WO2000034308A2 - Protein transduction system and methods of use thereof - Google Patents
Protein transduction system and methods of use thereof Download PDFInfo
- Publication number
- WO2000034308A2 WO2000034308A2 PCT/US1999/029289 US9929289W WO0034308A2 WO 2000034308 A2 WO2000034308 A2 WO 2000034308A2 US 9929289 W US9929289 W US 9929289W WO 0034308 A2 WO0034308 A2 WO 0034308A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- protein
- tat
- cells
- domain
- protein transduction
- Prior art date
Links
- 0 CC1**CC1 Chemical compound CC1**CC1 0.000 description 4
- GDOPTJXRTPNYNR-UHFFFAOYSA-N CC1CCCC1 Chemical compound CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16311—Human Immunodeficiency Virus, HIV concerning HIV regulatory proteins
- C12N2740/16322—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
Definitions
- the present invention relates to a protein transduction system that selectively kills or injures diseased or pathogen-infected cells by introducing into the cells a fusion protein comprising a protein transduction domain and a cytotoxic domain.
- the cytotoxic domain is capable of being specifically activated in cells exhibiting a unique characteristic.
- Further provided are specified transduction domains that enhance transduction capacity of the fusion protein.
- the present invention can be used as an anti-pathogen system for killing or injuring cells infected by one or more pathogenic viruses or plasmodia.
- the present invention can also be applied to any human disease involving the expression of a cellular protease specifically in the diseased cell type and no other cells.
- other cell specific properties can also be exploited to target specific cell types, such as high levels of heavy metals, DNA damage, uncontrolled cell division, etc.
- pathogens infect mammals, particularly primates such as humans.
- certain viruses, bacteria, fungi, yeasts, worms, plasmodia, and protozoa are recognized human pathogens. See e.g., Harrison's Principles of Internal Medicine, 12 th ed. McGraw-Hill, Inc. (1991).
- Pathogens often kill or injure cells by mechanisms that manifest morphological characteristics. For example, pathogen-infected cells undergoing apoptosis or necrosis exhibit readily identifiable cellular changes.
- cell proteins and particularly enzymes involved in apoptosis.
- caspases i.e. cysteinyl aspartate-specific proteases
- C. elegans ced-3 and granzyme B have been implicated in apoptosis. Nucleic acid sequences encoding several capsases and proteolytic substrates for same are known.
- caspase-3 i.e. CPP32
- caspase-3 has been particularly well-studied. See e.g., Thompson, C. B. Science, 267:1456 (1995); and Walker, N.P.C. et al. Cell, 78:343 (1994).
- necrosis is thought to follow expression of certain DNA viruses such as herpes viruses.
- Pathogens often induce synthesis of certain proteins, particularly enzymes such as proteases. It is likely that nearly all pathogens require one or more specific proteases to complete a productive infection. For example, it is believed that the following exemplary human pathogens require expression of at least one pathogen- specific protease: cytomegalovirus (CMV), herpes simplex virus, e.g., type-1 (HSV- 1); hepatitis virus, e.g., type C (HCV); certain plasmodia, e.g., P.
- CMV cytomegalovirus
- HSV- 1 herpes simplex virus
- HCV- 1 hepatitis virus
- HCV hepatitis virus
- certain plasmodia e.g., P.
- human immunodeficiency virus type 1 HAV-1, also referred to as HTLV-III, LAV or HTLV- III/LAV
- human immunodeficiency virus type 2 HAV-2
- Kaposi's sarcoma- associated herpes virus KSHV or human herpes virus 8
- yellow fever virus certain flaviviruses and rhinovirus.
- proteases are encoded by the pathogen itself.
- the proteases are often referred to as pathogen-specific proteases.
- CMV, HCV, HIV-1, HIV-2, KSHV, and P. falciparum are representative of pathogens that encode pathogen-specific proteases.
- These proteases serve a variety of functions and can be nearly indispensable for a productive infection.
- pathogen specific proteases such as serine-type proteinases encoded by HCV, aspartic proteases (i.e. plasmepsins I and II) encoded by P. falciparum, and a maturational protease encoded by HSV-1. See e.g., Dilanni, C. L. et al., J. Biol. Chem., 268:2048 (1993); and Francis, S.E. et al., EMBOJ., 13:306 (1994).
- inducible expression of certain host cell proteases is believed to modulate productive infection by other pathogens.
- These host cell proteases are sometimes referred to as inducible host cell proteases.
- bacterial infection of eukaryotes such as certain plants can induce expression of normally quiescent host cell proteases. Induction of the host cell proteases may be an attempt to damage the pathogen, thereby protecting the host cell from infection.
- HIV viruses Infection by HIV viruses has attracted substantial attention. There is now almost universal agreement that the human family of these retroviruses are the etiological agent of acquired immune deficiency syndrome (AIDS) and related disorders. Productive infection by nearly all HIV viruses requires expression of certain HlV-specific proteases. See, for example, Barre-Sinoussi et al., Science, 220:868-871 (1983); Gallo et al., Science, 224:500-503 (1984).
- RT reverse transcriptase
- cytotoxin For example, methods that use a cytotoxin to kill cells have not always been successful.
- One explanation may relate to pleiotropic effects reported for many intracellular cytotoxins. Those effects can often complicate analysis of cell killing.
- many gene constructs that encode a cytotoxin can exhibit undesirably high basal activities inside host cells. These problems can produce what is known as "leaky" cytotoxin expression, leading to death of infected and non-infected cells.
- TAT has been reported to transactivate certain HIV genes and it is believed to be essential for productive infection by most human HIV retroviruses.
- the TAT protein has been used to bring certain types of fusion proteins into cells. This process is generally referred to as transduction. See U.S. Pat. No. 5,652,122 to Frankel et al.; and Chen, L.L. et al., Anal. Biochem., 227:168 (1995).
- TAT or TAT fragments may confer certain biological characteristics to the fusion proteins. Some of these characteristics and particularly nuclear localization and RNA binding may not always be desirable. In particular, there has been concern that many TAT fusion proteins may be difficult to position outside the nucleus or away from RNA. See e.g., Dang et al, J. Biol. Chem., 264:18109 (1989); Calnan, BJ. et al., Genes Dev., 5:201 (1991) for a discussion of TAT-associated properties.
- an anti-pathogen system that exhibits high transduction efficiency and can specifically deliver a cytotoxin to pathogen infected cells. It would be further desirable to have an anti-pathogen system that can deliver the cytotoxin as an essentially inactive molecule that can be activated by pathogen infected cells.
- the present invention relates to a method to transduce proteins into a cell and to selectively injure or kill cells exhibiting a unique characteristic.
- the protein transduction system includes a fusion molecule that comprises a transduction domain and a cytotoxic domain genetically and hence covalently linked together as an in-frame fusion molecule.
- the invention further relates to transduction domains that enhance the transduction efficiency of the fusion molecules.
- the system can be used to specifically injure or kill pathogen-infected cells.
- the anti-pathogen system is essentially inactive in uninfected cells but it is specifically activated in cells infected by the pathogen. Further provided are methods of using the anti-pathogen system to treat infection by a pathogen and particularly human pathogens such as certain viruses and plasmodia.
- the present invention can also be applied to any human disease involving the expression of a cellular protease specifically in the diseased cell type and no other cells. Moreover, other cell specific properties can also be exploited to target specific cell types, such as high levels of heavy metals, DNA damage, uncontrolled cell division, etc.
- Preferred use of the anti-pathogen system entails that the pathogen infection induce at least one pathogen specific protease.
- that protease is capable of specifically cleaving a target amino acid sequence.
- the target amino acid sequence is one component of the fusion molecule and it is sometimes referred to herein as a protease recognition or cleavage site.
- Specific cleavage of the protease recognition site cleaves the fusion molecule, generally at or near the cytotoxin domain, to form a cytotoxin.
- the cytotoxin so formed is specifically capable of killing or injuring cells infected by the pathogen.
- the present anti-pathogen system links formation of the cytotoxin to presence of the pathogen-induced protease, thereby providing highly focused cytotoxic action to infected cells. Formation of the cytotoxin is minimized or eliminated in uninfected cells and in infected cells that keep the pathogen inactive.
- the anti-pathogen system is therefore capable of effectively and specifically discriminating between productively infected and uninfected cells.
- the present anti-pathogen system has a number of important advantages. For example, it can be readily manipulated to respond to changes in pathogen serotype. That is, the anti-pathogen system can be specifically tailored to kill or injure cells infected by one or more pathogen strains. In contrast, prior methods of blocking infection and especially drug-based methods are not usually designed to respond to changes in pathogen serotype. This deficiency often results in uncontrolled growth of drug-resistant pathogen strains. As will become more apparent from the discussion that follows, the anti-pathogen system has capacity to harness production of one or more pathogen-induced protease to kill or injure cells infected by the pathogen serotype.
- the anti- pathogen system is particularly useful against emergence of HIV serotypes.
- many patients infected by HIV manifest several viral strains.
- Conventional drug-based therapies usually attempt to block activity of an HIV enzyme such as RT or an HIV protease.
- the clinical outcome of such treatment is often emergence of a spectrum of HIV serotypes.
- the HIV serotypes can develop partial or even complete resistance to the therapies.
- Even so-called "cocktail" therapies employing multiple anti-HIV drugs have been problematic.
- the anti -pathogen system of the present invention is highly flexible and can be adapted to kill or injure cells that produce the HIV serotypes by employing HIV proteases.
- the anti-pathogen system is also formulated to meet an increase in the activity of those HIV proteases or an increase in the number of infected cells with enhanced activation of the system.
- the flexibility of the present anti -pathogen system arises in part because it can be tailored to kill or injure cells infected by nearly any number of HIV serotypes.
- This feature is highly useful in several respects. For example, it provides a specific method of fighting an HIV infection in a single patient without resorting to administration of potentially harmful or ineffective drugs.
- the anti-pathogen system can be formatted to be effective at nanomoler doses or less. This low level of anti-viral activity is significantly lower than many present drug-based therapies. This feature of the invention positively impacts patient tolerance for the anti-pathogen system.
- present anti-pathogen system is fully compatible with recognized anti-HIV therapies such as those using a "cocktail" format (i.e. combination of anti- HIV drugs) to kill or injure infected cells.
- a "cocktail” format i.e. combination of anti- HIV drugs
- the anti-pathogen system is employed to reduce or eliminate emergence of HIV serotypes by exploiting the HIV protease produced by the virus.
- the present anti -pathogen system is capable of transducing unexpectedly large fusion molecules into cells.
- the anti-pathogen system accommodates misfolded (i.e. partially or completely unfolded) fusion molecules and provides for efficient transduction of those molecules into cells.
- the anti-pathogen system is compatible with misfolded fusion molecules having a molecular weight in the range of about 1 to about 500 kDa or more. The anti-pathogen system therefore is widely applicable to transducing a large spectrum of fusion molecules into cells.
- misfolded fusion proteins used in accord with this invention significantly enhance transduction efficiency sometimes by as much as about 10 fold or greater.
- misfolding the fusion proteins it has been found that it is possible to optimize the amount of the fusion molecules inside cells. Preparation and storage of the fusion molecules are also positively impacted by the misfolding.
- the present anti-pathogen system is flexible. For example, it is not limited to any particular type of pathogen or cell provided that the pathogen is capable of inducing at least one specified protease in that cell.
- the protease can be a pathogen-induced or host cell induced protease that is specifically induced (i.e. synthesized or activated) in response to the infection.
- the specified protease must be capable of cleaving the protease recognition site on the fusion molecule to activate the cytotoxin.
- the present anti-pathogen system and methods of using same can be used in vitro or in vivo. Further, the order or number of components of the fusion molecule are not important so long as each component on the molecule is operatively linked and can perform specified functions for which it is intended.
- the cytotoxin produced by the anti-pathogen system is preferably selected to kill or injure infected cells in the presence of one or more of cell proteases and usually the pathogen- or host cell- induced proteases.
- the cytotoxin can kill at least about 20%, 25%, 50%, 75%, 80%, or 90% of the cells and preferably up to about 95%), 98%) or 100% of the cells infected by the pathogen as assayed by standard cell viability tests.
- a preferred viability test is a standard Trypan Blue exclusion assay although other assays may be used as needed. It is also preferred that the cytotoxin activity be limited to cells in which it is produced.
- the present anti-pathogen system includes an in-frame fusion molecule.
- the fusion can be accomplished by conventional recombinant nucleic acid methods. If desired, the fusion can also be achieved by chemically linking the transducing protein to the cytotoxic domain according to conventional methods described below.
- the transduction domain of the fusion molecule can be nearly any synthetic or naturally-occurring amino acid sequence that can transduce or assist in the transduction of the fusion molecule.
- transduction can be achieved in accord with the invention by use of a protein sequence and particularly an HIV TAT protein or fragment thereof that is covalently linked to the fusion molecule.
- the transducing protein can be the Antennapedia homeodomain or the HSV VP22 sequence, or suitable transducing fragments thereof such as those known in the field.
- transducing amino acid sequence will be guided by several parameters including the extent of transduction desired. Preferred sequences will be capable of transducing at least about 20%, 25%, 50%, 75%, 80% or 90% of the cells of interest, more preferably at least about 95%>, 98%>% and up to about 100% of the cells. Transduction efficiency, typically expressed as the percentage of transduced cells, can be determined by several conventional methods such as those specific microscopical methods discussed below (e.g., flow cytometric analysis).
- transducing sequences will manifest cell entry and exit rates (sometimes referred to as ki and k 2> respectively) that favor at least picomolar amounts of the fusion molecule in the cell.
- the entry and exit rates of the amino acid sequence can be readily determined or at least approximated by standard kinetic analysis using detectably-labeled fusion molecules.
- the ratio of the entry rate to the exit rate will be in the range of from between about 5 to about 100 up to about 1000.
- transducing amino acid sequences that include at least a peptide featuring substantial alpha-helicity. It has been discovered that transduction is optimized when the transducing amino acid sequence exhibits significant alpha-helicity. Also preferred are those sequences having basic amino acid residues that are substantially aligned along at least one face of the peptide. Typically such preferred transduction sequences are synthetic protein or peptide sequences.
- transducing amino acid sequences are referred to as class I transducing domains or like term and include a strong alpha helical structure with a trace of arginine (Arg) residues down the helical cylinder.
- the class I transducing domain is a peptide that is represented by the following general formula: Bl -X ⁇ -X 2 -X 3 -B -X 4 -X 5 -B 3 ; wherein Bj, B 2; and B 3 are each independently a basic amino acid, the same or different; and Xi, X 2) X 3 , X 4 and X 5 are each independently an alpha-helix enhancing amino acid the same or different.
- the class I transducing peptide is represented by the following general formula: B ] -X ⁇ -X 2 -B 2 -B 3 -X 3 -X -B 4 ; wherein Bj, B 2 , B 3 , and B 4 are each independently a basic amino acid, the same or different; and Xi, X 2 , X 3 , and X 4 are each independently an alpha-helix enhancing amino acid the same or different.
- transducing peptides are often referred to herein as "class II" domains or like terms. These domains generally require basic residues, e.g., lysine (Lys) or arginine (Arg), preferably arginine (Arg), and further including at least one proline (Pro) residue sufficient to introduce "kinks" into the domain.
- lysine Lys
- Arg arginine
- Pro proline
- the class II domain is a peptide represented by the following sequence: X-X-R-X-(P/X)-(B/X)-B-(P/X)-X-B-(B/X), wherein X is any alpha helical promoting residue, preferably alanine; P/X is either proline or X as previously defined; B is a basic amino acid residue, e.g., arginine (Arg) or lysine (Lys), preferably arginine (Arg); R is arginine (Arg) and B/X is either B or X as defined above.
- Additional transducing sequences in accord with this invention include a TAT fragment that comprises at least amino acids 49 to 56 of TAT up to about the full- length TAT sequence.
- a preferred TAT fragment includes one or more amino acid changes sufficient to increase the alpha-helicity of that fragment.
- the amino acid changes introduced will involve adding a recognized alpha-helix enhancing amino acid.
- the amino acid changes will involve removing one or more amino acids from the TAT fragment that impede alpha helix formation or stability.
- the TAT fragment will include at least one amino acid substitution with an alpha-helix enhancing amino acid.
- the TAT fragment will be made by standard peptide synthesis techniques although recombinant DNA approaches may be preferred in some cases.
- Additional transduction proteins of this invention include the TAT fragment in which the TAT 49-56 sequence has been modified so that at least two basic amino acids in the sequence are substantially aligned along at least one face of the TAT fragment and preferably the TAT 49-56 sequence. In one embodiment, that alignment is achieved by making at least one specified amino acid addition or substitution to the TAT 49-56 sequence.
- Illustrative TAT fragments include at least one specified amino acid substitution in at least amino acids 49-56 of TAT which substitution aligns the basic amino acid residues of the 49-56 sequence along at least one face of the segment and preferably the TAT 49-56 sequence.
- Additional transduction proteins in accord with this invention include the TAT fragment in which the TAT 49-56 sequence includes at least one substitution with an alpha-helix enhancing amino acid.
- the substitution is selected so that at least two basic amino acid residues in the TAT fragment are substantially aligned along at least one face of that TAT fragment.
- the substitution is chosen so that at least two basic amino acid residues in the TAT 49- 56 sequence are substantially aligned along at least one face of that sequence.
- chimeric transducing proteins that include parts of at least two different transducing proteins.
- chimeric transducing proteins can be formed by fusing two different TAT fragments, e.g., one from HIV-1 and the other from HIV-2.
- other transducing proteins can be formed by fusing a desired transducing protein to heterologous amino acid sequences such as 6XHis, (sometimes referred to as "HIS"), EE, HA or Myc.
- the fusion molecule of the present invention also includes a fused cytotoxic domain.
- the cytotoxic domain includes a potentially toxic molecule and one or more specified protease cleavage sites.
- potentially toxic is meant that the molecule is not significantly cytotoxic to infected or non- infected cells (preferably less than about 30%, 20%, 10%, 5%, 3%, or 2% cell mortality as assayed by standard cell viability tests. More preferred is 1% or less cell mortality) when present as part of the cytotoxic domain.
- the protease cleavage sites are capable of being specifically cleaved by one or more than one of the proteases induced by the pathogen infection.
- the protease cleavage sites are selected to remain essentially uncleaved in uninfected cells, thereby maintaining the cytotoxic domain in an inactive state. These protease cleavage sites may also be selected to remain essentially uncleaved in cells in which the pathogen is inactive. However, in the presence of a specified pathogen-induced or host cell induced protease, the protease cleavage sites are specifically cleaved to produce a cytotoxin from the potentially toxic molecule. That is, cleavage of the protease sites releases the cytotoxic domain from the fusion molecule, thereby forming an active cytotoxin.
- the one or more protease cleavage sites are generally positioned in the cytotoxic domain to optimize release of all or part of the domain from the fusion protein and to enhance formation of the cytotoxin.
- protease cleavage sites are selected so as not to be cleaved by a protease normally associated with an uninfected cell.
- proteases have been generically referred to as "housekeeping" proteases and are well known.
- Protease cleavage sites are sometimes referred to herein as "pathogen-specific" cleavage sites to denote capacity to be specifically cleaved by one or more proteases induced by the pathogen infection.
- the protease cleavage sites are "responsive" to a pathogen (or more than one pathogen) insofar as cleavage of those sites releases the cytotoxin domain from the fusion molecule, thereby activating the cytotoxin.
- the cytotoxic domain can include one or more of a variety of potentially toxic molecules provided that it can be released from the fusion molecule as discussed.
- An illustrative cytotoxic domain for use in the fusion molecules includes an immature enzyme. These immature enzyme is sometimes referred to as zymogen, proenzyme, preproenzyme or simply as "pre-" "pre-pro” or "pro-” forms of more mature enzyme.
- Preferred zymogens can be specifically activated to a cytotoxin (i.e. a cytotoxic enzyme) by site-specific proteolysis at one or more naturally-occurring protease cleavage sites on the zymogen.
- the zymogens can be further processed in some instances by self-proteolysis.
- a cytotoxic domain that includes a preferred zymogen will include one or more specified protease cleavage sites that have been added within and/or around the zymogen.
- the cleavage sites are optionally positioned to facilitate release and processing of the zymogen to a mature or more mature cytotoxic enzyme.
- the addition of the protease cleavage sites to the zymogens can be supplative with respect to the naturally-occurring protease cleavage sites in that zymogen.
- the cleavage sites be substituted for one or more of the naturally-occurring cleavage sites.
- the substituted protease cleavage sites in the zymogen are capable of being specifically cleaved by one or more pathogen-specific proteases.
- zymogens are suitable for inclusion in the cytotoxic domain as discussed below. Active forms of those zymogens generally include bacterial toxins and particularly exotoxins, plant toxins, and invertebrate toxins including conotoxins, snake and spider toxins.
- cytotoxic domains include known proteins with potential to exert genetically dominant characteristics. That is, the proteins can be specifically cleaved from the fusion protein and can subsequently override one or more cell functions such as cell replication. In this embodiment, the potentially dominant protein must not manifest the dominant characteristic (sometimes known as a dominant phenotype) until that protein is released from the fusion protein. Examples of potentially dominant proteins in accord with the invention include proteins that inhibit cell replication such as the retinoblastoma protein (Rb), pl6 and p53.
- Rb retinoblastoma protein
- cytotoxic domains include essentially inactive enzymes that have capacity to convert certain nucleosides or analogs thereof into a cytotoxin.
- the cytotoxic domain will include one or more specified protease cleavage sites, that is preferably positioned to release the inactive enzyme from the fusion protein. Following the release, the enzyme converts the nucleoside or analog thereof into a cytotoxin. Examples of such enzymes include viral thymidine kinase and nucleoside deaminases such as cytosine deaminase.
- cytotoxic domains comprising catalytically active fragments of the enzymes such as those generally known in the field.
- the present anti-pathogen system provides a number of additional important advantages.
- the anti-pathogen system unexpectedly accommodates misfolded fusion proteins.
- that feature has been found to substantially boost levels of the fusion protein inside cells.
- a corresponding increase in the amount of administered fusion protein is not required.
- transduction of misfolded fusion molecules requires modest numbers of molecules and only a few of those need be refolded to manifest an effective cytotoxic effect.
- certain preferred fusion proteins such as those described below in Examples 5-6, only about 10 to 100 correctly refolded fusion proteins are needed to kill or injure infected cells.
- the present invention can decrease or even eliminate the need to concentrate large number of cytotoxic molecules inside cells to achieve significant anti-pathogen activity.
- activity of the present anti-pathogen system is enhanced in many cases by mass action. More particularly, it has been found that specific cleavage of the cytotoxic domain can draw additional fusion molecules into infected cells. This feature can be particularly advantageous for those fusion proteins that include cytotoxic domains that are preferably administered in sub-optimal doses. In such instances, the fusion protein is specifically concentrated in infected cells, thereby increasing levels of the cytotoxin to lethal or near lethal levels. Importantly, the cytotoxin remains at sub-optimal levels in uninfected cells.
- fusion proteins of the invention that include the TAT fragment described above.
- the cytotoxic domain of a protein fused to the TAT fragment need not be directed to the cell nucleus or to RNA.
- the present fusion molecules are formatted to separate the cytotoxic domain from the TAT fragment inside infected cells, thereby avoiding unnecessary concentration of the protein in the nucleus or with RNA. It is recognized that in uninfected cells, such fusion proteins may be directed to the nucleus or to RNA.
- differential localization of the fusion protein in infected and non-infected cells can provide means of distinguishing such cells from one another, e.g., by inspection.
- the anti -pathogen system of the invention can also positively impact certain drug-based anti-pathogen therapies. More specifically, cells infected by retroviruses and particularly HIV can harbor infectious particles for long periods of time, sometimes months or even years. Over this time, retroviruses can develop substantial resistance to most drugs, sometimes by changing one or only a few genomic sequences. It has been recognized that once the retroviruses become resistant to one class of drugs, such viruses can become resistant to a spectrum of drugs. Thus, therapies using drug-based approaches are generally inflexible and do not readily adapt to presence of resistant viruses. Related concerns have been raised with respect to development of other resistant pathogen strains such as certain plasmodia.
- the present anti-pathogen system kills or injures cells infected by pathogens regardless of pathogen capacity to acquire drug resistance. It is believed that development of drug resistant pathogens and particularly drug resistant HIV strains, is nearly impossible with the present anti-pathogen system due to the large number of protease cleavage sites that the system can accommodate. As an illustrative example, HIV virus has been reported to have about 8 to 10 such cleavage sites. In order to develop substantial resistance against the anti-pathogen system, which system could include one or more of these sites, that virus would have to modify those cleavage sites as well as the corresponding viral protease.
- use of the present anti-pathogen system is expected to significantly reduce or even eliminate the presence of many pathogen resistant strains and particularly certain drug resistant HIV strains.
- the anti-pathogen system of the invention is compatible with a variety of drug-based therapies.
- the anti-pathogen system can be used as a sole active agent or in combination with one or more therapeutic drugs, e.g. to minimize or eliminate pathogens and particularly drug resistant pathogen strains.
- the invention also provides nucleic acid sequences encoding the fusion proteins, particularly extrachromosomal DNA sequences organized as an autonomously replicating DNA vector.
- the invention also provides methods for suppressing or eliminating infection by one or more pathogens in a mammal, particularly a primate such as a human.
- the methods more specifically include administering a therapeutically effective amount of the present anti-pathogen system.
- the methods further include treatment of a mammal that suffers from or is susceptible to infection by one or pathogens.
- Preferred methods according to the invention for suppressing or eliminating infection by the one or more pathogens include providing the anti-pathogen system as an aerosol and administering same, e.g., through nasal or oral routes.
- Particularly contemplated are modes of administration which are specifically designed to administer the anti-pathogen system to lung tissue so as to facilitate contact with lung epithelia and enhance transfer into the bloodstream.
- the cell infected by one or more pathogens may be a cell maintained in culture, e.g., an immortalized cell line or primary culture of cells or tissue; or the cell can be part of a tissue or organ in vivo (e.g., lung).
- the present anti-pathogen system can be used in vitro and in vivo as needed.
- the invention also provides substantially pure fusion molecules and particularly fusion proteins that in addition to the aforementioned transduction and cytotoxic domains may also include other components as needed. These components can be covalently or non-covalently linked thereto and may particularly include one or more polypeptide sequences.
- An added polypeptide sequence will sometimes be referred to herein as protein identification or purification "tag". Exemplary of such tags are EE, 6Xhis, HA and MYC.
- the fusion proteins described herein by provided in misfolded form although in some instances it may be desirable to use properly folded fusion proteins.
- the misfolded fusion proteins are typically purified by chromatographic approaches that can be tailored if needed to purify a desired fusion molecule from cell components that naturally accompany it. Typically, the approaches involve isolation of inclusion bodies from suitable host cells, denaturation of misfolded fusion proteins, and use of conventional chromatographic methods to purify the fusion molecules.
- Expression of the misfolded fusion proteins in the inclusion bodies has several advantages including protecting the misfolded fusion protein from degradation by host cell proteases. In addition, by providing the fusion proteins in misfolded form, time-consuming and costly protein refolding techniques are avoided.
- the methods include expressing desired fusion molecules in suitable host cells, culturing the cells, and purifying the fusion molecules therefrom to obtain substantially pure fusion molecules.
- the methods can be used to express and purify a desired fusion protein on a large-scale (i.e. in at least milligram quantities) from a variety of implementations including roller bottles, spinner flasks, tissue culture plates, bioreactor, or a fermentor.
- the present methods for isolating and purifying the fusion proteins of the invention are highly useful. For example, for a fusion protein exhibiting a desired killing or injuring activity, it is very useful to have methods for expressing and purifying the fusion proteins. It is particularly useful to have methods that can produce the fusion proteins in large quantities, so that the fusion molecule can be made as one component of a kit suitable for medical, research, home or commercial use. Further, it is useful to have large-scale quantities of the fusion proteins available to simplify structural analysis, as well as for further purification and/or testing if desired.
- the invention also features in vitro and in vivo screens to detect compounds with therapeutic capacity to modulate and preferably inhibit, proteins and especially proteases induced by a pathogen infection.
- one method generally comprises infecting a desired cell with a pathogen, contacting the cell with a fusion protein of the invention, transducing the fusion protein, adding the compound to the cells and detecting cells killed or injured by the fusion protein. Efficacy of a particular compound can be readily evaluated by determining the extent of cell killing or injury as a function of concentration of the added compound. Further provided are methods of suppressing a pathogen infection in a mammal, particularly a primate such as a human, comprising administering to the mammal a therapeutically effective amount of the anti-pathogen system.
- the fusion protein includes a covalently linked protein transduction domain and a cytotoxic domain.
- the method includes transducing the fusion protein into cells of the mammal, cleaving the fusion protein sufficient to release the cytotoxic domain from the fusion protein, concentrating the cytotoxic domain in the cells; and producing a cytotoxin sufficient to suppress the pathogen infection in the mammal.
- pathogens include but are not limited to retroviruses, herpesviruses, viruses capable of causing influenza or hepatitis; and plasmodia capable of causing malaria.
- Preferred cytotoxic domains and cytotoxins are described in more detail below.
- a prodrug is administered (e.g., a suitable nucleoside or analog thereof) and a cytotoxin is produced by contacting the prodrug with the concentrated cytotoxic domain.
- fusion proteins that include covalently linked in sequence: 1) A TAT segment and particularly a protein transducing fragment thereof, and 2) a pathogen induced or host cell induced protease, e.g., HIV protease; or a catalytically active fragment thereof.
- an anti-pathogen system wherein the fusion protein comprises covalently linked in sequence: 1) a transduction domain, 2) a first zymogen subunit, 3) a protease cleavage site, and 4) a second zymogen subunit.
- the transduction domain is TAT
- the first zymogen subunit is p5 Bid
- the protease cleavage site is an HIV protease cleavage site
- the second zymogen subunit is pl5 Bid.
- the invention also provides an anti-pathogen system, wherein the fusion protein comprises covalently linked in sequence: 1) a transduction domain, 2) a first protease cleavage site, 3) first zymogen subunit, 3) a second protease cleavage site, and 4) a second zymogen subunit.
- an anti-pathogen system wherein the transduction domain is TAT, the first protease cleavage site is an HIV p7-pl protease cleavage site, the first zymogen subunit is pi 7 caspase-3, the second protease cleavage site is an HIV pl7-p24 protease cleavage site, and the second zymogen subunit is pl2 caspase-3.
- the present invention also provides a method of killing an HIV-infected cell.
- the method includes contacting the cell with an effective dose of a fusion protein, wherein the fusion protein comprises covalently linked in sequence: 1) a transduction domain, 2) a first zymogen subunit, 3) a protease cleavage site, and 4) a second zymogen subunit; or 1) a transduction domain, 2) a first protease cleavage site, 3) first zymogen subunit, 3) a second protease cleavage site, and 4) a second zymogen subunit.
- the fusion protein can be administered in vitro or in vivo as needed.
- the fusion protein can be administered in vivo to a mammal in need of such treatment, e.g., a primate and particularly a human patient infected by the HIV virus.
- Methods of the present invention can also be applied to human diseases. Indeed, any human disease involving the expression of a cellular protease specifically in the diseased cell type and no other cells can be exploited to specifically kill that cell. Moreover, other cell specific properties can also be exploited to target specific cell types, such as high levels of heavy metals, DNA damage, uncontrolled cell division, etc.
- Prostate cancer is an illustrative example of the flexibility of the present invention for treatment of non-pathogen related human diseases.
- Prostate cells express specific cellular proteases, such as Prostate Specific Antigen (PSA), and also have a 1000-fold elevated level of the heavy metal Zinc compared to rest of the human body. Importantly, both of these attributes are maintained in prostate cancer cells. Moreover, as a model system, after the patient has had children, the prostate is not required for any bodily function. Thus, all prostate cells, malignant or not, may be cleared from the body without loss of viability to the patient.
- PSA Prostate Specific Antigen
- PSA is a sub-family member of the kallikrein family of cellular proteases (Lilja et al., 1985; Watt et al.); however, it has a chymotrypsin-type of substrate specificity that distinguishes it from other kallikrein family members as well as chymotrypsin and trypsin (Christensson et al.; Lilja et al. 1989).
- PSA is specifically synthesized by prostate cells and secreted into the lumen of the prostate. The function of PSA is to cleave gel forming proteins present in the seminal fluid, such as Sg I & II (Lilja et al. 1985).
- PSA Due to the tight cell-cell junctions in the prostate, PSA never leaks into or is detected in the blood stream. However, during rapid growth of prostate tumors, the junctions are looser and allow for low level release of PSA into the blood stream. In addition, metastasis of prostate tumor cells results in the further release and detection of PSA in the blood stream. As with exploiting pathogen specific proteases to discriminate and kill infected cells, PSA is 1.) specifically expressed in malignant prostate cells and 2.) has a specific substrate specificity or cleavage site. Thus, PSA is an excellent example of a human disease that can be targeted by transducible killing proteins.
- PSA activated transducible killing molecule can take several forms, as can pathogen activated killing molecules, as previously discussed.
- PSA present in excretory vesicles can be utilized to activate the transduced zymogen intracellularly into a killing form as outlined above.
- extracellular PSA can be utilized to activate a zymogen that then transduces into the nearest cell, i.e. a prostate cell, and induces apoptosis.
- the 1000-fold excess of Zinc in prostate cells could also be exploited by transduction of an inactive protein that requires a high concentration of Zn for dimerization and hence, activation.
- Such examples include utilizing dimerization domains of cellular transcription factors (Tx F) and dimerization domain of HPV E7 protein.
- Tx F cellular transcription factors
- the Caspase-3 pl7 and pl2 domains can be engineered to have terminal tags of E7 or Tx F dimerization domains. Requisite dimerization of transduced Caspase-3 pl7 and pl2 subunits would be dependent on dimerization of E7/ Tx F domains via coordination of Zn above a threshold concentration. Therefore, apoptotic induction would only be achieved when the E7/Tf F domains were dimerized by Zn. Such a transducible killing molecule would therefore be specifically activated only in cells containing high levels of Zn, such as prostate cells.
- the protein transduction system of the present invention can also be used to target and kill cancer cells in general based on their high proliferative activity as compared to normal cells.
- tumors containing the wild type p53 tumor suppressor protein respond significantly better to traditional anti-tumor regimens such as radiation and chemotherapy than tumors containing mutant p53 (Lowe et al.).
- p53 status is currently the single most significant determinant for patient outcome after treatment.
- introducing wild type p53 into tumors should restore the sensitivity of these tumors to traditional anti-cancer therapies and, importantly, may allow for significant reductions in the amount of anti-cancer therapy required to kill the tumor cells.
- TAT-p53 1-364 transducible version of p53
- TAT-p53 1-364 protein resulted in specific cell death of the tumor cells, while normal cells showed minimal toxicity.
- TAT-pl6 or TAT-p27 Transduction of additional anti-cell cycle tumor suppressor proteins, such as TAT-pl6 or TAT-p27, in combination with TAT-p53 proteins will likely synergize to further increase the killing.
- TAT-p53 proteins in combination with traditional small molecule chemotherapeutics that induce DNA damage will likely result in a further activation of the transduced p53 and hence, a synergy.
- Figure 1 is a plasmid map of pTAT/pTAT-HA.
- Figure 2 shows nucleotide and amino acid sequences of pTAT linker and pTAT HA linker.
- a minimal TAT domain is in bold.
- Underlined sequence designates the minimal TAT domain flanked by glycine residues.
- FIGS. 3A-D are drawings depicting illustrative DNA vectors according to the invention based on the pTAT/pTAT-HA plasmid.
- HIS denotes optional addition of a 6XHIS tag
- protein transduction domain PTD
- HIV protease- RT cleavage site HIV protease- RT cleavage site
- HSV TK herpes simplex virus thymidine kinase
- Lg large caspase-3 domain
- Sm small caspase-3 domain
- HIV pl7-p24 protease cleavage site HIV 2
- pl6 mutant or wild-type pl6 protein
- Figure 4 is a schematic drawing outlining cell killing with a fusion protein comprising an enzyme capable of converting a prodrug into an active drug.
- HIV ⁇ j2 is defined in Figs. 3A-D above.
- FIG. 5 is a schematic drawing showing one method of constructing a TAT- CPP32 fusion protein according the invention.
- Figure 6 A is a bar graph showing percentage of viable cells after transduction of various TAT fusion proteins and treatment with anti-HIV drug.
- Figure 6B is a table showing percentages of viable cells (under column 2) used in the bar graph of Figure 6A.
- FIG. 7 is a drawing showing helical wheel projections of preferred transduction proteins of this invention.
- TAT (47-57) refers to amino acids 47 to 57 of the TAT peptide (SEQ ID NO:2).
- SFD refers to specified transduction domain sequences.
- relative intracellular concentration in Figure 7 refers to the intracellular amount of transduced peptide sequence relative to the TAT peptide.
- Figures 8A-C are drawings illustrating various protein constructs.
- Figure 8A is a diagram of the Bid protein highlighting the p5 and pi 5 domains. The caspase cleavage site at Arg 59 is shown.
- Figure 8B outlines the cloning of the TAT-p5-HIV- pl5 fusion protein.
- Figure 9A-E are drawings showing generation and transduction of TAT fusion proteins.
- Figure 9A shows the caspase 3 (Casp3) protein and various TAT/HIV fusion proteins made using the Casp3 pi 7 and pl2 domains.
- Figures 9B-E are graphs showing FACS analysis of various fluorescein (FITC) labeled TAT fusion proteins.
- FITC fluorescein
- Figures 10A-B are representations of immunoblots showing in vivo processing of various TAT fusion proteins in Jurkat T cells.
- the immunoblots were probed with anti-pl6 ( Figure 10A) or anti-Caspase-3 antibody ( Figure 10B).
- Figures 11 A-B are graphs showing activation of TAT-Casp3 and apoptotic induction in cotransduced cells.
- Figure 11 A shows cell viability following transduction with various TAT fusion proteins along with the HIV protease inhibitor Ritonavir (Rit).
- Figure 1 IB illustrates cell viability following transduction with various TAT fusion proteins.
- Figure 12A-B are graphs showing HIV protease activates TAT-CaspS ⁇ protein.
- Figure 12A shows results of TUNEL positive cells (apoptotic end-marker) using a TAT fusion protein.
- Figure 12 B shows results of a caspase-3 enzyme assay using a TAT fusion protein.
- Figure 13 is a graph illustrating specific killing of HIV infected cells.
- Figure 14 is a diagram illustrating the plasmid maps for pTAT-p53 WT and pTAT-p53 1-364.
- the full length wild-type (aa 1-393) and C terminal truncated (aa 1-364) forms of p53 ORF were isolated from plasmid pTW300 (R. Brachman, Washington University, St. Louis, MO, unpublished data) were ligated into pTAT-HA (Ezhevsky et al. and Nagahara et al.) resulting in plasmids pTAT-p53 WT and pTAT- p53 1-364.
- FIG. 15 shows a coomassie blue stained gel of TAT-p53 WT protein purified using a Ni-NTA column.
- TAT-p53 WT protein was expressed in E. coli cells in an insoluble form. The insoluble protein was pelleted and resuspended in 6 M GuHCl/20 mM HEPES/100 mM NaCl and sonicated. 1 mL fractions of TAT-p53 WT protein were eluted from a Ni-NTA column with increasing imidazole concentration, as indicated. A high level of TAT-p53 WT bound to the Ni-NTA column in the presence of 6 M GuHCl.
- Figures 16A-B are p53 immunoblots of purified TAT-p53 WT and purified
- TAT-p53 1-364 proteins eluted from a Ni-NTA column with increasing concentrations of imidazole (as indicated).
- Panel A shows the blot for TAT-p53 WT protein and
- panel B shows the blot for TAT-p53 1-364 protein.
- Figure 17 is a graph of cell viability of tumor cells (shaded bars) as compared to normal cells (unshaded bars) following 48 hours transduced TAT-p53 1-364 protein.
- Figure 18A-B shows the killing of tumor cells by forced cell cycle arrest due to transduction of Cdk inhibitor proteins.
- Panel A is a schematic diagram showing that upon treatment with a PTD-Cdk inhibitor protein fusion (TAT-p27, TAT-Cdk2- DN (dominant negative) and/or TAT-pl6) tumor cells are susceptible to cell cycle arrest mediated apoptosis, whereas normal cells are resistant.
- Panel B is a graph showing the % cell survival of tumor cells at 48 hours post administration of the indicated PTD-fusion protein.
- Figure 19 is a graph showing cell viability of tumor cells (shaded bars) vs. normal cells (unshaded bars) following 48 hours of transduced TAT-pl6 peptide.
- the present invention features a protein transduction system that exhibits high transduction efficiency and can be used to specifically kill or injure cells exhibiting a unique characteristic such as, e.g. pathogen infection, cellular proliferation, etc.
- the protein transduction or anti-pathogen system generally includes a fusion protein that includes a transduction domain fused to a cytotoxic domain as a genetic in-frame fusion protein.
- Preferred fusion proteins exhibit enhanced transduction efficiency as determined, e.g., by assays which follow.
- the transduction domain transduces the fusion protein into cells and once inside the cells, the cytotoxic domain is released from the fusion protein and forms a cytotoxin in the infected cells.
- function of the fusion protein has been specifically enhanced, e.g., by optimizing transduction domain structure and by misfolding the fusion molecule.
- Methods of the present invention can also be applied to human diseases. Indeed, any human disease involving the expression of a cellular protease specifically in the diseased cell type and no other cells can be exploited to specifically kill that cell. Moreover, other cell specific properties can also be exploited to target specific cell types, such as high levels of heavy metals.
- Preferred fusion proteins are capable of killing at least about 25%, 40%, 50%, 60%, or 70%, preferably 80%, 90%, and more preferably at least 95% up to 100% of the cells infected by the pathogen as assayed by standard cell viability tests discussed below.
- an "anti-pathogen system” includes one or more of the fusion molecules described herein as well as any additional components that may be added thereto such as those that may facilitate solublization, stability and/or activity including transduction efficiency.
- examples include but are not limited to a serum protein such as bovine serum albumin, a buffer such as phosphate buffered saline, or a pharmaceutically acceptable vehicle or stabilizer. See generally Reminington 's Pharmaceutical Sciences, infra, for a discussion of pharmaceutically acceptable vehicles, stabilizers, etc.
- a preferred anti-pathogen system includes from between about 1 to 3 and are preferably 1 fusion protein dissolved in a pharmaceutically acceptable carrier such as water or buffered saline.
- the anti-pathogen system is provided sterile.
- the anti-pathogen system can be administered as a sole active agent or in combination with one or more medicaments such as those specifically provided below.
- fusion molecule a transducing molecule and usually a protein or peptide sequence covalently linked (i.e. fused) to a cytotoxic domain by recombinant, chemical or other suitable method.
- the fusion molecule can be fused at one or several sites through a peptide linker sequence. That peptide sequence can include one or more sites for cleavage by a pathogen induced or host cell induced protease.
- the peptide linker may be used to assist in construction of the fusion molecule.
- the cytotoxic domain will usually include one potentially toxic molecule such a zymogen sometimes from between about 2 up to about 5 to 10 of such molecules.
- Specifically preferred fusion molecules are fusion proteins.
- components of the fusion proteins disclosed herein can be organized in nearly any fashion provided that the fusion protein has the function for which it was intended.
- each component of the fusion protein can be spaced from another component by at least one suitable peptide linker sequence if desired.
- the fusion proteins may include tags, e.g., to facilitate identification and/or purification of the fusion protein. More specific fusion proteins are described below.
- Preferred peptide linker sequences typically comprise up to about 20 or 30 amino acids, more preferably up to about 10 or 15 amino acids, and still more preferably from about 1 to 5 amino acids.
- the linker sequence is generally flexible so as not to hold the fusion molecule in a single rigid conformation.
- the linker sequence can be used, e.g., to space the DNA binding protein from the fused molecule.
- the peptide linker sequence can be positioned between the protein transduction domain and the cytotoxic domain, e.g., to chemically cross-link same and to provide molecular flexibility.
- the term "misfolded" as it relates to the fusion proteins is meant a protein that is partially or completely unfolded (i.e. denatured).
- a fusion protein can be partially or completely misfolded by contact with one or more chaotropic agents as discussed below.
- misfolded fusion proteins disclosed herein are representative of a high Gibbs free energy ( ⁇ G) form of the corresponding native protein.
- ⁇ G Gibbs free energy
- a native fusion protein is usually correctly folded, it is fully soluble in aqueous solution, and it has a relatively low ⁇ G. Accordingly, that native fusion protein is stable in most instances.
- misfolding can be detected by a variety of conventional biophysical techniques including optical rotation measurements using native (control) and misfolded molecules.
- preferred administration of the anti -pathogen system involves transduction of misfolded fusion proteins in vitro and in vivo. Without wishing to be bound to theory, it is believed that after transduction of the fusion protein into cells, misfolded fusion proteins are significantly refolded, e.g., by chaperones, sufficient to produce a fusion protein than can be activated in response to pathogen infection.
- the fusion molecule and particularly a fusion protein that is not readily sedimented under low G-force centrifugation (e.g. less than about 30,000 revolutions per minute in a standard centrifuge) from an aqueous buffer, e.g., cell media.
- the fusion molecule is soluble if it remains in aqueous solution at a temperature greater than about 5-37°C and at or near neutral pH in the presence of low or no concentration of an anionic or non-ionic detergent. Under these conditions, a soluble protein will often have a low sedimentation value e.g., less than about 10 to 50 svedberg units.
- Aqueous solutions referenced herein typically have a buffering compound to establish pH, typically within a pH range of about 5-9, and an ionic strength range between about 2mM and 500mM. Sometimes a protease inhibitor or mild non-ionic detergent is added. Additionally, a carrier protein may be added if desired such as bovine serum albumin (BSA) to a few mg/ml.
- BSA bovine serum albumin
- Exemplary aqueous buffers include standard phosphate buffered saline, tris-buffered saline, or other well known buffers and cell media formulations.
- polypeptide refers to any polymer preferably consisting essentially of any of the 20 natural amino acids regardless of its size.
- protein is often used in reference to relatively large proteins, and “peptide” is often used in reference to small polypeptides, use of these terms in the field often overlaps.
- polypeptide refers generally to proteins, polypeptides, and peptides unless otherwise noted.
- potentially toxic molecule an amino acid sequence such as a protein, polypeptide or peptide; a sugar or polysaccharide; a lipid or a glycolipid, glycoprotein, or lipoprotein that can produce the desired toxic effects as discussed herein.
- potentially toxic nucleic acids encoding a toxic or potentially toxic protein, polypeptide, or peptide.
- suitable molecules include regulatory factors, enzymes, antibodies, or drugs as well as DNA, RNA, and oligonucleotides.
- the potentially toxic molecule can be naturally-occurring or it can be synthesized from known components, e.g., by recombinant or chemical synthesis and can include heterologous components.
- a potentially toxic molecule is generally between about 0.1 to 100 KD or greater up to about 1000 KD, preferably between about 0.1, 0.2, 0.5, 1, 2, 5, 10, 20 ,30 and 50 KD as judged by standard molecule sizing techniques such as centrifugation or SDS-polyacrylamide gel electrophoresis.
- the term "cell” is intended to include any primary cell or immortalized cell line, any group of such cells as in, a tissue or an organ.
- the cells are of mammalian and particularly of human origin, and can be infected by one or more pathogens.
- a "host cell” in accord with the invention can be an infected cell or it can be a cell such as E. coli that can be used to propagate a nucleic acid described herein.
- the present anti-pathogen system is suitable for in vitro or in vivo use with a variety of cells that are infected or that may become infected by one or more pathogens.
- a cultured cell can be infected by a pathogen of a single serotype.
- the infected cell is then contacted by a specified fusion protein in vitro.
- the fusion protein is configured so that the cytotoxic domain is activated in the presence of one or more proteases induced by the pathogen infection.
- the cells are allowed to cleave the fusion protein for a time period of about up to about 2 to 24 hours, typically about 18 hours.
- the cells are washed in a suitable buffer or cell medium and then evaluated for viability.
- the time allotted for cell killing or injury by the fusion protein will vary with the particular cytotoxic domain chosen. However viability can often be assessed after about 2 to 6 hours up to about 24 hours. As will be explained in more detail below, cell viability can be readily measured and quantified by monitoring uptake of certain well-known dyes (e.g., trypan blue) or fluors.
- the present anti-pathogen system is also suitable for in vitro or in vivo treatment of a variety of human diseases in which expression of a cellular protease occurs specifically in the diseased cell type and in no other cells.
- other cell specific properties can also be exploited to target specific cell types, such as high levels of heavy metals, DNA damage, uncontrolled cell division, etc.
- Preferred embodiments of the invention provide methods for treating an animal bearing primary or secondary tumors including tumors in the breast, prostate, ovary, central nervous system, brain, colon, lung, skin, etc. or disseminated tumors such as leukemic cells etc.
- a preferred method for treating cancer is using a transducible form of the p53 protein.
- the fusion protein may comprise the full length p53 protein or it may comprise a portion of the p53 protein which is capable of acting as a tumor suppressor. Particularly preferred is the TAT-p53 1-364 fusion construct.
- the anti-pathogen system is flexible and can be provided in formats that are tailored for a specific use.
- the system can be provided with two fusion proteins in which the first fusion protein includes a transduction domain and a cytotoxic domain, and the second fusion protein includes a transducing domain and a pathogen-induced or host cell induced protease.
- Cells transduced by the fusion molecules of the present invention can be assayed for viability by standard methods.
- cell viability can be readily assayed by measuring DNA replication following or during transduction.
- a preferred assay involves cell uptake of one or more detectably-labeled nucleosides such as radiolabelled thymidine. The uptake can be conveniently measured by several conventional approaches including trichloroacetic acid (TCA) precipitation followed by scintillation counting.
- TCA trichloroacetic acid
- Other cell viability methods include well know trypan blue exclusion techniques.
- fusion molecules of the present invention are efficiently transduced into target cells or groups of such cells. Transduction efficiency can be monitored and quantified if desired by one or a combination of different strategies.
- one approach involves an in vitro assay that measures uptake of the fusion protein by the cell.
- the assay includes detectably-labeling the fusion protein with, e.g., a radioactive atom, fluorescent, phosphorescent, or luminescent tag (e.g., fluorescein, rhodamine or FITC) and then measuring uptake of the labeled fusion protein.
- the fusion protein can be labeled with an enzyme capable of forming a detectable label such as horseradish peroxidase, ⁇ -galactosidase, chloramphenicol acetyl transferase or luciferase.
- GFP green fluorescent protein
- Uptake can be measured by several conventional methods such as by quantifying labeled cells in a standard cell sorter (e.g., FACS), by fluorescence microscopy or by autoradiography. See generally Sambrook et al. and Ausubel et al. infra for disclosure relating to the assays.
- Preferred fusion proteins of the invention are capable of transducing at least about 20%, to 80%, and more preferably at least about 90%, 95%, 99% up to 100% of the total number of target cells as determined by any conventional methods for monitoring protein uptake by cells and particularly the FACS or related microscopical techniques.
- the total number of target cells can be estimated by standard techniques.
- the present invention pertains to fusion proteins and nucleic acids (e.g., DNA) encoding the fusion proteins.
- the term fusion protein is intended to describe at least two polypeptides, typically from different sources, which are operatively linked.
- the term "operatively linked" is intended to mean that the two polypeptides are connected in manner such that each polypeptide can serve its intended function. Typically, the two polypeptides are covalently attached through peptide bonds. As discussed, the two polypeptides may be separated by a peptide linker if desired.
- the fusion proteins described herein are preferably produced by standard recombinant DNA techniques.
- a DNA molecule encoding the first polypeptide can be ligated to another DNA molecule encoding the second polypeptide.
- the resultant hybrid DNA molecule can be expressed in a suitable host cell to produce the fusion protein.
- the DNA molecules are ligated to each other in a 5' to 3' orientation such that, after ligation, the translational frame of the encoded polypeptides is not altered (i.e., the DNA molecules are ligated to each other in-frame).
- the resulting DNA molecules encode an in-frame fusion protein.
- the components of the fusion protein can be organized in nearly any order provided each is capable of performing its intended function.
- the protein transduction domain is adjacent to a pathogen-specific protease cleavage site included within the cytotoxic domain.
- the cytotoxic domain can be flanked by pathogen-specific protease cleavage sites, one or both of which can also be adjacent to the protein transduction domain.
- the present invention also contemplates circular fusion proteins.
- Preferred cytotoxic domains including the pathogen-specific cleavage sites will have sizes conducive to the function for which those domains are intended.
- preferred cytotoxic domains can be at least about 0.1, 0.2, 0.5, 0.75, 1, 5, 10, 25, 30, 50, 100, 200, 500 kD, up to about 1000 kD or more. It should be apparent that the size of the cytotoxic domain usually dominates the size of the fusion protein.
- Preferred pathogen-specific cleavage sites will be between about 4 to about 30 or 40, preferably about 8 to about 20 and more preferably about 14 amino acids in length. See Table I, below.
- the pathogenic-specific protease cleavage sites can be made and fused to the cytotoxic domain by a variety of methods including well-known chemical cross-linking methods. See e.g., Means, G.E. and Feeney, R.E. (1974) in Chemical Modification of Proteins, Holden-Day. See also, S.S. Wong (1991) in Chemistry of Protein Conjugation and Cross-Linking, CRC Press. However it is generally preferred to use recombinant manipulations to make the in-frame fusion protein.
- a fusion molecule in accord with the invention can be organized in several ways.
- the C-terminus of the transduction domain is operatively linked to the N-terminus of the cytotoxic domain. That linkage can be achieved by recombinant methods if desired.
- the N-terminus of the transduction domain is linked to the C-terminus of the cytotoxic domain.
- the N-terminus of a first pathogen-specific protease cleavage site can be operatively linked to the C-terminus of the transduction domain and the C-terminus of the protease cleavage site can be operatively linked to the N-terminus of a potentially toxic molecule.
- the C-terminus of the cytotoxic domain can be linked to the N-terminus of a second pathogen-specific protease cleavage site the same or different from the first pathogen-specific site.
- the first and second pathogen-cleavage sites will be specifically cleaved by the same protease induced by the pathogen infection.
- one or more additional protease cleavage sites can be inserted into the potentially toxic molecule as needed.
- Preferred fusion proteins in accord with the present invention typically include operatively linked in sequence (N to C terminus): 1) a transduction domain/one or more pathogen-specific protease cleavage sites/and a potentially toxic molecule; 2) a transduction domain/a pathogen specific protease cleavage site/and a zymogen; and 3) a transduction domain/a first pathogen specific protease cleavage site/a first zymogen subunit/a second pathogen specific protease cleavage site/and a second zymogen subunit.
- one or more protein tags such as EE, HA, Myc, and polyhistidine, particularly 6Xhis, can be fused to the N-terminus of the transduction domains as desired, e.g., to improve solubility or the facilitate isolation and identification of the fusion protein. See Examples below.
- a polypeptide sequence to the fusion proteins to promote transport to a cell nucleus.
- Amino acid sequences which, when included in a protein, function to promote transport of the protein to the nucleus are known in the art and are termed nuclear localization signals (NLS). Nuclear localization signals typically are composed of a stretch of basic amino acids.
- the nuclear localization signal When attached to a heterologous protein (e.g., a fusion protein of the invention), the nuclear localization signal promotes transport of the protein to a cell nucleus.
- the nuclear localization signal is attached to a heterologous protein such that it is exposed on the protein surface and does not interfere with the function of the protein.
- the NLS is attached to one end of the protein, e.g. the N-terminus.
- the SV40 nuclear localization signal is a non-limiting example of an NLS that can be included in a fusion protein of the invention.
- the SV40 nuclear localization signal has the following amino acid sequence: Thr-Pro-Pro-Lys-Lys-Lys- Lys-Arg-Lys-Val (SEQ ID NO:3).
- a nucleic acid encoding the nuclear localization signal is spliced by standard recombinant DNA techniques in-frame to the nucleic acid encoding the fusion protein (e.g., at the 5
- a fusion protein of the invention is composed, in part, of a first polypeptide, sometimes referred to herein as a protein transduction domain, transduction domain, transducing protein, or "PTD", which provides for entry of the fusion protein into the cell.
- Peptides having the ability to provide entry of a coupled peptide into a cell include those mentioned previously such as TAT, Antennapedia homeodomain, referred to as "Penetratin” Ala-Lys-Ile-Trp-Phe-Gln- Asn-Arg-Arg-Met-Lys-Trp-Lys-Lys-Glu-Asn (SEQ ID NO:l) (Derossi et al., J. Bio. Chem., 269:10444 (1994)) and HSV VP22 (Elliot and O'Hare, Cell, 88:223 (1997)).
- TAT fragment that includes at least the TAT basic region (amino acids 49-57 of naturally-occurring TAT protein).
- TAT fragments can be between about 9, 10, 12, 15, 20 , 25, 30, or 50 amino acids in length up to about 86 amino acids in length.
- the TAT fragments preferably are deficient in the TAT cysteine-rich region (amino acids 22-36 of naturally-occurring TAT protein) and the TAT exon 2 encoded by a carboxy-terminal domain (amino acids 73-86 of naturally-occurring TAT protein).
- a TAT transduction domain has the following amino acid sequence: YGRKKRRQRRR (SEQ ID NO:2).
- That amino acid sequence will sometimes be referenced herein as a "minimal TAT sequence". See U. S. Pat. No. 5,674,980 and references cited therein for disclosure relating to TAT structure. See also Green, M. and Lowenstein, P. M. (1988) for the TAT sequence.
- the protein transduction domain of the fragment can be flanked by glycine residues to allow for free rotation. See e.g., Fig. 2 of the drawings.
- glycine residues to allow for free rotation.
- other amino acid sequences and particularly neutral and/or hydrophilic residues may be added to the TAT fragment as desired.
- Protein tags may be added to a TAT fragment such as those known in the field. Examples of such protein tags include 6XHis, HA, EE and Myc.
- the size of the modified TAT fragment will be at least 10, 12, 15, 20, 25, 30, 50, 100, 200, to about 500 amino acids in length.
- the transduction domain of the fusion protein can be obtained from any protein or portion thereof that can assist in the entry of the fusion protein into the cell.
- preferred proteins include, for example TAT, Antennapedia homeodomain and HSV VP22 as well as non-naturally-occurring sequences.
- the suitability of a synthetic protein transduction domain can be readily assessed, e.g., by simply testing a fusion protein to determine if the synthetic protein transduction domain enables entry of the fusion protein into cells as desired.
- synthetic protein or like term a non-naturally occurring amino acid sequence which is made be recombinant methods or methods involving chemical peptide synthesis.
- transducing TAT proteins Numerous variants of transducing TAT proteins have been described in the field. These variants can be used in accord with the present invention. See e.g., U.S. Pat. No. 5,652,122 which reports methods of making and using transducing TAT proteins, the disclosure of which is inco ⁇ orated by reference.
- transduction domains and particularly transducing proteins can be readily identified by conventional techniques.
- a candidate transduction domain such as a desired TAT fragment is fused to a desired cytotoxic domain using standard recombinant manipulations to form the in-frame fusion protein.
- the fusion protein is subsequently detectably-labeled with, e.g., a radioactive atom or fluorescent label such as FITC.
- the detectably-labeled fusion protein is then added to cells as described above and the levels of the fusion protein are measured.
- a preferred transduction domain will be capable of achieving an intracellular concentration of the fusion protein of between about 1 picomolar to about 100 micromolar, preferably about 50 picomolar to about 75 micromolar, and more preferably about 1 to about 100 nanomolar.
- transducing proteins are those obtained by targeted mutagenesis of known transducing proteins or fragments, e.g., TAT, VP22 or the
- the mutagenized transducing protein will exhibit at least about 2, 3, 4, 5, 10, 20, 30, 40 or 50 fold better transduction of a desired fusion protein when compared to that same fusion protein comprising a corresponding full-length transducing protein sequence.
- Preferred transduction proteins in accord with this invention are Class I amino acid sequences, preferably peptide sequences, that include at least a peptide represented by the following general formula: Bl -X ⁇ -X -X -B 2 -X -X 5 -B 3 ; wherein Bi, B > and B are each independently a basic amino acid, the same or different; and Xi, X 2 , X 3 , X 4 and X 5 are each independently an alpha-helix enhancing amino acid the same or different. Typically these sequences are synthetic.
- basic amino acid refers to an amino acid having a basic residue such as a primary, secondary or tertiary amine, or a cyclic group containing nitrogen ring member.
- Preferred basic amino acids are lysine (Lys) and arginine (Arg), with arginine being particularly preferred.
- Histidine (His) also can be a suitable basic amino acid.
- alpha-helix enhancing amino acid or like term is meant an amino acid which has a recognized tendency to form or stabilize an alpha-helix as measured by assays well-known in the field. See generally O'Neil, K.T. and DeGrado, W.F. (1990) Science 250: 646 and references cited therein for such an assay.
- Preferred alpha-helix enhancing amino acids include alanine (Ala), arginine (Arg), lysine (Lys), leucine (Leu), and methionine (Met).
- a particularly preferred alpha-helix enhancing amino acid is alanine.
- substantially alpha-helicity is meant that a particular peptide has a recognizable alpha-helical structure as determined, e.g., by a helical wheel diagram or other conventional means.
- the peptide is represented by the formula B]-X ⁇ -X 2 -X 3 - B 2 -X 4 -X -B 3; wherein at least one of Bi, B 2 ⁇ or B 3 is arginine, preferably all of Bi, B 2> and B 3 are arginine; and X ⁇ , X 2j X , X 4 and X 5 are each independently an alpha-helix enhancing amino acid the same or different.
- at least one of Xi, X 2> X 3 , X 4 or X 5 is an alanine, more preferably all of Xi, X 2 , X 3 , X 4 and X 5 are alanine.
- the peptide is represented by the formula B X ⁇ -X -X -B 2 -X 4 - X 5 -B 3 ; wherein Bj, B 2 , and B 3 are each independently a basic amino acid, the same or different; and at least X ] X 3 , X 4 or X 5 is alanine, preferably all of Xi, X 2 , X 3 , X and X 5 each is alanine.
- basic amino acid residues such as arginine are substantially aligned along at least one face of the peptide, typically along one face.
- transduction proteins in accord with this invention are synthetic amino acid sequences, preferably peptide sequences, that include at least a peptide represented by the following general formula: B ⁇ -X ⁇ -X -B 2 -B 3 -X 3 -X 4 -B 4 ; wherein B ⁇ , B ⁇ B 3> and B 4 are each independently a basic amino acid, the same or different; and Xi, X 2> X 3 , and X are each independently an alpha-helix enhancing amino acid the same or different.
- At least one of Bi, B > B 3 ⁇ or B 4 is arginine, preferably all of Bi, B 2> B 3> and B are arginine; and the X], X > X 3 , and X are each independently an alpha-helix enhancing amino acid the same or different.
- each of the B ] ; B 2j B 3 , and B 4 are independently a basic amino acid, the same or different; and at least one of X 1 ⁇ X 2> X 3 , or X 4 is an alanine, preferably all of Xi, X , X 3 , and X 4 are alanine residues.
- basic amino acid residues such as arginine are substantially aligned along at least one face of the peptide, typically along one face.
- substantially aligned basic amino acid residue By the term “substantial alignment" of a basic amino acid residue or like term is meant that the basic amino acid residue is positioned with respect to at least one other basic amino acid residue so that each residue is spaced from the other on a conceptualized alpha-helix by between about 3 to about 4 Angstroms, preferably about 3.6 Angstroms. Alignment can be performed by several conventional methods including inspection of standard helical wheel diagrams such as those shown below in Figure 7. Preferred transduction domains exhibit between about 2, 3, 4, 5, 6, or about 7 up to about 10 substantially aligned basic amino acid residues.
- More preferred transduction proteins of this invention include at least a peptide represented by the following specific peptide sequences: YARKARRQARR (SEQ ID NO:3), YARAAARQARA (SEQ ID NO:4), YARAARRAARR (SEQ ID NO:5), YARAARRAARA (SEQ ID NO:6), YARRRRRRR (SEQ ID NO:7), and YAAARRRRRRR (SEQ ID NO:8).
- transducing peptide sequences consisting of the following peptide sequences: YARKARRQARR (SEQ ID NO:3), YARAAARQARA (SEQ ID NO:4), YARAARRAARR (SEQ ID NO:5), YARAARRAARA (SEQ ID NO:6), YARRRRRRR (SEQ ID NO:7), and YAAARRRRRRR (SEQ ID NO:8).
- Additional transduction proteins of this invention are amino acid sequences, preferably synthetic sequences, that include at least one amino acid modification in at least amino acids 49 to 56 of TAT.
- the synthetic peptide sequences include at least amino acids 47 to 56, 48 to 56, 47 to 57, 48 to 57, or 49 to 57 of TAT which TAT sequence has been modified to increase the alpha- helicity of that TAT sequence relative to a suitable TAT control sequence.
- the TAT sequence includes at least one amino acid substitution with an alpha-helix enhancing amino acid such as alanine.
- Additional transduction proteins are amino acid sequences, preferably synthetic peptide sequences that include at least amino acids 47 to 56, 48 to 56, 47 to 57, 48 to 57, or 49 to 57 of TAT which TAT sequence has been modified so that two or more basic amino acids such as arginine are substantially aligned along at least one face of that TAT sequence.
- the alignment can be facilitated by a variety of approaches including visualizing the TAT sequence as an alpha-helix on a helical wheel. See Figure 7 which follows.
- transduction proteins of this invention are peptide sequences that include at least amino acids 49 to 56 of TAT, preferably 47 to 56, 48 to 56, 49 to 56, 47 to 57, 48 to 57, or 49 to 57 of TAT, in which the TAT sequence includes at least one amino acid substitution with an alpha-helix enhancing amino acid.
- the amino acid substitution is selected to align substantially two or more arginine residues along at least one face of that TAT sequence, preferably alone one face of the TAT sequence. In one embodiment, preferably about 2, 3, 4, or 5 arginine residues are substantially aligned along at least one face of the helix, more specifically along one face of the helix.
- amino acid residues up to about 6 amino acids residues in the TAT sequence can be substituted with an alanine residue to enhance alpha-helicity and to align the arginine residues on at least one face of the helix.
- Additional transduction proteins in accord with this invention include amino acid sequences that comprise at least a transducing portion of the Antp sequence (SEQ ID NO: 10; see Table 2 below), preferably the Antp sequence which has been modified along lines described above for specified TAT sequences.
- the modifications can include at least one suitable amino acid substitution, deletion or addition that has been selected to enhance the alpha-helicity of the transduction protein, to align basic amino acid residues (e.g., Arginine) along at least one face of the Antp sequence, or both.
- transduction proteins that include the Antp sequence (SEQ ID NO: 10) in which the Antp sequence has been modified to include at least one amino acid modification sufficient to increase transduction efficiency of the protein by between about 2, 5 or 10 up to 100 or more fold compared to a suitable control peptide, e.g., the Antp sequence (SEQ ID NO: 10).
- class II transducing amino acid sequence is a peptide represented by the following formula: X,-X 2 -R X 3 -(P/X 4 )-(B/X 5 )-B-(P/X 6 )-X 4 -B-(B/X 7 ), wherein each of Xi, X > X 3 ⁇ , X 5 , X 6> X is an alpha helical promoting residue the same or different; each of (P/X 4 ) and (P/X 6 ) are independently proline or an alpha helical promoting residue; B is a basic amino acid residue; (B/X 5 ) (B/X 7 ) are each independently B or an alpha helical promoting residue; and R is arginine (Arg).
- a preferred alpha helical promoting residue is alanine (Ala).
- Preferred basic amino acid residues are arginine (Arg), lysine (Lys), especially Arg.
- Particularly preferred class II transducing amino acid sequences include at least one proline residue, usually between from about one to three residues.
- transduction domains described herein will vary according to parameters such as intended use and transduction efficiency desired. Generally, the transduction domain will exhibit a molecular weight of between from about 1 , 2, 3, 5, 10, 20 to about 50 kDa as judged by SDS-PAGE gel electrophoresis or other suitable assay. Specifically preferred transduction domains are described more fully below and in the examples which follow.
- transduction efficiency assay the transduction efficiency is determined by reference to a control assay in which one or more suitable control molecules are transduced into cells in parallel with a desired transduction protein.
- transduction rate and intracellular amounts of a specified transduction domain are measured and compared to the control molecule.
- Illustrative control molecules suitable include amino acids 47 to 57 of TAT (SEQ ID NO: 1), amino acids 49 to 57 of TAT, and the Antp sequence (SEQ ID NO: 10).
- Two or more protein transduction domains of this invention can be covalently linked to a desired molecule to be transduced.
- the protein transduction domains can be linked in tandem or can be separated by at least one suitable peptide linker as desired.
- Preferred transduction proteins of this invention exhibit an increase in transduction efficiency of between about 5 to 10 up to 100 or more fold when compared to a suitable control sequence, e.g., the minimal TAT sequence or other suitable control molecule. Examples of preferred transduction assays are described below in Example 7.
- the transduction proteins of the present invention can be made by a variety of conventional methods.
- DNA coding for a desired transduction protein can be obtained by isolating DNA from natural sources or by known synthetic methods, e.g. the phosphate triester method. See, e.g., Oligonucleotide Synthesis, IRL Press (M. Gait, ed., 1984). Synthetic oligonucleotides also may be prepared using commercially available automated oligonucleotide synthesizers. The synthetic oligonucleotide encoding the transducing protein may be inserted into a variety of suitable vectors (e.g., pTAT Vector) and expressed in an appropriate host cell. See generally Ausubel et al. and Sambrook et al. infra.
- the transduction domain of the fusion protein is operatively linked to a cytotoxic domain (sometimes referred to herein as "CD").
- cytotoxic domain sometimes referred to herein as "CD"
- the function of the cytotoxic domain in this example is to produce a cytotoxin that can kill or injure infected cells under specified conditions.
- the cytotoxic domain is transduced into the cell as part of the fusion molecule, and it is specifically intended to be released from that fusion molecule in the presence of one or more specified proteases induced by the pathogen infection. In some instances, release of the cytotoxin will be accompanied by further processing or maturation by the hosting cell.
- a preferred method of operatively linking the transduction domain and the cytotoxic domain is to use a nucleic acid sequence which encodes same ligated together to form an in-frame genetic fusion protein.
- cytotoxic domains include potentially toxic molecules such as known zymogens.
- most zymogens exhibit insignificant catalytic activity.
- the zymogens are converted into mature enzymes.
- the conversion includes proteolysis
- the cleavage occurs at site specific locations in the zymogen.
- fragments released from the zymogen are themselves catalytically active and upon release, further process the immature enzyme to a less immature or fully mature form. Zymogens including such fragments are often referred to as autocatalytic enzymes.
- the fragments are devoid of significant catalytic activity and must be cleaved to form the mature enzyme.
- a particular catalytic fragment can be naturally-associated with the zymogen or it can be recombinantly added to zymogen in accord with standard techniques to form a heterologous zymogen.
- Naturally- occurring protease cleavage sites in the zymogen usually serve to demarcate subunits within the zymogen. These can be replaced or added to in accordance with methods discussed herein.
- zymogens for use in accord with this invention include those associated with apoptosis, particularly cysteinyl aspartate-specific proteinases (caspases) and particularly caspase-3 (CPP32, apopain, Yama), caspases-5 (ICE re ⁇ -III, TY), caspase-4(ICE re ⁇ -II TX, ICH-2), caspase-1 (ICE), caspase-7 (Mch3, ICE-LAP3, CMH-1), caspase-6 (Mch2), caspase-8 (MACH, FLICE, Mch5), caspase-10 (Mch4), caspase-2 (ICH-1), caspase-9 (ICH-LAP6, Mch6) and catalytically active fragments thereof that are relatively inert zymogen fragments.
- caspases cysteinyl aspartate-specific proteinases
- CPP32 caspase-3
- apopain Yama
- caspases-5 ICE re ⁇ -III, TY
- Caspase-3 has previously been shown to be a rubicon of apoptosis by cleavage of the inhibitor of caspase-activated DNAse (ICAD) resulting in the activation of CAD and ultimately cell death.
- ICAD caspase-activated DNAse
- the structure of the Casp3 zymogen is known to include a N' terminal Pro domain, followed by a caspase cleavage recognition site, then the pi 7 domain that contains the catalytic Cys residue, a second caspase cleavage site and finally the pl2 domain (see Fig. 9A).
- the zymogen form of Casp3 remains inactive; however, during apoptotic signaling, it is cleaved by upstream caspases, such as Caspase-8 in T cells, resulting in loss of the Pro domain and an active pl7:pl2 heterotetramer. See Woo, M. et al., (supra).
- Example 12 illustrates a specific inactivation of the HIV viral replication machinery to treat HIV infected cells.
- the strategy exploits the HIV Protease to kill the infected cell while leaving uninfected cells unharmed.
- a modified Caspase 3 protein, TAT-Casp3 was made. This fusion protein transduces into -100%) of infected and uninfected cells.
- TAT-Casp3 is only specifically activated by HIV Protease in infected cells, resulting in apoptosis, whereas in uninfected cells it remains in the inactive zymogen form. See Ratner, L.
- Example 12 shows production of a transducible, modified apoptotic promoting caspase-3 protein (i.e. TAT-Casp3), that substitutes HIV proteolytic cleavage sites for endogenous sites.
- TAT-Casp3 a transducible, modified apoptotic promoting caspase-3 protein
- the fusion molecule efficiently transduces into -100% of cells, but remains inactive in uninfected cells.
- TAT-Casp3 becomes processed into an active form by HIV protease resulting in apoptosis of the infected cell.
- this specific strategy is generally applicable and could be used to combat other pathogens encoding specific proteases, such as Hepatitis C virus, cytomegalovirus and malaria.
- An additionally preferred zymogen is granzyme B.
- Bid An additionally preferred zymogen is Bid.
- the Bid protein has been reported to be a 20kDa protein related to the Bcl2/Bax family of apoptotic regulatory proteins. See Luo et al. (1998) Cell 94: 481; Li et al. (1998) Cell 94: 491; Wang et al. (1996) Genes & Dev. (1996) 10: 2859.
- the murine Bid sequence can be found in GenBank, accession number: U75506; NID: gl669513. See Example 11.
- mass action enhances the activity of certain embodiments of the anti-pathogen system. More particularly, it is believed that it is possible to administer the anti-pathogen system in many instances at extremely low doses (i.e., nanomoler levels). This feature can be particularly advantageous as it can enhance cell (and patient) tolerance for the anti-pathogen system.
- cleavage of the cytotoxic domain appears to draw additional fusion molecules into infected cells, thereby resulting in specific concentration of the cytotoxic domain and the cytotoxin in those infected cells. That concentration can be particularly significant with some cytotoxins, particularly those that require a high concentration to exhibit an optimal effect.
- Illustrative examples of such cytotoxins include those obtained from zymogens of blood coagulation proteases such as thrombin and fibrin; trypsin, chymotrypsin, diphtheria toxin, ricin, shiga toxin, abrin, cholera toxin, saporin, pseudomonas exotoxin (PE), pokeweed antiviral protein, and gelonin.
- cytotoxic domains which include proteins and particularly enzymes such as certain kinases and nucleoside deaminases associated with necrosis.
- enzymes include viral thymidine kinases, e.g., HSV thymidine kinase, and cytosine deaminase, respectively, as well as catalytically active fragments thereof.
- zymogens include those active at the surface of pathogen-infected cells such as a phospholipase enzyme, particularly phospholipase C.
- Preferred zymogens and enzymes are generally capable of killing cells as determined by a suitable cell viability assay, e.g., Trypan blue exclusion. More preferred zymogens and enzymes have a molecular weights of between about 5, 10, 20, 30, 40, 50 kD up to about 100 to 500 kD or more as assayed by standard methods. The molecular weight can be determined by a number of conventional techniques such as SDS-PAGE gel electrophoresis, sedimentation centrifugation, and column chromatography.
- a particularly preferred zymogen is caspase-3 (CPP32, apopain, Yama) or a catalytically active fragment thereof. See Examples 5-6 below.
- HSV-1 thymidine kinase or a catalytically active fragment thereof. See Example 8 below.
- preparation of the fusion molecules of the invention includes conventional recombinant steps involving, e.g., polymerase chain amplification reactions (PCR), preparation of plasmid DNA, cleavage of DNA with restriction enzymes, preparation of oligonucleotides, ligation of DNA, isolation of mRNA, introduction of the DNA into a suitable cell, and culturing of the cell.
- PCR polymerase chain amplification reactions
- the fusion molecules can be isolated and purified using chaotropic agents and well known electrophoretic, centrifugation and chromatographic methods. See generally, Sambrook et al., Molecular Cloning: A Laboratory Manual (2nd ed.
- HSV-1 maturational protease and protease cleavage site has been described. See e.g. Hall, M.R.T. and W. Gibson, Virology, 227:160 (1997); the disclosure of which is inco ⁇ orated by reference.
- any of the above-referenced protease cleavage sites can be modified as desired (e.g., by site-specific mutagenesis) so long as the sites are specifically cleaved by the pathogen-specific protease for which they are intended.
- it may be useful to determine the minimal sequence necessary for specific proteolytic cleavage e.g., to optimize size and spatial considerations relating to the fusion protein.
- Such minimal sequences have been reported for many pathogen- specific protease cleavage sites.
- the minimal sequence for a desired proteolytic cleavage site can be readily obtained by mutagenesis, particularly deletion analysis and site specific mutagenesis (e.g., alanine scanning mutagenesis).
- the modified cleavage site can be readily assayed in a standard protease cleavage assay as described below.
- protease cleavage sites are specifically broken (i.e. hydrolyzed) by one or more proteases induced by a pathogen infection. That is, the protease cleavage sites are not broken by proteases that naturally occur in an infected or uninfected cell such as those proteases referred to as housekeeping proteases. Specific cleavage of those protease cleavage sites can be monitored by a variety of techniques including SDS- polyacrylamide gel electrophoretic methods.
- Preferred pathogen-specific protease cleavage sites include the HSV-1 protease cleavage sites pl7-p24 (SQVSQNY— PIVQNLQ; SEQ ID NO:9), p7-pl (CTERQN— FLGKIWP; SEQ ID NO: 10), and pr-RT (IGCTLNF— PISPIET; SEQ ID NO: 11). See Table I above and the Examples below.
- Particularly preferred fusion proteins include operatively linked in sequence
- TAT or a suitable transducing fragment thereof such as the minimal TAT sequence/ the pl7-p24 or protease-RT cleavage site/ HSV TK
- TAT or a suitable transducing fragment thereof such as the minimal TAT sequence/ the protease-RT cleavage site/ the large domain of CPP32/ the pl7-p24 cleavage site/ and the small subunit of CPP32
- TAT or a suitable transducing fragment thereof such as the minimal TAT sequence/ the pl7-p24 or protease-RT cleavage site/ and pl6 wild-type or mutant form thereof
- TAT or a suitable transducing fragment thereof such as the minimal TAT sequence/(p7-pl) protease change site/HIV protease.
- present anti-pathogen system is facilitated by providing the fusion proteins in a misfolded form.
- native fusion proteins when used in accord with the present anti-pathogen system, transduce much less efficiently than corresponding misfolded sequences.
- present fusion proteins be fully or partially denatured prior to use in the present anti-pathogen system.
- Methods for fully or partially denaturing proteins are well known and include treatment with recognized chaotropic agents such as urea, particularly about 6-8M urea, ⁇ - mercaptoethanol, DTT, SDS or other detergents, particularly ionic detergents.
- physical treatments capable of denaturing proteins and polypeptides such as heating or sonication.
- methods including one or more chaotropic agents and physical treatments are also envisioned.
- the fusion protein is introduced into the cell as a misfolded fusion protein.
- rate and quantity of fusion protein uptake into the cell is significantly enhanced when compared to the same fusion protein introduced into the same cells in a low energy and essentially native conformation.
- the invention further provides nucleic acid sequences and particularly DNA sequences that encode the present fusion proteins.
- the DNA sequence is carried by a vector suited for extrachromosomal replication such as a phage, virus, plasmid, phagemid, cosmid, YAC, or episome.
- a DNA vector that encodes a desired fusion protein can be used to facilitate preparative methods described herein and to obtain significant quantities of the fusion protein.
- the DNA sequence can be inserted into an appropriate expression vector, i.e., a vector that contains the necessary elements for the transcription and translation of the inserted protein-coding sequence.
- a variety of host-vector systems may be utilized to express the protein-coding sequence.
- mammalian cell systems infected with virus e.g., vaccinia virus, adenovirus, etc.
- insect cell systems infected with virus e.g., baculovirus
- microorganisms such as yeast containing yeast vectors, or bacteria transformed with bacteriophage DNA, plasmid DNA or cosmid DNA.
- any one of a number of suitable transcription and translation elements may be used. See generally Sambrook et al., supra and Ausubel et al. supra.
- a preferred DNA vector according to the invention comprises a nucleotide sequence linked by phosphodiester bonds comprising, in a 5' to 3' direction a first cloning site for introduction of a first nucleotide sequence encoding a protein transduction domain, operatively linked to a sequence encoding a cytotoxic domain.
- the encoded cytotoxic domain includes additional cloning sites for an encoded potentially toxic molecule such as a zymogen. It is further preferred that the cytotoxic domain include additional cloning sites for encoded protease cleavage sites.
- Figures 3A-C depict particularly preferred DNA vectors of the invention.
- the DNA vectors are derived from the pTAT/pTAT-HA vector illustrated in Fig. 1.
- Preferred nucleic acid linker sequences for use with the pTAT/pTAT-HA vector are shown in Fig. 2.
- each of the fusion protein components encoded by the DNA vector be provided in a "cassette" format.
- cassette is meant that each component can be readily substituted for another component by standard recombinant methods.
- a DNA vector configured in a cassette format is particularly desirable when the encoded fusion protein is to be used against pathogens that may have or have capacity to develop serotypes.
- certain pathogen serotypes may be associated with individual protease cleavage sites specific for that serotype.
- one or more existing protease cleavage sites in a DNA vector formatted as a cassette can be replaced with other pre-determined protease cleavage sites as needed.
- Particular protease cleavage sites can be selected in accord with presence of the pathogen in individual patients.
- DNA vectors according to the invention formatted as a cassette minimize or eliminate occurrence of pathogen serotypes during treatment of a mammal and particularly a human by providing means to add or replace fusion protein components as needed.
- the DNA vectors are specifically formatted to adapt to specific strains of the virus and future mutation of the virus by providing means to substitute new HIV proteolytic cleavage sites into the fusion protein.
- These sites can be readily determined in a patient by polymerase chain reaction (PCR) amplification of the DNA obtained from patient and DNA sequencing across the viral cleavage sites using standard oligonucleotide primers.
- PCR polymerase chain reaction
- oligonucleotide primers a variety of suitable oligonucleotide primers could be selected for the amplification in accord with published sequences.
- the new/altered cleavage site can then be inserted into a fusion protein, e.g., the pTAT-CPP32 bacterial expression vector described in the examples below, protein purified and misfolded and then administered to the patient in a relatively short time frame (about 3-4 weeks).
- a fusion protein e.g., the pTAT-CPP32 bacterial expression vector described in the examples below
- the present anti-pathogen system can thus serve as an effective "warning system” that can register changes in pathogen serotype in vitro or in vivo.
- development of pathogen serotypes will be evidenced by decreased killing or injuring by the anti-pathogen system.
- the ability to rapidly detect appearance of the genetically altered pathogen serotypes is particularly relevant to developing rational therapies and can be remedied, e.g., by modifying the fusion protein as described above and/or by implementing a "cocktail" therapy approach as described below.
- cloning site is intended to encompass at least one restriction endonuclease site. Typically, multiple different restriction endonuclease sites (e.g., a polylinker) are contained within the nucleic acid. It optimal positioning of cloning sites in a DNA vector facilitate the cassette format.
- the fusion proteins of the present invention can be separated and purified by appropriate combination of known techniques. These methods include, for example, methods utilizing solubility such as salt precipitation and solvent precipitation, methods utilizing the difference in molecular weight such as dialysis, ultra-filtration, gel-filtration, and SDS-polyacrylamide gel electrophoresis, methods utilizing a difference in electrical charge such as ion-exchange column chromatography, methods utilizing specific affinity such as affinity chromatograph, methods utilizing a difference in hydrophobicity such as reverse-phase high performance liquid chromatograph and methods utilizing a difference in isoelectric point, such as isoelectric focusing electrophoresis, metal affinity columns such as Ni-NTA. See generally Sambrook et al. and Ausubel et al. supra for disclosure relating to these methods.
- the fusion proteins of the present invention be substantially pure. That is, the fusion proteins have been isolated from cell substituents that naturally accompany it so that the fusion proteins are present preferably in at least 80% or 90% to 95% homogeneity (w/w). Fusion proteins having at least 98 to 99% homogeneity (w/w) are most preferred for many pharmaceutical, clinical and research applications.
- the fusion protein should be substantially free of contaminants for therapeutic applications.
- the soluble fusion proteins can be used therapeutically, or in performing in vitro or in vivo assays as disclosed herein. Substantial purity can be determined by a variety of standard techniques such as chromatography and gel electrophoresis.
- fusion proteins of the invention can be expressed in insoluble forms. That can avoid proteolytic degradation of the fusion protein, significantly increase protein yields and increase delivery of fusion protein into target cells.
- the insoluble protein can be purified by known procedures such as affinity chromatography or other methods as detailed above.
- a host cell can be used for preparative pu ⁇ oses to propagate nucleic acid encoding a desired fusion protein.
- a host cell can include a prokaryotic or eukaryotic cell in which production of the fusion protein is specifically intended.
- host cells specifically include yeast, fly, worm, plant, frog, mammalian cells and organs that are capable of propagating nucleic acid encoding the fusion.
- mammalian cell lines which can be used include CHO dhfr- cells (Urlaub and Chasm, Proc. Natl. Acad. Sci. USA, 77:4216 (1980)), 293 cells (Graham et a ⁇ ., JGen.
- Host cells capable of propagating nucleic acid encoding a desired fusion protein encompass non-mammalian eukaryotic cells as well, including insect (e.g., Sp. frugiperda), yeast (e.g., S. cerevisiae, S. pombe, P. pastoris., K. lactis, H. polymorpha; as generally reviewed by Fleer, R., Current Opinion in Biotechnology, 3(5):486496 (1992)), fungal and plant cells. Also contemplated are certain prokaryotes such as E. coli and Bacillus.
- Nucleic acid encoding a desired fusion protein can be introduced into a host cell by standard techniques for transfecting cells.
- transfecting or “transfection” is intended to encompass all conventional techniques for introducing nucleic acid into host cells, including calcium phosphate co-precipitation, DEAE- dextran-mediated transfection, lipofection, electroporation, microinjection, viral transduction and/or integration. Suitable methods for transfecting host cells can be found in Sambrook et al. supra, and other laboratory textbooks.
- the present invention further provides a production process for isolating a fusion protein of interest.
- a host cell e.g., a yeast, fungus, insect, bacterial or animal cell
- a nucleic acid encoding the protein of the interest operatively linked to a regulatory sequence
- the fusion protein of interest is isolated from harvested host cells or from the culture medium. Standard protein purification techniques can be used to isolate the protein of interest from the medium or from the harvested cells.
- the purification techniques can be used to express and purify a desired fusion protein on a large-scale (i.e. in at least milligram quantities) from a variety of implementations including roller bottles, spinner flasks, tissue culture plates, bioreactor, or a fermentor.
- misfolded fusion protein for use in accordance with the invention can be produced by a variety of methods.
- a desired fusion protein is expressed in suitable bacterial cells and then isolated from those cells as inclusion bodies.
- the fusion protein is subsequently denatured in a strong chaotropic agent such as about 6 to 8 M urea followed by chromatography on a first column to separate the fusion protein from other bacterial cell components which accompany it.
- the bound fusion protein is then eluted from the column by standard means followed by dialysis in a suitable buffer or additional chromatography on a second column to remove the urea.
- fusion protein that is at least partially denatured means that at least a portion of the protein sample, e.g. at least about 10, 15, 20, 30, 40, 50, 60, 70 or 75 percent of the total number of amino acid residues in a substantially pure fusion protein sample, is in a conformation other than lowest energy refolded conformation.
- a fusion protein misfolded into a mixture of conformations can then be transduced into desired cells.
- the fusion protein can be directly added to cultured cells or to media in which those cells are being propagated.
- the higher energy denatured forms of a fusion protein of the invention are able to adopt lower energy conformations that can be more easily transduced into a cell of interest.
- the protein in its favored folded conformation will necessarily exist in a low energy state, and will be unable to adopt the relatively higher energy and hence unstable conformations that will be more easily introduced into a cell.
- the invention thus provides methods of treatment against pathogen infections such as virus infections and diseases associated with viruses, which methods in general will comprise administration of a therapeutically effective amount of one or more of the fusion proteins discussed above to a mammal, particularly a human, suffering from or susceptible to the pathogen infection.
- the fusion proteins of the invention be useful to treat cells infected with a virus capable of causing an immunodeficiency disease, particularly in a human.
- the fusion proteins will be particularly useful to treat retroviral infection in cells and in a human, particularly HIV infected human cells.
- retroviral infections which may be treated in accordance with the invention include human retroviral infections such as HIV-1, HIV-2, and Human T-cell Lymphotropic Virus (HTLV) e.g. HTLV-I or HTLV-II infections.
- the invention also provides methods of treatment of other diseases caused by or otherwise associated with a virus such as influenza including influenza A and B as well as diseases associated with viruses of the he ⁇ es family, e.g., he ⁇ es simplex viruses (HSV) including he ⁇ es simplex 1 and 2 viruses (HSV 1, HSV 2), varicella zoster virus (VZV; shingles), human he ⁇ es virus 6, cytomegalovirus (CMV), Epstein-Barr virus (EBV), and other he ⁇ es virus infections such as feline he ⁇ es virus infections, and diseases associated with hepatitis viruses including hepatitis C viruses (HCV).
- HSV he ⁇ es simplex viruses
- HSV 1 and 2 viruses HSV 1, HSV 2 viruses
- VZV varicella zoster virus
- CMV cytomegalovirus
- EBV Epstein-Barr virus
- Examples of clinical conditions which are caused by such viruses include he ⁇ etic keratitis, he ⁇ etic encephalitis, cold sores and genital infections (caused by he ⁇ es simplex), chicken pox and shingles (caused by varicella zoster) and CMV-pneumonia and retinitis, particularly in immunocompromised patients including renal and bone marrow transplant patients and patients with Acquired Immune Deficiency Syndrome (AIDS).
- Epstein-Barr virus can cause infectious mononucleosis, and is also suggested as the causative agent of nasopharyngeal cancer, immunoblastic lymphoma and Burkitt's lymphoma.
- the pathogen may be present in a virulent, latent, or attenuated form. Also contemplated is a population of pathogens including a mixture of those forms. Examples of particular pathogens of interest are viruses, e.g., CMV, HSV-1, HCV, particularly HCV type-C, HIV-1, HIV-2, KSH, yellow fever virus, certain flaviviruses and rhinoviruses.
- the pathogen can be any one of those capable of causing malaria or a medical condition relating to same such as P. falciparum, P. vivax, P. ovale, or P. malariae.
- the plasmodia cause malaria or various medical complications relating to malaria.
- the invention can be used to treat an existing condition or it can be used prophylactically to prevent infection by one or more pathogens.
- the anti-pathogen system and especially the fusion proteins of the invention can be administered to cells in vivo or in vitro by one or a combination of strategies.
- the fusion proteins can be administered to primary or immortalized cells growing in culture in vitro by conventional cell culture techniques that generally include contacting the cells with the fusion protein and allowing the fusion protein to transduce through the cells for a specified period of time.
- cell media will be removed from the cells prior to the contact to increase fusion protein concentration.
- the fusion proteins can be administered to cells in vivo, for example, by using a specified delivery mechanism suitable for introduction of fusion proteins into those cells.
- a specified delivery mechanism suitable for introduction of fusion proteins into those cells will be guided by several considerations including the location of the cells, the degree of transduction needed to kill or injure cells infected by the pathogen, and the general health of the cells.
- the fusion proteins of the invention may be administered to a mammal, particularly a primate such as a human, using a variety of suitable routes including oral, topical (including transdermal, buccal or sublingual), nasal and parenteral (including intraperitoneal, subcutaneous, / «tr ⁇ venous, intradermal or intramuscular injection.
- suitable routes including oral, topical (including transdermal, buccal or sublingual), nasal and parenteral (including intraperitoneal, subcutaneous, / «tr ⁇ venous, intradermal or intramuscular injection.
- suitable routes including oral, topical (including transdermal, buccal or sublingual), nasal and parenteral (including intraperitoneal, subcutaneous, / «tr ⁇ venous, intradermal or intramuscular injection.
- the fusion proteins of the present invention can be administered as a sole active agent, in combination with one or more other fusion proteins as provided herein or in combination with other medicaments such as reverse transcriptase inhibitors such as a dideoxynucleoside including AZT, ddl, ddC, d4T, 3TC, FTC, DAPD, 1592U89 or CS92; TAT antagonists such as Ro 3-3335 and Ro 24-7429; and other agents such as 9-(2-hydroxyefhoxymefhyl) guanine (acyclovir), ganciclovir or penciclovir, interferon, e.g., alpha-interferon or interleukin II, or in conjunction with other immune modulation agents including bone marrow or lymphocyte transplants or other medications such as levamisol or thymosin which would increase lymphocyte numbers and/or function as is appropriate.
- reverse transcriptase inhibitors such as a dideoxynucleoside including AZT,
- Additional medicaments that can be co-administered with one or more fusion proteins of the invention include standard anti -malarial such as those disclosed in Goodman, G. et al. (1993), The Pharmacological Basis of Therapeutics, 8 th ed. McGraw-Hill Inc. pp. 978-998.
- Preferred anti -malarial drugs include chloroquine, chloroguanidine, pyrimethamine, mefloquine, primaquaine and quinine.
- fusion proteins of the invention may be administered alone, they also may be present as part of a pharmaceutical composition in mixture with conventional excipient, preferably a pharmaceutically acceptable organic or inorganic carrier substances that is generally suitable for oral or nasal delivery as mentioned previously. However, in some cases, other modes of administration may be indicated in which case the fusion protein can be combined with a vehicle suitable for parenteral, oral or other desired administration and which do not deleteriously react with the fusion proteins and are not deleterious to the recipient thereof.
- Suitable pharmaceutically acceptable carriers include but are not limited to water, salt solutions, alcohol, vegetable oils, polyethylene glycols, gelatin, lactose, amylose, magnesium stearate, talc, silicic acid, viscous paraffin, perfume oil, fatty acid monoglycerides and diglycerides, petroethral fatty acid esters, hydroxymethyl- cellulose, polyvinylpyrrolidone, etc.
- the pharmaceutical preparations can be sterilized and if desired mixed with auxiliary agents, e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, colorings, flavorings and/or aromatic substances and the like which do not deleteriously react with the fusion proteins.
- auxiliary agents e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, colorings, flavorings and/or aromatic substances and the like which do not deleteriously react with the fusion proteins.
- solutions preferably oily or aqueous solutions as well as suspensions, emulsions, or implants, including suppositories.
- Ampules are convenient unit dosages.
- tablets, dragees or capsules having talc and/or carbohydrate carrier binder or the like are particularly suitable, the carrier preferably being lactose and/or corn starch and/or potato starch.
- a syrup, elixir or the like can be used wherein a sweetened vehicle is employed.
- Sustained release compositions can be formulated including those wherein the active component is protected with differentially degradable coatings, e.g., by microencapsulation, multiple coatings, etc.
- Therapeutic fusion proteins of the invention also may be inco ⁇ orated into liposomes. The inco ⁇ oration can be carried out according to known liposome preparation procedures, e.g. sonication and extrusion.
- a suitable effective dose of one or more fusion proteins will be in the range of from 0.01 to 100 milligrams per kilogram of bodyweight of recipient per day, preferably in the range of from 0.1 to 50 milligrams per kilogram bodyweight of recipient per day, more preferably in the range of 1 to 20 milligrams per kilogram bodyweight of recipient per day.
- the desired dose is suitably administered once daily, or several sub-doses, e.g. 2 to 5 sub-doses, are administered at appropriate intervals through the day, or other appropriate schedule.
- a preferred mode of administration is in an aerosol format and particularly by nasal or oral routes.
- kits that include the components of the anti-pathogen system of the invention.
- a kit can be used to kill or injure cells infected by one or more pre-determined pathogens.
- the kit includes a carrier means having in close confinement therein at least two container means: a first container means which contains one or more fusion proteins of the invention, and an optional second container means which contains a recombinant vector that encodes the fusion proteins.
- the fusion protein is administered to cells, in vitro or in vivo in accordance with methods described above.
- the invention is widely applicable to a variety of situations where it is desirable to kill or injure cells infected by one or a combination of pathogens.
- the invention is also applicable to studying mechanisms of pathogen infection of eukaryotic cells such as those cells of plant, insect, or animal origin, e.g., as in cells from primates and other mammals such as domesticated animals including certain birds, dogs, cats, horses, sheep, cows and the like.
- the present invention can be used for protection of crops or foodstuffs against pathogen attack.
- the anti -pathogen system can be used to screen candidate compounds for therapeutic capacity to inhibit certain proteins and particularly pathogen-specific proteases in infected cells.
- a preferred screening method includes transducing the anti-pathogen into desired cells, preferably cultured cells including immortalized or primary cells; infecting the cells with a pathogen, adding a candidate compound with potential therapeutic capacity to inhibit a pathogen-specific protease, and testing the cells for resistance to the pathogen, e.g., by performing a conventional cell viability assay.
- the assay is usually compared to a baseline control to determine the effect of the compound of interest on the cell, e.g. the resulting phenotype.
- the candidate compound can be added before, during or after transducing the cells with the anti-pathogen system.
- the baseline control may be the cell before introduction of the fusion protein, the cell in which the fusion protein has not been introduced, or the cell in which the fusion protein is non- functional, e.g. has a non- functional transcription activator region.
- One or more pre-determined pathogens can be added to the cell either before, after or during administration of the compound.
- the candidate compound of interest can be one of several molecules, including cytokines, tumor suppressors, antibodies, receptors, muteins, fragments or portions of such proteins, and active RNA molecules, e.g., an antisense RNA molecule or ribozyme, or a drug.
- active RNA molecules e.g., an antisense RNA molecule or ribozyme, or a drug.
- a combinatorial library of derivatives of known HIV RT inhibitors such as AZT can be readily tested by the present methods.
- Preferred compounds according to the invention are capable of reducing cell killing by at least about 40%, 50%, 60%, 70%, preferably at least about 80%, and more preferably at least about 90% or greater as assayed by standard cell viability tests such as by a Trypan blue exclusion test.
- a preferred method of screening a candidate compound for therapeutic capacity to inhibit a pathogen-specific protease comprises:
- the protein transduction domain can be selected from TAT, Antennapedia homeodomain, HSV VP22; a suitable fragment thereof; or any non-naturally occurring sequences that are capable of transduction.
- the cytotoxic domain can include a caspase and one or more protease cleavage sites.
- DNA and protein sequences described herein can be obtained from a variety of public sources including those specifically mentioned.
- a preferred source is the National Center for Biotechnology Information (NCBI)- Genetic Sequence Data Bank (Genbank) at the National Library of Medicine, 38A, 8N05, Rockville Pike, Bethesda, MD 20894.
- Genbank is also available on the Internet at http://www.ncbi.nlm.nih.gov. See generally Benson, D.A. et al., Nucl Acids Res., 25:1 (1997) for a description of Genbank.
- a prefened plasmid for TAT fusion protein expression was prepared as follows. A map of that plasmid is depicted in Figure 1 of the drawings. Figure 2 shows a nucleotide sequence (SEQ ID NO: 12) and amino acid sequence (SEQ ID NO: 13) of the pTAT linker as well as a nucleotide sequence (SEQ ID NO: 14) and amino acid sequence (SEQ ID NO: 15) of the pTAT-HA linker.
- pTAT and pTAT-HA (tag) bacterial expression vectors were generated by inserting an oligonucleotide corresponding to the 11 amino acid TAT domain flanked by glycine residues to allow for free-bound rotation of the TAT domain (G- RKKRRQRRR-G) (SEQ ID NO: 16) into the BamHI site of pRSET-A (Invitrogen).
- a polylinker was added C terminal to the TAT domain (see Figure 1) by inserting a second oligonucleotide into the Ncol site (5' or N') and Eco RI site that contained NcoI-Kpnl-Agel-XhoI-Sphl-EcoRI cloning sites. This is followed by the remaining original polylinker of the pRSET-A plasmid that includes BstBI-Hind III sites.
- the pTAT-HA plasmid was made by inserting an oligonucleotide encoding the HA tag (YPYDVPDYA; SEQ ID NO: 17; see Figure 2) where sequence is bold) flanked by glycines into the Ncol site of pTAT.
- the 5' or N' Ncol site was inactivated leaving only the 3' or C to the HA tag followed by the above polylinker.
- the HA tag allows the detection of the fusion protein by immunoblot, immunoprecipitation or immunohistostaining by using 12CA5 anti -HA antibodies.
- the nucleotide and amino acid sequences of each linker are set forth in Figure 2.
- the pRSET-A backbone encodes ampicillin resistance, fl, ori, ColEl ori (plasmid replication) and the transcript is driven by a T7 RNA polymerase promoter.
- the TAT fusion proteins described below were purified from host cells and pu ⁇ osefully misfolded to enhance transduction. More specifically, the fusion proteins were purified by sonication of transfected BL21(DE3) pLysS cells (Novagen) obtained from a 5 hr 1 L culture. That culture was inoculated with 100ml from an overnight culture in 10 ml of buffer A (8M urea 20mM HEPES (pH 7.2 (100 mM NaCl) ). Cell lysates were resolved by centrifugation, loaded onto a Ni-NTA column (Qiagen) in buffer A plus 20mM imidazole.
- TAT fusion protein were eluted with a 1 M NaCl step, desalted on a PD-10 desalting column (Pharmacia) into PBS or 20 mM HEPES [pH 7.2]/137 mM NaCl and frozen in 10% glycerol at -80°C.
- TAT pi 6 fusion proteins including HIV protease cleavage sites were made according to the following method.
- This plasmid was made by inserting double stranded oligomers encoding the pl7-pl4 and p7-pl HIV cleavage sites into the Ncol site of pTAT and pTAT-HA.
- the cleavage consist 14 amino acids, 7 on each side of the HIV cleavage site.
- pl7-p24 site (57mer), positive strand:
- oligonucleotide corresponding to the HIV cleavage site pl7-p24 (SEQ ID NO: 18) or p7-pl (SEQ ID NO: 19) was fused to the pTAT vector described in Example 1 (3' to the PTD sites) to produce a pTAT-HIVi or pTAT-HIV 2 vector, respectively.
- the pTAT-HIV ⁇ vectors served as parental vectors for the constructs shown for example in Figs. 3A-C.
- a pi 6 protein cDNA sequence was fused to the pl7-p24 HIV cleavage site to produce an in- frame TAT-pl6 fusion protein cDNA (Fig. 3C).
- a second pi 6 fusion protein was made by fusing the pl6 cDNA to the pTAT-HIV 2 vector.
- the order of components in each vector construct (N 1 terminus to C-terminus) was: HIS-TAT-PTD-CLEAVAGE SITE-pl6-PROTEIN, whereby "cleavage site” denotes the pl7-p24 or p7-pl cleavage sites, respectively.
- the fusion proteins were each purified and misfolded according to the method described in Example 2 above.
- the TAT-pl6 cDNA vectors were propagated in DH5- ⁇ bacteria.
- Purified pi 6 fusion proteins were individually transduced into Jurkat T-cells infected by HIV. Methods for infecting the Jurkat T-cells with HIV and transducing fusion proteins are described in examples, which follow. After 4, 8 and 12 hours, the cells are analyzed for cleavage of the fusion protein by Western immunoblot analysis using a commercially available anti-P16 antibody (Santa Cruz). As a control, the pl6 cDNA was fused to the pTAT vector described in Example 1 to produce the vector shown in Fig. 3D (no HIV protease cleavage site). That vector encoded a pi 6 fusion protein fused to TAT that was not cleaved in the infected cells. However, efficient cleavage was observed with pi 6 fusion proteins encoded by vectors shown in Fig. 3C that contained the HIV cleavage sites.
- the TAT-HIV ⁇ > 2 pi 6 fusion proteins produced in Example 3 were purified and labeled with FITC as described above. About 35 to 45 nanomoler of the labeled fusion protein was then added to HIV-infected (NLHX strain) and uninfected Jurkat T-cells in accordance with Example 6 below. The transduced Jurkat T-cells were then analyzed by FACS. All (100%) of cells in a mixed population of HIV infected/uninfected cells were transduced with the pl6 protein at 1, 2, 4, 8 and 16 hours post-addition of the FITC conjugated fusion protein.
- the infected cells retained the cleaved substrate (the pi 6 portion) but uninfected controls lost all of the transduced protein (it transduced out) as determined by the continued presence of FITC-labeled pi 6 as analyzed by FACS.
- a human CPP32 cDNA ( Alnemri et al., J. Biol. Chem., 269:30761 (1994); Genbank Accession No. U13737) was generated by independently PCRing (i.e. performing a Polymerase Chain Amplification (PCR) step) the CPP32 pi 7 and pl2 domains, then adding these DNA fragments together and PCRing using the outside PCR primers.
- the protocol is outlined in Figure 5. This is called a double PCR cloning approach and is a common methodological approach to link to two independent DNA fragments together, as follows:
- the pi 7 domain of CPP32 cDNA was PCRed using the A (+ strand) and B(- strand) primers (see below) and the pi 2 domain by using B(+ strand) and C(- strand) primers.
- the A primer encodes the pl7-pl HIV cleavage site in-frame with the CPP32 pi 7 domain coding sequence and the B primer encodes the pl7-p24 HIV cleavage site.
- the two purified fragments were mixed together and PCRed using only the A(+ strand) and C(- strand) primers.
- the two DNA fragments base pair together because the 3' end of pi 7 PCRed domain contains the (-) strand of the 5' end of the pl2 PCRed domain.
- the resultant DNA fragment was digested with Xhol at the 5' end and EcoRI at the 3' end yielding an approximately 900 bp fragment. This fragment was cloned into the Xhol and EcoRI Sites of pTAT and pTAT-HA plasmids.
- the protein, TAT-CPP32 wild type, was produced in BL2I(DE3) cells as outlined above.
- HIS-TAT-PTD- HIV 2 - subunit of CPP32-HIV The order of components in the resulting construct ( N' terminus to C- terminus) was: HIS-TAT-PTD- HIV 2 - subunit of CPP32-HIV, - small subunit of CPP32.
- the vectors encoding the HIS-TAT-PTD- HIV 2 - large subunit of CPP32- HIV-small subunit of CPP32 cDNA fusion proteins were each propagated in DH5- ⁇ bacteria.
- the fusion proteins were expressed and purified as described in Example 2 above.
- the HIS-TAT-PTD-HIV 2 - large subunit of CPP32-HIV) small subunit of CPP32 fusion protein is referred to as "TAT-CPP32".
- Example 5 To show that the TAT-CPP32 fusion protein produced in Example 5 was capable of killing HIV-infected cells, the fusion protein was purified and detectably-
- TAT-CPP32 fusion protein * labeled with FITC as described above in Example 2.
- the labeled TAT-CPP32 fusion protein was tested by the following method.
- Jurkat T-cells were infected by HIV (strain NLHX; about 1 x 10 5 to 1 x 10 6 infectious virus per ml). The cells were propagated in RPMI media. Approximately 4 to 7 days after the infection, the media was removed from the plates and about 35 to 45 nanomoler of the TAT-CPP32 fusion protein was added to the cells. The cells were incubated with the fusion proteins for about 30 minutes to allow transduction into the cells. Using FACS analysis, it was found that about 100%> of the cells were transduced by the fusion protein. Subsequently, media was added back to the plates and after about 18 hours post-transduction, the cells were examined for cell killing using conventional trypan blue exclusion and microscopy.
- TAT-CPP32 TAT-CPP32 fusion protein specifically kills HIV-infected cells but does not kill the uninfected cells in the population.
- the protease inhibitor Ritonavir (Abbott) was added to infected cells after transduction of the fusion protein. Briefly, cells were infected and transduced as described in Example 6 above. Following the transduction, about 1 ⁇ g/ml Ritonavir was added to the cell media and allowed to incubate with the cells for about 18 hours. The cells were then assayed for cell killing by conventional trypan blue exclusion and microscopy.
- TAT-CPP32 was inactivated by mutating the catalytic cysteine at residue 163 to methionine.
- the TAT-CPP32 molecule made in Example 5 was mutagenized to change the catalytically active Cys residue (#163) in the active site to Met by site directed mutagenesis.
- the following double stranded oligomeric nucleotide was inserted into the Stul site (in the pi 7 domain at the 5' end of the insert) and Pstl site present in the pl7-p24 HIV cleavage site between the pl7 and pl2 domains in TAT-CPP32.
- the double stranded oligomer has a blunt end at the Stul 5' end and a 3' overhang at the Pstl 3' end.
- the fusion protein was referred to as "TAT-CPP32 mut "or "TAT-CPP32 mutant” to denote the mutated catalytic Cys residue at position 163 of the CPP32 fusion protein.
- the TAT-CPP32 mut fusion protein was purified and transduced into HIV- infected Jurkat T-cells as described above in Example 6. It was found that the fusion protein was not capable of killing the HIV-infected cells. In contrast, the results of Example 6 show that the TAT-CPP32 fusion protein (with wild-type catalytic Cys residue) specifically killed the HIV infected Jurkat cells.
- HIV replication generally requires the presence and specific activity of HIV protease to cleave and process viral polyproteins, such as gag and gag-pol, for maturation as part of its infective life cycle.
- Transduction of anti-HIV killing molecules into HIV infected cells undergoing HIV replication, but not uninfected cells, will result in the specific recognition of the engineered HIV cleavage sites in any anti-HIV killing molecule of the invention, converting it from the inactive protein into an active killing molecule.
- uninfected cells do not contain the HIV specific protease and therefore, although present in uninfected cells it will remain in its inactive form.
- any escaping packaged virus particles may contain an active anti-HIV killing molecule that could 1) kill the particle prior to infection of a new cell or 2) initiate apoptosis in the newly infected cell, if so it should occur prior to replication of any virus particles.
- Figure 4 outlines a method for killing HIV-infected Jurkat T-cells by transducing a fusion protein comprising TK into the cells and then contacting the transduced cells with a prodrug (Acyclovir (Glaxo Wellcome) ).
- TK released from the fusion protein converts the Acyclovir into an active killing molecule, thereby killing the infected cells.
- uninfected (control) cells are not harmed by transduction of the TK fusion protein and administration of the Acyclovir.
- the TAT-TK fusion protein was made by the following method.
- the HSV-1 TK sequence was obtained from Genbank (Accession No. J02224 ).
- PCR primers were generated that conesponded to the N' and C of TK.
- the DNA fragment was cut with Ncol and EcoRI and inserted into the Ncol and EcoRI sites of pTAT-(HIV p 17-p24 cleavage site) or pTAT-(HIV p7-p 1 cleavage site).
- TK reverse PCR primer 39MER: 5' GGC GGG CCG GGA ATT CTC AGT TAG CCT CCC CCA TCT CCC 3' (SEQ ID NO:26)
- the fusion proteins were each purified and misfolded as discussed above in Example 2.
- Jurkat T-cells were infected by HIV (strain NLHX) as described above in Example 6. Approximately 4 to 7 days after the infection, the media was removed from the plates and about 35 to 45 nanomoler of the TAT-TK fusion protein (pl7-p24 or p7-pl cleavage site) was added to the cells. The cells were incubated with the fusion proteins for about 30 minutes to allow transduction into the cells. Using FACS analysis, it was found that about 100%) of the cells were transduced by the fusion protein.
- Transduced cells were allowed to incubate for about 18 hours to allow buildup of TK cleaved from the TAT-TK fusion protein. After this time period, the cells were washed in media and allowed to incubate for a further 4 hours. At this point about 1 to 100 nanomoler Acyclovir was added to the plates. After about 3 days, infected and non-infected cells were examined for cell killing by conventional trypan blue exclusion and microscopy. It was found that approximately 100%> of the total number of infected cells were killed by administration of the TAT-TK fusion protein and acyclovir.
- the results show that the TK enzyme was specifically concentrated in infected cells. However, in uninfected cells, the TK enzyme was not concentrated; the TAT- TK fusion was found to be transduced back out of those cells after washing. Thus, it is believed that the HSV TK processed the prodrug into an active killing drug only in the cells where it is retained, the infected cells, and not in the normal cells due to the inability of human/mammalian TK to process the prodrug. The results thus demonstrate that the TAT-TK fusion protein is an effective anti-HIV killing molecule.
- an HSV cytosine deaminase cDNA can be readily substituted for the TK gene to provide specific killing or injuring of HSV infected cells in combination with certain nucleoside analogs known in the field.
- the TAT-TK and TAT-CPP32 fusion proteins specifically described can be administered to an HIV-infected patient either as an injectable or preferably via an inhalation device to deliver same to the lungs where it will transduce into the blood stream.
- the fusion proteins will transduce into all contacted cells (airway and lung tissue, blood cells, etc.) including those typically infected by HIV such as certain immune cells in the bloodstream.
- TAT-CPP32 co-transduction experiments were performed with an HIV protease fused to TAT. The goal was to co-transduce uninfected Jurkat T-cells with two fusion proteins, the first fusion protein including the cell killing molecule (TAT-CPP32) and the second fusion protein including the HIV protease (TAT- protease) for cleaving the first fusion protein.
- transducible HIV protease was constructed by PCR cloning
- protease from HIV NLHX strain into the pTAT-(p7-pl cleavage site) vector.
- PCR primers were synthesized conesponding to initiating ATG (methionine) of the protease and the translational termination site.
- the DNA fragment was inserted into pTAT -(p7-pl cleavage site) vector at the Ncol 57N' terminal end and the EcoRI site 37C terminal end.
- the protein was expressed from the plasmid, pTAT-(p7-pl)- Protease, in BL2I (DE3) cells and purified as described above.
- the fusion protein is refened to as "TAT-Protease.”
- TAT-Protease 50-100 nM
- TAT-CPP32 wild type 50-100 nM
- TAT-Protease TAT-CPP32 wild type and mutant proteins showed minimal cytotoxicity when added alone. See Figures 6 A and 6B.
- TAT-CPP32 wild type, but not mutant, plus TAT-protease resulted in a substantial loss of viable cells and hence, activation of the TAT-CPP32 wild type protein.
- the addition of the protease inhibitor to this experiment resulted in the loss of specific TAT-CPP32 wild type killing.
- activation of TAT-CPP32 requires the presence of HIV protease.
- the co-transduction method is generally applicable for killing or injuring cells that are not usually infected by HIV virus.
- examples of such cells include certain CD4 " (minus) immune cells and non-immune cells such as fibroblasts.
- the method is readily adapted to include other transducing fusion proteins described herein, e.g., specified TAT fusion proteins requiring administration of a prodrug (e.g., TAT-TK and Acyclovir).
- the following artificial (i.e. synthetic) peptides were made by conventional peptide synthesis as described above.
- a goal of this experiment was to produce transduction domains that could transduce more effectively as judged by the intracellular concentration in transduced cells.
- the transduction domains were tested against a suitable control, which typically was the "natural" TAT or an Antp transduction domain.
- a FITC group was synthetically attached to N-terminus of 100% of each peptide so that transduction rate and intracellular concentration of each peptide could be quantified at equilibrium.
- the TAT transduction domain is recognized to be alpha-helical. In each synthetic peptide sequence, an alpha-helix was modeled with varying amounts of Arg on one face.
- the synthetic peptides were transduced into Jurkat T-cells along lines described above in Example 4. As can be seen in Table 2, all of the synthetic peptides transduced into the cells. The data show that the synthetic peptides with the most favorable rate and intracellular peptide concentration had the highest probability of having alpha helical structure (compared to naturally-occurring TAT) due to the substituted Ala residues. Further, the best synthetic peptides had Arg residues aligned on a single surface of the helix as suggested by helical wheel diagrams. See Figure 7. In particular, the modified synthetic peptides represented by SEQ ID Nos. 3 to 8 exhibited about a 5 to 10 fold increase in intracellular concentrations when compared to naturally-occurring TAT (SEQ ID NO:l).
- the data indicate that it is possible to design synthetic peptides with enhanced transduction efficiency compared with TAT.
- the data show that it is possible to increase transduction efficiency of naturally-occumng TAT by increasing probability of alpha helical helix formation in the peptide and by aligning at least two Arg residues on a single peptide helical face.
- the synthetic peptide sequences shown in Table 2 can be used to increase the transduction efficiency of a variety of fused amino acids, e.g., addition of 2, 5, 10, 20, 50 and 100 amino acids to the synthetic peptide sequence.
- the synthetic peptide sequences can also be fused to protein sequences of about 10, 15, 20, 30, 50, or about 100, up to about 500 kD or greater. The resulting fusion proteins can be tested for an increase in transduction efficiency as described above.
- the naturally-occumng Antp peptide typically exhibits a slower transduction rate than the TAT peptide.
- naturally-occurring TAT and the synthetic peptides described above will often be prefened for transducing amino acid sequences and particularly large proteins into cells.
- Table 2 shows synthetic peptide sequences that result in the rapid transport by transduction across cellular membranes enhanced into cells.
- the data show that those peptides having 1) a strong alpha helical nature and 2) at least a face/surface that is covered by Arg. residues are the best transducing domains.
- Figure 7 below shows a helical wheel plot showing the placement of the residues.
- All of the synthetic peptides have a transduction rate close to that of TAT (47-57), but some result in an increase intracellular concentration.
- Particular peptide sequences have at least the face of the helix containing basic residue such as Arg.
- the pro-apoptotic protein Bid is a 20 kDa protein related to the Bcl2/Bax family of apoptotic regulatory proteins. Bid is present in a zymogen proform in the cytoplasm. Activation of cells to undergo apoptosis by signaling through receptors such as Fas results in activation of two separate pro-apoptotic cascades/pathways. Additionally, Caspase-8 activation results in direct cleavage of cytosolic p20 Bid at residue Asp59 (aspartic acid residue #59 in mouse and Asp ⁇ O in human).
- Bid exists as an inactive proform/zymogen that can be specifically activated by proteolytic cleavage resulting in apoptotic induction through a different pathway than Caspase-3 (CPP32) via the DNA degradation pathway.
- a transducible TAT-Bid protein can be made by adding TAT to the N' terminus and removing the endogenous Caspase cleavage site of Bid and replacing it with an HIV cleavage site (TAT-p5 Bid-HIV cleavage-pl5 Bid). The goal was to test the effectiveness of the fusion protein in killing HIV infected cells or cells expressing HIV Protease.
- a TAT-HIV cleavage-pl5 Bid protein can also be made to provide a comparison between the two transducible Bid proteins.
- the cloning strategy is outlined below and, as with the TAT-CPP32 protein, any pathogen protease cleavage site could be cloned into this killing protein.
- the HIV cleavage site is used in this example as a model system.
- killing by TAT-Bid may be more effective than TAT-CPP32 in some cell types/diseases or, more than likely, be complimentary to TAT-CPP32 such that co-transduction of both killing proteins may result in a synergistic effect leading to further killing of the infected cells and potentially at lower concentration levels.
- Cloning Strategy- Murine Bid was PCR amplified by utilization of the following DNA primers in which the end product results in Ncol (DNA cleavage site) - p5 Bid domain - HIV proteolytic cleavage site (on the encoded protein) - pi 5 Bid domain by performing a double PCR.
- a TAT-HIV cleavage -pl5 Bid is also described and under construction.
- the p5 domain is PCR amplified with primer IF and 2R and in a separate PCR reaction the pi 5 domain is PCRed with primer 2F and 4R.
- primer 2F and 4R These DNA fragments are purified, mixed together and hybridized via the common regions present in 2F and 2R which are present on the 3' and 5' ends of the respective DNA fragments. The ends of this DNA fragment are extended and a final PCR reaction is performed using only primers IF and 4R which selects for the full length DNA fragment. This is a common cloning technique. The full length fragment is then cloned/ligated into pTAT-HA by cleavage with Ncol at the 5' end and EcoRI at the 3' end.
- the resultant plasmid, pTAT-Bid was transformed into E. coli strain DH5 ⁇ and then into E. coli strain BL21(DE3)pLysS and the protein was purified as outlined for the TAT-CPP32 protein.
- the resultant protein will contain an HIV Protease cleavage site between the TAT-p5 and pi 5 domains and is designated TAT-p5-HIV-pl5 Bid.
- TAT-HIV-pl5 Bid can be constructed similarly to the above except only a single PCR reaction is required.
- the primer 3F contains an Ncol DNA cleavage site followed by the HIV proteolytic cleavage site and contains DNA sequence homology to the 5' end of the pi 5 Bid domain.
- the DNA fragment generated from the PCR reaction with primer 3F and primer 4R is digested with Ncol and EcoRI and cloned into the Ncol and EcoRI sites of pTAT-HA, as outlined above.
- Primer IF (87mer): CGC GCC ATG GGC GGC TCC CAG GTG TCA CAG AAC
- Primer 2F (52mer): TTC CTG GGC AAA ATC TGG CCA GGC GGC AGC CAG
- Primer 2R (46mer): GTT AGC CTG GCG TTC GGT GCA GCC TGT CTG CAG
- Primer 4R (71 mer): CGC GAA TTC TCA GTC AGC ATA GTC TGG GAG GTC ATA TGG ATAGCC GTC CAT CTC GTT TCT AAC CAA
- the HIV Protease-activated Caspase-3 protein was generated to specifically kill cells infected by the HIV virus.
- the fusion protein was made as follows:
- a modified Casp3 protein was made by deletion of two residues from the two endogenous caspase cleavage sites (Asp-Ser) on Casp3 and insertion of fourteen residues encompassing the HIV pl7-p24 gag cleavage site ("A" site) and a p7-pl cleavage site ("D" site) (Fig. 9A).
- Asp-Ser caspase cleavage sites
- D p7-pl cleavage site
- TCR- antigen induced cell death occurs from a late Gi phase cell cycle check point. Immunity 8: 57 (1998); Nagahara, H. et al., Highly efficient transduction of full length TAT fusion proteins directly into mammalian cells: p27 K ⁇ pl mediates cell migration. Nature Med. (in press) (1998); Vocero-Akbani, A., et al., Transaction of full length TAT fusion proteins directly into mammalian cells: analysis of TCR- activation induced cell death (AID). In Methods in Enzymology (ed. Reed, J. C.) (Academic Press, San Diego) (in press) (1998).
- bacterially produced, misfolded fusion proteins containing an in- frame N' terminal protein transduction domain from HIV TAT are capable of transducing in a rapid and concentration-dependent fashion into -100% of all target cell types, including: peripheral blood lymphocytes (PBL), all cells present in whole blood, diploid fibroblasts, fibrosarcoma cells, hepatocellular carcinoma cells and leukemic T cells.
- PBL peripheral blood lymphocytes
- all cells present in whole blood diploid fibroblasts
- fibrosarcoma cells hepatocellular carcinoma cells
- leukemic T cells leukemic T cells.
- the Pro domain of the modified Casp3 was removed and substituted with the TAT transduction domain resulting in TAT-Casp3WT fusion protein (Fig. 9A).
- a catalytically inactive TAT-Casp3 mutant protein was generated by substituting a Met residue for the Casp3 active site Cys63 residue (TAT-Casp3MUT).
- TAT-Casp3 proteins were conjugated to fluorescein (FITC), then added directly to the media of Jurkat T cells and analyzed by Flow Cytometry (FACS) (Figs. 9B-C). Both TAT-Casp3 WT and TAT-Casp3MUT proteins rapidly transduced into - 100% of cells, achieving maximum intracellular concentration in less than 20 min. In addition, based on the narcow peak width before and after addition of FITC labeled proteins, individual cells within the population contain near identical intracellular concentrations of TAT-Casp3-FITC protein.
- FITC fluorescein
- TAT-Casp3FITC proteins in both cytoplasmic and nuclear compartments and not merely attached to the cellular membrane.
- FACS analysis of transduced cells at equilibrium 1 hr post- addition of 3, 6 and 12 nM TAT-Casp3WT-FITC protein demonstrated a concentration-dependency for protein transduction (Fig. 9E).
- TAT-Casp3 proteins readily transduce into -100%) of all cells in a rapid and concentration-dependent fashion.
- a model substrate was made by inserting HIV proteolytic cleavage sites into a previously characterized TAT-pl6 fusion protein. See Ezhevsky, S. A. et al. , Lissy, N. A., et al., and Vocero-Akbani, A., et al. (supra).
- the HIV A cleavage site was inserted between the TAT and pl6 domains, yielding TAT-A-pl6 fusion protein (Fig. 9A).
- TAT-HIV Pr transducible HIV Protease
- TAT-A-pl6, TAT-16 proteins See Ezhevsky, S. A., et al., and Vocero-Akbani, A., et al. (supra) and TAT-HIV Pr protein (Fig. 9D) were found to rapidly transduced into -100% of cells.
- FIG. 9 A Diagram depicting HIV cleavage site sequences and domains of TAT fusion proteins.
- Figures 9B-D FACS kinetic analysis of fluorescein (FITC) labeled TAT-Casp3WT, TAT-Casp3MUT and TAT-HIV Pr proteins added to cells at 0, 20 and 30 min.
- Figure 9E FACS dose analysis of 3, 6, and 12 nM TAT-Casp3WT-FITC protein added to cells at I hr post-addition. Note rapid, concentration-dependent transduction of all FITC labeled protein into ⁇ 100%> of cells and near identical intracellular concentration within the population as measured by FACS peak width of control vs. transduced cells.
- p 16(-) Jurkat T cells were transduced with 100 nM TAT-Apl6 or control TAT-pl6 protein (no HIV cleavage site) alone or in combination with 50 nM TATHIV Pr fusion protein for 5 hr and analyzed by anti-pl6 immunoblot for in vivo cleavage at the HIV A proteolytic cleavage site (Fig. 10A).
- Co-transduction of TAT-A-pl6 protein substrate with TAT-HIV Pr resulted in specific substrate cleavage while control TAT-pl6 protein (no HIV cleavage site) was not cleaved.
- TAT-Casp3MUT protein was transduced in combination with TAT-HIV Pr protein into cells (Fig. 10B).
- Co-transduction of TAT-Casp3 with TAT-HIV Pr resulted in detection of specific cleavage of TAT-Casp3 at the HIV "A" site between the pi 7 and pl2 domains in an HIV Protease-dependent fashion, yielding a TAT-D sitepl7-A half site protein.
- FIG. 10A Cultures of pl6(-) Jurkat T cells were transduced with TAT-pl6 or TAT-A-pl6 substrate proteins in combination with TATHIV Pr proteins for 5 hr and subjected to anti-pl6 immunoblot analysis. Co-transduction of TATA-pl6 protein with TAT-HIV Pr protein resulted in specific cleavage at the HIV A site.
- WCE HepG2 whole cell lysate containing wild type endogenous pl6; A-pl6, cleaved TAT-A-16 product retaining the HIV half site on pl6.
- Figure 10B Figure 10B.
- TAT-D-pl7-A cleaved product of TAT-Casp3 containing the N' terminal HIV A half site.
- TAT-Casp3 protein In addition, the ability of TAT-Casp3 protein to induce apoptosis in cells co-transduced with TAT-HIV Pr protein was tested.
- Jurkat T cells were treated with 100 nM TAT-Casp3WT or TATCasp3MUT proteins alone or in combination with 50 nM TAT-HIV Pr protein and assayed for cell viability 16 hr post-treatment (Fig. 11A).
- Transduction of TAT-Casp3WT protein alone into cells demonstrated a minor level of cytotoxicity.
- co-transduction of TAT-Casp3WT with TATHIV Pr protein into cells resulted in marked cytotoxicity.
- TAT-Casp3 -dependent cell death demonstrated a linear killing curve with cellular death detected as early as 4 hr post-transduction (Fig. 1 IB).
- cytotoxicity occurs only in the presence of catalytically active TAT-Casp3WT protein and that activation of TAT-Casp3WT specifically requires active HIV Protease, consistent with HIV Protease cleavage of TAT-Casp3 (Fig. 10B).
- FIG. 11 A Cultures of Jurkat T cells were transduced with combinations of TAT-Casp3WT (WT), TAT-Casp3MUT (MUT) and TAT-HIV Protease (Pr) proteins for 16 hr and analyzed for cell viability.
- WT TAT-Casp3WT
- MUT TAT-Casp3MUT
- Pr TAT-HIV Protease
- TUNEL assay Molecular methods for the identification of apoptosis in tissues. J. Histotechnology 17: 261 (1994).
- Transduction of 100 nM TAT-Casp3 WT, 100 nM TAT-Casp3MUT or 50 nM TAT-HIV Pr proteins alone into cells showed only background levels of TUNEL positive cells (Fig. 12 A).
- co-transduction of TAT-Casp3WT with TAT-HIV Pr protein resulted in a marked increase in TUNEL positive cells.
- TAT-Casp3MUT with TAT-HIV Pr protein showed only background TUNEL positive cells (Fig. 12A). Activation of TAT-Casp3 was also assayed by its ability to cleave an artificial
- Jurkat T cells were treated with 100 nM TAT-Casp3WT or TAT- Casp3MUT proteins alone or in combination with 50 nM TAT-HIV Pr protein for 6 hr and then assayed for cleavage of DEVD-AFC by release of fluorescent AFC (Fig. 12B). See Xiang, J., et al., Bax-induced cell death may not require interleukin 1B- converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA 93: 14550 (1996).
- FIG. 12 A Cultures of Jurkat T cells were cotransduced with TAT-Casp3WT (WT) and TAT-HIV Pr (Pr) protein resulted in specific TUNEL positive cells, an apoptotic end-marker.
- WT TAT-Casp3WT
- Pr TAT-HIV Pr
- TAT-Casp3 WT Transduction of TAT-Casp3 WT, TAT-Casp3MUT or TAT-HIV Pr alone showed only background levels of caspase activity.
- the appearance of cells containing ⁇ 2N DNA content was detected in TAT-Casp3 and TAT-HIV Pr co-transduced cells, a classic hallmark of Caspase-3 induced apoptosis as opposed to necrosis. See Woo, M. et al. (supra).
- Jurkat T cells were infected for 7-14 days with the NLHX strain of HIV-I and examined microscopically for HIV cytopathic effects. See Westervelt, P., et al., Identification of a determinant within the HIV-1 surface envelope glycoprotein critical for productive infection of cultured primary monocytes. Proc. Natl. Acad. Sci USA 88: 3097 (1991). At the start of each transduction experiment approximately 50%) of the culture was HIV positive. HIV infected cultures were transduced for 16 hr with 100 nM TAT-Casp3WT or TATCasp3MUT protein and then assayed for cell viability (Fig. 13).
- TAT-Casp3WT protein Treatment of HIV infected cells with TAT-Casp3WT protein resulted in a dramatic loss of HIV positive cells from the cultures. In addition, we detected both the appearance of cells containing ⁇ 2N DNA content and cells with condensed nuclei in TAT-Casp3 treated cells. However, transduction of TATCasp3MUT protein showed negligible effects. To determine if TAT-Casp3WT induced apoptosis was dependent on active HIV Protease in the infected cultures, HIV infected cultures were pretreated with 1 ⁇ g/ml Ritonavir prior to transduction with 100 nM TAT-Casp3WT protein (Fig. 5).
- TAT-Casp3WT TAT-Casp3MUT
- MUT TAT-Casp3MUT
- Pretreatment of HIV infected cells with HIV Protease inhibitor Ritonavir protects infected cells from TAT-Casp3WT protein killing. Ctrl, control addition of PBS to cultures; abscissa, %> viability of HIV positive cells in the population at start of transduction; enor bars represent SD.
- the present example demonstrates a novel strategy to kill HIV infected cells.
- this strategy harnesses the HIV encoded Protease by utilizing a modified zymogen form of an apoptotic inducer, Casp3, combined with a protein transduction delivery system.
- the results show that the transduction of proteins into cells is a rapid, concentration-dependent process that targets ⁇ 100%> of cells.
- TAT-Casp3 protein remains inactive in uninfected cells and is specifically activated by HIV Protease-dependent cleavage in HIV infected cells. This degree of specificity suggests that killing HIV infected cells by such a strategy may result in a high therapeutic index in patients.
- TAT-Casp3 protein specifically kills HIV infected cells.
- selection for mutations that renders the HIV Protease resistant to a broad spectrum of inhibitors is a continuing and growing problem in combating the HIV epidemic.
- the approach provided in this example and elsewhere in this disclosure allows for the continual adaptability of
- TAT-Casp3 proteins to HIV strain proteolytic cleavage site variance and/or mutation.
- TAT-Casp3 proteins described herein can be used to combat other pathogens by manipulating the proteins to contain relevant pathogen specific protease cleavage sites.
- TAT-Casp3 For cytotoxicity of TAT-Casp3 on uninfected cells, 1 X10 6 cells were transduced with 100 nM TAT-Casp3WT, TAT-Casp3MUT and/or 50 nM TAT-HIV Pr proteins for 16 hr and assayed for viability by Trypan Blue exclusion and/or genomic DNA damage by TUNEL assay (Trevigen). Number of TUNEL positive cells reported as per high-powered microscopic field with four fields per experiment averaged.
- TAT-Casp3 activity was measured by incubation of 20 ⁇ g of whole cell lysate with 50 ⁇ M DEVD-AFC and fluorescent AFC formation measured on a FL500 microplate fluorescence reader (Bio-Tek) as described. See Xiang, J., et al., (supra). Cells were preincubated with 1 ⁇ g/ml Ritonavir (Abbott Labs) for 1 hr prior to transduction.
- TAT-Casp3 For cytotoxicity of TAT-Casp3 on infected cells, Jurkat cultures were infected with 100 ng p24 Ag equivalent NLHX HIV- 1 strain for 7-14 days, assayed microscopically for cytopathic effects, then replated at 1 x 10 6 /ml and transduced with 100 nM of TAT-Casp3WT or TAT-Cas ⁇ 3MUT proteins for 16 hr followed by exclusionary dye viability analysis. Infected cells were pretreated with 1 ⁇ g/ml Ritonavir for 24 hr prior to transduction with TAT-Casp3WT protein. See Xiang, J., et al., (supra).
- FACS Fluorescein conjugated TAT fusion proteins were added to Jurkat T cell culture media and 1 xlO 4 cells were analyzed by FACS as described. See Ezhevsky, S. A., et al. (supra).
- TAT fusion proteins- pTAT-A/D-pl6 expression vectors were constructed by inserting double stranded oligomeric nucleotides encoding 14 residues of the HIV pl7-p24 ("A") cleavage site (single amino acid code:
- pTAT-Casp3WT vector was constructed by independent PCR amplification of the pl7 and pl2 domains containing engineered HIV A and D cleavage sites (14 residues) into the primers (pl7-forward primer:
- pl7-reverse primer 5'
- p 1 2-forward primer 5'- GGCGGCTCCCAGGTGTCACAGAACTATCCAATCGTGCAGAACC TGCAGGGCGGTGTTGATGATGACATGGCG 3' (SEQ ID NO:34);
- pl2-reverse primer 5'-CGAGCTACGCGAATTCTTAGTGATAAAAATAGAGTTC 3'; (SEQ ID NO:35)
- pTAT-Casp3MUT vector was constructed by inserting a double stranded oligomeric nucleotide
- pTAT-HIV Pr vector was constructed by PCR cloning the HIV Protease gene from HXB2R HIV strain (forward primer: 5'
- TAT fusion proteins were purified by sonication of high expressing BL21(DE3)pLysS (Novagen) cells in 8 M urea, purified over a Ni-NTA column and misfolded on a Mono S column as described23 24.
- FITC conjugated TAT fusion proteins were generated by fluorescein isothiocyanate labeling (Pierce), followed by gel purification in PBS on an S-12 column attached to an FPLC (Pharmacia) or PD-10 desalting column (Pharmacia), then added directly to cells in culture media and analyzed by FACS or microscopy.
- Class II synthetic transduction domains also require basic residues, such as Arginine or Lysine, but preferably Arginine; however, the introduction of kinks in the secondary structure due to the inclusion of Proline residues distinguishes them from Class I domains.
- transducible killing proteins that target pathogen infected cells based on the specificity of pathogen encoded specific proteases can also be applied to human diseases. Indeed, any human disease involving the expression of a cellular protease specifically in the diseased cell type and no other cells can be exploited to specifically kill that cell. Moreover, other cell specific properties can also be exploited to target specific cell types, such as high levels of heavy metals.
- a good example is prostate cells that express specific cellular proteases, such as Prostate Specific Antigen (PSA), and also have a 1000-fold elevated level of the heavy metal Zinc compared to rest of the human body. Importantly, both of these attributes are maintained in prostate cancer cells. Moreover, as a model system, after the patient has had children, the prostate is not required for any bodily function. Thus, all prostate cells, malignant or not, may be cleared from the body without loss of viability to the patient.
- PSA Prostate Specific Antigen
- PSA is a sub-family member of the kallikrein family of cellular proteases
- PSA is specifically synthesized by prostate cells and secreted into the lumen of the prostate.
- the function of PSA is to cleave gel forming proteins present in the seminal fluid, such as Sg I & II (Lilja et al. 1985).
- PSA Due to the tight cell-cell junctions in the prostate, PSA never leaks into or is detected in the blood stream. However, during rapid growth of prostate tumors, the junctions are looser and allow for low level release of PSA into the blood stream. In addition, metastasis of prostate tumor cells results in the further release and detection of PSA in the blood stream. As with exploiting pathogen specific proteases to discriminate and kill infected cells, PSA is 1.) specifically expressed in malignant prostate cells and 2.) has a specific substrate specificity or cleavage site. Thus, PSA is an excellent example of a human disease that can be targeted by transducible killing proteins.
- PSA activated transducible killing molecule can take several forms, as can pathogen activated killing molecules (see examples from above).
- PSA present in excretory vesicles can be utilized to activate the transduced zymogen intracellularly into a killing form as outlined above.
- extracellular PSA can be utilized to activate a zymogen that then transduces into the nearest cell, i.e. a prostate cell, and induces apoptosis.
- PSA Activated Transducible Killing Proteins Examples of PSA Activated Transducible Killing Proteins:
- TAT-HSV TK-PSA Structure TAT/STD-UB-HSV TK-PSA cleavage-TK Inhibitory Domain
- Cleavage by PSA removes a C terminal inhibitory domain while still allowing transduction of the enzyme into the cytoplasm of the cell as outlined above.
- Cleavage by a second constitutive cellular protease such as Ubiquitin Protease- 1 (UBP- 1) between the transduction domain (TAT/STD)-Ubiquitin motif and HSV TK allows retention of HSV TK specifically in Prostate cells.
- a prodrug such as acyclovir for HSV TK, is specifically processed into a cytotoxic form by the captured HSV TK and remains inactive in other cells of the body.
- the 1000-fold excess of Zinc in prostate cells could also be exploited by transduction of an inactive protein that requires a high concentration of Zn for dimerization and hence, activation.
- Such examples include utilizing dimerization domains of cellular transcription factors (Tx F) and dimerization domain of HPV E7 protein.
- Tx F cellular transcription factors
- the Caspase-3 pl7 and pl2 domains can be engineered to have terminal tags of E7 or Tx F dimerization domains. Requisite dimerization of transduced Caspase-3 pl7 and pl2 subunits would be dependent on dimerization of E7/ Tx F domains via coordination of Zn above a threshold concentration. Therefore, apoptotic induction would only be achieved when the E7/Tf F domains were dimerized by Zn. Such a transducible killing molecule would therefore be specifically activated only in cells containing high levels of Zn, such as prostate cells.
- Example 15 Specific killing of tumor cells by transduction of p53 proteins and restoration of p53 function.
- tumors containing wild type p53 tumor suppressor protein respond significantly better to traditional anti-tumor regimens such as radiation and chemotherapy than tumors containing mutant p53 (Lowe et al.). Indeed, p53 status is cunently the single most significant determinant for patient outcome after treatment. Thus, introducing wild type p53 into tumors should restore the sensitivity of these tumors to traditional anti-cancer therapies and, importantly, may allow for significant reductions in the amount of anti-cancer therapy required to kill the tumor cells. This would provide a distinct advantage to the patient by avoiding high concentrations of the anti-cancer treatments that also result in non-specific killing of normal cells in the patient.
- the last 30 aa are a negative regulatory domain that sterically blocks the DNA binding domain (DBD) from making contact with DNA (refs. 1-4). Therefore, a TATp53 1-364 was generated by cloning the ORF of p53 aa 1-364 into pTAT-HA by standard molecular biology techniques. Plasmids encoding TAT-p53 WT and TAT-p53 1-364 proteins were transformed into BL21(DE3)LysS cells and fusion proteins were purified as described in Nagahara et al., (1998) using the 6x His leader present in the N' terminus of the fusion protein. However several key differences to the standard protocol were required to purify TAT-p53 proteins ( Figures 15 & 16).
- the bacterial cells were sonicated in PBS and the insoluble recombinant TAT-p53 protein was pelleted by centrifugation. The pellet was resuspended in 6 M GuHCl/20 mM HEPES/100 mM NaCl and sonicated again. This was performed due to the poor binding of TAT-p53 protein to Ni-NTA columns in 8 M urea.
- the initial PBS sonication step removes the soluble cellular proteins and any soluble TAT-p53 protein that is usually comprised of degradation products.
- the two step sonication results in dramatic increases of full length protein that is highly purified.
- TAT-p53 1-364 protein was added to 2.5 x 10 5 human Jurkat leukemic T cells and 2.5 x 10 5 normal human diploid fibroblasts at 50, 100 and 200 nM final concentration at 0 and 24 hours.
- One negative control of only PBS addition and the other using TAT-Bid MUT protein at 200 nM final concentration was included to control for non-specific protein transduction effects.
- the cells were assayed for cell viability by trypan blue exclusionary dye at 48 hours post-addition of the first administration (Figure 17). Addition of increasing concentrations of TAT-p53 1-364 protein resulted in specific cell death of the tumor cells, while normal cells treated with TAT-p53 1-364 protein showed minimal toxicity.
- the negative control of TAT-Bid MUT protein showed only background levels of toxicity.
- TAT-pl6 or TAT-p27 additional anti-cell cycle tumor suppressor proteins, such as TAT-pl6 or TAT-p27
- TAT-pl6 or TAT-p27 additional anti-cell cycle tumor suppressor proteins
- TAT-p27 additional anti-cell cycle tumor suppressor proteins
- TAT-pl6 or TAT-p27 transduction of TAT-p53 proteins in combination with traditional small molecule chemotherapeutics that induce DNA damage will likely result in a further activation of the transduced p53 and hence, a synergy.
- Example 16- Inappropriate cell cycle arrest in tumor cells by transduction of Cdk Inhibitors results in tumor cell specific apoptosis (cell death).
- Jurkat leukemic T cells were treated at 0 and 24 hours with 200 nM of transducible Cdk Inhibitors, TAT-pl6 (Ezhevsky et al., 1997), TAT-p27 (Nagahara et al., 1998) and TAT-Cdk2-DN (dominant-negative; Nagahara et al., 1998), and assayed for cell viability by trypan blue exclusionary dye at 48 hours post addition of the first administration (Figure 18).
- TAT-p27 resulted in >70% specific cell death of the tumor cells, while TAT-Cdk2DN killed 60% and TAT-pl6 killed 40%). Transduction of these proteins into control, normal diploid fibroblasts resulted in less than 5%o toxicity.
- Example 17 Killing tumor cells by transduction of small peptides that specifically bind cyclin-dependent kinases (Cdks) and non-specifically bind other unknown proteins.
- Cdks cyclin-dependent kinases
- Tumor cells may also be more sensitive to cell cycle anest induced by transduction of peptidyl mimetics of Cdk inhibitors, such as pl6.
- Cdk inhibitors such as pl6.
- Treatment of normal and non-tumorigenic immortalized cells with a transducible peptide that contains an N' terminal PTD followed by a glycine and 20 amino acids of pl6 results in Gl cell cycle anest when maintained at low concentrations ( ⁇ 100 ⁇ M) (see Gius et al., 1999).
Abstract
Description
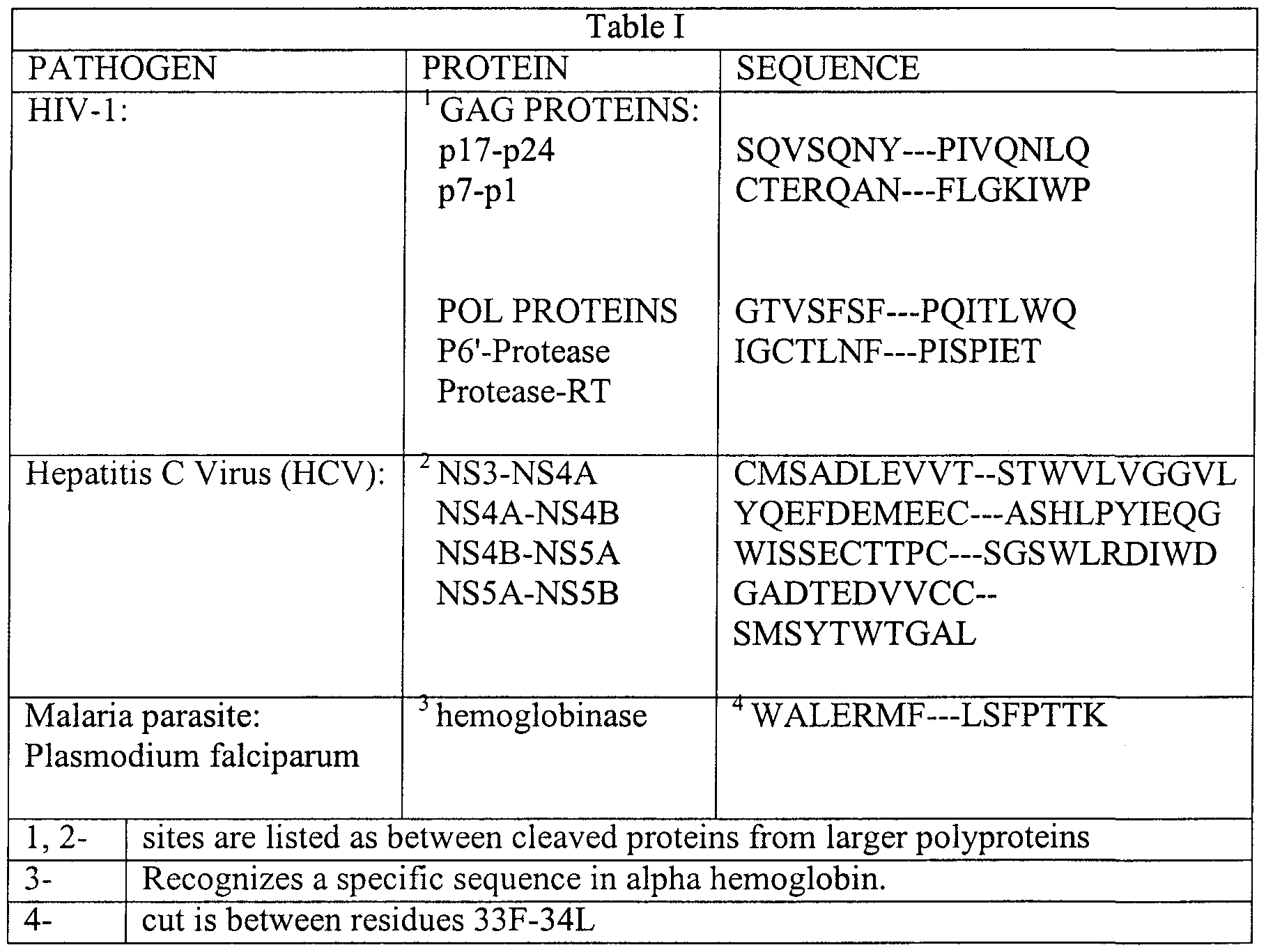
Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002354044A CA2354044A1 (en) | 1998-12-10 | 1999-12-10 | Protein transduction system and methods of use thereof |
| EP99966101A EP1137664A2 (en) | 1998-12-10 | 1999-12-10 | Protein transduction system and methods of use thereof |
| AU21728/00A AU2172800A (en) | 1998-12-10 | 1999-12-10 | Protein transduction system and methods of use thereof |
| JP2000586751A JP2002531113A (en) | 1998-12-10 | 1999-12-10 | Protein transduction system and methods of use |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11170198P | 1998-12-10 | 1998-12-10 | |
| US60/111,701 | 1998-12-10 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2000034308A2 true WO2000034308A2 (en) | 2000-06-15 |
| WO2000034308A3 WO2000034308A3 (en) | 2000-10-19 |
Family
ID=22340003
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/029289 WO2000034308A2 (en) | 1998-12-10 | 1999-12-10 | Protein transduction system and methods of use thereof |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1137664A2 (en) |
| JP (1) | JP2002531113A (en) |
| AU (1) | AU2172800A (en) |
| CA (1) | CA2354044A1 (en) |
| WO (1) | WO2000034308A2 (en) |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003020213A2 (en) * | 2001-08-30 | 2003-03-13 | Praecis Pharmaceuticals Incorporated | Methods and compositions for treating inflammatory disorders |
| WO2002055684A3 (en) * | 2001-01-09 | 2003-06-05 | University Of Iowa Research Foundation | Synthetic proteins containing a protein transduction domain |
| WO2003096986A2 (en) * | 2002-05-17 | 2003-11-27 | Washington University | Compositions and methods for treatment of wounds |
| WO2003106491A2 (en) * | 2002-06-18 | 2003-12-24 | Cepep Ab | Cell penetrating peptides |
| WO2005122675A2 (en) * | 2004-06-21 | 2005-12-29 | Quattromed As | Optimized recognition site of the alphavirus non-structural protease for tag removal and specific processing of recombinant proetins |
| WO2005084361A3 (en) * | 2004-03-03 | 2006-03-16 | Essentia Biosystems Inc | Multi-component biological transport systems |
| US7101844B2 (en) | 2002-03-29 | 2006-09-05 | Creagene, Inc. | Cytoplasmic transduction peptides and uses thereof |
| US7166692B2 (en) | 2003-03-04 | 2007-01-23 | Canbrex Bio Science Walkersville, Inc. | Intracellular delivery of small molecules, proteins, and nucleic acids |
| US7183066B2 (en) | 2002-09-27 | 2007-02-27 | Allergan, Inc. | Cell-based fluorescence resonance energy transfer (FRET) assays for clostridial toxins |
| US7374896B2 (en) | 2001-08-28 | 2008-05-20 | Allergan, Inc. | GFP-SNAP25 fluorescence release assay for botulinum neurotoxin protease activity |
| US7399607B2 (en) | 2004-09-22 | 2008-07-15 | Allergan, Inc. | Fluorescence polarization assays for determining clostridial toxin activity |
| EP2133362A1 (en) | 2003-07-25 | 2009-12-16 | Amgen, Inc | Antagonists and agonists of LDCAM and methods of use |
| WO2010105236A1 (en) | 2009-03-13 | 2010-09-16 | Allergan, Inc. | Immuno-based retargeted endopeptidase activity assays |
| WO2010105234A1 (en) | 2009-03-13 | 2010-09-16 | Allergan, Inc. | Cells useful for immuno-based botulinum toxin serotype a activity assays |
| US7807780B2 (en) | 2000-07-21 | 2010-10-05 | Revance Therapeutics, Inc. | Multi-component biological transport systems |
| US7846722B2 (en) | 2001-08-28 | 2010-12-07 | Allergan, Inc. | Luminescence resonance energy transfer (LRET) assays for clostridial toxin activity |
| EP2264458A1 (en) | 2005-04-05 | 2010-12-22 | Allergan, Inc. | Clostridial toxin activity assays |
| US20110071273A1 (en) * | 2001-05-18 | 2011-03-24 | Mayo Foundation For Medical Education And Research | Chimeric antigen-specific t cell-activating polypeptides |
| US8003753B2 (en) | 2001-08-28 | 2011-08-23 | Allergan, Inc. | Fret protease assays for clostridial toxins |
| US8022179B2 (en) | 2005-03-03 | 2011-09-20 | Revance Therapeutics, Inc. | Compositions and methods for topical application and transdermal delivery of an oligopeptide |
| EP2578601A1 (en) | 2008-03-14 | 2013-04-10 | Allergan, Inc. | Immuno-based botulinum toxin serotype A activity assays |
| US20130251727A1 (en) * | 2010-10-12 | 2013-09-26 | Arizona Biomedical Research Commission | Egfr-based peptides |
| WO2013165816A2 (en) | 2012-05-02 | 2013-11-07 | Merck Sharp & Dohme Corp. | SHORT INTERFERING NUCLEIC ACID (siNA) COMPOSITIONS |
| US20140004541A1 (en) * | 2011-03-11 | 2014-01-02 | Merz Pharma Gmbh & Co. Kgaa | Method for the determination of botulinum neurotoxin biological activity |
| US8623811B2 (en) | 2007-07-26 | 2014-01-07 | Revance Therapeutics, Inc. | Antimicrobial peptide, compositions, and methods of use |
| WO2014030780A1 (en) * | 2012-08-22 | 2014-02-27 | Mogam Biotechnology Research Institute | Screening and engineering method of super-stable immunoglobulin variable domains and their uses |
| US9724378B2 (en) | 2014-05-19 | 2017-08-08 | Samsung Electronics Co., Ltd. | Fusion protein comprising granzyme B and use thereof |
| EP3252068A2 (en) | 2009-10-12 | 2017-12-06 | Larry J. Smith | Methods and compositions for modulating gene expression using oligonucleotide based drugs administered in vivo or in vitro |
| US10066004B2 (en) | 2010-10-12 | 2018-09-04 | Arizona Cancer Therapeutics, Llc | EGFR-based inhibitor peptides for combinatorial inactivation of ERBB1, ERBB2, and ERBB3 |
| US10080786B2 (en) | 2005-03-03 | 2018-09-25 | Revance Therapeutics, Inc. | Methods for treating pain by topical application and transdermal delivery of botulinum toxin |
| US10172877B2 (en) | 2004-03-03 | 2019-01-08 | Revance Therapeutics, Inc. | Compositions and methods for topical diagnostic and therapeutic transport |
| US10201594B2 (en) | 2012-10-28 | 2019-02-12 | Revance Therapeutics, Inc. | Compositions and methods for safe treatment of rhinitis |
| US11484580B2 (en) | 2014-07-18 | 2022-11-01 | Revance Therapeutics, Inc. | Topical ocular preparation of botulinum toxin for use in ocular surface disease |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1641820B1 (en) * | 2003-06-30 | 2008-05-28 | Université de Lausanne | Rasgap derived peptide for selectively killing cancer cells |
| US9211248B2 (en) | 2004-03-03 | 2015-12-15 | Revance Therapeutics, Inc. | Compositions and methods for topical application and transdermal delivery of botulinum toxins |
| EP2801366A3 (en) * | 2004-09-02 | 2015-04-29 | Cognosci, Inc. | Improved apo E analogs and methods for their use |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0609580A1 (en) * | 1988-01-29 | 1994-08-10 | Chiron Corporation | Recombinant CMV neutralising proteins |
| EP0610046A2 (en) * | 1993-02-01 | 1994-08-10 | Bristol-Myers Squibb Company | Expression vectors encoding bispecific fusion proteins and methods of producing biologically active bispesific fusion proteins in a mammalian cell |
| US5652122A (en) * | 1989-12-21 | 1997-07-29 | Frankel; Alan | Nucleic acids encoding and methods of making tat-derived transport polypeptides |
| WO1999029721A1 (en) * | 1997-12-10 | 1999-06-17 | Washington University | Anti-pathogen system and methods of use thereof |
| WO1999055899A1 (en) * | 1998-04-28 | 1999-11-04 | Washington University | Methods for transducing fusion molecules |
-
1999
- 1999-12-10 WO PCT/US1999/029289 patent/WO2000034308A2/en not_active Application Discontinuation
- 1999-12-10 JP JP2000586751A patent/JP2002531113A/en active Pending
- 1999-12-10 CA CA002354044A patent/CA2354044A1/en not_active Abandoned
- 1999-12-10 EP EP99966101A patent/EP1137664A2/en not_active Withdrawn
- 1999-12-10 AU AU21728/00A patent/AU2172800A/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0609580A1 (en) * | 1988-01-29 | 1994-08-10 | Chiron Corporation | Recombinant CMV neutralising proteins |
| US5652122A (en) * | 1989-12-21 | 1997-07-29 | Frankel; Alan | Nucleic acids encoding and methods of making tat-derived transport polypeptides |
| EP0610046A2 (en) * | 1993-02-01 | 1994-08-10 | Bristol-Myers Squibb Company | Expression vectors encoding bispecific fusion proteins and methods of producing biologically active bispesific fusion proteins in a mammalian cell |
| WO1999029721A1 (en) * | 1997-12-10 | 1999-06-17 | Washington University | Anti-pathogen system and methods of use thereof |
| WO1999055899A1 (en) * | 1998-04-28 | 1999-11-04 | Washington University | Methods for transducing fusion molecules |
Non-Patent Citations (8)
| Title |
|---|
| BONIFACI N. ET AL.: 'Nuclear Translocation of an Exogenous Fusion Protein Containing HIV Tat Requires Unfolding' AIDS, vol. 9, no. 9, 1995, pages 995 - 1000, XP002927983 * |
| CHEN L.L. ET AL.: 'Increased Cellular Uptake of the Human Immunodeficiency Virus-1 tat Protein After Modification with Biotin' ANALYTICAL BIOCHEMISTRA, vol. 227, 1995, pages 168 - 175, XP002927982 * |
| GREEN ET AL.: 'mutational Amalysis of HIV-1 tat Minimal Domain Peptides: Identification of Trans-Dominant Mutants That Suppress HIV-LTR-Driven Gene Expression' CELL, vol. 58, 1989, pages 215 - 223, XP002927984 * |
| KOLESNITCHENKO V. ET AL.: 'A Major Human Immunodeficiency Virus Type 1-Initiated Killing Pathway Distinct from Apoptosis' JOURNAL OF VIROLOGY, vol. 71, no. 12, December 1997, pages 9753 - 9763, XP002927985 * |
| MOCHIZUKI ET AL.: 'High-titer human immunodeficiency virus type 1-based vector systems for gene delivery into nondividing cells' JOURNAL OF VIROLOGY, vol. 72, no. 11, November 1998, pages 8873 - 8883, XP002927986 * |
| NAGAHARA ET AL.: 'Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27kip1 induces cell migration' NATURE MEDICINE, vol. 4, no. 12, December 1998, pages 1449 - 1452, XP002927987 * |
| SCHARZE ET AL.: 'In vivo protein transduction: Delivery of a biologically active protein into the mouse' SCIENCE, vol. 285, no. 5433, 03 September 1999, pages 1569 - 1572, XP002927988 * |
| TALANIAN R.V. ET AL.: 'Granule-Mediated Killing: Pathways for Granzyme B-initiated Apoptosis' J. EXP. MED., vol. 186, no. 8, 20 October 1997, pages 1323 - 1331, XP002927981 * |
Cited By (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7807780B2 (en) | 2000-07-21 | 2010-10-05 | Revance Therapeutics, Inc. | Multi-component biological transport systems |
| WO2002055684A3 (en) * | 2001-01-09 | 2003-06-05 | University Of Iowa Research Foundation | Synthetic proteins containing a protein transduction domain |
| US20110071273A1 (en) * | 2001-05-18 | 2011-03-24 | Mayo Foundation For Medical Education And Research | Chimeric antigen-specific t cell-activating polypeptides |
| US8822182B2 (en) * | 2001-05-18 | 2014-09-02 | Mayo Foundation For Medical Education And Research | Chimeric antigen-specific T cell-activating polypeptides |
| US8053208B2 (en) | 2001-08-28 | 2011-11-08 | Allergan, Inc. | FRET protease assays for clostridial toxins |
| US8003753B2 (en) | 2001-08-28 | 2011-08-23 | Allergan, Inc. | Fret protease assays for clostridial toxins |
| US8022172B2 (en) | 2001-08-28 | 2011-09-20 | Allergan, Inc. | Luminescence resonance energy transfer (LRET) assays for clostridial toxin activity |
| US7838260B2 (en) | 2001-08-28 | 2010-11-23 | Allergan, Inc. | GFP-SNAP25 fluorescence release assay for botulinum toxin protease activity |
| US8013113B2 (en) | 2001-08-28 | 2011-09-06 | Allergan, Inc. | FRET protease assays for clostridial toxins |
| US8053209B2 (en) | 2001-08-28 | 2011-11-08 | Allergan, Inc. | FRET protease assays for clostridial toxins |
| US7846722B2 (en) | 2001-08-28 | 2010-12-07 | Allergan, Inc. | Luminescence resonance energy transfer (LRET) assays for clostridial toxin activity |
| US8048643B2 (en) | 2001-08-28 | 2011-11-01 | Allergan, Inc. | FRET protease assays for clostridial toxins |
| US7374896B2 (en) | 2001-08-28 | 2008-05-20 | Allergan, Inc. | GFP-SNAP25 fluorescence release assay for botulinum neurotoxin protease activity |
| WO2003020213A2 (en) * | 2001-08-30 | 2003-03-13 | Praecis Pharmaceuticals Incorporated | Methods and compositions for treating inflammatory disorders |
| WO2003020213A3 (en) * | 2001-08-30 | 2004-03-11 | Praecis Pharm Inc | Methods and compositions for treating inflammatory disorders |
| US7101844B2 (en) | 2002-03-29 | 2006-09-05 | Creagene, Inc. | Cytoplasmic transduction peptides and uses thereof |
| WO2003096986A2 (en) * | 2002-05-17 | 2003-11-27 | Washington University | Compositions and methods for treatment of wounds |
| WO2003096986A3 (en) * | 2002-05-17 | 2004-07-08 | Univ Washington | Compositions and methods for treatment of wounds |
| WO2003106491A3 (en) * | 2002-06-18 | 2004-12-23 | Cepep Ab | Cell penetrating peptides |
| JP2006514602A (en) * | 2002-06-18 | 2006-05-11 | シーイーピーイーピー エービー | Cell invasion peptide |
| WO2003106491A2 (en) * | 2002-06-18 | 2003-12-24 | Cepep Ab | Cell penetrating peptides |
| US7183066B2 (en) | 2002-09-27 | 2007-02-27 | Allergan, Inc. | Cell-based fluorescence resonance energy transfer (FRET) assays for clostridial toxins |
| US7749759B2 (en) | 2002-09-27 | 2010-07-06 | Allergan, Inc. | Cell compositions useful for cell-based FRET assays for clostridial toxins |
| US7420031B2 (en) | 2003-03-04 | 2008-09-02 | Lonza Walkersville, Inc. | Intracellular delivery of small molecules, proteins, and nucleic acids |
| US7166692B2 (en) | 2003-03-04 | 2007-01-23 | Canbrex Bio Science Walkersville, Inc. | Intracellular delivery of small molecules, proteins, and nucleic acids |
| EP2133362A1 (en) | 2003-07-25 | 2009-12-16 | Amgen, Inc | Antagonists and agonists of LDCAM and methods of use |
| US10172877B2 (en) | 2004-03-03 | 2019-01-08 | Revance Therapeutics, Inc. | Compositions and methods for topical diagnostic and therapeutic transport |
| WO2005084361A3 (en) * | 2004-03-03 | 2006-03-16 | Essentia Biosystems Inc | Multi-component biological transport systems |
| WO2005122675A3 (en) * | 2004-06-21 | 2006-04-27 | Quattromed As | Optimized recognition site of the alphavirus non-structural protease for tag removal and specific processing of recombinant proetins |
| WO2005122675A2 (en) * | 2004-06-21 | 2005-12-29 | Quattromed As | Optimized recognition site of the alphavirus non-structural protease for tag removal and specific processing of recombinant proetins |
| US7674601B2 (en) | 2004-09-22 | 2010-03-09 | Allergan, Inc. | Fluorescence polarization assays for determining clostridial toxin activity |
| US7638294B2 (en) | 2004-09-22 | 2009-12-29 | Allergan, Inc. | Fluorescence polarization assays for determining clostridial toxin activity |
| US7635574B2 (en) | 2004-09-22 | 2009-12-22 | Allergan, Inc. | Fluorescence polarization assays for determining clostridial toxin activity |
| US7632655B2 (en) | 2004-09-22 | 2009-12-15 | Allergan, Inc. | Fluorescence polarization assays for determining clostridial toxin activity |
| US7399607B2 (en) | 2004-09-22 | 2008-07-15 | Allergan, Inc. | Fluorescence polarization assays for determining clostridial toxin activity |
| US10744078B2 (en) | 2005-03-03 | 2020-08-18 | Revance Therapeutics, Inc. | Compositions and methods for topical application and transdermal delivery of botulinum toxins |
| US8022179B2 (en) | 2005-03-03 | 2011-09-20 | Revance Therapeutics, Inc. | Compositions and methods for topical application and transdermal delivery of an oligopeptide |
| US10080786B2 (en) | 2005-03-03 | 2018-09-25 | Revance Therapeutics, Inc. | Methods for treating pain by topical application and transdermal delivery of botulinum toxin |
| EP2264458A1 (en) | 2005-04-05 | 2010-12-22 | Allergan, Inc. | Clostridial toxin activity assays |
| US8623811B2 (en) | 2007-07-26 | 2014-01-07 | Revance Therapeutics, Inc. | Antimicrobial peptide, compositions, and methods of use |
| EP2578601A1 (en) | 2008-03-14 | 2013-04-10 | Allergan, Inc. | Immuno-based botulinum toxin serotype A activity assays |
| EP3851451A1 (en) | 2008-03-14 | 2021-07-21 | Allergan, Inc. | Immuno-based botulinum toxin serotype a activity assays |
| EP3556770A1 (en) | 2008-03-14 | 2019-10-23 | Allergan, Inc. | Immuno-based botulinum toxin serotype a activity assays |
| EP3031825A1 (en) | 2008-03-14 | 2016-06-15 | Allergan, Inc. | Immuno-based botulinum toxin serotype a activity assays |
| EP3858984A1 (en) | 2009-03-13 | 2021-08-04 | Allergan, Inc. | Cells useful for immuno-based botulinum toxin serotype a activity assays |
| EP3330372A1 (en) | 2009-03-13 | 2018-06-06 | Allergan, Inc. | Cells useful for immuno-based botulinum toxin serotype a activity assays |
| EP3281953A1 (en) | 2009-03-13 | 2018-02-14 | Allergan, Inc. | Immuno-based retargeted endopeptidase activity assays |
| EP2570809A1 (en) | 2009-03-13 | 2013-03-20 | Allergan, Inc. | Immuno-based retargeted endopeptidase activity assays |
| WO2010105236A1 (en) | 2009-03-13 | 2010-09-16 | Allergan, Inc. | Immuno-based retargeted endopeptidase activity assays |
| EP2568285A1 (en) | 2009-03-13 | 2013-03-13 | Allergan, Inc. | Immuno-based retargeted endopeptidase activity assays |
| WO2010105234A1 (en) | 2009-03-13 | 2010-09-16 | Allergan, Inc. | Cells useful for immuno-based botulinum toxin serotype a activity assays |
| EP4089169A1 (en) | 2009-10-12 | 2022-11-16 | Larry J. Smith | Methods and compositions for modulating gene expression using oligonucleotide based drugs administered in vivo or in vitro |
| EP3252068A2 (en) | 2009-10-12 | 2017-12-06 | Larry J. Smith | Methods and compositions for modulating gene expression using oligonucleotide based drugs administered in vivo or in vitro |
| US10066004B2 (en) | 2010-10-12 | 2018-09-04 | Arizona Cancer Therapeutics, Llc | EGFR-based inhibitor peptides for combinatorial inactivation of ERBB1, ERBB2, and ERBB3 |
| US20130251727A1 (en) * | 2010-10-12 | 2013-09-26 | Arizona Biomedical Research Commission | Egfr-based peptides |
| US9585938B2 (en) * | 2010-10-12 | 2017-03-07 | Arizona Cancer Therapeutics, Llc | EGFR-based peptides |
| US20140004541A1 (en) * | 2011-03-11 | 2014-01-02 | Merz Pharma Gmbh & Co. Kgaa | Method for the determination of botulinum neurotoxin biological activity |
| US9212355B2 (en) | 2011-03-11 | 2015-12-15 | Merz Pharma Gmbh & Co. Kgaa | Method for the determination of botulinum neurotoxin biological activity |
| US9096886B2 (en) * | 2011-03-11 | 2015-08-04 | Merz Pharma Gmbh & Co. Kgaa | Method for the determination of botulinum neurotoxin biological activity |
| EP3919620A1 (en) | 2012-05-02 | 2021-12-08 | Sirna Therapeutics, Inc. | Short interfering nucleic acid (sina) compositions |
| WO2013165816A2 (en) | 2012-05-02 | 2013-11-07 | Merck Sharp & Dohme Corp. | SHORT INTERFERING NUCLEIC ACID (siNA) COMPOSITIONS |
| US10078085B2 (en) | 2012-08-22 | 2018-09-18 | Mogam Biothechnology Institute | Screening and engineering method of super-stable immunoglobulin variable domains and their uses |
| WO2014030780A1 (en) * | 2012-08-22 | 2014-02-27 | Mogam Biotechnology Research Institute | Screening and engineering method of super-stable immunoglobulin variable domains and their uses |
| CN104769113A (en) * | 2012-08-22 | 2015-07-08 | 财团法人牧岩生命工学研究所 | Screening and engineering method of super-stable immunoglobulin variable domains and their uses |
| CN104769113B (en) * | 2012-08-22 | 2017-10-31 | 牧岩生命科学研究所 | The screening and remodeling method and its application in overstable immunoglobulin variable domain domain |
| US10201594B2 (en) | 2012-10-28 | 2019-02-12 | Revance Therapeutics, Inc. | Compositions and methods for safe treatment of rhinitis |
| US9724378B2 (en) | 2014-05-19 | 2017-08-08 | Samsung Electronics Co., Ltd. | Fusion protein comprising granzyme B and use thereof |
| US11484580B2 (en) | 2014-07-18 | 2022-11-01 | Revance Therapeutics, Inc. | Topical ocular preparation of botulinum toxin for use in ocular surface disease |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002531113A (en) | 2002-09-24 |
| WO2000034308A3 (en) | 2000-10-19 |
| EP1137664A2 (en) | 2001-10-04 |
| CA2354044A1 (en) | 2000-06-15 |
| AU2172800A (en) | 2000-06-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6221355B1 (en) | Anti-pathogen system and methods of use thereof | |
| EP1137664A2 (en) | Protein transduction system and methods of use thereof | |
| CA2364690A1 (en) | Novel transduction molecules and methods for using same | |
| US5674980A (en) | Fusion protein comprising tat-derived transport moiety | |
| US5804604A (en) | Tat-derived transport polypeptides and fusion proteins | |
| JP4522697B2 (en) | Protease detection system | |
| Vocero-Akbani et al. | Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein | |
| EP0656950B1 (en) | Tat-derived transport polypeptides | |
| AU658818B2 (en) | Method of delivering molecules into eukaryotic cells using tat protein conjugate | |
| AU711126B2 (en) | Protein targeting into HIV virions based on HIV-1 VPR fusion molecules | |
| Freund et al. | A possible regulation of negative factor (Nef) activity of human immunodeficiency virus type 1 by the viral protease | |
| CA2230925C (en) | Fusion protein delivery system and uses thereof | |
| JP2022549057A (en) | Conjugates for intracellular delivery of molecules | |
| CA2339850C (en) | A non-cytolytic recombinant hiv-1 vaccine | |
| Boross et al. | Drug targets in human T-lymphotropic virus type 1 (HTLV-1) infection | |
| JP2008500267A (en) | Interactive polypeptides containing heptapeptide patterns and cell permeation domains | |
| CA2236036A1 (en) | Signal-regulated, cleavage-mediated toxins | |
| AU743113C (en) | Fusion protein delivery system and uses thereof | |
| Badley | HIV Protease (PR) and Cell Death |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2354044 Country of ref document: CA Ref country code: CA Ref document number: 2354044 Kind code of ref document: A Format of ref document f/p: F |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 586751 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 21728/00 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999966101 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999966101 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999966101 Country of ref document: EP |