Description
NOVEL PROCESS FOR THE PREPARATION OF CILASTATIN
SODIUM SALT
Technical Field
[1] The present invention relates to novel process for preparing cilastatin sodium salt used as a dehydropeptidase 1 inhibitor.
Background Art [2] Cilasatin sodium salt i.e., [R-[R*, S*-(Z)]] -
7-[(2-amino-2-carboxyethylthio)-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino-2-hepa tenoic acid monosodium salt represented by following chemical formulae (1)1 has been used with imipenem in order to prevent its renal metabolism. Imipenem/cilastatin sodium is used as a potent broad spectrum antibacterial agent. [3] There have been several reports on the method for preparing a cilastatin sodium until now: for example, EP 48301 Bl discloses a method for the preparation of a cilastatin sodium salt using by Grignard reaction started from l-bromo-5-chloropentane (2') explained by following Reaction Scheme 1; Donald W.
Graham et al discloses a preparation method using ethyl- 1, 3-dithian-2-carboxylate as a starting material (Donald W. Graham et al, J. Med. Chem., 30, pplO74, 1987) etc. [4] [Reaction Scheme 1]
[5]
[6]
[7] [8] As shown in the above Reaction Scheme 1, l-bromo-5-chloropentane (2') is reacted with diethyl oxalate through Grignard reaction to afford ethyl 7-chloro-2-oxo-hepanoate (3')at the 1st step; ethyl 7-chloro-2-oxo-heptanoate (3') is reacted with (S)-2, 2-dimethylcyclopropanecarboxamide to obtain ethyl (Z )-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate (4') at the 2n step.
[9] However, present inventors has confirmed that considerable amount (about 10 to 13%) of (E)-form isomer thereof (7')was produced during the 2nd step as a reaction impurity by gas chromatography. The (E)-form isomer is further subjected to hydrolysis resulting in (E)-7-chloro-2-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (8')as shown in following Reaction Scheme 2.
[10]
[H] However, present inventors has confirmed that considerable amount (about 10 to 13%) of (E)-form isomer thereof (7') was produced during the 2nd step as an reaction impurities by gas chromatography as shown in following Reaction Scheme 2. The (E )-form isomer is further subjected to hydrolysis resulting in (E)-7-chloro-2-((S)-2, 2-dimethylcyclopropylcarboxamide)-2-heptanoic acid (8').
[12] [Reaction Scheme 2] [13]
[14] [15] There have been tried to solve the problems for example, the isomer impurity was removed by the acidification followed by recrystallization step or by adding cysteine to the reaction solution obtained in the 3r step at the above described 4 step, reacting with together to form (E)-7-(L-amino-2-carboxyethylthio)-2-((S )-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid and finally removing the reacted impurity by acidifying and heating step in the known preparation till now. However, the present inventors found that there remained unsolved problem such that the recrystallization yield of the product, i.e., (Z)-7-chloro-2-((S )-2,2-dimethylcyclopropylcarboxamide)-2-heptanoic acid was very poor because of the formed byproduct, i.e., (E
)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid in 3rd step and further the unknown impurity (10') and (S)-2,2-dimethylcyclopropanecarboxamide (H') were produced by acidifying and heating reaction solution at the above described the 4 step as shown in following Reaction Scheme 3 confirmed by HPLC analysis, which give rise to another difficulty in the purification of final products.
[16] [17] [Reaction Scheme 3] [18]
NH
(Z) and (E) m ix ture (91)
(105 0 15
[19] In addition to above described problems, present inventors have found that the cilastatin isolated through the above described 4th step consisting of eluting the cation exchange resin with ammonia solution, concentrating the eluate and solidifying with ethanol and diethyl ether exists in the form of its ammonium salt not free acid form as disclosed in the patent. Using an acid such as hydrochloric acid in order to obtain free acid accompany with unwanted formation of inorganic ammonium salt such as ammonium chloride, which could not afford high purity of cilastatin sodium salt in the end.
[20]
[21] Therefore, there have been tried to solve the above-described problems: for example, PCTAVO 0318544 (Al) discloses the isolation method using by neutral HP 20 resin column instead of cationic resin disclosed in EP 48301 Bl; PCTAVO 02094742 (Al) discloses the method for preparing cilastatin sodium salt (Ia) from cilastatin (6'), the disclosure of which cited documents are incorporated herein by reference.
[22]
[23] However, the above-described methods for preparing cilastatin using column chro¬ matographic process are not suitable for commercial mass production.
[24]
[25] The present inventors have made extensive researches to discover novel method for preparing cilastatin sodium salt with high yield and mass production and finally completed the invention by founding novel preparation for obtaining purposed cilastatin sodium salt; i.e., selectively hydrolyzing (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate, isolating (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt from the reaction mixture, adopting the cilastatin amine salt instead of free acid form disclosed in cited references and the use of sodium hydroxide and cationic exchange resin with pH control in order to obtain cilastatin sodium salt with high purity and high yield.
[26]
Disclosure of Invention Technical Problem
[27] Accordingly, in a preferred embodiment of the present invention, the present invention provides novel method for preparing cilastatin sodium salt used as a dehy- dropeptidase 1 inhibitor.
Technical Solution
[28] The present invention provides novel method for preparing (Z)-7-chloro-2-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt represented by general chemical formula (12) comprising the steps consisting of: selectively hy¬ drolyzing (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate represented by chemical formula (4) in reaction solvent under basic condition and removing un-reacted product remained in reaction solvent layer with washing organic solvent with controlling the pH of reaction solution with acid to be neutralization at the 1st step; concentrating remaining water layer, adding alcohol thereto with heating, stirring to the extent to dissolve the solid, filtering out un-dissolved salt, and con¬ centrating the filtrate under reduced pressure at the 2nd step; adding organic solvent to solidify the concentrate, filtrating the solution to isolate and purify (Z)-7-chloro-2-((S
)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt represented by chemical formula (12) at the final step.
[29]
[30]
[31]
[32] Wherein M+ is alkali metal salt. [33] Specifically, at the 1st step in the above-described reaction, the reaction solvent may be used water, lower alcohol such as methanol, ethanol and propanol, or the mixture thereof, preferably the mixture of water and methanol, water and ethanol or water and propanol may be preferably used, and the reaction is preferably to be finished where the ratio of (Z) and (E) isomer is 100-10: 1, more preferably 50-15: 1, most preferably 30-20: 1 to provide with selective hydrolysis. Strong base such as lithium hydroxide (LiOH), sodium hydroxide (NaOH), potassium hydroxide (KOH) etc may be preferably used and the organic solvent such as diisopropylether, dichloromethane, carbon tetrachloride, chloroform, hexane etc are preferable as a washing organic solvent. The pH of reaction solution ranging from 6 to 8, preferably about 7 may be used to neutralization by controlling with strong acid such as hydrochloric acid, sulfuric acid etc or weak acid such as acetic acid etc at the 1st step.
[34]
,nd [35] At the 2 step, the stirring with alcohol preferably is performed at the temperature ranging from about 10 to 80°C, preferably about 30 to 60°C, more preferably about
50°C, for the period ranging from 10 minutes to 24 hours, preferably 30 minutes to 1 hour and the lower alcohol such as methanol, ethanol and propanol, or mixture thereof may be preferably used to remove formed inorganic salt.
[36] At the final step, the lower alcohol such as methanol, ethanol and propanol, acetone, acetonitrile or the mixture thereof, preferably the mixture of water and alcohol, alcohol and acetonitrile, alcohol and acetone etc may be preferably used to isolate final product.
[37]
[38] Through above-described preparation method, (Z)-7-chloro-2-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid salt represented by following chemical formula (12) can be synthesized with high purity (more than 90%) and the method could provide the next step with reproducibility with quantitative supply in the synthesis of cilastatin amine salt (13).
[39]
[40] The M group of general chemical formula (12) includes all the alkali metal salt such as lithium salt, sodium salt, potassium salt and the like however the present invention does not intent to limit or define the kind of salt herein.
[41]
[42] In another preferred embodiment of the present invention, the present invention provides novel method for preparing cilastatin sodium salt represented by chemical formula (1) comprising the steps consisting of: reacting (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptanoic acid or the salt thereof represented by chemical formula (12) with cysteine in base solution at the 1st step; controlling the pH of the reaction solution obtain in step 1, concentrating, adsorbing the concentrate with cationic exchange resin, washing with water, eluting with amine solution to concentrate the elutes at the 2n step; dissolving the concentrates in recrystallization solvent and subjecting to recrystallization process by adding alcohol in a dropwise manner to afford pure cilastatin amine salt (13) at the 3r step; reacting cilastatin amine salt (13) with sodium hydroxide and controlling the pH with cationic exchange resin at the 4 step.
[43]
[44]
[45] Wherein R is a hydrogen atom or lower alkyl group. [46] The R group of general chemical formula (13) includes a hydrogen atom or C1-C4 alkyl group such as methyl, ethyl, propyl group etc preferably.
[47] [48] Specifically, at the 1st step in the above-described reaction, (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid or the salt thereof (12) dissolved in water, alcohol such as methanol, ethanol or propanol, or the mixture thereof may be reacted with catalyst such as sodium bromide, sodium iodide, potassium iodide, potassium bromide etc and cysteine in the presence of strong base such as sodium hydroxide, potassium hydroxide etc under nitrogen atmosphere at the temperature ranging from about 10 to 80°C, preferably about 30 to 60°C, more preferably about 55°C, for the period ranging from 1 hour to 24 hours, preferably 5 to 10 hours, more preferably about 8 hours.
[49] At the 2n step in the above-described reaction, the product obtained from 1st step is adjusted to the pH of the solution ranging from 3.0 to 7.0, preferably 5.0 to 5.5, con¬ centrated and the alcohol such as methanol, ethanol or propanol is added thereto. The solution is stirred at the temperature ranging from about 10 to 80°C, preferably about 30 to 60°C, more preferably about 50°C, for the period ranging from 10 minutes to 3
hours, preferably 30 minutes to 1.5 hours, more preferably about 1 hour, subjected to filtration to remove resulting inorganic salt and the filtrate is concentrated to the extent that total volume of solution is reduced to about 1/2. The concentrate is adsorbed with cationic exchange resin such as styrene strong acidic resin and the column is washed with water to the extent that the conductivity becomes less than 20μs, preferably lOμs and eluted with amine solution in the concentration ranging from 1 to 5 N, preferably 2N amine solution to concentrate the eluate. In case that ammonia water is used as an eluate, cilastatin ammonium salt (13) is formed. However, the cilastatin amine salt (13) of which salt form is varied according to the kind of amine salt may be formed if amine solution is used as an eluate.
[50]
[51] At the 3rd step, the water and ammonia water, or the mixture thereof may be used as a recrystallization solvent, preferably the solvent in the amount ranging from 1:3 to 2:1 (w/v) of the weight of cilastatin amine salt is added to the concentrate obtained in step 2 and alcohol such as ethanol, n-propanol, 2-propanol etc, preferably the solvent in the amount ranging from 1 : 10 to 1 :40 (w/v) of the weight of cilastatin amine salt is added. The recrystallization process is preferably performed at less than 100°C, preferably 5 to 97°C. In an alternative method to purify the concentrate, the above-described solvent is added to cilastatin concentrate, subjected to salting out method performed by refluxing for the period ranging from 2 to 3 hours, cooling, and filtrating the solution to obtain purposed cilastatin amine salt (13).
[52] At the 4 step, appropriate amount of sodium hydroxide dissolved in recrys¬ tallization solvent is added to cilastatin amine salt (13), stirred for the period ranging from 10 minutes to 1.5 hour, preferably about 30 minutes and concentrated in vaccuo at the temperature ranging from 30 to 70°C, preferably about 60°C. Distilled water is added to the concentrate, stirred and the pH of the solution is adjusted to 6.0-8.0, preferably about 7.0 with cationic exchange resin. The solution is filtered, and the filtrate is lyophilized to obtain purposed highly pure cilastatin sodium salt (1) with the purity of more than 99%.
[53]
[54] Through above-described process, highly pure cilastatin sodium salt can be obtained and the inventive method of the present invention is a novel method using nevel intermediate, i.e., cilastatin amine salt.
[55]
[56] In the other preferred embodiment of the present invention, the present invention provides novel intermediate represented by chemical formula (13).
[57]
[59] Wherein R is a hydrogen atom or lower alkyl group. [60] [61] The cilastatin sodium salt of the invention of formula (1) may be chemically synthesized by the methods which will be explained by following reaction schemes hereinafter, which are merely exemplary and in no way limit the invention. The reaction schemes show the representative method for preparing cilastatin sodium salt of the present invention, and the other method also may be modified by following the steps with appropriate modifications of reagents and starting materials, which are envisaged by those skilled in the art. Advantageous Effects
[62] The novel method of the present invention could prevent the production of (E)-isomer from the preparation of dehydropeptidase 1 inhibitor, i.e., (Z)-7-[chloro-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt and isolate the dehydropeptidase 1 inhibitor in situ providing simpler process with high yield and high purity. Best Mode for Carrying Out the Invention
[63] It will be apparent to those skilled in the art that various modification and variation can be made in the compositions, use and preparations of the present invention without departing from the spirit or scope of the invention.
[64] The present invention is more specifically explained by the following figures and examples. However, it should be understood that the present invention is not limited to these examples in any manner. Mode for the Invention
[65] The present invention is more specifically explained by the following examples. However, it should be understood that the present invention is not limited to these examples in any manner.
[66] Comparative Example 1: Preparation of ethyl (Z)-7-chloro-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoate (4)
[67]
[68] l-bromo-5-chloropentane (29 Ig, 1.57 mol) was reacted with diethyl oxalate
(206.5g) through Grignard reaction to obtain ethyl 7-chloro-2-oxo-heptanoate (3) and the compound (3) was reacted with (S)-2,2-dimethylcyclopropanecarboxamide to obtain ethyl (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptanoate (237g, 0.79 mol). The above-described step was performed by the procedure according to the procedure disclosed in EP 48301 (Bl).
[69]
[70] Example 1: Preparation of ethyl (Z)-7-chloro-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12)
[71]
[72] 1-1. ( Z V7-chloro-(YSV2. 2-dimethylcyclopropanecarboxamidoV2-heptanoic acid sodium salt
[73] The ethyl (Z)-7-chloro-2-((S
)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoate (237g, 0.79 mol) obtained in Comparative Example 1 was dissolved in 877ml of methanol and 1.8 L of sodium hydroxide solution (0.48 M) was added with stirring at room temperature. The reaction was finished when the area ratio of (Z) isomer and (E) isomer becomes 20: 1 by HPLC analysis and the un-reacted organic reagent was extracted with 490 ml of dichloromethane. The pH of the solution was adjusted to 7-8 with 3N HCl and the un- reacted organic reagent was extracted with 490 ml of dichloromethane again. The water layer was concentrated under reduced pressure and 650ml of ethanol was added and stirred until the solid had been dissolved at 50°C, for 30 minute to 1 hour. The un- dissolved solid was removed with filtration and the filtrate was concentrated under reduced pressure. 2.4 L of acetonitrile is added thereto and stirred to obtain 140.8g of ( Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12 ; 55% yield).
[74]
[75]
[76] m.p.: 219°C;
[77] 1H-NMR (D2O, 300MHz) δppm: 0.87 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.61 (m, 2H), 1.68 (dd, IH), 1.78 (m, 2H), 2.12 (m, 2H), 3.62 (t, 2H), 6.47 (t, IH);
[78]
13
[79] 13C ( -NMR (D2O, 300MHz) δppm: 19.47, 19.99, 22.55, 25.74, 26.75, 27.53, 29.44,
32.27, 46.11, 131.41, 136.52, 172.74, 174.62.
[80]
[81] [82]
[83] 1-2. ( Z V7-chloro-(YSV2. 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid
(12-D
[84] 140.8g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12) obtained from Example 1-1 was dissolved in 422 ml of distilled water. The pH of the solution was adjusted to 2.0-3.0 with 3N HCl, extracted with 592 ml of isopropylether two times and 59.2g of anhydrous magnesium sulfate was added to isopropylether layer, stirred and subjected to filtration. The filtrate was concentrated to afford 127.7g of (Z)-7-chloro-2-((S )-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (12-1, 98% yield).
[85]
[86] 1H-NMR (CDCl3, 300MHz) δppm: 0.83 (dd, IH), 1.19 (s, 7H), 1.44 (dd, IH), 1.19
(s, 3H), 1.64 (m, 2H), 1.81 (m, 2H), 2.21 (m, 2H), 3.54 (t, 2H), 6.78 (t, IH), 7.04 (br, IH);
[87]
13
[88] 13C ( -NMR (CDCl3, 300MHz) δppm: 18.69, 20.82, 22.86, 25.36, 27.03, 28.53,
29.27, 32.17, 44.60, 124.88, 139.49, 168.96, 170.15.
[89]
[90]
[91] 1-3. ( Z V7-chloro-((SV2. 2-dimethylcyclopropanecarboxamidoV2-heptenoic acid ammonium salt (12-2)
[92] 127.7g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (12-1) obtained from Example 1-2 was dissolved in 422 ml of EtOH. 100 ml of 25% ammonia water solution was added thereto, stirred and concentrated to obtain 135.6g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid ammonium salt (12-2, 100% yield).
[93]
[94] Example 2: Preparation of cilastatin ammonium salt (13-1)
[95] 4Og of (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12, 0.14 mol) obtained in Example 1-1 was dissolved in 120 ml of 0.48 M sodium hydroxide solution and 240 ml of ethanol and the mixture of 1.4g of NaBr (0.013 mol) and 25.3g of L-cysteineDHClDH) was added thereto, stirred at 55°C, for 8 hours.
[96] The pH of the reaction solution was adjusted to 5.5-5.0 with 3N HCl, concentrated and 800ml of methanol was added, stirred at 55°C for 1 hour and un-dissolved salt was filtered out. The filtrate was concentrated to the extent that the volume of total solution was reduced to about 1/2. The concentrate was adsorbed with cationic exchange resin
(PK208 model, Samyang Co.), washed with distilled water to the extent that the con¬ ductivity of the solution became less than lθμs(microsiemens), eluted with 2N ammonia water and the eluate was concentrated under the reduced pressure to give brown solid compound. The compound was dissolved in 40 ml of distilled water. 0.8 L of 2-propanol was added thereto and the solution was subjected to salting out method with reflux for 2 hours. The resulting solid was cooled and filtered to obtain 45.66g of cilastatin ammonium salt (13-1. 90% yield).
[97]
[98] m. p.: 161°C;
[99] Element Analysis: C16H29N3O5S (MW: 375.183): CaI. Q51.18; 7.78; N:11.19; Est.
C:51.01; H: 7.97; N: 11.04;
[100] MS m/z : 375 (M+, 49), 312(36), 97 (84.2), 69 (100);
[101] 1H-NMR (D2O, 300MHz) δppm: 0.87 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.62 (m, 5H), 2.1 l(q, 2H), 2.62 (t, 2H), 3.06 (m, 4H), 3.91 (dd, IH), 6.47 (t, IH);
13
[102] " (C-NMR (D2O, 300MHz) δppm: 19.49, 19.97, 22.53, 26.74, 27.44, 27.86, 29.09,
29.43, 31.94, 32.85, 54.44, 131.23, 136.83, 172.70, 173.71, 174.64.
[103]
[104] Example 3: Preparation of cilastatin ethylamine salt (13-2)
[105] 4Og of (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12-1, 0.15 mol) obtained in Example 1-2 was dissolved in 165 ml of 0.66 M sodium hydroxide solution and 330 ml of ethanol and the mixture of 1.5g of NaBr (0.015 mol) and 27.6g of L-cysteineDHClDH) was added thereto, stirred at 55°C, for 8 hours.
[106] The pH of the reaction solution was adjusted to 5.5-5.0 with 3N HCl, concentrated and 800ml of methanol was added, stirred at 55°C for 1 hour and un-dissolved salt was filtered out.. The filtrate was concentrated to the extent that the volume of total solution was reduced to about 1/2. The concentrate was adsorbed with cationic exchange resin (PK208 model, Samyang Co.), washed with distilled water to the extent that the conductivity of the solution became less than 10μs(microsiemens), eluted with 2N ethylamine water and the eluate was concentrated under the reduced pressure to give brown solid compound. The compound was dissolved in 40 ml of distilled water. 0.8 L of 2-propanol was added thereto and the solution was subjected to salting out method with reflux for 2 hours. The resulting solid was cooled and purified with filtration to obtain 49.38g of cilastatin ethylamine salt (13-2. 90% yield).
[107]
[108] 1H-NMR (D2O, 300MHz) δppm: 0.86 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.27 (t, 3H), 1.60 (m, 5H), 2.1 l(q, 2H), 2.62 (t, 2H), 3.06 (m, 4H), 3.91 (dd, IH), 6.47 (t, IH);
[109] 13C-NMR (D2O, 300MHz) δppm: 14.7, 21.57, 22.04, 24.63. 28.82, 29.52, 29.94,
31.16, 31.49, 34.00, 34.91, 37.78, 56.50, 133.27, 138.96, 174.75, 175.81, 176.74.
[HO]
[111] Example 4 : Purification of cilastatin ammonium salt
[112] 4-1. Purification using by water and ethanol
[113] 45.66g of cilastatin ammonium salt (13-1,0.12 mol) obtained in Example 2 was dissolved in 45.66 ml of distilled water and 1.3L of anhydrous ethanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 38.81g of cilastatin ammonium salt (Yield: 85%, Purity: 99.8%).
[114]
[115] 4-2. Purification using by ammonia water and propanol Q)
[116] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 50 ml of 25% ammonia water and 1.5L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 41.2g of cilastatin ammonium salt (Yield: 82.4%, Purity: 99.3%).
[117]
[118] 4-3. Purification using by ammonia water and propanol (1)
[119] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in
100 ml of 25% ammonia water and 2.0L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was purified with filtration to obtain 35.4g of cilastatin ammonium salt (Yield: 70.8%, Purity: 99.3%)
[120]
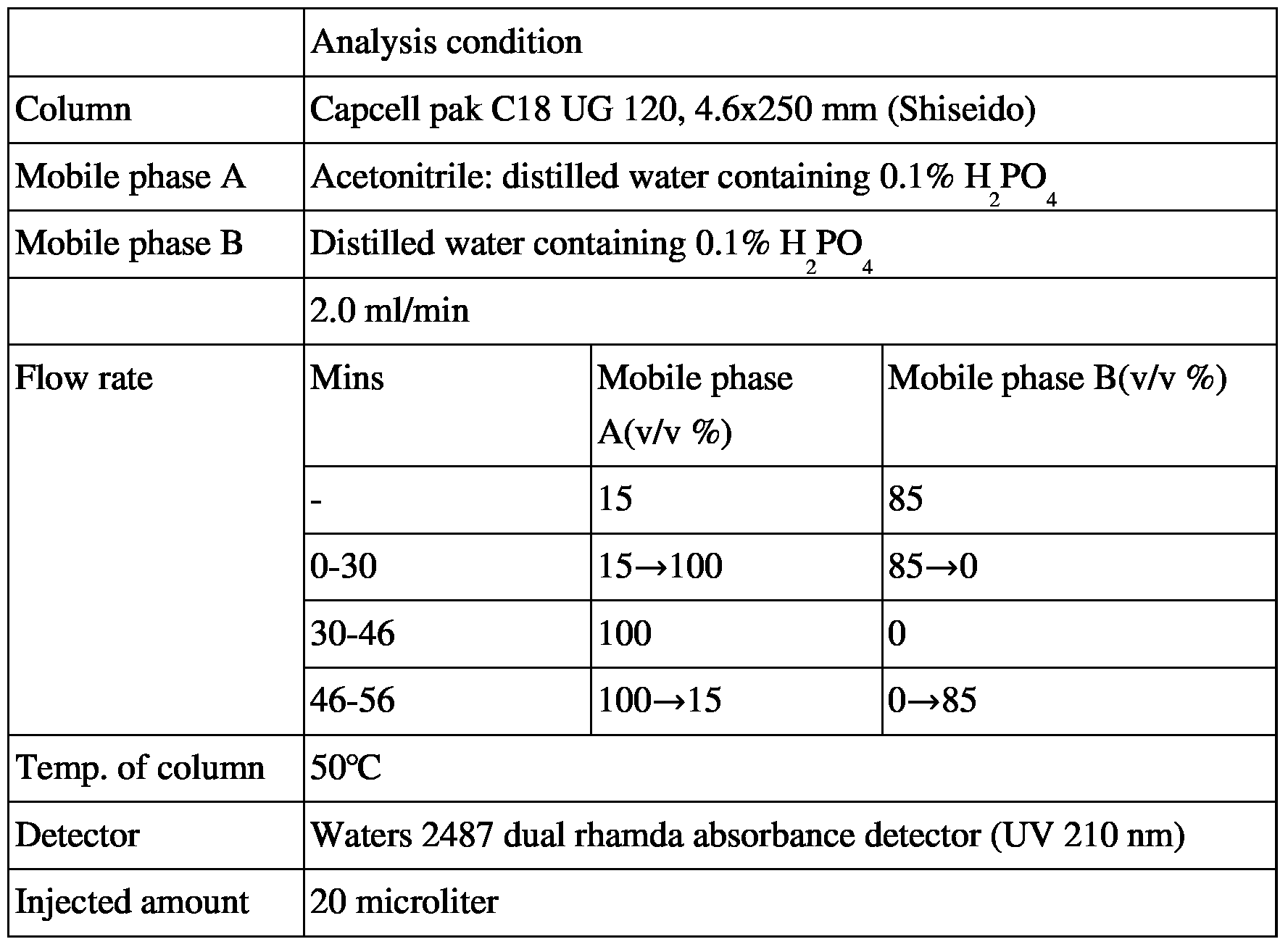
[121] 4-4. Purification using by ammonia water and ethanol
[122] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 50 ml of 25% ammonia water and 1.5 L of anhydrous ethanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 35.6g of cilastatin ammonium salt (Yield: 71.2%, Purity: 99.8%).
[123]
[124] 4-5. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (1)
[125] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 50ml of 4N ammonia water, and 2.0 L of 1 -propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 89.3g of cilastatin ammonium salt (Yield: 89.3%, Purity: 99.6%).
[126]
[127] 4-6. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (1)
[128] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 50ml of 2N ammonia water, and 2.0 L of 1-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 95.0g of cilastatin ammonium salt (Yield: 95.0%, Purity: 99.5%).
[129]
[130] 4-7. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (3)
[131] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 100 ml of distilled water and 50ml of 25% ammonia water, and 2.0 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 89.7g of cilastatin ammonium salt (Yield: 89.7%, Purity: 99.8%).
[132]
[133] 4-8. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (4)
[134] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 100ml of 25% ammonia water, and 3.0 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 80.Og of cilastatin ammonium salt (Yield: 80.0%, Purity: 99.7%).
[135]
[136] 4-9. Purification using by water and propanol
[137] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 100 ml of distilled water and 1.5 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 87.2g of cilastatin ammonium salt (Yield: 87.2%, Purity: 99.6%).
[138]
[139] Example 5: Preparation of cilastatin sodium salt
[140] 4.28g of sodium hydroxide (0.107 mol) was dissolved in 38.3 ml of distilled water and 191.5 ml of ethanol. 38.81g of cilastatin ammonium salt (0.1 mol) obtained in Example 4-1 was added thereto and stirred for 30 minutes. The solution was con¬ centrated under reduced pressure at 60°C and 153 ml of distilled water was added to the concentrate. The solution was stirred to dissolve the concentrate and the pH of the solution was adjusted to 7.0 using by cationic exchange resin and filtered. The filtrate was lyophilized to obtain high purity (99.4%) of cilastatin sodium salt.
[141]
[142] Experimental Example 1: Purity Determination
[143] The purity of cilastatin ammonium salt obtained in Example 4 was determined by
HPLC on condition as shown in Table 1 and the determined result was shown in Table 2.
[144] Table 1
[145] Table 2
[146]
Industrial Applicability
[147] The novel method of the present invention could prevent the formation of (E )-isomer from the preparation of novel intermediate for preparing cilastatin sodium, i.e., (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt and isolate the intermediate in situ providing simpler process with high yield and purity. Furthermore, it can provide with highly purified cilastatin sodium salt by isolating novel cilastatin amine salt and using sodium hydroxide and cationic exchange resin. Accordingly, the method can be very useful in preparing cilastatin sodium salt with high yield and high purity.