WO1993015120A1 - Verwendung von emulsionspfropfcopolymerisaten als filmbildner in kosmetischen und pharmazeutischen zubereitungen und verfahren zur herstellung der emulsionspfropfcopolymerisate - Google Patents
Verwendung von emulsionspfropfcopolymerisaten als filmbildner in kosmetischen und pharmazeutischen zubereitungen und verfahren zur herstellung der emulsionspfropfcopolymerisate Download PDFInfo
- Publication number
- WO1993015120A1 WO1993015120A1 PCT/EP1993/000118 EP9300118W WO9315120A1 WO 1993015120 A1 WO1993015120 A1 WO 1993015120A1 EP 9300118 W EP9300118 W EP 9300118W WO 9315120 A1 WO9315120 A1 WO 9315120A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- monomers
- emulsion graft
- grafted
- graft copolymers
- vinyl
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61Q—SPECIFIC USE OF COSMETICS OR SIMILAR TOILETRY PREPARATIONS
- A61Q5/00—Preparations for care of the hair
- A61Q5/06—Preparations for styling the hair, e.g. by temporary shaping or colouring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/72—Cosmetics or similar toiletry preparations characterised by the composition containing organic macromolecular compounds
- A61K8/91—Graft copolymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F271/00—Macromolecular compounds obtained by polymerising monomers on to polymers of nitrogen-containing monomers as defined in group C08F26/00
- C08F271/02—Macromolecular compounds obtained by polymerising monomers on to polymers of nitrogen-containing monomers as defined in group C08F26/00 on to polymers of monomers containing heterocyclic nitrogen
Definitions
- the present invention relates to the use of emulsion graft copolymers as film formers in cosmetic and pharmaceutical preparations.
- the invention further relates to an improved process for the preparation of these emulsion graft copolymers.
- film formers for cosmetic preparations, in particular for hair cosmetics.
- these are synthetically obtained polymers that are applied to the hair in dissolved form by rubbing or spraying on, where they leave a transparent, colorless film after the solvent has evaporated.
- the film produced on the hair is intended to solidify it elastically and impart elasticity to it without adversely affecting its natural fall.
- the film should adhere firmly to the hair and not peel off or dust off. It should be clear, shiny and not sensitive to moisture, so that the treated hairstyle remains dimensionally stable even in high air humidity, does not stick and binds dust.
- the polymer should be able to be washed out without residue using a commercially available shampoo.
- the sprayability of the film-forming solution requires sufficient solubility of the polymer in the solvents customary for hair treatment agents and, in the case of application as a spray from a pressure pack, the perfect miscibility of the solution with the blowing agents.
- US Pat. No. 3,594,344 (1) describes emulsion graft copolymers for hairspray formulations which have been prepared by grafting a monomer mixture of alkyl acrylates and at least 1% by weight glycidyl methacrylate onto polymeric N-vinyllactam.
- a disadvantage of these polymers However, ten is not only the toxicity of glycidyl methacrylate but also the high tendency to crosslink this bifunctional monomer. For example, this can lead to storage instability of the dispersion.
- the epoxy groups of the polymer can form a permanent chemical bond with the protein substance of the hair and the hair bottom. Such polymers cannot be washed out and are rejected as physiologically questionable.
- ÜS-A 3 770 683 (2) describes emulsion graft copolymers for hairspray formulations which are grafted onto polymeric N- by grafting a monomer mixture of (meth) acrylic esters and at least 0.5% by weight of an ethylenically unsaturated monocarboxylic acid. Vinyl lactam were produced.
- Such carboxyl-containing polymers however, have to be at least partially neutralized with bases in order to be washable again from the hair, for which purpose mostly amino alcohols are used.
- the scope for the degree of neutralization is relatively narrow; a small amount of base does not leave a washable film on the hair, with a higher proportion of base the film becomes soft and sensitive to moisture, which leads to strong dust binding and quickly makes the hairstyle unsightly.
- the use of acrylic acid also harbors the risk that homopolymer thickening from polyacrylic acid arises during the polymerization.
- the acrylic acid content can also lead to incompatibilities with cationic formulation components.
- the object of the present invention was therefore to replace the means known from the prior art with those which do not have the disadvantages described.
- the document (3) relates to (meth) acrylic esters as grafted monomers, the document (5) to a mixture of vinyl acetate and vinyl stearate and the document (4) to vinyl ester, although in the latter case only 5 up to 20% by weight of the vinyl pyrrolidone present in the resulting copolymer were used as the polymeric graft component.
- the mode of addition of the comonomers is carried out according to the customary technique, in which part is introduced and the rest is added continuously.
- emulsion graft copolymers made from polyvinyl lactams or water-soluble copolymers made from N-vinyl lactams and copolymerizable ethylenically unsaturated comonomers as the graft base and vinyl esters of C 2 to cis carboxylic acids or acrylic or methacrylic acid C Ci to Ci ⁇ alkyls or mixtures thereof can be found as grafted monomers as film formers in cosmetic and pharmaceutical preparations.
- the emulsion graft copolymers to be used according to the invention are particularly suitable as film formers in hair cosmetic preparations, especially in hair setting agents such as hair sprays, aqueous hair setting solutions, hair gels or hair foams, and as film formers, i.e. Binding or coating agents, for pharmaceutical slow-release formulations, coatings and matrix tablets.
- the emulsion graft copolymers to be used according to the invention are excellent film formers for the formulations of hair sprays and other hair fixatives. They can be added to bases and hygroscopes without further additions. Applying hydrophobizing agents to the hair, solidify it elastically, give it shine, adhere well, impart neither hygroscopicity nor sensitivity to moisture, show reduced stickiness at high atmospheric humidity and can be easily removed from the hair with commercially available hair washing agents.
- the emulsion graft copolymers can generally be applied directly to the hair as a dispersion, that is to say free of solvents and blowing agents, for example as a pump spray.
- Solid pharmaceutical forms with controlled release of the active ingredient through the use of a polymer are mostly used as matrix tablets, film tablets or coated pellets or granules with or without a hard gelatin shell. These polymers envelop the active ingredient and release it in gastric or intestinal juice according to the principle of slow diffusion through the polymer or erosion of the polymer as a function of the pH or independently of it. One goal must be to get sufficient release initially to reach the minimum drug concentration in the blood, followed by a slower drug release phase.
- the essentially water-insoluble emulsion graft copolymer described is used either as an aqueous dispersion, as a spray-dried or freeze-dried powder which is very easily and finely redispersible in water, or as a solution in an organic solvent.
- the technique of moist granulation of the active ingredient in a kneader or mixer or fluidized bed granulation is usually used in the production of the pharmaceutical matrix tablets or granules described.
- the dispersion of the emulsion graft copolymer can be used as a binder suspension, the desired concentration and viscosity of this dispersion being easy to set.
- the active ingredient can be mixed with the emulsion graft copolymer as a powder and this mixture can be granulated with a solvent, preferably water or an alcohol, using the abovementioned techniques. A combination of both methods can also be useful in some cases.
- a filler to the active ingredient, e.g.
- lactose starch or calcium hydrogen phosphate
- an anti-adhesive such as talc
- the active ingredient can be dissolved or finely dispersed in the aqueous dispersion according to the invention or in the organic solution of the emulsion graft copolymer.
- the coating of carrier material particles e.g. Sugar pearls
- this solution or dispersion in the usual way, e.g. in a fluidized bed until the desired amount of active ingredient has been applied.
- Another more or less strong coating of the pellets with a solution or dispersion of the Emulsions ⁇ graft copolymer without active ingredient is used to control the release of active ingredient from the pellets.
- an aqueous dispersion or an organic solution of the emulsion graft copolymer is used together with the additives customary for film coatings, e.g. Pigments, colored lacquers or talcum, applied in the coating pan, Accela-Cota, fluidized bed or comparable apparatus to the active substance-containing dragee cores.
- the additives customary for film coatings e.g. Pigments, colored lacquers or talcum
- the emulsion graft copolymer to be used according to the invention results in pH-independent control of the release of active substance, which is comparable to that of other previously used slow-release film formers, e.g. of an ethyl acrylate-methyl methacrylate copolymer is comparable or even better.
- the release profile of the active substance can be controlled by varying the amount of polymer used.
- N-vinyl lactams for the preparation of the graft base polyvinyl lactams or copolymers thereof 5-, 6- or 7-membered cyclic carboxylic acid amides of the general formula are particularly suitable
- n stands for the number 3, 4 or 5, e.g. 1-vinyl pyrrolidone, 1-vinyl-5-methyl pyrrolidone, 1-vinyl piperidone or N-vinyl ⁇ -caprolactam; 1-vinylpyrrolidone is preferred.
- Polymers made from various N-vinyl lactams can also be used as the graft base.
- polyvinyl lactams or water-soluble copolymers of N-vinyl lactams and acrylic acid or methacrylic acid C 1 -C 8 -alkyl esters which additionally contain hydroxyl groups or mono-C 1 -C 4 -alkylamino- or di as the graft base are used as the graft base -Ci to C -alkylamino groups can carry vinyl esters of C 2 - to Ci 8 ⁇ carboxylic acids, ethylenically unsaturated C - to C 5 -M0- no- or dicarboxylic acids or their anhydrides, mono-Ci- to Ci ⁇ -alkyl esters of ethylenically unsaturated C - to Cs-dicarboxylic acids, half-amides of ethylenically unsaturated C - to Cs-dicarboxylic acids, vinyl derivatives of nitrogen-containing heterocyclic compounds, N-mono-C ⁇ ⁇ to C
- methacrylic acid amides methacrylic acid amides, anionic comonomers with sulfo groups or cationic comonomers with quaternary ammonium groups.
- Suitable alcohol components in the acrylic acid or methacrylic acid ci to cis alkyl esters are in particular the usual lower alkanols and the fatty alcohols, e.g. Methanol, ethanol, n-propanol, iso-propanol, n-butanol, iso-butanol, tert-butanol, lauryl alcohol, myristyl alcohol, cetyl alcohol or stearyl alcohol.
- fatty alcohols e.g. Methanol, ethanol, n-propanol, iso-propanol, n-butanol, iso-butanol, tert-butanol, lauryl alcohol, my
- Methyl, ethyl, n-butyl and tert-butyl esters of acrylic acid and methacrylic acid are particularly preferred.
- Acrylic or methacrylic acid esters containing hydroxyl or amino groups in the alcohol radical include, for example, 2-hydroxyethyl ester, 3-hydroxypropyl ester, 4-hydroxybutyl ester, 2- (monomethylamino) ethyl ester, 2- (dimethylamino) ethyl ester, 2- (Diethylamino) ethyl ester, 4- (dimethylamino) butyl ester and 4- (diethylamino) butyl ester.
- Vinyl esters of C 2 - to C ⁇ s-carboxylic acids which are mainly saturated aliphatic C 2 - to Cis-monocarboxylic acids, are, for example, the vinyl esters of acetic acid, propionic acid, butyric acid, isobutyric acid, valeric acid, isovaleric acid To name pivalic acid, lauric acid, myristic acid, palmitic acid and stearic acid.
- Vinyl esters of C - to Cs-carboxylic acids are preferred, but in particular vinyl acetate.
- Suitable ethylenically unsaturated C to Cs mono- or dicarboxylic acids or their anhydrides are, for example, acrylic acid, methacrylic acid, crotonic acid, maleic acid, maleic anhydride, fumaric acid or itaconic acid.
- Examples of mono-Ci to Cis-alkyl esters of ethylenically unsaturated C to Cs dicarboxylic acids include Maleic acid monomethyl ester, maleic acid monoethyl ester, fumaric acid monomethyl ester, fumaric acid monoethyl ester or the two isomeric ren itaconic acid monomethyl esters.
- Suitable half-amides of ethylenically unsaturated C - to Cs-dicarboxylic acids are maleic acid half-amide, fumaric acid halamide or the two isomeric itaconic acid halamides.
- N-vinylimidazole, N-vinylimidazoline, N-vinylimidazolidine and 2-, 3- or 4-vinylpyridine are used in particular as vinyl derivatives of nitrogen-containing heterocyclic compounds.
- Suitable N-mono-C 1 -C 4 -alkyl or N, N-di-C 1 -C 8 -alkyl-acrylic acid or -methacrylic acid amides are, for example, N-methylacrylic acid amide, N, N-dimethylacrylic acid amide, N-methyl -methacrylic acid amide or N, N-dimethyl-methacrylic acid amide.
- Particularly suitable anionic comonomers with sulfo groups are acrylamido-C 1 -C 4 -alkyl sulfonic acids.
- Mainly ⁇ -tri-C ⁇ to C -alkylammonium-alkyl (meth) acrylate halides such as 2-trimethylammonium-ethyl methacrylate chloride serve as cationic comonomers.
- Copolymers of N-vinyl lactams and the specified co-monomers are usually obtained by copolymerization of 30 to 99% by weight, preferably 50 to 99% by weight, in particular 70 to 99% by weight, of the vinyl lactam with 1 to 70% by weight. %, preferably 1 to 50% by weight, in particular 1 to 30% by weight, of the comonomer.
- polyvinylpyrrolidone is used as the graft base.
- This is usually to be understood as a homopolymer of N-vinylpyrrolidone with a Fikentscher K value of 10 to 100, preferably 11 to 33, in each case measured in 1% strength by weight aqueous solution at 25 ° C.
- vinyl esters of C 2 to cis carboxylic acids and acrylic acid or methacrylic acid cis to C ⁇ s alkyl esters as described above are used as grafted monomers.
- the monomers mentioned to be grafted on can each be used alone or mixtures of different vinyl esters, mixtures of different acrylates or methacrylates or mixtures of vinyl esters and (meth) acrylates can be used.
- vinyl esters of C 2 -C 6 -carboxylic acids, in particular vinyl acetate are used as the only monomers to be grafted on.
- the grafted monomers should be the main constituent in the resulting graft polymer. It has proven to be advantageous to use the graft base and monomers to be grafted on in a weight ratio of 10:90 to 60:40, preferably 20:80 to 50:50, in particular 25:75 to 40:60.
- Emulsion graft copolymers are to be understood as meaning graft copolymers which have been prepared by emulsion polymerization.
- Graft copolymers are understood here to mean polymers which are obtained by grafting a monomer component consisting of one or more types of monomers onto a graft base which can be obtained by prepolymerizing one or more types of monomers. were manufactured. Part of the grafted-on monomer component can also be present in the resulting graft copolymer as isolated polymer chains without grafting having taken place.
- the present invention furthermore relates to an improved process for the preparation of emulsion graft copolymers from polyvinyl lactams or water-soluble copolymers from N-vinyl lactams and copolymerizable ethylenically unsaturated comonomers as the graft base and vinyl esters of C 2 -C 6 -carboxylic acids or acrylic acid or methacrylic acid -C ⁇ to C ⁇ s alkyl esters or mixtures thereof or mixtures of the esters mentioned with acrylic acid or methacrylic acid as grafted monomers by emulsion graft polymerization in water at a polymerization temperature of 40 to 100 ° C. in the presence of conventional water-soluble initiators and the usual ones for this Emulsifiers and protective colloids, which is characterized in that
- step (a) 3 to 8%, in particular 5 to 6% of the total amount of monomer to be grafted on are introduced in step (a), 42 to 48%, in particular 44 to 45% of the total amount in step (b) and in step ( c) the remaining 47 to 53%, in particular 50 to 51% of the total amount added.
- the reaction mixture is left in step (c) for 5 to 60 min, in particular 10 to 30 min, at the polymerization temperature.
- the rate of addition of the amount of monomer to be grafted on in step (b) should be higher than in step (d).
- Usual addition times on a laboratory scale are, for example, 60 to 80 min for step (b) and 120 to 180 min for step (d).
- step (a) Another important aspect is the specification of part of the initiator in step (a). This part of the initiator should make up a significant part of the total. It has proven to be particularly advantageous to use at least 50% of the total amount of initiator, in particular at least 54% of the total amount of initiator, in step (a).
- step (a) It also contributes to the success of the process according to the invention if the amount of initiator in step (a) is added at somewhat elevated temperatures. It is particularly advantageous to add the initiator between 30 ° C., in particular 35 ° C., and the polymerization temperature. The amount of initiator in step (a) can be carried out all at once, continuously lent during the heating or in portions at various temperatures.
- steps (b), (d) and (e) are comparatively uncritical. It can be carried out, for example, continuously or in portions at the beginning of the respective step.
- the emulsion graft polymerization according to the invention is carried out in water in which polyvinyl lactams are essentially soluble and the monomers used to be grafted on are essentially insoluble.
- the range from 60 to 90 ° C. is preferred as the polymerization temperature.
- polymerization is carried out under normal pressure, but it is also possible to work under pressure, in particular autogenous pressure.
- Hydrogen peroxide, organic peroxides and hydroperoxides if appropriate in combination with reducing compounds such as sodium bisulfite or sodium formaldehyde sulfoxylate, can be used as initiators which form free radicals, water-soluble azo compounds such as 2,2-azobis (2-amidino-propane) dihydrochloride, but above all inorganic peroxides such as the alkali metal or ammonium salts of peroxodisulfuric acid in amounts of at least 0.1% by weight, based on the total amount of monomer to be grafted on.
- reducing compounds such as sodium bisulfite or sodium formaldehyde sulfoxylate
- Particularly suitable emulsifiers and protective colloids which serve mainly as dispersants, are anionic surfactants such as the alkali metal salts of fatty acids (soaps) or the alkali metal salts of alkyl sulfates, for example sodium lauryl sulfate, but also nonionic and cationic emulsifiers or protective colloids, for example polyethylene oxides.
- anionic surfactants such as the alkali metal salts of fatty acids (soaps) or the alkali metal salts of alkyl sulfates, for example sodium lauryl sulfate
- nonionic and cationic emulsifiers or protective colloids for example polyethylene oxides.
- Polyoxyethylene-polyoxypropylene block copolymers and quaternary ammonium salts such as cetyltrimethylammonium bromide can be used.
- other conventional auxiliaries can be added to the reaction mixture, such as, for example, buffer substances
- a post-polymerization step (e) can be carried out after step (d) in order to complete the reaction.
- the reaction mixture is usually kept at the polymerization temperature for 1 to 5 hours.
- aqueous dispersions produced by the process according to the invention can be used as they are, if appropriate after setting a certain solids content by adding or distilling off water, or can be worked up, preferably by drying, to give free-flowing powders.
- the dispersion can be dried by freeze-drying or preferably spray drying, optionally with the addition of spray aids and antiblocking agents.
- Spray drying is usually carried out in a manner known per se in spray towers, it being possible for the dispersion to be dried to be sprayed with the aid of atomizing disks or single or multi-component nozzles.
- the dispersion is dried at temperatures of approx. 90 to 130 ° C (inlet temperature) with hot gases, e.g. Nitrogen or air.
- One or more substances suitable for this purpose are used as spraying aids at temperatures of 60 ° C. or above.
- the spray auxiliaries can be added in amounts of up to 50% by weight, based on the graft polymer.
- Polymeric water-soluble substances in particular those with high degrees of polymerization, e.g. Polyvinyl alcohols, cellulose derivatives, condensation products from naphthalenesulfonic acids and formaldehyde, polyacrylic acids and polyacrylamides.
- the powder obtained in this way can be admixed with up to 30% by weight, based on the total weight of polymeric constituents, of conventional antiblocking agents such as colloidal silica gel, ground clays, talc or calcium carbonate.
- conventional antiblocking agents such as colloidal silica gel, ground clays, talc or calcium carbonate.
- the emulsion graft copolymers prepared by the process according to the invention can be used in the form of the primary dispersions of the powders obtained by drying and of the dispersions obtained by redispersing these powders in the construction sector, in the production of paints and coatings, of glues and of adhesives and especially as film formers in cosmetic and pharmaceutical preparations.
- the process according to the invention achieves highly stable polymer dispersions which are distinguished by a significantly lower viscosity than in the case of the dispersions known from the prior art.
- the Brookfield viscosity of the dispersions prepared according to the invention measured at a solids content of 30% by weight, is generally below 100 mPas, very often between 10 and 50 mPas. This considerably simplifies their use for films or coatings which are applied to the object to be coated by spraying, and in particular enables spray drying to be more cost-effective since there is no need for extensive thinning.
- the dispersion powder obtained in this way which is free-flowing and easily redispersible in water, can be stored as dry powder in a space- and weight-saving manner and without risk of loss of quality until the dispersion used as film former or coating agent is prepared. Even without the use of antiblocking agents, the powder generally practically does not block if stored for a long time.
- Feed 2 from 158.8 g of vinyl acetate was started immediately after and metered in over the course of 2 hours. After the end of the feed, 1 ml of initiator solution was added and the 3-hour postpolymerization was carried out at 80.degree. The resulting dispersion had a solids content of
- the dispersion set to 30% by weight of solids had a viscosity of 30 mPas, the light transmission was 85% and the minimum film-forming temperature (according to DIN 53 787) was 12 ° C.
- the dispersion had a solids content of 40% by weight.
- the dispersion adjusted to a solids content of 30% by weight, had a viscosity of 193 mPas and the light transmission was 88%.
- the viscous dispersion coagulated after about 3 weeks of storage.
- Feed 2 (202.4 g of vinyl acetate) was then added dropwise over the course of 2 hours, with another 0.5 ml of initiator solution being added after about 1 hour. After the feed had ended, a further 0.6 ml of initiator solution were added and the polymerization was continued for 2 hours.
- the dispersion adjusted to 30% by weight solids, had a viscosity of 16 mPas, the light transmission was 15%, the minimum film-forming temperature (according to DIN 53 787) was 8 ° C.
- the initial charge consisted of 600 g of distilled water, 90.0 g of the same polyvinylpyrrolidone as in Example 1, 20.2 g of vinyl acetate and 1.58 g of sodium lauryl sulfate.
- Feed 1 was 149.2 g of vinyl acetate and feed 2 was 171.3 g of vinyl acetate. It was polymerized analogously to Example 2 with ammonium peroxodisulfate as an initiator.
- the dispersion which was set at 30% by weight of solids, had a viscosity of 22.5 mPas and a light transmittance of 21%, and the minimum film-forming temperature (in accordance with DIN 53 787) was 15 ° C.
- Example 4 Example 4
- the initial charge consisted of 600 g of distilled water, 180 g of the same polyvinylpyrrolidone as in Example 1, 15.12 g of vinyl acetate and 1.58 g of sodium lauryl sulfate.
- Feed 1 was 118.3 g of vinyl acetate and feed 2 was 135.8 g of vinyl acetate. It was polymerized analogously to Example 2 with ammonium peroxodisulfate as an initiator.
- the dispersion which was set at 30% by weight of solids, had a viscosity of 40.4 mPas and a light transmittance of 92%.
- the initial charge consisted of 600 g of distilled water, 225 g of the same polyvinylpyrrolidone as in Example 1, 12.6 g of vinyl acetate and 1.58 g of sodium lauryl sulfate.
- Feed 1 was 98.6 g of vinyl acetate and feed 2 was 113.87 g of vinyl acetate. It was analogous to Example 2 with ammonium peroxodisulfate as
- Initiator polymerized however, the feed times were 80 minutes for feed 1 and 3 hours for feed 2.
- the dispersion set to 30 wt.% Solids had a viscosity of 39 mPas and a light transmission of 95.5%. on.
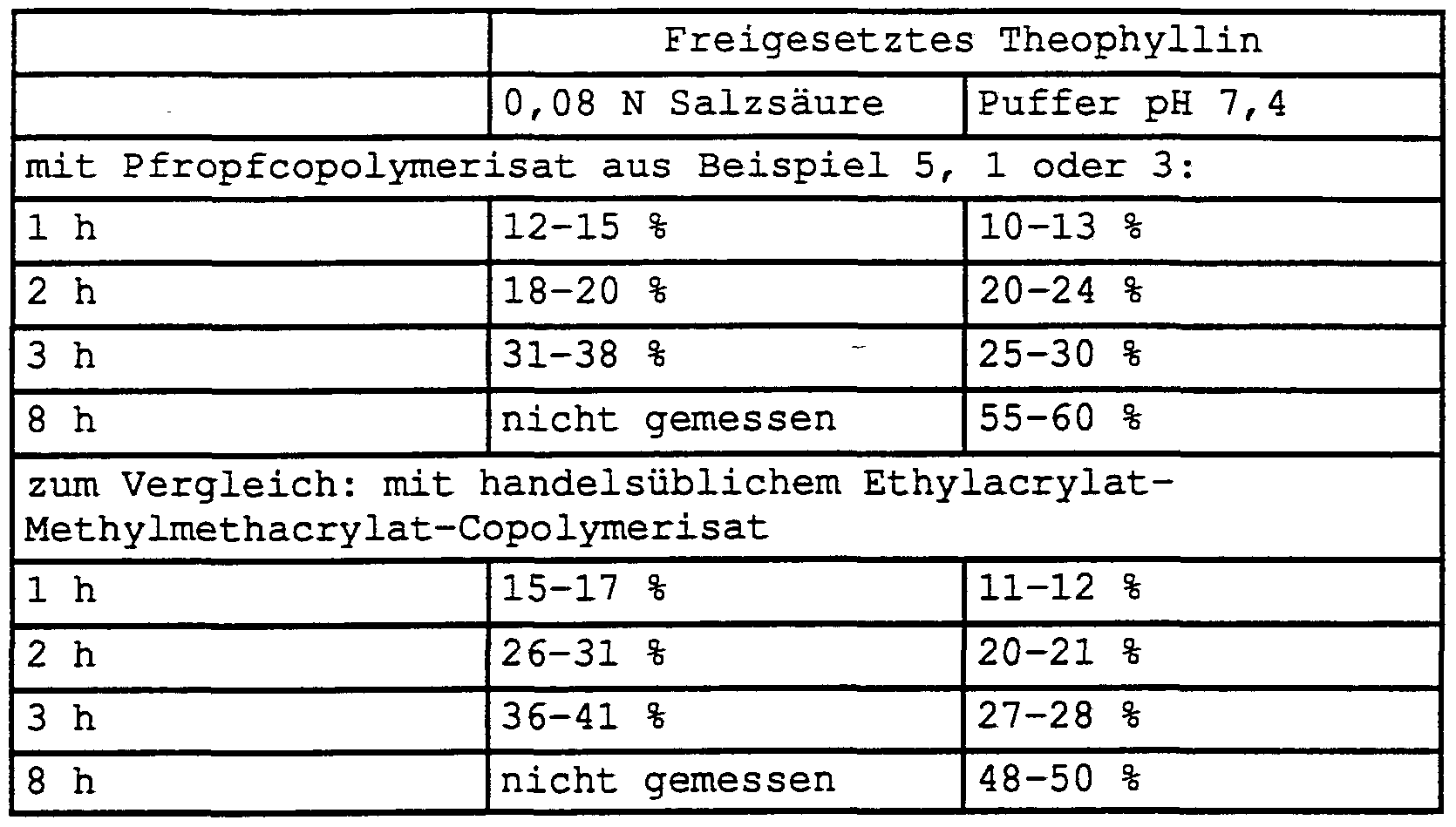
- Example 1 The dispersion from Example 1 was spray-dried (inlet temperature 110-120 ° C., outlet temperature 80-90 ° C.) and reconstituted by stirring in cold water to give a dispersion with a solids content of 15% by weight. This dispersion was applied as a pump spray with the addition of fragrances and flow aids. The resulting films were clear and shiny. Examples 7 to 9 (tablet matrix for theophylline)
- Mixture I was granulated with dispersion II, sieved, dried, mixed with III and compressed to tablets on a rotary machine with a low pressing force.
- the tablets obtained had the following physical properties:
Abstract
Description


Claims
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP5512903A JPH07503036A (ja) | 1992-01-28 | 1993-01-20 | エマルジョングラフト共重合体の化粧品,医薬品組成物用のフィルム形成剤としての利用,及びエマルジョングラフト共重合体の製造方法 |
| EP93903203A EP0624169A1 (de) | 1992-01-28 | 1993-01-20 | Verwendung von emulsionspfropfcopolymerisaten als filmbildner in haarfestigungsmitteln und verfahren zur herstellung der emulsionspfropfcopolymerisate |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19924202193 DE4202193A1 (de) | 1992-01-28 | 1992-01-28 | Verwendung von emulsionspfropfcopolymerisaten als filmbildner in kosmetischen und pharmazeutischen zubereitungen und verfahren zur herstellung der emulsionspfropfcopolymerisate |
| DEP4202193.6 | 1992-01-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1993015120A1 true WO1993015120A1 (de) | 1993-08-05 |
Family
ID=6450353
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1993/000118 WO1993015120A1 (de) | 1992-01-28 | 1993-01-20 | Verwendung von emulsionspfropfcopolymerisaten als filmbildner in kosmetischen und pharmazeutischen zubereitungen und verfahren zur herstellung der emulsionspfropfcopolymerisate |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0624169A1 (de) |
| JP (1) | JPH07503036A (de) |
| CA (1) | CA2127373A1 (de) |
| DE (1) | DE4202193A1 (de) |
| WO (1) | WO1993015120A1 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6107397A (en) * | 1997-03-24 | 2000-08-22 | Basf Aktiengesellschaft | Aqueous copolymer dispersions of water-soluble monomers with N-vinyl groups and hydrophobic monomers |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19853046A1 (de) * | 1998-11-18 | 2000-05-25 | Basf Ag | Wasserlösliche oder wasserdispergierbare Pfropfcopolymerisate auf der Basis eines Polyvinyllactams, deren Herstellung und Verwendung |
| JP6210999B2 (ja) * | 2011-12-21 | 2017-10-11 | ルブリゾル アドバンスド マテリアルズ, インコーポレイテッド | アクリルポリマーを調製する方法およびそれにより製造された生成物 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1174991B (de) * | 1962-03-21 | 1964-07-30 | Hoechst Ag | Verfahren zur Herstellung von Polyvinylalkohol |
| US3468832A (en) * | 1966-11-17 | 1969-09-23 | Gaf Corp | Stable aqueous emulsions |
| US3652481A (en) * | 1970-01-29 | 1972-03-28 | Gaf Corp | Stable aqueous emulsions of graft copolymers |
| US3770683A (en) * | 1972-02-14 | 1973-11-06 | Gaf Corp | Graft copolymers of poly(vinylpyrrolidone) with acrylic acid and acrylic ester |
-
1992
- 1992-01-28 DE DE19924202193 patent/DE4202193A1/de not_active Withdrawn
-
1993
- 1993-01-20 CA CA 2127373 patent/CA2127373A1/en not_active Abandoned
- 1993-01-20 EP EP93903203A patent/EP0624169A1/de not_active Withdrawn
- 1993-01-20 JP JP5512903A patent/JPH07503036A/ja active Pending
- 1993-01-20 WO PCT/EP1993/000118 patent/WO1993015120A1/de not_active Application Discontinuation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1174991B (de) * | 1962-03-21 | 1964-07-30 | Hoechst Ag | Verfahren zur Herstellung von Polyvinylalkohol |
| US3468832A (en) * | 1966-11-17 | 1969-09-23 | Gaf Corp | Stable aqueous emulsions |
| US3652481A (en) * | 1970-01-29 | 1972-03-28 | Gaf Corp | Stable aqueous emulsions of graft copolymers |
| US3770683A (en) * | 1972-02-14 | 1973-11-06 | Gaf Corp | Graft copolymers of poly(vinylpyrrolidone) with acrylic acid and acrylic ester |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6107397A (en) * | 1997-03-24 | 2000-08-22 | Basf Aktiengesellschaft | Aqueous copolymer dispersions of water-soluble monomers with N-vinyl groups and hydrophobic monomers |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2127373A1 (en) | 1993-08-05 |
| JPH07503036A (ja) | 1995-03-30 |
| DE4202193A1 (de) | 1993-07-29 |
| EP0624169A1 (de) | 1994-11-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0545209B1 (de) | Redispergierbares Dispersionspulver aus N-Vinylpyrrolidon-Vinylacetat-Copolymerisat, dessen Herstellung und Verwendung | |
| EP0575930B1 (de) | Verfahren zur Herstellung von festen pharmazeutischen Retardformen | |
| EP0058765B1 (de) | In Magensaft lösliche oder quellbare Überzugsmasse und ihre Verwendung in einem Verfahren zum Überziehen von Arzneiformen | |
| EP0181515B1 (de) | Verfahren zur Herstellung einer wässrigen Überzugsmitteldispersion und ihre Verwendung zum Überziehen von Arzneimitteln | |
| EP0152038B1 (de) | Arzneimittelüberzug | |
| EP1571164B1 (de) | Verwendung wässriger Polymerdispersionen auf Basis von Alkyl(meth)-aycrylaten | |
| EP0088951A2 (de) | Verfahren zum Überziehen von Arzneiformen mittels eines in Wasser dispergierten Überzugsmittels | |
| EP0704208A2 (de) | Überzugs- und Bindemittel für Arzneiformen sowie damit hergestellte Arzneiform | |
| EP0783535B1 (de) | Verfahren zur herstellung wässriger lösungen von poly(n-vinyl-epsilon-caprolactam) und ihre verwendung | |
| EP0868910A2 (de) | Verwendung von redispergierbaren Polymerpulvern oder Polymergranulaten als Bindemittel zur Herstellung von festen pharmazeutischen Darreichungsformen | |
| DE19617716A1 (de) | In wäßriger Lösung redispergierbare Polymerpulver | |
| EP1325044B1 (de) | Wässrige vinylacetat polymerdispersion | |
| EP0867455A2 (de) | Wässrige Copolymerisatdispersionen aus wasserlöslichen Monomeren mit N-Vinylgruppen und hydrophoben Monomeren | |
| EP1924633B1 (de) | Wässrige polyvinylacetatdispersionen mit hoher scherstabilität | |
| EP1195394B1 (de) | (Co-)Polymerisate von Hydroxyalkyl(meth)acrylaten, Verfahren zu deren Herstellung sowie deren Verwendung in pharmazeutischen Darreichungsformen | |
| EP1263813A1 (de) | Verfahren zur einstellung der teilchengrösse von popcornpolymeren während der popcornpolymerisation | |
| WO1993015120A1 (de) | Verwendung von emulsionspfropfcopolymerisaten als filmbildner in kosmetischen und pharmazeutischen zubereitungen und verfahren zur herstellung der emulsionspfropfcopolymerisate | |
| EP0536595B1 (de) | Verfahren zum Überziehen von Arzneiformen | |
| EP0979649B1 (de) | Verwendung von N-Vinylpyrrolidon-und Vinylacetat-haltigen Copolymeren als Matrix zur Herstellung von festen, oralen, pharmazeutischen und kosmetischen Zubereitungen | |
| EP1669374B1 (de) | Verfahren zur Herstellung wässriger Sekundärdispersionen von in Wasser nicht löslichen Polymeren | |
| DE19712247A1 (de) | Wässrige Copolymerisatdispersionen aus wasserlöslichen Monomeren mit N-Vinylgruppen und hydrophoben Monomeren | |
| DE10048888A1 (de) | Wässrige Polymerdispersion | |
| DE10122786A1 (de) | Wässrige Polymerdispersion | |
| DE2612889B1 (de) | UEberzugsmittel fuer Arzneiformlinge | |
| DE19748545A1 (de) | Wässrige Copolymerisatdispersionen aus wasserlöslichen Monomeren mit N-Vinylgruppen und hydrophoben Monomeren |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CA JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1993903203 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2127373 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 1994 256624 Country of ref document: US Date of ref document: 19940728 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993903203 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993903203 Country of ref document: EP |