PROCESS FOR THE PREPARATION OF ARYLOXY PROPANOLAMINES
BACKGROUND OF THE INVENTION
Aryloxy propanolamines are widely known as beta 3 agonists. Beta 3-agonists have many uses, including as drugs for the treatment of obesity and Type II diabetes and as feed additives for livestock to aid in the production of leaner meat. The structure of aryloxypropanolamine is shown below:
Ar represents an aryl group. Generally, aryloxy propanolamines with beta 3 activity have a substituted amine group .
Some of the more active aryloxy propanolamines, referred to herein as "aryloxy propanolamine amides" ''are substituted with a 2- [4- (2-yl-2- methylpropyl ) phenoxy] pyridinecarboxamide group (see, for example, WO 97/10822 to Bell et al . , WO 98/09625 to Crowell et al . , U.S. Patent No. 5,808,080 and U.S. Patent No. 6,046,227, the entire teachings of which are incorporated herein by reference) . The structure of an aryloxy propanolamine amide is shown below:
Aryloxy propanolamines, including aryloxy propanolamine amides, have generally been prepared from epoxide precursors which are known to be bacterial mutagens, see, for example, WO 97/10825. The commercial potential of aryloxy propanolamine amides as pharmaceutical products or as food additives for livestock is critically dependent upon the development of processes for their preparation which
eliminate these mutagenic epoxides from the final product. In addition, such processes must be economical, efficient and amenable to large-scale production of bulk quantities.
SUMMARY OF THE INVENTION
It has now been found that salts of novel aryloxy propanolamines in which the amine group is substituted with an alkyl 2- [4- (2-yl-2-methylpropyl)phenoxy]pyridine- carboxylate group (hereinafter "aryloxy propanolamine esters") can be readily crystallized in high yield and in high purity, substantially free (typically less than 1 ppm) of epoxide precursors (Example 42) . An example of an aryloxy propanolamine ester is shown below:
It has also been found that the corresponding carboxylic acids of aryloxy propanolamine esters (hereinafter "aryloxy propanolamine carboxylic acids") can be isolated by precipitation at their isoelectric point in high yield and high purity (Examples 28 and 32) . A further discovery reported herein is that both aryloxy propanolamine esters and carboxylic acids can be prepared in high yield from readily accessible starting materials and can be readily amidated to produce beta-3 active aryloxy propanolamine amides. Based on these discoveries, novel methods of preparing aryloxy propanolamine amides and novel intermediates in the preparation of aryloxy propanolamine amides are disclosed herein.
One embodiment of the present invention is a method of preparing an aryloxy propanolamine ester represented by Structural Formula (I):
and quarternary ammonium salts thereof.
Ri is an aryl group or a substituted aryl group. Examples of suitable aryl groups for Ri are shown in Structural Formulas (VII) -(XX). In a preferred embodiment, Ri is not a carbazolyl or substituted carbazolyl group. In another preferred embodiment, Ri is a substituted or unsubstituted monocyclic or bicylic heteroaryl group or a substituted or unsubstituted tricylic heteroaryl group with at least two ring nitrogen atoms. More preferably, Ri is a substituted or unsubstituted indolyl group, e.g., a 4-indolyl group.
R2 is a Cl to C6 straight or branched chain alkyl group or a C7 to C9 substituted or unsubstituted aralkyl group. Preferably, R2 is methyl or ethyl.
The method comprises the step of reacting an epoxide starting material with an amine starting material. The epoxide starting material is represented by Structural Formula (II) and the amine starting material is represented by Structural Formula (III):
Ri and R in Structural Formulas (II) and (III) are as defined in Structural Formula (I) ; and reacting the aryloxy propanolamine ester with an acid to form a quarternary ammonium salt of the aryloxy propanolamine ester; and optionally crystallizing the ammonium salt.
Another embodiment of the present invention is a method of preparing an aryloxy propanolamine amide represented by Structural Formula (IV) :
Ri is as defined for Structural Formula (I);
R and R5 are independently -H, a C1-C6 straight or branched chain alkyl group or, taken together with the nitrogen atom to which they are bonded, a non-aromatic heterocyclic group. Preferably, R and R5 are both -H.
The method comprises the step of amidating an aryloxy propanolamine ester quarternary ammonium salt represented by Structural Formula (I) with an amine NHR4R5; where R4 and R5 are as described for Structural Formula (IV) .
Another embodiment of the present invention is a method of preparing an aryloxy propanolamine carboxylic acid or a carboxylate salt thereof from an aryloxy propanolamine ester represented by Structural Formula (I) . The aryloxy propanolamine carboxylic acid is represented by Structural Formula (V) :
where Ri is as described for Structural Formula (I) .
The method comprises the step of hydrolyzing the -COOR2 group of the aryloxy propanolamine ester or quarternary ammonium salt thereof; and isolating the aryloxy propanolamine carboxylic acid or carboxylate salt by: a) precipitating the aryloxy propanolamine carboxylic acid out of an aqueous basic or acidic solution by adjusting
the pH of the solution to the isoelectric point of the aryloxy propanolamine carboxylic acid; or b) absorbing the aryloxy propanolamine carboxylic acid or carboxylate salt onto an ion exchange resin, washing the ion exchange resin with a solvent suitable for dissolving impurities, and eluting the aryloxy propanolamine carboxylic acid or carboxylate salt from the ion exchange resin.
Preferred carboxylate salts of the compound represented by Structural Formula (V) include alkali metal carboxylate salts such as the sodium carboxylate salt, the potassium carboxylate salt and the lithium carboxylate salt.
Another embodiment of the present invention is a method of preparing an aryloxy propanolamine amide represented by Structural Formula (IV) . The method comprises the step of amidating an aryloxy propanolamine carboxylic acid represented by Structural Formula (V) or a carboxylate salt thereof with NHR4R5. Preferably, the aryloxy propanolamine carboxylic acid or carboxylate salt is reacted with NHR4R5/ a peptide coupling agent and a non-nucleophilic base. R4 and R5 are as described above for Structural Formula (IV) .
Yet another embodiment of the present invention is a compound represented by Structural Formula (VI):
and quarternary ammonium salts of the compound represented by Structural Formula (VI) are also included; where R
6 is -COOR
8; and
R7 is an aryl group or a substituted aryl group. Examples of suitable aryl groups for R7 are shown in Structural Formulas (VII) -(XX). In a preferred embodiment, R7 is not a carbazolyl or substituted carbazolyl group. In
another preferred embodiment, R7 is a substituted or unsubstituted bicylic heteroaryl group or a substituted or unsubstituted tricylic heteroaryl group with at least two ring nitrogen atoms. More preferably, R7 is a substituted or unsubstituted indolyl group, e.g., a 4-indolyl group;
R8 is a C1-C6 straight or branched chain alkyl group, a C7-C9 substituted or unsubstituted aralkyl group. Preferably, R8 is methyl or ethyl.
In yet another preferred embodiment of the present invention, R6 and R7 in the compound represented by Structural Formula (VI) are defined as follows:
Re is -COOH or a metal carboxylate. Preferred metal carboxylates include alkali metal carboxylates such as the sodium carboxylate, the potassium carboxylate and the lithium carboxylate.
R7 is an aryl group or a substituted aryl group, provided that R is not a carbazolyl or substituted carbazolyl group. Alternatively, R7 is a substituted or unsubstituted bicylic heteroaryl group or a substituted or unsubstituted tricylic heteroaryl group with at least two ring nitrogen atoms. Examples of suitable aryl groups for R7 are shown in Structural Formulas (VII) -(XII), (XIV) - (XV) and (XVII) -(XX). Preferably, R7 is a substituted or unsubstituted indolyl group, e.g., a 4-indolyl group. The reactions of the present invention can be combined to synthesize beta-3 active aryloxy propanolamine esters, acids, and amides from readily available starting materials. The reactions proceed in high yield and allow the preparation of a highly pure final product, substantially free of the mutagenic epoxide precursors used in their preparation .
DETAILED DESCRIPTION OF THE INVENTION
Aryl groups include carbocyclic aromatic groups such as phenyl, 1-naphthyl, 2-naphthyl, 1-anthracyl and 2-anthracyl, and heteroaryl groups such as N-imidazolyl , 2-imidazolyl, 2-thienyl, 3-thienyl, 2-furanyl, 3-furanyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 2-pyranyl, 3-pyranyl, 3-pyrazolyl, 4-pyrazolyl, 5-pyrazolyl, 2-pyrazinyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2- oxazolyl, 4-oxazolyl and 5-oxazolyl.
Aryl groups also include fused polycyclic heteroaryl ring systems in which a carbocyclic aromatic ring or heteroaryl ring is fused to one or more other heteroaryl rings. Two rings are "fused" when both rings contain the same two ring adjacent ring atoms. Examples include 1- benzimidazolinyl, 2-benzimidazolonyl, 1-benzimidthioazolinyl, 2-benzimidthioazolonyl, 1- carbazolyl, 2-carbazolyl, 3-carbazolyl, 4-carbazolyl, 3- indazolyl, 4-indazolyl, 5-indazolyl, 6-indazolyl, 2-benzothienyl, 3-benzothienyl, 2-benzofuranyl, 3-benzofuranyl, 2-indolyl, 3-indolyl, 2-quinolinyl, 3-quinolinyl, 2-benzothiazolyl, 2-benzooxazolyl, 2-benzimidazolyl, 2-quinolinyl, 3-quinolinyl, 1-isoquinolinyl , 3-quinolinyl, 1-isoindolyl and 3-isoindolyl .
Examples of preferred monocyclic heteroaryl groups and fused polycyclic heteroaryl groups for Ri are shown below in Structural Formulas (VII) - (XIII) :
Rings A-I are independently substituted or unsubstituted.
Ai and A are independently carbon or nitrogen
A3 and A4 are independently -0- , -S-, -NH- , -CH2-, -N(C1-C6 straight of branched alkyl group)- (e.g., -N(CH3)- or -N(CH2CH3)-) . n is 1 or 2.
Examples of more preferred monocyclic heteroaryl groups and fused polycyclic heteroaryl groups for Ri are shown below in Structural Formulas (XIV) - (XX) :
(XIV) (XV) (XVI)
Y is as described above.
An aliphatic group refers to a straight, branched chain or cyclic hydrocarbon having from one to about twenty carbon atoms . The hydrocarbon can be saturated or can have one or more units of unsaturation.
A substituted aralkyl group has one or more substituents on the aryl moiety of the aralkyl group. Non-aromatic heterocyclic rings are non-aromatic carbocyclic rings which include one, two or three heteroatoms selected from nitrogen, oxygen and sulfur in the ring that will afford a stable structure. The ring can be five, six, seven or eight-membered. Examples include 2-tetrahydrofuranyl , 3-tetrahydrofuranyl,
2-tetrahyrothiophenyl, 3-tetrahyrothiophenyl, 2-morpholino, 3-morpholino, 4-morpholino, 2-thiomorpholino, 3-thiomorpholino, 4-thiomorpholino, 1-pyrrolidinyl, 2-pyrrolidinyl, 3-pyrrolidinyl, 1-piperazinyl, 2- piperazinyl, 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 4-piperidinyl and 4-thiazolidinyl .
Suitable substituents for an aliphatic group, aryl group (carbocyclic and heteroaryl), non-aromatic heterocyclic ring or benzyl group are those which are substantially unreactive under the reaction conditions described herein and which cause minimal side reactions. Examples of suitable substituents include, for example, -OH, halogen (-Br, -Cl, -I and -F) , -OR, -OCONH2, -OCONHR, -OCONR2, -NHCOOR, -NRCOOR, -CN, -N02, -NHR, -NR2, -COR, -CONH , -CONHR, -CONR2, -SR, -S(0)R, -S02R, and -NH-C (=NH) -NH2. R is C1-C6
alkyl, benzyl or phenyl. A substituted non-aromatic heterocyclic ring can also have =0, -S , =NH or =NR as a substituent. A substituted aliphatic, substituted aryl, substituted non-aromatic heterocyclic ring or substituted benzyl group can have one, two or three substituents. For example, Rings A-I can have zero, one, two or three more substituents other than those specifically depicted in Structural Formulas (VII) - (XIII) .
Substituent groups such as primary amines, esters and aldehydes which react under the reaction conditions described herein can also be used, provided they are first converted to a protected form. Suitable protecting groups are known to those skilled in the art and are disclosed in Green and Wuts, "Protecting Groups in Organic Synthesis ", John Wiley and Sons, 1991, the teachings of which are incorporated herein by reference.
Because of the amine moiety present in the aryloxy propanolamines disclosed herein, ammonium salts of these compounds can be prepared by reacting with a suitable acid. Acids commonly employed to form such salts include inorganic acids such as hydrochloric, hydrobromic, hydroiodic, sulf ric and phosphoric acid, as well as organic acids such as para-toluenesulfonic, methanesulfonic, oxalic, para- bromophenylsulfonic, carbonic, succinic, citric, benzoic, acetic acid, and related inorganic and organic acids. Such pharmaceutically acceptable salts thus include sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, phosphate, mono-hydrogenphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate, chloride, bromide, iodide, acetate, propionate, decanoate, caprylate, formate, isobutyrate, heptanoate, propiolate, oxalate, malonate, succinate,
suberate, sebacate, fumarate, maleate, 2-butyne-l,4 dioate, 3-hexyne-2, 5-dioate, benzoate, chlorobenzoate, hydroxybenzoate, methoxybenzoate, phthalate, xylenesulfonate, phenylacetate, phenylpropionate, phenylbutyrate, citrate, lactate, hippurate, β-hydroxybutyrate, glycolate, maleate, tartrate, methanesulfonate, propanesulfonate, naphthalene-1-sulfonate, naphthalene-2-sulfonate and mandelate salts. Carboxylate salts can be formed from the aryloxy propanolamines disclosed herein which have a carboxylic acid functional group by reacting with a suitable base. Such salts include those derived from inorganic bases such as ammonium and alkali and alkaline earth metal hydroxides, carbonates, bicarbonates, and the like, as well as salts derived from basic organic amines such as aliphatic and aromatic amines, aliphatic diamines, hydroxy alkylamines, and the like. Such bases useful in preparing the salts of this invention thus include ammonium hydroxide, potassium carbonate, sodium bicarbonate, calcium hydroxide, sodium hydroxide, lithium hydroxide, potassium hydroxide, methylamine, diethylamine, ethylenediamine, cyclohexylamine, ethanolamine and the like.
In the structural formulas depicted herein, the bond by which a chemical group or moiety is connected to the remainder of the molecule or compound is indicated by the following symbol:
For example, the' corresponding symbol in Structural Formulas (VII) -(XX) indicates the bond by which the depicted aryl group is connected to the 3-oxygen atom of the compound represented by Structural Formulas (I)-(V).
As used herein, "crystallizing" refers to providing a solvent or solvent mixture in which the aryloxy propanolamine compound is highly soluble and in which a quarternary ammonium salt(s) thereof is insoluble or only slightly soluble. The compound is dissolved in the solvent or solvent mixture and then converted to the quarternary ammonium salt by the addition of at least one equivalent of the appropriate acid, after which the quarternary ammonium salt precipitates. To minimize contamination of the precipitated product, between about 1.0 and about 1.1 equivalents of acid are preferably used. Impurities present in the aryloxy propanolamine are preferably highly soluble in the solvent or solvent mixture, resulting in a precipitated salt which is purified relative to the free base prior to precipitation. More preferably, the precipitated compound is crystalline.
Isolation by acidic (or basic) extraction refers to dissolving a compound with a basic functional group such as an amine (or a compound with an acidic functional group, such as a carboxylic acid) in aqueous or alcoholic acid (or aqueous or alcoholic base, in the case of a compound with an acidic functional group) . The aqueous solution can then be washed with organic solvents that are not miscible with water to remove organic impurities. The pH of the solution is then adjusted to the isoelectric point of the compound, thereby precipitating the compound or allowing its extraction into an organic solvent.
Isolation by precipitation at the isoelectric point refers to adjusting the pH of an aqueous solution of a compound with both an acidic and basic functional group
(e.g., an amino acid) to its isoelectric point, thereby causing the compound to precipitate from solution. "Isoelectric pH" is the pH at which the compound is electrically neutral and therefore least soluble in aqueous
solution. Preferably, impurities present in the compound are highly soluble at the isoelectric point. As a result, the precipitated compound is purified relative to the compound prior to precipitation. More preferably, the precipitated compound is crystalline.
Optionally, a compound with both an acidic and basic functional group can be isolated by both by acidic (or basic) extraction and by precipitation at its isoelectric point . The present invention includes solvates of the compounds of Structural Formula I and the physiologically acceptable salts thereof. A particular compound of the present invention or a physiologically acceptable salt thereof may form solvates with water or common organic solvents. Such solvates are included within the scope of compounds of the present invention.
The preparation of the aryloxy propanolamine ester represented by Structural Formula (I) from the epoxide and amine represented by Structural Formulas (II) and (III), respectively, is generally carried out in a solvent at room temperature to the reflux temperature of the reaction mixture. Temperatures of 40-140 °C are generally preferred. The solvents that may be used in the reaction include: alcoholic solvents, such as methanol, ethanol or isopropanol, (with the preferred solvent corresponding to the particular ester being used in the reaction to prevent transesterification) ; aromatic solvents, such as benzene, toluene, xylenes, chlorobenzene, dichlorobenzene, other haloaromatics, nitrobenzene, benzonitrile, or trifluoromethylbenzene; or dipolar aprotic solvents, such as dimethylsulfoxide (DMSO) , N,N-dimethylacetamide (DMAC) or N,N-dimethylformamide; or other solvents where the reagents are soluble and the temperature can be elevated to the range previously described. Equimolar amounts of epoxide and amine
can be used. Alternatively, up to about a five fold excess of one reagent is used. Preferably, however, between about 0.9 to about 2.0 equivalents of amine relative to epoxide is preferred. Examples of specific conditions for preparing aryloxy propanolamine esters by this method are provided in Examples 6, 10, 17 and 18.
Aryloxy propanolamine esters, prepared as described above, can be isolated by any suitable means, including by converting to a suitable quarternary ammonium salt and crystallizing, as described above.
For the aryloxy propanolamine ester represented by Structural Formula (XXI), examples of suitable ammonium salts include the 4-hydroxybenzoate salt, the phthalate salt, the 2-hydroxynicotinate salt, the glycolate salt and the terephthalate .salt . The ammonium 4-hydroxybenzoate salt is preferred. Ethyl acetate is an example of a suitable crystallization solvent.
For the aryloxy propanolamine ester represented by Structural Formula (XXII) , examples of suitable quarternary ammonium salts include the 4-hydroxybenzoate salt. Examples of suitable crystallization solvents include ethyl acetate, anisole, chlorobenzene, 1, 2-dichloroethane, ethyl butyrate, ethyl formate, dimethoxyethane, ethanol, ethyl acetate, isobutyl butyrate, toluene and mixtures thereof. Preferred solvents include ethanol and mixtures of ethanol with chlorobenzene, anisole or dichloromethane .
(XXII) Examples of suitable quarternary ammonium salts for the aryloxy propanolamine ester represented by Structural Formula (XXIII) include the quarternary ammonium salicylate salt, the quarternary ammonium glycolate salt, the quarternary ammonium benzoate salt or the quarternary ammonium naphthoate salt of the aryloxy propanolamine. The quarternary ammonium salicyclate salt is preferred. Examples of suitable crystallization solvents include ethyl acetate, iso-propyl acetate, acetonitrile, methanol and ethanol. Preferred solvents include ethyl acetate for the ammonium salicylate salt, acetonitrile for the ammonium glycolate and naphthoate salts, and iso-propyl acetate for the ammonium benzoate salt.
(XXIII) Examples of suitable quarternary ammonium salts for the aryloxy propanolamine ester represented by Structural
Formula (XIV) include the quarternary ammonium benzoate salt. Examples of suitable crystallization solvents include iso-propyl acetate.
(XIV) Amine starting materials represented by Structural Formula (III) can be prepared by reacting a phenoxide salt of the alkylamino phenol represented by Structural Formula (XXV) with a 2-halopyridine ester represented by Structural Formula (XXVI) :
R2 in Structural Formula (XXVI) is as described for Structural Formula (I) . X is a halo group. The coupling reaction and the preparation of the compound represented by Structural Formula (XXV) can be carried out according to procedures described in the aforementioned WO 97/10822 to Bell et al . , WO 98/09625 to Crowell et al . , U.S. Patent No. 5,808,080 and U.S. Patent No. 6,046,227 and is described in detail in Examples 1, 2, 3 and 4.
Generally, the reaction is carried out by mixing the alkylamino phenol with a base in the presence of the
2-halopryidine ester. Equimolar amounts of the starting materials are preferably used. However, molar excesses up to about five or ten fold of one starting material relative to the other can be used. The coupling is performed by mixing the amino phenol with a base in a suitable solvent or solvent system. The reaction can be carried out at temperatures as low as room temperature, but is preferably carried out by heating the mixture at reflux while
azeotropically removing water formed during the deprotonation step. The 2-halopyridine ester is then added and the reaction continued until the reaction is complete. Suitable solvents include dipolar aprotic, ethereal, and aromatic solvents, as well as combinations thereof.
Dipolar aprotic solvents include solvents such as DMSO, N,N- dimethylacetamide, N-methylpyrrolidinone, 1, 3-dimethyl-2- imidazolindinone (DMI) , 1, 3-dimethyl-3 , 4 , 5 , 6-tetrahydro- 2 ( 1H) -pyrimidinone (DMPU) with a lower boiling solvent to azeotropically remove water such as benzene, toluene, isooctane, xylenes or other solvents capable of forming binary azeotropes with water but which are inert under the reaction conditions. Other suitable compounds for azeotropic removal of water may be found in Advances In Chemistry Series 116 - Azeotropic Data III, American
Chemical Society: Washington D.C., 1973. Ethereal solvents include tetrahydrofuran, dioxane and 1 , 2-dimethoxyethane. Aromatic solvents include benzene, toluene, chlorobenzene, anisole and 1, 2-dichlorobenzene . Preferred is the use of an aromatic solvent such as chlorobenzene containing 0.1-10 equivalents of a dipolar aprotic solvent such as N,N- dimethylacetamide . Suitable bases include alkali metal alkoxides, such as alkali metal methoxides, ethoxides and tert-butoxides (preferably corresponding to the alkyl group of the ester to prevent transesterification) , alkali metal hydroxides, such as lithium hydroxide, sodium hydroxide and potassium hydroxide, or alkali metal carbonates, such as Na2C03 or K2C03. Although excesses of base up to at least about ten fold can be used, preferred is the use of stoichiometric quantities of hydroxide or alkoxide bases or 1.5-5 equivalents of K2C03. Specific conditions for carrying out this reaction are provided in Examples 3, 4, 14 and 15.
The preparation of the epoxide starting material represented by Structural Formula (II) can be carried out by
reacting RiOH with (2S) - (+) -glycidyl 3-nitrobenzenesulfonate in an inert solvent (e.g., acetone, methyl ethyl ketone, methyl isobutylketone, dimethyl sulfoxide, N,N- dimethylacetamide (DMAC) or DMF) and in the presence of a base. Although about equimolar amounts of the starting materials are preferred, molar excess up to about five to about ten of one starting material relative to the other can also be used. Suitable bases include non-nucleophilic bases such as potassium carbonate, sodium carbonate and alkali metal alkoxides; potassium carbonate is preferred. Although an excess of base can be used, between about 1.05 and about 1.50 equivalents of K2CO3 are preferred. The reaction is carried out at temperatures ranging from about ambient temperature to about 40 °C, preferably at about 30 °C. Other sulfonate esters can also be used (e.g., tosylate, nosylate or mesylate) as well as halides such as epibromohydrin or epichlorohydrin. The nosylate is preferred.
The amidation of the aryloxy propanolamine ester quarternary salt represented by Structural Formula (I) to form the aryloxy propanolamine amide represented by Structural Formula (IV) (hereinafter the "amidation reaction") can be carried out by any suitable means for amidating an ester, e.g., by procedures disclosed in March, "Advanced Organic Chemistry" , 3rd edition, John Wiley & Sons (1985), pages 375-76; Larock, R.C., Comprehensive Organic Transformations, VCH: New York, 1989, pp. 987-988, and references cited therein. The entire relevant teachings of these references are incorporated herein by reference.
Preferably, the amidation reaction is carried out in an alcoholic solvent, a dipolar aprotic solvent (e.g., DMSO, DMF or DMAC) or a polar aprotic solvent (e.g., acetonitrile). Methanol is a preferred solvent.
Preferred temperatures and pressures for the amidation reaction range from room temperature to about 70° C and from
atmospheric pressure to about 100 psi, respectively. To increase the reaction rate and yield, the amidation can be carried out at elevated temperatures and pressures, e.g., about 35° C to about 55 °C, and about 20 psi to about 80 psi, respectively.
Equimolar amounts of amine and aryloxy propanolamine ester can be used. Generally, however, the reaction is carried out with up to about a 50 fold excess of amine relative to the aryloxy propanolamine ester, and preferably with between about 2 and about 20 equivalents of amine.
Optionally, the amidation reaction can be carried out in the presence of one or more reagents known to accelerate the reaction rate and/or decrease the formation of byproducts in the amidation reaction when added in substoichiometric, stoichiometric, or superstioichiometric amounts. Such reagents are referred to herein as an "amidation catalyst". Typically, between about zero and about six equivalents of an amidation catalyst are used, depending on the starting materials. Examples of suitable amidation catalysts include bases such as alkoxides (e.g., lithium methoxide, sodium methoxide, magnesium ethoxide and potassium t-butoxide) . Other amidation catalysts include nucleophilic salts such as potassium iodide and sodium cyanide and bidentate reagents which can act both as proton donors and proton acceptors. Examples include potassium phosphate monobasic, 2-hydroxypyridine, 4-hydroxypyridine, cytidine, 2-hydroxypryrimidine. Combinations of catalysts can also be used, e.g., 2-hydroxypyridine in combination with an alkoxide base. Examples include three equivalents of 2-hydroxypyridine in combination with one equivalent of lithium methoxide, sodium methoxide, potassium tert- butoxide, potassium methoxide or magnesium ethoxide.
The preparation of aryloxy propanolamine amide (XXVII) from aryloxy propanolamine ester or quarternary ammonium
salt thereof (XXI) or (XXII) and ammonia in methanol can generally be carried out without the addition of an amidation catalyst. Specific conditions for this reaction are described in Example 12.
(XXVII) However, the reaction between aryloxy propanolamine ester (XXIII) or (XXIV) and ammonia in methanol to form the aryloxy propanolamine represented by Structural Formula (XXVIII) typically requires at least 72 hours and the use of elevated temperatures and pressures to go to completion, for example 45° C and 50 psi (Example 25) . As a consequence, this reaction is generally carried out in the presence of an amidation catalyst (Example 26) . 2-Hydroxypyridine/lithium methoxide is a preferred catalyst for the preparation of this amide. Preferred quantities of lithium methoxide in the reaction range from between about 0.7 and about 1.5 equivalents, more preferably between about 1.0 and about 1.2 equivalents relative to the aryloxy propanolamine ester. Preferred quantities of 2-hydroxypyridine range from between about 2.3 and about 4.3 equivalents, more preferably between about 3.0 and about 3.6 equivalents relative to the aryloxy propanolamine ester. Use of the preferred catalyst allows the reaction temperature to be lowered to between about 20° C and about 30° C. Specific conditions for carrying out this reaction are described in Examples 25 and 26.
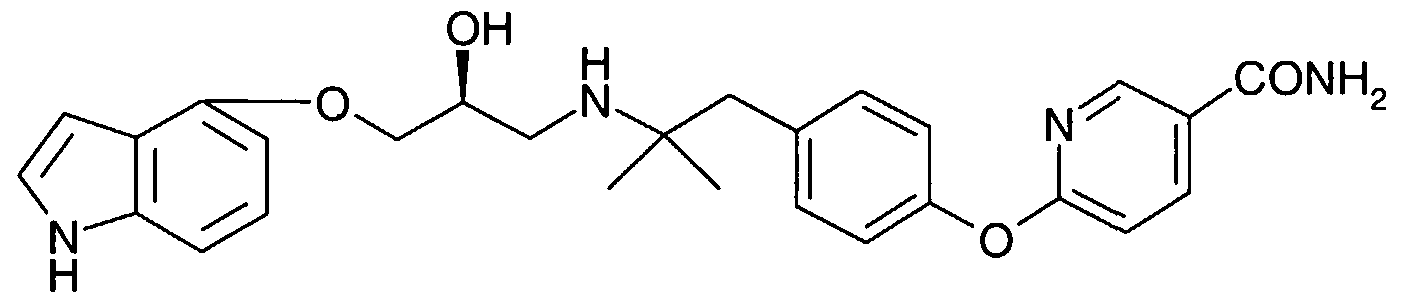
(XXVIII) Aryloxy propanolamine amides, prepared as described above, can be isolated by any suitable means, including by converting to a suitable quarternary ammonium salt and crystallizing, as described above.
For the aryloxy propanolamine amide represented by Structural Formula (XXVII), preferred quarternary ammonium salts include the quarternary ammonium acetate salt, the quarternary ammonium chloride salt, the quarternary ammonium oxalate salt and the quarternary ammonium 4-hydroxybenzoate salt. Of these, the quarternary ammonium acetate salt is more preferred. Methyl ethyl ketone is the preferred crystallization solvent for the ammonium acetate salt, and 7:1 ethyl acetate/ethanol is also a preferred crystallization solvent for this salt. Ethanol is a preferred crystallization solvent for the quarternary ammonium chloride salt; ethanol and methanol are preferred crystallization solvents for the quarternary ammonium oxalate salt; and 2:1 ethyl acetate/ethanol is a preferred crystallization solvent for the quarternary ammonium 4- hydroxybenzoate salt.
For the aryloxy propanolamine amide represented by Structural Formula (XXVIII) , examples of suitable quarternary ammonium salts include the quarternary ammonium glycolate salt, the quarternary ammonium benzoate salt, the para-hydroxybenzoate salt and the quarternary ammonium para- toluate salt. The quarternary ammonium glycolate salt is preferred. Examples of suitable crystallization solvents for ammonium glycolate salt include methanol, ethanol, ethanol/ethyl acetate mixtures and methanol/ethanol
mixtures . Ethanol is preferred. Examples of suitable crystallization solvents ' include isopropanol and ethanol/ethyl acetate mixtures for the benzoic acid salt; ethyl acetate for the para-hydroxybenzoic acid salt; and ethanol/ethyl acetate mixtures for the toluic acid salt. The hydrolysis of the aryloxy propanolamine ester or quarternary ammonium salt thereof represented by Structural Formula (I) to form the carboxylic acid represented by Structural Formula (V) or the carboxylate salt thereof can be carried out by any suitable means, including by procedures disclosed in Larock, R. C. Comprehensive Organic Transformations; VCH: New York, 1989, pp. 981-985; March, "Advanced Organic Chemistry" , 3rd edition, John Wiley & Sons (1985), pages 375-76 and references cited therein, the entire relevant teachings of which are incorporated herein by reference. This reaction is referred to herein as the "hydrolysis reaction" .
Preferably, the hydrolysis reaction is carried out in alcoholic base such as lithium hydroxide, sodium hydroxide or potassium hydroxide in methanol or ethanol. Methanolic sodium hydroxide is preferred. The hydrolysis can be carried out with one equivalent of base relative to aryloxy propanolamine ester. Alternatively, an excess of base, for example, an excess up to about ten to about twenty fold can be used to form the carboxylate salt. Preferably, the excess of base is between about zero and about 30 percent. Generally, the reaction temperature varies between room temperature and the reflux temperature of the solvent, and is typically typically 40-70 °C. The carboxylic acid product (or carboxylate salt thereof) is isolated by conventional means, for example, by removal of the solvent in vacuo .
The aryloxy propanolamine carboxylic acid or carboxylate salt thereof can be isolated by basic (or acidic) extraction, e.g., by dissolving in aqueous base
(e.g., aqueous sodium hydroxide, potassium hydroxide or lithium hydroxide) , and/or by precipitation at its isoelectric point. Preferably, the aqueous solution is extracted with one or more organic solvents which are not miscible with water to remove impurities. The pH of the aqueous solution can then adjusted to its isoelectric point with aqueous acid (e.g., aqueous HCl, H2S0 , acetic acid or a sulfonic acid) , thereby causing the aryloxy propanolamine carboxylic acid to precipitate. It will be appreciated the isolation also performs a purification of the desired precipitate .
The aryloxy propanolamine carboxylic acids or carboxylate salts thereof can also be isolated by using either cationic ion-exchange resins, such as as AMB 15 (H form) , 50WX2-400 (H form) , or IR50S (H form) and eluting with an alcoholic solution of an alkali metal acetate, such as 6% (w/v) sodium acetate in methanol; or by using anionic ion-exchange resins, such as IRA900 (chloride form) or A21 (acetate form) and eluting with a basic solution of water and either alcohol or acetonitrile. It will be appreciated the isolation also performs a purification of the desired eluate .
Examples of specific conditions for the hydrolysis of an aryloxy propanolamine ester or quarternary ammonium salt thereof represented by Structural Formula (I) and the isolation (and purification) of the resulting carboxylic acid product or carboxylate salt are provided Examples 27- 32.
The amidation of the aryloxy propanolamine carboxylic acid represented by Structural Formula (V) to form the aryloxy propanolamine amide represented by Structural Formula (IV) is carried by any suitable means, including by methods disclosed in Bodansky, M. ; Bodansky, A. "The Practice of Peptide Synthesis", Springer-Verlag: New York,
1984; and "The Peptides : Analysis, Synthesis, Biology"; Gross, E., Meienhofer, J., Eds.; Academic Press, Inc.: New York; Vol. 1; 1979, the entire relevant teachings of which are incorporated herein by reference. Preferably, this amidation is carried out by reacting the aryloxy propanolamine carboxylic acid with NHRR5 in the presence of a peptide coupling agent and a non-nucleophilic base. R and Rs are as described above in Structural Formula (IV) . The purification of the aryloxy propanolamine amide is described above.
Peptide coupling agents are well known in the art and react with carboxylic acid functional groups to convert hydroxyl portion of the carboxylic acid into a good leaving group. Thus, the hydroxyl portion of the carboxylic acid is more easily displaced by nucleophiles after reaction with a peptide coupling agent than in its absence. Examples of peptide coupling, carboxylic acid activating agents include 2-chloro-4, 6-dimethoxy-l, 3, 5-triazine (CDMT) , carbonyldiimidazole and related reagents, 1, 1-carbodiimide reagents such as 1, 3-dicyclohexylcarbodiimide and mixed anhydrides such as those described in "The Practice of Peptide Synthesis" and "The Peptides: Analysis, Synthesis, Biology", cited above. CDMT is a preferred peptide coupling agent . Non-nucleophilic bases are also well known in the art. Except for acid base reactions, non-nucleophilic bases are substantially unreactive during the reaction between a peptide coupling agent and a carboxylic acid. Examples of non-nucleophilic bases include tertiary bases such as triethyl amine, ethyl diisopropyl amine and N-methyl morpholine (ΝMM) . Other non-nucleophilic bases include DBU and DBΝ. ΝMM is a preferred non-nucleophilic base.
The amidation of the aryloxy propanolamine carboxylic acid or carboxylate salt can be carried out using equimolar
amounts of the aryloxy propanolamine carboxylic acid (or salt thereof), NHR3R4, peptide coupling agent and non- nucleophilic base, or an excess of any one or more of the reagents up to about five to about ten fold relative to the aryloxy propanolamine. Preferably, an excess of between zero and about 20% of each reagent is used relative to the aryloxy propanolamine carboxylic acid. Generally, the reaction is carried out between 0° C and about 50° C, preferably at ambient temperature. Suitable solvents include ethereal solvents (e.g., diethyl ether, tetrahydrofuran, glyme and 1, 4-dioxane) , dipolar aprotic solvents (e.g., dimethyl sulfoxide, and dimethyl formamide) and polar aprotic solvents (e.g., acetonitrile and nitromethane) . Ethyl acetate and dimethylformamide are preferred solvents. An example of specific conditions for the amidation of an aryloxy propanolamine carboxylic acid represented by Structural Formula (V) and the purification of the resulting product is provided in Example 33.
Except as otherwise described herein, reaction concentrations of the reaction described herein generally range from about 0.005 M to about 5.0 M, typically from about 0.1 M to about 1.0 M.
Scheme 1 is a synthetic schematic showing the preparation of the aryloxy propanolamine amide quarternary ammonium acetate salt represented by Structural Formula (XXVII) from readily available starting materials. The synthesis uses the methods of the present invention and goes through an aryloxy propanolamine ester quarternary ammonium parahydroxybenzoate salt synthetic intermediate.
Scheme 2 is a synthetic schematic showing the preparation of the aryloxy propanolamine amide quarternary ammonium chloride salt represented by Structural Formula (XXVII) from readily available starting materials. The synthesis uses the methods of the present invention and goes through an aryloxy propanolamine ester quarternary ammonium parahydroxybenzoate salt synthetic intermediate.
The individual optically active isomers of the compounds prepared by the present invention may be prepared from their respective optically active precursors by the procedures described above, or by resolving the racemic mixtures. This resolution can be carried out by derivatization with a chiral reagent followed by chromatography or by repeated crystallization. Removal of the chiral auxiliary by standard methods affords substantially optically pure isomers of the compounds of the present invention or their precursors. Further details regarding resolutions can be obtained in Jacques, et al . , Enantiomers, Race ates, and Resolutions, John Wiley & Sons, 1981. The compounds employed as initial starting materials in the synthesis of the compounds of this invention are well known and, to the extent not commercially available, are readily synthesized by standard procedures commonly employed by those of ordinary skill in the art. The invention is illustrated by the following examples which are not intended to be limiting in any way.
EXEMPLIFICATION
Example 1 - Preparation of 4- (2-Methyl-2-Nitropropyl) Phenol
Potassium tert-butoxide (29.6 grams, 264 mmol) was added to a solution of 2-nitropropane (260 L, 2.90 mol) and. 4-hydroxybenzyl alcohol (65.0 grams, 524 mmol) in diglyme (260 mL) at room temperature with mechanical stirring. During the addition the reaction temperature increased from 25 °C to 39 °C. The reaction mixture was heated to reflux
and stirred for 6 hours at ca . 137 °C, using a Dean-Stark trap to remove the water as it was formed (total volume of distillate 28 mL, 7 mL aqueous phase) . After cooling to room temperature, deionized H20 (325 mL) and ethyl acetate (520 mL) were added to the reaction solution. The phases were separated and the organic phase was washed with deionized H20 (2 x 325 mL) . The organic phase was concentrated by rotary evaporation at 78 °C to give 181.2 grams of an oil. This oil was dissolved in methanol (65 mL) for use in the next reaction. The concentration of the resulting solution was determined by 1H NMR analysis to be 56.3% by weight (99.6% yield) .
Example 2 - Preparation of the Acetate Salt of 4- (2-Amino-2-
Methylpropyl ) phenol
To a N2-degassed solution of 4- (2-methyl-2- nitropropyl) phenol (45.0 grams, 230 mmol) in MeOH (450 mL) was added 5% Pd/C (13.5 grams of 50% water-wet catalyst, 15% by weight on a dry-basis) . The mixture was pressurized to 35-40 psi with hydrogen and heated to 60 °C with vigorous agitation. When the reaction was complete (ca. 6 hours), the mixture was cooled to room temperature and the catalyst was carefully removed by filtration through Hy-Flow filter aid. The cake was washed with 50 °C methanol (135 mL) and the combined filtrates were concentrated by rotary evaporation to a net weight of ca . 120 grams. The concentrate was diluted with ethyl acetate (500 mL) and a solution of acetic acid (14.2 grams, 235 mmol) in ethyl acetate (250 mL) was added to the resulting solution over 30 minutes. The resulting slurry was stirred for 2 hours at room temperature. The slurry was filtered and the solid was washed with ethyl acetate (2 x 100 mL) . The product was
vacuu -dried at 65 °C/5 Torr for 24 hours to give 46.5 grams (89.6%) of a white crystalline solid, mp 209-215.9 (dec).
Example 3 Synthesis of 2- [4- (2-amino-2- methylpropy1 ) phenoxy] -3-pyridinecarboxy1ic acid, methyl ester
N, N-dimethylacetamide (600 mL) , isooctane (167 mL) , 4- (2-amino-2-methylpropyl)phenol (66.7 grams, 0.212 mol), and powdered K2C03 (61.5 grams, 0.445 mol, 300 mesh) were combined. The mixture was brought to reflux for 2 hours during which time H0 was removed via a Dean-Stark trap. The mixture was cooled to below reflux and methyl 2- chloronicotinate (40.0 grams, 0.233 mol) was added over 10 minutes. The system was brought back to reflux for 3 hours. The solution was cooled and filtered through Celite. The solvents were removed by rotary evaporation at 80 °C/5 Torr. The resulting thick oil was dissolved in methyl tert-butyl ether (450 mL, MTBE) and extracted with H20 (400 mL) . The organic layer was extracted with a combination of H0 (400 mL) and 1 Ν HCl (24 mL) . The aqueous layer was extracted with MTBE (200 mL) and then combined with MTBE (400 mL) and brought to pH 12 with 1 Ν ΝaOH. The organic layer was extracted with a saturated solution of ΝaCl (200 mL) and the solvents were removed by rotary evaporation to give 50.0 grams (97.6% purity, 76.6% yield) as a light yellow oil. Anal. Calcd for Cι7H20Ν2O3 : C, 67.98; H, 6.71; N, 9.33. Found: C, 67.79; H, 6.69; N, 9.31.
Example 4 - Synthesis of 2- [4- (2-amino-2- methylpropyl)phenoxy] -3-pyridinecarboxylic acid', ethyl ester.
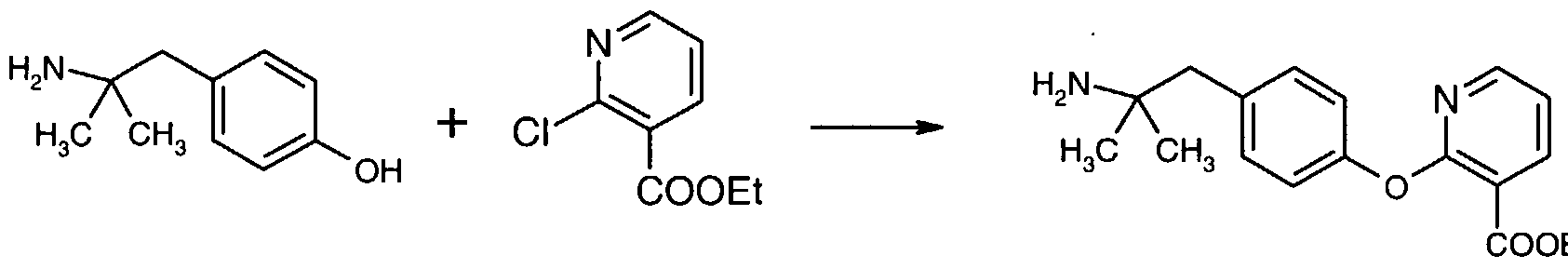
Et 4- ( 2-Amino-2-methylpropyl) phenol (55.18 grams, 244.9 mmol) was added to 5.05 N KOH (97.2 mmol, 2.0 equiv). The mixture was warmed to 50 °C to give a homogeneous yellow solution. Chlorobenzene (1104 mL) and N,N-dimethylacetamide (10.7 grams, 122 mmol) were added and the mixture was heated to reflux (ca. 100 °C) . The water was removed azeotropically via a Dean-Stark trap. At ca . 125 °C a solid began to form. When the pot temperature reached 132 °C the water had been removed and the reaction mixture was a thick but stirrable slurry (mechanical stirring required) . The Dean-Stark trap was removed and an additional 100 mL of chlorobenzene was removed and discarded. Dry chlorobenzene (50 mL) was added to the slurry, followed by ethyl 2-chloronicotinate (50.0 grams, 269 mmol) in chlorobenzene (50 mL) . The slurry was heated at reflux until the reaction was complete (ca. 24 hours) . As the reaction progressed the slurry thinned and became beige in color. After cooling to room temperature, water (385 mL) and 1 N NaOH (25 mL, 0.1 equiv) were added to the mixture and the phases were separated. The organic phase was washed with water (285 mL) and the solution was concentrated to a net weight of 700 grams (9.83% potency by HPLC, 89% yield) for use in the next reaction.
Example 5 - Preparation of 2- [4- (2-amino-2- methylpropyl)phenoxy] -3-pyridinecarboxylic acid, ethyl ester, acetic acid salt.
Ethyl 2- [4- ( 2-amino-2-methylpropyl) phenoxy] -3- pyridinecarboxylate (10.3 grams, 32.8 mmol) was dissolved in ethyl acetate (52 mL) and heptane (41 mL) and the solution was heated to reflux. Acetic acid (1.97 grams, 38.8 mmol) was added, the solution was seeded, and cooled slowly to room temperature. After 30 minutes at room temperature, the slurry was cooled to 0 °C and stirred for 1.5 hours. The product was filtered, washed with cold 1:1 ethyl acetate/heptane (20 mL) , and vacuum-dried at 50 °C for 18 hours to give 10.29 g (97% purity, 81% yield), mp 122.9- 124.5 °C.
Example 6 - Preparation of (5) -2- [4- [2- [2-hydroxy-3- (1H- indol-4-yloxy)propylamino] -2-methylpropyl] - phenoxy] -3-pyridinecarboxylic acid, methyl ester.
Methanol (250 mL) , 4- [ (2S) -oxiranylmethoxy] -iH-indole (17.6 grams, 93.2 mmol) and methyl 2- [4- (2-amino-2- methylpropyl) phenoxy] -3-pyridinecarboxylate (49.0 grams,
163.1 mmol) were combined and heated at 70 °C for 24 hours. The amount of methanol was reduced by rotary evaporation until a total weight of 110 grams remained. Twice, ethyl acetate (150 mL) was added and the solvents were removed by
rotary evaporation until a total of 120 grams remained. Ethyl acetate (460 mL) and H20 (300 mL) were added and the pH was adjusted to 6.8 with glacial acetic acid (5.3 mL) . After separation the organic layer was extracted with H20 (100 mL) . The solvents were removed by rotary evaporation to give 45.6 grams (86.5% purity, 89% yield) as a beige foam. This material was used without further purification for the salt formations, vida infra . A portion of the glycolic acid salt was free based to give purified ester for conversion to the corresponding amide and for characterization. For this purpose methylene chloride (125 mL) , ethyl acetate (25 mL) , and the glycolate salt (10.5 grams, 18.6 mmol) were combined. 1 N NaOH (100 mL) was added and the slurry was stirred until two translucent layers persisted. The organic layer was separated, washed with a saturated solution of NaCl (50 mL) , then dried over MgS0 . After filtration, removal of the solvents by rotary evaporation afforded 9.1 grams (98.6% purity, 99% yield) as a white foam, mp 48-53 °C . Anal. Calcd for C28H3ιN305: C, 68.69; H, 6.38; N, 8.58. Found: C, 67.52; H, 6.39; N, 8.44.
Example 7 - General screening procedure for preparation of carboxylic acid salts of methyl ( S) -2- [4- [2- [2- hydroxy-3- (lH-indol-4-yloxy)propylamino] -2- methylpropyl] -phenoxy] -3-pyridinecarboxylate.
For each screening, methyl { S) -2 - [4- [2- [2-hydroxy-3- (lH-indol-4-yloxy) propylamino] -2 -methylpropyl] -phenoxy] -3- pyridinecarboxylate (100.0 mg, 0.204 mmol , 86.5% purity, vide supra) was dissolved in ethyl acetate (1.0 mL) . To each was added 1.0 equivalent of one of the carboxylic acids listed below. After stirring at 80 °C for 30 minutes and then overnight at 24 °C, the solids were isolated by centrifuge without washing. The yield and purity for each screening experiment are given below:
Carboxylic Acid Methyl ester Yield salt (%)
(HPLC Area %)
4-hydroxybenzoic acid 95.5 95.1 phthalic acid 92.1 88.3
2-hydroxynicotinic acid 91.8 98.8 glycolic acid 94.1 83.0 terephthalic acid 92.4 85.2
Example 8 - Preparation of (S) -2- [4- [2- [2-hydroxy-3- (1H- indol-4-yloxy) propylamino] -2-methylpropyl] - phenoxy] -3-pyridinecarboxylic acid, methyl ester, 2-hydroxyacetic acid salt. Methyl ( S) -2- [4- [2- [2-hydroxy-3- (lff-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylate (2.21 grams, 4.51 mmol, 86.5% purity, vide supra) was combined with ethanol (0.5 mL) and placed in a 70 °C bath. After a clear viscous material was obtained, ethyl acetate (15.0 mL) was added. Glycolic acid (0.343 grams, 4.51 mmol) was dissolved in ethanol (0.5 mL) and ethyl acetate (1.0 mL) at 50 °C . The glycolic acid solution was added to the solution, rinsing with ethyl acetate (6.0 mL) . The solution was seeded and its temperature lowered to 45 °C. After stirring overnight, the temperature was lowered to 24 °C and the beige mixture was stirred an additional 24 hours. The solid was isolated by vacuum filtration, using the filtrate to facilitate the transfer, and washed 3 times with ethyl acetate (3.0 mL) . After drying 3 hours at 50 °C/5 Torr, 1.70 grams (97.5% purity, 84% yield) was obtained as a white solid, mp 122-125 °C.
Anal. Calcd for C30H35N3O6 : C, 63.71; H, 6.24; N, 7.42. Found: C, 62.94; H, 6.10; N, 7.33.
Example 9 - ( S) -2- [4- [2- [2-hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylic acid, methyl ester, 4- hydroxybenzoic acid salt Methyl (S) -2- [4- [2- [2-hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylate (526 mg, 1.08 mmol, 86.5% purity, vide supra) , 4-hydroxybenzoic acid (129 mg, 1.08 mmol), and ethyl acetate (5.3 mL) were combined. The mixture was brought to reflux giving a yellow solution. Reflux was continued for 1.5 hours during which time a white precipitate formed. The heat was turned off and over 1 hour the temperature decreased to 37 °C, after which the bath was removed. After 2 hours additional stirring, the solid was isolated by vacuum filtration, using the filtrate to facilitate in the transfer, and washed twice with ethyl acetate (1.5 mL; 1.0 mL) . After drying overnight at 50 °C/5 Torr, 488 mg (94.8% purity, 88 % yield) was obtained as a white solid, mp 147- 149 °C. Anal. Calcd for C35H37N308: C, 66.97; H, 5.94; N, 6.69. Found: C, 66.52; H, 5.92; N, 6.33.
Example 10 - ( S) -2- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylic acid, ethyl ester, 4- hydroxybenzoic acid salt 4- [ (2S)-0xiranylmethoxy]-lH-indole (9.00 grams, 47.6 mmol) was added to a solution of ethyl 2- [4- (2-amino-2- methylpropyl) -phenoxy] -3-pyridinecarboxylate (162.8 grams of a 10.1% w/w solution in chlorobenzene, 52.3 mmol) and the resulting solution was heated at reflux for 37 hours. When the epoxide had been consumed, the solution was cooled to 80
°C, and a 50 °C solution of 4-hydroxybenzoic acid (6.57 grams, 47.6 mmol) in 2B-3 ethanol (34 grams) was added in one portion. The homogeneous solution was seeded at 70 °C and cooled slowly to room temperature with stirring. After stirring for 1 hour at 0 °C the slurry was filtered, washed with chlorobenzene (3 x 50 mL) , and vacuum-dried at 70 °C for 18 hours to give 20.82 grams (68% yield) of product as an off-white solid, mp 172.4-175 °C. Anal. Calcd for C29H33N305 • C7H603: C, 67.38; H, 6.12; N, 6.54. Found: C, 67.18; H, 6.07; N, 6.77.
Example 11 - (5) -2- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylic acid, ethyl ester The 4-hydroxybenzoic acid salt of the title compound
(17.1 grams, 26.6 mmol) was added with stirring to a mixture of methyl tert-butyl ether (200 mL, MTBE) and 1 N NaOH (75 mL) . When all of the initial solid was dissolved a small amount of a dark green solid remained which was easily removed by filtration (Whatman #1 paper) . The organic phase was washed with brine (2 x 30 mL) and dried over anhydrous MgS0 . The drying agent was removed by filtration. The MTBE solvent was exchanged with methanol by concentrating the solution using rotary evaporation, redissolving the residue in MeOH, and reconcentrating again. This process was repeated and the residue was dissolved in anhydrous methanol and used directly in the subsequent reactions.
Example - 12 (S) -2- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -
3-pyridinecarboxamide Methanol (10.0 mL) and methyl ( S) -2- [4- [2- [2-hydroxy-3- (lϋ'-indol-4-yloxy)propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylate (2.00 grams, 4.08 mmol) were combined.
The vessel was then pressurized and vented 3 times with 25 psig ammonia. The internal vessel pressure was then brought to 50 psig with ammonia and the yellow solution stirred at 40 °C for 19 hours. The solvent and ammonia were removed by rotary evaporation to give 1.87 grams (97% yield) as an off-white solid, mp 75-78 °C. Anal. Calcd for C30H27NO4: C, 68.34; H, 6.37; N, 11.81. Found: C, 66.69; H, 6.29; N, 11.71.
Example 13 (S) -2- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxamide, acetic acid salt
A solution of ethyl ( S) -2 - [4- [2- [2-hydroxy-3- (lH-indol- 4-yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylate (13.2 grams, 26.1 mmol) in methanol (66 mL) was poured into a vessel that was then pressurized and vented 3 times with 25 psig ammonia. The internal vessel pressure was then brought to 50 psig with ammonia and the yellow solution stirred at 24 °C for 18 hours. The solvent and ammonia were removed by rotary evaporation until 25 grams remained in the flask. Ethanol (100 mL, 2B-3) was added and the solvent again removed by rotary evaporation until 26 grams remained. This addition of ethanol and
evaporation was repeated 3 more times for the solvent exchange into ethanol and removal of ammonia. After the last evaporation, the contents of the flask weighed 25.0 grams and was taken to be the theoretical 12.5 g of freebase and 12.5 grams of ethanol. Ethyl acetate (87.7 mL) and H0 (1.0 mL) were added and the solution was brought to reflux. Acetic acid (1.73 grams, 28.8 mmol) was added and the solution was seeded. After 1 hour, heating was removed from the white mixture. After stirring at 24 °C for 1 hour, the white solid was collected by vacuum-filtration and washed twice with 7:1, ethyl acetate: ethanol (20 mL) and once with with 7:1 ethyl acetate : ethanol (10 mL) . Vacuum drying overnight at 50 °C/5 Torr gave 11.6 g (96.9% purity, 97% yield) as a white solid, mp 157-158 °C. Anal. Calcd for C7H30NO4: C, 65.15; H, 6.41; N, 10.48. Found: C, 65.01; H, 6.28; N, 10.26.
Example 14 - 6- [4- (2-Amino-2-methylpropyl) phenoxy] -3- pyridinecarboxylic acid, methyl ester
4- (2-Amino-2-methylpropyl) phenol (3.00 grams, 18.2 mmol), methyl 6-chloronicotinate (3.27 grams, 19.1 mmol), powdered K2C03 (3.76 grams, 27.2 mmol, 300 mesh), N,N- dimethylacetamide (60 mL, DMAC), and toluene (15 mL) were combined and heated to reflux. The water formed during the reaction was removed azeotropically via a Dean-Stark trap. After about 2 hours the internal temperature reached 154 °C and the reaction was complete. The slurry was cooled to 30 °C and filtered. The filter cake was washed with DMAC and the combined organic fractions concentrated by rotary
evaporation at 75 °C . The resulting oil was dissolved in ethyl acetate (50 mL) , and washed with water (30 mL) . The phases were separated, and the aqueous phase was extracted with ethyl acetate (20 mL) after some saturated NaCl solution (10 mL) was added to facilitate phase separation. The combined organic fractions were washed with water (2 x 30 mL) and a saturated solution of NaCl (30 mL) , then dried over Na2S0 . After filtration the solution was concentrated to give 4.60 grams (95% purity by HPLC, 80% yield) of a tan flocculent solid, mp 112.4-114.9 °C. Anal. Calcd for
Cι7H20N2O3: C, 67.98; H, 6.71; N, 9.33. Found: C, 67.81; H, 6.63; N, 9.24.
Example 15 - 6- [4- (2-Amino-2-methylpropyl) phenoxy] -3- pyridinecarboxylic acid, ethyl ester
4- (2-Amino-2-methylpropyl) henol acetic acid salt (45.1 grams, 0.200 mol), powdered K2C03 (58.1 grams, 0.420 mol, 300 mesh) , and isooctane (140 mL) were added sequentially to
N,N-dimethylacetamide (500 mL, DMAC) , and the resulting mixture was heated to reflux under a Dean-Stark trap until water production almost ceased (ca. 2 hours, 121 °C internal temp). A solution of ethyl 6-chloronicotinate (39.0 grams, 0.210 mol) in DMAC (50 mL) was added over 15 minutes and the reaction was heated at reflux, while continuing to remove any water or alcohol as formed until the reaction was deemed complete by HPLC (ca. 2 hours) . The slurry was cooled to room temperature and filtered through a 1" pad of Celite, and the cake was rinsed with methyl tert-butyl ether (2 x 75 mL, MTBE) . The filtrate was then concentrated by rotary
evaporation to a net weight of ca. 134 grams. The resulting oil was dissolved in MTBE (315 mL) and washed with water (315 mL) . The product was extracted from the organic phase with 1 N HC1 (220 mL) and water (140 mL) . Ethyl acetate (315 mL) was added to the aqueous phase and the mixture basified with a solution of Na2C03 (1.2 equiv in 135 mL H0) while stirring. Caution must be exercised to avoid foaming. The phases were separated and the organic phase was washed with 10% w/w solution of NaCl (135 mL) . The solvent was removed by rotary evaporation, the oil was dissolved in methanol
(135 mL) , and the solvent was concentrated again. The solid was dried overnight at room temperature to afford 61.0 grams (97% purity by HPLC, 94% yield) which was used without further purification, mp 65.0-67.3 °C.
Example 16 - 6- [4- (2-Amino-2-methylpropyl) phenoxy] -3- pyridinecarboxylic acid, methyl ester (via transesterification of ethyl ester)
A solution of sodium methoxide (1.04 grams, 19.2 mmol) in methanol (30 mL) was added to a solution of ethyl 6- [4-
(2-amino-2-methylpropyl) phenoxy] -3-pyridinecarboxylate (60.5 grams, 192 mmol) in methanol (250 mL) . The solution was heated for 2 hours at 35 °C until equilibrium was achieved. Acetic acid (1.39 grams, 23.1 mmol) was added to neutralize the sodium methoxide, which is deleterious in the next step. The solution was cooled to room temperature and used as a solution (277.2 grams net weight) in the next step. A yield of 96% was determined by concentration and analysis of an aliquot. HPLC shows a 98:2 ratio of methyl to ethyl esters, respectively.
Example 17 - ( S) -6- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxylic acid, ethyl ester
4- [ (2S) -Oxiranylmethoxy] -lH-indole (1.00 grams, 5.28 mmol) was added to a solution of ethyl 6- [4- (2-amino-2- methylpropyl) phenoxy] -3-pyridinecarboxylate (3.32 grams, 10.6 mmol) in 2B-3 ethanol (30 mL) and the resulting solution was heated at reflux for 8 hours. The solution was concentrated by rotary evaporation and the tacky solid dissolved in ethyl acetate (50 mL) . The excess starting amine was removed by sequential extraction with 5.5 mL 1 N HC1/15 mL water, water (2 x 10 mL) , saturated NaCl solution (10 mL) , and an acidic solution of saturated brine (9 mL saturated NaCl, 3 mL 1 N HC1) . It was critical to keep the pH ca . 7.0-7.2 during the extractions to give the best partitioning of the starting amine from the product. The solution was dried over Na2S0 . After filtration, the solvent was removed by rotary evaporation and the product was purified by flash chromatography (110 grams Si02, 350 mm column, 95:5 CHC13/Methanol (7 x 100 mL) followed by 95:5:0.5 CHCl3/Methanol/NH40H (9 x 100 mL) to give 1.96 grams (74%) of an amorphous foam. Anal. Calcd for C2gH33N305: C, 69.17; H, 6.61; N, 8.34. Found: C, 69.16; H, 6.48; N, 8.22.
Example 18 - ( S) -6- [ 4- [ 2 - [ 2 -Hydroxy-3 - ( lff-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxylic acid, methyl ester, 2- hydroxybenzoic acid salt 4- [ (2S) -Oxiranylmethoxy] -lH-indole (21.0 grams, 111 mmol) was added to a solution of methyl 6- [4- (2-amino-2- methylpropyl) phenoxy] -3-pyridinecarboxylate (58.3 grams, 194 mmol) in methanol (291 mL) prepared via the transesterification process (and thus contains ca . 2% of the corresponding ethyl ester) . The solution was heated at reflux for 17 hours until the epoxide was consumed. One half of the solution was retained and the other half was worked up as follows. The solvent was removed by rotary evaporation (net weight 79.5 grams) and the oil was dissolved in ethyl acetate (100 grams) . The slightly turbid solution was concentrated to a net weight of 94.9 grams, and diluted with additional ethyl acetate (210 mL) . Water (190 mL) and glacial acetic acid (2.00 g) were added and the mixture stirred for 10 minutes. The pH should be ca . 7.2. The phases were separated and the organic phase was extracted with dilute acetic acid solution containing some NaCl (100 mL H20, 0.50 grams acetic acid, 10 mL saturated NaCl solution) . The organic phase was washed sequentially with H20 (100 mL) , 1 N NaOH (20 mL) , and a saturated solution of NaCl (20 mL) . The solution was concentrated by rotary evaporation, dissolved in ethyl acetate (200 mL) and filtered to remove a small amount of solid. The solution was concentrated to a net weight of 198 grams and heated to 55 °C. Salicylic acid (7.65 grams, 55.4 mmol) was added, then the solution was seeded and stirred for 1.5 hours at 75
°C. The slurry was cooled slowly and was stirred overnight at room temperature. The solid was isolated by filtration and the cake was washed with ethyl acetate (2 x 50 mL) . The product was vacuum-dried at 50 °C/5 Torr for 36 hours to
give 25.11 grams (98.3% purity (contains 1.4% ethyl ester), 72% yield) of a white solid, 167.2-169.0 °C. Anal. Calcd for C35H37N308: C, 66.97; H, 5.94; N, 6.69. Found: C, 66.80; H, 5.91; N, 6.85.
Example 19 - Recovery of methyl 6- [4- (2-amino-2- methylpropyl ) phenoxy] -3-pyridinecarboxylate for recycle Methylene chloride (100 mL) was added to the combined acidic extracts from the above procedure and the pH adjusted to 10 with 5 N NaOH (8 mL) . The phases were separated, then the aqueous phase was adjusted to pH 10 and extracted with CH2C12 (100 mL) . The combined organic fractions were washed with H20 (2 x 50 mL) and were dried with Na2S04. After filtration, the solution was concentrated to a solid by rotary evaporation and vacuum-dried at 50 °C/5 Torr overnight to give 13.2 grams (95% purity, 102% yield). Anal. Calcd for Cι7H20N2O3 : C, 67.98; H, 6.71; N, 9.33. Found: C, 68.06; H, 6.95; N, 9.32.
Example 20 - General procedure for the preparation of methyl (S)-6-[4-[2- [2-hydroxy-3- (lff-indol-4- yloxy) propylamino] -2-methylpropyl] phenoxy] -3- pyridinecarboxylate via transesterification of the ethyl ester in methanol
A solution of the ethyl ester (100 mg, 0.20 mmol) in methanol (1 mL) was treated with 10 mol% of a base such as sodium methoxide, potassium carbonate, or lithium methoxide for 1-3 hours at 25 °C to 40 °C until equilbrium was established (ca. >99:1 methyl ester/ethyl ester). The base was then neutralized with acetic acid and the solvent removed by rotary evaporation. The crude product can be purified as the 2-hydroxybenzoic acid salt, as described above .
Example 21 - ( S) - 6 - [ 4- [ 2 - [ 2 -Hydroxy-3 - ( lH-indol-4 - yloxy) propylamino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxylic acid, methyl ester, 2- naphthoic acid salt
2-Naphthoic acid (351.2 mg, 2.04 mmol) was added to a solution of the methyl ester free base (1.00 grams, 2.04 mmol) in acetonitrile (10.0 mL) at ca. 60 °C. The mixture was cooled slowly to room temperature and stirred for 1 hour at 25 °C and 1 hour at 0 °C. The product was filtered, washed with acetonitrile, and dried to afford 0.98 grams (97% purity, 77% yield) of a white solid.
Example 22 - ( S) -6- [4- [2- [2-Hydroxy-3- (lff-indol-4- yloxy) ropylamino] -2-methylpropyl] -phenoxy] -
3-pyridinecarboxylic acid, methyl ester, 2- hydroxyacetic acid salt Glycolic acid (155.1 mg, 2.04 mmol) was added to a solution of the methyl ester free base (1.00 grams, 2.04 mmol) in acetonitrile (10.0 mL) at ca . 60 °C. The mixture was cooled slowly to room temperature and stirred for 1 hour at 25 °C and 1 hour at 0 °C . The product was filtered, washed with acetonitrile, and dried to afford 1.06 grams (95% purity, 94% yield) of a white solid, mp 169.5-171.7 °C. Anal. Calcd for C28H3ιN305»C2H403 : C, 63.71; H, 6.24; N, 7.43. Found: C, 63.15; H, 6.20; N, 7.71.
Example 23 - ( S) -6- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxylic acid, methyl ester, benzoic acid salt Benzoic acid (4.46 grams, 36.5 mmol) was added to a solution of the methyl ester free base (18.8 grams, 38.4 mmol) in iso-propyl acetate (180 mL) at ca . 40 °C. The
mixture was seeded, cooled slowly to room temperature, and stirred for 48 hours at 25 °C and 1 hour at 0 °C. The product was filtered, washed with cold iso-propyl acetate (50 mL) , and dried to afford 14.1 grams (98% purity, 60% yield) of a white solid, mp 148.6-152.0 °C. Anal. Calcd for C28H3ιN3 05»C7H602: C, 68.72; H, 6.10; N, 6.87. Found: C, 68.22; H, 6.00; N, 7.12
Example 24 - (S) -6- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylic acid, ethyl ester, benzoic acid salt Benzoic acid (0.244 grams, 2.00 mmol) was added to a solution of the ethyl ester free base (1.00 grams, 1.99 mmol) in iso-propyl acetate (12.5mL) at ca . 50 °C. The mixture was cooled slowly to room temperature, and stirred for 18 hours at 25 °C and 2 hours at 0 °C. The product was filtered, washed with cold iso-propyl acetate (3 mL) , and dried to afford 0.52 grams (42% yield) of a white solid, mp 139.0-144.0 °C. Anal. Calcd for C29H33N305»C7H602 : C, 69.10; H, 6.28; N, 6.72. Found: C, 68.75; H, 6.53; N, 6.73.
Example 25 - (S) -6- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxamide
Methanol (20.0 mL) and methyl ( S) -6- [4- [2- [ [2-hydroxy- 3- ( lH-indol-4-yloxy) propyl ] amino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxylate (4.50 grams, 9.19 mmol) were combined. The vessel was purged and vented 3 times and then brought to 30 psig with ammonia. The pressurized solution was stirred at 24 °C for 84 hours. The solvent and ammonia were removed by rotary evaporation. The beige foam was further dried overnight at 50 °C/5 Torr to give 4.22 grams (97% purity, 97% yield) as a beige solid, mp 83-85 °C. Anal. Calcd for C27H30NO : C, 68.34; H, 6.37; N, 11.81; 0, 13.49. Found: C, 69.27; H, 6.36; N, 11.82; 0, 13.60.
Example 26 - ( S) -6- [4- [2- [2-Hydroxy-3- (lff-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxamide
Methanol (10.0 mL) , 2-hydroxypyridine (2.40 grams, 25.3 mmol) and lithium methoxide (0.320 grams, 8.43 mmol) were stirred together until an amber solution was obtained. Methanol (9.8 mL) was used to assist in adding methyl (S)-6- [4- [2- [2-hydroxy-3- ( lJf-indol-4-yloxy) propylamino] -2- methylpropyl] -phenoxy] -3-pyridinecarboxylate (3.75 grams, 7.66 mmol) . The vessel was purged and vented 3 times and then brought to 50 psig with ammonia. The pressurized solution was stirred at 24 °C for 38 hours. The solvent and ammonia were removed by rotary evaporation. The beige foam was dissolved in ethyl acetate (100 mL) and extracted sequentially with 1 N NaOH (100 mL) , H20 (100 mL) and a saturated solution of NaCl (100 mL) . The organic layer was dried over MgS04 and, after filtration, the solvents were removed by rotary evaporation. Further drying overnight at 45 °C/5 Torr gave 3.54 grams (99 % yield) as a beige solid, mp 74-78 °C. HRMS Calcd 475.2345, found 475.2341.
Example 27 - (S) -6- [4- [2- [2-Hydroxy-3- ( lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylic acid
Methyl (5)-6-[4-[2- [2-hydroxy-3- (lff-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylate (1.00 grams, 2.04 mmol) was added to a solution of 1 N NaOH (5.11 mL, 5.11 mmol) in methanol (10 mL) . The solution was heated for 1 hour at 60 °C until the reaction was complete by HPLC. The solution was neutralized with 1 N HCl while maintaining the temperature at ca . 35 °C. Since some tacky solid formed, the temperature was increased to 40 °C, where the tacky material redissolved and a white precipitate formed. The mixture was stirred for 20 minutes at 55 °C, 60 minutes at 25. °C, and 60 minutes at 0 °C. The slurry was filtered and the cake was washed with cold 1:1 methanol/water . The product vacuum-dried at 50 °C to give 0.91 grams (94% yield) of a white powder, mp 243 (dec) . Anal. Calcd for C27H29N305 : C, 68.20; H, 6.15; N, 8.84. Found: C, 68.17; H, 6.16; N, 8.83.
Example 28 - (S) -6- [4- [2- [2-Hydroxy-3- (lff-indol-4- yloxy) ropylamino] -2-methylpropyl] -phenoxy]
3-pyridinecarboxylic acid
Ethyl ( S) -6- [4- [2- [2-hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylate (1.00 grams, 1.99 mmol) was added to a solution of 1 N NaOH (2.48 mL, 2.48 mmol) in 2B-3 ethanol (10 mL) . The solution was heated for 1 hour at 70 °C until the reaction was complete by HPLC. The solution was neutralized with 1 N HCl (ca. 2.48 mL) while maintaining the temperature at 50 °C. The mixture was seeded and stirred over the weekend at room temperature. The slurry was filtered and the product was vacuum-dried at 50 °C to give 0.74 grams (84% yield) of a white powder, mp 243 (dec). Anal. Calcd for C27H29N305 : C, 68.20; H, 6.15; N, 8.84. Found: C, 66.57; H, 6.54; N, 8.68. Elemental analysis was inaccurate due to partial hydration of the solid.
Example 29 ( S) -6- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] 3-pyridinecarboxylic acid, potassium salt
Potassium trimethylsilylsilanolate (32.1 mg, 90% purity, 2.05 mmol) was added to a solution of methyl (S)-6- [4- [2- [2-hydroxy-3- ( lH-indol-4-yloxy) propylamino] -2- methylpropyl] -phenoxy] -3-pyridinecarboxylate (100.3 mg, 0.205 mmol) in THF (2 mL) . The solution was purged with N2, wrapped in foil and stirred at 25 °C for 18 hours. Analysis by HPLC showed the reaction was complete. The solution was filtered through Millipore 0.5 μ LCR filter and concentrated to dryness to give a gray-white powder. The solid was triturated with CH2C1 and vacuum-dried at 50 °C to give 91.8 mg of a gray-white powder. No attempt was made to purify the salt.
Example 30 - ( S) -6- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) ropylamino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxylic acid, sodium salt
A I M solution of sodium trimethylsilylsilanolate (0.22 mL, 0.22 mmol) in THF was added to a solution of methyl (_?)- 6- [4- [2- [2-hydroxy-3- (lH-indol-4-yloxy) propylamino] -2- methylpropyl] -phenoxy] -3-pyridinecarboxylate (99.9 mg, 0.204 mmol) in THF (1.8 mL) . The solution was purged with N , wrapped in foil and stirred at 25 °C for 24 hours. Analysis by HPLC showed the reaction was 75% complete. The mixture was concentrated and the resulting solid was triturated with CH2C12 to remove unreacted starting material and NaOTMS. The remaining solid was vacuum-dried at 50 °C to give 67.2 mg (66% yield) of a gray-white powder. No attempt was made to purify the salt.
Example 31 - (S) -2- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylic acid
A solution of 1 N NaOH (0.77 mL, 0.77 mmol) was added to a solution of methyl ( S) -2 - [4- [2- [2-hydroxy-3- (lff-indol- 4-yloxy) propylamino] -2-methylpropyl] phenoxy] -3- pyridinecarboxylate (300.0 mg, 0.613 mmol) in methanol (3.0 mL) . The solution was heated for 1 hour at 60 °C until the reaction was complete. At the same temperature, water (1.6 mL) and 1 N HCl (ca. 0.77 mL, 0.77 mmol) were added sequentially until the pH was 7-8. The solution was removed from the oil bath and upon cooling the product precipitated. The solid was isolated by filtration and the cake was washed with water (2 x 3 mL) . The product was vacuum-dried at 50 °C overnight to give 250 mg (86% yield) of a white powder, mp 243 dec. Anal. Calcd for C27H29N305 : C, 68.20; H, 6.15; N, 8.84. Found: C, 67.61; H, 6.06; N, 8.68.
Example 32 - ( S) -2 - [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) ropylamino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxylic acid
A solution of 1 N NaOH (26.3 mL, 26.3 mmol) was added to a solution of ethyl (S) -2- [4- [2- [2-hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] phenoxy] -3- pyridinecarboxylate (10.6 grams, 21.1 mmol) in methanol (110 mL) . The solution was heated for 4 hours at 60 °C until the reaction was complete. At the same temperature, water (80 mL) and 1 N HCl (ca. 26 mL, 26 mmol) were added sequentially until the pH was 7-8. The solution was removed from the oil bath and upon cooling the product precipitated. The solid was isolated by filtration and the cake was washed with water (2 x 100 mL) . The product was vacuum-dried at 50 °C overnight to give 8.11 grams (81% yield) of a white powder, mp 243 dec. Anal. Calcd for C27H29N305 : C, 68.20; H, 6.15; N, 8.84. Found: C, 67.81; H, 6.13; N, 8.72.
Example 33 6- (4- [2- [ ( (2S) -3-carbazol-4-yloxy-2- hydroxypropy1 ) amino] -2- methylpropyl ] phenoxy) pyridine-3 -carboxamide
N, N-Dimethylformamide (2.0 mL) and 6- (4- [2- [ ( (2S) -3- carbazol-4-yloxy-2-hydroxypropyl) a ino] -2- methylpropyl ] phenoxy) -3-pyridinecarboxylic acid (223.0 mg, 0.424 mmol) were combined at 24 °C. After adding 6-chloro- 2 , 4-dimethoxy-l, 3 , 5-triazine (93.1 mg, 0.530 mmol), the yellow solution was cooled to 0 °C. 4-Methylmorpholine (47.2 mg, 0.467 mmol) was added and the solution was further cooled to -68 °C. Ammonia gas was lightly passed over the solution surface for 3 seconds during which time a 5 °C exotherm occurred. Cooling was removed and 2 layers were established by adding ethyl acetate (7.0 mL) and H0 (4.0 mL) . The organic layer was extracted with 1 Ν ΝaOH (5.0 mL) , washed with a saturated solution of ΝaCl (5.0 mL) , and then dried over MgS04. After filtration, the solvents were removed by rotary evaporation. Further drying overnight at 45 °C/5 Torr gave 150.6 mg (95.0% purity, 65% yield) as a beige solid, mp 94-98 °C. HRMS Calcd 525.2502, found 525.2491.
Example 34 - 6- (4- [2- [ ( (2S) -3-carbazol-4-yloxy-2- hydroxypropyl) amino] -2- methylpropyl] phenoxy) pyridine-3 -carboxamide, glycolic acid salt
The free base of the title compound (500 mg, about 1 mmol) was dissolved in 3.0 ml absolute ethanol with warming to 35-40°C. Glycolic acid (0.076 g, 1 mmol), dissolved in 1.0 ml of ethanol, was added to the reaction mixture with a 1.0 ml ethanol rinse in. Crystallization began as the addition finished. This mixture was stirred at 38-40°C for 90 minutes. The heat was turned off and stirring continued 90 minutes at ambient temperature. The crystals were filtered and washed with 3.5 ml of ethanol. This large volume of wash was needed to remove the crystals from the flask. Less may be used on a larger scale. Vacuum drying at 55°C for 18 hours and without heat for two days left 1.1% EtOH by NMR. m.p. = 171-1722C. Yield was an estimated 90% uncorrected for solvents. HPLC analysis gave an uncorrected purity >99%.
HPLC analysis conducted as follows: Gradient system - Solvent A: ACN; Solvent B: pH 2 phosphate buffer (1.0 liter DI H2O, 1.0 g KH2PO4, 3.0 ml cone. H3PO4). Gradient curve: 20%A/80%B to 80%A/20%B over 20 min. Flow rate: 2 ml/min. Column: Zorbax RX C18, 4.6 x 250 mm, heated to 40°C. UV detector wavelength: 240 nm.
!H NMR (300 MHz, DMSO-d6): 11.1 (s, 1H) , 8.64 (d, J = 2.6 Hz, 1H) , 8.26 (dd, J = 8.7, 2.6 Hz, 1H) , 8.05 (br s, 1H) , 7.48 (br s, 1H) , 7.26 (d, J = 8.7 Hz, 2H) , 7.21 (t, J = 2.6 Hz, 1H) , 7.1-6.95 (m, 5H) , 6.49 (m, 2H) , 6.25 (br s,
3H) , 4.13 (m, 3H) , 3.73 (s, 2H) , 3.13 (d, J = 11.3 Hz, 1H) , 2.96 (dd, J = 11.3, 7.7 Hz, 1H) , 2.86 (s, 2H) , 1.14 (br s, 6H) ppm.
13C NMR (75 MHz, DMSO-dg): 175.1, 165.8, 164.6, 152.0, 151.9, 147.5, 139.4, 137.3, 133.6, 131.7, 125.2, 123.5,
121.7, 120.7, 118.4, 110.6, 105.0, 100.0, 98.3, 70.1, 67.3, 60.6, 55.8, 44.6, 43.7, 24.5 ppm; IR (KBr) 1086, 1250, 1291, 1374, 1484, 1508, 1568, 1588, 1598, 1610, 1622, 1684, 3182, 3303 cm-1.
MS(FIA) m/z 475; UV (EtOH) 265 (15884), 242 (17682), 217 (51352) nm(epsilon) ; OR (MeOH) -10.71(589) sp. rot. (nm) .
EA calculated for C27H30 4O4 ^2^03 : C, 63.26; H,/ 6.22; N, 10.18. Found: C, 65.34; H, 5.70; N, 10.93.
Example 35 - 6- (4- [2- [ ( (2S) -3-carbazol-4-yloxy-2- hydroxypropy1 ) amino] -2- methylpropyl] phenoxy) pyridine-3-carboxamide, benzoic acid salt
A sample of free base of the title compound (2.373 grams, 5 mmol) was dissolved in 2.5 ml absolute ethanol/35 ml ethyl acetate with warming to 35-40°C. Benzoic acid (0.611 grams, 5 mmol) was dissolved in 8 ml ethyl acetate and this solution added to the free base solution with a 4.5 ml ethyl acetate rinse in. The resulting clear solution was seeded and stirred at 38-40°C until the flask filled with crystals, about 1 to 2 hours. The heat was turned off and the mixture stirred overnight at ambient temperature. The crystals were filtered and washed with 27 ml of 5% ethanol in ethyl acetate. Vacuum drying at 55°C removed all the ethanol but left 2.3% ethyl acetate by NMR. Vacuum drying at 65°C reduced the ethyl acetate to 1.55% by NMR. M.p.=107- 1102C. Yield was an estimated 83%. HPLC analysis gave an uncorrected purity >99%.
Example 36 - 6- (4- [2- [ ( (2S) -3-carbazol-4-yloxy-2- hydroxypropyl ) amino] -2- methylpropyl ] phenoxy) pyridine-3 -carboxamide, para-toluic acid salt
A sample of the free base of the title compound (0.475 grams, 1 mmol) was dissolved in 0.25 ml absolute EtOH/7 ml ethyl acetate with warming to 35-40°C. p-Toluic acid (0.611 grams, 5 mmol) was dissolved in 0.25 ml ethanol/2 ml ethyl
acetate and this solution added to the free base solution with a 0.5 ml ethyl acetate rinse in. The resulting clear solution was seeded and stirred at 38-40°C until the flask filled with crystals, after about 1 hour. The heat was turned off and the mixture stirred 3 hours at ambient temperature. The crystals were filtered and washed with 5.5 ml of 5% ethanol in ethyl acetate. Vacuum drying at 50°C for 22 hours removed all the ethanol and left < 0.05% ethyl acetate by NMR. M.p. =107-1102C . Yield was an estimated 80% uncorrected for solvents. HPLC analysis gave an uncorrected purity >99%.
Example 37 - 6- (4- [2- [ ( (2S) -3-carbazol-4-yloxy-2- hydroxypropyl ) amino] -2- methylpropyl] henoxy) pyridine-3-carboxamide,
4-hydroxybenzoic acid salt
The free base of the title compound (2.37 grams, 5 mmol) and 4-hydroxybenzoic acid (0.69 grams, 5 mmol) were dissolved in 15 ml absolute ethanol at ambient temperature. The resulting clear solution was seeded with 4- hydroxybenzoate salt and stirred at ambient temperature. After 20 minutes the crystallization mixture became very thick and was diluted with an additional 15 ml of ethanol. This mixture was stirred 2 hours at ambient temperature.
The crystals were filtered and washed with 7 ml of ethanol. A 700 mg sample of the undried crystals was set aside for X- ray analysis. The remainder was vacuum dried at 65°C, 20 hours, to 0.11% ethanol and then at 50°C, 16 hours, to 0.08% ethanol by NMR. Powder X-rays showed undried and dried crystals are very different polymorphs . Dried crystal m.p.=130-135eC. Yield was estimated at about 81%. HPLC analysis gave an uncorrected purity > 99%.
Example 38 (S) -6- [4- [2- [ [2-Hydroxy-3- ( lH-carbazol-4- yloxy) ropyl] amino] -2-methylpropyl] -phenoxy] 3-pyridinecarboxamide
The title compound was prepared from methyl (S)-6-[4- [2- [ [2-hydroxy-3- ( lH-carbazol-4-yloxy) propyl] amino] -2- methylpropyl] -phenoxy] -3-pyridinecarboxylate according to procedures described in Example 25.
Example 39 - ( S) -2 - [4- [2- [2-Hydroxy-3- ( lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylic acid, ethyl ester, 4- hydroxybenzoic acid salt
4- [ (2S) -Oxiranylmethoxy]-lH-indole (8.00 grams, 42.3 mmol was added to ethyl 2- [4- (2-amino-2 -methylpropyl) - phenoxy] -3-pyridinecarboxylate (14.4 grams, 46.5 mmol) in 2B-3 ethanol (80 mL) and the resulting solution was heated at reflux for 20 hours until the epoxide had been consumed. The solution was cooled to 70-75 °C, and a 70-75 °C solution of 4-hydroxybenzoic acid (5.9 grams, 42.3 mmol) in 2B-3 ethanol (20 mL) was added in one portion. The homogeneous solution was seeded and stirring continued at 70-75 °C for 1 hour. The mixture was cooled to 26 °C and stirred for 1
hour, then cooled to 5 °C and stirred for an additional hour. The solid was collected by filtration, washed with 2B-3 ethanol (45 mL) , and vacuum-dried at 50 °C for 45 hours to give 21.8 grams (80.4% yield) of product as an off-white solid, mp 174-176 °C .
Example 40 - ( S) -2- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylic acid, ethyl ester To the 4-hydroxybenzoic acid salt of the title compound (40.0 grams, 62.3 mmol) were added with stirring methyl tert-butyl ether (350 mL, MTBE) and methyl alcohol (20 mL) . Stirring was continued for 90 minutes until the solid was well dispersed, then IN sodium hydroxide (160 mL) and deionized water (40 mL) were added. Once the solid was dissolved the layers were separated, and the organic phase was extracted twice with a solution of deionized water (120 mL) and sodium chloride (8.0 g) . After cooling to 0-5 °C and filtering through glass paper the organic layer was concentrated to 80 mL at 40 °C and 24-28 inches Hg. Methyl alcohol (160 mL) was added and the volume again reduced to 80 mL . The total volume was brought back to 240 mL with methyl alcohol and this solution was used directly in the subsequent reactions.
Example 41- (S) -2- [4- [2- [2-Hydroxy-3- (lH-indol-4- yloxy) propylamino] -2-methylpropyl] -phenoxy] - 3-pyridinecarboxamide, acetic acid salt A solution of ethyl ( S) -2- [4- [2- [2-hydroxy-3- (ltf-indol- 4-yloxy) propylamino] -2-methylpropyl] -phenoxy] -3- pyridinecarboxylate (31.4 grams, 62.3 mmol) in methyl alcohol (264 mL) was pressurized and vented 3 times with 5 psig ammonia. The internal vessel pressure was then brought to 5 psig with ammonia and the yellow solution stirred at 40
°C for 22 hours. The volume was concentrated to 60 mL at 30-40 °C and 25 inches Hg. Methyl ethyl ketone (450 mL, MEK) was added, and the volume reduced to 60 mL at 40-50 °C and 25 inches Hg. MEK was again added (200 mL) and the volume reduced to 60 mL . At 15-20 °C the contents were brought back to a total volume 300 mL using MEK, filtered using a 20 micron fritted glass filter funnel, then rinsed with MEK to give a total filtrate volume of 310 mL . The solution was heated to 65 °C and a solution of glacial acetic acid (3.75 g, 62.4 mmol) in MEK (15 mL) at 65 °C was added. After seeding the mixture was stirred at 65 °C for 90 minutes, then allowed to cool to 25 °C with stirring over 14 hours. The mixture was cooled to 3 °C and stirred for 1 hour. The slurry was collected by filtration, washed with MEK (90 mL) , and vacuum-dried at 70 °C for 46 hours to give 30.3 grams (90.9% yield) of product as an off-white solid, mp 156-158 °C.
Example 42 - Determination of the amount of 4-[(2S)- oxiranylmethoxy] -lH-indole in aryloxy propanolamines Reference standard solutions containing 0.2, 0.45, 2.0, 4.5 μg/mL of 4- [ (2S) -oxiranylmethoxy] -lH-indole in methanol were prepared. Sample solutions containing 50 mg/mL of the sample to be tested in methanol were prepared. Spiked samples were prepared by using the appropriate standard solution as the sample diluent.
The sample and references solutions were then analyzed by HPLC using the following conditions:
1) Column: 25 cm x 4.6 mm I.D. Zorbax SB-C18, 5 μm 2) Flow rate: 1.0 mL/min 3) Gradient elution:
Time (min) %A: MeOH w/ ACN %B: Buffer
0 15 85
25 85 15
35 85 15
37 15 85
47 15 85
4)UV detector: 220 nm 5) Column temperature: 30 °C 6) Injection volume: 10 μL; rinse with several volumes of methanol
Blank and calibration standard solutions were injected onto the column and the peak areas from the UV detector for each run were recorded. The samples and sample spikes were injected onto the column and the peak areas recorded for each run.
A least-squares linear regression of each standard peak response was carried out to obtain the y-intercept and slope for 4- [ (2S) -oxiranylmethoxy] -lH-indole. y = mx + b where y = peak area response of 4-[(2S)- oxiranylmethoxy] -lH-indole; x = concentration (μg/mL) of standard b = intercept m = slope or response
The amounts of 4- [ (2S) -oxiranylmethoxy] -lH-indole in the solution aliquots were calculated as follows:
((peak area - blank area) - intercept) μg/ mL = slope
The μg/g values of 4- [ (2S) -oxiranylmethoxy] -lH-indole in the samples were calculated as follows:
μg/g = (aliquot amount, μg/mL) / (sample cone, mg/mL) * 1000
The sample spike recovery was calculated as follows:
„ (meas spike aliquot - spiked amt) . _. Λ
% recovery = - - x 100 meas sample aliquot
Salts formed by the procedure described in Example 10 were analyzed and typically had less than 1 ppm of epoxide; in all lots analyzed the amount of epoxide in the salt has been <10ppm. The freebase of Example 18 was also analyzed according to the procedure described above and found to have less than 1 ppm epoxide.
EQUIVALENTS While this invention has been particularly shown and described with references to preferred embodiments thereof, it will be understood by those skilled in the art that various changes in form and details may be made therein without departing from the spirit and scope of the invention as defined by the appended claims. Those skilled in the art will recognize or be able to ascertain using no more than routine experimentation, many equivalents to the specific embodiments of the invention described specifically herein. Such equivalents are intended to be encompassed in the scope of the claims.



































































































