NOVEL ΪSOFORMS OF THE HUMAN ESTROGEN RECEPTOR-α
The present invention relates to novel isoforms of the human estrogen receptor-α (hER-α).
More particularly, the invention provides a polynucleotide sequence encoding newly identified hER-α splice variants, the encoded polypeptides, the use of such polynucleotides and polypeptides, a method for modulating the action of such polypeptides and a method for identifying molecules which modulate the actions of such polypeptides.
Estrogen receptors are important for normal development and various important physiological functions. Alterations in the expression of the ERs have been associated with pathogenesis of a number of diseases such as cancer, atherosclerosis and osteoporosis. To date, two estrogen receptors (ER-α and β), encoded by different genes, have been identified. These receptors belong to a large family of ligand- activated transcription factors, whose members regulate gene expression by interaction with cognate DNA sequences called responsive elements. ER proteins are composed of five structural domains, each having a unique function in ligand binding, gene promoter activation and activation with other factors of the general transcription apparatus. A transcriptional activation function 1 (AF 1) has been located in the A/B domain and functions as a hormone independent activator of transcription of estrogen- responsive genes such as pS2 and c-fos. In addition, it is also involved in growth factor interactions with ER-α signaling pathways. This domain has been hypothesized to activate target genes by associating with components of the core transcriptional machinery, such as co-activators and repressors. The DNA-binding domain (DBD) corresponds to region C. The D region allows ERs to alter conformation after ligand binding. It also contains nuclear localization signals as well as sequences required for dimerization of the receptor. The E region contains the ligand-binding domain (LBD), which is responsible for ligand-dependent activation of transcription. Additionally, a
hormone-dependent transcription activating function (AF2) is present in the LBD. Finally, the C-terminal F region, in addition to region E, influences AF2 via modulating the magnitude of gene transcription, but the relative contribution shows cell and promoter specificity. It is known that the estrogen receptor-α gene in human (Flouriot et al.), chicken
(Griffin et al.) and mouse, contains several promoters which are essential for the process of protein expression to be initiated. Differential promoter usage and alternative splicing events can lead to ER-α transcripts with differential transcription activity, which may help to potentiate the diverse action of estrogen through a single gene. Human and chicken ER genes generate several mRNA variants (A-F hER-α mRNAs and A-D cER-α mRNAs) by alternative splicing of upstream exons to a common splice site upstream of the translation start site. Those transcripts differ in their 5- untranslated region, therefore resulting in the translation of the same estrogen receptor protein of 66 kDa size. First data that the standard ER-α protein is in fact not the only and main isoform expressed in all tissues were obtained from the chicken system. The existence of two forms of the chicken ER-α protein (cER-α), the previously reported receptor (cER-α form I) which has a size of 66 kDa and a new form, referred to as cER-α isoform II (61 kDa in size) has been discovered. This form is lacking the 42 amino acids at its N- terminus present in the full length cER-α form I. Whereas the 66 kDa protein is the translation product of several cER-α mRNAs (Al-D), the cER-α isoform II is encoded by a new class of mRNA, which is transcribed in vivo from a specific promoter mapping the region of the previously assigned translation start site of the cER-α gene. SI nuclease mapping analysis reveals that cER-α mRNA form II is liver enriched. Both receptor forms I and II differ in their ability to modulate estrogen target gene expression in a promoter and cell-type specific manner. Whereas cER-α form I activates or represses in a strictly estrogen-dependent manner, the truncated form is characterized by a partial transactivation or repression activity in the absence
of its ligands. Sequence comparison of the N-terminal coding region of different vertebrate ER-α reveals a conservation of the translation start site of cER-α form II in other oviparous species (rainbow trout and xenopus leavis) but not in mammals (Griffin et al.). Analyzing human tissues and cell lines, the inventors have now found that the human estrogen receptor-α can exist in two alternative forms, which are generated by alternative splicing of the receptor gene and have a molecular weight of 46 and 50 kDa.
First evidence of this existence came from RT-PCR and S 1 nuclease mapping analysis showing the presence of hER-α transcripts with a deletion of exon 1. Those shorter transcripts are generated by splicing events from two upstream exons 1C or 1E/F directly to a splice acceptor site of exon 2 at position +685. This position is upstream from an internal ATG located within exon 2 (+753/758) of the hER-α coding region. In vitro translation of these two novel hER-α mRNA splice variants give rise to new ER-α proteins with the molecular size of 46 kDa (when exon 1E/F splices into exon 2) and 50 kDa (-missing the A/B domain -splicing of exon 1C to exon 2), respectively. Western blot analysis using hER antibodies mapping different domains of the receptor show the expression of both receptors (MW 66 kDa and 46 kDa) in MCF-7 cells. The differential expression of hER-α variants plays an important role in human bone. PCR analysis and SI analysis of primary human osteoblasts shows that the F- promoter is the predominantly used in bone cells. Southern blotting revealed two amplification products, a 1.8 kb fragment, corresponding to the full length receptor, when exon IF splices to the common splice site at position +163, but also a shorter PCR product. This product is due to a splicing event with the splice donor site located in exon 1E/F and a splice acceptor site located at position +753/758 in exon 2, which also leads to the expression of the hER protein variant of 46 kDa.
According to a first aspect, the invention provides a nucleic acid molecule
encoding a hER-α isoform having the sequence of SEQ ID No.: 1 or 2. The term " nucleic acid molecule" , as used herein, comprises nucleic acid sequences which are degenerative to the above nucleic acid sequences as well as deletion, addition or insertion sequence variants. Furthermore, the invention relates to polynucleotides which hybridize to the herein above described sequences under stringent conditions.
The meaning of " stringent conditions" is well known to those skilled in the art and is explained for example in Sambrook, "Molecular Cloning, A Laboratory Manual", Cold Spring Harbor Laboratory, N.Y. (1989), or in Higgins and Hames (Eds) "Nucleic acid hybridization, a practical approach" IRL Press Oxford, Washington DC (1985).
Stringent conditions for hybridation or wash are for example 0.1 x SSC, 0.1% SDS at 65°c.
The present invention also relates to a vector comprising the above mentioned nucleic acid molecule. The vector may be a plasmid, cosmid, virus, bacteriophage or any other vector which is conventionally used in genetic engineering and which, besides the DNA sequence encoding the hER-α isoforms, may comprise additional control elements, like expression control elements, suitable for eukaryotic or prokaryotic expression, including a promoter, translation initiation codon, translation and insertion site and marker genes for the selection of said vector in a suitable host cell and under suitable conditions.
A further object of the invention is a host cell transfected or transformed with the vector of the invention or a non-human host carrying the vector of the invention. The host cell may be any prokaryotic or eukaryotic cell, whereas the non-human host is preferably a mammal. According to a further aspect, the invention is directed to a protein isoform of hER-α having the amino acid sequence of SEQ ID No 3 or 4. The invention further comprises variants of the polypeptides of SEQ ID No 3 and 4, which differ from those polypeptides for conservative amino acid substitutions which involve the replacement
of a residue with another of similar characteristics, without altering the whole protein function.
Sequence variants have an identity of at least 60%, preferably 70%, more preferably 90% with the sequences of SEQ. ID No. 3 and 4. A considerable amount of evidence indicates that the different isoforms of the estrogen receptor, namely the 46 kDa, the 50 kDa and the 66kDa human ER proteins, have different activities. In particular, a number of transfection experiments shows that the 46 kDa transcript and the 50 kDa protein act in a manner which is antagonistic to that of the Estrogen Receptor in the presence of Estradiol in certain tissues and with certain promotors (Fig. 5 and 6). The experiments further demonstrate that the new protein isoform hER46 can transactivate promoters containing EREs in Hela cells in an E2-dependent manner whereas in HepG2 cells, activation of reporter gene transcription by this isoform is not detectable. Cotransfection experiments in HepG2 cells using plasmid constructs coding for the hER66 and hER46 demonstrated that hER46 can act as a trans-dominant repressor of activation in a concentration- dependent manner (Fig.6).
The distribution of the isoforms in different tissues has been investigated using RT-PCR analysis or SI nuclease mapping analysis (Fig. 3).
SI analysis of bone using a specific probe which maps to this region shows that splicing of exon 1E/F to exon 2 is detectable as a protected fragment in primary osteoblast RNA and in bone marrow RNA, where the signal consisted of approximately 50% of the total protected hER-α mRNA, but it is absent in SaOs RNA. Immunoprecipitation experiments demonstrate that both proteins are expressed in primary osteoblasts showing a ratio hER66 to hER46 of approximately 50%, whereas in MCF-7 cells, the 46 kDa protein represents approximately 10% of total
ER-α protein. The high expression level of the shorter ER protein variant compared to the full length receptor protein in bone cells might be explained by the additional expression of the 46 kDa isoform from the internal ATG due to leakiness in the start
of translation, as mentioned above. Since in osteoblasts both mechanisms can lead to the expression of a shorter protein of exactly the same size, it is difficult to distinguish whether one mechanism is more important in the formation of the hER46 protein than the other. According to a further aspect, the invention provides antibodies that recognize and bind to the hER-α protein isoforms herein disclosed. Such antibodies include, but are not limited to, polyclonal, monoclonal, chimeric, single chain, humanized, bispecific antibodies, or fragments such as Fv, Fab, Fab', or F(ab')2.
Techniques for producing and processing polyclonal antibodies are known in the art and are described, for example, in Mayer and Walker, eds., " Immunochemical Methods in Cell and Molecular Biology" , Academic Press, London (1987).
In a preferred embodiment, the antibodies can selectively distinguish the different isoforms of the receptor, and are raised to polypeptides corresponding to the N-terminal region of the protein. The antibodies of the invention are useful not only as modulator of receptor-estrogen interaction, but also in immunoassays to detect hER-α isoforms or in their purification.
A further object of the invention is a method for identifying molecules which bind to and activate or inhibit the hER-α isoforms. In a preferred embodiment, such a method is used to screen those compounds that selectively distinguish between the different receptor isoforms. The method will measure the ability of the test compound to block interaction of a ligand, preferably an estrogen, with the receptor, and in general comprises the step of a) incubating the receptor with the candidate molecule, b) adding a receptor ligand, c) determining whether the candidate molecule modulates the binding of the receptor to its ligand. As an example, a cellular or a subcellular preparation expressing the receptor isoform could be incubated with labeled ligand in the presence of the candidate molecule. The ability of this molecule to block this interaction could then be measured. In a typical competition assay, for example, an appropriate radio-labelled
ligand and a potential agonist/antagonist may be added to a cell preparation containing the receptor under examination, or to a recombinant form of the receptor, under suitable conditions for a competitive inhibition assay, whereby the effectiveness of the candidate molecule to compete with the radiolabelled ligand for receptor binding is measured as the number of ligand molecules bound to the receptor. Alternatively, candidate molecules may be screened for their ability to augment or inhibit dimerization of the receptor isoform or to augment or inhibit the receptor from assuming the activated state, for example using a two hybrid system. Furthermore, molecules disrupting the ligand/receptor interaction may be identified using the recently described inverse two hybrid technique (Vidal M. et al., Proc. Natl. Aca. Sci. USA 93, 1996, 10315-10320). A functional assay may be designed in which the response to the test compound is measured in terms of DNA synthesis activation, for example by determining the expression of a reporter gene which is under the control of a nucleic acid sequence responsive to the specific receptor isoform. The identification of molecules modulating the activity of the receptors herein disclosed allows to prepare therapeutical agents which are indicated for a number of important clinical conditions, including cancer, osteoporosis and other bone disorders, Alzheimer's disease as well as cardiovascular diseases.
Another embodiment of the present invention is the use of the coding sequences of hER-α isoforms for quantitatively determining the levels of corresponding mRNAs present in cell or tissue preparations and the subsequent use of these data in diagnosis and prognosis of diseases. Methods for quantitatively determining mRNA levels of hER-α isoforms include Northern blotting, in situ hybridization, nucleic acid hybridization, RT-PCR and SI protection analysis. In a further embodiment, the invention comprises the quantitative determination of the protein isoforms expressed in different tissues by in vitro immunoassay of a tissue sample using specific polyclonal or monoclonal antibodies. The quantitative determination of differential expression of the receptor isoforms may be useful to provide a diagnosis or prognosis
of a particular pathological condition and to establish a proper therapeutic protocol. For example, it may be used to determine the nature of a cancer, the cancer progression in relation to treatments such as irradiation, chemotherapy or surgery, the metastatic nature of the cancer, or to monitor the response of the tumor to the therapeutic treatment and to provide a prognosis for the patient concerning the course of the disease.
The polypeptides, polynucleotides and agonist/inhibitors of the present invention may be used in combination with a suitable pharmaceutical carrier. Thus, the compositions of the invention comprise a therapeutically or prophylactically effective amount of the active substance, and a pharmaceutically acceptable carrier or excipient.
As examples, carriers may be selected from saline, buffered saline, dextrose, water, glycerol, ethanol, and combination thereof. The pharmaceutical compositions may be administered in a convenient manner such as by the oral, topical, intravenous, intraperitoneal, intramuscular, subcutaneous, intranasal or intradermal routes. FIGURES
Fig. 1. Existence of novel liver enriched cER-α transcripts whose 5' ends are located downstream of the previously assigned translation start site
(A) Schematic representation of the cER-α transcripts. The original cER-α mRNA is encoded by eight exons labeled 1-8. The position of the two initiator methionines (ATG1- codon 1 and ATG2- codon 42) and the termination codon (TAA) are indicated. The division of the cER-α protein into six regions, A-F, together with the DNA - (region C) and hormone - (region E) binding domains are shown directly above the cDNA. The location of the splice acceptor site at +154 in exon 1A is also marked. Three alternative upstream 5' non coding exons (B, C and D) splice to this position giving rise to cER-α mRNA isoforms IB- ID. Probe A (from -169 to + 318),
B (from +1298 to +1678)' and C (from +158 to +892) used for SI nuclease mapping are indicated. For primer extension analysis, a primer complementary to positions +360 to +572 of the cER-α cDNA was used. (B) Preliminary evidence for a new cER-
α mRNA (A2) using SI nuclease mapping analysis. 100 μg of total RNA from laying hen oviduct and liver was hybridized to a labeled probe C and treated with SI nuclease. Yeast RNA was used as a negative control. The SI nuclease resistant hybrids were separated on a 4 % polyacrylamide gel adjacent to DNA molecular weight markers and free probe. The position of the two cER-α mRNA A2 transcription start sites are indicated on the right side of the figure. Also indicated are the relative positions of ATG 1 and ATG2. (C) Mapping of the cER-α transcription initiation sites by primer extension. 50 μg of total RNA from laying hen oviduct and liver was hybridized to the long primer, treated with reverse transcriptase and the extension products were separated on a sequencing gel. The transcription start sites of Al and A2 cER-α mRNAs are indicated. Also indicated are the relative positions of ATG 1 and ATG2. (D) The pattern of distribution and the relative levels of the two classes of cER-α transcripts, Al-D cER-α mRNAs and A2 cER-α mRNAs, were determined by SI nuclease mapping assays using total RNA from various sources as indicated at the top of each lane. M and F indicates male and female samples, respectively. Yeast
RNA was used as negative control. Protected fragments are marked with arrows. The relative abundance of the two classes of cER-α mRNA transcripts are shown below each lane of Fig. ID. The values were calculated from the densitometric scanning of the protected fragments obtained after SI nuclease analysis and expressed as the percentage of the total cER-α mRNA expressed in the oviduct. +/- indicates that a weak expression of cER-α mRNA was observed in a minority of the analyzed RNA samples.
Fig. 2. Two protein isoforms I and II of cER-α gene are produced in vitro and in vivo (A) Schematic representation of the cDNAs inserted within the expression vectors pSG cER-α I, pSG cER-α II and HEO and encoding the cER-α form I, cER- α form II and hER-α proteins. The position of the two initiator methionines (ATG1- codon 1 and ATG2- codon 42) and the termination codon (TAA) are indicated. In the
expression vector pSG cER-α II, the cER-α sequences between nucleotides +158 and +308 were not included. Therefore, the sequences preceding ATG2 are noncoding. (B) pSG cER-α I, pSG cER-α II and HEO plasmids were in vitro transcribed and translated by rabbit reticulocyte lysate in the presence of
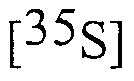
methionine. Translation products were resolved on a 10 % SDS-polyacrylamide gel and sized relative to the migration of prestained molecular size markers. (C) Nuclear protein extracts from chicken oviduct and liver were separated in parallel on the same gel and then subjected to immunoblotting with the H 222 antibody. Immunoreactive bands 66 and 61 KDa in size were visualized by ECL. Fig. 3. Evidence for an alternative splicing event at exon 2 acceptor splice site of the hER-α gene.
Panel A: Experimental design for 1A hER-α mRNAs detection, indicating the location and the size of the single-stranded probe A and each protected fragment obtained after SI digestion of probe/hER-α mRNA hybrids. Probe A (from +617 to +1538) was specific for normal hER-a transcripts (A/F hER-α mRNAs) but was also able to partially protect 1A hER-α mRNA isoforms up to the splice acceptor site position of exon 2. Open boxes indicate the unique (1A-1F) and common (1-8) exons encoding each normal hER-α mRNA isoforms. The position of the initiator methionine (ATG) and the termination codon (TGA) are indicated. The division of the hER-a protein into six regions, A-F, are shown directly above the cDNA.
Panel B: 30 μg of total RNA from MCF7 cells and 30 μg of Yeast RNA used as a negative control were hybridized to the labeled SI probes A, treated with SI nuclease and the resistant hybrids were separated on a sequencing gel as described in Experimental Procedure. The undigested probe is shown in a separate lane. Fig. 4. Exon IE is alternatively spliced to exon 1A or exon 2.
Panel A: Schematic representation of the RT-PCR experiment designed to identify 1A hER-α mRNAs. Open boxes indicate the unique (1A-1F) and the two first common (1A, 2) exons encoding each hER-α mRNA isoforms. Approximate locations
of primers are shown by short arrows. Primer IV, located in exon 2, was used to prime hER-α cDNA synthesis by revers transcriptase. Primers Al-Fl which are specific for each hER-α cDNA 5' region, were used in a round of PCR amplification with primer V which is nested to primer IV in exon 2. The oligonucleotide probes PI and P2, respectively from exon 1A and 2, were used to confirm the specificity of the PCR products as well as the exon 1 A deletion for some hER-a transcripts.
Panel B: The hER-α cDNA isoforms were amplified as described above, using total RNA from MCF7. PCR products were electrophoresed through an agarose gel and transferred by southern blot to a membrane which was then hybridized with the oligonucleotide probes PI and P2 as described in Experimental Procedures. Positions of migration of the molecular size marker are shown on the left side of the figure.
Panel C: The sequence of the PCR products from lane E or F which did not hybridize to the oligonucleotide probe PI but hybridized to P2 probe revealed that they contain the donor site of exon IE joined to the acceptor site of exon 2. Fig. 5. E/F and E/F 1A hER-α mRNA isoform distribution analysis.
Panel A: RT-PCR analysis. Open boxes indicate the unique (IE or IF) and common (part of IE and 1A-8) exons encoding E/F hER-α mRNA isoforms. Approximate locations of primer are shown by short arrows. Primer I, located in the 3' untranslated region of exon 8, was used to prime hER-α cDNA synthesis by reverse transcriptase, using total RNA from various sources as indicated at the top of each lane. Yeast total RNA was used as a negative control. Primer E/Fl which is specific for both E and F hER-a cDNA 5' regions (in the common part of exon IE), was then used in a first round of PCR amplification with primer II which is nested to primer I in exon 8. A second round of PCR reaction was performed with a specific (E/F2) and common (III) nested primers. An oligonucleotide probe from exon 2 was used to confirm the specificity of the PCR products. Positions of migration of the molecular size markers are shown on the left side of the figure.
Panel B, C and D: SI nuclease mapping analysis. The SI nuclease mapping
assays of E/F and E/F 1A hER-α mRNA isoforms were performed as described in Materials and Methods, with the single-stranded probes F (panel B), F 1A (panel C) and E 1 A (panel D) and using 30 μg of total RNA from various sources as indicated at the top of each lane. Yeast total RNA was used as negative control. The location and the size of each single-stranded probe (F, F 1A and E 1A) and each protected fragment obtained after SI digestion of the probe/hER-α mRNA hybrids are indicated. Each probe was specific for one hER-α transcript (for example, F 1A hER mRNA) but was also able to partially protect the other hER-α mRNA isoforms [for example, (-E/F 1 A) hER mRNA] up to the splice site positions. The probes were designed to contain vector sequence in their extremity (denoted by the thinner black line) in order to discriminate between undigested probes (>) and specific protected fragments.
Fig. 6. E/F 1 A hER-α mRNA isoforms encode a 46 kDa protein which lacks the A/B domain present in the 66 kDa form.
Panel A: Schematic representation of the cDNAs inserted within the expression vector pSG5 which gave rise to pSG hER-α 66 (HEO) and pSG hER-α 46. The position of the initiator methionine for the 66 kDa hER-α, the initiator methionine for the 46 kDa hER-α and the termination codon (TGA) are indicated. The division of the hER-α protein (66 kDa) into six regions, A-F, together with the DNA - (region C) and hormone - (region E) binding domains ar shown directly above the cDNAs. Also showed are the epitopes recognized by the anti-hER antibodies HC20, H226 and H222, used in panel B. HC20 is a polyclonal antibody and H226 an H222 are monoclonal antibodies.
Panel B: pSG hER-α 66 and pSG hER-α 46 plasmids were in vitro transcribed and translated in rabbit reticulocyte lysate. The obtained translation products as well as 40 μg of whole cell extracts from MCF7 (ER positive breast cancer cell line),
MDA-MB-435 (ER negative breast cancer cell line) and HeLa (ER negative cell line) were resolved on a 10 % SDS-polyacrylamide gel and then subjected to immunoblotting with the HC20, H226 and H222 antibodies. Immunoreactive bands 66
and 46 KDa in size were visualized by ECL.
Fig. 7. The 66 and 46 kDa hER-α differ in their ability to modulate estrogen target gene expression.
HeLa and HepG2 cells were transiently transfected with 5 μg of the reporter plasmid coll73(APl)-LUC or (ERE)2-tk-LUC, together with 0.5 μg of the expression vector pSG5, pSG hER-a 66 (HEO) or pSG hER-α 46. Cells were treated with or without estradiol (10-8 M) for 48 before being assayed for luciferase activity. Results are expressed as a percentage of the reporter gene activity measured in presence of the expression vector pSG hER-α 66 and E2. Luciferase activities were normalized using the internal reference control EF-la-CAT. Values correspond to the average of at least three separate transfection experiments ± SEM.
Fig. 8. The 46 kDa hER-α is a dominant negative form in HepG2 cells, suppressing the activity of the 66 kDa hER-α.
HepG2 cells were transiently transfected with 5 mg of the reporter plasmid (ERE)2-tk-LU( together with either 0.5 mg of the expression vector pSG5, 0.5 μg of pSG hER-α 46 or 0.5 μg of pSG hER-α 66 (HEO) with increasing concentration of pSG hER-α 46 (0 to 4 μg). Cells were treated with 10-8 M estradiol for 48 h before being assayed for luciferase activity. Results are expressed as a percentage of the reporter gene activity measured in presence of the expression vector pSG hER-α 66. Luciferase activities were normalized using the internal reference control EF- 1 a- CAT.
Fig. 9. Panel A: Immunoprecipitation of estrogen receptor proteins in human primary osteoblasts demonstrate the expression of a 66 kDa ER-α protein as a result of the translation start at position +233 in exonl . A second translational product of 46 kDa size results from a splicing event of an upstream exon (1F/E) directly to position
+685 in exon 2, which is located upstream from a internal ATG (+753/758). The positions of the initiator methionine (ATG) and the termination codon (TGA) are indicated. An isoform with a molecular weight of 39 kDa which was also precipitated
using H222 monoclonal antibody has to be further characterized. In vitro transcribed and translated pSG5 expression vectors encoding either the full length hER-α (IVT66) or the 46 kDa isoform (IVT46) in the presence of 35-S methionine were used as controls. Panel B: In vitro translation of 50 kDa protein isoform. Examination of exon 1C of the hER-α gene demonstrated the beginning of an open reading frame which is in frame to the remainder of the hER-α open reading frame when spliced to the acceptor site at position +685 in exon 2. In order to investigate, whether this in frame ATG could function as a translation initiation codon for a shorter hER-α protein isoform of a predicted size of 50 kDa cDNA was inserted into pSG5 expression vector. Western blotting revealed that this splicing event leads to the expression of a hER-α protein of 50 kDa size as well as to the expression of the hER-α 46 protein isoform due to the leakiness in the translation initiation. IVT66 and IVT46 were used as controls. EXPERIMENTAL PROCEDURES. Cells and tissues
All cell lines were maintained in DMEM (Gibco, BRL, Eggenstein, Germany) supplemented with 10% fetal calf serum (FCS, Gibco BRL), penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37°Cin a 5% incubator. Human primary osteoblasts were isolated from bone tissues of patients that underwent hip or knee operations under sterile conditions. The outgrowing osteoblast cells were cultivated in DMEM + 20% FCS, characterized by staining for alkaline phosphatase (AP kit, Sigma, Deisenhofen, Germany) and taken for experiments until passage 3. RNA isolation and RT-PCR (Fig. 2) Expression of different hER-α isoforms was examined using reverse transcription of RNA followed by PCR and Southern Blotting. Total RNA was isolated from MCF-7 cells using TRIzol reagent (Gibco, BRL) as described by the manufacturer. Reverse transcription was performed using 1 μg of total RNA, an oligonucleotide primer (I) from exon 8, located within the 3' untranslated region (3'
UTR) of hER-α (5' TTGGCTAAAGTGGTGCATGATGAGG) instead of an oligo (dT) primer and 50 U of Expand reverse transcriptase (Roche Diagnostics, Mannheim, Germany) following the protocol of the supplier. Two microlitres of the reaction were then used for two rounds of 35 cycles PCR amplification. The 5' primers and nested primers used for the amplification of hER isoforms A, B, C, E and F were:
Al (5' CTCGCGTGTCGGCGGGACAT SEQ ID No. 5) and A2 (5' GCTGCGTCGCCTCTAACCTC SEQ ID No 6), Bl (5' CTGGCCGTGAAACTCAGCCT SEQ ID No 7) and B2 (5' ATCCAGCAGCGACGACAAGT SEQ ID No 8), Cl (5' TCTCTCGGCCCTTGACTTCT SEQ ID No 9) and
C2 (5' CAAGCCCATGGAACATTTCTG SEQ ID No 10), El (5' AGCCTCAAATATCTCCAAAATCT SEQ ID No 11) and E2 (5'AATTATATTCTGTAGCTACCAAAGAAG SEQ ID No 12), FI (5' TTCTATAGCATAAGAAGACAG SEQ ID No 13) and F2 (5' GAGTGATAATCTTC SEQ ID No 14), respectively. The 3' primer V
(5 TTATCTGAACCGTGTGGGAG SEQ ID No 15) was chosen within exon 2 of the hER-α gene. A nested primer IV (5' CGTGAAGTACGACATGTCTAC SEQ ID No 16) was selected upstream of primer V. the Expand Long Template PCR system (Roche Diagnostics) was used for amplification as recommended by the manufacturer. Five microlitres of each reaction were analyzed on a 1% agarose gel. Southern Blotting (Fig. 2)
After separation on agarose gels, PCR products were transferred to nylon membranes (Hybond N+, Amersham, Arlington Heights, IL) with 20x saline sodium citrate (SSC) as transfer solution. The membranes were incubated in a prehybridization buffer containing 6x SSC, 5x Denhardt's solution, 0.05% sodium pyrophosphate, 100 μg salmon sperm DNA, and 0.5% SDS at 37°C for lh. then, the membranes were hybridized in 6xSSC, lxDenhart's solution, 0.05% sodium pyrophosphate, 100 μg/ml yeast t-RNA with an oligonucleotide probe selected in exon
1 (1) and exon 2 (P2), respectively, which had been end-labeled using T4 polynucleotide kinase and [γ-32P] ATP (3000 Ci/mmol, Amersham)). The most stringent wash was carried out for 20 min at 55°C in 6xSSC, 0.05% sodium pyrophosphate. The specific PCR products were visualized by exposing the membranes to an x-ray film.
SI nuclease assay (Fig 1, Fig 3)
A modified SI nuclease mapping procedure was followed as described by Flouriot et al. The method involves the use of biotinylated single-stranded DNA probes by extension from a specific primer by extension from a specific primer by the T7 DNA polymerase in the presence of [α32-P] deoxy-CTP (3000 Ci/mmol). These probes are then hybridized with the appropriate RNA sample and subjected to an SI nuclease digestion. In order to prepare the templates for the different probes, RT-PCR reactions were performed. The PCR products were subcloned upstream of T7 primer and downstream of Ml 3 reverse primer in the TA cloning vector pCRTM2.1 (Invitrogen, San Diego, CA). a PCR reaction was then performed using a biotinylated forward primer together with a reverse primer either from vector (Ml 3) or hER- α coding region. Biotinylated PCR products were bound to streptavidin-coated magnetic beads (Dynal, Great Neck, NY) as recommended by the manufacturer and the nonbiotinylated DNA strands were removed using magnetic separation, 105 cpm probe was coprecipitated with 30-100 μg total RNA and then dissolved in 20 μl hybridization buffer (80% formamide, 40 mM piperazine-N,N'bis[2-ethane sulfonic acid] (Pipes, pH 6.4), 400 mM NaCl, 1 mM EDTA (pH 8)), denatured at 70°C for 10 min, and hybridized overnight at 55°C. The SI digestions were carried out for lh at 30°C and after precipitation the samples were separated on 4% polyacrylamide-urea gels.
ER-α expression vector preparation (fig 4, Fig. 7)
To create the expression vector pSGhER46, PCR was used to amplify cDNA of the hER coding region from position +519 up to +1788 using the plasmid pSG5HEO
(kindly provided by P. Chambon). In order to construct pSG5hER50, a specific primer (5 CTCTCGGCCCTTGACTTCTGCCAAATTCAGATAATCGACGCCAGGG SEQ ID No 17) was designed, mapping the hER-α isoform C and extending directly into exon 2. The reverse primer started in exon 2 and extended into exon 1C (5' CCACCCTGGCGTCGATTATCTGAATTTGGCAGAAGTCAAGGGCCGAGAGA SEQ ID No 18). An additional PCR resulted in amplification of exon 1C directly liked to position +452 in exon 2 up to position +1788 in exon 8. The primers were designed to introduce BamHI restriction sites at the ends of the PCR products. The amplified fragments were directionally cloned into the polylinker of pSG5 expression plasmid downstream of S V40 promoter.
In vitro transcription and translation (FIG. 4, Fig. 7)
In vitro transcription and translation was accomplished with the TNT coupled Reticulocyte Lysate system from Promega (Madison , WI, USA) following the manufacturers protocol. The expression vector pSG5HEO, pSGhER50 and pSGhER46 were used as templated for transcription with T7 RNA polymerase followed by translation to generate human ER-α proteins (hER66, hER50 and hER46).
Western Blot analysis (Fig. 4, Fig 7B)
Total cell lysates (40 μg) and five microlitres of the in vitro transcription and translation mix were run on a 10% SDS-PAGE gel as outlined by Laemmli (2) and electrotransferred to Optitran membrane (Schleicher & Schuell, Dassel, Germany). The membrane was blocked in TBST (10 mM Tris, pH 7.4, 0.5 M NaCl, and 0.5% Tween 20), containing 5% non fat dry milk powder. The membrane was incubated with primary antibody (1 μg/ml) in TBST for lh at room temperature. Primary antibodies used were rat monoclonal antibodies H222, directed against the hormone- binding domain of hER-α, and H226, directed against the A/B domain of hER-α (both kindly provided by G. Greene, University of Chicago) as well as a rabbit polyclonal antiserum HC20 (Santa Cruz Biotechnology, Santa Cruz, CA), which mapped both
isotopes. Incubation with secondary peroxidase-conjugated goat-anti rat (H222, H226) or peroxidase-coupled goat anti-rabbit (HC20) was performed under the same conditions. ER-α proteins were visualized by chemiluminescence using the ECL system from Amersham according to the manufacturers instructions. Immunoprecipitation (Fig. 7A)
Human primary osteoblasts were cultivated in 15 cm TC-plated in DMEM + 10% FCS until subconfluency. After twice washing with PBS, the cells were incubated in methionin-free DMEM + 10% methionin-free FCS for 10 hours. Then, 1000 μCi radiolabeled Pro-mix (35S-methionin/35S-cystein-rnix. Amersham) were added per plate and the cells were incubated overnight at 37°C and 5% C02. After washing with cold PBS, the cells were harvested and lysed in 1 ml RIPA-buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) for 30 min at 4°C. The lysate and the in vitro transcribed and translated hER46 and hER66, which served as positive controls, were incubated with an unspecific antibody for lh (anti-α-Actin, 1 μg/ml, Roche Diagnostics), followed by binding to
5% protein- A-sepharose (Pharmacia Biotech, Freiburg, Germany) and centrifuged for 5 min at 10000 rpm. Surnatants were incubated with a hER-α specific antibody (H222, 1 μg/ml) and then linked to sepharose for lh at 4°C. After extensive washes, Laemmli-buffer was added to the precipitates and the samples were boiled for 5 min. After centrifugation, the supernatants were separated on a 10% PAGE gel, dried and then subjected to autoradiography.
Transient transfection (Fig. 5, Fig. 6)
For transfection experiments, cells were plated into 6 cm dishes (Nunc, Wiesbaden, Germany) at a density of lxlO3 cells/plate and grown in DMEM media supplemented with 10% FCS. After 3 days, the cells were washed with lxPBS and the medium was replaced by phenol-red free DMEM + 2.5% charcoal stripped, E2-free FCS. After additional 24 hours, transient transfections were carried out using the calcium precipitate method. Transfections were performed using 5 μg of luciferase
reporter plasmid containing estrogen responsive elements (EREtkLuc) (Fig. 5 and 6) or an AP-1 site (AP-l(coll73)-Luc) (Fig 5) together with 0.25 μg of a CAT plasmid as a correction for transfection efficiency and 0.5 μg of pSG5 expression plasmid coding for hER66 (pSG5HEO) or hER46 (pSGhER46). For competition experiments, pSG5HEO was kept at a concentration of 5.0 μg to 0.006 μg while contemporaneously pSGhER46 was added at increasing dilutions comprised in the range 4 μg - 0.006 μ.g After overnight incubation, the transfection media was removed, the cells were washed twice with PBS, and 3 ml of phenol-red free DMEM supplemented with 2.5% charcoal stripped FCS and 10"10M estradiol were added. After 48 hours cells were harvested and luciferase assays and CAT ELISAs were performed using commercial kits (Roche Diagnostics).