconjugation. These modified sugars can include modifying groups, such as water-soluble polymers, i.e. poly(ethylene glycol), methoxy-poly(ethylene glycol). Modification of the IFN-α peptide can improve the stability and retention time of IFN-α in a patient's circulation, and/or reduce the antigenicity of IFN-α.
[0111] The methods of the invention make it possible to assemble IFN-α conjugates that have a substantially homogeneous derivatization or conjugation pattern. The enzymes used in the invention are generally selective for a particular amino acid residue, combination of amino acid residues, or particular glycosyl residue of the IFN-α peptide. The methods are also practical for large-scale production of IFN-α conjugates. Thus, the methods of the invention provide a practical means for large-scale preparation of glycopeptides having preselected, uniform derivatization patterns.
[0112] The present invention also provides conjugates of glycosylated and unglycosylated peptides with increased therapeutic half-life due to, for example, reduced clearance rate, or reduced rate of uptake by the immune or reticuloendothelial system (RES). Moreover, the methods of the invention provide a means for masking antigenic determinants on peptides, thus reducing or eliminating a host immune response against the peptide. Selective attachment of targeting agents can also be used to target a peptide to a particular tissue or cell surface receptor that is specific for the particular targeting agent.
The Compositions
The Conjugates
[0113] In a first aspect, the present invention provides a conjugate between a selected modifying group and an IFN-α peptide. The present invention also encompasses a method for the modification of the glycan structure on IFN-α, providing a conjugate between interferon alpha (IFN-α) and a modifying group.
[0114] The link between the peptide and the modifying moiety includes a glycosyl linking group interposed between the peptide and the selected moiety. As discussed herein,
the selected modifying moiety is essentially any species that can be attached to a saccharide unit, resulting in a "modified sugar" that is recognized by an appropriate transferase enzyme, which appends the modified sugar onto the peptide, or a glycosyl residue attached thereto. The saccharide component of the modified sugar, when interposed between the peptide and a selected moiety, becomes a "glycosyl linking group," e.g., an "intact glycosyl linking group." The glycosyl linking group is formed from any mono- or oligosaccharide that, after modification with the modifying group, is a substrate for an enzyme that adds the modified sugar to an amino acid or glycosyl residue of a peptide.
[0115] The glycosyl linking group can be, or can include, a saccharide moiety that is degradatively modified before or during the addition of the modifying group. For example, the glycosyl linking group can be derived from a saccharide residue that is produced by oxidative degradation of an intact saccharide to the corresponding aldehyde, e.g., via the action of metaperiodate, and subsequently converted to a Schiff base with an appropriate amine, which is then reduced to the corresponding amine.
[0116] The conjugates of the invention typically correspond to the general structure:
in which the symbols a, b, c, d and s represent a positive, non-zero integer; and t is either 0 or a positive integer. The "agent" is a therapeutic agent, a bioactive agent, a detectable label, water-soluble moiety (e.g., PEG, m-PEG, PPG, and m-PPG) or the like. The "agent" can be a peptide, e.g. , enzyme, antibody, antigen, etc. The linker can be any of a wide array of linking groups, infra. Alternatively, the linker may be a single bond or a "zero order linker."
[0117] In an exemplary embodiment, the selected modifying group is a water-soluble polymer, e.g., m-PEG. The water-soluble polymer is covalently attached to the peptide via a glycosyl linking group. The glycosyl linking group is covalently attached to an amino acid residue or a glycosyl residue of the peptide. The invention also provides conjugates in which an amino acid residue and a glycosyl residue are modified with a glycosyl linking group.
[0118] An exemplary water-soluble polymer is poly(ethylene glycol), e.g., methoxy- poly (ethylene glycol). The poly(ethylene glycol) used in the present invention is not restricted to any particular form or molecular weight range. For unbranched poly(ethylene
glycol) molecules the molecular weight is preferably between 500 and 100,000. A molecular weight of 2000-60,000 is preferably used and preferably of from about 5,000 to about 30,000.
[0119] In another embodiment the poly(ethylene glycol) is a branched PEG having more than one PEG moiety attached. Examples of branched PEGs are described in U.S. Pat. No. 5,932,462; U.S. Pat. No. 5,342,940; U.S. Pat. No. 5,643,575; U.S. Pat. No. 5,919,455; U.S. Pat. No. 6,113,906; U.S. Pat. No. 5,183,660; WO 02/09766; Kodera Y., Bioconjugate Chemistry 5: 283-288 (1994); and Yamasaki et al, Agric. Biol. Chem., 52: 2125-2127, 1998. In a preferred embodiment the molecular weight of each poly(ethylene glycol) of the branched PEG is less than or equal to 40,000 daltons.
[0120] In addition to providing conjugates that are formed through an enzymatically added glycosyl linking group, the present invention provides conjugates that are highly homogenous in their substitution patterns. Using the methods of the invention, it is possible to form peptide conjugates in which essentially all of the modified sugar moieties across a population of conjugates of the invention are attached to a structurally identical amino acid or glycosyl residue. Thus, in a second aspect, the invention provides a peptide conjugate having a population of water-soluble polymer moieties, which are covalently bound to the peptide through a glycosyl linking group, e.g., an intact glycosyl linking group. In a preferred conjugate of the invention, essentially each member of the population is bound via the glycosyl linking group to a glycosyl residue of the peptide, and each glycosyl residue of the peptide to which the glycosyl linking group is attached has the same structure.
[0121] Also provided is a peptide conjugate having a population of water-soluble polymer moieties covalently bound thereto through a glycosyl linking group. In a preferred embodiment, essentially every member of the population of water soluble polymer moieties is bound to an amino acid residue of the peptide via a glycosyl linking group, and each amino acid residue having a glycosyl linking group attached thereto has the same structure.
[0122] The present invention also provides conjugates analogous to those described above in which the peptide is conjugated to a therapeutic moiety, diagnostic moiety, targeting moiety, toxin moiety or the like via an intact glycosyl linking group. Each of the above- recited moieties can be a small molecule, natural polymer (e.g., polypeptide) or synthetic polymer. When the modifying moiety is attached to a sialic acid, it is generally preferred that the modifying moiety is substantially non-fluorescent.
[0123] Essentially any IFN-α peptide or agent, having any sequence, is of use as the peptide component of the conjugates of the present invention. IFN-α is an antiviral glycoprotein that, in humans, is secreted by human primary fibroblasts after induction with virus or double-stranded RNA. IFN-α is a member of a family of approximately twenty peptides of approximately 18kDa. IFN-α is known as a Type I interferon, which bind to the same cellular receptor and elicit similar responses. Type I IFNs inhibit viral replication, increase the lytic potential of NK cells, modulate MHC molecule expression, and inhibit cellular proliferation, among other things. Type I IFN has been used as a therapy for viral infections, particularly hepatitis viruses, and as a therapy for multiple sclerosis. For references relevant to interferon-α, see, Asano, et ah, Eur. J. Cancer, 27(Suppl 4):S21-S25 (1991); Nagy, et al, Anticancer Research, 8(3):467-470 (1988); Dron, et al, J. Biol. Regul Homeost. Agents, 3(1):13-19 (1989); Habib, et al, Am. Surg., 67(3):257-260 (3/2001); and Sugyiama, et al, Eur. J. Biochem., 217:921-927 (1993).
[0124] The present invention further includes a method for remodeling and/or modifying IFN-α. Current compositions of IFN-α are, as described above, useful compounds for both the modulation of aberrant immunological responses and as a therapy for a variety of diseases. However, they are hampered by decreased potency and function, and a limited half- life in the body as compared to natural cytokines including the natural complement of glycosylation.
[0125] IFN-α has been cloned and sequenced. In an exemplary embodiment, IFN-α has an amino acid sequence according to SEQ ID NO:1 and SEQ ID NO:2 (FIG. 1). The present invention is in no way limited to the sequences set forth herein. One of skill in the art will readily appreciate that many variants of IFN-α exist both naturally and as engineered derivatives.
[0126] As discussed above, the conjugates of the invention are formed by the enzymatic attachment of a modified sugar to the glycosylated or unglycosylated IFN-α peptide. The modified sugar, when interposed between the IFN-α peptide and the modifying group on the sugar becomes what may be referred to herein e.g. , as an "intact glycosyl linking group." Using the exquisite selectivity of enzymes, such as glycosyltransferases, the present method provides peptides that bear a desired group at one or more specific locations. Thus, according to the present invention, a modified sugar is attached directly to a selected locus on the IFN-α peptide chain or, alternatively, the modified sugar is appended onto a carbohydrate
moiety of a glycopeptide. Peptides in which modified sugars are bound to both a glycopeptide carbohydrate and directly to an amino acid residue of the IFN-α peptide backbone are also within the scope of the present invention.
[0127] In contrast to known chemical and enzymatic peptide elaboration strategies, the methods of the invention make it possible to assemble peptides and glycopeptides that have a substantially homogeneous derivatization pattern; the enzymes used in the invention are generally selective for a particular amino acid residue or combination of amino acid residues of the IFN-α peptide. The methods are also practical for large-scale production of modified peptides and glycopeptides. Thus, the methods of the invention provide a practical means for large-scale preparation of glycopeptides having preselected uniform derivatization patterns. The methods are particularly well suited for modification of therapeutic peptides, including but not limited to, glycopeptides that are incompletely glycosylated during production in cell culture cells (e.g., mammalian cells, insect cells, plant cells, fungal cells, yeast cells, or prokaryotic cells) or transgenic plants or animals.
[0128] The present invention also provides conjugates of glycosylated and unglycosylated IFN-α peptides with increased therapeutic half-life due to, for example, reduced clearance rate, or reduced rate of uptake by the immune or reticuloendothelial system (RES). Moreover, the methods of the invention provide a means for masking antigenic determinants on peptides, thus reducing or eliminating a host immune response against the peptide. Selective attachment of targeting agents can also be used to target a peptide to a particular tissue or cell surface receptor that is specific for the particular targeting agent.
[0129] In an exemplary embodiment, neither the amino nor the carboxy terminus of the IFN-α peptide is derivatized with a polymeric modifying moiety.
[0130] The peptides of the invention include at least one O-linked or N-linked glycosylation site, which is glycosylated with a glycosyl residue that includes a polymeric modifying moiety, e.g., a PEG moiety. In an exemplary embodiment, the PEG is covalently attached to the peptide via an intact glycosyl linking group. The glycosyl linking group is covalently attached to either an amino acid residue or a glycosyl residue of the peptide. Alternatively, the glycosyl linking group is attached to one or more glycosyl units of a glycopeptide. The invention also provides conjugates in which a glycosyl linking group is attached to both an amino acid residue and a glycosyl residue.
[0131] The PEG moiety is attached to an intact glycosyl linker directly, or via a non- glycosyl linker, e.g., substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl.
[0132] In an exemplary embodiment, the invention utilizes a modified sugar amine that has the formula:
in which J is a glycosyl moiety (e.g., a nucleotide sugar), L is a bond or a linker and R
1 is the modifying group, e.g., a polymeric modifying moiety. Exemplary bonds are those that are formed between an NH
2 moiety on the glycosyl moiety and a group of complementary reactivity on the modifying group. For example, when R
1 includes a carboxylic acid moiety, this moiety may be activated and coupled with the NH
2 moiety on the glycosyl residue affording a bond having the structure NHC(O)R
1. J is preferably a glycosyl moiety that is "intact", not having been degraded by exposure to conditions that cleave the pyranose or furanose structure, e.g. oxidative conditions, e.g., sodium periodate.
[0133] Exemplary linkers include alkyl and heteroalkyl moieties. The linkers include linking groups, for example acyl-based linking groups, e.g., -C(O)NH-, -OC(O)NH-, and the like. The linking groups are bonds formed between components of the species of the invention, e.g., between the glycosyl moiety and the linker (L), or between the linker and the modifying group (R1). Other exemplary linking groups are ethers, thioethers and amines. For example, in one embodiment, the linker is an amino acid residue, such as a glycine residue. The carboxylic acid moiety of the glycine is converted to the corresponding amide by reaction with an amine on the glycosyl residue, and the amine of the glycine is converted to the corresponding amide or urethane by reaction with an activated carboxylic acid or carbonate of the modifying group.
[0134] Another exemplary linker is a PEG moiety, e.g., a PEG moiety that is functionalized with an amino acid residue. The PEG linker is conjugated to the glycosyl group through the amino acid residue at one PEG terminus and bound to R1 through the other PEG terminus. Alternatively, the amino acid residue is bound to R1 and the PEG terminus, which is not bound to the amino acid, is bound to the glycosyl group.
[0135] An exemplary species of NH-L-R1 has the formula:
-NH{C(O)(CH2)aNH}s{C(O)(CH2)b(OCH2CH2)cO(CH2)dNH}tR1 5 in which the indices s and t are independently 0 or 1. The indices a, b and d are independently integers from 0 to 20, and c is an integer from 1 to 2500. Other similar linkers are based on species in which an -NH moiety is replaced by another group, for example, -S, -O or -CH2. As those of skill will appreciate one or more of the bracketed moieties corresponding to indices s and t can be replaced with a substituted or unsubstituted alkyl or heteroalkyl moiety.
[0136] More particularly, the invention utilizes compounds in which NH-L-R1 is: NHC(O)(CH2)aNHC(O)(CH2)b(OCH2CH2)cO(CH2)dNHR1,
NHC(O)(CH2)b(OCH2CH2)cO(CH2)dNHR1 , NHC(O)O(CH2)b(OCH2CH2)cO(CH2)dNHR1 , NH(CH2)aNHC(O)(CH2)b(OCH2CH2)cO(CH2)dNHR1 5 NHC(O)(CH2)aNHR1, NH(CH2)ONHR1, and NHR1. In these formulae, the indices a, b and d are independently selected from the integers from 0 to 20, preferably from 1 to 5. The index c is an integer from 1 to about 2500.
[0137] In an exemplary embodiment, c is selected such that the PEG moiety is approximately 1 kD, 5 kD, 10, kD, 15 kD, 20 kD, 25 kD, 30 kD, 35 kD, 40 kD, 45 kD, 50 kD, 55 kD, or 60 IcD.
[0138] In the discussion that follows, the invention is illustrated by reference to the use of selected derivatives of furanose and pyranose. Those of skill in the art will recognize that the focus of the discussion is for clarity of illustration and that the structures and compositions set forth are generally applicable across the genus of saccharide groups, modified saccharide groups, activated modified saccharide groups and conjugates of modified saccharide groups.
[0139] In an exemplary embodiment, the invention provides a glycopeptide that is conjugated to a polymeric modifying moiety through an intact glycosyl linking group having a formula that is selected from:
In Formula I R
2 is H, CH
2OR
7, COOR
7 or OR
7, in which R
7 represents H, substituted or unsubstituted alkyl or substituted or unsubstituted heteroalkyl. When COOR
7 is a carboxylic acid or carboxylate, both forms are represented by the designation of the single structure COO
" or COOH. In Formulae I and II, the symbols R
3, R
4, R
5, R
6 and R
6' independently represent H, substituted or unsubstituted alkyl, OR
8, NHC(O)R
9. The index d is O or 1. R
8 and R
9 are independently selected from H, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, sialic acid or polysialic acid. At least one of R
3, R
4, R
5, R
6 or R
6 includes the polymeric modifying moiety e.g., PEG, linked through a bond or a linking group. In an exemplary embodiment, R
6 and R
6 , together with the carbon to which they are attached are components of the pyruvyl side chain of sialic acid. In a further exemplary embodiment, this side chain is functionalized with the polymeric modifying moiety. In another exemplary embodiment, R
6 and R
6 , together with the carbon to which they are attached are components of the side chain of sialic acid and the polymeric modifying moiety is a component of R
5.
[0140] In a further exemplary embodiment, the polymeric modifying moiety is bound to the sugar core, generally through a heteroatom, e.g, nitrogen, on the core through a linker, L, as shown below:
(R1)w— L |
R1 is the polymeric moiety and L is selected from a bond and a linking group. The index w represents an integer selected from 1-6, preferably 1-3 and more preferably 1-2. Exemplary linking groups include substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl moieties and sialic acid. An exemplary component of the linker is an acyl moiety.
[0141] An exemplary compound according to the invention has a structure according to Formulae I or II, in which at least one of R2, R3, R4, R5, R6 or R6' has the formula:
I- NH — L — R1
[0142] In another example according to this embodiment at least one of R2, R3, R4, R5, R6 or R6 has the formula:
I NHC(O)(CH2)S— NHC(O) R1
in which s is an integer from O to 20 and R1 is a linear polymeric modifying moiety.
[0143] In an exemplary embodiment, the polymeric modifying moiety-linker construct is a branched structure that includes two or more polymeric chains attached to central moiety. In this embodiment, the construct has the formula:
(R1V L ≥ ς in which R1 and L are as discussed above and w' is an integer from 2 to 6, preferably from 2 to 4 and more preferably from 2 to 3.
[0144] When L is a bond it is formed between a reactive functional group on a precursor of R1 and a reactive functional group of complementary reactivity on the saccharyl core. When L is a non-zero order linker, a precursor of L can be in place on the glycosyl moiety prior to reaction with the R1 precursor. Alternatively, the precursors of R1 and L can be incorporated into a preformed cassette that is subsequently attached to the glycosyl moiety. As set forth herein, the selection and preparation of precursors with appropriate reactive functional groups is within the ability of those skilled in the art. Moreover, coupling the precursors proceeds by chemistry that is well understood in the art.
[0145] In an exemplary embodiment, L is a linking group that is formed from an amino acid, or small peptide (e.g., 1-4 amino acid residues) providing a modified sugar in which the polymeric modifying moiety is attached through a substituted alkyl linker. Exemplary linkers include glycine, lysine, serine and cysteine. The PEG moiety can be attached to the amine moiety of the linker through an amide or urethane bond. The PEG is linked to the sulfur or oxygen atoms of cysteine and serine through thioether or ether bonds, respectively.
[0146] In an exemplary embodiment, R5 includes the polymeric modifying moiety. In another exemplary embodiment, R5 includes both the polymeric modifying moiety and a linker, L, joining the modifying moiety to the remainder of the molecule. As discussed above, L can be a linear or branched structure. Similarly, the polymeric modifying can be branched or linear.
[0147] In one embodiment, the present invention provides an IFN-α conjugate including the moiety:
wherein D is a member selected from -OH and R
1 -L-HN-; G is a member selected from H and R
1 -L- and -C(O)(C
1-C
6)alkyl; R
1 is a moiety including a straight-chain or branched ρoly(ethylene glycol) residue; and L is a linker, e.g., a bond ("zero order"), substituted or unsubstituted alkyl and substituted or unsubstituted heteroalkyl. In exemplary embodiments, when D is OH, G is R
1 -L-, and when G is -C(O)(C
1-C
6)alkyl
5 D is R
1 -L-NH-.
[0148] In another exemplary embodiment, the invention provides a conjugate formed between a modified sugar of the invention and a substrate IFN-α peptide. In this embodiment, the sugar moiety of the modified sugar becomes a glycosyl linking group interposed between the peptide substrate and the modifying group. An exemplary glycosyl linking group is an intact glycosyl linking group, in which the glycosyl moiety or moieties forming the linking group are not degraded by chemical (e.g., sodium metaperiodate) or enzymatic (e.g., oxidase) processes. Selected conjugates of the invention include a modifying group that is attached to the amine moiety of an amino-saccharide, e.g., mannosamine, glucosamine, galactosamine, sialic acid etc. Exemplary modifying group- intact glycosyl linking group cassettes according to this motif are based on a sialic acid structure, such as those having the formulae:
[0149] In the formulae above, R1 and L are as described above. Further detail about the structure of exemplary R1 groups is provided below.
[0150] In still a further exemplary embodiment, the conjugate is formed between a substrate IFN-α and a saccharyl moiety in which the modifying group is attached through a linker at the 6-carbon position of the saccharyl moiety. Thus, illustrative conjugates according to this embodiment have the formula:
in which the radicals are as discussed above. Such saccharyl moieties include, without limitation, glucose, glucosamine, N-acetyl-glucosamine, galactose, galactosamine, N-acetyl- galactosamine, mannose, mannosamine, N-acetyl-mannosamine, and the like.
[0151] Due to the versatility of the methods available for modifying glycosyl residues on a therapeutic peptide such as IFN-α, the glycosyl structures on the peptide conjugates of the invention can have substantially any structure. Moreover, the glycans can be O-linked or N- linked. As exemplified in the discussion below, each of the pyranose and furanose derivatives discussed above can be a component of a glycosyl moiety of a peptide.
[0152] The invention provides an IFN-α conjugate that includes a glycosyl group having the formula:
[0153] In other embodiments, the group has the formula:
[0154] In a still further exemplary embodiment, the group has the formula:
[0155] In yet another embodiment, the group has the formula:
in which the index p represents and integer from 1 to 10; and c is either O or 1.
[0156] In another exemplary embodiment, the IFN-α peptide includes at least one glycosyl linking group including a substructure having the formula:
-(GIcNAc Gal)p R15 ; and I (GIcNAc Gal)p Sia R15
R15 is the modified sialyl residue. The index p is an integer from 1 to 10.
[0157] In an exemplary embodiment, an IFN-α conjugate of the invention includes at least one N-linked glycosyl residue selected from the glycosyl residues set forth below:
"/x/yy (Fuc)t Man (GIcNAc-GaOp-R15'
AA — GIcNAc — GIcNAc — Man
■su\n Man
v/w> (Fuc), Man
I I
AA — GIcNAc — GIcNAc — Man
,ΛΛΛ Man (GIcNAc- Gal)p-R15'
1^ (Fuc), Man (GIcNAc- Gal)p-R15'
AA — GIcNAc — GIcNAc — Man ^w Man (GIcNAc- Gal)p-R15'
Man (GIcNAc-GaOp-R15'
"™ (Fuc),
AA — GIcNAc — GIcNAc — Man
Man (GIcNAc-GaOp-R15' ; and
(GIcNAc-GaOp-R16'
GIcNAc-GaOp-R15'
Man (GIcNAc-GaOp-R15'
^ (Fuc),
AA — GIcNAc — GIcNAc — Man
Man (GIcNAc-GaOp-R15'
(GIcNAc-GaOp-R15'
[0158] In the formulae above, the index t is 0 or 1 and the index p is an integer from 1 to 10. Each symbol R15 independently can represent H, OH (e.g., GaI-OH), a sialyl moiety, a polymer modified sialyl moiety (i.e., glycosyl linking group-polymeric modifying moiety (Sia-L-R1)) or a sialyl moiety to which is bound a polymer modified sialyl moiety (e.g., Sia-Sia-L-R1) ("Sia-Siap"). Exemplary polymer modified saccharyl moieties have a structure according to Formulae I and II and can include either linear or branched modifying groups (such as PEG). An exemplary IFN-α conjugate of the invention will include at least one
glycan having a R15 that includes a structure according to Formulae I or II. The oxygen, with the open valence, of Formulae I and II is preferably attached through a glycosidic linkage to a carbon of a Gal or GaINAc moiety. In a further exemplary embodiment, the oxygen is attached to the carbon at position 3 of a galactose residue. In an exemplary embodiment, the modified sialic acid is linked α2,3-to the galactose residue. In another exemplary embodiment, the sialic acid is linked α2,6-to the galactose residue. In another exemplary embodiment, the amino acid residue is an asparagine residue.
[0159] In another exemplary embodiment, the invention provides an IFN-α conjugate that includes a glycosyl linking group, such as those set forth above, that is covalently attached to an amino acid residue of the peptide. In one embodiment according to this motif, the glycosyl linking moiety is linked to a galactose residue through a Sia residue:
? GaI Sia Sia L R1
An exemplary species according to this motif is prepared by conjugating Sia-L-R1 to a terminal sialic acid of a glycan using an enzyme that forms Sia-Sia bonds, e.g., CST-II, ST8Sia-π, ST8Sia-III and ST8Sia-IV.
[0160] In another exemplary embodiment, the glycans have a formula that is selected from the group:
^Υ" (Fuc), Man
A IA — G IIcNAc — GIcNAc — M I an vΛJ Iv, M Ian GIcNAc — GaI R15 ',
v/w" (FUC)
1 Man GIcNAc — GaI R
15'
AA — GIcNAc — GIcNAc — Man ;
^ΛΛ^ M Ian GIcNAc — GaI — R15
and combinations thereof.
[0161] The glycans of this group generally correspond to those found on an IFN-α peptide that is produced by bacterial (E.coli) cells, mammalian cells, or insect (e.g., Sf-9) cells,
following remodeling according to the methods set forth herein. For example insect-derived IFN-α that is expressed with a tri-mannosyl core is subsequently contacted with a GIcNAc donor and a GIcNAc transferase and a Gal donor and a Gal transferase. Appending GIcNAc and Gal to the tri-mannosyl core is accomplished in either two steps or a single step. A modified sialic acid is added to at least one branch of the glycosyl moiety as discussed herein. Those Gal moieties that are not functionalized with the modified sialic acid are optionally "capped" by reaction with a sialic acid donor in the presence of a sialyl transferase.
[0162] In an exemplary embodiment, at least 60% of terminal Gal moieties in a population of peptides is capped with sialic acid, preferably at least 70%, more preferably, at least 80%, still more preferably at least 90% and even more preferably at least 95%, 96%, 97%, 98% or 99% are capped with sialic acid.
[0163] In each of the formulae above, RI5/R15' is as discussed above. Moreover, an exemplary modified IFN-α conjugate of the invention will include at least one glycan with an R15/R15' moiety having a structure according to Formulae I or II.
[0164] In an exemplary embodiment, the glycosyl linking moiety has a formula which is a member selected from:
in which b is 0 or 1. The index s represents and integer from 1 to 10; and f represents and integer from 1 to 2500. In another exemplary embodiment, s is 1; b is 0 and f is an integer from about 200 to about 500. In another exemplary embodiment, s is 1 ; b is 0 and f is an integer from about 400 to about 500. In an exemplary embodiment, a PEG moiety is selected that has a molecular weight of about 20 kDa, or about 30 kDa, or about 40 kDa, or about 50 kDa, or about 60 kDa. In any of the N-linked structures of glycans herein, the GaINAc can be bound to a Sia. In an exemplary embodiment, Q is selected from H and CH3. In another exemplary embodiment, the amino acid residue is a member selected from asparagine and lysine. In another exemplary embodiment, wherein the amino acid residue is a member selected from serine and threonine. In another exemplary embodiment, the IFN-α peptide has the amino acid sequence of SEQ ID NO.l and SEQ ID NO:2 (FIG. 1). In a still further preferred embodiment, the IFN-α peptide has the amino acid sequence of SEQ ID NO:1 or SEQ ID NO:2 and the glycosyl linking group is directly or indirectly attached to Thr106.
[0165] In another exemplary embodiment, the IFN-α is derived from insect cells, and the peptide is remodeled by adding GIcNAc and Gal to the mannose core and glycoPEGylated using a sialic acid bearing a linear PEG moiety, affording an IFN-α conjugate that includes at least one moiety having the formula:
in which s represents and integer from 1 to 10; and f represents and integer from 1 to 2500. Those of skill in the art will appreciate that these structures can be produced by remodeling and/or glycoPEGylating IFN peptides expressed in systems other than insect cells.
[0166] As discussed herein, the PEG of use in the conjugates of the invention can be linear or branched. An exemplary precursor of use to form the branched conjugates according to this embodiment of the invention has the formula:
R16-X2 X5-C-X3' R17-x4 (V).
[0167] The branched polymer species according to this formula are essentially pure water-soluble polymers. X3 is a moiety that includes an ionizable, e.g., OH, COOH, H2PO4, HSO3, HPO3, and salts thereof, etc.) or other reactive functional group, e.g., infra. C is carbon. X5 is preferably a non-reactive group (e.g., H, unsubstituted alkyl, unsubstituted heteroalkyl), and can be a polymeric arm. R16 and R17 are independently selected polymeric arms, e.g., nonpeptidic, nonreactive polymeric arms (e.g., PEG)). X2 and X4 are linkage fragments that are preferably essentially non-reactive under physiological conditions, which may be the same or different. An exemplary linker includes neither aromatic nor ester moieties. Alternatively, these linkages can include one or more moiety that is designed to degrade under physiologically relevant conditions, e.g., esters, disulfides, etc. X2 and X4 join polymeric arms R16 and R17 to C. When X3 is reacted with a reactive functional group of complementary reactivity on a linker, sugar or linker-sugar cassette, X3 is converted to a component of linkage fragment X3.
[0168] Exemplary linkage fragments for X2, X3 and X4 are independently selected and include S, SC(O)NH, HNC(O)S, SC(O)O, 0, NH, NHC(O), (O)CNH and NHC(O)O, and OC(O)NH, CH2S, CH2O , CH2CH2O, CH2CH2S, (CH2)00, (CH2)OS or (CH2)0Y'-PEG wherein, Y' is S, NH, NHC(O), C(O)NH, NHC(O)O, OC(O)NH, or 0 and o is an integer from 1 to 50. In an exemplary embodiment, the linkage fragments X2 and X4 are different linkage fragments.
[0169] In an exemplary embodiment, the precursor (V), or an activated derivative thereof, is reacted with, and thereby bound to a sugar, an activated sugar or a sugar nucleotide through a reaction between X3 and a group of complementary reactivity on the sugar moiety, e.g., an amine. Alternatively, X3 reacts with a reactive functional group on a precursor to linker, L. One or more of R2, R3, R4, R5 R6 or R6 of Formulae I and II can include the branched polymeric modifying moiety, or this moiety bound through L.
[0170] In an exemplary embodiment, the moiety:
is the linker arm, L. In this embodiment, an exemplary linker is derived from a natural or unnatural amino acid, amino acid analogue or amino acid mimetic, or a small peptide formed from one or more such species. For example, certain branched polymers found in the compounds of the invention have the formula:
[0171] X
a is a linkage fragment that is formed by the reaction of a reactive functional group, e.g., X
3 , on a precursor of the branched polymeric modifying moiety and a reactive functional group on the sugar moiety, or a precursor to a linker. For example, when X
3 is a carboxylic acid, it can be activated and bound directly to an amine group pendent from an amino-saccharide (e.g., Sia, GaINH
2, GIcNH
2, ManNH
2, etc.), forming an X
a that is an amide. Additional exemplary reactive functional groups and activated precursors are
described hereinbelow. The index c represents an integer from 1 to 10. The other symbols have the same identity as those discussed above.
[0172] In another exemplary embodiment, Xa is a linking moiety formed with another linker:
£ xa — 1_1 Xb £
in which X is a second linkage fragment and is independently selected from those groups set forth for Xa, and, similar to L, L1 is a bond, substituted or unsubstituted alkyl or substituted or unsubstituted heteroalkyl.
[0173] Exemplary species for Xa and Xb include S, SC(O)NH3 HNC(O)S, SC(O)O, O, NH, NHC(O), C(O)NH and NHC(O)O, and OC(O)NH.
[0174] In another exemplary embodiment, X is a peptide bond to R17, which is an amino acid, di-peptide (e.g.,, Lys-Lys) or tri-peptide (E.G., Lys-Lys-Lys) in which the alpha-amine moiety(ies) and/or side chain heteroatom(s) are modified with a polymeric modifying moiety.
[0175] In a further exemplary embodiment, the conjugates of the invention include a moiety, e.g., an R15/R15 'moiety that has a formula that is selected from:
in which the identity of the radicals represented by the various symbols is the same as that discussed hereinabove. L
a is a bond or a linker as discussed above for L and L
1, e.g., substituted or unsubstituted alkyl or substituted or unsubstituted heteroalkyl moiety. In an exemplary embodiment, L
a is a moiety of the side chain of sialic acid that is functionalized with the polymeric modifying moiety as shown. Exemplary L
a moieties include substituted or unsubstituted alkyl chains that include one or more OH or NH
2.
[0176] In yet another exemplary embodiment, the invention provides conjugates having a moiety, e.g., an R
15/R
15' moiety with formula:
The identity of the radicals represented by the various symbols is the same as that discussed hereinabove. As those of skill will appreciate, the linker arm in Formulae VII, VIII5 IX and X is equally applicable to other modified sugars set forth herein. In exemplary embodiment, the species of Formulae VII, VIII, IX and X are the R15 moieties attached to the glycan structures set forth herein.
[0177] In yet another exemplary embodiment, the IFN-α conjugate includes an R15 moiety with- the formula:
in which the identities of the radicals are as discussed above. An exemplary species for L
a is -(CH
2)
jC(O)NH(CH
2)
hC(O)NH-, in which h and j are independently selected integers from 0 to 10. A further exemplary species is -C(O)NH-.
[0178] The embodiments of the invention set forth above are further exemplified by reference to species in which the polymer is a water-soluble polymer, particularly pόly(ethylene glycol) ("PEG"), e.g., methoxy-poly(ethylene glycol). Those of skill will appreciate that the focus in the sections that follow is for clarity of illustration and the various motifs set forth using PEG as an exemplary polymer are equally applicable to species in which a polymer other than PEG is utilized.
[0179] PEG of any molecular weight, e.g., 1 kDa, 2 kDa, 5 kDa, 10 kDa, 15 kDa, 20 kDa, 30 kDa, 35 kDa, 40 kDa, 45 kDa, 50 kDa, 55 kDa or 60 kDa is of use in the present invention.
[0180] In an exemplary embodiment, the R15 moiety has a formula that is a member selected from the group:
In each of the structures above, the linker fragment -NH(CH2)a- can be present or absent. The integers a, b and d are independently selected from 1 to 10, preferably from 1 to 5 and more preferably from 2 to 4. The integer c is selected from 1 to 2500, preferably from 1 to 100 and more preferably from 2 to 10.
[0181] In other exemplary embodiments, the conjugate includes an R15 moiety selected from the group:
[0182] In each of the formulae above, the indices e and f are independently selected from the integers from 1 to 2500. In further exemplary embodiments, e and f are selected to provide a PEG moiety that is about 1 kD, 2 kD, 10 kD, 15 kD, 20 kD, 25 kD, 30 kD, 35 kD, 40 IcD, 45 kD, 50 kD, 55 kD, or 60 kD. The symbol Q represents substituted or unsubstituted alkyl (e.g., C1-C6 alkyl, e.g., methyl), substituted or unsubstituted heteroalkyl or H.
[0183] In another exemplary embodiment, the glycosyl group has the formula which is a member selected from:
and
[0184] In each of the formulae above, the indices e and f are independently selected from the integers from 1 to 2500. In further exemplary embodiments, e and fare selected to provide a PEG moiety that is about 1 kD, 2 kD, 10 kD, 15 kD, 20 IdD, 25 kD, 30 kD, 35 kD, 40 kD, 45 kD, 50 kD, 55 kD, or 60 kD. The symbol Q represents substituted or unsubstituted alkyl (e.g., C1-C6 alkyl, e.g., methyl), substituted or unsubstituted heteroalkyl or H. In an
exemplary embodiment, AA is serine or threonine. In another exemplary embodiment, AA is Thr106 from SEQ ID NO: 1 or SEQ ID NO: 2.
[0185] Exemplary branched polymers of use in the conjugates set forth herein include, e.g. :
or di-lysine (Lys-Lys) peptides, e.g.:
or tri-lysine peptides (Lys-Lys-Lys), e.g.:
In each of the figures above, e, f, f and f" represent integers independently selected from 1 to 2500. The indices q, q' and q" represent integers independently selected from 1 to 20.
[0186] Exemplary IFN-α conjugates include a glycosyl moiety selected from the formulae:
; and
[0187] For example:
in which L
a is a bond or a linker as described herein; the index t represents 0 or 1 ; and the index c represents 0 or 1. Each of these groups can be included as components of the mono-, bi-, tri- and tetra-antennary saccharide structures set forth above. In a preferred embodiment,
the amino acid residue is a member selected from asparagine and lysine. In another preferred embodiment, the amino acid residue is a member selected from serine and threonine. In another preferred embodiment, the amino acid residue is Thr
106. In yet another preferred embodiment, the peptide has the amino acid sequence of SEQ. ID NO:1 or SEQ. ID NO:2 (FIG. 1).
[0188] In a further exemplary embodiment, the invention utilizes modified sugars in which the 6-hydroxyl position of the sugar is converted to the corresponding amine moiety, which bears a linker-modifying group cassette such as those set forth above. Exemplary saccharyl groups that can be used as the core of these modified sugars include Gal, GaINAc, GIc, GIcNAc, Fuc, XyI, Man, and the like. A representative modified sugar according to this embodiment has the formula:
in which R
π-R
14 are members independently selected from H, OH, C(O)CH
3, NH, and NH C(O)CH
3. R
10 is a link to another glycosyl residue (-O-glycosyl) or to an amino acid of the IFN-α peptide (-NH-(G-CSF)). R
14 is OR
1, NHR
1 or NH-L-R
1. R
1 and NH-L-R
1 are as described above.
[0189] Selected conjugates according to this motif are based on mannose, galactose or glucose, or on species having the stereochemistry of mannose, galactose or glucose. The general formulae of these conjugates are:
[0190] As discussed above, the invention provides saccharides bearing a modifying group, activated analogues of these species and conjugates formed between species such as peptides and lipids and a modified saccharide of the invention.
Modified Sugars
[0191] The present invention uses modified sugars and modified sugar nucleotides to form conjugates of the modified sugars. In modified sugar compounds of use in the invention, the sugar moiety is preferably a saccharide, a deoxy-saccharide, an amino-saccharide, or an N- acyl saccharide. The term "saccharide" and its equivalents, "saccharyl," "sugar," and "glycosyl" refer to monomers, dimers, oligomers and polymers. The sugar moiety is also functionalized with a modifying group. The modifying group is conjugated to the sugar moiety, typically, through conjugation with an amine, sulfhydryl or hydroxyl, e.g., primary hydroxyl, moiety on the sugar. In an exemplary embodiment, the modifying group is attached through an amine moiety on the sugar, e.g., through an amide, a urethane or a urea that is formed through the reaction of the amine with a reactive derivative of the modifying group.
[0192] Any sugar can be utilized as the sugar core of the glycosyl linking group of the conjugates of the invention. Exemplary sugar cores that are useful in forming the compositions of the invention include, but are not limited to, glucose, galactose, mannose, fucose, and sialic acid. Other useful sugars include amino sugars such as glucosamine, galactosamine, mannosamine, the 5-amine analogue of sialic acid and the like. The sugar core can be a structure found in nature or it can be modified to provide a site for conjugating the modifying group. For example, in one embodiment, the invention provides a sialic acid derivative in which the 9-hydroxy moiety is replaced with an amine. The amine is readily derivatized with an activated analogue of a selected modifying group.
[0193] Exemplary modified sugars are modified with water-soluble or water-insoluble polymers. Examples of useful polymer are further exemplified below.
Water-Soluble Polymers [0194] Many water-soluble polymers are known to those of skill in the art and are useful in practicing the present invention. The term water-soluble polymer encompasses species such as saccharides (e.g., dextran, amylose, hyalouronic acid, poly(sialic acid), heparans, heparins, etc.); poly (amino acids), e.g., poly(asρartic acid) and poly(glutamic acid); nucleic acids; synthetic polymers (e.g., poly(acrylic acid), poly(ethers), e.g., poly(ethylene glycol); peptides, proteins, and the like. The present invention may be practiced with any water- soluble polymer with the sole limitation that the polymer must include a point at which the remainder of the conjugate can be attached.
[0195] Methods for activation of polymers can also be found in WO 94/17039, U.S. Pat. No. 5,324,844, WO 94/18247, WO 94/04193, U.S. Pat. No. 5,219,564, U.S. Pat. No. 5,122,614, WO 90/13540, U.S. Pat. No. 5,281,698, and more WO 93/15189, and for conjugation between activated polymers and peptides, e.g. Coagulation Factor VIII (WO 94/15625), hemoglobin (WO 94/09027), oxygen carrying molecule (U.S. Pat. No.
4,412,989), ribonuclease and superoxide dismutase (Veronese at al, App. Biochem. Biotech. 11: 141-45 (1985)).
[0196] Preferred water-soluble polymers are those in which a substantial proportion of the polymer molecules in a sample of the polymer are of approximately the same molecular weight; such polymers are "homodisperse."
[0197] The present invention is further illustrated by reference to a poly(ethylene glycol) conjugate. Several reviews and monographs on the functionalization and conjugation of PEG are available. See, for example, Harris, Macronol. Chem. Phys. C25: 325-373 (1985); Scouten, Methods in Enzymology 135: 30-65 (1987); Wong et al, Enzyme Microb. Technol. 14: 866-874 (1992); Delgado et al, Critical Reviews in Therapeutic Drug Carrier Systems 9: 249-304 (1992); Zalipsky, Bioconjugate Chem. 6: 150-165 (1995); and Bhadra, et al, Pharmazie, 57:5-29 (2002). Routes for preparing reactive PEG molecules and forming conjugates using the reactive molecules are known in the art. For example, U.S. Patent No. 5,672,662 discloses a water soluble and isolatable conjugate of an active ester of a polymer acid selected from linear or branched poly(alkylene oxides), poly(oxyethylated polyols), poly(olefmic alcohols), and poly(acrylomorpholine).
[0198] U.S. Patent No. 6,376,604 sets forth a method for preparing a water-soluble 1-benzotriazolylcarbonate ester of a water-soluble and non-peptidic polymer by reacting a terminal hydroxyl of the polymer with di(l-benzotriazoyl)carbonate in an organic solvent. The active ester is used to form conjugates with a biologically active agent such as a protein or peptide.
[0199] WO 99/45964 describes a conjugate including a biologically active agent and an activated water soluble polymer including a polymer backbone having at least one terminus linked to the polymer backbone through a stable linkage, wherein at least one terminus includes a branching moiety having proximal reactive groups linked to the branching moiety, in which the biologically active agent is linked to at least one of the proximal reactive groups. Other branched polyethylene glycols) are described in WO 96/21469, U.S. Patent No.
5,932,462 describes a conjugate formed with a branched PEG molecule that includes a branched terminus that includes reactive functional groups. The free reactive groups are available to react with a biologically active species, such as a protein or peptide, forming conjugates between the poly(ethylene glycol) and the biologically active species. U.S. Patent No. 5,446,090 describes a bifunctional PEG linker and its use in forming conjugates having a peptide at each of the PEG linker termini.
[0200] Conjugates that include degradable PEG linkages are described in WO 99/34833 ; and WO 99/14259, as well as in U.S. Patent No. 6,348,558. Such degradable linkages are applicable in the present invention.
[0201] The art-recognized methods of polymer activation set forth above are of use in the context of the present invention in the formation of the branched polymers set forth herein and also for the conjugation of these branched polymers to other species, e.g., sugars, sugar nucleotides and the like.
[0202] The modified sugars are prepared by reacting the glycosyl core (or a linker on the core) with a polymeric modifying moiety (or a linker on the polymeric modifying moiety). The discussion that follows provides examples of selected polymeric modifying moieties of use in the invention. For example, representative polymeric modifying moieties include structures that are based on side chain-containing amino acids, e.g., serine, cysteine, lysine, and small peptides, e.g., lys-lys. Exemplary structures include:
HO' Y "S (CH2CH2O)8CH3 ; Hθ' (CH2CH2O)8CH3
NHC(O)CH
2CH
2(OCH
2CH
2)(OCH
3
H
3 Ha
Those of skill will appreciate that the free amine in the di-lysine structures can also be pegylated through an amdie or urethane bond with a PEG moiety.
[0203] In yet another embodiment, the branched PEG moiety is based upon a tri-lysine peptide. The tri-lysine can be mono-, di-, tri-, or tetra-PEG-ylated. Exemplary species according to this embodiment have the formulae:
in which e, f and f are independently selected integers from 1 to 2500; and q, q' and q" are independently selected integers from 1 to 20.
[0204] As will be apparent to those of skill, the branched polymers of use in the invention include variations on the themes set forth above. For example the di-lysine-PEG conjugate shown above can include three polymeric subunits, the third bonded to the α-amine shown as unmodified in the structure above. Similarly, the use of a tri-lysine functionalized with three or four polymeric subunits labeled with the polymeric modifying moiety in a desired manner is within the scope of the invention.
[0205] The polymeric modifying moieties can be activated for reaction with the glycosyl core. Exemplary structures of activated species (e.g., carbonates and active esters) include:
[0206] Other activating, or leaving groups, appropriate for activating linear and branched PEGs of use in preparing the compounds set forth herein include, but are not limited to the species:
PEG molecules that are activated with these and other species and methods of making the activated PEGs are set forth in WO 04/083259.
[0207] Those of skill in the art will appreciate that one or more of the m-PEG arms of the branched polymers shown above can be replaced by a PEG moiety with a different terminus, e.g., OH, COOH, NH2, C2-Ci0-alkyl, etc. Moreover, the structures above are readily modified by inserting alkyl linkers (or removing carbon atoms) between the α-carbon atom and the functional group of the amino acid side chain. Thus, "homo" derivatives and higher homologues, as well as lower homologues are within the scope of cores for branched PEGs of use in the present invention.
[0208] The branched PEG species set forth herein are readily prepared by methods such as that set forth in the scheme below:
in which X
d is O or S and r is an integer from 1 to 5. The indices e and f are independently selected integers from 1 to 2500. In an exemplary embodiment, one or both of these indices are selected such that the polymer is about 10 kD, 15 kD or 20 kD in molecular weight.
[0209] Thus, according to this scheme, a natural or unnatural amino acid is contacted with an activated m-PEG derivative, in this case the tosylate, forming 1 by alkylating the side- chain heteroatom Xd. The mono-functionalize m-PEG amino acid is submitted to N- acylation conditions with a reactive m-PEG derivative, thereby assembling branched m-PEG 2. As one of skill will appreciate, the tosylate leaving group can be replaced with any suitable leaving group, e.g., halogen, mesylate, triflate, etc. Similarly, the reactive carbonate utilized to acylate the amine can be replaced with an active ester, e.g., N- hydroxysuccinimide, etc., or the acid can be activated in situ using a dehydrating agent such as dicyclohexylcarbodiimide, carbonyldiimidazole, etc.
[0210] In other exemplary embodiments, the urea moiety is replaced by a group such as a amide.
[0211] In an illustrative embodiment, the modified sugar is sialic acid and selected modified sugar compounds of use in the invention have the formulae:
The indices a, b and d are integers from O to 20. The index c is an integer from 1 to 2500. The structures set forth above can be components of R
15.
[0212] In another illustrative embodiment, a primary hydroxyl moiety of the sugar is functionalized with the modifying group. For example, the 9-hydroxyl of sialic acid can be converted to the corresponding amine and functionalized to provide a compound according to the invention. Formulae according to this embodiment include:
The structures set forth above can be components of R15/R15 .
[0213] As those of skill in the art will appreciate, the sialic acid moiety in the exemplary compounds above can be replaced with any other amino-saccharide including, but not limited to, glucosamine, galactosamine, mannosamine, their N-acyl derivatives, and the like.
[0214] Although the present invention is exemplified in the preceding sections by reference to PEG, as those of skill will appreciate, an array of polymeric modifying moieties is of use in the compounds and methods set forth herein.
[0215] In selected embodiments, R1 or L-R1 is a branched PEG, for example, one of the species set forth above. Illustrative modified sugars according to this embodiment include:
in which X
4 is a bond or O. In each of the structures above, the alkylamine linker -(CH
2)
aNH- can be present or absent. The structures set forth above can be components of R
15/R
15'.
[0216] As discussed herein, the polymer-modified sialic acids of use in the invention may also be linear structures. Thus, the invention provides for conjugates that include a sialic acid moiety derived from a structure such as:
in which q and e are as discussed above.
Water-Insoluble Polymers
[0217] In another embodiment, analogous to those discussed above, the modified sugars include a water-insoluble polymer, rather than a water-soluble polymer. The conjugates of the invention may also include one or more water-insoluble polymers. This embodiment of the invention is illustrated by the use of the conjugate as a vehicle with which to deliver a therapeutic peptide in a controlled manner. Polymeric drug delivery systems are known in the art. See, for example, Dunn et al, Eds. POLYMERIC DRUGS AND DRUG DELIVERY SYSTEMS, ACS Symposium Series Vol. 469, American Chemical Society, Washington, D. C. 1991. Those of skill in the art will appreciate that substantially any known drug delivery system is applicable to the conjugates of the present invention.
[0218] The motifs forth above for R1, L-R1, R15, R15' and other radicals are equally applicable to water-insoluble polymers, which may be incorporated into the linear and branched structures without limitation utilizing chemistry readily accessible to those of skill in the art. Similarly, the incorporation of these species into any of the modified sugars discussed herein is within the scope of the present invention. Accordingly, the invention provides conjugates containing, and for the use of to prepare such conjugates, sialic acid and other sugar moieties modified with a linear or branched water-insoluble polymers, and activated analogues of the modified sialic acid species (e.g., CMP-Sia-(water insoluble polymer)).
[0219] Representative water-insoluble polymers include, but are not limited to, polyphosphazines, poly( vinyl alcohols), polyamides, polycarbonates, polyalkylenes, polyacrylamides, polyalkylene glycols, polyalkylene oxides, polyalkylene terephthalates, polyvinyl ethers, polyvinyl esters, polyvinyl halides, polyvinylpyrrolidone, polyglycolides, polysiloxanes, polyurethanes, poly(methyl methacrylate), poly(ethyl methacrylate), poly(butyl methacrylate), poly(isobutyl methacrylate), poly(hexyl methacrylate), poly(isodecyl methacrylate), poly(lauryl methacrylate), poly(phenyl methacrylate), poly(methyl acrylate), poly(isopropyl acrylate), poly(isobutyl acrylate), poly(octadecyl acrylate) polyethylene, polypropylene, poly(ethylene glycol), poly(ethylene oxide), poly (ethylene terephthalate), poly(vinyl acetate), polyvinyl chloride, polystyrene, polyvinyl pyrrolidone, pluronics and polyvinylphenol and copolymers thereof.
[0220] Synthetically modified natural polymers of use in conjugates of the invention include, but are not limited to, alkyl celluloses, hydroxyalkyl celluloses, cellulose ethers, cellulose esters, and nitrocelluloses. Particularly preferred members of the broad classes of
synthetically modified natural polymers include, but are not limited to, methyl cellulose, ethyl cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose, hydroxybutyl methyl cellulose, cellulose acetate, cellulose propionate, cellulose acetate butyrate, cellulose acetate phthalate, carboxymethyl cellulose, cellulose triacetate, cellulose sulfate sodium salt, and polymers of acrylic and methacrylic esters and alginic acid.
[0221] These and the other polymers discussed herein can be readily obtained from commercial sources such as Sigma Chemical Co. (St. Louis, MO.), Polysciences (Warrenton, PA.), Aldrich (Milwaukee, WL), Fluka (Ronkonkoma, NY), and BioRad (Richmond, CA), or else synthesized from monomers obtained from these suppliers using standard techniques.
[0222] Representative biodegradable polymers of use in the conjugates of the invention include, but are not limited to, polylactides, polyglycolides and copolymers thereof, poly(ethylene terephthalate), poly(butyric acid), poly(valeric acid), poly(lactide-co- caprolactone), poly(lactide-co-glycolide), polyanhydrides, polyorthoesters, blends and copolymers thereof. Of particular use are compositions that form gels, such as those including collagen, pluronics and the like.
[0223] The polymers of use in the invention include "hybrid' polymers that include water- insoluble materials having within at least a portion of their structure, a bioresorbable molecule. An example of such a polymer is one that includes a water-insoluble copolymer, which has a bioresorbable region, a hydrophilic region and a plurality of crosslinkable functional groups per polymer chain.
[0224] For purposes of the present invention, "water-insoluble materials" includes materials that are substantially insoluble in water or water-containing environments. Thus, although certain regions or segments of the copolymer may be hydrophilic or even water- soluble, the polymer molecule, as a whole, does not to any substantial measure dissolve in water.
[0225] For purposes of the present invention, the term "bioresorbable molecule" includes a region that is capable of being metabolized or broken down and resorbed and/or eliminated through normal excretory routes by the body. Such metabolites or break down products are preferably substantially non-toxic to the body.
[0226] The bioresorbable region may be either hydrophobic or hydrophilic, so long as the copolymer composition as a whole is not rendered water-soluble. Thus, the bioresorbable
region is selected based on the preference that the polymer, as a whole, remains water- insoluble. Accordingly, the relative properties, i.e., the kinds of functional groups contained by, and the relative proportions of the bioresorbable region, and the hydrophilic region are selected to ensure that useful bioresorbable compositions remain water-insoluble.
[0227] Exemplary resorbable polymers include, for example, synthetically produced resorbable block copolymers of poly(α-hydroxy-carboxylic acid)/poly(oxyalkylene, (see, Cohn et al., U.S. Patent No. 4,826,945). These copolymers are not crosslinked and are water- soluble so that the body can excrete the degraded block copolymer compositions. See, Younes et al., JBiomed. Mater. Res. 21: 1301-1316 (1987); and Cohn et al, JBiomed. Mater. Res. 22: 993-1009 (1988).
[0228] Presently preferred bioresorbable polymers include one or more components selected from poly(esters), poly(hydroxy acids), poly(lactones), poly(amides), poly(ester- amides), poly (amino acids), poly(anhydrides), poly(orthoesters), poly(carbonates), poly(phosphazines), poly(phosphoesters), poly(thioesters), polysaccharides and mixtures thereof. More preferably still, the biosresorbable polymer includes a poly(hydroxy) acid component. Of the poly(hydroxy) acids, polylactic acid, polyglycolic acid, polycaproic acid, polybutyric acid, polyvaleric acid and copolymers and mixtures thereof are preferred.
[0229] In addition to forming fragments that are absorbed in vivo ("bioresorbed"), preferred polymeric coatings for use in the methods of the invention can also form an excretable and/or metabolizable fragment.
[0230] Higher order copolymers can also be used in the present invention. For example, Casey et al., U.S. Patent No. 4,438,253, which issued on March 20, 1984, discloses tri-block copolymers produced from the transesterification of poly(glycolic acid) and an hydroxyl- ended poly(alkylene glycol). Such compositions are disclosed for use as resorbable monofilament sutures. The flexibility of such compositions is controlled by the incorporation of an aromatic orthocarbonate, such as tetra-p-tolyl orthocarbonate into the copolymer structure.
[0231] Other polymers based on lactic and/or glycolic acids can also be utilized. For example, Spinu, U.S. Patent No. 5,202,413, which issued on April 13, 1993, discloses biodegradable multi-block copolymers having sequentially ordered blocks of polylactide and/or polyglycolide produced by ring-opening polymerization of lactide and/or glycolide
onto either an oligomeric diol or a diamine residue followed by chain extension with a di- functional compound, such as, a diisocyanate, diacylchloride or dichlorosilane.
[0232] Bioresorbable regions of coatings useful in the present invention can be designed to be hydrolytically and/or enzymatically cleavable. For purposes of the present invention, "hydrolytically cleavable" refers to the susceptibility of the copolymer, especially the bioresorbable region, to hydrolysis in water or a water-containing environment. Similarly, "enzymatically cleavable" as used herein refers to the susceptibility of the copolymer, especially the bioresorbable region, to cleavage by endogenous or exogenous enzymes.
[0233] When placed within the body, the hydrophilic region can be processed into excretable and/or metabolizable fragments. Thus, the hydrophilic region can include, for example, polyethers, polyalkylene oxides, polyols, poly(vinyl pyrrolidine), poly(vinyl alcohol), poly(alkyl oxazolines), polysaccharides, carbohydrates, peptides, proteins and copolymers and mixtures thereof. Furthermore, the hydrophilic region can also be, for example, a poly(alkylene) oxide. Such poly(alkylene) oxides can include, for example, poly(ethylene) oxide, poly(propylene) oxide and mixtures and copolymers thereof.
[0234] Polymers that are components of hydrogels are also useful in the present invention. Hydrogels are polymeric materials that are capable of absorbing relatively large quantities of water. Examples of hydrogel forming compounds include, but are not limited to, polyacrylic acids, sodium carboxymethylcellulose, polyvinyl alcohol, polyvinyl pyrrolidine, gelatin, carrageenan and other polysaccharides, hydroxyethylenemethacrylic acid (HEMA), as well as derivatives thereof, and the like. Hydrogels can be produced that are stable, biodegradable and bioresorbable. Moreover, hydrogel compositions can include subunits that exhibit one or more of these properties.
[0235] Bio-compatible hydrogel compositions whose integrity can be controlled through crosslinking are known and are presently preferred for use in the methods of the invention. For example, Hubbell et ah, U.S. Patent Nos. 5,410,016, which issued on April 25, 1995 and 5,529,914, which issued on June 25, 1996, disclose water-soluble systems, which are crosslinked block copolymers having a water-soluble central block segment sandwiched between two hydrolytically labile extensions. Such copolymers are further end-capped with photopolymerizable acrylate functionalities. When crosslinked, these systems become hydrogels. The water soluble central block of such copolymers can include poly(ethylene glycol); whereas, the hydrolytically labile extensions can be a poly(α-hydroxy acid), such as
polyglycolic acid or polylactic acid. See, Sawhney et al, Macromolecules 26: 581-587 (1993).
[0236] In another preferred embodiment, the gel is a thermoreversible gel. Thermoreversible gels including components, such as pluronics, collagen, gelatin, hyalouronic acid, polysaccharides, polyurethane hydrogel, polyurethane-urea hydrogel and combinations thereof are presently preferred.
[0237] In yet another exemplary embodiment, the conjugate of the invention includes a component of a liposome. Liposomes can be prepared according to methods known to those skilled in the art, for example, as described in Eppstein et ah, U.S. Patent No. 4,522,811. For example, liposome formulations may be prepared by dissolving appropriate lipid(s) (such as stearoyl phosphatidyl ethanolamine, stearoyl phosphatidyl choline, arachadoyl phosphatidyl choline, and cholesterol) in an inorganic solvent that is then evaporated, leaving behind a thin film of dried lipid on the surface of the container. An aqueous solution of the active compound or its pharmaceutically acceptable salt is then introduced into the container. The container is then swirled by hand to free lipid material from the sides of the container and to disperse lipid aggregates, thereby forming the liposomal suspension.
[0238] The above-recited microparticles and methods of preparing the microparticles are offered by way of example and they are not intended to define the scope of microparticles of use in the present invention. It will be apparent to those of skill in the art that an array of microparticles, fabricated by different methods, is of use in the present invention.
[0239] The structural formats discussed above in the context of the water-soluble polymers, both straight-chain and branched are generally applicable with respect to the water- insoluble polymers as well. Thus, for example, the cysteine, serine, dilysine, and trilysine branching cores can be functionalized with two water-insoluble polymer moieties. The methods used to produce these species are generally closely analogous to those used to produce the water-soluble polymers.
The Methods
[0240] In addition to the conjugates discussed above, the present invention provides methods for preparing these and other conjugates. Moreover, the invention provides methods of preventing, curing or ameliorating a disease state by administering a conjugate of the invention to a subject at risk of developing the disease or a subject that has the disease.
[0241] In another exemplary embodiment, the invention provides a method of preparing an IFN-α conjugate, the method includes (a) contacting a substrate IFN-α peptide including a glycosyl moiety selected from:
> GaINAc ζ GaI (Sia)c
^ ; and ^ with a PEG-sialic acid donor having the formula:
wherein the index c is O or 1 ; (b) contacting the IFN-α peptide and the PEG-sialic acid donor with an enzyme that transfers PEG-sialic acid from the donor onto the glycosyl moiety, under conditions appropriate for the transfer.
[0242] In another exemplary embodiment, prior to step (a): there is a step involving expressing the substrate IFN-α peptide in a suitable host. In another exemplary embodiment, the host is selected from a bacterial cell, an insect cell and a mammalian cell. In another exemplary embodiment, the host is a bacterial cell and the bacterial cell is an E. coli cell line. In another exemplary embodiment, the host is an insect cell and the insect cell is a Spodopterafrugiperda cell line.
[0243] In exemplary embodiments, the conjugate is formed between a polymeric modifying moiety and a glycosylated or non-glycosylated peptide. The polymer is conjugated to the peptide via a glycosyl linking group, which is interposed between, and covalently linked to both the peptide (or glycosyl residue) and the modifying group (e.g., water-soluble polymer). The method includes contacting the peptide with a mixture containing a modified sugar and an enzyme, e.g., a glycosyltransferase that conjugates the modified sugar to the substrate. The reaction is conducted under conditions appropriate to form a covalent bond between the modified sugar and the peptide. The sugar moiety of the modified sugar is preferably selected from nucleotide sugars.
[0244] In an exemplary embodiment, the modified sugar, such as those set forth above, is activated as the corresponding nucleotide sugars. Exemplary sugar nucleotides that are used
in the present invention in their modified form include nucleotide mono-, di- or triphosphates or analogs thereof. In a preferred embodiment, the modified sugar nucleotide is selected from a UDP-glycoside, CMP-glycoside, or a GDP-glycoside. Even more preferably, the sugar nucleotide portion of the modified sugar nucleotide is selected from UDP-galactose, UDP-galactosamine, UDP-glucose, UDP-glucosamine, GDP-mannose, GDP-fucose, CMP- sialic acid, or CMP-NeuAc. In an exemplary embodiment, the nucleotide phosphate is attached to C-I.
[0245] Thus, in an illustrative embodiment in which the glycosyl moiety is sialic acid, the method of the invention utilizes compounds having the formulae:
in which L-R
1 is as discussed above, and L
1 -R
1 represents a linker bound to the modifying group. As with L, exemplary linker species according to L
1 include a bond, alkyl or heteroalkyl moieties.
[0246] Moreover, as discussed above, the present invention provides for the use of nucleotide sugars that are modified with a water-soluble (or -insoluble) polymer, which is either straight-chain or branched. For example, compounds having the formula shown below are of use to prepare conjugates within the scope of the present invention:
in which X
4 is O or a bond.
[0247] The invention also provides for the use of sugar nucleotides modified with L-R1 at the 6-carbon position. Exemplary species according to this embodiment include:
in which the R groups, and L, represent moieties as discussed above. The index "y" is 0, 1 or 2. In an exemplary embodiment, L is a bond between NH and R
1. The base is a nucleic acid base.
[0248] Exemplary nucleotide sugars of use in the invention in which the carbon at the 6- position is modified include species having the stereochemistry of GDP mannose, e.g.:
in which X
5 is a bond or O. The index i represents 0 or 1. The index a represents an integer from 1 to 20. The indices e and f independently represent integers from 1 to 2500. Q, as discussed above, is H or substituted or unsubstituted C
1-C
6 alkyl. As those of skill will
appreciate, the serine derivative, in which S is replaced with O also falls within this general motif.
[0249] In a still further exemplary embodiment, the invention provides a conjugate in which the modified sugar is based on the stereochemistry of UDP galactose. An exemplary nucleotide sugar of use in this invention has the structure:
Those of skill will appreciate that other linear and brached polymer species discussed herein can take the place of the exemplary PEG moiety.
[0250] In another exemplary embodiment, the nucleotide sugar is based on the stereochemistry of glucose. Exemplary species according to this embodiment have the formulae:
Those of skill will appreciate that other linear and brached polymer species discussed herein can take the place of the exemplary PEG moiety.
[0251] In general, the sugar moiety or sugar moiety-linker cassette and the PEG or PEG- linker cassette groups are linked together through the use of reactive groups, which are typically transformed by the linking process into a new organic functional group or unreactive species. The sugar reactive functional group(s), is located at any position on the sugar moiety. Reactive groups and classes of reactions useful in practicing the present invention are generally those that are well known in the art of bioconjugate chemistry.
Currently favored classes of reactions available with reactive sugar moieties are those, which proceed under relatively mild conditions. These include, but are not limited to nucleophilic substitutions {e.g., reactions of amines and alcohols with acyl halides, active esters), electrophilic substitutions {e.g., enamine reactions) and additions to carbon-carbon and carbon-heteroatom multiple bonds {e.g. , Michael reaction, Diels- Alder addition). These and other useful reactions are discussed in, for example, March, ADVANCED ORGANIC CHEMISTRY, 3rd Ed., John Wiley & Sons, New York, 1985; Hermanson, BIOCONJUGATE TECHNIQUES, Academic Press, San Diego, 1996; and Feeney et al, MODIFICATION OF
PROTEINS; Advances in Chemistry Series, Vol. 198, American Chemical Society, Washington, D.C., 1982.
[0252] Useful reactive functional groups pendent from a sugar nucleus or modifying group include, but are not limited to:
(a) carboxyl groups and various derivatives thereof including, but not limited to,
N-hydroxysuccinimide esters, N-hydroxybenztriazole esters, acid halides, acyl imidazoles, thioesters, p-nitrophenyl esters, alkyl, alkenyl, alkynyl and aromatic esters;
(b) hydroxyl groups, which can be converted to, e.g., esters, ethers, aldehydes, etc. (c) haloalkyl groups, wherein the halide can be later displaced with a nucleophilic group such as, for example, an amine, a carboxylate anion, thiol anion, carbanion, or an alkoxide ion, thereby resulting in the covalent attachment of a new group at the functional group of the halogen atom;
(d) dienophile groups, which are capable of participating in Diels-Alder reactions such as, for example, maleimido groups;
(e) aldehyde or ketone groups, such that subsequent derivatization is possible via formation of carbonyl derivatives such as, for example, imines, hydrazones, semicarbazones or oximes, or via such mechanisms as Grignard addition or alkyllithium addition; (f) sulfonyl halide groups for subsequent reaction with amines, for example, to form sulfonamides; (g) thiol groups, which can be, for example, converted to disulfides or reacted with acyl halides;
(h) amine or sulfhydryl groups, which can be, for example, acylated, alkylated or oxidized;
(i) alkenes, which can undergo, for example, cycloadditions, acylation, Michael addition, etc; and Q) epoxides, which can react with, for example, amines and hydroxyl compounds.
[0253] The reactive functional groups can be chosen such that they do not participate in, or interfere with, the reactions necessary to assemble the reactive sugar nucleus or modifying group. Alternatively, a reactive functional group can be protected from participating in the reaction by the presence of a protecting group. Those of skill in the art understand how to
protect a particular functional group such that it does not interfere with a chosen set of reaction conditions. For examples of useful protecting groups, see, for example, Greene et al, PROTECTIVE GROUPS IN ORGANIC SYNTHESIS, John Wiley & Sons, New York, 1991.
[0254] In the discussion that follows, a number of specific examples of modified sugars that are useful in practicing the present invention are set forth. In the exemplary embodiments, a sialic acid derivative is utilized as the sugar nucleus to which the modifying group is attached. The focus of the discussion on sialic acid derivatives is for clarity of illustration only and should not be construed to limit the scope of the invention. Those of skill in the art will appreciate that a variety of other sugar moieties can be activated and derivatized in a manner analogous to that set forth using sialic acid as an example. For example, numerous methods are available for modifying galactose, glucose, N- acetylgalactosamine and fucose to name a few sugar substrates, which are readily modified by art recognized methods. See, for example, Elhalabi et al., Curr. Med. Chem. 6: 93 (1999); and Schafer et al., J. Org. Chem. 65: 24 (2000)).
[0255] In an exemplary embodiment, the modified sugar is based upon a 6-amino-N- acetyl-glycosyl moiety. As shown in FIG. 4 for N-acetylgalactosamine, the 6-amino-sugar moiety is readily prepared by standard methods.
[0256] In the scheme above, the index n represents an integer from 1 to 2500. In an exemplary embodiment, this index is selected such that the polymer is about 10 kD, 15 kD or 20 kD in molecular weight. The symbol "A" represents an activating group, e.g., a halo, a component of an activated ester (e.g., a N-hydroxysuccinimide ester), a component of a carbonate (e.g., p-nitrophenyl carbonate) and the like. Those of skill in the art will appreciate that other PEG-amide nucleotide sugars are readily prepared by this and analogous methods.
[0257] The acceptor peptide is typically synthesized de novo, or recombinantly expressed in a prokaryotic cell (e.g. , bacterial cell, such as E. coli) or in a eukaryotic cell such as a mammalian, yeast, insect, fungal or plant cell. The peptide can be either a full-length protein or a fragment. Moreover, the peptide can be a wild type or mutated peptide. In an exemplary embodiment, the peptide includes a mutation that adds one or more N- or O-linked glycosylation sites to the peptide sequence.
[0258] The method of the invention also provides for modification of incompletely glycosylated peptides that are produced recombinantly. Many recombinantly produced glycoproteins are incompletely glycosylated, exposing carbohydrate residues that may have
undesirable properties, e.g., immunogenicity, recognition by the RES. Employing a modified sugar in a method of the invention, the peptide can be simultaneously further glycosylated and derivatized with, e.g., a water-soluble polymer, therapeutic agent, or the like. The sugar moiety of the modified sugar can be the residue that would properly be conjugated to the acceptor in a fully glycosylated peptide, or another sugar moiety with desirable properties.
[0259] Those of skill will appreciate that the invention can be practiced using substantially any peptide or glycopeptide from any source. Exemplary peptides with which the invention can be practiced are set forth in WO 03/031464, and the references set forth therein.
[0260] The present invention contemplates the use of IFN-α peptides in which the attachment site of the glycan chain(s) on the peptide have been altered from that of the native peptide. These IFN-α peptides can be altered to create or eliminate N-linked glycosylation sites. IFN-α can also be altered to create or eliminate N-linked glycosylation sites. It is possible for an IFN-α peptide to have one or more N-linked glycosylation sites created or eliminated and one or more 0-linked glycosylation sites created or eliminated.
The Peptides
[0261] IFN-α has been cloned and sequenced. In an exemplary embodiment, IFN-α has an amino acid sequence according to SEQ. ID NO.l or SEQ. ID NO:2 (FIG. 1). The present invention is in no way limited to the sequences set forth herein. One of skill in the art will readily appreciate that many variants of IFN-α exist both naturally and as engineered derivatives. Examples of modified IFN-α are well known in the art (see Table 1), and are described in, for example. U.S. Pat. Nos. 4,966,843, 5,376,567, 5,795,779 describe IFN-α-61 and IFN-α-76. U.S. Patent Nos. 4,748,233 and 4,695,543 describe IFN-α gx-1, whereas U.S. Patent No. 4,975,276 describes IFN-α-54. In addition, U.S. Patent Nos. 4,695,623, 4,897,471, 5,661,009, and 5,541,293 describe a consensus IFN-α sequence. Additional examples of IFN-α peptides are found in U.S. Pat. App. No. 60/620,927 ("Branched PEG Remodeling and Glycosylation of Interferon Alpha", filed Oct. 21, 2004) and PCT App. No. PCT/US2005/000799 ("O-linked Glycosylation of Peptides", filed Jan. 10, 2005). This list of IFN-α and variants thereof is not exhaustive, and one of skill in the art will readily understand that the present invention encompasses IFN-α molecules, derivatives, and variants known or to be discovered in the future. IFN-α sequences can also contain a methionine at the 1- position; in these cases, the amino acid references in this application are adjusted
accordingly. For example, for SEQ. ID NO:1 or SEQ. ID NO:2, Thr106 would be understood to be Thr107 if a methionine were at the 1 -position.
Table 1. IFN-α Isoforms. α type AA characteristic
Ib V114
2a K23; H34
2b R23; H34
2c R23; R34
4a A51; E114
4b T51; V114
7a M132; K159; G161
7b M132; Q159; R161
7c T132; K159; G161
8a V98; L"; C100; D101; R161
8b S98; C"; V100; M101; R161
8c S98; C"; V100; M101; D161Δ(162-166)
10a S8; L89
10b T8; I89
14a F152; Q159; R161
14b F152; K159; G161
14c L152; Q159; R161
17a P34; S55; I161
17b H34; S55; I161
17c H34; S55; R161
17d H34; P55; R161
21a M96
21b L96
[0262] Methods of expressing interferons, such as IFN-α, in recombinant cells are well known in the art, and is easily accomplished using techniques described in, for example U.S. Patent No. 4,966,843, and in Sambrook et al. (2001, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York) and Ausubel et al. (1997, Current Protocols in Molecular Biology, Green & Wiley, New York). Assays to determine the
biological activity of a Type I IFN modified by the present invention will be well known to the skilled artisan. For example, the assay described in Rubinstein et al., (1981, Journal of Virology 37:755-758) is commonly used to determine the effect of an Type I IFN by measuring the cytopathic effects of viral infection on a population of cells. This method is only one of many known in the art for assaying the biological function of a Type I IFN.
Creation of N-linked glycosylation sites
[0263] Typically, N-linked glycan chains are linked to the primary peptide structure at asparagine residues where the asparagine residue is within an amino acid sequence that is recognized by a membrane-bound glycosyltransferase in the endoplasmic reticulum (ER). Typically, the recognition site on the primary peptide structure is the sequence asparagine-X- serine/threonine where X can be any amino acid except proline and aspartic acid. While this recognition site is typical, the invention further encompasses peptides that have N-linked glycan chains at other recognition sites where the N-linked chains are added using natural or recombinant glycosyltransferases.
[0264] Since the recognition site for N-linked glycosylation of a peptide is known, it is within the skill of persons in the art to create mutated primary peptide sequences wherein a native N-linked glycosylation recognition site is created. Most simply, an asparagine residue can be added to the primary sequence of the peptide thereby adding an attachment site for a glycan. For example, a native site with the sequence of leucine-serine-serine can be modified to asparagine-serine-serine, thus adding a N-linked glycosylation site at this position.
[0265] In the case of N-linked glycosylation sites including residues other than the typical recognition sites described above, the skilled artisan can determine the sequence and residues required for recognition by the appropriate glycosyltransferase, and then mutate at least one residue so the appropriate glycosyltransferase recognizes that site. In other words, it is well within the skill of the artisan to manipulate the primary sequence of a peptide such that N- linked glycosylation sites are created, thereby generating a peptide having an altered glycosylation pattern. The invention should therefore not be construed to be limited to any primary peptide sequence provided herein as the sole sequence for glycan remodeling, but rather should be construed to include any and all peptide sequences suitable for glycan remodeling, glycoconjugation, glycoPEGylation and the like.
[0266] To create a mutant peptide, the nucleic acid sequence encoding the primary sequence of the peptide is altered so that native codons encoding native amino acid residues
are mutated to generate a codon encoding another amino acid residue. Techniques for altering nucleic acid sequence are common in the art and are described for example in any well-known molecular biology manual.
[0267] In addition, the nucleic acid encoding a primary peptide structure can be synthesized in vitro, using standard techniques. For example, a nucleic acid molecule can be synthesized in a "gene machine" using protocols such as the phosphoramidite method. If chemically- synthesized double stranded DNA is required for an application such as the synthesis of a nucleic acid or a fragment thereof, then each complementary strand is synthesized separately. The production of short nucleic acids (60 to 80 base pairs) is technically straightforward and can be accomplished by synthesizing the complementary strands and then annealing them. For the production of longer nucleic acids (>300 base pairs), special strategies may be required, because the coupling efficiency of each cycle during chemical DNA synthesis is seldom 100%. To overcome this problem, synthetic genes (double- stranded) are assembled in modular form from single-stranded fragments that are from 20 to 100 nucleotides in length. For reviews on polynucleotide synthesis, see, for example, Glick and Pasternak (Molecular Biotechnology, Principles and Applications of Recombinant DNA, 1994, ASM Press), Itakura et al. (1984, Annu. Rev. Biochem. 53:323), and Climie et al. (1990, Proc. Nat'lAcad. ScL USA 87:633).
[0268] Additionally, changes in the nucleic acid sequence encoding the peptide can be made by site-directed mutagenesis. As will be appreciated, this technique typically employs a phage vector existing in both a single stranded and double stranded form. Typical vectors useful in site-directed mutagenesis include vectors such as the Ml 3 phage. These phage are readily available and their use is generally well known to those skilled in the art. Double stranded plasmids are also routinely employed in site-directed mutagenesis which eliminates the step of transferring the nucleic acid of interest from a plasmid to a phage.
[0269] In general, site-directed mutagenesis is performed by first obtaining a single- stranded vector or melting the two strands of a double stranded vector including within its sequence a DNA sequence which encodes the desired peptide. An oligonucleotide primer bearing the desired mutated sequence is prepared generally synthetically. This primer is then annealed with the single-stranded vector, and subjected to DNA polymerizing enzymes such as E. coli polymerase I Klenow fragment, in order to complete the synthesis of the mutation- bearing strand. Thus, a heteroduplex is formed wherein one strand encodes the original non-
mutated sequence and the second strand bears the desired mutation. This heteroduplex vector is then used to transform or transfect appropriate cells, such as E. coli cells, and clones are selected which include recombinant vectors bearing the mutated sequence arrangement. A genetic selection scheme was devised by Kunkel et al. (1987, Kunkel et al., Methods Enzymol. 154:367-382) to enrich for clones incorporating the mutagenic oligonucleotide. Alternatively, the use of PCR™ with commercially available thermostable enzymes such as Taq polymerase may be used to incorporate a mutagenic oligonucleotide primer into an amplified DNA fragment that can then be cloned into an appropriate cloning or expression vector. The PCR™-mediated mutagenesis procedures of Tomic et al. (1990, Nucl. Acids Res., 12:1656) and Upender et al. (1995, Biotechniques, 18:29-31) provide two examples of such protocols. A PCR™ employing a thermostable ligase in addition to a thermostable polymerase may also be used to incorporate a phosphorylated mutagenic oligonucleotide into an amplified DNA fragment that may then be cloned into an appropriate cloning or expression vector. The mutagenesis procedure described by Michael (1994, Biotechniques 16:410-412) provides an example of one such protocol.
[0270] Not all Asn-X-Ser/Thr sequences are N-glycosylated suggesting the context in which the motif is presented is important. In another approach, libraries of mutant peptides having novel N-linked consensus sites are created in order to identify novel N-linked sites that are glycosylated in vivo and are beneficial to the activity, stability or other characteristics of the peptide.
[0271] As noted previously, the consensus sequence for the addition of N-linked glycan chains in glycoproteins is Asn-X-Ser/Thr where X can be any amino acid. The nucleotide sequence encoding the amino acid two positions to the carboxyl terminal side of the Asn may be mutated to encode a Ser and/or Thr residue using standard procedures known to those of ordinary skill in the art. As stated above not all Asn-X-Ser/Thr sites are modified by the addition of glycans. Therefore, each recombinant mutated glycoprotein must be expressed in a fungal, yeast or animal or mammalian expression system and analyzed for the addition of an N-linked glycan chain. The techniques for the characterization of glycosylation sites are well known to one skilled in the art. Further, the biological function of the mutated recombinant glycoprotein can be determined using assays standard for the particular protein being examined. Thus, it becomes a simple matter to manipulate the primary sequence of a peptide and identify novel glycosylation sites contained therein, and further determine the effect of the novel site on the biological activity of the peptide.
[0272] In an alternative embodiment, the nucleotide sequence encoding the amino acid two positions to the amino terminal side of Ser/Thr residues may be mutated to encode an Asn using standard procedures known to those of ordinary skill in the art. The procedures to determine whether a novel glycosylation site has been created and the effect of this site on the biological activity of the peptide are described above.
Creation or elimination of O-linked glycosylation sites
[0273] O-linked glycosylation refers to the attachment of one or more sugars {e.g., N- acetylgalactosamine, galactose, GaINAc-GaI, mannose, GIcNAc, glucose, fucose or xylose) to the hydroxy side chain of a hydroxyamino acid, preferably serine or threonine, although unusual or non-natural amino acids, e.g., 5-hydroxyproline or 5-hydroxylysine may also be used. The addition of an O-linked glycosylation site to a peptide is conveniently accomplished by altering the primary amino acid sequence of the peptide such that it contains one or more additional O-linked glycosylation sites compared with the beginning primary amino acid sequence of the peptide. The addition of an O-linked glycosylation site to the peptide may also be accomplished by incorporation of one or more amino acid species into the peptide which includes an -OH group, preferably serine or threonine residues, within the sequence of the peptide, such that the OH group is accessible and available for O-linked glycosylation. Similar to the discussion of alteration of N-linked glycosylation sites in a peptide, the primary amino acid sequence of the peptide is preferably altered at the nucleotide level. Specific nucleotides in the DNA sequence encoding the peptide may be altered such that a desired amino acid is encoded by the sequence. Mutation(s) in DNA are preferably made using methods known in the art, such as the techniques of phosphoramidite method DNA synthesis and site-directed mutagenesis described above.
[0274] Alternatively, the nucleotide sequence encoding a putative site for O-linked glycan addition can be added to the DNA molecule in one or several copies to either 5' or the 3' end of the molecule. The altered DNA sequence is then expressed in any one of a fungal, yeast, or animal or mammalian expression system and analyzed for the addition of the sequence to the peptide and whether or not this sequence is a functional O-linked glycosylation site. Briefly, a synthetic peptide acceptor sequence is introduced at either the 5' or 3' end of the nucleotide molecule. In principle, the addition of this type of sequence is less disruptive to the resulting glycoprotein when expressed in a suitable expression system. The altered DNA is then expressed in CHO cells or other suitable expression system and the proteins expressed
thereby are examined for the presence of an O-linked glycosylation site. In addition, the presence or absence of glycan chains can be determined.
[0275] In yet another approach, advantageous sites for new O-linked sites may be found in a peptide by creating libraries of the peptide containing various new O-linked sites. For example, the consensus amino acid sequence for N-acetylgalactosamine addition by an N- acetylgalactosaminyltransferase depends on the specific transferase used. The amino acid sequence of a peptide may be scanned to identify contiguous groups of amino acids that can be mutated to generate potential sites for addition of O-linked glycan chains. These mutations can be generated using standard procedures known to those of ordinary skill in the art as described previously. In order to determine if any discovered glycosylation site is actually glycosylated, each recombinant mutated peptide is then expressed in a suitable expression system and is subsequently analyzed for the addition of the site and/or the presence of an O-linked glycan chain.
[0276] A variety of IFN-α conjugates in which the IFN-α peptide is a mutant are also encompassed by the scope of the invention. An O-linked glycosylation site similar to that of interferon alpha-2 can be incorporated into any interferon alpha protein at the same relative position. This can be performed by aligning the amino acid sequence of interest with the IFN-alpha-2β sequence (10-20 amino acids long) and modifying the amino acid sequence to incorporate the glycosylation site. Mutation with any amino acid, deletion or insertion can be used to create the site. Exemplary mutants maintain as high an homology as possible with the IFN-alpha-2 sequence in this region with an emphasis on the T at position 106 (shown below in bold). An example of how this is performed is shown below.
Alignments of Interferon alpha's in the NCBI Protein Database GI# AA# AA Sequence Name IFN-a-2β 1 CVIQGVGVTETPLMKEDSIL 20 (SEQ ID NO:X)
124449 98 117 IFN-alpha 2 (a,b,c)
20178265 99 ....E...E N 118 IFN-alpha 14
124453 99 ....E...E N 118 IFN-alpha 10
585316 99 ....E..ME N 118 IFN-alpha 17 124442 99 ....E...E N.. F.. 118 IFN-alpha 7
124438 99 ....E...E NV.... 118 IFN-alpha 4
417188 99 ..M.E... I . S...Y 118 IFN-alpha 8
20178289 99 ....E...E NV 118 IFN-alpha 21
124457 99 .MM.E...ED....NV.... 118 IFN-alpha 5 124463 99 ..T.E...E. IA..N 118 IFN-alpha 16
124460 99 ..M.E.W.GG....N 118 IFN-alpha 6
124455 99 ..M.EER.G NA.... 118 IFN-alpha 1/13
[0277] Glycosylation/Glyco-PEG-ylation occurs at T106 (IFN-alpha-2). Protein numbering begins with the first amino acid after removal of the protein leader sequence of the naturally expressed pre-pro form.
[0278] Interferon alpha mutations to introduce 0-Linked Glycosylation Sites in IFN- alpha' s that lack this site.
GI# AA# AA Sequence Name
IFN-a-2β I CVIQGVGVTETPLMKEDSIL 20 (SEQ ID NO:X)
124449 98 117 IFN-alpha 2 (a,b,c)
20178265 99 ,E. ,T. .N. 118 IFN-alpha 14 (E107T)
20178265 99 .G. .T. ,N. 118 IFN-alpha 14 (E103G;
E107T)
124453 99 ,E. .T. .N. 118 IFN-alpha 10 (Eiυ/T)
124453 99 G. .T. ,N. 118 IFN-alpha 10 (E 1i0U3JG, ;
E107T)
585316 99 ....E..MT N 118 IFN-alpha 17 (E107T)
585316 99 ....E..VT N 118 IFN-alpha 17 (ME107VT)
585316 99 ....G..MT N 118 IFN-alpha 17 (E103G; E107T)
124442 99 ....E...T N.. F.. 118 IFN-alpha 7 (E107T)
124442 99 ....G...T N.. F.. 118 IFN-alpha 7 (E103G;
E107T)
124438 99 ....E...T NV 118 IFN-alpha 4 (E107T)
124438 99 G...T NV 118 IFN-alpha 4 (E103G;
E107T)
417188 99 ,M. ,E. ,T. ,Y. 118 IFN-alpha 8 (I107T)
417188 99 ,M..G. ,T, ,y. 118 IFN-alpha 8 (E103G;
I107T)
20178289 99 ,E. ,T. .NV. 118 IFN-alpha 21 (E 1X0U7'rT)
20178289 99 ,G. .T. .NV. 118 IFN-alpha 21 (E 110U3JG, ;
E107T)
124457 99 .MM. E. ,TD. .NV. 118 IFN-alpha 5 (E107T)
124457 99 .MM. E. TE. .NV. 118 IFN-alpha 5 (ED108TE)
124457 99 .MM. G. .TD. .NV. 118 IFN-alpha 5 (E103G;
E107T)
124463 99 ,T.E, .T. IP..N. 118 IFN-alpha 16
(E107T; A110 P)
124463 99 T. E. , T. TP..N. 118 IFN-alpha 16
124463 99 .T. G. . T . TP .. N . 118 IFN-alpha 16
(E103G ;E107T ,-IA110TP)
124460 99 • M.E. W. TG. .N. 118 IFN-alpha 6 (G107T) 124460 99 ,M.E, G. TG. .N. 118 IFN-alpha 6 (W105G;G1 124460 99 M. G G. TE. .N. 118 IFN-alpha 6
(E103G;W105G;GG 108 TE)
124455 .M. EER. T. .NA. 118 IFN-alpha 1/13 (G107T)
124455 99 .M. EEG. T. .NA. 118 IFN-alpha 1/13
( R105G ;G1C T)
124455 99 .M.GVG.T NA.... 118 IFN-alpha 1/13
(EER105GVG; G 107 T) [0279] The GI numbers in the above table, except the first number 124449, refer to those of the unmodified wild-type proteins.
[0280] The O-linked glycosylation site can be created in any interferon alpha isoform by placing a T or S at the appropriate amino acid site as shown above. The substitution is T as shown in the above table. The amino acid sequences between the various interferon alpha forms are similar. Any amino acid mutation, insertion, deletion can be made in this region as long as the T or S is at the appropriate position for glycosylation/glyco-PEG-ylation relative to P109 (IFN-alpha-2) in the alignment sequence shown above.
[0281] Moreover, in addition to peptides, the methods of the present invention can be practiced with other biological structures (e.g., glycolipids, lipids, sphingoids, ceramides, whole cells, and the like, containing a glycosylation site).
[0282] Addition of glycosylation sites to a peptide or other structure is conveniently accomplished by altering the amino acid sequence such that it contains one or more glycosylation sites. The addition may also be made by the incorporation of one or more species presenting an -OH group, preferably serine or threonine residues, within the sequence of the peptide (for O-linked glycosylation sites). The addition may be made by mutation or by full chemical synthesis of the peptide. The peptide amino acid sequence is preferably altered through changes at the DNA level, particularly by mutating the DNA encoding the peptide at preselected bases such that codons are generated that will translate into the desired amino acids. The DNA mutation(s) are preferably made using methods known in the art.
[0283] In an exemplary embodiment, the glycosylation site is added by shuffling polynucleotides. Polynucleotides encoding a candidate peptide can be modulated with DNA shuffling protocols. DNA shuffling is a process of recursive recombination and mutation, performed by random fragmentation of a pool of related genes, followed by reassembly of the fragments by a polymerase chain reaction-like process. See, e.g., Stemmer, Proc. Natl. Acad. ScI USA 91 :10747-10751 (1994); Stemmer, Nature 370:389-391 (1994); and U.S. Patent Nos. 5,605,793, 5,837,458, 5,830,721 and 5,811,238.
[0284] Exemplary peptides with which the present invention can be practiced, methods of adding or removing glycosylation sites, and adding or removing glycosyl structures or substructures are described in detail in WO03/031464 and related U.S. and PCT applications.
[0285] The present invention also takes advantage of adding to (or removing from) a peptide one or more selected glycosyl residues, after which a modified sugar is conjugated to at least one of the selected glycosyl residues of the peptide. The present embodiment is useful, for example, when it is desired to conjugate the modified sugar to a selected glycosyl
residue that is either not present on a peptide or is not present in a desired amount. Thus, prior to coupling a modified sugar to a peptide, the selected glycosyl residue is conjugated to the peptide by enzymatic or chemical coupling. In another embodiment, the glycosylation pattern of a glycopeptide is altered prior to the conjugation of the modified sugar by the removal of a carbohydrate residue from the glycopeptide. See, for example WO 98/31826.
[0286] Addition or removal of any carbohydrate moieties present on the glycopeptide is accomplished either chemically or enzymatically. An exemplary chemical deglycosylation is brought about by exposure of the polypeptide variant to the compound trifluoromethanesulfonic acid, or an equivalent compound. This treatment results in the cleavage of most or all sugars except the linking sugar (N-acetylglucosamine or N- acetylgalactosamine), while leaving the peptide intact. Chemical deglycosylation is described by Hakimuddin et al, Arch. Biochem. Biophys. 259: 52 (1987) and by Edge et al, Anal. Biochem. 118: 131 (1981). Enzymatic cleavage of carbohydrate moieties on polypeptide variants can be achieved by the use of a variety of endo- and exo-glycosidases as described by Thotakura et al, Meth Enzymol 138: 350 (1987).
[0287] In an exemplary embodiment, the peptide is essentially completely desialylated with neuraminidase prior to performing glycoconjugation or remodeling steps on the peptide. Following the glycoconjugation or remodeling, the peptide is optionally re-sialylated using a sialy transferase. In an exemplary embodiment, the re-sialylation occurs at essentially each (e.g., >80%, preferably greater than 85%, greater than 90%, preferably greater than 95% and more preferably greater than 96%, 97%, 98% or 99%) terminal saccharyl acceptor in a population of sialyl acceptors. In a preferred embodiment, the saccharide has a substantially uniform sialylation pattern (i.e., substantially uniform glycosylation pattern).
[0288] Chemical addition of glycosyl moieties is carried out by any art-recognized method. Enzymatic addition of sugar moieties is preferably achieved using a modification of the methods set forth herein, substituting native glycosyl units for the modified sugars used in the invention. Other methods of adding sugar moieties are disclosed in U.S. Patent No. 5,876,980, 6,030,815, 5,728,554, and 5,922,577.
[0289] Exemplary attachment points for selected glycosyl residue include, but are not limited to: (a) consensus sites for N-linked glycosylation, and sites for O-linked glycosylation; (b) terminal glycosyl moieties that are acceptors for a glycosyltransferase; (c) arginine, asparagine and histidine; (d) free carboxyl groups; (e) free sulfhydryl groups such as
those of cysteine; (f) free hydroxyl groups such as those of serine, threonine, or hydroxyproline; (g) aromatic residues such as those of phenylalanine, tyrosine, or tryptophan; or (h) the amide group of glutamine. Exemplary methods of use in the present invention are described in WO 87/05330 published Sep. 11, 1987, and in Aplin and Wriston, CRC CRIT. REV. BIOCHEM., pp. 259-306 (1981).
[0290] In one embodiment, the invention provides a method for linking two or more peptides through a linking group. The linking group is of any useful structure and may be selected from straight- and branched-chain structures. Preferably, each terminus of the linker, which is attached to a peptide, includes a modified sugar (i.e., a nascent intact glycosyl linking group).
[0291] In an exemplary method of the invention, two peptides are linked together via a linker moiety that includes a polymeric (e.g., PEG linker). The construct conforms to the general structure set forth in the cartoon above. As described herein, the construct of the invention includes two intact glycosyl linking groups (i.e., s + 1 = 1). The focus on a PEG linker that includes two glycosyl groups is for purposes of clarity and should not be interpreted as limiting the identity of linker arms of use in this embodiment of the invention.
[0292] Thus, a PEG moiety is functionalized at a first terminus with a first glycosyl unit and at a second terminus with a second glycosyl unit. The first and second glycosyl units are preferably substrates for different transferases, allowing orthogonal attachment of the first and second peptides to the first and second glycosyl units, respectively. In practice, the (glycosyl)1-PEG-(glycosyl)2 linker is contacted with the first peptide and a first transferase for which the first glycosyl unit is a substrate, thereby forming (peptide)1-(glycosyl)1-PEG-(glycosyl)2. Transferase and/or unreacted peptide is then optionally removed from the reaction mixture. The second peptide and a second transferase for which the second glycosyl unit is a substrate are added to the
(peptide) -(glycosyl) -PEG-(glycosyl) conjugate, forming
(peptide)1 -(glycosyl) 1-PEG-(glycosyl)2-(peptide)2 ; at least one of the glycosyl residues is either directly or indirectly O-linked. Those of skill in the art will appreciate that the method outlined above is also applicable to forming conjugates between more than two peptides by, for example, the use of a branched PEG, dendrimer, poly(amino acid), polysaccharide or the like.
[0293] In an exemplary embodiment, the peptide that is modified by a method of the invention is a glycopeptide that is produced in mammalian cells (e.g., CHO cells) or in a transgenic animal and thus, contains N- and/or O-linked oligosaccharide chains, which are incompletely sialylated. The oligosaccharide chains of the glycopeptide lacking a sialic acid and containing a terminal galactose residue can be PEGylated, PPGylated or otherwise modified with a modified sialic acid.
[0294] In Scheme 1 , the amino glycoside 1, is treated with the active ester of a protected amino acid (e.g., glycine) derivative, converting the sugar amine residue into the corresponding protected amino acid amide adduct. The adduct is treated with an aldolase to form α-hydroxy carboxylate 2. Compound 2 is converted to the corresponding CMP derivative by the action of CMP-SA synthetase, followed by catalytic hydrogenation of the CMP derivative to produce compound 3. The amine introduced via formation of the glycine adduct is utilized as a locus of PEG attachment by reacting compound 3 with an activated PEG or PPG derivative (e.g., PEG-C(O)NHS, PEG-OC(O)O-p-nitrophenyl), producing species such as 4 or 5, respectively.
Scheme 1 CTP
CMP-SA-5-NHCOCH2NH — C(O)O-PEG 5
Conjugation of Modified Sugars to Peptides
[0295] The PEG modified sugars are conjugated to a glycosylated or non-glycosylated peptide using an appropriate enzyme to mediate the conjugation. Preferably, the concentrations of the modified donor sugar(s), enzyme(s) and acceptor peptide(s) are selected such that glycosylation proceeds until the acceptor is consumed. The considerations
discussed below, while set forth in the context of a sialyltransferase, are generally applicable to other glycosyltransferase reactions.
[0296] A number of methods of using glycosyltransferases to synthesize desired oligosaccharide structures are known and are generally applicable to the instant invention. Exemplary methods are described, for instance, WO 96/32491, Ito et ah, Pure Appl. Chem. 65: 753 (1993), U.S. Pat. Nos. 5,352,670, 5,374,541, 5,545,553, commonly owned U.S. Pat. Nos. 6,399,336, and 6,440,703, and commonly assigned published PCT applications, WO 03/031464, WO 04/033651, WO 04/099231, which are incorporated herein by reference.
[0297] The present invention is practiced using a single glycosyltransferase or a combination of glycosyltransferases. For example, one can use a combination of a sialyltransferase and a galactosyltransferase. In those embodiments using more than one enzyme, the enzymes and substrates are preferably combined in an initial reaction mixture, or the enzymes and reagents for a second enzymatic reaction are added to the reaction medium once the first enzymatic reaction is complete or nearly complete. By conducting two enzymatic reactions in sequence in a single vessel, overall yields are improved over procedures in which an intermediate species is isolated. Moreover, cleanup and disposal of extra solvents and by-products is reduced.
[0298] In a preferred embodiment, each of the first and second enzyme is a glycosyltransferase (e.g., Core-1 and/or Core-2 glycosyltransferases). In another preferred embodiment, one enzyme is an endoglycosidase, e.g., an endoglycosidase mutated to run synthetically, rather than degradatively. In an additional preferred embodiment, more than two enzymes are used to assemble the modified glycoprotein of the invention. The enzymes are used to alter a saccharide structure on the peptide at any point either before or after the addition of the modified sugar to the peptide.
[0299] In another embodiment, the method makes use of one or more exo- or endoglycosidase. The glycosidase is typically a mutant, which is engineered to form glycosyl bonds rather than rupture them. The mutant glycanase typically includes a substitution of an amino acid residue for an active site acidic amino acid residue. For example, when the endoglycanase is endo-H, the substituted active site residues will typically be Asp at position 130, GIu at position 132 or a combination thereof. The amino acids are generally replaced with serine, alanine, asparagine, or glutamine.
[0300] The mutant enzyme catalyzes the reaction, usually by a synthesis step that is analogous to the reverse reaction of the endoglycanase hydrolysis step. In these embodiments, the glycosyl donor molecule (e.g., a desired oligo- or mono-saccharide structure) contains a leaving group and the reaction proceeds with the addition of the donor molecule to a GIcNAc residue on the protein. For example, the leaving group can be a halogen, such as fluoride. In other embodiments, the leaving group is a Asn, or a Asn- peptide moiety. In further embodiments, the GIcNAc residue on the glycosyl donor molecule is modified. For example, the GIcNAc residue may include a 1,2 oxazoline moiety.
[0301] In a preferred embodiment, each of the enzymes utilized to produce a conjugate of the invention are present in a catalytic amount. The catalytic amount of a particular enzyme varies according to the concentration of that enzyme's substrate as well as to reaction conditions such as temperature, time and pH value. Means for determining the catalytic amount for a given enzyme under preselected substrate concentrations and reaction conditions are well known to those of skill in the art.
[0302] The temperature at which an above process is carried out can range from just above freezing to the temperature at which the most sensitive enzyme denatures. Preferred temperature ranges are about 0 0C to about 55 0C, and more preferably about 20 ° C to about 37 0C. In another exemplary embodiment, one or more components of the present method are conducted at an elevated temperature using a thermophilic enzyme.
[0303] The reaction mixture is maintained for a period of time sufficient for the acceptor to be glycosylated, thereby forming the desired conjugate. Some of the conjugate can often be detected after a few h, with recoverable amounts usually being obtained within 24 h or less. Those of skill in the art understand that the rate of reaction is dependent on a number of variable factors (e.g, enzyme concentration, donor concentration, acceptor concentration, temperature, solvent volume), which are optimized for a selected system.
[0304] The glycosylation and/or glycoPEGylation reaction can be performed at pH's between 6-7.5 and at temperatures of 30-320C while maintaining similar reaction rates and conversion yields. These reactions can be performed sequentially or simultaneously in one pot with little effect on overall glycoPEGylation yields. Enzyme amounts used in these studies were typically 10 to 15 mU for GalNAc-T2 and 63 to 69 mU for ST6GalNAc-l for addition of two sugars to 1 mg of IFN-alpha-2b and 10 to 15 mU for GalNAc-T2, 30 mU core-l-β3-galactosyltransferase-l (core- 1 -GaI-Tl) and 69 to 9OmU for ST3Gal2 for the
addition of three sugars to 1 mg of IFN-alpha-2b. Typical reaction times were beween 24-48 hours. The PEG's were all introduced onto the protein by the use of CMP-SA-PEG using either linear (20 and 30 KDa) or branched (40 and 60 KDa) PEG. Typical amounts of this reagent used during glycoPEGylation were between 0.2 and 0.25 niM and 3-5 mole equivalents.
[0305] The present invention also provides for the industrial-scale production of modified peptides. As used herein, an industrial scale generally produces at least one gram of finished, purified conjugate.
[0306] A surprising feature of the instant method is the ability to efficiently run one or more of these synthetic reactions, adding a glycosyl or modified glycosyl moiety, in the same vessel. The reactions can be run with all of the reactants necessary for two or more glycosyl additions present in the vessel. Alternatively, the reactants necessary for one or more glycosyl addition can be present in the vessel and, when the reaction(s) is judged sufficiently complete, the reactants necessary for at least one more glycosyl or modified glycosyl addition are incorporated into the existing reaction mixture, with no prior purification of the mixture to which they are added. This feature greatly facilitates the applicability of the instant methods to the production of large (industrial) quantities of the glycosylated peptide substrates and the conjugates of these substrates.
[0307] In the discussion that follows, the invention is exemplified by the conjugation of modified sialic acid moieties to a glycosylated peptide. The exemplary modified sialic acid is labeled with PEG. The focus of the following discussion on the use of PEG-modified sialic acid and glycosylated peptides is for clarity of illustration and is not intended to imply that the invention is limited to the conjugation of these two partners. One of skill understands that the discussion is generally applicable to the additions of modified glycosyl moieties other than sialic acid. Moreover, the discussion is equally applicable to the modification of a glycosyl unit with agents other than PEG including other PEG moieties, therapeutic moieties, and biomolecules.
[0308] An enzymatic approach can be used for the selective introduction of PEGylated or PPGylated carbohydrates onto a peptide or glycopeptide. The method utilizes modified sugars containing PEG, PPG, or a masked reactive functional group, and is combined with the appropriate glycosyltransferase or glycosynthase. By selecting the glycosyltransferase that will make the desired carbohydrate linkage and utilizing the modified sugar as the donor
substrate, the PEG or PPG can be introduced directly onto the peptide backbone, onto existing sugar residues of a glycopeptide or onto sugar residues that have been added to a peptide.
[0309] In an exemplary embodiment, an acceptor for a sialyltransferase is present on the peptide to be modified either as a naturally occurring structure or it is placed there recombinantly, enzymatically or chemically. Suitable acceptors, include, for example, galactosyl acceptors such as Galβ 1,4GIcNAc, Gal βl, 4GaIN Ac, Galβ 1,3 GaINAc, lacto-N- tetraose, Galβ 1,3 GIcN Ac, Galβl,3Ara, Galβ 1,6GIcNAc, Galβ 1,4GIc (lactose), and other acceptors known to those of skill in the art {see, e.g., Paulson et ah, J. Biol Chem, 253: 5617- 5624 (1978)). Exemplary sialyltransferases are set forth herein.
[0310] In one embodiment, an acceptor for the sialyltransferase is present on the glycopeptide to be modified upon in vivo synthesis of the glycopeptide. Such glycopeptides can be sialylated using the claimed methods without prior modification of the glycosylation pattern of the glycopeptide. Alternatively, the methods of the invention can be used to sialylate a peptide that does not include a suitable acceptor; one first modifies the peptide to include an acceptor by methods known to those of skill in the art. In an exemplary embodiment, a GaINAc residue is added by the action of a GaINAc transferase.
[0311] In an exemplary embodiment, the galactosyl acceptor is assembled by attaching a galactose residue to an appropriate acceptor linked to the peptide, e.g., a GaINAc. The method includes incubating the peptide to be modified with a reaction mixture that contains a suitable amount of a galactosyltransferase (e.g., Galβ 1,3 or Galβ 1,4), and a suitable galactosyl donor (e.g., UDP-galactose). The reaction is allowed to proceed substantially to completion or, alternatively, the reaction is terminated when a preselected amount of the galactose residue is added. Other methods of assembling a selected saccharide acceptor will be apparent to those of skill in the art.
[0312] In yet another embodiment, glycopeptide-linked oligosaccharides are first "trimmed," either in whole or in part, to expose either an acceptor for the sialyltransferase or a moiety to which one or more appropriate residues can be added to obtain a suitable acceptor. Enzymes such as glycosyltransferases and endoglycosidases (see, for example U.S. Patent No. 5,716,812) are useful for the attaching and trimming reactions. In another embodiment of this method, the sialic acid moieties of the peptide are essentially completely
removed (e.g., at least 90, at least 95 or at least 99%), exposing an acceptor for a modified sialic acid.
[0313] In the discussion that follows, the method of the invention is exemplified by the use of modified sugars having a PEG moiety attached thereto. The focus of the discussion is for clarity of illustration. Those of skill will appreciate that the discussion is equally relevant to those embodiments in which the modified sugar bears a therapeutic moiety, biomolecule or the like.
[0314] In an exemplary embodiment of the invention in which a carbohydrate residue is "trimmed" prior to the addition of the modified sugar high mannose is trimmed back to the first generation biantennary structure. A modified sugar bearing a PEG moiety is conjugated to one or more of the sugar residues exposed by the "trimming back." In one example, a PEG moiety is added via a GIcNAc moiety conjugated to the PEG moiety. The modified GIcNAc is attached to one or both of the terminal mannose residues of the biantennary structure. Alternatively, an unmodified GIcNAc can be added to one or both of the termini of the branched species.
[0315] In another exemplary embodiment, a PEG moiety is added to one or both of the terminal mannose residues of the biantennary structure via a modified sugar having a galactose residue, which is conjugated to a GIcNAc residue added onto the terminal mannose residues. Alternatively, an unmodified Gal can be added to one or both terminal GIcNAc residues.
[0316] In yet a further example, a PEG moiety is added onto a Gal or a GaINAc residue using a modified sialic acid such as those discussed above.
[0317] In another exemplary embodiment, a high mannose structure is "trimmed back" to the mannose from which the biantennary structure branches. In one example, a PEG moiety is added via a GIcNAc modified with the polymer. Alternatively, an unmodified GIcNAc is added to the mannose, followed by a Gal with an attached PEG moiety. In yet another embodiment, unmodified GIcNAc and Gal residues are sequentially added to the mannose, followed by a sialic acid moiety modified with a PEG moiety.
[0318] A high mannose structure can also be trimmed back to the elementary tri-mannosyl core.
[0319] In a further exemplary embodiment, high mannose is "trimmed back" to the GIcNAc to which the first mannose is attached. The GIcNAc is conjugated to a Gal residue bearing a PEG moiety. Alternatively, an unmodified Gal is added to the GIcNAc, followed by the addition of a sialic acid modified with a water-soluble sugar. In yet a further example, the terminal GIcNAc is conjugated with Gal and the GIcNAc is subsequently fucosylated with a modified fucose bearing a PEG moiety.
[0320] High mannose may also be trimmed back to the first GIcNAc attached to the Asn of the peptide. In one example, the GIcNAc of the GlcNAc-(Fuc)a residue is conjugated wit ha GIcNAc bearing a water soluble polymer. In another example, the GIcNAc of the GlcNAc-(Fuc)a residue is modified with Gal, which bears a water soluble polymer. In a still further embodiment, the GIcNAc is modified with Gal, followed by conjugation to the Gal of a sialic acid modified with a PEG moiety.
[0321] Other exemplary embodiments are set forth in commonly owned U.S. Patent application Publications: 20040132640; 20040063911; 20040137557; U.S. Patent application Nos: 10/369,979; 10/410,913; 10/360,770; 10/410,945 and PCT/US02/32263 each of which is incorporated herein by reference.
[0322] The Examples set forth above provide an illustration of the power of the methods set forth herein. Using the methods described herein, it is possible to "trim back" and build up a carbohydrate residue of substantially any desired structure. The modified sugar can be added to the termini of the carbohydrate moiety as set forth above, or it can be intermediate between the peptide core and the terminus of the carbohydrate.
[0323] In an exemplary embodiment, an existing sialic acid is removed from a glycopeptide using a sialidase, thereby unmasking all or most of the underlying galactosyl residues. Alternatively, a peptide or glycopeptide is labeled with galactose residues, or an oligosaccharide residue that terminates in a galactose unit. Following the exposure of or addition of the galactose residues, an appropriate sialyltransferase is used to add a modified sialic acid.
[0324] In another exemplary embodiment, an enzyme that transfers sialic acid onto sialic acid is utilized. This method can be practiced without treating a sialylated glycan with a sialidase to expose glycan residues beneath the sialic acid. An exemplary polymer-modified sialic acid is a sialic acid modified with poly(ethylene glycol). Other exemplary enzymes that add sialic acid and modified sialic acid moieties onto glycans that include a sialic acid
residue or exchange an existing sialic acid residue on a glycan for these species include ST3Gal3, CST-II5 ST8Sia-II, ST8Sia-III and ST8Sia-IV.
[0325] In yet a further approach, a masked reactive functionality is present on the sialic acid. The masked reactive group is preferably unaffected by the conditions used to attach the modified sialic acid to the IFN-α. After the covalent attachment of the modified sialic acid to the peptide, the mask is removed and the peptide is conjugated with an agent such as PEG. The agent is conjugated to the peptide in a specific manner by its reaction with the unmasked reactive group on the modified sugar residue.
[0326] Any modified sugar can be used with its appropriate glycosyltransferase, depending on the terminal sugars of the oligosaccharide side chains of the glycopeptide. As discussed above, the terminal sugar of the glycopeptide required for introduction of the PEGylated structure can be introduced naturally during expression or it can be produced post expression using the appropriate glycosidase(s), glycosyltransferase(s) or mix of glycosidase(s) and glycosyltransferase(s) .
[0327] In a further exemplary embodiment, UDP-galactose-PEG is reacted with β 1 ,4- galactosyltransferase, thereby transferring the modified galactose to the appropriate terminal N-acetylglucosamine structure. The terminal GIcNAc residues on the glycopeptide may be produced during expression, as may occur in such expression systems as mammalian, insect, plant or fungus, but also can be produced by treating the glycopeptide with a sialidase and/or glycosidase and/or glycosyltransferase, as required.
[0328] In another exemplary embodiment, a GIcNAc transferase, such as GNTl -5, is utilized to transfer PEGylated-GlcNAc to a terminal mannose residue on a glycopeptide. In a still further exemplary embodiment, an the N- and/or O-linked glycan structures are enzymatically removed from a glycopeptide to expose an amino acid or a terminal glycosyl residue that is subsequently conjugated with the modified sugar. For example, an endoglycanase is used to remove the N-linked structures of a glycopeptide to expose a terminal GIcNAc as a GlcNAc-linked-Asn on the glycopeptide. UDP-GaI-PEG and the appropriate galactosyltransferase is used to introduce the PEG-galactose functionality onto the exposed GIcNAc.
[0329] In an alternative embodiment, the modified sugar is added directly to the peptide backbone using a glycosyltransferase known to transfer sugar residues to the peptide backbone. Exemplary glycosyltransferases useful in practicing the present invention include,
but are not limited to, GaINAc transferases (GaINAc Tl -14), GIcNAc transferases, fucosyltransferases, glucosyltransferases, xylosyltransferases, mannosyltransferases and the like. Use of this approach allows the direct addition of modified sugars onto peptides that lack any carbohydrates or, alternatively, onto existing glycopeptides. In both cases, the addition of the modified sugar occurs at specific positions on the peptide backbone as defined by the substrate specificity of the glycosyltransferase and not in a random manner as occurs during modification of a protein's peptide backbone using chemical methods. An array of agents can be introduced into proteins or glycopeptides that lack the glycosyltransferase substrate peptide sequence by engineering the appropriate amino acid sequence into the polypeptide chain.
[0330] In each of the exemplary embodiments set forth above, one or more additional chemical or enzymatic modification steps can be utilized following the conjugation of the modified sugar to the peptide. In an exemplary embodiment, an enzyme (e.g., fucosyltransferase) is used to append a glycosyl unit (e.g., fucose) onto the terminal modified sugar attached to the peptide. In another example, an enzymatic reaction is utilized to "cap" sites to which the modified sugar failed to conjugate. Alternatively, a chemical reaction is utilized to alter the structure of the conjugated modified sugar. For example, the conjugated modified sugar is reacted with agents that stabilize or destabilize its linkage with the peptide component to which the modified sugar is attached. In another example, a component of the modified sugar is deprotected following its conjugation to the peptide. One of skill will appreciate that there is an array of enzymatic and chemical procedures that are useful in the methods of the invention at a stage after the modified sugar is conjugated to the peptide. Further elaboration of the modified sugar-peptide conjugate is within the scope of the invention.
[0331] Enzymes and reaction conditions for preparing the conjugates of the present invention are discussed in detail in the parent of the instant application as well as co-owned published PCT patent applications WO 03/031464, WO 04/033651, WO 04/099231.
[0332] In a selected embodiment, an IFN-α peptide, expressed in insect cells, is remodeled such that glycans on the remodeled glycopeptide include a GlcNAc-Gal glycosyl residue. The addition of GIcNAc and Gal can occur as separate reactions or as a single reaction in a single vessel. In this example, GlcNAc-transferase I and Gal-transferase I are used. The modified sialyl moiety is added using ST3 GaI-III.
[0333] In another embodiment, the addition of GIcNAc, Gal and modified Sia can also occur in a single reaction vessel, using the enzymes set forth above. Each of the enzymatic remodeling and glycoPEGylation steps are carried out individually.
[0334] When the peptide is expressed in mammalian cells, different methods are of use. In one embodiment, the peptide is conjugated without need for remodeling prior to conjugation by contacting the peptide with a sialyltransferase that transfers the modified sialic acid directly onto a sialic acid on the peptide forming Sia-Sia-L-R1, or exchanges a sialic acid on the peptide for the modified sialic acid, forming Sia-L-R1. An exemplary enzyme of use in this method is CST-II. Other enzymes that add sialic acid to sialic acid are known to those of skill in the art and examples of such enzymes are set forth the figures appended hereto.
[0335] In yet another method of preparing the conjugates of the invention, the peptide expressed in a mammalian system is desialylated using a sialidase. The exposed Gal residue is sialylated with a modified sialic acid using a sialyltransferase specific for O-linked glycans, providing an IFN-α conjugate with an O-linked modified glycan. The desialylated, modified IFN-α peptide is optionally partially or fully re-sialylated by using a sialyltransferase such as ST3Gaim.
[0336] In another aspect, the invention provides a method of making a PEGylated IFN-α conjugate of the invention. The method includes: (a) contacting a substrate IFN-α peptide including a glycosyl group selected from:
-GaINAc -GaI- ■(Sia)a
5 ; and *> with a PEG-sialic acid donor having the formula:
and an enzyme that transfers PEG-sialic acid from the donor onto a member selected from the GaINAc, Gal and the Sia of the glycosyl group, under conditions appropriate for the transfer. An exemplary modified sialic acid donor is CMP-sialic acid modified, through a linker
moiety, with a polymer, e.g., a straight chain or branched poly(ethylene glycol) moiety. As discussed herein, the peptide is optionally glycosylated with GaINAc and/or Gal and/or Sia ("Remodeled") prior to attaching the modified sugar. The remodeling steps can occur in sequence in the same vessel without purification of the glycosylated peptide between steps. Alternatively, following one or more remodeling step, the glycosylated peptide can be purified prior to submitting it to the next glycosylation or glycPEGylation step.
[0337] As illustrated in the examples and discussed further below, placement of an acceptor moiety for the PEG-sugar is accomplished in any desired number of steps. For example, in one embodiment, the addition of GaINAc to the peptide can be followed by a second step in which the PEG-sugar is conjugated to the GaINAc in the same reaction vessel. Alternatively, these two steps can be carried out in a single vessel approximately simultaneously.
[0338] In an exemplary embodiment, the PEG-sialic acid donor has the formula:
[0339] In another exemplary emodiment, the invention provides a method including:
(a) contacting a substrate IFN-α peptide including a glycosyl moiety selected from :
-GaINAc (GaI)n -GaI- -(SIa)0
with a PEG-sialic acid donor having the formula:
wherein the index c is 0 or 1, and the index is r is 0 or 1; (b) contacting the IFN-α peptide and the PEG-sialic acid donor with an enzyme that transfers PEG-sialic acid from the donor onto the glycosyl moiety, under conditions appropriate for the transfer.
[0340] In another exemplary embodiment, prior to step (a): there is a step involving expressing the substrate IFN-α peptide in a suitable host.
[0341] In a further exemplary embodiment, the IFN-α peptide is expressed in an appropriate expression system prior to being glycoPEGylated or remodeled. Exemplary expression systems include Sf-9/baculovirus and Chinese Hamster Ovary (CHO) cells.
[0342] Methods of expressing IFN in recombinant cells are well known in the art, and is easily accomplished using techniques described in, for example U.S. Patent No. 4,966,843, and in Sambrook et al. (2001, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York) and Ausubel et al. (1997, Current Protocols in Molecular Biology, Green & Wiley, New York). Assays to determine the biological activity of a Type I IFN modified by the present invention will be well known to the skilled artisan. For example, the assay described in Rubinstein et al., (1981, Journal of Virology 37:755-758) is commonly used to determine the effect of a Type I IFN by measuring the cytopathic effects of viral infection on a population of cells. This method is only one of many known in the art for assaying the biological function of a Type IFN.
[0343] The IFN-α conjugates may be administered to a patient selected from the group consisting of a patient having hairy cell leukemia, a patient having malignant melanoma, a patient having follicular lymphoma, a patient having condylomata acuminata, a patient having AIDS-related Kaposi's sarcoma, a patient having Hepatitis C, a patient having Hepatitis B, a patient having a human papilloma virus infection, a patient having Chronic Myeloid Leukemia (CML), a patient having chronic phase Philadelphia chromosome (Ph) positive Chronic Myelogenous Leukemia, a patient having non-Hodgkin's lymphoma (NHL), a patient having lymphoma, a patient having bladder cancer, and a patient having renal cancer. Preferably, the patient is a mammal. Preferably, the patient is a human patient.
[0344] In another exemplary embodiment, the invention provides a method of treating a condition in a mammal, wherein the condition is a member selected from inhibiting viral replication, increasing the lytic potential of NK cells, modulating MHC molecule expression, and inhibiting cellular proliferation the method including administering to the mammal an IFN-α conjugate. In another exemplary embodiment, the invention provides a method of
treating infection in a subject in need thereof, the method including the step of administering to the subject an amount of an IFN-α conjugate, effective to ameliorate the condition in the subject.
Purification of IFN-α Conjugates [0345] After the enzymatic reaction, the IFN-α conjugates can be used without purification. However, it is usually preferred to recover the product and one or more of the intermediates, e.g., nucleotide sugars, branched and linear PEG species, modified sugars and modified nucleotide sugars. A purification strategy can employ a combination of IEX (SP sepharose) and SEC (Superdex 200). The formulation buffer can be selected based on previous studies with IFN-α and all protein products can be stored frozen. The reaction products can be characterized with MALDI, peptide mapping and SDS-PAGE gel analysis (colloidal blue and silver stain).
[0346] Standard, well-known techniques for recovery of glycosylated saccharides such as thin or thick layer chromatography, column chromatography, ion exchange chromatography, or membrane filtration can be used. It is preferred to use membrane filtration, more preferably utilizing a reverse osmotic membrane, or one or more column chromatographic techniques for the recovery as is discussed hereinafter and in the literature cited herein. For instance, membrane filtration wherein the membranes have molecular weight cutoff of about 3000 to about 10,000 can be used to remove proteins such as glycosyl transferases.
[0347] If the peptide is produced intracellularly, as a first step, the particulate debris, either host cells or lysed fragments, is removed. Following glycoPEGylation, the PEGylated peptide is purified by art-recognized methods, for example, by centrifugation or ultrafiltration; optionally, the protein may be concentrated with a commercially available protein concentration filter, followed by separating the polypeptide variant from other impurities by one or more steps selected from immunoaffinity chromatography, ion-exchange column fractionation (e.g., on diethylaminoethyl (DEAE) or matrices containing carboxymethyl or sulfopropyl groups), chromatography on Blue-Sepharose, CM Blue- Sepharose, MONO-Q, MONO-S, lentil lectin-Sepharose, WGA-Sepharose, Con A- Sepharose, Ether Toyopearl, Butyl Toyopearl, Phenyl Toyopearl, or protein A Sepharose, SDS-PAGE chromatography, silica chromatography, chromatofocusing, reverse phase HPLC (e.g., silica gel with appended aliphatic groups), gel filtration using, e.g., Sephadex molecular
sieve or size-exclusion chromatography, chromatography on columns that selectively bind the polypeptide, and ethanol or ammonium sulfate precipitation.
[0348] Modified glycopeptides produced in culture are usually isolated by initial extraction from cells, enzymes, etc., followed by one or more concentration, salting-out, aqueous ion- exchange, or size-exclusion chromatography steps. Additionally, the modified glycoprotein may be purified by affinity chromatography. Finally, HPLC may be employed for final purification steps.
[0349] A protease inhibitor, e.g. , methylsulfonylfluoride (PMSF) may be included in any of the foregoing steps to inhibit proteolysis and antibiotics or preservatives may be included to prevent the growth of adventitious contaminants.
[0350] Within another embodiment, supernatants from systems which produce the modified glycopeptide of the invention are first concentrated using a commercially available protein concentration filter, for example, an Amicon or Millipore Pellicon ultrafiltration unit. Following the concentration step, the concentrate may be applied to a suitable purification matrix. For example, a suitable affinity matrix may include a ligand for the peptide, a lectin or antibody molecule bound to a suitable support. Alternatively, an anion-exchange resin may be employed, for example, a matrix or substrate having pendant DEAE groups. Suitable matrices include acrylamide, agarose, dextran, cellulose, or other types commonly employed in protein purification. Alternatively, a cation-exchange step may be employed. Suitable cation exchangers include various insoluble matrices including sulfopropyl or carboxymethyl groups. Sulfopropyl groups are particularly preferred.
[0351] Other methods of use in purification include size exclusion chromatography (SEC), hydroxyapatite chromatography, hydrophobic interaction chromatography and chromatography on Blue Sepharose. These and other useful methods are illustrated in co- assigned U.S. Provisional Patent No. 60/678,822, filed May 6, 2005.
[0352] One or more RP-HPLC steps employing hydrophobic RP-HPLC media, e.g., silica gel having pendant methyl or other aliphatic groups, may be employed to further purify a polypeptide conjugate composition. Some or all of the foregoing purification steps, in various combinations, can also be employed to provide a homogeneous or essentially homogeneous modified glycoprotein.
[0353] The modified glycopeptide of the invention resulting from a large-scale fermentation may be purified by methods analogous to those disclosed by Urdal et al, J. Chromatog. 296: 171 (1984). This reference describes two sequential, RP-HPLC steps for purification of recombinant human IL-2 on a preparative HPLC column. Alternatively, techniques such as affinity chromatography may be utilized to purify the modified glycoprotein.
[0354] In an exemplary embodiment, IFN-α can be purified on a SP Sepharose column (HiTrap HP, FF, 5 mL, Amersham) was connected to a Varian HPLC system and the absorbance at 280 nm monitored. The column can be washed with NaCl in sodium acetate at an appropriate pH. The running buffer can also contain an emulsifying agent, e.g.,
Polysorbate 80. Gradient elution is of use to purify the conjugates of the invention. For example the product can be eluted using the following gradient: 0-15 min, 25mM NaAc, pH 4.5, 0.005% Polysorbate 80; 15-35 min, 0-0.6 M NaCl in 25mM NaAc, pH 4.5, 0.005% Polysorbate 80; 35-45 min, 1.0 M NaCl in 25mMNaAc, pH 4.5, 0.005% Polysorbate 80. Fractions can be collected and concentrated to about 0.4 mL with a Centricon centrifugal filter, 5 KDa MWCO for analysis and further purification. Samples can be stored at 40C. The methods, and modifications thereof, set forth in commonly owned, co-assigned U.S. Provisional Patent No. 60/665,588, filed March 24, 2005, are also of use to purify the instant conjugates.
Pharmaceutical Compositions
[0355] In another aspect, the invention provides a pharmaceutical composition. The pharmaceutical composition includes a pharmaceutically acceptable diluent and a covalent conjugate between a non-naturally-occurring, PEG moiety, therapeutic moiety or biomolecule and a glycosylated or non-glycosylated peptide. The polymer, therapeutic moiety or biomolecule is conjugated to the peptide via an intact glycosyl linking group interposed between and covalently linked to both the peptide and the polymer, therapeutic moiety or biomolecule.
[0356] Pharmaceutical compositions of the invention are suitable for use in a variety of drug delivery systems. Suitable formulations for use in the present invention are found in Remington's Pharmaceutical Sciences, Mace Publishing Company, Philadelphia, PA, 17th ed. (1985). For a brief review of methods for drug delivery, see, Langer, Science 249:1527- 1533 (1990).
[0357] The pharmaceutical compositions may be formulated for any appropriate manner of administration, including for example, topical, oral, nasal, intravenous, intracranial, intraperitoneal, subcutaneous or intramuscular administration. For parenteral administration, such as subcutaneous injection, the carrier preferably includes water, saline, alcohol, a fat, a wax or a buffer. For oral administration, any of the above carriers or a solid carrier, such as mannitol, lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose, glucose, sucrose, and magnesium carbonate, may be employed. Biodegradable microspheres (e.g., polylactate polyglycolate) may also be employed as carriers for the pharmaceutical compositions of this invention. Suitable biodegradable microspheres are disclosed, for example, in U.S. Patent Nos. 4,897,268 and 5,075,109.
[0358] Commonly, the pharmaceutical compositions are administered parenterally, e.g. , intravenously. Thus, the invention provides compositions for parenteral administration that include the compound dissolved or suspended in an acceptable carrier, preferably an aqueous carrier, e.g., water, buffered water, saline, PBS and the like. The compositions may contain pharmaceutically acceptable auxiliary substances as required to approximate physiological conditions, such as pH adjusting and buffering agents, tonicity adjusting agents, wetting agents, detergents and the like.
[0359] These compositions may be sterilized by conventional sterilization techniques, or may be sterile filtered. The resulting aqueous solutions may be packaged for use as is, or lyophilized, the lyophilized preparation being combined with a sterile aqueous carrier prior to administration. The pH of the preparations typically will be between 3 and 11, more preferably from 5 to 9 and most preferably from 7 and 8.
[0360] In some embodiments the glycopeptides of the invention can be incorporated into liposomes formed from standard vesicle-forming lipids. A variety of methods are available for preparing liposomes, as described in, e.g., Szoka et al., Ann. Rev. Biophys. Bioeng. 9: 467 (1980), U.S. Pat. Nos. 4,235,871, 4,501,728 and 4,837,028. The targeting of liposomes using a variety of targeting agents (e.g., the sialyl galactosides of the invention) is well known in the art (see, e.g., U.S. Patent Nos. 4,957,773 and 4,603,044).
[0361] Standard methods for coupling targeting agents to liposomes can be used. These methods generally involve incorporation into liposomes of lipid components, such as phosphatidylethanolamine, which can be activated for attachment of targeting agents, or derivatized lipophilic compounds, such as lipid-derivatized glycopeptides of the invention.
[0362] Targeting mechanisms generally require that the targeting agents be positioned on the surface of the liposome in such a manner that the target moieties are available for interaction with the target, for example, a cell surface receptor. The carbohydrates of the invention may be attached to a lipid molecule before the liposome is formed using methods known to those of skill in the art (e.g., alkylation or acylation of a hydroxyl group present on the carbohydrate with a long chain alkyl halide or with a fatty acid, respectively). Alternatively, the liposome may be fashioned in such a way that a connector portion is first incorporated into the membrane at the time of forming the membrane. The connector portion must have a lipophilic portion, which is firmly embedded and anchored in the membrane. It must also have a reactive portion, which is chemically available on the aqueous surface of the liposome. The reactive portion is selected so that it will be chemically suitable to form a stable chemical bond with the targeting agent or carbohydrate, which is added later. In some cases it is possible to attach the target agent to the connector molecule directly, but in most instances it is more suitable to use a third molecule to act as a chemical bridge, thus linking the connector molecule which is in the membrane with the target agent or carbohydrate which is extended, three dimensionally, off of the vesicle surface.
[0363] The compounds prepared by the methods of the invention may also find use as diagnostic reagents. For example, labeled compounds can be used to locate areas of inflammation or tumor metastasis in a patient suspected of having an inflammation. For this use, the compounds can be labeled with 1251, 14C, or tritium.
[0364] The active ingredient used in the pharmaceutical compositions of the present invention is glycoPEGylated IFN-α and its derivatives having the biological properties of stimulating granulocyte production. Preferably, the IFN-α conjugate of the present invention is administered parenterally (e.g. IV, IM, SC or IP). Effective dosages are expected to vary considerably depending on the condition being treated and the route of administration but are expected to be in the range of about 0.1 (~7U) to 100 (~7000U) μg/kg body weight of the active material. Preferable doses for treatment of anemic conditions are about 50 to about 300 Units/kg three times a week. Because the present invention provides a IFN-α conjugate with an enhanced in vivo residence time, the stated dosages are optionally lowered when a composition of the invention is administered.
[0365] Preparative methods for species of use in preparing the compositions of the invention are generally set forth in various patent publications, e.g., US 20040137557; WO
04/083258; and WO 04/033651. The following examples are provided to illustrate the conjugates, and methods and of the present invention, but not to limit the claimed invention.
[0366] The following examples are provided to illustrate the conjugates, and methods and of the present invention, but not to limit the claimed invention.
EXAMPLES
EXAMPLE 1
1.1 Preparation of CMP-Sialic Acid-PEG
[0367] The CMP-Sialic Acid-branched PEG compounds, and their linear PEG analogues, employed in the production of IFN-α conjugates of the invention are readily synthesized. An example of such as synthesis is shown in FIG. 4.
[0368] The PEG component of the sialic acid-PEG compounds can be branched or linear. An example of the synthesis of the branched PEG component is shown below.
Preparation of Cysteine-PEG2 ( 2 )
[0369] Potassium hydroxide (84.2 mg, 1.5 mmol, as a powder) was added to a solution of L-cysteine (93.7 mg, 0.75 mmol) in anhydrous methanol (20 L) under argon. The mixture was stirred at room temperature for 30 min, and then mPEG-O-tosylate of molecular mass 20 kilodalton (Ts; 1.0 g, 0.05 mmol) was added in several portions over 2 hours. The mixture was stirred at room temperature for 5 days, and concentrated by rotary evaporation. The residue was diluted with water (30 rnL), and stirred at room temperature for 2 h to destroy any excess 20 kilodalton mPEG- O-tosylate. The solution was then neutralized with acetic acid, the pH adjusted to pH 5.0 and loaded onto a reversed phase chromatography (C- 18 silica) column. The column was eluted with a gradient of methanol/water (the product elutes at about 70% methanol), product elution monitored by evaporative light scattering, and the
appropriate fractions collected and diluted with water (500 mL). This solution was chromatographed (ion exchange, XK 50 Q, BIG Beads, 300 ml, hydroxide form; gradient of water to water/acetic acid-0.75N) and the pH of the appropriate fractions lowered to 6.0 with acetic acid. This solution was then captured on a reversed phase column (C- 18 silica) and eluted with a gradient of methanol/water as described above. The product fractions were pooled, concentrated, redissolved in water and freeze-dried to afford 453 mg (44%) of a white solid (1). Structural data for the compound were as follows: 1H-NMR (500 MHz; D2O) δ 2.83 (t, 2H, 0-C-CH2-S), 3.05 (q, IH, S-CHH-CHN), 3.18 (q, IH5 (q, IH, S-CHH- CHN), 3.38 (s, 3H, CH3O), 3.7 (t, OCH2CH2O), 3.95 (q, IH, CHN). The purity of the product was confirmed by SDS PAGE.
b. Synthesis of 2 (Cysteine-PEG2)
[0370] Triethylamine (-0.5 mL) was added dropwise to a solution of compound 1 (440 mg, 22 μmol) dissolved in anhydrous CH2Cl2 (30 mL) until the solution was basic. A solution of 20 kilodalton mPEG-O-p-nitrophenyl carbonate (660 mg, 33 μmol) and N- hydroxy succinimide (3.6 mg, 30.8 μmol) in CH2Cl2 (20 mL) was added in several portions over 1 h at rt. The reaction mixture was stirred at room temperature for 24 h. The solvent was then removed by rotary evaporation, the residue was dissolved in water (100 mL), and the pH adjusted to 9.5 with 1.0 N NaOH. The basic solution was stirred at room temperature for 2 h and was then neutralized with acetic acid to a pH 7.0. The solution was then loaded onto a reversed phase chromatography (C-18 silica) column. The column was eluted with a gradient of methanol/water (the product elutes at about 70% methanol), product elution monitored by evaporative light scattering, and the appropriate fractions collected and diluted with water (500 mL). This solution was chromatographed (ion exchange, XK 50 Q, BIG Beads, 300 mL, hydroxide form; gradient of water to water/acetic acid-0.75N) and the pH of the appropriate fractions lowered to 6.0 with acetic acid. This solution was then captured on a reversed phase column (C- 18 silica) and eluted with a gradient of methanol/water as described above. The product fractions were pooled, concentrated, redissolved in water and freeze-dried to afford 575 mg (70 %) of a white solid (2). Structural data for the compound were as follows: 1H-NMR (500 MHz; D2O) δ 2.83 (t, 2H, 0-C-CH2-S), 2.95 (t, 2H, O-C- CH2-S), 3.12 (q, IH, S-CHH-CHN), 3.39 (s, 3H CH3O), 3.71 (t, OCH2CH2O). The purity of the product was confirmed by SDS PAGE.
EXAMPLE 2
2.1 Preparation of Interferon-ateha-lb-GalNAc-SA-P EG-IOKDa
[0371] The IFN-alpha-2b (2 mL, 4.0 mg, 0.2 micromoles) was buffer exchanged twice with 10 mL of Washing Buffer and then concentrated to a volume of 0.3 mL using a Centricon centrifugal filter, 5 KDa MWCO. The IFN-alpha-2b was reconstituted from the spin cartridge using 2.88 mL of Reaction Buffer and then UDP-GaINAc (12 micromoles, 0.15 mL of an 80 mM solution in Reaction Buffer), GalNAc-T2 (0.06 mL, 58 mU), CMP-SA-PEG- 20KDa (17.5 mg, 0.875 micromoles dissolved in 0.75 mL of Reaction Buffer, 0.22 mM final reaction concentration), and STόGalNAcl (0.06 mL, 258 mU) were added to the reaction mixture to bring the total reaction volume to 4.0 mL. The reaction was incubated at 320C for 40 hours under a slow rotary movement and was monitored by SDS PAGE at 0 h and 40 h. The product, interferon-alpha-2b-GalNAc-SA-PEG-20KDa, was analyzed by MALDI (FIG. 9), peptide map (FIG. 10) and SDS-PAGE (colliodal blue and sliver stain) (FIG. 11 and 12).
[0372] The identity of the rest of the products in Example 2 were similarly confirmed by MALDI, peptide map, and SDS-PAGE.
2.2 Preparation of Interferon-alpha^b-GalNAc-SA-PEGSOKDa
[0373] The IFN-alpha-2b (2 mL, 4.0 mg, 0.2 micromoles) was buffer exchanged twice with 10 mL of Washing Buffer and then concentrated to 0.3 mL using a Centricon centrifugal filter, 5 KDa MWCO. The IFN-alpha-2b was reconstituted from the spin cartridge using 2.98 mL of Reaction Buffer and then UDP-GaINAc (12 micromoles, 0.15 mL of an 80 mM solution in Reaction Buffer), GalNAc-T2 (0.06 mL, 58 mU), CMP-SA-PEG-30K (26.3 mg, 0.875 micromoles in 0.75 mL of Reaction Buffer), and ST6GalNAcl (0.06 mL, 258 mU) were added to the reaction mixture to bring the total volume to 4.0 mL. The reaction was incubated at 320C for 40 hours under a slow rotary movement and the reaction monitored by SDS PAGE at 0 h and 40 h.
2.3 Preparation of Interferon-alpha-2b-GalNAc-Gal-SA-PEG-40KDa
[0374] The IFN-alpha-2b (2.5 mL, 5.0 mg, 0.25 micromoles) was concentrated to 0.1 mL using a Centricon centrifugal filter, 5 KDa MWCO. The concentrated sample was diluted with 5 mL of Washing Buffer and concentrated again to 0.1 mL. The IFN-alpha-2b was reconstituted from the spin cartridge using 1.9 mL of Reaction Buffer and UDP-GaINAc (15 micromoles, 0.10 mL of a 150 mM solution in Reaction Buffer), GalNAc-T2 (0.05 mL, 50 mU), UDP-Galactose (15 micromoles, 0.1 mL of 150 mM solution in Reaction Buffer), core-
1 -GaI-Tl (0.3 niL, 150 mU), CMP-SA-PEG-40KDa (50 mg, 1.25 micromoles dissolved in 1.95 mL Rxn Buffer, 0.25 mM final reaction concentration), and ST3Gal2 (0.51 rnL, 450 mU) were added to the reaction mixture to bring the total reaction volume to 5.0 mL. The reaction was incubated at 320C for 40 hours under a slow rotary movement and was monitored by SDS PAGE at 0 h and 40 h.
2.4 Preparation of Interferon-alpha^b-GalNAc-SA-PEG-WKDa
[0375] The IFN-alpha-2b (5.0 mL, 5.0 mg, 0.25 micromoles) was buffer exchanged twice with 10 mL of Washing Buffer and then concentrated to 0.3 mL using a Centricon centrifugal filter, 5 KDa MWCO. The IFN-alpha-2b was reconstituted from the spin cartridge using 2.61 mL of Reaction Buffer and then UDP-GaINAc (15 micromoles, 0.19 mL of an 80 mM
Reaction Buffer solution), GalNAc-T2 (0.07 mL, 67 mU), CMP-SA-PEG-40 KDa (40 mg, 1.0 micromoles dissolved in 1.75 mL of Reaction Buffer, 0.2 mM final reaction concentration), and ST6GalNAcl (0.08 mL, 344 mU) were added to the reaction mixture to bring the total volume to 5.0 mL. The reaction was incubated at 320C for 40 hours under a slow rotary movement and was monitored by SDS PAGE at 0 h and 40 h.
2.5 Preparation of Interferon-alpha^b-GalNAc-Gal-SA-PEG-όOKDa
[0376] The IFN-alpha-2b (3.2 mg, 0.17 micromoles) was reconstituted with 0.64 mL of Reaction Buffer and UDP-GaINAc (7.5 micromoles, 0.08 mL of a 50 mM solution in Reaction Buffer), GalNAc-T2 (0.032 mL, 32 mU), UDP-Galactose (12 micromoles, 0.08 mL of a 150 mM solution in Reaction Buffer), core-1 -GaI-Tl (0.2 mL, 100 mU), CMP-S A-PEG- 60 KDa (32 mg, 0.53 micromoles dissolved in 1.6 mL of Reaction Buffer, 0.17 mM final reaction concentration), and ST3Gal2 (0.24 mL, 220 mU) were added to the reaction mixture to bring the total volume to 3.2 mL. The reaction mixture was incubated at 320C for 40 hours under a slow rotary movement and was monitored by SDS PAGE gel electrophoresis at time points of O h and 4O h.
EXAMPLE 3
Preparaing the IFN-a conjugate formulations
[0377] All of the IFN-α conjugates were formulated in two steps, first by buffer exchange using spin filters, and second adjusting the formulation buffer volume to provide the desired protein concentration of between 50 to 100 mcg/mL. The formulation buffer was PBS, pH 7.06, 2.5% mannitol, and 0.01% Polysorbate 80. If necessary, endotoxin was removed from the formulated IFN-α conjugate by an endotoxin removal column of polymyxin. The IFN-α
conjugate was then sterile filtered (0.2 micro) into vials and stored frozen, although the IFN-α conjugate was stable for periods of days to weeks at 4 0C. These conjugates were then used in the assays described below.
EXAMPLE 4 Viral Inhibition Assay
[0378] The NewLab assay was used to test the PEGylated compound's ability to inhibit viral growth. The NewLab antiviral activity assay measures the inhibition of cytopathic effects of encephalomyocarditis virus (EMCV) in the Hep-2C cell line. The assay determined the relative potency of the glycoPEGylated INF-alpha-2b samples and chemoPEGylated IFN-α compared to the IFN-alpha-2b reference standard.
ChemoPEGylated IFN-α (containing PEG-40 KDa) and IFN-α-GalNAc-Gal-SA-PEG-40 KDa conjugates showed very similar potency. Dose response curves are shown in FIG. 13.
EXAMPLE 5
Cell Anti-Proliferation Assay [0379] An in vitro anti-proliferation assay was used to detect cell based effects of glycoPEGylated variants of IFN-alpha 2b. This assay is utilized as a quick method for determining whether compounds are active and to establish the relative potencies of the glycoPEGylated products. The dose response curves from which the antiproliferative specific activities were calculated are shown in FIG. 14.
EXAMPLE 6
6.1 Rat Pharmacokinetic Data a. Intravenous (iv) Pharmacokinetics
[0380] Following intravenous injection of IFN-alpha-2b, as well as chemoPEGylated IFN- α and IFN-α conjugates, clearance was characterized by an initial rapid phase, followed after 12 hours by a much slower phase FIG. 15. The major differences among the chemoPEGylated IFN-α and the IFN-α conjugate were mainly due to the rate and extent of clearance from plasma during the initial 8 hour period. IFN-alpha-2b showed a rapid drop in concentration during the initial 2 hour period. IFN-alpha-2b-GalNAc-SA-PEG conjugates prepared with linear 20 KDa or 30 KDa PEG exhibited slower clearance rates during the initial 8 hour period inversely related to the size of attached PEG. The clearance of chemoPEGylated IFN-α (covalently linked branched 40 KDa PEG) was significantly slower
that that observed for IFN-alpha with no PEG or with linear 20 KDa or 30KDa. The two IFN-α conjugates containing a single branched 40KDa PEG moiety linked to the glycan chain at Thr106 both were cleared more slowly than chemoPEGylated IFN-α, the slowest clearance profile belonging to the construct with IFN-alρha-2b-GalNAc-SA-PEG (40 KDa).
b. Subcutaneous (sc) Pharmacokinetics
[0381] Following subcutaneous injection of IFN-alpha, there was an initial decline in concentration from the first time (5 min) point until 4 hours, after which the very low concentration of IFN-alpha showed a further gradual decline FIG. 16. IFN-α conjugates prepared with linear 20KDa or 30KDa PEG reached concentration maxima at 8 hours, whereas the two IFN-α conjugates with branched 40KDa PEG, and the chemoPEGylated
IFN-α (covalently linked branched 40 KDa PEG) reached concentration maxima at 24 hours. The maximum concentration values increased in direct relation to the size of the attached PEG moiety, with nearly identical profiles for chemoPEGylated IFN-α and IFN-alpha-2b- GalNAc-Gal-SA-PEG-40 KDa and somewhat higher concentration values observed for IFN- alpha-2b-GalNAc-SA-PEG-40 KDa at all points after 2 hours.
EXAMPLE 7
Monkey Pharmacokinetic Data
[0382] Pharmacokinetic data was obtained from monkeys. The subjects used were young adult Cynomolgus monkeys between 3.5-6.5 years old. They were matched for sex and body weight: 3.3-3.6 kg for males; 2.3-3.1 kg for females. All of the selected monkeys completed the study. A single subcutaneous dose of the compound was given to each monkey and the size of the dosage was linked to body weight: 100 meg of the compound per Kg monkey body weight.
[0383] At certain time intervals, blood was drawn from the monkeys. 2 mL of blood were drawn for each interval, for a total of 26 draws. The results of this study are presented in the table below and in FIG. 17.
Pharmacokinetic Determinants
[0384] The effect of the compounds on 2' -5' oligoadenylate syntethase was also measured, the results of this study are presented in the table below and in FIG. 18.
Pharmacod namic Determinants
[0385] It is understood that the examples and embodiments described herein are for illustrative purposes only and that various modifications or changes in light thereof will be suggested to persons skilled in the art and are to be included within the spirit and purview of this application and scope of the appended claims. All publications, patents, and patent applications cited herein are hereby incorporated by reference in their entirety for all purposes.