"ASSAYS FOR KDR/FLK- 1 RECEPTOR TYROSINE KINASE INHIBITORS"
1. FIELD OF THE INVENTION
The present invention relates to methods for the identification and use of compounds capable of selectively and potently inhibiting the signal transduction of a specific receptor tyrosine kinase, and more particularly to compounds selectively inhibiting the enzymatic function of a VEGF receptor, e . g. , the KDR/FLK-1 receptor, and the compounds so identified.
2. BACKGROUND OF THE INVENTION
Receptor Tyrosine Kinases . Receptor tyrosine kinases (RTKs) comprise a large family of transmembrane receptors for polypeptide growth factors with diverse biological activities. The intrinsic function of RTKs is activated upon ligand binding, which results in phosphorylation of the receptor and multiple cellular substrates, and subsequently in a variety of cellular responses. Ullrich and Schlessinger, 1990, Cell 51:203-212.
As has been reported, RTKs, as well as, more generally, protein tyrosine kinases, play an important role in the control of cell growth and differentiation (for review, see, Schlessinger and Ullrich, 1992, Neuron 5:383-391) . This is reflected in the observation that aberrant expression or mutations in members of the RTK family lead to either uncontrolled cell proliferation { e. g. , malignant tumor growth) or to defects in key developmental processes.
Consequently, the biomedical community has expended significant resources to discover the specific
biological role of members of the RTK family, their function in differentiation processes, including their involvement in tumorigenesis and in other diseases, the biochemical mechanisms underlying their signal transduction pathways activated upon ligand stimulation and the development of novel antineoplastic drugs.
At present, at least nineteen (19) distinct RTK subfamilies have been identified. One RTK subfamily is believed to be comprised of the KDR/FLK-1 receptor, the fetal liver kinase 4 (FLK-4) receptor and the fms- like tyrosine 1 (flt-1) receptor. Each of these receptors was initially believed to be receptors for hematopoietic growth factors. The KDR/FLK-1 Receptor and VEGF. Normal vasculogenesis and angiogenesis play important roles in a variety of physiological processes such as embryonic development, wound healing, organ regeneration and female reproductive processes such as follicle development in the corpus luteum during ovulation and placental growth after pregnancy. Folkman and Shing, 1992, J. Biological Chem. 267:10931-34. However, many diseases are driven by persistent unregulated or inappropriate angiogenesis. For example, in arthritis, new capillary blood vessels invade the joint and destroy the cartilage. In diabetes, new capillaries in the retina invade the vitreous, bleed and cause blindness. Folkman, 1987, in: Congress of Thrombosis and Haemostasis (Verstraete, et. al , eds.), Leuven University Press, Leuven, pp.583-596. Ocular neovascularization is the most common cause of blindness and dominates approximately twenty (20) eye diseases.
Moreover, vasculogenesis and/or angiogenesis have been associated with the growth of malignant solid tumors and metastasis. A tumor must continuously
stimulate the growth of new capillary blood vessels for the tumor itself to grow. Furthermore, the new blood vessels embedded in a tumor provide a gateway for tumor cells to enter the circulation and to metastasize to distant sites in the body. Folkman, 1990, J. Na tl . Cancer Inst . 82 : 4 - 6 ; Klagsbrunn and Soker, 1993, Current Biology 3:699-702; Folkman, 1991, J. Natl . , Cancer Ins t . 92:4-6; Weidner et al . , 1991, New Engl . J. Med. 324 : 1 - 5 . Several polypeptides with in vi tro endothelial cell growth promoting activity have been identified. Examples include acidic and basic fibroblastic growth factor (aFGF, bFGF) , vascular endothelial growth factor (VEGF) and placental growth factor. Unlike aFGF and bFGF, VEGF has recently been reported to be an endothelial cell specific mitogen. Ferrara and Henzel, 1989, Biochem. Biophys . Res . Comm . 261:851- 858; Vaisman et al . , 1990, J". Biol . Chem . 265: 19461- 19566. Thus, the identification of the specific receptors to which VEGF binds is an important advancement in the understanding of the regulation of endothelial cell proliferation. Two structurally closely related RTKs have been identified to bind VEGF with high affinity: the flt-1 receptor (Shibuya et al . , 1990, Oncogene 5:519-524; De Vries et al . , 1992, Science 255:989-991) and the KDR/FLK-1 receptor, discussed in the U.S. Patent Application No. 08/193,829. Consequently, it had been surmised that these RTKs may have a role in the modulation and regulation of endothelial cell proliferation.
Evidence, such as the disclosure set forth in copending U.S. Application Serial No. 08/193,829, strongly suggests that VEGF is not only responsible for endothelial cell proliferation, but also is a prime regulator of normal
and pathological angiogenesis . See generally, Klagsburn and Soker, 1993, Current Biology 3:699-702; Houck et al . , 1992, J. Biol . Chem. 267: 26031-26037. Moreover, it has been shown that KDR/FLK-1 and flt-1 are abundantly expressed in the proliferating endothelial cells of a growing tumor, but not in the surrounding quiescent endothelial cells. Plate et al . , 1992, Nature 359:845-848; Shweiki et al . , 1992, Nature 359:843-845. Identification Of Agonists And Antagonists To The KDR/FLK-1 Receptor. In view of the deduced importance of RTKs in the control, regulation and modulation of endothelial cell proliferation and potentially vasculogenesis and/or angiogenesis, many attempts have been made to identify RTK "inhibitors" using a variety of approaches. These include the use of mutant ligands (U.S. Patent No. 4,966,849) ; soluble receptors and antibodies (Application No. WO 94/10202; Kendall and Thomas, 1994, Proc . Natl . Acad . Sci . USA 90 : 10705 - 10709; Kim et al . , 1993, Nature 362:841-844) ; and R A ligands (Jellinek et al . , 1994, Biochemistry 33:10450- 10456) . However, so far, none of these efforts have resulted in the identification or isolation of inhibitors useful for therapeutic applications. Furthermore, tyrosine kinase inhibitors (WO
94/03427; WO 92/21660; WO 91/15495; WO 94/14808; U.S. Patent No. 5,330,992; Mariani et al . , 1994, Proc . Am . Assoc . Cancer Res . 35:2268), and inhibitors acting on receptor tyrosine kinase signal transduction pathways, such as protein kinase C inhibitors have been identified (Schuchter et al . , 1991, Cancer Res . 52:682-687) ; Takano et al . , 1993, Mol . Bio . Cell 4:358A; Kinsella et al . , 1992, Exp. Cell Res . 199 : 56 - 62; Wright et al . , 1992, J. Cellular Phys . 152:448- 57) .
More recently, attempts have been made to identify small molecules which act as tyrosine kinase inhibitors. For example, bis monocyclic, bicyclic or heterocyclic aryl compounds (PCT WO 92/20642) , vinylene-azaindole derivatives (PCT WO 94/14808) and 1-cycloproppyl-4-pyridyl-quinolones (U.S. Patent No. 5,330,992) have been described generally as tyrosine kinase inhibitors. Styryl compounds (U.S. Patent No. 5,217,999) , styryl-substituted pyridyl compounds (U.S. Patent No. 5,302,606) , certain quinazoline derivatives (EP Application No. 0 566 266 Al) , seleoindoles and selenides (PCT WO 94/03427) , tricyclic polyhydroxylic compounds (PCT WO 92/21660) and benzylphosphonic acid compounds (PCT WO 91/15495) have been described as compounds for use as tyrosine kinase inhibitors for use in the treatment of cancer. None of these compounds, however, have been demonstrated to selectively act on the enzymatic function of a specific receptor tyrosine kinase, such as the KDR/FLK-1 receptor. Indeed, because in a substantial percentage of tumors, overexpression of various RTKs had been shown to dictate the uncontrolled proliferation of the tumor cells these compounds have been identified with the objective to inhibit receptor tyrosine kinase function in general. Likewise, these compounds have not been associated with the selective inhibition of specific receptor tyrosine kinases, or the inhibition of vasculogenesis and/or angiogenesis. Consequently, there is an unmet need for the identification and generation of effective small compounds which selectively inhibit the signal transduction of the KDR/FLK-1 receptor in order to effectively and specifically suppress vasculogenesis.
3. SUMMARY OF THE INVENTION
The present invention, for the first time, provides for a process to systematically and rationally identify compounds targeting specific biomolecules in order to affect a particular physiological process. More particularly, the present invention relates to a process for producing a compound that inhibits vasculogenesis and/or angiogenesis comprising screening a plurality of test compounds to identify a subset of compounds which selectively and potently inhibits the VEGF receptor. The present invention relates to a process for the identification of compounds that are both highly potent and highly selective for inhibiting the activity of a VEGF receptor, e. g. , KDR/FLK-l, in order to inhibit vasculogenesis and/or angiogenesis. More specifically, the invention relates to an assay cascade comprised of several "filter steps" of increasing selectivity, which identify a limited subset of candidate compounds affecting the VEGF receptor on the molecular level . The assays comprising these filter steps are preferrably high- throughput type assays.
A "high-throughput" cellular assay is employed as the first filter step to identify subset candidate compounds inhibiting VEGF-induced tyrosine phosphorylation of KDR/FLK-l in cultured cells. Controls are designed to determine the candidate compounds' selectivity for inhibition of KDR/FLK-l relative to other receptor tyrosine kinases, for example, the IGF-l-R or the EGF-R. Subset candidate compounds with a IC50 of <50 μM, and more preferably <10 μM, and with a selectivity for KDR/FLK-l of twofold, and more preferably fivefold, relative to control receptors are further pursued. All other
"negative" compounds which do not meet these criteria are discarded.
In the second filter step a bioresponse assay is employed to determine the efficacy of the KDR/FLK-l inhibitors in cultured endothelial cells. In one embodiment of the method of the invention, the human umbilical vein endothelial cell line HUV-EC-C is employed. HUV-EC cells express the KDR/FLK-l receptor, and upon VEGF stimulation, a mitotic response can be measured. Controls are designed to determine the selectivity of inhibition of the compounds against the VEGF-induced bioresponse relative to the bioresponse mediated by other receptor tyrosine kinases expressed in the cultured endothelial cells, for example the receptor for aFGF may be compared. Subset candidate compounds with a efficacy of <50 μM, preferably <10 μM, and with a selectivity of inhibition twofold, preferably threefold relative to a control receptor, are determined to be "positive". Compounds identified as "positive" are saved for further testing; the "negative" compounds are removed.
Optionally, a biochemical assay may be employed as a "filter" step in order to test the subset candidate compounds' effect on the VEGF-induced autophosphorylation of KDR/FLK-l in a cell free system. More specifically, the compounds identified as "positive" in the above cellular assay may be tested in a defined in vi tro assay to confirm that the inhibitory effects are due to direct interaction of the subset candidate compounds with KDR/FLK-l rather than the result of any secondary effects in the cell. The subset candidate compounds identified as "positive" are again saved for the next "filter." All of the candidate compounds which survive as "positives" through the above described filter steps
are then tested for toxicity by determination of the LD10 value and further for efficacy an in vivo model for angiogenesis. Suitable in vivo models are those for metastasis and or diabetic retinopathy. The exemplary model described herein is a subcutaneous tumor xenograph model. The compounds of the invention are those which are determined to inhibit tumor angiogenesis in this model, indicated by inhibition of the tumor growth by at least 30% relative to untreated controls, at a dose smaller than the LD10.
Compounds of this invention include, for example, derivatives of quinazoline, quinoxiline, substituted aniline, indoline, isoxazoles, acrylonitrile and phenyl-acrylonitrile compounds. The compounds of the present invention, however, are not limited to certain chemical classes, as they are defined by the process of identification and their so determined physiological activity.
The present invention is further directed to pharmaceutical compositions comprising a pharmaceutically effective amount of the compounds of the invention and a pharmaceutically acceptable carrier or excipient. Such compositions are believed to specifically inhibit the KDR/FLK-l receptor by, e . g. , inhibiting its catalytic activity, affinity to ATP or ability to interact with a substrate, and thus will be useful in inhibition of diseases related to vasculogenesis and/or angiogenesis, including diabetes, arthritis, and cancer. Finally, the present invention is also directed to methods for treating diseases related to pathological or inappropriate vasculogenesis and/or angiogenesis, including but not limited to diabetes, diabetic retinopathy, rheumatoid arthritis, hemangioma and cancer and more particularly cancer related to solid cell tumor growth ( e . g. , glioblastoma, melanoma
and Kaposi's sarcoma and ovarian, lung, mammary, prostate, pancreatic, colon and epidermoid carcinoma) .
. DEFINITIONS The following terms, in singular and plural, are be intended to have, for the purpose of this invention, the following meaning.
"Pharmaceutically acceptable acid addition salt" refers to those salts which retain the biological effectiveness and properties of the free bases and which are obtained by reaction with inorganic acids such as hydrochloric acid, hydrobromie acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like.
"Selectivity for", "selective inhibition of", or "selectively inhibit (ing) " an VEGF receptor, for example the KDR/FLK-l receptor refers to the preferential inhibitory ability of a compound against a VEGF receptor compared to a receptor that belong to another RTK subfamily, such as for example the EGF-R. "VEGF recptor" refers to a transmembrane protein with tyrosine kinase activity that binds VEGF, including KDR/FLK-l and flt-1.
5. DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to processes for the identification of compounds capable of selectively and potently inhibiting the activity of a VEGF receptor, e . g. KDR/FLK-l, useful in the treatment of diseases related to unregulated or inappropriate angiogenesis and/or vasculogenesis and the compounds so produced. More particularly, the present invention is based, in part, on the recent discovery that the KDR/FLK-l tyrosine kinase receptor is expressed on the surface of endothelial cells and may
play a role in endothelial cell growth, which is one of the crucial steps involved in vasculogenesis and/or angiogenesis. See, copending U.S. Patent Application No. 08/193,829, filed February 9, 1994, incorporated by reference in its entirety herein. The invention is also based on the identification of VEGF as a high affinity ligand of KDR/FLK-l and the characterization of KDR/FLK-l as a regulator of vasculogenesis and/or angiogenesis rather than a hematopoietic receptor. Thus, the surmised role of VEGF in endothelial cell proliferation and migration during angiogenesis and vasculogenesis indicates an important role for the KDR/FLK-l in these processes.
The invention is further based on the observation that an array of severe diseases, such as diabetic retinopathy (Folkman, 1987, in Xlth Congress of Thrombosis and Haemostasis (Verstraeta et al . , eds.) pp. 583-596, Leuven University Press, Leuven) and arthritis, as well as malignant tumor growth involve, as an essential requirement for the disease to develop, uncontrolled or inappropriate angiogenesis. See e . g. , Folkman, 1971, N. Engl . J. Med . 285:1182- 1186. For example, in arthritis, new capillaries invade the joint and destroy the cartilage; in at least twenty (20) ocular diseases, including diabetic retinopathy, new and bleeding capillaries cause blindness .
Moreover, a tumor must continuously stimulate the growth of new blood vessels for the tumor itself to grow. Also metastasis of malignant tumors has been directly associated with vasculogenesis and/or angiogenesis, as new blood vessels embedded in a tumor provide a gateway for tumor cells to enter the circulation in order to manifest at distant sites in the body.
While not wishing to be bound by any particular theory, although it is believed that the compounds of the present invention act on the endothelial cells forming new blood vessels during vasculogensis and/or angiogenesis, the compounds may also act directly on the tumors cells.
For purposes of this application, although the nomenclature of the human and murine counterparts of the generic "FLK-1" receptor differ slightly, they are, in many respects, interchangeable. Models which rely upon the FLK-1 receptor therefore are directly applicable to understanding the KDR receptor. The murine receptor, FLK-1, and its human counterpart, KDR, share a sequence homology of 93.4% within the intracellular domain which is responsible for the enzymatic activity and resulting signal transduction. Likewise, murine FLK-1 binds human VEGF with the same affinity as mouse VEGF, and accordingly, is activated by the ligand derived from either species. Millauer et al . , 1993, Cell 72:835-846; Quinn et al . , 1993, Proc . Na tl . Acad . Sci . USA 90:7533-7537. FLK-1 also associates with and subsequently tyrosine phosphorylates human RTK substrates (e.g., PLC-γ or p85) when coexpressed in 293 cells (human embryonal kidney fibroblasts) .
Use of the murine FLK-1 receptor in methods to identify compounds which regulate the signal transduction pathway are directly applicable to the identification of compounds which may be used to regulate the human signal transduction pathway, and more specifically, activity related to the KDR receptor. Angiogenesis is a very complex process involving the invasion of endothelial cells into the nonvascularized tissue. No in vi tro model exists which mimics exactly this multistep process comprising the degradation of the basal membrane surrounding the
endothelial cells, migration into the perivascular stroma and eventually proliferation and formation of the new vascular sprout. However, in vivo mouse and rat animal models have been demonstrated to be of excellent value for the examination of the clinical potential of agents acting on the KDR/FLK-l induced signal transduction pathway.
In sum, the receptors to which VEGF specifically binds are an important and powerful therapeutical target for the regulation and modulation of vasculogenesis and/or angiogenesis and a variety of severe diseases which involve abnormal cellular growth caused by such processes. Plowman et al . , 1994, DN&P 7:334-339. More particularly, the KDR/FLK-l receptor's high specificity and role in the neovascularization make it a very distinct and powerful target for therapeutic approaches for the treatment of cancer and other diseases which involve the uncontrolled or inappropriate formation of blood vessels.
This invention is therefore directed to the a process designed to systematically and rationally identify compounds which inhibit vasculogenesis and/or angiogenesis by selectively targeting and inhibiting VEGF receptor, e . g . , KDR/FLK-l, on a molecular level. The compounds produced by the process of the invention may inhibit the KDR/FLK-l receptor by, e . g. , inhibiting its catalytic activity, affinity to ATP or ability to interact with a substrate.
5.1. The Assay Cascade For The Identification Of Compounds Selectively Inhibiting The KDR/FLK-l Receptor Signal Transduction
The present invention, for the first time, provides for a process to systematically and rationally identify compounds targeting a VEGF receptor, e . g. , KDR/FLK-l, in order to affect its
physiological functionality, i . e . , vasculogenesis and/or angiogenesis. More specifically, the process of the invention allows one to produce, by way of identification, compounds selectively targeting KDR/FLK-l for the treatment of diseases related to uncontrolled or inappropriate vasculogenis and/or angiogenesis.
More specifically, the invention relates to a cascade of assays including filter steps of increasing stringency for the identification of compounds which selectively and potently inhibit KDR/FLK-l tyrosine kinase signal transduction. Using the process of this invention allows one to produce highly potent inhibitors which are highly specific for KDR/FLK-l as molecular target. Acting on a specifically defined molecular target involved in the regulation of vasculogenesis and/or angiogenesis, the compounds and their pharmaceutical compositions containing them should have superior therapeutic value while having fewer undefined side effects than drugs identified by traditional means and screening methods.
The steps involved in cascades of assays are designed as "filter steps" of increasing stringency. For example, the first filter steps are cellular and/or biochemical high-throughput screening assays, respectively, allowing the identification of subset candidate compounds selectively targeting and potently inhibiting KDR/FLK-l on the molecular level. The next filter step tests the subset candidate compounds' efficacy and selectivity of inhibition against a VEGF- induced bioresponse in cultured endothelial cells. Finally, a last filter step, the compounds are tested in an in vivo model of angiogenesis to select compounds that are both efficacious and relatively non-toxic as well as having an acceptable pharmacological profile. Models for diabetic
retinopathy or metastasis, both of which are well known in the art, would be appropriate. The present disclosure describes the use of a mouse subcutaneous xenograft model designed to identify compounds inhibiting tumor vasculogenesis and/or angiogenesis.
5.1.1. High Throughput Cellular Assay For The Identification Of KDR/FLK-l Inhibitors
A first "filter step" of the assay cascade, is designed to allow for high-throughput testing of compounds from any source to identify molecules having an inhibitory effect on KDR/FLK-l autophosphorylation.
For example, a cellular assays may be developed to screen for subset candidate compounds which inhibit KDR/FLK-l autophosphorylation, for example using cultured cells expressing high levels of KDR/FLK-l cDNA.
In a specific embodiment of the invention, NIH 3T3/FLK-1 cells are seeded in 96 well cell culture plates, grown to suitable density and then incubated with a test candidate compound. Subsequently, the cells are stimulated with VEGF as previously described (see, copending U.S. Patent Application 08/485,323, filed June 7, 1995) , and lysed. The level of tyrosine phosphorylation is determined via Enzyme Linked Immunoabsorbent Assays (ELISA) and compared with controls not treated with candidate compounds, and controls not stimulated with VEGF, respectively.
The ELISA for detection and measure of the degree of tyrosine kinase activity may generally be conducted according to protocols known in the art, which are described in, for example, Voller et al . , 1980, "Enzyme-Linked Immunosorbent Assay," In: Manual of Clinical Immunology, 2d ed. , edited by Rose and Friedman, pp 359-371 (Am. Soc. Of Microbiology, Washington, D.C.) and in the co-pending U.S. Patent
Application No. 08/279,321, filed July 22, 1994, incorporated herein by reference in its entirety.
For example, the ELISA plates may be coated with a suitable antibody against KDR/FLK-l, such as L4 or E38, see, Section 6.7. Lysates of stimulated and non- stimulated cells are added and incubated under suitable conditions. After washing, the ELISA plates are incubated with anti-phosphotyrosine antibody, linked to a detection entity, e . g. , biotin. In the specific embodiments, the biotinylated anti- phosphotyrosine antibody 4G10 (UBI, Catalog No. 16- 103) is used. The plates are washed, the assay developed, and the phosphotyrosine levels of the different wells determined on an ELISA reader, for example a Dynatech MR5000.
Alternatively, the order of the antibodies may be reversed: Anti-phosphotyrosine antibody may be used as a first antibody for coating the ELISA plate, and antibodies against KDR/FLK-l, linked to a detection entity, for example biotin, may be used as the second antibody.
The first assay round serves to quickly eliminate inactive compounds reducing the time and cost of the process. Those subset candidate compounds which inhibit the tyrosine phosphorylation of KDR/FLK-l by 50% compared to a control are identified and retested using the same assay, however, in the second round the cells are incubated with various concentrations of the subset candidate compounds to determine the IC50 value . In order to identify compounds selectively inhibiting KDR/FLK-l, parallel assays may be performed to determine the subset candidate compounds' effect on the ligand-induced autophosphorylation of NIH 3T3 cells transfected with a control receptor tyrosine kinase, for example the EGF-R or the IGF-R.
Subset candidate compounds with an IC50 <50 μM, preferably <10 μM, and with a twofold, preferably fivefold selectivity for inhibition of KDR/FLK-l relative to the control receptor are determined "positive" and further pursued. Test compounds which do not meet this standard are removed.
5.1.2. Bioresponse Assay For The
Determination Of The Inhibitor's Efficacy In A Biological System
As a second "filter" step, a bioresponse assay is employed to determine the efficacy of the KDR/FLK-l inhibitors in a more physiologically relevant manner, herein the effect on cultured endothelial cells. A number of endothelial cell lines have been described in the literature which may be employed for this assay. For example, bovine aortic endothelial (BAE) cells and human umbilical vein endothelial (HUV-EC) cells have been shown to express the KDR/FLK-l receptor as well as a suitable control receptor, the receptor for aFGF. If such cultured endothelial cells are starved by growth factor depletion, a mitogenic effect may be induced and measured upon stimulation with VEGF. A similar mitotic response may be induced by aFGF.
In a preferred embodiment, HUV-EC cells are seeded in tissue culture flasks and grown to a suitable density. After starving, i.e., depletion of growth factors, the cells are incubated with different concentrations of subset candidate compounds. Subsequently, the cells are stimulated with VEGF or aFGF, respectively, and the induced mitogenic effect is measured by determination of, for example, 3H- thymidine incorporation into the DNA. The subset candidate compounds' selectivity for inhibition of the VEGF-induced bioresponse is determined by comparison
with the subset candidate compounds' effect on the AFGF-induced bioresponse.
Subset candidate compounds with an IC50 of <50 μM, preferably <10 μM, and with a selectivity of 5 inhibition against KDR/FLK-l of twofold, preferably threefold relative to the control, are determined to be "positive" and are further pursued. The "negative" test compounds removed.
10 5.1.3. Biochemical Assay For The
Determination Of The Specificity Of Inhibition The Subset Candidate Compound For KDR/FLK-l Receptor On The Molecular Level
As an additional "filter", a defined in vi tro
._ assay may be employed to confirm that the inhibitory effects of the subset candidate compounds determined in the first filter cellular assay described in Section 5.1.1. are due to direct interaction of the subset candidate compound with KDR/FLK-l rather than
__ the result of secondary effects occurring in the cells. Specifically, a biochemical assay may be employed to test the subset candidate compounds' effect on the VEGF-induced autophosphorylation of isolated KDR/FLK-l in vi tro . The biochemical assay is
__ an optional step in the assay cascade of this invention; it is simply designed to confirm that KDR/FLK-l is in fact the direct molecular target of the inhibitor.
For example, NIH 3T3/FLK-1 cells, or any other
-0 cultured cells expressing KDR/FLK-l, are lysed, and aliquots of the lysates are distributed on ELISA plates which have been coated with KDR/FLK-1-specific antibodies. After incubation for a suitable time under suitable conditions, the plates are washed to
_5 remove the unbound proteins and the subset candidate compounds are added to the wells. Subsequently, the
receptor kinase reaction is induced by addition of kinase buffer containing MnCl2. Mn2+ ions are known to stimulate enzymatic activity of receptor tyrosine kinases and thus receptor autophosphorylation. The plates are then washed, and incubated with an anti- phosphotyrosine antibody, linked to a detection entity, e . g. , biotin. In preferred embodiments, biotinylated anti-phosphotyrosine antibody 4G10 is used (UBI, Catalog No. 16-103) . The plates are washed, the assay developed, and the phosphotyrosine levels of the different wells determined on an ELISA reader, for example a Dynatech MR5000. Subset candidate compounds determined to inhibit the in vi tro autophosphorylation are further pursued, the "negative" ones are removed.
As the biochemical assays, as true for the cellular assay described herein, may be adopted as a initial high-throughput screening assay, the biochemical assay may employed in a ddition of, or as substitute of, the cellular assay described as the
"first filter". Preferably, however, both assays are used to verify the selectivity and potency of a test compound for KDR/FLK-l as the molecular target on two independent assay levels .
5.1.4. Determination Of Toxicity Of The Compounds
Therapeutic compounds should be more potent in inhibiting receptor tyrosine kinase activity than in exerting a cytotoxic effect. The tolerance of the subset candidate compounds may be assessed by determination by assessment of the LD50 value, i.e., the lethal dose for 50% of a group of animals.
Various doses of the test compound, estimated to cover the range from 0 to 100% lethality, are administered to several groups of animals. The mortality in each
group within a fixed period of time is determined and used to construct a curve relating factual mortality to dose. Determination of LD50 values is further described in Fabian et al . , 1993, Regulatory Toxicology and Pharmacology 18:206-213; Paumgartten et al . , 1989, Brazilian J. Med . Biol . Res . 22:987-991.
5.1.5. Determination Of The Compounds Efficacy In Vivo In A Mouse Subcutaneous Xenograft Tumor Model
The subset candidate compounds which have been determined to effectively and selectively inhibit the signal transduction induced by VEGF and mediated by KDR/FLK-l are further analyzed in an in vivo model relevant to the process of vascularization and/or angiogenesis. In general, suitable models include, but are not limited to, in vivo tumor models, tumor invasion models, retinopathy models, etc.
In a specific emdodiment of the invention, a mouse xenograft tumor model is employed as angiogenesis model. In general, tumor cell lines are implanted subcutaneously in nude mice. Subsequently, the animals are treated with the candidate compounds to determine their effect on tumor angiogenesis and tumor growth. In general, maximum dose of compound which is administered to the animal is the amount which has been determined as the LD10 value, i.e., the dose lethal for 10% of the test animals. The xenografts are measured regularly, preferably at least every three days. At the end of the experiment, the tumors are resected to be immunologically and histologically examined to determine of the subset candidate compounds' inhibitory effect on the tumor angiogenesis.
In a specific embodiment of the invention, EPH-4 (Reichmann et al . , 1989, J. Biol . Cell . 108:1127-1138) cells transfected with VEGF (EPH-4/VEGF cells) are
employed as test tumors. Wild type EPH-4 cells are immortalized epithelial cells which do not grow as tumor when implanted subcutaneously in nude mice. EPH-4/VEGF cells which have been engineered to express high levels of VEGF, however, have been shown to grow as highly vascularized subcutaneous tumors in test animals. In contrast to other tumor cells, which produce VEGF only after induction, for example by hypoxia and the hypoxia induced factor (Liu et al . , 1995, Circ. Res . 77:638-643), EPH4/VEGF cells produce VEGF constitutively. Accordingly, they are extremely potent inducers of tumor angiogenesis and as such provide for a very stringent in vivo test system. Those subset candidate compounds which are determined to inhibit the EPH-4/VEGF tumor growth by at least 30% relative to untreated controls at a doses smaller than the LD10/ are the desired compounds of this invention.
5.1.6. Summary Of The Experimental
Results
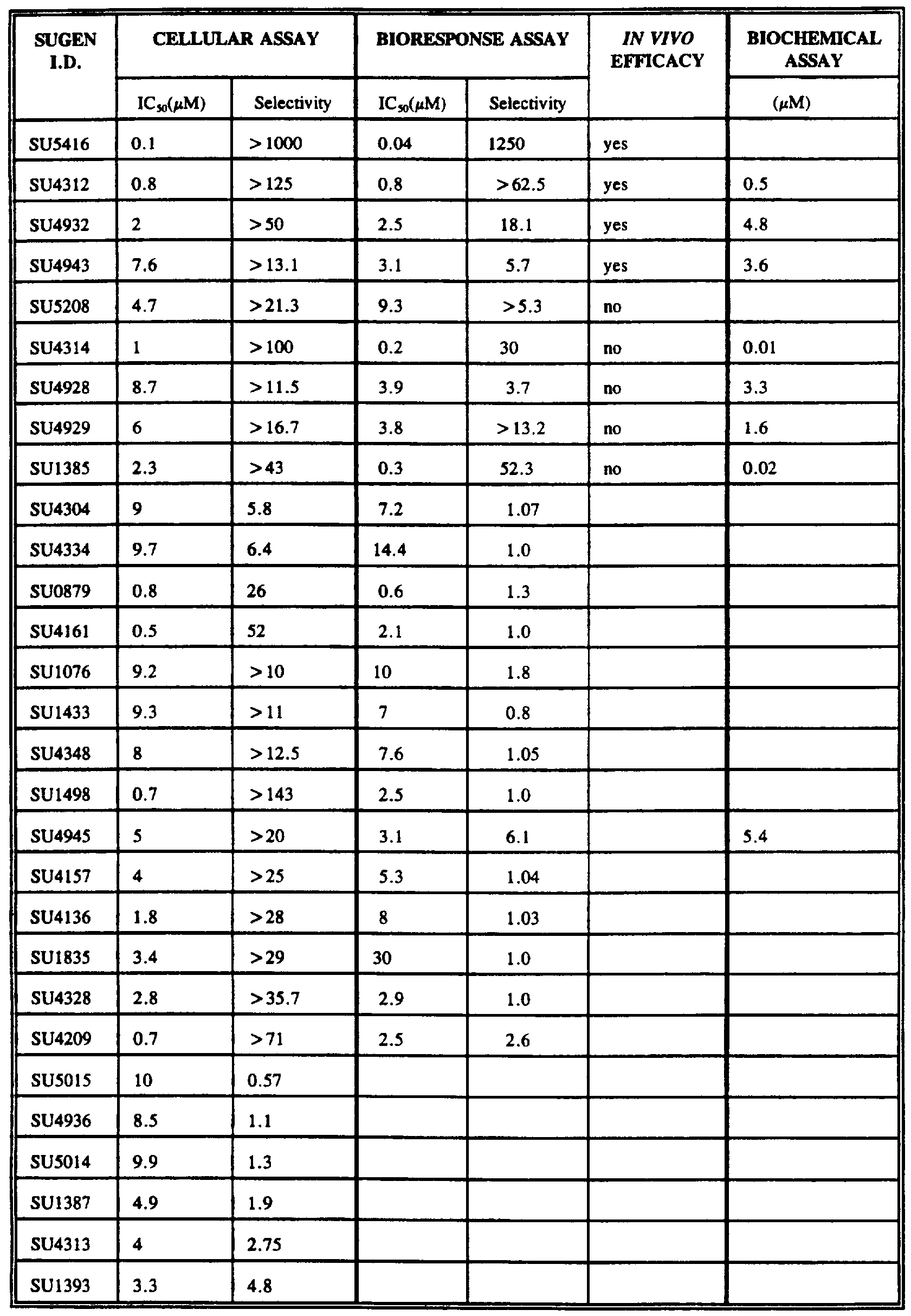
TABLE I summarizes the compounds identified in each "filter" of the assay cascade of the invention.
TABLE I COMPOUND SELECTION THROUGH ASSAY CASCADE COMPRISING FILTERS OF INCREASING STRINGENCY
The test candidate compounds employed for the process of this invention may be obtained from any commercial source, including Aldrich (1001 West St. Paul Ave., Milwaukee, WI 53233), Sigma Chemical (P.O. Box 14508, St. Louis, MO 63178), Fluka Chemie AG (Industriestrasse 25, CH-9471 Buchs, Switzerland (Fluka Chemical Corp. 980 South 2nd Street, Ronkonkoma, NY 11779)) , Eastman Chemical Company, Fine Chemicals (P.O Box 431, Kingsport, TN 37662) ,
Boehringer Mannheim GmbH (Sandhofer Strasse 116, D- 68298 Mannheim) , Takasago (4 Volvo Drive, Rockleigh, NJ 07647) , SST Corporation (635 Brighton Road, Clifton, NJ 07012) , Ferro (111 West Irene Road, Zachary, LA 70791) , Riedel-deHaen Aktiengesellschaft (P.O. Box D-30918, Seelze, Germany) , PPG Industries Inc., Fine Chemicals (One PPG Place, 34th Floor, Pittsburgh, PA 15272) . Further any kind of natural products may be screened using the assay cascade of the invention, including microbial, fungal or plant extracts.
5.3. Compounds Identified Using The Process Of The Invention
Using the process of this invention, a number of compounds selectively targeting and inhibiting the signal transduction induced by VEGF and mediated by KDR/FLK-l for the inhibition of vasculogenesis and/or angiogenesis in vivo have been identified. The compounds identified by the process of the invention include, for example, derivatives of quinazoline, quinoxiline, substituted aniline, indoline, isoxazoles, acrylonitrile and phenylacrylonitrile compounds. The compounds of the present invention, however, are not limited to any particular chemical structure, as they are solely defined by the assay
cascade of the invention, which allows, for the first time, to systematically and rationally identify highly potent and highly selective inhibitors targeting the activity of KDR/FLK-l on a molecular level. In addition to the above compounds and their pharmaceutically acceptable salts, the invention is further directed, where applicable, to solvated as well as unsolvated forms of the compounds ( e . g. hydrated forms) produced and identified by the process of the invention.
5.4. Indications
The compounds identified by the methods of the present invention are believed to bind to and specifically inhibit the KDR/FLK-l receptor by, e.g., inhibiting its catalytic activity, affinity for ATP or ability to interact with a substrate.
Thus, pharmaceutical compositions comprising a therapeutically effective amount of a compound identified by the process of the invention will be useful for the treatment of diseases driven by persistent unregulated angiogenesis. For example, in arthritis, new capillary blood vessels invade the joint and destroy the cartilage. Further, in at least twenty (20) diseases relating to ocular neovascularization, as for example diabetes mellitus, new and bleeding capillaries cause blindness.
Moreover, vasculogenesis and/or angiogenesis has been associated with the growth of malignant solid tumors and metastasis. Indeed, a tumor must continuously stimulate the growth of new capillary blood vessels for the tumor itself to grow. Furthermore, the new blood vessels embedded in a tumor provide a gateway for tumor cells to enter the circulation and to metastasize to distant sites in the body. Thus, the compounds and pharmaceutical
compositions identified by the process of the present invention may be included in methods for treating, among other diseases, diabetic retinopathy and other diseases related to ocular neovacularization, 5 arthritis, glioma, melanoma, Kaposi's sarcoma, psoriasis, hemangioma and ovarian, breast, lung, pancreatic, prostate, colon and epidermoid cancer. Thus, in general, the disorders which may be treated with the compounds and compositions, and 0 pharmaceutical formulations identified by the process of the invention generally refer to angiogenic and vasculogenic disorders resulting in or caused by inappropriate proliferation of blood vessels.
5 5.5. Pharmaceutical Formulations And Routes Of Administration
The identified compounds can be administered to a human patient alone or in pharmaceutical compositions where they are is mixed with suitable carriers or 0 excipient (s) at therapeutically effective doses to treat or ameliorate a variety of disorders. A therapeutically effective dose further refers to that amount of the compound sufficient to result in amelioration of symptoms as determined in a decrease 5 of vasculogenesis and/or angiogenesis. Techniques for formulation and administration of the compounds of the instant application may be found in "Remington's
Pharmaceutical Sciences," Mack Publishing Co., Easton,
PA, latest edition.
30
5.5.1. Routes Of Administration.
Suitable routes of administration may, for example, include oral, rectal, transmucosal, or intestinal administration; parenteral delivery,
__ including intramuscular, subcutaneous, intramedullary injections, as well as intrathecal, direct
intraventricular, intravenous, intraperitoneal, intranasal, or intraocular injections.
Alternately, one may administer a compound of the invention in a local rather than systemic manner, for example, via injection of the compound directly into a solid tumor, often in a depot, or in a sustained release formulation.
Furthermore, one may administer the drug via a targeted drug delivery system, for example, in a liposome coated with tumor-specific antibody. The liposomes will be targeted to and taken up selectively by the tumor.
5.5.2. Composition/Formulation The pharmaceutical compositions of the present invention may be manufactured by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or lyophilizing processes. Pharmaceutical compositions for use in accordance with the present invention thus may be formulated in a conventional manner using one or more physiologically acceptable carriers comprising excipients and auxiliaries which facilitate processing of the active compounds into preparations which can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen.
For injection, the agents of the invention may be formulated in aqueous solutions, preferably in physiologically compatible buffers such as Hanks' s solution, Ringer's solution, or physiological saline buffer. For transmucosal administration, penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art.
For oral administration, the compounds can be formulated readily by combining the active compounds with pharmaceutically acceptable carriers well known in the art . Such carriers enable the compounds of the invention to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a patient to be treated. Pharmaceutical preparations for oral use can be obtained as a solid excipient, optionally grinding a resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores. Suitable excipients include fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP) . If desired, disintegrating agents may be added, such as the cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
Dragee cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures. Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
Pharmaceutical preparations which can be used orally include push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. The
push-fit capsules can contain the active ingredients in admixture with fillers such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers. In soft capsules, the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols. In addition, stabilizers may be added. All formulations for oral administration should be in dosages suitable for such administration.
For buccal administration, he compositions may take the form of tablets or lozenges formulated in conventional manner.
For administration by inhalation, the compounds for use according to the present invention are conveniently delivered in the form of an aerosol spray presentation from pressurized packs or a nebulizer, with the use of a suitable propellant, e . g. , dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In the case of a pressurized aerosol the dosage unit may be determined by providing a valve to deliver a metered amount. Capsules and cartridges of, e . g. , gelatin, for use in an inhaler or insufflator, may be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch.
The compounds may be formulated for parenteral administration by injection, e. g. , by bolus injection or continuous infusion. Formulations for injection may be presented in unit dosage form, e . g. , in ampoules or in multi-dose containers, with an added preservative. The compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
Pharmaceutical formulations for parenteral administration include aqueous solutions of the active compounds in water-soluble form. Additionally, suspensions of the active compounds may be prepared as appropriate oily injection suspensions. Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes. Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the suspension may also contain suitable stabilizers or agents which increase the solubility of the compounds to allow for the preparation of highly concentrated solutions.
Alternatively, the active ingredient may be in powder form for constitution with a suitable vehicle, such as sterile pyrogen-free water, before use.
The compounds may also be formulated in rectal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter or other glycerides . In addition to the formulations described previously, the compounds may also be formulated as a depot preparation. Such long acting formulations may be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection. Thus, for example, the compounds may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
A pharmaceutical carrier for the hydrophobic compounds of the invention is a cosolvent system comprising benzyl alcohol, a nonpolar surfactant, a water-miscible organic polymer, and an aqueous phase.
The cosolvent system may be the VPD co-solvent system. VPD is a solution of 3% w/v benzyl alcohol, 8% w/v of the nonpolar surfactant polysorbate 80, and 65% w/v polyethylene glycol 300, made up to volume in absolute ethanol. The VPD co-solvent system (VPD:5W) consists of VPD diluted 1:1 with a 5% dextrose in water solution. This co-solvent system dissolves hydrophobic compounds well, and itself produces low toxicity upon systemic administration. Naturally, the proportions of a co-solvent system may be varied considerably without destroying its solubility and toxicity characteristics. Furthermore, the identity of the co-solvent components may be varied: for example, other low-toxicity nonpolar surfactants may be used instead of polysorbate 80; the fraction size of polyethylene glycol may be varied; other biocompatible polymers may replace polyethylene glycol, e . g. , polyvinyl pyrrolidone; and other sugars or polysaccharides may be substituted for dextrose. Alternatively, other delivery systems for hydrophobic pharmaceutical compounds may be employed. Liposomes and emulsions are well known examples of delivery vehicles or carriers for hydrophobic drugs. Certain organic solvents such as dimethylsulfoxide also may be employed, although usually with a greater toxicity.
Additionally, the compounds may be delivered using a sustained-release system, such as semipermeable matrices of solid hydrophobic polymers containing the therapeutic agent. Various sustained- release materials have been established and are well known by those skilled in the art. Sustained-release capsules may, depending on their chemical nature, release the compounds for a few weeks up to over 100 days.
Depending on the chemical nature and the biological stability of the therapeutic reagent, additional strategies for protein stabilization may be employed. The pharmaceutical compositions also may comprise suitable solid or gel phase carriers or excipients. Examples of such carriers or excipients include but are not limited to calcium carbonate, calcium phosphate, various sugars, starches, cellulose derivatives, gelatin, and polymers such as polyethylene glycols.
Many of the KDR/FLK-l inhibiting compounds of the invention may be provided as salts with pharmaceutically compatible counterions. Pharmaceutically compatible salts may be formed with many acids, including but not limited to hydrochloric, sulfuric, acetic, lactic, tartaric, malic, succinic, etc. Salts tend to be more soluble in aqueous or other protonic solvents that are the corresponding free base forms.
5.5.3. Effective Dosage.
Pharmaceutical compositions suitable for use in the present invention include compositions wherein the active ingredients are contained in an effective amount to achieve its intended purpose. More specifically, a therapeutically effective amount means an amount effective to prevent development of or to alleviate the existing symptoms of the subject being treated. Determination of the effective amounts is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
For any compound used in the method of the invention, the therapeutically effective dose can be estimated initially from cell culture assays. For
example, a dose can be formulated in animal models to achieve a circulating concentration range that includes the IC50 as determined in cell culture ( i . e . , the concentration of the test compound which achieves a half-maximal inhibition of the KDR/FLK-l activity) . Such information can be used to more accurately determine useful doses in humans.
A therapeutically effective dose refers to that amount of the compound that results in amelioration of symptoms or a prolongation of survival in a patient. Toxicity and therapeutic efficacy of such compounds can be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e . g. , for determining the LD50 (the dose lethal to 50% of the population) and the ED50 (the dose therapeutically effective in 50% of the population) . The dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio between LD50 and ED50. Compounds which exhibit high therapeutic indices are preferred.
The data obtained from these cell culture assays and animal studies can be used in formulating a range of dosage for use in human. The dosage of such compounds lies preferably within a range of circulating concentrations that include the ED50 with little or no toxicity. The dosage may vary within this range depending upon the dosage form employed and the route of administration utilized. The exact formulation, route of administration and dosage can be chosen by the individual physician in view of the patient's condition. (See, e . g. , Fingl et al . , 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 pl) .
Dosage amount and interval may be adjusted individually to provide plasma levels of the active moiety which are sufficient to maintain the kinase
modulating effects, or minimal effective concentration (MEC) . The MEC will vary for each compound but can be estimated from in vi tro data; e.g., the concentration necessary to achieve 50-90% inhibition of the kinase using the assays described herein. Dosages necessary to achieve the MEC will depend on individual characteristics and route of administration. However, HPLC assays or bioassays can be used to determine plasma concentrations. Dosage intervals can also be determined using MEC value. Compounds should be administered using a regimen which maintains plasma levels above the MEC for 10-90% of the time, preferably between 30-90% and most preferably between 50-90%. In cases of local administration or selective uptake, the effective local concentration of the drug may not be related to plasma concentration.
The amount of composition administered will, of course, be dependent on the subject being treated, on the subject's weight, the severity of the affliction, the manner of administration and the judgment of the prescribing physician.
5.5.4. Packaging The compositions may, if desired, be presented in a pack or dispenser device which may contain one or more unit dosage forms containing the active ingredient . The pack may for example comprise metal or plastic foil, such as a blister pack. The pack or dispenser device may be accompanied by instructions for administration. Compositions comprising a compound of the invention formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labelled for treatment of an indicated condition. Suitable conditions indicated on the label may include inhibition of
angiogenesis, treatment of a tumor, treatment of arthritis, diabetes, and the like.
6. EXAMPLES
The following assays and animal models may be used to identify compounds which selectively inhibit the KDR/FLK-l receptor signal transduction. More specifically, high-throughput cellular and biochemical assays are designed for the preselection of subset candidate compounds having inhibitory activity on KDR/FLK-l. Subset candidate compounds identified as positive by these "filters" are then evaluated in bioresponse assays and animal models for their selectivity for inhibition of VEGF-induced and KDR/FLK-l-mediated signal transduction and for their in vivo efficacy.
6.1. Cellular Assays For The Initial Screening Of The Acquired Test Compounds The following assay has been developed to provide for the high throughput testing of test candidate compounds for the identification of potent KDR/FLK-l- specific inhibitors. NIH 3T3 cells expressing FLK-1 (NIH 3T3/FLK-1 cells) are seeded and grown on 96 well plates and incubated with the test candidate compounds or extracts. Control cells, e . g. , NIH 3T3 cells expressing EGF-R (NIH 3T3/EGF-R) are treated identically. The cells are stimulated with the corresponding ligand, VEGF and EGF, respectively, lysed, and receptor tyrosine phosphorylation is determined via Enzyme Linked Immunosorbent Assays (ELISA) .
In general, the ELISA for detection and measurement of the presence of tyrosine kinase activity may be conducted according to protocols known in the art, which are described in, for example,
Voller et al . , 1980, "Enzyme-Linked Immunosorbent Assay," In: Manual of Clinical Immunology, 2d ed . , edited by Rose and Friedman, pp 359-371 Am. Soc. Of Microbiology, Washington, D.C. The objective of the following protocol is to provide a consistent method for measuring phosphotyrosine levels of FLK-1 receptors isolated from lysates of FLK-l/NIH cells.
Selectivity of inhibition of the subset candidate compounds for KDR/FLK-l relative to control receptor (here the EGF-R) is determined. Compounds having an IC50 of <50 μM, preferably <10 μM, and further a selectivity of twofold, preferably fivefold, for KDR/FLK-l are "positive" and are further pursued
6.1.1. Reagents And Supplies
1. Corning 96-well ELISA plates (Corning, Catalog No. 25805-96) .
2. PBS (Phosphate Buffered Saline) ; Formulation: 2.7 mM KCl; 1.1 mM KH2P04; 0.5 mM MgCl2 (anhydrous) ; 138 mM NaCl; 8.1 mM Na2HP04.
3. HNTG Buffer; Formulation: 20 mM ptkHEPES buffer pH 7.5; 150 mM NaCl; 0.2% Triton X-100; 10% Glycerol .
4. EDTA (0.5 M pH 7.0) . 5. Na3V04 (0.1 M pH 10.0) .
6. Na4P207 (0.2 M) .
7. DMEM (Dulbecco' s Modified Eagle Medium) with lx high glucose, L-Glutamine (Catalog No. 11965-050) .
8. FBS (Fetal Bovine Serum) ; CS (Calf Serum) . 9. L-Glutamine (200 mM stock) .
10. Growth media: DMEM 10% heat inactivated FBS (10% CS)+ 2 mM L-Glutamine.
11. Starvation media: DMEM 0.1% FBS (0.1% CS)+ 2 mM L-Glutamine. 12. NIH 3T3/FLK-1 cells, NIH 3T3/EGF-R cells, grown in growth media in 5% C02 at 37°C.
13. VEGF: 10 μg/ml in Milli-Q water stored at - 20°C (Peprotech, -20°C) .
14. EGF: stock concentration = 16.5 μM; EGF 201, TOYOBO, Co., Ltd. Japan. 15. 05-101 (UBI) (a monoclonal antibody recognizing an EGFR extracellular domain) .
16. Anti-FLK-ID monoclonal antibody: Produced and purified by the Biochemistry laboratory, Sugen Inc. 17. 4G10 Biotin-conjugated anti-phosphotyrosine (UBI, Catalog No. 16-103) .
18. Solution A+B: (Vector Laboratories, Burlingame, California, Catalog No. PK-6100.)
19. ABTS solution; Formulation: 100 mM Citric acid (anhydrous) ; 250 mM Na2HP04, pH 4.0; 0.5 mg/ml
ABTS (2,2-azino-bis (3-ethyl benzthiazoline-6-sulfonic acid) . Keep solution in dark at 4°C until ready to use.
20. Hydrogen peroxide 30% (Fisher, Catalog No. H325) .
21. ABTS/H202; Formulation: 15 ml ABTS solution; 8 μl H202.
22. 0.1 M Na2C03, pH 9.6.
23. TBSW Buffer (Tris buffered Saline with Tween-20) ; Formulation: 50 mM Tris pH 7.2; 150 mM
NaCl; 0.1% Tween 20.
24. 20% Ethanolamine Stock, pH 7.0.
25. Corning 96-well round bottom cell culture plates (Corning, Catalog No. 25850) . 26. Nunc Polyproplene 96-well V bottom plates.
27. HNTG*; Formulation: HNTG plus 5 mM NaV04; 5 mM EDTA; 2 mM NaP207.
28. TBSW/0.5% Ethanolamine; Formulation: TBSW buffer plus 0.5% Ethanolamine.
6.1.2. Procedure A. Coating And Blocking Of ELISA Plates .
1. Corning ELISA plates are coated with 4.0 μg FLK-1D or EGF-R (05-101) antibody/well, respectively, in 0.1 M Na2C03 at a final volume per well of 100 μl over night at 4°C. Plates can be used for at least three days when kept at 4°C.
2. Unbound antibody is removed from wells by inverting plate. Plates are washed 3x with TBSW/0.5% Ethanolamine.
3. Plates are blocked by incubation with 200 μl TBSW/0.5% Ethanalmine per well while shaking at room temperature for 30 min.
B. Growth And Seeding Of FLK-1/NIH Cells
1 . NIH 3T3/FLK-1 and NIH 3T3/EGF-R cells, respectively, are plated in a 15 cm dish with 30 ml growth media and grown to 90-100% confluence. Subsequently, cells are harvested by trypsinization (3 ml trypsin/EDTA per 15 cm dish) . Trypsinization is stopped by addition of 10 ml growth medium. Then, the cells are sedi ented by centrifugation for 5 min at 2,000x g, and the cells resuspended in growth media to yield a final dilution of 25,000 cells/100 μl . 2. NIH 3T3/FLK-1 and NIH 3T3/EGF-R cells, respectively, are seeded into Corning 96 cell plate at 25,000 cells/well in 100 μl growth media.
3. Cells are grown for 1-2 days at 37°C, 5% C02.
4. Cells are washed with 100 μl of PBS per well.
5. The PBS is removed and 100 μl of Starvation medium are added to each well. The cells are incubated overnight at 37°C, 5% C02.
C. Assay Procedure.
1 . The compound/extract stocks at a concentration of about lOmM are diluted 1:10 in a polypropylene 96-well plate using DMEM. DMSO is diluted 1:10 for control wells.
2. The starvation media is removed from the wells of the cell plates and 90 μl of DMEM are added to each well.
3. 10 μl of diluted compound/extract and controls are added to the wells. The final drug dilution and DMSO dilution is 1:100. . The cells are incubated with diluted compounds/extracts for 2 hours at 37°C, 5% C02.
5. NIH 3T3/FLK-1 cells are stimulated with 50 μl/well of 3 mM Na3V04 and 0.3 μl/ml VEGF in DMEM
(final concentration of 1 mM Na3V04 and 100 ng/ml VEGF) for 8 minutes at 37°C. Control NIH 3T3/FLK-1 cells are incubated with 50 μl/well of 3 mM Na3V04 only. NIH 3T3/EGF-R cells are stimulated with EGF (and optionally with Na3V04) at a final concentration of 25 nM (and 1 mM) , respectively. Control NIH 3T3/EGF-R cells are incubated with 50 μl/well of 3 mM Na3V04 accordingly.
6. After incubation with/without the respective ligands, the supematants are aspirated and the cell plates washed lx with PBS.
7. Cells are lysed in 100 μl of HNTG* by incubation on ice for 5 minutes.
8. The ELISA plates are washed 3x as described above in step A.2.
9. The lysates of stimulated and non- stimulated cells are transferred from cell plates to the corresponding wells of the ELISA plates by repeated aspiration and dispensing while scraping the sides of each cell well. The ELISA plates are
incubated with the lysates for 2 hours at 4°C while shaking.
10. The ELISA plates are washed 3x as described in step A.2. 11. 100 μl/ biotinylated 4G10, diluted
1:10000 in TBSW/0.5% Ethanolamine are added to each well and incubated for 30 minutes while shaking.
12. At the same time as step 11, A+B reagents are mixed at a dilution of 1:5000 in TBSW/0.5% Ethanolamine. The solution is incubated at room temperature until step 13 is completed.
13. The ELISA plates are washed 3x as described in step A.2..
14. Per well, 100 μl of the A+B reagent mixture are added and incubated while shaking for 30 minutes .
15. The ELISA plates are washed 3x with TBST and twice with water.
16. ABTS/H202 solution is prepared 5 minutes prior to use.
17. The ELISA plate is developed by addition of 100 μl/well of the ABTS/H202 solution. The plates are incubated for approximately 10 minutes and then read on a Dynatech MR5000 at 410 nm against a reference filter of 630 nm.
6.1.3. Experimental Results From Cellular Assays
Subset candidate compounds obtained with the cellular assay are summarized in TABLE II.
TABLE II
SELECTIVITY AND POTENCY OF COMPOUNDS IDENTIFIED IN
HIGH-THROUGHPUT CELLULAR ASSAY
333333333333333333333333333333333
6.2. Biochemical Assay For Determination Of The Compounds Specificity For KDR/FLK-l
The following biochemical assay is designed to confirm a subset candidate compound's inhibitory effect on the KDR/FLK-l receptor on a molecular level.
More specifically, the following assay is designed to measure receptor autophosphorylation on isolated receptors in a well-defined in vi tro system, where secondary effects/influences of other cellular molecules can be excluded. A subset of candidate compounds obtained from the cellular assar are tested in the biochemical assay. Compounds determined as
"positive" in the following assay are compounds which in fact specifically inhibit the autophosphorylation of KDR/FLK-l.
6.2.1. Reagents And Supplies
1. 15 cm tissue culture dishes
2. NIH 3T3/FLK-1 cells: NIH fibroblast line overexpressing human FLK-1 clone 3 Sugen, Inc. (obtained from the Max Planck Institute, Martinsried, Germany) .
3. Growth medium: DMEM plus heat inactivated 10% FBS and 2 mM Glutamine, Gibco-BRL, Gaithersburg, USA.
4. Starvation medium: DMEM plus 0.5% heat- inactivated FBS, 2 mM Glutamine (Gibco-BRL, Gaithersburg, USA) .
5. Corning 96-well ELISA plates (Corning, Catalog No. 25805-96) .
6. L4 or E 38: Monoclonal antibody specific for FLK-1, purified by Protein A-agarose affinity chromatography; Sugen, Inc.
7. PBS (Dulbecco' s Phosphate-Buffered Saline) , pH 7.2 (Gibco, Catalog No. 450-1300EB) . Formulation:
2.7 mM KCl; 1.1 mM KH2P04; 0.5 mM MgCI2 (anhydrous) ; 138 mM NaCl; 8.1 M Na2HP04) .
8. HNTG buffer; Formulation: 20 M Hepes/HCI buffer, pH 7.2, 150 mM NaCl, 0.5% Triton X-100, 10% glycerol, 1 mM PMSF, 5 mg/1 Aprotinin.
9. Biorad Protein Assay Solution (Biorad, Hercules, CA; Catalog No. 500-0006) .
10. Blocking Buffer; Formulation: 5 % carnation instant dry milk in PBS. 11. TBST; Formulation: 50 mM Tris/HCI, 150 mM NaCl, 0.1% Triton X-100.
12. Kinase Buffer; Formulation: 25 mM Hepes/CI pH 7.0, 100 mM NaCl, 10 mM MnCl2 and 2 % Glycerol.
13. Stop solution: 50 mM EDTA. 14. Biotinylated 4G10, specific for phosphotyrosine (UBI, Lake Placid, NY; Catalog No. 16- 103) .
15. ABC kit (Vector Laboratories, Burlingame, CA; Catalog No. 4000) . 16. DMSO (Sigma, Catalog No. D-2650) .
17. NUNC 96-well V bottom polypropylene plates for compounds (Applied Scientific, Catalog No. AS- 72092) .
18. ABTS Solution; Formulation: 100 mM Citric Acid (anhydrous) ; 250 mM Na2HP04 pH 4.0; 0.5 mg/ml
(2,2' -azino-bis (3-ethylbenzthiazoline-6-sulfonic acid) (Sigma, Catalog No. A-1888) . (Solution should be kept in the dark at 4°C until ready to use.)
19. Hydrogen peroxide, 30% solution (Fisher, Catalog No. H325) . (Should be stored in the dark at
4°C until ready to use.)
20. ABTS/H202; Formulation: 15 ml ABTS solution and 2 μl H202. Prepare 5 minutes before use and leave at room temperature. 21. ATP (Sigma, Catalog No. A-7699) .
6.2.2. Procedures
A. Cell Growth And Lysate Preparation.
1. Cells are seeded into growth medium and grown for 2-3 days to 90-100% confluency at 37°C and 5% C02. Cells should not exceed passage No. 20.
2. The medium is removed and the cells washed twice with PBS, then lysed with HNTG lysis buffer. All lysates are collected and votexed for 20 seconds.
3. The soluble material is removed by centrifugation (5 min, 10,000x g) .
4. The protein concentration is determined according to Bradford.
5. The lysates are divided into 1 mg aliquots and the tubes quick-freezed in a dry ice/ethanol mixture. The tubes are stored at -80°C.
B . Test Plate And Drug Plate Preparation.
1 . Corning 96-well ELISA plates are coated by incubation with 5 μg/well of purified L4 or E 38 in 100 μl coating buffer overnight at 4°C. Plates can be used for one week when kept at 4°C.
2. The unbound proteins are removed from the wells by inverting the plates to remove the liquid. The plates are patted on a paper towel to remove excess liquid and bubbles.
3. The plates are blocked by incubation with 100 μl blocking buffer per well for 45 minutes while shaking on a microtiter plate shaker.
4. The blocking buffer is removed and the ELISA plates washed three time with TBST. The plates are patted on a paper towel to remove excess liquid and bubbles.
5. The cleared lysate from NIH 3T3/FLK-1 cells are thawed, 75 μg of lysate/well are added, and incubated for 3 hours at 4°C while shaking the plate. Alternatively, the incubation can be performed
overnight, provided the temperature is between 4-0°C. Overnight incubation results in a higher delta in the ELISA.
6. The unbound proteins are removed from wells by inverting the plates. The plates are washed four times with TBST and the plates are patted on a paper towel to remove excess liquid and bubbles.
7. 80 μl of kinase buffer are added to the wells. 8. The compounds (10 mM, dissolved in 100 % DMSO) are diluted 20-fold into wells of a polypropylene plate filled with TBST plus 1% DMSO.
9. 10 μl of the pre-diluted compounds are added to the ELISA wells containing immobilized FLK-1 and mixed. The volume is now 90 μl . Control wells receive no drug.
10. The kinase reaction is started by addition of 10 μl 0.3 mM ATP per well. Negative controls receive distilled water. 11. The plates are incubated for 60 min at room temperature while shaking on a microtiter plate shaker.
12. The liquid is aspirated and the ELISA plate washed four times with TBST. The plate is patted on a paper towel to remove excess liquid.
13. 100 μl of 1:10,000 fold diluted biotinylated 4G10 is added to all wells and the mixture incubate for 45 minutes while shaking the plates. At the same time as the 4G10 addition, solution A+B is added (50 μl each) to 10 ml TBST.
14. The supernatant is aspirated and the plates washed five times as described in step 6.
15. 100 μl of the pre-formed complex (solution A+B in TBST) are added to all wells and incubated for 45 min while shaking the plates.
16. The supernatant is aspirated and the plates are washed five times as described in step 6.
17. 100 μl of the ABST/H202 solution are added to each well. The plates are incubated 10-15 minutes
5 while shaking the plates.
18. The bubbles are removed using a stream of air.
19. The plates are read on a Dynatech MR5000 ELISA reader at 410 nM against a reference filter of
10 630 nM.
6.2.3. Experimental Results From Biochemical Assays
Subset candidate compounds obtained with the
,_ biochemical assay are summarized in TABLE III. 15
20
25
30
35
TABLE III POTENCY OF TARGET SPECIFIC COMPOUNDS IDENTIFIED IN
BIOCHEMICAL ASSAY
SUGEN Inhibition in I.D. Biochem
FLK-IR
Kinase
Assay
(μM)
SU4928 3.3
SU4943 3.6
SU4945 5.4
SU4932 4.8
SU4314 0.01
SU1385 0.02
SU4929 1.6
SU4312 0.5
6.3. Determination Of The Bioresponse Via FLK-1 Assay Using HUV-EC-C Cells
The following protocol has been designed to test the efficacy of anti-FLK-1 inhibitors in an in vitro assay using a human umbilical vein endothelial cell line (HUV-EC-C) . HUV-EC-C express the KDR/FLK-l receptor and respond to induction with VEGF with cell proliferation, and are therefore an excellent system in order to determine the subset candidate compound's effect on VEGF-induced bioresponse. As aFGF induces proliferation in HUV-EC cells as well, the subset candidate compounds' effect on the aFGF-induced bioresponse is measured in control experiments in order to determine the subset candidate compounds' selectivity inhibition for KDR/FLK-l. Subset candidate compounds inhibiting the VEGF-induced bioresponse of HUV-EC-C at an IC50 <10 μM at a threefold selectivity relative to the bioresponse induced by aFGF, are tested further.
PATENT
6.3.1. Procedure
A. Day 1 .
1. HUV-EC-C cells (human umbilical vein endothelial cells, American Type Culture Collection; catalogue no. 1730 CRL) are washed twice with
Dulbecco' s phosphate-buffered saline (D-PBS; obtained from Gibco BRL; catalogue no. 14190-029) and then trypsinized with 0.05% trypsin-EDTA in non-enzymatic cell dissociation solution (Sigma Chemical Company; catalogue no. C-1544) . After cells have detached from the flask, they are resuspended in D-PBS and transferred to a 50 ml sterile centrifuge tube (Fisher Scientific; catalogue no. 05-539-6) .
2. The cells are sedimented by centrifugation, the supernatant is aspirated. The cells resuspended and washed twice or three times with D-PBS. Finally, the cells are resuspended in about 1 ml assay medium/15 cm2 of tissue culture flask. Assay medium consists of F12K medium (Gibco BRL; catalogue no. 21127-014) + 0.5% heat-inactivated fetal bovine serum. The cells are counted with a Coulter Counter®v Coulter Electronics, Inc.) ; the cell suspension is diluted in assay medium to the cells to obtain a concentration of 0.8-l.OxlO5 cells/ml. 3. The cells are seeded in 96-well flat-bottom plates at 100 μl/well or 0.8-1.0xl04 cells/well and incubated 37°C, 5% C02 for about 24 hours.
B . DAY 2 . 1. The compounds are adjusted to lOOx working stocks. Typically, the compounds are in 20 mM stocks, however, some are at other concentrations. At 20 mM, the compounds are diluted 1:100 to arrive at 200 μM, which is a 4x concentration (i.e., it comprises 1/4 of the total volume of the well and ultimately will be 50 μM) . If the compounds are dissolved in DMSO, they
should not be diluted <1:50, because higher concentrations of DMSO might kill the HUV-EC-C cells. 2. For each candidate compound a total amount 90 μl/well are needed for seven (7) wells, i.e., three (3) well for VEGF and aFGF, respectively, and one (1) , well for non-ligand media control.
3. Since the compounds are in 1:100 DMSO:assay medium, a diluent of the same DMSO:assay medium ratio needs to be made for the compound titrations to keep the DMSO concentration constant when diluting the compound.
4. The compounds are titrated as follows: a. For better results, 96-well round- bottom plates are used to do the compound titrations. b. 90 μl/well of compound is added per in the well of the top row (row A) of designated plates. In a typical experiment, two compounds can be assayed/plate (3 columns x 2 ligands (VEGF, aFGF) x 2 drugs) . In the top well of the columns for control without, 90 μl/well of the compound are added as well, c. 60 μl/well of the DMSO:assay medium diluent are added to the rest of the rows of the plates (row B-H) where compound had been added to row A. d. Three-fold dilutions are made by pipetting 30 μl of the 90 μl/well in row A into row B. 30 μl of the 90 μl/well in row B into row C, and so on down to row G. In the end, the additional 30 μl/well in row G are removed. No compounds are added to row H which is left as the no-drug control. At the end of this pipetting cascade, each well A-G should contain 60 μl of compound at different concentrations. For example, if the compound stock solution was 20 mM and diluted 1:100 to 200 μM, giving 50 μM final concentration, the dilutions yield compound
concentrations as follows: 50, 16.6, 5.5, 1.8, 0.6, 0.2, 0.07, and 0 μM. e. 50 μl/well of the 60 μl per well are transferred to the assay plate and incubated for 2-3 h at 37°C.
5. Ligand (VEGF and aFGF) and media control is added as follows: a. For each compound tested, 1.2 ml of VEGF and aFGF will be needed (50 μl/well, 24 wells total per compound) . b. For VEGF, the stock is 10 μg/ml, and the final concentration in the assay is 20 ng/ml. As the volume of VEGF is 1/4 that of the total assay volume (as true for the compounds) , a 4x concentration is needed (80 ng/ml) . c. For aFGF, the stock is 10 μg/ml (as with VEGF) , and the final concentration in the assay is 0.25 ng/ml. Since the volume of aFGF is 1/4 that of the total assay volume (as true for the compounds and VEGF) , a 4x concentration is needed (1 ng/ml) . d. 50 μl of VEGF are added to columns 1-3 and 7-9; add 50 μl of aFGF to columns 4-6 and 10-12, except the media control wells, which get 50 μl/ml of assay medium. e. The plates are incubated overnight (20-
24 h) at 37°C.
C . DAY 3 .
1 . 1 μCi 3H-thymidine/we11 (10 μl/well of 100 μCi/ml solution stock solution) are added and incubated overnight (20-24 h) at 37°C.
D . DAY 4 .
1. The plates are frozen overnight at -20°C.
E. DAY 5.
1. The plates are thawed and the cells harvested with Tomtec 96-well plate harvester and applied to filter mats. The 3H-thymidine incorporation is determined using the betaplate liquid scintillation counter.
6.3.2. Experimental Results From HUV-EC-C Cell Assays
Subset candidate compounds obtained with the bioresponse assay are summarized in TABLE IV.
TABLE IV SELECTIVITY AND POTENCY OF COMPOUNDS IDENTIFIED IN
BIORESPONSE ASSAY
SUGEN HUVEC HUVEC Specificity I.D. VEGF FGF Factor Thy incorp Thy incorp
IC,o
(μM) (μM)
SU4928 3.9 14.5 3.7
SU4943 3.1 17.7 5.7
SU4945 3.1 18.8 6.1
SU4932 2.5 45.2 18.1
SU4314 0.2 6 30
SU1385 0.3 15.7 52.3
SU5416 0.04 50 1250
SU4929 3.8 >50 > 13.2
SU5208 9.3 >50 >5.3
SU4312 0.8 >50 > 62.5 3 FOLD OR GREATER
SU1433 7 5.6 0.8
SU4136 8 8.2 1.03
SU4157 5.3 5.5 1.04
SU4348 7.6 8.3 1.05
SU4304 7.2 7.7 1.07
SU0879 0.6 0.8 1.3
SU1076 10 18.3 1.8
SU4209 2.5 6.5 2.6
SU1498 2.5 2.6 1.0
SU1835 30 31 1.0
SU4161 2.1 2.0 1.0
SU4328 2.9 3.0 1.0
SU4334 14.4 14.6 1.0
6.4. In Vivo Animal Models All subset candidate compounds which have been determined to effectively and selectively inhibit the
signal transduction induced by VEGF and mediated by KDR/FLK-l are further analyzed in an in vivo mouse xenograft tumor model .
More specifically, EPH-4 cells transfected with VEGF (EPH-4/VEGF cell) propagated in tissue culture flasks. 3x IO6 cells in PBS are implanted in 100 μM subutaneously in the hind flank of nude mice. Dosing of subset candidate compounds is initiated on the following day with daily i.p. injections (in 50 - 100 μl) . The growth of the implanted tumor cells is monitored and the size determined twice a week using calipers. Subset candidate compounds which inhibit tumor angiogenesis in vivo as indicated by inhibition of the tumor growth by at least 30% relative to untreated controls, at a doses smaller than the LD10 are identified. At the end of the experiment, the tumors are resected and further examined immunologically and histologically to determine the candidate compounds' inhibitory effect on tumor angiogenesis.
6.4.1. Experimental Results From The Mouse Subcutaneous Xenograft Assays
Subset candidate compounds obtained with the subcutaneous xenograft model are summarized in TABLE V.
TABLE V IN VIVO EFFICACY OF COMPOUNDS
SUGEN Efficacy I.D.
SU4943 yes***
SU4932 yes***
SU5416 yes***
SU4312 yes***
SU4928 no
SU4929 no
SU5208 no
SU4945 •
SU4314 no
SU1385 no
6.5. Tumor Invasion Model
The following tumor invasion model has been developed and may be used for the evaluation of therapeutic value and efficacy of the compounds identified to selectively inhibit KDR/FLK-l receptor by the cascade of screening assays of the invention.
6.5.1. Procedure
8 week old nude mice (female) (Simonsen Inc.) was used as experimental animals. Implantation of tumor cells was performed in a laminar flow hood. For anesthesia, Xylazine/Ketamine Cocktail (100 mg/kg ketamine and 5 mg/kg) are administered intraperitoneally. A midline incision is done to expose the abdominal cavity (approximately 1.5 cm in length) to inject IO7 tumor cells in a volume of 100 μl medium. The cells are injected either into the
duodenal lobe of the pancreas or in the serosa of the colon. The peritoneum and muscles are closed with a 5-0 silk continuous suture and the skin by gluing with Vetbond and with at least two (5-0 silk) interrupted sutures. Animals are observed daily.
6.5.2. Analysis
After 2-6 weeks, depending on gross observations of the animals, the mice are sacrificed, and the local tumor metastases, to various organs (lung, liver, brain, stomach, spleen, heart, muscle) are excised and analyzed (measurements of tumor size, grade of invasion, immunochemistry, and in situ hybridization) .
6.6. Production Of FLK-1-Specific Monoclonal Antibodies
A. Antigen Production .
A fragment encoding for the 523 bp C-terminal fragment of FLK-1 was constructed into a pGEX3X vector. XLI-Blue Electroporation-competent cells (Stratagene No. 200228) were transformed with the "FLK-lD-GST(pGex-3X FLK-1-523 (D) ) construct by electroporation. Expression was induced by IPTG induction. The fusion protein was purified to homogeneity by Glutathione agarose affinity chromatography, then the purified protein dialyzed against PBS, concentrated by ultra filtration and stored at - 80°C.
B . Immunization .
Balb C mice were immunized with the above described antigen according to standard procedures. See, Harlow and Lane, 1988.
C. Antisera Testing.
After the initial immunization and boosts with antigen, the mice were bled and the serum tested in the FLK-1 D-GST ELISA.
D. FLK-1 D-GST -ELISA.
Purified FLK-1 D-GST or GST control alone were coated to ELISA plates according to standard procedures. Antisera were tested for cross-reactivity with antigen by incubating serially diluted antisera with antigen, followed by HRP-conjugated Goat-anti mouse antiserum.
E. Fusion. Splenocyte/Myeloma cell fusion was performed and hybridoma cells were cultured according to standard procedures .
F. Screening Of Supematants . Supematants were screened using the FLK-ID-GST ELISA. Pools that tested positive in the ELISA were subcloned. In the next round of screening, only pools were selected, which cross-related with FLK-ID-GST and not with GST-controls.
G. Testing Hybridoma Supematants With Full Length FLK-1 In Western Blotting.
Positive pools were tested in Western blotting using lysates from NIH 3T3/FLK-1 clone 3 (NIH 3T3 cells transfected with FLK-1) . Pools which specifically recognized the FLK-1 receptor protein were further subcloned.
H. Testing Hybridoma Supematants Wi th Full Length FLK-1 In Immunoprecipi tation.
Positive pools were tested in immunoprecipitation using lysates from NIH 3T3 cells/FLK-1 clone 3. Positive pools were further subcloned. All clones were tested in immunoprecipitation experiments comparing lysates from non-stimulated vs. VEGF- stimulated NIH 3T3/FLK-1 cells. Western blots were probed with anti-phosphotyrosine specific antiserum. Immunoprecipitates from all clones showed ligand dependent tyrosine phosphorylation of FLK-1.
I. Final Evaluation Of FLK-1 Specific Monoclonal Antibodies . Antibodies from FLK-1 specific clones were purified by Protein A/G agarose affinity chromatography according to standard procedures.
Purified antibodies were coated to ELISA plates and were tested for the ability to capture FLK-1 from lysates of VEGF-stimulated and non-stimulated NIH
3T3/FLK-1 cells. Clones were obtained which fulfilled these criteria ( eg. , L4 and E 38) .
The present invention is not to be limited in scope by the exemplified embodiments which are intended as illustrations of single aspects of the invention. Indeed, various modifications of the invention in addition to those described herein will become apparent to those skilled in the art from the foregoing description and accompanying drawings. Such modifications are intended to fall within the scope of the appended claims.
All references cited herein are hereby incorporated by reference in their entirety.