WO2005100343A1 - Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel - Google Patents
Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel Download PDFInfo
- Publication number
- WO2005100343A1 WO2005100343A1 PCT/EP2005/003741 EP2005003741W WO2005100343A1 WO 2005100343 A1 WO2005100343 A1 WO 2005100343A1 EP 2005003741 W EP2005003741 W EP 2005003741W WO 2005100343 A1 WO2005100343 A1 WO 2005100343A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oxo
- chloro
- amino
- piperidine
- diazepin
- Prior art date
Links
- 0 *CC(CC(*)=O)C(N(*)*)=O Chemical compound *CC(CC(*)=O)C(N(*)*)=O 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
- C07D451/04—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof with hetero atoms directly attached in position 3 of the 8-azabicyclo [3.2.1] octane or in position 7 of the 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
Definitions
- the present invention relates to the CGRP antagonists of the general formula
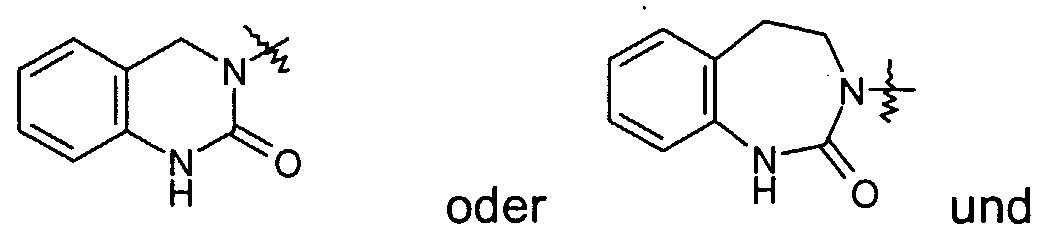
- A is a radical of the formula
- X is an oxygen atom, a methylene or NH group
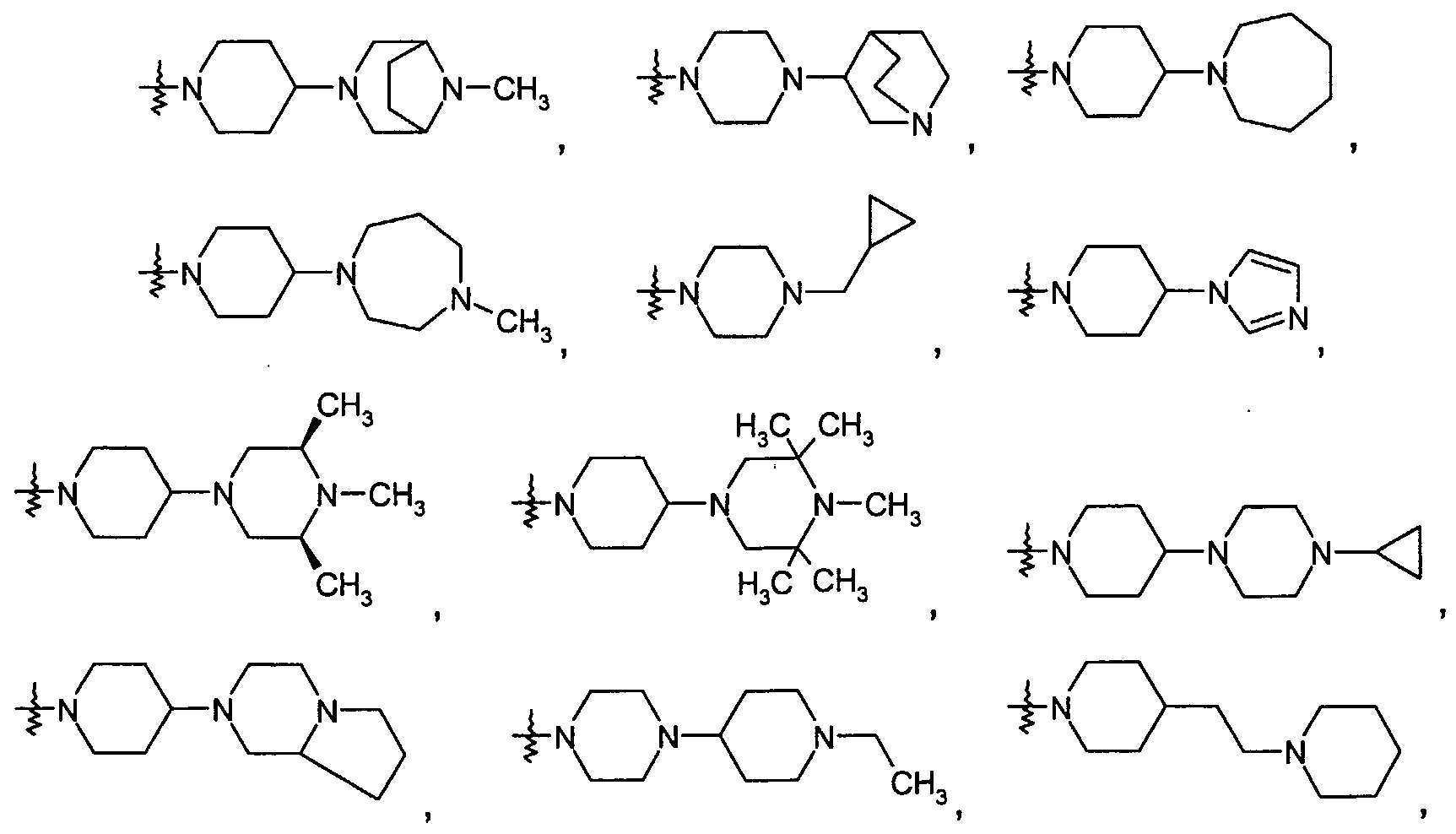
- R 1 is a radical of the formula
- NO. 2 Ro3 is a radical of the formula
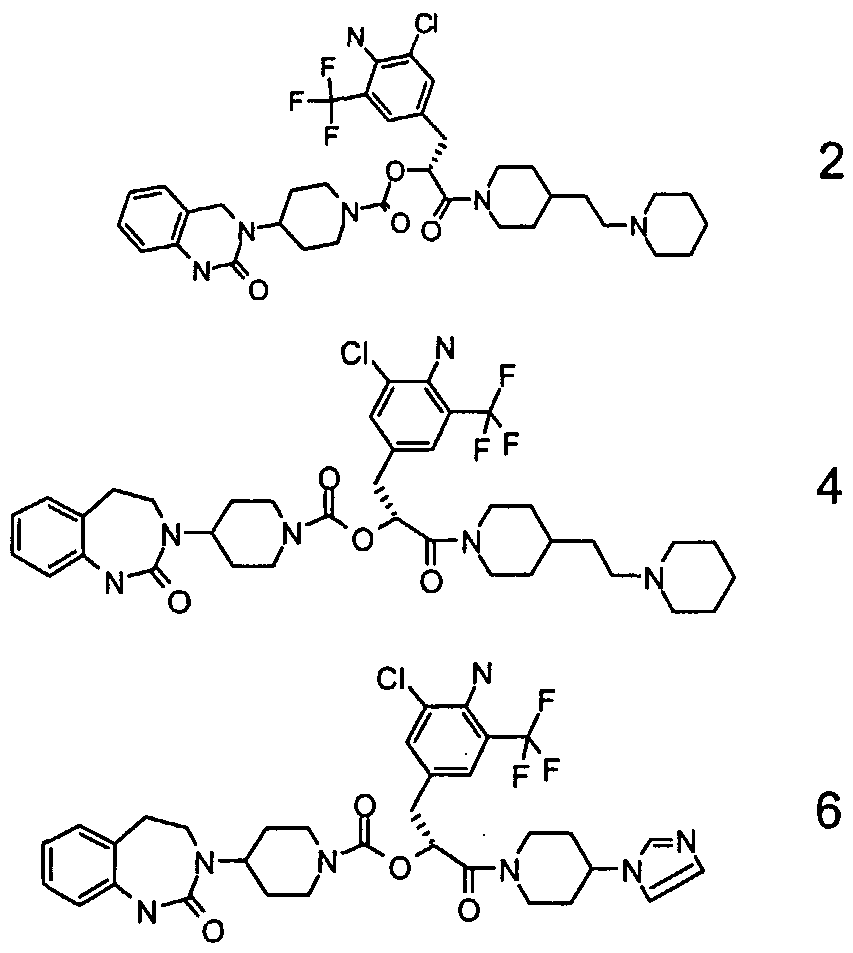
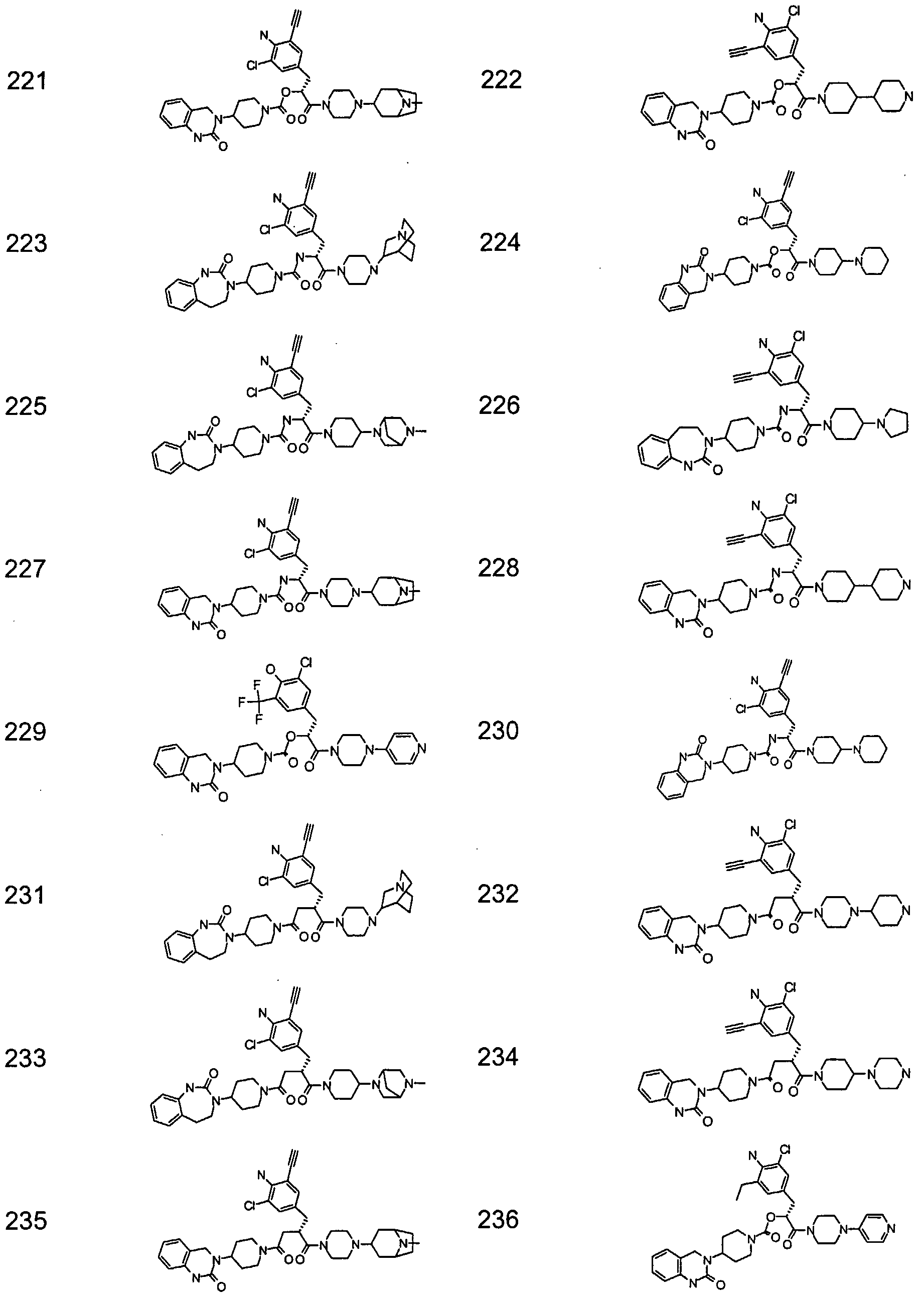
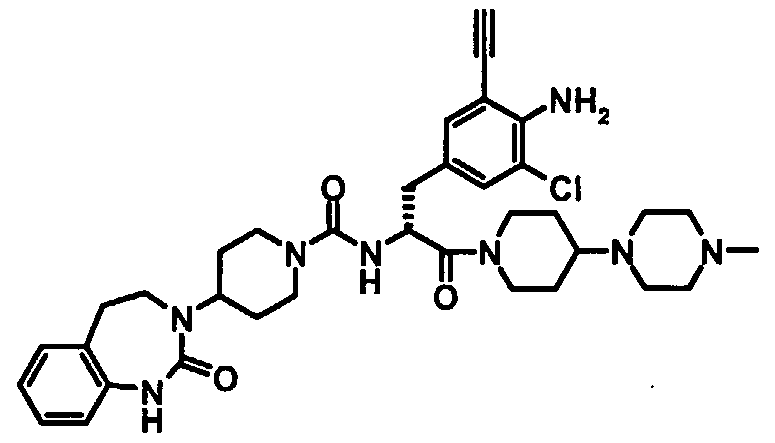
- Particularly preferred compounds of the above general formula (I) are, for example, the following:
- the compounds of general formula (I) are prepared by methods known in principle. The following processes have proven particularly suitable for the preparation of the compounds of general formula (I) according to the invention:
- a nucleofugic group which may be identical or different, preferably the phenoxy, 1H-imidazol-1-yl, 1 / - / - 1, 2,4-triazol-1-yl, trichloro-methoxy- or the 2, ⁇ -dioxopyrrolidin-1-yloxy group, with the proviso that X represents the NH group, or
- a nucleofuge group which may be the same or different, preferably the chlorine atom, the p-nitrophenoxy or trichloromethoxy group, with the proviso that X represents the oxygen atom,
- R 2 and R 3 are defined as mentioned above, with the proviso that R 2 and R 3 contain no further free, unprotected, primary or secondary aliphatic amino function.
- An optionally present in the radical -NR 2 R 3 additionally present primary or secondary amino function is provided in each case with a suitable protective group.
- the two-stage reactions in principle are generally carried out as a one-pot process, preferably in such a way that in the first stage one of the two components (III) or (V) with equimolar amounts of the carbonic acid derivative of the general formula (IV) in a brings appropriate solvent at a lower temperature to the reaction, then at least equimolar amounts of the other component (III) or (V) is added and the reaction is terminated at a higher temperature.
- the reactions with bis (trichloromethyl) carbonate are preferably in the presence of at least 2 equivalents (based on bis (trichloromethyl) carbonate) of a tertiary base, for example of triethylamine, ⁇ / ethyldiisopropylamine, pyridine, 1, ⁇ -diaza- bicyclo [4.3.0] non- ⁇ -ene, 1, 4-diazabicyclo [2.2.2] octane or 1, 8-diazabicyclo [ ⁇ .4.0] -un-dec-7-ene.
- a tertiary base for example of triethylamine, ⁇ / ethyldiisopropylamine, pyridine, 1, ⁇ -diaza- bicyclo [4.3.0] non- ⁇ -ene, 1, 4-diazabicyclo [2.2.2] octane or 1, 8-diazabicyclo [ ⁇ .4.0] -un-dec-7-
- Suitable solvents which should be anhydrous are, for example, tetrahydrofuran, dioxane, dimethylformamide, dimethylacetamide, .alpha.-methyl-2-pyrrolidone, 1,3-dimethyl-2-imidazolidinone or acetonitrile, when bismuth is used.
- (Trichloromethyl) - carbonate as carbonyl anhydrous chlorinated hydrocarbons, for example dichloromethane, 1, 2-dichloroethane or trichlorethylene, are preferred.
- the Reaction temperatures are for the first reaction stage between -30 ° C and + 2 ⁇ ° C, preferably - ⁇ ° C and + 10 ° C, for the second reaction stage between + 1 ⁇ ° C and the boiling temperature of the solvent used, preferably between + 20 ° C. and + 70 ° C
- HA Staab and W. Rohr "Syntheses with Heterocyclic Amides (Azolides)", Newer Methods of Preparative Organic Chemistry, Volume V, pp 63-93, Verlag Chemie, Weinheim / Bergstr., 1967; P. Majer and RS Randad, J. Org. Chem., ⁇ 9, 1937-1938 (1994), K. Takeda, Y.
- X is the methylene group and A and R 1 to R 3 are defined as mentioned above, with the proviso that no further free, unprotected, primary or secondary aliphatic amino function is contained:
- R 1 has the meanings mentioned above.
- the coupling is preferably carried out using methods known from peptide chemistry (see eg Houben-Weyl, Methoden der Organischen Chemie, Vol. Dicyclohexylcarbodiimide (DCC), diisopropylcarbodiimide (DIC) or ethyl- (3-dimethylaminopropyl) -carbodiimide, 0- (1 - / - benzotriazol-1-yl) - / V, / V- [beta] -tetramethyluronium hexafluorophosphate (HBTU) or tetrafluoroborate (TBTU) or 1 / - / - benzotriazole-1-yl-oxy-tris- (dimethylamino) phosphonium hexafluorophosphate (BOP).
- DCC Dicyclohexylcarbodiimide
- DIC diisopropylcarbodiimide
- the reaction rate can be increased.
- the couplings are usually with equimolar proportions of the coupling components and the coupling reagent in solvents such as dichloromethane, tetrahydrofuran, acetonitrile, dimethylformamide (DMF), dimethylacetamide (DMA), ⁇ / -methylpyrrolidone (NMP) or mixtures thereof and at temperatures between -30 ° C and + 30 ° C, preferably -20 ° C and + 25 ° C performed.
- DMF dimethylformamide
- DMA dimethylacetamide
- NMP ⁇ / -methylpyrrolidone
- als / -Ethyldiisopropylamin (Hünig base) is preferred as an additional auxiliary base.
- the mixed anhydride is obtained from the coupling of the carboxylic acid of the general formula (VI) and the carbonic monoisobutylester.
- the preparation of this mixed anhydride and the coupling with amines is carried out in a one-pot process, using the abovementioned solvents and in Temperatures between -20 and + 25 ° C, preferably 0 ° C and + 2 ⁇ ° C.
- R 2 and R 3 are defined as mentioned above, with the proviso that R 2 and R 3 contain no free, unprotected, primary or secondary amine, and Nu is a leaving group, for example a halogen atom, such as the chlorine, Bromine or iodine atom, an alkylsulfonyloxy group having 1 to 10 carbon atoms in the alkyl moiety, an optionally substituted by chlorine or bromine atoms, by methyl or nitro groups mono-, di- or trisubstituted phenylsulfonyloxy or Naphthylsulfonyloxy distr, where the substituents may be identical or different, one 1r7-imidazol-1-yl, a 1H-pyrazol-1-yl, optionally substituted by one or two methyl groups in the carbon skeleton, a 1H-1, 2,4-triazol-1-yl, 1H-1,2, 3-triazol-1-yl, 1H-1,2,
- R 1 is defined as mentioned above.
- auxiliary bases are alkali metal and alkaline earth metal hydroxides, for example sodium hydroxide, potassium hydroxide or barium hydroxide, alkali metal carbonates, eg. Sodium carbonate, potassium carbonate or cesium carbonate, alkali acetates, e.g.
- Sodium or potassium acetate, and tertiary amines for example pyridine, 2,4,6-tri-methylpyridine, quinoline, triethylamine, / V-ethyl-diisopropylamine, ⁇ / -ethyl-dicyclohexyl-amine, 1, 4-di-azabicyclo [ 2.2.2] octane or 1, 8-diazabicyclo [5.4.0] undec-7-ene, as solvent, for example, dichloromethane, tetrahydrofuran, 1, 4-dioxane, acetonitrile, dimethylformamide, dimethylacetamide, ⁇ / -methylpyrrolidone or mixtures thereof into consideration; If alkali metal or alkaline earth metal hydroxides, alkali metal carbonates or acetates are used as auxiliary bases, water may also be added to the reaction mixture as cosolvent.
- A, X and R 1 are as defined above, with an amine of the general formula HNR 2 R 3 , in which R 2 and R 3 are as defined above, with the proviso that they are no further free, unprotected, primary or secondary aliphatic amino function.
- the coupling is preferably carried out using methods known from peptide chemistry (see, for example, Houben-Weyl, Methods of Organic Chemistry, Vol. 15/2), for example carbodiimides, such as, for example, dicyclohexylcarbodiimide (DCC), diisopropylcarbodiimide (DIC) or ethyl (3-dimethylaminopropyl) carbodiimide, 0- (1 / - benzotriazol-1-yl) - ⁇ /, / V- ⁇ / ', ⁇ / , tetramethyluronium hexafluorophosphate ( HBTU) or tetrafluoroborate (TBTU) or 1 H-benzotriazole-1-yl-oxy-tris- (dimethylamino) phosphonium hexafluorophosphate (BOP).
- DCC dicyclohexylcarbodiimide
- DI diisopropylcarbod
- the reaction rate can be increased by adding 1-hydroxybenzotriazole (HOBt) or 3-hydroxy-4-oxo-3,4-dihydro-1,2,3-benzotriazine (HOOBt).
- the couplings are usually with equimolar proportions of the coupling components and the coupling reagent in solvents such as dichloromethane, tetrahydrofuran, acetone nitrile, dimethylformamide (DMF), dimethylacetamide (DMA), ⁇ / -methylpyrrolidone (NMP) or mixtures thereof and at temperatures between -30 and + 30 ° C, preferably -20 and + 25 ° C performed.
- solvents such as dichloromethane, tetrahydrofuran, acetone nitrile, dimethylformamide (DMF), dimethylacetamide (DMA), ⁇ / -methylpyrrolidone (NMP) or mixtures thereof and at temperatures between -30 and + 30 ° C, preferably -20 and
- Formula (I) is the so-called “anhydride” method (see also: M. Bodanszky, “Peptide Chemistry", Springer-Verlag 1988, p.8- ⁇ 9, M. Bodanszky, “Phnciples of Peptide Synthesis ", Springer-Verlag 1984, pp. 21-27), the” mixed anhydride method "being preferred in the variant according to Vaughan (JR Vaughan Jr., J. Amer. Chem. Soc. 73, 3 ⁇ 47 (1951).
- the mixed anhydride is obtained from the carboxylic acid of general formula (VIII) to be coupled and monoisobutyl carbonate
- Anhydride and the coupling with the amines of the general formula HNR 2 R 3 is carried out in a one-pot process, using the abovementioned solvents and at temperatures between -20 ° C and + 25 ° C, preferably between 0 ° C and + 25 ° C.
- A, X and R 1 are as defined above and Nu is a leaving group, for example a halogen atom, such as the chlorine, bromine or iodine atom, an alkylsulfonyloxy group having 1 to 10 carbon atoms in the alkyl moiety, one optionally substituted by chlorine or bromine atoms, by methyl or nitro groups mono-, di- or trisubstituted phenylsulfonyloxy or naphthylsulfonyloxy group, where the substituents may be identical or different, a 1H-imidazol-1-yl, a 1H-pyrazol-1-yl optionally substituted by one or two methyl groups in the carbon skeleton, a 1H-1, 2,4-triazol-1-yl, 1 / - / - 1, 2,3-triazol-1-yl, 1H-1, 2,3,4-tetrazol-1-yl -, a vinyl, propargyl,
- auxiliary bases are alkali metal and alkaline earth metal hydroxides, for example sodium hydroxide, potassium hydroxide or barium hydroxide, alkali metal carbonates, eg. Sodium carbonate, potassium carbonate or cesium carbonate, alkali acetates, e.g.
- Sodium or potassium acetate, and tertiary amines for example pyridine, 2,4,6-T ⁇ methylpyridin, quinoline, T ethylamine, ⁇ / -Ethyldiisopropylamin, ⁇ / -Ethyldicyclohexylamin, 1, 4-di-azabicyclo [2.2.2] octane or 1 , 8-diazabicyclo [ ⁇ .4.0] undec-7-ene, as solvent, for example, dichloromethane, tetrahydrofuran, 1, 4-dioxane, acetonitrile, dimethylformamide, dimethylacetamide, ⁇ / -methylpyrrolidone or mixtures thereof into consideration; If alkali metal or alkaline earth metal hydroxides, alkali metal carbonates or acetates are used as auxiliary bases, water may also be added to the reaction mixture as cosolvent.
- solvent for example, dichloromethane,
- novel compounds of the general formula (I) according to the invention contain one or more chiral centers. For example, if there are two chiral centers, then the compounds can be in the form of two diastereomeric antipode pairs.
- the invention includes the individual isomers as well as theirs Mixtures.
- the separation of the respective diastereomers is possible due to their different physicochemical properties, e.g. by fractional crystallization from suitable solvents, by high pressure liquid or column chromatography using chiral or preferably achiral stationary phases.
- racemates covered by the general formula (I) succeeds, for example, by HPLC on suitable chiral stationary phases (eg Chiral AGP, Chiralpak AD). Racemates which contain a basic function can also be resolved via the diastereomeric, optically active salts which, when reacted with an optically active acid, for example (+) - or (-) - tartaric acid, (+) - or (-) - Diacetyltartaric acid, (+) - or (-) - monomethyl tartrate or (+) - camphorsulfonic arise.
- an optically active acid for example (+) - or (-) - tartaric acid, (+) - or (-) - Diacetyltartaric acid, (+) - or (-) - monomethyl tartrate or (+) - camphorsulfonic arise.
- the racemic compound of the general formula (I) is reacted with one of the above-mentioned optically active acids in an equimolar amount in a solvent, and the resulting crystalline diastereomeric optically active salts are separated by utilizing their various solubilities.
- This reaction can be carried out in any kind of solvents as long as they have a sufficient difference in the solubility of the salts.
- each of the optically active salts is dissolved in water, carefully neutralized with a base, such as sodium carbonate or potassium carbonate, or with a suitable acid, for example, with dilute hydrochloric acid or aqueous methanesulfonic acid, thereby giving the corresponding free compound in the (+) - or ( -) - Form received.
- a base such as sodium carbonate or potassium carbonate
- a suitable acid for example, with dilute hydrochloric acid or aqueous methanesulfonic acid
- the phenyalanine derivatives necessary for the preparation of the optically pure compounds of the general formula (V) can be prepared from the compounds of the general formula
- A is defined as mentioned above and R is an unbranched alkyl group, preferably the methyl or ethyl group, are prepared by racemate resolution.
- This racemate resolution can be carried out by enzymatic methods, wherein only one enantiomer of the racemate is transformed and the resulting mixture is then separated by means of physicochemical methods, preferably by chromatographic methods.
- a suitable enzyme system for this step is the enzyme Alcalase 2.4 L FG (Novozymes A / S; DK 2880 Bagsvaerd).
- the compounds of the general formula (X) can then be converted into the enantiomerically pure compounds of the general formula (V) using methods which are familiar to peptide chemists. be transferred.
- the compounds of the general formula (XI) can be obtained by diazotization of compounds of the general formula (X) with a suitable diazotization reagent, preferably sodium nitrite in an acidic medium.
- a suitable diazotization reagent preferably sodium nitrite in an acidic medium.
- the resulting diastereomeric products can then be separated by physicochemical methods, preferably by chromatographic methods.
- the hydrolytic cleavage of the chiral auxiliary, coupling with amines of the general formula HNR 2 R 3 and cleavage of the benzyl protecting group also provides access to enantiomerically pure hydroxycarboxylic acid compounds of the general formula (V).
- the starting compounds of the general formula (VI) are obtained, for example, by reaction of amines of the general formula HNR 2 R 3 with 2- (alkoxycarbonylmethyl) -3-aryl-propanoic acids and subsequent hydrolytic cleavage of the alkyl group.
- the required 2- (alkoxycarbonylmethyl) -3-aryl-propanoic acids can be prepared in analogy to methods known from the literature (David A. Evans, Leester D. Wu, John JM Wiener, Jeffrey S. Johnson, David HB Ripin and Jason S. Tedrow, J. Org. Chem 64, 6411-6417 [1999]; Saul G. Cohen and Aleksander Milovanovic, J. Am. Chem. Soc.
- Carboxylic acids of the general formula (VIII) can be prepared according to the processes specified in WO 98/11128 from generally available starting materials.
- the compounds of the general formula (I) obtained may, provided they contain suitable basic functions, be converted, in particular for pharmaceutical applications, into their physiologically acceptable salts with inorganic or organic acids.
- suitable acids are hydrochloric, hydrobromic, phosphoric, nitric, sulfuric, methanesulfonic, ethanesulfonic, benzenesulfonic, p-toluenesulfonic, acetic, fumaric, succinic, lactic, mandelic, malic, citric, tartaric and maleic acids.
- the present invention relates to racemates, provided that the compounds of the general formula (I) have only one chiral element.
- the application also includes the individual pairs of diastereomeric antipodes or mixtures thereof which are present when more than one chiral element is present in the compounds of general formula (I) and the individual optically active enantiomers constituting the racemates mentioned.
- Also included in the subject matter of this invention are the compounds according to the invention, including their salts, in which one or more hydrogen atoms are replaced by deuterium.
- novel compounds of the general formula (I) and their physiologically tolerable salts have valuable pharmacological properties which are based on their selective CGRP antagonistic properties.
- Another object of the invention are these compounds containing drugs, their use and their preparation.
- the above new compounds and their physiologically acceptable salts have CGRP antagonistic properties and show good affinities in CGRP receptor binding studies.
- the compounds have CGRP-antagonistic properties in the pharmacological test systems described below.
- SK-N-MC cells are cultured in "Dulbecco's modified Eagle Medium”. The medium of confluent cultures is removed. The cells are washed twice with PBS
- Buffer (Gibco 041-04190 M) washed, by adding PBS buffer, mixed with
- the cells are centrifuged twice at 100 xg and resuspended in BSS. After determination of the
- Tris buffer (10 mM Tris, ⁇ O mM NaCl, ⁇ mM MgCl 2 , 1 mM EDTA, pH 7.40), supplemented with 1% bovine serum albumin and 0.1% bacitracin, recentrifuged and resuspended (1 ml / 1,000,000 cells). The homogenate is frozen at -80 ° C.
- the membrane preparations are stable under these conditions for more than 6 weeks.
- the homogenate is diluted 1:10 with assay buffer ( ⁇ OmM Tris, 110mM NaCl, ⁇ mM MgCl 2 , 1mM EDTA, pH 7.40) and homogenized for 30 seconds with an Ultra-Turrax. 230 ⁇ l of the homogenate are incubated for 180 minutes at room temperature with ⁇ O pM 125 l-iodotyrosyl-calcitonin gene-related peptides (Amersham) and increasing concentrations of the test substances in one Total volume of 2 ⁇ 0 ul incubated. Incubation is terminated by rapid filtration through polyethyleneimine (0.1%) treated GF / B glass fiber filters using a cell harvester. The protein bound radioactivity is determined using a gamma counter. Non-specific binding is defined as the bound radioactivity after the presence of 1 ⁇ M human CGRP-alpha during the incubation.
- assay buffer ⁇ OmM Tris, 110mM NaCl, ⁇ mM MgC
- concentration-binding curves The analysis of the concentration-binding curves is carried out by means of a computer-aided non-linear curve fitting.
- SK-N-MC cells (1 million cells) are washed twice with 2 ⁇ 0 ⁇ l of incubation buffer (Hanks ' HEPES, 1 mM 3-isobutyl-1-methylxanthine, 1% BSA, pH 7.4) and at 37 ° C for 16 minutes pre-incubated. After addition of CGRP (10 .mu.l) as agonist in increasing concentrations (10 ⁇ 11 to 10 ⁇ 6 M) or in addition of substance in 3 to 4 different concentrations is incubated again for 1 ⁇ minutes.
- Intracellular cAMP is then extracted by adding 20 ⁇ l 1M HCl and centrifuging (2000 x g, 4 ° C for 1 NASA minutes). The supernatants are frozen in liquid nitrogen and stored at -20 ° C.
- the cAMP contents of the samples are determined by means of radioimmunoassay (Amersham) and the pA 2 values of antagonistic substances are determined graphically.
- the compounds of the invention show in the described in vitro test model CGRP-antagonistic properties in a dosage range between 10 -12 to 10 -5 M. Because of their pharmacological properties, the compounds according to the invention and their salts with physiologically tolerated acids are thus suitable for the acute and prophylactic treatment of headaches, in particular migraine or cluster headache. Furthermore, the compounds according to the invention also have a positive influence on the following diseases:
- Non-insulin-dependent diabetes mellitus (“NIDDM”), complex regional pain syndrome (CRPS1), cardiovascular diseases, morphine tolerance, clostritis toxin-related diarrheal diseases, skin disorders, in particular thermal and radiation-induced skin damage, including sunburn, inflammatory diseases, e.g. inflammatory joint diseases (arthritis), neurogenic inflammations of the oral mucosa, inflammatory lung diseases, allergic rhinitis, asthma, diseases associated with excessive vasodilation and consequent reduced tissue perfusion, e.g. Shock and sepsis.
- the compounds according to the invention have a soothing effect on pain conditions in general.
- the dosage required to achieve a corresponding effect is advantageously 0.01 to 3 mg / kg body weight, preferably 0.01 to 1 mg / kg body weight, given by oral or subcutaneous administration, 0.01 to 20 mg / kg body weight, preferably 0.1 to 10 mg / kg when given orally Body weight, and by nasal or inhalative administration 0.01 to 10 mg / kg body weight, preferably 0.1 to 10 mg / kg body weight, in each case 1 to 3 times a day.
- the compounds according to the invention can be administered either alone or optionally in combination with other active substances for the treatment of migraine intravenously, subcutaneously, intramuscularly, intrarectally, intranasally, by inhalation, transdermally or orally, in particular aerosol formulations being suitable for inhalation.
- the combinations may be administered either simultaneously or sequentially.
- active ingredient classes include angiotensin II receptor antagonists, ⁇ -agonists and ⁇ -antagonists, ⁇ -HTi B / i D agonists, AMPA antagonists, weak analgesics, antidepressants, antiemetics, anticonvulsants, antimuscuric agents, ⁇ -blockers, calcium antagonists.
- Antagonists corticosteroids, ergot alkaloids, histamine H1 receptor antagonists, neurokinin antagonists, neuroleptics, non-steroidal anti-inflammatory drugs, NO synthase inhibitors, prokinetic agents, selective serotonin reuptake inhibitors or other antimigraine agents associated with one or more inert conventional carriers and / or diluents, for example with corn starch, lactose, cane sugar, microcrystalline cellulose, magnesium stearate, polyvinylpyrrolidone, citric acid, tartaric acid, water, water / ethanol, water / glycerol, water / sorbitol, water / polyethylene glycol, propylene glycol, cetylstearyl alcohol, carboxymethylcellulose or fatty substances such as hard fat or their suitable mixtures, can be incorporated into common pharmaceutical preparations such as tablets, dragees, capsules, powders, suspensions, solutions, metered aerosols or suppositories.
- the non-steroidal anti-inflammatory drugs aceclofenac, acemetacin, acetylsalicylic acid, azathioprine, diclofenac, diflunisal, fenbufen, fenoprofen, flurbiprofen, ibuprofen, indometacin, ketoprofen, leflunomide, lornoxicam, mefenamic acid, naproxen, are therefore used as further active substances.
- the dose for these active substances is expediently 1 / ⁇ of the usually recommended lowest dosage up to 1/1 of the normally recommended dosage, thus for example 20 to 100 mg of sumatriptan.
- Another object of the invention is the use of the compounds of the invention as valuable tools for generating and purifying (affinity chromatography) of antibodies and, after appropriate radioactive labeling, for example by tritiation of suitable precursors, for example by catalytic hydrogenation with trithium or replacement of halogen atoms by tritium, in RIA and ELISA assays and as diagnostic and analytical tools in neurotransmitter research.
- IR, 1 H-NMR and / or mass spectra are available for the compounds prepared.
- Rr values are determined using DC Finished Plates Kieselgel 60 F254 (E. Merck, Darmstadt, Item No. 1.06714) with no chamber saturation.
- the R f values determined under the name Alox are determined using TLC plates alumina 60 F2 ⁇ 4 (E. Merck, Darmstadt, Article No. 1,06713) without chamber saturation.
- the ratios indicated for the flow agents relate to volume units of the respective solvents.
- the indicated volume units at NH 3 refer to a concentrated solution of NH 3 in water.
- the acid, base and salt solutions used in the work-up of the reaction solutions are aqueous systems of the indicated
- HPLC data are measured under the following parameters:
- Analytical column Zorbax column (Agilent Technologies), SB (stable bond) - C18; 3. ⁇ ⁇ m; 4.6 x 7 ⁇ mm; Column temperature: 30 ° C; Flow: 0.8 mL / min; Injection volume: ⁇ ⁇ L; Detection at 2 ⁇ 4 nm
- Preparative HPLC purifications generally use the same gradients used in the collection of analytical HPLC data.
- the collection of products is mass-controlled, the product-containing fractions are combined and freeze-dried.
- the residue was treated with 200 ml of aqueous 1 molar sodium hydroxide solution and 150 ml of TBME and stirred well.
- the aqueous phase was separated, acidified with 2 molar hydrochloric acid solution with stirring and extracted twice with 250 ml of TBME each time.
- the combined organic phases were filtered through charcoal and concentrated under reduced pressure.
- the residue was heated to boiling with 500 ml of water and the hot solution was filtered through Celite. The precipitate formed at room temperature was filtered off with suction and dried at 65 ° C. in a circulating air drying cabinet.
- the mixture was stirred for 1 hour at RT, the THF i.vac. diluted with 100 mL of water and added dropwise with ice cooling 1 M aqueous hydrochloric acid solution until acidic reaction.
- the precipitated substance was filtered, washed with water and dried in air.
- reaction mixture was concentrated under reduced pressure and taken up in ethyl acetate.
- the insoluble precipitate was filtered off with suction and the filtrate was concentrated under reduced pressure.
- the residue was purified by column chromatography.
- reaction solution was hydrogenated at RT for 50 h with 50 bar hydrogen.
- the reaction mixture was concentrated under reduced pressure, the residue was dissolved in 100 ml of EtOAc and washed twice with 70 ml of 2-molar hydrochloric acid.
- the organic phase was then extracted three times with 15% potassium carbonate solution, the combined aqueous phases with conc. Hydrochloric acid and extracted twice with EtOAc.
- the combined organic phases were dried over sodium sulfate, filtered and concentrated under reduced pressure. Yield: 1.7 g (74% of theory)
- Composition 1 capsule for powder inhalation contains: active substance 1.0 mg
- the active ingredient is ground to the particle size required for inhalation.
- the ground active substance is mixed homogeneously with the milk sugar. The mixture is filled into hard gelatin capsules.
- Composition 1 stroke contains:
- the active substance and benzalkonium chloride are dissolved in water and filled into Respimat® cartridges.
- composition 1 vial contains: Active ingredient 0.1 g
- Active substance sodium chloride and benzalkonium chloride are dissolved in water.
- 1 stroke contains: active substance 1.0 mg
- micronised active substance is homogeneously suspended in the mixture of lecithin and propellant gas.
- the suspension is filled into a pressure vessel with metering valve.
- the active substance and the excipients are dissolved in water and in a corresponding
- Dissolve glycofurol and glucose in water for injections Wfl
- Add human serum albumin Dissolve active ingredient with heating
- fill with Wfl to batch volume Fill into ampoules under nitrogen fumigation.
- Disodium hydrogen phosphate Na 2 HPO 4 > 2H 2 O 2 mg
- Dissolve polysorbate 80 sodium chloride, monopotassium dihydrogen phosphate and disodium hydrogen phosphate in water for injections (Wfl); Add human serum albumin; Dissolve active ingredient with heating; fill with Wfl to batch volume; fill in ampoules.
- Dissolve mannitol in water for injections Wfl
- Wfl water for injections
- Human serum albumin Dissolve active ingredient with heating
- fill with Wfl to batch volume to fill in vials; freeze-dry.
- Polysorbate 80 Tween 80 20 mg Mannitol 200 mg
- Example IX Tablets containing 20 mg of active substance
- composition Active substance 20 mg
- composition Active substance 10 mg
- Dissolve mannitol in water for injections Wfl
- Wfl water for injections
- Human serum albumin Dissolve active ingredient with heating
- fill with Wfl to batch volume Fill into ampoules under nitrogen fumigation.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Dermatology (AREA)
- Neurosurgery (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Rheumatology (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description



























































Claims













Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007507723A JP2007532600A (ja) | 2004-04-15 | 2005-04-09 | 選択されたcgrpアンタゴニスト、その製造方法及びその薬物としての使用 |
| CA002562526A CA2562526A1 (en) | 2004-04-15 | 2005-04-09 | Selected cgrp antagonists, method for producing the same and the use thereof as drugs |
| DE502005010105T DE502005010105D1 (de) | 2004-04-15 | 2005-04-09 | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| EP05731650A EP1737842B1 (de) | 2004-04-15 | 2005-04-09 | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| AT05731650T ATE478066T1 (de) | 2004-04-15 | 2005-04-09 | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102004018795.9 | 2004-04-15 | ||
| DE102004018795A DE102004018795A1 (de) | 2004-04-15 | 2004-04-15 | Ausgewählte CGRP-Antagonisten, Verfahren zu deren Herstellung sowie deren Verwendung als Arzneimittel |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005100343A1 true WO2005100343A1 (de) | 2005-10-27 |
Family
ID=34964247
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2005/003741 WO2005100343A1 (de) | 2004-04-15 | 2005-04-09 | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
Country Status (11)
| Country | Link |
|---|---|
| EP (1) | EP1737842B1 (de) |
| JP (1) | JP2007532600A (de) |
| AR (1) | AR049883A1 (de) |
| AT (1) | ATE478066T1 (de) |
| CA (1) | CA2562526A1 (de) |
| DE (2) | DE102004018795A1 (de) |
| ES (1) | ES2348638T3 (de) |
| PE (1) | PE20060176A1 (de) |
| TW (1) | TW200603799A (de) |
| UY (1) | UY28848A1 (de) |
| WO (1) | WO2005100343A1 (de) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006072413A1 (de) * | 2004-12-29 | 2006-07-13 | Boehringer Ingelheim International Gmbh | Verwendung ausgewählter cgrp-antagonisten in kombination mit anderen arzneistoffen gegen migräne für die behandlung von migräne |
| WO2007020261A2 (de) * | 2005-08-17 | 2007-02-22 | Boehringer Ingelheim International Gmbh | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| WO2007045672A1 (de) * | 2005-10-21 | 2007-04-26 | Boehringer Ingelheim International Gmbh | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| US7220862B2 (en) | 2002-06-05 | 2007-05-22 | Bristol-Myers Squibb Company | Calcitonin gene related peptide receptor antagonists |
| US7384931B2 (en) | 2004-11-03 | 2008-06-10 | Bristol-Myers Squibb Company | Constrained compounds as CGRP-receptor antagonists |
| US7384930B2 (en) | 2004-11-03 | 2008-06-10 | Bristol-Myers Squibb Company | Constrained compounds as CGRP-receptor antagonists |
| US7449586B2 (en) | 2004-12-03 | 2008-11-11 | Bristol-Myers Squibb Company | Processes for the preparation of CGRP-receptor antagonists and intermediates thereof |
| US7479488B2 (en) | 2004-03-29 | 2009-01-20 | Boehringer Ingelheim International Gmbh | Selected CGRP—antagonists, process for preparing them and their use as pharmaceutical compositions |
| WO2009021942A1 (de) * | 2007-08-13 | 2009-02-19 | Boehringer Ingelheim International Gmbh | Neues herstellverfahren |
| US7569578B2 (en) | 2003-12-05 | 2009-08-04 | Bristol-Meyers Squibb Company | Heterocyclic anti-migraine agents |
| US7595312B2 (en) | 2002-10-25 | 2009-09-29 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Selected CGRP antagonists, processes for preparing them and their use as pharmaceutical compositions |
| US7696195B2 (en) | 2004-04-22 | 2010-04-13 | Boehringer Ingelheim International Gmbh | Selected CGRP-antagonists, process for preparing them and their use as pharmaceutical compositions |
| US7700589B2 (en) | 2002-10-25 | 2010-04-20 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | CGRP antagonists |
| US7772244B2 (en) | 2004-03-29 | 2010-08-10 | Bristol-Myers Squibb Company | Therapeutic agents for the treatment of migraine |
| US7834007B2 (en) | 2005-08-25 | 2010-11-16 | Bristol-Myers Squibb Company | CGRP antagonists |
| US7842808B2 (en) | 2002-06-05 | 2010-11-30 | Bristol-Myers Squibb Company | Anti-migraine spirocycles |
| US8168592B2 (en) | 2005-10-21 | 2012-05-01 | Amgen Inc. | CGRP peptide antagonists and conjugates |
| US8450327B2 (en) | 2007-10-18 | 2013-05-28 | Boehringer Ingelheim International Gmbh | CGRP antagonists |
| US8629137B2 (en) | 2007-10-18 | 2014-01-14 | Boehringer Ingelheim International Gmbh | CGRP antagonists |
| US8829006B2 (en) | 2007-11-22 | 2014-09-09 | Boehringer Ingelheim International Gmbh | Compounds |
| TWI722035B (zh) * | 2015-10-30 | 2021-03-21 | 英商黑普達斯醫療公司 | Cgrp受體拮抗劑 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102004063752A1 (de) * | 2004-12-29 | 2006-07-13 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Verwendung ausgewählter CGRP-Antagonisten zur Bekämpfung menopausaler Hitzewallungen |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998011128A1 (de) | 1996-09-10 | 1998-03-19 | Dr. Karl Thomae Gmbh | Abgewandelte aminosäuren, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung |
| WO2001032649A1 (de) | 1999-10-29 | 2001-05-10 | Boehringer Ingelheim Pharma Kg | Arylalkane, arylalkene und aryl-azaalkane, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung |
| WO2003070753A1 (de) * | 2002-02-19 | 2003-08-28 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Verfahren zur herstellung eines pulverinhalativums enthaltend ein salz des cgrp-antagonisten bibn4096 |
| WO2004000289A2 (de) * | 2002-06-19 | 2003-12-31 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Zubereitungen zur intranasalen applikation ausgewählter, von aminosäuren abgeleiteter grp-antagonisten |
| WO2004037811A1 (de) | 2002-10-25 | 2004-05-06 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| WO2004037810A1 (de) | 2002-10-25 | 2004-05-06 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| WO2004063171A1 (de) | 2003-01-14 | 2004-07-29 | Boehringer Ingelheim International Gmbh | N- (1-benzyl-2-oxo-2- (1-piperazinyl) ethyl) -1-piperidincarboxamid-derivate und verwandte verbindungen als cgrp-antagonisten zur behandlung von kopfschmerzen |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2487976C (en) * | 2002-06-05 | 2011-07-26 | Bristol-Myers Squibb Company | Calcitonin gene related peptide receptor antagonists |
| US7161780B2 (en) * | 2003-02-03 | 2007-01-09 | Leviton Manufacturing Co., Inc. | Circuit interrupting device with single throw, double mode button for test-reset function |
-
2004
- 2004-04-15 DE DE102004018795A patent/DE102004018795A1/de not_active Withdrawn
-
2005
- 2005-04-09 CA CA002562526A patent/CA2562526A1/en not_active Abandoned
- 2005-04-09 ES ES05731650T patent/ES2348638T3/es active Active
- 2005-04-09 WO PCT/EP2005/003741 patent/WO2005100343A1/de not_active Application Discontinuation
- 2005-04-09 AT AT05731650T patent/ATE478066T1/de active
- 2005-04-09 DE DE502005010105T patent/DE502005010105D1/de active Active
- 2005-04-09 EP EP05731650A patent/EP1737842B1/de active Active
- 2005-04-09 JP JP2007507723A patent/JP2007532600A/ja active Pending
- 2005-04-12 UY UY28848A patent/UY28848A1/es not_active Application Discontinuation
- 2005-04-13 PE PE2005000410A patent/PE20060176A1/es not_active Application Discontinuation
- 2005-04-14 TW TW094111775A patent/TW200603799A/zh unknown
- 2005-04-15 AR ARP050101488A patent/AR049883A1/es unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998011128A1 (de) | 1996-09-10 | 1998-03-19 | Dr. Karl Thomae Gmbh | Abgewandelte aminosäuren, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung |
| WO2001032649A1 (de) | 1999-10-29 | 2001-05-10 | Boehringer Ingelheim Pharma Kg | Arylalkane, arylalkene und aryl-azaalkane, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung |
| WO2003070753A1 (de) * | 2002-02-19 | 2003-08-28 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Verfahren zur herstellung eines pulverinhalativums enthaltend ein salz des cgrp-antagonisten bibn4096 |
| WO2004000289A2 (de) * | 2002-06-19 | 2003-12-31 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Zubereitungen zur intranasalen applikation ausgewählter, von aminosäuren abgeleiteter grp-antagonisten |
| WO2004037811A1 (de) | 2002-10-25 | 2004-05-06 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| WO2004037810A1 (de) | 2002-10-25 | 2004-05-06 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| WO2004063171A1 (de) | 2003-01-14 | 2004-07-29 | Boehringer Ingelheim International Gmbh | N- (1-benzyl-2-oxo-2- (1-piperazinyl) ethyl) -1-piperidincarboxamid-derivate und verwandte verbindungen als cgrp-antagonisten zur behandlung von kopfschmerzen |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7754732B2 (en) | 2002-06-05 | 2010-07-13 | Bristol-Myers Squibb Company | Spirocyclic anti-migraine compounds |
| US7842808B2 (en) | 2002-06-05 | 2010-11-30 | Bristol-Myers Squibb Company | Anti-migraine spirocycles |
| US7220862B2 (en) | 2002-06-05 | 2007-05-22 | Bristol-Myers Squibb Company | Calcitonin gene related peptide receptor antagonists |
| US7314883B2 (en) | 2002-06-05 | 2008-01-01 | Bristol-Myers Squibb Company | Anti-migraine treatments |
| US7700589B2 (en) | 2002-10-25 | 2010-04-20 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | CGRP antagonists |
| US7595312B2 (en) | 2002-10-25 | 2009-09-29 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Selected CGRP antagonists, processes for preparing them and their use as pharmaceutical compositions |
| US7569578B2 (en) | 2003-12-05 | 2009-08-04 | Bristol-Meyers Squibb Company | Heterocyclic anti-migraine agents |
| US7479488B2 (en) | 2004-03-29 | 2009-01-20 | Boehringer Ingelheim International Gmbh | Selected CGRP—antagonists, process for preparing them and their use as pharmaceutical compositions |
| US7700598B2 (en) | 2004-03-29 | 2010-04-20 | Boehringer Ingelheim International Gmbh | Selected CGRP-antagonists, process for preparing them and their use as pharmaceutical compositions |
| US7772244B2 (en) | 2004-03-29 | 2010-08-10 | Bristol-Myers Squibb Company | Therapeutic agents for the treatment of migraine |
| US7696195B2 (en) | 2004-04-22 | 2010-04-13 | Boehringer Ingelheim International Gmbh | Selected CGRP-antagonists, process for preparing them and their use as pharmaceutical compositions |
| US7384930B2 (en) | 2004-11-03 | 2008-06-10 | Bristol-Myers Squibb Company | Constrained compounds as CGRP-receptor antagonists |
| US7384931B2 (en) | 2004-11-03 | 2008-06-10 | Bristol-Myers Squibb Company | Constrained compounds as CGRP-receptor antagonists |
| US7544680B2 (en) | 2004-11-03 | 2009-06-09 | Bristol-Myers Squibb Company | Constrained compounds as CGRP-receptor antagonists |
| US7449586B2 (en) | 2004-12-03 | 2008-11-11 | Bristol-Myers Squibb Company | Processes for the preparation of CGRP-receptor antagonists and intermediates thereof |
| WO2006072413A1 (de) * | 2004-12-29 | 2006-07-13 | Boehringer Ingelheim International Gmbh | Verwendung ausgewählter cgrp-antagonisten in kombination mit anderen arzneistoffen gegen migräne für die behandlung von migräne |
| WO2007020261A2 (de) * | 2005-08-17 | 2007-02-22 | Boehringer Ingelheim International Gmbh | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| JP4852607B2 (ja) * | 2005-08-17 | 2012-01-11 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 選ばれたcgrpアンタゴニスト、それらの調製方法及び薬物としてのそれらの使用 |
| US7858622B2 (en) | 2005-08-17 | 2010-12-28 | Boehringer Ingelheim International Gmbh | Selected CGRP antagonists, processes for preparing them and their use as pharmaceutical compositions |
| WO2007020261A3 (de) * | 2005-08-17 | 2008-08-21 | Boehringer Ingelheim Int | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| JP2011256200A (ja) * | 2005-08-17 | 2011-12-22 | Boehringer Ingelheim Internatl Gmbh | 選ばれたcgrpアンタゴニスト、それらの調製方法及び薬物としてのそれらの使用 |
| US7579341B2 (en) | 2005-08-17 | 2009-08-25 | Boehringer Ingelheim International Gmbh | Selected CGRP antagonists, processes for preparing them and their use as pharmaceutical compositions |
| US7834007B2 (en) | 2005-08-25 | 2010-11-16 | Bristol-Myers Squibb Company | CGRP antagonists |
| WO2007045672A1 (de) * | 2005-10-21 | 2007-04-26 | Boehringer Ingelheim International Gmbh | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel |
| US7625886B2 (en) | 2005-10-21 | 2009-12-01 | Boehringer Ingelheim International Gmbh | Selected CGRP antagonists, processes for preparing them and their use as pharmaceutical compositions |
| US8168592B2 (en) | 2005-10-21 | 2012-05-01 | Amgen Inc. | CGRP peptide antagonists and conjugates |
| JP2010535837A (ja) * | 2007-08-13 | 2010-11-25 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 新規調製方法 |
| WO2009021942A1 (de) * | 2007-08-13 | 2009-02-19 | Boehringer Ingelheim International Gmbh | Neues herstellverfahren |
| US8222402B2 (en) | 2007-08-13 | 2012-07-17 | Boehringer Ingelheim International Gmbh | Preparation process |
| US8450327B2 (en) | 2007-10-18 | 2013-05-28 | Boehringer Ingelheim International Gmbh | CGRP antagonists |
| US8629137B2 (en) | 2007-10-18 | 2014-01-14 | Boehringer Ingelheim International Gmbh | CGRP antagonists |
| US8829006B2 (en) | 2007-11-22 | 2014-09-09 | Boehringer Ingelheim International Gmbh | Compounds |
| TWI722035B (zh) * | 2015-10-30 | 2021-03-21 | 英商黑普達斯醫療公司 | Cgrp受體拮抗劑 |
Also Published As
| Publication number | Publication date |
|---|---|
| ES2348638T3 (es) | 2010-12-10 |
| JP2007532600A (ja) | 2007-11-15 |
| DE502005010105D1 (de) | 2010-09-30 |
| DE102004018795A1 (de) | 2005-10-27 |
| PE20060176A1 (es) | 2006-04-18 |
| TW200603799A (en) | 2006-02-01 |
| AR049883A1 (es) | 2006-09-13 |
| CA2562526A1 (en) | 2005-10-27 |
| EP1737842A1 (de) | 2007-01-03 |
| EP1737842B1 (de) | 2010-08-18 |
| ATE478066T1 (de) | 2010-09-15 |
| UY28848A1 (es) | 2005-11-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2005100343A1 (de) | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel | |
| EP1587795B1 (de) | N- (1-benzyl-2-oxo-2- (1-piperazinyl) ethyl) -1-piperidincarboxamid-derivate und verwandte verbindungen als cgrp-antagonisten zur behandlung von kopfschmerzen | |
| EP1558600B1 (de) | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel | |
| EP1732917B1 (de) | Ausgewahlte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel | |
| EP1737864A1 (de) | Ausgewaehlte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel | |
| EP1722792A1 (de) | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel | |
| DE10250082A1 (de) | Ausgewählte CGRP-Antagonisten, Verfahren zu deren Herstellung sowie deren Verwendung als Arzneimittel | |
| WO2005100352A1 (de) | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel | |
| EP2438067B1 (de) | Spirolactame als cgrp-antagonisten | |
| EP1863799A1 (de) | Cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel | |
| WO2003076432A1 (de) | Benzodiazepin-substituierte piperdine zur verwendung in der behandlung von cardiovaskulären erkrankungen | |
| DE102004019492A1 (de) | Ausgewählte CGRP-Antagonisten, Verfahren zu deren Herstellung sowie deren Verwendung als Arzneimittel | |
| DE19963868A1 (de) | Neue substituierte Piperidine, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung | |
| DE19952147A1 (de) | Neue Cyclopropane, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung | |
| EP1917256A2 (de) | Ausgewählte cgrp-antagonisten, verfahren zu deren herstellung sowie deren verwendung als arzneimittel | |
| US7439237B2 (en) | Selected CGRP-antagonists, process for preparing them and their use as pharmaceutical compositions | |
| EP1863791B1 (de) | 2-oxo-1,2,4,5-tetrahydro-1,3-benzdiazepin-3-yl-piperidine als cgrp-antagonisten | |
| DE102004010254A1 (de) | Ausgewählte CGRP-Antagonisten, Verfahren zu deren Herstellung sowie deren Verwendung als Arzneimittel | |
| DE102004028751A1 (de) | Ausgewählte CGRP-Antagonisten, Verfahren zu deren Herstellung sowie deren Verwendung als Arzneimittel |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005731650 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2562526 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007507723 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005731650 Country of ref document: EP |