WO2003016270A2 - Selective estrogen receptor modulators - Google Patents
Selective estrogen receptor modulators Download PDFInfo
- Publication number
- WO2003016270A2 WO2003016270A2 PCT/US2002/025394 US0225394W WO03016270A2 WO 2003016270 A2 WO2003016270 A2 WO 2003016270A2 US 0225394 W US0225394 W US 0225394W WO 03016270 A2 WO03016270 A2 WO 03016270A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- acryloyl
- diphenyl
- enyl
- dihydro
- Prior art date
Links
- 0 *CC(c1ccc(C=C*)cc1)c1ccccc1* Chemical compound *CC(c1ccc(C=C*)cc1)c1ccccc1* 0.000 description 2
- BXBZZSDWMDXCKP-JPXVSUISSA-N CC/C(/c1ccccc1)=C(\c1ccccc1)/c1ccc(/C=C/C(NS(c2cc(C(F)(F)F)ccc2)(=O)=O)=O)cc1 Chemical compound CC/C(/c1ccccc1)=C(\c1ccccc1)/c1ccc(/C=C/C(NS(c2cc(C(F)(F)F)ccc2)(=O)=O)=O)cc1 BXBZZSDWMDXCKP-JPXVSUISSA-N 0.000 description 1
- TZJOAPVUOGSUGW-MYMNJIRVSA-N CC/C(/c1ccccc1)=C(\c1ccccc1)/c1ccc(/C=C/C(NS(c2nc3ccccc3[s]2)(=O)=O)=O)cc1 Chemical compound CC/C(/c1ccccc1)=C(\c1ccccc1)/c1ccc(/C=C/C(NS(c2nc3ccccc3[s]2)(=O)=O)=O)cc1 TZJOAPVUOGSUGW-MYMNJIRVSA-N 0.000 description 1
- JMOWFWKQINJIOZ-KNTRCKAVSA-N CS(NC(/C=C/c(cc1)ccc1C(c1c(CCC2)cccc1)=C2c1ccccc1)=O)(=O)=O Chemical compound CS(NC(/C=C/c(cc1)ccc1C(c1c(CCC2)cccc1)=C2c1ccccc1)=O)(=O)=O JMOWFWKQINJIOZ-KNTRCKAVSA-N 0.000 description 1
- XOWVSXCPJHZAFV-OBGWFSINSA-N O=C(/C=C/c(cc1)ccc1C(c1c(CCC2)cccc1)=C2c1ccccc1)NS(C(F)(F)F)(=O)=O Chemical compound O=C(/C=C/c(cc1)ccc1C(c1c(CCC2)cccc1)=C2c1ccccc1)NS(C(F)(F)F)(=O)=O XOWVSXCPJHZAFV-OBGWFSINSA-N 0.000 description 1
- UHOVQNZJYSORNB-UHFFFAOYSA-N c1ccccc1 Chemical compound c1ccccc1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D257/04—Five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/30—Oestrogens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/32—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms having cyano groups bound to acyclic carbon atoms of a carbon skeleton containing at least one six-membered aromatic ring
- C07C255/34—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms having cyano groups bound to acyclic carbon atoms of a carbon skeleton containing at least one six-membered aromatic ring with cyano groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by unsaturated carbon chains
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/50—Compounds containing any of the groups, X being a hetero atom, Y being any atom
- C07C311/51—Y being a hydrogen or a carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/487—Separation; Purification; Stabilisation; Use of additives by treatment giving rise to chemical modification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C57/00—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms
- C07C57/46—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms containing six-membered aromatic rings and other rings, e.g. cyclohexylphenylacetic acid
- C07C57/50—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms containing six-membered aromatic rings and other rings, e.g. cyclohexylphenylacetic acid containing condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/42—Unsaturated compounds containing hydroxy or O-metal groups
- C07C59/54—Unsaturated compounds containing hydroxy or O-metal groups containing six-membered aromatic rings and other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/58—Unsaturated compounds containing ether groups, groups, groups, or groups
- C07C59/72—Unsaturated compounds containing ether groups, groups, groups, or groups containing six-membered aromatic rings and other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/612—Esters of carboxylic acids having a carboxyl group bound to an acyclic carbon atom and having a six-membered aromatic ring in the acid moiety
- C07C69/618—Esters of carboxylic acids having a carboxyl group bound to an acyclic carbon atom and having a six-membered aromatic ring in the acid moiety having unsaturation outside the six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/734—Ethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/68—Benzothiazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D277/70—Sulfur atoms
- C07D277/76—Sulfur atoms attached to a second hetero atom
- C07D277/80—Sulfur atoms attached to a second hetero atom to a nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/58—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/34—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/10—Systems containing only non-condensed rings with a five-membered ring the ring being unsaturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/10—One of the condensed rings being a six-membered aromatic ring the other ring being six-membered, e.g. tetraline
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/12—One of the condensed rings being a six-membered aromatic ring the other ring being at least seven-membered
Definitions
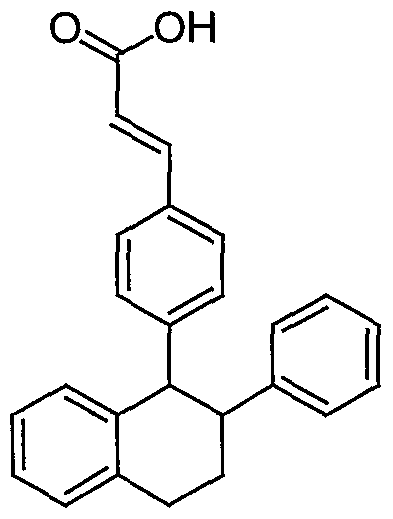
- This invention pertains to triphenylethylene derivatives, such as, 3- ⁇ 4- [6- (3-Methoxy-phenyl) -8, 9-dihydro-7H- benzocyclohepten-5-yl] -phenyl ⁇ -acrylic acid, as selective estrogen receptor modulators.
- This invention also provides methods for the treatment and/or prevention of estrogen stimulated diseases in mammals including breast, uterine, ovarian, prostrate and colon cancer, osteoporosis, cardiovascular disease, and benign proliferative disorders, as well as pharmaceutical compositions of the compounds of the present invention.
- SERMs selective estrogen receptor modifiers
- tamoxifen is a partial estrogen receptor agonist/antagonist that produces objective responses in approximately 50% of the patients.
- tamoxifen is a partial estrogen receptor agonist/antagonist that produces objective responses in approximately 50% of the patients.
- 100% of patients who take tamoxifen eventually relapse with tamoxifen- resistant tumors.
- Approximately 50% of the patients that fail tamoxifen treatment will respond to a subsequent hormonal manipulation therapy such as castration, aromatase inhibitors, or other SERMs.
- the second line therapies for hormonal manipulation therapy of metastatic breast cancer represent a substantial unmet need because no single agent has become the treatment of choice for patients who fail tamoxifen therapy.
- the ideal agent would be a medication that induces regression of metastatic breast cancer lesions in women who have previously responded to tamixofen therapy.
- the present invention is directed to novel, highly soluble salt forms of the compound 3- [4 [ (1, 2-diphenyl-but-l-enyl) -phenyl] -acrylic acid, which is described in U.S. Patent Number 5,681,835, the contents of which are herein incorporated by reference in their entirety.
- SERMs modulate the proliferation of uterine tissue, skeletal bone density, and cardiovascular health, including plasma cholesterol levels.
- estrogen stimulates breast and endometrial tissue proliferation, enhances bone density, and lowers plasma cholesterol.
- Many SERMs are bifunctional in that they antagonize some of these functions while stimulating others.
- tamoxifen which is a partial agonist/antagonist at the estrogen receptor inhibits estrogen-induced breast cancer cell proliferation but stimulates endometrial tissue growth and prevents bone loss .
- Estrogens are an important class of steroidal hormones that stimulate the development and maintenance of fundamental sexual characteristics in humans.
- estrogens have been found useful in the treatment of certain medical conditions and diseases.
- estradiol a steroid hormone produced by the ovary, is useful in the treatment of osteoporosis, cardiovascular disease, premenstrual syndrome, vasomotor symptoms associated with menopause, atrophic vaginitis,
- HRT Hormone replacement therapy
- ER Upon binding ligand, ER undergoes a conformational change initiating a cascade of events leading ultimately to its association with specific regulatory regions within target genes (O'Malley et al . , Hormone Research 47:1-26 (1991)). The ensuing effect on transcription is influenced by the cell and promoter context of the DNA-bound receptor (Tora et al . Cell 59:471-487 (1989) (Tasset et al . , Cell 62:1177-1181 (1990); McDonnell et all Mol. Endocrinol. 9:659-669 (1995); Tzuker an et al . Mol. Endocrinol . 8:21-30 (1994)). It is in this manner that the physiological ER-agonist, extradiol, exerts its biological activity in the reproductive, skeletal and cardiovascular systems (Clark and Peck, Female Sex Steroids :Receptors and
- Tamoxifen functions as an antagonist in most ER-positive tumors of the breast and ovum, but displays a paradoxical agonist activity in bone and the cardiovascular system and partial agonist activity in the uterus (Kedar et al . Lancet 343:1318-
- the agonist/antagonist activity of the ER-tamoxifen complex is influenced by cell context. This important observation is in apparent contradiction to longstanding models that hold that ER only exists in the cell in an active or an inactive state (Clark and Peck, Female Sex Steroids :Receptors and Functions (eds) Monographs on Endocrinology, Springer-Verlag, New York (1979)). It indicates instead that different ligands acting through the same receptor can manifest different biologies in different cells.
- Tamoxifen as well as a structurally similar compound known as raloxifene have been developed for the treatment and/or prevention of osteoporosis, cardiovascular disease and breast cancer in addition to the treatment and/or prevention of a variety of other disease states. Both compounds have been shown to exhibit an osteoprotective effect on bone mineral density combined with a positive effect on plasma cholesterol levels and a greatly reduced incidence of breast and uterine cancer.
- tamoxifen and raloxifene both have unacceptable levels of life-threatening side effects such as endometrial cancer and hepatocellular carcinoma.
- the likely mechanism for the cell selective agonist/antagonist activity of tamoxifen has been determined using an in vi tro approach (Tora et al .
- ER contains two activation domains, AF-1 (Activation Function-1) and AF-2, which permit its interaction with the transcription apparatus.
- AF-1 Activation Function-1
- AF-2 Activation Function-2
- the relative contribution of these AFs to overall ER efficacy differs from cell to cell (Tora et al . , Cell 59:477-487 (1989); McDonnell et al . , Mol. Endocrinol. 9@65-9-669 (1995); Tzukerman et al., Mol. Endocrinol. 8:21-30 (1994)).
- Estradiol was determined to function as both an AF-1 and an AF-2 agonist, in that it exhibited maximal activity regardless of which AF was dominant in a given cellular environment.
- Tamoxifen functions as an AF-2 antagonist, inhibiting ER activity in cells where AF-2 is required or is the dominant activator (Tora et al . , Cell 59:477-487 (1989); McDonnell et al., Mol. Endocrinol. 9:659-669 (1995); Tzukerman et al . , Mol. Endocrinol. 8:21-30 (1994)).
- tamoxifen functions as an agonist when AF-1 alone is required (McDonnell et al .
- the present invention describes compounds represented by Formula (I) :
- R 1 -R 5 are defined below.
- the present invention is also directed to pharmaceutical compositions comprising one or more compounds of Formula (I) .
- the present invention is directed to methods for the treatment and/or prevention of estrogen stimulated diseases including breast, uterine, ovarian, prostate and colon cancer, osteoporosis, cardiovascular disease, and benign proliferative disorders, comprising: administering to a host in need of such treatment a therapeutically effective amount of a compound of Formula (I) .
- the present invention further provides methods of modulating the estrogen receptor in a patient comprising the step of administering to the patient a therapeutically effective amount of a compound of Formula (I) .
- the present invention further provides pharmaceutical compositions including compounds of Formula (I) and a pharmaceutically acceptable carrier.
- This invention pertains to compounds of Formula (I) as selective estrogen receptor modulators:
- R 2 is selected from the group consisting of H, C ⁇ _ s alkyl and halo;
- R 3 is selected from the group consisting of H, C, 8 alkyl, C, 8 alkylenyl, halo, or CN;
- R 2 and R 3 together with the atoms to which they are attached, form a six- or seven-membered ring structure where one or more of the atoms forming the ring may be oxygen;
- R 4 is selected from the group consisting of H, OH, C ⁇ alkyl, OC ⁇ alkyl and halo;
- R 5 is selected from the group consisting of H, OH, CN, nitro, C 1.8 alkyl, OC ⁇ alkyl and halo;
- R 6 is selected from the group of H, OH, CN, OC ⁇ alkyl methyl, ethyl, propyl and butyl;
- R 7 is selected from the group consisting of H, aryl, C ⁇ g alkyl, OH, and OC ⁇ g alkyl;
- R 8 is selected from the group consisting of aryl, C ⁇ g alkyl, OH, and OC 1 _ 8 alkyl, wherein said R s is optionally substituted with 1 to 2 substituents selected from halo, nitro, OH, CN, C 1 _ i alkyl , 0C X _ 4 alkyl , NH 2 , and NHC (0) OC (CH 3 ) 3 ;
- X is selected from the group consisting of 0 or NH, wherein when X is O, R ⁇ is other than OH; and,
- the broken line represents an optional double bond.
- compounds of Formula (1) include:
- compositions for the treatment and/or prevention of breast, uterine, ovarian, prostrate and colon cancer, osteoporosis, cardiovascular disease, and benign proliferative disorders include any of the above compounds and a pharmaceutically acceptable carrier.
- Another embodiment of the present invention provides a method of treating breast, uterine, ovarian, prostate and colon cancer, osteoporosis, cardiovascular disease, endometriosis, uterine fibroid, Alzheimer's disease, macular degeneration, urinary incontinence, type II diabetes, and benign proliferative disorders, comprising: administering to a host in need of such treatment a therapeutically effective amount of a compound of Formula (I) .
- the present invention further provides methods of modulating the estrogen receptor in a patient comprising the step of administering to the patient a therapeutically effective amount of a compound of Formula (I) . Also included in the present invention are compounds of Formula (II) :
- R 1 is selected from the group consisting of
- R 6 is selected from the group of H, methyl, ethyl, propyl and butyl;
- R 7 is selected from the group consisting of aryl and C ⁇ g alkyl, optionally substituted with one or more substituent groups;
- R , R , R and R are the same or different, and are independently selected from the group consisting of: H, C ⁇ alkyl, C 2.8 alkenyl, C 2.8 alkynyl, aryl, N0 2 , NH 2 , OH, OC ⁇ - 8 alkyl, CHO, COOH, halo and CN, wherein said R 8 is optionally substituted with 1 to 2 substituents selected from halo, nitro, OH, CN, C ⁇ _ alkyl, OC ⁇ _ alkyl, NH 2 , and NHC(0)OC(CH 3 ) 3 .
- the compounds of the present invention may contain an asymmetrically substituted carbon atom, and may be isolated in optically active or racemic forms. It is well known in the art how to prepare optically active forms, such as by resolution of racemic forms or by synthesis from optically active starting materials. All chiral, diastereomeric, racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomer form is specifically indicated.
- alkyl is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms.
- alkyl include, but are not limited to, methyl, ethyl, n-propyl, i-propyl, n-butyl , s-butyl, t-butyl, n-pentyl, and s-pentyl.
- haloalkyl refers to an alkyl substituted with one or more halogens.
- linear and cyclic heteroalkyl are defined in accordance with the term “alkyl” with the suitable replacement of carbon atoms with some other atom such as nitrogen or sulfur which would render a chemically stable species.
- halo or halogen as used herein refer to fluoro, chloro, bromo and iodo .
- aryl is intended to mean an aromatic cyclic or bicyclic ring structure containing from 5 to 13 ring atoms, including compounds, such as, for example phenyl, indanyl and naphthyl .
- aryl is intended to include both unsubstituted and substituted aryl groups, the latter referring to aryl moieties having one or more hydrogen substituents replaced by, for example, halogen, hydroxyl, carbonyl, alkoxy, keto, ester, ether, cyano, phosphoryl, amino, imino, amido, sulfhydryl, alkythio, thioester, sulfonyl, nitro, and/or heterocyclo .
- haloaryl refers to an aryl mono, di or tri substituted with halogen atoms.
- cycloalkyl As used herein, the terms “cycloalkyl” “bicycloalkyl” “carbocycle” or “carbocyclic residue” are intended to mean any stable 3- to 7-membered monocyclic or bicyclic or 7- to 13-membered bicyclic or tricyclic, any of which may be saturated, partially unsaturated, or aromatic.
- carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, adamantyl, cyclooctyl, ; [3.3.0]bicyclooctane, [4.3.0]bicyclononane, [4.4.0]bicyclodecane (decalin) , [2.2.2]bicyclooctane, fluorenyl, phenyl, naphthyl, indanyl, adamantyl, or tetrahydronaphthyl (tetralin) .
- heterocycle or “heterocyclic system” or “heterocyclyl” is intended to mean a stable 5- to 7- membered monocyclic or bicyclic or 7- to 10-membered bicyclic heterocyclic ring which is partially unsaturated or unsaturated (aromatic) , and which consists of carbon atoms and from 1 to 4 heteroatoms independently selected from the group consisting of N, O and S and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the nitrogen and sulfur heteroatoms may optionally be oxidized.
- the heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom which results in a stable structure.
- heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable. If specifically noted, a nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and 0 atoms in the heterocycle exceeds 1, then these heteroatoms are not adjacent to one another. It is preferred that the total number of S and 0 atoms in the heterocycle is not more than 1.
- aromatic heterocyclic system or “heteroaryl” is intended to mean a stable 5- to 7- membered monocyclic or bicyclic or 7- to 10-membered bicyclic heterocyclic aromatic ring which consists of carbon atoms and from 1 to 4 heterotams independently selected from the group consisting of N, 0 and S. It is preferred that the total number of S and 0 atoms in the aromatic heterocycle is not more than 1.
- heterocycles include, but are not limited to, lH-indazole, 2-pyrrolidonyl, 2H, 6H-1, 5, 2-dithiazinyl, 2H- pyrrolyl, 3H-indolyl, 4-piperidonyl, 4aH-carbazole, 4H- quinolizinyl, 6H-1, 2 , 5-thiadiazinyl, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazalonyl, carbazolyl, 4aH " -carbazolyl, b-carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydr
- oxazolyl oxazolidinylperimidinyl, phenanthridinyl , phenanthrolinyl, phenarsazinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, pteridinyl, piperidonyl,
- Preferred heterocycles include, but are not limited to, pyridinyl, piperidinyl, furanyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, indolyl, benzimidazolyl, li ⁇ -indazolyl, oxazolidinyl, benzotriazolyl, benzisoxazolyl, oxindolyl, benzoxazolinyl, or isatinoyl. Also included are fused ring and spiro compounds containing, for example, the above heterocycles.
- heteroaryl further includes a 5-membered or 6- membered heterocyclic aromatic group that can optionally carry a fused benzene ring and that can be unsubstituted or substituted.
- pharmaceutically acceptable salts refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, meglumine, lysine, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disul onic, oxalic, isethionic, and the like.
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods.
- such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred.
- Lists of suitable salts are found in Remington ' s Pharmaceutical Sciences, 18th ed. , Mack Publishing Company, Easton, PA, 1990, p. 1445, the disclosure of which is hereby incorporated by reference in it's entirety as though set forth in full.
- phrases "pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication commensurate with a reasonable benefit/risk ratio.
- Prodrugs are intended to include any covalently bonded carriers which release an active parent drug of the present invention in vivo when such prodrug is administered to a mammalian subject. Since prodrugs are known to enhance numerous desirable qualities of pharmaceuticals (i.e., solubility, bioavailability, manufacturing, etc.) the compounds of the present invention may be delivered in prodrug form. Thus, the present invention is intended to cover prodrugs of the presently claimed compounds, methods of delivering the same, and compositions containing the same. Prodrugs of the present invention are prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound.
- Prodrugs include compounds of the present invention wherein a hydroxy, amino, or sulfhydryl group is bonded to any group that, when the prodrug of the present invention is administered to a mammalian subject, it cleaves to form a free hydroxyl, free amino, or free sulfydryl group, respectively.
- Examples of prodrugs include, but are not limited to, acetate, formate, and benzoate derivatives of alcohol and amine functional groups in the compounds of the present invention.
- Substituted is intended to indicate that one or more hydrogens on the atom indicated in the expression using “substituted” is replaced with a selection from the indicated group(s), provided that the indicated atom's normal valency is not exceeded, and that the substitution results in a stable compound.
- “substitutent group” refers to R 1 , R 2 , R 3 , R 4 , R 5 , R ⁇ , and R 7 or any group selected from the group consisting of -N0 2 , -NH 2 , -COOH, - CHO, OH, alhoxy keto, -S0 2 , halogen, hydrogen, -CN and aryl.
- anti cancer or "anti- proliferative" agent includes, but is not limited to, altretamine, busulfan, chlorambucil, cyclophosphamide, ifosfamide, mechlorethamine, melphalan, thiotepa, cladribine, fluorouracil, floxuridine, gemcitabine, thioguanine, pentostatin, methotrexate, 6-mercaptopurine, cytarabine, carmustine, lomustine, streptozotocin, carboplatin, cisplatin, oxaliplatin, iproplatin, tetraplatin, lobaplatin, JM216, JM335, fludarabine, aminoglutethimide, flutamide, goserelin, leuprolide, megestrol acetate, cyproterone acetate, tamoxifen, anastrozole, bicalutamide,
- the term "host” refers to mammals including humans .
- the selective estrogen receptor modulator compounds of this invention can be administered as treatment for or prevention of cancer or other disease states by any means that produces contact of the active agent with the agent's site of action in the body of a mammal. They can be administered by any conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in combination with other compounds according to the present invention and/or other therapeutic agents, such as anti-cancer or anti-proliferative agents. When used in combination, the therapeutic agents may be administered together or separately so long as the therapeutic agents, or their active metabolites, are present in the host during an overlapping time period. The therapeutic agents can be administered alone, but preferably are administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
- the dosage administered will, of course, vary depending upon known factors, such as the pharmacodynamic characteristics of the particular agent and its mode and route of administration; the age, health and weight of the recipient; the nature and extent of the symptoms; the kind of concurrent treatment; the frequency of treatment; and the effect desired.
- a daily dosage of active ingredient can be expected to be about 0.001 to about 1000 milligrams per kilogram of body weight, with the preferred dose being about 0.1 to about 30 mg/kg.
- compositions suitable for administration contain from about 1 mg to about 100 mg of active ingredient per unit.
- the active ingredient will ordinarily be present in an amount of about 0.5- 95% by weight based on the total weight of the composition.
- the active ingredient can be administered orally in solid dosage forms, such as capsules, tablets and powders, or in liquid dosage forms, such as elixirs, syrups and suspensions. It can also be administered parenterally, in sterile liquid dosage forms .
- Gelatin capsules contain the active ingredient and powdered carriers, such as lactose, starch, cellulose derivatives, magnesium stearate, stearic acid, and the like. Similar diluents can be used to make compressed tablets. Both tablets and capsules can be manufactured as sustained release products to provide for continuous release of medication over a period of hours. Compressed tablets can be sugar coated or film coated to mask any unpleasant taste and protect the tablet from the atmosphere, or enteric coated for selective disintegration in the gastrointestinal tract. Liquid dosage forms for oral administration can contain coloring and flavoring to increase patient acceptance.
- water a suitable oil, saline, aqueous dextrose (glucose) , and related sugar solutions and glycols such as propylene glycol or polyethylene glycols are suitable carriers for parenteral solutions .
- Solutions for parenteral administration preferably contain a water soluble salt of the active ingredient, suitable stabilizing agents, and if necessary, buffer substances.
- Anti-oxidizing agents such as sodium bisulfite, sodium sulfite, or ascorbic acid, either alone or combined, are suitable stabilizing agents.
- citric acid and its salts, and sodium EDTA are also used.
- parenteral solutions can contain preservatives, such as benzalkonium chloride, methyl- or propyl-paraben and chlorobutanol .
- preservatives such as benzalkonium chloride, methyl- or propyl-paraben and chlorobutanol .
- Suitable pharmaceutical carriers are described in Remington ' s Pharmaceutical Sciences, 18th ed. , Mack Publishing Company, Easton, PA, 1990, a standard reference text in this field, the disclosure of which is hereby incorporated by reference.
- the compounds of the present invention can be prepared in a number of ways well known to one skilled in the art of organic synthesis.
- the compounds of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or variations thereon as appreciated by those skilled in the art. Preferred methods include, but are not limited to, those described below. All references cited herein are hereby incorporated in their entirety herein by reference.
- novel compounds of this invention may be prepared using the reactions and techniques described in this section.
- the reactions are performed in solvents appropriate to the reagents and materials employed and are suitable for the transformations being effected.
- all proposed reaction conditions including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment and workup procedures, are chosen to be the conditions standard for that reaction, which should be readily recognized by one skilled in the art.
- the functionality present on various portions of the molecule must be compatible with the reagents and reactions proposed.
- Isolation of the desired compounds of this invention can be achieved using standard chromatographic techniques known to those skilled in the art .
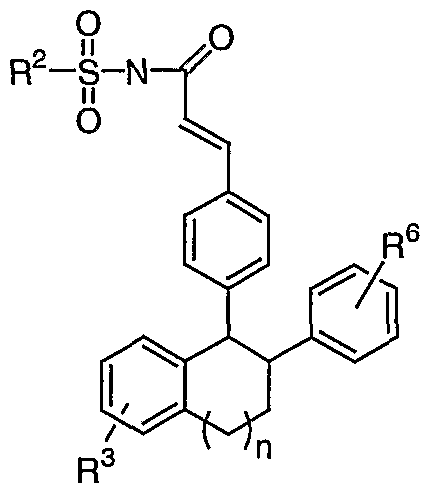
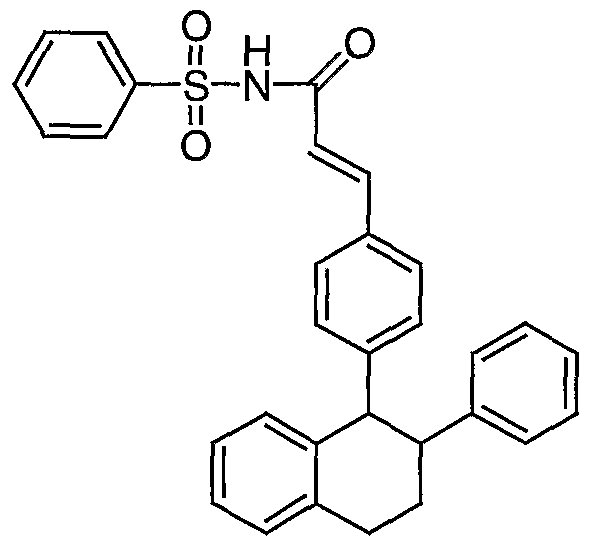
- Conversion of substituted acrylic acid I to an acylsulfonamide II employs a coupling reaction with a sulfonamide R2S02NH2 (Scheme 1) .
- the coupling reaction is performed in the presence of DMAP and 1- (3-dimethyl- aminopropyl) -3-ethylcarbodiimide hydrochloride (EDC).
- EDC 1- (3-dimethyl- aminopropyl) -3-ethylcarbodiimide hydrochloride
- Other reagents, besides EDC, which can be employed in this coupling reaction include HATU, TBTU, BOP, pyBOP, EDC, GDI, and DCC.
- Suitable solvents for the coupling reaction include CH.C1, .
- Solvents for the Heck reaction are well known to the skilled artisan and include amides (e.g., dimethylformamide, dimethylacetamide, N-methylpyrrolidone) , nitriles (e.g., acetonitrile, propionitrile, butyronitrile) , ethers (e.g., dimethoxyethane , tetrahydrofuran) , and hydrocarbons (e.g., cyclohexane, benzene, toluene, xylene)
- amides e.g., dimethylformamide, dimethylacetamide, N-methylpyrrolidone
- nitriles e.g., acetonitrile, propionitrile, butyronitrile
- ethers e.g., dimethoxyethane , tetrahydrofuran
- hydrocarbons e.g., cyclohexane, benzene,
- Palladium catalysts suitable for the Heck coupling reaction include, but are not limited to, Pd(OAc)2, PdCl2 (CH3CN) 2 or Pd2 (dba)3.
- Bases which are useful in the Heck reaction include alkaline metal carbonates and hydrogen carbonates, alkaline metal acetates, alkaline metal phosphates, and tertiary amines.
- bases for the Heck coupling reaction include TEA, PMP (1,2,2, 6, 6-pentamethylpiperidine) , Na2C03 , Ag2C03 , or NaHC03.
- Preferred bases are tertiary amines .
- the catalyst may be obtained as a solid and separated off by filtration.
- the crude product is freed of the solvent or the solvents and is subsequently purified by methods known to those skilled in the art e.g. by chromatography.
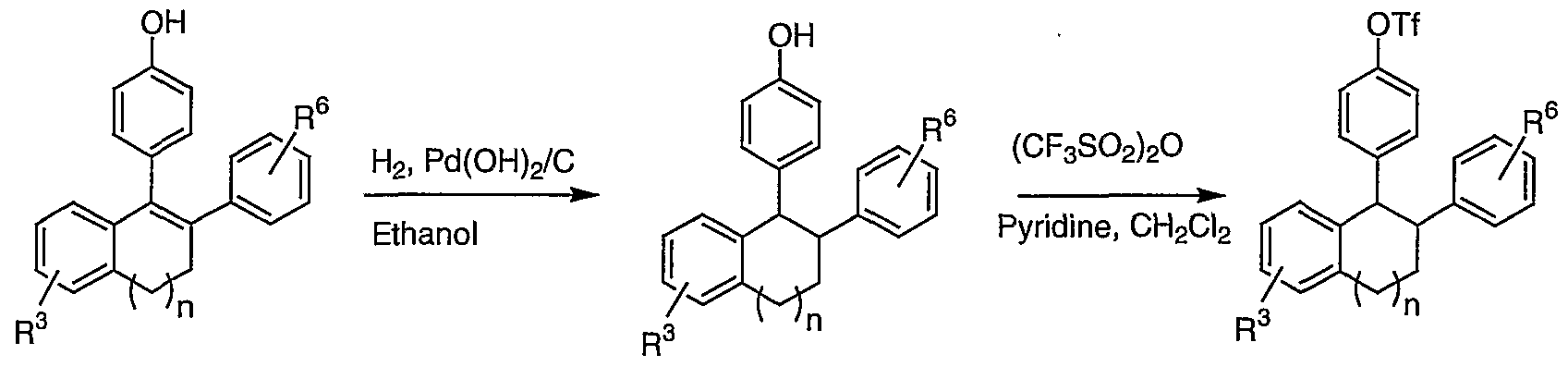
- Halogenation of benzocyclohexene or dihydro benzocycloheptene IV can be performed by employing a variety of electrophilic halogenating reagents such as pyridinium bromide perbromide or NBS to afford halo benzocyclohexene and halo dihydro benzocycloheptene V.
- Halogenation reactions are well known to those skilled in the art and are described in the chemical literature. See for example: Larock, R. C. Comprehensive Organic Transformations, VCH Publishers, New York, 1989.
- a Suzuki coupling of a halo benzocyclohexene or a halo dihydro benzocycloheptene V with a boronic acid (R ⁇ -Ph) B (OH) 2 in the presence of a palladium catalyst and a base provides bis- aryl substituted benzocyclohexene or bis-aryl substituted dihydro benzocycloheptene VI.
- Suitable solvents for this coupling include, but not limited to, THF, H2O, DMSO, Et2 ⁇ , toluene, DMF, dioxane and ethanol, i-PrOH, or a combination of two or more of these solvents.
- the reaction is carried out in the presence of a palladium catalyst, for example,
- Suitable bases for the Suzuki coupling reaction include TEA, KOH, TlOH, Na2C ⁇ 3, CS2CO3, and K2CO3. See, for example, Miyaura, N. , Suzuki, A. Chem. Rev. 1995, 95, 2457-2483; Suzuki, A., J. Organometallic Chem . 1999, 576, 147-168; and Suzuki, A. in Metal -catalyzed Cross-coupling Reactions, Diederich, F., and Stang, P.J., Eds.; Wiley-VCH: New York, 1998, pp. 49-97.
- Suitable bases include hydroxides or carbonates in an ether, alcohol, or aqueous alcohol or aqueous ether solvent systems. See Larock, R. C, Comprehensive Organic Transformations, VCH Publishers, New York, 1989.
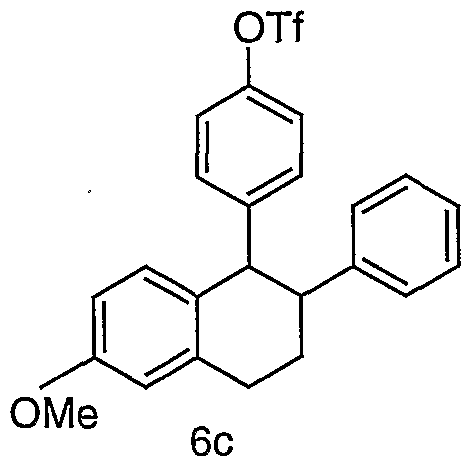
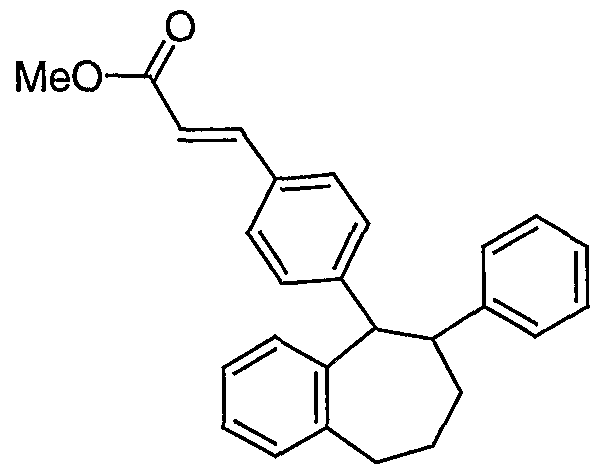
- Benzopyran XIII can be prepared from substituted 2H- chromene IX using a reaction sequence similar to that of Scheme 2 with several modifications.
- One is that the halogenation and suzuki coupling steps are not needed in the synthesis of benzopyran XIII since the phenyl bearing substituent R ⁇ is already present in the starting material 2H-chromene IX.
- the second modification is that a reaction to convert the OH group 2H-chromene IX to an tosylate, mesylate or triflate is needed for a subsequent coupling reaction.
- the preparation of benzopyran XIII from substituted 2H-chromene IX takes four steps: 1) preparation of aryl-X coupling partner for a subsequent Heck reaction, e.g.
- trifluoro-methanesulfonyl ester 2, Heck coupling reaction, 3) hydrolysis, and 4) sulfonylation.
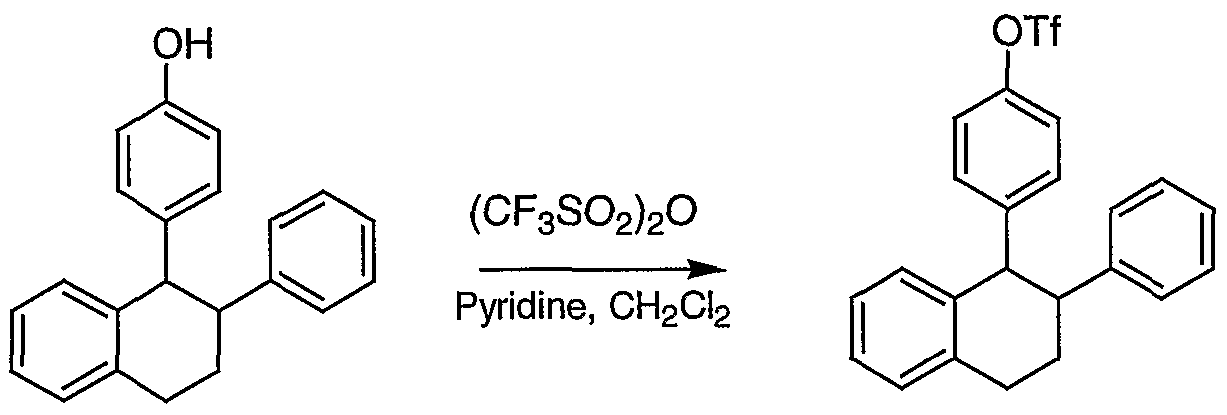
- Preparation of trifluoromethanesulfonyl ester X from phenol IX is well known in the literature. See: Larock, R. C. Comprehensive Organic Transformations, VCH Publishers, New York, 1989. Subsequent transformations of aryl-OTf X to Benzopyran XIII (steps 2-4) can be accomplished using the method described in Scheme 2.
- Tetrazole XV is prepared from benzaldehyde XIII in two steps.
- the first step is a 1,2-addition of acetonitrile to benzaldehyde XIII followed by elimination of water to provide cinnamonitrile XIV.
- a base such as KOH is used to deprotonate the hydrogen of the methyl group of acetonitrile in this reaction.
- the second step involves an addition of an azide to cinnamonitrile XIV to form tetrazole XV in the presence of a Lewis acid catalyst.
- compound XV may be further functionalized by methylation of the tetrazole group. Such N- methylation is well known in the art.
- Dihydro benzocycloheptenyl tetrazole XIX can be prepared from dihydro benzocycloheptenyl phenol XVI in three steps. Step 1) preparation of triflate XVII; step 2) Heck coupling, and step 3) tetrazole formation.
- the preparation of tetrazole XIX involves the addition of an azide to cinnamonitrile XVIII in the presence of a Lewis acid catalyst.
- compound XIX may be further functionalized by methylation of the tetrazole group . See Scheme 4.
- n 1 ,2
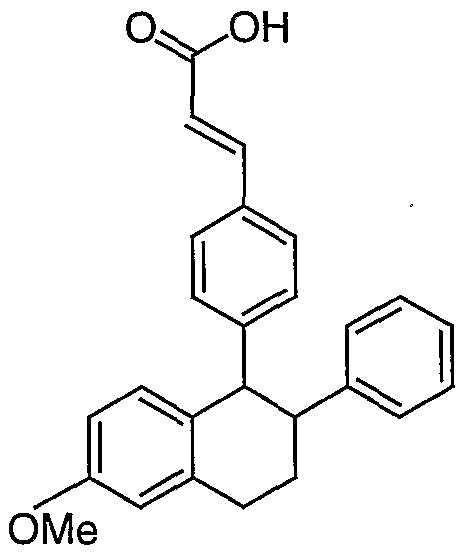
- ester (2d) (4.2 g, 11.0 mmol) in methanol (370 mL) and THF (225 mL) was added IN KOH (188 mL) . After stirring overnight the mixture was heated to 50°C for 0.5 h and was allowed to cool to rt . After stirring 2h the solvent was partially removed under reduced pressure, the mixture was acidified with IN HCl, and was extracted with EtOAc. The combined organic layers were washed with water, brine and were dried (MgS0 4 ) .
- Example le To a solution of la (0.20 g, 0.564 mmol) in CH 2 C1 2 (3 mL) were added trifluoromethanesulfonamide (0.236 g, 1.58 mmol), 4-dimethylaminopyridine (0.104 g, 0.851 mmol), and l-[3- (dimet ylamino)propyl] -3-ethylcarbodiimide hydrochloride (0.162 g, 0.851 mmol). After stirring at room temperature for 24 hours, PS-Trisamine resin (0.600 mg) was added and stirring continued for one hour. The resin was collected by vacuum filtration and washed with 5% MeOH in CH 2 C1 2 .
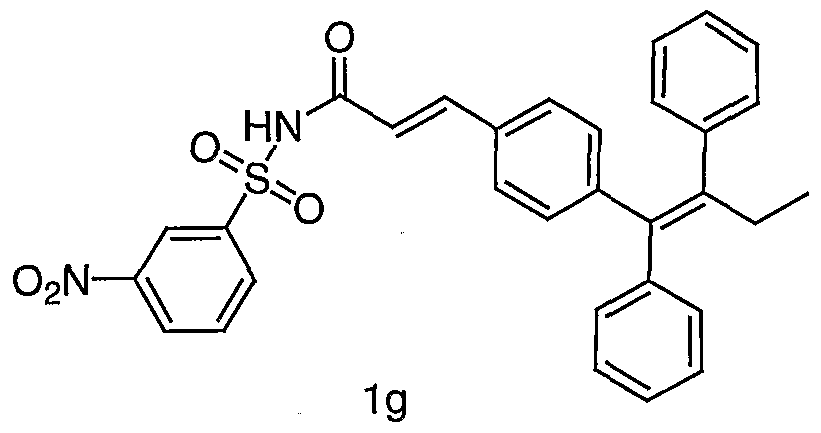
- Example lg Prepared from the coupling of la and 3- nitrobenzenesulfonamide by the method described in Procedure 1,
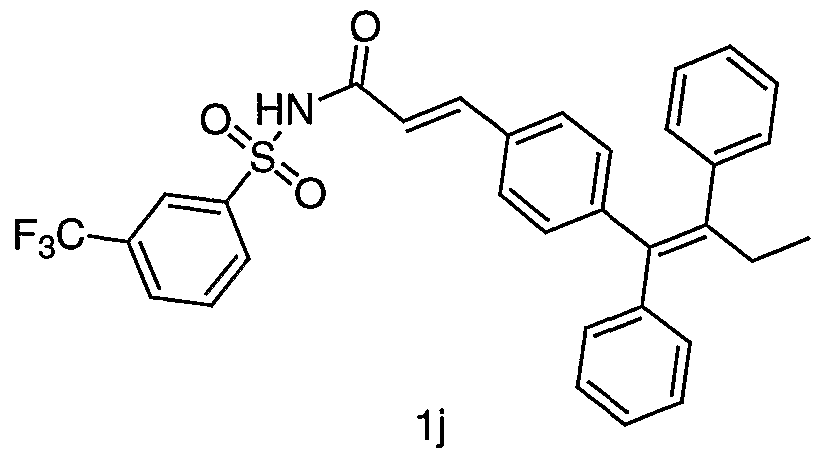
- Example 1j Prepared from the coupling of la and 3-
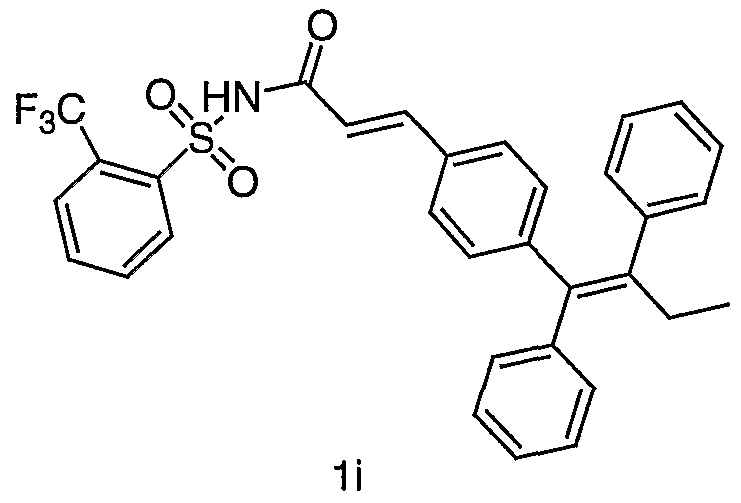
- Example lp Prepared from the coupling of la and o- toluenesulfonamide by the method described in Procedure 1,
- Example Is Prepared from the coupling of la and p- carboxybenzenesulfonamide methyl ester (Synlett, 1997, 375) by the method described in Procedure 1, Method B. Yield
- Example lu Prepared from the coupling of la and 3- methoxybenzenesulfonamide (Synlett, 1997, 375) by the method described in Procedure 1, Method B. Yield (27 %) ; X H NMR (d 6 -DMS0) ⁇ 12.18 (br s, IH) , 7.53-7.07 (m, 17H) ,
- Example lv Prepared from It by treating with boron tribromide (3 eq) in CH 2 Cl 2 at room temperature. Yield (29 %) ;
- Example lw Prepared from lu by treating with boron tribromide (3 eq) in CH 2 C1 2 at room temperature. Yield (57%) ;
- Example lx Prepared from the coupling of la and 3- chloropropanesulfonamide (Synlett, 1997, 375) by the method described in Procedure 1, Method A. Yield (80%) ;
- X H NMR (d 6 - DMSO) ⁇ 11.82 (br s, IH) , 7.48 (d, J 15.7 Hz, IH) , 7.40-7.09
- Example Iz Prepared from the coupling of lx and piperidine (15 eq) in tetrahydrofuran at reflux. The product precipitated from a mixture of ethyl acetate and IN HCl as the HCl salt.
- Example lcc Prepared from the coupling of laa with N,N- dimethylglycme (2.8 eq) using 4-dimethylaminopyridine (1.5 eq) and 1- [3- (dimethylamino)propyl] -3-ethylcarbodiimide hydrochloride (1.5 eq) in CH 2 C1 2 at room temperature. The product was isolated as the hydrochloride salt. Yield (28%) ;
- Example l h Prepared from the coupling of la and 2,2,2- trifluoroethanesulfonamide (Synlett, 1997, 375) by the method described in Procedure 1, Method A. Yield (34%); *H NMR (d 6 -
- Example Iii Prepared from the coupling of la and 2- thiophenesulfonamide (Synlett, 1997, 375) by the method described in Procedure 1, Method A. Yield (27%); H NMR (d 6 ⁇
- Example ljj Prepared from the coupling of la and [ (4- trifluoromethylphenyl) methyl] sulfonamide (Synlett, 1997, 375) by the method described in Procedure 1, Method B. Yield
- Example lkk Prepared from the coupling of Iff and methylamine hydrochloride (2.5 eq) in the presence of BOP reagent (2.2 eq) and 4-methylmorpholine (1.5 eq) using N,N- dimethylformamide as solvent.
- Compound 7a is prepared according to the procedure set forth on pages 138-144 of Volume 10 J.Med. Chem 1967.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurosurgery (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Urology & Nephrology (AREA)
- Biomedical Technology (AREA)
- Rheumatology (AREA)
- Neurology (AREA)
- Hospice & Palliative Care (AREA)
- Hematology (AREA)
- Emergency Medicine (AREA)
- Cardiology (AREA)
- Reproductive Health (AREA)
- Obesity (AREA)
- Psychiatry (AREA)
- Heart & Thoracic Surgery (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyrane Compounds (AREA)
Abstract
Description





















































































Claims








Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HU0500573A HUP0500573A2 (en) | 2001-08-11 | 2002-08-09 | Triphenyl-ethylene derivatives as selective estrogen receptor modulators and pharmaceutical compositions containing the same |
| JP2003521198A JP2005528320A (en) | 2001-08-11 | 2002-08-09 | Selective estrogen receptor modulator |
| SK60-2004A SK602004A3 (en) | 2001-08-11 | 2002-08-09 | Selective estrogen receptor modulators |
| AU2002323098A AU2002323098A1 (en) | 2001-08-11 | 2002-08-09 | Selective estrogen receptor modulators |
| EEP200400061A EE200400061A (en) | 2001-08-11 | 2002-08-09 | Triphenylethylene Derivatives as Selective Estrogen Receptor Modulators and Pharmaceutical Compositions Containing Them |
| EP02757058A EP1417169A2 (en) | 2001-08-11 | 2002-08-09 | Selective estrogen receptor modulators |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US31146601P | 2001-08-11 | 2001-08-11 | |
| US60/311,466 | 2001-08-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2003016270A2 true WO2003016270A2 (en) | 2003-02-27 |
| WO2003016270A3 WO2003016270A3 (en) | 2003-11-06 |
Family
ID=23206988
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2002/025394 WO2003016270A2 (en) | 2001-08-11 | 2002-08-09 | Selective estrogen receptor modulators |
Country Status (11)
| Country | Link |
|---|---|
| US (3) | US6927224B2 (en) |
| EP (1) | EP1417169A2 (en) |
| JP (1) | JP2005528320A (en) |
| AU (1) | AU2002323098A1 (en) |
| BG (1) | BG108561A (en) |
| CZ (1) | CZ2004220A3 (en) |
| EE (1) | EE200400061A (en) |
| HU (1) | HUP0500573A2 (en) |
| PL (1) | PL374215A1 (en) |
| SK (1) | SK602004A3 (en) |
| WO (1) | WO2003016270A2 (en) |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005033056A2 (en) * | 2003-10-08 | 2005-04-14 | Smithkline Beecham Corporation | Triphenylethylene compounds as selective estrogen receptor modulators |
| US7067551B2 (en) | 2000-09-01 | 2006-06-27 | Novartis Ag | Deacetylase inhibitors |
| US7560589B2 (en) | 2003-07-28 | 2009-07-14 | Smithkline Beecham Corporation | Cycloalkylidene compounds as modulators of estrogen receptor |
| US7601855B2 (en) | 2004-09-21 | 2009-10-13 | Novogen Research Pty Ltd | Substituted chroman derivatives, medicaments and use in therapy |
| WO2010107474A1 (en) * | 2009-03-16 | 2010-09-23 | The Research Foundation Of State University Of New York | Antiestrogens for breast cancer therapy |
| US8080675B2 (en) | 2004-09-21 | 2011-12-20 | Marshall Edwards, Inc. | Chroman derivatives, medicaments and use in therapy |
| GB2483736A (en) * | 2010-09-16 | 2012-03-21 | Aragon Pharmaceuticals Inc | Estrogen receptor modulators and use thereof |
| WO2013142266A1 (en) | 2012-03-20 | 2013-09-26 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| US8703810B2 (en) | 2010-06-10 | 2014-04-22 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| US8853423B2 (en) | 2010-06-17 | 2014-10-07 | Seragon Pharmaceuticals, Inc. | Indane estrogen receptor modulators and uses thereof |
| CN104211695A (en) * | 2013-06-04 | 2014-12-17 | 中国医学科学院医药生物技术研究所 | New use of group of carbamyl phenylsulfonyl compounds |
| US9187460B2 (en) | 2011-12-14 | 2015-11-17 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| CN105121413A (en) * | 2013-03-14 | 2015-12-02 | 赛拉根医药股份有限公司 | Polycyclic estrogen receptor modulators and uses thereof |
| US9663484B2 (en) | 2010-11-01 | 2017-05-30 | Mei Pharma, Inc. | Isoflavonoid compounds and methods for the treatment of cancer |
| WO2019023481A1 (en) * | 2017-07-28 | 2019-01-31 | Zeno Royalties & Milestones, LLC | Acrylic acid analogs |
| WO2019169001A1 (en) * | 2018-02-27 | 2019-09-06 | Artax Biopharma Inc. | Chromene derivatives as inhibitors of tcr-nck interaction |
| US10570090B2 (en) | 2016-02-15 | 2020-02-25 | Sanofi | Substituted 6,7-dihydro-5H-benzo[7]annulene compounds, processes for their preparation and therapeutic uses thereof |
| US10624878B2 (en) | 2015-10-01 | 2020-04-21 | Olema Pharmaceuticals, Inc. | Tetrahydro-1H-pyrido [3,4-b]indole anti-estrogenic drugs |
| CN112105607A (en) * | 2018-01-22 | 2020-12-18 | 雷迪厄斯制药公司 | Estrogen receptor modulating compounds |
| US10980774B2 (en) | 2015-02-02 | 2021-04-20 | Mei Pharma, Inc. | Combination therapies |
| EP3659307A4 (en) * | 2017-07-28 | 2021-09-22 | Yale University | Anticancer Drugs and Methods of Making and Using Same |
| US11149031B2 (en) | 2016-11-17 | 2021-10-19 | Sanofi | Substituted N-(3-fluoropropyl)-pyrrolidine compounds, processes for their preparation and therapeutic uses thereof |
| US11260057B2 (en) | 2017-07-24 | 2022-03-01 | Sanofi | Combination comprising palbociclib and 6-(2,4-dichlorophenyl)-5-[4-[(3S)-1-(3-fluoropropyl)pyrrolidin-3-yl]oxyphenyl]-8,9-dihydro-7H-benzo[7] annulene-2-carboxylic acid and its use for the treatment of cancer |
| US11713296B2 (en) | 2018-09-07 | 2023-08-01 | Sanofi | Salts of methyl 6-(2,4-dichlorophenyl)-5-[4-[(3S)-l-(3-fluoropropyl)pyrrolidin-3-yl]oxyphenyl]-8,9-dihydro-7H-benzo[7]annulene-2-carboxylate and preparation process thereof |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6180731A (en) * | 1984-09-28 | 1986-04-24 | Toshiba Corp | Method of manufacture valve |
| US7208526B2 (en) * | 2005-05-20 | 2007-04-24 | Hoffmann-La Roche Inc. | Styrylsulfonamides |
| EP2048126A1 (en) * | 2007-10-11 | 2009-04-15 | Bayer Schering Pharma AG | Benzocycloheptane derivatives as selectively active oestrogens |
| NZ591955A (en) | 2007-10-16 | 2011-10-28 | Repros Therapeutics Inc | Trans-clomiphene for diabetes mellitus type 2 |
| US20090215733A1 (en) * | 2008-02-26 | 2009-08-27 | Michael Charles Scally | Therapy for the prophylaxis or treatment of adverse body composition changes and/or decreased muscle strength after androgen or gnrh analogue intake |
| DE102010030538A1 (en) * | 2010-06-25 | 2011-12-29 | Bayer Schering Pharma Aktiengesellschaft | 6,7-Dihydro-5H-benzo [7] annulene derivatives, process for their preparation, pharmaceutical preparations containing them and their use for the preparation of medicaments |
| DE102011087987A1 (en) | 2011-12-08 | 2013-06-13 | Bayer Intellectual Property Gmbh | 6,7-Dihydro-5H-benzo [7] annulene derivatives, process for their preparation, pharmaceutical preparations containing them and their use for the preparation of medicaments |
| ES2681023T3 (en) | 2012-02-29 | 2018-09-11 | Repros Therapeutics Inc. | Combination therapy to treat androgen deficit |
| WO2014008159A1 (en) * | 2012-07-03 | 2014-01-09 | The University Of North Carolina At Chapel Hill | Selective estrogen receptor degraders for treatment of tamoxifen resistant tumors |
| CN105061316B (en) * | 2015-07-17 | 2017-12-22 | 苏州大学 | Thick cyclics, preparation method and purposes |
| EP3373967B9 (en) | 2015-11-10 | 2023-10-04 | Paracrine Therapeutics AB | Treatment of er-negative breast cancer with an pdgf-cc inhibitor and an anti estrogen |
| CN111032627B (en) * | 2017-09-25 | 2023-10-20 | 罗欣健康科技发展(北京)有限公司 | Crystal form of estrogen receptor inhibitor and preparation method thereof |
| TW202313558A (en) * | 2021-07-08 | 2023-04-01 | 大陸商勤浩醫藥(蘇州)有限公司 | Benzo-seven-membered heterocyclic compound and application thereof |
| WO2023023531A1 (en) * | 2021-08-20 | 2023-02-23 | Biotheryx, Inc. | Estrogen receptor degraders, pharmaceutical compositions, and therapeutic applications |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5681835A (en) * | 1994-04-25 | 1997-10-28 | Glaxo Wellcome Inc. | Non-steroidal ligands for the estrogen receptor |
| WO1999008682A1 (en) * | 1997-08-15 | 1999-02-25 | Duke University | A method of preventing or treating estrogen-dependent diseases and disorders |
| US6005003A (en) * | 1998-05-06 | 1999-12-21 | Hoechst Marion Roussel | Derivatives of dihydro or tetrahydronaphthalene, and the pharmaceutical compositions containing them |
| WO2001026651A2 (en) * | 1999-10-14 | 2001-04-19 | Endorecherche, Inc. | Selective estrogen receptor modulators in the treatment or reduction of the risk of acquiring hypertension, cardiovascular diseases, and insulin resistance |
| WO2001077055A2 (en) * | 2000-04-05 | 2001-10-18 | Bristol-Myers Squibb Pharma Company | Specific salt forms of triphenylethylene derivatives as selective estrogen receptor modulators |
| WO2001077057A2 (en) * | 2000-04-05 | 2001-10-18 | Dupont Pharmaceuticals Company | Halogenated triphenylethylene derivatives as selective estrogen receptor modulators |
-
2002
- 2002-08-09 CZ CZ2004220A patent/CZ2004220A3/en unknown
- 2002-08-09 US US10/216,253 patent/US6927224B2/en not_active Expired - Lifetime
- 2002-08-09 EP EP02757058A patent/EP1417169A2/en not_active Withdrawn
- 2002-08-09 WO PCT/US2002/025394 patent/WO2003016270A2/en not_active Application Discontinuation
- 2002-08-09 EE EEP200400061A patent/EE200400061A/en unknown
- 2002-08-09 SK SK60-2004A patent/SK602004A3/en not_active Application Discontinuation
- 2002-08-09 AU AU2002323098A patent/AU2002323098A1/en not_active Abandoned
- 2002-08-09 PL PL02374215A patent/PL374215A1/en not_active Application Discontinuation
- 2002-08-09 JP JP2003521198A patent/JP2005528320A/en not_active Withdrawn
- 2002-08-09 HU HU0500573A patent/HUP0500573A2/en unknown
-
2004
- 2004-02-03 BG BG108561A patent/BG108561A/en unknown
-
2005
- 2005-06-15 US US11/152,888 patent/US7045540B2/en not_active Expired - Lifetime
- 2005-06-15 US US11/152,891 patent/US7323587B2/en not_active Expired - Lifetime
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5681835A (en) * | 1994-04-25 | 1997-10-28 | Glaxo Wellcome Inc. | Non-steroidal ligands for the estrogen receptor |
| WO1999008682A1 (en) * | 1997-08-15 | 1999-02-25 | Duke University | A method of preventing or treating estrogen-dependent diseases and disorders |
| US6005003A (en) * | 1998-05-06 | 1999-12-21 | Hoechst Marion Roussel | Derivatives of dihydro or tetrahydronaphthalene, and the pharmaceutical compositions containing them |
| WO2001026651A2 (en) * | 1999-10-14 | 2001-04-19 | Endorecherche, Inc. | Selective estrogen receptor modulators in the treatment or reduction of the risk of acquiring hypertension, cardiovascular diseases, and insulin resistance |
| WO2001077055A2 (en) * | 2000-04-05 | 2001-10-18 | Bristol-Myers Squibb Pharma Company | Specific salt forms of triphenylethylene derivatives as selective estrogen receptor modulators |
| WO2001077057A2 (en) * | 2000-04-05 | 2001-10-18 | Dupont Pharmaceuticals Company | Halogenated triphenylethylene derivatives as selective estrogen receptor modulators |
Non-Patent Citations (2)
| Title |
|---|
| DATABASE CAPLUS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; Database accession no. 121:34956 XP002222761 -& TIMOTHY M. WILSON ET AL: "3-[4-(1,2-Diphenylbut-1-enyl)phenylÜacryl ic Acid: A Non-Steroidal Estrogen with Functional Selectivity for Bone over Uterus in Rats" J.MED.CHEM., vol. 37, no. 11, 1994, pages 1550-1552, XP002091552 * |
| THOMAS S. SCANLAN: "Differential SERM activation of the estrogen receptors (ERalpha and ERbeta) at AP-1 sites" CHEMISTRY & BIOLOGY, vol. 8, no. 5, 2001, pages 427-436, XP002220838 * |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7067551B2 (en) | 2000-09-01 | 2006-06-27 | Novartis Ag | Deacetylase inhibitors |
| US7799828B2 (en) | 2003-07-28 | 2010-09-21 | Glaxosmithkline Llc | Cycloalkylidene compounds as modulators of estrogen receptor |
| US7560589B2 (en) | 2003-07-28 | 2009-07-14 | Smithkline Beecham Corporation | Cycloalkylidene compounds as modulators of estrogen receptor |
| US7569601B2 (en) | 2003-07-28 | 2009-08-04 | Smithkline Beecham Corporation | Cycloalkylidene compounds as modulators of estrogen receptor |
| WO2005033056A3 (en) * | 2003-10-08 | 2005-06-23 | Smithkline Beecham Corp | Triphenylethylene compounds as selective estrogen receptor modulators |
| US7442833B2 (en) | 2003-10-08 | 2008-10-28 | Smithkline Beecham Corporation | Triphenylethylene compounds as selective estrogen receptor modulators |
| WO2005033056A2 (en) * | 2003-10-08 | 2005-04-14 | Smithkline Beecham Corporation | Triphenylethylene compounds as selective estrogen receptor modulators |
| US9138478B2 (en) | 2004-09-21 | 2015-09-22 | Mei Pharma, Inc. | Substituted chroman derivatives, medicaments and use in therapy |
| US8697891B2 (en) | 2004-09-21 | 2014-04-15 | Marshall Edwards, Inc. | Substituted chroman derivatives, medicaments and use in therapy |
| US8080675B2 (en) | 2004-09-21 | 2011-12-20 | Marshall Edwards, Inc. | Chroman derivatives, medicaments and use in therapy |
| US8084628B2 (en) | 2004-09-21 | 2011-12-27 | Marshall Edwards, Inc. | Substituted chroman derivatives, medicaments and use in therapy |
| US9381186B2 (en) | 2004-09-21 | 2016-07-05 | Mei Pharma, Inc. | Substituted chroman derivatives, medicaments and use in therapy |
| US9198895B2 (en) | 2004-09-21 | 2015-12-01 | Mei Pharma, Inc. | Chroman derivatives, medicaments and use in therapy |
| US7601855B2 (en) | 2004-09-21 | 2009-10-13 | Novogen Research Pty Ltd | Substituted chroman derivatives, medicaments and use in therapy |
| US8461361B2 (en) | 2004-09-21 | 2013-06-11 | Marshall Edwards, Inc. | Chroman derivatives, medicaments and use in therapy |
| US8957109B2 (en) | 2004-09-21 | 2015-02-17 | Mei Pharma, Inc. | Chroman derivatives, medicaments and use in therapy |
| WO2010107474A1 (en) * | 2009-03-16 | 2010-09-23 | The Research Foundation Of State University Of New York | Antiestrogens for breast cancer therapy |
| US9078871B2 (en) | 2010-06-10 | 2015-07-14 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| US8703810B2 (en) | 2010-06-10 | 2014-04-22 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| US8853423B2 (en) | 2010-06-17 | 2014-10-07 | Seragon Pharmaceuticals, Inc. | Indane estrogen receptor modulators and uses thereof |
| EP2735562A1 (en) * | 2010-09-16 | 2014-05-28 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| US8299112B2 (en) | 2010-09-16 | 2012-10-30 | Aragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| US9399646B2 (en) | 2010-09-16 | 2016-07-26 | Genentech, Inc. | Estrogen receptor modulators and uses thereof |
| EP2616445A2 (en) * | 2010-09-16 | 2013-07-24 | Aragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| GB2483736A (en) * | 2010-09-16 | 2012-03-21 | Aragon Pharmaceuticals Inc | Estrogen receptor modulators and use thereof |
| CN103189361A (en) * | 2010-09-16 | 2013-07-03 | 亚拉冈制药公司 | Estrogen receptor modulators and use thereof |
| US8455534B2 (en) | 2010-09-16 | 2013-06-04 | Aragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| GB2483736B (en) * | 2010-09-16 | 2012-08-29 | Aragon Pharmaceuticals Inc | Estrogen receptor modulators and uses thereof |
| CN103189361B (en) * | 2010-09-16 | 2015-09-30 | 塞拉根制药公司 | Estrogenic agents and uses thereof |
| EP2616445A4 (en) * | 2010-09-16 | 2014-01-15 | Seragon Pharmaceuticals Inc | Estrogen receptor modulators and uses thereof |
| US10973799B2 (en) | 2010-11-01 | 2021-04-13 | Mei Pharma, Inc. | Isoflavonoid compositions and methods for the treatment of cancer |
| US10369132B2 (en) | 2010-11-01 | 2019-08-06 | Mei Pharma, Inc. | Isoflavonoid compositions and methods for the treatment of cancer |
| US9663484B2 (en) | 2010-11-01 | 2017-05-30 | Mei Pharma, Inc. | Isoflavonoid compounds and methods for the treatment of cancer |
| US10105346B2 (en) | 2010-11-01 | 2018-10-23 | Mei Pharma, Inc. | Isoflavonoid compounds and methods for the treatment of cancer |
| US11583514B2 (en) | 2010-11-01 | 2023-02-21 | Mei Pharma, Inc. | Isoflavonoid compounds and methods for the treatment of cancer |
| US11723893B2 (en) | 2010-11-01 | 2023-08-15 | Mei Pharma, Inc. | Isoflavonoid compositions and methods for the treatment of cancer |
| US9981936B2 (en) | 2010-11-01 | 2018-05-29 | Mei Pharma, Inc. | Isoflavonoid compositions and methods for the treatment of cancer |
| US9708283B2 (en) | 2010-11-01 | 2017-07-18 | Mei Pharma, Inc. | Isoflavonoid compositions and methods for the treatment of cancer |
| US9193714B2 (en) | 2011-12-14 | 2015-11-24 | Seragon Pharmaceuticals, Inc. | Fluorinated estrogen receptor modulators and uses thereof |
| US9187460B2 (en) | 2011-12-14 | 2015-11-17 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| EP2828243A4 (en) * | 2012-03-20 | 2015-10-28 | Seragon Pharmaceuticals Inc | Estrogen receptor modulators and uses thereof |
| US9499538B2 (en) | 2012-03-20 | 2016-11-22 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| WO2013142266A1 (en) | 2012-03-20 | 2013-09-26 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| CN105121413B (en) * | 2013-03-14 | 2018-09-14 | 赛拉根医药股份有限公司 | Polycyclic estrogen receptor modulators and uses thereof |
| CN105121413A (en) * | 2013-03-14 | 2015-12-02 | 赛拉根医药股份有限公司 | Polycyclic estrogen receptor modulators and uses thereof |
| CN104211695A (en) * | 2013-06-04 | 2014-12-17 | 中国医学科学院医药生物技术研究所 | New use of group of carbamyl phenylsulfonyl compounds |
| CN104211695B (en) * | 2013-06-04 | 2017-04-12 | 中国医学科学院医药生物技术研究所 | Use of group of carbamyl phenylsulfonyl compounds |
| US10980774B2 (en) | 2015-02-02 | 2021-04-20 | Mei Pharma, Inc. | Combination therapies |
| US11229630B2 (en) | 2015-10-01 | 2022-01-25 | Olema Pharmaceuticals, Inc. | Tetrahydro-1H-pyrido [3,4-b]indole anti-estrogenic drugs |
| US11672785B2 (en) | 2015-10-01 | 2023-06-13 | Olema Pharmaceuticals, Inc. | Tetrahydro-1H-pyrido [3,4-b]indole anti-estrogenic drugs |
| US10624878B2 (en) | 2015-10-01 | 2020-04-21 | Olema Pharmaceuticals, Inc. | Tetrahydro-1H-pyrido [3,4-b]indole anti-estrogenic drugs |
| US11214541B2 (en) | 2016-02-15 | 2022-01-04 | Sanofi | Substituted 6,7-dihydro-5H-benzo[7]annulene compounds, processes for their preparation and therapeutic uses thereof |
| US10570090B2 (en) | 2016-02-15 | 2020-02-25 | Sanofi | Substituted 6,7-dihydro-5H-benzo[7]annulene compounds, processes for their preparation and therapeutic uses thereof |
| US11149031B2 (en) | 2016-11-17 | 2021-10-19 | Sanofi | Substituted N-(3-fluoropropyl)-pyrrolidine compounds, processes for their preparation and therapeutic uses thereof |
| US11260057B2 (en) | 2017-07-24 | 2022-03-01 | Sanofi | Combination comprising palbociclib and 6-(2,4-dichlorophenyl)-5-[4-[(3S)-1-(3-fluoropropyl)pyrrolidin-3-yl]oxyphenyl]-8,9-dihydro-7H-benzo[7] annulene-2-carboxylic acid and its use for the treatment of cancer |
| WO2019023481A1 (en) * | 2017-07-28 | 2019-01-31 | Zeno Royalties & Milestones, LLC | Acrylic acid analogs |
| EP3659307A4 (en) * | 2017-07-28 | 2021-09-22 | Yale University | Anticancer Drugs and Methods of Making and Using Same |
| CN112105607A (en) * | 2018-01-22 | 2020-12-18 | 雷迪厄斯制药公司 | Estrogen receptor modulating compounds |
| US11008310B2 (en) | 2018-02-27 | 2021-05-18 | Artax Biopharma Inc. | Chromene derivatives as inhibitors of TCR-Nck interaction |
| WO2019169001A1 (en) * | 2018-02-27 | 2019-09-06 | Artax Biopharma Inc. | Chromene derivatives as inhibitors of tcr-nck interaction |
| US10696663B2 (en) | 2018-02-27 | 2020-06-30 | Artax Biopharma Inc. | Chromene derivatives as inhibitors of TCR-NCK interaction |
| CN112189009A (en) * | 2018-02-27 | 2021-01-05 | 阿塔克斯生物制药有限公司 | Chromene derivatives as inhibitors of TCR-NCK interaction |
| AU2019229258B2 (en) * | 2018-02-27 | 2023-09-14 | Artax Biopharma Inc. | Chromene derivatives as inhibitors of TCR-Nck interaction |
| CN112189009B (en) * | 2018-02-27 | 2023-09-29 | 阿塔克斯生物制药有限公司 | Chromene derivatives as inhibitors of TCR-NCK interactions |
| US11807633B2 (en) | 2018-02-27 | 2023-11-07 | Artax Biopharma Inc. | Chromene derivatives as inhibitors of TCR-Nck interaction |
| US11713296B2 (en) | 2018-09-07 | 2023-08-01 | Sanofi | Salts of methyl 6-(2,4-dichlorophenyl)-5-[4-[(3S)-l-(3-fluoropropyl)pyrrolidin-3-yl]oxyphenyl]-8,9-dihydro-7H-benzo[7]annulene-2-carboxylate and preparation process thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US6927224B2 (en) | 2005-08-09 |
| PL374215A1 (en) | 2005-10-03 |
| CZ2004220A3 (en) | 2004-06-16 |
| BG108561A (en) | 2005-04-30 |
| AU2002323098A1 (en) | 2003-03-03 |
| EE200400061A (en) | 2004-04-15 |
| US20030105148A1 (en) | 2003-06-05 |
| EP1417169A2 (en) | 2004-05-12 |
| US20050267183A1 (en) | 2005-12-01 |
| US7323587B2 (en) | 2008-01-29 |
| JP2005528320A (en) | 2005-09-22 |
| US20050245602A1 (en) | 2005-11-03 |
| WO2003016270A3 (en) | 2003-11-06 |
| SK602004A3 (en) | 2004-11-03 |
| US7045540B2 (en) | 2006-05-16 |
| HUP0500573A2 (en) | 2005-11-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6927224B2 (en) | Selective estrogen receptor modulators | |
| JP4072208B2 (en) | Trisubstituted phenyl derivatives with biological activity of retinoid agonist, antagonist or inverse agonist type | |
| EP1268389B1 (en) | Halogenated triphenylethylene derivatives as selective estrogen receptor modulators | |
| NZ545032A (en) | Cycloalkylidene compounds as modulators of estrogen receptor | |
| WO1996008483A1 (en) | Gallic acid derivatives, method for their preparation and their use as drugs | |
| EP0524846A1 (en) | 2-(Piperidin-1-yl)ethanol derivatives, their preparation and their use in therapeutics | |
| Carpino et al. | Benz [f] indene | |
| US6417394B2 (en) | Specific salt forms of triphenylethylene derivatives as selective estrogen receptor modulators | |
| EP0338895B1 (en) | Heteroarotinoid derivatives, their processes of preparation and pharmaceutical compositions containing them | |
| AU2001253191A1 (en) | Specific salt forms of triphenylethylene derivatives as selective estrogen receptor modulators | |
| Manchand et al. | Synthesis of the leukotriene antagonist ablukast | |
| EP0100257B1 (en) | Aminoalkyl naphthalene derivatives, their salts, process for their preparation and the therapeutical use of these derivatives and salts | |
| CN100532353C (en) | Preparation process of (Z)-3'-amino-3,4,4', 5-tetramethoxyl stilbene | |
| EP3459947B1 (en) | Novel chiral phase-transfer catalyst, and method for preparing alpha-amino acid by using the same | |
| JP3030347B2 (en) | Monoalkylation method | |
| CN115806521A (en) | Monofluoromethyl etoricoxib and preparation method thereof | |
| CH650255A5 (en) | IMIDAZOLE DERIVATIVES, THEIR PREPARATION PROCESS AND PHARMACEUTICAL COMPOSITION CONTAINING THEM. | |
| CH446379A (en) | Process for the preparation of substituted acethydroxamic acids | |
| JPS6330895B2 (en) | ||
| CH637378A5 (en) | CYCLOALKYLALKYLIC BENZAMIDES. | |
| JPS62181244A (en) | 4(4(4-chlorophenyl)butoxy)phenylmethylamine derivative, acid addition salt and production thereof | |
| BE842763A (en) | NEW PHOSPHINYL DERIVATIVES AND METHODS FOR PREPARING THESE DERIVATIVES |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BY BZ CA CH CN CO CR CU CZ DE DM DZ EC EE ES FI GB GD GE GH HR HU ID IL IN IS JP KE KG KP KR LC LK LR LS LT LU LV MA MD MG MN MW MX MZ NO NZ OM PH PL PT RU SD SE SG SI SK SL TJ TM TN TR TZ UA UG UZ VC VN YU ZA ZM Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ UG ZM ZW AM AZ BY KG KZ RU TJ TM AT BE BG CH CY CZ DK EE ES FI FR GB GR IE IT LU MC PT SE SK TR BF BJ CF CG CI GA GN GQ GW ML MR NE SN TD TG Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LU MC NL PT SE SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 602004 Country of ref document: SK |
|
| ENP | Entry into the national phase |
Ref document number: 10856102 Country of ref document: BG Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 108561 Country of ref document: BG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003521198 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2004-220 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 374215 Country of ref document: PL Ref document number: 2002757058 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002757058 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2004-220 Country of ref document: CZ |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2002757058 Country of ref document: EP |