WO2001044258A1 - Novel heterocycles - Google Patents
Novel heterocycles Download PDFInfo
- Publication number
- WO2001044258A1 WO2001044258A1 PCT/US2000/034487 US0034487W WO0144258A1 WO 2001044258 A1 WO2001044258 A1 WO 2001044258A1 US 0034487 W US0034487 W US 0034487W WO 0144258 A1 WO0144258 A1 WO 0144258A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- independently
- substituted
- occurrence
- absent
- compound
- Prior art date
Links
- 125000000623 heterocyclic group Chemical group 0.000 title claims description 146
- 150000001875 compounds Chemical class 0.000 claims abstract description 254
- 125000000217 alkyl group Chemical group 0.000 claims description 210
- 125000003118 aryl group Chemical group 0.000 claims description 147
- 238000000034 method Methods 0.000 claims description 122
- 125000001072 heteroaryl group Chemical group 0.000 claims description 120
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 82
- -1 sulfonate ester Chemical class 0.000 claims description 81
- 125000003342 alkenyl group Chemical group 0.000 claims description 78
- 125000001424 substituent group Chemical group 0.000 claims description 75
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 71
- 229910052739 hydrogen Inorganic materials 0.000 claims description 65
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 claims description 65
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 62
- 125000005842 heteroatom Chemical group 0.000 claims description 60
- 239000001257 hydrogen Substances 0.000 claims description 60
- 229910052771 Terbium Inorganic materials 0.000 claims description 58
- 125000004429 atom Chemical group 0.000 claims description 45
- 229910052736 halogen Inorganic materials 0.000 claims description 45
- 150000002367 halogens Chemical class 0.000 claims description 45
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 42
- 229910052760 oxygen Inorganic materials 0.000 claims description 41
- HSFWRNGVRCDJHI-UHFFFAOYSA-N Acetylene Chemical compound C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 40
- 210000002997 osteoclast Anatomy 0.000 claims description 40
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 39
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 39
- 208000020084 Bone disease Diseases 0.000 claims description 36
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical class C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 claims description 36
- 229910052757 nitrogen Inorganic materials 0.000 claims description 35
- 229910052717 sulfur Inorganic materials 0.000 claims description 31
- 239000008194 pharmaceutical composition Substances 0.000 claims description 29
- 238000011282 treatment Methods 0.000 claims description 26
- 125000004076 pyridyl group Chemical group 0.000 claims description 25
- 125000005741 alkyl alkenyl group Chemical group 0.000 claims description 23
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 claims description 23
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 23
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 21
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 20
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 20
- 150000003839 salts Chemical class 0.000 claims description 19
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 claims description 18
- 125000003368 amide group Chemical group 0.000 claims description 17
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 claims description 17
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 16
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 16
- 125000001589 carboacyl group Chemical group 0.000 claims description 15
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 15
- 125000003367 polycyclic group Polymers 0.000 claims description 15
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 14
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 13
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 13
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 claims description 11
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 11
- 125000003396 thiol group Chemical group [H]S* 0.000 claims description 11
- 150000001409 amidines Chemical class 0.000 claims description 10
- 125000002619 bicyclic group Chemical group 0.000 claims description 10
- 229910052799 carbon Inorganic materials 0.000 claims description 10
- 150000002576 ketones Chemical class 0.000 claims description 10
- ACVYVLVWPXVTIT-UHFFFAOYSA-M phosphinate Chemical compound [O-][PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-M 0.000 claims description 10
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 claims description 9
- 150000001408 amides Chemical class 0.000 claims description 9
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 claims description 9
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 8
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 claims description 8
- 239000004202 carbamide Substances 0.000 claims description 8
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 8
- 230000002265 prevention Effects 0.000 claims description 8
- 125000004442 acylamino group Chemical group 0.000 claims description 6
- 125000003375 sulfoxide group Chemical group 0.000 claims description 6
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 claims description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 5
- 125000001931 aliphatic group Chemical group 0.000 claims description 5
- 125000003739 carbamimidoyl group Chemical group C(N)(=N)* 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 4
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 3
- 125000005213 alkyl heteroaryl group Chemical group 0.000 claims description 3
- 239000005977 Ethylene Substances 0.000 claims 3
- 210000000988 bone and bone Anatomy 0.000 abstract description 53
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 133
- 239000000203 mixture Substances 0.000 description 101
- 239000007787 solid Substances 0.000 description 85
- 239000000243 solution Substances 0.000 description 79
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 74
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 59
- 235000019439 ethyl acetate Nutrition 0.000 description 57
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 54
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 47
- 238000006243 chemical reaction Methods 0.000 description 46
- 125000004122 cyclic group Chemical group 0.000 description 43
- 150000002431 hydrogen Chemical class 0.000 description 43
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 41
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 41
- 239000011541 reaction mixture Substances 0.000 description 41
- 239000000047 product Substances 0.000 description 36
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 35
- 239000000741 silica gel Substances 0.000 description 34
- 229910002027 silica gel Inorganic materials 0.000 description 34
- 229960001866 silicon dioxide Drugs 0.000 description 34
- 230000000694 effects Effects 0.000 description 33
- 239000003814 drug Substances 0.000 description 31
- 238000003786 synthesis reaction Methods 0.000 description 29
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical group CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 28
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 28
- 230000015572 biosynthetic process Effects 0.000 description 28
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 27
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 25
- 239000003795 chemical substances by application Substances 0.000 description 25
- 230000005764 inhibitory process Effects 0.000 description 25
- 239000010410 layer Substances 0.000 description 25
- 239000011347 resin Substances 0.000 description 25
- 229920005989 resin Polymers 0.000 description 25
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 24
- 239000012267 brine Substances 0.000 description 24
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 24
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 23
- 230000008685 targeting Effects 0.000 description 23
- 0 CNC1N(*)c2nc(N*)ncc2C=C1* Chemical compound CNC1N(*)c2nc(N*)ncc2C=C1* 0.000 description 22
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 22
- 229910052938 sodium sulfate Inorganic materials 0.000 description 22
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 22
- 230000024279 bone resorption Effects 0.000 description 21
- 239000000284 extract Substances 0.000 description 21
- 239000002904 solvent Substances 0.000 description 21
- 208000006386 Bone Resorption Diseases 0.000 description 20
- 239000012044 organic layer Substances 0.000 description 20
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 238000003818 flash chromatography Methods 0.000 description 17
- 102000009076 src-Family Kinases Human genes 0.000 description 17
- 108010087686 src-Family Kinases Proteins 0.000 description 17
- 125000000304 alkynyl group Chemical group 0.000 description 16
- 210000004027 cell Anatomy 0.000 description 16
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 16
- 239000003921 oil Substances 0.000 description 16
- 235000019198 oils Nutrition 0.000 description 16
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 15
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 15
- 150000002148 esters Chemical class 0.000 description 15
- 238000004007 reversed phase HPLC Methods 0.000 description 15
- 229940124597 therapeutic agent Drugs 0.000 description 15
- 239000007832 Na2SO4 Substances 0.000 description 14
- 208000001132 Osteoporosis Diseases 0.000 description 14
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 14
- 239000011324 bead Substances 0.000 description 14
- 238000004128 high performance liquid chromatography Methods 0.000 description 14
- 239000007788 liquid Substances 0.000 description 14
- 239000007790 solid phase Substances 0.000 description 14
- 239000002253 acid Substances 0.000 description 13
- 125000005518 carboxamido group Chemical group 0.000 description 13
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 13
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 12
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 12
- 239000000872 buffer Substances 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 12
- 239000003153 chemical reaction reagent Substances 0.000 description 12
- 238000010828 elution Methods 0.000 description 12
- 125000001183 hydrocarbyl group Chemical group 0.000 description 12
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 11
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 11
- 125000003545 alkoxy group Chemical group 0.000 description 11
- 235000019441 ethanol Nutrition 0.000 description 11
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 11
- 239000000463 material Substances 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 10
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 10
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 10
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 10
- 239000000969 carrier Substances 0.000 description 10
- 208000035475 disorder Diseases 0.000 description 10
- 230000006870 function Effects 0.000 description 10
- 239000003112 inhibitor Substances 0.000 description 10
- 229910052740 iodine Inorganic materials 0.000 description 10
- 210000000963 osteoblast Anatomy 0.000 description 10
- 239000001301 oxygen Chemical class 0.000 description 10
- 239000012071 phase Substances 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- 229940002612 prodrug Drugs 0.000 description 10
- 239000000651 prodrug Substances 0.000 description 10
- 239000011593 sulfur Chemical class 0.000 description 10
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical class [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 9
- 125000004414 alkyl thio group Chemical group 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical class [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 238000001816 cooling Methods 0.000 description 9
- 239000013058 crude material Substances 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 9
- 108090000765 processed proteins & peptides Proteins 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 9
- AMQIPHZFLIDOCB-UHFFFAOYSA-N 3-(2-hydroxyethyl)phenol Chemical compound OCCC1=CC=CC(O)=C1 AMQIPHZFLIDOCB-UHFFFAOYSA-N 0.000 description 8
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 8
- 108091000080 Phosphotransferase Proteins 0.000 description 8
- 102100032709 Potassium-transporting ATPase alpha chain 2 Human genes 0.000 description 8
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 8
- 108010083204 Proton Pumps Proteins 0.000 description 8
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 8
- 150000001299 aldehydes Chemical class 0.000 description 8
- 125000005119 alkyl cycloalkyl group Chemical group 0.000 description 8
- 230000004097 bone metabolism Effects 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 125000004404 heteroalkyl group Chemical group 0.000 description 8
- 229940043355 kinase inhibitor Drugs 0.000 description 8
- 238000001819 mass spectrum Methods 0.000 description 8
- 102000020233 phosphotransferase Human genes 0.000 description 8
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- 150000003457 sulfones Chemical class 0.000 description 8
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 7
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 7
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 238000013459 approach Methods 0.000 description 7
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 7
- 125000002837 carbocyclic group Chemical group 0.000 description 7
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 7
- 239000012216 imaging agent Substances 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 230000002401 inhibitory effect Effects 0.000 description 7
- 125000005647 linker group Chemical group 0.000 description 7
- 125000004430 oxygen atom Chemical group O* 0.000 description 7
- 230000026731 phosphorylation Effects 0.000 description 7
- 238000006366 phosphorylation reaction Methods 0.000 description 7
- 108090000623 proteins and genes Proteins 0.000 description 7
- 102000004169 proteins and genes Human genes 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 7
- 238000004679 31P NMR spectroscopy Methods 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 6
- 150000001412 amines Chemical class 0.000 description 6
- 230000008878 coupling Effects 0.000 description 6
- 238000010168 coupling process Methods 0.000 description 6
- 238000005859 coupling reaction Methods 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 235000019341 magnesium sulphate Nutrition 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- 150000003212 purines Chemical class 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 125000004434 sulfur atom Chemical group 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 6
- UNRIYCIDCQDGQE-UHFFFAOYSA-N 6-chloro-2-fluoro-7h-purine Chemical compound FC1=NC(Cl)=C2NC=NC2=N1 UNRIYCIDCQDGQE-UHFFFAOYSA-N 0.000 description 5
- 102000004171 Cathepsin K Human genes 0.000 description 5
- 108090000625 Cathepsin K Proteins 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 238000010936 aqueous wash Methods 0.000 description 5
- 229910052786 argon Inorganic materials 0.000 description 5
- 150000001499 aryl bromides Chemical class 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 description 5
- 238000003776 cleavage reaction Methods 0.000 description 5
- 210000004268 dentin Anatomy 0.000 description 5
- 238000004108 freeze drying Methods 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 150000002894 organic compounds Chemical class 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 230000007017 scission Effects 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229960001407 sodium bicarbonate Drugs 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 241000894007 species Species 0.000 description 5
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- 108010049047 Echinocandins Proteins 0.000 description 4
- 241000282414 Homo sapiens Species 0.000 description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 4
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 4
- 230000002159 abnormal effect Effects 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- COJRWHSKVYUZHQ-UHFFFAOYSA-N alpha-methyl-3-hydroxybenzyl alcohol Natural products CC(O)C1=CC=CC(O)=C1 COJRWHSKVYUZHQ-UHFFFAOYSA-N 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 239000001913 cellulose Substances 0.000 description 4
- 229920002678 cellulose Polymers 0.000 description 4
- 235000010980 cellulose Nutrition 0.000 description 4
- 235000014113 dietary fatty acids Nutrition 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 239000000194 fatty acid Substances 0.000 description 4
- 229930195729 fatty acid Natural products 0.000 description 4
- 235000011187 glycerol Nutrition 0.000 description 4
- 125000004475 heteroaralkyl group Chemical group 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- 229910052500 inorganic mineral Inorganic materials 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 235000010755 mineral Nutrition 0.000 description 4
- 239000011707 mineral Substances 0.000 description 4
- 150000002829 nitrogen Chemical class 0.000 description 4
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 102000004196 processed proteins & peptides Human genes 0.000 description 4
- 238000011321 prophylaxis Methods 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 4
- 238000010532 solid phase synthesis reaction Methods 0.000 description 4
- 125000000547 substituted alkyl group Chemical group 0.000 description 4
- 125000003107 substituted aryl group Chemical group 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 125000004001 thioalkyl group Chemical group 0.000 description 4
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 4
- DIOHEXPTUTVCNX-UHFFFAOYSA-N 1,1,1-trifluoro-n-phenyl-n-(trifluoromethylsulfonyl)methanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)N(S(=O)(=O)C(F)(F)F)C1=CC=CC=C1 DIOHEXPTUTVCNX-UHFFFAOYSA-N 0.000 description 3
- TWHDMOASZWUGTG-UHFFFAOYSA-N 3-[3,4-bis(diethoxyphosphoryl)phenyl]propanoic acid Chemical compound CCOP(=O)(OCC)C1=CC=C(CCC(O)=O)C=C1P(=O)(OCC)OCC TWHDMOASZWUGTG-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 229940122361 Bisphosphonate Drugs 0.000 description 3
- SUYVBPMJNFQYHR-UHFFFAOYSA-N CCOC(CCCN)([PH2]=O)P(=O)(OCC)OCC Chemical compound CCOC(CCCN)([PH2]=O)P(=O)(OCC)OCC SUYVBPMJNFQYHR-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- KBHCPIJKJQNHPN-UHFFFAOYSA-N N=NP(O)=O Chemical group N=NP(O)=O KBHCPIJKJQNHPN-UHFFFAOYSA-N 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- 102000001253 Protein Kinase Human genes 0.000 description 3
- 206010037211 Psychomotor hyperactivity Diseases 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 3
- 108010090804 Streptavidin Proteins 0.000 description 3
- 239000004098 Tetracycline Substances 0.000 description 3
- 230000005856 abnormality Effects 0.000 description 3
- 229960000583 acetic acid Drugs 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 239000003708 ampul Substances 0.000 description 3
- 150000001540 azides Chemical group 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- XSCHRSMBECNVNS-UHFFFAOYSA-N benzopyrazine Natural products N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 3
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 3
- 150000004663 bisphosphonates Chemical class 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 125000000392 cycloalkenyl group Chemical group 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- XZEHJCKDEUHHJY-UHFFFAOYSA-N diethyl 4-bromopyridine-2,6-dicarboxylate Chemical compound CCOC(=O)C1=CC(Br)=CC(C(=O)OCC)=N1 XZEHJCKDEUHHJY-UHFFFAOYSA-N 0.000 description 3
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 239000012634 fragment Substances 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 229910052588 hydroxylapatite Inorganic materials 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 3
- 230000030991 negative regulation of bone resorption Effects 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 230000011164 ossification Effects 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 description 3
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 229960004063 propylene glycol Drugs 0.000 description 3
- 108060006633 protein kinase Proteins 0.000 description 3
- 229940126409 proton pump inhibitor Drugs 0.000 description 3
- 239000000612 proton pump inhibitor Substances 0.000 description 3
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 3
- OYRRZWATULMEPF-UHFFFAOYSA-N pyrimidin-4-amine Chemical compound NC1=CC=NC=N1 OYRRZWATULMEPF-UHFFFAOYSA-N 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 238000002821 scintillation proximity assay Methods 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 3
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 235000019364 tetracycline Nutrition 0.000 description 3
- 150000003522 tetracyclines Chemical class 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- 239000003053 toxin Substances 0.000 description 3
- 231100000765 toxin Toxicity 0.000 description 3
- 108700012359 toxins Proteins 0.000 description 3
- BDZBKCUKTQZUTL-UHFFFAOYSA-N triethyl phosphite Chemical compound CCOP(OCC)OCC BDZBKCUKTQZUTL-UHFFFAOYSA-N 0.000 description 3
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 3
- WVLBCYQITXONBZ-UHFFFAOYSA-N trimethyl phosphate Chemical compound COP(=O)(OC)OC WVLBCYQITXONBZ-UHFFFAOYSA-N 0.000 description 3
- IPTXXSZUISGKCJ-UHFFFAOYSA-N 1-bromo-4-(diethoxyphosphorylmethyl)benzene Chemical compound CCOP(=O)(OCC)CC1=CC=C(Br)C=C1 IPTXXSZUISGKCJ-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 description 2
- UVIMLVOKKVNPHX-UHFFFAOYSA-N 4-[4-amino-5-(3-methoxyphenyl)pyrrolo[2,3-d]pyrimidin-7-yl]benzoic acid Chemical compound COC1=CC=CC(C=2C3=C(N)N=CN=C3N(C=3C=CC(=CC=3)C(O)=O)C=2)=C1 UVIMLVOKKVNPHX-UHFFFAOYSA-N 0.000 description 2
- KWMFMAPHZAIIEB-UHFFFAOYSA-N 4-bromo-2-diethoxyphosphorylpyridine Chemical compound CCOP(=O)(OCC)C1=CC(Br)=CC=N1 KWMFMAPHZAIIEB-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- WUJLOBZQUKCCNH-UHFFFAOYSA-N 5-bromo-2-diethoxyphosphorylpyridine Chemical compound CCOP(=O)(OCC)C1=CC=C(Br)C=N1 WUJLOBZQUKCCNH-UHFFFAOYSA-N 0.000 description 2
- AISKFDFVSSENFX-UHFFFAOYSA-N 6-chloro-2-fluoro-9-propan-2-ylpurine Chemical compound N1=C(F)N=C2N(C(C)C)C=NC2=C1Cl AISKFDFVSSENFX-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 208000014311 Cushing syndrome Diseases 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- 208000029725 Metabolic bone disease Diseases 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 108091034117 Oligonucleotide Proteins 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 208000010191 Osteitis Deformans Diseases 0.000 description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 208000027868 Paget disease Diseases 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- 241000283222 Physeter catodon Species 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 102100030852 Run domain Beclin-1-interacting and cysteine-rich domain-containing protein Human genes 0.000 description 2
- 229940124639 Selective inhibitor Drugs 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 102000007591 Tartrate-Resistant Acid Phosphatase Human genes 0.000 description 2
- 108010032050 Tartrate-Resistant Acid Phosphatase Proteins 0.000 description 2
- AGIPULZXANWTHI-UHFFFAOYSA-N [3,4-bis(diethoxyphosphoryl)phenyl]methanamine Chemical compound CCOP(=O)(OCC)C1=CC=C(CN)C=C1P(=O)(OCC)OCC AGIPULZXANWTHI-UHFFFAOYSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 230000001464 adherent effect Effects 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 229940062527 alendronate Drugs 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 239000000908 ammonium hydroxide Substances 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 239000003443 antiviral agent Substances 0.000 description 2
- 229940121357 antivirals Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- ISAOCJYIOMOJEB-UHFFFAOYSA-N benzoin Chemical compound C=1C=CC=CC=1C(O)C(=O)C1=CC=CC=C1 ISAOCJYIOMOJEB-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 150000005829 chemical entities Chemical class 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 238000007906 compression Methods 0.000 description 2
- 230000006835 compression Effects 0.000 description 2
- 238000002591 computed tomography Methods 0.000 description 2
- 235000008504 concentrate Nutrition 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 239000002872 contrast media Substances 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 2
- 239000006184 cosolvent Substances 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- LXCYSACZTOKNNS-UHFFFAOYSA-N diethoxy(oxo)phosphanium Chemical compound CCO[P+](=O)OCC LXCYSACZTOKNNS-UHFFFAOYSA-N 0.000 description 2
- APSXRHUXMFYHMQ-UHFFFAOYSA-N diethoxyphosphorylmethyl-ethoxy-oxophosphanium Chemical compound CCO[P+](=O)CP(=O)(OCC)OCC APSXRHUXMFYHMQ-UHFFFAOYSA-N 0.000 description 2
- LGTLXDJOAJDFLR-UHFFFAOYSA-N diethyl chlorophosphate Chemical compound CCOP(Cl)(=O)OCC LGTLXDJOAJDFLR-UHFFFAOYSA-N 0.000 description 2
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 239000002532 enzyme inhibitor Substances 0.000 description 2
- 239000002024 ethyl acetate extract Substances 0.000 description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 239000000835 fiber Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 229910001385 heavy metal Inorganic materials 0.000 description 2
- 239000003667 hormone antagonist Substances 0.000 description 2
- 201000000916 idiopathic juvenile osteoporosis Diseases 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 150000003949 imides Chemical class 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000000099 in vitro assay Methods 0.000 description 2
- 238000005462 in vivo assay Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 208000027202 mammary Paget disease Diseases 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 210000004379 membrane Anatomy 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- QMUOSMPEACAQFZ-UHFFFAOYSA-N n-(3-chlorophenyl)-2-fluoro-9-propan-2-ylpurin-6-amine Chemical compound N1=C(F)N=C2N(C(C)C)C=NC2=C1NC1=CC=CC(Cl)=C1 QMUOSMPEACAQFZ-UHFFFAOYSA-N 0.000 description 2
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 2
- 125000005151 nonafluorobutanesulfonyl group Chemical group FC(C(C(S(=O)(=O)*)(F)F)(F)F)(C(F)(F)F)F 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 230000005298 paramagnetic effect Effects 0.000 description 2
- 230000000737 periodic effect Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- RDOWQLZANAYVLL-UHFFFAOYSA-N phenanthridine Chemical compound C1=CC=C2C3=CC=CC=C3C=NC2=C1 RDOWQLZANAYVLL-UHFFFAOYSA-N 0.000 description 2
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 239000001488 sodium phosphate Substances 0.000 description 2
- 229910000162 sodium phosphate Inorganic materials 0.000 description 2
- 235000019345 sodium thiosulphate Nutrition 0.000 description 2
- 125000006850 spacer group Chemical group 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008174 sterile solution Substances 0.000 description 2
- 230000003637 steroidlike Effects 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- MBDFUYZNHQSWRD-UHFFFAOYSA-N tert-butyl n-[2-(3,4-dihydroxyphenyl)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCC1=CC=C(O)C(O)=C1 MBDFUYZNHQSWRD-UHFFFAOYSA-N 0.000 description 2
- BUEFFVNTSVYZBM-UHFFFAOYSA-N tert-butyl n-[[3,4-bis(diethoxyphosphoryl)phenyl]methyl]carbamate Chemical compound CCOP(=O)(OCC)C1=CC=C(CNC(=O)OC(C)(C)C)C=C1P(=O)(OCC)OCC BUEFFVNTSVYZBM-UHFFFAOYSA-N 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 229960002180 tetracycline Drugs 0.000 description 2
- 229930101283 tetracycline Natural products 0.000 description 2
- 150000007970 thio esters Chemical class 0.000 description 2
- 229930192474 thiophene Natural products 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 150000003626 triacylglycerols Chemical class 0.000 description 2
- 150000003852 triazoles Chemical class 0.000 description 2
- 229940086542 triethylamine Drugs 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 2
- 239000011800 void material Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- WWJFFVUVFNBJTN-UIBIZFFUSA-N (2S)-2-[[(2S,3S,4S)-2-amino-4-hydroxy-4-(5-hydroxypyridin-2-yl)-3-methylbutanoyl]amino]-2-[(2R,3S,4S,5R)-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]acetic acid Chemical class C[C@@H]([C@H](N)C(=O)N[C@@H]([C@H]1O[C@H]([C@@H](O)[C@@H]1O)n1ccc(=O)[nH]c1=O)C(O)=O)[C@H](O)c1ccc(O)cn1 WWJFFVUVFNBJTN-UIBIZFFUSA-N 0.000 description 1
- YKFCISHFRZHKHY-NGQGLHOPSA-N (2s)-2-amino-3-(3,4-dihydroxyphenyl)-2-methylpropanoic acid;trihydrate Chemical compound O.O.O.OC(=O)[C@](N)(C)CC1=CC=C(O)C(O)=C1.OC(=O)[C@](N)(C)CC1=CC=C(O)C(O)=C1 YKFCISHFRZHKHY-NGQGLHOPSA-N 0.000 description 1
- UGBUVVBCVYWCSO-JDNUQWHUSA-N (2s,3s,4r,5r)-3-[(2-amino-3-methylpentanoyl)amino]-5-(6-aminopurin-9-yl)-4-hydroxyoxolane-2-carboxylic acid Chemical compound O1[C@H](C(O)=O)[C@@H](NC(=O)C(N)C(C)CC)[C@@H](O)[C@@H]1N1C2=NC=NC(N)=C2N=C1 UGBUVVBCVYWCSO-JDNUQWHUSA-N 0.000 description 1
- HJBGZJMKTOMQRR-UHFFFAOYSA-N (3-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC(C=O)=C1 HJBGZJMKTOMQRR-UHFFFAOYSA-N 0.000 description 1
- BIWQNIMLAISTBV-UHFFFAOYSA-N (4-methylphenyl)boronic acid Chemical compound CC1=CC=C(B(O)O)C=C1 BIWQNIMLAISTBV-UHFFFAOYSA-N 0.000 description 1
- NSOITZIGPJTCMQ-KCXFZFQESA-N (4r,7s,10s,13s,16s,19s,22r)-22-[[(2s,3s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-1-[(2s)-2-[[(2s,3s)-2-[[(2s)-6-amino-2-[[(2s,3s)-2-[[(2s)-2-[[2-[[2-[[(2s)-2-[[(2s)-2-amino-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]acetyl]amino]-4-methylpentanoyl]a Chemical compound C([C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]1C(N[C@@H](C)C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CSSC1)C(O)=O)[C@@H](C)O)C(C)C)=O)C1=CC=CC=C1 NSOITZIGPJTCMQ-KCXFZFQESA-N 0.000 description 1
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 1
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 description 1
- OXDSKEQSEGDAFN-UHFFFAOYSA-N 1,1,1-trifluoro-n-phenylmethanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)NC1=CC=CC=C1 OXDSKEQSEGDAFN-UHFFFAOYSA-N 0.000 description 1
- CXWGKAYMVASWDQ-UHFFFAOYSA-N 1,2-dithiane Chemical compound C1CCSSC1 CXWGKAYMVASWDQ-UHFFFAOYSA-N 0.000 description 1
- FLBAYUMRQUHISI-UHFFFAOYSA-N 1,8-naphthyridine Chemical compound N1=CC=CC2=CC=CN=C21 FLBAYUMRQUHISI-UHFFFAOYSA-N 0.000 description 1
- QKIIAXXFKBPWFZ-UHFFFAOYSA-N 1-[bis(diethoxyphosphoryl)methyl]-4-bromobenzene Chemical compound CCOP(=O)(OCC)C(P(=O)(OCC)OCC)C1=CC=C(Br)C=C1 QKIIAXXFKBPWFZ-UHFFFAOYSA-N 0.000 description 1
- NYYLZXREFNYPKB-UHFFFAOYSA-N 1-[ethoxy(methyl)phosphoryl]oxyethane Chemical compound CCOP(C)(=O)OCC NYYLZXREFNYPKB-UHFFFAOYSA-N 0.000 description 1
- YLRBJYMANQKEAW-UHFFFAOYSA-N 1-bromo-4-(bromomethyl)benzene Chemical compound BrCC1=CC=C(Br)C=C1 YLRBJYMANQKEAW-UHFFFAOYSA-N 0.000 description 1
- AXTGDCSMTYGJND-UHFFFAOYSA-N 1-dodecylazepan-2-one Chemical compound CCCCCCCCCCCCN1CCCCCC1=O AXTGDCSMTYGJND-UHFFFAOYSA-N 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- TZMSYXZUNZXBOL-UHFFFAOYSA-N 10H-phenoxazine Chemical compound C1=CC=C2NC3=CC=CC=C3OC2=C1 TZMSYXZUNZXBOL-UHFFFAOYSA-N 0.000 description 1
- RSXFZXJOBQZOOM-INBIVCPUSA-N 143591-04-2 Chemical compound O([C@@H]\1COC(=O)C[C@H](C2=CC=C(C(=N2)Cl)O[C@@H]2[C@@H](O[C@@H]3O[C@@H](C)[C@H](O)[C@](C)(O)C3)[C@]34O[C@H]3C#C/C=C/1C#CC4=C2)NC(=O)C=1C(O)=CC2=CC(OC(C)C)=C(C(=C2C=1)OC)OC)[C@H]1C[C@H](O)[C@H](N(C)C)[C@H](C)O1 RSXFZXJOBQZOOM-INBIVCPUSA-N 0.000 description 1
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- WVXRAFOPTSTNLL-NKWVEPMBSA-N 2',3'-dideoxyadenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1CC[C@@H](CO)O1 WVXRAFOPTSTNLL-NKWVEPMBSA-N 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- ZHXUWDPHUQHFOV-UHFFFAOYSA-N 2,5-dibromopyridine Chemical compound BrC1=CC=C(Br)N=C1 ZHXUWDPHUQHFOV-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- SXGZJKUKBWWHRA-UHFFFAOYSA-N 2-(N-morpholiniumyl)ethanesulfonate Chemical compound [O-]S(=O)(=O)CC[NH+]1CCOCC1 SXGZJKUKBWWHRA-UHFFFAOYSA-N 0.000 description 1
- IMSODMZESSGVBE-UHFFFAOYSA-N 2-Oxazoline Chemical compound C1CN=CO1 IMSODMZESSGVBE-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- UXGVMFHEKMGWMA-UHFFFAOYSA-N 2-benzofuran Chemical compound C1=CC=CC2=COC=C21 UXGVMFHEKMGWMA-UHFFFAOYSA-N 0.000 description 1
- SGUAFYQXFOLMHL-UHFFFAOYSA-N 2-hydroxy-5-{1-hydroxy-2-[(4-phenylbutan-2-yl)amino]ethyl}benzamide Chemical compound C=1C=C(O)C(C(N)=O)=CC=1C(O)CNC(C)CCC1=CC=CC=C1 SGUAFYQXFOLMHL-UHFFFAOYSA-N 0.000 description 1
- JAWKOSRQDZQJMU-UHFFFAOYSA-N 2-methylpropan-2-ol;toluene Chemical compound CC(C)(C)O.CC1=CC=CC=C1 JAWKOSRQDZQJMU-UHFFFAOYSA-N 0.000 description 1
- FUBFWTUFPGFHOJ-UHFFFAOYSA-N 2-nitrofuran Chemical class [O-][N+](=O)C1=CC=CO1 FUBFWTUFPGFHOJ-UHFFFAOYSA-N 0.000 description 1
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- DTYYGFIFNKZDAP-UHFFFAOYSA-N 3-(3,4-diphosphonophenyl)propanoic acid Chemical compound OC(=O)CCC1=CC=C(P(O)(O)=O)C(P(O)(O)=O)=C1 DTYYGFIFNKZDAP-UHFFFAOYSA-N 0.000 description 1
- GJAZSXGQOOAJQE-UHFFFAOYSA-N 3-[2-[tert-butyl(dimethyl)silyl]oxyethyl]phenol Chemical compound CC(C)(C)[Si](C)(C)OCCC1=CC=CC(O)=C1 GJAZSXGQOOAJQE-UHFFFAOYSA-N 0.000 description 1
- PIVIYQXUQRKVNG-UHFFFAOYSA-N 3-[4-amino-5-(3-methoxyphenyl)pyrrolo[2,3-d]pyrimidin-7-yl]benzoic acid Chemical compound COC1=CC=CC(C=2C3=C(N)N=CN=C3N(C=3C=C(C=CC=3)C(O)=O)C=2)=C1 PIVIYQXUQRKVNG-UHFFFAOYSA-N 0.000 description 1
- PSUXTZLDBVEZTD-UHFFFAOYSA-N 3-bromopropoxymethylbenzene Chemical compound BrCCCOCC1=CC=CC=C1 PSUXTZLDBVEZTD-UHFFFAOYSA-N 0.000 description 1
- PNPCRKVUWYDDST-UHFFFAOYSA-N 3-chloroaniline Chemical compound NC1=CC=CC(Cl)=C1 PNPCRKVUWYDDST-UHFFFAOYSA-N 0.000 description 1
- WHNPOQXWAMXPTA-UHFFFAOYSA-N 3-methylbut-2-enamide Chemical compound CC(C)=CC(N)=O WHNPOQXWAMXPTA-UHFFFAOYSA-N 0.000 description 1
- ZVAYUUUQOCPZCZ-UHFFFAOYSA-N 4-(diethoxyphosphorylmethyl)aniline Chemical compound CCOP(=O)(OCC)CC1=CC=C(N)C=C1 ZVAYUUUQOCPZCZ-UHFFFAOYSA-N 0.000 description 1
- WWMDQMZNHXGEKI-UHFFFAOYSA-N 4-[4-amino-5-(4-methylphenyl)-4a,7a-dihydropyrrolo[2,3-d]pyrimidin-7-yl]pyridine-2,6-dicarboxylic acid Chemical compound C1=CC(C)=CC=C1C1=CN(C=2C=C(N=C(C=2)C(O)=O)C(O)=O)C2C1C(N)=NC=N2 WWMDQMZNHXGEKI-UHFFFAOYSA-N 0.000 description 1
- RKYWAGNCLSZGRF-UHFFFAOYSA-N 4-chloro-5-iodo-6,7-dihydropyrrolo[3,2-d]pyrimidine Chemical compound ClC1=NC=NC2=C1N(I)CC2 RKYWAGNCLSZGRF-UHFFFAOYSA-N 0.000 description 1
- KJHYDYATVCGFKG-UHFFFAOYSA-N 4-chloro-5-iodo-7-propan-2-ylpyrrolo[2,3-d]pyrimidine Chemical compound N1=CN=C2N(C(C)C)C=C(I)C2=C1Cl KJHYDYATVCGFKG-UHFFFAOYSA-N 0.000 description 1
- CBWBJFJMNBPWAL-UHFFFAOYSA-N 4-chloro-5-iodo-7h-pyrrolo[2,3-d]pyrimidine Chemical compound ClC1=NC=NC2=C1C(I)=CN2 CBWBJFJMNBPWAL-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- GDRVFDDBLLKWRI-UHFFFAOYSA-N 4H-quinolizine Chemical compound C1=CC=CN2CC=CC=C21 GDRVFDDBLLKWRI-UHFFFAOYSA-N 0.000 description 1
- VZYMZAMQIGDUJW-UHFFFAOYSA-N 5-iodopyrrolo[3,2-d]pyrimidine Chemical compound C1=NC=C2N(I)C=CC2=N1 VZYMZAMQIGDUJW-UHFFFAOYSA-N 0.000 description 1
- KDOPAZIWBAHVJB-UHFFFAOYSA-N 5h-pyrrolo[3,2-d]pyrimidine Chemical compound C1=NC=C2NC=CC2=N1 KDOPAZIWBAHVJB-UHFFFAOYSA-N 0.000 description 1
- RYYIULNRIVUMTQ-UHFFFAOYSA-N 6-chloroguanine Chemical compound NC1=NC(Cl)=C2N=CNC2=N1 RYYIULNRIVUMTQ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- JJTNLWSCFYERCK-UHFFFAOYSA-N 7h-pyrrolo[2,3-d]pyrimidine Chemical compound N1=CN=C2NC=CC2=C1 JJTNLWSCFYERCK-UHFFFAOYSA-N 0.000 description 1
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical compound C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- 208000000819 Adrenocortical Hyperfunction Diseases 0.000 description 1
- 229930195730 Aflatoxin Natural products 0.000 description 1
- XWIYFDMXXLINPU-UHFFFAOYSA-N Aflatoxin G Chemical compound O=C1OCCC2=C1C(=O)OC1=C2C(OC)=CC2=C1C1C=COC1O2 XWIYFDMXXLINPU-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 108010083590 Apoproteins Proteins 0.000 description 1
- 102000006410 Apoproteins Human genes 0.000 description 1
- 235000003911 Arachis Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 108010001478 Bacitracin Proteins 0.000 description 1
- 206010065687 Bone loss Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- UBBKJMZDXKBEJZ-UHFFFAOYSA-N C(C1=CC=CC=C1)OCCCC([PH2]=O)(OCC)P(O)(O)=O Chemical compound C(C1=CC=CC=C1)OCCCC([PH2]=O)(OCC)P(O)(O)=O UBBKJMZDXKBEJZ-UHFFFAOYSA-N 0.000 description 1
- VHIBOFWCGOAFJE-UHFFFAOYSA-N C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.Cl.Cl.[Fe+2] Chemical compound C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.Cl.Cl.[Fe+2] VHIBOFWCGOAFJE-UHFFFAOYSA-N 0.000 description 1
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 1
- NQRORYWLJHSFEI-UHFFFAOYSA-N CC(CC=C1)(C=C1C(C1C(CC2)=NC=NC1(C)N)=CC2c1cc(CNCCCC2OC(C[O](O)(=O)=O)C=C2O)ccc1)OC Chemical compound CC(CC=C1)(C=C1C(C1C(CC2)=NC=NC1(C)N)=CC2c1cc(CNCCCC2OC(C[O](O)(=O)=O)C=C2O)ccc1)OC NQRORYWLJHSFEI-UHFFFAOYSA-N 0.000 description 1
- LHXPIJVXJXUMEI-UHFFFAOYSA-N CCOC(CCCOCc1ccccc1)([PH2]=O)P(=O)(OCC)OCC Chemical compound CCOC(CCCOCc1ccccc1)([PH2]=O)P(=O)(OCC)OCC LHXPIJVXJXUMEI-UHFFFAOYSA-N 0.000 description 1
- IOVSHOREAGHTBZ-UHFFFAOYSA-N CCOC([PH2]=O)(c1ccc(N)cc1)P(=O)(OCC)OCC Chemical compound CCOC([PH2]=O)(c1ccc(N)cc1)P(=O)(OCC)OCC IOVSHOREAGHTBZ-UHFFFAOYSA-N 0.000 description 1
- 101150012716 CDK1 gene Proteins 0.000 description 1
- ZCXUMDMLUGOCPU-UHFFFAOYSA-N COc1cc(-c(c2c(N)ncnc22)c[n]2-c(cc2)ccc2C(N)=O)ccc1 Chemical compound COc1cc(-c(c2c(N)ncnc22)c[n]2-c(cc2)ccc2C(N)=O)ccc1 ZCXUMDMLUGOCPU-UHFFFAOYSA-N 0.000 description 1
- 102000055006 Calcitonin Human genes 0.000 description 1
- 108060001064 Calcitonin Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 108050004290 Cecropin Proteins 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 1
- GJSURZIOUXUGAL-UHFFFAOYSA-N Clonidine Chemical compound ClC1=CC=CC(Cl)=C1NC1=NCCN1 GJSURZIOUXUGAL-UHFFFAOYSA-N 0.000 description 1
- 208000023890 Complex Regional Pain Syndromes Diseases 0.000 description 1
- 241000252095 Congridae Species 0.000 description 1
- 108010069514 Cyclic Peptides Proteins 0.000 description 1
- 102000001189 Cyclic Peptides Human genes 0.000 description 1
- 108010037414 Cytoskeletal Proteins Proteins 0.000 description 1
- 102000010831 Cytoskeletal Proteins Human genes 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 108010002069 Defensins Proteins 0.000 description 1
- 102000000541 Defensins Human genes 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 101100244111 Dictyostelium discoideum stlA gene Proteins 0.000 description 1
- BXZVVICBKDXVGW-NKWVEPMBSA-N Didanosine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(NC=NC2=O)=C2N=C1 BXZVVICBKDXVGW-NKWVEPMBSA-N 0.000 description 1
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 1
- CTENFNNZBMHDDG-UHFFFAOYSA-N Dopamine hydrochloride Chemical compound Cl.NCCC1=CC=C(O)C(O)=C1 CTENFNNZBMHDDG-UHFFFAOYSA-N 0.000 description 1
- 101710164770 Drosomycin Proteins 0.000 description 1
- 229930193152 Dynemicin Natural products 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 208000002197 Ehlers-Danlos syndrome Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 101100059559 Emericella nidulans (strain FGSC A4 / ATCC 38163 / CBS 112.46 / NRRL 194 / M139) nimX gene Proteins 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 229930189413 Esperamicin Natural products 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- 241000400611 Eucalyptus deanei Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 229910052688 Gadolinium Inorganic materials 0.000 description 1
- 208000015872 Gaucher disease Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 229940122957 Histamine H2 receptor antagonist Drugs 0.000 description 1
- 101000761509 Homo sapiens Cathepsin K Proteins 0.000 description 1
- 101000635799 Homo sapiens Run domain Beclin-1-interacting and cysteine-rich domain-containing protein Proteins 0.000 description 1
- 206010020365 Homocystinuria Diseases 0.000 description 1
- 206010020564 Hyperadrenocorticism Diseases 0.000 description 1
- 208000037147 Hypercalcaemia Diseases 0.000 description 1
- 201000002980 Hyperparathyroidism Diseases 0.000 description 1
- 206010020707 Hyperparathyroidism primary Diseases 0.000 description 1
- 206010020850 Hyperthyroidism Diseases 0.000 description 1
- 206010058359 Hypogonadism Diseases 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 101800004361 Lactoferricin-B Proteins 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000030136 Marchiafava-Bignami Disease Diseases 0.000 description 1
- 208000001826 Marfan syndrome Diseases 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 208000008948 Menkes Kinky Hair Syndrome Diseases 0.000 description 1
- 208000012583 Menkes disease Diseases 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- DBTDEFJAFBUGPP-UHFFFAOYSA-N Methanethial Chemical compound S=C DBTDEFJAFBUGPP-UHFFFAOYSA-N 0.000 description 1
- QXKHYNVANLEOEG-UHFFFAOYSA-N Methoxsalen Chemical compound C1=CC(=O)OC2=C1C=C1C=COC1=C2OC QXKHYNVANLEOEG-UHFFFAOYSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 101100185019 Mycobacterium bovis (strain ATCC BAA-935 / AF2122/97) pks15/1 gene Proteins 0.000 description 1
- 229930184499 Nikkomycin Natural products 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 206010031243 Osteogenesis imperfecta Diseases 0.000 description 1
- 101150084980 PKS1 gene Proteins 0.000 description 1
- 102000003982 Parathyroid hormone Human genes 0.000 description 1
- 108090000445 Parathyroid hormone Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 108010040201 Polymyxins Proteins 0.000 description 1
- 229930182764 Polyoxin Natural products 0.000 description 1
- 201000000981 Primary Hyperparathyroidism Diseases 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 229930194194 Rubratoxin Natural products 0.000 description 1
- WBTCZXYOKNRFQX-UHFFFAOYSA-N S1(=O)(=O)NC1=O Chemical group S1(=O)(=O)NC1=O WBTCZXYOKNRFQX-UHFFFAOYSA-N 0.000 description 1
- 102000001332 SRC Human genes 0.000 description 1
- 108060006706 SRC Proteins 0.000 description 1
- 101100136769 Sarocladium schorii aspks1 gene Proteins 0.000 description 1
- 208000005770 Secondary Hyperparathyroidism Diseases 0.000 description 1
- 206010039984 Senile osteoporosis Diseases 0.000 description 1
- 206010072610 Skeletal dysplasia Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 239000005708 Sodium hypochlorite Substances 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 244000028419 Styrax benzoin Species 0.000 description 1
- 235000000126 Styrax benzoin Nutrition 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 235000008411 Sumatra benzointree Nutrition 0.000 description 1
- ZQVJBRJGDVZANE-UHFFFAOYSA-N Syringomycin Natural products N1C(=O)C(CCCN=C(N)N)NC(=O)C(CCN)NC(=O)C(CCN)NC(=O)C(CO)NC(=O)C(NC(=O)CC(O)CCCCCCCCC)COC(=O)C(C(O)CCl)NC(=O)C(C(O)C(O)=O)NC(=O)C(=CC)NC(=O)C1CC1=CC=CC=C1 ZQVJBRJGDVZANE-UHFFFAOYSA-N 0.000 description 1
- 201000008736 Systemic mastocytosis Diseases 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 244000269722 Thea sinensis Species 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical class [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 101800003783 Tritrpticin Proteins 0.000 description 1
- 208000026928 Turner syndrome Diseases 0.000 description 1
- 239000003875 Wang resin Substances 0.000 description 1
- PCWZKQSKUXXDDJ-UHFFFAOYSA-N Xanthotoxin Natural products COCc1c2OC(=O)C=Cc2cc3ccoc13 PCWZKQSKUXXDDJ-UHFFFAOYSA-N 0.000 description 1
- 101500009721 Xenopus laevis Magainin-2 Proteins 0.000 description 1
- 101100273808 Xenopus laevis cdk1-b gene Proteins 0.000 description 1
- WREGKURFCTUGRC-POYBYMJQSA-N Zalcitabine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)CC1 WREGKURFCTUGRC-POYBYMJQSA-N 0.000 description 1
- 101710095824 Zeamatin Proteins 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- NERFNHBZJXXFGY-UHFFFAOYSA-N [4-[(4-methylphenyl)methoxy]phenyl]methanol Chemical compound C1=CC(C)=CC=C1COC1=CC=C(CO)C=C1 NERFNHBZJXXFGY-UHFFFAOYSA-N 0.000 description 1
- SHXZXRGRQZTMSQ-UHFFFAOYSA-N [4-[[(2-methylpropan-2-yl)oxycarbonylamino]methyl]-2-(trifluoromethylsulfonyloxy)phenyl] trifluoromethanesulfonate Chemical compound CC(C)(C)OC(=O)NCC1=CC=C(OS(=O)(=O)C(F)(F)F)C(OS(=O)(=O)C(F)(F)F)=C1 SHXZXRGRQZTMSQ-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- FBCLKBXYZRAXNA-PDIPHZEPSA-N aculeacin A Chemical class C1([C@H](O)[C@@H](O)[C@H]2C(=O)N[C@H](C(=O)N3C[C@H](C)[C@H](O)[C@H]3C(=O)N[C@H](O)[C@H](O)C[C@@H](C(N[C@H](C(=O)N3C[C@H](O)C[C@H]3C(=O)N2)[C@@H](C)O)=O)NC(=O)CCCCCCCCCCCCCCC)[C@H](O)CC(N)=O)=CC=C(O)C=C1 FBCLKBXYZRAXNA-PDIPHZEPSA-N 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 238000005377 adsorption chromatography Methods 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000005409 aflatoxin Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 229960004538 alprazolam Drugs 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229920003180 amino resin Polymers 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- 229940072174 amphenicols Drugs 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 229940051880 analgesics and antipyretics pyrazolones Drugs 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000003257 anti-anginal effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000000123 anti-resoprtive effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 229940124345 antianginal agent Drugs 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000001961 anticonvulsive agent Substances 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 229960002274 atenolol Drugs 0.000 description 1
- MNFORVFSTILPAW-UHFFFAOYSA-N azetidin-2-one Chemical class O=C1CCN1 MNFORVFSTILPAW-UHFFFAOYSA-N 0.000 description 1
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 1
- 229960003071 bacitracin Drugs 0.000 description 1
- 229930184125 bacitracin Natural products 0.000 description 1
- CLKOFPXJLQSYAH-ABRJDSQDSA-N bacitracin A Chemical compound C1SC([C@@H](N)[C@@H](C)CC)=N[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]1C(=O)N[C@H](CCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2N=CNC=2)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)NCCCC1 CLKOFPXJLQSYAH-ABRJDSQDSA-N 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- 229960002130 benzoin Drugs 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000002805 bone matrix Anatomy 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 150000001639 boron compounds Chemical class 0.000 description 1
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 1
- ZDIGNSYAACHWNL-UHFFFAOYSA-N brompheniramine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=C(Br)C=C1 ZDIGNSYAACHWNL-UHFFFAOYSA-N 0.000 description 1
- 229960000725 brompheniramine Drugs 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 description 1
- 229960004015 calcitonin Drugs 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- 239000003560 cancer drug Substances 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- 229940041011 carbapenems Drugs 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 229940097217 cardiac glycoside Drugs 0.000 description 1
- 239000002368 cardiac glycoside Substances 0.000 description 1
- 239000002327 cardiovascular agent Substances 0.000 description 1
- 229940125692 cardiovascular agent Drugs 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 229940083181 centrally acting adntiadrenergic agent methyldopa Drugs 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 150000001793 charged compounds Chemical group 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- SOYKEARSMXGVTM-UHFFFAOYSA-N chlorphenamine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=C(Cl)C=C1 SOYKEARSMXGVTM-UHFFFAOYSA-N 0.000 description 1
- 229960003291 chlorphenamine Drugs 0.000 description 1
- QZHPTGXQGDFGEN-UHFFFAOYSA-N chromene Chemical compound C1=CC=C2C=C[CH]OC2=C1 QZHPTGXQGDFGEN-UHFFFAOYSA-N 0.000 description 1
- 229960001380 cimetidine Drugs 0.000 description 1
- CCGSUNCLSOWKJO-UHFFFAOYSA-N cimetidine Chemical compound N#CNC(=N/C)\NCCSCC1=NC=N[C]1C CCGSUNCLSOWKJO-UHFFFAOYSA-N 0.000 description 1
- WCZVZNOTHYJIEI-UHFFFAOYSA-N cinnoline Chemical compound N1=NC=CC2=CC=CC=C21 WCZVZNOTHYJIEI-UHFFFAOYSA-N 0.000 description 1
- VDUWPHTZYNWKRN-UHFFFAOYSA-N cinoxacin Chemical compound C1=C2N(CC)N=C(C(O)=O)C(=O)C2=CC2=C1OCO2 VDUWPHTZYNWKRN-UHFFFAOYSA-N 0.000 description 1
- 229960004621 cinoxacin Drugs 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- ACSIXWWBWUQEHA-UHFFFAOYSA-N clodronic acid Chemical compound OP(O)(=O)C(Cl)(Cl)P(O)(O)=O ACSIXWWBWUQEHA-UHFFFAOYSA-N 0.000 description 1
- 229960002896 clonidine Drugs 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 239000005515 coenzyme Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 238000011970 concomitant therapy Methods 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 230000003436 cytoskeletal effect Effects 0.000 description 1
- 210000004292 cytoskeleton Anatomy 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000002354 daily effect Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000017858 demethylation Effects 0.000 description 1
- 238000010520 demethylation reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000009795 derivation Methods 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- YFHLIDBAPTWLGU-CTKMSOPVSA-N dermaseptin Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(O)=O)[C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCSC)NC(=O)[C@@H](NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)N)[C@@H](C)O)[C@@H](C)O)C1=CN=CN1 YFHLIDBAPTWLGU-CTKMSOPVSA-N 0.000 description 1
- 108090000454 dermaseptin Proteins 0.000 description 1
- 229940049701 dermaseptin Drugs 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 description 1
- 239000002027 dichloromethane extract Substances 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 1
- 229960005156 digoxin Drugs 0.000 description 1
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- LOZWAPSEEHRYPG-UHFFFAOYSA-N dithiane Natural products C1CSCCS1 LOZWAPSEEHRYPG-UHFFFAOYSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 229940125532 enzyme inhibitor Drugs 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 239000003172 expectorant agent Substances 0.000 description 1
- 230000003419 expectorant effect Effects 0.000 description 1
- 229940066493 expectorants Drugs 0.000 description 1
- 229960001596 famotidine Drugs 0.000 description 1
- XUFQPHANEAPEMJ-UHFFFAOYSA-N famotidine Chemical compound NC(N)=NC1=NC(CSCCC(N)=NS(N)(=O)=O)=CS1 XUFQPHANEAPEMJ-UHFFFAOYSA-N 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229930003935 flavonoid Natural products 0.000 description 1
- 150000002215 flavonoids Chemical class 0.000 description 1
- 235000017173 flavonoids Nutrition 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 238000001506 fluorescence spectroscopy Methods 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- JKFAIQOWCVVSKC-UHFFFAOYSA-N furazan Chemical compound C=1C=NON=1 JKFAIQOWCVVSKC-UHFFFAOYSA-N 0.000 description 1
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 229960005150 glycerol Drugs 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 235000019382 gum benzoic Nutrition 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- XLYOFNOQVPJJNP-ZSJDYOACSA-N heavy water Substances [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 150000002390 heteroarenes Chemical class 0.000 description 1
- 238000012203 high throughput assay Methods 0.000 description 1
- 239000012510 hollow fiber Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 208000008750 humoral hypercalcemia of malignancy Diseases 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 230000000148 hypercalcaemia Effects 0.000 description 1
- 208000030915 hypercalcemia disease Diseases 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 238000010191 image analysis Methods 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000012750 in vivo screening Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- USSYUMHVHQSYNA-SLDJZXPVSA-N indolicidin Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N1CCC[C@H]1C(=O)N[C@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(N)=O)CC1=CNC2=CC=CC=C12 USSYUMHVHQSYNA-SLDJZXPVSA-N 0.000 description 1
- HOBCFUWDNJPFHB-UHFFFAOYSA-N indolizine Chemical compound C1=CC=CN2C=CC=C21 HOBCFUWDNJPFHB-UHFFFAOYSA-N 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 230000002608 insulinlike Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 210000004347 intestinal mucosa Anatomy 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- FMKOJHQHASLBPH-UHFFFAOYSA-N isopropyl iodide Chemical compound CC(C)I FMKOJHQHASLBPH-UHFFFAOYSA-N 0.000 description 1
- XUGNVMKQXJXZCD-UHFFFAOYSA-N isopropyl palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(C)C XUGNVMKQXJXZCD-UHFFFAOYSA-N 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- CFFMZOZGXDAXHP-HOKBLYKWSA-N lactoferricin Chemical compound C([C@H](NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C(C)C)NC(=O)[C@@H]1CSSC[C@@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCCN)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](C)C(=O)N2CCC[C@H]2C(=O)N[C@@H](CO)C(=O)N[C@H](C(N[C@H](C(=O)N1)[C@@H](C)O)=O)[C@@H](C)CC)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(O)=O)C1=CC=CC=C1 CFFMZOZGXDAXHP-HOKBLYKWSA-N 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- QDLAGTHXVHQKRE-UHFFFAOYSA-N lichenxanthone Natural products COC1=CC(O)=C2C(=O)C3=C(C)C=C(OC)C=C3OC2=C1 QDLAGTHXVHQKRE-UHFFFAOYSA-N 0.000 description 1
- 229940041028 lincosamides Drugs 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 239000003120 macrolide antibiotic agent Substances 0.000 description 1
- 229940041033 macrolides Drugs 0.000 description 1
- MGIUUAHJVPPFEV-ABXDCCGRSA-N magainin ii Chemical compound C([C@H](NC(=O)[C@H](CCCCN)NC(=O)CNC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(O)=O)C1=CC=CC=C1 MGIUUAHJVPPFEV-ABXDCCGRSA-N 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 238000002595 magnetic resonance imaging Methods 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- AWIJRPNMLHPLNC-UHFFFAOYSA-N methanethioic s-acid Chemical compound SC=O AWIJRPNMLHPLNC-UHFFFAOYSA-N 0.000 description 1
- 229960004469 methoxsalen Drugs 0.000 description 1
- SQBBOVROCFXYBN-UHFFFAOYSA-N methoxypsoralen Natural products C1=C2OC(=O)C(OC)=CC2=CC2=C1OC=C2 SQBBOVROCFXYBN-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical compound CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229940041009 monobactams Drugs 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 210000002200 mouth mucosa Anatomy 0.000 description 1
- 238000003541 multi-stage reaction Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- DILRJUIACXKSQE-UHFFFAOYSA-N n',n'-dimethylethane-1,2-diamine Chemical compound CN(C)CCN DILRJUIACXKSQE-UHFFFAOYSA-N 0.000 description 1
- AWEROZFZHJVVKB-UHFFFAOYSA-N n-(3-chlorophenyl)-2-fluoro-9-methylpurin-6-amine Chemical compound N1=C(F)N=C2N(C)C=NC2=C1NC1=CC=CC(Cl)=C1 AWEROZFZHJVVKB-UHFFFAOYSA-N 0.000 description 1
- QQFIHWSNVOXCEY-UHFFFAOYSA-N n-(3-iodophenyl)-2,2-dimethylpropanamide Chemical compound CC(C)(C)C(=O)NC1=CC=CC(I)=C1 QQFIHWSNVOXCEY-UHFFFAOYSA-N 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 1
- 238000011587 new zealand white rabbit Methods 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- SGXXNSQHWDMGGP-IZZDOVSWSA-N nizatidine Chemical compound [O-][N+](=O)\C=C(/NC)NCCSCC1=CSC(CN(C)C)=N1 SGXXNSQHWDMGGP-IZZDOVSWSA-N 0.000 description 1
- 229960004872 nizatidine Drugs 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- OGJPXUAPXNRGGI-UHFFFAOYSA-N norfloxacin Chemical compound C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 OGJPXUAPXNRGGI-UHFFFAOYSA-N 0.000 description 1
- 229960001180 norfloxacin Drugs 0.000 description 1
- 150000007523 nucleic acids Chemical group 0.000 description 1
- 235000020939 nutritional additive Nutrition 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000001599 osteoclastic effect Effects 0.000 description 1
- 230000001009 osteoporotic effect Effects 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- ADIMAYPTOBDMTL-UHFFFAOYSA-N oxazepam Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(O)N=C1C1=CC=CC=C1 ADIMAYPTOBDMTL-UHFFFAOYSA-N 0.000 description 1
- 229960004535 oxazepam Drugs 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 239000000199 parathyroid hormone Substances 0.000 description 1
- 229960001319 parathyroid hormone Drugs 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- KQXKVJAGOJTNJS-HNNXBMFYSA-N penbutolol Chemical compound CC(C)(C)NC[C@H](O)COC1=CC=CC=C1C1CCCC1 KQXKVJAGOJTNJS-HNNXBMFYSA-N 0.000 description 1
- 229960002035 penbutolol Drugs 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 150000002960 penicillins Chemical class 0.000 description 1
- 231100000435 percutaneous penetration Toxicity 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 229950000688 phenothiazine Drugs 0.000 description 1
- GJSGGHOYGKMUPT-UHFFFAOYSA-N phenoxathiine Chemical compound C1=CC=C2OC3=CC=CC=C3SC2=C1 GJSGGHOYGKMUPT-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- LFSXCDWNBUNEEM-UHFFFAOYSA-N phthalazine Chemical compound C1=NN=CC2=CC=CC=C21 LFSXCDWNBUNEEM-UHFFFAOYSA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- PHUTUTUABXHXLW-UHFFFAOYSA-N pindolol Chemical compound CC(C)NCC(O)COC1=CC=CC2=NC=C[C]12 PHUTUTUABXHXLW-UHFFFAOYSA-N 0.000 description 1
- 229960002508 pindolol Drugs 0.000 description 1
- 239000000902 placebo Substances 0.000 description 1
- 229940068196 placebo Drugs 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 150000004291 polyenes Chemical class 0.000 description 1
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 229920006316 polyvinylpyrrolidine Polymers 0.000 description 1
- 208000001685 postmenopausal osteoporosis Diseases 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- IENZQIKPVFGBNW-UHFFFAOYSA-N prazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1=CC=CO1 IENZQIKPVFGBNW-UHFFFAOYSA-N 0.000 description 1
- 229960001289 prazosin Drugs 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 201000009395 primary hyperaldosteronism Diseases 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- CPNGPNLZQNNVQM-UHFFFAOYSA-N pteridine Chemical compound N1=CN=CC2=NC=CN=C21 CPNGPNLZQNNVQM-UHFFFAOYSA-N 0.000 description 1
- 201000010108 pycnodysostosis Diseases 0.000 description 1
- JEXVQSWXXUJEMA-UHFFFAOYSA-N pyrazol-3-one Chemical class O=C1C=CN=N1 JEXVQSWXXUJEMA-UHFFFAOYSA-N 0.000 description 1
- 150000003217 pyrazoles Chemical class 0.000 description 1
- YAAWASYJIRZXSZ-UHFFFAOYSA-N pyrimidine-2,4-diamine Chemical class NC1=CC=NC(N)=N1 YAAWASYJIRZXSZ-UHFFFAOYSA-N 0.000 description 1
- 150000004040 pyrrolidinones Chemical class 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 1
- 150000007660 quinolones Chemical class 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000003439 radiotherapeutic effect Effects 0.000 description 1
- 108010047804 ranalexin Proteins 0.000 description 1
- VMXUWOKSQNHOCA-LCYFTJDESA-N ranitidine Chemical compound [O-][N+](=O)/C=C(/NC)NCCSCC1=CC=C(CN(C)C)O1 VMXUWOKSQNHOCA-LCYFTJDESA-N 0.000 description 1
- 229960000620 ranitidine Drugs 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- MYFATKRONKHHQL-UHFFFAOYSA-N rhodamine 123 Chemical compound [Cl-].COC(=O)C1=CC=CC=C1C1=C2C=CC(=[NH2+])C=C2OC2=CC(N)=CC=C21 MYFATKRONKHHQL-UHFFFAOYSA-N 0.000 description 1
- 229960000329 ribavirin Drugs 0.000 description 1
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 238000011808 rodent model Methods 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940058287 salicylic acid derivative anticestodals Drugs 0.000 description 1
- 150000003872 salicylic acid derivatives Chemical class 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 210000001991 scapula Anatomy 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 1
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 1
- 238000000526 short-path distillation Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 239000004289 sodium hydrogen sulphite Substances 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000008227 sterile water for injection Substances 0.000 description 1
- 229930002534 steroid glycoside Natural products 0.000 description 1
- 150000008143 steroidal glycosides Chemical class 0.000 description 1
- 239000000021 stimulant Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 229940079488 strontium ranelate Drugs 0.000 description 1
- KVJIIORUFXGTFN-UHFFFAOYSA-L strontium;5-[bis(carboxymethyl)amino]-3-(carboxymethyl)-4-cyanothiophene-2-carboxylate Chemical compound [Sr+2].OC(=O)CN(CC(O)=O)C=1SC(C([O-])=O)=C(CC(O)=O)C=1C#N.OC(=O)CN(CC(O)=O)C=1SC(C([O-])=O)=C(CC(O)=O)C=1C#N KVJIIORUFXGTFN-UHFFFAOYSA-L 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 150000008053 sultones Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- IMCGHZIGRANKHV-AJNGGQMLSA-N tert-butyl (3s,5s)-2-oxo-5-[(2s,4s)-5-oxo-4-propan-2-yloxolan-2-yl]-3-propan-2-ylpyrrolidine-1-carboxylate Chemical compound O1C(=O)[C@H](C(C)C)C[C@H]1[C@H]1N(C(=O)OC(C)(C)C)C(=O)[C@H](C(C)C)C1 IMCGHZIGRANKHV-AJNGGQMLSA-N 0.000 description 1
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 1
- WHWMOMRHHQLBQQ-UHFFFAOYSA-N tert-butyl 4-hydroxybenzoate Chemical compound CC(C)(C)OC(=O)C1=CC=C(O)C=C1 WHWMOMRHHQLBQQ-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- XREGVKDMOATJAW-UHFFFAOYSA-N tert-butyl n-[(3,4-dihydroxyphenyl)methyl]carbamate Chemical compound CC(C)(C)OC(=O)NCC1=CC=C(O)C(O)=C1 XREGVKDMOATJAW-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 229940040944 tetracyclines Drugs 0.000 description 1
- RAOIDOHSFRTOEL-UHFFFAOYSA-N tetrahydrothiophene Chemical compound C1CCSC1 RAOIDOHSFRTOEL-UHFFFAOYSA-N 0.000 description 1
- 108010032153 thanatin Proteins 0.000 description 1
- GVIJJXMXTUZIOD-UHFFFAOYSA-N thianthrene Chemical compound C1=CC=C2SC3=CC=CC=C3SC2=C1 GVIJJXMXTUZIOD-UHFFFAOYSA-N 0.000 description 1
- CBDKQYKMCICBOF-UHFFFAOYSA-N thiazoline Chemical compound C1CN=CS1 CBDKQYKMCICBOF-UHFFFAOYSA-N 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 1
- QERYCTSHXKAMIS-UHFFFAOYSA-N thiophene-2-carboxylic acid Chemical compound OC(=O)C1=CC=CS1 QERYCTSHXKAMIS-UHFFFAOYSA-N 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- 210000002303 tibia Anatomy 0.000 description 1
- 229960004605 timolol Drugs 0.000 description 1
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 229950003937 tolonium Drugs 0.000 description 1
- HNONEKILPDHFOL-UHFFFAOYSA-M tolonium chloride Chemical compound [Cl-].C1=C(C)C(N)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 HNONEKILPDHFOL-UHFFFAOYSA-M 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- 239000003204 tranquilizing agent Substances 0.000 description 1
- 230000002936 tranquilizing effect Effects 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- ZGYICYBLPGRURT-UHFFFAOYSA-N tri(propan-2-yl)silicon Chemical compound CC(C)[Si](C(C)C)C(C)C ZGYICYBLPGRURT-UHFFFAOYSA-N 0.000 description 1
- JOFWLTCLBGQGBO-UHFFFAOYSA-N triazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1Cl JOFWLTCLBGQGBO-UHFFFAOYSA-N 0.000 description 1
- 229960003386 triazolam Drugs 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- FTKYRNHHOBRIOY-HQUBJAAMSA-N tritrptcin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](N)C(C)C)C1=CC=CC=C1 FTKYRNHHOBRIOY-HQUBJAAMSA-N 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 210000000689 upper leg Anatomy 0.000 description 1
- LSGOVYNHVSXFFJ-UHFFFAOYSA-N vanadate(3-) Chemical compound [O-][V]([O-])([O-])=O LSGOVYNHVSXFFJ-UHFFFAOYSA-N 0.000 description 1
- 229940124549 vasodilator Drugs 0.000 description 1
- 239000003071 vasodilator agent Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229960000523 zalcitabine Drugs 0.000 description 1
- 150000003952 β-lactams Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/548—Phosphates or phosphonates, e.g. bone-seeking
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4003—Esters thereof the acid moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4006—Esters of acyclic acids which can have further substituents on alkyl
- C07F9/4012—Esters of acyclic acids which can have further substituents on alkyl substituted by B, Si, P or a metal
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4003—Esters thereof the acid moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4025—Esters of poly(thio)phosphonic acids
- C07F9/405—Esters of poly(thio)phosphonic acids containing nitrogen substituent, e.g. N.....H or N-hydrocarbon group which can be substituted by halogen or nitro(so), N.....O, N.....S, N.....C(=X)- (X =O, S), N.....N, N...C(=X)...N (X =O, S)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
Definitions
- the osteoclast uses a proton pump (an ATP-dependent transport of protons) to achieve this acidification and thus any therapeutic inhibition of the osteoclast proton pump should lead to a decrease in bone loss or turnover.
- proton pump an ATP-dependent transport of protons
- many novel therapeutics developed to reduce bone resorption have focused on the inhibition of the proton pump to prevent osteoclast activity and excessive bone resorption.
- Src protein kinases play a cruical role in osteoclastic function, and it has been shown in different cell types that phosphorylation by Src, and related kinases, of proteins proposed to participate or regulate the cytoskeletal architecture is one important requirement for their proper function (see, for example, Missbach et al., "A Novel Inhibitor of the Tyrosine Kinase Src Suppresses Phosphorylation of Its Major Cellular Substrates and Reduces Bone Resorption In Vitro and in Rodent Models In Vivo," Bone 1999, 24, 437-449).
- osteoporosis many of the existing therapeutics that have been developed for the treatment of bone disorders such as osteoporosis are thought to act by inhibiting osteoclast activity.
- estrogens, bisphosphonates, calcitonin, flavonoids, and selective estrogen receptor modulators are believed to act by the inhibition of osteoclast activity.
- novel therapeutics have been developed to promote a fast increase in bone mineral content by promoting osteoblast activity.
- Such examples include peptides from the parathyroid hormone family, strontium ranelate, and growth hormone and insulin-like growth response (see, for example, Reginster et al. "Promising New Agents in Osteoporosis," Drugs R & D 1999, 3, 195-201).
- a significant problem of many of these therapetic agents is that they are not specific enough for bone tissue and thus may lead to unwanted adverse side effects.
- a subject compound has the structure of Formula (I):
- L and K independently, are absent or represent -M n -Y-M p -;
- X, Y, and Z independently, are absent or represent NR, O, or S;
- He represents a heterocycle, preferably a nitrogen-containing heterocycle
- R 4 independently for each occurrence represents H or lower alkyl, preferably H or C C 3 lower alkyl.
- Tb is selected from i, v, vi, vii, viii, and ix
- Z is preferably absent.
- Tb is selected from ii, iii, and iv
- Z may be absent or represent O or NR.
- Tb is selected from x, xi, xii, xiii, xiv, and xv
- Z may be absent or represent O or NR, preferably being absent.
- Cy represents a carbocycle or a nitrogen-bearing heterocycle. Cy is preferably uncharged.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0]-bicyclononane) ring system.
- Cy represents a fused ring system, one substituent of L and K (or He or Tb) may be attached to one of the two rings, and the other substituent to the other of the two rings.
- Cy is phenyl.
- R 3 represents a bond to K
- R 2 is selected from hydrogen, alkyl, alkenyl, alkynyl, aralkyl, cycloalkyl, heteroaryl, heterocyclyl, cycloalkyl, polycyclyl, alkyl alkenyl, alkyl alkynyl, and alkanoyl
- Ri is selected from hydrogen, halogen, aryl, and heteroaryl.
- Ri is a substituted aryl moiety selected from of monohaloaryl, dihaloaryl, monomethylaryl, and dimethylaryl.
- at least one of Ri, R 2 , and R 3 , other than the bond to K, represents an aryl or heteroaryl substituent, preferably a substituted or unsubstituted phenyl.
- Tb is represented by vii or viii
- the compound is free of hydrolyzable linkages.
- L and K do not comprise nitrogen.
- He represents a heterocyclic bicycle.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- R 4 represents H for all occurrences.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0]-bicyclononane) ring system.
- Cy represents a fused ring system
- one substituent of L and K (or He or Tb) may be attached to one of the two rings, and the other substituent to the other of the two rings.
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0]-bicyclononane) ring system.
- Cy represents a fused ring system
- one substituent of L and K (or He or Tb) may be attached to one of the two rings, and the other substituent to the other of the two rings.
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0]-bicyclononane) ring system.
- Cy represents a fused ring system
- one substituent of L and K (or He or Tb) may be attached to one of the two rings, and the other substituent to the other of the two rings
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- K does not include an amide bond, is free of carbonyls, is free of amine substituents, or is free of nitrogen atoms.
- He represents a heterocyclic bicycle.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- the compound does not include a hydrolyzable linkage.
- R 4 represents H for all occurrences.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0]- bicyclononane) ring system.
- Cy represents a fused ring system
- one substituent of L and K may be attached to one of the two rings, and the other substituent to the other of the two rings.
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- Tb is represented by ii, iii, iv, or v
- the compound does not include a hydrolyzable linkage.
- He represents a heterocyclic bicycle.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- R 4 represents H for all occurrences.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0] -bicyclononane) ring system.
- Cy represents a fused ring system
- one substituent of L and K may be attached to one of the two rings, and the other substituent to the other of the two rings.
- K is directly attached to a heteroatom of He.
- K is absent.
- Cy is preferably uncharged, and/or L-Cy-K is preferably free of hydrolyzable linkages. In certain embodiments, the compound is free of hydrolyzable linkages.
- Cy is preferably selected from aryl, carbocyclic, nitrogen-containing heterocyclic, and nitrogen-containing heteroaryl groups, and preferably does not include S or O atoms in the ring structure. In preferred embodiments, Cy contains 0 or 1 heteroatoms.
- He represents a heterocyclic bicycle. In certain embodiments, He represents a bicyclic heteroaryl structure. In certain embodiments, He is selected from xvi, xvii, xviii, xix, and xx.
- 1 ⁇ represents H for all occurrences.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0] -bicyclononane) ring system.
- Cy represents a fused ring system
- one substituent of L and K may be attached to one of the two rings, and the other substituent to the other of the two rings.
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- 1 ⁇ represents H for all occurrences.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0]-bicyclononane) ring system.
- Cy represents a fused ring system
- one substituent of L and K may be attached to one of the two rings, and the other substituent to the other of the two rings.
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- 1 ⁇ represents H for all occurrences.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0] -bicyclononane) ring system.
- one substituent of L and K may be attached to one of the two rings, and the other substituent to the other of the two rings.
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- Tb is selected from i and ix, and K is absent or represents -Y-, such as -NH-.
- He is selected from xvi, xvii, xviii, xix, and xx.
- Cy is aryl or heteroaryl, preferably aryl.
- He is further substituted with an aryl group, e.g., at a position of Ri, R 2 , and R 3 not substituted with K.
- each of Rj, R 2 , and R 3 represents a hydrocarbon substituent.
- L represents alkyl, alkyl- Y-alkyl or alkyl- Y-acyl, wherein Y is preferably NR, such as NH or NMe.
- He, X, K, Z, and Tb are as defined above.
- Tb is selected from x, xi, xii, xiii, xiv, and xv.
- Z may be absent or represent O or NR, preferably being absent.
- R represents H for all occurrences.
- the compound is free of hydrolyzable linkages.
- Hydrolyzable linkages as the term is used herein, are saturated (sp -hybridized) carbons bound to two heteroatoms, of which at least one is selected from S, N, or O.
- K represents alkyl- Y-alkyl, alkyl- Y-acyl, or alkyl.
- He represents a bicyclic structure, preferably including heteroatoms in both rings.
- the ring(s) of He consist of C and N atoms.
- He includes at least one aryl substituent.
- R 2 represents a bond to K
- Ri and R 3 are selected, independently, from hydrogen, alkyl, cycloalkyl, aryl, heterocyclyl, heteroaryl, aralkyl, heteroalkyl, heteroaralkyl, alkyl alkenyl, alkyl alkynyl, alkyl cycloalkyl, and alkyl heterocyclyl.
- Ri is alkyl or branched alkyl and R 3 is aryl, heteroaryl, or cycloalkyl.
- R 3 is selected from monohaloaryl, dihaloaryl, monohaloheteroaryl, dihaloheteroaryl, monohalocycloalkyl, or dihalocycloalkyl.
- at least one of Ri, R 2 , and R 3 , other than the bond to K, represents an aryl or heteroaryl substituent, preferably a substituted or unsubstituted phenyl.
- Tb is represented by the moiety xiii
- that moiety is not present in another portion of the compound, e.g., He is not xiii, etc.
- K does not include an amide bond, or is preferably free of nitrogen atoms.
- He represents a heterocyclic bicycle.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- the compound does not include a hydrolyzable linkage.
- R represents H for all occurrences.
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- K does not include an amide bond, is free of carbonyls, is free of amine substituents, or is free of nitrogen atoms.
- the compound does not include a hydrolyzable linkage.
- He represents a heterocyclic bicycle.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- I represents H for all occurrences.
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- Tb is represented by ii, iii, iv, or v
- the compound does not include a hydrolyzable linkage.
- He represents a heterocyclic bicycle.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- R represents H for all occurrences.
- K is directly attached to a heteroatom of He. In certain embodiments, K is absent.
- the compound is preferably free of hydrolyzable linkages. In certain embodiments, the compound is free of hydrolyzable linkages. In certain embodiments, He represents a heterocyclic bicycle. In certain embodiments, He represents a bicyclic heteroaryl structure. In certain embodiments, He is selected from xvi, xvii, xviii, xix, and xx. In certain embodiments, 1 ⁇ represents H for all occurrences. In embodiments wherein X is absent, K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- He represents a heterocyclic bicycle. In certain embodiments, He represents a bicyclic heteroaryl structure. In certain embodiments, He is selected from xvi, xvii, xviii, xix, and xx. In certain embodiments, R4 represents H for all occurrences. In embodiments wherein X is absent, K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- Tb is xii
- K is absent or represents -
- Y- such as -NH-.
- He is selected from xvi, xvii, xviii, xix, and xx.
- He is further substituted with an aryl group, e.g., at a position of Rj, R 2 , and R 3 not substituted with K.
- each of R ⁇ , R , and R 3 represents a hydrocarbon substituent.
- compositions comprising any one of the compounds of the present invention, or a pharmaceutically acceptable derivative thereof, and one or more pharmaceutically acceptable excipients.
- compounds of the invention, or compositions containing such compounds are administered to cells or to animals, preferably to a mammal in need therof, as a method for treating bone disorders.
- inventive methods using a pharmaceutical composition comprising a bone targeted compound which is capable of inhibiting bone resorption.
- method using a pharmaceutical composition comprising a bone targeted compound that specifically acts as a Src kinase inhibitor.
- a bone targeted compound of the present invention having a pay load attached thereto that is capable of treating bone disorders by other means.
- inventive methods using a pharmaceutical composition comprising a bone targeting moiety that, alone, is capable of effecting treatment of bone disorders by inhibiting bone resorption or by other means.
- pharmaceutically acceptable derivatives of the foregoing compounds, where the phrase "pharmaceutically acceptable derivative” denotes any pharmaceutically acceptable salt, ester, or salt of such ester, of an inventive compound, or any other adduct or derivative which, upon administration to a patient, is capable of providing (directly or indirectly) a compound as otherwise described herein, or a metabolite or residue thereof, preferably one which is capable of inhibiting bone resorption.
- Pharmaceutically acceptable derivatives thus include among others pro-drugs.
- a pro-drug is a derivative of a compound, usually with significantly reduced pharmacological activity, which contains an additional moiety which is susceptible to removal in vivo yielding the parent molecule as the pharmacologically active species.
- pro-drug is an ester which is cleaved in vivo to yield a compound of interest.
- Pro-drugs of a variety of compounds, and materials and methods for derivatizing the parent compounds to create the pro- drugs, are known and may be adapted to the present invention.
- One technique for providing a prodrug of a compound of the present invention is described generally in Niemi et al., J Med. Chem. 1999, 42, 5053-5058.
- inhibitors of bone resorption or “inhibition of osteoclast activity” or “inhibition of Src kinase activity” preferably refer to specific inhibition. Any of a variety of in vivo or in vitro assays may be employed to assess the ability of inventive compounds and compositions to treat or prevent bone disorders and/or other conditions, and in particular to inhibit bone resorption and/or to inhibit Src tyrosine phosphorylation (see, for example, the Exemplification section, which describes a useful rabbit osteoclast assay for studying effects on bone resorption, and a useful Src kinase inhibition assay).
- the observed effects on bone metabolism are selective in that the inventive compounds or compositions do not exert significant negative effects on biological processes other than bone metabolism, or specifically bone resporption or Src kinase activity.
- inventive compositions show specific inhibition of Src kinase activity as compared with the activity of non-Src kinanses, or kinases located at sites away from bone.
- such specific inhibition may result from specific localization of the inventive composition to bone sites, so that compositions delivered in vivo do not have the opportunity to inhibit processes that occur away from bone; in other cases, specific inhibition may be attributed to specific action of the inventive payload on the osteoclast activity or on Src kinase activity, as compared with other cells or kinases.
- payload includes therapeutic agents (e.g., a small molecule, a drug, a radiotherapeutic atom, etc.), detectable labels (e.g., fluorescent, radioactive, radiopaque, etc.), or any other moiety desired to be delivered to a site of action (e.g., a bone or other site suffering an abnormal condition).
- therapeutic agents e.g., a small molecule, a drug, a radiotherapeutic atom, etc.
- detectable labels e.g., fluorescent, radioactive, radiopaque, etc.
- any other moiety desired to be delivered to a site of action e.g., a bone or other site suffering an abnormal condition.
- a "small molecule” as the term is used herein refers to an organic molecule of less than about 2500 amu, preferably less than about 1000 amu.
- Subject shall mean a human or animal (e.g., rat, mouse, cow, pig, horse, sheep, monkey, cat, dog, goat, etc.).
- targeting moiety refers to any molecular structure which assists the inventive composition in localizing to a particular target area, entering a target cell(s), and/or binding to a target receptor.
- Preferred targeting moieties according to the present invention include bone targeting moieties, as described herein.
- a "therapeutic agent” shall mean an agent capable of having a biological effect on a host.
- Preferred therapeutic agents are capable of preventing and/or treating one or more symptoms of a bone disorder, such as a metabolic bone disorder.
- Other preferred therapeutic agents are capable of preventing or treating other bone disorders or related conditions. Examples of therapeutic agents considered to be within the scope of the present invention include boron-containing compounds (e.g.
- chemotherapeutic nucleotides drugs (e.g., antibiotics, antivirals, antifungals), enediynes (e.g., calicheamicins, esperamicins, dynemicin, neocarzinostatin chromophore, and kedarcidin chromophore), heavy metal complexes (e.g., cisplatin), hormone antagonists (e.g., tamoxifen), non-specific (non-antibody) proteins (e.g., sugar oligomers), oligonucleotides (e.g., antisense oligonucleotides that bind to a target nucleic acid sequence (e.g., mRNA sequence)), peptides, photodynamic agents (e.g., rhodamine 123), radionuclides (e.g., 1-131, Re-186, Re-188, Y-90, Bi-212,
- drugs e
- the therapeutic agent is a radionuclide, toxin, hormone antagonist, heavy metal complex, oligonucleotide, chemotherapeutic nucleotide, peptide, non-specific (non-antibody) protein, a boron compound or an enediyne.
- the therapeutic agent is a Src kinase inhibitor, capable of inhibiting the overactivity of osteoclasts.
- R group will generally have the structure which is recognized in the art as corresponding to R groups having that name.
- representative R groups as enumerated in the specification and claims of the present application are defined herein. These definitions are intended to supplement and illustrate, not preclude, the definitions known to those of skill in the art.
- alkyl refers to the radical of saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl substituted cycloalkyl groups, and cycloalkyl substituted alkyl groups.
- a straight chain or branched chain alkyl has 30 or fewer carbon atoms in its backbone (e.g., C1-C30 for straight chain, C3-C30 for branched chain), and more preferably 20 or fewer.
- preferred cycloalkyls have from 3-10 carbon atoms in their ring structure, and more preferably have 5, 6 or 7 carbons in the ring structure.
- alkyl (or “lower alkyl) as used throughout the specification, examples, and claims is intended to include both “unsubstituted alkyls” and “substituted alkyls”, the latter of which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone.
- the moieties substituted on the hydrocarbon chain can themselves be substituted, if appropriate.
- the substituents of a substituted alkyl may include substituted and unsubstituted forms of amino, azido, imino, amido, phosphoryl (including phosphonate and phosphinate), sulfonyl (including sulfate, sulfonamido, sulfamoyl and sulfonate), and silyl groups, as well as ethers, alkylthios, carbonyls (including ketones, aldehydes, carboxylates, and esters), -CF3, -CN and the like.
- Cycloalkyls can be further substituted with alkyls, alkenyls, alkoxys, alkylthios, aminoalkyls, carbonyl-substituted alkyls, -CF3, -CN, and the like.
- lower alkyl as used herein means an alkyl group, as defined above, but having from one to ten carbons, more preferably from one to six carbon atoms in its backbone structure. Likewise, “lower alkenyl” and “lower alkynyl” have similar chain lengths. Preferred alkyl groups are lower alkyls. In preferred embodiments, a substituent designated herein as alkyl is a lower alkyl.
- aryl groups having heteroatoms in the ring structure may also be referred to as "aryl heterocycles" or “heteroaromatics.”
- the aromatic ring can be substituted at one or more ring positions with such substituents as described above, for example, halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, -CF3, -CN, or the like.
- ortho, meta and para apply to 1,2-, 1,3- and 1,4-disubstituted benzenes, respectively.
- the names 1 ,2-dimethylbenzene and ortho- dimethylbenzene are synonymous.
- Heterocyclyl groups include, for example, thiophene, thianthrene, furan, pyran, isobenzofuran, chromene, xanthene, phenoxathiin, pyrrole, imidazole, pyrazole, isothiazole, isoxazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, pyrimidine, phenanthroline, phenazine, phenarsazine, phenothiazine, furazan, phenoxazine, pyrrolidine, o
- the heterocyclic ring can be substituted at one or more positions with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
- substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl,
- polycyclyl or “polycyclic group” refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls) in which two or more carbons are common to two adjoining rings, e.g., the rings are "fused rings". Rings that are joined through non-adjacent atoms are termed "bridged" rings.
- Each of the rings of the polycycle can be substituted with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
- substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl
- carrier refers to an aromatic or non-aromatic ring in which each atom of the ring is carbon.
- nitro means -NO2; the term “halogen” designates - F, -CI, -Br or -I; the term “sulfhydryl” means -SH; the term “hydroxyl” means -OH; and the term “sulfonyl” means -SO2-.
- amine and “amino” are art-recognized and refer to both unsubstituted and substituted amines, e.g., a moiety that can be represented by the general formula:
- R9, R ⁇ Q and R' ⁇ Q each independently represent a hydrogen, an alkyl, an alkenyl, -(CH2) m -R-8' or R9 a 10 " &10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure;
- Rg represents an aryl, a cycloalkyl, a cycloalkenyl, a heterocycle or a polycycle; and
- m is zero or an integer in the range of 1 to 8.
- RlO can be a carbonyl, e.g., RQ, RI O and the nitrogen together do not form an imide.
- RQ and Ri Q (and optionally R' IO) each independently represent a hydrogen, an alkyl, an alkenyl, or -(CH2) m -R8-
- alkylamine as used herein means an amine group, as defined above, having a substituted or unsubstituted alkyl attached thereto, i.e., at least one of RQ and Ri Q is an alkyl group.
- acylamino is art-recognized and refers to a moiety that can be represented by the general formula:
- R 9 is as defined above, and R' ⁇ ⁇ represents a hydrogen, an alkyl, an alkenyl or -(CH2)n_-R8, where m and Rg are as defined above.
- amino is art recognized as an amino-substituted carbonyl and includes a moiety that can be represented by the general formula:
- RQ, RJQ are as defined above.
- Preferred embodiments of the amide will not include imides which may be unstable.
- amidine is art-recognized as a group that can be represented by the general formula:
- alkylthio refers to an alkyl group, as defined above, having a sulfur radical attached thereto.
- the "alkylthio" moiety is represented by one of -S-alkyl, -S-alkenyl, -S-alkynyl, and -S-(CH2) m -R-8' wherein m and Rg are defined above.
- Representative alkylthio groups include methylthio, ethyl thio, and the like.
- carbonyl is art recognized and includes such moieties as can be represented by the general formula:
- alkoxyl or “alkoxy” as used herein refers to an alkyl group, as defined above, having an oxygen radical attached thereto. Representative alkoxyl groups include methoxy, ethoxy, propyloxy, tert-butoxy and the like.
- An “ether” is two hydrocarbons covalently linked by an oxygen.
- triflyl, tosyl, mesyl, and nonaflyl are art-recognized and refer to trifluoromethanesulfonyl, -toluenesulfonyl, methanesulfonyl, and nonafluorobutanesulfonyl groups, respectively.
- triflate, tosylate, mesylate, and nonaflate are art-recognized and refer to trifluoromethanesulfonate ester, p- toluenesulfonate ester, methanesulfonate ester, and nonafluorobutanesulfonate ester functional groups and molecules that contain said groups, respectively.
- Me, Et, Ph, Tf, Nf, Ts, and Ms represent methyl, ethyl, phenyl, trifluoromethanesulfonyl, nonafluorobutanesulfonyl, /.-toluenesulfonyl and methanesulfonyl, respectively.
- a more comprehensive list of the abbreviations utilized by organic chemists of ordinary skill in the art appears in the first issue of each volume of the Journal of Organic Chemistry; this list is typically presented in a table entitled Standard List of Abbreviations. The abbreviations contained in this list, and all abbreviations utilized by organic chemists of ordinary skill in the art are hereby incorporated by reference.
- R41 is as defined above.
- sulfonyl refers to a moiety that can be represented by the general formula:
- R44 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- sulfoxido refers to a moiety that can be represented by the general formula:
- R44 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclyl, aralkyl, or aryl.
- a "phosphoryl” can in general be represented by the formula:
- OR 46 wherein Q represented S or O, and R46 represents hydrogen, a lower alkyl or an aryl.
- R46 represents hydrogen, a lower alkyl or an aryl.
- each expression e.g. alkyl, m, n, etc., when it occurs more than once in any structure, is intended to be independent of its definition elsewhere in the same structure.
- substitution or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc.
- substituted is contemplated to include all permissible substituents of organic compounds.
- the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic substituents of organic compounds.
- Illustrative substituents include, for example, those described herein above.
- the permissible substituents can be one or more and the same or different for appropriate organic compounds.
- the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. This invention is not intended to be limited in any manner by the permissible substituents of organic compounds .
- protecting group means temporary substituents which protect a potentially reactive functional group from undesired chemical transformations.
- protecting groups include esters of carboxylic acids, silyl ethers of alcohols, and acetals and ketals of aldehydes and ketones, respectively.
- the field of protecting group chemistry has been reviewed (Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 2 nd ed.; Wiley: New York, 1991, incorporated herein by reference).
- Certain compounds of the present invention may exist in particular geometric or stereoisomeric forms.
- the present invention contemplates all such compounds, including cis- and tr ⁇ s-isomers, R- and S-enantiomers, diastereomers, (D)-isomers, (L)-isomers, the racemic mixtures thereof, and other mixtures thereof, as falling within the scope of the invention.
- Additional asymmetric carbon atoms may be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention. Isomeric mixtures containing any of a variety of isomer ratios may be utilized in accordance with the present invention.
- mixtures containing 50:50, 60:40, 70:30, 80:20, 90:10, 95:5, 96:4, 97:3, 98:2, 99:1, or 100:0 isomer ratios are all contemplated by the present invention.
- Those of ordinary skill in the art will readily appreciate that analogous ratios are contemplated for more complex isomer mixtures.
- a particular enantiomer of a compound of the present invention is desired, it may be prepared by asymmetric synthesis, or by derivation with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers.
- diastereomeric salts are formed with an appropriate optically-active acid or base, followed by resolution of the diastereomers thus formed by fractional crystallization or chromatographic means well known in the art, and subsequent recovery of the pure enantiomers.
- Contemplated equivalents of the compounds described above include compounds which otherwise correspond thereto, and which have the same general properties thereof (e.g., bone targeting agents), wherein one or more simple variations of substituents are made which do not adversely affect the efficacy of the compound in targeting bone.
- the compounds of the present invention may be prepared by the methods illustrated in the general reaction schemes as, for example, described below, or by modifications thereof, using readily available starting materials, reagents and conventional synthesis procedures. In these reactions, it is also possible to make use of variants which are in themselves known, but are not mentioned here.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0] -bicyclononane) ring system.
- Cy represents a fused ring system
- one substituent of L and K (or He or Tb) may be attached to one of the two rings, and the other substituent to the other of the two rings.
- K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- Cy represents a phenyl, pyridyl, cyclopentyl, cyclohexyl, or a fused bicyclic ring system, preferably having between 8 and 11 atoms, such as a fused cyclohexyl/cyclopentyl ([4.3.0]-bicyclononane) ring system.
- Cy represents a fused ring system
- one substituent of L and K may be attached to one of the two rings, and the other substituent to the other of the two rings.
- K is directly attached to a heteroatom of He.
- K is absent.
- Tb is selected from i and ix, and K is absent or represents -Y-, such as -NH-.
- He is selected from xvi, xvii, xviii, xix, and xx.
- Cy is aryl or heteroaryl, preferably aryl.
- He is further substituted with an aryl group, e.g., at a position of R ls R 2 , and R 3 not substituted with K.
- each of Ri, R 2 , and R 3 represents a hydrocarbon substituent.
- L represents alkyl, alkyl-Y-alkyl or alkyl- Y-acyl, wherein Y is preferably NR, such as NH or NMe.
- K-X-Hc is free of hydrolyzable linkages.
- Hydrolyzable linkages as the term is used herein, are saturated (sp -hybridized) carbons bound to two heteroatoms, of which at least one is selected from S, N, or O.
- He represents a bicyclic structure, preferably including heteroatoms in both rings.
- the ring(s) of He consist of C and N atoms.
- R 2 is selected from hydrogen, (CH 2 ) n Ph, where Ph is phenyl or substituted phenyl and n is 0, 1, 2, or 3; heteroaryl, cycloalkyl, C ⁇ -C 6 alkanoyl, C ⁇ -C alkyl, C 2 -C 6 alkenyl, and C 2 -C 6 alkynyl, where the alkyl, alkenyl and alkynyl groups may be substituted by NR R ⁇ , phenyl, thioalkyl, alkyloxy, hydroxy, carboxy, halogen, cycloalkyl, and where R 5 and R ⁇ are independently hydrogen, C ⁇ - C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, (CH 2 ) n Ph where Ph is phenyl and n is 0, 1, 2, or 3; cycloalkyl, heteroaryl, and R 5 and R ⁇ taken together with the
- At least one of Ri, R 2 , or R 3 represents a bond to K
- R 3 if not a bond to K, is selected from hydrogen or alkyl
- R 2 if not a bond to K, is selected from alkyl, cycloalkyl, alkyl alkenyl, alkyl alkynyl
- R l5 if not a bond to K, is selected from hydrogen, alkyl, cycloalkyl, aryl, heteroaryl, heterocyclyl, aralkyl, heteroalkyl, alkyl alkenyl, alkyl alkynyl, alkyl cycloalkyl, or alkyl heterocyclyl.
- at least one of Ri, R 2 , and R 3 other than the bond to K, represents an aryl or heteroaryl substituent, preferably a substituted or unsubstituted phenyl.
- Tb is represented by vii or viii
- K-X-Hc is free of hydrolyzable linkages.
- K does not comprise nitrogen.
- He represents a heterocyclic bicycle.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- R represents H for all occurrences.
- K is preferably directly attached to a heteroatom of He.
- K is absent.
- Tb is represented by xi, xii, xiv, or xv.
- K does not include an amide bond, or are preferably free of nitrogen atoms.
- He represents a heterocyclic bicycle.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- K-X-Hc does not include a hydrolyzable linkage.
- ILj represents H for all occurrences.
- K is preferably directly attached to a heteroatom of He.
- K is absent.
- K-X-Hc does not include a hydrolyzable linkage.
- He represents a heterocyclic bicycle.
- He represents a bicyclic heteroaryl structure.
- He is selected from xvi, xvii, xviii, xix, and xx.
- 1 ⁇ represents H for all occurrences.
- K is directly attached to a heteroatom of He.
- K is absent.
- He represents a heterocyclic bicycle. In certain embodiments, He represents a bicyclic heteroaryl structure. In certain embodiments, He is selected from xvi, xvii, xviii, xix, and xx. In certain embodiments, R 4 represents H for all occurrences. In embodiments wherein X is absent, K is preferably directly attached to a heteroatom of He. In certain embodiments, K is absent.
- Tb is selected from i and ix, and K is branched or unbranched alkyl.
- He is selected from xvi, xvii, xviii, xix, and xx.
- He is further substituted with an aryl group, e.g., at a position of Ri, R 2 , and R 3 not substituted with K.
- each of R 1 ; R 2 , and R 3 represents a hydrocarbon substituent.
- Tb is xii, and K is absent or represents - Y-, such as -NH-.
- He is selected from xvi, xvii, xviii, xix, and xx.
- He is further substituted with an aryl group, e.g., at a position of Rj, R 2 , and R 3 not substituted with K.
- each of Ri, R 2 , and R 3 represents a hydrocarbon substituent.
- Tb is x, and K represents alkyl or -Y-.
- He is selected from xvi, xvii, xviii, xix, and xx.
- He is further substituted with an aryl group, e.g., at a position of Ri, R 2 , and R 3 not substituted with K.
- each of Ri, R 2 , and R 3 represents a hydrocarbon substituent.
- a subject compound has the structure of formula III: Tb-L-V, wherein Tb, R, and L are as defined above, and
- V represents OR, NR 2 , or SR.
- Tb is selected from i, ii, iii, iv, v, vi, vii, viii, and ix
- L is preferably not absent, and even more preferably represents alkyl.
- Tb represents i, ii, iii, or iv, preferably i.
- Tb represents v or vi.
- Tb represents vii or viii.
- Tb represents ix.
- V represents NR 2 . In certain embodiments, all occurrences of R in V are H.
- a subject compound has the structure of Formula IV:
- Tb-Cy-L-V wherein Tb, Cy, L, and V are as defined above, and Tb is selected from i, ii, iii, iv, v, vi, vii, viii, and ix.
- Tb represents i, ii, iii, or iv, preferably i.
- Tb represents v or vi.
- Tb represents vii or viii.
- Tb represents ix.
- L is absent.
- Cy represents a phenyl ring.
- V represents NR 2 .
- all occurrences of R in V are H.
- L represents lower alkyl or is absent.
- a suitable leaving group e.g., a moiety whose conjugate acid, UH, has a pKa lower than 5, preferably lower than 0.
- Tb is selected from i, ii, iii, iv, v, vi, vii, viii, and ix
- L is preferably not absent, and even more preferably represents alkyl.
- Tb represents i, ii, iii, or iv, preferably i.
- Tb represents v or vi.
- Tb represents vii or viii.
- Tb represents ix.
- Tb is selected from x, xi, xii, xiii, xiv, xv, xxi, xxii, xxiiv, and xxv. In certain embodiments, Tb is selected from xi, xii, xiv, or xv. In certain embodiments, Tb is x. In certain embodiments, Tb is xiii. In certain embodiments, V represents NR 2 . In certain embodiments, Tb is xxi, xxii, xxiii, xxiv, or xxv. In certain embodiments, Tb is xx.
- Tb may have a structure selected from:
- each occurence of Y is independently a covalent bond, -O-, -S-, or -N(Rj) 2 , wherein Rj, for each occurence, is independently hydrogen, aliphatic, heteroaliphatic, aryl, heteroaryl, alkylaryl, or alkylheteroaryl;
- Re represents from 0-3 substituents selected from halogen, lower alkyl, lower alkenyl, aryl, heteroaryl, carbonyl, thiocarbonyl, ketone, aldehyde, amino, acylamino, amido, amidino, cyano, nitro, azido, sulfonyl, sulfoxido, sulfate, sulfonate, sulfamoyl, sulfonamido, phosphoryl, phosphonate, phosphinate, -(CH 2 )palkyl, -(CH 2 )palkenyl, - (CH 2 ) p alkynyl, -(CH 2 ) p aryl, -(CH 2 ) p aralkyl, -(CH 2 ) p OH, -(CH 2 ) p O-lower alkyl, - (CH 2 ) p O-lower alkenyl
- M, where it occurs in Tb is selected from CH 2 , CHJ and CJ 2 , wherein J represents a halogen, such as F, CI, Br, or I, preferably F or CI, even more preferably F.
- J represents a halogen, such as F, CI, Br, or I, preferably F or CI, even more preferably F.
- Re is selected from lower alkyl, hydrophilic groups, and lower alkyl substituted with hydrophilic groups.
- Representative hydrophilic groups include hydroxyl, sulfhydryl, amino, amido, carboxyl, sulfonate, phosphonate, and salts thereof.
- Tb has the structure xxxxii:
- A represents a group selected from GPO 3 (R 4 ) 2 , GCO 2 R , and GSO ⁇ ;
- B represents a group selected from NH 2 , OH, GPO 3 (P M ) 2 , GCO 2 R J , and GSO ⁇ ; and G is absent or represents a linkage of one or two atoms, such as CF 2 , CH 2 , O, S, NR, CH 2 S, CH 2 NR, CH 2 O, etc.
- Tb has the structure xxxxiv or xxxxv:
- C represents H, R ⁇ , NH 2 , OH, GPO 3 (R 4 ) 2 , G ⁇ I ⁇ , or GSO ⁇ , and in xxxxiv, any one occurrence of A or B is present, and the other occurrences may represent a bond to Z, H, or R 6 as desired.
- the pyridyl ring may bear one or more additional e substituents.
- Tb represents a heteroaryl, preferably a nitrogen-containing heteroaryl, bearing one B substituent, preferably an A substituent, or two B substituents, and optionally including one or more R substituents.
- the heteroaryl group is selected from thiazoline, oxazoline, pyrrole, pyrazole, imidazole, pyridine, pyrazine, pyridazine, and pyrimidine, preferably pyridine, pyrazole, pyrazine, and pyrimidine.
- inventive compounds may be prepared by any available method.
- solid-phase syntheses will be modified versions of the compounds described herein that allow their attachment to a solid support.
- the present invention also contemplates particularly preferred because it enables the use of more rapid split and pool techniques to generate larger libraries (e.g., greater than 10,000 members) more easily.
- solid phase parallel synthesis techniques also can be utilized, such as those described in U.S. Patents 5,712,171 and 5,736,412, incorporated herein by reference.
- a solid support for the purposes of this invention, is defined as an insoluble material to which compounds are attached during a synthesis sequence.
- the use of a solid support is advantageous for the synthesis of libraries because the isolation of support-bound reaction products can be accomplished simply by washing away reagents from the support-bound material and therefore the reaction can be driven to completion by the use of excess reagents. Additionally, the use of a solid support also enables the use of specific encoding techniques to "track" the identity of the inventive compounds in the library.
- a solid support can be any material which is an insoluble matrix and can have a rigid or semi-rigid surface.
- Exemplary solid supports include, but are not limited to, pellets, disks, capillaries, hollow fibers, needles, pins, solid fibers, cellulose beads, pore-glass beads, silica gels, polystyrene beads optionally cross-linked with divinylbenzene, grafted co-poly beads, poly-acrylamide beads, latex beads, dimethylacrylamide beads optionally crosslinked with N-N'-bis- acryloylethylenediamine, and glass particles coated with a hydrophobic polymer.
- a Tentagel amino resin a composite of 1) a polystyrene bead crosslinked with divinylbenzene and 2) PEG (polyethylene glycol), is employed for use in the present invention.
- Tentagel is a particularly useful solid support because it provides a versatile support for use in on- bead or off-bead assays, and it also undergoes excellent swelling in solvents ranging from toluene to water.
- the compounds of the present invention may be attached directly to the solid support or may be attached to the solid support through a linking reagent. Direct attachment to the solid support may be useful if it is desired not to detach the library member from the solid support. For example, for direct on-bead analysis of biological/pharmacological activitiy or analysis of the compound structure, a stronger interaction between the library member and the solid support may be desirable.
- linking reagent may be useful if more facile cleavage of the inventive library members from the solid support is desired.
- any linking reagent used in the present invention may comprise a single linking molecule, or alternatively may comprise a linking molecule and one or more spacer molecules.
- a spacer molecule is particularly useful when the particular reaction conditions require that the linking molecule be separated from the library member, or if additional distance between the solid support/linking unit and the library member is desired.
- photocleavable linkers are employed to attach the solid phase resin to the component. Photocleavable linkers are particularly advantageous for the presently claimed invention because of the ability to use these linkers in in vivo screening strategies.
- Exemplary photocleavable linkers include, but are not limited to ortho-Nitrobenzyl photolinkers and dithiane protected benzoin photolinkers.
- One of ordinary skill in the art will readily appreciate that the method of the present invention is not limited to the use of photocleavable linkers; rather other linkers may be employed, preferably those that are capable of delivering the desired compounds in vivo.
- the synthesis of libraries of bone targeting agents is performed using established combinatorial methods for solution phase, solid phase, or a combination of solution phase and solid phase synthesis techniques.
- the synthesis of combinatorial libraries is well known in the art and has been reviewed (see, e.g., "Combinatorial Chemistry", Chemical and Engineering News, Feb. 24, 1997, p. 43; Thompson, L.A., Ellman, J.A., Chem. Rev. 1996, 96, 555, incorporated herein by reference.)
- One of ordinary skill in the art will realize that the choice of method will depend upon the specific number of compounds to be synthesized, the specific reaction chemistry, and the availability of specific instrumentation, such as robotic instrumentation for the preparation and analysis of the inventive libraries.
- the reactions to be performed on the inventive scaffolds to generate the libraries are selected for their ability to proceed in high yield, and in a stereoselective fashion, if applicable.
- libraries are generated using a solution phase technique.
- Traditional advantages of solution phase techniques for the synthesis of combinatorial libraries include the availability of a much wider range of organic reactions, and the relative ease with which products can be characterized.
- a parallel synthesis technique is utilized, in which all of the products are assembled separately in their own reaction vessels.
- a microtitre plate containing n rows and m columns of tiny wells which are capable of holding a few milliliters of the solvent in which the reaction will occur is utilized. It is possible to then use n variants of reactant A, and m variants of reactant B, to obtain n x m variants, in n x m wells.
- reactant A and m variants of reactant B, to obtain n x m variants, in n x m wells.
- a solid phase synthesis technique in which the desired scaffold structures are attached to the solid phase directly or though a linking unit, as discussed above.
- Advantages of solid phase techniques include the ability to more easily conduct multi-step reactions and the ability to drive reactions to completion because excess reagents can be utilized and the unreacted reagent washed away.
- One of the most significant advantages of solid phase synthesis is the ability to use a technique called "split and pool", in addition to the parallel synthesis technique, develped by Furka. (Furka et al., Abstr. 14th Int. Congr. Biochem., Prague, Czechoslovakia, 1988, 5, 47; Furka et al., Int. J. Pept.
- the solid support scaffolds can be divided into n vessels, where n represents the number species of reagent A to be reacted with the scaffold structures. After reaction, the contents from n vessels are combined and then split into m vessels, where m represents the number of species of reagent B to be reacted with the scaffold structures. This procedure is repeated until the desired number of reagents is reacted with the scaffold structures to yield the inventive library.
- solid phase techniques in the present invention may also include the use of a specific encoding technique.
- Specific encoding techniques have been reviewed by Czarnik. (Czarnik, A.W., Current Opinion in Chemical Biology, 1997, 1, 60)
- an encoding technique involves the use of a particular "identifiying agent" attached to the solid support, which enables the determination of the structure of a specific library member without reference to its spatial coordinates.
- an encoding technique involves the use of a particular "identifiying agent" attached to the solid support, which enables the determination of the structure of a specific library member without reference to its spatial coordinates.
- an encoding technique involves the use of a particular "identifiying agent" attached to the solid support, which enables the determination of the structure of a specific library member without reference to its spatial coordinates.
- an encoding technique involves the use of a particular "identifiying agent" attached to the solid support, which enables the determination of the structure of a specific library member without reference to its spatial coordinates.
- Examples of alternative encoding techniques that can be utilized in the present invention include, but are not limited to, spatial encoding techniques, graphical encoding techniques, including the "tea bag” method, chemical encoding methods, and spectrophotometric encoding methods.
- Spatial encoding refers to recording a reaction's history based on its location.
- Graphical encoding techniques involve the coding of each synthesis platform to permit the generation of a relational database.
- Examples of preferred spectrophotometic encoding methods include the use of mass spectroscopy, fluorescence emission, and nuclear magnetic resonance spectroscopy.
- chemical encoding methods are utilized, which uses the structure of the reaction product to code for its identity. Decoding using this method can be performed on the solid phase or off of the solid phase.
- One of ordinary skill in the art will realize that the particular encoding method to be used in the present invention must be selected based upon the number of library members desired, and the reaction chemistry employed.
- Subsequent characterization of the library members, or individual compounds, can be performed using standard analytical techniques, such as mass spectrometry, Nuclear Magnetic Resonance Spectroscopy, and gas chromatrography.
- specific assay techniques such as those described herein, may be utilized to test the ability of compounds to function as Src kinase inhibitors. In certain preferred embodiments, high throughput assay techniques are utilized.
- the compounds of the present invention are useful in the selective treatment or prevention of bone disorders. These compounds or pharmaceutical compositions may effect treatment via inhibition of osteoclast activity, promotion of osteoblast activity, or promotion or inhibition of other cellular events necessary for healthy bone metabolism. In certain preferred embodiments, these compounds are useful for the treatment or prevention of diseases and conditions associated with bone metabolic disorders such as osteoclast overactivity. In still other preferred embodiments, the compounds of the present invention are targeted Src kinase inhibitors and thus inhibit bone resorption by osteoclasts.
- the present invention therefore provides a method for the treatment, prophylaxis, and/or prevention of bone and other related disorders which method comprises the administration of an effective non-toxic amount of an inventive compound, or a pharmaceutically composition thereof.
- inventive compounds effect treatment via several mechanisms, (i.e. inhibition of osteoclast activity, promotion of osteoblast activity, or regulation of other cellular events necessary for healthy bone metabolism), in certain preferred embodiments, these compounds are selective inhibitors of osteoclast activity.
- the present invention provides an inhibitor of mammalian osteoclasts, for example any one of the compounds of the present invention or a pharmaceutical composition thereof.
- the present invention provides compounds or pharmaceutical compositions that are selective Src kinase inhibitors.
- the method of present invention comprises providing any one of the compounds of the present invention or a pharmaceutically composition thereof, for use in the treatment of and/or prophylaxis of osteoporosis and related osteopenic diseases.
- the present invention also contemplates the use of bone targeting agents alone for the treatment of bone disorders, preferably by the selective inhibition of osteoclast activity.
- the present invention also contemplates the treatment and prophylaxis or prevention of Paget' s disease, hypercalcemia associated with bone neoplasms and other types of osteoporotic diseases and related disorders, including but not limited to involutional osteoporosis, Type I or postmenopausal osteoporosis, Type II or senile osteoporosis, juvenile osteoporosis, idiopathic osteoporosis, endocrine abnormality, hyperthyroidism, hypogonadism, ovarian agensis or Turner's syndrome, hyperadrenocortogni or Cushing's syndrome, hyperparathyroidism, bone marrow abnormalities, multiple myeloma and related disorders, systemic mastocytosis, disseminated carcinoma, Gaucher's disease, connective tissue abnormalities, osteogenesis imperfect
- the present invention contemplates the use of alternate payloads to achieve desired therapeutic effects.
- the targeted constructs of the present invention may include any of a wide variety of chemical entities to be delivered to the target site or into target cells.
- the payloads may be categorized as therapeutic agents (such as Src kinase inhibitors) or imaging agents. Imaging agents comprise those payloads which are detectable, e.g., by emitting light, radioactive emissions, or chemical signals, by absorbing radiation (e.g., x-rays), or by otherwise changing a characteristic of treated cells relative to untreated cells.
- Therapeutic agents include payloads which are biologically active, preferably by countering an abnormal condition of the bone targeted site (e.g., tumor or infection).
- a therapeutic agent useful in a targeted compound may be any of a number of chemical entities, e.g., an enzyme, drug, radionuclide, enzyme inhibitor, etc.
- moieties useful as therapeutic agents include inhibitors of osteoclast activity such as Src kinase inhibitors, cathespin inhibitors, or proton pump inhibitors, amino acids and their derivatives; analgesics such as acetaminophen, aspirin, and ibuprofen; antifungal agents including: allyamines, imidazoles, polyenes, and triazoles; antigens and antibodies thereto; antihistamines such as chlorpheniramine and brompheniramine; antihypertensive agents such as clonidine, methyldopa, prazosin, verapamil, nifedipine, captopril, and enalapril; antiinflammatory agents including non-steroidal agents, such as aminoarylcarboxylic acid derivatives, arylacetic acid derivative
- acyclovir dideoxy -cytidine, - adenosine, or -inosine, interferons, amantadine, ribavirin); beta-blockers such as propranolol, metoprolol, atenolol, labetolol, timolol, penbutolol, and pindolol; cancer drugs including chemotherapeutic agents; cardiovascular agents including cardiac glycosides, antianginals and vasodilators; coenzymes; enzymes; enzyme inhibitors; expectorants; glycoproteins; H-2 antagonists such as nizatidine, cimetidine, famotidine, and ranitidine; haptens and antibodies thereto; hormones, lipids, liposomes; protein analogs in which at least one non-peptide linkage replaces a peptide linkage; phospholipids; prostaglandins; radionuclides (e.g.
- toxins such as aflatoxin, digoxin, rubratoxin, and xanthotoxin
- tranquilizers such as diazepam, chordiazepoxide, oxazepam, alprazolam, and triazolam
- vitamins and mineral and nutritional additives for other therapeutic agents, see, e.g., the Merck Index.
- the present invention contemplates agents that are useful for treating or preventing the progression of a
- the compounds and compositions of the present invention are also inhibitors of cathepsin K, and thus can be used to treat bone disorders.
- cathepsin K it has been discovered that osteoclasts contain large quantities of cathepsin K and it has been suggested that cathepsin K thus plays an important role in bone resorption (Smith et al., Exp. Opin. Ther. Patents 1999, 9, 683-694 and references cited therein).
- the enzyme is active over a pH range (3.5-4.0) consistent with the pH of the local bone environment during resorption, that a cathepsin K antisense nucleotide was effective in the inhibition of osteoclast bone resorption, and that there is a link between mutations in the human cathepsin K gene and the rare skeletal dysplasia, pycnodysostosis, a disease characterized by abnormal bone resorption (see, Smith et al.).
- compounds of the present that are cathepsin K inhibitors would be useful as agents for the inhibition of bone resorption and thus could be used to treat osteoporosis or other disorders resulting from abnormal bone resorption.
- Bone-targeted compounds can alternatively or additionally be labeled with any of a variety of imaging agents which are known in the art and which will depend to some extent on the means used to detect or monitor the compound in vivo or in vitro.
- Preferred imaging agents for performing positron emission tomography (PET) and single photon emission computer tomography (SPECT) include F-18, Tc-99m, and 1-123.
- Preferred imaging agents for magnetic resonance imaging (MRI) include an appropriate atom with unpaired spin electrons or a free radical.
- the payload comprises a moiety such as a radionuclide or paramagnetic contrast agent, fluorescent or chemiluminescent label, or other type of detectable marker.
- the imaging agents described above may contain any label in accordance with the invention. Highly specific and sensitive labels are provided by radionuclides, which can then be detected using positron emission tomography (PET) or Single Photon Emission
- the imaging agent of the invention contains a radionuclide selected from the group consisting of I, I, I , 99m Tc, 18 F, 68 Ga, 67 Ga, 72 As, 89 Zr, 64 Cu, 62 Cu, u l In, 203 Pb, I98 Hg, ⁇ C, 97 Ru, and 201 T1 or a paramagnetic contrast agent, such as gadolinium, cobalt, nickel, manganese, and iron.
- SPECT Computed Tomography
- the compounds of the present invention are useful in the treatment of bone disorders, preferably imbalances in bone metabolism, such as overactivity of bone resorption.
- the compounds of the present invention are useful as inhibitors of Src tyrosine phosphorylation.
- the compounds of the present invention When used for therapeutic and/or prophylactic administration, they can exist in free form, or, where appropriate, in salt form.
- Pharmceutically acceptable salts of many types of compounds and their preparation are well-known to those of skill in the art.
- the pharmaceutically acceptable salts of compounds of this invention include the conventional non-toxic salts or the quaternary ammonium salts of such compounds which are formed, for example, from inorganic or organic acids of bases.
- the compounds of the invention may form hydrates or solvates. It is known to those of skill in the art that charged compounds form hydrated species when lyophilized with water, or form solvated species when concentrated in solution with an appropriate organic solvent.
- compositions comprising a therapeutically (or prophylactically) effective amount of the compound, and a pharmaceutically acceptable carrier or excipient.
- Carriers include, e.g., saline, buffered saline, dextrose, water, glycerol, ethanol, and combinations thereof, and are discussed in greater detail below.
- the composition if desired, can also contain minor amounts of wetting or emulsifying agents, or pH buffering agents.
- the composition can be a liquid solution, suspension, emulsion, tablet, pill, capsule, sustained release forrmulation, or powder.
- the compsition can be formulated as a suppository, with traditional binders and carriers such as triglycerides.
- Oral formulation can include standard carriers such as pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate, etc. Formulation may involve mixing, granulating and compressing or dissolving the ingredients as appropriate to the desired preparation.
- the pharmaceutical carrier may be, for example, either a solid or liquid.
- Illustrative solid carriers include lactose, terra alba, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and the like.
- a solid carrier can include one or more substances which may also act as flavoring agents, lubricants, solubilizers, suspending agents, fillers, glidannts, compression aids, binders or tablet- disintegrating agents; it can also be an encapsulating material.
- the carrier is a finely divided solid which is in admixture with the finely divided active ingredient.
- the active ingredient is mixed with a carrier having the necessary compression properties in suitable proportions and compacted in the shape and size desired.
- the powders and tablets preferably contain up to 99% of the active ingredient.
- suitable solid carriers include, for example, calcium phosphate, magnesium stearate, talc, sugars, lactose, dextrin, starch, gelatin, cellulose, methyl cellulose, sodium carboxymethyl cellulose, polyvinylpyrrolidine, low melting waxes and ion exchange resins.
- Illustrative liquid carriers include syrup, peanut oil, olive oil, water, et.
- Liquid carriers are used in preparing solutions, suspensions, emulsions, syrups, elixirs and pressurized compositions.
- the active ingredient can be dissolved or suspended in a pharmceutically acceptable oils or fats.
- the liquid carrier can contain other suitable pharmaceutical additives such as solubilizers, emulsifiers, buffers, preservatives, sweeteners, flavoring aents, suspending agents, thickening agents, colors, viscosity regulators, stabilizers or osmo-regulators.
- suitable examples of liquid carriers for oral and parenteral administration include water (partially containing additives as above, e.g.
- cellulose derivatives preferably sodium carboxymethyl cellulose solution
- alcohols including monohydric alcohols and polyhydric alcohols, e.g. glycols) and their derivatives, and oils (e.g. fractionated coconut oil and arachis oil).
- the carrier can also be an oily ester such as ethyl oleate and isopropyl myristate.
- Sterile liquid carders are useful in sterile liquid form compositions for parenteral administration.
- the liquid carrier for pressurized compositions can be halogenated hydrocarbon or other pharmaceutically propellant.
- Liquid pharmaceutical compositions which are sterile solutions or suspensions can be utilized by, for example, intramuscular, intraperitoneal or subcutaneous injection. Sterile solutions can also be administered intravenously.
- the compound can also be administered orally either in liquid or solid composition form.
- the carrier or excipient may include time delay material well known to the art, such as glyceryl monostearate or glyceryl distearate along or with a wax, ethylcellulose, hydroxypropylmethylcellulose, methylmethacrylate and the like.
- time delay material such as glyceryl monostearate or glyceryl distearate along or with a wax, ethylcellulose, hydroxypropylmethylcellulose, methylmethacrylate and the like.
- Tween 80 in PHOSAL PG-50 phospholipid concentrate with 1 ,2-propylene glycol, A. Nattermann & Cie. GmbH
- PHOSAL PG-50 phospholipid concentrate with 1 ,2-propylene glycol, A. Nattermann & Cie. GmbH
- a solid carrier the preparation can be tableted, placed in a hard gelatin capsule in powder or pellet form or in the form of a troche or lozenge.
- the amount of solid carrier will vary widely but preferably will be from about 25 mg to about lg.
- a liquid carrier the preparation will be in the form of a syrup, emulsion, soft gelatin capsule, sterile injectible solution or suspension in an ampule or vial or nonaqueous liquid suspension.
- a pharmaceutically acceptable salt of the compound may be dissolved in an aqueous solution or an organic or inorganic acid, such as a 0.3 M solution of succinic acid or citric acid.
- acidic derivatives can be dissolved in suitable basic solutions. If a soluble salt form is not available, the compound is dissolved in a suitable cosolvent or combinations thereof.
- suitable cosolvents include, but are not limited to, alcohol, propylene glycol, polyethylene glycol 300, polysorbate 80, glycerin, polyoxyethylated fatty acids, fatty alcohols or glycerin hydroxy fatty acids esters and the like in concentrations ranging from 0-60% of the total volume.
- Various delivery systems are know and can be used to administer the compound, or the various formulations thereof, including tablets, capsules, injectable solutions, encapsulation in liposomes, microparticles, microcapsules, etc.
- Methods of introduction include but are not limited to dermal, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal, pulmonary, epidural, ocular and (as is usually preferred) oral routes.
- the compound may be administered by any convenient or otherwise appropriate route, for example by infusion or bolus injection, by abso ⁇ tion through epithelial or mucocutaneous linings (e.g. oral mucosa, rectal and intestinal mucosa, etc.) and may be administered together with other biologically active agents. Administration can be systemic or local.
- preferred routes are oral, nasal or via a bronchial aerosol or nebulizer.
- compositions for intravenous administration are solutions in sterile isotonic aqueous buffer.
- the composition may also include a solubilizing agent and a local anesthetic to ease pain at the side of the injection.
- the ingredients are supplied either separately or mixed together in unit dosage form, for example, as a lyophilized powder or water free concentrate in a hermetically sealed container such as an ampoule or sachette indicating the quantitity of active agent.
- a hermetically sealed container such as an ampoule or sachette indicating the quantitity of active agent.
- the composition is to be administered by infusion, it can be dispensed with an infusion bottle containing sterile pharmaceutical grade water or saline.
- an ampoule of sterile water for injection or saline can be provided so that the ingredients may be mixed prior to administration.
- Topical carriers include liquid petroleum, isopropyl palmitate, polyethylene glycol, ethanol (95%), polyoxyethylene monolaurate (5%) in water, or sodium lauryl sulfate (5%) in water.
- Other materials such as anti-oxidants, humectants, viscosity stabilizers, and similar agents may be added as necessary.
- Percutaneous penetration enhancers such as Azone may also be included.
- Scheme 5 describes the synthesis of compound 15 starting from 5- iodopyrrolo-pyrimidine (J.Med.Chem., 1990).
- 6-Chloro-2-fluoro-9H-purine (517.7 mg, 3 mmol), 2-propanol (198.3 mg, 3.3 mmol), PPh 3 (866 mg, 3.3 mmol) was mixed under N 2 in a 50 mL round-bottom flask at 0 °C.
- DEAD (575 mg, 3.3 mmol) was added via syringe dropwise to the mixture. The temperature was raised to r. t. and the mixture was stirred overnight. Sovent was removed in vacuo and the resulting residue was chromatographed on silica gel (C ⁇ 2 Cl 2 /EtOAc, 4:1, Rf 0.62). The product was obtained as a white solid (411 mg, 64 %).
- pyrimidin- 1 - yl)-butyl ester (0.85 g, 2.5 mmol), and LiOH » H 2 O (0.25 g, 5.96 mmol) were dissolved in THF (3 mL)/H 2 O ( 10 mL) and heated to 70 °C for 2 h. After cooling, the mixture was dumped into water and extracted with EtOAc. The combined extracts were washed with water, dried over magnesium sulfate, and concentrated to a yellow solid which was used without purification in the next reaction (0.20 g, 27%).
- the reaction mixture was then diluted with half saturated aq NH 4 C1 (20 mL).
- the mixture was further acidified by the addition of a small amount of 6 Naq HCl, and then extracted with EtOAc.
- the extract was washed with H 2 O (10 mL) and brine (10 mL).
- the aqueous washes were reextracted once with EtOAc, and the combined extracts were dried over MgSO 4 and concentrated.
- the crude material was purified by flash chromatography on silica gel. Elution with 5:1 hexanes-EtOAc followed by 3:1 hexanes-EtOAc afforded 1.32 g (94%) of the desired phosphonate as a light yellow oil.
- Phenol 9 was converted to x-D in the same fashion as previously described ( -
- the RV was cooled to 0 °C (Julabo chiller) and then added, under an atmosphere of N 2 , 2.0 mL (1.44 mmol) of a 0.72 M solution of DEAD (diethyl azodicarboxylate) in THF.
- the resin mixture was warmed, while agitating, to ambient temperature over 2 h and then agitated for an additional 20 h, upon which the
- the RV was cooled to 0 °C (Julabo chiller) and then added, under an atmosphere of N 2 , 2.0 mL (1.44 mmol) of a 0.72 M solution of DEAD (diethyl azodicarboxylate) in THF.
- the resin mixture was warmed, while agitating, to ambient temperature over 1.5 h and then agitated for an additional 22 h, upon which the RV was drained and the resin washed successively with THF (5x5.0 mL), DMA (5x5.0 mL), CH 2 C1 2 (5x5.0 mL), Et 2 O (2x5.0 mL), CH 2 C1 2 (1 5.0 mL), Et 2 O (1x5.0 mL), and CH 2 C1 2 (2x5.0 mL).
- the sealed RV was heated at 110 °C for 16 h, upon which the RV was cooled to ambient temperature, drained, and the resin washed successively with DMA (5x5.0 mL), CH 2 C1 (5x5.0 mL), Et 2 O (2x5.0 mL), CH 2 C1 2 (1x5.0 mL), Et 2 O (1x5.0 mL), and CH 2 CI 2 (2x5.0 mL). Excess solvent was removed via N2 flow overnight to provide the aminated purine resin lc. The following analytical data was obtained upon cleavage of lc (3-5 mg) with 30% TFA/CH 2 C1 2 ( ⁇ 5 min): m/z 592 (M+H).
- the sealed RV was heated at 110 °C for 16 h, upon which the heat was turned off, the RV drained immediately, and the resin washed (while still hot) successively with DMA (5x5.0 mL), C ⁇ 2CI2 (5x5.0 mL, at ambient temperature), Et 2 O (2x5.0 mL), CH 2 C1 2 (1x5.0 mL), Et 2 O (1x5.0 mL), and CH 2 C1 2 (2x5.0 mL). Excess solvent was removed via N 2 flow overnight to provide the bis-aminated purine resin.
- Example 22 The white solid was isolated as a TFA salt: m/z 602 (M+ ⁇ )
- Example 22 The product was isolated as a white solid: m/z 549 (M+H)
- Example 22 The product was obtained as a white solid: m/z 533 (M+ ⁇ )
- Example 22 The product was obtained as a white solid: m/z 581 (M+ ⁇ )
- Method C Carboxylic acid (0.25 mmol) 19/22 was taken up in DMF (5 mL) and cooled in ice. HATU (0.5 mmol) was then added followed by the bone-targeting amines A-D and ethyl diisopropyl amine (0.5 mmol). The reaction mixture was stirred at ambient temp, for 2 days. DMF was removed in vacuo and the residue was taken up in ethyl acetate. Ethyl acetate layer was washed with sodium bicarbonate (10%) followed by 10%) citric acid and then water. Organic extract was dried over sodium sulphate and concentrated and purified by chromatography using methylene chloride/methanol (5 - 10%).
- Buffers include: (a) Kinase buffer which contains MES 30 mM pH 6.8, MgCl 2 10 mM, Orthovanadate 0.25 mM, PMSF 0.1 mM, and DTT ImM; (b) ATP buffer which contains ATP 5 mM in MgCl 2 50 mM buffer (stock solution). Note that before each use dilute in MES to 100 ⁇ M (5X solution) add 100 ⁇ Ci/mL [ 33 P]ATP; and (c) PBS Stop buffer which contains ATP 0.1 mM, EDTA 40 mM, Triton 0.1%. Streptavidin beads are suspended at 3.3 mg/ml in stop buffer and mixed by shaking.
- Hydroxyapatite is the principal mineral component of bone. Hydroxyapatite adsorption chromatography is used as an assay to evaluate the bone-targeting potential of both individual bone-targeting moieties ("monomers”) and of pharmaceuticals inco ⁇ orating bone-targeting groups.
- the bone targeting moiety may be provided as a pro-drug with the formula:
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Physical Education & Sports Medicine (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Biochemistry (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Biophysics (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description








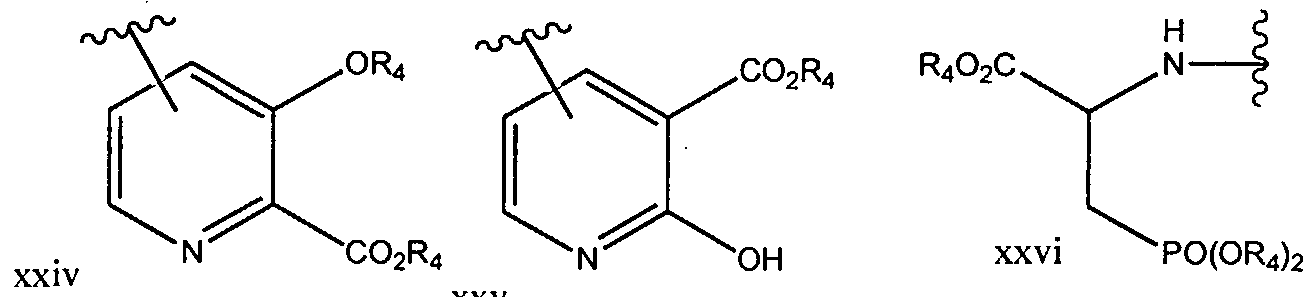

































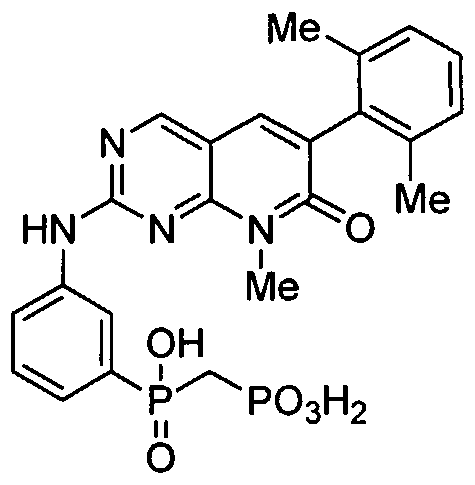









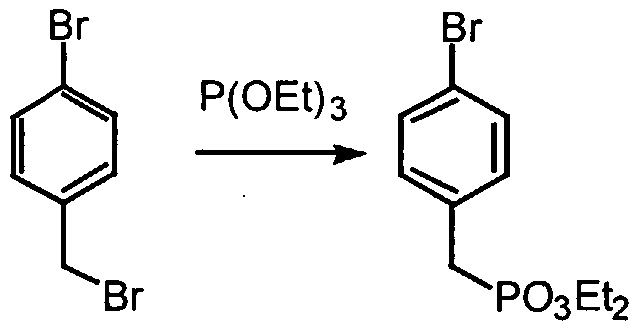











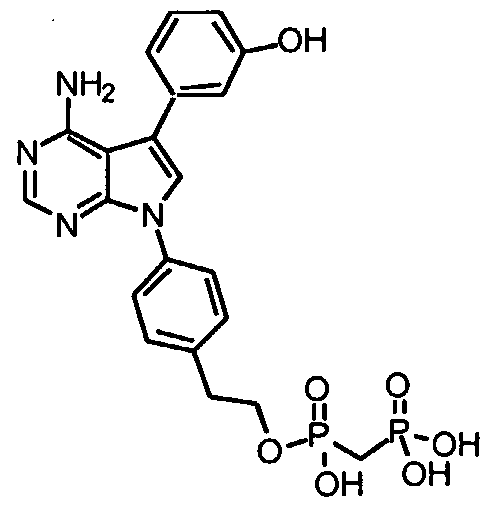





















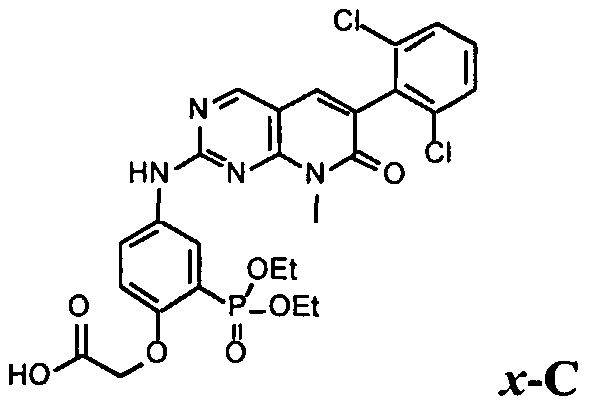


































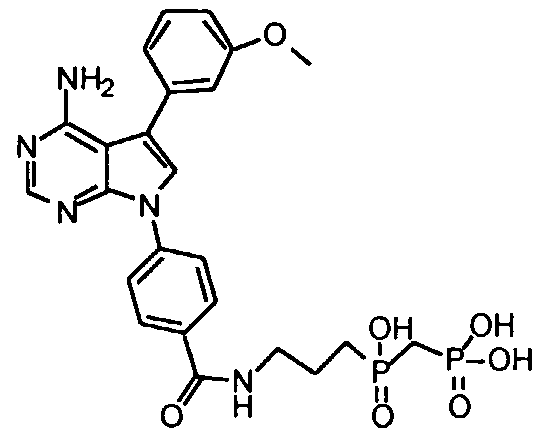















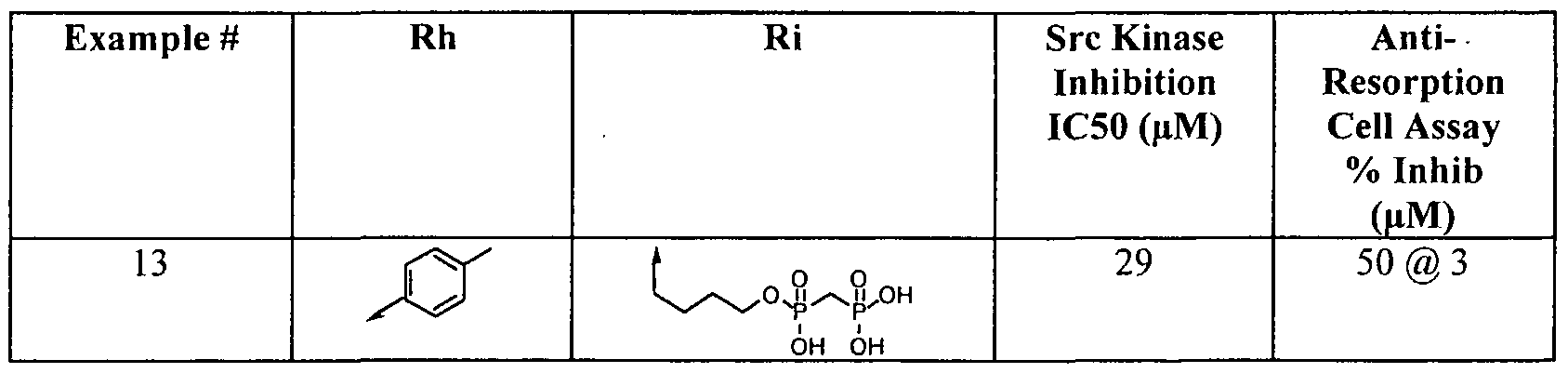




Claims



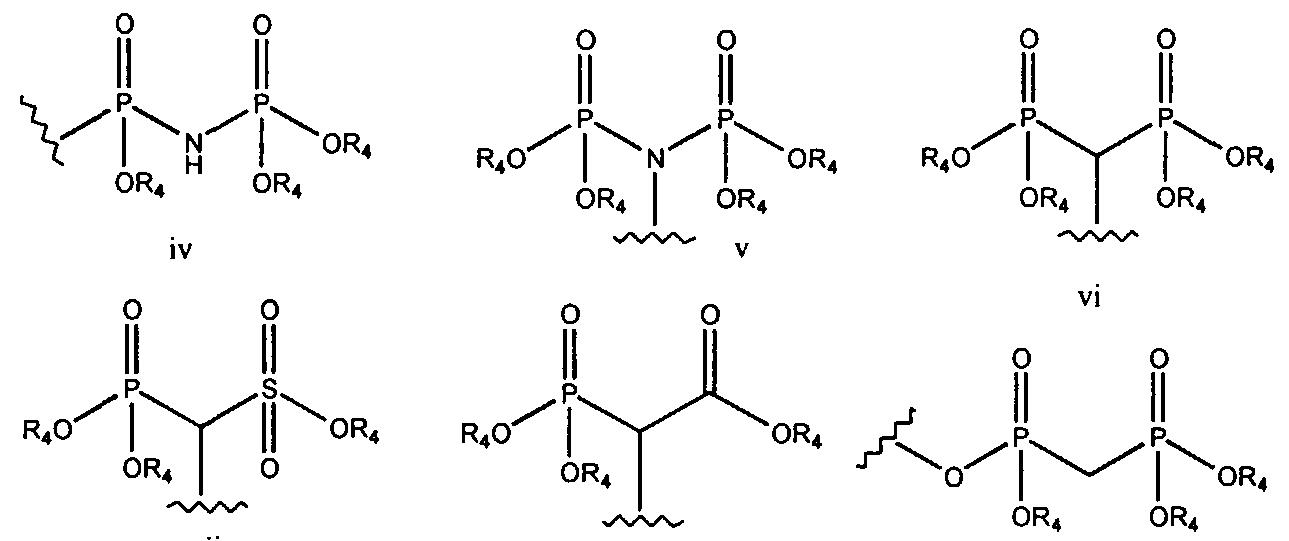







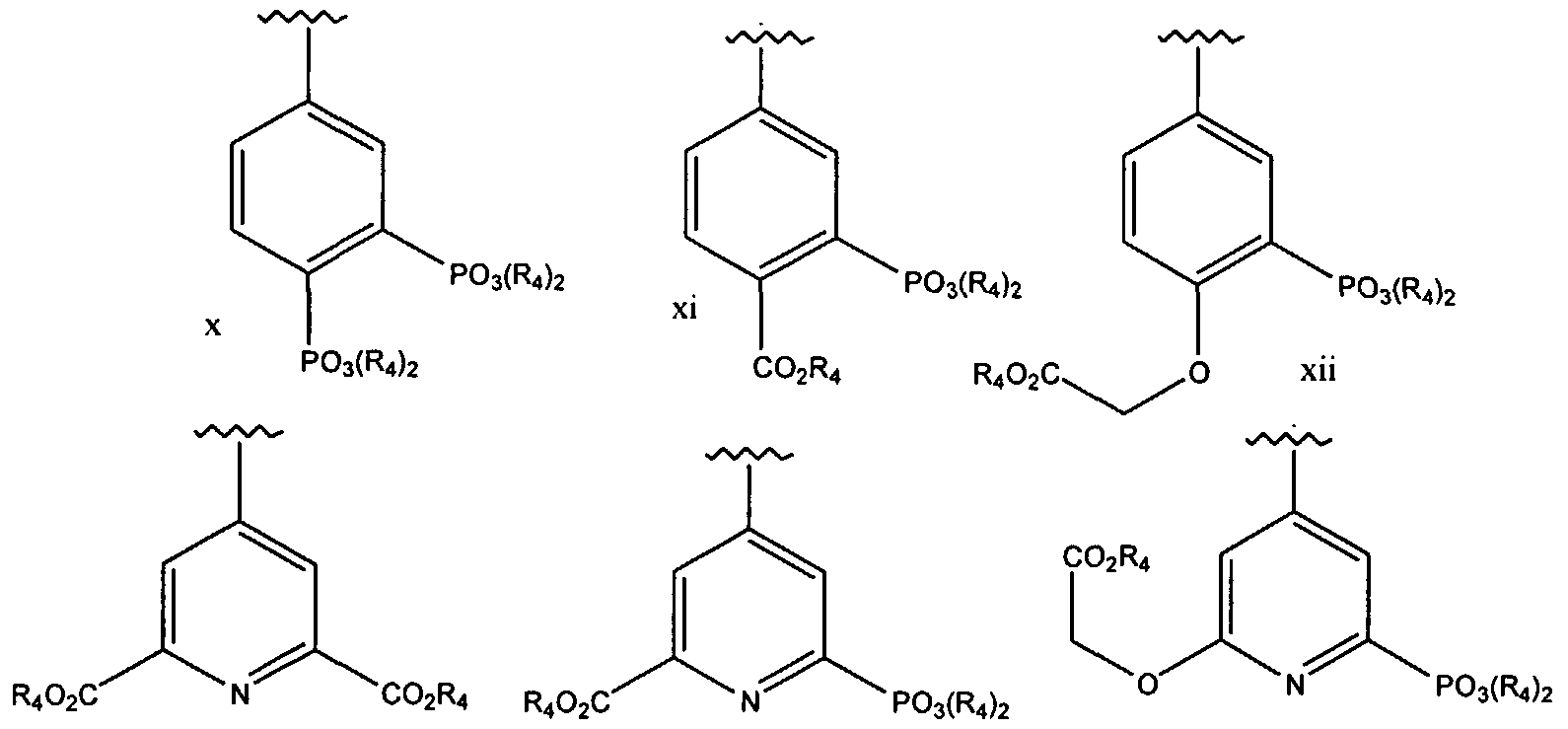









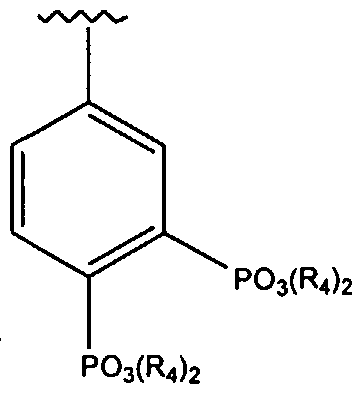


















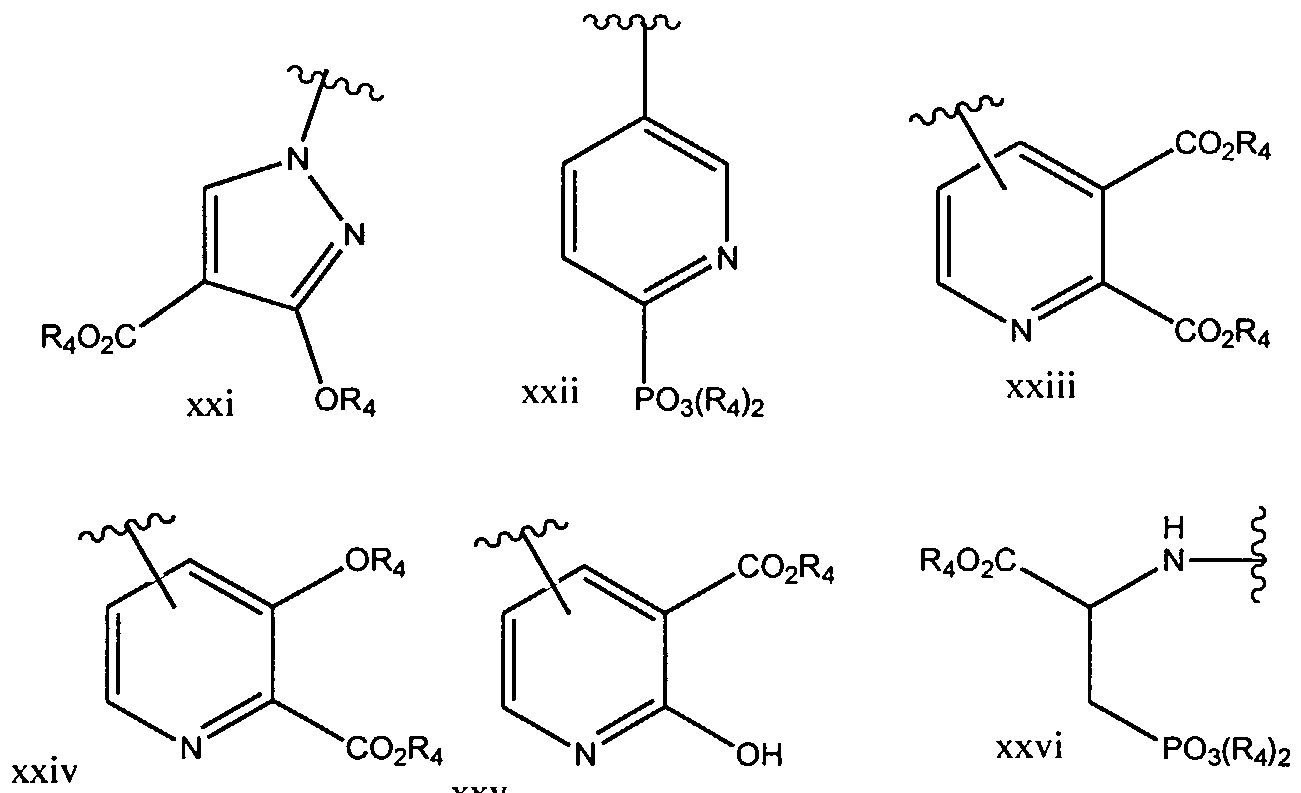


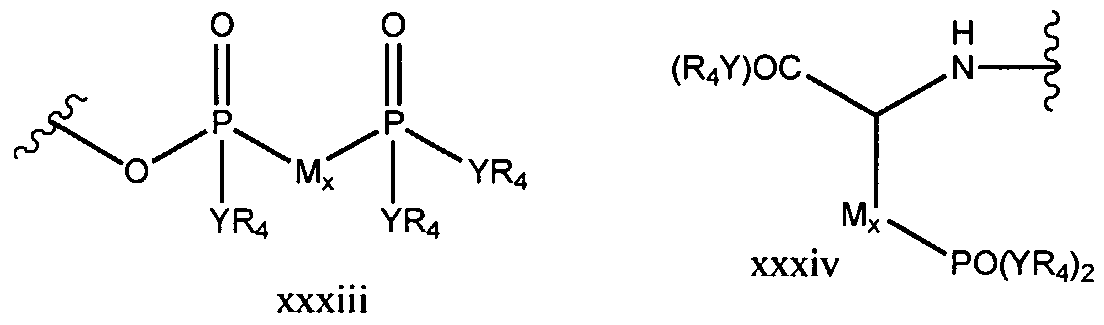





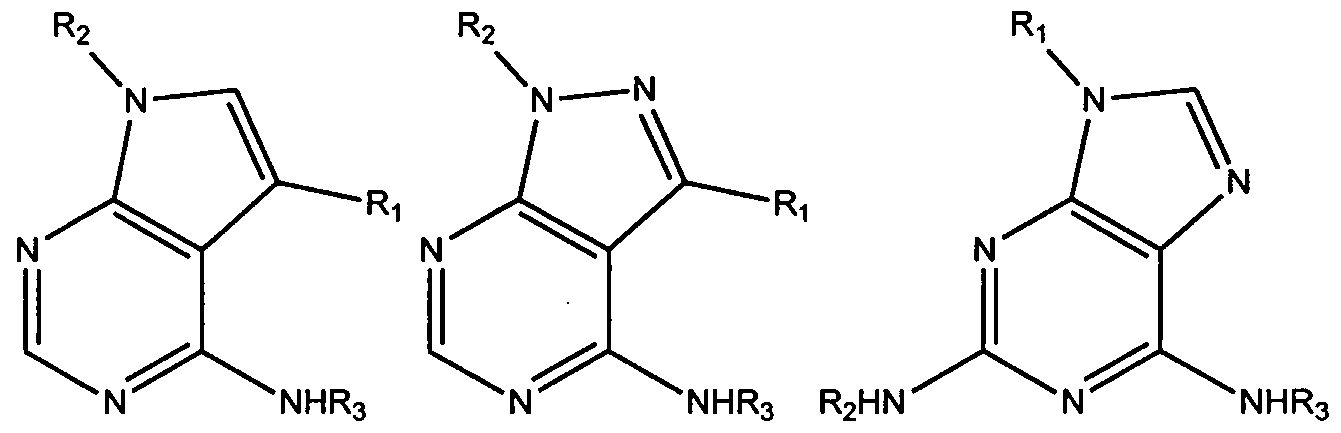
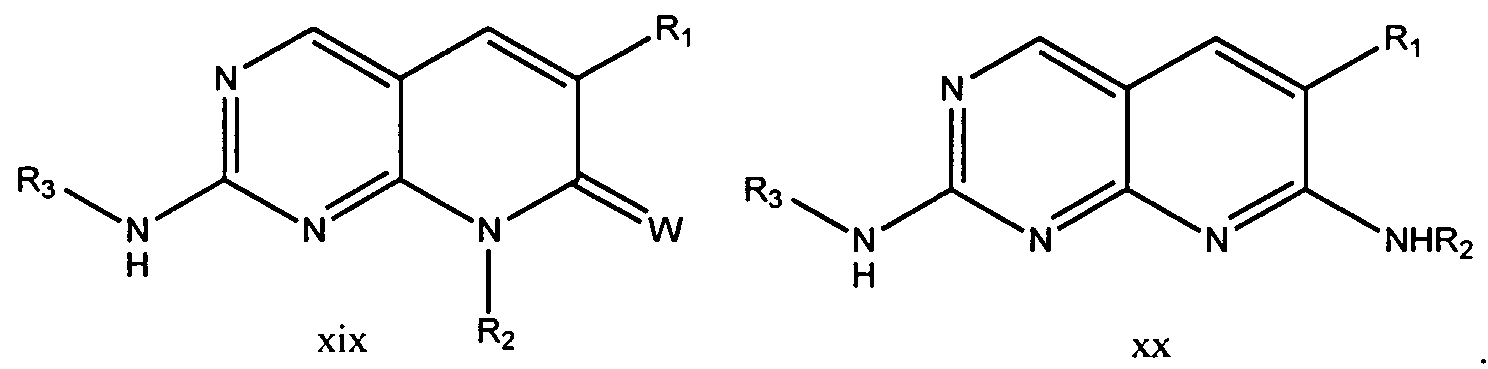




























Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU24397/01A AU2439701A (en) | 1999-12-17 | 2000-12-18 | Novel heterocycles |
| EP00988160A EP1246829A1 (en) | 1999-12-17 | 2000-12-18 | Novel heterocycles |
| IL15005900A IL150059A0 (en) | 1999-12-17 | 2000-12-18 | Novel heterocycles |
| JP2001544748A JP2003532632A (en) | 1999-12-17 | 2000-12-18 | New heterocycle |
| CA002394650A CA2394650A1 (en) | 1999-12-17 | 2000-12-18 | Novel heterocycles |
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17251099P | 1999-12-17 | 1999-12-17 | |
| US17216199P | 1999-12-17 | 1999-12-17 | |
| US60/172,510 | 1999-12-17 | ||
| US60/172,161 | 1999-12-17 | ||
| US24078800P | 2000-10-16 | 2000-10-16 | |
| US60/240,788 | 2000-10-16 | ||
| US09/740,653 US20020132819A1 (en) | 1999-12-17 | 2000-12-18 | Novel purinse |
| US09/741,619 | 2000-12-18 | ||
| US09/740,619 US6420384B2 (en) | 1999-12-17 | 2000-12-18 | Proton pump inhibitors |
| US09/740,653 | 2000-12-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001044258A1 true WO2001044258A1 (en) | 2001-06-21 |
Family
ID=27538857
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2000/034487 WO2001044258A1 (en) | 1999-12-17 | 2000-12-18 | Novel heterocycles |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP1246829A1 (en) |
| JP (1) | JP2003532632A (en) |
| AU (1) | AU2439701A (en) |
| CA (1) | CA2394650A1 (en) |
| IL (1) | IL150059A0 (en) |
| WO (1) | WO2001044258A1 (en) |
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003000187A2 (en) * | 2001-06-21 | 2003-01-03 | Ariad Pharmaceuticals, Inc. | Novel pyrazolo-and pyrrolo-pyrimidines and uses thereof |
| WO2003000011A2 (en) * | 2001-06-21 | 2003-01-03 | Ariad Pharmaceuticals, Inc. | Novel pyridopyrimidines and uses thereof |
| WO2003000270A1 (en) * | 2001-06-21 | 2003-01-03 | Ariad Pharmaceuticals, Inc. | Novel pyridopyrimidones and uses thereof |
| WO2004041823A1 (en) * | 2002-11-04 | 2004-05-21 | F. Hoffmann-La Roche Ag | Novel amino-substituted dihydropyrimido[4,5-d]pyrimidinone derivatives, their manufacture and use as pharmaceutical agents |
| WO2004063195A1 (en) * | 2003-01-03 | 2004-07-29 | Sloan-Kettering Institute For Cancer Research | Pyridopyrimidine kinase inhibitors |
| US6900316B2 (en) | 2000-08-04 | 2005-05-31 | Warner-Lamber Company | Process for preparing 2-(4-pyridyl)amino-6-dialkyloxyphenyl-pyrido(2,3-d)pyrimidin-7-ones |
| US7384937B2 (en) | 2002-11-06 | 2008-06-10 | Bristol-Myers Squibb Co. | Fused heterocyclic compounds and use thereof |
| US7514444B2 (en) | 2006-09-22 | 2009-04-07 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US7625880B2 (en) | 2006-01-13 | 2009-12-01 | Pharmacyclics, Inc. | Inhibitors of tyrosine kinases and uses thereof |
| US7659394B2 (en) | 2004-04-30 | 2010-02-09 | Pfizer Inc | Substituted morpholine compounds for the treatment of central nervous system disorders |
| WO2010019511A2 (en) | 2008-08-13 | 2010-02-18 | Targanta Therapeutics Corp. | Phosphonated rifamycins and uses thereof for the prevention and treatment of bone and joint infections |
| US8372970B2 (en) | 2009-10-09 | 2013-02-12 | Afraxis, Inc. | 8-ethyl-6-(aryl)pyrido[2,3-D]pyrimidin-7(8H)-ones for the treatment of CNS disorders |
| US8377946B1 (en) | 2011-12-30 | 2013-02-19 | Pharmacyclics, Inc. | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| WO2013032527A1 (en) | 2011-09-02 | 2013-03-07 | The Regents Of The University Of California | Llp2a-bisphosphonate conjugates for osteoporosis treatment |
| US20130252966A1 (en) * | 2010-06-09 | 2013-09-26 | Afraxis, Inc. | 6-(sulfonylaryl)pyrido[2,3-d]pyrimidin-7(8h)-ones for the treatment of cns disorders |
| US8674095B2 (en) | 2008-12-19 | 2014-03-18 | Afraxis Holdings, Inc. | Compounds for treating neuropsychiatric conditions |
| US8754090B2 (en) | 2010-06-03 | 2014-06-17 | Pharmacyclics, Inc. | Use of inhibitors of bruton's tyrosine kinase (Btk) |
| US8846664B2 (en) | 2008-11-12 | 2014-09-30 | Ariad Pharmaceuticals, Inc. | Pyrazinopyrazines and derivatives as kinase inhibitors |
| US8883803B2 (en) | 2008-07-16 | 2014-11-11 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase for the treatment of solid tumors |
| US8940750B2 (en) | 2007-03-28 | 2015-01-27 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US8987233B2 (en) | 2006-11-03 | 2015-03-24 | Pharmacyclics, Inc. | Bruton's tyrosine kinase activity probe and method of using |
| US9096604B2 (en) | 2012-11-15 | 2015-08-04 | Pharmacyclics, Inc. | Pyrrolopyrimidine compounds as kinase inhibitors |
| CN104829620A (en) * | 2015-04-08 | 2015-08-12 | 重庆华邦胜凯制药有限公司 | Method for preparing aminopyrrolo[2,3-d]pyrimidine derivatives |
| US9119884B2 (en) | 2010-09-02 | 2015-09-01 | The Regents Of The University Of California | LLP2A-bisphosphonate conjugates for osteoporosis treatment |
| US9296753B2 (en) | 2012-06-04 | 2016-03-29 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US9415050B2 (en) | 2013-08-12 | 2016-08-16 | Pharmacyclics Llc | Methods for the treatment of HER2 amplified cancer |
| US9421208B2 (en) | 2013-08-02 | 2016-08-23 | Pharmacyclics Llc | Methods for the treatment of solid tumors |
| US9533991B2 (en) | 2014-08-01 | 2017-01-03 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US9545407B2 (en) | 2014-08-07 | 2017-01-17 | Pharmacyclics Llc | Formulations of a bruton's tyrosine kinase inhibitor |
| US9593137B2 (en) | 2011-12-22 | 2017-03-14 | Geron Corporation | Guanine analogs as telomerase substrates and telomere length affectors |
| US9624224B2 (en) | 2013-09-30 | 2017-04-18 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US9655857B2 (en) | 2015-03-03 | 2017-05-23 | Pharmacyclics Llc | Pharmaceutical formulations of a Bruton's tyrosine kinase inhibitor |
| US9862722B2 (en) | 2011-07-13 | 2018-01-09 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US9885086B2 (en) | 2014-03-20 | 2018-02-06 | Pharmacyclics Llc | Phospholipase C gamma 2 and resistance associated mutations |
| US10463668B2 (en) | 2013-10-25 | 2019-11-05 | Pharmacyclics Llc | Methods of treating and preventing graft versus host disease |
| US10899884B2 (en) | 2018-11-29 | 2021-01-26 | International Business Machines Corporation | Flame-retardant polyetheretherketone-based compounds |
| US10954567B2 (en) | 2012-07-24 | 2021-03-23 | Pharmacyclics Llc | Mutations associated with resistance to inhibitors of Bruton's Tyrosine Kinase (BTK) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006138590A1 (en) * | 2005-06-17 | 2006-12-28 | The Board Of Regents Of The University Of Texas System | Inhibition of osteolytic lesions by src kinase inhibitors |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0084822A2 (en) * | 1982-01-27 | 1983-08-03 | Schering Aktiengesellschaft | Diphosphonic acid derivatives and pharmaceutical preparations containing them |
| EP0085321A2 (en) * | 1982-01-27 | 1983-08-10 | Schering Aktiengesellschaft | Diphosphonic acid derivatives and pharmaceutical preparations containing them |
| EP0170228A1 (en) * | 1984-08-02 | 1986-02-05 | Roche Diagnostics GmbH | Diphosphonic-acid derivatives, process for their preparation and medicines containing them |
| EP0186405A2 (en) * | 1984-12-21 | 1986-07-02 | The Procter & Gamble Company | Pharmaceutical compositions containing geminal diphosphonates |
| EP0304962A2 (en) * | 1984-12-21 | 1989-03-01 | The Procter & Gamble Company | Novel diphosphonates and use thereof in pharmaceutical compositions |
| WO1994009017A1 (en) * | 1992-10-09 | 1994-04-28 | The Upjohn Company | Pyrimidine bisphosphonate esters and (alkoxymethylphosphinyl)alkyl phosphonic acids as anti-inflammatories |
| US5866556A (en) * | 1994-06-09 | 1999-02-02 | Leiras Oy | Pyridinylbisphosphonates for use as a therapeutical agent |
-
2000
- 2000-12-18 IL IL15005900A patent/IL150059A0/en unknown
- 2000-12-18 JP JP2001544748A patent/JP2003532632A/en not_active Withdrawn
- 2000-12-18 EP EP00988160A patent/EP1246829A1/en not_active Withdrawn
- 2000-12-18 AU AU24397/01A patent/AU2439701A/en not_active Abandoned
- 2000-12-18 WO PCT/US2000/034487 patent/WO2001044258A1/en not_active Application Discontinuation
- 2000-12-18 CA CA002394650A patent/CA2394650A1/en not_active Abandoned
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0084822A2 (en) * | 1982-01-27 | 1983-08-03 | Schering Aktiengesellschaft | Diphosphonic acid derivatives and pharmaceutical preparations containing them |
| EP0085321A2 (en) * | 1982-01-27 | 1983-08-10 | Schering Aktiengesellschaft | Diphosphonic acid derivatives and pharmaceutical preparations containing them |
| EP0170228A1 (en) * | 1984-08-02 | 1986-02-05 | Roche Diagnostics GmbH | Diphosphonic-acid derivatives, process for their preparation and medicines containing them |
| EP0186405A2 (en) * | 1984-12-21 | 1986-07-02 | The Procter & Gamble Company | Pharmaceutical compositions containing geminal diphosphonates |
| EP0304962A2 (en) * | 1984-12-21 | 1989-03-01 | The Procter & Gamble Company | Novel diphosphonates and use thereof in pharmaceutical compositions |
| WO1994009017A1 (en) * | 1992-10-09 | 1994-04-28 | The Upjohn Company | Pyrimidine bisphosphonate esters and (alkoxymethylphosphinyl)alkyl phosphonic acids as anti-inflammatories |
| US5866556A (en) * | 1994-06-09 | 1999-02-02 | Leiras Oy | Pyridinylbisphosphonates for use as a therapeutical agent |
Cited By (131)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6900316B2 (en) | 2000-08-04 | 2005-05-31 | Warner-Lamber Company | Process for preparing 2-(4-pyridyl)amino-6-dialkyloxyphenyl-pyrido(2,3-d)pyrimidin-7-ones |
| WO2003000187A3 (en) * | 2001-06-21 | 2004-08-05 | Ariad Pharma Inc | Novel pyrazolo-and pyrrolo-pyrimidines and uses thereof |
| WO2003000270A1 (en) * | 2001-06-21 | 2003-01-03 | Ariad Pharmaceuticals, Inc. | Novel pyridopyrimidones and uses thereof |
| WO2003000011A3 (en) * | 2001-06-21 | 2003-03-20 | Ariad Pharma Inc | Novel pyridopyrimidines and uses thereof |
| WO2003000011A2 (en) * | 2001-06-21 | 2003-01-03 | Ariad Pharmaceuticals, Inc. | Novel pyridopyrimidines and uses thereof |
| WO2003000187A2 (en) * | 2001-06-21 | 2003-01-03 | Ariad Pharmaceuticals, Inc. | Novel pyrazolo-and pyrrolo-pyrimidines and uses thereof |
| WO2004041823A1 (en) * | 2002-11-04 | 2004-05-21 | F. Hoffmann-La Roche Ag | Novel amino-substituted dihydropyrimido[4,5-d]pyrimidinone derivatives, their manufacture and use as pharmaceutical agents |
| US7091345B2 (en) | 2002-11-04 | 2006-08-15 | Hoffmann-La Roche Inc. | Amino-substituted dihydropyrimido[4,5-D]pyrimidinone derivatives |
| US7981881B2 (en) | 2002-11-06 | 2011-07-19 | Bristol-Myers Squibb Company | Fused heterocyclic compounds and use thereof |
| US7384937B2 (en) | 2002-11-06 | 2008-06-10 | Bristol-Myers Squibb Co. | Fused heterocyclic compounds and use thereof |
| WO2004063195A1 (en) * | 2003-01-03 | 2004-07-29 | Sloan-Kettering Institute For Cancer Research | Pyridopyrimidine kinase inhibitors |
| US7659394B2 (en) | 2004-04-30 | 2010-02-09 | Pfizer Inc | Substituted morpholine compounds for the treatment of central nervous system disorders |
| US7625880B2 (en) | 2006-01-13 | 2009-12-01 | Pharmacyclics, Inc. | Inhibitors of tyrosine kinases and uses thereof |
| US8067395B2 (en) | 2006-01-13 | 2011-11-29 | Pharmacyclics, Inc. | Inhibitors of tyrosine kinases and uses thereof |
| US8658653B2 (en) | 2006-09-22 | 2014-02-25 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8741908B2 (en) | 2006-09-22 | 2014-06-03 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US7960396B2 (en) | 2006-09-22 | 2011-06-14 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US7732454B2 (en) | 2006-09-22 | 2010-06-08 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8008309B2 (en) | 2006-09-22 | 2011-08-30 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US9133202B2 (en) | 2006-09-22 | 2015-09-15 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8088781B2 (en) | 2006-09-22 | 2012-01-03 | Pharmacyclics, Inc. | Inhibitors of brutons tyrosine kinase |
| US8158786B2 (en) | 2006-09-22 | 2012-04-17 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8232280B2 (en) | 2006-09-22 | 2012-07-31 | Pharmacyclics, Inc. | Inhibitors of bruton'S tyrosine kinase |
| US8236812B2 (en) | 2006-09-22 | 2012-08-07 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US9133198B2 (en) | 2006-09-22 | 2015-09-15 | Pharmacyclics Llc | Inhibitors of bruton'S tyrosine kinase |
| US9127012B2 (en) | 2006-09-22 | 2015-09-08 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US7514444B2 (en) | 2006-09-22 | 2009-04-07 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US8399470B2 (en) | 2006-09-22 | 2013-03-19 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US8476284B2 (en) | 2006-09-22 | 2013-07-02 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8497277B2 (en) | 2006-09-22 | 2013-07-30 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8501751B2 (en) | 2006-09-22 | 2013-08-06 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US9409911B2 (en) | 2006-09-22 | 2016-08-09 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| US8552010B2 (en) | 2006-09-22 | 2013-10-08 | Pharmacyclics, Inc. | Inhibitors of Bruton'S tyrosine kinase |
| US8563563B2 (en) | 2006-09-22 | 2013-10-22 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US9133201B2 (en) | 2006-09-22 | 2015-09-15 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US9181257B2 (en) | 2006-09-22 | 2015-11-10 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US8691546B2 (en) | 2006-09-22 | 2014-04-08 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8697711B2 (en) | 2006-09-22 | 2014-04-15 | Pharmacyclics, Inc. | Inhibitors of bruton'S tyrosine kinase |
| US8703780B2 (en) | 2006-09-22 | 2014-04-22 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8735403B2 (en) | 2006-09-22 | 2014-05-27 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8735404B2 (en) | 2006-09-22 | 2014-05-27 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US7825118B2 (en) | 2006-09-22 | 2010-11-02 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US8748439B2 (en) | 2006-09-22 | 2014-06-10 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8748438B2 (en) | 2006-09-22 | 2014-06-10 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US9193735B2 (en) | 2006-09-22 | 2015-11-24 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US8754091B2 (en) | 2006-09-22 | 2014-06-17 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US8759516B2 (en) | 2006-09-22 | 2014-06-24 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US9266893B2 (en) | 2006-09-22 | 2016-02-23 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US9206189B2 (en) | 2006-09-22 | 2015-12-08 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| US8883435B2 (en) | 2006-09-22 | 2014-11-11 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8975266B2 (en) | 2006-09-22 | 2015-03-10 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US9212185B2 (en) | 2006-09-22 | 2015-12-15 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US8957079B2 (en) | 2006-09-22 | 2015-02-17 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8952015B2 (en) | 2006-09-22 | 2015-02-10 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase |
| US8987233B2 (en) | 2006-11-03 | 2015-03-24 | Pharmacyclics, Inc. | Bruton's tyrosine kinase activity probe and method of using |
| US8940750B2 (en) | 2007-03-28 | 2015-01-27 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US9079908B2 (en) | 2007-03-28 | 2015-07-14 | Pharmacyclics, Inc. | Inhibitors of Bruton'S tyrosine kinase |
| US9181263B2 (en) | 2007-03-28 | 2015-11-10 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| US9139591B2 (en) | 2007-03-28 | 2015-09-22 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| US9556182B2 (en) | 2007-03-28 | 2017-01-31 | Pharmacylics LLC | Inhibitors of Bruton's tyrosine kinase |
| US9795605B2 (en) | 2008-07-16 | 2017-10-24 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase for the treatment of solid tumors |
| US8883803B2 (en) | 2008-07-16 | 2014-11-11 | Pharmacyclics, Inc. | Inhibitors of Bruton's tyrosine kinase for the treatment of solid tumors |
| US9278100B2 (en) | 2008-07-16 | 2016-03-08 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors |
| US9107924B2 (en) | 2008-07-16 | 2015-08-18 | Pharmacyclics, Inc. | Inhibitors of Bruton'S tyrosine kinase for the treatment of solid tumors |
| WO2010019511A2 (en) | 2008-08-13 | 2010-02-18 | Targanta Therapeutics Corp. | Phosphonated rifamycins and uses thereof for the prevention and treatment of bone and joint infections |
| US8846664B2 (en) | 2008-11-12 | 2014-09-30 | Ariad Pharmaceuticals, Inc. | Pyrazinopyrazines and derivatives as kinase inhibitors |
| US8674095B2 (en) | 2008-12-19 | 2014-03-18 | Afraxis Holdings, Inc. | Compounds for treating neuropsychiatric conditions |
| US8372970B2 (en) | 2009-10-09 | 2013-02-12 | Afraxis, Inc. | 8-ethyl-6-(aryl)pyrido[2,3-D]pyrimidin-7(8H)-ones for the treatment of CNS disorders |
| US9125889B2 (en) | 2010-06-03 | 2015-09-08 | Pharmacyclics, Inc. | Use of inhibitors of Bruton's tyrosine kinase (Btk) |
| US10478439B2 (en) | 2010-06-03 | 2019-11-19 | Pharmacyclics Llc | Use of inhibitors of bruton's tyrosine kinase (Btk) |
| US11672803B2 (en) | 2010-06-03 | 2023-06-13 | Pharmacyclics Llc | Use of inhibitors of Brutons tyrosine kinase (Btk) |
| US9801883B2 (en) | 2010-06-03 | 2017-10-31 | Pharmacyclics Llc | Use of inhibitors of bruton's tyrosine kinase (Btk) |
| US8999999B2 (en) | 2010-06-03 | 2015-04-07 | Pharmacyclics, Inc. | Use of inhibitors of Bruton's tyrosine kinase (Btk) |
| US9814721B2 (en) | 2010-06-03 | 2017-11-14 | Pharmacyclics Llc | Use of inhibitors of bruton'S tyrosine kinase (BTK) |
| US10004745B2 (en) | 2010-06-03 | 2018-06-26 | Pharmacyclics Llc | Use of inhibitors of Bruton'S tyrosine kinase (Btk) |
| US10004746B2 (en) | 2010-06-03 | 2018-06-26 | Pharmacyclics Llc | Use of inhibitors of Bruton's tyrosine kinase (Btk) |
| US9801881B2 (en) | 2010-06-03 | 2017-10-31 | Pharmacyclics Llc | Use of inhibitors of bruton's tyrosine kinase (BTK) |
| US8754090B2 (en) | 2010-06-03 | 2014-06-17 | Pharmacyclics, Inc. | Use of inhibitors of bruton's tyrosine kinase (Btk) |
| US10751342B2 (en) | 2010-06-03 | 2020-08-25 | Pharmacyclics Llc | Use of inhibitors of Bruton's tyrosine kinase (Btk) |
| US10016435B2 (en) | 2010-06-03 | 2018-07-10 | Pharmacyclics Llc | Use of inhibitors of Bruton's tyrosine kinase (Btk) |
| US10653696B2 (en) | 2010-06-03 | 2020-05-19 | Pharmacyclics Llc | Use of inhibitors of bruton's tyrosine kinase (BTK) |
| US20130252966A1 (en) * | 2010-06-09 | 2013-09-26 | Afraxis, Inc. | 6-(sulfonylaryl)pyrido[2,3-d]pyrimidin-7(8h)-ones for the treatment of cns disorders |
| US8912203B2 (en) * | 2010-06-09 | 2014-12-16 | Afraxis Holdings, Inc. | 6-(sulfonylaryl)pyrido[2,3-D]pyrimidin-7(8H)-ones for the treatment of CNS disorders |
| US10494401B2 (en) | 2010-09-02 | 2019-12-03 | The Regents Of The University Of California | LLP2A-bisphosphonate conjugates for osteoporosis treatment |
| US9119884B2 (en) | 2010-09-02 | 2015-09-01 | The Regents Of The University Of California | LLP2A-bisphosphonate conjugates for osteoporosis treatment |
| US9561256B2 (en) | 2010-09-02 | 2017-02-07 | The Regents Of The University Of California | LLP2A-bisphosphonate conjugates for osteoporosis treatment |
| US9862722B2 (en) | 2011-07-13 | 2018-01-09 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| EP3453711A1 (en) * | 2011-09-02 | 2019-03-13 | The Regents of the University of California | Llp2a- bisphosphonate conjugates for osteoporosis treatment |
| EP2753626A1 (en) | 2011-09-02 | 2014-07-16 | The Regents of The University of California | Llp2a-bisphosphonate conjugates for osteoporosis treatment |
| WO2013032527A1 (en) | 2011-09-02 | 2013-03-07 | The Regents Of The University Of California | Llp2a-bisphosphonate conjugates for osteoporosis treatment |
| EP2753626A4 (en) * | 2011-09-02 | 2015-03-25 | Univ California | Llp2a-bisphosphonate conjugates for osteoporosis treatment |
| US10035814B2 (en) | 2011-12-22 | 2018-07-31 | Geron Corporation | Guanine analogs as telomerase substrates and telomere length affectors |
| US9593137B2 (en) | 2011-12-22 | 2017-03-14 | Geron Corporation | Guanine analogs as telomerase substrates and telomere length affectors |
| US10562926B2 (en) | 2011-12-22 | 2020-02-18 | Geron Corporation | Guanine analogs as telomerase substrates and telomere length affectors |
| US11279720B2 (en) | 2011-12-22 | 2022-03-22 | Geron Corporation | Guanine analogs as telomerase substrates and telomere length affectors |
| US9546172B2 (en) | 2011-12-30 | 2017-01-17 | Pharmacyclics Llc | Pyrazolo[3,4-d]pyrimidine and pyrazolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| US8377946B1 (en) | 2011-12-30 | 2013-02-19 | Pharmacyclics, Inc. | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| US9273051B2 (en) | 2011-12-30 | 2016-03-01 | Pharmacyclics Llc | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| US10266540B2 (en) | 2012-06-04 | 2019-04-23 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US10752634B2 (en) | 2012-06-04 | 2020-08-25 | Pharmacyclics Llc | Crystalline forms of a brutons tyrosine kinase inhibitor |
| US9725455B1 (en) | 2012-06-04 | 2017-08-08 | Pharmacyclics Llc | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| US10961251B1 (en) | 2012-06-04 | 2021-03-30 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US9296753B2 (en) | 2012-06-04 | 2016-03-29 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US9828383B1 (en) | 2012-06-04 | 2017-11-28 | Pharmacyclic s LLC | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| US9713617B2 (en) | 2012-06-04 | 2017-07-25 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US10294231B2 (en) | 2012-06-04 | 2019-05-21 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US10294232B2 (en) | 2012-06-04 | 2019-05-21 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US9540382B2 (en) | 2012-06-04 | 2017-01-10 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US10125140B1 (en) | 2012-06-04 | 2018-11-13 | Pharmacyclics Llc | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| US10065968B2 (en) | 2012-06-04 | 2018-09-04 | Pharmacyclics Llc | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| US10106548B2 (en) | 2012-06-04 | 2018-10-23 | Pharmacyclics Llc | Crystalline forms of a Bruton's tyrosine kinase inhibitor |
| US10954567B2 (en) | 2012-07-24 | 2021-03-23 | Pharmacyclics Llc | Mutations associated with resistance to inhibitors of Bruton's Tyrosine Kinase (BTK) |
| US9540385B2 (en) | 2012-11-15 | 2017-01-10 | Pharmacyclics Llc | Pyrrolopyrimidine compounds as kinase inhibitors |
| US9096604B2 (en) | 2012-11-15 | 2015-08-04 | Pharmacyclics, Inc. | Pyrrolopyrimidine compounds as kinase inhibitors |
| US9421208B2 (en) | 2013-08-02 | 2016-08-23 | Pharmacyclics Llc | Methods for the treatment of solid tumors |
| US10016434B2 (en) | 2013-08-12 | 2018-07-10 | Pharmacyclics Llc | Methods for the treatment of HER2 amplified cancer |
| US9415050B2 (en) | 2013-08-12 | 2016-08-16 | Pharmacyclics Llc | Methods for the treatment of HER2 amplified cancer |
| US9724349B2 (en) | 2013-08-12 | 2017-08-08 | Pharmacyclics Llc | Methods for the treatment of HER2 amplified cancer |
| US9624224B2 (en) | 2013-09-30 | 2017-04-18 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US10463668B2 (en) | 2013-10-25 | 2019-11-05 | Pharmacyclics Llc | Methods of treating and preventing graft versus host disease |
| US10695350B2 (en) | 2013-10-25 | 2020-06-30 | Pharmacyclics Llc | Methods of treating and preventing graft versus host disease |
| US9885086B2 (en) | 2014-03-20 | 2018-02-06 | Pharmacyclics Llc | Phospholipase C gamma 2 and resistance associated mutations |
| US9533991B2 (en) | 2014-08-01 | 2017-01-03 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US20180028537A1 (en) | 2014-08-07 | 2018-02-01 | Pharmacyclics Llc | Novel Formulations of a Bruton's Tyrosine Kinase Inhibitor |
| US9545407B2 (en) | 2014-08-07 | 2017-01-17 | Pharmacyclics Llc | Formulations of a bruton's tyrosine kinase inhibitor |
| US10010507B1 (en) | 2015-03-03 | 2018-07-03 | Pharmacyclics Llc | Pharmaceutical formulations of a bruton's tyrosine kinase inhibitor |
| US10828259B2 (en) | 2015-03-03 | 2020-11-10 | Pharmacyclics Llc | Pharmaceutical formulations of a Bruton's tyrosine kinase inhibitor |
| US9655857B2 (en) | 2015-03-03 | 2017-05-23 | Pharmacyclics Llc | Pharmaceutical formulations of a Bruton's tyrosine kinase inhibitor |
| US10213386B2 (en) | 2015-03-03 | 2019-02-26 | Pharmacyclics Llc | Pharmaceutical formulations of a Bruton's tyrosine kinase inhibitor |
| CN104829620A (en) * | 2015-04-08 | 2015-08-12 | 重庆华邦胜凯制药有限公司 | Method for preparing aminopyrrolo[2,3-d]pyrimidine derivatives |
| US10899884B2 (en) | 2018-11-29 | 2021-01-26 | International Business Machines Corporation | Flame-retardant polyetheretherketone-based compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2003532632A (en) | 2003-11-05 |
| EP1246829A1 (en) | 2002-10-09 |
| IL150059A0 (en) | 2002-12-01 |
| AU2439701A (en) | 2001-06-25 |
| CA2394650A1 (en) | 2001-06-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20020103161A1 (en) | Novel heterocycles | |
| WO2001044258A1 (en) | Novel heterocycles | |
| US7645747B2 (en) | Therapeutic phosphonate compounds | |
| TWI389908B (en) | Antiviral compounds | |
| US6214812B1 (en) | Bisphosphonate conjugates and methods of making and using the same | |
| CA2394654A1 (en) | Proton pump inhibitors | |
| KR20070087266A (en) | Pi-3 kinase inhibitor prodrugs | |
| KR101327635B1 (en) | Phosphonated rifamycins and uses thereof for the prevention and treatment of bone and joint infections | |
| EA011399B1 (en) | Tricyclic compounds – hiv-integrase inhibitor, methods for inhibition thereof (variants), a pharmaceutical composition based thereon, a process for making thereof, and methods for use thereof in the treatment of diseases | |
| TW200408645A (en) | Non nucleoside reverse transcriptase inhibitors | |
| EP1259520B1 (en) | Novel purines | |
| EP1244679B1 (en) | Purine derivatives | |
| KR20110079813A (en) | Imidazopyridinyl bisphosphonates | |
| EP2678039B1 (en) | Bisphosphonate-prodrugs | |
| US7598246B2 (en) | Bisphosphonate conjugates and methods of making and using the same | |
| US6896871B2 (en) | Biphosphonate conjugates and methods of making and using the same | |
| PL180705B1 (en) | Pyridyl biphposphonates for use as therapeutic agents | |
| CA3003741A1 (en) | Bortezomib conjugates and methods using same | |
| US7432277B2 (en) | Phosphorus-containing macrocycles | |
| US20040002479A1 (en) | Peptide analogues and uses thereof | |
| JP2003516986A (en) | Substituted bisindole ylmaleimide for cell growth inhibition |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 24397/01 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 150059 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2394650 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2001 544748 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000988160 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000988160 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2000988160 Country of ref document: EP |