PROCESS FOR THE MANUFACTURE OF GEFITINIB
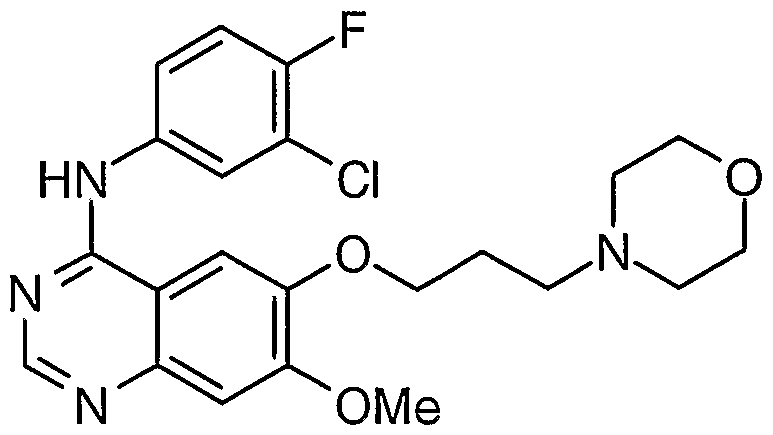
The present invention relates to improved chemical processes and intermediates useful in the manufacture of compounds that possess pharmacological activity. In particular, the present invention relates to chemical processes and intermediates useful in the manufacture of quinazoline derivatives, or pharmaceutically- acceptable salts thereof, which possess anti-proliferative properties and are useful in the treatment or prevention of cancers in a warm-blooded ammal such as man. The invention also relates to processes for the manufacture of said intermediates and to processes for the manufacture of such quinazoline derivatives utilising said intermediates. In particular, the present invention relates to a chemical process useful in the manufacture of the quinazoline derivative 4-(3'-c oro-4'-fluoroani1rno)- 7-methoxy-6-(3-morphojjj opropoxy)quiiiazoline which compound is disclosed in Example 1 of International Patent Application WO 96/33980. That compound is an inhibitor of the epidermal growth factor receptor (EGFR) family of tyrosine kinase enzymes such as erbBl and possesses anti-proliferative activity such as anti-cancer activity and, accordingly, is useful in methods of treatment of proliferative disease such as cancer in the human or animal body. That compound has the structure of Formula I

I and is now known as Iressa (registered trade mark) and gefitinib (Unites States Adopted Name) and by way of the code number ZD1839 and Chemical Abstracts Registry Number 184475-35-2. Two routes for preparing the compound of Formula I are disclosed in International Patent Application WO 96/33980. Each route involves the use of the compound 4-(3'-c oro-4'-fluoroarilino)-6-hydroxy-7-methoxyqujj azoline as an intermediate with the formation of the 3-morpholinopropoxy side-chain at the 6-position occurring toward the end of the syntheses. These existing routes are satisfactory for the synthesis of relatively small
amounts of the compound of Formula I but they involve linear rather than convergent syntheses, each requiring the use of multiple chromato graphic purification steps and the isolation of a substantial number of intermediates. As such, the overall yields of these syntheses are not high. There is therefore a need for a more efficient synthesis of the compound of 5 Formula I suitable for use to make larger quantities of that compound. The new synthesis should not involve costly and time-consuming chromatograpMc purification procedures. A further route for preparing the compound of Formula I is disclosed in International Patent Application WO 2004/024703. We have now devised a further suitable process for the manufacture of the compound of
10 Formula I that is based on a chemical transformation that is known as a Dimroth rearrangement reaction (as reviewed, for example, by Wahren, M: Z Chem. 1969, 9(1), 241). The new process is advantageous in that it allows the final product to be made in high quality and yield on a large scale. The process is more convergent than the previous routes and allows a substantial reduction in the number of intermediates that must be isolated. This provides
15 significant advantages of time and cost. Chromatographic purification procedures are not required. According to the invention, processes are provided for the manufacture of key intermediates that may be used in the preparation of the compound of Formula I. A rearrangement reaction has been disclosed for the preparation of pyrrolo[2,3--i]pyrijmidine derivatives (Organic Process Research & Development, 2001, 5,
20 581-586 and International Patent Application WO 98/43973). It was stated in the journal article that the conversion of the compound 3-(3-chlorophenyl)-5,6-dimethyl- 4H-pyrrolo[2,3--7Tpyrir dme-4-imine (A) to provide 4-(3-c orophenylamino)-5,6-dimethyl- 7H-ρyrrolo[2,3-6flpyrimidine (B) was not truly a rearrangement but rather a hydrolysis of the pyrimidine ring at the N3-C2 bond with subsequent rotation of 180° around the C9-C4 bond,
25 and ring closure. It was further stated that water would be required for the isomerisation to occur.
(A) (B)
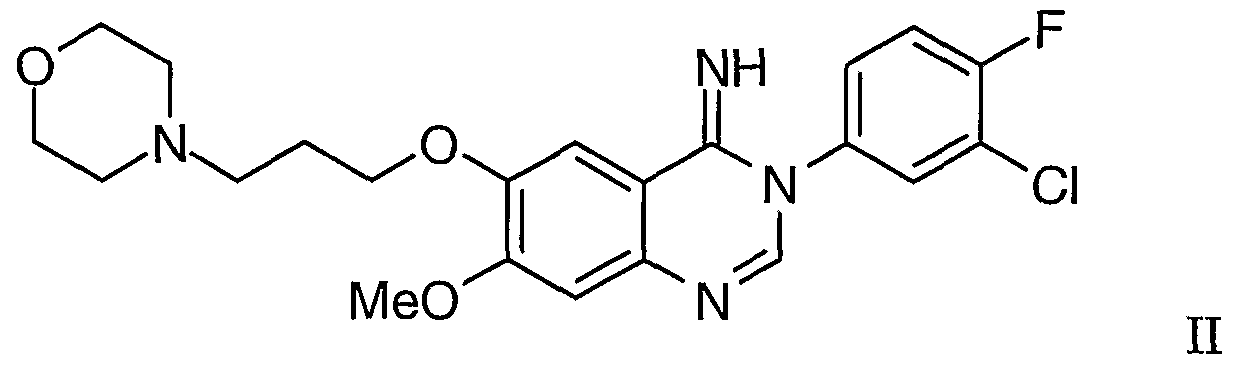
According to a first aspect of the present invention, there is provided a process for the rnanufacture of 4-(3'-cMoro-4'-fluoroarjαljjio)-7-m of Formula I
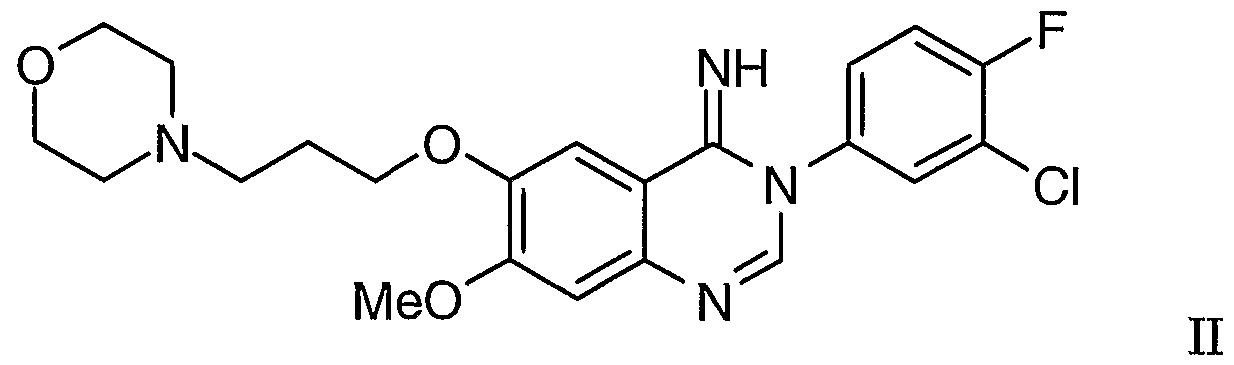
or a pharmaceutically-acceptable salt thereof which comprises the rearrangement reaction optionally in the presence of a suitable catalyst of 3-(3'-chloro-4,-fluorophenyl)-7-methoxy- 6-(3-morpholinopropoxy)-3,4-dihydroquinazθjjj -4-jjmine of Formula II

whereafter a compound of Formula I obtained in the form of a salt may be converted into the free compound using a conventional procedure and the compound of Formula I may be converted into a pharmaceutically-acceptable salt using a conventional procedure. The reaction may conveniently be carried out by any of the many procedures known for such a transformation which is known as a Dimroth rearrangement reaction. For example, the rearrangement reaction may be carried out by heating the imine of Formula II to an elevated temperature. Conveniently, the imine of Formula II is mixed with a suitable solvent or diluent or a mixture thereof and heated to an elevated temperature. A suitable elevated temperature is, for example, a temperature in the range, for example, 40 to 250°C. A suitable solvent or diluent is, for example, an aromatic solvent such as toluene, a xylene, cumene or chlorobenzene. A further suitable solvent or diluent is a polar aprotic solvent such as acetonitrile, propionitrile, butyronitrile, ethyl acetate, tetrahydrofuran or 1,4-dioxan or a dipolar aprotic solvent such as N,N-dimemylformamide, N,N-dimethylacetamide, N-methylpyrro in-2-one or dimethylsulphoxide. A further suitable solvent or diluent is water or a polar protic solvent such as a primary, secondary or tertiary
alkyl alcohol, for example, methanol, ethanol, a butanol or pentanol. Mixtures of such suitable solvents or diluents may be used. Conveniently, the reaction temperature is the reflux temperature of the reaction solvent or diluent or mixture thereof. For example, when the solvent or diluent is an aromatic solvent such as toluene, a xylene or cumene, or a mixture thereof, the reaction temperature is in the range, for example, 80 to 250°C, conveniently in the range, for example, 100 to 170°C or in the range, for example, 100 to 140°C, more conveniently at or near 110°C or at or near 130°C. In one aspect of the invention, the rearrangement reaction is carried out under substantially anliydrous conditions. In that case, a convenient solvent or diluent is, for example, an aromatic solvent such as toluene or a xylene, or mixtures thereof. Conveniently, the rearrangement reaction may be carried out in the presence of a suitable catalyst such as an acidic catalyst. A suitable acidic catalyst is, for example, an inorganic or organic acid. A suitable inorganic acid catalyst is, for example, hydrochloric, hydrobromic or sulphuric acid. A suitable organic acid catalyst is, for example, a (l-8C)alkanoic acid such as formic, acetic, trifluoroacetic, propionic or butyric acid, a heteroarylcarboxylic acid such as a pyridine carboxylic acid, for example pyridine-4-carboxylic acid or nicotinic acid, a (l-8C)alkanesulphonic acid such as methanesulphonic acid or an arylsulphonic acid such as benzenesulphonic acid or a toluenesulphonic acid. An alternative suitable organic acid catalyst is, for example, a salt of a suitable organic acid catalyst as defined hereinbefore with an organic amine base, for example with pyridine, 2,6-lutidine, collidine, 4-dimethylaminopyridine, triethylamine, morpholine, N-methylmorpholine or diazabicyclo[5.4.0]undec-7-ene. Conveniently the acidic catalyst is formic acid, acetic acid or 4-toluenesulphonic acid. When the rearrangement reaction is carried out under substantially anhydrous conditions, a suitable catalyst is an acidic catalyst such as an inorganic or organic acid. A suitable inorganic acid catalyst is, for example, hydrogen chloride or hydrogen bromide. A suitable organic acid catalyst is, for example, a (l-8C)alkanoic acid such as formic, acetic, trifluoroacetic, propionic or butyric acid, a (l-8C)alkanesulphonic acid such as methanesulphonic acid or an arylsulphonic acid such as benzenesulphonic acid or a toluenesulphonic acid. Conveniently the acidic catalyst is formic acid, acetic acid or 4-toluenesulphonic acid.
The compound of Formula I may be obtained from this process in the form of the free base or alternatively it may be obtained in the form of an acid addition salt such as a 4-toluenesulphonic acid salt. When it is desired to obtain the free base from the salt, the salt may be treated with a suitable base, for example, an organic amine base such as, for example, pyridine, 2,6-lutidine, collidine, 4-dimethylaminopyridine, triethylamine, mo holine,
N-me yjjmorpholine or diazabicyclo[5.4.0]undec-7-ene, or, for example, an alkali or ajjkaline earth metal carbonate or hydroxide, for example sodium carbonate, potassium carbonate, calcium carbonate, sodium hydroxide or potassium hydroxide. A suitable pharmaceutically-acceptable salt of a compound of Formula I is, for example, an acid-addition salt of a compound of Formula I, for example an acid-addition salt with an inorganic or organic acid such as hydrochloric, hydrobromic, sulphuric, trifluoroacetic, citric or maleic acid. The imine of Formula II is a novel compound that forms a further feature of the present invention. The imine of Formula II may be prepared by the displacement reaction optionally in the presence of a suitable catalyst of 2-aπmo-4-methoxy-5-(3-morρholinopropoxy)benzonitrile of Formula III
with an hriine of Formula IV
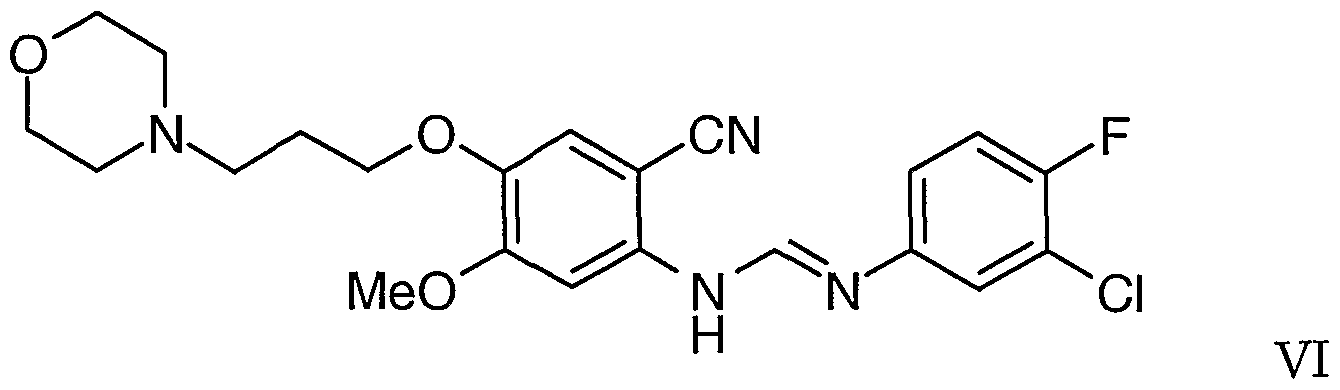
wherein Z is a suitable displaceable group and the in situ cyclisation of the intermediate so formed. The intermediate that cyclises to provide the imine of Formula II is believed to be the compound N-(3-chloro-4-fluorophenyl)-N'-[2-cyano-5-methoxy- 4-(3-morpholinopropoxy)phenyl]formarrιidine of Formula VI
or a tautomer thereof. It will be appreciated that an imine of Formula IV may exist in the form of two geometric isomers or as a mixture thereof (with the Z and phenyl groups on the same side or on opposite sides of the imine bond). A 'wavy' bond to the H atom has been used to indicate that Formula IV represents each of the geometric isomers. A suitable displaceable group Z is, for example, a hydroxy, alkoxy, optionally substituted aryloxy, amino, alkylarnino, diajj ylamino or optionally substituted arylamino group, for example a hydroxy, methoxy, optionally substituted phenoxy, amino, methylamino, dimethylamino or optionally substituted anilino group. The displacement and cyclisation reactions may be carried out in the presence of a suitable catalyst such as an acidic catalyst. A suitable acidic catalyst is, for example, an inorganic or organic acid. A suitable inorganic acid catalyst is, for example, hydrochloric, hydrobromic or sulphuric acid. A suitable organic acid catalyst is, for example, a (l-8C)alkanoic acid such as formic, acetic, trifluoroacetic, propionic or butyric acid, a heteroarylcarboxylic acid such as a pyridine carboxylic acid, for example pyridine-4-carboxylic acid or nicotinic acid, a (l-8C)alkanesulphonic acid such as methanesulphonic acid, an arylsulphonic acid such as benzenesulphonic acid or a toluenesulphonic acid. An alternative suitable organic acid catalyst is, for example, a salt of a suitable organic acid catalyst as defined hereinbefore with an organic amine base, for example with pyridine, 2,6-lutidine, collidine, 4-dimethylaminopyridine, triethylamine, morpholine, N-methylmorpholine or diazabicyclo[5.4.0]undec-7-ene. Conveniently the acidic catalyst is formic acid, acetic acid or 4-toluenesulphonic acid. The displacement and cyclisation reactions may be carried out in the presence of a suitable solvent or diluent, for example in an aromatic solvent such as toluene, a xylene, cumene or chlorobenzene, in a polar aprotic solvent such as acetonitrile, propionitrile, butyronitrile, ethyl acetate, tetrahydrofuran or 1,4-dioxan or a dipolar aprotic solvent such as N,N-dimethyjjforrrj ride, N,N-dimemylacetamide, N-methylpyrrolidin-2-one or
dimethylsulphoxide. A further suitable solvent or diluent is water or a polar protic solvent such as a primary, secondary or tertiary alkyl alcohol, for example, methanol, ethanol, a butanol or pentanol. Mixtures of such suitable solvents or diluents may be used. Conveniently, the reaction is carried out in an aromatic solvent, for example toluene or a xylene, at a temperature in the range, for example, 30 to 150°C, conveniently in the range, for example, 80 to 150°C, more conveniently at or near 110°C or at or near 130°C. In one aspect of the invention, the displacement and cyclisation reactions are carried out under substantially anhydrous conditions. In that case, a convenient solvent or diluent is, for example, an aromatic solvent such as toluene or a xylene, or mixtures thereof. The imine of Formula IV may be prepared by conventional procedures, for example by the reaction optionally in the presence of a suitable catalyst of 3-cMoro-4-fluoroaruIine with formic acid or a reactive derivative thereof. Suitable reactive derivatives of formic acid include, but are not lii ited to, esters, amides and alkyl orthoformates. For example, a suitable reactive derivative of formic acid is, for example, a trialkyl orthoformate, for example triethyl orthoformate; a formic acid amide such as formamide or N,N-dimemylformamide; a mixed anhydride, for example an anhydride formed by the reaction of formic acid and a chloroformate such as isobutyl chloroformate; the product of the reaction of formic acid with a carbodiimide such as dicyclohexylcarbodiimide; or the product of the reaction of formic acid with a mixture of an azo compound such as diethyl or di-te/ -butyl azodicarboxylate and a phosphine such as triphenylphosphine. Conveniently, the reaction is carried out in an aromatic solvent, for example toluene or a xylene, at a temperature in the range, for example, 30 to 150°C, conveniently in the range, for example, 60 to 120°C and volatile side products are distilled off to allow the internal reaction temperature to rise to the boiling point of the solvent. A suitable catalyst for the preparation of an imine of Formula IV is, for example, an inorganic or organic acid. A suitable inorganic acid catalyst is, for example, hydrochloric, hydrobromic or sulphuric acid. A suitable organic acid catalyst is, for example, a (l-8C)alkanoic acid such as formic, acetic, trifluoroacetic, propionic or butyric acid, a heteroarylcarboxylic acid such as a pyridine carboxylic acid, for example pyridine-4-carboxylic acid or nicotinic acid, a (l-8C)alkanesulphonic acid such as methanesulphonic acid or an arylsulphonic acid such as benzenesulphonic acid or a toluenesulphonic acid. An alternative suitable organic acid catalyst is, for example, a salt of a suitable organic acid catalyst as defined
hereinbefore with an organic amine base, for example with pyridine, 2,6-lutidine, collidine, 4-dimethylaminopyridine, triethylarnine, morpholine, N-methylmorpholine or diazabicyclo[5.4.0]undec-7-ene. Conveniently the acidic catalyst is formic acid or acetic acid. Conveniently, N,N'-bis-(3-cHoro-4-fluorophenyl)formamidine of Formula V
is used as a particular example of an imine of Formula IV. The formamidine of Formula V may be prepared by conventional procedures, for example by the reaction of 3-chloro- 4-fluoroarjiline with formic acid or a reactive derivative thereof, for example a trialkyl orthoformate, in the presence of an acid catalyst, for example formic acid or acetic acid, in an aromatic solvent, for example toluene or a xylene, at a temperature in the range, for example, 30 to 150°C, conveniently at or near 110°C or at or near 130°C. Conveniently, the intermediate of the Formula II is not isolated as such, but is directly subjected to the rearrangement reaction. Thereby, the compound of Formula I may be manufactured from the compound of Formula III in a one-pot procedure. According to this aspect of the invention there is provided a one pot process for the manufacture of
4-(3'-cHoro-4'-fluoroarjjjjmo)-7-meto of Formula I
I or a pharmaceutically-acceptable salt thereof, which comprises the reaction optionally in the presence of a suitable catalyst of 2-amino-4-methoxy-5-(3-morpholinopropoxy)benzorιitrile of Formula III
III
with N,N'-bis-(3-cHoro-4-fluoroρhenyl)foraιamidine of Formula V
to give N-(3-chloro-4-fluorophenyl)-N'-[2-cyano-5-methoxy- 4-(3-morpholinopropoxy)phenyl]formamidine of Formula VI
or a tautomer thereof which cyclises to give 3-(3'-chloro-4'-fluorophenyl)-7-methoxy- 6-(3-morphojjjiopropoxy)-3,4-dihydroqujj azolm-4-jj ine of Formula II
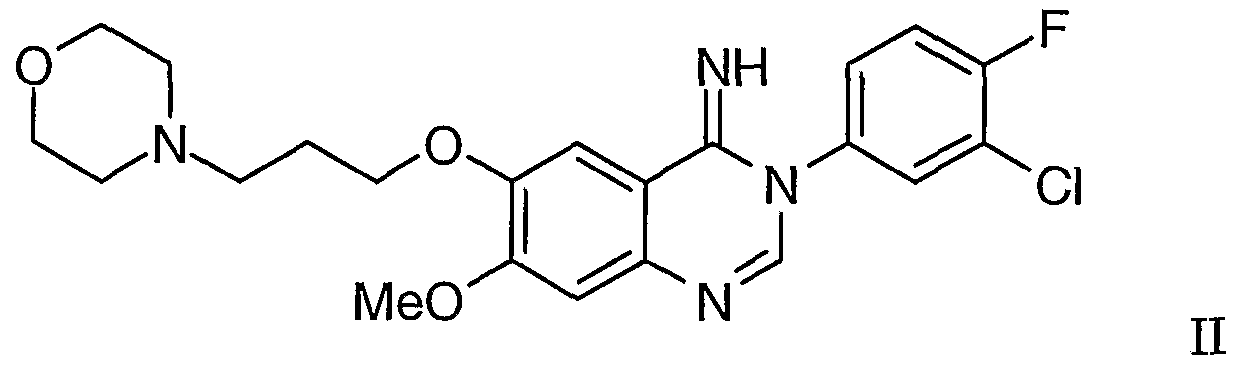
which on heating to an elevated temperature undergoes a rearrangement reaction to provide the compound of Formula I; whereafter a compound of Formula I obtained in the form of a salt may be converted into the free compound using a conventional procedure and the compound of Formula I may be converted into a pharmaceutically-acceptable salt using a conventional procedure. In one aspect of the invention, the conversions from the compound of Formula III to the compound of Formula I are carried out under substantially anliydrous conditions. In that case, a convenient solvent or diluent is, for example, an aromatic solvent such as toluene or a xylene, or mixtures thereof. In another aspect of the invention, the conversions from the compound of Formula III to the compound of Formula I are carried out in the presence of a suitable catalyst such as an acidic catalyst as defined hereinbefore. Conveniently the conversions are carried out under substantially anhydrous conditions using formic acid, acetic acid or 4-toluenesulphonic acid as acidic catalyst and toluene or xylene as the reaction solvent or diluent and the reaction mixtui-e is heated at a temperature in the range, for example of 80 to 150°C, more conveniently at or near 110°C or at or near 130°C.
The compound 2-amjno-4-methoxy-5-(3-morpholinopropoxy)benzonitrile of Formula III may be prepared using the following procedures :- (a) the coupling of 3-hydroxy-4-methoxybenzonitrile of Formula VII
VII with a 3-morpholinopropane derivative of Formula VIII
VIII wherein Z is a displaceable group as defined hereinbefore to give 4-methoxy- 3-(3-rno hθjjn.opropoxy)berjzonitrιle of Formula IX
IX (b) the nitration of the compound of Formula IX to give 4-methoxy- 5-(3-morphθjjnopropoxy)-2-nitrobenzoιιitrile of Formula X

and (c) the reduction of 4-methoxy-5-(3-morphoIinopropoxy)-2-rntroberrzonitrile of Formula X to give 2-amino-4-methoxy-5-(3-morpholinopropoxy)berjzonitrile of Formula III. For process step (a), the coupling step may conveniently be an alkylation reaction which may be carried out by any of the many procedures known for such a transformation. For an alkylation reaction, a suitable displaceable group Z is any of the displaceable groups defined hereinbefore, for example, a halogeno, alkoxy, aryloxy or sulphonyloxy group, for example a chloro, bromo, methoxy, phenoxy, methanesulphonyloxy or 4-toluenesulphonyloxy group. The alkylation may be carrried out, for example, in the presence of a suitable base and in a suitable
inert solvent or diluent and at a temperature in the range, for example, 10 to 150°C, conveniently at or near 80°C. A suitable base is, for example, an organic amine base such as, for example, pyridine, 2,6-lutidine, collidine, 4-djj ethylaminopyridine, triethylamine, morpholine, N-methylmorpholine or diazabicyclo[5.4.0]undec-7-ene, or, for example, an alkali or alkaline earth metal carbonate or hydroxide, for example sodium carbonate, potassium carbonate, calcium carbonate, sodium hydroxide or potassium hydroxide, or, for example, an alkali metal hydride, for example sodium hydride. A suitable inert solvent or diluent, for example an alcohol such as methanol, ethanol or isopropanol, a halogenated solvent such as methylene chloride, chloroform or carbon tetrachloride, an aromatic solvent such as toluene, a polar aprotic solvent such as acetonitrile, ethyl acetate, tetrahydrofuran or 1,4-dioxan or a dipolar aprotic solvent such as N,N-dimethylfθΩiιamide, N,N-dimethylacetamide, N-methylpyrrolidin-2-one or dimethylsulphoxide. Alternatively, for process step (a), the coupling step may conveniently be a dehydration reaction which may be carried out in the presence of a suitable dehydrating agent. For a dehydration reaction, a suitable displaceable group Z is, for example, a hydroxy group. A suitable dehydrating agent is, for example, a carbodiήnide reagent such as dicyclohexylcarbodiimide or l-(3-dimethylaminopropyl)-3-ethylcarboduιrιide or a mixture of an azo compound such as diethyl or di-tert-butyl azodicarboxylate and a phosphine such as tπphenylphosphine. The dehydration reaction is conveniently carried out in the presence of a suitable inert solvent or diluent, for example a halogenated solvent such as methylene chloride, chloroform or carbon tetrachloride and at a temperature in the range, for example, 10 to 150°C, conveniently at or near ambient temperature. Conveniently, the coupling reaction is carried out as an alkylation reaction in the presence of an alkali metal carbonate such as potassium carbonate, in a dipolar aprotic solvent such as N,N-dimethylformamide and at a temperature in the range, for example, 40 to 120°C, conveniently at or near 80°C. For process step (b), the nitration step may conveniently be carried out by any of the many procedures known for such a transformation. Conveniently, the nitration may be carried out using concentrated nitric acid, optionally in the presence of concentrated sulphuric acid and optionally in the presence of a polar protic solvent such as acetic acid, at a temperature in the
range, for example, 0 to 80°C, conveniently at or near ambient temperature. Conveniently, the sulphuric acid concentration is greater than 50% (weight/weight % with water), preferably about 70%. On completion of the reaction, the reaction mixture is neutralised with an aqueous base such as sodium or ammonium hydroxide solution and the compound of Formula X is extracted into an organic solvent. Conveniently, the intermediate of Formula IX is not isolated as such but is prepared and used as a solution in an organic solvent. For process step (c), the reduction may conveniently be carried out by any of the many procedures known for such a transformation. The reduction may be carried out, for example, by the hydrogenation of a solution of the nitro compound in the presence of a suitable metal catalyst such as palladium or platinum on an inert carrier such as carbon and/or barium sulphate and in an inert solvent or diluent such as water, a polar protic solvent such as methanol or ethanol or a polar aprotic solvent such as ethyl acetate. A further suitable reducing agent is, for example, an activated metal such as activated iron (produced by washing iron powder with a dilute solution of an acid such as hydrochloric, hydrobromic, sulphuric or acetic acid). Thus, for example, the reduction may be carried out by heating a mixture of the nitro compound and the activated metal in a suitable solvent or diluent such as a polar protic solvent or a mixture of water and an alcohol, for example methanol or ethanol, at a temperature in the range, for example, 30 to 150°C, conveniently at or near 70°C. Further suitable reaction conditions include, for example, the use of ammonium formate or hydrogen gas in the presence of a catalyst, for example a metallic catalyst such as palladium-on-carbon. Conveniently, the reduction is carried out in the presence of a water-soluble inorganic reducing agent such as sodium dithionite and at a temperature in the range, for example, 20 to 100°C, conveniently at or near 50°C. Alternatively, the imine of Formula II may be prepared by the displacement reaction optionally in the presence of a suitable catalyst of 3-c oro-4-fluoroarjiline with an imine of Formula XI
wherein Z is a suitable displaceable group as defined hereinbefore to give N-(3-chloro- 4-fluorophenyl)-N'-[2-cyano-5-methoxy-4-(3-morphojjjiopropoxy)phenyl]formanjidine of Formula VI
or a tautomer thereof which cyclises to give 3-(3'-chloro-4'-fluorophenyl)-7-methoxy- 6-(3-mo holjiopropoxy)-3,4-dihydroqujnazolm-4-imine of Formula II
The imine of Formula XI may be prepared by conventional procedures, for example by the reaction optionally in the presence of a suitable catalyst of 2-amino-4-methoxy- 5-(3-morpholinopiOpoxy)beτιzorLitrile of Formula III with formic acid or a reactive derivative thereof. Suitable reactive derivatives of formic acid include, but are not limited to, esters, amides and alkyl orthoformates. For example, a suitable reactive derivative of formic acid is, for example, a trialkyl orthoformate, for example triethyl orthoformate; a formic acid amide such as forrmmide or N,N-dimethylfoπnamide; a mixed anhydride, for example an anhydride formed by the reaction of formic acid and a chloroformate such as isobutyl chloroformate; the product of the reaction of formic acid with a carbodiimide such as dicyclohexylcarbodumide; or the product of the reaction of formic acid with a mixture of an azo compound such as diethyl or di-te/ -butyl azodicarboxylate and a phosphine such as triphenylphosphine. Conveniently, the reaction is carried out in an aromatic solvent, for example toluene or a xylene, at a temperature in the range, for example, 30 to 150°C, conveniently in the range, for example, 60 to 120°C and volatile side products are distilled off to allow the internal reaction temperature to rise to the boiling point of the solvent. A suitable catalyst for the preparation of an imine of Formula XI is, for example, an inorganic or organic acid. A suitable inorganic acid catalyst is, for example, hydrochloric, hydrobromic or sulphuric acid. A suitable organic acid catalyst is, for example, a
(l-8C)alkanoic acid such as formic, acetic, trifluoroacetic, propionic or butyric acid, a heteroarylcarboxylic acid such as a pyridine carboxylic acid, for example pyridine-4-carboxylic acid or nicotinic acid, a (l-8C)aIkanesulphonic acid such as methanesulphonic acid or an arylsulphonic acid such as benzenesulphonic acid or a toluenesulphonic acid. An alternative suitable organic acid catalyst is, for example, a salt of a suitable organic acid catalyst as defined hereinbefore with an organic amine base, for example with pyridine, 2,6-lutidine, collidine, 4-dimethylaminopyridine, triethyla ine, morpholine, N-methylmorpholine or diazabicyclo[5.4.0]undec-7-ene. Conveniently the acidic catalyst is formic acid or acetic acid. The invention is further illustrated, but not liinited, by the following Examples in which, unless otherwise stated, in general :- (i) operations were carried out at ambient temperature, i.e. in the range 17 to 25°C and under an atmosphere of an inert gas such as nitrogen or argon unless otherwise stated; (ii) in general, the course of reactions was followed by thin layer chromatography (TLC) and/or analytical high pressure liquid chromatography (HPLC); the reaction times that are given are not necessarily the rj jnimum attainable; (iii) when necessary, organic solutions were dried over anhydrous magnesium sulphate, work-up procedures were carried out after removal of residual solids by filtration, evaporations were carried out by rotary evaporation in vacuo; (iv) yields, where present, are not necessarily the maximum attainable, and, when necessary, reactions were repeated if a larger amount of the reaction product was required; (v) in general, the structures of reaction products were confirmed by nuclear magnetic resonance (NMR) and/or mass spectral techniques; electrospray mass spectral data were obtained using a Waters ZMD or Waters ZQ LC/mass spectrometer acquiring both positive and negative ion data, generally, only ions relating to the parent structure are reported; proton NMR chemical shift values were measured on the delta scale using a Bruker Spectrospin DPX300 spectrometer operating at a field strength of 300 MHz; the following abbreviations have been used: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad; (vi) intermediates were not necessarily fully purified but their structures and purity were assessed by TLC, analytical HPLC, infra-red (IR) and/or NMR analysis; (vϋ) unless otherwise stated, column chromatography (by the flash procedure) and medium pressure liquid chromatography (MPLC) were performed on Merck Kieselgel silica (Art. 9385); and
(viϋ) preparative HPLC was performed on C 18 reversed-phase silica, for example on a Waters 'Xterra' preparative reversed-phase column (5 microns silica, 19 mm diameter, 100 mm length) using decreasingly polar mixtures as eluent, for example decreasingly polar mixtures of water (containing 1% acetic acid or 1% aqueous ammonium hydroxide (d=0.88) and acetonitrile.
Example 1 4-(3'-cMoro-4'-fluoroaιnlino)-7-methoxy-6-(3-mon}hotooproOθxy)αιιinazoliιιe N,N'-bis-(3-CMoro-4-fluorophenyl)formamidine (0.49 g) and 2-amino-4-methoxy- 5-(3-morpholinopropoxy)beιιzonitrile (0.44 g) were slurried in a mixture of toluene (6.5 ml) and 4-toluenesulphonic acid (0.014 g). The reaction was heated to about 110°C for 96 hours. The mixture was cooled to ambient temperature over 1 hour and then stirred for 16 hours. The resultant solid was isolated by filtration and washed with toluene (3 x 1.5 ml) and dried in vacuo to give a pale pink solid (0.48 g). There was thus obtained the title compound; mp. 194-198°C. The N,N'-bis-(3-cHoro-4-fluorophenyl)forτnamidine starting material was prepared as follows :- Triethyl orthoformate (7.6 g) was added to a stirred solution of 3-chloro- 4-fluoro aniline (14.6 g), acetic acid (0.6 g) and toluene (44 ml) at ambient temperature. The reaction mixture was heated to 80°C and the resultant ethanol was removed under Dean-Stark conditions. The temperature of the reaction mixture was increased to 110°C to maintain a steady azeo trope. After 1.5 hours no further ethanol distillate was collected and the solution was cooled to ambient temperature. The solution was evaporated. The resultant solid was slurried in toluene (4 4 ml) and the toluene was evaporated. There was thus obtained the title compound as a pale brown solid (13.6 g); H NMR Spectrum: (DMSOd6) 6.92 (s, 2H), 7.07- 7.18 (m, 4H), 7.97 (s, 1H), NH not observed; 1 C NMR Spectrum: (DMSOd6) 117.06 (d), 118.76 (d), 120.96, 121.51 (d), 148.63, 153.62, 156.07: Mass Spectrum: M+H+ 301. The 2-amino-4-methoxy-5-(3-morpholinopropoxy)benzoιιitrile starting material as prepared as follows: 3-Hydroxy-4-methoxybenzaldehyde (36.7 kg) and sodium formate (30.6 kg) were added to formic acid (96%, 204 kg) and the resultant mixture was heated to approximately 85 °C. Hydroxylamine sulphate (21.6 kg) was added in eight equal portions at 30 minute intervals and the mixture was heated to 85°C for 5 hours. The resultant mixture was cooled to approximately 25°C and added to a solution of sodium chloride (140 kg) in water (700 litres). The resultant solid was collected by filtration, washed with water and dried to give 3-hydroxy- 4-methoxybenzonitrile (34 kg, 94%; Chemical Abstracts Registry Number 52805-46-6). A mixture of 3-hydroxy-4-methoxybenzonitrile (34.3 kg), potassium carbonate (54.5 kg) and N,N-dimethyjformamide (226 kg) was stirred and heated to approximately 85 °C.
A toluene solution (91.4 kg) containing N-(3-cHoropropyl)morpholine (41.1 kg) was added to the heated mixture and the resultant rnixture was heated to about 85°C for an additional 10 hours. The bulk of the N,N-dimethyjformamide was removed by vacuum distillation and the residue was diluted with water (286 litres). The aqueous mixture was extracted with three portions of a 1: 1 mixture of heptane and ethyl acetate (239 litres,
139 litres and 139 litres respectively). The combined organic layers were washed with water, concentrated to approximately 150 litres by vacuum distillation and diluted with glacial acetic acid (133 kg). Additional solvent was removed by vacuum distillation. Addition of glacial acetic acid (84 kg) provided a solution of 4-methoxy-3-(3-morpholjnopropoxy)benzonitrile (62.5 kg) in acetic acid (122 kg) which was used without further purification. [A portion of the 4-methoxy-3-(3-morpholinopiOpoxy)benzorjitrile was isolated using the following procedure :- A sample of the heptane and ethyl acetate solution was washed with water and evaporated to leave an oil. The oil was partitioned between tert-butyl methyl ether and water. The organic solution was dried over magnesium sulphate and evaporated. The residue was triturated under heptane and the resultant solid was isolated and dried at ambient temperature. There was thus obtained 4-methoxy-3-(3-morpholinopiOpoxy)benzorιitrile, rap. 52.4°C; NMR Spectrum: (DMSOd6) 1.87 (m, 2H), 2.38 (t, 4H), 2.38 (t, 2H), 3.57 (t, 4H), 3.84 (s, 3H), 4.05 (t, 2H), 7.11 (d, 1H), 7.39 (s, 1H), 7.42 (s, 1H); Mass Spectrum M+H+ 277.] Whilst the temperature of the reaction mixture was cooled to less than or equal to 20°C, a solution of 4-methoxy-3-(3-morpholinopropoxy)benzonitrile (62.5 kg) in acetic acid (122 kg) was added to a stirred rnixture of sulphuric acid (70%, 245 kg) and nitric acid (70%, 31 kg). After 2 hours, additional nitric acid (3.4 kg) was added and the resultant mixture was stirred as approximately 20°C for 50 hours. The mixture so obtained was added to water (1115 litres) and the resultant mixture was warmed to 30-35 °C. The mixture was basified to about pHl 1 by the addition of concentrated sodium hydroxide liquor. The reaction mixture was extracted with three portions of methylene chloride (679 kg, 272 kg and 272 kg respectively) and the combined organic extracts were filtered to remove particulate matter. The methylene chloride was removed by a sequence of distillation/addition steps involving the addition of six equal portions of ethyl acetate (total 840 litres). There was thus obtained a warmed (65°C) solution of the reaction product in ethyl acetate (360 litres). The solution was cooled to 5°C and the precipitate was isolated by filtration. There was thus obtained
4-methoxy-5-(3-mo hθjjnopropoxy)-2-rn^roberj^zorjitrile (56.4 kg), p. 127°C; NMR Spectrum (DMSOd6) 1.92 (m, 2H), 2.36 (m, 4H), 2.41 (t, 2H), 3.58 (m, 4H), 3.98 (s, 3H), 4.24 (t, 2H), 7.69 (s, 1H), 7.86 (s, 1H); Mass Spectrum: M+H+ 322. Sodium ditliionite (89%, 81.4 kg) was added to a stirred slurry of 4-methoxy- 5-(3-morphθjjnopropoxy)-2-nitrobenzonitrile (48.8 kg) in water (867 litres) and the resultant mixture was heated to 50°C for approximately 2 hours to complete the reaction. The temperature of the reaction mixture was raised to approximately 70°C and a concentrated aqueous hydrochloric acid solution (36%, 270 kg) was added over 3 hours. The resultant mixture was cooled to 20-25°C and sodium hydroxide liquor (47%, 303.7 kg) was added whilst stirring of the reaction mixture was continued. The reaction mixture was extracted with two portions of methylene chloride (1082 kg and 541 kg respectively) and the combined organic extracts were washed with water (510 litres). The organic phase was evaporated to give 2- amino-4-methoxy-5-(3-morpholjnopropoxy)benzoτjitrile (46.3 kg, 99% yield); mp. 87.5°C; NMR Spectrum: (DMSOd6) 1.79 (m, 2H), 2.36 (t, 4H), 2.36 (t, 2H), 3.56 (t, 4H), 3.73 (s, 3H), 3.86 (t, 2H), 5.66 (br s, 2H), 6.4 (s, 1H), 6.89 (s, 1H); Mass Spectrum: M+H+ 292. The N-(3-cMoropropyl)morpholine solution used as a starting material was obtained as follows :- A mixture of morpholine (178.5 kg) and toluene (560 litres) was stirred and warmed to approximately 77°C. l-Bromo-3-chloropropane (147 kg) was added slowly over approximately 2 hours and the resultant mixture was heated at approximately 77 °C for a further 20 hours. The mixture was cooled to ambient temperature and diluted with additional toluene (293 litres). The mixture was extracted with dilute aqueous hydrochloric acid solution (18%, 206 kg). The aqueous layer was separated, basified to pH9-10 by the addition of concentrated aqueous sodium hydroxide solution and extracted with toluene (250 litres). The resultant toluene layer was concentrated by distillation until a distillate having b.p. 56°C at 0.065 bar was obtained. There was thus obtained a toluene solution (129 kg) containing N-(3-cMoropropyl)mo holine (58 kg; Chemical Abstracts Registry Number 7357-67-7) that was used without further purification.
Exainple 2 4-(3'-cMoro-4'-fluoroaιιiMno)-7-methoxy-6-(3-ιnoiOhoKnopropoxy)αuinazoIine A mixture of 2-amino-4-methoxy-5-(3-mo hθjjnopropoxy)berjsonitrile (39.3 g), N,N'-bis-(3-cMoro-4-fluorophenyl)formarrιidine (44.6 g), formic acid (1.02 ml) and xylene (588 ml) was heated to 130°C for 9 hours. The mixture was allowed to cool to ambient temperature over 10 hours. The resultant solid was isolated by filtration, washed with xylene (2 x 80 ml) and dried in vacuo at 45°C for 16 hours. There was thus obtained the title compound as a pale brown solid (51.39 g); mp. 194-198°C.
Example 3
3-(3'-chloro-4'-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)- 3,4-d vdroαuinazoHn-4-iιrjine (of Formula II) The procedure described in Example 2 was repeated except that the reaction was heated to 110°C for only 24 hours before being cooled to ambient temperature. The crude reaction mixture was purified using preparative HPLC in order to isolate a sample of the imine intermediate. A Waters 'Xterra' MS C8 preparative reversed-phase column (5 microns silica, 19 mm diameter, 50 mm length) was used with an injection volume of 0.4 ml of a solution containing an estimated 40 mg of the imine was used. A solvent gradient ranging from a 19: 1 mixture of a 0.2% aqueous ammonium hydroxide solution and acetonitrile to a 1:1 mixture of water and acetonitrile was used as eluent. Under these conditions, the retention time of the imine was 2.71 minutes. There was thus obtained the title compound, the structure of which was confirmed H and 13C NMR spectroscopy (using approximately 3 mg of the material dissolved in CDC13) based on the following assignment of signals (chemical shift values for the 1H spectrum were referenced to tetramethylsilane and chemical shift values for the 13C spectrum were referenced to solvent at 77.0 ppπϊ).
Additional data were acquired by way of a Nuclear Overhauser Enhancement (NOE) experiment using a Varian Unitylnova 400 MHz spectrometer and by way of proton and carbon Heteronuclear Single Quantum Coherence (HSQC) and Heteronuclear Multiple Bond Correlation (HMBC) experiments using a Varian Unitylnova 500 MHz spectrometer. The NOE experiment showed correlations from H 12 to H10 and fromH5 to H7 that allowed the identification of the aromatic singlets for H7 and H10. Analysis of the HSQC and HMBC spectra enabled a full assignment for the compound to be made. All correlations observed were consistent with the 'imine' structure of the intermediate. Of the eight quaternary carbons in the molecule (at positions 6, 11, 8, 9, 14, 15, 17 and 18), C6 and Cll correlated with the aliphatic protons at H5 and H12 respectively, C8 and C9 gave correlations to H10, and
C15, C17 and C18 showed fluorine coupling and/or correlations to the aromatic protons H16, H19 and H20. C14 showed a chemical shift of 154.3 ppm which is close to that expected for such an imine carbon. In addition, proton HI 3 showed a correlation to C14 which demonstrated that these two atoms are connected through four or fewer bonds. IR Spectral data (cm1) :- 3310 (broad w); 2955 (w); 1637 (s); 1612 (m); 1496 (s);
1448 (s); 1409 (w); 1360 (w); 1325 (w); 1267 (s) ; 1226 (m); 1175 (w); 1100 (s); 1069 (m); 1035 (w); 990 (w); 961 (w); 862 (s); 845 (s); 827; 762; 726. There was no absorption band at around 2200-2400 cm"1 that would be expected if a CN group were present. Hence the isolated reaction intermediate was assigned the structure of the πriine of Formula II rather than the benzonitrile of Formula VI.