WO1998023621A1 - Cephalosporin derivatives - Google Patents
Cephalosporin derivativesInfo
- Publication number
- WO1998023621A1 WO1998023621A1 PCT/US1997/021785 US9721785W WO9823621A1 WO 1998023621 A1 WO1998023621 A1 WO 1998023621A1 US 9721785 W US9721785 W US 9721785W WO 9823621 A1 WO9823621 A1 WO 9823621A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- hydrogen
- compound
- halogen
- Prior art date
Links
- 0 C*(C)(C(C=C)=CC=C)NC Chemical compound C*(C)(C(C=C)=CC=C)NC 0.000 description 4
- IHCMMZHZWHUTKM-UHFFFAOYSA-N CCCc(c(Cl)c1)cc(Cl)c1SC Chemical compound CCCc(c(Cl)c1)cc(Cl)c1SC IHCMMZHZWHUTKM-UHFFFAOYSA-N 0.000 description 1
- REQOXZPUPOPNRQ-UHFFFAOYSA-N CSc1cc(Cl)c(CCC(NCC(O)=O)=O)cc1Cl Chemical compound CSc1cc(Cl)c(CCC(NCC(O)=O)=O)cc1Cl REQOXZPUPOPNRQ-UHFFFAOYSA-N 0.000 description 1
- XLTIFEBCUFTIBF-GUXUPKRYSA-N C[C@H](C(C1O)N)N1/C(/C)=C(/C)\CS Chemical compound C[C@H](C(C1O)N)N1/C(/C)=C(/C)\CS XLTIFEBCUFTIBF-GUXUPKRYSA-N 0.000 description 1
- MNTAOVIUCGXCRW-UHFFFAOYSA-N Cc(c(Cl)c1)cc(Cl)c1SC Chemical compound Cc(c(Cl)c1)cc(Cl)c1SC MNTAOVIUCGXCRW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D501/14—Compounds having a nitrogen atom directly attached in position 7
- C07D501/16—Compounds having a nitrogen atom directly attached in position 7 with a double bond between positions 2 and 3
- C07D501/20—7-Acylaminocephalosporanic or substituted 7-acylaminocephalosporanic acids in which the acyl radicals are derived from carboxylic acids
- C07D501/24—7-Acylaminocephalosporanic or substituted 7-acylaminocephalosporanic acids in which the acyl radicals are derived from carboxylic acids with hydrocarbon radicals, substituted by hetero atoms or hetero rings, attached in position 3
- C07D501/36—Methylene radicals, substituted by sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention is directed to new cephem derivatives represented by the general formula
- Q is an optionally substituted pyridinium group connected to the sulfur atom via a ring carbon atom;
- X is halogen;
- Y is hydrogen or halogen;
- A is C0 2 H, P0 3 H 2 , S0 3 H or tetrazole;
- L 1 is a furan group, a thiophene group, a C 2 -C 10 alkyl group, or a C 2 -C 10 alkyl group interrupted by one or more groups independently selected from vinyl, S, SO, S0 2 , S0 2 NH,
- n is 0 or 1; and R 1 is hydrogen or a carboxyl-protecting group; or pharmaceutically acceptable salts and /or prodrugs thereof.
- the derivatives are gram-positive antibacterial agents especially useful in the treatment of diseases caused by methicillin-resistant Staphylococcus aureus (also referred to below as MRSA or methicillin-resistant S. aureus).
- the literature discloses a vast number of cephem derivatives having a wide variety of C-3 and C-7 substituents.
- R j is hydrogen or chloro
- R 2 is hydroxy or amino
- Z is oxygen or sulfur
- A is acetoxy or N-pyridinium
- M is hydrogen, pharmaceutically acceptable non-toxic cations or an anionic charge when A is N-pyridinium.
- EP 638,574 Al discloses cephem derivatives of the general formula
- Z -H, halogen, -OH, -C 5 O-alkyl, -OCH 2 CONH 2 , -OCONH 2 , -OS0 2 NH 2 , -OCH 2 CN, -NH 2 either as such or
- amides of benzene and toluene derivatives -N0 2 , -NO, -CHO, -CH 2 OH, -COOH, -SH, -SOH, -S0 2 H, -S0 3 H, -S-alkyl where the alkyl residue is -C 8 , -CF 3 ;
- R -H, -OH, - -O-alkyl with the alkyl residue possibly containing halogens, acid functionalities either free or salified with alkaline or alkaline earth metals, basic functions such as -OCH 2 CH 2 NH 2 , -OCH 2 CH 2 NH-CH 3 , -
- A -S-, -O-, -CH 2 -, -SO-, S0 2 -;
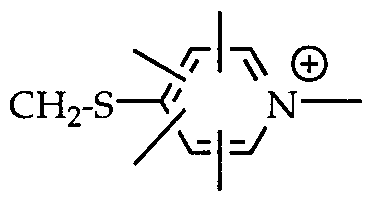
- R 2 is an alkyl or aryl radical -CH 2 -OCOCH 3 , -CH 2 -S (CH 2 ) 2 -N-CH
- C-3 substituents employed in the compounds of the present invention are known in the cephem art, but have not previously been combined with the C-7 substituents of the present invention. Applicants have discovered that the combination of C-3 and C-7 substituents provided in the compounds of the present invention unexpectedly gives the desired solubility, activity and toxicity profile needed for commercially viable anti-MRSA cephem products.
- the present invention provides a novel series of cephem derivatives of the general formula
- Q is an optionally substituted pyridinium group connected to the sulfur atom via a ring carbon atom;
- X is halogen;
- Y is hydrogen or halogen;
- A is C0 2 H, P0 3 H 2 , S0 3 H or tetrazole;
- L 1 is a furan group, a thiophene group, a C 2 -C 10 alkyl group, or a C 2 -C 10 alkyl group interrupted by one or more groups independently selected from vinyl, S, SO, S0 2 , S0 2 NH,
- the compounds of formula I are antibacterial agents useful in the treatment of infections in humans and other animals caused by a variety of gram-positive bacteria, particularly methicillin-resistant S aureus.
- the present invention provides novel cephem derivatives of general formula I above which are antibacterial agents useful in the treatment of infectious diseases in humans and other animals.
- the compounds exhibit good activity against a wide variety of gram-positive microorganisms, e.g. S. pneumoniae, S. pyogenes, S. aureus. E. faecalis, S. epidermidis and S. hemolyticus. and are particularly useful against strains of methicillin-resistant S. aureus.
- X is halogen
- Y is hydrogen or halogen
- A is C0 2 H, P0 3 H 2 , S0 3 H
- L 1 is a furan group, a thiophene group, a C 2 -C 10 alkyl group, or a C 2 -C 10 alkyl group interrupted by one or more (preferably 1 or 2) groups
- n 0 or 1.
- X is halogen
- Y is hydrogen or halogen
- A is C0 2 H, P0 3 H 2 , S0 3 H or tetrazole
- L 1 is a furan group, a thiophene group, a C 2 -C 10 alkyl group, or a C 2 -C 10 alkyl group interrupted by one or more groups independently selected from vinyl, S, SO, S0 2 , S0 2 NH, o H
- n is 0 or 1;
- R 3 and R 4 are each independently selected from hydrogen or - C 6 alkyl;
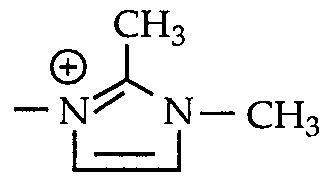
- R 2 is hydrogen, NH 2 , pyrrolidinyl, C j -C 8 alkyl, C 3 -C 6 cycloalkyl, C 2 - C 6 alkyl substituted by one or more substituents independently selected from OH, NR 5 R 6 in which R s and R 6 are each independently hydrogen or C ⁇ -C 6 alkyl, C0 2 H, morpholinyl, morpholinyl quaternized by a - alkyl
- X is halogen
- Y is hydrogen or halogen
- A is C0 2 H, P0 3 H 2 , S0 3 H or tetrazole
- L 1 is a furan group, a thiophene group, a C 2 -C 10 alkyl group or a C 2 -C 10 alkyl group interrupted by one or two groups independently
- n 0 or 1;
- R 3 and R 4 are each independently selected from hydrogen or -
- R 2 is hydrogen, NH 2 , pyrrolidinyl, C ⁇ -C 6 alkyl, C ⁇ -C 6 alkyl substituted by one or two substituents independently selected from OH,
- R 5 and R 6 are each independently hydrogen or -Cg alkyl, C0 2 H, morpholinyl, morpholinyl quaternized by a C -C 6 alkyl group, oxo, halogen, S0 3 H, P0 3 H 2 , tetrazolyl,
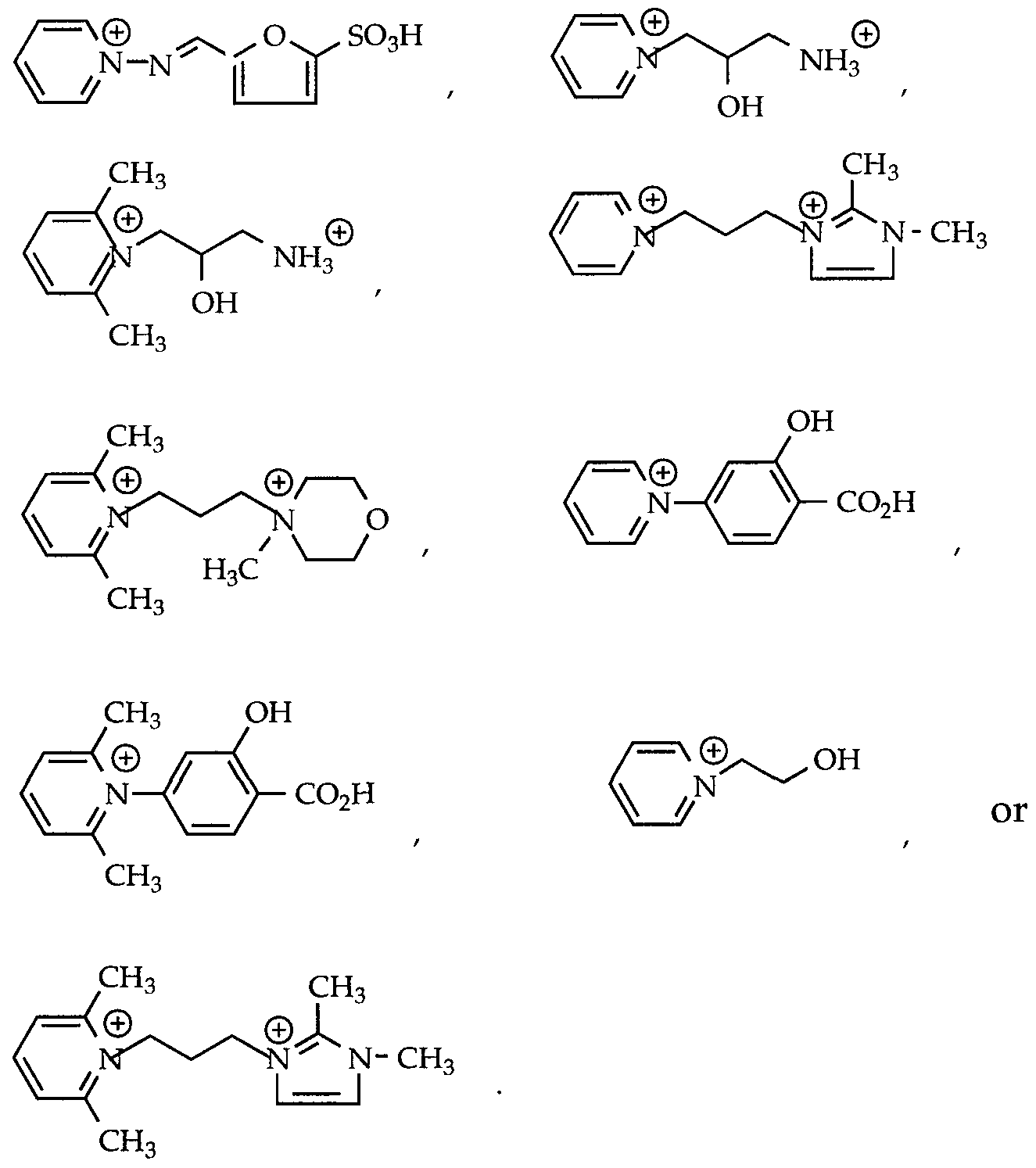
- R 7 is a furan or thiophene radical optionally substituted by a -C0 2 H or -S0 3 H group, phenyl or phenyl substituted by 1-2 substituents independently selected from OH, NR 5 R 6 in which R 5 and R 6 are as defined
- R 1 is hydrogen or a carboxyl-protecting group
- Halogen includes chloro, bromo, fluoro and iodo, and is preferably chloro or bromo and most preferably chloro.
- the aliphatic "alkyl” groups may be straight or branched chains containing the specified number of carbon atoms.
- salts as used herein is intended to include the nontoxic acid addition salts with inorganic or organic acids, e.g. salts with acids such as hydrochloric, phosphoric, sulfuric, maleic, acetic, citric, succinic, benzoic, fumaric, mandelic, p- toluenesulfonic, methanesulfonic, ascorbic, lactic, gluconic, trifluoracetic, hydroiodic, hydrobromic, and the like.
- acids such as hydrochloric, phosphoric, sulfuric, maleic, acetic, citric, succinic, benzoic, fumaric, mandelic, p- toluenesulfonic, methanesulfonic, ascorbic, lactic, gluconic, trifluoracetic, hydroiodic, hydrobromic, and the like.
- acids such as hydrochloric, phosphoric, sulfuric, maleic, acetic, citric, succinic, benzo
- alkali metal salts particularly sodium or potassium
- alkaline earth metal salts particularly calcium or magnesium
- suitable organic bases such as lower alkylamines (methylamine, ethylamine, cyclohexylamine, and the like) or with substituted lower alkylamines (e.g. hydroxyl-substituted alkylamines such as diethanolamine, triethanolamine or tris-(hydroxymethyl)amino- methane), or with bases such as piperidine or morpholine, are also intended to be encompassed by the term "pharmaceutically acceptable salt".
- the counter anion X may be selected so as to provide pharmaceutically acceptable salts for therapeutic administration.
- the carboxyl-protecting group R 1 is intended to include readily removable ester groups which have been conventionally employed to block a carboxyl group during the reaction steps used to prepare the compounds of the present invention and which can be removed by methods which do not result in any appreciable destruction of the remaining portion of the molecule, e.g. by chemical or enzymatic hydrolysis, treatment with chemical reducing agents under mild conditions, irradiation with ultraviolet light or catalytic hydrogenation.
- protecting groups include benzhydryl, p-nitrobenzyl, 2- naphthylmethyl, allyl, benzyl, p-methoxybenzyl, trichloroethyl, silyl such as trimethylsilyl, phenacyl, acetonyl, o-nitrobenzyl, 4-pyridylmethyl and C j -Cg alkyl such as methyl, ethyl or t-butyl. Included within such protecting groups are those which are hydrolyzed under physiological conditions such as pivaloyloxymethyl, acetoxymethyl, phthalidyl,
- hydroxyl-protecting groups may be employed, for example, to increase the solubility of a cephem derivative.
- ester "prodrugs" of this type are compounds wherein one or more hydroxy substituent groups are converted to sulfate (-OS0 3 H) or phosphate (-OP0 3 H 2 ) groups.
- the compounds of the present invention can be made by conventional methods.
- a suitable procedure is summarized by the following reaction scheme:
- Acid intermediate VI is then coupled with a suitable cephem intermediate having a 3-substituent leaving group.
- the leaving group may be acetoxy or halogen.
- the cephem intermediate is the 3- chloromethyl cephem V, but other suitable cephem intermediates with equivalent leaving groups at the 3-position could also be employed.
- the cephem intermediate V may be acylated with VI or a reactive derivative thereof by conventional acylation procedures well-known in the cephalosporin art to give N-acylated intermediate IV.
- the free arylthioacetic acid e.g.
- acylating agent VI may also be employed in the form of equivalent acylating derivatives such as an acid anhydride, mixed anhydride, activated ester, or acid halide.
- the cephem intermediate preferably has the carboxyl group protected by a conventional carboxyl- protecting group which can be readily removed. Examples of such protecting groups are discussed above and include benzyl, 4-nitrobenzyl, 1,1 dimethylethyl, 4-methoxybenzyl, diphenylmethyl, allyl, and the like. Other examples of suitable protecting groups are disclosed in Protective Groups in Organic Synthesis, 2nd Ed., Theodora W.
- intermediate V may be acylated with acid VI in the presence of dicyclohexylcarbodiimide and in an inert solvent such as tetrahydrofuran or dichloromethane.
- the reaction temperature is typically between -20 °C and 50 °C.
- insoluble material is removed by filtration, the filtrate is concentrated, and the residue is treated with a relatively non-polar solvent such as diethyl ether or ethyl acetate resulting in precipitation of the desired product.
- acid VI may be
- Cephem IV is typically isolated after aqueous work-up and evaporation of
- cephem acid IV is obtained upon treatment of IV with trifluoroacetic acid neat or in an inert solvent such as methylene chloride.
- a reagent such as anisole may also be employed to scavenge the liberated ester protecting group.
- the reaction temperature is usually at or below room temperature.
- the deprotection may also be carried out by treatment with other protic acids such as hydrochloric acid in a solvent such as methanol.
- the final product is typically isolated by precipitation or crystallization.
- the primary amine may be in the form of a zwitterion in examples where there is a free acid group present in the molecule.
- a base such as sodium hydroxide, sodium bicarbonate, or pyridine is added to form the free amine in situ.
- the product may be isolated as its sodium salt by evaporation of volatile solvents, followed by trituration with a solvent such as diethyl ether or ethyl acetate.
- reaction mixture may be acidified and extracted with an organic solvent to afford the product as the free carboxylic acid.
- carboxylate group is protected as an ester
- the amine may be free or present as an acid salt.
- a base such as sodium hydroxide, sodium bicarbonate, or pyridine is added to form the free amine in situ.
- the product is typically isolated by precipitation or by reversed phase column chromatography following removal of volatile solvents.
- thiopyridone intermediates of formula III may have an amine functional group protected as the t-butyloxycarbamate. Suitable protecting groups and methods for their removal are illustrated, for example, in Protective Groups in Organic Synthesis, 2nd Ed., Theodora W. Greene (John Wiley & Sons, 1991). It is intended that such "protected" intermediates and end- products are included within the scope of the present disclosure and claims.
- the desired end-product of formula I may be recovered either as the zwitterion or in the form of a pharmaceutically acceptable acid addition salt, e.g. by addition of the appropriate acid such as HC1, HI or methanesulfonic acid to the zwitterion.
- R! is hydrogen or an anionic charge, or a pharmaceutically acceptable salt thereof, may be converted by conventional procedures to a corresponding
- Rl is hydrogen, an anionic charge or a physiologically hydrolyzable carboxyl-protecting group, or the
- pharmaceutically acceptable salts or prodrugs thereof are potent antibiotics active against many gram-positive bacteria. While they may be used, for example, as animal feed additives for promotion of growth, as
- preservatives for food as bactericides in industrial applications, for example in water-based paint and in the white water of paper mills to inhibit the growth of harmful bacteria, and as disinfectants for destroying or inhibiting the growth of harmful bacteria on medical and dental equipment, they are especially useful in the treatment of infectious disease in humans and other animals caused by the gram-positive bacteria sensitive to the new derivatives. Because of their excellent activity against MRSA organisms, they are particularly useful in the treatment of infections resulting from such bacteria.
- compositions comprising, in
- the compounds may be administered by a variety of means, for example, orally, topically or parenterally (intravenous or
- compositions may be in
- compositions for injection the compositions for injection, the
- preferred route of delivery may be prepared in unit dose form in ampules
- compositions may be in ready-to- use form or in powder form for reconstitution at the time of delivery
- a suitable vehicle such as sterile water.
- the dosage to be administered depends, to a large extent, on the particular compound being used, the particular composition formulated, the route of administration, the nature and condition of the host and the particular situs and organism being treated. Selection of the particular preferred dosage and route of application, then, is left to the discretion of the physician or veterinarian. In general, however, the compounds may be administered parenterally or orally to mammalian hosts in an amount of from about 50 mg/day to about 20 g/day. Administration is generally carried out in divided doses, e.g., three to four times a day, analogous to dosing with a cephalosporin such as cefotaxime.
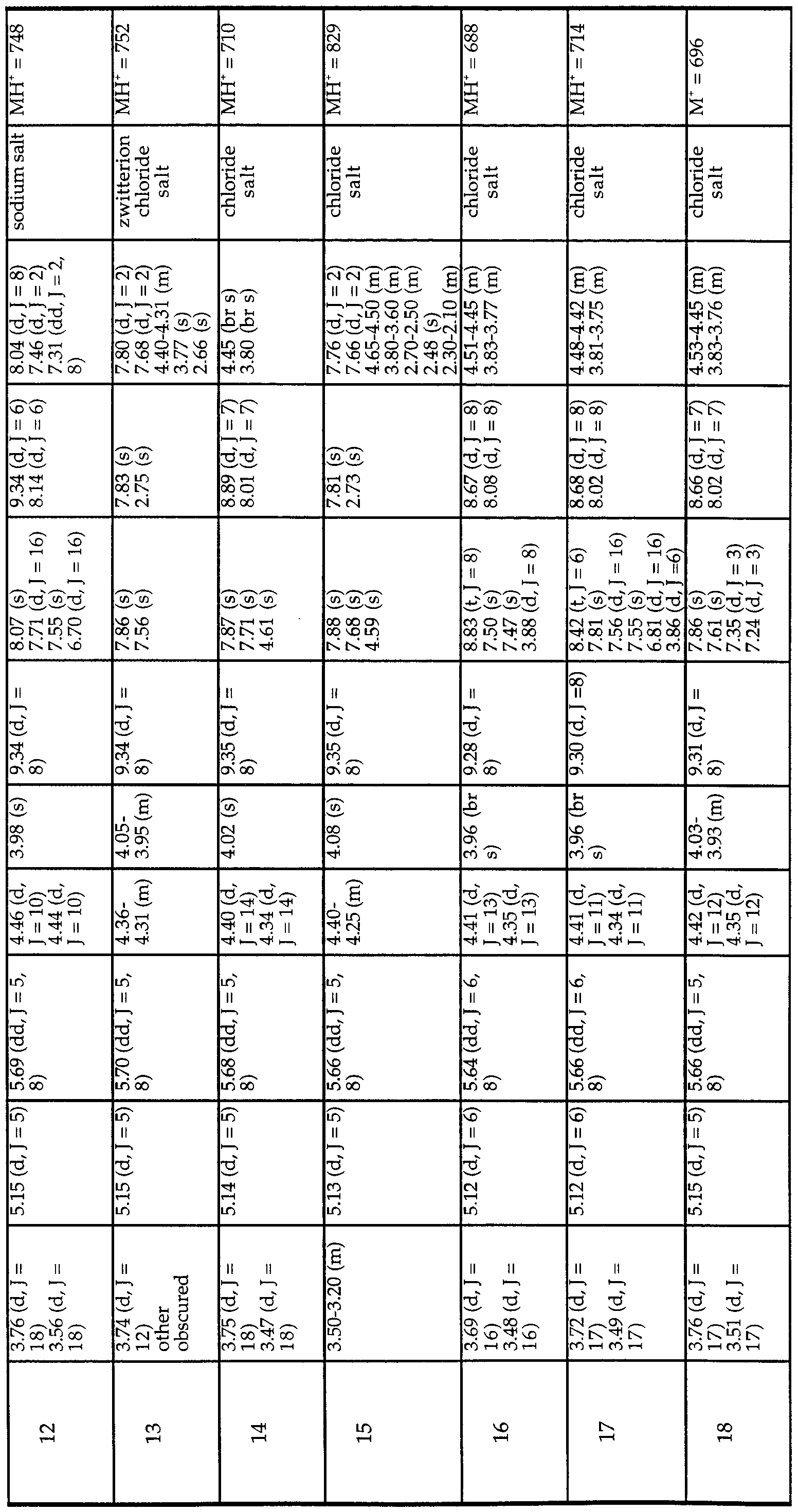
- MIC Minimum Inhibitory Concentrations
- NCLS Mueller-Hinton medium
- inoculate contained approximately 5 x 10 ⁇ cfu/ml and the plates were
- the MIC was defined as the lowest drug concentration that prevented visible growth.
- Organisms The test organism, MRSA strain A27223 used to generate systemic infection in mice, is grown on two large Brain Heart Infusion Agar plates. On each plate, 0.5 ml of frozen stock culture is plated out.
- mice are infected
- mice are used as controls.
- Mice Mice used are male ICR mice. The average weight of the animals is
- Drug preparation and treatment Compounds are tested at 4 dose levels, (25, 6.25, 1.56, and 0.39 mg/kg) and prepared in 5% cremophor, unless otherwise specified. Vancomycin is used as the control compound, and is dosed at 6.25, 1.56, 0.39, and 0.098 mg/kg. It is prepared in 0.1M phosphate buffer. There are five infected mice per dose level, and they are treated with 0.2 ml of the test compound, preferably by intramuscular injection. Treatment begins 15 minutes and 2 hours post-infection.
- Test duration A PD50 (the dose of drug given which protects 50% of
- mice from mortality experiment runs for 5 days. During this time, mortality of mice are checked every day and deaths are recorded. The cumulative mortality at each dose level is used to calculate a PD50 value
- mice are sacrificed at the end of day 5 by
- Ph 3 P triphenylphosphine
- TMS tetramethylsilane
- Analytical thin-layer chromatography was carried out on precoated silica gel plates (60F-254) and visualized using UV light, iodine vapors, and/or staining by heating with methanolic phosphomolybdic acid.
- Column chromatography also referred to as flash chromatography, was performed in a glass column using finely divided silica gel at pressures somewhat above atmospheric pressure with the indicated solvents. Reversed-phase analytical thin-layer chromatography was
- Reversed-phase column chromatography was performed in a glass column using Baker Octadecyl (Cis), 40 mm.
- N-Methylmorpholine (3.90 mL, 0.036 mol) is added, and then the mixture is cooled to 0°C.
- Ester 7 (1.03 g., 3.35 mmol) is dissolved in 200 mL CH2CI2 and 100
- ester 10 as a pale yellow oil (2.56 g., 9.10 mmol; 70% yield) which solidifies overnight in the refrigerator to
- ester 10 (2.5 g., 11.3 mmol) is converted to diester 11 (3.20 g., 9.12 mmol; 81% yield). Diester 11 was obtained as a white solid by chromatography on silica gel using 80% methylene chloride /hexane.
- Dicyclohexylcarbodiimide (0.866 g., 4.20 mmol) is added, followed by tert- butyl glycine (0.550 g., 4.20 mmol), and the mixture stirred at room temperature for 2 hours.
- Ether is added to the flask, and the solids removed by filtration. The filtrate is evaporated to yield 1.96 g. of crude material. Chromatography on silica using 80% CH2CI2 /hexane, followed
- 2,4,5-Trichlorothiophenol (35.0 g., 0.166 mol) is dissolved in 500 mL methylene chloride and cooled to 0°C.
- Triethylamine (22.0 g., 0.217 mol) is added, followed by a solution tert-butyl bromoacetate (35.1 g., 0.180 mol)
- Ester 15 (51 g., 0.156 mol; 94%) as obtained is of suitable purity for subsequent reactions.
- Ester 15 (50.0 g., 0.153 mol) is dissolved in 500 mL chloroform and cooled to 0°C.
- m-Chloroperoxybenzoic acid 50-60% from Aldrich, 48.0 g., 0.140-0.168 mol
- the ice bath is removed and stirring continued for 2.5 hours at room temperature.
- the solids were removed by filtration, and the filtrate washed with dilute aqueous NaHS ⁇ 3, 5% aqueous Na2C ⁇ 3, saturated aqueous NaHC ⁇ 3, and
- sulfoxide 16 (35.0 g., 105 mmol) is converted to diester 17 (32.5 g., 78.7 mmol; 75% yield).
- diester 17 (32.5 g., 78.7 mmol; 75% yield).
- the compound is isolated as a white solid after chromatography on silica using methylene chloride then 3% methanol /methylene chloride.
- Diester 17 (28.0 g., 67.8 mmol) is dissolved in 500 mL acetone. Sodium iodide (48.7 g., 325 mmol) is added, followed by trifluoroacetic anhydride (40.0 g., 191 mmol) over 5 minutes. After stirring at room temperature for 1 hour, the solvents are evaporated. Methylene chloride is added and evaporated twice. The residue is taken up in methylene chloride and washed with aqueous NaHS ⁇ 3 solution (3X), water, and
- Ester 15 (7.00 g., 21.4 mmol) is dissolved in 40 mL chloroform and treated with m-chloroperoxybenzoic acid (-60% from Aldrich, 12.0 g., -42 mmol). After stirring for 1 hour at room temperature, the solids are removed by filtration, and the filtrate is washed with dilute aqueous NaHS ⁇ 3, 5% aqueous Na2C ⁇ 3, saturated aqueous NaHC ⁇ 3, and then
- 2,5-Dichlorophenol (20.4 g., 0.125 mol) is placed in a 1L round bottom flask equipped with a large egg-shaped stir bar and is dissolved in
- reaction is diluted with CH2CI2 (-200 ml) and filtered through
- Compound 26 is obtained as a yellow crystalline solid (13.0 g., 36.0 mmol; 33% yield from 2,5-dichlorophenol).
- the solution is heated to reflux for 6 hours.
- the mixture is diluted with
- stannane 31 (1.70 g., 3.60 mmol) and aryl iodide 28 (1.03 g., 2.73 mmol) are converted to diester 32 (0.920 g., 2.13 mmol; 78% yield as a light yellow solid).
- 2,4,5-Trichloroiodobenzene (25 g., 81.3 mmol) is dissolved in 80 mL DMF.
- tert-Butyl acrylate (48 mL, 328 mmol), tributylamine (58 mL, 243 mmol), triphenylphosphine (4.08 g., 15.5 mmol), and palladium acetate (3.23 g., 14.4 mmol) are added, and the mixture heated to 80°C for three hours.
- the solvents are evaporated and the residue is partitioned with EtOAc and water.
- the aqueous phase is extracted with EtOAc, and the combined organic phase is washed with brine, dried (MgS ⁇ 4), and
- the sodium salt of methyl mercaptoacetate is best made fresh before use. Approximately 30 mL of methyl mercaptoacetate is dissolved in -250 mL THF. One equivalent of 5 N NaOH is added slowly in pipetfulls, and the mixture allowed to stir for 5 minutes. The solvents are removed in vacuo (including water) and the sticky solid is co-evaporated with ether (-200 mLs) and then dry THF (2 x 200 mLs). The solid is
- Diester 3 (4.40 g., 0.012 mol) is dissolved in 30 mL THF. To this
- Acid 1 is obtained as a tan solid (3.80
- Cephem amine V (15.04 g., 0.035 mmol) is suspended under a nitrogen atmosphere in 65 mL THF. A solution of DCC in methylene chloride (1M, 36.2 mL, 0.036 mmol) is added, and the mixture allowed to stir for 5 minutes. Acid 1 (13.15 g., 0.035 mmol) is added and the mixture is stirred for 1.5 hours. Ether (-30 mL) is added, and the solids (mostly DCU) are filtered off. The red-colored filtrate is evaporated to -25-30 mL and ether and pentane are added to precipitate the cephem product. The solid cephem is collected, washed with ether, and dried under vacuum to afford diester IV" (14.2 g., 0.019 mmol; 54% yield).
- Diester IV (0.760 g., 1.00 mmol) is dissolved in 4 mL CH2CI2 and
- Diacid IV is obtained (0.420 g., 0.780 mmol; 78% yield) as a light yellow solid.
- Diacid IV (0.780 g., 1.45 mmol) is dissolved in 3 mL methanol and 8 mL CH2O2.
- Thiopyridone III' (0.395 g., 1.45 mmol) is added, and the mixture is
- Cephem IA' (0.605 g., 0.747 mmol) is suspended in 3 mL CH2C12,
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Cephalosporin Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description





































































Claims






































Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP97950724A EP0966472A1 (en) | 1996-11-27 | 1997-11-21 | Cephalosporin derivatives |
| IL12943797A IL129437A0 (en) | 1996-11-27 | 1997-11-21 | Cephalosporin derivatives |
| NZ335892A NZ335892A (en) | 1996-11-27 | 1997-11-21 | Cephalosporin derivatives for treating bacterial infections |
| BR9713350-7A BR9713350A (en) | 1996-11-27 | 1997-11-21 | Cephalosporin derivatives |
| CZ991853A CZ185399A3 (en) | 1996-11-27 | 1997-11-21 | Cephalosporin derivative and pharmaceutical composition containing thereof |
| CA002272717A CA2272717A1 (en) | 1996-11-27 | 1997-11-21 | Cephalosporin derivatives |
| JP52485698A JP2001506607A (en) | 1996-11-27 | 1997-11-21 | Cephalosporin derivative |
| AU53652/98A AU734948B2 (en) | 1996-11-27 | 1997-11-21 | Cephalosporin derivatives |
| NO992526A NO992526D0 (en) | 1996-11-27 | 1999-05-26 | Cephalosporin derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US3404696P | 1996-11-27 | 1996-11-27 | |
| US60/034,046 | 1996-11-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998023621A1 true WO1998023621A1 (en) | 1998-06-04 |
Family
ID=21873971
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1997/021785 WO1998023621A1 (en) | 1996-11-27 | 1997-11-21 | Cephalosporin derivatives |
Country Status (17)
| Country | Link |
|---|---|
| EP (1) | EP0966472A1 (en) |
| JP (1) | JP2001506607A (en) |
| KR (1) | KR20000057283A (en) |
| CN (1) | CN1238775A (en) |
| AR (1) | AR010650A1 (en) |
| AU (1) | AU734948B2 (en) |
| BR (1) | BR9713350A (en) |
| CA (1) | CA2272717A1 (en) |
| CZ (1) | CZ185399A3 (en) |
| HU (1) | HUP9903621A3 (en) |
| IL (1) | IL129437A0 (en) |
| NO (1) | NO992526D0 (en) |
| NZ (1) | NZ335892A (en) |
| PL (1) | PL334558A1 (en) |
| TW (1) | TW414796B (en) |
| WO (1) | WO1998023621A1 (en) |
| ZA (1) | ZA9710662B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6265394B1 (en) * | 1997-07-31 | 2001-07-24 | Bristol-Myers Squibb Company | Bis quaternary MRSA cephem derivatives |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20020085180A (en) * | 2001-05-07 | 2002-11-16 | 주식회사 엘지생명과학 | Novel cephalosporin antibiotics and process for preparing same |
| KR20020085176A (en) * | 2001-05-07 | 2002-11-16 | 주식회사 엘지생명과학 | Novel cephalosporin antibiotics and process for preparing same |
| KR20020085181A (en) * | 2001-05-07 | 2002-11-16 | 주식회사 엘지생명과학 | Novel cephalosporin antibiotics and process for preparing same |
| KR20020085178A (en) * | 2001-05-07 | 2002-11-16 | 주식회사 엘지생명과학 | Novel cephalosporin antibiotics and process for preparing same |
| KR20030071311A (en) * | 2002-02-28 | 2003-09-03 | 주식회사 엘지생명과학 | Novel cephalosporin compounds and process for preparing the same |
| US8895587B2 (en) * | 2011-05-18 | 2014-11-25 | Syngenta Participations Ag | Insecticidal compounds based on arylthioacetamide derivatives |
| CN105131018B (en) * | 2015-09-23 | 2017-12-26 | 浙江华方药业股份有限公司 | A kind of preparation method of ceftezole acid |
| CN114957056A (en) * | 2021-02-20 | 2022-08-30 | 帕潘纳(北京)科技有限公司 | Process for preparing methyl 3-methyl-2-chloro-4-methylsulfonylbenzoate and intermediates thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5620969A (en) * | 1995-04-25 | 1997-04-15 | Bristol-Myers Squibb Company | Cephalosporin derviatives |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3345366A (en) * | 1967-02-15 | 1967-10-03 | American Cyanamid Co | Substituted 7-acetylamino cephalosporanic acids |
| US4148997A (en) * | 1977-11-07 | 1979-04-10 | Yeda Research And Development Co., Ltd. | 7-[Sulfomethyl)phenyl]acetamidocephalosporin derivatives |
| US5567698A (en) * | 1995-02-15 | 1996-10-22 | Bristol-Myers Squibb Company | Pyridinium thiomethyl substituted chepholosporin derivatives |
| AU1278197A (en) * | 1995-12-20 | 1997-07-14 | Bristol-Myers Squibb Company | Cephalosporin derivatives |
-
1997
- 1997-11-19 TW TW086117285A patent/TW414796B/en not_active IP Right Cessation
- 1997-11-21 HU HU9903621A patent/HUP9903621A3/en unknown
- 1997-11-21 CN CN97199930A patent/CN1238775A/en active Pending
- 1997-11-21 KR KR1019990704689A patent/KR20000057283A/en not_active Application Discontinuation
- 1997-11-21 WO PCT/US1997/021785 patent/WO1998023621A1/en not_active Application Discontinuation
- 1997-11-21 BR BR9713350-7A patent/BR9713350A/en unknown
- 1997-11-21 CA CA002272717A patent/CA2272717A1/en not_active Abandoned
- 1997-11-21 EP EP97950724A patent/EP0966472A1/en not_active Ceased
- 1997-11-21 IL IL12943797A patent/IL129437A0/en unknown
- 1997-11-21 AU AU53652/98A patent/AU734948B2/en not_active Ceased
- 1997-11-21 PL PL97334558A patent/PL334558A1/en unknown
- 1997-11-21 JP JP52485698A patent/JP2001506607A/en active Pending
- 1997-11-21 CZ CZ991853A patent/CZ185399A3/en unknown
- 1997-11-21 NZ NZ335892A patent/NZ335892A/en unknown
- 1997-11-26 AR ARP970105544A patent/AR010650A1/en unknown
- 1997-11-26 ZA ZA9710662A patent/ZA9710662B/en unknown
-
1999
- 1999-05-26 NO NO992526A patent/NO992526D0/en not_active Application Discontinuation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5620969A (en) * | 1995-04-25 | 1997-04-15 | Bristol-Myers Squibb Company | Cephalosporin derviatives |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6265394B1 (en) * | 1997-07-31 | 2001-07-24 | Bristol-Myers Squibb Company | Bis quaternary MRSA cephem derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2001506607A (en) | 2001-05-22 |
| TW414796B (en) | 2000-12-11 |
| PL334558A1 (en) | 2000-03-13 |
| KR20000057283A (en) | 2000-09-15 |
| HUP9903621A3 (en) | 2001-05-28 |
| AR010650A1 (en) | 2000-06-28 |
| NZ335892A (en) | 2001-07-27 |
| ZA9710662B (en) | 1999-05-26 |
| CA2272717A1 (en) | 1998-06-04 |
| AU734948B2 (en) | 2001-06-28 |
| BR9713350A (en) | 2000-01-25 |
| EP0966472A1 (en) | 1999-12-29 |
| NO992526L (en) | 1999-05-26 |
| NO992526D0 (en) | 1999-05-26 |
| EP0966472A4 (en) | 1999-12-29 |
| IL129437A0 (en) | 2000-02-17 |
| HUP9903621A2 (en) | 2000-04-28 |
| CZ185399A3 (en) | 1999-08-11 |
| CN1238775A (en) | 1999-12-15 |
| AU5365298A (en) | 1998-06-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5859256A (en) | Cephalosporin antibiotics | |
| US6265394B1 (en) | Bis quaternary MRSA cephem derivatives | |
| KR20000005238A (en) | Cephem compound and drug containing the compound | |
| EP0966472A1 (en) | Cephalosporin derivatives | |
| US5567698A (en) | Pyridinium thiomethyl substituted chepholosporin derivatives | |
| US5559108A (en) | Cephalosporin derivatives | |
| US5620969A (en) | Cephalosporin derviatives | |
| US6025352A (en) | Cephalosporin antibiotics | |
| US6093712A (en) | Cephalosporin derivatives | |
| US5734047A (en) | Cephalosporin derivatives | |
| US20020049191A1 (en) | Cephalosporin derivatives | |
| US6030965A (en) | Cephalosporin antibiotics | |
| WO1997037997A1 (en) | Cephalosporin derivatives | |
| MXPA99004823A (en) | Cephalosporin derivatives | |
| RU2172317C2 (en) | Cephalosporin derivatives, antibacterial composition containing thereof, derivatives of 2-aminothiazoles as intermediates, and method of preparation thereof | |
| WO1999006048A1 (en) | Bis quaternary mrsa cephem derivatives | |
| EP1059293A1 (en) | Novel amino chlorothiazole compounds | |
| NZ500512A (en) | Cephalosporin for treating beta-lactam antibiotic resistant bacteria (such as S. aureus, E. faecium or E. faecalis) or PBP2a-producing bacteria |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 97199930.9 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH HU IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH KE LS MW SD SZ UG ZW AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1997950724 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 335892 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1999-1853 Country of ref document: CZ Ref document number: PA/a/1999/004823 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2272717 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 53652/98 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 1998 524856 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019997004689 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1999-1853 Country of ref document: CZ |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997950724 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019997004689 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 53652/98 Country of ref document: AU |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1997950724 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1997950724 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1019997004689 Country of ref document: KR |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV1999-1853 Country of ref document: CZ |