WO1993009185A1 - Long wavelength chemically reactive dipyrrometheneboron difluoride dyes and conjugates - Google Patents
Long wavelength chemically reactive dipyrrometheneboron difluoride dyes and conjugates Download PDFInfo
- Publication number
- WO1993009185A1 WO1993009185A1 PCT/US1992/008350 US9208350W WO9309185A1 WO 1993009185 A1 WO1993009185 A1 WO 1993009185A1 US 9208350 W US9208350 W US 9208350W WO 9309185 A1 WO9309185 A1 WO 9309185A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- bathochromic
- arh
- ester
- Prior art date
Links
- 239000000975 dye Substances 0.000 title claims abstract description 111
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 60
- 125000001424 substituent group Chemical group 0.000 claims abstract description 57
- 125000003118 aryl group Chemical group 0.000 claims abstract description 32
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 28
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 22
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 22
- 239000001257 hydrogen Substances 0.000 claims abstract description 22
- 125000000524 functional group Chemical group 0.000 claims abstract description 21
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 20
- 102000004169 proteins and genes Human genes 0.000 claims abstract description 18
- 239000007850 fluorescent dye Substances 0.000 claims abstract description 16
- 239000003446 ligand Substances 0.000 claims abstract description 16
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 11
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 11
- 150000002367 halogens Chemical class 0.000 claims abstract description 11
- 125000003710 aryl alkyl group Chemical group 0.000 claims abstract description 10
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 claims abstract description 10
- 125000000962 organic group Chemical group 0.000 claims abstract description 7
- 230000009870 specific binding Effects 0.000 claims abstract description 5
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 99
- -1 succinimidyl ester Chemical class 0.000 claims description 52
- 150000001875 compounds Chemical class 0.000 claims description 40
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 30
- 108091034117 Oligonucleotide Proteins 0.000 claims description 17
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 16
- 150000002148 esters Chemical class 0.000 claims description 15
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 15
- 150000002431 hydrogen Chemical class 0.000 claims description 13
- 125000003545 alkoxy group Chemical group 0.000 claims description 12
- 239000002773 nucleotide Substances 0.000 claims description 12
- 125000003729 nucleotide group Chemical group 0.000 claims description 12
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims description 10
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 10
- 150000001408 amides Chemical class 0.000 claims description 10
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 claims description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 9
- 239000002253 acid Substances 0.000 claims description 9
- 229940079593 drug Drugs 0.000 claims description 9
- 239000003814 drug Substances 0.000 claims description 9
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 claims description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 7
- 150000004820 halides Chemical class 0.000 claims description 7
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 7
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 7
- 150000001540 azides Chemical class 0.000 claims description 6
- 238000006862 quantum yield reaction Methods 0.000 claims description 6
- MNCMBBIFTVWHIP-UHFFFAOYSA-N 1-anthracen-9-yl-2,2,2-trifluoroethanone Chemical group C1=CC=C2C(C(=O)C(F)(F)F)=C(C=CC=C3)C3=CC2=C1 MNCMBBIFTVWHIP-UHFFFAOYSA-N 0.000 claims description 5
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 claims description 5
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 claims description 5
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 claims description 5
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 claims description 5
- 150000008064 anhydrides Chemical class 0.000 claims description 5
- 150000003857 carboxamides Chemical class 0.000 claims description 5
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims description 5
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 claims description 5
- 150000002540 isothiocyanates Chemical class 0.000 claims description 5
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 claims description 5
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 claims description 5
- 229930192474 thiophene Natural products 0.000 claims description 5
- 239000012948 isocyanate Substances 0.000 claims description 4
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 4
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 claims description 3
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 claims description 3
- 150000001732 carboxylic acid derivatives Chemical group 0.000 claims description 3
- 230000000295 complement effect Effects 0.000 claims description 3
- 150000002463 imidates Chemical class 0.000 claims description 3
- 150000002513 isocyanates Chemical class 0.000 claims description 3
- 150000003461 sulfonyl halides Chemical class 0.000 claims description 3
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 claims description 2
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 claims 4
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 claims 4
- 150000001350 alkyl halides Chemical class 0.000 claims 2
- 125000000732 arylene group Chemical group 0.000 claims 2
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 claims 1
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 claims 1
- 238000010521 absorption reaction Methods 0.000 abstract description 19
- 239000000463 material Substances 0.000 abstract description 9
- 239000000126 substance Substances 0.000 abstract description 9
- 150000001720 carbohydrates Chemical class 0.000 abstract description 7
- 235000014633 carbohydrates Nutrition 0.000 abstract description 7
- 108020004707 nucleic acids Proteins 0.000 abstract description 6
- 150000007523 nucleic acids Chemical class 0.000 abstract description 6
- 102000039446 nucleic acids Human genes 0.000 abstract description 6
- 125000004435 hydrogen atom Chemical class [H]* 0.000 abstract 1
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 116
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 42
- 239000000243 solution Substances 0.000 description 31
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 30
- 239000000203 mixture Substances 0.000 description 27
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 24
- 230000015572 biosynthetic process Effects 0.000 description 23
- 239000011541 reaction mixture Substances 0.000 description 23
- 239000000562 conjugate Substances 0.000 description 22
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 20
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 20
- 238000003786 synthesis reaction Methods 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- 238000000034 method Methods 0.000 description 16
- 230000003595 spectral effect Effects 0.000 description 16
- 238000006243 chemical reaction Methods 0.000 description 15
- 239000000741 silica gel Substances 0.000 description 15
- 229910002027 silica gel Inorganic materials 0.000 description 15
- 239000002904 solvent Substances 0.000 description 15
- 238000004587 chromatography analysis Methods 0.000 description 14
- 239000000985 reactive dye Substances 0.000 description 14
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 12
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 11
- 239000012043 crude product Substances 0.000 description 11
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- 229940126214 compound 3 Drugs 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- 150000003573 thiols Chemical class 0.000 description 9
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 8
- 150000001336 alkenes Chemical group 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- 230000005284 excitation Effects 0.000 description 8
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 7
- 150000001298 alcohols Chemical class 0.000 description 7
- 125000005842 heteroatom Chemical group 0.000 description 7
- UBOXXEGSGUIRAJ-UHFFFAOYSA-N methyl 3-(1h-pyrrol-2-yl)propanoate Chemical compound COC(=O)CCC1=CC=CN1 UBOXXEGSGUIRAJ-UHFFFAOYSA-N 0.000 description 7
- 238000010898 silica gel chromatography Methods 0.000 description 7
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 229940125782 compound 2 Drugs 0.000 description 6
- 238000001514 detection method Methods 0.000 description 6
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- 238000005874 Vilsmeier-Haack formylation reaction Methods 0.000 description 5
- 230000008033 biological extinction Effects 0.000 description 5
- 238000005286 illumination Methods 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 150000002989 phenols Chemical class 0.000 description 5
- 230000009257 reactivity Effects 0.000 description 5
- ULUNQYODBKLBOE-UHFFFAOYSA-N 2-(1h-pyrrol-2-yl)-1h-pyrrole Chemical compound C1=CNC(C=2NC=CC=2)=C1 ULUNQYODBKLBOE-UHFFFAOYSA-N 0.000 description 4
- 238000002835 absorbance Methods 0.000 description 4
- 150000001299 aldehydes Chemical class 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 4
- 150000001735 carboxylic acids Chemical class 0.000 description 4
- 230000021615 conjugation Effects 0.000 description 4
- FHVUFHQVCSNBMO-UHFFFAOYSA-N methyl 2-[4-(1h-pyrrol-2-yl)phenoxy]acetate Chemical compound C1=CC(OCC(=O)OC)=CC=C1C1=CC=CN1 FHVUFHQVCSNBMO-UHFFFAOYSA-N 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- ZSKGQVFRTSEPJT-UHFFFAOYSA-N pyrrole-2-carboxaldehyde Chemical compound O=CC1=CC=CN1 ZSKGQVFRTSEPJT-UHFFFAOYSA-N 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 3
- XCJMGRZQOXNTRE-UHFFFAOYSA-N 2-thiophen-2-yl-1h-pyrrole Chemical compound C1=CNC(C=2SC=CC=2)=C1 XCJMGRZQOXNTRE-UHFFFAOYSA-N 0.000 description 3
- IBWXCAJDNOHNLF-UHFFFAOYSA-N 5-(furan-2-yl)-1h-pyrrole-2-carbaldehyde Chemical compound N1C(C=O)=CC=C1C1=CC=CO1 IBWXCAJDNOHNLF-UHFFFAOYSA-N 0.000 description 3
- XFJBGINZIMNZBW-CRAIPNDOSA-N 5-chloro-2-[4-[(1r,2s)-2-[2-(5-methylsulfonylpyridin-2-yl)oxyethyl]cyclopropyl]piperidin-1-yl]pyrimidine Chemical compound N1=CC(S(=O)(=O)C)=CC=C1OCC[C@H]1[C@@H](C2CCN(CC2)C=2N=CC(Cl)=CN=2)C1 XFJBGINZIMNZBW-CRAIPNDOSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 229910015900 BF3 Inorganic materials 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 3
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 3
- 238000000862 absorption spectrum Methods 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 229910052796 boron Inorganic materials 0.000 description 3
- 229940098773 bovine serum albumin Drugs 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 229940125904 compound 1 Drugs 0.000 description 3
- 229940125936 compound 42 Drugs 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 238000011033 desalting Methods 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 238000000295 emission spectrum Methods 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- XELZGAJCZANUQH-UHFFFAOYSA-N methyl 1-acetylthieno[3,2-c]pyrazole-5-carboxylate Chemical compound CC(=O)N1N=CC2=C1C=C(C(=O)OC)S2 XELZGAJCZANUQH-UHFFFAOYSA-N 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000000376 reactant Substances 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 125000006850 spacer group Chemical group 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- OVTCUIZCVUGJHS-VQHVLOKHSA-N trans-dipyrrin Chemical compound C=1C=CNC=1/C=C1\C=CC=N1 OVTCUIZCVUGJHS-VQHVLOKHSA-N 0.000 description 3
- AVBGNFCMKJOFIN-UHFFFAOYSA-N triethylammonium acetate Chemical compound CC(O)=O.CCN(CC)CC AVBGNFCMKJOFIN-UHFFFAOYSA-N 0.000 description 3
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- GCTFTMWXZFLTRR-GFCCVEGCSA-N (2r)-2-amino-n-[3-(difluoromethoxy)-4-(1,3-oxazol-5-yl)phenyl]-4-methylpentanamide Chemical compound FC(F)OC1=CC(NC(=O)[C@H](N)CC(C)C)=CC=C1C1=CN=CO1 GCTFTMWXZFLTRR-GFCCVEGCSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- QXOGPTXQGKQSJT-UHFFFAOYSA-N 1-amino-4-[4-(3,4-dimethylphenyl)sulfanylanilino]-9,10-dioxoanthracene-2-sulfonic acid Chemical compound Cc1ccc(Sc2ccc(Nc3cc(c(N)c4C(=O)c5ccccc5C(=O)c34)S(O)(=O)=O)cc2)cc1C QXOGPTXQGKQSJT-UHFFFAOYSA-N 0.000 description 2
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical compound C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 description 2
- BYBZIJZNTITRID-UHFFFAOYSA-N 2-(furan-2-yl)-1h-pyrrole Chemical compound C1=CNC(C=2OC=CC=2)=C1 BYBZIJZNTITRID-UHFFFAOYSA-N 0.000 description 2
- SYNPRNNJJLRHTI-UHFFFAOYSA-N 2-(hydroxymethyl)butane-1,4-diol Chemical compound OCCC(CO)CO SYNPRNNJJLRHTI-UHFFFAOYSA-N 0.000 description 2
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 2
- KXJJSRMUFORMAT-UHFFFAOYSA-N 5-thiophen-2-yl-1h-pyrrole-2-carbaldehyde Chemical compound N1C(C=O)=CC=C1C1=CC=CS1 KXJJSRMUFORMAT-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 108090001008 Avidin Proteins 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 2
- YDNKGFDKKRUKPY-JHOUSYSJSA-N C16 ceramide Natural products CCCCCCCCCCCCCCCC(=O)N[C@@H](CO)[C@H](O)C=CCCCCCCCCCCCCC YDNKGFDKKRUKPY-JHOUSYSJSA-N 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 2
- CRJGESKKUOMBCT-VQTJNVASSA-N N-acetylsphinganine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@H](CO)NC(C)=O CRJGESKKUOMBCT-VQTJNVASSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 229940106189 ceramide Drugs 0.000 description 2
- ZVEQCJWYRWKARO-UHFFFAOYSA-N ceramide Natural products CCCCCCCCCCCCCCC(O)C(=O)NC(CO)C(O)C=CCCC=C(C)CCCCCCCCC ZVEQCJWYRWKARO-UHFFFAOYSA-N 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 229940125833 compound 23 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 229940127113 compound 57 Drugs 0.000 description 2
- 229940125900 compound 59 Drugs 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- QPUSANCBJDDXSA-UHFFFAOYSA-N ethanethiol;sodium Chemical compound [Na].CCS QPUSANCBJDDXSA-UHFFFAOYSA-N 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 238000001215 fluorescent labelling Methods 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- CPBQJMYROZQQJC-UHFFFAOYSA-N helium neon Chemical compound [He].[Ne] CPBQJMYROZQQJC-UHFFFAOYSA-N 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- 229910052743 krypton Inorganic materials 0.000 description 2
- DNNSSWSSYDEUBZ-UHFFFAOYSA-N krypton atom Chemical compound [Kr] DNNSSWSSYDEUBZ-UHFFFAOYSA-N 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- YDCHPLOFQATIDS-UHFFFAOYSA-N methyl 2-bromoacetate Chemical compound COC(=O)CBr YDCHPLOFQATIDS-UHFFFAOYSA-N 0.000 description 2
- NLAHDEYGLDURJZ-RMKNXTFCSA-N methyl 4-[(e)-2-(1h-pyrrol-2-yl)ethenyl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1\C=C\C1=CC=CN1 NLAHDEYGLDURJZ-RMKNXTFCSA-N 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- DYGBNAYFDZEYBA-UHFFFAOYSA-N n-(cyclopropylmethyl)-2-[4-(4-methoxybenzoyl)piperidin-1-yl]-n-[(4-oxo-1,5,7,8-tetrahydropyrano[4,3-d]pyrimidin-2-yl)methyl]acetamide Chemical compound C1=CC(OC)=CC=C1C(=O)C1CCN(CC(=O)N(CC2CC2)CC=2NC(=O)C=3COCCC=3N=2)CC1 DYGBNAYFDZEYBA-UHFFFAOYSA-N 0.000 description 2
- 229920005615 natural polymer Polymers 0.000 description 2
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 2
- VVGIYYKRAMHVLU-UHFFFAOYSA-N newbouldiamide Natural products CCCCCCCCCCCCCCCCCCCC(O)C(O)C(O)C(CO)NC(=O)CCCCCCCCCCCCCCCCC VVGIYYKRAMHVLU-UHFFFAOYSA-N 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 230000008520 organization Effects 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229920001282 polysaccharide Polymers 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 150000003233 pyrroles Chemical class 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- WWUZIQQURGPMPG-KRWOKUGFSA-N sphingosine Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)CO WWUZIQQURGPMPG-KRWOKUGFSA-N 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 150000003871 sulfonates Chemical class 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- 229920001059 synthetic polymer Polymers 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- WWUZIQQURGPMPG-UHFFFAOYSA-N (-)-D-erythro-Sphingosine Natural products CCCCCCCCCCCCCC=CC(O)C(N)CO WWUZIQQURGPMPG-UHFFFAOYSA-N 0.000 description 1
- TYKASZBHFXBROF-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-(2,5-dioxopyrrol-1-yl)acetate Chemical compound O=C1CCC(=O)N1OC(=O)CN1C(=O)C=CC1=O TYKASZBHFXBROF-UHFFFAOYSA-N 0.000 description 1
- HJVJLAOFAYPFHL-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-pyridin-2-ylpropanedithioate Chemical compound C=1C=CC=NC=1C(C)C(=S)SN1C(=O)CCC1=O HJVJLAOFAYPFHL-UHFFFAOYSA-N 0.000 description 1
- LWAVGNJLLQSNNN-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-azidobenzoate Chemical compound C1=CC(N=[N+]=[N-])=CC=C1C(=O)ON1C(=O)CCC1=O LWAVGNJLLQSNNN-UHFFFAOYSA-N 0.000 description 1
- VHYRHFNOWKMCHQ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-formylbenzoate Chemical compound C1=CC(C=O)=CC=C1C(=O)ON1C(=O)CCC1=O VHYRHFNOWKMCHQ-UHFFFAOYSA-N 0.000 description 1
- YXMISKNUHHOXFT-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) prop-2-enoate Chemical compound C=CC(=O)ON1C(=O)CCC1=O YXMISKNUHHOXFT-UHFFFAOYSA-N 0.000 description 1
- RBNSZWOCWHGHMR-UHFFFAOYSA-N (2-iodoacetyl) 2-iodoacetate Chemical compound ICC(=O)OC(=O)CI RBNSZWOCWHGHMR-UHFFFAOYSA-N 0.000 description 1
- QWHLASPBRRZDEV-VFQQELCFSA-N (2r,3s,4r,5r)-6-amino-2,3,4,5-tetrahydroxyhexanal;hydrochloride Chemical compound Cl.NC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O QWHLASPBRRZDEV-VFQQELCFSA-N 0.000 description 1
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 1
- XZMLMTGOWVWIRV-DUGSHLAESA-N (2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-formamidohexanoyl]amino]-4-methylpentanoyl]amino]-3-phenylpropanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]hexanoic acid Chemical compound C([C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC=O)CCCC)C(=O)N[C@@H](CCCC)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CCCCN)C(O)=O)C1=CC=CC=C1 XZMLMTGOWVWIRV-DUGSHLAESA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- QIVUCLWGARAQIO-OLIXTKCUSA-N (3s)-n-[(3s,5s,6r)-6-methyl-2-oxo-1-(2,2,2-trifluoroethyl)-5-(2,3,6-trifluorophenyl)piperidin-3-yl]-2-oxospiro[1h-pyrrolo[2,3-b]pyridine-3,6'-5,7-dihydrocyclopenta[b]pyridine]-3'-carboxamide Chemical compound C1([C@H]2[C@H](N(C(=O)[C@@H](NC(=O)C=3C=C4C[C@]5(CC4=NC=3)C3=CC=CN=C3NC5=O)C2)CC(F)(F)F)C)=C(F)C=CC(F)=C1F QIVUCLWGARAQIO-OLIXTKCUSA-N 0.000 description 1
- IOPFJNFBBPRZAZ-KPKJPENVSA-N (5e)-5-(2-phenylethylidene)pyrrole-2-carbaldehyde Chemical compound C1=CC(C=O)=N\C1=C\CC1=CC=CC=C1 IOPFJNFBBPRZAZ-KPKJPENVSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- 125000000355 1,3-benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- KEQGZUUPPQEDPF-UHFFFAOYSA-N 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione Chemical compound CC1(C)N(Cl)C(=O)N(Cl)C1=O KEQGZUUPPQEDPF-UHFFFAOYSA-N 0.000 description 1
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- GJFNRSDCSTVPCJ-UHFFFAOYSA-N 1,8-bis(dimethylamino)naphthalene Chemical compound C1=CC(N(C)C)=C2C(N(C)C)=CC=CC2=C1 GJFNRSDCSTVPCJ-UHFFFAOYSA-N 0.000 description 1
- NFDXQGNDWIPXQL-UHFFFAOYSA-N 1-cyclooctyldiazocane Chemical compound C1CCCCCCC1N1NCCCCCC1 NFDXQGNDWIPXQL-UHFFFAOYSA-N 0.000 description 1
- 150000003923 2,5-pyrrolediones Chemical class 0.000 description 1
- LRMKNDMDNZWNPB-UHFFFAOYSA-N 2-(4-methoxyphenyl)-1h-pyrrole Chemical compound C1=CC(OC)=CC=C1C1=CC=CN1 LRMKNDMDNZWNPB-UHFFFAOYSA-N 0.000 description 1
- AVQPFZCDRFCTQL-QPJJXVBHSA-N 2-[(e)-2-(4-methoxyphenyl)ethenyl]-1h-pyrrole Chemical compound C1=CC(OC)=CC=C1\C=C\C1=CC=CN1 AVQPFZCDRFCTQL-QPJJXVBHSA-N 0.000 description 1
- LFOIDLOIBZFWDO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenylmethoxyphenyl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(C=1)=CC=CC=1OCC1=CC=CC=C1 LFOIDLOIBZFWDO-UHFFFAOYSA-N 0.000 description 1
- RDFZYUOHJBXMJA-UHFFFAOYSA-N 3,5-dimethyl-1h-pyrrole-2-carbaldehyde Chemical compound CC1=CC(C)=C(C=O)N1 RDFZYUOHJBXMJA-UHFFFAOYSA-N 0.000 description 1
- WADSJYLPJPTMLN-UHFFFAOYSA-N 3-(cycloundecen-1-yl)-1,2-diazacycloundec-2-ene Chemical compound C1CCCCCCCCC=C1C1=NNCCCCCCCC1 WADSJYLPJPTMLN-UHFFFAOYSA-N 0.000 description 1
- RGUKYNXWOWSRET-UHFFFAOYSA-N 4-pyrrolidin-1-ylpyridine Chemical compound C1CCCN1C1=CC=NC=C1 RGUKYNXWOWSRET-UHFFFAOYSA-N 0.000 description 1
- LYRBCXWNTUSZEQ-KBXRYBNXSA-N 5-[(1e,3e)-4-phenylbuta-1,3-dienyl]-1h-pyrrole-2-carbaldehyde Chemical compound N1C(C=O)=CC=C1\C=C\C=C\C1=CC=CC=C1 LYRBCXWNTUSZEQ-KBXRYBNXSA-N 0.000 description 1
- LCFMUZCRNBJGSP-VOTSOKGWSA-N 5-[(e)-2-phenylethenyl]-1h-pyrrole-2-carbaldehyde Chemical compound N1C(C=O)=CC=C1\C=C\C1=CC=CC=C1 LCFMUZCRNBJGSP-VOTSOKGWSA-N 0.000 description 1
- FSRMODOJIHTULE-NSCUHMNNSA-N 5-[(e)-prop-1-enyl]-1h-pyrrole-2-carbaldehyde Chemical compound C\C=C\C1=CC=C(C=O)N1 FSRMODOJIHTULE-NSCUHMNNSA-N 0.000 description 1
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 229940126639 Compound 33 Drugs 0.000 description 1
- 108010062580 Concanavalin A Proteins 0.000 description 1
- 102000009024 Epidermal Growth Factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 108010055358 F-chemotactic peptide Proteins 0.000 description 1
- 238000005727 Friedel-Crafts reaction Methods 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 108010046516 Wheat Germ Agglutinins Proteins 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 1
- BVSZTAUOUPODLX-BQYQJAHWSA-N [4-[(e)-2-(1h-pyrrol-2-yl)ethenyl]phenyl]methanol Chemical compound C1=CC(CO)=CC=C1\C=C\C1=CC=CN1 BVSZTAUOUPODLX-BQYQJAHWSA-N 0.000 description 1
- 150000003926 acrylamides Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 235000013405 beer Nutrition 0.000 description 1
- 238000005842 biochemical reaction Methods 0.000 description 1
- 230000009141 biological interaction Effects 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- KQNZDYYTLMIZCT-KQPMLPITSA-N brefeldin A Chemical compound O[C@@H]1\C=C\C(=O)O[C@@H](C)CCC\C=C\[C@@H]2C[C@H](O)C[C@H]21 KQNZDYYTLMIZCT-KQPMLPITSA-N 0.000 description 1
- JUMGSHROWPPKFX-UHFFFAOYSA-N brefeldin-A Natural products CC1CCCC=CC2(C)CC(O)CC2(C)C(O)C=CC(=O)O1 JUMGSHROWPPKFX-UHFFFAOYSA-N 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 210000003850 cellular structure Anatomy 0.000 description 1
- XTHPWXDJESJLNJ-UHFFFAOYSA-N chlorosulfonic acid Substances OS(Cl)(=O)=O XTHPWXDJESJLNJ-UHFFFAOYSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940127271 compound 49 Drugs 0.000 description 1
- 229940126545 compound 53 Drugs 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- MGNCLNQXLYJVJD-UHFFFAOYSA-N cyanuric chloride Chemical compound ClC1=NC(Cl)=NC(Cl)=N1 MGNCLNQXLYJVJD-UHFFFAOYSA-N 0.000 description 1
- VMKJWLXVLHBJNK-UHFFFAOYSA-N cyanuric fluoride Chemical compound FC1=NC(F)=NC(F)=N1 VMKJWLXVLHBJNK-UHFFFAOYSA-N 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 239000012973 diazabicyclooctane Substances 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 125000002541 furyl group Chemical class 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229940042795 hydrazides for tuberculosis treatment Drugs 0.000 description 1
- 150000002429 hydrazines Chemical class 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 230000000984 immunochemical effect Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical compound NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000000990 laser dye Substances 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 230000031700 light absorption Effects 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Chemical class 0.000 description 1
- FWODYBSMOMHLDY-SNAWJCMRSA-N methyl (e)-3-(1h-pyrrol-2-yl)prop-2-enoate Chemical compound COC(=O)\C=C\C1=CC=CN1 FWODYBSMOMHLDY-SNAWJCMRSA-N 0.000 description 1
- NTNUDYROPUKXNA-UHFFFAOYSA-N methyl 2-(triphenyl-$l^{5}-phosphanylidene)acetate Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=CC(=O)OC)C1=CC=CC=C1 NTNUDYROPUKXNA-UHFFFAOYSA-N 0.000 description 1
- IXZMPXRMPSVTKP-UHFFFAOYSA-N methyl 2-[4-(5-formyl-1h-pyrrol-2-yl)phenoxy]acetate Chemical compound C1=CC(OCC(=O)OC)=CC=C1C1=CC=C(C=O)N1 IXZMPXRMPSVTKP-UHFFFAOYSA-N 0.000 description 1
- MKHOUYKOJXYACI-QPJJXVBHSA-N methyl 2-[4-[(e)-2-(1h-pyrrol-2-yl)ethenyl]phenoxy]acetate Chemical compound C1=CC(OCC(=O)OC)=CC=C1\C=C\C1=CC=CN1 MKHOUYKOJXYACI-QPJJXVBHSA-N 0.000 description 1
- SLRVYSPSQNAGND-UHFFFAOYSA-N methyl 3-(5-formyl-1h-pyrrol-2-yl)propanoate Chemical compound COC(=O)CCC1=CC=C(C=O)N1 SLRVYSPSQNAGND-UHFFFAOYSA-N 0.000 description 1
- HNNUQHJWFIPTLJ-UHFFFAOYSA-N methyl 4-(2-bromoethyl)benzoate Chemical compound COC(=O)C1=CC=C(CCBr)C=C1 HNNUQHJWFIPTLJ-UHFFFAOYSA-N 0.000 description 1
- RYWXQMBUVGUWTP-AATRIKPKSA-N methyl 4-methyl-2-[(e)-2-(1h-pyrrol-2-yl)ethenyl]-1,3-oxazole-5-carboxylate Chemical compound CC1=C(C(=O)OC)OC(\C=C\C=2NC=CC=2)=N1 RYWXQMBUVGUWTP-AATRIKPKSA-N 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- YCJZWBZJSYLMPB-UHFFFAOYSA-N n-(2-chloropyrimidin-4-yl)-2,5-dimethyl-1-phenylimidazole-4-carboxamide Chemical compound CC=1N(C=2C=CC=CC=2)C(C)=NC=1C(=O)NC1=CC=NC(Cl)=N1 YCJZWBZJSYLMPB-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 238000006396 nitration reaction Methods 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- XULSCZPZVQIMFM-IPZQJPLYSA-N odevixibat Chemical compound C12=CC(SC)=C(OCC(=O)N[C@@H](C(=O)N[C@@H](CC)C(O)=O)C=3C=CC(O)=CC=3)C=C2S(=O)(=O)NC(CCCC)(CCCC)CN1C1=CC=CC=C1 XULSCZPZVQIMFM-IPZQJPLYSA-N 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000000863 peptide conjugate Substances 0.000 description 1
- 239000000575 pesticide Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 239000008055 phosphate buffer solution Substances 0.000 description 1
- 150000008300 phosphoramidites Chemical class 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000012460 protein solution Substances 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 229940043267 rhodamine b Drugs 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 238000006277 sulfonation reaction Methods 0.000 description 1
- 150000003463 sulfur Chemical class 0.000 description 1
- OBTWBSRJZRCYQV-UHFFFAOYSA-N sulfuryl difluoride Chemical class FS(F)(=O)=O OBTWBSRJZRCYQV-UHFFFAOYSA-N 0.000 description 1
- 229920002994 synthetic fiber Polymers 0.000 description 1
- ABZLKHKQJHEPAX-UHFFFAOYSA-N tetramethylrhodamine Chemical class C=12C=CC(N(C)C)=CC2=[O+]C2=CC(N(C)C)=CC=C2C=1C1=CC=CC=C1C([O-])=O ABZLKHKQJHEPAX-UHFFFAOYSA-N 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 125000001544 thienyl group Chemical class 0.000 description 1
- 238000006177 thiolation reaction Methods 0.000 description 1
- ZWZVWGITAAIFPS-UHFFFAOYSA-N thiophosgene Chemical compound ClC(Cl)=S ZWZVWGITAAIFPS-UHFFFAOYSA-N 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 1
- SWGJCIMEBVHMTA-UHFFFAOYSA-K trisodium;6-oxido-4-sulfo-5-[(4-sulfonatonaphthalen-1-yl)diazenyl]naphthalene-2-sulfonate Chemical compound [Na+].[Na+].[Na+].C1=CC=C2C(N=NC3=C4C(=CC(=CC4=CC=C3O)S([O-])(=O)=O)S([O-])(=O)=O)=CC=C(S([O-])(=O)=O)C2=C1 SWGJCIMEBVHMTA-UHFFFAOYSA-K 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 238000011179 visual inspection Methods 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 229910052724 xenon Inorganic materials 0.000 description 1
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H13/00—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids
- C07H13/02—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids
- C07H13/04—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids having the esterifying carboxyl radicals attached to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/022—Boron compounds without C-boron linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B57/00—Other synthetic dyes of known constitution
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/06—Luminescent, e.g. electroluminescent, chemiluminescent materials containing organic luminescent materials
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/531—Production of immunochemical test materials
- G01N33/532—Production of labelled immunochemicals
- G01N33/533—Production of labelled immunochemicals with fluorescent label
Definitions
- This invention relates to chemically reactive fluorescent dyes that can be attached to ligands and the resulting fluorescently labeled ligands.
- the invention relates to chemically reactive dyes that are derivatives of dipyrrometheneboron difluoride dyes (derivatives of 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene dyes) that have an absorption maximum at a wavelength longer than about 525 nm.
- These reactive dyes are used for the fluorescent labeling of nucleic acids, nucleotides, carbohydrates, drugs, polymers, proteins, and other biologically derived or synthetic chemical materials.
- Fluorescent dyes have many uses and are known to be particularly suitable for biological applications in which the high detectability of fluorescence is required. Fluorescent dyes are used to impart both visible color and fluorescence to other materials. Many applications utilize chemically reactive fluorescent dyes by chemically attaching the dye to reactive sites on a wide variety of materials such as cells, tissues, proteins, antibodies, enzymes, drugs, hormones, lipids, nucleotides, nucleic acids, or natural or synthetic polymers to make fluorescent conjugates. With these synthetic probes, ligands are frequently used to confer a specificity for a biochemical reaction that is to be observed and the fluorescent dye provides the means of detection or quantitation of the interaction.
- Fluorescence useful for such applications is generally initiated by absorption of light from an external, relatively concentrated light source.
- the sensitivity of these applications is improved by having dyes that have high absorbance of the exciting light and high fluorescence quantum yield.
- the applications are furthermore improved by having dyes that resist photobleacbing by the exciting light and that have spectral wavelengths in a range that avoids the background from contaminants that may be present in the samples.
- dyes whose fluorescence is not quenched by water since most biological measurements are made in aqueous solution.
- Certain lasers are particularly useful as a concentrated light source for the excitation of fluorescence.
- the argon laser has been the most common light source for excitation of fluorescence, with principal output at 488 nm and 514 nm.
- other lasers are increasingly used, such as helium-neon lasers that can be selected to have maximum output at either 543 nm, 594 nm, or 633 nm; the krypton laser which has significant output at 568 nm and 647 nm; and light emitting diodes which are available at this time with output commonly above 660 nm; resulting in increased demand for longer wavelength fluorescent dyes.
- a number of dyes that have previously been found to be fluorescent do not have significant absorbance at desired longer excitation wavelengths. Many also have other characteristics which interfere with or limit their usefulness. For example, many known fluorescent dyes are significantly quenched in aqueous solution or are unstable during the illumination.
- Dyes derived from dipyrrometheneboron difluoride have many desirable characteristics. Simple alkyl derivatives of the fluorophore 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene have been described by Treibs & Kreuzer, Difluorboryl-komplexe von di- und tripyrrylmethenen. LIEBIGS ANNALEN CHEM.
- the emission of the alkyl derivatives of 4,4-difluoro-4-bora-3a,4a-diaza-s- indacene fluorescent dyes clearly overlaps that of fluorescein.
- the overlap allows the alkyl derivatives of dipyrrometheneboron difluoride to be used with the same optical equipment as used with fluorescein- based dyes without modification of the excitation sources or optical filters.
- the fluorescence of the known class of alkyl-substituted 4,4- difluoro-4-bora-3a,4a-diaza-s-indacenes is not readily suitable for detection in combination with fluorescein or for use in applications where excitation by longer wavelength sources such as the helium-neon or krypton lasers or light emitting diodes is required.
- the '339 patent discloses some dyes with an absorption maxim ⁇ m of greater than 525 nm (Table 5), the '339 patent is neither enabling nor prior art for the invention of the subject long wavelength reactive derivatives of dipyrrometheneboron difluoride dyes. Of the longer wavelength dyes listed, all were measured in chloroform which slightly enhances the spectral absorption and shifts the emission maxima to longer wavelength by about 10 nm (compared to methanol).
- the '339 patent also discloses a non-reactive dye having an absorption maximum greater than 600 nm (cmpd. 27) which has been found to have an undesirably low quantum yield and very broad absorption and emission spectral band widths.
- U.S. Patent 4,916,711 to Boyer, et al. (1990) discloses a method of using derivatives of 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene dyes, in particular symmetrical alkyl and sulfonated alkyl derivatives, as laser dyes.
- the '711 patent also discloses a multitude of possible alternatives for substituents of the basic tricyclic structure which can be used for the patented method.
- the '711 patent is neither enabling nor prior art for the invention of long wavelength reactive derivatives of dipyrrometheneboron difluoride dyes.
- the dyes described in the '711 patent are not reactive dyes.
- the '711 patent neither recognizes nor recites the effect or advantage of substituents that enhance the long wavelength fluorescence properties of such dyes.
- novel dyes described in this invention contain one or more substituent groups coupled to the 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene ("BDI") fluorophore resulting in significant absorbance and fluorescence at desired longer wavelengths.
- the dyes of this invention also have the chemical reactivity necessary for conjugation to the reactive sites commonly found in biomolecules, drugs, and natural and synthetic polymers.
- the reactive dyes of this invention are combined with a variety of materials to form novel dye conjugates.
- novel dye conjugates are desirable for use in combination with conjugates formed from other fluorescent dyes such as fluorescein or alkyl-substituted 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene dyes in that the fluorophore of the novel conjugates can be both selectively excited and detected because of their spectral shift to longer wavelengths, particularly an absorption maximum at greater than 525 nm and an emission maximum at greater than 550 nm.
- the new reactive dyes have the advantage over other dyes that absorb at these wavelengths, of being electrically neutral, photostable and being, in most cases, highly fluorescent in both organic and aqueous solution with relatively narrow absorption and emission spectra.
- Figure 1 is a general reaction scheme for synthesis of long wavelength reactive BDI dyes.
- the general method consists of a formation of pyrromethene intermediates followed by cyclization with boron trifiuoride in the presence of base to give substituted dipyrrometheneboron diifluoridedyes.
- Figure 2 is a graph of the relative spectral separations of selected examples of long wavelength reactive BDI dyes in comparison with an alkyl-substituted BDI dye, in pH 7 phosphate buffer solution.
- Figure 3 is a graph showing the relatively narrow emission band width of a selected long wavelength reactive BDI dye in comparison with another known dye emitting at the same wavelength (in methanol, excited at 540 nm).
- Figure 4 is a graph showing the increased photostability of representative long wavelength reactive BDI dyes in comparison with an alkyl-substituted BDI dye, in CH 3 CN solution.
- This invention describes novel fluorescent dyes that are reactive derivatives of the 4,4-difluoro-4- bora-3a,4a-diaza-s-indacene (“BDI”) fluorophore that can be chemically reacted to bond with the reactive sites present in many materials to form novel fluorescent conj000tes.
- BDI 4,4-difluoro-4- bora-3a,4a-diaza-s-indacene
- the BDI dyes generally have the structure of formula I:
- At least one of the substituents R 1 -R 7 on the BDI fluorophore is -L m G where G is a reactive functional group that allows the BDI fluorophore to be coupled to other synthetic or natural molecules.
- G is a reactive functional group that allows the BDI fluorophore to be coupled to other synthetic or natural molecules.
- the most common reactive functional groups are carboxylic acids and derivatives thereof including succinimidyl esters, acyl azides, anhydrides, and acid halides.
- reactive groups include acrylamides, alcohols, aldehydes, amines, azides, imido esters, sulfonate esters, haloacetamides, alkyl and aryl halides, sulfonyl halides, hydrazines, isocyanates, isothiocyanates, and maleimides.
- Table 1 lists common reactive functional groups and the reactive sites with which they most commonly react. The tabulation is not all inclusive since with the approprate choice of solvent, temperature and catalysts, other functional groups can be made to react.
- the spacer L is a substituted or unsubstituted. alkyl (containing 1-5 carbons) or aryl group.
- L is a linking bathochromic moiety i.e. a bathochromic moiety that links the fluorophore and a reactive functional group such that the reactive functional group is a substituent on the linking bathochromic moiety.
- L when present, is a straight chain unsubstituted alkyl group that contains 1-4 carbons, an unsubstituted phenyl group, or a linking bathochromic moiety.
- At least one of the substituents R 1 -R 7 contains a bathochromic moiety.
- a bathochromic moiety shifts the fluorescence of the compound toward the red part of the spectrum and gives the fluorophore an absorption maximum of greater than about 525 nm.
- the bathochromic moiety also gives the fluorophore an emission maximum greater than about 550 nm.
- the bathochromic moiety is an unsaturated organic group such as a substituted or unsubstituted ethenyl, dienyl, trienyl or heteroaryl group.
- a bathochromic moiety may be present as a substituent separate from the reactive functional group (separate bathochromic moiety). At least one, and as many as six, of the substituents, can be separate bathochromic moieties any of which may be the same or different. A bathochromic moiety may also or alternatively be included in the spacer (L) connecting the reactive functional group to the BDI fluorophore (linking bathochromic moiety). One or more bathochromic moieties may be included only as linking moieties, or only separately, or combinations thereof, but a bathochromic moiety must be included in at least one of the substituents R 1 -R 7 .
- bathochromic moieties there may be as many as seven bathochromic moieties, some or all of which may each be the same or different.
- the dye compound contains seven bathochromic moieties, at least one bathochromic moiety and as many a seven contain(s) a reactive functional group.
- at least one of the groups R 1 , R 2 , R 3 or R 4 contains a bathochromic moiety, which may contain a reactive functional group as a substituent.
- the bathochromic moiety is preferably present as a substituent separate from the reactive group.
- the bathochromic moiety is directly attached to the BDI fluorophore by a covalent bond.
- heteroaryl in one embodiment, there is a single bathochromic moiety that is heteroaryl.
- heteroaryl as used throughout this document, means an aromatic heterocyclic substituent. When a heteroaryl group is present as a substituent, it is an aromatic heterocyclic moiety that contains at least one and as many as three hetero atom (a non-carbon atom formmg the ring structure) that is N, O, or S.
- the heteroaryl group can contain one, two, or three hetero atoms.
- the heteroaryl group can be a single ring structure or a fused two- or three-ring structure.
- a ring can be a 5- or 6- membered ring.
- heteroaryl substituents are pyrrole, thiophene, or furan (single ring, single hetero atom), or oxazole, isoxazole, oxadiazole, or imidazole (single ring, multiple hetero atoms).
- the heteroaryl group is a multi-ring structure contaimng one or more hetero atoms; for example, the heteroaryl substituent is benzoxazole, benzothiazole, or benzimidazole (multi-ring, multiple hetero atoms), or benzofuran or indole (multi-ring, single hetero atom).
- heteroaryl includes substituted derivatives of the heteroaryl substituents, such as alkyl-, aryl-, arylalkyl- or heteroaryl-substituted pyrrolyl, furyl or thienyl.
- the bathochromic heteroaryl group may also contain a reactive functional group as a substituent.
- the single bathochromic moiety is an alkenyl group.
- the alkenyl group is ethenyl, dienyl or trienyl.
- the alkenyl group is unsubstituted or contains substituents, which may be the same or different that are hydrogen, halogen, alkyl (containing 1-5 carbon atoms), cyano, carboxylate ester, carboxamide, aryl or heteroaryl, or polyethenyl to form a conjugated dienyl or trienyl substituent, which in turn may be substituted with substituents, which may be the same or different, that are hydrogen, halogen, alkyl (containing 1-5 carbon atoms), cyano, ester, amide, aryl or heteroaryl groups.
- the bathochromic alkenyl group may also contain a reactive functional group as a substituent.
- the fluorophore has 2-4 bathochromic substituents, which may be the same or different, that are substituted or unsubstituted ethenyl, dienyl, trienyl, or heteroaryl groups.
- One or two of the bathochromic substituents may contain reactive functional groups, which may be the same or different, as substituents.
- the remainder of the substituents R 1 -R 7 that are neither reactive functional groups nor bathochromic substituents, which remaining substituents may be the same or different, are hydrogen, halogen, cycloalkyl or alkyl (contaimng 1-5 carbons), aryl, arylalkyl (the alkyl portions of which contain 1-5 carbon atoms), or sulfo, alone or in combination, as long as they do not destroy the reactivity of the dyes.
- the remaining substituents are hydrogen.
- Preferred aryl groups in any of the described substituents are phenyl, 1-naphthyl, 2-naphthyl, 1- pyrenyl, 9-anthryl, and their alkoxy substituted derivatives wherein the alkyl portions of such derivatives have less than 5 carbon atoms.
- Any aryl or heteroaryl group in any substituent may be further substituted one or more times by alkyl (containing 1-5 carbons); or alkoxy groups, the alkyl portions of which have less than 5 carbon atoms; or combinations thereof.
- Any alkyl group in any substituent may be further substituted by an ester or amide substituent.
- Table 2 contains a sample of representative long wavelength reactive dyes and their key precursors.
- Table 3 describes physical properties of selected dyes from Table 2.
- the additional electronic conjugation that results from incorporation of bathochromic moieties into the BDI fluorophore results in new long wavelength reactive BDI dyes that have spectral properties that are significantly shifted from those of the related alkyl-substituted fluorophores, thus permitting their use in multi-color fluorescence applications in combination with fluorescein or alkyl- substituted BDI dyes.
- this wavelength shift is usually accompanied by an increase in photostability of the long wavelength reactive BDI dyes and in most cases by an increase in the extinction coefficient of the long wavelength reactive BDI dyes relative to the alkyl-substituted BDI dyes.
- Table 5 lists the spectral properties of selected dyes from Table 2.
- ⁇ Compound R is presented as an example of alkyl-substituted dipyrrometheneboron difluoride dyes for comparison.
- the integrated fluorescent intensity is also corrected for variation of incident excitation light intensity with wavelength by recording spectra in ratio mode relative to a rhodamine B/ethylene glycol quantum counter solution.
- the novel dyes have an absorption maximum at greater than about 525 nm and, preferably, an emission maximum at greater than about 550 nm (see Table 3). These spectral properties distinguish the long wavelength reactive BDI dyes from the related alkyl-substituted BDI dyes. As indicated in Table 5 below, the absorption and emission spectra of the new reactive BDI dyes are shifted to significantly longer wavelengths as compared to the alkyl-substituted dyes.
- the long wavelength reactive BDI dyes also demonstrate improved photostability (Table 5, Figure 4) relative to the alkyl-substituted dyes; with high extinction coefficients, generally greater than 80,000 cm -1 M -1 (Table 5).
- the dyes of this invention also have a substantial quantum yield, i.e. greater than 0.1 as measured in methanol (Table 5), which distinguishes these dyes from previously disclosed BDI derivatives having an absorption maximum greater than about 525 nm.
- Suitable substituents R 1 to R 7 (Fig. 1) on the pyrrole intermediates include, but are not limited to, hydrogen, alkyl, cycloalkyl, aryl, arylalkyl, heteroaryl and alkenyl.
- the general method consists of an acid catalyzed condensation of a 2-acylpyrrole or appropriately substituted 2-acylpyrrole with pyrrole or substituted pyrrole having a hydrogen on the 2-position used in approximately stoichiometric proportions to give a pyrromethene intermediate.
- there are two alternative routes whose choice depends primarily on the availability or ease of synthesis of the 2- acylpyrrole reactants.
- Suitable acids include, but are not limited to, hydrogen halides, metal salts typically used in Friedel-Craft reactions such as zinc halides, and non-metallic, electron deficient Lewis acids such as boron halides, halides of sulfur acids and phosphorous oxy chloride in that such acids contain elements or groups of elements capable of forming an anionic counterion.
- boron halides, halides of sulfur acids and phosphorous oxy chloride in that such acids contain elements or groups of elements capable of forming an anionic counterion.
- phosphorous oxychloride since its use results in moderate to good yields of pyrromethene intermediates.
- Cyclization of the heterocyclic ring with formation of the BDI dye is completed by addition of boron trifluoride in combination with a suitable base.
- Boron trifluoride is preferably used as one of its ether complexes due to the ease of handling of these complexes rather than the gaseous reagent.
- Suitable bases include, but are not limited to, trimethylamine, triethylamine, N,N-dnsopropylethylamine, N,N,N',N'-tetramethylethylenediamine, 1,8-bis(dimethylamino)naphthalene, diazabicyclooctane, diazabicycloundecene, 4-dimethylaminopyridine, 4-pyrrolidinopyridine and other similar bases.
- the long wavelength reactive BDI dyes may be further modified by sulfonation, nitration, alkylation, acylation, halogenation and other reactions by methods known in the art, such as described in Example 24 and by U.S. Patent 4,916,711 to Boyer, et al. (1990) (incorporated by reference); Worries, et al. , A novel water-soluble fluorescent probe: Synthesis, luminescence and biological properties of the sodium salt of the 4-sulfonato-3,3',5.5'-tetramethyl-2,2'-pyrromethen-1, 1'-BF 2 complex, RECL. TRAV. CHIM. PAYS-BAS 104, 288 (1985).
- the resulting products are generally soluble in organic solvents.
- Aqueous solubility can be obtained by adding appropriate water solubilization groups that include sulfonates, carboxylates, ammonium and hydroxyl residues to the dyes.
- the solid dyes are easily purified by techniques of chromatography and/or crystallization from a suitable solvent.
- the chemical structures of representative examples of long wavelength reactive BDI dyes have been confirmed by nuclear magnetic resonance spectroscopy (Table 4).
- the new long wavelength chemically reactive BDI dyes can be used to fluorescently label a wide variety of other materials, including cells, tissues, carbohydrates, drugs, lipids, nucleic acids, nucleotides, polymers, oligonucleotides, proteins, toxins, viruses, vitamins, and other biologically derived or synthetic chemical materials.
- the novel BDI dyes are particularly useful for the fluorescent labeling of members of specific binding pairs (sbp) to form dye conjugates which can be used to detect biological interactions.
- the sbp member can be a ligand or a receptor.
- the term ligand means any organic compound for which a receptor naturally exists or can be prepared.
- a receptor is any compound or composition capable of recognizing a spatial or polar organization of a molecule, e.g. epitopic or determinant site.
- Ligands for which naturally occurring receptors exist include natural and synthetic proteins, including avidin and streptavidin, antibodies, enzymes, and hormones; nucleotides and natural or synthetic oligonucleotides, including primers for RNA and single- and double-stranded DNA; lipids; polysaccharides and carbohydrates; and a variety of drugs, including therapeutic drugs and drugs of abuse; and pesticides.
- Ligands and receptors are complementary members of a specific binding pair, each sbp member having an area on the surface or in a cavity which specifically binds to and is complementary with a particular spatial and polar organization of the other.
- One embodiment of the invention is a method of preparing a fluorescent conjugate comprising combining a BDI dye of formula I with the sbp member being labeled.
- Another embodiment of the invention is a sbp member labeled with a new reactive BDI dye.
- the sbp member is a ligand, more preferably the ligand is a nucleotide, oligonucleotide, peptide, protein, lipid polysaccharide or carbohydrate.
- the new BDI dyes of this reaction are combined with a sbp member of interest which sbp member is capable of reacting with the reactive group (G) on the BDI fluorophore.
- a sbp member of interest which sbp member is capable of reacting with the reactive group (G) on the BDI fluorophore.
- any sbp member containing one or more of the reactive sites listed in Table 1 can be labeled using the new dyes.
- Preferred reactive sites are amines, thiols or carboxylic acids.
- the sbp member may be monovalent (containing a single reactive site) or polyvalent (containing multiple reactive sites).
- the starting material for nucleic acid conjugates is an oligonucleotide (oligo) which has incorporated a reactive residue, typically an amine or thiol, to form a derivatized oligo.
- oligo oligonucleotide
- the reactive residue is attached through a carbon chain spacer of a variety of lengths at either a substituted nucleotide base or the 5' end of the oligo.
- the thiol- or amine-derivatized oligos are typically commercially available with a carbon chain spacer of about 2-12 carbons.
- the reactive BDI dye is coupled directly to the sbp member.
- the sbp member is a ligand that has an amino substituent and the reactive dye has a carboxyl substituent
- the ligand and fluorophore may be directly conjugated to each other by procedures known in the art; for example, an active ester technique.
- the sbp member is a protein, it is less likely to require derivatization to add additional reactive sites.
- other groups can be introduced such as by thiolation with succinimidyl 2-pyridyldithiopropionate (SPDP).
- SPDP succinimidyl 2-pyridyldithiopropionate
- a sbp member can be labeled with the BDI dyes by using procedures such as those illustrated in Examples 25-30.
- the derivatized or reactive sbp member is solubilized in an appropriate solvent.
- the conditions required for solubilizing sbp members are generally known in the art.
- the dye is solubilized in an appropriate solvent that is mutually miscible with the solvent that is used to solubilize the sbp member.
- appropriate solvents include, for example, methanol, ethanol, dimethylformamide (DMF), dirnethylsulfoxide (DMSO), and acetone.
- appropriate solvents include, for example, DMF, DMSO, chloroform, toluene, and many other organic solvents.
- the temperature at which the reaction for preparing the conjugates of this invention proceeds is not critical.
- the temperature should be one which is sufficient to initiate and maintain the reaction, without compromising the stability or activity of the sbp member or its conjugate. Generally, for convenience and economy, room temperature ( ⁇ 25°C) is sufficient.
- the ratio of reactants is not narrowly critical.
- the ratio of reactants is not narrowly critical.
- 50-100 fold excess of dye has been found to be appropriate.
- the combined reactants, dye and sbp member, are incubated for sufficient time to allow the conjugation to occur, which may range from about 10 minutes to 24 hours, more typically from about 30 minutes to about 12 hours.
- the conjugate is then purified using standard purification techniques, for example, using chromatography, crystallization, etc.
- the dye-conjugated ligands are useful in a wide variety of areas as biochemical tools for the detection, identification, and measurement of biological compounds, particularly immunochemically reactive components such as those found in body fluids, cells, and cell components.
- the labeled proteins can indicate the presence and amount of certain antigens or identify the site of certain immunochemical reactions, using known assay methods and imaging techniques for detection of the fluorescence signal.
- the labeled nucleotides and oligonucleotides can be used in research efforts directed at determining the presence or sequence of nucleic acids in genes, using known gene mapping techniques.
- EXAMPLE 1 4,4-Difluoro-3-(2-methoxycarbonylethyl)-5-(2-thienyl)-4-bora-3a,4a-diaza-s-indacene (Compound 1): To a solution of 500 mg (2.82 mmol) of 2-formyl-5-(2-thienyl)pyrroleand 435 mg (2.84 mmol) of 2-(2-methoxycarbonylethyl) pyrrole in 50 mL of dichloromethane is added 265 ⁇ L (2.84 mmol) of phosphorus oxychloride.
- reaction mixture is stirred at room temperature for 16 hours and then 2.0 mL (11.5 mmol) of N,N-diisopropylethylamine is added, followed by the addition of 1.5 mL (11.5 mmol) of boron trifluoride etherate. After the whole reaction mixture is stirred at room temperature for 2 hours, it is washed with two 50 mL portions of brine. The organic layer is separated, dried over anhydrous sodium sulfate and concentrated under reduced pressure to give a dark brown solid. The crude product is purified by chromatography on silica gel with chloroform as eluant to give 380 mg (52%) of Compound 1 as a dark purple solid.
- the 2-formyl-5-(2-thienyl)pyrrole is prepared from 2-(2-thienyl)pyrrole by the Vilsmeier-Haack formylation, according to R.M. Silverstein et al., ORG. SYNTH. COLL. Vol. IV, p. 831.
- the 2-(2-thienyl)pyrrole needed for this synthesis is prepared in a similar manner as described in Kruse, et al., HETEROCYCLES 26, 3141 (1987).
- the 2-(2-methoxycarbonylethyl)pyrrole is prepared as follows: A solution of pyrrole-2-carboxaldehyde (3.9 g, 41 mmol) and (carbomethoxymethylene)-triphenylphosphorane (14 g, 42 mmol) in 160 mL of dry benzene is heated at reflux for 18 hours under a nitrogen atmosphere. After cooling, the reaction mixture is concentrated under vacuum and the resulting oil is purified by silica gel column chromatography, eluting with a mixture of ethyl acetate and hexane (1:9) to give 5.11 g of a mixture of cis- and trans-methyl 2-(2-pyrrolyl)acrylate.
- EXAMPLE 2 4,4-Difluoro-3-(2-methoxycarbonylethyl)-5-(2-furyl)-4-bora-3a,4a-diaza-s-indacene (Compound 5): This is prepared in the same manner as described in Example 1 from 2-(2-methoxycarbonylethyl)pyrrole (240 mg, 1.56 mmol) and 2-formyl-5-(2-furyl)pyrrole (250 mg, 1.55 mmol). Compound 5 (295 mg, 55 % yield) is obtained as a dark purple solid.
- the 2-formyl-5-(2-furyl)pyrrole is prepared from 2-(2-furyl)pyrrole by the Vilsmeier-Haack formylation as described in ORG. SYNTH. COLL. Vol. IV, p. 831.
- the 2-(2-furyl)pyrrole needed for this synthesis is prepared in a similar manner as described in HETEROCYCLES 26, 3141 (1987).
- EXAMPLE 3 4,4-Difluoro-3-(2-methoxycarbonylethyl)-5-(2-pyrrolyl)-4-bora-3a,4a-diaza-s-indacene (Compound 7): To a solution of 150 mg (1.13 mmol) of 2,2'-bipyrrole and 205 mg (1.13 mmol) of 2- (2- methoxycarbonylethyl)-5-formylpyrrole in 10 mL of dichloromethane is added 110 ⁇ L (1.18 mmol) of phosphorus oxychloride.
- the reaction mixture is stirred at room temperature for 12 hours and then is added 800 ⁇ L (4.59 mmol) of N,N-diisopropylethylamine, followed by the addition of 560 ⁇ L (4.55 mmol) of boron trifluoride etherate. After the whole mixture is stirred at room temperature for 2 hours, it is washed with two 10 mL portions of brine. The organic layer is separated, dried over anhydrous sodium sulfate and concentrated under reduced pressure to give a dark red solid. The crude product is purified by chromatography on silica gel with a mixture of chloroform and hexane (1:1) as eluant to give 125 mg (32%) of Compound 7 as a dark purple solid.
- the 2,2'-bipyrrole is prepared as described in Rappaport et al., J. AM. CHEM. SOC., 84, 2178
- the 2-(2-methoxycarbonylethyl)-5-formyl ⁇ yrrole is prepared from 2-(2- methoxycarbonylethyl)pyrrole by the Vilsmeier-Haack formylation according to ORG. SYNTH. COLL. Vol. IV, p 831.
- EXAMPLE4 4, 4-Difluoro-1,3-dimethyl-2-(2-methoxycarbonylethyl)-5-(2-pyrroly l)-4-bora-3a,4a-diaza-s- indacene (Compound 10): This is prepared in the same method as described in Example 3 from 2,2'- bipyrrole (500 mg, 3.78 mmol) and 2.4-dimethyl-5-formyl-3-(2-methoxycarbonylethyl)pyrrole(795 mg, 3.80 mmol). Compound 10 (760 mg, 54%) is obtained as a dark purple solid.
- the 2,4-dimethyl-3-(2-methoxycarbonylethyl)pyrroleis prepared as described in Bullock, et al., J. CHEM. SOC. 1420 (1958).
- EXAMPLE5 4,4-Difluoro-1,3-dirnemyl-8-(2-meraoxycarbonylethyl)-5-(2-thienyl)-4-bora-3a,4a-diaza-s--indacene
- Compound 12 To a solution of 500 mg (2.39 mmol) of methyl 4-[2-(3,5-dimethyl)pyrrolyl]-4-oxobutanoate and 355 mg (2.38 mmol) of 2-(2-thienyl)pyrrole in 30 mL of dichloromethane is added 230 ⁇ L (2.47 mmol) of phosphorous oxychloride. The reaction mixture is heated under reflux in a nitrogen atmosphere for 2 days.
- EXAMPLE 6 4,4-Difluoro-3-[4-(methoxycarbonylmethoxy)phenyl]-5-(2-thienyl)-4-bora-3a,4a-diaza-s- indacene
- Compound 14 This is prepared in the same method as described in Example 1 from 2-[4-(methoxycarbonylmethoxy)phenyl]pyrrole (100 mg, 0.43 mmol) and 2-formyl-5-(2-thienyl)pyrrole (75 mg, 0.42 mmol). Compound 14 (68 mg, 37%) is obtained as a dark purple solid.
- the 2-[4-(methoxycarbonylmethoxy)phenyl]pyrrole needed for this synthesis is prepared as follows: To a suspension of 1.0 g (11.9 mmol) of ethanethiol, sodium salt in 10 mL of dry DMF is added 2.0 g (11.5 mmol) of 2-(4-methoxyphenyl)pyrrole [prepared in a similar method as described in HETEROCYCLES 26, 3141 (1987)] while the reaction mixture is stirred in an ice bath under nitrogen atmosphere. It is then stirred at 110°C for 4 hours. After the reaction mixture is cooled to room temperature, 1.82 g (11.9 mmol) of methyl bromoacetate is added.
- the Compounds 17, 22, 25, and 39 are prepared by the reaction of 2-formyl-5-(2-furyl)pyrrole; 2-formyl-5-[(E)-2-phenylethen-1-yl]pyrrole2-formyl-5-[(E,E)-4-phenyl-1,3-butadien-1-yl]pyrrole2,-formyl-5-[(E)-propen-1-yl]pyrrole, respectively, with2-[4-(methoxycarbonylmethoxy)phenyl]pyrroleas described in Example 6.
- Compound 19 is prepared by the reaction of 2,2'-bipyrrole with 2-formyl-5-[4-(methoxycarbonylmethoxy)phenyl]pyrrole, which is obtained by the Vilsmeier-Haack formylation of 2-[4-(methoxycarbonylmethoxy)phenyl]pyrrole, as described in Example 6.
- EXAMPLE 7 4,4-Difluoro-3-[(E)-2-[4-(methoxycarbonylmethoxy)phenyl]ethen-1-yl]-5-[(E)-2-phenylethen-1-yl]-4-bora-3a,4a-diaza-s-indacene
- Compound 23 This is prepared in the same manner as described in Example 1 from 2-formyl-5-[(E)-phenylethen-1-yI]pyrrole(75 mg, 0.38 mmol) and 2-[(E)-2-[(4-methoxycarbonylmethoxy)phenyl]ethen-1-yl]pyrrole(100 mg, 0.39 mmol).
- the Compound 23 (59 mg, 32%) is obtained as a dark blue solid.
- the 2-[(E)-2-[4-(methoxycarbonylmethoxy)phenyl]ethen-1-yl]pyrrole needed for this synthesis is prepared in the similar way as described in Example 6 by the reaction of 2-[(E)-2-(4-methoxyphenyl)ethen-1-yl]pyrrole with ethanethiol, sodium salt, and methyl bromoacetate in DMF.
- Compounds 27, 38, and 44 are prepared by the reaction of 2-formyl-5-[(E,E)-4-phenyl-1,3-butadien-1-yl]pyrrole, 2-formyl-5-[(E)-propen-1-yl]pyrrole and 2-formyl-5-[(E)-2-(1-naphthyl)ethen-1-yl)pyrrole, respectively, with 2-(2-methoxycarbonylethyl)-pyrrole as described in Example 8.
- Compound 33 is prepared from 2-[(E)-2-carbomethoxyethen-1-yl]pyrrole and 3 ,5-diphenylpyrrole-2- cafboxaldehyde in the similar way as described in Example 8.
- EXAMPLE 10 Succinimidyl ester (Compound 3): To a solution of 650 mg (1.88 mmol) of Compound 2 in 70 mL of tetrahydrofuran is added 230 mg (1.99 mmol) of N-hydroxysuccinimide followed by addition of 410 mg (1.99 mmol) of N.N'-dicyclohexylcarbodiimide. The mixture is stirred at room temperature for 24 hours. The resulting precipitate is removed by filtration and the filtrate is evaporated to dryness under reduced pressure to give a crude product. It is purified by chromatography on silica gel with chloroform as eluant to give 715 mg (86 %) of Compound 3 as a dark purple solid.
- EXAMPLE 11 Hydrazide (Compound 4): To a solution of 10 ⁇ L of anhydrous hydrazine in 1 mL of chloroform is added 25 mg of Compound 3 and the mixture is stirred at room temperature for 30 minutes. The reaction mixture is diluted with 15 mL of chloroform, washed with water, dried over anhydrous sodium sulfate and concentrated under reduced pressure to give a crude product. It is purified by silica gel column chromatography with 2% methanol in chloroform as eluant to give 21 mg (98 %) of Compound 4.
- EXAMPLE 12 Amine (Compound 48): To a solution of 150 ⁇ L (2.24 mmol) of ethylenediamine in 2 mL of chloroform is added a solution of 100 mg (0.22 mmol) of Compound 47 in 3 mL of chloroform dropwise over a period of 10 minutes and the mixture is stirred at room temperature for 1 hour. It is then diluted with 15 mL of chloroform, washed with water (3 ⁇ 20 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to give a crude product. It is purified by chromatography on silica gel with 10% methanol in chloroform as eluant to give 75 mg (85%) of the amine 48.
- Compound 61 is prepared from Compound 3 by the same method as described in Example 12.
- EXAMPLE 13 lodoacetamide (Compound 49): To a suspension of 20 mg (0.05 mmol) of Compound 48 in 3 mL of chloroform is added 10 ⁇ L of triethylamine, followed by addition of 12 mg (0.05 mmol) of iodoacetic anhydride and the mixture is stirred at room temperature for 2 hours. After evaporation under reduced pressure, it is purified by silica gel column chromatography with 2% methanol in chloroform as eluant to give 19 mg (67%) of the iodoacetamide 49.
- Compound 62 is prepared from Compound 61 by the same method as described in Example 13.
- EXAMPLE 14 Maleimide (Compound 50): To a suspension of 20 mg (0.05 mmol) of Compound 48 in 3 mL of chloroform is added 10 ⁇ L of chloroform is added 10 ⁇ L of triethylamine, followed by addition of 13 mg (0.05) of succinimidyl maleimidylacetate and the mixture is stirred at room temperature for 2 hours. It is concentrated and purified by column chromatography on silica gel with 2% methanol in chloroform as eluant to give 18 mg (67%) of the maleimide 50.
- EXAMPLE 15 Aldehyde (Compound 51): To a suspension of 20 mg (0.05 mmol) of Compound 48 in 3 mL of chloroform is added 10 ⁇ L of triethylamine, followed by addition of 12 mg (0.05 mmol) of succinimidyl p-formylbenzoate and the mixture is stirred at room temperature for 2 hours. It is concentrated and purified by column chromatography on silica gel with 2 % methanol in chloroform as eluant to give 22 mg (83 %) of the aldehyde 51.
- EXAMPLE 16 Azide (Compound 52): To a suspension of 20 mg (0.05 mmol) of Compound 48 in 3 mL of chloroform is added 10 ⁇ L of triethylamine, followed by addition of 13 mg (0.05 mmol) of succinimidyl p-azidobenzoate. After stirring at room temperature for 2 hours, it is concentrated under reduced pressure and purified by column chromatography on silica gel eluting with 1% methanol in chloroform to give 17 mg (63 %) of the azide 52.
- EXAMPLE 17 Acrylamide (Compound 53): To a suspension of 20 mg (0.05 mmol) of Compound 48 in 3 mL of chloroform is added 10 ⁇ L of triethylamine, followed by addition of 9 mg (0.05 mmol) of succinimidyl acrylate and the mixture is stirred at room temperature for 2 hours. After concentration under reduced pressure, it is purified by silica gel column chromatography with 1% methanol in chloroform as eluant to give 18 mg (79%) of the acrylamide 53.
- EXAMPLE 18 Alcohol (Compound 42): To a solution of 125 mg (1.01 mmol) of 3,5-dimethylpyrrole- 2-carboxaldehyde and 200 mg (1.00 mmol) of 2-[(E)-2-(4-hydroxymethylphenyl)ethen-1-yl]pyrrolein 15 mL of dichloromethane is added 100 ⁇ L (1.07 mmol) of phosphorus oxychloride. The reaction mixture is stirred at room temperature for 24 hours and then is added 700 ⁇ L (4.02 mmol) of N,N- diisopropylethylamine, followed by addition of 490 ⁇ L (3.98 mmol) of boron trifluoride etherate.
- the compound 2-[(E)-2-(4-hydroxymethylphenyl)ethen-1-yl]pyrrole needed for this synthesis is prepared as follows: To a solution of 1.0 g (4.4 mmol) of 2-[(E)-2-(4-carbomethoxvphenyl)ethen-1-yl]pyrrole in 20 mL of dry tetrahydrofuran is added 167 mg (4.4 mmol) of lithium aluminum hydride portionwise while the reaction mixture is stirred at 0°C under nitrogen atmosphere. After addition is complete, it is stirred at room temperature for 15 hours. It is then cooled with an ice-water bath and the excess lithium aluminum hydride is destroyed by the slow addition of 2 mL water.
- EXAMPLE 19 Halide (Compound 43): To a solution of 20 mg (0.06 mmol) of Compound 42 in 5 mL of dichloromethane is added one drop of triethylamine, followed by the addition of 5 mg (0.03 mmol) of cyanuric chloride. The mixture is stirred at room temperature for 1 hour. It is then subjected to silica gel column chromatography with 1: 1 hexane/chloroform as eluant to give 15 mg (71%) of the halide 43.
- EXAMPLE 20 Isothiocyanate (Compound 54): To a suspension of 30 mg (0.07 mmol) of Compound 48 in 5 mL of chloroform is added 20 ⁇ L of triethylamine, followed by the addition of 10 ⁇ L (0.13 mmol) of thiophosgene and the mixture is stirred at room temperature for 1 hour. It is concentrated under reduced pressure and the resulting residue is purified by column chromatography on silica gel with chloroform as eluant to give 22 mg (66%) of the isothiocyanate 54.
- EXAMPLE 21 Anhydride (Compound 55): To a suspension of 30 mg (0.09 mmol) of Compound 2 in 5 mL of chloroform is added 15 ⁇ L of triethylamine, followed by addition of 10 ⁇ L of ethyl chloroformate and the mixture is stirred at room temperature for 1 hour. Without isolation of the anhydride, Compound 55, it is characterized as its n-butyl amide, Compound 58, by addition of 15 ⁇ L of n-butylamine to the reaction mixture. The crude residue isolated by evaporation of the reaction mixture is purified by column chromatography on silica gel with chloroform as eluant to yield 29 mg (85%) of the Compound 58.
- EXAMPLE 22 Acyl azide (Compound 56): To a suspension of 30 mg (0.09 mmol) of Compound 2 in 5 mL of chloroform is added 15 ⁇ L of triethylamine, followed by the addition of 20 ⁇ L (0.09 mmol) of diphenylphosphoryl azide and the reaction mixture is stirred at room temperature for 3 hours. It is concentrated under reduced pressure and purified by column chromatography on silica gel, eluting with chloroform to give 17 mg (53 %) of the acyl azide 56.
- EXAMPLE 24 Sulfonyl Chloride (Compound 60): To an ice-cooled solution of 30 mg (0.08 mmol) of 4,4-difluoro-3,5-bis[(E)-2-phenylethen-1-yl]-4-bora-3a,4a-diaza-s-indacene(which is prepared from 2-[(E)-2-phenylethen-1-yl]pyrroleand 2-formyl-5-[(E)-2-phenylethen-1-yl]pyrrole as described in Example 1) in 3 mL of dichloromethane is added 12 ⁇ L (0.18 mmol) of chlorosulfonic acid and the mixture is stirred at 0°C for 1 ⁇ 2 hours under nitrogen atmosphere.
- the resulting precipitate is collected by filtration, dissolved in 1 mL of water and the solution is treated with 20 mg of sodium bicarbonate.
- the crude product is purified by chromatography over lipophilic Sephadex using water for eiution and the desired fractions are combined and lyophilized to give 12 mg of Compound 59.
- To 300 ⁇ L of thionyl chloride is added 30 ⁇ L of dry dimethylformamide and the mixture is stirred at room temperature for 10 minutes.
- To the above solution is added 10 mg of Compound 59 and the mixture is stirred at room temperature for 2 hours.
- the resulting reaction mixture is treated with 10 mL of chloroform. It is poured into 10 mL of ice water and shaken vigorously for a few minutes.
- EXAMPLE 26 Nucleotide Conjugate: To a solution of 2 mg of 5-(3-aminoallyl)-2'-deoxyuridine-5'- triphosphate, ammonium salt (Sigma Chemical Co., St. Louis, MO) in 200 ⁇ L of water is added a solution of 2 mg of Compound 3 in 200 ⁇ L of acetonitrile, followed by addition of 5 ⁇ L of triethylamine. After the mixture is stirred at room temperature for 3 hours, it is purified by chromatography over lipophilic Sephadex using water for elution.
- ammonium salt Sigma Chemical Co., St. Louis, MO
- EXAMPLE 27 Oligonucleotide Conjugate: The 5'-amine-derivatized oligonucleotide of sequence XTGTAAAACGACGGCCAGT is prepared by automated solid phase synthesis on a ABI 380A using the standard 1 ⁇ M synthesis cycle, where X, the last phosphoramidite (5' end), is the commercially available C6 aminolink-TFA obtained from Glen Research Inc., Sterling, VA. After the standard cleavage from the solid phase column and deblocking in concentrated aqueous ammonia (5 hours at 55°C), the oligonucleotide is concentrated on a vortex evaporator to dryness in a 15 mL plastic centrifuge tube.
- the oligonucleotide is desalted with a Bio-Rad 10DG desalting column, Bio-Rad Labs, Richmond, CA, using the standard procedure.
- the oligonucleotide is aliquoted into convenient 10 ODU portions in 2 mL microcentrifuge tubes and evaporated on a speed-vac.
- the 5'-amine-derivatized oligonucleotide, 10 ODU (350 ⁇ g, 0.055 ⁇ M) dissolved in 250 ⁇ L of 0.5 M NaHCO 3 /Na 2 CO 3 pH 9 aqueous buffer in the microcentrifuge tube.
- the compound 16, 2 mg (3.8 ⁇ M), is dissolved in 200 ⁇ L of DMF and added to the oligonucleotide solution. This is shaken by hand and allowed to sit for 16 hours. 1 mL of 0.1 M TEAA is added and the solution is passed through a Bio-Rad 10DG desalting column substituting 0.1 M TEAA for deionized water in the standard procedure. The second 2 mL fraction is collected and evaporated in a 15 mL centrifuge tube on a vortex evaporator.
- the labeled oligonucleotide is then HPLC purified on a 220 mm ⁇ 10 mm 300 A C8 reverse phase column, Rainin Instrument Co., Woburn, MA, using the following gradient: Solvent A - 0.1 M TEAA , Solvent B - acetonitrile. Ramp solvent B from 15% to 95% over 40 minutes then hold at 95% for 5 minutes. Detection is with a Waters 490 dual wavelength UV-Vis detector monitoring 254 nm and 590 nm. The appropriate peak is collected and evaporated to give 2.3 ODU (80 ⁇ g, 0.013 ⁇ M) 24% yield.
- Example 28 Protein Conjugates: The succinimidyl esters, Compounds 3 and 16, are dissolved to give a concentration of 10 mg dye/mL DMF. Bovine serum albumin (BSA) or other proteins are dissolved to a concentration of approximately 10 mg protein /mL in 0.1 M sodium bicarbonate. Amicon GH-25 desalting media in phosphate buffered saline (PBS) and 1.5 M hydroxylamine pH 8 are freshly prepared. The appropriate amount of each dye in DMF corresponding to a molar ratio of 10 (dye/protein) is slowly added to the separate protein solutions with stirring. Each reaction is incubated at room temperature for one hour and is then terminated by the addition of hydroxylamine to a final concentration of 0.15 M.
- BSA bovine serum albumin
- PBS phosphate buffered saline
- the protein conjugates are separated from free unreacted dye in the two solutions by gel filtration on a GH-25 column equilibrated in PBS.
- the initial, protem-containing fractions from each solution are pooled and the degree of substitution is determined from the extinction coefficient for each dye at its absorption maximum (559 nm for Compound 3 and 590 nm for Compound 16).
- An approximate degree of substitution of 3 (dye/BSA) is determined for both cases.
- the fluorescence of each final protein conjugate is approximately 10% of the free dye.
- Example 29 Peptide Conjugate: To a suspension of 5 mg of N-formyl-Nle-Leu-Phe-Nle-Tyr-Lys (Bachem. Inc., Torrance, CA) in 500 ⁇ L of dimethylformamide is added 3 ⁇ L of triethylamine followed by the addition of 3 mg of Compound 3 and the whole reaction mixture is stirred at room temperature for 30 minutes. The reaction mixture is purified by chromatography over lipophilic Sephadex using water for elution. The desired fractions are combined and lyophilized to give 4 mg of the fluorescent hexapeptide.
- Example 30 Carbohydrate Conjugate: To a solution of 5 mg of 6-amino-6-deoxy-D-glucose hydrochloride (U.S. Biochemical Corp., Cleveland, OH) in 0.5 mL of water and 1 mL of tetrahydrofuran is added 5 ⁇ L of triethylamine followed by the addition of 10 mg of Compound 3. After the mixture is stirred at room temperature for 1 hour, it is purified by chromatography over silica gel using 10% methanol in chloroform. From the desired combined fractions 12 mg of a fluorescent carbohydrate is obtained: m.p.
- n-butylamine reacts to form a new product with 3, 9, 16, 21, 32, 35, 47, 54, 55, 56, and 57, that 2-mercaptoethanol reacts with 43, 49, 50, and 62 that acetic anhydride reacts with 4, 42, 48, and 61, that acetone reacts with 4, that hydrazine reacts with 51, and that brefeldin A reacts with 2 in the presence of N,N'-dicyclohexylcarbodiimide to give an ester. Furthermore, it is demonstrated that the esters such as 1 can react with hydrazine to give hydrazides such as 4.
- EXAMPLE 32 Spectral Characterization Of The Dyes: 1 H NMR spectra are measured using a General Electric QE-400 MHz spectrometer for solutions in CDCl 3 (unless otherwise stated), with tetramethylsilane (TMS) as an internal standard. Chemical shifts are given in ppm from TMS and splitting patterns are designated as: s, singlet; d, doublet; t, triplet; m, multiplet. Results of spectral data for representative new dyes are given in Table 4.
- Absorption spectra are obtained using an IBM 9429 UV/visible spectrophotometer by dissolving the dye at a concentration of approximately 5 ⁇ 10 -6 M in an appropriate solvent including but not limited to methanol, chloroform, acetonitrile, acetone or hexane. Extinction coefficients (e) of the dyes at their absorption maxima are determined by standard Beer's law calculations. Results of the spectral determination for representative new dyes are tabulated in Table 3 and Table 5.
- Fluorescence of new long wavelength reactive dipyrrometheneboron difluoride dyes is determined using a Perkin-Elmer Model 650-40 fluorescence spectrophotometer equipped with a Perlrin-Elmer/Hitachi 057 X-Y recorder by dissolving the dye at a concentration of above 1 ⁇ 10 -10 M (optimum concentration ranges, 10 -6 ⁇ 10 -7 M) in an appropriate solvent including but not limited to methanol, water, ethanol, acetonitrile, acetone, chloroform, toluene or hexane. Results of the spectral determination for representative new dyes are summarized in Table 3 and Table 5.
- EXAMPLE 33 Photobleaching Studies: Photostability measurements of new long wavelength reactive BDI dyes are conducted in acetonitrile solution with continuous illumination by a 250 watt xenon arc lamp with 20 nm slit width at the corresponding excitation maxima. After thirty minutes of continuous illumination while stirring at room temperature in a cuvette of a fluorometer, emission intensity data is collected with 2.5 nm slit width at the emission maxima of individual samples.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Medicinal Chemistry (AREA)
- Microbiology (AREA)
- Cell Biology (AREA)
- Food Science & Technology (AREA)
- Materials Engineering (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
Description


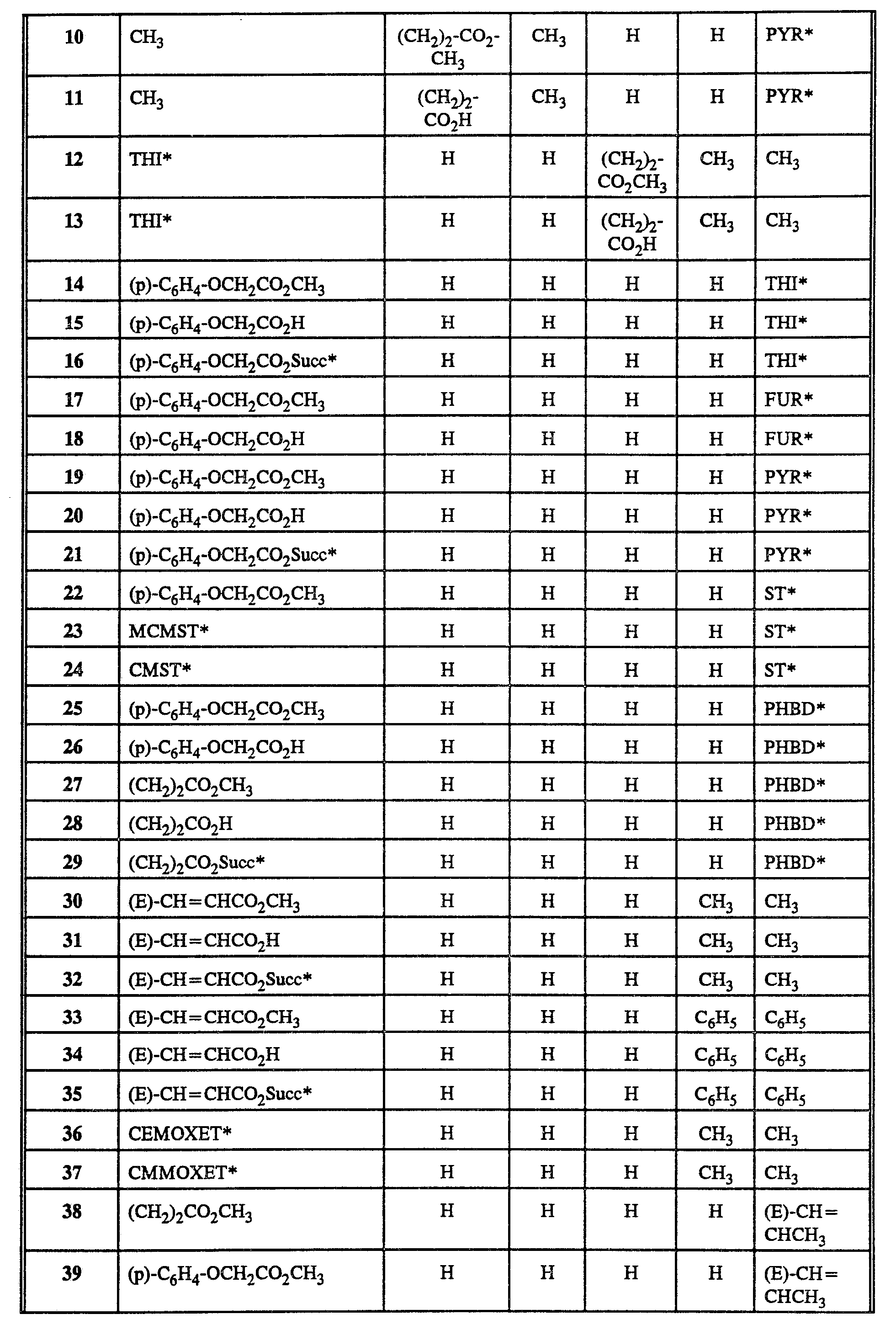




Claims


Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE69223985T DE69223985T2 (en) | 1991-11-01 | 1992-09-30 | IN THE LONG WAVELENGTH RANGE ABSORBING CHEMICALLY REACTIVE DIPYROMETHENBORDIFLUOR DYES AND THEIR CONJUGATES |
| EP92922004A EP0612336B1 (en) | 1991-11-01 | 1992-09-30 | Long wavelength chemically reactive dipyrrometheneboron difluoride dyes and conjugates |
| CA002122627A CA2122627C (en) | 1991-11-01 | 1992-09-30 | Long wavelength chemically reactive dipyrrometheneboron difluoride dyes and conjugates |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US786,767 | 1991-11-01 | ||
| US07/786,767 US5274113A (en) | 1991-11-01 | 1991-11-01 | Long wavelength chemically reactive dipyrrometheneboron difluoride dyes and conjugates |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1993009185A1 true WO1993009185A1 (en) | 1993-05-13 |
Family
ID=25139533
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1992/008350 WO1993009185A1 (en) | 1991-11-01 | 1992-09-30 | Long wavelength chemically reactive dipyrrometheneboron difluoride dyes and conjugates |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US5274113A (en) |
| EP (1) | EP0612336B1 (en) |
| AT (1) | ATE161871T1 (en) |
| CA (1) | CA2122627C (en) |
| DE (1) | DE69223985T2 (en) |
| WO (1) | WO1993009185A1 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0684950A1 (en) * | 1993-02-19 | 1995-12-06 | Bo-Dekk Ventures, Ltd. | Chemical compounds useful in photodynamic therapy and production of laser light |
| EP0747448A2 (en) * | 1995-06-07 | 1996-12-11 | Carnegie-Mellon University | Rigidized monomethine cyanine dyes |
| EP0822544A1 (en) * | 1996-07-29 | 1998-02-04 | Mitsui Toatsu Chemicals, Incorporated | Optical recording medium |
| WO1998013524A1 (en) * | 1996-09-27 | 1998-04-02 | Laboratory Of Molecular Biophotonics | Probes for detecting polynucleotides and detection method |
| US5852191A (en) * | 1995-06-07 | 1998-12-22 | Carnegie Mellon University | Rigidized monomethine cyanines |
| WO1999011813A2 (en) * | 1997-09-04 | 1999-03-11 | Bayer Corporation | Oligonucleotide probes bearing quenchable fluorescent labels, and methods of use thereof |
| US6277984B1 (en) | 1995-06-07 | 2001-08-21 | Carnegie Mellon University | Monomethine cyanines rigidized by a two-carbon chain |
| WO2003004569A1 (en) * | 2001-07-02 | 2003-01-16 | Arctic Diagnostics Oy | Method for increasing hydrophilicity of fluorescent label compounds |
| WO2003005030A1 (en) * | 2001-07-02 | 2003-01-16 | Arctic Diagnostics Oy | Two-photon absorbing dipyrrometheneboron difluoride dyes and their applications |
| US7425553B2 (en) | 2003-05-30 | 2008-09-16 | Gemin X Pharmaceuticals Canada Inc. | Triheterocyclic compounds, compositions, and methods for treating cancer or viral diseases |
| US7910617B2 (en) | 2004-02-24 | 2011-03-22 | Sankyo Company, Limited | Method for suppressing the number of peripheral blood lymphocytes using an amino alcohol compound |
| US8067396B2 (en) | 2002-01-11 | 2011-11-29 | Sankyo Company, Limited | Amino alcohol compounds or phosphonic acid derivatives thereof |
| WO2012118444A1 (en) * | 2011-03-01 | 2012-09-07 | Agency For Science, Technology And Research | Novel compounds with photoluminescnce properties and applications thereof |
| WO2017139689A1 (en) * | 2016-02-12 | 2017-08-17 | Oregon Health & Science University | Derivatives of bodipy |
Families Citing this family (303)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5648270A (en) * | 1995-02-06 | 1997-07-15 | Molecular Probes, Inc. | Methods of sensing with fluorescent conjugates of metal-chelating nitrogen heterocycles |
| US5433896A (en) * | 1994-05-20 | 1995-07-18 | Molecular Probes, Inc. | Dibenzopyrrometheneboron difluoride dyes |
| US5723218A (en) * | 1990-04-16 | 1998-03-03 | Molecular Probes, Inc. | Dipyrrometheneboron difluoride labeled flourescent microparticles |
| US5338854A (en) * | 1991-02-13 | 1994-08-16 | Molecular Probes, Inc. | Fluorescent fatty acids derived from dipyrrometheneboron difluoride dyes |
| US7322927B2 (en) | 1993-09-24 | 2008-01-29 | Biosite, Inc. | Hybrid phthalocyanine derivatives and their uses |
| US7083984B2 (en) | 1993-09-24 | 2006-08-01 | Biosite, Inc. | Hybrid phthalocyanine derivatives and their uses |
| US5824799A (en) * | 1993-09-24 | 1998-10-20 | Biosite Diagnostics Incorporated | Hybrid phthalocyanine derivatives and their uses |
| US6210941B1 (en) | 1997-06-27 | 2001-04-03 | The Trustees Of Boston University | Methods for the detection and isolation of proteins |
| US5643722A (en) | 1994-05-11 | 1997-07-01 | Trustees Of Boston University | Methods for the detection and isolation of proteins |
| US5693679A (en) * | 1995-04-04 | 1997-12-02 | Advanced Bioconcept, Inc. | Fluorescent somatostatin |
| US5824772A (en) * | 1995-04-04 | 1998-10-20 | Advanced Bioconcept, Inc. | Fluorescent somatostatin |
| US6127134A (en) * | 1995-04-20 | 2000-10-03 | Carnegie Mellon University | Difference gel electrophoresis using matched multiple dyes |
| US6426190B1 (en) * | 1995-04-20 | 2002-07-30 | Carnegie Mellon University | Difference detection methods using matched multiple dyes |
| US6008373A (en) | 1995-06-07 | 1999-12-28 | Carnegie Mellon University | Fluorescent labeling complexes with large stokes shift formed by coupling together cyanine and other fluorochromes capable of resonance energy transfer |
| US5614386A (en) * | 1995-06-23 | 1997-03-25 | Baylor College Of Medicine | Alternative dye-labeled primers for automated DNA sequencing |
| US5994063A (en) * | 1995-06-23 | 1999-11-30 | Metzker; Michael L. | Substituted 4,4-difluoro-4-bora-3A,4A-diaza-s-indacene compounds for homogenous amplification/detection assays |
| WO1997000967A1 (en) * | 1995-06-23 | 1997-01-09 | Baylor College Of Medicine | Alternative dye-labeled primers, ribonucleotides, deoxyribonucleotides, and dideoxyribonucleotides for automated dna analysis and homogeneous amplification/detection assays |
| US5728529A (en) * | 1995-06-23 | 1998-03-17 | Baylor College Of Medicine | Alternative dye-labeled ribonucleotides, deoxyribonucleotides, and dideoxyribonucleotides for automated DNA analysis |
| US6821952B1 (en) | 1995-07-20 | 2004-11-23 | Perkinelmer Las, Inc. | Fluorescent vasoactive intestinal peptide (VIP) |
| US5869689A (en) * | 1995-10-17 | 1999-02-09 | Molecular Probes, Inc | Stains for acidic organelles |
| US5952236A (en) * | 1995-10-26 | 1999-09-14 | Thompson; Richard B. | Enzyme-based fluorescence biosensor for chemical analysis |
| US7253557B2 (en) * | 1996-02-08 | 2007-08-07 | Bright Solutions, Inc. | Light source provided with a housing enclosing voltage regulator means and method of manufacturing thereof |
| US6590220B1 (en) * | 1996-02-08 | 2003-07-08 | Bright Solutions, Inc. | Leak detection lamp |
| US6005113A (en) * | 1996-05-15 | 1999-12-21 | Molecular Probes, Inc. | Long wavelength dyes for infrared tracing |
| US5719031A (en) * | 1996-08-14 | 1998-02-17 | Molecular Probes, Inc. | Dye labeled polymers as reagents for measuring polymer degradation |
| US5786219A (en) * | 1996-10-28 | 1998-07-28 | Molecular Probes, Inc. | Microspheres with fluorescent spherical zones |
| US5773236A (en) * | 1997-04-25 | 1998-06-30 | Molecule Probes, Inc. | Assay for glutathiane transferase using polyhaloaryl-substituted reporter molecules |
| AT405461B (en) * | 1997-05-30 | 1999-08-25 | Avl List Gmbh | LUMINESCENT INDICATOR |
| US7208528B1 (en) | 1997-07-15 | 2007-04-24 | Mediquest Therapeutics, Inc. | Polyamine analogues as therapeutic and diagnostic agents |
| US6646149B1 (en) | 1997-07-15 | 2003-11-11 | Nicolaas M. J. Vermeulin | Polyamine analogues as therapeutic and diagnostic agents |
| WO1999003823A2 (en) | 1997-07-15 | 1999-01-28 | Oridigm Corporation | Novel polyamine analogues as therapeutic and diagnostic agents |
| US6106999A (en) * | 1997-08-12 | 2000-08-22 | Mitsui Chemicals | Photosensitizer, visible light curable resin composition using the same, and use of the composition |
| US7745142B2 (en) | 1997-09-15 | 2010-06-29 | Molecular Devices Corporation | Molecular modification assays |
| US20050147534A1 (en) | 1998-05-05 | 2005-07-07 | Massachusetts Institute Of Technology | Emissive sensors and devices incorporating these sensors |
| WO1999057222A1 (en) | 1998-05-05 | 1999-11-11 | Massachusetts Institute Of Technology | Emissive polymers and devices incorporating these polymers |
| US6372512B1 (en) | 1998-09-16 | 2002-04-16 | Resolution Sciences Corporation | Combined en bloc staining and embedding process |
| WO2000023594A1 (en) * | 1998-10-22 | 2000-04-27 | The General Hospital Corporation | BIOACTIVE PEPTIDES AND PEPTIDE DERIVATIVES OF PARATHYROID HORMONE (PTH) AND PARATHYROID HORMONE-RELATED PEPTIDE (PTHrP) |
| EP1975243B1 (en) * | 1998-12-07 | 2010-03-24 | Duke University | BODIPY aminoacetaldehyde diethyl acetal |
| DE60001531T2 (en) | 1999-04-23 | 2003-10-02 | Molecular Probes, Inc. | XANTHENE DYES AND THEIR USE AS LUMINESCENT EXTINGUISHING COMPOUNDS |
| US6653126B1 (en) | 1999-06-04 | 2003-11-25 | Massachusetts Insititute Of Technology | Compositions and methods for the screening of compounds to enhance or reduce apoptosis |
| AU709242B3 (en) | 1999-06-21 | 1999-08-26 | Patricia Catherine Hastie | An educational device |
| ES2161152B1 (en) * | 1999-07-09 | 2002-07-01 | Consejo Superior Investigacion | Set of polymers incorporating chromophores consists of solid state laser materials with copolymers in solid matrix |
| ATE349548T1 (en) * | 1999-08-25 | 2007-01-15 | Ambergen Inc | METHOD FOR DETECTION, ANALYSIS AND ISOLATION OF RELEASED PROTEINS |
| US6306628B1 (en) * | 1999-08-25 | 2001-10-23 | Ambergen, Incorporated | Methods for the detection, analysis and isolation of Nascent proteins |
| US6323186B1 (en) | 1999-09-17 | 2001-11-27 | Molecular Probes, Inc. | Phosphate-bound polyazaindacene derivatives of nucleotides |
| AU7734800A (en) * | 1999-09-29 | 2001-04-30 | General Hospital Corporation, The | Polypeptide derivatives of parathyroid hormone (pth) |
| ATE456575T1 (en) | 1999-10-18 | 2010-02-15 | Prince Henrys Inst Med Res | IMMUNE-INTERACTIVE FRAGMENTS OF THE ALPHA-C SUBUNIT OF INHIBIN |
| US6376669B1 (en) | 1999-11-05 | 2002-04-23 | 3M Innovative Properties Company | Dye labeled imidazoquinoline compounds |
| US6916925B1 (en) | 1999-11-05 | 2005-07-12 | 3M Innovative Properties Co. | Dye labeled imidazoquinoline compounds |
| US20010051348A1 (en) * | 2000-01-28 | 2001-12-13 | Lee Chee Wee | Novel ligands and methods for preparing same |
| US6323337B1 (en) | 2000-05-12 | 2001-11-27 | Molecular Probes, Inc. | Quenching oligonucleotides |
| GB0012229D0 (en) | 2000-05-19 | 2000-07-12 | Devgen Nv | Lipid uptake assays |
| US7252932B1 (en) | 2000-08-23 | 2007-08-07 | Ambergen, Inc. | Methods for the detection, analysis and isolation of nascent proteins |
| CA2420149C (en) * | 2000-08-24 | 2006-10-31 | Bio-Rad Laboratories, Inc. | Dry powder formulation for protein labeling |
| US20020049986A1 (en) * | 2000-10-10 | 2002-04-25 | Steven Farber | High throughput genetic screening of lipid and cholesterol processing using fluorescent compounds |
| US7579463B2 (en) * | 2000-12-20 | 2009-08-25 | Life Technologies Corporation | Crown ether derivatives |
| US6962992B2 (en) | 2000-12-20 | 2005-11-08 | Molecullar Probes, Inc. | Crown ether derivatives |
| USRE43327E1 (en) | 2001-01-08 | 2012-04-24 | Aminex Therapeutics, Inc. | Hydrophobic polyamine analogs and methods for their use |
| US6916492B2 (en) | 2001-03-30 | 2005-07-12 | Council Of Scientific & Industrial Research | Natural nontoxic multicolor fluorescent protein dye from a marine invertebrate, compositions containing the said dye and its uses |
| US6689391B2 (en) | 2001-03-30 | 2004-02-10 | Council Of Scientific & Industrial Research | Natural non-polar fluorescent dye from a non-bioluminescent marine invertebrate, compositions containing the said dye and its uses |
| BR0210149A (en) * | 2001-05-09 | 2004-06-29 | Newbiotics Inc | Peptide deformylase activated prodrugs |
| AU2002339843B2 (en) * | 2001-07-23 | 2007-12-06 | The General Hospital Corporation | Conformationally constrained parathyroid hormone (PTH) analogs |
| US6956122B2 (en) * | 2001-09-05 | 2005-10-18 | Council Of Scientific & Industrial Research | Multiple fluorescent natural dye compound from a marine organism |
| AU2002367886B8 (en) | 2001-10-12 | 2008-08-14 | Perkinelmer Las, Inc. | Compilations of nucleic acids and arrays and methods of using them |
| US20050069962A1 (en) * | 2001-10-12 | 2005-03-31 | Archer Robert M | Antibody complexes and methods for immunolabeling |
| US8323903B2 (en) * | 2001-10-12 | 2012-12-04 | Life Technologies Corporation | Antibody complexes and methods for immunolabeling |
| US7439346B2 (en) | 2001-10-12 | 2008-10-21 | Perkinelmer Las Inc. | Nucleic acids arrays and methods of use therefor |
| US6972326B2 (en) | 2001-12-03 | 2005-12-06 | Molecular Probes, Inc. | Labeling of immobilized proteins using dipyrrometheneboron difluoride dyes |
| WO2003066812A2 (en) * | 2002-02-05 | 2003-08-14 | Baylor College Of Medecine | Substituted 4,4-difluoro-4-bora-3a, 4a-diaza-s-indacene compounds for 8-color dna sequencing |
| US6916621B2 (en) | 2002-03-27 | 2005-07-12 | Spectral Genomics, Inc. | Methods for array-based comparitive binding assays |
| US7150967B2 (en) | 2002-04-08 | 2006-12-19 | University Of Medicine & Dentistry Of New Jersey | Fluorescent tags for amino acid and nucleic acid analysis |
| US20050096254A1 (en) * | 2002-04-18 | 2005-05-05 | Sergeeva Maria V. | Peptide deformylase activated prodrugs |
| US7445894B2 (en) | 2002-05-03 | 2008-11-04 | Molecular Probes, Inc. | Compositions and methods for detection and isolation of phosphorylated molecules |
| EP1546118A4 (en) | 2002-05-03 | 2010-08-04 | Molecular Probes Inc | Compositions and methods for detection and isolation of phosphorylated molecules |
| US20040171034A1 (en) * | 2002-05-03 | 2004-09-02 | Brian Agnew | Compositions and methods for detection and isolation of phosphorylated molecules |
| US20040087842A1 (en) * | 2002-05-30 | 2004-05-06 | Lakowicz Joseph R. | Fluorescent probes for saccharrides |
| WO2004003179A1 (en) | 2002-06-27 | 2004-01-08 | The University Of Queensland | Differentiation modulating agents and uses therefor |
| DK1545599T3 (en) | 2002-07-26 | 2015-08-03 | Inst Medical W & E Hall | IMMUNOGENIC PREPARATIONS AND DIAGNOSTIC AND THERAPEUTIC APPLICATIONS THEREOF |
| US6774758B2 (en) * | 2002-09-11 | 2004-08-10 | Kalyan P. Gokhale | Low harmonic rectifier circuit |
| AU2003270619A1 (en) * | 2002-09-12 | 2004-04-30 | Molecular Probes, Inc. | Site-specific labeling of affinity tags in fusion proteins |
| US8791053B2 (en) * | 2002-09-27 | 2014-07-29 | Mpm-Holding Aps | Spatially encoded polymer matrix |
| AU2003290779A1 (en) * | 2002-11-14 | 2004-06-03 | Celmed Oncology (Usa), Inc. | Peptide deformylase activated prodrugs |
| US20040101822A1 (en) | 2002-11-26 | 2004-05-27 | Ulrich Wiesner | Fluorescent silica-based nanoparticles |
| US7795220B2 (en) * | 2003-03-19 | 2010-09-14 | The General Hospital Corporation | Conformationally constrained parathyroid hormones with alpha-helix stabilizers |
| AU2003901325A0 (en) | 2003-03-21 | 2003-04-03 | Stephen Locarnini | Therapeutic, prophylactic and diagnostic agents |
| US20070043512A1 (en) * | 2003-03-26 | 2007-02-22 | Michael Rolph | Therapeutic and prophylactic compositions and uses therefor |
| GB0307559D0 (en) * | 2003-04-02 | 2003-05-07 | Univ Nottingham | Fluorescently tagged ligands |
| US8445702B2 (en) * | 2003-05-05 | 2013-05-21 | Life Technologies Corporation | Zinc binding compounds and their method of use |
| US20050250214A1 (en) * | 2004-05-05 | 2005-11-10 | Gee Kyle R | Zinc binding compounds and their method of use |
| US7642064B2 (en) * | 2003-06-24 | 2010-01-05 | Ventana Medical Systems, Inc. | Enzyme-catalyzed metal deposition for the enhanced detection of analytes of interest |
| EP1653985A4 (en) | 2003-07-17 | 2009-08-05 | Gen Hospital Corp | Conformationally constrained parathyroid hormone (pth) analogs |
| CA2445420A1 (en) | 2003-07-29 | 2005-01-29 | Invitrogen Corporation | Kinase and phosphatase assays |
| US7727752B2 (en) | 2003-07-29 | 2010-06-01 | Life Technologies Corporation | Kinase and phosphatase assays |
| US7619059B2 (en) | 2003-07-29 | 2009-11-17 | Life Technologies Corporation | Bimolecular optical probes |
| US7271265B2 (en) * | 2003-08-11 | 2007-09-18 | Invitrogen Corporation | Cyanine compounds and their application as quenching compounds |
| WO2005018556A2 (en) | 2003-08-12 | 2005-03-03 | 3M Innovative Properties Company | Hydroxylamine substituted imidazo-containing compounds |
| ES2406730T3 (en) | 2003-08-27 | 2013-06-07 | 3M Innovative Properties Company | Imidazoquinolines substituted with aryloxy and arylalkylenoxy |
| AU2004270201A1 (en) | 2003-09-05 | 2005-03-17 | 3M Innovative Properties Company | Treatment for CD5+ B cell lymphoma |
| US7282339B2 (en) * | 2003-09-17 | 2007-10-16 | Invitrogen Corporation | Competitive immunoassay |
| US7125945B2 (en) * | 2003-09-19 | 2006-10-24 | Varian, Inc. | Functionalized polymer for oligonucleotide purification |
| US7544697B2 (en) | 2003-10-03 | 2009-06-09 | Coley Pharmaceutical Group, Inc. | Pyrazolopyridines and analogs thereof |
| CA2540541C (en) | 2003-10-03 | 2012-03-27 | 3M Innovative Properties Company | Alkoxy substituted imidazoquinolines |
| EP1685129A4 (en) | 2003-11-14 | 2008-10-22 | 3M Innovative Properties Co | Oxime substituted imidazo ring compounds |
| WO2005048945A2 (en) | 2003-11-14 | 2005-06-02 | 3M Innovative Properties Company | Hydroxylamine substituted imidazo ring compounds |
| US20050214807A1 (en) * | 2003-11-19 | 2005-09-29 | Iain Johnson | Environmental sensitive fluorogenic compounds and their application for singlet oxygen and protein detection |
| JP4891088B2 (en) | 2003-11-25 | 2012-03-07 | スリーエム イノベイティブ プロパティズ カンパニー | Substituted imidazo ring systems and methods |
| US7776529B2 (en) | 2003-12-05 | 2010-08-17 | Life Technologies Corporation | Methine-substituted cyanine dye compounds |
| EP1720944B1 (en) | 2003-12-05 | 2013-07-17 | Life Technologies Corporation | Cyanine dye compounds |
| US6903214B1 (en) * | 2003-12-11 | 2005-06-07 | Eastman Kodak Company | Synthesis of bis(azinyl)amine-BF2 complex |
| EP1701784A1 (en) * | 2003-12-22 | 2006-09-20 | VersaMatrix A/S | Identification of encoded beads |
| EP1699553A2 (en) * | 2003-12-22 | 2006-09-13 | VersaMatrix A/S | Apparatus and methods for analysis and sorting of particles such as polymer beads |
| JP2007517035A (en) | 2003-12-29 | 2007-06-28 | スリーエム イノベイティブ プロパティズ カンパニー | Arylalkenyl and arylalkynyl substituted imidazoquinolines |
| CA2551399A1 (en) | 2003-12-30 | 2005-07-21 | 3M Innovative Properties Company | Imidazoquinolinyl, imidazopyridinyl, and imidazonaphthyridinyl sulfonamides |
| US20050227309A1 (en) | 2004-01-21 | 2005-10-13 | Corry Schuyler B | Optically-detectable enzyme substrates and their method of use |
| DK1729824T3 (en) | 2004-03-02 | 2009-11-02 | Cellectar Inc | Phospholipid analogues for the treatment of cancers |
| US8540968B2 (en) | 2004-03-02 | 2013-09-24 | Cellectar, Inc. | Phospholipid ether analogs as agents for detecting and locating cancer, and methods thereof |
| CA2559863A1 (en) | 2004-03-24 | 2005-10-13 | 3M Innovative Properties Company | Amide substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines |
| EP2527447A1 (en) | 2004-06-03 | 2012-11-28 | Athlomics Pty Ltd | Agents and methods for diagnosing stress |
| US8017779B2 (en) | 2004-06-15 | 2011-09-13 | 3M Innovative Properties Company | Nitrogen containing heterocyclyl substituted imidazoquinolines and imidazonaphthyridines |
| US8026366B2 (en) | 2004-06-18 | 2011-09-27 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted thiazoloquinolines and thiazolonaphthyridines |
| US7915281B2 (en) | 2004-06-18 | 2011-03-29 | 3M Innovative Properties Company | Isoxazole, dihydroisoxazole, and oxadiazole substituted imidazo ring compounds and method |
| WO2006038923A2 (en) | 2004-06-18 | 2006-04-13 | 3M Innovative Properties Company | Aryl substituted imidazonaphthyridines |
| WO2006007664A1 (en) * | 2004-07-22 | 2006-01-26 | Genomics Research Partners Pty Ltd | Agents and methods for diagnosing osteoarthritis |
| WO2006023231A2 (en) | 2004-07-27 | 2006-03-02 | Molecular Probes, Inc. | Fluorescent metal ion indicators with large stokes shift |
| US7807407B2 (en) * | 2004-07-30 | 2010-10-05 | Ambergen, Inc. | Detection of truncation mutations by mass spectrometry |
| WO2006034081A2 (en) * | 2004-09-17 | 2006-03-30 | Massachusetts Institute Of Technology | Polymers for analyte detection |
| AU2005326708C1 (en) | 2004-12-30 | 2012-08-30 | 3M Innovative Properties Company | Substituted chiral fused [1,2]imidazo[4,5-c] ring compounds |
| AU2005322898B2 (en) | 2004-12-30 | 2011-11-24 | 3M Innovative Properties Company | Chiral fused (1,2)imidazo(4,5-c) ring compounds |
| CA2597092A1 (en) | 2005-02-04 | 2006-08-10 | Coley Pharmaceutical Group, Inc. | Aqueous gel formulations containing immune reponse modifiers |
| WO2006086634A2 (en) | 2005-02-11 | 2006-08-17 | Coley Pharmaceutical Group, Inc. | Oxime and hydroxylamine substituted imidazo[4,5-c] ring compounds and methods |
| FR2882056B1 (en) * | 2005-02-15 | 2007-06-01 | Centre Nat Rech Scient | UNSATURATED BOROCARBON DIPYRROMETHENES-BORE |
| CA2602590A1 (en) | 2005-04-01 | 2006-10-12 | Coley Pharmaceutical Group, Inc. | 1-substituted pyrazolo (3,4-c) ring compounds as modulators of cytokine biosynthesis for the treatment of viral infections and neoplastic diseases |
| EP1869043A2 (en) | 2005-04-01 | 2007-12-26 | Coley Pharmaceutical Group, Inc. | Pyrazolopyridine-1,4-diamines and analogs thereof |
| US8084001B2 (en) * | 2005-05-02 | 2011-12-27 | Cornell Research Foundation, Inc. | Photoluminescent silica-based sensors and methods of use |
| JP5306811B2 (en) | 2005-05-11 | 2013-10-02 | ライフ テクノロジーズ コーポレーション | Fluorescent chemicals with high selectivity to double-stranded DNA and their use |
| US20110045991A1 (en) * | 2005-06-23 | 2011-02-24 | Sadanand Gite | Methods for the Detection of Colorectal Cancer |
| EP1910565A4 (en) | 2005-07-07 | 2009-10-28 | Athlomics Pty Ltd | Polynucleotide marker genes and their expression, for diagnosis of endotoxemia |
| US9216212B2 (en) * | 2005-08-05 | 2015-12-22 | University Of Massachusetts | Virus-like particles as vaccines for paramyxovirus |
| US7951384B2 (en) * | 2005-08-05 | 2011-05-31 | University Of Massachusetts | Virus-like particles as vaccines for paramyxovirus |
| WO2007030521A1 (en) * | 2005-09-06 | 2007-03-15 | Invitrogen Corporation | Control of chemical modification |
| US8093012B2 (en) * | 2005-10-13 | 2012-01-10 | Aureon Laboratories, Inc. | Multiplex in situ immunohistochemical analysis |
| US7521577B2 (en) * | 2006-01-12 | 2009-04-21 | Molecular Probes, Inc. | Heavy metal binding compounds and their method of use |
| US20070196860A1 (en) * | 2006-01-18 | 2007-08-23 | Invitrogen Corporation | Methods for Measuring Real Time Kinase Activity |
| EP1984724A4 (en) | 2006-01-24 | 2015-07-08 | Life Technologies Corp | Device and methods for quantifying analytes |
| EP1987361A4 (en) * | 2006-01-30 | 2009-03-04 | Invitrogen Corp | Compositions and methods for detecting and quantifying toxic substances in disease states |
| ATE551067T1 (en) | 2006-02-03 | 2012-04-15 | Crc For Asthma And Airways Ltd | TUMSTATIN FOR THE TREATMENT OF DISEASES RELATED TO REMODELING OF RESPIRATORY TISSUE |
| US20070249014A1 (en) | 2006-02-10 | 2007-10-25 | Invitrogen Corporation | Labeling and detection of post translationally modified proteins |
| US8114636B2 (en) | 2006-02-10 | 2012-02-14 | Life Technologies Corporation | Labeling and detection of nucleic acids |
| US8562802B1 (en) | 2006-02-13 | 2013-10-22 | Life Technologies Corporation | Transilluminator base and scanner for imaging fluorescent gels, charging devices and portable electrophoresis systems |
| CA2643991A1 (en) | 2006-03-23 | 2007-11-29 | Absorber Ab | Blood group antigens of different types for diagnostic and therapeutic applications |
| EP2029799B1 (en) | 2006-04-20 | 2012-08-22 | Becton, Dickinson and Company | Thermostable proteins and methods of making and using thereof |
| US20090186366A1 (en) * | 2006-06-28 | 2009-07-23 | Ge Healthcare Bio-Sciences Ab | Method of detecting interactions between protein components |
| WO2008008432A2 (en) | 2006-07-12 | 2008-01-17 | Coley Pharmaceutical Group, Inc. | Substituted chiral fused( 1,2) imidazo (4,5-c) ring compounds and methods |
| JP2009545320A (en) * | 2006-08-04 | 2009-12-24 | ザ ジェネラル ホスピタル コーポレイション | Polypeptide derivative of parathyroid hormone (PTH) |
| WO2008042289A2 (en) | 2006-09-29 | 2008-04-10 | Massachusetts Institute Of Technology | Polymer synthetic technique |
| US8802447B2 (en) | 2006-10-05 | 2014-08-12 | Massachusetts Institute Of Technology | Emissive compositions with internal standard and related techniques |
| EP2064290B1 (en) | 2006-10-27 | 2013-10-09 | Life Technologies Corporation | Fluorogenic ph sensitive dyes and their method of use |
| US20090215189A1 (en) | 2006-10-27 | 2009-08-27 | Massachusetts Institute Of Technology | Sensor of species including toxins and chemical warfare agents |
| US8586743B2 (en) * | 2007-01-30 | 2013-11-19 | Life Technologies Corporation | Labeling reagents and methods of their use |
| US8044212B2 (en) * | 2007-04-30 | 2011-10-25 | Hewlett-Packard Development Company, L.P. | Reconfigurable molecules and molecular switches, sensors, and dyes employing the same |
| US8513451B2 (en) * | 2007-05-30 | 2013-08-20 | Hee Chol Kang | Fluorescent phospholipase A2 indicators |
| AU2008282805B2 (en) | 2007-08-01 | 2014-05-01 | Chugai Pharmaceutical Co., Ltd. | Screening methods using G-protein coupled receptors and related compositions |
| DE102007059752A1 (en) | 2007-12-10 | 2009-06-18 | Bayer Schering Pharma Aktiengesellschaft | Functionalized solid polymer nanoparticles containing epothilones |
| US10996218B2 (en) | 2008-03-11 | 2021-05-04 | Ournextbaby Llc | Methods for chemotaxis / redox driven separation of X and Y chromosome bearing sperm and their insemination in gender specific menstrual cycles |
| CN104263816B (en) | 2008-05-16 | 2018-10-19 | 生命技术公司 | Double label method for measuring cell Proliferation |
| FR2940653B1 (en) | 2008-12-29 | 2011-02-04 | Centre Nat Rech Scient | NOVEL FLUORESCENT DIPYRROMETHEN-BOROSUBSTITUTE COMPOUNDS AND THEIR USE FOR DIAGNOSIS |
| EP2400990A2 (en) | 2009-02-26 | 2012-01-04 | OSI Pharmaceuticals, LLC | In situ methods for monitoring the emt status of tumor cells in vivo |
| WO2010144788A2 (en) | 2009-06-12 | 2010-12-16 | Cellectar, Inc. | Ether and alkyl phospholipid compounds for treating cancer and imaging and detection of cancer stem cells |
| PL3223013T3 (en) | 2009-07-02 | 2019-07-31 | Sloan-Kettering Institute For Cancer Research | Fluorescent silica-based nanoparticles |
| GB0915664D0 (en) | 2009-09-08 | 2009-10-07 | Enigma Diagnostics Ltd | Reaction method |
| KR101590220B1 (en) | 2009-12-31 | 2016-01-29 | 벤타나 메디컬 시스템즈, 인코포레이티드 | Methods for producing uniquely specific nucleic acid probes |
| WO2011106495A1 (en) | 2010-02-26 | 2011-09-01 | Ventana Medical Systems, Inc. | Cytogenic analysis of metaphase chromosomes |
| ES2606012T3 (en) | 2010-02-26 | 2017-03-17 | Ventana Medical Systems, Inc. | In situ hybridization with PolyTag probes |
| WO2011109678A1 (en) * | 2010-03-05 | 2011-09-09 | Angstrom Pharmaceuticals, Inc. | Modulation of intracellular signaling |
| US8974651B2 (en) | 2010-04-17 | 2015-03-10 | C.C. Imex | Illuminator for visualization of fluorophores |
| RU2604809C2 (en) | 2010-05-13 | 2016-12-10 | Дзе Дженерал Хоспитал Корпорейшн | Parathyroid hormone analogs and uses thereof |
| JP2011241160A (en) * | 2010-05-17 | 2011-12-01 | Yamamoto Chem Inc | Color conversion material, composition including the material, color conversion optical part using the composition, and light-emitting element using the color conversion optical part |
| US8623324B2 (en) | 2010-07-21 | 2014-01-07 | Aat Bioquest Inc. | Luminescent dyes with a water-soluble intramolecular bridge and their biological conjugates |
| CN103354748B (en) | 2010-08-04 | 2016-09-28 | 麦克法兰博尼特医学健康研究公司 | The hepatitis C virus protein modified |
| WO2012022734A2 (en) | 2010-08-16 | 2012-02-23 | Medimmune Limited | Anti-icam-1 antibodies and methods of use |
| WO2012036691A1 (en) | 2010-09-16 | 2012-03-22 | Urobiologics Llc | Use of female mammal's urine for determination of fetal gender related characteristics |
| EP2686438B1 (en) | 2011-03-14 | 2018-04-18 | Ventana Medical Systems, Inc. | A method of analyzing chromosomal translocations and a system therefor |
| CN103649335B (en) | 2011-05-04 | 2015-11-25 | Htg分子诊断有限公司 | Quantitative nucleic acid enzyme protection measures the improvement of (QNPA) and order-checking (QNPS) |
| WO2013006195A1 (en) | 2011-07-01 | 2013-01-10 | Htg Molecular Diagnostics, Inc. | Methods of detecting gene fusions |
| WO2013057586A1 (en) | 2011-10-19 | 2013-04-25 | Oslo Universitetssykehus Hf | Compositions and methods for producing soluble t - cell receptors |
| CN102516799B (en) * | 2011-12-12 | 2015-06-03 | 安徽师范大学 | Novel long-wavelength boron tripyrrole fluorescent dye and synthesizing method thereof |
| WO2013108126A2 (en) | 2012-01-16 | 2013-07-25 | University Of Oslo | Methyltransferases and uses thereof |
| CN102702774B (en) * | 2012-04-11 | 2013-12-25 | 安徽师范大学 | Near infrared fluoro-boron dipyrrole fluorescent dyes and synthesis method thereof |
| WO2013167387A1 (en) | 2012-05-10 | 2013-11-14 | Ventana Medical Systems, Inc. | Uniquely specific probes for pten, pik3ca, met, top2a, and mdm2 |
| ES2648176T3 (en) | 2012-07-12 | 2017-12-28 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting survival time and response to treatment of a patient suffering from solid cancer with a hallmark of at least 7 genes |
| WO2014048942A1 (en) | 2012-09-25 | 2014-04-03 | Ventana Medical Systems, Inc. | Probes for pten, pik3ca, met, and top2a, and method for using the probes |
| US20150246146A1 (en) | 2012-10-25 | 2015-09-03 | Life Technologies Corporation | Methods and compositions for enzyme-mediated site-specific radiolabeling of glycoproteins |
| CN103194085A (en) * | 2013-04-03 | 2013-07-10 | 天津理工大学 | Novel BODIPY flourescent dye with adjustable emission wavelength and preparation method thereof |
| WO2015036405A1 (en) | 2013-09-10 | 2015-03-19 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for diagnosing and treating basal cell carcinoma |
| WO2015050959A1 (en) | 2013-10-01 | 2015-04-09 | Yale University | Anti-kit antibodies and methods of use thereof |
| ES2837392T3 (en) | 2013-10-02 | 2021-06-30 | Medimmune Llc | Neutralizing anti-influenza A antibodies and their uses |
| US9504405B2 (en) | 2013-10-23 | 2016-11-29 | Verily Life Sciences Llc | Spatial modulation of magnetic particles in vasculature by external magnetic field |
| US10542918B2 (en) | 2013-10-23 | 2020-01-28 | Verily Life Sciences Llc | Modulation of a response signal to distinguish between analyte and background signals |
| US20160265036A1 (en) | 2013-11-05 | 2016-09-15 | Htg Molecular Diagnostics, Inc. | Methods for detecting nucleic acids |
| CA2940118C (en) | 2014-02-24 | 2023-05-23 | Ventana Medical Systems, Inc. | Automated rna detection using labeled 2'-o-methyl rna oligonucleotide probes and signal amplification systems |
| US9835587B2 (en) | 2014-04-01 | 2017-12-05 | C.C. Imex | Electrophoresis running tank assembly |
| EP3148591B1 (en) | 2014-05-29 | 2020-03-11 | Memorial Sloan Kettering Cancer Center | Nanoparticle drug conjugates |
| EP3009147A1 (en) | 2014-10-16 | 2016-04-20 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for treating resistant glioblastoma |
| EP3242690B1 (en) | 2015-01-06 | 2020-09-16 | De Haas, Anthony H. | Near-infrared fluorescent surgical dye markers |
| WO2016113233A1 (en) | 2015-01-12 | 2016-07-21 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the diagnosis of pancreatic cancer |
| US9861710B1 (en) | 2015-01-16 | 2018-01-09 | Verily Life Sciences Llc | Composite particles, methods, and in vivo diagnostic system |
| US10028659B2 (en) | 2015-03-26 | 2018-07-24 | Verily Life Sciences Llc | Aptamer-based sensors, implantable devices and detection system |
| US10736972B2 (en) | 2015-05-29 | 2020-08-11 | Memorial Sloan Kettering Cancer Center | Methods of treatment using ultrasmall nanoparticles to induce cell death of nutrient-deprived cancer cells via ferroptosis |
| WO2017029391A1 (en) | 2015-08-20 | 2017-02-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New method for treating cancer |
| WO2017055319A1 (en) | 2015-09-29 | 2017-04-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for quantifying the population of b cells in a tissue sample |
| WO2017055326A1 (en) | 2015-09-29 | 2017-04-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for quantifying the population of myeloid dendritic cells in a tissue sample |
| WO2017055324A1 (en) | 2015-09-29 | 2017-04-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for quantifying the population of cells of monocytic origin in a tissue sample |
| WO2017055320A1 (en) | 2015-09-29 | 2017-04-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for quantifying the population of cytotoxic lymphocytes in a tissue sample |
| WO2017055325A1 (en) | 2015-09-29 | 2017-04-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for quantifying the population of nk cells in a tissue sample |
| WO2017055321A1 (en) | 2015-09-29 | 2017-04-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for quantifying the population of fibroblasts in a tissue sample |
| WO2017055327A1 (en) | 2015-09-29 | 2017-04-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for quantifying the population of endothelial cells in a tissue sample |
| WO2017055322A1 (en) | 2015-09-29 | 2017-04-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for quantifying the population of neutrophils in a tissue sample |
| WO2017060397A1 (en) | 2015-10-09 | 2017-04-13 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting the survival time of subjects suffering from melanoma metastases |
| WO2017067944A1 (en) | 2015-10-19 | 2017-04-27 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting the survival time of subjects suffering from triple negative breast cancer |
| CA3004504A1 (en) | 2015-11-06 | 2017-05-11 | Ventana Medical Systems, Inc. | Tissue homogenisation for representative diagnostics |
| US10793909B2 (en) | 2015-11-10 | 2020-10-06 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Methods for predicting the survival time of patients with decompensated alcoholic cirrhosis |
| US10760135B2 (en) | 2016-01-13 | 2020-09-01 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Methods for predicting pancreatic cancer treatment response |
| WO2017123647A1 (en) | 2016-01-15 | 2017-07-20 | Quantapore, Inc. | Optically-based nanopore analysis with reduced background |
| AU2017217248B2 (en) | 2016-02-11 | 2023-05-18 | Peter Maccallum Cancer Institute | Method of diagnosis |
| HUE052620T2 (en) | 2016-03-25 | 2021-05-28 | Aminex Therapeutics Inc | Bioavailable polyamines |
| EP3443126B1 (en) | 2016-04-15 | 2023-11-15 | Icahn School of Medicine at Mount Sinai | Tissue profiling using multiplexed immunohistochemical consecutive staining |
| WO2017182834A1 (en) | 2016-04-19 | 2017-10-26 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New method for treating resistant glioblastoma |
| JP7133473B2 (en) | 2016-05-20 | 2022-09-08 | クアンタム-エスアイ インコーポレイテッド | Labeled Nucleotide Compositions and Methods for Determining the Sequence of Nucleic Acids |
| US20190292259A1 (en) | 2016-05-24 | 2019-09-26 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Methods and pharmaceutical compositions for the treatment of non small cell lung cancer (nsclc) that coexists with chronic obstructive pulmonary disease (copd) |
| JP2019520071A (en) | 2016-06-14 | 2019-07-18 | アンスティチュ ナショナル ドゥ ラ サンテ エ ドゥ ラ ルシェルシュ メディカル | Method for predicting treatment response of acute severe colitis |
| WO2018011107A1 (en) | 2016-07-11 | 2018-01-18 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of er-alpha 46 in methods and kits for assessing the status of breast cancer |
| WO2018011166A2 (en) | 2016-07-12 | 2018-01-18 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for quantifying the population of myeloid dendritic cells in a tissue sample |
| JP2019528065A (en) | 2016-08-19 | 2019-10-10 | クアンタポール, インコーポレイテッド | Optically based nanopore sequencing using quenchers |
| WO2018046738A1 (en) | 2016-09-12 | 2018-03-15 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting the survival time of patients suffering from cancer |
| WO2018046736A1 (en) | 2016-09-12 | 2018-03-15 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting the survival time of patients suffering from cancer |
| WO2018054960A1 (en) | 2016-09-21 | 2018-03-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting and treating resistance to chemotherapy in npm-alk(+) alcl |
| WO2018055080A1 (en) | 2016-09-22 | 2018-03-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for reprograming immune environment in a subject in need thereof |
| WO2018122245A1 (en) | 2016-12-28 | 2018-07-05 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods of predicting the survival time of patients suffering from cms3 colorectal cancer |
| WO2018122249A1 (en) | 2016-12-28 | 2018-07-05 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting the survival time of patients suffering from a microsatellite stable colorectal cancer |
| WO2018146239A1 (en) | 2017-02-10 | 2018-08-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Biomarker for outcome in aml patients |
| WO2018162404A1 (en) | 2017-03-06 | 2018-09-13 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Biomarker for outcome in aml patients |
| WO2018172540A1 (en) | 2017-03-24 | 2018-09-27 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method to predict the progression of alzheimer's disease |
| WO2018178171A1 (en) | 2017-03-29 | 2018-10-04 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for assessing pregnancy outcome |
| US20210108253A1 (en) | 2017-03-30 | 2021-04-15 | Life Technologies Corporation | Quantification of ngs dna by adapter sequence |
| WO2018189215A1 (en) | 2017-04-12 | 2018-10-18 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for predicting the survival time of a patient suffering from hepatocellular carcinoma |
| CA3061429A1 (en) | 2017-05-05 | 2018-11-08 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Microtubule-targeting drugs as immune checkpoint inhibitors and methods of screening novel immune checkpoint inhibitors for the treatment of cancers and infectious diseases |
| CA3064253A1 (en) | 2017-05-25 | 2018-11-29 | Memorial Sloan Kettering Cancer Center | Ultrasmall nanoparticles labeled with zirconium-89 and methods thereof |
| US10640545B2 (en) | 2017-07-03 | 2020-05-05 | The Governors Of The Univ Of Alberta Tec Edmonton | Apelin peptides and uses thereof |
| WO2019038219A1 (en) | 2017-08-21 | 2019-02-28 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New prognostic method of pancreatic cancer |
| WO2019043138A1 (en) | 2017-09-01 | 2019-03-07 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for predicting the outcome of a cancer |
| US11579147B2 (en) | 2017-09-25 | 2023-02-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of VNN1 as a biomarker and a therapeutic target in sarcomas |
| WO2019077123A1 (en) | 2017-10-20 | 2019-04-25 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and kits for determining whether a subject has or is at risk of having of an autoimmune myopathy |
| US11691141B2 (en) | 2017-11-13 | 2023-07-04 | Roche Sequencing Solutions, Inc. | Devices for sample analysis using epitachophoresis |
| CN108484650B (en) * | 2018-02-07 | 2020-09-25 | 四川大学 | 3, 5-dibenzoimidazolyl-8-p-methylphenyl fluoroboron fluorescent derivative and preparation method and application thereof |
| CN108191902A (en) * | 2018-02-07 | 2018-06-22 | 四川大学 | Glimmering analog derivative of a kind of 3,5- bisbenzimidazoles base -8- thienyl fluorine boron and its preparation method and application |
| CN108409763B (en) * | 2018-02-07 | 2020-08-11 | 四川大学 | 3, 5-dibenzoimidazolyl-8-p-cyanophenyl fluoroboron fluorescent derivative and preparation method and application thereof |
| WO2019207030A1 (en) | 2018-04-26 | 2019-10-31 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting a response with an immune checkpoint inhibitor in a patient suffering from a lung cancer |
| WO2019229489A1 (en) | 2018-05-31 | 2019-12-05 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of mir-146a-5p and mir-186 as biomarkers of osteoarthritis |
| WO2020089428A1 (en) | 2018-11-02 | 2020-05-07 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New prognostic method of pancreatic cancer |
| WO2020089432A1 (en) | 2018-11-02 | 2020-05-07 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New prognostic method of pancreatic cancer |
| US20220073626A1 (en) | 2019-01-03 | 2022-03-10 | Institut National De La Santé Et De La Recheche Médicale (Inserm) | Methods and pharmaceutical compositions for enhancing cd8+ t cell-dependent immune responses in subjects suffering from cancer |
| WO2020148349A1 (en) | 2019-01-16 | 2020-07-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Variants of erythroferrone and their use |
| EP3924520A1 (en) | 2019-02-13 | 2021-12-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and compositions for selecting a cancer treatment in a subject suffering from cancer |
| WO2020182932A1 (en) | 2019-03-13 | 2020-09-17 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New gene signatures for predicting survival time in patients suffering from renal cell carcinoma |
| WO2020193740A1 (en) | 2019-03-28 | 2020-10-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New strategy for treating pancreatic cancer |
| JP2022527972A (en) | 2019-04-02 | 2022-06-07 | アンスティチュ ナショナル ドゥ ラ サンテ エ ドゥ ラ ルシェルシュ メディカル | How to predict and prevent cancer in patients with premalignant lesions |
| US20220244245A1 (en) | 2019-04-15 | 2022-08-04 | INSERM (Institut National de la Santé et de la Recherche Médicale) | A method of profiling the energetic metabolism of a population of cells |
| US20220211741A1 (en) | 2019-04-18 | 2022-07-07 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment and prognosis of cancer |
| WO2020216832A1 (en) | 2019-04-24 | 2020-10-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for predicting the response of antipsychotic drugs |
| WO2020229521A1 (en) | 2019-05-14 | 2020-11-19 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for inhibiting or reducing bacterial biofilms on a surface |
| US20220325268A1 (en) | 2019-05-14 | 2022-10-13 | Roche Sequencing Solutions, Inc | Devices and methods for sample analysis |
| WO2021001539A1 (en) | 2019-07-04 | 2021-01-07 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New strategy to detect and treat eosinophilic fasciitis |
| EP4025712A1 (en) | 2019-09-05 | 2022-07-13 | Institut National de la Santé et de la Recherche Médicale (INSERM) | Method of treatment and pronostic of acute myeloid leukemia |
| WO2021063968A1 (en) | 2019-09-30 | 2021-04-08 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and composition for diagnosing chronic obstructive pulmonary disease |
| WO2021074391A1 (en) | 2019-10-17 | 2021-04-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for diagnosing nasal intestinal type adenocarcinomas |
| US20230113705A1 (en) | 2020-02-28 | 2023-04-13 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for diagnosing, prognosing and managing treatment of breast cancer |
| US20230086718A1 (en) | 2020-03-20 | 2023-03-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for predicting the survival time of a patient suffering from a cancer |
| WO2021250106A1 (en) | 2020-06-10 | 2021-12-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for treating and prognosing cancer like glioblastoma |
| EP4168006A1 (en) | 2020-06-18 | 2023-04-26 | Institut National de la Santé et de la Recherche Médicale (INSERM) | New strategy for treating pancreatic cancer |
| KR20230034333A (en) | 2020-07-02 | 2023-03-09 | 라이프 테크놀로지스 코포레이션 | Trinucleotide Cap Analogues, Preparation and Uses Thereof |
| WO2022018163A1 (en) | 2020-07-22 | 2022-01-27 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for predicting survival time in patients suffering from cancer |
| US20230304932A1 (en) | 2020-07-23 | 2023-09-28 | Life Technologies Corporation | Compositions, systems and methods for biological analysis involving energy transfer dye conjugates and analytes comprising the same |
| KR20230056683A (en) | 2020-07-23 | 2023-04-27 | 라이프 테크놀로지스 코포레이션 | Energy Transfer Dye Conjugates for Use in Biological Assays |
| WO2022064049A1 (en) | 2020-09-28 | 2022-03-31 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for diagnosing brucella infection |
| WO2022072586A1 (en) | 2020-09-30 | 2022-04-07 | Aminex Therapeutics, Inc. | Combination drug substance of polyamine transport inhibitor and dfmo |
| US20230383350A1 (en) | 2020-10-20 | 2023-11-30 | Institut National De La Sante Et De La Recherche Medicale | Method for predicting the response to tnf inhibitors |
| CN116917502A (en) | 2020-11-06 | 2023-10-20 | Inserm(法国国家健康医学研究院) | Methods of diagnosing and treating polycystic ovary syndrome (PCOS) |
| WO2022135753A1 (en) | 2020-12-21 | 2022-06-30 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for prognosis the humoral response of a subject prior to vaccination |
| WO2022136252A1 (en) | 2020-12-21 | 2022-06-30 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for prognosis the humoral response of a subject prior to vaccination |
| WO2022152698A1 (en) | 2021-01-12 | 2022-07-21 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of npdk-d to evaluate cancer prognosis |
| WO2022171611A1 (en) | 2021-02-09 | 2022-08-18 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New method to pronostic lung cancer |
| EP4308934A1 (en) | 2021-03-17 | 2024-01-24 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for diagnosing pancreatic cancer |
| WO2022207566A1 (en) | 2021-03-29 | 2022-10-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New method to evaluate pancreatic cancer prognosis |
| EP4326903A1 (en) | 2021-04-23 | 2024-02-28 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Methods and compositions for treating cell senescence accumulation related disease |
| WO2022263451A1 (en) | 2021-06-15 | 2022-12-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Peptides derived from the spike protein of sars-cov-2 and uses thereof for diagnosis and vaccine purposes |
| WO2023280790A1 (en) | 2021-07-05 | 2023-01-12 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Gene signatures for predicting survival time in patients suffering from renal cell carcinoma |
| CN113527347B (en) * | 2021-07-15 | 2023-12-08 | 淮阴工学院 | Near infrared fluorescence labeling fatty acid and preparation method thereof |
| CN117897454A (en) | 2021-07-21 | 2024-04-16 | 生命技术公司 | Dibenzoxanthene quencher, use and preparation method |
| WO2023089159A1 (en) | 2021-11-22 | 2023-05-25 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New strategy targeting stroma/tumor cell crosstalk to treat a cancer |
| WO2023144303A1 (en) | 2022-01-31 | 2023-08-03 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Cd38 as a biomarker and biotarget in t-cell lymphomas |
| WO2023152133A1 (en) | 2022-02-08 | 2023-08-17 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for diagnosing colorectal cancer |
| WO2024061930A1 (en) | 2022-09-22 | 2024-03-28 | Institut National de la Santé et de la Recherche Médicale | New method to treat and diagnose peripheral t-cell lymphoma (ptcl) |
| WO2024115935A1 (en) | 2022-11-29 | 2024-06-06 | Inserm | Methods for the treatment of b-cell lymphoma using cd39 inhibitors |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4774339A (en) * | 1987-08-10 | 1988-09-27 | Molecular Probes, Inc. | Chemically reactive dipyrrometheneboron difluoride dyes |
| EP0361936A2 (en) * | 1988-09-29 | 1990-04-04 | Joseph H. Boyer | Lasing compositions and methods for using the same |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0329184A3 (en) * | 1988-02-19 | 1990-05-23 | Neorx Corporation | Antimers and antimeric conjugation |
| US5248782A (en) * | 1990-12-18 | 1993-09-28 | Molecular Probes, Inc. | Long wavelength heteroaryl-substituted dipyrrometheneboron difluoride dyes |
| US5187288A (en) * | 1991-05-22 | 1993-02-16 | Molecular Probes, Inc. | Ethenyl-substituted dipyrrometheneboron difluoride dyes and their synthesis |
-
1991
- 1991-11-01 US US07/786,767 patent/US5274113A/en not_active Expired - Lifetime
-
1992
- 1992-09-30 CA CA002122627A patent/CA2122627C/en not_active Expired - Fee Related
- 1992-09-30 EP EP92922004A patent/EP0612336B1/en not_active Expired - Lifetime
- 1992-09-30 WO PCT/US1992/008350 patent/WO1993009185A1/en active IP Right Grant
- 1992-09-30 DE DE69223985T patent/DE69223985T2/en not_active Expired - Lifetime
- 1992-09-30 AT AT92922004T patent/ATE161871T1/en active
-
1993
- 1993-04-08 US US08/045,758 patent/US5451663A/en not_active Expired - Lifetime
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4774339A (en) * | 1987-08-10 | 1988-09-27 | Molecular Probes, Inc. | Chemically reactive dipyrrometheneboron difluoride dyes |
| EP0361936A2 (en) * | 1988-09-29 | 1990-04-04 | Joseph H. Boyer | Lasing compositions and methods for using the same |
Non-Patent Citations (7)
| Title |
|---|
| BRAIN RESEARCH vol. 547, no. 2, 3 May 1991, UNITED STATES pages 199 - 207 A. BARTON; H.C. KANG; M.S. RINAUDO; F.J. MONSMA; R.M. STEWART-FRAM; J.A. MACINKO; R.P. HAUGLAND; M.A. ARIANO; D.R. SIBLEY 'Multiple fluorescent ligands for dopamine receptors. I. Pharmacological characterization and receptor selectivity' * |
| BULLOCK ET AL., J. CHEM. SOC., 1958, pages 1420 |
| HETEROCYCLES, vol. 26, 1987, pages 3141 |
| JOURNAL OF NEUROCHEMISTRY vol. 52, no. 5, May 1989, NEW YORK pages 1641 - 1644 F.J. MONSMA; A.C. BARTON; H.C. KANG; D.L. BRASSARD; R.P. HAUGLAND; D.R. SIBLEY 'Characterization of Novel Fluorescent Ligands with High Affinity for D1 and D2 Dopaminergic Receptors' * |
| ORG. SYNTH. COLL., vol. IV, pages 831 |
| RAPPAPORT ET AL., J. AM. CHEM. SOC., vol. 84, 1962, pages 2178 |
| THE JOURNAL OF CELL BIOLOGY vol. 113, no. 6, 6 June 1991, UNITED STATES pages 1267 - 1279 R.E. PAGANO; O.C. MARTIN; H.C. KANG; R.P. HAUGLAND 'A Novel Fluorescent Ceramid Analogue for Studying Membrane Traffic in Animal Cells: Accumulation at the Golgi Apparatus Results in Altered Spectral Properties of the Sphingolipid Precursor' * |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0684950A4 (en) * | 1993-02-19 | 1997-10-15 | Bo Dekk Ventures Ltd | Chemical compounds useful in photodynamic therapy and production of laser light. |
| EP0684950A1 (en) * | 1993-02-19 | 1995-12-06 | Bo-Dekk Ventures, Ltd. | Chemical compounds useful in photodynamic therapy and production of laser light |
| US6277984B1 (en) | 1995-06-07 | 2001-08-21 | Carnegie Mellon University | Monomethine cyanines rigidized by a two-carbon chain |
| EP0747448A2 (en) * | 1995-06-07 | 1996-12-11 | Carnegie-Mellon University | Rigidized monomethine cyanine dyes |
| EP0747448A3 (en) * | 1995-06-07 | 1997-03-05 | Univ Carnegie Mellon | Rigidized monomethine cyanine dyes |
| US5852191A (en) * | 1995-06-07 | 1998-12-22 | Carnegie Mellon University | Rigidized monomethine cyanines |
| US6207464B1 (en) | 1995-06-07 | 2001-03-27 | Carnegie Mellon University | Rigidized monomethine cyanines |
| EP0822544A1 (en) * | 1996-07-29 | 1998-02-04 | Mitsui Toatsu Chemicals, Incorporated | Optical recording medium |
| US5948593A (en) * | 1996-07-29 | 1999-09-07 | Mitsui Chemicals, Inc. | Optical recording medium |
| US6060606A (en) * | 1996-07-29 | 2000-05-09 | Mitsui Chemicals, Inc. | Dipyrromethene metal chelate compounds |
| WO1998013524A1 (en) * | 1996-09-27 | 1998-04-02 | Laboratory Of Molecular Biophotonics | Probes for detecting polynucleotides and detection method |
| US6284462B1 (en) | 1996-09-27 | 2001-09-04 | Laboratory Of Molecular Biophotonics | Probes and methods for polynucleotide detection |
| US6465175B2 (en) | 1997-09-04 | 2002-10-15 | Bayer Corporation | Oligonucleotide probes bearing quenchable fluorescent labels, and methods of use thereof |
| WO1999011813A2 (en) * | 1997-09-04 | 1999-03-11 | Bayer Corporation | Oligonucleotide probes bearing quenchable fluorescent labels, and methods of use thereof |
| WO1999011813A3 (en) * | 1997-09-04 | 1999-05-06 | Chiron Diagnostics Corp | Oligonucleotide probes bearing quenchable fluorescent labels, and methods of use thereof |
| WO2003004569A1 (en) * | 2001-07-02 | 2003-01-16 | Arctic Diagnostics Oy | Method for increasing hydrophilicity of fluorescent label compounds |
| WO2003005030A1 (en) * | 2001-07-02 | 2003-01-16 | Arctic Diagnostics Oy | Two-photon absorbing dipyrrometheneboron difluoride dyes and their applications |
| US7198958B2 (en) | 2001-07-02 | 2007-04-03 | Arctic Diagnostics Oy | Method for increasing hydrophilicity of fluorescent label compounds |
| US7763439B2 (en) | 2001-07-02 | 2010-07-27 | Arctic Diagnostics Oy | Two-photon absorbing dipyrrometheneboron difluoride dyes and their applications |
| US8067396B2 (en) | 2002-01-11 | 2011-11-29 | Sankyo Company, Limited | Amino alcohol compounds or phosphonic acid derivatives thereof |
| US8101650B2 (en) | 2002-01-11 | 2012-01-24 | Daiichi Sankyo Company, Limited | Method for treating a immunology-related disease |
| US8420638B2 (en) | 2003-05-30 | 2013-04-16 | Gemin X Pharmaceuticals Canada Inc. | Triheterocyclic compounds and compositions thereof |
| US7709477B2 (en) | 2003-05-30 | 2010-05-04 | Gemin X Pharmaceuticals Canada Inc. | Methods for treating cancer |
| US7425553B2 (en) | 2003-05-30 | 2008-09-16 | Gemin X Pharmaceuticals Canada Inc. | Triheterocyclic compounds, compositions, and methods for treating cancer or viral diseases |
| US7910617B2 (en) | 2004-02-24 | 2011-03-22 | Sankyo Company, Limited | Method for suppressing the number of peripheral blood lymphocytes using an amino alcohol compound |
| WO2012118444A1 (en) * | 2011-03-01 | 2012-09-07 | Agency For Science, Technology And Research | Novel compounds with photoluminescnce properties and applications thereof |
| CN103502253A (en) * | 2011-03-01 | 2014-01-08 | 新加坡科技研究局 | Novel compounds with photoluminescnce properties and applications thereof |
| WO2017139689A1 (en) * | 2016-02-12 | 2017-08-17 | Oregon Health & Science University | Derivatives of bodipy |
| US9751897B1 (en) | 2016-02-12 | 2017-09-05 | Oregon Health & Science University | Derivatives of Bodipy |
Also Published As
| Publication number | Publication date |
|---|---|
| US5274113A (en) | 1993-12-28 |
| CA2122627A1 (en) | 1993-05-13 |
| EP0612336B1 (en) | 1998-01-07 |
| ATE161871T1 (en) | 1998-01-15 |
| DE69223985D1 (en) | 1998-02-12 |
| EP0612336A1 (en) | 1994-08-31 |
| US5451663A (en) | 1995-09-19 |
| DE69223985T2 (en) | 1998-07-16 |
| CA2122627C (en) | 2002-06-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5274113A (en) | Long wavelength chemically reactive dipyrrometheneboron difluoride dyes and conjugates | |
| US4774339A (en) | Chemically reactive dipyrrometheneboron difluoride dyes | |
| US7408062B2 (en) | Rigidized trimethine cyanine dyes | |
| US5433896A (en) | Dibenzopyrrometheneboron difluoride dyes | |
| US5132432A (en) | Chemically reactive pyrenyloxy sulfonic acid dyes | |
| US6686145B1 (en) | Rigidized trimethine cyanine dyes | |
| JP5008044B2 (en) | Energy transfer assays and reagents | |
| AU717569B2 (en) | Fluorinated xanthene derivatives | |
| US5442045A (en) | Biological conjugates of fluorescent rhodol dyes | |
| US5808044A (en) | Indocarbocyanine and benzindocarbocyanine phosphoramidites | |
| WO1998049176A1 (en) | Glycoconjugated fluorescent labeling reagents | |
| GB2319250A (en) | Derivatives of 6,8-difluoro-7-hydroxycoumarin | |
| KR20240031282A (en) | Compound based cyanine, labeling dye, kit and contrast medium composition for biomolecule comprising the same | |
| EP2360211A1 (en) | Fluorescently-labelled target components | |
| EP0747448B1 (en) | Rigidized monomethine cyanine dyes | |
| US6828159B1 (en) | Carbopyronine fluorescent dyes | |
| JP4098839B2 (en) | Indocyanine compounds | |
| US7351829B2 (en) | Compounds on the basis of 2- and 4-chromenylidene-merocyanines respectively, and their use | |
| WO2002068537A2 (en) | Fluorescent dye | |
| US20120016128A1 (en) | Rhodamine lactone phosphoramidites and polymers | |
| EP3865544A1 (en) | Fluorescent rhodamine dyes with enhanced cell permeability | |
| Gu et al. | Synthesis and spectral properties of 6′-triazolyl-dihydroxanthene-hemicyanine fused near-infrared dyes | |
| JP7479667B2 (en) | Compound having a sulfonylaniline skeleton or a salt thereof, or organic fluorescent material having the same | |
| KR102682109B1 (en) | Novel compounds and their applications | |
| KR102260233B1 (en) | Compound based merocyanine, labeling dye, kit and contrast medium composition for biomolecule comprising the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CA JP |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2122627 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1992922004 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1992922004 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1992922004 Country of ref document: EP |