WO2005121149A1 - Furanopyrimidine compounds effective as potassium channel inhibitors - Google Patents
Furanopyrimidine compounds effective as potassium channel inhibitors Download PDFInfo
- Publication number
- WO2005121149A1 WO2005121149A1 PCT/GB2005/002318 GB2005002318W WO2005121149A1 WO 2005121149 A1 WO2005121149 A1 WO 2005121149A1 GB 2005002318 W GB2005002318 W GB 2005002318W WO 2005121149 A1 WO2005121149 A1 WO 2005121149A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- furo
- phenyl
- ylmethyl
- Prior art date
Links
- 0 CCOC=Nc([o]c(*)c1C)c1C#N Chemical compound CCOC=Nc([o]c(*)c1C)c1C#N 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
Definitions
- the present invention relates to furanopyrimidine compounds which are potassium channel inhibitors.
- Pharmaceutical compositions comprising the compounds and their use in the treatment of arrythmia are also provided.
- Ion channels are proteins that span the lipid bilayer of the cell membrane and provide an aqueous pathway through which specific ions such as Na + , K + , Ca + and CI " can pass (Herbert, 1998). Potassium channels represent the largest and most diverse sub-group of ion channels and they play a central role in regulating the membrane potential and controlling cellular excitability (Armstrong & Hille, 1998). Potassium channels have been categorized into gene families based on their amino acid sequence and their biophysical properties (for nomenclature see Gutman et al, 2003).
- potassium channels which modulate potassium channels have multiple therapeutic applications in several disease areas including cardiovascular, neuronal, auditory, renal, metabolic and cell proliferation (Shieh et al., 2000; Ford et al., 2002). More specifically potassium channels such as Kv4.3, Kir2.1, hERG, KVLQTl/minK, and Kvl.5 are involved in the repolarisation phase of the action potential in cardiac atrial myocytes. These potassium channel subtypes have been associated with cardiovascular diseases and disorders including long QT syndrome, hypertrophy, ventricular fibrillation, and atrial fibrillation, all of which can cause cardiac failure and fatality (Marban, 2002).
- cardiovascular diseases and disorders including long QT syndrome, hypertrophy, ventricular fibrillation, and atrial fibrillation, all of which can cause cardiac failure and fatality (Marban, 2002).
- Kvl.5 The human delayed rectifier voltage gated potassium channel subunit, Kvl.5, is exclusively expressed in atrial myocytes and is believed to offer therapeutic opportunities for the management of atrial fibrillation for several different reasons (see review of Brendel and Peukert, 2002): (i) There is evidence that Kvl.5 underlies the cardiac ultrarapid delayed rectifier (Kv (ur) ) physiological current in humans due to similar biophysical and pharmacological properties (Wang et al, 1993; and Fedida et al., 1993). This has been supported with antisense oligonucleotides to Kvl.5 which have been shown to reduce Kv (ur) amplitude in human atrial myocytes (Feng et al, 1997).
- the novel benzopyran derivative, NIP- 142 blocks Kvl.5 channels, prolongs the atrial refractory period and terminates atrial fibrillation and flutter in in vivo canine models (Matsuda et al, 2001), and S9947 inhibited Kvl.5 stably expressed in both Xenopus oocytes and Chinese hamster ovary (CHO) cells and Kv (ur) in native rat and human cardiac myocytes (Bachmann et al, 2001).
- Furanopyrimidines have been reported to be useful as folate inhibitors, anti-histamines, muscle relaxants and agrochemicals. Furanopyrimidines have not previously been reported as useful agents for modulating ion channels.
- Furanopyrimidines substituted in the 5 -position with an alkyl group have been identified as pesticides and fungicides.
- EP459611 discloses a family of 4-phenylethyl derivatives
- EP196524 discloses a series of 4-phenoxypropyl derivatives.
- Furanopyrimidines substituted with an alkyl group at the 5-position have also been disclosed as having muscle-relaxant activity (JP48081893 and DEI 817146).
- This invention provides compounds that are potassium channel inhibitors. Specifically, furanopyrimidines with an aromatic or heteroaromatic substituent at the 5-position are disclosed. These compounds are particularly useful for inhibiting potassium channels Kvl.5 or Kv (ur) , which are known targets for the treatment of cardiac arrhythmia in the atria such as atrial fibrillation (Nattel et al, 1999; Wang et al, 1993).
- This invention is not limited to treating cardiac arrythmias, the compounds also being useful to treat other diseases which require potassium channel inhibition (e.g. Shieh et al, 2000; Ford et al, 2002).
- the present invention provides a compound of formula I.
- Rl is aryl, heteroaryl, cycloalkyl or alkyl
- R2 is H, alkyl, nitro, -CO 2 R7, CONR4R5 or halo
- R3 is H, NR4R5, NC(O)R8, halo, trifluoromethyl, alkyl, nitrile or alkoxy
- R4 and R5 may be the same or different, and may be H, alkyl, aryl, heteroaryl or cycloalkyl; or R4 and R5 may together form a saturated, unsaturated or partially saturated 4 to 7 member ring, wherein said ring may optionally comprise one or more further heteroatoms selected from N, O or S;
- X is O, S or NR6;
- R6 is H or alkyl;
- R7 is hydrogen, methyl or ethyl;
- R8 is methyl or ethyl;
- L is (CH 2 ) n , where n is 1, 2 or 3
- alkyl group or moiety may be unsubstituted or substituted at any position. Typically, it is unsubstituted or carries one or two substituents. Suitable substituents include halogen, cyano, nitro, NR9R10, alkoxy, hydroxyl, unsubstituted aryl, unsubstituted heteroaryl, CO 2 R7, C(O)NR9R10, NC(O)R8 and SO 2 NR9R10.
- an aryl group is typically a C 6 -C 10 aryl group such as phenyl or napthyl.
- a preferred aryl group is phenyl.
- An aryl group may be unsubstituted or substituted at any position. Typically, it carries 1, 2, 3 or 4 substituents. Suitable substituents include cyano, halogen, nitro, trifluoromethyl, alkyl, alkyl thio, alkoxy, NR9R10, CO 2 R7, C(O)NR9R10, NC(O)R8 and SO 2 NR9R10 and hydroxyl.
- a heterocyclic group is a heteroaryl group, typically a 5- to 10- membered aromatic ring, such as a 5- or 6- membered ring, containing at least one heteroatom selected from O, S and N.
- heteroaryl groups include pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, furanyl, thienyl, pyrazolidinyl, pyrrolyl and pyrazolyl groups.
- Preferred heteroaryl groups are furanyl, thienyl and pyridyl.
- polycyclic heterocycles include indolyl, benzofuranyl, benzothiophenyl and benzodioxolyl.
- Non-aryl heterocyclic groups are also included, such as tetrahydrofuranyl or pyrrolidinyl.
- a heterocyclic group may be unsubstituted or substituted at any position.
- Suitable substituents include cyano, nitro, halogen, alkyl, alkylthio, alkoxy, NR9R10, CO 2 R7, C(O)NR9R10, NC(O)R8 and SO 2 NR9R10 and hydroxyl.
- R9 and R10 can be the same or different, and may be selected from H, unsubstituted alkyl, unsubstituted aryl, unsubstituted heteroaryl, unsubstituted cycloalkyl, aminoethyl, methylaminoethyl, dimethylaminoethyl, hydroxyethyl, alkoxyethyl, or R9 and R10 may together form a saturated, unsaturated or partially saturated 4 to 7 member ring.
- the ring may optionally comprise one, two, or three further heteroatoms.
- alkoxy means C ⁇ -3 alkoxy
- cycloalkyl means C 3 _ 6 cycloalkyl
- halogen means CI, F, Br, or I, preferably CI, F or Br.
- Preferred compounds of formula I are those wherein Rl is aryl or heteroaryl, R2 is H or alkyl, R3 is H, NR4R5, alkoxy or alkyl, X is O or NR6, R6 is H, n is 1 or 2 and Y is alkyl, cycloalkyl, aryl or heteroaryl.
- More preferred compounds of formula I are those wherein Rl is aryl or heteroaryl, R2 is H or methyl, R3 is H, NR4R5, alkoxy or alkyl, X is NR6, R6 is H, n is 1 and Y is aryl or heteroaryl.
- Y is phenyl, furanyl, thienyl or pyridyl. More preferably Y is optionally substituted phenyl, furan-2-yl or pyridine-2-yl.
- Preferred compounds include: 5-Phenyl-N-(pyridin-2'-ylmethyl)furo[2,3-d]pyrimidin-4-amine,
- a suitable nucleophile HX-L-Y is an alcohol, preferably ethanol. If a solvent is present the reaction is carried out at the reflux temperature of the solvent. Optionally the reaction may be carried out in a microwave reactor.
- a compound of formula II may be obtained from a compound of formula III by reaction with a trialkyl orthoformate in a suitable solvent or no solvent, and with heating.
- the trialkyl orthoformate is triethyl orthoformate.
- a solvent is present. Suitable solvents include acetic anhydride. When a solvent is present the reaction is carried out at reflux temperature.
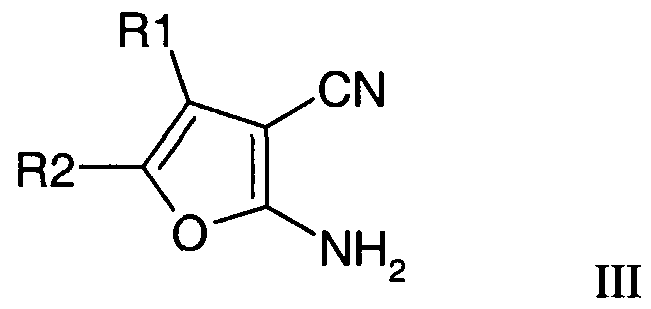
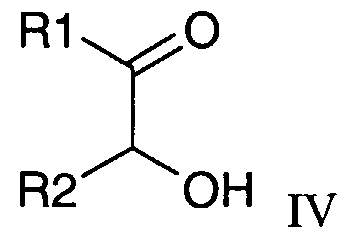
- Compounds of formula III may be obtained by the reaction of a compound of formula IV with malononitrile.
- the reaction may be carried out in the presence of a suitable solvent and a base.
- a suitable solvent is ethanol and the reaction is carried out under reflux conditions.
- a base is present.
- Suitable bases include hindered organic amines such as triethylamine.
- a compound of formula IV can be prepared by reaction of a compound of formula V under oxidative conditions.
- Preferred oxidizing agents include [& ⁇ ' .y(trifluoroacetoxy) iodo] benzene.
- the reaction is preferably carried out in the presence of a solvent and an organic acid.
- Suitable solvents include acetonitrile.
- Suitable organic acids include trifluoroacetic acid. When a solvent is present the reaction is carried out at reflux temperature.
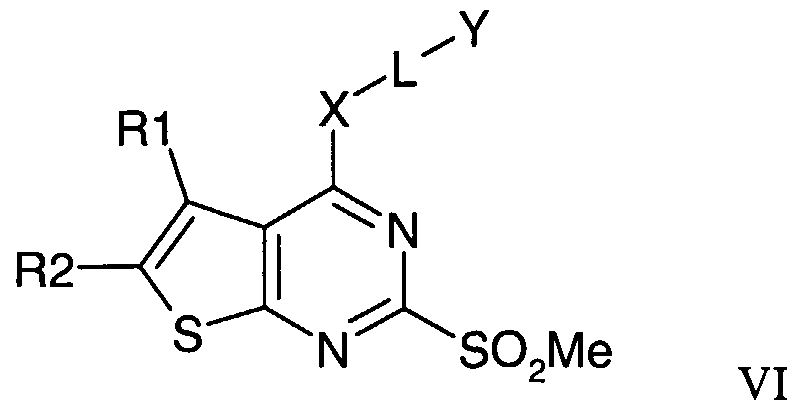
- a suitable nucleophile R3H is reacted with a compound of formula VI.
- the reaction is carried out in the presence of a base and a solvent.
- the reaction may be carried out in a microwave reactor.
- a compound of formula VI is conveniently prepared from a compound of formula VII.
- Suitable reaction conditions include the use of a nucleophile HX-L-Y in the presence of a base and a solvent.
- the base is a hindered organic amine base such as triethylamine and the solvent is an alcohol such as ethanol.
- a compound of formula VII may be prepared by oxidation of a compound of formula VIII. Suitable conditions include the use of a peroxide reagent in an organic acid.
- a peroxide reagent is hydrogen peroxide and the organic acid is acetic acid.
- a compound of formula VIII may be prepared by reaction of a compound of formula IX under alkylating conditions.
- Suitable conditions include the use of a methyl iodide, under basic conditions.
- the basic conditions comprise a metal hydroxide in a mixture of alcohol and water.
- the alcohol is ethanol
- the metal hydroxide is potassium hydroxide
- the alkyl halide is methyl iodide.
- a compound of formula IX may be conveniently prepared by reaction of a compound of formula III with potassium ethyl xanthate.
- the reaction is carried out in alcohol.
- the alcohol is butanol.
- a compound of formula X is reacted with a suitable nucleophile HX-L- Y.
- the reaction is carried out in the presence of a base and in a solvent.
- the base is preferably an organic amine base such as triethylamine and the solvent is preferably an alcohol such as ethanol.
- the reaction is carried out at the reflux temperature of the solvent. Alternatively the reaction may be carried out with microwave heating.
- a compound of formula X may be prepared by reaction of a compound of formula XI with a suitable chlorinating reagent.
- a suitable chlorinating reagent is thionyl chloride, phosphorous oxychloride or diphenylphosphinic chloride.
- the reaction is carried out in the presence of a base such as an amine base.
- bases include triethylamine and diethylaniline.
- the base may also serve as the solvent.
- the reaction is carried out at 60-100°C.
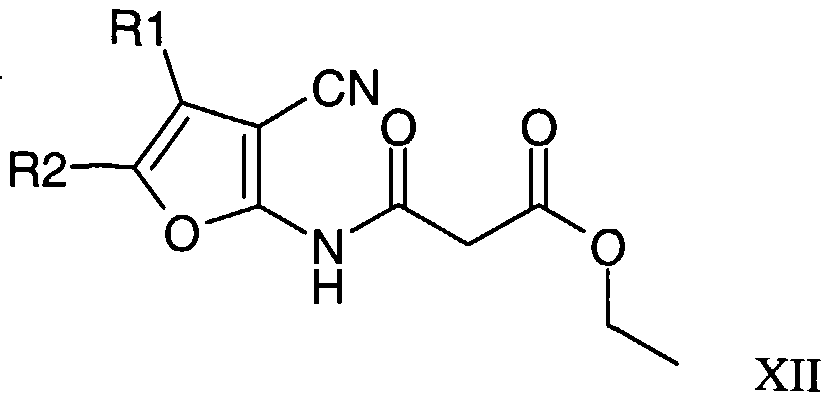
- Compounds of formula XI may be prepared by reaction of a compound of formula XII under acidic conditions in a solvent.
- the acid is an inorganic acid such as hydrochloric acid and the solvent is an organic solvent such as dioxane. The reaction is carried out at or below the reflux temperature of the solvent.
- compounds of formula I wherein R3 is a carboethoxy group may undergo functional group transformation of the ester moiety using methods familiar to those skilled in the art. In a preferred instance such compounds may undergo amidation by reaction with an alkyl or dialkylamine. In another preferred instance compounds of formula I wherein R3 is a 1-hydroxyethyl group can be prepared by the reaction with a reducing agent such as diisobutylaluminium hydride or lithium aluminium hydride. In a further instance compounds of formula I wherein R3 is a carboethoxy group may be reacted with a dialkyl carbonate under basic conditions to provide a compound of formula I wherein R3 is a dialkylmalonyl group. Such compounds may be reduced, preferably with a reducing agent such as diisobutylaluminium hydride or lithium aluminium hydride, to provide compounds of formula I wherein R3 is a propanediol group.
- a reducing agent such as diisobutylaluminium
- Suitable starting materials include:
- the compounds of the invention are useful in the treatment of various conditions.
- the present invention provides a pharmaceutical formulation comprising at least one compound of the invention and optionally one or more excipients, carriers or diluents, wherein said compound has the formula:
- Rl is aryl, heteroaryl, cycloalkyl or alkyl
- R2 is H, alkyl, nitro, -CO 2 R7, CONR4R5 or halo
- R3 is H, NR4R5, NC(O)R8, halo, trifluoromethyl, alkyl, nitrile or alkoxy
- R4 and R5 may be the same or different, and may be H, alkyl, aryl, heteroaryl or cycloalkyl; or R4 and R5 may together form a saturated, unsaturated or partially saturated 4 to 7 member ring, wherein said ring may optionally comprise one or more further heteroatoms selected from N, O or S;
- X is O, S or NR6;
- R6 is H or alkyl;
- R7 is hydrogen, methyl or ethyl;
- R8 is methyl or ethyl;
- L is (CH 2 ) n , where n is 1, 2 or 3
- compositions of the invention may be presented in unit dose forms containing a predetermined amount of each active ingredient per dose.
- a unit may be adapted to provide 5-100mg/day of the compound, preferably either 5-15mg/day, 10-30mg/day, 25- 50mg/day 40-80mg/day or 60-100mg/day.
- doses in the range 100-lOOOmg/day are provided, preferably either 100-400mg/day, 300-600mg/day or 500-lOOOmg/day.
- Such doses can be provided in a single dose or as a number of discrete doses. The ultimate dose will of course depend on the condition being treated, the route of administration and the age, weight and condition of the patient and will be at the doctor's discretion.
- compositions of the invention may be adapted for administration by any appropriate route, for example by the oral (including buccal or sublingual), rectal, nasal, topical
- formulations may be prepared by any method known in the art of pharmacy, for example by bringing into association the active ingredient with the carrier(s) or excipient(s).
- compositions adapted for oral administration may be presented as discrete units such as capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids; edible foams or whips; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
- compositions adapted for transdermal administration may be presented as discrete patches intended to remain in intimate contact with the epidermis of the recipient for a prolonged period of time.
- the active ingredient may be delivered from the patch by iontophoresis as generally described in Pharmaceutical Research, 3(6), 318 (1986).
- compositions adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- the formulations are preferably applied as a topical ointment or cream.
- the active ingredient may be employed with either a paraffinic or a water- miscible ointment base.
- the active ingredient may be formulated in a cream with an oil-in-water cream base or a water-in-oil base.
- compositions adapted for topical administration to the eye include eye drops wherein the active ingredient is dissolved or suspended in a suitable carrier, especially an aqueous solvent.
- compositions adapted for topical administration in the mouth include lozenges, pastilles and mouth washes.
- Pharmaceutical formulations adapted for rectal administration may be presented as suppositories or enemas.
- compositions adapted for nasal administration wherein the carrier is a solid include a coarse powder having a particle size for example in the range 20 to 500 microns which is administered in the manner in which snuff is taken, i.e. by rapid inhalation through the nasal passage from a container of the powder held close up to the nose.
- Suitable formulations wherein the carrier is a liquid, for administration as a nasal spray or as nasal drops, include aqueous or oil solutions of the active ingredient.
- Fine particle dusts or mists which may be generated by means of various types of metered dose pressurised aerosols, nebulizers or insufflators.
- compositions adapted for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations.
- compositions adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the formulations may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
- Preferred unit dosage formulations are those containing a daily dose or sub-dose, as herein above recited, or an appropriate fraction thereof, of an active ingredient.
- formulations may also include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavouring agents.
- the present invention provides a compound, or a pharmaceutical composition comprising said compound for use in medicine, wherein said compound has the formula:
- Rl is aryl, heteroaryl, cycloalkyl or alkyl
- R2 is H, alkyl, nitro, CO 2 R7, CONR4R5 or halo
- R3 is H, NR4R5, NC(O)R8, halo, trifluoromethyl, alkyl, nitrile or alkoxy
- R4 and R5 may be the same or different, and may be H, alkyl, aryl, heteroaryl or cycloalkyl; or R4 and R5 may together form a saturated, unsaturated or partially saturated 4 to 7 member ring, wherein said ring may optionally comprise one or more further heteroatoms selected from N, O or S;
- X is O, S or NR6;
- R6 is H or alkyl;
- R7 is hydrogen, methyl or ethyl;
- R8 is methyl or ethyl;
- L is (CH 2 ) n , where n is 1, 2 or 3; and
- the compound is a compound of the first aspect.
- compositions of the invention can be used to treat conditions which require inhibition of potassium channels, for example in the treatment of arrythmia.
- the present invention provides:
- a method of treating or preventing a disorder which requires potassium channel inhibition comprising administering to a subject an effective amount of at least one compound or of a pharmaceutical composition comprising said at least one compound and optionally one or more excipients, diluents and/or carriers wherein said compound has the formula:
- Rl is aryl, heteroaryl, cycloalkyl or alkyl
- R2 is H, alkyl, nitro, CO 2 R7, CONR4R5 or halo
- R3 is H, NR4R5, NC(O)R8, halo, trifluoromethyl, alkyl, nitrile or alkoxy
- R4 and R5 may be the same or different, and may be H, alkyl, aryl, heteroaryl or cycloalkyl; or R4 and R5 may together form a saturated, unsaturated or partially saturated 4 to 7 member ring, wherein said ring may optionally comprise one or more further heteroatoms selected from N, O or S;
- X is O, S or NR6;
- R6 is H or alkyl;
- R7 is hydrogen, methyl or ethyl;
- R8 is methyl or ethyl;
- L is (CH 2 ) n , where n is 1, 2 or 3; and
- Rl is aryl, heteroaryl, cycloalkyl or alkyl
- R2 is H, alkyl, nitro, CO 2 R7, CONR4R5 or halo
- R3 is H, NR4R5, NC(O)R8, halo, trifluoromethyl, alkyl, nitrile or alkoxy
- R4 and R5 may be the same or different, and may be H, alkyl, aryl, heteroaryl or cycloalkyl; or R4 and R5 may together form a saturated, unsaturated or partially saturated 4 to 7 member ring, wherein said ring may optionally comprise one or more further heteroatoms selected from N, O or S;
- X is O, S or NR6;
- R6 is H or alkyl;
- R7 is hydrogen, methyl or ethyl;
- R8 is methyl or ethyl;
- L is (CH 2 ) n , where n is 1, 2 or 3; and
- the medicament is for use in the treatment or prevention of arrhythmia.
- the compounds are compounds of the first aspect.
- Example 1 l-(4-Fluorophenyl)-2-hydroxyethanone Under a nitrogen atmosphere, a stirred solution of 4'-fluoroacetophenone (1.15g, 8.32mmol) in acetonitrile (42ml) was treated with [fcw(trifluoroacetoxy)iodo]benzene (7.16g, 16.6mmol) followed by water (8.3ml) and trifluoroacetic acid (1.3ml). The resultant solution was heated under reflux for 2hr before standing overnight at ambient temperature. The solvent was then removed in vacuo; water (30ml) was added and the mixture extracted with dichloromethane (3x30ml).
- Example 27 5-Phenyl-lH-furo[2,3-d]pyrimidine-2,4-dithione A stirred mixture of 2-amino-4-phenyl-furan-3-carbonitrile (5.0g, 0.027mol) and potassium ethyl xanthate (4.5g, 0.027mol) in butan-1-ol (25ml) was heated for 2hr at 110°C. After cooling to ambient temperature, the solid was filtered off, washed with a little butan-1-ol and dissolved in water (100ml). The solution was then acidified with glacial acetic acid to give a light-brown precipitate. This was filtered off, washed with water and dried in vacuo to give the crude 5-phenyl-lH-furo[2,3-d]pyrimidine-2,4- dithione (3.0g) which was used without further purification.
- Example Compound 31 2,4-Bis-methylsulfanyl-6-methyl-5-phenyl-lH-furo[2,3-d]pyrimidine
- Example Compound 32 2,4-Bis-methylsulfanyl-5-(4-fluorophenyl)-6-methyl-lH-furo[2,3- d]pyrimidine
- Example 36 (2-Methanesulfonyl-5-phenyl-furo[2,3d]pyrimidin-4-yl)-pyridin-2-ylmethylamine
- Example Compound 37 (2-Methanesulfonyl-5-phenyl-furo[2,3-d]pyrimidin-4-yl)-(6-methylpyridin-2- ylmethyl)-amine 38 Furan-2-ylmethyl-(2-methanesulfonyl-5-phenyl-furo[2,3-d]pyrimidin-4-yl)- amine 39 (2-Methanesulfonyl-6-methyl-5-phenyl-furo[2,3-d]pyrimidin-4-yl)-pyridin-2- ylmethylamine 40 [5-(4-Fluorophenyl)-2-methanesulfonyl-6-methyl-furo[2,3-d]pyrimidin-4-yl]- pyridin-2-ylmethylamine
- Example Compound 42 (2-Morpholin-4-yl-5-phenyl-furo[2,3-d]pyrimidin-4-yl)-puridin-2-ylmethyl- amine 43 2-((2-Hydroxyethyl)- ⁇ 5-Phenyl-4-[(pyridine-2-ylmethyl)-amino]-furo[2,3- d]pyrimidin-2-yl ⁇ -amino)-ethanol 44 2- ⁇ 5-Phenyl-4-[(pyridin-2-ylmethyl)-amino]furo[2,3-d]pyrimidin-2- yl amino ⁇ -propane- 1 ,3-diol 45 2-((2-Hydroxyethyl)- ⁇ 5 -(4-fluorophenyl)-4- [(pyridine-2-ylmethyl)-amino] - furo[2,3-d]pyrimidin-2-yl ⁇ -amino)-ethanol
- Ethanol (1ml) was saturated with dimethylamine and (2-methanesulfonyl-5-phenyl- furo[2,3d]pyrimidin-4-yl)-pyridin-2-ylmethylamine (50mg, 0.132mmol) was added.
- the mixture was then stirred and heated by microwave irradiation at a pressure of 250psi for 30mins, the temperature reaching a maximum of 138°C.
- the resultant solution was then diluted with water (50ml) and extracted with dichloromethane (3x50ml).
- Example Compound 47 N ,N -Dimethyl-N -(6-methyl-pyridin-2-ylmethyl)-5-phenyl-furo[2,3- d]pyrimidine-2,4-diamine 48 N -Furan-2-ylmethyl-N ,N -dimethyl-5-phenyl-furo[2,3-d]pyrimidine-2,4- diamine 49 6,N ⁇ 2 ,N. ⁇ 2 -Trimethyl-5-phenyl-N4-pyridin-2-ylmethyl-furo[2,3-d]pyrimidine- 2,4-diamine 50 5-(4-Fluorophenyl)-6,N 2 ,N 2 -trimethyl-N 4 -pyridin-2-ylmethyl-furo[2,3- d]pyrimidine-2,4-diamine
- Example 51 [2-(2-Methoxyethoxy)-5-phenyl-furo[2,3-d]pyrimidin-4-yl]-pyridin-2-ylmethylamine
- 2-methoxyethanol (1ml) was treated with 60% sodium hydride (lOmg) followed, after lOmins, by (2-methanesulfonyl-5-phenyl-furo[2,3d]pyrimidin-4-yl)-pyridin-2- ylmethylamine (50mg, 0.132mmol) and heated by microwave irradiation for 30mins at 150°C.
- the resultant solution was then diluted with water (50ml) and extracted with dichloromethane (3x50ml).
- Ethyl malonyl chloride (3.8ml, 29.89mmol) was added dropwise to a solution of 2- amino-4-phenyl-3-furonitrile (5g, 27.17mmol) and triethylamine (4.2ml, 29.89 mmol) in THF (30ml) maintaining temperature below 5°C under nitrogen. This was allowed to warm up to room temperature and stirred for 2hr. The reaction mixture was diluted with water and extracted with DCM. The organic layers were combined and washed with brine and dried over MgSO . The solution was filtered and solvent removed under reduced pressure. The residue was purified by column chromatography using DCM isolating an orange oil (3.7g ,46%).
- Ethyl (4-oxo-5-phenyl-3,4-dihydrofuro[2,3-d]pyrimin-2-yl)acetate (827mg, 2.79mmol) was added to a solution of phosphorus oxychloride (13ml, 139.5mmol) and diethyl aniline (4.4ml, 27.8mmol) and heated to 65C and stirred for 2.5hr. Phosphorous oxychloride was removed under reduced pressure and the residue was diluted with DCM. The organic layer was washed with water twice and then with saturated sodium hydrogen carbonate solution and finally with brine. The solution was dried over MgSO and filtered. Solvent was removed under reduced pressure and the residue was purified by column chromatography (20-100% DCM/40°-60° petrol) to provide an orange oil (lOOmg).
- Example 55 Ethyl ⁇ 5-phenyl-4-[(pyridine-2-ylmethyl)amino]furo[2,3-d]pyrimidin-2-yl ⁇ -acetate 2-Aminomethyl pyridine (36microL, 0.347 mmol) was added to a solution of ethyl (4- chloro-5-phenylfuro[2,3-d]pyrimin-2-yl)acetate (lOOmg, 0.316mmol) and triethylamine (48microL, 0.347mmol) in ethanol (5ml) and heated to reflux for 4hr. The reaction mixture was cooled and diluted with water. The aqueous mixture was extracted three times with DCM.
- DMEM Dulbecco's Modified Eagle media
- PCS Fetal Calf Serum
- PCS Fetal Calf Serum
- 20 ⁇ l/ml penicillin
- streptomycin 5000/ ⁇ g/ml
- lO ⁇ l/ml [lOOx] glutamine lO ⁇ l/ml [lOOx] glutamine
- blasticidin 7.5 ⁇ g/ml
- the external bathing solution contained (in mM): 150 NaCl, 10 KC1, 100 Potassium Gluconate, 3 MgCl 2 , 1 CaCl 2 , 10 HEPES, pH 7.4.
- Patch pipettes were filled with an electrode solution of composition (in mM): 160 KC1, 0.5 MgCl 2 , 10 HEPES, 1 EGTA, pH 7.4 with KOH.
- Compounds were dissolved in DMSO (100 %) and made up in the external bather at a concentration of l ⁇ M. All experiments were conducted at room temperature (22-24°C).
- the lOO ⁇ l cell resuspension solution was then stored on the bench at 4°C (using a Peltier-based temperature control device) until used.
- a length of capillary glass (1B150F-4, WPI) was dipped into the cell suspension solution, such that ⁇ 3 cm column of fluid was taken up by capillary action.
- An Ag/AgCl wire was dropped into the non-dipped end of the capillary also.
- the outside of the solution-filled end of the capillary was then dried and the capillary was loaded into the AutoPatchTM.
- Borosilicate glass patch pipettes (from 1.5mm OD, thin-walled filamented, GC150-TF capillary glass, Harvard) were pulled using a DMZ pipette puller (Zeitz Instruments), and were back-filled using the internal pipette solution, being careful that no bubbles remain at the tip or in the body of the pipette. Patch pipettes typically had resistances of 2.3-3.5 M ⁇ . Once filled, the pipette tip and a proportion of the shaft ( ⁇ 15 mm) were dipped into Sigmacote (Sigma). The recording pipette was then loaded into the AutoPatchTM. Automated patch-clamping was initiated by the operator, but thereafter AutoPatch.exe continued the experiment providing that pre-set conditions and criteria were satisfied.
- Kvl.5 channel electrophysiology data for the representative compounds described within are given in the table below.
- Antonov et al Synthesis of heterocyclic compounds based on adducts of polyhaloalkanes with unsaturated systems. 6. Transformations of the trichloroethyl group in 2-methyl-3-(2,2,2-trichloroethyl)-4-(R-amino)furo[2,3-d]pyrimidines, their isomers and some of their precursors.
- Belenkii et al. Synthesis of heterocycles based on products of addition of polyhaloalkanes to unsaturated systems. 4. Synthesis of substituted furo[2,3- d]pyrimidines.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description


















Claims



Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007526555A JP4955557B2 (en) | 2004-06-10 | 2005-06-10 | Furanopyrimidine compounds effective as potassium channel inhibitors |
| BRPI0511917-0A BRPI0511917A (en) | 2004-06-10 | 2005-06-10 | pharmaceutically acceptable compound or salts thereof, process for preparing a compound, pharmaceutical composition, method for treating or preventing a disorder requiring potassium channel inhibition, and use of a compound |
| MXPA06014256A MXPA06014256A (en) | 2004-06-10 | 2005-06-10 | Furanopyrimidine compounds effective as potassium channel inhibitors. |
| AU2005252440A AU2005252440A1 (en) | 2004-06-10 | 2005-06-10 | Furanopyrimidine compounds effective as potassium channel inhibitors |
| EP05751879A EP1758909A1 (en) | 2004-06-10 | 2005-06-10 | Furanopyrimidine compounds effective as potassium channel inhibitors |
| CA002568304A CA2568304A1 (en) | 2004-06-10 | 2005-06-10 | Furanopyrimidine compounds effective as potassium channel inhibitors |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US57835004P | 2004-06-10 | 2004-06-10 | |
| US60/578,350 | 2004-06-10 | ||
| GBGB0412986.2A GB0412986D0 (en) | 2004-06-10 | 2004-06-10 | Compounds |
| GB0412986.2 | 2004-06-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005121149A1 true WO2005121149A1 (en) | 2005-12-22 |
Family
ID=35094157
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2005/002318 WO2005121149A1 (en) | 2004-06-10 | 2005-06-10 | Furanopyrimidine compounds effective as potassium channel inhibitors |
Country Status (8)
| Country | Link |
|---|---|
| EP (1) | EP1758909A1 (en) |
| JP (1) | JP4955557B2 (en) |
| KR (1) | KR20070055486A (en) |
| AU (1) | AU2005252440A1 (en) |
| BR (1) | BRPI0511917A (en) |
| CA (1) | CA2568304A1 (en) |
| MX (1) | MXPA06014256A (en) |
| WO (1) | WO2005121149A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102007019691A1 (en) | 2007-04-26 | 2008-10-30 | Bayer Healthcare Ag | Use of acyclically substituted furopyrimidine derivatives for the treatment of pulmonary arterial hypertension |
| DE102007019690A1 (en) | 2007-04-26 | 2008-10-30 | Bayer Healthcare Ag | Use of cyclic substituted furopyrimidine derivatives for the treatment of pulmonary arterial hypertension |
| DE102007027799A1 (en) | 2007-06-16 | 2008-12-18 | Bayer Healthcare Ag | Substituted furopyrimidines and their use |
| DE102007027800A1 (en) | 2007-06-16 | 2008-12-18 | Bayer Healthcare Ag | Substituted bicyclic heteroaryl compounds and their use |
| DE102007054786A1 (en) | 2007-11-16 | 2009-05-20 | Bayer Healthcare Ag | Trisubstituted Furopyrimidines and their Use |
| US8183246B2 (en) | 2005-12-21 | 2012-05-22 | Bayer Intellectual Property Gmbh | Acyclically substituted furopyrimidine derivatives and use thereof |
| US8324222B2 (en) | 2005-12-21 | 2012-12-04 | Bayer Intellectual Property Gmbh | Cyclically substituted furopyrimidine derivatives and use thereof |
| WO2013072694A1 (en) | 2011-11-15 | 2013-05-23 | Xention Limited | Thieno- and furo - pyrimidines and pyridines, useful as potassium channel inhibitors |
| WO2013112932A1 (en) | 2012-01-27 | 2013-08-01 | Gilead Sciences, Inc. | Combination therapies using late sodium ion channel blockers and potassium ion channel blockers |
| US8575184B2 (en) | 2009-09-03 | 2013-11-05 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| WO2014134419A1 (en) | 2013-03-01 | 2014-09-04 | Gilead Sciences, Inc. | Use of ikach blockers for the treatment of cardiac diseases |
| US9695192B2 (en) | 2011-07-01 | 2017-07-04 | Gilead Sciences, Inc. | Fused heterocyclic compounds as ion channel modulators |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0459611A2 (en) * | 1990-03-30 | 1991-12-04 | Dowelanco | Furopyrimidine derivatives |
| WO2001046155A1 (en) * | 1999-12-21 | 2001-06-28 | Icagen, Inc. | Potassium channel inhibitors |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA782928B (en) * | 1977-05-27 | 1979-05-30 | Ici Ltd | Pesticidal compounds and compositions |
| AU754204B2 (en) * | 1998-09-01 | 2002-11-07 | Bristol-Myers Squibb Company | Potassium channel inhibitors and method |
| DE10141212A1 (en) * | 2001-08-22 | 2003-03-06 | Bayer Ag | New 4-aminofuropyrimidines and their use |
-
2005
- 2005-06-10 MX MXPA06014256A patent/MXPA06014256A/en not_active Application Discontinuation
- 2005-06-10 KR KR1020077000590A patent/KR20070055486A/en not_active Application Discontinuation
- 2005-06-10 JP JP2007526555A patent/JP4955557B2/en not_active Expired - Fee Related
- 2005-06-10 EP EP05751879A patent/EP1758909A1/en not_active Withdrawn
- 2005-06-10 CA CA002568304A patent/CA2568304A1/en not_active Abandoned
- 2005-06-10 WO PCT/GB2005/002318 patent/WO2005121149A1/en active Application Filing
- 2005-06-10 BR BRPI0511917-0A patent/BRPI0511917A/en not_active IP Right Cessation
- 2005-06-10 AU AU2005252440A patent/AU2005252440A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0459611A2 (en) * | 1990-03-30 | 1991-12-04 | Dowelanco | Furopyrimidine derivatives |
| WO2001046155A1 (en) * | 1999-12-21 | 2001-06-28 | Icagen, Inc. | Potassium channel inhibitors |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI405764B (en) * | 2005-12-21 | 2013-08-21 | 拜耳智慧財產有限公司 | Novel, acyclically substituted furopyrimidine derivatives and use thereof |
| US8324222B2 (en) | 2005-12-21 | 2012-12-04 | Bayer Intellectual Property Gmbh | Cyclically substituted furopyrimidine derivatives and use thereof |
| US8183246B2 (en) | 2005-12-21 | 2012-05-22 | Bayer Intellectual Property Gmbh | Acyclically substituted furopyrimidine derivatives and use thereof |
| WO2008131858A3 (en) * | 2007-04-26 | 2009-07-16 | Bayer Schering Pharma Ag | Use of acyclically substituted furopyrimidine derivatives for treating pulmonary arterial hypertonia |
| WO2008131859A3 (en) * | 2007-04-26 | 2009-07-16 | Bayer Schering Pharma Ag | Use of cyclically substituted furopyrimidine derivatives for treating pulmonary arterial hypertonia |
| WO2008131859A2 (en) * | 2007-04-26 | 2008-11-06 | Bayer Schering Pharma Aktiengesellschaft | Use of cyclically substituted furopyrimidine derivatives for treating pulmonary arterial hypertonia |
| WO2008131858A2 (en) * | 2007-04-26 | 2008-11-06 | Bayer Schering Pharma Aktiengesellschaft | Use of acyclically substituted furopyrimidine derivatives for treating pulmonary arterial hypertonia |
| DE102007019690A1 (en) | 2007-04-26 | 2008-10-30 | Bayer Healthcare Ag | Use of cyclic substituted furopyrimidine derivatives for the treatment of pulmonary arterial hypertension |
| DE102007019691A1 (en) | 2007-04-26 | 2008-10-30 | Bayer Healthcare Ag | Use of acyclically substituted furopyrimidine derivatives for the treatment of pulmonary arterial hypertension |
| DE102007027799A1 (en) | 2007-06-16 | 2008-12-18 | Bayer Healthcare Ag | Substituted furopyrimidines and their use |
| DE102007027800A1 (en) | 2007-06-16 | 2008-12-18 | Bayer Healthcare Ag | Substituted bicyclic heteroaryl compounds and their use |
| WO2008155017A1 (en) * | 2007-06-16 | 2008-12-24 | Bayer Schering Pharma Aktiengesellschaft | Substituted bicyclic heteroaryl compounds for the treatment of cardiovascular diseases |
| DE102007054786A1 (en) | 2007-11-16 | 2009-05-20 | Bayer Healthcare Ag | Trisubstituted Furopyrimidines and their Use |
| US9458114B2 (en) | 2009-09-03 | 2016-10-04 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US8575184B2 (en) | 2009-09-03 | 2013-11-05 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US9822096B2 (en) | 2009-09-03 | 2017-11-21 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US10214511B2 (en) | 2009-09-03 | 2019-02-26 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US10676460B2 (en) | 2009-09-03 | 2020-06-09 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US11008306B2 (en) | 2009-09-03 | 2021-05-18 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US9695192B2 (en) | 2011-07-01 | 2017-07-04 | Gilead Sciences, Inc. | Fused heterocyclic compounds as ion channel modulators |
| JP2014533259A (en) * | 2011-11-15 | 2014-12-11 | ゼンション・リミテッドXention Limited | Thieno and furopyrimidine and pyridine useful as potassium channel inhibitors |
| WO2013072694A1 (en) | 2011-11-15 | 2013-05-23 | Xention Limited | Thieno- and furo - pyrimidines and pyridines, useful as potassium channel inhibitors |
| WO2013112932A1 (en) | 2012-01-27 | 2013-08-01 | Gilead Sciences, Inc. | Combination therapies using late sodium ion channel blockers and potassium ion channel blockers |
| WO2014134419A1 (en) | 2013-03-01 | 2014-09-04 | Gilead Sciences, Inc. | Use of ikach blockers for the treatment of cardiac diseases |
Also Published As
| Publication number | Publication date |
|---|---|
| MXPA06014256A (en) | 2007-03-12 |
| CA2568304A1 (en) | 2005-12-22 |
| JP4955557B2 (en) | 2012-06-20 |
| BRPI0511917A (en) | 2008-01-15 |
| JP2008501773A (en) | 2008-01-24 |
| AU2005252440A1 (en) | 2005-12-22 |
| EP1758909A1 (en) | 2007-03-07 |
| KR20070055486A (en) | 2007-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7456187B2 (en) | Furanopyrimidine compounds as potassium ion channel inhibitors | |
| EP1758909A1 (en) | Furanopyrimidine compounds effective as potassium channel inhibitors | |
| Ford et al. | Compounds | |
| US8022076B2 (en) | Substituted thieno[2,3-d]pyrimidines as potassium channel inhibitors | |
| CA2528848C (en) | Thienopyrimidine derivatives as potassium channel inhibitors | |
| EP1879899B1 (en) | Thienopyridine derivatives as potassium channel inhibitors | |
| ZA200610042B (en) | Levodopa prodrugs, and compositions and uses thereof | |
| US9216992B2 (en) | Thieno[3,2-c]pyridine potassium channel inhibitors | |
| US8193215B2 (en) | Thieno[2 3-b]pyridines as potassium channel inhibitors | |
| WO2005092858A2 (en) | Alpha aryl or heteroaryl methyl beta piperidino propanamide compounds as orl1-receptor antagonist | |
| AU2012235882B2 (en) | Thieno [2, 3 -d] pyrimidine derivatives and their use to treat arrhythmia |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2568304 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 7134/DELNP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005252440 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/10142 Country of ref document: ZA Ref document number: 200610142 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/014256 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580018876.8 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007526555 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2005252440 Country of ref document: AU Date of ref document: 20050610 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005751879 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005252440 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077000590 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005751879 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0511917 Country of ref document: BR |