WO2005025583A2 - Tlr7 ligands for the treatment of hepatitis c - Google Patents
Tlr7 ligands for the treatment of hepatitis c Download PDFInfo
- Publication number
- WO2005025583A2 WO2005025583A2 PCT/US2004/028236 US2004028236W WO2005025583A2 WO 2005025583 A2 WO2005025583 A2 WO 2005025583A2 US 2004028236 W US2004028236 W US 2004028236W WO 2005025583 A2 WO2005025583 A2 WO 2005025583A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- aryl
- tlr7 ligand
- substituted
- prodrug
- Prior art date
Links
- 0 *c1c(*)[n]([C@@]([C@@]2O)O[C@@](CO)[C@@]2O)c(N=C(N)N2)c1C2=O Chemical compound *c1c(*)[n]([C@@]([C@@]2O)O[C@@](CO)[C@@]2O)c(N=C(N)N2)c1C2=O 0.000 description 10
- VDCRFBBZFHHYGT-IOSLPCCCSA-N C=CCN(C1=C(N2[C@@H]([C@@H]3O)O[C@H](CO)[C@H]3O)N=C(N)NC1=O)C2=O Chemical compound C=CCN(C1=C(N2[C@@H]([C@@H]3O)O[C@H](CO)[C@H]3O)N=C(N)NC1=O)C2=O VDCRFBBZFHHYGT-IOSLPCCCSA-N 0.000 description 1
- BUIKJNWRANGGHG-PFHPDILHSA-N CC(C)C(C(OC[C@H]([C@H]([C@H]1O)O)O[C@H]1N(C(N=C(N)NC1=O)=C1S1)C1=O)=O)N Chemical compound CC(C)C(C(OC[C@H]([C@H]([C@H]1O)O)O[C@H]1N(C(N=C(N)NC1=O)=C1S1)C1=O)=O)N BUIKJNWRANGGHG-PFHPDILHSA-N 0.000 description 1
- OGCSVEVSYHYLOO-LSCFUAHRSA-N CC(OC[C@H]([C@H]([C@H]1OC(C)=O)OC(C)=O)O[C@H]1N(C(N=C(N)NC1=O)=C1N1CC=C)C1=O)=O Chemical compound CC(OC[C@H]([C@H]([C@H]1OC(C)=O)OC(C)=O)O[C@H]1N(C(N=C(N)NC1=O)=C1N1CC=C)C1=O)=O OGCSVEVSYHYLOO-LSCFUAHRSA-N 0.000 description 1
- FOUUTBBXBSSXEE-LSCFUAHRSA-N CC(OC[C@H]([C@H]([C@H]1OC(C)=O)OC(C)=O)O[C@H]1N(c1nc(N)nc(Cl)c1N1CC=C)C1=O)=O Chemical compound CC(OC[C@H]([C@H]([C@H]1OC(C)=O)OC(C)=O)O[C@H]1N(c1nc(N)nc(Cl)c1N1CC=C)C1=O)=O FOUUTBBXBSSXEE-LSCFUAHRSA-N 0.000 description 1
- AXSHOJYKARNQTQ-KQYNXXCUSA-N CC1(C)O[C@H]2[C@H]([n]3c(Br)nc4c3N=C(N)NC4=O)O[C@H](CO)[C@H]2O1 Chemical compound CC1(C)O[C@H]2[C@H]([n]3c(Br)nc4c3N=C(N)NC4=O)O[C@H](CO)[C@H]2O1 AXSHOJYKARNQTQ-KQYNXXCUSA-N 0.000 description 1
- ULYHIGSJUNODHA-UHFFFAOYSA-N CCCCCOC(Nc1nc(cccc2)c2c2c1nc[n]2CC(C)C)=O Chemical compound CCCCCOC(Nc1nc(cccc2)c2c2c1nc[n]2CC(C)C)=O ULYHIGSJUNODHA-UHFFFAOYSA-N 0.000 description 1
- YKUSFJFHUCAXOG-SDBHATRESA-N CCOC(N(C)COc(nc(N)nc1N2[C@@H]([C@@H]3O)O[C@H](CO)[C@H]3O)c1N(CC=C)C2=O)=O Chemical compound CCOC(N(C)COc(nc(N)nc1N2[C@@H]([C@@H]3O)O[C@H](CO)[C@H]3O)c1N(CC=C)C2=O)=O YKUSFJFHUCAXOG-SDBHATRESA-N 0.000 description 1
- QZNHXDIAIUAXLP-UHFFFAOYSA-N CCOC(Oc([n](Cc1ccccc1)c1n2)nc1c(N)nc2OCCOC)=O Chemical compound CCOC(Oc([n](Cc1ccccc1)c1n2)nc1c(N)nc2OCCOC)=O QZNHXDIAIUAXLP-UHFFFAOYSA-N 0.000 description 1
- ASUCSHXLTWZYBA-UMMCILCDSA-N NC(NC1=O)=Nc([n]2[C@@H]([C@@H]3O)O[C@H](CO)[C@H]3O)c1nc2Br Chemical compound NC(NC1=O)=Nc([n]2[C@@H]([C@@H]3O)O[C@H](CO)[C@H]3O)c1nc2Br ASUCSHXLTWZYBA-UMMCILCDSA-N 0.000 description 1
- BBLQSWGGUCEGTH-ZIYNGMLESA-N Nc(nc1N2[C@@H]([C@@H]3O)O[C@H](CO)[C@H]3O)ncc1SC2=O Chemical compound Nc(nc1N2[C@@H]([C@@H]3O)O[C@H](CO)[C@H]3O)ncc1SC2=O BBLQSWGGUCEGTH-ZIYNGMLESA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
- A61K31/708—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid having oxo groups directly attached to the purine ring system, e.g. guanosine, guanylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
- A61K31/522—Purines, e.g. adenine having oxo groups directly attached to the heterocyclic ring, e.g. hypoxanthine, guanine, acyclovir
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- This invention relates to methods for treating or preventing hepatitis C virus infections in mammals using Toll-Like Receptor (TLR)7 ligands and prodrugs thereof. More particularly, this invention relates to methods of orally administering a therapeutically effective amount of one or more prodrugs of TLR7 ligands for the treatment or prevention of hepatitis C viral infection. Oral administration of these TLR7 immunomodulating ligands and prodrugs thereof to a mammal provides therapeutically effective amounts and reduced undesirable side effects.
- TLR7 Toll-Like Receptor
- TLRs Toll-Like Receptors
- TLRs detect PAMPs (pathogen-associated molecular patterns) and stimulate immune cells via the MyD88-dependent interleukin 1 receptor (IL-1R)-TLR signaling pathway, which leads to activation of the transcription factor NF- B2.
- IL-1R MyD88-dependent interleukin 1 receptor
- TLRl to TLR10 Ten functional family members of TLRs (TLRl to TLR10) have been identified in humans. Akira S. et al, Nature Immunol, 2, 675-680 (2001). TLR2, TLR4, and TLR5 are crucial for the recognition of peptidoglycan, lipopolysacharide, and flagellin. Hayashi, F. et al, Nature, 410, 1099-1103 (2001). TLR6 associates with TLR2 and recognizes lipoproteins from mycoplasma.
- TLR9 detects bacterial DNA containing unmethylated CpG motifs and TLR3 activates immune cells in response to double-stranded RNA. Hemmi, H. et al, Nature, 408, 740-745 (2000).
- D- and L-purine nucleosides have been the subject of considerable research the past two decades. See, e.g., Reitz et al, J. Med. Chem., 37, 3561-78 (1994); Michael et al, J. Med. Chem., 36, 3431-36 (1993) (immunomodulatory guanosine analogs having substituents at the7-and/or 8-positions); PatentNo. 5,821,236 to Krenitsky et al. (disclosing 6-alkoxy derivatives of arabinofuranosyl purine derivatives that are useful for tumor therapy); U.S. Patent No. 5,041,426 to Robins et al.
- TLR7 ligands A number of compounds known to be immunostimulants have recently been identified in the literature as TLR7 ligands, see, e.g., Heil et al, Eur. J. Immunol, 33(11), 2987-97 (2003), Lore et al, J. Immunol, 171(8), 4320-8 (2003), Nagase et al, J. Immunol, 171(8), 3977-82 (2003), Mohty et al, J. Immunol, 171(7), 3385-93 (2003), Pinhal-Enfield, et al, Am. J. Pathol, 163(2), 711-21 (2003), Doxsee et al, J.
- TLR7 ligands are known to stimulate immune responses in vitro and in animal species, and this has led to testing of the uses of these compounds for several therapeutic uses, including antiviral and cancer therapies.
- These compounds have been characterized as analogs or derivatives of a) guanosine, b) imidazoquinoline, and c) pyrimidine. See Akira, Current Opinion, 15, 5-11 (2003).
- One member (imiquimod) of the imidazoquinoline chemical class has been found effective for treating topical genital infections by papilloma virus.
- a second member of the imidazoquinoline class, resiquimod has been tested for the treatment of HCV, but this compound failed to show anti-HCV effect at tolerated oral doses.
- TLR7 ligands for the treatment of immunological disease and viral infections; see, e.g., U.S. Patent Nos. 5,041,426 and 4,880,784 to Robins etal. (3- ⁇ -D-ribofuranosylthiazolo[4,5-d]pyridimines demonstrating significant immunoactivity, including murine spleen cell proliferation and in vivo activity against Semliki Forest virus); United States Patent Application Publication No. US 2003/0199461 and WO 03/045968 to Averett et al.
- immunomodulatory nucleosides have relatively poor oral tolerability when compared to that of the intravenous route.
- the gastrointestinal tract presents a particular tolerability barrier to immunologic agents by virtue of the large amount of immune tissue associated with the intestinal wall (i.e., gut).
- gut the intestinal wall
- the immune tissue also may become preferentially affected after oral administration of immunomodulatory compounds because of the high local concentrations of the administered compound in the gut. This leads to undesirable side effects, for example in the case of immune activating agents there is observed gastroenteritis and localized hemorrhagic effects.
- a solution to the problem of effective oral delivery of immunomodulators is not evident in the literature. Available evidence indicates that systemic levels of administered drugs in this class have been limited by gastrointestinal toxicities arising after low oral doses. Therefore there remains a need for immunomodulating TLR7 ligands that have improved oral availability and reduced gastrointestinal irritancy.
- This invention encompasses novel methods for the treatment or prevention of hepatitis C viral infection, and novel pharmaceutical compositions which utilize TLR7 ligands or pharmaceutically acceptable salts, hydrates, metabolites or stereoisomers thereof.
- the invention encompasses a method of treating or preventing a hepatitis C virus infection in a patient in need thereof comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand or a pharmaceutically acceptable salt, hydrate, metabolite or stereoisomer thereof or a pharmaceutically acceptable salt or hydrate of said stereoisomer.
- the invention encompasses a method of treating or preventing a hepatitis C virus infection in a patient in need thereof comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand selected from analogs and derivatives of a) guanosine b) imidazoquinoline c) adenine, and d) pyrimidine.
- a TLR7 ligand selected from analogs and derivatives of a) guanosine b) imidazoquinoline c) adenine, and d) pyrimidine.
- the invention encompasses a method of treating or preventing a hepatitis C virus infection in a patient in need thereof comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand selected from
- each R 1 is H, or a substituted or unsubstituted alkyl, alkenyl, or alkynyl, which may be interrupted by one or more 0, S, or N heteroatoms, or a substituted or unsubstituted aryl or heteroaryl;
- R 2 is H, OH, SH, halo, or a substituted or unsubstituted alkyl, alkenyl, or alkynyl, which may be interrupted by one or more O, S, or N heteroatoms, or a substituted or unsubstituted
- R 3 is H, OH, or SH, or a substituted or unsubstituted alkyl, alkenyl, alkynyl, aryl, heteroaryl, -O-(alkyl), -O-(aryl), -O-(heteroaryl), -S-(alkyl), -S-(aryl), -S-(heteroaryl),
- X is O or S
- Y is H, halo, OH, OR 4 , SH, SR 4 , or a substituted or unsubstituted alkyl or aryl;
- Z is H, halo, OH, OR 4 , SH, or SR 4 ; or a pharmaceutically acceptable salt, hydrate, metabolite or stereoisomer thereof or a pharmaceutically acceptable salt or hydrate of said stereoisomer.
- the invention encompasses a method of treating or preventing a hepatitis C virus infection in a patient in need thereof comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand selected from Formula la, lb, Ic, Id, Ie, If, Ig, and Ih, wherein R 1 is H or a substituted or unsubstituted alkyl, alkenyl, or alkynyl; R 2 is H, OH, halo, or a substituted or unsubstituted alkyl, alkenyl, or alkynyl, or -CH 2 -0-(alkyl); R 3 is H, OH, or SH, or a substituted or unsubstituted -O-(alkyl), -S-(alkyl), or -NH(alkyl); X is O or S; Y is H, halo, OH, OR 4 , SH, or
- the invention encompasses a method of treating or preventing a hepatitis C virus infection in a patient in need thereof comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand selected from
- the invention encompasses a method for treating or preventing hepatitis C virus infection in a mammal in need thereof, preferably in a human in need thereof.
- the invention encompasses a method for treating or preventing hepatitis C virus infection in a patient in need thereof, comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand and a pharmaceutically acceptable excipient, carrier, or vehicle.
- the invention encompasses a method for treating or preventing hepatitis C virus infection in a patient in need thereof, comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand orally, mucosally, topically or transdermally.
- the invention encompasses a method for treating or preventing hepatitis C virus infection in a patient in need thereof, comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand parenterally.
- the invention encompasses a method for treating or preventing hepatitis C virus infection in a patient in need thereof, comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand and an additional therapeutic agent, preferably an additional antiviral or immunomodulatory agent.
- the invention also encompasses pharmaceutical compositions suitable for parenteral administration to a patient comprising a therapeutically or pharmaceutically acceptable amount of a TLR7 ligand of the invention in a sterile form; pharmaceutical compositions suitable for oral administration to a patient comprising a therapeutically or pharmaceutically acceptable amount of a TLR7 ligand of the invention, wherein such compositions are formulated to reduce exposure of the subepithelial immune anatomy to the TLR7 ligand while improving systemic absorption of the TLR7 ligand; pharmaceutical compositions suitable for mucosal administration to a patient comprising a therapeutically or pharmaceutically acceptable amount of a TLR7 ligand of the invention, wherein such compositions are formulated to reduce exposure of the subepithelial immune anatomy to the TLR7 ligand while improving systemic absorption of the TLR7 ligand; and pharmaceutical compositions suitable for topical administration to a patient comprising a therapeutically or pharmaceutically acceptable amount of aTLR7 ligand of the invention, wherein such compositions
- each of these compositions is in single unit dosage form and comprising an amount of active ingredient sufficient to treat or prevent human infection by hepatitis C virus.
- the invention encompasses a pharmaceutical composition comprising a TLR7 ligand selected from analogs and derivatives of a) guanosine, b) imidazoquinoline, c) adenine, and d) pyrimidine.
- a TLR7 ligand selected from analogs and derivatives of a) guanosine, b) imidazoquinoline, c) adenine, and d) pyrimidine.
- the invention encompasses a pharmaceutical composition comprising a TLR7 ligand selected from Formulas la, lb, Ic, Id, Ie, If, Ig, and
- TLR7 Ligand Prodrugs This invention also encompasses novel methods for the treatment or prevention of hepatitis C viral infection, and novel pharmaceutical compositions which utilize TLR7 ligand prodrugs or pharmaceutically acceptable salts, hydrates, metabolites or stereoisomers thereof.
- This invention also encompasses novel methods of treating diseases responsive to immuno therapy with immunologic agents, comprising orally administering a TLR7 ligand prodrug to a patient in need of immuno therapy, wherein the TLR7 prodrug achieves a therapeutically effective plasma concentration of the TLR7 ligand in the patient.
- the invention encompasses a method of treating a hepatitis C virus infection in a patient comprising orally administering to the patient a TLR7 ligand prodrug or a pharmaceutically acceptable salt, hydrate, or stereoisomer thereof, wherein the oral administration of the TLR7 ligand prodrug achieves a therapeutically effective plasma concentration of the TLR7 ligand while reducing undesirable side effects associated with TLR7 ligands.
- the TLR7 ligand prodrug is a masked TLR7 ligand prodrug.
- the invention also encompasses a method of treating diseases responsive to immuno therapy while reducing undesirable side effects associated with immunologic agents, comprising orally administering a TLR7 ligand prodrug to a patient in need of immuno therapy, wherein the TLR7 prodrug achieves a therapeutically effective plasma concentration of the TLR7 ligand in the patient.
- the TLR7 ligand prodrug is a masked TLR7 ligand prodrug.
- the oral administration of the TLR7 ligand prodrug improves the in vivo bioavailability of the TLR7 ligand.
- the oral administration of the TLR7 ligand prodrug achieves an in vivo effective plasma concentration of the TLR7 ligand that is 10% to 500% of the effective in vivo exposure obtained upon oral administration of the TLR7 ligand alone.
- the oral administration of the masked TLR7 ligand prodrug achieves an in vivo effective plasma concentration of the TLR7 ligand that is 50%> to 200% of the effective in vivo exposure obtained upon oral administration of the TLR7 ligand alone.
- the oral administration of the TLR7 ligand prodrug reduces adverse side effects.
- the side effect comprises gastrointestinal irritancy, wherein gastrointestinal irritancy comprises hemorrhage, lesions, and emesis.
- the TLR7 ligand prodrug improves oral availability by at least 25% and reduces gastrointestinal irritancy by at least 50% in a patient relative to the oral administration of the TLR7 ligand alone. In another embodiment, the TLR7 ligand prodrug improves oral availability by at least 50% and reduces gastrointestinal irritancy by such that other toxicities become limiting in a patient relative to the oral administration of the TLR7 ligand alone.
- the TLR7 ligand prodrug achieves a therapeutically effective plasma concentration that is 25% to 200% of the effective in vivo concentration of the TLR7 ligand in a patient after oral administration, with minimal gastrointestinal irritancy.
- the methods of the invention encompass administering to a patient in need thereof a therapeutically or prophylactically effective amount of a prodrug of a TLR7 ligand selected from analogs and derivatives of a) guanosine, b) imidazoquinoline, c) adenine, and d) pyrimidine.
- the methods of the invention encompass administering to a patient in need thereof a therapeutically or prophylactically effective amount of a prodrug of a TLR7 ligand selected from analogs and derivatives of a) guanosine, b) imidazoquinoline, c) adenine, and d) pyrimidine, wherein the prodrug is an (a) amide, carbamate, or amidine moiety after conversion of a TLR7 ligand amine substituent,
- the methods of the invention encompass administering to a patient in need thereof a therapeutically or prophylactically effective amount of a prodrug of a TLR7 ligand selected from lid He Ilf
- each R 1 is H, or a substituted or unsubstituted alkyl, alkenyl, or alkynyl, which may be interrupted by one or more O, S, or N heteroatoms, or a substituted or unsubstituted aryl or heteroaryl;
- R 2 is H, OH, SH, halo, or a substituted or unsubstituted alkyl, alkenyl, or alkynyl, which may be interrupted by one or more O, S, or N heteroatoms, or a substituted or unsubstituted
- R 3 is H, OH, or SH, or a substituted or unsubstituted alkyl, alkenyl, alkynyl, aryl, heteroaryl, -O-(alkyl), -O-(aryl), -0-(heteroaryl), -S-(alkyl), -S-(aryl), -S-(heteroaryl), -
- R 4 is a substituted or unsubstituted alkyl
- R 5 is independently H, -C(0)(C ⁇ 8 alkyl), or a racemic, L-, or D- amino acid group -C(0)CHNH 2 R y ;
- R 6 is H, OR 10 , or NCR 1 ⁇
- R 7 is independently H or a substituted or unsubstituted -C(0)(C ⁇ - ⁇ 8 alkyl) or -C(0) 2 (C ⁇ - isalkyl);
- R 8 is H, -OH, -O-(alkyl), -OC0 2 (C ri8 alkyl), -OC(0)(C ⁇ 8 alkyl), or a racemic, L-, or D- amino acid group -OC(0)CHNH 2 R 1 ;
- R 9 is H, or a substituted or unsubstituted alkyl, C(0)CH(d -6 alkyl)NH 2 , or -C(O)CH(CH 2 - aryl)NH 2 ;
- R 10 is independently H, C w alkyl, C 3-7 alkenyl, C 3 . 7 alkynyl, -(CR 12 R 13 ) t (C 6 -C 10 aryl),
- R 1 ' is independently H, Cj.6 alkyl, C 3 -C ⁇ o cycloalkyl, or together with nitrogen forms a 5- or
- R 12 and R 13 are independently H, C ⁇ -6 alkyl, C 2-6 alkenyl, or C 2-6 alkynyl;
- R 14 is H, C 1-6 alkyl, or -CH 2 -aryl
- X is O or S
- Y is H, halo, OH, OR 4 , SH, SR 4 , or a substituted or unsubstituted alkyl or aryl;
- Z is H, halo, OH, OR 4 , SH, or SR 4 ; or a pharmaceutically acceptable salt, hydrate, metabolite or stereoisomer thereof or a pharmaceutically acceptable salt or hydrate of said stereoisomer.
- the invention encompasses a method of treating or preventing a hepatitis C virus infection in a patient in need thereof comprising administering to the patient a therapeutically or prophylactically effective amount of a TLR7 ligand selected from Formula Ila, lib, lie, lid, He, Ilf, Ilg, and Ilh, wherein R 1 is H or a substituted or unsubstituted alkyl, alkenyl, or alkynyl; R 2 is H, OH, halo, or a substituted or unsubstituted alkyl, alkenyl, or alkynyl, or -CH 2 -O-(alkyl); R 3 is H, OH, or SH, or a substituted or unsubstituted -O-(alkyl), -S-(alkyl), or -NH(alkyl); R 5 is independently H, -C(0)(Ci- 18 alkyl), or a racemic, L-
- the invention encompasses a method of treating or preventing a hepatitis C virus infection in a patient in need thereof comprising administering to the patient a therapeutically or prophylactically effective amount of a prodrug of a TLR7 ligand selected from
- the TLR7 ligand prodrug is an amino acid ester prodrug of the TLR7 ligand.
- the amino acid ester prodrug of the TLR7 ligand is a valyl ester.
- R is not a racemic, L-, or D- amino acid group -C(0)CHNH 2 R .
- R is not a racemic, L-, or D- amino acid group -C(0)CHNH 2 R 9 when the TLR7 ligand prodrug is selected from a compound of Formula Ilh.
- the invention encompasses a method for treating or preventing hepatitis C virus infection in a patient in need thereof, comprising administering to the patient a therapeutically or prophylactically effective amount of a prodrug of a TLR7 ligand and a pharmaceutically acceptable excipient, carrier, or vehicle.
- the invention encompasses a method for treating or preventing hepatitis C virus infection in a patient in need thereof, comprising administering to the patient a therapeutically or prophylactically effective amount of a prodrug of a TLR7 ligand and an additional therapeutic agent, preferably an additional antiviral or immunomodulatory agent.
- the invention also encompasses pharmaceutical compositions suitable for parenteral administration to a patient comprising a therapeutically or pharmaceutically acceptable amount a prodrug of a TLR7 ligand of the invention in a sterile form; pharmaceutical compositions suitable for parenteral administration to a patient comprising a therapeutically or pharmaceutically acceptable amount of a prodrug of a TLR7 ligand of the invention; pharmaceutical compositions suitable for mucosal administration to a patient comprising a therapeutically or pharmaceutically acceptable amount of a prodrug of a TLR7 ligand of the invention; and pharmaceutical compositions suitable for topical administration to a patient comprising a therapeutically or pharmaceutically acceptable amount of a prodrug of a TLR7 ligand of the invention.
- each of these compositions is in single unit dosage form and comprising an amount of active ingredient sufficient to treat or prevent human infection by hepatitis C virus.
- the invention encompasses a pharmaceutical composition
- a pharmaceutical composition comprising a prodrug of a TLR7 ligand selected from Formula Ila, lib, lie, lid, He, Ilf, Ilg, and Ilh, or a pharmaceutically acceptable salt, hydrate or stereoisomer thereof or a pharmaceutically acceptable salt or hydrate of said stereoisomer.
- TLR7 ligand prodrugs are useful as immune system enhancers and have certain immune system properties including modulation, mitogenicity, augmentation, and/or potentiation or they are intermediates for compounds that have these properties.
- the compounds are expected to express effects after administration to a mammal on at least one of the cell populations characterized as the natural killer cells, macrophages, dendritic cells, and lymphocyte cells of the immune system of a host. Because of these properties they are useful as an anti-infective including, but not limited to antiviral agents, and as antitumor agents or as intermediates for the same. They can be used to treat an affected host by serving as the active ingredients of suitable pharmaceutical compositions. [0048] In one aspect of the invention, TLR7 ligand prodrugs are utilized to treat the full range of viral diseases in mammals by administering to the mammal a therapeutically effective amount of the compounds.
- Viral diseases contemplated to be treated with TLR7 ligand prodrugs include acute and chronic infections caused by both RNA and DNA viruses. Without limiting in any way the range of viral infections that may be treated, TLR7 ligand prodrugs are particularly useful in the treatment of infections caused by adenovirus, cytomegalovirus, hepatitis A virus (HAV), hepatitis B virus (HBV), flaviviruses including Yellow Fever virus, hepacivirus including hepatitis C virus (HCV), herpes simplex type 1 and 2, herpes zoster, human herpesvirus 6, human immunodeficiency virus (HIV), human papilloma virus (HPV), influenza A virus, influenza B virus, measles, parainfluenza virus, pestivirus, poliovirus, poxvirus (including smallpox and monkeypox virus), rhinovirus, coronovirus, respiratory syncytial virus (RSV), multiple families of viruses that cause hemorrhagic fever
- TLR7 ligand prodrugs are utilized to treat bacterial, fungal, and protozoal infections in mammals by administering to the mammal a therapeutically effective amount of the prodrugs.
- the full range of pathogenic microorganisms is contemplated to be treatable by the TLR7 ligand prodrugs of the present invention, including without limitation those organisms that are resistant to antibiotics.
- the ability of TLR7 ligand prodrugs to activate multiple components of the immune system bypasses resistance mechanisms commonly found to reduce susceptibility to antibiotics, and thus treatment of infections in a mammal caused by such resistant microorganisms by TLR7 ligand prodrugs is a particular utility of the present invention.
- TLR7 ligand prodrugs are utilized to treat tumors in mammals by administering to the mammal a therapeutically effective amount of the prodrugs.
- Tumors or cancers contemplated to be treated include both those arising from aberrations in normal cellular processes as well as those caused by virus, and the effect may involve inhibiting the spread of cancerous cells, accelerating the killing of cancerous cells, inhibiting transformation of virus-infected cells to a neoplastic state, inhibiting the spread of viruses from transformed cells to other normal cells, and/or arresting the growth of virus- transformed cells.
- a method of treating a mammal comprises administering a therapeutically and/or prophylactically effective amount of a pharmaceutical containing a TLR7 ligand prodrug of the invention.
- the effect may relate to modulation of some portion of the mammal's immune system, especially modulation of cytokine activities of Thl and Th2, including but not restricted to the interleukin family, e.g., IL-1 through IL-12, and other cytokines such as TNF alpha, and interferons including interferon alpha, interferon beta, and interferon gamma, and their downsteam effectors.
- cytokine activities of Thl and Th2 including but not restricted to the interleukin family, e.g., IL-1 through IL-12, and other cytokines such as TNF alpha, and interferons including interferon alpha, interferon beta, and interferon gamma, and their downsteam effectors.
- the modulation may include stimulation of both Thl and Th2, suppression of both Thl and Th2, stimulation of either Thl or Th2 and suppression of the other, or a bimodal modulation in which one effect on Thl/Th2 levels (such as generalized suppression) occurs at a high concentration, while another effect (such as stimulation of either Thl or Th2 and suppression of the other) occurs at a lower concentration.
- pharmaceutical compositions containing a prodrug of a TLR7 ligand are administered in therapeutically effective doses to a mammal that is receiving immunomodulatory drugs not included in this invention.
- the doses of the immunomodulatory drug are reduced below their customary effective dose, to reduce adverse effects.
- the immunomodulatory drug is used at its customary dose, but with an improved therapeutic effect when a prodrug of a TLR7 ligand is also administered.
- compositions containing a prodrug of a TLR7 ligand are administered in a therapeutically effective dose to a mammal that is receiving anti-infective drugs not included in this invention.
- the pharmaceutical compositions containing a prodrug of a TLR7 ligand are administered in a therapeutically effective dose with anti-infective drug(s) that act directly upon the infectious agent to inhibit the growth of or kill the infectious agent.
- Figure 1 is a graphical depiction of plasma levels of isatoribine and interferon alpha in mice.
- Figure 2 is a graphical depiction of Viral Load Changes in HCV infected
- nucleoside refers to a compound composed of any pentose or modified pentose moiety attached to a specific position of a heterocycle or to the natural position of a purine (9-position) or pyrimidine (1 -position) or to the equivalent position in an analog.
- purine refers to nitrogenous bicyclic heterocycles.
- D-nucleosides refers to the nucleoside compounds that have a D- ribose sugar moiety (e.g., Adenosine).
- L-nucleosides refers to the nucleoside compounds that have a L- ribose sugar moiety.
- immunomodulator refers to natural or synthetic products capable of modifying the normal or aberrant immune system through stimulation or suppression.
- NOAEL is the No Observed Adverse Event Level, which is a toxicology term for the dose of drug that results in no significant toxicity under the specified conditions of dose level, frequency, duration in a selected species.
- Ligand means a low molecular weight molecule capable of binding to a biologic receptor. A ligand may be either an agonist or an antagonist, or may have no effect.
- An "agonist” is a ligand that, upon binding, stimulates the receptor to exert a biologic response that is consistent with the normal biologic activity of the receptor.
- An “antagonist” is a ligand that, upon binding, causes the receptor to not exert the normal biologic activity of the receptor.
- the term "mammal” includes both animals and humans.
- preventing refers to the ability of a compound or composition of the invention to prevent a disease identified herein in mammals diagnosed as having the disease or who are at risk of developing such disease. The term also encompasses preventing further progression of the disease in mammals who are already suffering from or have symptoms of such disease.
- treating refers to: (i) preventing a disease, disorder, or condition from occurring in a mammal that may be predisposed to the disease, disorder and/or condition, but has not yet been diagnosed as having it; (ii) inhibiting the disease, disorder, or condition, i.e., arresting its development; and (iii) relieving the disease, disorder, or condition, i.e., causing regression of the disease, disorder, and/or condition.
- the terms “ ⁇ ” and “ ⁇ ” indicate the specific stereochemical configuration of a substituent at an asymmetric carbon atom in a chemical structure as drawn.
- the terms “patient” or “subject” mean an animal (e.g., cow, horse, sheep, pig, chicken, turkey, quail, cat, dog, mouse, rat, rabbit, guinea pig, etc.) or a mammal, including chimeric and transgenic animals and mammals.
- the term “patient” or “subject” preferably means a monkey or a human, most preferably a human.
- the patient or subject is infected by or exposed to the hepatitis C virus.
- the patient is a human infant (age 0-2), child (age 2-17), adolescent (age 12-17), adult (age 18 and up) or geriatric (age 70 and up) patient.
- the patient includes immunocompromised patients such as HIV positive patients, cancer patients, patients undergoing immunotherapy or chemotherapy.
- the patient is a healthy individual, i.e., not displaying symptoms of other viral infections.
- a "therapeutically effective amount” refers to an amount of the
- TLR7 ligand or prodrug of a TLR7 ligand of the invention sufficient to provide a benefit in the treatment or prevention of viral disease, to delay or minimize symptoms associated with viral infection or viral-induced disease, or to cure or ameliorate the disease or infection or cause thereof.
- a therapeutically effective amount means an amount sufficient to provide a therapeutic benefit in vivo. Used in connection with an amount of a compound of the invention, the term preferably encompasses a non-toxic amount that improves overall therapy, reduces or avoids symptoms or causes of disease, or enhances the therapeutic efficacy of or synergies with another therapeutic agent.
- prophylactically effective amount refers to an amount of a compound of the invention or other active ingredient sufficient to result in the prevention of infection, recurrence or spread of viral infection.
- a prophylactically effective amount may refer to an amount sufficient to prevent initial infection or the recurrence or spread of the infection or a disease associated with the infection.
- the term preferably encompasses a non-toxic amount that improves overall prophylaxis or enhances the prophylactic efficacy of or synergizes with another prophylactic or therapeutic agent.
- in combination refers to the use of more than one prophylactic and/or therapeutic agents simultaneously or sequentially and in a manner that their respective effects are additive or synergistic.
- the term "pharmaceutically acceptable salts” refer to salts prepared from pharmaceutically acceptable non- toxic acids or bases including inorganic acids and bases and organic acids and bases. If the inventive TLR7 ligand prodrug is a base, the desired pharmaceutically acceptable salt may be prepared by any suitable method available in the art, for example, treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, or with an organic acid, such as acetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, a pyranosidyl acid, such as glucuronic acid or galacturonic acid, an alpha-hydroxy acid, such as citric acid or tartaric acid, an amino acid, such as aspartic acid or glutamic acid, an aromatic acid, such as benzoic acid or c
- the desired pharmaceutically acceptable salt may be prepared by any suitable method, for example, treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary or tertiary), an alkali metal hydroxide or alkaline earth metal hydroxide, or the like.
- suitable salts include organic salts derived from amino acids, such as glycine and arginine, ammonia, primary, secondary, and tertiary amines, and cyclic amines, such as piperidine, morpholine and piperazine, and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium.
- prodrug is intended to mean any chemical entity that after administration is converted via metabolic actions or solvolysis to a different chemical entity that retains biological activity.
- TLR7 ligand prodrug is intended to mean any chemical entity that after administration is converted via metabolic actions or solvolysis to a different chemical entity that retains biological activity and that is a ligand for TLR7.
- a TLR7 ligand prodrug may itself be a ligand for TLR7, or it may be "masked” in that it does not function efficiently as a TLR7 ligand.
- TLR7 ligand prodrug is intended to mean any chemical entity that after administration is converted via metabolic actions or solvolysis to a different chemical entity that retains biological activity and that is a ligand for TLR7, and where the administered chemical entity is a less efficient ligand for TLR7 than the chemical entity arising from metabolic conversion or solvolysis.
- a pharmaceutically active metabolite is intended to mean a pharmacologically active product produced through metabolism in the body of a specified compound or salt thereof. After entry into the body, most drugs are substrates for chemical reactions that may change their physical properties and biologic effects. These metabolic conversions, which usually affect the polarity of the TLR7 ligand, alter the way in which drugs are distributed in and excreted from the body. However, in some cases, metabolism of a drug is required for therapeutic effect. For example, many anticancer drugs of the anti- metabolite class must be converted to their active forms after they have been transported into a cancer cell.
- alkyl means a saturated straight chain or branched non-cyclic hydrocarbon having from 1 to 20 carbon atoms, preferably 1-10 carbon atoms and most preferably 1-4 carbon atoms.
- saturated straight chain alkyls include -methyl, -ethyl, -n-propyl, -n-butyl, -n-pentyl, -n- hexyl, -n-heptyl, -n-octyl, -n-nonyl and -n-decyl; while saturated branched alkyls include - isopropyl, -sec-butyl, -isobutyl, -tert-butyl, -isopentyl, 2-methylbutyl, 3-methylbutyl, 2- methylpentyl, 3-methylpentyl, 4-methylpentyl, 2-methylhexyl, 3-methylhexyl, 4- methylhexyl, 5-methylhexyl, 2,3-dimethylbutyl, 2,3-dimethylpentyl, 2,4-dimethylpentyl, 2,3-dimethylhexyl, 2,4-dimethyl
- aryl means a carbocyclic aromatic ring containing from 5 to 14 ring atoms.
- the ring atoms of a carbocyclic aryl group are all carbon atoms.
- Aryl ring structures include compounds having one or more ring structures such as mono-, bi-, or tricyclic compounds as well as benzo- fused carbocyclic moieties such as 5,6,7,8-tetrahydronaphthyl and the like.
- the aryl group is a monocyclic ring or bicyclic ring.
- aryl groups include phenyl, tolyl, anthracenyl, fluorenyl, indenyl, azulenyl, phenanthrenyl and naphthyl.
- a carbocyclic aryl group can be unsubstituted or substituted.
- substituted means that the specified group or moiety bears one or more substituents.
- unsubstituted means that the specified group bears no substituents.
- a "substituted alkyl” or “substituted aryl” is substituted by one or more substituents including halogen (F, CI, Br, or I), lower alkyl (Ci- ⁇ ), -OH, -N0 2 , -CN, -C0 2 H, -O-lower alkyl, -aryl, -aryl-lower alkyl, -CO 2 CH 3 , -CONH 2 , -OCH 2 CONH 2 , -NH 2 , - SO 2 NH 2 , haloalkyl (e.g., -CF 3 , -CH 2 CF 3 ), -O-haloalkyl (e.g., -OCF 3 , -OCHF 2 ), and the
- stereomerically pure means a composition that comprises one stereoisomer of a compound and is substantially free of other stereoisomers of that compound.
- a stereomerically pure compound having one chiral center will be substantially free of the opposite enantiomer of the compound.
- a typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of other stereoisomers of the compound, more preferably greater than about 90% by weight of one stereoisomer of the compound and less than about 10% by weight of the other stereoisomers of the compound, even more preferably greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, and most preferably greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound. Since many of the compounds of the invention comprise saccharides which can exist in either the D or L forms, the invention encompasses either or both D and L sugars.
- a stereomerically pure D sugar will be substantially free of the L form.
- the use of L forms of a TLR7 ligand will be substantially free of the D form.
- the methods and compositions disclosed herein include in an alternative embodiment the use of such levorotatory sugars or polymers made therefrom.
- the compounds of the invention may exhibit the phenomenon of tautomerism. While Formulas I and II cannot expressly depict all possible tautomeric forms, it is to be understood that Formula I is intended to represent any tautomeric form of the depicted compound and are not to be limited merely to a specific compound form depicted by the formula drawings. For example, it is understood that regardless of whether or not the substituents are shown in their enol or their keto form, they represent the same compound (as shown in the Formula Ila example below).
- TLR7 ligands include, but are not limited to (1) guanosine analogs, such as 7-deazaguanosine and related compounds, including but not limited to those described in Townsend, J. Heterocyclic Chem, 13, 1363 (1976), and Seela, et al, Chem. Ber., 114(10), 3395-3402 (1981); 7-allyl, 8-oxo-guanosine (loxorabine) and related compounds, including but not limited to those described in Reitz, et al, J. Med. Chem, 37, 3561-3578 (1994); 7-methyl, 9-deazaguanosine and related compounds including, but not limited to, those described in Girgis et al, J.
- guanosine analogs such as 7-deazaguanosine and related compounds, including but not limited to those described in Townsend, J. Heterocyclic Chem, 13, 1363 (1976), and Seela, et al, Chem. Ber., 114
- WO 94/17043 l-isobutyl-lH-imidazo[4,5-c]quinolin-4-ylamine (resiquimoid) as described in International Patent Application Publication No. WO 94/17043 and United States Patent Application Nos. 10/357,777 (United States Patent Application Publication No. US 2003/0195209), 10/357,733 (United States Patent Application Publication No. US 2003/0186949), 10/358,017(United States Patent Application Publication No. US 2003/0176458), 10/357,995 (United States Patent Application Publication No. US 2003/0162806), 10/165,222 (United States Patent Application Publication No.
- TLR7 ligands can be readily identified by known screening methods. See, e.g., Hirota et al, J. Med. Chem., 45, 5419-5422 (2002); and Akira S. et al, Immunology Letters, 85, 85-95 (2003). Using a variant of one of these known screening methods (as described in Section 6.1), analogs and derivatives of adenine were also identified as TLR7 ligands. Adenine derivatives known in the art are described in European Patent Application Publication Nos. EP 1 035 123, EP 1 043 021, and EP 0 882 727; United States Patent No. 6,376,501; United States Patent No. 6,329,381; United States Patent No.6,028,076, and United States Patent Application Publication No. US 2003/0162806.
- TLR7 ligands of Formulas Ia-Ih can be synthesized using methods known to one of skill in the art, particularly in light of the references and patents listed above.
- the TLR7 ligand prodrugs of the invention are prepared by making an (a) amide, carbamate, or amidine moiety after conversion of aTLR7 ligand amine substituent, (b) ester, carbonate, carbamate, ether, imidate, acetal, or ketal moiety after conversion of a TLR7 ligand alcohol substituent, (c) acetal or ketal moiety after conversion of a TLR7 ligand amine substituent, (d) imidate moiety after conversion of a TLR7 ligand carbonyl of an amido substituent, (e) deoxygenated moiety after conversion of a TLR7 ligand oxo substituent of pyrimidine or guanosine, or (f) amine.
- TLR7 ligand prodrugs are prepared by either (1) converting an hydroxyl (OH) substituents of the TLR7 ligand into an amino acid ester, or (2) making an amine substituent of the TLR7 ligand into an amide or carbamate.
- the process for preparing prodrugs is well known in the art and is described by Burger's Medicinal Chemistry and Drug Chemistry, 1, 172-178, 949-982 (1995). See also Bertolini et al, J. Med. Chem., 40, 2011-2016 (1997); Shan, et al, J. Pharm. Sci., 86 (7), 765-767; Bagshawe, Drug Dev.
- Schemes 1-18 show a general procedure to prepare representative compounds of Formula II.
- Schemes 1-6 describe how 5 '-amino acid esters can be synthesized from analogs and derivatives of guanosine.
- the 2',3 '-hydroxyl groups of the ⁇ -D-ribose moiety of Formulaa la, lb, Id, Ie, or Ih can first protected, preferably with an acetonide as shown for 2, 6, 10, or 14.
- the free 5 '-hydroxyl can then be subjected to a variety of esterification methods with a N-protected amino acid to form 3, 7, 11, or 15.
- the nitrogen of the amino acid ester and the 2',3 '-hydroxyls of the ribose unit can then be subjected to various deprotection conditions, preferably concurrently, followed by salt formation of the free amine of the amino acid ester as illustrated for 4, 8, 12, or 16.
- Schemes 7 and 8 describe how carbamates and carbonates can be synthesized from analogs and derivatives of adenine.
- Schemes 9 and 10 describes how carbamates and can be synthesized from imidazoquinoline analogs.
- the amino group of a derivative of Formula Ic can be subjected to a variety of conditions with carbonates, pyrocarbonates or chloroformates to form carbamates.
- Scheme 14 shows a general procedure for preparing 7-allyl-2-Amino-9- ⁇ -D- ribofuranosyl-7,9-dihydro-purin-8-one.
- Scheme 15 shows a general procedure for preparing 7-allyl-2-Amino-6- ethoxy-9- ⁇ -D-ribofuranosyl-7,9-dihydro-purin-8-one.
- 40 can be subjected to a variety of conditions to convert the carbonyl at the C-6 position to various imido-ethers, including but not limited to ethyl, as shown for 44.
- the 2', 3 ',5 '-hydroxyls of the ribose unit are then subjected to appropriate deprotection conditions, to produce 45.
- Compound 45 can further be appropriately modified if so desired.
- Scheme 16 describes how ethers can be synthesized from analogs and derivatives of adenine.
- the adenine derivative can be halogenated at C-8.
- the halogen can then be displaced with an appropriate alkoxide to form derivatives such as 64.
- Scheme 17 shows a general procedure for preparing 7-allyl-2-Amino-6- substituted alkoxy-9- ⁇ -D-ribofuranosyl-7,9-dihydro-purin-8-ones.
- Scheme 17 70 71 a) Et 3 SiCI, imidazole, DMF, rt b) oA/ OH , polymer supported PPh 3 , DEAD, THF, rt c) HF-NE.3, CH 3 OH, rt
- the hydroxyl groups on ribose of 17 can be protected as silyl ethers.
- the carbonyl at the C-6 position of 69 can be subjected to a variety of conditions to convert the carbonyl to various imido-ethers, including but not limited to the ether of 4-hydroxymethyl-5-methyl-[l,3]dioxol-2-one, as shown for 70.
- the 2',3',5'- hydroxyls of the ribose unit are then subjected to appropriate deprotection conditions, to produce 71.
- Scheme 18 shows a general procedure for preparing 7-allyl-2-Amino-6- substituted alkoxy-9- ⁇ -D-ribofuranosyl-7,9-dihydro-purin-8-ones.
- Scheme 18 69 72 73 a) H0CH 2 N(CH 3 )C0 2 Et, polymer supported PPh 3 , DEAD, THF, rt b) HF-NEt 3 , CH 3 OH, rt
- the carbonyl at the C-6 position of 69 can be subjected to a variety of conditions to convert the carbonyl to various imido-ethers, including but not limited to the ether of N-methyl-N-(hydroxymethyl)urethane, as shown for 72.
- the 2',3',5'-hydroxyls of the ribose unit are then subjected to appropriate deprotection conditions, to produce 73.
- Compound 73 can further be appropriately modified if so desired.
- the present invention provides methods for treating or preventing a hepatitis
- the present invention further provides methods for introducing a therapeutically effective amount of a TLR7 ligand or a prodrug thereof, or combination of such ligands and prodrugs into the blood stream of a patient in the treatment and/or prevention of hepatitis C viral infections.
- TLR7 ligand prodrug of the invention or a pharmaceutically acceptable salt, solvate, hydrate, or stereoisomer thereof in the acute or chronic treatment or prevention of an infection will vary, however, with the nature and severity of the infection, and the route by which the active ingredient is administered.
- the dose, and in some cases the dose frequency, will also vary according to the infection to be treated, the age, body weight, and response of the individual patient. Suitable dosing regimens can be readily selected by those skilled in the art with due consideration of such factors.
- the methods of the present invention are particularly well suited for human patients.
- the methods and doses of the present invention can be useful for immunocompromised patients including, but not limited to cancer patients, HIV infected patients, and patients with an immunodegenerative disease.
- the methods can be useful for immunocompromised patients currently in a state of remission.
- the methods and doses of the present invention are also useful for patients undergoing other antiviral treatments.
- the prevention methods of the present invention are particularly useful for patients at risk of viral infection.
- These patients include, but are not limited to health care workers, e.g., doctors, nurses, hospice care givers; military personnel; teachers; childcare workers; patients traveling to, or living in, foreign locales, in particular third world locales including social aid workers, missionaries, and foreign diplomats.
- the methods and compositions include the treatment of refractory patients or patients resistant to treatment such as resistance to viral polymerase inhibitors, protease inhibitors, etc.
- Toxicity and efficacy of the compounds of the invention can be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50 (the dose lethal to 50% of the population) and the ED50 (the dose therapeutically effective in 50% of the population).
- the dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio LD 50 ED50.
- the data obtained from the cell culture assays and animal studies can be used in formulating a range of dosage of the compounds for use in humans.
- the dosage of such compounds lie preferably within a range of circulating concentrations that include the ED50 with little or no toxicity.
- the dosage may vary within this range depending upon the dosage form employed and the route of administration utilized.
- the therapeutically effective dose can be estimated initially from cell culture assays.
- a dose may be formulated in animal models to achieve a circulating plasma concentration range that includes the IC 50 (i.e., the concentration of the test compound that achieves a half-maximal inhibition of symptoms) as determined in cell culture; alternatively, the dose of the TLR7 ligand prodrug may be formulated in animal models to achieve a circulating plasma concentration range of the TLR7 ligand that corresponds to the concentration required to achieve a fixed magnitude of response.
- IC 50 i.e., the concentration of the test compound that achieves a half-maximal inhibition of symptoms
- the dose of the TLR7 ligand prodrug may be formulated in animal models to achieve a circulating plasma concentration range of the TLR7 ligand that corresponds to the concentration required to achieve a fixed magnitude of response.
- Such information can be used to more accurately determine useful doses in humans.
- Levels in plasma may be measured, for example, by high performance liquid chromatography.
- the protocols and compositions of the invention are preferably tested in vitro, and then in vivo, for the desired therapeutic or prophylactic activity, prior to use in humans.
- in vitro assays which can be used to determine whether administration of a specific therapeutic protocol is indicated, include in vitro cell culture assays in which cells that are responsive to the effects of the TLR7 ligands are exposed to the ligand and the magnitude of response is measured by an appropriate technique. The assessment of the TLR7 ligand potency is then evaluated with respect to the TLR7 ligand prodrug potency, and the degree of conversion of the TLR7 ligand prodrug.
- Compounds for use in methods of the invention can be tested in suitable animal model systems prior to testing in humans, including but not limited to in rats, mice, chicken, cows, monkeys, rabbits, hamsters, etc. The compounds can then be used in the appropriate clinical trials.
- the magnitude of a prophylactic or therapeutic dose of a prodrug of a TLR7 ligand of the invention or a pharmaceutically acceptable salt, solvate, hydrate, or stereoisomer thereof in the acute or chronic treatment or prevention of an infection or condition will vary with the nature and severity of the infection, and the route by which the active ingredient is administered.
- the dose, and perhaps the dose frequency will also vary according to the infection to be treated, the age, body weight, and response of the individual patient.
- Suitable dosing regimens can be readily selected by those skilled in the art with due consideration of such factors.
- the dose administered depends upon the specific compound to be used, and the weight and condition of the patient.
- the dose may differ for various particular TLR7 ligands prodrugs; suitable doses can be predicted on the basis of the aforementioned in vitro measurements, in particular by use of such measurements of the TLR7 ligand to which the TLR7 ligand prodrug is related, and on the basis of animal studies, such that smaller doses will be suitable for those TLR7 ligand prodrugs that show effectiveness at lower concentrations than other TLR7 ligand prodrugs when measured in the systems described or referenced herein.
- the dose per day is in the range of from about 0.001 to 100 mg/kg, preferably about 1 to 25 mg/kg, more preferably about 5 to 15 mg/kg.
- about 0.1 mg to about 15 g per day is administered in about one to four divisions a day, preferably 100 mg to 12 g per day, more preferably from 100 mg to 8000 mg per day.
- compounds such as prodrugs of 3- ⁇ -D-ribofuranosylthiazolo[4,5- d]pyrimidines from 200 mg to 8000 mg per day is administered in about one to four divisions a day.
- the recommended daily dose ran can be administered in cycles as single agents or in combination with other therapeutic agents.
- the daily dose is administered in a single dose or in equally divided doses.
- the recommended daily dose can be administered once time per week, two times per week, three times per week, four times per week or five times per week.
- the compounds of the invention are administered to provide systemic distribution of the compound within the patient.
- the compounds of the invention are administered to produce a systemic effect in the body.
- the compounds of the invention are administered via oral, mucosal (including sublingual, buccal, rectal, nasal, or vaginal), parenteral (including subcutaneous, intramuscular, bolus injection, intraarterial, or intravenous), transdermal, or topical administration.
- the compounds of the invention are administered via mucosal (including sublingual, buccal, rectal, nasal, or vaginal), parenteral (including subcutaneous, intramuscular, bolus injection, intraarterial, or intravenous), transdermal, or topical administration.
- the compounds of the invention are administered via oral administration.
- the compounds of the invention are not administered via oral administration.
- Specific methods of the invention further comprise the administration of an additional therapeutic agent (i.e., a therapeutic agent other than a compound of the invention).
- an additional therapeutic agent i.e., a therapeutic agent other than a compound of the invention.
- the compounds of the invention can be used in combination with at least one other therapeutic agent.
- Therapeutic agents include, but are not limited to antibiotics, antiemetic agents, antidepressants, and antifungal agents, anti-inflammatory agents, antiviral agents, anticancer agents, immunomodulatory agents, ⁇ -interferons, alkylating agents, hormones or cytokines.
- the invention encompasses the administration of an additional therapeutic agent that is HCV specific or demonstrates anti-HCV activity.
- the TLR7 ligands prodrugs of the invention can be administered or formulated in combination with antibiotics.
- they can be formulated with a macrolide (e.g., tobramycin (Tobi®)), a cephalosporin (e.g., cephalexin (Keflex®), cephradine (Velosef®), cefuroxime (Ceftin®), cefprozil (Cefzil®), cefaclor (Ceclor®), cefixime (Suprax®) or cefadroxil (Duricef®)), a clarithromycin (e.g., clarithromycin (Biaxin®)), an erythromycin (e.g., erythromycin (EMycin®)), a penicillin (e.g., penicillin V (V-Cillin K® or Pen Vee K®)) or a quinolone (e.g., ofloxacin (Floxin®), cipr
- the TLR7 ligand prodrugs of the invention can also be administered or formulated in combination with an antiemetic agent.
- Suitable antiemetic agents include, but are not limited to, metoclopromide, domperidone, prochlorperazine, promethazine, chlorpromazine, trimethobenzamide, ondansetron, granisetron, hydroxyzine, acethylleucine monoethanolamine, alizapride, azasetron, benzquinamide, bietanautine, bromopride, buclizine, clebopride, cyclizine, dimenhydrinate, diphenidol, dolasetron, meclizine, methallatal, metopimazine, nabilone, oxyperndyl, pipamazine, scopolamine, sulpiride, tetrahydrocannabinols, thiethylperazine, thioproperazine,
- the TLR7 ligand prodrugs of the invention can be administered or formulated in combination with an antidepressant.
- Suitable antidepressants include, but are not limited to, binedaline, caroxazone, citalopram, dimethazan, fencamine, indalpine, indeloxazine hydrocholoride, nefopam, nomifensine, oxitriptan, oxypertine, paroxetine, sertraline, thiazesim, trazodone, benmoxine, iproclozide, iproniazid, isocarboxazid, nialamide, octamoxin, phenelzine, cotinine, rolicyprine, rolipram, maprotiline, metralindole, mianserin, mirtazepine, adinazolam, amitriptyline, amitriptylinoxide, amoxa
- TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with an antifungal agent.
- Suitable antifungal agents include but are not limited to amphotericin B, itraconazole, ketoconazole, fluconazole, intrathecal, flucytosine, miconazole, butoconazole, clotrimazole, nystatin, terconazole, tioconazole, ciclopirox, econazole, haloprogrin, naftifine, terbinafine, undecylenate, and griseofuldin.
- the TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with an anti-inflammatory agent.
- Useful anti- inflammatory agents include, but are not limited to, non-steroidal anti-inflammatory drugs such as salicylic acid, acetylsalicylic acid, methyl salicylate, diflumsal, salsalate, olsalazine, sulfasalazine, acetaminophen, indomethacin, sulindac, etodolac, mefenamic acid, meclofenamate sodium, tolmetin, ketorolac, dichlofenac, ibuprofen, naproxen, naproxen sodium, fenoprofen, ketoprofen, flurbinprofen, oxaprozin, piroxicam, meloxicam, ampiroxicam, droxicam, pivoxicam, tenoxicam, nabumetome
- TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with another antiviral agent.
- Useful antiviral agents include, but are not limited to, protease inhibitors, nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors and nucleoside analogs.
- the antiviral agents include but are not limited to zidovudine, acyclovir, gangcyclovir, vidarabine, idoxuridine, trifluridine, levovirin, viramidine and ribavirin, as well as foscarnet, amantadine, rimantadine, saquinavir, indinavir, amprenavir, lopinavir, ritonavir, the alpha-interferons, beta-interferons, gamma-interferons, adefovir, clevudine, entecavir, and pleconaril.
- the TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with an immunomodulatory agent.
- Immunomodulatory agents include, but are not limited to, methothrexate, leflunomide, cyclophosphamide, cyclosporine A, mycophenolate mofetil, rapamycin (sirolimus), mizoribine, deoxyspergualin, brequinar, malononitriloamindes (e.g., leflunamide), T cell receptor modulators, and cytokine receptor modulators, peptide mimetics, and antibodies (e.g., human, humanized, chimeric, monoclonal, polyclonal, Fvs, ScFvs, Fab or F(ab)2 fragments or epitope binding fragments), nucleic acid molecules (e.g., antisense nucleic acid molecules and triple helices), small molecules, organic compounds, and inorganic compounds.
- T cell receptor modulators include, but are not limited to, anti-T cell receptor antibodies (e.g., anti-CD4 antibodies (e.g., cM-T412 (Boeringer), IDEC- CE9.1® (IDEC and SKB), mAB 4162W94, Orthoclone and OKTcdr4a (Janssen-Cilag)), anti-CD3 antibodies (e.g., Nuvion (Product Design Labs), OKT3 (Johnson & Johnson), or Rituxan (IDEC)), anti-CD5 antibodies (e.g., an anti-CD5 ricin-linked immunoconjugate), anti-CD7 antibodies (e.g., CHH-380 (Novartis)), anti-CD8 antibodies, anti-CD40 ligand monoclonal antibodies (e.g., IDEC-131 (IDEC)), anti-CD52 antibodies (e.g., CAMPATH IH (Ilex)), anti-CD2 antibodies, anti-CDlla antibodies (e.g.,
- cytokine receptor modulators include, but are not limited to, soluble cytokine receptors (e.g., the extracellular domain of a TNF- ⁇ receptor or a fragment thereof, the extracellular domain of an IL-l ⁇ receptor or a fragment thereof, and the extracellular domain of an IL-6 receptor or a fragment thereof), cytokines or fragments thereof (e.g., interleukin (IL)-2, IL- 3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12, IL-15, TNF- ⁇ , interferon (IFN)- ⁇ , IFN- ⁇ , IFN- ⁇ , and GM-CSF), anti-cytokine receptor antibodies (e.g., anti-IFN receptor antibodies, anti-IL-2 receptor antibodies (e.g., Zenapax (Protein Design Labs)), anti-IL-4 receptor antibodies, anti-IL-6 receptor antibodies, anti-IL-10 receptor antibodies, and anti- IL-12 receptor
- TLR7 ligands or TLR ligand prodrugs of the invention can be administered or formulated in combination with an agent which inhibits viral enzymes, including but not limited to inhibitors of HCV protease, such as BILN 2061 and inhibitors of NS5b polymerase such as NM107 and its prodrug NM283 (Idenix Pharmaceuticals, Inc., Cambridge, MA).
- an agent which inhibits viral enzymes including but not limited to inhibitors of HCV protease, such as BILN 2061 and inhibitors of NS5b polymerase such as NM107 and its prodrug NM283 (Idenix Pharmaceuticals, Inc., Cambridge, MA).
- TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with an agent which inhibits HCV polymerase such as those described in Wu, Curr Drug Targets Infect Disord. 2003;3(3):207-19 or in combination with compounds that inhibit the helicase function of the virus such as those described in Bretner M, et al Nucleosides Nucleotides Nucleic Acids. 2003;22(5-8):1531, or with inhibitors of other HCV specific targets such as those described in Zhang X. IDrugs. 2002;5(2): 154-8.
- an agent which inhibits HCV polymerase such as those described in Wu, Curr Drug Targets Infect Disord. 2003;3(3):207-19 or in combination with compounds that inhibit the helicase function of the virus such as those described in Bretner M, et al Nucleosides Nucleotides Nucleic Acids. 2003;22(5-8):1531, or with inhibitors of other HCV specific targets such as those described
- the TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with an agent that inhibits viral replication.
- the TLR7 ligands or TLR ligand prodrugs of the invention can be administered or formulated in combination with cytokines.
- cytokines include, but are not limited to, inter leukin-2 (IL-2), inter leukin-3 (IL-3), interleukin-4 (IL-4), interleukin-5 (IL-5), interleukin-6 (IL-6), interleukin-7 (IL-7), interleukin-9 (IL-9), interleukin- 10 (IL-10), interleukin- 12 (IL-12), interleukin 15 (IL-15), interleukin 18 (IL-18), platelet derived growth factor (PDGF), erythropoietin (Epo), epidermal growth factor (EGF), fibroblast growth factor (FGF), granulocyte macrophage stimulating factor (GMCSF), granulocyte colony stimulating factor (G-CSF), macrophage colony stimulating factor (M-CSF), prolactin, and interferon (IFN), e.g., IFN-alpha, and IFN-gamma).
- PDGF platelet derived growth factor
- Epo epidermal growth factor
- FGF
- the TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with hormones.
- hormones include, but are not limited to, luteinizing hormone releasing hormone (LHRH), growth hormone (GH), growth hormone releasing hormone, ACTH, somatostatin, somatotropin, somatomedin, parathyroid hormone, hypothalamic releasing factors, insulin, glucagon, enkephalins, vasopressin, calcitonin, heparin, low molecular weight heparins, heparinoids, synthetic and natural opioids, insulin thyroid stimulating hormones, and endorphins.
- LHRH luteinizing hormone releasing hormone
- GH growth hormone
- ACTH ACTH
- somatostatin somatotropin
- somatomedin parathyroid hormone
- hypothalamic releasing factors insulin
- glucagon enkephalins
- vasopressin vasopressin
- TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with ⁇ -interferons which include, but are not limited to, interferon beta- la, interferon beta- lb.
- TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with ⁇ -interferons which include, but are not limited to, interferon alpha- 1, interferon alpha-2a (roferon), interferon alpha-2b, intron, Peg- Intron, Pegasys, consensus interferon (infergen) and albuferon.
- ⁇ -interferons include, but are not limited to, interferon alpha- 1, interferon alpha-2a (roferon), interferon alpha-2b, intron, Peg- Intron, Pegasys, consensus interferon (infergen) and albuferon.
- the TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with an absorption enhancer, particularly those which target the lymphatic system, including, but not limited to sodium glycocholate; sodium caprate; N-lauryl- ⁇ -D-maltopyranoside; EDTA; mixed micelle; and those reported in Muranishi Crit. Rev. Ther. Drug Carrier Syst, 7-1-33, which is hereby incorporated by reference in its entirety. Other known absorption enhancers can also be used.
- the invention also encompasses a pharmaceutical composition comprising one or more TLR7 ligand prodrugs of the invention and one or more absorption enhancers.
- the TLR7 ligands or TLR7 ligand prodrugs of the invention can be administered or formulated in combination with an alkylating agent.
- alkylating agents include, but are not limited to nitrogen mustards, ethylenimines, methylmelamines, alkyl sulfonates, nitrosoureas, triazenes, mechlorethamine, cyclophosphamide, ifosfamide, melphalan, chlorambucil, hexamethylmelaine, thiotepa, busulfan, carmustine, streptozocin, dacarbazine and temozolomide.
- the compounds of the invention and the other therapeutics agent can act additively or, more preferably, synergistically.
- a composition comprising a compound of the invention is administered concurrently with the administration of another therapeutic agent, which can be part of the same composition or in a different composition from that comprising the compounds of the invention.
- a compound of the invention is administered prior to or subsequent to administration of another therapeutic agent.
- a compound of the invention is administered to a patient who has not previously undergone or is not currently undergoing treatment with another therapeutic agent, particularly an antiviral agent.
- the methods of the invention comprise the administration of one or more TLR7 ligands or TLR7 ligand prodrugs of the invention without an additional therapeutic agent.
- PHARMACEUTICAL COMPOSITIONS AND DOSAGE FORMS Pharmaceutical compositions and single unit dosage forms comprising a TLR7 ligand or prodrug of the invention, or a pharmaceutically acceptable salt, hydrate or stereoisomer thereof, are also encompassed by the invention.
- Individual dosage forms of the invention may be suitable for oral, mucosal (including sublingual, buccal, rectal, nasal, or vaginal), parenteral (including subcutaneous, intramuscular, bolus injection, intraarterial, or intravenous), transdermal, or topical administration.
- Pharmaceutical compositions and dosage forms of the invention typically also comprise one or more pharmaceutically acceptable excipients. Sterile dosage forms are also contemplated.
- composition encompassed by this embodiment includes a TLR7 ligand or prodrug of the invention, or a pharmaceutically acceptable salt, hydrate or stereoisomer thereof, and at least one additional therapeutic agent.
- additional therapeutic agents include, but are not limited to, those listed above in section 5.2.2.
- composition, shape, and type of dosage forms of the invention will typically vary depending on their use.
- a dosage form used in the acute treatment of a disease or a related disease may contain larger amounts of one or more of the active ingredients it comprises than a dosage form used in the chronic treatment of the same disease.
- a parenteral dosage form may contain smaller amounts of one or more of the active ingredients it comprises than an oral dosage form used to treat the same disease or disorder.
- dosage forms include, but are not limited to: tablets; caplets; capsules, such as soft elastic gelatin capsules; cachets; troches; lozenges; dispersions; suppositories; ointments; cataplasms (poultices); pastes; powders; dressings; creams; plasters; solutions; patches; aerosols (e.g., nasal sprays or inhalers); gels; liquid dosage forms suitable for oral or mucosal administration to a patient, including suspensions (e.g., aqueous or non-aqueous liquid suspensions, oil-in-water emulsions, or a water-in-oil liquid emulsions), solutions, and elixirs; liquid dosage forms suitable for parenteral administration to a patient; and sterile solids (e.g., crystalline or amorphous solids) that can be reconstituted to provide liquid dosage forms suitable for parenteral administration to a patient.
- suspensions e.g., aqueous
- Typical pharmaceutical compositions and dosage forms comprise one or more carriers, excipients or diluents.
- Suitable excipients are well known to those skilled in the art of pharmacy, and non-limiting examples of suitable excipients are provided herein. Whether a particular excipient is suitable for incorporation into a pharmaceutical composition or dosage form depends on a variety of factors well known in the art including, but not limited to, the way in which the dosage form will be administered to a patient.
- oral dosage forms such as tablets may contain excipients not suited for use in parenteral dosage forms. The suitability of a particular excipient may also depend on the specific active ingredients in the dosage form.
- This invention further encompasses anhydrous pharmaceutical compositions and dosage forms comprising active ingredients, since water can facilitate the degradation of some compounds.
- water e.g., 5%
- water is widely accepted in the pharmaceutical arts as a means of simulating long-term storage in order to determine characteristics such as shelf-life or the stability of formulations over time. See, e.g., Jens T. Carstensen, Drug Stability: Principles & Practice, 2d. Ed., Marcel Dekker, NY, NY, 1995, pp. 379-80.
- water and heat accelerate the decomposition of some compounds.
- the effect of water on a formulation can be of great significance since moisture and/or humidity are commonly encountered during manufacture, handling, packaging, storage, shipment, and use of formulations.
- Anhydrous pharmaceutical compositions and dosage forms of the invention can be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions.
- anhydrous pharmaceutical composition should be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions are preferably packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits. Examples of suitable packaging include, but are not limited to, hermetically sealed foils, plastics, unit dose containers (e.g., vials), blister packs, and strip packs.
- compositions and dosage forms that comprise one or more compounds that reduce the rate by which an active ingredient will decompose.
- Such compounds which are referred to herein as “stabilizers,” include, but are not limited to, antioxidants such as ascorbic acid, pH buffers, or salt buffers.
- antioxidants such as ascorbic acid, pH buffers, or salt buffers.
- the amounts and specific types of active ingredients in a dosage form may differ depending on factors such as, but not limited to, the route by which it is to be administered to patients.
- typical dosage forms of the invention comprise TLR7 ligand prodrugs of the invention, or a pharmaceutically acceptable salt, hydrate, or stereoisomers thereof comprise 0.1 mg to 1500 mg per unit to provide doses of about 0.01 to 200 mg/kg per day.
- Oral Dosage Forms comprise TLR7 ligand prodrugs of the invention, or a pharmaceutically acceptable salt, hydrate, or stereoisomers thereof comprise 0.1 mg to 1500 mg per unit to provide doses of about 0.01 to 200 mg/kg per day.
- compositions of the invention that are suitable for oral administration can be presented as discrete dosage forms, such as, but are not limited to, tablets (e.g., chewable tablets), caplets, capsules, and liquids (e.g., flavored syrups).
- dosage forms contain predetermined amounts of active ingredients, and may be prepared by methods of pharmacy well known to those skilled in the art. See generally, Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA (1990).
- Typical oral dosage forms of the invention are prepared by combining the active ingredient(s) in an intimate admixture with at least one excipient according to conventional pharmaceutical compounding techniques. Excipients can take a wide variety of forms depending on the form of preparation desired for administration.
- excipients suitable for use in oral liquid or aerosol dosage forms include, but are not limited to, water, glycols, oils, alcohols, flavoring agents, preservatives, and coloring agents.
- excipients suitable for use in solid oral dosage forms include, but are not limited to, starches, sugars, micro-crystalline cellulose, diluents, granulating agents, lubricants, binders, and disintegrating agents.
- tablets and capsules represent the most advantageous oral dosage unit forms, in which case solid excipients are employed. If desired, tablets can be coated by standard aqueous or nonaqueous techniques.
- Such dosage forms can be prepared by any of the methods of pharmacy.
- pharmaceutical compositions and dosage forms are prepared by uniformly and intimately admixing the active ingredients with liquid carriers, finely divided solid carriers, or both, and then shaping the product into the desired presentation if necessary.
- a tablet can be prepared by compression or molding.
- Compressed tablets can be prepared by compressing in a suitable machine the active ingredients in a free-flowing form such as powder or granules, optionally mixed with an excipient. Molded tablets can be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- excipients that can be used in oral dosage forms of the invention include, but are not limited to, binders, fillers, disintegrants, and lubricants.
- Binders suitable for use inpharmaceutical compositions and dosage forms include, but are not limited to, corn starch, potato starch, or other starches, gelatin, natural and synthetic gums such as acacia, sodium alginate, alginic acid, other alginates, powdered tragacanth, guar gum, cellulose and its derivatives (e.g., ethyl cellulose, cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethyl cellulose), polyvinyl pyrrolidone, methyl cellulose, pre-gelatinized starch, hydroxypropyl methyl cellulose, (e.g., Nos. 2208, 2906, 2910), microcrystalline cellulose, and mixtures thereof.
- fillers suitable for use in the pharmaceutical compositions and dosage forms disclosed herein include, but are not limited to, talc, calcium carbonate (e.g., granules or powder), microcrystalline cellulose, powdered cellulose, dextrates, kaolin, mannitol, silicic acid, sorbitol, starch, pre-gelatinized starch, and mixtures thereof.
- the binder or filler in pharmaceutical compositions of the invention is typically present in from about 50 to about 99 weight percent of the pharmaceutical composition or dosage form.
- Suitable forms of microcrystalline cellulose include, but are not limited to, the materials sold as AVICEL-PH-101, AVICEL-PH-103 AVICEL RC-581, AVICEL-PH- 105 (available from FMC Corporation, American Viscose Division, Avicel Sales, Marcus Hook, PA), and mixtures thereof.
- a specific binder is a mixture of microcrystalline cellulose and sodium carboxymethyl cellulose sold as AVICEL RC-581.
- Suitable anhydrous or low moisture excipients or additives include AVICEL-PH-103TM and Starch 1500 LM.
- Disintegrants are used in the compositions of the invention to provide tablets that disintegrate when exposed to an aqueous environment. Tablets that contain too much disintegrant may disintegrate in storage, while those that contain too little may not disintegrate at a desired rate or under the desired conditions. Thus, a sufficient amount of disintegrant that is neither too much nor too little to detrimentally alter the release of the active ingredients should be used to form solid oral dosage forms of the invention.
- the amount of disintegrant used varies based upon the type of formulation, and is readily discernible to those of ordinary skill in the art. Typical pharmaceutical compositions comprise from about 0.5 to about 15 weight percent of disintegrant, specifically from about 1 to about 5 weight percent of disintegrant.
- Disintegrants that can be used in pharmaceutical compositions and dosage forms of the invention include, but are not limited to, agar-agar, alginic acid, calcium carbonate, microcrystalline cellulose, croscarmellose sodium, crospovidone, polacrilin potassium, sodium starch glycolate, potato or tapioca starch, pre-gelatinized starch, other starches, clays, other algins, other celluloses, gums, and mixtures thereof.
- Lubricants that can be used in pharmaceutical compositions and dosage forms of the invention include, but are not limited to, calcium stearate, magnesium stearate, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, stearic acid, sodium lauryl sulfate, talc, hydrogenated vegetable oil (e.g., peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil, corn oil, and soybean oil), zinc stearate, ethyl oleate, ethyl laureate, agar, and mixtures thereof.
- calcium stearate e.g., magnesium stearate, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, stearic acid, sodium lauryl sulfate, talc
- hydrogenated vegetable oil e.g., peanut oil, cottonseed oil
- Additional lubricants include, for example, a syloid silica gel (AEROSIL 200, manufactured by W.R. Grace Co. of Baltimore, MD), a coagulated aerosol of synthetic silica (marketed by Degussa Co. of Piano, TX), CAB-O-SIL (a pyrogenic silicon dioxide product sold by Cabot Co. of Boston, MA), and mixtures thereof. If used at all, lubricants are typically used in an amount of less than about 1 weight percent of the pharmaceutical compositions or dosage forms into which they are incorporated.
- AEROSIL 200 manufactured by W.R. Grace Co. of Baltimore, MD
- a coagulated aerosol of synthetic silica marketed by Degussa Co. of Piano, TX
- CAB-O-SIL a pyrogenic silicon dioxide product sold by Cabot Co. of Boston, MA
- lubricants are typically used in an amount of less than about 1 weight percent of the pharmaceutical compositions or dosage forms into which they are incorporated.
- Active ingredients of the invention can be administered by controlled release means or by delivery devices that are well known to those of ordinary skill in the art. Examples include, but are not limited to, those described in U.S. Patent Nos.: 3,845,770; 3,916,899; 3,536,809; 3,598,123; and 4,008,719, 5,674,533, 5,059,595, 5,591,767, 5,120,548, 5,073,543, 5,639,476, 5,354,556, and 5,733,566, each of which is incorporated herein by reference.
- Such dosage forms can be used to provide slow or controlled-release of one or more active ingredients using, for example, hydropropylmethyl cellulose, other polymer matrices, gels, permeable membranes, osmotic systems, multilayer coatings, microparticles, liposomes, microspheres, or a combination thereof to provide the desired release profile in varying proportions.
- Suitable controlled-release formulations known to those of ordinary skill in the art, including those described herein, can be readily selected for use with the active ingredients of the invention.
- the invention thus encompasses single unit dosage forms suitable for oral administration such as, but not limited to, tablets, capsules, gelcaps, and caplets that are adapted for controlled-release.
- controlled-release pharmaceutical products have a common goal of improving drug therapy over that achieved by their non-controlled counterparts.
- the use of an optimally designed controlled-release preparation in medical treatment is characterized by a minimum of drug substance being employed to cure or control the condition in a minimum amount of time.
- Advantages of controlled-release formulations include extended activity of the drug, reduced dosage frequency, and increased patient compliance.
- controlled-release formulations can be used to affect the time of onset of action or other characteristics, such as blood levels of the drug, and can thus affect the occurrence of side (e.g., adverse) effects.
- Controlled-release formulations are designed to initially release an amount of drug (active ingredient) that promptly produces the desired therapeutic effect, and gradually and continually release other amounts of drug to maintain this level of therapeutic or prophylactic effect over an extended period of time. In order to maintain this constant level of drug in the body, the drug must be released from the dosage form at a rate that will replace the amount of drug being metabolized and excreted from the body. Controlled-release of an active ingredient can be stimulated by various conditions including, but not limited to, pH, temperature, enzymes, water, or other physiological conditions or compounds.
- Parenteral dosage forms can be administered to patients by various routes including, but not limited to, subcutaneous, intravenous (including bolus injection), intramuscular, and intraarterial. Because their administration typically bypasses patients' natural defenses against contaminants, parenteral dosage forms are preferably sterile or capable of being sterilized prior to administration to a patient. Examples of parenteral dosage forms include, but are not limited to, solutions ready for injection, dry and/or lyophylized products ready to be dissolved or suspended in a pharmaceutically acceptable vehicle for injection (reconstitutable powders), suspensions ready for injection, and emulsions.
- Suitable vehicles that can be used to provide parenteral dosage forms of the invention are well known to those skilled in the art. Examples include, but are not limited to: Water for Injection USP; aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride Injection, and Lactated Ringer's Injection; water-miscible vehicles such as, but not limited to, ethyl alcohol, polyethylene glycol, and polypropylene glycol; and non-aqueous vehicles such as, but not limited to, corn oil, cottonseed oil, peanut oil, sesame oil, ethyl oleate, isopropyl myristate, and benzyl benzoate.
- water for Injection USP Water for Injection USP
- aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride
- Transdermal dosage forms include "reservoir type” or “matrix type” patches, which can be applied to the skin and worn for a specific period of time to permit the penetration of a desired amount of active ingredients.
- Suitable excipients e.g., carriers and diluents
- other materials that can be used to provide transdermal and topical dosage forms encompassed by this invention are well known to those skilled in the pharmaceutical arts, and depend on the particular tissue to which a given pharmaceutical composition or dosage form will be applied.
- typical excipients include, but are not limited to, water, acetone, ethanol, ethylene glycol, propylene glycol, butane- 1, 3 -diol, isopropyl myristate, isopropyl palmitate, mineral oil, and mixtures thereof.
- penetration enhancers can be used to assist in delivering the active ingredients to the tissue.
- Suitable penetration enhancers include, but are not limited to: acetone; various alcohols such as ethanol, oleyl, and tetrahydrofuryl; alkyl sulfoxides such as dimethyl sulfoxide; dimethyl acetamide; dimethyl formamide; polyethylene glycol; pyrrolidones such as polyvinylpyrrolidone; Kollidon grades (Povidone, Polyvidone); urea; and various water-soluble or insoluble sugar esters such as Tween 80 (polysorbate 80) and Span 60 (sorbitan monostearate).
- the pH of a pharmaceutical composition or dosage form, or of the tissue to which the pharmaceutical composition or dosage form is applied may also be adjusted to improve delivery of one or more active ingredients.
- the polarity of a solvent carrier, its ionic strength, or tonicity can be adjusted to improve delivery.
- Compounds such as stearates can also be added to pharmaceutical compositions or dosage forms to advantageously alter the hydrophilicity or lipophilicity of one or more active ingredients so as to improve delivery.
- stearates can serve as a lipid vehicle for the formulation, as an emulsifying agent or surfactant, and as a delivery-enhancing or penetration-enhancing agent.
- Different salts, hydrates or solvates of the active ingredients can be used to further adjust the properties of the resulting composition.
- Topical dosage forms of the invention include, but are not limited to, creams, lotions, ointments, gels, solutions, emulsions, suspensions, or other forms known to one of skill in the art. See, e.g., Remington's Pharmaceutical Sciences, 18th eds., Mack Publishing, Easton PA (1990); and Introduction to Pharmaceutical Dosage Forms, 4th ed., Lea & Febiger, Philadelphia (1985).
- Suitable excipients e.g., carriers and diluents
- other materials that can be used to provide transdermal and topical dosage forms encompassed by this invention are well known to those skilled in the pharmaceutical arts, and depend on the particular tissue to which a given pharmaceutical composition or dosage form will be applied.
- typical excipients include, but are not limited to, water, acetone, ethanol, ethylene glycol, propylene glycol, butane- 1, 3 -diol, isopropyl myristate, isopropyl palmitate, mineral oil, and mixtures thereof.
- penetration enhancers can be used to assist in delivering the active ingredients to the tissue.
- Suitable penetration enhancers include, but are not limited to: acetone; various alcohols such as ethanol, oleyl, and tetrahydrofuryl; alkyl sulfoxides such as dimethyl sulfoxide; dimethyl acetamide; dimethyl formamide; polyethylene glycol; pyrrolidones such as polyvinylpyrrolidone; Kollidon grades (Povidone, Polyvidone); urea; and various water-soluble or insoluble sugar esters such as Tween 80 (polysorbate 80) and Span 60 (sorbitan monostearate).
- Tween 80 polysorbate 80
- Span 60 sorbitan monostearate
- Mucosal dosage fo ⁇ ns of the invention include, but are not limited to, ophthalmic solutions, sprays and aerosols, or other forms known to one of skill in the art. See, e.g., Remington's Pharmaceutical Sciences, 18th eds., Mack Publishing, Easton PA (1990); and Introduction to Pharmaceutical Dosage Forms, 4th ed., Lea & Febiger, Philadelphia (1985). Dosage forms suitable for treating mucosal tissues within the oral cavity can be formulated as mouthwashes or as oral gels.
- the aerosol comprises a carrier. In another embodiment, the aerosol is carrier free.
- TLR7 ligands or TLR7 ligand prodrugs of the invention may also be administered directly to the lung by inhalation.
- a TLR7 ligand can be conveniently delivered to the lung by a number of different devices.
- a Metered Dose Inhaler which utilizes canisters that contain a suitable low boiling propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas can be used to deliver a TLR7 ligand directly to the lung.
- MDI devices are available from a number of suppliers such as 3M Corporation, Aventis, Boehringer Ingleheim, Forest Laboratories, Glaxo- Wellcome, Schering Plough and Vectura.
- DPI Dry Powder Inhaler
- DPI devices typically use a mechanism such as a burst of gas to create a cloud of dry powder inside a container, which can then be inhaled by the patient.
- DPI devices are also well known in the art and can be purchased from a number of vendors which include, for example, Fisons, Glaxo- Wellcome, Inhale Therapeutic Systems, ML Laboratories, Qdose and Vectura.
- MDDPI multiple dose DPI
- MDDPI devices are available from companies such as AstraZeneca, Glaxo Wellcome, IV AX, Schering Plough, SkyePharma and Vectura.
- capsules and cartridges of gelatin for use in an inhaler or insufflator can be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch for these systems.
- a liquid spray device supplied, for example, by Aradigm Corporation.
- Liquid spray systems use extremely small nozzle holes to aerosolize liquid drug formulations that can then be directly inhaled into the lung.
- a nebulizer device is used to deliver TLR7 ligands or TLR ligand prodrugs to the lung. Nebulizers create aerosols from liquid drug formulations by using, for example, ultrasonic energy to form fine particles that can be readily inhaled (See e.g., Verschoyle et al, British J.
- nebulizers include devices supplied by Sheffield/Systemic Pulmonary Delivery Ltd, Aventis, and Batelle Pulmonary Therapeutics. See U.S. Pat. Nos. 5,954,047; 5,950,619; 5,970,974, which are herein incorporated by reference.
- an electrohydrodynamic (“EHD”) aerosol device is used to deliver TLR7 ligands and TLR7 ligand prodrugs to the lung.
- EHD aerosol devices use electrical energy to aerosolize liquid drug solutions or suspensions (see, e.g., Noakes et al, U.S. Pat. No. 4,765,539; Coffee, U.S. Pat. No., 4,962,885; Coffee, PCT Application, WO 94/12285; Coffee, PCT Application, WO 94/14543; Coffee, PCT Application, WO 95/26234, Coffee, PCT Application, WO 95/26235, Coffee, PCT Application, WO 95/32807, which are herein incorporated by reference).
- the electrochemical properties of the TLR7 ligands and TLR7 ligand prodrugs formulation may be important parameters to optimize when delivering this drug to the lung with an EHD aerosol device and such optimization is routinely performed by one of skill in the art.
- EHD aerosol devices may more efficiently delivery drugs to the lung than existing pulmonary delivery technologies.
- Other methods of intra-pulmonary delivery of TLR7 ligand and TLR7 ligand prodrugs will be known to the skilled artisan and are within the scope of the invention.
- Liquid drug formulations suitable for use with nebulizers and liquid spray devices and EHD aerosol devices will typically include a TLR7 ligand or TLR7 ligand prodrug with a pharmaceutically acceptable carrier.
- the pharmaceutically acceptable carrier is a liquid such as alcohol, water, polyethylene glycol or a perfiuorocarbon.
- another material may be added to alter the aerosol properties of the solution or suspension of the TLR7 ligand or prodrug of a TLR7 ligand.
- this material is liquid such as an alcohol, glycol, polyglycol or a fatty acid.
- a TLR7 ligand or prodrug of a TLR7 ligand can also be formulated in rectal or vaginal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter or other glycerides.
- a TLR7 ligand or TLR7 ligand prodrug can also be formulated as a depot preparation.
- Such long acting formulations can be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection.
- the compounds can be formulated with suitable polymeric or hydrophobic materials (for example, as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- Liposomes and emulsions are well known examples of delivery vehicles that can be used to deliver TLR7 ligands and TLR7 ligand prodrugs. Certain organic solvents such as dimethylsulfoxide can also be employed, although usually at the cost of greater toxicity.
- a TLR7 ligand or prodrug of a TLR7 ligand can also be delivered in a controlled release system.
- a pump can be used (Sefton, CRC Crit. Ref Biomed Eng., 1987, 14, 201; Buchwald et al, Surgery, 1980, 88, 507; Saudek et al, N. Engl. J. Med., 1989, 321, 574).
- polymeric materials can be used (see Medical Applications of Controlled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Fla. (1974); Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, J. Macromol. Sci. Rev. Macromol. Chem., 1983, 23, 61; see also Levy et al, Science, 1985, 228, 190; During et al, Ann. Neurol., 1989,25,351; Howard et al, 1989, J. Neurosurg. 71, 105).
- a controlled-release system can be placed in proximity of the target of the compounds of the invention, e.g., the lung, thus requiring only a fraction of the systemic dose (see, e.g., Goodson, in Medical Applications of Controlled Release, supra, vol. 2, pp. 115 (1984)).
- Other controlled-release system can be used (see, e.g. Langer, Science, 1990, 249, 1527).
- Suitable excipients e.g., carriers and diluents
- excipients include, but are not limited to, water, ethanol, ethylene glycol, propylene glycol, butane-l,3-diol, isopropyl myristate, isopropyl palmitate, mineral oil, and mixtures thereof, which are non-toxic and pharmaceutically acceptable.
- additional ingredients are well known in the art. See, e.g., Remington's Pharmaceutical Sciences, 18th eds., Mack Publishing, Easton PA (1990).
- the pH of a pharmaceutical composition or dosage form, or of the tissue to which the pharmaceutical composition or dosage form is applied can also be adjusted to improve delivery of one or more active ingredients.
- the polarity of a solvent carrier, its ionic strength, or tonicity can be adjusted to improve delivery.
- Compounds such as stearates can also be added to pharmaceutical compositions or dosage forms to advantageously alter the hydrophilicity or lipophilicity of one or more active ingredients so as to improve delivery.
- stearates can serve as a lipid vehicle for the formulation, as an emulsifying agent or surfactant, and as a delivery-enhancing or penetration-enhancing agent.
- Different salts, hydrates or solvates of the active ingredients can be used to further adjust the properties of the resulting composition.
- the invention provides a pharmaceutical pack or kit comprising one or more containers comprising a TLR7 ligand prodrug useful for the treatment or prevention of a Hepatitis C virus infection.
- the invention provides a pharmaceutical pack or kit comprising one or more containers comprising a TLR7 ligand prodrug useful for the treatment or prevention of a Hepatitis C virus infection and one or more containers comprising an additional therapeutic agent, including but not limited to those listed in section 5.2.2 above, in particular an antiviral agent, an interferon, an agent which inhibits viral enzymes, or an agent which inhibits viral replication, preferably the additional therapeutic agent is HCV specific or demonstrates anti-HCV activity.
- the invention also provides a pharmaceutical pack or kit comprising one or more containers comprising one or more of the ingredients of the pharmaceutical compositions of the invention.
- a pharmaceutical pack or kit comprising one or more containers comprising one or more of the ingredients of the pharmaceutical compositions of the invention.
- Optionally associated with such container(s) can be a notice in the form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals or biological products, which notice reflects approval by the agency of manufacture, use or sale for human administration.
- ASSAYS ASSAYS
- TLR7 ligands, TLR ligand prodrugs, compositions and dosage forms of the invention can be tested in vitro or in vivo by a variety of methods known in the art to test activity. See, for example, the methods discussed below and used throughout the examples.
- a range of assays for the purpose of evaluating TLR7 activity are available, and are described in the following publications, each of which is incorporated-by-reference: Hirota et al, J. Med. Chem., 45, 5419-5422 (2002); and Akira S. et al, Immunology Letters, 85, 85-95 (2003).
- one system useful for the assay of TLR7 ligands is where the gene for TLR7 is cloned by methods known to those of skill in the art and transfected into an appropriate cell type such that TLR7 is expressed and coupled to a NFkB-luciferase reporter plasmid.
- TLR7 ligand prodrugs in cell culture results in a measurable luminescence signal. See, e.g., Lee et al, Proc. Nat. Acad. USA, 100, 6646-6651 (2003); Hemmi et al, Nat. Immunol, 3, 196-200 (2002); and Jurk et al, Nat. Immunol, 3, 499 (2002) (wherein the Lee et al, Hemmi et al, and Jurk et al, references are all incorporated herein by reference).
- PBMC peripheral blood mononuclear cells
- TLR7 ligand prodrug for a predetermined interval (e.g., 2 hours to 24 hours), followed by measurement of immunologic activity.
- immunologic activity may include induction of the synthesis of cytokines, which can be measured in the culture supernate by ELISA assay of the cytokine protein, such as a Type 1 interferon (interferon alpha, interferon beta) or Type 2 interferon (interferon gamma).
- the PBMC can be harvested after incubation with the candidate TLR7 ligand prodrug, the PBMC RNA extracted, and the level of induction of immune system genes determined by RNAse protection assays of the extracted RNA.
- Genes typically induced include 2'5'-OAS, or interferon gamma, but a range of cytokines can be measured. See, e.g., Hirota et al, J. Med. Chem., 45, 5419-5422 (2002).
- RNAse protection assays are a method known in the art wherein RNA analytes are quantitated by first hybridizing them to a radioalabeled RNA sequence specific for the analyte RNA, followed by digestion with enzymes that selectively degrade single stranded RNA. The sample is then submitted to gel electrophoresis under conditions that resolve the hybridized, protected double stranded RNA. The gel is then autoradiographed to reveal the location and intensity of the bands. These can be quantitated by methods well known in the art. Multiple analyte RNA species can be simultaneously assayed, if the protected fragments are sufficiently different in size to allow their separation by gel electrophoresis.
- RNAse protection assays can be performed as follows:
- RNA is purified from PMBC pellets using the "RNAeasy" kit (Qiagen), according to the manufacturer's instructions. Template sets may be obtained from PharMingen (BD Biosciences); a useful set that is commercially available from this supplier contains materials allowing the assay of TNF-a, IL12 p35, IP10, IL-la, IL-lb, IL-6, Interferon gamma, and the control RNA species L32 and GAPDH. This template set contains DNA that is appropriate for synthesis of the proper RNA probe for each of the listed genes.
- Probe synthesis is performed using the PharMingen in vitro transcription kit provided in the kit.
- the reaction contains RNase inhibitor; transcription buffer; 50 ng of the tempate set; 0.1375 mM of each of rGTP, rCTP, rATP; 0.003 mM rUTP;10 mM DTT, 0.010 mCi of [alpha 32P] UTP, and 20 Units of T7 RNA polymerase in a volume of 20 microliters.
- the reaction mixture is incubated for one hour at 37 °C, and then stopped by addition of 2 units of RNAse free DNAse, with an additional incubation of 30 minutes at 37 °C.
- RNA probes synthesized in this incubation are extracted once with 5.2 mM EDTA, 25 ⁇ l of Tris Saturated phenol, 25 ⁇ L of chloroform, and 4 ⁇ g of yeast tRNA, and then extracted with 50 ⁇ L of chloroform.
- the RNA is precipitated by addition of 50 ⁇ L of 4M ammonium acetate and 250 ⁇ L of ice-cold 100% ethanol, and after incubation at -80 °C for 30 minutes the preparation is centrifuged at high speed for 30 minutes. The pellet is washed in 100 % ethanol, and after removal of the ethanol the probe is resuspended and used in the RNAse protection incubation.
- RNAse protection assay uses the probe material prepared above and
- RNA extracted from PBMC The PBMC RNA is washed in 100% ethanol, quantitated by absorbance at 260 nm.
- the RPA is performed as described in the protocol provided in the PharMingen RiboQuant kit.
- Eight ⁇ L of the PBMC RNA samples are mixed with the 2 ⁇ L of the probe set in a thin-walled PCR tube, mixed well, briefly centrifuged and then overlaid with mineral oil. The tube is then placed in a 90 °C PCR block, cooled to 56 °C, and incubated for 16 hours. The samples are then cooled to 37 °C and RNAse A and RNAse TI are added.
- RNA is extracted with phenol-chloroform, and then precipitated from the phenol-chloroform with ammonium acetate-ethanol.
- the pellet is washed with ethanol and resuspended in buffer for electrophoresis.
- the samples are analysed by gel electrophoresis by methods well known in molecular biology. [00197] A number of assays may be employed in accordance with the present invention in order to determine the degree of anti-viral activity of a compound of the invention such as cell culture, animal models, and administration to human subjects.
- the assays described herein may be used to assay viral growth over time to determine the growth characteristics of a virus in the presence of a compound of the invention.
- a virus and a compound of the invention are administered to animal subjects susceptible to infection with the virus.
- the incidence, severity, length, virus load, mortality rate of infection, etc. can be compared to the incidence, severity, length, virus load, mortality rate of infection, etc. observed when subjects are administered the virus alone (in the absence of a compound of the invention).
- Anti-virus activity of the compound of the invention is demonstrated by a decrease in incidence, severity, length, virus load, mortality rate of infection, etc. in the presence of the compound of the invention.
- the virus and the compound of the invention are administered to the animal subject at the same time. In another specific embodiment, the virus is administered to the animal subject before the compound of the invention. In another specific embodiment, the compound of the invention is administered to the animal subject before the virus.
- the growth rate of the virus can be tested by sampling biological fluids/clinical samples (e.g., nasal aspirate, throat swab, sputum, broncho-alveolar lavage, urine, saliva, blood, or serum) from human or animal subjects at multiple time points post-infection either in the presence or absence of a compound of the invention and measuring levels of virus.
- biological fluids/clinical samples e.g., nasal aspirate, throat swab, sputum, broncho-alveolar lavage, urine, saliva, blood, or serum
- the growth rate of a virus is assayed by assessing the presence of virus in a sample after growth in cell culture, growth on a permissible growth medium, or growth in subject using any method well-known in the art, for example, but not limited to, immunoassay (e.g., ELISA; for discussion regarding ELISAs see, e.g., Ausubel et al, eds, 1994, Current Protocols in Molecular Biology, Vol.
- immunoassay e.g., ELISA; for discussion regarding ELISAs see, e.g., Ausubel et al, eds, 1994, Current Protocols in Molecular Biology, Vol.
- viral titers can be determined by obtaining biological fluids/clinical samples from infected cells or an infected subject, preparing a serial dilution of the sample and infecting a monolayer of cells that are susceptible to infection with the virus (e.g. primary cells, transformed cell lines, patient tissue samples, etc) at a dilution of the virus that allows for the emergence of single plaques. The plaques can then be counted and the viral titer expressed as plaque forming units per milliliter of sample.
- the growth rate of a virus in a subject can be estimated by the titer of antibodies against the virus in the subject.
- Antibody serum titer can be determined by any method well-known in the art, for example, but not limited to, the amount of antibody or antibody fragment in serum samples can be quantitated by, e.g., ELISA.
- in vivo activity of a TLR7 ligand or prodrug of a TLR7 ligand can be determined by directly administering the compound to a test animal, collecting biological fluids (e.g., nasal aspirate, throat swab, sputum, broncho-alveolar lavage, urine, saliva, blood, or serum) and testing the fluid for anti-virus activity.
- biological fluids e.g., nasal aspirate, throat swab, sputum, broncho-alveolar lavage, urine, saliva, blood, or serum
- samples to be assayed for virus levels are biological fluids/clinical samples (e.g., nasal aspirate, throat swab, sputum, broncho-alveolar lavage, urine, saliva, blood, or serum)
- the samples may or may not contain in tact cells.
- Samples from subjects containing intact cells can be directly processed, whereas isolates without intact cells may or may not be first cultured on a permissive cell line (e.g. primary cells, transformed cell lines, patient tissue samples, etc) or growth medium (e.g., LB broth/agar, YT broth/agar, blood agar, etc.).
- a permissive cell line e.g. primary cells, transformed cell lines, patient tissue samples, etc
- growth medium e.g., LB broth/agar, YT broth/agar, blood agar, etc.
- Cell suspensions can be cleared by centrifugation at, e.g., 300xg for 5 minutes at room temperature, followed by a PBS, pH 7.4 (Ca++ and Mg++ free) wash under the same conditions.
- Cell pellets can be resuspended in a small volume of PBS for analysis.
- a compound of the invention is administered to a human subject infected with a virus.
- the incidence, severity, length, viral load, mortality rate of infection, etc. can be compared to the incidence, severity, length, viral load, mortality rate of infection, etc. observed in human subjects infected with a virus in the absence of a compound of the invention or in the presence of a placebo.
- Anti-viral activity of the compound of the invention is demonstrated by a decrease in incidence, severity, length, viral load, mortality rate of infection, etc. in the presence of the compound of the invention. Any method known in the art can be used to determine anti-viral activity in a subject such as those described previously.
- in vivo activity of a TLR7 ligand or prodrug of TLR7 ligand can be determined by directly administering the compound to an animal or human subject, collecting biological fluids/clinical samples (e.g., nasal aspirate, throat swab, sputum, broncho-alveolar lavage, urine, saliva, blood, or serum) and testing the biological fluids/clinical samples for anti-viral activity (e.g., by addition to cells in culture in the presence of the virus).
- biological fluids/clinical samples e.g., nasal aspirate, throat swab, sputum, broncho-alveolar lavage, urine, saliva, blood, or serum
- TLR7 Ligand Identification There are three known chemical classes of TLR7 ligands: guanosines, imidazoquinolines, and pyrimidmes (see Section 5.2). As described above, additional TLR7 ligands are readily identified by known screening methods. For example, adenine analogs and derivatives were identified as TLR ligands by using the following screening procedure. See Tables 1 and 2. A stable HEK293-hTLR7 cell line was obtained from Invivogen (San Diego, California), transfected with pNiFty2-Luc, an NF-kB inducible luciferase reporter plasmid (Invivogen) and (dual) stable transfectants selected.
- the resultant dual (hTLR7/pNiFty2- Luc) cell lines were functionally tested by response to loxoribine and isatoribine as measured by fold luciferase induction relative to a no drug control.
- the C23 line was chosen due to its satisfactory response and sensitivity profile with these (and other) TLR7 agonists.
- the biological rationale which connects TLR7 engagement and NF-kB activation has met with widespread acceptance (for a review, see Akira S. et al, Immunol.
- Isatoribine investigational drug product was supplied as a 1 mg/mL solution in sterile normal saline contained in a 50 mL vial. Isatoribine was administered in humans by intravenous infusion once daily for 7 days, at 200, 400, 600 or 800 mg per dose. All doses were administered by constant rate infusion over a 60-minute period, except the 800 mg dose was administered over an 80-minute period.
- the flow rate for each dose was as follows: 3.33 mL/min for the 200 mg dose; 6.67 mL/min for the 400 mg dose; 8.33 mL/min for the 500 mg dose; or 10.0 mL/min for the 600 mg and 800 mg dose. [00213] Four to twelve patients were enrolled in each of the dose groups (200 mg,
- Plasma HCV RNA was determined at baseline (an average of 2 pre-treatment measurements taken on Day -1 or pre-treatment and on Day 1) and once daily prior to the start of the first daily isatoribine intravenous infusion on Days 2 through 7 for these daily (x7 days) dosing groups. See Figure 2.
- the viral load was measured by the branched DNA method (VersantTM v3.0 bDNA assay, Bayer Diagnostics).
- the maximum change from the pre-treatment baseline was estimated using log-transformed values.
- Plasma HCV RNA decreased over the course of isatoribine treatment, with the larger changes generally occurring in patients who received the higher daily doses (Figure 2).
- HCV Replicon-Based Viral Bioassav It has been demonstrated that HCV replicons are highly sensitive to the inhibitory effects of interferon- ⁇ and interferon- ⁇ . Therefore the HCV replicon becomes a very useful system for measuring the amount of biologically active interferons in supematants from human PBMCs stimulated with a TLR7 agonist. A quantitative assay was developed which is based on measuring the activity of the luciferase reporter gene that was integrated into an HCV replicon. By using this system, interferons from TLR7 agonist- treated PBMCs were measured and their inhibitory activity was assessed using the luciferase reporter replicon.
- Human PBMCs isolated from a healthy donor were placed in replicate cell culture wells (5 x 10 6 cells per well).
- the PBMCs are incubated in the absence of test compounds at 37 °C in a humidified atmosphere containing 5% CO 2 for 24 hours to allow stabilization to the culture conditions, and then the TLR7 ligand or a drug free control is added to replicate wells containing PBMCs from the same donor.
- the concentrations of TLR7 ligand may be varied to suit the particular experiment, and the PBMC cultures are then incubated at 37 °C in a humidified atmosphere containing 5% C0 for eight hours.
- Samples of cell culture supernatant are taken at the eight hour time point (or twenty four hour time point in case of Loxoribine and its prodrugs) from TLR7 ligand treated and control wells and are assayed for interferon-alpha production by ELISA.
- Supematants from compound-treated cells and a no-drug control were diluted at 1:10, 1:100, and 1:1000 in RPMI media and applied to a 96-well plate of Huh7 hepatocyte cells containing the luciferase reporter replicon. Cells were incubated for 48 hours at 37 °C in a tissue culture incubator.
- the 96-well plates are washed 2X with PBS and are lysed with passive lysis buffer (Promega). Plates are shaken at room temperature for 20 minutes and standard luciferase assay reagent (Promega) is added to each well by injection and the plate is read on an Lmax luminometer (Molecular Devices). The raw relative light units are converted to a percent inhibition that is compared to the no-drug control wells to determine the level of inhibition observed in the replicon assay.
- the estimated maximal concentration of interferons required to inhibit HCV replicon replication was determined to be at a 1:10 dilution of supernatant of PBMC-stimulated cells which fell within our test range of the dilution series. For all TLR7 agonists tested, 100% inhibition was observed on the luciferase reporter replicon system at the 1:10 dilution.
- Tables 3-8 represents the inhibition of the HCV replicon system by the supernatant collected after exposure of PBMC cells to the compound at an initial concentration for the incubation time indicated and diluted as specified in the first column ("PBMC exposed to compound"). A supernatant collected from PBMC cells non-exposed to the compound and diluted as specified in the first column was used as a control ("Blank supernatant"). The PBMC cells were isolated from a single blood donor as specified. Table 3: Antiviral Effect of Isatoribine in the in vitro HCV Replicon Bioassav No. 1 Incubation time: 8 hours Initial concentration: 100 ⁇ M Blood donor number: FL72035
- TLR7 Ligand Prodrugs Compounds of the invention can be synthesized using the methodology described in Schemes 1-18 above. Unless otherwise indicated all temperatures are set forth in degrees Celsius and all parts and percentages are by weight. Reagents are purchased from commercial suppliers such as Aldrich Chemical Company or Lancaster Synthesis Ltd. and are used without further purification unless otherwise indicated. Tetrahydrofuran (THF) and N,N-dimethylforamide (DMF) are purchased from Aldrich in Sure Seal bottles and used as received. Unless otherwise indicated, the following solvents and reagents are distilled under a blanket of dry nitrogen.
- THF and Et 2 0 are distilled from Na- benzophenone ketyl; CH 2 C1 2 (DCM), diisopropylamine, pyridine and Et 3 N are distilled from CaH 2 ; MeCN is distilled first from P 0 5 , then from CaH 2 ; MeOH is distilled from Mg; PhMe, EtOAc and i-PrOAc are distilled from CaH 2 ; TFAA was purified via simple atmospheric distillation under dry argon.
- the reactions set forth are done generally under a positive pressure of argon at an ambient temperature (unless otherwise stated) in anhydrous solvents, and the reaction flasks are fitted with rubber septa for the introduction of substrates and reagents via syringe.
- NMR spectra is recorded on a Varian Mercury- VX400 instrument operating at 400 MHz and 13 C-NMR spectra are recorded operating at 75 MHz.
- NMR spectra are obtained as CDC1 3 solutions (reported in ppm), using chloroform as the reference standard (7.27 ppm and 77.00 ppm), CD 3 OD (3.4 and 4.8 ppm and 49.3 ppm), DMSO-de, or internally tetramethylsilane (0.00 ppm) when appropriate. Other NMR solvents are used as needed.
- Infrared (IR) spectra are recorded on a Thermo Nicolet Avatar 370 FT-IR as neat oils or solids, and when given are reported in wave numbers (cm "1 ). Mass spectra reported (+)-ES Thermo Finnegan LCQ LC/MS conducted by the Analytical Chemistry Department at Anadys Pharmaceuticals, Inc. Elemental analyses are conducted by the Atlantic Microlab, Inc. in Norcross, GA or by NuMega, in San Diego, CA. Melting points (mp) are determined on an open capillary apparatus, and are uncorrected.
- Step I Preparation of 7-Allyl-2-amino-9-(2 ',3 ',5'-tri-0-acetyl- ⁇ -D-ribofuranosyl)-7,9- dihydro-lH-purine-6, 8-dione (40)
- a heterogeneous mixture of 7-allyl-2-amino-9- ⁇ -D-ribofuranosyl-7,9- dihydro-lH-purine-6,8-dione 17 (1.00 g, 2.95 mmol, prepared according to Reitz et al, JMC, 37, 3561-3578 (1994)), DMAP (0.036 mg, 0.29 mmol) and NEt 3 (2.05 mL, 14.74 mmol) was stirred in dry acetonitrile (25 mL). Acetic anhydride (0.862 mL, 9.13 mmol) was added slowly to the suspension and the reaction mixture was stirred at ambient temperature for 16 h.
- Step 2 Preparation of7-Allyl-2-amino-6-chloro-9-(2 ',3 ',5 '-tri-O-acetyl- ⁇ -D- ribofuranosyl)-7,9-dihydro-purin-8-one (41) [00226]
- Compound 40 (0.65 g, 1.39 mmol) was dissolved in phosphorus oxychloride
- Step 3 Preparation of7-Allyl-2-amino-9-(2 ',3 ',5'-tri-0-acetyl- ⁇ -D-ribofuranosyl)-7,9- dihydro-purin-8-one (42)
- Step 4 Preparation of7-Allyl-2-amino-9- ⁇ -D-ribofuranosyl-7,9-dihydro-purin-8-one (43) [00228] To a solution of 42 (0.13 g, 0.29 mmol) in methanol (4 mL) was added solid
- Step 2 Preparation of7-Allyl-2-amino-6-ethoxy-9- ⁇ -D-ribofuranosyl-7,9-dihydro-purin-8- one (45)
- Step 1 Preparation of4-(2-Amino-5-bromo-6-phenyl-pyrimidin-4-yloxymethyl)-5-methyl- [l,3]dioxol-2-one (46)
- Step 1 Preparation of(5-Bromo-6-oxo-4-phenyl-l,6-dihydro-pyrimidin-2-yl)-carbamic acid ethyl ester (36)
- NEt 3 (0.14 mL, 0.99 mmol
- diethyl pyrocarbonate (0.27 mL, 1.89 mmol).
- the reaction mixture was maintained at 65 °C for 20 h.
- the solvent was removed and the residue treated with DCM.
- the resulting mixture was filtered to remove the remaining starting material 35 and the filtrate washed with aqueous NaHC0 3 , brine and dried (MgSO ).
- Step 1 Preparation of(5-Bromo-6-oxo-4-phenyl-l,6-dihydro-pyrimidin-2-yl)-carbamic acid pentyl ester (49)
- Step 1 Preparation of (1 -Isobutyl-1 H-imidazo[4,5-c]quinolin-4-yl)-carbamic acid pentyl ester (34)
- Step I Preparation of (1 -Isobutyl-1 H-imidazo[4,5-c]quinolin-4-yl)-carbamic acid ethyl ester (50)
- Step I Preparation of Carbonic acid6-amino-9-benzyl-2-(2-methoxy-ethoxy)-9H-purin-8- yl ester ethyl ester (51)
- Examples 11-20 were prepared from 6-amino-9-benzyl-2-(2-methoxy- ethoxy)-9H-purin-8-ol, 29 and the appropriate chloroformate according to the procedure described in Example 10.
- Step 1 Preparation of7-Allyl-2-amino-9-(2 ',3 '-0-isopropylidene- ⁇ -D-ribofuranosyl)-7,9- d ⁇ hydro-1 H-purine-6, 8-dione (66)
- Step 2 Preparation of 7-Allyl-2-amino-9-(2 ',3 '-O-isopropylidene-5 '-N-tert- butoxycarbonyl-L-valinyl)- ⁇ -D ⁇ ribofuranosyl)-7,9-dihydro-lH-purine-6,8-dione (67) [00254] A mixture of 66 (0.13 g, 0.34 mmol), BOC-Valine (0.08 g, 0.36 mmol), EDC
- Step 3 7-Allyl-2-Amino-9-(5'-0-L-valinyl- ⁇ -D-ribofuranosyl)-7,9-dihydro-lH-purine-6,8- dione (68)
- Example 25 7-AHyI ⁇ 2-amino-9- ⁇ -D-ribofuranosyl-6-(5-methyl-2-oxo-[l,3]dioxol-4- yImethoxy)-7,9-dihydro-purin-8-one (71)
- Step I Preparation of7-Allyl-2-amino-9-(2 ',3 ',5'-tris-0-triethylsilanyl- ⁇ -D- ribofuranosyl) - 7, 9-dihydro-lH-purine-6, 8-dione (69)
- Step 2 Preparation of7-Allyl-2-amino-9-(2 ',3 ',5 '-tris-O-triethylsilanyl- ⁇ -D- ribofuranosyl)-6-(5-methyl-2-oxo-[l,3]dioxol-4-ylmethoxy)-7,9-dihydro-purin-8-one (70) [00257]
- compound 70 was prepared from compound 69 and 4-hydroxymethyl-5-methyl-[l,3]dioxol-2-one (prepared according to the procedure of Alepegiani, Syn.
- Step 3 Preparation of7-Allyl-2-amino-9- ⁇ -D-ribofuranosyl-6-(5-methyl-2-oxo-[l,3]dioxol- 4-ylmethoxy)-7, 9-dihydro-purin-8-one (71) [00258] To a solution of 70 (9.0 mg, 0.012 mmol) in MeOH (1.5 mL) was added
- Step I Preparation o (7-Allyl-2-amino-9-f2 '3 ',5 '-tris-O-triethylsilanyl- ⁇ -D- r/t> «r ⁇ R ⁇ 5 '/)-8-oxo-8,9-dihydro-7H-purin-6-yloxymethyl)-methyl-carbamic acid ethyl ester (72)
- Step 2 Preparation of(7-Allyl-2-amino-9- ⁇ -D-ribofuranosyl-8-oxo-8,9-dihydro-7H-purin-
- Step I Preparation of 5-Amino-3-(2 ' ,3 '-O-isopropylidene- ⁇ -D-ribofuranosyl) thiazolo[4,5-d]pyrimidine-2, 7-dione (22)
- Step 2 Preparation of 5-Amino-3-(2 ',3 '-O-isopropylidene -5 '-N-tert- butoxycarbonyl-L-valinyl)- ⁇ -D-ribofuranosyl)-thiazolo[4,5-d]pyrimidine-2,7-dione (23) [00262] To a " solution of N-butoxycarbonyl-(L)-valine (671 mg, 2.81 mmol) in THF
- Step 3 Preparation of5-Amino-3-(5'-0-L-valinyl- ⁇ -D-ribofuranosyl)thiazolo[4,5- dJpyrimidine-2, 7-dione Dihydrochloride (24)
- a stream of HCl gas was passed through a bubbler of concentrated H 2 S0 , and subsequently directed (via fritted dispersion tube) into a 250 mL 3-neck Morton flask containing dry isopropyl acetate (80 mL) at 0 °C until a saturated solution was obtained.
- a solution of the Boc-amino acid ester from Step 2 above (5.53 g, 9.95 mmol) in isopropyl acetate (30 mL), resulting in the formation of a white solid precipitate within 5 min.
- the reaction mixture was warmed to room temperature, then stirred 12 h.
- the heterogeneous reaction mixture was diluted with dry toluene (100 mL). Filtration using a medium pore scintered glass funnel under N 2 provided an off-white, amorphous solid. Trituration of the solid in dry THF was followed by filtration and vacuum drying at 65 °C, affording 3.677 g (81%) of the title compound 24 as a white solid: mp 166-68 °C (dec); !
- Step I Preparation of 5-Acetylamino-3-(2 ',3 ',5'-tri-0-acetyl- ⁇ -D- ribofuranosyl)thiazolo[4,5-d]pyrimidine-2,7(6H)-dione (74)
- Step 1 Preparation of S-Amino-3-(2 ',3 ' ,5 '-tri-0-acetyl- ⁇ -D-ribofuranosyl)thiazolo[4,5- dJpyrimidine-2, 7(6H)-dione (75)
- Step 1 Preparation of 5-Acetylamino-7-ethoxy-3-(2 ',3 ',5 '-tri-O-acetyl- ⁇ -D-ribofuranosyl)- thiazolo[4, 5-d]pyrimidin-2-one (76)
- step 2 the title compound was prepared from 76 in 65% yield as a white solid: ! H NMR (400 MHz, - 6 -DMSO) ⁇ 6.87 (s, 2H), 5.85
- Step 1 Preparation of 5-Acetylamino-7-methoxy-3-(2 ',3 ',5 '-tri-O-acetyl- ⁇ -D- r ⁇ bofuranosyl) -thiazolo[4, 5-d]pyrimidin-2-one (78) "[00268]
- step 1 77 was prepared from 74 and methanol in 65% yield as a white foam: MS (+)-ES [M+H] + 499.
- R f 0.5 (75% Ethyl acetate-CHCl 3 ).
- Step I Preparation of 5-Amino-3-(2 ',3 ',5'-tris-0-triethylsilanyl- ⁇ -D-ribofuranosyl)- thiazolo[4,5-d]pyrimidin-2, 7-dione (80)
- Step 2 Preparation of 5-Amino-3-(2 ',3 ',5'-tris-0-triethylsilanyl- ⁇ -D-ribofuranosyl)- 2,3- dihydro-thiazolo[4,5-d]pyrimidin-7-yloxymethyl)-carbamic acid ethyl ester (81) [00271] ' in a manner similar to Step 1 of Example 2, compound 81 was prepared from
- Step 3 Preparation of(5-Amino-2-oxo-3- ⁇ -D-ribofuranosyl-2,3-dihydro-thiazolo[4,5- d]pyrimidin-7-yloxymethyl)-carbamic acid ethyl ester (82)
- Step 1 Preparation of (5-Amino-2-oxo-3-(2 ',3 ',5 '-tri-0-acetyl- ⁇ -D-ribofuranosyl)-2,3- dihydro-thiazolo[4,5-d]pyrimidin-7-yloxymethyl)-methyl-carbamic acid ethyl ester (83) [00273] In a manner similar to Example 2, step 1, compound 83 was prepared from
- Example 34 5- Amino-7-(5-methyl-2-oxo- [1 ,3] ioxol-4-ylmethoxy)-3-(2 ' ,3' ,5' -tri-O- acetyl- ⁇ -D-ribofuranosyl)-thiazolo[4,5- ⁇ /]pyri ⁇ nidin-2-one (85)
- Step I Preparation of 5-Amino-7-(5-methyl-2-oxo-[l ,3] dioxol-4-ylmethoxy)-3-(2 ',3 ',5'- tri-0-acetyl- ⁇ -D-ribofuranosyl)-thiazolo[4,5-d]pyrimidin-2-one (85) [00275] To a solution of triacetate 75 (1.55 g, 3.50 mmol) in THF (50 mL) at 0 °C was added polymer supported-triphenylphosphine (4.95 g, 10.50 mmol, Argonaut).
- Step I Preparation of 5-Amino-7-(5-methyl-2-oxo-[l ,3] dioxol-4-ylmethoxy)-3-(2 ',3 ',5 '- tris-0-triethylsilanyl- ⁇ -D-ribofuranosyl)-thiazolo[4,5-d]pyrimidin-2-one (86)
- Step 2 Preparation of 5-Amino-7-(5-methyl-2-oxo-[l ,3] dioxol-4-ylmethoxy)-3- ⁇ -D- ribofuranosyl-thiazolo[4, 5-d]pyrimidin-2-one (87)
- Step I Preparation of 5-Amino-7-thioxo-3-(2 ',3 ',5 '-tri-O-acetyl- ⁇ -D-ribofuranosyl)- thiazolo[4, 5-d]pyrimidin-2-one (88)
- Step 2 Preparation of 5-Amino-3-(2 ',3 ',5 '-tri-0-acetyl- ⁇ -D-ribofuranosyl)-3H- thiazolo[4, 5-d]pyrimidin-2-one (89)
- Step 1 Preparation of 5-Amino-3-(5 '-0-tert-butyl-dimethylsilanyl- ⁇ -D-ribofuranosyl)-3H- thiazolo[4, 5-d]pyrimidin-2-one (91)
- Step 2 Preparation of 5-Amino-3-(2 ',3 '-di-O-acetyl, 5 '-O-tert-butyl-dimethylsilanyl- ⁇ -D- ribofuranosyl)-3H-thia ⁇ olo [4, 5-d]pyrimidin-2-one (92)
- Step I Preparation of [2 -Ethoxymethyl- 1 -(2-hydroxy-2-methyl-propyl)-l H-imidazo[4,5- [00284]
- the title compound from l-(4-amino-2-ethoxymethyl-imidazo[4,5- c]quinolin-l-yl)-2-methyl-propan-2-ol (38) (prepared according to the procedure given in
- PBMC peripheral blood mononuclear cells
- TLR7 ligand and TLR7 ligand prodrug are added to separate wells containing PBMC from the same donor; untreated controls are included.
- concentrations of TLR7 ligand and TLR7 ligand prodrug may be varied to suit the particular experiment, and the PBMC cultures are then incubated at 37 °C in a humidified atmosphere containing 5% C0 for a period of time ranging from two hours to 48 hours. Samples of cell culture supernate media are taken during the incubation. These are assayed for cytokine production by ELISA.
- TLR7 ligand and TLR7 ligand prodrug remaining at the end of the incubation may be assayed by LC-MS. Cytokine production is calculated relative to production in the isatoribine control, following subtraction of the cytokine production in untreated controls. The cytokine results are compared to determine the extent that the TLR7 ligand is more active than the corresponding TLR7 ligand prodrug.
- the TLR7 ligand prodrug may be deemed a "masked" TLR7 ligand prodrug.
- the magnitude of reduction in cytokine production that constitutes "masking" may be as little as a 25%> reduction relative to the parent TLR, since this would afford a corresponding increase in administered dose for a given level of tolerability.
- Tables 9 through 14 provide data illustrating that TLR7 ligands of multiple chemical classes can be masked.
- the examples shown demonstrate substantial masking relative to the parent TLR7 ligand.
- the chemical substitutions shown are exemplary, and in no way restrictive of the invention, since additional chemical substitutions may also exhibit masking and are contemplated in the invention.
- Masking can be achieved by introduction of substitutions at a range of locations on any TLR7 ligand, and as shown can incorporate a variety of chemical linkages. It will be appreciated that the preferred substitution and linkage may vary for different parent TLR7 ligands.
- Table 9 Masking of Isatoribine Prodrugs
- PBMC assay The results of the PBMC assay (Table 9) show the amount of INFa released after exposure of the parent compound and its prodrugs for either 8 hours (val-isatoribine, 24) or 24 hours (other prodrugs) at the initial concentration of 100 ⁇ M. The amount of the released INFa was normalized to that induced by 100 ⁇ M of isotoribine at 100 ⁇ M in the same blood donor with the same exposure time. Table 10: Masking of Loxoribine Prodrugs
- PBMC assay The results of the PBMC assay (Table 11) show the amount of INFa released after exposure of the parent compound and its prodrugs for 24 hours at the initial concentration 100 ⁇ M. The amount of the released INFa was normalized to that induced by 100 ⁇ M of isatoribine at 100 ⁇ M in the same blood donor with the same exposure time. Table 12: Masking of Resiquimod Prodrugs
- PBMC assay The results of the PBMC assay (Table 12) show the amount of INFa released after exposure of the parent compound and its prodrugs for 24 hours at the initial concentrations of either 1 or 100 ⁇ M. The amount of the released INFa was normalized to that induced by 100 ⁇ M of isatoribine at 100 ⁇ M in the same blood donor with the same exposure time. Table 13: Masking of Bropirimine Prodrugs
- PBMC assay The results of the PBMC assay (Table 13) show the amount of INFa released after exposure of the parent compound and its prodrugs for 24 hours at the initial concentration of 100 ⁇ M. The amount of the released INFa was normalized to that induced by 100 ⁇ M of Isatoribine at 100 ⁇ M in the same blood donor with the same exposure time. Table 14: Masking of Adenine Prodrugs
- adenine prodrugs can be demonstrated in a PBMC assay.
- the results of the PBMC assay show the amount of INFa released after exposure of the parent compound and its prodrugs for 24 hours at different initial concentrations specified in the table.
- the amount of the released INFa was normalized to that induced by 100 ⁇ M of isatoribine at 100 ⁇ M in the same blood donor with the same exposure time.
- TLR7 ligand prodrugs can also be assessed in vitro for their conversion to the active parent TLR7 ligand. This can be measured by incubation of the prodrug in blood, plasma, or in a cell culture of hepatocytes. At selected time intervals, samples are taken to determine the amount of prodrug remaining and the amount of TLR7 ligand produced. Such determinations are readily made by use of analytical tools known in the art such as LC-MS. The determination of the extent of conversion of a masked TLR7 ligand prodrug to the parent TLR7 ligand is useful in interpreting data wherein masking is apparent at shorter times but diminishes upon long incubations in the PBMC assay described herein.
- the rate of conversion of the prodrug to the TLR7 ligand may be determined to ensure that the cytokine results arise predominately from exposure to prodrug rather than from exposure to TLR7 ligand generated by rapid conversion of the prodrug under the conditions of the experiment.
- TLR7 ligand prodrugs can be assessed by performing studies in vivo.
- the candidate prodrugs are administered by oral administration to mice, rats, monkeys, and/or dogs, and blood samples are taken at selected intervals.
- the blood samples are analysed for both the prodrug and the desired TLR7 ligand. Additional blood or liver samples may be analyzed for the presence of interferons and other cytokines that indicate functional activation of the TLR7 pathway in vivo. Desired candidates will demonstrate a blood exposure to the prodrug and also demonstrate a blood exposure to the desired TLR7 ligand of about 10% to 99%> of the applied dose, as measured on a molar basis.
- TLR7 ligand prodrug val-isatoribine 24
- TLR7 ligand prodrug val-isatoribine 24
- FIG. 10/305,061 incorporated herein by reference in its entirety.
- the normal mouse provides a useful system for the assessment of the degree to which the inventions described herein provide material improvement in the oral delivery of 21 (isatoribine). Not only can one measure the plasma concentrations of isatoribine arising from oral administration of the said prodrug(s) but also the extensive immunological research conducted in the mouse has provided reagents suitable for measuring the levels of interferon alpha, a cytokine of interest reflecting one of the desired biologic activities of isatoribine.
- Table 16 records the results of assays for murine interferon alpha in the plasma of mice that first were dosed with bicarbonate and then four hours later were dosed orally with isatoribine, formulated in bicarbonate, at a level of 50 mg/kg. Interferon was reported in the plasma from four mice, including two that had received the bicarbonate vehicle dose. All the values reported in this experiment were low, and the reported interferon levels were not consistently reported for all three mice assessed at each time point, suggesting that these signals may be artifacts arising from measurement near the lower limits of the assay.
- Table 17 records the results of assays for murine interferon alpha in the plasma of mice that were dosed orally with val-isatoribine, dissolved in bicarbonate, at a dose that is equivalent to 50 mg/kg of isatoribine on a molar basis. It is evident that interferon was readily measurable at 1.0 hour, 1.5 hours, and 2.0 hours after dosing. Interferon was detected in all mice assayed at a given time point, indicating the reliability of the effect following val-isatoribine administration. Thus a single administration of val- isatoribine was superior to either a single dose or a repeated dose of isatoribine.
- Table 17 Plasma Concentration (pg/mD of Interferon Alpha (Mu-IFN- ⁇ ) in Mice Following a Single 73.0 mg/kg Dose of Val-Isatoribine Time, h Individual Value Mean SD
- Formulation 2 Val-isatoribine in phosphate buffered saline, 1.62 and 6.48 mg/mL, equivalent to 1 and 4 mg/mL of isatoribine on a molar basis.
- Four male and four female adult beagle dogs weighing between 15 to 27 kg and approximately 1-2 years old were used at the beginning of the study. The animals were divided into 2 groups of 2 males and 2 females each. The test material was administered by gavage on Days 1 and 8, allowing a 7 day washout period between administrations. Blood samples (2 mL) were collected from each animal at predose, 15, 30 minutes, 1, 2, 3, 4, 6, 8 and 10 hours into lithium heparin tubes after each dosing.
- the plasma was frozen at -70 °C until analysis.
- the plasma was analyzed for isatoribine by an HPLC -MS/MS assay.
- the pharmacokinetic parameters for isatoribine arising from isatoribine or val-isatoribine in each dog are summarized in Tables 18 and 19.
- the ratios for the key pharmacokinetic parameters defining the maximum concentration (Cmax) and total exposure as measured by the area under the time-concentration curve (AUC) for the prodrug and the bicarbonate solution at the 50 mg/kg dose are summarized in Table 20.
- the Cmax ratio was 2.98 ⁇ 0.695
- the AUC ratio was 2.38 ⁇ 0.485.
- the maximum concentrations of isatoribine achieved after oral dosing are at least doubled, and the systemic exposure to isatoribine is enhanced by approximately 2-fold following oral administration of the prodrug val- isatoribine, compared to isatoribine itself, at both 10 and 50 mg/kg dose.
- Table 18 Pharmacokinetic Parameters of Isatoribine in Dogs dosed at 50 mg/kg
- Dog 3532828 Cmax Dog 3532828 Cmax, ng/mL 6134.7 15987.9 Tmax, h 0.50 0.50 AUC(O-inf), ng-h/mL 12069.7 32987.0 T 1 ⁇ , h 1.4 1.6
- the prodrug val-isatoribine is preferred for several reasons. First, the prodrug is easily formulated to provide a high proportion of active agent. This results in small capsule sizes for a given dose, which is an advantage for an oral product. Second, at the doses tested, val-isatoribine provides plasma levels of isatoribine that are well within the range desirable for biologic effect after oral administration, which is not the case for isatoribine itself. Cynomolgus Monkey
- the plasma samples were analyzed for a prodrug and a parent compound using well-known LCMS/MS quantitation techniques by triple quadrupole instruments, i.e Sciex API3000.
- the quantitation results for the parent compound delivered by oral administration of the parent compound itself or by its prodrug administered orally were used to calculate the area-under-the-cureve (AUC) values from time zeto to 24 hours (PO AUC0- 24h).
- AUC area-under-the-cureve
- TLR7 ligand prodrugs of the invention also demonstrate unexpected and greatly reduced toxicology effects, and in particular reduced GI irritancy.
- GI gastrointestinal
- TLR7 ligand prodrugs offer the prospect of masking the active structure as the agent passes through lymphoid tissue lining the gut, which should minimize activation of this tissue and thereby reduce GI irritancy.
- Robins et al. have shown that elimination of the 5'-hydroxyl of isatoribine nucleoside eliminates activity. See Robins et al, Adv. Enzyme Regul, 29, 97-121 (1989).
- the compound was administered as a solution, by gavage or by intravenous infusion. Multiple parameters were assessed, as is customary in a toxicology study. In the studies providing higher potential exposure to isatoribine, the plasma concentration of isatoribine was assessed by a LC/MS method. The notable GI findings were graded and are listed in Table 27.
- Intravenously administered isatoribine resulted in emesis and/or loose stools as a common finding in dogs; this effect occurred at substantially higher applied doses than orally administered isatoribine. No lesions were seen in the GI tract either at necropsy or histopathologic evaluation of tissues. The GI toxicity did not affect the NOAEL, which was established as 12.5 mg/kg on the basis of other findings.
- Orally administered val-isatoribine demonstrated a toxicology profile similar to intravenously administered isatoribine. At higher applied doses, emesis and loose stools were observed. No GI lesions were found, although this was a focus of evaluation in this study. As for intravenously administered isatoribine, the NOAEL was established on the basis of other findings. The correspondence of observed toxicity to systemic exposures of isatoribine is of interest in this study; the threshold of isatoribine AUC for observation of emesis and loose stools is similar for intravenously administered isatoribine and orally administered val-isatoribine (Table 27).
- Table 28 illustrates a batch formulation and a single dose unit formulation containing 100 mg of val-isatoribine.
- microcrystalline cellulose, croscarmellose sodium, and val-isatoribine components are passed through a #30 mesh screen (about 430 ⁇ to about 655 ⁇ ).
- the Pluronic F-68® (manufactured by JRH Biosciences, Inc. of Lenexa, KS) surfactant is passed through a #20 mesh screen (about 457 ⁇ to about 1041 ⁇ ).
- the Pluronic F-68® surfactant and 0.5 kgs of croscarmellose sodium are loaded into a 16 qt. twin shell tumble blender and are mixed for about 5 minutes. The mix is then transferred to a 3 cubic foot twin shell tumble blender where the microcrystalline cellulose is added and blended for about 5 minutes.
- the thalidomide is added and blended for an additional 25 minutes.
- This pre-blend is passed through a roller compactor with a hammer mill attached at the discharge of the roller compactor and moved back to the tumble blender.
- the remaining croscarmellose sodium and magnesium stearate is added to the tumble blender and blended for about 3 minutes.
- the final mixture is compressed on a rotary tablet press with 250 mg per tablet (200,000 tablet batch size).
- Ill - 6.7 Mucosal Composition A concentrate is prepared by combining isatoribine, and a 12.6 kg portion of the trichloromonofluoromethane in a sealed stainless steel vessel equipped with a high shear mixer. Mixing is carried out for about 20 minutes. The bulk suspension is then prepared in the sealed vessel by combining the concentrate with the balance of the propellants in a bulk product tank that is temperature controlled to 21° to 27°C. and pressure controlled to 2.8 to 4.0 BAR. 17 ml aerosol containers which have a metered valve which is designed to provide 100 inhalations of the composition of the invention.
- Each container is provided with the following: val-isatoribine 0.0120 g trichloromonofluoromethane 1.6960 g dichlorodifluoromethane 3.7028g dichlorotetrafluoroethane 1.5766 g total 7.0000 g
- the intravenous formulation is prepared by reconstituting a compound of the invention with an appropriate liquid medium, such as water for injection (WFI) or a 5% dextrose solution.
- a desired concentration of the intravenous formulation can be obtained by reconstituting an appropriate amount of a compound of the invention with an appropriate volume of liquid medium.
- a desired concentration of the intravenous formulation provides a therapeutically effective amount of a compound of the invention to the patient, preferably a mammal, more preferably a human, in need of the intravenous pharmaceutical formulation and maintains a therapeutically effective level of a compound of the invention in the patient.
- the dose which is therapeutically effective will depend on the rate at which the intravenous formulation is delivered to the patient and the concentration of the intravenous formulation.
- one vial containing a composition e.g., 50 mg of a compound of the invention per vial
- a 5% dextrose solution 14 ml of 5% dextrose solution per vial
- the reconstituted solution is incorporated into a dextrose solution in an infusion bag and q.s. to 50 mL, resulting in a solution containing 1 mg/ml of a compound of the invention suitable for intravenous infusion administration.
- the preferred concentration of a compound of the invention in the liquid medium, in the infusion bag is about 0.001 to about 3 mg/ml, preferably about 0.75 to about 1 mg/ml.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Virology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Saccharide Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description
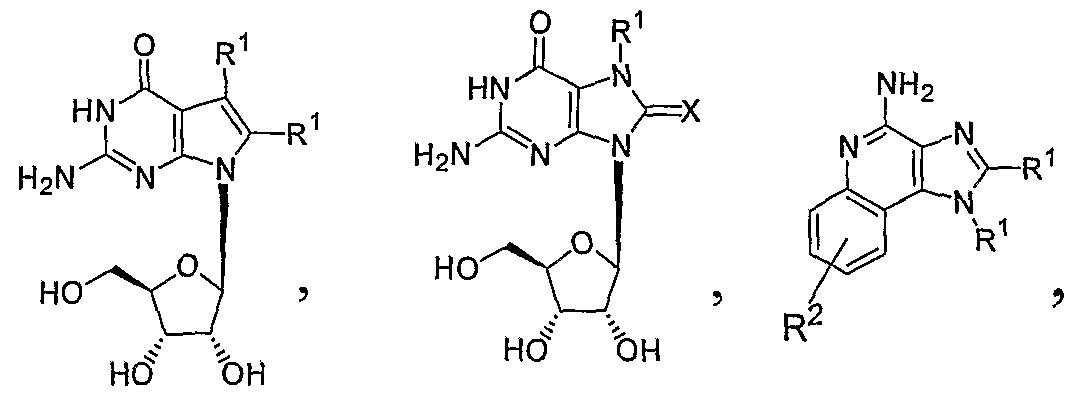









































































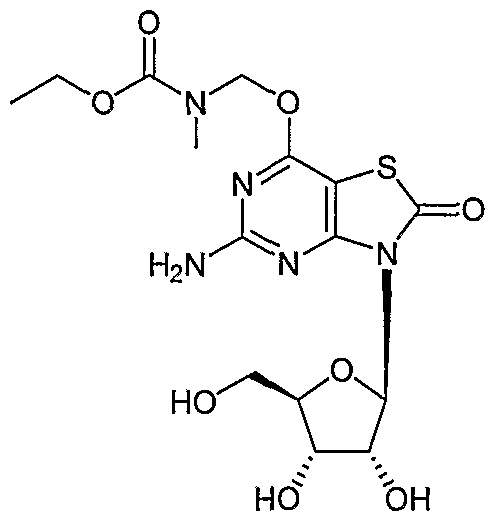


















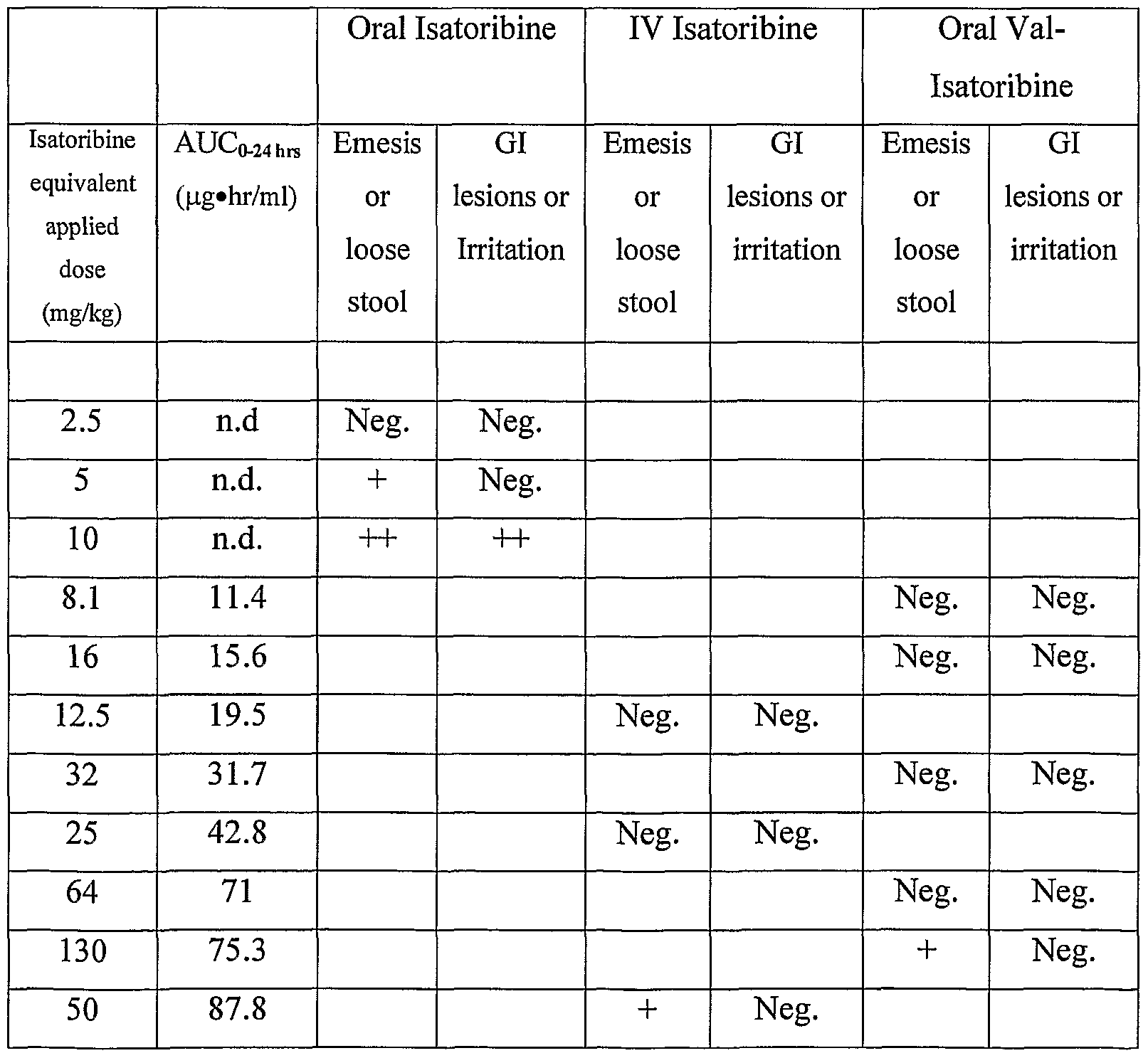


Claims















Priority Applications (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2537450A CA2537450C (en) | 2003-09-05 | 2004-09-01 | Administration of tlr7 ligands and prodrugs thereof for treatment of infection by hepatitis c virus |
| DE602004026891T DE602004026891D1 (en) | 2003-09-05 | 2004-09-01 | TLR7 LIGANDS FOR THE TREATMENT OF HEPATITIS C |
| EP04782670A EP1667694B1 (en) | 2003-09-05 | 2004-09-01 | Tlr7 ligands for the treatment of hepatitis c |
| MXPA06002309A MXPA06002309A (en) | 2003-09-05 | 2004-09-01 | Tlr7 ligands for the treatment of hepatitis c. |
| EA200600540A EA200600540A1 (en) | 2003-09-05 | 2004-09-01 | INTRODUCTION OF TLR7 LIGANDS AND THEIR TREATMENTS FOR THE TREATMENT OF HEPATITIS C VIRUS INFECTION |
| BRPI0414045-1A BRPI0414045A (en) | 2003-09-05 | 2004-09-01 | administration of tlr7 ligands and prodrugs thereof for the treatment of hepatitis C virus infection |
| AU2004271972A AU2004271972B2 (en) | 2003-09-05 | 2004-09-01 | TLR7 ligands for the treatment of hepatitis C |
| AT04782670T ATE465742T1 (en) | 2003-09-05 | 2004-09-01 | TLR7 LIGANDS FOR TREATING HEPATITIS C |
| AP2006003542A AP2006003542A0 (en) | 2003-09-05 | 2004-09-01 | Administration of TLR7 ligands and prodrugs for treatment of infection by hepatitis C virus. |
| CN2004800255325A CN1845745B (en) | 2003-09-05 | 2004-09-01 | Administration of TLR7 ligands and prodrugs thereof for treatment of infection by hepatitis c virus |
| JP2006525384A JP2007504232A (en) | 2003-09-05 | 2004-09-01 | Administration of TLR7 ligand and prodrug thereof for the treatment of hepatitis C virus infection |
| NZ545536A NZ545536A (en) | 2003-09-05 | 2004-09-01 | TLR7 ligands for the treatment of hepatitis C |
| IL173890A IL173890A (en) | 2003-09-05 | 2006-02-23 | Pharmaceutical composition for the treatment of hepatitis c virus infection comprising a tlr7 ligand or a masked tlr7 ligand prodrug and uses of the ligand or prodrug |
| TNP2006000076A TNSN06076A1 (en) | 2003-09-05 | 2006-03-06 | Administration of tlr7 ligands and prodrugs thereof for treatment of infection by hepatitis c virus |
| NO20061183A NO20061183L (en) | 2003-09-05 | 2006-03-14 | Administration of TLR7 ligands and their prodrugs for the treatment of infection with hepatitis C virus |
| EC2006006465A ECSP066465A (en) | 2003-09-05 | 2006-03-30 | ADMINISTRATION OF TLR7 LIGANDS AND SIMILAR DRUGS FOR THE TREATMENT OF HEPATITIS C VIRUS INFECTIONS |
| HK06112114.0A HK1091410A1 (en) | 2003-09-05 | 2006-11-03 | Tlr7 ligands for the treatment of hepatitis c |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US50033903P | 2003-09-05 | 2003-09-05 | |
| US60/500,339 | 2003-09-05 | ||
| US51899603P | 2003-11-10 | 2003-11-10 | |
| US51899703P | 2003-11-10 | 2003-11-10 | |
| US60/518,996 | 2003-11-10 | ||
| US60/518,997 | 2003-11-10 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005025583A2 true WO2005025583A2 (en) | 2005-03-24 |
| WO2005025583A3 WO2005025583A3 (en) | 2005-05-19 |
Family
ID=34317460
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/028236 WO2005025583A2 (en) | 2003-09-05 | 2004-09-01 | Tlr7 ligands for the treatment of hepatitis c |
Country Status (26)
| Country | Link |
|---|---|
| US (4) | US7576068B2 (en) |
| EP (1) | EP1667694B1 (en) |
| JP (1) | JP2007504232A (en) |
| KR (1) | KR20060096415A (en) |
| AP (1) | AP2006003542A0 (en) |
| AT (1) | ATE465742T1 (en) |
| AU (1) | AU2004271972B2 (en) |
| BR (1) | BRPI0414045A (en) |
| CA (1) | CA2537450C (en) |
| CR (1) | CR8273A (en) |
| DE (1) | DE602004026891D1 (en) |
| EA (1) | EA200600540A1 (en) |
| EC (1) | ECSP066465A (en) |
| ES (1) | ES2342069T4 (en) |
| GE (1) | GEP20084545B (en) |
| HK (1) | HK1091410A1 (en) |
| IL (1) | IL173890A (en) |
| MA (1) | MA28082A1 (en) |
| MX (1) | MXPA06002309A (en) |
| NO (1) | NO20061183L (en) |
| NZ (1) | NZ545536A (en) |
| OA (1) | OA13310A (en) |
| SG (1) | SG146637A1 (en) |
| TN (1) | TNSN06076A1 (en) |
| TW (1) | TWI347189B (en) |
| WO (1) | WO2005025583A2 (en) |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1550662A1 (en) * | 2002-09-27 | 2005-07-06 | Sumitomo Pharmaceuticals Company, Limited | Novel adenine compound and use thereof |
| WO2006117670A1 (en) * | 2005-05-04 | 2006-11-09 | Pfizer Limited | 2-amido-6-amino-8-oxopurine derivatives as toll-like receptor modulators for the treatment of cancer and viral infections, such as hepatitis c |
| WO2007038720A2 (en) * | 2005-09-27 | 2007-04-05 | Coley Pharmaceutical Gmbh | Modulation of tlr-mediated immune responses using adaptor oligonucleotides |
| WO2007093901A1 (en) * | 2006-02-17 | 2007-08-23 | Pfizer Limited | 3 -deazapurine derivatives as tlr7 modulators |
| EP1951914A2 (en) * | 2005-11-09 | 2008-08-06 | Cheng Si Yuan (China-International) Hepatitis Research Foundation | Diagnostic and therapeutic methods and agents |
| JP2009501546A (en) * | 2005-07-18 | 2009-01-22 | ノバルティス アーゲー | A small animal model for HCV replication |
| JP2009504803A (en) * | 2005-08-22 | 2009-02-05 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | TLR agonist |
| WO2010055340A1 (en) * | 2008-11-12 | 2010-05-20 | Ludwig Institute For Cancer Research | Use of inkt or tlr agonists for protecting against or treating a disease such as acute infection or cancer |
| JP2010516802A (en) * | 2007-01-31 | 2010-05-20 | チョンシー ユー | Positively charged water-soluble prodrugs of 1H-imidazo [4,5-c] quinolin-4-amine and related compounds with very high skin permeability |
| JP2010522151A (en) * | 2007-03-19 | 2010-07-01 | アストラゼネカ・アクチエボラーグ | 9-Substituted-8-oxo-adenine compounds as TOLL-like receptor (TLR7) modulators |
| WO2010116248A1 (en) | 2009-04-10 | 2010-10-14 | Novartis Ag | Organic compounds and their uses |
| WO2010115981A1 (en) | 2009-04-10 | 2010-10-14 | Novartis Ag | 7-azadispiro [3.0.4.1] decane-8-carboxamides as hepatitis c virus inhibitors |
| US7998492B2 (en) | 2002-10-29 | 2011-08-16 | Coley Pharmaceutical Group, Inc. | Methods and products related to treatment and prevention of hepatitis C virus infection |
| US8012964B2 (en) | 2004-03-26 | 2011-09-06 | Dainippon Sumitomo Pharma Co., Ltd. | 9-substituted 8-oxoadenine compound |
| US8067426B2 (en) | 2008-08-11 | 2011-11-29 | Glaxosmithkline Llc | 6-amino-purin-8-one compounds |
| US8138172B2 (en) | 2006-07-05 | 2012-03-20 | Astrazeneca Ab | 8-oxoadenine derivatives acting as modulators of TLR7 |
| WO2012048235A1 (en) | 2010-10-08 | 2012-04-12 | Novartis Ag | Vitamin e formulations of sulfamide ns3 inhibitors |
| EP2518079A2 (en) | 2006-04-11 | 2012-10-31 | Novartis AG | HCV/HIV inhibitors and their uses |
| US8357374B2 (en) | 2007-02-07 | 2013-01-22 | The Regents Of The University Of California | Conjugates of synthetic TLR agonists and uses therefor |
| US8466159B2 (en) | 2011-10-21 | 2013-06-18 | Abbvie Inc. | Methods for treating HCV |
| US8492386B2 (en) | 2011-10-21 | 2013-07-23 | Abbvie Inc. | Methods for treating HCV |
| US8563717B2 (en) | 2008-08-11 | 2013-10-22 | Glaxosmithkline Llc | Adenine derivatives |
| US8575181B2 (en) | 2008-08-11 | 2013-11-05 | Glaxosmithkline Llc | Purine derivatives for use in the treatment of allergic, inflammatory and infectious diseases |
| US8575340B2 (en) | 2010-02-10 | 2013-11-05 | Glaxosmithkline Llc | Purine derivatives and their pharmaceutical uses |
| EP2700638A1 (en) * | 2006-05-31 | 2014-02-26 | The Regents Of the University of California | Purine analogs |
| US8729088B2 (en) | 2009-02-11 | 2014-05-20 | The Regents Of The University Of California | Toll-like receptor modulators and treatment of diseases |
| US8802684B2 (en) | 2008-08-11 | 2014-08-12 | Glaxosmithkline Llc | Adenine derivatives |
| US8809265B2 (en) | 2011-10-21 | 2014-08-19 | Abbvie Inc. | Methods for treating HCV |
| US8853176B2 (en) | 2011-10-21 | 2014-10-07 | Abbvie Inc. | Methods for treating HCV |
| US8895570B2 (en) | 2010-12-17 | 2014-11-25 | Astrazeneca Ab | Purine derivatives |
| US9045472B2 (en) | 2010-12-16 | 2015-06-02 | Astrazeneca Ab | Imidazoquinoline compounds |
| US9050319B2 (en) | 2010-04-30 | 2015-06-09 | Telormedix, Sa | Phospholipid drug analogs |
| US9066940B2 (en) | 2009-02-06 | 2015-06-30 | Telormedix, Sa | Pharmaceutical compositions comprising imidazoquinolin(amines) and derivatives thereof suitable for local administration |
| US9173936B2 (en) | 2010-04-30 | 2015-11-03 | Telormedix Sa | Phospholipid drug analogs |
| US9428512B2 (en) | 2012-11-20 | 2016-08-30 | Glaxosmithkline Llc | Compounds |
| US9441008B2 (en) | 2014-12-08 | 2016-09-13 | Hoffmann-La Roche Inc. | 3-substituted 5-amino-6H-thiazolo[4,5-D]pyrimidine-2,7-dione compounds for the treatment and prophylaxis of virus infection |
| US9452217B2 (en) | 2013-06-22 | 2016-09-27 | Nitor Therapeutics | Methods for potentiating immune response for the treatment of infectious diseases and cancer |
| US9540383B2 (en) | 2012-11-20 | 2017-01-10 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9550785B2 (en) | 2012-11-20 | 2017-01-24 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9555036B2 (en) | 2012-08-24 | 2017-01-31 | Glaxosmithkline Llc | Pyrazolopyrimidine compounds |
| WO2017189978A1 (en) | 2016-04-28 | 2017-11-02 | Emory University | Alkyne containing nucleotide and nucleoside therapeutic compositions and uses related thereto |
| WO2018095426A1 (en) | 2016-11-28 | 2018-05-31 | 江苏恒瑞医药股份有限公司 | Pyrazolo-heteroaryl derivative, preparation method and medical use thereof |
| US10065973B2 (en) | 2015-05-12 | 2018-09-04 | Hoffmann-La Roche Inc. | Substituted aminothiazolopyrimidinedione for the treatment and prophylaxis of virus infection |
| US10112946B2 (en) | 2011-07-22 | 2018-10-30 | Glaxosmithkline Llc | Composition |
| WO2018210298A1 (en) | 2017-05-18 | 2018-11-22 | 江苏恒瑞医药股份有限公司 | Heteroaryl-pyrazole derivative, and preparation method therefor and medical application thereof |
| US10183954B2 (en) | 2015-05-08 | 2019-01-22 | Hoffmann-La Roche Inc. | Oxathiolane carboxylic acids and derivatives for the treatment and prophylaxis of virus infection |
| WO2019223773A1 (en) | 2018-05-25 | 2019-11-28 | 江苏恒瑞医药股份有限公司 | Crystal form of hydrochloride of pyrazoloheteroaryl derivative and preparation method |
| US10533005B2 (en) | 2017-02-17 | 2020-01-14 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10533007B2 (en) | 2016-04-19 | 2020-01-14 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10556903B2 (en) | 2016-04-19 | 2020-02-11 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10590146B2 (en) | 2015-06-30 | 2020-03-17 | Hoffmann-La Roche Inc. | Substituted aminothiazolopyrimidinediones |
| US10703755B2 (en) | 2016-04-26 | 2020-07-07 | Sumitomo Dainippon Pharma Co., Ltd. | Substituted purine derivative |
| US10780105B2 (en) | 2014-06-24 | 2020-09-22 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10780180B2 (en) | 2014-01-10 | 2020-09-22 | Birdie Biopharmaceuticals, Inc. | Compounds and compositions for immunotherapy |
| US11046781B2 (en) | 2016-01-07 | 2021-06-29 | Birdie Biopharmaceuticals, Inc. | Anti-HER2 combinations for treating tumors |
| US11053240B2 (en) | 2017-04-27 | 2021-07-06 | Birdie Biopharmaceuticals, Inc. | 2-amino-quinoline derivatives |
| US11130812B2 (en) | 2014-09-01 | 2021-09-28 | Birdie Biopharmaceuticals, Inc. | Anti PD-L1 conjugates for treating tumors |
| US11136397B2 (en) | 2016-01-07 | 2021-10-05 | Birdie Pharmaceuticals, Inc. | Anti-EGFR combinations for treating tumors |
| US11142534B2 (en) | 2017-01-06 | 2021-10-12 | Hoffmann-La Roche Inc. | Process for the preparation of 3-substituted 5-amino-6H-thiazolo[4,5-d]pyrimidine-2,7-dione compounds |
| US11220552B2 (en) | 2016-01-07 | 2022-01-11 | Birdie Biopharmaceuticals, Inc. | Anti-CD20 combinations for treating tumors |
| US11279761B2 (en) | 2014-07-09 | 2022-03-22 | Birdie Biopharmaceuticals, Inc. | Anti-PD-L1 combinations for treating tumors |
| US11517567B2 (en) | 2017-06-23 | 2022-12-06 | Birdie Biopharmaceuticals, Inc. | Pharmaceutical compositions |
| US11697851B2 (en) | 2016-05-24 | 2023-07-11 | The Regents Of The University Of California | Early ovarian cancer detection diagnostic test based on mRNA isoforms |
| US11771699B2 (en) | 2015-03-16 | 2023-10-03 | Hoffmann-La Roche Inc. | Combined treatment with a TLR7 agonist and an HBV capsid assembly inhibitor |
| EP4003369A4 (en) * | 2019-07-27 | 2023-10-18 | Brii Biosciences, Inc. | Adenosine derivative and pharmaceutical composition comprising the same |
| US11890297B2 (en) | 2021-01-25 | 2024-02-06 | Brii Biosciences, Inc. | Combination therapy for HIV with adenosine derivative and capsid inhibitors |
Families Citing this family (151)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6207646B1 (en) * | 1994-07-15 | 2001-03-27 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| US20030026782A1 (en) * | 1995-02-07 | 2003-02-06 | Arthur M. Krieg | Immunomodulatory oligonucleotides |
| UA67760C2 (en) * | 1997-12-11 | 2004-07-15 | Міннесота Майнінг Енд Мануфакчурінг Компані | Imidazonaphthyridines and use thereof to induce the biosynthesis of cytokines |
| EP1176966B1 (en) * | 1999-04-12 | 2013-04-03 | THE GOVERNMENT OF THE UNITED STATES OF AMERICA, as represented by THE SECRETARY, DEPARTMENT OF HEALTH AND HUMAN SERVICES | Oligodeoxynucleotide and its use to induce an immune response |
| US6331539B1 (en) * | 1999-06-10 | 2001-12-18 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| US20030129251A1 (en) | 2000-03-10 | 2003-07-10 | Gary Van Nest | Biodegradable immunomodulatory formulations and methods for use thereof |
| US6545016B1 (en) | 2000-12-08 | 2003-04-08 | 3M Innovative Properties Company | Amide substituted imidazopyridines |
| UA74852C2 (en) * | 2000-12-08 | 2006-02-15 | 3M Innovative Properties Co | Urea-substituted imidazoquinoline ethers |
| US6664265B2 (en) | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US6667312B2 (en) * | 2000-12-08 | 2003-12-23 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| US6664264B2 (en) * | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| US6677347B2 (en) * | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamido ether substituted imidazoquinolines |
| CA2598144A1 (en) * | 2000-12-08 | 2006-08-31 | 3M Innovative Properties Company | Compositions and methods for targeted delivery of immune response modifiers |
| US6660735B2 (en) * | 2000-12-08 | 2003-12-09 | 3M Innovative Properties Company | Urea substituted imidazoquinoline ethers |
| JP4598389B2 (en) * | 2001-06-21 | 2010-12-15 | ダイナバックス テクノロジーズ コーポレイション | Chimeric immunomodulatory compounds and methods of use thereof |
| US7321033B2 (en) * | 2001-11-27 | 2008-01-22 | Anadys Pharmaceuticals, Inc. | 3-B-D-ribofuranosylthiazolo [4,5-d] pyrimidine nucleosides and uses thereof |
| NZ537054A (en) | 2002-06-07 | 2006-10-27 | 3M Innovative Properties Co | Ether substituted imidazopyridines |
| WO2004058759A1 (en) | 2002-12-20 | 2004-07-15 | 3M Innovative Properties Company | Aryl / hetaryl substituted imidazoquinolines |
| US20040265351A1 (en) | 2003-04-10 | 2004-12-30 | Miller Richard L. | Methods and compositions for enhancing immune response |
| WO2004096144A2 (en) * | 2003-04-28 | 2004-11-11 | 3M Innovative Properties Company | Compositions and methods for induction of opioid receptors |
| MXPA06001669A (en) * | 2003-08-12 | 2006-04-28 | 3M Innovative Properties Co | Oxime substituted imidazo-containing compounds. |
| CA2535338C (en) * | 2003-08-14 | 2013-05-28 | 3M Innovative Properties Company | Substituted 1h-imidazo[4,5-c]pyridin-4-amines,1h-imidazo[4,5-c]quinolin -4-amines and 1h-imidazo[4,5-c]naphthyridin-4-amines as immune response modifiers |
| US8961477B2 (en) * | 2003-08-25 | 2015-02-24 | 3M Innovative Properties Company | Delivery of immune response modifier compounds |
| US7897597B2 (en) | 2003-08-27 | 2011-03-01 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted imidazoquinolines |
| WO2005023190A2 (en) * | 2003-09-05 | 2005-03-17 | 3M Innovative Properties Company | Treatment for cd5+ b cell lymphoma |
| US7615539B2 (en) * | 2003-09-25 | 2009-11-10 | Coley Pharmaceutical Group, Inc. | Nucleic acid-lipophilic conjugates |
| US20090075980A1 (en) * | 2003-10-03 | 2009-03-19 | Coley Pharmaceutical Group, Inc. | Pyrazolopyridines and Analogs Thereof |
| CA2540598C (en) | 2003-10-03 | 2013-09-24 | 3M Innovative Properties Company | Pyrazolopyridines and analogs thereof |
| US8871782B2 (en) | 2003-10-03 | 2014-10-28 | 3M Innovative Properties Company | Alkoxy substituted imidazoquinolines |
| US7544697B2 (en) * | 2003-10-03 | 2009-06-09 | Coley Pharmaceutical Group, Inc. | Pyrazolopyridines and analogs thereof |
| CA2545825A1 (en) | 2003-11-14 | 2005-06-02 | 3M Innovative Properties Company | Hydroxylamine substituted imidazo ring compounds |
| CA2545774A1 (en) * | 2003-11-14 | 2005-06-02 | 3M Innovative Properties Company | Oxime substituted imidazo ring compounds |
| AU2004293078B2 (en) * | 2003-11-25 | 2012-01-19 | 3M Innovative Properties Company | Substituted imidazo ring systems and methods |
| WO2005051324A2 (en) * | 2003-11-25 | 2005-06-09 | 3M Innovative Properties Company | Hydroxylamine and oxime substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
| US20050226878A1 (en) * | 2003-12-02 | 2005-10-13 | 3M Innovative Properties Company | Therapeutic combinations and methods including IRM compounds |
| AU2004315771A1 (en) * | 2003-12-04 | 2005-08-25 | 3M Innovative Properties Company | Sulfone substituted imidazo ring ethers |
| CA2552101A1 (en) * | 2003-12-29 | 2005-07-21 | 3M Innovative Properties Company | Piperazine, [1,4]diazepane, [1,4]diazocane, and [1,5]diazocane fused imidazo ring compounds |
| EP1701955A1 (en) * | 2003-12-29 | 2006-09-20 | 3M Innovative Properties Company | Arylalkenyl and arylalkynyl substituted imidazoquinolines |
| CA2551399A1 (en) * | 2003-12-30 | 2005-07-21 | 3M Innovative Properties Company | Imidazoquinolinyl, imidazopyridinyl, and imidazonaphthyridinyl sulfonamides |
| EP1730143A2 (en) * | 2004-03-24 | 2006-12-13 | 3M Innovative Properties Company | Amide substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines |
| CN101426524A (en) * | 2004-04-28 | 2009-05-06 | 3M创新有限公司 | Compositions and methods for mucosal vaccination |
| US20050267145A1 (en) * | 2004-05-28 | 2005-12-01 | Merrill Bryon A | Treatment for lung cancer |
| WO2005123079A2 (en) * | 2004-06-14 | 2005-12-29 | 3M Innovative Properties Company | Urea substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines |
| WO2005123080A2 (en) * | 2004-06-15 | 2005-12-29 | 3M Innovative Properties Company | Nitrogen-containing heterocyclyl substituted imidazoquinolines and imidazonaphthyridines |
| US7884207B2 (en) * | 2004-06-18 | 2011-02-08 | 3M Innovative Properties Company | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
| US7915281B2 (en) * | 2004-06-18 | 2011-03-29 | 3M Innovative Properties Company | Isoxazole, dihydroisoxazole, and oxadiazole substituted imidazo ring compounds and method |
| WO2006009826A1 (en) * | 2004-06-18 | 2006-01-26 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted thiazoloquinolines and thiazolonaphthyridines |
| US8541438B2 (en) | 2004-06-18 | 2013-09-24 | 3M Innovative Properties Company | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
| WO2006038923A2 (en) * | 2004-06-18 | 2006-04-13 | 3M Innovative Properties Company | Aryl substituted imidazonaphthyridines |
| WO2006026470A2 (en) * | 2004-08-27 | 2006-03-09 | 3M Innovative Properties Company | Hiv immunostimulatory compositions |
| WO2006026760A2 (en) * | 2004-09-02 | 2006-03-09 | 3M Innovative Properties Company | 1-amino imidazo-containing compounds and methods |
| CA2578741C (en) * | 2004-09-02 | 2014-01-14 | 3M Innovative Properties Company | 1-alkoxy 1h-imidazo ring systems and methods |
| EP1784180A4 (en) | 2004-09-02 | 2009-07-22 | 3M Innovative Properties Co | 2-amino 1h imidazo ring systems and methods |
| EP1804583A4 (en) * | 2004-10-08 | 2009-05-20 | 3M Innovative Properties Co | Adjuvant for dna vaccines |
| WO2006063072A2 (en) * | 2004-12-08 | 2006-06-15 | 3M Innovative Properties Company | Immunomodulatory compositions, combinations and methods |
| JP2008526751A (en) * | 2004-12-30 | 2008-07-24 | 武田薬品工業株式会社 | 1- (2-methylpropyl) -1H-imidazo [4,5-C] [1,5] naphthyridin-4-amineethanesulfonate and 1- (2-methylpropyl) -1H-imidazo [4,5- C] [1,5] naphthyridin-4-amine methanesulfonate |
| EP1830876B1 (en) * | 2004-12-30 | 2015-04-08 | Meda AB | Use of imiquimod for the treatment of cutaneous metastases derived from a breast cancer tumor |
| US8436176B2 (en) * | 2004-12-30 | 2013-05-07 | Medicis Pharmaceutical Corporation | Process for preparing 2-methyl-1-(2-methylpropyl)-1H-imidazo[4,5-c][1,5]naphthyridin-4-amine |
| JP5313502B2 (en) | 2004-12-30 | 2013-10-09 | スリーエム イノベイティブ プロパティズ カンパニー | Substituted chiral condensed [1,2] imidazo [4,5-c] cyclic compounds |
| AU2005322898B2 (en) | 2004-12-30 | 2011-11-24 | 3M Innovative Properties Company | Chiral fused (1,2)imidazo(4,5-c) ring compounds |
| AU2006210392A1 (en) | 2005-02-04 | 2006-08-10 | Coley Pharmaceutical Group, Inc. | Aqueous gel formulations containing immune response modifiers |
| CA2597324C (en) | 2005-02-09 | 2015-06-30 | Coley Pharmaceutical Group, Inc. | Alkyloxy substituted thiazoloquinolines and thiazolonaphthyridines |
| JP2008530252A (en) | 2005-02-09 | 2008-08-07 | コーリー ファーマシューティカル グループ,インコーポレイテッド | Thiazolo [4,5-c] ring compounds and methods substituted with oximes and hydroxylamines |
| EP1845988A2 (en) | 2005-02-11 | 2007-10-24 | 3M Innovative Properties Company | Substituted imidazoquinolines and imidazonaphthyridines |
| JP2008530113A (en) | 2005-02-11 | 2008-08-07 | コーリー ファーマシューティカル グループ,インコーポレイテッド | Oxime and hydroxyramine substituted imidazo [4,5-c] ring compounds and methods |
| EP1851220A2 (en) | 2005-02-23 | 2007-11-07 | 3M Innovative Properties Company | Hydroxyalkyl substituted imidazonaphthyridines |
| AU2006223634A1 (en) | 2005-02-23 | 2006-09-21 | Coley Pharmaceutical Group, Inc. | Hydroxyalkyl substituted imidazoquinolines |
| JP2008538203A (en) | 2005-02-23 | 2008-10-16 | コーリー ファーマシューティカル グループ,インコーポレイテッド | A method for preferentially inducing biosynthesis of interferon |
| JP2008531567A (en) | 2005-02-23 | 2008-08-14 | コーリー ファーマシューティカル グループ,インコーポレイテッド | Hydroxyalkyl-substituted imidazoquinoline compounds and methods |
| CN101175493A (en) | 2005-03-14 | 2008-05-07 | 3M创新有限公司 | Method of treating actinic keratosis |
| AU2006232377A1 (en) | 2005-04-01 | 2006-10-12 | Coley Pharmaceutical Group, Inc. | Pyrazolopyridine-1,4-diamines and analogs thereof |
| WO2006107851A1 (en) | 2005-04-01 | 2006-10-12 | Coley Pharmaceutical Group, Inc. | 1-substituted pyrazolo (3,4-c) ring compounds as modulators of cytokine biosynthesis for the treatment of viral infections and neoplastic diseases |
| JP2008539252A (en) * | 2005-04-25 | 2008-11-13 | スリーエム イノベイティブ プロパティズ カンパニー | Immune activation composition |
| ZA200803029B (en) * | 2005-09-09 | 2009-02-25 | Coley Pharm Group Inc | Amide and carbamate derivatives of alkyl substituted /V-[4-(4-amino-1H-imidazo[4,5-c] quinolin-1-yl)butyl] methane-sulfonamides and methods |
| US8476292B2 (en) | 2005-09-09 | 2013-07-02 | 3M Innovative Properties Company | Amide and carbamate derivatives of N-{2-[4-amino-2-(ethoxymethyl)-1H-imidazo[4,5-c] quinolin-1-Yl]-1,1-dimethylethyl}methanesulfonamide and methods |
| EP1948173B1 (en) | 2005-11-04 | 2013-07-17 | 3M Innovative Properties Company | Hydroxy and alkoxy substituted 1h-imidazoquinolines and methods |
| CA2630463C (en) | 2005-11-21 | 2015-01-06 | Anadys Pharmaceuticals, Inc. | Novel process for the preparation of 5-amino-3h-thiazolo[4,5-d]pyrimidin-2-one |
| US8951528B2 (en) | 2006-02-22 | 2015-02-10 | 3M Innovative Properties Company | Immune response modifier conjugates |
| WO2007106854A2 (en) | 2006-03-15 | 2007-09-20 | Coley Pharmaceutical Group, Inc. | Hydroxy and alkoxy substituted 1h-imidazonaphthyridines and methods |
| AU2006342608A1 (en) * | 2006-04-25 | 2007-11-01 | Intercell Ag | HCV vaccinations |
| EP2023722A4 (en) * | 2006-05-15 | 2012-01-18 | Univ Kentucky | Toll-like receptor (tlr) stimulation for ocular angiogenesis and macular degeneration |
| CL2007001427A1 (en) * | 2006-05-22 | 2008-05-16 | Novartis Ag | 5-AMINO-3- MALEATE SALT (2 ', 3'-DI-O-ACETYL-BETA-D-RIBOFURANOSIL) -3H-TIAZOLO [4,5-D] PIRIMIDIN-2-ONA; PREPARATION PROCEDURE; PHARMACEUTICAL COMPOSITION THAT INCLUDES SUCH COMPOUND; AND USE OF THE COMPOUND FOR THE TREATMENT OF A PO INFECTION |
| JP5345527B2 (en) * | 2006-06-22 | 2013-11-20 | アナディス ファーマシューティカルズ インク | Prodrug 5-amino-3- (3'-deoxy-β-D-ribofuranosyl) -thiazolo [4,5-d] pyrimidine-2,7-dione |
| US7906506B2 (en) * | 2006-07-12 | 2011-03-15 | 3M Innovative Properties Company | Substituted chiral fused [1,2] imidazo [4,5-c] ring compounds and methods |
| DE602007012881D1 (en) * | 2006-07-18 | 2011-04-14 | Anadys Pharmaceuticals Inc | Carbonat- und carbamat-prodrugs von thiazolo ä4,5-dü-pyrimidinen |
| WO2008030511A2 (en) | 2006-09-06 | 2008-03-13 | Coley Pharmaceuticial Group, Inc. | Substituted 3,4,6,7-tetrahydro-5h, 1,2a,4a,8-tetraazacyclopenta[cd]phenalenes |
| BRPI0717741A2 (en) * | 2006-10-17 | 2014-04-08 | Anadys Pharmaceuticals Inc | PREPARATION METHODS, METHOD OF PREPARATION, SUBSTITUTED SULFONIL COMPOUND REDUCTION METHOD, PHARMACEUTICAL COMPOSITION TREATMENT OR DISEASE PREVENTION |
| TW200831105A (en) * | 2006-12-14 | 2008-08-01 | Astrazeneca Ab | Novel compounds |
| US20080149123A1 (en) * | 2006-12-22 | 2008-06-26 | Mckay William D | Particulate material dispensing hairbrush with combination bristles |
| CL2008000496A1 (en) * | 2007-02-19 | 2008-09-22 | Smithkline Beecham Corp | COMPOUNDS DERIVED FROM PURINA; PROCEDURE FOR PREPARATION OF SUCH COMPOUNDS; PHARMACEUTICAL COMPOSITION THAT INCLUDES SUCH COMPOUNDS; AND ITS USE TO TREAT INFECTIOUS DISEASES, CANCER, ALLERGIES AND OTHER INFLAMMATORY AFFECTIONS. |
| WO2008114006A1 (en) * | 2007-03-19 | 2008-09-25 | Astrazeneca Ab | 9-substituted-8-oxo-adenine compounds as toll-like receptor (tlr7) modulators |
| PE20081887A1 (en) * | 2007-03-20 | 2009-01-16 | Dainippon Sumitomo Pharma Co | NEW ADENINE COMPOUND |
| EP2138497A4 (en) * | 2007-03-20 | 2012-01-04 | Dainippon Sumitomo Pharma Co | Novel adenine compound |
| WO2008135791A1 (en) * | 2007-05-08 | 2008-11-13 | Astrazeneca Ab | Imidazoquinolines with immuno-modulating properties |
| US20110166092A1 (en) * | 2007-08-20 | 2011-07-07 | Anadys Pharmaceuticals, Inc. | Dosing methods for treating disease |
| PE20091236A1 (en) * | 2007-11-22 | 2009-09-16 | Astrazeneca Ab | PYRIMIDINE DERIVATIVES AS IMMUNOMODULATORS OF TLR7 |
| PE20091156A1 (en) * | 2007-12-17 | 2009-09-03 | Astrazeneca Ab | SALTS OF (3 - {[[3- (6-AMINO-2-BUTOXY-8-OXO-7,8-DIHIDRO-9H-PURIN-9-IL) PROPYL] (3-MORFOLIN-4-ILPROPIL) AMINO] METHYL} PHENYL) METHYL ACETATE |
| EA201001264A1 (en) * | 2008-02-07 | 2011-04-29 | Дзе Регентс Оф Дзе Юниверсити Оф Калифорния | METHOD FOR TREATING URINARY BUBBLE DISEASES WITH TLR7 Activator |
| WO2010050889A1 (en) * | 2008-10-31 | 2010-05-06 | Moberg Derma Ab | Topical composition comprising a combination of at least two penetration enhancing agents |
| ES2623794T3 (en) | 2008-12-09 | 2017-07-12 | Gilead Sciences, Inc. | Intermediates for the preparation of toll receptor modulators |
| MX2011006088A (en) | 2008-12-09 | 2011-06-21 | Coley Pharm Group Inc | Immunostimulatory oligonucleotides. |
| US8552165B2 (en) * | 2008-12-09 | 2013-10-08 | Heather Davis | Immunostimulatory oligonucleotides |
| WO2010091265A1 (en) * | 2009-02-05 | 2010-08-12 | Yale University | Compounds and methods for treating viral encephalitis |
| CN102482233A (en) | 2009-05-21 | 2012-05-30 | 阿斯利康(瑞典)有限公司 | Novel Pyrimidine Derivatives And Their Use In The Treatment Of Cancer And Further Diseases |
| GB0908772D0 (en) * | 2009-05-21 | 2009-07-01 | Astrazeneca Ab | New salts 756 |
| WO2011068233A1 (en) * | 2009-12-03 | 2011-06-09 | Dainippon Sumitomo Pharma Co., Ltd. | Imidazoquinolines which act via toll - like receptors (tlr) |
| WO2011101332A1 (en) | 2010-02-16 | 2011-08-25 | Proyecto De Biomedicina Cima, S.L. | Compositions based on the fibronectin extracellular domain a for the treatment of melanoma |
| EP3222621B1 (en) | 2010-08-17 | 2023-03-08 | 3M Innovative Properties Company | Lipidated immune response modifier compound and its medical use |
| US9422250B2 (en) | 2011-04-08 | 2016-08-23 | Janssen Sciences Ireland Uc | Pyrimidine derivatives for the treatment of viral infections |
| JP6415979B2 (en) | 2011-06-03 | 2018-10-31 | スリーエム イノベイティブ プロパティズ カンパニー | Hydrazino 1H-imidazoquinolin-4-amine and complexes prepared therefrom |
| MX347240B (en) | 2011-06-03 | 2017-04-20 | 3M Innovative Properties Co | Heterobifunctional linkers with polyethylene glycol segments and immune response modifier conjugates made therefrom. |
| PT2776439T (en) | 2011-11-09 | 2018-11-02 | Janssen Sciences Ireland Uc | Purine derivatives for the treatment of viral infections |
| AR090699A1 (en) * | 2012-04-18 | 2014-12-03 | Biocryst Pharm Inc | INHIBITING COMPOUNDS OF VIRAL POLYMERASE RNA ACTIVITY |
| CN104302628B (en) | 2012-05-18 | 2017-06-23 | 大日本住友制药株式会社 | Carboxylic acid compound |
| WO2014004704A1 (en) * | 2012-06-26 | 2014-01-03 | California Institute Of Technology | Systems and methods for implementing bulk metallic glass-based macroscale gears |
| AU2013288600B2 (en) | 2012-07-13 | 2017-06-29 | Janssen Sciences Ireland Uc | Macrocyclic purines for the treatment of viral infections |
| CN103566377A (en) | 2012-07-18 | 2014-02-12 | 上海博笛生物科技有限公司 | Targeted immunotherapy for cancer |
| KR102217111B1 (en) | 2012-10-10 | 2021-02-18 | 얀센 사이언시즈 아일랜드 언리미티드 컴퍼니 | Pyrrolo[3,2-d]pyrimidine derivatives for the treatment of viral infections and other diseases |
| NZ706226A (en) | 2012-11-16 | 2019-09-27 | Janssen Sciences Ireland Uc | Heterocyclic substituted 2-amino-quinazoline derivatives for the treatment of viral infections |
| EP2958900B1 (en) | 2013-02-21 | 2019-04-10 | Janssen Sciences Ireland Unlimited Company | 2-aminopyrimidine derivatives for the treatment of viral infections |
| EA202090547A3 (en) * | 2013-03-29 | 2020-12-30 | Янссен Сайенсиз Айрлэнд Юси | MACROCYCLIC DEASE-OXIPURINS FOR TREATMENT OF VIRAL INFECTIONS |
| ES2657283T3 (en) | 2013-05-24 | 2018-03-02 | Janssen Sciences Ireland Uc | Pyridone derivatives for the treatment of viral infections and additional diseases |
| SG10201803331PA (en) | 2013-06-27 | 2018-06-28 | Janssen Sciences Ireland Uc | Pyrrolo[3,2-d]pyrimidine derivatives for the treatment of viral infections and other diseases |
| BR112016001570B1 (en) | 2013-07-30 | 2020-12-15 | Janssen Sciences Ireland Uc | TIENO DERIVATIVES [3,2-D] PYRIMIDINES AND PHARMACEUTICAL COMPOSITION THAT UNDERSTANDS THEM FOR THE TREATMENT OF VIRAL INFECTIONS |
| MA39898B1 (en) * | 2014-04-22 | 2020-08-31 | Hoffmann La Roche | 4-amino-imidazoquinoline compounds |
| CN105233291A (en) * | 2014-07-09 | 2016-01-13 | 博笛生物科技有限公司 | Combined therapy composition and combined therapy method for treating cancers |
| CA2954056C (en) | 2014-07-11 | 2020-04-28 | Gilead Sciences, Inc. | Modulators of toll-like receptors for the treatment of hiv |
| JP2017526730A (en) | 2014-09-16 | 2017-09-14 | ギリアード サイエンシーズ, インコーポレイテッド | Solid form of Toll-like receptor modulator |
| KR101729236B1 (en) | 2015-06-01 | 2017-04-21 | (주)노터스생명과학 | TLR7 agonist agent for treatment and prevention of liver disease |
| SG11201811448RA (en) | 2016-07-01 | 2019-01-30 | Janssen Sciences Ireland Unlimited Co | Dihydropyranopyrimidines for the treatment of viral infections |
| KR20160141683A (en) | 2016-09-21 | 2016-12-09 | (주)노터스생명과학 | TLR7 agonist agent for treatment and prevention of liver disease |
| AU2017335205B2 (en) | 2016-09-29 | 2021-11-04 | Janssen Sciences Ireland Unlimited Company | Pyrimidine prodrugs for the treatment of viral infections and further diseases |
| KR20170010887A (en) | 2017-01-20 | 2017-02-01 | (주)노터스생명과학 | TLR7 agonist agent for treatment and prevention of liver disease |
| WO2018162450A1 (en) | 2017-03-06 | 2018-09-13 | Fundación Para La Investigación Médica Aplicada | New inmunostimulatory compositions comprising an entity of cold inducible rna-binding protein with an antigen for the activation of dendritic cells |
| CN111511740B (en) | 2017-12-20 | 2023-05-16 | 3M创新有限公司 | Amide substituted imidazo [4,5-C ] quinoline compounds with branched linking groups for use as immune response modifiers |
| CN108558864B (en) * | 2018-01-30 | 2020-07-03 | 中国医学科学院药用植物研究所 | Acylated derivative of resiquimod, preparation method and application |
| TW201945003A (en) | 2018-03-01 | 2019-12-01 | 愛爾蘭商健生科學愛爾蘭無限公司 | 2,4-diaminoquinazoline derivatives and medical uses thereof |
| BR112020023866A2 (en) | 2018-05-25 | 2021-04-06 | Primmune Therapeutics, Inc. | TLR7 AGONISTS |
| WO2020040327A1 (en) * | 2018-08-23 | 2020-02-27 | 광주과학기술원 | Use of ciclopirox for inhibiting hbv core assembly |
| WO2020051356A1 (en) * | 2018-09-07 | 2020-03-12 | Birdie Biopharmaceuticals, Inc. | Imidazoquinoline compounds and uses thereof |
| KR20220150879A (en) | 2019-11-26 | 2022-11-11 | 프리뮨 테라퓨틱스, 인크. | TLR7 agonist |
| AU2020407871A1 (en) | 2019-12-20 | 2022-06-30 | Nammi Therapeutics, Inc. | Formulated and/or co-formulated liposome compositions containing toll-like receptor ("TLR") agonist prodrugs useful in the treatment of cancer and methods thereof |
| WO2021150613A1 (en) | 2020-01-20 | 2021-07-29 | Incyte Corporation | Spiro compounds as inhibitors of kras |
| CA3174304A1 (en) * | 2020-04-02 | 2021-10-07 | Ashish Kumar Pathak | Novel 2-pyrimidone analogs as potent antiviral agents against alphaviruses |
| US11739102B2 (en) | 2020-05-13 | 2023-08-29 | Incyte Corporation | Fused pyrimidine compounds as KRAS inhibitors |
| US11999752B2 (en) | 2020-08-28 | 2024-06-04 | Incyte Corporation | Vinyl imidazole compounds as inhibitors of KRAS |
| WO2022072783A1 (en) | 2020-10-02 | 2022-04-07 | Incyte Corporation | Bicyclic dione compounds as inhibitors of kras |
| US12077539B2 (en) | 2021-03-22 | 2024-09-03 | Incyte Corporation | Imidazole and triazole KRAS inhibitors |
| WO2023049697A1 (en) | 2021-09-21 | 2023-03-30 | Incyte Corporation | Hetero-tricyclic compounds as inhibitors of kras |
| WO2023056421A1 (en) | 2021-10-01 | 2023-04-06 | Incyte Corporation | Pyrazoloquinoline kras inhibitors |
| KR20240101561A (en) | 2021-10-14 | 2024-07-02 | 인사이트 코포레이션 | Quinoline compounds as inhibitors of KRAS |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4880784A (en) * | 1987-12-21 | 1989-11-14 | Brigham Young University | Antiviral methods utilizing ribofuranosylthiazolo[4,5-d]pyrimdine derivatives |
| US5041426A (en) * | 1987-12-21 | 1991-08-20 | Brigham Young University | Immune system enhancing 3-β-d-ribofuranosylthiazolo[4,5-d]pyridimine nucleosides and nucleotides |
| WO2003045968A1 (en) * | 2001-11-27 | 2003-06-05 | Anadys Pharmaceuticals, Inc. | 3-β-D-RIBOFURANOSYLTHIAZOLO[4,5-d]PYRIDIMINE NUCLEOSIDES AND USES THEREOF |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4643992A (en) * | 1982-11-09 | 1987-02-17 | Scripps Clinic And Research Foundation | Modulation of animal cellular responses with compositions containing 8-substituted guanine derivatives |
| US4539205A (en) * | 1982-11-09 | 1985-09-03 | Scripps Clinic And Research Foundation | Modulation of animal cellular responses with compositions containing 8-substituted guanine derivatives |
| US5011828A (en) * | 1985-11-15 | 1991-04-30 | Michael Goodman | Immunostimulating guanine derivatives, compositions and methods |
| GB9015914D0 (en) * | 1990-07-19 | 1990-09-05 | Wellcome Found | Heterocyclic compounds |
| AU5165793A (en) | 1992-10-01 | 1994-04-26 | Mcneilab, Inc. | Derivatives of 7,8-disubstituted guanosines |
| US5395937A (en) | 1993-01-29 | 1995-03-07 | Minnesota Mining And Manufacturing Company | Process for preparing quinoline amines |
| US5994321A (en) * | 1993-12-20 | 1999-11-30 | Aronex Pharmaceuticals, Inc. | Antiviral guanine analogs |
| AU698419B2 (en) * | 1996-07-03 | 1998-10-29 | Dainippon Sumitomo Pharma Co., Ltd. | A novel purine derivative |
| KR100518903B1 (en) | 1996-10-25 | 2005-10-06 | 미네소타 마이닝 앤드 매뉴팩춰링 캄파니 | Immune response modifier compounds for treatment of the th2 mediated and related diseases |
| CA2311742C (en) | 1997-11-28 | 2009-06-16 | Sumitomo Pharmaceuticals Co., Ltd. | 6-amino-9-benzyl-8-hydroxypurine derivatives |
| JPH11180981A (en) * | 1997-12-19 | 1999-07-06 | Sumitomo Pharmaceut Co Ltd | Heterocyclic derivative |
| TW572758B (en) | 1997-12-22 | 2004-01-21 | Sumitomo Pharma | Type 2 helper T cell-selective immune response inhibitors comprising purine derivatives |
| JP4189048B2 (en) * | 1997-12-26 | 2008-12-03 | 大日本住友製薬株式会社 | Heterocyclic compounds |
| MY164523A (en) * | 2000-05-23 | 2017-12-29 | Univ Degli Studi Cagliari | Methods and compositions for treating hepatitis c virus |
| US6664264B2 (en) * | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| US6667312B2 (en) * | 2000-12-08 | 2003-12-23 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| US6545017B1 (en) * | 2000-12-08 | 2003-04-08 | 3M Innovative Properties Company | Urea substituted imidazopyridines |
| US6545016B1 (en) * | 2000-12-08 | 2003-04-08 | 3M Innovative Properties Company | Amide substituted imidazopyridines |
| US6664260B2 (en) * | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Heterocyclic ether substituted imidazoquinolines |
| JP4331944B2 (en) | 2001-04-17 | 2009-09-16 | 大日本住友製薬株式会社 | New adenine derivatives |
| US20050148538A1 (en) * | 2001-07-27 | 2005-07-07 | John Hadden | Adjuvant formulations for bacterial and virus vaccines and method of making same |
| WO2003026589A2 (en) * | 2001-09-28 | 2003-04-03 | Idenix (Cayman) Limited | Methods and compositions for treating hepatitis c virus using 4'-modified nucleosides |
| EP1501850A2 (en) * | 2002-05-06 | 2005-02-02 | Genelabs Technologies, Inc. | Nucleoside derivatives for treating hepatitis c virus infection |
| WO2005016235A2 (en) * | 2003-04-14 | 2005-02-24 | The Regents Of The University Of California | Combined use of impdh inhibitors with toll-like receptor agonists |
-
2004
- 2004-09-01 AU AU2004271972A patent/AU2004271972B2/en not_active Ceased
- 2004-09-01 EP EP04782670A patent/EP1667694B1/en not_active Expired - Lifetime
- 2004-09-01 ES ES04782670T patent/ES2342069T4/en not_active Expired - Lifetime
- 2004-09-01 AT AT04782670T patent/ATE465742T1/en not_active IP Right Cessation
- 2004-09-01 JP JP2006525384A patent/JP2007504232A/en active Pending
- 2004-09-01 MX MXPA06002309A patent/MXPA06002309A/en active IP Right Grant
- 2004-09-01 OA OA1200600076A patent/OA13310A/en unknown
- 2004-09-01 AP AP2006003542A patent/AP2006003542A0/en unknown
- 2004-09-01 DE DE602004026891T patent/DE602004026891D1/en not_active Expired - Lifetime
- 2004-09-01 GE GEAP20049332A patent/GEP20084545B/en unknown
- 2004-09-01 BR BRPI0414045-1A patent/BRPI0414045A/en not_active IP Right Cessation
- 2004-09-01 NZ NZ545536A patent/NZ545536A/en not_active IP Right Cessation
- 2004-09-01 US US10/931,130 patent/US7576068B2/en not_active Expired - Fee Related
- 2004-09-01 EA EA200600540A patent/EA200600540A1/en unknown
- 2004-09-01 WO PCT/US2004/028236 patent/WO2005025583A2/en active Search and Examination
- 2004-09-01 CA CA2537450A patent/CA2537450C/en not_active Expired - Fee Related
- 2004-09-01 SG SG200806816-5A patent/SG146637A1/en unknown
- 2004-09-01 KR KR1020067004593A patent/KR20060096415A/en not_active Application Discontinuation
- 2004-09-03 TW TW093126782A patent/TWI347189B/en not_active IP Right Cessation
-
2006
- 2006-02-23 IL IL173890A patent/IL173890A/en not_active IP Right Cessation
- 2006-03-03 CR CR8273A patent/CR8273A/en not_active Application Discontinuation
- 2006-03-06 TN TNP2006000076A patent/TNSN06076A1/en unknown
- 2006-03-14 NO NO20061183A patent/NO20061183L/en not_active Application Discontinuation
- 2006-03-30 EC EC2006006465A patent/ECSP066465A/en unknown
- 2006-04-03 MA MA28909A patent/MA28082A1/en unknown
- 2006-11-03 HK HK06112114.0A patent/HK1091410A1/en not_active IP Right Cessation
-
2009
- 2009-06-29 US US12/493,737 patent/US7858637B2/en not_active Expired - Fee Related
- 2009-06-30 US US12/493,708 patent/US8034802B2/en not_active Expired - Fee Related
-
2011
- 2011-10-07 US US13/269,099 patent/US8211863B2/en not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4880784A (en) * | 1987-12-21 | 1989-11-14 | Brigham Young University | Antiviral methods utilizing ribofuranosylthiazolo[4,5-d]pyrimdine derivatives |
| US5041426A (en) * | 1987-12-21 | 1991-08-20 | Brigham Young University | Immune system enhancing 3-β-d-ribofuranosylthiazolo[4,5-d]pyridimine nucleosides and nucleotides |
| WO2003045968A1 (en) * | 2001-11-27 | 2003-06-05 | Anadys Pharmaceuticals, Inc. | 3-β-D-RIBOFURANOSYLTHIAZOLO[4,5-d]PYRIDIMINE NUCLEOSIDES AND USES THEREOF |
Non-Patent Citations (7)
| Title |
|---|
| LEE, J. ET AL: "Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: Activation of Toll-like receptor 7" PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES, vol. 100, no. 11, 27 May 2003 (2003-05-27), pages 6646-6651, XP002307483 * |
| MEALY, N.E.: "ANA-971" DRUGS OF THE FUTURE, vol. 29, no. 5, 2004, page 507, XP009040480 * |
| MEALY, N.E.: "Isatoribine" DRUGS OF THE FUTURE, vol. 29, no. 5, 2004, page 526, XP009040479 * |
| MICHAEL, M.A. ET AL.: "Alkylpurines as Immunepotentiating Agents: Synthesis and Antiviral Activity of Certain Alkylguanines" JOURNAL OF MEDICINAL CHEMISTRY, vol. 36, no. 22, 1993, pages 3431-3436, XP002307482 cited in the application * |
| RANEY. A.K.; HAMATAKE, R.K.; HONG, Z.: "HEP DART 2003: Frontiers in drug development for viral hepatitis" EXPERT OPINION ON INVESTIGATIONAL DRUGS, vol. 13, no. 3, 2004, pages 289-293, XP001203944 * |
| REITZ, A.B. ET AL.: "Small-Molecule Immunostimulants. Synthesis and Activity of 7,8-Disubstituted Guanosines and Structurally Related Compounds" J. MED. CHEM., vol. 37, no. 21, 1994, pages 3561-3578, XP002316151 cited in the application * |
| RHODES, J.: "Discovery of immunopotentiatory drugs: current and future strategies" CLIN. EXP. IMMUNOL., vol. 130, 2002, pages 363-369, XP002316152 * |
Cited By (117)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7754728B2 (en) | 2002-09-27 | 2010-07-13 | Dainippon Sumitomo Pharma Co., Ltd. | Adenine compound and use thereof |
| EP1550662A4 (en) * | 2002-09-27 | 2007-03-07 | Dainippon Sumitomo Pharma Co | Novel adenine compound and use thereof |
| US8148371B2 (en) | 2002-09-27 | 2012-04-03 | Dainippon Sumitomo Pharma Co., Ltd. | Adenine compound and use thereof |
| EP1550662A1 (en) * | 2002-09-27 | 2005-07-06 | Sumitomo Pharmaceuticals Company, Limited | Novel adenine compound and use thereof |
| US7998492B2 (en) | 2002-10-29 | 2011-08-16 | Coley Pharmaceutical Group, Inc. | Methods and products related to treatment and prevention of hepatitis C virus infection |
| US8969362B2 (en) | 2004-03-26 | 2015-03-03 | Astrazeneca Aktiebolag | 9-substituted 8-oxoadenine compound |
| US8012964B2 (en) | 2004-03-26 | 2011-09-06 | Dainippon Sumitomo Pharma Co., Ltd. | 9-substituted 8-oxoadenine compound |
| US8575180B2 (en) | 2004-03-26 | 2013-11-05 | Astrazeneca Aktiebolag | 9-substituted 8-oxoadenine compound |
| WO2006117670A1 (en) * | 2005-05-04 | 2006-11-09 | Pfizer Limited | 2-amido-6-amino-8-oxopurine derivatives as toll-like receptor modulators for the treatment of cancer and viral infections, such as hepatitis c |
| US7642350B2 (en) | 2005-05-04 | 2010-01-05 | Pfizer Limited | Purine derivatives |
| EP2614709A1 (en) * | 2005-07-18 | 2013-07-17 | Novartis AG | Small animal model for HCV replication |
| JP2009501546A (en) * | 2005-07-18 | 2009-01-22 | ノバルティス アーゲー | A small animal model for HCV replication |
| US9359360B2 (en) | 2005-08-22 | 2016-06-07 | The Regents Of The University Of California | TLR agonists |
| JP2013040209A (en) * | 2005-08-22 | 2013-02-28 | Regents Of The Univ Of California | Tlr agonist |
| JP2009504803A (en) * | 2005-08-22 | 2009-02-05 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | TLR agonist |
| WO2007038720A3 (en) * | 2005-09-27 | 2007-06-21 | Coley Pharm Gmbh | Modulation of tlr-mediated immune responses using adaptor oligonucleotides |
| WO2007038720A2 (en) * | 2005-09-27 | 2007-04-05 | Coley Pharmaceutical Gmbh | Modulation of tlr-mediated immune responses using adaptor oligonucleotides |
| EP1951914A2 (en) * | 2005-11-09 | 2008-08-06 | Cheng Si Yuan (China-International) Hepatitis Research Foundation | Diagnostic and therapeutic methods and agents |
| EP1951914A4 (en) * | 2005-11-09 | 2009-11-04 | Cheng Si Yuan China Internatio | Diagnostic and therapeutic methods and agents |
| US7691877B2 (en) | 2006-02-17 | 2010-04-06 | Pfizer Inc. | Pharmaceuticals |
| WO2007093901A1 (en) * | 2006-02-17 | 2007-08-23 | Pfizer Limited | 3 -deazapurine derivatives as tlr7 modulators |
| NL2000480C2 (en) * | 2006-02-17 | 2012-09-17 | Pfizer Ltd | NEW PHARMACEUTICAL FUNDS. |
| EP2518079A2 (en) | 2006-04-11 | 2012-10-31 | Novartis AG | HCV/HIV inhibitors and their uses |
| EP2700638A1 (en) * | 2006-05-31 | 2014-02-26 | The Regents Of the University of California | Purine analogs |
| US8846697B2 (en) | 2006-05-31 | 2014-09-30 | The Regents Of The University Of California | Purine analogs |
| US8138172B2 (en) | 2006-07-05 | 2012-03-20 | Astrazeneca Ab | 8-oxoadenine derivatives acting as modulators of TLR7 |
| US8349866B2 (en) | 2007-01-31 | 2013-01-08 | Chongxi Yu | High penetration prodrug compositions of 1H-imidazo[4,5-C]quinolin-4-amines and 1H-imidazo[4,5-C]quinolin-4-amine-related compounds |
| JP2010516802A (en) * | 2007-01-31 | 2010-05-20 | チョンシー ユー | Positively charged water-soluble prodrugs of 1H-imidazo [4,5-c] quinolin-4-amine and related compounds with very high skin permeability |
| US9567329B2 (en) | 2007-01-31 | 2017-02-14 | Techfields Pharma Co., Ltd. | High penetration prodrug compositions of 1H-imidazo[4,5-c]quinolin-4-amines and 1H-imidazo[4,5-c]quinolin-4-amine-related compounds and uses thereof |
| US8357374B2 (en) | 2007-02-07 | 2013-01-22 | The Regents Of The University Of California | Conjugates of synthetic TLR agonists and uses therefor |
| US9050376B2 (en) | 2007-02-07 | 2015-06-09 | The Regents Of The University Of California | Conjugates of synthetic TLR agonists and uses therefor |
| US8790655B2 (en) | 2007-02-07 | 2014-07-29 | The Regents Of The University Of California | Conjugates of synthetic TLR agonists and uses therefor |
| JP2010522151A (en) * | 2007-03-19 | 2010-07-01 | アストラゼネカ・アクチエボラーグ | 9-Substituted-8-oxo-adenine compounds as TOLL-like receptor (TLR7) modulators |
| US9233962B2 (en) | 2008-08-11 | 2016-01-12 | Glaxosmithkline Llc | Purine derivatives for use in the treatment of allergic, inflammatory and infectious diseases |
| US9346806B2 (en) | 2008-08-11 | 2016-05-24 | Glaxosmithkline Llc | 6-amino-purin-8-one compounds |
| US8563717B2 (en) | 2008-08-11 | 2013-10-22 | Glaxosmithkline Llc | Adenine derivatives |
| US10117873B2 (en) | 2008-08-11 | 2018-11-06 | Glaxosmithkline Llc | 6-amino-purin-8-one compounds |
| US8575181B2 (en) | 2008-08-11 | 2013-11-05 | Glaxosmithkline Llc | Purine derivatives for use in the treatment of allergic, inflammatory and infectious diseases |
| US8765772B2 (en) | 2008-08-11 | 2014-07-01 | Glaxosmithkline Llc | Purine derivatives for use in the treatment of allergic, inflammatory and infectious diseases |
| US9877968B2 (en) | 2008-08-11 | 2018-01-30 | Glaxosmithkline Llc | 6-amino-purin-8-one compounds |
| US8802684B2 (en) | 2008-08-11 | 2014-08-12 | Glaxosmithkline Llc | Adenine derivatives |
| US8067426B2 (en) | 2008-08-11 | 2011-11-29 | Glaxosmithkline Llc | 6-amino-purin-8-one compounds |
| WO2010055340A1 (en) * | 2008-11-12 | 2010-05-20 | Ludwig Institute For Cancer Research | Use of inkt or tlr agonists for protecting against or treating a disease such as acute infection or cancer |
| US9066940B2 (en) | 2009-02-06 | 2015-06-30 | Telormedix, Sa | Pharmaceutical compositions comprising imidazoquinolin(amines) and derivatives thereof suitable for local administration |
| US8729088B2 (en) | 2009-02-11 | 2014-05-20 | The Regents Of The University Of California | Toll-like receptor modulators and treatment of diseases |
| WO2010116248A1 (en) | 2009-04-10 | 2010-10-14 | Novartis Ag | Organic compounds and their uses |
| WO2010115981A1 (en) | 2009-04-10 | 2010-10-14 | Novartis Ag | 7-azadispiro [3.0.4.1] decane-8-carboxamides as hepatitis c virus inhibitors |
| US8575340B2 (en) | 2010-02-10 | 2013-11-05 | Glaxosmithkline Llc | Purine derivatives and their pharmaceutical uses |
| US9180183B2 (en) | 2010-04-30 | 2015-11-10 | Telormedix Sa | Phospholipid drug analogs |
| US9173935B2 (en) | 2010-04-30 | 2015-11-03 | Telormedix Sa | Phospholipid drug analogs |
| US9173936B2 (en) | 2010-04-30 | 2015-11-03 | Telormedix Sa | Phospholipid drug analogs |
| US9050319B2 (en) | 2010-04-30 | 2015-06-09 | Telormedix, Sa | Phospholipid drug analogs |
| WO2012048235A1 (en) | 2010-10-08 | 2012-04-12 | Novartis Ag | Vitamin e formulations of sulfamide ns3 inhibitors |
| US9045472B2 (en) | 2010-12-16 | 2015-06-02 | Astrazeneca Ab | Imidazoquinoline compounds |
| US8895570B2 (en) | 2010-12-17 | 2014-11-25 | Astrazeneca Ab | Purine derivatives |
| US10112946B2 (en) | 2011-07-22 | 2018-10-30 | Glaxosmithkline Llc | Composition |
| US8809265B2 (en) | 2011-10-21 | 2014-08-19 | Abbvie Inc. | Methods for treating HCV |
| US8853176B2 (en) | 2011-10-21 | 2014-10-07 | Abbvie Inc. | Methods for treating HCV |
| US8492386B2 (en) | 2011-10-21 | 2013-07-23 | Abbvie Inc. | Methods for treating HCV |
| US8969357B2 (en) | 2011-10-21 | 2015-03-03 | Abbvie Inc. | Methods for treating HCV |
| US8466159B2 (en) | 2011-10-21 | 2013-06-18 | Abbvie Inc. | Methods for treating HCV |
| US8993578B2 (en) | 2011-10-21 | 2015-03-31 | Abbvie Inc. | Methods for treating HCV |
| US9452194B2 (en) | 2011-10-21 | 2016-09-27 | Abbvie Inc. | Methods for treating HCV |
| US8680106B2 (en) | 2011-10-21 | 2014-03-25 | AbbVic Inc. | Methods for treating HCV |
| US8685984B2 (en) | 2011-10-21 | 2014-04-01 | Abbvie Inc. | Methods for treating HCV |
| US10022442B2 (en) | 2012-08-24 | 2018-07-17 | Glaxosmithkline Llc | Pyrazolopyrimidine compounds |
| US9662336B2 (en) | 2012-08-24 | 2017-05-30 | Glaxosmithkline Llc | Pyrazolopyrimidine compounds |
| US9555036B2 (en) | 2012-08-24 | 2017-01-31 | Glaxosmithkline Llc | Pyrazolopyrimidine compounds |
| US9428512B2 (en) | 2012-11-20 | 2016-08-30 | Glaxosmithkline Llc | Compounds |
| US9550785B2 (en) | 2012-11-20 | 2017-01-24 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9907847B2 (en) | 2012-11-20 | 2018-03-06 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9540383B2 (en) | 2012-11-20 | 2017-01-10 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9452217B2 (en) | 2013-06-22 | 2016-09-27 | Nitor Therapeutics | Methods for potentiating immune response for the treatment of infectious diseases and cancer |
| US9616129B2 (en) | 2013-06-22 | 2017-04-11 | Nitor Therapeutics | Compositions and methods for potentiating immune response, enhancing immunotherapy, and increasing vaccine potency |
| US11786604B2 (en) | 2014-01-10 | 2023-10-17 | Birdie Biopharmaceuticals, Inc. | Compounds and compositions for treating HER2 positive tumors |
| US11633495B2 (en) | 2014-01-10 | 2023-04-25 | Birdie Biopharmaceuticals, Inc. | Compounds and compositions for immunotherapy |
| US11633494B2 (en) | 2014-01-10 | 2023-04-25 | Birdie Biopharmaceuticals, Inc. | Compounds and compositions for immunotherapy |
| US10780180B2 (en) | 2014-01-10 | 2020-09-22 | Birdie Biopharmaceuticals, Inc. | Compounds and compositions for immunotherapy |
| US11590155B2 (en) | 2014-06-24 | 2023-02-28 | Janssen Pharmaceutica Nv | Substituted nucleosides, nucleotides and analogs thereof |
| US10780105B2 (en) | 2014-06-24 | 2020-09-22 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US11279761B2 (en) | 2014-07-09 | 2022-03-22 | Birdie Biopharmaceuticals, Inc. | Anti-PD-L1 combinations for treating tumors |
| US11130812B2 (en) | 2014-09-01 | 2021-09-28 | Birdie Biopharmaceuticals, Inc. | Anti PD-L1 conjugates for treating tumors |
| US10040815B2 (en) | 2014-12-08 | 2018-08-07 | Hoffmann-La Roche Inc. | 3-substituted 5-amino-6H-thiazolo[4,5-d]pyrimidine-2,7-dione compounds for the treatment and prophylaxis of virus infection |
| US11180525B2 (en) | 2014-12-08 | 2021-11-23 | Hoffmann-La Roche, Inc. | Process for making 3-substituted 5-amino-6H-thiazolo[4,5-d]pyrimidine-2, 7-dione compounds useful for the treatment of virus infection |
| US10618929B2 (en) | 2014-12-08 | 2020-04-14 | Hoffmann-La Roche Inc. | 3-substituted 5-amino-6H-thiazolo[4,5-D]pyrimidine-2, 7-dione compounds for the treatment and prophylaxis of virus infection |
| US9441008B2 (en) | 2014-12-08 | 2016-09-13 | Hoffmann-La Roche Inc. | 3-substituted 5-amino-6H-thiazolo[4,5-D]pyrimidine-2,7-dione compounds for the treatment and prophylaxis of virus infection |
| US11771699B2 (en) | 2015-03-16 | 2023-10-03 | Hoffmann-La Roche Inc. | Combined treatment with a TLR7 agonist and an HBV capsid assembly inhibitor |
| US10183954B2 (en) | 2015-05-08 | 2019-01-22 | Hoffmann-La Roche Inc. | Oxathiolane carboxylic acids and derivatives for the treatment and prophylaxis of virus infection |
| US10065973B2 (en) | 2015-05-12 | 2018-09-04 | Hoffmann-La Roche Inc. | Substituted aminothiazolopyrimidinedione for the treatment and prophylaxis of virus infection |
| US10975098B2 (en) | 2015-06-30 | 2021-04-13 | Hoffmann-La Roche Inc. | Substituted aminothiazolopyrimidinediones for the treatment of virus infection |
| US10590146B2 (en) | 2015-06-30 | 2020-03-17 | Hoffmann-La Roche Inc. | Substituted aminothiazolopyrimidinediones |
| US11136397B2 (en) | 2016-01-07 | 2021-10-05 | Birdie Pharmaceuticals, Inc. | Anti-EGFR combinations for treating tumors |
| US11702476B2 (en) | 2016-01-07 | 2023-07-18 | Birdie Biopharmaceuticals, Inc. | Anti-EGFR combinations for treating tumors |
| US11046781B2 (en) | 2016-01-07 | 2021-06-29 | Birdie Biopharmaceuticals, Inc. | Anti-HER2 combinations for treating tumors |
| US11220552B2 (en) | 2016-01-07 | 2022-01-11 | Birdie Biopharmaceuticals, Inc. | Anti-CD20 combinations for treating tumors |
| US10556903B2 (en) | 2016-04-19 | 2020-02-11 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10533007B2 (en) | 2016-04-19 | 2020-01-14 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10703755B2 (en) | 2016-04-26 | 2020-07-07 | Sumitomo Dainippon Pharma Co., Ltd. | Substituted purine derivative |
| WO2017189978A1 (en) | 2016-04-28 | 2017-11-02 | Emory University | Alkyne containing nucleotide and nucleoside therapeutic compositions and uses related thereto |
| US11192914B2 (en) | 2016-04-28 | 2021-12-07 | Emory University | Alkyne containing nucleotide and nucleoside therapeutic compositions and uses related thereto |
| US11697851B2 (en) | 2016-05-24 | 2023-07-11 | The Regents Of The University Of California | Early ovarian cancer detection diagnostic test based on mRNA isoforms |
| US11117898B2 (en) | 2016-11-28 | 2021-09-14 | Jiangsu Hengrui Medicine Co., Ltd. | Pyrazolo-heteroaryl derivative, preparation method and medical use thereof |
| WO2018095426A1 (en) | 2016-11-28 | 2018-05-31 | 江苏恒瑞医药股份有限公司 | Pyrazolo-heteroaryl derivative, preparation method and medical use thereof |
| US11142534B2 (en) | 2017-01-06 | 2021-10-12 | Hoffmann-La Roche Inc. | Process for the preparation of 3-substituted 5-amino-6H-thiazolo[4,5-d]pyrimidine-2,7-dione compounds |
| US11827632B2 (en) | 2017-02-17 | 2023-11-28 | Innate Tumor Immunity, Inc | NLRP3 modulators |
| US10533005B2 (en) | 2017-02-17 | 2020-01-14 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US11053240B2 (en) | 2017-04-27 | 2021-07-06 | Birdie Biopharmaceuticals, Inc. | 2-amino-quinoline derivatives |
| US11834448B2 (en) | 2017-04-27 | 2023-12-05 | Birdie Biopharmaceuticals, Inc. | 2-amino-quinoline derivatives |
| EP3636646A4 (en) * | 2017-05-18 | 2020-12-09 | Jiangsu Hengrui Medicine Co., Ltd. | Heteroaryl-pyrazole derivative, and preparation method therefor and medical application thereof |
| US11111249B2 (en) | 2017-05-18 | 2021-09-07 | Jiangsu Hengrui Medicine Co., Ltd. | Heteroaryl-pyrazole derivative, and preparation method therefor and medical application thereof |
| WO2018210298A1 (en) | 2017-05-18 | 2018-11-22 | 江苏恒瑞医药股份有限公司 | Heteroaryl-pyrazole derivative, and preparation method therefor and medical application thereof |
| CN110177793A (en) * | 2017-05-18 | 2019-08-27 | 江苏恒瑞医药股份有限公司 | Heteroaryl and pyrazole derivatives, preparation method and its application in medicine |
| CN110177793B (en) * | 2017-05-18 | 2021-12-21 | 江苏恒瑞医药股份有限公司 | Heteroaryl pyrazole derivative, preparation method and application thereof in medicine |
| US11517567B2 (en) | 2017-06-23 | 2022-12-06 | Birdie Biopharmaceuticals, Inc. | Pharmaceutical compositions |
| WO2019223773A1 (en) | 2018-05-25 | 2019-11-28 | 江苏恒瑞医药股份有限公司 | Crystal form of hydrochloride of pyrazoloheteroaryl derivative and preparation method |
| EP4003369A4 (en) * | 2019-07-27 | 2023-10-18 | Brii Biosciences, Inc. | Adenosine derivative and pharmaceutical composition comprising the same |
| US11890297B2 (en) | 2021-01-25 | 2024-02-06 | Brii Biosciences, Inc. | Combination therapy for HIV with adenosine derivative and capsid inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1667694B1 (en) | Tlr7 ligands for the treatment of hepatitis c | |
| US7321033B2 (en) | 3-B-D-ribofuranosylthiazolo [4,5-d] pyrimidine nucleosides and uses thereof | |
| TWI407956B (en) | 3,5-disubstituted and 3,5,7-trisubstituted-3h-oxazolo and 3h-thiazolo[4,5-d]pyrimidin-2-one compounds and prodrugs thereof | |
| JP5225991B2 (en) | Carbonate and carbamate prodrugs of thiazolo [4,5-d] pyrimidine | |
| TWI418561B (en) | Prodrugs of 5-amino-3-(3'-deoxy-β-d-ribofuranosyl)-thiazolo[4,5-d]pyrimidin-2,7-dione | |
| EP1758916B1 (en) | 3-BETA-D-RIBOFURANOSYLTHIAZOLO[4,5-d]PYRIDIMINE NUCLEOSIDES AND USES THEREOF | |
| ZA200601632B (en) | Administration of TLR7 ligands and prodrugs thereof for treatment of infection by hepatitis C virus | |
| KR100880298B1 (en) | 3-?-D-RIBOFURANOSYLTHIAZOLO[4,5-d]PYRIDIMINE NUCLEOSIDES AND USES THEREOF | |
| Averett et al. | 3-Beta-D-Ribofuranosylthiazolo[ 4, 5-D] Pyridimine Nucleosides and Uses Thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480025532.5 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BW BY BZ CA CH CN CO CR CU CZ DK DM DZ EC EE EG ES FI GB GD GE GM HR HU ID IL IN IS JP KE KG KP KZ LC LK LR LS LT LU LV MA MD MK MN MW MX MZ NA NI NO NZ PG PH PL PT RO RU SC SD SE SG SK SY TJ TM TN TR TT TZ UA UG US UZ VN YU ZA ZM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SZ TZ UG ZM ZW AM AZ BY KG MD RU TJ TM AT BE BG CH CY DE DK EE ES FI FR GB GR HU IE IT MC NL PL PT RO SE SI SK TR BF CF CG CI CM GA GN GQ GW ML MR SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 173890 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004271972 Country of ref document: AU Ref document number: 545536 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/002309 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2537450 Country of ref document: CA Ref document number: 12006500425 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006525384 Country of ref document: JP Ref document number: CR2006-008273 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020067004593 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 419.06 Country of ref document: BZ |
|
| ENP | Entry into the national phase |
Ref document number: 2004271972 Country of ref document: AU Date of ref document: 20040901 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004271972 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2004000135 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004782670 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9332 Country of ref document: GE Ref document number: 06032880 Country of ref document: CO Ref document number: 06032753 Country of ref document: CO Ref document number: 200600540 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20060111 Country of ref document: UZ Ref document number: 1200600533 Country of ref document: VN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004782670 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067004593 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0414045 Country of ref document: BR |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) |