WO2000047759A1 - Stereoselective synthesis of nucleoside analogues - Google Patents
Stereoselective synthesis of nucleoside analogues Download PDFInfo
- Publication number
- WO2000047759A1 WO2000047759A1 PCT/CA2000/000144 CA0000144W WO0047759A1 WO 2000047759 A1 WO2000047759 A1 WO 2000047759A1 CA 0000144 W CA0000144 W CA 0000144W WO 0047759 A1 WO0047759 A1 WO 0047759A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- protease
- group
- anomer
- mixture
- dioxolane
- Prior art date
Links
- 0 CC(C)(OC1)O[C@@]1C(O*)=O Chemical compound CC(C)(OC1)O[C@@]1C(O*)=O 0.000 description 3
- GQQJXYZMNVLTRX-IQLKVPPVSA-N COC([C@@H]1OC(C(COC(c2ccccc2)=O)N)OC1)=O Chemical compound COC([C@@H]1OC(C(COC(c2ccccc2)=O)N)OC1)=O GQQJXYZMNVLTRX-IQLKVPPVSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P41/00—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture
- C12P41/003—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture by ester formation, lactone formation or the inverse reactions
- C12P41/005—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture by ester formation, lactone formation or the inverse reactions by esterification of carboxylic acid groups in the enantiomers or the inverse reaction
Definitions
- the present invention relates generally to a novel method for the preparation of nucleoside analogues and their precursors and more particularly to a method of preparing a nucleoside analogue by the use of specific enzymes to stereoselectively produce dioxolane nucleoside analogues or their precursors.
- the pharmacological activity of pharmaceutical compounds depend mainly on their interaction with biological matrices (drug targets) , such as proteins (receptors and enzymes) , nucleic acids (DNA and RNA) and biomembranes (phospholipids and glycolipids) . All these drug targets have complex three-dimensional structures which are capable of binding specifically to the drug in only one of the many possible arrangements in the three- dimensional space. It is the three-dimensional structure of the drug target that in part, determines which of the potential drug is bound within its cavity and with what affinity.
- drug targets such as proteins (receptors and enzymes)
- DNA and RNA nucleic acids
- biomembranes phospholipids and glycolipids
- Stereoisomers are compounds with identical chemical composition and atom connectivity (i.e. same constitution), but different arrangements of the atoms in space (i.e. different configurations). Stereoisomers are classified according to the number of chiral centers in each molecule and the spatial arrangement of the chiral center.
- Chiral centers of organic molecules include chiral carbon atoms which have four different substituents connected thereto and arranged in a generally tetrahedral configuration.
- the chirality of molecules that are the subject of the present application refer to chirality created by chiral atoms and not chiral bonds. The following discussions will be limited to chirality created at one or more chiral carbon atoms which have four different substituents bound to each of the four different binding sites of the carbon.
- stereoisomers When a molecule has a single chiral carbon, there are two stereoisomers that are mirror images of each other. This pair of isomers is termed enantiomers or an enantiomeric pair. When there are two chiral carbon atoms, there are four stereoisomers and two pairs of mirror images or enantiomers. A stereoisomer which is not a mirror image of another stereoisomer is a diastereoisomer.
- Cyclic sugars can be designated as a particular anomer depending upon the stereochemical configuration.
- the term "anomer” designates the spatial arrangement of cyclic sugars or analogues or derivatives thereof that have two chiral centers in a five or six member ring.
- the anomeric designation defines the relative configuration of the two chiral centers relative to a hypothetical plane defined by the ring.
- the chiral centers typically have two substituents outside the ring. One substituent is a H.
- the other substituent is a larger moiety such as a hydroxyl , methoxyl , purine or pyrimidine base, carboxyl, etc.
- each chiral center i When the two larger constituents on each chiral center i are on the same side of the plane in the ring, they are defined as a ⁇ -anomer (cis-isomer) . When two larger moieties are on opposite sides of the plane in the ring, they are defined as the ⁇ -anomer (trans-isomer) .
- An anomer is a type of diastereoisomer .
- chirality may affect biological activity or toxicity, it is important from the point of view of drug development to evaluate the physiological effect of each isomer. Frequently, one stereoisomer is considerably more active than the other. In other situations, the non-active isomer may inhibit the activity of the more active form. In some instances, the less preferred stereoisomer may be equally potent but have greater toxicity than the preferred stereoisomer. In each of these instances, the therapeutic effect of a drug can be increased if the single most preferred stereoisomer is administered in higher purity.
- dioxolane nucleoside analogues 3 ' -oxa-substituted 2 ' , 3 ' -dideoxynucleoside analogues. These compounds have two chiral centers corresponding to the substituted carbons 2 and 4 of the dioxolane ring (C2 and C4 respectively) . Thus each compound can exist as four different stereoisomers depending on the position of both substituents with respect to the dioxolane ring.
- stereoisomers of a dioxolane nucleoside analogue are represented by the following diagrams where the letter B represents a purine or pyrimidine base or an analogue or derivative of a purine or pyrimidine base as defined herewith.
- the C2 carbon in each of the above formula is the carbon atom in the ring that connects the methyloxy group to the dioxolane ring.
- the C4 carbon is the carbon atom in each of the above formula that connects the base (B) substituent to the dioxolane ring.
- the four stereoisomers represented above correspond to two pairs of enantiomers.
- the ⁇ -anomers represent one set of enantiomers, the ⁇ -L enantiomer and the ⁇ -D enantiomer.
- the -anomers represent the other set of enantiomers, the ⁇ -L enantiomer and the ⁇ -D enantiomer.
- Compounds with D-configuration have an outward directed methyloxy group when the ring is oriented in the plane of the page with the oxygen in the first position (03) at the top of ' the page with the carbon in the two position (C2) on the left side as illustrated above. This is also represented by the designation (2R) .
- Compounds having an L-configuration has inward directed methyloxy group when the ring is oriented in the plane of the page with the 03 oxygen at the top of the page with the C2 carbon on the left side as illustrated above. This is also represented by the designation (2S) .
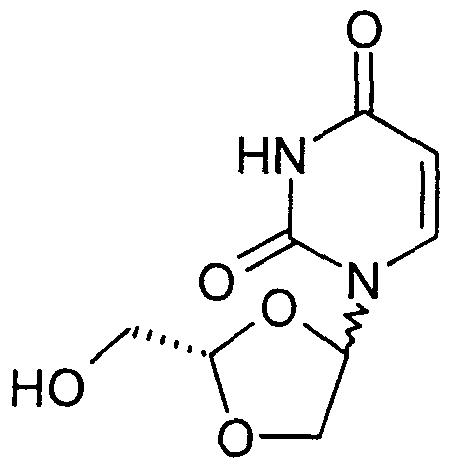
- dioxolane nucleoside analogues have been identified to have antiviral and anticancer activity.
- 9- ( ⁇ -D-2-hydroxymethyl-l , 3 -dioxolan-4-yl) -2- aminopurine ( ⁇ -D-DAPD) and its metabolite 9- ( ⁇ -D-2- hydroxymethyl-1, 3-dioxolan-4-yl) -guanine ( ⁇ -D-DXG) have been reported to have potent and selective activity against human immunodeficiency virus (HIV) and hepatitis B virus (HBV) (Rajagopalan et al . , Antiviral Chem. Chemother.
- 1- ( ⁇ -L-2- hydroxymethyl -1 , 3-dioxolan-4-yl) -thymine (Dioxolane- T) (Norbeck et al . , Tetrahedron Lett., 1989,30, 6263-66) possess anti-HIV and anti-HBV activity.
- ⁇ -L-OddC is the first nucleoside analogue with an L-configuration shown to have anticancer activity. Since stereoisomers of dioxolane nucleosides usually have different biological activities and toxicity, obtaining the pure therapeutically active isomer becomes crucial. i
- the present invention provides a novel process for making dioxolane nucleoside analogues with a high degree of steric purity, greater efficiency and higher yields.
- the present invention provides a process for making dioxolane nucleoside analogues with a high degree of steric purity which includes the use of certain hydrolytic enzymes for separating ⁇ and ⁇ anomers from an anomeric mixture represented by the following formula A or formula B:
- W is benzyl or benzoyl ;
- R x is selected from the group consisting of C 1-6 alkyl and C 6-15 aryl .
- the process involves the step of hydrolyzing the mixture of compounds represented by fc ⁇ mula A and formula B with an enzyme selected from the group consisting of cholesterol esterase, ESL-001-02, horse liver esterase, bovine pancreatic protease, ⁇ -chymotrypsin, protease from Strep tomyces caespi tosis, substilisin from Bacillus licheniformis, protease from Aspergillus oryzae, proteinase from Bacillus licheniformis , protease from Strep tomyces griseus, acylase from Aspergillus elleus , proteinase from Bacillus subtilis, ESL-001-05, pronase protease from Streptomyces griseus, lipase from Rhizopus arrhizus, lipoprotein lipase from Pseudomonas species type B, lipase from Pseudomonas cepacia and
- the process stereoselectively hydrolyses predominantly one anomer to form a product where Rj of formula A and formula B is replaced with H.
- the other anomer remains substantially unhydrolysed.
- the process also comprises separating the hydrolyzed product from unhydrolysed starting material.
- the process of one embodiment further includes the step of stereoselectively replacing the functional group at i the C4 position of the dioxolane (e.g. COORi) with a purinyl, pyrimidinyl or analogue or derivative thereof to produce a dioxolane nucleoside analogue that has a high degree of steric purity.
- a purinyl, pyrimidinyl or analogue or derivative thereof to produce a dioxolane nucleoside analogue that has a high degree of steric purity.
- the step of hydrolyzing results in a starting material having an anomeric purity of at least 80%. According to another embodiment, the step of hydrolyzing results in a starting material having an anomeric purity of at least 90%. In yet another embodiment, the step of hydrolyzing results in a starting material having an anomeric purity of at least 95%. In an additional embodiment, the step of hydrolyzing results in a starting material having an anomeric purity of at least 98%.
- the step of hydrolyzing results in a product having an anomeric purity of at least 80%. According to another embodiment, the step of hydrolyzing results in a product having an anomeric purity of at least 90%. In yet another embodiment, the step of hydrolyzing results in a product having an anomeric purity of at least 95%. In an additional embodiment, the step of hydrolyzing results in a product having an anomeric purity of at least 98%.
- W of formula A or formula B is benzyl and the enzyme is selected from the group consisting of cholesterol esterase, ESL-001-02, horse liver esterase, bovine pancreatic protease, ⁇ - chymotrypsin, protease from Streptomyces caespi tosis, substilisin from Bacillus licheniformis .
- the enzyme is ⁇ -chymotrypsin.
- the enzyme is bovine pancreatic protease.
- W of formula A and formula B is benzoyl and the enzyme is selected from the group consisting of protease from Aspergill us oryzae, proteinase from Bacillus licheniformis, subtilisin from Bacillus licheniformis , protease from Strep tomyces griseus, acylase from Aspergillus melleus, proteinase from Bacillus subtilis, ESL-001-05, pronase protease from Streptomyces griseus, lipase from Rhizopus arrhizus, lipopr ⁇ tein lipase from Pseudomonas species type B, bacterial proteinase, lipase from Pseudomonas cepacia .
- the enzyme is selected from the group consisting of protease from Aspergill us oryzae, proteinase from Bacillus licheniformis, subtilisin from Bacillus licheniformis , protea
- the enzyme is selected from the group consisting of Aspergillus oryzae protease, proteinase from Bacillus licheniformis , subtilisin from Bacillus licheniformis , protease from Streptomyces griseus, pronase protease from Streptomyces griseus, and lipase from Rhizopus arrhizus .
- the enzyme is selected from the group comprising Aspergillus oryzae and proteinase from Bacillus licheni formi s .
- the ⁇ -anomer is the predominant product. In another embodiment, the ⁇ -anomer is the predominant product.
- the ⁇ -L- enantiomer is the predominant product. In an additional embodiment, the ⁇ -D-enantiomer is the predominant product. In yet another embodiment, the ⁇ -L-enantiomer is the predominant product. In an additional embodiment, the ⁇ -D-enantiomer is the predominant product.
- the invention is a process for stereoselectively preparing a dioxolane nucleoside analogue by separating ⁇ and ⁇ -anomers from an anomeric mixture represented by formula A or formula B according to one of the above embodiments.
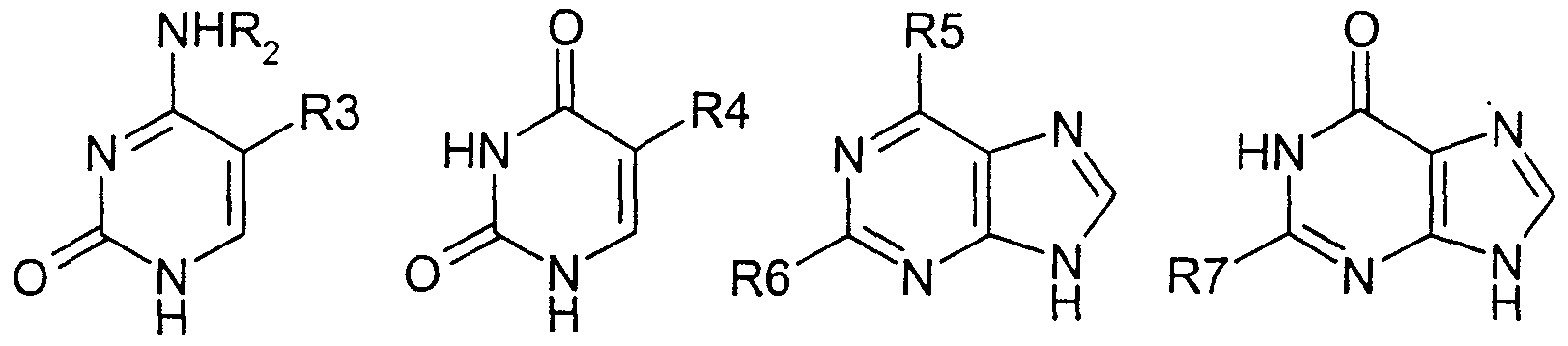
- the process further includes the step of stereoselectively replacing the functional group at the C4 position (COORi) with a purinyl or pyrimidinyl or analogue or derivative selected from the group consisting of:
- R 2/ R 9 and Rn are independently selected from the group consisting of hydrogen, C ⁇ -6 alkyl, C ⁇ - 6 acyl and R 8 C(0) wherein R 8 is hydrogen or d -6 alkyl.
- R 3 , R 4 and R 10 are each independently selected from the group consisting of hydrogen, C 1-6 alkyl, bromine, chlorine, fluorine, iodine and CF 3 ; and
- R 5 R 6 and R 7 are each independently selected from the group consisting of hydrogen, bromine, chlorine, fluorine, iodine, amino, hydroxyl and C 3-6 cycloalkylamino.
- the process results in the production of a stereochemical isomer of the dioxolane nucleoside analogue .
- the process further includes the step of stereoselectively replacing the functional group at the C4 position (COORi) with a purinyl or pyrimidinyl or derivative selected from the group consisting of:
- R 2 is selected from the group consisting of hydrogen, C ⁇ -6 alkyl, C 1-6 acyl and R 8 C(0) wherein R 8 is hydrogen or C ⁇ -6 alkyl.
- R 3 and R 4 are each independently selected from the group consisting of hydrogen, C ⁇ -6 alkyl, bromine, chlorine, fluorine, iodine and CF 3 ; and R 5 , R ⁇ and R 7 are each independently selected from the group consisting of hydrogen, bromine, chlorine, fluorine, iodine, amino, hydroxyl and C 3 . 6 cycloalkylamino.
- the process results in the production of a stereochemical isomer of a dioxolane nucleoside analogue.
- the process further includes the step of stereoselectively replacing the functional group at the C4 position (COORi) with a pyrimidinyl or analogue or derivative selected from the group consisting of:
- R 9 and Rn are independently selected from the group consisting of hydrogen, C ⁇ _ 6 alkyl, C 1-6 acyl and R 8 C(0) .
- R ⁇ 0 is selected from the group consisting of hydrogen, C ⁇ -6 alkyl, bromine, chlorine, fluorine, iodine and CF 3 . The process results in the production of a stereochemical isomer of a dioxolane nucleoside analogue.
- the process comprises stereoselectively preparing a dioxolane nucleoside analogue by separating ⁇ and ex anomers from an anomeric mixture represented by formula A or formula B according to one of the above embodiments and further comprises stereoselectively replacing the functional group at the
- the process comprises making a dioxolane nucleoside analogue by separating a compound according to formula A or formula B.
- the process includes stereoselectively replacing the R group with a purinyl or pyrimidinyl moiety or analogue or derivative thereof by acylating the second mixture to produce an acylated second mixture.
- This embodiment also includes the step of glycosylating the acetylated second mixture with a purine or pyrimidine base or analogue or derivative thereof and a Lewis Acid to produce a dioxolane nucleoside analogue.
- the present invention involves a high yield process of separating ⁇ and ⁇ anomers from an anomeric mixture of dioxolane nucleoside analogue precursors which provides higher yield and greater efficiency.
- this method is used in the production of dioxolane nucleoside analogues having a high degree of anomeric purity at lower cost.
- another aspect of the present invention involves synthesizing starting material having a higher degree of anomeric purity.
- the present invention provides a process of preparing dioxolane nucleoside analogues having a predominant ⁇ -L- configuration using enzymes, namely hydrolases.
- the procedure improves overall yield and has relatively few steps, thereby improving overall efficiency.
- the process involves the following steps.
- W is benzyl or benzoyl
- R x is selected from the group consisting of H, C -6 alkyl and C 6- i5 aryl .
- the mixture is hydrolyzed with an enzyme selected from the group consisting of cholesterol esterase, ESL-001-02, horse liver esterase, bovine pancreatic protease, -chymotrypsin, protease from Strep tomyces caespi tosis , substilisin from Bacillus li cheniformis , protease from Aspergillus oryzae, proteinase from Bacillus licheniformis , protease from Streptomyces griseus , acylase from Aspergill us elleus, proteinase from Bacillus subtilis, ESL-001-05, pronase protease from Streptomyces griseus, lipase from Rhizopus arrhizus, lipoprotein lipase from
- the hydrolyzing step stereoselectively hydrolyzes the ⁇ -anomer of the mixture of either formula A or formula B.
- the result is an unhydrolysed ⁇ -anomer.
- the ⁇ -anomer can be separated easily from the ⁇ -anomer. If an anomeric mixture of the compound of formula A is selected, the result is the production of the compound of formula C and formula D:
- the mixture (C) / (D) or (E) / (F) is then subjected to oxidative decarboxylation which replaces the R x group with an acyl moiety. It is then glycosylated with a purine or pyrimidine base or analogue or derivative thereof in the presence of a Lewis Acid.
- the final step produces a dioxolane nucleoside analogue in the ⁇ -L configuration for the mixture (C)/(D) and a dioxolane nucleoside analogue in the ⁇ -D configuration for the mixture (E)/(F) .
- Nucleoside is defined as any compound which consists of a purine or pyrimidine base, linked to a pentose sugar.
- Dioxolane nucleoside analogue is defined as any compound containing a dioxolane ring as defined hereinafter linked to a purine or pyrimidine base or analogue or derivative thereof.
- a "dioxolane ring” is any substituted or unsubstituted five member monocyclic ring that has an oxygen in the 1 and 3 positions of the ring as illustrated below:
- Purine or pyrimidine base is defined as the naturally occurring purine or pyrimidine bases adenine, guanine, cytosine, thymine and uracil.
- a purine or pyrimidine that is a moiety is a purinyl or pyrimidinyl, respectively.
- Alkyl is defined as a substituted or unsubstituted, saturated or unsaturated, straight chain, branched chain or carbocyclic moiety, wherein the straight chain, branched chain or carbocyclic moiety can be optionally interrupted by one or more heteroatoms (such as oxygen, nitrogen or sulfur) .
- a substituted alkyl is substituted with a halogen (F, Cl , Br, I), hydroxyl, amino or C 6-20 aryl .
- Aryl is defined as a carbocyclic moiety which can be optionally substituted or interrupted by one heteroatom (such as oxygen, nitrogen or sulfur) and containing at least one benzenoid-type ring (such as phenyl and naphthyl) .
- Carbocyclic moiety is defined as a substituted or unsubstituted, saturated or unsaturated, C 3-6 cycloalkyl wherein a substituted cycloalkyl is substituted with a C ⁇ -6 alkyl, halogen (i.e. F. Cl , Br, I), amino, carbonyl (i.e. COOH) , or N0 2 .
- a “derivative" of a purine or pyrimidine base refers to one of the following structures:
- pyrimidine H wherein one or more of the pyrimidine H are substituted with substituents that are known in the art .
- the bonds represented by a broken line are optional and are present only in cases which require the bond to complete the valence of the ring atom.
- Substitutents bound to the ring members by a single bond include but are not limited to halogen such as F, Cl, Br, I; an akyl such as lower akyls; aryl; cyano carbamoyl ; amino including primary, secondary and tertiary amino; and hydroxyl groups.
- Analogue of a purine or pyrimidine base refers to any derivative of purine or pyrimidine bases that is further modified by substituting one or more carbon in the ring structure with a nitrogen.
- Steposelective enzymes are defined as enzymes which participate as catalysts in reactions that selectively yield one specific stereoisomer over other stereoisomers.
- “Anomeric purity” is defined as the amount of a particular anomer of a compound divided by the total amount of all anomers of that compound present in the mixture multiplied by 100%.
- Alkoxy is defined as an alkyl group, wherein the alkyl group is covalently bonded to an adjacent element through an oxygen atom (such as methoxy and ethoxy) .
- Alkoxycarbonyl is defined as an alkoxy group attached to the adjacent group of a carbonyl.
- Acyl is defined as a radical derived from a carboxylic acid, substituted (by a halogen, C 6 - 2 o aryl or C 1-6 alkyl) or unsubstituted by replacement of the -OH group.
- an acyl radical may be aliphatic or aromatic, substituted (by halogen, C -6 alkoxyalkyl, nitro or 0 2 ) or unsubstituted, and whatever the structure of the rest of the molecule may be, the properties of the functional group remain essentially the same (such as acetyl , propionyl , isobutanoyl, pivaloyl , hexanoyl , trifluoroacetyl , chloroacetyl and cyclohexanoyl) .
- Alkoxyalkyl is defined as an alkoxy group attached to i the adjacent group by an alkyl group (such as methoxymethyl) .
- acyloxy is defined as an acyl group attached to the adjacent group by an oxygen atom.
- one embodiment of the present invention is a process for separating ⁇ and ⁇ anomers from an anomeric mixture represented by the following formula A or formula B:
- W is benzyl or benzoyl and Ri is selected from the group consisting of C ⁇ -6 alkyl and C fi - ⁇ 5 aryl .
- the process stereoselectively hydrolyses predominantly the ⁇ -anomer to form a product where R x of formula A and formula B is replaced with H.
- the ⁇ -anomer remains substantially unhydrolyzed.
- the process also comprises separating the hydrolyzed product from unhydrolyzed starting material.
- Scheme 1 depicts the manufacture of a mixture that includes formula A or B.
- An oxyacetaldehyde represented by formula 1A (wherein W is benzyl or benzoyl) is reacted with 1, 3 -dioxolane-4- (4R) carboxylic acid-2 , 2 -dimethyl -methyl ester (formula IB) in approximately equimolar proportions.
- the dioxolane of formula IB has a chiral center at the C4 carbon.
- the reaction occurs in a toluene solvent.
- the mixture is heated to 58°C.
- the catalyst, PTSA is added.
- the mixture is heated to a temperature between 64-67°C.
- a vacuum is applied at 70 kPa, and the reaction proceeds for 40 minutes. Traces of solvent are then removed by high vacuum.
- the catalyst is removed by filtration using a 1:1 ratio of HexanerEtOAc as an eluent .
- the preferred filter is a silica gel pad.
- the resulting product is a crude oil containing a mixture of the compounds of formula 1C and ID, wherein the ratio is 2:1 of (1C:1D) respectively.
- the reaction of the compound of formula 1A with the compound of formula IB is done in the presence of catalyst in an amount between about 1.0 wt% and 10.0 wt% of the starting material.
- the amount of catalyst is between about 2.5 wt% and about 5.5 wt% of the starting materials.
- the amount of catalyst is between about 3.0 wt% and about 5.0 wt%.
- the amount of catalyst is between about 3.5%. and about 5.5%.
- the amount of catalyst is between about 2.5 wt% and about 7.5 wt%.
- the amount of catalyst is about 5.0 wt%.
- the reaction of the compound of formula 1A with the compound of formula IB is done at a temperature ranging from about 40°C to about 80°C. In another embodiment of the present invention, the temperature ranges from about 50°C to about 75°C. In still another embodiment, the temperature ranges from about 60°C to about 70°C. In an additional embodiment, the temperature ranges from about 65°C to about 79°C.
- the reaction time between the compound of formula 1A and the compound of formula IB corresponds to a period ranging from about 30 minutes to about 2 hours. In yet another embodiment, the period ranges from about 30 minutes to about 1 hour. In still another embodiment, the period ranges from about 30 minutes to about 50 minutes.
- a starting material can be selected to have a (4S) or (4R) stereochemistry.
- the resulting product is an anomeric mixture favoring the ⁇ -L configuration over the ⁇ -L configuration.
- the starting material represented by formula IB (4S) is selected and shown below:
- the reaction proceeds according to the principles described above.
- the resulting product will have an anomeric purity of the ⁇ -L anomer over the ⁇ -L anomer of greater than 55%, preferably 60% and more preferably 65%.
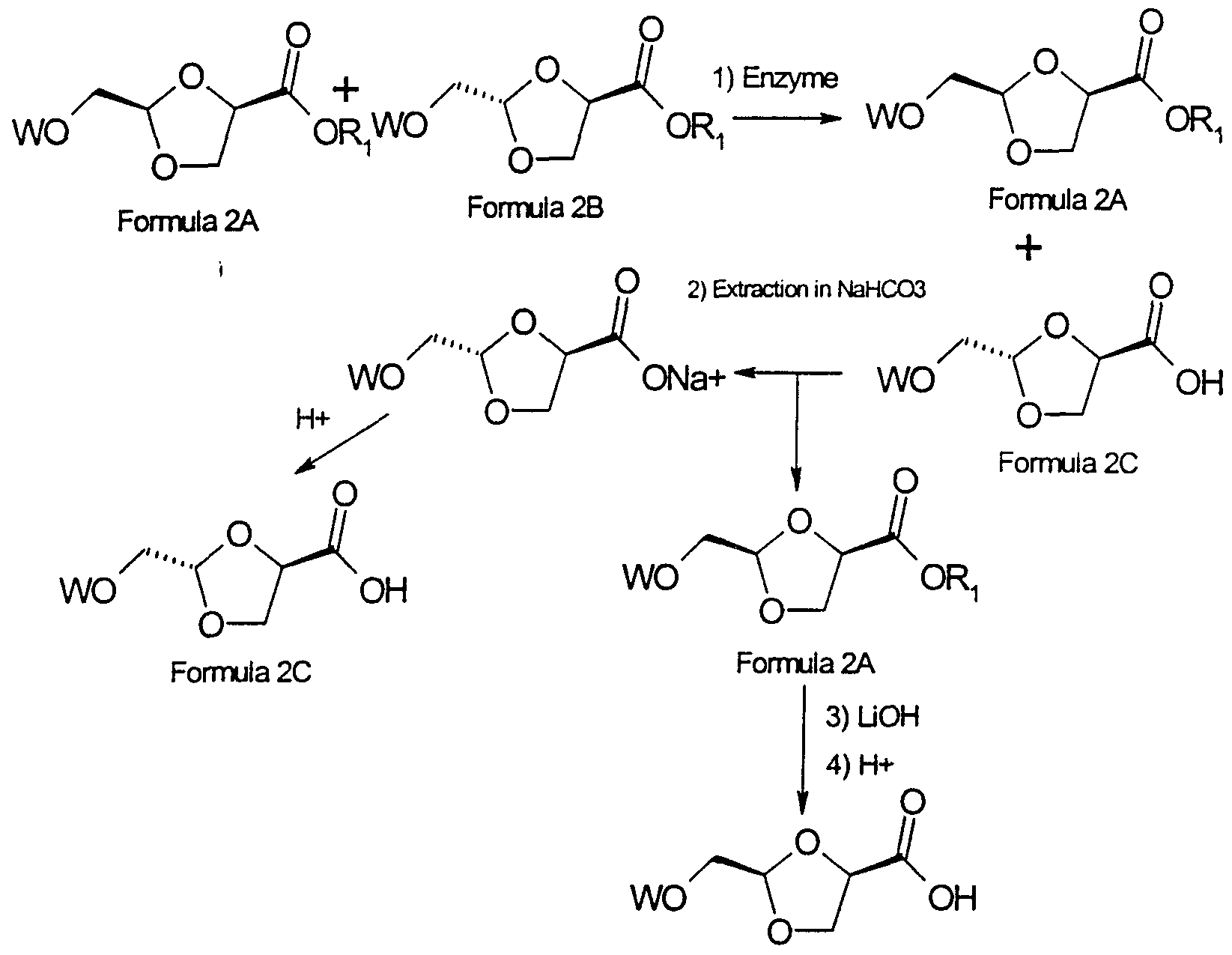
- the present invention is a method of separating ⁇ -anomers from ⁇ -anomers according to the following Scheme 2:
- a mixture of anomers is obtained as represented by formula 2A or formula 2B.
- a mixture represented by formula 2A or formula 2B can be obtained according to the reaction described above or according to any method known in the art .
- the reaction is prepared as follows: A portion of the material represented by formula 2A and formula 2B is weighed into a reaction vessel. For a small scale reaction, about 0.001 mmol of the mixture of formula 2A and formula 2B is added to about 46 mL of a 5 mmol solution of BES buffer (for a final concentration of about 2 mM in substrate) . For a preparative scale reaction, about 0.8 mmol of the mixture is added to about 10 mL of buffer (for a final concentration of about 80 mM in substrate) .
- the pH of the buffer should be between 7.0 and 7.5 and preferably 7.2.
- the enzyme is selected from the group consisting of cholesterol esterase, ESL-001-02, horse liver esterase, bovine pancreatic protease, ⁇ -chymotrypsin, protease from
- Stre oiTjyces caespi tosis substilisin from Bacillus licheniformis , protease from Aspergillus oryzae, proteinase from Bacillus licheniformis, protease from Streptomyces griseus, acylase from Aspergillus melleus, proteinase from Bacillus subtilis, ESL-001-5, pronase protease from Streptomyces griseus, lipase from Rhizopus arrhizus, lipoprotein lipase from Pseudomonas species type B, lipase from Pseudomonas cepacia and bacterial proteinase .
- Bovine cholesterol esterase was purchased from Genzyme (Cambridge, MA) ; ESL-001-02 from Diversa Corp. (San Diego, CA) ; Horse liver esterase and subtilisin from Bacillus licheniformis from Fluka Chemie (Oakville, ON); Bovine pancreas protease type 1, ⁇ - chymotrypsin and Streptomyces caespi tosis from Sigma- Aldrich (Oakville, ON) .
- the enzyme is selected from the group consisting of protease from Aspergillus oryzae, proteinase from Bacillus licheniformis, subtilisin from Bacillus licheniformis , protease from Streptomyces griseus, acylase from Aspergillus melleus , proteinase from Bacillus subtilis, ESL-001-005, pronase protease from Streptomyces griseus , lipase from Rhizopus arrhi zus , lipoprotein lipase from Pseudomonas species type B, bacterial proteinase and lipase from Pseudomonas cepacia .
- the selection of one of these enzymes is preferred according to this embodiment when the oxyacetaldehyde represented by the compound of formula 1A in Scheme 1 is selected to be benzoyloxyacetaldehyde .
- the oxyacetaldehyde represented by the compound of formula 1A is benzoyloxyacetaldehyde.
- the enzyme is selected from the group consisting of protease from Aspergillus oryzae, proteinase from Bacillus licheniformis , subtilisin from Bacillus licheniformis , pronase protease from Streptomyces griseus, and lipase from Rhizopus arrhizus .
- the enzyme is selected from the group consisting of protease from Aspergillus oryzae and proteinase from Bacillus licheniformis .
- the enzyme is selected from the group consisting of cholesterol esterase, ESL-001-02, horse liver esterase, bovine pancreatic protease, ⁇ - chymotrypsin, protease from Streptomyces caespi tosis and substilisin from Bacillus licheniformis .
- the selection of one of these enzymes is preferred according to this embodiment when the oxyacetaldehyde represented by the compound of formula 1A is benzyloxyacetaldehyde .
- the stereospecific enzyme selected is then added to begin the hydrolysis reaction.
- the enzymatic reaction hydrolyzes primarily the ⁇ -anomer by replacing the R x group of the ⁇ -anomer of the compound of formula 2B with H to form the compound of formula 2C.
- the amount of the enzyme added can be determined according to principles known by any person of ordinary skill in the art. According to another embodiment, about 500 ⁇ L was added to begin the reaction.
- the rate and degree of hydrolysis was monitored by a pH-stat according to principles known in the art,. As the compound of formula 2B is hydrolyzed, the pH of the mixture decreases. Thus, the change in pH as monitored by a pH stat corresponds to the completeness of the reaction.
- the reaction time is allowed to proceed longer than the optimal reaction time, the ⁇ -anomer may be converted resulting in lower anomeric purity of the final product. If the reaction time is too short, less than optimal amount of the ⁇ -anomer is converted resulting in a lower anomeric purity of the remaining unhydrolyzed reactant. According to one embodiment, the reaction is allowed to proceed until 43% completion. It will be appreciated by a person of ordinary skill in the art that the exact degree of completion may change depending upon the reactant used, the enzyme used and other principles known to a person of ordinary skill in the art.
- the ester starting material is separated from the hydrolyzed acid product when the desired endpoint is reached.
- the ester starting material and the hydrolysed product are separated by increasing the pH of the solution to more than pH 7.0 with bicarbonate solution and extracting with ethyl acetate (for example, 3 x 20 mL) .
- the unhydrolyzed starting material is extracted in the ethyl acetate and the hydrolyzed product remains in salt form in the aqueous solution.
- the pH of the solution is then adjusted to pH 2.
- the hydrolyzed product is further extracted with ethyl acetate (for example, 3 x 20 mL) .
- the reactants and the products are dried with MgS0 4 , filtered and concentrated in-vacuo .
- the unhydrolyzed product can be hydrolyzed by procedures known in the art such as reaction with LiOH followed by acidification.
- the anomeric purity of the hydrolyzed and separated ⁇ -anomer is considerable. Furthermore, the anomeric purity of the unhydrolyzed and separated ⁇ -anomer is also considerable. In one embodiment, the anomeric purity of the respective separated ⁇ -anomer and/or the ⁇ -anomer is greater than 80%. In another embodiment, the anomeric purity of the respective separated ⁇ -anomer and/or the ⁇ -anomer is greater than 90%. In another embodiment, the anomeric purity of the respective separated ⁇ -anomer and/or the ⁇ - anomer is greater than 95%. In another embodiment, the anomeric purity of the respective separated ⁇ -anomer and/or the ⁇ -anomer is greater than 98%.
- the ⁇ -anomer represented by formula 2E is hydrolyzed.
- the result is the separation of the hydrolyzed ⁇ -anomer represented by formula 2F from the unhydrolyzed ⁇ -anomer represented by formula 2D.
- the ⁇ -anomer containing both D and L enantiomers is hydrolyzed.
- the result is the separation of the hydrolyzed ⁇ -anomer containing both D and L enantiomers from the unhydrolyzed ⁇ -anomer containing both D and L enantiomers.
- the resulting dioxolane ring can be linked with a purine or pyrimidine base or analogue or derivative.
- a purine or pyrimidine base or analogue or derivative there are several examples known by skilled artisan on how to link a purine or pyrimidine base or analogue or derivative to the dioxolane ring.
- PCT Publ No.
- WO/97/21706 by Mansour et al . describes one method of stereoselectively attaching the purine or pyrimidine base or analogue or derivative to a dioxolane ring.
- WO/97/21706 is incorporated herein fully by reference.
- the starting material is an acylated dioxolane ring.
- the starting material of the procedure disclosed in WO/97/21706 can be obtained by oxidative decarboxylation of a product of Scheme 2 discussed above. Oxidative decarboxylation destroys the stereochemistry of the C4 carbon while preserving the stereochemistry of the C2 carbon.
- the oxidative decarboxylation step occurs after the hydrolysis step of Scheme 2.
- a compound having the desired stereochemistry on the C2 carbon is selected.
- it is dissolved in between about 2.5 and about 4.0 mL of acetonitrile.
- between about 3.0 and about 3.5 mL of acetonitrile was added for each mmol of compound.
- between about 3.3 and about 3.4 mL of acetonitrile was added for each mmol of compound.
- 0.12 mL of pyridine was added. In another embodiment, between about 0.09 and about 0.11 mL of pyridine was added for each mmol of compound. In yet another embodiment, approximately 0.1 mL of pyridine was added for each mmol of compound.
- the mixture was stirred for 18 hours at room temperature. Then, the mixture was poured into a saturated solution of NaHC0 3 . Between approximately 2.0 and 3.0 mL of NaHC0 3 were used for each mmol of compound. In one embodiment, between about 2.5 mL and about 2.7 mL, and more preferably about 2.6 mL of NaHC0 3 was used for each mmol of compound. The solution was then stirred for an additional 30 minutes. The organic layer was separated from the aqueous layer by four extractions of ethyl acetate. Extracts were combined, dried on anhydrous Na 2 S0 4 and evaporated under a vacuum.
- the crude can be further purified by chromatography on silica gel using a gradient of 0-15% ethyl acetate in hexane .
- the oxidative decarboxylation step is followed by glycosylation.
- the glycosylation is represented by the following Scheme 3.
- the first step in the glycosylation procedure is to obtain a compound with the desired stereospecificity at the C2 carbon.
- a compound having an S stereochemistry at the C2 carbon, as represented by the compound of formula 3A is preferred.
- the result is that a higher ratio of the ⁇ -L anomer is in the product 3C.
- compound having an R stereochemistry at the C2 carbon is preferred.
- the result is a product that has a higher ratio of the ⁇ -D anomer in the final product.
- the compound of formula 3A is reacted with an iodosilane to produce the compound of formula 3B.
- the iodosilane is iodotrimethylsilane .
- the iodosilane is diiodosilane .
- the temperature is preferably between 0 °C and -78°C prior to glycosylation with silylated pyrimidine or purine base or analogue or derivative thereof.
- the iodo intermediate represented by formula 3B is then dissolved in dichloromethane and is cooled down to between 0° C and -78° C.
- a purine or pyrimidine base or analogue or derivative thereof is then selected.
- the purine or pyrimidine base or analogue or derivative thereof is selected from the following group:
- R, R 9 and R Xi are each independently selected from the group consisting of hydrogen, C ⁇ -6 alkyl, C ⁇ -6 acyl and
- R 8 C(0) wherein R 8 is hydrogen or C ⁇ -6 alkyl
- R 3 , R 4 and R 10 are each independently selected from the group consisting of hydrogen, C ⁇ - 6 alkyl, bromine, chlorine, fluorine, iodine and CF 3 ;
- R 5 , R 6 and R 7 are each independently selected from the group consisting of hydrogen, bromine, chlorine, fluorine, iodine, amino, hydroxyl and C 3-6 cycloalkylamino .
- the purine or pyrimidine base or derivative is selected from the group consisting of:
- R 2 is selected from the group consisting of hydrogen, C 1-6 alkyl, C 1-6 acyl and R 8 C(0) wherein R 8 is hydrogen or C 1-6 alkyl.
- R 3 and R 4 are each independently selected from the group consisting of hydrogen, C ⁇ -6 alkyl, bromine, chlorine, fluorine, iodine and CF 3 ; and R 5 , R 6 and R 7 are each independently selected from the group consisting of hydrogen, bromine, chlorine, fluorine, iodine, amino, hydroxyl and C 3-6 cycloalkylamino.
- the purine or pyrimidine base or analogue or derivative thereof is selected from the group consisting of:
- R 9 and R ⁇ are independently selected from the group consisting of hydrogen, C ⁇ -6 alkyl, C ⁇ -6 acyl and R 8 C(0) .
- Rio is selected from the group consisting of hydrogen, C ⁇ . 6 alkyl, bromine, chlorine, fluorine, iodine and CF 3 .
- the purine or pyrimidine or analogue or derivative thereof is persylated by a sylating agent and ammonium sulphate followed by evaporation of HMDS to form a persylated purine or pyrimidine base or analogue or derivative thereof herein referred to as the persylated base and designated as P in Scheme 3.
- the sylating agent is selected from the group consisting of 1, 1, 1, 3 , 3 , 3-hexamethyldisilazane, trimethylsilyl triflate, t-butyldimethylsilyl triflate or trimethylsilyl chloride.
- the sylating agent is 1 , 1 , 1 , 3 , 3 , 3 , -hexamethyldisilazane .
- the persylated base P was dissolved in 30 mL of dichloromethane and was added to the iodo intermediate represented by formula 3B.
- the reaction mixture was maintained at between 0 and -78°C for 1.5 hours then poured onto aqueous sodium bicarbonate and extracted with dichloromethane (2x25 mL) .
- the organic phase was dried over sodium sulphate to obtain the compound of formula 3C.
- the B represents a moiety of the purine or pyrimidine base or analogue or derivative thereof which was persylated in the above step to form P.
- the compound of formula 3C was removed by filtration and the solvent was evaporated in-vacuo to produce a crude mixture.
- the product represented by formula 3C has predominantly a 4S configuration at the C4 carbon with an anomeric purity of 80%.
- the starting material is a compound represented by formula 3A
- the product forms predominantly the ⁇ -L enantiomer having an anomeric purity of 80%.
- the compound of formula 3C is deprotected to produce the compound of formula 3D. This can be accomplished by dissolving a compound represented by formula 3C in ethanol and then adding cyclohexene and palladium oxide.
- the deprotection step can also be done by other methods which are well known by those skilled in the art.
- the reaction mixture is refluxed for 7 hours. It is then cooled and filtered to remove solids. The solvents are removed from the filtrate by vacuum distillation.
- the product represented by formula 3D is purified by flash chromatography on silica-gel (5% MEOH in ethylacetate) .
- the deprotection step can also be done by other methods that are well known by a person skilled in
- the alcohol is of the formula RiOH wherein R x is a C -4 alkyl .
- the alcohol is methanol or ethanol . The resulting solid was removed by filtration.
- the clear solution was stirred at room temperature for 30-40 hours, the alcohol removed by vacuum to yield a D-glycerate in the form of a yellow viscous syrup.
- the D-glycerate is then reacted with between about 0.9-1.1 eq of a dialkyl acetal at a temperature of about 85-95°C.
- suitable dialkyl acetals include benzoyloxyacetaldehyde dialkyl acetal and benzyloxyacetaldehyde dialkyl acetal .
- suitable alkyls for the dialkyl acetal is methyl and ethyl .
- PTSA is added.
- the reaction mixture is kept under vacuum at a temperature between 85-95°C for 2-3 hours.
- the mixture is then cooled to room temperature, diluted with ethylacetate (250 mL) and poured onto saturated sodium bicarbonate solution (250 mL) under stirring.
- the organic phase is separated and the aqueous phase concentrated, purified on a silica gel column eluting with 5-10% ethylacetate/hexanes to yield the desired dioxolane as a light yellow oil (about 59%) with ⁇ / ⁇ ratio of 2:1 or higher.
- the reactants of step 3 of Scheme 4 can be substituted by corresponding reactants of Scheme 1.
- the D-glycerate represented by Formula 4C is replaced with an 1,3 dioxolane-4 - (4R) carboxylic acid-2,2- dimethyl alkyl ester represented by Formula IB according to one embodiment.
- the dialkyl acetal represented by Formula 4D is replaced with an oxyaldehyde represented by Formula 1A.
- the starting material of Scheme 4 is L-Serine which produces an end product having an S-configuration at the C4 carbon of the dioxolane ring.
- the L-glycerate of Step 3 can be replaced with an 1,3 dioxolane-4 - (4S) carboxylic acid-2 , 2 -dimethyl alkyl ester to produce an end product having predominantly an S-configuration at the C4 carbon of the resulting dioxolane ring.
- Example 1 Enzyme catalyzed hydrolytic resolution of the dioxolane methyl ester.
- ⁇ - Chymotrypsin 500 ⁇ L of a 5mg/mL BES buffer, pH 7.2 solution, ⁇ .019 units by 4-nitrophenylacetate assay
- a pH-stat which maintained the pH at 7 by automatic titration with 0.0981 mmol NaOH.
- the reaction was terminated at 43% conversion for high anomeric purity as determined by Sih's equations for recycling (Chen. C.S.; Fujimoto, Y.; Girdaukas, G.; Sih, C.J., J.AM. Chem. Soc .
- the product acid is obtained from example 1 after it was dried with MgS0 4 , filtered and concentrated in-vacuo. It is analyzed for purity by NMR. The ⁇ -anomer is isolated with high anomeric purity.
- Example 4 Enzymatic Resolution of ⁇ -anomer with Protease from Asvercrillus oryzae .
- Examples 1-2 were followed using protease from Aspergillus oryzae as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2-(S)- benzoyloxymethyl) -4 -carboxylic acid-1 , 3 -dioxolane methyl ester) .
- the result is a ⁇ -anomer that has high anomeric purity.
- Example 5 Enzymatic Resolution of ⁇ -anomer with Protease from Asvercrillus oryzae .
- Example 6 Enzymatic Resolution of ⁇ -anomer with Proteinase from Bacillus licheniformis .
- Examples 1-2 were followed using proteinase from bacillus licheniformis as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2-(S)- benzoyloxymethyl) -4 -carboxylic acid-1, 3 -dioxalane methyl ester) .
- the result is a ⁇ -anomer that has high anomeric purity.
- Example 7 Enzymatic Resolution of ⁇ -anomer with Proteinase from Bacillus licheniformis .
- the product acid is obtained from Example 6 after it is dried with MgS0 4 , filtered and concentrated in -vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 8 Enzymatic Resolution of ⁇ -anomer with Subtilisin from Bacillus licheniformis .
- Example 9 Enzymatic Resolution of ⁇ -anomer with Subtilisin from Bacillus licheniformis .
- the product acid is obtained from Example 8 after it is dried with MgS0 4 , filtered and concentrated in -vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 10 Enzymatic Resolution of ⁇ -anomer with Protease from 5treptoi ⁇ yceg griseus .
- Examples 1-2 were followed using protease from streptomyces griseus as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2-(S)- benzoyloxymethyl) -4 -carboxylic acid-1 , 3 -dioxolane methyl ester) .
- the result is a ⁇ -anomer that has high anomeric purity.
- Example 11 Enzymatic Resolution of ⁇ -anomer with Protease from Streptomyces griseus .
- the product acid is obtained from Example 10 after it is dried with MgS0 4 , filtered and concentrated in -vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 12 Enzymatic Resolution of ⁇ -anomer with Acylase from AsOergillus melleus .
- Examples 1-2 were followed using acylase from aspergillus melleus as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2- (S) -benzoyloxymethyl ) -4 - carboxylic acid-1 , 3 -dioxolane methyl ester). The result is a ⁇ -anomer that has high anomeric purity.
- Example 13 Enzymatic Resolution of ⁇ -anomer with Acylase from Aspergillus melleus .
- the product acid is obtained from Example 12 after it is dried with MgS0 4 , filtered and concentrated in -vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 14 Enzymatic Resolution of ⁇ -anomer with Proteinase ⁇ from Bacillus subtilis .
- Examples 1-2 were followed using proteinase from bacillus subtilis as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2-(S)- benzoyloxymethyl) -4 -carboxylic acid-1 , 3 -dioxolane methyl ester) .
- the result is a ⁇ -anomer that has high anomeric purity.
- Example 15 Enzymatic Resolution of ⁇ -anomer with Proteinase from Bacillus subtilis .
- the product acid is obtained from Example 14 after it is dried with MgS0 , filtered and concentrated in -vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 16 Enzymatic Resolution of ⁇ -anomer with ESL- 001-05
- Examples 1-2 were followed using diversa clonezymes #5 as the enzyme to separate a 2:1 ( ⁇ .- ⁇ ) anomeric mixture of (2- (S) -benzoyloxymethyl) -4 -carboxylic acid-1, 3 -dioxolane methyl ester).
- the result is a ⁇ - anomer that has high anomeric purity.
- the product acid is obtained from Example 16 after it is dried with MgS0 4 , filtered and concentrated in -vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 18 Enzymatic Resolution of ⁇ -anomer with Pronase protease from Streptomyces griseus .
- Examples 1-2 were followed using pronase from streptomyces as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2- (S) -benzoyloxymethyl) -4 -carboxylic acid-1, 3 -dioxolane methyl ester).
- the result is a ⁇ - anomer that has high anomeric purity.
- Example 19 Enzymatic Resolution of ⁇ -anomer with Pronase protease from Streptomyces griseus .
- the product acid is obtained from Example 18 after it is dried with MgS0 , filtered and concentrated in-vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 20 Enzymatic Resolution of ⁇ -anomer with Lipase from Rhizopus arrhizus .
- Example 21 Enzymatic Resolution of ⁇ -anomer with Lipase from Rhizopus arrhizus .
- the product acid is obtained from Example 20 after it is dried with MgS0 4 , filtered and concentrated in -vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 22 Enzymatic Resolution of ⁇ -anomer with Lipoprotein Lipase from Pseudomonas Species Type B.
- Examples 1-2 were followed using lipoprotein lipase from pseudomonas sp. type B as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2- (S) -benzoyloxymethyl) -4-carboxylic acid-1, 3-dioxolane methyl ester) .
- the result is a ⁇ -anomer that has high purity.
- Example 23 Enzymatic Resolution of ⁇ -anomer with Lipoprotein lipase from Pseudomonas Species Type B.
- the product acid is obtained from Example 22 after it is dried with MgS0 4 , filtered and concentrated in-vacuo. The ⁇ -anomer is isolated with high anomeric purity.
- Example 24 Enzymatic Resolution of ⁇ -anomer with Bacterial Proteinase.
- Examples 1-2 were followed using bacterial proteinase as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2- (S) -benzoyloxymethyl) -4- carboxylic acid-1 , 3 -dioxolane methyl ester).
- the result is a ⁇ -anomer that has high anomeric purity.
- Example 25 Enzymatic Resolution of ⁇ -anomer with Bacterial Proteinase.
- Example 26 Enzymatic Resolution of ⁇ -anomer with Lipase from Pseudomonas cepacia .
- Examples 1-2 were followed using lipase from pseudomonas cepacia as the enzyme to separate a 2 : 1 ( ⁇ : ⁇ ) anomeric mixture of (2- (S) -benzoyloxymethyl) -4- carboxylic acid-1 , 3 -diaxolane methyl ester).
- the result is a ⁇ -anomer that has high anomeric purity.
- Example 27 Enzymatic Resolution of ⁇ -anomer with Lipase from Pseudomonas cepacia .
- the product acid is obtained from Example 26 after it is dried with MgS0 4 , filtered and concentrated in-vacuo. The ⁇ -anomer is isolated with high anomeric purity.
- Example 28 Enzymatic Resolution of ⁇ -anomer with Cholesterol esterase.
- Examples 1-2 were followed using cholesterol esterase as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2- (S) -benzyloxymethyl) -4- carboxylic acid-1, 3 -diaxolane methyl ester).
- the result is a ⁇ -anomer that has high anomeric purity.
- the product acid is obtained from Example 28 after it is dried with MgS0 4 , filtered and concentrated in -vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 30 Enzymatic Resolution of ⁇ -anomer with ESL-001-02.
- Examples 1-2 were followed using ESL- 001-02 as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2- (S) -benzyloxymethyl) -4-carboxylic acid-1, 3- dioxolane methyl ester) .
- the result is a ⁇ -anomer that has high anomeric purity.
- the product acid is obtained from Example 30 after it is dried with MgS0 , filtered and concentrated in-vacuo.
- the ⁇ -anomer is isolated with high anomeric purity.
- Examples 1-2 were followed using horse liver esterase as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2- (S) -benzyloxymethyl) -4-carboxylic acid-1, 3 -diaxolane methyl ester).
- the result is a ⁇ - anomer that has high anomeric purity.
- Example 33 Enzymatic Resolution of ⁇ -anomer with Horse Liver Esterase.
- the product acid is obtained from Example 32 after it is dried with MgS0 4 , filtered and concentrated in-vacuo. The ⁇ -anomer is isolated with high anomeric purity.
- Example 34 Enzymatic Resolution of ⁇ -anomer with Bovine Pancreatic Protease.
- Examples 1-2 were followed using bovine pancreatic protease as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2- (S) -benzyloxymethyl) -4-carboxylic acid-1, 3-dioxolane methyl ester).
- the result is a ⁇ - anomer that has high anomeric purity.
- the product acid is obtained from Example 34 after it is dried with MgS0 4 , filtered and concentrated in-vacuo .
- the ⁇ -anomer is isolated with high anomeric purity.
- Example 36 Enzymatic Resolution of ⁇ -anomer Protease from Streptomyces caespitosus .
- Examples 1-2 were followed using protease from streptomyces caespitosis as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2-(S)- benzyloxymethyl) -4-carboxylic acid-1 , 3 -dioxolane methyl ester) .
- the result is a ⁇ -anomer that has high anomeric purity.
- Example 37 Enzymatic Resolution of ⁇ -anomer with Protease from Strep tomyces caespi tosus .
- the product acid is obtained from Example 36 after it is dried with MgS0 4 , filtered and concentrated in -vacuo. The ⁇ -anomer is isolated with high anomeric purity.
- Example 38 Enzymatic Resolution of ⁇ -anomer with Substilisin from Bacillus licheniformis .
- Examples 1-2 were followed using substilisin from bacillus licheniformis as the enzyme to separate a 2:1 ( ⁇ : ⁇ ) anomeric mixture of (2-(S)- benzyloxymethyl) -4-carboxylic acid-1 , 3-dioxolane methylester) .
- the result is a ⁇ -anomer that has high anomeric purity.
- Example 39 Enzymatic Resolution of ⁇ -anomer with Substilisin from Bacillus licheniformis .
- Example 40 Preparation of 2 - (S) -Benzyloxymethyl -4- (R) - iodo-1, 3-dioxolane and 2- (S) -Benzyloxymethyl -4- (S) -iodo- 1.3-dioxolane (Compound 40).
- examples 44-46 illustrate a method of preparing the starting material of example 1 (2-(S)- benzyloxymethyl -4 -carboxylic acid-1 , 3-dioxolane methyl ester) .
- Example 46 Preparation of 2- (R, S) -benzoyloxymethyl-4-R- methylcarboxylate-1, 3-dioxolane.
- Example 47 illustrates a large scale version of the enzymatic separation of Example 1.
- Example 47 Large scale preparation of ⁇ 2-(R)- benzoyloxymethyl-1, 3-dioxolane-4- (R) -methylcarboxylate.
- total volume 46 mL total volume 46 mL
- the pH was constantly monitored and maintained between 7.1 and 7.2 while the temperature was kept at 30°C. After six hours, the mixture was extracted with EtOAc (1X80 mL) . Then, the organic layer was separated, and the aqueous layer was extracted with EtOAc (2X50mL) . Combined organic layers were washed with saturated NaHC0 3
- Example 48 Preparation of ⁇ 2- (R) -benzoyloxymethyl-1.3- dioxolane-4- (R) -carboxylic acid.
- aqueous phase was acidified by 30% H 2 S0 (9.5 mL) under tight pH-meter control (initial pH:8.36 to 3.02) then extracted with DCM (4X60mL) .
- the organic phases were combined and the solvent removed under vacuum to furnish a light green syrup (14.26 g) which was kept under vacuum overnight.
- Example 49 Preparation of ⁇ 2- (R) -benzoyloxymethyl -4- (R. S) -methylcarboxylate-1 , 3-dioxolane .
- the filtrate was again filtered through a small pad of celite (about 1 inch) to remove the black lead salts to yield a pale yellow mixture.
- the organic phase was separated and the aqueous phase was extracted with ethylacetate (4X2L) .
- the combined organic phase was concentrated, and the oil obtained was co-evaporated with toluene (3X2L) to yield a brown syrup .
- This syrup (374 g) was further purified by filtering through a small pad of silica gel (1 g crude; 2 g silica), eluting with 3.5 L of the solvent mixture (ethyl acetate: hexanes 8:2) to yield 332,3 g (74%) of pure product. This last filtration step is optional.
- Example 50 Preparation of 9- (2 - (R) -benzyloxymethyl-l , 3- dioxolan-4-yl) -6-chloro-2-amino purine (Compound 50a) and 9- (2- (R) -benzyloxymethyl- 1, 3 -dioxolan-4 -yl) -6- iodo-2- amino purine (Compound 50b) .
- the desired final product is the same compound but with opposite stereochemistry (i.e. a 2:1 mixture of ⁇ : ⁇ stereoisomers in the L-configuration) .
- opposite stereochemistry i.e. a 2:1 mixture of ⁇ : ⁇ stereoisomers in the L-configuration
- the sugar 2 - (R) -benzyloxymethyl -4 -carboxyl - 1 , 3 -dioxolane is replaced with 2- (S) -benzyloxymethyl -4 -carboxyl-1, 3- dioxolane .
- Example 52 Preparation of 9- (2- (R) -hydroxymethyl-1, 3- dioxolan-4-yl) -6- (N-eyelopropyl) amino-2 -amino purine (Compound 52) .
- the ⁇ / ⁇ isomers were separated by chromatography on silica gel using DCM/MeOH as eluent to yield 1.18 g (70% ⁇ isomer) .
- Example 53 Preparation of 9- (2- (R) -hydroxymethyl-1, 3- dioxolan-4-yl-6- (N-2-cyclopropyl-2 -aminomethoxyl) -2-amino purine (Compound 53).
- Example 54 Preparation of 9- (2- (S) -hydroxymethyl-1, 3 - dioxolan-4 -yl) -2-amino purine (Compound 54).
- Example 50 The procedure of Example 50 was performed. Thereafter 6.3 g of Compound 50 was subject to hydrogenation conditions under 50 psi of hydrogen over 10% Pd/c in 300 mL of ethanol containing 100 mL of triethylamine . After 3 hours of shaking, the catalyst was removed by filtration. Then the sovent was evaporated to yield a solid which was recrystallised to from ethanol -ether to give about 4 g of Compound 54 having about a 2 : 1 mixture of ⁇ : ⁇ stereoisomers in the L-configuration.
- the desired final product is the same compound but with opposite stereochemistry (i.e. about a
- Example 50 The procedures set forth in Examples 50 and 51 were performed. However, when following the steps of Example 50, the 1 equivalent of the silated 2 -amino- 6- chloropurine is replaced with 1 equivalent of silated 6- aminopurine. The result is a yield of 9-(2-(S)- hydroxymethyl-1, 3 -dioxolan-4 -yl) -6-amino purine having a ⁇ : ⁇ ratio of about 2:1.
- the desired final product is the same compound but with opposite stereochemistry (i.e. a 2:1 mixture of ⁇ : ⁇ stereoisomers in the D-configuration) .
- opposite stereochemistry i.e. a 2:1 mixture of ⁇ : ⁇ stereoisomers in the D-configuration
- Example 50 The procedure of Example 50 was performed. Thereafter, 6 g of Compound 50 was dissolved in 0.9 L of methanol saturated at 0°C with dry ammonia and the solution is heated in a steel bomb to 105°C to 110°C for 16 hours. The solution was evaporated to dryness and the residue purified by chromatography on silica gel using chloroform-methanol (4:1) as the eluent to give about 3g of crude Compound 56. The product can be recrystallised from methanol -ether to yield purified Compound 56 having a ⁇ : ⁇ ratio of about 2:1.
- Example 50 The procedure of Example 50 was performed. Thereafter, about 6 g of Compound 50 was dissolved in a mixture of 200 mL of methanol, 50 mL of water and 10 g of NaoH . The solution was heated under reflux for 5 hours after which time it was diluted with 300 mL of water and excess pyridinium sulfonate resin. The slurry was filtered, the resin washed with water and the combined aqueous filtrates were evaporated to dryness in vacuo to leave a residue which was taken up in 50% aqueous methanol. The solution was treated with activated charcoal, filtered and the filtrate evaporated to dryness in vacuo to give a solic residue that was recrystallized from ethanol water to yield pure compound 57 having a ⁇ : ⁇ ratio of about 2:1.
- Example 58 Preparation of 9-(2-(S) hvdroxymethyl-1, 3- dioxolan-4-yl) -2 -oxo-4 -amino- 5-methyl pyrimidine (Compound 58) .
- Example 50 The procedure of Example 50 was performed followed by the procedure of Example 52. However, when following the steps of Example 50, the 1 equivalent of the silated 2- amino-6-chloropurine is replaced with 1 equivalent of silated 2 -oxo-4 -amino- 5 -methyl -pyrimidine . The result is a yield of 9- (2- (S) -hydroxymethyl-1 , 3 -dioxolan-4 -yl) -2- oxo-4 -amino- 5 -methyl pyrimidine having a ⁇ : ⁇ ratio of about 2:1.
- Example 59 Preparation of 9-(2-(S) hydroxymethyl-1, 3- dioxolan-4-yl) -2 -oxo-4-amino-5-fluoro pyrimidine (Compound 59) .
- Example 50 The procedure of Example 50 was performed followed by the procedure of Example 52. However, when following the steps of Example 50, the 1 equivalent of the silated 2- amino-6-chloropurine is replaced with 1 equivalent of silated 2-oxo-4-amino-5-fluoro-pyrimidine . The result is a yield of 9-(2-(S) hydroxymethyl-1 , 3-dioxolan-4 -yl) -2- oxo-4-amino-5-fluoro pyrimidine having a ⁇ : ⁇ ratio of about 2:1.
- Example 50 The procedure of Example 50 was performed followed by the procedure of Example 52. However, when following the steps of Example 50, the 1 equivalent of the silated 2- amino-6-chloropurine is replaced with 1 equivalent of silated 2, 4-dioxo pyrimidine. The result is a yield of 9- (2- (S) -hydroxymethyl-1, 3-dioxolan-4-yl) -2 , -dioxo pyrimidine having a ⁇ .- ⁇ ratio of about 2:1.
- Example 61 Preparation of 9-(2-(S) hydroxymethyl-1.3 - dioxolan-4-yl) -2 , 4-dioxo-5-methyl pyrimidine (Compound 61) .
- Example 50 The procedure of Example 50 was performed followed by the procedure of Example 52. However, when following the steps of Example 50, the 1 equivalent of the silated 2- amino-6-chloropurine is replaced with 1 equivalent of silated 2 , 4-dioxo-5-methyl pyrimidine. The result is a yield of 9-(2-(S) hydroxymethyl-1 , 3-dioxolan-4-yl) -2 , 4- dioxo-5-methyl pyrimidine having a ⁇ : ⁇ ratio of about 2:1.
- the desired final product is the same compound but with opposite stereochemistry (i.e. a 2:1 mixture of ⁇ : ⁇ stereoisomers in the D-configuration) .
- opposite stereochemistry i.e. a 2:1 mixture of ⁇ : ⁇ stereoisomers in the D-configuration
Abstract
Description








































Claims



Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP00904756A EP1151133A1 (en) | 1999-02-11 | 2000-02-11 | Stereoselective synthesis of nucleoside analogues |
| AU26532/00A AU2653200A (en) | 1999-02-11 | 2000-02-11 | Stereoselective synthesis of nucleoside analogues |
| CA002362570A CA2362570A1 (en) | 1999-02-11 | 2000-02-11 | Stereoselective synthesis of nucleoside analogues |
| JP2000598654A JP2002538780A (en) | 1999-02-11 | 2000-02-11 | Stereoselective synthesis of nucleoside analogs |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11975699P | 1999-02-11 | 1999-02-11 | |
| US60/119,756 | 1999-02-11 | ||
| US11988599P | 1999-02-12 | 1999-02-12 | |
| US60/119,885 | 1999-02-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000047759A1 true WO2000047759A1 (en) | 2000-08-17 |
Family
ID=26817660
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CA2000/000144 WO2000047759A1 (en) | 1999-02-11 | 2000-02-11 | Stereoselective synthesis of nucleoside analogues |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1151133A1 (en) |
| JP (1) | JP2002538780A (en) |
| AU (1) | AU2653200A (en) |
| CA (1) | CA2362570A1 (en) |
| WO (1) | WO2000047759A1 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001058894A1 (en) * | 2000-02-11 | 2001-08-16 | Shire Biochem Inc. | Stereoselective synthesis of nucleoside analogues |
| WO2002051852A1 (en) * | 2000-12-27 | 2002-07-04 | Mitsui Chemicals, Inc. | Process for producing nonnatural saccharide derivative |
| US6566365B1 (en) | 1999-11-04 | 2003-05-20 | Biochem Pharma Inc. | Method for the treatment of Flaviviridea viral infection using nucleoside analogues |
| US6583149B1 (en) | 1999-09-24 | 2003-06-24 | Biochem Pharma Inc. | Method for the treatment or prevention of viral infection using nucleoside analogues |
| WO2004048590A1 (en) * | 2002-11-18 | 2004-06-10 | Shire Biochem Inc. | Stereoselective process for the production of dioxolane nucleoside analogues |
| US7560550B2 (en) * | 2003-07-31 | 2009-07-14 | Rfs Pharma, Llc | Method for the production of OH protected[4-(2.6-diamino-9H-purine-9-yl)-1.3-dioxolane-2-yl]methanol derivatives |
| US7785839B2 (en) | 2004-02-03 | 2010-08-31 | Emory University | Methods to manufacture 1,3-dioxolane nucleosides |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DD277698A1 (en) * | 1988-12-06 | 1990-04-11 | Ve Forschungszentrum Biotechno | PROCESS FOR PREPARING SUBSTITUTED (S) -1,3-DIOXOLAN-4-CARBOXYLIC ACID ESTERS FROM THE RACEMATES |
| US5190867A (en) * | 1986-05-08 | 1993-03-02 | Shell International Petroleum Company, Ltd. | Process for the preparation of R-2,2-R1,R2 -1,3-dioxolane-4-methanol |
| US5276151A (en) * | 1990-02-01 | 1994-01-04 | Emory University | Method of synthesis of 1,3-dioxolane nucleosides |
| US5283346A (en) * | 1989-03-23 | 1994-02-01 | Hoffmann-La Roche Inc. | Dioxolanes |
| WO1997021706A1 (en) * | 1995-12-14 | 1997-06-19 | Biochem Pharma Inc. | METHOD AND COMPOSITIONS FOR THE SYNTHESIS OF DIOXOLANE NUCLEOSIDES WITH β-CONFIGURATION |
-
2000
- 2000-02-11 AU AU26532/00A patent/AU2653200A/en not_active Abandoned
- 2000-02-11 WO PCT/CA2000/000144 patent/WO2000047759A1/en not_active Application Discontinuation
- 2000-02-11 JP JP2000598654A patent/JP2002538780A/en active Pending
- 2000-02-11 EP EP00904756A patent/EP1151133A1/en not_active Withdrawn
- 2000-02-11 CA CA002362570A patent/CA2362570A1/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5190867A (en) * | 1986-05-08 | 1993-03-02 | Shell International Petroleum Company, Ltd. | Process for the preparation of R-2,2-R1,R2 -1,3-dioxolane-4-methanol |
| DD277698A1 (en) * | 1988-12-06 | 1990-04-11 | Ve Forschungszentrum Biotechno | PROCESS FOR PREPARING SUBSTITUTED (S) -1,3-DIOXOLAN-4-CARBOXYLIC ACID ESTERS FROM THE RACEMATES |
| US5283346A (en) * | 1989-03-23 | 1994-02-01 | Hoffmann-La Roche Inc. | Dioxolanes |
| US5276151A (en) * | 1990-02-01 | 1994-01-04 | Emory University | Method of synthesis of 1,3-dioxolane nucleosides |
| WO1997021706A1 (en) * | 1995-12-14 | 1997-06-19 | Biochem Pharma Inc. | METHOD AND COMPOSITIONS FOR THE SYNTHESIS OF DIOXOLANE NUCLEOSIDES WITH β-CONFIGURATION |
Non-Patent Citations (1)
| Title |
|---|
| JANES, LANA E. ET AL: "Protease -Mediated Separation of Cis and Trans Diastereomers of 2(R,S)-benzyloxymethyl-4(S)-carboxylic Acid 1,3- Dioxolane Methy Ester: Intermediates for the Synthesis of Dioxolane Nucleosides", J. ORG. CHEM. (1999), 64(25), 9019-9029, 19 November 1999 (1999-11-19), XP002137411 * |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6964968B2 (en) | 1999-09-24 | 2005-11-15 | Shire Biochem, Inc. | Method for the treatment or prevention of viral infection using nucleoside analogues |
| US6583149B1 (en) | 1999-09-24 | 2003-06-24 | Biochem Pharma Inc. | Method for the treatment or prevention of viral infection using nucleoside analogues |
| US6566365B1 (en) | 1999-11-04 | 2003-05-20 | Biochem Pharma Inc. | Method for the treatment of Flaviviridea viral infection using nucleoside analogues |
| US6541625B2 (en) | 2000-02-11 | 2003-04-01 | Biochem Pharma, Inc. | Stereoselective synthesis of nucleoside analogues |
| US6743910B2 (en) | 2000-02-11 | 2004-06-01 | Biochem Pharma, Inc. | Stereoselective synthesis of nucleoside analogues |
| WO2001058894A1 (en) * | 2000-02-11 | 2001-08-16 | Shire Biochem Inc. | Stereoselective synthesis of nucleoside analogues |
| WO2002051852A1 (en) * | 2000-12-27 | 2002-07-04 | Mitsui Chemicals, Inc. | Process for producing nonnatural saccharide derivative |
| WO2004048590A1 (en) * | 2002-11-18 | 2004-06-10 | Shire Biochem Inc. | Stereoselective process for the production of dioxolane nucleoside analogues |
| JP2006506096A (en) * | 2002-11-18 | 2006-02-23 | シェア バイオケム インコーポレーテッド | Stereoselective process for the preparation of dioxolane nucleoside analogues |
| AU2003285230B2 (en) * | 2002-11-18 | 2010-04-22 | Takeda Pharmaceutical Company Limited | Stereoselective process for the production of dioxolane nucleoside analogues |
| US7955835B2 (en) | 2002-11-18 | 2011-06-07 | Shire Canada Inc. | Stereoselective process for the production of dioxolane nucleoside analogues |
| US7560550B2 (en) * | 2003-07-31 | 2009-07-14 | Rfs Pharma, Llc | Method for the production of OH protected[4-(2.6-diamino-9H-purine-9-yl)-1.3-dioxolane-2-yl]methanol derivatives |
| US7785839B2 (en) | 2004-02-03 | 2010-08-31 | Emory University | Methods to manufacture 1,3-dioxolane nucleosides |
| US8420354B2 (en) | 2004-02-03 | 2013-04-16 | Emory University | Methods to manufacture 1,3-dioxolane nucleosides |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002538780A (en) | 2002-11-19 |
| AU2653200A (en) | 2000-08-29 |
| EP1151133A1 (en) | 2001-11-07 |
| CA2362570A1 (en) | 2000-08-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100381705B1 (en) | Method for the Stereoselective Synthesis of BCH-189 and Related Compounds, and New Analogs of BCH-189 | |
| CN1070191A (en) | 1, the 3-oxathiolane nucleoside analogues | |
| EP0182024A2 (en) | Purine derivatives and their pharmaceutical use | |
| US6541625B2 (en) | Stereoselective synthesis of nucleoside analogues | |
| WO2000047759A1 (en) | Stereoselective synthesis of nucleoside analogues | |
| KR100851515B1 (en) | Method for producing 2-azetidinone derivative | |
| US8420354B2 (en) | Methods to manufacture 1,3-dioxolane nucleosides | |
| US6677120B2 (en) | Building blocks for the solution phase synthesis of oligonucleotides | |
| JP4658807B2 (en) | Process for producing α-1-phosphorylated 2-deoxy-2-fluoroarabinoside and 2'-deoxy-2'-fluoro-β-D-arabinonucleoside | |
| EP0869185B9 (en) | Production of optically active sphingoid compound | |
| JP3010382B2 (en) | Method for producing (R) -2-propoxybenzene derivative | |
| JP3545442B2 (en) | Method for producing optically active 4- (2-halo-1-hydroxyethyl) -2-trifluoromethylthiazole | |
| EP0747487A2 (en) | Method for production of optically active (+)-4,4,4-trifluoro-3-(indole-3-)butyric acid | |
| JP3173850B2 (en) | Method for producing optically active inositol derivative | |
| JPH0690790A (en) | Production of optically active 1,4-dihydropyridine-3,5-dicarboxilic acid monoester derivative | |
| JPH0588114B2 (en) | ||
| JPH10248592A (en) | Production of optically active 2-benzylsuccinic acid and its derivative | |
| JP2000109455A (en) | Compound having accelerating action on expression of apolipoprotein ai gene | |
| JPH07231794A (en) | Preparation of penem kind |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2362570 Country of ref document: CA Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2000 598654 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000904756 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000904756 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09890283 Country of ref document: US |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2000904756 Country of ref document: EP |