WO2002018339A2 - Compounds useful as insecticides, compounds useful as acaricides, and processes to use and make same - Google Patents
Compounds useful as insecticides, compounds useful as acaricides, and processes to use and make same Download PDFInfo
- Publication number
- WO2002018339A2 WO2002018339A2 PCT/US2001/026777 US0126777W WO0218339A2 WO 2002018339 A2 WO2002018339 A2 WO 2002018339A2 US 0126777 W US0126777 W US 0126777W WO 0218339 A2 WO0218339 A2 WO 0218339A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- group
- halo
- compound
- mmol
- Prior art date
Links
- 0 C*C(C(C)Oc(cn1)ccc1Cl)=O Chemical compound C*C(C(C)Oc(cn1)ccc1Cl)=O 0.000 description 2
- WSONBNOEPFNTFW-UHFFFAOYSA-N CC(C(NC)=O)Oc(cc1)cnc1Br Chemical compound CC(C(NC)=O)Oc(cc1)cnc1Br WSONBNOEPFNTFW-UHFFFAOYSA-N 0.000 description 1
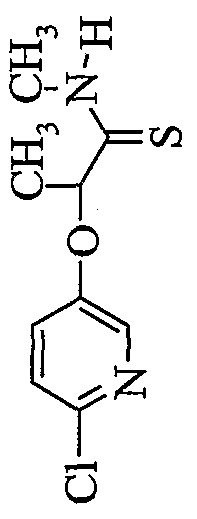
- QVSTVLVAOMRVAP-UHFFFAOYSA-N CC(C(NC)=S)Oc(cc1)cnc1Cl Chemical compound CC(C(NC)=S)Oc(cc1)cnc1Cl QVSTVLVAOMRVAP-UHFFFAOYSA-N 0.000 description 1
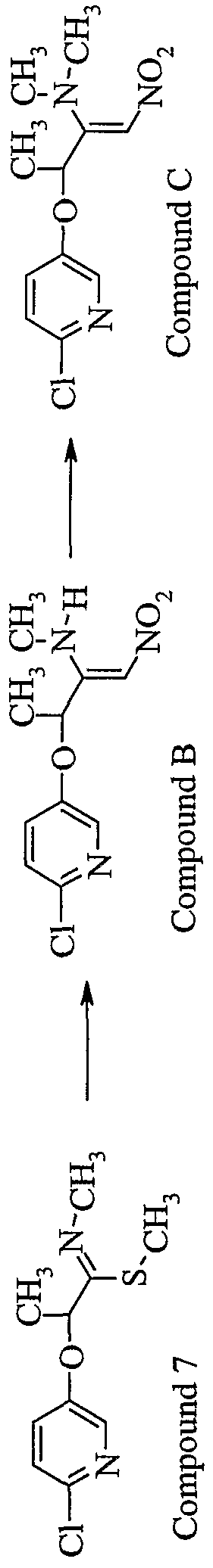
- CEUPTQMQOKWVFN-YFHOEESVSA-N CCN/C(/C(C)Oc(cc1)cnc1Cl)=C\[N+]([O-])=O Chemical compound CCN/C(/C(C)Oc(cc1)cnc1Cl)=C\[N+]([O-])=O CEUPTQMQOKWVFN-YFHOEESVSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/34—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with one nitrogen atom as the only ring hetero atom
- A01N43/40—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with one nitrogen atom as the only ring hetero atom six-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/65—One oxygen atom attached in position 3 or 5
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/14—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- This invention provides compounds that are useful as insecticides and compounds that are useful as acaricides, and processes of making and using these compounds.
- A represents a five or six membered heterocyclic ring containing at least one heteroatom selected from the group consisting of an oxygen, sulfur, or nitrogen.
- a six membered heterocyclic ring is used. It is even more preferred when such six membered heterocyclic ring contains one or two nitrogen atoms as the heteroatoms.
- This heterocyclic ring may be substituted by one or more substituents selected from the group consisting of fluorine, chlorine, bromine, iodine, C MO alkyl, halo C ⁇ -10 alkyl, nitro, cyano, C MO alkoxy, C MO alkylthio, C MO alkylsulfinyl, C MO alkylsulfonyl, C O alkenyl, halo C O alkoxy, halo C MO alkylthio, halo C MO alkenyl, acylamino, haloacylamino, CM O alkoxycarbonyl, C MO alkynyl, amino, CM O alkylamino, C O dialkylamino, C 3 . ⁇ 2 cycloalkyl, CM O alkoxyalkyl, acyl, formyl, C ⁇ - ⁇ 2 aryl, mono-or poly substituted C 6- ⁇ 2 aryl, heteroaryl, and mono-or poly substitute
- the C MO is a C] -6
- the heterocyclic ring is substituted, it is preferred when it is mono- substituted with either methyl, ethyl, fluoro, chloro, or bromo.
- the substituent is ortho to a heteroatom.
- E is selected from the group consisting of O, SO Mask where n is 0-2, NH, and NX where X is selected from the group consisting of CM O alkyl or halo C MO alkyl. Currently, it is preferred when E is O.
- J and R are independently selected from the group consisting of H, CM O alkyl, CMO alkenyl, C O alkynyl, halo CM O alkyl, and CM O alkoxyalkyl. Currently, it is preferred when J and R are H.
- G and T are independently selected from the group consisting of H, C MO alkyl, CM O alkenyl, C MO alkynyl, halo C MO alkyl, and CMO alkoxyalkyl. Currently, it is preferred when G and T are methyl or ethyl.
- G and T can also be joined together by a single bond, or through a connecting bridge, where such connecting bridge is selected from the group consisting of CH 2 , CHCH3, C(CH 3 ) 2 , CH(halo C M0 alkyl), C(halo C 0 alkyl) 2 , CHF, CF 2 , O, SO n where n is 0-2, NH, and NX where X is selected from the group consisting of CM O alkyl or halo CM O alkyl.
- the connecting bridge is a single bond or a CH 2 .
- the compounds of the invention are useful for the control of insects, mites, and aphids. Therefore, the present invention also is directed to a method for inhibiting an insect, mite, or aphid which comprises applying to a locus of the insect or mite an insect- or mite-inhibiting amount of a compound of Formula One.
- these compounds control insects in the order Homoptera, including the families Aphididae (aphids), Aleyrodidae (whiteflies), Delphacidae (planthoppers), and Cicadellidae (leafhoppers). They also control insects in the order Coleoptera (beetles), including the family Chrysomelidae (leaf beetles).
- the compounds are useful for reducing populations of insects and mites and are useful in a method of inhibiting an insect or mite population which comprises applying to a locus of the insect or mite an effective insect- or mite-inactivating amount of a compound of Formula One.
- insects or mites is a term used herein to refer to the environment in which the insects or mites live or where their eggs are present, including the air surrounding them, the food they eat, or objects or materials which they contact.
- plant-ingesting insects or mites can be controlled by applying the active compound to plant parts that the insects or mites eat, particularly the foliage.
- Soil- inhabiting insects such as termites can be controlled by applying the active compound to the soil that the insects move through.
- Insects such as fleas that infest animals can be controlled by applying the active compound to the animal that is infested. It is contemplated that the compounds might also be useful to protect textiles, paper, stored grain, or seeds by applying an active compound to such substance.
- inhibiting an insect or mite refers to a decrease in the numbers of living insects or mites, or a decrease in the number of viable insect or mite eggs.
- the extent of reduction accomplished by a compound depends, of course, upon the application rate of the compound, the particular compound used, and the target insect or mite species. At least an inactivating amount should be used.
- the terms "insect-inactivating amount” and “mite-inactivating amount” are used to describe the amount, which is sufficient to cause a measurable reduction in the treated insect or mite, population. Generally an amount in the range from about 1 to about 1000 ppm by weight active compound is used.
- the present invention is directed to a method for inhibiting a mite or aphid which comprises applying to a plant an effective mite- or aphid- inactivating amount of a compound of Formula One.
- compositions which comprise a compound of this invention and a phytologically-acceptable inert carrier.
- the compositions are either concentrated formulations which are dispersed in water for application, or are dust or granular formulations which are applied without further treatment.
- the compositions are prepared according to procedures and formulae which are conventional in the agricultural chemical art, but which are novel and important because ofthe presence therein ofthe compounds of this invention.
- the dispersions in which the compounds are applied are most often aqueous suspensions or emulsions prepared from concentrated formulations ofthe compounds.
- Such water-soluble, water-suspendable or emulsifiable formulations are either solids, usually known as wettable powders, or liquids usually known as emulsifiable concentrates or aqueous suspensions.
- Wettable powders which may be compacted to form water dispersible granules, comprise an intimate mixture of the active compound, an inert carrier, and surfactants.
- the concentration ofthe active compound is usually from about 10% to about 90% by weight.
- the inert carrier is usually chosen from among the attapulgite clays, the montmorillonite clays, the diatomaceous earths, or the purified silicates.
- Effective surfactants comprising from about 0.5% to about 10% of the wettable powder, are found among the sulfonated lignins, the condensed naphthalenesulfonates, the naphthalenesulfonates, the alkylbenzenesulfonates, the alkyl sulfates, and nonionic surfactants such as ethylene oxide adducts of alkyl phenols.
- Emulsifiable concentrates of the compounds comprise a convenient concentration of a compound, such as from about 50 to about 500 grams per liter of liquid, equivalent to about 10% to about 50%, dissolved in an inert carrier which is either a water miscible solvent or a mixture of water-immiscible organic solvent and emulslfiers.
- a compound such as from about 50 to about 500 grams per liter of liquid, equivalent to about 10% to about 50%, dissolved in an inert carrier which is either a water miscible solvent or a mixture of water-immiscible organic solvent and emulslfiers.
- Useful organic solvents include aromatics, especially the xylenes, and the petroleum fractions, especially the high-boiling naphthalenic and olefmic portions of petroleum such as heavy aromatic naphtha.
- organic solvents may also be used, such as the terpenic solvents including rosin derivatives, aliphatic ketones such as cyclohexanone, and complex alcohols such as 2-ethoxyethanol.
- Suitable emulsif ⁇ ers for emulsifiable concentrates are chosen from conventional nonionic surfactants, such as those discussed above.
- Aqueous suspensions comprise suspensions of water-insoluble compounds of this invention, dispersed in an aqueous vehicle at a concentration in the range from about 5% to about 50% by weight.
- Suspensions are prepared by finely grinding the compound, and vigorously mixing it into a vehicle comprised of water and surfactants chosen from the same types discussed above.
- Inert ingredients such as inorganic salts and synthetic or natural gums, may also be added, to increase the density and viscosity ofthe aqueous vehicle. It is often most effective to grind and mix the compound at the same time by preparing the aqueous mixture, and homogenizing it in an implement such as a sand mill, ball mill, or piston-type homogenizer.
- the compounds may also be applied as granular compositions, which are particularly useful for applications to the soil.
- Granular compositions usually contain from about 0.5%) to about 10% by weight of the compound, dispersed in an inert carrier which consists entirely or in large part of clay or a similar inexpensive substance.
- Such compositions are usually prepared by dissolving the compound in a suitable solvent and applying it to a granular carrier which has been pre-formed to the appropriate particle size, in the range of from about 0.5 to 3 mm.
- Such compositions may also be formulated by making a dough or paste of the carrier and compound and crushing and drying to obtain the desired granular particle size.
- Dusts containing the compounds are prepared simply by intimately mixing the compound in powdered form with a suitable dusty agricultural carrier, such as kaolin clay, ground volcanic rock, and the like. Dusts can suitably contain from about 1 % to about 10%) ofthe compound.
- a suitable dusty agricultural carrier such as kaolin clay, ground volcanic rock, and the like. Dusts can suitably contain from about 1 % to about 10%) ofthe compound.
- the active compositions may contain adjuvant surfactants to enhance deposition, wetting and penetration ofthe compositions onto the target crop and organism. These adjuvant surfactants may optionally be employed as a component of the formulation or as a tank mix. The amount of adjuvant surfactant will vary from 0.01 percent to 1.0 percent v/v based on a spray- volume of water, preferably 0.05 to 0.5 percent.
- Suitable adjuvant surfactants include ethoxylated nonyl phenols, ethoxylated synthetic or natural alcohols, salts of the esters of sulphosuccinic acids, ethoxylated organosilicones, ethoxylated fatty amines, crop oil concentrates containing high molecular weight paraffinic oils and blends of surfactants with mineral and vegetable oils. It is equally practical, when desirable for any reason, to apply the compound in the form of a solution in an appropriate organic solvent, usually a bland petroleum oil, such as the spray oils, which are widely used in agricultural chemistry.
- Insecticides and acaricides are generally applied in the form of a dispersion of the active ingredient in a liquid carrier. It is conventional to refer to application rates in terms of the concentration of active ingredient in the carrier. The most widely used carrier is water.
- the compounds of the invention can also be applied in the form of an aerosol composition.
- the active compound is dissolved or dispersed in an inert carrier, which is a pressure-generating propellant mixture.
- the aerosol composition is packaged in a container from which the mixture is dispensed through an atomizing valve.
- Propellant mixtures comprise either low-boiling halocarbons, which may be mixed with organic solvents, or aqueous suspensions pressurized with inert gases or gaseous hydrocarbons.
- the actual amount of compound to be applied to loci of insects, mites, and aphids is not critical and can readily be determined by those skilled in the art in view of the examples above. In general, concentrations of from- 10 ppm to 5000 ppm by weight of compound are expected to provide good control. With many of the compounds, concentrations of from 100 to 1500 ppm will suffice.
- the locus to which a compound is applied can be any locus inhabited by an insect or arachnid, for example, vegetable crops, fruit and nut trees, grape vines, and ornamental plants.
- Another aspect ofthe invention is a method of protecting a plant from insects which comprises treating plant seed prior to planting it, treating soil where plant seed is to be planted, or treating soil at the roots of a plant after it is planted, with an effective amount of a compound of Formula One.
- inventive compounds can be broadened by adding other, for example insecticidally, acaricidally, and/or nematocidally active, ingredients.
- inventive compounds can suitably be combined with the compounds of the invention:
- organophosphorus compounds such as acephate, azinphosmethyl, cadusafos, chlorethoxyfos, chlorpyrifos, coumaphos, dematon, demeton-S-methyl, diazinon, dichlorvos, dimethoate, EPN, erthoate, ethoprophos, etrimfos, fenamiphos, fenitrothion, fensulfothion, fenthion, fonofos, formothion, fosthiazate, heptenophos, malathion, methamidophos, methyl parathion, mevinphos, monocrotophos, parathion, phorate, phosalone, phosmet, phosphamidon, phosphocarb, phoxim, profenofos, propaphos, propetamphos, prothiofos, pyrimiphos-methyl, pyrimipho
- carbamates such as aldicarb, bendiocarb, benfuracarb, bensultap, BPMC, butoxycarbocim, carbaryl, carbofuran, carbosulfan, cloethocarb, ethiofencarb, fenobucarb, furathiocarb, methiocarb, isoprocarb, methomyl, oxamyl, pirimicarb, promecarb, propoxur, thiodicarb, and thiofurox;
- pyrethroids such as acrinathrin, allethrin, beta-cyfluthrin, bifenthrin, bioresmethrin, cyfluthrin; cyhalothrin; lambda-cyhalothrin; gamma-cyhalothrin, cypermethrin; alpha- cypermethrin; zeta-cypermethrin; deltamethrin, esfenvalerate, fenvalerate, fenfluthrin, fenpropathrin, flucythrinate, fiumethrin, fluvalinate, tau-fluvalinate, halfenprox, permethrin, protrifenbute, resmethrin, silafluofen, tefluthrin, tetramethrin, tralomethrin, fish safe pyrethroids for example ethofen
- acylureas other types of insect growth regulators and insect hormone analogs such as buprofezin, chromfenozide, chlorfluazuron, diflubenzuron, fenoxycarb, flufenoxuron, halofenozide, hexaflumuron, hydroprene, leufenuron, methoprene, methoxyfenozide, novaluron, pyriproxyfen, teflubenzuron and tebufenozide, N-[3,5-dichloro-2-fluoro-4- (1,1 ,2,3 ,3 ,3-hexafluoropropoxy)phenyl]-N' (2,6-difluorobenzoyl)urea; neonicotnioids and other nicotinics such as acetamiprid, AKD-1022, cartap, TI-435, clothiamidin, MTI-446, dinotefuran, imida
- avermectins such as avermectins, milbemycins,or spinosyns for example such as abamectin, ivermectin, milbemycin, emamectin benzoate and spinosad; and
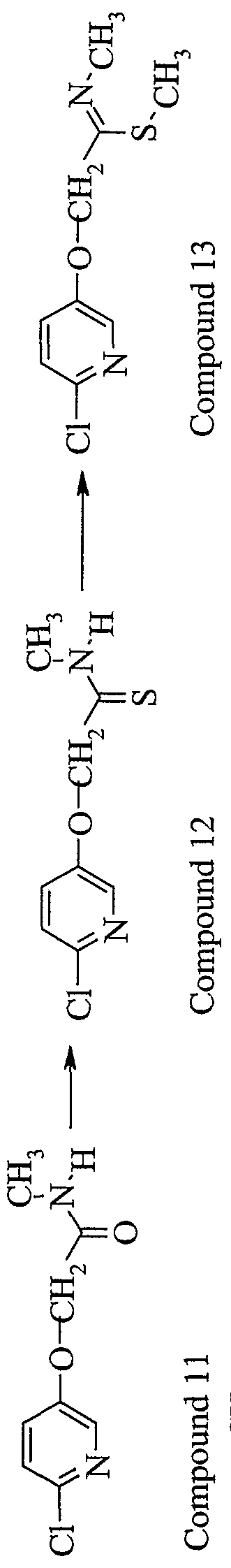
- Hastelloy autoclave was loaded with compound 21 (6.80 g, 60J mmol), compound 4 (8.8 g, 72.2 mmol), potassium carbonate (10.0 g, 72.2 mmol) and acetonitrile (100 mL).
- the vessel was purged with nitrogen, then pressurized to 50 psi with nitrogen and heated at 150°C for 12 hr. After cooling, the solvent was removed in vacuo, the light brown residue taken up in methylene chloride (200 mL) and washed with dilute aqueous sodium hydroxide (2 x 100 mL), removing the majority of color.
- the organic layer was washed with brine (100 mL), dried over Na 2 SO 4 and concentrated in vacuo leaving 10.2 g (86%>) of desired product as a beige powder.
- reaction mixture was washed with water, 0.50 M NaOH and dried over Na 2 SO 4 .
- the solvent was removed in vacuo leaving 19.4 g (93.0%o) of desired product.
- Trituration with hot methyl cyclohexane afforded 18.5 g (88.7%) of off-white crystalline product.
- the residue was purified by column chromatography, using 15%) ethyl acetate/ hexanes as the eluents. Fractions containing the crude product were combined and concentrated in vacuo. The crude product was further purified by preparative TLC, using 50% ethyl acetate/ dichloromethane as the eluents. The silica containing the desired product was collected, and the material removed from the silica using ethyl acetate as the eluent. The material was filtered, dried over MgSO 4 and concentrated in vacuo to afford 0.040 mg (5.2%) ofthe desired product.
- the mixture was poured over 40 ml of ice water, and the pH was adjusted to 7 with 1 ⁇ HCl.
- the mixture was extracted with 2 - 75 ml portions of dichloromethane.
- the extracts were dried over MgSO 4 and concentrated in vacuo.
- the residue was purified by column chromatography using 5% methanol/ dichloromethane as eluents. Fractions containing the desired product were combined, and the solvent removed in vacuo to obtain 0.010 g (1.8%)) ofthe desired product as pale yellow crystals.
- the mixture was diluted with 100 ml of dichloromethane.
- the solution was washed with 2 - 100 ml portions of brine, followed by 2 - 100 ml portions of water.
- the organic phase was dried over MgSO 4 and concentrated in vacuo.
- the residue was purified by column chromatography using first 100%) dichloromethane, then 5% methanol/ dichloromethane as eluents. Fractions containing the desired product were combined, and the solvent removed in vacuo to obtain crude product.
- the material was further purified using preparative HPLC (40%> water/ acetonitrile, flow rate 8 ml/min). The fractions containing product were collected and concentrated in vacuo to obtain 0.055 g ofthe desired product as an off-white solid.
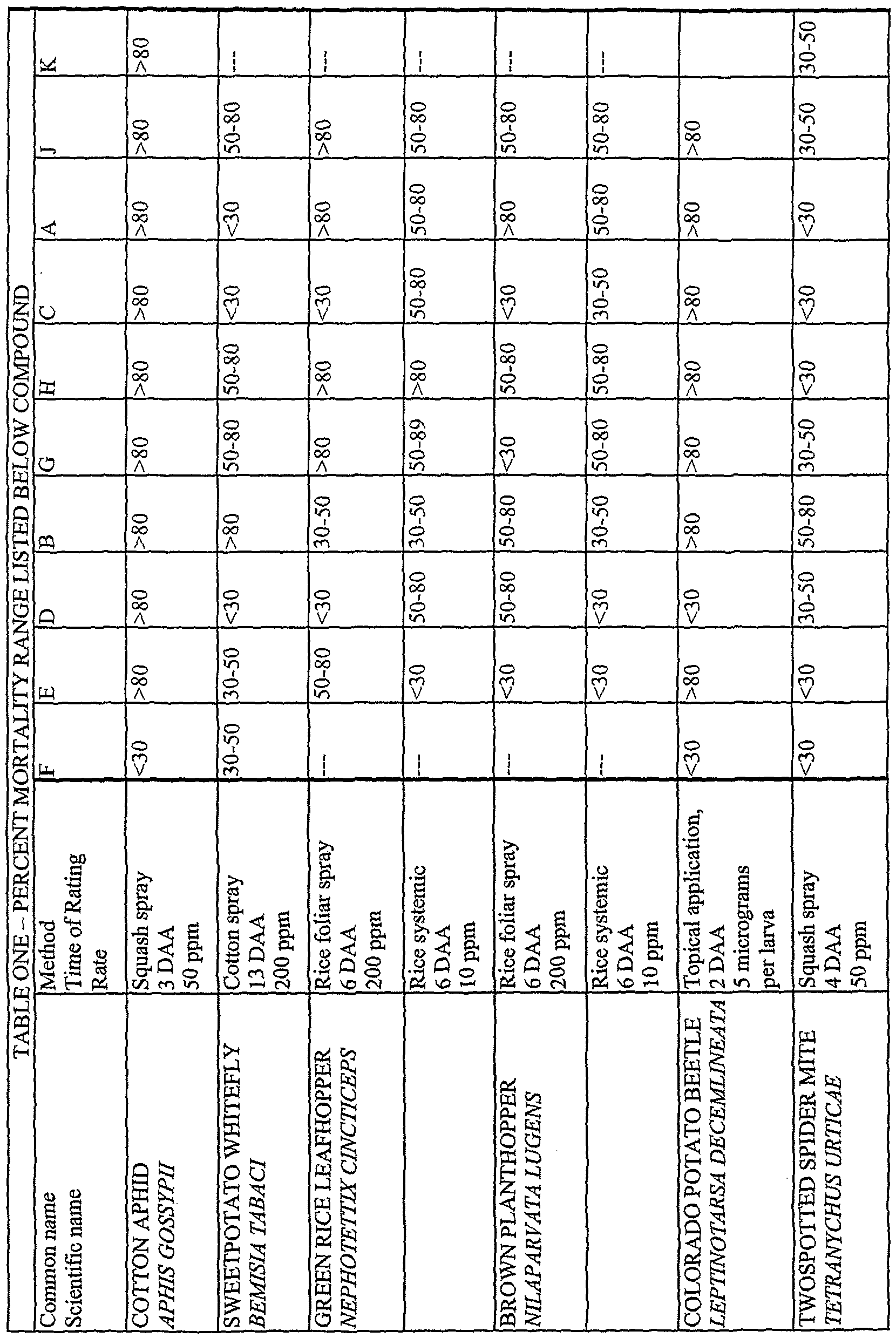
- Cotton Aphid (Aphis gossypii) - Squash Spray Method Yellow crookneck squash, Cucurbita pepo, is planted in 3 inch pots and placed in a greenhouse. Plants are watered regularly for 5 to 7 days until they reach the first emergent leaf stage. Plants are then trimmed to a single cotyledon.
- the squash assay consists of four squash plants per treatment with each plant cotyledon considered a replicate. Four additional plants are used as a control treatment (receiving solvent blank application only). Twenty-four hours prior to application, a leaf section of heavily infested squash plant from the aphid colony is placed onto each cotyledon, allowing a mixed population of A.
- the pre-infested squash cotyledons are sprayed on both the upper and lower surfaces using an airbrush sprayer set at 2 psi.
- Formulation is an aqueous solution containing 5% solvent and 0.025% Tween 20 surfactant to yield a concentration of 50 ppm of the test compound.
- Plants are sprayed to runoff. Tests are held in ambient laboratory temperatures for three days. At 3 days after application (DAA) the number of live aphids are counted with the aid of a dissecting microscope. The number of live aphids in the treatment is compared to the number of live aphids in the solvent blank-treated controls and percent mortality is calculated.
- Either mixed-age mobile mites or mite nymphs are transferred to 5 to 7 day old squash plants trimmed to a single cotyledon.
- Four mite-infested plants per rate are sprayed to runoff with a 50 ppm solution of test compound using a hand syringe equipped with a spray nozzle.
- Eight solvent blank-treated plants are held as negative controls. Plants are held at ambient temperature and humidity in the laboratory and then graded at 4 days after application. The number of dead mites in each treatment is compared to the number dead in the controls and percent mortality is calculated.
- Sweetpotato Whitefly (Bemisia tabaci) - Cotton Spray Method Technical materials are dissolved in a mixture of 90:10 acetone: ethanol; this is then diluted in water containing 0.05%> v/v Tween 20 surfactant to produce a spray eggs on the plants for 2 to 3 days, solution containing 200 ppm of the test compound.
- Four week-old cotton (Gossypium hirsutum) plants are trimmed to the first two true leaves and B. tabaci adults are allowed to lay eggs on the leaves over a 48 hour period. Solutions of the test compounds are applied to both sides of each cotton leaf using a hand syringe equipped with spray nozzle. A total of four leaves are treated with test compound, eight leaves are treated with a solvent blank control. After 12 to 14 days, the number of live whitefly nymphs on the treated plants are counted and compared to the number in the control treatment and percent mortality is calculated.
- test compound is dissolved in acetone, making a 10,000 ppm solution.
- test compound is dissolved in acetone and further diluted in water to make a 200 ppm solution.
- Several four week-old rice seedlings are placed in glass cylinder cages, supported by plastic mesh, 4 glass cylinders are used in each treatment.
- 0.5 ml of test compound solution is sprayed onto the rice seedlings in each glass cylinder.
- 5 laboratory-reared third instar nymphs of either brown planthopper or green leafhopper are introduced into the glass cylinder cages.
- the cylinders are held in a growth chamber at 28° C and 75% relative humidity, with a photoperiod of 14 hours. The number of dead insects is counted 6 days after application and percent mortality is calculated.
- Topical method for insecticidal activity against Colorado potato beetle (Leptinotarsa decemlineata)
- the test compound is dissolved in acetone to yield a concentration of 5 micrograms per microliter.
- One microliter of this solution is pipetted on to the dorsal surface of third instar E. decemlineata larvae, achieving a dose of 5 micrograms per larva.
- Six larvae are treated with each solution.
- the larvae are placed on potato (Solarium tubersum) foliage and held at ambient temperature and humidity in the laboratory for 2 days. After 2 days, the number of dead larvae are counted and percent mortality is calculated.
- Green Peach Aphid Bioassay Plant Preparation and Infestation Head cabbage seedlings, (Brassica oleracea capitat), at the 2-4 leaf stage, approximately 12 days old, are infested with all stages of Green peach aphid (Myzus persic ⁇ e) by shaking heavily infested, colony, leaf sections above the cabbage seedlings 4 days prior to the application of the test material. The aphids moved to the succulent plant material and settled to feed predominantly on the underside of the leaves. The plants are examined for good infestation prior to application of experimental compounds.
- Spray Solution Preparation Technical material of each experimental compound is dissolved at 1 mg/ml in 90:10 acetone:alcohol, then diluted in tap water containing 0.05% Tween 20. Additional serial dilutions are made to yield subsequent solutions of 50, 12.5, 3.13, 0.78, 0.195 and 0.049 ppm.
- Tests are held in a holding room for 72 hours at approximately 74°F and 40° relative humidity, 24 hour photoperiod prior to grading. Tests are graded 3 days after application by assessing the live aphid count (all non-winged stages) on the underside of each leaf using a dissecting binocular microscope. Live count results are used to calculate a percent control based on comparison to the aphid population on the solvent blank controls.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Environmental Sciences (AREA)
- Engineering & Computer Science (AREA)
- Dentistry (AREA)
- General Health & Medical Sciences (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Plant Pathology (AREA)
- Pest Control & Pesticides (AREA)
- Agronomy & Crop Science (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description






























































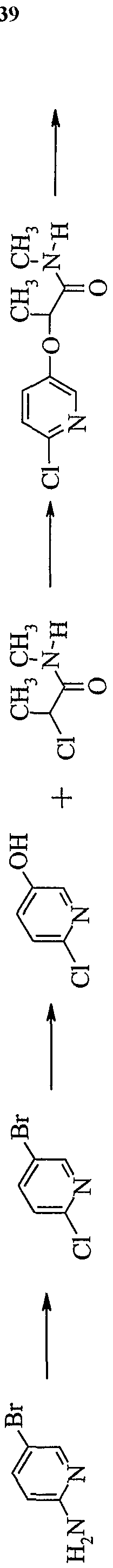

Claims
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002523457A JP2004521077A (en) | 2000-08-30 | 2001-08-28 | Compounds useful as insecticides, compounds useful as acaricides, uses thereof and methods for producing the same |
| AU2001286839A AU2001286839A1 (en) | 2000-08-30 | 2001-08-28 | Compounds useful as insecticides, compounds useful as acaricides, and processes to use and make same |
| EP01966314A EP1313704A2 (en) | 2000-08-30 | 2001-08-28 | Compounds useful as insecticides, compounds useful as acaricides, and processes to use and make same |
| KR10-2003-7002946A KR20030029878A (en) | 2000-08-30 | 2001-08-28 | Compounds useful as insecticides, compounds useful as acaricides, and processes to use and make same |
| BR0113633-0A BR0113633A (en) | 2000-08-30 | 2001-08-28 | Compounds useful as insecticides, compounds useful as acaricides and processes for using and obtaining them |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US22911000P | 2000-08-30 | 2000-08-30 | |
| US60/229,110 | 2000-08-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002018339A2 true WO2002018339A2 (en) | 2002-03-07 |
| WO2002018339A3 WO2002018339A3 (en) | 2002-05-16 |
Family
ID=22859873
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2001/026777 WO2002018339A2 (en) | 2000-08-30 | 2001-08-28 | Compounds useful as insecticides, compounds useful as acaricides, and processes to use and make same |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP1313704A2 (en) |
| JP (1) | JP2004521077A (en) |
| KR (1) | KR20030029878A (en) |
| CN (1) | CN1501917A (en) |
| AU (1) | AU2001286839A1 (en) |
| BR (1) | BR0113633A (en) |
| WO (1) | WO2002018339A2 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7053088B2 (en) | 2002-05-22 | 2006-05-30 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7144888B2 (en) | 2002-08-08 | 2006-12-05 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7301022B2 (en) | 2005-02-15 | 2007-11-27 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7511044B2 (en) | 2004-02-11 | 2009-03-31 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7534798B2 (en) | 2004-02-11 | 2009-05-19 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| WO2017087619A1 (en) | 2015-11-17 | 2017-05-26 | Viamet Pharmaceuticals, Inc. | 4-((6-(2,4-difluorophenyl)-1,1-difluoro-2-hydroxy-3-(1h-1,2,4-triazol-1-yl)propyl)pyridin-3-yl)oxy)benzonitrile and processes of preparation |
| KR20220069696A (en) * | 2020-11-20 | 2022-05-27 | 주식회사 엘피엔 | Method for synthesis of ligand for preparation of organometallic compounds |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20180101342A (en) * | 2015-11-17 | 2018-09-12 | 다우 아그로사이언시즈 엘엘씨 | 4 - ((6- (2- (2,4-difluorophenyl) -1,1-difluoro-2-hydroxy- ) Propyl) pyridin-3-yl) oxy) benzonitrile and the preparation method |
| CN108882709A (en) * | 2015-11-17 | 2018-11-23 | 美国陶氏益农公司 | 4- ((6- (the fluoro- 2- oxoethyl of 2- (2,4 difluorobenzene base) -1,1- two) pyridin-3-yl) oxygroup) benzonitrile and preparation method |
| CN118324677B (en) * | 2024-04-12 | 2024-10-01 | 江苏三吉利化工股份有限公司 | Butyl sulfide residual killing agent method for synthesizing wei |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2321522A1 (en) * | 1972-05-04 | 1973-11-15 | Shell Int Research | INSECTICIDES FOR CONTROL OF CATERPILLAR CAUSES BY LEAVES |
| GB1425763A (en) * | 1972-05-04 | 1976-02-18 | Shell Int Research | 2-nitromethylene- piperidines and their use as insecticides |
| EP0376279A2 (en) * | 1988-12-27 | 1990-07-04 | Takeda Chemical Industries, Ltd. | Guanidine derivatives, their production and insecticides |
| EP0375907A1 (en) * | 1988-11-29 | 1990-07-04 | Nihon Bayer Agrochem K.K. | Insecticidally active nitro compounds |
| WO1994024124A1 (en) * | 1993-04-08 | 1994-10-27 | Ciba-Geigy Ag | Novel 2-nitromethylidene/2-cyanimino/2-nitro-imino-pyrrolidines and piperidines, intermediates, and their use as pesticides |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59161347A (en) * | 1983-03-07 | 1984-09-12 | Sagami Chem Res Center | 3-amino-2-alkenenitrile and its preparation |
-
2001
- 2001-08-28 EP EP01966314A patent/EP1313704A2/en not_active Withdrawn
- 2001-08-28 AU AU2001286839A patent/AU2001286839A1/en not_active Abandoned
- 2001-08-28 KR KR10-2003-7002946A patent/KR20030029878A/en not_active Application Discontinuation
- 2001-08-28 JP JP2002523457A patent/JP2004521077A/en active Pending
- 2001-08-28 CN CNA018149138A patent/CN1501917A/en active Pending
- 2001-08-28 BR BR0113633-0A patent/BR0113633A/en not_active Application Discontinuation
- 2001-08-28 WO PCT/US2001/026777 patent/WO2002018339A2/en not_active Application Discontinuation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2321522A1 (en) * | 1972-05-04 | 1973-11-15 | Shell Int Research | INSECTICIDES FOR CONTROL OF CATERPILLAR CAUSES BY LEAVES |
| GB1425763A (en) * | 1972-05-04 | 1976-02-18 | Shell Int Research | 2-nitromethylene- piperidines and their use as insecticides |
| EP0375907A1 (en) * | 1988-11-29 | 1990-07-04 | Nihon Bayer Agrochem K.K. | Insecticidally active nitro compounds |
| EP0376279A2 (en) * | 1988-12-27 | 1990-07-04 | Takeda Chemical Industries, Ltd. | Guanidine derivatives, their production and insecticides |
| WO1994024124A1 (en) * | 1993-04-08 | 1994-10-27 | Ciba-Geigy Ag | Novel 2-nitromethylidene/2-cyanimino/2-nitro-imino-pyrrolidines and piperidines, intermediates, and their use as pesticides |
Non-Patent Citations (9)
| Title |
|---|
| ARTYOMOV V A ET AL: "N-Cyanochloroacetamidine - a Convenient Reagent for the Regioselective Synthesis of Fused Diaminopyrimidines." TETRAHEDRON., vol. 52, no. 3, 1996, pages 1011-1026, XP002190927 ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM., NL ISSN: 0040-4020 * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; Database accession no. 7147006 XP002190930 & KHIM. GETEROTSIKL. SOEDIN., no. 1, 1994, pages 122-132, * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; Database accession no. 7148988 XP002190928 & KHIM. GETEROTSIKL. SOEDIN., no. 1, 1994, pages 122-132, * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; Database accession no. 7150892 XP002190929 & KHIM. GETEROTSIKL. SOEDIN., no. 1, 1994, pages 122-132, * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; Database accession no. 804240 XP002190934 & BOLL. CHIM. FARM., vol. 118, 1979, pages 661-666, * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; Database accession no. 812614 XP002190935 & BOLL. CHIM. FARM., vol. 118, 1979, pages 661-666, * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; Database accession no. 814098 XP002190931 & BOLL. CHIM. FARM., vol. 118, 1979, pages 661-666, * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; Database accession no. 817142 XP002190932 & BOLL. CHIM. FARM., vol. 118, 1979, pages 661-666, * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; retrieved from 793074 XP002190933 & BOLL. CHIM. FARM., vol. 118, 1979, pages 661-666, * |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7524874B2 (en) | 2002-05-22 | 2009-04-28 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7053088B2 (en) | 2002-05-22 | 2006-05-30 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7396831B2 (en) | 2002-05-22 | 2008-07-08 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7144888B2 (en) | 2002-08-08 | 2006-12-05 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7148221B2 (en) | 2002-08-08 | 2006-12-12 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7332511B2 (en) | 2002-08-08 | 2008-02-19 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US8227469B2 (en) | 2004-02-11 | 2012-07-24 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7511044B2 (en) | 2004-02-11 | 2009-03-31 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7534798B2 (en) | 2004-02-11 | 2009-05-19 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| US7301022B2 (en) | 2005-02-15 | 2007-11-27 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| WO2017087619A1 (en) | 2015-11-17 | 2017-05-26 | Viamet Pharmaceuticals, Inc. | 4-((6-(2,4-difluorophenyl)-1,1-difluoro-2-hydroxy-3-(1h-1,2,4-triazol-1-yl)propyl)pyridin-3-yl)oxy)benzonitrile and processes of preparation |
| CN108882708A (en) * | 2015-11-17 | 2018-11-23 | 美国陶氏益农公司 | 4- ((6- (fluoro- 2- hydroxyl -3- (1H-1,2,4- triazol-1-yl) propyl of 2- (2,4 difluorobenzene base) -1,1- two) pyridin-3-yl) oxygroup) benzonitrile and preparation method |
| US10513506B2 (en) | 2015-11-17 | 2019-12-24 | Dow Agrosciences Llc | 4-((6-(2-(2,4-difluorophenyl)-1,1-difluoro-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl)pyridin-3-yl and processes of preparation |
| KR20220069696A (en) * | 2020-11-20 | 2022-05-27 | 주식회사 엘피엔 | Method for synthesis of ligand for preparation of organometallic compounds |
| KR102467497B1 (en) | 2020-11-20 | 2022-11-15 | 주식회사 엘피엔 | Method for synthesis of ligand for preparation of organometallic compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| BR0113633A (en) | 2005-06-07 |
| WO2002018339A3 (en) | 2002-05-16 |
| CN1501917A (en) | 2004-06-02 |
| JP2004521077A (en) | 2004-07-15 |
| EP1313704A2 (en) | 2003-05-28 |
| KR20030029878A (en) | 2003-04-16 |
| AU2001286839A1 (en) | 2002-03-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105431414A (en) | 4-membered ring carboxamides used as nematicides | |
| EP0539588A1 (en) | Amine derivative | |
| JP2010533662A (en) | New insecticide | |
| US7902233B2 (en) | Compounds useful as pesticides | |
| WO2002018339A2 (en) | Compounds useful as insecticides, compounds useful as acaricides, and processes to use and make same | |
| JP2002532497A (en) | 4-trifluoromethyl-3-oxadiazolylpyridine, a process for its preparation, a medicament containing the compound and its use as a pesticide | |
| KR20230145066A (en) | Substituted Isoxazoline Derivatives | |
| JP2005509017A (en) | Dihalopropene compounds, processes for their preparation, compositions containing them and their use as pesticides | |
| JP2004505967A (en) | Heterocyclylalkylazole derivatives and their use as pesticides | |
| EA019508B1 (en) | Insecticidal compounds | |
| JPH054966A (en) | Amine derivative, production thereof and insecticide | |
| JP2003506360A (en) | Trifluoromethylpyri (mi) zine carboxamide | |
| JPH0853464A (en) | Microbicide | |
| US4994473A (en) | Pyridyl containing insecticides | |
| JP2003502413A (en) | Pesticide active tetrazine derivative | |
| US5137886A (en) | Insecticidal compounds | |
| JP2517992B2 (en) | Pyridylpyrimidine derivative and plant disease controlling agent containing the same | |
| US7375122B2 (en) | Compounds useful as pesticides | |
| MXPA02006318A (en) | Azolylalkyl azole derivatives, a method for their production and their use as pesticides. | |
| US20060058359A1 (en) | Compounds useful as pesticides | |
| JP2003507477A (en) | Derivatives of trifluoromethylpyri (mi) zine | |
| JP2734692B2 (en) | A cyanoacetamide derivative and a plant disease controlling agent containing the same as an active ingredient. | |
| JP2007519639A (en) | Heterocyclic allyl derivatives | |
| JP2803076B2 (en) | Phenoxyalkylamine derivatives, their preparation and pest control agents | |
| WO2024061665A1 (en) | N-(3-(aminomethyl)-phenyl)-5-(4-phenyl)-5-(trifluoromethyl)-4,5-dihydroisoxazol-3-amine derivatives and similar compounds as pesticides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2001966314 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020037002946 Country of ref document: KR Ref document number: 2002523457 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 018149138 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020037002946 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001966314 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2001966314 Country of ref document: EP |