WO2004112687A2 - Antiviral acylguanidine compounds and methods - Google Patents
Antiviral acylguanidine compounds and methods Download PDFInfo
- Publication number
- WO2004112687A2 WO2004112687A2 PCT/AU2004/000866 AU2004000866W WO2004112687A2 WO 2004112687 A2 WO2004112687 A2 WO 2004112687A2 AU 2004000866 W AU2004000866 W AU 2004000866W WO 2004112687 A2 WO2004112687 A2 WO 2004112687A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- guanidine
- cinnamoylguanidine
- amiloride
- trans
- hydrochloride
- Prior art date
Links
- 0 CCC(N=C1C(N=C(*)N)=O)=C(N(C)CC(C)C)N(C)C1N Chemical compound CCC(N=C1C(N=C(*)N)=O)=C(N(C)CC(C)C)N(C)C1N 0.000 description 8
- NGGXACLSAZXJGM-UHFFFAOYSA-N CC(N=C(N)N)=O Chemical compound CC(N=C(N)N)=O NGGXACLSAZXJGM-UHFFFAOYSA-N 0.000 description 1
- AUCKYMGUOXTEQV-UHFFFAOYSA-N CCN(C(C)C)c1c(C2CC2)nc(C(N=C(N)N)=O)c(N)n1 Chemical compound CCN(C(C)C)c1c(C2CC2)nc(C(N=C(N)N)=O)c(N)n1 AUCKYMGUOXTEQV-UHFFFAOYSA-N 0.000 description 1
- MMVUASXGHTWYSF-VOTSOKGWSA-N Cc1c(/C=C/C(NC(N)=N)=O)cccc1 Chemical compound Cc1c(/C=C/C(NC(N)=N)=O)cccc1 MMVUASXGHTWYSF-VOTSOKGWSA-N 0.000 description 1
- JQNHSSXOJOMZCK-UHFFFAOYSA-N N=C(NC(CCc1cccc([N+]([O-])=O)c1)=O)NI Chemical compound N=C(NC(CCc1cccc([N+]([O-])=O)c1)=O)NI JQNHSSXOJOMZCK-UHFFFAOYSA-N 0.000 description 1
- FYOXCRMHHBYILR-KBXRYBNXSA-N NC(N)=NC(/C=C/C=C/c1ccccc1)=O Chemical compound NC(N)=NC(/C=C/C=C/c1ccccc1)=O FYOXCRMHHBYILR-KBXRYBNXSA-N 0.000 description 1
- HNQFEGDVNFRIBW-UHFFFAOYSA-N NC(N)=NC(c1cccc2c1CCC=C2)=O Chemical compound NC(N)=NC(c1cccc2c1CCC=C2)=O HNQFEGDVNFRIBW-UHFFFAOYSA-N 0.000 description 1
- UGJNJXYDMZXDLY-DHZHZOJOSA-N NC(NC(/C=C/c(cc1)ccc1-c1ccccc1)=O)=N Chemical compound NC(NC(/C=C/c(cc1)ccc1-c1ccccc1)=O)=N UGJNJXYDMZXDLY-DHZHZOJOSA-N 0.000 description 1
- FUCGZHDPARMCKE-UHFFFAOYSA-N NC(NC(C1=Cc2ccccc2C2C1=CCCC2)=O)=N Chemical compound NC(NC(C1=Cc2ccccc2C2C1=CCCC2)=O)=N FUCGZHDPARMCKE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D241/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with nitrogen atoms directly attached to ring carbon atoms
- C07D241/28—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with nitrogen atoms directly attached to ring carbon atoms in which said hetero-bound carbon atoms have double bonds to oxygen, sulfur or nitrogen atoms
- C07D241/34—(Amino-pyrazine carbonamido) guanidines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/46—Acylated amino or imino radicals by carboxylic acids, or sulfur or nitrogen analogues thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/15—Oximes (>C=N—O—); Hydrazines (>N—N<); Hydrazones (>N—N=) ; Imines (C—N=C)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4406—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 3, e.g. zimeldine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C279/00—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C279/20—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups containing any of the groups, X being a hetero atom, Y being any atom, e.g. acylguanidines
- C07C279/22—Y being a hydrogen or a carbon atom, e.g. benzoylguanidines
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to methods for retarding, reducing or otherwise inhibiting viral growth and/or functional activity.
- the invention also relates to compounds and compositions suitable for use in the methods.
- PCT/AU99/00872 describes the use of compounds 5-(N,N- hexamethylene)-amil ⁇ ide and 5-(N,N-dime1hyl)-amiloride in the treatment of HIV infection.
- HCV Hepatitis C virus
- Coronaviruses (Order Nid ⁇ vir les. family Coronaviridae, Genus Coronavirus) are enveloped positive-stranded RNA viruses that bud from the endoplasmic reticulum- Golgi intermediate compartment or the er ⁇ -Golgi network (Fischer, Stegen et al. 1998; Maeda, Maeda et al. 1999i Corse and Machamer 2000; Maeda, Repass et al. 2001 ; Ruo and Masters 2003)
- Animal coronaviruses can cause respiratory, gastrointestinal, neurological, or hepatic diseases in their host (Peiris, Lai et al. 2003).
- Several animal coronavirus are significant veterinary pathogens (Rota, Oberste et al. 2003).
- Severe acute respiratory syndrome is caused by a newly identified virus.
- SARS is a respiratory illness that has recently been reported in Asia, North America, and Europe (Peiris, Lai et al.2003).
- the causative agent of SARS was identified as a coronavirus. (Drosten, Gunther et al.2003; Ksiazek, Erd an et al. 2003; Peiris, Lai et al.2003).
- the World Health Organization reports that the cumulative number of reported probable cases of SARS from 1 November 2002 to the 11 th July 2003 is 8,437 with 813 deaths, nearly a 10% death rate. It is believed that SARS will not be eradicated, but will cause seasonal epidemics like the.cold or influenza viruses (Vogel 2003).
- the inventors have surprisingly found that certain compounds that fall under the classification of substituted acylguanidines have antiviral activity against viruses from a range of different virus families, Without intending to be bound by any $ particular theory or mechanism of action, and despite current dogma, it appears possible that viral replication can be retarded by inhibiting or otherwise down- regulating the activity of ion channels expressed in the host cell.
- the negative impact of the compounds of the present invention on viral replication may be mediated by the inhibition or otherwise down-regulation of a membrane ion channel 0 relied upon by the virus for repHcation.
- This membrane ion channel may be a viral membrane ion channel (exogenous to the host cell) or a host cell ion channel induced as a result of viral infection (endogenous to the host cell).
- the compounds f the present invention may inhibit Vpu or p7 function and thereby inhibit the continuation of the respective HIV or HCV life cycle.
- the SARS virus encodes an E protein which is shown for the first time, by the present inventors, to act as an ion channel.
- E proteins are present in other coronaviruses, the compounds, compositions and methods of the present invention would have utility in the inhibition and/or treatment of infections by other coronaviruses.
- the present invention is concerned with novel antiviral compounds that fall under the classification of substituted acylguanidines.
- a first aspect of the present invention provides an acylguanidine with antiviral activity.
- the present invention provides an antiviral compound of Formula I
- R4 7 wherein t -1 ⁇ are independently aromatic groups, heteroaromatic groups, alkylaromatic groups, alkylheteroaromatic groups, alkenylaromatic groups, alkenylheteroaromatic groups, cycloalkylaromatic groups, cycloalkylheteroaromatic groups, aryloxyalkyl groups, heteroaryloxyalkyl groups, said groups are mono or polycyclic, and are optionally substituted with one or more substitutents independently selected from hydrogen, hydroxy, nitro, halo, amino, substituted amino, alkyl-substituted ammo, cycloalkyl-substituted amino, aryl-substituted amino, Ci.
- the present invention provides an antiviral compound of Formula I
- R 2 , 3 and R are independently hydrogen
- X hydrogen, hydroxy, nitro, halo, Ci-ealkyl, halo-substituted Ci. ⁇ alkyloxy, phenyl, C t -ealkeneyl, C 3 . 6 cycloalkeneyl, C ⁇ alkeneoxy, or benzo; ⁇ Be, R ⁇ ,,Rfl f I , e, Rf.
- the compounds of the invention include the following: 5-(N,N-hcxamethylene)a iloride comprising the structure
- EIPA 5-(N-ethyl-N-isopropyl)amiloride
- N-Benzyl -N'rtS.S-diamino- ⁇ -chioro-pyizme ⁇ -carbony ⁇ -guamdine comprising the structure
- N-amidino-3,5-diamino-6-phemyl-2-pyrazinecarboxamide comprising the structure
- N-amidino-3-am o-5-phenyl-6-chloro-2-pyrazinecarboxamide comprising the structure
- Bodipy-FL Amiloride comprising the structure
- N-(2-napthoyl)-N'-phenylguanidine comprising the structure
- N,N'-bis(l-napthoyl)guanidme comprising the structure
- N-Cinnamoyl-N' N'-dimethylguanidine comprising the structure
- N,N'-Bis(amidino)napthalene-2,6-dica boxamide comprising the structure
- N,N'-Bis(3-phenyl ⁇ ro ⁇ aaoyl)guamdine comprising the structure
- the compounds of the invention are capable of reducing, retarding or otherwise inhibiting viral growth and/or replication.
- the antiviral activity of the compounds of the invention is against viruses such as those belonging to the Lentivirus family, and the Coronovirus family family of viruses.
- viruses such as Human Immunodeficiency Virus (HIV), Severe Acute Respiratory Syndrome virus (SARS), Mouse Hepatitis virus (), and Hepatitis C virus (HCV).
- HIV Human Immunodeficiency Virus
- SARS Severe Acute Respiratory Syndrome virus
- HCV Hepatitis C virus
- a pharmaceutical composition comprising an antiviral compound according to any one of the first, second or third aspects, and optionally one or more pharmaceutical acceptable carriers or derivatives, wherein said compound is capable of reducing, retarding or otherwise inhibiting viral growth and/or replication.
- the antiviral activity of the compounds of the invention is against viruses such as those belonging to the Lentivirus family, and the Coronovirus family of viruses.
- viruses such as Human Immunodeficiency Virus (HIV), Severe Acute Respiratory Syndrome virus (SARS), Human Coronavirus 229E, Human Coronavirus OC43, Mouse Hepatitis virus (MHV), Bovine Coronavirus (BCV), Porcine Respiratory Coronavirus (PRCV), Hepatitis C virus (HCV) and Equine Arteritis Virus (EAV).
- viruses such as Human Immunodeficiency Virus (HIV), Severe Acute Respiratory Syndrome virus (SARS), Human Coronavirus 229E, Human Coronavirus OC43, Mouse Hepatitis virus (MHV), Bovine Coronavirus (BCV), Porcine Respiratory Coronavirus (PRCV), Hepatitis C virus (HCV) and Equine Arteritis Virus (EAV).
- Coronaviruses hich can be inhibited or their infections treated by the compounds of the invention are those listed in Table 1.
- compositions of the invention may further comprise one or more known antiviral compounds or molecules.
- a method for reducing, retarding or otherwise inhibiting growth and/or replication of a virus comprising contacting a cell infected with said virus or exposed to said virus with a compound according to any one of the first, second or third aspects.
- the virus is from the Lentivirus family, or the Coronavirus family.
- the virus is Human Immunodeficiency Virus (HIV), Severe Respiratory Syndrome virus (SARS), Human Coronavirus 229E, Human Coronavirus OC43, Mouse Hepatitis virus (MHV), Bovine Coronavirus (BCV), Porcine Respiratory Coronavirus (PRCV), Mouse Hepatitis virus (MHV), Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV).
- HIV Human Immunodeficiency Virus
- SARS Severe Respiratory Syndrome virus
- Human Coronavirus 229E Human Coronavirus OC43
- MHV Mouse Hepatitis virus
- BCV Bovine Coronavirus
- PRCV Porcine Respiratory Coronavirus
- MHV Mouse Hepatitis virus
- HCV Hepatitis C virus
- EAV Equine Arteritis Virus
- the virus is HW-1, HIV-2, the SARS virus, Coronaviruse 229E, Coronavirus OC43, PRCV, BCV, HCV, or EAV.
- Coronaviruses which can be inhibited or their infections treated by the compounds of the invention are those listed in Table 1 ,
- a method for preventing the infection of a cell exposed to a virus comprising contacting said cell with a compound according to any one of the first, second or third aspects.
- the virus is from the Lentivirus family, or the Coronavirus family. More preferably, the virus is Human Immunodeficiency Virus (HIV), Severe Respiratory Syndrome virus (SARS), Human Coronavirus 229E, Human Coronavirus OC43, Mouse Hepatitis virus (MHV), Bovine Coronavirus (BCV), Porcine Respiratory Coronavirus (PRCV), Mouse Hepatitis virus (MHV), Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV). Most preferably, the virus is HJV-1, HIV-2, the SARS virus, Coronaviruse 229E, Coronavirus OC43, PRCV, BCV, HCV, EAV.
- HCV Human Immunodeficiency Virus
- SARS Severe Respiratory Syndrome virus
- MHV Mouse Hepatitis virus
- BCV Bovine Coronavirus
- PRCV Porcine Respiratory Coronavirus
- HCV Hepatitis C virus
- EAV Equine Arteritis Virus
- the virus is HJ
- Coronaviruses which can be inhibited or their infections treated by the compounds of the invention are those listed in Table 1.
- a method for the therapeutic or prophylactic treatment of a subject infected with or exposed to a virus comprising the administration of a compound according to any one of the first, second or third aspects, to a subject in need of said treatment.
- infection with a virus or exposure to a virus occurs with viruses belonging to the Lentivirus family, or the Coronovirus family. More preferably, infection or exposure occurs with HIV, SARS, Human Coronavirus 229E, Human Coronavirus OC43, Mouse Hepatitis virus (MHV), Bovine Coronavirus (BCV), Porcine Respiratory Coronavirus (PRCV), Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV). Most preferably, infection or exposure occurs with HlV-1, HIV-2, SARS, Human Coronavirus 229E, Human Coronavirus OC43, Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV). Other Coronaviruses which can be inhibited or their infections treated by the compounds of the invention are those listed in Table 1.
- the subject of the viral inhibition is generally a mammal such as but not limited to human, primate, livestock animal (e.g. sheep, cow, horse, donkey, pig), companion animal (e.g. dog, cat), laboratory test animal (e.g. mouse, rabbit, rat, guinea pig, hamster), captive wild animal (e.g. fox, deer).
- livestock animal e.g. sheep, cow, horse, donkey, pig
- companion animal e.g. dog, cat
- laboratory test animal e.g. mouse, rabbit, rat, guinea pig, hamster
- captive wild animal e.g. fox, deer.
- the subject is a primate, or horse.
- the subject is a human.
- a method of down regulating a membrane ion channel functional activity in a cell infected with a virus comprismg contacting said cell with a compound according to any one of the first, second or third aspects,
- the membrane ion channel may be endogenous to the cell or exogenous to the celL
- the membrane ion channel of which functional activity is down ⁇ egulated is that which Lentiviruses, and Coronaviruses utilise for mediating viral - replication and include, for example, the HIV membrane ion channel Vp , the HCV membrane ion channel P7, the Coronavirus E protein membrane ion channel, and the .
- infection with a virus or exposure to a virus occurs with viruses belonging to the Lentivirus family, or the Coronovirus family. More preferably, infection or exposure occurs with HIV, SARS, Human Coronavirus 229E, Human Coronavirus OC43, Mouse Hepatitis virus (MHV), Bovine Coronavirus (BCV), Porcine Respiratory Coronavirus (PRCV), Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV). Most preferably, infection or exposure occurs with HIV-1, HIV-2, SARS, Human Coronavirus 229E, Human Coronavirus OC43, Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV).
- a method of reducing, retarding or otherwise inhibiting growth and/or replication of a virus that has infected a cell comprising contacting said infected cell with a compound according to any one of the first, second or third aspects, wherein said compound down regulates functional activity of a membrane ion channel derived from said virus and expressed in said infected cell.
- infection occurs with a virus belonging to tiie Lentivirus family, or the Coronovirus family.
- infection or exposure occurs withHTV, SARS, Human Coronavirus 229E, Human Coronavirus OC43, Mouse Hepatitis virus (MHV), Bovine Coronavirus (BCV), Porcine Respiratory Coronavirus (PRCV), Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV).
- HTV Human Coronavirus 229E
- Human Coronavirus OC43 Mouse Hepatitis virus (MHV)
- BCV Bovine Coronavirus
- PRCV Porcine Respiratory Coronavirus
- HCV Hepatitis C virus
- EAV Equine Arteritis Virus
- the membrane ion channel of which functional activity is down regulated is that which Lentiviruses, and Coronaviruses utilise for mediating viral replication and include, for example, the HIV membrane ion channel Vpu , the HCV membrane ion channel VI, and the Coronavirus E protein membrane ion channel.
- the present invention provides a method of reducing, retarding or otherwise inhibiting growth and or replication of a virus that has infected a cell in a mammal, said method comprising administering to said mammal a compound according to any one of the first, second or third aspects, or a pharmaceutical composition according to the fourth aspect, wherein said compound or said composition down regulates functional activity of a membrane ion channel expressed in said infected cell
- infection occurs with a virus belonging to the Lentivirus family, or the Coronovirus family. More preferably, infection or exposure occurs with HIV, SARS, Human Coronavirus 229E, Human Coronavirus OC43, Mouse Hepatitis virus (MHV), Bovine Coronavirus (BCV), Porcine Respiratory Coronavirus (PRCV), Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV). Most preferably, infection or exposure occurs with HIV-1, HIV-2, SARS, Human Coronavirus 229E, Human Coronavirus OC43, Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV).
- HIV-1 HIV-2, SARS, Human Coronavirus 229E, Human Coronavirus OC43, Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV).
- the membrane ion channel of which functional activity is down regulated is that which Lentiviruses, and Coronaviruses utilise for mediating viral replication and include, for example, the HIV membrane ion channel Vpu , the HCV membrane ion channel P7, and the Coronavirus E protein membrane ion channel.
- the subject of the viral inhibition is generally a mammal such as but not limited to human, primate, livestock animal (e.g. sheep, cow, horse, donkey, pig), companion animal (e.g. dog, cat), laboratory test animal (e.g. mouse, rabbit, rat, guinea pig, hamster), captive wild animal (e.g. fox, deer).
- livestock animal e.g. sheep, cow, horse, donkey, pig
- companion animal e.g. dog, cat
- laboratory test animal e.g. mouse, rabbit, rat, guinea pig, hamster
- captive wild animal e.g. fox, deer
- the subjec is a primate, or horse.
- the subject is a human.
- the present invention provides a method for the therapeutic or prophylactic treatment of a subject infected with or exposed to a virus comprismg administering to said subject a compound according to any one of the first, second or third aspects, or a pharmaceutical composition according to the fourth aspect, wherein said compound or said composition down-regulates functional activity of a membrane ion channel derived from said virus.
- infection occurs with a virus belonging to the Lentivirus family, or the Coronovirus family of viruses. More preferably, infection or exposure occurs with HIV, SARS, Human Coronavirus 229E, Human Coronavirus OC43, Mouse Hepatitis virus (MHV), Bovine Coronavirus (BCV), Porcine Respiratory Coronavirus (PRCV), Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV). Most preferably, infection or exposure occurs with EtfV-1, HTV-2, SARS " , Human Coronavirus 229E, Human Coronavirus OC43, Hepatitis C virus (HCV), or Equine Arteritis Virus (EAV). Other Coronaviruses which can be inhibited or their infections treated by the compounds of the invention are those listed in Table 1.
- the membrane ion channel of which functional activity is down regulated is that which Lentiviruses, and Coronaviruses utilise for mediating viral replication and include, for example, the HTV membrane ion channel Vpu , the HCV membrane ion channel P7, and the Coronavirus E protein membrane ion channel,.
- the subject of the viral inhibition is generally a mammal such as but not limited to human, primate, livestock animal (e.g. sheep, cow, horse, donkey, pig), companion animal (e.g. dog, cat), laboratory test animal (e.g. mouse, rabbit, rat, guinea pig, hamster), captive wild animal (e.g. fox, deer).
- livestock animal e.g. sheep, cow, horse, donkey, pig
- companion animal e.g. dog, cat
- laboratory test animal e.g. mouse, rabbit, rat, guinea pig, hamster
- captive wild animal e.g. fox, deer.
- the subject is a primate, or horse.
- the subject is a human.
- the invention provides an antiviral compound selected from the group consisting of:
- Bodipy-FL amiloride 3-hydroxy-5-hexamefhyleneimino-amiloride
- the present invention provides a pharmaceutical composition comprising a compound according to the twelfth aspect, and optionally one or more pharmaceutical acceptable carriers or derivatives.
- the pharmaceutical composition may further comprise one or more known antiviral compounds or molecules.
- Figure 1 is a schematic representation of plasmids used for expression of Vpu in E. coli.
- A The amino acid sequence ( ⁇ 400 > 1) encoded by the vpu open reading frame (ORF) generated by PCR from an HIV- 1 strain HXB2 cDNA clone.
- the vpu ORF was cloned in-frame at the 3' end of the GST gene in p2GEX to generate p2GEXVpu (B). It was subsequently cloned into pPL451 to produce the plasmid pPL + Vpu (Q.
- FIG. 2 is a photographic representation of the expression and purification of Vpu in E. coli.
- A Western blotting after SDS-PAGE was used to detect expressed Vpu in E. coli extracts. Lanes 1-4 contain samples, at various stages of purity, of Vpu expressed from p2GEXVpu: lane 1, GST-Vpu fusion protein isolated by glutathione-agarose affinity chromatography; lane 2, Vpu liberated from the fusion protein by treatment with thrombin; lane 3, Vpu purified by HPLC anion exchange chromatography; lane 4, Vpu after passage through the im unoaffinity column. Lanes 5 and 6, membrane vesicles prepared from 42'C induced cells containing pPL+Vpu or pPL451 , respectively. B. Silver stained SDS-PAGE gelt lane 1, Vpu purified by HPLC anion exchange chromatography; lane 2, Vpu after passage through the immunoaffinity column.
- Figure 3 is a graphical representation of ion channel activity observed after exposure of lipid bilayers to aliquots containing purified Vpu.
- the CIS chamber contained 500mM NaCl and the TRANS chamber contained 50mM NaCl; both solutions were buffered at pH 6.0 with 10 mM MES.
- B shows a current versus voltage curve generated from data similar to that shown in A.
- Figure 4 is a photographic representation of bacterial cross-feeding assays.
- Met * , Pro auxotrophic strain was used to seed a soft agar overlay.
- Plates A and B contain minimal drop-out medium minus proline; in plate C the medium was minus methionine.
- the discs labelled P and M contained added proline or methionine, respectively.
- the discs labelled C and V were inoculated with Met + , Pro + E, coli cells containing the plasmids ⁇ PL451 orpPL+Vpu, respectively.
- FIG. 5 is a graphical representation of the screening of drugs for potential Vpu channel blockers. The photograph shows a section of a minimal medium-lacking adenine - agarose plate onto which a lawn of XLrl-blue E. coli cells containing the Vpu expression plasmid pPLVpu has been seeded.
- Numbers 6-11 are located at the sites of application of various drugs being tested, which were applied in 3 ⁇ l drops and allowed to soak into the agarose. The plate was then incubated at 37°C for 4Shr prior to being photographed. The background grey shade corresponds to areas of no bacterial growth.
- the bright circular area around " 10 " represents bacterial cell growth as a result of application of adenine at that location (positive control).
- the smaller halo of bacterial growth around "9” is due to the application of 5-(HN- he ⁇ amethylene)-amiloride at that location.
- FIG. 6 S RS E protein ion channel activity observed in NaCl solutions after exposure of lipid bi ⁇ ayer to 3-10 ⁇ g of E protein.
- the CIS chamber contained 50mM NaCl in 5mM HEPES buffer pH 7.2
- the TRANS chamber contained 500mM NaCl in 5mM HEPES buffer pH 7.2.
- the CIS chamber was earthed and the TRANS chamber was held at various potentials between -100 to +1 OOmV.
- FIG. 7 SARS E protein ion channel activity observed in NaCl solutions after exposure of lipid bilayer to 3-10 ⁇ g of E protein.
- the CIS chamber contained 50n ⁇ M NaCl in 5mM HEPES buffer pH 7.2
- the TRANS chamber contained 500mM NaCl in 5mM HEPES buffer pH 7.2.
- the CIS chamber was earthed and the TRANS chamber was held at various potentials between -100 to +1 OOmV.
- Cinnamoylgua ⁇ idine inhibits SARS E protein ion channel activity in NaCl solution.
- A Representative currents at holding potential of-40mV, Scale bar is 30QmS and 5pA.
- B All points histogram at holding potential of - 4QmV.
- C Average current (pA), before formation of E protein ion channel, E protein ion channel activity and after addition of 1 OO ⁇ M Bit036.
- Figure 10 Part A shows raw currents generated by the 229E-E protein ion channel in a planar lipid bilayer. The top trace shows current activity prior to drug addition and the lower trace shows the effect of addition of lOO ⁇ M cinnamoylguanidine on channel activity.
- Part B is a graphical representation of the average current flowing across the bilayer (in arbitrary units), before and after addition of cinnamoylguanidine.
- Figure 11 MHV E protein Ion channel activity in Hpid bilayers NaCl solutions.
- Part A shows raw currents generated by the MHV-E protein ion channel in a planar lipid bilayer.
- the top trace shows current activity prior to drug addition and the lower trace shows the effect of addition of lOO ⁇ M cinnamoylguanidine on channel activity
- Part B is a graphical representation of the average current flowing across the bilayer (in arbitrary units), before and after addition of cinnamoylguanidine.
- the present invention is based, in part, on the surprising determination that certain compounds that fall under the classification of substituted acylguanidines have antiviral activity against viruses from a range of different virus families.
- the negative impact of the compounds of the present invention on viral replication may be mediated by the inhibition or otherwise down-regulation of a membrane ion channel rehed upon by the virus for replication.
- This membrane ion channel may be a viral membrane ion channel (exogenous to the host cell) or a host cell ion channel induced as a result of viral infection (endogenous to the host cell).
- the compounds of the present invention may inhibit Vpu or p7 function and thereby inhibit the continuation of the respective HIV or HCV life cycle.
- the SARS virus encodes an E protein which is shown for the first time, by the present inventors, to act as an ion channel.
- E proteins are present in other coronaviruses, the compounds, compositions and methods of the present invention would have utility in the inhibition and/or treatment of infections by other coronaviruses.
- While the present invention is concerned with novel antiviral compounds falling under the classification of substituted acylguanidines, it does not include in its scope the use of compounds 5-(N,N-hexamethylene)amiloride and 5-(N,N-dimethyl)- amiloride for retarding, reducing or otherwise inhibiting viral growth and/or functional activity of HIV. It will be understood by those skilled in the art that the compounds of the invention may be administered in the form of a composition or formulation comprising pharmaceutically acceptable carriers and excipients.
- compositions of the invention may further comprise one or more known antiviral compounds or molecules.
- the known antiviral compounds are selected from the group consisting of Vidarabine, Acyclovir, Ganciclovir, Valgan clovir, Valacyclovir, Cidofovir, Fa ciclovir, Ribavirin, Amantadine, Rimantadine, Interferon, Oseltamivir, Palivizumab, Rimantadine, Zanamivir, nucleoside-analog reverse transcriptase inhibitors (NRTI) such as Zidovudine, Didanosine, Zalcitabine, Stavudine, Lamivudine and Abacavir, non- nucleoside reverse transcriptase inhibitors (NNRIT) such as Nevirapine, Delavirdine and Efav ⁇ renz, protease inhibitors such as Saquinavir, Ritonavir, Indinavir, Nelfinavir, Amprenavir, and
- Canine enteric coronavirus (strain INSAVC-l)
- Canine enteric coronavirus (strain K378)
- Feline enteric coronavirus (strain 79-1683)
- Bovine coronavirus (strain LSU-94LSS-0S1)
- Bovine coronavirus (STRAIN LY-138)
- Bovine coronavirus (strain O .-0514-3)
- Bovine coronavirus (STRAIN QUEBEC)
- Bovine enteric coronavirus (strain 98TXSF-110-ENT)
- Murine coronavirus strain DVBVt
- Murine hepatitis virus (strain A59)
- Murine hepatitis virus (strain S)
- Murine hepatitis virus strain l Murine hepatitis virus strain l
- Murine hepatitis virus strain 2 Murine hepatitis virus strain 2
- Murine hepatitis virus strain 3 Murine hepatitis virus strain 3
- Murine hepatitis virus strain 4 Murine hepatitis virus strain 4.
- Porcme hemagglutinating encephalomyelitis virus (strain IAF-404) Puffinosis virus
- Rat coronavirus (strain 681)
- Rat coronavirus (strain NJ)
- Bovine respiratory coronavirus (strain 98TXSF-110-LUN)
- the present invention extends to ion channels which may function by means such as passive, osmotic, active or exchange transport.
- the ion channel may be formed by intracellular or extracellular means.
- the ion channel maybe an ion channel which is naturally formed by a cell to facilitate its normal functioning.
- the ion channel ma be formed by extracellular means.
- Extracellular means would include, for example, the formation of ion channels due to introduced chemicals, drugs or other agents such as ionophores or due to the functional activity of viral proteins encoded by a virus which has entered a cell.
- the ion channels which are the subject of certain embodiments of the present invention facilitate the transport of ions across membranes.
- Said membrane may be any membrane and is not limited to Ihe outer cell wall plasma membrane.
- membrane encompasses the membrane surrounding any cellular organelle, such as the Golgi apparatus and endoplasmic reticulum, the outer cell membrane, the membrane surrounding any foreign antigen which is located within the cell (for example, a viral envelope) or the membrane of a foreign organism which is located extracellularly.
- the membrane is typically, but not necessarily, composed of a fluid lipid bilayer.
- the subject ion channel may be of any structure.
- the Vpu ion channel is foiflied by Vpu which is an integral membrane protein encoded by HTV-1 which associates with, for example, the Golgi and endoplasmic reticulum membranes of infected cells.
- Vpu ion channels is a reference to all related ion channels for example P7 HCV and M2 cf influenza and the like.
- HAV HTV
- SARS SARS
- Coronavirus coronavirus
- HCV HCV
- references to the "functional activity" of an ion channel should be understood as / a reference to any one or more of the functions which an ion channel performs or is involved in.
- the Vpu protein encoded ion channel in addition to facilitating the transportation of Na + , K + , CI * and P0 3" , also plays a role in the degradation of the CD4 molecule in the endoplasmic reticulum.
- the Vpu protein encoded ion channel is also thought to play a role in mediating the HIV life cycle.
- the present invention is not limited to treating HIV infection via the mechanism of inhibiting the HIV life cycle and, in particular, HIV replication. Rather, the present invention should be understood to encompass any mechanism by which the compounds of the present invention exert fheir anti-viral activity and may include inhibition of HIV viability or functional activity. This also applies to HCV, Coronaviruses, and to other viruses.
- Ion channel mediation of viral replication may be by direct or indirect means. Said ion channel mediation is by direct means if the ion channel interacts directly with the virion at any one or more of its life cycle stages. Said ion channel mediation is indirect if it interacts with a molecule other than those of the virion, which other molecule either directly or indirectly modulates any one or more aspects or stages of the viral life cycle. Accordingly, the method of the present invention encompasses the mediation of viral replication via the induction of a cascade of steps which lead to the mediation of any one or more aspects or stages of the viral life cycle.
- references to "down-regulating' ion channel functional activity should be understood as a reference to the partial or complete inhibition of any one or more aspects of said activity by both direct and indirect mechanisms.
- a suitable agent may interact directly with an ion channel to prevent replication of a virus or, alternatively, may act indirectly to prevent said replication by, for example, interacting with a molecule other than an ion channel.
- a further alternative is that said other molecule interacts with and inhibits the activity of the ion channel.
- a "cell” infected with a virus should be understood as a reference to any cell, prokaryotic or eukaryotic, which has been infected with a virus. This includes, for example, immortal or primary cell lines, bacterial cultures and cells in situ.
- the preferred infected cells would be macrophages monocytes or hepatocytes/lymphoid cells infected with either HIV or HCV respectively.
- the compounds of the present invention are thought to inhibit viral replication or virion release from cells by causing ion channels, namely VPU of HIV, the E protein of SARS and other Coronaviruses, or P7 of HCV to become blocked.
- the present invention encompasses antiviral compounds that are substituted acylguanidines.
- the present invention also includes the use of compounds 5-(N,N- hexamethylene)amiloride and 5-(N,N-dimethyl)-amiloride in the control of viral replication and/or growth other than HTV.
- the subject of the viral inhibition is generally a mammal such as but not limited to human, primate, livestock animal (e.g. sheep, cow, horse, donkey, pig), companion animal (e.g. dog, cat), laboratory test animal (e.g. mouse, rabbit, rat, guinea pig, hamster), captive wild animal (e.g. fox, deer).
- livestock animal e.g. sheep, cow, horse, donkey, pig
- companion animal e.g. dog, cat
- laboratory test animal e.g. mouse, rabbit, rat, guinea pig, hamster
- captive wild animal e.g. fox, deer
- the method of the present invention is useful in the treatment and prophylaxis of viral infection such as, for example, but not limited to HTV infection, HCV infection and other viral infections.
- the antiviral activity may be effected in subjects known to be infected with HIV in order to prevent replication of HIV thereby preventing the onset of AIDS.
- the method of the present invention may be used to reduce serum viral load or to alleviate viral infection symptoms.
- antiviral treatment may be effected in subjects known to be infected with, for example, HCV, in order to prevent replication of HCV, thereby preventing the further hepatocyte involvement and the ultimate degeneration of liver tissue.
- the method of the present invention may be particularly useful either in the early stages of viral infection to prevent the establishment of a viral reservoir in affected cells or as a prophylactic treatment to be applied immediately prior to or for a period after exposure to a possible source of virus.
- terin "prophylaxis” may be considered as reducing the severity of onset of a particular condition. Therapy may also reduce the severity of an existing condition or the frequency of acute attacks.
- more than one compound or composition may be co-administered with one or more other compounds, such as known anti-viral compounds or molecules.
- co- administered is meant simultaneous administration in the same formulation or in two different formulations via the same or different routes or sequential administration by the same or different routes.
- sequential administration is meant a time difference of from seconds, minutes, hours or days between the administration of the two or more separate compounds.
- the subject antiviral compounds may be administered in any order.
- Routes of administration include but are not limited to intravenously, intraperitionealy, subcutaneously, intracranialy, intradermally, intramuscularly, intraocularly, intrathecaly, intracerebrally, intranasally, transmucosally, by infusion, orally, rectally, via iv drip, patch and implant. Intravenous routes are particularly preferred.
- compositions suitable for injectable use include sterile aqueous solutions (where water soluble) and sterile powders for the extemporaneous preparation of sterile injectable solutions.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol and liquid polyethylene glycol, and the like), suitable mixtures thereof and vegetable oils.
- the prevention of the action of microorganisms can be brought about by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thi ⁇ nerosal and the like. In many cases, it will be preferable to include isotonic agents, for example, sugars or sodium chloride.
- Prolonged absorption of the injectable compositions can be brought about by the use in the compositions of agents delaying absorption, for example, aluminum monostearate and gelatin.
- Sterile injectable solutions are prepared by incorporating the active compounds in the required amount in the appropriate solvent with various of the other ingredients enumerated above, as required, followed by, for example, filter sterilization or sterilization by other appropriate means.
- Dispersions are also contemplated and these may be prepared by incorporating the various sterilized active ingredients into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above.
- a preferred method of preparation includes vacuum drying and the fieeze-drying technique which yield a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution.
- the active ingredients may be orally administered, for example, with an inert diluent or with an assimilable edible carrier, or it may be enclosed in hard or soft shell gelatin capsule, or it may be compressed into tablets.
- the active compound may be incorporated with excipients and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like.
- compositions and preparations should contain at least 0.01 % by weight, more preferably 0.1 % by weight, even more preferably 1% by weight of active compound.
- the percentage of the compositions and preparations may, of course, be varied and may conveniently be between about 1 to about 99%, more preferably about 2 to about 90 %, even more preferably about 5 to about 80% of the weight of the unit.
- the amount of active compound in such therapeutically useful compositions in such that a suitable dosage will be obtained.
- Preferred compositions or preparations according to the present invention are prepared so that an oral dosage unit form contains between about 0.1 ng and 2000 mg of active compound.
- the tablets, troches, pills, capsules and the like may also contain the components as listed hereafter: A binder such as gum, acacia, com starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid and the like; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin may be added or a flavouring agent such as peppermint, oil of wintergreen, or cherry flavouring.
- a binder such as gum, acacia, com starch or gelatin
- excipients such as dicalcium phosphate
- a disintegrating agent such as corn starch, potato starch, alginic acid and the like
- a lubricant such as magnesium stearate
- a sweetening agent such as sucrose, lactose or saccharin
- a flavouring agent such as peppermint, oil of wintergreen, or
- tablets, pills, or capsules may be coated with shellac, sugar or both.
- a Syrup or elixir may contain the active compound, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and flavouring such as cherry or orange flavour. Any material used in preparing any dosage unit form should be pharmaceutically pure and substantially non-toxic in the amounts employed.
- the active compound(s) may be incorporated into sustained-release preparations and formulations.
- the present invention also extends to forms suitable for topical application such as creams, lotions and gels.
- the anti-clotting peptides may need to be modified to permit penetration of the surface barrier.
- Procedures for the preparation of dosage unit forms and topical preparations are readily available to those skilled in the art from texts such as Pharmaceutical Handbook. A Martind ⁇ le Companion Volume Ed. Ainley Wade Nineteenth Edition The Pharmaceutical Press London, CRC Handbook of Chemistry and Physics Ed. Robert C. Weast Ph D. CRC Press Inc.; Goodman and G ⁇ lm n's; The Pharmacological basis ofTherapeutics. Ninth Ed McGraw Hill; Remington- and The Science and Practice of Pharmacy. Nineteenth Ed. Ed. Alfonso R. Gennaro Mack Publishing Co. Easton Pennsylvania.
- Pharmaceutically acceptable carriers and/or diluents include any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like.
- the use of such media and agents for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, use thereof in the therapeutic compositions is contemplated.
- Supplementary active ingredients can also be incorporated into the compositions. It is especially advantageous to formulate parenteral compositions in dosage unit form for ease of administration and uniformity of dosage.
- Dosage unit form refers to physically discrete units suited as unitary dosages for the mammalian subjects to be treated; each unit containing a predetermined quantity of active material calculated.to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- the specification for the novel dosage unit forms of the invention are dictated by and directly dependent on (a) the unique characteristics of the active material and the particular therapeutic effect to be achieved and (b) the limitations inherent in the art of compounding.
- Effective amounts contemplated by the present invention will vary depending on the severity of the pain and the health and age of the recipient. In general terms, effective amounts may vary from 0.01 ng/kg body weight to about 100 g kg body weight.
- Alternative amounts include for about 0. 1 ng kg body weight about 100 mg kg body weight or from 1.0 ng/kg body weight to about 80 mg kg body weight.
- the subject of the viral inhibition is generally a mammal such as but not limited to human, primate, livestock animal (e.g. sheep, cow, horse, donkey, pig), companion animal (e.g. dog, cat), laboratory test animal (e.g. mouse, rabbit, rat, guinea pig, hamster), captive wild animal (e.g. fox, deer).
- livestock animal e.g. sheep, cow, horse, donkey, pig
- companion animal e.g. dog, cat
- laboratory test animal e.g. mouse, rabbit, rat, guinea pig, hamster
- captive wild animal e.g. fox, deer.
- the subject is a human or primate. Most preferably, the subject is a human.
- the methods of the present invention is useful in the treatment and prophylaxis of viral infection such as, for example, but not limited to HTV infection, HCV infection and other viral infections.
- the antiviral activity may be. effected in subjects known to be infected with HIV in order to prevent replication of HIV thereby preventing the onset of AIDS.
- the methods of the present invention may be used to reduce serum viral load or to alleviate viral infection symptoms.
- antiviral treatment may be effected in subjects known to be infected with, for example, HCV, in order to prevent replication of HCV, thereby preventing the further hepatocyte involvement and the ultimate degeneration of liver tissue.
- the methods of the present invention may be particularly useful either in the early stages of viral infection to prevent the establishment of a viral reservoir in affected cells or as a prophylactic treatment to be applied immediately prior to or for a period after exposure to a possible source of virus.
- the compounds of the present invention may be made from the corresponding acid chlorides or methyl esters as shown in Scheme 1. Both of these methods are well described in the literature.
- cDNA fragments for the various viral proteins listed in Table 2 were obtained either by PCR amplification from a parental virus genome clone, or by direct chemical synthesis of the polynucleotide sequence.
- Vpu open reading frame encoding Vpu (Fig la) was amplified by PCR from a cDNA clone of an Nde I fragment of the HIV-1 genome (isolate HXB2, McFarlane Burnet Centre, Melbourne, Australia) as follows: Native PfU DNA polymerase (Stratagene; 0.035 U/ D) was chosen to catalyse the PCR reaction to minimise possible PCR introduced errors by virtue of the enzyme's proofreading activity.
- the 5 sense, primer AGTAGGATCCATGCAACCTATACC ( ⁇ 400 > 2) introduces a BamHl site (underlined) for cloning in-frame with the 3' end of the GST gene in p2GEX (41).
- This primer also repairs the start codon (bold T replaces a Q of the vpu gene which is a threonine codon in the HXB2 isolate.
- TCTGGAATTLTACAGATCAT CAAC ( ⁇ 400 > 3) introduces an EcoRl site (underlined) to the other end of the PCR product to facilitate cloning.
- the 268bp fragment was purified, digested with BamHl and EcoRl and ligated to p2GEX prepared by digestion with the same two enzymes.
- the resultant recombinant plasmid is illustrated in Fig lb.
- the entireVpu open reading frame and the BamHl and EcoRl ligation sites were sequenced by cycle sequencing, using the Applied Biosystems dye-terminator kit, to confirm the DNA sequence.
- Other cDNAs were synthesised for us using state of the art methods by GenScript Corporation (New Jersey, USA). Codon sequences were optimised for expression in bacterial, insect or mammalian cells, as appropriate. Restriction endonuclease enzyme recognition sites were incorportated at the 5' and 3' ends of the synthetic cDNAs to facilitate cloning into plasmid expression vectors, ⁇ cDNA3.1, pFastBac and pPL451 for expression of the encoded virus proteins in mammalian, insect or bacterial cells, respectively.
- p2GEXVpu was first digested with BamHl and the 5' base overhang was filled in the Klenow DNA poly erase in the presence of dNTPs.
- the , Vpu-encoding fragment was then liberated by digestion with EcoRl, purified from an agarose gel and ligated into pPL451 which had been digested with Hpal and EcoRl.
- Western blots subsequently confirmed that the pPLVpu construct (Fig lc) expressed Vpu after induction of cultures at 42°C to inactivate the cI857 repressor of the PR and PL promoters.
- E. coli strain XU-blue cells containing p2GEXVpu were grown at 30°C with vigorous aeration in LB medium supplemented with glucose (6g L) and ampicillin (50mg L) to a density of approximately 250 Klett units, at which time IPTG was added to a final concentration of O.OhnM and growth was continued for a further 4hr.
- the final culture density was approximately 280 Klett units.
- the osmotically sensitised cells were pelleted at 12,000g and resuspended to the original volume in water to burst the cells.
- the suspension was then made up to lxMTEBS/DTT using a lOx buffer stock and the ghosts were isolated by centrifugation and resuspended in MTPBS/DTT to which was then sequentially added glycerol (to 20 % wt vol) and CHAPS (to 2 % wt/vol) to give a final volume of one quarter the original volume. This mixture was stirred on ice for 1 hr and then centrifuged at 400,000g for lhr to remove insoluble material.
- the GST-Vpu fusion protein was purified from the detergent extract by affinity chromatography on a glutathione agarose resin (Sigma). The resin was thoroughly washed in 50mM Tris pH 7.5 containing glycerol (5 %), DTT (ImM), and CHAPS (0.5 %) (Buffer A) and then the Vpu portion of the fusion protein was liberated and eluted from the resin-bound GST by treatment of a 50% (v v) suspension of the beads with human thrombin (lOOU/ml; 37°C for lhr). PMSF . (0.5mM) was added to the eluant to eliminate any remaining thrombm activity.
- This Vpu fraction was further purified on a column of MA7Q anion exchange resin attached to a BioRad HPLC and eluted with a linear NaCl gradient (0-2M) in buffer A.
- the Vpu was purified to homogeneity - as determined on silver stained gels - on an immunoaffinity column as follows: HPLC fractions containing Vpu were desalted on a NAP 25 column (Pharmacia) into buffer A and then mixed with the antibody- agarose beads for lhr at room temperature. The beads were washed thoroughly and Vpu was eluted by increasing the salt concentration to 2M. Protein was quantitated using the BioRad dye binding assay.
- the plasmid p2GEXVpu (Fig. 1) was constructed to create an in-frame gene fbsion between the GST and Vpu open-reading frames. This system enabled IPTG-inducible expression of the Vpu polypeptide fUsed to the C-te ⁇ ninus of GST and allowed purification of the fusion protein by affinity chromatography on glutathione agarose. Optimal levels of GST-Vpu expression were obtained by growing the cultures at 30°C to a cell density of approximately 250-300 Klett units and inducing with low levels of IPTG (O.Ol M).
- a combined cellular fraction containing the cell debris and plasma membrane was prepared by lysozyme treatment of the induced cells followed by a low-speed centrifugation. Approximately 50% of the GST-Vpu protein could be solubilised from this fraction using the zwitterionic detergent CHAPS. Affinity chromatography using glutathione-agarose beads was used to enrich the fusion protein and thrombm was used to cleave the fusion protein at the high affinity thrombin site between the fusion partners, liberating Vpu (Fig. 2A). In fractions eluted from the anion exchange column Vpu was the major protein visible on silver stained gels (Fig.2B, lane 1).
- Vpu was purified to apparent homogeneity on an immunoaffinity column (Fig.2B, lane 2).
- the N-terminal amino acid sequence of the protein band (excised from SDS-PAGE gels) corresponding to the immunodetected protem confirmed its identity as Vpu.
- Proteoliposomes containing Vpu were prepared by the detergent dilution method (New, 1990). A mixture of lipids (PEiPC:PS; 5:3:2; lmg total lipid) dissolved in chloroform was dried under a stream of nitrogen gas and resuspended in 0.1 ml of potassium phosphate buffer (50mM pH 7.4) containing DTT (ImM). A 25 ⁇ l aliquot containing purified Vpu was added, followed by octylglucoside to a final concentration of 1.25 % (wt vol).
- This mixture was subject to three rounds of freezing in liquid nitrogen, thawing and sonication in a bath type sonicator (20-30 sec) and was then rapidly diluted into 200 volumes of the potassium phosphate buffer.
- Proteoliposomes were collected by centrifugation at 400,000g for lhr and resuspended in approximately 1 0 ⁇ l of phosphate buffer.
- Vpu was tested for its ability to induce channel activity in planar lipid bilayers using standard techniques as described elsewhere (Miller, 1986; and Piller et al, 1996).
- the solutions in the CIS and TRANS chambers were separated by a DelrinTM plastic wall containing a small circular hole of approximately lOO ⁇ m diameter across which a lipid bilayer was painted so as to form a high resistance electrical seal.
- Bilayers were painted from a mixture (8:2) of palmitoyl-oleoly- phosphatidyl-ethanolamine and pahnitoyl-oleolyphosphatidyl-choline (Avanti Polar L ⁇ pids, Alabaster, Alabama) in n-decane.
- the solutions in the two chambers contained MES buffer (lOmM, pH 6.0) to which various NaCl or KCl concentrations were added. Currents were recorded with an AxopatchTM 200 amplifier. The electrical potential between the two chambers could be manipulated between +/-200mV (TRANS relative to grounded CIS). Aliquots containing Vpu were added to the CIS chamber either as a detergent solution or after incorporation of the protein into phospholipid vesicles. The chamber was stined until currents were observed.
- Example 10 Vpu Forms Ion Channels in Lipid Bilayers.
- Channel activity was observed in over 40 individual experiments with Vpu samples prepared from five independent purifications. In different experiments, the amplitude of the currents varied over a large range and, again, seemed to approximately correlate with the amount of protein added. The smallest and largest channels measured had conductances of 14 pS and 280 pS, respectively. The channels were consistently smaller when lipid vesicles containing Vpu were prepared and fused to the bilayer rather than when purified protein in detergent solution was added. This may be because the former method included treatment with high concentrations of detergent and a dilution step that may have favoured the breakdown of large aggregates into monomers.
- This bio-assay is based on the observation that expression of Vpu in E. coli results in an active Vpu channel located in the plasmalemma that dissipates the transmembrane sodium gradient.
- a sodium dependent co-transporter for example proline or adenine
- metabolites whose accumulation within the cells is mediated by a sodium dependent co-transporter (for example proline or adenine) leak out of the cell faster than they can be synfhesised so that the metabolites' intracellular levels become limiting for growth of the cell.
- a sodium dependent co-transporter for example proline or adenine
- the vpu open-reading frame was cloned into the plasmid pPL451 to create the recombinant plasmid pPL-Vpu (Fig. lb).
- the strong P and P lambda promoters are used to drive expression of Vpu under control of the temperature sensitive cl857 represser, such that when grown at 30°C expression is tightly repressed and can be induced by raising the temperature to between 37*C and 42 ⁇ C.
- the temperature sensitive cl857 represser such that when grown at 30°C expression is tightly repressed and can be induced by raising the temperature to between 37*C and 42 ⁇ C.
- cells containing pPL-Vpu grew when incubated at 30°C and 37°C but not at 42°C, while control strains grew well at 42°C.
- the plasma membrane fraction was prepared and western blotting, using an antibody that specifically binds to the C-terminus of Vpu, detected a single band at approximately l ⁇ fcDa, indicating that Vpu was expressed an associated with the membranes (Fig. 2A, lane 5).
- Example 14 E.Coli Cells Expressing Vpn Require Adenine in the External Medium for Growth. It was observed that, due to an uncharacterised mutation in the adenine synthesis pathway, growth of E. coli cells of the XLI-blue strain expressing Vpu at 37°C was dependant on the presence of adenine in the medium.
- Vpu N-terminal peptide (residues 1- 32) dissolved in trifluoroethanol was added to the CIS chamber of the bilayer apparatus and the solutions was stirred until ion currents were observed, indicating incorporation of one or more Vpu ion channels into the bilayer. After recording the channel activity for a few minutes, drugs were added to the solutions in the CIS and TRANS chambers - with stirring - to a final concentration of lOO ⁇ M.
- Channel activity was then recorded for at least a further three minutes and the effect of drug addition on ion current was determined by comparing the channel activity before and after drug addition.
- drug effect was classified into four categories: “Stong block”, if current was inhibited approximately 90-100%; “weak block”, approx. 50-90% inhibition; “partial block”, ⁇ 50%; and “no effect”.
- Stong block if current was inhibited approximately 90-100%
- “weak block” approx. 50-90% inhibition
- Partial block ⁇ 50%
- no effect Experiments were disregarded if currents larger than ⁇ 50pA were generated after . addition of Vpu N-peptide because in such cases it is possible that non-native peptide aggregates contribute to bilayer breakdown. Such aggregates, by virtue of their disorganized structure may not be specifically blocked by the drugs at the concentrations tested.
- Example 16 Compound Screening using the Bacterial Bio-Assav for the Vpu protein.
- halos of growth around the site of application of particular drugs - as described in example 14— were given a score between zero and six reflecting the size and density of the zone of bacterial cell growth. Scores greater than 3 represent strong inhibition of the Vpu protein; scores between 1.5 and 3 . represent moderate inhibition and scores between 0.01 and 1.5 represent fair inhibition.
- Table 4 lists the scores for inhibition of Vpu protein in the bacterial bio-assay.
- Human monocytes were isolated from peripheral blood and cultured either for 24hr (one day old monocytes) or for 7 days to allow differentiation into monocyte derived acrophages (MDM). These cells were then exposed to cell-free preparations of HIV isolates and allowed to absorb for 2hr before complete aspiration of the medium, washing once with virus-free medium and resuspension in fresh medium. The cells were exposed to various concentration of compound either 24 hr prior to infection or after infection. Subsequent HIV replication, at various times after infection, was compared in cells exposed to drugs and in cells not exposed to drugs (controls). The progression and extent of viral replication was assayed using either an HIV DNA PCR method (Fear et al, 1998) or an ELISA method to quantitate ⁇ 24 in culture supernatants (Kelly et al, 1998).
- Table 5 provides examples of results obtained using this assay and test antiviral compounds.
- Example IS SARS Coronavirus.
- a peptide corresponding to the full-length SARS-CoV (isolate Tor2 and Urbani) E protein (MYSFVSEETGTLIVNSVLLFLAFVVi iVTLA ⁇ LTAlRLCA YCCNIVNVSLVKPTVYVYSRVKNLNSSEGVPDLLV) and a second peptide comprising the first 40 amino acids of the full length E protein which correspond to the transmembrane domain (MYSFVSEETGTLIVNSVLLFLAFVVF LLVTLAILTALRLC) were synthesized manually using FMOC chemistry and solid phase peptide synthesis The synthesis was done at the Biomolecular Resource Facility (John Curtin School of Medical Research, ANU, Australia) using a Symphony 11 Peptide Synthesiser from Protein Technologies fcic.(Tucson, AZ, USA) according to the manufacturers instructions.
- Example 19 Peptide purification Mass spectral analysis of the synthetic peptide revealed that the preparation contained significant amounts of material with lower na/z ratio than expected for the full-length product. The majority of these are presumably truncated peptides generated during the peptide synthesis process. To enrich the full-length E protein, the following procedure was used, which relies on differential solubility of the smaller molecules and full-length peptide. The crude preparation was suspended at 12 mg ml in 70% CH3CN, 0.1 %TFA and droneexed for 10 minutes. This suspension was centrifiiged at 10,000g for 10 minutes at 20°C. The supernatant was discarded and the insoluble fractions was extracted with 70% CH 3 CN, 0.1 % TFA, as above, two .
- the SARS virus E protein was resuspended at lmg/ l in 2,2,2-trifluoroethanoL
- the SARS virus E protein's ability to form ion channels was tested on a Warner (Warner instruments, Inc. 1125 Dixwell Avenue, Hamden, CT 06 14) bilayer rig as follows; A lipidmix of 3:1:1, l-Palmitoyl-2-oleolyi phosphatidyl Ethanolamine: l-Pahnitoyl-2- oleolyl phosphatidyl Serine: l-Pahnitoyl-2-oleolyl phosphatidyl choline in CHC1 3 was dried under 2 gas and resuspended to 50mg ml in n-decane, Bilayers were painted across a circular hole of approximately lOO ⁇ m diameter in a DelrinTM cup separating aqueous solution in the CIS and TRANS chambers.
- the CIS chamber contained a solution of 500mM NaCl or KCl, in a 5mM HEPES buffer pH 7.2
- the TRANS chamber contained a solution of 50mM NaCl or KCl, in a 5mM HEPES buffer pH 7.2.
- Silver electrodes coated in chloride with 2% agarose bridges are placed in the CIS and TRANS chamber solutions.
- the SARS E protein full-length or N- terminal peptides (3-1 Ou ) were added to the CIS chamber, which was st ⁇ ed until channel activity was detected.
- the CIS chamber was earthed and the TRANS chamber was held at various holding potentials ranging between +100 to -lOOmV.
- cinnamoylguanidine (Bit036), a compound which was shown in earlier experiments to be antiviral and to inhibit ion channel proteins from other viruses.
- Example 20 Polvacrylamide gel electrophoresis Purified E protein was dissolved to 1 mg/ml, 5 mg/ml and 10 mg ml in, 6 M
- the purified synthetic peptide was reconstituted into planar lipid bilayers' (21).
- 3 ⁇ g of SARS full-length E protein was added to the CIS chamber, while stirring.
- This CIS chamber contained 500 mM NaCl and the TRANS chamber contained 50 mM NaCl.
- ion currents due to SARS E protein ion channel activity were observed after about 5 -15 minutes of stirring. Activity was detected more rapidly and reliably with a holding potential of approximately —1 OOmV across the bilayer. Currents recorded at -lOOmV, (A) and at -60mV (B) in one of these experiments are shown in Figure 6.
- Figure 7a shows typical current traces recorded over a range of potentials in NaCl solutions.
- the IV curve shows that at the lower voltages the average current flow across the bilayer is small but at higher potentials there is an increase in average current across the bilayer, resulting in a non-linear IV relationship.
- the average reversal potential was +48.3 + 2.3 mV (mean + 1SEM), indicating that the channels were about 37 times more permeable to Na+ ion than to CI " ions.
- the reversal potential is close to the Na+ equilibrium potential (+53mV), therefore the channel is selective for Na+ ions.
- SARS E protein N-terminal peptide also formed ion channels in KCl solution that were similarly selective for K+ ions compared to the full-length E protein.
- the average channel reversal potential was +39.5 ⁇ 3.6 V, therefore the channel is about 11 times more permeable to + ions than CI " ions.
- SDS-PAGE of the purified full-length E protein peptide showed bands corresponding to the full-length E protein (Data not shown). Larger bands of varying size up to about 20 kDa were detected, suggesting that SARS E protein may form homo- oligomers.
- Example 21 SARS E protein ion channel is blocked by cinnamoylguanidine and other compounds
- the average current across the bilayer was reduced to baseline by 1 OO ⁇ M cinnamoylguanidine.
- 100 to 200 ⁇ M ci ⁇ namoulguanidine reduced the average current across the bilayer about 4 fold'.
- 100 to 200 ⁇ M cinnamoylguanidine blocked channels formed by full-length E protein in KCl solutions.
- E protein Ion channels were about 37 times more selective for Na+ ions over Cl-ions and about 7.2 times more selective for K+ ions over CI- ions. In over 60 experiments the Na+ conductance of the E protein ion channel varied from as low as 26 pS to as high as 164 pS.
- the SARS virus full length E protem ion channel activity and N-terminal domain E protem ion channel activity on planar lipid bilayers in NaCl and KCl solutions was inhibited by addition of between lOO ⁇ M to 200 ⁇ M cinnamoylguanidine to the CIS chamber. Inhibition or partial, inhibition of the E protein ion channel activity by cinnamoylguanidine has been observed in seven independent experiments in NaCl solution and four independent experiments in KCl solution.
- coronaviruses encode an E protein with a hydrophobic N-terminus transmembrane domain therefore all coronaviruses E proteins could form ion channels on planar lipid bilayers. This indicates that the E protein could be a suitable target for antiviral drugs and potentially stop the spread of coronavirus from infected host cells. Drugs that block the E protein ion channel could be effective antiviral therapy for the treatment of several significant human and veterinary coronavirus diseases including SARS and the common cold.
- Example 22 Bacterial Bio-Assav for Screening Potential SARS-CoV E protein Ion Channel-Blocking Drugs. SARS-CoV E protein Ion Channel inhibits Bacterial Cell growth.
- a bio-assay of SARS-CoV E protem function in bacterial cells was developed.
- a synthetic cDNA fragment encoding SARS-CoV E protein was cloned into the expression plasmid ⁇ PL451, creating a vector in which E protein expression is . temperature inducible, as described in Example 4.
- Inhibition of the growth of E.coli cells expressing E protein at 37°C was observed as an indicator of ⁇ 7 ion channel function dissipating the normal Na+ gradient maintained by the bacterial cells.
- Example 23 Compound Screening using the Bacterial Bio-Assay for SARS coronavirus E protein.
- Table 7 lists the scores for inhibition of SARS-CoV E protein in the bacterial bio- assay.
- Example 24 SARS Antiviral Assay for testing compounds against replication of SARS co ⁇ mavir ⁇ s (SARS-CoV).
- Example 25 Effect of compounds in SARS CoV antiviral assay
- Table 8 provides Virus titration data presented as % of a control (SARS CoV grown for 48 hours in the absence of compounds).
- a peptide corresponding to the full-length 229E-E protein (sequence: MFLKLVDDHALVVIWLLWCVVLIVILLVC ⁇ KLIKLCFTCHMFCNRTVYG ⁇
- KNVYHIYQSYMHIDPFPKRVIDF accession number NP_073554
- the synthesis was done at the Biomolecular Resource Facility (John Curtin School of Medical Research, ANU, Australia) using a Symphony* 1 Peptide Synthesiser from Protein Technologies Inc.(Woburn, MS, USA) according to the manufacturers instructions to give C-terminal amides, the coupling was done with HBTU and hydroxybenzotriazole in N-methylpyrrolidone. Each of the synthesis cycles used double coupling and a 4-fold excess of the amino acids. Temporary ⁇ -N Fmoc- protecting groups were removed using 20% piperidine in DMF.
- the crude synthetic peptide was purified using the ProteoPIusTM kit (Qbiogene inc. CA), following manufactures instructions. Briefly, the peptides were diluted in loading buffer (60mM Tris-HCl pH 8.3, 6M urea, 5% SDS, 10% glycerol, 0.2% Bromophenol blue, + 100 mM ⁇ -tnercaptoethanol) and run on 4-20% gradient polyacrylamide gels (Gradipore, NSW, Australia) in tris-glycine electrophoresis buffer (25 mM Tris, 250 M glycine, 0.1% SDS). The peptides were stained with gel code blue (Promega, NSW) and the bands corresponding to the full-length peptide were excised out of the gel.
- loading buffer 60mM Tris-HCl pH 8.3, 6M urea, 5% SDS, 10% glycerol, 0.2% Bromophenol blue, + 100 mM ⁇ -tnercaptoethanol
- the gel slice was transferred to the ProteoPLUSTM tube and filled with tris-glycine electrophoresis buffer.
- the tubes were emerged in tris-glycine electrophoresis buffer and subjected to 100 volts for approximately 1 hour. The polarity of the electric current was reversed for 1 minute to increase the amount of protein recovered.
- the peptides were harvested and centrifuged at 13, 000 rp for 1 minute. The purified peptides were dried in a Speedvac and the weight of the final product was used to calculate the yield.
- Examnle 27.229E-E protein forms ion channels in planar lipid bflavers.
- Lipid bilayer studies were performed as described elsewhere (Sunstrom, 1996; Miller, 1 86).
- the lipid mixture was painted onto an aperture of 150-200 ⁇ m in the wall of a 1 ml deidn cup, The aperture separates two chambers, cis and trans, both containing salt solutions at different concentrations.
- the cis chamber was connected to ground and the trans chamber to the input of an Axopatch 200 amplifier. Normally the cis chamber contained either 500mMNaClor500mM Cl an thetrans 50 mMNaCl or50mMKCl.
- the bilayer formation was monitored electrically by the amplitude of the current pulse generated by a current ramp. The potentials were measured in the trans chamber with respect to the cis.
- the synthetic peptide was added to the cis chamber and stirred until channel activity was seen. The currents were filtered at 1000 Hz, digitized at 5000 Hz and steered on magnetic disk.
- the 229E E synthetic peptide was dissolved in 2,2,2-trifluorethanol (TFE) at 0.05mg ml to 1 mg ml. 10 ⁇ l of this was added to the cis chamber (1ml aqueous volume) of the bilayer apparatus, which was stirred via a magnetic "flea". Ionic currents, indicating channel activity in the bilayer, were typically detected within 15- 30 min. After channels were detected the holding potential across the bilayer was varied between -l ⁇ OmV and +100mV to characterise the size and polarity of current flow and enable the reversal potential to be determined.
- TFE 2,2,2-trifluorethanol
- the graph is a representative plot of average bilayer current (pA; y-axis) versus holding potential (mV; x-axis).
- Example 28 Chemical compounds inhibit the ion channel activity of the 229E E protein synthetic peptide.
- Compound stock solutions were typically prepared at 500 mM in DMSO. This solution was further diluted to 50 M, or lower concentration in 50% DMSO/50% methanol and 2 ⁇ l of the appropriately diluted compound was added to the cis and/or trans chambers to yield the desired final concentration.
- Example. 29 Bacterial Bio-Assay for Screening Potential 229E-CoV E protein Ion Channel-Blocking Drugs.
- 229E-CoV E-protein Ion Channel inhibits Bacterial Cell growth.
- a bio-assay of 229E-CoV E-protein function in bacterial cells was developed.
- a synthetic cDNA fragment encoding 229E-CoV E-protein was cloned into the expression plasmid ⁇ PL451, creating a vector in which E protein expression is temperature inducible, as described in Example 4.
- Inhibition of the growth of Kcoli cells expressing E protein at 37°C was observed as an indicator of p7 ion channel function dissipating the normal Na+ gradient maintained by the bacterial cells.
- Example 30 Compound Screening using the Bacterial Biq-Assay for 229E-CoV E-protein.
- Table 9 list the scores for inhibition of 229E-CoV E-protein in the bacterial bio-assay. Table 9
- Example 31 Antiviral Assay for testing compounds against replication of human coronavirus 229E (229E1
- an ELISA assay was developed measuring the release of the viral N-protein into culture supernatants from monolayers of OC43- infected MRC-5 cells (human lung fibroblasts ;ATCC CCL-171): First, a virus working stock was prepared by amplification in MRC-5 cells. This was then used to infect confluent monolayers of MRC-5 cells grown in 6-well tissue culture plates by exposure to the virus at an MOI of approx. 0.01 pfu/cell for 1 hour at 35°C in 5%C ⁇ 2 .
- the infective inoculum was removed and replaced with fresh medium (DMEM supplemented with 10% fetal calf serum) containing various test concentrations of compounds or the appropriate level of solvent used for the compounds (control). Plates were subsequently incubated at 35°C (in 5% CO 2 ) for 5 days post infection, after which time culture supernatant was harvested and cellular debris removed by centrifugation at 5000 x g for 10 minutes. For N-antigen detection, lOO ⁇ l samples of clarified culture supernatant were added to duplicate wells of a 96-well Maxi-Sorb plate; lOO ⁇ l of RIPA buffer was added per well with mixing and the plate was covered and incubated at 4°C overnight to enable protein binding to the plastic wells.
- fresh medium DMEM supplemented with 10% fetal calf serum
- Example 34 Mouse Hepatitis Virus (MHV).
- VLSPSIYLYDRSKQLYKYYNEEMRLPLLEVDDI accession number NP_068673
- Each of the synthesis cycles used double coupling and a 4-fold excess of the amino acids.
- Temporary ⁇ -N F oc- protecting groups were removed using 20% piperidine in DMF.
- the crude synthetic peptide was purified using the ProteoPlusTM kit (Qbiogene inc. CA), following manufactures instructions. Briefly, the peptides were diluted in loading buffer (60mM Tris-HCl H 8.3, 6M urea, 5% SDS, 10% glycerol, 0.2% Bromophenol blue, ⁇ 100 mM ⁇ -mercaptoethanol) and run on 4-20% gradient polyacrylamide gels (Gradipore, NSW, Australia) in tris-glycine electrophoresis buffer (25 mM Tris, 250 M glycine, 0.1 % SDS). The peptides were stained with gel code blue (Promega, NSW) and the bands corresponding to the full-length peptide were excised out of the gel.
- loading buffer 60mM Tris-HCl H 8.3, 6M urea, 5% SDS, 10% glycerol,
- the gel slice was transferred to the ProteoPLUSTM tube and filled with tris- glycine electrophoresis buffer.
- the tubes were emerged in tris-glycine electrophoresis buffer and subjected to 100 volts for approximately 1 hour.
- the polarity of the electric curreait was reversed for 1 minute to increase the amount of protein recovered.
- the peptides were harvested and centrifiiged at 13, 000 rp for 1 minute.
- the purified peptides were dried in a Speedvac and the weight of the final product was used to calculate the yield.
- Example 35 MHV-E protein forms ion channels in planar lipid bilayers.
- the cis chamber was connected to ground and (he trans chamber to the input of an Axopatch 200 amplifier. Normally the cis chamber contained either 500 mM NaCl or 500mM KCl and the trans 50 mM NaCl or 50mM KCl. The bilayer formation was monitored electrically by the amplitude of the current pulse generated by a current ramp. The potentials were measured in the trans chamber with respect to the cis. The synthetic peptide was added to the cis chamber and stirred until channel activity was seen. The currents were filtered at 1000 Hz, digitized at 5000 Hz and stored on magnetic disk.
- the MHV E synthetic peptide was dissolved in 2,2,2-trifluorethanol (TFE) at 0.05mg ml to 1 mg ml. 10 ⁇ l of this was added to the cis chamber (1ml aqueous volume) of the bilayer apparatus, which was stirred via a magnetic "flea". Ionic currents, indicating channel activity in the bilayer, were typically detected within 15- 30 min. After channels were detected the holding potential across the bilayer was varied between -lOOmV and +100mV to characterise the size and polarity of current flow and enable the reversal potential to be determined.
- TFE 2,2,2-trifluorethanol
- Figure 11 shows examples of raw current data for the MHV E ion channel at various holding potentials (cis relative to trans) in asymmetrical NaCl solutions (500/50 M).
- the graph is a representative plot of average bilayer current (pA; y- axis) versus holding potential (mV; x-axis).
- Example 36 Chemical compounds inhibit the ion channel actrvity of the MHV F. protein synthetic peptide.
- Example 37 Bacterial Bio-Assay for Screening Potential MHV E-protein Ion Channel-Blocking Drugs.
- MHV E-protein Ion Channel inhibits Bacterial Cell growth.
- a bio-assay of MHV E-protein function in bacterial cells was developed.
- a synthetic cDNA fragment encoding MHV E-protein was cloned into the expression plasmid pPL451, creating a vector in which E protein expression is temperature inducible, as described in Example 4.
- Inhibition of the growth of E.coli cells expressing E protein at 37°C was observed as an indicator of p7 ion channel function dissipating the normal Na+ radient maintained by the bacterial cells.
- Example 38 Compound Screening using the Bacterial Bio-Assav for MHV E protein.
- Table 12 lists the scores for inhibition of MHV E protein in the bacterial bio-assay.
- Example 39 MHV Antiviral Assay for testing compounds against replication of mouse hepatitis virus (MHV).
- the infective inoculum was removed and replaced with fresh medium (DMEM supplemented with 10% horse serum) containing various test concentrations of compounds or the appropriate level of solvent used for the compounds (control). Plates were subsequently incubated at 37°C (in 5% CO2) for 16 - 24 hours post infection, after which time culture supernatant was removed and the cells were stained with 0.1% crystal violet solution in 20% ethanol for 10 minutes. Plaques were counted in all wells and the percentage reduction in plaque number compared to solvent control was calculated. Measurements were performed in duplicate to quadruplicate wells.
- Example 40 Effect of compounds in MHV antiviral assay.
- Table 13 provides the results obtained from this study.
- Example 41 Porcine Respiratory Coronavims (PRCV) Antiviral Assay for testing compounds against replication of porcine respiratory coronavirns fPRCV).
- PRCV Porcine Respiratory Coronavims
- luM, luM, lOuM and 20uM Either duplicates or quadruplicates were performed at each concentration. Controls were performed where the same volume of solvent was. added to the overlay. The overlay was allowed to set at room temp for 20 ins. The plates were then incubated at 37°C for 2 days. The monolayers were then fixed and stained overnight at room temperature by adding Iml/well of 0.5% methylene blue, 4% formaldehyde. Overlay agarose and stain was then rinsed off to visualize stained and fixed monolayer
- the viral supernatant was removed and 2ml well of overlay containing 1% Seaplaque agarose in lx MEM, 5% FCS was added to each well.
- Compounds to be tested were added to the overlay mixture by diluting the compounds from a 0.5M frozen stock to a concentration so that the same volume of compound/solvent would be added to the overlay for each concentration of compound. The volume of compound/solvent never exceeded 0.07% of the volume of the overlay.
- the solvent used to dissolve compounds was DMSO and raiethanol mixed in equal proportions.
- Compounds were tested for anti-plaque forming activity at four concentrations, O.luM, luM, lOuM and 20uM. Quadruplicates were performed at each concentration.
- Controls were performed where the same volume of solvent was added to the overlay.
- the overlay was allowed to set at room temp for 20 mins.
- the plates were then incubated at 37°C for 7 days.
- the monolayers were then fixed and stained by adding Iml well of 0.5% methylene blue, 4% formaldehyde.
- a peptide mimicking the protein P7 encoded by the hepatitis C virus (HCV) 5 was synthesised having the following amino acid sequence:
- phosphatidylserme and palmitoyl-oleoyl-phosphatidylcholine 5:3:2) (Avanti Polar Lipids, Alabaster, Alabama) was used.
- the lipid mixture was painted onto an aperture of 150-200 m in the wall of a 1 ml dehrin cup.
- the aperture separates two chambers, s and trans, both containing salt solutions at different concentrations.
- the cis chamber was connected to ground and the trans chamber to the input of an Axopatch
- the cis chamber contained 500 mM KCl and the trans 50 mM KCl.
- the bilayer formation was monitored electrically by the amplitude of the current pulse generated by a current ramp. The potentials were measured in the trans chamber with respect to the cis.
- the protein was added to the cis chamber and stirred until channel activity was seen. The currents were filtered at 1000 Hz, digitized at 0 2000 Hz and stored on magnetic disk.
- the P7 peptide was dissolved in 2,2,2- trifluorethanol (TFE) at lGmg ml. 10 ul of this was added to the cis chamber of the bilayer which was stirred. Channel activity was seen within 15-20 min.
- TFE 2,2,2- trifluorethanol
- the channels formed by the P7 peptide were blocked by 5-(N,N- hexamethylene) amiloride (HMA),
- cDp7.coli Two cDNA fragments, each encoding the same polypeptide corresponding to the amino acid sequence of the HCV-la p7 protein, were synthesised commercially by GeneScript.
- the two cDNAs differed in nucleotide sequence such that in one cDNA (“cDp7.coli") the codons were optimised for expression of the p7 protein in . E.coli while in the other cDNA ( ⁇ cDp7,mar ⁇ )" codons were biased for expression in mammalian cell lines.
- cD ⁇ 7.coli was cloned into the plasmid pPL451 as a BamHI/EcoRI fragment for expression in E.coli.
- cDp7.mam was cloned into vectors (for example, pcDNA3.1 vaccinia virus, pfasfBac-1) for expression of ⁇ 7 in mammalian cell lines.
- Example 47 Role of p7 in enhancement of Gag VLP Budding.
- VLP virus-like particles
- Example 48 Assay of the ability of compounds to inhibit p7 ion channel functional activity.
- Example 33 The two methods of detecting p7 ion channel functional activity, described in Examples 33-35, were employed to assay the ability of compounds to inhibit the p7 channel.
- compounds were tested for their abihty to inhibit p7 channel activity in planar lipid bilayers.
- Example 35 compounds were tested for their ability to reduce the number ofVLPs released from cells expressing both p7 and HTV-l Gag. Example 49.
- HCV D7 Ion Channel inhibits Bacterial Cell growth.
- a bio-assay of p7treatment in bacterial cells was developed.
- the p7-encoding synthetic cDNA fragment cDp7.coli was cloned into the expression plasmid pPL451, creating the vector pPLp7, in which p7 expression is temperature inducible, as described in Example 4.
- Inhibition of the growth of E.coli cells expressing p7 at 37'C was observed as an indicator of p7 ion channel function dissipating the normal Na+ gradient maintained by the bacterial cells.
- Example 50 Compound Screening using the Bacterial Bio-Assav for HCV p7 protein.
- Table 16 lists the scores for inhibition of HCV ⁇ 7 protein in the bacterial bio-assay.
- Phenamil methanesulfonate salt 0.5 /1
- EAV equine arteritis virus
- strain Bucyrus ATCC VR-796
- ATCC CCL-10 EAV infected BHK-21 cells
- a virus stock was amplified in RK-13 cells (ATCC CCL-37) and this was then used to infect confluent monolayers of BHK-21 cells grown in 6-well tissue .
- Example 54 Incorporation of Dengue M virus protein into lipid bilayers. Lipid bilayer studies were performed as described elsewhere (Sunstrom, 1996;
- the cis chamber contained 500 M KCl and the trans 50 mM KCl.
- the bilayer formation was monitored electrically by the amplitude of the current pulse generated by a current ramp. The potentials were measured in the trans chamber with respect to the cis.
- the protein was added to the cis chamber and stirred until channel activity was seen. The currents were filtered at 1000 Hz, digitized at 5000 Hz and stored on magnetic disk.
- DMVC dengue virus M protein C-terminal peptide
- TFE 2,2,2- trifluorethanol
- E ⁇ am ⁇ le 55 Hexamethylene amiloride (HMA) to inhibits ion channel activity of the dengue virus M protein C-termfnal peptide.
- HMA Hexamethylene amiloride
- Solutions of 50 mM HMA were prepared by first making a 500 inM solution in DMSO. This solution was further diluted to 50 mM HMA using 0.1 M HCI.2 ⁇ l of the 50 mM HMA was added to the cis chamber after channel activity was seen. The cis chamber contained 1 ml of solution making the final concentration of HMA 100 ⁇ M.
- Example 56 Antiviral Assay for testing compounds against Effects of Dengne flavivirns against cvtoproliferation.
- the cultures were allowed to grow for 7 days and then Alamar Blue, a fluorescent dye that measures the metabolism of the cultures (red/ox), was added to each culture and the fluorescence value for each culture was measured.
- the negative control without experimental compound or virus was fixed at 100%.
- the positive controls and the cultures with compound were scored by calculating their average fluorescence as a percentage of the negative control. At least six replicate wells were measured for each experimental condition.
- Example 58 Positive correlation between Bacterial Assay and Anti-viral Assays
- the p24-antige ⁇ data for twelve compounds representing various substituted acylguanidines was compared with the activity scores obtained for those compounds in the Vpu bacterial assay.
- the data from each assay was initially rank ordered for effectiveness.
- the rank order for the Vpu bacterial assay was determined from all activity scores, the highest score indicating the greatest effectiveness.
- the rank order for the anti-HIV-1 assay was determined based on the overall average value of p24 antigen measured in culture sup ⁇ matants at all of the drug concentrations tested, with the lowest score indicating the greatest effectiveness.
- the two rank orders generated ware then compared statistically by generating the Spearman's Rank correlation coefficient.
- Table 19a Comparison of Rank order of efficacy of 12 substituted acylguanidines in the Vpu bacterial assay and anti-HTV assay.
- MHV plaque reduction activity data for 96 compounds screened were sorted from greatest to least percent plaque reduction and rank orders were assigned to the list of compounds. This was performed for the data generated by exposure to both lO ⁇ M and 1 ⁇ M concentrations of the compounds, giving rise to two rank order lists. Similarly, a rank order list was generated for the MHVE bacterial bioassay scores for the same 96 compounds. Where one or more compounds had the same score, the rank values for that group were averaged.
- the rank order comparison of 96 compounds assayed in the bacterial bioassay and the antiviral assay show that MHVE bacterial assay rank order for the compounds tested is significantly positively correlated with the rank orders generated by the MHV plaque reduction assay.
- the significant correlation between the assays is highly indicative that either assay may be utilised to identity compounds that may be useful.
- the bacterial assay may thereby be a useful tool in screening for compounds that exhibit anti-viral activity.
- 229E plaque reduction activity data for 97 compounds screened against 2.5 ⁇ M compound concentration were sorted from greatest to least percent plaque reduction and rank orders were assigned to the list of compounds.
- a rank order list was generated for the 229E E bacterial bioassay scores for the same 97 compounds. Where one or more compounds had the same score, the rank values for that group were averaged.
- Wcstcrvclt P., Hcnkcl, T., Trowbridgc, D.B., Orcnstcin, J., Heuser, , Gendclman,
Abstract
Description




















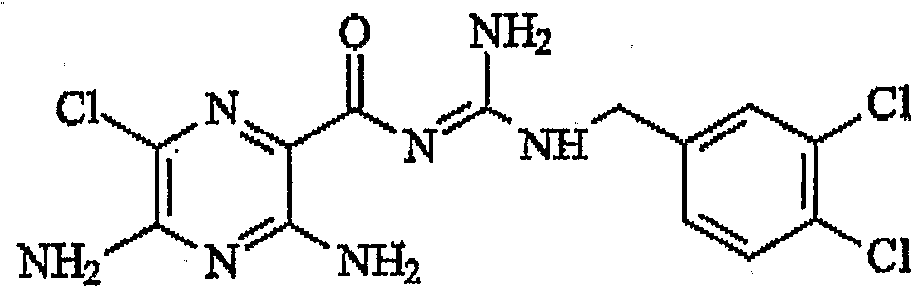































































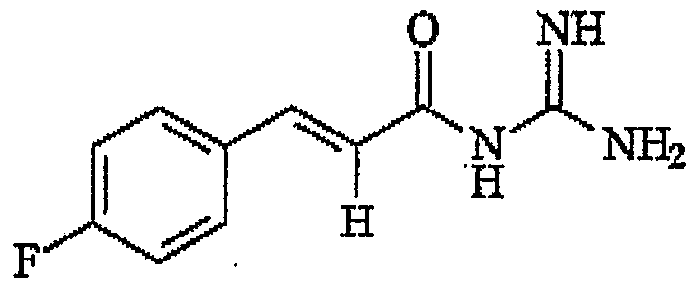




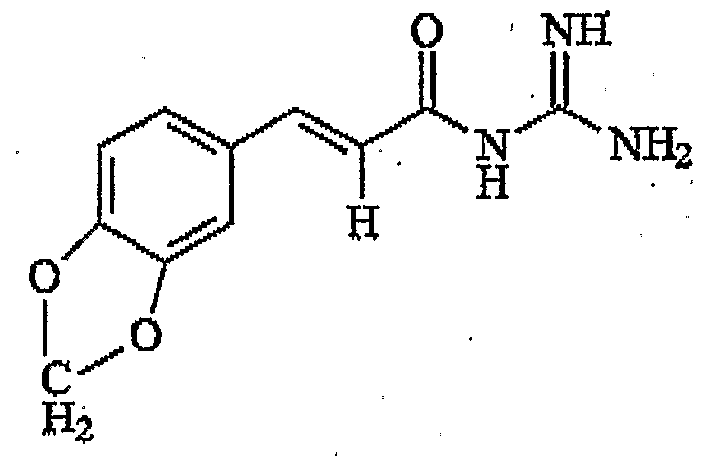





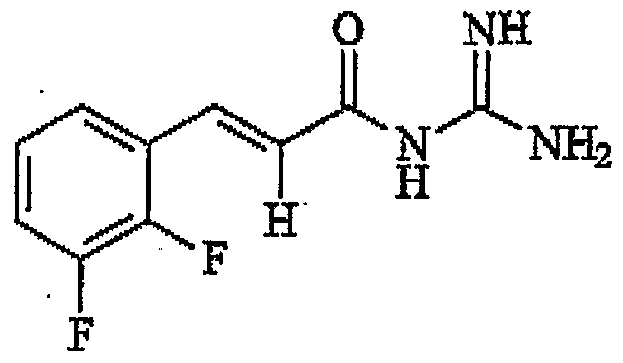


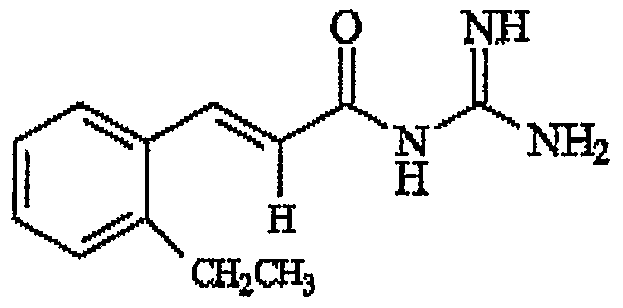









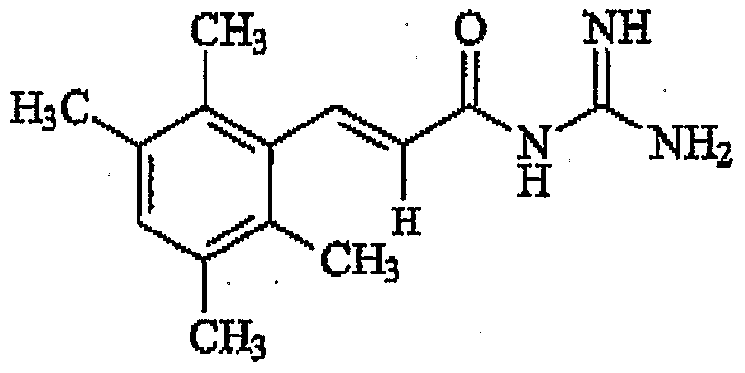
















































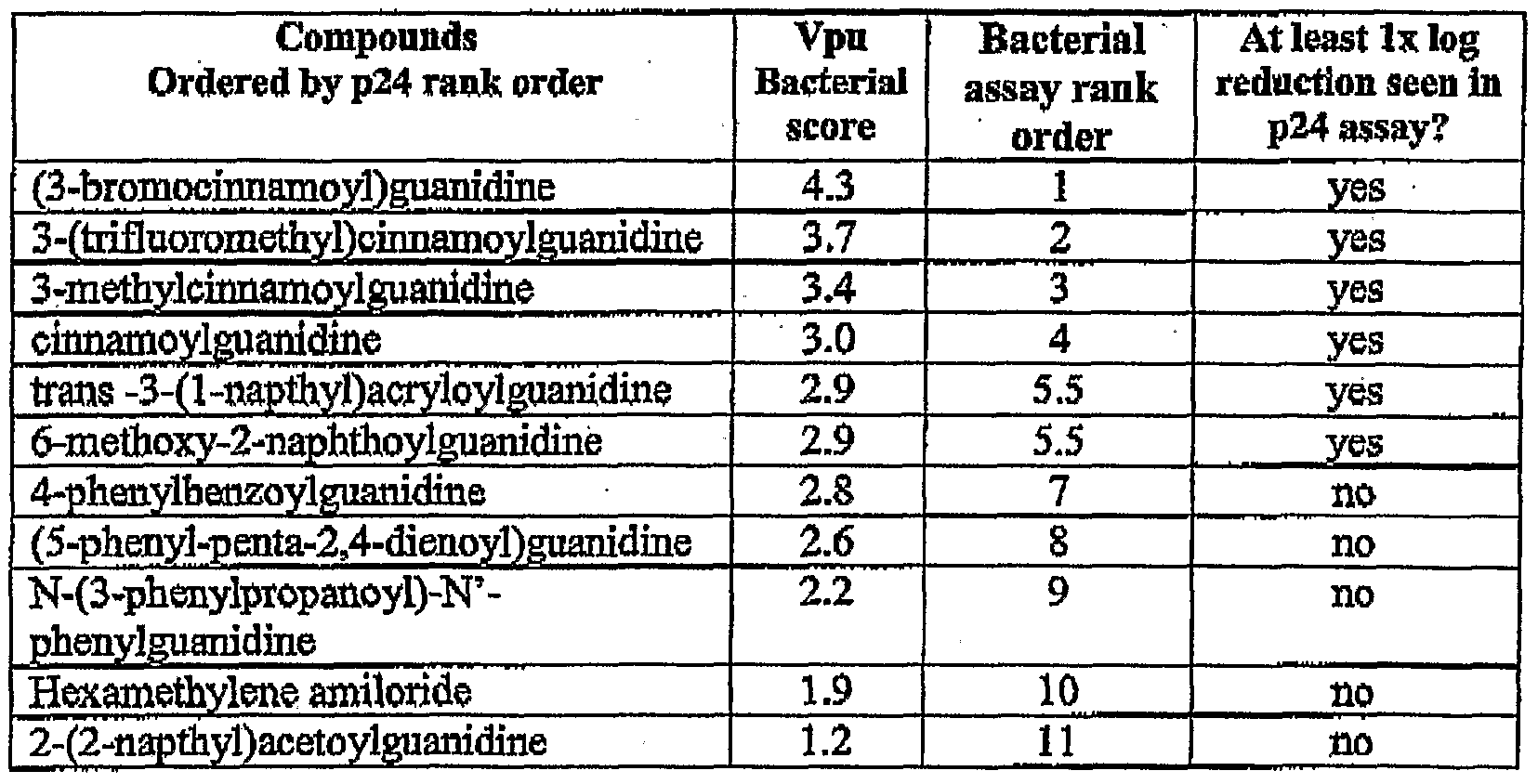


Claims











Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/562,296 US20070099968A1 (en) | 2004-06-26 | 2004-06-24 | Antiviral compounds and methods |
| NZ544671A NZ544671A (en) | 2003-06-26 | 2004-06-26 | Antiviral compounds and methods |
| CN2004800240974A CN101111475B (en) | 2003-06-26 | 2004-06-26 | Antiviral compounds and methods |
| CA2529949A CA2529949C (en) | 2003-06-26 | 2004-06-26 | Antiviral compounds and methods |
| AU2004248859A AU2004248859C1 (en) | 2003-06-26 | 2004-06-26 | Antiviral acylguanidine compounds and methods |
| EP13164504.6A EP2617709B8 (en) | 2003-06-26 | 2004-06-26 | Guanidine derivatives as antiviral agents |
| JP2006515560A JP5030587B2 (en) | 2003-06-26 | 2004-06-26 | Antiviral acylguanidine compounds and methods |
| BRPI0411900A BRPI0411900B8 (en) | 2003-06-26 | 2004-06-26 | compounds and pharmaceutical compositions comprising them |
| KR1020057024860A KR101153254B1 (en) | 2003-06-26 | 2004-06-26 | Antiviral compounds and methods |
| EP04737487A EP1646371A4 (en) | 2003-06-26 | 2004-06-26 | Antiviral compounds and methods |
| US13/553,239 US20130035328A1 (en) | 2003-06-26 | 2012-07-19 | Antiviral compounds and methods |
| US14/615,616 US20150313909A1 (en) | 2003-06-26 | 2015-02-06 | Antiviral compounds and methods |
| US15/602,526 US10472332B2 (en) | 2003-06-26 | 2017-05-23 | Antiviral compounds and methods |
| US16/554,990 US11192863B2 (en) | 2003-06-26 | 2019-08-29 | Antiviral compounds and methods |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2003903251 | 2003-06-26 | ||
| AU2003903251A AU2003903251A0 (en) | 2003-06-26 | 2003-06-26 | Antiviral compounds and methods |
| AU2003903850A AU2003903850A0 (en) | 2003-07-25 | 2003-07-25 | Anti-coronavirus compounds and methods |
| AU2003903850 | 2003-07-25 | ||
| AU2003904692 | 2003-08-29 | ||
| AU2003904692A AU2003904692A0 (en) | 2003-08-29 | Anti-flavivirus compounds and methods | |
| AU2004902902 | 2004-05-31 | ||
| AU2004902902A AU2004902902A0 (en) | 2004-05-31 | Anti-flavivirus compounds and methods |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/562,296 A-371-Of-International US20070099968A1 (en) | 2004-06-26 | 2004-06-24 | Antiviral compounds and methods |
| US13/553,239 Continuation US20130035328A1 (en) | 2003-06-26 | 2012-07-19 | Antiviral compounds and methods |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004112687A2 true WO2004112687A2 (en) | 2004-12-29 |
| WO2004112687A3 WO2004112687A3 (en) | 2007-07-12 |
Family
ID=33545379
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/AU2004/000866 WO2004112687A2 (en) | 2003-06-26 | 2004-06-26 | Antiviral acylguanidine compounds and methods |
Country Status (8)
| Country | Link |
|---|---|
| US (4) | US20130035328A1 (en) |
| EP (2) | EP2617709B8 (en) |
| JP (1) | JP5030587B2 (en) |
| KR (1) | KR101153254B1 (en) |
| BR (1) | BRPI0411900B8 (en) |
| CA (1) | CA2529949C (en) |
| NZ (1) | NZ544671A (en) |
| WO (1) | WO2004112687A2 (en) |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006135978A1 (en) * | 2005-06-24 | 2006-12-28 | Biotron Limited | Antiviral compounds and methods |
| WO2007071400A1 (en) | 2005-12-22 | 2007-06-28 | Novartis Ag | Pyrazine derivatives as epithelial sodium channel blocker |
| WO2009018609A1 (en) * | 2007-08-03 | 2009-02-12 | Biotrom Limited | Hepatitis c antiviral compositions and methods |
| JP2009520728A (en) * | 2005-12-22 | 2009-05-28 | ノバルティス アクチエンゲゼルシャフト | Pyrazinoylguanidine compounds useful for the treatment of inflammation or allergic conditions |
| US7745442B2 (en) * | 2003-08-20 | 2010-06-29 | Parion Sciences, Inc. | Methods of reducing risk of infection from pathogens |
| US20110217265A1 (en) * | 2008-09-23 | 2011-09-08 | Glenn Jeffrey S | Screening for Inhibitors of HCV Amphipathic Helix (AH) Function |
| AU2013205388B2 (en) * | 2005-06-24 | 2016-05-05 | Biotron Limited | Antiviral compounds and methods |
| AU2013219202B2 (en) * | 2007-08-03 | 2016-12-08 | Biotron Limited | Hepatitis C antiviral compositions and methods |
| WO2018081863A1 (en) * | 2016-11-04 | 2018-05-11 | University Of Wollongong | 6-SUBSTITUTED DERIVATIVES OF HEXAMETHYLENE AMILORIDE AS INHIBITORS OF uPA AND USES THEREOF |
| US11007208B2 (en) | 2015-09-16 | 2021-05-18 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| WO2021154687A1 (en) | 2020-01-27 | 2021-08-05 | Gilead Sciences, Inc. | Methods for treating sars cov-2 infections |
| WO2021207049A1 (en) | 2020-04-06 | 2021-10-14 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carbanucleoside analogs |
| WO2021262826A2 (en) | 2020-06-24 | 2021-12-30 | Gilead Sciences, Inc. | 1'-cyano nucleoside analogs and uses thereof |
| US11260070B2 (en) | 2017-03-14 | 2022-03-01 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| EP3960165A1 (en) * | 2020-08-25 | 2022-03-02 | Fondation EspeRare | Nhe-1 inhibitors for the treatment of coronavirus infections |
| WO2022046631A1 (en) | 2020-08-24 | 2022-03-03 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| WO2022047065A2 (en) | 2020-08-27 | 2022-03-03 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11266666B2 (en) | 2014-10-29 | 2022-03-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US11266681B2 (en) | 2017-07-11 | 2022-03-08 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| WO2022081973A1 (en) | 2020-10-16 | 2022-04-21 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| US11492353B2 (en) | 2010-07-22 | 2022-11-08 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US11491169B2 (en) | 2020-05-29 | 2022-11-08 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| WO2022251318A1 (en) | 2021-05-26 | 2022-12-01 | Gilead Sciences, Inc. | Phospholipid formulations of 1'-cyano substituted carba-nucleoside analogs |
| WO2023023527A1 (en) | 2021-08-18 | 2023-02-23 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| US11597742B2 (en) | 2017-05-01 | 2023-03-07 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy) (phenoxy) phosphoryl)amino)propanoate |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| WO2023168194A1 (en) | 2022-03-03 | 2023-09-07 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2023168254A1 (en) | 2022-03-03 | 2023-09-07 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2023167938A1 (en) | 2022-03-02 | 2023-09-07 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023167944A1 (en) | 2022-03-02 | 2023-09-07 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023239665A1 (en) | 2022-06-06 | 2023-12-14 | Gilead Sciences, Inc. | Methods for treatment of viral infections including sars-cov-2 |
| WO2024006461A1 (en) | 2022-06-30 | 2024-01-04 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
| WO2024006376A1 (en) | 2022-06-29 | 2024-01-04 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
| EP4125373A4 (en) * | 2020-03-30 | 2024-04-17 | Univ Jefferson | Compounds, compositions, and methods for treating, ameliorating, or preventing viral infections |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004112687A2 (en) | 2003-06-26 | 2004-12-29 | Biotron Limited | Antiviral acylguanidine compounds and methods |
| WO2022040321A1 (en) * | 2020-08-19 | 2022-02-24 | Natreon, Inc. | Protection against coronavirus infection by extracts and extract components |
| WO2023012329A1 (en) | 2021-08-06 | 2023-02-09 | Intervet International B.V. | Method of treating veterinary viral diseases |
| WO2023167055A1 (en) * | 2022-03-03 | 2023-09-07 | 日本曹達株式会社 | Antiviral agent |
Family Cites Families (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2734904A (en) * | 1956-02-14 | Xcxnhxc-nh | ||
| NL299931A (en) * | 1962-10-30 | |||
| FR1435379A (en) * | 1964-07-30 | 1966-04-15 | American Cyanamid Co | Process for stabilizing rigid polyvinyl chloride against the effects of light |
| US3527758A (en) * | 1967-04-13 | 1970-09-08 | Merck & Co Inc | Process for the preparation of pyrazinoylguanidines from a pyrazinoic azide and a guanidine |
| DE2006186A1 (en) | 1970-02-11 | 1971-08-19 | Huebner Vamag | Method for inserting a seat ring for the shut-off element of a valve |
| DE2531343A1 (en) * | 1975-07-14 | 1977-02-10 | Henkel & Cie Gmbh | Antimicrobial compsns. contg. substd. benzoyl phenyl guanidines - prepd. from opt. substd. benzoyl cyanamides and substd. anilines |
| DD200618A1 (en) * | 1981-10-05 | 1983-05-25 | Manfred Augustin | PROCESS FOR THE PREPARATION OF NEW N-AROYL AND HETAROYLIMIDES |
| US4496573A (en) * | 1982-08-24 | 1985-01-29 | William H. Rorer, Inc. | 1-Pyridylmethyl-3-acyl guanidines |
| US4894376A (en) * | 1988-02-26 | 1990-01-16 | Trustees Of The University Of Pennsylvania | Methods of treating diseases characterized by hyperexcitability of neurons |
| JPH0789859A (en) * | 1990-04-05 | 1995-04-04 | Celtrix Pharmaceut Inc | Pharmaceutic composition containing pyrazine derivative and curing method for inhibiting neogenesis of new blood vessel |
| IN177137B (en) * | 1992-12-11 | 1996-11-16 | Hoechst India | |
| DE4301739A1 (en) * | 1993-01-22 | 1994-07-28 | Sauer Professor Dr Gerhard | Treatment of primary tumours and viral diseases |
| JPH0725768A (en) * | 1993-07-09 | 1995-01-27 | Mitsubishi Chem Corp | Suppressing agent for hyperplasia of vascular endosporium |
| DE4325822A1 (en) * | 1993-07-31 | 1995-02-02 | Hoechst Ag | Substituted benzoylguanidines, process for their preparation, their use as medicament or diagnostic agent, and medicament containing them |
| TW415937B (en) * | 1994-01-25 | 2000-12-21 | Hoechst Ag | Phenyl-substituted alkylcarboxylic acid guanidides bearing perfluoroalkyl groups, process for their preparation, their use as a medicament or diagnostic, and medicament containing them |
| DE4415873A1 (en) * | 1994-05-05 | 1995-11-09 | Hoechst Ag | Substituted bicyclic heteroaroylguanidines, process for their preparation, their use as medicament or diagnostic agent and medicament containing them |
| DE4421536A1 (en) * | 1994-06-20 | 1995-12-21 | Hoechst Ag | Phenyl-substituted alkenylcarboxylic acid guanidines bearing perfluoroalkyl groups, processes for their preparation, their use as medicaments or diagnostic agents, and medicaments containing them |
| JPH08225513A (en) * | 1994-12-21 | 1996-09-03 | Kanebo Ltd | Naphthoylguanidine derivative |
| AT408330B (en) | 1995-01-27 | 2001-10-25 | Colop Stempelerzeugung Skopek | SELF-COLORING STAMP |
| ATE183498T1 (en) * | 1995-04-18 | 1999-09-15 | Hoechst Ag | SUBSTITUTED INDENOYLGUANIDINES WITH ANTIARRHYTHMIC AND CARDIOPROTECTIVE EFFECTS |
| DE19518796A1 (en) * | 1995-05-22 | 1996-11-28 | Hoechst Ag | Fluorophenyl-substituted alkenylcarboxylic acid guanidines, processes for their preparation, their use as medicaments or diagnostic agents and medicaments containing them |
| DE19527305A1 (en) * | 1995-07-26 | 1997-01-30 | Hoechst Ag | Substituted cinnamic acid guanidides, process for their preparation, their use as a medicament or diagnostic agent and medicament containing them |
| US5628984A (en) | 1995-07-31 | 1997-05-13 | University Of North Carolina At Chapel Hill | Method of detecting lung disease |
| JPH0967332A (en) * | 1995-08-29 | 1997-03-11 | Yamanouchi Pharmaceut Co Ltd | N-(2-propenoyl)guanidine derivative |
| DE19621482A1 (en) * | 1996-05-29 | 1997-12-04 | Hoechst Ag | Substituted 1-naphthoylguanidines, processes for their preparation, their use as medicaments or diagnostic agents and medicaments containing them |
| DE19622222A1 (en) * | 1996-06-03 | 1997-12-04 | Hoechst Ag | Use of sodium=proton exchange inhibitor |
| AUPO545297A0 (en) * | 1997-03-04 | 1997-03-27 | Fujisawa Pharmaceutical Co., Ltd. | Guanidine derivatives |
| IL126276A0 (en) * | 1997-09-24 | 1999-05-09 | Hoechst Marion Roussel De Gmbh | The use of an inhibitor of the na+/H+ exchanger for the production of a medicament for the treatment or prophylaxis of disorders of the central nervous system |
| US7041702B1 (en) * | 1997-10-21 | 2006-05-09 | Scion Pharmaceuticals, Inc. | Pharmaceutically active compounds and methods of use |
| KR20010031065A (en) * | 1997-10-21 | 2001-04-16 | 케임브리지 뉴로사이언스, 인코포레이티드 | Pharmaceutically active compounds and methods of use |
| JP2002508507A (en) * | 1997-12-18 | 2002-03-19 | セプラコア インコーポレーテッド | A method for simultaneous identification of novel biological targets and drug discovery lead structures |
| US6011059A (en) * | 1997-12-24 | 2000-01-04 | Bristol-Myers Squibb Company | Acyl guanidine sodium/proton exchange inhibitors and method |
| AUPP646498A0 (en) * | 1998-10-12 | 1998-11-05 | Australian National University, The | A method of modulating ion channel functional activity |
| DE19849722A1 (en) * | 1998-10-28 | 2000-05-04 | Aventis Pharma Gmbh | Substituted phenyl-alkenoylguanidines, processes for their preparation, their use as medicaments or diagnostic agents and medicaments containing them |
| DE19859727A1 (en) * | 1998-12-23 | 2000-06-29 | Aventis Pharma Gmbh | The use of inhibitors of the sodium-hydrogen exchanger for the manufacture of a medicament for the prevention of age-related organ dysfunctions, age-related illnesses for the prolongation of life |
| AUPQ223999A0 (en) * | 1999-08-16 | 1999-09-09 | University Of Sydney, The | Intracellular feedback controls in the diagnosis and treatment of human disease |
| AUPS022802A0 (en) | 2002-01-31 | 2002-02-21 | Macfarlane Burnet Institute For Medical Research And Public Health Limited, The | Anti-viral compounds |
| WO2004112687A2 (en) | 2003-06-26 | 2004-12-29 | Biotron Limited | Antiviral acylguanidine compounds and methods |
| DK1902017T3 (en) * | 2005-06-24 | 2014-06-30 | Biotron Ltd | Antiviral acylguanidine compounds |
-
2004
- 2004-06-26 WO PCT/AU2004/000866 patent/WO2004112687A2/en active Search and Examination
- 2004-06-26 NZ NZ544671A patent/NZ544671A/en unknown
- 2004-06-26 EP EP13164504.6A patent/EP2617709B8/en active Active
- 2004-06-26 KR KR1020057024860A patent/KR101153254B1/en active IP Right Grant
- 2004-06-26 EP EP04737487A patent/EP1646371A4/en not_active Withdrawn
- 2004-06-26 JP JP2006515560A patent/JP5030587B2/en active Active
- 2004-06-26 BR BRPI0411900A patent/BRPI0411900B8/en active IP Right Grant
- 2004-06-26 CA CA2529949A patent/CA2529949C/en active Active
-
2012
- 2012-07-19 US US13/553,239 patent/US20130035328A1/en not_active Abandoned
-
2015
- 2015-02-06 US US14/615,616 patent/US20150313909A1/en not_active Abandoned
-
2017
- 2017-05-23 US US15/602,526 patent/US10472332B2/en active Active
-
2019
- 2019-08-29 US US16/554,990 patent/US11192863B2/en active Active
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1646371A4 * |
Cited By (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7745442B2 (en) * | 2003-08-20 | 2010-06-29 | Parion Sciences, Inc. | Methods of reducing risk of infection from pathogens |
| AU2013205388B2 (en) * | 2005-06-24 | 2016-05-05 | Biotron Limited | Antiviral compounds and methods |
| JP2008543886A (en) * | 2005-06-24 | 2008-12-04 | バイオトロン リミテッド | Antiviral compounds and methods |
| US9440926B2 (en) | 2005-06-24 | 2016-09-13 | Biotron Limited | Antiviral compounds and methods |
| US10683263B2 (en) | 2005-06-24 | 2020-06-16 | Biotron Limited | Antiviral compounds and methods |
| AU2006261593B2 (en) * | 2005-06-24 | 2013-01-24 | Biotron Limited | Antiviral compounds and methods |
| JP2013049676A (en) * | 2005-06-24 | 2013-03-14 | Biotron Ltd | Antiviral compound and method |
| CN101208297B (en) * | 2005-06-24 | 2013-04-24 | 拜伊特朗有限公司 | Antiviral compounds and methods |
| WO2006135978A1 (en) * | 2005-06-24 | 2006-12-28 | Biotron Limited | Antiviral compounds and methods |
| EP2826770A1 (en) | 2005-06-24 | 2015-01-21 | Biotron Limited | Acylguanidine compounds with antiviral activity |
| US8669280B2 (en) | 2005-06-24 | 2014-03-11 | Biotron Limited | Antiviral compounds and methods |
| JP2009520728A (en) * | 2005-12-22 | 2009-05-28 | ノバルティス アクチエンゲゼルシャフト | Pyrazinoylguanidine compounds useful for the treatment of inflammation or allergic conditions |
| WO2007071400A1 (en) | 2005-12-22 | 2007-06-28 | Novartis Ag | Pyrazine derivatives as epithelial sodium channel blocker |
| AU2008286240B2 (en) * | 2007-08-03 | 2013-05-23 | Biotron Limited | Hepatitis C antiviral compositions and methods |
| EP2659885A1 (en) * | 2007-08-03 | 2013-11-06 | Biotron Limited | Hepatitis C Antiviral Compositions and Methods |
| AU2013219202B2 (en) * | 2007-08-03 | 2016-12-08 | Biotron Limited | Hepatitis C antiviral compositions and methods |
| WO2009018609A1 (en) * | 2007-08-03 | 2009-02-12 | Biotrom Limited | Hepatitis c antiviral compositions and methods |
| US20110217265A1 (en) * | 2008-09-23 | 2011-09-08 | Glenn Jeffrey S | Screening for Inhibitors of HCV Amphipathic Helix (AH) Function |
| US11492353B2 (en) | 2010-07-22 | 2022-11-08 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US11266666B2 (en) | 2014-10-29 | 2022-03-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US11344565B2 (en) | 2014-10-29 | 2022-05-31 | Gilead Sciences, Inc. | Methods for the preparation of ribosides |
| US11382926B2 (en) | 2015-09-16 | 2022-07-12 | Gilead Sciences, Inc. | Methods for treating Arenaviridae and Coronaviridae virus infections |
| US11007208B2 (en) | 2015-09-16 | 2021-05-18 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| WO2018081863A1 (en) * | 2016-11-04 | 2018-05-11 | University Of Wollongong | 6-SUBSTITUTED DERIVATIVES OF HEXAMETHYLENE AMILORIDE AS INHIBITORS OF uPA AND USES THEREOF |
| US11260070B2 (en) | 2017-03-14 | 2022-03-01 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| US11597742B2 (en) | 2017-05-01 | 2023-03-07 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy) (phenoxy) phosphoryl)amino)propanoate |
| US11266681B2 (en) | 2017-07-11 | 2022-03-08 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US11660307B2 (en) | 2020-01-27 | 2023-05-30 | Gilead Sciences, Inc. | Methods for treating SARS CoV-2 infections |
| WO2021154687A1 (en) | 2020-01-27 | 2021-08-05 | Gilead Sciences, Inc. | Methods for treating sars cov-2 infections |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| EP4125373A4 (en) * | 2020-03-30 | 2024-04-17 | Univ Jefferson | Compounds, compositions, and methods for treating, ameliorating, or preventing viral infections |
| US11701372B2 (en) | 2020-04-06 | 2023-07-18 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carba-nucleoside analogs |
| WO2021207049A1 (en) | 2020-04-06 | 2021-10-14 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carbanucleoside analogs |
| US11903953B2 (en) | 2020-05-29 | 2024-02-20 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11491169B2 (en) | 2020-05-29 | 2022-11-08 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| WO2021262826A2 (en) | 2020-06-24 | 2021-12-30 | Gilead Sciences, Inc. | 1'-cyano nucleoside analogs and uses thereof |
| US11939347B2 (en) | 2020-06-24 | 2024-03-26 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and uses thereof |
| WO2022046631A1 (en) | 2020-08-24 | 2022-03-03 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| EP3960165A1 (en) * | 2020-08-25 | 2022-03-02 | Fondation EspeRare | Nhe-1 inhibitors for the treatment of coronavirus infections |
| WO2022043343A1 (en) * | 2020-08-25 | 2022-03-03 | Fondation Esperare | Nhe-1 inhibitors for the treatment of coronavirus infections |
| US11814406B2 (en) | 2020-08-27 | 2023-11-14 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11926645B2 (en) | 2020-08-27 | 2024-03-12 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2022047065A2 (en) | 2020-08-27 | 2022-03-03 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2022081973A1 (en) | 2020-10-16 | 2022-04-21 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| WO2022251318A1 (en) | 2021-05-26 | 2022-12-01 | Gilead Sciences, Inc. | Phospholipid formulations of 1'-cyano substituted carba-nucleoside analogs |
| WO2023023527A1 (en) | 2021-08-18 | 2023-02-23 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| WO2023167944A1 (en) | 2022-03-02 | 2023-09-07 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11780844B2 (en) | 2022-03-02 | 2023-10-10 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023167938A1 (en) | 2022-03-02 | 2023-09-07 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023168254A1 (en) | 2022-03-03 | 2023-09-07 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2023168194A1 (en) | 2022-03-03 | 2023-09-07 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2023239665A1 (en) | 2022-06-06 | 2023-12-14 | Gilead Sciences, Inc. | Methods for treatment of viral infections including sars-cov-2 |
| WO2024006376A1 (en) | 2022-06-29 | 2024-01-04 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
| WO2024006461A1 (en) | 2022-06-30 | 2024-01-04 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US11192863B2 (en) | 2021-12-07 |
| US20170260147A1 (en) | 2017-09-14 |
| CA2529949A1 (en) | 2004-12-29 |
| EP1646371A2 (en) | 2006-04-19 |
| KR101153254B1 (en) | 2012-07-02 |
| NZ544671A (en) | 2009-02-28 |
| BRPI0411900B1 (en) | 2018-11-21 |
| EP1646371A4 (en) | 2010-09-22 |
| EP2617709A1 (en) | 2013-07-24 |
| WO2004112687A3 (en) | 2007-07-12 |
| BRPI0411900A (en) | 2006-09-19 |
| EP2617709B8 (en) | 2022-12-21 |
| US20190389816A1 (en) | 2019-12-26 |
| KR20060103086A (en) | 2006-09-28 |
| JP5030587B2 (en) | 2012-09-19 |
| BRPI0411900B8 (en) | 2021-05-25 |
| US20150313909A1 (en) | 2015-11-05 |
| JP2007508234A (en) | 2007-04-05 |
| EP2617709B1 (en) | 2022-11-16 |
| US20130035328A1 (en) | 2013-02-07 |
| US10472332B2 (en) | 2019-11-12 |
| CA2529949C (en) | 2013-08-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11192863B2 (en) | Antiviral compounds and methods | |
| ES2339306T3 (en) | USEFUL GUANILHYDRAZONES IN THE TREATMENT OF DISEASES ASSOCIATED WITH THE ACTIVATION OF CELLS T. | |
| CN102858157A (en) | Combination pharmaceutical agents as inhibitors of hcv replication | |
| CN102958525B (en) | Antiviral compounds | |
| US11357771B2 (en) | Methods of preventing or treating flavivirus virus infections and methods of inhibiting the entry of flvivirus, enterovirus or lentivirus into host cells | |
| KR20190093600A (en) | Antibacterial peptide | |
| US20070099968A1 (en) | Antiviral compounds and methods | |
| US11135219B2 (en) | Methods of treating or preventing Zika virus infection | |
| AU2004248859B2 (en) | Antiviral acylguanidine compounds and methods | |
| JP2009242247A (en) | Human immunodeficiency virus infection inhibitor and aids-treating or preventing drug | |
| CN101111475B (en) | Antiviral compounds and methods | |
| KR20200012894A (en) | Peptides and Uses thereof as Antiviral Agents | |
| US20070161550A1 (en) | Antiviral compositions which inhibit paramyxovirus infection | |
| EP0684818B1 (en) | Method of reversing resistance of hiv-1 strains to zidovudine | |
| US11331371B2 (en) | Labyrinthopeptins as anti-viral agents | |
| US20230086360A1 (en) | Antimicrobial Peptides | |
| WO2018189566A1 (en) | Use of a chemical compound as a therapeutic agent | |
| WO2024040156A2 (en) | Novel cyclic gamma aapeptide pan-coronavirus inhibitor and method of treating coronavirus infection | |
| KR20110100759A (en) | Peptide compound having an inhibition activity for orientia tsutsugamushi infection or proliferation and pharmaceutical composition for treating scrub typhus using the same | |
| CN115710303A (en) | Polypeptide derivative for resisting H1N1 influenza virus and application | |
| MXPA99007077A (en) | Quinoxaline in triple combination with protease inhibitors and reverse transcriptase inhibitors as medicines for treating aids | |
| EP0879055A1 (en) | Inhibition of viral replication |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480024097.4 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2529949 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006515560 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007099968 Country of ref document: US Ref document number: 10562296 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020057024860 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004248859 Country of ref document: AU Ref document number: 544671 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 159/KOLNP/2006 Country of ref document: IN Ref document number: 00159/KOLNP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/00650 Country of ref document: ZA Ref document number: 200600650 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004737487 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2004248859 Country of ref document: AU Date of ref document: 20040626 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004248859 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004737487 Country of ref document: EP |
|
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: PI0411900 Country of ref document: BR |
|
| WWP | Wipo information: published in national office |
Ref document number: 10562296 Country of ref document: US |