WO1994000125A1 - 17-ALKYLKETONE STEROIDS USEFUL AS 5-α-REDUCTASE INHIBITORS - Google Patents
17-ALKYLKETONE STEROIDS USEFUL AS 5-α-REDUCTASE INHIBITORS Download PDFInfo
- Publication number
- WO1994000125A1 WO1994000125A1 PCT/US1993/006238 US9306238W WO9400125A1 WO 1994000125 A1 WO1994000125 A1 WO 1994000125A1 US 9306238 W US9306238 W US 9306238W WO 9400125 A1 WO9400125 A1 WO 9400125A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- estra
- triene
- pharmaceutically acceptable
- Prior art date
Links
- 0 *c1c[n]c2c1cccc2 Chemical compound *c1c[n]c2c1cccc2 0.000 description 4
- PDSOIUHLYKBZLZ-VRSOYXCKSA-N C[C@](CC1)(C(CC2)C(CCc3c4)C1c3ccc4O)[C@H]2C#N Chemical compound C[C@](CC1)(C(CC2)C(CCc3c4)C1c3ccc4O)[C@H]2C#N PDSOIUHLYKBZLZ-VRSOYXCKSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/565—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids not substituted in position 17 beta by a carbon atom, e.g. estrane, estradiol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J41/00—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring
- C07J41/0033—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005
- C07J41/0094—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 containing nitrile radicals, including thiocyanide radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/575—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of three or more carbon atoms, e.g. cholane, cholestane, ergosterol, sitosterol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J3/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by one carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J3/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by one carbon atom
- C07J3/005—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by one carbon atom the carbon atom being part of a carboxylic function
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
- C07J31/006—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring not covered by C07J31/003
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J7/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms
- C07J7/0005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21
- C07J7/001—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group
- C07J7/0015—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group not substituted in position 17 alfa
- C07J7/002—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group not substituted in position 17 alfa not substituted in position 16
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J7/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms
- C07J7/0005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21
- C07J7/0065—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by an OH group free esterified or etherified
- C07J7/007—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by an OH group free esterified or etherified not substituted in position 17 alfa
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J9/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J9/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane
- C07J9/005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane containing a carboxylic function directly attached or attached by a chain containing only carbon atoms to the cyclopenta[a]hydrophenanthrene skeleton
Definitions
- 17-alkylketone steroids useful as 5- ⁇ -reductase inhibitors.
- the present invention relates to certain novel 17 ⁇ and 17ß-alkylketone-3-carboxy aromatic A ring steroidal compounds, pharmaceutical compositions containing these compounds, and methods for using these compounds to inhibit steroid 5- ⁇ -reductase isozyme 1 and steroid 5- ⁇ -reductase isozyme 2. Also invented are novel intermediates and processes useful in preparing these compounds.
- the class of steroidal hormones known as androgens is responsible for the physical characteristics that differentiate males from females. Of the several organs that produce androgens, the testes produce these hormones in the greatest amounts. Centers in the brain exert primary control over the level of androgen production. Numerous physical manifestations and disease states result when ineffective control results in excessive androgen hormone production. For example, acne vulgaris, seborrhea, female hirsutism, male pattern baldness and prostate diseases such as benign prostatic hypertropy arc correlated with elevated androgen levels.
- Testosterone is the principal androgen secreted by the testes and is the primary androgenic steroid in the plasma of males. It now is known that 5- ⁇ - reduced androgens are the active hormones in some tissues such as the prostate and sebaceous gland. Circulating testosterone thus serves as a prohormone for dihydrotestosterone (DHT), its 5- ⁇ -reduced analogue, in these tissues but not in others such as muscle and testes.
- DHT dihydrotestosterone
- Steroid 5- ⁇ -reductase is a Nicotinamide Adenine dinucleotide Phosphate(NADPH)dependent enzyme that converts testosterone to DHT.
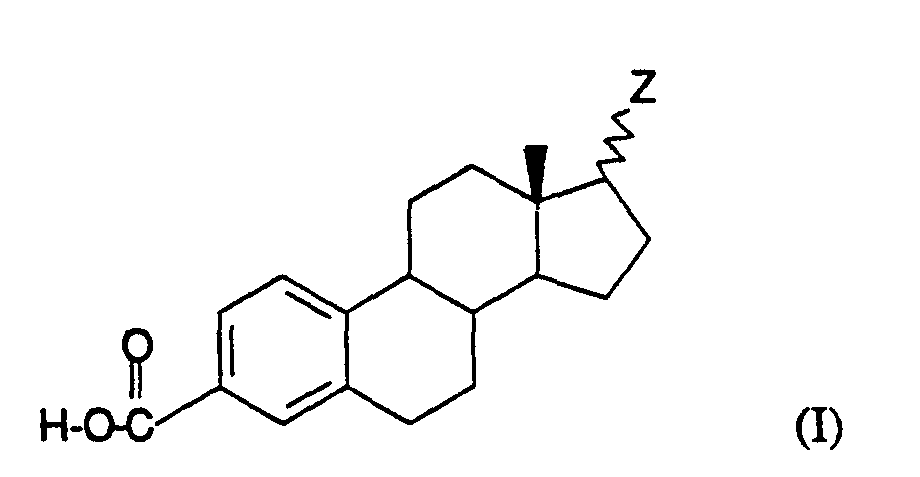
- This invention relates to a compound of the formula I:
- R is C 1-20 linear or branched, saturated or unsaturated alkyl and pharmaceutically acceptable salts, hydrates, solvates and esters thereof.
- the invention also is a method for simultaneously inhibiting 5- ⁇ -reductase isozyme 1 and 5- ⁇ -reductase isozyme 2 activity in mammals, including humans, that comprises administering to a subject an effective amount of a presently invented 5- ⁇ -reductase inhibiting compounds.
- novel intermediates and novel processes useful in preparing the presently invented dual 5- ⁇ -reductase inhibiting compounds include pharmaceutical compositions comprising a pharmaceutical carrier and compounds useful in the methods of the invention. Also included in the present invention are methods of co-administering the presnetiy invented dual 5- ⁇ -reductase inhibiting compounds with further active ingredients.
- R is C 1-20 linear or branched, saturated or unsaturated alkyl and pharmaceutically acceptable salts, hydrates, solvates and esters thereof.
- R is C 1-20 linear or branched, saturated or unsaturated alkyl and pharmaceutically acceptable salts, hydrates, solvates and esters thereof.
- Preferred among the presently invented Formula II compounds are those having Formula III
- R 2 is C 1-8 linear or branched alkyl and pharmaceutically acceptable salts, hydrates solvates and esters thereof.
- Preferred among Formula (III) compounds are those in which R 2 is methyl, ethyl, propyl, 3-methylbutyl, isopropyl, n-butyl, isobutyl, 1-methylpropyl, t-butyl, pentyl, 1,1-dimethylpropyl, 2,2-dimethylpropyl, octyl or 3,3-dimethylbutyl.
- Formula (HI) compounds are those in which R 2 is 1-methylpropyl, n-butyl, isopropyl, n-pentyl, 3-methylbutyl, 2,2-dimethylpropyl, t-butyl, 1,1-dimethylpropyl, isobutyl n-octyl, tert-pentyl, n-propyl, methyl or 3-3-dimethylbutyl.
- alkyl C l-n alkyl and derivatives thereof and in all carbon chains as used herein, unless otherwise defined, is meant a C l-n linear or branched carbon chain having 1 to n carbons.
- alkyl and derivatives thereof as used herein include: methyl, ethyl, propyl, 3-methylbutyl, isopropyl, n-butyl, isobutyl, 1-methylpropyl, t-butyl, n-pentyl, 1,1-dimethylpropyl, 2,2-dimethylpropyl, n-octyl, tert-pentyl and 3,3-dimethylbutyl.
- treating is meant prophylatic or therapeutic therapy.
- isobutyl as used herein, is meant -CH 2 CH(CH 3 ) 2 .
- metal-catalyzed coupling reaction as used herein is meant that the prepared 3-trifluoromethyl sulfonate or 3-fluorosulfonate compound is reacted in a suitable organic solvent, preferably toluene, dimethylformamide or THF with a base, preferably a tertiaryamine base such as triethylamine, pyridine or
- tributylamine a phosphine such as bis(diphenylphosphino)alkane, preferably 1,3 bis(diphenylphosphi ⁇ o)propane or tri-o-tolyphosphine, or a C 1-6 alkOH
- a metal catalyst preferably a palladium catalyst such as palladium (II) acetate, palladium (II) chloride or bis(triphenylphosphine) palladium II acetate, and a coupling reagent.
- Coupled reagent as used herein is meant a compound which is capable of reacting with an aryl radical to form a carboxylic acid substituent.
- Carbon monoxide is a preferred coupling reagnet, which when added to the metal-catalyzed coupling reaction, as described herein, yields the desired carboxylic acid group.
- esters can be employed, for example methyl, ethyl, pivaloyloxymethyl, and the like, and those esters known in the art for modifying solubility or hydrolysis characteristics for use as sustained release or prodrug formulations.
- ⁇ -receptor antagonist refers to a known class of alpha-andrenergic receptor antagonist comounds, such as described in Lafferty, et al. U.S. Patent No. 4,963,547, which are utilized in treating vascular disorders such as diabetes, cardiovascular disease, benign prostatic hypertrophy and ocular hypertension.
- Preferred alpha-andrenergic receptor antagonists for use in the compositions and methods of the invention include amsulosin, terazosin, doxazosin, alfuzosin, indoramin, prazosin and 7-chloro-2-ethyl-3,4,5,6-tetrahydro-4-methylthieno[4,3,2-ef][3]-benzapine.
- amsulosin as used herein is meant a compound of the structure
- amsulosin is designated as (-)-(R)-5-[2-[[2-(O-ethoxyphenoxy)ethyl]amino]propyl]-2-methoxybenzenesulfonamide.
- terazosin as used herein is meant a compound of the structure
- terazosin is designated as 1-(4-amino-6,7-dimethoxy-2 quinazolinyl)-4-[(tetrahydro-2-furoyl)carbonyl]piperazine.
- Terazosin is disclosed in U.S. Patent Number 4,251,532.
- doxazosin as used herein is meant a compound of the structure
- doxazosin is designated as 1-(4-amino-6,7-dimethoxy-2-quinazolinyl)-4-[(2,3-dihydro-1,4-benzodioxin-2-yl)carbonyl]-piperazine.
- alfuzosin as used herein is meant a compound of the structure
- Chemically alfuzosin is designated as N-[3-[(4-amino-6,7-dimethoxy-2- quinazolinyl)methylamino]propyl] tetrahydro-2-furancarboxamide.
- indoramin as used herein is meant a compound fo the structure
- prazosin as used herein is meant a compound of the structure
- Chemically prazosin is designated as 1-(4-amino-6,7-dimethoxy-2-quinazolinyl)-4-(2-furanylcarbonyl)piperazine.
- Prazosin is disclosed in U.S. Patent Number 3,511,836.
- alpha-andrenergic receptor antagonist a compound other than one specifically referred to herein is a alpha-andrenergic receptor antagonist by utilizing the assay described in Lafferty I. Thus, all such compounds are included within the scope of the term "alpha-andrenergic receptor antagonist" as used herein.
- minoxidil as used herein is meant the compound of the structure:
- Minoxidil is the active ingredient in Rogaine ® which is sold as topical solution for stimulating hair growth by the Upjohn Company, Kalamazoo,
- aromatase inhibitor refers to a known class of compounds, steroidal and non-steroidal, which prevent the conversion of androgens to estrogens, such as described in Gormley et al. International Publication Number WO 92/18132. Aromatase inhibitors are disclosed in Gormley et al. as having utility in treating benign prostatic hyperplasia when used in combination with a 5- ⁇ -reductase inhibitor.
- a preferred aromatase inhibitor for use in the compositions and methods of the invention 4-(5,6,7,8-tetrahydroirnidazo-[1,5- ⁇ ]pyridin-5-yl)benzonitrile (fadrazole). Fadrazole is disclosed in U.S. Patent No. 4,728,645. Additionally, all compounds disclosed in Gormley, et al. International Publication No. WO
- aromatase inhibitors as used herein.
- said 5- ⁇ -reductase inhibitor can be co-administered with said further active ingredient or ingredients.
- co-administering and derivatives thereof as used herein is meant either simultaneous administration or any manner of separate sequential administration of a 5- ⁇ -reductase inhibiting compound, as described herein, and a further active ingredient or ingredients, such as other compounds known to treat the disease states of acne vulgaris, seborrhea, female hirsutism, male pattern baldness, benign prostate hypertrophy or prostatic adenocarcinoma or compounds known to have utility when used in combination with 5- ⁇ -reductase inhibitors.
- the administration is not simultaneous, the compounds are administered in a close time proximity to each other.
- the compounds are administered in the same dosage form, e.g. one compound may be administered topically and another compound may be administered orally.
- novel compounds of Formula (II) of the present invention can be prepared by methods outlined in schemes 1-4 below and in the Examples from known and readily available estrone which has the formula:
- Scheme I outlines formation of Formula II compounds.
- compound (b) is prepared from compound (a) according to the procedure of Baldwin, et al., J. Chem. Soc. (c), 1968, 2283-2289.
- Compound (b) is then stirred in an appropriate organic solvent, preferably methanol, with a base, preferably sodium hydroxide, and then acidified to yield compound (c).
- Compound (c) is next treated with a Grignard reagent, described hereinbelow, or a lithium reagent in an appropriate organic solvent, preferably tetrahydrof uran or diethylether solvent, preferably at reflux temperature to yield formula (d) compounds.
- a base preferably 2,5-di-t-butyl-3-methylpyridine in an appropriate organic solvent, preferably dichloromethane
- Formula (f) compounds are prepared by reacting a formula(e) compound in a metal catalyzed coupling reaction.
- a formula (e) compound dissolved in dimethylformide (DMF) an organic base preferably, triethylamine, a phosphine, preferably bis(diphenylphosphin ⁇ )propane, a palladium(II) compound, preferably, palladium(II) acetate, and a C 1-6 alkyl alcohol (C 1-6 alkOH), followed by addition of carbon monoxide (CO).
- a suitable base preferably potassium carbonate
- Scheme II outlines formation of Formula II compounds.
- the starting materials in Scheme II are formula (d) compounds prepared as described in Scheme I.
- a formula (d) compound and a base preferably 2,5-di-t-butyl-3-methyl-pyridine in an appropriate organic solvent, preferably dichloromethane, is cooled to -20°C to 20°C, preferably 0°, and reacted with fluorosulfonic anhydride to form compounds (h).
- Formula (f) compounds are prepared by reacting a Formula (h) compound in a metal-catalyzed coupling reaction.
- a Formula (h) compound is dissolved in dimethylformide (DMF) an organic base preferably triethylamine, a phosphine preferably bis (diphenylphospine)propane, a palladium(II) compound, preferably, palladium(II) acetate, and a C 1-6 alkyl alcohol ( C 1-6 alkOH), followed by addition of carbon monoxide (CO).
- DMF dimethylformide
- an organic base preferably triethylamine
- a phosphine preferably bis (diphenylphospine)propane
- a palladium(II) compound preferably, palladium(II) acetate
- C 1-6 alkOH C 1-6 alkyl alcohol
- CO carbon monoxide
- Scheme III outlines formation of Formula II compounds.
- R 1 is CF 3 O 2 SO- or FO 2 SO-.
- the 3-hydroxyl acid (i) is converted to the 3-trifluoromethylsulfonylate or 3-fluorosulfonylate derivative (j) (step A) by treating (i) with trifluoromethylsulfonyl anhydride or fluorosulfonic anhydride and an amine base, such as pyridine, preferably 2,5 di-t-butyl-3-methyl-pyridine, in an appropriate organic solvent, preferably dichloromethane at about -20°C to 20°C, preferably 0°.
- an amine base such as pyridine, preferably 2,5 di-t-butyl-3-methyl-pyridine
- the activated ester (k) is produced (step B) by treating (j) with 2,2-dithiopyridyl and triphenylphosphine in an appropriate organic solvent solution preferably, tetrahydrofuran/toluene at room temperature for about 8-14 hours.
- the 17-acyl derivative (1) is produced (step C) by treating (k) with a Grignard reagent, described hereinbelow, in tetrahydrofuran or diethyl ether solvent, at a temperature of about -50 to -70°C, for 1-16 hours.
- the 3-alkyl ester (f) is produced (step D) by treating (1) under carbonylation conditions, preferably by bubbling carbon monoxide gas through a solution of (1) in an appropriate organic solvent, preferably methanol, containing palladium acetate catalyst, triphenylphosphine, and a tertiary organic amine preferably triethylamine at about room temperature for 1-16 hours.
- an appropriate organic solvent preferably methanol, containing palladium acetate catalyst, triphenylphosphine, and a tertiary organic amine preferably triethylamine
- Compounds (g) can also be produced (step G) by treating (1) under carboxylation conditions, preferably by bubbling carbon monoxide gas through a solution of 0) in an an appropriate non-alcoholic solvent, preferably DMSO, containing a palladium catalyst, preferably palladium (II) diacetate and 1,1-Bis(diphenylphosphino)ferrocene (DPPF); and a base, preferably potassium acetate, preferably at increased temperatures.
- an an appropriate non-alcoholic solvent preferably DMSO
- a palladium catalyst preferably palladium (II) diacetate and 1,1-Bis(diphenylphosphino)ferrocene (DPPF)
- DPPF 1,1-Bis(diphenylphosphino)ferrocene
- Route 2 involves converting the starting steroidal acid (i) to the 3-trifluoromethylsulfonylate or the 3-fluorosulfonylate derivative (j) by the above-described step A; carbonylating (j) to (m) by step D; forming the activated 2-pyridylthio ester (n) by step B; forming the 17-acyl compound (f) by step C; and hydrolyzing the 3-ester to the 3 acid final product (g) by step F.
- Route 3 involves converting the starting acid (i) to the activated ester (o) by the above-described step B; forming the 17-acyl compound (d) by reacting (o) by the above described step C; converting (d) to the 3-trifluoromethylsulfonylate or 3-fluorosulfonylate derivative (1) by the above-described step A; and converting (l) to the final product (g) by the above described step G or by the above-described step
- propylmagnesium bromide preferably propyl
- a Formula (c) compound and a base preferably 2,5-di-t-butyl-3-methyl-pyridine in an appropriate organic solvent, preferably dichloromethane, is cooled to -20°C to 20°C, preferably 0°C, and reacted with a trihaloalkyl sulfonic anhydride, preferably trifluoromethanesulfonic anhydride to form compounds (p).
- a base preferably 2,5-di-t-butyl-3-methyl-pyridine in an appropriate organic solvent, preferably dichloromethane
- Formula (q) compounds are prepared by reacting a Formula (p) compound in a metal catalyzed coupling reaction.
- a Formula (p) compound dissolved in dimethylformide (DMF) and organic base preferably, triethylamine, a phosphine, preferably bis(diphenylphosphino)propane, a palladium(II) compound, preferably, palladium(II) acetate, and a C 1 -C 6 alkyl alcohol (C 1 -C 6 alkOH), followed by addition of carbon monoxide (CO).
- Formula (q) compounds are reacted with a reducing agent, preferably diisobutylaluminum hydride, to yield Formula (r) compounds.
- Formula (s) compounds are produced by treating Formula (r) compounds with a grignard reagent (as described in Scheme III) or a lithium reagent in tetrahydrofuran or diethylether solvent, at a temperature of about -50° to -70°C, for 1-16 hours.
- a grignard reagent as described in Scheme III
- a lithium reagent in tetrahydrofuran or diethylether solvent
- Formula (g) compounds are prepared by oxidation of Formula (s) compounds.
- oxidation will utilize a Jones reagent or
- Grignard reagents of the type, XMgR, (where R is C 1-20 linear or branched, saturated or unsaturated alkyl) for use in preparing all of the species included within the scope of this invention, are available or can be made readily by one skilled in the art.
- Formula I compounds in which Z is in the ⁇ position are prepared from compounds which contain the corresponding ⁇ substituent by the General Method below.
- a base such as a hydroxide or alkoxide base, preferably sodium hydroxide, potassium hydroxide or sodium methoxide, at a temperature over 100°C preferably at reflux temperatures to yield the
- dimethyl sulfoxide or other non-reactive high boiling solvents are preferred when the starting 17ß dual 5 ⁇ -reductase inhibiting steroidal compound contains reactive substituents or reactive unsaturated bonds that are, for example, subject to nucleophilic attack and ethylene glycol, or other reactive high boiling solvents can be used when the reactivity of the substituents or any unsaturated bonds of the starting 17ß dual 5 ⁇ -reductase inhibiting steroidal compound is not a consideration.
- R is C 1 -20 linear or branched, saturated or unsaturated alkyl and pharmaceutically acceptable salts, hydrates, solvates and esters thereof.
- Preferred among the presently invented ketone reduction products described above are the secondary alcohols wherein the substituent is in the ß position.
- Particularly preferred among the presently invented ketone reduction products described above are 17ß-(1-hydroxyethyl)-estra-1,3,5(10)-triene-3-carboxylic acid and 17ß-(1-hydroxybutyl)-estra-1,3,5(10)-triene-3-carboyxlic acid.
- These compounds can be made by conventional sodium borohydride reduction of the carbonyl attached to R without epimerization of the 17 substituent or reducing the carboxyl in Ring A or the aromatic A ring.
- the borohydride reduction can be carried out in e.g. water or aqueous methanol, at a temperature of room temperature to 50°C and the product then isolated and purified by conventional means.
- the compounds are also active as dual inhibitors of 5-alpha reductase.
- solvent or "appropriate solvent” as used herein and the in the claims is meant a solvent such as methylene chloride, ethylene chloride,
- dimethylformamide (DMF), hexane, water, pyridine, quinoline or ethanol.
- R is C 1-20 linear or branched, saturated or unsaturated alkyl
- R 4 is fluorosulfonyloxy.
- R is C 1-20 linear or branched, saturated or unsaturated alkyl and pharmaceutically acceptable salts, hydrates, solvates and esters thereof comprises reacting a compound of the formula
- R is as described above with fluorosulfonic anhydride and a base, preferably, 2,5-t-butyl-3-methyl-pyridine, in a solvent, preferably dichloromethane, to form a compound of the formula
- R is as described above and subsequently reacting said compound in a metal-catalyzed coupling reaction in the presence of an approrpirate coupling reagent, preferably, carbon monoxide followed by an optional, if applicable, hydrolysis reaction to form a compound of formula II, and thereafter optionally forming a pharmaceutically acceptable salt, hydrate, solvate or ester thereof.
- an approrpirate coupling reagent preferably, carbon monoxide followed by an optional, if applicable, hydrolysis reaction to form a compound of formula II, and thereafter optionally forming a pharmaceutically acceptable salt, hydrate, solvate or ester thereof.
- the presently invented pharmaceutically active compounds are potent dual inhibitors of steroid 5- ⁇ -reductase activity, they have therapeutic utility in treating diseases and conditions wherein decreases in DHT activity produces the desired therapeutic effect.
- diseases and conditions include acne vulgaris, seborrhea, female hirsutism, male pattern baldness, prostate diseases such as benign prostatic hypertrophy, and prostatic adenocarcinoma.
- Chinese hamster ovary (CHO) cells containing expressed, recombinant human steroid 5 ⁇ -reductase isoenzyme 1 were homogenized in 20 mM potassium phosphate, pH 6.5, buffer containing 0.33 M sucrose, 1 mM
- the suspended paniculate solution was stored at -80°C.
- Frozen human prostates were thawed and minced into small pieces ( Brinkmann Polytron (Sybron Corp., Westbury, New York). The solution was sonicated for 3 to 5 minutes with a Sonifier (Branson Sonic Power Co.) followed by hand homogenization in a Dounce hand homogenizer. Prostatic particles were obtained by differential centrifugation at 600 or 1000 ⁇ g for 20 minutes and 140,000 ⁇ g for 60 minutes at 4°C. The pellet obtained from the 140,000 ⁇ g centrifugation was washed with 5 to 10 tissue volumes of the buffer described above and centrifuged at 140,000 ⁇ g. The resulting pellet was suspended in buffer B and the particulate suspension was stored at -80°C.
- Chinese hamster ovary (CHO) cells containing expressed, recombinant human steroid 5- ⁇ -reductase isozyme 2 were homogenized in 20 mM potassium phosphate, pH 6.5, buffer containing 0.33 M sucrose, ImM dithiothreitol, and 50 ⁇ M NADPH (buffer A) using a Douce hand homogenizer.
- Membrane particulates containing the recombinant human enzyme were isolated by centrifugation
- NADPH buffer B
- Assays for human steroid 5 ⁇ -reductase isoenzyme 1 were conducted with a sample of the recombinant protein expressed in CHO cells in 50 mM phosphate buffer, pH 7.5 while assays of isoenzyme 2 were conducted with a suspension of human prostatic particulates and/or recombinant protein expressed in CHO cells in 50 mM citrate buffer at pH 5.0.
- the radiochemical content in the bands of the substrate and the products was determined with a BIOSCAN Imaging Scanner (Bioscan Inc., Washington, D.C.). The percent of recovered radiolabel converted to product was calculated, from which enzyme activity was determined. All incubations were conducted such that no more than 20% of the substrate (testosterone) was consumed.
- the experimentally obtained data was computer fit to a linear function by plotting the reciprocal of the enzyme activity (l/velocity) against the variable inhibitor concentration; apparent inhibition constants (Ki app) were determined by the Dixon analysis (Dixon, M. (1953).
- Solid or liquid pharmaceutical carriers are employed.
- Solid carriers include, starch, lactose, calcium sulfate dihydrate, terra alba, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, and stearic acid.
- Liquid carriers include syrup, peanut oil, olive oil, saline, and water.
- the carrier or diluent may include any prolonged release material, such as glyceryl
- the preparation will peferably be in the form of a syrup, elixir, emulsion, soft gelatin capsule, sterile injectable liquid such as an ampoule, or an aqueous or nonaqueous liquid suspension.
- the pharmaceutical preparations are made following conventional techniques of a pharmaceutical chemist involving mixing, granulating, and compressing, when necessary, for tablet forms, or mixing, filling and dissolving the ingreidents, as appropriate, to give the desired oral or parenteral products.
- Doses of the presently invented pharmaceutically active compounds in a pharmaceutical dosage unit as described above will be an efficacious, nontoxic quantity preferably selected from the range of
- the selected dose is administered preferably from 1-6 times daily, orally or parenterally.
- Preferred forms of parenteral administration include topically, rectally, transdermally, by injection and continuously by infusion.
- Oral dosage units for human administration preferably contain from 1 to 500 mg of active compound.
- Oral administration, which uses lower dosages is preferred.
- Parenteral administration, at high dosages, however, also can be used when safe and convenient for the patient.
- the method of this invention of inhibiting steroid 5- ⁇ -reductase isozyme 1 and steroid 5- ⁇ -reductase isozyme 2 activity in mammals, including humans, comprises administering to a subject in need of such inhibition an effective dual inhibiting amount of a compound of the present invention.
- the invention also provides for the use of a compound of Formula (I) or a compound of Formula (IV) in the manufacture of a medicament for use in the dual inhibition of steroid 5- ⁇ -reductase.
- the invention also provides for a pharmaceutical composition for use in the treatment of benign prostate hypertrophy which comprises a compound of Formula I or a compound of Formula (IV) and a pharmaceutically acceptable carrier.
- the invention also provides for a pharmaceutical composition for use in the treatment of prostatic adenocarcinoma which comprises a compound of Formula I or a compound of Formula (IV) and a pharmaceutically acceptable carrier.
- the invention also provides for a process for preparing a pharmaceutical composition containing a pharmaceutically acceptable carrier or diluent and a compound of Formula I or a compound of Formula (IV) which comprises bringing the compound of Formula I or the compound of Formula (IV) into association with the pharmaceutically acceptable carrier or diluent.
- the pharmaceutically active compounds of the present invention can be co-administered with further active ingredients, such as other compounds known to treat the disease states of acne vulgaris, sebonhea, female hirsutism, male pattern baldness, benign prostate hypertrophy or prostatic adenocarcinoma or compounds known to have utility when used in combination with 5- ⁇ -reductase inhibitors.
- further active ingredients such as other compounds known to treat the disease states of acne vulgaris, sebonhea, female hirsutism, male pattern baldness, benign prostate hypertrophy or prostatic adenocarcinoma or compounds known to have utility when used in combination with 5- ⁇ -reductase inhibitors.
- Particularly preferred is the co-administration of a dual 5- ⁇ -reductase inhibitor, as disclosed herein, and minoxidil for use in the treatment of male pattern baldness.
- Particularly preferred is the co-administration of a dual 5 ⁇ -reductase inhibitor, as disclosed herein, and a ⁇ -
- a mixture of 17 ⁇ -isobutylcarbonyl-estra-1,3,5(10)-triene-3-fluorosulfonate 1,3-bis(diphenylphosphino)propane, palladium diacetate, triethylamine, methanol, DMSO and 1,2-dichloroethane is heated and vigorously stirred for 5 h at 80°C under an atmosphere of carbon monoxide. After cooling to ambient temperature, the resulting mixture is diluted with dichloromethane. The organic phase is thoroughly washed with water, dried (MgSO 4 ) and evaporated to dryness.
- Example 5 The title compound was prepared according to Example 5 (ii) by substituting trifluoromethyl-17 ⁇ -(ten-pentylcarbonyl)-estra-1,3,5(10)-triene-3-sulfonate, as prepared in Example 6 (i), for trifluoromethyl-17 ⁇ -(octylcarbonyl)-estra-1,3,5(10)-triene-3-sulfonate. mp 199-200°C.
- Example 5 The title compound was prepared according to Example 5 (ii) by substituting trifluoromethyl- 17 ⁇ -(2,2-dimethylpropylcarbonyl)-estra- 1 ,3,5( 10)-triene-3-sulfonate, as prepared in Example 7 (i), for trifluoromethyl- 17 ⁇ -(octylcarbonyl)estra-1,3,5(10)-triene-3-sulfonate. mp 210°C.
- Example 8 Corresponding to Scheme IV
- Methyl-17 ⁇ -cyano-estra-1,3,5(10)-triene-3-carboxylate (0.8 g) was dissolved in 30 ml of toluene and treated with DIBAL (6 ml, 1M). The mixture was stirred at room temp under argon for 2.5 hours. The mixture was then poured into 50 ml of 5% H 2 SO 4 and the mixture was stirred for 1 hour, filited, dried, and concentrated. The residue was chromatographed (silica gel, eluting with 20% EtOAc in hexane) to provide 424 mg of the title compound, mp 146-150°C.
- An oral dosage form for administering Formula I comounds is produced by screening, mixing, and filling into hard gelatin capsules the ingredients in the proportions shown in Table 1, below.
- sucrose, calcium sulfate dihydrate and Formula (I) compound shown in Table II below are mixed and granulated in the proportions shown with a 10% gelatin solution.
- the wet granules are screened, dried, mixed with the starch, talc and stearic acid, screened and compressed into a tablet.
- 17ß-Isobutylcarbonyl-estra-1,3,5(10)-triene-3-carboxylic acid 75 mg, is dispursed in 25 ml of normal saline to prepare an injectable preparation.
Abstract
Description

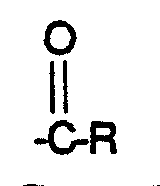

























Claims



















Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP6502660A JPH08500821A (en) | 1992-06-30 | 1993-06-30 | 17-Alkyl ketone steroids useful as 5-α-reductase inhibitors |
| SK1599-94A SK159994A3 (en) | 1992-06-30 | 1993-06-30 | 17-alkylketone steroids useful as 5-alpha-reductase inhibitors |
| EP93915499A EP0651643A4 (en) | 1992-06-30 | 1993-06-30 | 17-ALKYLKETONE STEROIDS USEFUL AS 5--g(a)-REDUCTASE INHIBITORS. |
| AU45459/93A AU4545993A (en) | 1992-06-30 | 1993-06-30 | 17-alkylketone steroids useful as 5-alpha-reductase inhibitors |
| BR9306748A BR9306748A (en) | 1992-06-30 | 1993-06-30 | 17-alkylacetone steroids useful as 5-reductase inhibitors |
| NO945074A NO945074L (en) | 1992-06-30 | 1994-12-29 | 17-Alkylethone steroids useful as 5-itators |
| BG99313A BG99313A (en) | 1992-06-30 | 1994-12-29 | 117-alkylketonesteroids as 5- -reductase inhibitors |
| KR1019940704802A KR950702118A (en) | 1992-06-30 | 1994-12-29 | 17-Alkylketone steroids useful as 5-α-reductase inhibitors |
| FI946189A FI946189A0 (en) | 1992-06-30 | 1994-12-30 | 17-alkyl ketone steroids useful as 5-alpha-reductase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9213835.3 | 1992-06-30 | ||
| GB929213835A GB9213835D0 (en) | 1992-06-30 | 1992-06-30 | Compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994000125A1 true WO1994000125A1 (en) | 1994-01-06 |
Family
ID=10717920
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1993/006238 WO1994000125A1 (en) | 1992-06-30 | 1993-06-30 | 17-ALKYLKETONE STEROIDS USEFUL AS 5-α-REDUCTASE INHIBITORS |
Country Status (23)
| Country | Link |
|---|---|
| EP (1) | EP0651643A4 (en) |
| JP (1) | JPH08500821A (en) |
| KR (1) | KR950702118A (en) |
| CN (1) | CN1095382A (en) |
| AP (1) | AP412A (en) |
| AU (1) | AU4545993A (en) |
| BG (1) | BG99313A (en) |
| BR (1) | BR9306748A (en) |
| CA (1) | CA2138956A1 (en) |
| CZ (1) | CZ334694A3 (en) |
| FI (1) | FI946189A0 (en) |
| GB (1) | GB9213835D0 (en) |
| HU (1) | HUT69409A (en) |
| IL (1) | IL106158A0 (en) |
| MA (1) | MA22927A1 (en) |
| MX (1) | MX9303970A (en) |
| NO (1) | NO945074L (en) |
| OA (1) | OA10122A (en) |
| RU (1) | RU94046378A (en) |
| SI (1) | SI9300350A (en) |
| SK (1) | SK159994A3 (en) |
| WO (1) | WO1994000125A1 (en) |
| ZA (1) | ZA934645B (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5510351A (en) * | 1992-05-20 | 1996-04-23 | Merck & Co., Inc. | Delta-17 and delta-20 olefinic and saturated 17 beta-substituted 4-aza-5 alpha-androstan-ones as 5 alpha reductase inhibitors useful in the prevention and treatment of hyperandrogenic disorders |
| US5543417A (en) * | 1994-10-21 | 1996-08-06 | Merck & Co., Inc. | Combination method of treating acne using 4-AZA-5α-cholestan-ones and 4-AZA-5α-androstan-ones as selective 5α-reductase inhibitors with anti-bacterial, keratolytic, or anti-inflammatory agents |
| US5939570A (en) * | 1991-01-07 | 1999-08-17 | Pherin Corporation | Estrenes for inducing hypothalamic effects |
| WO2012110768A1 (en) | 2011-02-18 | 2012-08-23 | The University Of Birmingham | Therapeutic uses of diarylalkanes such as mitotane |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015129095A (en) | 2013-12-19 | 2015-07-16 | ロレアル | composition |
| CN114805462A (en) * | 2018-02-11 | 2022-07-29 | 江苏豪森药业集团有限公司 | Steroid derivative regulator and preparation method and application thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4954446A (en) * | 1988-05-25 | 1990-09-04 | Smithkline Beecham Corporation | Aromatic steroid 5-α-reductase inhibitors |
-
1992
- 1992-06-30 GB GB929213835A patent/GB9213835D0/en active Pending
-
1993
- 1993-06-26 ZA ZA934645A patent/ZA934645B/en unknown
- 1993-06-28 MA MA23217A patent/MA22927A1/en unknown
- 1993-06-28 IL IL106158A patent/IL106158A0/en unknown
- 1993-06-30 SK SK1599-94A patent/SK159994A3/en unknown
- 1993-06-30 EP EP93915499A patent/EP0651643A4/en not_active Withdrawn
- 1993-06-30 CN CN93109540A patent/CN1095382A/en active Pending
- 1993-06-30 HU HU9403833A patent/HUT69409A/en unknown
- 1993-06-30 CZ CZ943346A patent/CZ334694A3/en unknown
- 1993-06-30 AP APAP/P/1993/000542A patent/AP412A/en active
- 1993-06-30 RU RU94046378/04A patent/RU94046378A/en unknown
- 1993-06-30 MX MX9303970A patent/MX9303970A/en unknown
- 1993-06-30 WO PCT/US1993/006238 patent/WO1994000125A1/en not_active Application Discontinuation
- 1993-06-30 AU AU45459/93A patent/AU4545993A/en not_active Abandoned
- 1993-06-30 JP JP6502660A patent/JPH08500821A/en active Pending
- 1993-06-30 BR BR9306748A patent/BR9306748A/en not_active Application Discontinuation
- 1993-06-30 SI SI9300350A patent/SI9300350A/en unknown
- 1993-06-30 CA CA002138956A patent/CA2138956A1/en not_active Abandoned
-
1994
- 1994-12-29 BG BG99313A patent/BG99313A/en unknown
- 1994-12-29 OA OA60599A patent/OA10122A/en unknown
- 1994-12-29 NO NO945074A patent/NO945074L/en unknown
- 1994-12-29 KR KR1019940704802A patent/KR950702118A/en not_active Application Discontinuation
- 1994-12-30 FI FI946189A patent/FI946189A0/en not_active Application Discontinuation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4954446A (en) * | 1988-05-25 | 1990-09-04 | Smithkline Beecham Corporation | Aromatic steroid 5-α-reductase inhibitors |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5939570A (en) * | 1991-01-07 | 1999-08-17 | Pherin Corporation | Estrenes for inducing hypothalamic effects |
| US6352980B1 (en) | 1991-01-07 | 2002-03-05 | Pherin Pharmaceuticals, Inc. | Estrenes for inducting hypothalamic effects |
| US5510351A (en) * | 1992-05-20 | 1996-04-23 | Merck & Co., Inc. | Delta-17 and delta-20 olefinic and saturated 17 beta-substituted 4-aza-5 alpha-androstan-ones as 5 alpha reductase inhibitors useful in the prevention and treatment of hyperandrogenic disorders |
| US5760045A (en) * | 1992-05-20 | 1998-06-02 | Merck & Co., Inc. | Delta-17 and delta-20 olefinic and saturated 17β-substituted-4-aza-5α-androstan-3-ones as 5α-reductase inhibitors useful in the prevention and treatment of hyperandrogenic disorders |
| US5543417A (en) * | 1994-10-21 | 1996-08-06 | Merck & Co., Inc. | Combination method of treating acne using 4-AZA-5α-cholestan-ones and 4-AZA-5α-androstan-ones as selective 5α-reductase inhibitors with anti-bacterial, keratolytic, or anti-inflammatory agents |
| WO2012110768A1 (en) | 2011-02-18 | 2012-08-23 | The University Of Birmingham | Therapeutic uses of diarylalkanes such as mitotane |
Also Published As
| Publication number | Publication date |
|---|---|
| SI9300350A (en) | 1993-12-31 |
| NO945074L (en) | 1995-02-16 |
| AU4545993A (en) | 1994-01-24 |
| EP0651643A1 (en) | 1995-05-10 |
| CA2138956A1 (en) | 1994-01-06 |
| MX9303970A (en) | 1994-04-29 |
| HUT69409A (en) | 1995-09-28 |
| MA22927A1 (en) | 1994-04-01 |
| FI946189A (en) | 1994-12-30 |
| OA10122A (en) | 1996-12-18 |
| BR9306748A (en) | 1998-12-08 |
| CN1095382A (en) | 1994-11-23 |
| RU94046378A (en) | 1996-10-10 |
| FI946189A0 (en) | 1994-12-30 |
| GB9213835D0 (en) | 1992-08-12 |
| CZ334694A3 (en) | 1995-10-18 |
| IL106158A0 (en) | 1993-10-20 |
| BG99313A (en) | 1995-09-29 |
| AP9300542A0 (en) | 1993-07-31 |
| AP412A (en) | 1995-09-29 |
| NO945074D0 (en) | 1994-12-29 |
| SK159994A3 (en) | 1996-01-10 |
| JPH08500821A (en) | 1996-01-30 |
| ZA934645B (en) | 1994-01-25 |
| EP0651643A4 (en) | 1995-10-04 |
| KR950702118A (en) | 1995-06-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA1319362C (en) | 4-substituted androstendione derivatives and process for their preparation | |
| JP2675418B2 (en) | Aromatic steroid 5-α-reductase inhibitor | |
| US5032586A (en) | 7-keto or hydroxy 3,5-diene steroids as inhibitors of steroid 5-alpha reductase | |
| US5041433A (en) | 11-keto or hydroxy 3,5-diene steroids as inhibitors of steriod 5-α-reductase | |
| US5527806A (en) | 17α and 17β substituted acyl 4 aza steroids | |
| US5137882A (en) | Steroidal 3-acetic acid derivatives as 5-alpha-reductase inhibitors | |
| EP0673251A1 (en) | 17 substituted acyl-3-carboxy 3,5-diene steroidals as 5-alpha-reductase inhibitors | |
| AP412A (en) | 17a and 17B-Alkylketone-3-carboxyaromatic a ring steroidal compounds and pharmaceutical compositions containing these compounds. | |
| JPH05194582A (en) | 17 beta-acyl-3-carboxyandrosta-3,5-diene as testosterone 5alpha-reductase inhibitor | |
| US5641877A (en) | 17-α and 17-β substituted acyl-3-carboxy-3, 5-dienes and use in inhibiting 5-α-reductase | |
| US5618806A (en) | 17α and 17β-substituted estra-1,3,5(10)-triene-3-carbboxlic acid | |
| US5683995A (en) | 17 substituted acyl-3-carboxy 3,5-diene steroidals as α-reductase inhibitors | |
| WO1995021185A1 (en) | Acyl 3-carboxy aromatic a ring steroid as 5-alpha reductase inhibitors | |
| WO1994000121A1 (en) | 17-acyl-4-aza-steroid-3-one as 5-alpha-reductase inhibitors | |
| WO1995028413A1 (en) | 17β-SUBSTITUTED 3-CARBOXY STEROIDS THAT INHIBIT 5-α-REDUCTASE | |
| JPH08503474A (en) | Use in the inhibition of 17-alpha and 17-beta substituted acyl-3-carboxy-3,5-dienes and 5-alpha-reductases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR BY CA CZ FI HU JP KP KR KZ LK MG MN MW NO NZ PL RO RU SD SK UA US VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2138956 Country of ref document: CA Ref document number: 253968 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 159994 Country of ref document: SK |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 94-02119 Country of ref document: RO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1993915499 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1994-3346 Country of ref document: CZ Ref document number: 946189 Country of ref document: FI |
|
| ENP | Entry into the national phase |
Ref document number: 1995 351327 Country of ref document: US Date of ref document: 19950209 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993915499 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1994-3346 Country of ref document: CZ |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV1994-3346 Country of ref document: CZ |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993915499 Country of ref document: EP |