WO1993018160A1 - Anti-viral fusion peptides - Google Patents
Anti-viral fusion peptides Download PDFInfo
- Publication number
- WO1993018160A1 WO1993018160A1 PCT/GB1993/000505 GB9300505W WO9318160A1 WO 1993018160 A1 WO1993018160 A1 WO 1993018160A1 GB 9300505 W GB9300505 W GB 9300505W WO 9318160 A1 WO9318160 A1 WO 9318160A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- peptide
- fusion
- derived
- binding
- hiv
- Prior art date
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims abstract description 278
- 102000004196 processed proteins & peptides Human genes 0.000 title claims abstract description 72
- 230000004927 fusion Effects 0.000 title claims description 90
- 230000000840 anti-viral effect Effects 0.000 title description 3
- 238000009739 binding Methods 0.000 claims abstract description 112
- 241000700605 Viruses Species 0.000 claims abstract description 45
- 210000004369 blood Anatomy 0.000 claims abstract description 36
- 239000008280 blood Substances 0.000 claims abstract description 36
- 201000004792 malaria Diseases 0.000 claims abstract description 32
- 210000003936 merozoite Anatomy 0.000 claims abstract description 28
- 230000003612 virological effect Effects 0.000 claims abstract description 25
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 claims description 142
- 241000725303 Human immunodeficiency virus Species 0.000 claims description 56
- 210000003743 erythrocyte Anatomy 0.000 claims description 45
- 108010041397 CD4 Antigens Proteins 0.000 claims description 43
- 108091005250 Glycophorins Proteins 0.000 claims description 39
- 150000001413 amino acids Chemical class 0.000 claims description 38
- 239000003795 chemical substances by application Substances 0.000 claims description 37
- 102000028180 Glycophorins Human genes 0.000 claims description 36
- 210000004899 c-terminal region Anatomy 0.000 claims description 34
- 125000000539 amino acid group Chemical group 0.000 claims description 27
- 239000003814 drug Substances 0.000 claims description 27
- 108090000623 proteins and genes Proteins 0.000 claims description 26
- 238000005304 joining Methods 0.000 claims description 20
- 238000011282 treatment Methods 0.000 claims description 18
- 101001001336 Homo sapiens Guanylate-binding protein 1 Proteins 0.000 claims description 17
- 102100035688 Guanylate-binding protein 1 Human genes 0.000 claims description 15
- 244000045947 parasite Species 0.000 claims description 13
- 229960005486 vaccine Drugs 0.000 claims description 13
- 208000031886 HIV Infections Diseases 0.000 claims description 12
- 108010033276 Peptide Fragments Proteins 0.000 claims description 12
- 102000007079 Peptide Fragments Human genes 0.000 claims description 12
- 230000001225 therapeutic effect Effects 0.000 claims description 11
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 10
- 101710094214 Duffy receptor Proteins 0.000 claims description 8
- 239000003433 contraceptive agent Substances 0.000 claims description 8
- 101100067996 Mus musculus Gbp1 gene Proteins 0.000 claims description 7
- 102000005962 receptors Human genes 0.000 claims description 7
- 108020003175 receptors Proteins 0.000 claims description 7
- OBMZMSLWNNWEJA-XNCRXQDQSA-N C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 Chemical compound C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 OBMZMSLWNNWEJA-XNCRXQDQSA-N 0.000 claims description 6
- 101710176384 Peptide 1 Proteins 0.000 claims description 6
- 239000006071 cream Substances 0.000 claims description 6
- 238000012360 testing method Methods 0.000 claims description 6
- 208000037357 HIV infectious disease Diseases 0.000 claims description 5
- 241000713772 Human immunodeficiency virus 1 Species 0.000 claims description 5
- 239000006260 foam Substances 0.000 claims description 5
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 claims description 5
- 238000011002 quantification Methods 0.000 claims description 5
- 241000894007 species Species 0.000 claims description 5
- VZQHRKZCAZCACO-PYJNHQTQSA-N (2s)-2-[[(2s)-2-[2-[[(2s)-2-[[(2s)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]propanoyl]amino]prop-2-enoylamino]-3-methylbutanoyl]amino]propanoic acid Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](C(C)C)NC(=O)C(=C)NC(=O)[C@H](C)NC(=O)[C@@H](N)CCCNC(N)=N VZQHRKZCAZCACO-PYJNHQTQSA-N 0.000 claims description 4
- 108060003951 Immunoglobulin Proteins 0.000 claims description 4
- 241000224016 Plasmodium Species 0.000 claims description 4
- 101900073696 Plasmodium vivax Duffy receptor Proteins 0.000 claims description 4
- 230000005540 biological transmission Effects 0.000 claims description 4
- 229940124558 contraceptive agent Drugs 0.000 claims description 4
- 230000002254 contraceptive effect Effects 0.000 claims description 4
- 239000003889 eye drop Substances 0.000 claims description 4
- 229940012356 eye drops Drugs 0.000 claims description 4
- 102000018358 immunoglobulin Human genes 0.000 claims description 4
- 101710203310 Apical membrane antigen 1 Proteins 0.000 claims description 3
- 108020004707 nucleic acids Proteins 0.000 claims description 3
- 102000039446 nucleic acids Human genes 0.000 claims description 3
- 230000002265 prevention Effects 0.000 claims description 3
- 101710110110 Glycophorin-binding protein Proteins 0.000 claims description 2
- 101100005713 Homo sapiens CD4 gene Proteins 0.000 claims description 2
- 241000224017 Plasmodium berghei Species 0.000 claims description 2
- 241000224024 Plasmodium chabaudi Species 0.000 claims description 2
- 230000009286 beneficial effect Effects 0.000 claims description 2
- 230000003111 delayed effect Effects 0.000 claims description 2
- 229910052757 nitrogen Inorganic materials 0.000 claims description 2
- 101710145634 Antigen 1 Proteins 0.000 claims 2
- 108010057081 Merozoite Surface Protein 1 Proteins 0.000 claims 2
- 208000002672 hepatitis B Diseases 0.000 claims 2
- 208000005176 Hepatitis C Diseases 0.000 claims 1
- 208000005331 Hepatitis D Diseases 0.000 claims 1
- 241000223992 Plasmodium gallinaceum Species 0.000 claims 1
- 108070000030 Viral receptors Proteins 0.000 claims 1
- 238000001514 detection method Methods 0.000 claims 1
- 235000015110 jellies Nutrition 0.000 claims 1
- 239000008274 jelly Substances 0.000 claims 1
- 239000007921 spray Substances 0.000 claims 1
- 210000004027 cell Anatomy 0.000 abstract description 100
- 238000000034 method Methods 0.000 description 52
- 239000012634 fragment Substances 0.000 description 37
- 108091028043 Nucleic acid sequence Proteins 0.000 description 24
- 238000004519 manufacturing process Methods 0.000 description 20
- 239000002245 particle Substances 0.000 description 18
- 208000030507 AIDS Diseases 0.000 description 16
- 108020004414 DNA Proteins 0.000 description 15
- 239000000427 antigen Substances 0.000 description 15
- 102000036639 antigens Human genes 0.000 description 15
- 108091007433 antigens Proteins 0.000 description 15
- 241000282414 Homo sapiens Species 0.000 description 14
- 125000003275 alpha amino acid group Chemical group 0.000 description 14
- 230000000670 limiting effect Effects 0.000 description 14
- 208000015181 infectious disease Diseases 0.000 description 13
- 239000000047 product Substances 0.000 description 13
- YSMPVONNIWLJML-FXQIFTODSA-N Ala-Asp-Pro Chemical compound C[C@H](N)C(=O)N[C@@H](CC(O)=O)C(=O)N1CCC[C@H]1C(O)=O YSMPVONNIWLJML-FXQIFTODSA-N 0.000 description 12
- 101100228463 Plasmodium falciparum (isolate FCBR / Columbia) GBPH gene Proteins 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- UFRXVQGGPNSJRY-CYDGBPFRSA-N Ile-Met-Arg Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CCSC)C(=O)N[C@H](C(O)=O)CCCN=C(N)N UFRXVQGGPNSJRY-CYDGBPFRSA-N 0.000 description 10
- 108010013835 arginine glutamate Proteins 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 108010043277 recombinant soluble CD4 Proteins 0.000 description 10
- HVKAAUOFFTUSAA-XDTLVQLUSA-N Glu-Tyr-Ala Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](C)C(O)=O HVKAAUOFFTUSAA-XDTLVQLUSA-N 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 238000004587 chromatography analysis Methods 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 230000006870 function Effects 0.000 description 9
- 239000000499 gel Substances 0.000 description 9
- 244000052769 pathogen Species 0.000 description 9
- 229920000642 polymer Polymers 0.000 description 9
- 239000000126 substance Substances 0.000 description 9
- 238000006467 substitution reaction Methods 0.000 description 9
- 108010056243 alanylalanine Proteins 0.000 description 8
- 108010041407 alanylaspartic acid Proteins 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 108010044348 lysyl-glutamyl-aspartic acid Proteins 0.000 description 8
- 241000223810 Plasmodium vivax Species 0.000 description 7
- 238000013459 approach Methods 0.000 description 7
- 108010068380 arginylarginine Proteins 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 230000007969 cellular immunity Effects 0.000 description 7
- 238000010367 cloning Methods 0.000 description 7
- 125000005647 linker group Chemical group 0.000 description 7
- 230000007246 mechanism Effects 0.000 description 7
- 239000000523 sample Substances 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- RZVVKNIACROXRM-ZLUOBGJFSA-N Asn-Ala-Asp Chemical compound C[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)O)NC(=O)[C@H](CC(=O)N)N RZVVKNIACROXRM-ZLUOBGJFSA-N 0.000 description 6
- 241000894006 Bacteria Species 0.000 description 6
- -1 Boc-amino Chemical group 0.000 description 6
- WTMZXOPHTIVFCP-QEWYBTABSA-N Glu-Ile-Phe Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 WTMZXOPHTIVFCP-QEWYBTABSA-N 0.000 description 6
- NTHIHAUEXVTXQG-KKUMJFAQSA-N Glu-Tyr-Arg Chemical compound C1=CC(=CC=C1C[C@@H](C(=O)N[C@@H](CCCN=C(N)N)C(=O)O)NC(=O)[C@H](CCC(=O)O)N)O NTHIHAUEXVTXQG-KKUMJFAQSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- AEDWWMMHUGYIFD-HJGDQZAQSA-N Leu-Thr-Asn Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(O)=O AEDWWMMHUGYIFD-HJGDQZAQSA-N 0.000 description 6
- OWRUUFUVXFREBD-KKUMJFAQSA-N Lys-His-Leu Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC(C)C)C(O)=O OWRUUFUVXFREBD-KKUMJFAQSA-N 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- 241000223960 Plasmodium falciparum Species 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 210000000805 cytoplasm Anatomy 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 239000012528 membrane Substances 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- 102000004169 proteins and genes Human genes 0.000 description 6
- DCGLNNVKIZXQOJ-FXQIFTODSA-N Arg-Asn-Ala Chemical compound C[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CCCN=C(N)N)N DCGLNNVKIZXQOJ-FXQIFTODSA-N 0.000 description 5
- FBODFHMLALOPHP-GUBZILKMSA-N Asn-Lys-Glu Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(O)=O FBODFHMLALOPHP-GUBZILKMSA-N 0.000 description 5
- BWJZSLQJNBSUPM-FXQIFTODSA-N Asp-Pro-Asn Chemical compound OC(=O)C[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CC(N)=O)C(O)=O BWJZSLQJNBSUPM-FXQIFTODSA-N 0.000 description 5
- AHWRSSLYSGLBGD-CIUDSAMLSA-N Asp-Pro-Glu Chemical compound OC(=O)C[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(O)=O)C(O)=O AHWRSSLYSGLBGD-CIUDSAMLSA-N 0.000 description 5
- 239000004971 Cross linker Substances 0.000 description 5
- 208000012266 Needlestick injury Diseases 0.000 description 5
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 5
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 5
- JXKMXEBNZCKSDY-JIOCBJNQSA-N Thr-Asp-Pro Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)N1CCC[C@@H]1C(=O)O)N)O JXKMXEBNZCKSDY-JIOCBJNQSA-N 0.000 description 5
- BRPKEERLGYNCNC-NHCYSSNCSA-N Val-Glu-Arg Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H](C(O)=O)CCCN=C(N)N BRPKEERLGYNCNC-NHCYSSNCSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 208000007502 anemia Diseases 0.000 description 5
- 210000000612 antigen-presenting cell Anatomy 0.000 description 5
- 238000005119 centrifugation Methods 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 210000000987 immune system Anatomy 0.000 description 5
- 238000002156 mixing Methods 0.000 description 5
- 230000001717 pathogenic effect Effects 0.000 description 5
- 108010070643 prolylglutamic acid Proteins 0.000 description 5
- 230000004224 protection Effects 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- 229940124597 therapeutic agent Drugs 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- JDHOJQJMWBKHDB-CIUDSAMLSA-N Asp-Asn-Lys Chemical compound C(CCN)C[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CC(=O)O)N JDHOJQJMWBKHDB-CIUDSAMLSA-N 0.000 description 4
- YDJVIBMKAMQPPP-LAEOZQHASA-N Asp-Glu-Val Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(O)=O YDJVIBMKAMQPPP-LAEOZQHASA-N 0.000 description 4
- ZRUBWRCKIVDCFS-XPCJQDJLSA-N Asp-Leu-Thr-Ser Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(O)=O ZRUBWRCKIVDCFS-XPCJQDJLSA-N 0.000 description 4
- WOMUDRVDJMHTCV-DCAQKATOSA-N Glu-Arg-Arg Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O WOMUDRVDJMHTCV-DCAQKATOSA-N 0.000 description 4
- 241000238631 Hexapoda Species 0.000 description 4
- 108091092878 Microsatellite Proteins 0.000 description 4
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- PQLXHSACXPGWPD-GSSVUCPTSA-N Thr-Asn-Thr Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O PQLXHSACXPGWPD-GSSVUCPTSA-N 0.000 description 4
- GYKDRHDMGQUZPU-MGHWNKPDSA-N Tyr-Lys-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC1=CC=C(C=C1)O)N GYKDRHDMGQUZPU-MGHWNKPDSA-N 0.000 description 4
- 239000008186 active pharmaceutical agent Substances 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 210000000170 cell membrane Anatomy 0.000 description 4
- 238000009833 condensation Methods 0.000 description 4
- 230000005494 condensation Effects 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 238000000605 extraction Methods 0.000 description 4
- 230000036541 health Effects 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 230000009545 invasion Effects 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 150000007523 nucleic acids Chemical class 0.000 description 4
- 238000005498 polishing Methods 0.000 description 4
- GKBMIFPNPOSTHB-BJBKLNMKSA-N recombinant soluble cd4 Chemical compound NC(=O)C[C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CS)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(O)=O GKBMIFPNPOSTHB-BJBKLNMKSA-N 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 210000000952 spleen Anatomy 0.000 description 4
- 241000701447 unidentified baculovirus Species 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 102220479950 Alkaline phosphatase, germ cell type_D63A_mutation Human genes 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- 241000255925 Diptera Species 0.000 description 3
- 208000035874 Excoriation Diseases 0.000 description 3
- NJCALAAIGREHDR-WDCWCFNPSA-N Glu-Leu-Thr Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O NJCALAAIGREHDR-WDCWCFNPSA-N 0.000 description 3
- LZEUDRYSAZAJIO-AUTRQRHGSA-N Glu-Val-Glu Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O LZEUDRYSAZAJIO-AUTRQRHGSA-N 0.000 description 3
- 206010018910 Haemolysis Diseases 0.000 description 3
- OQDLKDUVMTUPPG-AVGNSLFASA-N His-Leu-Glu Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O OQDLKDUVMTUPPG-AVGNSLFASA-N 0.000 description 3
- LDFWDDVELNOGII-MXAVVETBSA-N His-Lys-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC1=CN=CN1)N LDFWDDVELNOGII-MXAVVETBSA-N 0.000 description 3
- ZXFRGTAIIZHNHG-AJNGGQMLSA-N Lys-Ile-Leu Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(C)C)C(=O)O)NC(=O)[C@H](CCCCN)N ZXFRGTAIIZHNHG-AJNGGQMLSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 206010057249 Phagocytosis Diseases 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 230000024932 T cell mediated immunity Effects 0.000 description 3
- GFZQWWDXJVGEMW-ULQDDVLXSA-N Tyr-Arg-Lys Chemical compound C1=CC(=CC=C1C[C@@H](C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCCCN)C(=O)O)N)O GFZQWWDXJVGEMW-ULQDDVLXSA-N 0.000 description 3
- 208000027418 Wounds and injury Diseases 0.000 description 3
- 238000005299 abrasion Methods 0.000 description 3
- 238000001042 affinity chromatography Methods 0.000 description 3
- 230000036436 anti-hiv Effects 0.000 description 3
- 108010093581 aspartyl-proline Proteins 0.000 description 3
- 108010038633 aspartylglutamate Proteins 0.000 description 3
- 230000004888 barrier function Effects 0.000 description 3
- SQVRNKJHWKZAKO-UHFFFAOYSA-N beta-N-Acetyl-D-neuraminic acid Natural products CC(=O)NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO SQVRNKJHWKZAKO-UHFFFAOYSA-N 0.000 description 3
- 238000012512 characterization method Methods 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 230000001086 cytosolic effect Effects 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 230000034994 death Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 210000003617 erythrocyte membrane Anatomy 0.000 description 3
- 239000013604 expression vector Substances 0.000 description 3
- 238000002523 gelfiltration Methods 0.000 description 3
- 108010049041 glutamylalanine Proteins 0.000 description 3
- 230000008588 hemolysis Effects 0.000 description 3
- 230000002209 hydrophobic effect Effects 0.000 description 3
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 210000003000 inclusion body Anatomy 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 238000003780 insertion Methods 0.000 description 3
- 230000037431 insertion Effects 0.000 description 3
- 210000003292 kidney cell Anatomy 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 3
- 230000008782 phagocytosis Effects 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 239000013612 plasmid Substances 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 239000012521 purified sample Substances 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 108010026333 seryl-proline Proteins 0.000 description 3
- SQVRNKJHWKZAKO-OQPLDHBCSA-N sialic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)OC1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-OQPLDHBCSA-N 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 2
- OSJPPGNTCRNQQC-UWTATZPHSA-N 3-phospho-D-glyceric acid Chemical compound OC(=O)[C@H](O)COP(O)(O)=O OSJPPGNTCRNQQC-UWTATZPHSA-N 0.000 description 2
- XVVOVPFMILMHPX-ZLUOBGJFSA-N Asn-Asp-Asp Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(O)=O)C(O)=O XVVOVPFMILMHPX-ZLUOBGJFSA-N 0.000 description 2
- BHQQRVARKXWXPP-ACZMJKKPSA-N Asn-Asp-Glu Chemical compound C(CC(=O)O)[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CC(=O)N)N BHQQRVARKXWXPP-ACZMJKKPSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 241000238586 Cirripedia Species 0.000 description 2
- 241000723655 Cowpea mosaic virus Species 0.000 description 2
- OABOXRPGTFRBFZ-IMJSIDKUSA-N Cys-Cys Chemical compound SC[C@H](N)C(=O)N[C@@H](CS)C(O)=O OABOXRPGTFRBFZ-IMJSIDKUSA-N 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 101710196841 Erythrocyte-binding antigen 175 Proteins 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 206010061598 Immunodeficiency Diseases 0.000 description 2
- 208000029462 Immunodeficiency disease Diseases 0.000 description 2
- 108091092195 Intron Proteins 0.000 description 2
- 241000880493 Leptailurus serval Species 0.000 description 2
- ILDSIMPXNFWKLH-KATARQTJSA-N Leu-Thr-Ser Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(O)=O ILDSIMPXNFWKLH-KATARQTJSA-N 0.000 description 2
- 102000003960 Ligases Human genes 0.000 description 2
- 108090000364 Ligases Proteins 0.000 description 2
- 241000282560 Macaca mulatta Species 0.000 description 2
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- SITLTJHOQZFJGG-UHFFFAOYSA-N N-L-alpha-glutamyl-L-valine Natural products CC(C)C(C(O)=O)NC(=O)C(N)CCC(O)=O SITLTJHOQZFJGG-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 101000895911 Plasmodium falciparum (isolate Camp / Malaysia) Erythrocyte-binding antigen 175 Proteins 0.000 description 2
- 241000223801 Plasmodium knowlesi Species 0.000 description 2
- SMCHPSMKAFIERP-FXQIFTODSA-N Pro-Asn-Asp Chemical compound OC(=O)C[C@@H](C(O)=O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@@H]1CCCN1 SMCHPSMKAFIERP-FXQIFTODSA-N 0.000 description 2
- GHPQVUYZQQGEDA-BIIVOSGPSA-N Ser-Asp-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CC(=O)O)NC(=O)[C@H](CO)N)C(=O)O GHPQVUYZQQGEDA-BIIVOSGPSA-N 0.000 description 2
- BTPAWKABYQMKKN-LKXGYXEUSA-N Ser-Asp-Thr Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O BTPAWKABYQMKKN-LKXGYXEUSA-N 0.000 description 2
- PPCZVWHJWJFTFN-ZLUOBGJFSA-N Ser-Ser-Asp Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(O)=O PPCZVWHJWJFTFN-ZLUOBGJFSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- 239000012317 TBTU Substances 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- FWTFAZKJORVTIR-VZFHVOOUSA-N Thr-Ser-Ala Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(O)=O FWTFAZKJORVTIR-VZFHVOOUSA-N 0.000 description 2
- VCXWRWYFJLXITF-AUTRQRHGSA-N Tyr-Ala-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 VCXWRWYFJLXITF-AUTRQRHGSA-N 0.000 description 2
- PZTZYZUTCPZWJH-FXQIFTODSA-N Val-Ser-Ser Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)O)N PZTZYZUTCPZWJH-FXQIFTODSA-N 0.000 description 2
- 206010058874 Viraemia Diseases 0.000 description 2
- 206010052428 Wound Diseases 0.000 description 2
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 101150115889 al gene Proteins 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 238000004873 anchoring Methods 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 239000010836 blood and blood product Substances 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- 108010004073 cysteinylcysteine Proteins 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- FSXRLASFHBWESK-UHFFFAOYSA-N dipeptide phenylalanyl-tyrosine Natural products C=1C=C(O)C=CC=1CC(C(O)=O)NC(=O)C(N)CC1=CC=CC=C1 FSXRLASFHBWESK-UHFFFAOYSA-N 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- QQFBQBDINHJDMN-UHFFFAOYSA-N ethyl 2-trimethylsilylacetate Chemical compound CCOC(=O)C[Si](C)(C)C QQFBQBDINHJDMN-UHFFFAOYSA-N 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 108010073628 glutamyl-valyl-phenylalanine Proteins 0.000 description 2
- 230000013595 glycosylation Effects 0.000 description 2
- 238000006206 glycosylation reaction Methods 0.000 description 2
- 108010015792 glycyllysine Proteins 0.000 description 2
- 238000011194 good manufacturing practice Methods 0.000 description 2
- 238000005534 hematocrit Methods 0.000 description 2
- 108010092114 histidylphenylalanine Proteins 0.000 description 2
- 230000036737 immune function Effects 0.000 description 2
- 230000007813 immunodeficiency Effects 0.000 description 2
- 230000002163 immunogen Effects 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000005342 ion exchange Methods 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 239000006210 lotion Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 108010009298 lysylglutamic acid Proteins 0.000 description 2
- 108010054155 lysyllysine Proteins 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 230000008774 maternal effect Effects 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 229920002401 polyacrylamide Polymers 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000000630 rising effect Effects 0.000 description 2
- 102220095193 rs745439506 Human genes 0.000 description 2
- 210000003079 salivary gland Anatomy 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 125000006850 spacer group Chemical group 0.000 description 2
- 238000010911 splenectomy Methods 0.000 description 2
- 229910021653 sulphate ion Inorganic materials 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 108010061238 threonyl-glycine Proteins 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 239000013598 vector Substances 0.000 description 2
- 238000012795 verification Methods 0.000 description 2
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- VVSASNKOFCZVES-UHFFFAOYSA-N 1,3-dimethyl-1,3-diazinane-2,4,6-trione Chemical compound CN1C(=O)CC(=O)N(C)C1=O VVSASNKOFCZVES-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- WJMFXQBNYLYADA-UHFFFAOYSA-N 1-(3,4-dihydroxyphenyl)-6,7-dihydroxy-1,2-dihydronaphthalene-2,3-dicarboxylic acid Chemical compound C12=CC(O)=C(O)C=C2C=C(C(O)=O)C(C(=O)O)C1C1=CC=C(O)C(O)=C1 WJMFXQBNYLYADA-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- AVFZOVWCLRSYKC-UHFFFAOYSA-N 1-methylpyrrolidine Chemical compound CN1CCCC1 AVFZOVWCLRSYKC-UHFFFAOYSA-N 0.000 description 1
- GZCWLCBFPRFLKL-UHFFFAOYSA-N 1-prop-2-ynoxypropan-2-ol Chemical compound CC(O)COCC#C GZCWLCBFPRFLKL-UHFFFAOYSA-N 0.000 description 1
- UAIUNKRWKOVEES-UHFFFAOYSA-N 3,3',5,5'-tetramethylbenzidine Chemical compound CC1=C(N)C(C)=CC(C=2C=C(C)C(N)=C(C)C=2)=C1 UAIUNKRWKOVEES-UHFFFAOYSA-N 0.000 description 1
- FTSFSHHUBPYPIE-UHFFFAOYSA-N 5-(azidomethyl)-1h-pyrimidine-2,4-dione Chemical compound [N-]=[N+]=NCC1=CNC(=O)NC1=O FTSFSHHUBPYPIE-UHFFFAOYSA-N 0.000 description 1
- 102000013563 Acid Phosphatase Human genes 0.000 description 1
- 108010051457 Acid Phosphatase Proteins 0.000 description 1
- NWVVKQZOVSTDBQ-CIUDSAMLSA-N Ala-Glu-Arg Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O NWVVKQZOVSTDBQ-CIUDSAMLSA-N 0.000 description 1
- CWEAKSWWKHGTRJ-BQBZGAKWSA-N Ala-Gly-Met Chemical compound [H]N[C@@H](C)C(=O)NCC(=O)N[C@@H](CCSC)C(O)=O CWEAKSWWKHGTRJ-BQBZGAKWSA-N 0.000 description 1
- CCDFBRZVTDDJNM-GUBZILKMSA-N Ala-Leu-Glu Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O CCDFBRZVTDDJNM-GUBZILKMSA-N 0.000 description 1
- OYJCVIGKMXUVKB-GARJFASQSA-N Ala-Leu-Pro Chemical compound C[C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N1CCC[C@@H]1C(=O)O)N OYJCVIGKMXUVKB-GARJFASQSA-N 0.000 description 1
- OINVDEKBKBCPLX-JXUBOQSCSA-N Ala-Lys-Thr Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O OINVDEKBKBCPLX-JXUBOQSCSA-N 0.000 description 1
- MDNAVFBZPROEHO-UHFFFAOYSA-N Ala-Lys-Val Natural products CC(C)C(C(O)=O)NC(=O)C(NC(=O)C(C)N)CCCCN MDNAVFBZPROEHO-UHFFFAOYSA-N 0.000 description 1
- KLALXKYLOMZDQT-ZLUOBGJFSA-N Ala-Ser-Asn Chemical compound C[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CC(N)=O KLALXKYLOMZDQT-ZLUOBGJFSA-N 0.000 description 1
- NCQMBSJGJMYKCK-ZLUOBGJFSA-N Ala-Ser-Ser Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(O)=O NCQMBSJGJMYKCK-ZLUOBGJFSA-N 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 241000252073 Anguilliformes Species 0.000 description 1
- OGUPCHKBOKJFMA-SRVKXCTJSA-N Arg-Glu-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)CCCN=C(N)N OGUPCHKBOKJFMA-SRVKXCTJSA-N 0.000 description 1
- DIIGDGJKTMLQQW-IHRRRGAJSA-N Arg-Lys-His Chemical compound C1=C(NC=N1)C[C@@H](C(=O)O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCN=C(N)N)N DIIGDGJKTMLQQW-IHRRRGAJSA-N 0.000 description 1
- KMFPQTITXUKJOV-DCAQKATOSA-N Arg-Ser-Leu Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(O)=O KMFPQTITXUKJOV-DCAQKATOSA-N 0.000 description 1
- QLSRIZIDQXDQHK-RCWTZXSCSA-N Arg-Val-Thr Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O QLSRIZIDQXDQHK-RCWTZXSCSA-N 0.000 description 1
- 102100026376 Artemin Human genes 0.000 description 1
- LJUOLNXOWSWGKF-ACZMJKKPSA-N Asn-Asn-Glu Chemical compound C(CC(=O)O)[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CC(=O)N)N LJUOLNXOWSWGKF-ACZMJKKPSA-N 0.000 description 1
- YVXRYLVELQYAEQ-SRVKXCTJSA-N Asn-Leu-Lys Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CC(=O)N)N YVXRYLVELQYAEQ-SRVKXCTJSA-N 0.000 description 1
- NLDNNZKUSLAYFW-NHCYSSNCSA-N Asn-Lys-Val Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(O)=O NLDNNZKUSLAYFW-NHCYSSNCSA-N 0.000 description 1
- YUOXLJYVSZYPBJ-CIUDSAMLSA-N Asn-Pro-Glu Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(O)=O)C(O)=O YUOXLJYVSZYPBJ-CIUDSAMLSA-N 0.000 description 1
- HPASIOLTWSNMFB-OLHMAJIHSA-N Asn-Thr-Asp Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(O)=O HPASIOLTWSNMFB-OLHMAJIHSA-N 0.000 description 1
- OERMIMJQPQUIPK-FXQIFTODSA-N Asp-Arg-Ala Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(O)=O OERMIMJQPQUIPK-FXQIFTODSA-N 0.000 description 1
- UAXIKORUDGGIGA-DCAQKATOSA-N Asp-Pro-Lys Chemical compound C1C[C@H](N(C1)C(=O)[C@H](CC(=O)O)N)C(=O)N[C@@H](CCCCN)C(=O)O UAXIKORUDGGIGA-DCAQKATOSA-N 0.000 description 1
- ZBYLEBZCVKLPCY-FXQIFTODSA-N Asp-Ser-Arg Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O ZBYLEBZCVKLPCY-FXQIFTODSA-N 0.000 description 1
- KGHLGJAXYSVNJP-WHFBIAKZSA-N Asp-Ser-Gly Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CO)C(=O)NCC(O)=O KGHLGJAXYSVNJP-WHFBIAKZSA-N 0.000 description 1
- ITGFVUYOLWBPQW-KKHAAJSZSA-N Asp-Thr-Val Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(O)=O ITGFVUYOLWBPQW-KKHAAJSZSA-N 0.000 description 1
- 206010003645 Atopy Diseases 0.000 description 1
- 241000701822 Bovine papillomavirus Species 0.000 description 1
- 108091028026 C-DNA Proteins 0.000 description 1
- 108010041884 CD4 Immunoadhesins Proteins 0.000 description 1
- 101100234822 Caenorhabditis elegans ltd-1 gene Proteins 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 241000282552 Chlorocebus aethiops Species 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- GEEXORWTBTUOHC-FXQIFTODSA-N Cys-Arg-Ser Chemical compound C(C[C@@H](C(=O)N[C@@H](CO)C(=O)O)NC(=O)[C@H](CS)N)CN=C(N)N GEEXORWTBTUOHC-FXQIFTODSA-N 0.000 description 1
- WVLZTXGTNGHPBO-SRVKXCTJSA-N Cys-Leu-Leu Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O WVLZTXGTNGHPBO-SRVKXCTJSA-N 0.000 description 1
- NRVQLLDIJJEIIZ-VZFHVOOUSA-N Cys-Thr-Ala Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](C)C(=O)O)NC(=O)[C@H](CS)N)O NRVQLLDIJJEIIZ-VZFHVOOUSA-N 0.000 description 1
- KFYPRIGJTICABD-XGEHTFHBSA-N Cys-Thr-Val Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](C(C)C)C(=O)O)NC(=O)[C@H](CS)N)O KFYPRIGJTICABD-XGEHTFHBSA-N 0.000 description 1
- 101710088194 Dehydrogenase Proteins 0.000 description 1
- 241000243694 Diodia vein chlorosis virus Species 0.000 description 1
- 102100029108 Elongation factor 1-alpha 2 Human genes 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102220508665 Ephrin type-A receptor 4_Q40A_mutation Human genes 0.000 description 1
- 108010074604 Epoetin Alfa Proteins 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- 241001539473 Euphoria Species 0.000 description 1
- 206010015535 Euphoric mood Diseases 0.000 description 1
- 102000001690 Factor VIII Human genes 0.000 description 1
- 108010054218 Factor VIII Proteins 0.000 description 1
- 208000036119 Frailty Diseases 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 101150048348 GP41 gene Proteins 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- JJKKWYQVHRUSDG-GUBZILKMSA-N Glu-Ala-Lys Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(O)=O JJKKWYQVHRUSDG-GUBZILKMSA-N 0.000 description 1
- RJONUNZIMUXUOI-GUBZILKMSA-N Glu-Asn-Lys Chemical compound C(CCN)C[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CCC(=O)O)N RJONUNZIMUXUOI-GUBZILKMSA-N 0.000 description 1
- JVSBYEDSSRZQGV-GUBZILKMSA-N Glu-Asp-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](N)CCC(O)=O JVSBYEDSSRZQGV-GUBZILKMSA-N 0.000 description 1
- QJCKNLPMTPXXEM-AUTRQRHGSA-N Glu-Glu-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)CCC(O)=O QJCKNLPMTPXXEM-AUTRQRHGSA-N 0.000 description 1
- LSPKYLAFTPBWIL-BYPYZUCNSA-N Glu-Gly Chemical compound OC(=O)CC[C@H](N)C(=O)NCC(O)=O LSPKYLAFTPBWIL-BYPYZUCNSA-N 0.000 description 1
- DVLZZEPUNFEUBW-AVGNSLFASA-N Glu-His-Leu Chemical compound CC(C)C[C@@H](C(=O)O)NC(=O)[C@H](CC1=CN=CN1)NC(=O)[C@H](CCC(=O)O)N DVLZZEPUNFEUBW-AVGNSLFASA-N 0.000 description 1
- ZIYGTCDTJJCDDP-JYJNAYRXSA-N Glu-Phe-Lys Chemical compound C1=CC=C(C=C1)C[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CCC(=O)O)N ZIYGTCDTJJCDDP-JYJNAYRXSA-N 0.000 description 1
- ALMBZBOCGSVSAI-ACZMJKKPSA-N Glu-Ser-Asn Chemical compound C(CC(=O)O)[C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(=O)N)C(=O)O)N ALMBZBOCGSVSAI-ACZMJKKPSA-N 0.000 description 1
- YQAQQKPWFOBSMU-WDCWCFNPSA-N Glu-Thr-Leu Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(O)=O YQAQQKPWFOBSMU-WDCWCFNPSA-N 0.000 description 1
- UZWUBBRJWFTHTD-LAEOZQHASA-N Glu-Val-Asn Chemical compound NC(=O)C[C@@H](C(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](N)CCC(O)=O UZWUBBRJWFTHTD-LAEOZQHASA-N 0.000 description 1
- ZYRXTRTUCAVNBQ-GVXVVHGQSA-N Glu-Val-Lys Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CCC(=O)O)N ZYRXTRTUCAVNBQ-GVXVVHGQSA-N 0.000 description 1
- FVGOGEGGQLNZGH-DZKIICNBSA-N Glu-Val-Phe Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 FVGOGEGGQLNZGH-DZKIICNBSA-N 0.000 description 1
- 108010070600 Glucose-6-phosphate isomerase Proteins 0.000 description 1
- 102000005731 Glucose-6-phosphate isomerase Human genes 0.000 description 1
- FUESBOMYALLFNI-VKHMYHEASA-N Gly-Asn Chemical compound NCC(=O)N[C@H](C(O)=O)CC(N)=O FUESBOMYALLFNI-VKHMYHEASA-N 0.000 description 1
- QITBQGJOXQYMOA-ZETCQYMHSA-N Gly-Gly-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)CNC(=O)CN QITBQGJOXQYMOA-ZETCQYMHSA-N 0.000 description 1
- MHXKHKWHPNETGG-QWRGUYRKSA-N Gly-Lys-Leu Chemical compound [H]NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O MHXKHKWHPNETGG-QWRGUYRKSA-N 0.000 description 1
- HAOUOFNNJJLVNS-BQBZGAKWSA-N Gly-Pro-Ser Chemical compound NCC(=O)N1CCC[C@H]1C(=O)N[C@@H](CO)C(O)=O HAOUOFNNJJLVNS-BQBZGAKWSA-N 0.000 description 1
- JSLVAHYTAJJEQH-QWRGUYRKSA-N Gly-Ser-Phe Chemical compound NCC(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 JSLVAHYTAJJEQH-QWRGUYRKSA-N 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 101710110781 Guanylate-binding protein 1 Proteins 0.000 description 1
- 108010083930 HIV Receptors Proteins 0.000 description 1
- 102000006481 HIV Receptors Human genes 0.000 description 1
- 206010061188 Haematotoxicity Diseases 0.000 description 1
- 102000005548 Hexokinase Human genes 0.000 description 1
- 108700040460 Hexokinases Proteins 0.000 description 1
- YAALVYQFVJNXIV-KKUMJFAQSA-N His-Leu-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CC1=CN=CN1 YAALVYQFVJNXIV-KKUMJFAQSA-N 0.000 description 1
- 101000657452 Homo sapiens Rotatin Proteins 0.000 description 1
- 101000639987 Homo sapiens Stearoyl-CoA desaturase 5 Proteins 0.000 description 1
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 description 1
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 1
- 206010064091 Iatrogenic infection Diseases 0.000 description 1
- TZCGZYWNIDZZMR-NAKRPEOUSA-N Ile-Arg-Ala Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](C)C(=O)O)N TZCGZYWNIDZZMR-NAKRPEOUSA-N 0.000 description 1
- TZCGZYWNIDZZMR-UHFFFAOYSA-N Ile-Arg-Ala Natural products CCC(C)C(N)C(=O)NC(C(=O)NC(C)C(O)=O)CCCN=C(N)N TZCGZYWNIDZZMR-UHFFFAOYSA-N 0.000 description 1
- BGZIJZJBXRVBGJ-SXTJYALSSA-N Ile-Asp-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)O)N BGZIJZJBXRVBGJ-SXTJYALSSA-N 0.000 description 1
- KIMHKBDJQQYLHU-PEFMBERDSA-N Ile-Glu-Asp Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC(=O)O)C(=O)O)N KIMHKBDJQQYLHU-PEFMBERDSA-N 0.000 description 1
- CSQNHSGHAPRGPQ-YTFOTSKYSA-N Ile-Ile-Lys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)O)N CSQNHSGHAPRGPQ-YTFOTSKYSA-N 0.000 description 1
- PHRWFSFCNJPWRO-PPCPHDFISA-N Ile-Leu-Thr Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)O)N PHRWFSFCNJPWRO-PPCPHDFISA-N 0.000 description 1
- FFAUOCITXBMRBT-YTFOTSKYSA-N Ile-Lys-Ile Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O FFAUOCITXBMRBT-YTFOTSKYSA-N 0.000 description 1
- UDBPXJNOEWDBDF-XUXIUFHCSA-N Ile-Lys-Val Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)O)N UDBPXJNOEWDBDF-XUXIUFHCSA-N 0.000 description 1
- USXAYNCLFSUSBA-MGHWNKPDSA-N Ile-Phe-His Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CC2=CN=CN2)C(=O)O)N USXAYNCLFSUSBA-MGHWNKPDSA-N 0.000 description 1
- VZSDQFZFTCVEGF-ZEWNOJEFSA-N Ile-Phe-Tyr Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](Cc1ccc(O)cc1)C(O)=O VZSDQFZFTCVEGF-ZEWNOJEFSA-N 0.000 description 1
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 description 1
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 description 1
- 206010062016 Immunosuppression Diseases 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 102000004195 Isomerases Human genes 0.000 description 1
- 108090000769 Isomerases Proteins 0.000 description 1
- 241000255777 Lepidoptera Species 0.000 description 1
- XIRYQRLFHWWWTC-QEJZJMRPSA-N Leu-Ala-Phe Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 XIRYQRLFHWWWTC-QEJZJMRPSA-N 0.000 description 1
- WGNOPSQMIQERPK-UHFFFAOYSA-N Leu-Asn-Pro Natural products CC(C)CC(N)C(=O)NC(CC(=O)N)C(=O)N1CCCC1C(=O)O WGNOPSQMIQERPK-UHFFFAOYSA-N 0.000 description 1
- HPBCTWSUJOGJSH-MNXVOIDGSA-N Leu-Glu-Ile Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O HPBCTWSUJOGJSH-MNXVOIDGSA-N 0.000 description 1
- QVFGXCVIXXBFHO-AVGNSLFASA-N Leu-Glu-Leu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(O)=O QVFGXCVIXXBFHO-AVGNSLFASA-N 0.000 description 1
- WQWSMEOYXJTFRU-GUBZILKMSA-N Leu-Glu-Ser Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(O)=O WQWSMEOYXJTFRU-GUBZILKMSA-N 0.000 description 1
- FMEICTQWUKNAGC-YUMQZZPRSA-N Leu-Gly-Asn Chemical compound [H]N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(O)=O FMEICTQWUKNAGC-YUMQZZPRSA-N 0.000 description 1
- CSFVADKICPDRRF-KKUMJFAQSA-N Leu-His-Leu Chemical compound CC(C)C[C@H]([NH3+])C(=O)N[C@H](C(=O)N[C@@H](CC(C)C)C([O-])=O)CC1=CN=CN1 CSFVADKICPDRRF-KKUMJFAQSA-N 0.000 description 1
- RXGLHDWAZQECBI-SRVKXCTJSA-N Leu-Leu-Ser Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(O)=O RXGLHDWAZQECBI-SRVKXCTJSA-N 0.000 description 1
- VUZMPNMNJBGOKE-IHRRRGAJSA-N Leu-Leu-Val Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(O)=O VUZMPNMNJBGOKE-IHRRRGAJSA-N 0.000 description 1
- BGZCJDGBBUUBHA-KKUMJFAQSA-N Leu-Lys-Leu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O BGZCJDGBBUUBHA-KKUMJFAQSA-N 0.000 description 1
- PWPBLZXWFXJFHE-RHYQMDGZSA-N Leu-Pro-Thr Chemical compound CC(C)C[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)O)C(O)=O PWPBLZXWFXJFHE-RHYQMDGZSA-N 0.000 description 1
- ZJZNLRVCZWUONM-JXUBOQSCSA-N Leu-Thr-Ala Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C)C(O)=O ZJZNLRVCZWUONM-JXUBOQSCSA-N 0.000 description 1
- QWWPYKKLXWOITQ-VOAKCMCISA-N Leu-Thr-Leu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@H](C(O)=O)CC(C)C QWWPYKKLXWOITQ-VOAKCMCISA-N 0.000 description 1
- DAYQSYGBCUKVKT-VOAKCMCISA-N Leu-Thr-Lys Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(O)=O DAYQSYGBCUKVKT-VOAKCMCISA-N 0.000 description 1
- WPIKRJDRQVFRHP-TUSQITKMSA-N Leu-Trp-Trp Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(O)=O WPIKRJDRQVFRHP-TUSQITKMSA-N 0.000 description 1
- KVSBQLNBMUPADA-AVGNSLFASA-N Leu-Val-Val Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(O)=O KVSBQLNBMUPADA-AVGNSLFASA-N 0.000 description 1
- UWKNTTJNVSYXPC-CIUDSAMLSA-N Lys-Ala-Ser Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CCCCN UWKNTTJNVSYXPC-CIUDSAMLSA-N 0.000 description 1
- FACUGMGEFUEBTI-SRVKXCTJSA-N Lys-Asn-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)CCCCN FACUGMGEFUEBTI-SRVKXCTJSA-N 0.000 description 1
- DGWXCIORNLWGGG-CIUDSAMLSA-N Lys-Asn-Ser Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(O)=O DGWXCIORNLWGGG-CIUDSAMLSA-N 0.000 description 1
- LLSUNJYOSCOOEB-GUBZILKMSA-N Lys-Glu-Asp Chemical compound NCCCC[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(O)=O LLSUNJYOSCOOEB-GUBZILKMSA-N 0.000 description 1
- GCMWRRQAKQXDED-IUCAKERBSA-N Lys-Glu-Gly Chemical compound [NH3+]CCCC[C@H]([NH3+])C(=O)N[C@@H](CCC([O-])=O)C(=O)NCC([O-])=O GCMWRRQAKQXDED-IUCAKERBSA-N 0.000 description 1
- ULUQBUKAPDUKOC-GVXVVHGQSA-N Lys-Glu-Val Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(O)=O ULUQBUKAPDUKOC-GVXVVHGQSA-N 0.000 description 1
- NJNRBRKHOWSGMN-SRVKXCTJSA-N Lys-Leu-Asn Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(O)=O NJNRBRKHOWSGMN-SRVKXCTJSA-N 0.000 description 1
- YPLVCBKEPJPBDQ-MELADBBJSA-N Lys-Leu-Pro Chemical compound CC(C)C[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CCCCN)N YPLVCBKEPJPBDQ-MELADBBJSA-N 0.000 description 1
- OIQSIMFSVLLWBX-VOAKCMCISA-N Lys-Leu-Thr Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O OIQSIMFSVLLWBX-VOAKCMCISA-N 0.000 description 1
- YUAXTFMFMOIMAM-QWRGUYRKSA-N Lys-Lys-Gly Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)NCC(O)=O YUAXTFMFMOIMAM-QWRGUYRKSA-N 0.000 description 1
- YDDDRTIPNTWGIG-SRVKXCTJSA-N Lys-Lys-Ser Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(O)=O YDDDRTIPNTWGIG-SRVKXCTJSA-N 0.000 description 1
- QQPSCXKFDSORFT-IHRRRGAJSA-N Lys-Lys-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](N)CCCCN QQPSCXKFDSORFT-IHRRRGAJSA-N 0.000 description 1
- YTJFXEDRUOQGSP-DCAQKATOSA-N Lys-Pro-Ser Chemical compound [H]N[C@@H](CCCCN)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CO)C(O)=O YTJFXEDRUOQGSP-DCAQKATOSA-N 0.000 description 1
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 1
- DLAFCQWUMFMZSN-GUBZILKMSA-N Met-Arg-Ala Chemical compound CSCC[C@H](N)C(=O)N[C@H](C(=O)N[C@@H](C)C(O)=O)CCCN=C(N)N DLAFCQWUMFMZSN-GUBZILKMSA-N 0.000 description 1
- CTVJSFRHUOSCQQ-DCAQKATOSA-N Met-Arg-Glu Chemical compound CSCC[C@H](N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(O)=O CTVJSFRHUOSCQQ-DCAQKATOSA-N 0.000 description 1
- JACAKCWAOHKQBV-UWVGGRQHSA-N Met-Gly-Lys Chemical compound CSCC[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CCCCN JACAKCWAOHKQBV-UWVGGRQHSA-N 0.000 description 1
- DBXMFHGGHMXYHY-DCAQKATOSA-N Met-Leu-Ser Chemical compound CSCC[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(O)=O DBXMFHGGHMXYHY-DCAQKATOSA-N 0.000 description 1
- 241001439204 Microphorus Species 0.000 description 1
- 241000713333 Mouse mammary tumor virus Species 0.000 description 1
- 102000016943 Muramidase Human genes 0.000 description 1
- 108010014251 Muramidase Proteins 0.000 description 1
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 1
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 1
- YBAFDPFAUTYYRW-UHFFFAOYSA-N N-L-alpha-glutamyl-L-leucine Natural products CC(C)CC(C(O)=O)NC(=O)C(N)CCC(O)=O YBAFDPFAUTYYRW-UHFFFAOYSA-N 0.000 description 1
- SQVRNKJHWKZAKO-PFQGKNLYSA-N N-acetyl-beta-neuraminic acid Chemical group CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-PFQGKNLYSA-N 0.000 description 1
- KZNQNBZMBZJQJO-UHFFFAOYSA-N N-glycyl-L-proline Natural products NCC(=O)N1CCCC1C(O)=O KZNQNBZMBZJQJO-UHFFFAOYSA-N 0.000 description 1
- 108010002311 N-glycylglutamic acid Proteins 0.000 description 1
- AHVYPIQETPWLSZ-UHFFFAOYSA-N N-methyl-pyrrolidine Natural products CN1CC=CC1 AHVYPIQETPWLSZ-UHFFFAOYSA-N 0.000 description 1
- 101000800755 Naja oxiana Alpha-elapitoxin-Nno2a Proteins 0.000 description 1
- BQVUABVGYYSDCJ-UHFFFAOYSA-N Nalpha-L-Leucyl-L-tryptophan Natural products C1=CC=C2C(CC(NC(=O)C(N)CC(C)C)C(O)=O)=CNC2=C1 BQVUABVGYYSDCJ-UHFFFAOYSA-N 0.000 description 1
- 101000706020 Nicotiana tabacum Pathogenesis-related protein R minor form Proteins 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 102100035593 POU domain, class 2, transcription factor 1 Human genes 0.000 description 1
- 101710084414 POU domain, class 2, transcription factor 1 Proteins 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- RIYZXJVARWJLKS-KKUMJFAQSA-N Phe-Asp-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 RIYZXJVARWJLKS-KKUMJFAQSA-N 0.000 description 1
- APJPXSFJBMMOLW-KBPBESRZSA-N Phe-Gly-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)CNC(=O)[C@@H](N)CC1=CC=CC=C1 APJPXSFJBMMOLW-KBPBESRZSA-N 0.000 description 1
- YZJKNDCEPDDIDA-BZSNNMDCSA-N Phe-His-Lys Chemical compound C([C@@H](C(=O)N[C@@H](CCCCN)C(O)=O)NC(=O)[C@@H](N)CC=1C=CC=CC=1)C1=CN=CN1 YZJKNDCEPDDIDA-BZSNNMDCSA-N 0.000 description 1
- QEFHBVDWKFFKQI-PMVMPFDFSA-N Phe-His-Trp Chemical compound [H]N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(O)=O QEFHBVDWKFFKQI-PMVMPFDFSA-N 0.000 description 1
- WWPAHTZOWURIMR-ULQDDVLXSA-N Phe-Pro-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@@H](N)CC1=CC=CC=C1 WWPAHTZOWURIMR-ULQDDVLXSA-N 0.000 description 1
- 239000005922 Phosphane Substances 0.000 description 1
- 108010069341 Phosphofructokinases Proteins 0.000 description 1
- 102000001105 Phosphofructokinases Human genes 0.000 description 1
- 102000012288 Phosphopyruvate Hydratase Human genes 0.000 description 1
- 108010022181 Phosphopyruvate Hydratase Proteins 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 208000007452 Plasmacytoma Diseases 0.000 description 1
- 241000223821 Plasmodium malariae Species 0.000 description 1
- 241001505293 Plasmodium ovale Species 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- 241001505332 Polyomavirus sp. Species 0.000 description 1
- 208000031951 Primary immunodeficiency Diseases 0.000 description 1
- BNBBNGZZKQUWCD-IUCAKERBSA-N Pro-Arg-Gly Chemical compound NC(N)=NCCC[C@@H](C(=O)NCC(O)=O)NC(=O)[C@@H]1CCCN1 BNBBNGZZKQUWCD-IUCAKERBSA-N 0.000 description 1
- NMELOOXSGDRBRU-YUMQZZPRSA-N Pro-Glu-Gly Chemical compound OC(=O)CNC(=O)[C@H](CCC(=O)O)NC(=O)[C@@H]1CCCN1 NMELOOXSGDRBRU-YUMQZZPRSA-N 0.000 description 1
- RMODQFBNDDENCP-IHRRRGAJSA-N Pro-Lys-Leu Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O RMODQFBNDDENCP-IHRRRGAJSA-N 0.000 description 1
- LEIKGVHQTKHOLM-IUCAKERBSA-N Pro-Pro-Gly Chemical compound OC(=O)CNC(=O)[C@@H]1CCCN1C(=O)[C@H]1NCCC1 LEIKGVHQTKHOLM-IUCAKERBSA-N 0.000 description 1
- RMJZWERKFFNNNS-XGEHTFHBSA-N Pro-Thr-Ser Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(O)=O RMJZWERKFFNNNS-XGEHTFHBSA-N 0.000 description 1
- FIODMZKLZFLYQP-GUBZILKMSA-N Pro-Val-Ser Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CO)C(O)=O FIODMZKLZFLYQP-GUBZILKMSA-N 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108010011939 Pyruvate Decarboxylase Proteins 0.000 description 1
- 102000013009 Pyruvate Kinase Human genes 0.000 description 1
- 108020005115 Pyruvate Kinase Proteins 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 239000008156 Ringer's lactate solution Substances 0.000 description 1
- 102100034742 Rotatin Human genes 0.000 description 1
- 108010077895 Sarcosine Proteins 0.000 description 1
- 239000012506 Sephacryl® Substances 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- OOKCGAYXSNJBGQ-ZLUOBGJFSA-N Ser-Asn-Asn Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O OOKCGAYXSNJBGQ-ZLUOBGJFSA-N 0.000 description 1
- MMAPOBOTRUVNKJ-ZLUOBGJFSA-N Ser-Asp-Ser Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)O)NC(=O)[C@H](CO)N)C(=O)O MMAPOBOTRUVNKJ-ZLUOBGJFSA-N 0.000 description 1
- BPMRXBZYPGYPJN-WHFBIAKZSA-N Ser-Gly-Asn Chemical compound [H]N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(O)=O BPMRXBZYPGYPJN-WHFBIAKZSA-N 0.000 description 1
- MIJWOJAXARLEHA-WDSKDSINSA-N Ser-Gly-Glu Chemical compound OC[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CCC(O)=O MIJWOJAXARLEHA-WDSKDSINSA-N 0.000 description 1
- NFDYGNFETJVMSE-BQBZGAKWSA-N Ser-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H](N)CO NFDYGNFETJVMSE-BQBZGAKWSA-N 0.000 description 1
- FPCGZYMRFFIYIH-CIUDSAMLSA-N Ser-Lys-Ser Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(O)=O FPCGZYMRFFIYIH-CIUDSAMLSA-N 0.000 description 1
- RWDVVSKYZBNDCO-MELADBBJSA-N Ser-Phe-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CC2=CC=CC=C2)NC(=O)[C@H](CO)N)C(=O)O RWDVVSKYZBNDCO-MELADBBJSA-N 0.000 description 1
- CUXJENOFJXOSOZ-BIIVOSGPSA-N Ser-Ser-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CO)NC(=O)[C@H](CO)N)C(=O)O CUXJENOFJXOSOZ-BIIVOSGPSA-N 0.000 description 1
- RXUOAOOZIWABBW-XGEHTFHBSA-N Ser-Thr-Arg Chemical compound OC[C@H](N)C(=O)N[C@@H]([C@H](O)C)C(=O)N[C@H](C(O)=O)CCCN=C(N)N RXUOAOOZIWABBW-XGEHTFHBSA-N 0.000 description 1
- LGIMRDKGABDMBN-DCAQKATOSA-N Ser-Val-Lys Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CO)N LGIMRDKGABDMBN-DCAQKATOSA-N 0.000 description 1
- 102220525770 Sodium channel subunit beta-3_Q89L_mutation Human genes 0.000 description 1
- 102100033930 Stearoyl-CoA desaturase 5 Human genes 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- 239000012505 Superdex™ Substances 0.000 description 1
- 241001185310 Symbiotes <prokaryote> Species 0.000 description 1
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 1
- 101150104425 T4 gene Proteins 0.000 description 1
- DFTCYYILCSQGIZ-GCJQMDKQSA-N Thr-Ala-Asn Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(O)=O DFTCYYILCSQGIZ-GCJQMDKQSA-N 0.000 description 1
- DHPPWTOLRWYIDS-XKBZYTNZSA-N Thr-Cys-Glu Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(O)=O)C(O)=O DHPPWTOLRWYIDS-XKBZYTNZSA-N 0.000 description 1
- NCXVJIQMWSGRHY-KXNHARMFSA-N Thr-Leu-Pro Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N1CCC[C@@H]1C(=O)O)N)O NCXVJIQMWSGRHY-KXNHARMFSA-N 0.000 description 1
- YOOAQCZYZHGUAZ-KATARQTJSA-N Thr-Leu-Ser Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(O)=O YOOAQCZYZHGUAZ-KATARQTJSA-N 0.000 description 1
- VRUFCJZQDACGLH-UVOCVTCTSA-N Thr-Leu-Thr Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O VRUFCJZQDACGLH-UVOCVTCTSA-N 0.000 description 1
- ZEJBJDHSQPOVJV-UAXMHLISSA-N Thr-Trp-Thr Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(=O)N[C@@H]([C@@H](C)O)C(O)=O ZEJBJDHSQPOVJV-UAXMHLISSA-N 0.000 description 1
- KPMIQCXJDVKWKO-IFFSRLJSSA-N Thr-Val-Glu Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O KPMIQCXJDVKWKO-IFFSRLJSSA-N 0.000 description 1
- 102100023935 Transmembrane glycoprotein NMB Human genes 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- 102000005924 Triose-Phosphate Isomerase Human genes 0.000 description 1
- 108700015934 Triose-phosphate isomerases Proteins 0.000 description 1
- YDTKYBHPRULROG-LTHWPDAASA-N Trp-Ile-Thr Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H]([C@@H](C)O)C(=O)O)NC(=O)[C@H](CC1=CNC2=CC=CC=C21)N YDTKYBHPRULROG-LTHWPDAASA-N 0.000 description 1
- GSCPHMSPGQSZJT-JYBASQMISA-N Trp-Ser-Thr Chemical compound C[C@H]([C@@H](C(=O)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC1=CNC2=CC=CC=C21)N)O GSCPHMSPGQSZJT-JYBASQMISA-N 0.000 description 1
- NMOIRIIIUVELLY-WDSOQIARSA-N Trp-Val-Leu Chemical compound C1=CC=C2C(C[C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CC(C)C)C(O)=O)C(C)C)=CNC2=C1 NMOIRIIIUVELLY-WDSOQIARSA-N 0.000 description 1
- DLZKEQQWXODGGZ-KWQFWETISA-N Tyr-Ala-Gly Chemical compound OC(=O)CNC(=O)[C@H](C)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 DLZKEQQWXODGGZ-KWQFWETISA-N 0.000 description 1
- JJNXZIPLIXIGBX-HJPIBITLSA-N Tyr-Ile-Cys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CC1=CC=C(C=C1)O)N JJNXZIPLIXIGBX-HJPIBITLSA-N 0.000 description 1
- HSRXSKHRSXRCFC-WDSKDSINSA-N Val-Ala Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](C)C(O)=O HSRXSKHRSXRCFC-WDSKDSINSA-N 0.000 description 1
- FOADDSDHGRFUOC-DZKIICNBSA-N Val-Glu-Phe Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)O)N FOADDSDHGRFUOC-DZKIICNBSA-N 0.000 description 1
- UMPVMAYCLYMYGA-ONGXEEELSA-N Val-Leu-Gly Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)NCC(O)=O UMPVMAYCLYMYGA-ONGXEEELSA-N 0.000 description 1
- WLHIIWDIDLQTKP-IHRRRGAJSA-N Val-Leu-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)C(C)C WLHIIWDIDLQTKP-IHRRRGAJSA-N 0.000 description 1
- BTWMICVCQLKKNR-DCAQKATOSA-N Val-Leu-Ser Chemical compound CC(C)[C@H]([NH3+])C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C([O-])=O BTWMICVCQLKKNR-DCAQKATOSA-N 0.000 description 1
- UXODSMTVPWXHBT-ULQDDVLXSA-N Val-Phe-His Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CC2=CN=CN2)C(=O)O)N UXODSMTVPWXHBT-ULQDDVLXSA-N 0.000 description 1
- QTPQHINADBYBNA-DCAQKATOSA-N Val-Ser-Lys Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CCCCN QTPQHINADBYBNA-DCAQKATOSA-N 0.000 description 1
- UVHFONIHVHLDDQ-IFFSRLJSSA-N Val-Thr-Glu Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)O)NC(=O)[C@H](C(C)C)N)O UVHFONIHVHLDDQ-IFFSRLJSSA-N 0.000 description 1
- QHSSPPHOHJSTML-HOCLYGCPSA-N Val-Trp-Gly Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CC1=CNC2=CC=CC=C21)C(=O)NCC(=O)O)N QHSSPPHOHJSTML-HOCLYGCPSA-N 0.000 description 1
- RLVTVHSDKHBFQP-ULQDDVLXSA-N Val-Tyr-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)CC1=CC=C(O)C=C1 RLVTVHSDKHBFQP-ULQDDVLXSA-N 0.000 description 1
- MOJFVLVTLZDQGW-AVGNSLFASA-N Val-Val-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](N)C(C)C MOJFVLVTLZDQGW-AVGNSLFASA-N 0.000 description 1
- 244000042314 Vigna unguiculata Species 0.000 description 1
- MJYGPNBYCMSMFL-UHFFFAOYSA-N [O].C1CCOC1 Chemical compound [O].C1CCOC1 MJYGPNBYCMSMFL-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 108010011559 alanylphenylalanine Proteins 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- KOSRFJWDECSPRO-UHFFFAOYSA-N alpha-L-glutamyl-L-glutamic acid Natural products OC(=O)CCC(N)C(=O)NC(CCC(O)=O)C(O)=O KOSRFJWDECSPRO-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 244000000054 animal parasite Species 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 238000005571 anion exchange chromatography Methods 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000010100 anticoagulation Effects 0.000 description 1
- 229940019748 antifibrinolytic proteinase inhibitors Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 210000001106 artificial yeast chromosome Anatomy 0.000 description 1
- 206010003549 asthenia Diseases 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000003416 augmentation Effects 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 102000023732 binding proteins Human genes 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 229940125691 blood product Drugs 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 244000078885 bloodborne pathogen Species 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000008366 buffered solution Substances 0.000 description 1
- 230000003139 buffering effect Effects 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000005277 cation exchange chromatography Methods 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 239000002458 cell surface marker Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 208000019065 cervical carcinoma Diseases 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- QBPFLULOKWLNNW-UHFFFAOYSA-N chrysazin Chemical compound O=C1C2=CC=CC(O)=C2C(=O)C2=C1C=CC=C2O QBPFLULOKWLNNW-UHFFFAOYSA-N 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000003750 conditioning effect Effects 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 230000008094 contradictory effect Effects 0.000 description 1
- 238000009295 crossflow filtration Methods 0.000 description 1
- OOTFVKOQINZBBF-UHFFFAOYSA-N cystamine Chemical compound CCSSCCN OOTFVKOQINZBBF-UHFFFAOYSA-N 0.000 description 1
- 229940099500 cystamine Drugs 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 239000013578 denaturing buffer Substances 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 238000011026 diafiltration Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000011038 discontinuous diafiltration by volume reduction Methods 0.000 description 1
- 231100000676 disease causative agent Toxicity 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000005430 electron energy loss spectroscopy Methods 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940089118 epogen Drugs 0.000 description 1
- 230000003628 erosive effect Effects 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- 210000004700 fetal blood Anatomy 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 210000003754 fetus Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 108010063718 gamma-glutamylaspartic acid Proteins 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000001641 gel filtration chromatography Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 230000002414 glycolytic effect Effects 0.000 description 1
- 108010074027 glycyl-seryl-phenylalanine Proteins 0.000 description 1
- 108010089804 glycyl-threonine Proteins 0.000 description 1
- 108010077515 glycylproline Proteins 0.000 description 1
- 231100000226 haematotoxicity Toxicity 0.000 description 1
- 210000002443 helper t lymphocyte Anatomy 0.000 description 1
- 108010025306 histidylleucine Proteins 0.000 description 1
- 238000000265 homogenisation Methods 0.000 description 1
- 230000000652 homosexual effect Effects 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 230000008105 immune reaction Effects 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 230000002584 immunomodulator Effects 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 208000033065 inborn errors of immunity Diseases 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 230000001678 irradiating effect Effects 0.000 description 1
- 108010078274 isoleucylvaline Proteins 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 238000002386 leaching Methods 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 108010071185 leucyl-alanine Proteins 0.000 description 1
- 108010034529 leucyl-lysine Proteins 0.000 description 1
- 108010091871 leucylmethionine Proteins 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 210000005229 liver cell Anatomy 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 231100000183 lymphotoxic Toxicity 0.000 description 1
- 230000001917 lymphotoxic effect Effects 0.000 description 1
- 239000004325 lysozyme Substances 0.000 description 1
- 229960000274 lysozyme Drugs 0.000 description 1
- 235000010335 lysozyme Nutrition 0.000 description 1
- 108010064235 lysylglycine Proteins 0.000 description 1
- 210000003593 megakaryocyte Anatomy 0.000 description 1
- 210000004914 menses Anatomy 0.000 description 1
- 230000002175 menstrual effect Effects 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 108010034507 methionyltryptophan Proteins 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 210000000865 mononuclear phagocyte system Anatomy 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 101800002712 p27 Proteins 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 239000000123 paper Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 238000010587 phase diagram Methods 0.000 description 1
- 108010051242 phenylalanylserine Proteins 0.000 description 1
- 229910000064 phosphane Inorganic materials 0.000 description 1
- 229940080469 phosphocellulose Drugs 0.000 description 1
- 150000008300 phosphoramidites Chemical class 0.000 description 1
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical compound OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 229940118768 plasmodium malariae Drugs 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 230000000063 preceeding effect Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 208000028529 primary immunodeficiency disease Diseases 0.000 description 1
- 108010029020 prolylglycine Proteins 0.000 description 1
- 108010090894 prolylleucine Proteins 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000017610 release of virus from host Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 239000012508 resin bead Substances 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 229940043230 sarcosine Drugs 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 210000000717 sertoli cell Anatomy 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 239000013605 shuttle vector Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 230000003019 stabilising effect Effects 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 108091007466 transmembrane glycoproteins Proteins 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 108010005834 tyrosyl-alanyl-glycine Proteins 0.000 description 1
- 108010051110 tyrosyl-lysine Proteins 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 230000007485 viral shedding Effects 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
- 206010048282 zoonosis Diseases 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/70514—CD4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/44—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from protozoa
- C07K14/445—Plasmodium
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2123/00—Preparations for testing in vivo
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/02—Fusion polypeptide containing a localisation/targetting motif containing a signal sequence
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/32—Fusion polypeptide fusions with soluble part of a cell surface receptor, "decoy receptors"
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/33—Fusion polypeptide fusions for targeting to specific cell types, e.g. tissue specific targeting, targeting of a bacterial subspecies
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the field of this invention relates to protein molecules in particular hybrid protein molecules for the prevention of treatment of human beings afflicted with AIDS and diseases caused by Human Immuno Deficiency Viruses especially types 1 and 2; and other blood borne pathogens.
- the AIDS syndrome is widely believed by the majority of Engineers (with a few exceptions) to be the end result of infection with Human immuno Deficiency Virus type 1 or 2.
- life expectancy is short and death occurs usually within two years - death resulting from overwhelming infection and/or neoplastic diseases provoked by a profound immuno deficiency .
- the causative agent of this condition the HIV virus was proposed by Montagnier L and by Barre- Sinoussi F et al 1 983, Science 220, 868-870 and slightly later by Gallo R et al 1 983, Science 220 865-867.
- the present disclosure provides a means of improving the safety of blood products in one step.
- the mechanism of HIV infection involves the binding of viral GP1 20 or envelope peptide to the CD4 peptide molecule.
- the CD4 protein is a 433 amino acid transmembrane glycoprotein which is found predominantly of the surface of T4 helper cells. It is also found on the surface of the mega karyocytes, monocytes, macrophages and other cells.
- the CD4 molecule has three parts; an extracellular part which is divided into 4 variable domains V1 being responsible for the binding of HIV GP1 20; a short transmembrane region and an intracellular domain.
- the CD4 molecule is part of the Immunoglobuiin super family. That CD4 was the receptor for the virus was shown by Dalglaoise et al 1 984. "The CD4 antigen is an essential component of the receptor for the AIDS retrovirus" Nature; 21 2: 763-766. Also Madden P J Dalglaoise A G McDougall J S et al "The T4 gene encloses the AIDS virus receptor and is expressed in the immune system and the brain" Cell: 47, 333- 348. Maddon P et al Cell: Vol 47, 333-348 have shown that transformed mouse cells expressing the CD4 molecule possess the capabilities of binding the HIV virus though lack the ability to support the HIV infection at a cytoplasmic level.
- the remarkable specificity of the virus for the CD4 molecule has lead to the attempts by many workers to utilise CD4 based peptides as therapeutic agents.
- the binding affinity for the GP120 with CD4 lies in the region of 3nM. Many efforts have been made to utilise this high specificity.
- the affinity is 20-25 fold less, however the binding strength as described is such that even a 25 fold reduction of affinity would not reduce the usefulness of therapeutic molecules based on CD4 and for that reaon the binding reaction between GP1 20 and CD4 remains a logical target for anti HIV pharmaceuticals.
- CD4 based therapeutic molecules should contain amino acids at least from the first 1 1 7 amino acids of the CD4 sequence.
- the present disclosure proposes that clinical disappointment with the T4 agents, such as disclosed in PCT WO89/01 9140 relates more to the short half life of the peptides in plasma and to the fact that 72 binding sites exist on a single virus, rather than due to lack of affinity between the agent and the virus itself. So great is the affinity between CD4 molecule and the GP1 20 molecule that even affinities 1 000 fold less than those seen in vitro should be pharmaceutically effective.
- a soluble CD4 as a HIV therapy
- other workers have advanced alternatives in the hope of utilising the GP1 20-CD4 interaction. Examples of some of these alternatives are fusion peptides where a CD4 component is fused to another peptide having a different function.
- Capon D J et al disclosed the use of CD4 fused to the FC component of the human Igh antibodies. The objective being to mobilise the compliment system and thereby destroy the virus particle by the compliment cascade. Capon refers to these new molecules as immuno adhesives. A possible disadvantage associated with this approach is the necessity of a fully functional compliment system, unlikely to the case in advanced HIV infections. See Capon et al "Designing CD4 Immunoadhesins for AIDS Therapy" Nature Vol 337.
- the molecular machines of the present disclosure do not require a functional immune system and may be effective in cases where no immune function exists.
- AZT anti-viral AZT
- ARC and AIDS Richman et al The toxicity of the Azidothymine", New England of Medicine, 31 7; p 192-1 97, 1 987. So profound is the anaemia provoked by this agent that many patients require frequent blood transfusions or treatment with Epogen (R), a novel genetically engineered erythropotein.
- the molecular machines of the present disclosure may be used in combination with AZT or other treatments and will augment the effectiveness of blood transfusions given to patients receiving AZT who are suffering from severe anaemia.
- Other agents acting on the reverse transscriptase such as DDC or DDI suffer high side effect profiles also and are toxic compounds in their own right.
- agents acting at other sites such as glycosylation are known to exhibit severe toxic effects on the host.
- the agents of the present disclosures are anticipitated to have lower toxic effects on the host than existing treatment modalities.
- the agents may be used alone or in combination with existing treatments.
- a virus immobilised in this way would be harmless and be carried around the circulation on the red cell surface to await final phagocytosis and digestion at the end of the red cells life (usually 1 20 days from formation). It is thought that many antigens and pathogens are cleared from circulation by this mechanism rather than by direct phagocytosis and the like so frequently discussed in immunology texts. Accordingly, Taylor R P proposed a technique to utilise this long forgotten mechanism. He suggested the use of hybrid antibodies capable of binding both to the HIV virus and to the compliment 1 receptor molecule of the red cell surface. The adherence of virus particles to the red cell surface membrane in this way would clear the blood stream of free virus particles and free GP1 20. Thereby attempting to achieve Nelson's proposition that one day red blood cells could be harnessed to rid civilization of infection.
- the peptide machines of the present disclosure provide totally different mechanisms to achieve Nelson's dream ie to bring about the binding of HIV virus particles to the red cell surface membrane.
- the agents of the present disclosure carry the additional advantage in that antibodies to the compliment peptides and red cell surface CR 1 are not a feature. This would be an advantage in that the risk of destabilising an already unbalanced immune system is not expected to be a feature of the molecules of the present disclosure.
- the Third World or underdeveloped world is no stranger to epidemics and has prior to the development of AIDS and HIV been ravaged by infectious disease; the most serious on a global scale is probably malaria.
- Various forms of importance to humans are Plasmodium Falciparum, Plasmodium Vivax, Plasmodium Ovale, Plasmodium Malariae. This serious parasite infests red blood cells.
- the life cycle is complex and consists in part of a short life cycle in the salivary gland of mosquitoes. Following i ⁇ nocuiation into a human being, the malaria organism migrates to the liver and to the red blood cell. It multiplies forming further merozoites. Merozoites may then proceed to infect other red blood cells, or may find their way into another mosquito where they replicate in the mosquito salivary gland and later proceed to infect another human being.
- the course of malaria is a variable one and may be characterised by a short acute Hlness which can bring death in a matter of hours; or a longer more chronic illness associated with debility and anaemia.
- Other forms of malaria infect animals such as plasmodium Knowlesi which is known to infect the Rhesus monkey; and many other zooanotic forms.
- the preferred location for the malaria parasite is within the red cells of the infected host and for much of its life span it lives i ⁇ tracellularly protected from the host immune system .
- glycophorin A molecule is a highly glycolated peptide.
- Pasvol has suggested that glycophorin binding peptide binds to the region of glycophorin close to the iipid bi-layer.
- Pasvol G et al “Inhibition of Malaria Parasite Invasion by Monoclonical antibodies against glycophorin A correlates with a reduction in red cell membrane deformality.” Blood 74, No 5, Oct 1 985, 1836-1 843. Debate continues within the literature as to the requirement for sialic acid on the glycophorin molecule to effect invasion by merozoites.
- glycophorin binding protein For several years a peptide called the glycophorin binding protein was believed to be the primary peptide responsible for binding merozoites to erythrocytes.
- a gene coding for GBP was isloated by M Ravetch and Kochan J and disclosed in Science Vol 227, p 1 593-1 596, 29 March 1 985 and incorportated herein fully by reference.
- GBP 1 30 is characterised by a tandem repeated sequence believed to be the site of erythrocyte binding, "A tandem repeated sequence determines the binding domatin for an erythrocyte receptor binding protein of plasmodium falciparum". Kochan J, Perkins M and Ravetch J Cell, Vol 44, 689-696 March 1986. See figure 2 p 691 which also discloses the full sequences and genetic code of the GBP 1 30 molecule and is incorporated fully herein by reference.
- EBA 1 75 ( t is known that the EBA 1 75 molecule has an affinity for ofigosaccharides which are found on the surface of the red cell molecule. However a problem arises in that the EBA 175 molecule does not bind effectively with the malaria merozoite parasite. Therefore it is thought that the EBA 1 75 serves a function as a bridge.
- This disclosure proposes an alternative mechanism in that the EBA 1 75 molecule is responsible for bringing the merozoite close to the erythrocyte membrane surface, thereafter GBP 130 drags the merozoite closer still by binding with the base of the glycophorin A peptide; thus bringing the Iipid bilayer of the malaria parasite into approximation with the Iipid bilayer of the red cell membrane and thereby allowing the incorportation of the parasite into the erythrocyte itself.
- This disclosure suggests the merozoite is winched into the RBC cytoplasm.
- EBA 1 75 The genetic sequence and the peptide sequence of EBA 1 75 was disclosed by Kim Lee Sim, Orlandi P et at "Primary structure of the 1 75 K plasmodium falciparum erythrocyte binding antigen and identification of a peptide which elicits antibodies that inhibit malaria merozoite invasion" See J Cell Biology Vol 1 1 1 (1 990) p 1877-1884 figure 2 of P 1880 for the sequence of amino acids and DNA sequence.
- nucleotide sequence and amino acid sequence of one form of the peptide GPBH is dislcosed by Dagmar Nolte et al in the Journal of Molecular and Biochemical Parasitology 49, (1991 ) p 253-264 see figure 2 of p 257 incorporated herein fully by reference. See also figure 3 of p 258 the same journal and paper which lists a comparison between GBP 1 30 and GBPH. Binding and entry of merozoites into RBC's probably involves several peptides or several alternatives as fail safes for the organism.
- Erythrocyte binding using different peptides and surface molecules is exhibited by other species of the malaria parasite in particular the Plasmodium Vivax organism.
- This organism can infect only persons expressing the Duffy marker.
- the Duffy antigen is a red cell surface marker and is one of many blood group markers and carried by a percentage of the population. Persons not expressing Duffy antigens are therefore immune from infection by Plasmodium Vivax.
- the Plasmodium Vivax merozoite expresses a Duffy binding receptor molecule the P.
- vivax Duffy receptor cloned and sequenced by Xiangdang Fang and disclosed in Molecular and Biochemical Parasitology 44 (1 991 ) p 1 25-1 32 see especially figure 1 of p 1 27 for the genetic sequence and amino acid sequence. Similar to Plasmodium Vivax is Pladmodium Knowlesi which also uses the Duffy antigen. This organism parasitises Rhesus monkeys. Also in the same Journal, same figure, same page is listed the genetic sequence of Plasmodium Knowlesi Duffy receptor molecule which may find a use in the machines of the present disclosure.
- the machines of the present invention consists of hybrid peptides formed by the fusion of malaria derived peptide sequences sufficient to exhibit erythrocyte membrane affinity fused to a CD4 derived peptide sequence sufficient to demonstrate affinity for the HIV virus.
- the restriction map and peptide sequence of the CD4 molecule are disclosed by Madden et al Proceedings National Acad Science USA 84; 1987' p 1955-1959 see especially figures 1 and 2 incorporated herein fully by reference. 1Z Also Madden et al Cell Vol 42, P 93-104 August 1985 especially figure 6 of P 97.
- CD4 peptide components will not be restricted to segments of the naturally occurring CD4 molecule.
- the malaria derived peptide fragments may be any that are capable of binding to the red cell membrane and one is especially directed to those referenced herein before.
- the present disclosure provides novel hybrid or fusion peptides having a minimum of two different peptide components possessing different functionality.
- One component preferably the CD4 molecule or segment or derivative thereof which is capable of binding to an HIV virus is fused or joined to a malaria derived peptide or derivative or fragment thereof which is capable of binding to a red cell membrane.
- the fusion peptide will in one aspect bind the free HIV virus to a red cell producing possible therapeutics benefits of:
- hybrid molecules such as:-
- Figure 1 (a) depicts free HIV virus particles drawn to and immobilised on.a red cell (RBC).
- RBC red cell
- Figure Kb depicts the formation of a novel vaccine within the host blood stream utilising HIV components (live virus) derived from the host rather than provided from external sources as would be usual; for vaccines.
- the molecular machines of the present disclosure are prepared by the conjugation of at least two separate components into a single larger molecular- entity or conjugate peptide.
- a minimum of one HIV binding component is joined to a minimum of one RBC ⁇ peptide.
- a minimum of one HIV binding component is joined to a minimum of one RBC binding component.
- the components may be joined directly or indirectly to a functional sequence or cross linkers.
- the simplest form of a molecular machine will consist of a HIV binding peptide and an RBC binding peptide fused by peptide bonds giving the following structure:
- the functional areas of the machines are preferably situated towards either end. Therefore the middle of the molecule and the site of fusion is not critical.
- the junctional areas of the CD4 molecule b) the Fc segment of an immunoglobulin; c) or any macromolecuie or cross linker or part thereof.
- the interposed segment may or may not retain functionally of its own but must not interfere with the function of HIV binding and RBC binding.
- the preferred HIV binding component is a CD4 peptide sequence all or part thereof retaining HIV binding.
- a CD4 therapeutic agent such as CD4-Fc retaining a funtionai HIV binding moiety would also provide a suitable HIV binding component. In that event, the Fc fragment would become a spacer or peptide cross linker between the CD4 segment and the RBC binding peptide.
- CD4-Fc peptide or any CD4- P where P is a non specific peptide sequence falls, within the scope and spirit of this invention regardless of whether the middle component retains functionality or not.
- One is directed to WO 89/02922 Genetech Daniai J Capon for a description of immunoadehesions comprising eg CD4-Fc fusion peptides which may be used in place of CD4; and is incorporated fully herein by reference.
- the envisaged mode of action of the therapeutic machines of the present disclosure is that one component of the molecular machines, the CD4 derived component will bind with the HIV virus GP1 20 molecule or free GP120 whilst the merozoite derived peptide segment will bind with the erythrocyte membrane surface. Either glycophorin A, B, C or to the Duffy antigen where a Duffy receptor peptide is used.
- the outcome of this binding will be to immobilise the virus particle onto the surface of the red cell much like a barnacle to the surface of a ship, and thereby neutralise it.
- the peptides of the present disclosure will find uses as molecules to be injected prior to surgery, molecules to be injected prior to delivery, molecules to be mixed with blood transfusions (whole blood) to be given to AIDS victims thereby to improve the therapeutic effectiveness of tranfusions and molecules to reduce the high viral titre levels believed to be associated with symptomatic AIDS and molecules for prophylaxis generally.
- a further benefit of the machines of the present disclosure is the possibility of incorporation of the virus into the red cell cytoplasm.
- Red cells lack a nucleus and a virus once incorporated into a red cell cytoplasm is immobilised and neutralised forever.
- a peptides of the present disclosure will range from 0.01 mg/kg to 10 mg/kg.
- the agents of the present disclosure may also find a use in diagnostic testing and viral titre quantification.
- the molecular machines of the present disclosure may act beneficially in further ways.
- the GP120 binding component (CD4 derived sequence) may bind the molecular machines to infected cells expressing GP1 20. Expression of GP1 20 by infected cells occurs prior to viral shedding
- APC antigen presenting cell
- Live viruses are processed differently to non infectious particles by APC's.
- Live viral antigens are digested in the cytoplasm of the APC (cytoplasmic pathway) and digested particles presented on the APC surface in association with class 1 MHC markers provoke a cellular immune response (Whitton J L Oldstone M B
- a Class 1 MHC can present endogenous peptide to cytotoxic lymphocytes J Exp Med 1 989 1 70: 1051 -56).
- the molecular machines of this invention provide a vaccine of a particularly novel variety in that:
- the vaccine as administered contains no components derived from the pathogen target of the vaccine (no HIV component), eg rGP1 20;
- the vaccine obtains its pathogen derived component direct from the host and modifies host produced live virus
- vaccines should usually contain material derived from the target pathogen.
- This disclosure teaches vaccines which do not provide target pathogen-derived material in that the material derived from the target pathogen of the vaccine is provided later in the host.
- the precise mechanism for the generation of cellular immunity is not understood. It is a further unproven postulate of this disclosure that cellular immunity generally proceeds from the processing of pathogens bound to erythrocytes when the said pathogens are engulfed with their red cell symbiot in the liver, bone marrow or spleen. Possible proof of this hypothesis lie in the following facts:
- the molecular machines of the present disclosure in one aspect are prepared and packaged in lypholised form and stored in refridgerated conditions.
- the lypholised fusion peptides are reconstituted by means of injectable solvents such as injectable preparations of normal saline or dextrose 5% or the like together with buffers and other stabilising ingredients incorporated as necessary.
- the reconstituted molecular machines are administered to patients by either the Direct or Indirect method.
- Direct method involves the intravenous or even intra muscular administration of the reconstituted injectable solution of the molecular machines of amounts usually not exceeding 1 ,000 mg of agent per ml.
- the agents may be infused slowly by means of an intravenous drip or when safe to do so or if necessary by bolus injection.
- the slower administration rates provide more even mixing of agent with red cells therefore labelling many more red cells.
- Indirect methods of administration envisage the mixing of the reconstituted agents with red cells outisde the body.
- the agents may be added to blood transfusion units all or part prior to administration or the agents may be mixed with a quantity of the patients own blood.
- Ten mis of the patients own blood is mixed with citrated buffer and thereafter with the reconstituted molecular machines.
- a degree of mechanical agitation is provided.
- the treated blood is now reintroduced into a suitable vein, slowly at first to observe for untoward reactions. The procedures above may be repeated until side effects such as haemolysis or idiosyncratic reactions prevent their further use. It is envisaged that the agent will be administered as courses of treatment on a 3 to 4 monthly basis.
- the agents are not necessarily restricted to lypholised forms. It has recently been shown that following liquid storage recombinant peptides perform better than freeze dried preparations, in some cases this was demonstrated for IFN see Pearlamn and Tue Nguyen J Pharm Phamacol 1 992, 44 (suppl 1 ): 1 78-1 85.
- the agents may be formulated in slow release forms allowing the gradual leaching of active peptide molecular machines over a time period. Examples of such methods are the incorporation of the agents into biodegradable polymers. Such polymers could be injected as subdermal 'rice grains' or form part of an intrauterine contraceptive device.
- Intrauterine contraceptive devices impregnated with the agents of the present disclosure would slowly leak the agents towards the cervix. Cerival erosions are known to be an area of major importance in women's vulnerability to AIDS. Indeed the heavier menses associated with lUD's would leach more of the peptides from the polymer and by targetting menstrual blood or on their own provide a useful degree of protection against the virus. Such a use would be very helpful in the Third World.
- novel peptide agents of the present disclosure may by incorporated into creams and gels or foams for use as topical preparations. Such compositions may be used in conjunction with:
- barrier contraceptives in place of existing lubricating agents or with lubricating agents generally
- the said creams, gels or foams may also be used to dress wounds, cuts or open sores or as contraceptive agents or in conjunction with contraceptives especially barrier methods. In the case of HIV positive patients such sores would become reduced sources of infection for others.
- the agents of the present disclosure are formulated into drops, especially eyedrops.
- eye splashes are a frequent occurrence.
- Theatre staff although strongly warned to use eye protection often fail to do so for reasons of discomfort, haste or carelessness.
- Existing eyewash preaprations do not contain specific anti-viral components.
- the present agents are formulated as surgical eyedrops or eye washes useful as emergency decontaminants of splashed eyes during operative procedures.
- a measured volume of blood or fluid to be assayed is mixed with a known quantity of molecular machines; usually 1 ml of sample blood with 20 ug of CD4-merozoite fusion peptides. following a period of incubation, agitating the sample so as to bring red cells into contact with the virus, the sample is further incubated with a fluorescent labelled anti HIV GP120, anti GP1 60 or anti GP41 antibody or cocktail thereof. Separation is carried out by centrifugation or filtration. After adequate washing of cells by appropriate buffer, cell fluorescence is measured in a cell sorter.
- a fluorescent labelled anti HIV GP120, anti GP1 60 or anti GP41 antibody or cocktail thereof Separation is carried out by centrifugation or filtration. After adequate washing of cells by appropriate buffer, cell fluorescence is measured in a cell sorter.
- fusion peptides of the present disclosure may be manufactured by a wide variety of manufacturing methods and strategies; which may be applied in various combinations.
- the fusion peptides may be manufactured by means of the separate manufacture of components such as components A and B and subsequent fusion of A and B invitro using various means eg by chemical linkers. Peptide sequences even greater than 1 50 amino aicds long may be manufactured entirely synthetically invitro. Accordingly where component A or B of the fusion peptide is less than 1 50 amino acids in length this component may be made synthetically as a single component. Where component A or B is very long component A or B may be manufactured synthetically in smaller fragments to be bonded chemically there after invitro, the said bonding to be preferably peptide bonds and achieved by methods such as segment condensation or by other methods known to the art.
- the two peptide components A and B may also be fused using chemical linkers or by disulphide bonds as disclosed by Chan et al 1 977, J Bio Chem 252; 1 51 5-1 522.
- Miskimins et al 1 979 Bio Chemi Biophys Ref Conse 1 991 ; 143-1 51 discloses a fusion peptide formed using cystamine as the cross linking agent.
- Cawley et al 1 980 Cell 22, 563-70 discloses the use or hetrobifunctional cross linking agents to form a hybrid molecule consisting of RICIN A joined to epiderminal groth factor EGF.
- Other useful strategies for fusing peptide sequences employ linking groups such as the chelating agent DTPA.
- Detachment of the peptide from the HYCRAM (R) support employs palladium tatra-kis (triphynyl-phosphane) a catalyst in a suitable solvent such as 50% (v/v) dimethylsuphoxide with dimethyl formamide; N-methylpyrrolidine, tetrahydrofuran and water. Oxygen tetrahydrofuran must be excluded. Acceptor molecules, morpholine, dimedine or N, N'-di methylbarbiturate may be added to take up the allyl ⁇ c group.
- the Ddz-/t-butyl amino acid protections are easier to cleave using with 1 -5% (v/v) trifluroacetic acid in dichloromethane a process taking 10 to 30 minutes or by means of the more environmental friendly acetic acid or dioxane containing 1 % (w/v) HCL gas.
- the other useful protocol is the Fmoc-/t-butyl strategy. Cleavage of F moc can be achieved using 20-50% (v/v) piperidine/dimethyl formamide. Deprotection can be monitored in both cases photometrically.
- Boc-; Fmoc-; or Ddz-amino acid derivatives may employ the inexpensive (DCC) diclohexylcarbodi-imide.
- DCC inexpensive diclohexylcarbodi-imide.
- HOBT N-hydroxybenzotriazole
- activating agents are the Castro Reagent or BOP; Benzotriazole-l-yl-oxy-tris (dimethyl amino) phosphonium hexa fluorophosphate; one is directed to CASTRO B et al ( 1 957) Tetrahedron Lett 14, 1 21 9; and TBTU the Knorr reagent, Benzotriazole-l-yl-oxy-l, 1 , 3, 3-tetramethyluronium tetrafluoroborate one is directed to Knorr R et al ( 1 989) Tetrahedron Lett 30, 1 927. Fragment condensation can be achieved using the BOP or the TBTU reagent with HOBT in excess. Protected peptides may also be in excess, however solvents and excesses can often be recycled.
- a typical production process involves either the separate synthesis of peptide sequences by their expression in suitable hosts, and their subsequent purification and fusion; or chemical synthesis such as on a solid substrate for example by the sequential addition of amino acid residues or peptide fragments which are protected, the protection of the amino acid residues as required and the subsequent reacting of the peptide chains with linking agents before removing the peptide chains from the said solid substrates and the final purification by the various means is such as reverese phase chromatography; or any combination of the above.
- a fused gene is a genetic sequence which codes for both components of the hybrid component molecule.
- One is directed to Murphy United States Patent 4,675,382 for a detailed disclosure of the use of fused genes in the manufacture of hybrid peptides having the components MSH or Melanocyte Stimulating Hormone fused to dipcheria A toxins.
- peptide fragments may be manufactured by DNA cloning and expression in suitable hosts and recovery with subsequent condensation in vitro.
- cloned sequences useful for the production of fusion peptides will have the transmembrane domain and the c ⁇ toplasmic domain sequences removed.
- DNA may be made by the chemical synthesis of DNA polymer fragments using phosphotriester, phosphite or phosphoramidite chemistry.
- solid phase techniques one is directed to Chemical and Enzymatic Synthesis of Gene Fragments - A Laboratory manual ed H G Gassen and L Lang, Verlag Chemiee, Weinheim 1 982; and Gait M J et al Nucleic Acids Research 1 982, 10, 6243; Spoat B S et al Tetrehedron Letters 1 980, 21 , 71 9; Matteuci M D et al J of American Chemical Society 1981 , 103, 31 85; S ⁇ nha N D et al Nucleic Acids Research 1 984, 1 2, 4539; Adams S P et al J American Chemical Society ' 1 983, 1 95; 661 ; and Matthes H ' W D et al Embro Journal 1984 , 3, 801 ; and are incorporated herein fully by reference.
- Reverse transceptase techniques may also be used to generate a complimentary c DNA strand by means of the reverse transciption of malaria parasite derived mRNA. Kits are available for this purpose.
- the DNA fragments may be ligated by either blunt-ended or staggered-ended termini using restriction enzymes; digestion; filling in as required; and treatment with alkali and phosphatase for protection and subsequent ligation with suitable ligases.
- leader sequences may be chosen from the many available.
- the cloning of the DNA sequences of the hybrid peptides of this invention may take place in prokaryotes such as E col ⁇ for example K1 2 strain 294 or E coli B or E coli XI 776 by way of non limiting examples or by means of the polymerase chain reaction.
- prokaryotes such as E col ⁇ for example K1 2 strain 294 or E coli B or E coli XI 776 by way of non limiting examples or by means of the polymerase chain reaction.
- Subseqent expression of the hybrid peptides may take place in any host cell, but preferably mammalian host cells.
- Other useful cells are fungi, yeasts, insects and prokaryotes.
- Signals suited to the chosen host cell are chosen as appropriate, in the case of prokaryotes one can chose from a large group including alkaline phosphatase and the like.
- prokaryotes are used to express the hybrid peptides, then they are transformed by an expression vector usually a plasmid such as PBR322 into which the DNA encoding the fusion peptide or fragment has been ligated such a plasmid will also feature suitable marker sequences, promoters and Shine-Dalgarno sequences may be chosen as appropriate.
- a prokaryote host such as E coli may be transformed by treatment using a solution of CaCI 2 as described by Cohen et al PNAS 1 973, j59; 21 10 or by treatment with a solution comprising a mixture of RbCI, Mncl 2 , potassium acetate and glycerol and then subsequently with 3 - (N-morpholino) - propene-sulphonic acid and RbCI and glycerol.
- the plasmid YRp7 as the expression vector may be used.
- the chosen host is a yeast such as Saccharomyces cerevisiae
- the plasmid YRp7 as the expression vector may be used.
- One is directed to Stinchcomb et al Nature 282, 39, (1 979), Kingsman et al Gene 7; 141 (1 979); Tschemper et al. Gene 10; 1 75 (1980).
- yeasts such as saccharoyces cerevisiae large segements such as 230 kilobases may be cloned successfully using yeast artificial chromosomes such as pYAC-4; Tony Triglia and David Kemp, Molecular and Biochemical Parasitology, 44, 1 991 , 207-21 2.
- yeast artificial chromosomes such as pYAC-4; Tony Triglia and David Kemp, Molecular and Biochemical Parasitology, 44, 1 991 , 207-21 2.
- yeast artificial chromosomes such as pYAC-4; Tony Triglia and David Kemp, Molecular and Biochemical Parasitology, 44, 1 991 , 207-21 2.
- promoters are available for use in yeast cell expression systems and include by way on non limiting examples 3-phosphoglycerate kinase and one is directed to Hitzman et al J Biol Chem 255; 2073 (1 980); also enolase, giyceraldehyde-3- phosphate dehydroginase, hexokinase, pyruvate decarboxylase, phosphofructokinase, gluocse-6-phosphate isomerase, 3-phosphoglycerate mutase, pyruvate kinase, triose phosphate isomerase, phospho glucose isomerase ad glucok ⁇ naise being glycolytic enzymes and one is directed to Hess et al J Adv Enzyme Reg X, 1 49 ( 1 968) and to Holland Biochemistry 1 7: 4900; 91 978.
- promoters suitable for yeast expression systems include the promoter regions for alcohol deyhydrognase 2, 1 00 cytocrome C, acid phosphatase, also mettallothionei ⁇ s. glycoraldehyde-3-phosphate dehydrogenase and others one is directed to H ⁇ tzman R et al European patent publication No 73, 657A.
- a suitable vector would be Baculovirus.
- Baculovirus Such a system would contain the target peptide encoding sequence linked to a baculovirus promoter within a shuttle vector with sufficient baculovirus DNA flanking the target peptide encoding sequence to permit recombination.
- Insect larvae can also be used to produce transformed insect cells, particularly Heiiothis v ⁇ rescens caterpillars and one is direced to PCT/WO/88/02030 Miller at al.
- cowpea plant provides a suitable expression system.
- CPMV cowpea mosaic virus
- mammalian cells are the chosen hosts for expression, these cells may be grown invitro in tissue culture or suitable bioreactors or in vivo in animals.
- Vectors useful for mammalian cells host systems involve the use of DNA derived from animal viruses such as SV40 virus; retroviruses such as RSV, MMTV, MOMLV, baculovirus, Vaccinovirus, Adenov ⁇ rus, polyoma or bovine papilloma virus.
- Promoters suitable for mammalian cells systems may be chosen from the many available. One is directed to Friers et al Nature 273; 1 1 3 (1987) and Greenway P J et al Gene 1 8; 353-360 ( 1 982) and Okayamah Mol Cell Biol 3; 280; 1 ; 1 983 by way of example.
- suitable enhancers may be chosen from the many available.
- One is directed to Laimins L et al PNAS 78; 993 (1 981 ) and Lusky M L et al Mol Cell Bio 3; 1 108 (1983) and Banerji J L et al Cell 33; 729 (1 983) and Osbourne T F et al Mol Cell Bio 4; 1293 (1984).
- Some techniques useful for the introduction of the expression vector in to the host cell involve protoplast fusion, calcium phosphate precipitation, electroporation and other techniques as mentioned herein before.
- the expression of the fusion peptide or fragment by the host may be confirmed by using an anti CD4 antibody such as a complimentary probe.
- an anti CD4 antibody such as a complimentary probe.
- One is directed to Dalgleish et al Nature 31 2; 763-766 (1 984), Klatzman et al Immunol Today 7; 291 -297 1986; McDougal et al J Immunol 1 53 1 35; 31 51 -31 62 1 985 and McDougal et al J Immunol 1 37 2937-2944 ( 1 986) and to verification experiments mentioned herein after.
- Mammalian host cells suitable for the expression of fragments of, or in some cases the entire fusion peptides may now be chosen from the large number now available such as VERO or CHO-K1 , the hybridoma SP2 ⁇ 0-AG 1 4 ot the myloma cell line P3x63Sgh or their P3xSgh derivatives also J558L, cells.
- eukaryotic host cells include COS cells; human embryonic kidney cells; mouse plasmacytoma cells; mouse sertoli cells; baby hamster kidney cells 9BHK cells); Chinese hamster ovary cells - DKFR (CHO); monkey kidney cells; African green monkey cells; human cervical carcinoma cells (HELA cells); human liver cells, and mouse mammary tumour cells by way of non limiting examples.
- the chosen host cells will preferably express the minimum levels proteases within their cytoplasm. It will also be appreciated that amino acid sequence variations of the peptide sequences involved may be insertions, subst ⁇ tutional or deletional variations involving single amino acid residues or peptide fragments; combinations of or multiples of such variations, or strain or species variation.
- the purpose of such variations may be to increase the affinity of the components, to improve stability, to reduce the cost of preparation or to increase the half life, or to lesson the severity of side effects such as atopic reactions.
- the final form of the molecular machines of the present disclosure may involve any combination of substitutions, delations or insertion of amino acid sequences, provided the binding ability of the epitopes or peptide sequences are retained.
- glycosylation variations ie variants completely unglycolated, variants having glycosylated amino acids other than those glycosylated in the natural peptides or variants having a greater number of amino acid residues glycosylated or fewer glycosylated residues or residues glycosylated byoligosaccharides other than those oligosaccharides usually associated with the said sequences.
- the peptides are recovered and purified by means known to the ordinary skilled artisan.
- Such methods may include acid extraction, ethanol precipitation using amonium sulphate, anion or cation exchange chromatography, phosphocellulose chromatography, amino affinity chromatography, hydroxyappetite chromatography and reverse phase chromatography.
- Extraction may be accomplished using sonication techniques; or solid sheartechniques - on a small scale.
- Lysozyme based techniques are expensive. More usually in the case of bacterial hosts homogenisers or liquid shear techniques are employed.
- a product is expressed as an inclusion body cell paste may be solublised by solvents such as 8M - guanidinium.
- Centrifugation provides the removal cellular debris, and continuous flow centrifuges are preferred for large scale operations.
- cross flow filtration using flat or tubular membranes and high shear forces may provide a useful alternative to centrifugation.
- Precipitation using amonium sulphate, organic solvents, polyethylene glycol or other polymers can be used to acomplish this step; with the addition of some variety of chromatography usually absorptive chromotagraphy techniques employing ion exchange, hydrophobic or bioaffinity interactions; followed by washing and desorption; chromatography is not restricted to columns by may take place in membranes or even in spirally wound cartridges.
- the rationalist approach seeks to produce conditions favouring the native state while at the same time keeping intermediates in solution.
- a problem may arise in connection with disulphide bridges which may form in non- native configurations.
- a way around this problem is to oxidise disulphides under denaturing conditions, using gel filtration remove covalent aggregates and thereafter dilute the product in a non denaturing buffer.
- the peptide which collapses into an amorphous tangle often rearranges itself into the native form.
- Highly resolving chromatography is the preferred technique for final purification.
- columns are preferred and techniques such as gel filtration, ion exchange, hydrophobic interaction or affinity chromatography may be used alone or in combination as dictated by economies of scale.
- Ion exchange chromtography techniques are very useful in early purification stages and can deal with large volumes at great speed, producing yields of high resolution.
- Hydrophobic interaction chromatography provides both high resolution and high speed even at large batch volumes.
- Bio affinity chromatography produces the highest resolution, at high speed, but batch size may require curtailment. This technique is an ideal late stage technique.
- Particle size is decreasing from the 90 um of traditional gels to approximately 40 um for newer gels such as Sepharose HR (R); Sephacryl HR (R); Superdex (R) (Pharmcia - LKB) or Fractogel (R) (Toso-Haas).
- newer gels such as Sepharose HR (R); Sephacryl HR (R); Superdex (R) (Pharmcia - LKB) or Fractogel (R) (Toso-Haas).
- the first chromatography column will involve large diameter packings circa 100 um which are often chosen so that they can by resanitised by sodium hydroxide to reduce costs. Low resolution steps to be followed by higher reoslution steps until the desired product purity is obtained.
- a typical strategy might involve:
- Step One Hydrophobic interaction chromatography at low ionic strength Purpose to absorb proteinases. Target peptides not absorbed and collected. Alternatively use proteinase inhibitors.
- Step Two Rerun through hlc column adding salts to bind the target peptide to the hlc column.
- the objective is volume reduction.
- Alternatively employ ultra filtration as described. Use a step gradient to elute the target peptide.
- Stage Three A step gradient to a low ionic stength buffer will remove remaining salts.
- Alternatively employ polyethyleneimine precipitation, centrifugation and diafiltration.
- Product polishing involves the removal of polymers of the product and other pyrogens.
- compositions for product polishing involve additional gel filtration with or without a buffer exchange step; treatment with alyhdrogel (aluminim hydroxide) or treatment with specific lecithins or anion exchanges.
- alyhdrogel aluminim hydroxide
- the hybrid peptides may be combined with carriers, binders, preservatives, antioxidants, antimcrobial agents and the like to formulate a pharmaceutical composition; and one is directed to the British and American Pharmacopoea incorporated herein by reference.
- intravenous administration of injectable compositions will be a principle route of administration of the hybrid peptides.
- other compositions for administration include microphores, lyosomes or sustained release formulations, replantable or microcapsular sustained release matrices; intra muscular administration may also be used.
- the hybrid peptides may also be components of capsules, tablets, creams, gels, lotions, drops, patches or may be administered where appropriate by any other of the known means of administration of a drug to a human where appropriate.
- Hydro phobic solvents may be used for intravenous or parentral administration in some circumstances.
- solvents are propylene glycol, polythylene glycol, vegetable oils, organic esters.
- Aqueous carriers which are preferred are by way of example water for injection, emulsions or suspensions, saline or buffers, alcoholic aqueous solutions, sodium chloride solutions, ringers lactate or dextrose.
- the manufacturing process only finishes with the secure packaging of the peptides in a suitable vacuum or atmosphere in suitable containers usually vials of glass or polymers with suitable stoppers.
- the said glass or polymer vials are then housed in a sturdy housing of cardboard or polymer after appropriate sealing.
- the cardboard housing will ideally be further wrapped in a clear polymer such as cellulose and provided with a seal to disclose tampering. All relevant information such as product name, date of manufacture, batch number and date of expriy and the like should be clearly visible and printed on the vials and housing as required by law and good manufacturing practice.
- the products should also be accompanied by leaflets, indicating material facts relevant to their use such as dosage, side effects, contra indications and the like it will be appreciated that the manufacturing process should take place in an appropriately designed facility, the floors, walls and ceilings of which should be finished to a high standard. Product flow must be arranged to reduce risk of mix up. Water systems employed should meet the US Pharmacopia specifications for pure water and be of injection quality in final purification stages. Compressed air and nitrogen must be filtered through C.2 u inch filters and be free of oil.
- Final purification stages should ideally take place under laminate flow hoods or equivalent in suitably clean rooms.
- Exemplary embodiment number 1 is directed to a molecular machine or hybrid protein which comprises a peptide sequence derived from the CD4 molecule or fragment thereof which possesses the capability of binding to HIV GP1 20, fused to a peptide sequence or fragment thereof derived from the merozoite glycophorin binding peptide GBP1 30.
- the peptide sequence of the CD4 molecule providing derived fragments useful for the preparation for the first component was disclosed by Paul Jay Maddon et al in Cell, Vol 42, 93-104, August 1985. See Figure 6 page 97; incorporated fully herin by reference.
- the second component peptide of the hybrid peptide of the present embodiment may be derived from the peptide sequence of GBP1 30 glycophorin binding peptide 130 as disclosed by Jarema Kochan et al in Cell Vol 44, 689-696 March 14 1 986. See figure 2 page 691 and incorporated fully herein by reference.
- the peptide sequence consisting of 1 -371 of the CD4 molecule is fused at the C terminal end to the N terminal end of a peptide sequence derived from the GBP130 molecule amino acid residues 201 -774 of the GBP molecule or a fragment thereof.
- val tyr lys lys glu gly glu gin val glu phe ser phe pro leu ala phe thr val glu lys leu thr gly
- This exemplary embodiment is also directed to a protein gene which comprises a DNA sequence which codes for the CD4 molecule or fragment thereof which binds HIV GP1 20 fused to a DNA sequence which encodes the glyphorin 1 30 or fragment thereof which possesses the capability of binding to red cells.
- DNA sequences suitable for forming the cloned DNA of the fusion peptide may be found in the disclosure of Paul Jay Maddon Cell, Vol 42 93-1 04, August 1 985 see Figure 6 page 97. Some or all of the DNA sequence coding for the CD4 molecule as disclosed may be fused to the DNA sequence conditioning for the DNA glyphorin binding peptide. It may be preferable to include the leader sequence of the CD4 molecule. It may also be preferable to delete the transmembrane and cytoplasmic domains. A DNA sequence encoding the glycophorin peptide 1 30 and suitable for the fabrication.
- Suitable sites of fusion might be in the region of site 733 ie Leu AAA or in the vicinity of site 736 ie val, GTT or in the vicinity of Site 739 ie Ser TCT in the case of GBP1 30; and in the region of site 1 257 for the CD4 derived sequence.
- CD4 DNA sequence is restricted at the BspM1 site position 514 and fused to some or all of the DNA sequence coding for GBP1-30 as disclosed above or restricted at other convenient sites
- the DNA sequence for the CD4 component is also obtained by restricting the DNA sequence encoding for the CD4 peptide component as previously disclosed at the Pvu 1 1 site, position 691 and joined to the cloned DNA encoding the glycophorin binding peptide 130 fragment as previously described or smaller fragments thereof, restricted as desired.
- Fusion will be carried out by suitable l ⁇ gases and gene fragments, even those gene fragments encoding additional amino acids or sequences of amino acids may be inserted to enable the ligation provided the inserted segments do not interfere with the functionality of either the CD4 derived peptide or the GBP1 30 derived peptide segment.
- the hybrid peptide will have 2 components; the first being a CD4 derived peptide sequence comprising amino acids 1 -1 77 ie + 1 gin - 1 77 val fused to a glyophorin binding protein (GBP) 1 30 derived peptide sequence such as Leu 226- Ala 774 or Thr 227 Ala 274, fusion to take place at Leu 226 or Thr 227 by way of example fusion to consist of a peptide bond formed between CD4 amino acid 1 77 and the glycophrin binding peptide 1 30 amino acid 226 or 227 fused at the C terminal end of the CD4 derived peptide and the N terminal end of the GBP1 30 peptide sequence.
- the said sequences and the enumeration of the said sequences to be found in the sources mentioned herein before.
- This variation of the embodiment contains only the tandem repeat sequence of the GBP- 1 30 molecule.
- a peptide sequence comprising the first 1 1 7 amino acids of the CD4 molecule or fragment thereof are fused C terminally to a peptide sequence of the glycophorin binding peptide GBP1 30.
- a suitable site of fusion would be Ala 370 N terminally to provide an amino acid lead sequence to be followed by the tandem repeat sequence beginning with Leu 376; the sequence terminating at Asp 425. It is appreciated that smaller sections of such a tandem sequence could be used as can smaller sections of the CD4 peptide sequence.
- the example is intended to be illustrative rather than limiting. 1 1 77 370..376 425
- the CD4 component peptide may be fused at the N terminal end to the C terminal end of the GBP1 30 derived peptide sequence. It was shown by P Yeh et al PNAS March 1 992 P1 904- 1 908 that functionality is not lost when CD4 is fused N terminally. EXEMPLARY EMBODIMENT NUMBER 2
- This exemplary embodiment is directed to a molecular machine or hydrbid peptide characterised by the fusion of some or all of the amino acid peptide sequence, amino acid residues 1-371 of the CD4 molecule fused at the C terminal end to a peptide sequence derived from the glycophorin binding peptide homologue molecule.
- Amino acid resides 70-427 or segments thereof of the glyophorin binding peptide homolgue(s).
- the amino acid sequence of the CD4 molecule has been disclosed by Paul Jay Maddon, the reference is given in Exemplary Embodiment No.1.
- glycophorin binding peptide homologue amino acid sequence is disclosed by Dagmar Nolte et al in Molecular and Biochem Parasitology 49, (1991) page 253-264 figure 2 page 257, incorporated fully herein by reference, gin val-ser ser
- 1-371 70-427 in one aspect is;
- This example is also directed to a molecular machine or fusion peptide which is characterised by the fusion of CD4 peptide sequence to a glycophorin binding peptide homologue sequence.
- the CD4 derived sequence comprises some or all of the amino acids from number 1-177 of the CD4 molecule.
- the sequence numbering and source of CD4 peptide sequence has been referenced herein before, see exemplary embodiment No. 1.
- glycophorin binding homologue amino acid residues 70-427 or some or part thereof.
- the source of glycophorin binding peptide homologue being the sequence disclosed by Dagmar Nolte (full reference given above).
- This example is also directed to a molecular machine or hybrid peptide formed by the fusion of an amino acid peptide sequence derived from the CD4 molecule amino acid residues 1 -371 or fragment thereof fused C terminally to a peptide sequence derived from the glycophorin binding peptide homologue fused N terminally at amino acid 109; the sequence comprising amino acids 1 09-427 some or part thereof.
- the sequencing of CD4 and glycophorin peptide binding homologue as already referenced.
- This example is also directed to a molecular machine or hybrid peptide which comprises the fusion of an amino sequence derived from the CD4 molecule the sequence as previously disclosed and consists of amino acid residues 1 -1 77 or fragments thereof fused C terminally to a tandem repeat sequence(s) derived from the glycophorin peptide binding homologue molecule as disclosed herein before.
- a sequence may consist of amino acid residues 230-268. It will be appreciated that an even shorter fragment could be used. It will be also appreciated that one or more of the tandem repeat sequences could be included. It will be further appreciated that the tandem repeat sequence could be shortened to an even smaller fragment containing only the RBC binding epitopes; and that this smaller fragment could be repeated.
- glycophorin binding peptide homologue molecules whose sequences are known but not as yet disclosed in the public domain could provide peptide sequences - tandem repeat units - fulfilling the functions of GBP130 or the GBPH sequences as disclosed above "in the same way” without departing from the scope and spirit of the invention.
- This example is further directed to a fused gene coding for a hybrid peptide comprising a amino acid sequence derived from the CD4 molecule joined to an amino acid sequence derived from the glycophorin binding peptide homologue molecule.
- a typical fused gene might consist of;
- Ligation to take place using ligases and additional linker segments by conventional means, known to the skilled artisan.
- This example is further directed to a fusion gene consisting of a DNA sequence encoding a peptide derived from the CD4 molecule truncated at the Pvull site 691 fused to a single tandem repeat encoding DNA sequence derived from glycophorin binding peptide homologue gene as disclosed by Nolte as referenced herin before or part thereof.
- This example is directed to a molecular machine or hybrid peptide formed by the fusion of a peptide sequence derived from the CD4 molecule or fragment thereof; the amino acid resides 1 -371 fused at their C terminal end to a peptide sequence derived from the EBA 175 antigen otherwise known as the erythrocyte binding antigen 175; amino acid residues 20-1435 all or part thereof.
- some or all ie fragments of the component peptide sequences may be used to fabricate the hybrid peptide.
- CD4 is truncated at 371 and EBA-175 truncated at 19 ie excluding the hydrophobic sequence. CD4 after Maddon. EBA-175 after Kim Lee Sim J Cell Biology Vol 111 1990, 1880
- 1-371 20-1435 in one aspect would be:
- This example is also directed to a molecular machine or fusion peptide comprising the fusion of a peptide sequence derived from CD4 consisting of the first 177 amino acid residues or fragment peptide sequence thereof; amino acid 1-177 fused at the C terminal end to the N terminal end of a peptide sequence derived from EBA 175 amino acid 1062-1103.
- This particular peptide sequence is also named EBA peptide 4 and has the ability to inhibit the binding of EBA 175 to red blood cells.
- Also included would be fused genes formed by the fusion of DNA sequences encoding the CD4 molecule fused to Sequences encoding the erythrocyte binding antigen 175 or its derived epeptide EBA peptide 4.
- the DNA sequences of the EBA peptide number 4 and EBA 175 molecules may be obtained from the Journal of Cell Biology Vol 111 1990, p 1880 in the article by J Kim Lee Sim et al in the same journal.
- the CD4 component may be fused at the N terminal end to the
- the P Vivax Duffy Receptor molecule is joined to the CD4 receptor molecule both truncated at their transmembrane extrecellular domain regions.
- amino acid sequence of CD4 as presented in this embodiment is the same as for Maddon P N A Sci 1987 but in reverse order.
- the CD4 peptide segment may be fused at the N terminal end to the C terminal of the P Vivax Duffy peptide, or at the C terminal end of the CD4 molecule to the N terminal of the P Vivax Duffy Receptor; by peptide bonds.
- This embodiment is also directed to a molecular machine or hybrid peptide comprosing the fusion or joining together of two amino acid sequences; one derived from the CD4 molecule and consisting some of a fragment of amino acid or peptide sequence derived from plasmodium Vivax Duffy Receptor molecule or fragment thereof; amino acid sequence 23-1051 joined at the C terminal ends which will require that the component sequences be expressed separately in separate hosts and fused subsequently.
- J is a joining segment or cross linker in one aspect
- This example is directed to a molecular machine or hybrid peptide formed by the fusion of a peptide sequence derived from the P Knowlesi Duffy Receptor binding molecule as disclosed by Xiangdong Fang in Molecular and Biochemical Parasitology 44, (1991) 125-132 see figure 1 P 127.
- the choice of peptide fragment is non- critical provided it contains erythrocyte binding ability and the method of joining to the CD4 component is also non critical, examples of typical methods of joining peptides are given -in the preceding examples.
- the fusion may also be by peptide bonds N terminal of CD4 component to C terminal of P Knowlesi or N terminal of P Knowlesi component to C terminal of CD4 component
- This example is directed to a molecular machine or hybrid peptide formed by the fusion of a peptide sequence derived from the expression of the PMMSA gene or precursor to the major merozoite surface antigen genes as disclosed by M Gregory Peterson in Molecular and Biochemical Parasitology, 27 (1988) P291 -302.
- One is directed to p294 and p295 figure 3.
- nucleic acid residues 177-5169 all or part fused at the C terminal end to the C terminal end of the __3 CD4 derived peptide sequence such as formed from the expression of nucleic acid residues 1 14-1260 or fragment thereof.
- the fusion at the C terminal end being accomplished by suitable joining sequences chemical ligands or sulphidryl bonds or other means.
- cys residues be added as required as described in exemplary embodiment number 14; the site of insertion is non critical providing binding function is maintained.
- Exemplary embodiment number 7 contemplates joining sequences suitable for joining two peptide fragments at the C terminal ends.
- the joining sequences could be a straight peptide chain which will have cys amino acid residues added to form sulphidryl bonds with cys residues also inserted into the peptide sequences to be joined.
- Other methods of ligation are also envisaged.
- a suitable straight segment, but by no means the only suitable straight segment may be found in the joining chain amino acid resodues 95-1 1 2 inclusive of the CD4 molecule; the sequence disclosed by Paul Jay Maddon and already referenced herein before.
- 'Cys' residues may be inserted at any site for example between amino acid 97 and 98 and between 107 and 108 to name but two possible sites.
- the CD4 molecule is part of the immunoglobulin superfamily.
- the immunoglobulin superfamily provide many examples of straight and bent chains with cys already inserted for the purposes of such bonding.
- Exemplary embodiment number 8 illustrates how a joining sequence J can be used to join an EBA1 75 fragment with a GBP1 30 fragment and both to be joined to a CD4 derived peptide or variations already listed.
- the J peptide sequence may be similar to that already provided or have additional cys residues added.
- This example envisages molecular machined formed by the fusion of ZOOANOTIC MEROZOITE derived peptide fragments whose function in nature is to bind with non human red blood cells, fused to CD4 segments in the manner already suggested.
- Useful sources of peptides are Plasmodium Chabaudi Plasmodium Yeolii Plasmodium Berghei Plasmodium Gallinacium Plasmodium Cyanomogli
- a sample of human blood to be tested is mixed with a known quantity of animal blood treated with the molecular machines of example 9.
- the sample is agitated and thereafter the animal RBC removed selectively by anti-animal RBC antibodies bound to argarose or polyacrylamide gels or the like.
- Fluorescent labelled anti HIV antibody is then employed demonstrate virus particles adherent to the animal red cell. Quantification may then take place by automated systems in the usual way.
- Purified samples of the molecular machines in amounts ranging from 0.1 ug to 1 ug are used to coat the walls of ELISA plates and are thereafter incubated with preparations of 0KT4A mAb (Ortho Diagnostics); the activity of the 0KT4A are first altered by incubation with varying amounts of soluble CD4 preparations. A labelled anti-mouse serum is then used to assess the amount of bound mouse antibody.
- Purified samples of the molecular machines in amounts ranging from 0.1 ug to 1 ug are used to coat the walls of ELISA plates and are thereafter incubated with monoclonal antibodies directed against the erythrocyte binding peptide component. Binding is revealed by goat anti mouse antibodies the said goat antibodies to be labelled by conventional means.
- a buffered solution containing 100,000 human red cells is incubated with 20 ug of fusion peptides.
- the washed red cells are thereafter incubated with 1 x 10" M of 0KT4A (Ortho Diagnostics).
- the RBC's are washed again with appropriate isotonic buffer and incubated with peroxidase labelled goat anti mouse IGG serum (BIOYSIS). Further washing and the addition of 3, 3', 5, 5' - tetramethylbenzidine permits assessment of binding by the measurement of absorbance at 600 rim.
- BIOYSIS peroxidase labelled goat anti mouse IGG serum
- a known quantity of a purified sample of fusion peptide 0.1 ug to 1 ug in increments of x 2 are incubated with 100,000 RBC's in isotonic buffer solution.
- 1 25-1 labelled 0KT4A or otherwise labelled 0KT4A are added at set times 1 min, 2 min, 4, 8 , 1 6, 30 min and 1 hour; the red cells arecentrifuged through Percoll 2 minutes after the addition of 1 25-1 labelled 0KT4A. Accordingly both the optimum ratio of RBC to molecular machines can be calculated together with the binding time to saturation of each incremental dose of fusion peptides.
- 50 ml samples of blood are obtained from HIV positive and AIDS patients and divided into of 10 ml after appropriate buffering and anticoagulation.
- One 1 0 ml sample from each patient is assayed for viral titre levels by conventional means.
- the remaining 4 samples of 10 ml of blood from each patient are treated by molecular machines of exemplary embodiments 1 ,2,3,4,5,6 and 8 in amounts ranging from 0.1 ug to 1 mg and thereafter assayed for viral titre levels by conventional means. Additional smaples are centrifuged and the packed cells rediluted and assayed for viral titre levels separate from the plasma also assayed.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Toxicology (AREA)
- Genetics & Genomics (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Cell Biology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Description
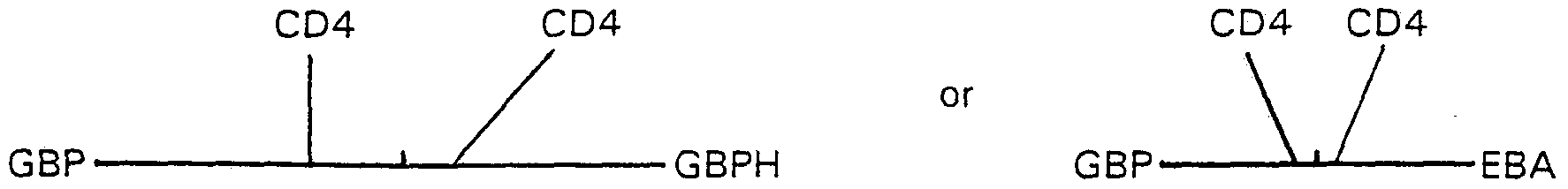







Claims
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/307,742 US7585508B1 (en) | 1992-03-11 | 1993-03-10 | Fusion proteins comprising CD4 and the malaria parasite merozoite glycophorin binding protein 130 (GBP-130) |
| DE69329145T DE69329145D1 (en) | 1992-03-11 | 1993-03-10 | Anti-viral fusion peptides |
| AU36447/93A AU3644793A (en) | 1992-03-11 | 1993-03-10 | Anti-viral fusion peptides |
| EP93905560A EP0630407B1 (en) | 1992-03-11 | 1993-03-10 | Anti-viral fusion peptides |
| US12/583,780 US20110082075A1 (en) | 1992-03-11 | 2009-08-27 | Anti-viral fusion peptides |
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9205276.0 | 1992-03-11 | ||
| GB929205276A GB9205276D0 (en) | 1992-03-11 | 1992-03-11 | Anti-viral fusion peptides |
| GB9214481.5 | 1992-07-08 | ||
| GB929214481A GB9214481D0 (en) | 1992-07-08 | 1992-07-08 | Imaging compositions |
| GB929215829A GB9215829D0 (en) | 1992-07-24 | 1992-07-24 | Anti-viral fusion peptides |
| GB9215829.4 | 1992-07-24 | ||
| GB929219562A GB9219562D0 (en) | 1992-03-11 | 1992-09-16 | Anti-viral peptides |
| GB9219562.7 | 1992-09-16 | ||
| GB939304311A GB9304311D0 (en) | 1992-03-11 | 1993-03-03 | Anti-viral fusion peptides |
| GB9304311.5 | 1993-03-03 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/583,780 Division US20110082075A1 (en) | 1992-03-11 | 2009-08-27 | Anti-viral fusion peptides |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1993018160A1 true WO1993018160A1 (en) | 1993-09-16 |
Family
ID=27517106
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB1993/000505 WO1993018160A1 (en) | 1992-03-11 | 1993-03-10 | Anti-viral fusion peptides |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP0630407B1 (en) |
| DE (1) | DE69329145D1 (en) |
| WO (1) | WO1993018160A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995006737A1 (en) * | 1993-09-03 | 1995-03-09 | Kenneth Francis Prendergast | Glycophorin binding protein (gbp130) fusion compositions |
| WO1995007353A2 (en) * | 1993-09-10 | 1995-03-16 | The Government Of The United States Of America, Represented By The Secretary Of The Department Of Health And Human Services | Binding domains from plasmodium vivax and plasmodium falciparum erythrocyte binding proteins |
| US5993827A (en) * | 1993-09-10 | 1999-11-30 | The United States Of America As Represented By The Secretary, Department Of Health And Human Services | Binding domains from plasmodium vivax and plasmodium falciparum erythrocyte binding proteins |
| US6410695B1 (en) | 1995-02-21 | 2002-06-25 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Individual medicament dosing conjugate |
| US20060018912A1 (en) * | 2004-05-28 | 2006-01-26 | University Of Massachusetts | Snares for pathogenic or infectious agents and uses related thereto |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9517257B2 (en) | 2010-08-10 | 2016-12-13 | Ecole Polytechnique Federale De Lausanne (Epfl) | Erythrocyte-binding therapeutics |
| US9850296B2 (en) | 2010-08-10 | 2017-12-26 | Ecole Polytechnique Federale De Lausanne (Epfl) | Erythrocyte-binding therapeutics |
| AU2011289579B2 (en) | 2010-08-10 | 2016-11-17 | Ecole Polytechnique Federale De Lausanne | Erythrocyte-binding therapeutics |
| US10953101B2 (en) | 2014-02-21 | 2021-03-23 | École Polytechnique Fédérale De Lausanne (Epfl) | Glycotargeting therapeutics |
| US10046056B2 (en) | 2014-02-21 | 2018-08-14 | École Polytechnique Fédérale De Lausanne (Epfl) | Glycotargeting therapeutics |
| US10946079B2 (en) | 2014-02-21 | 2021-03-16 | Ecole Polytechnique Federale De Lausanne | Glycotargeting therapeutics |
| BR112016019274A2 (en) | 2014-02-21 | 2017-10-10 | Anokion Sa | glyco-oriented therapeutic agents |
| EP3638296A1 (en) | 2017-06-16 | 2020-04-22 | The University Of Chicago | Compositions and methods for inducing immune tolerance |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0298280A2 (en) * | 1987-06-30 | 1989-01-11 | HAPGOOD, C.V., a Netherlands Antilles Limited Partnership | Animal derived cell with antigenic protein introduced therein |
| WO1989002922A1 (en) * | 1987-10-02 | 1989-04-06 | Genentech, Inc. | Adheson variants |
-
1993
- 1993-03-10 WO PCT/GB1993/000505 patent/WO1993018160A1/en active IP Right Grant
- 1993-03-10 DE DE69329145T patent/DE69329145D1/en not_active Expired - Lifetime
- 1993-03-10 EP EP93905560A patent/EP0630407B1/en not_active Expired - Lifetime
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0298280A2 (en) * | 1987-06-30 | 1989-01-11 | HAPGOOD, C.V., a Netherlands Antilles Limited Partnership | Animal derived cell with antigenic protein introduced therein |
| WO1989002922A1 (en) * | 1987-10-02 | 1989-04-06 | Genentech, Inc. | Adheson variants |
Non-Patent Citations (2)
| Title |
|---|
| MOL. BIOCHEM. PARASITOL. vol. 49, no. 2, 1991, ELSEVIER, AMSTERDAM, NY,US; pages 253 - 264 D. NOLTE ET AL. 'A Plasmodium falciparum blood stage antigen highly homologous to the glycophorin binding protein GBP' cited in the application * |
| PROC. NATL. ACAD SCI. vol. 88, no. 8, 15 April 1991, NATL. ACAD SCI., WASHINGTON,DC,US; pages 3305 - 3309 R.P.TAYLOR ET AL. 'Use of heteropolymeric monoclonal antibodies to attach antigens to the C3b receptor of human erythrocytes: A potential therapeutic treatment' cited in the application * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995006737A1 (en) * | 1993-09-03 | 1995-03-09 | Kenneth Francis Prendergast | Glycophorin binding protein (gbp130) fusion compositions |
| WO1995007353A2 (en) * | 1993-09-10 | 1995-03-16 | The Government Of The United States Of America, Represented By The Secretary Of The Department Of Health And Human Services | Binding domains from plasmodium vivax and plasmodium falciparum erythrocyte binding proteins |
| WO1995007353A3 (en) * | 1993-09-10 | 1995-06-01 | Us Health | Binding domains from plasmodium vivax and plasmodium falciparum erythrocyte binding proteins |
| US5993827A (en) * | 1993-09-10 | 1999-11-30 | The United States Of America As Represented By The Secretary, Department Of Health And Human Services | Binding domains from plasmodium vivax and plasmodium falciparum erythrocyte binding proteins |
| US6392026B1 (en) | 1993-09-10 | 2002-05-21 | The United States Of America As Represented By The Department Of Health And Human Services | Binding domains from plasmodium vivax and plasmodium falciparum erythrocyte binding proteins |
| US6962987B2 (en) | 1993-09-10 | 2005-11-08 | The United States Of America As Represented By The Department Of Health And Human Services | Binding domains from Plasmodium vivax and Plasmodium falciparum erythrocyte binding proteins |
| US6410695B1 (en) | 1995-02-21 | 2002-06-25 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Individual medicament dosing conjugate |
| US20060018912A1 (en) * | 2004-05-28 | 2006-01-26 | University Of Massachusetts | Snares for pathogenic or infectious agents and uses related thereto |
| US9493538B2 (en) * | 2004-05-28 | 2016-11-15 | University Of Massachusetts | Snares for pathogenic or infectious agents and uses related thereto |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0630407B1 (en) | 2000-08-02 |
| EP0630407A1 (en) | 1994-12-28 |
| DE69329145D1 (en) | 2000-09-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20110082075A1 (en) | Anti-viral fusion peptides | |
| CN108137708B (en) | Human factor IX fusion protein and preparation method thereof and purposes | |
| US5817767A (en) | Synergistic composition of CD4-based protein and anti-HIV-1 antibody, and methods of using same | |
| Larkin et al. | Oligosaccharide-mediated interactions of the envelope glycoprotein gp120 of HIV-1 that are independent of CD4 recognition | |
| Ezekowitz et al. | The structure and function of vertebrate mannose lectin-like proteins | |
| US6043347A (en) | Compositions and methods for treating viral infections | |
| EP0630407B1 (en) | Anti-viral fusion peptides | |
| JPH05503009A (en) | Fusion protein consisting of a ligand-binding protein and a stable plasma protein | |
| ZA200006385B (en) | Antigenic complex comprising immunostimulatory peptide, CD4, and chemokine receptor domain for HIV treatment and immune disorders. | |
| EP0465633A1 (en) | C4 binding protein fusion proteins | |
| EP1340769B1 (en) | CD4-gamma2 and CD4-IgG2 chimeras | |
| IL102092A (en) | Use of recombinant hiv envelope protein in medicament for treating hiv and therapeutic composition containing the agglomerated protein | |
| CN104356220B (en) | A kind of have AntiHIV1 RT activity Aedes albopictus albumen of anticoagulant function and preparation method thereof | |
| JP2004509621A (en) | Ligand / receptor-specific converters that redirect antibodies to receptors on pathogens | |
| AU2006200454B2 (en) | Compositions and methods for treating viral infections | |
| Taverna et al. | A multi-mode chromatographic method for the comparison of the N-glycosylation of a recombinant HIV envelope glycoprotein (gp160s-MN/LAI) purified by two different processes | |
| JP3364181B2 (en) | Human immunodeficiency virus envelope polypeptide and its antibody | |
| US7070991B2 (en) | Cells expressing a CD4-IgG2 chimeric heterotetramer | |
| Liu | Natural and biotech‐derived therapeutic proteins: What is the future? | |
| CA2065029A1 (en) | Therapeutic compositions; methods for treatment of hiv infections | |
| AU2004201321A1 (en) | Compositions and methods for treating viral infections | |
| AU2006200455A1 (en) | Compositions and methods for treating viral infections |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AT AU BB BG BR CA CH CZ DE DK ES FI GB HU JP KP KR LK LU MG MN MW NL NO NZ PL PT RO RU SD SE SK UA US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 08307742 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1993905560 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993905560 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| NENP | Non-entry into the national phase |
Ref country code: CA |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1993905560 Country of ref document: EP |