WO2004048317A1 - Substituted amides active at the cannabinoid-1 receptor - Google Patents
Substituted amides active at the cannabinoid-1 receptor Download PDFInfo
- Publication number
- WO2004048317A1 WO2004048317A1 PCT/US2003/007039 US0307039W WO2004048317A1 WO 2004048317 A1 WO2004048317 A1 WO 2004048317A1 US 0307039 W US0307039 W US 0307039W WO 2004048317 A1 WO2004048317 A1 WO 2004048317A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- trifluoromethyl
- methylpropanamide
- pyridyloxy
- chlorophenyl
- methyl
- Prior art date
Links
- VPDAKGOKLCCXMI-UHFFFAOYSA-N CC(C(Cc(cc1)ccc1Cl)N(CC1)c2c1cccc2)N Chemical compound CC(C(Cc(cc1)ccc1Cl)N(CC1)c2c1cccc2)N VPDAKGOKLCCXMI-UHFFFAOYSA-N 0.000 description 1
- PRCDFCBCHLBJAE-UHFFFAOYSA-N CC(C(Cc(cc1)ccc1Cl)c1cccnc1)N Chemical compound CC(C(Cc(cc1)ccc1Cl)c1cccnc1)N PRCDFCBCHLBJAE-UHFFFAOYSA-N 0.000 description 1
- ZUTDVFRJNNKHND-UHFFFAOYSA-N CC(C)(C(O)=O)Oc1ccc(C(F)(F)F)cn1 Chemical compound CC(C)(C(O)=O)Oc1ccc(C(F)(F)F)cn1 ZUTDVFRJNNKHND-UHFFFAOYSA-N 0.000 description 1
- NARGBGXBGLUPLB-UHFFFAOYSA-N CC(C)(C(O)=O)Oc1nccc(C(F)(F)F)c1 Chemical compound CC(C)(C(O)=O)Oc1nccc(C(F)(F)F)c1 NARGBGXBGLUPLB-UHFFFAOYSA-N 0.000 description 1
- JLTDJTHDQAWBAV-UHFFFAOYSA-N CN(C)c1ccccc1 Chemical compound CN(C)c1ccccc1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/64—One oxygen atom attached in position 2 or 6
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C235/06—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C235/18—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated having at least one of the singly-bound oxygen atoms further bound to a carbon atom of a six-membered aromatic ring, e.g. phenoxyacetamides
- C07C235/20—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated having at least one of the singly-bound oxygen atoms further bound to a carbon atom of a six-membered aromatic ring, e.g. phenoxyacetamides having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
- C07C237/06—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/40—Acylated substituent nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/89—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B50/00—Methods of creating libraries, e.g. combinatorial synthesis
- C40B50/08—Liquid phase synthesis, i.e. wherein all library building blocks are in liquid phase or in solution during library creation; Particular methods of cleavage from the liquid support
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Definitions
- Marijuana (Cannabis sativa L.) and its derivatives have been used for centuries for medicinal and recreational purposes.
- a major active ingredient in marijuana and hashish has been determined to be ⁇ 9-tetrahydrocannabinol ( ⁇ 9-THC).
- ⁇ 9-THC ⁇ 9-tetrahydrocannabinol
- CBl and CB2 G-protein coupled receptors
- the CBl receptor is primarily found in the central and peripheral nervous systems and to a lesser extent in several peripheral organs.
- the CB2 receptor is found primarily in lymphoid tissues and cells.
- mice The genes for the respective cannabinoid receptors have each been disrupted in mice.
- the CBl-/- receptor knockout mice appeared normal and fertile. They were resistant to the effects of ⁇ 9-THC and demonstrated a strong reduction in the reinforcing properties of morphine and the severity of withdrawal syndrome. They also demonstrated reduced motor activity and hypoalgesia. Excessive exposure to ⁇ 9-THC can lead to overeating, psychosis, hypothermia, memory loss, and sedation.
- CBl modulator characterized as an inverse agonist or an antagonist, N-(l ⁇ piperidinyl)-5 ⁇ (4-chlorophenyl)-l-(2,4-dichlorophenyl)-4- methylpyrazole-3-carboxamide (SR141716A), in clinical trials for treatment of eating disorders at this time.
- CBl modulator characterized as an inverse agonist or an antagonist, N-(l ⁇ piperidinyl)-5 ⁇ (4-chlorophenyl)-l-(2,4-dichlorophenyl)-4- methylpyrazole-3-carboxamide (SR141716A)
- CBl modulators that have pharmacokinetic and pharmacodynamic properties suitable for use as human pharmaceuticals.
- CBl receptor modulators such as CBl inverse agonists
- CBl inverse agonists presynaptic cannabinoid CBl receptors mediate the inhibition of noradrenaline release (in the guinea pig lung) (Europ. J. of Pharmacology, 2001, 431 (2), 237-244).
- CBl receptor modulators Treatment of cirrhosis of the liver with CBl receptor modulators is supported by the finding that a CBl receptor modulator will reverse the low blood pressure observed in rats with carbon tetrachloride-induced liver cirrhosis and will lower the elevated mesenteric blood flow and portal vein pressure (Nature Medicine, 2001, 7 (7), 827-832).
- PCT Application Nos. WO98/37061, WO00/10967, and WO00/10968 disclose diaryl ether sulfonamides having activity against the cannabinoid receptors.
- PCT Application Nos. WO97/29079 and WO99/02499 disclose alkoxy-isoindolones and alkoxy-quinolones as having activity against the cannabinoid receptors.
- US Patent US 5,532,237 discloses N-benzoyl-indole derivatives having activity against the cannabinoid receptors.
- PCT publication WO 01/58869 discloses pyrazoles, pyrroles and imidazole cannabinoid receptor modulatorsuseful for treating respiratory and non- respiratory leukocyte activation-associated disorders.
- PCT publications WO 01/64632, 01/64633, and 01/64634 assigned to Aventis are directed to azetidine derivatives as cannabinoid antagonists.
- the compounds of the present invention are modulators of the Cannabinoid-1 (CBl) receptor and are useful in the treatment, prevention and suppression of diseases mediated by the Cannabinoid-1 (CBl) receptor.
- compounds of the present invention are antagonists or inverse agonists of the CBl receptor.
- the invention is concerned with the use of these compounds to modulate the Cannabinoid-1 (CBl) receptor.
- compounds of the present invention are useful as centrally acting drugs in the treatment of psychosis, memory deficits, cognitive disorders, migraine, neuropathy, neuro-inflammatory disorders including multiple sclerosis and Guillain-Barre syndrome and the inflammatory sequelae of viral encephalitis, cerebral vascular accidents, and head trauma, anxiety disorders, stress, epilepsy, Parkinson's disease, movement disorders, and schizophrenia.
- the compounds are also useful for the treatment of substance abuse disorders, particularly to opiates, alcohol, marijuana, and nicotine.
- the compounds are also useful for the treatment of eating disorders by inhibiting excessive food intake and the resulting obesity and complications associated therewith.
- the compounds are also useful for the treatment of constipation and chronic intestinal pseudo-obstruction, as well as for the treatment of asthma, and ci ⁇ hosis of the liver.
- the present invention is concerned with novel substituted amides of the general Formula I :
- (I) and pharmaceutically acceptable salts thereof which are antagonists and/or inverse agonists of the Cannabinoid-1 (CBl) receptor and are useful in the treatment, prevention and suppression of diseases mediated by the Cannabinoid-1 (CBl) receptor.
- the invention is concerned with the use of these novel compounds to selectively antagonize the Cannabinoid-1 (CBl) receptor.
- compounds of the present invention are useful as centrally acting drugs in the treatment of psychosis, memory deficits, cognitive disorders, migraine, neuropathy, neuro-inflammatory disorders including multiple sclerosis and Guillain-Ba ⁇ e syndrome and the inflammatory sequelae of viral encephalitis, cerebral vascular accidents, and head trauma, anxiety disorders, stress, epilepsy, Parkinson's disease, movement disorders, and schizophrenia.
- the compounds are also useful for the treatment of substance abuse disorders, particularly to opiates, alcohol, marijuana, and nicotine, including smoking cessation.
- the compounds are also useful for the treatment of obesity or eating disorders associated with excessive food intake and complications associated therewith.
- the compounds are also useful for the treatment of constipation and chronic intestinal pseudo-obstruction.
- the compounds are also useful for the treatment of cirrhosis of the liver.
- the compounds are also useful for the treatment of asthma.
- the present invention is also concerned with treatment of these conditions, and the use of compounds of the present invention for manufacture of a medicament useful in treating these conditions.
- the present invention is also concerned with treatment of these conditions through a combination of compounds of formula I and other currently available pharmaceuticals.
- the invention is also concerned with novel compounds of structural formula I.
- the invention is also concerned with pharmaceutical formulations comprising one of the compounds as an active ingredient.
- the invention is further concerned with processes for preparing the compounds of this invention.
- R2 is selected from:
- each cycloalkyl, aryl and heteroaryl is optionally substituted with one to three substituents independently selected from Rb; each R a is independently selected from:
- each R D is independently selected from: (1) halogen,
- R c is independently selected from:
- each R c may be unsubstituted or substituted with one to three substituents selected from Rh;
- Rd is independently selected from:
- each Rd may be unsubstituted or substituted with one to three substituents selected from R n ; each R n is independently selected from: (1) halogen, (2) Ci- 3 alkyl,
- Rl is selected from:
- each aryl and heteroaryl is optionally substituted with one or two substitutents independently selected from Rb, and each pyridyl is optionally present as the N-oxide.
- Rl is selected from:
- Rl is 5-cyano-3- pyridyl.
- R2 is selected from: (1) Ci-6alkyl,
- R is selected from:
- R2 is selected from: (1) 2-methylpropyl,
- each R a is independently selected from:
- each R a is independently selected from:
- each Rb is independently selected from: (1) halogen,
- each Rb is independently selected from:
- each Rb is independently selected from: (1) fluoro, (2) chloro,
- each R c is independently selected from: (1) hydrogen,
- each R c may be unsubstituted or substituted with a substituent selected from Rh.
- R c is phenyl.
- Rd is selected from: (1) C4_6cycloalkyl
- Rd may be unsubstituted or substituted with one or two substituents selected from Rh.
- Rd is selected from:
- Rd is selected from:
- Rd is 5-trifluoromethyl-2-pyridyl.
- each Rh is independently selected from:
- each Rh is independently selected from:
- novel compounds which may be employed in the methods, uses and compositions of the present invention, include:
- Alkyl as well as other groups having the prefix “alk”, such as alkoxy, alkanoyl, means carbon chains which may be linear or branched or combinations thereof.
- alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl, and the like.
- Cycloalkyl means mono- or bicyclic or bridged saturated carbocyclic rings, each of which having from 3 to 10 carbon atoms
- cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and the like.
- Aryl means mono- or bicyclic aromatic rings containing only carbon atoms. Examples of aryl include phenyl, naphthyl, and the like.
- Heteroaryl means a mono- or bicyclic aromatic ring containing at least one heteroatom selected from N, O and S, with each ring containing 5 to 6 atoms.
- heteroaryl include py ⁇ olyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyrimidyl, pyridazinyl, pyrazinyl, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzofuranyl, benzothiophenyl, furo(2,3-b)pyridyl, quinolyl, indolyl, isoquinolyl, imidazothiazolyl, and the like.
- Cycloheteroalkyl means mono- or bicyclic or bridged saturated rings containing at least one heteroatom selected from N, S and O, each of said ring having from 3 to 10 atoms in which the point of attachment may be carbon or nitrogen.
- the term also includes monocyclic heterocycle fused to an aryl or heteroaryl group in which the point of attachment is on the non-aromatic portion.
- Examples of “cycloheteroalkyl” include indolyl, azaindolyl and the like. The cycloheteroalkyl ring may be substituted on the ring carbons and/or the ring nitrogens.
- Halogen includes fluorine, chlorine, bromine and iodine.
- substituted shall be deemed to include multiple degrees of substitution by a named substitutent. Where multiple substituent moieties are disclosed or claimed, the substituted compound can be independently substituted by one or more of the disclosed or claimed substituent moieties, singly or plurally. By independently substituted, it is meant that the (two or more) substituents can be the same or different.
- Compounds of Formula I may contain one or more asymmetric centers and can thus occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers. The present invention is meant to comprehend all such isomeric forms of the compounds of Formula I.
- Tautomers are defined as compounds that undergo rapid proton shifts from one atom of the compound to another atom of the compound. Some of the compounds described herein may exist as tautomers with different points of attachment of hydrogen. Such an example may be a ketone and its enol form known as keto-enol tautomers. The individual tautomers as well as mixture thereof are encompassed with compounds of Formula I.
- Compounds of the Formula I may be separated into diastereoisomeric pairs of enantiomers by, for example, fractional crystallization from a suitable solvent, for example MeOH or EtOAc or a mixture thereof.
- a suitable solvent for example MeOH or EtOAc or a mixture thereof.
- the pair of enantiomers thus obtained may be separated into individual stereoisomers by conventional means, for example by the use of an optically active amine as a resolving agent or on a chiral HPLC column.
- any enantiomer of a compound of the general Formula I may be obtained by stereospecific synthesis using optically pure starting materials or reagents of known configuration.
- Racemic mixtures can be separated into their individual enantiomers by any of a number of conventional methods. These include chiral chromatography, derivatization with a chiral auxiliary followed by separation by chromatography or crystallization, and fractional crystallization of diastereomeric salts.
- crystalline forms for compounds of the present invention may exist as polymorphs and as such are intended to be included in the present invention.
- some of the compounds of the instant invention may form solvates with water or common organic solvents. Such solvates are encompassed within the scope of this invention.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occu ⁇ ing substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- pharmaceutically acceptable salt further includes all acceptable salts such as acetate, lactobionate, benzenesulfonate, laurate, benzoate, malate, bicarbonate, maleate, bisulfate, mandelate, bitartrate, mesylate, borate, methylbromide, bromide, methylnitrate, calcium edetate, methylsulfate, camsylate, mucate, carbonate, napsylate, chloride, nitrate, clavulanate, N-methylglucamine, citrate, ammonium salt, dihydrochloride, oleate, edetate, oxalate, edisylate, pamoate (embonate), estolate, palmitate, esylate, pantothenate, fumarate, phosphate/diphosphate, gluceptate, polygalacturonate, gluconate, salicylate, glutamate, stearate, glycollyl
- Compounds of the present invention are modulators of the CBl receptor.
- the compounds of structural formula I are antagonists or inverse agonists of the CBl receptor.
- An "agonist” is a compound (hormone, neurotransmitter or synthetic compound) which binds to a receptor, inducing a conformational change in the receptor which, in turn, produces a response such as contraction, relaxation, secretion, change in enzyme activity, etc. similar to that elicited by the physiologically relevant agonist ligand(s) for that receptor.
- An "antagonist” is a compound which attenuates the effect of an agonist.
- An “inverse agonist” is a compound which acts on a receptor but produces the opposite effect produced by the agonist of the particular receptor.
- Compounds of this invention are modulators of the CBl receptor and as such are useful as centrally acting drugs in the treatment of psychosis, memory deficits, cognitive disorders, migraine, neuropathy, neuro-inflammatory disorders including multiple sclerosis and Guillain-Barre syndrome and the inflammatory sequelae of viral encephalitis, cerebral vascular accidents, and head trauma, anxiety disorders, stress, epilepsy, Parkinson's disease, movement disorders, and schizophrenia.
- the compounds are also useful for the treatment of substance abuse disorders, particularly to opiates, alcohol, marijuana, and nicotine.
- the compounds are also useful for the treatment of obesity or eating disorders associated with excessive food intake and complications associated therewith.
- the compounds are also useful for the treatment of constipation and chronic intestinal pseudo-obstruction.
- the compounds are also useful for the treatment of cirrhosis of the liver.
- the compounds are also useful for the treatment of asthma.
- administering should be understood to mean providing a compound of the invention or a prodrug of a compound of the invention to the individual in need of treatment.
- the administration of the compound of structural formula I in order to practice the present methods of therapy is carried out by administering an effective amount of the compound of structural formula I to the patient in need of such treatment or prophylaxis.
- the need for a prophylactic administration according to the methods of the present invention is determined via the use of well known risk factors.
- the effective amount of an individual compound is determined, in the final analysis, by the physician in charge of the case, but depends on factors such as the exact disease to be treated, the severity of the disease and other diseases or conditions from which the patient suffers, the chosen route of administration other drugs and treatments which the patient may concomitantly require, and other factors in the physician's judgment.
- the utilities of the present compounds in these diseases or disorders may be demonstrated in animal disease models that have been reported in the literature.
- mice The following are examples of such animal disease models: a) suppression of food intake and resultant weight loss in rats (Life Sciences 1998, 63, 113-117); b) reduction of sweet food intake in marmosets (Behavioural Pharm. 1998, 9, 179-181); c) reduction of sucrose and ethanol intake in mice (Psychopharm. 1997, 132, 104- 106); d) increased motor activity and place conditioning in rats (Psychopharm. 1998, 135, 324-332; Psychopharmacol 2000, 151: 25-30); e) spontaneous locomotor activity in mice (J. Pharm. Exp. Ther. 1996, 277, 586-594); f) reduction in opiate self- administration in mice (Sci.
- prophylactic or therapeutic dose of a compound of Formula I will, of course, vary with the nature of the severity of the condition to be treated and with the particular compound of Formula I and its route of administration. It will also vary according to the age, weight and response of the individual patient. In general, the daily dose range lie within the range of from about 0.001 mg to about 100 mg per kg body weight of a mammal, preferably 0.01 mg to about 50 mg per kg, and most preferably 0.1 to 10 mg per kg, in single or divided doses. On the other hand, it may be necessary to use dosages outside these limits in some cases.
- a suitable dosage range is from about 0.001 mg to about 25 mg (preferably from 0.01 mg to about 1 mg) of a compound of Foimula I per kg of body weight per day and for preventive use from about 0.1 mg to about 100 mg (preferably from about 1 mg to about 100 mg and more preferably from about 1 mg to about 10 mg) of a compound of Formula I per kg of body weight per day.
- a suitable dosage range is, e.g. from about 0.01 mg to about 1000 mg of a compound of Formula I per day, preferably from about 0.1 mg to about 10 mg per day.
- the compositions are preferably provided in the form of tablets containing from 0.01 to 1,000 mg, preferably 0.01, 0.05, 0.1, 0.5, 1, 2.5, 5, 10, 15, 20, 25, 30, 40, 50, 100, 250, 500, 750 or 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- compositions which comprises a compound of Formula I and a pharmaceutically acceptable carrier.
- composition is intended to encompass a product comprising the active ingredient(s), and the inert ingredient(s) (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of Formula I, additional active ingredient(s), and pharmaceutically acceptable excipients.
- Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dosage of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed.
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
- compositions of the present invention comprise a compound of Formula I as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
- the compounds of the present invention are conveniently delivered in the form of an aerosol spray presentation from pressurized packs or nebulizers.
- the compounds may also be delivered as powders which may be formulated and the powder composition may be inhaled with the aid of an insufflation powder inhaler device.
- the preferred delivery systems for inhalation are metered dose inhalation (MDI) aerosol, which may be formulated as a suspension or solution of a compound of Formula I in suitable propellants, such as fluorocarbons or hydrocarbons and dry powder inhalation (DPI) aerosol, which may be formulated as a dry powder of a compound of Formula I with or without additional excipients.
- MDI metered dose inhalation
- DPI dry powder inhalation
- Suitable topical formulations of a compound of fo ⁇ nula I include transdermal devices, aerosols, creams, solutions, ointments, gels, lotions, dusting powders, and the like.
- the topical pharmaceutical compositions containing the compounds of the present invention ordinarily include about 0.005% to 5% by weight of the active compound in admixture with a pharmaceutically acceptable vehicle.
- Transdermal skin patches useful for administering the compounds of the present inveniton include those well known to those of ordinary skill in that art. To be administered in the form of a transdermal delivery system, the dosage administration will, of course, be continuous rather than intermittent throughout the dosage regimen.
- the compounds of Formula I can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous).
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as, for example, suspensions, elixirs and solutions; or carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, capsules and tablets, with the solid oral preparations being prefe ⁇ ed over the liquid preparations. Because of their ease of administration, tablets and capsules represent the most advantageous oral dosage unit form in which case solid pharmaceutical carriers are obviously employed. If desired, tablets may be coated by standard aqueous or nonaqueous techniques.
- compositions of the present invention suitable for oral administration may be presented as discrete units such as capsules (including timed release and sustained release formulations), pills, cachets, powders, granules or tablets each containing a predetermined amount of the active ingredient, as a powder or granules or as a solution or a suspension in an aqueous liquid, a non-aqueous liquid, an oil-in-water emulsion or a water-in-oil liquid emulsion, incluidng elixirs, tinctures, solutions, suspensions, syrups and emulsions.
- Such compositions may be prepared by any of the methods of pharmacy but all methods include the step of bringing into association the active ingredient with the carrier which constitutes one or more necessary ingredients.
- compositions are prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product into the desired presentation.
- a tablet may be prepared by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine, the active ingredient in a free-flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent. Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent.
- each tablet contains from 0.01 to 1,000 mg, particularly 0.01, 0.05, 0.1, 0.5, 1, 2.5, 3, 5, 6, 10, 15, 25, 50, 75, 100, 125, 150, 175, 180, 200, 225, 500, 750 and 1,000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated
- each cachet or capsule contains from about 0.01 to 1,000 mg, particularly 0.01, 0.05, 0.1, 0.5, 1.0, 2.5, 3, 5, 6, 10, 15, 25, 50, 75, 100, 125, 150, 175, 180, 200, 225, 500, 750 and 1,000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- Additional suitable means of administration of the compounds of the present invention include injection, intravenous bolus or infusion, intraperitoneal, subcutaneous, intramuscular and topical, with or without occlusion.
- Exemplifying the invention is a pharmaceutical composition comprising any of the compounds described above and a pharmaceutically acceptable carrier. Also exemplifying the invention is a phannaceutical composition made by combining any of the compounds described above and a pharmaceutically acceptable carrier.
- An illustration of the invention is a process for malting a pharmaceutical composition comprising combining any of the compounds described above and a pharmaceutically acceptable carrier.
- the dose may be administered in a single daily dose or the total daily dosage may be administered in divided doses of two, three or four times daily. Furthermore, based on the properties of the individual compound selected for administration, the dose may be administered less frequently, e.g., weekly, twice weekly, monthly, etc. The unit dosage will, of course, be co ⁇ espondingly larger for the less frequent administration.
- Compound of Formula I 25 Microcrystalline Cellulose 415 Povidone 14.0
- Compounds of Formula I may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which compounds of Formula I are useful. Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I.
- a pharmaceutical composition containing such other drugs in addition to the compound of Formula I is prefe ⁇ ed.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of Formula I.
- Examples of other active ingredients that may be combined with a compound of Formula I include, but are not limited to: antipsychotic agents, cognition enhancing agents, anti-migraine agents, anti-asthmatic agents, antiinflammatory agents, axiolytics, anti-Parkinson's agents, anti-epileptics, anorectic agents, and serotonin reuptake inhibitors, and other anti-obesity agents which may be administered separately or in the same pharmaceutical compositions. It will be appreciated that for the treatment or prevention of eating disorders, including obesity, bulimia nervosa and compulsive eating disorders, a compound of the present invention may be used in conjunction with other anorectic agents.
- the present invention also provides a method for the treatment or prevention of eating disorders, which method comprises administration to a patient in need of such treatment an amount of a compound of the present invention and an amount of an anorectic agent, such that together they give effective relief.
- "Obesity” is a condition in which there is an excess of body fat. The operational definition of obesity is based on the Body Mass Index (BMI), which is calculated as body weight per height in meters squared (kg/m2).
- BMI Body Mass Index
- “Obesity” refers to a condition whereby an otherwise healthy subject has a Body Mass Index (BMI) greater than or equal to 30 kg/m2, or a condition whereby a subject with at least one co- morbidity has a BMI greater than or equal to 27 kg/m2.
- An “obese subject” is an otherwise healthy subject with a Body Mass Index (BMI) greater than or equal to 30 kg/m2 or a subject with at least one co-morbidity with a BMI greater than or equal to 27 kg/m2.
- BMI Body Mass Index
- a “subject at risk for obesity” is an otherwise healthy subject with a BMI of 25 kg/m2 to less than 30 kg/m2 or a subject with at least one co-morbidity with a BMI of 25 kg/m2 to less than 27 kg/m2.
- BMI Body Mass Index
- “obesity” refers to a condition whereby a subject with at least one obesity-induced or obesity-related co- morbidity that requires weight reduction or that would be improved by weight reduction, has a BMI greater than or equal to 25 kg/m2.
- an “obese subject” refers to a subject with at least one obesity-induced or obesity-related co-morbidity that requires weight reduction or that would be improved by weight reduction, with a BMI greater than or equal to 25 kg/m2.
- a "subject at risk for obesity” is a subject with a BMI of greater than 23 kg/m2 to less than 25 kg/m.2.
- obesity is meant to encompass all of the above definitions of obesity.
- Obesity-induced or obesity-related co-morbidities include, but are not limited to, diabetes, non-insulin dependent diabetes mellitus - type 2, impaired glucose tolerance, impaired fasting glucose, insulin resistance syndrome, dyslipidemia, hypertension, hyperuricacidemia, gout, coronary artery disease, myocardial infarction, angina pectoris sleep apnea syndrome, Pickwickian syndrome, fatty liver; cerebral infarction, cerebral thrombosis, transient ischemic attack, orthopedic disorders, arthritis deformans, lumbodynia, emmeniopathy, and infertility.
- co-morbidities include: hypertension, hyperlipidemia, dyslipidemia, glucose intolerance, cardiovascular disease, sleep apnea, diabetes mellitus, and other obesity-related conditions.
- Treatment refers to the administration of the compounds or compositions of the present invention to reduce or maintain the body weight of an obese subject.
- One outcome of treatment may be reducing the body weight of an obese subject relative to that subject's body weight immediately before the administration of the compounds or compositions of the present invention.
- Another outcome of treatment may be preventing regain of body weight previously lost as a result of diet, exercise, or pharmacotherapy.
- Another outcome of treatment may be decreasing the occurrence of and or the severity of obesity-related diseases.
- the treatment may suitably result in a reduction in food or calorie intake by the subject, including a reduction in total food intake, or a reduction of intake of specific components of the diet such as carbohydrates or fats; and/or the inhibition of nutrient absorption; and/or the inhibition of the reduction of metabolic rate; and in weight reduction in patients in need thereof.
- the treatment may also result in an alteration of metabolic rate, such as an increase in metabolic rate, rather than or in addition to an inhibition of the reduction of metabolic rate; and/or in minimization of the metabolic resistance that normally results from weight loss.
- "Prevention” refers to the administration of the compounds or compositions of the present invention to reduce or maintain the body weight of a subject at risk for obesity.
- One outcome of prevention may be reducing the body weight of a subject at risk for obesity relative to that subject's body weight immediately before the administration of the compounds or compositions of the present invention. Another outcome of prevention may be preventing body weight regain of body weight previously lost as a result of diet, exercise, or pharmacotherapy. Another outcome of prevention may be preventing obesity from occurring if the treatment is administered prior to the onset of obesity in a subject at risk for obesity. Another outcome of prevention may be decreasing the occu ⁇ ence and/or severity of_obesity-related disorders if the treatment is administered prior to the onset of obesity in a subject at risk for obesity.
- such treatment may prevent the occu ⁇ ence, progression or severity of obesity-related disorders, such as, but not limited to, arteriosclerosis, Type II diabetes, polycystic ovarian disease, cardiovascular diseases, osteoarthritis, dermatological disorders, hypertension, insulin resistance, hypercholesterolemia, hypertriglyceridemia, and cholelithiasis.
- Obesity-related disorders are associated with, caused by, or result from obesity.
- obesity-related disorders include overeating and bulimia, hypertension, diabetes, elevated plasma insulin concentrations and insulin resistance, dyslipidemias, hyperlipidemia, endometrial, breast, prostate and colon cancer, osteoarthritis, obstructive sleep apnea, cholelithiasis, gallstones, heart disease, abnormal heart rhythms and arrythmias, myocardial infarction, congestive heart failure, coronary heart disease, sudden death, stroke, polycystic ovarian disease, craniopharyngioma, the Prader-Willi Syndrome, Frohlich's syndrome, GH-deficient subjects, normal variant short stature, Turner's syndrome, and other pathological conditions showing reduced metabolic activity or a decrease in resting energy expenditure as a percentage of total fat-free mass, e.g, children with acute lymphoblastic leukemia.
- obesity-related disorders are metabolic syndrome, also known as syndrome X, insulin resistance syndrome, sexual and reproductive dysfunction, such as infertility, hypogonadism in males and hirsutism in females, gastrointestinal motility disorders, such as obesity-related gastro-esophageal reflux, respiratory disorders, such as obesity-hypoventilation syndrome (Pickwickian syndrome), cardiovascular disorders, inflammation, such as systemic inflammation of the vasculature, arteriosclerosis, hypercholesterolemia, hyperuricaemia, lower back pain, gallbladder disease, gout, and kidney cancer.
- the compositions of the present invention are also useful for reducing the risk of secondary outcomes of obesity, such as reducing the risk of left ventricular hypertrophy.
- diabetes includes both insulin- dependent diabetes mellitus (i.e., LDDM, also known as type I diabetes) and non- insulin-dependent diabetes mellitus (i.e., NIDDM, also known as Type II diabetes.
- Type I diabetes, or insulin-dependent diabetes is the result of an absolute deficiency of insulin, the hormone which regulates glucose utilization.
- Type II diabetes, or insulin-independent diabetes i.e., non-insulin-dependent diabetes mellitus
- Most of the Type II diabetics are also obese.
- the compounds and compositions of the present invention are useful for treating both Type I and Type II diabetes.
- the compounds and compositions are especially effective for treating Type II diabetes.
- the compounds and compositions of the present invention are also useful for treating and/or preventing gestational diabetes mellitus.
- substance abuse disorders includes substance dependence or abuse with or without physiological dependence.
- the substances associated with these disorders are: alcohol, amphetamines (or amphetamine-like substances), caffeine, cannabis, cocaine, hallucinogens, inhalants, marijuana, nicotine, opioids, phencyclidine (or phencyclidine-like compounds), sedative-hypnotics or benzodiazepines, and other (or unknown) substances and combinations of all of the above.
- the term "substance abuse disorders” includes drug withdrawal disorders such as alcohol withdrawal with or without perceptual disturbances; alcohol withdrawal delirium; amphetamine withdrawal; cocaine withdrawal; nicotine withdrawal; opioid withdrawal; sedative, hypnotic or anxiolytic withdrawal with or without perceptual disturbances; sedative, hypnotic or anxiolytic withdrawal delirium; and withdrawal symptoms due to other substances. It will be appreciated that reference to treatment of nicotine withdrawal includes the treatment of symptoms associated with smoking cessation.
- substance abuse disorders include substance-induced anxiety disorder with onset during withdrawal; substance-induced mood disorder with onset during withdrawal; and substance-induced sleep disorder with onset during withdrawal.
- a combination of a conventional antipsychotic drug with a CBl receptor modulator may provide an enhanced effect in the treatment of mania. Such a combination would be expected to provide for a rapid onset of action to treat a manic episode thereby enabling prescription on an "as needed basis”. Furthermore, such a combination may enable a lower dose of the antispychotic agent to be used without compromising the efficacy of the antipsychotic agent, thereby minimizing the risk of adverse side-effects.
- a yet further advantage of such a combination is that, due to the action of the CB 1 receptor modulator, adverse side-effects caused by the antipsychotic agent such as acute dystonias, dyskinesias, akathesia and tremor may be reduced or prevented.
- the present invention also provides a method for the treatment or prevention of mania, which method comprises administration to a patient in need of such treatment or at risk of developing mania of an amount of a CBl receptor modulator and an amount of an antipsychotic agent, such that together they give effective relief. It will be appreciated that the CB 1 receptor modulator and the antipsychotic agent may be present as a combined preparation for simultaneous, separate or sequential use for the treatment or prevention of mania.
- the CBl receptor modulator and the antipsychotic agent may be in the same pharmaceutically acceptable carrier and therefore administered simultaneously. They may be in separate pharmaceutical carriers such as conventional oral dosage forms which are taken simultaneously.
- the term “combination” also refers to the case where the compounds are provided in separate dosage forms and are administered sequentially. Therefore, by way of example, the antipsychotic agent may be administered as a tablet and then, within a reasonable period of time, the CBl receptor modulator may be administered either as an oral dosage form such as a tablet or a fast-dissolving oral dosage form.
- a fast-dissolving oral formulation is meant, an oral delivery form which when placed on the tongue of a patient, dissolves within about 10 seconds.
- a combination of a conventional antipsychotic drug with a CBl receptor modulator may provide an enhanced effect in the treatment of schizophrenic disorders. Such a combination would be expected to provide for a rapid onset of action to treat schizophrenic symptoms thereby enabling prescription on an "as needed basis". Furthermore, such a combination may enable a lower dose of the CNS agent to be used without compromising the efficacy of the antipsychotic agent, thereby minimizing the risk of adverse side-effects.
- a yet further advantage of such a combination is that, due to the action of the CB 1 receptor modulator, adverse side-effects caused by the antipsychotic agent such as acute dystonias, dyskinesias, akathesia and tremor may be reduced or prevented.
- a combination of a conventional anti- asthmatic drug with a CBl receptor modulator may provide an enhanced effect in the treatment of asthma.
- the present invention also provides a method for the treatment or prevention of asthma, which method comprises administration to a patient in need of such treatment an amount of a compound of the present invention and an amount of an anti-asthmatic agent, such that together they give effective relief.
- the method of treatment of this invention comprises a method of modulating the CBl receptor and treating CBl receptor mediated diseases by administering to a patient in need of such treatment a non-toxic therapeutically effective amount of a compound of this invention that selectively antagonizes the CBl receptor in preference to the other CB or G-protein coupled receptors.
- terapéuticaally effective amount means the amount the compound of structural formula I that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician, which includes alleviation of the symptoms of the disorder being treated.
- the novel methods of treatment of this invention are for disorders known to those skilled in the art.
- the term “mammal” includes humans.
- Step A 3-(4-Chlorophenyl)-2-phenylpropanoic acid, methyl ester.
- methyl phenylacetate (12 g, 80 mmol) and 4-chlorobenzyl bromide (16 g, 80 mmol) in 250 mLanhydrous THF at -78°C was added sodium hexamethyldisilazide (1 M in THF, 80 mL, 80 mmol) (potassium hexamethyldisilazide in toluene may be used with similar results).
- the reaction was allowed to warm to room temperature overnight.
- Step B 3-(4-Chlorophenyl)-2-phenylpropanoic acid.
- Step A To a mixture of methyl 3-(4-chlorophenyl)-2-phenylpropionate (Step A, 20 g, 74 mmol) in acetonitrile (100 mL) and water (100 mL) was added lithium hydroxide monohydrate (8.8 g, 0.21 mol). After stirring at room temperature for 3 days, the volatile materials were removed by concentrating on a rotary evaporator and the residue was partitioned between water (300 mL) and hexane/ether (1:1, 200 mL).
- Step E 4-(4-Chlorophenyl)-3-phenyl-2-butanol.
- Step D To a solution of 4-(4-chlorophenyl)-3-phenyl-2-butanone (Step D, 13 g, 50 mmol) in MeOH (100 mL) at 0 °C was added sodium borohydride (3.8 g, 100 mmol). After stirring at 0°C for 30 min, the reaction was quenched by addition of 2 M hydrochloric acid (50 mL). The volatile materials were removed by concentrating on a rotary evaporator and the residue partitioned between water (100 mL) and EtOAc (200 mL). The organic layer was separated and the aqueous layer extracted with EtOAc (2 x 200 mL).
- Step F 4-(4-Chlorophenyl)-2-methanesulfonyloxy-3-phenylbutane.
- 4-(4-chlorophenyl)-3-phenyl-2-butanol (Step E, faster eluting isomer, 9.0 g, 34 mmol) in EtOAc (100 mL) at 0°C was added triethyl amine (dried over activated molecular sieves, 5.8 mL. 42 mmol) and methanesulfonyl chloride (3.0 mL, 38 mmol).
- Step F To a solution of 4-(4-chlorophenyl)-2-methanesulfonyloxy-3-phenylbutane (Step F, 12 g, 34 mmol) in DMF (50 mL) was added sodium azide (11 g, 0.17 mol). After stirring at 120°C for 1 h, the reaction mixture was poured into water (200 mL), and the product was extracted with ether (2 x 100 mL). The combined organic extracts were washed with water, dried over MgSO4, filtered and concentrated to dryness, and the residue was purified on a silica gel column eluting with hexane to give the title compound.
- Step H 2-(N-tert-Butoxycarbonyl)amino-4-(4-chlorophenyl)-3-phenylbutane
- 2-azido-4-(4-chlorophenyl)-3-phenylbutane (Step G, 7.0 g, 24 mmol) in EtOAc (150 mL) was added di (tert-butyl) dicarbonate (8.0 g, 37 mmol) and platinum dioxide (0.50 g, 2.2 mmol).
- the mixture was degassed and filled with hydrogen with a balloon.
- Step I ⁇ -r3-(4-Chlorophenyl)-2-phenyl-l-methylpropy ⁇ -amine hydrochloride (Diastereomer ). 2-(N-tert-butoxycarbonyl)amino-4-(4-chlorophenyl)-3-phenylbutane (Step H, 7.0 g, 24 mmol) was treated with a saturated solution of hydrogen chloride in EtOAc (100 mL) at room temperature for 30 min (4 M hydrogen chloride in dioxane may be used with similar results). The mixture was concentrated to dryness to give the title compound.
- Step A The product of Step A (4-(4-chlorophenyl)-3(S)-phenyl-2(R)-butanol, 1.8 g, 7.0 mmol) was converted to the title compound following the steps described in Reference Example 3, Steps F-I, except hydrogen chloride in dioxane (4 M) was used in place of hydrogen chloride in EtOAc.
- Steps F-I except hydrogen chloride in dioxane (4 M) was used in place of hydrogen chloride in EtOAc.
- !H ⁇ MR 500 MHz, CD3OD): ⁇ 7.35-6.98 (m, 9H), 3.62 (m, IH), 3.20 (dd, IH), 3.05 (m, IH), 2.98 (dd, IH), 1.19 (d, 3H).
- LCMS m/e 260 (M + H) + (2.3 min).
- Step A 4-(4-Chlorophenyl)-3-pyridyl-2-butanone.
- Step B N-r3-(4-chlorophenyl)-2-(3-pyridyl)-l-methylpropyn-amine, hydrochloride (mixture of diastereomers ⁇ / ⁇ 10:1).
- the product of Step A (4-(4-chlorophenyl)-3-pyridyl-2-butanone) (14 g, 57 mmol) was converted to the title compound following the procedure described in Reference Example 3, Steps E-I.
- Step B Benzyl 2-(2-Pyridyloxy -2-methylbutanoate.
- Step B N- 1 " 3 -(4-Chlorophenyl)-2-(3 -c yanophenyl)- 1 -methylprop yll amine hydrochloride (Diastereomer ⁇ )
- Oxalyl chloride (0.95 mL, 11 mmol) was added dropwise to a suspension of 2-(l- l,2,3-triazolyl))acetic acid (Step B, 1.27 g, 10 mmol) in 10 mL CH2CI2 containing 0.05 mL DMF. Vigorous effervescence was observed. This mixture was sti ⁇ ed at room temperature for 4 h and cooled to -78°C. A solution of N.O- dimethylhydroxylamine hydrochloride (1.2 g, 13 mmol) and diisopropylethyl amine (6.0 mL, 35 mmol) in 10 mL CH2CI2 was added slowly over 3 min.
- Step D N-Methoxy-N-methyl-3-(4-chlorophenyl)-2-(l-(l,2,3-triazolyl)) propionamide
- Lithium hexamethyldisilazide (lmolar in THF, 8.4 mL, 8.4 mmol) was added dropwise to a solution of N-methoxy-N-methyl-2-(l-(l,2,3-triazolyl))acetamide (Step C, 1.19 g, 7 mmol) in 15 mL THF at -78°C. After additional 30 min stirring, a solution of 4-chlorobenzyl bromide (1.65 g, 8 mmol) in 5 mL THF was added dropwise.
- Step D N-methoxy-N-methyl-3-(4-chlorophenyl)-2-(l -(1,2,3- triazolyl)propionamide was converted to the title compound following the procedures described in Reference Example 3, Step D-G.
- lH NMR 400 MHz, CDCI3: ⁇ 1.219-1.246 (d's 3H), 3.253-4.754 (m, 4H0, 6.866-7.299 (d's, 4H), 7.313, 7.618, 7.63, & 7.706 (s's, 2H).
- Step B N-r3-(4-Chlorophenyl)-2-(3-methylphenyl)-l-methylpropyllamine hydrochloride (Diastereomer )
- Step B 4-(5-Chloro-2-pyridyl)-3(S)-phenyl-2(R)-butanol.
- Step A To a solution of 5-chloro-2-methylpyridine (Step A, 1.1 g, 8.7 mmol) in 15 mL anhydrous ether was added phenyl lithium (1.8 M in cyclohexane/ether, 7.2 mL, 13 mmol) at 0°C, and the reaction was sti ⁇ ed at room temperature for 30 min. The resulting mixture was cooled back to 0°C, and was added (lR,2R)-l-phenylpropylene oxide (2.3 g, 17 mmol), and the reaction was allowed to warm to room temperature overnight. The reaction mixture was partitioned between EtOAc (100 mL) and water (100 mL).
- Step D N-r3-(5-Chloro-2-pyridyl)-2(Sl-phenyl-l(S)-methylpropyllamine, hydrochloride
- the product of Step C (0.20 g, 0.70 mmol) was converted to the title compound following the procedure described in Reference Example 3, Steps H-I, except hydrogen chloride in dioxane (4 M) was used in place of hydrogen chloride in EtOAc.
- iH ⁇ MR 500 MHz, CD3OD: ⁇ 8.75 (d, IH), 8.19 (dd, IH), 7.55 (d, IH), 7.4-7.2
- Step C 3-(3-Bromophenyl)-4-(5-chloro-2-pyridyl)-2-butanol
- 3-(3-bromophenyl)-4-(5-chloro-2-pyridyl)-2-butanone (Step B, 6.7 g, 20 mmol) in 50 mL anhydrous THF at -78°C was added lithium tri(sec- butyl)borohydride (1.0 M in THF, 30 mL, 30 mmol), and the reaction was allowed to warm to room temperature overnight.
- the reaction was cooled to 0°C, and was carefully added 2 M hydrochloric acid (50 mL), and the resulting mixture was partitioned between hexane (200 mL) and water (200 mL). The aqueous layer was separated and the organic layer extracted with 2 M hydrochloric acid (2 x 100 mL). The combined aqueous extracts were neutralized with 5 N aqueous sodium hydroxide (pH > 12), and was extracted with EtOAc (2x200 mL). The combined extracts were dried over anhydrous sodium sulfate, filtered, and concentrated to dryness to afford the title compound.
- Step D N-r2-(3-Bromophenyl)-3-(5-chloro-2-pyridyl)-l-methylpropyl1amine, hydrochloride
- the product of Step C (5.9 g, 17 mmol) was converted to the title compound following the procedure described in Reference Example 18, Steps C-D.
- Step C N-r2-(5-Bromo-3-pyridyl)-3-(4-chlorophenyl)-l-methylpropyllamine hydrochloride (Diastereomer ) The title compound was prepared following the procedure described for Reference Example 4, Step B. LC-MS: m/e 339 (M + H) + (2.5 min).
- Step B N-r3-(4-ChloiOphenyl)-2-(5-cvano-2-pyridyl)-l-methylpropyllamine hydrochloride (Diastereomer / ⁇ 5:1)
- the title compound was prepared following the procedure described for Reference Example 5 substituting 3-pyridylacetone with 5-cyano-3-pyridylacetone (Step A).
- Step B N-r3-(4-Chlorophenyl)-2-(5-chloro-3-pyridyl)-l-methylpropyllamine hydrochloride (Diastereomer )
- the title compound was prepared following the procedure described for Reference Example 20, Step B-C substituting 5-bromo-3-pyridylacetone with 5-chloro-3- pyridylacetone at Step B.
- LC-MS m/e 295 (M + H) + (1.9 min).
- Step A Ethyl 3-(4-chlorophenyl)-2-indolin-N-ylpropanoate.
- l.lg LiOH H2 ⁇ 26.25 mmol
- DMF 20 mL
- 4 angstrom molecular sieves 4 angstrom molecular sieves.
- room temperature 2.8 mL (25mmol) indoline was added dropwise.
- room temperature 2.9 mL (26.25 mmol) ethyl bromoacetate was added dropwise.
- the solid material was filtered and the residue was washed with copious amounts of EtOAc.
- Step B N,O-dimethyl-3-(4-chlorophenyl)-2-indolin-N-ylpropanamide.
- Step C 4-(4-chlorophenyl)-3-indolin-N-ylbutan-2-one.
- a 1-M solution of CH3MgBr in THF was added dropwise to a stirring solution of N,O-dimethyl-3-(4- chlorophenyl)-2-indolinylpropanamide (from Step B, 965 mg) in 25 mL anhydrous THF.
- the solution was sti ⁇ ed for 4 h while being allowed to warm to room temperature. Then approximately 20 mL water were added.
- the mixture was extract three times with 50 mL ether.
- the combined extracts were dried over MgSO4.
- Step D 4-(4-chlorophenyl)-3-indolin-N-ylbutan-2-one methoxime.
- This compound was prepared in an analogous manner to Reference Example 28.
- the aqueous layer was separated and neutralized to pH 7 with 2N hydrochloric acid and extracted again into CH2CI2.
- the combined organic washes were dried with MgSO4 and concentrated.
- the crude material was purified by column chromatography on silica gel eluting from 0-10% EtOAc/hexane to give the title compound.
- Step C 4-(4-Methylphenyl)-3-phenylbutan-2-amine
- a solution of the 4-(4-methylphenyl)-3-phenylbutan-2-one (308 mg, 1.29 mmol) in 7M ammonia in MeOH (5 mL) and acetic acid (3 mL) was added sodium cyanoborohydride (130 mg, 2.06 mmol) and the reaction sti ⁇ ed at room temperature overnight.
- the reaction was quenched by pouring into 2M sodium carbonate solution and extracted into EtOAc.
- the aqueous layer was salted and re-extracted.
- the combined organic extracts were dried over MgSO4 and concentrated to give the title compound as a mixture of 4 isomers which was used without further purification.
- LCMS m/e 240 (M + H) + (2.22 min).
- Step B 2-(lH-l,2,3-Benzotriazol-l-yl)-3-(4-chlorophenyl)-N-methoxy-N- methyl-propanamide.
- 2-(lH-.l,2,3-benzotriazol-l-yl)-N-methoxy-N- methylacetamide in 15 mL anhydrous THF at -78 °C, 10 mL (10 mmol) of IM lithium bis(trimethylsilyl)amide was added dropwise. After stirring for 25 min, a solution of 2.06 g (10 mmol) of 4-chlorobenzyl bromide in 2 mL anhydrous THF was added.
- Step D 2-(lH-l,2,3-Benzotriazol-l-yl)-3-(4-chlorophenyl)-l-methyl propylamine
- 2-(lH-l,2,3-benzotriazol-l-yl)-3-(4- chlorophenyl)-butan-2-one in 8.5 mL (60 mmol) of 7N ammonia in MeOH at 0 °C, 4 mL (964 mmol) of glacial acetic acid was added followed by 410 mg (6.5 mmol) of sodium cyanoborohydride.
- Example 34 substituting thiophene-3-acetic acid for 2-(lH-l,2,3-benzotriazol-l- yl)acetic acid in Step A.
- Step B 3-(3-Cyanophenyl)-4-cvclobutyl-butan-2-one
- a solution of 1.45 g (9.07 mmol) of l-(3-cyanophenyl)acetone in 18 mL acetonitrile 1.1 mL (9.5 mmol) cyclobutyl bromide and 5.91 g (18.1 mmol) cesium carbonate were added.
- the filtrate was partitioned between water and EtOAc and the aqueous layer was extracted with EtOAc.
- the combined organic layer was washed with brine, dried and concentrated.
- Diastereomer ⁇ lH ⁇ MR (500 MHz, CD3OD): ⁇ 7.16 (d, 2H), 7.14 (d, 2H), 7.09 (d, 2H), 6.99 (d, 2H), 6.88 (d, 2H), 6.64 (d, 2H), 4.33 (m, IH), 3.12 (dd, IH), 3.03 (ddd, IH), 2.74 (dd, IH), 1.36 (s, 3H), 1.30 (d, 3H), 1.30 (s, 3H).
- LC-MS m/e 490(M + H) + (4.7 min).
- Examples 4-7 (Table 2) were prepared following the procedures described in Examples 2 and 3 substituting 2-amino-3,4-bis(4-chlorophenyl)butane hydrochloride salt with the appropriate amines from the Reference Examples and 2- (4-chlorophenyloxy)-2-methylpropionic acid with the appropriate acids from the Reference Examples.
- commercial acids or acyl chlorides were employed, and N-diisopropyl-ethylamine may be used in place of N- methylmorpholine with similar results.
- the diastereomer designations ( ⁇ or ⁇ ) co ⁇ espond to designations of the starting amines.
- N-r2,3-Bis(4-Chlorophenyl)-l-methylpropyn-2-(4-chlorophenyloxy)-2- methylpropanamide (Diastereomer , Enantiomers A and B).
- Example 60 1.0 g in hexane (3 mL)/ethanol (7 mL) was loaded onto a Chiralpak AD column (2 cm x 25 cm), which was eluted with 5% ethanol in hexane (flow rate 9 mL/min, 500 ⁇ L per injection) to give the two pure enantiomers.
- Example 18 (Table 4) was prepared following the procedures described in Examples 2-3 employing N-[3-(4-chlorophenyl)-2(S)-phenyl-l(S)- methylpropyl] -amine, hydrochloride from Reference Example 4 coupled to the appropriate carboxylic acid. Table 4. Single enantiomeric compounds prepared with N-[3-(4-chloro ⁇ henyl)-2(S)- phenyl-l(S)-methylpropyl]-amine, hydrochloride from Reference Example 4.
- N-r2,3-Bis(4-chlorophenyl)-l-methylpropyl1-2-(4-chlorophenylamino)-2- methylpropanamide N-r2,3-Bis(4-chlorophenyl)-l-methylpropyl1-2-(4-chlorophenylamino)-2- methylpropanamide.
- 2-amino-3,4-bis(4-chlorophenyl)butane hydrochloride salt Diastereomer ⁇ , Section I, Reference Example 1, 0.31 g, 0.94 mmol
- 2-(4- chlorophenylamino)-2-methylpropionic acid (0.20 g, 0.94 mmol) in 5 mL CH2CI2 was added N-methylmorpholine (0.41 mL, 3.5 mmol) and tris(pyrrolindinyl)phosphonium hexafluorophosphate (0.73 g, 1.4 mmol).
- Examples 30-33 were prepared from N-[3-(4-chlorophenyl)- 2(S)-phenyl-l(S)-methylpropyl]amine, hydrochloride (Reference Example 4) or N-[3- (5-chloro-2-pyridyl)-2(S)-phenyl-l(S)-methylpropyl]amine, hydrochloride (Reference Example 18) and the appropriate carboxylic acid following the procedures described in Examples 2-3 (via an acyl chloride intermediate) or Example 19 (with a coupling reagent),
- Examples 34-39 were prepared from the appropriate amine and acid of Reference Examples following the procedures described in Examples 2-3 (via an acyl chloride intermediate) or Example 19 (with a coupling reagent).
- Examples 41-52 (Table 11) were isolated as single enantiomers from the co ⁇ esponding racemic material (Table 10) following the procedures described in Examples 8-9 with appropriate modifications of (1) the eluent composition (4-15% ethanol/hexane), (2) flow rate (6-9 mL/min) and (3) injection volume (200 to 2000 ⁇ L).
- Examples 53-56 (Table 12) were isolated as diastereomers as indicated (Isomer A or B) on silica gel chromatography columns. The single enantiomers noted were separated on the chiral AD column noted above.
- Example 57 Prepared as in Example 57 only using 3-[2-amino-l-(4- fluorobenzyl)propyl]benzonitrile (Reference example 33) as the amine component to give the title compound as a mixture of 4 isomers.
- the diastereomers were separated by HPLC on a Zorbax RxSi column eluting 96% hexane: 4% ethanol at 20 mL/min with retention times of: less polar diastereomer eluted at 11.75 minutes;
- Binding affinity determination is based on recombinant human CBl receptor expressed in Chinese Hamster Ovary (CHO) cells (Felder et al, Mol.
- Binding buffer contains 50mM Tris-HCl, pH7.4, 2.5 mM EDTA, 5mM MgCl2, 0.5mg/mL fatty acid free bovine serum albumin and protease inhibitors (Cat#P8340, from Sigma). To initiate the binding reaction, 5 ⁇ l of radioligand solution is added, the mixture is incubated with gentle shaking on a shaker for 1.5 h at 30°C.
- binding is terminated by using 96-well harvester and filtering through GF/C filter presoaked in 0.05% polyethylenimine.
- the bound radiolabel is quantitated using scintillation counter. Apparent binding affinities for various compounds are calculated from IC50 values (DeBlasi et al., Trends Pharmacol Sci 10: 227-229, 1989).
- the binding assay for CB2 receptor is done similarly with recombinant human CB2 receptor expressed in CHO cells.
- CBl receptor The functional activation of CBl receptor is based on recombinant human CBl receptor expressed in CHO cells (Felder et al, Mol. Pharmacol. 48: 443- 450, 1995).
- 50 ul of CB1-CHO cell suspension are mixed with test compound and 70 ul assay buffer containing 0.34 mM 3-isobutyl-l-methylxanthine and 5.1 uM of forskolin in 96-well plates.
- the assay buffer is comprised of Earle's Balanced Salt Solution supplemented with 5 mM MgCl2, 1 mM glutamine, 10 mM HEPES, and 1 mg/mL bovine serum albumin.
- the mixture is incubated at room temperature for 30 minutes, and terminated by adding 30uL/well of 0.5M HCl.
- the total intracellular cAMP level is quantitated using the New England Nuclear Flashplate and cAMP radioimmunoassay kit.
- the reaction mixture also contains 0.5 nM of the agonist CP55940, and the reversal of the
- CP55940 effect is quantitated.
- a series of dose response curves for CP55940 is perfoimed with increasing concentration of the test compound in each of the dose response curves.
- the functional assay for the CB2 receptor is done similarly with recombinant human CB2 receptor expressed in CHO cells.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Psychiatry (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Pulmonology (AREA)
- Molecular Biology (AREA)
- Structural Engineering (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Diabetes (AREA)
- Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Child & Adolescent Psychology (AREA)
- Addiction (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyridine Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pyrane Compounds (AREA)
Abstract
Description













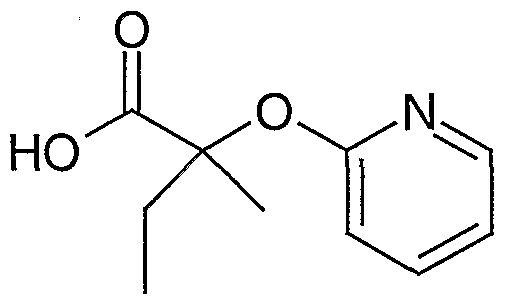













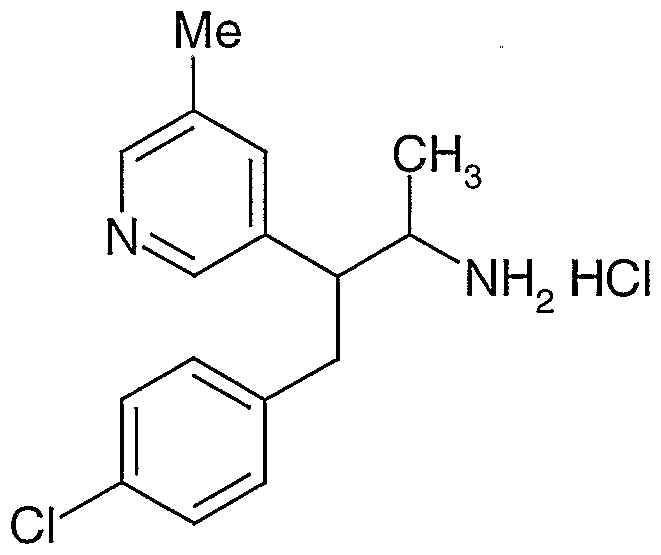














































Claims



Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR0308349-7A BR0308349A (en) | 2002-11-22 | 2003-03-07 | Compound, composition, and use of a compound |
| YUP-791/04A RS79104A (en) | 2002-11-22 | 2003-03-07 | Substituted amides active at the cannabinoid-1 receptor |
| EA200401066A EA007747B1 (en) | 2002-11-22 | 2003-03-07 | Submitted amides active at the cannabinoid-1 receptor |
| AU2003218005A AU2003218005A1 (en) | 2002-11-22 | 2003-03-07 | Substituted amides active at the cannabinoid-1 receptor |
| MXPA04008748A MXPA04008748A (en) | 2002-11-22 | 2003-03-07 | Substituted amides active at the cannabinoid-1 receptor. |
| KR1020047014299A KR100748380B1 (en) | 2002-11-22 | 2003-03-07 | Substituted amides active at the cannabinoid-1 receptor |
| CNA03805678XA CN1639112A (en) | 2002-11-22 | 2003-03-07 | Substituted amides active at the cannabinoid-1 receptor |
| IL16382403A IL163824A0 (en) | 2002-11-22 | 2003-03-07 | Substituted amides active at the cannabinoid-1 receptor |
| UA20040907424A UA76590C2 (en) | 2002-11-22 | 2003-07-03 | Substituted amides acting on receptor of cannabinoid-1 |
| IS7411A IS7411A (en) | 2002-11-22 | 2004-08-19 | Substituted amides active at the cannabinoid-1 receptor |
| TNP2004000176A TNSN04176A1 (en) | 2002-11-22 | 2004-09-10 | Substituted amides active at the cannabinoid-1 receptor |
| NO20043803A NO20043803L (en) | 2002-11-22 | 2004-09-10 | Substituted amides active in the cannabinoid 1 receptor |
| HR20040823A HRP20040823A2 (en) | 2002-11-22 | 2004-09-10 | Substituted amides active at the cannabinoid-1 receptor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US42841502P | 2002-11-22 | 2002-11-22 | |
| US60/428,415 | 2002-11-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004048317A1 true WO2004048317A1 (en) | 2004-06-10 |
Family
ID=32393400
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2003/007039 WO2004048317A1 (en) | 2002-11-22 | 2003-03-07 | Substituted amides active at the cannabinoid-1 receptor |
Country Status (25)
| Country | Link |
|---|---|
| KR (1) | KR100748380B1 (en) |
| CN (1) | CN1639112A (en) |
| AR (1) | AR038948A1 (en) |
| AU (1) | AU2003218005A1 (en) |
| BR (1) | BR0308349A (en) |
| CR (1) | CR7432A (en) |
| DO (1) | DOP2003000609A (en) |
| EA (1) | EA007747B1 (en) |
| EC (1) | ECSP045289A (en) |
| GE (1) | GEP20074208B (en) |
| HR (1) | HRP20040823A2 (en) |
| IL (1) | IL163824A0 (en) |
| IS (1) | IS7411A (en) |
| JO (1) | JO2482B1 (en) |
| MA (1) | MA27185A1 (en) |
| MX (1) | MXPA04008748A (en) |
| MY (1) | MY134457A (en) |
| NO (1) | NO20043803L (en) |
| PE (1) | PE20040599A1 (en) |
| PL (1) | PL200328B1 (en) |
| RS (1) | RS79104A (en) |
| TN (1) | TNSN04176A1 (en) |
| TW (1) | TW200408620A (en) |
| UA (1) | UA76590C2 (en) |
| WO (1) | WO2004048317A1 (en) |
Cited By (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1623741A2 (en) | 2004-07-22 | 2006-02-08 | Cadila Healthcare Ltd. | Cannabinoid receptor ligands for hair growth modulation |
| EP1641493A1 (en) * | 2003-06-30 | 2006-04-05 | Merck & Co., Inc. | Radiolabeled cannabinoid-1 receptor modulators |
| WO2006035760A1 (en) * | 2004-09-27 | 2006-04-06 | Santen Pharmaceutical Co., Ltd. | Drug for treating skin disease |
| WO2006035759A1 (en) * | 2004-09-27 | 2006-04-06 | Santen Pharmaceutical Co., Ltd. | Drug for treating respiratory disease |
| WO2006043518A1 (en) * | 2004-10-18 | 2006-04-27 | Santen Pharmaceutical Co., Ltd. | Drug for treating neurological disease |
| EP1682494A1 (en) * | 2003-10-30 | 2006-07-26 | Merck & Co., Inc. | Aralkyl amines as cannabinoid receptor modulators |
| US7091216B2 (en) | 2002-08-02 | 2006-08-15 | Merck & Co., Inc. | Substituted furo[2,3-b]pyridine derivatives |
| EP1765771A2 (en) * | 2004-07-08 | 2007-03-28 | Merck & Co., Inc. | Formation of tetra-substituted enamides and stereoselective reduction thereof |
| WO2007041052A2 (en) | 2005-09-29 | 2007-04-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| WO2007062314A2 (en) | 2005-11-23 | 2007-05-31 | Bristol-Myers Squibb Company | Heterocyclic cetp inhibitors |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2006119260A3 (en) * | 2005-05-02 | 2008-02-28 | Merck & Co Inc | Combination of dipeptidyl peptidase-iv inhibitor and a cannabinoid cb1 receptor antagonist for the treatment of diabetes and obesity |
| US7348456B2 (en) | 2002-12-19 | 2008-03-25 | Merck & Co., Inc. | Substituted amides |
| WO2008059335A1 (en) | 2006-11-13 | 2008-05-22 | Pfizer Products Inc. | Diaryl, dipyridinyl and aryl-pyridinyl derivatives and uses thereof |
| WO2008070496A2 (en) | 2006-12-01 | 2008-06-12 | Bristol-Myers Squibb Company | N- ( (3-benzyl) -2, 2- (bis-phenyl) -propan-1-amine derivatives as cetp inhibitors for the treatment of atherosclerosis and cardiovascular diseases |
| WO2008120653A1 (en) | 2007-04-02 | 2008-10-09 | Banyu Pharmaceutical Co., Ltd. | Indoledione derivative |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| EP2078729A1 (en) | 2005-02-04 | 2009-07-15 | Pfizer Products Inc. | PPY agonists and uses thereof |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| US7649002B2 (en) | 2004-02-04 | 2010-01-19 | Pfizer Inc | (3,5-dimethylpiperidin-1yl)(4-phenylpyrrolidin-3-yl)methanone derivatives as MCR4 agonists |
| WO2010051236A1 (en) | 2008-10-30 | 2010-05-06 | Merck Sharp & Dohme Corp. | Isonicotinamide orexin receptor antagonists |
| WO2010056717A1 (en) | 2008-11-17 | 2010-05-20 | Merck Sharp & Dohme Corp. | Substituted bicyclic amines for the treatment of diabetes |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| US7816534B2 (en) | 2002-03-12 | 2010-10-19 | Merck Sharp & Dohme Corp. | Substituted amides |
| WO2011011508A1 (en) | 2009-07-23 | 2011-01-27 | Schering Corporation | Benzo-fused oxazepine compounds as stearoyl-coenzyme a delta-9 desaturase inhibitors |
| WO2011011506A1 (en) | 2009-07-23 | 2011-01-27 | Schering Corporation | Spirocyclic oxazepine compounds as stearoyl-coenzyme a delta-9 desaturase inhibitors |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011137024A1 (en) | 2010-04-26 | 2011-11-03 | Merck Sharp & Dohme Corp. | Novel spiropiperidine prolylcarboxypeptidase inhibitors |
| WO2011143057A1 (en) | 2010-05-11 | 2011-11-17 | Merck Sharp & Dohme Corp. | Novel prolylcarboxypeptidase inhibitors |
| EP2392567A1 (en) | 2005-10-21 | 2011-12-07 | Bristol-Myers Squibb Company | Benzothiazine derivatives and their use as lxr modulators |
| WO2011156246A1 (en) | 2010-06-11 | 2011-12-15 | Merck Sharp & Dohme Corp. | Novel prolylcarboxypeptidase inhibitors |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| US8138188B2 (en) | 2006-02-23 | 2012-03-20 | Pfizer Inc. | Melanocortin type 4 receptor agonist piperidinoylpyrrolidines |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2013059222A1 (en) | 2011-10-19 | 2013-04-25 | Merck Sharp & Dohme Corp. | 2-pyridyloxy-4-nitrile orexin receptor antagonists |
| US8592403B2 (en) | 2008-08-06 | 2013-11-26 | Pfizer Limited | Diazepine and diazocane compounds as MC4 agonists |
| EP2698157A1 (en) | 2006-09-22 | 2014-02-19 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| WO2020167706A1 (en) | 2019-02-13 | 2020-08-20 | Merck Sharp & Dohme Corp. | 5-alkyl pyrrolidine orexin receptor agonists |
| WO2021026047A1 (en) | 2019-08-08 | 2021-02-11 | Merck Sharp & Dohme Corp. | Heteroaryl pyrrolidine and piperidine orexin receptor agonists |
| WO2022040070A1 (en) | 2020-08-18 | 2022-02-24 | Merck Sharp & Dohme Corp. | Bicycloheptane pyrrolidine orexin receptor agonists |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998041519A1 (en) * | 1997-03-18 | 1998-09-24 | Smithkline Beecham Corporation | Novel cannabinoid receptor agonists |
| WO2002068388A2 (en) * | 2001-02-28 | 2002-09-06 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin-4 receptor agonists |
-
2003
- 2003-02-11 MY MYPI20030464A patent/MY134457A/en unknown
- 2003-02-18 TW TW092103282A patent/TW200408620A/en unknown
- 2003-03-03 JO JO200320A patent/JO2482B1/en active
- 2003-03-07 RS YUP-791/04A patent/RS79104A/en unknown
- 2003-03-07 PL PL373656A patent/PL200328B1/en not_active IP Right Cessation
- 2003-03-07 PE PE2003000230A patent/PE20040599A1/en not_active Application Discontinuation
- 2003-03-07 IL IL16382403A patent/IL163824A0/en unknown
- 2003-03-07 MX MXPA04008748A patent/MXPA04008748A/en active IP Right Grant
- 2003-03-07 AU AU2003218005A patent/AU2003218005A1/en not_active Abandoned
- 2003-03-07 CN CNA03805678XA patent/CN1639112A/en active Pending
- 2003-03-07 KR KR1020047014299A patent/KR100748380B1/en not_active IP Right Cessation
- 2003-03-07 WO PCT/US2003/007039 patent/WO2004048317A1/en not_active Application Discontinuation
- 2003-03-07 EA EA200401066A patent/EA007747B1/en not_active IP Right Cessation
- 2003-03-07 BR BR0308349-7A patent/BR0308349A/en not_active IP Right Cessation
- 2003-03-07 GE GEAP8404A patent/GEP20074208B/en unknown
- 2003-03-12 DO DO2003000609A patent/DOP2003000609A/en unknown
- 2003-03-12 AR ARP030100857A patent/AR038948A1/en unknown
- 2003-07-03 UA UA20040907424A patent/UA76590C2/en unknown
-
2004
- 2004-08-19 IS IS7411A patent/IS7411A/en unknown
- 2004-08-24 CR CR7432A patent/CR7432A/en unknown
- 2004-09-10 MA MA27851A patent/MA27185A1/en unknown
- 2004-09-10 HR HR20040823A patent/HRP20040823A2/en not_active Application Discontinuation
- 2004-09-10 EC EC2004005289A patent/ECSP045289A/en unknown
- 2004-09-10 TN TNP2004000176A patent/TNSN04176A1/en unknown
- 2004-09-10 NO NO20043803A patent/NO20043803L/en not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998041519A1 (en) * | 1997-03-18 | 1998-09-24 | Smithkline Beecham Corporation | Novel cannabinoid receptor agonists |
| WO2002068388A2 (en) * | 2001-02-28 | 2002-09-06 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin-4 receptor agonists |
Cited By (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7816534B2 (en) | 2002-03-12 | 2010-10-19 | Merck Sharp & Dohme Corp. | Substituted amides |
| US7091216B2 (en) | 2002-08-02 | 2006-08-15 | Merck & Co., Inc. | Substituted furo[2,3-b]pyridine derivatives |
| US7576239B2 (en) | 2002-12-19 | 2009-08-18 | Merck & Co., Inc. | Substituted amides |
| US7348456B2 (en) | 2002-12-19 | 2008-03-25 | Merck & Co., Inc. | Substituted amides |
| EP1641493A1 (en) * | 2003-06-30 | 2006-04-05 | Merck & Co., Inc. | Radiolabeled cannabinoid-1 receptor modulators |
| EP1641493A4 (en) * | 2003-06-30 | 2007-10-31 | Merck & Co Inc | Radiolabeled cannabinoid-1 receptor modulators |
| US7390835B2 (en) | 2003-10-30 | 2008-06-24 | Merck & Co., Inc. | Aralkyl amines as cannabinoid receptor modulators |
| EP1682494A1 (en) * | 2003-10-30 | 2006-07-26 | Merck & Co., Inc. | Aralkyl amines as cannabinoid receptor modulators |
| EP1682494A4 (en) * | 2003-10-30 | 2006-11-08 | Merck & Co Inc | Aralkyl amines as cannabinoid receptor modulators |
| US7649002B2 (en) | 2004-02-04 | 2010-01-19 | Pfizer Inc | (3,5-dimethylpiperidin-1yl)(4-phenylpyrrolidin-3-yl)methanone derivatives as MCR4 agonists |
| EP1765771A4 (en) * | 2004-07-08 | 2008-04-02 | Merck & Co Inc | Formation of tetra-substituted enamides and stereoselective reduction thereof |
| EP1765771A2 (en) * | 2004-07-08 | 2007-03-28 | Merck & Co., Inc. | Formation of tetra-substituted enamides and stereoselective reduction thereof |
| US7629470B2 (en) | 2004-07-08 | 2009-12-08 | Merck & Co., Inc. | Formation of tetra-substituted enamides and stereoselective reduction thereof |
| EP1623741A2 (en) | 2004-07-22 | 2006-02-08 | Cadila Healthcare Ltd. | Cannabinoid receptor ligands for hair growth modulation |
| WO2006035760A1 (en) * | 2004-09-27 | 2006-04-06 | Santen Pharmaceutical Co., Ltd. | Drug for treating skin disease |
| WO2006035759A1 (en) * | 2004-09-27 | 2006-04-06 | Santen Pharmaceutical Co., Ltd. | Drug for treating respiratory disease |
| WO2006043518A1 (en) * | 2004-10-18 | 2006-04-27 | Santen Pharmaceutical Co., Ltd. | Drug for treating neurological disease |
| EP2078729A1 (en) | 2005-02-04 | 2009-07-15 | Pfizer Products Inc. | PPY agonists and uses thereof |
| WO2006119260A3 (en) * | 2005-05-02 | 2008-02-28 | Merck & Co Inc | Combination of dipeptidyl peptidase-iv inhibitor and a cannabinoid cb1 receptor antagonist for the treatment of diabetes and obesity |
| WO2007041052A2 (en) | 2005-09-29 | 2007-04-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| EP2392567A1 (en) | 2005-10-21 | 2011-12-07 | Bristol-Myers Squibb Company | Benzothiazine derivatives and their use as lxr modulators |
| WO2007062314A2 (en) | 2005-11-23 | 2007-05-31 | Bristol-Myers Squibb Company | Heterocyclic cetp inhibitors |
| US8138188B2 (en) | 2006-02-23 | 2012-03-20 | Pfizer Inc. | Melanocortin type 4 receptor agonist piperidinoylpyrrolidines |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| EP2698157A1 (en) | 2006-09-22 | 2014-02-19 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| EP2946778A1 (en) | 2006-09-22 | 2015-11-25 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| WO2008059335A1 (en) | 2006-11-13 | 2008-05-22 | Pfizer Products Inc. | Diaryl, dipyridinyl and aryl-pyridinyl derivatives and uses thereof |
| WO2008070496A2 (en) | 2006-12-01 | 2008-06-12 | Bristol-Myers Squibb Company | N- ( (3-benzyl) -2, 2- (bis-phenyl) -propan-1-amine derivatives as cetp inhibitors for the treatment of atherosclerosis and cardiovascular diseases |
| WO2008120653A1 (en) | 2007-04-02 | 2008-10-09 | Banyu Pharmaceutical Co., Ltd. | Indoledione derivative |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| US8592403B2 (en) | 2008-08-06 | 2013-11-26 | Pfizer Limited | Diazepine and diazocane compounds as MC4 agonists |
| WO2010051236A1 (en) | 2008-10-30 | 2010-05-06 | Merck Sharp & Dohme Corp. | Isonicotinamide orexin receptor antagonists |
| WO2010056717A1 (en) | 2008-11-17 | 2010-05-20 | Merck Sharp & Dohme Corp. | Substituted bicyclic amines for the treatment of diabetes |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011011506A1 (en) | 2009-07-23 | 2011-01-27 | Schering Corporation | Spirocyclic oxazepine compounds as stearoyl-coenzyme a delta-9 desaturase inhibitors |
| WO2011011508A1 (en) | 2009-07-23 | 2011-01-27 | Schering Corporation | Benzo-fused oxazepine compounds as stearoyl-coenzyme a delta-9 desaturase inhibitors |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011137024A1 (en) | 2010-04-26 | 2011-11-03 | Merck Sharp & Dohme Corp. | Novel spiropiperidine prolylcarboxypeptidase inhibitors |
| WO2011143057A1 (en) | 2010-05-11 | 2011-11-17 | Merck Sharp & Dohme Corp. | Novel prolylcarboxypeptidase inhibitors |
| WO2011156246A1 (en) | 2010-06-11 | 2011-12-15 | Merck Sharp & Dohme Corp. | Novel prolylcarboxypeptidase inhibitors |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013059222A1 (en) | 2011-10-19 | 2013-04-25 | Merck Sharp & Dohme Corp. | 2-pyridyloxy-4-nitrile orexin receptor antagonists |
| WO2020167706A1 (en) | 2019-02-13 | 2020-08-20 | Merck Sharp & Dohme Corp. | 5-alkyl pyrrolidine orexin receptor agonists |
| WO2021026047A1 (en) | 2019-08-08 | 2021-02-11 | Merck Sharp & Dohme Corp. | Heteroaryl pyrrolidine and piperidine orexin receptor agonists |
| WO2022040070A1 (en) | 2020-08-18 | 2022-02-24 | Merck Sharp & Dohme Corp. | Bicycloheptane pyrrolidine orexin receptor agonists |
Also Published As
| Publication number | Publication date |
|---|---|
| AR038948A1 (en) | 2005-02-02 |
| CN1639112A (en) | 2005-07-13 |
| EA007747B1 (en) | 2006-12-29 |
| MA27185A1 (en) | 2005-01-03 |
| KR20050083563A (en) | 2005-08-26 |
| TW200408620A (en) | 2004-06-01 |
| EA200401066A1 (en) | 2005-04-28 |
| HRP20040823A2 (en) | 2005-06-30 |
| TNSN04176A1 (en) | 2007-03-12 |
| DOP2003000609A (en) | 2004-06-15 |
| RS79104A (en) | 2007-02-05 |
| UA76590C2 (en) | 2006-08-15 |
| GEP20074208B (en) | 2007-10-10 |
| KR100748380B1 (en) | 2007-08-10 |
| ECSP045289A (en) | 2004-10-26 |
| PL373656A1 (en) | 2005-09-05 |
| BR0308349A (en) | 2005-01-25 |
| MXPA04008748A (en) | 2004-12-06 |
| NO20043803L (en) | 2005-05-24 |
| IL163824A0 (en) | 2005-12-18 |
| AU2003218005A1 (en) | 2004-06-18 |
| PL200328B1 (en) | 2008-12-31 |
| MY134457A (en) | 2007-12-31 |
| JO2482B1 (en) | 2009-01-20 |
| CR7432A (en) | 2005-10-05 |
| IS7411A (en) | 2004-08-19 |
| PE20040599A1 (en) | 2004-09-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004048317A1 (en) | Substituted amides active at the cannabinoid-1 receptor | |
| JP4719469B2 (en) | Substituted amides | |
| JP4459629B2 (en) | Bicyclic amide | |
| JP3813152B2 (en) | Substituted amides | |
| AU2003215024B2 (en) | Spirocyclic amides as cannabinoid receptor modulators | |
| US20050154202A1 (en) | Substituted aryl amides | |
| AU2003218068B9 (en) | Substituted amides | |
| AU2007201276B2 (en) | Substituted amides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200400838 Country of ref document: VN Ref document number: P-791/04 Country of ref document: YU |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: CR2004-007432 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 375.04 Country of ref document: BZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 163824 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1972/CHENP/2004 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2004/008748 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref country code: GE Ref document number: GE P |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20040823A Country of ref document: HR Ref document number: 373656 Country of ref document: PL Ref document number: 8404 Country of ref document: GE Ref document number: 2003805678X Country of ref document: CN Ref document number: 1020047014299 Country of ref document: KR Ref document number: 200401066 Country of ref document: EA |
|
| WWG | Wipo information: grant in national office |
Ref document number: 375.04 Country of ref document: BZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 375.04 Country of ref document: BZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020047014299 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: JP |