WO2003064397A1 - Indazole compounds useful as protein kinase inhibitors - Google Patents
Indazole compounds useful as protein kinase inhibitors Download PDFInfo
- Publication number
- WO2003064397A1 WO2003064397A1 PCT/US2003/002096 US0302096W WO03064397A1 WO 2003064397 A1 WO2003064397 A1 WO 2003064397A1 US 0302096 W US0302096 W US 0302096W WO 03064397 A1 WO03064397 A1 WO 03064397A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- independently selected
- nitrogen
- sulfur
- oxygen
- membered saturated
- Prior art date
Links
- 0 CCCNc(c(C)c1)ccc1NC(*)C(c1cccc(Cl)c1)NCCNCc1ccccc1 Chemical compound CCCNc(c(C)c1)ccc1NC(*)C(c1cccc(Cl)c1)NCCNCc1ccccc1 0.000 description 9
- JSWZMMDZKUMYAQ-UHFFFAOYSA-N CC(C)(C)OC(CC(C(OC)=O)c(cc1Cl)ccc1Cl)=O Chemical compound CC(C)(C)OC(CC(C(OC)=O)c(cc1Cl)ccc1Cl)=O JSWZMMDZKUMYAQ-UHFFFAOYSA-N 0.000 description 1
- WINVUZDEQYPFRR-UHFFFAOYSA-N CC(c1cc(NC(C(CCN)c2ccc[o]2)=O)ccc1N)=[N-] Chemical compound CC(c1cc(NC(C(CCN)c2ccc[o]2)=O)ccc1N)=[N-] WINVUZDEQYPFRR-UHFFFAOYSA-N 0.000 description 1
- IWBANJDJGQLUEG-UHFFFAOYSA-N CC(c1cc(NC(C(CCN)c2ccccc2)=O)ccc1N)=N Chemical compound CC(c1cc(NC(C(CCN)c2ccccc2)=O)ccc1N)=N IWBANJDJGQLUEG-UHFFFAOYSA-N 0.000 description 1
- ILKSKUIESJBWBY-UHFFFAOYSA-N CC1=NCCc(cc2)c1cc2NC(CCC(c(cccc1)c1F)N)=O Chemical compound CC1=NCCc(cc2)c1cc2NC(CCC(c(cccc1)c1F)N)=O ILKSKUIESJBWBY-UHFFFAOYSA-N 0.000 description 1
- KGZURDDWPDUJNQ-UHFFFAOYSA-N CC1C(OC)=CC=CC1C(CCN)C(Nc1cc(C(Cc2cccc(C(N)=O)c2)NCC2)c2cc1)=O Chemical compound CC1C(OC)=CC=CC1C(CCN)C(Nc1cc(C(Cc2cccc(C(N)=O)c2)NCC2)c2cc1)=O KGZURDDWPDUJNQ-UHFFFAOYSA-N 0.000 description 1
- DMRGRBJWDYMGHH-UHFFFAOYSA-N CC1C=CC=CC1C(CCN)C(Nc(cc1)cc(C(C(NCc2ccccc2)O)=[N-])c1N)=O Chemical compound CC1C=CC=CC1C(CCN)C(Nc(cc1)cc(C(C(NCc2ccccc2)O)=[N-])c1N)=O DMRGRBJWDYMGHH-UHFFFAOYSA-N 0.000 description 1
- RFRUPCCGHCRINA-UHFFFAOYSA-N CCC(Cc1c2)Nc1ccc2NC(CCOc(cc1)ccc1Cl)=O Chemical compound CCC(Cc1c2)Nc1ccc2NC(CCOc(cc1)ccc1Cl)=O RFRUPCCGHCRINA-UHFFFAOYSA-N 0.000 description 1
- NLBWCVDHELZMKN-UHFFFAOYSA-N CCCc(c(C(C)=N)c1)ccc1NC(C(CCN)c1c[s]cc1)=O Chemical compound CCCc(c(C(C)=N)c1)ccc1NC(C(CCN)c1c[s]cc1)=O NLBWCVDHELZMKN-UHFFFAOYSA-N 0.000 description 1
- OIQHKQNFQRNAMG-UHFFFAOYSA-N CCCc(c(C=N)cc(NC(C(CCN)c(cc1Cl)ccc1Cl)=O)c1)c1-c1ccccc1 Chemical compound CCCc(c(C=N)cc(NC(C(CCN)c(cc1Cl)ccc1Cl)=O)c1)c1-c1ccccc1 OIQHKQNFQRNAMG-UHFFFAOYSA-N 0.000 description 1
- AUHNCYVOPYUVLL-UHFFFAOYSA-N CCCc(ccc(NC(C(CCN)c1ccccc1)=O)c1)c1C(NC1C=CC=C(C)C1C)=N Chemical compound CCCc(ccc(NC(C(CCN)c1ccccc1)=O)c1)c1C(NC1C=CC=C(C)C1C)=N AUHNCYVOPYUVLL-UHFFFAOYSA-N 0.000 description 1
- ZKODZVFUDPXZOP-UHFFFAOYSA-N CCc1cc2cc(NC(C(c3cc(Cl)ccc3)NCCc3ncccc3)=O)ccc2[nH]1 Chemical compound CCc1cc2cc(NC(C(c3cc(Cl)ccc3)NCCc3ncccc3)=O)ccc2[nH]1 ZKODZVFUDPXZOP-UHFFFAOYSA-N 0.000 description 1
- HPCKVNDHAMPMQF-UHFFFAOYSA-N CCc1cc2cc(NC(CCCc3ccccc3)=O)ccc2[nH]1 Chemical compound CCc1cc2cc(NC(CCCc3ccccc3)=O)ccc2[nH]1 HPCKVNDHAMPMQF-UHFFFAOYSA-N 0.000 description 1
- RKZXAILXEAOHKQ-UHFFFAOYSA-N CCc1cc2cc(NC(Cc3ccccc3)=O)ccc2[nH]1 Chemical compound CCc1cc2cc(NC(Cc3ccccc3)=O)ccc2[nH]1 RKZXAILXEAOHKQ-UHFFFAOYSA-N 0.000 description 1
- DZOOTKRESCUXKU-UHFFFAOYSA-N NCCC(C(Nc(cc1C=N)cc(-c2ccccc2Cl)c1N)=O)c(cc1Cl)ccc1Cl Chemical compound NCCC(C(Nc(cc1C=N)cc(-c2ccccc2Cl)c1N)=O)c(cc1Cl)ccc1Cl DZOOTKRESCUXKU-UHFFFAOYSA-N 0.000 description 1
- AZFVQGLLSIXVIO-UHFFFAOYSA-N NCCC(C(Nc(cc1C=N)ccc1N)=O)c(cccc1)c1F Chemical compound NCCC(C(Nc(cc1C=N)ccc1N)=O)c(cccc1)c1F AZFVQGLLSIXVIO-UHFFFAOYSA-N 0.000 description 1
- FIQWLCYWGRJEHX-UHFFFAOYSA-N O=C(C(c1cccc(Cl)c1)NCc1ncccc1)Nc1ccc2NNCc2c1 Chemical compound O=C(C(c1cccc(Cl)c1)NCc1ncccc1)Nc1ccc2NNCc2c1 FIQWLCYWGRJEHX-UHFFFAOYSA-N 0.000 description 1
- XYUAXZDQSDTCHO-UHFFFAOYSA-N O=S(c1ccccc1)=O Chemical compound O=S(c1ccccc1)=O XYUAXZDQSDTCHO-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/416—1,2-Diazoles condensed with carbocyclic ring systems, e.g. indazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4162—1,2-Diazoles condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention is in the field of medicinal chemistry and relates to compounds that are protein kinase inhibitors, compositions containing such compounds and methods of use. More particularly, the compounds are inhibitors of AKT, PKA, PDK1, p70S6K, and ROCK kinases and are useful for treating diseases, such as cancer.
- Protein kinases mediate intracellular signal transduction. They do this by effecting a phosphoryl transfer from a nucleoside triphosphate to a protein acceptor that is involved in a signaling pathway.
- kinases and pathways through which extracellular and other stimuli cause a variety of cellular responses to occur inside the cell. Examples of such stimuli include environmental and chemical stress signals (e.g. osmotic shock, heat shock, ultraviolet radiation, bacterial endotoxin, H 2 O 2 ), cytokines (e.g. interleukin-1 (IL-1) and tumor necrosis factor (TNF- ⁇ )), and growth factors (e.g.
- environmental and chemical stress signals e.g. osmotic shock, heat shock, ultraviolet radiation, bacterial endotoxin, H 2 O 2
- cytokines e.g. interleukin-1 (IL-1) and tumor necrosis factor (TNF- ⁇ )
- growth factors e.g.
- GM-CSF granulocyte macrophage-colony-stimulating factor
- FGF fibroblast growth factor
- An extracellular stimulus may effect one or more cellular responses related to cell growth, migration, differentiation, secretion of hormones, activation of transcription factors, muscle contraction, glucose metabolism, control of protein synthesis and regulation of cell cycle.
- diseases include autoimmune diseases, inflammatory diseases, neurological and neurodegenerative diseases, cancer, cardiovascular diseases, allergies and asthma, Alzheimer's disease or hormone-related diseases. Accordingly, there has been a substantial effort in medicinal chemistry to find protein kinase inhibitors that are effective as therapeutic agents. A challenge has been to find protein kinase inhibitors that act in a selective manner. Since there are numerable protein kinases that are involved in a variety of cellular responses, non-selective inhibitors may lead to unwanted side effects.
- AKT also known as PKB or Rac-PK beta
- PKB a serine/threonine protein kinase
- AKT comprises an N-terminal pleckstrin homology (PH) domain, a kinase domain and a C-terminal "tail" region.
- the PH domain binds 3-phosphoinositides, which are synthesized by phosphatidyl inositol 3- kinase (PI3K) upon stimulation by growth factors such as platelet derived growth factor (PDGF), nerve growth factor (NGF) and insulin-like growth factor (IGF-1) [(Kulik et al., Mol. Cell. Biol, 17, pp. 1595-1606, 1997); (Hemmings, B.A., Science, 275, pp. 628-630, 1997)].
- PDGF platelet derived growth factor
- NGF nerve growth factor
- IGF-1 insulin-like growth factor
- Lipid binding to the PH domain promotes translocation of AKT to the plasma membrane and facilitates phosphorylation by another PH-domain-containing protein kinases, PDK1 at Thr308, Thr309, and Thr305 for the AKT isoforms 1, 2 and 3, respectively.
- a second, as of yet unknown, kinase is required for the phosphorylation of Ser473, Ser474 or Ser472 in the C-terminal tails of AKT-1, -2 and -3 respectively, in order to yield a fully activated AKT enzyme.
- AKT mediates several functions within the cell including the metabolic effects of insulin (Calera, M.R. et al., J. Biol. Chem., 273, pp. 7201-7204, 1998), induction of differentiation and/or proliferation, protein synthesisans stress responses (Alessi, D.R. et al., Curr. Opin. Genet. Dev., 8, pp. 55-62, 1998).
- Manifestations of altered AKT regulation appear in both injury and disease, the most important role being in cancer. The first account of AKT was in association with human ovarian carcinomas where expression of AKT was found to be amplified in 15% of cases (Cheng, J.Q. et al., Proc.
- PKA also known as cAMP-dependent protein kinase
- PKA has been shown to regulate many vital functions including energy metabolism, gene transcription, proliferation, differentiation, reproductive function, secretion, neuronal activity, memory, contractility and motility (Beebe, S.J., Semin. Cancer Biol, 5, pp. 285-294, 1994).
- PKA is a tetrameric holoenzyme, which contains two catalytic subunits bound to a homodimeric regulatory subunit (which acts to inhibit the catalytic sub-units).
- Rho-associated coiled-coil forming kinase (Ishizaki, T. et al., EMBO J., 15, pp. 1885-1893, 1996) is a 160 kDa serine/threonine kinase that activates the small G-protein RhoA.
- ROCK has been implicated in numerous diseases including hypertension [(Chitaley, et al., Curr. Hypertens. Rep. 2001 Apr., 3(2), pp.139-144); (Uehata, M. et al., Nature, 389, pp. 990-994, 1997)], erectile dysfunction (Chitaley, K. et al., Nature Medicine, 7, pp.
- angiogenesis (Uchida, S. et al., Biochem. Biophys. Res. Commun., 269 (2) , pp. 633-40, 2000), neuroregeneration (Bito, H. et al., Neuron, 26, pp. 431-441, 2000), metastasis [(Takamura, M. et al., Hepatology, 33, pp. 577-581, 2001); (Genda, T. et al., Hepatology, 30, pp. 1027-1036, 1999)], glaucoma (Rao, et al, Invest. Ophthalmol. Vis. Sci. , 42, pp.
- the ribosomal protein kinases p70S6K-l and -2 are members of the AGC subfamily of protein kinases that consists of, amongst others, PKB and MSK.
- the p70S6 kinases catalyze the phosphorylation and subsequent activation of the ribosomal protein S6, which has been implicated in the translational up-regulation of mRNAs coding for the components of protein synthetic apparatus.
- mRNAs contain an oligopyrimidine tract at their 5' transcriptional start site, termed a 5TOP, which has been shown to be essential for their regulation at the translational level (Nolarevic, S. et al., Prog. Nucleic Acid Res. Mol. Biol. 65, pp 101-186, 2001).
- p70 S6K dependent S6 phosphorylation is stimulated in response to a variety of hormones and growth factors primarily via the PI3K pathway (Coffer, P.J. et al., Biochem. Biophys. Res.
- rapamycin acts to inhibit p70S6K activity and blocks protein synthesis, specifically as a result of a down-regulation of translation of these mR ⁇ A's encoding ribosomal proteins (Kuo, C.J. et al., Nature, 358, pp 70-73, 1992).
- PDKl In vitro PDKl catalyses the phosphorylation of Thr252 in the activation loop of the p70 catalytic domain, which is indispensable for p70 activity (Alessi, D.R., Curr. Biol, 8, pp 69-81, 1998).
- the use of rapamycin and gene deletion studies of dp70S6K from Drosophila and p70S6Kl from mouse have established the central role p70 plays in both cell growth and proliferation signaling.
- the 3-phosphoinositide-dependent protein kinase- 1 plays a key role in regulating the activity of a number of kinases belonging to the AGC subfamily of protein kinases (Alessi, D. et al., Biochem. Soc. Trans, 29, pp. 1, 2001). These include isoforms of protein kinase B (PKB, also known as AKT), p70 ribosomal S6 kinase (S6K) (Avruch, J. et al., prog. Mol. Subcell. Biol, 2001, 26, pp. 115, 2001), and p90 ribosomal S6 kinase (Frodin, M.
- PKT protein kinase B
- S6K p70 ribosomal S6 kinase
- PDKl mediated signaling is activated in response to insulin and growth factors and as a consequence of attachment of the cell to the extracellular matrix (integrin signaling). Once activated these enzymes mediate many diverse cellular events by phosphorylating key regulatory proteins that play important roles controlling processes such as cell survival, growth, proliferation and glucose regulation [(Lawlor, M.A. et al., J. Cell Sci. , 114, pp. 2903-2910, 2001), (Lawlor, M.A. et al., EMBO J. , 21, pp. 3728-3738, 2002)].
- PDKl is a 556 amino acid protein, with an N-terminal catalytic domain and a C-terminal pleckstrin homology (PH) domain, which activates its substrates by phosphorylating these kinases at their activation loop (Belham, C. et al., Curr. Biol. , 9, pp. R93-R96, 1999).
- Many human cancers including prostate and NSCL have elevated PDKl signaling pathway function resulting from a number of distinct genetic events such as PTEN mutations or over-expression of certain key regulatory proteins [(Graff, J.R., Expert Opin. Ther. Targets, 6, pp. 103-113, 2002), (Brognard, J., et al, Cancer Res.
- PDKl is a critical mediator of the PI3K signalling pathway, which regulates a multitude of cellular function including growth, proliferation and survival. Consequently inhibition of this pathway could affect four or more of the six defining requirements for cancer progression, as such it is anticipated that a PDKl inhibitor will have an effect on the growth of a very wide range of human cancers.
- PI3K pathway activity has been directly associated with the development of a number of human caners, progression to an aggressive refractory state (acquired resistance to chemotherapies) and poor prognosis.
- This increased activity has been attributed to a series of key events including decreased activity of negative pathway regulators such as the phosphatase PTEN, activating mutations of positive pathway regulators such as Ras, and overexpression of components of the pathway itself such as PKB, examples include: brain (gliomas), breast, colon, head and neck, kidney, lung, liver, melanoma, ovarian, pancreatic, prostate, sarcoma, thyroid [(Teng, D.H. et al., Cancer Res.
- Neoplasia, 3, pp. 278, 2001 ovarian [(Hayakawa, J. et al., Cancer Res. , 60, pp. 5988-5994, 2000), Neoplasia, 3, pp. 278, 2001)], breast ⁇ Mol. Cancer Ther., 1, pp. 707, 2002), colon [ ⁇ Neoplasia, 3, pp. 278, 2001), (Arico, S. et al., /. Biol. Chem., 277, pp. 27613-27621, 2002)], cervical (Neoplasia, 3, pp. 278, 2001), prostate [ ⁇ Endocrinology, 142, pp.
- V , V , V , R , and R are as defined below.
- These compounds, and pharmaceutically acceptable compositions thereof, are useful for treating or lessening the severity of a variety of disorders, including allergic disorders such as asthma, inflammatory disease, proliferative disorders, and neurological disorders.
- the present invention relates to a compound of formula I:
- R 1 is selected from halogen, CN, N(R 4 ) 2 , T-R, or T-Ar; each T is independently selected from a valence bond or a C 1-6 alkylidene chain, wherein up to two methylene units of T are optionally, and independently, replaced by -O-, -N(R)-, -S-, -N(R)C(O)-, -C(O)N(R)-, -C(O)-, or -SO 2 -; each R is independently selected from hydrogen or an optionally substituted C 1-6 aliphatic group, or: two R groups on the same nitrogen, taken together with the nitrogen atom attached thereto, form a 5-7 membered saturated, partially unsaturated, or aromatic ring having 1-3 heteroatoms independently selected from nitrogen, oxygen, or sulfur; R 2 is selected from Q-Ar, Q-N(R 5 ) 2 , or Q-C(R)(Q
- R and R 3 optionally form a 5-7 membered saturated or partially unsaturated ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur; each Q is independently selected from a valence bond or a C 1-4 alkylidene chain; each Ar is independently an optionally substituted ring selected from a 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an 8-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur;
- R 3 is selected from R', Ar 1 , Q-OR 5 , Q-OC(O)R 5 , Q-CONHR 5 , Q-OC(O)NHR 5 , Q-SR 5 , Q-N(R 4 ) 2 , N(R)(Q-Ar), N(R)C(O)Q-N(R 4 ) 2 , or N(R)Q-N(R 4 ) 2 ;
- R' is an optionally substituted C 1-6 aliphatic group
- each R 4 is independently selected from R, COR 5 , CO 2 R 5 , CON(R 5 ) 2 , SO 2 R 5 , SO 2 N(R 5 ) 2 , or Ar 1
- each R 5 is independently selected from R or Ar;
- V 1 , V 2 and V 3 are each independently selected from nitrogen or C(R 6 ); each R 6 is independently selected from R, Ar 1 , halogen, CN, NO 2 , OR, SR, N(R 4 ) 2 ,
- each Ar 1 is independently selected from an optionally substituted 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur; provided that: when V 1 , V 2 , and V 3 are each CH, T is a valence bond, and R 2 is Q-C(R)(Q-Ar)R 3 , wherein Ar is an optionally substituted phenyl ring, then R is other than Q-OR or C(O)NH 2 ; and when V 1 , V 2 , and V 3 are each CH and R 1
- substituted or unsubstituted Unless otherwise indicated, an optionally substituted group may have a substituent at each substitutable position of the group, and each substitution is independent of the other.
- aliphatic or "aliphatic group” as used herein means a straight-chain or branched C ⁇ -C 12 hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation, or a monocyclic C 3 -C 8 hydrocarbon or bicyclic C 8 -C 12 hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic (also referred to herein as “carbocycle” or “cycloalkyl”), that has a single point of attachment to the rest of the molecule wherein any individual ring in said bicyclic ring system has 3-7 members.
- suitable aliphatic groups include, but are not limited to, linear or branched or alkyl, alkenyl, alkynyl groups and hybrids thereof such as (cycloalkyl)alkyl, (cycloalkenyl)alkyl or (cycloalkyl)alkenyl.
- alkyl used alone or as part of a larger moiety includes both straight and branched chains containing one to twelve carbon atoms.
- alkenyl and “alkynyl” used alone or as part of a larger moiety shall include both straight and branched chains containing two to twelve carbon atoms.
- haloalkyl means alkyl, alkenyl or alkoxy, as the case may be, substituted with one or more halogen atoms.
- halogen means F, Cl, Br, or I.
- heteroatom means nitrogen, oxygen, or sulfur and includes any oxidized form of nitrogen and sulfur, and the quaternized form of any basic nitrogen.
- nitrogen includes a substitutable nitrogen of a heterocyclic ring.
- the nitrogen in a saturated or partially unsaturated ring having 0-4 heteroatoms selected from oxygen, sulfur or nitrogen, the nitrogen may be N (as in 3,4-dihydro-2H-pyrrolyl), N ⁇ (as in pyrrolidinyl) or NR + (as in N-substituted pyrrolidinyl).
- aryl used alone or as part of a larger moiety as in “aralkyl”, “aralkoxy”, or “aryloxyalkyl”, refers to monocyclic, bicyclic and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains 3 to 7 ring members.
- aryl may be used interchangeably with the term “aryl ring”.
- heterocycle means non-aromatic, monocyclic, bicyclic or tricyclic ring systems having five to fourteen ring members in which one or more ring members is a heteroatom, wherein each ring in the system contains 3 to 7 ring members.
- heteroaryl used alone or as part of a larger moiety as in “heteroaralkyl” or “heteroarylalkoxy”, refers to monocyclic, bicyclic and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic, at least one ring in the system contains one or more heteroatoms, and wherein each ring in the system contains 3 to 7 ring members.
- heteroaryl may be used interchangeably with the term “heteroaryl ring” or the term “heteroaromatic”.
- An aryl (including aralkyl, aralkoxy, aryloxyalkyl and the like) or heteroaryl (including heteroaralkyl and heteroarylalkoxy and the like) group may contain one or more substituents.
- Suitable substituents on the unsaturated carbon atom of an aryl, heteroaryl, aralkyl, or heteroaralkyl group are selected from halogen, oxo, N 3 , -R°, -OR°, -SR°, 1,2-methylene-dioxy, 1,2-ethylenedioxy, protected OH (such as acyloxy), phenyl (Ph), Ph substituted with R°, -O(Ph), O-(Ph) substituted with R°, -CH 2 (Ph), -CH 2 (Ph) substituted with R°, -CH 2 CH 2 (Ph), -CH 2 CH 2 (Ph) substituted with R°, -NO 2 , -CN, -N(R°) 2 , -NR°C(O)R°, -NR°C(O)N(R°) 2 , -NR°CO 2 R°, -NR°NR°C(O)R°, -NR°NR°C(O
- Substituents on the aliphatic group of R° are selected from NH 2 , NH(C ⁇ - aliphatic), N ⁇ i ⁇ aliphatic) 2 , halogen, C 1-4 aliphatic, OH, O-(C ⁇ - aliphatic), NO 2 , CN, CO 2 H, CO 2 (C 1- aliphatic), -O(halo C 1- aliphatic), or halo C 1-4 aliphatic.
- An aliphatic group or a non-aromatic heterocyclic ring may contain one or more substituents.
- Substituents on the aliphatic group of R are selected from NH 2 , NH( - aliphatic), N( .
- aliphatic 2 , halogen, - aliphatic, OH, O-(C 1- aliphatic), NO 2 , CN, CO 2 H, CO 2 (C 1- aliphatic), -O(halo C 1-4 aliphatic), or halo C 1- aliphatic.
- Substituents on the aliphatic group or the phenyl ring of R + are selected from NH 2 , NH(C 1-4 aliphatic), N(C 1-4 aliphatic) 2 , halogen, C 1- aliphatic, OH, O-(C 1- aliphatic), NO 2 , CN, CO 2 H, CO 2 (C 1-4 aliphatic), -O(halo C 1-4 aliphatic), or halo C 1- aliphatic.
- alkylidene chain refers to a straight or branched carbon chain that may be fully saturated or have one or more units of unsaturation and has two points of connection to the rest of the molecule.
- the compounds of this invention are limited to those that are chemically feasible and stable. Therefore, a combination of substituents or variables in the compounds described above is permissible only if such a combination results in a stable or chemically feasible compound.
- a stable compound or chemically feasible compound is one in which the chemical structure is not substantially altered when kept at a temperature of 40 °C or less, in the absence of moisture or other chemically reactive conditions, for at least a week.
- structures depicted herein are also meant to include all stereochemical forms of the structure; i.e., the R and S configurations for each asymmetric center. Therefore, single stereochemical isomers as well as enantiomeric and diastereomeric mixtures of the present compounds are within the scope of the invention.
- structures depicted herein are also meant to include compounds which differ only in the presence of one or more isotopically enriched atoms. For example, compounds having the present structures except for the replacement of a hydrogen by a deuterium or tritium, or the replacement of a carbon by a 13 C- or 1 C- enriched carbon are within the scope of this invention.
- the present invention relates to a compound of formula I wherein V is N, V is CH, and V is CH.
- Preferred compounds of formula I include those wherein V 1 is C-R 6 , V 2 is CH, and V 3 is CH or N.
- Another preferred embodiment of the present invention relates to a compound of formula I wherein V 1 is C-R 6 , V 2 is CH, and V 3 is N.
- Another preferred embodiment of the present invention relates to a compound of formula I wherein V 1 is C-R 6 , V 2 is CH, and V 3 is CH.
- the present invention relates to a compound of formula la wherein V 2 is CH and V 3 is N.
- the present invention relates to a
- R 1 groups of formula I or la include hydrogen, halogen, CN, N(R 4 ) 2 , and optionally substituted C 1-6 aliphatic.
- R 1 groups include chloro, bromo, fluoro, NH 2 , NHMe, NHEt, NH-(optionally substituted phenyl), NH-cyclohexyl, NHCH 2 (optionally substituted phenyl), NHC(O)(optionally substituted phenyl), NHC(O)NH(optionally substituted phenyl), NHC(O)CH 2 (optionally substituted phenyl), NHC(O)CH 2 CH 2 (optionally substituted phenyl), N(R)C(O)(optionally substituted phenyl), NHC(O)naphthyl, NHC(O)thienyl, NRC(O)thienyl, SC(O)thienyl, CH 2 C(O)thienyl, NHC(O)naph
- the optional substituents of the phenyl rings of R 1 of formula I or la when present, are optionally substituted R°, halogen, nitro, CN, OR 0 , SR°, N(R°) 2 , SO 2 R°, C(O)R°, C(O)OR, and C(O)N(R°) 2 , wherein each R° is as defined supra.
- Examples of such groups include chloro, bromo, fluoro, CN, nitro, OMe, OPh, OCF 3 , OCH 2 Ph, OEt, SCHF 2 , methyl, ethyl, isopropyl, propyl, vinyl, CF 3 , acetylenyl, CH 2 Ph, CH 2 NH 2 , CH 2 N(Et) 2 , CH 2 morpholin-4-yl, CH 2 piperdin-l-yl, CH 2 imidazol-l-yl, CH 2 ⁇ iperazin-l-yl, C(O)NH 2 , C(O)Me, SO 2 Me, NHEt, and NHMe.
- R 1 of formula I or la is T-Ar
- preferred Ar groups are selected from an optionally substituted 5-6 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Ar rings include optionally substituted phenyl, thienyl, furan, and pyridyl rings.
- T moieties of the T- Ar group of R 1 are selected from a valence bond, -N(R)C(O)-, -NH-, -NHCH 2 -, -NHSO 2 -, -CH 2 NH-, -SC(O)-, -CH 2 C(O)-, -C ⁇ C-, -CH 2 - or -CH 2 CH 2 -. More preferred T moieties of the T-Ar group of R 1 are selected from -NHC(O)-, -NH-, -NHCH 2 -, -CH 2 -, -C ⁇ C-, or -CH 2 CH 2 -.
- T moieties of the T-Ar group of R 1 are selected from -N(R)C(O)-, -NH-, or -NHCH 2 -.
- Preferred substituents on the Ar group when present, include fluoro and CF 3 , Me, Et, iPr, vinyl, acetylene, Ar, Cl, CF 3 , nitro, CN, OMe, OPh, OCF 3 , SO 2 NH2, C(O)OEt, C(O)OH, CH 2 CO 2 H, CH 2 CH 2 CO 2 H, CH 2 NH 2 and C(O)NH 2 , thienyl, oxazolyl, isoxazolyl, and tetrazolyl.
- Preferred Q groups of formula I or la are selected from a valence bond, -CH 2 -, or -CH 2 CH 2 -.
- preferred Ar groups are an optionally substituted ring selected from a 5-6 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or a 9-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- monocyclic rings include phenyl, pyridyl, pyrimidinyl, pyridonyl, furanyl, tetrazolyl, thienyl, cyclopentyl, cyclohexyl, and cycloheptyl.
- bicyclic rings examples include benzo[l,3]dioxolyl, indan-1-onyl, naphthyl, benzothiophenyl, 2,3-dihydro-lH-isoindolyl, indanyl, benzofuranyl, and indolyl.
- preferred substituents on the Ar ring of R 2 include R°, halogen, oxo, OR 0 , phenyl, optionally substituted dialkylamino, haloalkyl, C(O)R°, N ⁇ C(O)R, or SR°.
- preferred substituents include chloro, bromo, fluoro, OH, OMe, NHC(O)CH 3 , OEt, C(O)phenyl, Ophenyl, N(CH 2 CH 2 C1) 2 , N(Me) 2 , CF 3 , and SCF 3 .
- Other examples of preferred Ar groups of formula I or la also include those shown in Table 1 below.
- R 2 group of formula I or la is Q-C(R)(Q-Ar)R 3
- preferred R 3 groups include R', Q-OR 5 , Q-N(R 4 ) 2 , Ar 1 , N(R)C(O)Q-N(R 4 ) 2 , and N(R)Q-N(R 4 ) 2 .
- R 3 groups include CH 2 OH, OH, NH 2 , CH 2 NH 2 , CH 2 NHMe, CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , CH 2 CH 2 NHMe, CH 2 CH 2 N(Me) 2 , CH 2 C(Me) 2 NH 2 , CH 2 C(Me) 2 CHMe, NHC0 2 t-butyl, phenyl, cyclopentyl, methyl, ethyl, isopropyl, cyclopropyl, NH(CH 2 ) 3 NH 2 , NH(CH 2 ) 2 NH 2 , NH(CH 2 ) 2 NHEt, NHCH 2 pyridyl, NHSO 2 phenyl, NHC(O)CH 2 C(O)Ot-butyl, NHC(O)CH 2 NH 3 , and NHCH 2 -imidazol-4-yl.
- the R group of formula I or la is selected from OH, NH 2 , CH 2 NH 2 , CH 2 NHMe, CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , CH 2 CH 2 NHMe, CH 2 CH 2 N(Me) 2 , CH 2 C(Me) 2 NH 2 , CH 2 C(Me) 2 CHMe, NHCO 2 t-butyl, phenyl, NH(CH 2 ) 3 NH 2 , NH(CH 2 ) 2 NH 2 , NH(CH 2 ) 2 NHEt, NHCH 2 pyridyl, NHSO 2 ⁇ henyl, NHC(O)CH 2 C(O)Ot- butyl, NHC(O)CH 2 NH 3 , and NHCH 2 -imidazol-4-yl.
- the R 3 group of formula I or la is selected from CH CH NH 2 .
- Preferred rings formed by the R and R 3 moieties of the Q-C(R)(Q-Ar)R 3 group of R are selected from a 5-6 membered saturated ring having 0-2 heteroatoms independently selected from nitrogen, oxygen, or sulfur. Examples of such rings formed by R and R include piperidinyl, pyrrolidinyl, piperazinyl, morpholinyl, and thiomorpholinyl.
- prefeixed Ar groups of the Q-C(R)(Q-Ar)R 3 moiety are selected from an optionally substituted 5-6 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an optionally substituted 9-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- monocyclic rings include phenyl, pyridyl, furanyl, pyridone, and thienyl.
- bicyclic rings examples include benzo[l,3]dioxolyl, naphthyl, indanyl, and indolyl.
- preferred substituents on the Ar ring of the Q-C(R)(Q-Ar)R group of R 2 include R°, halogen, OR 0 , phenyl, N(R°) 2 , NHC(O)R°, or SR°.
- R 6 groups of formula I or la when present, are selected from halogen, R, and Ar 1 .
- R 6 groups of formula I or la when present, are selected from halogen, optionally substituted C 1-4 aliphatic, or an optionally substituted 5-6 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- R 6 groups of formula I or la when present, are selected from halogen, optionally substituted C 1-4 aliphatic, or an optionally substituted 5-6 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- examples of such groups include chloro, bromo, methyl, ethyl, t-butyl, cyclopropyl, isopropyl, phenyl, and pyridyl.
- the present invention relates to a compound of formula I':
- R 1 is selected from halogen, CN, N(R 4 ) 2 , T-R, or T'-Ar;
- T is selected from a valence bond or a C ⁇ -6 alkylidene chain, wherein up to two methylene units of T are optionally, and independently, replaced by -O-, -N(R)-, -S-,
- T' is a C 1-6 alkylidene chain, wherein up to two methylene units of T' are optionally, and independently, replaced by -O-, -N(R)-, -S-, -N(R)C(O)-, -C(O)N(R)-, -C(O)-, or
- each R is independently selected from hydrogen or an optionally substituted C 1-6 aliphatic group, or: two R groups on the same nitrogen, taken together with the nitrogen atom attached thereto, form a 5-7 membered saturated, partially unsaturated, or aromatic ring having 1-3 heteroatoms independently selected from nitrogen, oxygen, or sulfur;
- R 2 is selected from Q-Ar, Q-N(R 5 ) 2 , or Q-C(R)(Q-Ar)R 3 , wherein:
- R and R 3 optionally form a 5-7 membered saturated or partially unsaturated ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur; each Q is independently selected from a valence bond or a C 1-4 alkylidene chain; each Ar is independently an optionally substituted ring selected from a 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an 8-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having 0-
- R 3 is selected from R', Ar 1 , Q-OR 5 , Q-OC(O)R 5 , Q-CONHR 5 , Q-OC(O)NHR 5 , Q-SR 5 ,
- R' is an optionally substituted C 1-6 aliphatic group
- each R 4 is independently selected from R, COR 5 , CO 2 R 5 , CON(R 5 ) 2 , SO 2 R 5 , SO 2 N(R 5 ) 2 , or Ar 1
- each R 5 is independently selected from R or Ar
- V 1 , V 2 and V 3 are each independently selected from nitrogen or C(R 6 );
- each R 6 is independently selected from R, Ar 1 , halogen, CN, NO 2 , OR, SR, N(R 4 ) 2 ,
- N(R)COR N(R)CON(R 4 ) 2 , N(R)C(O)OR, CON(R 4 ) 2 , OC(O)N(R 4 ) 2 , CO 2 R, OC(O)R,
- each Ar is independently selected from an optionally substituted 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur; provided that: when V 1 , V 2 , and V 3 are each CH and R 1 is hydrogen, then R 2 is Q-C(R)(Q-Ar)R 3 , wherein R 3 is other than R', Q-OC(O)R 5 , or OCH 2 phenyl.
- Preferred R and R groups of formula I' are those described above for compounds of formulae I and la.
- preferred T' groups of formula I' are selected from -NHC(O)-, -NH-, -NHCH 2 -, -NHSO 2 -, -CH 2 NH-, -CH 2 -, -C ⁇ C-, or -CH 2 CH 2 -.
- More preferred T' groups of formula I' are selected from -NHC(O)-, -NH-, -NHCH 2 -, -NHSO 2 -, or -CH NH-.
- the present invention relates to a compound of formula lb:
- Preferred R 1 groups of formula lb include those described above for compounds of formula I and la.
- V 1 , V 2 , and V 3 groups of formula lb are the preferred V 1 , V 2 , and V 3 groups set forth for compounds of formula I, supra.
- Preferred Q groups of formula lb include those described above for compounds of formula I and la.
- Preferred Ar groups of formula lb include an optionally substituted ring selected from a 5-6 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or a 9-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- monocyclic rings include phenyl, pyridyl, thienyl, furanyl, cyclopentyl, cyclohexyl, and cycloheptyl.
- bicyclic rings examples include benzo[l,3]dioxolyl, indan-1-onyl, naphthyl, benzothiophenyl, 2,3-dihydro-lH-isoindolyl, indanyl, benzofuranyl, and indolyl.
- preferred substituents on the Ar group of formula lb include R°, halogen, OR 0 , phenyl, optionally substituted dialkylamino, haloalkyl, C(O)R°, or SR°.
- substituents examples include tetrazolyl, oxazolyl, isoxazolyl, chloro, bromo, fluoro, O ⁇ , OMe, OEt, C(O)phenyl, Ophenyl, N(C ⁇ 2 C ⁇ 2 C1) 2 , N(Me) 2 , CF 3 , and SCF 3 .
- R 3 groups of formula lb include R', Q-OR 5 , Q-N(R 4 ) 2 , Ar 1 , N(R)C(O)Q-N(R 4 ) 2 , and N(R)Q-N(R 4 ) 2 .
- R 3 groups include CH 2 OH, OH, NH 2 , CH 2 NH 2 , CH 2 NHMe, CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , CH 2 CH 2 NHMe, CH 2 C(Me) 2 NH 2 , CH 2 C(Me) 2 CHMe, CH 2 CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , NHCO 2 t-butyl, phenyl, cyclopentyl, methyl, ethyl, isopropyl, cyclopropyl, NH(CH 2 ) 3 NH 2 , NH(CH 2 ) 2 NH 2 , NH(CH 2 ) 2 NHEt, NHCH 2 pyridyl, NHSO 2 ⁇ henyl, NHC(O)CH 2 C(O)Ot- butyl, NHC(O)CH 2 NH 3 , and NHCH 2 -imidazol-4-yl.
- the R group of formula lb is selected from OH, NH , CH 2 NH 2 , CH 2 NHMe, CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , CH 2 CH 2 NHMe, CH 2 CH 2 N(Me) 2 , CH 2 C(Me) 2 NH 2 , CH 2 C(Me) 2 CHMe, NHCO 2 t-butyl, phenyl, NH(CH 2 ) 3 NH 2 , NH(CH 2 ) 2 NH 2 , NH(CH 2 ) 2 NHEt, NHCH 2 pyridyl, NHSO 2 phenyl, NHC(O)CH 2 C(O)Ot- butyl, NHC(O)CH 2 NH 3 , and NHCH 2 -imidazol-4-yl.
- the R 3 group of formula lb is selected from CH 2 CH 2 NH 2 .
- Preferred rings formed by the R and R 3 moieties of the Q-C(R)(Q-Ar)R 3 group of formula lb are selected from a 5-6 membered saturated ring having 0-2 heteroatoms independently selected from nitrogen, oxygen, or sulfur. Examples of such rings formed by R and R 3 include piperidinyl, pyrrolidinyl, piperazinyl, morpholinyl, and thiomorpholinyl.
- Another embodiment of the present invention relates to a compound of formula Ila:
- R 1 is selected from halogen, CN, N(R 4 ) 2 , or T-R;
- T is selected from a valence bond or a C 1-6 alkylidene chain, wherein up to two methylene units of T are optionally, and independently, replaced by -O-, -N(R)-, -S-,
- each R is independently selected from hydrogen or an optionally substituted C 1-6 aliphatic group, or: two R groups on the same nitrogen, taken together with the nitrogen atom attached thereto, form a 5-7 membered saturated, partially unsaturated, or aromatic ring having 1-3 heteroatoms independently selected from nitrogen, oxygen, or sulfur;
- R 2 is Q-C(R)(Q-Ar)R 3 , wherein:
- R and R optionally form a 5-7 membered saturated or partially unsaturated ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur; each Q is independently selected from a valence bond or a C 1-4 alkylidene chain; each Ar is independently an optionally substituted ring selected from a 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an 8-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having 0-
- R 3 is selected from R', Ar 1 , Q-OR 5 , Q-OC(O)R 5 , Q-CONHR 5 , Q-OC(O)NHR 5 , Q-SR 5 ,
- each RR 44 is independently selected from R, COR, CO 2 R, CON(R) 2 , SO 2 R, SO 2 N(R) 2 , or Ar 1 ; each ti RR 55 ; is independently selected from R or Ar:
- V , V and V are each independently selected from nitrogen or C(R ); each R 6 is independently selected from R, Ar 1 , halogen, CN, NO , OR, SR, N(R 4 ) 2 ,
- each Ar 1 is independently selected from an optionally substituted 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur; provided that when R 1 is hydrogen then R 3 is other than R', Q-OC(O)R 5 , or OCH 2 phenyl.
- R 1 groups of formula Ila include halogen, N(R 4 ) 2 , and optionally substituted C 1-6 aliphatic.
- examples of such groups include chloro, bromo, fluoro, NH 2 , NHMe, NHEt, NH-cyclohexyl, methyl, ethyl, propyl, isopropyl, cyclopropyl, acetylenyl, and t-butyl.
- V 1 , V 2 , and V 3 groups of formula Ila are the preferred V 1 , V 2 , and V 3 groups set forth for compounds of formula I, supra.
- Preferred Q groups of formula Ila are selected from a valence bond, -CH 2 -, or -CH 2 CH 2 -.
- R 3 groups of formula Ila include R', Q-OR 5 , Q-N(R 4 ) 2 , Ar 1 , N(R)C(O)Q-N(R 4 ) 2 , and N(R)Q-N(R 4 ) 2 .
- R 3 groups include CH 2 OH, OH, NH 2 , CH 2 NH 2 , CH 2 NHMe, CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , CH 2 CH 2 NHMe, CH 2 C(Me) 2 NH 2 , CH 2 C(Me) 2 CHMe, CH 2 CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , NHCO 2 t-butyl, phenyl, cyclopentyl, methyl, ethyl, isopropyl, cyclopropyl, NH(CH 2 ) 3 NH 2 , NH(CH 2 ) 2 NH 2 , NH(CH 2 ) 2 NHEt, NHCH 2 pyridyl, NHSO 2 phenyl, NHC(O)CH 2 C(O)Ot- butyl, NHC(O)CH 2 NH 3 , and NHCH 2 -imidazol-4-yl.
- the R 3 group of formula Ila is selected from OH, NH 2 , CH 2 NH 2 , CH 2 NHMe, CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , CH 2 CH 2 NHMe, CH 2 CH 2 N(Me) 2 , CH 2 C(Me) 2 NH 2 , CH 2 C(Me) 2 CHMe, NHCO 2 t-butyl, phenyl, NH(CH 2 ) 3 NH 2 , NH(CH 2 ) 2 NH 2 , NH(CH 2 ) 2 NHEt, NHCH 2 pyridyl, NHSO 2 phenyl, NHC(O)CH 2 C(0)Ot- butyl, NHC(O)CH 2 NH 3 , and NHCH 2 -imidazol-4-yl.
- the R 3 group of formula Ila is selected from CH 2 CH 2 NH 2 .
- Preferred rings formed by the R and R moieties of R of formula Ila are selected from a 5-6 membered saturated ring having 0-2 heteroatoms independently selected from nitrogen, oxygen, or sulfur. Examples of such rings formed by R and R include piperidinyl, pyrrolidinyl, piperazinyl, morpholinyl, and thiomorpholinyl. O 03/064397
- Preferred Ar groups of R of formula Ila are selected from an optionally substituted 5-6 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an optionally substituted 9-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- monocyclic rings include phenyl, pyridyl, furanyl, and thienyl.
- Examples of such bicyclic rings include benzo[l,3]dioxolyl, naphthyl, indanyl, and indolyl.
- preferred substituents on the Ar ring of the Q-C(R)(Q-Ar)R 3 group of R 2 of formula Ila include R°, halogen, OR 0 , phenyl, N(R°) 2 , NHC(O)R°, or SR°.
- substituents on the Ar ring of the Q-C(R)(Q-Ar)R 3 group of R 2 of formula Ila include R°, halogen, OR 0 , phenyl, N(R°) 2 , NHC(O)R°, or SR°.
- examples of such groups include fluoro, chloro, bromo, CF , OH, OMe, OPh, OCH 2 PH, SMe, NH 2 , NHC(O)Me, methyl, ethyl, isopropyl, isobutyl, and cyclopropyl.
- Another embodiment relates to a compound of formula lib:
- R 1 is T-Ar; each T is independently selected from a valence bond or a C 1-6 alkylidene chain, wherein up to two methylene units of T are optionally, and independently, replaced by -O-, -N(R)-, -S-, -N(R)C(O)-, -C(O)N(R)-, -C(O)-, or -SO 2 -; each R is independently selected from hydrogen or an optionally substituted C 1-6 aliphatic group, or: two R groups on the same nitrogen, taken together with the nitrogen atom attached thereto, form a 5-7 membered saturated, partially unsaturated, or aromatic ring having 1-3 heteroatoms independently selected from nitrogen, oxygen, or sulfur; R 2 is Q-C(R)(Q-Ar)R 3 , wherein:
- R and R optionally form a 5-7 membered saturated or partially unsaturated ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur; each Q is independently selected from a valence bond or a C 1-4 alkylidene chain; O 03/064397
- each Ar is independently an optionally substituted ring selected from a 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an 8-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having 0- 4 heteroatoms independently selected from nitrogen, oxygen, or sulfur;
- R 3 is selected from R', Ar 1 , Q-OR 5 , Q-OC(O)R 5 , Q-CONHR 5 , Q-OC(O)NHR 5 , Q-SR 5 , Q-N(R 4 ) 2 , N(R)(Q-Ar), N(R)C(O)Q-N(R 4 ) 2 , or N(R)Q-N(R 4 ) 2 ;
- R' is an optionally substituted C 1-6 aliphatic group
- each R 4 is independently selected from R, COR 5 , CO 2 R 5 , CON(R 5 ) 2 , SO 2 R 5 , SO 2 N(R 5 ) 2 , or Ar 1
- each R 5 is independently selected from R or Ar;
- V , V and V are each independently selected from nitrogen or C(R ); each R 6 is independently selected from R, Ar 1 , halogen, CN, NO 2 , OR, SR, N(R 4 ) 2 ,
- N(R)COR N(R)CON(R 4 ) 2 , N(R)C(O)OR, CON(R 4 ) 2 , OC(O)N(R 4 ) 2 , CO 2 R, OC(O)R,
- each Ar 1 is independently selected from an optionally substituted 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur;
- V 1 9 9 provided that when V , V , and V are each CH,T is a valence bond, and R is Q-C(R)(Q- Ar)R , wherein Ar is an optionally substituted phenyl ring, then R is other than Q-OR 5 or C(0)NH 2 .
- Preferred V 1 , V 2 , and V 3 groups of formula lib are those set forth for compounds of formula I, supra.
- Preferred Ar groups of R 1 of formula lib are selected from an optionally substituted 5-6 membered aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Preferred T moieties of the T-Ar group of R 1 of formula lib are selected from a valence bond, -NHC(O)-, -NH-, -NHCH 2 -, -NHSO 2 -, -CH 2 NH-, -C ⁇ C-, -CH 2 - or -CH 2 CH 2 -.
- T moieties of the T-Ar group of R 1 are selected from -NHC(O)-, -NH-, -NHCH 2 -, -CH 2 - or -CH 2 CH 2 -.
- R 1 groups of formula lib include NHCH 2 (optionally substituted phenyl), NHC(O)(optionally substituted phenyl), NHC(O)NH(o ⁇ tionally substituted phenyl), NHC(O)CH 2 (optionally substituted phenyl), NHC(O)CH 2 CH 2 (optionally substituted phenyl), NHC(O)(optionally substituted phenyl), NHC(O)naphthyl, NHC(O)thienyl, NHC(O)pyridyl, NHC(O)furanyl, methyl, ethyl, propyl, isopropyl, cyclopropyl, acetylenyl, and t-butyl.
- Preferred substituents on the Ar group of R 1 of formula lib when present, include R°, halogen, nitro, CN, OR 0 , SR°, N(R°) 2 , SO 2 R°, C(O)R°, C(O)OR, and C(O)N(R°) 2 , wherein each R° is as defined supra.
- Examples of such groups include chloro, bromo, fluoro, CN, nitro, OMe, OPh, OCF 3 , OCH 2 Ph, OEt, SCHF 2 , methyl, ethyl, isopropyl, propyl, vinyl, CF 3 , acetylenyl, CH 2 Ph, CH 2 NH 2 , CH 2 N(Et) 2 , CH 2 morpholin-4- yl, CH 2 piperdin-l-yl, CH 2 imidazol-l-yl, CH 2 piperazin-l-yl, C(O)NH 2 , C(O)Me, SO 2 Me, NHEt, and NHMe.
- Preferred Q groups of formula lib are those set forth above for compounds of formula I and lb.
- R 3 groups of formula lib include R', Q-OR 5 , Q-N(R 4 ) 2 , Ar 1 , N(R)C(O)Q-N(R 4 ) 2 , and N(R)Q-N(R 4 ) 2 .
- R 3 groups include CH 2 OH, OH, NH 2 , CH 2 NH 2 , CH 2 NHMe, CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , CH 2 CH 2 NHMe, CH 2 C(Me) 2 NH 2 , CH 2 C(Me) 2 CHMe, CH 2 CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , NHCO 2 t-butyl, phenyl, cyclopentyl, methyl, ethyl, isopropyl, cyclopropyl, NH(CH 2 ) 3 NH , NH(CH 2 ) 2 NH 2 , NH(CH 2 ) 2 NHEt, NHCH 2 pyridyl, NHSO 2 phenyl, NHC(O)CH 2 C(O)Ot- butyl, NHC(O)CH 2 NH 3 , and NHCH 2 -imidazol-4-yl.
- the R 3 group of formula lib is selected from CH 2 NHMe, CH 2 N(Me) 2 , CH 2 CH 2 NH 2 , CH 2 CH 2 NHMe, CH 2 CH 2 N(Me) 2 , CH 2 C(Me) 2 NH 2 , CH 2 C(Me) 2 CHMe, NHCO 2 t-butyl, phenyl, NH(CH 2 ) 3 NH 2 , NH(CH 2 ) 2 NH 2 , NH(CH 2 ) 2 NHEt, NHCH 2 pyridyl, NHSO 2 phenyl, NHC(O)CH 2 C(O)Ot-butyl, NHC(O)CH 2 NH 3 , and NHCH 2 -imidazol-4-yl.
- the R 3 group of formula lib is selected from CH 2 CH 2 NH 2 .
- Preferred rings formed by the R and R 3 moieties of R 2 of formula lib are selected from a 5-6 membered saturated ring having 0-2 heteroatoms independently selected from nitrogen, oxygen, or sulfur. Examples of such rings formed by R and R include piperidinyl, pyrrolidinyl, piperazinyl, morpholinyl, and thiomorpholinyl.
- Preferred Ar groups of R 2 of formula lib are selected from an optionally substituted 5-6 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an optionally substituted 9-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- monocyclic rings include phenyl, pyridyl, pyrimidinyl, pyridonyl, furanyl, tetrazolyl, thienyl, cyclopentyl, cyclohexyl, and cycloheptyl.
- bicyclic rings examples include benzo[l,3]dioxolyl, indan-1-onyl, naphthyl, benzothiophenyl, 2,3-dihydro-lH-isoindolyl, indanyl, benzofuranyl, and indolyl.
- preferred substituents on the Ar ring of the Q-C(R)(Q-Ar)R 3 group of R 2 of formula lib include R°, halogen, OR 0 , phenyl, N(R°) 2 , N ⁇ C(O)R°, or SR°.
- the present invention relates to a compound of formula III:
- V 1 , V 2 , V 3 , R 1 , R 3 , Q, and Ar groups of formula III are those set forth above for compounds of formula I or lb.
- the present invention relates to a compound of formula IV:
- V 1 , V 2 , V 3 , R 1 , R 3 , Q, and Ar groups of formula IV are those set forth above for compounds of formula I or lb.
- the present invention relates to a compound of formula V:
- each R is independently selected from hydrogen or an optionally substituted C 1-6 aliphatic group, or: two R groups on the same nitrogen, taken together with the nitrogen atom attached thereto, form a 5-7 membered saturated, partially unsaturated, or aromatic ring having 1-3 heteroatoms independently selected from nitrogen, oxygen, or sulfur;
- R 2 is Q-C(R)(Q-Ar)R 3 , wherein:
- R and R optionally form a 5-7 membered saturated or partially unsaturated ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur; each Q is independently selected from a valence bond or a C 1-4 alkylidene chain; each Ar is independently an optionally substituted ring selected from a 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an 8-10 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring having
- R 3 is selected from R', Ar 1 , Q-OR 5 , Q-OC(O)R 5 , Q-CONHR 5 , Q-OC(O)NHR 5 , Q-SR 5 ,
- R' is an optionally substituted C 1-6 aliphatic group
- each R 4 is independently selected from R, COR 5 , CO 2 R 5 , CON(R 5 ) 2 , SO 2 R 5 , SO 2 N(R 5 ) 2 , or Ar 1
- each R is independently selected from R or Ar
- V 1 , V 2 and V 3 are each independently selected from nitrogen or C(R 6 );
- each R 6 is independently selected from R, Ar 1 , halogen, CN, NO 2 , OR, SR, N(R 4 ) 2 ,
- N(R)COR N(R)CON(R 4 ) 2 , N(R)C(O)OR, CON(R 4 ) 2 , OC(O)N(R 4 ) 2 , CO 2 R, OC(O)R,
- each Ar 1 is independently selected from an optionally substituted 5-7 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- V 1 , V 2 , and V 3 groups of formula V are those set forth for compounds of formula I or lb, supra.
- R 2 groups of formula V are those set forth for compounds of formula
- reduction of the nitro group of compound 2 can be achieved by treating 2 with hydrogen gas in the presence of Pd/C by methods substantially similar to those described by Boyer, et al, J Chem. Res. Miniprint, 1990, 11, 2601.
- Another alternative method for achieving the reduction of the nitro group of compound 2 is by hydrolysis using a method substantially similar to that described by Lee, et al, Synthesis, 2001, 1, 81.
- Scheme III above shows a method for preparing compounds of formula I where R 1 is halogen.
- 5-nitro-lH-indazole (5) may be chlorinated to afford 3- chloro-5-nitro-lH-indazole (6) using the methods described by v. Auwers, et al, Justus Liebigs Ann. Chem., 1927, 451, 295.
- the nitroindazole 5 can be treated with N-chlorosucciniimide to form a 3-chloro nitroindazole 6.
- the reduction of 6 to form the amino compound 7 may be achieved by following the methods described by Boyer, et al, J Chem. Res. Miniprint, 1990, 11, 2601.
- Scheme N above shows a general method for preparing ⁇ -hydroxy acids 12 used for preparing compounds of formula lb, where R 3 is OH, according to the methods described in Scheme IV.
- the formation of the ⁇ -hydroxy ester compound 11 from 9 and 10 was achieved by methods substantially similar to those described by Hernandez, et al, J. Org. Chem., 1995, 60, 2683.
- the oxaziridine reagent 10 can be prepared according to the procedure described by Davies, et al, J. Org. Chem., 1988, 53, 2087.
- Scheme NI shows a general method for preparing compounds of formula lb where R 3 is a variety of amino groups from compounds of formula lb where R 3 is a hydroxy group as described above in Scheme V.
- Compound 13 may be treated with methanesulfonyl chloride and pyridine in THF to afford the mesyl derivative 14. The mesyl group may then be displaced by the desired amino group to afford compound 15. Removal of the Boc protecting group provides compound 16.
- Scheme VLI Scheme VLI
- Scheme VII above shows a method for preparing carboxylic acid intermediates useful for preparing compounds of formula lb where R 3 is an amino group. This method may be used to prepare compounds of formula lb where R is a variety of ammo groups of formula N(R 4 ) 2 , N(R)COT n N(R 4 ) 2 , or N(R)T n N(R 4 ) 2 . Each of the above steps is well known to one of skill in the art. Carboxylic acid compound 20 may then be coupled to the amino-indazole according to Scheme IV to afford compounds of formula lb.
- Reagents (a), I 2 , KOH, DMF, rt.
- Scheme VIII above shows the preparation of 3-iodo-5-nitroindazole (21) from 5-nitroindazole (5) according to methods substantially similar to that described in published PCT application number WO 02/10137.
- Reagents (a) Br 2 , AcOH, reflux.
- Scheme LX above shows a method for the preparation of 3-bromo-5- nitroindazole, by a method substantially similar to that described by Benchidimi, et al, J
- Scheme LX above shows a general method for the preparation of compounds of formula I where R 1 is an alkynyl group.
- the bromoindazole (22) is coupled with propyne (23), by the Sonograshira coupling method, to afford 5-nitro-3-prop-l-ynyl-lH- indazole (24).
- propyne 23
- Sonograshira coupling method to afford 5-nitro-3-prop-l-ynyl-lH- indazole (24).
- Reagents (a) NaNO 2 , AcOH, reflux.
- Scheme XI above shows a general method for the preparation of 7-chloro-5- nitroindazole (28) by treating 2-chloro-6-methyl-4-nitro-phenylamine (27) with sodium nitrate in the presence of acetic acid.
- the indazole ring is formed at step (b) by treating intermediate (30) with sodium nitrate and acetic acid at reflux.
- Reagents (i) LDA, -78°C (ii) ICH 2 CN, -78°C to ambient temperature, THF; (b)H 2 , PtO 2 , HCI, MeOH;(C) 8M HCI, reflux; (d) (Boc) 2 O, Na 2 CO 3 , aq. THF, ambient temperature.
- Scheme XLV above shows a general method for preparing the protected amino acid intermediates (36) useful for preparing compounds of formula lb where R 3 is T n N(R 4 ) 2 .
- the cyano compound (33) is prepared by treating the ester (17) with lithiumdiisopropylamide (LDA) at -78°C then adding iodoacetonitrile.
- LDA lithiumdiisopropylamide
- the nitrile is reduced using hydrogen in the presence of a platinum catalyst by a method substantially similar to that described by Prager, et al, Aust. J. Chem., 1997, 50, 813.
- the resulting amine (34) is hydrolyzed to form the acid compounds 35.
- amino groups is then protected with a BOC group by treating 35 with BOC-anhydride in the presence of aqueous sodium carbonate in tetrahydrofuran.
- Other amino protecting groups are well known in the art and are described in detail in Protecting Groups in Organic Synthesis, Theodora W. Greene and Peter G. M. Wuts, 1991, published by John Wiley and Sons.
- Reagents (a) tert-Butylacrylate, KO'Bu, THF, -78°C to rt; (b) TFA, DCM; (c) DPP A, PbMe, rt; (d) 'BuOH, SnCl 2 (cat), 80°C, (e) LiOH, aq. THF.
- Scheme XV above shows an alternative method for preparing the protected amino acid intermediates (36) useful for preparing compounds of formula lb where R is T n ⁇ (R 4 ) 2 .
- Michael addition of tert-butylacrylate to the anion of ester 17 affords the diester 37.
- the tert-butyl ester of compound 37 is selectively cleaved to afford the acid intermediate 38.
- the mono-ester 38 is then treated sequentially with diphenylphosphorylazide and tert-butanol to afford the BOC-protected amino ester 39. Hydrolysis of ester affords the desired protected amino acid intermediate (36).
- Scheme XVI above shows a general method for preparing compounds of the present invention where V 3 is N.
- the pyridopyrazole intermediate 41 useful for the preparation of compounds of the present invention where V 3 is N, is prepared from the amino-protected pyridine compound 40 by methods substantially similar to those described by Foster, H. E. et. al., J. Chem. Soc, Perkin Trans 1, 1973, 2901.
- Scheme XVI above shows a general method for preparing compounds of the present invention wherein a methylene unit of the T moiety of the R 1 group of formula I is replaced by either -O- or -S-.
- the formation of the indazole 42 was achieved by methods substantially similar to those described by Pfannstiel, K. et. al, Berl942, 75B, 1096 and Vicente, J et al, Heterocycles, 1997, 45 (1), 129.
- Indazole 45 was synthesized from compound 42 following a procedure outlined by Kuroda, T et al in JP50130759.
- Scheme XVIII above shows a general scheme for preparing compounds of formula I where V 1 is nitrogen by methods substantially similar to that described by Fanta, Org. Synth. Coll., 4, 844.
- Scheme XLX shows a general scheme for preparing compounds of formula I where V 1 is nitrogen by methods substantially similar to that described by Fanta, Org. Synth. Coll., 4, 844.
- PKA, PDKl, p70S6K, or ROCK kinase may be assayed in vitro, in vivo or in a cell line according to methods known in the art.
- In vitro assays include assays that determine inhibition of either the phosphorylation activity or ATPase activity of activated AKT, PKA, PDKl, p70S6K, or ROCK. Alternate in vitro assays quantitate the ability of the inhibitor to bind to AKT, PKA, PDKl, p70S6K, or ROCK.
- Inhibitor binding may be measured by radiolabelling the inhibitor prior to binding, isolating the inhibitor/ AKT, inhibitor/PKA, inhibitor/PDKl, inhibitor/p70S6K, or inhibitor/ROCK complex and determining the amount of radiolabel bound.
- inhibitor binding may be determined by running a competition experiment where compounds are incubated with AKT, PKA, PDKl, p70S6K, or ROCK bound to known radioligands.
- Detailed conditions for assaying a compound utilized in this invention as an inhibitor of AKT, PKA, PDKl, p70S6K, or ROCK kinase are set forth in the Examples below.
- the invention provides a composition comprising a compound of this invention or a pharmaceutically acceptable derivative thereof and a pharmaceutically acceptable carrier, adjuvant, or vehicle.
- the amount of compound in the compositions of this invention is such that is effective to measurably inhibit a protein kinase, particularly AKT, PKA, PDKl, p70S6K, or ROCK kinase, in a biological sample or in a patient.
- the composition of this invention is formulated for administration to a patient in need of such composition.
- the composition of this invention is formulated for oral administration to a patient.
- patient as used herein, means an animal, preferably a mammal, and most preferably a human.
- compositions of this invention refers to a non-toxic carrier, adjuvant, or vehicle that does not destroy the pharmacological activity of the compound with which it is formulated.
- Pharmaceutically acceptable carriers, adjuvants or vehicles that may be used in the compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxyprop
- the term "measurably inhibit”, as used herein means a measurable change in AKT, PKA, PDKl, p70S6K, or ROCK activity between a sample comprising said composition and a AKT, PKA, PDKl, p70S6K, or ROCK kinase and an equivalent sample comprising AKT, PKA, PDKl, p70S6K, or ROCK kinase in the absence of said composition.
- a "pharmaceutically acceptable salt” means any non-toxic salt or salt of an ester of a compound of this invention that, upon administration to a recipient, is capable of providing, either directly or indirectly, a compound of this invention or an inhibitorily active metabolite or residue thereof.
- the term “inhibitorily active metabolite or residue thereof” means that a metabolite or residue thereof is also an inhibitor of a AKT, PKA, PDKl, p70S6K, or ROCK family kinase.
- Pharmaceutically acceptable salts of the compounds of this invention include those derived from pharmaceutically acceptable inorganic and organic acids and bases.
- Suitable acid salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptanoate, glycerophosphate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oxalate, palmoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, prop
- Salts derived from appropriate bases include alkali metal (e.g., sodium and potassium), alkaline earth metal (e.g., magnesium), ammonium and N + (C 1- alkyl) salts.
- alkali metal e.g., sodium and potassium
- alkaline earth metal e.g., magnesium
- ammonium and N + (C 1- alkyl) salts e.g., sodium and potassium
- compositions of the present invention may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally or via an implanted reservoir.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- the compositions are administered orally, intraperitoneally or intravenously.
- Sterile injectable forms of the compositions of this invention may be aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non- toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3- butanediol.
- a non- toxic parenterally-acceptable diluent or solvent for example as a solution in 1,3- butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or di-glycerides.
- Fatty acids such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, such as carboxymethyl cellulose or similar dispersing agents that are commonly used in the formulation of pharmaceutically acceptable dosage forms including emulsions and suspensions.
- Other commonly used surfactants such as Tweens, Spans and other emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
- compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, aqueous suspensions or solutions.
- carriers commonly used include lactose and corn starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried cornstarch.
- aqueous suspensions are required for oral use, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening, flavoring or coloring agents may also be added.
- the pharmaceutically acceptable compositions of this invention may be administered in the form of suppositories for rectal administration. These can be prepared by mixing the agent with a suitable non-irritating excipient that is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug. Such materials include cocoa butter, beeswax and polyethylene glycols.
- a suitable non-irritating excipient that is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug.
- Such materials include cocoa butter, beeswax and polyethylene glycols.
- the pharmaceutically acceptable compositions of this invention may also be administered topically, especially when the target of treatment includes areas or organs readily accessible by topical application, including diseases of the eye, the skin, or the lower intestinal tract. Suitable topical formulations are readily prepared for each of these areas or organs.
- Topical application for the lower intestinal tract can be effected in a rectal suppository formulation (see above) or in a suitable enema formulation. Topically- transdermal patches may also be used.
- the pharmaceutically acceptable compositions may be formulated in a suitable ointment containing the active component suspended or dissolved in one or more carriers.
- Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene, polyoxypropylene compound, emulsifying wax and water.
- the pharmaceutically acceptable compositions can be formulated in a suitable lotion or cream containing the active components suspended or dissolved in one or more pharmaceutically acceptable carriers.
- Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the pharmaceutically acceptable compositions may be formulated as micronized suspensions in isotonic, pH adjusted sterile saline, or, preferably, as solutions in isotonic, pH adjusted sterile saline, either with or without a preservative such as benzylalkonium chloride.
- the pharmaceutically acceptable compositions may be formulated in an ointment such as petrolatum.
- compositions of this invention may also be administered by nasal aerosol or inhalation.
- Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other conventional solubilizing or dispersing agents.
- compositions of this invention are formulated for oral administration.
- amount of the compounds of the present invention that may be combined with the carrier materials to produce a composition in a single dosage form will vary depending upon the host treated, the particular mode of administration.
- the compositions should be formulated so that a dosage of between 0.01 - 100 mg/kg body weight/day of the inhibitor can be administered to a patient receiving these compositions.
- a specific dosage and treatment regimen for any particular patient will depend upon a variety of factors, including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, rate of excretion, drag combination, and the judgment of the treating physician and the severity of the particular disease being treated.
- the amount of a compound of the present invention in the composition will also depend upon the particular compound in the composition.
- additional therapeutic agents which are normally administered to treat or prevent that condition, may also be present in the compositions of this invention.
- additional therapeutic agents that are normally administered to treat or prevent a particular disease, or condition are known as "appropriate for the disease, or condition, being treated”.
- chemotherapeutic agents or other anti-proliferative agents may be combined with the compounds of this invention to treat proliferative diseases and cancer.
- known chemotherapeutic agents include, but are not limited to, GleevecTM, adriamycin, dexamethasone, vincristine, cyclophosphamide, fluorouracil, topotecan, taxol, interferons, and platinum derivatives.
- agents the inhibitors of this invention may also be combined with include, without limitation: treatments for Alzheimer's Disease such as Aricept ® and Excelon ® ; treatments for Parkinson's Disease such as L-DOPA/carbidopa, entacapone, ropinrole, pramipexole, bromocriptine, pergolide, trihexephendyl, and amantadine; agents for treating Multiple Sclerosis (MS) such as beta interferon (e.g., Avonex ® and Rebif ® ), Copaxone ® , and mitoxantrone; treatments for asthma such as albuterol and Singulair ® ; agents for treating schizophrenia such as zyprexa, risperdal, seroquel, and haloperidol; anti-inflammatory agents such as corticosteroids, TNF blockers, LL-1 RA, azathioprine, cyclophosphamide, and sulfasalazine; immunomodulates, anti-inflammatory agents
- the amount of additional therapeutic agent present in the compositions of this invention will be no more than the amount that would normally be administered in a composition comprising that therapeutic agent as the only active agent.
- the amount of additional therapeutic agent in the presently disclosed compositions will range from about 50% to 100% of the amount normally present in a composition comprising that agent as the only therapeutically active agent.
- the invention relates to a method of inhibiting AKT, PKA, PDKl, p70S6K, or ROCK kinase activity in a biological sample comprising the step of contacting said biological sample with a compound of this invention, or a composition comprising said compound.
- the method comprises the step of contacting said biological sample with a preferred compound of the present invention, as described herein supra.
- biological sample includes, without limitation, cell cultures or extracts thereof; biopsied material obtained from a mammal or extracts thereof; and blood, saliva, urine, feces, semen, tears, or other body fluids or extracts thereof.
- Inhibition of AKT, PKA, PDKl , p70S6K, or ROCK kinase activity in a biological sample is useful for a variety of purposes that are known to one of skill in the art. Examples of such purposes include, but are not limited to, blood transfusion, organ- transplantation, biological specimen storage, and biological assays.
- Another aspect of this invention relates to a method for treating an AKT-, PKA-, PDK1-, p70S6K-, or ROCK-mediated disease in a patient, which method comprises administering to a patient in need thereof, a therapeutically effective amount of a compound of the present invention, or a pharmaceutically acceptable composition comprising said compound.
- the invention relates to administering a compound of formula I', or a pharmaceutically acceptable composition comprising said compound.
- a more preferred embodiment relates to administering a preferred compound of formula F, as described herein supra, or a pharmaceutically acceptable composition comprising said compound .
- the present invention relates to a method for treating an AKT-, PKA-, PDK1-, p70S6K-, or ROCK-mediated disease in a patient, which method comprises administering to a patient in need thereof, a therapeutically effective amount of a compound of formula Ila, lib, or V, or a pharmaceutically acceptable composition comprising said compound.
- said method comprises administering to a patient in need thereof, a therapeutically effective amount of a preferred compound of formula Ila, lib, or V, as described herein supra, or a pharmaceutically acceptable composition comprising said compound.
- the present invention relates to a method for treating an AKT-, PKA-, PDK1-, p70S6K-, or ROCK-mediated disease in a patient, which method comprises administering to a patient in need thereof, a therapeutically effective amount of a compound of formula III or IV, or a pharmaceutically acceptable composition comprising said compound.
- said method comprises administering to a patient in need thereof, a therapeutically effective amount of a preferred compound of formula III, or IV, as described herein supra, or a pharmaceutically acceptable composition comprising said compound.
- the invention provides a method for treating or lessening the severity of an AKT-mediated disease or condition in a patient comprising the step of administering to said patient a composition according to the present invention.
- the term "AKT-mediated condition” or “disease”, as used herein, means any disease or other deleterious condition in which AKT is known to play a role.
- the term “AKT-mediated condition” or “disease” also means those diseases or conditions that are alleviated by treatment with an AKT inhibitor.
- AKT-mediated diseases or conditions include, but are not limited to, proliferative disorders, cancer, cardiovascular disorders, rheumatoid arthritis, and neurodegenerative disorders.
- said cancer is selected from pancreatic, prostate, or ovarian cancer.
- the invention provides a method for treating or lessening the severity of a PKA-mediated disease or condition in a patient comprising the step of administering to said patient a composition according to the present invention.
- PKA-mediated condition or “disease”, as used herein, means any disease or other deleterious condition in which PKA is known to play a role.
- PKA-mediated condition or “disease” also means those diseases or conditions that are alleviated by treatment with a PKA inhibitor.
- PKA-mediated diseases or conditions include, but are not limited to, proliferative disorders and cancer.
- the invention provides a method for treating or lessening the severity of a PDKl-mediated disease or condition in a patient comprising the step of administering to said patient a composition according to the present invention.
- the invention provides a method for treating or lessening the severity of an PDKl-mediated disease or condition in a patient comprising the step of administering to said patient a composition according to the present invention.
- PDKl-mediated condition or “disease”, as used herein, means any disease or other deleterious condition in which PDKl is known to play a role.
- PDKl-mediated condition or “disease” also means those diseases or conditions that are alleviated by treatment with a PDKl inhibitor.
- PDKl-mediated diseases or conditions include, but are not limited to, proliferative disorders, and cancer.
- said cancer is selected from pancreatic, prostate, or ovarian cancer.
- the invention provides a method for treating or lessening the severity of a p70S6K-mediated disease or condition in a patient comprising the step of administering to said patient a composition according to the present invention.
- p70S6K-mediated condition or “disease”, as used herein, means any disease or other deleterious condition in which p70S6K is known to play a role.
- the term “p70S6K-mediated condition” or “disease” also means those diseases or conditions that are alleviated by treatment with a p70S6K inhibitor.
- p70S6K-mediated diseases or conditions include, but are not limited to, proliferative disorders, such as cancer and tuberous sclerosis.
- the invention provides a method for treating or lessening the severity of a ROCK-mediated disease or condition in a patient comprising the step of administering to said patient a composition according to the present invention.
- ROCK -mediated condition or “disease”, as used herein, means any disease or other deleterious condition in which ROCK is known to play a role.
- ROCK -mediated condition or “disease” also means those diseases or conditions that are alleviated by treatment with a ROCK inhibitor. Such conditions include, without limitation, hypertension, angina pectoris, cerebrovascular contraction, asthma, peripheral circulation disorder, premature birth, cancer, arteriosclerosis, spasm, retinopathy, inflammatory disorders, autoimmune disorders, AIDS, and osteoporosis.
- the present invention relates to a method for treating or lessening the severity of a disease or condition selected from a proliferative disorder, a cardiac disorder, an inflammatory disorder, an autoimmune disorder, a viral disease, or a bone disorder, wherein said method comprises the step of administering an effective amount of a compound of the present invention.
- said method comprises the step of administering an effective amount of a preferred compound of the present invention.
- the present invention relates to a method for treating or lessening the severity of a disease or condition selected from cancer, rheumatoid arthritis, asthma, HIN, angina pectoris, peripheral circulation disorder, hypertension, arteriosclerosis, or osteoporosis.
- a disease or condition selected from cancer, rheumatoid arthritis, asthma, HIN, angina pectoris, peripheral circulation disorder, hypertension, arteriosclerosis, or osteoporosis.
- the present invention relates to a method for treating or lessening the severity of a cancer.
- the present invention relates to a method for treating or lessening the severity of a cancer selected from brain (gliomas), breast, colon, head and neck, kidney, lung, liver, melanoma, ovarian, pancreatic, prostate, sarcoma, or thyroid.
- a cancer selected from brain (gliomas), breast, colon, head and neck, kidney, lung, liver, melanoma, ovarian, pancreatic, prostate, sarcoma, or thyroid.
- the present invention relates to a method for treating or lessening the severity of pancreatic, prostate, or ovarian cancer..
- the methods of this invention that utilize compositions that do not contain an additional therapeutic agent comprise the additional step of separately administering to said patient an additional therapeutic agent. When these additional therapeutic agents are administered separately they may be administered to the patient prior to, sequentially with or following administration of the compositions of this invention.
- the compounds of this invention or pharmaceutical compositions thereof may also be incorporated into compositions for coating an implantable medical device, such as prostheses, artificial valves, vascular grafts, stents and catheters.
- an implantable medical device such as prostheses, artificial valves, vascular grafts, stents and catheters.
- Vascular stents for example, have been used to overcome restenosis (re-narrowing of the vessel wall after injury).
- patients using stents or other implantable devices risk clot formation or platelet activation. These unwanted effects may be prevented or mitigated by pre-coating the device with a pharmaceutically acceptable composition comprising a compound of this invention.
- Suitable coatings and the general preparation of coated implantable devices are described in US Patents 6,099,562; 5,886,026; and 5,304,121.
- the coatings are typically biocompatible polymeric materials such as a hydrogel polymer, polymethyldisiloxane, polycaprolactone, polyethylene glycol, polylactic acid, ethylene vinyl acetate, and mixtures thereof.
- the coatings may optionally be further covered by a suitable topcoat of fluorosilicone, polysaccarides, polyethylene glycol, phospholipids or combinations thereof to impart controlled release characteristics in the composition.
- Implantable devices coated with a compound of this invention are another embodiment of the present invention.
- R t (min) refers to the HPLC retention time, in minutes, associated with the compound. Unless otherwise indicated, the HPLC method utilized to obtain the reported retention time is as follows:
- Example 2 [00160] We have prepared other compounds of formula I by methods substantially similar to those described in Example 1. The characterization data for these compounds is summarized in Table 2 below and includes HPLC, LC/MS (observed) and 1H NMR data.
- 5-[2-(3-Chloro-phenyl)-2-methanesulfonyloxy-acetyIamino]-indazole-l-carboxylic acid tert-butyl ester 5-Amino-indazole-l -carboxylic acid tert-butyl ester was prepared following the procedure outlined by S.J. Brickner, WO9002744.
- Example 6 [00162] We have prepared other compounds of formula I by methods substantially similar to those described in Examples 1, 3, 4, and 5. The characterization data for these compounds is summarized in Table 3 below and includes HPLC, LC/MS (observed) and 1H NMR data.
- Example 9 We have prepared other compounds of formula I by methods substantially similar to those described in Examples 7 and 8. The characterization data for these compounds is summarized in Table 4 below and includes HPLC, LC/MS (observed) and 1H NMR data.
- 3-Amino-5-nitroindazole-l-carboxylic acid tert-butyl ester 600 mg, 2 mmol was dissolved in dry pyridine (15 mL) under nitrogen. The solution was cooled on an ice bath and 3-chlorobenzoyl chloride (0.3 mL, 2 mmol) added. After 6 hours, the mixture was diluted with EtOAc, washed with IM hydrochloric acid solution (x3) and brine then dried over magnesium sulfate and concentrated to a solid.
- 3-iodo-l-(2-methoxyethoxymethyl)-5-nitro-lff-indazole To a solution of 3-iodo-5- nitro-lH-indazole (lO.Og, 34.6 mmol) in T ⁇ F (50 ml) was added sodium bis(trimethylsilyl)amide (IM in T ⁇ F, 48.4 mmol, 48.4 ml) and the solution stirred at room temperature for 20 minutes. 2-methoxyethoxymethyl chloride (4.9g, 39.1 mmol, 4.5 ml) was added and the solution stirred at room temperature for 15 hours.
- iodoacetonitrile (3.5 mL, 49 mmol) was added rapidly to the reaction mixture.
- the reaction mixture was allowed to warm to 0°C and ammonium chloride solution added (10 mL, saturated aqueous).
- the reaction mixture was concentrated and the residue partitioned between EtOAc and brine.
- the aqueous phase was extracted with EtOAc and the combined organics were dried over magnesium sulfate then concentrated to afford a black oil.
- 2-(3,4-Dichloro-phenyI)-succinic acid 1-methyl ester Trifluoroacetic acid (100 ml) was added to a mixture of 2-(3,4-dichloro-phenyl)-succinic acid 4-tert-butyl ester 1- methyl ester (23.64 g, 0.071 mol) and dichloromethane (100 ml). The reaction mixture was stirred at room temperature for 3 hours then concentrated in vacuo. The crude mixture was kept under vacuo for several hours before being used without further purification in the next step. ).
- Example 40 We have prepared other compounds of formula I by methods substantially similar to those described in Examples 1 through 39.
- the characterization data for these compounds is summarized in Table 5 below and includes HPLC, LC/MS (observed) and 1H NMR data.
- Example 41 We have prepared other compounds of formula I by methods substantially similar to those described in Examples 1 through 39 and by the general synthetic Schemes I-XN.
- the characterization data for these compounds is summarized in Table 6 below and includes HPLC, LC/MS (observed), LR, and 1H ⁇ MR data.
- 1H ⁇ MR data is summarized in Table 6 below wherein 1H ⁇ MR data was obtained at 400 MHz in deuterated DMSO, unless otherwise indicated, and was found to be consistent with structure.
- Compound numbers correspond to the compound numbers listed in Table 1. Table 6. Characterization data for Selected Compounds of Formula I
- Example 42 [00170] We have prepared other compounds of formula I by methods substantially similar to those described in Examples 1 through 39 and by the general synthetic Schemes I-NII.
- the characterization data for these compounds is summarized in Table 7 below and includes HPLC, LC/MS (observed), IR, and 1H ⁇ MR data.
- 1H ⁇ MR data is summarized in Table 7 below wherein 1H ⁇ MR data was obtained at 400 MHz in deuterated DMSO, unless otherwise indicated, and was found to be consistent with structure.
- Compound numbers correspond to the compound numbers listed in Table 1.
- Example 43 AKT-3 Inhibition Assay [00172] Compounds were screened for their ability to inhibit AKT using a standard coupled enzyme assay (Fox et al., Protein Sci., (1998) 7, 2249). Assays were carried out in a mixture of 100 mM HEPES 7.5, 10 mM MgC12, 25 mM NaCI , 1 mM DTT and 3% DMSO. Final substrate concentrations in the assay were 170 ⁇ M ATP (Sigma Chemicals) and 200 ⁇ M peptide (RPRAATF, American Peptide, Sunnyvale, CA). Assays were carried out at 30 °C and 45 nM AKT.
- the plate was pre-incubated for about 10 minutes at 30°C and the reaction initiated by addition of 10 ⁇ l of enzyme (final concentration 45 nM) and 1 mM DTT. Rates of reaction were obtained using a Molecular Devices SpectraMax Plus plate reader over a 15 minute read time at 30°C. Compounds showing greater than 50% inhibition versus standard wells containing the assay mixture and DMSO without test compound were titrated to determine IC 50 values.
- Ki values between 1.0 and 10.0 ⁇ M for AKT-3 (Compound numbers correspond to the compound numbers listed in Table 1.): 1-5, 1-16, 1-35, 1-40, 1-43, 1-48 through 1-51, 1-53 through 1-56, 1-58, 1-63, 1-68, 1-71, 1-72, 1-76, 1-77, 1-78, 1-83, and 1-85, 1-107, 1-111, 1-113, 1-114, 1-123, 1-128, 1-137, 1-142, 1-150 through 1-152, 1-154, 1-156 through 1-159, 1-176, 1-191, 1-192, 1-235, 1-236, 1-241, 1-250, 1-255, 1-259, 1-1017, 1-1018, 1-1023, 1-1028, 1-1038, 1-1039, 1-1041, 1-1048, 1-1050 through 1-1052, 1-1055 and 1-1056.
- Example 44 PDK-1 Inhibition Assay Compounds were screened for their ability to inhibit PDK-1 using a radioactive-phosphate incorporation assay (Pitt and Lee, J. Biomol. Screen., (1996) 1, 47). Assays were carried out in a mixture of 100 mM HEPES (pH 7.5), 10 mM MgCl 2 , 25 mM NaCI , 2 mM DTT. Final substrate concentrations in the assay were 40 ⁇ M ATP (Sigma Chemicals) and 65 ⁇ M peptide (PDKtide, Upstate, Lake Placid, NY). Assays were carried out at 30 °C and 25 nM PDK-1 in the presence of -27.5 nCi/ ⁇ L of [ ⁇ -
- P] ATP (Amersham Pharmacia Biotech, Amersham, UK).
- An assay stock buffer solution was prepared containing all of the reagents listed above, with the exception of ATP, and the test compound of interest. 15 ⁇ l of the stock solution was placed in a 96 well plate followed by addition of 1 ⁇ l of 0.5 mM DMSO stock containing the test compound (final compound concentration 25 ⁇ M, final DMSO concentration 5%). The plate was preincubated for about 10 minutes at 30°C and the reaction initiated by addition of 4 ⁇ l ATP (final concentration 40 ⁇ M).
- Example 45 ROCK Inhibition Assay Compounds were screened for their ability to inhibit ROCK using a standard coupled enzyme assay (Fox et al (1998) Protein Sci 1, 2249). Reactions were carried out in 100 mM HEPES pH 7.5, 10 mM MgC12, 25 mM NaCI , 1 mM DTT and 1.5% DMSO. Final substrate concentrations in the assay were 13 ⁇ M ATP (Sigma chemicals) and 200 ⁇ M peptide (KKRNRTLSV, American Peptide, Sunnyvale, CA). Assays were carried out at 30 °C and 200 nM ROCK.
- An assay stock buffer solution was prepared containing all of the reagents listed above, with the exception of ROCK, DTT and the test compound of interest.
- 56 ⁇ l of the test reaction was placed in a 384 well plate followed by addition of 1 ⁇ l of 2 mM DMSO stock containing the test compound (final compound concentration 30 ⁇ M).
- the plate was preincubated for about 10 minutes at 30 °C and the reaction initiated by addition of 10 ⁇ l of enzyme (final concentration 100 nM). Rates of reaction were obtained using a BioRad Ultramark plate reader (Hercules, CA) over a 5 minute read time at 30°C.
- EXAMPLE 46 PKA Inhibition Assay Compounds were screened for their ability to inhibit PKA using a standard coupled enzyme assay (Fox et al., Protein Sci., (1998) 7, 2249). Assays were carried out in a mixture of 100 mM HEPES 7.5, 10 mM MgC12, 25 mM NaCI , 1 mM DTT and 3% DMSO. Final substrate concentrations in the assay were 50 ⁇ M ATP (Sigma Chemicals) and 80 ⁇ M peptide (Kemptide, American Peptide, Sunnyvale, CA). Assays were carried out at 30 °C and 18 nM PKA.
- An assay stock buffer solution was prepared containing all of the reagents listed above, with the exception of ATP, and the test compound of interest. 55 ⁇ l of the stock solution was placed in a 96 well plate followed by addition of 2 ⁇ l of DMSO stock containing serial dilutions of the test compound (typically starting from a final concentration of 5 ⁇ M). The plate was preincubated for 10 minutes at 30°C and the reaction initiated by addition of 5 ⁇ l of ATP (final concentration 50 ⁇ M). Initial reaction rates were determined with a Molecular Devices SpectraMax Plus plate reader over a 15 minute time course. IC50 and Ki data were calculated from non-linear regression analysis using the Prism software package (GraphPad Prism version 3.0a for Macintosh, GraphPad Software, San Diego California, USA).
- the spots were left to soak for at least 5 minutes, prior to wash steps (4 x 200 ⁇ L lOOmM phosphoric acid, 0.01% Tween- 20). After drying, 20 ⁇ L Optiphase 'SuperMix' liquid scintillation cocktail (Perkin Elmer) was added to the well prior to scintillation counting (1450 Microbeta Liquid Scintillation Counter, Wallac).
Abstract
Description



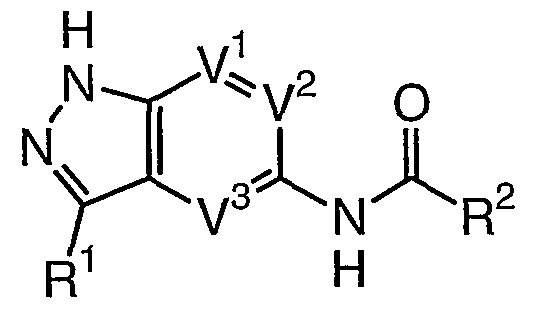



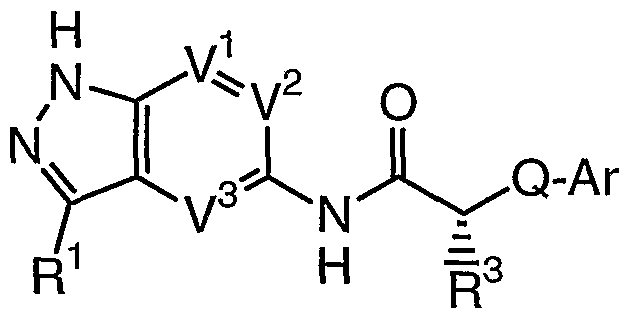




































































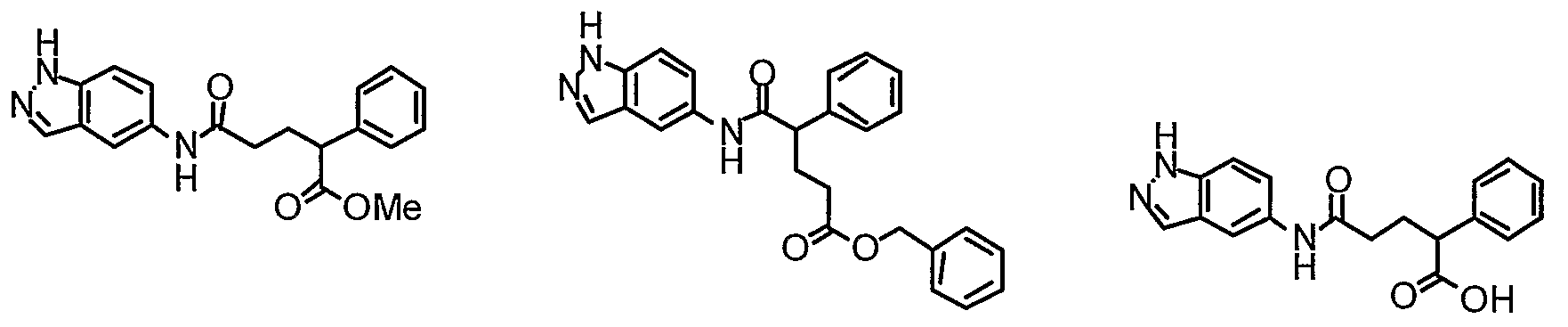




























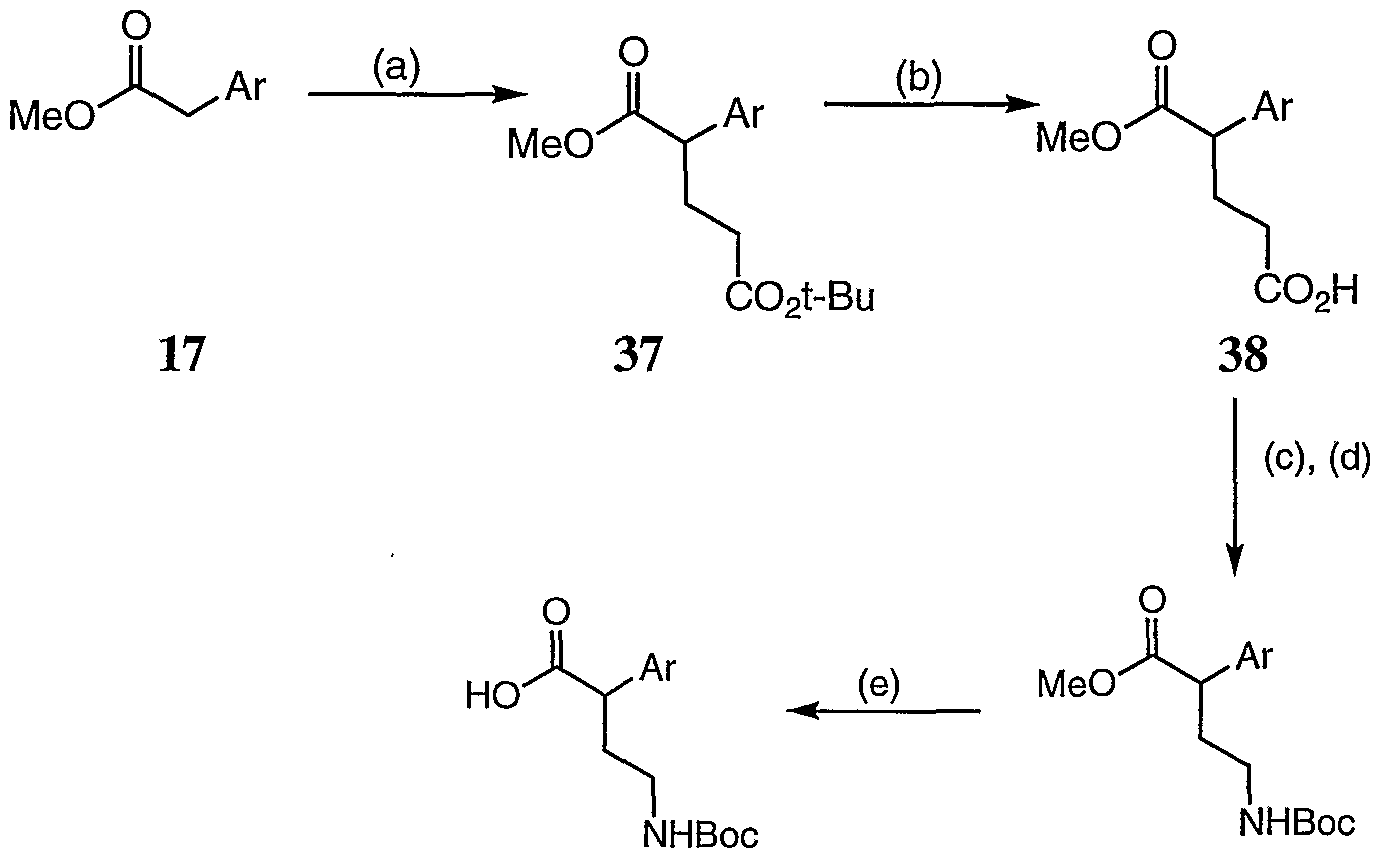
































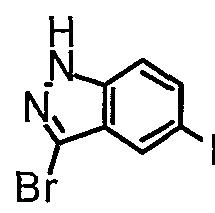










































Claims























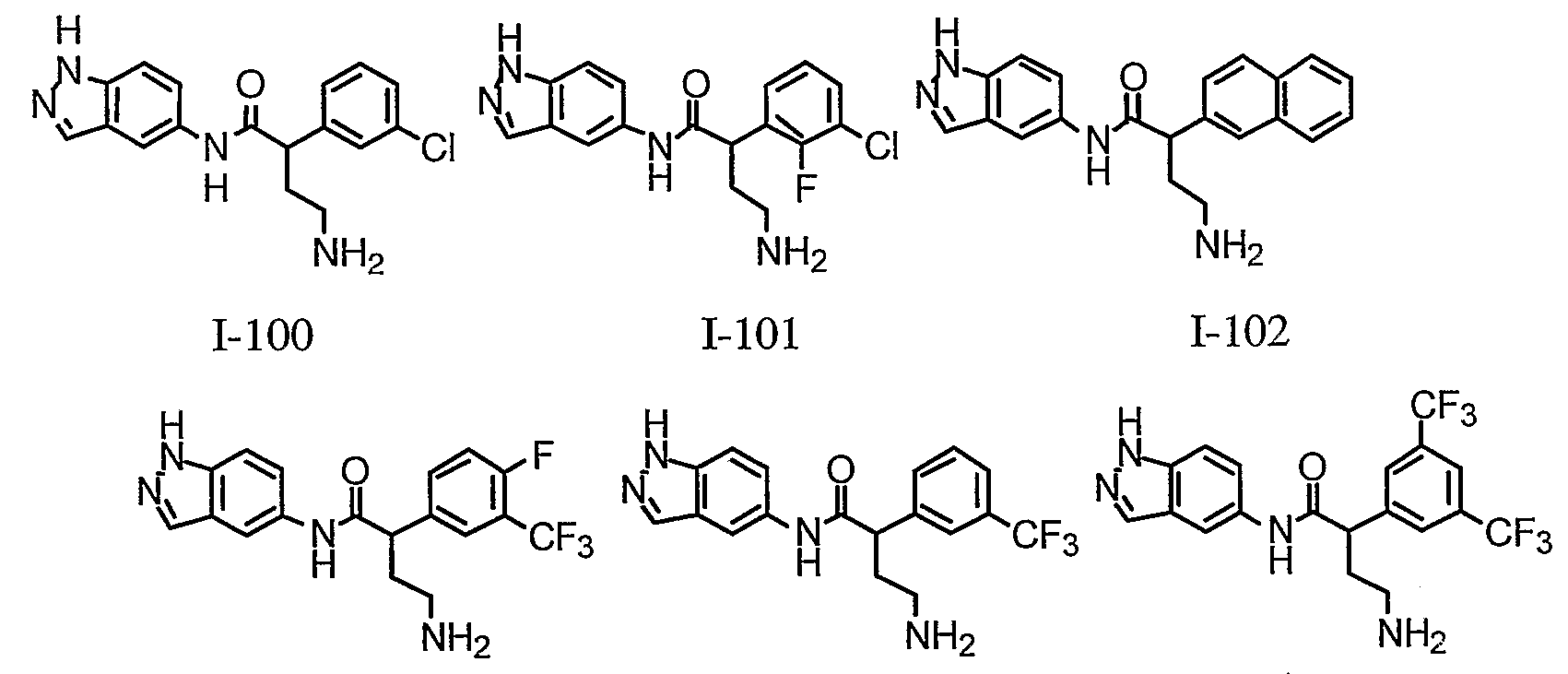










































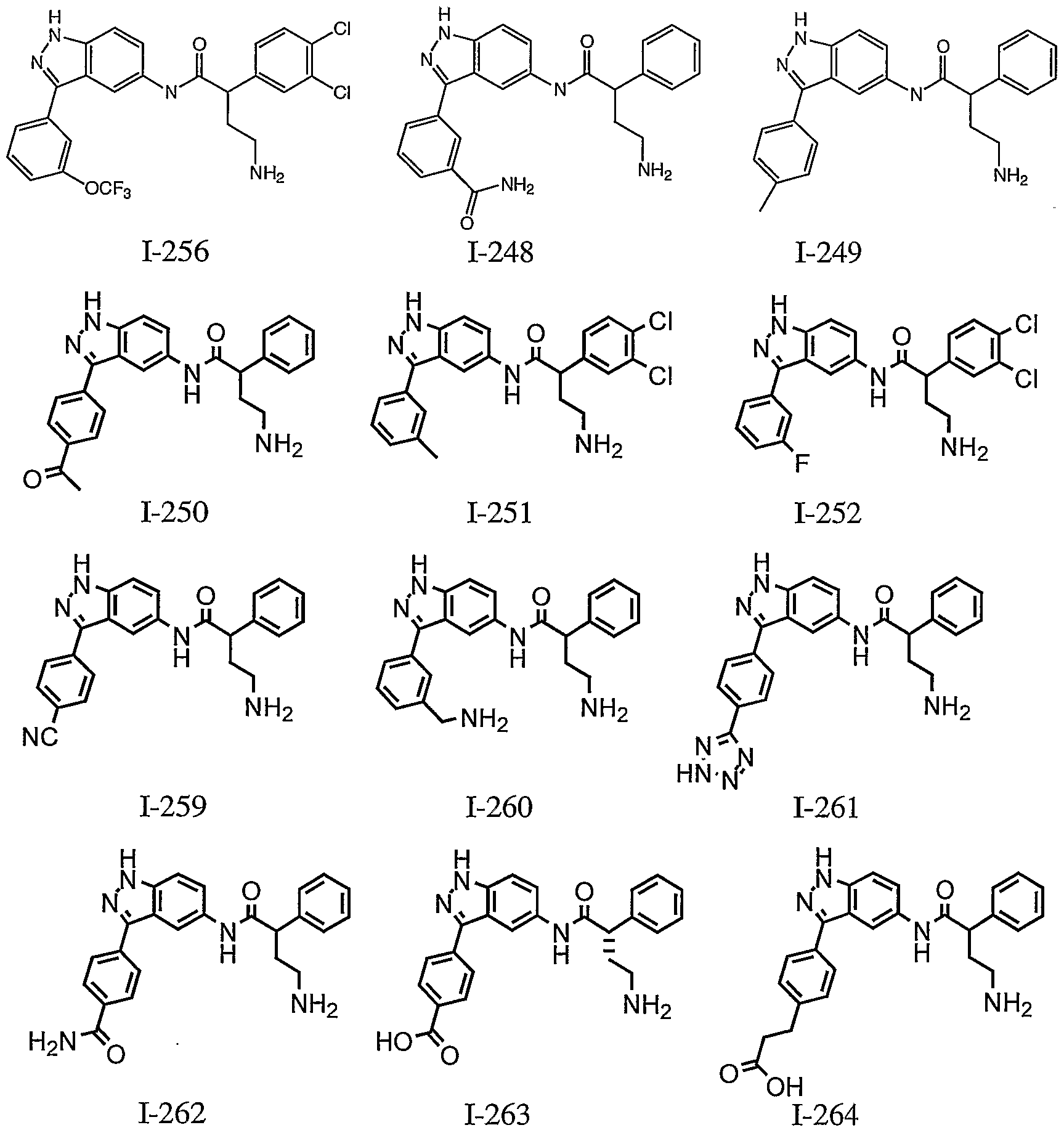














Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03708869A EP1467972A1 (en) | 2002-01-25 | 2003-01-23 | Indazole compounds useful as protein kinase inhibitors |
| AU2003212833A AU2003212833C1 (en) | 2002-01-25 | 2003-01-23 | Indazole compounds useful as protein kinase inhibitors |
| KR10-2004-7011500A KR20040078676A (en) | 2002-01-25 | 2003-01-23 | Indazole compounds useful as protein kinase inhibitors |
| MXPA04007126A MXPA04007126A (en) | 2002-01-25 | 2003-01-23 | Indazole compounds useful as protein kinase inhibitors. |
| CN038056127A CN1812973B (en) | 2002-01-25 | 2003-01-23 | Indazole compounds useful as protein kinase inhibitors |
| CA2473986A CA2473986C (en) | 2002-01-25 | 2003-01-23 | Indazole compounds useful as protein kinase inhibitors |
| JP2003564020A JP4718118B2 (en) | 2002-01-25 | 2003-01-23 | Indazole compounds useful as protein kinase inhibitors |
| IL163117A IL163117A (en) | 2002-01-25 | 2004-07-20 | Indazole derivatives and pharmaceutical compositions containing the same |
| NO20043531A NO328041B1 (en) | 2002-01-25 | 2004-08-24 | Indazole Compounds Useful as Protein Kinase Inhibitors, and Their Use for the Preparation of Medications |
| HK07101170.3A HK1096385A1 (en) | 2002-01-25 | 2007-02-01 | Indazole compounds useful as protein kinase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US35159702P | 2002-01-25 | 2002-01-25 | |
| US60/351,597 | 2002-01-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003064397A1 true WO2003064397A1 (en) | 2003-08-07 |
Family
ID=27663008
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2003/002096 WO2003064397A1 (en) | 2002-01-25 | 2003-01-23 | Indazole compounds useful as protein kinase inhibitors |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US7041687B2 (en) |
| EP (3) | EP2295413A1 (en) |
| JP (2) | JP4718118B2 (en) |
| KR (2) | KR20040078676A (en) |
| CN (1) | CN1812973B (en) |
| AR (1) | AR038202A1 (en) |
| AU (1) | AU2003212833C1 (en) |
| CA (1) | CA2473986C (en) |
| HK (1) | HK1096385A1 (en) |
| IL (1) | IL163117A (en) |
| MX (1) | MXPA04007126A (en) |
| NO (1) | NO328041B1 (en) |
| PL (1) | PL371512A1 (en) |
| RU (1) | RU2004125852A (en) |
| TW (1) | TW200306819A (en) |
| WO (1) | WO2003064397A1 (en) |
Cited By (99)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1487436A1 (en) * | 2002-03-08 | 2004-12-22 | Signal Pharmaceuticals, Inc. | Combination therapy for treating, preventing or managing proliferative disorders and cancers |
| WO2005014554A1 (en) * | 2003-08-08 | 2005-02-17 | Astex Therapeutics Limited | 1h-indazole-3-carboxamide compounds as mapkap kinase modulators |
| EP1512401A1 (en) * | 2003-09-03 | 2005-03-09 | Yung Shin Pharmaceutical Ind. Co., Ltd. | Enhancement of bone growth and inhibition of bone resorption |
| WO2005040116A2 (en) * | 2003-10-24 | 2005-05-06 | Schering Aktiengesellschaft | Indolinone derivatives and their use in treating disease-states such as cancer |
| FR2864084A1 (en) * | 2003-12-17 | 2005-06-24 | Aventis Pharma Sa | New organophosphorus derivatives of indazole, useful for treating e.g. cancer, are modulators of kinase activity, particularly of Tie2 and Aurora-2 |
| WO2005077912A1 (en) * | 2004-02-12 | 2005-08-25 | Mitsubishi Pharma Corporation | Indazole compound and pharmaceutical use thereof |
| WO2005123688A2 (en) * | 2004-06-15 | 2005-12-29 | Merck Patent Gmbh | 3-aminoindazoles |
| WO2006088088A1 (en) * | 2005-02-16 | 2006-08-24 | Astellas Pharma Inc. | Pain remedy containing rock inhibitor |
| WO2007058626A1 (en) * | 2005-11-16 | 2007-05-24 | S*Bio Pte Ltd | Indazole compounds |
| JP2007520463A (en) * | 2003-12-17 | 2007-07-26 | エスジーエックス ファーマシューティカルズ,インコーポレイティド | Bicyclic pyrazolo-condensed compounds as protein kinase modulators |
| JP2007523186A (en) * | 2004-02-20 | 2007-08-16 | スミスクライン・ビーチャム・コーポレイション | New compounds |
| WO2007117607A2 (en) | 2006-04-06 | 2007-10-18 | Novartis Ag | Quinazolines for pdk1 inhibition |
| WO2008009335A2 (en) * | 2006-07-18 | 2008-01-24 | Merck Patent Gmbh | Aminoindazole urea derivatives |
| JP2008508304A (en) * | 2004-07-27 | 2008-03-21 | エスジーエックス ファーマシューティカルズ、インコーポレイテッド | Fused ring heterocyclic kinase regulator |
| US7378532B2 (en) | 2004-03-26 | 2008-05-27 | Yung Shin Pharmaceutical Ind. Co., Ltd. | Fused pyrazolyl compound |
| WO2008086854A1 (en) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | 5-([1,3,4] oxadiazol-2-yl)-1h-indazol and 5-([1,3,4] thiadiazol-2-yl)-1h-indazol derivatives as sgk inhibitors for the treatment of diabetes |
| WO2008089307A2 (en) * | 2007-01-18 | 2008-07-24 | Lexicon Pharmaceuticals, Inc. | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| US7429609B2 (en) * | 2002-05-31 | 2008-09-30 | Eisai R & D Management Co., Ltd. | Pyrazole compound and medicinal composition containing the same |
| DE102007022565A1 (en) | 2007-05-14 | 2008-11-20 | Merck Patent Gmbh | Heterocyclic indazole derivatives |
| WO2009008992A2 (en) * | 2007-07-06 | 2009-01-15 | Osi Pharmaceuticals Inc. | Combination anti-cancer therapy comprising an inhibitor of both mtorc1 and mt0rc2 |
| WO2009147187A1 (en) * | 2008-06-05 | 2009-12-10 | Glaxo Group Limited | 4-carboxamide indazole derivatives useful as inhibitors of p13-kinases |
| EP2134175A1 (en) * | 2007-03-29 | 2009-12-23 | Smithkline Beecham Corporation | Inhibitors of akt activity |
| DE102008038221A1 (en) | 2008-08-18 | 2010-02-25 | Merck Patent Gmbh | 7-azaindole derivatives |
| WO2010020305A1 (en) | 2008-08-18 | 2010-02-25 | Merck Patent Gmbh | Oxadiazole derivatives for treating diabetes |
| DE102008038222A1 (en) | 2008-08-18 | 2010-02-25 | Merck Patent Gmbh | Indazol-5-carboxylic acid derivatives |
| US7795245B2 (en) | 2005-05-20 | 2010-09-14 | Atlantos Pharmaceuticals Holding, Inc. | Heterobicyclic metalloprotease inhibitors |
| WO2011017009A1 (en) | 2009-08-07 | 2011-02-10 | Merck Patent Gmbh | Novel azaheterocyclic compounds |
| US8003651B2 (en) | 2006-07-06 | 2011-08-23 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8063050B2 (en) | 2006-07-06 | 2011-11-22 | Array Biopharma Inc. | Hydroxylated and methoxylated pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| WO2012016001A1 (en) | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| WO2012013282A1 (en) | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides as inhibitors of the kinase p70s6k |
| WO2012069146A1 (en) | 2010-11-24 | 2012-05-31 | Merck Patent Gmbh | Quinazoline carboxamide azetidines |
| US8268994B2 (en) | 2004-07-27 | 2012-09-18 | Sgx Pharmaceuticals, Inc. | Fused ring heterocycle kinase modulators |
| US8329701B2 (en) | 2006-07-06 | 2012-12-11 | Array Biopharma Inc. | Dihydrofuro pyrimidines as AKT protein kinase inhibitors |
| US8377937B2 (en) | 2007-07-05 | 2013-02-19 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| WO2013040044A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| WO2013040059A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Novel imidazole amines as modulators of kinase activity |
| US8524751B2 (en) | 2009-03-09 | 2013-09-03 | GlaxoSmithKline Intellecutual Property Development | 4-oxadiazol-2-YL-indazoles as inhibitors of P13 kinases |
| US8536169B2 (en) | 2008-06-05 | 2013-09-17 | Glaxo Group Limited | Compounds |
| US8575162B2 (en) | 2009-04-30 | 2013-11-05 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8618097B2 (en) | 2007-07-05 | 2013-12-31 | Array Biopharma, Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8637532B2 (en) | 2009-02-11 | 2014-01-28 | Merck Patent Gmbh | Amino azaheterocyclic carboxamides |
| US8658635B2 (en) | 2008-06-05 | 2014-02-25 | Glaxosmithkline Intellectual Property Development Limited | Benzpyrazol derivatives as inhibitors of PI3 kinases |
| US8680114B2 (en) | 2003-11-21 | 2014-03-25 | Array Biopharma, Inc. | AKT protein kinase inhibitors |
| WO2014078637A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel heterocyclic derivatives as modulators of kinase activity |
| WO2014078634A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel imidazol-piperidinyl derivatives as modulators of kinase activity |
| WO2014085528A1 (en) | 2012-11-29 | 2014-06-05 | Merck Patent Gmbh | Azaquinazoline carboxamide derivatives |
| US8765743B2 (en) | 2008-06-05 | 2014-07-01 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8822524B2 (en) | 2006-12-07 | 2014-09-02 | University Of South Florida | Substrate-mimetic Akt inhibitor |
| US8835434B2 (en) | 2008-01-09 | 2014-09-16 | Array Biopharma, Inc. | Hydroxylated pyrimidyl cyclopentanes as akt protein kinase inhibitors |
| WO2014143612A1 (en) | 2013-03-11 | 2014-09-18 | Merck Patent Gmbh | 6-[4-(1 h-imidazol-2-yl)piperidin-1 -yl]pyrimidin-4-amine derivatives as modulators of kinase activity |
| US8846683B2 (en) | 2007-07-05 | 2014-09-30 | Array Biopharma, Inc. | Pyrimidyl cyclopentanes as Akt protein kinase inhibitors |
| US8846673B2 (en) | 2009-08-11 | 2014-09-30 | Bristol-Myers Squibb Company | Azaindazoles as kinase inhibitors and use thereof |
| US8853216B2 (en) | 2008-01-09 | 2014-10-07 | Array Biopharma, Inc. | Hydroxylated pyrimidyl cyclopentane as AKT protein kinase inhibitor |
| WO2015041533A1 (en) | 2013-09-20 | 2015-03-26 | Stichting Het Nederlands Kanker Instituut | Rock in combination with mapk-pathway |
| US8993576B2 (en) | 2010-10-27 | 2015-03-31 | Glaxo Group Limited | 6-(1H-indol-4-yl)-4-(5-{[4-1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1H-indazole hemi succinate salt, polymorphs and pharmaceutical compositions thereof |
| US8993574B2 (en) | 2008-04-24 | 2015-03-31 | F2G Ltd | Pyrrole antifungal agents |
| EP2766352A4 (en) * | 2011-10-12 | 2015-06-03 | Univ Health Network Uhn | Indazole compounds as kinase inhibitors and method of treating cancer with same |
| US9150549B2 (en) | 2011-04-01 | 2015-10-06 | Genentech, Inc. | Combinations of AKT inhibitor compounds and erlotinib, and methods of use |
| WO2015149909A1 (en) | 2014-04-03 | 2015-10-08 | Merck Patent Gmbh | Combinations of cancer therapeutics |
| EP2424857A4 (en) * | 2009-05-01 | 2015-11-11 | Aerie Pharmaceuticals Inc | Dual-action inhibitors and methods of using same |
| WO2015200920A1 (en) | 2014-06-27 | 2015-12-30 | The Regents Of The University Of California | Cultured mammalian limbal stem cells, methods for generating the same, and uses thereof |
| US9303040B2 (en) | 2006-07-06 | 2016-04-05 | Array Biopharma Inc. | Substituted piperazines as AKT inhibitors |
| US9365518B2 (en) | 2007-01-10 | 2016-06-14 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US9409886B2 (en) | 2007-07-05 | 2016-08-09 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US9415043B2 (en) | 2013-03-15 | 2016-08-16 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| WO2016161192A1 (en) | 2015-04-03 | 2016-10-06 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US9512101B2 (en) | 2008-07-25 | 2016-12-06 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| WO2017015152A1 (en) | 2015-07-17 | 2017-01-26 | Memorial Sloan-Kettering Cancer Center | Combination therapy using pdk1 and pi3k inhibitors |
| US9643927B1 (en) | 2015-11-17 | 2017-05-09 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| US9682082B2 (en) | 2011-04-01 | 2017-06-20 | Genentech, Inc. | Combinations of AKT and MEK inhibitor compounds, and methods of use |
| US10066201B2 (en) | 2015-09-11 | 2018-09-04 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US10080753B2 (en) | 2011-12-22 | 2018-09-25 | Merck Patent Gmbh | Heterocyclic carboxamides as modulators of kinase activity |
| US10100285B2 (en) | 2015-04-03 | 2018-10-16 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| WO2018194885A1 (en) * | 2017-04-18 | 2018-10-25 | Eli Lilly And Company | Phenyl-2-hydroxy-acetylamino-2-methyl-phenyl compounds |
| WO2018234354A1 (en) * | 2017-06-20 | 2018-12-27 | Grünenthal GmbH | Novel substituted 3-indole and 3-indazole compounds as phosphodiesterase inhibitors |
| US10201524B2 (en) | 2014-11-21 | 2019-02-12 | F2G Limited | Antifungal agents |
| WO2019161162A1 (en) * | 2018-02-16 | 2019-08-22 | Constellation Pharmaceuticals, Inc. | P300/cbp hat inhibitors |
| EP3553168A1 (en) | 2010-11-12 | 2019-10-16 | Georgetown University | Immortalization of epithelial cells and methods of use |
| EP3165523B1 (en) * | 2014-07-01 | 2020-01-08 | Gwangju Institute of Science and Technology | Composition for inducing cell reprogramming |
| US10550087B2 (en) | 2015-11-17 | 2020-02-04 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| WO2020041065A1 (en) | 2018-08-20 | 2020-02-27 | Propagenix Inc. | Epithelial cell spheroids |
| WO2020047229A1 (en) | 2018-08-29 | 2020-03-05 | University Of Massachusetts | Inhibition of protein kinases to treat friedreich ataxia |
| US10624882B2 (en) | 2006-09-20 | 2020-04-21 | Aerie Pharmaceuticals, Inc. | Rho kinase inhibitors |
| WO2020084580A1 (en) | 2018-10-26 | 2020-04-30 | Novartis Ag | Methods and compositions for ocular cell therapy |
| US10858339B2 (en) | 2017-03-31 | 2020-12-08 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| WO2021021893A1 (en) * | 2019-07-29 | 2021-02-04 | Constellation Pharmaceuticals, Inc. | Compounds for use in treating neurological disorders |
| US10973821B2 (en) | 2016-05-25 | 2021-04-13 | F2G Limited | Pharmaceutical formulation |
| WO2021220132A1 (en) | 2020-04-27 | 2021-11-04 | Novartis Ag | Methods and compositions for ocular cell therapy |
| EP3901156A4 (en) * | 2020-02-21 | 2022-03-30 | Vivavision (Shanghai) Ltd | Nitrooxyderivative of rock kinase inhibitor |
| WO2022101459A1 (en) | 2020-11-16 | 2022-05-19 | Merck Patent Gmbh | Kinase inhibitor combinations for cancer treatment |
| US11389441B2 (en) | 2016-08-31 | 2022-07-19 | Aerie Pharmaceuticals, Inc. | Ophthalmic compositions |
| US11414384B2 (en) | 2018-02-16 | 2022-08-16 | Constellation Pharmaceuticals, Inc. | P300/CBP hat inhibitors |
| US11427563B2 (en) | 2018-09-14 | 2022-08-30 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US11479533B2 (en) | 2017-07-21 | 2022-10-25 | Kadmon Corporation, Llc | Inhibitors of Rho associated coiled-coil containing protein kinase |
| WO2023067394A2 (en) | 2021-10-22 | 2023-04-27 | Evia Life Sciences Inc. | Methods for making extracellular vesicles, and compositions and methods of use thereof |
| WO2023069949A1 (en) | 2021-10-18 | 2023-04-27 | Evia Life Sciences Inc. | Compositions and methods of use thereof for treating liver fibrosis |
| RU2804280C2 (en) * | 2017-04-18 | 2023-09-26 | Эли Лилли Энд Компани | Phenyl-2-hydroxy-acetylamino-2-methyl-phenyl compounds |
| US11819503B2 (en) | 2019-04-23 | 2023-11-21 | F2G Ltd | Method of treating coccidioides infection |
Families Citing this family (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050070567A1 (en) * | 2002-08-12 | 2005-03-31 | The Regents Of The University Of Michigan | Diagnosis and treatment of diseases arising from defects in the tuberous sclerosis pathway |
| JP2005535332A (en) * | 2002-08-12 | 2005-11-24 | ザ リージェンツ オブ ザ ユニバーシティ オブ ミシガン | Diagnosis and treatment of tuberous sclerosis |
| TW200523262A (en) * | 2003-07-29 | 2005-07-16 | Smithkline Beecham Corp | Inhibitors of AKT activity |
| CA2539549A1 (en) | 2003-09-23 | 2005-04-07 | Vertex Pharmaceuticals Incorporated | Pyrazolopyrrole derivatives as protein kinase inhibitors |
| US7625890B2 (en) | 2005-11-10 | 2009-12-01 | Smithkline Beecham Corp. | Substituted imidazo[4,5-c]pyridine compounds as Akt inhibitors |
| EP2385053B1 (en) | 2005-11-17 | 2013-10-02 | OSI Pharmaceuticals, Inc. | Intermediates for the preparation of fused bicyclic mTOR inhibitors |
| AR060316A1 (en) | 2006-01-17 | 2008-06-11 | Vertex Pharma | UTILITY AZAINDOLS AS INHIBITORS OF JANUS QUINASAS |
| GB0606429D0 (en) * | 2006-03-30 | 2006-05-10 | Novartis Ag | Organic compounds |
| US20070286864A1 (en) * | 2006-06-09 | 2007-12-13 | Buck Elizabeth A | Combined treatment with an EGFR kinase inhibitor and an agent that sensitizes tumor cells to the effects of EGFR kinase inhibitors |
| UA95641C2 (en) * | 2006-07-06 | 2011-08-25 | Эррей Биофарма Инк. | Hydroxylated cyclopenta [d] pyrimidines as akt protein kinase inhibitors |
| US20080207671A1 (en) * | 2006-07-31 | 2008-08-28 | The Regents Of The University Of Michigan | Diagnosis and treatment of diseases arising from defects in the tuberous sclerosis pathway |
| WO2008061109A2 (en) * | 2006-11-15 | 2008-05-22 | Forest Laboratories Holdings Limited | Indazole derivatives useful as melanin concentrating receptor ligands |
| WO2008079346A1 (en) * | 2006-12-21 | 2008-07-03 | Vertex Pharmaceuticals Incorporated | 5-cyan0-4- (pyrrolo [2, 3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| US20080188461A1 (en) * | 2007-02-01 | 2008-08-07 | Regents Of The University Of Michigan | Compositions and methods for detecting, preventing and treating seizures and seizure related disorders |
| UY30892A1 (en) | 2007-02-07 | 2008-09-02 | Smithkline Beckman Corp | AKT ACTIVITY INHIBITORS |
| JP5323834B2 (en) * | 2007-08-17 | 2013-10-23 | エルジー・ライフ・サイエンシーズ・リミテッド | Indole compounds as cell necrosis inhibitors |
| MY155320A (en) | 2007-10-05 | 2015-09-30 | Acucela Inc | Alkoxy compounds for disease treatment |
| EP2262498A2 (en) * | 2008-03-10 | 2010-12-22 | Vertex Pharmceuticals Incorporated | Pyrimidines and pyridines useful as inhibitors of protein kinases |
| US8158656B2 (en) * | 2008-05-16 | 2012-04-17 | Shenzhen Chipscreen Biosciences Ltd. | 2-indolinone derivatives as multi-target protein kinase inhibitors and histone deacetylase inhibitors |
| US20110129455A1 (en) * | 2008-06-26 | 2011-06-02 | Hong Lin | Inhibitors of akt activity |
| WO2010017047A1 (en) * | 2008-08-05 | 2010-02-11 | Merck & Co., Inc. | Therapeutic compounds |
| JP5743897B2 (en) * | 2008-11-20 | 2015-07-01 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニーGlaxoSmithKline LLC | Compound |
| SG172926A1 (en) * | 2009-01-30 | 2011-08-29 | Glaxosmithkline Llc | Crystalline n-{(1-s)-2-amino-1-[(3-fluorophenyl)methyl]ethyl}-5-chloro-4-(4-chloro-1-methyl-1h-pyrazol-5-yl)-2-thiophenecarboxamide hydrochloride |
| WO2010120854A1 (en) * | 2009-04-15 | 2010-10-21 | Glaxosmithkline Llc | Chemical compounds |
| US8211901B2 (en) | 2009-05-22 | 2012-07-03 | Shenzhen Chipscreen Biosciences Ltd. | Naphthamide derivatives as multi-target protein kinase inhibitors and histone deacetylase inhibitors |
| CN101906076B (en) | 2009-06-04 | 2013-03-13 | 深圳微芯生物科技有限责任公司 | Naphthaline amide derivative serving as protein kinase inhibitor and histone deacetylase inhibitor and preparation method and application thereof |
| PE20120508A1 (en) | 2009-06-17 | 2012-05-09 | Vertex Pharma | INHIBITORS OF THE REPLICATION OF FLU VIRUSES |
| US8487102B2 (en) * | 2010-04-20 | 2013-07-16 | Hoffmann-La Roche Inc. | Pyrrazolopyridine compounds as dual NK1/NK3 receptor antagonists |
| WO2012033525A2 (en) * | 2010-09-08 | 2012-03-15 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | P53-mdm2 antagonists |
| CN103492381A (en) | 2010-12-16 | 2014-01-01 | 沃泰克斯药物股份有限公司 | Inhibitors of influenza viruses replication |
| UA118010C2 (en) | 2011-08-01 | 2018-11-12 | Вертекс Фармасьютікалз Інкорпорейтед | INFLUENCES OF INFLUENZA VIRUS REPLICATION |
| WO2013104829A1 (en) * | 2012-01-13 | 2013-07-18 | Medeia Therapeutics Ltd | Novel arylamide derivatives having antiandrogenic properties |
| US8980934B2 (en) | 2012-06-22 | 2015-03-17 | University Health Network | Kinase inhibitors and method of treating cancer with same |
| WO2014056083A1 (en) * | 2012-10-12 | 2014-04-17 | University Health Network | Kinase inhibitors and method of treating cancer with same |
| CN112472699A (en) | 2013-07-26 | 2021-03-12 | 种族肿瘤学公司 | Combination methods for improving the therapeutic benefit of bisantrene and derivatives |
| CN103570624B (en) * | 2013-08-06 | 2016-07-06 | 安徽世华化工有限公司 | The synthesis technique of 3-bromo-5-nitro-1H-indazole |
| SG10201804021TA (en) | 2013-11-13 | 2018-07-30 | Vertex Pharma | Methods of preparing inhibitors of influenza viruses replication |
| EP3068776B1 (en) | 2013-11-13 | 2019-05-29 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| WO2016183116A1 (en) | 2015-05-13 | 2016-11-17 | Vertex Pharmaceuticals Incorporated | Methods of preparing inhibitors of influenza viruses replication |
| JP6857617B2 (en) | 2015-05-13 | 2021-04-14 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Influenza virus replication inhibitor |
| GB201511382D0 (en) | 2015-06-29 | 2015-08-12 | Imp Innovations Ltd | Novel compounds and their use in therapy |
| EP3652164B1 (en) * | 2017-07-12 | 2023-06-21 | Bristol-Myers Squibb Company | Phenylacetamides as inhibitors of rock |
| WO2019018853A1 (en) * | 2017-07-21 | 2019-01-24 | Kadmon Corporation, Llc | Inhibitors of rho associated coiled-coil containing protein kinase |
| TWI777325B (en) * | 2019-12-31 | 2022-09-11 | 財團法人工業技術研究院 | Β-amino acid derivative, kinase inhibitor and pharmaceutical composition containing the same, and use of the same |
| CN114605329B (en) * | 2022-03-28 | 2024-01-26 | 河南中医药大学 | Substituted indazole carboxamides or substituted azaindazole carboxamides FLT3 inhibitors and uses thereof |
| CN116891460A (en) * | 2023-07-12 | 2023-10-17 | 浙江大学 | Indazole derivative or pharmaceutically acceptable salt thereof and application thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001056988A1 (en) * | 2000-02-01 | 2001-08-09 | Kirin Beer Kabushiki Kaisha | Nitrogen-containing compounds having kinase inhibitory activity and drugs containing the same |
| WO2001085719A1 (en) * | 2000-05-09 | 2001-11-15 | Schering Aktiengesellschaft | Ortho-substituted anthranilic acid amides and their use as medicaments |
| US20020103229A1 (en) * | 2000-07-31 | 2002-08-01 | Bhagwat Shripad S. | Indazole derivatives as JNK inhibitors and compositions and methods related thereto |
| WO2002083648A1 (en) * | 2001-04-16 | 2002-10-24 | Eisai Co., Ltd. | Novel 1h-indazole compound |
| WO2002100833A1 (en) * | 2001-06-12 | 2002-12-19 | Sumitomo Pharmaceuticals Company, Limited | Rho KINASE INHIBITORS |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5640715B2 (en) | 1974-04-01 | 1981-09-22 | ||
| ES2157934T3 (en) | 1988-09-15 | 2001-09-01 | Upjohn Co | FENIL-5-BETA-AMIDOMETILOXAZOLIDIN-2-WAVES REPLACED WITH NITROGEN IN POSITION 3. |
| US5304121A (en) | 1990-12-28 | 1994-04-19 | Boston Scientific Corporation | Drug delivery system making use of a hydrogel polymer coating |
| US5716981A (en) | 1993-07-19 | 1998-02-10 | Angiogenesis Technologies, Inc. | Anti-angiogenic compositions and methods of use |
| US6099562A (en) | 1996-06-13 | 2000-08-08 | Schneider (Usa) Inc. | Drug coating with topcoat |
| US6638980B1 (en) | 1999-05-24 | 2003-10-28 | Millennium Pharmaceuticals, Inc. | Inhibitors of factor Xa |
| AU5414000A (en) | 1999-06-14 | 2001-01-02 | Eli Lilly And Company | Compounds |
-
2003
- 2003-01-07 TW TW092100235A patent/TW200306819A/en unknown
- 2003-01-21 AR ARP030100171A patent/AR038202A1/en unknown
- 2003-01-23 KR KR10-2004-7011500A patent/KR20040078676A/en active IP Right Grant
- 2003-01-23 EP EP10181668A patent/EP2295413A1/en not_active Withdrawn
- 2003-01-23 RU RU2004125852/04A patent/RU2004125852A/en not_active Application Discontinuation
- 2003-01-23 WO PCT/US2003/002096 patent/WO2003064397A1/en active Application Filing
- 2003-01-23 CN CN038056127A patent/CN1812973B/en not_active Expired - Fee Related
- 2003-01-23 EP EP03708869A patent/EP1467972A1/en not_active Withdrawn
- 2003-01-23 EP EP10181562A patent/EP2295412A1/en not_active Withdrawn
- 2003-01-23 KR KR1020107022397A patent/KR20100119902A/en not_active Application Discontinuation
- 2003-01-23 MX MXPA04007126A patent/MXPA04007126A/en active IP Right Grant
- 2003-01-23 CA CA2473986A patent/CA2473986C/en not_active Expired - Fee Related
- 2003-01-23 JP JP2003564020A patent/JP4718118B2/en not_active Expired - Fee Related
- 2003-01-23 PL PL03371512A patent/PL371512A1/en not_active Application Discontinuation
- 2003-01-23 AU AU2003212833A patent/AU2003212833C1/en not_active Ceased
- 2003-01-23 US US10/350,806 patent/US7041687B2/en not_active Expired - Fee Related
-
2004
- 2004-07-20 IL IL163117A patent/IL163117A/en not_active IP Right Cessation
- 2004-08-24 NO NO20043531A patent/NO328041B1/en not_active IP Right Cessation
-
2006
- 2006-01-06 JP JP2006001918A patent/JP2006176530A/en active Pending
-
2007
- 2007-02-01 HK HK07101170.3A patent/HK1096385A1/en not_active IP Right Cessation
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001056988A1 (en) * | 2000-02-01 | 2001-08-09 | Kirin Beer Kabushiki Kaisha | Nitrogen-containing compounds having kinase inhibitory activity and drugs containing the same |
| EP1256574A1 (en) * | 2000-02-01 | 2002-11-13 | Kirin Beer Kabushiki Kaisha | Nitrogen-containing compounds having kinase inhibitory activity and drugs containing the same |
| WO2001085719A1 (en) * | 2000-05-09 | 2001-11-15 | Schering Aktiengesellschaft | Ortho-substituted anthranilic acid amides and their use as medicaments |
| US20020103229A1 (en) * | 2000-07-31 | 2002-08-01 | Bhagwat Shripad S. | Indazole derivatives as JNK inhibitors and compositions and methods related thereto |
| WO2002083648A1 (en) * | 2001-04-16 | 2002-10-24 | Eisai Co., Ltd. | Novel 1h-indazole compound |
| WO2002100833A1 (en) * | 2001-06-12 | 2002-12-19 | Sumitomo Pharmaceuticals Company, Limited | Rho KINASE INHIBITORS |
Non-Patent Citations (1)
| Title |
|---|
| NICOLAIDES, ERNEST D. ET AL: "Potential antiviral agents. Carbobenzoxy di- and tripeptides active against measles and herpes viruses", JOURNAL OF MEDICINAL CHEMISTRY (1968), 11(1), 74-9, XP002243849 * |
Cited By (208)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1487436A4 (en) * | 2002-03-08 | 2009-06-03 | Signal Pharm Inc | Combination therapy for treating, preventing or managing proliferative disorders and cancers |
| EP1487436A1 (en) * | 2002-03-08 | 2004-12-22 | Signal Pharmaceuticals, Inc. | Combination therapy for treating, preventing or managing proliferative disorders and cancers |
| US7429609B2 (en) * | 2002-05-31 | 2008-09-30 | Eisai R & D Management Co., Ltd. | Pyrazole compound and medicinal composition containing the same |
| WO2005014554A1 (en) * | 2003-08-08 | 2005-02-17 | Astex Therapeutics Limited | 1h-indazole-3-carboxamide compounds as mapkap kinase modulators |
| EP1512401A1 (en) * | 2003-09-03 | 2005-03-09 | Yung Shin Pharmaceutical Ind. Co., Ltd. | Enhancement of bone growth and inhibition of bone resorption |
| US7160916B2 (en) | 2003-09-03 | 2007-01-09 | Yung Shin Pharmaceutical Ind. Co., Ltd. | Method of increasing bone density or treating osteoporosis |
| WO2005040116A2 (en) * | 2003-10-24 | 2005-05-06 | Schering Aktiengesellschaft | Indolinone derivatives and their use in treating disease-states such as cancer |
| US7105563B2 (en) | 2003-10-24 | 2006-09-12 | Schering Aktiengesellschaft | Indolinone derivatives and their use in treating disease-states such as cancer |
| WO2005040116A3 (en) * | 2003-10-24 | 2005-06-16 | Schering Ag | Indolinone derivatives and their use in treating disease-states such as cancer |
| US8680114B2 (en) | 2003-11-21 | 2014-03-25 | Array Biopharma, Inc. | AKT protein kinase inhibitors |
| US7491710B2 (en) | 2003-12-17 | 2009-02-17 | Aventis Pharma S.A. | Organophosphorus derivatives of indazoles and use thereof as medicinal products |
| WO2005058923A1 (en) * | 2003-12-17 | 2005-06-30 | Aventis Pharma S.A. | Organophosphorous indazole derivatives and use thereof as protein kinase inhibitors |
| FR2864084A1 (en) * | 2003-12-17 | 2005-06-24 | Aventis Pharma Sa | New organophosphorus derivatives of indazole, useful for treating e.g. cancer, are modulators of kinase activity, particularly of Tie2 and Aurora-2 |
| JP4728965B2 (en) * | 2003-12-17 | 2011-07-20 | エスジーエックス ファーマシューティカルズ、インコーポレイテッド | Bicyclic pyrazolo-condensed compounds as protein kinase modulators |
| JP2007520463A (en) * | 2003-12-17 | 2007-07-26 | エスジーエックス ファーマシューティカルズ,インコーポレイティド | Bicyclic pyrazolo-condensed compounds as protein kinase modulators |
| WO2005077912A1 (en) * | 2004-02-12 | 2005-08-25 | Mitsubishi Pharma Corporation | Indazole compound and pharmaceutical use thereof |
| US7994196B2 (en) | 2004-02-12 | 2011-08-09 | Mitsubishi Tanabe Pharma Corporation | Indazole compound and pharmaceutical use thereof |
| JPWO2005077912A1 (en) * | 2004-02-12 | 2007-10-18 | 三菱ウェルファーマ株式会社 | Indazole compounds and their pharmaceutical use |
| JP4734119B2 (en) * | 2004-02-12 | 2011-07-27 | 田辺三菱製薬株式会社 | Indazole compounds and their pharmaceutical uses |
| JP2007523186A (en) * | 2004-02-20 | 2007-08-16 | スミスクライン・ビーチャム・コーポレイション | New compounds |
| JP4857128B2 (en) * | 2004-02-20 | 2012-01-18 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | New compounds |
| US7378532B2 (en) | 2004-03-26 | 2008-05-27 | Yung Shin Pharmaceutical Ind. Co., Ltd. | Fused pyrazolyl compound |
| WO2005123688A3 (en) * | 2004-06-15 | 2006-02-23 | Merck Patent Gmbh | 3-aminoindazoles |
| WO2005123688A2 (en) * | 2004-06-15 | 2005-12-29 | Merck Patent Gmbh | 3-aminoindazoles |
| JP2008502610A (en) * | 2004-06-15 | 2008-01-31 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフトング | 3-aminoindazole |
| US7872039B2 (en) * | 2004-06-15 | 2011-01-18 | Merck Patent Gesellschaft | 3-aminoindazoles |
| AU2005254617B2 (en) * | 2004-06-15 | 2011-05-26 | Merck Patent Gmbh | 3-aminoindazoles |
| JP2008508304A (en) * | 2004-07-27 | 2008-03-21 | エスジーエックス ファーマシューティカルズ、インコーポレイテッド | Fused ring heterocyclic kinase regulator |
| US8268994B2 (en) | 2004-07-27 | 2012-09-18 | Sgx Pharmaceuticals, Inc. | Fused ring heterocycle kinase modulators |
| WO2006088088A1 (en) * | 2005-02-16 | 2006-08-24 | Astellas Pharma Inc. | Pain remedy containing rock inhibitor |
| US8835441B2 (en) | 2005-05-20 | 2014-09-16 | Amgen Inc. | Heterobicyclic metalloprotease inhibitors |
| US7795245B2 (en) | 2005-05-20 | 2010-09-14 | Atlantos Pharmaceuticals Holding, Inc. | Heterobicyclic metalloprotease inhibitors |
| WO2007058626A1 (en) * | 2005-11-16 | 2007-05-24 | S*Bio Pte Ltd | Indazole compounds |
| WO2007117607A2 (en) | 2006-04-06 | 2007-10-18 | Novartis Ag | Quinazolines for pdk1 inhibition |
| WO2007117607A3 (en) * | 2006-04-06 | 2007-12-21 | Novartis Vaccines & Diagnostic | Quinazolines for pdk1 inhibition |
| US7932262B2 (en) | 2006-04-06 | 2011-04-26 | Novartis Ag | Quinazolines for PDK1 inhibition |
| US9303040B2 (en) | 2006-07-06 | 2016-04-05 | Array Biopharma Inc. | Substituted piperazines as AKT inhibitors |
| US8329701B2 (en) | 2006-07-06 | 2012-12-11 | Array Biopharma Inc. | Dihydrofuro pyrimidines as AKT protein kinase inhibitors |
| US8853199B2 (en) | 2006-07-06 | 2014-10-07 | Array Biopharma, Inc. | Hydroxylated and methoxylated pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8846681B2 (en) | 2006-07-06 | 2014-09-30 | Array Biopharma, Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US9359340B2 (en) | 2006-07-06 | 2016-06-07 | Array Biopharma Inc. | Hydroxylated and methoxylated pyrimidyl cyclopentanes as Akt protein kinase inhibitors |
| US8063050B2 (en) | 2006-07-06 | 2011-11-22 | Array Biopharma Inc. | Hydroxylated and methoxylated pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8003651B2 (en) | 2006-07-06 | 2011-08-23 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| WO2008009335A3 (en) * | 2006-07-18 | 2008-02-28 | Merck Patent Gmbh | Aminoindazole urea derivatives |
| US8207210B2 (en) | 2006-07-18 | 2012-06-26 | Merck Patent Gmbh | Aminoindazolylurea derivatives |
| WO2008009335A2 (en) * | 2006-07-18 | 2008-01-24 | Merck Patent Gmbh | Aminoindazole urea derivatives |
| US10624882B2 (en) | 2006-09-20 | 2020-04-21 | Aerie Pharmaceuticals, Inc. | Rho kinase inhibitors |
| US8822524B2 (en) | 2006-12-07 | 2014-09-02 | University Of South Florida | Substrate-mimetic Akt inhibitor |
| US9365518B2 (en) | 2007-01-10 | 2016-06-14 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US10472327B2 (en) | 2007-01-10 | 2019-11-12 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US9890123B2 (en) | 2007-01-10 | 2018-02-13 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US10899714B2 (en) | 2007-01-10 | 2021-01-26 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| WO2008089307A3 (en) * | 2007-01-18 | 2008-11-06 | Lexicon Pharmaceuticals Inc | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| WO2008086854A1 (en) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | 5-([1,3,4] oxadiazol-2-yl)-1h-indazol and 5-([1,3,4] thiadiazol-2-yl)-1h-indazol derivatives as sgk inhibitors for the treatment of diabetes |
| WO2008089307A2 (en) * | 2007-01-18 | 2008-07-24 | Lexicon Pharmaceuticals, Inc. | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| DE102007002717A1 (en) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | Heterocyclic indazole derivatives |
| EP2134175A4 (en) * | 2007-03-29 | 2012-01-11 | Glaxosmithkline Llc | Inhibitors of akt activity |
| EP2134175A1 (en) * | 2007-03-29 | 2009-12-23 | Smithkline Beecham Corporation | Inhibitors of akt activity |
| DE102007022565A1 (en) | 2007-05-14 | 2008-11-20 | Merck Patent Gmbh | Heterocyclic indazole derivatives |
| US9409886B2 (en) | 2007-07-05 | 2016-08-09 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8377937B2 (en) | 2007-07-05 | 2013-02-19 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8618097B2 (en) | 2007-07-05 | 2013-12-31 | Array Biopharma, Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8846683B2 (en) | 2007-07-05 | 2014-09-30 | Array Biopharma, Inc. | Pyrimidyl cyclopentanes as Akt protein kinase inhibitors |
| WO2009008992A3 (en) * | 2007-07-06 | 2009-02-26 | Osi Pharm Inc | Combination anti-cancer therapy comprising an inhibitor of both mtorc1 and mt0rc2 |
| WO2009008992A2 (en) * | 2007-07-06 | 2009-01-15 | Osi Pharmaceuticals Inc. | Combination anti-cancer therapy comprising an inhibitor of both mtorc1 and mt0rc2 |
| US8835434B2 (en) | 2008-01-09 | 2014-09-16 | Array Biopharma, Inc. | Hydroxylated pyrimidyl cyclopentanes as akt protein kinase inhibitors |
| US8853216B2 (en) | 2008-01-09 | 2014-10-07 | Array Biopharma, Inc. | Hydroxylated pyrimidyl cyclopentane as AKT protein kinase inhibitor |
| US9452168B2 (en) | 2008-04-24 | 2016-09-27 | F2G Ltd | Pyrrole antifungal agents |
| US8993574B2 (en) | 2008-04-24 | 2015-03-31 | F2G Ltd | Pyrrole antifungal agents |
| US8536169B2 (en) | 2008-06-05 | 2013-09-17 | Glaxo Group Limited | Compounds |
| US8765743B2 (en) | 2008-06-05 | 2014-07-01 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| WO2009147187A1 (en) * | 2008-06-05 | 2009-12-10 | Glaxo Group Limited | 4-carboxamide indazole derivatives useful as inhibitors of p13-kinases |
| US8658635B2 (en) | 2008-06-05 | 2014-02-25 | Glaxosmithkline Intellectual Property Development Limited | Benzpyrazol derivatives as inhibitors of PI3 kinases |
| US20110112070A1 (en) * | 2008-06-05 | 2011-05-12 | Ian Robert Baldwin | 4-carboxamide indazole derivatives useful as inhibitors of p13-kinases |
| US8163743B2 (en) * | 2008-06-05 | 2012-04-24 | GlaxoGroupLimited | 4-carboxamide indazole derivatives useful as inhibitors of PI3-kinases |
| US10532993B2 (en) | 2008-07-25 | 2020-01-14 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US10882840B2 (en) | 2008-07-25 | 2021-01-05 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US10112920B2 (en) | 2008-07-25 | 2018-10-30 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US9884840B2 (en) | 2008-07-25 | 2018-02-06 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US11021456B2 (en) | 2008-07-25 | 2021-06-01 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US9512101B2 (en) | 2008-07-25 | 2016-12-06 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| DE102008038222A1 (en) | 2008-08-18 | 2010-02-25 | Merck Patent Gmbh | Indazol-5-carboxylic acid derivatives |
| DE102008038220A1 (en) | 2008-08-18 | 2010-02-25 | Merck Patent Gmbh | oxadiazole |
| WO2010020305A1 (en) | 2008-08-18 | 2010-02-25 | Merck Patent Gmbh | Oxadiazole derivatives for treating diabetes |
| DE102008038221A1 (en) | 2008-08-18 | 2010-02-25 | Merck Patent Gmbh | 7-azaindole derivatives |
| US8637532B2 (en) | 2009-02-11 | 2014-01-28 | Merck Patent Gmbh | Amino azaheterocyclic carboxamides |
| US9040560B2 (en) | 2009-02-11 | 2015-05-26 | Merck Patent Gmbh | Amino azaheterocyclic carboxamides |
| US8524751B2 (en) | 2009-03-09 | 2013-09-03 | GlaxoSmithKline Intellecutual Property Development | 4-oxadiazol-2-YL-indazoles as inhibitors of P13 kinases |
| US10383879B2 (en) | 2009-04-30 | 2019-08-20 | Glaxo Group Limited | Compounds |
| US8575162B2 (en) | 2009-04-30 | 2013-11-05 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8586583B2 (en) | 2009-04-30 | 2013-11-19 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US10946025B2 (en) | 2009-04-30 | 2021-03-16 | Glaxo Group Limited | Compounds |
| US8580797B2 (en) | 2009-04-30 | 2013-11-12 | Glaxo Smith Kline Intellectual Property Development Limited | Compounds |
| US8609657B2 (en) | 2009-04-30 | 2013-12-17 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8586590B2 (en) | 2009-04-30 | 2013-11-19 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US10624898B2 (en) | 2009-04-30 | 2020-04-21 | Glaxo Group Limited | Compounds |
| EP2424857A4 (en) * | 2009-05-01 | 2015-11-11 | Aerie Pharmaceuticals Inc | Dual-action inhibitors and methods of using same |
| US11028081B2 (en) | 2009-05-01 | 2021-06-08 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US11618748B2 (en) | 2009-05-01 | 2023-04-04 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US10654844B2 (en) | 2009-05-01 | 2020-05-19 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US10316029B2 (en) | 2009-05-01 | 2019-06-11 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US10174017B2 (en) | 2009-05-01 | 2019-01-08 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US9951059B2 (en) | 2009-05-01 | 2018-04-24 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US9023847B2 (en) | 2009-08-07 | 2015-05-05 | Merck Patent Gmbh | Azaheterocyclic compounds |
| WO2011017009A1 (en) | 2009-08-07 | 2011-02-10 | Merck Patent Gmbh | Novel azaheterocyclic compounds |
| US8846673B2 (en) | 2009-08-11 | 2014-09-30 | Bristol-Myers Squibb Company | Azaindazoles as kinase inhibitors and use thereof |
| WO2012016001A1 (en) | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| WO2012013282A1 (en) | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides as inhibitors of the kinase p70s6k |
| US9586938B2 (en) | 2010-07-29 | 2017-03-07 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| US10087166B2 (en) | 2010-07-29 | 2018-10-02 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| US8710044B2 (en) | 2010-07-29 | 2014-04-29 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides |
| US8993576B2 (en) | 2010-10-27 | 2015-03-31 | Glaxo Group Limited | 6-(1H-indol-4-yl)-4-(5-{[4-1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1H-indazole hemi succinate salt, polymorphs and pharmaceutical compositions thereof |
| EP3553168A1 (en) | 2010-11-12 | 2019-10-16 | Georgetown University | Immortalization of epithelial cells and methods of use |
| WO2012069146A1 (en) | 2010-11-24 | 2012-05-31 | Merck Patent Gmbh | Quinazoline carboxamide azetidines |
| US9610289B2 (en) | 2011-04-01 | 2017-04-04 | Genentech, Inc. | Combinations of AKT inhibitor compounds and erlotinib, and methods of use |
| US9346789B2 (en) | 2011-04-01 | 2016-05-24 | Genentech, Inc. | Combinations of AKT inhibitor compounds and abiraterone, and methods of use |
| US10092567B2 (en) | 2011-04-01 | 2018-10-09 | Genentech, Inc. | Combinations of AKT inhibitor compounds and chemotherapeutic agents, and methods of use |
| US9150548B2 (en) | 2011-04-01 | 2015-10-06 | Genentech, Inc. | Combinations of AKT inhibitor compounds and vemurafenib, and methods of use |
| US9150549B2 (en) | 2011-04-01 | 2015-10-06 | Genentech, Inc. | Combinations of AKT inhibitor compounds and erlotinib, and methods of use |
| US9717730B2 (en) | 2011-04-01 | 2017-08-01 | Genentech, Inc. | Combinations of AKT inhibitor compounds and chemotherapeutic agents, and methods of use |
| US9682082B2 (en) | 2011-04-01 | 2017-06-20 | Genentech, Inc. | Combinations of AKT and MEK inhibitor compounds, and methods of use |
| US9321760B2 (en) | 2011-09-12 | 2016-04-26 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| US10080750B2 (en) | 2011-09-12 | 2018-09-25 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| WO2013040059A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Novel imidazole amines as modulators of kinase activity |
| WO2013040044A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| US9145392B2 (en) | 2011-09-12 | 2015-09-29 | Merck Patent Gmbh | Imidazole amines as modulators of kinase activity |
| US9662330B2 (en) | 2011-09-12 | 2017-05-30 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| EP2766352A4 (en) * | 2011-10-12 | 2015-06-03 | Univ Health Network Uhn | Indazole compounds as kinase inhibitors and method of treating cancer with same |
| US9580390B2 (en) | 2011-10-12 | 2017-02-28 | University Health Network | Indazole compounds as kinase inhibitors and method of treating cancer with same |
| US10080753B2 (en) | 2011-12-22 | 2018-09-25 | Merck Patent Gmbh | Heterocyclic carboxamides as modulators of kinase activity |
| US10287300B2 (en) | 2012-11-16 | 2019-05-14 | Merck Patent Gmbh | Heterocyclic derivatives as modulators of kinase activity |
| US9315517B2 (en) | 2012-11-16 | 2016-04-19 | Merck Patent Gmbh | Imidazol-piperidinyl derivatives as modulators of kinase activity |
| EP3299362A1 (en) | 2012-11-16 | 2018-03-28 | Merck Patent GmbH | Imidazol-piperidinyl derivatives as modulators of kinase activity |
| WO2014078637A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel heterocyclic derivatives as modulators of kinase activity |
| US9580443B2 (en) | 2012-11-16 | 2017-02-28 | Merck Patent Gmbh | Heterocyclic derivatives as modulators of kinase activity |
| US9850255B2 (en) | 2012-11-16 | 2017-12-26 | Merck Patent Gmbh | Heterocyclic derivatives as modulators of kinase activity |
| WO2014078634A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel imidazol-piperidinyl derivatives as modulators of kinase activity |
| US9981925B2 (en) | 2012-11-29 | 2018-05-29 | Merck Patent Gmbh | Substituted benzo[d][1,2,3]triazines as p70S6K inhibitors |
| WO2014085528A1 (en) | 2012-11-29 | 2014-06-05 | Merck Patent Gmbh | Azaquinazoline carboxamide derivatives |
| US10233160B2 (en) | 2012-11-29 | 2019-03-19 | Merck Patent Gmbh | Substituted pyrido[3,4-d]pyrimidines and pyrido[4,3-d]pyrimidines as p70S6K inhibitors |
| US9440968B2 (en) | 2012-11-29 | 2016-09-13 | Merck Patent Gmbh | Substituted pyrido[3,2-d]pyrimidines for treating cancer |
| WO2014143612A1 (en) | 2013-03-11 | 2014-09-18 | Merck Patent Gmbh | 6-[4-(1 h-imidazol-2-yl)piperidin-1 -yl]pyrimidin-4-amine derivatives as modulators of kinase activity |
| US9867826B2 (en) | 2013-03-11 | 2018-01-16 | Merck Patent Gmbh | Heterocycles as modulators of kinase activity |
| US9458134B2 (en) | 2013-03-11 | 2016-10-04 | Merck Patent Gmbh | Heterocycles as modulators of kinase activity |
| US11185538B2 (en) | 2013-03-15 | 2021-11-30 | Aerie Pharmaceuticals, Inc. | Compositions for treating glaucoma or reducing intraocular pressure |
| US10568878B2 (en) | 2013-03-15 | 2020-02-25 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US9993470B2 (en) | 2013-03-15 | 2018-06-12 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US9931336B2 (en) | 2013-03-15 | 2018-04-03 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US11197853B2 (en) | 2013-03-15 | 2021-12-14 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US9849122B2 (en) | 2013-03-15 | 2017-12-26 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US11020385B2 (en) | 2013-03-15 | 2021-06-01 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US10588901B2 (en) | 2013-03-15 | 2020-03-17 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US9415043B2 (en) | 2013-03-15 | 2016-08-16 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| WO2015041533A1 (en) | 2013-09-20 | 2015-03-26 | Stichting Het Nederlands Kanker Instituut | Rock in combination with mapk-pathway |
| WO2015149909A1 (en) | 2014-04-03 | 2015-10-08 | Merck Patent Gmbh | Combinations of cancer therapeutics |
| WO2015200920A1 (en) | 2014-06-27 | 2015-12-30 | The Regents Of The University Of California | Cultured mammalian limbal stem cells, methods for generating the same, and uses thereof |
| EP3165523B1 (en) * | 2014-07-01 | 2020-01-08 | Gwangju Institute of Science and Technology | Composition for inducing cell reprogramming |
| US10201524B2 (en) | 2014-11-21 | 2019-02-12 | F2G Limited | Antifungal agents |
| US11065228B2 (en) | 2014-11-21 | 2021-07-20 | F2G Limited | Antifungal agents |
| US10596150B2 (en) | 2014-11-21 | 2020-03-24 | F2G Limited | Antifungal agents |
| WO2016161192A1 (en) | 2015-04-03 | 2016-10-06 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US11680246B2 (en) | 2015-04-03 | 2023-06-20 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US10100285B2 (en) | 2015-04-03 | 2018-10-16 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US10119121B2 (en) | 2015-04-03 | 2018-11-06 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US9963680B2 (en) | 2015-04-03 | 2018-05-08 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US10711250B2 (en) | 2015-04-03 | 2020-07-14 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US10711251B2 (en) | 2015-04-03 | 2020-07-14 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US9790471B2 (en) | 2015-04-03 | 2017-10-17 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| EP3822339A1 (en) | 2015-04-03 | 2021-05-19 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US11696924B2 (en) | 2015-07-17 | 2023-07-11 | Memorial Sloan-Kettering Cancer Center | Combination therapy using PDK1 and PI3K inhibitors |
| WO2017015152A1 (en) | 2015-07-17 | 2017-01-26 | Memorial Sloan-Kettering Cancer Center | Combination therapy using pdk1 and pi3k inhibitors |
| EP3892717A1 (en) | 2015-09-11 | 2021-10-13 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US10066201B2 (en) | 2015-09-11 | 2018-09-04 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US10514163B2 (en) | 2015-09-11 | 2019-12-24 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US11060715B2 (en) | 2015-09-11 | 2021-07-13 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| EP3892718A1 (en) | 2015-09-11 | 2021-10-13 | Propagenix Inc. | Ex vivo proliferation of epithelial cells |
| US9643927B1 (en) | 2015-11-17 | 2017-05-09 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| US10550087B2 (en) | 2015-11-17 | 2020-02-04 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| US10973821B2 (en) | 2016-05-25 | 2021-04-13 | F2G Limited | Pharmaceutical formulation |
| US11707460B2 (en) | 2016-08-31 | 2023-07-25 | Aerie Pharmaceuticals, Inc. | Ophthalmic compositions |
| US11389441B2 (en) | 2016-08-31 | 2022-07-19 | Aerie Pharmaceuticals, Inc. | Ophthalmic compositions |
| US11590123B2 (en) | 2016-08-31 | 2023-02-28 | Aerie Pharmaceuticals, Inc. | Ophthalmic compositions |
| US10858339B2 (en) | 2017-03-31 | 2020-12-08 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US11312700B2 (en) | 2017-03-31 | 2022-04-26 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US11655214B2 (en) | 2017-04-18 | 2023-05-23 | Eli Lilly And Company | Phenyl-2-hydroxy-acetylamino-2-methyl-phenyl compounds for the treatment of pancreatic cancer |
| EP4039675A1 (en) * | 2017-04-18 | 2022-08-10 | Eli Lilly and Company | Phenyl-2-hydroxy-acetylamino-2-methyl-phenyl compounds |
| WO2018194885A1 (en) * | 2017-04-18 | 2018-10-25 | Eli Lilly And Company | Phenyl-2-hydroxy-acetylamino-2-methyl-phenyl compounds |
| KR102575246B1 (en) | 2017-04-18 | 2023-09-06 | 일라이 릴리 앤드 캄파니 | Phenyl-2-hydroxy-acetylamino-2-methyl-phenyl compound |
| RU2804280C2 (en) * | 2017-04-18 | 2023-09-26 | Эли Лилли Энд Компани | Phenyl-2-hydroxy-acetylamino-2-methyl-phenyl compounds |
| KR20190140966A (en) * | 2017-04-18 | 2019-12-20 | 일라이 릴리 앤드 캄파니 | Phenyl-2-hydroxy-acetylamino-2-methyl-phenyl compound |
| WO2018234354A1 (en) * | 2017-06-20 | 2018-12-27 | Grünenthal GmbH | Novel substituted 3-indole and 3-indazole compounds as phosphodiesterase inhibitors |
| US11479533B2 (en) | 2017-07-21 | 2022-10-25 | Kadmon Corporation, Llc | Inhibitors of Rho associated coiled-coil containing protein kinase |
| CN112218857A (en) * | 2018-02-16 | 2021-01-12 | 星座制药公司 | P300/CBP HAT inhibitors and methods of use thereof |
| US11414384B2 (en) | 2018-02-16 | 2022-08-16 | Constellation Pharmaceuticals, Inc. | P300/CBP hat inhibitors |
| US11274090B2 (en) | 2018-02-16 | 2022-03-15 | Constellation Pharmaceuticals, Inc. | P300/CBP HAT inhibitors |
| WO2019161162A1 (en) * | 2018-02-16 | 2019-08-22 | Constellation Pharmaceuticals, Inc. | P300/cbp hat inhibitors |
| WO2020041065A1 (en) | 2018-08-20 | 2020-02-27 | Propagenix Inc. | Epithelial cell spheroids |
| WO2020047229A1 (en) | 2018-08-29 | 2020-03-05 | University Of Massachusetts | Inhibition of protein kinases to treat friedreich ataxia |
| US11427563B2 (en) | 2018-09-14 | 2022-08-30 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US11891376B2 (en) | 2018-09-14 | 2024-02-06 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| WO2020084580A1 (en) | 2018-10-26 | 2020-04-30 | Novartis Ag | Methods and compositions for ocular cell therapy |
| US11819503B2 (en) | 2019-04-23 | 2023-11-21 | F2G Ltd | Method of treating coccidioides infection |
| WO2021021893A1 (en) * | 2019-07-29 | 2021-02-04 | Constellation Pharmaceuticals, Inc. | Compounds for use in treating neurological disorders |
| EP3901156A4 (en) * | 2020-02-21 | 2022-03-30 | Vivavision (Shanghai) Ltd | Nitrooxyderivative of rock kinase inhibitor |
| WO2021220132A1 (en) | 2020-04-27 | 2021-11-04 | Novartis Ag | Methods and compositions for ocular cell therapy |
| WO2022101459A1 (en) | 2020-11-16 | 2022-05-19 | Merck Patent Gmbh | Kinase inhibitor combinations for cancer treatment |
| WO2023069949A1 (en) | 2021-10-18 | 2023-04-27 | Evia Life Sciences Inc. | Compositions and methods of use thereof for treating liver fibrosis |
| WO2023067394A2 (en) | 2021-10-22 | 2023-04-27 | Evia Life Sciences Inc. | Methods for making extracellular vesicles, and compositions and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1812973B (en) | 2010-12-22 |
| TW200306819A (en) | 2003-12-01 |
| US20040009968A1 (en) | 2004-01-15 |
| KR20100119902A (en) | 2010-11-11 |
| RU2004125852A (en) | 2006-01-20 |
| EP2295413A1 (en) | 2011-03-16 |
| MXPA04007126A (en) | 2005-03-31 |
| US7041687B2 (en) | 2006-05-09 |
| KR20040078676A (en) | 2004-09-10 |
| NO20043531L (en) | 2004-10-25 |
| EP1467972A1 (en) | 2004-10-20 |
| JP4718118B2 (en) | 2011-07-06 |
| AU2003212833B2 (en) | 2008-02-21 |
| CN1812973A (en) | 2006-08-02 |
| AR038202A1 (en) | 2005-01-05 |
| HK1096385A1 (en) | 2007-06-01 |
| NO328041B1 (en) | 2009-11-16 |
| PL371512A1 (en) | 2005-06-27 |
| JP2006176530A (en) | 2006-07-06 |
| JP2005524631A (en) | 2005-08-18 |
| IL163117A (en) | 2010-05-31 |
| EP2295412A1 (en) | 2011-03-16 |
| CA2473986A1 (en) | 2003-08-07 |
| CA2473986C (en) | 2012-01-17 |
| AU2003212833C1 (en) | 2010-12-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1467972A1 (en) | Indazole compounds useful as protein kinase inhibitors | |
| AU2003212833A1 (en) | Indazole compounds useful as protein kinase inhibitors | |
| RU2423361C2 (en) | Aminopyrimidines as kinase inhibitors | |
| AU2002364536B2 (en) | Pyrimidine-based compounds useful as GSK-3 inhibitors | |
| AU2004212421B2 (en) | Heteroaryl substituted pyrolls useful as inhibitors of protein kinases | |
| EP1660487B1 (en) | Pyrrole compositions useful as inhibitors of c-met | |
| WO2003040131A1 (en) | Aminopyrimidines and pyridines | |
| EP1399440A1 (en) | 5-(2-aminopyrimidin-4-yl)benzisoxazoles as protein kinase inhibitors | |
| EP1501829A1 (en) | Thiadiazoles or oxadiazoles and their use as inhibitors of jak protein kinase | |
| US9527832B2 (en) | Substituted condensed pyrimidine compounds | |
| WO2018191146A1 (en) | Heteroaryl rheb inhibitors and uses thereof | |
| AU2004276341B2 (en) | Pyrazolopyrrole derivatives as protein kinase inhibitors | |
| KR101674695B1 (en) | Pyridazinone derivatives | |
| WO2014146246A1 (en) | Cycloalkyl nitrile pyrazolo pyridones as janus kinase inhibitors | |
| WO2004072029A2 (en) | Pyrazolopyridazines useful as inhibitors of protein kinases | |
| AU2008202214A1 (en) | Indazole Compounds Useful as Protein Kinase Inhibitors | |
| AU2013201630B2 (en) | Aminopyrimidines useful as kinase inhibitors | |
| AU2016203378A1 (en) | Aminopyrimidines useful as kinase inhibitors | |
| AU2002350657A1 (en) | Aminopyrimidines and pyridines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003212833 Country of ref document: AU Ref document number: 163117 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2473986 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003564020 Country of ref document: JP Ref document number: 2003708869 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2004/007126 Country of ref document: MX Ref document number: KR Ref document number: 1020047011500 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004125852 Country of ref document: RU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20038056127 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003708869 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020107022397 Country of ref document: KR |